

EN LAS
ENFERMEDADES PULMONARES
INTERSTICIALES DIFUSAS
Dra. Ana Boldova Loscertales
Dr. José Antonio Ros Lucas
MANUAL DE CONSULTA EN LAS ENFERMEDADES
PULMONARES
INTERSTICIALES DIFUSAS
Dra. Ana Boldova Loscertales
Dr. José Antonio Ros Lucas


Patrocinado por:

Boehringer Ingelheim financia la difusión, la edición y la maquetación del libro, pero en ningún caso participa en la creación del contenido, del que son responsables los autores y el GEEPID de SEPAR.
ISBN: 978-84-127307-4-6
DEPÓSITO LEGAL: B 12017-2024
Editado y coordinado por RESPIRA-FUNDACIÓN ESPAÑOLA DEL PULMÓN-SEPAR. Reservados todos los derechos. Ninguna parte de esta publicación puede ser reproducida ni transmitida en ninguna forma o medio alguno, electrónico o mecánico, incluyendo las fotocopias, grabaciones o cualquier sistema de recuperación de almacenaje de información, sin el permiso escrito del titular del copyright. 2024
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES
INTERSTICIALES DIFUSAS
COORDINADORES DEL MANUAL
Dra. Ana Boldova Loscertales
Servicio de Neumología. Hospital Royo Villanova. Zaragoza.
Dr. José Antonio Ros Lucas
Servicio de Neumología. Hospital Clínico Universitario Virgen de la Arrixaca. Murcia.
COORDINADORES DE LOS CAPÍTULOS
Dr. Orlando Acosta Fernández
Servicio de Neumología. Hospital Universitario de Canarias. La Laguna. Santa Cruz de Tenerife.
D. Xavier Alsina Restoy
Enfermero de práctica avanzada en enfermedades intersticiales y funcionalismo pulmonar. Hospital Clínic de Barcelona.
Dra. Eva Cabrera Cesa
Servicio de Neumología. Hospital Virgen de la Victoria. Málaga.
Dr. Esteban Cano Jiménez
Servicio de Neumología. Hospital Lucus Augusti. Lugo.
Dr. Álvaro Casanova Espinosa
Servicio de Neumología. Hospital Universitario del Henares. Coslada. Madrid.
Dr. Diego M. Castillo Villegas
Servicio de Neumología. Hospital de la Santa Creu i Sant Pau. Barcelona.
Dr. Sergio Curi Chércoles
Servicio de Neumología. Hospital Universitario de Navarra. Pamplona.
Dra. Raquel García Sevila
Servicio de Neumología. Hospital General Universitario Dr. Balmis. Alicante.
Dr. Raúl Godoy Mayoral
Servicio de Neumología. Complejo Hospitalario Universitario de Albacete.
Dr. David Iturbe Fernández
Servicio de Neumología. Hospital Universitario Marqués de Valdecilla. Santander.
Dra. Rosalía Laporta Hernández
Servicio de Neumología. Hospital Universitario Puerta de Hierro. Majadahonda. Madrid.
Dra. Belén López-Muñiz Ballesteros
Servicio de Neumología. Hospital Infanta Leonor. Madrid.
Dra. María Molina Molina
Unidad Funcional de Intersticio Pulmonar. Servicio de Neumología. Hospital Universitario de Bellvitge. Hospitalet de Llobregat. Barcelona.
Dra. María Asunción Nieto Barbero
Servicio de Neumología. Hospital Clínico San Carlos. Madrid.
Dra. Belén Núñez Sánchez
Servicio de Neumología. Hospital Universitario Son Espases. Palma de Mallorca.
Dr. Íñigo Ojanguren Arráez
Servicio de Neumología. Hospital Universitario Vall d´Hebron. Barcelona.
Dra. Teresa Peña Miguel
Servicio de Neumología. Hospital Universitario de Burgos.
Dra. Raquel Pérez Rojo
Servicio de Neumología. Hospital Universitario 12 de Octubre. Madrid.
Dra. María Teresa Rio Ramírez
Servicio de Neumología. Hospital Universitario de Getafe. Madrid.
Dra. María Jesús Rodríguez Nieto
Servicio de Neumología. Hospital Universitario Fundación Jiménez Díaz. Madrid.
Dr. José Antonio Rodríguez Portal
Servicio de Neumología. Hospital Universitario Virgen del Rocío. Sevilla.
Dra. Ana Dolores Romero Ortiz
Servicio de Neumología. Hospital Virgen de las Nieves. Granada.
Dra. Miren Begoñe Salinas Lasa
Servicio de Neumología. Hospital de Galdakao-Usansolo. Galdakao.
Dr. Jacobo Sellares Torres
Servicio de Neumología. Hospital Clínic de Barcelona.
Dra. Claudia Valenzuela
Servicio de Neumología. Hospital Universitario de La Princesa. Madrid.
Dra. Ana Villar Gómez
Servicio de Neumología. Hospital Universitario Vall d´Hebron. Barcelona.
AUTORES DE LOS CAPÍTULOS
Dr. Mario Nicolás Albani Pérez
Neumología. Hospital Obispo Polanco. Teruel.
Dra. Nuria Albacar Ingla
Servicio de Neumología. Hospital Clínic de Barcelona.
Dr. Miguel Arias Guillén
Servicio de Neumología. Hospital Universitario Central de Asturias. Oviedo.
Dña. María José Beceiro Padreño
Enfermera unidad de EPID. Hospital Universitario de La Princesa. Madrid.
Dr. Jaume Bordás Martínez
Servicio de Neumología. Hospital General de Granollers. Barcelona.
Dra. Olaia Bronte Moreno
Servicio de Neumología. Hospital de Galdakao. Bilbao.
Dra. Elena Cabezas Pastor
Servicio de Neumología. Hospital Universitario Fundación Jiménez Díaz. Madrid.
Dr. Oswaldo Antonio Caguana Vélez
Servicio de Neumología. Hospital del Mar. Barcelona.
Dra. Cristina Caupena Auledas
Servicio de Neumología. Parc Sanitari Sant Joan de Déu de Sant Boi de Llobregat. Barcelona.
Dra. María Churruca Arrospide
Servicio de Neumología. Hospital Universitario Puerta de Hierro. Madrid.
Dra. Dolores del Puerto García
Servicio de Neumología. Hospital San Pedro. Logroño.
Dr. José Antonio Delgado Torralbo
Servicio Neumología. Hospital Virgen de la Macarena. Sevilla.
Dr. Diego Durán Barata
Servicio de Neumología. Hospital Universitario de Getafe. Madrid.
Dr. David Espejo Castellanos
Servicio de Neumología. Hospital Universitario Vall d´Hebron. Barcelona.
Dra. Koral Fernández de Roitegui Pérez
Servicio de Neumología. Hospital Universitario de Araba. Vitoria.
Dr. Diego Ferrer Pargada
Servicio de Neumología. Hospital Universitario de Marqués de Valdecilla. Santander.
Dr. Joel Rodrigo Francesqui Candela
Servicio de Neumología. Hospital de la Santa Creu i Sant Pau. Barcelona.
Dra. Yojana García Carrascal
Servicio de Neumología. Hospital Universitario Miguel Servet. Zaragoza.
Dra. Beatriz García Pulido
Neumología. Hospital Grande Covián, Arriondas. Asturias.
Dr. Ignacio Gayá García-Manso
Servicio de Neumología. Hospital General Universitario Dr. Balmis. Alicante.
Dr. Whalter Iván Girón Matute
Servicio de Neumología. Hospital General Universitario Gregorio Marañón. Madrid.
Dra. Marta Hernández Argudo
Servicio de Neumología. Hospital Universitario de Bellvitge. L’Hospitalet de Llobregat. Barcelona.
Dra. Fernanda Hernández González
Servicio de Neumología. Hospital Clínic de Barcelona.
Dra. Sheila Izquierdo Cuervo
Servicio de Neumología. Hospital Universitario Marqués de Valdecilla. Santander.
Dra. Ana Jaureguizar Oriol
Servicio de Neumología. Hospital Ramón y Cajal. Madrid.
Dr. David Eugenio Jerves Donoso
Servicio de Neumología. Hospital Santa Bárbara. Soria.
Dra. Aylaf Latif Essa
Servicio de Neumología. Hospital General Universitario Gregorio Marañón. Madrid.
Dr. Francisco León Román
Servicio de Neumología. Hospital Recoletas Campo Grande. Valladolid.
Dra. Ana Belén Llanos González
Servicio de Neumología. Hospital Universitario de Canarias. La Laguna. Santa Cruz de Tenerife.
Dra. Ángeles López Bauza
Servicio de Neumología. Hospital Universitario Virgen del Rocío. Sevilla.
Dra. Cecilia López Ramírez
Servicio de Neumología. Hospital Universitario Virgen del Rocío. Sevilla.
Dr. Juan Margallo Iribarnegaray
Servicio de Neumología. Hospital Universitario 12 de Octubre. Madrid.
Dr. Pablo Mariscal Aguilar
Servicio de Neumología. Hospital Universitario La Paz. Madrid.
Dra. Andrea Marín González
Neumología. Complejo Hospital Universitario de Ourense.
Dra. Elisa Martínez Besteiro
Servicio de Neumología. Hospital Universitario de La Princesa. Madrid.
Dra. Cristina Matesanz López
Servicio de Neumología. Hospital Clínico San Carlos. Madrid.
Dra. Paloma Millán Billi
Servicio de Neumología. Hospital Universitario Germans Trias i Pujol. Badalona. Barcelona.
Dra. Celia Montaño Montaño
Servicio de Neumología. Hospital Universitario de Bellvitge. Hospitalet de Llobregat. Barcelona.
Dra. Alba Mulet Arabí
Servicio de Neumología. Hospital Clínico Universitario de Valencia.
Dra. Pilar Muñoz Zara
Servicio de Neumología. Hospital Universitario de Jerez de la Frontera. Cádiz.
Dra. María Belén Noboa Sevilla
Servicio de Neumología. Hospital Metropolitano. Quito. Ecuador.
Dra. Laura Nuñez García
Servicio de Neumología. Hospital Universitario Fundación Jiménez Díaz. Madrid.
Dra. Marta Orta Caamaño
Servicio de Neumología. Hospital General Universitario de Ciudad Real. Ciudad Real.
Dr. Joy Selene Osorio Chávez
Servicio de Neumología. Hospital Universitario Marqués de Valdecilla. Santander.
Dra. Eli Nancy Pérez Rodas
Servicio de Neumología. Hospital Municipal de Badalona y Hopsital Clínic de Barcelona.
Dra. María Florencia Pilia
Servicio de Neumología. Hospital Universitario Vall d´Hebron. Barcelona.
Dr. Miguel Henrique Reyes Cotes
Neumología. Hospital del Noroeste. Caravaca de la Cruz. Murcia.
Dr. Juan Rigual Bobillo
Servicio de Neumología. Hospital Ramón y Cajal. Madrid.
Dra. Inés Ruiz Gemar
Servicio de Neumología. Hospital Universitario Parc Taulí. Sabadell. Barcelona.
Dra. Lirios Sacristan Bou
Servicio de Neumología. Hospital General Universitario de Ciudad Real.
Dr. Martín Nikolai Sánchez Mitacc
Servicio de Neumología. Hospital Daniel Alcides Carrion. Perú.
Dra. Candela Serra
Servicio de Neumología. Consorci Hospitalari de Vic. Barcelona.
Dra. Elena Solana Martínez
Servicio de Neumología. Hospital Clínico Universitario Virgen de la Arrixaca. Murcia.
D. Rodrigo Torres Castro
Fisioterapeuta Respiratorio. Hospital Clínic de Barcelona.
ABREVIATURAS
1STS: Prueba de sedestación y bipedestación durante 1 minuto.
ABPA: Aspergilosis Broncopulmonar Alérgica.
ACSM: American College of Sports Medicine.
ADE: Amplitud de Distribución Eritrocitario.
AFOP: Neumonía Fibrinosa y Organizada Aguda.
ANA: Anticuerpos Antinucleares.
ANCA: Anticuerpos Anticitoplasma de Neutrófilos.
Anti-CCP: Anti-péptido citrulinado.
Anti-MDA-5: Anti-melanoma differentiation-associated gene 5.
Anti-Ro/SS-A: Anti-Sjögren’s syndrome related antigen A.
Anti-tRNA sintetasa: Anti sintetasa del ácido ribonucleico de transferencia.
AOS: Apnea Obstructiva del Sueño.
AR: Artritis Reumatoide.
ATS: American Thoracic Society.
AZA: Azatioprina.
BCG: Bacilo de Calmette-Guérin.
BO: Bronquiolitis Obliterante.
BPQ: Biopsia Pulmonar Quirúrgica.
BPTBc: Biopsia Pulmonar Transbronquial Convencional.
BR-EPID: Bronquioitis Respiratoria asociada a EPID.
BTB: Biopsia Transbronquial.
CCD: Cateterismo Cardiaco Derecho.
CMD: Comité Multidisciplinar.
CMV: Citomegalovirus.
CLAD: Disfunción Crónica del Aloinjerto Pulmonar.
CPAP: Presión Positiva Continua.
CPFE: Combinación Fibrosis Pulmonar Enfisema.
CPI: Índice Fisiológico Compuesto.
CPK: Createninkinasa.
CPT: Capacidad Pulmonar Total.
CT90%: Tiempo con saturación de oxihemoglobina por debajo del 90%.
CTB: Biopsia Transbronquial con Criosonda.
CVRS: Calidad de Vida Relacionada con la Salud.
CYC: Ciclofodfamida.
DAD: Daño Alveolar Difuso.
DI: Dosis de Inducción.
DLco: Difusión de Monóxido de Carbono.
DM: Dermatomiositis.
EAS: Enfermedad Autoinmune Sistémica.
EA: Exacerbación Aguda.
ECA: Ensayos Clínicos Aleatorizados.
ECG: Electrocardiograma.
ECMO: Membrana extracorpórea.
EGFR: Receptor del Factor de Crecimiento Epidérmico.
EICH: Enfermedad Injerto Contra Huésped.
EP: Enfermedad Profesional.
EPID: Enfermedad Pulmonar Intersticial Difusa.
EPIRP: Enfermedades Pulmonares Intersticiales Rápidamente Progresivas.
EPOC: Enfermedad Pulmonar Obstructiva Crónica.
EPQD: Enfermedad Pulmonar Quística Difusa.
EREA: Enfermedad Respiratoria Exacerbada por Aspirina.
ERS: European Respiratori Society.
ES: Esclerosis Sistémica.
ET: Esclerosis Tuberosa.
ETC: Enfermedad del Tejido Conectivo.
ETV: Enfermedad Tromboembólica Venosa.
EUVAS: Grupo Europeo de Estudio de las Vasculitis.
e.v.: Endovenoso.
EVALI: E-cigarette, or Vaping, product use Associated Lung Injury.
FAME: Fármacos Antirreumáticos Moduladores de Enfermedad.
FBC: Fibrobroncoscopia.
FCFV: Filamentos Continuos de Fibra de Vidrio.
FENO: Óxido Nitríco Exhalado.
F-EPID: EPID Fibrosante.
FEV1: Volumen espirado en el primer segundo.
FIAT: Fibrosis Intersticial Asociada a Tabaco.
FGF: Factor de crecimiento fibroblástico.
FLCN: Gen de la foliculina.
FMA: Fibras Minerales Artificiales.
FMP: Fibrosis Masiva Progresiva.
FPI: Fibrosis Pulmonar Idiopática.
FPP: Fibrosis Pulmonar Progresiva.
FQ: Fibrosis Quística.
FR: Factor Reumatoide.
FRC: Capacidad Residual Funcional.
FVC: Capacidad Vital Forzada.
GCI: Glucocorticoides inhalados.
GNAF: Gafas Nasales de Alto Flujo.
GEMA: Grupo Español de Manejo del Asma.
GEPA: Granulomatosis Eosinofílica con Poliangeítis.
GINA: Iniciativa Global para el Asma.
GM-CSF: Factor estimulante de colonias de granulocitos y macrófagos.
GPA: Granulomatosis con poliangeítis.
HAD: Hemorragia Alveolar Difusa.
HAP: Hipertensión Arterial Pulmonar.
HPCL: Histiocitosis Pulmonar de Células de Langerhans.
HTP: Hipertensión Pulmonar.
ICIs: Inhibidores de puntos de control inmunitario.
IECA: Inhibidores de la Enzima de Conversión de la Angiotensina.
IgA: Inmunoglobulina A.
IgE: Inmunoglobilina E.
IgG: Inmunoglobulina G.
IL: Interleuquina.
ILA: Interstitial Lung Anormalities.
IMC: Índice de Masa Corporal.
IPAF (Inglés): Interstitial Pneumonia with Autoinmune Features.
ISHLT: International Society for Heart and Lung Trasplantation.
Kco: Constante de Krogh.
KL-6: Glicoproteina Krebs von den Lungen-6.
MAP: Microlitiasis Alveolar Pulmonar.
MII: Miopatías Inflamatorias Idiopáticas.
MMF: Micofenolato Mofetil.
MPO: Mieloperodixasa.
mTOR: Mammalian target of rapamycin.
MTX: Metotrexato.
NEA: Neumonía Eosinófila Aguda.
NEC: Neumonía Eosinófila Crónica.
NIA: Neumonía Intersticial Aguda.
NID: Neumonía Intersticial Descamativa.
NIL: Neumonía Intersticial Linfoide.
NH: Neumonitis por Hipersensibilidad.
NHF: Neumonitis por Hipersensibilidad Fibrótica.
NINE: Neumonía Intersticial No Específica.
NIU: Neumonía Intersticial Usual.
NO: Neumonía Organizada.
NOC: Neumonía Organizada Criptogénica.
NR: Lesión pulmonar inducida por radiación.
LAM: Linfangioleiomiomatosis.
LBA: Lavado Broncoalveolar.
LEF: Leflunomida.
LES: Lupus Eritematoso Sistémico.
LDH: Lactato Deshidrogenosa.
OCD: Oxigenoterapia Crónica Domiciliaria.
ODI: Índice de Desaturación de Oxígeno.
OTLD: Oxigenoterapia de Larga Duración.
PAAF: Punción Aspiración con Aguja Fina.
PAM: Poliangeítis Microscópica.
PAP: Proteinosis Alveolar Primaria.
PCR: Proteina C Reactiva.
PDGF: Factor de crecimiento derivado de las plaquetas.
PERDS: Síndrome de dificultad respiratoria periinjerto.
PFR: Pruebas de Función Respiratorias.
PjP: Pneumocistis jirovecii.
PM: Polimiositis.
PM6M: Prueba de Marcha de 6 Minutos.
PPFE: Fibroelastosis Pleuroparenquimatosa.
PR3: Proteinasa 3.
PRR: Neumonitis por Recuerdo de Radiación.
RAS: Síndrome restrictivo del injerto.
RGE: Reflujo Gastroesofágico.
RMN: Resonancia Magnética Nuclear.
RR: Rehabilitación Respiratoria.
RT: Radioterapia.
RTX: Rituximab.
RVP: Resistencias Vasculares Pulmonares.
SDRA: Síndrome de Distrés Respiratorio del Adulto.
SNI: Síndrome de Neumonía Idiopática.
SBO: Síndrome de Bronquiolitis Obliterante.
SBRT: Terapia de radiación corporal estereotáxica.
SSc: Esclerosis Sistémica.
TAC: Tomografía Axial Computarizada.
TBNA: Transbronquial Needle Aspiration.
TCAR: Tomografía Computarizada de Alta Resolución.
TEP: Tromboembolismo de Pulmón.
THC: Tetrahidrocannabinol.
TLC: Capacidad Pulmonar Total.
Tmax: Tiempo (en minutos) en el que un fármaco alcanza su concentración máxima.
TNF: Factor de Necrosis Tumoral.
TPH: Trasplante de Progenitores Hematopoyéticos.
TRALI: Lesión pulmonar aguda relacionada con transfusiones.
TSC: Genes del complejo de la Esclerosis Tuberosa.
TVP: Trombosis Venosa Profunda.
TxT: Trasplante Pulmonar.
UCP: Unidad de Cuidados Paliativos.
VAA: Vasculitis Asociados a ANCA.
VATS: Biopsia Pulmonar Vídeo Asistida.
VC: Capacidad Vital.
VEGF: Factor de Crecimiento del Endotelio Vascular.
VHS: Virus Herpes Simple.
VIH: Virus de la Inmunodeficiencia Humana.
VMI: Ventilación Mecánica Invasiva.
VMNI: Ventilación Mecánica No Invasiva.
VO2max: Consumo Máximo de Oxígeno.
VR: Volumen Residual.
VRS: Virus Respiratorio Sincitial.
VSG: Velocidad de Sedimentación Globular.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
ÍNDICE
Capítulo 1. Introducción y clasificación de las Enfermedades Pulmonares Intersticiales Difusas. 20
Capítulo 2. Algoritmo diagnóstico en las EPID: principales pruebas complementarias. 36
Capítulo 3. Técnicas diagnósticas invasivas en las EPID. 50
Capítulo 4. Fibrosis Pulmonar Idiopática. 65
Capítulo 5. Genética y EPID. Fibrosis Pulmonar Familiar. 82
Capítulo 6. Enfermedad pulmonar intersticial difusa idiopática no FPI. 90
Capítulo 7. Sarcoidosis y otras enfermedades granulomatosas. 100
Capítulo 8. Neumonitis por hipersensibilidad. 119
Capítulo 9. Síndrome Combinado de Fibrosis Pulmonar /Enfisema y EPID asociada a tabaco. 137
Capítulo 10. EPID asociadas a conectivopatías. 153
Capítulo 11. EPID con características autoinmunes (IPAF). 171
Capítulo 12. Vasculitis y hemorragias alveolares. 182
Capítulo 13. Enfermedades intersticiales de origen ocupacional. 200
Capítulo 14. Enfermedades quísticas pulmonares. 216
Capítulo 15. Neumonía organizada. 234
Capítulo 16. Enfermedad pulmonar intersticial producida por fármacos y radioterapia. 247
Capítulo 17. EPID secundarias: infecciones y enfermedad injerto contra huésped (EICH) . 266
Capítulo 18. EPID raras: proteinosis alveolar, microdiálisis, amiloidosis, EPID por alteración del surfactante pulmonar EPID por alteraciones del surfactante pulmonar). 278
Capítulo 19. Eosinofilias pulmonares. 291
Capítulo 20. Fibrosis Pulmonar Progresiva. 308
Capítulo 21. Comorbilidades.
318
Capítulo 22. Exacerbaciones y complicaciones de las EPID y EPID rápidamente progresivas. 326
Capítulo 23. Tratamientos no farmacológicos. Oxigenoterapia y rehabilitación cardiopulmonar en pacientes con EPID. 338
Capítulo 24. Tratamientos farmacológicos: antifibróticos e inmunosupresores (dosis y efectos secundarios) 352
Capítulo 25. Indicaciones de derivación de las epid a trasplante pulmonar. 371
Capítulo 26. Final de vida. Paliativos en EPID. 387
AGRADECIMIENTOS
Este Manual supone un ambicioso reto del Grupo Emergente de EPID (GEEPID), en el que se ha recogido una actualización de las enfermedades intersticiales pulmonares, y nos va a servir de gran ayuda en la labor asistencial de nuestras consultas, para unificar el diagnóstico y tratamiento de estas complejas patologías.
Ana Boldova y José Antonio Ros, como coordinadores del Grupo GEEPID de SEPAR, queremos agradecer:
- En primer lugar, a todos los coordinadores que nos han ayudado en la revisión de cada uno de los capítulos; grandes expertos en el ámbito nacional del manejo de las enfermedades intersticiales, pilar fundamental para llegar a la meta de la publicación de este este manual de revisión de consulta de EPID.
- A los miembros del GEEPID, por su entusiasmo que nos demuestran en cada una de las jornadas compartidas con ellos, por su crecimiento personal y profesional, y que, desde el primer momento, con actitud vehemente y con su trabajo en equipo, respondieron ante este gran reto, para poder desarrollar el primer manual de EPID.
- A SEPAR y al Área de EPID, por la confianza depositada en nosotros, para seguir coordinando este maravilloso grupo de jóvenes, tan involucrados en la patología intersticial y con tantas ganas de aprender y publicar.
- A Boehringer Ingelheim: sin su apoyo y ayuda al GEEPID, ningún proyecto podría salir adelante, incluida la publicación de este manual.
Les agradecemos que, desde un primer momento, mostraron un gran interés en que se llevara a cabo este libro del que ya disponemos.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES
INTERSTICIALES DIFUSAS
INTRODUCCIÓN Y CLASIFICACIÓN DE LAS
ENFERMEDADES PULMONARES
INTERSTICIALES DIFUSAS
Autores
Mario Nicolás Albani
David Eugenio Jerves Donoso
Coordinadora
Teresa Peña Miguel
1. INTRODUCCIÓN
Las enfermedades intersticiales pulmonares difusas (EPID) forman un grupo heterogéneo de enfermedades que afectan el intersticio pulmonar y, en algunos casos, las vías aéreas respiratorias, y que comparten unas características comunes, como la infiltración celular inflamatoria o la fibrosis, caracterizada esta última por el engrosamiento de los tabiques alveolares, la proliferación de los fibroblastos, el depósito de colágeno en la matriz extracelular, o pueden coexistir ambas en un paciente con la misma enfermedad, y todas ellas afectan de forma difusa el intersticio.¹
Las EPID abarcan más de 200 enfermedades diferentes que muestran una variación considerable en términos de curso clínico, tratamiento y pronóstico. En algunas, la recuperación puede ser completa. Otras pueden curarse, pero con secuelas, mientras que otras aún evolucionan de forma progresiva hacia la insuficiencia respiratoria y la muerte o el trasplante.
El diagnóstico² de estas enfermedades es complejo, por lo que el Comité Multidisciplinar (CMD) ha cobrado una importancia cada vez mayor en los últimos veinte años, siendo en el momento actual imprescindible para el diagnóstico y manejo de estas entidades. Dicho comité debe estar formado, como mínimo, por un neumólogo, un
radiólogo y un anatomopatólogo, y puede incluir profesionales de otras especialidades, como reumatología, medicina interna, farmacia, enfermería y psicología.
Para un diagnóstico correcto, se requiere una exhaustiva historia clínica, que puede orientar el diagnóstico en un 30% de los casos. La edad y el sexo son importantes, ya que determinadas enfermedades son más frecuentes en función del sexo y de la edad. Se debe conocer si el paciente está expuesto al tabaquismo, su actividad laboral, sus aficiones, las características de su hogar, los fármacos que utiliza, si está sometido a radioterapia, etc., además de antecedentes como enfermedades autoinmunes sistémicas (EAS), que cursan con frecuencia con una enfermedad intersticial.
La clínica puede ser aguda o de meses de evolución. Estas enfermedades cursan con síntomas muy inespecíficos, como disnea y tos, que son los más frecuentes. Debemos interrogar, además, sobre otra sintomatología, como la asociada a otras patologías, como las EAS, ya que en ocasiones es la primera manifestación. A la exploración, el paciente suele presentar crepitantes secos en bases y, en algunos casos, puede presentar dedos en palillo de tambor o manos de mecánico, que nos pueden sugerir una EAS.
La TCAR es la técnica diagnóstica imprescindible que va a confirmar nuestra sospecha. En las últimas décadas, ha habido grandes avances desde el punto de vista técnico que han permitido mejorar las imágenes, como con la definición de patrones radiológicos, lo que ha permitido una mejor clasificación de las EPID. Hay nuevas técnicas prometedoras, como el CALIPER, que son más precisas para la cuantificación del grado de fibrosis, que será de utilidad tanto en el diagnóstico como para valorar la progresión.
Los datos de laboratorio básicos nos ayudarán a orientar el proceso, como eosinófilos, función renal y hepática, CPK, aldolasa, VSG, determinaciones de IgG específicas (para hongos y aves), determinaciones serológicas como ANA, factor reumatoide, ac anticitrulinados y patrón de miositis, ECA, VEG-D en determinadas patologías como EA, neumonitis por hipersensibilidad (NH), sarcoidosis y linfangioleiomiomatosis (LAM).
Como veremos en otros capítulos, el Comité Multidisciplinar debe valorar la realización de otras técnicas, como la fibrobroncoscopia, la biopsia pulmonar quirúrgica o la criobiopsia, en función del riesgo-beneficio.
El BAL se debe realizar en el estudio con poblaciones y subpoblaciones linfocitarias. Estas pueden ayudarnos a diagnosticar determinadas enfermedades, como neumonías eosinofílicas, proteinosis, histiocitosis, NH y sarcoidosis, y a descartar infecciones.
En la actualidad, no está totalmente definido en qué casos está indicado realizar una biopsia pulmonar quirúrgica o una criobiopsia, por lo que la elección de una u otra técnica depende de la experiencia del centro realizador. Los futuros estudios definirán cómo se posicionarán ambas técnicas en el algoritmo diagnóstico.
A día de hoy, no se dispone de biomarcadores para el diagnóstico en la práctica clínica, pero se está investigando determinados marcadores que pueden ayudar en un futuro para el diagnóstico diferencial de la FPI y otras EPID, como el antígeno de Krebs von den Lungen (KL6), las proteínas del surfactante A y D (SP-A y SP-D); la metaloproteasa de la matriz (MMP, la más estudiada de las cuales es la MMP7), la periostina y la proteína similar a la lixil oxidasa² (LOXL2), factores de crecimiento, citocinas (CCL18) y células circulantes del sistema inmunológico. También otras como la calprotectina y algunas quimiocinas, como CCL2 o CXCL13.³
A pesar de no ser diagnóstica, la función pulmonar es importante para conocer el pronóstico y la evolución, siendo un marcador de progresión en estas patologías. Habitualmente, estas patologías cursan con patrón restrictivo, pero algunas, como sarcoidosis, LAM, silicosis, histiocitosis o neumonía eosinofílica, pueden tener alteración obstructiva, mientras que la mayoría presentan disminución de la DLco.
La patogenia de las EPID es variable, según la enfermedad subyacente. De forma general, se pueden dividir en las EPID en que predomina la inflamación y las EPID en que predomina la fibrosis. La FPI es de causa desconocida, es el prototipo de enfermedad fibrótica y su evolución es progresiva. Otras EPID pueden ser desencadenadas por un agente causal, conocido o no. En algunos casos, a pesar de tener como base patogénica inicial la inflamación, y a pesar del tratamiento correcto, pueden evolucionar a fibrosis y progresar como la FPI.4,5
Una gran parte de EPID tendrán una combinación de inflamación y fibrosis, y la efectividad de la terapia inmunomoduladora varía según las condiciones. Es más probable que ésta estabilice y/o ralentice la progresión de la enfermedad, como puede ocurrir en la neumonía intersticial no específica (NINE), la EPID asociada a enfermedades del tejido conectivo y subconjuntos de fibrosis crónica en neumonitis por hipersensibilidad (NH).
Las EPID relacionadas con otras exposiciones ocupacionales son causadas por la inhalación y retención en los pulmones de diversos polvos. Las más comunes de estas afecciones son la asbestosis (fibras de amianto) y la silicosis (dióxido de silicio cristalino libre o sílice). También exposición a la agricultura, a la ganadería, a polvo metálico, carbón, arena y polvo resultante de la preparación de prótesis dentales.³
Dentro de las EPID, la FPI, el prototipo más frecuente en la mayoría de las series, es de una evolución clínica progresiva hacia la insuficiencia respiratoria y la muerte. Gracias a la aparición de los tratamientos antifibróticos, se ralentiza su progresión. En la FPI se produce una acumulación descontrolada de matriz extracelular en el parénquima pulmonar, que progresa afectando a más unidades alveolo-intersticiales, por lo que el pulmón pierde su estructura y su función. La FPI es de causa desconocida, pero hay una serie de factores predisponentes, como son:
1. Factores genéticos (en los casos de enfermedades intersticiales familiares), especialmente en los genes encargados del mantenimiento en la longitud telomérica transcriptasa inversa de la telomerasa (TERT), componente de ARN de la telomerasa (TERC), regulador del alargamiento de telómeros helicasa 1 (RTEL1), los genes de la proteína C (SFTPC) y A2 (SFTPA2) del surfactante, y el polimorfismo rs35705950 en la región promotora del gen que codifica la mucina de las vías respiratorias (MUC5B).
2. Senescencia celular (en la división celular y apoptosis), a mayor edad, mayor riesgo; se cree que aquí juega un papel muy importante la desregulación de las células alveolares epiteliales tipo 2 (CAE-2).
3. Microbioma, algunos estudios sugieren que la alteración de la microbiota, aumenta el riesgo; otros factores, como tabaquismo, exposición ocupacional y ambiental, polvos orgánicos, metales, minerales, madera y asbesto, también lo aumentan.
Otro de los grupos importantes dentro de las EPID son las asociadas a enfermedades autoinmunes sistémicas (EAS), como es la artritis reumatoide (AR), y esta se produce por una alteración en la citrulinación de las células estructurales, predisponiendo al desarrollo de una serie de autoanticuerpos, mediante la activación del sistema adaptativo del sistema inmunológico, liberando una variedad de citoquinas, incluyendo TNF, interleucina (IL)-1, IL-6 y prostaglandinas.
La esclerosis sistémica y las miopatías, comparten similitudes con la patogénesis de artritis reumatoide, pero se caracterizan por diferentes autoanticuerpos.
En las enfermedades granulomatosas, se forman por macrófagos agregados, que a menudo se fusionan para formar grandes células multinucleadas y más del 90% de las personas padecen afectación pulmonar. El típico granuloma sarcoide contiene células T colaboradoras CD4+ y está estrechamente rodeado de células T reguladoras empaquetadas, fibroblastos y células B, lo que sugiere que la activación inmune innata y adaptativa contribuye al desarrollo de la enfermedad.
Las enfermedades del tejido conectivo (ETC) asociadas con EPID pueden tener un fenotipo fibrosante. Entre ellas se cuentan AR, esclerodermia (SSc), miopatía inflamatoria idiopática (polimiositis y dermatomiositis), síndrome de Sjögren, lupus eritematoso sistémico y ETC mixtas. De estas afecciones, la SSc y la AR se asocian con mayor frecuencia con la EPID.5
La neumonitis por hipersensibilidad se produce por la exposición repetida a una amplia gama de antígenos potenciales. También se caracteriza por una inflamación granulomatosa. La forma no fibrótica de la enfermedad está mediada por la formación de complejos inmunes, mientras que la de tipo fibrótica se caracteriza por células alveolares y dendríticas. Estas presentan antígenos a los linfocitos T, que sesgan hacia un fenotipo
T-helper-1 a través de la polarización por citocinas, como IL-12 e IFN-γ.
La linfangioleiomiomatosis es una enfermedad pulmonar quística que afecta a las mujeres y se produce por la activación de la señalización mTOR en células musculares.
La proteinosis alveolar pulmonar se caracteriza por la acumulación de lípidos y macrófagos, resultando de una falla del GM-CSF.
La histiocitosis de células de Langerhans es una enfermedad pulmonar quística causada por la proliferación de un subconjunto de células dendríticas, debido a mutaciones somáticas en BRAF y genes MAPK.1
2. EPIDEMIOLOGÍA
Los estudios de incidencia y prevalencia de los que disponemos son heterogéneos, difíciles de comparar porque utilizan distinta metodología, distintas fuentes y diferente terminología para las enfermedades. Sin embargo, conocer la epidemiología de las EPID es importante para conocer la carga de estas o para ver las diferencias entre distintas regiones, lo que nos puede ayudar a caracterizar mejor los factores de riesgo en función de las posibles exposiciones y variables genéticas.
En una publicación reciente, Bhavika Kaul y col.6 revisaron 17 estudios que utilizaban metodologías diferentes. Examinaron la incidencia, la prevalencia y las frecuencias relativas de EPI. Según su estudio, la incidencia de EPI osciló entre 1 y 31,5 por 100.000 personas-año y la prevalencia osciló entre 6,3 y 71 por 100.000 personas. En América del Norte y en Europa, la fibrosis pulmonar idiopática y la sarcoidosis fueron las EPID más prevalentes, mientras que la frecuencia relativa de neumonitis por hipersensibilidad fue mayor en Asia, particularmente en India (10,7-47,3%) y Pakistán (12,6%). La frecuencia relativa de la EPID por enfermedad del tejido conectivo demostró la mayor variabilidad geográfica, desde el 7,5% de los casos en Bélgica hasta el 33,3% de los casos en Canadá y el 34,8% de los casos en Arabia Saudita.
En la figura 1 vemos la frecuencia relativa del subtipo de enfermedad pulmonar intersticial por área geográfica, fibrosis pulmonar idiopática (FPI), enfermedad del tejido conectivo (CTD), neumonitis por hipersensibilidad (NH).
En España, un estudio multicéntrico llevado a cabo entre 2000 y 2001 observó una incidencia estimada de EPID de 7,6 por 100.000 personas-año. La FPI fue el subtipo de EPID más común (38,6%), seguida de sarcoidosis (14,9%), EPID-EA (10%) y NH (6,6%), mientras que un 5% de los casos eran inclasificables. Entre la cohorte EPIDEA, la artritis reumatoide fue la etiología más común, y la causa más común de NH fue dedicarse al cuidado de aves. 7
En la región de los Ríos de Chile, en el período comprendido entre 2018 y 2019 se reclutaron 339 casos, de los cuales el 64% fueron mujeres, con una edad media de 71 ±10, siendo la prevalencia de 84 por cada 100.000 habitantes.7
En niños hay menos estudios y la prevalencia es mucho menor. En un estudio realizado en niños en Alemania, la incidencia de casos nuevos fue de 1,32/100.000 de niños y año, y la prevalencia probablemente es inferior a 1/100.000, siendo la edad de inicio más frecuente el primer año de vida, en contraste con la prevalencia en adultos, de 60-80/100.000.9
En la mayoría de los estudios más recientes, la edad media es superior a los 65 años, en la mayoría, tanto las EPID como las FPI y ocupacionales son más frecuentes en varones, y las asociadas a enfermedades del tejido conectivo son más frecuentes en mujeres y en edades más jóvenes.
En esta revisión6 se ha visto que hay una importante heterogeneidad geográfica en la epidemiología de estas enfermedades. Esto puede ser debido a diferencias reales basadas en la demografía, en las exposiciones o genéticas de las poblaciones, pero también a las diferencias metodológicas en el reclutamiento de pacientes y clasificación de enfermedades. Conocer mejor la epidemiología de las EPID a nivel regional nos ayudaría a conocer mejor los factores de riesgo y, por tanto, podríamos intervenir en estos. Por otro lado, conocer la carga de morbilidad y mortalidad de estas enfermedades ayudaría a una mejor distribución de los recursos por parte de los sistemas sanitarios. En consecuencia, se necesitan estudios epidemiológicos diseñados para recoger de forma estandarizada la incidencia y la prevalencia de estas enfermedades utilizando una clasificación igual en todos los países.
Un estudio reciente realizado en Europa, entre 2014 y 2018, el PERSEIDS,10 es interesante por ser el primer estudio que evalúa la epidemiología y la progresión de las EPID en seis países europeos. Su objetivo fue estimar la incidencia/prevalencia de enfermedades pulmonares intersticiales (EPID), enfermedades pulmonares intersticiales fibrosantes (EPID-F), fibrosis pulmonar idiopática (FPI), EPID asociada a esclerosis sistémica (EPID-ES) y otras F no relacionadas con FPI, EPID y sus formas fibrosantes progresivas (FPP). El estudio se realizó en dos fases. En la primera, se recogieron los casos de EPID mencionados y se estimó la incidencia/prevalencia. Para las EPID-F sin FPI, también se determinó el porcentaje relativo de subtipos. En la segunda fase, se revisó un subconjunto de casos EPID-F sin FPI para determinar el porcentaje de casos con FPP y el patrón similar a la neumonía intersticial (NIU). Se calculó un porcentaje medio ponderado de progresión para cada país y se utilizó para extrapolar la incidencia/ prevalencia de EPID fibrosantes progresivas.
En 2018, la incidencia/105 personas-año osciló entre 9,4 y 83,6 (EPID), 7,7 y 76,2 (EPID-F), 0,4 y 10,3 (FPI), 6,6 y 71,7 (EPID-F sin FPI) y 0,3 y 1,5 (EPID-ES); y la prevalencia/105 personas osciló entre 33,6 y 247,4 (EPID), 26,7 y 236,8 (EPID-F), 2,8 y 31,0 (FPI), 22,3 y 205,8 (EPID-F sin FPI) y 1,4 y 10,1 (EPID-ES). Entre las F-EPID no relacionadas con FPI, la sarcoidosis fue el subtipo más frecuente. El comportamiento
de la PF y el patrón similar a la UIP estuvieron presentes en un tercio de los casos de EPID-F sin FPI cada uno, y la neumonitis por hipersensibilidad mostró el mayor porcentaje de progresión. La incidencia de EPID-F osciló entre 2,1 y 14,5/105 personas-año, y la prevalencia, entre 6,9 y 78,0/105 personas.
En este estudio, la incidencia y la prevalencia de las EPID fueron mayores y más variables entre países de lo publicado en estudios anteriores en los mismos países. En aproximadamente un tercio de los casos, las EPID-F no FPI mostraron un comportamiento de FPP, y casi la mitad de ellos (43%) tenían un patrón similar a NIU. La prevalencia e incidencia de las EPID-F oscilaron entre 2,1 y 14,5/105 personas-año y 6,9-78,0/105 personas, y las diferencias entre países probablemente se explican por la distribución relativa de los subtipos de EPID-F sin FPI y sus diferentes tasas de progresión (Figura 2).
3. CLASIFICACIÓN
Las enfermedades intersticiales difusas se han clasificado en grupos: a) neumonías intersticiales idiopáticas; b) de causa conocida o asociadas a un proceso bien definido; y c) EPID primarias o asociadas a otros procesos no bien definidos (Tabla I).10-11
Hasta la publicación de la Clasificación de Consenso Multidisciplinario Internacional de las Neumonías Intersticiales Idiopáticas de la ATS/ ERS de 2002,11 las distintas clasificaciones de las que disponíamos eran histológicas, la de Liebow y Carrington (1969), la Müller y Colby (1997) y la de Katzenstein (1997), si bien estas procedían únicamente de pacientes con muestra histológica, cuando en una gran parte de estos pacientes no se disponía de ella.
Con el objetivo de introducir los nuevos avances radiológicos (TCAR) y ante la necesidad de estandarizar la nomenclatura y los criterios diagnósticos, se realiza esta clasificación de las neumonías intersticiales idiopáticas, se introduce el término de trabajo multidisciplinar (neumólogos, radiólogos y patólogos se reúnen para la realización de esta clasificación), se introduce el término de patrones, tanto radiológicos como histológicos, y se establece el término de «proceso diagnóstico dinámico», mientras que el diagnóstico final se establece por consenso entre las tres especialidades implicadas con un diagnóstico clínico, radiológico y patológico, en el caso que se disponga de biopsia. En esta clasificación se proponen 7 entidades: fibrosis pulmonar idiopática (FPI), neumonía intersticial no específica (NINE), neumonía organizada criptogénica (NOC), neumonía intersticial aguda (NIA), enfermedad pulmonar intersticial asociada a bronquiolitis respiratoria (BR-EPID, neumonía intersticial descamativa (NID) y neumonía intersticial linfoide (NIL), mientras que la NINE queda como una entidad provisional, porque, según refieren, no hay datos suficientes en la literatura como para considerarla una entidad específica.
En 2013 se realiza una Actualización de la Clasificación ATS/ERS de 2002, a la luz de los nuevos datos de la literatura. En esta revisión de la clasificación NII se conservan las principales entidades, pero hay algunos cambios importantes.
Se elimina la alveolitis fibrosante criptogénica, dejando la fibrosis pulmonar idiopática (FPI) como único término clínico para el diagnóstico de esta entidad; la neumonía intersticial idiopática no específica (NINE) ahora se acepta como una entidad clínica definitiva y se elimina término «provisional», las NII más frecuentes se diferencian de las NII raras y de los casos inclasificables. Se reconocen patrones histológicos raros de neumonía fibrinosa y organizada aguda (AFOP) y neumonías intersticiales con distribución broncocéntrica (Tabla II).
En esta guía,12 se propone una clasificación en la que las principales NII se agrupan según la evolución en fibrosantes crónicas FPI, NINE relacionadas con el tabaquismo (bronquiolitis respiratoria-enfermedad pulmonar intersticial, BR-EPID) y neumonía intersticial descamativa y NII agudas/subagudas (neumonía organizada criptogénica (NOC) y neumonía intersticial aguda (NIA) y se propone una clasificación en función del comportamiento clínico de la enfermedad (Tabla III).
Por último, se revisan las características moleculares y genéticas, que a día de hoy todavía no se usan en la práctica clínica, pero que en un futuro nos ayudarán a diagnosticar estas entidades.
En los últimos años, se han publicado numerosos estudios y revisiones sobre el comportamiento de las EPID fibrosantes, no FPI, ya que en algunas enfermedades, que se han iniciado como inflamatorias en un porcentaje de pacientes, evolucionan a fibrosantes, y estas pueden progresar a pesar de tratamientos inmunomoduladores hacia una fibrosis, con un comportamiento similar a la FPI.
Estas enfermedades presentan progresión a pesar del tratamiento adecuado de la enfermedad de base, que es lo que hemos denominado con distintos términos como EPID fibrosantes sin FPI (EPID-F). Estas pueden tener un pronóstico similar al de la FPI cuando hay progresión y/o neumonía intersticial usual (NIU). De los subtipos de EPID-F sin FPI, la EPID asociada a esclerosis sistémica (EPID-ES) es de particular interés, ya que se presenta temprano en el curso de la enfermedad.5,12 En la figura 3, dentro de las distintas EPID, se señalan en color las enfermedades que con mayor frecuencia presentan FP y en qué proporción pueden presentar FPP.
En 2022, se publica las última guía internacional,12 que nace de la necesidad de consensuar una terminología común para designar todos de la misma forma y para definir unos criterios comunes consensuados para definir la progresión, denominando Fibrosis Pulmonar Progresiva (FPP), siempre teniendo en cuenta que no es una enfermedad en sí misma sino una característica de un grupo de EPID fibrosantes que progresan. En esta guía se ha definido también los criterios de progresión. Dichos criterios de progresión se definen en la tabla IV.
BIBLIOGRAFÍA
1. Wijsenbeek M, Suzuki A, Maher TM. Interstitial lung diseases. Lancet. 2022;400(10354):769786.
2. López Ramírez C, López Bauza A, De Benito Zorrero E, Rodríguez Portal JA. «Neumopatías Intersticiales: Concepto, clasificación, manifestaciones clínicas y aproximación diagnóstica», en Manual de Neumología y Cirugía Torácica, 4ª ed.
3. Margallo Iribarnegaray J, Churruca Arrospide M, Matesanz Lopez C, Perez Rojo R. Interstitial Lung Disease. Open Respir Arch. 2023;5(2):100248.
4. Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung Dideases. NEngl J Mede. 2020;383(10):958-968.
5. Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150).
6. Kaul B, Cottin V, Collard HR, Valenzuela C. Variability in Global Prevalence of Interstitial Lung Disease Front. Med. 04 November 2021. Sec. Pulmonary Medicine Volume 8.
7. Xaubet A, Ancochea J, Morell F, Rodríguez-Arias JM, Villena V, Blanquer R, et al. Informe sobre la incidencia de las enfermedades pulmonares intersticiales en España. Sarcoidosis Vasc Enfermedad Pulmonar Difusa. 2004;21:64-70.
8. Castillo-Orellana P, Toro-Munoz N, Barria-Pailaquilen RM. Prevalence and lethality of interstitial pulmonary diseases in the Region of Los Rios, Chile. Rev Med Chil. 2022;150(2):154162.
9. Moreno Galdó A, De Mir Messa I, Liñán Cortés S. Neumopatía intersticial. Sospecha clínica y abordaje. Pediatría AEd, editor, 2017. p. 221-325.
10. Hilberg O, Hoffmann-Vold AM, Smith V, Bouros D, Kilpelainen M, Guiot J, et al. Epidemiology of interstitial lung diseases and their progressive-fibrosing behaviour in six European countries. ERJ Open Res. 2022;8(1).
11. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial. Am J Respir Crit Care Med. 2002:277304.
12. Travis WD, Costabel U, Hansell DM, King TE, Jr., Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733-748.
13. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):18-47.
14. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnóstico de la fibrosis pulmonar idiopática. Una guía de práctica clínica oficial de ATS/ERS/JRS/ALAT. Am J Respir Crit Care Med. 2018;198:44-68. doi: 10.1164/rccm.201807-1255ST.
TABLAS Y FIGURAS
Tabla I. American Thoracic Society/European Respiratory Society. International Multidisciplinary Consensus Classification of the Idiopathic Interstitial. Am J Respir Crit Care Med. 2002;15

Tabla II. Travis WD et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013

Tabla III. Neumonias intersticiales idiopáticas: clasificación según el comportamiento de la enfermedad

Travis WD et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013
Tabla IV. Raghu G. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults An Official ATS/ERS/JRS/ALAT.14 Clinical Practice Guideline. 2022

1. Kaul et al. Variability in Global Prevalence of ILD. Frontiers in Medicine, 2021

Figura


Figura 2. Hilbert O et al. ERJ Open Research. 2022
Figura 3. Raghu G, Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults An Official ATS/ERS/JRS/ALAT.13 Clinical Practice Guideline. 2022

Figura 3. Interstitial lung diseases (ILDs) manifesting progressive pulmonary fibrosis (PPF), developed using consensus by discussion. The haded area represents the estimated proportion of patients with various types of ILD who manifest PPF. Note that idiopathic pulmonary fibrosis (IPF) is not included in the figure, because it is excluded from the definition of PPF. While virtually all patients with IPF will manifest disease progression similar to PPF, the proportion of patients with ILDs other than IPF who manifest PPF is based on the consensus of opinions and the perception of the international committee. There are no data to provide the exact or estimated proportion of patients manifesting PPF in ILDs, other than IPF. *The committee acknowledges that eosinophilic pneumonia of unknown cause was not included in the IIP classification. †Myositis includes PM/ DM/antisynthetase syndrome, which may be amyopathic. ‡Although respiratory bronchiolitis interstitial lung disease (RBILD) is acknowledged to be a consequence of exposure to cigarette smoke in virtually all patients with RBILD, RBILD and desquamative interstitial pneumonia (DIP) often coexist. Although DIP is also related to exposure to cigarette smoke in a majority of patients, DIP is also seen in some patients with connective tissue disease, without exposure to cigarette smoke, and without a known cause. Antifibrotic treatment is indicated for patients diagnosed with IPF (3). Antifibrotic treatment of the other types of ILD upon manifesting PPF is as suggested/recommended in this guideline. AFOP = acute fibrinous and organizing pneumonia; AIP = acute interstitial pneumonia; COP = cryptogenic organizing pneumonia; DM= dermatomyositis; HP = hypersensitivity pneumonitis; iDIP = idiopathic DIP; IIP = idiopathic interstitial pneumonia; iLIP = idiopathic lymphoid interstitial pneumonia; iNSIP = idiopathic nonspecific interstitial pneumonia; iPPFE = idiopathic pleuroparenchymal fibroelastosis; LAM= lymphangioleiomyomatosis; LCH= Langerhans cell histiocytosis; MCTD=mixed connective tissue disease; PAP = pulmonary alveolar proteinosis; PM= polymyositis; RA = rheumatoid arthritis; SLE = systemic lupus erythematosus; SSc = systemic sclerosis.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES
INTERSTICIALES DIFUSAS
ALGORITMO DIAGNÓSTICO EN
LAS EPID:
PRINCIPALES PRUEBAS
COMPLEMENTARIAS
Autores
Pilar Muñoz Zara
José Antonio Delgado Torralbo
Coordinadora
Ana Dolores Romero Ortiz
1. INTRODUCCIÓN
Las enfermedades pulmonares intersticiales difusas (EPID) constituyen un grupo heterogéneo de entidades que afectan a la estructura alveolo-intersticial y presentan manifestaciones clínicas, funcionales respiratorias y radiológicas similares. Para un diagnóstico correcto, se debe combinar información procedente de datos clínicos y analíticos, pruebas de función respiratoria y una variedad de pruebas diagnósticas, invasivas y no invasivas. Un enfoque multidisciplinario que involucre a neumólogos, radiólogos, patólogos y cirujanos torácicos es esencial para realizar un diagnóstico definitivo. Hay que recordar que el diagnóstico de estas entidades es dinámico y requiere la incorporación de información aportada por la evolución clínica a lo largo del tiempo que pueda orientar el diagnóstico. A pesar de una investigación exhaustiva, no se puede realizar un diagnóstico definitivo en aproximadamente el 15-20% de los casos. Se trata de las denominadas enfermedades intersticiales inclasificables.¹
2. ANAMNESIS
Sigue siendo crucial y el punto de partida para la orientación diagnóstica, y debe incluir los puntos que se especifican a continuación:
1. Presentación de los síntomas
- Forma aguda (días o varias semanas): puede ocurrir en la neumonitis eosinofílica aguda (NEA) secundaria a la ingestión de toxinas o fármacos, la neumonitis por hipersensibilidad (NH), la hemorragia alveolar, la neumonía asociada con lupus eritematoso sistémico (LES) o la neumonía intersticial aguda (NIA).
- Forma subaguda (semanas o meses): esta es la forma de presentación de la mayoría de las EPID. La neumonía intersticial asociada a sarcoidosis, la neumonía organizada criptogénica (NOC), algunas neumonitis por hipersensibilidad y algunas enfermedades autoinmunes sistémicas o polimiositis suelen presentarse de esta manera.
- Forma crónica (durante meses o años): ocurre en la FPI, la NH, la neumoconiosis, la histiocitosis de células de Langerhans (HPCL) y algunas enfermedades intersticiales asociadas con la artritis reumatoide (AR) o la esclerodermia.
- Forma episódica: en algunos casos, como vasculitis, algunos tipos de NH, neumonitis eosinofílica crónica (NEC) o incluso neumonía organizada (NO), la presentación es por brotes, con periodos libres de síntomas entre ellos.
2. Sexo
La FPI o la EPID asociada a la artritis reumatoide son más comunes en los hombres. En cambio, la neumonía intersticial no específica (NINE) en la esclerodermia ocurre con mayor frecuencia en mujeres. La linfangioleiomiomatosis (LAM) solo afecta a mujeres en edad fértil. Debido a los antecedentes ocupacionales, las neumoconiosis (silicosis, asbestosis) ocurren con mayor frecuencia en hombres. No hay diferencias de género en la HPCL.
3. Edad
La FPI suele iniciarse entre la sexta y la séptima década de edad, pero en el caso de la FPI familiar puede desarrollarse a edades más tempranas. La sarcoidosis y la LAM ocurren en personas de entre 20 y 40 años. La afectación intersticial de las enfermedades autoinmunes sistémicas suele ocurrir en la cuarta y quinta décadas de vida. Algunos casos de neumonía eosinofílica aguda pueden observarse en personas muy jóvenes después de empezar a fumar.
4. Antecedentes familiares
Hay antecedentes familiares en las EPID asociadas a neurofibromatosis y esclerosis tuberosa, que también ocurre con su asociación con la LAM. En algunos casos de sarcoidosis y otras enfermedades raras, como el síndrome de Hermansky-Pudlak y la microlitiasis alveolar (una herencia autosómica recesiva), existe agregación familiar. En la FPI, hasta un 15-20% de los casos tienen antecedentes familiares.
5. Historia laboral / Exposición medio-ambiental
De especial interés en neumopatías intersticiales por inhalación de polvos inorgánicos (neumoconiosis) u orgánicos (NH). Es importante registrar las diferentes ocupaciones del paciente, la duración de cada una y si el paciente empleó protección respiratoria. A veces, la exposición ocurre durante actividades de ocio (cuidado de aves, saunas, pintura, jardinería, piscinas climatizadas, etc.).
6. Tabaco
Muchas enfermedades, como la FPI, son más comunes entre los fumadores. En otros casos, como HPCL, NID, BR-ILD y una combinación de fibrosis pulmonar y enfisema, la enfermedad ocurre casi exclusivamente en fumadores. Algunas hemorragias alveolares están asociadas con el tabaquismo. La neumonía eosinofílica aguda (NEA) suele ir acompañada de antecedentes recientes de tabaquismo o de un mayor número de cigarrillos fumados. La NH y la sarcoidosis son menos comunes entre los fumadores.
7.Tratamientos actuales y previos
Registrar todos los fármacos que está tomando o ha tomado el paciente, la posología y el inicio del tratamiento, ya que muchos de ellos son responsables del desarrollo de EPID. Existe una amplia lista de fármacos susceptibles de ocasionar EPID, que pueden consultarse en el sitio web pneumotox.com.
8. Otras enfermedades presentes
Por ejemplo, enfermedades autoinmunes.
3. MANIFESTACIONES CLÍNICAS Y EXPLORACIÓN FÍSICA
La mayoría de las enfermedades intersticiales presentan síntomas inespecíficos. La sospecha clínica se suele iniciar ante un cuadro de disnea de esfuerzo y tos seca de varios meses de evolución. En raras ocasiones, el diagnóstico es casual en un paciente asintomático.
Respecto a los signos físicos, también son inespecíficos para su diagnóstico. Incluso, en muchas de estas enfermedades la exploración puede ser normal, sobre todo en fases iniciales. Sin embargo, en otros, casos la presencia de crepitantes teleinspiratorios de tipo «velcro» en la auscultación pulmonar puede ser el primer signo que alarme sobre la existencia de enfermedad pulmonar intersticial. Pueden aparecer acropaquias y, en fases avanzadas, cianosis central, taquipnea, taquicardia o refuerzo del 2º tono cardiaco (si existe ya hipertensión pulmonar secundaria). Existen signos menos frecuentes, como las sibilancias en entidades centradas en la vía aérea distal, como es el caso de la bronquiolitis en la granulomatosis eosinofílica con poliangitis (GEPA) o la NH. En esta última, son característicos unos sonidos con tonalidad más ruda conocidos en la literatura anglosajona como squeaks, unos chirridos que son bastante característicos.²
Las EPID asociadas a enfermedades autoinmunes sistémicas pueden presentar, además de los síntomas respiratorios, síntomas y signos extrapulmonares, como fiebre, afectación ocular, cutánea o articular y fenómeno de Raynaud.³ (Tabla I).
4. LABORATORIO
Los resultados analíticos pueden ayudar al diagnóstico de exclusión. Se debe realizar un hemograma completo, velocidad de sedimentación globular (VSG), estudio de coagulación, bioquímica (glucosa, función renal, sodio, potasio, calcio, enzimas hepáticas, lactato deshidrogenasa (LDH), proteína C reactiva (PCR), enzima convertidora de angiotensina (ECA), creatinkinasa (CPK), aldolasa y estudio autoinmune que incluya factor reumatoide, anticuerpos anticitrulina (CCP), anticuerpos antinucleares (ANA), anticuerpos anticitoplasmáticos (ANCA) (Figura 1). En el seguimiento del paciente con EPID, si aparecen síntomas o signos que sugieran una posible enfermedad autoinmune se repetirá el estudio de autoinmunidad. Además, se llevará a cabo un sistemático de orina elemental y de 24 horas. Estas determinaciones resultan de utilidad en las EPID asociadas con enfermedad autoinmune o vasculitis sistémicas. En la NH es útil medir la IgG específica contra ciertos antígenos, como hongos (Aspergillus, Penicillium) y aquellos antígenos que se encuentran en las plumas de las aves (loros, periquitos o palomas). Su presencia indica exposición, pero no enfermedad, y su ausencia no la descarta. En el diagnóstico de LAM, los niveles elevados del factor de crecimiento endotelial sanguíneo (VEGF-D) >800 pg/ml son muy específicos y pueden evitar la biopsia, pero esta determinación no se realiza de forma rutinaria en la mayoría de los laboratorios.
5. FUNCIÓN PULMONAR
Las pruebas de función pulmonar no solo son útiles para orientar el diagnóstico, sino también para monitorizar la evolución y valorar el pronóstico. Se suele encontrar un patrón restrictivo y una disminución de la difusión de CO (DLco). La TLC (capacidad pulmonar total), la FVC (capacidad vital forzada) y la DLco están disminuidas. El cociente FEV1/FVC se encuentra normal o elevado. No obstante, un estudio funcional respiratorio normal no excluye el diagnóstico de EPID. Sin embargo, entidades como la GEPA, neumonías eosinofílicas, HCL, LAM, NH o silicosis pueden presentar una alteración ventilatoria obstructiva. La prueba de marcha de 6 minutos proporciona información sobre el nivel de actividad física, mientras que la presencia de desaturación de oxígeno (<88%) durante el ejercicio puede influir en el pronóstico y ayudar a determinar la necesidad de oxigenoterapia.4
6. RADIOLOGÍA
1. Radiografía simple de tórax
Esta es la primera prueba radiológica que se debe realizar cuando se investiga una EPID, si bien es cierto que sus resultados son inespecíficos. La sensibilidad es baja porque a menudo puede ser normal en las primeras etapas. En otros casos, puede conducir a un diagnóstico. Resulta de utilidad para valorar la distribución de las lesiones, monitorizar su evolución y detectar complicaciones. El hallazgo más frecuente es un patrón intersticial bilateral, pero también se puede observar un patrón alveolar en la NO, NIA, neumonía intersticial linfocítica (NIL), NH, proteinosis alveolar o eosinofilias pulmonares. Se puede encontrar neumotórax como primera manifestación de la LAM o la HPCL. El derrame pleural sucede en enfermedades asociadas a enfermedades autoinmunes, como LES, AR y LAM (quilotórax). Es importante la presencia de adenopatías hiliares y mediastínicas, como en la sarcoidosis.
2. Tomografía computarizada de alta resolución (TCAR)
La TCAR es la técnica radiológica más sensible en el estudio de las EPID. Permite detectar la enfermedad en pacientes con radiografía simple normal, identificar patrones radiológicos, así como valorar su distribución, extensión y naturaleza, lo que la convierte de gran utilidad para seleccionar el área pulmonar más adecuada para la práctica de técnicas invasivas (lavado broncoalveolar, biopsia transbronquial, punción adenopática, biopsia quirúrgica). En el caso de la FPI, la presencia de un patrón radiológico de neumonía intersticial usual (NIU) constituye un criterio diagnóstico, con unas características radiológicas definidas5 (Tabla II), si bien es cierto que este patrón se puede presentar en otras EPID, como en las asociadas a enfermedades autoinmunes, NH o toxicidad farmacológica. Los hallazgos de la TCAR en otras EPID son orientativos, por lo que se precisará la valoración del resto de datos clínicos, funcionales e histológicos para obtener un diagnóstico definitivo (Tabla III).
7. MÉTODOS INVASIVOS
1. Lavado broncoalveolar
El lavado broncoalveolar (LBA) es una muestra obtenida durante la realización de una broncoscopia flexible y constituida por células y fluidos procedentes de la vía aérea distal y del alveolo6 que permite el contaje, el tipo celular y los cultivos de virus, bacterias y hongos (Tabla IV). El procedimiento se realiza mediante la instilación de entre 150 y 200 ml de solución salina isotónica, generalmente en bolos de entre 20 y 50 ml, a través del canal de trabajo del broncoscopio, tras encajarlo en el bronquio seleccionado y procediendo a la aspiración suave del contenido para evitar el colapso de las paredes bronquiales.
El LBA de un individuo sano contiene menos de 150.000-200.000 células/ml, de las cuales, el 80-90% son macrófagos, el 5-10% son linfocitos, con una relación CD4/CD8
entre 1 y 2, menos del 5% son neutrófilos, menos del 2% son eosinófilos y menos del 5% son células bronquiales.7
Las subpoblaciones linfocitarias orientan el enfoque diagnóstico: un predominio linfocítico se encuentra en la sarcoidosis, la neumonitis por hipersensibilidad o la silicosis. La neutrofilia es frecuente en la FPI, la asbestosis, la neumonitis asociada a enfermedad del tejido conectivo o una infección bacteriana. La presencia de eosinófilos se muestra en la neumonía eosinofílica y en la GEPA. Finalmente, la visualización de macrófagos CD1 positivos es típica de la HPCL. Con respecto al cociente CD4/CD8, es igualmente de interés en el camino hacia el diagnóstico, siendo este elevado (>3,5) en la sarcoidosis y disminuido en la NH.8 En ocasiones, los hallazgos en el BAL proporcionan el diagnóstico definitivo, por observación de elementos patognomónicos y algunas veces del agente causal: quistes de Pneumocystis jirovecii; tinción de Gomori Grocott en neumocistosis; células de Langerhans y antígenos de membrana CD1 >5% y gránulos de Birbeck en la HPCL; aspecto lechoso y acumulación de una sustancia lipoproteinácea en la proteinosis alveolar; macrófagos cargados de lípidos en la neumonía lipoidea; presencia de células neoplásicas en la linfangitis carcinomatosa y los cuerpos de amianto en la asbestosis son algunos ejemplos. Pero, a pesar de todo ello, el análisis del LBA sigue siendo en gran medida inespecífico, además de no haber mostrado utilidad en el seguimiento y en la respuesta a tratamiento de las EPID.
2. Biopsia bronquial y transbronquial (BTB)
La biopsia, obtenida a través de pinzas insertadas en el canal de trabajo del broncoscopio que permiten la extracción de muestras, facilita el estudio de estructuras linfáticas, la mucosa, submucosa bronquial, bronquiolos terminales y algunos alveolos adyacentes. El diagnóstico definitivo y específico de las EPID requiere, en muchos casos, el análisis histológico del parénquima pulmonar. Se recomienda la obtención de entre 3 y 6 muestras para mejorar el rendimiento. La rentabilidad diagnóstica de este procedimiento es, en promedio, del 40%, un porcentaje que mejora sensiblemente si la distribución de la lesión es peribronquiolar y perilinfática (sarcoidosis, linfangitis carcinomatosa, NOC, LAM). Sin embargo, una distribución perivascular y heterogénea como en FPI, NINE, vasculitis o HPCL limita el diagnóstico.9 Además, esta técnica, dificulta la visualización tisular, debido al tamaño reducido de las biopsias y a los artefactos y a la distorsión de la arquitectura, secundarios al aplastamiento propiciado por la pinza. Como efectos no deseados, pueden producirse neumotórax (5%) y hemoptisis en algunos casos.
3. Biopsia transbronquial con criosonda o criobiopsia (CTB)
La criobiopsia transbronquial surge con el objetivo de mejorar el rendimiento diagnóstico de la BTB. Gracias a la congelación entre -75 °C y -89 °C durante 5 o 6 segundos del tejido sobre el que se aplica la sonda, se consigue obtener muestras de mayor dimensión (entre 40 y 50 mm2) y con una histología mejor preservada, lo que se traduce en un aumento de la rentabilidad diagnóstica. Por primera vez, en la actualización de la guía de práctica clínica de la ATS/ERS/JRS/ALAT de la FPI y de la fibrosis pulmonar progresiva se menciona la criobiopsia como una alternativa válida al estándar de oro hasta el momento, que ha sido la biopsia pulmonar quirúrgica10 (Figura 2), siempre
llevada a cabo en centros con experiencia en su realización. En diversos estudios, se ha medido la rentabilidad promedio de la técnica, concretamente en las enfermedades pulmonares intersticiales, que asciende a un 77,5%, lo que supone una importante mejora respecto a la BTB convencional. En cuanto a la seguridad y a las complicaciones, el neumotórax es la complicación más frecuentemente asociada, aunque su tasa varía considerablemente dependiendo del estudio analizado, desde menos de un 1% hasta un 30%. En un metanálisis que incluyó 15 ensayos con un total de 994 pacientes, la tasa promedio fue de un 10%. Los mismos resultados fueron confirmados en un metanálisis más reciente. Los factores de riesgo relacionados con esta complicación fueron una histología compatible con FPI, la reticulación fibrótica en la TCAR y la toma de biopsias cercana a la pleura. El sangrado es otra complicación común,8 que por lo general se controla vía endoscópica. Aunque no existe literatura específica al respecto, se consideran contraindicaciones absolutas la plaquetopenia por debajo de 50.000, las alteraciones en la coagulación y relativas, la hipertensión pulmonar o el deterioro grave de la función respiratoria (FVC <50% o DLCO < 35%).10
4. Ecobroncoscopia lineal (EBUS-TBNA)
Es una técnica endoscópica guiada bajo control ecográfico que permite la exploración y la punción de adenopatías mediastínicas e hiliares, así como de masas que contacten directamente con las paredes de las vías respiratorias de gran calibre (tráquea y bronquios principales). Para la obtención de muestras, se utiliza una aguja fina con aspiración (PAAF), lo que permite únicamente la obtención de muestras citológicas. Su uso en las neumopatías intersticiales se fundamenta en aquellas patologías que cursan con afectación de los ganglios linfáticos, como las micobacteriosis, los síndromes linfoproliferativos o la sarcoidosis, siendo en esta especialmente rentable.8
5. Biopsia pulmonar quirúrgica
La biopsia pulmonar por videotoracoscopia ha sido considerada el estándar de oro para el diagnóstico histopatológico del tipo específico de neumopatía intersticial y la mejor guía para el tratamiento.8,10 En un metanálisis en el que se incluyeron 43 estudios, la biopsia pulmonar presentó una rentabilidad diagnóstica del 93,5%, frente al 80,7% de la criobiopsia.10 No obstante, y a pesar de la diferencia, se considera que el impacto real de la técnica de la biopsia debe discernirse en el marco de una evaluación multidisciplinaria del caso. La tasa de mortalidad de la biopsia quirúrgica es del 1,7%, más del doble que la CTB. Esto, al igual que las complicaciones, puede verse influido por las comorbilidades del paciente, por el estado de inmunodepresión y por el grado de deterioro de la función pulmonar.
BIBLIOGRAFÍA
1. British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee, B Bradley, H M Branley, J J Egan, et al. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63(V):1-58.
2. Lopez C, Lopez A, De Benito E, Rodriguez JA. Capítulo 37: Neumopatías intersticiales: concepto, clasificación, manifestaciones clínicas y aproximación diagnóstica. Manual de Neumología y Cirugía Torácica, 4ª ed., 2021.
3. Denton CP, Wells AU, Coghlan JG. Mayor lung complications of systemic sclerosis. Nat Rev Rheumatol. 2018;14(9):511-527.
4. Du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: Test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184:1382-1389.
5. Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138-153.
6. Costabel U, Guzmán J. Bronchoalveolar lavage. En: King TE, Schwarz MI (eds.). Interstitial lung disease, 4ª ed., Hamilton: B.C. Decker, 2003; 114-133.
7. The BAL Cooperative Group Steering Committee. Bronchoalveolar lavage constituents in healthy individuals, idiopathic pulmonary fibrosis and selected comparison groups. Am RevRespirDis. 1990;141:169-202.
8. Duysinx B, Guiot J, Pellegrini I, et al. Techniques diagnostiques invasives des pneumopathies interstitielles diffuses. RevMedLiege. 2018;73(3):147-155.
9. Shim HS, Park MS, Park IK. Histopathologic findings of transbronchial biopsy in usual interstitial pneumonia. PatholInt. 2010;60:373-377.
10. Rodrigues I, Gomes RE, Coutinho LM, et al. Diagnostic yield and safety of transbronchial lung cryobiopsy and surgical lung biopsy in interstitial lung diseases: a systematic review and meta-analysis. EurRespir Rev. 2022;31(166):210280.
TABLAS Y FIGURAS
Tabla I. Manifestaciones sistémicas en las EPID

Tabla II. Criterios de NIU en TCAR

Tabla III. Patrones radiológicos más frecuentes en la TCAR en algunas EPID

Tabla IV. Características del lavado broncoalveolar en las principales EPID

Figura 1. Algoritmo sugerido para la evaluación analítica inicial del paciente con EPID. Tomado y modificado de T. Bahmer et al.: The use of auto-antibody testing in the evaluation of interstitial lung disease (ILD). A practical approach for the pulmonologist. Respiratory Medicine, 2016

Figura 2. Algoritmo diagnóstico en enfermedad pulmonar intersticial difusa. Tomado y modificado de: Cotiin V, Valenzuela C. Diagnostic approach of fibrosing interstitial lung diseases of unknown origin. Presse Med. 2020

MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
TÉCNICAS DIAGNÓSTICAS
INVASIVAS EN LA EPID
Autores
Martín Nikolai Sánchez Mitacc
Marta Hernández Argudo
Coordinador
Sergio Curi Chércoles
1. INTRODUCCIÓN
En muchas ocasiones, el diagnóstico de las enfermedades pulmonares intersticiales difusas (EPID) constituye un reto para los clínicos. En los casos en que la radiología no es diagnóstica tras una anamnesis y una exploración exhaustivas, o en que haya discrepancias clinico-radiológicas, las técnicas diagnósticas invasivas pueden ayudar para llegar a un diagnóstico certero.
Según las guías clínicas de la ERS/ATS publicadas en 2022, debemos recurrir a las técnicas diagnósticas invasivas en aquellos casos en que no se haya identificado por clínica o analítica (autoinmunidad, precipitinas, etc.) la posible causa de la EPID, o cuando una tomografía computarizada de alta resolución (TCAR) muestra un patrón distinto al de neumonía intersticial usual (NIU o PROBABLE NIU).¹
A continuación, describiremos las principales técnicas invasivas disponibles en la actualidad explicando en qué consisten, sus principales indicaciones, sus contraindicaciones y sus posibles complicaciones.
2. LAVADO BRONCOALVEOLAR (LBA)
Consiste en instilar entre 20 y 50 ml de suero fisiológico en bolos (volumen total de 150-300 ml) una vez enclavados en el bronquio escogido previamente, que se recoge
en varias alícuotas, idealmente de forma manual. El aspecto macroscópico puede orientar el diagnóstico diferencial, ya que, en condiciones normales, deberíamos obtener un líquido claro, muy similar al instilado. Recuperar alícuotas cada vez más hemorrágicas confirmaría, en un contexto clínico radiológico adecuado, el diagnóstico de hemorragia alveolar, mientras que un aspecto lechoso es característico de la proteinosis alveolar debido a su alto contenido lipoproteináceo. Es frecuente recuperar moldes de moco en pacientes que asocian bronquiectasias. El análisis microscópico nos permite obtener un estudio citológico de las células predominantes (neutrófilos, linfocitos, eosinófilos...), así como las subpoblaciones linfocitarias (CD3, CD4, CD8, cociente CD4/CD8), que refleja los mecanismos inmunitarios e inflamatorios que se están sucediendo a nivel alveolar e intersticial.² En los sujetos sanos, el LBA está constituido por un 90% de macrófagos, un 5-10% de linfocitos y poca cantidad de neutrófilos y eosinófilos. Dentro de los linfocitos T, distinguimos entre los CD4 y CD8 con una relación normal entre 1,5 y 2,1.
Por un lado, hay una serie de patologías en las que los hallazgos del LBA nos permiten realizar el diagnóstico directamente, siendo orientativo en otras.² (Tabla I)
Los hallazgos más característicos de cada enfermedad se detallan en las siguientes tablas.
• Enfermedades según su celularidad en el LBA (Tabla II)
• Diagnóstico diferencial según relación CD4/CD8 (Tabla III)
Ante la sospecha de sarcoidosis, un cociente CD4/CD8 > 3,5 es muy sugestivo de la misma. Por otro lado, en el caso de sospechar una neumonitis por hipersensibilidad, una linfocitosis >30% es también muy sugestiva.³
En el caso de una EPID de inicio agudo (definida como aparición de síntomas <4 semanas en pacientes sin neumopatía previa, sin SDRA ni factor de riesgo claro como un shock séptico o un traumatismo), así como en la exacerbación de EPID, el LBA también tiene un papel importante, ya que nos permite descartar causas infecciosas y orienta al diagnóstico de la EPID según la celularidad (predominio linfocitario, hemorragia alveolar, eosinofilia…). Por ejemplo, una linfocitosis elevada junto con el hallazgo de células plasmáticas en un paciente con exposición de riesgo conocida puede ser diagnóstico de neumonitis por hipersensibilidad.4
3. BIOPSIA PULMONAR TRANSBRONQUIAL CONVENCIONAL CON PINZAS O FORCEPS (BPTBc)
Es una técnica endoscópica que nos permite obtener muestras del parénquima pulmonar mediante una pinza de biopsia. El objetivo principal de utilizar esta técnica en la EPID es obtener un diagnóstico definitivo evitando otros procedimientos más agresivos,
establecer un pronóstico o decidir el tratamiento. Hay algunas indicaciones específicas dentro de las EPID, y va a depender de la distribución y de la zona afectada. Las probabilidades aumentan si hay nódulos centrolobulillares, patrón en vidrio esmerilado o si existe afectación peribroncovascular y linfangítica.
Esta técnica tiene una amplia gama de indicaciones y su rendimiento diagnóstico es mayor en enfermedades granulomatosas (lo que la hace útil en sarcoidosis de estadios II-IV), linfangitis, daño alveolar difuso, algunas infecciones, proteinosis alveolar y neumonía eosinófila. Con menor frecuencia, puede ser diagnóstica en vasculitis, amiloidosis, linfangioleiomiomatosis (LAM) e histiocitosis pulmonar de células de Langerhans (HPCL). Por el contrario, sería de muy poca ayuda en el diagnóstico de enfermedades con afectación periférica al lobulillo pulmonar, o cuyo patrón morfológico requiera una visualización a poco aumento para determinar su arquitectura (patrón NIU), neumonía intersticial no especifica (NINE), neumonía intersticial descamativa (NID) o neumonía intersticial linfoidea (NIL).1,5
Es una técnica muy segura en la que se avanza el broncoscopio por el segmento elegido lo más distal posible, seguidamente se introduce una pinza de biopsia sin dientes a través del canal de trabajo hasta notar leve resistencia, se retira aproximadamente 1 cm, se abre la pinza, se avanza y cierra nuevamente, se extrae la pinza y se mantiene el broncoscopio enclavado para observar si hay sangrado, y se contiene la hemorragia en caso de presentarse.5
Un estudio demostró que en algunas entidades de enfermedad intersticial con lesiones localizadas o difusas en las que se realizó BPTBc, esta reportó un rendimiento diagnóstico del 55,2% en los pacientes con una lesión localizada y del 67% en aquellos con lesiones difusas. Está bien establecida la correlación entre el mayor número de muestras y su rendimiento, de modo que tomar más de cuatro biopsias aumentó el rendimiento del 52% a más del 70%.
El procedimiento puede ser ambulatorio. El porcentaje de complicaciones respiratorias, como neumotórax y hemorragia, suele ser variable entre las series, oscilando entre 1-6% y 1-4,4% respectivamente. Esto sugiere que la BPTBc es un procedimiento de diagnóstico útil, con una baja tasa de complicaciones.5
En pacientes con EPID fibrótica, exacerbación aguda y enfisema, aumenta la probabilidad de neumotórax, que es superior al 30% en algunas series. Las limitaciones de la prueba son el número de muestras, su pequeño tamaño (1-3 mm), la presencia de artefactos (pared bronquial, cartílago, coágulos, atelectasias), el aplastamiento por el fórceps y la poca presencia de tejido alveolar.
Según la guía de práctica clínica ATS/JRS/ALAT en neumonitis por hipersensibilidad (NH), la inclusión de BPTBc en el enfoque diagnóstico de estos pacientes incrementó
la probabilidad de llegar a un diagnóstico en comparación con un enfoque sin BPTBc (RR, 1,67; (95% CI, 1,21-2,30). Se reporta que, del total de muestras obtenidas en diferentes grupos estudiados (EPID, DAD y NH), la rentabilidad con BPTBc que permitió diagnóstico histopatológico en el grupo de EPID fue tan solo del 37% y que el rendimiento diagnóstico en el grupo de NH conocido no se pudo calcular; sin embargo, en el 41% de estos pacientes que se sometieron a BPTBc se confirmó que tenían NH. Hubo consenso general en que el rendimiento diagnóstico de BPTBc fue subóptimo, ya que solo la mitad de los procedimientos dieron como resultado un diagnóstico. La recomendación para pacientes con diagnóstico reciente de EPID, donde se incluye como diagnóstico diferencial NH no fibrótica, sugieren la realización de BPTBc, y si la sospecha es NH fibrótica, no hay recomendación a favor o en contra de BPTBc. La justificación se basa en la posibilidad de observar granulomas y que estos sean diagnósticos en la NH no fibrótica.6
4. CRIOBIOPSIA TRANSBRONQUIAL (CTB)
Consiste en la obtención de muestras parenquimatosas mediante una criosonda que se introduce a través del broncoscopio. Esta criosonda, una vez situada en el segmento bronquial de donde queremos obtener la muestra, permite congelar el parénquima y posteriormente extraerlo. Tal y como se muestra en la figura 1, y según las últimas guías ERS/ATS para el diagnóstico y tratamiento de la FPI y la EPID fibrosantes progresivas publicadas en el 2022, la criobiopsia pulmonar es una alternativa aceptable a la biopsia pulmonar quirúrgica en centros con la adecuada experiencia en la técnica.¹ Para un buen rendimiento diagnóstico, se deben obtener entre 3 y 5 muestras de tejido, obtenidas a más de 1 cm de la pleura para disminuir el riesgo de complicaciones.7 Con esto se obtiene una rentabilidad diagnóstica de entre el 50,6 y el 100%, frente al 25-65% que nos proporciona la biopsia transbronquial convencional.5 Gracias a esta técnica, se ha conseguido aumentar la rentabilidad diagnóstica, que es del 91% para la neumonitis por hipersensibilidad conocida o de alta sospecha y del 82% para el resto de EPID. Entre los pacientes con sospecha de NH o NH conocida en quienes se realizó un diagnóstico mediante CTB, se confirmó que el 100% (IC del 95%, 91,4-100%) tenían NH y ninguno tenía un diagnóstico alternativo de EPID o de no EPID.6
A pesar de sus ventajas en cuanto a rendimiento diagnóstico, para realizar la CTB se recomienda intubación orotraqueal con sedación profunda o anestesia general, así como el empleo de un balón de oclusión para detener un eventual sangrado y controlar la obtención de las muestras mediante fluoroscopia.5 Todo ello supone mayor complejidad y requiere más tiempo en comparación a las BPTBc. La técnica puede realizarse también de manera ambulatoria, tras permanecer de 3 a 4 horas en observación para descartar las principales complicaciones, que son el sangrado (1-12%) y el neumotórax (1,4-26%);5 por este motivo, se recomienda realizar una ecografía torácica al finalizar
el procedimiento. Un metanálisis llevado a cabo por la ERS/ATS halló que la incidencia global de neumotórax en la CTB es del 8%, y la de sangrado leve, del 30%.8 La incidencia de otras complicaciones, como las infecciosas, la mortalidad, el sangrado grave o el riesgo de exacerbación de EPID, es muy baja, cercana al 0%.8
Respecto a las contraindicaciones de la técnica, la edad no es una contraindicación absoluta, siempre que el paciente no tenga comorbilidades relevantes y no haya contraindicaciones para una anestesia general.7 Sin embargo, sí que se ha descrito que aquellos pacientes con peor función pulmonar, como capacidad vital forzada (FVC) <50%, capacidad de difusión de monóxido de carbono (DLco) <35% o sospecha de hipertensión pulmonar (presión arteria pulmonar por ecocardiografía >40-45 mmHg), tienen más riesgo de complicaciones, por lo que se consideran contraindicaciones relativas.7
Una de las principales limitaciones de la CTB se presenta a la hora de establecer un patrón histológico de NIU. Los criterios histológicos de NIU han sido establecidos en biopsia pulmonar quirúrgica (BPQ), aunque también pueden ser identificados en CTB. La presencia de pleura o septo será necesaria para el patrón de NIU, puesto que la distribución de la fibrosis es uno de los 4 requisitos. Por tanto, las criobiopsias que no contengan estas estructuras solo podrán establecer un patrón de «probable NIU». Sin embargo, la distribución parcheada y los focos de proliferación fibroblástica son fácilmente identificables mediante CTB.7No existen todavía guías clínicas que nos indiquen cuándo debemos obtener la muestra histológica mediante CTB o mediante BPQ. Sin embargo, existen documentos de consenso clínicos que nos ayudan a elegir una técnica u otra dependiendo de las características del paciente y del patrón radiológico que presenta en la TCAR. La tabla IV muestra una adaptación de la tabla propuesta por Castillo et al. en el 2020.7
5. ECOBRONCOSCOPIA (ENDOBRONCHIAL ULTRASOUND: EBUS) – TBNA (TRANSBRONCHIAL NEEDLE ASPIRATION)
Técnica que permite explorar los ganglios mediastínicos e hiliares mediante un broncoscopio que tiene incorporado una sonda de ecografía. Aunque se utiliza principalmente en el diagnóstico y el estadiaje del cáncer de pulmón, también puede ser útil para el diagnóstico de algunas EPID, principalmente en aquellas que presentan afectación ganglionar. Es de especial interés en la sarcoidosis, ya que la presencia de granulomas no necrotizantes en los ganglios nos permitirá hacer el diagnóstico de esta enfermedad, pudiéndonos ahorrar así la BPTB o la BPQ.9 El diagnóstico de sarcoidosis requiere la presencia de pruebas compatibles, características clínicas y radiológicas, junto con la demostración de granulomas de células epitelioides no caseificantes, después de la exclusión de otras causas conocidas de inflamación granulomatosa. El muestreo de tejido suele ser útil, y se comienza con el método de menor riesgo y menos invasivo. Sin embargo, la mayoría de los casos nuevos de sarcoidosis carecen de estos hallazgos
y se realizan pruebas invasivas desde el inicio. Las técnicas endoscópicas usadas son BPTB, biopsia transbronquial con aguja (TBNA) y EBUS-TBNA. Se ha establecido la superioridad del muestreo de ganglios linfáticos guiado por EBUS sobre la BPTB en la sarcoidosis pulmonar,9,10 pero se sugiere que la combinación de técnicas aumenta el rendimiento del diagnóstico.
6. CRIOBIOPSIA MEDIASTÍNICA TRANSBRONQUIAL GUIADA POR ECOBRONCOSCOPIA (CRYOEBUS)
De interés creciente es la técnica de criobiopsia mediastínica transbronquial, en la que se combina la EBUS para localizar los ganglios o lesiones a estudiar, pero en la que las muestras se obtienen gracias a una sonda de criobiopsia, lo que permite obtener muestras histológicas más representativas. Esta técnica podría resultar especialmente útil en el diagnóstico diferencial de los procesos linfoproliferativos con afectación ganglionar mediastínica y/o hiliar en los que con la EBUS-TBNA hay un bajo rendimiento diagnóstico.11 Recientemente, un grupo español ha introducido un método sencillo de cuatro pasos (procedimiento Ariza-Pallarés) que prescinde del bisturí de alta frecuencia (de elevado coste) y utiliza una aguja con punta de corona de calibre 22G para crear un «túnel» (siempre bajo control ecográfico) por el que a continuación se introduce la criosonda de 1,1 mm. La técnica CRYOEBUS es segura, por lo que el procedimiento se realiza en la sala de broncoscopias, con sedación consciente12 y de forma ambulatoria; si no hay complicaciones, el paciente puede ser dado de alta en el periodo de observación posterior al procedimiento.
7. BIOPSIA PULMONAR QUIRÚRGICA (BPQ)
La biopsia pulmonar por VATS (videoasistida) es considerada el estándar de oro para obtener tejido en pacientes con EPID no diagnosticada (Figura 2). Las indicaciones de biopsia pulmonar quirúrgica se realizan en el comité multidisciplinar, siendo actualmente la biopsia pulmonar videoasistida (VATS) la técnica más utilizada, elegida por encima de la biopsia a cielo abierto por toracotomía, ya que tiene el mismo rendimiento diagnóstico, pero con menor morbimortalidad.5
El procedimiento habitual requiere anestesia general y ventilación uni-pulmonar, consiguiendo el bloqueo del pulmón que va a ser manipulado. Para ello, se realiza una intubación bronquial selectiva mediante la colocación de un tubo de doble luz.5 Los sitios de biopsia para EPID deben seleccionarse cuidadosamente para evitar complicaciones postoperatorias como las que se observan en pacientes sometidos a biopsia del vértice del pulmón, que tuvieron una tasa significativamente mayor de neumotórax. Se debe evitar la biopsia en las zonas de panalización por su fibrosis establecida y su marcada distorsión arquitectural, siendo de elección las zonas de vidrio esmerilado. Se ha de-
mostrado la presencia de distintos patrones histológicos en los diferentes segmentos y lóbulos pulmonares. Esta heterogeneidad hace que se aconseje la biopsia de más de un lóbulo, lo que, sin embargo, aumenta el tiempo, el coste y las complicaciones. El tamaño de la muestra considerada como representativa para el diagnóstico es de 2 cm aproximadamente.5
La biopsia pulmonar por VATS se contraindica de forma absoluta en pacientes con antecedentes de IAM, angor inestable, arritmia cardiaca no controlada, antecedentes de neumonectomía, enfermedad avanzada asociada a uso de ventilación mecánica y presencia de adherencias pleurales que impidan el colapso pulmonar.5
Hay datos muy variables respecto a la morbimortalidad (0-34%) debido a la diferencia de los objetivos de investigación, la selección de casos (FPI versus otras EPID), el tipo de cirugía (cielo abierto versus VATS) y los resultados informados (mortalidad a 30 días y «postoperatoria»).13 Las complicaciones postoperatorias de la BPQ son un problema grave en los pacientes con EPID. Se ha informado que los pacientes con una DLCO <50% del valor previsto, que necesitan ventilación mecánica (VM) o que tienen inmunodeficiencia o hipertensión pulmonar, tienen un mayor riesgo de muerte después de una BPQ, por lo que es necesaria una buena elección de los pacientes. Se ha reportado mortalidad hospitalaria de 1,7% para los procedimientos electivos y del 16% para los procedimientos no electivos.13
La exacerbación aguda de la EPID tiene, generalmente, un pronóstico grave, y después de una BPQ es una de las principales causas de mortalidad. El procedimiento toracoscópico videoasistido (VATS) típico se realiza bajo anestesia general con ventilación mecánica, que se cree que pudiera desencadenar o empeorar la lesión pulmonar aguda y el síndrome de dificultad respiratoria aguda (SDRA). La VATS no intubada (AVATS) es otra opción para la biopsia pulmonar en la EPID, puesto que tiene menos complicaciones postoperatorias y tiempos de recuperación más rápidos al no utilizar relajantes musculares ni el tubo con tubo de doble luz, lo que evita las lesiones por ventilación con presión positiva.13
8. PROPUESTA DE ALGORITMO
Se ha demostrado la concordancia entre la interpretación diagnóstica de las muestras de CTB y las muestras de BPQ. Un estudio demostró una concordancia del 70,8%, que aumentó al 76,9% después del CMD. El análisis post hoc sugirió que la concordancia de CTB con BPQ mejora al tomar más muestras.14 Por el contrario, otros estudios reportan una baja concordancia diagnóstica (Figura 2). 15
BIBLIOGRAFÍA
1. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205:18-47.
2. Jara-Palomares L, Martín-Juan J, Gómez-Izquierdo L, Cayuela-Domínguez A, Rodríguez-Becerra E, Rodríguez-Panadero F. Hallazgos en el lavado broncoalveolar de pacientes con enfermedad pulmonar intersticial difusa. Estudio de una cohorte prospectiva de 562 pacientes. Archivos de Bronconeumología. 2009;45(3):111-117, ISSN 0300-2896, https:// doi.org/10.1016/j.arbres.2008.04.005.
3. Meyer KC. An Official American Thoracic Society Clinical Practice Guideline: The Clinical Utility of Bronchoalveolar Lavage Cellular Analysis in Interstitial Lung Disease. Am J Respir Crit Care Med. 2012:185(9):1004-1014.
4. Meyer KC, Raghu G. Bronchoalveolar lavage for the evaluation of interstitial lung disease: is it clinically useful? European Respiratory Journal. 2011;38(4):761-769; DOI: 10.1183/09031936.00069509.
5. Jimenez-Ruiz CA, Peces-Barba G, Moreno Balsalobre R, Plaza Moral V. 4ª edición de Manual SEPAR.
6. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Diagnosis of Hypersensitivity Pneumonitis in Adults: An Official ATS/JRS/ALAT Clinical Practice Guideline. American Journal of Respiratory and Critical Care Medicine. 2020;202:36-69.
7. Castillo D, Sánchez-Font A, Pajares V, Franquet T, Llatjós R, Sansano I, Sellarés J, Centeno C, Fibla JJ, Sánchez M, Ramírez J, Moreno A, Trujillo-Reyes JC, Barbeta E, Molina-Molina M, Torrego A. Propuesta multidisciplinar respecto al algoritmo diagnóstico de la fibrosis pulmonar idiopática: papel de la criobiopsia transbronquial. Archivos de Bronconeumología. 2020;56(2):99-105, ISSN 0300-2896, https://doi.org/10.1016/j.arbres.2019.07.001.
8. Kheir F, Uribe Becerra JP, Bissell B, et al. Transbronchial Lung Cryobiopsy in Patients with Interstitial Lung Disease: A Systematic Review. Ann Am Thorac Soc. 2022;19(7):1193-1202. doi:10.1513/AnnalsATS.202102-198OC.
9. Crouser ED, Maier LA, Wilson KC, Bonham CA, et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. American Journal of Respiratory and Critical Care Medicine. 2020;201(8).
10. Agarwal R, Aggarwal AN, Gupta D. Efficacy and safety of conventional transbronchial needle aspiration in sarcoidosis: a systematic review and meta-analysis. Respir Care. 2013;58(4):683-693. doi: 10.4187/respcare.02101. PMID: 23050747.
11. Botana-Rial M, Lojo-Rodriguez R, Leiro-Fernandez V, et al. Is the diagnostic yield of mediastinal lymph node cryobiopsy (cryo-EBUS) better for diagnosing mediastinal node involvement compared to endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA)? A systematic review. Respiratory Medicine. 2023;218.
12. Ariza-Prota MA, Pérez-Pallarés J, et al. Transbronchial Mediastinal Cryobiopsy in the Diagnosis of Mediastinal Lymph Nodes: A Case Series – How to do it. Archivos de Bronconeumología, 2022;58(10):718-721.
13. Hutchinson JP, Fogarty AW, McKeever TM, Richard B. Hubbard. In-Hospital Mortality after Surgical Lung Biopsy for Interstitial Lung Disease in the United States 2000 to 2011. J Respir Crit Care Med. 2016;193(10):1161-1167.
14. Troy LK, Grainge C, Corte TJ, Williamson JP, Vallely MP, Cooper WA, et al.; Cryobiopsy versus Open Lung Biopsy in the Diagnosis of Interstitial lung Disease Alliance (COLDICE) Investigators. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir Med. 2020;8:171-181.
15. Romagnoli M, Colby TV, Berthet J-P, Gamez AS, Mallet J-P, Serre I, et al. Poor concordance between sequential transbronchial lung cryobiopsy and surgical lung biopsy in the diagnosis of diffuse interstitial lung diseases. Am J Respir Crit Care Med. 2019;199:1249-1256.
16. Raj R, Raparia K, Lynch DA, Brown KK. Surgical Lung Biopsy for Interstitial Lung Diseases. Chest. 2017;151(5):1131-1140. doi: 10.1016/j.chest.2016.06.019. Epub 2016 Jul 26. PMID: 27471113.
TABLAS Y FIGURAS
Tabla I. Valor del BAL en las diferentes EPID.² Hallazgos en el lavado broncoalveolar de pacientes con enfermedad pulmonar intersticial difusa. Estudio de una cohorte prospectiva de 562 pacientes. Archivos de Bronconeumología. 2009;45(3):111-117, ISSN 0300-2896
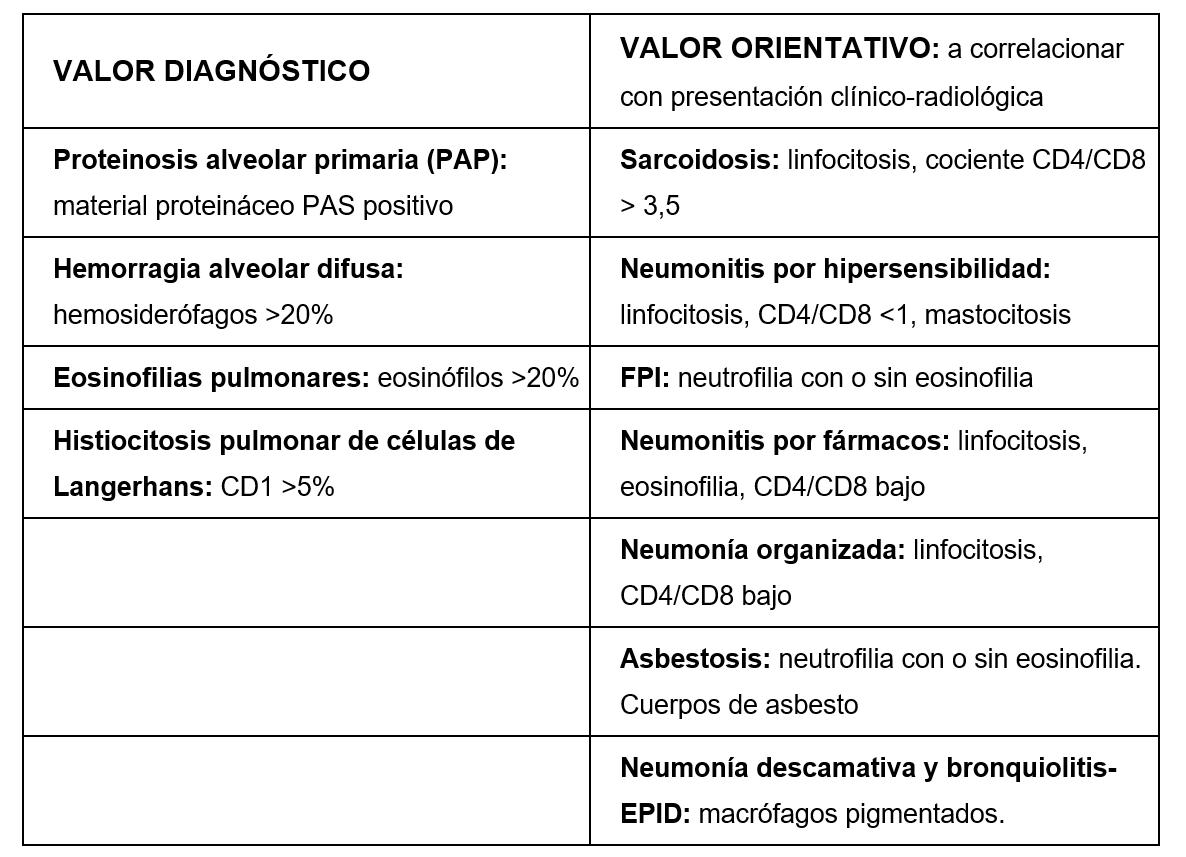
Tabla II. Adaptación de la tabla de «Summary of BAL cellular patterns in normal/ healthy adults nonsmokers and in patients with common interstitial lung diseases».³ An Official American Thoracic Society Clinical Practice Guideline: The Clinical Utility of Bronchoalveolar Lavage Cellular Analysis in Interstitial Lung Disease. Am J Respir Crit Care Med. 2012;185(9):1004-1014

Tabla III. Adaptación de la tabla de «Summary of BAL cellular patterns in normal/ healthy adults nonsmokers and in patients with common interstitial lung diseases».³ An Official American Thoracic Society Clinical Practice Guideline: The Clinical Utility of Bronchoalveolar Lavage Cellular Analysis in Interstitial Lung Disease. Am J Respir Crit Care Med. 2012;185(9):1004-1014

Tabla IV. Adaptación de la Tabla «Factores a considerar en cada paciente y centro a la hora de decidir entre criobiopsia transbronquial y biopsia pulmonar quirúrgica».5 Propuesta multidisciplinar respecto al algoritmo diagnóstico de la fibrosis pulmonar idiopática: papel de la criobiopsia transbronquial. Archivos de Bronconeumología. 4ª edición de Manual SEPAR

1. Algoritmo diagnóstico de la FPI según las guías ERS/ATS 2022

Figura
Figura 2. Sugerencia de algoritmo para el uso de técnicas invasivas en EPID.
Adaptación del algoritmo "Suggested algorithm outlining the role of surgical lung biopsy in interstitial lung disease" de Rishi Raj, Kirtee Raparia16 .

BPQ: Biopsia pulmonar quirúrgica, TCAR: Tomografía de alta resolución, PFR: Prueba funcional respiratoria
CMD: Comitè multidisciplinar, CTD: Enfermedad del tejido conectivo, FPI: Fibrosis pulmonar idiopática, NH: Neumonitis por hipersensibilidad, BPTBc: Biopsia pulmonar transbronquial convencional, BAL: Lavado broncoalveolar, CTB: Criobiopsia transbronquial, EBUS: Ultrasonido endobronquial.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
FIBROSIS PULMONAR IDIOPÁTICA
Autoras
María Churruca Arróspide
Elisa Martínez Besteiro
Coordinadora
Claudia Valenzuela
1. DEFINICIÓN
La fibrosis pulmonar idiopática (FPI), la forma más común de enfermedad pulmonar intersticial fibrosante, es de naturaleza crónica y progresiva. Tiene un pronóstico desfavorable y una media de supervivencia de 3 a 5 años desde el diagnóstico. No existe aún una causa identificada, por lo que se denomina idiopática.¹ Su diagnóstico, que tiene importantes implicaciones terapéuticas y pronósticas, se basa en la discusión multidisciplinar e integra la información clínica, radiológica e histológica -cuando está disponible-.²
2. EPIDEMIOLOGÍA. INCIDENCIA Y PREVALENCIA
La incidencia y la prevalencia de la FPI van en aumento y varían en función de la región y de la población estudiadas. Se estima una incidencia global de 1-13 casos por cada 100.000 habitantes y una prevalencia de 3-45 casos por cada 100.000 habitantes. En Europa, la incidencia y la prevalencia de la enfermedad oscilan entre 1-9 casos/100.000 habitantes y 10-40 casos/100.000 habitantes, respectivamente.²
La incidencia de la enfermedad aumenta con la edad y afecta mayoritariamente a varones entre la sexta y la séptima décadas de vida. En muy raras ocasiones, puede debutar en forma de agudización o en pacientes menores de 50 años, en cuyo caso siempre deberán sospecharse formas familiares de la enfermedad.¹
3. PRESENTACIÓN CLÍNICA
Debe considerarse el diagnóstico de FPI en individuos que presenten un cuadro clínico insidioso y progresivo de disnea de esfuerzo, tos, crepitantes teleinspiratorios bibasales en la exploración física y, en ocasiones, acropaquias asociadas.¹
El diagnóstico de la FPI requiere la exclusión de otras causas alternativas de enfermedad pulmonar intersticial (EPID), especialmente las secundarias a conectivopatías (ETC) y las relacionadas con exposiciones específicas.3
Es fundamental realizar una historia clínica detallada que recoja los antecedentes de posible exposición ambiental, a drogas o a fármacos neumotóxicos, y que ayude al despistaje de ETC. Aunque en muchas ocasiones tiene una implicación incierta en el proceso diagnóstico, es importante identificar la exposición a antígenos clínicamente significativos que hagan sospechar la posibilidad de neumonitis por hipersensibilidad fibrótica.4
HISTORIA CLÍNICA DETALLADA: ASPECTOS A TENER EN CUENTA4
- Datos generales de los síntomas respiratorios: Inicio, duración y gravedad.
- Despistaje de ETC: ¿Existen signos o síntomas extrapulmonares sugestivos de ETC? ¿Existen datos de autoinmunidad serológicos? Para responder a esta última pregunta, debe solicitar analítica completa que incluya panel de autoinmunidad.
- ¿Existe exposición clínicamente relevante a algún antígeno inhalado potencialmente relacionado con neumonitis por hipersensibilidad? Debe investigarse la duración de la exposición, la intensidad y la temporalidad (causa-efecto).
- Animales domésticos: especialmente aves.
- Exposiciones laborales: debe interrogarse acerca de trabajos con exposición a asbesto u otros neumotóxicos.
- Exposiciones domiciliarias: humedades, sistemas de aire acondicionado, piscinas, pozos, etc.
- Tratamientos recibidos: hierbas, vitaminas, suplementos, fármacos potencialmente neumotóxicos o drogas recreativas.
- Susceptibilidad genética: debe interrogarse acerca de antecedentes familiares de fibrosis pulmonar u otras EPID, cirrosis criptogenética, discrasias hematológicas, enfermedades autoinmunes, etc.
4. FACTORES DE RIESGO
Aunque la FPI es considerada una enfermedad de causa desconocida, se sabe que existen múltiples factores genéticos y ambientales que están asociados a su desarrollo.²
DEMOGRÁFICOS
- Edad: la incidencia y la prevalencia de la FPI aumentan con la edad, siendo más frecuente a partir de 60 años.
- Sexo masculino: en los registros y estudios disponibles, los hombres representan el 70% del total de pacientes con diagnóstico de FPI, si bien es cierto que se han reportado sesgos por género en muchos de ellos.
GENÉTICOS
- Relacionados con la defensa pulmonar del huésped:
∙ El gen MUC5B codifica para la proteína mucina 5B, una glicoproteína presente en la vía aérea que interviene en el aclaramiento mucociliar y la respuesta inmune innata. Si bien el mecanismo es aún poco conocido, el polimorfismo rs35705950 de este gen lleva a un aumento de la expresión de la proteína mucina 5B y, en consecuencia, a una disminución del aclaramiento mucociliar.
∙ Mutación en el gen de la proteína TOLL (TOLLIP): se ha demostrado que los individuos que presentan una expresión disminuida de estos receptores son más susceptibles al desarrollo de fibrosis pulmonar.
- Acortamiento telomérico: algunas variantes en los genes implicados en el mantenimiento de los telómeros (TERT, TERC, PARN, RTEL1, DKC Y TINF21) conllevan un acortamiento de los mismos y una senescencia celular acelerada que parece jugar un papel fundamental en los procesos de reparación del daño epitelial. Se asocian con formas de fibrosis pulmonar tanto familiares como esporádicas.
- Alteraciones de los genes implicados en la formación del surfactante (SFTPC, SFTPA2 y ABCA3): se encuentran en formas familiares de la enfermedad.
- Alteraciones de genes implicados en la adhesión celular e integridad epitelial (DSP y DPP9).
- Alteraciones de la señalización fibrótica (AKAP13; polimorfismo rs62025270).
AMBIENTALES
- Tabaquismo.
- Exposición laboral a neumotóxicos como polvo, asbesto, polvo de metal o madera.
- Contaminación ambiental: se ha relacionado con un aumento tanto de la incidencia como de la mortalidad en la FPI.
COMORBILIDADES
- Enfermedad por reflujo gastroesofágico (ERGE): varios estudios observacionales han notificado una asociación entre la presencia de ERGE y el desarrollo de fibrosis pulmonar, aunque sin llegar a establecer una relación causal definitiva.
- Apnea obstructiva del sueño (AOS): es una comorbilidad muy a menudo relacionada con la FPI; su presencia se ha propuesto como posible causa del desarrollo de fibrosis.
- Disbiosis pulmonar: la alteración de la microbiota pulmonar podría jugar un papel en el desarrollo de la fibrosis, aunque todavía no disponemos de clara evidencia al respecto.
- Infecciones virales: especialmente de la familia de los herpes virus, podrían jugar un papel en el desarrollo de la enfermedad y en las agudizaciones de la misma.
ANTECEDENTES FAMILIARES
- La existencia de antecedentes familiares de fibrosis pulmonar constituye un importante factor de riesgo para el desarrollo de la misma, así como para una mayor progresión y un peor pronóstico de la enfermedad. Los miembros de una misma familia pueden presentar hallazgos radiológicos, clínicos e histopatológicos diferentes no siempre compatibles con el diagnóstico de FPI. El término «fibrosis pulmonar familiar» engloba un complejo grupo de enfermedades fibróticas que resultan de factores heredados determinantes.²
5. DIAGNÓSTICO
El proceso diagnóstico y el manejo de los pacientes con EPID supone un reto constante en la práctica clínica habitual. Ha sido ampliamente demostrado que la valoración multidisciplinar de estos pacientes mejora el acuerdo interobservador tanto radiológico como anatomopatológico y, por tanto, constituye el estándar de oro en el diagnóstico de los pacientes con FPI1,5 (Figura 1).
En 2018, las sociedades ATS/ERS/JRS/ALAT definieron cuatro patrones radiológicos en el abordaje diagnóstico de la FPI (Tabla I).
La panalización consiste en espacios quísticos aéreos, con un diámetro de 3 a 10 mm, dotados de una pared bien definida y que suelen localizarse formando hileras subpleurales en las regiones basales y posteriores del pulmón, lo que en ocasiones dificulta el diagnóstico diferencial con otros hallazgos radiológicos a nivel subpleural, como el enfisema paraseptal o las bronquiolectasias por tracción.¹ La identificación de panal en el TCAR de tórax influye tanto en el diagnóstico como en el pronóstico de la enfermedad y constituye el resultado final de un continuo proceso de remodelado que va desde la
aparición de bronquiectasias por tracción hasta la transformación en quistes de panal por colapso de los septos fibrosados y dilatación de la vía aérea terminal. El patrón radiológico de NIU es un hallazgo típico de la FPI, pero no exclusivo de la misma, ya que puede encontrarse también en otras patologías, como la neumonitis por hipersensibilidad fibrótica, las EPID secundarias a conectivopatías o las derivadas de exposiciones laborales. La fibroelastosis pleuroparenquimatosa puede encontrarse en un 6-10% de los pacientes con FPI y se asocia a un rápido deterioro de la función pulmonar y a un mayor riesgo de neumotórax y neumomediastino, con el consiguiente peor pronóstico.6
Recientemente, con las últimas guías de la ATS/ERS/JRS/ALAT de 2022, se ha focalizado la atención en los patrones radiológicos de neumonía intersticial usual (NIU) y de probable NIU,6 que presentan un curso clínico y aproximación diagnóstica similares, y tienen una correlación altamente específica con un patrón de NIU histológico.
Por tanto, para el diagnóstico de FPI, después de descartar otras causas, en pacientes con un patrón radiológico de NIU se desaconseja la realización de técnicas diagnósticas invasivas para obtener muestras histológicas, al igual que en los pacientes con patrón radiológico de probable NIU que tengan un contexto clínico adecuado. Por otro lado, en los casos en que no se pueda establecer el diagnóstico en discusión multidisciplinar en base a los hallazgos clínicos y radiológicos (patrón indeterminado o alternativo para NIU), se deberá obtener de una muestra histológica para integrar dichos resultados en la discusión multidisciplinar6 (Figura 1).
Originalmente, los patrones histológicos que caracterizan a la FPI se describieron en base a hallazgos en muestras de biopsia pulmonar abierta o quirúrgica. Sin embargo, la biopsia transbronquial (BTB) por criobiopsia se ha planteado como una buena alternativa para el diagnóstico anatomopatológico de la FPI.¹ Respecto a esto, una revisión reciente de los estudios publicados hasta ahora ha permitido a las sociedades de patología respiratoria emitir una recomendación condicional a favor de la criobiopsia como una alternativa aceptable a la biopsia pulmonar quirúrgica, por los motivos que se enumeran a continuación.
- Tiene buena rentabilidad diagnóstica, mayor del 80% en combinación con la discusión en comité multidisciplinar. Esta rentabilidad puede aumentar hasta el 85% cuando se toman 3 o más muestras de tejido, y disminuye al 77% cuando se toman menos de este número.
- Presenta buen perfil de seguridad, siempre y cuando se realice en centros expertos y bajo protocolos estandarizados. La incidencia de neumotórax se sitúa en torno al 9%, y en torno al 30% en el caso del sangrado. No obstante, el sangrado grave, la mortalidad periprocedimiento, las exacerbaciones, las infecciones respiratorias y la fuga persistente son complicaciones rara vez encontradas.
- Es fundamental seleccionar al paciente adecuado considerando las siguientes contraindicaciones por su asociación con una elevada tasa de eventos adversos:
∙ Deterioro grave de la función pulmonar (FVC < 50%, DLco < 35%).
∙ Hipertensión pulmonar moderada o grave (presión sistólica en la arteria pulmonar >40 mmHg).
∙ Riesgo de sangrado no corregible.
∙ Hipoxemia significativa (pO2 55-60 mmHg).6,7
En el último consenso de la ATS/ERS/JRS/ALAT de 2022, los hallazgos histopatológicos característicos de los patrones de NIU y probable NIU fueron revisados y confirmados.1,5 El patrón histológico de NIU se caracteriza por desestructuración del parénquima pulmonar, fibrosis densa y parcheada de distribución predominantemente subpleural y/o paraseptal y el hallazgo típico de focos fibroblásticos. El patrón histológico de probable NIU se define, bien por la presencia única de panalización, bien por datos histológicos de patrón de NIU, sin llegar a ser definitivos, y en ausencia de otros hallazgos que sugieran otras alternativas diagnósticas.
Funcionalmente, la FPI se caracteriza por una alteración ventilatoria restrictiva con disminución de la capacidad vital forzada (FVC) y la capacidad pulmonar total (TLC), junto con un descenso de la capacidad de difusión de monóxido de carbono (DLco). Los valores espirométricos pueden ser normales en fases iniciales de la enfermedad.3
Por tanto, según las últimas guías publicadas,5 se puede establecer el diagnóstico de FPI cuando se cumplan los siguientes criterios:
- Exclusión de otras causas de EPID (exposición doméstica y/o ocupacional, conectivopatías, toxicidad por fármacos o drogas), y al menos uno de los siguientes: Patrón radiológico de NIU o probable NIU, en un contexto clínico adecuado y tras valoración del caso por un comité multidisciplinar.
- Combinación de patrón radiológico e histopatológico compatible en los pacientes en que se ha obtenido muestra de biopsia pulmonar (Tabla II). En la actualización de las guías, la combinación de un patrón radiológico sugestivo de una etiología alternativa con un patrón histológico de probable NIU se considera indeterminado para FPI, en lugar de no-FPI, en base a que la evolución de estos casos en ocasiones es muy similar a la de los pacientes con FPI (Tabla II).
6. AGUDIZACIÓN DE FPI
Una agudización o exacerbación de FPI² es un evento agudo de extrema relevancia en su historia natural. Inicialmente, se definió como un empeoramiento clínico agudo (de un mes de duración aproximadamente) y aparición de infiltrados radiológicos nuevos, que requería la exclusión de otras causas desencadenantes, como infecciones o aspi-
raciones de contenido gástrico. Posteriormente, en 2016 se actualiza la definición: se considera exacerbación a un deterioro respiratorio agudo, clínicamente significativo y asociado a infiltrados alveolares bilaterales y/o consolidaciones de nueva aparición en TCAR, de naturaleza no cardiogénica. Además, se admite que una exacerbación aguda además de «idiopática», pueda ser «desencadenada» por agentes externos o «triggers», tales como procesos infecciosos o broncoaspirativos, fármacos e intervenciones quirúrgicas, que generan un daño pulmonar agudo en muchas ocasiones indiferenciable de otros procesos idiopáticos.
Histológicamente, los hallazgos clínicos y radiológicos se correlacionan con un patrón de daño alveolar difuso o, menos probable, con un patrón de neumonía organizada encontrada en zonas de pulmón preservadas y alejadas de las regiones más fibróticas.
La incidencia de exacerbación reportada es muy variable. Se cree que aproximadamente el 5-10% de los pacientes con diagnóstico de FPI presentará una agudización anual, que será más común en aquellos con enfermedad avanzada y con mayor deterioro funcional.
Factores de riesgo de exacerbación aguda de FPI:
- Valores bajos de FVC o deterioro reciente de la misma.
- DLco <40% al diagnóstico.
- Caída de la distancia recorrida en el test de la marcha de los 6 minutos.
- Coexistencia de hipertensión pulmonar.
- Presencia de hipoxemia previa.
La exacerbación de FPI conlleva importantes implicaciones pronósticas. La mortalidad intrahospitalaria puede alcanzar el 50%, según los datos reportados por algunas series, con una media de supervivencia tras la agudización de 3-4 meses.
7. TRATAMIENTO
FÁRMACOS ANTIFIBRÓTICOS
Disponemos actualmente de dos fármacos antifibróticos aprobados para el tratamiento de la FPI8,9,11 (Tabla III).
Una vez establecido el diagnóstico de FPI, debe iniciarse el tratamiento antifibrótico tan pronto como sea posible. Podría plantearse una actitud expectante en pacientes asintomáticos o con mínima repercusión funcional, pero siempre valorando los riesgos y beneficios de dicha actitud. En cuanto a la elección del antifibrótico, se planteará de forma individual en función de riesgos, medicación concomitante y antecedentes del paciente9 (Figura 2).
8. PROGRESIÓN DE FPI Y CRITERIOS DE DERIVACIÓN PARA
TRASPLANTE
El curso clínico de la FPI es impredecible y muy variable de un paciente a otro. Algunos pacientes presentan una evolución lenta con pérdida progresiva de la función pulmonar, otros alternan periodos de estabilidad clínica con rápidos deterioros en contexto de exacerbaciones agudas, y un tercer grupo de pacientes pueden presentar progresión acelerada desde el diagnóstico y pronóstico infausto a corto-medio plazo. En cualquier caso, se trata de una enfermedad progresiva por naturaleza, entendiendo progresión como una disminución de la FVC, un empeoramiento de los síntomas respiratorios o una disminución de la capacidad de ejercicio, con el deterioro en la calidad de vida de los pacientes que ello conlleva.12 En este sentido, tal y como se detalla en la figura 2, la valoración funcional y radiológica supone una herramienta indispensable para el seguimiento de la evolución de la FPI, y debe hacer considerar la posibilidad de inclusión en ensayos clínicos independientemente del estadio de la enfermedad, así como la necesidad de derivación a una unidad de trasplante pulmonar.
La FPI es la primera causa de trasplante pulmonar entre las enfermedades respiratorias crónicas. Según los datos del registro de la Organización Nacional de Trasplantes (ONT), supuso el 38% de los trasplantes realizados de 2001 a 2021. En algunos pacientes, puede mejorar la calidad de vida y prolongar la supervivencia (50% a los 5 años del trasplante pulmonar).10 La derivación precoz de pacientes con un patrón radiológico y/o histológico típico o de probable NIU, en el momento del diagnóstico e incluso del inicio del tratamiento antifibrótico, es fundamental para evitar retrasos en la indicación del trasplante. Las necesidades de oxigenoterapia, así como la presencia de criterios de fibrosis progresiva apoyarán la derivación. En el caso de pacientes con diagnóstico de fibrosis pulmonar familiar, la recomendación de derivación precoz es aún más relevante, pues son pacientes en general afectos por manifestaciones extrapulmonares que requieren una evaluación más exhaustiva.
El momento de incluir a un paciente en lista de espera para trasplante pulmonar será individualizado y basado en los siguientes factores:
- Edad.
- Necesidad de oxigenoterapia en reposo o con los esfuerzos.
- Progresión de la enfermedad a los 6 meses (caída de la FVC de > 10%, de la DLCO > 10% y/o caída de la FVC> 5% asociada a progresión radiológica).
- Existencia de desaturación < 88% o una caída de > 50 metros en la distancia recorrida en el TM6M a los 6 meses.
- Asociación de hipertensión pulmonar (por ETT o cateterismo derecho).
- Ingresos por agudización, deterioro clínico o neumotórax.
Recientemente, se han publicado algunos estudios que han demostrado que la terapia antifibrótica no afecta a la cicatrización de la herida quirúrgica o de la anastomosis, ni tampoco aumenta el riesgo de sangrado, por lo que se mantendrán los fármacos hasta el mismo momento del trasplante.13
9. TEXTOS RECOMENDADOS
∙ CRITERIOS DE ISHLT DERIVACIÓN PARA TRASPLANTE PULMONAR:
Leard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, et al. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-79.
∙ GUÍA ESC/ERS 2022 SOBRE EL DIAGNÓSTICO Y TRATAMIENTO DE LA HIPERTENSIÓN PULMONAR
Humbert M, et al. 2022 ERC/ERJ Guidelines for diagnosis and treatment of Pulmonary Hypertention. Eur Respir J 2023 Jan 6; 61 (1) :2200879
BIBLIOGRAFÍA
1. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, et.al. American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018 Sep 1;198(5):e44-e68.
2. Podolanczuk AJ, Thomson CC, Remy-Jardin M, Richeldi L, Martinez FJ, Kolb M, Raghu, G. Idiopathic pulmonary fibrosis: state of the art for 2023. The European respiratory journal, 2023;61(4):2200957.
3. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378(19):18111823.
4. Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, Goldin JG, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018 Feb;6(2):138-153.
5. Chaudhuri N, Spencer L, Greaves M, Bishop P, Chaturvedi A, Leonard C. A Review of the Multidisciplinary Diagnosis of Interstitial Lung Diseases: A Retrospective Analysis in a Single UK Specialist Centre. J Clin Med. 2016;5(8):66.
6. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, el at. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022;205(9):18-47, https: //doi.org/10.1164/rccm.202202-0399ST.
7. Korevaar DA, Colella S, Fally M, Camuset J, Colby TV, Hagmeyer L, Hetzel J, et. al. European Respiratory Society guidelines on transbronchial lung cryobiopsy in the diagnosis of interstitial lung diseases. Eur Respir J. 2022;60(5):2200425. doi: 10.1183/13993003.004252022. PMID: 35710261.
8. Glass DS, Grossfeld D, Renna HA, Agarwala P, Spiegler P, DeLeon J, Reiss, AB. Idiopathic pulmonary fibrosis: Current and future treatment. The clinical respiratory journal, 2022;16(2), 84-96, https://doi.org/10.1111/crj.13466.
9. Xaubet A, Molina-Molina M, Acosta O, Bollo E, Castillo D, Fernandes-Fabrellas E, Rodríguez-Portal JA, Valenzuela C, Ancochea J. Normativa sobre el tratamiento farmacológico de la fibrosis pulmonar idiopática. Arch Bronconeumol. 2017;53(5):263-269.
10. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941-1952.
11. Ficha Técnica de OFEV®. Boehringer Ingelheim. Disponible en: https://cima.aemps.es/ cima/dochtml/ft/114979004/
12. Wuyts WA, Wijsenbeek M, Bondue B, Bouros D, Bresser P, Robalo Cordeiro C, Hilberg O, Magnusson J, Manali ED, Morais A, Papiris S, Shaker S, Veltkamp M, Bendstrup E. Idiopathic Pulmonary Fibrosis: Best Practice in Monitoring and Managing a Relentless Fibrotic Disease. Respiration. 2020;99(1):73-82. doi: 10.1159/000504763. Epub 2019 Dec 12. PMID: 31830755; PMCID: PMC6979429
13. Leard, L. E., Holm, A. M., Valapour, M., Glanville, A. R., Attawar, S., Aversa, M., Campos, S. V., Christon, L. M., Cypel, M., Dellgren, G., Hartwig, M. G., Kapnadak, S. G., Kolaitis, N. A., Kotloff, R. M., Patterson, C. M., Shlobin, O. A., Smith, P. J., Solé, A., Solomon, M., Weill, D., … Ramos, K. J. (2021). Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation, 40(11), 1349–1379. https://doi.org/10.1016/j.healun.2021.07.005
TABLAS Y FIGURAS
Tabla I. Patrones radiológicos

Adaptada de Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, el at. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022;205(9):18-47, https:// doi.org/10.1164/rccm.202202-0399ST.
Tabla II. Combinación de patrón radiológico e histopatológico compatible en los pacientes en que se ha obtenido muestra de biopsia pulmonar

Adaptada de Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, el at. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022;205(9):18-47, https:// doi.org/10.1164/rccm.202202-0399ST.
Tabla III. Fármacos antifibróticos aprobados para el tratamiento de la FPI8,9,11 (parte 1)

(Continua)
Tabla III. Fármacos antifibróticos aprobados para el tratamiento de la FPI8,9,11 (parte 2)

Figura 1. Algoritmo diagnóstico para la fibrosis pulmonar idiopática

Adaptada de Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, el at. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022;205(9):18-47, https:// doi.org/10.1164/rccm.202202-0399ST.
Figura 2. Algoritmo de manejo no farmacológico de la FPI

Adaptada de Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, el at. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022;205(9):18-47, https:// doi.org/10.1164/rccm.202202-0399ST.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
GENÉTICA
Y EPID. FIBROSIS PULMONAR FAMILIAR
Autores
Jaume Bordás Martínez
Juan Margallo Iribarnegaray
Coordinadora
Raquel Pérez Rojo
1. INTRODUCCIÓN
Existe una predisposición genética para el desarrollo de fibrosis pulmonar, tal y como sugiere el aumento de prevalencia de fibrosis pulmonar idiopática (FPI) entre los familiares de pacientes tambien afectos por enfermedad pulmonar intersticial difusa (EPID). El exponencial conocimiento en este campo hace suponer que a medida que acumulemos experiencia será una parte fundamental en el diagnóstico de estas enfermedades, pudiedo probablemente disminuir la necesidad de toma de muestras histológicas, con la consecuente disminución de complicaciones y exacerbaciones que se asocia a los estudios invasivos.
2. GENÉTICA Y EPID
Se han encontrado diferentes alteraciones genéticas relacionadas con el desarrollo de fibrosis pulmonar, fundamentalmente en FPI, pero también en otras enfermedades fibrosantes.¹ Estas mutaciones afectan sobre todo a genes relacionados con la regulación de los telómeros y del surfactante; las más frecuentes se muestran en la tabla I.
- Mutaciones relacionadas con la homeostasis de los telómeros. Los telómeros son regiones de ADN no codificante formadas por secuencias repetitivas de nucleótidos, localizadas en los extremos de los cromosomas y que mantienen su estabilidad. Con cada división celular, los telómeros se van acortando hasta llegar a un punto en que el ciclo celular se detiene, llegando a la senescencia.
El complejo de la telomerasa incluye varias proteínas que se encargan de la adición de nuevos nucleótidos en los telómeros para impedir su acortamiento, por lo que las mutaciones relacionadas con su función pueden llevar a un envejecimiento tisular precoz.² Las mutaciones de los genes que codifican para los diferentes componentes del complejo de la telomerasa (TERT, TERC, PARN…) son los que se encuentran más a menudo en pacientes con FPP.
- Mutaciones relacionadas con las proteínas del surfactante. El surfactante es un agente tensioactivo compuesto por fosfolípidos y diferentes proteínas, producido en las células alveolares epiteliales tipo 2. Diferentes mutaciones en los genes que codifican para estas proteínas se han relacionado con el desarrollo de fibrosis pulmonar. En adultos, las mutaciones más frecuentes son las producidas en los genes de la proteína A del surfactante (SP-A), la proteína C del surfactante (SP-C) y el miembro 3 de unión de la subfamilia A del casete de unión del ATP (ABCA3).
- Mutaciones de la mucina 5B. El gen MUC5B codifica para la mucina 5B, una proteína implicada en la producción de moco y que interviene en la respuesta inmunitaria del tracto respiratorio, cuya sobreexpresión se ha implicado en el desarrollo de fibrosis pulmonar. En concreto, la mutación en un polimorfismo de nucleótido simple (SNP) es el factor de riesgo más importante para el desarrollo de FPI, aunque curiosamente estos pacientes presentan una mejor supervivencia.
FIBROSIS PULMONAR
FAMILIAR
El último consenso de la European Respiratory Society (ERS)3 define la fibrosis pulmonar familiar (FPF) como la presencia de cualquier EPID fibrótica en al menos dos parientes consanguíneos de primer o segundo grado. El término fibrosis pulmonar genética, menos utilizado, se refiere a aquellos pacientes portadores de alguna mutación que causa EPID en ausencia de familiares con fibrosis pulmonar.
La FPF puede representar hasta un 10-20% de los casos de fibrosis pulmonar. La enfermedad suele ser fibrosante y progresiva, con unas manifestaciones clínicas similares a las de las formas esporádicas, aunque habitualmente los pacientes son más jóvenes. Desde un punto de vista radiológico, pueden aparecer todo tipo de patrones radiológicos, siendo el más frecuente el de neumonía intersticial usual (NIU).4
En la consulta, ¿cuándo debo sospecharla y quién se puede beneficiar del estudio genético? Dada la prevalencia de estas enfermedades y sus implicaciones tanto para el paciente como para sus familiares, es fundamental tratar de identificar los casos con un componente familiar.
Debido al fenómeno de anticipación genética, es frecuente que estas enfermedades debuten de forma precoz, por lo que siempre deberemos sospechar una FPF en casos
de EPID fibrosante en pacientes menores de 50 años. Es fundamental incorporar a la anamnesis una historia médica de los familiares focalizándonos en la búsqueda activa de casos de fibrosis pulmonar.3
Las mutaciones de los genes de la telomerasa se asocian, además de con fibrosis pulmonar, con una serie de manifestaciones extrapulmonares, que deberemos buscar activamente tanto en el paciente como en sus familiares. Las alteraciones hematológicas son frecuentes, por lo que hallazgos analíticos como anemia, macrocitosis, plaquetopenia o, más específicamente, antecedentes de leucemia o síndrome mielodisplásico deben hacernos sospechar una telomeropatía. La presencia de una enfermedad hepática, cuyas manifestaciones pueden variar desde alteraciones asintomáticas del perfil hepático hasta cirrosis criptogenética, se asocia también con mutaciones en la telomerasa.
Las recomendaciones de la ERS para ofrecer la realización de un estudio genético3 se muestran en la tabla II.
Una herramienta útil y relativamente sencilla de utilizar en consulta es la realización de un árbol genealógico prestando atención a la presencia de EPID y de otras manifestaciones extrapulmonares asociadas.
3. COMITÉ Y MANEJO MULTIDISCIPLINAR
La evaluación de la posible afectación genética, así como de sus implicaciones clínicas, terapéuticas y pronósticas, es compleja. Además, tiene implicaciones vitales, tanto para el paciente como para sus familiares. Por ello, los pacientes con afectación genética o agregación familiar deberían ser objeto de una evaluación individualizada por parte de un genetista clínico o consejero genético. Debido a la complejidad de estas enfermedades, debería plantearse la incorporación presencial o telemática de un genetista en los comités multidisciplinares, pudiendo contar también con el apoyo de otras especialidades, como hematología o digestivo, en casos de afectación multisistémica, así como la elaboración de un protocolo entre genetistas y neumólogos para la realización de estos estudios de forma estandarizada.¹
4. HISTORIA NATURAL. ¿QUÉ CURSO EVOLUTIVO ES EL MÁS FRECUENTE?
La evolución más habitual de los pacientes con FPF o fibrosis pulmonar genética es un curso crónico y fibrosante progresivo. En algunas cohortes, se ha descrito un declive más rápido de la función pulmonar que en casos de EPID fibrosante esporádica e incluso de FPI esporádica.5 Desde un punto de vista radiológico, se ha descrito que los pacientes con FPF con patrón de NIU y probable NIU presentaron una clara progresión en solo un año.4 Los pacientes con FPF tienen más exacerbaciones agudas y más riesgo de muerte o trasplante en comparación con pacientes con fibrosis pulmonar
esporádica (hasta un 80% más de riesgo de muerte que en FPI esporádica y el doble respecto a otras EPID fibrosantes).6 Además, la FPP presenta una mortalidad precoz, respecto a la FPI esporádica.7
5. MANEJO TERAPÉUTICO. ¿QUÉ DEBO CONSIDERAR EN SU TRATAMIENTO Y QUÉ ALTERNATIVAS TENGO?
Tratamiento antifibrótico
El tratamiento antifibrótico ha demostrado beneficios en pacientes con FPI y fibrosis pulmonar progresiva (FPP) tanto en ensayos clínicos como en la vida real, independientemente de la presencia de alteraciones genéticas asociadas a fibrosis pulmonar o FPF. Por ello, con la evidencia actual se debe iniciar tratamiento antifibrótico según las actuales guías internacionales8 independientemente de la condición de FPF o fibrosis pulmonar familiar, tal y como indica el posicionamiento de la Task Force de la ERS.3
Reforzando dicho enfoque, diversos estudios retrospectivos en la vida real y subanálisis de ensayos clínicos han evaluado cohortes de pacientes con y sin (o no estudiada) agregación familiar, portadores de alteraciones genéticas y/o acortamiento de la telomerasa. En los diversos estudios, el uso de tratamiento antifibrótico se ha asociado a una menor pérdida de la capacidad pulmonar e incluso de la mortalidad, con un perfil de seguridad, efectos adversos y tolerancia similar en las cohortes con FPF y fibrosis pulmonar genética.
Tratamiento inmunosupresor
La evidencia del uso de tratamiento inmunosupresor en fibrosis pulmonar genética y FPF es escasa y principalmente retrospectiva. Un subanálisis del estudio PANTHER-IPF reportó peores resultados de supervivencia, hospitalización y función pulmonar en los pacientes con FPI y acortamiento telomérico que fueron expuestos a tratamiento inmunosupresor. El uso de inmunosupresores tampoco mostró mejoría ni en el pronóstico ni en la supervivencia de pacientes con neumonitis por hipersensibilidad y acortamiento telomérico,9 ni en pacientes con EPID portadores de mutaciones PARN.10
Por todo ello, y teniendo en cuenta que tanto la inmunodeficiencia como las neoplasias hematológicas son potenciales manifestaciones de los pacientes con acortamiento telomérico, es recomendable limitar el uso de tratamiento inmunosupresor a aquellos pacientes con enfermedades subyacentes en que sea requerido como tratamiento de base, como en la fibrosis pulmonar asociada a enfermedades del tejido conectivo y la neumonitis por hipersensibilidad inflamatoria. En las exacerbaciones de EPID, el uso de glucocorticoides sigue siendo el tratamiento habitual.
Trasplante pulmonar
Las particularidades del trasplante pulmonar se abordan en el capítulo referente. No obstante, dentro de las particularidades de este capítulo, la literatura actual sugiere que estos pacientes presentan un mayor porcentaje de complicaciones en el perioperatorio y en la fase de recuperación postrasplante. Sin embargo, no se ha encontrado evidencia contundente de peores resultados en términos de supervivencia ni de rechazo a largo plazo. En el caso de los pacientes con acortamiento telomérico, se ha sugerido una inmunosupresión cuidadosamente monitorizada y ajustada.3,11
6. ESTUDIO DE FAMILIARES: SECUENCIACIÓN GENÉTICA, SCREENING Y SEGUIMIENTO
La herencia de las FPF generalmente es autosómica dominante, por lo que la probabilidad de transmisión de la mutación es del 50%, con una penetrancia variable. La Task Force de la ERS³ apoya la realización de estudio genético en familiares asintomáticos en casos de enfermedad monógenica. No obstante, esta información puede suponer un impacto psicológico importante, por lo que el paciente debe ser mayor de 18 años y estar bien informado antes de realizar el estudio genético.
Estas patologías pueden tener una fase preclínica amplia en la que se va desarrollando la enfermedad sin que aparezcan síntomas respiratorios. Aunque no existen guías ni una estandarización, la realización de un screening podría ser interesante para detectar la enfermedad de forma precoz. La Task Force de la ERS³ recomienda realizar una evaluación clínica periódica a todos los familiares asintomáticos de primer grado de pacientes con FPF y un TC torácico y unas pruebas de función respiratoria, por lo menos, cuando desarrollen síntomas respiratorios.
BIBLIOGRAFÍA
1. Borie R, Kannengiesser C, Gouya L, Dupin C, Amselem S, Ba I, et al. Pilot experience of multidisciplinary team discussion dedicated to inherited pulmonary fibrosis. Orphanet J Rare Dis. 2019;14(1):280.
2. Diaz de Leon A, Cronkhite JT, Katzenstein AL, Godwin JD, Raghu G, Glazer CS, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 2010;5(5):e10680.
3. Borie R, Kannengiesser C, Antoniou K, Bonella F, Crestani B, Fabre A, et al. European Respiratory Society statement on familial pulmonary fibrosis. Eur Respir J. 2023;61(3).
4. Bennett D, Mazzei MA, Squitieri NC, Bargagli E, Refini RM, Fossi A, et al. Familial pulmonary fibrosis: Clinical and radiological characteristics and progression analysis in different high resolution-CT patterns. Respir Med. 2017;126:75-83.
5. Guenther A, Krauss E, Tello S, Wagner J, Paul B, Kuhn S, et al. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):141.
6. Ravaglia C, Tomassetti S, Gurioli C, Piciucchi S, Dubini A, Gurioli C, et al. Features and outcome of familial idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(1):28-36.
7. Krauss E, Gehrken G, Drakopanagiotakis F, Tello S, Dartsch RC, Maurer O, et al. Clinical characteristics of patients with familial idiopathic pulmonary fibrosis (f-IPF). BMC Pulm Med. 2019;19(1):130.
8. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):18-47.
9. Adegunsoye A, Morisset J, Newton CA, Oldham JM, Vittinghoff E, Linderholm AL, et al. Leukocyte telomere length and mycophenolate therapy in chronic hypersensitivity pneumonitis. Eur Respir J. 2021;57(3).
10. Philippot Q, Kannengiesser C, Debray MP, Gauvain C, Ba I, Vieri M, et al. Interstitial lung diseases associated with mutations of poly(A)-specific ribonuclease: A multicentre retrospective study. Respirology. 2022;27(3):226-235.
11. Bordas-Martinez J.MJR, Mathot B.J., Seghers L., Galjaard R-J.H., Raaijmakers M. H.G.P. Aalbers A.M., Wijsembeek M., Molina-Molina M., Hellemons M.E. Outcomes of lung transplantation in patients with telomere-related forms of progressive fibrosing interstitial lung disease pulmonary fibrosis: A sistemyc review. JHLT Open. 2024;2024;3:100054.
TABLAS Y FIGURAS
Tabla I. Mutaciones genéticas más frecuentes en EPID. Adaptado de European Respiratory Society statement on familial pulmonary fibrosis³

TERT: telomerasa transcriptasa inversa; TERC: componente ARN de la telomerasa; RTEL1: regulador de la helicasa de elongación telómerica 1; PARN: ribonucleasa poliA específica; SPA1: proteína surfactante A1; SPA2: proteína surfactante A2; SPC: proteína surfactante C; ABCA3: miembro 3 de unión de la subfamilia A del casete de unión del ATP; AD: autosómica dominante.
Tabla II. Indicaciones para realización de estudio genético. Adaptado de European Respiratory Society statement on familial pulmonary fibrosis³

MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
ENFERMEDAD PULMONAR
INTERSTICIAL DIFUSA IDIOPÁTICA NO FPI
Autores
Sheila Izquierdo Cuervo
Joy Selene Osorio Chávez
Coordinador
David Iturbe Fernández
1. INTRODUCCIÓN
En la neumopatía intersticial, el término «idiopática» aparece a partir de 1970 con la FPI y va adquiriendo cada vez más relevancia hasta dar lugar a la clasificación de neumonía intersticial idiopática (NII) de las guías ATS/ERS de 2002 y 2013¹. Este perfil de patología hace referencia a patrones radiológicos e histológicos de daño pulmonar bien definidos, pero sin una caus a atribuible (Figura 1)(Tabla I).
2. NEUMONÍA INTERSTICIAL NO ESPECÍFICA (NINE) IDIOPÁTICA
La NINE fue descrita por primera vez en 1994 por Katzenstein y Fiorilli. Como entidad independiente de enfermedad pulmonar intersticial difusa (EPID) idiopática, se incorporó en la actualización de la ATS/ERS de 2013, cuando se retiró el término «provisional» que aparecía en la clasificación de 2002. Ocupa el segundo puesto en frecuencia dentro de las NII tras la FPI y se da hasta en el 25% de los casos.
- Cuadro clínico: el perfil más característico se da en mujer de mediana edad (de 40 a 50 años) sin historia tabáquica y con sintomatología insidiosa de tos seca y disnea de esfuerzo con o sin síndrome general. Limitación funcional típica: alteración ventilatoria restrictiva y disminución de la difusión, además de la aparición de hipoxemia a medida que la enfermedad progresa.
- Diagnóstico radiológico e histológico: patrón específico de NINE con características propias que permiten diferenciarlo del patrón de neumonía intersticial usual (NIU), como son la distribución simétrica y la presencia de vidrio deslustrado en la radiografía, la expansión homogénea por inflamación celular del intersticio con o sin fibrosis y la ausencia o la poca representación de panalización (Tabla II).
- Diagnóstico diferencial: el diagnóstico final es de exclusión y debe ser realizado por el comité multidisciplinar, descartando EPID secundaria a conectivopatía o a neumonía intersticial con características autoinmunes (IPAF) (asociaciones más frecuentes), neumonitis por hipersensibilidad (NH), EPID asociada a fármacos, radioterapia o inmunoterapia, fibrosis pulmonar familiar y síndrome de distrés respiratorio del adulto (SDRA).
- Tratamiento: en función de la gravedad y del tipo de NINE. Manejo conservador en casos leves/inflamatorios no progresivos. Corticoide sistémico y/o tratamiento inmunomodulador en NINE celular/mixta con repercusión clínica y funcional, y en casos de progresión con datos radiológicos y/o histológicos de fibrosis pulmonar, asociar tratamiento antifibrótico. Sin olvidarnos de la derivación precoz al centro trasplantador (capítulo 25 del manual).
- Pronóstico: la historia natural de la NINE es impredecible y variable, aunque el pronóstico es más favorable que en la FPI. La NINE asociada a conectivopatía es la de mejor pronóstico, mientras que la NINE fibrótica, sobre todo la idiopática, es la de peor pronóstico.²
3. BRONQUIOLITIS RESPIRATORIA ASOCIADA A EPID (BR-EPID)
La diferencia con la bronquiolitis respiratoria, hallazgo histológico incidental y frecuente en fumadores, es que la BR-EPID se acompaña de síntomas respiratorios, alteración de las pruebas de función respiratoria y, a veces, afectación fibrótica.
- Cuadro clínico: pacientes de entre 30 y 60 años con historia tabáquica relevante (índice acumulado > 30 paquetes-año) en el 100% de los casos. Clínica larvada de disnea y tos que en ocasiones es productiva. A nivel funcional, hay poca repercusión; puede presentar alteración ventilatoria mixta, con disminución de la difusión.
- Diagnóstico radiológico: nodulillos y/o vidrio deslustrado parcheado e irregular de distribución centrolobulillar difusa + engrosamiento de las paredes bronquiales ± enfisema pulmonar en lóbulos superiores.
- Diagnóstico histológico: no suele ser necesario. Acúmulo de macrófagos pigmentados (macrófagos «del fumador») que contienen pequeñas partículas granulares
marronáceas con la tinción de hematoxilina-eosina derivadas del tabaco, localizados en la luz de bronquiolos respiratorios, conductos alveolares y alveolos, y a veces, bronquiolo terminal. Se acompañan de inflamación ± fibrosis peribronquial.
- Diagnóstico diferencial con la NH subaguda/inflamatoria: El lavado broncoalveolar nos puede ayudar al presentarse con un predominio de macrófagos y sin linfocitosis. Además, este tipo de NH suele darse en no fumadores, por el papel antiinflamatorio «protector» del tabaco.
- Tratamiento: abandono del hábito tabáquico. Corticoide sistémico como segunda línea en cuadros que progresan a pesar de retirar el tabaco. Si el paciente también es EPOC, asociar broncodilatadores ± corticoide inhalado.
- Pronóstico: suele presentar una evolución favorable tras el cese tabáquico.3,4
4. NEUMONÍA INTERSTICIAL DESCAMATIVA (NID)
La NID fue descubierta por Liebow en 1965. Se confirmó que la nomenclatura era errónea tras comprobar que lo que se creía que eran células epiteliales descamadas intraalveolares son macrófagos con espacios de aire en su interior. La BR-EPID y la NID pertenecen a un espectro histológico común basado en el acúmulo de macrófagos, siendo la NID la fase más avanzada al presentar mayor extensión, localización difusa, a diferencia de la bronquiolocéntrica de la BR-EPID, y presencia de fibrosis pulmonar.
- Cuadro clínico: varón de mediana edad con alta carga tabáquica, aunque aprox. el 20% se da en no fumadores. No es rara la presencia de acropaquias. La clínica y la limitación funcional es más pronunciada que en la BR-EPI, destacando una mayor afectación del intercambio gaseoso (caída más pronunciada de la difusión y desaturación en el esfuerzo).
- Diagnóstico radiológico: vidrio deslustrado bilateral, simétrico y difuso en periferia de lóbulos inferiores; es frecuente la asociación con quistes pulmonares (dilataciones del bronquiolo y del conducto alveolar). Puede presentar fibrosis pulmonar con bronquiectasias de tracción y pérdida de volumen en lóbulos inferiores.
- Diagnóstico histológico: acúmulo extenso, difuso y uniforme de macrófagos pigmentados con citoplasma eosinofílico en el espacio alveolar + engrosamiento septal por inflamación mononuclear, a veces eosinófilos y células gigantes multinucleadas, además de depósito de colágeno e hiperplasia de neumocitos tipo 2. Arquitectura alveolar preservada.
- Diagnóstico diferencial: no fumadores/exfumadores/fumadores con bajo consumo tabáquico + macrófagos no pigmentados, descartar NID secundaria a enfermedades del tejido conectivo, exposición laboral (serrador, soldador o fundidor, bombero), otros tóxicos inhalados (marihuana, contaminación ambiental), fármacos e infecciones (receptores de trasplante y otros inmunodeprimidos).5
- Tratamiento: abandono del hábito tabáquico y corticoide sistémico. En casos más graves o que progresan a pesar del tratamiento inicial, se puede plantear asociar fármacos inmunomoduladores y/o antifibróticos, además de la derivación al centro trasplantador (capítulo 25 del manual).
- Pronóstico: a diferencia de la BR-EPID, un porcentaje pequeño de casos se resuelven o estabilizan tras el cese tabáquico, existiendo casos que progresan a un patrón de neumonía intersticial usual e insuficiencia respiratoria. Aun así, presenta un buen pronóstico a largo plazo.3,4
5. NEUMONÍA INTERSTICIAL LINFOIDE (NIL)
Entidad clinicopatológica en la que se produce una infiltración del intersticio pulmonar y los espacios alveolares por linfocitos, células plasmáticas e histiocitos.
- Cuadro clínico: más común en mujeres en la quinta década de la vida, pero puede debutar en cualquier rango de edad. Se ha asociado a diferentes tipos de enfermedades, como el VIH, conectivopatías como el síndrome de Sjögren, artritis reumatoide, lupus eritematoso sistémico, enfermedad celíaca, miastenia gravis, anemia perniciosa, procesos asociados con la producción de anticuerpos (inmunodeficiencia común variable), hepatitis activa crónica y cirrosis biliar.6 Los síntomas más frecuentes son tos y disnea de esfuerzo. Otros síntomas pueden ser fiebre, pérdida de peso, dolor torácico de características pleuríticas y artralgias. En la exploración física, destacan la presencia de crepitantes y las acropaquias, que solo se han descrito en el 10% de los pacientes.
- Diagnóstico radiológico: patrón de vidrio deslustrado, nódulos centrolobulillares mal definidos, engrosamiento del intersticio perilinfático, quistes intrapulmonares múltiples (uni o bilaterales) y adenopatías mediastínicas.
- Diagnóstico histológico: en el BAL podemos encontrar un recuento diferencial ≥ 50% linfocitos. Infiltrado intersticial difuso de las áreas afectadas con distribución septal, infiltrados compuestos predominantemente de células T, plasmáticas e histiocitos, además de hiperplasia linfoide (BALT).
- Diagnóstico diferencial: hiperplasia linfoide difusa, hiperplasia linfoide nodular, linfomas y los patrones de NO, NINE, neumonitis por hipersensibilidad y linfangioleiomiomatosis.
- Tratamiento: están indicados los corticoides sistémicos si los pacientes presentan deterioro de las pruebas de función respiratoria. En casos refractarios, se podrían utilizar cierto tipo de inmunosupresores, como ciclofosfamida, azatioprina, ciclosporina o rituximab.
- Pronóstico: la mediana de supervivencia varía entre los cinco y doce años. El riesgo de transformación en malignidad es bajo.7
6. NEUMONÍA INTERSTICIAL AGUDA (NIA)
Entidad clinicopatológica que se caracteriza por su rápida instauración, desde unos días hasta pocas semanas, con progresión hacia la insuficiencia respiratoria aguda grave con criterios de distrés respiratorio agudo y necesidad de ventilación mecánica en la mayoría de los casos.
- Cuadro clínico: la edad media de presentación es de 55 años, y no existe una clara predilección en cuanto a género. La mayoría de los pacientes presentan un cuadro clínico pseudogripal en el que predomina la tos típicamente irritativa acompañada de cefalea, mialgias y febrícula. La disnea, que aparece a los pocos días del inicio de estos síntomas, agrava críticamente al paciente que, además, muestra taquipnea, taquicardia y claros signos de hipoxia, como la cianosis.
- Diagnóstico radiológico:
a. Fase aguda o exudativa, caracterizada por áreas de vidrio deslustrado con patrón en mosaico que pueden consolidarse.
b. Fase tardía u organizativa, en la que se presenta alteración de la arquitectura pulmonar con bronquiectasias y bronquiolectasias por tracción, así como quistes, siendo las condensaciones sustituidas por vidrio deslustrado.
- Diagnóstico histológico: en el BAL, podemos encontrar un recuento diferencial ≥ 50% neutrófilos.
a. Fase exudativa: aparece en la primera semana tras el inicio; muestra edema alveolar e intersticial, membranas hialinas, hemorragia intraalveolar e inflamación aguda intersticial formada por células mononucleadas.
b. Fase de organización: comienza unas dos semanas después del inicio y muestra fibrosis laxa, mayoritariamente dentro de los septos alveolares, y una hiperplasia intensa con atipia de los neumocitos tipo 2. Suelen encontrarse trombos en arteriolas de pequeño y mediano calibre junto con metaplasia escamosa del epitelio bronquiolar.
- Diagnóstico diferencial: exacerbación de FPI, síndrome de distrés respiratorio en pacientes con enfermedades del tejido conectivo o de otra causa conocida (especialmente infecciones por Pneumocystis jiroveci y Legionella), neumonitis tóxicas.
- Tratamiento: basado en el tratamiento de soporte, a través de la oxigenación y/o ventilación mecánica. A pesar de la recomendación de iniciar tratamiento empírico con corticoides, a día de hoy no existe un beneficio claro en este grupo de pacientes. La oxigenación por membrana extracorpórea y/o el trasplante pulmonar podrían ser una alternativa en aquellos pacientes que no han respondido al tratamiento convencional.
- Pronóstico: presenta una mortalidad superior al 60% en los primeros 6 meses.8
6. FIBROELASTOSIS PLEUROPULMONAR (PPFE)
Entidad clínica patológica que se caracteriza por la afectación pleural y parenquimatosa de predominio en los lóbulos superiores, con fibrosis pleural y fibroelastosis subpleural. Algunos casos han sido identificados como idiopáticos, pero también ha sido asociada como complicación tras el trasplante de médula ósea, trasplante pulmonar, tratamiento con quimioterapia y algunas conectivopatías.
- Cuadro clínico: afecta con mayor frecuencia a adultos jóvenes (de tercera a cuarta década de la vida), con una media de edad de 57 años y es más frecuente en mujeres. La presentación más habitual es la disnea de esfuerzo y la tos seca de aparición insidiosa. La pérdida de peso también es un síntoma frecuentemente referido. La presencia de dolor pleurítico puede ser secundaria a un neumotórax espontáneo y puede ser el primer síntoma en algunos pacientes. Se puede decir que el neumotórax es una complicación característica en la historia natural en esta enfermedad. Puede ser idiopática o asociada a otras patologías.
- Diagnóstico radiológico: engrosamientos pleurales prominentes, asociados a signos de fibrosis (bronquiectasias de tracción, distorsión de la arquitectura pulmonar, pérdida de volumen), reticulación, más marcada en los lóbulos superiores. A medida que la enfermedad progresa, las opacidades reticulares y los signos de fibrosis se extienden al resto de los campos pulmonares.
- Diagnóstico histológico: incluye la presencia de fibrosis de pleura visceral en lóbulos superiores con marcada y homogénea fibrosis intraalveolar con elastosis alveolar/septal (> 80% de fibras elásticas); se observa también que el parénquima distal a la pleura está preservado, la presencia de infiltrado linfoplasmocitario desigual y algunos focos fibroblásticos.
- Diagnóstico diferencial: neumonitis por hipersensibilidad, sarcoidosis, infección por micobacterias no tuberculosas, neumoconiosis, neoplasia maligna.
- Tratamiento: En la actualidad, no existe ningún tratamiento que haya demostrado su eficacia en esta enfermedad. Los pacientes han sido tratados de forma empírica con corticoides, N-acetilcisteína y diversos inmunosupresores, sin evidencia clara de mejoría. La pirfenidona se ha utilizado en casos aislados de origen idiopático, con resultados anecdóticos variables.
- Pronóstico: datos heterogéneos, pues mientras en algunos casos se describe una evolución lentamente progresiva, en otros existe rápido deterioro clínico.9
BIBLIOGRAFÍA
1. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733-734.
2. Teoh AKY, Corte TJ. Non specific Interstitial Pneumonia. Semin Respir Crit Care Med. 2020;41(2):184-201.
3. Dawod YT, Cook NE, Graham WB, Madhani-Lovely F, Thao C. Smoking-associated interstitial lung disease: update and review. Expert Rev Respir Med. 2020;14(8):825-834.
4. Serrano Gotarredona MP, Navarro Herrero S, Gómez Izquierdo L, Rodríguez Portal JA. Smoking-related interstitial lung disease. Radiologia (Engl Ed). 2022;64(3):277-289.
5. Cottin V. Desquamative interstitial pneumonia: still orphan and not always benign. Eur Respir Rev. 2020;29(156):200183.
6. Panchabhai TS, Farver C, Highland KB. Lymphocytic Interstitial Pneumonia. Clin Chest Med. 2016;37(3):463-474, doi: 10.1016/j.ccm.2016.04.009, PMID: 27514593.
7. Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122(6):2150-2164, doi: 10.1378/chest.122.6.2150, PMID: 12475860.
8. Bouros D, Nicholson AC, Polychronopoulos V, Du Bois RM. Acute interstitial pneumonia. Eur Respir J. 2000;15(2):412-418, doi: 10.1034/j.1399-3003.2000.15b31.x, PMID: 10706513.
9. Chua F, Desai SR, Nicholson AG, Devaraj A, Renzoni E, Rice A, Wells AU. Pleuroparenchymal Fibroelastosis. A Review of Clinical, Radiological, and Pathological Characteristics. Ann Am Thorac Soc. 2019;16(11):1351-1359, doi: 10.1513/AnnalsATS.201902-181CME, PMID: 31425665; PMCID: PMC6945468.
TABLAS Y FIGURAS
Tabla I. Neumonía interstical ideopática mayor
NEUMONÍA INERSTICIAL IDIOPÁTICA MAYOR
CATEGRORÍA DIAGNÓTICO PATRÓN RADIOLÓGICO/ HISTOLÓGICO
FIBROSANTE CRÓNICA
RELACIONADA CON TABACO
AGUDA/SUBAGUDA
FIBROSIS PULMONAR IDIOPÁTICA
NEUMONÍA INTERSTICIAL NO ESPECÍFICA
BRONQUIOLITIS RESPIRATORIA - EPID
NEUMONÍA INTERSTICIAL DESCAMATIVA
NEUMONÍA ORGANIZADA CRITOGÉNICA
NEUMONÍA INTERSTICIAL AGUDA
NEUMONÍA INTERSTICIAL USUAL
NEUMONÍA INTERSTICIAL NO ESPECÍFICA
BRONQUIOLITIS RESPIRATORIA
NEUMONÍA INTERSTICIAL DESCAMATIVA
NEUMONÍA ORGANIZADA
DAÑO ALVEOLAR DIFUSO
Tabla II. Patrón radiológico e histológico de neumonía intersticial no específica

Figura 1. Clasificación de neumonía intersticial idiopática de la guía ATS/ERS de 2013
NEUMONÍAS INTERSTICIALES IDIOPÁTICAS
RARAS NIL FPI
BR-EPID
NIA
MAYORES
NEUMONIAS INTERSTICIALES IDEOPÁTICAS NO CLASIFICABLES
FIBROELASTOSIS PLEUROPULMONAR
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
SARCOIDOSIS Y OTRAS ENFERMEDADES GRANULOMATOSAS
Autores
Joel Francesqui
María Belén Noboa
Coordinador
Jacobo Sellarés
1. SARCOIDOSIS
1.1. DEFINICIÓN
La sarcoidosis es una enfermedad crónica y sistémica que afecta principalmente a los pulmones y se caracteriza por la formación de granulomas no necrotizantes, los cuales pueden afectar el funcionamiento normal de los órganos. Aunque la etiología de la sarcoidosis es aún desconocida, se sospecha que puede estar relacionada con una respuesta anormal del sistema inmunológico a ciertos estímulos ambientales y/o infecciosos en personas genéticamente susceptibles.1,2
1.2. EPIDEMIOLOGÍA
La sarcoidosis es una enfermedad rara, con una prevalencia variable según las diferentes regiones del mundo. Existen datos que respaldan que los pacientes afroamericanos experimentan una enfermedad pulmonar más grave con mayor afectación multiorgánica y peor pronóstico.2,3 La mayor incidencia de sarcoidosis se reporta en Escandinavia y América del Norte. La enfermedad puede afectar a personas de cualquier edad, pero a menudo se diagnostica entre los 20 y 40 años, sin una clara predominancia de género.
En España, la prevalencia es relativamente baja. Sin embargo, los datos pueden variar según las regiones y los estudios epidemiológicos, que hasta la fecha son escasos (Figura 1).2
1.3. ETIOLOGÍA Y FISIOPATOLOGÍA
Las características multifactoriales extensas y aún no plenamente conocidas de esta enfermedad se describen en la (Figura 1).
1.4. SARCOIDOSIS Y ENFERMEDAD TORÁCICA
La afectación torácica es la presentación más común y significativa de la enfermedad3 (Figura 2).
1.5. MANIFESTACIONES CLÍNICAS TORÁCICAS
Las principales manifestaciones clínicas se encuentran resumidas en la tabla I, mientras que las manifestaciones y clasificación clásica de la radiografía de tórax se encuentran resumidas en la tabla II.
La presentación clínica y la gravedad de la afectación torácica pueden variar considerablemente entre individuos. Es fundamental realizar una evaluación clínica completa, que incluya estudios de imagen, pruebas de función pulmonar y otros exámenes complementarios, para establecer un diagnóstico y tratamiento adecuado5 .
1.6. EXPLORACIONES COMPLEMENTARIAS
- Tomografía computarizada (TC) de tórax: Proporciona imágenes más detalladas con hallazgos característicos que permiten una evaluación más precisa de la extensión de la enfermedad.6
- PET-TC: Permite evaluar imágenes de alto contraste de enfermedad granulomatosa metabólica activa. Su uso resulta útil, especialmente en la evaluación de la actividad de la enfermedad en órganos específicos. Además, puede ayudar en el proceso de diagnóstico de la sarcoidosis valorando lesiones potencialmente biopsiables, especialmente cuando el resto de las pruebas son inconcluyentes.7
- Enzima convertidora de angiotensina (ECA): Aunque no es específico de la sarcoidosis, los niveles elevados de ECA pueden observarse en algunos casos. Existen nuevos biomarcadores séricos en investigación, con poca utilidad clínica hasta el momento por su variabilidad en especificidad y sensibilidad en diversos estudios.3
- Calcemia y calciuria: La sarcoidosis puede afectar el metabolismo del calcio resultando en niveles elevados en sangre y orina.
- Pruebas de función pulmonar: Habitualmente, se demuestra un patrón restrictivo con disminución de la difusión, en relación con la gravedad de la afectación parenquimatosa.
- Biopsia de pulmón o ganglios linfáticos: La confirmación diagnóstica de la sarcoidosis a menudo se realiza mediante la biopsia del tejido afectado en que se demuestre la presencia de granulomas no caseificantes.1,3,4
El diagnóstico de la sarcoidosis implica una combinación de hallazgos clínicos compatibles (pruebas de imagen, función pulmonar, analíticas), exclusión de otras enfermedades y confirmación histopatológica de granulomas no necrotizantes.
1.7. DIAGNÓSTICO DIFERENCIAL
- Enfermedades infecciosas granulomatosas: micobacterias, histoplasmosis.
- Granulomatosis pulmonares no infecciosas: beriliosis, enfermedad granulomatosa crónica, EPI asociada con inmunodeficiencia común variable (GLILD).
- Linfoma.
- Enfermedad pulmonar intersticial (EPI).
- Enfermedades autoinmunes.
- Histiocitosis de células de Langerhans (HCL).
- Vasculitis pulmonares. Granulomatosis eosinofílica (GEPA), granulomatosis con poliangeítis (GPA).
1.8. TRATAMIENTO
El tratamiento puede variar según la gravedad de la enfermedad, los órganos afectados y la respuesta individual al tratamiento. En algunos casos, la sarcoidosis puede autolimitarse, mientras que en otros la afectación tiende a cronificarse y requerir intervención médica.
La principal indicación de tratamiento a nivel pulmonar se da cuando la afectación parenquimatosa pone en peligro la función pulmonar o cuando se prevé que, por su extensión y agresividad, la enfermedad progrese.1,8
En general, la primera línea de tratamiento farmacológico son los glucocorticoides sistémicos a dosis no mayores de 20-30 mg/día de prednisona o su equivalente, durante un tiempo variable, aunque siempre limitado por sus efectos adversos, mientras que los inmunosupresores (metotrexate, azatioprina, micofenolato) y otros tratamientos biológicos (infliximab, adalimumab) se reservan como 2a y 3a línea de tratamientos para casos refractarios y graves.7
Es importante destacar que el tratamiento de la sarcoidosis debe ser individualizado y que la decisión de iniciar o cambiar el tratamiento debe basarse en la evaluación clínica. Además, dado que la sarcoidosis puede tener un curso variable y que la duración del tratamiento puede ser prolongada, se requiere un abordaje multidisciplinar del mismo9,8 (Figura 3).
1.9. SARCOIDOSIS PULMONAR AVANZADA
Es el término utilizado para referirnos a los estadios fibróticos pulmonares de la enferm9dad. El mecanismo por el cual la inflamación granulomatosa evoluciona a fibrosis en un subgrupo de pacientes es aún desconocido. Sin embargo, se ha propuesto que la fibrosis pudiera desencadenarse por una predisposición fibrótica inherente y/o por eventos profibróticos como respuesta anómala a la inflamación crónica descontrolada, implicando factores profibróticos como el TGF-β, interleucinas IL4, IL13 e IFN-alfa.
Manifestaciones clínicas
La presentación clínica se asemeja a la de la sarcoidosis no fibrótica e incluye disnea y tos persistente, así como producción de esputo purulento y/o hemoptisis (si se relaciona con bronquiectasias o infecciones fúngicas). En el examen físico, se encuentra crepitantes secos, acropaquia y sibilancias (en casos de fibrosis centrada en la vía aérea).1,5
Hallazgos radiográficos
Es importante reconocer tres aspectos relevantes del estadio fibrótico de la sarcoidosis (Tabla III y Figura 4).
Función pulmonar
Similar a lo que sucede en la sarcoidosis no fibrótica, las pruebas de función pulmonar muestran un patrón restrictivo con disminución de la difusión en la mayoría de los casos, aunque en un tercio de los pacientes se puede asociar una alteración obstructiva en relación a la afectación de la vía aérea.1,5
Histopatología
Se describen hallazgos de fibrosis asociados a infiltración granulomatosa, donde pueden encontrarse focos fibroblásticos y panalización, pero no bronquiolar o distal como en la NIU. En fases fibróticas muy tardías, los hallazgos podrían ser indistinguibles del patrón de NIU clásico.
Complicaciones
La mayoría de las complicaciones son secundarias al proceso fibrótico per se. Destaca la presencia de estenosis bronquial, bronquiectasias y neumotórax. Además, en estadios avanzados se puede asociar infecciones por Aspergillus, infecciones bacterianas, enfermedad tromboembólica venosa e hipertensión pulmonar asociada a sarcoidosis.
1.10. HIPERTENSIÓN PULMONAR ASOCIADA A SARCOIDOSIS (HP-SARCOIDOSIS)
La incidencia HP-sarcoidosis es de un 5%, con un mecanismo fisiopatológico multifactorial que la ubica en el grupo 5 de la clasificación de hipertensión pulmonar. La HP-sarcoidosis puede estar presente en el contexto de una hipertensión venosa pulmonar debida a una sarcoidosis cardiaca subyacente o a una cardiopatía no relacionada con la sarcoidosis. Asimismo, puede observarse en el contexto de una enfermedad tromboembólica crónica, por afectación granulomatosa directa de la vasculatura pulmonar o por la compresión de los ganglios linfáticos mediastínicos agrandados. Por último, la hipótesis más aceptada, es que sea asociada a la fibrosis pulmonar que causa distorsión de la vasculatura, así como hipoxia crónica y otras enfermedades pulmonares intersticiales fibróticas.5,10
El ecocardiograma es la prueba screening, aunque el cateterismo cardíaco derecho es el estándar de oro para el diagnóstico.10
Respecto al tratamiento, el uso de corticoides e inmunosupresores no ha demostrado un impacto real sobre la hemodinamia del paciente. Asimismo, los vasodilatadores pulmonares pueden empeorar la hipoxemia. Los inhibidores de endotelina, de fosfodiesterasa5 y los estimulantes de la guanilato ciclasa tampoco muestran datos concluyentes sobre su beneficio. En algunos casos, el uso de prostaciclinas parenterales (epoprostenol y treprostinil) mostraron una mejoría hemodinámica a largo plazo en monoterapia o en combinación.7,9
Por último, se encuentra en evaluación el uso de treprostinil inhalado como tratamiento de la HP asociada a neumopatía intersticial, lo cual puede ser un tratamiento promisorio para esta condición.
Otras opciones, como la angioplastia pulmonar con balón y la colocación de stents en la vasculatura pulmonar para tratar compresiones extrínsecas podrían ser útiles en algunos pacientes.5
Asimismo, en ausencia de contraindicaciones o comorbilidades graves, el trasplante pulmonar debe ser considerado en los casos de sarcoidosis terminal e HP-sarcoidosis. Es importante remarcar que este tipo de pacientes requieren un abordaje multidisciplinar y en centros de referencia.
1.11. ASPERGILOSIS ASOCIADA A SARCOIDOSIS
La aspergilosis pulmonar crónica complica la sarcoidosis fibroquística en aproximadamente el 2% de los casos. La presentación más frecuente son los aspergilomas intracavitarios. Se encuentran sobre todo en los lóbulos superiores, y aunque suelen permanecer clínicamente estables, pueden presentarse con hemoptisis, a veces poten-
cialmente mortal. Las cavidades preexistentes son el factor de riesgo más común para el desarrollo de aspergilomas en la sarcoidosis.5
El diagnóstico se apoya en la identificación de los hongos en las muestras de biopsia, lavado broncoalveolar o en cultivos de esputo repetidamente positivos. Las pruebas de anticuerpos IgG contra Aspergillus en suero suelen ser positivas, así como la presencia de 1, 3-beta-D-glucano.
No existe consenso respecto a cuándo y cómo tratar activamente a los pacientes con aspergiloma no complicado. En pacientes asintomáticos, la observación puede ser una opción válida. Se ha descrito el uso de voriconazol oral, aunque la evidencia es escasa. En caso de hemoptisis amenazante, está indicada la embolización de la arteria bronquial. En casos refractarios, la resección quirúrgica puede ser necesaria. En casos poco frecuentes de aspergilosis invasiva, el uso de antifúngicos sistémicos puede ayudar a controlar la enfermedad. La duración del tratamiento suele prolongarse un mínimo de seis meses. La instilación intracavitaria, guiada por TAC, de anfotericina u otros antifúngicos son opciones en situaciones inoperables, aunque con escasa evidencia científica.9
1.12.
PRINCIPALES AFECTACIONES EXTRATORÁCICAS
Las principales manifestaciones extratorácicas11 se encuentran resumidas en la tabla IV.
2. ENFERMEDAD PULMONAR INTERSTICIAL GRANULOMATOSA LINFOCÍTICA
(GLILD)
2.1. DEFINICIÓN
La enfermedad pulmonar granulomatosa linfocítica (GLILD por sus siglas en inglés) se define como una entidad clínico-radio-patológica distintiva que ocurre en pacientes con inmunodeficiencia común variable (IDCV), asociada con un infiltrado linfocítico y/o granulomatoso pulmonar, y en los que se ha excluido otras causas de afectación granulomatosa.12
Aunque su etiología es desconocida, se sospecha que la infección pulmonar por herpes virus humano 8 (HHV-8) y la sobreexpresión del factor de necrosis tumoral (TNF)-alfa pueden ser causantes y amplificadores de la enfermedad.12
Habitualmente, los pacientes con GLILD son asintomáticos respiratorios. La enfermedad se suele descubrir en evaluaciones de rutina con TAC de tórax de pacientes con IDCV. Sin embargo, en muchas ocasiones, las infecciones respiratorias derivadas del trastorno inmune de base son los que llevan a la realización de las exploraciones complementarias.12,14,15
También es frecuente encontrar asociada esplenomegalia, adenopatías difusas y afectación hepática.
2.2.
RADIOLOGÍA
El diagnóstico se sospecha con los hallazgos radiológicos del TACAR, donde es frecuente encontrar opacidades en vidrio deslustrado y lesiones micro y macro nodulares sólidas predominantes en lóbulos inferiores con adenopatías hiliares y/o mediastínicas.12
2.3.
HISTOLOGÍA
El diagnóstico definitivo requiere un estudio anatomopatológico, ya sea por VATS o por criobiopsia. Los hallazgos característicos de GLILD en una muestra de biopsia son:
- Afectación intersticial linfocítica.
- Granulomas no necrotizantes.
- Inflamación linfocítica peribronquiolar. Predominio de células T CD4+.
- Neumonía organizativa.
La combinación de estos hallazgos histológicos típicos apoya el diagnóstico.13
2.4. DIAGNÓSTICO DIFERENCIAL
- Sarcoidosis.
- EPID asociada a enfermedad de Sjögren y/o artritis reumatoide.
- Neumonía organizada.
- Linfomas de alto o bajo grado.
- Infecciones granulomatosas (micobacterias u hongos).
Respecto al diagnóstico diferencial, es importante remarcar las similitudes y diferencias entre la GLILD y la sarcoidosis (Tabla V). Ambas son procesos granulomatosos sistémicos no necrotizantes con afectación pulmonar. Sin embargo, existen ciertas características que marcan las diferencias entre ambas entidades.14
En la GLILD, la afectación pulmonar va acompañada de afectación ganglionar y esplénica en mayor porcentaje que en la sarcoidosis. Asimismo los niveles de IgG suelen estar marcadamente disminuidos en relación con la IDCV asociada a la GLILD, a diferencia de los valores habitualmente normales de la sarcoidosis, lo que va de la mano de la prevalencia de infecciones en los pacientes con GLILD.14,15
Además, la afectación pulmonar es predominante en lóbulos inferiores con nódulos de distintos tamaños y de distribución difusa, a diferencia de la sarcoidosis, que afecta
principalmente los lóbulos superiores con lesiones micronodulillares y de distribución peribronquial y perilinfática.
Otro dato relevante es la afectación de intersticial linfocítica y peribronquial con focos de bronquiolitis folicular, característica de la GLILD.14,15
2.5. TRATAMIENTO
Se recomienda suplementar con IgG endovenosa para mejorar los niveles séricos antes de asociar tratamiento inmunosupresor.
- Las alternativas de tratamiento inmunosupresor incluyen glucocorticoides, azatioprina, micofenolato, rituximab o combinaciones de ellos para casos refractarios.12
BIBLIOGRAFÍA
1. Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee. Am J RespirCrit Care Med. 1999;160(2):736-755, doi: 10.1164/ ajrccm.160.2.ats4-99.
2. Hena KM. Sarcoidosis Epidemiology: Race Matters. Front Immunol. 2020;11:537382, doi: 10.3389/fimmu.2020.537382.
3. Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: A Clinical Overview from Symptoms to Diagnosis. Cells. 2021;10(4):766, doi: 10.3390/cells10040766.
4. Keijsers RGM, Grutters JC. In Which Patients with Sarcoidosis Is FDG PET/CT Indicated? J Clin Med. 2020;9(3):890, doi:10.3390/jcm9030890.
5. Francesqui J, Kalra S. Carmona Eva. Pulmonary fibrosis in sarcoidosis. BRN Rev. 2021;7:125-136, doi: 10.23866/BRNRev:2021-M0066.
6. Criado E, Sánchez M, Ramírez J, et al. Pulmonary sarcoidosis: typical and atypical manifestations at high-resolution CT with pathologic correlation. Radiographics. 2010;30(6):15671586, doi: 10.1148/rg.306105512.
7. Sellarés J, Francesqui J, Llabres M, Hernandez-Gonzalez F, Baughman RP. Current treatment of sarcoidosis. CurrOpinPulm Med. 2020;26(5):591-597, doi:10.1097/ MCP.0000000000000720.
8. Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. 2021;58(6):2004079, doi: 10.1183/13993003.04079-2020.
9. Marrades P, Orozco JP, Francesqui J, Sellares J. Current treatment of sarcoidosis. Minerva RespirMed2023;62:126-134, doi: 10.23736/S2784-8477.23.02063-6.
10. Bandyopadhyay D, Humbert M. An update on sarcoidosis-associated pulmonary hypertension. CurrOpinPulmMed. 2020;26(5):582-590, doi: 10.1097/MCP.0000000000000701.
11. Brito-Zeron P, Sellarés J, P-ere-Alvarez R, et al. Avances en sarcoidosis. (2020). España: ICG Marge, SL.
12. Hurst JR, Verma N, Lowe D, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network Consensus Statement on the Definition, Diagnosis, and Management of Granulomatous-Lymphocytic Interstitial Lung Disease in Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. 2017;5(4):938-945, doi: 10.1016/j. jaip.2017.01.021.
13. Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency--histologic and immunohistochemical analyses of 16 cases. Hum Pathol. 2015;46(9):13061314, doi: 10.1016/j.humpath.2015.05.011.
14. Verbsky JW, Routes JM. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med. 2014;35(3):330-335, doi: 10.1055/s-00341376862.
15. Bouvry D, Mouthon L, Brillet PY, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J. 2013;41(1):115122, doi: 10.1183/09031936.00189011.
TABLAS Y FIGURAS
Tabla I. Manifestaciones clínicas torácicas de la sarcoidosis

Tabla II. Clasificación Radiológica de la Sarcoidosis (Estadios de Scadding) 1,3,4

Adaptado de Criado E, et al. Pulmonary sarcoidosis: typical and atypical manifestations at high-resolution CT with pathologic correlation. Radiographics. 2010;30(6):1567-1586.

Hallazgos radiológicos de sarcoidosis fibrótica. Adaptado de Criado E, et al. Pulmonary sarcoidosis: typical and atypical manifestations at high-resolution CT with pathologic correlation. Radiographics. 2010;30(6):1567-1586.
Tabla III. Hallazgos radiológicos de sarcoidosis fibrótica
Tabla IV. Afectación extratorácica en sarcoidosis (parte 1)

Tabla IV. Afectación extratorácica en sarcoidosis (parte 2)

Tabla V. Comparación entre GLILD y sarcoidosis

Adaptado de Verbsky JW, et al. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med. 2014;35(3):330-335.
Figura 1. Etiología y fisiopatología1,3
Acumulacion de células inmunológicas en diferentes tejidos, principalmente macrófagos que se agrupan en respuesta a la inflamación
Respuesta
Inmunológica Anormal:
Interacción entre Factores Ambientales e Imunológicos
Exposición a sustancias desconocidas
Exposiciones ocupacionales
Contaminantes ambientales
Partículas orgánicas
Factores Infecciosos
Mycobacterias
ETIOLOGÍA Y FISIOPATOLOGíA DE LA SARCOIDOSIS (MULTIFACTORIAL)
Interacción entre Factores Genéticos + Ambientales Bacterias Virus
Predisposición genética
FISIOPATOLOGÍA
Compleja interacción entre factores genéticos y ambientales que desencadenan una respuesta inmunológica anormal MULTISISTÉMICA
Activación
Inmunológica
Desregulada Formación de Granulomas
Fibrosis
Inflamación crónica
Mayor predisposición en ascendencia africana y afro-caribeña

Figura 2. Afectación torácica de la sarcoidosis
Figura 3. Manejo de la sarcoidosis1,7,8
Manejo específico en afectación multiorgánica
Manejo de síntomas y la prevención de complicaciones
Cuando no afecta significativamente la función de los órganos, puede ser apropiado adoptar un enfoque de observación sin tratamiento activo.
Manejo de Complicaciones Específicas
Cuidados de apoyo
Calcio y vitamina D
En casos de hipercalcemia asociada con sarcoidosis, evitar suplementos de calcio y Vit D y se prefiere tto con corticoides y/o hidroxicloroquina.
Observación y seguimiento
Primera línea para casos de moderados a graves, por sus propiedades antiinflamatorias y en la formación de granulomas. Se utilizan en dosis que se ajustan gradualmente.
Corticosteroides
Sarcodosis
Agentes biológicos
Inmunomoduladores
En casos más graves o refractarios, se pueden considerar agentes biológicos, como el infliximab o adalimumab, que afectan específicamente a ciertas moléculas del sistema inmunológico.
Cuando los corticosteroides no son suficientes o no son tolerados. Se consideran los inmunomoduladores para mediar la respuesta inmunológica y reducir la inflamación (azatioprina, metotrexato o micofenolato mofetilo).
Figura 4. Patrones radiológicos más frecuentes en sarcoidosis fibrótica

Adaptado de Francesqui J, et al. Pulmonary fibrosis in sarcoidosis. BRN Rev. 2021;7:125-136.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
NEUMONITIS POR HIPERSENSIBILIDAD
Autores
Oswaldo Antonio Caguana Vélez
María Florencia Pilia
Coordinadora
Ana Villar Gómez
1. INTRODUCCIÓN
1.1. DEFINICIÓN
La neumonitis por hipersensibilidad (NH) es una enfermedad inflamatoria y/o fibrosante que afecta el parénquima pulmonar y las vías respiratorias pequeñas. La NH es el resultado de una reacción inmunomediada provocada por un antígeno inhalado evidente u oculto en individuos susceptibles.¹
1.2. CLASIFICACIÓN
Históricamente, la NH se clasificaba como aguda, subaguda o crónica en función de la duración de los síntomas, pero en la actualidad esta clasificación se considera de escaso valor clínico debido a las dificultades para definir estas categorías y su falta de asociación con los resultados.²
Algunos pacientes presentan un curso benigno y se recuperan por completo una vez eliminada la exposición al agente causante, mientras que en otros pacientes la enfermedad avanza hacia insuficiencia respiratoria crónica, independientemente de si se ha clasificado como NH aguda, subaguda o crónica.¹
Dado que la presencia de fibrosis es un factor crítico en la determinación del pronóstico, las guías clínicas más recientes para el diagnóstico de la NH proponen que los pacientes se clasifiquen en dos categorías: NH no fibrótica (puramente inflamatoria) o NH fibrótica (inflamatoria mixta y fibrosa o puramente fibrosa).²
Este reciente enfoque refleja un acuerdo general, a saber, que la categorización en NH fibrótica o no fibrótica es un criterio más objetivo que puede reflejar la forma en que se manifiesta la enfermedad y que, posiblemente, guarda una relación más directa con la evolución clínica y otros desenlaces.¹
Asimismo, los pacientes en los que no se ha identificado exposición pero que, por lo demás, presentan características típicas de la NH, pueden describirse como "NH criptogénica" o "NH de causa indeterminada".
1.3. HISTORIA NATURAL Y PRONÓSTICO
La historia natural de la NH puede oscilar desde una mejoría inicial hasta un empeoramiento progresivo que en algunos casos puede llevar a complicaciones graves, como insuficiencia respiratoria, e incluso desenlaces fatales.
Los pacientes con NH no fibrótica que evitan la exposición continua al agente desencadenante pueden tener un pronóstico favorable con la posibilidad de estabilización o recuperación completa. Por otro lado, los pacientes con NH fibrótica, en particular aquellos con un patrón radiológico de neumonía intersticial usual (NIU), tienen una menor supervivencia.3 Otras características descritas asociadas con un mal pronóstico incluyen el tabaquismo, un volumen espiratorio forzado inicial más bajo, la ausencia de linfocitosis en el lavado broncoalveolar (LBA), la presencia de focos fibroblásticos en las muestras histológicas, la exposición persistente al agente desencadenante y/o la incapacidad para identificar dicho agente.3-6 Es fundamental destacar que en un 3050% de los casos evaluados en centros especializados en enfermedades pulmonares intersticiales no se logra identificar un agente desencadenante.5
1.4. EPIDEMIOLOGíA
La NH puede ocurrir a cualquier edad, aunque suele presentarse después de la cuarta década de vida.7 La prevalencia mundial de la NH es variable dependiendo del clima y la exposición ocupacional y ambiental,¹ lo que refleja la compleja interacción entre los factores de riesgo del huésped, el antígeno y el ambiente. Un amplio estudio realizado en Estados Unidos entre 2004 y 2013 estimó que la prevalencia en 1 año era entre 1,67 a 2,71 por cada 100.000 habitantes.8 La tasa de incidencia es fluctuante según los diferentes estudios. Se estima que la incidencia es de entre 0,3 y 0,9 por cada 100.000 individuos, aunque podría ser aún mayor según un estudio que informó que la enfermedad de los criadores de aves afecta a 4,9 de cada 100.000 individuos durante un período de 10 años o a 54,6 de cada 100.000 criadores de aves.¹
2. FISIOPATOLOGÍA
Los mecanismos inmunopatológicos propuestos para la NH involucran una interacción compleja de factores genéticos y ambientales. La NH se desarrolla en individuos susceptibles tras exposiciones repetidas a antígenos proteicos derivados de microorganismos, hongos o animales, así como polisacáridos o sustancias químicas no proteicas (ver Etiología). La exposición puede ser de origen laboral, doméstica o recreativa. En individuos sensibilizados, la respuesta inmunológica a un antígeno implica tanto respuestas humorales (anticuerpos IgG específicos) como celulares (células T helper o Th). Estas conducen a un patrón inflamatorio de predominio linfocítico e inflamación de tipo granulomatosa. Asimismo, la inflamación neutrofílica puede desempeñar un papel tanto al inicio de la enfermedad como durante la fibrosis subsiguiente, mientras que la función alterada de las células T reguladoras puede contribuir a la respuesta inmunológica exagerada. Se ha sugerido que un cambio de la actividad de células tipo de Th1 a Th2, junto con apoptosis epitelial aumentada y actividad anormal de fibroblastos, pueden contribuir al desarrollo de la fibrosis pulmonar. La susceptibilidad genética es un factor importante en el desarrollo de NH. En este contexto, se han identificado variantes específicas en genes relacionados con la inmunidad innata y adaptativa, especialmente en el complejo principal de histocompatibilidad clase II, así como polimorfismos en genes como MUC5B y mutaciones en genes relacionados con telómeros.1-9
3. ETIOLOGÍA
La etiología de la NH se resume en la tabla I.¹
4. DIAGNÓSTICO
El diagnóstico de NH puede ser un desafío y requiere una combinación de criterios clínicos, historial de la exposición, criterios radiológicos, linfocitosis en el lavado broncoalveolar (LBA) y, en algunos casos, datos histopatológicos, preferiblemente discutidos en un entorno multidisciplinario especializado en enfermedades pulmonares intersticiales.1-7
Según las directrices de la guía CHEST (Algoritmo 1), se puede establecer un diagnóstico seguro de NH en pacientes que cuentan con una exposición identificada y presentan un patrón característico en la tomografía computarizada de alta resolución (TCAR).7 Además, de acuerdo con las recomendaciones de la ATS/ERS/ALAT (Algoritmo 2), para confirmar el diagnóstico de NH se requieren pruebas que evidencien linfocitosis en el LBA.1 Sin embargo, la variabilidad de los diagnósticos depende del fenotipo clínico. Algunas pruebas, como la linfocitosis en el LBA, pueden ser menos sugerentes en la NH de carácter fibrótico. La evaluación no solo pretende confirmar un diagnóstico de NH
con alta confiabilidad, sino también caracterizar el fenotipo, ya sea inflamatorio, fibrótico o una combinación de ambos, lo que proporciona una guía valiosa para la elección del tratamiento y el pronóstico del paciente.7
4.1. Evaluación clínica y laboratorio
- Anamnesis: Para cualquier exposición potencial provocada, se debe tener en cuenta la duración, el alcance y la frecuencia de exposición y su relación con los síntomas.
- Manifestaciones clínicas: La disnea y la tos son los síntomas clínicos más comunes de NH. El examen físico revela con mayor frecuencia los chirping rales (sibilantes cortos mesoteleinspiratorios) y crepitantes y acropaquias en la forma fibrótica.²
- Laboratorio: Las precipitinas séricas detectan la presencia de anticuerpos IgG específicos en suero sanguíneo en respuesta a la exposición a ciertos alérgenos (esporas de hongos, aves, entre otros). Las pruebas de IgG específicas pueden ser útiles, pero carecen de valores definidos y no distinguen entre la sensibilización y la enfermedad. Podemos encontrar elevación de la velocidad de eritrosedimentación, de la proteína C reactiva, positividad del factor reumatoide y, en ocasiones, de los isotipos de inmunoglobulinas IgG, IgM o IgA, todo ello reflejo de un proceso inflamatorio agudo y/o crónico. La eosinofilia es rara y observamos neutrofilia en las formas agudas de la enfermedad o tras realizar una prueba de provocación bronquial específica.1,2,7
- Pruebas cutáneas específicas: Son pruebas en las que se administra por vía intradérmica el antígen específico a estudio y se realiza una lectura inmediata (a los 15 minutos); se considera que la prueba es positiva si se evidencia una pápula mayor de 10 mm.
4.2. Características radiológicas
El diagnóstico y la caracterización de la NH se basa en un protocolo de TCAR de tórax, similar al utilizado en la fibrosis pulmonar idiopática,10 que permite una detección precisa de las características radiológicas de la enfermedad. La TCAR se realiza en posición supina, con imágenes tomadas en inspiración profunda y espiración prolongada (TCAR en espiración) y permite evaluar el atrapamiento aéreo, un hallazgo importante en NH. Se distinguen dos patrones principales: NH no fibrótica y NH fibrótica.1,7 Las características radiológicas de los patrones no fibrótico y fibrótico se describen en las tabla II y figura 3
En resumen, los hallazgos de la TCAR son: 1) altamente sugestivos de NH, que categorizamos como "NH típica"; 2) menos sugestivos pero compatibles con NH, a los que
nos referimos como "compatibles con NH", o 3) "indeterminados para NH" cuando los hallazgos de TCAR no son sugestivos ni compatibles con características de NH.1,7
4.3. Lavado broncoalveolar (LBA)
La linfocitosis en el LBA es una característica común en la NH. Se define como un porcentaje de linfocitos en el LBA que supera el 20% (predominio CD8), índice CD4/ CD8 inferior a 1.9 En el caso de NH no fibrótica, el recuento de linfocitos puede ser aún más elevado y puede llegar a superar el 50%. Por otro lado, en la NH fibrótica, el porcentaje de linfocitos en el LBA suele ser menor y, en ocasiones, puede ser normal, especialmente en pacientes fumadores o en individuos de edad avanzada.1,7
4.4. Biopsia pulmonar e histología
Se debe evitar la biopsia pulmonar en los casos en que sea posible un diagnóstico definitivo de NH tras una discusión multidisciplinar de los hallazgos clínicos, los antecedentes de exposición, la radiología y la linfocitosis del LBA (Figura 2 y Figura 3).
Se emplean tres métodos para la obtención de tejido pulmonar: 1) biopsia transbronquial (BTB), con un rendimiento diagnóstico del 37%; 2) criobiopsia pulmonar transbronquial (CPTB), con un rendimiento diagnóstico del 82%, y 3) biopsia pulmonar quirúrgica (BPQ), con un rendimiento diagnóstico del 96%.²
Histología de NH no fibrótica: Se identifica 3 características claves: 1) una expansión broncocéntrica del intersticio con la presencia de linfocitos, células plasmáticas y ocasionalmente eosinófilos; 2) bronquiolitis celular crónica (obliterante), y 3) granulomas laxos peribronquiolares. La inflamación se caracteriza por grupos de macrófagos epitelioides y células multinucleadas, junto con células gigantes aisladas. La ausencia de granulomas o células gigantes multinucleadas aún se considera compatible con NH si las otras dos características están presentes.²
Histología de NH fibrótica: El estudio histológico puede asemejarse al de la NIU, que se observa en la fibrosis pulmonar idiopática (FPI). Se identifican 3 características claves: 1) fibrosis centrada en vía área, a menudo acompañada o no por metaplasia peribronquiolar generalizada; 2) neumonía intersticial con una formación fibrosante, que puede manifestarse como una variante de la neumonía intersticial no específica (NINE), NIU, fibrosis peribronquiolar aislada o una enfermedad pulmonar fibrosante que no encaja en una clasificación específica, y 3) granulomas mal formados (Figura 3). Al igual que en la NH no fibrótica, la ausencia de granulomas o células gigantes multinucleadas aún se considera compatible con la NH fibrótica si las otras características están presentes.²
4.5. Pruebas funcionales respiratorias
- Pruebas funcionales respiratorias: En particular, la capacidad vital forzada (FVC) y la capacidad de difusión para monóxido de carbono (DLco) son útiles para la evaluación de la NH. Estos parámetros pueden revelar la respuesta al tratamiento y predecir la progresión de la enfermedad, especialmente en casos de restricción y fibrosis. Además, se pueden identificar patrones obstructivos y capacidad de respuesta de las vías respiratorias en pacientes con bronquiolitis.²
- Prueba de broncoprovocación o inhalación específica:14 Esta prueba implica exponer al paciente a un antígeno sospechoso. La exposición se realiza en un ambiente controlado administrando el antígeno aerosolizado (suero de ave esterilizado, heno, etc.) al 1/100 (0,01 mg/ml o bien 10 µg/ml de proteínas). Según los resultados, se sigue un protocolo específico:
1. Si la prueba es negativa, se repite al día siguiente a una concentración de 1/10 (0,1 mg/ml o 100 µg/ml de proteínas). Si sigue siendo negativa, se repite durante 3 días consecutivos a la misma concentración para verificar si hay una disminución gradual de la función pulmonar.
2. Si la prueba sigue siendo negativa, se puede considerar una exposición directa al antígeno en el mismo laboratorio de pruebas funcionales respiratorias. Es importante destacar que, según las guías,1,7 no existe una indicación para su realización en el proceso diagnóstico de la NH. La ausencia de criterios universalmente validados para definir un resultado positivo, la falta de estandarización de la preparación del extracto antigénico y la necesidad de personal entrenado contribuyen a la complejidad en su realización y dificultan su inclusión dentro del algoritmo diagnóstico. Sin embargo, es una prueba que ha mostrado una sensibilidad y una especificidad de 85% y 86% respectivamente para el diagnóstico de NH por antígeno aviar y/o fúngico.14
Según Muñoz et al.,14 los criterios de positividad son:
1. Descenso de la FVC >15% o de la DLco>20%.
2. Descenso del 10-15% de la FVC más uno de los siguientes cambios: a) incremento de los leucocitos sanguíneos ≥ 20%; b) descenso ≥3% de la SatO2 en Hb; c) cambios evidentes en la radiografía de tórax; d) aumento de la temperatura >0,5 ºC; e) síntomas clínicos (tos, disnea, etc.).
3. Descenso de la FVC <10%, con tres o más de los criterios mencionados.
5. TRATAMIENTO
- Evitación antigénica: La identificación y evitación antigénica son fundamentales para mejorar la evolución de los pacientes afectos de NH. Se ha descrito que la incapacidad para identificar el agente se asocia de manera independiente con una supervivencia reducida.5
- Corticoesteroides: No existe un algoritmo establecido para el tratamiento farmacológico. Los pacientes con NH no fibrótica y una inflamación activa persistente, como opacidades en vidrio esmerilado en una TCAR, linfocitosis en el LBA (más del 20%) y/o inflamación granulomatosa en la biopsia pulmonar, tienen más probabilidades de responder a la terapia con glucocorticoides que los pacientes que no tienen estas características.11 Si bien puede existir una respuesta inicial a los corticoides, hay escasa evidencia de que el tratamiento corticoideo proporcione un beneficio a largo plazo o frene la progresión de la NH fibrótica.2
- Inmunosupresores: La ausencia de respuesta clínica o la progresión de la enfermedad a pesar del tratamiento con glucocorticoides puede motivar el uso de terapia inmunosupresora dirigida a suprimir las respuestas inflamatorias e inmunológicas en curso.12 El tratamiento disponible para la NH progresiva con cambios mixtos inflamatorios y fibrosos es el tratamiento inmunomodulador combinado (corticosteroides con azatioprina o micofenolato), aunque la evidencia de su utilidad se basa en estudios retrospectivos. No obstante, el tratamiento combinado puede estar asociado con menos efectos secundarios que la monoterapia de altas dosis de esteroides.9
- Antifibróticos: Se ha postulado que una vez que la NH se ha vuelto fibrótica es necesaria una terapia que inhiba las vías fisiopatológicas de la fibrosis para frenar su progresión independientemente del desencadenante inicial. En el ensayo INBUILD realizado en 663 pacientes con EPI fibróticas distintas de la FPI que cumplieron con los criterios de progresión de la EPI en los 2 años anteriores a pesar del tratamiento considerado apropiado en la práctica clínica, el nintedanib ralentizó la tasa de disminución de la FVC (mL/año) durante 52 semanas en un 57% en comparación con placebo. Los pacientes con NH fibrótica comprendieron 173 (26%) de los pacientes incluidos. Aunque el ensayo INBUILD no fue diseñado para estudiar enfermedades pulmonares intersticiales específicas, los análisis de subgrupos sugirieron que la tasa de declive de la FVC y los eventos adversos asociados con el nintedanib fueron homogéneos en todos los subgrupos.2 En la actualidad, los pacientes con NH fibrótica que cumplen criterios de fibrosis pulmonar progresiva (FPP), podrían ser candidatos a recibir tratamiento antifibrótico (ver capítulo FPP).
En conclusión, aunque la base de evidencia es insuficiente para definir un algoritmo de tratamiento para la NH, en la práctica clínica se puede aplicar un enfoque basado en el perfil de la enfermedad.
Oxigenoterapia: Se recomienda el uso de oxigenoterapia a largo plazo y oxígeno ambulatorio en pacientes con EPID e hipoxemia crónica grave en reposo, así como al esfuerzo. Sin embargo, no existe evidencia científica que demuestre su impacto en la supervivencia.
Trasplante pulmonar: La Sociedad Internacional de Trasplante de Pulmón y Corazón (ISHLT) recomienda en su último consenso de 2021:13
∙ Remisión para valoración a un centro de trasplante en el momento del diagnóstico, incluso si el paciente está iniciando la terapia, así como para los pacientes con patrón de NIU histopatológica o evidencia radiográfica de un patrón de NIU probable o definitivo.
∙ Cualquier forma de fibrosis pulmonar con FVC de <80% previsto o DLco <40% previsto.
∙ Cualquier forma de fibrosis pulmonar con evidencia de progresión o alguno de los siguientesen los últimos 2 años:
- Disminución relativa del FVC 10%.
- Disminución relativa de DLco 15%.
- Disminución relativa del FVC del 5 % en combinación con empeoramiento de los síntomas respiratorios o progresión radiográfica.
∙ Pacientes que requieran oxigenoterapia, ya sea en reposo o al esfuerzo.
Las intervenciones no farmacológicas, como la rehabilitación pulmonar, la aplicación de vacunas y la participación en grupos de pacientes, pueden constituir una parte importante del cuidado integral de pacientes con EPI progresiva. En algunos casos, el manejo de comorbilidades y el tratamiento paliativo pueden contribuir a mejorar la calidad de vida de los pacientes.²
BIBLIOGRAFÍA
1. Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vašáková M, Bargagli E, et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2020;202(3):3669, https://doi.org/10.1164/rccm.202005-2032ST
2. Hamblin M, Prosch H, Vašáková M. Diagnosis, course and management of hypersensitivity pneumonitis. European respiratory review : an official journal of the European Respiratory Society. 2022;31(163):210169, https://doi.org/10.1183/16000617.0169-2021
3. Chiba S, Tsuchiya K, Akashi T, Ishizuka M, Okamoto T, Furusawa H, Tateishi T, et al. Chronic Hypersensitivity Pneumonitis With a Usual Interstitial Pneumonia-Like Pattern: Correlation Between Histopathologic and Clinical Findings. Chest. 2016;149(6):1473-1481, https://doi. org/10.1016/j.chest.2015.12.030.
4. Ojanguren I, Morell F, Ramón MA, Villar A, Romero C, Cruz MJ, Muñoz, X. Long-term outcomes in chronic hypersensitivity pneumonitis. Allergy. 2019;74(5):944-952, https://doi. org/10.1111/all.13692.
5. Fernández Pérez ER, Swigris JJ, Forssén AV, Tourin O, Solomon JJ, Huie TJ, Olson AL, Brown KK. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144(5):1644-1651, https://doi.org/10.1378/ chest.12-2685
6. Wang P, Jones KD, Urisman A, Elicker BM, Urbania T, Johannson KA, Assayag D, et al. Pathologic Findings and Prognosis in a Large Prospective Cohort of Chronic Hypersensitivity Pneumonitis. Chest. 2017;152(3):502-509, https://doi.org/10.1016/j.chest.2017.02.011
7. Fernández Pérez ER, Travis WD, Lynch DA, Brown KK, Johannson KA, Selman M, Ryu JH, et al. Diagnosis and Evaluation of Hypersensitivity Pneumonitis: CHEST Guideline and Expert Panel Report. Chest. 2021;160(2):97-156, https://doi.org/10.1016/j.chest.2021.03.066.
8. Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of Hypersensitivity Pneumonitis among an Insured Population in the United States: A Claims-based Cohort Analysis. Annals of the American Thoracic Society. 2018;15(4):460469, https://doi.org/10.1513/AnnalsATS.201704-288OC.
9. Vašáková M, Selman M, Morell F, Sterclova M, Molina-Molina M, Raghu G. Hypersensitivity Pneumonitis: Current Concepts of Pathogenesis and Potential Targets for Treatment. American journal of respiratory and critical care medicine. 2019;200(3):301-308, https://doi. org/10.1164/rccm.201903-0541PP
10. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, et al. American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2018;198(5):44-68, https://doi.org/10.1164/rccm.201807-1255ST.
11. Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Martinez FJ, Flaherty KR. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Where We Stand and Where We Need to Go. American journal of respiratory and critical care medicine. 2017;196(6):690-699, https://doi.org/10.1164/rccm.201608-1675PP
12. Costabel U, Miyazaki Y, Pardo A, Koschel D, Bonella F, Spagnolo P, Guzman J, et al. Hypersensitivity pneumonitis. Nature reviews. Disease primers. 2020;06(1):65, https://doi. org/10.1038/s41572-020-0191-z.
13. Leard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, Campos SV, et al. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2021;40(11):1349-1379, https://doi.org/10.1016/j.healun.2021.07.005
14. Muñoz X, Sánchez-Ortiz M, Torres F, Villar A, Morell F, Cruz MJ. Diagnostic yield of specific inhalation challenge in hypersensitivity pneumonitis. Eur Respir J. 2014;44(6):1658-1665, https://doi:10.1183/09031936.00060714
TABLAS
Y FIGURAS
Tabla I. Fuentes de antígenos conocidos por causar neumonitis por hipersensibilidad. Adaptado de Raghu G, et al. Am J RespirCrit Care Med. 2020;202(3):36-69. (parte 1)

Tabla I. Fuentes de antígenos conocidos por causar neumonitis por hipersensibilidad. Adaptado de Raghu G, et al. Am J RespirCrit Care Med. 2020;202(3):36-69 (parte 2) (Continúa)

Tabla I. Fuentes de antígenos conocidos por causar neumonitis por hipersensibilidad. Adaptado de Raghu G, et al. Am J RespirCrit Care Med. 2020;202(3):36-69 (parte 3) (Continúa)

Tabla I. Fuentes de antígenos conocidos por causar neumonitis por hipersensibilidad. Adaptado de Raghu G, et al. Am J RespirCrit Care Med. 2020;202(3):36-69 (parte 4)

Tabla II. Características de la exploración por TCAR de tórax del patrón NH no fibrótica. Adaptado de Fernández Pérez, E. R. et al. Chest, 144(5):1644-1651

Figura / Algoritmo 1. Algoritmo para la evaluación diagnóstica de la neumonitis por hipersensibilidad (NH). Adaptado de Fernández Pérez, E. R. et al. Chest, 144(5):1644-1651

Figura / Algoritmo 2. Algoritmo para la evaluación diagnóstica de la neumonitis por hipersensibilidad (NH). Adaptado de Raghu G, et al. m Am J Respir Crit Care Med. 2020;202(3):36-69

Figura 3. Características de la exploración por TCAR de tórax del patrón NH fibrótica

MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
SÍNDROME COMBINADO DE FIBROSIS PULMONAR –ENFISEMA Y EPID ASOCIADA
A TABACO
Autoras
Celia Montaño Montaño
Marta Orta Caamaño
Coordinador
Eva Cabrera Cesar
1. INTRODUCCIÓN
El tabaquismo guarda una relación causal bien establecida con la enfermedad pulmonar obstructiva crónica (EPOC), el enfisema y el cáncer de pulmón. Sin embargo, la relación del tabaco con la enfermedad pulmonar intersticial (EPID) es menos conocida, a pesar de que se ha demostrado que el humo del tabaco puede desencadenar mecanismos de daño intersticial que resultan de distintas alteraciones patológicas y fibrosis pulmonar.¹El humo del tabaco conlleva una variedad de partículas tóxicas y sustancias químicas que inducen estímulos inflamatorios, genotóxicos, proliferativos y de otros tipos sobre las células pulmonares que intervienen tanto en las vías respiratorias como en el intercambio de gases. Entre los mecanismos patogénicos del tabaco se encuentra el daño epitelial directo y la activación celular endotelial que conduce a la secreción de citoquinas que reclutan células inflamatorias que se acumulan en las vías aéreas y en el intersticio pulmonar. El tabaco induce la senescencia celular del epitelio alveolar y, en determinados sujetos predispuestos, puede desencadenar una respuesta inflamatoria y de remodelado tisular, además de desencadenar factores profibróticos (TGF-b1 y PDGF-A y -B). En este contexto, hay que tener en cuenta que ciertos eventos patogénicos, como la disfunción de los telómeros y la predisposición a la senescencia celular, pueden proporcionar información sobre la susceptibilidad de algunos fumadores a desarrollar enfisema, cambios fibróticos o ambos.²
La EPI relacionada con el tabaco comprende un espectro heterogéneo de enfermedades pulmonares parenquimatosas, además del enfisema, que comparten rasgos histológicos, clínicos y radiológicos, y que precisan un abordaje multidisciplinar para su diagnóstico. Las manifestaciones histológicas oscilan desde alteraciones inflamatorias reversibles hasta enfisema y fibrosis, y en muchos casos se solapan.¹
En general, se podría clasificar a estas enfermedades en dos grupos según el grado de asociación o causalidad que guarden con el hábito tabáquico.¹
- Enfermedades con una relación causal establecida con el tabaco:
- Bronquiolitis respiratoria con EPI (BR-EPID)
- Neumonía intersticial descamativa (NID)
- Neumonía eosinófila aguda (NEA)
- Fibrosis intersticial asociada al tabaco (FIAT)
- Histiocitosis de células de Langerhans (Capítulo 14. Enfermedades quísticas)
- Enfermedades para las que el tabaco es un factor de riesgo junto con otros factores endógenos y exógenos:
- Fibrosis pulmonar idiopática (FPI) (Capítulo 4. Fibrosis pulmonar idiopática)
- Neumonía intersticial no específica (NINE) (Capítulo 6. EPID idiopáticas no FPI)
- Síndrome combinado fibrosis pulmonar y enfisema (CPFE)
Además, con el creciente uso de las tomografías computarizadas torácicas y el programa de detección de cáncer de pulmón ha aumentado significativamente la identificación de hallazgos intersticiales subclínicos o incidentales, que en inglés se conocen con las siglas ILA ( Interstitial Lung Abnormalities), cuyo impacto aún está por estudiar. Puede tratarse tanto de hallazgos inespecíficos como de la manifestación temprana de una EPID concreta en estado incipiente. Se describen como anomalías intersticiales bilaterales en áreas independientes del pulmón, con una extensión superior al 5%. No existe un punto de corte determinado en relación con la extensión para la distinción entre la ILA y la EPID, que se diferenciarán por la integración de síntomas y signos clínicos. El seguimiento de los pacientes con ILA mediante pruebas de imagen ha mostrado una persistencia de los hallazgos aproximadamente en el 60% de los casos y una progresión de los mismos entre un 20-43%, en un periodo de 6 años. La progresión de los hallazgos fibróticos se relacionó con la edad y el hábito de fumar. 3,4
A continuación, se realiza un breve resumen de cada una de las enfermedades intersticiales relacionadas con el tabaco.
2. SÍNDROME COMBINADO FIBROSIS PULMONAR-ENFISEMA (CPFE)
Se define por la coexistencia de fibrosis pulmonar, provocada por diferentes etiologías, y enfisema pulmonar. Tiene una prevalencia desconocida. La edad media de presentación es 65-70 años, y es más frecuente en varones y en fumadores, con un índice paquetes año (IPA) ≥40, aunque puede aparecer en pacientes nunca fumadores. Se recomienda el trabajo de Cottin V et al: Syndrome of Combined Pulmonary Fibrosis and Emphysema: An Official ATS/ERS/JRS/ALAT Research Statement. 2022. 5
Exposiciones y etiologías asociadas a CPFE 5,6:
a. Factores de riesgo y demografía: Tabaquismo, sexo masculino.
b. Enfermedades: Fibrosis pulmonar idiopática (FPI), enfermedades del tejido conectivo (ETC), vasculitis asociada a ANCA, neumonitis por hipersensibilidad (NH).
c. Exposición a inhalantes: Polvos de carbón, amianto, sílice y polvo mineral, otros inhalantes.
d. Variantes genéticas: Genes asociados con la telomerasa (TERT, RTEL1), genes relacionados con el surfactante (SFTPC, ABCA3), otros genes (Naf1, PEPD), polimorfismos genéticos, Genes MMP-9, TGF-β-1, gen AGER.
Clínica
La clínica más frecuente es la tos y la disnea, aunque es inespecífica.5,6
Características de la función pulmonar:
- FVC: ↓ o N (más conservada en comparación con FPI sola)
- FEV1: ↓o N
- FEV1/FVC: variable (N, ↓ o ↑)
- TLC: variable (N, ↓o↑)
- FRC: variable (N, ↓ o ↑)
- VR: variable (N,↓ o ↑)
- DLco: ↓ desproporcionadamente
- Kco: ↓ severa
- Saturación durante el ejercicio: ↑ severa
- VO2máx: ↓
Características radiológicas
Existe variedad de hallazgos en la tomografía computarizada de alta resolución (TCAR), descritos en la tabla I. Hay focos de enfisema centrolobulillar, paraseptal o panacinar asociado a fibrosis intersticial (reticulación y/u opacidades en vidrio deslustrado asociadas a panal de abeja y/o bronquiectasias de tracción), como se muestra en el caso ejemplo de la figura 1. 5-7
Los patrones clásicos de la TCAR pueden verse alterados cuando el enfisema y la fibrosis se superponen.6,7
Cuantificación visual del enfisema: establecer un porcentaje de afectación de enfisema respecto al volumen total pulmonar en TCAR de manera visual. Debe presentar un porcentaje > al 5%.5-7
Cuantificación de la fibrosis pulmonar: Presencia de fibrosis pulmonar de cualquier etiología. Se cuantifica por la suma de las opacidades en vidrio deslustrado (solo si están superpuestas por líneas reticulares o bronquiectasias de tracción), reticulación y panal de abeja.5-7
Hallazgos histológicos
No existen criterios histológicos para establecer el diagnóstico de CPFE. El patrón más frecuentemente informado es de neumonía intersticial usual (NIU), seguido de neumonía intersticial no específica fibrótica y neumonía intersticial descamativa.7
Comorbilidades asociadas
a. Hipertensión Pulmonar: La presentan entre un 15 y un 55% de los pacientes. Tienen peor supervivencia que aquellos pacientes con EPID o enfisema sin hipertensión pulmonar. Principalmente en aquellos casos con la presencia de hipertensión pulmonar desproporcionada para la afectación fibrótica o enfisematosa que presenten. No existe tratamiento específico, pero podría evaluarse, individualmente, el uso de sildenafilo o treprostinil en unidades especializadas en hipertensión pulmonar.5
b. Cáncer de pulmón: se describe en del 2 al 52% de los pacientes. Los más frecuentes son el cáncer de pulmón de células escamosas y el adenocarcinoma. Suele aparecer con más frecuencia en lóbulos inferiores. Presentan peor supervivencia que los pacientes con cáncer de pulmón sin CPFE.5
c. Exacerbación aguda de CPFE: los pacientes con CPFE tienen el mismo riesgo de exacerbar que los pacientes con FPI, pero tienen mejor pronóstico en su recuperación.5
Pronóstico
Los pacientes con CPFE tienen peor pronóstico y supervivencia que los pacientes con FPI o enfisema aislados.5
Los predictores de peor pronóstico y mortalidad son5:
a. Mayor edad.
b. Anticuerpos antinuclerares negativos.
c. Aumento de la amplitud de distribución eritrocitaria (ADE).
d. Descenso de la DLco.
e. Descenso de la FVC por debajo del 50% (por encima de este valor, no ha demostrado ser un parámetro predictor de muerte).
f. Caída de la FEV1 ≥ 10% en 12 meses.
g. Puntuación en el índice fisiológico compuesto (CPI) ≥ 45%.
h. Saturación de oxígeno en aire ambiente < 90%.
i. Necesidad de oxigenoterapia domiciliaria.
j. Presencia de patrón NIU, panal de abeja o gran extensión de fibrosis.
k. Presencia de hipertensión pulmonar, cáncer de pulmón o exacerbación aguda de CPFE.
Tratamiento
No existe un tratamiento específico para CPFE. La implantación de tratamiento se basará en la extrapolación del manejo indicado para el enfismea y la fibrosis pulmonar aisladamente.5,6
a. Medidas generales: Dejar de fumar, rehabilitación pulmonar, vacunación contra la gripe, neumococo y COVID-19, oxigenoterapia según recomendaciones y considerar trasplante pulmonar.
b. Fibrosis pulmonar: decisión individualizada sobre el inicio de antifibrótico en un comité multidisciplinar. Considerar iniciar antifibrótico al diagnóstico de FPI con enfisema o ante CPFE que cumpla criterios de fibrosis progresiva. Y se debe prestar especial atención al descenso de la DLco en el seguimiento, ya que la extensión del enfisema >15% se ha asociado con una FVC relativamente estable a lo largo del tiempo.
c. Enfisema pulmonar: considerar corticoides inhalados y broncodilatadores, según indicaciones para la EPOC.
d. Manejo de las comorbilidades cardiovaculares y de cáncer de pulmón de manera individualizada.
3. BRONQUIOLITIS RESPIRATORIA (BR) Y BRONQUIOLITIS RESPIRATORIA ASOCIADA A EPI (BR-EPI)
Datos demográficos
La BR es muy frecuente en fumadores, sin encontrar diferencia respecto a prevalencia según el sexo.2
Clínica
Se diferencia de la BR-EPI en que esta última presenta clínica respiratoria y en que algunos autores apoyan que tiene mayor fibrosis alveolar.1
Los síntomas son inespecíficos; disnea de esfuerzo y tos.
Función pulmonar
Patrón ventilatorio mixto o incluso predominantemente obstructivo.2
Hiperinsuflación.
Disminución de capacidad de difusión del monóxido de carbono (DLco) en algunos casos.
Características radiológicas
La TCAR muestra nódulos centrolobulillares y opacidades irregulares en vidrio deslustrado, con engrosamiento central de las paredes bronquiales, de predominio en lóbulos superiores. Es infrecuente encontrar bronquiectasias de tracción o panal.1
En pacientes muy fumadores, puede coexistir con enfisema centrolobulillar y áreas de atrapamiento aéreo.
Hallazgos histológicos
El diagnóstico de esta enfermedad en pacientes fumadores se basa en los hallazgos clínico-radiológicos y la presencia de macrófagos pigmentados en el lavado broncoalveolar (LBA). Estos casos pueden plantear el diagnóstico diferencial con la neumonitis por hipersensibilidad, que puede cursar también con nódulos centrolobulillares, opacidades en vidrio deslustrado y atrapamiento aéreo, pero el BAL se caracteriza por la presencia de linfocitosis.2,8
Histológicamente, se observan acúmulos de macrófagos pigmentados en bronquiolos terminales y espacios alveolares adyacentes sin evidencia de fibrosis, aunque se puede presentar fibrosis peribronquiolar leve en algunos casos.8
Tratamiento y pronóstico
La medida principal en esta y en todas las enfermedades que se desarrollarán en este capítulo es el abandono del hábito tabáquico, y aunque algunos signos son reversibles, la mejoría clínica y fisiológica es lenta e incompleta. Los macrófagos pigmentados pueden persistir décadas después de dejar de fumar.2
Los corticoides inhalados y los broncodilatadores se pueden considerar en pacientes con signos de atrapamiento aéreo, patrón ventilatorio obstructivo o enfisema.
Los corticoides sistémicos a menudo se prescriben en BR-EPID progresiva, aunque la evidencia es escasa y debe realizarse una evaluación individualizada en cada paciente.2
El pronóstico de los pacientes con BR-EPID es bueno, aunque las cifras de supervivencia disponibles están basadas en series de casos.2
4. NEUMONÍA INTERSTICIAL DESCAMATIVA (NID)
Datos demográficos
Suele debutar entre la quinta y sexta década de vida, con predominio en sexo masculino con una ratio 2:1.1,2
Se piensa que la NID y la BR-EPID forman parte del mismo espectro, pero la NID es más rara, la relación con el hábito tabáquico no es tan fuerte como en la BR-EPID y presenta mayor limitación clínica, funcional y peor pronóstico que la BR-EPID.1
A diferencia de la BR-EPID, que es exclusiva de fumadores, la NID también se ha descrito en no fumadores con relación a factores ambientales, exposición a polvos o fármacos, enfermedades de tejido conectivo e infecciones.1
Clínica
La forma de presentación clínica más frecuente es la disnea de esfuerzo y la tos seca persistente.2
Existen acropaquias en casi la mitad de los pacientes.2
Función pulmonar
El patrón ventilatorio restrictivo suele ser leve o, en ocasiones, estar ausente.
Disminución de la DLco en la mayoría de los pacientes.2
Características radiológicas
Principalmente son opacidades irregulares difusas en vidrio deslustrado que pueden ser bilaterales, de predominio periférico y en campos medios e inferiores (a diferencia de otras enfermedades pulmonares por inhalación de tóxicos que afectan a campos superiores).1
Se han descrito pequeños quistes dentro de las áreas de vidrio deslustrado, pero la reticulación y la panalización son inusuales.1
Se plantea diagnóstico diferencial con neumonía intersticial no específica (NINE) e infecciones atípicas, como neumonía por Pneumocystis jiroveci. La evolución de NID a fibrosis grave es poco probable, y en este caso, es más probable que estemos ante una NINE fibrosante.1
Hallazgos histológicos
Descrito por Liebow en 1965, pero el término se sigue utilizando a pesar de que se ha demostrado que no se trata de la descamación de células epiteliales sino de un acúmulo anómalo de macrófagos pigmentados en los espacios alveolares.2
Las características histológicas se asemejan a la BR-EPID, pero la clave para diferenciarlas es la distribución y la extensión de las lesiones, que en el caso de la BR-EPID es bronquiolocéntrica y en la NID es difusa. El LBA contiene igualmente gran número de macrófagos pigmentados, y puede haber un leve incremento del porcentaje de eosinófilos, inespecífico.1,9
Tratamiento y pronóstico
La NID generalmente se caracteriza por un curso estable. En algunos pacientes, puede haber mejoría espontánea cuando abandonan el hábito tabáquico.2
La evidencia de la corticoterapia oral es escasa.
5. NEUMONÍA EOSINÓFILA AGUDA (NEA)
Datos demográficos
Es rara. Afecta sobre todo a varones entre la segunda y cuarta década de la vida.1
Puede ser idiopática o secundaria a la exposición de ciertos fármacos y tóxicos inhalados, entre ellos el tabaco, con el que se ha demostrado una relación directa, tanto con su consumo como con la recaída tras la reanudación del hábito. Al menos dos tercios de los pacientes son fumadores.1
Clínica
Es una enfermedad aguda rápidamente progresiva que cursa con fiebre, disnea e insuficiencia respiratoria hipoxémica, que a menudo requiere ventilación mecánica.1
Función pulmonar
Por su carácter agudo, no suele realizarse.
Características radiológicas
Se manifiesta como opacidades pulmonares difusas consolidativas o en vidrio deslustrado asociadas a engrosamiento septal y en ocasiones derrame pleural uni o bilateral.1,2
Se plantea el diagnóstico diferencial con el distrés respiratorio, edema pulmonar, hemorragia alveolar y neumonías infecciosas graves bilaterales (estas últimas no suelen cursar con engrosamiento septal).1,2
El análisis del derrame pleural suele ser compatible con un exudado de predominio eosinofílico.2
Hallazgos histológicos
El diagnóstico es la suma de los hallazgos clínico-radiológicos descritos y la presencia de eosinofilia (>25% eosinófilos) en el LBA, con la exclusión de otras causas de eosinofilia pulmonar o infecciosas (parásitos, hongos).1,2
La NEA asociada al tabaco comparado con otras causas es más grave y menos probable que curse con eosinofilia periférica.2
El recuento de eosinófilos en sangre puede ser normal.9
Aunque la biopsia no es necesaria en la mayoría de los casos, el cuadro histopatológico corresponde a infiltración de eosinófilos en el intersticio y espacios alveolares con rasgos de daño alveolar difuso.1
Tratamiento y pronóstico
Se asocia con un buen pronóstico una vez que se realiza el diagnóstico y se inicia tratamiento con corticoides sistémicos.2,9
6. FIBROSIS INTERSTICIAL ASOCIADA AL TABACO (FIAT)
Datos demográficos
La FIAT fue descrita por primera vez a través de hallazgos objetivados en tejido pulmonar no neoplásico obtenido de resecciones pulmonares de pacientes fumadores con carcinoma pulmonar. 8,10
Es habitualmente un hallazgo incidental en el tejido pulmonar biopsiado sin necesaria traducción radiológica.1
Su importancia puede radicar en el hecho de que, a pesar de ser un cuadro incidental sin trascendencia clínica, es muy frecuente en fumadores, en los que puede coexistir con otras enfermedades intersticiales asociadas al tabaco o idiopáticas que sí pueden ser clínicamente relevantes y presentar patrón de neumonía intersticial usual (NIU) o NINE fibrosante. En casos de progresión clínica, limitación funcional y disminución de la DLco significativa, la biopsia puede llegar a ser imprescindible porque cambia el pronóstico.1
Clínica
Estos pacientes suelen encontrarse asintomáticos o presentar síntomas inespecíficos, como tos y disnea.2
Función pulmonar
Patrón ventilatorio obstructivo, restrictivo o mixto.1,2
Puede haber disminución de la DLco.1,2
Características radiológicas
No existen unas características radiológicas definidas, pues existen patrones variables en la TCAR. Se puede encontrar nodulillos y vidrio deslustrado (hallazgos de BR), enfisema, mínima reticulación, quistes aéreos de paredes finas o pequeños quistes en las zonas de reticulación que pueden confundirse con patrón NIU.1,2
Los quistes de la FIAT, a diferencia de la NIU, tienen preferencia por los lóbulos superiores y zonas medias de los inferiores, ligeramente separados de la superficie pleural. Las imágenes quísticas con puntos centrales, que representan restos de los vasos y bronquios distales, pueden ayudar a diferenciar el enfisema en la TCAR.1,2
Hallazgos histológicos
A diferencia del resto de enfermedades tratadas en el capítulo, hasta el momento la FIAT se considera una entidad cuyo diagnóstico es exclusivamente histológico.1,2
Se caracteriza por la presencia de fibrosis intersticial hialina que se asocia a enfisema y bronquiolitis respiratoria. Consiste en engrosamiento de los septos alveolares por depósito uniforme de colágeno denso y eosinofílico que puede entremezclarse con bandas de músculo liso hipertrófico, y adopta una distribución subpleural y peribronquial acompañada casi siempre de enfisema.2,8
Se pueden encontrar macrófagos pigmentados, indicador de bronquiolitis respiratoria o mínima inflamación crónica (a diferencia de la NINE, que sería mayor) junto a la fibrosis intersticial.8
La arquitectura pulmonar está conservada, no hay panalización ni focos fibroblásticos, a diferencia del patrón de NIU.2,8
Tratamiento y pronóstico
La evolución clínica de la FIAT es favorable, en general estable o con mínima progresión.2
El abandono del hábito tabáquico es la principal recomendación.2
En casos de enfermedad grave, se sugiere el tratamiento con corticoides, pero sin que existan suficientes datos que lo apoyen.2
En resumen (Tabla II), en este capítulo se muestran las diferentes enfermedades intersticiales que guardan relación con el tabaco. La clínica de todas ellas es muy inespecífica, siendo necesaria una TCAR en todos los pacientes, mientras que en algunas ocasiones se requiere muestra histológica. El tratamiento más efectivo es dejar de fumar y en ocasiones, aunque con escasa evidencia, los corticoides y/o los broncodilatadores.
BIBLIOGRAFÍA
1. Serrano Gotarredona MP, Navarro Herrero S, Gómez Izquierdo L, Rodriguez Portal JA. Smoking-related interstitial lung disease. Radiología 64 (2022): 277-289
2. Kumar A, Cherian SV, Vassallo R, Yi ES, Ryu JH. Current Concepts in Pathogenesis, Diagnosis, and Management of Smoking-Related Interstitial Lung Diseases. Chest. 2018 Aug;154(2):394-408
3. Hunninghake GM. Interstitial lung abnormalities: erecting fences in the path towards advanced pulmonary fibrosis. Thorax. 2019 May; 74(5):506-511.
4. Silva M, Milanese G, Sverzellati N. Interstitial lung abnormalities: prognostic stratification of subtle radiological findings. Curr Opin Pulm Med. 2018 Sep;24(5):432-439.
5. Cottin V, Selman M, Inoue Y, et al. Syndrome of Combined Pulmonary Fibrosis and Emphysema: An Official ATS/ERS/JRS/ALAT Research Statement. Am J Respir Crit Care Med. 2022;206(4).
6. Wells AU, Nicholson AG, Hansell DM. Challenges in pulmonary fibrosis: smoking-induced diffuse interstitial lung diseases. Thorax. BMJ Publishing Group Ltd; 2007 Oct;62(10):904–10.
7. Desai SR, Ryan SM, Colby TV. Smoking-related Interstitial Lung Diseases: Histopathological and Imaging Perspectives. Clinical Radiology. 2003 Apr;58(4):259–68.
8. Katzenstein AL. Smoking-related interstitial fibrosis (SRIF): pathologic findings and distinction from other chronic fibrosing lung diseases. J Clin Pathol. 2013 Oct;66(10):882-7.
9. Galvin JR, Franks TJ. Smoking-related lung disease. J Thorac Imaging. 2009 Nov;24(4):27484.
10. Wick MR. Pathologic features of smoking-related lung diseases, with emphasis on smoking-related interstitial fibrosis and a consideration of differential diagnoses. Semin Diagn Pathol. 2018;35:315-23.
TABLAS Y FIGURAS
Tabla I. Hallazgos radiológicos de la TCAR en CPFE

Tabla resumen modificada y traducida de la publicación ATS/ERS/JRS/ALAT Research Statement «Syndrome of Combined Pulmonary Fibrosis and Emphysema» (referencia bibliográfica 4)
Tabla II. Resumen de las EPID asociadas a tabaco mencionadas en este capítulo (tabla original, realizada a partir de las referencias bibliográficas referidas en el presente capítulo)
TRATAMIENTO
HISTOLOGICAS
RADIOLOGIAS
FUNCIÓN PULMONAR
CLÍNICAS
DEMOGRÁFICAS
CARACTERÍSTICAS

Figura 1. Paciente hombre de 67 años muy fumador con disnea a grandes esfuerzos diagnosticado de CPFE




Cortes axiales de TCAR. Se puede observar el importante enfisema paraseptal, centrolobulillar y panacinar, predominante en lóbulos superiores, junto con patrón fibrótico donde predomina el engrosamiento septal asociado a bronquiectasias de tracción. (Imagen original de nuestra consulta de EPID).
Figura 2. FIAT en paciente hombre de 68 años exfumador asintomático

Cortes axiales de TCAR muestran discreto enfisema centrolobulillar bilateral, opacidades en vidrio deslustrado asociado a leve reticulación de predominio subpleural bilateral y también de distribución peribronquial en lóbulo superior izquierdo y base derecha. Presenta además alguna zona de mayor hipoatenuación subpleural que podría corresponder a atrapamiento aéreo (sería necesario cortes en espiración para confirmación). (Imagen original de nuestra consulta de EPID).
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
10
EPID ASOCIADAS A CONECTIVOPATÍAS
Autores
Fernanda Hernández González
Paloma Millán Billi
Coordinador
Diego Castillo Villegas
1. INTRODUCCIÓN
Ante la presencia de una enfermedad pulmonar intersticial difusa (EPID), debemos realizar un estudio amplio y sistemático, con un extenso diagnóstico diferencial que incluya diferentes tipos de enfermedades autoinmunes sistémicas (EAS), ya que la EPID puede ser su principal o única expresión.
En general, la sintomatología común en todas ellas es la disnea y la tos seca, aunque también se debe realizar una búsqueda activa de signos y síntomas extrapulmonares. El diagnóstico siempre es multidisciplinar, y en él intervendrán especialistas en enfermedades autoinmunes sistémicas. El manejo de estos pacientes es complejo y requiere una interacción continua y activa de los diferentes especialistas para un seguimiento y tratamiento óptimos.
2. ENFERMEDAD
PULMONAR INTERSTICIAL DIFUSA ASOCIADA A ESCLEROSIS
SISTÉMICA
La esclerosis sistémica (ES) es una enfermedad caracterizada por un aumento de colágeno en los tejidos que provoca manifestaciones clínicas características y variables consistentes en engrosamiento de la piel (esclerodermia), alteraciones de varios sistemas (gastrointestinal, renal y musculoesquelético), fenómeno de Raynaud, así como hipertensión arterial pulmonar o EPID.
Existen diversas clasificaciones, aunque la más idónea considera cuatro subtipos de enfermedad (limitada, difusa, sin esclerodermia, preesclerodermia), ya que combina hallazgos clínicos en diferentes órganos, capilaroscopia e inmunología. (Tabla I).
Más del 50% de pacientes con EPID-ES presentan clínica respiratoria asociada y suelen tener un inicio precoz, especialmente en la forma difusa y en pacientes con anti-topoisomerasa. Así, en pacientes con sospecha clínica o diagnóstico confirmado de ES, según los criterios del American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR), se deberá realizar un cribado para descartar EPID subyacente.
Detección de la EPID-ES
Para la detección de la EPID-ES, se debe tener en cuenta lo siguiente:
Antecedentes
- Personales o familiares de ES, ya que existe un aumento de riesgo atribuido a polimorfismos en la región del HLA que parece depender del estado de autoanticuerpos (HLA-DRB111 confiere un riesgo considerable en pacientes con anticuerpos anti-topoisomerasa 1 o Scl-70, mientras que HLA-DRB1301 se asocia con un mayor riesgo en pacientes sin anticuerpos anti-topoisomerasa).
- Tóxicos o fármacos previos para realizar un diagnóstico diferencial con los síndromes esclerodermiformes.
Factores de riesgo EPID-ES
- Tabaquismo activo.
- Sexo masculino.
- Edad avanzada en el momento de presentación.
Síntomas extrapulmonares
- Fenómeno de Raynaud.
- Afectación cutánea (telangiectasias, engrosamiento y endurecimiento cutáneo, microstomía, úlceras digitales).
- Clínica gastrointestinal.
- Afectación renal (insuficiencia renal con o sin HTA, anemia hemolítica microangiopática).
Exploración física
- Crepitantes tipo velcro de predominio en las bases pulmonares.
- Presencia de signos extrapulmonares sugestivos de ES.
Exploraciones complementarias
- TACAR: El patrón más frecuente es la neumonía intersticial no específica (NINE). Se puede observar patrón de neumonía intersticial usual (NIU) de manera infrecuente. La extensión es importante para valorar el inicio del tratamiento.
- Pruebas funcionales respiratorias (PFR): es característico un patrón ventilatorio restrictivo con alteración de la difusión alveolar.
- Analítica: anticuerpos antinucleares (ANA), anti-topoisomerasa, anticentrómero, anti-RNA polimerasa III, y otros (antiTh/To, antiPM/Scl) son fundamentales.
- Otras exploraciones útiles: capilaroscopia, ecocardiograma, tránsito gastrointestinal (reflujo gastroesofágico puede empeorar la EPID-ES).
Pronóstico
Actualmente, la presencia de EPID es la causa más frecuente de muerte en pacientes con ES, con una prevalencia de hasta el 30% y una mortalidad a 10 años de hasta el 40%. La presencia de hipertensión pulmonar empeora el pronóstico de manera significativa. Así, los factores de peor pronóstico son:
- Descenso de la FVC > 10%.
- Descenso de la FVC entre 5-9% con disminución de DLCO > 15%.
- -20% fibrosis en TACAR.
- Ratio arteria pulmonar/aorta > 1:1.
- Patrón NIU.
Enfoque terapéutico
Se recomienda iniciar tratamiento en pacientes sintomáticos con una enfermedad extensa definida por: 1) TCAR con afectación de extensión indeterminada o < 20%; o 2) patrón restrictivo con FVC > 70%, así como un deterioro de la FVC > 10% durante el seguimiento, o de la DLco (siempre descartando otras causas).
Es importante minimizar las dosis de glucocorticoides por el riesgo de crisis renal esclerodérmica, aunque se permiten dosis iniciales de 10-15 mg diarios con reducción progresiva en 1 mes hasta 5-7,5 mg/día.
El micofenolato de mofetilo (1-1,5 g cada 12 horas durante 1-3 años) constituye la primera línea terapéutica por su buena tolerancia, con una efectividad similar a la ciclofosfamida oral. Como alternativa, puede utilizarse ciclofosfamida intravenosa (600 mg/m2/mes durante 6 meses) seguida de micofenolato o azatioprina (2-3 mg/kg/día).
El tratamiento con rituximab (1 g cada 2 semanas, 2 dosis) y el tocilizumab suelen utilizarse en casos refractarios. En casos de fibrosis pulmonar progresiva, se puede asociar tratamiento antifibrótico (nintedanib 150 mg/12 hs), y en casos avanzados a pesar de tratamiento óptimo, debe valorarse la opción de un trasplante pulmonar.
3. ENFERMEDAD PULMONAR INTERSTICIAL DIFUSA ASOCIADA A MIOPATÍAS INFLAMATORIAS IDIOPÁTICAS
Las miopatías inflamatorias idiopáticas (MII) son un grupo de enfermedades autoinmunes con afectación muscular, cutánea, articular y pulmonar que comparten la debilidad muscular en mayor o menor medida. Se caracteriza por presencia histopatológica de inflamación y necrosis muscular. Existen varias entidades: dermatomiositis, polimiositis, miopatía necrosante inmunomediada y miositis con cuerpos de inclusión (forma esporádica). Cualquiera de estas variantes puede asociarse a enfermedades autoinmunes sistémicas u órgano-específicas.
El diagnóstico de cada subtipo requiere recoger una combinación de manifestaciones clínicas detallando el tiempo de evolución, el patrón de afectación muscular y de otros órganos, el grado de elevación de enzimas musculares, la presencia de determinados anticuerpos y los hallazgos en resonancia magnética o electromiografía.
La prevalencia de EPID es variable (15-70%) dependiendo del tipo de anticuerpo concreto. Los casos con dermatomiositis con formas hipo o amiopáticas (afectación muscular leve o ausente) que presenten anticuerpos ant-MDA5, característicamente asocian un elevado riesgo de EPID grave y rápidamente progresiva.
El síndrome antisintetasa es considerado como una forma de solapamiento. Se acepta el diagnóstico en casos con presencia de anticuerpos antisintetasa, en especial anti-Jo-1, por ser el más frecuente, y al menos una manifestación mayor (miositis, artritis o EPID), aunque actualmente los criterios clasificatorios no están validados. Por tanto, la presencia de EPID es muy frecuente, ya que es uno de los componentes de la tríada clásica.
Detección de EPID
En la detección de EPID asociada a MII, se debe tener en consideración:
Síntomas extrapulmonares
- Debilidad.
- Disfagia.
- Eritema en heliotropo.
- Pápulas de Gottron.
- Fenómeno de Raynaud.
- Artritis/artralgias.
- Fiebre.
- Fisuras digitales distales (“manos de mecánico”).
- Síndrome constitucional.
Exploración física
- Crepitantes tipo velcro de predominio en las bases pulmonares
- Presencia de signos extrapulmonares sugestivos de MII
Exploraciones complementarias
- TACAR: El patrón más frecuente es la NINE, aunque se puede observar patrón NIU de manera infrecuente. Pueden presentar neumonía organizada o patrón de solapamiento NINE/neumonía organizada (NO).
- PFR: es característica una alteración restrictiva con alteración de la difusión alveolar. Se debe evaluar la afectación muscular diafragmática como posible causa de alteración de la FVC.
- Analítica: ANA (menos frecuente), anti-MDA5, anti-TIF1gamma, anti-Ro52, anti-RNA sintetasa (existen 8 tipos diferentes, aunque sólo 6 son detectables mediante ELISA, CLIA o inmunoblot: anti-Jo1, anti-PL12, anti-PL7, anti-OJ, anti-EJ, anti-KS), entre otros anticuerpos. Inespecíficos: Aldolasa, CPK, LDH, ALAT (GPT), ASAT (GOT).
- Otras exploraciones útiles: capilaroscopia, electromiograma, resonancia nuclear magnética de todo el cuerpo, biopsia muscular.
Enfoque terapéutico
Debido a que la mayor parte de la información parte de estudios retrospectivos, no existe un consenso respecto al tratamiento de la EPID-MII.
En los casos con formas rápidamente progresivas, se preferirá un abordaje agresivo inicial con bolus de metilprednisolona intravenosa (500 mg/día durante 3 días) seguidos de prednisona 1 mg/kg/día (o su equivalente de metilprednisolona) durante 1 mes con posterior pauta descendente, asociado a otros inmunosupresores (rituximab o ciclofosfamida intravenosa). También se debe valorar el uso de recambios plasmáticos con perfusión de inmunoglobulinas.
En el caso de formas más crónicas, se recomienda el uso de corticoterapia a dosis moderadas (0,5 mg/kg/día) con micofenolato de mofetilo (1 g/12 hs) o azatioprina (2 mg/kg/día). Otros tratamientos inmunosupresores anticalcineurínicos, como tacrólimus (2 mg/12 hs, controlando niveles séricos) o ciclosporina, pueden considerarse como alternativas, especialmente en el síndrome antisintetasa.
En pacientes con criterios de fibrosis pulmonar progresiva, se puede asociar tratamiento antifibrótico (nintedanib 150 mg/12 hs), y en casos avanzados a pesar de tratamiento óptimo, se contemplará el trasplante pulmonar.
4. ENFERMEDAD PULMONAR INTERSTICIAL DIFUSA ASOCIADA A ARTRITIS REUMATOIDE
La artritis reumatoide (AR) es una enfermedad crónica que se caracteriza principalmente por la inflamación de las articulaciones. Desde el punto de vista respiratorio, encontramos afectación intersticial, de vía aérea (bronquiectasias, bronquiolitis), pleural y, en menor medida, vascular.
La detección precoz de la AR en pacientes con EPID es fundamental, aunque es importante tener en cuenta que la manifestación pulmonar puede preceder en años a las manifestaciones articulares (10-20%) por lo que se debe re-interrogar de manera periódica sobre los síntomas, en especial en pacientes con EPID de etiología no establecida.
La incidencia y la prevalencia de la EPID-AR varían de un estudio a otro. Se ha descrito una prevalencia de EPID en pacientes con AR entre 12-56%.
Detección de EPID asociada a AR
La principal sintomatología respiratoria será la disnea de esfuerzo y la tos. En la detección de EPID asociada a AR se debe tener en cuenta:
Antecedentes
Personales o familiares de AR y otras enfermedades autoinmunes sistémicas. Está descrita la mutación del gen MUC5B tanto en los casos de AR como de EPID.
Factores de riesgo EPID-AR
- Consumo de tabaco.
- Sexo masculino.
- Edad (> 60años).
- Positividad para anticuerpos anti-péptidos citrulinados (Anti-CCP) y factor reumatoide (FR).
Síntomas extrapulmonares
- Dolor articular persistente, presencia de edema y rigidez, sobre todo en articulaciones pequeñas; normalmente, afectación simétrica.
- Duración de los síntomas.
- Rigidez matutina que dura más de 30 minutos.
- Deformación de articulaciones (en fases avanzadas).
Exploración física
- Detección de edema o deformidad de las articulaciones (sobre todo de manos).
- Búsqueda de otros hallazgos, como nódulos reumatoideos.
- Crepitantes tipo velcro de predominio en las bases pulmonares.
- Presencia de acropaquia.
Exploraciones complementarias
- TACAR: los patrones más frecuentes son la NIU y la NINE. Asimismo, se puede observar NO e incluso nódulos reumatoides. Se debe realizar diagnóstico diferencial con procesos infecciosos o neoplásicos.
- PFR: patrón ventilatorio restrictivo que se podrá acompañar de un descenso de la difusión alveolar. Patrón obstructivo o mixto en fumadores o con afectación de la vía aérea.
- Analítica: marcadores específicos: anti-CCP. No específicos: FR, PCR o VSG.
- Otras exploraciones útiles: radiografía de manos, ecografía para valoración del edema e inflamación articular.
Pronóstico
El riesgo de mortalidad es entre 3 y 10 veces mayor si se asocia a EPID. En el caso de los pacientes con EPID fibrosante, el diagnóstico de AR puede ampliar las opciones terapéuticas.
Los siguientes factores están relacionados a un peor pronóstico:
- La edad avanzada, el género masculino y la duración de la AR.
- La extensión de la afectación radiológica (> 20-30%) y el patrón NIU o probable NIU.
- FVC basal disminuida o descenso de la misma en el seguimiento.
- Distancia recorrida en el test de la marcha.
- Consumo tabáquico. Éste se relaciona con peor control de síntomas articulares.
Según las recomendaciones SER-SEPAR, el índice GAP y el fisiológico compuesto (CPI) tienen utilidad para predecir el riesgo de mortalidad.
Enfoque terapéutico
La decisión de inicio de tratamiento dependerá de la sintomatología y de la afectación radiológica y funcional. En pacientes asintomáticos desde el punto de vista respiratorio y con PFR estables, no será necesario el tratamiento específico para la EPID.
Según las recomendaciones de la SER-SEPAR en aquellos casos con criterio de tratamiento, se valorarán las opciones terapéuticas que se detallan a continuación.
Glucocorticoides: se pueden utilizar en los patrones con componente inflamatorio relevante (NINE no fibrótica, NO). Se recomienda la menor dosis (< 30 mg/día de prednisona) y el menor tiempo posibles, ya que su uso prolongado de > 7,5 mg/día aumenta el riesgo de infecciones graves y empeora el riesgo cardiovascular.
En caso de necesidad por gravedad inicial o exacerbación aguda, se puede valorar la administración de pulsos de metilprednisolona (3 días) con dosis variable, entre 125 y 250 mg al día o 500 mg/día en los casos más graves.
Metotrexato (MTX): fármaco con uso discutido en los últimos años por la posibilidad de neumonitis asociada. Según las recomendaciones SER-SEPAR, (Figura 1)
Leflunomida: No se ha demostrado que su uso se asocie a aumento del riesgo de EPID, salvo en población asiática, por lo que en nuestro entorno se puede utilizar como alternativa a MTX.
FAMEs biológicos: Se sugiere el uso tanto de abatacept como de rituximab (vigilar infecciones respiratorias o urinarias con su uso prolongado por el desarrollo de hipogammaglobulinemia).
Antifibróticos: Se recomienda el uso de nintedanib en los pacientes con criterios de fibrosis pulmonar progresiva a pesar de la optimización del tratamiento de la AR.
Valorar derivación precoz a unidades de trasplante pulmonar.
Tratamiento sintomático/paliativo.
5.
ENFERMEDAD
PULMONAR INTERSTICIAL DIFUSA ASOCIADA A SÍNDROME DE SJÖGREN PRIMARIO
El síndrome de Sjögren primario (pSS) es una enfermedad autoinmune sistémica crónica que se caracteriza principalmente por la infiltración linfocitaria glandular con sequedad ocular y bucal, fatiga y dolor articular. Se puede encontrar afectación sistémica en 30-40% de los pacientes (hepática, renal, neurológica). Es importante tener presente que el riesgo de linfoma de células B es entre 15 y 20 veces mayor en pacientes con SSp.
La afectación pulmonar ocurre en un 9-20% de los pacientes en forma de:
- EPID: la NINE es el patrón más frecuente. Puede coexistir con la afectación de vía aérea.
- Vía aérea: bronquiolitis folicular, hiperrespuesta bronquial y bronquiectasias (aumento de riesgo de infecciones).
- Tromboembolismo pulmonar, hipertensión pulmonar.
- Complicaciones: amiloidosis pulmonar (rara) y linfoma pulmonar.
El linfoma suele ser del tipo MALT y se pueden encontrar áreas consolidativas o nódulos pulmonares en la TACAR. En estos casos, debemos interrogar sobre síntomas B y realizar una biopsia pulmonar para confirmar la sospecha diagnóstica.
Detección del Síndrome de Sjögren asociada a EPID
La principal sintomatología respiratoria será la disnea de esfuerzo y la tos seca (muchas veces multifactorial). En la detección del Síndrome de Sjögren asociada a EPID, se debe tener en consideración los puntos que se detallan a continuación.
Antecedentes
- Personales o familiares de pSS.
- Enfermedades autoinmunes sistémicas.
- Infecciones víricas previas (citomegalovirus, VIH, hepatitis C).
- Fármacos que puedan causar síndrome seco (antidepresivos tricíclicos).
2. Síntomas extrapulmonares
- Oral: xerostomía, caries, queilitis angular y úlceras bucales.
- Ocular: xeroftalmia, fotofobia, queratitis ocular, úlceras, conjuntivitis, uveítis, escleritis, vasculitis retinal entre otros.
- Parotiditis o edema de glándulas salivales.
- Sequedad de órganos genitales, dispareunia.
- Afectación cutánea: sequedad, fenómeno de Raynaud, púrpura.
- Astenia, ansiedad o depresión.
- Artralgias y/o mialgias.
- Afectación neurológica (rara).
Exploración física
- Revisión de parótidas y alteraciones cutáneas.
- Crepitantes tipo velcro de predominio en las bases pulmonares.
- Sibilantes.
- Presencia de acropaquia (raro).
Exploraciones complementarias
- TACAR: realizar estudio con cortes en inspiración/espiración. El patrón más frecuente es la NINE (hasta un 45%), aunque podemos encontrar patrón de NIU y de NO. La neumonía intersticial linfoidea (NIL) es el patrón más específico, aunque no patognomónico.
- PFR: patrón ventilatorio restrictivo que se podrá acompañar de un descenso de la difusión alveolar. Patrón obstructivo o mixto si hay afectación de la vía aérea. Realizar prueba broncodilatadora para valorar la presencia de hiperrespuesta bronquial.
- Analítica: Marcadores específicos: anti-Ro60. No específicos: anti-Ro52, Anti-La, ANA, FR. Otros: anemia, leucopenia, hipergammaglobulinemia policlonal, crioglobulinas.
- Broncoscopia con lavado bronco-alveolar (LBA): linfocitosis (>20%) en hasta un 64% de los pacientes.
- Otras exploraciones útiles: test de Schirmer, biopsia de glándula salival menor, gammagrafía glándulas salivales, sialometría.
Criterios diagnósticos de pSS
El diagnóstico se realizará tras valoración conjunta por parte de médicos especialistas en enfermedades autoinmunes siguiendo los criterios diagnósticos de ACR-EULAR 2016 con una puntuación de 4 o más de la tabla II
Enfoque terapéutico
Se valorará inicio de tratamiento para EPID-pSS en pacientes sintomáticos, con alteración funcional y según la afectación radiológica. Se recomienda (Figura 2):
1. Primera línea:
- Corticoterapia con dosis inicial entre 0,5 y 1 mg/kg/día (se recomienda no superar los 60 mg/d).
- Micofenolato o azatioprina como inmunosupresores de mantenimiento y ahorradores de corticoides.
2. Segunda línea: rituximab, ciclosporina o tacrolimus.
3. Si cumple criterios de fibrosante progresiva, valorar nintedanib.
4. Valorar derivación precoz a unidades de trasplante pulmonar.
5. Tratamiento sintomático o paliativo.
6. ENFERMEDAD PULMONAR INTERSTICIAL DIFUSA ASOCIADA A OTRAS ENFERMEDADES DEL TEJIDO CONECTIVO
Lupus eritematoso sistémico (LES)
La afectación intersticial es muy poco frecuente en pacientes con LES, aunque se puede manifestar en forma de hemorragia alveolar difusa, neumonía intersticial aguda e insuficiencia respiratoria grave. En estos casos, será importante el diagnóstico diferencial con procesos infecciosos.
La afectación pleural es la manifestación más frecuente con derrame pleural en hasta un 60% de los pacientes y con el síndrome del pulmón encogido como manifestación poco frecuente. También podemos encontrar hipertensión pulmonar o enfermedad tromboembólica, sobre todo en asociación con el síndrome antifosfolipídico.
El tratamiento será dirigido a la enfermedad de base, sintomático en caso de hemoptisis y, si existe afectación fibrótica progresiva, plantear el uso de nintedanib.
Enfermedad mixta del tejido conectivo
Es una enfermedad que se caracteriza por la presencia de títulos elevados de anticuerpos anti-RNP y sintomatología similar al LES, esclerodermia y miositis. Podemos encontrar afectación pulmonar en el 80% de los pacientes, principalmente en forma de hipertensión pulmonar o EPID. Los hallazgos radiológicos son variables, siendo el más frecuente la NINE. El tratamiento se realizará con corticoterapia o fármacos inmunosupresores, siempre de manera multidisciplinar.
Conclusión
En la valoración inicial de todo paciente con EPID, se debe realizar una anamnesis dirigida a descartar enfermedades autoinmunes sistémicas subyacentes por su implicación terapéutica y pronóstica. La neumonía intersticial no específica (NINE) constituye el patrón radiológico más frecuente, aunque en algunas entidades existe predominancia de otros tipos de patrones. En la gran mayoría de los casos, no se requiere de biopsia pulmonar, excepto en aquellos en que existan dudas con otras causas alternativas. El manejo de estos pacientes es multidisciplinar, siendo el tratamiento más común los glucocorticoides y los fármacos inmunosupresores. El tratamiento antifibrótico con nintedanib se debe considerar en los casos con fibrosis pulmonar progresiva, y en algunos casos se debe realizar una derivación precoz a programas de trasplante pulmonar.
BIBLIOGRAFÍA
1. Otylia Kowal-Bielecka, Jaap Fransen, Jerome Avouac, Mike Becker, Agnieszka Kulak, Yannick Allanore, Oliver Distler, Philip Clements, Maurizio Cutolo, Laszlo Czirjak, Nemanja Damjanov, Francesco Del Galdo, Christopher P Denton, Jörg H W Distler, Ivan Foeldvari, Kim Figelstone, Marc Frerix, Daniel E Furst, Serena Guiducci, Nicolas Hunzelmann, Dinesh Khanna, Marco Matucci-Cerinic, Ariane L Herrick, Frank van den Hoogen, Jacob M van Laar, Gabriela Riemekasten, Richard Silver, Vanessa Smith, Alberto Sulli, Ingo Tarner, Alan Tyndall, Joep Welling, Frederic Wigley, Gabriele Valentini, Ulrich A Walker, Francesco Zulian, Ulf Müller-Ladner; EUSTAR CoauthorsUpdate of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017 Aug;76(8):1327-1339.
2. Cervera R, Espinosa G, Ramos-Casals M, Hernández-Rodríguez J, Prieto-González S, Espigol-Frigole G, Cid MC. Enfermedades autoinmunes sistémicas. Diagnóstico y tratamiento. Editorial Panamericana. ISBN9788491106524.
3. Van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, Matucci-Cerinic M, Naden RP, Medsger TA Jr, Carreira PE, Riemekasten G, Clements PJ, Denton CP, Distler O, Allanore Y, Furst DE, Gabrielli A, Mayes MD, van Laar JM, Seibold JR, Czirjak L, Steen VD, Inanc M, Kowal-Bielecka O, Müller-Ladner U, Valentini G, Veale DJ, Vonk MC, Walker UA, Chung L, Collier DH, Csuka ME, Fessler BJ, Guiducci S, Herrick A, Hsu VM, Jimenez S, Kahaleh B, Merkel PA, Sierakowski S, Silver RM, Simms RW, Varga J, Pope JE. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013 Nov;65(11):2737-47.
4. Apostolos Perelas, Richard M Silver, Andrea V Arrossi, Kristin B Highland. Systemic sclerosis-associated interstitial lung disease. Lancet Respir Med. 2020 Mar;8(3):304-320.
5. Ganesh Raghu, Martine Remy-Jardin, Luca Richeldi, Carey C Thomson, Yoshikazu Inoue, Takeshi Johkoh, Michael Kreuter, David A Lynch, Toby M Maher, Fernando J Martinez, Maria Molina-Molina, Jeffrey L Myers, Andrew G Nicholson, Christopher J Ryerson, Mary E Strek, Lauren K Troy, Marlies Wijsenbeek, Manoj J Mammen, Tanzib Hossain, Brittany D Bissell, Derrick D Herman, Stephanie M Hon, Fayez Kheir, Yet H Khor, Madalina Macrea, Katerina M Antoniou, Demosthenes Bouros, Ivette Buendia-Roldan, Fabian Caro, Bruno Crestani, Lawrence Ho, Julie Morisset, Amy L Olson, Anna Podolanczuk, Venerino Poletti, Moisés Selman, Thomas Ewing, Stephen Jones, Shandra L Knight, Marya Ghazipura, Kevin C Wilson. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022 May 1;205(9):e18-e47.
6. Meyer A, Scirè CA, Talarico R, Alexander T, Amoura Z, Avcin T, Barsotti S, Beretta L, Blagojevic J, Burmester G, Cavazzana I, Cherrin P, Damian L, Doria A, Fonseca JE, Furini F, Galetti I, Houssiau F, Krieg T, Maddalena L, Launay D, Campanilho-Marques R, Martin T, Matucci-Cerinic M, Moinzadeh P, Montecucco C, Moraes-Fontes MF, Mouthon L, Neri R, Paolino S, Piette Y, Rednic S, Tamirou F, Tincani A, Toplak N, Bombardieri S, Hachulla E, Mueller-Ladner U, Schneider M, Smith V, Vieira A, Cutolo M, Mosca M, Cavagna L. Idiopathic inflammatory myopathies: state of the art on clinical practice guidelines. RMD Open. 2019 Feb 26;4(Suppl 1):e000784.
7. Narváez J, Díaz Del Campo Fontecha P, Brito García N, Bonilla G, Aburto M, Castellví I, Cano-Jiménez E, Mena-Vázquez N, Nieto MA, Ortiz AM, Valenzuela C, Abad Hernández MÁ, Castrejón I, Correyero Plaza M, Francisco Hernández FM, Hernández Hernández MV, Rodríquez Portal JA. SER-SEPAR recommendations for the management of rheumatoid arthritis-related interstitial lung disease. Part 2: Treatment. Reumatol Clin (Engl Ed). 2022 Nov;18(9):501-512. doi: 10.1016/j.reumae.2022.03.004. Epub 2022 Sep 3. PMID: 36064885.
8. Luppi F, Sebastiani M, Silva M, Sverzellati N, Cavazza A, Salvarani C, Manfredi A. Interstitial lung disease in Sjögren's syndrome: a clinical review. Clin Exp Rheumatol. 2020 Jul-Aug;38 Suppl 126(4):291-300. Epub 2020 Oct 23. PMID: 33095142.
9. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ, Mariette X; International Sjögren's Syndrome Criteria Working Group. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren's Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol. 2017 Jan;69(1):35-45. doi: 10.1002/art.39859. Epub 2016 Oct 26. PMID: 27785888; PMCID: PMC56 50478.
10. Lee AS, Scofield RH, Hammitt KM, Gupta N, Thomas DE, Moua T, Ussavarungsi K, St Clair EW, Meehan R, Dunleavy K, Makara M, Carsons SE, Carteron NL; Consensus Expert Panel (CEP) Members. Consensus Guidelines for Evaluation and Management of Pulmonary Disease in Sjögren's. Chest. 2021 Feb;159(2):683-698. doi: 10.1016/j.chest.2020.10.011. Epub 2020 Oct 16. PMID: 33075377; PMCID: PMC8438162.
TABLAS Y FIGURAS
Tabla I. Hallazgos clínicos en diferentes órganos, capilaroscopia e inmunología

Tabla II. Diagnósticos de ACR-EULAR

Adaptado de Shiboski CH, et al. International Sjögren's Syndrome Criteria Working Group. 2016 American College of Rheumatology / European League Against Rheumatism Classification Criteria for Primary Sjögren's Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. ArthritisRheumatol. 2017.
Figura 1. Metotrexato (MTX)

Adaptado de Narváez et al. SER-SEPAR recommendations for the management of rheumatoid arthritis-related interstitial lung disease. Reumatol Clin (Engl Ed). 2022.
Figura 2. Algoritmo para el manejo según las recomendaciones SER-SEPAR
PACIENTES CON EPID ASOCIADA A AR
SITUACIÓN
PACIENTES CON CRITERIOS DE TRATAMIENTO
PACIENTES CON CRITERIOS DE TRATAMIENTO (evidencia de deterioro clínico, funcional y/o radioloógico)
• GLC orales (en casos graves: bolus de MTP± bolus de CYC).
• Control de la actividad de la AR:
- FAMESC: individualizar el uso de MTX¹.
- Si es necesario por severidad de la clinica articular o pulmonar añadir tratamiento biológico (RTXOABA).
Mejoría
Mejoría NO NIU
En caso de respuesta Inadecuada a RTXOABA valorar el uso de un Inhibidor de la IL-60 de un FAME sintético dirigido
Mantener tratamiento
• Añadir ABA o RTX si no lo tomaban (o valorar switching entre ellos).
- Si evidencia de EPID fibrosante progresiva añadir al tratamiento nintedanib, manteniendo el tratamiento de fondo (excepto GLC, cuyo uso se tendría que individualizar en esta fase).
-Si se decide ensayar inmunosupresores, se aconseja priorizar MMF.
Mantener tratamiento
Mejoría
• No GLC excepto en caso de exacerbación aguda de la EPID.
• Nintedanib si evidencia de EPID fibrosante progresiva.
• Control de la actividad de la AR:
- FAMESC: individualizar el uso de MTX¹
-Si es necesario por severidad de la clinica articular o pulmonar afadir tratamiento biológico (RTXOABA).
Mejoría Sí
• Añadir ABA RTX si no lo tomaban (o valorar switching entre ellos).
- Si se decide ensayar inmunosupresores, se aconseja prioritar MMF.
Mantener tratamiento NIU
Mantener tratamiento
Transplante pulmonar
Transplante pulmonar
Según Narváez et al. SER-SEPAR recommendations for the management of rheumatoid arthritis-related interstitial lung disease. Reumatol Clin (Engl Ed). 2022.

MMF
Adaptado de Lee AS et al. Consensus Guidelines for Evaluation and Management of Pulmonary Disease in Sjögren's. Chest. 2021.
Figura 3. Algoritmo para el manejo terapéutico
(micofenolato), AZA (azatioprina), RTX (rituximab).
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
11
EPID CON CARACTERÍSTICAS
AUTOINMUNES (IPAF)
Autores
Juan Rigual Bobillo
Ana Jaureguizar Oriol
Coordinadora
Belén López-Muñiz Ballesteros
1. INTRODUCCIÓN
Las enfermedades pulmonares intersticiales difusas (EPID) son un grupo heterogéneo de entidades clínico-patológicas (más de 200 descritas) con unas manifestaciones clínicas, radiológicas y funcionales comunes que afectan de forma difusa al parénquima pulmonar produciendo en él una inflamación y/o fibrosis con un comportamiento, una respuesta terapéutica y un pronóstico distintos. A día de hoy, las EPID se clasifican en 5 grupos: idiopáticas, autoinmunes, relacionadas con exposición, enfermedades quísticas, sarcoidosis y otras. El 30% de las EPID son secundarias a enfermedades del tejido conectivo (ETC) y forman un grupo en expansión debido a los avances diagnósticos que, en los últimos años, permiten una mejor caracterización de los pacientes.¹
La incidencia y la prevalencia de la EPID varían en las diferentes ETC. No obstante, en algunos casos los pacientes con EPID se presentan con ciertos rasgos propios de ETC, pero sin llegar a desarrollar una enfermedad autoinmune sistémica definida como tal. Estas neumopatías intersticiales, a las que se les presupone una base autoinmune, han recibido, a lo largo de las últimas décadas, diferentes nomenclaturas: «enfermedad del tejido conectivo indiferenciada», «enfermedad del tejido conectivo predominantemente pulmonar» o «EPID con rasgos autoinmunes».2-4 No obstante, la importante heterogeneidad que se encontraba detrás de la diferente terminología hacía muy difícil extraer cualquier tipo de conclusión en relación con este perfil de paciente. En el año 2015, la European Respiratory Society (ERS) y la American Thoracic Society (ATS) definen el
concepto actual y vigente de «Neumonía Intersticial con Características Autoinmunes» (Interstitial pneumonia with autoinmune features, IPAF) para clasificar a los pacientes que desarrollan una enfermedad pulmonar intersticial con rasgos de enfermedad autoinmune sistémica subyacente que no cumplen los criterios de una ETC en concreto.5 No obstante, es fundamental tener en consideración que esta definición surge en el ámbito de la investigación y con el objetivo de ser un apoyo clasificatorio, y no como unos criterios diagnósticos para una enfermedad concreta.
De hecho, hasta el momento, el concepto de IPAF es fuente de frecuentes debates, y realmente no existe una clara certeza de si esta situación clínica pertenece al conjunto de las EPID idiopáticas (dentro del grupo de las inclasificables) o de si, por el contrario, se debería englobar dentro de las afectaciones pulmonares intersticiales secundarias a ETC. Quizá el punto que más se aproxima a la realidad es aquel que sitúa la IPAF como una entidad clínica que se encuentra en la intersección de las enfermedades autoinmunes del tejido conectivo y las enfermedades pulmonares intersticiales idiopáticas (Figura 1).6
2. EPIDEMIOLOGÍA
Teniendo en cuenta la variabilidad terminológica utilizada y que el término IPAF como tal es un concepto relativamente novedoso, en la actualidad existen pocos informes epidemiológicos, mientras que los disponibles son variables. Se estima que los pacientes con IPAF constituyen entre el 7% y el 34% de todos los pacientes con EPID. La edad media de inicio son los 60-65 años, y aunque hay estudios que reportan una predominancia de mujeres nunca fumadoras, en líneas generales se acepta un equilibrio de frecuencias entre ambos sexos.7-9
3. ETIOLOGÍA Y PATOGÉNESIS
La etiología de IPAF no está completamente dilucidada. Se ha hipotetizado con la influencia de factores ambientales e inmunológicos. No obstante, es muy complicado determinar las vías etiopatogénicas implicadas en los pacientes con IPAF, ya que, como se explicaba anteriormente, esta definición no hace referencia a una enfermedad concreta, sino que es más bien una forma de clasificar a un determinado grupo de pacientes.
A pesar de ello, en los últimos años se han descrito algunas características diferenciales de los pacientes clasificados como IPAF en lo referente a la longitud de los telómeros de los leucocitos y el polimorfismo del MUC5B con respecto a los pacientes con fibrosis pulmonar idiopática (FPI) y EPID asociada a ETC.10 Estos hallazgos hacen pensar en factores genéticos implicados en las patogénesis de la IPAF, aunque son necesarios más estudios en este ámbito.
4. DIAGNÓSTICO
El diagnóstico de IPAF viene definido por una serie de criterios englobados en tres secciones o ámbitos: el ámbito clínico, el ámbito morfológico y el ámbito serológico. Estos criterios fueron establecidos por consenso de expertos en el año 2015, de manera multidisciplinar e internacional.5 Según estas recomendaciones, vigentes hoy en día, se podrá clasificar una EPID como IPAF siempre y cuando se trate de una enfermedad pulmonar intersticial presente en una tomografía de tórax de alta resolución (TCAR) o en una biopsia pulmonar quirúrgica, se hayan excluido diagnósticos alternativos, no se reúnan los criterios específicos de una ETC concreta y se cumpla como mínimo un criterio de dos ámbitos distintos (Tabla I).
En el ámbito clínico se incluyen algunos signos clínicos típicos de ETC. No obstante, su mera presencia aislada no permite un diagnóstico de ETC concreta. Estos criterios clínicos son: las «manos de mecánico» (entendidas como lesiones escamosas y fisuradas en la parte distal de los dedos), úlceras digitales distales, presencia de artritis inflamatoria o rigidez articular matinal, telangiectasias palmares, fenómeno de Raynaud y signo de Gottron o edema digital que aparece sin causa aparente.
El ámbito serológico hace referencia a la presencia de determinados anticuerpos específicos habitualmente relacionados con ETC. Se acepta como criterio serológico un título de anticuerpos antinucleares (ANA) mayores de 1:320 con patrón difuso u homogéneo, o en cualquier concentración si el patrón es patrón nucleolar o centromérico. También forman parte de este ámbito el factor reumatoide cuyo valor sea mayor del doble del límite superior de la normalidad, y los anticuerpos Anti-CCP, Anti-dsDNA, Anti-Ro (SS-A), Anti-La (SSB), Anti-ribonucleoproteina, Anti-Smith, Anti-topoisomerasa (Scl-70), Anti-tRNA sintetasa (Jo-1, PL-7, PL-12; EJ, OJ, KS, Zo, tRS) Anti-PM-Scl y Anti-MDA-5.
El ámbito morfológico hace referencia a los diferentes patrones radiológicos y anatomopatológicos y al componente multicompartimental. Entre los patrones radiológicos aceptados, se pueden encontrar la neumonía intersticial no específica (NINE), la neumonía organizada (NO), el solapamiento de ambas entidades (NINE-NO) y la neumonía intersticial linfoide (NIL). A nivel anatomopatológico, se incluyen los patrones de NINE, NO, solapamiento NINE-NO, la presencia de agregados linfoides intersticiales con centro germinal o la infiltración difusa linfoplasmocitaria (con o sin folículos linfoides). Como última sección del ámbito morfológico, se acepta la afectación multicompartimental. Se entiende como afectación multicompartimental la aparición paralela a la afectación pulmonar intersticial, de engrosamiento o derrame pleural, engrosamiento o derrame pericárdico, afectación intrínseca de la vía área en forma de obstrucción al flujo aéreo, bronquiolitis o bronquiectasias, o la presencia de vasculopatía pulmonar, siempre y cuando todos estos factores no tengan una etiología diferente y detectada.
Es fundamental tener en cuenta que, para aplicar correctamente estos criterios, es obligado el abordaje multidisciplinar y una estrecha colaboración de los diferentes especialistas que atienden al paciente con EPID. Son varias las dificultades que aparecen a la hora de clasificar a un paciente como IPAF en la práctica clínica habitual, así como las controversias que la aplicación de estos ámbitos implica. En primer lugar, las manifestaciones clínicas reconocidas dentro del ámbito clínico no son específicas ni únicas, si no que están extraídas de otras ETC, lo que en muchas ocasiones supone un gran reto a la hora de diferenciar una IPAF de una determinada ETC.
Este diagnóstico diferencial es especialmente complejo con el síndrome antisintetasa, por sus similitudes clínicas y serológicas. Tanto es así, que en la actualidad existen propuestas para modificar el concepto de IPAF y diferenciar entre IPAF con anticuerpos antisintetasa positivo y el resto. Por otra parte, aunque los anticuerpos ANCA han sido relacionados con ETC, no están aceptados como parte del ámbito serológico, debido a su mayor relación con el grupo de las vasculitis.
Uno de los puntos más controvertidos en lo referente al ámbito morfológico es la ausencia del patrón de neumonía intersticial usual (NIU), ya sea radiológico o anatomopatológico. No obstante, es importante aclarar que, aunque el patrón NIU no esté incluido ente los criterios morfológicos, su presencia no excluye la clasificación de IPAF, es decir, un paciente que padezca una determinada EPID con patrón NIU podrá ser clasificado como IPAF siempre y cuando cumpla al menos otros 2 criterios de 2 ámbitos diferentes.
5. TRATAMIENTO Y MANEJO
La evidencia científica en cuanto al tratamiento de la IPAF es escasa y se limita a series de casos. Como en otras EPID, el tratamiento no farmacológico incluye rehabilitación pulmonar, oxigenoterapia crónica domiciliaria en caso de insuficiencia respiratoria, vacunación/prevención de infecciones respiratorias y trasplante pulmonar en algunos casos graves.11
Dado que no es una enfermedad como tal, el tratamiento farmacológico específico se desconoce. No existen ensayos clínicos aleatorizados que apoyen el tratamiento inmunomodulador, pero en muchas ocasiones este tratamiento se extrapola del manejo farmacológico que se realiza habitualmente en pacientes con EPID secundaria a ETC.12 El micofenolato de mofetilo es un fármaco muy utilizado en las EPID secundarias a ETC, ya que ha demostrado ser seguro y su uso se ha asociado a una estabilización de la caída de función pulmonar. Se realizó un estudio de cohortes en pacientes con IPAF, en que el micofenolato de mofetilo parecía disminuir la caída de la capacidad vital forzada (FVC) y de la capacidad de difusión de monóxido de carbono (DLCO).13 También se ha descrito el empleo de otros fármacos habitualmente utilizados en EPID secundaria a ETC, como
la azatioprina, inhibidores de la calcineurina (ciclosporina o tacrolimus), ciclofosfamida o rituximab, aunque la evidencia es muy limitada. En un estudio realizado sobre pacientes con EPID inclasificable que incluía a un pequeño porcentaje de pacientes con IPAF, la ciclofosfamida en pulsos intravenosos podría estabilizar la pérdida de función pulmonar en los casos refractarios a corticoides.14 Por otro lado, el tratamiento para el reflujo gastroesofágico es relevante si el paciente tiene síntomas.11-12
Respecto al tratamiento antifibrótico en este perfil de paciente, un ensayo clínico aleatorizado en fase 2 que evaluó el empleo de pirfenidona en pacientes con EPID inclasificable progresiva incluyó a pacientes clasificados como IPAF que se encontraban en tratamiento con micofenolato. Sus resultados sugieren que la pirfenidona podría ser beneficiosa,15 aunque estos resultados no se confirmaron en el estudio posterior en fase 3. Por otro lado, el ensayo clínico INBUILD, que evaluó el efecto del nintedanib en pacientes con EPID fibrosante progresiva (FPP) entre los que de nuevo se incluyó a pacientes clasificados como IPAF, demostró que este fármaco reduce la caída de la FVC y presenta un aceptable grado de seguridad en este perfil de pacientes.16
6. EVOLUCIÓN Y PRONÓSTICO
La evolución de la IPAF es muy variable. En diferentes estudios se ha observado que alrededor de un 12%-16% de los pacientes terminarán desarrollando una determinada ETC a los 4 años de seguimiento. No obstante, en otros informes se plantea la posibilidad de que determinados pacientes clasificados como IPAF sean diagnosticados posteriormente de FPI.17
Los diferentes estudios de supervivencia realizados en pacientes que reunían criterios de IPAF son variables, mientras que sus resultados son controvertidos debido, principalmente, a sus discrepancias metodológicas. Por ello es muy complicado establecer correctamente el significado pronóstico de esta entidad. En general, se acepta que estos pacientes tienen mejor pronóstico que los pacientes con FPI. Sin embargo, la presencia de un patrón radiológico o histopatológico de NIU se asocia a peores resultados si se compara con los pacientes que desarrollan una afectación intersticial pulmonar en patrón de NINE. En un estudio retrospectivo, se observó una peor supervivencia en los pacientes con IPAF en comparación con los pacientes con EPID secundaria a ETC, pero mejor que los diagnosticados de FPI (Figura 2).9
7. PERSPECTIVAS FUTURAS
Dado que la descripción del término IPAF es reciente, existen todavía muchas dudas por resolver de cara al manejo clínico, como las posibles modificaciones en los factores que engloban los ámbitos diagnósticos, el papel de determinados anticuerpos como los ANCA o los anticuerpos antisintetasa, el valor del patrón radiológico o histológico de NIU
o la periodicidad con la que se deberían repetir los análisis autoinmunes serológicos o las pruebas de imagen radiológicas.
Otras cuestiones que requieren más respuestas son las referentes al manejo farmacológico, como el momento en que se debe iniciar el tratamiento o el papel de la inmunomodulación aislada o en terapia dual con el tratamiento antifibrótico.
Por otro lado, es vital detectar a los pacientes con peor pronóstico, para poder actuar de forma rápida y mejorar así los resultados. Es necesario realizar ensayos clínicos o estudios de alta calidad científica que avalen los tratamientos en pacientes con IPAF.
BIBLIOGRAFÍA
1. Lee CT, Oldham JM. Interstitial Pneumonia with Autoimmune Features: Overview of Proposed Criteria and Recent Cohort Characterization. Clin Pulm Med. 2017;24(5):191-196.
2. Fischer A, West SG, Swigris JJ, Brown KK, Du Bois RM. Connective Tissue Disease-Associated Interstitial Lung Disease. Chest. 2010;138(2):251-256.
3. Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B, et al. Idiopathic Nonspecific Interstitial Pneumonia: Lung Manifestation of Undifferentiated Connective Tissue Disease? Am J Respir Crit Care Med. 2007;176(7):691-697.
4. Vij R, Noth I, Strek ME. Autoimmune-Featured Interstitial Lung Disease. Chest. 2011;140(5):1292-1299.
5. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, Du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976-987.
6. Graney BA, Fischer A. Interstitial Pneumonia with Autoimmune Features. Ann Am Thorac Soc. mayo de 2019;16(5):525-533.
7. Ahmad K, Barba T, Gamondes D, Ginoux M, Khouatra C, Spagnolo P, et al. Interstitial pneumonia with autoimmune features: Clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir Med. 2017;123:56-62.
8. Chartrand S, Swigris JJ, Stanchev L, Lee JS, Brown KK, Fischer A. Clinical features and natural history of interstitial pneumonia with autoimmune features: A single center experience. Respir Med. 2016;119:150-154.
9. Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47(6):17671775.
10. Newton CA, Oldham JM, Ley B, Anand V, Adegunsoye A, Liu G, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J. 2019;53(4):1801641.
11. Fernandes L, Nasser M, Ahmad K, Cottin V. Interstitial Pneumonia With Autoimmune Features (IPAF). Front Med. 2019;6:209.
12. Mackintosh JA, Wells AU, Cottin V, Nicholson AG, Renzoni EA. Interstitial pneumonia with autoimmune features: challenges and controversies. Eur Respir Rev Off J Eur Respir Soc. 2021;30(162):210177.
13. McCoy SS, Mukadam Z, Meyer KC, Kanne JP, Meyer CA, Martin MD, et al. Mycophenolate therapy in interstitial pneumonia with autoimmune features: a cohort study. Ther Clin Risk Manag. 2018;14:2171-2181.
14. Wiertz IA, Van Moorsel CHM, Vorselaars ADM, Quanjel MJR, Grutters JC. Cyclophosphamide in steroid refractory unclassifiable idiopathic interstitial pneumonia and interstitial pneumonia with autoimmune features (IPAF). Eur Respir J. 2018;51(4):1702519.
15. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020;8(2):147-157.
16. 16. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med. 2019;381(18):1718-1727.
17. 17. Alevizos MK, Giles JT, Patel NM, Bernstein EJ. Risk of progression of interstitial pneumonia with autoimmune features to a systemic autoimmune rheumatic disease. Rheumatol Oxf Engl. 2020;59(6):1233-1240.
TABLAS Y FIGURAS
Tabla I. Criterios de clasificación IPAF (parte 1)

(Continúa)
Tabla I. Criterios de clasificación IPAF (parte 2)

Figura 1. Extraído y traducido de Graney et al.6

Figura 2. Curva de supervivencia Kaplan-Meier de pacientes con IPAF, FPI y EPID secundaria a ETC. Oldham et al.9

MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
12
VASCULITIS Y HEMORRAGIAS ALVEOLARES
Autoras
Elena Cabezas Pastor
Laura Núñez García
Coordinadora
María Jesús Rodríguez Nieto
1. VASCULITIS: CONCEPTO Y CLASIFICACIÓN. HEMORRAGIA ALVEOLAR
La afectación del tracto respiratorio es frecuente en las enfermedades sistémicas, especialmente en las colagenopatías y en las vasculitis. Las vasculitis sistémicas son un grupo heterogéneo de enfermedades caracterizadas por la inflamación en la pared de los vasos.1 Se ha descrito la presencia de un infiltrado inflamatorio, agudo o crónico, que lleva a la desestructuración de los vasos sanguíneos con disminución del flujo sanguíneo a los tejidos. Pueden afectar a uno o a varios órganos dependiendo del tamaño y de la localización de los vasos afectados, siendo más frecuente un carácter multisistémico. Según su origen, pueden ser vasculitis primarias, si el proceso inflamatorio se origina en la pared del vaso, o vasculitis secundarias, si derivan de procesos infecciosos o neoplásicos, colagenopatías o efectos adversos de fármacos o tóxicos.
La clasificación de las vasculitis sistémicas según el consenso de Chapel Hill 2012 (Tabla I) se basa en el tamaño de los vasos implicados: grandes, medianos y pequeños. 2
El aparato respiratorio puede verse afectado en las vasculitis sistémicas. El pulmón es un posible órgano diana de las vasculitis sistémicas, siendo las más frecuentes las asociadas a ANCA (Tabla II). Las vasculitis pulmonares comprenden un grupo heterogéneo de enfermedades y se asocian frecuentemente con la presencia de afectación sistémica, pudiendo haber un solapamiento de síntomas y signos con otras enfermedades, por lo que el diagnóstico no es fácil. Hay que incluir las vasculitis de pequeños vasos
en el diagnóstico diferencial de las enfermedades pulmonares intersticiales difusas. Es importante el diagnóstico y el tratamiento precoces en estas enfermedades, ya que son potencialmente mortales.
Todas las vasculitis tienen un claro componente inmunopatogénico. En las muestras histopatológicas, las vasculitis se manifiestan como una granulomatosis de las paredes alveolares, con capilaritis (infiltrado neutrofílico de capilares y vénulas) o como una hemorragia alveolar difusa.
Hemorragia alveolar difusa
La afectación pulmonar de las vasculitis puede desarrollarse en cualquier vaso, independientemente del tamaño. La capilaritis pulmonar es la vasculitis de la microvasculatura pulmonar. Es frecuente encontrarla en las vasculitis que presentan ANCA o enfermedades autoinmunes sistémicas. La complicación más grave de la capilaritis pulmonar, consecuencia del daño de la microcirculación, es la hemorragia alveolar pulmonar. Se define por la presencia de sangre en el espacio alveolar, como consecuencia de una alteración existente en la pared de los alveolos, asociada o no con una capilaritis pulmonar (casi siempre expresión de una vasculitis subyacente).3 La lesión del septo alveolar puede ser idiopática o secundaria a infecciones, tóxicos, hiperpresión en el lecho vascular, o por anomalías de los capilares en enfermedades de base inmunológica (los depósitos de inmunocomplejos, los ANCA, anticuerpos antimembrana basal).
La hemorragia pulmonar, la manifestación más grave de las vasculitis asociadas a ANCA, es más frecuente en la poliangeítis microscópica (PAM) y en la granulomatosis con poliangeítis. Con menor frecuencia, se asocia a granulomatosis eosinofílica con poliangeítis (Churg Strauss), al síndrome de Goodpasture, a la púrpura de Schönlein-Henoch, a la arteritis de Takayasu, a la arteritis de células gigantes, a la crioglobulinemia, a la poliarteritis nodosa o a la enfermedad de Behcet.
La hemorragia alveolar difusa es un síndrome caracterizado por la presencia de hemoptisis (aunque puede no estar presente), anemia ferropénica, insuficiencia respiratoria aguda e infiltrados alveolares pulmonares de aparición brusca. Otros síntomas más inespecíficos que pueden estar presentes son el dolor torácico, la tos y la fiebre.
La radiografía de tórax puede ser normal o mostrar infiltrados alveolares localizados o difusos, siendo una prueba inespecífica. Los hallazgos se confirman con la realización de tomografía computarizada de tórax.4 Las pruebas de función respiratoria se caracterizan por un aumento de la capacidad de difusión del monóxido de carbono (DLco), ya que el monóxido de carbono empleado en el test de la difusión se une a la hemoglobina intraalveolar acumulada. Se debe realizar una fibrobroncoscopia con lavado broncoalveolar (LBA), ya que ayuda a descartar otras patologías y a establecer el diagnóstico. Es muy típica la presencia de hemosiderófagos en el LBA, ya que los macrófagos alveolares fagocitan los eritrocitos que fluyen a través de la lesión en la
membrana basal del endotelio vascular hasta el alvéolo. La presencia de más de un 20% de hemosiderófagos es diagnóstica de hemorragia alveolar pulmonar. En la biopsia pulmonar del síndrome de hemorragia alveolar se pueden encontrar tres patrones histopatológicos diferentes: la capilaritis alveolar, el daño alveolar difuso y la hemorragia pulmonar blanda.
El abordaje terapéutico de los pacientes con hemorragia alveolar pulmonar depende de la gravedad de la misma, así como de las manifestaciones extrapulmonares del cuadro de vasculitis. Es una complicación potencialmente mortal. En los casos de gravedad, puede ser necesario ingresar al paciente en una unidad de cuidados intensivos con soporte respiratorio.
2. VASCULITIS CON AFECTACIÓN PULMONAR
2.1 Vasculitis ANCA +
La asociación entre la enfermedad pulmonar intersticial (EPID) y los anticuerpos anticitoplasma de neutrófilos (ANCA) o la vasculitis asociada a ANCA (VAA) ha sido cada vez más reconocida en los últimos años. Las VAA son un grupo heterogéneo de vasculitis sistémicas que afectan principalmente a los vasos sanguíneos pequeños. Este conjunto comprende tres síndromes clínicos distintos: la poliangeítis microscópica (PAM), la granulomatosis con poliangeítis (GPA) y la granulomatosis eosinofílica con poliangeítis (GEPA).
De todas ellas, la PAM es la que más se relaciona con el desarrollo de EPID. Además, se han documentado casos de pacientes con EPID y positividad para ANCA sin presentar otras manifestaciones de vasculitis sistémica, en especial aquellos con anticuerposMPO+.
2.1.1 Granulomatosis con poliangeítis (GPA)
La granulomatosis con poliangeítis (GPA) es una vasculitis necrotizante que involucra vasos de pequeño y mediano calibre. Afecta principalmente a los tractos respiratorios superior e inferior, así como a la vasculatura renal. Con una prevalencia aproximada de 25-150 casos por millón de habitantes, la GPA se diagnostica comúnmente entre los 55 y 65 años, siendo ligeramente más frecuente en hombres y en la población caucásica.
La etiología de la GPA y la poliangeítis microscópica (PAM) aún es desconocida, pero se ha explorado su relación con factores genéticos, infecciosos y ambientales. Se especula que la exposición a antígenos inhalados o propios de las vías respiratorias puede desencadenar un proceso autoinmune y generar inflamación vascular y el desarrollo de ANCA contra antígenos de polimorfonucleares y monocitos.
Manifestaciones clínicas
La presentación clínica de la enfermedad es variable y se divide en formas limitadas y extendidas. La forma limitada afecta principalmente a mujeres jóvenes e involucra el tracto respiratorio superior, pero sin afectación renal. En cambio, la forma extendida, más común, afecta principalmente los tractos respiratorios superior e inferior y el sistema renal. En la mayoría de los casos (90%), se presenta tempranamente afectación en la vía aérea superior, con rinorrea, pansinusitis y ulceraciones orales o nasales, pudiendo causar deformidad nasal (en silla de montar) y perforación del tabique.
El tracto respiratorio inferior queda afectado en el 70-90% de los pacientes, siendo excepcional su afectación sin compromiso del tracto respiratorio superior. Los síntomas incluyen tos, disnea, dolor torácico pleurítico, estridor o hemoptisis, asociados a diversas manifestaciones:
- Nódulos pulmonares: es la afectación más común; en general, miden entre 1 y 10 cm de tamaño; pueden cavitarse y presentar el signo del halo.
- Enfermedad intersticial pulmonar: en especial en forma de neumonía intersticial usual (43%), aunque también se ha asociado a fibroenfisema (20%) y a la neumonía intersticial no específica.
- Masas traqueales/bronquiales.
- Ulceraciones en la vía aérea que provocan estenosis subglótica, malacia o bronquiectasias.
- Hemorragia alveolar, de forma aguda o subaguda, con anemización y aparición de infiltrados alveolares en las pruebas de imagen.
- Derrame pleural (10%), generalmente unilateral y de pequeño tamaño.
- Estenosis arterial pulmonar: muy poco frecuente.
La afectación renal se da en el 60-80% de los pacientes, especialmente en los primeros dos años de evolución y tras el desarrollo de la afectación respiratoria. Se manifiesta en forma de glomerulonefritis necrotizante progresiva, que evoluciona a insuficiencia renal insidiosa o fulminante. Otras manifestaciones incluyen síntomas articulares (70%), oculares (50-80%), cardíacos (10-40%), neurológicos (25-40%), cutáneos (30%) o digestivos. Las principales características clínicas de la GPA y la PAM se resumen en la tabla III.
Diagnóstico
El diagnóstico se basa en tres pilares fundamentales: clínica compatible, datos serológicos e histología típica. La determinación de ANCA debe realizarse en cualquier paciente que presente síntomas sugestivos de vasculitis. La GPA se asocia principalmente con anticuerpos PR3-ANCA+ (65-75% de los casos). La negatividad de los anticuerpos no excluye el diagnóstico, puesto que al menos el 10% de los pacientes son ANCA negativos.
Encontraremos igualmente datos analíticos de inflamación: proteína C reactiva (PCR) elevada, neutrofilia y/o anemia, en especial en aquellos pacientes con hemorragia alveolar. Es igualmente imprescindible cuantificar los niveles de creatinina y descartar proteinuria en orina.
La biopsia de un órgano afectado para confirmar el diagnóstico no debe retrasar el inicio del tratamiento en un paciente con alta sospecha clínica de GPA. El órgano a biopsiar dependerá de los síntomas del paciente. En aquellos con afectación renal, la sensibilidad de la biopsia es superior al 90%. La gravedad de los hallazgos es generalmente paralela a la gravedad de los síntomas, que van desde la glomerulonefritis focal y segmentaria leve en pacientes con hematuria asintomática y función renal normal hasta la glomerulonefritis necrotizante difusa y semilunar, en pacientes con lesión renal aguda. En algunos casos, pueden observarse cambios granulomatosos.
En caso de plantear una biopsia pulmonar, la biopsia quirúrgica tiene una sensibilidad de casi el 90%, mientras que la sensibilidad de las biopsias transbronquiales es solo del 40%. En las biopsias del tracto respiratorio superior y en las biopsias pulmonares, el hallazgo típico es la inflamación aguda y crónica, ya sea con capilaritis o, menos comúnmente, con características granulomatosas.
2.1.2 Poliangeítis microscópica (PAM)
La poliangeítis microscópica es una vasculitis necrotizante sistémica con escasos depósitos inmunes, o sin ellos. Tiene una prevalencia entre 10 y 50 casos por millón, especialmente entre los 40 y 50 años, afecta de forma similar a ambos sexos y es algo más frecuente en la población asiática.
Manifestaciones clínicas
Las principales manifestaciones clínicas de la PAM se centran en el riñón y en el pulmón. Puede haber síntomas prodrómicos, como mialgias, artralgias o síndrome constitucional, que pueden preceder al resto de manifestaciones hasta en dos años.
La afectación renal es la más frecuente (80-90%), en forma de glomerulonefritis necrotizante segmentaria rápidamente progresiva. La afectación pulmonar (60%) ocasiona
principalmente hemorragia alveolar por capilaritis. Puede ocasionar también afectación intersticial, especialmente con patrón de NIU radiológico. No suele afectarse, sin embargo, la vía aérea superior.
Otras manifestaciones que pueden producirse son:
- Neurológica (30%): mononeuritis múltiple, pérdida de audición, hemorragia o infarto cerebral.
- Cutánea (33%): secundaria a vasculitis leucocitoclástica, que puede producir púrpura y úlceras orales.
- Digestiva (30%): dolor abdominal, náuseas, vómitos o diarrea.
- Ocular (20%): epiescleritis, uveítis o vasculitis retiniana.
Diagnóstico
De nuevo, el diagnóstico debe ser clínico, serológico e histológico. La presencia de anticuerpos ANCA la distingue de otras vasculitis. En la mayoría de los casos, son PR3-ANCA+ (55-65%), aunque, igual que en la GPA, su negatividad no excluye el diagnóstico.
A nivel histológico, se describen lesiones necrotizantes similares a las de GPA. En el riñón, la lesión más frecuente es, como se ha comentado, una glomerulonefritis necrotizante, con necrosis fibrinoide segmentaría, rotura de la pared de los capilares y formación de semilunas. Sin embargo, a diferencia de la GPA, no se observan granulomas. A nivel pulmonar, el hallazgo histológico típico es la capilaritis pulmonar.5-6
2.1.3. Granulomatosis eosinofílica con poliangeítis (GEPA)
La granulomatosis eosinofílica con poliangeítis (GEPA) es una vasculitis que afecta a vasos de pequeño y mediano calibre. El órgano más comúnmente afectado es el pulmón, seguido de la piel, aunque puede afectar a cualquier órgano o sistema. La edad media en el momento del diagnóstico es de 50 años.
Manifestaciones clínicas
La sintomatología de la GEPA se desarrolla característicamente de manera secuencial, aunque estas fases no están siempre claramente definidas:
- Fase prodrómica: se inicia comúnmente en la segunda y tercera década de la vida. Se caracteriza por enfermedad atópica, rinosinusitis alérgica, poliposis nasal y asma. El asma es la característica clínica cardinal de la GEPA y está presente en más del 90% de los casos. Suele preceder en 8-10 años a la fase vasculítica.
- Fase eosinofílica: incluye eosinofilia en sangre periférica e infiltración eosinofílica en múltiples órganos, especialmente el pulmón y el tracto gastrointestinal (dolor abdominal, diarrea y hemorragia gastrointestinal). Aproximadamente, el 50-70%
de los pacientes presentan opacidades pulmonares con eosinofilia, derrame pleural (a menudo eosinofílico) y nódulos, que rara vez son cavitados. La hemorragia alveolar es rara.
- Fase vasculítica: potencialmente grave. Se caracteriza por una vasculitis sistémica de los vasos medianos y pequeños, que a menudo se asocia con granulomatosis vascular y extravascular. Ocasiona síntomas generales (fiebre, malestar general y pérdida de peso) y lesiones cutáneas en forma de nódulos subcutáneos dolorosos en las superficies extensoras de los brazos, manos y piernas.
Pueden verse también afectados otros órganos y sistemas. La afectación cardíaca es una de las manifestaciones más graves de la GEPA y representa aproximadamente la mitad de las muertes atribuibles a la enfermedad. Incluye insuficiencia cardíaca, pericarditis y anomalías del ritmo cardiaco. Es más frecuente en pacientes con recuentos elevados de eosinófilos en el momento del diagnóstico.
A nivel neurológico, es característica la mononeuritis múltiple, que se observa hasta en el 75% de los pacientes, normalmente asociada a anticuerpos ANCA+. La afectación renal, sin embargo, no es tan común y varía de un estudio a otro.
Diagnóstico
Siempre se deberá descartar la GEPA en pacientes con asma no controlada a pesar de tratamiento adecuado. Es característico el recuento elevado de eosinófilos (generalmente 5.000-9.000 eosinófilos/microL), aunque en ocasiones pasa desapercibido como consecuencia del tratamiento previo con corticoides. Es especialmente relevante la cuantificación de IgE (elevada en el 75% de los casos) y cuyos niveles varían según la actividad de la enfermedad. Los ANCA se encuentran entre el 30 y el 60% de los pacientes con GEPA, en su mayoría MPO+. Los ANCA PR-3+ son inusuales.
El LBA se realiza habitualmente en pacientes con opacidades pulmonares en las pruebas de imagen. El líquido del LBA presenta habitualmente un recuento elevado de eosinófilos (superior al 30%), aunque no es específico de la enfermedad.
A nivel histológico, siempre se deberá optar por el lugar menos invasivo para biopsiar. En caso de existir lesiones cutáneas, la biopsia de las mismas suele revelar granulomas. La citología nasal muestra eosinofilia en el 60% de los casos, lo que apoya el diagnóstico, pero no es un criterio en sí mismo.
La biopsia pulmonar se reserva para los casos en los que ninguna de las localizaciones extrapulmonares afectadas es adecuada. En general, se prefiere la biopsia quirúrgica frente a la biopsia transbronquial. La histología pulmonar en la GEPA puede mostrar bronquitis asmática, neumonía eosinofílica, granulomas extravasculares o vasculitis (que afecta a arterias, venas o capilares).8
2.1.4. EPID con anticuerpos ANCA+
Como comentábamos al inicio, la gran mayoría de los casos de EPID se manifiestan en el contexto de la presencia de anticuerpos antimieloperoxidasa positivos (MPO+). La fibrosis pulmonar y la positividad de los ANCA pueden presentarse tanto con afectación sistémica como de manera independiente, aunque aún no se comprenden completamente los mecanismos patogénicos que vinculan los ANCA con el desarrollo de fibrosis pulmonar.
El patrón histológico típico es el de la neumonía intersticial usual (NIU), aunque también se asocia al patrón de neumonía intersticial no específica (NINE) y a otras características atípicas, como la bronquiolitis. La EPID en el contexto de vasculitis asociada a ANCA comporta por sí misma peor pronóstico, por lo que es fundamental la identificación temprana y el tratamiento oportuno de estos pacientes.
Además, se ha documentado la conversión a ANCA+ en individuos inicialmente diagnosticados con fibrosis pulmonar idiopática (FPI), desarrollando sintomatología de vasculitis sistémica en algunos casos. Sin embargo, persisten incertidumbres en cuanto a las diferencias fundamentales entre la EPID secundaria a vasculitis asociada a ANCA y la neumonitis intersticial idiopática con positividad aislada para ANCA.9
2.2. Vasculitis pulmonar asociada a enfermedades sistémicas
2.2.1. Vasculitis lúpica
Condiciona episodios de hemorragia alveolar. Es una complicación infrecuente, pero en ocasiones mortal. Los síntomas principales son la disnea y la hemoptisis, acompañada de una caída de la hemoglobina, que generalmente ocurre en el contexto de una afectación multiorgánica del LES. La histopatología muestra una hemorragia intraalveoar y macrófagos cargados de hemosiderina, aunque puede haber microvasculitis con infiltrado inflamatorio y necrosis de los tabiques alveolares.
2.2.2. Vasculitis reumatoidea
Es una complicación poco común, pero grave. Ocurre típicamente en pacientes con AR seropositivas y erosiva de larga evolución, con manifestaciones extraarticulares. A nivel pulmonar, afecta vasos tanto a nivel precipitar como postcapilar, con posterior desarrollo de necrosis y obstrucción vascular. Al igual que en el LES, la presentación clínica típica es en forma de hemorragia alveolar. Además, como otras enfermedades del tejido conectivo, puede ocasionar hipertensión arterial pulmonar (HAP).
3. OTRAS VASCULITIS CON AFECTACIÓN PULMONAR
INFRECUENTE
3.1.Arteritis de células gigantes
Es una vasculitis granulomatosa que afecta a los vasos de gran calibre, como la aorta, las carótidas y las arterias pulmonares. Aparece en personas mayores de edad y es frecuente que se asocie con la presencia de polimialgia reumática. Suele cursar con cefalea, pérdida de visión, fiebre, astenia, y dolor localizado en los músculos masticatorios. La afectación pulmonar es infrecuente, aunque puede producir disnea, tos seca y dolor torácico pleurítico. En la radiografía de tórax, es posible observar derrame pleural, infiltrados intersticiales y, en los casos más graves, infiltrados alveolares debidos a hemorragia pulmonar. La lesión más típica es el aneurisma, que suele ser múltiple, y que puede detectarse mediante diversas pruebas de imagen. Si se produce disección, la mortalidad es alta. El estudio histopatológico muestra datos de una vasculitis, con inflamación en la adventicia y la capa media de grandes vasos, con presencia de células gigantes y necrosis fibrinoide con destrucción de la pared vascular.
3.2. Arteritis de Takayasu
Es una vasculitis granulomatosa que afecta también a los grandes vasos. Al contrario que la arteritis de la temporal, se observa más en personas jóvenes, sobre todo mujeres. Las manifestaciones clínicas son muy variadas: mareos, síncopes, pérdida de visión, insuficiencia aórtica, arritmias cardiacas. La exploración física en esta enfermedad es fundamental, ya que puede sospecharse en un paciente joven con hipertensión arterial y asimetría en la exploración de los pulsos periféricos (“enfermedad sin pulso”). Los síntomas respiratorios más comunes son la tos, la disnea de esfuerzo y la hemoptisis.
Los hallazgos radiológicos y la histopatología son similares a la descrita en la arteritis de células gigantes, aunque en esta predomina la destrucción de fibras musculares lisas de la capa media de las arterias y posteriormente pueden aparecer fenómenos trombóticos con obliteración de la luz del vaso y fibrosis. Al igual que en la de células gigantes, la resonancia magnética y la tomografía con emisión de positrones (PET) han demostrado gran utilidad para el diagnóstico, así como para monitorizar la actividad de la enfermedad.
3.3. Poliarteritis nodosa
Es una vasculitis de arterias de mediano calibre, arteriolas, capilares o vénulas. Es poco frecuente la afectación del sistema respiratorio. Los síntomas respiratorios más comunes son la tos, la disnea de esfuerzo y la hemoptisis. En algún caso, se ha descrito la presencia de hemorragia alveolar pulmonar asociada a virus de la hepatitis B, así como de infiltrados intersticiales difusos. También se ha documentado algún caso con nódulos pulmonares no asociados a ANCA. Es más frecuente que se afecten las arterias bronquiales que las arterias pulmonares.
3.4. Crioglobulinemia
La crioglobulinemia es una vasculitis que afecta a vasos de pequeño tamaño. Se asocia a la presencia en suero de crioglobulinas, y en muchos casos se asocia virus de la hepatitis C. La afectación más frecuente es la cutánea, seguida de la renal, en forma de glomerulonefritis. La afectación pulmonar es infrecuente, y suele ser leve. Los pacientes presentan tos, disnea de esfuerzo o dolor torácico pleurítico. Es menos habitual el desarrollo de hemorragia alveolar pulmonar. La radiografía de tórax puede ser normal, o también encontrar infiltrados pulmonares, derrame pleural o datos de fibrosis. Cuando hay afectación pulmonar, en las pruebas de función predomina el patrón restrictivo con difusión de CO disminuida. Es característico que el lavado broncoalveolar muestre un aumento de linfocitos T, consecuencia de la alveolitis subyacente.
3.5. Enfermedad de Schönlein-Henoch
Es una vasculitis que afecta a vasos de pequeño calibre debida al depósito de inmunocomplejos formados por inmunoglobulina A (IgA). Ocurre fundamentalmente en la infancia, aunque puede observarse a cualquier edad. Las afectaciones más frecuentes son la cutánea, la renal y la intestinal. En los adultos, se expresa como una glomerulonefritis que se acompaña de una púrpura palpable, que corresponde a una vasculitis leucocitoclástica. La afectación pulmonar es muy poco frecuente, aunque se han descrito casos de neumonía intersticial usual, así como casos más graves de hemorragia alveolar pulmonar. En casos de hemorragia alveolar, la difusión de CO está aumentada. En la histopatología se observan depósitos de IgA además de necrosis de las paredes de los capilares pulmonares.
3.6.
Enfermedad de Behcet
Es una vasculitis que se caracteriza por presentar aftas orales y genitales, problemas oculares (el más frecuente, la uveítis) y lesiones cutáneas. La afectación pulmonar es infrecuente, pero puede producir aneurismas en las arterias pulmonares y tromboembolismo pulmonar. Histopatológicamente, suele corresponder a una vasculitis de pequeños vasos con depósitos de inmunocomplejo de inmunoglobulina (IgG) y de complemento. También puede afectar a grandes vasos, en cuyo caso pueden formarse aneurismas en las arterias bronquiales que al romperse originan hemoptisis, en ocasiones masiva. La hemoptisis también puede deberse a un infarto pulmonar causado por una obstrucción arterial.
4. TRATAMIENTO Y PRONÓSTICO
La base del tratamiento de las vasculitis con afectación pulmonar son los corticoesteroides y otros agentes inmunosupresores.10 Estos tratamientos pueden tener efectos adversos, por lo que deben utilizarse en función del grado de afectación de la enfer-
medad para evitar posibles toxicidades. Para clasificar el grado de actividad de estas enfermedades, se han propuesto diversos sistemas. El Grupo Europeo de Estudio de las Vasculitis (EUVAS) ha propuesto una clasificación basada en 5 niveles (Tabla IV): enfermedad localizada, enfermedad precoz generalizada, enfermedad generalizada, enfermedad grave y enfermedad refractaria. Se emplearán tratamientos más agresivos cuanto mayor sea el grado de actividad o de gravedad de la enfermedad. Con la introducción del tratamiento inmunosupresor, la mortalidad a 5 años de los pacientes con vasculitis ha disminuido del 50 al 12% desde los años setenta. Es importante el diagnóstico precoz de estas enfermedades porque estos tratamientos han mejorado el pronóstico de estos pacientes.
El tratamiento de las vasculitis pulmonares se basa en una fase de inducción (entre 3 y 6 meses) que trata de controlar la actividad de la vasculitis y alcanzar un estado de remisión en unas semanas, seguido de una fase de mantenimiento (entre 12 y 24 meses) para prevenir las recidivas.
4.1 Tratamiento de inducción
Como tratamiento de inducción de primera línea en enfermedad generalizada activa, habitualmente se utilizan los corticoides (1-2 mg/kg/día) asociados a ciclofosfamida oral o intravenosa (2 mg/kg/día, máximo 200 mg/día).
En enfermedad localizada o generalizada precoz (sin síntomas sistémicos ni afectación renal), se prefiere utilizar metotrexato (20-25 mg/kg/semana oral o parenteral) a ciclofosfamida con buenos resultados (remisión en el 70% de los casos).
El tratamiento con rituximab (anticuerpo monoclonal anti-CD20) es una alternativa a la ciclofosfamida en el tratamiento de las vasculitis ANCA positivas generalizadas. Existen dos estudios RAVE (Rituximab for the Treatment of Wegener’s Granulomatosis and Microscopic Polyangiitis) y RITUXVAS (estudio internacional, randomizado, abierto, en el que se compara tratamiento con rituximab versus tratamiento estándar con ciclofosfamida en pacientes con vasculitis generalizadas ANCA positivos). En ambos estudios, la dosis de rituximab es de 375 mg/m2 del área corporal una vez a la semana en cuatro infusiones. En ambos estudios, se concluyó que el tratamiento con rituximab fue no inferior al tratamiento con ciclofosfamida. El tratamiento con rituximab también está indicado en enfermedad refractaria y en recidivas.
En pacientes con enfermedad grave, con hemorragia alveolar, o con deterioro rápido de la función renal, el tratamiento recomendado se basa en la combinación de corticoesteroides en dosis más altas (como, por ejemplo, bolos de 250-500 mg metilprednisolona intravenosa al día), ciclofosfamida intravenosa y plasmaféresis. La plasmaféresis es un tratamiento adyuvante a la inmunosupresión durante la fase aguda en las vasculitis con afectación moderada-grave, así como en los casos de enfermedad refractaria.
4.2 Tratamiento de mantenimiento
El tratamiento se prolonga entre 12 y 24 meses para evitar recaídas. Se basa en corticoides, inicialmente 1 mg/kg/día, con una disminución lenta y progresiva de dosis hasta 5-10 mg/día o cese (no hay protocolo establecido), más azatioprina (de elección, dosis recomendada 2 mg/kg/día). Otras alternativas son: metotrexato, si existe intolerancia a la azatioprina y el filtrado glomerular es mayor de 30 ml/min, o micofenolato de mofetilo, si existe intolerancia a azatioprina y el filtrado glomerular es menor de 30 ml/min).
El micofenolato de mofetilo y la leflunomida han demostrado ser menos eficaces que la azatioprina para prevenir las recaídas.
Otros tratamientos son fármacos antifactor de necrosis tumoral e inmunoglobulinas intravenosas. El uso de inmunoglobulinas intravenosas, junto con el tratamiento con corticoesteroides e inmunosupresores, puede ser recomendable en pacientes con una recidiva de su vasculitis.
Además, en pacientes con vasculitis ANCA+ y afectación pulmonar tipo neumonía intersticial usual (NIU) o con criterios de fibrosis pulmonar progresiva, podría plantearse añadir tratamiento antifibrótico (estudio INBUILD). El inicio de estas terapias deberá valorarse de manera multidisciplinar incluyendo a neumólogos, reumatólogos, radiólogos y patólogos, para seleccionar de forma cuidadosa los pacientes en los que el componente fibrótico sea predominante.
En el caso de la GEPA, una alternativa terapéutica es el mepolizumab. Este anticuerpo anti-IL5 bloquea la unión con su receptor en el eosinófilo disminuyendo la producción y la supervivencia del mismo. El mepolizumab demostró ser un tratamiento efectivo en pacientes con GEPA con dosis subcutáneas de 300 mg al mes. Estudios posteriores han demostrado que con dosis inferiores (100 mg al mes) también se obtienen buenos resultados, disminuyendo el número de eosinófilos periféricos, con mejoría de las manifestaciones pulmonares y extrapulmonares, permitiendo reducir la dosis de corticoides e incluso su retirada.
5. CONCLUSIONES
Las vasculitis sistémicas pueden producir afectación pulmonar, siendo las más frecuentes las asociadas a ANCA. Por ello, las vasculitis de pequeño vaso se deben incluir en el diagnóstico diferencial de las enfermedades pulmonares intersticiales difusas. Su diagnóstico no es fácil, ya que las presentaciones clínicas, radiológicas e histopatológicas son variadas. Puede haber solapamiento de síntomas y signos con otras enfermedades. Son potencialmente graves e incluso mortales, por lo que su diagnóstico y su tratamiento precoces son fundamentales, ya que el tratamiento con corticoides e inmunosupresores ha demostrado un mejor pronóstico de estos pacientes y disminución de la mortalidad.
BIBLIOGRAFÍA
1. Martín-Suñé N, Ríos-Blanco JJ. Afectación pulmonar de las vasculitis. ArchBronconeumol. 2012;48:410-418.
2. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
3. Nieto Babero MA, García Asenjo JA Síndrome de hemorragia alveolar difusa. Nuemologiaclínica. Madrid: Elsevier; 2010, p. 428-438.
4. Castañer E, Alguersuari A, Gallardo X, Andreu M, Pallardó Y, Mata JM, et al. When to suspect pulmonary vasculitis: radiologic and clinical clues. RadiogrRevPublRadiolSoc N Am Inc. 2010;30(1):33-53.
5. Ross C, Makhzoum JP, Pagnoux C. Updates in ANCA-associated vasculitis. Eur J Rheumatol. 2022;9(3):153-166.
6. Pagnoux C. Microscopic polyangiitis and ranulomatosis with polyangiitis. En: Wuyts WA, Cottin V, Spagnolo P, et al. [eds.]. Pulmonary Manifestations of Systemic Diseases (ERS monograph). Sheffield: European Respiratory Society; 2019, p. 153-172.
7. Nappi E, De Santis M, Paoletti G, Pelaia C, Terenghi F, Pini D, Ciccarelli M, et al. New Onset of Eosinophilic Granulomatosis with Polyangiitis Following mRNA-Based COVID-19 Vaccine. Vaccines. 2022;10:716, https://doi.org/10.3390/ vaccines10050716.
8. Groh M, Pagnoux C, Baldini C, Bel E, Bottero P, Cottin V, Dalhoff K, et al. Eeosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA). Consensus Task Force recommendations for evaluation and management. Eur J Internal Med. 2015;(26):545-553.
9. Kadura S, Raghu G. Antineutrophil cytoplasmic antibody-associated interstitial lung disease: a review. eURrESPIRrEV. 2021;30:210123.
10. Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis. 2016;75(9):1583-1594.
TABLAS
Y FIGURAS
Tabla I. Clasificación de las vasculitis sistémicas según el tamaño de los vasos implicados²

II. Afectación pulmonar y vasculitis

Tabla
Tabla III. Principales características y diferencias entre la granulomatosis con poliangeítis (GPA) y la poliangeítis microscópica (PAM)6
Manifestaciones clínicas
Síndrome constitucional 70-100% 55-80%
Piel (púrpura) 10-50% 35-60%
VAS
Pulmón
50-95% Raro
60-80%: nódulos cavitados, hemorragia alveolar
60%: hemorragia alveolar
Asma No No
Renal
60-80%: glomeruonefritis pauciinmune
80-90% glomerulonefritis pauciinmune
Neuropatía periférica 25% 30%
Otras
Síntomas oculares, meningitis, trombosis venosa
Hallazgos
analíticos
Síntomas oculares, trombosis venosa (<10%)
Marcadores de inflamación PCR elevada, neutrofilia PCR elevada, neutrofilia
ANCA+ 90%: PR3+ (c-ANCA) 60-80% MPO + (p-ANCA)
Histología
Granulomas, vasculitis necrotizante de pequeño vaso
Vasculitis necrotizante de pequeño vaso
Tabla IV. Clasificación de la vasculitis según gravedad EUVAS (Grupo Europeo de Estudio de las Vasculitis)


Figura 1. Extraído y traducido de Graney et al.6
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
ENFERMEDADES
INTERSTICIALES DE ORIGEN
OCUPACIONAL
Autoras
Olaia Bronte
Koral Fernández de Roitegui Pérez
Coordinador
Iñigo Ojanguren Arraz
INTRODUCCIÓN
En el marco de las enfermedades de origen ocupacional, es conveniente destacar el concepto de “Enfermedad Profesional” (EP) como la contraída como consecuencia del trabajo ejecutado por cuenta ajena en las actividades que se especifiquen en el listado de EP vigente. Si bien diversas entidades pueden darse en el marco del ámbito ocupacional y fuera de él, este capítulo se centrará exclusivamente en las producidas por la inhalación del agente en el medio ocupacional y desarrollo de EPID.
1. NEUMOCONIOSIS
Las neumoconiosis son un grupo heterogéneo de enfermedades pulmonares intersticiales difusas (EPID) de causa conocida, producidas por la inhalación de polvos inorgánicos (mineral o metálico) en el ámbito ocupacional. Son enfermedades que pueden evitarse tomando las medidas preventivas adecuadas. El primer paso en la prevención es conocer el agente causal, por lo que la historia clínica laboral es fundamental. La silicosis, la neumoconiosis del polvo de carbón y la asbestosis son las neumoconiosis más frecuentes en nuestro medio. (Tabla I)
1.1. Silicosis
Es la neumoconiosis producida por la inhalación de dióxido de sílice (SiO2), denominado comúnmente sílice libre. Es la enfermedad respiratoria crónica de origen ocupacional más frecuente en el mundo. (Figura 1) 13
Clínica
Existen 3 formas clínicas de silicosis: la silicosis crónica, la acelerada y la aguda.
La silicosis crónica se clasifica a su vez en forma simple, complicada y fibrosis difusa. La silicosis crónica simple se caracteriza por su desarrollo a partir de los 10 años de exposición, forma nódulos < 1cm y suele presentar poca repercusión tanto clínica como funcional. En la forma crónica complicada, que aparece tras una exposición de más de 15 años, se forman masas de > 1 cm de fibrosis masiva progresiva (FMP) y produce síntomas neumológicos y afectación funcional respiratoria. Por último, la forma crónica en forma de fibrosis difusa aparece también a partir de los 10 años de exposición y presenta un patrón radiológico reticular.¹
La silicosis acelerada es la que se desarrolla en menos de 10 años y con una exposición más intensa; forma más rápidamente nódulos y masas que coalescen. La forma aguda o silicoproteinosis es menos frecuente, se produce tras una exposición corta (meses) pero intensa y produce un patrón alveolar con rápido deterioro respiratorio.
Diagnóstico
Se basa en la conjunción del antecedente de exposición, la presentación radiológica y clínica característica y la exclusión de otros datos que sugieran otra enfermedad. Cuando no se reúnen todas las características típicas, es necesario obtener muestra histológica para su confirmación, a través de criobiopsia pulmonar o biopsia quirúrgica. El lavado broncoalveolar (LBA) puede contribuir a excluir otras causas, pero habitualmente es inespecífico.
Radiología
A nivel radiológico, la silicosis simple se manifiesta por la presencia de un patrón nodular de 1-10 mm, de distribución difusa y predominio en lóbulos superiores y medios y en zonas posteriores y subpleurales. En la forma complicada, las masas de < 1 cm suelen ser bilaterales y con predominio en segmentos posteriores de lóbulos superiores y pueden producir retracción con atelectasias, enfisema y distorsión de la arquitectura. A nivel radiológico, la acelerada es semejante con más rapidez en la evolución, mientras que la forma aguda se caracteriza por consolidaciones alveolares bilaterales con rápida progresión en meses.
Tratamiento
No existe tratamiento que consiga revertir o modificar el curso de la enfermedad, habitualmente progresiva. Las investigaciones actuales sugieren el uso de antifibróticos en su evolución fibrosante. El trasplante pulmonar uni o bipulmonar puede ser el único tratamiento final en algunos pacientes con afectación severa de la enfermedad, siendo
los criterios de derivación y de inclusión en lista los recogidas en el último consenso de 2021 de trasplante ISHLT (International Society for Heart and Lung Trasplantation).¹¹ En las EPID en general, el momento de derivación a la unidad de referencia de trasplante pulmonar debe realizarse en el momento del diagnóstico, incluso si el paciente se encuentra iniciando el diagnóstico en el caso de que presente un patrón radiológico de NIU o de probable NIU. También, en cualquier forma en la que la FVC sea < 80% y/o la DLco < 40%, si en el seguimiento a lo largo de dos años se produce una pérdida relativa del 10% de la FVC, del 15% de la DLco o del 5% de la FVC junto con empeoramiento clínico o radiológico. Si el paciente requiere oxígeno suplementario en cualquier momento también será criterio de derivación. El momento de inclusión en lista de espera activa se indica cuando el paciente, a lo largo del seguimiento de 6 meses, presente una caída absoluta de > 10% de la FVC, > 10% de la DLco o > 5% de la FVC con progresión radiológica. También cuando en el test de la marcha de los 6 minutos presente una desaturación < 88% o una pérdida de > 50 m en el seguimiento en 6 meses. Y por último, cuando presente hipertensión pulmonar u hospitalización por empeoramiento, agudización o neumotórax. Los factores que ensombrecen el pronóstico son la duración y la intensidad de la exposición y la infección tuberculosa concomitante.
1.2. Neumoconiosis del polvo de carbón
Es la neumoconiosis por inhalación de polvo de carbón que presenta gran similitud clínica y radiológica con la silicosis.
Agente causal y mecanismo patogénico
El polvo generado en la explotación y el transporte del carbón puede inhalarse de forma casi aislada y producir la neumoconiosis del polvo de carbón o, junto con sílice libre, una neumoconiosis de polvo mixto en función del porcentaje de partículas de cada uno.
Clínica
Forma nodular y evolución hacia fibrosis masiva progresiva (FMP). Existe una entidad definida que ocurre en mineros del carbón con artritis reumatoide, conformando el síndrome de Caplan con formación de nódulos necrobióticos de distribución periférica.
Diagnóstico
Al igual que en la silicosis, el diagnóstico se lleva a cabo con la historia de exposición y el cuadro clínico típico junto con la exclusión de otras enfermedades.
Radiología
Nódulos centroacinares al azar confluyentes en masas de > 1 cm, pudiendo desarrollarse y apreciar FPM.
Tratamiento
No existe tratamiento dirigido.
1.3. Enfermedades pleuropulmonares producidas por asbesto
Los términos “amianto” y “asbesto” hacen referencia a una familia de minerales fibrosos con gran capacidad aislante, resistencia al fuego y capaces de soportar grandes temperaturas. El desarrollo de la enfermedad depende de la intensidad y del tiempo de exposición, además de las características físico-químicas de la fibra inhalada; requiere, por lo general, exposiciones elevadas. La presencia de derrame pleural benigno, placas pleurales calcificadas, fibrosis y atelectasia redonda solo implican exposición, y es lo que se denomina asbestosis benigna. A su vez, la inhalación de asbesto también puede originar carcinoma bronco-pulmonar y mesotelioma.
1.3.1. Asbestosis
Es la fibrosis pulmonar intersticial producida por la inhalación de fibras de asbesto. (Figura 2)
Clínica
La enfermedad aparece tras un periodo de latencia después de una exposición de al menos 10 años. Al igual que otras fibrosis pulmonares, se manifiesta con disnea de esfuerzo, tos seca, crepitantes de tipo velcro, acropaquías y patrón restrictivo.
Diagnóstico
Requiere historia de exposición con un periodo de latencia de 10 a 15 años y la presencia de patrón intersticial compatible, junto con alguno de los hallazgos característicos o confirmación histológica de la presencia de fibras de amianto.
Radiología
En la Rx simple de tórax, se suele apreciar un patrón reticular bilateral de predominio en campos inferiores con desarrollo de quistes de panal. En la TCAR, se puede apreciar un patrón NIU en fases avanzadas, pero además se pueden encontrar hallazgos característicos, como líneas curvilíneas paralelas a la pleural a 1 cm, bandas parenquimatosas perpendiculares procedentes de pleura y placas pleurales.
Tratamiento
No existe tratamiento dirigido; se ha iniciado el estudio del uso de nintedanib en casos progresivos.
1.3.2. Afectación pleural
Es la afectación pleural benigna por asbesto incluye el derrame pleural benigno, las placas pleurales calcificadas, la fibrosis pleural y la atelectasia redonda.
2. FIBRAS MINERALES ARTIFICIALES Y OTRAS SILICOSIS:
FIBRA DE VIDRIO, FIBRAS MINERALES, FIBRAS CERÁMICAS
El término “fibras minerales artificiales” (FMA) es el nombre genérico de una amplia variedad de materiales fibrosos procesados a partir de materia inorgánica (mayormente, silicatos u óxidos inorgánicos).²
Clasificación de las FMA
Hay cuatro grupos: 1) Filamentos continuos de fibra de vidrio (FCFV), de diámetro uniforme, más gruesos que las lanas; se fragmentan en fibras cortas y gruesas no respirables); 2) Lanas minerales (masas de fibras entrelazadas, de longitud y diámetro variables; existen tres clases diferenciadas: lana de vidrio, lana de roca y lana de escoria); 3) Fibras cerámicas refractarias (FCR) (mezcla de aluminio, sílice y otros óxidos refractarios; de 1,2-3,5 µm de diámetro y longitud variable); 4) Otras fibras.
Clínica
Mayoritariamente asintomática y con pruebas funcionales normales (a pesar de presentar alteraciones radiológicas) salvo en exposiciones a corto plazo, con síntomas cutáneos y/o de vía respiratoria superior.
Radiología
Radiografía de tórax (RxTx): pequeños nódulos en lóbulos inferiores, múltiples quistes y áreas de baja atenuación en campos superiores. TC tórax: nódulos bilaterales en lóbulos inferiores y engrosamiento pleural con/sin calcificación.
2.1. Neumoconiosis granulomatosas: beriliosis
El berilio es un metal rígido y ligero que se extrae por electroforesis del berilo o bertrandita.
2.1.1. Ocupaciones de riesgo
Extracción y metalurgia de berilio; industria aeroespacial, nuclear y electrónica; manufactura de aleación con cobre, aluminio, níquel, zinc; fundiciones; laboratorios dentales, fabricación de productos cerámicos, vidrio, porcelanas y productos altamente refractarios; fabricación de barras de control de reactores nucleares; pulido de gemas.³
2.1.2. Enfermedad por berilio aguda
Debido a la regulación de los niveles de exposición al berilio, los casos de beriliosis aguda son infrecuentes, se producen en contexto de accidentes laborales con exposición a dosis elevadas.
Clínica
Forma fulminante (días) o insidiosa (semanas) dependiendo de la intensidad de la exposición.
Tratamiento
Evitar la exposición a berilio. Si fuese necesario se puede plantear usa de esteroides, oxígeno o soporte ventilatorio en casos avanzados.
2.3. Beriliosis crónica
Puede desarrollarse años después del cese de la exposición. El tiempo medio entre el comienzo de la exposición y el desarrollo de la enfermedad es de 10 años (4 meses40 años).
Clínica
Insidiosa e inespecífica (tos seca, disnea y dolor torácico) acompañada de síntomas constitucionales (astenia, sudoración nocturna y pérdida de peso). Pueden aparecer acropaquías y/o crepitantes en los casos de fibrosis establecida. Forma subclínica: cada vez más frecuente por el cribado de la sensibilización en trabajadores expuestos.
Diagnóstico
Radiología
RxTx patrón intersticial retículo-nodulillar en campos medios y superiores, linfadenopatías hiliares bilaterales. Formas progresivas: bronquiectasias por tracción, bullas y engrosamiento pleural. TCAR: nodulillos peribroncovasculares, engrosamiento septal y de paredes bronquiales, adenopatías e infiltrados en vidrio deslustrado. En casos de fibrosis: bronquiectasias de tracción y panal.
Anatomía patológica
LBA: linfocitosis (>20%). Histología: granulomas no caseificantes y/o infiltrado intersticial mononuclear.
Test de proliferación de linfocitos contra el berilio (BeLPT)
Efectúa el diagnóstico de sensibilización al berilio mediante la demostración de una respuesta inmune celular mediada por linfocitos T, en sangre periférica o LBA. Es la
prueba de referencia para el cribado de sensibilización en trabajadores expuestos asintomáticos.
Tratamiento
Cese de la exposición y, si fuese necesario, esteroides sistémicos.
3. NEUMOCONIOSIS PRODUCIDAS POR OTROS METALES: SIDEROSIS, ESTANNOSIS, BARITOSIS
La siderosis, la estannosis y la baritosis son neumoconiosis producidas por la inhalación de polvos inorgánicos inertes, dado que no se ha evidenciado que tengan capacidad fibrogénica o repercusión clínico-funcional en la mayoría de casos, aunque puedan observarse alteraciones radiológicas muy llamativas.
3.1. Siderosis
Producida por depósito pulmonar de polvo o humos de hierro metálico y óxido de hierro, sobre todo en soldaduras u otros procesos de fusión del hierro. Otras ocupaciones de riesgo: minería, fundición, fabricación del acero, pulido de metales con óxido de hierro y pigmento ocre.
3.2. Estannosis
Producida por depósito pulmonar de óxido de estaño. Muy poco frecuente por su limitada exposición. Ocupaciones de riesgo: operaciones de purificación, fundición, extracción minera, soldadura, cerámica, esmaltados y pulido de granito.
3.3. Baritosis
Producida por depósito de polvo de bario (más común en su forma sulfatada o baritina). Ocupaciones de riesgo: fabricación de pinturas, textiles, cementos, cerámica, vidrio, electrónica, jabón; perforación de pozos (gas y petróleo); industria papelera y agente de contraste en radiología.
4. OTRAS ENTIDADES OCUPACIONALES ASOCIADAS A EPID
4.1. Fibrosis por nanomateriales
Durante las últimas décadas, la capacidad de manipular materiales hasta la nanoescala se ha incrementado de forma exponencial. Así, En este sentido, para elaborar diferentes materiales han comenzado a emplearse partículas de menos de 100 nm con base de carbono (nanotubos, nanocables, etc.), con base de metal (oro, plata, titanio, zinc, etc.) y con base biológica (liposomas, virus, etc.). En contexto de la estructura basal variable (carbono, metal o base biológica) de las nanopartículas, no es posible predecir la naturaleza de la toxicidad que pueden producir de forma genérica.
Los nanotubos de carbono se utilizan en áreas diversas, como la electrónica, la ingeniería o la medicina debido a propiedades como la conductividad eléctrica o su fuerza mecánica. Además, es relevante el hecho de que estos nanotubos pueden presentar una única pared o pueden ser multilaminares. La inhalación de estas partículas puede estimular la producción de TGF-��1, factor de crecimiento de plaquetas, dando lugar a reacciones granulomatosas y fibróticas en el pulmón.4
4.2. Neumonitis por petróleo
La combustión del petróleo y sus derivados son una causa conocida de enfermedad pulmonar. En estos procesos, se generan humos y partículas potencialmente cancerígenos y capaces de producir diversos efectos tóxicos, pudiendo dar lugar a neumonitis aguda, entre otras manifestaciones, en trabajadores expuestos en el sector de la minería o el transporte.Los hallazgos radiológicos y la histopatología son similares a la descrita en la arteritis de células gigantes, aunque en esta predomina la destrucción de fibras musculares lisas de la capa media de las arterias y posteriormente pueden aparecer fenómenos trombóticos con obliteración de la luz del vaso y fibrosis. Al igual que en la de células gigantes, la resonancia magnética y la tomografía con emisión de positrones (PET) han demostrado gran utilidad para el diagnóstico, así como para monitorizar la actividad de la enfermedad.
4.3. EPID por estaño / óxido de indio
El óxido de indio es un metal relativamente raro cuyo uso se ha incrementado de forma importante los últimos años en Japón y otros países asiáticos, especialmente en relación con la elaboración de pantallas planas, pantallas de cristal líquido, pantallas táctiles y otros dispositivos electrónicos. La exposición al óxido de indio tiene lugar durante su fabricación. Se han descrito distintas entidades englobadas dentro las EPID, como las fibrosis inespecíficas o la proteinosis alveolar.
Los 10 primeros casos fueron descritos en 2010 en los Estados Unidos de América en una serie en la que 7 pacientes presentaron fibrosis pulmonar y enfisema en relación con la exposición a óxido de indio y otros 3 pacientes presentaron proteinosis alveolar.
Todos los pacientes presentaron síntomas inespecíficos compatibles con tos y disnea, si bien en los pacientes con un patrón de tomografía computarizada de fibrosis sin signos de tipicidad predominó una disminución del test de transferencia del CO y fibrosis en la histología, y en los pacientes que mostraron un TC de tórax compatible con patrón de empedrado, la DLCO fue aún más baja y la histología mostró un exudado proteinaceo compatible con proteinosis alveolar pulmonar (PAP).5
4.4. EPID secundaria a microfibras
La exposición ocupacional a polímeros sintéticos de microfibras puede dar lugar a una afectación pulmonar en forma de enfermedades pulmonares intersticiales difusas. Las
fibras sintéticas de 0,2-5 mm de nylon, poliéster y el polipropileno, entre otros materiales, se emplean en el proceso de elaboración de plásticos, telas, tapicerías, alfombras, redes de pesca, etc. Los operarios expuestos a estas partículas respirables pueden presentar reacciones inflamatorias pulmonares mediadas por citoquinas (TNF-ala, IL-8, etc.) secretadas por macrófagos, lo que en algunos casos conduce a patología de la vía aérea, como bronquiolitis, panbronquiolitis e incluso EPID tras exposiciones intensas.6
4.5. Neumonía organizada por colorantes (síndrome de ardystil)
El síndrome de Ardystil hace referencia a una forma de EPID secundaria a la exposición masiva a un colorante, la acramina, de trabajadores textiles. Los primeros casos fueron descritos en Valencia (España) en 1994, en una fábrica textil con el nombre de “Ardystil”, en la que 22 trabajadores presentaron síntomas respiratorios en forma de disnea y tos. Las tomografías computarizadas de tórax realizadas mostraron hallazgos compatibles con neumonía organizada, mientras que en los casos en que se realizó una biopsia pulmonar, la histología mostró hallazgos de neumonía organizada. No se conoce con certeza el mecanismo patogénico involucrado, si bien se ha especulado con la posibilidad de un insulto pulmonar directa secundaria a la toxicidad por la acramina. Las formas de presentación descritas en los trabajadores expuestos han sido muy variables, con un espectro de patología que va desde síntomas leves inespecíficos a casos de fibrosis avanzadas que han conducido al fallecimiento.7
4.6. Inhalaciones agudas
Las inhalaciones agudas de gases, humos, polvos inorgánicos o vapores tienen lugar generalmente en el contexto de accidentes laborales o catástrofes. En este sentido, en función de la naturaleza de la sustancia inhalada se ha descrito un amplio repertorio de manifestaciones pulmonares, tanto a nivel de la vía aérea próxima como distal, en las que cabría destacar la bronquiolitis, la neumonía organizada, la neumonitis química o el distrés respiratorio.
5. Otros factores asociados a EPID
5.1. Calidad del aire
La calidad del aire depende de la presencia en mayor o menor medida de contaminantes en la atmosfera con potencial impacto negativo sobre la salud humana, el medio ambiente y otros sistemas. Los principales contaminantes con riesgos para la salud son el dióxido de azufre (SO2), los óxidos de nitrógeno (NOx), el ozono troposférico (O3), el monóxido de carbono (CO), el benceno y las partículas en suspensión (PM10 y PM2,5).
Los valores máximos permitidos de los principales contaminantes están regulados por ley (Directiva 2008/50/CE del Parlamento Europeo). La OMS, en su guía sobre la calidad del aire, establece recomendaciones sobre valores de contaminantes del aire (última actualización 2021) en base a la evidencia disponible (Tabla II).
La contaminación del aire (CA) es un factor de riesgo para el desarrollo y el empeoramiento de la mayoría de las enfermedades respiratorias. Sin embargo, los estudios que han examinado los efectos de la CA en las enfermedades pulmonares intersticiales son limitados y se centran en la fibrosis pulmonar idiopática (FPI). Varios mecanismos patogénicos apoyan el efecto deletéreo y posiblemente fibrosante de la CA. La exposición a CA induce estrés oxidativo, inflamación crónica y acortamiento de los telómeros, todos ellos mecanismos implicados en la fibrogénesis.
Los estudios sobre el impacto de la CA en la FPI sugieren que el nivel de exposición a contaminantes puede influir su incidencia (NO2), progresión (definida como disminución de la función pulmonar (FVC)) (PM10)) en el desarrollo de exacerbaciones agudas (O3 y NO2) y muerte (PM10 y PM2.5). Por otro lado, también se ha asociado la exposición a NO2 con mayor prevalencia de EPID subclínica, y el incremento de 10 µg/m3 de PM2,5, con mayor riesgo de desarrollar neumonitis por hipersensibilidad. Sin embargo, son necesarios más estudios para caracterizar y comprender el impacto de la CA en las enfermedades pulmonares intersticiales.8-9
5.2. VAPEO
La lesión pulmonar asociada al uso de cigarrillos electrónicos o vapeo (EVALI, por sus siglas en inglés: E-cigarette, or Vaping, product use Associated Lung Injury) es una enfermedad respiratoria aguda o subaguda potencialmente grave y, en algunos casos, mortal. Vapear es el proceso de inhalar un aerosol creado al calentar una sustancia como la nicotina o el tetrahidrocannabinol (THC) con un dispositivo electrónico, como un cigarrillo electrónico. En EEUU, se han notificado más de 2.000 casos, de los que dos tercios son hombres, el 80 % son menores de 35 años y la incidencia ha disminuido desde el primer brote reportado en verano de 2019. El factor de riesgo clave para EVALI es el uso de un cigarrillo electrónico o producto similar; pero no se ha identificado ningún constituyente común a todos los casos. Las posibles toxinas candidatas incluyen el THC y el acetato de vitamina E, aunque se han implicado otros compuestos, como la nicotina y otros aditivos desconocidos.
Clínica
La duración media del intervalo entre la exposición y la aparición de síntomas es de 6 días (de 0 días a 2 meses). Síntomas: disnea, tos, dolor torácico y hemoptisis. Aproximadamente, un tercio puede progresar a insuficiencia respiratoria aguda que requiere soporte ventilatorio. El 85 % asocia fiebre y síntomas gastrointestinales (náuseas, vómitos, diarrea y dolor abdominal).
Diagnóstico
No se han establecido criterios diagnósticos formales. Los criterios propuestos para la definición de caso incluyen: (1) Uso de un cigarrillo electrónico o producto relacionado en los 90 días anteriores; (2) Opacidades pulmonares (RxTx/TC); (3) Exclusión
de infección respiratoria (negatividad de PCR para virus respiratorios, test serológicos, hemocultivos, cultivo de esputo, lavado broncoalveolar y pruebas de detección de infecciones oportunistas relacionadas con el VIH); (4) Ausencia de un diagnóstico alternativo. Diagnóstico diferencial: neumonía adquirida en la comunidad (incluida la COVID-19), neumonía eosinofílica aguda, neumonía organizada, neumonía lipoidea y hemorragia alveolar difusa.10
Tratamiento
Se desconoce el tratamiento óptimo de EVALI. Sin embargo, en los casos reportados, se han empleado: (a) antibioticoterapia empírica precoz: con cobertura para patógenos más probables, en espera de los resultados de la evaluación inicial; (b) glucocorticoides sistémicos: su eficacia es desconocida; no se recomiendan de rutina. Indicación: EVALI con progresión clínica e hipoxemia. Dosis inicial: 0,5-1 mg/kg/día de metilprednisolona (o equivalente) con disminución gradual durante 5-10 días; (c) soporte ventilatorio: con el objetivo de optimizar la oxigenación y, en los casos más graves, la ventilación invasiva o no invasiva.
Pronóstico
Variable, muchos casos se resuelven por completo, sin embargo, por causas no conocidas, un pequeño número progresa hasta la muerte.
BIBLIOGRAFÍA
1. Li T, Yang X, Xu H, Liu H. Early Identification, Accurate Diagnosis, and Treatment of Silicosis. Can Respir J. 2022;3769134.
2. Diego Roza C, Cruz Carmona MJ, Fernández Álvarez R, Ferrer Sancho J, Marín Martínez B, Martínez González C, Rodríguez Portal JA, Romero Valero F, Villena Garrido V. Recommendations for the Diagnosis and Management of Asbestos-Related Pleural and Pulmonary Disease. Arch Bronconeumol. 2017;53(8):437-442.
3. Mayer A, Hamzeh N. Beryllium and other metal-induced lung disease. CurrOpinPulm Med. 2015;21(2):178-184.
4. Bonner JC, et al. Nanoparticles as a potential cause of pleural and interstitial lung disease. Proc Am Thorac Soc. 2010;7(2):138-141.
5. Cummings KJ, Nakano M, Omae K, Takeuchi K, Chonan T, Xiao YL, Harley RA, et al. Indium lung disease. Chest. 2012;141(6):1512-1521.
6. Atis S, Tutluoglu B, Levent E, Ozturk C, Tunaci A, Sahin K, Saral A, Oktay I, Kanik A, Nemery B. The respiratory effects of occupational polypropylene flock exposure. Eur Respir J. 2005;25(1):110-117.
7. Solé A, Cordero PJ, Morales P, Martínez ME, Vera F, Moya C. Epidemic outbreak of interstitial lung disease in aerographics textile workers –the "Ardystil syndrome": a first year follow up. Thorax. 1996;51(1):94-95.
8. World Health Organization. WHO global air quality guidelines: particulate matter (PM2. 5 and PM10), ozone, nitrogen dioxide, sulfur dioxide and carbon monoxide. World Health Organization, 2021.
9. Harari S, Raghu G, Caminati A, Cruciani M, Franchini M, Mannucci P. Fibrotic interstitial lung diseases and air pollution: a systematic literature review. Eur Respir Rev. 2020;29(157):200093.
10. Hayes D, Board A, Calfee CS, Ellington S, Pollack LA, Kathuria H, Eakin MN, et al. Pulmonary and Critical Care Considerations for e-Cigarette, or Vaping, Product Use-Associated Lung Injury. Chest. 2022;162(1):256-264.
11. Leard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, Campos SV, et al. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-1379.
TABLAS Y FIGURAS
Tabla I. Según el RD 1299/2006, por el que se aprueba la lista de enfermedades profesionales por el sistema de la seguridad social modificado en 2018, Anexo I Grupo 4: Enfermedades profesionales causadas por la inhalación de sustancias o agentes (Parte 1)
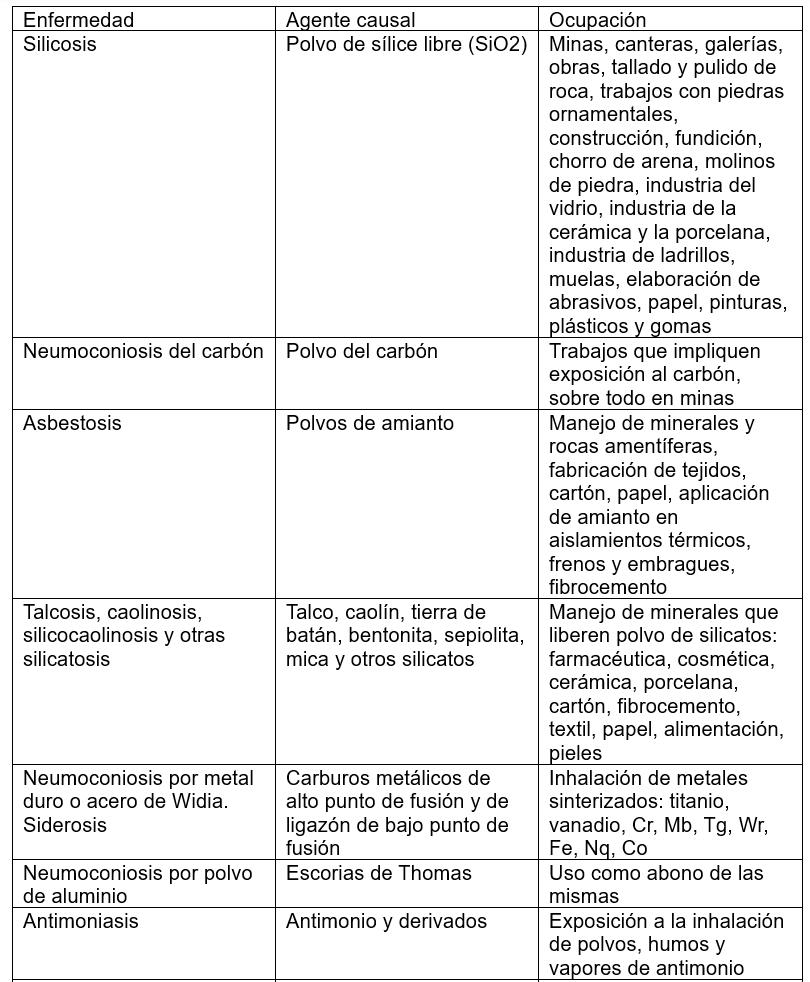
(Continúa)
Tabla I. Según el RD 1299/2006, por el que se aprueba la lista de enfermedades profesionales por el sistema de la seguridad social modificado en 2018, Anexo I Grupo 4: Enfermedades profesionales causadas por la inhalación de sustancias o agentes (Parte 2)

Tabla II. Valores recomendados de los contaminantes del aire por la UE y por la OMS. Fuente: elaboración propia


Figura 1. Silicosis Complicada
Figura 2.TCAR de asbestosis
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
14
ENFERMEDADES QUÍSTICAS
PULMONARES
Autores
Dolores del Puerto García
Pablo Mariscal Aguilar
Coordinador
Álvaro Casanova Espinosa
INTRODUCCIÓN
El quiste pulmonar es un espacio parenquimatoso pulmonar lleno de aire, rodeado de paredes finas (<2 mm), bien definidas y de baja atenuación en el TAC.1
La enfermedad pulmonar quística difusa (EPQD) debe distinguirse radiológicamente de otras entidades que pueden simular un quiste, como la bulla, las bronquiectasias quísticas, la cavidad, el enfisema, el “panal de abeja” y el neumatocele. En la tabla I se muestra la clasificación de las EPQD.
En este capítulo, nos centraremos en las enfermedades intersticiales quísticas difusas del pulmón, haciendo hincapié en la LAM y en la HPCL.
1. LINFANGIOLEIOMIOMATOSIS (LAM)
La LAM es una enfermedad neoplásica de baja prevalencia que afecta a mujeres en edad fértil.
Existen dos formas, la LAM esporádica (3,3-7,7 casos por millón mujeres) y la LAM asociada a esclerosis tuberosa (ET) (30-40% de mujeres con ET).
La base de la enfermedad es la proliferación de células musculares lisas atípicas, denominadas “células LAM”, que se acumulan en el intersticio pulmonar y alrededor de las estructuras broncovasculares. Se debe a una alteración funcional o mutación en los genes del complejo de la ET (TSC), que codifican dos proteínas: hamartina (TSC1)
y tuberina (TSC2). La pérdida de estas proteínas conlleva una activación anómala del centro regulador mTOR (mammalian Target of Rapamycin), dando lugar a un crecimiento celular descontrolado.3
Manifestaciones clínicas
- Afectación pulmonar: el síntoma principal es la disnea. Son frecuentes los neumotórax recidivantes. Menos frecuentes son el derrame pleural quiloso (quilotórax) y la hemoptisis.
- Afectación extrapulmonar: angiomiolipomas renales, ascitis quilosa, derrame pericárdico, quiluria y linfangioleiomiomas abdominales.
Diagnóstico
- Sospecha clínica. Mujer joven con disnea progresiva, limitación crónica al flujo aéreo en la espirometría e historia de neumotórax recurrente.
- Radiología. La radiografía de tórax suele ser normal o mostrar un patrón intersticial con lesiones quísticas. En la TCAR, se aprecian quistes distribuidos de forma difusa y homogénea por todo el parénquima pulmonar de un tamaño que oscila entre los pocos milímetros y los 2 cm (Figura 1). A nivel abdominal (ecografía abdomen, RMN o TC abdominal), se pueden observar angiomiolipomas renales, liquido ascítico, linfadenopatías y linfangioleiomiomas abdominales.
- Función pulmonar. La alteración más frecuente es el patrón obstructivo con disminución de la capacidad de difusión.
- Factor de crecimiento del endotelio vascular (VEGF-D). Un nivel en sangre de VEGF-D superior a 800 pg/ml se considera un criterio diagnóstico de LAM. La sensibilidad y la especificidad de este marcador son de un 71% y un 100%, respectivamente.
- Histología. Mediante biopsia transbronquial (convencional o criosonda) por fibrobroncoscopia o biopsia quirúrgica, se visualizan quistes pulmonares y “células LAM” que se tiñen positivamente con actina, miosina, desmina y el anticuerpo HMB-45. La muestra histológica o citológica no es imprescindible para llegar al diagnóstico.
- En la tabla II, se muestran los criterios diagnósticos de LAM propuestos por la ATS/JRS en el año 2017. En la figura 2, se describe una aproximación diagnóstica ante la sospecha clínica de LAM.
Tratamiento
- Medidas generales: vacunación para la gripe, neumococo y COVID, deshabituación tabáquica, evitar anticonceptivos, rehabilitación pulmonar y oxigenoterapia en caso de hipoxemia en reposo o durante el ejercicio.
- Tratamiento broncodilatador: en pacientes con obstrucción pulmonar, principalmente si se demuestra reversibilidad.
- Inhibidores de mTOR (sirolimus, everolimus). Son la base del tratamiento farmacológico de la LAM5. El sirolimus está indicado en pacientes sintomáticos con FEV¹ < 70%, evidencia de enfermedad progresiva o en casos de derrame pleural quiloso. Se inicia a una dosis de 1 o 2 mg/día y se ajusta según niveles del fármaco (niveles en sangre: 5-15 ng/ml). Se recomienda solicitar los niveles del fármaco al mes de iniciar el tratamiento y posteriormente a intervalos de 3 meses, pudiéndose ampliar hasta 6-12 meses según estabilidad. Los efectos secundarios más habituales son: diarrea, dislipemia, náuseas, acné y mucositis.
- Tratamiento hormonal (ooforectomía, progesterona, tamoxifeno y análogos de la hormona liberadora de gonadotropinas). La normativa de la ATS/JRS establece una recomendación en contra del uso de progestágenos en la LAM, condicionada a una baja calidad de la evidencia.
- Trasplante pulmonar: según las recomendaciones del consenso internacional de trasplante de corazón y pulmón de 2021, se debe derivar al centro de referencia de trasplante pulmonar cuando se cumpla cualquiera de las siguientes circunstancias a pesar de terapia con inhibidores de mTOR:
• Alteración grave de la función pulmonar (FEV1 < 30% predicho)
• Disnea NYHA III o IV
• Hipoxemia en reposo
• Hipertensión pulmonar
• Neumotórax recidivante.
- Fármacos en investigación: hidroxicloroquina, letrozol, simvastatina, saracatinib, nintedanib, loratadina, resveratrol.
Recomendaciones tras el diagnóstico
- Embarazo: las pacientes con LAM que deseen quedarse embarazadas deberán ser informadas de los riesgos asociados al embarazo, que incluyen: incremento del riesgo de neumotórax y quilotórax, progresión de la enfermedad pulmonar o sangrado de los angiomiolipomas. La decisión final será de la propia paciente.
Aunque no se conocen factores específicos que predigan este riesgo, en parte va a depender de la reserva pulmonar de la madre. De forma individualizada, en pacientes con alteración funcional grave se podría desaconsejar el embarazo.
- Deportes: actividades como el buceo, paracaidismo o puenting están desaconsejadas por el riesgo aumentado de neumotórax. Se recomienda fomentar una actividad física regular, como caminar, yoga u otras prácticas deportivas cardiosaludables.
- Viajes en avión: los cambios de la presión atmosférica asociados a los vuelos en avión pueden producir una expansión de los quistes pulmonares y un incremento del riesgo de neumotórax. Se estima una tasa de riesgo de 1 a 2 episodios por cada 100 vuelos. Aunque se debe alertar de esta posibilidad, las guías internaciones y la mayoría de expertos en LAM no desaconsejan volar en avión a las pacientes con LAM. En algunos casos, puede ser necesario prescribir oxígeno suplementario durante el vuelo.
2. HISTIOCITOSIS PULMONAR DE CÉLULAS DE LANGERHANS (HPCL)
La HPCL es una enfermedad rara (1-2 casos/1.000.000 individuos) caracterizada por la proliferación anormal de células dendríticas y macrófagos pertenecientes al sistema mononuclear fagocítico.6
Aunque puede afectar a diferentes órganos (piel, hueso, sistema nervioso central, tiroides, ganglios linfáticos) en formas uni o multisistémicas de la enfermedad, en este capítulo solo nos referiremos a la HPCL.
Manifestaciones clínicas
- Afectación pulmonar. Los síntomas más comunes son disnea, tos seca, dolor torácico pleurítico, pérdida de peso y fiebre. La mitad de los pacientes están asintomáticos en las fases iniciales. Es frecuente el neumotórax (25% de los casos).
- Afectación extrapulmonar. Se observan en el 20% de los casos. Incluyen lesiones óseas, diabetes insípida y lesiones cutáneas. Las lesiones óseas quísticas pueden ser un hallazgo incidental o causar dolor localizado con o sin fracturas. La afectación hipotalámica puede dar lugar a diabetes insípida y se asocia con un peor pronóstico. Las lesiones cutáneas consisten en pápulas de color marrón a violáceo, así como lesiones eccematoides o seborreicas.
Diagnóstico
- Sospecha clínica. Varón joven, fumador o exfumador, con disnea de esfuerzo, historia previa de neumotórax recurrente, diabetes insípida y dolor óseo.
- Radiología. En la radiografía de tórax, se visualiza un infiltrado intersticial bilateral, de predominio en campos medios y superiores. En la TCAR de tórax, destaca la presencia de nódulos irregulares, a veces de forma estrellada, de 1 a 10 mm de tamaño con una disposición central alrededor de los bronquiolos. Pueden cavitarse o desarrollar áreas radiolúcidas en su interior (Figura 3). A medida que la enfermedad progresa, aparecen quistes (2-20 mm de diámetro), áreas de reticulación, lesiones de enfisema y distorsión fibroquística. Las bases pulmonares y los ángulos costofrénicos suelen estar respetados.7
- Función pulmonar. Se pueden encontrar diferentes patrones: obstructivo, restrictivo o mixto, según el estadio de la enfermedad y una disminución de la DLCO.
- Histología. En un contexto clínico apropiado, el hallazgo de células CD1a y CD207 en el LBA y la BTB es diagnóstico de HPCL. Sin embargo, la sensibilidad de estas pruebas es baja. El diagnóstico definitivo requiere, a veces, la realización de una biopsia pulmonar. La identificación de las células dendríticas similares a Langerhans se realiza mediante la tinción inmunohistoquímica para CD1a, CD207 y S100. Estas células expresan muy a menudo el antígeno CD1a en su superficie, una característica que las distingue de otras células de origen histiocítico. La proteína Langerina (CD207) también se tiñe y suele asociarse con CD1a en la HPCL. La tinción de la proteína S100 intracelular, aunque menos específica, también se observa en algunas células de la HPCL.6-8
- Detección de mutaciones en el gen BRAF (como la variante BRAF V600E, y otras mutaciones en la vía de señalización MAPK). Puede ser de utilidad por su implicación terapéutica futura.
Se puede realizar una tomografía por emisión de positrones con fluorodesoxiglucosa (FDG-PET) si los síntomas sugieren enfermedad extrapulmonar.
Diagnóstico diferencial
El diagnóstico diferencial de HPCL incluye la LAM, la neumonía intersticial linfoide, el síndrome de Birt-Hogg-Dubé y la sarcoidosis. Para los pacientes con opacidades reticulares, nodulares o en vidrio esmerilado en la TCAR sin cambios quísticos, el diagnóstico diferencial es amplio e incluye neumonitis por hipersensibilidad y otras neumonías intersticiales idiopáticas, como la neumonía intersticial no específica (NINE).7
Tratamiento
- Medidas generales. El abandono del tabaquismo es una medida fundamental. Ha demostrado mejoría clínica, funcional y radiológica, sobre todo en las fases iniciales de la enfermedad.
- Corticoides orales. No se disponen de ensayos controlados. Útiles en pacientes con enfermedad nodular, aunque su eficacia es limitada. Prednisona a dosis de 0,25 a 0,5 mg/kg/día o 30 mg/día inicialmente con una disminución gradual durante seis meses. 6-8
- Agentes quimioterápicos. La cladribina, la citarabina y la vinblastina se han ensayado en algunos pacientes con mejoría en estudios observacionales. Se precisan ensayos clínicos controlados para establecer una recomendación específica.6-8
- Terapia dirigida a mutaciones de la vía MAPK. Se exploran opciones de terapia dirigida, como BRAF, V600E y MAPK2K1 con vemurafenib, dabrafenib o trametinib. Su eficacia clínica aún no está claramente demostrada.6-8
- Trasplante de pulmón. Es una opción para pacientes con enfermedad pulmonar avanzada y progresiva.
Pronóstico
La historia natural de la enfermedad es variable, aunque la supervivencia generalmente es buena. Se estima una supervivencia mayor del 90% a los 10 años. Algunos pacientes pueden experimentar una remisión espontánea de los síntomas. Solo un 5% de los pacientes desarrolla una enfermedad pulmonar fibrótica progresiva, complicación grave y potencialmente mortal de la enfermedad.
3. NEUMONÍA INTERSTICIAL LINFOCÍTICA
La neumonía intersticial linfocítica (NIL) es una forma poco común de EPID que se caracteriza por la infiltración de linfocitos, células plasmáticas y otros componentes linforreticulares en los espacios alveolares e intersticiales del pulmón. Aunque se considera benigna, la NIL plantea desafíos diagnósticos y terapéuticos significativos.
Asímismo, la NIL es considerada una variante de la hiperplasia linfoide pulmonar, que se distingue por un predominio de alteraciones intersticiales en el tejido pulmonar. En la mayoría de los casos, la NIL está asociada con otras condiciones médicas, como la infección por VIH (principalmente en niños), la enfermedad de Sjögren, la artritis reumatoide, el lupus eritematoso sistémico, la artritis idiopática juvenil, la enfermedad celíaca, la miastenia gravis, la anemia perniciosa, la hepatitis crónica activa y la cirrosis biliar. En menos del 20% de los casos, se presenta como una enfermedad idiopática, sin una causa subyacente clara.9
Por otro lado, la NIL puede ser resultado de la afectación pulmonar de los siguientes procesos: pseudolinfoma, granulomatosis linfomatoide, linfoma angioinmunoblástico de células T (linfadenopatía angioinmunoblástica) y linfoma pulmonar primario.
Manifestaciones clínicas
- Afectación pulmonar: la NIL suele presentarse en personas de mediana edad, con una mayor prevalencia en mujeres. Los síntomas son insidiosos, con disnea de esfuerzo progresiva, tos seca y ocasionalmente síntomas sistémicos como fiebre, pérdida de peso, artralgias y fatiga. Los estertores crepitantes son comunes en la auscultación y, en algunos casos, se pueden observar acropaquias.
- Afectación extrapulmonar: las manifestaciones extrapulmonares ayudan a sospechar una causa subyacente. Destacan la hipertrofia de la glándula parótida, artritis, hepatoesplenomegalia y linfadenopatía. La anemia y la hipergammaglobulinemia también son hallazgos frecuentes.
Diagnóstico
- Función pulmonar: Al igual que el resto de EPID, la función pulmonar suele mostrar un patrón restrictivo con disminución de la DLco. La DLco es el parámetro de función pulmonar más sensible en estos casos.
- Radiología: En la radiografía de tórax, se pueden observar opacidades reticulares de predominio en bases. La TCAR es útil para determinar la extensión y ubicación de la afectación pulmonar, definir la anatomía hiliar y detectar afectación pleural. Los hallazgos característicos son la presencia de un patrón intersticial difuso en vidrio esmerilado, principalmente en las bases pulmonares, junto con nódulos centrolobulillares de pequeño tamaño y engrosamiento intersticial peribroncovascular y subpleural. Los quistes pulmonares se presentan entre el 68 y el 82% de los casos de NIL, a diferencia de la neumonía intersticial no específica (NINE) y el linfoma pulmonar, donde son raros. Estos quistes suelen ser de pared fina, de distribución perivascular o subpleural, y varían en tamaño entre 1 y 30 mm (Figura 4).
- El engrosamiento pleural y el derrame pleural son poco frecuentes en la NIL, al igual que la linfadenopatía en las regiones hiliar y mediastínica, lo que sugeriría la presencia de un proceso subyacente de naturaleza maligna.
- Histología: El LBA es útil para descartar infecciones, enfermedades ocupacionales, neumonía eosinófila crónica y malignidad. Suele haber una linfocitosis, hallazgo inespecífico que puede aparecer en otras entidades, como la neumonitis por hipersensibilidad, la sarcoidosis, la NINE, la NOC y la enfermedad crónica por berilio.
El diagnóstico definitivo de la NIL requiere una biopsia pulmonar que muestre una infiltración extensa de los septos alveolares por linfocitos, células plasmáticas e histiocitos.
Es importante destacar que el infiltrado linfocitario en la NIL es policlonal, a diferencia de las lesiones linfoepiteliales o la destrucción alveolar que se observan en el linfoma.9
Diagnóstico diferencial
- Linfoma pulmonar primario: es un tipo de tumor que se origina en el tejido linfático del pulmón. Puede causar síntomas similares a la NIL, como disnea y tos.
- Granulomatosis linfomatoidea: enfermedad rara que involucra la formación de granulomas en el tejido pulmonar, que no suelen encontrarse en la NIL.
- Bronquiolitis folicular: afección caracterizada por inflamación de los bronquiolos y puede llevar a la formación de nódulos foliculares en el tejido pulmonar.
- Amiloidosis pulmonar: patología en la que se acumulan proteínas anormales (amiloides) en los tejidos, incluidos los pulmones. Puede causar síntomas pulmonares similares a la NIL.
- Neumonitis por hipersensibilidad: reacción inflamatoria pulmonar debida a la exposición a partículas o agentes ambientales, como polvo o esporas de hongos o antígenos aviares. Los síntomas y los hallazgos en el LBA (linfocitosis) pueden coincidir.
- Enfermedad por IgG4: a nivel radiológico, la TCAR muestra opacidades redondeadas, vidrio esmerilado difuso y nódulos sólidos. Los síntomas de sequedad ocular y bucal, que pueden confundirse con la enfermedad de Sjögren, son comunes. Aproximadamente el 85% de los pacientes presentan niveles séricos elevados de IgG4. A diferencia de la NIL, la citometría de flujo suele mostrar un aumento de plasmablastos y una tinción positiva de IgG4 en estudios histopatológicos.
- Neumonía por Pneumocystis jirovecii y citomegalovirus en pacientes VIH+: estas infecciones oportunistas pueden manifestarse clínica y radiológicamente de forma similar a la NIL.
Tratamiento
No se han realizado ensayos clínicos controlados para la NIL. La información sobre el tratamiento se deriva de informes de casos y series de casos. La indicación depende de la gravedad de los síntomas, el grado de deterioro funcional y la presencia de enfermedades subyacentes, como enfermedades reumáticas, inmunodeficiencia o infección por VIH.
La mayoría de los pacientes responde favorablemente al tratamiento con glucocorticoides orales, y algunos pueden requerir un agente inmunosupresor adicional.
En las formas de NIL asociada a una enfermedad autoinmune, como la enfermedad de
Sjögren, la artritis reumatoide o el lupus eritematoso sistémico, se recomienda iniciar el tratamiento ante un deterioro de la función pulmonar.
Para el tratamiento inicial de los pacientes con NIL sintomática y deterioro clínico o funcional (FVC < 70% o caída de la FVC y/o DLco > 10%), se recomienda comenzar con glucocorticoides sistémicos: prednisona oral o su equivalente (0,25 a 0,5 mg/kg/ día), que se continúa durante de 8 a 12 semanas. Se debe evaluar la respuesta y, si es favorable, se reduce gradualmente la dosis durante de 6 a 8 semanas. La duración del tratamiento es variable, generalmente entre 6 meses y un año.
En casos de enfermedad refractaria, cuando los pacientes no responden adecuadamente al tratamiento con glucocorticoides o experimentan efectos secundarios, se puede considerar la adición de otro agente inmunosupresor, como azatioprina, ciclofosfamida, ciclosporina o rituximab.9
Para pacientes con NIL idiopática, el enfoque de tratamiento es similar al de la NIL asociada a enfermedades reumáticas. Se recomienda realizar una evaluación cuidadosa antes de iniciar el tratamiento en casos con síntomas mínimos y enfermedad leve.
Pronóstico
La historia natural y el pronóstico de la NIL no se conocen con exactitud. Algunos pacientes experimentan una resolución espontánea. En algunos casos, la NIL responde favorablemente al tratamiento con glucocorticoides u otros agentes inmunosupresores. También existe el riesgo de que la enfermedad progrese a un linfoma o a una fibrosis pulmonar y desarrolle insuficiencia respiratoria.9
La variabilidad del pronóstico se debe a factores como la causa subyacente, la gravedad de la enfermedad y la respuesta o no al tratamiento.
4. SÍNDROME DE BIRT-HOGG-DUBÉ (SÍNDROME DE BHD)
Se trata de una enfermedad autosómica dominante causada por variantes poligénicas que afectan al gen de la foliculina (FLCN) ubicado en el cromosoma 17p11.2.
Se trata de un síndrome con manifestaciones a nivel cutáneo, pulmonar y renal.
Manifestaciones clínicas
- Afectación cutánea: fibrofoliculomas cutáneos, sobre todo en cabeza y cuello. Se observan en el 90% de los pacientes.
- Afectación pulmonar: se manifiesta radiológicamente por quistes pulmonares (de un tamaño variable, entre 0,2 y 7,8 cm). Pueden tener diferentes formas (redondos, ovalados y lentiformes) y ser de localización predominantemente basal y subpleural y a lo largo del mediastino. Alrededor del 40% de los pacientes pueden desarrollar un neumotórax.
- Afectación renal: mayor riesgo de tumores renales. Aunque la penetrancia del
cáncer renal es relativamente baja, los pacientes con síndrome de BHD tienen un riesgo siete veces mayor de sufrir tumores renales en comparación con la población general. Ante un diagnóstico de Síndrome de BHD es necesario realizar un estudio abdominal y llevar a cabo un seguimiento periódico para detectar y tratar precozmente estos tumores.
Diagnóstico
Los criterios propuestos por el Consorcio Europeo para el diagnóstico del síndrome
Birt-Hogg-Dubé se exponen en la tabla III. Para establecer el diagnóstico, el paciente debe cumplir al menos un criterio mayor o 2 criterios menores.
Tratamiento
No existe un tratamiento específico a nivel pulmonar. Se recomienda el abandono del tabaquismo en el paciente fumador y el tratamiento de las complicaciones como el neumotórax.
BIBLIOGRAFÍA
1. Raoof S, Bondalapati P, Vydyula R, Ryu JH, Gupta N, Raoof S, et al. Cystic lung diseases. Algorithmic approach. Chest 2016; 150: 945-965
2. Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse cystic lung disease. Part I. Am J Respir Crit Care Med 2015; 191: 1354-1366
3. McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, et al. Official American Thoracic Society/Japanese Respiratory Society clinical practice guidelines: lymphangioleiomyomatosis diagnosis and management. Am J Respir Crit Care Med 2016; 194: 748-761
4. Gupta N, Finlay GA, Kotloff RM, Strange C, Wilson KC et al. Lymphangioleiomyomatosis Diagnosis and Management: High-Resolution Chest Computed Tomography, Transbronchial Lung Biopsy, and Pleural Disease Management. An Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guideline. Am J Respir Crit Care Med 2017; 196: 1337-1348.
5. McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 2011; 364: 1595-606
6. Vassallo R, Harari S, Tazi A. Current understanding and management of pulmonary Langerhans cell histiocytosis. Thorax 2017; 72:937.
7. Benattia A, Bugnet E, Walter-Petrich A, de Margerie-Mellon C, Meignin V, Seguin-Givelet A, et al. Long-term outcomes of adult pulmonary Langerhans cell histiocytosis: a prospective cohort. Eur Respir J 2022; 59.
8. Lin HT, Wikenheiser-Brokamp KA, Udstuen G, Jones B, McCormack FX. Marked Improvement in Soft Tissue and CNS Manifestations of Adult Langerhans Cell Histiocytosis on Targeted MEK Inhibitor Therapy. Chest 2023; 163:e53.
9. Panchabhai TS, Farver C, Highland KB. Lymphocytic Interstitial Pneumonia. Clin Chest Med 2016; 37:463.
10. Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S, Ungari S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol 2009; 10:1199-206.
TABLAS
Y FIGURAS
Tabla I. Clasificación de las enfermedades pulmonares quísticas difusas (EPQD)²

Tabla II. Criterios diagnósticos de LAM de la ATS/JRS5

Tabla III. Criterios diagnósticos del Síndrome Birt-Hugg-Dubé10


Figura 1. TCAR de tórax de una paciente con LAM
Figura 2. Algoritmo diagnóstico para LAM (adaptado de Gupta N. y colaboradores)5
Sospecha de LAM esporádica basada en 1 o más características:
- Mujer joven con disnea progresiva.
- Neumotórax espontáneo
- Derrame quiloso
- PFR con obstrucción, y/o DLCO disminuida
¿Cumplen los quistes las características en TACAR?
Quistes de paredes finas y bien definidas, de tamaño uniforme, difusos, bilaterales.
¿Existen datos clínicos o entre las pruebas complementarias sugerente de esclerosis tuberosa?
LAM-Esclerosis tuberosa
Diagnóstico clínico LAM esporádica.
Solicitar TAC abdomen/pelvis y niveles de VEGDF en sangre
Realizar toracocentesis si existe DP.
¿Alguna característica presente?
Diagnóstico clínico LAM esporádica.
Angiomiolipoma renal / linfagioleiomioma VEGDF> 800 pg/ml. Derrame quiloso.
Considerar diagnóstico alternativo.
Tras tomar muestras de biopsia de la forma menos invasiva: ¿existe diagnóstico anatomopatológico de LAM?
Realizar biopsia pulmonar: ¿existe diagnóstico anatomopatológico de LAM?
Diagnóstico alternativo. Diagnóstico definitivo de LAM esporádica.

Figura 3. . TCAR de un paciente con HPCL

Figura 4. TCAR de un paciente con Neumonía intersticial linfocítica
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
15
NEUMONÍA ORGANIZADA
Autores
Ignacio Gayá García-Manso
Alba Mulet Arabí
Coordinadora
Raquel García Sevila
1. DEFINICIÓN
La neumonía organizada (NO) es un patrón histológico caracterizado por la excesiva proliferación de tejido de granulación dentro de la vía aérea y en los conductos alveolares asociado a inflamación crónica rodeando al alveolo. Se trata de una reacción de reparación tisular pulmonar tras una lesión. Este patrón constituye la base histológica de una entidad clínica, la neumonía organizada, que se clasifica como una enfermedad pulmonar intersticial difusa, que surge de una lesión en la pared alveolar y, generalmente, se caracteriza por la presencia de infiltrados pulmonares periféricos y por la buena respuesta al tratamiento con corticoides sistémicos.
Puede presentarse de forma idiopática (neumonía organizada criptogénica o NOC) o como respuesta a un daño pulmonar específico conocido, y también se puede observar como un hallazgo histopatológico en distintos contextos clínicos. La neumonía organizada criptogénica no tiene una causa conocida, y dentro de las enfermedades intersticiales se clasifica como una forma de neumonía intersticial idiopática (NII).¹ Además de la forma idiopática, puede ser secundaria a otras causas, como enfermedades del tejido conectivo, fármacos, neoplasias y otras neumonías intersticiales.
Anteriormente, esta entidad se conocía como bronquiolitis obliterante con neumonía organizada (BONO), pero su denominación se modificó para evitar la confusión con entidades cuya afectación es únicamente bronquiolar, como la bronquiolitis respiratoria asociada a trasplantes.
2. EPIDEMIOLOGÍA
La NOC es una enfermedad rara cuya incidencia y prevalencia es desconocida. En comparación con series antiguas, las series actuales muestran una menor incidencia, probablemente por mejor diagnóstico de las causas secundarias a las que se asocia. Una serie retrospectiva mostró una incidencia anual de NOC de 1,1 casos por cada 100.000 habitantes en un periodo de 20 años.² En un hospital de Canadá, se describió una prevalencia de 6,7 casos por 100.000 ingresos hospitalarios.³ En estudios sobre registros de enfermedad intersticial difusa, la prevalencia de NOC varía entre el 5%4 y el 10%; este último dato, procede de un estudio realizado en España en 2004.5
En general, la enfermedad se inicia en la 5ª o 6ª década de la vida. La incidencia es ligeramente superior en hombres (52%) que en mujeres, y es excepcional en niños. No se ha demostrado una relación clara con el tabaquismo activo o con antecedentes de tabaquismo, ya que, según algunas series, el 54% de los pacientes con NOC nunca habían fumado y menos del 15% de los fumadores y exfumadores declaraban estar fumando en el momento del diagnóstico del episodio de neumonía organizada.6
3. PATOGENIA
La neumonía organizada es un proceso inflamatorio y fibroproliferativo reversible que no altera la arquitectura pulmonar, como sí ocurre en otras EPID. Se produce inicialmente una lesión epitelial alveolar con necrosis de células epiteliales, que ocasiona una fuga de proteínas plasmáticas, produciéndose formación de fibrina y migración de células inflamatorias al espacio alveolar (macrófagos, linfocitos, neutrófilos, algunos eosinófilos, y, ocasionalmente, células plasmáticas y mastocitos).
A continuación, estas células proliferan y se diferencian en miofibroblastos, con posterior formación de las áreas fibroinflamatorias conocidas como cuerpos de Masson. Estas estructuras se entremezclan con el tejido conectivo rico en colágeno, fibronectina, procolágeno y proteoglicanos, y se produce una desregulación de factores de crecimiento del endotelio, fibroblastos y metaloproteasas, con aumento de la actividad angiogénica del nuevo tejido intraluminal formado de neumonía organizada.
Posteriormente, las células inflamatorias y los depósitos de fibrina comienzan a desaparecer de los brotes alveolares y se sustituyen por miofibroblastos organizados en anillos concéntricos. Los brotes intraalveolares se remodelan en intersticio, y se forman glóbulos de colágeno que fagocitan el colágeno y se rodean de nuevas células alveolares epiteliales que desarrollan de nuevo la membrana basal epitelial. Estas células proliferan y restablecen la continuidad de la membrana capilar alveolar, recuperando la integridad y la función de la unidad alveolar. En muy pocos pacientes, este proceso de
reparación no es completo, y se produce inicialmente un patrón fibrosante de NO, que evoluciona varios meses después a un patrón de neumonía intersticial no específica (NINE). También puede quedar un patrón de neumonía organizada cicatricial si falla la reabsorción de los cuerpos alveolares y la formación de glóbulos de colágeno, quedando algunas secuelas visibles en las pruebas de imagen.
Se ha sugerido relación entre NO y microaspiraciones de contenido gástrico en pacientes con enfermedad por reflujo gastroesofágico, y también se han relacionado otros factores ambientales, sin demostrar clara asociación o mecanismo patogénico.
4. PRESENTACIÓN CLÍNICA
La sintomatología habitual sugestiva de NOC es tos (71%), disnea (62%), fiebre (45%) y malestar, habitualmente de poco tiempo de evolución, de semanas o meses. El 75% de los pacientes presentan una sintomatología de menos de 2 meses de duración. La mitad de los casos se inician de forma aguda, como un cuadro pseudogripal con fiebre, malestar, disnea y tos. Se sospecha frecuentemente ante infiltrados pulmonares que sugieren neumonía infecciosa, pero que no se resuelven con antibioterapia o se presentan de forma recurrente. Son frecuentes los crepitantes a la auscultación pulmonar (el 60% de los casos), mientras que la presencia de sibilantes y acropaquias es muy infrecuente.
En ocasiones, la neumonía organizada puede aparecer como la primera manifestación de una enfermedad subyacente, como las enfermedades del tejido conectivo. En menos del 15% de los casos, la NOC se puede presentar de forma focal, como una lesión única que puede ser indistinguible radiológicamente de una neoplasia. En estos casos, habitualmente el paciente permanece asintomático.
5. CAUSAS / ETIOLOGÍA
Además de la NOC, que se define por ser idiopática, existen muchas otras entidades o procesos que se asocian a la aparición de neumonía organizada secundaria. Las causas a las que se asocia son muy variadas, algunas de ellas sencillas de diagnosticar y otras más difíciles, por lo que algunos autores defienden que la mayor parte de las NOC no son realmente idiopáticas, sino que probablemente sean secundarias a otro proceso que no ha podido ser identificado correctamente.
En la tabla I, se muestran las principales causas de neumonía organizada secundaria descritas en la literatura.
6. DIAGNÓSTICO
6.1. Evaluación
La neumonía organizada (NO) es una entidad rara, cuyo diagnóstico se realiza por exclusión. Precisa un abordaje multidisciplinar, siendo siempre necesaria una buena historia clínica y pruebas de radiología (recomendándose una tomografía computerizada). Según la seguridad diagnóstica, se empezará con tratamiento empírico, se realizará un LBA (lavado broncoalveolar) o se solicitará una biopsia.7
Historia clínica
Preguntar sobre infecciones respiratorias recientes, enfermedades sistémicas o síntomas de estas y revisar tratamientos susceptibles de producir NO secundaria.
Tests de laboratorio
Se suele observar elevación de reactantes de fase aguda, sin ningún hallazgo específico. Siempre deberemos solicitar perfil de autoinmunidad, puesto que la presentación de la NO puede preceder a varias conectivopatías que se asociarían a esta entidad.
Pruebas de función pulmonar
Suelen mostrar un patrón restrictivo y una DLCO disminuida, pero también pueden ser normales, según la extensión.
Estudios radiológicos
El hallazgo típico en la RX de tórax son las opacidades bilaterales parcheadas o difusas.
La TC de tórax (TCAR) nos sirve para objetivar mejor la gravedad del proceso. El patrón característico son consolidaciones periféricas multifocales (puede haber broncograma aéreo). Puede ser uni o bilateral y suele haber un predominio subpleural y en campos inferiores. Sin embargo, también se puede observar opacidades en vidrio deslustrado, nódulos, un patrón lineal o en bandas. También se puede objetivar el signo del halo reverso: una consolidación con un aclaramiento central, en vidrio deslustrado (hallazgo muy específico, pero poco frecuente).
La característica principal de estos infiltrados es que son migratorios.
Si la forma de presentación radiológica es con otro patrón asociado (NINE en la mayoría de los casos), habría que buscar siempre una causa secundaria, pero no hay ningún hallazgo característico que diferencie la NO criptogénica de la secundaria. (Figura 1)
Lavado broncoalveolar (LBA)
Sus hallazgos no son diagnósticos, pero es una herramienta clave, ya que ayuda a excluir otras entidades (infecciones, otras enfermedades intersticiales con LBA caracte-
rístico, como la neumonía eosinofílica). Habitualmente, tiene un predominio linfocitario (del 20-50%), con eosinofilia en algunos casos (del 5 al 25%) y con una proporción baja de neutrófilos (5 al 10%). El cociente CD4/CD8 está disminuido.
Biopsia
Únicamente se realizará cuando la posibilidad de un diagnóstico alternativo sea elevada o cuando no existe mejoría a pesar del tratamiento empírico. La decisión de la biopsia se deberá tomar en el comité multidisciplinar.
Métodos de biopsia
- Biopsia transbronquial (BTB): Muestra de menos tamaño, pero con menos riesgos. Suele ser rentable para el diagnóstico de NO.
- Criobiopsia: En centros con experiencia estaría indicada, ya que consigue muestras más grandes y representativas.
- Biopsia quirúrgica: Mediante videotoracoscopia o biopsia pulmonar quirúrgica.
6.2. Diagnóstico diferencial
- Neumonía infecciosa / Infección atípica. Muchas veces, los pacientes son diagnosticados de neumonía infecciosa, y el diagnóstico de NO ocurre ante la ausencia de mejoría con el tratamiento antibiótico.
- Neumonía eosinofílica. El conteo de eosinófilos elevados en el lavado broncoalveolar es diagnóstico de esta enfermedad, por lo que, si es normal, descartamos esta opción diagnóstica.
- Enfermedades granulomatosas. Tuberculosis, infección por micobacterias atípicas, sarcoidosis, beriliosis, neumonitis por hipersensibilidad (para excluirla es necesaria una buena anamnesis preguntando posibles agentes etiológicos).
- Vasculitis. Se deben solicitar ANCA en el estudio inicial.
- Adenocarcinoma mucinoso, linfoma pulmonar. En las formas más raras de presentación, como una lesión focal solitaria. Precisaría biopsia para el diagnóstico, ya que puede dar un falso positivo en el PET.
- Hemorragia alveolar. El LBA sería diagnóstico, con más de 20% de siderófagos.
- EVALI (daño pulmonar asociado al uso de cigarros electrónicos o vapeadores). Importancia de la historia clínica. En algunos pacientes, se precisa una biopsia pulmonar para confirmar el diagnóstico (daño alveolar difuso).
El diagnóstico definitivo requiere la demostración de características histopatológicas típicas en un paciente con clínica y patrón radiológico compatibles.
6.3. Diagnóstico histopatológico
Para su diagnóstico histológico, existen distintos elementos clave sugestivos de la enfermedad. También se pueden identificar cambios que sugieran que la NO es secundaria a otra enfermedad subyacente, como la presencia de inflamación crónica intersticial, agregados linfoides (toxicidad por fármacos o enfermedad de colágeno), atipia de neumocitos (neumonitis por radioterapia) (Tabla II).
Neumonía organizada fibrinosa aguda. Se trata de una variante histológica asociada a un proceso clínico de daño pulmonar agudo. Puede ser idiopática o asociada a otros procesos, como la neumonitis por hipersensibilidad, infecciones, toxicidad por fármacos o conectivopatías, entre otros. Histológicamente, se caracteriza por un depósito alveolar de fibrina. Estos pacientes suelen tener un mayor índice de recaídas (Figura 2).8
7. TRATAMIENTO
El tratamiento de la neumonía organizada no ha sido estudiado en ensayos clínicos, así que su evidencia se basa en la práctica clínica.9
La necesidad de iniciar tratamiento o no va a depender de la gravedad de los síntomas, la función pulmonar, la extensión radiológica y la rapidez de la progresión (Figura 3).
Se ha estudiado el uso de los macrólidos en los pacientes con enfermedad leve en los que se prefiere evitar el uso de corticoides. El beneficio de este tratamiento se relaciona con su efecto antiinflamatorio. Sin embargo, hay menos evidencia de su efectividad respecto a los corticoides.
Tratamiento para pacientes con enfermedad grave
Para los pacientes con enfermedad rápidamente progresiva o con insuficiencia respiratoria.
- Bolos de metilprednisolona 500 mg - 1 g al día durante 3-5 días y revalorar: Siempre que el paciente requiera soporte de oxigenoterapia importante.
- En caso de no mejoría a pesar de los bolos, se indicaría ciclofosfamida IV (sobre todo para pacientes en UCI/VNI) o azatioprina.
Una vez se consiga la estabilización clínica, se debería hacer una transición al tratamiento con prednisona a 0,5-1 mg/kg (con un máximo de 60 mg/día). Posteriormente, se irá descendiendo siguiendo el algoritmo de la enfermedad moderada/leve.
8. PRONÓSTICO
Dos tercios de los pacientes tratados con corticoides consiguen una normalización radiológica y mejoría clínica.
En general, la gravedad de la enfermedad en el momento del diagnóstico se relaciona con la posibilidad de recaídas posteriores.10
Factores de riesgo de recaídas
- Hipoxemia al diagnóstico
- Extensión radiológica
- Grado de afectación de las pruebas de función pulmonar
- Enfermedades subyacentes
- Retraso en el inicio del tratamiento
En los estudios retrospectivos de pacientes con NO, la causa de muerte debido a la enfermedad es inferior al 10%, con una supervivencia a los 5 años de más del 90%. Suele ser más frecuente la mortalidad por esta causa en los pacientes con una enfermedad subyacente que en los pacientes con neumonía criptogénica.
BIBLIOGRAFÍA
1. Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG, et al. An official American Thoracic Society / European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J RespirCrit Care Med. 2013;188(6):733-748.
2. Gudmundsson G, Sveinsson O, Isaksson HJ, Jonsson S, Frodadottir H, Aspelund T. Epidemiology of organising pneumonia in Iceland. Thorax. 2006;61(9):805-808.
3. Alasaly K, Muller N, Ostrow DN, Champion P, FitzGerald JM. Cryptogenic organizing pneumonia. A report of 25 cases and a review of the literature. Medicine (Baltimore). 1995;74(4):201-211.
4. Karakatsani A, Papakosta D, Rapti A, Antoniou KM, Dimadi M, Markopoulou A, et al. Epidemiology of interstitial lung diseases in Greece. Respir Med. 2009;103(8):1122-1129.
5. Xaubet A, Ancochea J, Morell F, Rodriguez-Arias JM, Villena V, Blanquer R, et al. Report on the incidence of interstitial lung diseases in Spain. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21(1):64-70.
6. Zhang Y, Li N, Li Q, Zhou Y, Zhang F, Chen T, et al. Analysis of the clinical characteristics of 176 patients with pathologically confirmed cryptogenic organizing pneumonia. Ann Transl Med. 2020;8(12):763.
7. Cherian SV, Patel D, Machnicki S, Naidich D, Stover D, Travis WD, et al. Algorithmic Approach to the Diagnosis of Organizing Pneumonia: A Correlation of Clinical, Radiologic, and Pathologic Features. Chest. 2022;162(1):156-178.
8. Beasley MB, Franks TJ, Galvin JR, Gochuico B, Travis WD. Acute fibrinous and organizing pneumonia: a histological pattern of lung injury and possible variant of diffuse alveolar damage. Arch Pathol Lab Med. 2002;126(9):1064-1070.
9. King TE, Lee JS. Cryptogenic Organizing Pneumonia. N Engl J Med. 2022;386(11):10581069.
10. Barroso E, Hernández L, Gil J, Garcia R, Aranda I, Romero, S. Idiopathic organizing pneumonia: a relapsing disease. 19 years of experience in a hospital setting. Respiration. 2007:74(6):624-631.
TABLAS Y FIGURAS
Tabla I. Adaptada de King TE, Lee JS. Cryptogenic Organizing Pneumonia. N Engl J Med. 2022;386(11)

Tabla II. Elementos Clave en el Diagnóstico Histológico

1. Imágenes de TCAR compatibles con NO.


Imagen superior: Infiltrados multifocales con broncograma aéreo.
Imagen inferior: Consolidaciones bibasales y periféricas en vidrio deslustrado.
Figura
Figura 2. Hallazgos histológicos característicos de neumonía organizada


En la imagen superior, se muestran los hallazgos histológicos característicos de neumonía organizada, con formación de cuerpos de Masson parcialmente colagenizados. En la imagen inferior, se visualizan, a mayor aumento, hallazgos muestra de neumonía organizada en fase más avanzada, con alteración de la estructura alveolar normal, reemplazada por fibrosis.
Figura 3. Adaptada de King TE, Lee JS. Cryptogenic Organizing Pneumonia.
N Engl J Med. 2022;386(11)

MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
ENFERMEDAD PULMONAR
1. INTRODUCCIÓN
INTERSTICIAL PRODUCIDA POR FÁRMACOS Y RADIOTERAPIA
Autoras
Cristina Matesanz López
Aylaf Latif Essa
Coordinadora
María Teresa Río Ramírez
Actualmente, la enfermedad pulmonar intersticial provocada por fármacos va en aumento, al igual que el número de fármacos que se relacionan con su desarrollo. Esto es debido, fundamentalmente, a la aparición de nuevos tratamientos dirigidos a las enfermedades reumatológicas y oncológicas, entre otras.
Hasta el momento, se han descrito más de 1.300¹ fármacos con potencial de producir toxicidad pulmonar. Todos ellos están recogidos y se pueden consultar en la web de Pneumotox (www.pneumotox.com).
2. EPIDEMIOLOGÍA
La incidencia de este tipo de EPID es difícil de estimar. Varía según distintos factores, como el tipo de fármaco, la dosis, los antecedentes personales, el sexo del paciente e, incluso, la raza, debido a las diferencias en el metabolismo. No obstante, si tenemos en cuenta estudios recientes, la frecuencia oscila entre el 2 y el 5%, en función de la fuente revisada.² 16
3. FACTORES DE RIESGO
En función del fármaco, se han descrito distintos factores de riesgo. Entre los más importantes, destacan la edad avanzada, la existencia previa de EPID o de enfermedad pulmonar, el tabaquismo activo, la dosis, el sexo masculino o la enfermedad de base. Es importante destacar la influencia de los factores genéticos. Por ejemplo, en el caso del metotrexato (MTX), el HLA-A*31:01 se ha relacionado con un aumento del riesgo de neumonitis. En el caso de la gemcitabina y el erlotinib, esta relación se ha encontrado con el HLA-DRB1*04:05.
4. CLÍNICA
La enfermedad pulmonar intersticial provocada por fármacos se manifiesta con una variedad de patrones clínicos que van desde síntomas respiratorios leves hasta insuficiencia respiratoria rápidamente progresiva y la muerte. En la mayoría de los casos, no existen características clínicas, de laboratorio, radiológicas o patológicas patognomónicas, y el diagnóstico de neumotoxicidad se sospecha en presencia de exposición a un fármaco que se sabe causante de toxicidad pulmonar, tras excluir causas alternativas de afectación intersticial, como insuficiencia cardiaca, infecciones o linfangitis.
5. MÉTODOS DIAGNÓSTICOS
Además de la historia clínica, la radiología es clave para orientar el diagnóstico. En gran número de casos, se observan infiltrados en la radiografía de tórax, aunque no siempre es así y se precisa la tomografía axial computarizada (TAC) de tórax, que es una prueba más sensible para la detección de EPID inducida por fármacos. Aun así, los hallazgos no son específicos, puesto que los patrones observados pueden apreciarse en otras EPID. La siguiente tabla muestra un resumen de los principales patrones radiológicos observados en estos casos (Tabla I).
Funcionalmente, se suele observar disminución de la capacidad vital forzada en la espirometría y disminución de la capacidad de difusión de monóxido de carbono.
La analítica puede mostrar eosinofilia periférica, sin ser un dato obligado en todos los casos.
La broncoscopia es otra técnica que puede ser de ayuda, sobre todo para descartar otros diagnósticos alternativos, como las infecciones o las neoplasias. El lavado broncoalveolar (LBA) suele mostrar una alveolitis linfocitaria con predominancia de las células CD8+, aunque en el caso del MTX o del sirolimus, pueden incrementarse las células CD4+. En caso de que se produzca una hemorragia alveolar o una neumonía eosinofílica, este LBA también será diagnóstico.
6. GRUPOS FARMACOLÓGICOS QUE CON MAYOR FRE-
CUENCIA PRODUCEN NEUMOTOXICIDAD
6.1. ANTIARRÍTMICOS
Amiodarona
La amiodarona es un antiarrítmico con alto contenido en yodo. Tiene una gran afinidad lipídica y es un potente inhibidor de la fosfolipasa, que induce a la acumulación de lípidos en los tejidos. Tras su metabolización hepática, se amplifica su capacidad de acumulación tisular, incluyendo al pulmón, lo que promueve su potencial toxicidad.
La incidencia de toxicidad pulmonar es del 1,2-8,8%, con mortalidad variable, entre el 3-37%, según las series.
Son factores de riesgo para el desarrollo de toxicidad: una dosis acumulada elevada (dosis superiores a los 400 mg diarios), duración superior a dos meses, edad avanzada, sexo masculino e historia previa de patología respiratoria.
El cuadro puede presentarse de forma aguda o subaguda. De forma excepcional, en el contexto de cirugía cardiaca o pulmonar, se ha descrito un desarrollo hiperagudo en forma de distrés respiratorio. En la forma aguda, se puede observar un cuadro brusco de disnea en el que se desarrolla insuficiencia respiratoria. Sin embargo, la forma más frecuente es la subaguda.
Histológicamente, se caracteriza por la presencia de macrófagos alveolares con citoplasma espumoso, cargados de lípidos y también de un infiltrado inflamatorio con linfocitos y leucocitos. En el LBA, se puede obtener un aumento de neutrófilos, de linfocitos o de ambos, con un predominio de linfocitos CD8. También se pueden ver macrófagos con citoplasma espumoso típicos, que sólo indican exposición y no toxicidad, pero su ausencia hace improbable el diagnóstico.
Flecainida
Es un agente antiarrítmico clase Ic. En caso de uso crónico, tiene capacidad de acumularse a nivel pulmonar y puede producir neumotoxicidad, aunque esta es muy infrecuente.
6.2. ANTIBIÓTICOS
Nitrofurantoina
Es un antibiótico empleado, fundamentalmente, en infecciones urinarias. La mayoría de los casos se presentan de forma aguda, tras una o dos semanas de tratamiento, y es el resultado de una respuesta de hipersensibilidad tipo I o III. En la TCAR, habitualmente se observa vidrio deslustrado bilateral y consolidaciones, con predominio de eosinófilos en LBA.
No obstante, también existe una forma crónica, tras tratamientos prolongados. Tiene un curso más insidioso con un cuadro similar a la fibrosis pulmonar idiopática.
6.3. ANTINEOPLÁSICOS
6.3.1.
QUIMIOTERAPIA
Bleomicina
Las bleomicinas son una familia de glicopéptidos aislados del hongo Streptomyces verticillus. Es un agente antineoplásico empleado habitualmente en el tratamiento del linfoma de Hodgkin y tumores de células germinales.
Su uso está limitado por la alta incidencia de toxicidad pulmonar que es dosis-dependiente, con incidencias que van del 3-5% al 20%, según si las dosis utilizadas son mayores de 300 mg y 500 mg, respectivamente.
La bleomicina genera un daño directo, a través de radicales libres que alteran el ciclo celular, y libera factores de crecimiento que participan como quimioatrayentes de células inflamatorias y fibroblastos. Se sospecha que tanto el pulmón como la piel son dos órganos “diana de su toxicidad” por carecer de una enzima que la inhibe denominada “enzima hidrolasa de bleomicina”.
A nivel radiológico, el daño alveolar difuso producido se asocia con consolidaciones parenquimatosas y opacidades en vidrio deslustrado. Es común observar predominio bibasal y bilateral. En algunas ocasiones, aparecen nódulos pulmonares, que simulan metástasis. Cuando el proceso es crónico, se puede observar un patrón fibrótico con reticulación, bronquiectasias de tracción, pérdida de volumen y panal de abeja.
Histológicamente, lo más frecuente es la infiltración de células mononucleares, la proliferación de fibroblastos y la fibrosis. Se ha descrito neumonía organizada en este contexto. El LBA suele mostrar la presencia de alveolitis polimorfonuclear.
Gemcitabina
La gemcitabina es un análogo de la pirimidina. Se emplea para tratar multitud de cánceres, entre los que se encuentra el cáncer de pulmón de células no pequeñas. No está clara la relación entre la toxicidad y la dosis. La incidencia de neumotoxicidad varía entre el 0,7% y el 13% en análisis retrospectivos. Se han informado toxicidades pulmonares debidas a la gemcitabina, que incluyen broncoespasmo, síndrome de fuga capilar, edema pulmonar no cardiogénico, reacción de hipersensibilidad, síndrome de dificultad respiratoria aguda, daño alveolar difuso, derrame pleural y neumonitis intersticial crónica, e incluso enfermedad venooclusiva.
Los taxanos son un conjunto de medicamentos antineoplásicos que impiden el crecimiento celular al bloquear la mitosis. Juegan un papel importante en el cáncer de mama, pulmón y ovario.
La neumonitis relacionada con el apaclitaxel fue descrita por primera vez en 1995, y se atribuye tanto a daños directos como a inmunomediados. Presenta una incidencia muy variable que oscila entre porcentajes inferiores al 1% y superiores al 10%. Esta puede verse aumentada por la combinación con otros agentes citotóxicos, y concretamente con su asociación con gemcitabina o radiación.
Los hallazgos radiológicos suelen implicar vidrio deslustrado e infiltrados parenquimatosos.
Terapias dirigidas al EGFR
Los inhibidores de EGFR, bloquean la actividad de una proteína denominada receptor del factor de crecimiento epidérmico (EGFR), una proteína transmembrana de la familia de los receptores de factores de crecimiento tipo tirosina cinasa. En este grupo de tratamientos, podemos encontrar el cetuximab, panitumumab, erlotinib y gefitinib.
Estos fármacos son empleados, fundamentalmente, para el tratamiento del cáncer de mama, colorrectal y de células no pequeñas de pulmón. El EGFR se relaciona con la reparación del epitelio bronquioalveolar, por lo que su inhibición puede ser la causa de la toxicidad pulmonar.
La incidencia de EPID en estos casos es baja, aunque la mortalidad puede llegar a ser elevada, con una mediana de 100 días de tratamiento hasta su desarrollo. Un estudio japonés describió tasas de mortalidad de entre el 41,6% y el 51,3% en pacientes tratados con cetuximab y panitumumab, respectivamente.
Inhibidores mTOR
La vía de mTOR, una proteína, es una vía de señalización molecular básica en la regulación del metabolismo, la proliferación y la diferenciación celular. Por ello, su bloqueo se emplea como tratamiento de enfermedades como el cáncer renal y los tumores neuroendocrinos o para prevenir el rechazo de trasplante de órgano sólido.
El sirolimus, everolimus y temsirolimus se han relacionado con toxicidad pulmonar, aunque su fisiopatología es poco conocida. Un metanálisis de 2.233 pacientes con cáncer tratados con everolimus describió una incidencia del 10% de neumonitis por el fármaco, de las que un 2,4% eran graves. No se apreció ninguna diferencia en función del sexo ni de la duración del tratamiento. Puede aparecer tanto al inicio como tras varios años de tratamiento.
A nivel radiológico, en muchas ocasiones la radiografía de tórax no muestra alteraciones, siendo necesaria una TCAR de tórax. Habitualmente, se observa vidrio deslustrado, aunque también se han descrito infiltrados periféricos con posterior patrón histológico de neumonía organizada. También se han descrito casos de fibrosis pulmonar.
6.3.2. INMUNOTERAPIA
La inmunoterapia facilita y mejora la respuesta inmunitaria del propio organismo. Dentro de la misma, destaca la terapia con inhibidores de puntos de control inmunitario (ICIs), la cual se divide en dos grandes grupos en función de su diana de actuación: los anti CTLA-4 y los anti PD/PD-L 1/2.
Dentro de los fármacos anti-PD-1/PD-L1, en la actualidad disponemos de nivolumab, pembrolizumab, atezolizumab, avelumab, durvalumab, cemiplimab, dostarlimab y retifanilimab. Se ha descrito una mayor incidencia de neumonitis en pacientes con estos tratamientos que con anti-CTLA-4, como el ipilimumab.
No obstante, sí se ha descrito un claro aumento de la incidencia al recibir un tratamiento combinado respecto a la monoterapia (10 % y 3 %, respectivamente, P < 0,001), al igual que un peor pronóstico. También se ha observado un aumento del riesgo de neumonitis en función del cáncer de base, siendo la incidencia mayor en cáncer de pulmón no microcítico y carcinoma de células renales respecto al melanoma.
El tiempo para la aparición de neumonitis relacionada con la inmunoterapia puede variar. La mediana se encuentra en 3 meses, con un rango de 2 a 24 meses, aunque el desarrollo es más rápido con tratamiento combinado que con monoterapia.
Lo más común es observar opacidades en vidrio deslustrado o infiltrados parcheados nodulares, fundamentalmente en lóbulos inferiores. A diferencia de otro tipo de toxicidades revisadas previamente, en las neumonitis por inmunoterapia es más frecuente observar una afectación focal y no difusa. Los síntomas suelen consistir en disnea, tos, fiebre y dolor torácico, además de insuficiencia respiratoria en los casos más graves.
Además de los hallazgos típicos de neumonitis, también se puede desarrollar derrame pleural o reacciones granulomatosas de tipo sarcoideo, con afectación nodular y ganglionar, lo cual puede llegar a confundirse con progresión de la enfermedad. En estos casos, la biopsia puede ser de gran ayuda.
6.4. TRATAMIENTOS INMUNOSUPRESORES
Metotrexato
El metotrexato (MTX) es un fármaco inmunosupresor conocido por ser el principal fármaco antirreumático modificador de enfermedad (FAME) en la artritis reumatoide (AR). No obstante, también se emplea para otras enfermedades, como la psoriasis o la sarcoidosis.
La neumonitis aguda por MTX suele ocurrir durante el primer año de tratamiento y está causada por un mecanismo de hipersensibilidad al fármaco. No depende de la dosis total acumulada.
Como factores de riesgo para su aparición, se han señalado la edad superior a 60 años, el uso previo de otros FAME, la presencia de diabetes mellitus insulinodependiente, la hipoalbuminemia y el antecedente de EPID previa.
El patrón radiológico que suele observarse es de NINE o daño pulmonar agudo. En estos casos, el LBA mostrará un predominio de linfocitos CD4 con incremento del índice CD4/ CD8. La presencia de eosinofilia es rara.
Aunque la neumonitis aguda por MTX es una realidad clínica, está sobrediagnosticada.
De hecho, en un estudio de cohortes, no se observó que el MTX aumentase el riesgo de EPID en AR,4 mientras que en estos pacientes con AR parece retrasar la aparición de la EPID y mejorar su pronóstico.5
Sulfasalazina
Es una sulfonamida empleada fundamentalmente en enfermedad inflamatoria intestinal y AR, por su función antiinflamatoria e inmunoduladora. Tras metabolizarse en intestino delgado, genera un metabolito que puede producir toxicidad pulmonar.
Lo más frecuente es observar disnea progresiva y tos. Puede producir infiltrados a nivel radiológico, al igual que eosinofilia periférica o pulmonar.
Leflunomida
La leflunomida (LEF) es un inmunomodulador con propiedades antiinflamatorias y antiproliferativas empleado fundamentalmente en AR y psoriasis.
La mayor parte de los datos de los que disponemos sobre su incidencia y prevalencia se han descrito en población japonesa. En estos estudios, la prevalencia de EPID inducida o exacerbada por LEF fue del 1%-1,2%.
La neumonitis por LEF suele desarrollarse durante los primeros cinco meses desde el inicio del tratamiento. Los principales factores de riesgo para su desarrollo son la administración de una dosis de carga de 100 mg/día durante 3 días, el tabaquismo, el bajo peso y el antecedente de EPID previa.
6.5. TRATAMIENTOS BIOLÓGICOS
Los tratamientos biológicos se producen o se extraen a partir de, como su propio nombre indica, una fuente biológica, como fluidos, tejidos humanos o animales o microorganismos.
Dentro de este grupo, los fármacos inhibidores del factor de crecimiento tumoral (anti-TNF), como el etanercept o el inflimiximab, son los que presentan mayor incidencia y prevalencia de EPID. No obstante, la EPID derivada del uso de terapias biológicas es rara, aunque se desconoce su incidencia exacta.
Como factores de peor pronóstico, se han observado la edad avanzada, la presencia previa de EPID y el uso concomitante de otro tipo de tratamientos inmunosupresores.
La aparición de infiltrados pulmonares bilaterales en la radiografía de tórax es la forma más frecuente de presentación, aunque también se han descrito casos sarcoidosis-like, sobre todo con los anti-TNF, especialmente el etanercept. Esto obliga a la toma de biopsias, puesto que este fármaco presenta riesgo de reactivación tuberculosa.
Además, los anti-TNF pueden favorecer el desarrollo de ANA y, aunque la mayoría de los pacientes son asintomáticos, se han descrito casos de vasculitis y síndromes lupus-like.
El rituximab, un fármaco anti-CD20, también se ha relacionado con el desarrollo de EPID, aunque en menor medida. Los casos descritos fueron fundamentalmente en pacientes que recibían el tratamiento por una neoplasia subyacente y se produjeron en los primeros tres meses tras el inicio del tratamiento.
Tratamiento y pronóstico
La base del tratamiento es la interrupción del fármaco. A pesar de que es frecuente asociar corticoides en casos de cierta gravedad, la evidencia sobre su eficacia es escasa, al igual que sobre la dosis necesaria y la duración mínima de tratamiento. La respuesta y el pronóstico son variables, desde la escasa o nula respuesta en los pacientes con daño alveolar difuso hasta la buena evolución en los patrones de neumonía organizada.
Los pacientes con patrón fibrótico que cumplan criterios de fibrosis pulmonar progresiva según las guías ATS6 podrían beneficiarse del uso del nintedanib como tratamiento antifibrótico.
Mención especial merece la inmunoterapia, puesto que existen guías de práctica clínica específicas dirigidas al tratamiento de la toxicidad por estos fármacos en función de la gravedad del cuadro (Tabla II).
6.6 ENFERMEDAD PULMONAR INTERSTICIAL PRODUCIDA POR RADIOTERAPIA
Durante muchos años, la radioterapia (RT) ha desempeñado un papel fundamental en los tratamientos contra el cáncer aplicados con intención curativa o paliativa además de como tratamiento coadyuvante a la quimioterapia o cirugía. La afectación pulmonar por RT sigue observándose actualmente en pacientes que reciben irradiación para tratamiento de cáncer de mama, cáncer de pulmón, enfermedades hematológicas y otro tipo de tumores de la cavidad torácica. En general, la radiación produce cambios, no sólo en la zona irradiada, sino también en zonas más alejadas.
Epidemiología
La incidencia de lesión pulmonar inducida por radiación (NR) es difícil de conocer, ya que varía en función de la técnica utilizada, pauta y dosificación del tratamiento y volumen de pulmón irradiado. Existen datos discordantes de incidencia en función de si la neumonitis es asintomática con afectación radiológica, encontrándose en el 45% de los pacientes, o si se considera solamente para neumonitis sintomática, en la que se describe incidencia del 10-15% de los casos.
En las últimas décadas, la incidencia de complicaciones pulmonares se encuentra en descenso debido a la mejora de la aplicación de la radiación, como la terapia de radiación corporal estereotáxica (SBRT) o de la radioterapia estereotáxica ablativa (SABR) que se aplica para el cáncer de pulmón en etapas tempranas en candidatos no quirúrgicos, así como para la enfermedad pulmonar oligometastásica de cualquier tumor sólido.
Factores de riesgo
Existen múltiples factores que pueden influir en el desarrollo de NR: unos dependen del tipo de tratamiento, mientras que otros se deben a la sinergia con otros tratamientos concomitantes y también a las características del propio paciente. En la tabla III se resumen los principales factores de riesgo.8,9
Fisiopatología
Las alteraciones histológicas producidas por la irradiación en el pulmón se pueden agrupar en 5 fases.8,9
- Fase inicial. Empieza en las primeras horas o días después del tratamiento y generalmente es asintomática. Se produce un daño vascular que induce la infiltración de células leucocitarias, la apoptosis de neumocitos tipo I y el edema pulmonar por aumento de la permeabilidad vascular.
- Fase tardía. Se caracteriza por disfunción ciliar e hipersecreción glandular bronquial.
- Fase exudativa. Ocurre a las 3-12 semanas después de la exposición a la RT. Se producen cambios degenerativos en el epitelio y endotelio bronquial con obstrucción de capilares y microtrombosis. Se produce la formación de membranas hialinas y la proliferación de neumocitos tipo II.
- Cuarta fase o fase intermedia. Se caracteriza por el depósito de colágeno, fibroblastos y engrosamiento del intersticio con resolución del exudado alveolar y la disolución de las membranas hialinas.
- Fase fibrótica. Suele ocurrir a partir de los 6 meses de tratamiento y puede progresar a lo largo de los años. Se produce un incremento de los miofibroblastos y un extenso depósito de colágeno en el intersticio y alveolos, lo que conlleva el colapso del espacio alveolar y la reducción del volumen pulmonar.
Manifestaciones clínicas y radiológicas
La gravedad de la NR varía desde hallazgos radiológicos sin síntomas hasta una enfermedad potencialmente mortal.
En general, la clínica consiste en tos seca, disnea de esfuerzo, fiebre elevada en casos más graves, además de clínica inespecífica, como malestar general y cuadro constitucional.
Comprende varias formas radiológicas de presentación que pueden incluir cualquier patrón radiológico:10
- Neumonía intersticial aguda/síndrome de dificultad respiratoria aguda (SDRA)
- Neumonía organizada
- Neumonía intersticial inespecífica
- Neumonitis por hipersensibilidad.
Neumonitis por recuerdo de radiación
La neumonitis por recuerdo de radiación (PRR) es una respuesta inflamatoria aguda en un pulmón previamente irradiado después de la administración de agentes antineoplásicos sistémicos. Tiene su inicio después de la exposición al agente desencadenante, pero puede ocurrir en cualquier momento durante el tratamiento. El tiempo transcurrido desde el inicio del tratamiento hasta la presencia de PRR varía de horas a años, y la gravedad no se correlaciona con el intervalo de tiempo entre la radioterapia y el tratamiento antineoplásico. Los taxanos y las antraciclinas son los fármacos más relacionados con la PRR. Los anticuerpos monoclonales, como el nivolumab o el durvalumab, se han asociado a casos muy graves de NR en pacientes con carcinoma de pulmón de células no pequeñas.
Neumonía eosinofílica
Se han descrito casos de mujeres asmáticas tratadas con radioterapia por cáncer de mama.
Diagnóstico
El diagnóstico se establece con la combinación de la clínica, tiempo transcurrido entre la clínica y la exposición, imágenes radiológicas compatibles y tras excluir otras causas. Generalmente, se recomiendan las pruebas que se detallan a continuación.
Analítica: No hay datos específicos. Puede observarse aumento de reactantes de fase aguda, como leucocitosis, VSG y proteína C reactiva. Se recomienda solicitar coagulación para descartar hemorragia pulmonar, además de pro-BNP y cultivos para descartar otras causas de infiltrados pulmonares.
Pruebas de imagen: La TACAR es la prueba de elección, ya que tiene más sensibilidad que la radiografía de tórax.
Pruebas de función pulmonar: Se suele observar un patrón restrictivo con disminución de la capacidad de difusión (DLCO) debido al daño del tejido pulmonar intersticial tras la RT que afecta el intercambio gaseoso. El test de la marcha puede ser una herramienta útil para valorar la evolución de la enfermedad.
Broncoscopia: en la mayoría de los pacientes, se realiza lavado broncoalveolar para excluir otros diagnósticos diferenciales. En pacientes con manifestaciones radiológicos atípicas, se podría plantear realizar biopsia bronquial, aunque en general no suele ser necesaria para confirmar el diagnóstico.
Tratamiento
Las recomendaciones de expertos aconsejan el uso de corticoides orales en casos sintomáticos con afectación aguda/subaguda. En aquellos con fibrosis establecida, al no existir un componente inflamatorio no se recomienda su uso. En pacientes asintomáticos u oligosintomáticos, puede presentarse resolución espontánea, y generalmente no se inicia el uso de esteroides a menos que empeore la clínica o se produzca una caída de la función pulmonar superior al 10%. Estudios aislados han mostrado la eficacia del uso de inmunosupresores, como la azatioprina y la ciclosporina.9,10
Nuevas opciones terapéuticas
En los últimos años, se han ensayado diferentes fármacos para la prevención del daño por radioterapia.9,10
Pentoxifilina. Tiene propiedades inmunomoduladoras y antiinflamatorias al inhibir el TNF-alfa. La toma de una dosis de 400 mg tres veces al día durante 8 semanas podría reducir la clínica y la fibrosis.
Vitamina E. El tratamiento combinado de pentoxifilina con vitamina E durante 6 meses podría mejorar de forma notable la fibrosis pulmonar.
Aminofostina. Es un agente radioprotector que actúa a través de la captación de radicales libres.
Otros. Los inhibidores de la enzima de conversión de la angiotensina (IECA) mostraron la reducción de fibrosis por irradiación en ratas, pero en humanos hay poca evidencia.
Colchicina, penicilamina y estatina. Estos fármacos pueden tener el potencial de modificar la progresión de la fibrosis.
Nintedanib. Recientemente, ha surgido como una forma prometedora de prevención y tratamiento de la neumonitis por radioterapia, sobre todo en aquellas con un componente importante de fibrosis, ya que mostró beneficios sobre todo en cuanto al enlentecimiento de la caída de la función pulmonar (Figura 1).
BIBLIOGRAFÍA
1. www.pneumotox.com.
2. Skeoch S, Weatherley N, Swift AJ, Oldroyd A, Johns C, Hayton C, et al. Drug-Induced Interstitial Lung Disease: A Systematic Review. J Clin Med. 2018;7(10):356.
3. Spagnolo P, Bonniaud P, Rossi G, Sverzellati N, Cottin V. Drug-induced interstitial lung disease. Eur Respir J. 2022;60(4):2102776.
4. Ibfelt EH, Jacobsen RK, Kopp TI, Cordtz RL, Jakobsen AS, Seersholm N, et al. Methotrexate and risk of interstitial lung disease and respiratory failure in rheumatoid arthritis: a nationwide population-based study. Rheumatology. 2021;60(1):346-352.
5. Narváez J, Díaz Del Campo Fontecha P, Brito García N, Bonilla G, Aburto M, Castellví I, et al. Recomendaciones SER-SEPAR para el manejo de la enfermedad pulmonar intersticial difusa asociada a la artritis reumatoide. Parte 2: tratamiento. Reumatología Clínica. 2022;18(9):501-512.
6. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):18-47.
7. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM et al. National Comprehensive Cancer Network. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 201810;36(17):1714-1768.
8. Hanania A, Mainwaring Walker, Ghebre Y. Radiation-Induced Lung Injury: Assessment and Management. Chest. 2019;156(1):150-162.
9. Arroyo-Hernandez M, Maldonado F, Lozano-Ruiz F. Radiation-induced lung injury: current evidence. BMC pulmonary medicine 2021;21(1):9.
10. Konkol M, Sniatala P, Milecki P. Radiation-induced lung injury - what we do in the era modern radiotherapy?. Rep Pract Oncol Radiother. 2022;27(3):552-565.
TABLAS Y FIGURAS
Tabla I. Resumen de las posibles afectaciones radiológicas inducidas por fármacos

MTX: metotrexato. BCG: bacilo de Calmette-Guérin. VEGF: factor de crecimiento endotelial vascular. Fuente: Spagnolo P, Bonniaud P, Rossi G, Sverzellati N, Cottin V. Drug-induced interstitial lung disease. EurRespir J. 2022 27;60(4):2102776

Tabla II. Resumen del tratamiento de la neumonitis por inmunoterapia según sus grados (parte 1) (Continúa)
Tabla II. Resumen del tratamiento de la neumonitis por inmunoterapia según sus grados (parte 2)

Resumen del tratamiento de la neumonitis por inmunoterapia según sus grados. TAC: tomografía axial computarizada. EFR: exploración funcional respiratoria. LBA: lavado broncoalveolar. Rx: radiografía. IV: intravenoso. Fuente: Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. National Comprehensive Cancer Network. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018 10;36(17):1714-1768.
Tabla III. Factores de riesgo de lesión pulmonar inducida por radiación

Tabla IV. Clasificación radiológica de la lesión pulmonar inducida por radiación

Figura 1. Algoritmo de manejo y tratamiento de la enfermedad pulmonar intersticial producida por radioterapia. Fuente: Arroyo-Hernandez M, Maldonado F, Lozano-Ruiz F. Radiation-induced lung injury: current evidence. BMC Pulm Med. 2021;21(1):9.

MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
EPID SECUNDARIAS:
INFECCIONES Y ENFERMEDAD INJERTO CONTRA HUÉSPED (EICH)
Autoras
Ángela López Bauzá
Cecilia López Ramírez
Coordinador
José Antonio Rodríguez Portal
1. INTRODUCCIÓN
La enfermedad injerto contra huésped (EICH) es una complicación potencialmente grave en pacientes sometidos a trasplante de progenitores hematopoyéticos (TPH). El TPH tiene como función principal la sustitución de un sistema hematopoyético alterado por otro con células progenitoras hematopoyéticas capaces de autorregenerarse y diferenciarse en las diferentes líneas celulares.¹ En la actualidad, la sangre periférica es la fuente de células madre más utilizada, aunque pueden obtenerse de médula ósea o de sangre del cordón umbilical. Existen varios tipos de trasplantes: singénicos, autólogos y alogénicos. Difieren entre sí por la técnica utilizada, la fuente del donante y las complicaciones asociadas.¹
El manejo de la EICH con afectación intersticial pulmonar y de las infecciones concomitantes es un desafío clínico complejo que requiere un enfoque multidisciplinario. En este capítulo, proporcionaremos herramientas prácticas y pautas para optimizar la identificación y el tratamiento de estos pacientes.
2. CLASIFICACIÓN DE LAS COMPLICACIONES PULMONARES POSTERIORES AL TPH
Las complicaciones relacionadas con el trasplante pueden clasificarse, en función de su tiempo de aparición y en relación con la secuencia de recuperación del sistema inmune postrasplante, en tres fases, que se describen a continuación.² 17
- Fase neutropénica (0-30 días). Abarca los 30 primeros días postrasplante. En esta fase, aumenta el riesgo de bacteriemia por bacterias grampositivas y gramnegativas, reactivación del virus del herpes simple (VHS) e infecciones fúngicas que involucran especies de Candida e incluso aspergilosis invasiva debido a la neutropenia prolongada y a la alteración de la barrera mucocutánea. Entre las complicaciones pulmonares no infecciosas, están la hemorragia alveolar difusa (HAD), la neumonitis por fármacos, el edema pulmonar o el Sd. perinjerto.
- Fase precoz o temprana (30-100 días). Engloba desde la recuperación neutrofílica hasta 100 días postrasplante. En esta fase, aunque ya se ha producido una reconstitución de neutrófilos, persiste un deterioro de la inmunidad celular y humoral y continúa existiendo riesgo de infecciones por microorganismo oportunistas y mayor susceptibilidad a los virus respiratorios comunes, como la influenza, el virus respiratorio sincitial (VRS) y el adenovirus. Entre las complicaciones no infecciosas, destacan la EICH aguda (EICHa) o el síndrome de neumonía idiopática (SNI), que es un síndrome clínico grave que abarca varias entidades que se caracterizan por un extenso daño alveolar.
- Fase tardía. Desde los 100 días del TPH en adelante, cuando las células B y las células T CD4 se recuperan lentamente y predominan las complicaciones no infecciosas, como la bronquiolitis obliterante o el síndrome de bronquiolitis obliterante (BO y SBO, respectivamente), la fibroelastosis pleuroparenquimatosa (PPFE) o el trastorno linfoproliferativo post TPH.
3. COMPLICACIONES PULMONARES INFECCIOSAS
Antiguamente, las complicaciones más frecuentes eran infecciosas, hecho que ha cambiado en los últimos años debido principalmente a la optimización de la profilaxis antimicrobiana, que ha llevado a un aumento relativo en la incidencia de complicaciones pulmonares no infecciosas. Pese a esto, dadas las alteraciones del sistema inmune en el post TPH, las complicaciones pulmonares infecciosas deben considerarse siempre la primera opción durante este período.
En cuanto a las complicaciones infecciosas, existen muchos factores que se han asociado con un aumento significativo del riesgo de infección post TPH, como la neutropenia, la linfopenia, la hipogammaglobulinemia, la lesión de la barrera mucosa, la EICH, así como el tipo de régimen de acondicionamiento, la fuente de células madre, el tipo de donante y la profilaxis de la EICH, entre otros.³
La TCAR de tórax es más sensible que la radiografía de tórax y puede proporcionar información útil junto a la broncoscopia con LBA. Otras pruebas no invasivas, como la PCR nasal de virus respiratorios, el galactamanano en sangre o en LBA y/o PCR de CMV, han demostrado ser útiles para la identificación precoz de infecciones en estos pacientes.4
La mayoría de los patrones radiológicos no son específicos, incluso pueden coexistir varios procesos (infecciosos y no infecciosos). Los patrones más comunes en las infecciones pulmonares agudas son la atenuación en vidrio deslustrado (VD), el árbol en brote, las consolidaciones y los nódulos (Figura 1). En pacientes con EICHc, fase en que el sistema inmune ya está recuperado, se observan infecciones similares al resto de la población y las secundarias al tratamiento inmunosupresor en pacientes con EICHc (Tabla I).
4. COMPLICACIONES PULMONARES NO INFECCIOSAS
Las complicaciones pulmonares no infecciosas de aparición tardía son comunes principalmente después del TPH alogénico. Se desarrollan hasta en el 20% de los pacientes y son más frecuentes durante los primeros dos años posteriores al trasplante. De todas ellas, la más habitual es el SBO. Los factores asociados con un mayor riesgo de complicaciones pulmonares no infecciosas incluyen la edad avanzada, la presencia de EICH, el régimen de acondicionamiento, la fuente de células madre y la enfermedad pulmonar subyacente.4 Las pruebas de función respiratoria (PFR) previas al trasplante, incluidos el volumen espiratorio forzado en primer segundo (FEV1) y la capacidad de difusión del monóxido de carbono (DLco) se utilizan para identificar a los pacientes con alto riesgo de desarrollar complicaciones pulmonares, insuficiencia respiratoria y/o mortalidad después del TPH.6
Para la correcta identificación de las complicaciones pulmonares posteriores al TPH alogénico, es importante que todos los pacientes sean evaluados antes del trasplante mediante anamnesis, examen físico, función pulmonar y radiografía de tórax. En pacientes de edad avanzada, fumadores o con una evaluación inicial anormal, puede estar indicada la TCAR de tórax.4 En el período post TPH tardío (>100 días post TPH), las complicaciones pulmonares crónicas no infecciosas adquieren más importancia. Se recomienda monitorizar cuidadosamente a los pacientes después del TPH. La presencia de un nuevo patrón obstructivo en comparación con los valores iniciales sugerirá, en ausencia de infección, SBO, mientras que un nuevo hallazgo restrictivo requerirá más estudios que permitan detectar precozmente una posible complicación pulmonar no infecciosa asociada al TPH. Los hallazgos de atrapamiento aéreo en la TCAR de tórax (patrón de mosaico), el engrosamiento de las vías respiratorias distales o bronquiectasias orientan al SBO, mientras que otras complicaciones pulmonares no infecciosas distintas del SBO se manifiestan radiológicamente, en general con opacidades multilobares persistentes (vidrio esmerilado, consolidación, pequeños cambios lineales y reticulares), como veremos con detalle más adelante.
4.1 Bronquiolitis obliterante o síndrome de bronquiolitis obliterante
La bronquiolitis obliterante (BO) o síndrome de bronquiolitis obliterante (SBO) es la única manifestación tardía considerada propia de la EICH pulmonar, una complicación grave y multisistémica del TPH alogénico. El SBO se caracteriza por una limitación del flujo de aire de nueva aparición después de un TPH alogénico. El análisis histológico muestra inflamación y fibrosis concéntrica que conduce a una obliteración de la luz bronquiolar. En general, se desarrolla entre 100 días y 2 años después del trasplante, pero se ha documentado su incidencia más allá de 5 años. Para confirmar el diagnóstico de BO, se requeriría una biopsia pulmonar quirúrgica, pero puesto que rara vez puede realizarse, esta entidad se denomina comúnmente SBO y se diagnostica en base a criterios espirométricos y radiográficos cuando se cumplen los siguientes criterios:6
1. FEV1/VC <0,7 o el percentil 5 del previsto.
2. FEV1 <75% del previsto con una disminución ≥10% en menos de 2 años.
3. Ausencia de infección en el tracto respiratorio validada por clínica, radiografías y TC de tórax y/o cultivos microbiológicos (aspiración sinusal, cultivo de esputo, lavado broncoalveolar, etc.).
4. Una de las siguientes:
a. Evidencia de atrapamiento aéreo en los cortes de espiración, engrosamiento de pequeña vía aérea o bronquiectasias en la TCAR (Figura 2).
b. Evidencia de atrapamiento aéreo por PFR: VR (volumen residual) > 120% del previsto o VR/TLC elevado.
El SBO presenta síntomas inespecíficos, como tos, disnea y sibilancias espiratorias, aunque pueden ser asintomáticos durante las primeras etapas. Para su diagnóstico, se recomiendan PFR previas al trasplante y 100 días después del trasplante, para continuar el seguimiento a intervalos de 3-6 meses durante los primeros dos años y después del diagnóstico inicial de EICHc. Además, se recomienda TCAR de tórax en fase espiratoria al diagnóstico y si existe caída funcional y/o empeoramiento sintomático que justifique dicha indicación;8 no obstante, se recomienda individualizar la monitorización según cada caso.
El tratamiento del SBO sigue siendo un desafío. En 2020, la European Society for Blood and Marrow Transplantation publicó un documento de consenso en el que se recogen las recomendaciones de profilaxis y tratamiento de la EICH y que recomienda el llamado régimen FAM de fluticasona inhalada 440 µg dos veces al día, azitromicina 250 mg tres veces por semana y montelukast 10 mg al día, como tratamiento inicial asociado a corticoides sistémicos con descenso rápido durante el primer mes del diagnóstico.9
Otros tratamientos utilizados incluyen la fotoaféresis extracorpórea (FEC), los inhibidores de la tirosina quinasa, el ibrutinib y el ruxolitinib, con resultados variables y sin datos respecto a su eficacia para el SBO, mientras que el trasplante de pulmón es la única opción de tratamiento final tras 5 años de remisión completa. Por otro lado, se está considerando también el uso de medicación antifibrótica para el tratamiento del SBO debido a su eficacia para retardar la fibrosis pulmonar idiopática.
Recientemente, se ha publicado una guía de práctica clínica sobre el tratamiento de la EICHc pulmonar (SBO) en adultos llevado a cabo por la Sociedad Europea de Respiratorio (ERS) y la Sociedad Europea de Trasplante de Sangre y Médula (EBMT).8 Basándose en la evidencia clínica actual, los expertos sugieren el uso de corticoides inhalados con o sin LABA, además del régimen inmunosupresor convencional en pacientes con SBO (baja certeza de la evidencia); los corticoides inhalados, como la fluticasona, la azitromicina y/o el montelukast además del régimen inmunosupresor convencional pueden ser útiles en estos pacientes (certeza de la evidencia muy baja). El ruxolitinib y el imatinib pueden ser beneficiosos para pacientes con fenotipo SBO por una EICHc, aunque con una certeza de evidencia muy baja (recomendación condicional para la intervención o la comparación), mientras que no sugieren el uso del ibrutinib en estos pacientes, dado que no existen estudios que evalúen la eficacia del ibrutinib en pacientes con SBO.8 En un estudio prospectivo con imatinib, el FEV1 se estabilizó en la mayoría de los pacientes después de comenzar el tratamiento con imatinib, y además permitió reducir la dosis de corticoides sistémicos en estos pacientes. El ruxolitinib ha demostrado ser eficaz en la EICH extrapulmonar; sin embargo en dos de tres estudios retrospectivos se observó estabilización e incluso mejoría del FEV1. El belumosudil podría ser útil en pacientes con SBO. En dos estudios, se ha observado una mejoría del FEV1 y de la sintomatología respiratoria en pacientes con EICH crónica avanzada, pero dada la evidencia limitada, hay que interpretar estos hallazgos con cautela. El panel de expertos de la guía sugiere su uso como una recomendación condicional, dada que la calidad de la evidencia es muy baja. La FVC ha demostrado mejorar y/o estabilizar la función pulmonar en cuatro estudios prospectivos, y por ello el panel de expertos sugiere usarlo asociado a su tratamiento inmunosupresor convencional (recomendación condicional, certeza de evidencia muy baja). Por último, el trasplante de pulmón es el único tratamiento curativo en personas con enfermedad pulmonar terminal. Varios estudios han documentado una buena supervivencia general a largo plazo para adultos seleccionados con EICHc pulmonar, por lo que el panel de expertos sugiere esta opción terapéutica, dado que es la única que salva vidas (recomendación condicional, certeza de evidencia muy baja).8
4.2. Síndrome de neumonía idiopática
La American Thoracic Society (ATS) ha propuesto el término “síndrome de neumonía idiopática” (SNI) para denominar varios síndromes clínicos que muy a menudo reflejan una lesión pulmonar generalizada en ausencia de una infección concurrente, sobrecarga hídrica, insuficiencia cardiaca o renal (Tabla II).10
El SNI ocurre tanto en pacientes con TPH alogénico como autólogo. Se ha intentado clasificar el SNI en entidades específicas (Tabla III) basándose en gran parte en los sitios anatómicos de inflamación y disfunción.10
4.3 Síndrome de dificultad respiratoria periinjerto
El síndrome de dificultad respiratoria periinjerto (PERDS, por sus siglas en inglés, Perigraft respiratory distress syndrome) es un trastorno difuso de fuga capilar que ocurre de forma aguda en un subgrupo de pacientes con síndrome de injerto (SI), complicación que se ha asociado tanto a TPH alogénico como a TPH autogénico. Se define por la presencia de insuficiencia respiratoria hipoxémica e infiltrados pulmonares bilaterales, y no puede ser explicado por la presencia de una insuficiencia cardíaca y/o infección. Se considera un subtipo dentro del espectro de SIN.10
Para el diagnóstico de PERDS, se han propuesto varios criterios diagnósticos. En la práctica habitual, los criterios de Spitzer11 (Tabla IV) son los más utilizados.
Las manifestaciones clínicas y las pruebas complementarias son inespecíficas, lo que dificulta distinguirlo de otras entidades. En la TCAR, es frecuente el engrosamiento de los septos interlobulillares, junto a áreas de vidrio deslustrado de distribución predominantemente perihiliar y bilateral, con o sin derrame pleural asociado. Generalmente es autolimitado, con un pronóstico generalmente bueno con tratamiento precoz. El PERDS responde muy bien al tratamiento con corticoides a dosis altas (1-2 mg/kg de metilprednisolona durante 3 días), seguido de una disminución rápida, con una tasa de respuesta de al menos el 90% y una mortalidad de solo entre el 7% y el 10%. Otros autores recomiendan dosis de dexametasona de 4 mg durante 3 días con reducción posterior para los pacientes con SI menos grave, mientras que los bolos de metilprednisolona de 1 mg/kg se reservan para los pacientes con SI grave o de riesgo vital.
4.4 Neumonía organizada
La neumonía organizada (NO) puede aparecer tanto tras TPH alogénico como TPH autogénico, generalmente entre 2 y 15 meses después del TPH.12 Clínicamente, puede presentarse con fiebre, disnea y tos seca, en general con restricción pulmonar en las PFR y consolidaciones uni o bilaterales irregulares multifocales, a menudo con broncograma aéreo, que pueden ir acompañadas de imágenes en vidrio deslustrado en la TCAR.
La realización de un LBA permite excluir otras etiologías como las infecciones, los procesos neoplásicos o hemorragia alveolar, entre otras, mostrando una alveolitis linfocitaria. Las muestras histológicas obtenidas mediante biopsia se caracterizan por un exudado fibroso en los espacios alveolares, que se expande hacia los conductos alveolares y los bronquiolos respiratorios acompañado de un infiltrado inflamatorio polimórfico compuesto por macrófagos, células plasmáticas, linfocitos y eosinófilos.
La NO no suele tener un buen pronóstico. El tratamiento con corticosteroides es el estándar de oro. Sin embargo, la dosis y la duración del tratamiento no están bien establecidas, con dosis que oscilan entre 0,5 y 1 mg/kg/día durante al menos cuatro semanas, seguidos de una reducción gradual en los siguientes de 6 a 12 meses, según la literatura publicada. Con la suspensión y/o reducción de la dosis de tratamiento, la recaída es común. En estos casos, se recomienda retomar la dosis mínima eficaz durante mayor tiempo. Los macrólidos pueden ser útiles en algunos pacientes con manifestaciones leves.
4.5 Hemorragia alveolar difusa
La hemorragia alveolar difusa (HAD) se define como el retorno del líquido del LBA progresivamente más sanguinolento o un recuento de macrófagos cargados de hemosiderina ≥20% en los pacientes que han recibido un TPH en ausencia de infección, con una rápida evolución hacia insuficiencia respiratoria.13
La incidencia oscila entre el 1% y el 21% en el TPH autogénico y entre el 2 y el 17% en el TPH alogénico, produciéndose entre 11 y 21 días post TPH. Los factores de riesgo para el desarrollo de HAD incluyen los regímenes mieloablativos, la irradiación corporal total (total body irradiation, TBI) mieloablativa, la fuente de cordón umbilical, la edad avanzada y el retraso o el fallo del injerto.13
Clínicamente, se manifiesta como disnea progresiva, hipoxia, tos, consolidación difusa en la radiografía de tórax y retorno de líquido sanguinolento en el LBA. Aunque la presentación más común de HAD en la población no trasplantada consiste en hemoptisis, disnea y anemia por déficit de hierro, la hemoptisis franca en los pacientes trasplantados es rara.
Radiológicamente, la TCAR presenta opacidades en vidrio deslustrado de ubicación predominantemente central y multilobar, con engrosamiento septal superpuesto, con o sin patrón de “crazy paving”.
El pronóstico de la HAD es muy pobre, con una elevada mortalidad que oscila entre el 64 y el 100% según las series. Debido principalmente a la etiopatogenia desconocida, el tratamiento continúa siendo empírico. De forma habitual, se utilizan corticoides sistémicos, con necesidad de ingreso en la unidad de cuidados intensivos y ventilación mecánica. De forma general, se recomiendan dosis que varían entre los 500 mg y los 2 g/día o 30 mg/kg/día de metilprednisolona intravenosa (iv) durante de 3 a 5 días, seguidos de una reducción gradual durante 4 semanas. Entre otras medidas sugeridas en el tratamiento de la HAD, se han propuesto terapias procoagulantes, como el factor VIIa recombinante, y antagonistas de citocinas, como el etanercept y la ciclofosfamida, con resultados variables, por lo que no se utilizan de forma rutinaria como estándar de tratamiento.4
BIBLIOGRAFÍA
1. Duarte RF, Labopin M, Bader P, et al. Indications for haematopoietic stem cell transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2019. Bone Marrow Transplant. 2019;54(10):1525-1552.
2. Fraebel J, Engelhardt BG, Kim TK. Noninfectious Pulmonary Complications after Hematopoietic Stem Cell Transplantation. Transplant Cell Ther. 2023;29(2):82-93.
3. Akhmedov M. Infectious complications in allogeneic hematopoietic cell transplant recipients: Review of transplant-related risk factors and current state of prophylaxis. Clin Transplant. 2021;35(2):14172.
4. Haider S, Durairajan N, Soubani AO. Noninfectious pulmonary complications of haematopoietic stem cell transplantation. Eur Respir Rev. 2020;29(156):190119, doi:10.1183/16000617.0119-2019.
5. Vista de Infecciones pulmonares en pacientes trasplantados de precursores hematopoyéticos y órganos sólidos. Secuencia temporal y diagnóstico diferencial [Internet]. Espacio-seram.com. [consultado el 8 de mayo de 2024]. Disponible en: https://piper.espacio-seram. com/index.php/seram/article/view/7879/6345.
6. Cheng GS. Pulmonary Function and Pretransplant Evaluation of the Hematopoietic Cell Transplant Candidate. Clin Chest Med. 2017;38(2):307-316.
7. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2015;21(3):389401.
8. Bos S, Murray J, Marchetti M, Cheng G-S, Bergeron A, Wolff D, et al. ERS/EBMT clinical practice guidelines on treatment of pulmonary chronic graft-versus-host disease in adults. Eur Respir J. 2024;63(3):2301727 [consultado el 8 de mayo de 2024]. Disponible en: https://pubmed.ncbi.nlm.nih.gov/38485149.
9. Penack O, Marchetti M, Ruutu T, et al. Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: updated consensus recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol. 2020;7(2):157-167.
10. Panoskaltsis-Mortari A, Griese M, Madtes DK, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med. 2011;183(9):1262-1279.
11. Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001;27(9):893-898.
12. Yoshihara S, Yanik G, Cooke KR, et al. Bronchiolitis obliterans syndrome (BOS), bronchiolitis obliterans organizing pneumonia (BOOP), and other late-onset noninfectious pulmonary complications following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2007;13(7):749-759.
13. Afessa B, Tefferi A, Litzow MR et al. Diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med. 2002;166(5):641-645.
TABLAS
Y FIGURAS
Tabla I. Infecciones pulmonares en pacientes con TPH. Secuencia temporal característica asociada al período posterior al TPH y al estado inmune5

Tabla II. Definición de la SNI según la ATS10

Tabla III. Espectro clínico de la lesión pulmonar después del TPH10

Tabla IV. Criterios diagnósticos de Spitzer et al. para el SI.

Se requieren 3 criterios mayores o dos mayores y uno menor o más para hacer un diagnóstico del SI11
Figura 1. Algoritmo de abordaje diagnóstico inicial de pacientes sometidos a TPH con síntomas respiratorios.

TCAR: tomografía computarizada de alta resolución; BAL: lavado broncoalveolar; PERDS: síndrome de dificultad respiratoria periinjerto; DAH: hemorragia alveolar difusa; COP: neumonía organizada criptogénica; AIP: neumonitis intersticial aguda; SDRA: síndrome de dificultad respiratoria aguda; PFT: prueba de función pulmonar.4
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
EPID RARAS: PROTEINOSIS
ALVEOLAR, MICROLIATIASIS, AMILOIDOSIS, EPID POR
ALTERACIÓN DEL SURFACTANTE
PULMONAR
Autoras
Lirios Sacristán Bou Diego Durán Barata
Coordinador
Raúl Godoy Mayoral
Dentro del grupo de enfermedades pulmonares intersticiales se incluyen las enfermedades por depósito en el intersticio o en espacios alveolares de constituyentes endógenos (amiloide, surfactante o calcio) en cantidad suficiente para deformar la estructura y alterar la función del pulmón o las vías respiratorias. Se describen cuatro entidades nosológicas:
- Proteinosis Alveolar.
- Microliatiasis Alveolar Pulmonar.
- Amiloidosis.
- EPID por alteración del surfactante pulmonar.
1. PROTEINOSIS ALVEOLAR (PAP)
Consiste en la acumulación de material lipoproteináceo del surfactante en los espacios alveolares y los bronquiolos terminales. Hay 3 formas clínicas principales, con diferentes mecanismos de disfunción de los macrófagos alveolares:
1. Primaria o autoinmune (aPAP) (90% de los casos): Interrupción de la señalización del factor estimulante de colonias de granulocitos y macrófagos (GM-CSF), habitualmente de origen autoinmune (causada por niveles elevados de autoanticuerpos frente a GM-CSF), que ocasiona que los macrófagos tengan un funcionamiento defectuoso: Catabolismo aumentado y déficit del aclaramiento del surfactante.
2. Secundaria: Se desencadena una reducción de número o de la función macrofágica a consecuencia de enfermedades hematológicas, infecciones, inhalación de tóxicos, inmunodeficiencias o inflamación crónica.
3. Congénita: Mutaciones en los genes CSF2RA o CSF2RB que codifican subunidades del receptor de GM-CSF. Afecta casi exclusivamente a niños con una frecuencia del 2%.
La PAP tiene una incidencia de 1 caso por cada 2 millones de habitantes. Suele debutar entre los 30 y 50 años, con predominio en sexo masculino, y se relaciona con el tabaquismo. Cursa con clínica inespecífica (disnea de esfuerzo, tos y febrícula prolongada). Hasta un tercio de los pacientes pueden estar asintomáticos. La analítica no muestra alteraciones reseñables. En radiografía es típico observar infiltrados alveolares perihiliares simétricos, similar a un edema agudo de pulmón. En la tomografía computerizada de alta resolución (TCAR) se suele observar patrón inespecífico en empedrado (crazy paving), que consiste en un patrón lineal-reticular con vidrio deslustrado sobrepuesto (imitando al pavimento de las calles europeas de la Edad Media).
Para el estudio diagnóstico de la PAP está indicado realizar broncoscopia con lavado broncoalveolar (LBA), esperando encontrar material lechoso PAS positivo (material proteico extracelular), fundamental para excluir infecciones (nocardia, micobacterias y hongos). En la actualidad, rara vez se precisa biopsia pulmonar quirúrgica para la confirmación diagnóstica.
En agosto de 2024 es publicado el task force report “European Respiratory Society guidelines for the Diagnosis and Management of Pulmonary Alveolar Proteinosis”¹, en donde se propone el siguiente algoritmo de diagnóstico:
1. En primer lugar, si se sospecha PAP ante hallazgos radiológicos típicos e historia compatible, con o sin LBA, se debe valorar la presencia de autoanticuerpos antiGM-CSF, con cuya confirmación se establecerá el diagnóstico de PAP autoinmune (aPAP).
2. Los individuos con títulos normales de autoanticuerpos que padezcan previamente una enfermedad de base que pudiera desencadenar PAP serán diagnosticados como PAP secundaria.
3. Si no se identifica ninguna causa subyacente, se estudiarán los niveles séricos de GM-CSF:
a. En caso cifras elevadas se deberán realizar pruebas genéticas para estudiar posibles mutaciones de los genes CSF2RA y/o CSF2RB (que codifican subunidades del receptor de GM-CSF), lo que permitirá establecer el diagnóstico de PAP hereditaria.
b. Ante niveles séricos normales de GM-CSF se habrá de continuar con un estudio orientado a determinar mutaciones genéticas que den lugar a alteraciones del surfactante.
4. Finalmente, ante ausencia de mutaciones genéticas relevantes, se valorará la necesidad de hacer biopsia pulmonar transbronquial o biopsia pulmonar quirúrgica para un examen histopatológico del parénquima, con lo cual podría situarse al paciente en alguno de los grupos previos, o bien diagnosticarlo de PAP no clasificable.
Una vez realizado el diagnóstico, en la misma guía de agosto de 2024 de la ERS¹ se definen 3 conceptos para el manejo terapéutico de la PAP:
- PAP activa: Tras excluir causas alternativas o complicaciones como infecciones respiratorias, embolia pulmonar, hipertensión pulmonar e insuficiencia cardíaca congestiva, presencia de: A) Síntomas continuos o progresivos como disnea, tos, producción de esputo, dolor en el pecho, pérdida de peso, y/o B) Disminución de la función pulmonar en la capacidad vital forzada (FVC) o capacidad de difusión de monóxido de carbono (DLco), y/o C) Hipoxemia medida mediante gases en sangre arterial (PaO2, SaO2, AaDO2), y/o D) infiltrados característicos de PAP nuevos o que empeoran en la TCAR, incluidos, entre otros, vidrio esmerilado y crazy paving.
- Gravedad de la PAP: Se propone continuar empleando la escala de puntuación Disease Severity Score (DSS) basada en síntomas y niveles de PaO2 que se viene usando como referencia desde 2008. Las categorías se establecen como:
1 = asintomático y PaO2 ≥ 70 mm Hg
2 = sintomático y PaO2 ≥ 70 mmHg
3 = 60 ≤ PaO2 < 70 mm Hg
4 = 50 ≤ PaO2 < 60 mm Hg
5 = PaO2 < 50 mm Hg.
Los pacientes pueden estratificarse en leves (DSS 1-2), moderados (DSS 3) y graves (DSS 4-5).
- Progresión de la enfermedad: No existe una definición estándar, sin embargo, se considera que se trata del empeoramiento de los síntomas respiratorios, la disminución de las pruebas de función pulmonar (FVC, DLco) y la aparición o empeo-
ramiento de insuficiencia respiratoria. No hay establecidos umbrales específicos para las alteraciones de los resultados de pruebas funcionales y gasométricas que definan progresión de la enfermedad. La reducción del intervalo de tiempo entre lavados broncoalveolares terapéuticos se ha utilizado como un indicador de progresión. Se debe confirmar esta situación mediante TACAR, que además aporta información acerca de otros posibles procesos concomitantes. La fibrosis pulmonar puede aparecer hasta en un 20% de los casos de PAP y se debe considerar como un signo de enfermedad progresiva.
Hasta un 25% de los casos llegan a remitir de forma espontánea. Se instaurará tratamiento con los objetivos de disminuir y mejorar los síntomas, favorecer la remisión, o conseguir estabilización a largo plazo.
No existe un protocolo estandarizado para el manejo terapéutico de la PAP, y la guía de agosto de 2024 de la ERS¹ recomienda que estos pacientes sean remitidos a centros con experiencia previa en esta patología.
1. Habitualmente se realiza LBA terapéutico bilateral, de 10-15 lavados con intervalos de 6-12 meses. Se aprecia buena respuesta a este tratamiento especialmente en aPAP con deterioro funcional y del intercambio gaseoso.
2. Otra línea terapéutica, menos invasiva y que puede lograr aumento del intervalo de tiempo entre lavados -concepto empleado frecuentemente como indicador de estabilidad-, es la administración inhalada de GM-CSF:
a. Sargramostim 125 μg /12 horas a semanas alternas durante 24 semanas2.
b. Molgramostim 300 μg /24 horas a semanas alternas durante 24 semanas.
La vía inhalatoria tiene mayor seguridad de uso, menores costes y menores efectos secundarios que el LBA terapéutico.
3. Actualmente, se sugiere el uso de rituximab para pacientes con aPAP confirmada sintomáticos a pesar de LBA terapéutico o tratamiento inhalado con GM-CSF y que requieren oxígeno suplementario. La seguridad de rituximab no se ha evaluado suficientemente en la aPAP, sin embargo hay datos extrapolables de enfermedades como la artritis reumatoide que abogarían por el beneficio de su empleo en dos dosis de 1000 mg distanciadas 6 meses.
4. La plasmaféresis se emplea cuando el requerimiento de oxígeno llega a ser FiO2 > 35% y persisten síntomas a pesar de tratamientos previos.
5. No existen criterios estándar que definan la respuesta al tratamiento, pero ante fracaso manifiesto se ha de valorar la opción del trasplante pulmonar.
6. Respecto al tratamiento de las complicaciones de la PAP, cabe destacar la importancia de la antibioterapia frente a las infecciones, tanto por patógenos ha-
bituales (Streptococcus, Haemophilus y Enterobacterias) como por oportunistas (Micobacterias, Nocardia, Actinomyces, Aspergillus o Cryptococcus). Además de su alta incidencia, las infecciones suponen un factor de recurrencia de episodios de empeoramiento sintomático y funcional en pacientes con PAP.
La PAP refractaria se puede definir como la persistencia o empeoramiento de los síntomas respiratorios e infiltrados radiológicos en la TCAR, además de deterioro de la función pulmonar y del intercambio gaseoso, a pesar de tratamiento adecuado, y haber excluido complicaciones postintervención como motivo de fracaso terapéutico. Se ha utilizado como indicador de falta de respuesta al tratamiento la necesidad de reducir el intervalo de tiempo entre dos LBA terapéuticos consecutivos.
En pacientes sintomáticos, la proteinosis alveolar pulmonar tiene una supervivencia global del 95% a los 5 años (Tabla I).
2. MICROLITIASIS ALVEOLAR PULMONAR (MAP)
Es un trastorno genético de transmisión autosómica recesiva debido a pérdida de función del gen SLC34A que codifica una proteína transportadora de fosfato dependiente de sodio que al ser defectuosa causa el depósito generalizado de cristales de fosfato de calcio en los espacios alveolares.
Tiene una alta relación con la consanguinidad. Un rasgo distintivo de la enfermedad es la discrepancia entre síntomas percibidos en el momento del diagnóstico en comparación con la apariencia extensa, similar a una tormenta de arena, con múltiples nódulos calcificados (microlitos) en la radiografía de tórax y en el TCAR. La sospecha inicial de MAP en las pruebas de imagen debe ir seguida de pruebas genéticas para verificar el diagnóstico e identificar la variante causante de la enfermedad. Cuando no es posible la realización de estudios genéticos, el diagnóstico se puede realizar mediante técnicas invasivas, mediante biopsia transbronquial, criobiopsia, o una biopsia pulmonar quirúrgica.
En familias con antecedentes de MAP, se debe ofrecer asesoramiento genético, así como pruebas preimplantacionales/prenatales si es necesario. No existe un tratamiento definitivo y, en algunos casos, la MAP puede progresar a una enfermedad pulmonar grave con insuficiencia respiratoria asociada y fallecimiento por este motivo. Se debe ofrecer siempre que se pueda tratamiento preventivo con vacunas, oxigenoterapia cuando sea necesario y, en casos avanzandos, en gente joven, se debe valorar la posibilidad del transplante pulmonar3 .
3. AMILOIDOSIS
Enfermedad sistémica que se origina por el depósito en tejido extracelular de material amiloide. Los depósitos de amiloide son fibrillas altamente ordenadas compuestas de subunidades de bajo peso molecular de una variedad de proteínas, describiéndose más de 30 implicadas en la aparición de distintas variantes de amiloidosis.
La clasificación de la amiloidosis sistémica se basa en los distintos tipos de proteínas, lo cual establecerá la afectación orgánica y la manifestación clínica (Tabla 2). Las variantes más frecuentes son la amiloidosis AL (cadenas ligeras de inmunoglobulinas), la ATTR (inestabilidad de la transtirretina) y la AA (trastornos inflamatorios crónicos4 . La afectación pulmonar es más frecuente en los tipos AL y AA, aunque raramente produce síntomas. La forma de presentación es variable, siendo las más típicas las formas nodular, traqueobronquial, alveolar, vascular y linfática5. Los pacientes con amiloidosis y afectación bronquial pueden presentar disnea, tos, hemoptisis, derrame pleural, neumonía e hipertensión pulmonar6 .
En la radiografía de tórax o tomografía computerizada (TC), los hallazgos pueden ser variables.
- En pacientes con amiloidosis localizada es frecuente observar grandes nódulos pulmonares cuyo tamaño oscila entre 8 mm y 3 cm, y pueden ser múltiples o solitarios (60% de los casos), y presentar bordes lisos o lobulados y de localización subpleural o periférica, pudiendo calcificar en el 20% de los casos.
- En pacientes con amiloidosis sistémica puede observarse un patrón reticular o reticulonodular debido a la afectación difusa – conocida como amiloidosis difusa parenquimatosa-. Es raro observar un patrón micronodular.
- Otros hallazgos pueden ser: engrosamiento de la pared bronquial, quistes, adenopatías, derrame pleural, atelectasia y opacidades de vidrio esmerilado (Imagen a/Figura 1).
Dada la inespecificidad de los hallazgos, se debe realizar un amplio diagnóstico diferencial que incluya neoplasias, infecciones, enfermedades intersticiales y enfermedades granulomatosas (Figura 1).
Dado que la imagen radiológica es inespecífica, con frecuencia, es preciso tomar muestras histológicas para realizar un diagnóstico definitivo, La tinción de estas con rojo congo muestra depósitos de color rojizo o naranja intenso; sin embargo, la verdadera positividad es la birrefringencia verde que presenta dicroísmo en el microscopio con luz polarizada5
El tratamiento, dada la baja incidencia y, por tanto, la dificultad de realizar ensayos clínicos se ha basado en tratamientos del mieloma múltiple y queda representado en la figura 2. Tiene mal pronóstico dada su elevada morbimortalidad, por lo que, es esencial diagnosticar y tratar adecuadamente la enfermedad7 .
4. EPID POR ALTERACIÓN DEL SURFACTANTE PULMONAR
Es conocido que existe una sustancia tensioactiva que tiene la capacidad de reducir la resistencia requerida para la aireación de los pulmones, conocida como surfactante pulmonar. Las proteínas asociadas al surfactante A y D tienen importancia en la defensa pulmonar, pero las proteínas hidrópicas B y C permiten la función del surfactante. Además, la ATP binding cassette, sub-family A, member 3 (ABCA3) es una proteína cuya función es el transporte de fosfolípidos y proteínas del surfactante transmembrana. Con ello, el surfactante permite reducir la tensión superficial en los alvéolos de los pulmones y en la interfaz aire/líquido, por lo tanto, es fundamental no sólo para reducir el trabajo respiratorio, sino que también previene el colapso alveolar al final de la espiración que, de otro modo, conduciría a atelectasia.
Esta sustancia es un producto de las células alveolares tipo 2 formada por una compleja mezcla de lípidos (90%) y proteínas (10%) que comienza a secretarse en los alvéolos a partir de las 24 semanas de edad gestacional.
Los trastornos de las proteínas surfactantes son un grupo raro de enfermedades causadas por mutaciones genéticas de los genes SFTPB, SFTPC, ABCA3 y TTF1.
La mutación del gen SFTPB en forma de herencia autosómica recesiva en el cromosoma 2 ocurre en el 70% de los pacientes con déficit de proteína surfactante B y conduce a una composición anormal del surfactante y a un deterioro de la función que resulta en un aumento de la tensión superficial alveolar.
- El gen SFTPC se localiza en el cromosoma 8 y sus mutaciones ocurren en el 25% de los pacientes con déficit de la proteína surfactante C.
- La mutación del gen ABCA3 se produce con herencia autosómica recesiva al producirse una alteración en el cromosoma 16, y es la causa más frecuente de déficit de proteína del surfactante.
La presentación clínica de las alteraciones de la proteína surfactante es variable, y según la mutación producida, el tiempo de aparición también variará. El déficit de la proteína surfactante B se ha identificado como causa de distrés respiratorio en neonatos, pero no se han identificado en pacientes con enfermedad intersticial difusa en el adulto. Esta afectación progresa rápidamente y produce insuficiencia respiratoria y muerte dentro de los primeros 3 a 6 meses de vida a pesar del tratamiento médico óp-
timo. Una supervivencia más larga podría atribuirse a un fenotipo leve de la enfermedad que permite la producción parcial de proteína B.
En cambio, el déficit de la proteína C puede presentarse en diferentes grupos de edad, con predominio en el final de la infancia, aunque también se ha podido observar en pacientes entre la quinta y sexta década de la vida. Por último, las mutaciones en el gen ABCA3 puede producir tos, disnea, tolerancia reducida al esfuerzo y deformidades de la pared torácica, sobre todo en la infancia tardía o en la edad adulta temprana.
La enfermedad de la membrana hialina es una de las enfermedades asociadas a alteraciones del surfactante. Suele producirse en recién nacidos prematuros y aparecer dentro de las primeras 48 a 72 horas en forma de distrés respiratorio. El diagnóstico se realiza con criterios clínicos, con la aparición de frecuencia respiratoria anormal, aleteo nasal, tiraje y asincronía torácico-abdominal con o sin cianosis. El tratamiento consiste en iniciar terapia de reemplazo de surfactante para recién nacidos prematuros y administración de corticosteroides oxigenoterapia8
BIBLIOGRAFÍA
1. McCarthy C, Bonella F, O'Callaghan M, Dupin C, Alfaro T, Fally M, Borie R, Campo I, Cottin V, Fabre A, Griese M, Hadchouel A, Jouneau S, Kokosi M, Manali E, Prosch H, Trapnell B, Veltkamp M, Wang T, Toews I, Mathioudakis A, Bendstrup E. European Respiratory Society guidelines for the Diagnosis and Management of Pulmonary Alveolar Proteinosis. Eur Respir J. 2024 Aug 15:2400725. doi: 10.1183/13993003.00725-2024. Epub ahead of print. PMID: 39147411.
2. Tazawa R, Ueda T, Abe M, Tatsumi K, Eda R, Kondoh S, Morimoto K, Tanaka T, Yamaguchi E, Takahashi A, Oda M, Ishii H, Izumi S, Sugiyama H, Nakagawa A, Tomii K, Suzuki M, Konno S, Ohkouchi S, Tode N, Handa T, Hirai T, Inoue Y, Arai T, Asakawa K, Sakagami T, Hashimoto A, Tanaka T, Takada T, Mikami A, Kitamura N, Nakata K. Inhaled GM-CSF for Pulmonary Alveolar Proteinosis. N Engl J Med. 2019 Sep 5;381(10):923-932. doi: 10.1056/NEJMoa1816216. PMID: 31483963.
3. Enemark A, Jönsson ÅLM, Kronborg-White S, Bendstrup E. Pulmonary Alveolar Microlithiasis - A Review. Yale J Biol Med. 2021 Dec 29;94(4):637-644.
4. Baqir M, Roden AC, Moua T. Amyloid in the Lung. Semin Respir Crit Care Med. 2020 Apr;41(2):299-310.
5. Howard M, Ireton J, Daniels F, et al. Pulmonary presentations of amyloidosis. Respirol 2001, 6:61-4.
6. Khoor A, Colby TV. Amyloidosis of the lung. Arch Pathol Lab Med 2017;141(02):247–254
7. Milani P, Basset M, Russo F, Foli A, Palladini G, Merlini G (2017). The lung in amyloidosis. Eur Respir Rev 26:170046
8. Singh J, Jaffe A, Schultz A, Selvadurai H. Surfactant protein disorders in childhood interstitial lung disease. Eur J Pediatr. 2021 Sep;180(9):2711-2721.
TABLAS Y FIGURAS
Tabla I. Resumen del manejo diagnóstico y terapéutico de la PAP. Elaboración propia

Tabla II. Clasificación de amiloidosis sistémica

Figura 1. Tomografía computarizada.

a y c, nódulo pulmonar solitario en lóbulo superior izquierdo. b y d, opacidades en vidrio deslustrado, áreas de consolidación y nódulos con distribución perilinfática.

Abreviaturas: ATCM: autotrasplante de células madre; dara-BCD: daratumumab, bortezomib, ciclofosfamida y dexametasona.
Figura 2. Algoritmo terapéutico amiloidosis pulmonar
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
19
EOSINOFILIAS PULMONARES
Autores
Elena Solana Martínez
Miguel Henrique Reyes Cotes
Coordinador
Belén Núñez Sánchez
INTRODUCCIÓN
Grupo de enfermedades que se caracterizan por infiltración de intersticio pulmonar y espacios alveolares por eosinófilos conservando la arquitectura pulmonar.¹
Podemos encontrar:
- Eosinofilia periférica: eosinófilos en sangre > 0,5 x 109/L* (500/mcL).
- Hipereosinofilia periférica: eosinófilos en sangre > 1,5 x 109/L* (1500/mcL).
- Eosinofilia alveolar: ≥ 25% eosinófilos en el lavado broncoalveolar (LBA), en general, mayor que 40%.
*En 2 determinaciones analíticas separadas en intervalo de 1 mes o más.
El diagnóstico de estas enfermedades se basa principalmente en la presencia de unas características clínico-radiológicas determinadas y en la demostración de eosinofilia alveolar. El LBA es fundamental para confirmar el diagnóstico, mientras que la biopsia pulmonar generalmente no es necesaria. Se pueden clasificar de diferentes formas según: a) la forma de inicio (aguda/crónica), b) si se identifica agente causal o no y c) si presenta manifestaciones sistémicas o está limitada al pulmón (Tabla I).
Estas enfermedades tienen en común una adecuada respuesta a corticoides sistémicos y curación sin secuelas en la mayoría de los casos, aunque las recaídas pueden ser frecuentes. Las terapias dirigidas a la IL-5 cobran cada vez mayor importancia.
1. ENFERMEDADES PULMONARES
EOSINOFÍLICAS DE CAU-
SA DESCONOCIDA
1.1 Neumonías eosinofílicas idiopáticas solitarias
1.1.1 Neumonía eosinofílica idiopática crónica (NEC)
Epidemiología
- Rara (<3% de todas las EPID).
- Eosinofilia pulmonar más frecuente en áreas no tropicales.
- Predominio mujeres (2:1), edad media 45 años.
- Relación con asma (hasta en 2/3 casos) o atopia (en la mitad de los casos, en forma de alergia medicamentosa, poliposis nasal, urticaria y/o eczema).
- No relación con tabaquismo, al contrario que en la neumonía eosinofílica idiopática aguda (NEA).
Clínica
- Cuadro progresivo o subagudo (semanas-meses) de disnea y tos (más frecuentes), en asociación con síntomas generales como fatiga, malestar, fiebre, anorexia, sudores nocturnos y pérdida de peso.
- El asma suele preceder a la enfermedad, y ocasionalmente puede coincidir al diagnóstico (15%).
- Pueden existir manifestaciones extratorácicas (derrame pericárdico, artralgias o elevación de transaminasas) aunque en este caso se debe considerar granulomatosis eosinofílica con poliangeítis (GEPA).
Laboratorio
- Se caracteriza por eosinofilia periférica (generalmente >5000-6000/mcL).
- BAL con eosinófilos >25% (generalmente >40%).
- Estos valores pueden ser normales si el paciente está recibiendo tratamiento con corticoides sistémicos.
- Aumento de PCR e IgE.
Radiología
- Infiltrados alveolares bilaterales con márgenes mal definidos. Predominio periférico y en lóbulos superiores. Imagen típica en “negativo fotográfico” de edema agudo de pulmón (25% de los casos).
- Migración espontánea de infiltrados en el 25% de los casos.
- En TCAR de tórax: consolidaciones u opacidades en vidrio deslustrado confluentes, bilaterales, en lóbulos superiores y subpleurales.
- Menos frecuente: engrosamiento septal, bandas subpleurales, nódulos centrolobulillares, adenopatías mediastínicas o derrame pleural leve.
Diagnóstico
- Por regla general, no requiere biopsia para su diagnóstico.
- Exclusión de otras: neumonía eosinofílica aguda, aspergilosis broncopulmonar alérgica, neumonía eosinofílica secundaria a tóxicos, GEPA y neumonía organizada criptogenética.
- Pruebas función respiratoria: 50% patrón restrictivo, 50% patrón obstructivo. Descenso DLco.
- Los criterios diagnósticos aparecen en la tabla II.
Tratamiento
- Resolución espontánea en 10%.
- Corticoides orales: no hay dosis ni duración establecida.
- Respuesta rápida en 48 h y resolución en 1 semana.
- Prednisona 0,5 mg/kg/día 2 semanas, seguido de 0,25 mg/kg/día 2 semanas. Reducción progresiva hasta completar aproximadamente 6 meses.
- Podría plantearse el uso de fármacos anti IL-5 (mepolizumab y benralizumab) en caso de recaídas frecuentes o asma grave, discutiendo cada caso de forma individualizada.
Pronóstico
- Recaídas en una proporción >50% en la disminución o suspensión de corticoides. Buena respuesta a la reanudación con 20 mg de prednisona.
- No suele haber secuelas, pero se recomienda seguimiento a largo plazo (aproximadamente el 10% puede desarrollar obstrucción crónica al flujo aéreo a pesar del tratamiento correcto).
1.1.2 Neumonía eosinofílica idiopática aguda (NEA)²
Epidemiología
- Varones, alrededor de 30 años.
- Se asocia al tabaco (2/3 de los casos), también a otros contaminantes inhalados.
- No relación con asma.
Clínica
- Inicio agudo (días) de disnea, fiebre, tos, dolor torácico pleurítico y mialgias.
- Suele existir un retraso al diagnóstico entre 7 días y 1 mes desde el inicio de síntomas.
- Suele evolucionar a insuficiencia respiratoria aguda (cumple criterios de distrés) con necesidad de ventilación mecánica.
Laboratorio
- Los eosinófilos en sangre suelen ser normales al inicio (diagnóstico erróneo como neumonía infecciosa) y posteriormente se elevan.
- Es clave la eosinofilia en LBA >40%.
- Puede presentar eosinofilia en líquido pleural.
- Cultivos del LBA estériles.
Radiología
- Rx de tórax: infiltrados bilaterales con opacidades alveolo-intersticiales.
- TCAR de tórax: nódulos en vidrio deslustrado mal definidos (100%), engrosamiento septal interlobular (90%), derrame pleural bilateral (76%) y consolidación (55%).
Diagnóstico
- El LBA es obligatorio para confirmar la presencia de eosinofilia alveolar y excluir infección.
- Diagnóstico de exclusión: neumonía bacteriana o viral, menos frecuente hongos (Aspergillus fumigatus o coccidioidomicosis), exposición a fármacos y SDRA.
- Puede producirse tras un trasplante alogénico de células madre.
- Pruebas de función respiratoria: restricción leve, descenso DLco e hipoxemia significativa.
- Los criterios diagnósticos aparecen en la tabla II
Tratamiento
- Corticoides sistémicos, generalmente durante 2 semanas.
- Prednisona oral 30 mg/día.
- En insuficiencia respiratoria aguda: metilprednisolona iv 1-2 mg/kg al día.
Pronóstico
- Recuperación clínica a las 72 h y respuesta radiológica/funcional en un mes.
- No suelen producirse recaídas.
- Deshabituación tabáquica fundamental por su papel etiológico.
1.2 Neumonía eosinofílica en síndromes sistémicos
1.2.1 Granulomatosis eosinofílica con poliangeítis (GEPA)³
Esta enfermedad se desarrolla en profundidad en el tema 12.
- Enfermedad sistémica.
- Vasculitis necrotizante asociada a ANCA (más frecuente p-ANCA).
- Afecta a vasos de pequeño y mediano calibre.
- Se caracteriza por presentar asma, neumonía eosinofílica y rinosinusitis.
- Cualquier órgano puede verse afectado; los más comunes: pulmón y piel.
- En el diagnóstico diferencial de eosinofilias pulmonares, las manifestaciones extratorácicas deben hacernos sospechar esta enfermedad.
1.2.2 Síndromes hipereosinofílicos idiopáticos¹-²
Epidemiología
- Grupo heterogéneo de enfermedades de origen hematológico.
- Varones (9:1) entre 20-50 años.
Clínica
- Manifestaciones respiratorias (49%), en general leves, rara vez neumonía eosinofílica, tos (24%), disnea (16%) y síntomas asmáticos (25%).
- Afección hematológica (100%), afección cardiovascular (58%), cutáneas (56%), neurológicas (54%), esplénicas (43%), hepáticas (30%), oculares (23%), y gastrointestinales (23%).
Laboratorio
- Eosinofilia ≥1500/mcL en dos determinaciones analíticas (separadas por un intervalo de 1 mes o más).
- No tiene relación con ANCA.
Radiología
- TC tórax: infiltrados intersticiales, opacidades en vidrio deslustrado, pequeños nódulos, derrame pleural.
- Debe hacerse diagnóstico diferencial con edema agudo de pulmón por afectación cardiaca de la enfermedad.
Diagnóstico
- Definido por tres criterios:
1. Eosinofilia persistente ≥1500/mcL y/o hipereosinofilia tisular definida por: 1) porcentaje de eosinófilos >20% en médula ósea y/o, 2) extensa infiltración tisular por eosinófilos, y/o, 3) marcado depósito de gránulos de proteínas de eosinófilos.
2. Daño/disfunción órgano diana.
3. Se deben descartar otras causas de eosinofilia (como medicamentos).
Tratamiento
- Prednisona 0,5 mg/kg/día (<50% de respuesta).
- Alternativas de tratamiento: hidroxiurea, ciclosporina e interferón.
- Imatinib: nueva opción terapéutica para pacientes con la variante mieloproliferativa.
- En casos de síndrome hipereosinofílico no controlado sin una causa secundaria no hematológica identificable: mepolizumab 300 mg cada 4 semanas.
2. ENFERMEDADES PULMONARES EOSINOFÍLICAS DE CAUSA CONOCIDA
2.1 Aspergilosis broncopulmonar alérgica (ABPA)
La ABPA es una reacción de hipersensibilidad compleja en respuesta a la colonización de las vías aéreas por el hongo Aspergillus fumigatus.
Epidemiología
- Ocurre casi exclusivamente en sujetos con enfermedad bronquial crónica. En asmáticos (1-2%), con fibrosis quística (7-10%)1 y raramente en EPOC, inmunocomprometidos.2,4
Clínica
- Tos crónica, disnea, expectoración con tapones mucosos marronáceos, fiebre y rinitis crónica.
- Curso crónico con exacerbaciones. No necesariamente sigue las 5 etapas clásicas (Tabla III).
Laboratorio
- Eosinofilia periférica >1000/mcL (durante las exacerbaciones) y/o elevación de la IgE total.
- IgE específica o IgG (precipitina) o prueba cutánea positiva a Aspergillus fumigatus.
- Cultivos de esputo + Pseudomonas aeruginosa, Staphylococcus aureus, Aspergillus fumigatus y/o micobacterias no tuberculosas.
Radiología
- Según la etapa de la enfermedad, se pueden observar daño de las vías aéreas, atelectasias por tapones mucosos, bronquiectasias, fibrosis y signos de cor pulmonale (Tabla III).
- Signo del “dedo del guante” (25%): impactación mucosa bronquial que irradia desde el hilio hacia la periferia.
Diagnóstico6
- Presencia de todos los siguientes:
1. Asma.
2. IgE específica Aspergillus > 0,35 KUA/L.
3. IgE total en suero > 500 UI/ml.
- Al menos 2 de los siguientes:
1. IgG específica A. fumigatus > 27 mgA/L.
2. Presencia de bronquiectasias en TC tórax.
3. Eosinofilia periférica > 500/mcL.
Tratamiento
- Convencional para la enfermedad de base, por ejemplo, en el asma con corticoides, broncodilatadores inhalados y otros en función de la gravedad.
- En agudizaciones, prednisona oral a 0,5 mg/kg/día 2 semanas e ir reduciendo (5 mg cada 2 semanas) hasta completar 3 meses (pauta corta) o hasta 12 meses (pauta larga) en función de gravedad.1,2,7 En pacientes con síntomas frecuentes y evidencia de daño pulmonar progresivo, se puede mantener la dosis mínima posible de forma crónica.
- En exacerbaciones ABPA refractarias, valorar altas dosis de bolos de metilprednisolona ev.2,7,8
- Se recomienda emplear antifúngico –el más usado es el itraconazol– durante 1632 semanas. Alternativa: voriconazol, posaconazol y anfotericina nebulizada en caso de intolerancia o de fracaso con itraconazol.
- En ocasiones, se han usado macrólidos como coadyuvantes antiinflamatorios.
- Se ha descrito el uso de anticuerpos monoclonales para el tratamiento de ABPA grave, tanto anti-IgE (omalizumab) como anti-IL5 (mepolizumab) y anti-IL4 y 13 (dupilumab).7,8
Pronóstico
- Dependerá de la etapa clínica, pero por lo general es de buen pronóstico.
- Tiene riesgo de agudizaciones, sobreinfecciones de bronquiectasias y atelectasias por tapones mucosos. En etapa avanzada, fibrosis pulmonar, riesgo de cor pulmonale e insuficiencia respiratoria.
2.2 Neumonías eosinofílicas de origen parasitario
Epidemiología
- Principal causa de neumonía eosinofílica en el mundo, menos común en Europa y Norteamérica.2,8,9
- La afectación pulmonar aguda se llama eosinofilia pulmonar simple o síndrome de Löffler (1932). Se diferencia en “primario”, que no tiene claro agente desencadenante, y “secundario”, que se relaciona con parásitos o fármacos.
- La mayoría de los casos de Sd. de Löffler secundario a parásitos se relacionan con helmintos, siendo el más frecuente a nivel mundial el Ascaris lumbricoides.² En España, tenemos el Ascaris vermicularis y, ocasionalmente, el Trichinella spiralis.9
Clínica
- Cuadro usualmente leve. Pueden presentar tos, sibilancias, en ocasiones disnea, fiebre y dolor retroesternal. En caso de hidatidosis: hemoptisis y vómica.
- En inmunodeprimidos, un síndrome de hiperinfección parasitaria grave puede ser mortal (Strongyloides).
Laboratorio
- Eosinofilia periférica.
- IgE sérica elevada e IgG específica frente al parásito.
- Parásitos en heces.
- En la eosinofilia tropical, hipereosinofilia >3300/mcL e hiperIgE >5000 UI/ml.9
Radiología
- En el síndrome de Löffler: Infiltrados pulmonares uni o bilaterales migratorios, alveolointersticiales, periféricos con base pleural, que suelen resolverse en <1 mes.
- En la eosinofilia tropical: infiltrados migratorios pulmonares, segmentario o miliar grueso, de predominio basal y medio, con resolución con tratamiento específico.
- En hidatidosis pulmonar: imagen quística pulmonar predomina basal derecha.1,8
Diagnóstico
- Según la correlación clínica y en función de la sospecha epidemiológica.
- Se debe realizar estudio de parásitos en heces de forma seriada (recomendable tres muestras separadas, una cada 15 días), serología y valorar respuesta al tratamiento.
Tratamiento
- Rara vez requieren corticoides, resolviéndose de forma espontánea en el caso del Löffler parasitario. El antiparasitario debe ser el específico y realizar control erradicador.
Pronóstico
- Suele ser bueno, autolimitado en el síndrome de Löffler y reversible tras el tratamiento específico en gran parte del resto de parasitosis.
- En el caso de la hidatidosis pulmonar, el quiste pulmonar podría precisar exéresis quirúrgica.
2.3 Neumonías eosinofílicas de otras causas infecciosas
- Relacionada con infecciones fúngicas (coccidiodomicosis, basidiobolomicosis, paracoccidiodomicosis) y con tuberculosis.
- El tratamiento es el específico: antibioterapia o antifúngico. En ocasiones, corticoides a dosis bajas.
2.4 Neumonías eosinofílicas inducidas por fármacos
Epidemiología
- Se debe realizar una investigación de los medicamentos que ha tomado el paciente en las semanas o días previos al inicio del cuadro clínico.
- Existe un amplio número de drogas relacionadas (www.pneumotox.com). La causalidad se ha establecido para al menos 20 fármacos, así como para radioterapia, drogas y venenos (Tabla V).
Clínica
- Variada, desde cuadros clínicos leves hasta cuadros graves en forma de neumonía eosinofílica aguda.
- Presencia de rash cutáneo y derrame pleural incrementan la probabilidad diagnóstica.
Tratamiento
- El manejo consiste en suspender el agente causal y, en ocasiones es necesario el uso de corticoterapia.
Pronóstico
- Suele ser favorable tras suspender el tratamiento.
3. ENFERMEDADES
EOSINOFÍLICAS DE LA VÍA AÉREA
3.1 Asma eosinofílica
Epidemiología
- Los pacientes suelen tener >20 años; algunos presentan rinosinusitis crónica y/o poliposis nasal. Un subgrupo (7%) intolerancia a AINES, enfermedad respiratoria exacerbada por aspirina (EREA).
- Los pacientes con neumonía eosinofílica crónica y GEPA suelen tener historia de asma eosinofílica.
Clínica
- Suelen tener peores síntomas y mayor riesgo de exacerbaciones.
Laboratorio
- Eosinofilia en esputo >3%, en sangre periférica >300 /mcL (si >1500/mcL, se denomina asma hipereosinofílica) y eosinofilia en biopsias bronquiales. Elevación de IL-5, IgE total y del óxido nítrico exhalado (FENO).8
Radiología
- No suelen observarse infiltrados. En algunos casos, atelectasias por tapón mucoso, y en casos graves, atrapamiento aéreo, con TC patrón en mosaico (destacable en espiración) con disminución de la vascularización en zonas de atrapamiento aéreo, lo que la diferencia del vidrio deslustrado de las EPID.
Diagnóstico
- Según las guías GINA y GEMA 5.3, precisa una correlación clínica con pruebas funcionales. Entre ellas, test broncodilatador positivo, FENO elevado, test de broncoprovocación.2
Tratamiento
- El tratamiento es el habitual para el asma. En casos graves, se puede emplear anti-IL-5 (mepolizumab, reslizumab, benralizumab) anti IL4-13 (dupilumab), anti-TSLP (tezepelumab).2
Pronóstico
- Por lo general, es de buen pronóstico, aunque variable, de intermitente a persistente grave.
3.2
Bronquitis eosinofílica
La bronquitis eosinofílica es una enfermedad caracterizada por tos crónica e inflamación bronquial eosinofílica, muy similar a la que se observa en el asma. No obstante, a diferencia del asma, no cursa con obstrucción variable al flujo aéreo ni hiperreactividad bronquial. Es una causa relativamente frecuente de tos crónica aislada, por lo que es muy importante tenerla en consideración en el diagnóstico diferencial de la tos crónica. Se ha relacionado con la exposición a fármacos, acrilatos, humos de soldadura, formaldehído, isocianato o harina. La radiología de tórax es normal. El tratamiento son los corticoides inhalados. En algunas ocasiones, puede causar obstrucción irreversible al flujo aéreo con o sin asma.10
3.3 Hipereosinofilia idiopática con bronquiolitis constrictiva (u obliterante)
Es una enfermedad que se caracteriza por tos, disnea y sibilantes. Acompañada de aumento de eosinofilia periférica (>1000/mcL) y/o >25% eosinófilos en BAL. Presenta obstrucción crónica al flujo aéreo. En la TC, presenta signos de bronquiolitis (nódulos centrolobulillares y opacidades ramificadas). Puede ser idiopática o relacionada con asma, ABPA, GEPA o inducida por fármacos (minociclina, sulfasalacina). El tratamiento son los corticoides orales; en algunos casos, se ha utilizado mepolizumab.
4. SÍNDROMES PULMONARES CON POSIBLE EOSINOFILIA
La eosinofilia puede manifestarse en otras enfermedades en las que la neumonía eosinofílica no es predominante: granulomatosis broncocéntrica, vasculitis sistémica eosinofílica idiopática, determinados casos de neumonías intersticiales idiopáticas, especialmente neumonía intersticial descamativa, fibrosis pulmonar idiopática, histiocitosis de células de Langerhans y sarcoidosis, entre otros.²
BIBLIOGRAFÍA
1. García Río F, Hidalgo Molina A, Arnedillo Muñoz A. Manual de Neumología y Cirugía Torácica. Sección VI. Enfermedades Pulmonares Intersticiales e Inflamatorias Difusas. [Internet]. 2023. Disponible en: https://separ.wademi.com/neumo/contenido.php?id_se=20.
2. Cottin V. Eosinophilic Lung Diseases. Immunol Allergy Clin N Am. 2023:289-322.
3. Rosenberg C, Khoury P. Approach to Eosinophilia Presenting With Pulmonary Symptoms. Chest. 2021;159(2):507-516.
4. Sisodia J, Bajaj T. Allergic Bronchopulmonary Aspergillosis. StatPearls (Internet) Treasure Island (FL) [Internet]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK542329/.
5. Martínez Caballero A, Córdoba Guzmán A, Robin Rada Escobar, Araújo Navarro M. Síndrome de pulmón hipereosinofílico: Revisión a un viejo problema. Revista Colombiana de Neumología. 2010;22(3):74-88.
6. Saxena P, Hansraj C, Valliappan M, Singh Sehgal I, Dhooria S, Thurai Prasad K, et al. Which Are the Optimal Criteria for the Diagnosis of Allergic Bronchopulmonary Aspergillosis? A Latent Class Analysis. Elsevier. 2021;9(1):328-335.
7. Ferran Morell (dir.). Pneumologica. Pautas, exploraciones complementarias y datos en medicina respiratoria. 12.a ed. Pulso ediciones, 2022, p. 45-47.
8. Jameson JL, Fauci AS, Kasper DL, et al. Principios de medicina interna. "Aparato Respiratorio". 20a ed. Mc Graw Hill, 2020, p. 98-101.
9. Álvarez-Sala Walter JL, Casan Clará P, Rodríguez de Castro F, Rodríguez Hermosa JL, Villena Garrido V. Neumología Clínica. Elsevier. 2010, p. 254-260 y 358-366.
10. Cottin V. Neumopatías eosinofílicas. En: Murray JF, Nadel JA. Tratado de Medicina Respiratoria. 7 ed. 2023. p. 1322-1342.
TABLAS Y FIGURAS
Tabla I. Clasificación de los síndromes eosinofílicos pulmonares

Fuente: García Río F, Hidalgo Molina A, Arnedillo Muñoz A. Manual de Neumología y Cirugía Torácica. Sección VI. Enfermedades Pulmonares Intersticiales e Inflamatorias Difusas, 2023. Disponible en: https://separ.wademi.com/neumo/contenido.php?id_se=2
Tabla II. Criterios diagnósticos y diferencias entre la neumonía eosinofílica crónica y aguda

Fuente: Cottin V. Eosinophilic Lung Diseases. Immunol Allergy Clin N Am. 2023;289-322.
Tabla III. Etapas clínicas de la ABPA

Martínez Caballero A, Córdoba Guzmán A, Robin Rada Escobar, Araújo Navarro M. Síndrome de pulmón hipereosinofílico: Revisión a un viejo problema. Revista Colombiana de Neumología. 2010;22(3):74-88.
Tabla IV. Fármacos y terapias relacionados con neumonías eosinofílicas

Fuente: Jameson JL, Fauci AS, Kasper DL, et al. Principios de medicina interna. Aparato Respiratorio. 20a ed. Mc Graw Hill; 2020. p. 98-101. Álvarez-Sala Walter JL, Casan Clará P, Rodríguez de Castro F, Rodríguez Hermosa JL, Villena Garrido V. Neumología Clínica. ELSEVIER; 2010. p. 254-260, 358-366
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
FIBROSIS PULMONAR
PROGRESIVA
Autores
David Espejo Castellanos
Candela Serra
Coordinadora
María Molina Molina
1. INTRODUCCIÓN
Las enfermedades pulmonares intersticiales difusas (EPID) son un grupo diverso de enfermedades definidas por la inflamación y la fibrosis del parénquima pulmonar. En el caso de las EPID fibrosantes distintas a la fibrosis pulmonar idiopática (FPI), es imprescindible identificar cuáles de ellas presentarán un comportamiento progresivo con peor pronóstico o fibrosis pulmonar progresiva (FPP).¹
Actualmente, la FPP se establece en base a 3 factores; empeoramiento de los síntomas respiratorios, evidencia de progresión fisiológica y/o progresión radiológica de la enfermedad. En base a la evidencia científica reciente, los pacientes con fibrosis pulmonar no FPI que presenten FPP a pesar de la primera línea de tratamiento, generalmente corticosteroides e inmunosupresores, se beneficiarían de la introducción de tratamiento antifibrótico.²
2. EPIDEMIOLOGÍA
En los últimos años, se han publicado varios estudios de cohortes que investigan la prevalencia de la progresión de las distintas EPID fibróticas no FPI. Los estudios muestran heterogeneidad en los resultados, probablemente debido a distintos aspectos, como las diferentes metodologías para establecer el diagnóstico de progresión, la clasificación de la enfermedad y el reclutamiento de pacientes. En 2022, Valenzuela y Cottin² publicaron
una revisión de los diferentes estudios de prevalencia de EPID fibrótica no FPI, tanto progresiva como no progresiva. La prevalencia media en base a los diferentes estudios se resume en la figura 1.
3. CRITERIOS DIAGNÓSTICOS
En el año 2019, el estudio INBUILD tuvo como objetivo determinar la eficacia y la seguridad del nintedanib en pacientes con EPID fibrótica no FPI que presentaban progresión en función de unos criterios de inclusión clínicos, funcionales y radiológicos.3 Paralelamente, en el estudio RELIEF, cuyo objetivo fue determinar la eficacia y la seguridad de la pirfenidona en pacientes con un perfil similar, se emplearon únicamente criterios de función pulmonar.4 Teniendo en cuenta los datos en la práctica clínica con este tipo de pacientes, en 2020 se desarrollaron unas consideraciones evolutivas a valorar durante el seguimiento para determinar qué pacientes presentaban EPID fibrótica progresiva (Tabla I).5
En 2022, se desarrolló la primera guía de práctica clínica para FPP a través del consenso entre las principales sociedades científicas en el ámbito de las enfermedades respiratorias (ATS/ERS/JRS/ALAT). En dicha guía, se consensuó la terminología y la definición para identificar a estos pacientes, así como los criterios clínicos, funcionales y radiológicos a través de un comité formado por expertos en EPID, metodólogos y representantes de pacientes, siguiendo una metodología GRADE (Tabla II).¹
Para decidir los valores funcionales que determinaran progresión fibrótica, se utilizaron los datos publicados de pacientes con FPI y EPID fibrótica no FPI progresiva, teniendo en cuenta que el comportamiento evolutivo en pacientes con FPP es similar a la FPI. Una disminución absoluta de la FVC >5% se considera clínicamente relevante, dado que asocia peor pronóstico y mayor mortalidad en FPI. En caso de la DLco corregida por Hb, el deterioro igual o mayor del 10% es un predictor de mortalidad en pacientes con EPID. Siempre hay que excluir otras causas que puedan justificar el empeoramiento de la función pulmonar. Especialmente en caso de empeoramiento clínico con disminución aislada de la DLco, se sugiere comprobar mediante TCAR de tórax el incremento fibrótico.
Con respecto a la determinación de la progresión de la fibrosis pulmonar en la TCAR, normalmente se realiza mediante la evaluación visual con la TCAR inicial y las TCAR de seguimiento por parte de un radiólogo experto en patología torácica. Esta revisión se basa en el porcentaje del volumen pulmonar que contiene características fibróticas en las zonas pulmonares superiores, medias e inferiores entre las diferentes exploraciones. De cara al futuro, se han introducido nuevas técnicas de evaluación radiológica cuantitativa de la progresión, incluyendo los programas de cuantificación automática, que permiten una medida más objetiva y reproducible, aunque solo se podrán recomendar en caso de existir mayor evidencia científica en la práctica clínica real.¹
4. TRATAMIENTO
El tratamiento antifibrótico en EPID fibrótica no FPI debe considerarse cuando hay progresión con el tratamiento adecuado acorde a la EPID de base, que en algunos casos puede ser la inmunosupresión, es decir, cuando se cumplen criterios de FPP.6
No hay tratamientos estandarizados para las distintas EPID, por lo que el tratamiento de base debe valorarse en cada caso en particular. Como ejemplo, en la neumonitis por hipersensibilidad fibrótica (NHF) el tratamiento dependerá de la posibilidad de evitar la exposición al antígeno, de la gravedad en el momento del diagnóstico, de la presencia de inflamación concurrente en la TCAR, de la presencia de patrón NIU, de la linfocitosis en el BAL, de las comorbilidades y de las preferencias del paciente.6
Se debe insistir en la importancia del diagnóstico de la EPID de base, con interrogatorio dirigido que valore exposiciones de riesgo, TCAR sin contraste con cortes finos que pueda contar con cortes espiratorios si se creen necesarios, analítica con autoanticuerpos e IgG específicas, espirometría, DLco, LBA y muestra histológica si procede. Estos resultados se deben valorar en un comité multidisciplinar para un diagnóstico lo más próximo posible al de certeza y un tratamiento óptimo inicial.7 También se deberá valorar el riesgo de FPP para realizar un seguimiento más estrecho y así detectar la necesidad de tratamiento antifibrótico sin demoras. En este sentido, se han descrito algunos factores de riesgo de progresión, que se muestran en la tabla III 6
En el ensayo clínico fase III INBUILD, randomizado, doble ciego, que incluyó a 663 pacientes con EPID fibrótica en progresión en los últimos 24 meses, se demostró una reducción significativa de la caída de la FVC en pacientes tratados con nintedanib a dosis de 150 mg cada 12 horas.3 La caída de la FVC es un predictor de mortalidad validado en EPID fibróticas como NHF, EPID asociada a artritis reumatoide (AR) o a esclerosis sistémica (ES).7
Se debe tener en cuenta que en el INBUILD no se incluyó a pacientes con tratamiento biológico o inmunosupresor, excepto pacientes con AR, y el uso concomitante de corticoides orales permitido fue a una dosis máxima de 20 mg/día. Las EPID más frecuentemente incluidas fueros NHF (26,1% de los pacientes) y asociada a enfermedad autoinmune sistémica (EAS) (en el 25,6%). En base a los resultados del ensayo clínico, análisis post-hoc y metanálisis, la guía internacional de FPI/FPP más reciente recomienda el uso de nintedanib en pacientes que cumplen criterios de FPP.1,8 En un estudio posterior del INBUILD, se describió que el nintedanib se asociaba a una menor progresión de la EPID o muerte, que era aún más evidente en patrón NIU (HR 0,69, IC 95% 0,53-0,91; nominal p = 0,009). También se vio una diferencia significativa en la presencia de agudizaciones o muerte en el grupo tratado con nintedanib vs placebo, que también era más evidente en el grupo con patrón NIU (HR 0,62, IC 95% 0,39-0,97;
p=0,03).9 Desde julio de 2022, este fármaco se encuentra financiado para FPP en el territorio español.
En el estudio fase IIb RELIEF, se valoró el uso de pirfenidona en pacientes de diferentes centros de Alemania que presentaban EPID fibrótica progresiva, pero el ensayo finalizó anticipadamente por el lento reclutamiento. Sin embargo, se realizaron imputaciones por la pérdida de datos y el análisis primario favoreció la rama con pirfenidona a 24 semanas.4 En el estudio fase II de pirfenidona en EPID fibrótica no clasificable, el objetivo primario basado en el cambio de FVC medido por espirómetro domiciliario a las 24 semanas no fue significativo, pero sí que lo fue al analizar la caída media de la FVC mediante la espirometría hospitalaria.10,11 En la última guía internacional de FPI/FPP, se reconoció que hacían falta más ensayos clínicos con pirfenidona para recomendar su uso en FPP.¹ El uso de pirfenidona ha sido también evaluado asociado a micofenolato mofetilo en EPID no clasificable o asociada a esclerosis sistémica. En EPID fibrótica asociada a esclerosis sistémica, podría haber beneficio en pacientes seleccionados.8
Actualmente, no existe evidencia sobre el efecto del tratamiento antifibrótico aislado en pacientes con FPP con patrón NIU, como podrían ser pacientes con artritis reumatoide o con neumonitis por hipersensibilidad fibrótica.6 Un grupo internacional de expertos sugiere el tratamiento inicial con un inmunosupresor si requiere y en un segundo tiempo, a los 3-6 meses, valorar el inicio de antifibrótico, para valorar si persiste la progresión a pesar del tratamiento instaurado. También se debe valorar si se requiere aumento del tratamiento inmunosupresor previo al antifibrótico o si, por el contrario, se debe discontinuar el tratamiento inmunosupresor ante falta de beneficio e intolerancia, al iniciar el antifibrótico.6 No hay evidencia científica que permita especificar mejor el tratamiento inmunosupresor, la dosis o el tiempo necesario para cada EPID fibrótica no FPI, pero una revisión de expertos sugiere valorar su retirada en pacientes con patrón NIU, edad avanzada, infecciones recurrentes y falta de evidencia de mejoría con efectos adversos asociados.6
A pesar de que se considera seguro el uso de antifibróticos con inmunosupresión, teniendo mecanismos de hepatotoxicidad diferentes, se recomienda el control de la función hepática y el hemograma de forma más frecuente en este grupo de pacientes, y se sugiere un control mensual los primeros 3 meses y luego cada 3 meses o basado en cada caso en particular.6
En cuanto al tratamiento integral, además del tratamiento antifibrótico se debe valorar la rehabilitación pulmonar en el curso de la FPP, con el objetivo de mejorar la calidad de vida de estos pacientes. El tratamiento sintomático o la paliación de síntomas deben realizarse teniendo en cuenta las prioridades del paciente, valorando el uso de opioides y otros fármacos que permiten reducir la disnea y la tos.6 A medida que avanza la enfermedad, el uso combinado de fármacos y el trabajo conjunto con los equipos
de paliativos pueden optimizar el manejo sintomático del paciente. La oxigenoterapia suplementaria se debe valorar en pacientes con desaturación al esfuerzo con el objetivo de mejorar la calidad de vida de estos pacientes. No se debe olvidar la vacunación como una opción para prevenir infecciones graves y posibles agudizaciones. Se recomienda realizar la detección de comorbilidades, como pueden ser las apneas del sueño, para realizar tratamiento si corresponde. Se debe recordar la referencia a programa de trasplante pulmonar a los pacientes que no tengan contraindicaciones, antes de estadios finales.
BIBLIOGRAFÍA
1. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Antoniou KM, Bissell BD, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):1847.
2. Valenzuela C, Cottin V. Epidemiology and real-life experience in progressive pulmonary fibrosis. Current Opinion in Pulmonary Medicine. 2022;28(5):407-413.
3. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. New England Journal of Medicine. 2019;381(18):17181727.
4. Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. 2021;9(5):476-486.
5. George PM, Spagnolo P, Kreuter M, Altinisik G, Bonifazi M, Martinez FJ, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities [Internet]. 2020. Disponible en: www.thelancet.com/respiratory.
6. Rajan SK, Cottin V, Dhar R, Danoff S, Flaherty KR, Brown KK, et al. Progressive pulmonary fibrosis: an expert group consensus statement. European Respiratory Journal. 2023;61(3).
7. Molina-Molina M, Buendia-Roldan I, Castillo D, Caro F, Valenzuela C, Selman M. Diagnostic and Therapeutic Developments in Progressive Pulmonary Fibrosis. Archivos de Bronconeumología. Sociedad Española de Neumología y Cirugía Torácica (SEPAR); 2022;58:418-424.
8. Ghazipura M, Mammen MJ, Herman DD, Hon SM, Bissell BD, Macrea M, et al. Nintedanib in Progressive Pulmonary Fibrosis A Systematic Review and Meta-Analysis. Ann Am Thorac Soc. 2022;19(6):1040-1049.
9. Flaherty KR, Wells AU, Cottin V, Devaraj A, Inoue Y, Richeldi L, et al. Nintedanib in progressive interstitial lung diseases: data from the whole INBUILD trial. European Respiratory Journal. 2022;59(3):2004538.
10. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020;8(2):147-157.
11. Ghazipura M, Mammen MJ, Bissell BD, Macrea M, Herman DD, Hon SM, et al. Pirfenidone in Progressive Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Annals of the American Thoracic Society. 2022;19(6):1030-1039.
TABLAS Y FIGURAS
Tabla I. Criterios para evaluar progresión según ensayo clínico INBUILD, RELIEF y consideraciones de expertos según la práctica clínica del 20203-5

TCAR: tomografía computarizada de alta resolución; FVC: capacidad vital forzada; DLco: capacidad de difusión del CO
Tabla II. Criterios diagnósticos de FPP

FPP: fibrosis pulmonar progresiva; FVC: capacidad vital forzada, DLco: capacidad de transferencia de monóxido de carbono; Hb: hemoglobina.
*Tabla adaptada de Raghu et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 20221;205(9):18-47.
Tabla III. Factores de riesgo de progresión en EPID

Extraída y modificada de Rajan SK et al.6 CCP: péptido citrulinado cíclico, DLco: capacidad de difusión de monóxido de carbono, FVC: capacidad vital forzada, IMC: índice de masa corporal, Kco: capacidad de difusión de monóxido de carbono ajustado al volumen alveolar, MUC5B: gen de la mucina 5B, ND: no disponible, NINE: neumonía intersticial no específica, NIU: neumonía intersticial usual, TCAR: tomografía computarizada, TLD: disfunción de los telómeros. *Solo OR disponible en la literatura
Figura
1. Prevalencia de EPID fibrótica FPP y no FPP y subtipos

Adaptado de Valenzuela C, Cottin V. Epidemiology and real-life experience in progressive pulmonary fibrosis. CurrOpinPulmMed. 2022;28(5):407-413. NH: neumonitis por hipersensibilidad
EPID-EAS: enfermedad pulmonar intersticial relacionada con enfermedad autoinmune. IPAF: neumonía intersticial con características autoinmunes. NINE: neumonía intersticial no específica. NII: neumonía intersticial idiopática.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
COMORBILIDADES
Autores
Francisco León Román
Walther Iván Girón Matute
Coordinadora
María Asunción Nieto Barbero
1. INTRODUCCIÓN
Las comorbilidades -como cáncer de pulmón, reflujo gastroesofágico, enfisema, infecciones, hipertensión pulmonar, tromboembolia de pulmón, apnea obstructiva del sueño, entre otras- pueden manifestarse antes, durante y después del diagnóstico de la enfermedad pulmonar intersticial difusa. Su correcta identificación y su tratamiento podrían mejorar la calidad de vida, reducir la progresión y disminuir la mortalidad asociada en algunos pacientes.1-10
2. CÁNCER DE PULMÓN
Las enfermedades pulmonares intersticiales, como la fibrosis pulmonar idiopática (FPI) y el cáncer de pulmón, presentan mecanismos patogénicos comunes, y también un factor de riesgo equivalente, que es el tabaquismo. Debido a esto, los pacientes con enfermedades intersticiales tienen mayor riesgo que la población general de desarrollar un carcinoma broncogénico de forma independiente. La frecuencia de esta enfermedad aumenta con la evolución de las enfermedades intersticiales, que son más frecuentes, como era de esperar, en varones, fumadores y de edad avanzada. Dicho carcinoma broncogénico se localiza en las zonas periféricas. Su subtipo histológico más frecuente es el carcinoma epidermoide. Esto sugiere que dicha ubicación concuerda con la distribución de lesiones fibróticas y, a su vez, que el proceso fibrótico puede desempeñar un papel en el desarrollo del cáncer.
El cáncer de pulmón se manifiesta a menudo como una lesión similar a una masa en o cerca de áreas de fibrosis. Además, el riesgo asociado a las técnicas diagnósticas y terapéuticas invasivas dificulta el manejo de estos pacientes. A esto se suma una mayor incidencia de exacerbaciones agudas en estos pacientes en relación a dichas técnicas diagnósticas así como a los tratamientos quirúrgicos, quimioterapia o radioterapia. El cáncer de pulmón también puede desarrollarse en enfermedades del tejido conectivo y es raro en sarcoidosis, lo que probablemente refleja la correlación inversa entre sarcoidosis y tabaquismo.
El dilema para el médico surge cuando estas enfermedades, cada una con muy mal pronóstico, han sido diagnosticadas a la vez en el mismo paciente. La supervivencia media después del diagnóstico es peor cuando se presentan ambas que en el caso de una enfermedad intersticial aislada o un cáncer de pulmón solo.¹
3. REFLUJO GASTROESOFÁGICO
La presencia del reflujo gastroesofágico (RGE) junto con microaspiraciones en pacientes con enfermedades intersticiales y fibrosis pulmonar se ha estudiado principalmente como factor causal de dichas enfermedades y, a la vez, agravante o responsable de las exacerbaciones agudas. La prevalencia informada de RGE es muy variable: la prevalencia de RGE distal puede ir del 67% al 88%, y la proximal, del 30% al 74%. Las microaspiraciones debidas al reflujo provocan daño al epitelio alveolar con inicio del proceso de fibrosis pulmonar.
En los pacientes con enfermedades intersticiales, el RGE se presenta frecuentemente de forma poco expresiva con ausencia de su sintomatología característica, haciendo que la historia clínica sea poco sensible para su detección. En consecuencia, se deben utilizar otros métodos diagnósticos, como la medición de pH en 24 horas o la medición de pepsina y ácidos biliares en el lavado broncoalveolar (LBA).
El RGE es una comorbilidad importante en las enfermedades intersticiales porque tiene implicaciones tanto para la patogenia como para la calidad de vida del paciente. Los potenciales beneficios para la calidad de vida justifican el tratamiento antiácido para el RGE sintomático, especialmente en pacientes con alteración importante del sueño debido a episodios de despertar con sensación de asfixia.
El tratamiento incluye el estilo de vida, es decir, cambios como comidas escasas y frecuentes, elevar la cabecera de la cama sobre bloques, evitar acostarse en decúbito supino durante 3 o 4 h después de comer, evitar el ajo, la cebolla, los alimentos muy condimentados o el exceso de té, café o alcohol. El tratamiento de esta enfermedad involucra inhibidores de la bomba de protones, antagonistas de los receptores H2 y agentes procinéticos que favorezcan el vaciamiento gástrico. En casos más graves, puede ser necesaria una funduplicatura laparoscópica en caso de reflujo sintomático resistente a dosis altas.²
4. ENFISEMA
En 2005, la combinación de fibrosis pulmonar y enfisema (CPFE) aparece como entidad clínica definida por primera vez. Su prevalencia es muy variable. Oscila entre un tercio y la mitad de los casos con fibrosis pulmonar. Los pacientes incluidos se caracterizaban por ser fumadores o exfumadores con importante disnea de esfuerzo y exploración física en que destacaban los crepitantes secos teleinspiratorios y las acropaquias. Los hallazgos tomográficos incluyen enfisema centrolobulillar y paraseptal en lóbulos superiores y enfermedad parenquimatosa pulmonar, sugestiva de fibrosis pulmonar, en los lóbulos inferiores, acompañado de opacidades reticulares, bronquiectasias de tracción y quistes de panal, junto con vidrio deslustrado en aproximadamente dos tercios de los casos.3
Funcionalmente, estos pacientes presentan una relación entre el volumen espiratorio forzado del primer segundo (FEV1) respecto a la capacidad vital forzada mayor de 0,7 y una capacidad pulmonar total en rangos casi normales junto con una capacidad de difusión del monóxido de carbono (DLco) gravemente disminuida. Además, presentan marcada hipoxemia durante el ejercicio. La prevalencia de hipertensión pulmonar se da en casi la mitad de los casos, que presentan mayor riesgo de desarrollar carcinoma broncogénico.
Actualmente, no existe un tratamiento específico para la CPFE. Se recomienda el abandono tabáquico lo antes posible y el uso de oxigenoterapia crónica domiciliaria en los pacientes con insuficiencia respiratoria en reposo. Los broncodilatadores son utilizados en los pacientes con obstrucción al flujo aéreo.4
5. INFECCIONES
Las infecciones pulmonares agudas ocupan un puesto importante en la rápida progresión de las enfermedades intersticiales. Tanto los virus como las bacterias se han asociado con la progresión de estas. Como la infección respiratoria aguda puede causar un rápido deterioro de una enfermedad intersticial subyacente, es frecuente administrar una terapia temprana con antibióticos de amplio espectro.
La infección aguda puede causar daño alveolar difuso (DAD) a nivel respiratorio, y a su vez, ser un desencadenante de una exacerbación aguda. Existe una mayor incidencia de las exacerbaciones durante los meses de invierno y primavera, coincidiendo con una mayor prevalencia de infecciones. Los pacientes con enfermedades intersticiales y DAD en asociación con infección no difieren en las pruebas de función pulmonar, los hallazgos de la tomografía computarizada de alta resolución (TCAR), el recuento diferencial de células del LBA o la mortalidad en comparación con los pacientes que presentan exacerbaciones respiratorias de las enfermedades intersticiales de otra causa. Esta
observación es importante, ya que el uso de LBA para diagnosticar dicha infección aparentemente aumenta el riesgo en pacientes con función pulmonar ya deteriorada y puede no agregar información útil en términos de tratamiento.
Hay que destacar que en las enfermedades intersticiales (EPID) asociadas a enfermedades del tejido conectivo (ETC), en las que el tratamiento inmunosupresor es la piedra angular del tratamiento, la distinción entre exacerbaciones de la EPID e infección aguda u oportunista tiene implicaciones importantes en el tratamiento. Por ejemplo, la EPID de aparición aguda en pacientes con artritis reumatoide en tratamiento con agentes biológicos suele deberse a una infección por Pneumocystis jirovecii. Es difícil determinar si se debe aumentar o disminuir el nivel de inmunosupresión e instaurar un tratamiento intensivo para la infección, adaptado a microorganismos cultivados. Debido a esto, en dichos casos el LBA juega un papel importante. En pacientes gravemente afectados, se debe considerar el ingreso en una unidad de cuidados intensivos para realizar el LBA con fácil acceso a ventilación mecánica.5
6. HIPERTENSIÓN PULMONAR
La hipertensión pulmonar es una comorbilidad frecuente en los pacientes con EPID. La gravedad de la hipertensión pulmonar se correlaciona directamente con la gravedad y la progresión de la EPID.6 Los síntomas son inespecíficos y pueden superponerse a los síntomas de la enfermedad de base, siendo justificados principalmente por la disfunción del ventrículo derecho7. La disnea durante el ejercicio, palpitaciones, hemoptisis, síncope, edema periférico, ingurgitación yugular y hepatomegalia son hallazgos habituales.7 La hipertensión pulmonar se identifica en alrededor de 8-15% de los pacientes con FPI evaluados inicialmente, y se incrementa hasta el 60% en casos de enfermedad pulmonar terminal.7
Diagnóstico
El ECG, la BNP/NT-proBNP, las pruebas de función respiratoria, la PM6M, el ecocardiograma, los hallazgos en TCAR de tórax y la ergoespirometría son pruebas complementarias no invasivas que pueden apoyar el diagnóstico de hipertensión pulmonar.6 La hipertensión pulmonar suele caracterizarse por una disminución de la DLco desproporcionada respecto a la FVC y/o FEV1, la hipoxemia en el reposo y durante el ejercicio, el incremento del diámetro de la arteria pulmonar y el aumento de la presión arterial pulmonar sistólica estimada mediante ecocardiograma.6-7 El cateterismo cardíaco derecho (CCD) es la prueba de elección para confirmar el diagnóstico de hipertensión pulmonar. Sin embargo, no se recomienda de forma rutinaria debido al riesgo asociado; el CCD se emplea únicamente para la valoración de trasplante pulmonar, ensayos clínicos y si su realización modifica el manejo terapéutico.7 Las EPID forman parte de la hipertensión pulmonar causada por enfermedades pulmonares e hipoxemia (grupo 3). En el
CCD, se identificará una presión arterial pulmonar (PAP) media >20 mmHg, presión de enclavamiento pulmonar (PEP) ≤15 mmHg y una resistencia vascular pulmonar (RVP) >2 UW.7 En estudios recientes, la RVP >5 UW se asoció con hipertensión pulmonar grave en pacientes con EPID, que empeoró la sintomatología, el pronóstico y la supervivencia.7
Tratamiento
El manejo de la hipertensión pulmonar del grupo 3 se basa en optimizar el tratamiento de la EPID, incluyendo programas de rehabilitación respiratoria, oxigenoterapia crónica domiciliaria y/o ventilación mecánica no invasiva si cumple los criterios establecidos.7 Los fármacos utilizados para el tratamiento de la hipertensión pulmonar del grupo 1 no se recomiendan de forma habitual para el manejo de la hipertensión pulmonar del grupo 3.6,7 Los estudios con ambrisentan y riociguat se asociaron con deterioro clínico en pacientes con EPID. Sin embargo, no existe evidencia sólida para recomendar a favor o en contra el uso de los fármacos inhibidores de la fosfodiesterasa5, debido a los resultados contradictorios obtenidos.7 El treprostinil inhalado demostró una mejoría de la capacidad de ejercicio en pacientes con hipertensión pulmonar asociada a EPID. 7 Finalmente, los pacientes deberán remitirse a valoración de trasplante pulmonar en caso de ser candidatos y a centros expertos si presentan una hipertensión pulmonar grave que no se justifique del todo por la afectación parenquimatosa.6,7
7. TROMBOEMBOLIA DE PULMÓN
Los pacientes con EPID presentan un riesgo incrementado de manifestar la enfermedad tromboembólica venosa (ETV), que comprende la trombosis venosa profunda (TVP) y la tromboembolia de pulmón (TEP).8 La inmovilización por la propia enfermedad, las exacerbaciones que requieren ingreso hospitalario, el estado de procoagulante y el cáncer, sobre todo de pulmón, podrían explicar la aparición de la ETV en EPID.6,7 La enfermedad cerebrovascular isquémica, el cáncer y la cardiopatía isquémica se asociaron a un mayor riesgo de ETV en pacientes con FPI.8 El diagnóstico y el tratamiento de la ETV en la EPID no cambian respecto a las guías actuales. La angiotomografía computarizada (angioTC) de tórax es el estudio de elección para diagnosticar la TEP; la gammagrafía V/Q se empleará como segunda opción en casos de alergia a contrastes iodados o aclaramiento de creatinina <30 mL/min, considerando que la afectación marcada por la EPID puede interferir con los resultados obtenidos.6 Posteriormente, se evaluará la estratificación del riesgo para el manejo. La ecografía doppler de miembros inferiores se realizará ante la sospecha de TVP. Los anticoagulantes orales de acción directa, la heparina de bajo peso molecular y los antagonistas de la vitamina K pueden emplearse en el tratamiento de la ETV en EPID; se recomienda el uso de heparina de bajo peso molecular en dosis profilácticas durante el ingreso hospitalario.6
8. ENFERMEDADES CARDIOVASCULARES
Los pacientes con EPID que presenten una disnea desproporcionada a la patología pulmonar requerirán una valoración cardiológica integral. La propia EPID, su tratamiento y la hipoxia crónica favorecen el desarrollo de cardiopatía, arritmias e insuficiencia cardíaca.6 Por otro lado, la cardiopatía isquémica es una comorbilidad prevalente en los pacientes con EPID que afecta en el 68% de los casos a pacientes con FPI.9 En un estudio, se identificó que las EPID, sobre todo la FPI, se asocian de forma independiente al riesgo de presentar infarto de miocardio y cardiopatía isquémica, siendo prioritaria la evaluación cardiovascular temprana en individuos de entre 60-69 años.9 Además, demuestran un riesgo incrementado de infarto de miocardio en mujeres con FPI y pacientes menores de 50 años diagnosticados de sarcoidosis pulmonar.9 La sarcoidosis, la artritis reumatoide, las miopatías inflamatorias y la esclerodermia, entre otras patologías autoinmunes, se relacionan con insuficiencia cardiaca, miocarditis, valvulopatías, bloqueos de rama y otras alteraciones en el electrocardiograma.6 Por tanto, el electrocardiograma, el ecocardiograma, la resonancia magnética cardiaca y el FDG-PET/CT son las alternativas actuales para la correcta evaluación cardíaca.6 El manejo dependerá de la optimización del tratamiento de la EPID de base y, a su vez, de la patología cardiaca relacionada, controlando de forma adecuada los factores de riesgo compartidos entre ambas entidades.
9. APNEA OBSTRUCTIVA DEL SUEÑO
La apnea obstructiva del sueño (AOS) es una comorbilidad que afecta a 3 de cada 4 pacientes diagnosticados de EPID. En concreto, la FPI es la patología que más se relaciona con AOS.10 La fisiopatología de la AOS en las EPID se podría justificar por el colapso de la vía aérea superior, que genera un incremento de las presiones torácicas, la hipoxia intermitente de la propia AOS y el RGE.10 Los síntomas de AOS en EPID son inespecíficos, mientras que las escalas empleadas se correlacionan erróneamente con los resultados de la gravedad de la AOS medida mediante polisomnografía.6 Además, en los estudios realizados no se ha logrado determinar si la AOS es un marcador de mal pronóstico en la EPID o si su efecto podría empeorar únicamente la hipertensión pulmonar asociada a la hipoxemia.6 El índice de desaturación de oxígeno (ODI) y el tiempo con saturación de oxihemoglobina por debajo del 90% (CT90%) alterados por la AOS repercuten en la calidad del sueño y empeoran la desaturación nocturna en los pacientes con EPID.10 Actualmente, pese a la repercusión clínica, no existen criterios establecidos para el screening de AOS en EPID. Los autores recomiendan un estudio mediante polisomnografía en casos de sospecha clínica.6 La presión positiva continua en las vías respiratorias (CPAP) es el tratamiento de elección para la AOS según los criterios establecidos, y mejora la actividad diaria y la calidad de vida y del sueño en
pacientes con AOS y FPI.10 Por tanto, su efecto puede extrapolarse a las otras EPID. Los humidificadores añadidos a la CPAP podrían tener un efecto positivo en la tos irritativa de las EPID.6 Finalmente, es fundamental valorar los posibles efectos adversos de los medicamentos en el sueño. Los corticoides utilizados habitualmente en las EPID ocasionan insomnio y múltiples despertares nocturnos.6
10. DEPRESIÓN
En un estudio, se evidenció que más del 20% de los pacientes con EPID tienen depresión clínicamente relevante.6 Los corticoides, la gravedad de la disnea, el dolor, la limitación funcional y las alteraciones del sueño se han asociado con depresión en la EPID.6 La depresión y la ansiedad impactan incluso en la adherencia a los tratamientos y, por tanto, deberá ser evaluada de forma rutinaria en las consultas de EPID.6 La rehabilitación respiratoria tiene un rol fundamental en el manejo de la disnea, la capacidad funcional y el manejo sintomático, además de proporcionar un mejor control de la depresión y la ansiedad.6
BIBLIOGRAFÍA
1. Tomassetti S, Gurioli C, Ryu JH, et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest. 2015;147:157-164.
2. Savarino E, Carbone R, Marabotto E, et al. Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients. Eur Respir J. 2013;42:1322-1331.
3. Bollo De Miguel E, Baloira-Villar A, Martínez-González C. Complicaciones y comorbilidades. En: Ancochea J, Xaubet A, Agüero R. Fibrosis Pulmonar idiopática. 1ª ed. Editorial Respira, Barcelona, 2015. p. 215-231.
4. Antoniou KM, Margaritopoulos GA, Goh NS, et al. Combined pulmonary fibrosis and emphysema in scleroderma lung disease has a major confounding effect on lung physiology and screening for pulmonary hypertension. Arthritis Rheumatol. 2016;68:1004-1012.
5. Wootton SC, Kim DS, Kondoh Y, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:1-30.
6. Margaritopoulos GA, Antoniou KM, Wells AU. Comorbidities in interstitial lung diseases. EurRespir Rev. 2017;26(143):160027.
7. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, et al; ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. EurRespir J. 2023;61(1):2200879.
8. Lee JH, Lee HH, Park HJ, Kim S, Kim YJ, Lee JS, Kim HC. Venous thromboembolism in patients with idiopathic pulmonary fibrosis, based on nationwide claim data. TherAdvRespirDis. 2023;17:17534666231155772.
9. Clarson LE, Bajpai R, Whittle R, Belcher J, Abdul Sultan A, Kwok CS, Welsh V, Mamas M, Mallen CD. Interstitial lung disease is a risk factor for ischaemic heart disease and myocardial infarction. Heart. 2020;106(12):916-922.
10. Li D, Wang B, Liu Y, Wang H. Prevalence and impact of comorbid obstructive sleep apnoea in diffuse parenchymal lung diseases. PLoSOne. 2021;16(2):e0246878.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
EXACERBACIONES Y COMPLICACIONES DE LAS EPID Y EPID RÁPIDAMENTE PROGRESIVAS
Autoras
Andrea Martín González
Beatriz García Pulido
Coordinador
Esteban Cano Jiménez
INTRODUCCIÓN
La definición de la exacerbación aguda de las EPID (EA-EPID) ha evolucionado a lo largo del tiempo, y sus características aún no están recogidas de manera uniforme en la literatura. Aunque se ha estudiado la etiopatogenia y las principales características en la fibrosis pulmonar idiopática (FPI), se han extrapolado dichos conocimientos al resto de las EPID.
De acuerdo con los criterios de EA-FPI actualizados en 20161, se define la EA-EPID como un deterioro respiratorio agudo (menos de 1 mes de duración), clínicamente significativo, con presencia de infiltrados radiológicos difusos, y que no se justifica exclusivamente por insuficiencia cardíaca o sobrecarga hídrica (y en el caso de enfermedades reumáticas, tampoco por el uso FAMES).
1. EPIDEMIOLOGÍA
La tasa de incidencia de la EA-EPID es muy variable. En la FPI, la incidencia anual es del 5 al 19%.1 En otras EPID fibróticas, la incidencia es más baja que en la FPI, siendo más frecuentes las que tienen un patrón histológico o radiológico de neumonía intersticial usual (NIU).1,2
El impacto de la EA-EPID en la morbimortalidad de los pacientes es muy elevado. Según las series publicadas, la mortalidad posterior a una exacerbación es también muy
variable y se sitúa entre el 33 y el 83%, con tasas de mortalidad hospitalaria del 50 al 100% en EPID asociadas a enfermedades del tejido conectivo (ETC-EPID), y del 75 al 100% en pacientes con neumonitis por hipersensibilidad.2
2. ETIOLOGÍA
La etiología de la EA en las EPID sigue siendo incierta. Se desconoce si la EA representa una progresión de la EPID subyacente o una respuesta aberrante a agresiones externas. En ocasiones, puede ser la primera manifestación de la enfermedad. En la FPI, se cree que puede estar relacionada con una disfunción biológica intrínseca del pulmón que hace que los individuos con FPI sean más susceptibles a las agresiones externas que los individuos sin FPI, lo que conduce a una lesión pulmonar aguda y a un daño alveolar difuso.3
Se han identificado posibles desencadenantes que pueden precipitar la EA-EPID, como infecciones respiratorias (infecciones virales), fármacos, broncoaspiración de contenido gástrico, contaminación ambiental o procedimientos diagnósticos y quirúrgicos (hiperoxia y ventilación mecánica). En las ETC-EPID, cabe distinguir que en los desencadenantes de exacerbación se incluye el tratamiento con FAMES debido a las reacciones de hipersensibilidad que pueden presentar los pacientes tratados con esta familia de fármacos.
3. FACTORES DE RIESGO
Por lo general, la EA-EPID es más frecuente en pacientes con enfermedad avanzada (inversamente proporcional a la CVF y DLCO), en los no fumadores y en la EPID con un patrón histológico o radiológico de neumonía intersticial usual (NIU).3,4 Un nivel sérico elevado de la glicoproteína Krebs von Lungen-6 (KL-6) se ha asociado con un mayor riesgo de EA.3
4. DIAGNÓSTICO
El diagnóstico de la EA-EPID se basa en la forma de presentación clínica (deterioro agudo e identificación de posibles desencadenantes) y los hallazgos radiológicos, siendo fundamental la exclusión de otras causas de deterioro respiratorio (sobre todo causas extra parenquimatosas, sobrecarga hídrica, etc.) (Tabla I).
En caso de no cumplirse los cuatro criterios, debemos etiquetar el cuadro de “sospecha de EA-EPID”.
TC de tórax (TCAR)
La aparición reciente de áreas de consolidación, la presencia de vidrio deslustrado, ya sea de manera bilateral y difusa o en combinación, superpuestas a un patrón intersticial
preexistente, resulta fundamental para el diagnóstico. En ausencia de una tomografía computarizada de tórax que muestre estas alteraciones alveolares recién aparecidas en una radiografía simple de tórax (incluso en el contexto clínico apropiado) no es posible establecer un diagnóstico definitivo, sino que se trata de un diagnóstico de “sospecha de EA-EPID”.
Con esta prueba radiológica también excluiremos otras causas, como embolia pulmonar (si se realiza el protocolo de Angiotac), y causas extra parenquimatosas, como neumotórax o derrame pleural.4
Pruebas de laboratorio
Se solicitan esencialmente para descartar otras causas del deterioro respiratorio, sobre todo infecciosas. Están indicadas la obtención de muestras microbiológicas (esputo, hemocultivos, antígenos de orina para Legionella y Streptococcus) y realización de PCR para detección de infección vírica.
La procalcitonina sérica puede ser un marcador útil para diferenciar la neumonía bacteriana de la EA-EPID, ya que también en esta puede haber elevación de reactantes de fase aguda asociados.5
En pacientes sin diagnóstico previo de EPID, se puede considerar la realización de una analítica con autoinmunidad con el fin de identificar EPID relacionada con enfermedad del tejido conectivo o síndromes de hemorragia alveolar pulmonar. Los dos últimos tienen más probabilidades de responder a altas dosis de inmunosupresión.
Broncoscopia
La realización de estudios broncoscopios para obtener muestras microbiológicas es controvertida debido a la gravedad del cuadro y a la posible situación de insuficiencia respiratoria del paciente. Por lo tanto, siempre habrá que valorar la relación riesgo-beneficio. Es de especial interés su realización si el paciente está con tratamiento inmunosupresor.
Las muestras de lavado bronco alveolar (LBA) presentan un recuento celular de predominio neutrofílico, lo que probablemente refleja la presencia de daño alveolar difuso en los tejidos y no permite diferenciarlo de la etiología infecciosa.5
En la figura 1 se presenta un algoritmo de actuación ante sospecha de EA-EPID.
5. TRATAMIENTO
Soporte ventilatorio
Consiste en la corrección de la hipoxemia mediante la suplementación de oxígeno a través de diferentes dispositivos: gafas nasales de alto flujo (GNAF), ventilación mecáni-
ca no invasiva (VMNI), ventilación mecánica invasiva (VMI) o membrana de oxigenación extracorpórea (ECMO).
Respecto a las GNAF, se ha visto que reducen el trabajo respiratorio, además de ser un dispositivo muy bien tolerado y cómodo para el paciente.4
En cuanto a la VMNI, existe menos evidencia de su uso en la EA-EPID, pues los estudios que la han evaluado son de poca calidad, la mayoría son retrospectivos y con pequeño tamaño muestral.4
La VMI se prefiere evitar en estos pacientes, ya que sus pulmones tienen una alteración de la V/Q y escasa compliancia, lo que los hace más propensos a sufrir un daño alveolar agudo y distrés respiratorio, siendo la mortalidad registrada de algunos estudios de casi el 100%.6 Sin embargo, esto es un punto que debe valorarse individualmente y de forma multidisciplinar, y en pacientes en espera de trasplante pulmonar puede ser una terapia puente hacia el mismo.4
En los casos de refractariedad, el empleo de ECMO veno-venosa puede ser una opción. Existen pocos estudios al respecto en EA-EPID, siendo empleada también como terapia puente en pacientes subsidiarios de trasplante pulmonar.4
Inmunosupresores
La elección de un tratamiento u otro debe realizarse de forma individualizada y con implicación multidisciplinar.
Aunque existe una recomendación débil para el uso habitual de esteroides sistémicos en pacientes con EA-EPID debido a la ausencia de evidencia científica al respecto, se emplean de forma habitual. En cuanto a la dosis y duración, no existe una pauta establecida. Suele utilizarse metilprednisolona en bolo, inicialmente a una dosis de 500-1000 mg al día durante 3 días, y en caso de respuesta, se hará un descenso gradual posterior a dosis.4 Los esteroides a dosis altas deben usarse con precaución en pacientes con SSc-ILD debido al riesgo de crisis renal SSc asociado.
También hay series de casos que han evaluado el papel de la ciclofosfamida o la ciclosporina en combinación con corticoesteroides, así como el rituximab en combinación con plasmaféresis.4
Recientemente, se están llevando a cabo varios ensayos clínicos (STRIVE-IPF y Europe Exchange-IPF) que emplean la combinación de plasmaféresis, rituximab, inmunoglobulinas intravenosas y corticoides, de cuyos resultados todavía no disponemos.4
Antifibróticos
Respecto al nintedanib y la pirfenidona, no existe un consenso respecto a su uso en EAEPID. Se ha visto que hasta el 80% de los facultativos continúan empleándolos durante la hospitalización por EA-EPID, y hasta el 66% inician la prescripción de los mismos.4
Antibioterapia
A menudo, se administran antibióticos empíricos de amplio espectro a pacientes con EA-EPID y sospecha de sobreinfección, además de valorar la adición de terapia antiviral durante períodos de mayor riesgo (por ejemplo, oseltamivir durante la temporada de influenza).
Debemos tener en cuenta que muchos de estos pacientes están inmunodeprimidos, por lo que debemos valorar también la cobertura empírica de gérmenes como Aspergillus o Pneumocystis en caso de estar contraindicada la videobroncoscopia.4
Antiácidos
Puesto que las microaspiraciones de contenido gástrico pueden jugar un papel en las EA-EPID, se opta por el mantenimiento de los mismos o su inicio en caso de tratamiento con corticoides o riesgo asociado de úlcera de estrés.4
Trasplante pulmonar
Las enfermedades pulmonares intersticiales son la causa más frecuente de trasplante pulmonar, especialmente la FPI, teniendo los pacientes una mediana de supervivencia de entre 5 y 7 años. Existen numerosas contraindicaciones para el mismo, como problemas psicosociales o una importante afectación extrapulmonar. Esta última puede darse en los pacientes con EA-EPID ingresados en UCI, ya que el empleo de VMI o ECMO se asocia a mayores complicaciones post trasplante. En los casos de pacientes ya incluidos en lista de espera de trasplante en el momento de la EA, la ECMO puede ser empleada como terapia puente.4
En cualquier caso, siempre debemos evaluar cada supuesto de forma individualizada y dentro de un equipo multidisciplinar, junto con la familia y el propio paciente.
Otras consideraciones
En casos de sospecha de EA-EPID exposicionales, se recomienda identificar y eliminar la posible exposición a los agentes tóxicos causantes caso por caso, particularmente en casos de neumonitis por hipersensibilidad.4
Dado el mal pronóstico de los pacientes con EA-EPID ingresados en UCI, es importante poner en marcha este recurso sanitario lo antes posible. Además, es importante que el paciente deje reflejadas sus voluntades anticipadas, así como las instrucciones del final de la vida.
6. PRONÓSTICO
Hasta el 46% de las muertes de pacientes con FPI están precedidas por una exacerbación. La mediana de supervivencia tras ella es de 3-4 meses, siendo la insuficiencia
respiratoria aguda grave la causa de mortalidad intrahospitalaria hasta en el 50% de los casos.
Dicho pronóstico es peor en los pacientes con CVF y DLCO basal más bajas, TC con mayor extensión de afectación aguda, peor oxigenación y LBA con mayor porcentaje de neutrofilia y linfocitosis.1,4
7. PREVENCIÓN
En la prevención de las EA-EPID, es importante la vacunación contra influenza y neumococo, el lavado de manos y la evitación de contacto con personas que presenten sintomatología respiratoria sugestiva de un proceso vírico (por ej.). También, la evitación de irritantes y contaminantes en el aire.
El reflujo gastroesofágico puede ser un factor de riesgo potencial para la progresión de la FPI, no estando claro si el tratamiento antiácido tiene un efecto preventivo sobre el desarrollo de EA-EPID.
Respecto a los tratamientos farmacológicos, se ha visto en el estudio INBUILD que el nintedanib parece retrasar el tiempo hasta el primer EA en pacientes con FPI.7
8. COMPLICACIONES Y COMORBILIDADES EN LA EPID
El curso clínico de las enfermedades pulmonares intersticiales puede ser variable, y en él intervienen varios factores. Además de las EA-EPID, las principales complicaciones y comorbilidades son las que se detallan a continuación.
- Reflujo gastroesofágico: presente en hasta el 87% de los pacientes. No está claro si es consecuencia de la distensión esofágica ante un pulmón fibrótico o causa del daño epitelial de la propia EPID o desencadenante de una exacerbación.
- Hipertensión pulmonar: definida como una presión media de la arteria pulmonar ≥20 mmHg (medida mediante cateterismo cardiaco derecho. En el caso de la asociada a colagenopatía, se encuadra en el grupo 1, y en el caso de la asociada a FPI, en el grupo 3. En cualquiera de ellos, existiría además una presión de enclavamiento de la arteria pulmonar ≤ 15 mmHg y unas resistencias vasculares periféricas elevadas (>2WU). Existe especial riesgo de su desarrollo en pacientes con síndrome combinado enfisema-fibrosis y enfermedad intersticial asociada a esclerodermia.
- Apnea obstructiva del sueño (AOS): hasta la mitad de los pacientes presentan una AOS moderada-grave asociada. Debe realizarse poligrafía respiratoria en aquellos casos en que se sospeche.
- Neumotórax/neumomediastino: incidencia del 11%.
- Carcinoma broncogénico: hasta cinco veces más frecuente en pacientes con EPID.
- Enfermedades cardiovasculares: arritmias, insuficiencia cardíaca.
- Hipotiroidismo.
- DM tipo 2.
- Ansiedad y depresión.
9. ENFERMEDADES PULMONARES INTERSTICIALES RÁPIDAMENTE PROGRESIVAS (EPIRP)
Las EPIRP se definen como un cuadro clínico de menos de 1 mes de evolución consistente en un empeoramiento radiológico, mayor disnea e insuficiencia respiratoria aguda. Dentro de ellas, distinguimos las que se presentan asociadas a anticuerpos contra la proteína 5 asociada a la diferenciación del melanoma (anti-MDA5), y dentro de la familia de miositis inflamatorias, las asociadas a acortamiento telomérico y formas familiares.
Los autoanticuerpos contra la proteína 5 asociada a la diferenciación del melanoma (anti-MDA5) se han encontrado entre el 13 y el 30% de los pacientes con miopatías inflamatorias (sobre todo a dermatomiositis y a la forma amiopática). Se asocia a enfermedad pulmonar intersticial que característicamente puede presentar una rápida progresión. Esta entidad es más frecuente en asiáticos (30-50%), y debemos tenerla en cuenta siempre que tengamos un paciente con insuficiencia respiratoria aguda grave y un patrón intersticial bilateral, aunque no existan manifestaciones cutáneas o musculares asociadas. En los casos en que se llega a realizar una biopsia pulmonar, los hallazgos son compatibles con un daño alveolar difuso.8
En cuanto a los biomarcadores, se ha visto que los niveles de anticuerpos anti-MDA5 disminuyen cuando existe remisión de la enfermedad y aumentan cuando existe una exacerbación, por lo que se correlacionan con la actividad de la misma.
Existen además otros marcadores que pueden ser de utilidad en esta patología, como son proteínas, anticuerpos y células (Tabla II). Dentro de ellos, hemos de destacar, por tener mayor disponibilidad en la mayoría de los centros, la ferritina y la lactato deshidrogenasa (LDH).
Los niveles elevados de ferritina >1000 μg/L se asocian con un peor pronóstico, siendo niveles ≥1250 μg/L un factor independiente de mortalidad a los 6 meses.
Los niveles de LDH >300 IU/L se asocian con afectación pulmonar intersticial grave.
Otro marcador a destacar, aunque no ampliamente disponible en todos los centros, es
la determinación de glicoproteína KL6, que podría emplearse para monitorizar la respuesta al tratamiento. Se ha visto que unos niveles ≥1000 U/ml son un factor independiente de mortalidad en estos pacientes. Si los niveles se mantienen invariables durante las cuatro primeras semanas tras el inicio de tratamiento inmunosupresor, sugieren refractariedad al tratamiento.9
Respecto al tratamiento, existe un régimen intensivo basado en corticoides intravenosos a dosis elevadas, ciclosporina oral y/o pulsos de ciclofosfamida intravenosa. También se ha descrito el empleo de la combinación de tofacinib con rituximab o hemoperfusión con polimixina B.11
BIBLIOGRAFÍA
1. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, Lee JS, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care Med. 2016;194(3):265-275.
2. Kolb M, Bondue B, Pesci A, Miyazaki Y, Woo Song J, Bhatt YN, Huggins JT, et al. Exacerbaciones agudas de enfermedades pulmonares intersticiales fibrosantes progresivas. Revista Respiratoria Europea. 2018;27:180071.
3. Song JW, Hong SB, Lim CM, Koh Y, Kim, DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. The European Respiratory Journal: Official Journal of the European Society for Clinical Respiratory Physiology. 2011;37(2):356-363.
4. Hayat Syed MK, Bruck O, Kumar A, Surani S. Acute exacerbation of interstitial lung disease in the intensive care unit: Principles of diagnostic evaluation and management. World Journal of Critical Care Medicine, 2023;12(3):153-164.
5. Polke M, Kondoh Y, Wijsenbeek M, Cottin V, Walsh SLF, Collard HR, Chaudhuri N, et al. (2021). Management of acute exacerbation of idiopathic pulmonary fibrosis in specialised and non-specialised ILD centres around the world. Frontiers in Medicine. 2021;8:699644.
6. Molina-Molina M, Ramon Badia J, Marín-Arguedas A, Xaubet A, José Santos M, María Nicolás J, Ferrer M, Torres A. (2003). Características clínicas y pronóstico de los pacientes con fibrosis pulmonar que ingresan en cuidados intensivos por insuficiencia respiratoria. Análisis de 20 casos. Medicina clínica. 2023;121(2):63-67.
7. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, et al. Nintedanib in progressive fibrosing interstitial lung diseases. The New England Journal of Medicine. 2019;381(18):1718-1727.
8. Amundson WH, Racilab E, Allen T, Dincer HE, Tomic R, Bhargava M, Perlman DM, Kim HJ. Acute exacerbation of interstitial lung disease after procedures. Respiratory Medicine 2019:150:30-37.
9. Luppi F, Sebastiani M, Salvarani C, Bendstrup E, Manfredi A. Acute exacerbation of interstitial lung disease associated with rheumatic disease. Nat Rev Rheumatol. 2022;18(2):8596, doi: 10.1038/s41584-021-00721-z.
10. Li X, Liu Y, Cheng L, Huang Y, Yan S, Li H, Zhan H, Li Y. Roles of biomarkers in anti-MDA5-positive dermatomyositis, associated interstitial lung disease, and rapidly progressive interstitial lung disease. J Clin Lab Anal. 2022;36(11):24726.
11. Kenyon S, Careless D, Navaratnam V, Reddy T. Rapidly progressive interstitial lung disease complicated by pulmonary interstitial emphysema in anti-MDA5 amyotrophic dermatomyositis. Thorax. 2023;78(9):946-947.
12. Chicot M, Valenzuela C, Rodríguez DA. Enfermedad pulmonar intersticial rápidamente progresiva sin afectación cutánea asociada a anticuerpos anti-MDA5. Medicina clínica. 2021;156(8):413-414, https://doi.org/10.1016/j.medcli.2020.01.012.
TABLAS Y FIGURAS
Tabla I. Criterios diagnósticos de EA-EPID
Criterios diagnósticos
- Diagnóstico previo o concurrente de FPI.
- Deterioro agudo o empeoramiento del grado de disnea de < 1 mes de evolución.
- Hallazgo en TC de nuevos infiltrados en vidrio deslustrado o consolidaciones superpuestas al patrón preexistente.
- Deterioro no completamente explicado por insuficiencia cardíaca o sobrecarga hídrica.
Tabla inspirada en la de Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJL, Lee JS, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. American Journal of Respiratory and Critical Care Medicine. 2016;194(3):265-275. https:// doi.org/10.1164/rccm.201604-0801ci.
Figura I. Algoritmo de actuación ante sospecha de EA-EPID
Deterioro agudo respiratorio de EPI (<1mes)
¿Causa extra-parenquimatosa? SÍ
¿Nuevos infiltrados bilaterales vidrio deslustrado o consolidación en TC?
No agudización (dx alternativo; neumotorax, derrame pleural, TEP)
Agudización EPI Causa desencadenante (infección, aspiración...) Idiopática
No agudización (dx alternativo: aspiración, toxicidad fármacos, IC)
Figura inspirada en la de Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJL, Lee JS, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. American Journal of Respiratory and Critical Care Medicine. 2016;194(3):265-275. https:// doi.org/10.1164/rccm.201604-0801ci.

*Considerar solo en caso de pacientes candidatos o en lista de espera de trasplante pulmonar.
Figura inspirada en la que figura en Hayat Syed MK, Bruck O, Kumar A, Surani S. Acute exacerbation of interstitial lung disease in the intensive care unit: Principles of diagnostic evaluation and management. World Journal of Critical Care Medicine. 2023;12(3):153-164. https://doi. org/10.5492/wjccm.v12.i3.153.
Figura 1. Algoritmo de tratamiento de la EA-EPID
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
TRATAMIENTOS NO FARMACOLÓGICOS.
OXIGENOTERAPIA Y REHABILITACIÓN
CARDIOPULMONAR EN PACIENTES CON EPID
Autores
Eli Nancy Pérez Rodas
Yojana García Carrascal
Rodrigo Torres Castro
Mª José Beceiro Padreño
Coordinador
Xavier Alsina Restoy
INTRODUCCIÓN
Las enfermedades intersticiales pulmonares difusas (EPID) forman un grupo heterogéneo de patologías que afectan el intersticio pulmonar, el tejido que rodea y sostiene los alvéolos. Estas enfermedades se caracterizan por la inflamación y fibrosis del intersticio, lo que conduce a la disfunción pulmonar progresiva y a la dificultad respiratoria. Entre las EPID más comunes, se encuentran la fibrosis pulmonar idiopática (FPI), la sarcoidosis, la neumonitis por hipersensibilidad y la fibrosis quística.
La oxigenoterapia desempeña un papel crucial en el manejo de las EPID, especialmente en los pacientes que presentan hipoxemia o bajos niveles de oxígeno en sangre. El suministro de oxígeno suplementario ayuda a mejorar la oxigenación tisular y a reducir la disnea o dificultad respiratoria, y puede contribuir a mejorar la calidad de vida y la capacidad funcional en estos pacientes. La oxigenoterapia puede ser administrada de forma continua o intermitente, según las necesidades individuales de cada paciente y la gravedad de la enfermedad.
Por otro lado, la rehabilitación respiratoria también juega un papel importante en el manejo integral de las EPID. Este programa de cuidados combina ejercicios físicos específicos para mejorar la capacidad de ejercicio y la función pulmonar, educación sobre la enfermedad para fomentar el autocuidado y el manejo de los síntomas respiratorios, técnicas de manejo de la respiración para optimizar la ventilación pulmonar y apoyo psicológico para ayudar a los pacientes a adaptarse a su enfermedad y mejorar su calidad de vida.
La rehabilitación respiratoria en pacientes con EPID puede contribuir a reducir la disnea, mejorar la tolerancia al ejercicio, optimizar la función pulmonar y reducir la frecuencia de exacerbaciones y hospitalizaciones. Además, fomenta la adopción de hábitos de vida saludables, como la actividad física regular y una alimentación equilibrada, que son fundamentales para el manejo y control de las EPID.
Tanto la oxigenoterapia como la rehabilitación respiratoria son dos componentes esenciales en el manejo integral de las enfermedades intersticiales pulmonares difusas. Estos tratamientos pueden mejorar la oxigenación tisular, aliviar los síntomas respiratorios, optimizar la función pulmonar y mejorar la calidad de vida de los pacientes afectados. Es fundamental que estos tratamientos sean prescritos y supervisados por un equipo multidisciplinario de profesionales de la salud especializados en el manejo de las enfermedades respiratorias para garantizar su eficacia y seguridad.
1. OXIGENOTERAPIA EN ENFERMEDADES PULMONARES INTERSTICIALES
Desde 1965, cuando el Dr. Petty elaboro el primer prototipo de tanque portátil con oxígeno líquido en seis pacientes con hipoxia, el oxígeno (O2) se convirtió en el pilar terapéutico para muchos pacientes con enfermedad pulmonar crónica.
Gran parte de los pacientes con EPID deberán iniciar oxigenoterapia, ya que este amplio grupo de enfermedades llevan a la fibrosis pulmonar (FP), siendo la disnea uno de sus síntomas más frecuentes, tanto en estadios avanzados como al hacer un esfuerzo o durante una exacerbación aguda.
La disnea se explica por un alto grado de deterioro de la difusión y alteración de la relación ventilación/perfusión (V/Q), que lleva a los pacientes a la disminución de la saturación capilar periférica de oxígeno (SpO2), a hipoxemia moderada (un SpO2 del 88-93% o una presión arterial de oxígeno de 56-60 mmHg (7,5-7,8 kPa)) o a hipoxemia grave (SpO2< 88% o PaO2 < 55 mmHg (7,3 kPa)), lo que está asociado a un mal pronóstico.1
En la indicación de la oxigenoterapia se debe diferenciar distintos escenarios: si estamos ante un paciente con insuficiencia respiratoria aguda, ya sea en contexto de agudización de su enfermedad, manifestación o empeoramiento de comorbilidades, como
la hipertensión pulmonar; o si estamos ante un paciente con insuficiencia respiratoria crónica por un estadio evolucionado de una EPID y/o de una comorbilidad subyacente. Además, debemos valorar si el tratamiento se va a iniciar a nivel hospitalario o ambulatorio.
1.1. Oxigenoterapia ambulatoria (OA)
La hipoxemia en reposo se manifiesta en estadios avanzados en EPID y está asociada a disnea limitante y disminución de la supervivencia a menos de 1 año.²
No hay ensayos clínicos aleatorizados (ECA) en oxigenoterapia de larga duración (uso ≥ 15 horas) (OTLD) que muestre mejoría de la supervivencia, lo que llevó a extrapolar los datos de la enfermedad pulmonar obstructiva crónica (EPOC) como un papel beneficioso del O2 en pacientes con EPID.
Todas las principales directrices internacionales recomiendan la OTLD en pacientes con hipoxemia diurna grave con evidencia de daño orgánico hipóxico, incluida la hipertensión pulmonar (HTP) y/o la insuficiencia cardiaca derecha. No se sabe con certeza si la OTLD evita los resultados clínicos adversos, como la hospitalización o las exacerbaciones agudas1,3 (Tabla I).
1.2. Oxigenoterapia al esfuerzo
En las EPID, es preciso realizar pruebas periódicas para valorar la desaturación, como la prueba de marcha de 6 minutos (PM6M), que es una prueba de ejercicio ampliamente utilizada. La desaturación >88% es el umbral más utilizado para la prescripción de OA. Esta permite monitorizar la progresión de la enfermedad y la necesidad de oxígeno suplementario1,3 (Tabla II).
1.3. Oxigenoterapia en hipoxemia nocturna
La hipoxemia nocturna se define como >10% de tiempo total de sueño con SpO2 <90%. En las EPID, puede producirse en ausencia de hipoxemia diurna en reposo, y parece asociarse a peores resultados. Se necesitan más estudios para comprender el impacto de los diferentes grados de hipoxemia nocturna en el pronóstico, y si su corrección se asocia a beneficios. Las directrices actuales no abordan la oxigenoterapia nocturna en la EPID.
1.4. Disnea
Puesto que la disnea es un síntoma complejo con muchas causas, actualmente no existe ninguna recomendación para prescribir oxígeno suplementario para su tratamiento en ausencia de hipoxemia.
La etiología de la EA en las EPID sigue siendo incierta. Se desconoce si la EA representa una progresión de la EPID subyacente o una respuesta aberrante a agresiones externas.
1.5. Pruebas diagnósticas
- Prueba de marcha de seis minutos (PM6M). Es una prueba submáxima de esfuerzo cardiorrespiratorio. Se usa para medir la capacidad de ejercicio del paciente. Además, muestra la capacidad de respuesta al cambio con la administración aguda de oxígeno y se utiliza para valorar el flujo de oxígeno necesario para corregir la hipoxemia. La PM6M requiere tiempo y espacio, con un descanso de 30 minutos si se realizan dos pruebas y un pasillo de 30 metros.
- Prueba de sedestación y bipedestación durante 1 minuto (1STS siglas en inglés). Se han observado correlaciones significativas entre la distancia caminada durante la PM6M y el número de repeticiones en la prueba de 1STS, similitudes en la captación máxima de oxígeno, la SpO2 nadir y la frecuencia de desaturación, lo que sugiere que la 1STS puede ofrecer una alternativa más sencilla a la PM6M, con un potencial uso en área primaria y en consultas externas. Sin embargo, no se ha demostrado la capacidad de respuesta a la administración aguda de oxígeno.
- Puntuación DeOx. Es un puntaje (0 hasta 2) que predice la desaturación en pacientes con EPID. Un puntaje de 2 predijo con éxito la desaturación de esfuerzo en el 78,5% de 300 pacientes de una cohorte. Podría utilizarse en entornos con limitación de tiempo (Tabla II).4
En el contexto agudo, se dispone de diferentes tipos de oxigenoterapia, como cánulas nasales, mascarillas faciales para oxígeno de alta concentración, como la máscara de reservorio y la máscara de Venturi, gafas nasales de alto flujo (GNAF), además del uso de VMNI (ventilación mecánica no invasiva). Con respecto a estos dos últimos sistemas, cabe mencionar algunas anotaciones.
- Gafas nasales de altos flujo (GNAF)
Las GNAF podrían ser una alternativa a la oxigenoterapia convencional en pacientes con EPID que requieran una alta concentración de oxígeno para corregir la hipoxemia, controlar la disnea y la taquipnea. Sin embargo, las pruebas son muy escasas y se necesitan ECA para establecer la eficacia real de este método de administración de oxígeno.
- Ventilación mecánica no invasiva
La VMNI puede desempeñar un papel principalmente en el contexto crónico en lo que respecta al impacto sobre la calidad de vida de los pacientes desde la paliación hasta los programas de rehabilitación. Sin embargo, dado el limitado número de estudios y la baja calidad de las pruebas disponibles, se necesitan estudios futuros para dilucidar su mejor aplicación.
- Ventilación mecánica invasiva
Las directrices actuales de la FPI (fibrosis pulmonar idiopática) recomiendan que la mayoría de los pacientes con insuficiencia respiratoria no reciban VMI.
1.6. Seguimiento
Aunque parece que la oxigenoterapia suplementaria puede mejorar la disnea, la tolerancia al ejercicio y la calidad de vida de algunos pacientes, esto puede verse contrarrestado por problemas relacionados con el uso real del equipo de oxígeno o por barreras funcionales. Por ende, los profesionales sanitarios deben proporcionar educación, seguimiento y una monitorización adecuada de los efectos secundarios para garantizar la seguridad, la eficacia y el uso correcto del tratamiento. Además, los pacientes deben acudir a su proveedor de equipos de oxigenoterapia con cualquier pregunta o duda sobre el sistema que están utilizando.
2. LA REHABILITACIÓN CARDIORRESPIRATORIA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
2.1. Introducción
En los últimos años, la importancia de la rehabilitación respiratoria (RR) en el tratamiento de las enfermedades respiratorias ha sido exponencial, con evidencia científica contundente de los beneficios alcanzados.5
La RR constituye una parte importante del tratamiento no farmacológico de los pacientes con enfermedades pulmonares crónicas y progresivas; su eficacia se ha demostrado no únicamente en la enfermedad pulmonar obstructiva crónica (EPOC), sino también en otras enfermedades, como el asma bronquial, las bronquiectasias, la enfermedad pulmonar intersticial difusa (EPID), la hipertensión pulmonar (HTP), etc.5,6 La RR está claramente reconocida como parte del tratamiento de las enfermedades respiratorias crónicas, como demuestran las guías de práctica clínica de la EPOC7 y la guía ESC/ERS 2022 sobre el diagnóstico y el tratamiento de la HTP.8
La RR es una intervención que centra su eficacia en mejorar la disnea, la capacidad de esfuerzo y la calidad de vida relacionada con la salud (CVRS), con un alto nivel de evidencia en paciente con EPID.9
2.2. Definición
La American Thoracic Society (ATS) y la European Respiratory Society (ERS) han definido la RR como: «una intervención integral basada en una minuciosa evaluación del paciente seguida de terapias diseñadas a medida, que incluyen, pero sin limitarse a ellos, el entrenamiento muscular, la educación y los cambios en los hábitos de vida, con el fin de mejorar la condición física y psicológica de las personas con enfermedad respiratoria crónica y fomentar la adherencia a conductas para mejorar la salud a largo plazo».5 Esta definición contempla no solo la EPOC, sino también enfermedades respiratorias crónicas, incorporando una gran variedad de estrategias terapéuticas centradas en el ejercicio y la educación.10
2.3. Componentes de los programas de RR
Los componentes fundamentales de los programas de RR son el ejercicio físico asociando el entrenamiento de las extremidades superiores e inferiores, combinando el entrenamiento de fuerza con el de resistencia, la educación centrada en el conocimiento de la enfermedad y su tratamiento, la fisioterapia respiratoria; siendo aconsejable también contemplar la terapia ocupacional, el soporte psicosocial y la intervención nutricional.10 Por otro lado, el entrenamiento específico de los músculos respiratorios debería añadirse a un entrenamiento general únicamente cuando los pacientes presenten debilidad muscular respiratoria.
Para alcanzar el máximo rendimiento sin riesgos, es imprescindible una evaluación por parte del equipo de RR y una prescripción del programa totalmente personalizada según las necesidades y posibilidades de cada paciente.10 La evidencia científica ha demostrado que la RR es más eficaz si se inicia lo antes posible11 y que es importante buscar estrategias que consigan mantener sus efectos a largo plazo, fundamentalmente incidiendo en los hábitos de vida y favoreciendo la actividad física.
Los programas de RR deben ser supervisados, por simples y autónomos que sean. Generalmente, se llevan a cabo en el medio hospitalario, ya sea en el paciente hospitalizado o en régimen ambulatorio, aunque se pueden alcanzar beneficios similares cuando se realizan en el domicilio.12
El consenso de la ATS/ERS sugiere que el mantenimiento de los beneficios a lo largo del tiempo tiene una relación directa con la duración del programa y recomienda un mínimo de 20 sesiones de entrenamiento, o bien de 8 a 12 semanas de programa.10
La terapia suplementaria con oxígeno durante el entrenamiento al ejercicio debe ser considerada en 2 situaciones: los pacientes que presentan hipoxemia en reposo (portadores de oxigenoterapia) y los que presentan una saturación de la oxihemoglobina (SpO2) <90% al esfuerzo.5
2.4. Rehabilitación respiratoria y enfermedad pulmonar intersticial difusa
En pacientes con EPID, la RR constituye una parte importante del tratamiento no farmacológico.13 La disminución de la capacidad de ejercicio constituye una característica cardinal de las EPID y a menudo está vinculada con disnea de esfuerzo, la cual puede manifestarse de forma significativa.5 Los individuos que presentan una mayor reducción de la capacidad de ejercicio presentan una peor CVRS.14 Además, se ha observado que una capacidad de ejercicio deficiente se asocia con una supervivencia más desfavorable.15 En la actualidad, la evidencia disponible en algunas EPID recomienda la RR como tratamiento efectivo en la mejoría de los síntomas y seguro.5
Dado que las EPID son un grupo heterogéneo de enfermedades y que algunas de ellas progresivas, la certeza de la evidencia sobre RR en EPID es moderada, debido a la es-
casez de ensayos clínicos randomizados. No obstante, las guías clínicas actuales hacen una recomendación alta para realizar RR en las EPID.5
Evidencia reciente ha mostrado que la distancia recorrida en 6 minutos (PM6M) aumentó después de la RR en comparación con el grupo de control en cerca de 40 metros, manteniéndose los efectos en el seguimiento de 6 a 12 meses.9 Los resultados también han sido favorables en otras evaluaciones de la capacidad de ejercicio, como el trabajo máximo o el consumo de oxígeno pico.9 Los resultados de la calidad de vida y de mejoría de la disnea han sido similares. En cuanto a hospitalización o mortalidad, no se han observado efectos al realizar RR.9
2.5. Fibrosis pulmonar idiopática (FPI), asbestosis, EPID asociada a conectivopatías y otras EPID
El grupo de Vainshelboim et al. demostró los beneficios de la RR en pacientes con FPI, obteniendo resultados significativos en la tolerancia al ejercicio, fuerza de miembros inferiores, disnea y calidad de vida.16
En el ECA realizado por el Dowman et al,,17 que incluía a pacientes con FPI, asbestosis, EPID relacionada con enfermedades del tejido conectivo y otras EPID, se analizan la eficacia de un programa de rehabilitación. Los resultados muestran que se producen mejoras mayores en la prueba de la marcha de los 6 minutos (PM6M), los cuestionarios de CVRS y la disnea en asbestosis y FPI en comparación con EPID relacionada con enfermedades del tejido conectivo, sin alcanzar significación estadística. Tras 6 meses de tratamiento, el grupo que obtuvo mayor beneficio fue el que tenía más síntomas (sobre todo disnea) y aquellos que habían iniciado el programa de rehabilitación con una afectación funcional moderada (PM6M inicial más baja). Se observaron mayores ganancias en aquellos cuya prescripción de ejercicio progresó con éxito según el protocolo. A los 6 meses, las mejoras sostenidas en el PM6M y los síntomas se asociaron con una mejor función pulmonar inicial y menos HTP. Por tanto, concluyen que el entrenamiento con ejercicios es eficaz en pacientes de todo tipo de EPID, con beneficios clínicamente significativos en la asbestosis y la FPI. La progresión exitosa del ejercicio maximiza las mejoras y los efectos sostenidos del tratamiento y favorecen a los pacientes con enfermedad más leve.10 Los autores resaltan la necesidad de iniciar programas de rehabilitación de manera precoz en el curso de la enfermedad, para que los beneficios obtenidos se mantengan en el tiempo.
Holland et al.18 también demostraron en su ECA que en los pacientes con FPI y otras EPID diferentes a FPI los programas de RR mejoran el PM6M, la puntuación de la disnea y los CRDQ, pero estos beneficios no se mantienen en el tiempo si no se continúa con un entrenamiento físico frecuente.
2.6. Sarcoidosis
El grupo de trabajo de Alsina-Restoy et al.19 ha publicado recientemente una revisión sistemática de RR en sarcoidosis. Un total de 5 artículos y 184 pacientes fueron analizados. Los programas de RR incluían entrenamiento de los músculos inspiratorios, programa de incentivo a la actividad física, telerehabilitación y ejercicio de componentes múltiples. Esta revisión sistemática encontró una mejora significativa de la capacidad de ejercicio y de la disnea en pacientes con sarcoidosis.19
Otros estudios, como el de Guber et al.20, qué valoró la RR en pacientes con diferentes etapas de gravedad de sarcoidosis, realizaron un programa de RR de 12 semanas en 52 sujetos, valoraron la capacidad máxima de ejercicio definida como VO2max mediante la prueba de ejercicio cardiopulmonar, la función pulmonar, el PM6M, cuestionarios de CVRS, antes y después de 6 meses de seguimiento de RR. El programa de RR aumentó significativamente el VO2máx, también mejoraron las puntuaciones de las escalas de la disnea y los cuestionarios de CVRS. El impacto de la RR sobre el VO2máx fue más pronunciado en sujetos con afectación del parénquima pulmonar. El aumento del VO2máx se correlacionó con la gravedad inicial de la enfermedad. Los sujetos con FEV1/FVC <70% y un KCO superior al 80% previsto mostraron una mayor mejoría en TM6M. A los 6 meses de seguimiento, las puntuaciones de VO2max, TM6M y cuestionarios de calidad de vida se mantuvieron estables, lo que sugiere efectos duraderos de la RR.
2.7.
Recomendaciones
El entrenamiento aeróbico es esencial en la rehabilitación pulmonar, utilizando principalmente la bicicleta ergométrica y la cinta rodante, aunque también se pueden emplear otros métodos como la caminata o el uso de escaleras.10 Se siguen las recomendaciones del American College of Sports Medicine (ACSM) para programas de 3 a 5 sesiones semanales de 20 a 60 minutos con intensidad moderada a alta, aumentando gradualmente la duración e intensidad.21 En los casos en que los pacientes no pueden alcanzar intensidades altas debido a síntomas como disnea o fatiga, se prefieren entrenamientos interválicos de alta intensidad.22
Es importante comenzar las primeras sesiones de entrenamiento centrándose en la seguridad y la comodidad del paciente, especialmente para los sedentarios o con poca experiencia en ejercicio. Se pueden usar índices subjetivos, como el índice de Borg, para prescribir ejercicio, evitando el uso de la frecuencia cardíaca en pacientes con alteraciones del sistema nervioso autónomo o comorbilidades cardiovasculares. Durante el ejercicio, se debe monitorear la saturación de oxígeno y la frecuencia cardíaca para garantizar la seguridad, utilizando oxígeno suplementario si es necesario.5 Se recomienda aumentar progresivamente la carga de trabajo cada dos o tres sesiones para mejorar el rendimiento del entrenamiento.
2.8. Conclusión
Las EPID son un grupo heterogéneo de enfermedades. La evidencia disponible muestra que se pueden realizar con seguridad programas de RR basados en ejercicio físico, educación, fisioterapia respiratoria, terapia ocupacional, soporte psicosocial e intervención nutricional. Los pacientes incluidos en estos programas conseguirán mejorar la capacidad de ejercicio, la disnea y la CVRS. Por eso, la RR debe formar parte del tratamiento no farmacológico de la EPID.
BIBLIOGRAFÍA
1
1. Jacobs SS, Krishnan JA, Lederer DJ, Ghazipura M, Hossain T, Tan AYM, et al. Home Oxygen Therapy for Adults with Chronic Lung Disease. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;202(10):121-141.
2. Khor YH, Gutman L, Abu Hussein N, Johannson KA, Glaspole IN, Guler SA, et al. Incidence and Prognostic Significance of Hypoxemia in Fibrotic Interstitial Lung Disease. Chest. 2021;160(3):994-1005.
3. Lim RK, Humphreys C, Morisset J, Holland AE, Johannson KA. Oxygen in patients with fibrotic interstitial lung disease: an international Delphi survey. Eur Respir J. 2019;54(2):1900421.
4. Cordeiro R, Nunes A, Smith O, Renzoni EA. Oxygen in interstitial lung diseases. Breathe. 2023;19(1):220271.
5. Rochester CL, Alison JA, Carlin B, Jenkins AR, Cox NS, Bauldoff G, Bhatt SP, et al. Pulmonary Rehabilitation for Adults with Chronic Respiratory Disease: An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2023;208(4):7-26.
6. Osadnik CR, Gleeson C, McDonald VM, Holland AE. Pulmonary rehabilitation versus usual care for adults with asthma. Cochrane Database Syst Rev. 2022;8(8):CD013485.
7. Venkatesan P. GOLD COPD report: 2024 update. Lancet Respir Med. 2024;12(1):15-16. doi: 10.1016/S2213-2600(23)00461-7, PMID: 38061380.
8. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, et al. ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61(1):2200879.
9. Dowman L, Hill CJ, May A, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst Rev. 2021;2(2):CD006322.
10. Spruit MA, Singh SJ, Garvey Ch, ZuWallack R. Nici L., Rochester C, et al. An Official American Thoracic Society/European Respiratory Society Statement: Key concepts and Advances in Pulmonary Rehabilitation. Am J Respir Crit Care Med. 2013;188(8):13-64, doi: 10.1164/ rccm.201309-1634ST.
11. Lu HY, Chen CF, Lee DL, Tsai YJ, Lin PC. Effects of Early Pulmonary Rehabilitation on Hospitalized Patients with Acute Exacerbation of Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis. Int J Chron Obstruct Pulmon Dis. 2023;18:881-893.
12. Stafinski T, Nagase FI, Avdagovska M, Stickland MK, Menon D. Effectiveness of home-based pulmonary rehabilitation programs for patients with chronic obstructive pulmonary disease (COPD): systematic review. BMC Health Serv Res. 2022;22(1):557.
13. Wytrychowski K, Hans-Wytrychowska A, Piesiak P, Majewska-Pulsakowska M, Rożek-Piechura K. Pulmonary rehabilitation in interstitial lung diseases: A review of the literature. Adv Clin Exp Med. 2020;29(2):257-264.
14. Chang JA, Curtis JR, Patrick DL, Raghu G. Assessment of health-related quality of life in patients with interstitial lung disease. Chest. 1999;116(5):1175-1182.
15. Lederer DJ, Arcasoy SM, Wilt JS, D'Ovidio F, Sonett JR, Kawut SM. Six-minute-walk distance predicts waiting list survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(6):659-664.
16. Vainshelboim B, Oliveira J, Fox BD, Soreck Y, Fruchter O, Kramer MR. Long-Term Effects of a 12-Week Exercise Training Program on Clinical Outcomes in Idiopathic Pulmonary Fibrosis. Lung. 2015;193(3):345-354.
17. Dowman LM, McDonald CF, Hill C, Lee AL, Barker K, Boote Cl, Glaspole A, et al. The evidence of benefits of exercise training in interstitial lung disease: a randomized controlled trial. Thorax. 2017;72(7):610-619.
18. Holland a E, Hill CJ, Conron M, Munro P, McDonald CF. Short term improvement in exercise capacity and symptoms following exercise training in interstitial lung disease. Thorax. 2008;63(6):549-554.
19. Alsina-Restoy X, Torres-Castro R, Caballería E, Gimeno Santos E, Solis-Navarro L, Francesqui J, Hernandez-Gonzalez F, Ramos-Casals M, Blanco I, Sellarés J. Pulmonary rehabilitation in sarcoidosis: A systematic review and meta-analysis. Respiratory Medicine. 2023;219:107432, https://doi.org/10.1016/j.rmed.2023.107432.
20. Guber E, Wand O, Epstein Shochet G, Romem A, Shitrit D. The short- and longterm impact of pulmonary rehabilitation in subjects with sarcoidosis: a prospective study and review of the literature, Respiration. 2021;100:423-431, https://doi. org/10.1159/000514917.
21. American College of Sports Medicine. ACSM's guidelines for exercise testing and prescription. Lippincott Williams & Wilkins, 2013.
22. Sawyer A, Cavalheri V, Hill K. Effects of high intensity interval training on exercise capacity in people with chronic pulmonary conditions: a narrative review. BMC SportsSciMedRehabil. 2020;12:22.
2
1. Ventura V, Viani M, Bianchi F, D’Alessandro M, Sestini P, Bargagli E. Effect of Ambulatory Oxygen on the Respiratory Pattern during the 6 Min Walking Test in Patients with Interstitial Lung Diseases. Biomedicines. 2023;11(7):1834.
2. Viani M, Ventura V, Bianchi F, D’Alessandro M, Bergantini L, Sestini P, et al. Oxygen Therapy during Exercise in Patients with Interstitial Lung Diseases. Biomolecules. 2022;12(5):717.
3. Khor YH, Smith DJF, Johannson KA, Renzoni E. Oxygen for interstitial lung diseases. Current Opinion in Pulmonary Medicine. 2020;26(5):464-469.
4. Faverio P, De Giacomi F, Bonaiti G, Stainer A, Sardella L, Pellegrino G, et al. Management of Chronic Respiratory Failure in Interstitial Lung Diseases: Overview and Clinical Insights. Int J MedSci. 2019;16(7):967-980.
5. Faverio P, De Giacomi F, Sardella L, Fiorentino G, Carone M, Salerno F, et al. Management of acute respiratory failure in interstitial lung diseases: overview and clinical insights. BMC Pulm Med. 2018;18:70.
6. Cuerpo S, Palomo M, Francesqui J, Alsina X, Hernández C, et al. Home Oxygen Monitoring in Patients with Interstitial Lung Disease. 2022;19(3):493-497.
7. Oishi K, Matsunaga K, Asami-Noyama M, Yamamoto T, Hisamoto Y, Fujii T, et al. The 1-minute sit-to-stand test to detect desaturation during 6-minute walk test in interstitial lung disease. NPJ Prim Care Respir Med. 2022;32:5.
8. Khor YH, Gutman L, Abu Hussein N, Johannson KA, Glaspole IN, Guler SA, et al. Incidence and Prognostic Significance of Hypoxemia in Fibrotic Interstitial Lung Disease. Chest. 2021;160(3):994-1005.
9. Alfieri V, Crisafulli E, Visca D, Chong WH, Stock C, Mori L, et al. Physiological predictors of exertional oxygen desaturation in patients with fibrotic interstitial lung disease. Eur Respir J. 2020;55(2):1901681.
10. Schaeffer MR, Molgat-Seon Y, Ryerson CJ, Guenette JA. Supplemental oxygen for the management of dyspnea in interstitial lung disease. Current Opinion in Supportive & Palliative Care. 2019;13(3):174-178.
11. Visca D, Mori L, Tsipouri V, Fleming S, Firouzi A, Bonini M, et al. Effect of ambulatory oxygen on quality of life for patients with fibrotic lung disease (AmbOx): a prospective, open-label, mixed-method, crossover randomised controlled trial. The Lancet Respiratory Medicine. 2018;6(10):759-770.
12. Carpagnano GE. Supplementary oxygen in healthy subjects and those with COPD increases oxidative stress and airway inflammation. Thorax. 2004;59(12):1016-1019.
TABLAS Y FIGURAS
Tabla I. Resumen indicaciones de oxigenoterapia ambulatoria en EPID

Abreviaturas: OTLD: oxigenoterapia de larga duración: >15 horas; SpO2: saturación capilar periférica de oxígeno; HTP: hipertensión pulmonar; PaO2: presión arterial de oxígeno. Basado en referencias bibliográficas 1 y 3.
Tabla II. Pruebas diagnósticas para evaluar la saturación al esfuerzo en EPID

Abreviaturas: PM6M: prueba de marcha de 6 minutos; prueba de sedestación y bipedestación durante 1 minuto (1STS siglas en inglés); SpO2: saturación capilar periférica de oxígeno; DLCO: prueba de difusión de monóxido de carbono. Basado en referencia bibliográfica 4.
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES
INTERSTICIALES DIFUSAS
TRATAMIENTOS FARMACOLÓGICOS: ANTIFIBRÓTICOS E INMUNOSUPRESORES
(DOSIS Y EFECTOS SECUNDARIOS)
Autoras
Ana Belén Llanos González
Nuria Albacar Ingla
Coordinador
Orlando Acosta Fernández
1. INTRODUCCIÓN
Las enfermedades pulmonares intersticiales difusas (EPID) configuran un amplio grupo de enfermedades respiratorias de naturaleza diversa y etiopatogenia compleja en las que coexisten, en intensidad variable, fenómenos inflamatorios y fibróticos pulmonares que, si bien inciden especialmente en el espacio intersticial pulmonar, también pueden afectar a la vía aérea y a la vasculatura. El tratamiento de este amplio espectro de enfermedades, cuyos comportamientos evolutivos y respuestas terapéuticas son heterogéneos, se ha basado, hasta el momento actual, en el uso de fármacos con acción antiinflamatoria-inmunomoduladora y medicación antifibrótica.
A continuación, se repasarán los estudios que avalan el uso de estos fármacos y algunos aspectos terapéuticos del uso de antifibróticos e inmunosupresores en las principales EPID.
2. ANTIFIBRÓTICOS
2.1 Ensayos que avalan su uso
En 2014, la FDA aprobó los antifibróticos pirfenidona y nintedanib para el tratamiento de la fibrosis pulmonar idiopática (FPI), que demostraron reducir la disminución de la capacidad vital forzada (FVC) y el riesgo de exacerbación aguda y de hospitalizaciones 1,2, tal y como pusieron de manifiesto los ensayos clínicos de fase III INPULSIS-1 e INPULSIS-2 realizados con nintedanib versus placebo y los estudios CAPACITY y ASCEND con pirfenidona versus placebo3,4
Los estudios CAPACITY y ASCEND fueron ensayos en fase III doble ciego randomizados y controlados con placebo para valorar la eficacia y la seguridad de la pirfenidona frente a placebo en pacientes con FPI y deterioro funcional leve-moderado que demostraron que el fármaco de ensayo era capaz de reducir el deterioro que experimentaban estos pacientes a lo largo del tiempo y la supervivencia libre de progresión. Cuando se analizaban de forma conjunta los datos arrojados por ambos ensayos, se demostró igualmente que los pacientes bajo tratamiento con pirfenidona presentaban una menor mortalidad a las 52 semanas que los que recibían placebo.
Los estudios INPULSIS fueron también ensayos randomizados, doble ciego y controlados con placebo en los que se valoró la eficacia y la seguridad del nintedanib en pacientes con FPI cuya FVC era superior al 50%. En estos estudios, se puso de manifiesto que el fármaco de estudio era capaz de reducir el deterioro funcional que sufren estos pacientes a lo largo del tiempo y de reducir la probabilidad de exacerbaciones. Cuando se analizaron de forma conjunta los datos de los estudios INPULSIS junto a su precedente en fase II (estudio TOMORROW), se apreció que el nintedanib podía reducir el riesgo asociado de mortalidad.
Posteriores ensayos clínicos controlados han demostrado que el nintedanib es también eficaz en el tratamiento de las EPID fibróticas con carácter progresivo y en la EPID secundaria a esclerosis sistémica (ES-EPID), al compartir vías patogénicas y biológicas similares a la FPI. Entre estos ensayos clínicos, destacan el SENSCIS5 y el INBUILD6 . El ensayo SENSCIS fue un ensayo amplio que evaluó la eficacia del nintedanib frente a placebo en pacientes con EPID secundaria a esclerosis sistémica (ES-EPID), y reconoció que el grupo de tratamiento redujo significativamente menos la pérdida de FVC que el grupo placebo a lo largo del año de estudio. De forma similar, el ensayo INBUILD, realizado en este caso con pacientes con EPID fibrosantes progresivas no FPI, permitieron demostrar igualmente un efecto significativamente protector de la caída de FVC a lo largo del año de estudio frente a placebo. La reducción de la disminución anual de la FVC fue similar a la tasa observada en los ensayos INPULSIS, lo que apoya un efecto biológico similar. Los pacientes con EPID fibróticas no FPI incluidos en el estudio
fueron clasificados en cinco grupos: neumonitis por hipersensibilidad, neumonía intersticial idiopática no específica, EPID inclasificable, EPID relacionadas con enfermedades autoinmunes y otras EPID fibrosantes. En un análisis post-hoc, no se observaron diferencias significativas en cuanto a eficacia entre los subgrupos de EPID incluidos en el ensayo.
En cuanto a la pirfenidona, se han realizado varios ensayos clínicos que han pretendido valorar su eficacia en EPID no FPI, si bien la calidad de evidencia es menor. Entre los principales estudios, destacan el UILD, el RELIEF y el TRAIL1, que se comentan a continuación6 .
El ensayo UILD, realizado en pacientes con EPID fibrótica progresiva inclasificables, pretendió demostrar la eficacia del fármaco respecto a placebo haciendo uso de espirometrías domiciliarias. El análisis estuvo limitado por la variabilidad intraindividual en la espirometría domiciliaria, aunque haciendo uso de FVC de espirometría hospitalaria (variable secundaria) pudo demostrarse una reducción de la disminución de la FVC en el grupo de tratamiento.
El ensayo RELIEF se realizó sobre pacientes con EPID fibróticas con deterioro funcional progresivo a pesar del tratamiento convencional, y si bien se advirtieron indicios de un menor deterioro en la FVC en el grupo de estudio respecto a placebo, el ensayo debió ser finalizado prematuramente por el lento reclutamiento y por la falta de poder estadístico, de modo que estos resultados deben interpretarse con cautela.
Por último, el ensayo TRAIL1 sobre pacientes con EPID y artritis reumatoide (AR) comparó el efecto sobre la evolución de FVC haciendo uso de pirfenidona o placebo, sin mostrar diferencias significativas entre los dos grupos. Este estudio también debió pararse antes de lo previsto por la lentitud del reclutamiento y la pandemia de COVID-19.
Del mismo modo, se han llevado a cabo otros estudios con cohortes prospectivas y unicéntricas en EPID asociadas a enfermedades autoinmunes (ES-EPID) e incluso con pacientes afectados por el síndrome de Hermansky-Pudlak, en los que se ha evaluado la variación de la FVC en los tratados con pirfenidona versus placebo, sin llegar a resultados concluyentes7
Por último, también se han llevado a cabo ensayos clínicos aleatorizados en los que se investigó el tratamiento antifibrótico combinado con nintedanib y pirfenidona versus nintedanib en monoterapia (INJOURNEY), en los que se ha destacado la seguridad y la tolerabilidad en el grupo de terapia combinada. (Tabla I).
2.2. Antifibróticos. Aspectos terapéuticos
NINTEDANIB
El nintedanib es un inhibidor triple de receptores de tirosinquinasa con actividad antifactor de crecimiento derivado de plaquetas, antifactor de crecimiento de fibroblastos y
antifactor de crecimiento vascular endotelial, todos ellos relacionados con mecanismos profibrogénicos. Está indicado en adultos con FPI, EPID fibrosantes progresivas y ESEPID.
La dosis recomendada de nintedanib es de 150 mg cada 12 horas administrada por vía oral, siendo la diarrea el efecto adverso más frecuente, cuya intensidad suele ser de leve a moderada; en no más del 5% de los casos llega a ser de intensidad tal que obliga a la retirada del tratamiento. La reducción de dosis a 100 mg cada 12 horas es una opción clínica válida hasta el control de síntomas, momento en que deberá intentarse restablecer la dosis óptima. El uso de antidiarreicos como la loperamida y la introducción en la dieta de harina de algarroba puede minimizar la clínica señalada. Otros efectos adversos reseñables con nintedanib son la hiporexia y la pérdida de peso, náuseas, vómitos y alteraciones de la función hepática.
Las principales contraindicaciones del nintedanib son la hipersensibilidad al fármaco, al cacahuete, a la soja o algún excipiente (triglicéridos de cadena media, grasa dura, lecitina (soja) [E322], gelatina, glicerol (85 %), dióxido de titanio [E171], óxido de hierro rojo [E172], óxido de hierro amarillo [E172])8. Asimismo, se debe tener precaución con su empleo en pacientes con diátesis hemorrágica, insuficiencia hepática grave, anticoagulación crónica o cirugía abdominal reciente.
La administración conjunta con ketoconazol, eritromicina o ciclosporina aumenta la exposición al nintedanib. Por el contrario, el uso conjunto con rifampicina, carbamazepina o fenitoína la reduce. Debe evitarse su uso durante el embarazo o quedar embarazada hasta tres meses después de la suspensión del fármaco ante la potencialidad de daño fetal.
PIRFENIDONA
La pirfenidona es un fármaco con propiedades antiinflamatorias y antifibróticas que inhibe la proliferación fibroblástica y la síntesis de citoquinas y de proteínas profibrogénicas. Está indicada en pacientes adultos con FPI.
La dosis de pirfenidona utilizada en los ensayos pivotales fue de 2403 mg/día divididos en tres tomas que deberán ser coincidentes con las comidas. Los efectos adversos suelen ser de grado leve, y en no más del 5% de los casos hay la necesidad de retirar el fármaco. Los efectos adversos más frecuentes son de tipo gastrointestinal, fundamentalmente dispepsia, náuseas, anorexia y pérdida de peso, así como rash cutáneo por fotosensibilidad y alteraciones de función hepática. De igual modo que con el nintedanib, si las circunstancias clínicas lo exigieran, la dosis puede reducirse o suspenderse transitoriamente hasta que cesen o se minimicen los efectos adversos, momento en que deberá reconsiderarse una escalada hasta dosis plena.
El uso de procinéticos o inhibidores de la bomba de protones puede ayudar a controlar dichos síntomas. Hay que tener presente que el omeprazol interactúa con la pirfenidona, por lo que se aconseja el uso de pantoprazol o esomeprazol.
Para minimizar las reacciones cutáneas debidas a la fotosensibilidad que produce, se recomienda el uso de cremas solares con alto nivel de protección y cubrir, en lo posible, las zonas más expuestas con la vestimenta o complemento apropiado.
Las principales contraindicaciones para el uso de pirfenidona son la hipersensibilidad al fármaco o sus excipientes, el uso concomitante de fluvoxamina y la hepatopatía o nefropatía grave y el antecedente de angioedema con el fármaco9. No hay datos sobre su uso en embarazadas. La administración conjunta con ciprofloxacino, amiodarona, propafenona, fluconazol, fluoxetina o paroxetina aumentan la exposición de la pirfenidona, por lo que se recomienda hacer un ajuste a la baja en la dosificación de la misma. Por el contrario, el uso de rifampicina o el uso concomitante de tabaco reducen su eficacia. El consumo de zumo de pomelo debe evitarse durante su uso.
2.3. Ensayos clínicos con antibróticos en curso
Actualmente, hay varios ensayos clínicos en curso para evaluar el empleo de los antifibróticos en las EPID6. Destacan los que evalúan la eficacia del nintedanib en pacientes con neumoconiosis (NCT05067517, NCT04161014), miositis (NCT05335278), linfangioleiomiomatosis (NCT03062943) e incluso con bronquiolitis obliterante (BOS) tras un trasplante alogénico de células hematopoyéticas (ensayo NINBOST2018, NCT03805477) o para evaluar la eficacia del antifibrótico sobre la reducción del FEV1 en pacientes con BOS tras un trasplante pulmonar (ensayo INFINITx-BOS, NCT03283007).
Por otra parte, hay cuatro ensayos clínicos en curso para evaluar la eficacia en pacientes post-COVID-19 con alteraciones intersticiales tras la infección (NCT04856111, NCT04619680, NCT04541680 y NCT04338802).
A su vez, aunque la pirfenidona sólo ha sido aprobada para el tratamiento de la FPI, existen varios ensayos clínicos con dicho antifibrótico en diferentes EPID, como, por ejemplo, los afectados por neumoconiosis, asbestosis, enfermedad de los trabajadores del carbón y silicosis (NCT05288179, NCT0513345, NCT05118256 y NCT04461587).
También hay tres ensayos en pacientes con neumonitis por hipersensibilidad (NCT02958917, NCT04675619, NCT02496182), uno que evalúa la eficacia de este antifibrótico en el síndrome de Hermansky-Pudlak (NCT04193592), varios que estudian sus efectos en las EPID asociadas a enfermedad autoinmune ((NCT05505409, NCT04928586, NCT03857854, NCT03856853, NCT03221257 y NCT03385668) y en pacientes con fibrosis post-COVID19 (NCT04607928), entre otros. Al mismo tiempo, tal como ocurre con el nintedanib, existen ensayos que evalúan la seguridad y la tolerabilidad de la pirfenidona en pacientes con BOS (ensayo STOP-BOS NCT03315741 tras un
trasplante de células hematopoyéticas y estudio EPOS NCT02262299 en receptores de trasplante de pulmón con BOS).
3. INMUNOSUPRESORES
3.1 Introducción
En el abordaje de las EPID, se presenta una diversidad de opciones terapéuticas inmunomoduladoras, especialmente en las distintas FPI debido a su componente inflamatorio. En estas, la inmunosupresión emerge como el pilar fundamental del tratamiento, con niveles variables de evidencia para diversos agentes inmunomoduladores, cada uno adaptado según las diferentes condiciones de cada EPID específica.
Antes de prescribir un tratamiento inmunosupresor, es crucial evaluar la naturaleza y la gravedad de la enfermedad, considerar el perfil individual del paciente, incluidas las comorbilidades y los factores de riesgo. Un enfoque personalizado, basado en criterios de clasificación y monitorización continua contribuye a la seguridad y eficacia del tratamiento.
3.2 Aspectos generales a considerar para prescribir cualquier inmunosupresor
El principal riesgo que existe al prescribir cualquier tratamiento inmunosupresor son las infecciones, por lo que es imperativo realizar una evaluación inmunológica basal del paciente. Este proceso incluye la verificación de la ausencia de riesgos previos de la inmunidad, así como la detección de cualquier infección activa o latente que podría intensificarse al inducir inmunodepresión.
Según el grado de inmunosupresión que generemos, hay que tener en cuenta la exclusión previa de tuberculosis, la evaluación de la presencia de virus de la hepatitis B o C o comprobar la inmunidad frente el virus de la varicela zóster, entre otras posibles infecciones oportunistas. Además, se debe garantizar que el calendario de vacunación del paciente esté al día, a fin de minimizar los riesgos asociados a la supresión inmunológica. Cuando se utilizan dos o más agentes durante un periodo superior a dos meses, generalmente se proporciona profilaxis contra la neumonía por Pneumocystis jirovecii (PjP).
Por otro lado, hay que tener presente los potenciales efectos adversos organoespecíficos según el tratamiento que estemos manejando. La vigilancia estrecha con análisis de sangre nos permitirá la detección temprana de estos problemas y así poder hacer ajustes en el tratamiento antes de que se desarrollen complicaciones más graves, así como garantizar la seguridad y la eficacia del tratamiento.
A continuación, se describen los mecanismos de acción de las diferentes terapias inmunomoduladoras usadas más frecuentemente, analizando sus indicaciones, dosis, efectos adversos y consideraciones a tener en cuenta al prescribirlos (Tabla II).
3.3 Corticosteroides
Mecanismo de acción: Inhiben la expresión de genes proinflamatorios y modulan las respuestas inmunitarias.
Indicaciones: Son en la mayoría de las guías clínicas el tratamiento de primera línea en gran parte de las EPID no FPI (Tabla II)10. Además, también son el tratamiento de elección de las EPID agresivas con insuficiencia respiratoria hipoxémica grave con opacidades en vidrio deslustrado difuso bilateral o en las exacerbaciones de cualquier neumopatía fibrosante.
Dosis: La presentación más utilizada vía oral es la prednisona. Sus dosis varían según la enfermedad específica y la gravedad de condición. Una aproximación común es iniciar prednisona oral a 0,5-1 mg/kg durante un periodo limitado para lograr mejoría o al menos estabilización de la enfermedad, y luego, 8-10 semanas después, introducir un agente ahorrador de esteroides para evitar efectos secundarios a largo plazo. La prednisona se reduce gradualmente a una dosis menor y, eventualmente, se suspende por completo si se logra la estabilización. Las dosis pueden variar desde 5 mg de prednisona como tratamiento de mantenimiento hasta dosis de 10 mg/kg de metilprednisolona endovenosa (ev) durante 3 días consecutivos en caso de exacerbaciones graves.
Efectos adversos: Dependen de la dosis y el tiempo de exposición del fármaco. Es importante tener en cuenta que pueden ocasionar osteoporosis, hipertensión, aumento de peso, hiperglicemia, trastornos psiquiátricos y trastornos del sueño, entre otros.
Consideraciones: Su duración debe ser lo más corta posible debido a los efectos adversos a largo plazo. Si se prevé un tratamiento largo, hay que considerar suplementos de calcio y/o vitamina D o bifosfonatos para prevenir la osteoporosis, la indicación de insulinoterapia o de antihipertensivos, además de valorar la necesidad de profilaxis para la prevención de la PjP (en el caso de requerir prednisona o equivalente con dosis de ≥ 60 mg/día o incluso de ≥ 25 mg/día de manera prolongada). Se recomienda administrarlos a primera hora del día.
3.4
Micofenolato de mofetilo (MMF)
Mecanismo de acción: Citostático sobre linfocitos T y B.
Indicaciones: Se suele utilizar como fármaco para ahorrar altas dosis de corticoides de manera prolongada y así poder evitar sus efectos adversos a largo plazo. Sin embargo, cada vez más, este se inicia desde el principio, especialmente cuando la enfermedad es grave. Destaca su empleo en las ES-EPID donde se establece la eficacia de la monoterapia y donde los corticoticosteroides pueden causar una crisis renal (Tabla III).10,11
Dosis: La habitual está entre 1-1,5 g/12h, aunque las dosis pueden variar según la tolerancia y la respuesta del paciente. En presencia de insuficiencia renal grave, se aconseja evitar dosis superiores a 1 g/12h.
Efectos adversos: Los más frecuentes son los gastrointestinales (náuseas, diarreas, dolor abdominal). Otros frecuentes son: taquicardia, afectación de la tensión arterial, toxicidad hematológica, hepatopancreática o renal. A mayor dosis o duración del tratamiento, mayor riesgo de tumores malignos (en especial melanomas y linfomas).
Consideraciones: Requiere especial precaución su uso en paciente con infecciones o neoplasias activas. Si hay alto riesgo de melanoma, hay que evitar la exposición directa a la luz solar.
3.5 Azatioprina (AZA)
Mecanismo de acción: Supresión de los linfocitos T y, en menor medida, los B.
Indicaciones: Se suele utilizar como fármaco ahorrador de corticoides con las mismas consideraciones que el MMF. Se utiliza principalmente en combinación con corticoides como tratamiento de primera línea de las EPID relacionadas con enfermedades del tejido conectivo (CTD), entre otras (Tabla III)10,11. Al igual que el MMF, puede utilizarse sin prednisona cuando se haya confirmado la ausencia de progresión rápida o cuando existan contraindicaciones significativas para el uso de esteroides.
Dosis: La típica es de 1-3 mg/kg/24h administrada en dosis divididas. Estas varían según la respuesta del paciente y su tolerabilidad.
Efectos adversos: Gastrointestinales, depresión medular, hepatotoxicidad, erupción cutánea, síndrome de hipersensibilidad, riesgo aumentado de neoplasias, como el cáncer de piel, entre otros.
Consideraciones: Antes de prescribirla, se aconseja repasar detenidamente el tratamiento habitual para descartar potenciales interacciones. Está contraindicada en caso de insuficiencia hepática grave. Se recomienda evitar la exposición solar sin protección.
3.6 Inhibidores de la calcineurina: ciclosporina y tacrolimus
Mecanismo de acción: Bloquean la acción de los linfocitos T.
Indicaciones: De segunda línea en la EPID asociada a Sjögren con deterioro moderado-grave12 .
Dosis: De ciclosporina: 2,5-5 mg/kg/12h. De tacrolimus: 0,075-0,15 mg/kg/24h, debe ajustarse la dosis óptima evaluando sus niveles en sangre.
Efectos adversos: De ciclosporina: nefrotoxicidad principalmente, neurotoxicidad, hipertensión, hiperglucemia, hirsutismo e hiperplasia gingival. De tacrolimus: nefrotoxicidad, neurotoxicidad, hipertensión, hiperglucemia, entre otros. También aumentan el riesgo de tumores malignos.
Consideraciones: Las contraindicaciones para la ciclosporina son la insuficiencia renal o la hipertensión arterial no controlada, así como el uso de otros inmunosupresores o radioterapia. Con el tacrolimus, hay que tener especial atención en insuficiencia renal o hepática graves. Hay que evitar la exposición solar sin protección.
3.7 Metotrexato (MTX)
Mecanismo de acción: Antagonista del ácido fólico que inhibe la replicación celular.
Indicaciones: Segunda línea en sarcoidosis y EPID asociadas a artritis reumatoide (AREPID) (Tabla III).10,13,14
Dosis: Se administran vía parenteral entre 7,25-25 mg/semana.
Efectos adversos: Son más frecuentes con tratamientos prolongados, y la mayoría reversibles si se detectan tempranamente. Se han descrito síntomas como estomatitis o úlcera bucal (las primeras 24-48h), síntomas gastrointestinales, hepatotoxicidad, astenia, toxicidad medular, entre otros. La toxicidad pulmonar puede cursar con disnea o tos, pero es poco frecuente.
Consideraciones: Hay que administrar ácido folínico en la primera hora tras la administración de MTX, ya que neutraliza los efectos tóxicos inmediatos en el sistema hematopoyético. Las dosis de ácido folínico dependerán de las de MTX. Está contraindicado en pacientes con niveles de bilirrubina >5 mg/dl y aclaramiento de creatinina <20 ml/min; si este está entre 20-50 ml/min, hay que administrar el 50% de la dosis recomendada.
3.8 Ciclofosfamida (CYC)
Mecanismo de acción: Supresor de la proliferación de células T y B.
Indicaciones: Fármaco de segunda o tercera línea (Tabla III). Uso especialmente en casos graves, progresivos y que requieran hospitalización.
Dosis: La dosis es variable y se indica en intervalos específicos según su vía de administración: 1-2 mg/kg/día oral (mensual) o 500-750 mg/m2 ev (mensual) sin exceder los 20 g, ya que aumenta el riesgo de cáncer de vejiga. Aun así, es inusual superar los 12 g en el tratamiento de las EPID.
Efectos adversos: El uso oral a largo plazo se asocia a mayores efectos adversos. Los más frecuentes son síntomas gastrointestinales, toxicidad en la médula ósea (sobre todo leucopenia o neutropenia, siendo el valor nadir 10-14 días después de un pulso ev; estos niveles pueden responder con el ajuste de la dosis), nefro y hepatotoxicidad, cistitis hemorrágica, aumento del riesgo de neoplasias, toxicidad gonadal, entre otros.
Consideraciones: Se prefiere el uso endovenoso en lugar del oral dado el perfil de efectos secundarios. Para reducir el riesgo de cistitis hemorrágica y de cáncer de vejiga, se recomiendan 250-500 ml de solución salina por vía endovenosa antes y después de la
infusión, así como una buena hidratación en las siguientes 72 horas. La administración concomitante de ondansetron reduce la frecuencia de los vómitos. Se debe monitorizar con analítica cada 15 días aproximadamente. Durante su uso, se recomienda realizar profilaxis para la PjP.
3.9 Rituximab (RTX)
Mecanismo de acción: Anticuerpo monoclonal anti-células B.
Indicaciones: Utilizado en neumonitis por hipersensibilidad (NH) fibrosante refractaria a otros tratamientos y otras EPI asociadas a enfermedad autoinmune (Tabla III)10,15 .
Dosis: Su administración es hospitalaria. La dosis habitual es 1 g ev en los días 1 y 15. Posteriormente, hay que valorar, según la respuesta al tratamiento, su dosificación en el tiempo, habitualmente, cada 6-12 meses.
Efectos adversos: Los más frecuentes son las reacciones transfusionales, mediadas por liberación de citoquinas que, en la mayoría de los casos, ocurren durante la primera infusión. Otros efectos adversos son los gastrointestinales, astenia, prurito o mareo, la hematotoxicidad o las reacciones de hipersensibilidad. Estas últimas son menos frecuentes y suelen darse en los primeros minutos de la infusión.
Consideraciones: Se recomienda hacer premedicación con antihistamínicos, antipiréticos/analgésicos y corticoterapia para evitar la incidencia y la gravedad de posibles reacciones transfusionales. Cuando estas aparecen, generalmente revierten al disminuir la velocidad o suspender la infusión. En la mayoría de los casos, una vez resueltos los síntomas, la perfusión se puede reanudar a un 50% de la velocidad anterior. Se necesita una monitorización estrecha de la función inmunológica, ya que pueden generar una hipogammaglobulinemia grave. Tiene contraindicaciones en pacientes con inmunodeficiencia grave y, en casos de artritis reumatoide con enfermedades cardíacas graves no controladas.
3.10 Infliximab
Mecanismo de acción: Anticuerpo monoclonal anti-TNF alfa.
Indicaciones: En casos de sarcoidosis con enfermedad pulmonar refractaria (Tabla III).10,13
Dosis: Su administración es hospitalaria. La dosis habitual es de 3-5 mg/kg ev en las semanas 0, 2 y 6. Posteriormente, cada 4-8 semanas.
Efectos adversos: El más importante son las reacciones de hipersensibilidad. Aunque también pueden generar alteraciones gastrointestinales, hepato o hematotoxicidad, cefalea, astenia, alteración de la presión arterial y palpitaciones, entre otros.
Consideraciones: Se conseja premedicación con antihistamínicos, cortisona y/o paracetamol para prevenir efectos leves y pasajeros. En caso de una reacción grave, no se
puede volver a administrar dicho fármaco. La administración concomitante de inmunomoduladores (MTX) se ha asociado a una reducción en la frecuencia de reacciones a la perfusión. Hay que tener especial precaución en aquellos que padezcan insuficiencia cardíaca, porque puede ser agravada por el fármaco.
3.11 Tocilizumab
Mecanismo de acción: Anticuerpo monoclonal anti-IL6.
Indicaciones: Opción de tercera o cuarta línea, sobre todo en EPID asociadas a esclerosis sistémica temprana o a enfermedades del tejido conectivo (Tabla III).10,11
Dosis: Su administración es hospitalaria. La dosis recomendada es de 162 mg administrados por vía subcutánea o intravenosa cada 2 semanas.
Efectos adversos: Son frecuentes las gastritis o aftas orales, la hepato o hematotoxicidad (leuco, neutro y trombocitopenia), hipertensión arterial, hipercolesterolemia, conjuntivitis, cefalea, mareo o erupciones cutáneas.
Consideraciones: Durante los 6 primeros meses, deberá evaluarse la función hepática cada 4-8 semanas, y posteriormente, cada 12 semanas. Según el aumento de los niveles de transaminasas, hay que reducir su dosis..
3.12
Conclusiones
El tratamiento inmunosupresor en las EPID es esencial, especialmente en casos no FPI, debido a su componente inflamatorio. Antes de prescribirlo, la evaluación integral, considerando la naturaleza y gravedad de la enfermedad, así como comorbilidades y potenciales riesgos, contribuye a una terapia personalizada y efectiva.
El enfoque personalizado, la vigilancia constante y la consideración cuidadosa de cada agente inmunomodulador son fundamentales para optimizar el tratamiento y mejorar la calidad de vida de estos pacientes.
4. TRATAMIENTO DE LAS EPID DURANTE EL EMBARAZO Y LA LACTANCIA
Los corticoides no cruzan con facilidad la barrera placentaria, de modo que el feto está expuesto a concentraciones mucho menores que la madre. En todo caso, en cuanto a la prednisona, la dosis materna durante el embarazo debería reducirse a niveles idealmente inferiores a 10 mg/día. La lactancia durante el uso de esteroides es segura.
El uso de la azatioprina durante el embarazo se acepta en aquellas situaciones en las que no existan otras alternativas terapéuticas viables. Existen datos contradictorios acerca de su posible contribución a la prematuridad o al bajo crecimiento intrauterino. Además, también se ha relacionado con alteraciones fetales congénitas, linfopenia o
pacitopenia. Su riesgo de generar inmunosupresión en el lactante hace que no sea aconsejable en la lactancia.
En general, se desaconseja el uso del rituximab durante el embarazo, si bien en los casos estrictamente necesarios puede administrarse con bajo riesgo para el feto, especialmente durante el primer trimestre. Durante la lactancia, puede ser usado sin aparentes riesgos para el recién nacido.
El metotrexato, el micofenolato, la ciclofosfamida, así como los antifibróticos actualmente disponibles (pirfenidona y nintedanib), se contraindican tanto durante el embarazo como durante la lactancia.
Al no disponer de estudios bien controlados sobre la seguridad del Tocilizumab en embarazo y lactancia se recomienda su suspensión o valorar el riesgo-beneficio de su uso.
Se sabe que el infliximab atraviesa la placenta y se ha detectado en el suero de recién nacidos hasta 6 meses después teniendo mayor riesgo de infección. En la lactancia materna sus niveles son bajos (5%). Aun así se aconseja precuaución en ambas situaciones y valorar su suspensión si hay alternativas más seguras16 .
Ver tabla III de resumen sobre los tratamientos farmacológicos en los EPID.
BIBLIOGRAFÍA
1. Richeldi L, Du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014;370:20712082.
2. Petnak T, Lertjitbanjong P, Thongprayoon C, Moua T. Impact of Antifibrotic Therapy on Mortality and Acute Exacerbation in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Chest. 2021;60(5):1751-1763.
3. Amati F, Spagnolo P, Oldham JM, Ryerson CJ, Stainer A, Gramegna A, Mantero M, et al. Trea- table traits in interstitial lung diseases: A call to action. Lancet Respir. Med. 2023;11(2):125- 128.
4. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022;205(9):18-47.
5. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M, Clerisme-Beaty E, Stowasser S, Tetzlaff K, Kuwana M, Maher TM; SENSCIS Trial Investigators. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med. 2019 Jun 27;380(26):2518-2528. doi: 10.1056/NEJMoa1903076
6. Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, Moua T, et al.; INBUILD trial investigators. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. 2020;8(5):453-460.
7. Amati F, Stainer A, Polelli V, Mantero M, Gramegna A, Blasi F, Aliberti S. Efficacy of Pirfenidone and Nintedanib in Interstitial Lung Diseases Other than Idiopathic Pulmonary Fibrosis: A Systematic Review. Int J Mol Sci. 2023;24(9):7849.
8. Ficha Técnica de OFEV®. Boehringer Ingelheim. Disponible en: https://cima.aemps.es/ cima/dochtml/ft/114979004/
9. Ficha Técnica de ESBRIET®. ROCHE. Disponible en: https://cima.aemps.es/cima/dochtml/ ft/111667018/FT_111667018.html
10. Van Den Bosch L, Luppi F, Mura M, Ferrara G. Immunomodulatory treatment for interstitial lung disease. Therapeutic Advances in Respiratory Disease. 2022;16:1-16., doi: 10.1177/17534666221117002.
11. Moua T, Petnak T, Charokopos A, Baqir M, Ryu JH. Challenges in the Diagnosis and Management of Fibrotic Hypersensitivity Pneumonitis: A practical Review of Current Approaches. Journal of Clinical Medicine. 2022;11(6):1473, doi:10.3390/jcm11061473.
12. Higuero JP, Memon A, Hinchcliff M. Learnings from clinical trials in patients with connective tissue disease-associated interstitial lung disease. Athritis Research & Therapy. 2023;25:118, doi:10.1186/s13075-023-03090-y.
13. Lee AS, Scofield RH, Hammitt KM, Gupta N, Thomas DE, Moua T, Ussavarungsi K, et al. Consensus Guidelines for Evaluation and Management of Pulmonary Disease in Sjögren’s. Chest. 2021;159(2):683-698, doi: 10.1016/j.chest.2020.10.011.
14. Gupta R, Judson MA, Baughman RP. Manegement of Advanced Pulmonary Sarcoidosis. American Journal of Respiratory and Critical Care Medicine. 2022;205(5):503-514, doi: 10.1164/rccm.202110-2145Cl.
15. Koudri G, Solomon JJ. Identification, Monitoring, and Management of Rheumathoid Arthritis. Associated Interstitial Lung Disease. 2023;12:2072-2076.
16. Grant-Orser A, Metcalfe A, Pope J, Johannson K. Pregnancy considerations for patients with Interstitial Lung Disease. Chest. 2022;162(5):1093-1105.
TABLAS Y FIGURAS
Tabla I. Tratamientos antifibróticos


Tabla II. Enfoque sugerido para el tratamiento por EPID
Tabla III. Tabla resumen: tratamientos farmacológicos en las EPID (parte 1) (Continúa)

Tabla III. Tabla resumen: tratamientos farmacológicos en las EPID (parte 2)

Tabla III. Tabla resumen: tratamientos farmacológicos en las EPID (parte 3)

MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
25
INDICACIONES DE DERIVACIÓN DE LAS EPID A TRASPLANTE PULMONAR
Autores
Miguel Arias Guillén
Diego Ferrer Pargada
Coordinadora
Rosalía Laporta Hernández
1. INDICACIONES GENERALES
El trasplante de pulmón (TxP) es una de las opciones de tratamiento a considerar en pacientes con enfermedad pulmonar avanzada. El campo del TxP está en crecimiento, y si se excluyen los efectos negativos de la pandemia de COVID-19, la Sociedad Internacional de Trasplante de Corazón y Pulmón (ISHLT) ha informado de un aumento gradual en todo el mundo del volumen de trasplantes durante las últimas 3 décadas hasta aproximadamente 4.500 procedimientos anuales.1,2
Esta tendencia se produce a nivel mundial, si bien el aumento ha sido más pronunciado fuera de América del Norte y Europa, representando el 8,5% del total de trasplantes de pulmón.2 Además, los receptores sobreviven cada vez más tiempo después del TxP. Así pues, con una población creciente de pacientes pretrasplante y postrasplante en todo el mundo, existe una necesidad no solo de neumólogos expertos en trasplante pulmonar, sino también de otros especialistas, para entender el proceso de selección y derivación de estos pacientes.
Las directrices de la ISHLT recomiendan que el TxP se considere solo en candidatos con una enfermedad pulmonar crónica en su última fase que reúnan los siguientes criterios:(Tabla I)3,4
∙ Alta probabilidad (>50%) de fallecer en 2 años sin el TxP.
∙ Alta probabilidad (>80%) de sobrevivir a los 90 días del TxP.
∙ Alta probabilidad (>80%) de sobrevivir a los 5 años del TxP teniendo en cuenta su condición general.
∙ Ausencia de otras posibles opciones terapéuticas.
2. CONTRAINDICACIONES ABSOLUTAS DEL TRASPLANTE PULMONAR
En la tabla II, se describen las condiciones asociadas a alto riesgo de mortalidad, por lo que la mayoría de los programas de trasplante no consideran la inclusión en lista de espera de este tipo de pacientes, salvo en circunstancias excepcionales.4
3. FACTORES DE RIESGO PARA EL TRASPLANTE PULMONAR
3.1. Factores de riesgo elevado o sustancialmente elevado
Los pacientes que presenten estos factores deberán ser considerados solo en centros con experiencia en el trasplante de candidatos con dicha condición (Tabla III). 4
- Probablemente, en ocasiones se desconozca o no se tengan datos que respalden la realización de un trasplante en los candidatos con alguno de estos factores. Sin embargo, la suma de varios de ellos posiblemente multiplique de forma significativa la probabilidad de un mal resultado tras el trasplante.
- Siempre que se detecte la presencia de alguno de estos factores de riesgo y sea posible, se deberán optimizar al máximo las condiciones modificables asociadas.
4. PECULIARIDADES DEL PACIENTE CON EPID PARA EL TRASPLANTE PULMONAR
Factores de riesgo sobre los resultados postrasplante en candidatos con EPID:
∙ Edad avanzada
∙ Sobrepeso
∙ Disfunción telomérica
∙ Cirugía torácica previa
∙ Limitada situación funcional, desacondicionamiento, fragilidad
∙ Reflujo gastroesofágico
∙ Enfermedad arterioesclerótica
∙ Manifestaciones extrapulmonares de las enfermedades del tejido conectivo
∙ Uso de corticoides y otros inmunosupresores
∙ Exacerbaciones agudas
∙ Necesidad de ventilación mecánica.
4.1. Edad avanzada
La edad sigue siendo un criterio de selección controvertido para el TxP. Basándose en los principios éticos de justicia y utilidad, la comunidad internacional de trasplantes ha expresado en general una preferencia por priorizar la asignación de los órganos a pacientes más jóvenes,4 y esto puede ser particularmente importante en programas y países con mayor escasez de donantes.
En la literatura publicada hasta el momento, se observa una peor supervivencia en los receptores con edades superiores a 65-70 años.1 Además, los receptores de edad avanzada parecen tener un mayor riesgo de desarrollar procesos malignos, enfermedad vascular, toxicidad en relación con los medicamentos y deterioro cognitivo.8-9 Las guías previas clasificaban la edad >65 años con baja reserva fisiológica como una contraindicación relativa al TxP, y también identificaban a los pacientes >75 años como candidatos poco probables al TxP.10 La literatura reciente es más esperanzadora para los pacientes de mayor edad, particularmente en lo que respecta a los resultados de TxP a corto plazo, en que la supervivencia al año después del trasplante parece comparable entre receptores de 70 a 79 años y 60 a 69 años. Con una población que envejece y una experiencia cada vez mayor, la proporción de trasplantes de pulmón realizada en pacientes de edad avanzada está aumentando. Los datos a nivel global muestran un aumento en la proporción de TxP en los receptores >65 años, que pasaron del 2,6% en 2004 al 17% en 2016.
Se necesitan más datos para estratificar mejor el riesgo de los pacientes de mayor edad y FPI sometidos a trasplante. Esto es especialmente importante en la era de los tratamientos antifibróticos, ya que el enlentecimiento de la progresión de la enfermedad puede retrasar la necesidad de TxP en un subconjunto de pacientes y, a su vez, conducir a una modificación de los límites de edad programáticos para el trasplante.
En última instancia, la ISHLT no respalda ningún límite de edad superior absoluto para TxP, de modo que la posibilidad del TxP en pacientes de edad avanzada sigue siendo una decisión específica del centro.4
4.2. Manejo de la obesidad y el desacondicionamiento físico
El sobrepeso es común en pacientes con EPI que se aproximan al TxP. La evidencia actual respalda un mayor riesgo de disfunción primaria del injerto y mortalidad pos-
trasplante en receptores obesos en comparación con candidatos con un BMI normal o con sobrepeso.
El asesoramiento nutricional y la pérdida de peso de forma adecuada pueden aumentar la probabilidad de éxito y mejorar las opciones para un TxP, al tiempo que también mejoran potencialmente los síntomas previos al trasplante.
El desacondicionamiento físico, la capacidad funcional, la fragilidad y la sarcopenia afectan a la trayectoria previa y posterior al trasplante. Todos parecen ser comunes en los pacientes con EPID en fase avanzada, pudiendo demostrarse fragilidad en aproximadamente el 30% de los casos de candidatos con enfermedad pulmonar restrictiva. La fragilidad y el deterioro del estado funcional se consideran factores de riesgo para malos resultados para el TxP, así, en los pacientes con EPID avanzada, está justificada una evaluación exhaustiva del acondicionamiento y del estado funcional.
La rehabilitación pulmonar se ha asociado con mejoría de los síntomas previos al trasplante, calidad de vida y 6MWT, junto a unos resultados más favorables postrasplante. Se debe considerar en todos los pacientes con EPID avanzada y puede ser necesario para el TxP.
4.3. Disfunción telomérica
En el pretrasplante, el acortamiento de los telómeros se asocia con una edad de presentación más temprana y un fenotipo progresivo4 y predice una mala respuesta a los inmunosupresores en la FPI.5
La longitud reducida de los telómeros se ha relacionado con peores resultados postrasplante. Entre 86 pacientes trasplantados por EPID, un estudio demostró que la longitud de los telómeros antes del trasplante <percentil 10 era asociado de forma independiente con una peor supervivencia (IR 10,9, IC 95% 2,7–44,8; p = 0,001), mayor riesgo de disfunción primaria del injerto, anomalías hematológicas y CLAD.
La realización de pruebas de longitud de los telómeros y las mutaciones asociadas en candidatos con EPID pueden ser particularmente útiles en pacientes de inicio temprano o EPI familiar.
Se necesitan más datos para determinar si es mejor integrar las pruebas como rutina y cuándo hacerlo en la evaluación, pero puede ayudar a determinar el pronóstico previo al trasplante, estratificar el riesgo de los candidatos y predecir las complicaciones posteriores al trasplante.
4.4. Consideraciones en relación con cirugía del tórax previa
La cirugía de tórax previa, particularmente la pleurodesis, se asocia con mayor pérdida de sangre y mayor morbilidad postoperatoria temprana, como disfunción renal y dis-
función primaria del injerto. La revisión de la literatura más reciente demuestra que, aunque hasta en el 45% de los trasplantes de pulmón los receptores se han sometido a procedimientos cardiotorácicos previos, no se ha observado ninguna diferencia en la supervivencia del trasplante de pulmón. Un TxP previo, especialmente en los pacientes que han desarrollado un síndrome de aloinjerto restrictivo, confiere un mayor riesgo de peor supervivencia.
En cuanto al neumotórax, cuando sea posible, su manejo en un paciente que puede ser un potencial TxP debe ser discutido con el centro trasplantador. Y en cuanto a la pleurodesis, aunque se prefiere evitar la pleurodesis con talco, no se considera una contraindicación absoluta y se le debe proporcionar la atención inmediata más adecuada.
4.5. Cirugía antirreflujo en RGE
Se ha demostrado RGE en hasta el 90% de las personas con FPI y se le ha implicado en la patogénesis de las EPID. El impacto del RGE puede verse amplificado en los pacientes con enfermedades con dismotilidad gastrointestinal coexistente, siendo la más notable la esclerosis sistémica.
Generalmente, los programas de trasplante exigen la evaluación del reflujo y de la motilidad esofágica durante la evaluación para el TxP debido a su asociación con infección, rechazo agudo y disfunción crónica del injerto. Se ha demostrado que la funduplicatura mitiga estos riesgos en el pre o postrasplante temprano. Aunque la cirugía previa al trasplante probablemente conlleva mayores riesgos en comparación con los pacientes con EPID menos grave, se ha demostrado que es seguro en pacientes bien seleccionados que se acercan al TxP, incluidos en una cohorte con una FVC media del 50% predicho.
4.6. Exacerbaciones agudas previas al trasplante
La mayor parte de la literatura sobre las exacerbaciones agudas de las EPID se refiere a la FPI (EA-FPI). El pronóstico de la EA-FPI es malo, con mortalidad hospitalaria de casi el 50% y hasta el 90% en aquellos que requieren ventilación mecánica,6 junto con una mediana de supervivencia de solo de 3 a 4 meses.
En el contexto del TxP, una EA-FPI también se asocia con peores resultados. Un análisis realizado en 2018 en 89 pacientes con FPI incluidos en lista mostró una supervivencia postrasplante a 1 y 3 años del 71% y el 60% respectivamente para los trasplantados en EA-FPI, en comparación con el 94% y el 90% en aquellos con enfermedad estable antes del trasplante. Este y otros estudios han documentado una asociación entre valores altos en la escala de priorización en lista (LAS) en el momento del trasplante y peores resultados postrasplante, lo que refuerza aún más la necesidad de derivar oportunamente para un TxP en las EPID.
Durante una exacerbación aguda, se requiere una comunicación estrecha con el centro trasplantador para revisar la posible inclusión en lista.
El desarrollo en tecnologías y la mayor experiencia en soporte respiratorio, como el puente de soporte vital extracorpóreo (ECMO), se utiliza cada vez más para mejorar los resultados.
4.7. Riesgos relativos asociados con las farmacoterapias para las EPID
Algunos tratamientos específicos pueden afectar los riesgos perioperatorios en pacientes con EPID que se acercan al TxP. Los corticosteroides usados de forma crónica y en dosis altas pueden aumentar las complicaciones de la cicatrización de heridas y el riesgo de dehiscencia anastomótica. Un estudio7 demostró una mayor mortalidad entre 62 pacientes (38% con EPID) tratados con dosis de prednisona previamente al trasplante >0,42 mg/kg-1/día-1 .
Otros datos sugieren que dosis de corticoesteroides <40 mg o 0,3 mg/kg-1/día-1 probablemente sean seguras. En función de los riesgos potenciales, muchos programas de TxP consideran que dosis de mantenimiento de prednisona de >20 mg/día-1 son una contraindicación (excluyendo dosis aumentadas para las exacerbaciones agudas).
No hay datos sobre el impacto de otros agentes inmunomoduladores sobre los resultados del TxP, pero, en general, es mejor limitar los fármacos inmunosupresores para la EPID al nivel de dosis efectiva más baja mientras un paciente está en la lista para trasplante.
Los estudios más recientes no han demostrado diferencias en cuanto a supervivencia, dehiscencia de anastomosis u otras complicaciones entre pacientes con EPID tratados con pirfenidona y tratados con nintedanib antes del trasplante. El uso de antifibróticos previo al día del TxP no se considera una contraindicación.
5. CRITERIOS DE DERIVACIÓN E INCLUSIÓN EN LISTA DE TRASPLANTE DE PULMÓN EN PACIENTES CON ENFERMEDAD PULMONAR INTERSTICIAL DIFUSA (EPID)
Se debe considerar la recomendación o inclusión en la lista si el paciente cumple algún criterio de los que se detallan a continuación.
5.1. Remitir al programa de trasplante pulmonar
- Al diagnóstico de NIU: histológico o radiológico.
- Cualquier forma de fibrosis pulmonar con FVC <80% o DLco 40%.
- Cualquier forma de fibrosis pulmonar con:
∙ Descenso relativo FVC del 10%.
∙ Descenso relativo FVC del 5% + progresión radiológica o clínica.
∙ Descenso relativo DLco del 15%.
- Necesidad de O2 con ejercicio o continuo.
- EPID de origen inflamatorio: progresión clínica o radiológica.
- Enfermedades del tejido conectivo, remitir al diagnóstico.
- Fibrosis pulmonar familiar, remitir al diagnóstico.
Descenso relativo FVC: (FVC inicial–FVC final)/FVC inicial
5.2. Inclusión en lista de espera
- Cualquier forma de fibrosis pulmonar con:
∙ Descenso absoluto FVC del 10%.
∙ Descenso absoluto FVC del 5% + progresión radiológica.
∙ Descenso absoluto DLco del 15%.
- Test 6 min: SaO2 <88% o >50m en 6 meses.
- Hipertensión pulmonar (HTP).
- Hospitalización por progresión, neumotórax o exacerbación.
Descenso absoluto FVC: FVC inicial – FVC final
6. RECOMENDACIONES PARA LOS CANDIDATOS CON ENFERMEDADES ESPECÍFICAS
6.1. Linfangioleiomiomatosis (LAM)
La LAM, una indicación muy rara de trasplante de pulmón, supone el 0,9% de los trasplantes realizados a nivel mundial entre 1995 y 2018.
Las indicaciones para remitir pacientes con LAM a un programa de trasplante pulmonar son:
- Deterioro de la función pulmonar con FEV1 <30%.
- Disnea de esfuerzo con grado III-IV de la NYHA.
- Hipoxemia en reposo.
- Hipertensión pulmonar.
- Neumotórax refractario.
- Para la inclusión en lista, el paciente debe presentar progresión en cualquiera de los criterios previos pese al uso de terapia mTOR.
- Una de las características de la LAM es el alto porcentaje de neumotórax, que aparece hasta en un 70% de los pacientes. El manejo con pleurodesis para evitar recidivas es habitual, algo que puede tener sus consecuencias en un futuro trasplante. La presencia de una pleurodesis previa puede aumentar el riesgo de sangrado durante y después de una cirugía de trasplante de pulmón. Esto ha sido demostrado en series de casos en que los pacientes que habían sido sometidos a procedimientos pleurales previos tenían mayor riesgo de sangrado durante y tras la cirugía de un trasplante de pulmón. En estos mismos pacientes, no se observó una mayor mortalidad, ni una mayor estancia hospitalaria pese a un mayor número de sangrados. La posibilidad de que un paciente requiera un trasplante pulmonar en un futuro no debe impedir la realización de una pleurodesis en caso de neumotórax espontáneo. En la actualidad, los expertos recomiendan una pleurodesis mecánica y no química como primera opción, pero con una evidencia escasa.8
- En pacientes con LAM, se prefiere la realización de trasplantes bipulmonares, pues existen estudios que indican mejores supervivencias de este tipo de trasplante en comparación con el unipulmonar. Además, en pacientes con LAM, con un trasplante unipulmonar es posible la aparición de quilotórax y neumotórax en el pulmón nativo.
- Según las recomendaciones de la ISHLT, se recomienda mantener la terapia con I-mTOR utilizada en este tipo de pacientes mientras se encuentran en lista de espera, con cese de la misma en el momento del trasplante.
6.2. EPID asociadas a conectivopatías (ETC-EPID)
El TxP se ha utilizado para enfermedades pulmonares avanzadas en pacientes con artritis reumatoide, enfermedad mixta del tejido conectivo, esclerosis sistémica, síndrome de Sjögren, lupus eritematoso sistémico, polimiositis/dermatomiositis/síndrome antisintetasa y vasculitis pulmonar. Aunque los datos son limitados en ETC-ILD, varios estudios de cohortes informan que la supervivencia postrasplante en estos pacientes es similar a la FPI, sin diferencias en la disfunción primaria del injerto o las tasas de rechazo.
Se ha demostrado que el estado funcional mejora, y son raros los trastornos recurrentes o progresivos no pulmonares de la enfermedad después del TxP.
Esclerosis sistémica
Algunos pacientes pueden ser considerados no elegibles para TxP debido a los riesgos de aspiración. Varios estudios muestran una supervivencia postrasplante similar a la FPI sin mayor riesgo de disfunción crónica del aloinjerto pulmonar (CLAD).
Debe reconocerse que los estudios disponibles sobre el TxP para las ETC-EPID son retrospectivos y están limitados por sesgos de selección. En este contexto, la ISHLT considera que las ETC son un factor de riesgo para peores resultados y recomienda detección de enfermedades extrapulmonares en colaboración con el departamento de reumatología cuando sea posible.4
Es necesario controlar la actividad de la enfermedad previa al trasplante en los niveles más bajos posibles para reducir los riesgos de enfermedad infecciosa perioperatoria, garantizando al mismo tiempo que cualquier enfermedad irreversible no represente una amenaza excesiva para los resultados postrasplante o la posibilidad de rehabilitación.
La recurrencia a nivel extrapulmonar y pulmonar tras el trasplante de pulmón es algo poco frecuente y que responde a tratamientos dirigidos.
La esclerosis sistémica con dismotilidad esofágica grave puede considerarse una contraindicación en algunos centros; la evaluación de la deglución y la función esofágica es crucial. Los pacientes también deben ser valorados para detectar engrosamiento de la piel, ulceración digital, insuficiencia renal, insuficiencia cardíaca y miocardiopatía.
Artritis reumatoide y trasplante pulmonar
En los estudios disponibles hasta la fecha, los pacientes trasplantados por EPID secundaria a artritis reumatoide no presentan diferencias significativas de supervivencia a los 1, 2 y 5 años en comparación con pacientes con FPI y otras EPID secundarias a enfermedades autoinmunes.
Miopatías inflamatorias y trasplante pulmonar
La cantidad de trasplantes por dermatomiositis es baja, lo que probablemente se deba a que esta enfermedad se asocia de forma frecuente con tumores malignos. La evidencia que hay hasta el momento indica que los pacientes trasplantados por miopatías inflamatorias tienen supervivencias similares, sin diferencias significativas si los comparamos con FPI o con otras afectaciones intersticiales que no tengan que ver con FPI. Dentro de las diferentes formas de afectación en las miopatías inflamatorias, los pacientes con afectación pulmonar, en la piel y con necesidad de varias líneas de inmunosupresión previa al trasplante tienen peores resultados. No se han observado recurrencias a nivel pulmonar tras el trasplante, pero sí a nivel muscular y de la piel.9
Lupus eritematoso sistémico y trasplante pulmonar
Existe poca literatura en cuanto a pacientes trasplantados por EPID o hipertensión pulmonar secundarias a lupus. En lo poco que hay descrito, los pacientes presentaban supervivencias y complicaciones aceptables y similares a otros trasplantados. Por ello, los pacientes con una afectación por lupus eritematoso sistémico que sean cuidadosamente seleccionados pueden tener resultados exitosos con un trasplante pulmonar.
Vasculitis ANCA positivo y trasplante de pulmón
La evidencia del trasplante en este tipo de enfermedades se basa únicamente en casos clínicos aislados que han presentado buenos resultados. Su presencia no es una contraindicación absoluta, pero la valoración de este tipo de pacientes debe ser cuidadosa y con gran atención a la situación renal.9
6.3. Consideraciones en otras EPID, incluida la sarcoidosis
El TxP es una opción para otras EPID, incluida la histiocitosis de células de Langerhans, EPID de origen ocupacional, incluyendo la silicosis, fibroelastosis pleuroparenquimatosa (PPFE), neumonitis por hipersensibilidad fibrótica y sarcoidosis.
La sarcoidosis representa el 2,4% de todos los trasplantes de pulmón a nivel mundial entre 1995 y 2018. Esta enfermedad se aborda de forma similar a otras afectaciones intersticiales. En la valoración pretrasplante, se debe ser muy cuidadoso en el estudio de la afectación de otros órganos, como el corazón con la presencia de enfermedad infiltrativa o alteraciones en la conducción. La disfunción grave cardiaca más pulmonar puede llevar a la necesidad de un trasplante de corazón y pulmón. La sarcoidosis constituye el 1,9% de los trasplantes combinados de corazón y pulmón a nivel mundial desde 1998 a 2018.
A la hora de enviar a un paciente con sarcoidosis a la unidad de trasplante de referencia, la ISHLT recomienda seguir las indicaciones generales de las enfermedades pulmonares intersticiales difusas. En la evidencia que hay hasta el momento, parece que los trasplantes de pulmón por sarcoidosis tienen resultados similares a los que hay en población general. No obstante, se ha descrito que la presencia de mayor fibrosis antes del trasplante estaba relacionada con una menor supervivencia tras el mismo.10
La recaída tras el trasplante varía según diferentes estudios entre el 14% y el 30%.
La PPFE, presentada como entidad propia o coexistiendo con otros patrones de EPID puede ser particularmente dificultosa quirúrgicamente, con adherencias densas que a menudo conducen a lesiones torácicas, contracción y aumento del riesgo de hemorragia.
6.4. Síndrome combinado de fibrosis pulmonar-enfisema
- Este síndrome asocia distintas características clínicas y fisiológicas, y lleva a un curso clínico más agresivo que la fibrosis pulmonar idiopática (FPI). La prevalen-
cia de este síndrome dentro de las EPID está en torno al 8% según diferentes estudios. La presencia de hipertensión pulmonar asociada es más frecuente en este tipo de pacientes y conlleva un peor pronóstico, por lo que es importante su búsqueda activa en el seguimiento.
- En cuanto a los resultados tras el trasplante, existe poca literatura relacionada. Uno de los principales estudios indica que los pacientes que fueron trasplantados por un síndrome de combinación EPID-enfisema eran más propensos a la disfunción primaria del injerto y a la disfunción crónica en comparación con los pacientes trasplantados por FPI, sin que ello conllevase diferencias significativas en la supervivencia (Figura 1).
BIBLIOGRAFÍA
1. Chambers DC, Cherikh WS, Harhay MO, Hayes D, Hsich E, Khush KK, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult lung and heart-lung transplantation Report-2019; Focus theme: Donor and recipient size match. J Heart Lung Transplant. 2019;38(10):1042-1055.
2. Chambers DC, Perch M, Zuckermann A, Cherikh WS, Harhay MO, Hayes D, Jr., et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-eighth adult lung transplantation report - 2021; Focus on recipient characteristics. J Heart Lung Transplant. 2021;40(10):1060-1072.
3. Kapnadak SG, Raghu G. Lung transplantation for interstitial lung disease. Eur Respir Rev. 2021;30(161).
4. Leard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, et al. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-1379.
5. Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710-1720.
6. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824.
7. McAnally KJ, Valentine VG, LaPlace SG, McFadden PM, Seoane L, Taylor DE. Effect of pre-transplantation prednisone on survival after lung transplantation. J Heart Lung Transplant. 2006;25(1):67-74.
8. Krishnan Warrior, Daniel F. Dilling, Lung transplantation for lymphangioleiomyomatosis. J Heart Lung Transplant. 2023;42(1):40-52, ISSN 1053-2498, https://doi.org/10.1016/j. healun.2022.09.021.
9. Zhang N, Liu S, Zhang Z, Liu Y, Mi L, Xu K. Lung Transplantation: A Viable Option for Connective Tissue Disease? Arthritis Care Res (Hoboken). 2023;75(11):2389-2398, doi: 10.1002/ acr.25133, PMID: 37052523.
10. Le Pavec J, Valeyre D, Gazengel P, Holm AM, Schultz HH, Perch M, Le Borgne A, et al. Lung transplantation for sarcoidosis: outcome and prognostic factors. Eur Respir J. 2021;58(2):2003358.
TABLAS Y FIGURAS
Tabla I. Indicaciones más frecuentes de trasplante de pulmón según periodos de inclusión * (% de TxP a nivel mundial según informe de ISHLT)

Abreviaturas: FQ: fibrosis quística. EPOC: enfermedad pulmonar obstructiva crónica. EPID: enfermedad pulmonar intersticial difusa. HP: hipertensión pulmonar. ISHLT: Sociedad Internacional de Trasplante de Pulmón y Corazón.
* Datos de Chambers DC, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult lung and heart-lung transplantation Report-2019; Focus theme: Donor and recipient size match; J Heart Lung Transplant. 2019;38(10):1042-1055.
** Incluye déficit de alfa-1-antitripsina.
*** Incluye fibrosis pulmonar idiopática.
Tabla II. Contraindicaciones. Leard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, et al. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-1379


Leard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, et al. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-1379.
Tabla III. Factores de riesgo

Figura 1. Pasos para derivar a valoración de trasplante pulmonar
MANUAL DE CONSULTA EN LAS ENFERMEDADES PULMONARES INTERSTICIALES DIFUSAS
FINAL DE VIDA. PALIATIVOS EN EPID
Autoras
Inés Ruiz Gemar
Cristina Caupena Auledas
Coordinadora
Miren Begoñe Salinas Lasa
1. INTRODUCCIÓN
Consideramos enfermedad terminal a esa enfermedad avanzada, progresiva e incurable donde ya se han utilizado todos los recursos y posibilidades razonables de respuesta a un tratamiento específico1 .
Las enfermedades terminales suelen contener síntomas múltiples, intensos y cambiantes que producen un gran impacto en el paciente que la sufre. Pero además también produce un gran impacto emocional en la familia de ese paciente por lo que el equipo terapéutico debe abordar, tratar y acompañar tanto al paciente como a los miembros que rodean a ese paciente1,2 .
Las enfermedades respiratorias crónicas avanzadas son procesos crónicos, progresivos e incurables, causan insuficiencia respiratoria, síntomas variados, déficit funcional, alta dependencia y frecuentación de los servicios sanitarios. El manejo de los pacientes con enfermedades pulmonares intersticiales difusas (EPID) y en particular, aquellas que son fibrosantes progresivas, exige mucha atención al final de la vida. Algunos pacientes son diagnosticados demasiado tarde para hacer un tratamiento específico, y todo lo que se puede hacer es ofrecer apoyo para el manejo de los síntomas, mediante las Unidades de Cuidados Paliativos (UCP). Sólo el 10-20% de los pacientes con EPID son atendidos en la UCP3 .
Se sugiere comenzar los cuidados paliativos tan pronto como los pacientes estén sintomáticos y los síntomas no ceden a pesar de tratamiento específico4. El principal objetivo de los cuidados paliativos es mejorar la calidad de vida del paciente. Sin embargo, los cuidados paliativos están enfocados a pacientes en fase avanzada, es decir, a aquellos que no respondan al tratamiento específico y en los que se espera, por lo tanto, que la enfermedad provoque su fallecimiento.
Para poder cumplir este objetivo es primordial informar al paciente desde su diagnóstico de la enfermedad, del curso, pronóstico y evolución de dicha enfermedad e introducir el concepto de cuidados paliativos para tratar los síntomas más invalidantes de dicha enfermedad5
Para que una UCP funcione y pueda cumplir su objetivo es importante conocer el concepto de equipo multidisciplinar6 formado por:
1. Neumólogo
2. Médico de cuidados paliativos
3. Equipo de Hospitalización domiciliaria
4. Equipo de atención primaria
Con este tipo de modelo multidisciplinar se puede garantizar la continuidad asistencial del paciente, el control de síntomas y evitar los ingresos. Es decir, con esto podemos mejorar la atención de los pacientes y optimizar el uso del sistema sanitario (Tabla 1).4
Para mejorar la calidad de vida de los pacientes, debemos tratar los síntomas más invalidantes (tos y disnea). Para los pacientes, la disnea, es sobre todo el síntoma más incapacitante y prevalente (5). Llegado el momento, el tratamiento propio de la enfermedad, el que puede mejorar al mismo tiempo los síntomas y el pronóstico, ya no es suficiente para controlar la disnea y se hace necesario el tratamiento sintomático de la misma. Es necesario, por tanto, que los médicos involucrados comuniquen adecuadamente a los pacientes y familiares, el pronóstico y evolución de la enfermedad y los deriven tempranamente a programas de cuidados paliativos1,6,7 .
Para mantener una buena calidad de vida en cada paciente, es importante realizar una valoración inicial de su situación sintomática, funcional, socio-familiar, conocimiento del diagnóstico-pronóstico, adaptación psico-emocional, existencia de voluntades anticipadas y decisión sobre últimos días y monitorizar las necesidades personales en el curso de la enfermedad en función de las preferencias, cultura y religión de cada paciente4 .
Otro objetivo importante para llevar a cabo en estas unidades es la planificación avanzada de la atención, donde se incluye la discusión sobre las limitaciones del tratamiento y las preferencias sobre la muerte, el poder programar/decidir sobre su final de vida,
su muerte. Es importante considerar desde el principio las expectativas del paciente ya que los beneficios de los tratamientos pueden ser escasos en la mayoría de ellos. Se debe asegurar que los pacientes, sus familiares y los cuidadores tengan acceso a toda la gama de servicios ofrecidos por los equipos de cuidados paliativos, garantizando la necesaria colaboración entre los profesionales de salud involucrados.
2. TRATAMIENTO ESPECÍFICO DE LOS SÍNTOMAS
2.1. TOS
La tos seca es un síntoma muy frecuente en la enfermedad pulmonar7. A menudo empeora con el esfuerzo, dificulta el descanso nocturno y suele ser refractaria al tratamiento médico afectando drásticamente a la calidad de vida8
Algunos pacientes con EPID pueden presentar tos causada por la propia EPID, por otras patologías (TEP, infecciones pulmonares, Insuficiencia cardíaca) o por complicaciones (HTP, cáncer, RGE…). Por esta razón se sugiere hacer un buen diagnóstico de la causa de la tos para decidir el tratamiento adecuado, ya que si es secundaria a un proceso tratable y no se ha tratado debe tratarse primero la causa de la tos8 .
Medidas generales
- Fisioterapia respiratoria si el paciente presenta secreciones.
- Cambios posturales si hay presencia de reflujo gastroesofágico (incorporar la cabecera de la cama unos 20-30 cm).
Tratamiento paliativo
TOS
- Dextrometorfano: 15-30 mg/6-8 h vo.
- Codeína: 30-60 mg/4-6 h vo.
- Corticoides inhalados: por ejemplo, budesonida a dosis 400-800 mcg al día divididas en dos tomas.
- Dexametasona: 2-4 mg/6-8 h (a dosis altas, reduce la irritación) o prednisona a dosis bajas (5-10 mg/día).
- Morfina: iniciar tratamiento con dosis bajas (5 mg/4 h vo o 10 mg de formulación retardada/12 h oral). Si tomaba morfina previamente, aumentar un 50%.
- Valorar nebulizaciones con anestésicos (lidocaína).
* Si se prescriben opioides, hay que valorar la necesidad de iniciar tratamiento antiemético y para evitar el estreñimiento.
TOS PRODUCTIVA
- Nebulización de suero salino con terbutalina.
- Bromuro de ipratropio (efecto anticolinérgico).
- Buscapina 10-20 mg/6-8h v.o/s.c (en paciente incapaz de toser).
- Los mucolíticos tienen escasa efectividad.
2.2.
DISNEA
La disnea es el síntoma más prevalente e incapacitante.
Medidas no farmacológicas o generales
- Compañía tranquilizadora.
- Aire fresco sobre la cara.
- Ejercicios respiratorios (respiración diafragmática y espiración con labios semi ocluidos).
- Técnicas de relajación.
- Posición confortable.
- Adaptación del estilo de vida (actividades vida diaria, barreras arquitectónicas, etc.)
Medidas farmacológicas
Opioides: Es el tratamiento farmacológico básico de la disnea en el paciente muy sintomático y/o terminal. Existen diferentes tipos de opioides y vías de administración por lo que la elección del tratamiento debe individualizarse en cada caso (Tabla II).9
En el paciente en situación de últimos días y necesidad de sedación paliativa es preferible la administración endovenosa y/o subcutánea mientras que en el resto de pacientes que no se encuentren en esta situación pero presenten mal control sintomático se recomienda valorar la administración oral, sublingual o transmucosa9
El tiempo de acción del fármaco varía en función de la vía de toma, presentando un mecanismo de acción más rápido cuanto menor es la absorción intestinal y por lo tanto el paso hepático (transmucoso > sublingual >oral).
Pautas de administración más habituales (Tabla III)
- Morfina de liberación rápida: (Sulfato o clorhidrato de morfina oral: 2,5-5 mg cada 4 h vo). Incrementos cada 2-3 días del 30-50 % de la dosis diaria, hasta el control de la disnea y a continuación pasar a Morfina de liberación retardada (MST).
- No altera los parámetros gasométricos ni de función pulmonar10 .
- En ausencia de dolor, dosis superiores a 15 mg cada 4 horas no aportan beneficios.
- En pacientes que ya tomaban previamente morfina para tratamiento de dolor, se aconseja aumentar la dosis hasta un 50% para el control de la disnea.
- La morfina nebulizada NO SIRVE para el tratamiento de la disnea en pacientes con enfermedad intersticial11 .
- Dosis de rescate/crisis aguda: opioides de liberación rápida a un 10 % de la dosis total diaria.
Morfina de liberación prolongada (MST): iniciar con 10mg/día (repartido en dos dosis, cada 12 horas) e incrementar dosis lentamente (10mg/día)10 .
- Dosis de rescate/crisis aguda: morfina oral de liberación rápida o fentanilo (transmucosos, sublingual o intranasal). Si más de tres rescates al día, aumentar dosis
- Fentanilo: para el tratamiento de fondo de los paciente con EPID se suelen preferir parches de liberación prolongada ya que estos pacientes suelen estar tomando bastantes fármacos orales (antifibróticos, corticoides orales, ansiolíticos, además del tto de comorbilidades asociadas). Habitualmente se inicia con dosis bajas, la inferior es la de 12 mcg pero como este parche se puede partir, se inicia con dosis de 6 mcg y en función del control de los síntomas se irá aumentando a dosis superiores hasta alcanzar 100 mcg.
Otra vía de administración sería la transmucosa (nasal o sublingual). Esta vía se usa preferiblemente como tratamiento para las crisis agudas o como tratamiento preventivo. Es sabido que la administración nasal tiene ventajas12, como su fácil administración, el rápido inicio de acción, alta biodisponibilidad y el buen control de la disnea. Se prefiere el fentanilo nasal con pectina ya que aumenta la adherencia a las mucosas y evita que parte del fármaco pase por vía oral, evitándose así el paso hepático del fármaco13 .
A pesar de la falta de consenso sobre el uso y el tipo de opioide a utilizar en la disnea en el paciente avanzado, diferentes sociedades respiratorias recomiendan el uso de la morfina, por sus beneficios y ausencia de efectos secundarios reseñables a las dosis habituales. Según experiencia del Hospital Universitario de Basurto (Bilbao), los fentanilos transdérmicos de liberación mantenida para la disnea basal y el fentanilo nasal en pectina o fentanilo sublingual para la disnea episódica, son una buena opción de tratamiento12
Fármacos adyuvantes
Clorpromazina: fármacos de segunda línea cuando no pueden utilizarse opioides, o añadido a estos.
- Prometazina: 25 mg/8-12 h o a demanda.
- Clorpromazina: 7.5- 25 mg vo o sc /6-8 h (habitualmente una dosis de 25mg noche) o a demanda. Tiene efecto ansiolítico y sedante y algunos autores sugieren que puede actuar directamente sobre el nivel de percepción de la disnea.
Benzodiazepinas: su utilidad se reduce al alivio de la ansiedad y los ataques de pánico respiratorio. No actúan sobre el mecanismo de la disnea
- Diazepam: oral o sublingual (5-10 mg/8-12h)
- Lorazepam: 0,5-1 mg/4-12 h o a demanda
- Midazolam: subcutáneo (5-10 mg/24 h) o intravenosa (2,5-10 mg/24h), de elección en la crisis de pánico respiratorio
Oxigenoterapia: Precisa de la objetivación previa de hipoxemia severa y de la demostración de la eficacia en la mejora del síntoma. No es imprescindible en estas situaciones 14 .
Talidomida. Este fármaco aparece en la guía del tratamiento de la FPI del 2013. Se usa en cuidados paliativos de otras enfermedades y en otros países. En España no se tiene experiencia sobre el uso de este fármaco, en la literatura no se mencionan dosis y no hay referencia de su utilización por sus elevados efectos secundarios.(Tabla III)
Efectos secundarios más frecuentes de los opioides
Manejo de los efectos adversos:
- Reducción de dosis o supresión del fármaco
- Cambio de la vía de administración (ver anexo: tabla conversión de opioides)
- Rotación de opioide (ver anexo: tabla de conversión de opioides)
- Tratamiento sintomático de los efectos adversos: valorar pautar desde el inicio tratamiento laxante +/- antiemético (metoclopramida) por la elevada frecuencia de aparición de efectos adversos gastrointestinales.
2.3 SÍNDROME ANSIOSO-DEPRESIVO
El diagnóstico de una enfermedad de este tipo produce una clínica ansiosa-depresiva tanto al paciente como a sus familiares directos. Para poder tratar esto se requiere de una psicoterapéutica importante proporcionada ya sea por el ámbito hospitalario como por atención primaria.
A parte de poder tratar esta parte con fármacos antidepresivos y ansiolíticos también hay que poder abordar por parte del equipo multidisciplinar todas las inquietudes que afectan al propio paciente y a sus cuidadores/familiares4 .
2.4 SITUACIÓN DE ÚLTIMOS DÍAS DE VIDA
Es importante procurar un ambiente óptimo y, siempre y cuando sea posible, respetar las preferencias del paciente sobre su final de vida. En estos días es muy importante mantener una estrecha relación entre el equipo multidisciplinar-paciente-cuidador y así poder detectar las descompensaciones precozmente para así poder ajustar el tratamiento para disminuir la intensidad de la crisis, pudiendo mantenerse en su domicilio si es la voluntad del paciente y las circunstancias lo permitan2 .
En esta fase hay que mantener los tratamientos previos y añadir aquellos que alivian la disnea más intensa que se manifiesta en esta fase final de vida, el fentanilo de acción rápida es un valioso instrumento en estos casos para controlar la disnea episódica.
Una vez llegados en esta fase introducimos el concepto de sedación paliativa, que es la disminución progresiva del nivel de conciencia del enfermo mediante la administración de los fármacos apropiados con el objetivo de evitar un sufrimiento intenso causado por uno o más síntomas refractarios de la EPID causante del final de vida2. Puede ser continua o intermitente y su profundidad se gradúa buscando el nivel de sedación mínimo que logre el alivio sintomático y el confort del paciente y familiares.
La sedación paliativa implica, para el enfermo, la decisión de renunciar a experimentar conscientemente la propia muerte y tiene también para su familia importantes efectos psicológicos y afectivos. Como previamente ya se ha comentado, esta decisión acerca la necesidad de disminuir el nivel de conciencia del enfermo como estrategia terapéutica en este punto de la enfermedad, ya ha sido deliberada y reflexionada, tanto por el enfermo como por la familia. Además, la sedación ha de estar siempre bien indicada y bien efectuada, siendo los elementos fundamentales el consentimiento, la administración de fármacos a dosis adecuadas, y la evaluación.
Los fármacos de elección en la sedación paliativa de las EPIDs son, por este orden, el midazolam, los neurolépticos sedativos (clorpromazina iv o levomepromazina sc, sobre todo si hay mucho delirium o fracaso del midazolam) y el fenobarbital im o sc en caso de haber más riesgo de convulsiones. La morfina en estas sedaciones también juega un papel muy importante sobre todo si el paciente ya la estaba recibiendo en esos momentos, no se debe retirar y se debe de ajustar la dosis para mantener en infusión continua2 .
Otro tipo de sedación paliativa es la sedación en la agonía2 , esta es la sedación paliativa que se utiliza cuando el enfermo se encuentra en sus últimos días u horas de vida para aliviar un sufrimiento intenso. En esta situación la sedación es continua y tan profunda como sea necesario para aliviar dicho sufrimiento.
En dicha sedación se utilizan, además de los fármacos previamente explicados, se suelen usar los anticolinérgicos como el n-butilbromuro de hioscina o la escopolamina bromhidrato, ambos ayudan a evitar las secreciones bronquiales.
Sedación subcutánea
Midazolam → Dosis de inducción (DI): 2,5-5 mg/4 h + rescates (DR) 2,5-5 mg,
Dosis de infusión continua: DI + DR en las primeras 24 h→ a pasar en bomba 24 h o mg/h (dosis total/24). Rescates de ⅙ parte de la dosis de infusión.
Levomepromazina → el objetivo de usarla es disminuir el 50% de la dosis requerida de midazolam. DI 12,5-25 mg/6-h. La dosis de infusión continua: suma de las DI de las primeras 24 horas (habitualmente unos 100 mg/día).
Sedación intravenosa
Midazolam → DI 1,5-3,5 mg en bolo/5 min hasta conseguir el confort del paciente, una vez determinada la DI se multiplica por 6 y esa será la dosis de infusión continua (en 24 h o mg/h).
Clorpromacina → DI 12,5-25 mg/6-8h. Dosis de mantenimiento 12,5-50 mg/6-8h como máximo 300 mg/día.
N-butilbromuro de hioscina → 20 a 40 mg/8h (IV o SC).
Escopolamina bromhidrato → 0,5 a 1 mg/4h (IV o SC).
Morfina → suma de todas las dosis necesarias durante el día (tanto de base como de rescate) para calcular la dosis de infusión.
3. CONCLUSIONES
Es imprescindible un inicio precoz del cuidado paliativo en los pacientes con EPIDs, igual que es imprescindible que estos pacientes conozcan el tipo de enfermedad que padecen y la evolución de ésta. Esta es una labor que debe hacer el especialista en neumología que los diagnostica y trata.
Es importante un tratamiento personalizado y que requiere de un equipo multidisciplinar, por lo que es esencial valorar el estado de la enfermedad, los factores pronósticos y las comorbilidades/complicaciones de cada paciente.
Los cuidados paliativos en las EPIDs suelen contribuir en el mejor control de los síntomas durante la evolución de la enfermedad y así mantener en lo posible la autonomía del paciente y su calidad de vida.
Es imprescindible la monitorización de los síntomas de los pacientes y la titulación de los fármacos utilizados para el manejo de síntomas como para el control de efectos secundarios.
Es esencial el tratamiento psicoterapéutico tanto del paciente como de los familiares/ cuidadores para poder optimizar/programar los últimos días de vida y la muerte del paciente.
BIBLIOGRAFÍA
1. Guía de sedación paliativa de la organización médica colegial (OMC). Sociedad Español de Cuidados Paliativos (SECPAL).
2. Daisy J.A. Janssen Et al. European Respiratory Society clinical practice guideline: Palliative care for people with COPD or interstitial lung disease. Eur Respir J 2023; 62: 2202014
3. Lindell KO, Liang Z, Hoffman LA, et al. Palliative care and location of death in decedents with idiopathic pulmonary fibrosis. Chest 2015; 147:423-429.
4. Michael Kreuter, Elisabeth Bendstrup, et al. Palliative care in interstitial lung disease: living well. Lancet Respiratory Medicine - DOI: http://dx.doi.org/10.1016/S2213-2600(17)30383-1
5. Deepa Ramadurai , Stephanie Corder, et al. Idiopathic pulmonary fibrosis: Educational needs of health-care providers, patients, and caregivers. Chronic Respiratory Disease. Volume 16: 1–8
6. Kathleen Oare Lindell, Mehdi Nouraie et al. Randomised clinical trial of an early palliative care intervention (SUPPORT) for patients with idiopathic pulmonary fibrosis (IPF) and their caregivers: protocol and key design considerations. Lindell KO, et al. BMJ Open Resp Res 2018;5:e000272. doi:10.1136/bmjresp-2017- 000272 1
7. Antoni Xaubet, Julio Ancochea, Elena Bollo, et al. Normativa SEPAR. Normativa sobre el diagnóstico y tratamiento de la fibrosis pulmonar idiopática. Arch Bronconeumol. 2013;49(8):343-53.
8. Birring SS, Kavanagh JE, Irwin RS, Keogh KA, Lim KG, Ryu JH; Collaborators. Treatment of Interstitial Lung Disease Associated Cough: CHEST Guideline and Expert Panel Report. Chest. 2018 Oct;154(4):904-917. doi: 10.1016/j.chest.2018.06.038. Epub 2018 Jul 20. PMID: 30036496.
9. Johnson MJ, Currow DC. Opioids for breathlessness: a narrative review. BMJ Support Palliat Care. 2020 Sep;10(3):287-295. doi: 10.1136/bmjspcare-2020- 002314. Epub 2020 Jul 3. PMID: 32620683.
10. Abernethy AP, Currow DC, Frith P, et al. Randomised, double blind, placebo centrolled crossover trial of sustaine reléase morphine for the management of refractory dyspnea. BMJ. 2003; 327:523-8.
11. Polosa R, Simidchiev A, Walters EH. Nebulised morphine for severe interstitial lung disease. Cochrane Database of Systematic Reviews 2002, Issue 3. Art. No.: CD002872. DOI: 10.1002/14651858.CD002872
12. María Ángeles Campo Artola; Santiago González de Etxabarri Otsoa; María José Arrizabalaga Arnaiz; Miren Begoñe Salinas Lasa; Jaione Aguirrezabal Rementeria; Manuel Virosta Allegue. Utilidad de los fentanilos en el tratamiento de la disnea en enfermedad pulmonar intersticial difusa. Medicina paliativa, ISSN 1134-248X, Vol. 27, Nº. Extra 1, 2020 (Ejemplar dedicado a: Número Especial Comunicaciones - XIII JORNADAS INTERNACIONALES DE LA SOCIEDAD ESPAÑOLA DE CUIDADOS PALIATIVOS), págs. 66-67.
13. Fallon M, Reale C, Davies A, Lux AE, Kumar K, Stachowiak A, Galvez R. Efficacy and safety of fentanyl pectin nasal spray compared with inmediated-release morphine sulfate tablets in the treatment of breakthrough cancer pain: a multicenter, randomized, controlled, double- blind, double-dummi multiple-crossover study. The journal of supportive oncology. 2011;9(6):224-231.
14. Booth S, Wade R, Johnson M, Kite S, Swannick M, Anderson H: The use of oxygen in the palliation of breathlessness. A report of the expert working group of the Scientific Committee of the Association of Palliative Medicine. Respir Med 2004; 98: pp. 66-77.
TABLAS Y FIGURAS
Tabla I. Ejemplificación de la mejora de la atención de los pacientes y la optimización de recursos sanitarios con la implementación del modelo multidisciplinar en UCP4

Tabla II. Tipos de opioides de administración oral, sublingual y transmucoso (principo activo, normbre comercial y presentación), tiempo de acción y características específicas a tener en cuenta a la hora de seleccionar el tratamiento. AEMS Fichas técnicas 2017

Tmáx: tiempo expresado en minutos que indica el momento en el que el fármaco alcanza su concentración máxima en sangre.
Tabla III. Pautas de administración de los fármacos utilizados más habiutalmente en el tratamiento de la disnea

Tabla IV. Efectos secundarios más frecuentes de los opioides y su tratamiento
AEMS Fichas técnicas 2017

* Tabla elaborada a partir de: Guía rápida de uso seguro de opioides en pacientes en situación terminal (consejería de salud).

Patrocinado por:
