JAHRGANG 33
HEFT 2
Mai 2024

JAHRGANG 33
HEFT 2
Mai 2024
JOURNAL OF PHARMACOLOGY AND THERAPY
Spinale Muskelatrophie: Klassifikation, Diagnostik und therapeutische Möglichkeiten
Risdiplam – das erste orale Medikament zur Therapie der spinalen Muskelatrophie kann den Mangel an SMN-Protein kompensieren
Gentherapie verbessert motorische Meilensteine bei spinaler Muskelatrophie
Chronische spontane Urtikaria: Omalizumab-Autoinjektor mit 300-mg-Dosis erleichtert die Selbstapplikation
Lower Urinary Tract Symptoms: App-basierte Therapielösung verbessert Symptome und Lebensqualität
Hereditäres Angioödem: Überzeugende Langzeitergebnisse der oralen Prophylaxe mit Berotralstat
Abemaciclib verbessert Gesamtüberleben beim HR+, HER2– Mammakarzinom
Omaveloxolon – das erste Arzneimittel zur Behandlung der Friedreich-Ataxie
S1P-Rezeptor-Modulator Etrasimod schließt Therapielücken bei Colitis ulcerosa und isolierter Proktitis
Evrysdi® bewahrt und verbessert Lebensqualität und Unabhängigkeit im Alltag1,2 Mit Evrysdi® bereit für mehr.
> 15.000
Patient:innen weltweit mit Evrysdi® behandelt3
Stand: Februar 2024






* Evrysdi® wird angewendet zur Behandlung der 5q-assoziierten spinalen Muskelatrophie (SMA) bei Patient:innen mit einer klinisch diagnostizierten SMA Typ 1, 2 oder 3 oder mit einer bis vier Kopien des SMN2 -Gens. 4 1. Oskoui M et al. J Neurol 2023;3:1–16. 2. Oskoui M et al. MDA 2023; Poster. 3. Roche Data on file. 4. Fachinformation Evrysdi®, Stand: Februar 2024. Evrysdi® 0,75 mg/ml Pulver zur Herstellung einer Lösung zum Einnehmen
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Bitte melden Sie Nebenwirkungen an die Roche Pharma AG unter grenzach.drug_safety@ roche.com oder Fax +49 7624/14-3183 oder an das Bundesinstitut für Arzneimittel und Medizinprodukte unter www.bfarm.de oder Fax: +49 228/207-5207.
Wirkstoff: Risdiplam. Zusammensetzung: 1 Flasche enthält 60 mg Risdiplam in 2 g Pulver zur Herstellung einer Lösung zum Einnehmen. 1 ml der rekonstituierten Lösung enthält 0,75 mg Risdiplam. Sonstige Bestandteile: Mannitol (E 421), Isomalt (Ph.Eur.) (E 953), Erdbeer-Aroma, Weinsäure (Ph.Eur.) (E 334), Natriumbenzoat (E 211), Macrogol 6000, Sucralose, Ascorbinsäure (E 300), Natriumedetat (Ph.Eur.). Anwendungsgebiet: Evrysdi wird angewendet zur Behandlung der 5q-assoziierten spinalen Muskelatrophie (SMA) bei Patienten mit einer klinisch diagnostizierten Typ 1-, Typ 2- oder Typ 3-SMA oder mit einer bis vier Kopien des SMN2-Gens. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Nebenwirkungen: Diarrhö, Ausschlag (umfasst Dermatitis, akneiforme Dermatitis, allergische Dermatitis, Erythem, Follikulitis, Ausschlag, erythematösen Ausschlag, makulopapulösen Ausschlag, papulösen Ausschlag), Kopfschmerzen, Pyrexie (einschließlich Hyperpyrexie), Übelkeit, Mundgeschwüre und aphthöse Geschwüre, Harnwegsinfektionen (einschließlich Zystitis), Arthralgie, kutane Vaskulitis. Warnhinweise: Pulver nicht einatmen. Hautkontakt mit dem Pulver und der rekonstituierten Lösung vermeiden. Enthält auch Natriumbenzoat (E 211) und Isomalt (Ph.Eur.) (E 953). Verschreibungspflichtig. Hinweise der Fachinformation beachten. Pharmazeutischer Unternehmer: Roche Registration GmbH, Grenzach-Wyhlen, DE. Weitere Informationen auf Anfrage erhältlich. Vertreter in Deutschland: Roche Pharma AG, Grenzach-Wyhlen. Stand der Information: Februar 2024.
Es ist noch gar nicht so lange her, da sorgte ein Lebensmittelskandal allenthalben für Entrüstung. Hatten doch einige besonders geschäftstüchtige Produzenten von Tiefkühlpizzen und Co. statt (teurem) Käse (billigen) „Analogkäse“ verwendet. Anders als „richtiger“ Käse, der das Ergebnis teilweise zeitintensiver natürlicher Prozesse (Milchsäurebakterien!) ist, lässt sich Analogkäse „labortechnisch konstruieren“. Inzwischen ist diese Art von Food Design salonfähig geworden. Die Zahl der Produkte, die deswegen besonders teuer sind, weil sie irgendetwas nicht enthalten, steigt zusehends, ist aber irgendwie vom Spektrum her limitiert (laktosefrei, glutenfrei).
Ungleich viel mehr Möglichkeiten tun sich auf, wenn das „frei von“ in Bezug gesetzt wird zu „tierischen Bestandteilen“. Und da feiert der billige Analogkäse als „veganer Käse“ ein ebenso wirtschaftlich sensationelles Revival wie Analogfleisch, Analogwurst, AnalogEiprodukte u.v.m. Alles, was nicht unmittelbar gesundheitsschädlich ist, findet Verwendung, Hauptsache es ist billig und hat Materialeigenschaften, die eine Ähnlichkeit in Konsistenz und/oder Textur vortäuschen. Emulgatoren, Stabilisatoren, „Gewürze“, Farbstoffe perfektionieren die Täuschung. Grundzutaten sind meist pflanzliches Protein, Proteinkonzentrat oder Proteinisolat. Soja, Bohnen und andere Hülsenfrüchte mit hohem Proteingehalt werden typischerweise hochverarbeitet. Grundsätzlich, finde ich, sollte man bei jeder Erörterung des Themas vegane Ernährung zwei Bereiche strikt trennen. Bereich 1 ist das ethisch-moralische (auch: umweltbewusste) Motiv, das jeden ehrt, der diesbezüglich bewusst durchs Leben geht und das für mich grundsätzlich ebenso sakrosankt ist wie z.B. jede persönliche Glaubensüberzeugung. Als Arzt interessiert mich aber der 2. Bereich, nämlich der ge-
sundheitliche Aspekt. Und da bin ich vor Kurzem über einen „Umbrella-Review“ im British Medical Journal gestolpert [1]. Natürlich habe ich bei „hochverarbeiteten Lebensmitteln“ sofort an Analogkäse, Tofuschnitzel und vegane Bratwürste gedacht. Das war zugegebenermaßen etwas voreingenommen. Greift man sich einen der wöchentlich den Briefkasten verstopfenden Prospekte einer der vier führenden Lebensmittelketten, so findet man typischerweise nur wenige naturbelassene oder wenig bearbeitete Lebensmittel. Das Gros der Angebote ist hochverarbeitet, vom Fruchtpudding über Dosenravioli bis hin eben zur Veggie-Bratwurst.
Der Umbrella-Review im BMJ konnte sich immerhin auf nicht weniger als 45 epidemiologische Datenanalysen mit fast 10 Millionen Studienteilnehmern stützen. Dabei fanden die Autoren „einen direkten Zusammenhang zwischen dem Konsum von hochverarbeiteten Lebensmitteln und 32 Gesundheitsparametern, u.a. Mortalität, maligne Erkrankungen sowie psychische, respiratorische, kardiovaskuläre, gastrointestinale und metabolische gesundheitliche Outcomes“.
Damit sollte die Frage nach den Auswirkungen hochverarbeiteter Lebensmittel auf Morbidität und Mortalität überzeugend beantwortet sein! Wirklich? Schon der Titel gibt einen Hinweis auf ein grundsätzliches Problem: Die Grundlage waren epidemiologische MetaAnalysen. Die epidemiologischen Studien regelhaft zu Grunde liegende Methodik ist folgende: Man befragt eine große Anzahl „Teil-

Prof. Dr. med. Karl-Ludwig Resch
nehmer“ (Auswahl und Zusammensetzung der Kohorte liegt im Ermessen der jeweiligen Studienleitung) und macht umfangreiche Untersuchungen zum Gesundheitsstatus. Nach einer gewissen Zeit (meist mehrere Jahre) erfolgt eine erneute Untersuchung. Für alle Teilnehmer mit neu aufgetretenen Gesundheitsstörungen greift man auf die Daten der Befragung zu Studienbeginn zurück und kann dann statistisch errechnen, ob sich die Anzahl der neu aufgetretenen Gesundheitsstörungen z.B. zwischen den Menschen, die zu Beginn angegeben hatten, viel oder wenig hochverarbeitete Lebensmittel zu konsumieren, signifikant unterscheidet. Ist das der Fall, kann der Konsum hochverarbeiteter Lebensmittel tatsächlich die Ursache für ein erhöhtes Krankheitsrisiko sein. Allerdings könnte der Konsum hochverarbeiteter Lebensmittel primär auch ein Indikator dafür sein, ob
Menschen ihre Gesundheit wichtig nehmen. Dann würden Menschen, denen ihre Gesundheit wichtig ist, sich wahrscheinlich auch mehr körperlich bewegen, weniger rauchen und/oder Alkohol konsumieren, hochwertigere Lebensmittel kaufen, sich mehr Mühe bei der Zubereitung geben, einfach achtsamer durchs Leben gehen. Ob das alles zusammen der Gesundheit zuträglich ist, dazu wird es wegen der exorbitanten Komplexität des Konstrukts zwar niemals einen Umbrella-Review geben. Dafür brauchen wir aber ganz sicher auch keine künstliche Intelligenz, vielmehr unsere eigene – und insbesondere das, was sich auch in der medizinischen Forschung zunehmend zum raren Gut entwickelt: gesunden Menschenverstand. Und das ist für mich die wichtigste Erkenntnis aus dem, formalmethodisch hochanspruchsvollen, für die Fragestellung eher untauglichen und damit am Ende überflüssigen Umbrella-Review. Um die Frage „vernünftig“ zu beantworten, muss man wohl nicht einmal Medizin studiert haben und erst recht kein Wissenschaftler sein. Deshalb konnte das schon vor fast 150 Jahren ein schwäbischer Landpfarrer, Sebastian Kneipp, ein aufmerksamer Beobachter mit einer besonders großen Portion gesundem Menschenverstand, treffend auf den Punkt bringen: „Lasst das Natürliche so natürlich wie möglich. Die Zubereitung der Speisen soll einfach und ungekünstelt sein. Je näher sie dem Zustande kommen, in welchem sie von der Natur geboten werden, desto gesünder sind sie“.
Karl-Ludwig Resch, Nürnberg
Quelle
Lane MM et al. Ultra-processed food exposure and adverse health outcomes: umbrella review of epidemiological meta-analyses. BMJ 2024;384:e077310. PMID: 38418082
Spinale Muskelatrophie: Klassifikation, Diagnostik und therapeutische Möglichkeiten 36 Brigitte Söllner
Risdiplam – das erste orale Medikament zur Therapie der spinalen Muskelatrophie kann den Mangel an SMN-Protein kompensieren 40
Gentherapie verbessert motorische Meilensteine bei spinaler Muskelatrophie
44
Chronische spontane Urtikaria: Omalizumab-Autoinjektor mit 300-mg-Dosis erleichtert die Selbstapplikation 46
Lower Urinary Tract Symptoms: App-basierte Therapielösung verbessert Symptome und Lebensqualität
Hereditäres Angioödem: Überzeugende Langzeitergebnisse der oralen Prophylaxe mit Berotralstat
47
50
Abemaciclib verbessert Gesamtüberleben beim HR+, HER2– Mammakarzinom 51
Omaveloxolon – das erste Arzneimittel zur Behandlung der Friedreich-Ataxie 52
S1P-Rezeptor-Modulator Etrasimod schließt Therapielücken bei Colitis ulcerosa und isolierter Proktitis 56
45, 63 Kongresse 60

Hinter einer Entwicklungsverzögerung bei Jungen kann mehr stecken. Helfen Sie Duchenne-Muskeldystrophie (DMD) auszuschließen!
WICHTIGE MERKMALE DER DMD:
• Kennzeichen der DMD sind progrediente Muskeldystrophie und Atrophie
• Unspezifische Frühsymptome können bereits im Säuglingsalter auftreten1
• Auftreten der Symptome im Alter von 2–5 Jahren2–4
SCHNELLE DIAGNOSE FÜR
IHRE PATIENTEN:
Zutreffen von Kriterium 1 und mindestens eines der Kriterien 2– 4 rechtfertigt ein selektives CK-Screening, um den Verdacht auf das Vorliegen einer DMD zu erhärten
Weitere Informationen finden Sie unter www.duchenne.de

1. Männliches Geschlecht (obligat)
2. Unspezifische Entwicklungsverzögerung (betrifft sowohl die Motorik, das Lernen als auch die Sprache)
3. Nichterreichen des freien Laufens mit 18 Monaten
4. Unklare Erhöhung der Transaminasen
Bei der 5q-assoziierten spinalen Muskelatrophie (SMA) handelt es sich um eine seltene autosomalrezessiv vererbte neuromuskuläre Erkrankung, die durch die Degeneration und den Verlust von Motoneuronen im Rückenmark und Hirnstamm charakterisiert ist und zu einem allmählichen Verlust der Bewegungsfähigkeit führt. Ursache ist ein Defekt im Gen SMN1, der dazu führt, dass das Protein Survival Moto Neuron (SMN), das für das Überleben der Mononeuronen essenziell ist, nicht mehr in ausreichender Menge produziert wird. Ein zweites, ebenfalls für SMN codierendes Gen, SMN2, ist zwar in mehreren Kopien vorhanden, produziert aber nur etwa 10 % intaktes Protein und kann daher den Verlust durch den Defekt im SMN1-Gen nicht komplett ausgleichen. Je nach Anzahl der SMN2-Genkopien verläuft die Erkrankung unterschiedlich schwer. Nach den maximal erreichbaren Fähigkeiten der Patienten wird unterschieden zwischen Non-Sittern, Sittern und Walkern. Ist die Diagnose durch einen Gentest gesichert, sollte unverzüglich mit der Behandlung begonnen werden. Neben Mitteln zur Linderung der Symptome stehen heute auch Medikamente zur Verfügung, die eine kausale Therapie ermöglichen: das Antisense-Oligonukleotid Nusinersen, die Gentherapie Onasemnogene Abeparvovec sowie der Spleißmodifikator Risdiplam.
Schlüsselwörter: spinale Muskelatrophie, Motoneuronerkrankungen, Bewegungsunfähigkeit, Gendefekt, Nusinersen, Onasemnogene Abeparvovec, Risdiplam
Söllner, Erlangen
Unter dem Formenkreis „Spinale Muskelatrophien“ wird eine Gruppe von genetisch bedingten Krankheiten zusammengefasst, bei denen es infolge eines Gendefekts zu einer fortschreitenden Degeneration von Alpha-Motoneuronen im Rückenmark und im unteren Hirnstamm kommt. Dadurch können elektrische Impulse vom Gehirn nicht mehr an die angeschlossenen Muskeln weitergeleitet werden. Die Folge ist eine progressive Muskelatrophie, die zu einem allmählichen Verlust der Bewegungsfähigkeit führt [1].
Defekte Gene als Ursache
Die Bezeichnung „spinale Muskelatrophie“ wird meist synonym für die am häufigsten (ca. 95 % aller Fälle*) auftretende Erkrankung aus diesem Formenkreis gebraucht, die autosomal-rezessive 5q-assoziierte spinale Muskelatrophie (SMA). Bei dieser Form liegt ein Defekt im SMN1-Gen vor, das auf dem Chromosom 5q lokalisiert ist und für das Protein
* In etwa 5 % der SMA-Fälle liegt eine Nicht-5q-SMA vor, die eine Gruppe seltener und atypischer genetisch heterogener Motoneuronerkrankungen umfasst. Sie werden durch genetische Mutationen außerhalb von Chromosom 5q verursacht und können in der Regel aufgrund ihrer klinischen Manifestation sicher von der 5q-SMA abgegrenzt werden [7, 8].
Prävalenz und Inzidenz
Die spinale Muskelatrophie wird durch ein defektes Gen auf Chromosom 5q ausgelöst. Da die Erkrankung rezessiv vererbt wird, müssen sowohl Mutter als auch Vater der Betroffenen eine defekte Genkopie aufweisen und diese jeweils weitergeben. Etwa jeder 50. Mensch trägt eine solche defekte Genkopie [2]. Weltweit erkrankt durchschnittlich etwa eines von 6.000 bis eines von 10.000 Neugeborenen an SMA [2]. Diese ist damit die häufigste autosomal-rezessive Erkrankung mit Todesfolge im Kleinkindalter [3]. In Deutschland leben etwa 2.000 Menschen mit SMA [4].
Survival Moto Neuron (SMN) codiert [5]. Dieses Protein ist für das Überleben von Motoneuronen von zentraler Bedeutung. Patienten mit SMA produzieren zu wenig SMNProtein, sodass Motoneuronen im Rückenmark zugrunde gehen. Da SMN auch an anderen grundlegenden zellulären Vorgängen wie z.B. an der Expression Proteincodierender Gene und an fast allen Prozessen des RNA-Metabolismus beteiligt ist, ist ein SMN-Mangel außer mit muskulären Defiziten bis hin zur vollständigen Bewegungs-
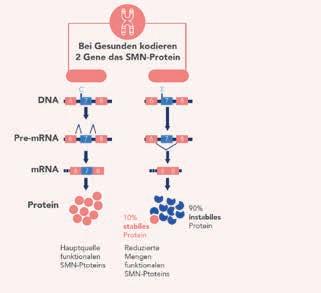
unfähigkeit und zur Notwendigkeit einer künstlichen Beatmung u.a. auch mit einer eingeschränkten Leberfunktion und kardialen Arrhythmien assoziiert, was je nach Schweregrad zu schwersten, lebensbedrohlichen Zuständen bis hin zum Tod führen kann [6]. Neben SMN1 gibt es aufgrund einer genomische Duplikation mit SMN2 noch ein weiteres Gen, das für das SMN-Protein codiert. Beide Gene unterscheiden sich in einer relevanten Basenpaarposition an Stelle 840: Ein voll funktionsfähiges SMN1-Gen enthält an dieser Stelle Cytosin, das SMN2Gen dagegen Thymin. Diese minimale Veränderung führt dazu, dass mit einer Wahrscheinlichkeit von 80 – 90 % in den SMN2-Transkripten das Exon 7 herausgeschnitten wird. Das daraus entstehende Protein SMNΔ7 ist verkürzt, funktionsuntüchtig und wird rasch abgebaut (Abb. 1) [3]. Von hoher Relevanz ist, dass jeder Mensch über mehrere Kopien des SMN2Gens verfügt, wobei die Anzahl zwischen 0 und 8 variiert. Da jede SMN2-Kopie immerhin etwa ca. 10 % funktionales SMN-Protein produzieren kann, steht bestenfalls eine kleine Menge funktionelles Protein zur Verfügung, das zwar die Lebensfähigkeit aufrechterhalten, das Fehlen von SMN1 jedoch nicht ausgleichen kann, sodass die Erkrankung kontinuierlich fortschreitet. Die Schwere der SMA hängt daher zum großen Teil von der Anzahl der intakten Kopien des SMN2-Gens ab, die die Funktion des defekten SMN1-Gens kompensieren können [3].
Klassifikation
Die SMA ist die häufigste Ursache für die genetisch bedingte Säug-
Eine C-T-Mutation der SplicingStelle bedingt, dass an SMN2 überwiegend SMN-Protein mit aberranter Struktur entsteht
Der Großteil des vom SMN2Gen produzierten Proteins ist instabil: SMN∆7-Protein wird rasch abgebaut
Abbildung 1: Einfluss der Gene SMN1 und SMN2 auf die Schwere der spinalen Muskelatrophie. Bei Gesunden produziert das Überlebens-Motoneuron-1-Gen SMN1 100 % funktionsfähiges SMN-Protein, während das SMN2-Gen etwa 10 % intaktes und etwa 90 % eines nicht funktionellen Proteins ohne Exon 7 (SMNΔ7) produziert. Bei SMA-Patienten geht das SMN1-Gen aufgrund von Mutationen oder Deletionen verloren. SMN2 bleibt bestehen und die geringe Menge an intaktem produziertem SMN reicht zum Überleben aus. Die Anzahl der SMN2-Kopien korreliert mit der Schwere der Erkrankung, wobei eine geringere Kopienzahl mit schwereren SMA-Typen verbunden ist [3].
lingssterblichkeit. In Deutschland ist etwa 1 von 7000 Neugeborenen betroffen [7]. Je nach Schweregrad der Erkrankung durchlaufen die Betroffenen zunächst eine unauffällige motorische Entwicklung, bevor sich die Erkrankung klinisch manifestiert und sich die motorischen Funktionen zunehmend verschlechtern. Nach den maximal erlangten motorischen Fähigkeiten (niemals Sitzen, Sitzen, Gehen) und dem Beginn der Erkrankung wurden die Patienten ursprünglich 3 Typen zugeordnet (Tab. 1) [5]. Bei dieser Klassifizierung der SMA sind die Übergänge zwischen den einzelnen Typen fließend. In der Praxis wird die Erkrankung heute anhand ihres aktuellen funktionel-
len Status unterteilt. Denn das hat den Vorteil, dass die Versorgung mit neuen Therapien optimal auf die Bedürfnisse der Menschen mit SMA angepasst werden kann [8]. In der 2020 revidierten Klassifikation werden die folgenden Typen unterschieden:
Non-Sitter haben das freie Sitzen nie erlernt oder wieder verloren. Sie haben eine stark ausgeprägte axiale und proximale Muskelschwäche und weisen eine zunehmend schwächere Gesichtsund Bulbärmuskulatur auf. Die Lungenfunktion ist häufig stark beeinträchtigt, die Betroffenen benötigen daher oft eine nicht invasive oder invasive Beatmung.
SMA Typ 1 Bei dieser schwersten Form der Erkrankung treten initiale klinische Anzeichen schon in den ersten 6 Lebensmonaten auf. Die Säuglinge haben bereits eine deutlich verminderte Muskelspannung („floppy infant“) und erreichen in ihrer motorischen Entwicklung nie die Fähigkeit, selbstständig zu sitzen. Die Lebenserwartung dieser Kinder ist stark eingeschränkt, zwei Drittel von ihnen versterben aufgrund einer respiratorischen Insuffizienz bereits in den ersten beiden Lebensjahren. Die SMA Typ 1 wurde erstmals von Werdnig und Hoffmann Ende des 19. Jahrhunderts beschrieben und auch nach ihnen benannt (Morbus Werdnig-Hoffmann, akute infantile SMA).
SMA Typ 2 Bei Kindern mit SMA Typ 2 manifestiert sich die Erkrankung in der Regel nach dem 6. und noch vor dem 18. Lebensmonat. Die Betroffenen können selbstständig sitzen, sind aber niemals in der Lage zu stehen oder zu gehen. Auch bei dieser Verlaufsform ist die Lebenserwartung deutlich reduziert. Die SMA Typ 2 wird auch als „chronische spätinfantile SMA“ oder als „intermediäre SMA“ bezeichnet.
SMA Typ 3 Diese weitere, langsamere Verlaufsform wird nach ihren Erstbeschreibern auch als Kugelberg-Welander-Form bezeichnet. Das Manifestationsalter liegt zwischen >18 Monaten und 30 Jahren. Die Betroffenen erreichen zwar die Gehfähigkeit, können diese aber im Verlauf wieder verlieren.
Tabelle 1: Ursprüngliche Klassifizierung der Phänotypen der SMA nach dem Beginn der Erkrankung und den maximal erlangten motorischen Fähigkeiten [5].
Außerdem können Kontrakturen und schwere Skoliosen operative Eingriffe nötig machen.
Sitter können für mindestens 10 Sekunden frei sitzen (aufrecht ohne Verwendung der Arme zur Unterstützung oder Balance, der Patient kann in die Sitzposition gebracht werden). Neben einer plegischen Beinmuskulatur besteht eine schwere Armschwäche, distale Muskeln sind besser erhalten. Typisch ist zudem eine Areflexie. Im Verlauf können sich Kontrakturen und Skoliosen entwickeln. Eine respiratorische Beteiligung ist geringer ausgeprägt als bei Non-Sittern, meist wird daher keine oder nur eine nächtliche nicht invasive Beatmung benötig.
Walker können über mindestens 10 Meter frei gehen (aufrecht mit gerader Wirbelsäule ohne Kontakt mit einem Objekt oder einer Person). Die Muskelschwäche ist bei diesen Betroffenen typischerweise proximal betont und in den unteren Extremitäten stärker ausgeprägt als in den oberen. Die Reflexe können in den oberen Extremitäten normal, abgeschwächt oder wie an den unteren Extremitäten nicht auslösbar sein. Walker verfügen über eine normale respiratorische
Funktion, in der Regel besteht keine faziale oder bulbäre Schwäche.
In Deutschland wird seit 2021 fast jedes Neugeborene routinemäßig auf das Vorliegen einer SMA untersucht [9]. Für diese Gentests werden nur wenige Tropfen Blut benötigt, sodass sie sich für jede Altersgruppe inklusive Säuglingen eignen. Bei einem positiven Ergebnis wird die Anzahl der SMN2Genkopien bestimmt. Im Zuge des Neugeborenenscreenings wird die homozygote Deletion des Exon 7 im SMN1-Gen detektiert, die in mehr als 95 % der Fälle für die SMA verantwortlich ist. Kinder mit Compound-Heterozygotie (< 5 %) werden nicht erkannt [9].
Eine wichtige Säule in der Behandlung von SMA-Patienten ist die langfristige, symptomorientierte, patientenindividuelle multidisziplinäre Versorgung. Sie beinhaltet insbesondere die Optimierung der Ernährungssituation, die Unterstützung der Respiration, Logopädie und physiotherapeutische Be-
gleitung sowie die orthopädische Hilfs- und Rehamittel-Versorgung [8].
Neben dieser Basistherapie zur Symptomlinderung stehen mittlerweile auch 3 krankheitsmodifizierende Therapieoptionen zur Verfügung:
Antisense-Oligonukleotid Nusinersen
Seit 2017 ist das Antisense-Oligonukleotid Nusinersen (Spinraza®) für die Behandlung von Patienten mit SMA zugelassen [10]. Die kurze synthetischen Nukleotidkette bindet selektiv an an einen regulierenden Abschnitt im SMN2-Gen und verhindert so, dass das Exon 7 beim Spleißen – dem Zusammensetzen der Messenger-RNA aus den einzelnen Exons – übersprungen wird [11]. Auf diese Weise kann vom SMN2-Gen mehr funktionsfähiges SMN-Protein produziert werden. Der Spleißmodulator reguliert indirekt die Genexpression – das Genom wird dadurch nicht verändert. Nusinersen muss nach einer initialen Anreicherungsphase in regelmäßigen Abständen per Lumbalpunktion in die Zerebrospinalflüssigkeit injiziert werden (4 Aufsättigungsdosen innerhalb
der ersten 2 Monate, danach in 4-monatigen Intervallen). Nusinersen ist indiziert zur Behandlung der 5q-assoziierten SMA [10].
Gentherapie Onasemnogene Abeparvovec
Neben Nusinersen ist seit 2020 in Europa auch die Gentherapie Onasemnogene Abeparvovec (Zolgensma®) für die Behandlung der SMA zugelassen [12]. Dabei wird eine funktionstüchtige Kopie des humanen SMN1-Gens mittels eines adeno-assoziierten viralen Vektors (scAAV9-SMN) in Zellen des Patienten eingeschleust [13]. Das scAAV9-SMN wird mit einer einmaligen intravenösen Injektion verabreicht und überwindet die Blut-Hirn-Schranke, um an die Zielzellen, insbesondere Motoneuronen im ZNS, zu gelangen. Das Genkonstrukt wird nicht ins menschliche Genom integriert. Sich nicht teilende Zellen (z.B. Neuronen) sollten lebenslänglich das Virus exprimieren, in sich teilenden Zellen, wird die VektorAnzahl mit jeder Zellteilung reduziert. Onasemnogene Abeparvovec ist indiziert zur Behandlung von Patienten mit 5q-assoziierter SMA mit einer biallelischen Mutation im SMN1-Gen und einer klinisch diagnostizierten SMA Typ 1 sowie von Betroffenen mit 5q-assoziierter SMA mit einer biallelischen Mutation im SMN1-Gen und bis zu 3 Kopien des SMN2-Gens [12].
Spleißmodifikator Risdiplam
2021 erhielt Risdiplam (Evrysdi®) die Zulassung in der EU zur Behandlung von Patienten ab 2
Monaten mit einer klinisch diagnostizierten Typ 1-, Typ 2- oder Typ 3-SMA oder mit 1 – 4 Kopien des SMN2-Gens. Im August 2023 folgte die Zulassungserweiterung für Kinder unter 2 Monaten [14]. Damit können nun Patienten aller Altersgruppen ab der Geburt mit Risdiplam behandelt werden [13]. Risdiplam korrigiert das Spleißen von SMN2, sodass das Exon-7 in das mRNA-Transkript eingeschlossen wird. Dies führt zu einer erhöhten Produktion von funktionellem und stabilem SMN-Protein. Im Gegensatz zu Nusinersen wird Risdiplam oral appliziert. Risdiplam zählt zur Gruppe der „Small Molecules“, die nach oraler Verabreichung potenziell eine systemische Wirkung entfalten und die Blut-Hirn-Schranke passieren können [14]. Derzeit unterliegt das Arzneimittel einer zusätzlichen Überwachung, um eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit zu ermöglichen.
Die 5q-spinale Muskelatrophie ist grundsätzlich behandelbar und sollte zeitnah erkannt werden. Von einer frühen Diagnose und einer rasch eingeleiteten Behandlung hängt gerade bei den schweren Verlaufsformen dieser autosomalrezessiv vererbten neuromuskulären Erkrankung der Erfolg der Therapie ab. Innovative Medikamente wie das Antisense-Oligonukleotid Nusinersen, die Gentherapie Onasemnogene Abeparvovec und der Spleißmodifikator Risdiplam ermöglichen erstmals eine kausale Therapie, von der in ersten Studien bereits zahlreiche Patienten profi-
tieren konnten (siehe dazu die Beiträge auf S. 40 und 44).
1 Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018; 28:103-115
2 Verhaart IEC et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 2017;12:124
3 Bowerman M et al. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis Model Mech 2017;10:943954
4 Servais L. Newly evolving phenotypes in neuromuscular diseases due to novel treatments. Präsentiert am ICNMD 2022
5 Kolb SJ et al. Spinal muscular atrophy. Neurol Clin 2015;33:831-846
6 Lauria F et al. SMN-primed ribosomes modulate the translation of transcripts related to spinal muscular atrophy. Nat Cell Biol 2020;22:1239-1251
7 Vill K et al. One year of newborn screening for SMA – results of a German pilot project. J Neuromuscul Dis 2019;6:503515
8 Kölbel H et al. S1-Leitlinie Spinale Muskelatrophie (SMA), Diagnostik und Therapie. AWMF-Register-Nr. 022-030. https://register.awmf.org/de/leitlinien/ detail/022-030
9 Müller-Felber W et al. From pilot project to nationwide screening in Germany. J Neuromuscul Dis 2023;10:55-65
10 Fachinformation Spinraza®; Stand: Januar 2022
11 Godfrey C et al. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol Med 2017;9:545-557
12 Fachinformation Zolgensma®; Stand: März 2023
13 Parente V et al. Advances in spinal muscular atrophy therapeutics. Ther Adv Neurol Disord 2018;11:17562856187545 01
14 Fachinformation Evrysdi®; Stand: August 2023
Anschrift der Verfasserin: Brigitte Söllner Medizinjournalistin und Wissenschaftliche Lektorin Lärchenweg 10 91058 Erlangen brigitte.soellner@online.de
Die spinale Muskelatrophie (SMA) zählt zu den häufigsten genetisch bedingten Todesursachen bei Säuglingen und Kleinkindern. In Deutschland ist etwa 1 von 7000 Neugeborenen betroffen [1]. Sie ist charakterisiert durch eine fortschreitende Degeneration der Alpha-Motoneuronen im Rückenmark und unteren Hirnstamm, was zu einem allmählichen Verlust der Bewegungsfähigkeit führt [2]. Lange Zeit konnte den betroffenen Kindern nur durch supportive, symptomlinderne Maßnahmen geholfen werden, erst seit wenigen Jahren gibt es Therapien, die an der Ursache der SMA, dem Gendefekt, ansetzen. Mit Risdiplam (Evrysdi® 0,75 mg/ml Pulver zur Herstellung einer Lösung zum Einnehmen) steht nun erstmals eine nicht invasive kausale Therapie zur Verfügung, die bereits ab der Geburt oral zuhause angewendet werden kann, was für die betroffenen Kinder von großem Vorteil ist [3].
Gendefekte führen zu einem Mangel an lebenswichtigem SMN-Protein
Ursache der SMA ist ein autosomal-rezessiv vererbter Defekt im Gen SMN1, das für das Protein Survival Moto Neuron (SMN) codiert. Dieses Protein ist für das Überleben der Mononeurone essenziell. Kann es nicht mehr in ausreichender Menge produziert werden, gehen die Motoneurone im Rückenmark zugrunde. Glücklicherweise gibt es noch ein zweites Gen, das für die Produktion von SMN codiert. Dieses SMN2-Gen kommt individuell in variabler Kopienzahl vor, kann jedoch nur etwa 10 % funktionierendes SMN-Protein bilden. Denn
aufgrund einer Punktmutation geht mit einer Wahrscheinlichkeit von 80 – 90% der codierende Genabschnitt Exon 7 des SMN2-Gens durch sog. alternatives Spleißen auf dem Weg der Übersetzung genetischer Information in das SMN-Protein verloren. Das dabei entstehende Protein SMNΔ7 ist verkürzt, funktionsuntüchtig und wird rasch abgebaut [4]. Das SMN2-Gen kann die fehlende Funktion des defekten SMN1Gens daher nur teilweise kompensieren, sodass die Erkrankung kontinuierlich fortschreitet. Dabei korreliert die Anzahl der intakten SMN2-Kopien mit der Schwere der Erkrankung: Bei Patienten mit der schwersten Form (Typ 1) finden sich nur 1 – 2 Kopien [3], wogegen es bei milderen SMAFormen (Typ 2 und 3) sehr häufig 3 – 4 Kopien sind [4]. Und genau das spielt beim Wirkmechanismus von Risdiplam eine wichtige Rolle.
Spleiß-Modifikator Risdiplam erhöht Expression von voll funktionsfähigem SMN
Risdiplam (Evrysdi®) ist ein Arzneimittel aus der Klasse der niedermolekularen Arzneistoffe
(Small Molecules), die die BlutHirn-Schranke passieren können. Es wurde 2021 in Deutschland zur Behandlung der 5q-assoziierten spinalen Muskelatrophie bei Patienten ab 2 Monaten mit einer klinisch diagnostizierten Typ-1-, Typ2- oder Typ-3-SMA oder mit 1 – 4 Kopien des SMN2-Gens zugelassen und wird oral eingenommen. Im August 2023 erhielt Risdiplam aufgrund der Ergebnisse einer Interimsanalyse der RAINBOWFISHStudie (s.u.) die Zulassungserweiterung für Kinder unter 2 Monaten und kann seitdem bereits ab der Geburt angewendet werden [3]. Die Lösung wird einmal täglich peroral nach einer Mahlzeit und immer ungefähr zur gleichen Uhrzeit mit einer wiederverwendbaren Applikationsspritze oder über eine Sonde zugeführt. Als Spleißmodifikator erhöht Risdiplam die Bildung von funktionsfähigem SMN-Protein auf Basis des SMN2-Gens sowohl im ZNS als auch in der Peripherie und kompensiert so den SMN1-Gendefekt. Dadurch wird bei den SMA-Patienten dem Untergang von Motoneuronen, dem fortschreitenden Muskelschwund sowie den Folgen des SMN-Proteinmangels im gesamten Körper entgegengewirkt.
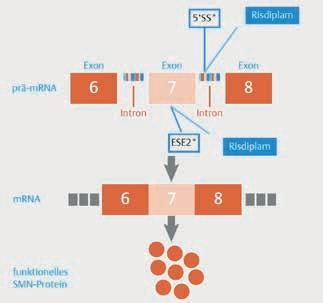
Abbildung 1: Wirkmechanismus von Risdiplam (Evrysdi®). Risdiplam korrigiert das Spleißen von SMN2, sodass das Exon 7 in das mRNA-Transkript eingeschlossen wird. Dies führt zu einer erhöhten Produktion von funktionellem und stabilem SMN-Protein [3].
Der Wirkstoff bindet an 2 Stellen der prä-mRNA: zum einen im Bereich der 5‘-Spleißstelle von Intron 7 und zum anderen an einer Verstärkersequenz im Exon [5]. Diese Bindung an 2 Stellen bewirkt, dass wichtige Kompo-
Sitzen ≥ 5 Sekunden* 2 24 Monate Behandlungsdauer 35/58 4 48 Monate Behandlungsdauer
%
Sitzen ≥ 5 S e kunden*
nenten des Spleißapparats, vor allem das U1-snRNP, besser binden können. In der Folge erhöht sich die Wahrscheinlichkeit, dass das Exon 7 in der mRNA des SMN2Gens eingeschlossen ist [5]. Damit können die Körperzellen
funktionsfähiges SMN-Protein in größerer Menge herstellen.
Studien zeigen hohe und konstante Wirksamkeit
Die Wirksamkeit und Sicherheit von Risdiplam werden derzeit in einem umfangreichen Studienprogramm geprüft:
FIREFISH-Studie
In die Phase-II/III-Zulassungsstudie FIREFISH [6] wurden 58 therapienaive Säuglinge im Alter von 1 – 7 Monaten mit bestätigter Diagnose einer SMA Typ 1 und mit 2 SMN2-Genkopien eingeschlossen. Teil 1 von FIREFISH war die Dosisfindungsphase der Studie, im zweiten Teil wurde die Wirksamkeit von Risdiplam geprüft. Endpunkt von Teil 2 war der Anteil der Patienten, die nach 12-monatiger Behandlung mindestens 5 Sekunden lang ohne Unterstützung sitzen konnten. Dies gelang 29 % der Kinder. Fast 93 % waren noch am Leben [6].
Nach 24 Monaten unter Behandlung mit Risdiplam konnten 60 % (35/58) der Säuglinge mindestens
Sitzen ≥ 30 Sekunden* Stehen* Gehen* 3/58 1/58 * ohne Un terstützung
Sitzen ≥ 30 Sekunden* Stehen*
Seite 1/3 37/58
Abbildung 2: Ergebnisse der FIREFISH-Studie mit 58 Säuglingen im Alter von 1 – 7 Monaten mit einer SMA Typ 1 und 2 SMN2-Genkopien unter der Therapie mit 1 täglich Risdiplam (Evrysdi®) [6].
zwischen 2 und 25 Jahren mit bestätigter Diagnose einer SMAb Typ 2 oder Typ 316
• doppelblinde, randomisierte, placebokontrollierte, zweiteilige, klinische Phase-2/3-Studie16 zum Therapiebeginn mit Risdiplam gemäß Motor Function Measure (MFM)-32Skalad. In den ersten 12 Monaten nahm der MFM-32-Gesamtscore verglichen mit dem Ausgangswert im Mittel um 2 Punkte zu.17 In den darauffolgenden Monaten konnten die Fortschritte, die unter der Risdiplam-Behandlung in Monat 12 beobachtet wurden, in Monat 48 beibehalten oder verbessert werden.17 Der MFM32-Gesamtscore war nach 12 bzw. 24 Monaten bei mehr als 50% der Behandelten signifikant und klinisch relevant um mindestens 3 Punkte höher als zu Beginn.17
MFM-32-Score (Motor Function Measure 32-items)
Arme bewegen Kopf bewegen
Hände/ Finger bewegen
Veränderung zur Baseline (± 95%-KI)
Evrysdi® (Monate 0–48)
Placebo (Monate 0–12)
Stehen/ Laufen 0612182430364248
Untersuchung (Monat)
Evrysdi® (n)
Placebo (n) 59575858
Die meisten der mit Risdiplam behandelten Säuglinge waren in Monat 12 in der Lage, selbstständig zu sitzen; viele konnten stehen und gehen (gemessen anhand des HINE-2-Tests)
2 SMN2-Kopien: Ergebnisse zu Monat 12 (n=8)*
Abbildung 3: Ergebnisse der placebokontrollierten SUNFISH-Studie bei Kindern und Jugendlichen im Alter zwischen 2 und 25 Jahren mit bestätigter Diagnose einer SMA Typ 2 oder Typ 3 [7].
RAINBOWFISH | vorläufige Ergebnisse4
STUDIENDESIGN
5 Sekunden sitzen, 40 % (23/58) sogar mindestens 30 Sekunden. Nach 48 Monaten Behandlungsdauer erhöhte sich der Anteil der Säuglinge, die mindestens 5 Sekunden sitzen konnten, auf 64 % (37/58) und der der Anteil der Kinder, die mindestens 30 Sekunden sitzen konnten, auf 62 % (36/58). Ohne Unterstützung stehen konnten nach 48 Monaten Behandlungsdauer 3 Kinder und ohne Unterstützung gehen konnte 1 Kind (Abb. 2).
Sitzen ohne Unterstützung Motorische Meilensteine innerhalb der WHO-Fenster für gesunde Kinder zu Monat 12
ERGEBNISSE
motorischen Fähigkeiten zu Monat 12 im Vergleich zum Therapiebeginn gemäß Motor Function Measure (MFM)-32-Skala.
• therapienaive, präsymptomatischec Neugeborene zwischen 0 und 6 Wochen mit SMAb gemäß genetischem Nachweis einer homozygoten Loss-of-Function-Mutation im SMN1-Gen18
Eigenständiges Stehen
• open-label, multizentrische, klinische Phase2-Studie18
7/8 Kindern (88 %) konnten sitzen, ohne Unterstützung
4/8 Kindern (50 %) konnten stehen, ohne Hilfsmittel
≥ 3 SMN2-Kopien: Ergebnisse zu (n=18)†
Die meisten der mit Risdiplam behandelten Säuglinge waren in Monat 12 in der Lage, selbstständig zu sitzen; viele konnten stehen und gehen (gemessen anhand des HINE-2-Tests)
Sitzen ohne Unterstützung Motorische Meilensteine innerhalb der WHO-Fenster für gesunde Kinder zu Monat 12
Eigenständiges Gehen
Eigenständiges Stehen
SUNFISH-Studie
Eigenständiges Gehen
Primärer Endpunkt: Anteil der Studienteilnehmer:innenf, die zu Monat 12 ohne Unterstützung sitzen können. Der primäre Endpunkt wurde erreicht. 7/8 Kindern (88%) mit 2 SMN2-Kopien konnten ohne Unterstützung sitzen und 17/18 Kindern (94%) mit mehr als 3 SMN2-Kopien konnten sitzen und sich dabei drehen. Viele Kinder konnten zudem stehen und gehen (gemessen anhand des HINE-2-Tests).
2 SMN2-Kopien: Ergebnisse zu Monat 12 (n=8)*
Balken stellen das 1. - 99. Perzentil für das Erreichen der motorischen Meilensteine auf der Grundlage der WHO-Studie zur motorischen Entwicklung von gesunden Kindern dar 1 1071038510010198 115113113112
1/8 Kindern (13 %) konnten gehen‡
≥ 3 SMN2-Kopien: Ergebnisse zu Monat 12 (n=18)†
369 121518 21 Alter (Monate)
7/8 Kindern (88 %) konnten sitzen, ohne Unterstützung
In der doppelblinden, randomisierten SUNFISH-Studie wurde die Wirksamkeit von Risdiplam bei 180 Patienten zwischen 2 und 25 Jahren mit spät einsetzender SMA (Typ 2 und 3) untersucht [7]. Sie erhielten 24 Monate lang randomisiert im Verhältnis 2:1 entweder Risdiplam oder Placebo. Primärer Endpunkt war die Änderung der
3 6 9 12 15 18 21 Alter (Monate)
In den ersten 12 Monaten nahm der MFM-32-Gesamtscore gegenüber dem Ausgangswert im Mittel um 2 Punkte zu. In den darauf folgenden Monaten konnten die Fortschritte, die unter der Risdiplam-Behandlung in Monat 12 beobachtet wurden, in Monat 48 beibehalten oder verbessert werden. Der MFM-32-Gesamtscore war nach 12 bzw. 24 Monaten bei mehr als 50 % der Behandelten signifikant und klinisch relevant um mindestens 3 Punkte höher als zu Beginn (Abb. 3) [7].
17/18 Kindern (94 %) konnten sitzen und sich dabei drehen
* Diese Gruppe umfasst 5 Kinder mit einer CMAP-Amplitude ≥1,5 mV und 3 mit einer CMAP-Amplitude <1,5 mV bei Studienbeginn.
† Ein Kind mit ≥4 Kopien war bei der Untersuchung in Monat 12 unkooperativ und wurde nicht getestet. Data cut-off: Feb 20, 2023.
‡ Ein Kind wurde zu Monat 12 nicht getestet
1. WHOMGRS. Acta Paediatr Suppl. 2006; 450:86–95
4/8 Kindern (50 %) konnten stehen, ohne Hilfsmittel
1/8 Kindern (13 %) konnten gehen‡
17/18 Kindern (94 %) konnten stehen
15/18 Kindern (83 %) konnten gehen
Balken stellen das 1. - 99. Perzentil für das Erreichen der motorischen Meilensteine auf der Grundlage der WHO-Studie zur motorischen Entwicklung von gesunden Kindern dar.1
* Diese Gruppe umfasst 5 Kinder mit einer CMAP-Amplitude ≥1,5 mV und 3 mit einer CMAP-Amplitude <1,5 mV bei Studienbeginn.
† Ein Kind mit ≥4 Kopien war bei der Untersuchung in Monat 12 unkooperativ und wurde nicht getestet. Data cut-off: Feb 20, 2023.
‡ Ein Kind wurde zu Monat 12 nicht getestet 1. WHOMGRS. Acta Paediatr Suppl. 2006; 450:86–95
In der noch laufenden offenen, einarmigen Phase-II-Studie RAINBOWFISH [8] werden die Wirksamkeit und Sicherheit von Risdiplam bei präsymptomati-
schen Säuglingen mit einer SMA Typ 1, Typ 2 oder Typ 3 oder mit 1 – 4 Kopien des SMN2-Gens untersucht, die ab Geburt bis zu einem Alter von 6 Wochen ihre erste Dosis erhalten haben. Diese wird einmal täglich oral verabreicht. Nach 12 Monaten Behandlung mit Risdiplam wurde der primäre Studienendpunkt erreicht: 88 % (7/8) der Säuglinge mit 2 SMN2-Kopien konnten mindestens 5 Sekunden lang frei sitzen. 94 % (17/18) der Säuglinge mit ≥3 SMN2-Kopien, konnten nach 12 Monaten frei sitzen und sich dabei drehen. Außerdem hatten viele Säuglinge auch Fortschritte bezüglich des Stehens und Laufens gemacht: Nach 12 Monaten Behandlung mit Risdiplam konnten 50 % (4/8) der Säuglinge mit 2 SMN2-Kopien sowie 94 % (17/18) der Säuglinge mit ≥3 SMN2-Kopien stehen und 83 % (15/18) der Säuglinge mit ≥3 SMN2-Kopien laufen (Abb. 4) [8]. Alle Kinder behielten die Fähigkeit zu schlucken und oral ge-
* Diese Gruppe umfasst 5 Kinder mit einer CMAP-Amplitude ≥1,5 mV und 3 mit einer CMAP-Amplitude <1,5 mV bei Studienbeginn. † Ein Kind mit ≥4 Kopien war bei der Untersuchung in Monat 12 unkooperativ und wurde nicht getestet. Data cut-off: Feb 20, 2023. ‡ Ein Kind wurde zu Monat 12 nicht getestet.
1. WHOMGRS. Acta Paediatr Suppl. 2006; 450:86–95
Seite 2/3
17/18 Kindern konnten sitzen dabei drehen
17/18 Kindern konnten stehen
15/18 Kindern konnten gehen
2 SMN2-Kopien: Ergebnisse zu Monat 12 (n=8)
7/8 Kindern (88 %) konnten sitzen, ohne Unterstützung
4/8 Kindern (50 %) konnten stehen, ohne Hilfsmittel
≥ 3 SMN2-Kopien: Ergebnisse zu Monat 12 (n=18)
17/18 Kindern (94 %) konnten sitzen und sich dabei drehen
17/18 Kindern (94 %) konnten stehen
füttert zu werden und zeigten die alterstypischen kognitiven Fähigkeiten gesunder Kinder, gemessen mit der BSID-III-Kognitionsskala. Kein Kind benötigte permanente Beatmung, kein Kind zeigte eine abnormale Sprachentwicklung. Die meisten Kinder mit 2 SMN2Kopien und einer Behandlungszeit von ≥12 Monaten erreichten CHOP-INTENDf-Scores nahe dem Maximalwert sowie motorische Meilensteine innerhalb der von der WHO für gesunde Kinder definierten Bereiche [8].
Brigitte Söllner, Erlangen
1/8 Kindern (13 %) konnten gehen
15/18 Kindern (83 %) konnten gehen
Abbildung 4: Ergebnisse der RAINBOWFISH-Studie bei insgesamt 12 Kindern nach einer 12-monatigen Behandlung mit Risdiplam (Evrysdi®). Besonders von der Therapie profitierten präsymptomatische Kinder mit ≥3 Kopien des SMN2-Gens [8].
Zulassungsrelevante
Die meisten Kinder mit 2 SMN2-Kopien und einer Behandlungszeit von ≥ 12 Monaten erreichten CHOP-INTENDf-Scores nahe dem Maximalwert sowie motorische Meilensteine innerhalb der von der WHO für gesunde Kinder definierte Bereiche.19,20
Von den 18 in die RAINBOWFISH-Studie eingeschlossenen präsymptomatischen Neugeborenen mit SMA wurden 6 Kinder, die 2 oder 3 Kopien des SMN2-Gens aufwiesen, über mindestens ein Jahr nachverfolgt. Nach einem Jahr Risdiplam-Therapie waren von diesen 6 Säuglingen alle in der Lage zu sitzen, 4 von 6 konnten stehen und 3 von 6 konnten ohne Hilfe gehen. Alle Kinder waren nach 12 Monaten am Leben und benötigten keine permanente Beatmung. Das Sicherheitsprofil von Risdiplam war dabei konsistent mit dem Sicherheitsprofil früherer Studien bei symptomatischen SMA-Patienten [9].
Aufgrund dieser Ergebnisse wurde die Zulassung von Evrysdi® im August 2023 auf die Behandlung Neugeborener erweitert. Seitdem können Patienten aller Altersgruppen ab der Geburt von der konstanten Wirksamkeit von Risdiplam profitieren [3].
Die UEs waren eher mit dem Alter der Kinder assoziiert als mit der zugrundeliegenden SMA. Es traten keine Todesfälle auf und es wurden keine therapiebedingten SUEs beobachtet, die zum Therapieabbruch geführt hätten.19
Literatur
1 Vill K et al. One year of newborn screening for SMA – results of a German pilot project. J Neuromuscul Dis 2019;6:503515
2 Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018;28:103-115
3 Fachinformation Evrysdi®, Stand: August 2023
4 Bowerman M et al. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis Model Mech 2017;10:943954
5 Sivaramakrishnan M et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun 2017;8:1476
6 Mazurkiewicz-Beldzinska M et al. FIREFISH Parts 1 and 2: 48-month efficacy and safety of risdiplam in Type 1 spinal muscular atrophy (SMA). Präsentation auf dem EAN, 01. Juli 2023
7 ClinicalTrials.gov, NCT02908685, https://clinicaltrials.gov/ct2/show/ NCT02908685
8 ClinicalTrials.gov, NCT03779334, https:// clinicaltrials.gov/ct2/show/NCT03779334
9 Finkel RS et al. MDA Clinical & Scientific Conference. 2022; Nashville, TN
Die spinale Muskelatrophie (SMA) ist eine seltene, genetisch bedingte neuromuskuläre Erkrankung und gehört zu den häufigsten genetisch bedingten Todesursachen bei Säuglingen. Verursacht durch das Fehlen eines funktionsfähigen SMN1-Gens kommt es bei der schwersten Form der SMA (Typ 1, der 60 % der Fälle ausmacht) zu einem raschen und irreversiblen Verlust von Motoneuronen, was die Muskelfunktionen, einschließlich Atmung, Schlucken und Grundbewegungen, beeinträchtigt. Unbehandelt führt die Erkrankung in mehr als 90 % aller Fälle innerhalb der ersten 2 Lebensjahre zur Notwendigkeit einer ständigen Beatmung oder zum Tod [1, 2].
Onasemnogen-Abeparvovec
Onasemnogen-Abeparvovec (Zolgensma® 2×10¹³ Vektorgenome/ml Infusionslösung) ist ein Gentherapeutikum, das das humane Survival-Motoneuron-(SMN-)Protein exprimiert und die bisher einzige Gentherapie zur Behandlung der SMA, die die zugrunde liegende genetische Ursache adressiert. Zolgensma® wird nur ein einziges Mal als intravenöse Infusion verabreicht und liefert eine funktionsfähige Kopie des menschlichen SMN-Gens, die das fehlende oder fehlerhafte Gen ersetzt, um durch eine anhaltende Expression des
SMN-Proteins das Fortschreiten der Erkrankung zu stoppen [3].
Onasemnogen-Abeparvovec hat Marktzulassungen in mehr als 51 Ländern erhalten. Mehr als 3.700 Patienten wurden bisher in klinischen Studien, kommerziell und im Rahmen von Härtefall-Programmen mit Onasemnogen-Abeparvovec behandelt [4].
Ergebnisse der SMART-Studie untermauern den klinischen Nutzen
Im Rahmen der diesjährigen Konferenz der Muscular Dystrophy Association (MDA) in Orlando, Florida, stellte Novartis aktuelle Daten der SMART-Studie vor, die die Sicherheit, Verträglichkeit und Wirksamkeit einer intravenösen Einmal-Infusion von Onasemnogen-Abeparvovec bei pädiatrischen Patienten mit symptomatischer SMA mit biallelischer Mutation im SMN1-Gen und einer beliebigen Kopienzahl des SMN2Gens untersucht. In die offene, einarmige Phase-IIIb-Studie wurden 24 Kinder mit heterogenen SMA-Phänotypen in 3 Gewichtsklassen (8,5 – 13 kg; >13 – 17 kg; >17 – 21 kg) im Alter von ~18 Monaten bis 9 Jahren aufgenommen. Die Studienpopulation unterscheidet sich damit durch das höhere Gewicht und Alter von vorherigen Untersuchungen. Erstmals wurden auch vorbehandelte Kinder eingeschlossen:
87,5 % der Studienteilnehmer wechselten von einer vorangegangenen krankheitsmodifizierenden Therapie mit Risdiplam oder Nusinersen zu der Einmal-Gentherapie. Die Studienergebnisse zeigen, dass nach 52 Wochen bei 95,8 % der Kinder motorische Meilensteine gegenüber dem Ausgangswert erhalten oder sogar verbessert waren:
• Der mittlere Anstieg der Gesamtpunktzahl des Revised Upper Limb Module (RULM) betrug 2 Punkte und der mittlere Anstieg der Gesamtpunktzahl der Hammersmith Functional Motor Scale – Expanded (HFMSE) betrug 3,7 Punkte. 4 Kinder wiesen in Woche 52 neue Entwicklungsmeilensteine auf.
• Nahezu alle Kinder (23/24, 95,8 %), die mit leichter Unterstützung sitzen konnten, behielten diesen Meilenstein auch in Woche 52 bei.
• 3 Patienten erreichten in Woche 52 den neuen Meilenstein des Stehens mit Unterstützung und ein Kind erreichte den neuen Meilenstein des Gehens mit Unterstützung.
• Alle Patienten (6/6, 100 %), die zu Studienbeginn gehen konnten, erhielten diesen Meilenstein bis zum Ende der Studie aufrecht.
Im Rahmen der Studie wurden keine neuen Sicherheitssignale beobachtet.
• Ein Anstieg der Transaminasen (ALT oder AST >3ULN)
wurde bei der Mehrzahl der Patienten (21/24, 87,5 %) beobachtet; eine vorübergehende Thrombozytopenie trat bei 17/24 (70,8 %) der Studienteilnehmer auf. Die betroffenen Kinder erhielten eine geeignete Behandlung und alle Fälle waren asymptomatisch.
Zulassungserweiterung:
Ryeqo® jetzt auch bei symptomatischer Endometriose nach Vorbehandlung einsetzbar
Für Patientinnen mit Endometriose, bei denen eine vorhergehende medikamentöse (z.B. mit Dienogest) oder chirurgische Behandlung nicht den erwünschten Erfolg hatte, nicht möglich war, zu unerwünschten Nebeneffekten oder einem Rezidiv führte, gibt es jetzt eine neue Therapieoption: Das Kombinationspräparat Ryeqo® (Relugolix 40 mg, Estradiol 1,0 mg und Norethisteronacetat 0,5 mg) kann jetzt auch zur Behandlung der symptomatischen Endometriose bei medizinisch oder chirurgisch vorbehandelten Patientinnen im reproduktionsfähigen Alter verordnet werden. Bereits seit Juli 2021 ist Ryeqo® für die Therapie von mäßigen bis starken Symptomen von Uterusmyomen bei erwachsenen Frauen im reproduktiven Alter zugelassen.
Signifikanter Rückgang von Dysmenorrhö und nicht menstruellen Unterbauchschmerzen
Basis für die Zulassungserweiterung von Ryeqo® waren die ran-
• Es wurden keine Fälle von akutem Leberversagen oder Bilirubinerhöhungen gemeldet. Die neuen klinischen Ergebnisse unterstreichen das Sicherheitsund Wirksamkeitsprofil der Gentherapie bei Kindern mit SMA und ergänzen die bisherigen Real-World-Daten zur Therapie mit
domisierten, doppelblinden und placebokontrollierten Phase-IIIStudien SPIRIT 1 und SPIRIT 2. Eingeschlossen wurden 1.261 Frauen im Alter zwischen 18 und 50 Jahren mit diagnostizierter Endometriose. Sie wurden zu gleichen Teilen auf 3 Therapiearme randomisiert und für einen Zeitraum von 24 Wochen wie folgt behandelt:
• Relugolix-Kombinationstherapie (Relugolix 40 mg, Estradiol 1,0 mg und Norethisteronacetat 0,5 mg) oder
• verzögerte Relugolix-Kombinationstherapie (12 Wochen Relugolix 40 mg als Monotherapie, gefolgt von 12 Wochen
Onasemnogen-Abeparvovec bei älteren und schwereren Kindern. Brigitte Söllner, Erlangen
Literatur
1 Anderton RS et al. Expert Rev Neurother 2015;15:895-908
2 Finkel RS et al. Neurology 2014;83:810817
3 Fachinformation Zolgensma®
4 Data on File. Novartis AG, 2023
Relugolix 40 mg, Estradiol 1,0 mg und Norethisteronacetat 0,5 mg) oder
• Placebo
Beide primären Endpunkte wurden erreicht: In beiden Studien kam es bei jeweils 75 % der Patientinnen zu einer signifikanten Verbesserung der Dysmenorrhö gegenüber 27 % (SPIRIT 1) und 30 % (SPIRIT 2) unter Placebo (jeweils p < 0,0001). Die nicht menstruellen Unterbauchschmerzen wurden bei 58 % der Patientinnen in SPIRIT 1 und bei 66 % in SPIRIT 2 reduziert (vs. 40 % bzw. 43 % unter Placebo; jeweils p < 0,0001).
E. W.Einzigartige Wirkstoffkombination
In der symptomatischen Endometriose-Therapie kommen neben einer begleitenden Schmerztherapie vor allem Gestagene, orale kombinierte Kontrazeptiva sowie Gonadotropin-Releasing-Hormon (GnRH)-Analoga infrage. Letztere spielen allerdings keine große Rolle, da sie wegen der starken Nebenwirkungen nur selten zum Einsatz kommen. Die Kombinationstherapie Ryeqo® wird 1 x täglich oral angewendet und beinhaltet 3 Wirkstoffe in einer Tablette: Zum einen den nicht peptidischen GnRH-Rezeptor-Antagonisten Relugolix (40 mg), der dosisabhängig die Freisetzung des luteinisierenden Hormons und des follikelstimulierenden Hormons und somit letztlich die Menge an in den Eierstöcken produzierten Estrogens und Progesterons vermindert. Zum anderen sind als Add-Back das Estrogen Estradiol (1,0 mg) und das synthetische Gestagen Norethisteronacetat (0,5 mg) enthalten, um mögliche Folgen eines Estrogenmangels zu reduzieren. Während Estradiol vasomotorische Symptome und Knochenschwund lindert, verringert Norethisteronacetat das estrogeninduzierte Risiko für eine Endometriumhyperplasie.
Seit November 2023 ist u.a. für die Behandlung der chronischen spontanen Urtikaria (csU) bei Patienten ab einem Alter von 12 Jahren ein neuer Autoinjektor (300 mg Injektionslösung im Fertigpen) für die Selbstapplikation von Omalizumab (Xolair®) zugelassen. Bereits seit 2014 darf Omalizumab für die Behandlung von csU angewendet werden und seit 2018 ist auch die Selbstapplikation in Form einer 150-mgSpritze möglich.
Der Autoinjektor kann die Selbstapplikation erleichtern, da die empfohlene Dosis von 300 mg Omalizumab alle 4 Wochen nun mit nur einer Injektion verabreicht werden kann. Novartis rechnet mit einer Marktverfügbarkeit ab Herbst 2024. Für einen optimierten Einsatz von Omalizumab im Klinikalltag führt das Unternehmen zudem eine neue Fertigspritze mit 300 mg Injektionslösung ein.
Studie bestätigt Bioäquivalenz gegenüber der 2 150 mg-Dosis per Fertigspritze
Die Zulassung des neuen 300-mgAutoinjektors basierte auf der randomisierten, Open-Label Parallelgruppen-Studie K12101 mit einer Laufzeit von 85 Tagen, welche die Bioäquivalenz einer einmaligen 300-mg-Dosis von Omalizumab gegenüber einer 300-mg-Dosis in Form von zwei 150-mg-Dosen verglich [2]. Hierfür wurden 193 gesunde Freiwillige gleichmäßig auf die Behandlungsgruppen Omalizumab 1 300 mg per Autoinjektor, 1 300 mg per Fertigspritze und 2 150 mg per Fertigspritze aufgeteilt. Die Omalizumab-Konzentration wurde mittels bestimmter Serumparameter gemessen, für die zu Studienbeginn zu erwarten-
de Werte festgelegt wurden. Die Ergebnisse bestätigten, dass die Konzentration bzw. Exposition von Omalizumab in allen Gruppen ähnlich war.
Omalizumab ist seit 2014 als Zusatztherapie für die Behandlung von csU bei Patienten ab 12 Jahren zugelassen, die nicht ausreichend auf H1-Antihistaminika ansprechen. Der humanisierte monoklo-
nale Anti-IgE-Antikörper bindet an IgE und senkt so die Menge an freiem IgE sowie die dadurch bedingten Effekte auf die zellulären Aktivierungsmechanismen. Damit unterdrückt es die durch Histamin induzierten Hautreaktionen und tieferen Schwellungen [1].
Die Zulassung von Omalizumab basierte auf den Studien ASTERIA I und II sowie GLACIAL. In den ASTERIA-Studien kam es unter der Therapie mit 300 mg Omalizumab alle 4 Wochen (bei gleichzeitiger Gabe von H1-Antihistaminika) zu einer Verringerung der
Die csU betrifft etwa 0,35 % der deutschen sowie rund 1,1 % der globalen Bevölkerung und stellt eine Erkrankung mit hoher Krankheitslast dar [6, 7]. Sie ist gekennzeichnet durch das wiederholte plötzliche Auftreten juckender Quaddeln und/oder tiefer Gewebeschwellungen (Angioödeme), die u.a. im Gesicht, am Hals, an den Händen und Füßen auftreten und über mehr als 6 Wochen anhalten können. Diese Symptome sowie die daraus möglicherweise resultierenden psychischen Probleme wie Depressionen oder Ängste können csU-Betroffene im Alltag erheblich einschränken und einen hohen Leidensdruck verursachen [8].
Therapieziel ist laut der europäischen Leitlinie [9] die vollständige Symptomfreiheit sowie der Schutz vor weiterer Krankheitsaktivität. Derzeit gibt es jedoch nur wenige wirksame Behandlungsmöglichkeiten für die csU, wobei über 50 % der Patienten mit der Erstbehandlung mit Antihistaminika keine vollständige Kontrolle erreichen [8]. Hier stehen Biologika wie Omalizumab als zusätzliche Therapieoption zur Verfügung.
Krankheitsaktivität, einer signifikanten Verbesserung des Juckens (Itch Severity Score) in Woche 12 sowie zu einer Überlegenheit in Bezug auf die Lebensqualität im Vergleich zu Placebo (p < 0,0001) [3]. Die Wirksamkeit und das gute Sicherheitsprofil von Omalizumab 300 mg alle 4 Wochen wurden durch die Ergebnisse der GLACIAL-Studie bestätigt [4]. Außerdem zeigte die Langzeitstudie XTEND, dass eine Omalizumab-Therapie über ein Jahr die Rückkehr der Symptome verhindert und eine Kontrolle der csU aufrechterhalten werden konnte [5].
BrigitteSöllner, Erlangen
Unter dem Begriff Lower Urinary Tract Symptoms (LUTS) werden Miktionsbeschwerden unterschiedlicher Art und Genese zusammengefasst; die häufigsten Symptome sind in Tabelle 1 aufgeführt. Mögliche Ursachen sind u.a. eine benigne Prostata-Hyperplasie (BPH), eine überaktive Blase (OAB), Infektionen, onkologische Erkrankungen und Blasensteine. Über alle Ausprägungen und Ätiologien hinweg leidet etwa jeder vierte Mann über 40 Jahre an LUTS, in der Altersgruppe der 70–80-Jährigen ist nahezu jeder zweite Mann betroffen [1].
Alle Ausprägungen von LUTS können die Lebensqualität erheb-
lich beeinträchtigen. In Abhängigkeit vom individuellen Beschwerdebild und der Schwere der Symptomatik gehen LUTS mit physischen Komorbiditäten wie erektiler Dysfunktion sowie sozialen Einschränkungen einher [2]. Viele Betroffene leiden unter Schlafstörungen infolge des nächtlichen Harndrangs [3], Symptome von Angststörungen und Depression treten bei 35,9 % bzw. 29,8 % der Männer mit LUTS auf [2].
DiGA Kranus Lutera schließt Therapielücke durch ganzheitlichen Behandlungsansatz
Je nach Ursache und Schweregrad der Symptome werden im Rahmen
Literatur
1 Fachinformation Xolair®, aktueller Stand
2 European Medicines Agency. Published online 2023. https://www.ema.europa.eu/ en/documents/variation-report/xolair-h-c606-x-0115-epar-assessment-report-extension_en.pdf
3 Casale TB et al. J Allergy Clin Immunol Pract 2015;3:743-750.e1
4 Kaplan A et al. J Allergy Clin Immunol 2013;132:101-109
5 Maurer M et al. J Allergy Clin Immunol 2018;141:1138-1139.e7
6 Peck G et al. Acta Derm Venereol 2021; 101:adv00433
7 Weller K et al. J Eur Acad Dermatol Venereol 2022;36:91-99
8 Maurer M et al. Allergy 2011;66:317-330
9 Zuberbier T et al. Published online 2022. https://register.awmf.org/assets/ guidelines/013-028l_S3_KlassifikationDiagnostik-Therapie-Urtikaria_2022-04. pdf
Speicherstörungen
• Pollakisurie
• Nykturie
• Dranginkontinenz
• Belastungsinkontinenz
Entleerungsstörungen
• Dysurie/Algurie
• Schwacher Harnstrahl
• Splitting & Spraying
• Intermittierende Miktion (geteilter oder schwer kontrollierbarer Harnstrahl)
• Verzögerte Miktion
• Straining (abdominale Muskulatur muss eingesetzt werden, um Wasser zu lassen)
• Terminales Tropfen
Postmiktionsstörungen
• Restharnempfinden
• Post-Miktionstropfen
Tabelle 1: Bei den Lower Urinary Tract Symptoms werden Speicher-, Entleerungs- und Postmiktionssymptome unterschieden. Häufig treten mehrere Symptome kombiniert auf [1].
Lebensstilmodifikationen
z.B. Meidung diuretisch wirkender Genussmittel (Kaffee, Alkohol, Nikotin) und Medikamente
Physiotherapeutische und mentale Übungen
z.B. Biofeedback-Übungen, Beckenbodentraining, Maßnahmen zur Stressreduktion
Pharmakotherapien, teilweise in Kombination
z.B. 5α-Reduktasehemmer (bei BPH), α1-Adrenorezeptorblocker, Phosphodiesterase5-Hemmer, Beta-3-Agonisten und Anticholinergika bzw. Muskarinrezeptor-Antagonisten (bei OAB)
Operative Therapie
Behandlung der BPH z.B. durch transurethrale Resektion der Prostata (TURP), Laserablation oder Prostatektomie, Entfernung von Blasensteinen
Tabelle 2: Bausteine der multimodalen Therapie der LUTS [1, 4].
der multimodalen Therapie der LUTS nichtmedikamentöse Behandlungen, pharmakotherapeutische und chirurgische Interventionen kombiniert (Tab. 2) [1, 4]. Da trotz dieser Behandlungsoptionen bei den Patienten oft noch ein hoher Leidensdrucks besteht, hat das Unternehmen Kranus Health eine innovative, App-basierte Therapielösung entwickelt, um die Betroffenen bei der Bewältigung ihrer Erkrankung zu unterstützen und die Symptome zu bessern. Ausgangspunkt war die Erkenntnis, dass nichtmedikamentöse Verfahren wie Beckenbodentraining, kognitive Verhaltenstherapie oder Achtsamkeitsübungen insbesondere bei Speichersymptomen zu einer signifikanten Reduktion der Beschwerden führen können. Entsprechend werden sie auch in europäischen urologischen Leitlinien im Rahmen einer multimodalen Therapiestrategie mit hoher wissenschaftlicher Evidenz empfohlen [4]. In der Praxis steht der Umsetzung dieser Empfehlung jedoch häufig der damit verbundene zeitliche und organisatorische Aufwand entgegen und sie werden deshalb in der Praxis häufig nicht angeboten [5]. Diese Versorgungslücke schließt die DiGA Kranus Lute-
ra, denn sie unterstützt zum einen Ärzte und Praxispersonal bei der Aufklärung sowie beim Monitoring von LUTS-Patienten. Zum anderen werden die Patienten durch Selbstmanagement-Strategien und Übungsangebote aktiv in die Therapie mit einbezogen und in ihrer Selbstwirksamkeit gestärkt [6]. Kranus Lutera ist die erste vom Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) zugelassene digitale Therapie zur Behandlung von LUTS und kann ab sofort Patienten mit häufigem Harndrang bei benigner Prostatahyperplasie (BPH) und/oder überaktiver Blase (OAB) zu Lasten der gesetzlichen Krankenkasse verordnet werden.
BEST-Studie belegt Wirksamkeit der App
Basis der Zulassung waren die Ergebnisse der prospektiven, einfach verblindeten BEST-Studie bei 237 erwachsenen Männern mit den Diagnosen BPH oder OAB. Sie wurden im Verhältnis 1:1 in 2 Gruppen randomisiert: Die Interventionsgruppe erhielt zusätzlich zu ihrer üblichen medizinischen Versorgung Zugang zur App-basierten Therapie Kranus Lutera. Die Kontrollgruppe bekam nur die übliche medizinische Versorgung.
Studienendpunkte waren die Verringerung der Prostata-assoziierten und Prostata-unabhängigen Symptomlast sowie die Verbesserung der krankheitsbezogenen Lebensqualität.
Die Effekte der App-basierten Intervention wurden mithilfe der validierten Fragebögen International Prostate Symptom Score (IPSS) und Overactive Bladder Questionnaire short form 1 und 2 (OABq-SF 1 und 2) gemessen.
Sowohl die Symptomlast als auch die Lebensqualität verbesserten sich bei den Nutzern der DiGA nach der 12-wöchigen Anwendung signifikant: Im Vergleich zur
Der Therapieschwerpunkt von Kranus Lutera liegt in der Behandlung von Speichersymptomen und zu einem geringeren Teil von Postmiktionssymptomen. Die Nutzer der App absolvieren eine 12-wöchige Therapie bestehend aus Beckenbodentraining, physiotherapeutischen Übungen, Blasentraining, kognitiver Verhaltenstherapie, mentalen Übungen, Kontrolle des akuten Harndrangs sowie Wissensvermittlung zur Erkrankung und Ernährung. Im Umfang der App ist darüber hinaus ein interaktives Miktionstagebuch enthalten. Die Übungen werden wöchentlich auf Basis des Patienten-Feedbacks individuell angepasst.
Verringerung der Prostata-assoziierten Symptomlast
IPSS
Verbesserung der Prostata-unabhängigen Symptomlast
OAB-q-SF 1
Interventionsgruppe (IG) Kontrollgruppe
Verbesserung der krankheitsbezogenen Lebensqualität
OAB-q-SF 2

Behandlungseffekt von -7 Punkten in der IG
* Analysen im FAS mittes J2R Imputation
Relative Verbesserung von 40,58 % in der IG
Relative Verbesserung von 46,27 % in der IG
Abbildung 1: Ergebnisse der BEST-Studie: Bei den Nutzern der DiGA Kranus Lutera verbesserten sich nach der 12-wöchigen Anwendung im Vergleich zur Kontrollgruppe sowohl die Symptomlast als auch die Lebensqualität signifikant (© Kranus Health).
Kontrollgruppe zeigte die Interventionsgruppe eine signifikante und klinisch bedeutsame Verbesserung des primären Endpunkts IPSS (Least Square Means[LSM]: –7,0 %-Punkte) und der sekundären Endpunkte OAB-q-SF Teil 1 (LSM: –18,6 %-Punkte) und Teil 2 (LSM: –11,2 %-Punkte) (Abb. 1). Beide Indikationsgruppen (BPH und OAB) profitierten in ähnlicher Weise von der App. Der Nutzen war unabhängig vom Alter und Schweregrad der Symptome und die Effektgröße übertraf die meis-
237 Patienten wurden randomisiert. Die demografischen Ausgangsdaten der beiden Gruppen waren ausgewogen. Im Vergleich zur Kontrollgruppe zeigte die Interventionsgruppe eine signifikante und klinisch bedeutsame Verbesserung der Symptomstärke anhand von IPSS und OABqSF 1 sowie der krankheitsbezogenen Lebensqualität nach OABqSF 2. Patienten profitierten unabhängig von Alter, Indikationsgruppe (BPH, OAB) und Schweregrad der Symptome.9,10
ten in medikamentösen Studien gemessenen Werte [7].
Brigitte Söllner, Erlangen
Literatur
Ausschlusskriterien/Kontraindikationen
3 Taylor BC et al. Prevalence, severity, and health correlates of lower urinary tract symptoms among older men: the MrOS study. Urology 2006;68:804-809
4 Gratzke C et al. EAU Guidelines on the assessment of non-neurogenic male lower urinary tract symptoms including benign prostatic obstruction. Eur Urol 2015; 67:1099-1109
Für der Verordnung von Kranus Lutera liegen keine absoluten Kontraindikationen vor. Relative Ausschlusskriterien sind:1
1 eMedpedia. Lower Urinary Tract Symptoms (LUTS). https://www.springermedizin.de/emedpedia/die-urologie/lower-urinary-tract-symptoms-luts?epediaDoi=10. 1007%2F978-3-642-41168-7_139
5 Albarqouni L et al. Self-Management for men with lower urinary tract symptoms: a systematic review and meta-analysis. Ann Fam Med 2021;19:157-167
Mit ICD-10-Klassifikation Ohne ICD-10-Klassifikation
• Wiederkehrende Unfähigkeit, die Blase ganz oder teilweise zu leeren (R33, Harnverhaltung)
2 Coyne KS et al. The impact of overactive bladder, incontinence and other lower urinary tract symptoms on quality of life, work productivity, sexuality and emotional well-being in men and women: results from the EPIC study. BJU Int 2008;101: 1388-1395
• Wiederkehrende Harnwegsinfektionen (N39.0, Harnwegsinfektion, Lokalisation nicht näher bezeichnet)
• Steine in der Harnblase (N21.0, Harnblasenkonkremente)
• Blut im Urin (N02. konservativ nicht beherrschbare, rezidivierende Makrohämaturien)
• Dilatation des oberen Harntraktes, eingeschränkte Nierenfunktion oder Niereninsuffizienz durch obstruktive Blasenentleerungsstörungen
6 Kranus Health GmbH. Kranus Lutera Gebrauchsanweisung. https://www.datocmsassets.com/82237/1701172632-gebrauchsanweisung_kranus_lutera_20231122_de.pdf
7 Kranus Health GmbH. Data on file. Stand März 2023
• Unfähigkeit physisch am Programm teilzunehmen
Patienten mit der seltenen Erkrankung Hereditäres Angioödem (HAE) profitieren unter der oralen Langzeitprophylaxe mit Berotralstat (Orladeyo®) neben der langfristigen Reduktion der HAE-Attacken auch von einer klinisch relevanten Verbesserung der krankheitsbezogenen Lebensqualität. Dies bestätigen die Ergebnisse der Langzeitanalyse der Studie APeX-2, die die Wirksamkeit und Sicherheit von Berotralstat über einen Zeitraum von 96 Wochen untersuchte [1].
Anhaltende Krankheitskontrolle bei guter Verträglichkeit
Als bisher einzige zielgerichtete orale Langzeitprophylaxe für HAE wurde Orladeyo® vor über 2 Jahren in der EU zugelassen [2]. Grundlage dafür waren u.a. die positiven Ergebnisse der dreiteiligen PhaseIII-Studie APeX-2, in deren 1. Teil zunächst eine anhaltende Krankheitskontrolle bei guter Verträglichkeit über 24 Wochen nachgewiesen wurde [3].
Im Rahmen der Open-Label-Extension wurden im Folgenden 81 HAE-Patienten analysiert, die in den Wochen 48 – 240 mit Berotralstat in der für die klinische Praxis in Europa zugelassenen Dosierung von 150 mg einmal täglich behandelt wurden. Die Sicherheit und Verträglichkeit von Berotralstat konnten bei diesen Patienten bestätigt werden. Es traten keine neuen Sicherheitssignale auf, die meisten unerwünschten Ereignisse waren mild bis moderat. Gastrointestinale unerwünschte Ereignisse, die direkt mit der Behandlung in Zusammenhang standen, wurden nur von 4,9 % der Patienten berichtet – und waren damit deutlich seltener als in den ersten 48 Behandlungswo-
chen in Teil 1 und 2 der Studie –und führten nur zu 2 Studienabbrüchen [1, 3].
Reduktion der HAE-Attacken um 90,8 %
Schon in den ersten beiden Studienteilen führte Berotralstat nach 24 Wochen zu einer signifikanten Reduktion der HAE-Attacken im Vergleich zu Placebo, die bis Woche 48 weiter zunahm [3, 4]. Die jetzt veröffentlichten Ergebnisse zeigen, dass die Patienten, die über 96 Wochen mit Berotralstat 150 mg behandelt wurden (n = 21), 90,8 % weniger HAE-Attacken hatten als zu Studienbeginn. Im dritten Studienteil hatten die Patienten aller Behandlungsgruppen durchschnittlich 93,1 % attackenfreie Tage, im Median waren 11 von 12 Monate komplett attackenfrei. Der Bedarf an Akutmedikation ging daher bei den Patienten, die für 96 Wochen mit einmal täglich 150 mg Berotralstat behandelt wurden, um 88,5 % zurück [1].
Verbesserung der HAE-bezogenen Lebensqualität und hohe Therapiezufriedenheit
Neben den direkten körperlichen Einschränkungen infolge der
Schwellungen belastet die ständige Sorge vor einer weiteren HAE-Attacke die Betroffenen in vielen Lebensbereichen [5, 6]. Nach 96 Wochen Behandlung mit Berotralstat zeigten alle analysierten Patienten in APeX-2 eine klinisch relevante Verbesserung der HAE-bezogenen Lebensqualität. Die größte Verbesserung erreichten dabei die Patienten, die über den gesamten Studienzeitraum mit Berotralstat behandelt wurden. Besonders hinsichtlich der Bewältigung von Alltagsituationen (Arbeit, körperliche Betätigung, Freizeit und soziale Beziehungen) berichteten die Patienten von einer signifikanten Verbesserung und waren durchweg mit dem Behandlungserfolg sehr zufrieden, zumal Berotralstat einfach oral einzunehmen und auch in der Langzeitprophylaxe gut verträglich ist.
Fabian Sandner, Nürnberg
Literatur
1 lergy Clin Immunol Pract 2024;12:733743
2 Fachinformation Orlandeyo®; Stand: Dezember 2022
3 Zuraw B et al. J Allergy Clin Immunol 2021;148:164-172
4 Wedner HJ et al. J Allergy Clin Immunol Pract 2021;9:2305-2314
5 Kaplan AP. World Allergy Organization J 2008;1:103-113
Postmenopausale Frauen mit Hormonrezeptor-positivem (HR+), HER2-negativem (HER2-) lokal fortgeschrittenem oder metastasiertem Brustkrebs erreichten mit der Zugabe des CDK4 & 6 Inhibitors Abemaciclib (Verzenios®) zu einem Aromatasehemmer (AI) eine Verbesserung des medianen Gesamtüberlebens (OS) um 13,1 Monate. Dies zeigt die finale Analyse der klinischen Studie MONARCH 3 nach einem medianen Follow-up von 8 Jahren, die im Rahmen des San Antonio Breast Cancer Symposiums 2023 vorgestellt wurde [1]. Verglichen mit einem alleinigen AI profitierten auch Frauen mit viszeralen Metastasen von der Kombination mit Abemaciclib mit einer Verlängerung des OS um 14,9 Monate.
Medianes Gesamtüberleben von mehr als 5,5 Jahren
In der finalen Analyse nach einem medianen Follow-up von 8 Jahren zeigte sich in der Intentionto-Treat-Population (ITT), dass Frauen im Verumarm ein medianes Gesamtüberleben (OS) von mehr als 5,5 Jahren erreicht hatten – eine Verbesserung von 13,1 Monaten gegenüber einer Monothe-
MONARCH 3
rapie mit einem AI (66,8 vs. 53,7 Monate). Da die Studie nicht für das OS gepowert war, waren diese Unterschiede statistisch nicht signifikant (HR: 0,804; 95%-KI: 0,637 – 1,015; p = 0,0664) [1].
Benefit bei Patientinnen mit viszeralen Metastasen
Ein Vorteil ergab sich auch für Frauen mit viszeralen Metastasen (53 % der Patientinnen in MONARCH 3) – einer Subgruppe mit einem erhöhten Risiko für einen Progress oder Tod, verglichen mit Patientinnen, bei denen kein solcher Befall vorliegt. Patientinnen mit viszeralen Metastasen erreichten ebenfalls ein medianes OS von mehr als 5 Jahren, mit einer Verlängerung um 14,9 Monate im Vergleich zur Kontrollgruppe (63,7 vs. 48,8 Monate). Auch dieser Unterschied war aufgrund der
Die Studie MONARCH 3 untersuchte den CDK4 & 6 Inhibitor Abemaciclib (Verzenios®) in Kombination mit einem Aromatasehemmer (AI) im Vergleich zu AI plus Placebo als endokrine Initialtherapie für postmenopausale Patientinnen mit HR+, HER2– lokal fortgeschrittenem oder metastasiertem Mammakarzinom. Die doppelblinde, placebokontrollierte Phase-III-Studie schloss 493 Patientinnen ein, die im fortgeschrittenen Setting noch nicht mit einer systemischen Therapie behandelt worden waren. Sie erhielten im Verhältnis 2:1 randomisiert entweder kontinuierlich über 28 Tage 2 × täglich 150 mg Abemaciclib sowie den AI Anastrozol oder Letrozol bzw. den AI plus Placebo. Primärer Endpunkt war das mediane progressionsfreie Überleben (PFS); sekundäre Endpunkte umfassten u.a. die Ansprechrate, die Dauer des Ansprechens, das Gesamtüberleben und die Sicherheit [2].
Underpowerung statistisch nicht signifikant (HR: 0,758; 95%-KI: 0,558 – 1,030; p = 0,0757) [1].
PFS-Vorteil bestätigt
Der Vorteil beim progressionsfreien Überleben (PFS), dem primären Endpunkt der MONARCH-3-Studie, blieb auch in der finalen Analyse erhalten (29,0 vs. 14,8 Monate; HR: 0,535; 95%-KI: 0,429 – 0,668; nominal p < 0,0001), wobei die 6-Jahres-Rate im AbemaciclibArm mit 23,3 % deutlich höher war als mit 4,3 % im Vergleichsarm. Die statistische Signifikanz des PFS wurde bereits in einer Zwischenanalyse im Jahr 2017 erreicht, was zur globalen Zulassung für diese Indikation im Jahr 2018 führte.
Die finale Analyse zeigte keine neuen Sicherheitssignale bei längerfristiger Anwendung [1]. Abemaciclib ist über alle zugelassenen Indikationen hinweg in Deutschland voll verschreibungsund erstattungsfähig.
Elisabeth Wilhelmi, München
Literatur
1 Goetz M et al. MONARCH 3: Final overall survival results of abemaciclib plus a nonsteroidal aromatase inhibitor as firstline therapy in patients with HR+, HER2advanced breast cancer. Poster presented at: San Antonio, USA, Oral LBA am 6. Dezember 2023
2 Goetz M et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol 2017;35:3638-3646
Mit Omaveloxolon (Skyclarys™) wurde im März 2024 die erste und bisher einzige Therapie für Menschen mit Friedreich-Ataxie (FA) ab einem Alter von 16 Jahren in Deutschland eingeführt [1]. Die FA ist eine seltene Multisystemerkrankung und die häufigste Form der erblichen Ataxien [2]. Leitsymptome sind Bewegungs- und Gleichgewichtsstörungen, Störungen der Sensorik, Verlust der Reflexe, undeutliche Sprache, Schluckschwierigkeiten, Skoliose und Hohlfuß (Pes cavus) [2]. Erste Symptome manifestieren sich in der Regel im Alter zwischen 5 und 25 Jahren. Neben den massiven körperlichen Einschränkungen entwickeln sich bei Betroffenen häufig auch Komorbiditäten wie eine hypertrophe Kardiomyopathie und Diabetes mellitus [2]. Diese breite klinische Manifestation der FA führt dazu, dass Alltagsaktivitäten in den meisten Fällen nur noch eingeschränkt ausgeübt werden können – die meisten Betroffenen sind 10 – 15 Jahre nach dem Auftreten erster Symptome auf einen Rollstuhl angewiesen [3, 4]. Außerdem geht die Krankheit mit einer erheblichen psychischen Belastung z.B. in Form einer Depression einher [2]. Insbesondere die kardialen Komplikationen sind für die schlechte Prognose von Menschen mit FA verantwortlich: Bei einer Lebenserwartung von durchschnittlich 37 Jahren versterben die Betroffenen am häufigsten an Herzinsuffizienz und kardialen Arrhythmien [3, 4].
Bislang beschränkte sich die Therapie der FA vor allem auf symptomorientierte Maßnahmen sowie die Behandlung von internistischen Begleiterkrankungen und Skelettdeformationen [5]. Mit Omaveloxolon lässt sich das Fortschreiten der Erkrankung nun signifikant verlangsamen und das Ausüben von Alltagsfähigkeiten verbessern, was für die Betroffenen eine spürbare Veränderung bedeuten kann [6, 7]. Insbesondere ein frühzeitiger Behandlungsbeginn kann für einen bestmöglichen Erhalt der Lebensqualität und Unabhängigkeit von Menschen mit FA entscheidend sein [8]. Zugelassen ist Omaveloxolon für die Behandlung der FA bei Erwachsenen und Jugendlichen ab 16 Jahren. Eingenommen wird das Arzneimittel einmal täglich oral auf nüchternen Magen. Die empfohlene Dosis beträgt 150 mg (3 Hartkapseln à 50 mg) [1].
Signifikante Verbesserung der motorischen und bulbären Funktionen
Relevant für die Zulassung von Skyclarys™ waren die Ergebnisse der randomisierten, doppelblinden und placebokontrollierten PhaseII-Studie MOXIe [6]. Sie schloss 103* Patienten im Alter zwischen 16 und 40 Jahren mit genetisch
* Der vollständige Analysedatensatz schloss 42 Personen im OmaveloxolonArm und 40 Personen im Placebo-Arm ein.
bestätigter FA ein, die auf der modifizierten Friedreichs Ataxia Rating Scale (mFARS) einen Wert zwischen 20 und 80 Punkten erreichten. Die Studienteilnehmer erhielten 1:1 randomisiert Omaveloxolon (150 mg einmal täglich) oder ein Placebo [6].
Als primärer Endpunkt wurde die Differenz im mFARS-Wert zwischen Therapiestart und Therapiewoche 48 im Vergleich zu Placebo definiert [6]**. Dieser Wert bietet die Möglichkeit, die fortschreitenden Auswirkungen der FA auf die motorischen und bulbären Funktionen von Menschen mit FA zu verfolgen, wobei geringere mFARS-Werte bessere Funktionen bedeuten [9].
Nach 48 Wochen hatten sich die motorischen und bulbären Funktionen der Teilnehmer unter Omaveloxolon signifikant gegenüber Placebo verbessert: Im Verumarm sank der mFARS-Wert zwischen Therapiestart und Therapiewoche 48 um −1,55 (±0,69) Punkte, während er in der Kontrollgruppe im selben Zeitraum um 0,85 (±0,64) Punkte zunahm (Abb. 1). Der beobachtete Unterschied von −2,40 Punkten (±0,96) zwischen Omaveloxolon- und Placebo-Arm war signifikant (p = 0,014) [6].
Omaveloxolon bewirkte dabei in allen Domänen des mFARS eine Verbesserung, d.h. in der Bulbärfunktion, der Koordination der oberen und der unteren Extremi-
** Vollständige Analysepopulation ohne Betroffene mit Pes cavus.
täten sowie in der Stabilität der aufrechten Körperhaltung [6].
Eine explorative Analyse von Subgruppen stratifiziert nach Alter, Geschlecht, Anzahl der GAA-Triplett-Repeats und der Gehfähigkeit lieferte Hinweise darauf, dass die Betroffenen unabhängig von diesen Parametern von einer Therapie mit Omaveloxolon in Form eines verzögerten Fortschreitens der FA profitierten [6].
Fortschreiten der Krankheit auch längerfristig verlangsamt
Wie die Ergebnisse einer Erweiterungsstudie von MOXIe [8] zeigen, lassen sich die motorischen und bulbären Funktionen unter Omaveloxolon auch längerfristig verbessern, wobei sich eine frühzeitige Behandlung als Vorteil erwies. In die offene zweiarmige Erweiterungsstudie wurden 34 Patienten aus dem OmaveloxolonArm und 39 Patienten aus dem Placebo-Arm eingeschlossen. Beide Gruppen erhielten ab Woche 52 Omaveloxolon 150 mg einmal täglich über 72 Wochen. Dabei blieb der mFARS-Vorteil in der Gruppe, die bereits bis Woche 48 Omaveloxolon erhalten hatte, gegenüber dem Vergleichsarm über diesen längeren Zeitraum bestehen (Δ −2,91 Punkte ±1,44) [8].
Klinisch relevante Verlangsamung der Progression gegenüber dem natürlichen Verlauf
Den Krankheitsverlauf von Patienten unter Omaveloxolon in der MOXIe-Verlängerungsstudie wurde im Rahmen einer explorativen Posthoc-Analyse mit dem von Teilnehmern an der FA-COMS-Studie verglichen. Dabei handelt es sich um
Veränderung des mFARS-Scores vs. Baseline
Omaveloxolon (n = 40)
Placebo (n = 42)
40404038363534 42 Omaveloxolon(n) Placebo(n)424241414141
Abbildung 1: In der MOXIe-Studie sank nach 48 Wochen Therapie mit Omaveloxolon (Skyclarys™) der mFARS-Score signifikant gegenüber Placebo [mod. nach [6]).
des mFARS-Scores vs. Baseline (95%-KI)
Omaveloxolon (Verlängerung der MOXIe-Studie)
Natürlicher Verlauf (FA-COMS)
Jahre
MOXIeVerl.(n)133 136 13610277 FA-COMS(n)10810383
0,0101
Abbildung 2: Wie ein Vergleich des Krankheitsverlaufs von Patienten aus der offenen Erweiterungsstudie von MOXIe und Patienten aus der FA-COMS-Studie über den Zeitraum von 3 Jahren zeigte, ließ sich durch die Therapie mit Omaveloxolon (Skyclarys™) die Krankheitsprogression gegenüber dem natürlichen Verlauf signifikant verlangsamen (mod. nach [10]).
eine große Studie zum natürlichen Verlauf der FA, in der über 1.350 Betroffene mit FA bis zu 15 Jahre lang beobachtet wurden. In diesem Vergleich wurden 136 Personen der MOXIe-Verlängerungsstudie gemäß Propensity-Score-Matching 136 entsprechenden Personen aus
der FA-COMS-Studie zugeordnet, in der über 1.350 Betroffene mit FA bis zu 15 Jahre lang beobachtet wurden [10]. Über einen Verlauf von 3 Jahren zeigten die Betroffenen unter Omaveloxolon konstant geringere Veränderungen im mFARS-Score (relativ zur BaseWochen
Die Friedreich-Ataxie (FA) ist die häufigste erbliche Ataxie [2]. Sie betrifft in Europa schätzungsweise 1 von 50.000 Menschen, weltweit geht man von etwa 15.000 Betroffenen aus [11, 12]. Bei der FA wird die Störung der Bewegungskoordination und Haltungsinnervation primär durch eine Degeneration des Hinterstrangs im Rückenmark, eine axonale Neuropathie und eine zerebelläre Störung verursacht [2, 13].
Der FA liegt ein Mangel an Frataxin zugrunde [2]. Dabei handelt es sich um ein Protein, das in den Mitochondrien an der Regulation unterschiedlicher essenzieller Prozesse beteiligt ist, u.a. der Bildung von Eisen-Schwefel-Clustern, der mitochondrialen Atmungskette, der Biosynthese des Häms, des EisenMetabolismus und der DNA-Reparatur [14]. Besteht ein Frataxin-Defizit, zieht das eine mitochondriale Dysfunktion nach sich, wodurch es zur vermehrten Bildung reaktiver Sauerstoffspezies und damit zu oxidativem Stress kommt [2, 15].
Frataxin wird ubiquitär exprimiert, wobei sich die höchste Frataxin-Expression in Geweben mit hoher Stoffwechselrate wie Herz und Rückenmark findet; eine Frataxin-Expression wurde auch in Kleinhirn, Leber, Pankreas und Muskeln nachgewiesen (Abb. 1) [15].
Von einem Frataxin-Mangel am stärksten betroffen sind u.a. sensorische Neuronen der Spinalganglien, Kardiomyozyten, Inselzellen der Bauchspeicheldrüse sowie Zellen des Gehirns und der Netzhaut [16]. Ursache für das Frataxin-Defizit bei Betroffenen mit FA ist eine GAA-Trinukleotid-Repeat-Expansion im Intron 1 des Frataxin-codierenden FXN-Gens [17]. Während im FXN-Gen gesunder Menschen normalerweise 5 bis etwa 60 GAA-Tripletts vorliegen, können Personen mit FA bis zu 1.700 solcher GAA-Tripletts aufweisen [18].
FXN-Genexpression bei Gesunden und genetische Veränderung bei Menschen mit FA, die zu einem Frataxin-Mangel und in der Folge zu oxidativem Stress führt (mod. nach [19]).
Eine höhere Anzahl von GAA-Tripletts korreliert mit einem früheren Erkrankungsbeginn, schweren Symptomen und einem beschleunigten Verlust der motorischen Funktion [20, 21]. Bei der überwiegenden Mehrheit der Menschen mit FA liegt die GAA-Triplett-Repeat-Expansion im FXN-Gen homozygot vor (Anteil: 96 %) [17]. Bei den übrigen 4 % liegt auf einem Allel eine Triplett-Expansion und auf dem anderen Allel eine Duplikation, Deletion, Insertion oder Punktmutation vor [17]. Die Vererbung der FA erfolgt autosomal-rezessiv, wobei die Häufigkeit heterozygoter Mutationsträger in Deutschland etwa 1:89 beträgt [2].
Omaveloxolon (Skyclarys™) wirkt oxidativem Stress, einem zentralen Pfeiler der FA-Pathophysiologie, entgegen. Oxidativer Stress beschreibt einen Überschuss an reaktiven Sauerstoffverbindungen in den Mitochondrien. Ursächlich dafür sind bei FA ein Mangel des mitochondrialen Proteins Frataxin sowie ein beeinträchtigter Nrf2-Signalpfad [7, 22, 23].
Nrf2 (Nuclear factor Erythroid 2-related Factor 2) ist ein Transkriptionsfaktor, der als zentraler Regulator der zellulären Redox-Homöostase fungiert [24]. Ist seine Aktivität erhöht, exprimieren Zellen vor allem zytoprotektive Proteine, die die Zelle vor oxidativen und inflammatorischen Schäden schützen [7]. Ist die Nrf2-Aktivität wie im Falle der FA beeinträchtigt, führt dies ebenfalls zu oxidativem Stress. Es gibt Hinweise, dass die Frataxin-Defizienz im Zusammenhang mit der eingeschränkten Nrf2-Aktivität steht [25].
Omaveloxolon bewirkt indirekt eine Aktivierung von Nrf2, indem es dessen negativen Regulator Keap1 (Kelch-like ECH-associated protein 1) bindet und hemmt [1, 23, 26]. Durch seinen Nrf2-induzierenden Effekt wirkt Omaveloxolon somit protektiv gegen den oxidativen Stress in Körperzellen von Menschen mit FA [7].
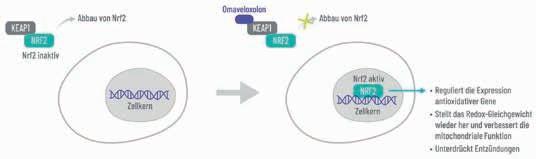
Abbildung 1: Schematische Darstellung des Wirkmechanismus von Omaveloxolon; adaptiert nach [1,8,12]
line) im Vergleich zu den Betroffenen aus der FA-COMS-Studie (Abb. 2). Konkret reduzierte die Behandlung mit Omaveloxolon das Fortschreiten der Erkrankung über 3 Jahre um 55 % gegenüber dem natürlichen Verlauf (Δ –3,61 Punkte nach 3 Jahren; p = 0,0001) [10].
Zulassungsstudie MOXIe
Gut handhabbares Nebenwirkungsprofil
unter Omaveloxolon zählten u.a. Kopfschmerz (37 %), erhöhte Alanin-Aminotransferase-Spiegel (37 %) und Übelkeit (33 %) [6]. Das Sicherheitsprofil in der Analyse der MOXIe-Studiendaten nach 48 Wochen war mit dem in der offenen Erweiterungsstudie nach 144 Wochen vergleichbar [6, 8].
Omaveloxolon bewirkt indirekt eine Aktivierung von Nrf2, indem es dessen negativen Regulator Keap1 (Kelch-like ECH-associated protein 1) bindet und hemmt (Abb. 1) [1,8,12]. Durch seinen Nrf2-induzierenden Effekt wirkt Omaveloxolon somit protektiv gegen den oxidativen Stress in Körperzellen von Menschen mit FA [3]
denken sollte. Nur durch eine enge interdisziplinäre Zusammenarbeit lässt sich die FA möglichst früh erkennen und die Diagnose mittels eines spezifischen Gentests sichern, sodass eine Therapie mit Omaveloxolon eingeleitet werden kann, die dazu beiträgt, dass den Patienten ihre Lebensqualität und Unabhängigkeit länger erhalten bleiben.
Studiendesign MOXIe war eine internationale, randomisierte, doppelblinde, placebokontrollierte Phase-IIStudie [2]. Sie schloss 103 * Patienten im Alter zwischen 16 und 40 Jahren mit genetisch bestätigter FA ein, die auf der modifizierten Friedreich’s Ataxia Rating Scale (mFARS) einen Wert zwischen 20 und 80 Punkten erreichten [2]. Die Studienteilnehmer erhielten 1:1randomisiert Omaveloxolon (150 mg einmal täglich) oder ein Placebo [2]
Primärer Endpunkt
Omaveloxolon erwies sich im Rahmen der MOXIe-Studie als gut verträglich und zeigte ein handhabbares Nebenwirkungsprofil [6]. Die meisten unerwünschten Ereignisse (UE) waren mild bis moderat. Zu den häufigsten UE
Fazit für die Praxis
Die Symptome der Friedreich-Ataxie können ziemlich unspezifisch sein, sodass der Arzt beim Auftreten mehrerer typischer Symptome auch an das Vorliegen dieser seltenen neuromuskulären Erkrankung
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Skyclarys™; Stand: 02/2024
2 Schulz JB et al. Nat Rev Neurol 2009; 5:222-224
Den primären Endpunkt bildete die Differenz im mFARS-Wert zwischen Therapiestart und Therapiewoche 48 im Vergleich zu Placebo [2]. † Der mFARS bietet die Möglichkeit, die fortschreitenden Auswirkungen der FA auf die motorischen und bulbären Funktionen von Menschen mit FA zu verfolgen, wobei geringere mFARS-Werte bessere Funktionen
3 Delatycki MB et al. J Child Neurol 2012; 27:1133-1137
4 Pandolfo M. J Neurol 2009;256 (Suppl. 1):3-8
5 Corben LA et al. Orphanet J Rare Dis 2022;17:415
6 Lynch DR et al. Ann Neurol 2021;89: 212-225
7 Abeti R et al. Front Cell Neurosci 2018; 12:188
8 Lynch DR et al. Mov Disord Off J Mov Disord Soc 2023;38:313-320
9 Etoom M et al. Healthcare 2022;10:2459
10 Lynch DR et al. Ann Clin Transl Neurol 2024;11:4-16
11 Becker E et al. Int J Biochem Cell Biol 2001;33:1-10
12 Friedreich’s Ataxia Research Alliance, What is FA?, https://www.curefa.org/ what-is-friedreichs-ataxia
13 Parkinson MH et al. J Neurochem 2013; 126:103-117
14 Maio N et al. Curr Opin Chem Biol 2020; 55:34-44
15 Gomes CM et al. Oxid Med Cell Longev 2013;2013:487534
16 Keita M et al. Neurodegener Dis Manag 2022;12:267-283
17 Galea CA et al. Ann Neurol 2016;79:485495
18 Barcia G et al. Eur J Med Genet 2018;61: 455-458
19 Gatchel JR et al. Nat Rev Genet 2005; 6:743-755
20 Dürr A et al. N Engl J Med 1996;335: 1169-1175
21 Metz G et al. Brain J Neurol 2013;136: 259-268
22 Cook A et al. Br Med Bull 2017;124:1930
23 Petrillo S et al. Int J Mol Sci 2019; 20:5211
24 Schmidlin CJ et al. Free Radic Biol Med 2019;134:702-707
25 Smith FM et al. Front Mol Biosci 2020; 7:569293
26 Saha S et al. Molecules 2020;25:5474
Seit Mitte Februar 2024 ist mit Etrasimod (Velsipity®) ein neuer Sphingosin-1-Phosphat (S1P)-Rezeptor-Modulator für die Behandlung der mittelschweren bis schweren aktiven Colitis ulcerosa (CU) in Europa zugelassen. Indiziert ist sie bei CU-Patienten, die auf eine konventionelle Therapie oder ein Biologikum unzureichend oder gar nicht angesprochen haben oder diese nicht vertragen. Etrasimod ist das erste und bislang einzige „Small Molecule“ für die Therapie der CU, von dem Patienten bereits ab 16 Jahren profitieren können [1]. Dank seiner besonderen Eigenschaften – starke Wirksamkeit, schnelle Symptomverbesserung bereits ab Tag 2, ein bislang über 2,5 Jahre gut untersuchtes Sicherheitsprofil, eine kurze Halbwertszeit sowie die orale Einnahme ohne Dosistitration [1, 2] – ist Etrasimod eine für den klinischen Alltag attraktive Medikation. Darüber hinaus ist Etrasimod die einzige innovative Therapie mit belegter klinischer Remission bei isolierter Proktitis [3]. Daher könnte der neue S1P-RezeptorModulator dazu beitragen, die bestehenden Lücken bei der Therapie der Colitis ulcerosa sowie der isolierten Proktitis weiter zu schließen.
Therapieziele werden oft nicht erreicht
Die Lücken in der Therapie der CU sind evident: Studiendaten zeigen, dass das in der aktualisierten S3-Leitlinie Colitis ulcerosa der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS) formulierte primäre Therapieziel – eine rasche klinische Remission und die Bewahrung einer langfristigen steroidfreien klinischen und endoskopischen Remission [4] –oft nicht erreicht wird.
Eine Analyse deutscher Krankenkassendaten kam zu dem Schluss, dass 75 % der CU-Patienten an einer unkontrollierten Erkrankung litten [5].
37 % der CU-Patienten mit 5-ASAErhaltungstherapie erlebten nach 6 – 12 Monaten erneut einen Schub [6]. Hinzu kommt, dass einer von 3 CU-Patienten unter den Symptomen einer isolierten Proktitis (IP) leidet [7]. Die Tendenz, die IP als eine leichtere Form der CU zu betrachten kann zu einer Untertherapie und damit letztlich zur Verschlechterung der Erkrankung führen [8]. Innovative und wirksame Therapieoptionen sind daher sowohl bei der CU als auch bei der IP vonnöten.
Internalisierung des Rezeptors
Ohne Velsipity:
Ohne Velsipity:
Recycling des Rezeptors
extrazellulär
intrazellulär
Internalisierung des Rezeptors
Abbildung 1: Wirkmechanismus der S1P-Rezeptor-Modulation [7, 11, 13, 14].
Etrasimod – eine orale Therapieoption mit innovativem Wirkmechanismus
In der Schleimhaut von CEDPatienten kommt es zu abnormen Immunreaktionen. Aktivierte TZellen schütten proinflammatorische Zytokine wie z.B. TNF-α, IL-4, IL-12, IL-17 und IL-23 aus, die Entzündungen verursachen und die epithelialen Barrieren beeinträchtigen. Als Reaktion auf Zellschäden werden in der Darmschleimhaut dendritische Zellen aktiviert, die in die Lymphknoten wandern, wo sie wiederum naive T- und B-Zellen aktivieren (primäre Reaktion). Diese wandern dann in die Darmmukosa und verursachen dort eine weitere Entzündung [9]. Einige dieser Lymphozyten verbleiben im Gewebe und werden bei erneuter Exposition gegenüber demselben Antigen reaktiviert (sekundäre Reaktion). Diese Mecha-
nismen halten die Entzündung bei CED aufrecht [10].
Der innovative Wirkansatz von Etrasimod zielt darauf ab, die Lymphozyten im Lymphknoten zurückzuhalten. Dies erfolgt durch die Modulation der Sphingosin-1-Phosphat (S1P)-Rezeptoren. Das Phospholipid S1P ist ein natürlich vorkommendes Signalmolekül, das an wichtigen immunologischen, inflammatorischen und kardiovaskulären Funktionen beteiligt ist.
Die unterschiedlichen physiologischen und pathophysiologischen Signale von S1P werden über die Rezeptoren S1P1 bis S1P5 vermittelt [11, 12]. S1P1, S1P4 und S1P5 sind an der Regulierung des Immunsystems beteiligt, indem sie die Migration oder Bewegung von Immunzellen wie T-Zellen, dendritischen Zellen und natürlichen Killerzellen beeinflussen. Eine Schlüsselrolle spielt dabei der Re-
Mit Velsipity:
Mit Velsipity:
Abbau des Rezeptors
zeptor S1P1, da er auch auf B- und T-Lymphozyten exprimiert wird und S1P durch Bindung an diesen Rezeptor deren Migration aus den Lymphknoten initiiert [12].
Indem Etrasimod an die S1P1-Rezeptoren bindet, induziert es die Internalisierung und den Abbau dieser Rezeptoren, sodass spezifische Lymphozyten-Subpopulationen in den Lymphknoten selektiv zurückgehalten werden (Abb. 1) [13]. Dadurch stehen weniger periphere Immunzellen zur Verfügung, die zu den Entzündungsherden im Darm gelangen können [14]. Dieser Prozess ist nach Absetzen von Etrasimod reversibel. In den beiden klinischen Studien ELEVATE UC 52 bzw. UC 12 kehrte die absolute Lymphozytenzahl bei 83 % bzw. 77 % der Patient innerhalb von 2 Wochen nach Beendigung der EtrasimodTherapie in den Normbereich zurück [2].

Abbildung 2: Ergebnisse für die Co-primären Endpunkte
Abbildung 2: Ergebnisse für die Co-primären Endpunkte der Studie ELEVATE UC 52, die klinische Remission in Woche 12 und Woche 52 [2].
Die Zulassung von Etrasimod basiert auf den Daten des klinischen Studienprogramms ELEVATE UC, bestehend aus den beiden unabhängigen randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien ELEVATE UC 12 und ELEVATE UC 52 [2]. Eingeschlossen wurden Patienten im Alter von 16 – 80 Jahren mit mittelschwerer bis schwerer aktiver CU, die auf eine konventionelle Therapie oder ein Biologikum unzureichend angesprochen haben, nicht mehr darauf ansprechen oder diese nicht vertragen haben. Auch Patienten mit isolierter Proktitis nahmen teil. Alle Studienteilnehmer erhielten randomisiert im Verhältnis 2:1 entweder Etrasimod 1 × täglich 2 mg oder Placebo. In die 12-wöchige Studie ELEVATE UC 12 zur Induktionstherapie wurden 354 Patienten eingeschlossen, ELEVATE UC 52 mit 433 Patienten umfasste eine 12-wöchige Induktion, gefolgt von 40 Wochen Erhaltungstherapie. Studienteilnehmer, bei denen zu Woche 12 keine Verbesserung oder eine Verschlechterung eintrat, konnten in eine offene
Extensionsstudie (Laufzeit bis zu 5 Jahren) wechseln. Nach 52 Wochen konnten alle Patienten in die Extensionsstudie wechseln [2]. Im Folgenden werden die Ergebnisse der 52-wöchigen Studie ELEVATE UC dargestellt.
Nach Abschluss der 12-wöchigen Induktionsphase erreichten 27 % der Patienten unter Etrasimod gegenüber 7 % unter Placebo eine klinische Remission. In Woche 52 war das bei 32 % in der EtrasimodGruppe und bei 7 % unter Placebo der Fall. Der Unterschied war jeweils signifikant (p < 0,0001) (Abb. 2) [2]. Auch bei den wichtigsten sekundären Endpunkten zeigte Etrasimod zu Woche 12 und zu Woche 52 eine überlegene Wirksamkeit gegenüber Placebo [2]:
• endoskopische Verbesserung: 35 % vs. 14 %
• symptomatische Remission: 46 % vs. 21 %
• endoskopische Verbesserung mit histologischer Remission: 21 % vs. 4 %
Primäres Therapieziel ist, dass die CU-Patienten eine anhaltende klinische Remission und eine steroidfreie klinische Remission errei-
Velsipity ®
chen. Zu Woche 52 wurden diese beiden sekundären Endpunkte erreicht [2]:
• anhaltende klinische Remission: 18 % vs. 2 %
• steroidfreie Remission (definiert als kein Gebrauch von Glukokortikoiden für mindestens 12 Wochen unabhängig von einer Glukokortikoid-Therapie zu Baseline): 32 % vs. 7 %
Alle Patienten, die sich zu Woche 52 in klinischer Remission befanden, waren gleichzeitig mindestens 12 Wochen lang steroidfrei. Somit erreichten 100 % der Patienten unter Etrasimod eine steroidfreie Remission [2].
Die Therapie mit Etrasimod führte außerdem zu einem raschen Wirkeintritt mit symptomatischer Remission zu Woche 2. Bereits zu diesem Zeitpunkt kam es bei einem signifikant größeren Teil der mit Etrasimod behandelten Patienten zu einer Abnahme von rektalen Blutungen und Stuhlfrequenz gegenüber Placebo [2].
Etwa 37 % der Patienten aus dem ELEVATE UC-Studienprogramm waren zuvor mit mindestens einem Biologikum oder JAK-Inhibitor (JAKi) behandelt worden. Die
Abbildung 3: Klinische Remission bei Patienten mit isolierter Proktitis [3].
3:
Abbildung 3: Klinische Remission bei Patienten mit isolierter Prokitis [3].
Studienergebnisse von ELEVATE UC 52 zeigten, dass Patienten, die Biologika-/JAKi-naiv waren noch etwas mehr von der Wirksamkeit von Etrasimod profitierten als jene, die mit Biologika-/JAKi-vorbehandelt waren. Es kann sich somit für CU-Patienten lohnen, Etrasimod bereits in der frühen Therapielinie, nach Versagen von konventionellen Behandlungsoptionen, einzusetzen.
In ELEVATE UC zeigte Etrasimod ein günstiges Sicherheitsprofil, das mit früheren Studien übereinstimmte. Bradykardien unter Etrasimod waren im Allgemeinen selten, mild, asymptomatisch und traten meist an Tag 1 mit der ersten Dosis auf. Auch Hypertonieereignisse traten selten auf und führten in keinem Fall zum Abbruch der Behandlung. Schwerwiegende Ereignisse von Bradykardie oder AV-Block wurden nicht berichtet. Im Vergleich zu Placebo kam es unter Etrasimod nicht zu einer Zu-
nahme von Infektionen. Es wurden keine Todesfälle, Malignome oder schwerwiegenden Leberschädigungen berichtet [2].
Gute Wirksamkeit auch bei isolierter Proktitis
In das ELEVATE-Studienprogramm wurden auch 64 Patienten mit isolierter Proktitis (Etrasimod n = 42, Placebo n = 22) eingeschlossen [2] In Woche 12 (ELEVATE UC 52 und UC 12) und Woche 52 (ELEVATE UC 52) erreichten in einer Post-hoc-Analyse 43 % und 44 % der Patienten unter Etrasimod den primären Endpunkt einer klinischen Remission (Placebo: 14 % und 11 %, Abb. 3) [3].
Auch bei den wichtigen sekundären Endpunkten – endoskopische Verbesserung, symptomatische Verbesserung und endoskopische Verbesserung mit histologischer Remission – war Etrasimod signifikant wirksamer als Placebo. Eine anhaltende klinische Remission sowie eine steroidfreie Remission
erreichten zu Woche 52 jeweils 30 % und 44 % unter Etrasimod (Placebo: in beiden Fällen 11 %) [3].
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Velsipity®; Stand: Februar 2024
2 Sandborn WJ et al. Lancet 2023;401: 1159-1171
3 Peyrin-Biroulet L et al. J Crohns Colitis 2023;17(Suppl 1):i536-i538
4 Aktualisierte S3-Leitlinie Colitis ulcerosa (Version 6.2) Januar 2024; AWMF-Registriernummer: 021-009
5 Bokemeyer B et al. Inflamm Bowel Dis 2022;28:1647-1657
6 Murray A et al. Cochrane Database Syst Rev 2020;8:CD000544
7 Ordás I et al. Lancet 2012;380:1606-1619
8 Gawel K et al. Scand J Gastroenterol 2022;57:1406-1411
9 Mann ER et al. World J Gastroenterol 2014;20:9653-9664
10 Wallace KL et al. World J Gastroenterol 2014;20:6-21
11 Choden T et al. Gastroenterol Hepatol (NY) 2022;18:265-271
12 Pérez-Jeldres T et al. Drugs 2021;81:9851002
13 Peyrin-Biroulet P et al. Autoimmmun Rev 2017;16:495-503
14 Danese S et al. J Crohns Colitis 2018; 12(Suppl 2):678-686
Levodopa-Äquivalenzdosis bei Parkinson-Medikamenten:
Überlegenes L-DopaEinsparpotenzial bei Safinamid 100 mg
Die Levodopa-Äquivalenzdosis von Parkinson-Medikamenten hilft bei notwendigen Therapieanpassungen die L-Dopa-Dosierung bedarfsgerecht zu adjustieren. Aktuelle Daten, die auf einem Symposium von Zambon beim Kongress der Deutschen Gesellschaft für Neurologie vorgestellt wurden, belegen ein überlegenes L-DopaEinsparpotenzial von 100 mg Safinamid (Xadago®) gegenüber herkömmlichen MAO-B-Hemmern.
L-Dopa-Therapie gezielt anpassen
Beim fortgeschrittenen ParkinsonSyndrom werden infolge einer Verschlechterung der Symptomatik häufig Therapieanpassungen notwendig. In dieser Phase wird je-
doch das therapeutische Fenster für die medikamentöse Behandlung zunehmend enger. Bei erforderlichen Dosissteigerungen von L-Dopa oder der Hinzunahme von Add-onMedikamenten gilt es zu vermeiden, dass überdosiert wird und sich Komplikationen wie Hyperkinesien entwickeln. Umgekehrt ist auch beim notwendigen Absetzen oder Reduzieren von Begleitmedikationen die L-Dopa-Therapie so anzupassen, dass der Patient weiterhin eine gute Beweglichkeit behält. Darauf wies Professor Wolfgang Jost, Wolfach, hin. „In diesen Fällen ist es von Nutzen, die Levodopa-Äquivalenzdosis der einzelnen Medikamente genau zu kennen“, betonte der Neurologe. „So kann die L-Dopa-Therapie gezielt angepasst werden, ohne dass das therapeutische Fenster verlassen wird“.
Safinamid besticht durch starke Wirksamkeit und hohe LevodopaÄquivalenzdosis
In neuen Untersuchungen wurde die Levodopa-Äquivalenzdosis
von verschiedenen ParkinsonMedikamenten ermittelt und eine entsprechende Hilfestellung angeboten. Jost stellte die aktuelle Arbeit von Cilia et al. vor, eine longitudinale retrospektive FallKontroll-Studie mit 500 Parkinson-Patienten, die die Wirksamkeit und Levodopa-Äquivalenzdosis von Safinamid in den Dosierungen 50 mg und 100 mg sowie von Rasagilin 1 mg untersuchte. Safinamid kann aufgrund seiner dualen Wirkweise sowohl motorische als auch nicht motorische Symptome verbessern und gilt daher beim fortgeschrittenen Morbus Parkinson als probate Add-on-Therapieoption.
Nach der Nachbeobachtungszeit von 8,8 – 10,1 Monaten wiesen die mit Safinamid 50 mg und 100 mg behandelten Patienten eine stärkere Verbesserung im UPDRS III Score (p < 0,001) und im UPDRS IV Score auf (p = 0,23) als die Patienten unter der Vergleichsmedikation Rasagilin bzw. in der Kontrollgruppe ohne MAO-BHemmer. Insgesamt entsprach die Wirksamkeit von Safinamid 50 mg
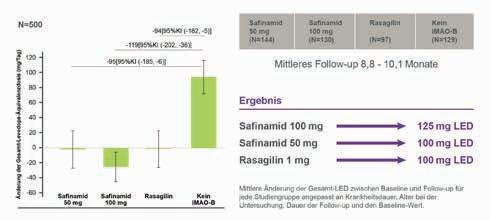
Abbildung 1: Levodopa-Äquivalenzdosis (LED) von Safinamid im Vergleich zu Rasagilin, Ergebnisse einer longitudinalen retrospektiven FallKontroll-Studie (mod. nach Cilia R et al. Mov Dis Clin Pract 2023;10:625-635).
und Rasagilin 1 mg in etwa einer Levodopa-Äquivalenzdosis von 100 mg. In der Dosierung von 100 mg erreichte Safinamid jedoch eine Levodopa-Äquivalenzdosis von 125 mg (Abb. 1). „Es gibt also einen Unterschied zwischen den beiden SafinamidDosierungen und auch einen Unterschied zwischen höher dosiertem Safinamid und Rasagilin, der bei der Dosisanpassung von L-Dopa berücksichtigt werden sollte“, riet Jost. „Aufgrund der überlegenen Wirkung von 100 mg Safinamid ergibt sich ein um 25 % höheres Einsparpotenzial bei L-Dopa. Wird die L-Dopa-Dosis entsprechend stärker reduziert, kann der Patient von den Effekten von Safinamid profitieren und gleichzeitig bleibt die Gefahr gering, dass das therapeutische Fenster verlassen wird“, fasste Jost zusammen.
Fabian Sandner, Nürnberg
Der schwelenden Neuroinflammation bei MS auf der Spur: Innovative Therapiestrategien mit BTKInhibitoren
Das Verständnis zum Krankheitsverlauf bei Multipler Sklerose (MS) erlebt derzeit einen Wandel. Anlässlich des 96. Kongresses der Deutschen Gesellschaft für Neurologie (DGN) diskutierten Experten die schleichende Zunahme der MS-Symptomatik unabhängig von Schüben, die auf schwelende neuroinflammatorische Prozesse im ZNS zurückgeführt wird. Als vielversprechender Ansatz zur Reduktion der schwelenden und akuten Neuroinflammation wurde u.a. der ZNS-gängige Hemmer der Bruton-
Tyrosinkinase (BTK) Tolebrutinib vorgestellt, der aktuell in einem umfassenden klinischen Phase-IIIStudienprogramm für alle Subtypen der MS untersucht wird.
PIRA – die Herausforderung in der MS-Therapie
„Die schubunabhängige Progression kann klinisch relevante Ausmaße annehmen“, berichtete Dr. Boris-Alexander Kallmann, Bamberg. „Viele Menschen mit MS erfahren auch unter Therapie eine schleichend größer werdende Zunahme ihrer Behinderung. Gut eingestellte Patienten berichten in meiner MS-Sprechstunde zum Beispiel, dass sie nicht mehr so schnell und weit wie früher Joggen können oder bei der Arbeit immer mehr Konzentrationsschwierigkeiten entwickeln.“ Neben der schubabhängigen Behinderungszunahme (Relapse-Associated Worsening, RAW) ist die schubunabhängige Progression (Progression Independent of Relapse Activity, PIRA) ein häufiges und relevantes Problem für Betroffene. Aktuell wird davon ausgegangen, dass auch bei Patienten mit schubförmiger MS (RMS) ein Großteil der Behinderungsprogression auf PIRA zurückgeführt werden kann. In einer groß angelegten Querschnitts-Befragung berichteten 88,9 % der Menschen mit ausgeprägter bis schwerer schubförmig-remittierender MS (Relapsing Remitting Multiple Sclerosis, RRMS), bereits Erfahrungen zu schubunabhängigen Verschlechterungen von Symptomen gemacht zu haben. „Daher besteht vor allem für Verlaufsformen mit hoher Krankheitsaktivität bei RMS und RRMS ein erheblicher therapeutischer Bedarf, um PIRA zu verhindern“, schlussfolgerte Kallmann.
Da die Progression von MS durch PIRA schon früh im Krankheitsverlauf beginnen kann, sollten Betroffene in regelmäßigen Abständen ihren Symptom-Status dokumentieren, empfahl der Experte.
BTK-Inhibitoren als neuer Wirkansatz
Um in Zukunft Prozesse der schwelenden Neuroinflammation im ZNS besser zu adressieren, sind für Professor Martin Kerschensteiner, München, BTK-Inhibitoren ein interessanter Ansatz. Zum einen, weil BTK-Inhibitoren wie Tolebrutinib gemäß den bisherigen Ergebnissen aus präklinischen Invivo- und In-vitro-Experimenten aller Wahrscheinlichkeit nach in ausreichenden Konzentrationen ZNS-gängig sind. Und zum anderen, weil BTK-Inhibitoren möglicherweise die entzündliche Aktivierung von B-Zellen, Makrophagen und Mikroglia und deren Wechselwirkungen herunterregulieren. Nach Einschätzung von Kerschensteiner könnten durch dieses Wirkprinzip die pathologischen Prozesse im Gehirn und insbesondere die schwelenden Entzündungsprozesse positiv moduliert werden. „Die bisherigen Befunde legen die Vermutung nahe, dass BTK-Inhibitoren wahrscheinlich relevante Zellen im ZNS erreichen und dort entzündliche Prozesse beeinflussen können“, schlussfolgerte der Experte.
Groß angelegtes Phase-IIIStudienprogramm zu Tolebrutinib
Professor Mathias Mäurer, Würzburg, stellte das umfassende Studienprogramm zum BTK-Inhibitor Tolebrutinib über das gesamte
Krankheitsspektrum der MS vor. Das Phase-III-Studienprogramm zu oralem Tolebrutinib wurde über die verschiedenen Verlaufsformen der MS hinweg sehr breit aufgestellt. So werden in der HERCULES-Studie Patienten mit nrSPMS (nicht schubförmiger sekundär progredienter MS), in der PERSEUS-Studie Patienten mit PPMS (primär progredienter MS) und in den beiden Zwillingsstudien GEMINI I & II Patienten mit RRMS untersucht.
Mit großer Spannung erwartet Mäurer vor allem die Ergebnisse aus der HERCULES-Studie. Nach seiner Einschätzung ist der therapeutische Bedarf bei nrSPMS besonders hoch, weil in dieser Phase der Erkrankung bisher noch keine wirklich überzeugende Modulation der Erkrankungsprogression gelungen ist. Im Rahmen von HERCULES werden derzeit 1131 therapienaive (35,1 %) und vorbehandelte (64,9 %) Patienten mit nrSPMS (mittleres Alter: 48,9 Jahre; 62 % weiblich) untersucht, die zu Baseline im Median einen EDSS-Score (Expanded Disability Status Scale) von 6,0 (Bereich 5,0 – 6,3) aufwiesen. „Die Rekrutierung zu den klinischen PhaseIII-Studien ist mittlerweile abgeschlossen. Die erwarteten Daten werden wichtige Einblicke zur Wirksamkeit und Sicherheit von Tolebrutinib geben“, so Mäurer.
Phase-II-Studiendaten deuten auf gute Verträglichkeit und Wirksamkeit hin
Erste Hinweise zur Wirksamkeit und Sicherheit liefern aktuelle 3-Jahres-Ergebnisse aus der Verlängerungsphase der Dosisfindungsstudie zu Tolebrutinib bei RRMS. „Bisher wurden keine
ernsten Sicherheitssignale im Laufe der Phase-II-Studie beobachtet“, berichtete Mäurer. Die Auswertungen deuteten bislang auf eine gute Verträglichkeit hin. Etwa 73 % der Behandelten (60 mg/Tag Tolebrutinib, oral) waren nach 2,5 Jahren schubfrei und mindestens 72 % entwickelten zwischen Woche 144 und Woche 96 beispielsweise keine neuen Gadolinium-anreichernden T1-gewichteten Läsionen. „Die bisherigen Daten sind erfreulich und machen Hoffnung, dass der Wirkansatz zur BTK-Inhibition einen therapeutischen Fortschritt bei MS mit sich bringen könnte“, schlussfolgerte der Experte.
Elisabeth Wilhelmi, München
Clensia zur Darmvorbereitung für die Koloskopie
Die Effektivität einer Koloskopie hängt u.a. von einer adäquaten Visualisierung ab, denn eine gute Sicht auf die Mukosa ist mitbestimmend für die Adenom-Detektionsrate (ADR) und die ZäkumIntubationsrate (ZIR). Mit Clensia ist jetzt ein Präparat zur Darmreinigung verfügbar, das als einziges in Deutschland den von der European Society of Gastrointestinal Endoscopy (ESGE) empfohlenen Entschäumer Simeticon enthält. Es ermöglicht – bei adhärenten Patienten – eine signifikant bessere Sicht als eine herkömmliche 2L-PEGASC-Lösung. Mit kostengünstigen Multipacks, patientenfreundlichem Limettengeschmack und praxisnahen Servicetools kann Clensia die Darmvorbereitungspalette attraktiv erweitern. Bei einem Pressegespräch machte Professor Peter Malfertheiner,
München, auf die vielfältigen Implikationen aufmerksam, die eine unzureichende Darmvorbereitung mit sich bringen kann. Neben den offensichtlichen Nachteilen wie einer geringeren ADR oder ZIR sind laut Malfertheiner auch praktische Aspekte wichtig: „Wenn Abläufe durcheinander geraten, weil einzelne Prozeduren länger dauern oder gar abgebrochen werden, ist das mehr als ärgerlich und belastend. Es ist kostspielig und organisatorisch aufwendig, und nicht wenige Patienten nehmen den Wiederholungstermin gar nicht wahr.“
Simeticon im Abführpräparat verbessert die Sicht auf die Mukosa
Nicht nur Stuhlreste, auch im Darm verbleibender Schaum kann die Sicht auf die Mukosa deutlich beeinträchtigen. Bei 32 – 57 % der Koloskopien sind noch Blasen oder Schaum zu beobachten (Abb. 1). Hier kann die Gabe von Simeticon einen entscheidenden Unterschied bedeuten: Ohne Simeticon sind allein zur Reinigung der Linse 4,6-mal so viele Wassersprühstöße notwendig, was mit einer deutlichen Ermüdung der Untersucher einhergeht: 2,97 ohne Simeticon vs. 1,31 mit Simeticon (auf einer Skala von 1 bis 10). „Alle Koloskopierenden, die von morgens bis spätnachmittags hochkonzentriert diese Untersuchungen durchführen, wissen, wie mühsam und lästig dieser zusätzliche Spülvorgang ist“, betonte Malfertheiner. Er verwies in diesem Zusammenhang auch darauf, dass die lokale Applikation von Simeticon zwar möglich ist, aber von der ESGE nicht empfohlen wird. „Die leider immer noch anzutreffende Praxis,
Abbildung 1: Clensia ermöglicht eine optimale Sicht auf die
Schaum nach Darmeinigung mit Clensia (© Alfasigma).
VivaScope 2500 – eine digitale Innovation in der Mohs-Chirurgie des Basalzellkarzinoms
Clensia
Clensia zählt zu den osmotisch wirksamen Laxanzien. Der primäre Wirkmechanismus beruht auf der osmotischen Wirkung von Macrogol 4000 (Polyethylenglykol, PEG), Natriumsulfat und Citraten, die dazu führt, dass Wasser im Dickdarm zurückgehalten wird. Dadurch werden feste Stuhlbestandteile vermehrt abtransportiert und eine abführende Wirkung erzielt, die letztlich zur Reinigung des Kolons führt.
Clensia ist als Pulver in 2 verschiedenen Beuteln verfügbar:
• Inhalt Beutel A: Macrogol 4000, Natriumsulfat und Simeticon
• Inhalt Beutel B: Natriumcitrat, Zitronensäure, Natriumchlorid und Kaliumchlorid
Dosierungsschema1
Für eine Einzelbehandlung muss der in 2 Liter Wasser aufgelöste Inhalt von jeweils 4 Beuteln A und B getrunken werden. Dabei sind zwei Einnahmeschemata möglich:
Für eine Einzelbehandlung muss der in 2 Liter Wasser aufgelöste Inhalt von jeweils 4 Beuteln A und B getrunken werden. Dabei sind 2 Einnahmeschemata möglich:

Wirkungsweise1
Simeticon über den Spülkanal zu applizieren, ist zwar eine Lösung, aber mit Simeticon im Abführpräparat heute meist vermeidbar.“
Sollte eine lokale Anwendung notwendig sein, ist lediglich der Arbeitskanal zulässig.
Sicherheit und Verträglichkeit
„Jetzt eine Alternative zu haben, Simeticon gleich mit der Darm-
vorbereitung oral zu geben, wird sicher gut angenommen. Wir können damit sowohl den Leitlinien als auch einer effizienten Praxisund Untersuchungsroutine nachkommen“, so das Fazit von Malfertheiner.
Clensia zählt zu den osmotisch wirksamen Laxanzien. Der primäre Wirkmechanismus beruht auf der osmotischen Wirkung von Macrogol 4000 (Polyethylenglykol, PEG), Natriumsulfat und Citraten, die dazu führt, dass Wasser im Dickdarm zurückgehalten wird. Dadurch werden feste Stuhlbestandteile vermehrt abtransportiert und eine abführende Wirkung erzielt, die letztlich zur Reinigung des Kolons führt.
• Günstiges Sicherheitsprofil und geringe Nebenwirkungsraten2,10
Elisabeth Wilhelmi, München• Die häufigsten Nebenwirkungen in kontrollierten klinischen Studien sowie nach Markteinführung sind gastrointestinale Beschwerden (abdominale Krämpfe, Blähungen, Übelkeit und Irritation der Analgegend), welche grundsätzlich häufig bei Darmreinigungen mit Mischungen aus Macrogol und Elektrolyten auftreten. Sie sind im Allgemeinen leicht ausgeprägt und klingen in der Regel rasch ab, wenn die Verabreichungsgeschwindigkeit gesenkt oder die Einnahme vorübergehend
Nach Angaben der Internationalen Agentur für Krebsforschung (IARC) steigt die Zahl der Krebserkrankungen weltweit und Krebsdiagnosen werden nach den Prognosen bis 2050 um 77 % auf 35 Millionen pro Jahr zunehmen. Kostengünstigere Behandlungsmethoden sind daher dringend notwendig. Eine wegweisende Studie unter der Leitung von Professor Jan Gutermuth vom Universitätsklinikum Brüssel beschreibt einen revolutionären Ansatz: den Einsatz der Ex Vivo Konfokalen Mikroskopie (EVCM) mit dem VivaScope 2500 im Rahmen der mikrographisch kontrollierten Chirurgie nach Mohs (MMS) zur Resektion des Basalzellkarzinoms (BCC). Gutermuth und sein Team konnten zeigen, dass durch den Einsatz dieser Technologie nicht nur eine präzisere, sondern auch schnellere Bewertung der Tumorausdehnung und Resektionsränder möglich ist. Die durchschnittliche Eingriffszeit wird deutlich verkürzt und es können 155 % mehr Mohs-Eingriffe pro Chirurg mit gleichzeitig weniger Personal durchgeführt werden. Die Patienten profitieren dadurch von kürzeren Wartezeiten auf Behandlungen, was schnellere Terminvergaben in den Kliniken ermöglicht, sowie von einer verbesserten Versorgung durch weniger Eingriffe und minimalinvasive Verfahren. Außerdem lassen sich durch den Einsatz des VivaScope 2500 in der Mohs-Chirurgie die Kosten für Kliniken um 57 % senken.
Das Ex-vivo-Mikroskop VivaScope 2500 –ein digitalisiertes Labor auf einem Wagen (© VivaScope GmbH).
Eingriffe werden deutlich kürzer
Die Mohs-Chirurgie gilt als die effektivste Technik zur Behandlung von Basalzellkarzinomen, einer der häufigsten Hautkrebsformen. Bisher ist dies jedoch für Kliniken sehr kostspielig. Für die Beurteilung der Schnittränder während der Hautkrebschirurgie wird bislang auf Gefrierschnitttechniken zurückgegriffen. Dies erfordert jedoch einen beträchtlichen personellen Aufwand und viel Zeit: Nach jedem Schnitt muss das Gewebe zur histopathologischen Analyse eingeschickt werden –Verzögerungen und gegebenenfalls zusätzliche Operationen sind die Folge.
Mit dem VivaScope kann das bei der Operation frisch entnommene Gewebe dagegen direkt gescannt und die Histologie der Schnittränder visualisiert und beurteilt werden. Dies dauert nur wenige
Minuten, sodass eine rasche Fortführung des Eingriffs möglich ist. Die Patienten können das Krankenhaus schon kurz nach dem EVCM-basierten Verfahren verlassen. Wie die Studie ergab, ließ sich durch den Einsatz des VivaScope 2500 die durchschnittliche Eingriffszeit pro Verfahren von 135 auf 75 Minuten reduzieren, was einer eindrucksvollen Senkung um 44 % entspricht.
Geringere Kosten bei hervorragenden Ergebnissen
Der Einsatz der Ex Vivo VivaScope Technologie führt außerdem dazu, dass weniger Personal benötigt und so die Kapazitätskostenrate (CCR) um die Hälfte reduziert wird, was die finanzielle Belastung des Gesundheitssystems verringert. Die Kosten für Personal, Operationssaal und Material gehen um 57 % zurück. Auch der Aufwand für das Erlernen der Technik ist gering: Es gibt etablierte Schulungsprogramme, die von Experten geleitet werden und es dem medizinischen Personal ermöglichen, sich in kurzer Zeit einzuarbeiten. Über die Jahre konnte mit dieser Methode die Anzahl der durchgeführten Prozeduren pro Vollzeit arbeitenden Chirurgen um 155 % gesteigert werden. Die Zeitersparnis muss aber nicht durch einen Qualitätsverlust erkauft werden. Denn die VivaScope 2500 Ex-Vivo-Technologie liefert H&E-ähnliche Schnittbilder, die aus 2 Komponenten generiert werden. 2 Laser unterschiedlicher Wellenlänge erzeugen 2 unterschiedliche Bilder, ein Fluoreszenzbild und ein Reflexionsbild. Beide Signale generieren zusammen pseudofarbige Bilder. Die Software des Geräts verwendet
Basalzellkarzinom; abgebildet mit dem VivaScope 2500 (links) und nach H&E-Färbung (rechts) (© VivaScope GmbH).
einen Algorithmus, um die erfassten Bildinformationen in Farben zu übersetzen, die H&E ähneln. Pathologen können diese Bilder aufgrund ihrer hohen Ähnlichkeit wie gewöhnliche H&E Bilder lesen und beurteilen – entweder vor Ort oder telemedizinisch aus der Ferne. Das untersuchte Gewebe bleibt durch den Eingriff unversehrt und kann für eine spätere histopathologische Analyse konserviert werden.
Dank der Genauigkeit der digitalen Bildgebung mit dem VivaScope 2500 konnte die Diskrepanz zwischen den histopathologischen Ergebnissen und denen der Ex Vivo VivaScope Technologie von 12 auf 2 % gesenkt werden. Insgesamt weist der Einsatz des VivaScope 2500 bemerkenswerte Kennzahlen auf, insbesondere bei der Erkennung von BCC-Resten in den Resektionsrändern während der MMS. Die hohen Zuverlässigkeitsraten der Sensitivität (96,6 %) und Spezifität (99 %) unterstreichen die Effektivität und Effizienz der Methode bei der Tumorerkennung.
B. S.
Die ROTE LISTE® bietet seit Jahrzehnten verlässliche, profunde Informationen rund um die in Deutschland verfügbaren Humanarzneimittel einschließlich EUZulassungen und bestimmter Medizinprodukte. Die ROTE LISTE® 2024 umfasst als Buchausgabe 1984 Druckseiten. Das Präparateverzeichnis orientiert sich an den Indikationen und ist in 88 Hauptgruppen aufgefächert. Innerhalb

Rote Liste 2024 Einzelausgabe, ISBN 9783-911149-00-6, 109,00 Euro, erhältlich ab April 2024 der Hauptgruppen erfolgt eine weitere Untergliederung in Unter-
Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin
Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
gruppen, die die Präparate alphabetisch sortiert enthalten. Damit ermöglicht die ROTE LISTE® ihren Nutzern einen raschen und umfassenden Überblick über das Arzneimittelspektrum in Deutschland. Spezielle Informationen zu den Wirkstoffgruppen, Medikamenten und Darreichungsformen sowie kurz gefasste Produktinformationen und Hintergrundinformationen für die Praxis in Sonderkapiteln geben Hilfestellung bei der Verordnung.
Titelbild: Krebsdiagnostik (Quelle iStock).
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden im Journal für Pharmakologie und Therapie personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz: HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM_W.R.
Roland Welker Austraße 7, 96114 Hirschaid Verlag PERFUSION GmbH Storchenweg 20 90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de E-Mail: perfusion@t-online.de
XADAGO ® – Zusatztherapie zu Levodopa für Ihre Parkinson-Patienten.4

-22%
XADAGO jetzt zum FestbetragSicher und wirtschaftlich verordnen
1. Borgohain R, et al. (Study 018) Two-year, randomized, controlled study of safinamide as add-on to Levodopa in mid to late Parkinson’s disease. Movement Disorders 2014; 29(10):1273-80. 2. Cattaneo C. et al. Long-term Efficacy of Safinamide on Parkinson’s Disease Chronic Pain. Adv Ther. 2018 Apr;35(4):515-522. 3. Borgohain R, et al. (Study 016) Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Movement Disorders 2014; 29(2):229–237. 4. XADAGO Fachinformation.
Xadago® 50 mg Filmtabletten; Xadago® 100 mg Filmtabletten. Wirkstoff: Safinamidmesilat (50 mg bzw. 100 mg Safinamid/Filmtablette). Sonst. Bestandteile: Mikrokristalline Cellulose, Crospovidon Typ A, Magnesiumstearat (Ph.Eur.), hochdisperses Siliciumdioxid; Hypromellose, Macrogol (6000), Titandioxid (E171), Eisen(III)-oxid (E172), Muscovit (E555). Anwendungsgebiet: Xadago ist indiziert für die Behandlung von erwachsenen Patienten mit idiopathischer Parkinson-Krankheit (PK) als Zusatztherapie zu einer stabilen Dosis Levodopa (L-Dopa) (als Monotherapie oder in Kombination mit anderen Parkinson-Arzneimitteln) bei Patienten im mittleren bis Spätstadium mit Fluktuationen. Gegenanzeigen: Bekannte Überempfindlichk. gegen Safinamidmesilat od. gegen einen sonst. Bestandteil, gleichz. Behandl. m. anderen Monoaminoxidase(MAO)-Hemmern, gleichz. Behandl. m. Pethidin, schwere Beeintr. d. Leberfunktion, Albinismus, Netzhautdegeneration, Uveitis, erbl. bedingte Retinopathie, schwere progressive diabet. Retinopathie. Nebenwirkungen: Die während der Behandlung am häufigsten berichtete Nebenwirkung von Safinamid war Dyskinesie, wenn es in Kombination mit L-Dopa allein od. in Kombination m. anderen PK-Arzneimitteln angewendet wurde. Impulskontrollstörungen; Spielsucht, gesteig. Libido, Hypersexualität, zwanghaftes Ausgeben v. Geld od. Kaufsucht, Essattacken (Binge-Eating) u. zwanghaftes Essen können b. Patienten während der Behandlung mit Dopamin-Agonisten u./od. anderen dopaminergen Arzneimitteln auftreten. Das Auftreten schwerw. Nebenwirkung. bei gleichzeit. Anw. von SSRIs, SNRIs, trizykl./ tetrazykl. Antidepressiva u. MAO-Hemmern ist bekannt, z.B. hypertensive Krise, malignes neurolept. Syndrom, Serotonin-Syndrom u. Hypotonie. Berichte über Wechselw. b. gleichzeit. Anw. v. MAO-Hemmern u. Sympathomimetika liegen vor. Häufig: Schlaflosigkeit; Dyskinesie, Somnolenz, Schwindel, Kopfschmerzen, ParkinsonKrankheit; Katarakt; orthost. Hypotonie; Übelkeit; Stürze. Gelegentlich: Harnwegsinfekt.; Basalzellkarzinom; Anämie, Leukopenie, Anomalie d. roten Blutk.; vermind. Appetit, Hypertriglyceridämie, erhöht. Appetit, Hypercholesterolämie, Hyperglykämie; Halluzinationen, Depressionen, anomale Träume, Angst, Verwirrtheitszust., Affektlabilität, gesteigerte Libido, Psychosen, Unruhe, Schlafstör.; Parästhesie, Gleichgewichtsstör., Hypoästhesie, Dystonie, Kopfbeschw., Dysarthrie, Synkopen, kognit. Stör.; Visustrübung, Skotom, Diplopie, Photophobie, Erkrank. d. Netzhaut, Konjunktivitis, Glaukom; Vertigo; Herzklopfen, Tachykardie, Sinusbradykardie, Arrhythmien; Hypertonie, Hypotonie, Krampfadern; Husten, Dyspnoe, Rhinorrhö; Verstopf., Dyspepsie, Erbrechen, Mundtrockenheit, Diarrhoe, Abdominalschmerzen, Gastritis, Flatulenz, abdominale Distension, Speichelhypersekretion, gastroösophageale Refluxkrankheit, aphthöse Stomatit.; Hyperhidr., allgem. Juckreiz, Photosensibilität, Erythem; Rückenschmerzen, Arthralgia, Muskelspasm., Muskelrigidität, Schmerzen i. d. Extremitäten, Muskelschwäche, Gefühl d. Schwere; Nykturie, Dysurie; erektile Dysfunkt.; Fatigue, Asthenie, Gangstör., peripheres Ödem, Schmerzen, Hitzegefühl; Gewichtszunahme, im Blut erhöht: Kreatinphosphokinase/Triglyceride/Blutzuckerspiegel/Harnstoff/alkalische Phosphatase/Bikarbonat/Kreatinin, EKG m. verl. QT-Zeit, anomaler Leberfunkt.test, anomale Urinanalysewerte, erhöht. od. erniedr. Blutdruck, anomaler ophthalmolog. Diagnosetest; Fußfraktur. Selten: Bronchopneumonie, Furunkel, Nasopharyngitis, Pyodermie, Rhinitis, Zahninfekt., Virusinfekt.; Akrochordon, melanozytärer Nävus, seborrhoische Keratose, Hautpapillom; Eosinophilie, Lymphopenie; Kachexie, Hyperkaliämie; Zwangsstör., Delirium, Desorientiertheit, Illusionen, impulsives Verhalten, Libidoverl., Zwangsgedanken, Paranoia, vorzeit. Ejakulation, Schlafattacken, soziale Phobie, Suizidged.; Koordinationsstör., Aufmerksamkeitsstör., Dysgeusie, Hyporeflexie, radikuläre Schmerzen, Restless-Legs-Syndr., Sedierung; Amblyopie, Chromatopsie, diabet. Retinopathie, Erythropsie, Augenblutung, Augenschmerzen, Augenlidödem, Hypermetropie, Keratitis, gesteig. Tränensekretion, Nachtblindheit, Papillenödem, Presbyopie, Strabismus; Myokardinf.; arterieller Spasm., Arteriosklerose, hypertens. Krise; Bronchospasm., Dysphonie, oropharyngeale Schmerzen und Spasm.; Magengeschwür, Würgen, Blutungen i. oberen Gastrointestinaltr.; Hyperbilirubinämie; Alopezie, Blasen, Kontaktdermatitis, Dermatose, Ekchymose, lichenoide Keratose, nächtl. Schwitzen, Hautschmerzen, Pigmentstör., Psoriasis, seborrh. Dermatit.; Spondylitis ankylosans, Flankenschmerzen, Gelenkschwell., Schmerzen d. Skelettmuskulatur, Myalgie, Nackenschmerzen, Osteoarthritis, Synovialzyste; Harndrang, Polyurie, Pyurie, verzög. Harnfluss; benigne Prostatahyperplasie, Erkrankungen d. Brust, Brustschmerzen; vermind. Arzneimittelwirksamk., Arzneimittelunverträglichk., Kältegefühl, Krankheitsgefühl, Pyrexie, Xerosis; i. Blut erniedr.: Kalzium/Kalium/Cholesterin, erhöhte Körpertemp., Herzgeräusche, anomales Belastungs-EKG, Hämatokrit erniedr., Hämoglobin erniedr., INR-Wert erniedr., Lymphozytenzahl erniedr., Blutplättchenzahl erniedr., Lipoprotein niederer Dichte erhöht; Prellungen, Fettembolien, Kopfverletzungen, Verletzungen des Mundes, Verletzungen des Skelettsystems; Spielsucht. Weitere Informationen siehe Fachinformation. Verschreibungspflichtig. Stand: September 2021. Pharmazeutischer Unternehmer: Zambon GmbH, Lietzenburger Straße 99, 10707 Berlin. Zulasssungsinhaber: Zambon S.p.A., Via Lillo del Duca 10, 20091 Bresso (MI), Italien