JAHRGANG 33
HEFT 1
Februar 2024

JAHRGANG 33
HEFT 1
Februar 2024
Endlich ein Fortschritt in der hormonfreien Behandlung von Wechseljahresbeschwerden!
ITA und SCS bereits frühzeitig in die multimodale Schmerztherapie integrieren
GOLD-Report 2024 und DGP-Positionspapier empfehlen RSV-Impfung bei COPD und anderen chronischen Grunderkrankungen
Ritlecitinib – ein Meilenstein in der Therapie der schweren Alopecia areata
Avelumab beim metastasierten Merkelzellkarzinom: Behandlungsstandard mit umfassender Evidenz
Lebrikizumab – eine neue Option zur Behandlung der mittelschweren bis schweren atopischen Dermatitis
Cemiplimab-Kombinationstherapie bereits ab Stadium IIIB/C des NSCLC möglich
Morbus Parkinson: Mit inhalativem Levodopa gastrointestinale Hürden umgehen
Rezidiviertes und refraktäres multiples Myelom: Bispezifischer Antikörper Elranatamab erweitert das Therapiespektrum
Daridorexant überzeugt in der Therapie der chronischen Insomnie
HOHE WIRKSAMKEIT ZUR PRÄVENTION VON RSVASSOZIIERTER LRTD 1, 2
94,6%
SEKUNDÄRER ENDPUNKT§ (95 % KI: 65,88; 99,87)
ERFAHREN SIE MEHR AUF GSKPRO.COM
Wie jeder Impfstoff schützt AREXVY möglicherweise nicht alle Geimpften vollständig.1
KI = Konfdenzintervall; LRTD = Erkrankungen der unteren Atemwege; RSV = Respiratorisches Synzytial-Virus

bei Patienten ab 60 Jahren mit mindestens einer relevanten Grunderkrankung* bei Erwachsenen ab 60 Jahren 82,6 % PRIMÄRER ENDPUNKT# (96,95 % KI: 57,89; 94,08)
# Auftreten von RSV-assoziierter LRTD: 7 Fälle von insgesamt 12.466 Patienten in der AREXVY-Gruppe und 40 Fälle von insgesamt 12.494 Patienten in der Placebo-Gruppe.1
* Relevante Grunderkrankungen1: Chronisch obstruktive Lungenerkrankung, Asthma, jede chronische respiratorische/pulmonale Erkrankung, chronische Herzinsuffzienz, Diabetes mellitus Typ 1 oder Typ 2 sowie fortgeschrittene Leber- oder Nierenerkrankungen (endokrin-metabolisch).
§ Auftreten von RSV-assoziierter LRTD: 1 Fall von insgesamt 4.937 Patienten in der AREXVY-Gruppe und 18 Fälle von insgesamt 4.861 Patienten in der Placebo-Gruppe.1
Diese Ergebnisse sind deskriptiv.2
1. Arexvy Fachinformation, Stand 06/2023 2. Papi A, et al. Respiratory syncytial virus prefusion F protein vaccine in older adults. N Engl J Med. 2023;388:595–608.
Wirkstoff: Arexvy Pulver und Suspension zur Herstellung einer Injektionssuspension, Respiratorischer Synzytial-Virus (RSV)-Impfstoff (rekombinant, adjuvantiert). Zusammensetzung: Nach der Rekonstitution enthält eine Dosis (0,5 ml): 120 µg RSVPreF3-Antigen, in der Präfusionskonformation stabilisiertes, rekombinantes Respiratorisches Synzytial-Virus-Glykoprotein F, hergestellt in immortalisierten Ovarialzellen des chinesischen Hamsters (CHO-Zellen) mittels rekombinanter DNA-Technologie; adjuvantiert mit AS01E, dieses enthält: 25 µg Pfanzenextrakt aus Quillaja saponaria Molina, Fraktion 21 (QS-21) und 25 µg 3-O-Desacyl-4’-monophosphoryl-Lipid A (MPL) aus Salmonella minnesota Sonstige Bestandteile: Pulver (RSVPreF3-Antigen): Trehalose-Dihydrat, Polysorbat 80 (E 433), Kaliumdihydrogenphosphat (E 340), Kaliummonohydrogenphosphat (E 340). Suspension (AS01E Adjuvanssystem): Colfosceriloleat (E 322), Cholesterol, Natriumchlorid, Natriummonohydrogenphosphat (E 339), Kaliumdihydrogenphosphat (E 340), Wasser für Injektionszwecke. Anwendungsgebiete: Arexvy ist indiziert zur aktiven Immunisierung von Erwachsenen im Alter von 60 Jahren und älter zur Prävention von durch das Respiratorische Synzytial-Virus verursachten Erkrankungen der unteren Atemwege (lower respiratory tract disease, LRTD). Gegenanzeigen: Überempfndlichkeit gegen die Wirkstoffe oder einen der genannten sonstigen Bestandteile. Nebenwirkungen: Sehr häufg: Myalgie, Arthralgie, Schmerzen an der Injektionsstelle, Ermüdung, Kopfschmerzen. Häufg: Erythem an der Injektionsstelle, Schwellung an der Injektionsstelle, Fieber, Schüttelfrost. Gelegentlich: Lymphadenopathie, Überempfndlichkeitsreaktionen (wie z. B. Hautausschlag), Übelkeit, Abdominalschmerz, Erbrechen, Jucken an der Injektionsstelle, Schmerz, Unwohlsein. Verschreibungspfichtig. Stand: Juni 2023. GlaxoSmithKline GmbH & Co. KG, 80700 München. de.gsk.com
Weitere Informationen über das Arzneimittel: Dosierung: Arexvy wird als Einzeldosis zu 0,5 ml ausschließlich intramuskulär, vorzugsweise in den M. deltoideus, injiziert. Die Notwendigkeit einer Auffrischimpfung mit einer weiteren Dosis ist nicht erwiesen. Wechselwirkungen: Verabreichung mit anderen Impfstoffen: Arexvy kann gleichzeitig mit einem saisonalen Grippeimpfstoff (quadrivalent, standarddosiert, nicht-adjuvantiert, inaktiviert) verabreicht werden. Bei gleichzeitiger Verabreichung mit anderen Impfstoffen sollte dies an unterschiedlichen Injektionsstellen erfolgen.
Weitere Warnhinweise laut Fachinformation: Der Impfstoff darf nicht intravasal oder intradermal verabreicht werden. Es kann als psychogene Reaktion auf die Nadelinjektion nach oder sogar vor einer Impfung zu einer Synkope (Ohnmacht) kommen. Die Sicherheit und Wirksamkeit von Arexvy bei Kindern und Jugendlichen sind nicht erwiesen. Bisher liegen keine Erfahrungen mit der Anwendung von Arexvy bei Schwangeren vor. Die Verabreichung während der Schwangerschaft und bei stillenden Frauen wird nicht empfohlen.
Weitere Informationen siehe Fachinformation
Nebenwirkungen melden Sie bitte ggf. bei der GSK-Hotline: 0800-1223355
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifzierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de zu melden.
Auswahl klinisch relevanter Sicherheitsinformationen zu Arexvy Gegenanzeigen: Überempfndlichkeit gegen die Wirkstoffe oder einen der genannten sonstigen Bestandteile. Warnhinweise: Der Impfstoff darf nicht intravasal oder intradermal verabreicht werden. Es kann als psychogene Reaktion auf die Nadelinjektion nach oder sogar vor einer Impfung zu einer Synkope (Ohnmacht) kommen. Die Sicherheit und Wirksamkeit von Arexvy bei Kindern und Jugendlichen sind nicht erwiesen. Die Verabreichung während der Schwangerschaft und bei stillenden Frauen wird nicht empfohlen. Nebenwirkungen: Lokalreaktionen an der Einstichstelle (Schmerzen, Erythem, Schwellung, Juckreiz) und systemische Symptome wie Kopfschmerzen, gastrointestinale Beschwerden, Muskel- und Gelenkschmerzen, Müdigkeit, Schüttelfrost, Fieber, Unwohlsein, Lymphadenopathien und Überempfndlichkeitsreaktionen. Für eine vollständige Aufistung der Kontraindikationen, Warnhinweise und Nebenwirkungen siehe Fachinformation
Eigentlich ist es ja logisch: Eine Störung der Gesundheit, die frühzeitig erkannt wird, kann nicht so viel Schaden anrichten wie eine, die erst viel später festgestellt wird. Was liegt also näher als ein regelmäßiger Gesundheits-Checkup? Deshalb hat jedes Mitglied einer Gesetzlichen Krankenkasse im Alter zwischen 18 und 34 Jahren Anspruch auf einen einmaligen „Check-up zur Erfassung von Gesundheitsrisiken und zur Früherkennung von häufg auftretenden Krankheiten, insbesondere von Herz-Kreislauf-Erkrankungen, Nierenerkrankungen und Diabetes mellitus“, ab 35 Jahren dann sogar alle 3 Jahre [1]. In Großbritannien etwa verfolgt man eine ähnliche Strategie [2].
Und weil diese Inklusivleistung in ihrem Umfang vorgegeben (und beschränkt) ist, bietet eine wachsende Zahl von Anbietern darüber hinausgehende Leistungen an. Google spuckte bei einer rezenten Suche mit den Begrifen „Gesundheit Checkup privat“ fast 700.000 Trefer aus. Privatpraxen und Privatkliniken beackern dabei mit teils schon vorhandenen technischen Ressourcen einen zusätzlichen Markt, private Präventionsinstitute mit z.T. animierenden Namen sprechen vor allem besonders Gesundheitsbewusste an bzw. Menschen mit hohen berufichen Leistungsanforderungen, also die sprichwörtlichen „Manager“. Und denen ist es nicht selten eine Menge Geld wert, sich „umfassend durchzuchecken lassen“, heißt, eine möglichst breite Palette von Untersuchungen zu absolvieren, um ein vermeintliches Gesundheitsproblem möglichst frühzeitig zu „entdecken“. Was kümmert da der Chor der Skeptiker, angeführt von einem Cochrane-Review [3], der zu dem Ergebnis kommt, dass ein solcher Rundumschlag, ein „General Health Check“, wohl nichts
Zählbares bringt („unlikely to be benefcial“). Wie denn das? Nun, der größte Teil der Untersuchungen, die über die Palette derer der Gesetzlichen Krankenkassen hinausgehen, betrift Gesundheitsstörungen, die in der Bevölkerung, epidemiologisch betrachtet, selten sind. Viel seltener jedenfalls als ein falsch positives Ergebnis, weil bei den meisten Untersuchungen der Normbereich so festgelegt ist, dass die Gefahr eines solchen Fehlalarms bei oder unter 5 % liegt. Das ist umgerechnet ein Zwanzigstel, was bedeutet, dass pro 20 Untersuchungen mit einem Fehlalarm zu rechnen ist. Bei 200 Parametern sind das also gut und gerne 10 falsche Alarme und damit eine hohe Wahrscheinlichkeit, dass weitere, dann oft auch mit Risiken verbundene diagnostische Abklärungen empfohlen und durchgeführt werden. Für das Beispiel Zöliakie (Inzidenz bei Erwachsenen etwa 1:5000) würde das bedeuten, dass auf jede tatsächliche Neudiagnose etwa 250 falsch positive Diagnosen kämen und damit 250-mal weitergehende Untersuchungen, nicht unwahrscheinlich auch jahrelange unnötige diätetische Einschränkungen etc. Als Geschäftsmodell ist ein solcher Rundumschlag allerdings extrem lukrativ. Frei nach dem Motto „Gefahr erkannt, Gefahr gebannt“ dürften sich die meisten Kunden trotz einer respektablen gesundheitlichen Mängelliste irgendwie erleichtert fühlen. Und die Anbieter können beherzt und beschwingt Empfehlungen geben, Supplemente verordnen

Prof. Dr. med. Karl-Ludwig Resch
u.v.m. Die Wahrscheinlichkeit ist groß, dass beim nächsten Check-up (der jetzt natürlich noch viel unerlässlicher ist) alles „geholfen hat“, da die statistische Wahrscheinlichkeit mit 5 % eher gering ist, dass bestimmte falsch positive Werte beim nächsten Mal erneut aufällig sind. Vielmehr werden dann andere Untersuchungen verdächtige Ergebnisse liefern – das Spiel beginnt von vorne. Also lieber keine Check-ups? Das gezielte Fahnden nach Risikofaktoren, also Faktoren, die das Auftreten einer bestimmten Erkrankung kausal fördern, kann durchaus unter dem Strich ein positives Nutzen-RisikoVerhältnis haben. Auf der Basis der bisher publizierten Studien ist davon auszugehen, dass die Koloskopie in regelmäßigen Intervallen wohl 69 % der neu auftretenden Kolonkarzinome verhindern und das Risiko eines
Endlich ein Fortschritt in der hormonfreien Behandlung von Wechseljahresbeschwerden! 4 Brigitte Söllner tödlichen Kolonkarzinoms um 88 % verringern kann [4]. Etliche häufge Erkrankungen (Beispiele s.o.) sind das Endergebnis einer langjährigen „ungesunden“ Lebensweise. Eine sorgfältige Anamnese und körperliche Untersuchung nähren einen entsprechenden Verdacht und daraus abgeleitete, gezielte weitere Untersuchungen sind wegen der hohen A-priori-Wahrscheinlichkeit eher selten falsch positiv.
Eine eigentlich allfällige Konsequenz aussagekräftiger Ergebnisse nicht zuletzt und gerade von Check-ups auf individuell erhöhte Risiken ist die praktische und individualisierte Arbeit an Lösungen, die über die üblichen „Empfehlungen“ hinausgehen und mit dem Lebensalltag bestmöglich kompatibel sind. Das inkludiert nicht zuletzt auch ein ergebnisofenes Austesten, fordert aber eine hohe Coaching-Kompetenz, da für jedes Problem neben den üblichen Standards möglichst profundes Wissen um adäquate Varianten und Alternativen im Coaching-Köcher stecken müssen.
Karl-Ludwig Resch, Nürnberg
ITA und SCS bereits frühzeitig in die multimodale Schmerztherapie integrieren 9
GOLD-Report 2024 und DGP-Positionspapier empfehlen RSV-Impfung bei COPD und anderen chronischen Grunderkrankungen 10
Ritlecitinib – ein Meilenstein in der Therapie der schweren Alopecia areata 13
Avelumab beim metastasierten Merkelzellkarzinom: Behandlungsstandard mit umfassender Evidenz 16
Lebrikizumab – eine neue Option zur Behandlung der mittelschweren bis schweren atopischen Dermatitis 18
Cemiplimab-Kombinationstherapie bereits ab Stadium IIIB/C des NSCLC möglich 21
Quellen
1 https://www.bundesgesundheitsministerium.de/checkup
2 Goodyear-Smith F. Government’s plans for universal health checks for people aged 40–75. Br Med J 2013;347:f4788
3 Krogsbøll LT, Jørgensen KJ, Gøtzsche PC. General health checks in adults for reducing morbidity and mortality from disease. Cochrane Database Syst Rev 2019;1:CD009009
4 Ladabaum U, Dominitz JA, Kahi C, Schoen RE. Strategies for colorectal cancer screening. Gastroenterol 2020;158:418-432
Morbus Parkinson: Mit inhalativem Levodopa gastrointestinale Hürden umgehen 22
Rezidiviertes und refraktäres multiples Myelom: Bispezifischer Antikörper Elranatamab erweitert das Therapiespektrum 24
Daridorexant überzeugt in der Therapie der chronischen Insomnie 26
Wissenswertes 8, 12, 25, 32 Kongresse 29
Direkter Umstieg nach MTX für PsO-Patient:innen
Überzeugendes Verträglichkeitsprofil1,5,7
Einfache orale Anwendung1
Otezla® schließt die Lücke.*
* Direkter Otezla®-Einsatz nach MTX und vor Biologika.1,2
1. Fachinformation Otezla® 2. Nast A et al. J Dtsch Dermatol Ges 2021;19(6):934–951. 3. Reich K et al. J Eur Acad Dermatol Venereol 2018;32:397–402. 4. Nash P et al. Ann Rheum Dis 2018;77:690–698. 5. Kavanaugh A et al. Arthritis Res Ther 2019;21:118. 6. Augustin M et al. EADV 2022; Poster P1624. 7. Crowley J et al. J Am Acad Dermatol 2017;77:310–317.e1.
Kurzinformation:
Otezla® 10 mg/20 mg/30 mg Filmtabletten. Wirkstof: Apremilast. Zusammensetzung: Jede Filmtablette enthält 10 mg/20 mg/30 mg Apremilast. Sonstige Bestandteile: Tablettenkern: mikrokristalline Cellulose, Lactose-Monohydrat, Croscarmellose-Natrium, Magnesiumstearat (Ph.Eur.). Filmüberzug: Poly(vinylalkohol), Titandioxid (E 171), Macrogol 3350, Talkum, Eisen(III)-oxid (E 172). Bei 20 mg zusätzlich: Eisen(III)-hydroxid-oxid x H2O (E 172); bei 30 mg zusätzlich: Eisen(III)-hydroxid-oxid x H2O (E 172), Eisen(II,III)-oxid (E 172). Anwendungsgebiete: Psoriasis-Arthritis: Otezla® ist allein oder in Kombination mit krankheitsmodifzierenden antirheumatischen Arzneimitteln (DMARDs) indiziert zur Behandlung der aktiven Psoriasis-Arthritis (PsA) bei erwachsenen Patienten, die auf eine vorangegangene DMARD-Therapie unzureichend angesprochen oder diese nicht vertragen haben. Psoriasis: Otezla® ist indiziert zur Behandlung der mittelschweren bis schweren chronischen Plaque-Psoriasis bei erwachsenen Patienten, die auf eine andere systemische Therapie, wie Ciclosporin oder Methotrexat oder Psoralen in Kombination mit UVA-Licht (PUVA), nicht angesprochen haben oder bei denen eine solche Therapie kontraindiziert ist oder die diese nicht vertragen haben. Behçet-Syndrom: Otezla® ist indiziert zur Behandlung von erwachsenen Patienten mit oralen Aphthen, die mit dem Behçet-Syndrom (BS) assoziiert sind und für die eine systemische Therapie infrage kommt. Warnhinweise: Bei neuen psychiatrischen Symptomen oder Verschlechterung bestehender Symptome oder Suizidgedanken/-versuch wird empfohlen, die Behandlung abzubrechen. Gegenanzeigen: Überempfndlichkeit gegen den Wirkstof oder einen der sonstigen Bestandteile. Schwangerschaft. Nebenwirkungen: Sehr häufg: Infektion der oberen Atemwege, Kopfschmerz, Diarrhoe, Übelkeit; Häufg: Bronchitis, Nasopharyngitis, verminderter Appetit, Schlafosigkeit, Depression, Migräne, Spannungskopfschmerz, Husten, Erbrechen, Dyspepsie, häufger Stuhlgang, Oberbauchschmerzen, gastroösophageale Refuxkrankheit, Rückenschmerzen, Fatigue; Gelegentlich: Überempfndlichkeit, Suizidgedanken und suizidales Verhalten, gastrointestinale Blutungen, Hautausschlag, Urtikaria, Gewichtsverlust; Nicht bekannt: Angioödem. Weitere Angaben: s. Fach- und Gebrauchsinformation. Verschreibungspfichtig. Stand der Information: April 2020. AMGEN Europe B.V., 4817 ZK Breda, Niederlande (örtlicher Vertreter Deutschland: AMGEN GmbH, 80992 München)
Für Frauen in den Wechseljahren beginnt nach dem Ende der reproduktiven Phase eine Zeit der hormonellen Umstellung, die häufg auch von klimakterischen Beschwerden begleitet ist. In Deutschland erleben Frauen die Menopause, also den Zeitpunkt der letzten Monatsblutung, in einem durchschnittlichen Alter von 51 Jahren [1]. Ab diesem Zeitpunkt spielen Wechseljahresbeschwerden in der Praxis eine zunehmende Rolle: Mit 45,3 % der abgerechneten Fälle sind sie die häufgste Diagnose bei Frauen über 50 Jahren in Deutschland [2]. Dabei gehören vasomotorische Symptome (VMS), auch bekannt als Hitzewallungen und/oder Nachtschweiß, zu den häufgsten Beschwerden – bis zu 80 % aller Frauen in den Wechseljahren erleben VMS, wobei diese Symptome einen gravierenden Einfuss auf die Aktivitäten des täglichen Lebens und auf die Lebensqualität der betroffenen Frauen haben [3]. Häufge VMS (VMS an ≥6 Tagen in den letzten 2 Wochen) dauern in der Regel bei der Hälfte der Frauen über 7 Jahre während der menopausalen Transition an [4]. Nach der letzten Monatsblutung halten VMS im Schnitt noch 4,5 Jahre an [4]. Als signifkante Risikofaktoren für Hitzewallungen konnten Adipositas, erhöhtes FSH vor der letzten Monatsblutung, erniedrigtes Estradiol und erhöhte Ängstlichkeit („anxiety“) identifziert werden [5].
Die Behandlungsmöglichkeiten der Wechseljahresbeschwerden sind trotz des hohen Bedarfs noch immer eingeschränkt: Neben der Hormonersatztherapie, die viele Frauen wegen des möglichen Krebsrisikos und anderer Nebenwirkungen ablehnen, werden zwar hormonfreie medikamentöse
Alternativen und verhaltenstherapeutische Maßnahmen angeboten, die in der Praxis aber oft nicht zum gewünschten Erfolg führen, da ihre Wirkung zu schwach oder nicht zielgerichtet ist und erst spät einsetzt. Ein neues hormonfreies Medikament gibt den von Wechseljahresbeschwerden geplagten Frauen nun Hoffnung: der Neurokinin-3-Rezeptor-Antagonist Fezolinetant (Veoza™). Er setzt direkt an der Ursache der vasomotorischen Wechseljahressymptome an – der Thermoregulation im Gehirn, die in den Wechseljahren aus dem Gleichgewicht gerät. Die beiden Zulassungsstudien bestätigen die Wirksamkeit und gute Verträglichkeit dieses innovativen Therapieansatzes [6, 7].
Zu den klinischen Zeichen und Befunden der VMS gehören Hitzewallungen und Schweißausbrüche, wobei die Zeitspanne, in der Frauen häufge Hitzewallungen (defniert als Hitzewallungen an mehr als 6 Tagen innerhalb der letzten 2 Wochen) wahrnehmen, durchschnittlich 7,4 Jahre umfasst [9]. Im Zusammenhang mit der Kern-
symptomatik kann es aber auch zu psychosozialen Begleitsymptomen kommen, die durch die VMS bedingt sind [3]:
• Schlafstörungen
• Stimmungsschwankungen
• soziale Einschränkungen bis zur sozialen Isolation
• Scham und Angstzustände
• Konzentrationsschwierigkeiten
• Energieverlust
• Einschränkungen des Sexuallebens
• Einschränkungen im Berufsleben
Zudem scheinen VMS unabhängig mit zahlreichen Indikatoren für ein erhöhtes kardiovaskuläres Risiko assoziiert zu sein sowie mit einem höheren Verlust an Knochenmasse und einem höheren Knochenumsatz. Außerdem scheinen VMS bei Frauen unterschiedlicher Ethnien auch unterschiedlich ausgeprägt zu sein: Afroamerikanische Frauen weisen wohl die längste Dauer von VMS auf, Japanerinnen und Chinesinnen hingegen die kürzeste [10]. Neben der Ethnizität können noch weitere Variablen die Dauer von VMS beeinfussen, so z.B. eine frühe menopausale Transition, anhaltender Stress und Symptome einer Depression beim erstmaligen Auftreten von VMS [10].
Ursächlich für die vasomotorischen Symptome ist die Thermoregulation im Gehirn, die in den Wechseljahren aus dem Gleichgewicht gerät
Während der Perimenopause schwankt der Spiegel des weiblichen Geschlechtshormons Östrogen zunächst und nimmt dann immer weiter ab. Im zentralen Nervensystem resultiert daraus eine Kaskade von zentralnervösen und peripheren Symptomen, zu denen auch die menopausalen Hitzewallungen und der Nachtschweiß gehören.
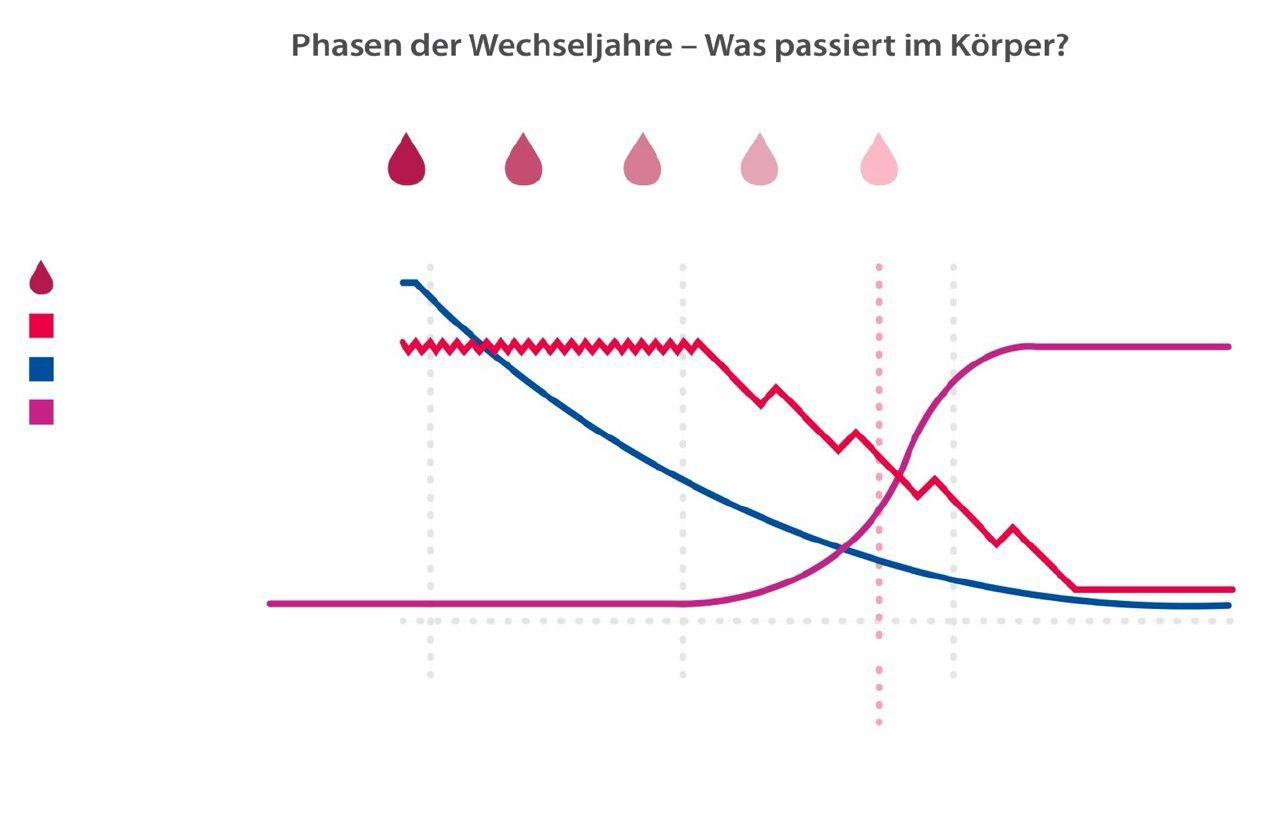
letzte
Regelblutung
Regelmäßigkeit des Zyklus Östrogen
Progesteron FSH
Prämenopause ab Anfang 40
Perimenopause ab Mitte 40
Menopause durchschnittlich im Alter von 51 Jahren
Postmenopause
Die Thermoregulation im Hypothalamus wird von Kisspeptin-/ Neurokinin-B/-Dynorphin-Neuronen (KNDy-Neuronen) gesteuert. Diese Neuronen werden einerseits durch das Neurokinin B (NKB) stimuliert, das dazu an den Neurokinin-Rezeptor 3 (NK3R) bindet, und andererseits durch Östrogene inhibiert. Infolge der sinkenden Östrogenspiegel in der Menopause entfällt diese Hemmung, was zu einer gesteigerten Stimulation des thermoregulatorischen Zentrums führt [8]. Dieses reagiert dadurch bereits auf kleinste Änderungen in der Körpertemperatur hypersensitiv mit einer verstärkten Aktivität bei der Wärmeabgabe, z.B. durch exzessives Schwitzen und Vasodilatation.
Die verschiedenen Phasen des menopausalen Übergangs werden überwiegend anhand klinischer Kriterien diagnostiziert. Hormonbestimmungen sind in der Regel nicht erforderlich. Der FSH-Spiegel kann allerdings bei Frauen zwischen dem 40. und 45. Lebensjahr bestimmt werden, wenn klimakte-
rische Symptome wie VMS bestehen [9].
Bisherige Therapiemöglichkeiten
Grundsätzlich stehen Frauen mit menopausalen Symptomen wie VMS nach den aktuellen Leitlinien 3 Behandlungsmodalitäten zur Verfügung [9]:
• hormonelle Behandlung
• nicht hormonelle Behandlung (z.B. Phytotherapeutika)
• nicht pharmakologische Interventionen (z.B. kognitive Verhaltenstherapie)
Die Hormonersatztherapie (HRT) ist bislang die effektivste Behandlung bei schweren vasomotorischen Beschwerden. Die Frequenz der Hitzewallungen pro Woche lässt sich durch jede Form der HRT um 75 % reduzieren. Die HRT wird entweder als ÖstrogenGestagen-Kombinationstherapie oder als Östrogen-Monotherapie eingesetzt, wobei eine EPT (estrogen-progestogen-therapy) mit adäquatem Gestagenanteil für nicht hysterektomierte Frauen und eine ET (estrogen therapy) bei Zustand nach Hysterektomie in Betracht gezogen werden sollte [9].
Allerdings wird die HRT in Verbindung mit der Entstehung hormonabhängiger Krebserkrankungen wie Brustkrebs gebracht und kann das Risiko für Schlaganfälle und Thromboembolien erhöhen [9]. Daher ist eine engmaschige Überwachung der Patientinnen, die eine HRT erhalten, nötig. Zudem lehnen viele Frauen eine Hormonbehandlung in den Wechseljahren grundsätzlich ab. Insbesondere die kontroverse Interpretation der Daten der WHI-Studie (Women‘s Health Initiative) aus dem Jahr 2002 führten zu einer kritischen Einstellung von Patientinnen sowie auch von Gynäkologen zur HRT [11]. Einer Umfrage unter knapp 10.000 deutschen Gynäkologen aus dem Jahr 2010 zufolge würden nur 9,1 % aller Frauenärzte ihren Patientinnen eine HRT als erste Behandlungsoption empfehlen [12].
Geringes Risiko für Schaden bzw. Therapieabbruch
Nutzen nachgewiesen
Nutzen möglich
Mittleres Risiko für Schaden bzw. Therapieabbruch
Abwarten bzw. Placebo, CBT (Achtsamkeit, kognitive und Verhaltenstherapie)
Cimicifuga (herbal preparation), Isoflavone, inkl. Phytoöstrogenreiche Ernährung, Rotklee, SEquol, Genistein, Rheum rhapontikum, Akupunktur, Johanniskraut
Nutzen unwahrscheinlich
Sport (3 - 6 Monate), Tiefenentspannung (4 - 12 Wochen), Vitamin E
Östrogene, Tibolon SSRI, SNRI, Gabapentin, Clonidin DHEA (Dehydroepiandrosteron), Raloxifen
Risiko für Schaden nicht ausreichend untersucht - - Chinesische Kräuter im Rahmen der TCM, Melatonin
Tabelle 1: Wirksamkeit und Risiken verschiedener Interventionen bei Hitzewallungen laut der aktuellen S3-Leitlinie „Peri- und Postmenopause – Diagnostik und Intervention“ [9].
Mittlerweile wurden die ursprünglichen Daten der WHI-Studie aber auch kritisiert (alte Patientinnenklientel, heute nicht mehr im Einsatz befndliche Hormonpräparate) und erneut ausgewertet. Dabei zeigte sich ein differenzierteres Bild des Nutzen-Risiko-Verhältnisses. Daher stellt die HRT heute – bei korrekter Indikationsstellung – einen wesentlichen Pfeiler in der Behandlung von Wechseljahresbeschwerden dar [9].
Nicht hormonelle medikamentöse Behandlung
Psychopharmaka wie SerotoninWiederaufnahmehemmer (SSRI), Serotonin-Noradrenalin-Wieder-
aufnahmehemmer (SNRI) und Gabapentin können eine Option für Frauen sein, bei denen eine Kontraindikation für die Hormonbehandlung besteht. Im Vergleich zur HRT sind sie allerdings weniger wirksam und werden in der aktuellen Leitlinie für die Therapie von VMS auch nicht empfohlen [9].
Phytotherapeutika spielen in der Behandlung von VMS ebenfalls eine Rolle, wobei die Sicherheit vieler Präparate wie Cimicifuga und Phytoöstrogenen ungewiss ist, da sich sowohl die Zubereitung der Präparate stark unterscheiden kann als auch Risiken aufgrund von Interaktionen mit anderen Arzneimitteln entstehen können [9].
Nicht pharmakologische Interventionen
Eine Verhaltenstherapie kann ebenfalls zur Linderung der vasomotorischen Symptomatik angeboten werden. Dabei wird sowohl durch die MBT (mindfulness based therapy) als auch die die CBT (cognitive behavioural therapy) die Beeinträchtigung durch Hitzewallungen und andere Beschwerden gelindert [9]. Tabelle 1 fasst die Bewertung der in der aktuellen S3-Leitlinie „Periund Postmenopause – Diagnostik und Intervention“ aufgeführten nicht hormonellen medikamentösen und nicht pharmakologischen Interventionen bei Hitzewallungen hinsichtlich Wirksamkeit und Risiken zusammen [9].
Neurokinin-3-Rezeptor-Antagonist
Fezolinetant – eine wirksame und gut verträgliche Alternative zur Hormonersatztherapie
Am 7. Dezember 2023 hat die Europäische Kommission Fezolinetant (Veoza™) für die Behandlung von moderaten bis schweren vasomotorischen Symptomen, die mit der Menopause assoziiert sind, zugelassen. Fezolinetant gehört zur Gruppe der Neurokinin-3-Rezeptor-(NKR3)-Antagonisten, die direkt in den Pathomechanismus der menopausalen Hitzewallungen und Schweißausbrüche eingreifen, indem sie die die neuronale Aktivität im thermoregulatorischen Zentrum des Gehirns im Hypothalamus beeinfussen [13]. Fezolinetant wirkt, indem es die Bindung von Neurokinin B an den NK3-Rezeptor blockiert. Dadurch „bremst“ es die in den Wechseljahren infolge der sinkenden Östrogenspiegel ungehemmte, durch Neurokinin-3 vermittelte Aktivierung der KNDy-Neurone im Thermoregulationszentrum, die zu einer verstärkten Wärmeabgabe führt. Auf diese Weise werden die Häufgkeit sowie Schwere von Hitzewallungen und nächtlichen Schweißausbrüchen reduziert. Die empfohlene Dosis beträgt 1 Tablette mit 45 mg einmal täglich [13].
Überzeugende Studienergebnisse
Die Wirksamkeit und Sicherheit von Fezolinetant wurden in den beiden randomisierten, placebokontrollierten und doppelblinden Phase-III-Studien SKYLIGHT 1 und SKYLIGHT 2 mit identischem Design nachgewiesen [6, 7]. In SKYLIGHT 1 wurden 527 Frauen im Alter zwischen 40 und 65
SKYLIGHT-2 Episoden mittelschwerer bis schwerer VMS pro Tag
Untersuchungswoche Placebo Fezolinetant 30 mg Fezolinetant 45 mg
Baseline 11,59 11,23 11,79 Woche 4 8,08 5,79* 5,67* Woche 12 6,73 4,80* 4,49*
Tabelle 2: Ergebnisse der Studie SKYLIGHT 2 für den primären Wirksamkeitsendpunkt, die statistisch signifkante Senkung der VMS-Frequenz pro Tag unter der Therapie mit 30 mg bzw. 45 mg Fezolinetant im Vergleich zu Placebo. * p > 0,001 [7].
Jahren mit mittelstarken bis starken Beschwerden und im Schnitt mindestens 7 Hitzewallungen pro Tag eingeschlossen. Sie wurden randomisiert einer 12-wöchigen Therapie mit Placebo, Fezolinetant 30 mg oder Fezolinetant 45 mg zugeteilt. Nach 2 Wochen schloss sich eine 40-wöchige Verlängerungsstudie an, für die die Placebo-Patientinnen in eine der beiden Verumgruppen randomisiert wurden [6].
Studienziel war eine statistisch signifkante Reduktion der Häufgkeit und der Intensität der vasomotorischen Symptome nach 4 und nach 12 Wochen im Vergleich zu Placebo. Dieser Endpunkt wurde mit beiden Fezolinetant-Dosierungen erreicht: Unter der Therapie mit der 30-mg-Dosis nahm die Häufgkeit von anfangs 10,7 Episoden pro Tag auf 5,4 nach 4 Wochen und auf 4,5 nach 12 Wochen ab. Unter der 45-mg-Dosis war ein Rückgang von 10,4 Episoden pro Tag auf 5,2 nach 4 Wochen und auf 4,1 nach 12 Wochen zu verzeichnen. Unter Placebo nahm die VMS-Rate dagegen von 10,5 Episoden pro Tag nur auf 7,3 nach 4 Wochen und und auf 6,9 nach 12 Wochen ab. Die Verbesserungen der Frequenz und Intensität von VMS unter der Therapie mit Fezolinetant waren bereits nach einer Woche signifkant. Der Effekt hielt außerdem bis zum Ende der Verlängerungsphase nach 52 Wochen an [6].
Nebenwirkungen waren unter Fezolinetant in den ersten 12 Wochen nicht häufger als unter Placebo (37 % unter 30 mg und 43 % unter 45 mg versus 45 % unter Placebo).
Relevante Anstiege der Leberenzyme ASAT oder ALT auf über das Dreifache des oberen Normwerts – eine potenzielle Nebenwirkung von NK3R-Antagonisten – wurden nur bei 2 Patientinnen unter der 30-mg-Dosis und einer Patientin in der Placebo-Gruppe festgestellt. Über die gesamten 52 Wochen kam es bei 9 Frauen unter Fezolinetant zu Leberwerterhöhungen [6].
Vergleichbare Ergebnisse erbrachte die Studie SKYLIGHT 2, die ebenfalls die Wirksamkeit und Sicherheit von 30 mg und 45 mg Fezolinetant mit der von Placebo bei 501 Patientinnen im Alter zwischen 40 und 65 Jahren mit mittelstarken bis starken Beschwerden und im Schnitt mindestens 7 Hitzewallungen pro Tag verglich [7]. Auch in dieser Studie war die Behandlung mit dem Neurokinin3-Rezeptor-Antagonisten in der 30- und 45-mg-Dosierung der Placebo-Behandlung überlegen (Tab. 2). Die Rate an unerwünschten Ereignissen unter den beiden Fezolinetant-Dosierungen war auch hier vergleichbar mit der unter Placebo beobachteten Nebenwirkungsrate: 40 % unter 30 mg und 36 % unter 45 mg Fezolinetant versus 32 % unter Placebo [7].
Der Fokus der Behandlung menopausaler Hitzewallungen und Schweißausbrüche lag bislang auf dem Ersatz von Östrogenen (HRT) sowie der Gabe von Phytotherapeutika oder homöopathischen Mitteln. Bei Letzteren war die Wirkung häufg zu schwach oder der Wirksamkeitsnachweis fehlte ganz. Die HRT dagegen war mit unerwünschten Risiken und Nebenwirkungen vergesellschaftet, sodass viele Frauen die Substitution von Östrogenen ablehnten. Der Neurokinin B-Antagonist Fezolinetant erscheint in dieser Hinsicht sicherer. Da er direkt in den Pathomechanismus der menopausalen Beschwerden eingreift, birgt er vermutlich weniger Risiken als eine HRT. Ein Vorteil gegenüber einer HRT ist auch, dass die Wirkung von Fezolinetant schon nach einer Woche – und damit in der Regel deutlich schneller – eintritt.
Gegen Übelkeit und Erbrechen in der Schwangerschaft:
Xonvea® 20 mg/20 mg Tabletten mit veränderter Wirkstofffreisetzung
Treten Übelkeit und Erbrechen in der Schwangerschaft auf, bedarf es einer möglichst raschen Linderung der Symptome. Mit Xonvea® 20 mg/20 mg steht nun betroffenen Frauen eine für diese Indikation zugelassene und wirksame Therapieoption in der bewährten Wirkstoffkombination aus 20 mg Doxylamin und 20 mg Pyridoxin mit veränderter Wirkstofffreisetzung zur Verfügung. Diese führt schon an Tag 1 zu höheren Wirkstoffspiegeln von Doxylamin und Pyridoxin im Vergleich zur bisherigen
Weitere Studien müssen zeigen, ob Frauen in der Menopause tatsächlich auch langfristig von der neuen Therapieoption proftieren.
1 Mann C et al. Auf gutem Weg durch die Wechseljahre. MMW Fortschr Med 2019;161:50-57
2 Krause L et al. Beratungs- und Behandlungsanlässe in gynäkologischen Praxen bei Frauen ab 50 Jahren. J Health Monitoring 2020;5(2)
3 Utian WH. Psychosocial and socioeconomic burden of vasomotor symptoms in menopause: a comprehensive review. Health Qual Life Outcomes 2005;3:47
4 Avis NE et al. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med 2015;175:531-539
5 Freeman EW et al. Risk of long-term hot flashes after natural menopause: evidence from the Penn Ovarian Aging Study cohort. Menopause 2014;21:924-932
6 Lederman S et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. The Lancet 2023;401: 1091-1102
7 Johnson K et al. Efficacy and safety of fezolinetant in moderate-to-severe vaso-
Formulierung. Die Wirkstoffspiegel werden nach Einnahme von Xonvea® 20 mg/20 mg bereits innerhalb von durchschnittlich 1 bzw. 1–2 Stunde(n) erreicht.
Reduktion der täglichen Tabletteneinnahme und des PUQE-Scores
Die Anfangsdosis von Xonvea® 20 mg/20 mg beträgt 1 Tablette abends. Bei anhaltenden Symptomen soll am 3. Tag zusätzlich eine Tablette morgens eingenommen werden. Im Vergleich zur 10 mg/10 mg-Formulierung nehmen Schwangere mit Xonvea® 20 mg/20 mg über einen typischen Behandlungszeitraum von 10 Wochen 137 Tabletten weniger ein. Die damit reduzierte tägliche Tab-
motor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metabol 2023;108:1981-1997
8 Stute P. Update NK3R-Antagonist Fezolinetant. Gynäkologische Endokrinologie 2021;19:163-164
9 S3-Leitlinie „Peri- und Postmenopause –Diagnostik und Interventionen“. Leitlinie der DGGG, SGGG und OEGGG (S3 Level, AW MF Register Nr. 015-062, Januar 2020). http://www.awmf.org/leitlinien/detail/ll/015-062.html
10 Gold EB et al. Longitudinal analysis of the association between vasomotor symptoms and race/ethnicity across the menopausal transition: study of women’s health across the nation. Am J Public Health 2006;96:1226-1235
11 Cagnacci A et al. The controversial history of hormone replacement therapy. Medicina (Kaunas) 2019; 55:602
12 Buhling KJ et al. Attitude of German gynecologists towards prescribing HRT before and after the WHI study. Climacteric 2012;15:326-331
13 Fachinformation VEOZA™; aktueller Stand
Anschrift der Verfasserin: Brigitte Söllner
Medizinjournalistin und Wissenschaftliche Lektorin
Lärchenweg 10 91058 Erlangen
E-Mail: brigitte.soellner@online.de
letteneinnahme von maximal 4 auf 2 kann eine Erleichterung für die Patientinnen darstellen und somit die Compliance verbessern. Die Steigerung der täglichen Dosis von 20 mg auf 40 mg Doxylamin/Pyridoxin führte in einer klinischen Studie zu einer signifkanten Reduktion des durchschnittlichen Pregnancy-Unique Quantifcation of Emesis and Nausea (PUQE)-Scores von 7,48 auf 6,06. Der PUQE-Score umfasst die Anzahl der täglichen Erbrechen-Episoden, die Anzahl der täglichen Würge-Episoden und die Dauer der täglichen Übelkeit in Stunden für einen GesamtScore von Symptomen, die von 3 (keine Symptome) bis 15 (am schwersten) bewertet wurden . S. M.
Patienten mit starken, chronischen Tumor- und NichtTumor-Schmerzen sind in ihrer Lebensqualität deutlich eingeschränkt. Ihre optimale Versorgung stellt die behandelnden Ärzte vor große Herausforderungen. Konservative Behandlungsstrategien mit stark wirksamen Opioiden, nichtsteroidalen Antiphlogistika, Antidepressiva und Antikonvulsiva schlagen bei diesen Patienten oft nicht mehr ausreichend an oder verursachen starke Nebenwirkungen und Abhängigkeiten. Eine effektive und nachhaltige Option bei diesen Patienten sind interventionelle Therapien mittels epiduraler Rückenmarkstimulation (SCS) und intrathekaler Analgesie (ITA), bei der der First-Pass-Effekt und die Blut-Hirn-Schranke umgangen werden. Im internationalen Vergleich wird die ITA in Deutschland aktuell aber nur selten und erst sehr spät eingesetzt, da zwar entsprechende Empfehlungen für die Anwendung der SCS in Deutschland existieren [1], nicht aber für die ITA. Dies steht im Kontrast zu den amerikanischen und europäischen Leitlinien [2–6], in denen ITA neben anderen Verfahren zum festen Bestandteil der Schmerztherapie gehört. Ein europäisches Konsensupapier zum intrathekalen Einsatz von Ziconotid (Prialt®) könnte dazu beitragen, dass die ITA auch hierzulande rechtzeitig im Management schwerer Schmerzen eingesetzt wird [7].
Opioid-bedingte Nebenwirkungen mit Ziconotid umgehen
Eine interventionelle Schmerztherapie kann die Weiterleitung von Informationen im und zum zentralen Nervensystem verändern, sodass chronische Schmerzen reduziert oder nicht mehr wahr-
frühzeitig
genommen werden und sich die Lebensqualität der betroffenen Patienten dadurch deutlich verbessert. Dies bestätigen die Langzeitdaten zur ITA mit Ziconotid, einem nichtopioiden N-Typ-Kalziumantagonisten, aus mehr als 15 Jahren Therapiepraxis [6, 8–12].
Das mit einer intrathekalen Pumpe verabreichte Ziconotid ist für alle Schmerzarten als Firstline-Therapie zugelassen und weist keine der opioidtypischen Nebenwirkungen wie Atemdepression, endokrinologische Störungen oder Toleranzentwicklung auf. Die Neubefüllung der Pumpe ist auch ambulant gut zu managen. Wichtig bei der Anwendung sind jedoch eine niedrige Startdosis und eine langsame Auftitrierung, um starke Nebenwirkungen zu vermeiden und das Nutzen-Risiko-Profl zu verbessern. Die Ermittlung der dafür benötigten, individuellen Dosis benötigt einige Zeit, die es sich aber lohnt zu investieren [8]. Der intrathekalen Einsatz von Ziconotid wurde 2021 von Matis et al. in einem euröpäischen Konsensuspapier evaluiert, wobei die Autoren insbesondere auf die fehlenden opioidbedingten Nebenwirkungen hinwiesen [7]. Langzeitdaten einer Fallserie, in der niedrigdosiertes Ziconotid als Erstlinientherapie mit einem im genannten Konsensuspapier entwickelten Stufenschema nach 1, 6, 24 und 30 Monaten eingesetzt wurde, zeigten eine bis zu 33%ige Schmerzreduktion (20 %, 30 %, 33 % und 31 %,
Angaben basierend auf einer visuellen Analogskala) [9]. Aktuelle Daten belegen, dass auch in Monat 42 keine abweichende Tendenz beobachtet werden konnte [12]. Daraus resultiert die Empfehlung, die ITA als Bestandteil eines multimodalen Schmerzkonzepts rechtzeitig und nicht erst als Ulitima Ratio einzusetzen, wenn konservative Behandlungsstrategien nicht mehr ausreichen, um eine Schmerzpersistenz zu unterbinden.
Brigitte Söllner, Erlangen
Literatur
1 Gillner S et al. DGS-PraxisLeitlinie Epidurale Rückenmarkstimulation zur Therapie chronischer Schmerzen. Version: 1.0 für Fachkreise, 2019
2 Fallon M et al. Ann Oncol 2018; 29:iv166–iv191
3 Haute Autorite de Sante (HAS). Analgesic management of refractory pain and sedative practices in adults: medical management in palliative care situations up to the end of life. 2020
4 Deer TR et al. Neuromodulation 2017; 20:96-132
5 Deer TR et al. Neuromodulation 2017; 20:133-154
6 Deer TR et al. 2017; 20:155-176
7 Matis G et al. Brain Behav 2021;Suppl 1:e02055
8 Fachinformation Prialt® 100 Mikrogramm/ml Infusionslösung; Stand: 08/ 2022
9 Matis G et al. Niedrig dosiertes Ziconotid als intrathekale Erstlinienmonotherapie zur Schmerzlinderung: Eine Fallserie von sechs Patienten mit 30-monatiger Langzeittherapie. Deutscher Schmerzkongress 2022. Poster-Nr. P05.048
10 Wallace MS et al. Anesth Analg 2008; 06:628-637
11 Webster LR et al. J Pain Symptom Manage 2009; 37:363-672
12 Matis G. Deutscher Schmerzkongress, Mannheim 2023
In ihren aktualisierten Empfehlungen zur Prävention und Therapie der chronisch obstruktiven Lungenerkrankung (COPD, chronic obstructive pulmonary disease) betont die „Global Initiative for Chronic Obstructive Lung Disease“ (GOLD) einmal mehr, dass das wichtigste Therapieziel die Senkung des Mortalitätsrisikos dieser Patienten ist [1]. Unabdingbar ist daher laut GOLD-Report 2024 ein proaktives Krankheitsmanagement, wobei Komorbiditäten und Exazerbationen frühzeitig erfasst werden müssen, um durch eine Therapieeskalation die Prognose des Patienten verbessern zu können. Da auch Infektionen mit dem Respiratorischen Synzytial-Virus (RSV) das Risiko für schwere Verläufe erhöht, empfehlt der GOLD-Report ausdrücklich, erwachsene COPD-Patienten ab 60 Jahren mit/ohne chronische Herz- und Lungenerkrankungen gegen eine RSV-Infektion zu impfen. Deutsche Fachgesellschaften empfehlen die RSV-Impfung sogar generell für alle Personen ab 60 Jahren [2].
Das Exazerbationsrisiko bei COPD minimieren
Die COPD ist weltweit die dritthäufgste Todesursache [3]. In Deutschland sind rund 6,8 Millionen Menschen betroffen, für 2030 werden 7,9 Millionen COPD-Patienten erwartet [4]. Wie schon in seiner letztjährigen Empfehlung betont auch der GOLD-Report 2024, dass das Risiko für Exazerbationen unbedingt minimiert werden muss, da Exazerbationen das Morbiditätsund Mortalitätsrisiko erhöhen [1]. GOLD fordert in diesem Zusammenhang eine höhere Effektivität der COPD-Therapie und einen stärkeren Fokus auf impfpräventive
Maßnahmen. Denn die Vorbeugung von Infektionskrankheiten durch Impfungen ist eine der wichtigsten prophylaktischen Maßnahmen gegen Exazerbationen. Da COPDPatienten aufgrund der Entzündungsaktivität in der Lunge ein geschwächtes Immunsystem haben, sind sie bei Infektionen einem höheren Risiko ausgesetzt [5]. Gerade saisonale respiratorische Virusinfektionen sind bei hospitalisierten COPD-Patienten mit einer um 50 % erhöhten Wahrscheinlichkeit für eine Aufnahme in die Intensivstation und mit einer um 90 % erhöhten Wahrscheinlichkeit für eine mechanische Beatmung assoziiert [6].
Durch RSV-Impfung die Patienten vor schweren Verläufen schützen
Im neuen Report spielen deshalb auch die Impfungen als Teil des Präventionskonzepts wieder eine Rolle, wobei die Gefahr durch Infektionen mit dem Respiratorischen Synzytial-Virus (RSV) erstmals hervorgehoben wird. Die Autoren des GOLD-Reports sprechen sich basierend auf den CDC-Empfehlungen für eine RSV-Impfung von Erwachsenen ab 60 Jahren mit/ohne chronische Herz- oder Lungenerkrankungen aus (Evidenz A*). Darüber hinaus
betonen sie das besonders hohe Risiko für schwere RSV-Verläufe bei Erwachsenen mit chronischen Herz- und Lungenkrankheiten, bei vorliegender Immunschwäche sowie bei Personen, die in Pfegeeinrichtungen leben [1].
Entgegen einer weit verbreiteten Meinung gefährden RSV-Infektionen nicht nur Neugeborene, Säuglinge und Kleinkinder, sondern können auch bei älteren und vorerkrankten Erwachsenen schwere Krankheitsverläufe wie Pneumonien sowie Komplikationen bestehender Erkrankungen auslösen [2, 7]. So ist eine COPD mit einem 3,5- bis 13,4-fach erhöhten Risiko für eine stationäre Behandlung aufgrund einer RSV-Infektion assoziiert. In ähnlicher Weise erhöhen auch andere Grunderkrankungen das Risiko für eine Klinikeinweisung nach einer RSV-Infektion deutlich, so z.B. Asthma (2,3- bis 2,5-fach erhöhtes Risiko), Diabetes (2,4- bis 6,4-fach erhöhtes Risiko), und koronare Herzkrankheiten (3,8bis 6,5-fach erhöhtes Risiko) [8].
* Erforderlich für den Evidenzgrad A sind qualitativ hochwertige Nachweise aus ≥2 klinischen Studien mit einer beträchtlichen Anzahl von Probanden oder eine einzige qualitativ hochwertige Studie mit einer beträchtlichen Anzahl von Probanden ohne jegliche Verzerrung. Die RSV-Impfung ist damit die einzige Impfempfehlung des Gold-Reports 2024 mit dem höchsten Evidenzgrad A.
Trelegy Ellipta ist eine Dreifach-Fixkombination aus folgenden Wirkstoffen:
• Fluticasonfuroat (FF), ein inhalatives Kortikosteroid (ICS)
• Umeclidiniumbromid (UMEC), ein Bronchodilatator und langwirksames Anticholinergikum (LAMA)
• Vilanterol (VI), ein langwirksamer Beta-2-Agonist (LABA)
Trelegy Ellipta ist angezeigt für die Erhaltungstherapie bei erwachsenen Patienten mit moderater bis schwerer chronisch obstruktiver Lungenerkrankung (COPD), die mit einer Kombination aus einem inhalativen Kortikosteroid und einem langwirksamen Beta-2-Agonisten (ICS + LABA) oder mit einer Kombination aus einem langwirksamen Beta-2-Agonisten und einem langwirksamen Muskarinrezeptor-Antagonisten (LABA + LAMA) nicht ausreichend eingestellt sind. Als initiale Therapie ist Trelegy Ellipta nicht zugelassen. Das Pulver wird mit dem Ellipta Inhalator appliziert [11].
Frühzeitig an eine Therapieeskalation denken
Laut GOLD-Report sollte bereits bei einer Punktezahl ≥10 im COPD Assessment Test sowie einem mMRC-Schweregrad ≥2 eine duale Kombinationstherapie aus LAMA/LABA eingeleitet werden, um die Progression der Erkrankung durch eine frühe wirkungsvolle Erhaltungstherapie zu stoppen [1]. Denn über 50 % der COPD-Patienten leiden bei einer Monotherapie weiter unter Atemnot [9]. Bei COPD-Patienten mit bestehender initialer Erhaltungstherapie und anhaltendem Risiko für Exazerbationen sowie bei einer Verschlechterung der Symptome oder einem Eosinophilen-Schwellenwert von ≥300 Zellen/μl empfehlen die Autoren den frühzeitigen Wechsel auf eine LAMA/ LABA/ICS-Dreifachtherapie. Dies verbessert, wie für FF/UMEC/VI (Trelegy Ellipta, vgl. Insert) belegt, die Lungenfunktion und reduziert nachweislich die Exazerbationsfrequenz [10]. Die bestehende Studienlage liefert außerdem eine Begründung für die Erwägung ei-
ner initialen Dreifachtherapie bei neu diagnostizierten Patienten mit entsprechend hoher Eosinophilenzahl (≥300 Zellen/μl). Weiterhin sollte die Therapie regelmäßig evaluiert werden, auch hinsichtlich der Mitarbeit des COPD-Patienten.
Die GOLD-Leitlinie empfehlt zur Steigerung der Therapieadhärenz Wirkstoffkombinationen, die mit einem einzigen Inhalator durchgeführt werden können. Hierzu sind geeignete Systeme auf dem Markt verfügbar.
Aktueller RSV-Impfappell der DGP
Anfang November 2023 wurde unabhängig von der internationalen GOLD-Leitlinie ein Positionspapier der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin (DGP) in Zusammenarbeit mit 10 weiteren medizinischen Fachgesellschaften publiziert, in dem die RSV-Impfung generell für alle Personen ab 60 Jahren empfohlen wird [2]. Insbesondere Erwachsene mit deutlich eingeschränkter Immunabwehr oder schweren Lungen- sowie HerzKreislauf-Vorerkrankungen soll-
Arexvy ist ein proteinbasierter RSV-Impfstoff bestehend aus dem gentechnisch hergestellten Antigen-Protein (RSVPreF3) und dem Adjuvans AS01E. Der Impfstoff ist seit Juni 2023 zur aktiven Immunisierung von Erwachsenen ab 60 Jahren zur Prävention einer durch RSV verursachten Erkrankung der unteren Atemwege zugelassen und seit August 2023 in Deutschland verfügbar. Arexvy wird als Einzeldosis zu 0,5 ml injiziert (vorzugsweise in den M. deltoideus) [13].
Zulassungsrelevant waren die Ergebnisse der Phase-III-Studie AReSVi-006 mit ca. 25.000 Teilnehmern, die dem Impfstoff eine hohe Wirksamkeit und gute Verträglichkeit bescheinigen. Eine Einzeldosis des adjuvantierten Impfstoffs erreichte in den Zulassungsstudien über alle geimpften Patienten eine Wirksamkeit von 82,6 % bezüglich der Prävention einer Erkrankung der unteren Atemwege. Für Patienten mit begleitenden Grunderkrankungen wurde ein Impfschutz von 94,6 % vor einer Erkrankung der unteren Atemwege beobachtet [13].
Bislang liegen Daten zu 2 aufeinanderfolgenden RSV-Saisons vor: Demnach ist eine Dosis des Impfstoffs wirksam gegen RSV-assoziierte Erkrankungen der unteren Atemwege für 2 volle RSV-Saisons und es gibt keine Hinweise für die Notwendigkeit einer Auffrischungsdosis nach 12 Monaten [14].
ten sich gegen eine RSV-Infektion impfen lassen.
Bereits zuvor hatte die Deutsche Gesellschaft für Hämatologie und medizinische Onkologie (DGHO) in einem eigenen Statement die RSV-Impfung für Patienten mit einer Immundefzienz ab 18 Jahren empfohlen [12]. Dazu gehören insbesondere Patienten mit einer immunsuppressiven Behandlung (Glukokortikoide, myelosuppressive Therapie bei malignen Erkrankungen, hämatopoetische Stammzelltransplantation, Transplantate solider Organe) sowie mit einer malignen hämatologischen Grundkrankheit wie Leukämie und multiplen Myelomen oder hereditären Immundefekten [12]. Entsprechend den Forderungen sollte die Sensibilisierung für Impfungen bei Ärzten verbessert werden – insbesondere bei Patienten mit COPD oder anderen pulmonalen bzw. kardiovaskulären Grunderkrankungen. So zeigte die RSVImpfung mit dem adjuvantierten Impfstoff Arexvy in einer großen Phase-III-Studie bei Menschen mit chronischen Grunderkrankungen und dadurch bedingter Immundefzienz eine Wirksamkeit von 94,6 % gegenüber einer Erkrankung der unteren Atemwege [13]. Gerade in den Wintermonaten sollten daher COPD-Patienten sowie ältere Menschen mit chronischen Grunderkrankungen ab 60 Jahren über das RSV-Risiko und eine mögliche Prävention vor dem hochansteckenden Virus informiert und einer Präventivimpfung zugeführt werden.
Brigitte Söllner, Erlangen
2 Deutsche Gesellschaft für Pneumologie und Beatmungsmedizin (DGP). Positionspapier zur RSV-Schutzimpfung bei besonders gefährdeten Patientinnen und Patienten der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin e.V. 02.11.2023. https://pneumologie.de/storage/app/media/uploaded-files/2023_ RSV-Impfung_DGP
3 World Health Organization. Chronic obstructive pulmonary disease (COPD). Key facts. https://www.who.int/news-room/ fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd)
4 Pritzkuleit R et al. Erkrankungszahlen in der Pneumologie – eine Projektion bis 2060. Pneumologie 2010;64:535-540
5 Simon S et al. The role of vaccination in COPD: influenza, SARS-CoV-2, pneumococcus, pertussis, RSV and varicella zoster virus. Eur Respir Rev 2023;32: 230034
6 Mulpuru S et al. Impact of respiratory viral infections on mortality and critical illness among hospitalized patients with chronic obstructive pulmonary disease. Influenza Other Respir Viruses 2022;16: 1172-1182
7 Robert Koch Institut (RKI). RKI-Ratgeber: RSV. https://www.rki.de/DE/Content/Infekt/EpidBull/Merkblaetter/Ratgeber_RSV.html
8 Branche AR et al. Incidence of respiratory syncytial virus infection among hospitalized adults, 2017–2020. Clin Infect Dis 2022:74:1004-1011
9 Dransfield MT et al. Disease severity and symptoms among patients receiving monotherapy for COPD. Prim Care Respir J 2011;20:46-53
10 Ismaila AS et al. Comparative efficacy of umeclidinium/vilanterol versus other bronchodilators for the treatment of chronic obstructive pulmonary disease: a network meta-analysis. Adv Ther 2022;39: 4961-5010
11 Fachinformation Trelegy Ellipta, aktueller Stand
12 Deutsche Gesellschaft für Hämatologie und medizinische Onkologie (DGHO). Empfehlung zur RSV-Schutzimpfung bei immundefizienten Patientinnen und Patienten mit hämatologischen und/oder onkologischen Erkrankungen (15.08.2023). https://www.dgho.de/aktuelles/news/ newsarchive/2023/download/rsv-impfung-20230815.pdf
13 Fachinformation Arexvy, aktueller Stand
Kindgerechte
Patientenbroschüre zur juvenilen idiopathischen Arthritis

In der von Pfzer herausgegebenen Patientenbroschüre „Prinzessin Emma kann wieder zaubern!“ wird am Beispiel von Prinzessin Emma beschrieben, wie der Diagnoseweg und die Therapie einer juvenilen idiopathischen Arthritis (JIA) aussehen können. Auf anschauliche Weise wird Kindern und ihren Eltern Wissenswertes zur Erkrankung vermittelt.
Die Geschichte macht Mut und zeigt, dass mit einer adäquaten Behandlung auch mit einer JIA-Erkrankung ein weitgehend normales Leben mit Spaß und Spielkameraden möglich ist.
Die 24-seitige Broschüre ist reich bebildert, zudem laden einfach verständliche Texte zum Vorlesen und Selberlesen ein. Sie wurde von Dr. med. Tobias Krickau, Kinderrheumatologe am Universitätsklinikum Erlangen, konzipiert und unter seiner fachlichen Beratung realisiert.
Literatur
1 Global Initiative for Chronic Obstructive Lung Disease (GOLD). 2024 GOLD Report. https://goldcopd.org/2024-gold-report/
14 GSK. Press release: GSK shares positive data for Arexvy, its respiratory syncytial virus (RSV) older adult vaccine, indicating protection over two RSV seasons (21.06.2023). https://www.gsk.com/engb/media/press-releases/gsk-shares-positive-data-for-arexvy-its-respiratory-syncytial-virus-older-adult-vaccine-indicating-protection-over-two-rsv-seasons/
Interessierte Ärzte können sich an ihren Pfzer-Außendienst wenden, um Exemplare der Broschüre für den Einsatz in ihrem Behandlungsalltag kostenfrei zu bestellen.
B. S.
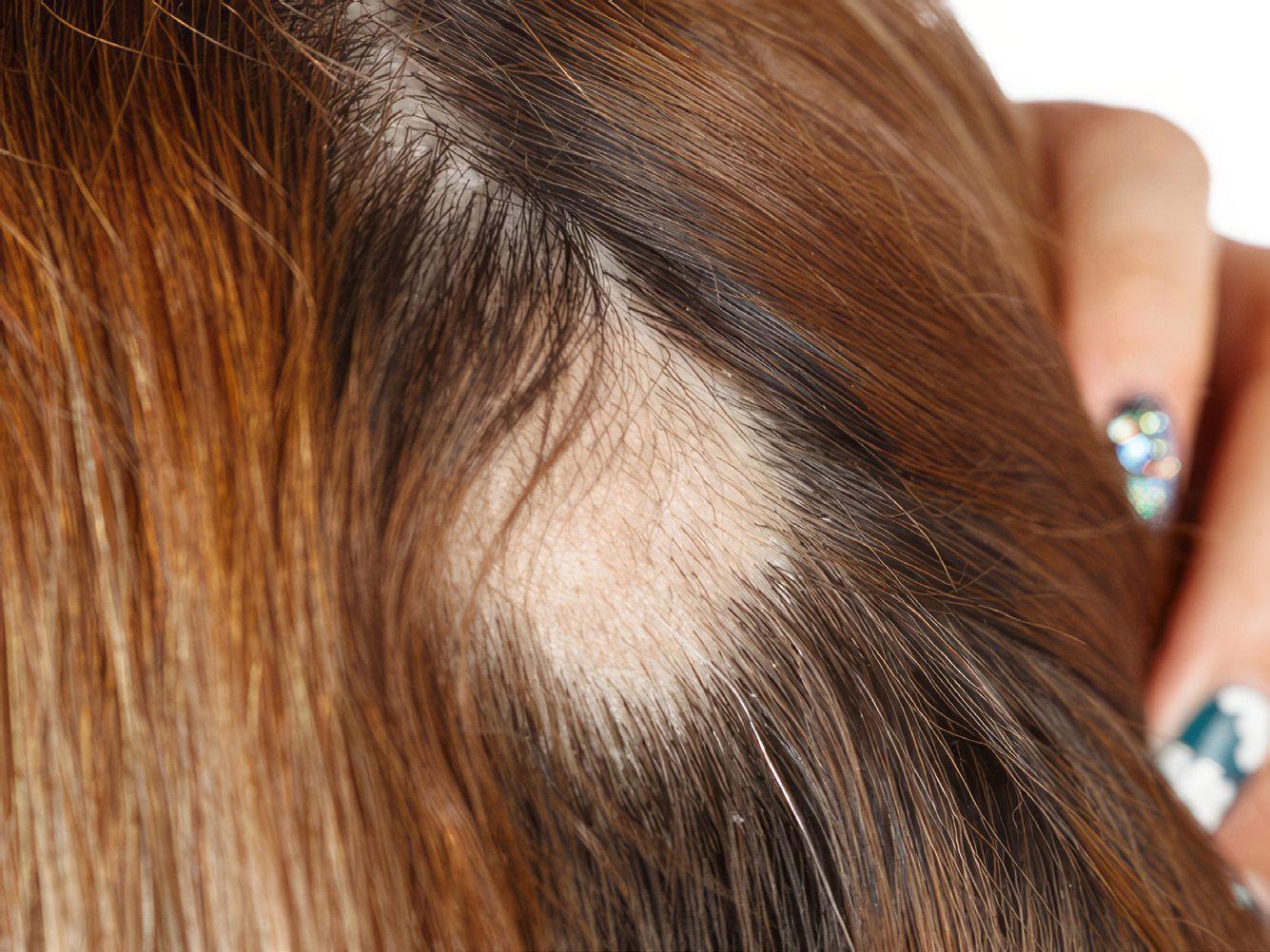
Die Alopecia areata ist durch einen plötzlich auftretenden, umschriebenen, oft kreisrunden Haarausfall gekennzeichnet (Abb. 1). Betroffen ist meist die behaarte Kopfhaut, der Haarausfall kann aber auch im Gesicht oder an anderen Körperstellen auftreten, manchmal fällt das gesamte Kopfhaar aus. Ursache ist eine Autoimmunreaktion gegen die in der Haut liegenden Haarbestandteile. Dabei greifen die körpereigenen Abwehrzellen die Haarfollikel an und lösen Entzündungsreaktionen aus, die zum Haarausfall führen und bei völliger Zerstörung der Haarfollikel das Haarwachstum sogar völlig unterbinden können. Möglicherweise erhöht eine genetisch bedingte Veranlagung das Risiko für eine derartige Autoimmunreaktion, denn die Alopecia tritt familiär gehäuft auf. Auch Angstzustände und anhaltender Stress können Auslöser für eine Alopecia areata sein [1, 2].
Der kreisrunde Haarausfall ist keine seltene Krankheit: Etwa 2 % der Bevölkerung erhalten diese Diagnose im Laufe des Lebens. In Deutschland leiden mehr als 1,5 Millionen vorwiegend jüngere Menschen an einer Alopecia areata. Männer und Frauen sind gleichermaßen betroffen, auch Kinder können an einer Alopecia areata erkranken [3].
Der Verlauf ist sehr unterschiedlich: In den meisten Fällen kommt es zur Spontanremission, d.h., die Alopezie verschwindet nach 6 – 12 Monaten von selbst und kommt nie wieder. Bei anderen Patienten wachsen die verlorenen Haare zwar ebenfalls nach, doch irgendwann tritt der kreisrunde Haar-
ausfall erneut auf. Im schlimmsten Fall tritt keine Besserung ein – etwa 20 – 30 % der Betroffenen leiden unter einem dauerhaften Haarverlust [4].
Der Haarverlust an sich ist für die Patienten nicht schädlich, er ist aber vor allem bei Kindern, Jugendlichen und jungen Erwachsenen mit enormen emotionalen und psychosozialen Belastungen verbunden. Da sich die Betroffenen wegen ihres Aussehens unwohl und unsicher fühlen, meiden sie soziale Kontakte. Mehrere Studien zeigen, dass Patienten mit Alopecia areata aufgrund der stigmatisierenden Erkrankung häufger unter Depressionen leiden [5]. In diesen Fällen ist eine medikamentöse Be-
handlung zu erwägen, vor allem bei einem schweren Verlauf, bei dem keine Spontanremission mehr zu erwarten ist.
Effektive Therapien werden dringend benötigt
Während die lokalisierte Form der Alopecia areata meist gut mit topischem oder intrakutan appliziertem Kortison behandelt werden kann, ist die Therapie der fortgeschrittenen Form oft eine große Herausforderung. Als klassisches Mittel der Wahl gilt hier systemisches Kortison, bei Therapieversagen wird Methotrexat empfohlen [6]. Allerdings sprechen nicht alle
Patienten (vor allem Kinder) auf diese steroidsparende Behandlung an und die Rezidivrate innerhalb eines Jahres liegt bei etwa 50 % [7]. Eine (umstrittene) Alternative ist die Immuntherapie, z.B. in Form des Kontaktallergens Diphenylcyclopropenon (DPCP). Dieses wird in steigenden Konzentrationen auf die betroffene Haut aufgetragen, bis eine milde Kontaktdermatitis ausgelöst wird, die immunregulatorische Mechanismen aktivieren bzw. verstärken soll. Jedoch wurden auch unter dieser Behandlung bei Kindern Rezidivraten mit über 85 % berichtet [8].
Bislang führt keine der bisherigen Therapien bei allen Patienten zu einer vollständigen Heilung – nach Ende bzw. Reduktion der Therapie kommt es teilweise zu hohen Rezidivraten. Um diese unbefriedigenden Behandlungsergebnisse bei der fortgeschrittenen Alopecia areata zu verbessern, wurde daher nach systemischen Therapien gesucht, die direkt in die Pathogenese dieser Autoimmunerkrankung eingreifen.
JAK-Inhibitoren als neue Behandlungsoption
Januskinasen (JAK) leiten Signale von Zytokinen und Wachstumsfaktoren weiter, die unter anderem eine wichtige Rolle bei immunologischen und infammatorischen Prozessen spielen. Die JAK-Enzymgruppe umfasst JAK1, JAK2, JAK3 und TYK2.
2022 wurde der JAK-Inhibitor Baricitinib* (Olumiant®) zur Thera-
* Die Kosten für die Behandlung mit Baricitinib und Ritlecitinib werden von den deutschen Krankenkassen nicht erstattet, da Paragraph 34 SGB V die Kostenübernahme von Arzneimitteln zur Verbesserung des Haarwuchses ausschließt.
pie der schweren Alopecia areata bei Erwachsenen zugelassen. Baricitinib hemmt die Januskinasen 1 und 2, die für die intrazelluläre Signalfortleitung sowohl von IFN-γ als auch IL-15 benötigt werden [9]. Seit Oktober 2023 ist mit Ritlecitinib* (Litfulo®) ein weiterer JAK-Inhibitor in der EU verfügbar. Ritlecitinib ist ebenfalls zur Behandlung von schweren Formen der Alopecia areata indiziert. Anders als Baricitinib ist es jedoch nicht nur für Erwachsene, sondern bereits ab einem Alter von 12 Jahren zugelassen, sodass auch Jugendliche von dieser neuen Therapiemöglichkeit proftieren können [10]. Ritlecitinib ist zudem der erste und einzige Wirkstoff, der selektiv und irreversibel die Januskinase 3 und die Tyrosinkinase hemmt, die auch in der Familie der Kinasen der hepatozellulären Karzinome (TEC) exprimiert wird. Durch die Blockade der beiden Enzyme wird die Signaltransduktion von Zytokinen gestört und die zytolytische Aktivität von natürlichen Killerzellen (NK-Zellen) und CD8+-T-Zellen vermindert. Damit werden die über JAK3 und die TEC-Familie vermittelten Signalwege unterbrochen, die maßgeblich an der Pathogenese der Alopecia areata beteiligt sind [10].
Die empfohlene Dosis von Litfulo® beträgt 50 mg (als Hartkapsel) einmal täglich. Bei Jugendlichen im Alter von 12 bis <18 Jahren ist keine Dosisanpassung erforderlich [10].
ALLEGRO-Studienprogramm belegt klinische Wirksamkeit von Ritlecitinib
Die Zulassung von Ritlecitinib basiert auf den Ergebnissen der randomisierten, doppelblinden,
placebokontrollierten Phase-IIb/ III-Studie ALLEGRO [11]. Eingeschlossen wurden 718 Patienten ab 12 Jahren mit einer Alopecia areata mit mindestens 50%igem Haarausfall auf der Kopfhaut, einer Alopecia totalis (Haarausfall auf der gesamten Kopfhaut) sowie einer Alopecia universalis (Haarausfall am ganzen Körper). Die Studie bestand aus einer placebokontrollierten Phase von 24 Wochen und einer Verlängerungsphase von weiteren 24 Wochen. Da in ALLEGRO verschiedene Dosierschemata untersucht wurden, erhielten letztlich 130 Studienteilnehmer randomisiert die zur Zulassung empfohlene Dosierung von 1 täglich 50 mg Ritlecitinib und 131 bekamen Placebo. Primärer Endpunkt war der Anteil der Patienten, die in Woche 24 einen SALT-Score (Severity of Alopecia Tool) von ≤10 erreichten (d.h. eine mindestens 90%ige Bedeckung der Kopfhaut mit Haaren). Wichtige sekundäre Endpunkte waren das Ansprechen in Bezug auf die „Patient‘s Global Impression of Change“ (PGI-C), ein SALT-Score von ≤20 (d.h. eine mindestens 80%ige Bedeckung der Kopfhaut mit Haaren) sowie Verbesserungen des Nachwachsens von Augenbrauen und/oder Wimpern in Woche 24.
Nach der 24-wöchigen Behandlung mit Ritlecitinib hatten sich die Symptome im Vergleich zu Placebo signifkant verbessert (Tab. 1):
• 13,4 % der Erwachsenen und Jugendlichen erreichten unter Ritlecitinib eine nahezu vollständige Remission, d.h. ein Nachwachsen der Haare auf der Kopfhaut von 90 % oder mehr (SALT-Score ≤10), in der Placebogruppe war dies nur bei 1,5 % der Fall.
• 23,0 % der Patienten der Ritlecitinib-Gruppe erreichten
Endpunkt
Ritlecitinib 50 mg einmal täglich (n = 30) % Responder
Placebo (n = 131) % Responder
Tabelle 1: Ergebnisse der ALLEGRO-Studie für die Wirksamkeit von Ritlecitinib (Litfulo®) im Vergleich zu Placebo nach 24 Wochen Therapie [10, 11].
SALT: Severity of Alopecia Tool (Skala zur Bewertung des Schweregrads der Alopezie), PGI-C: Patient’s Global Impression of Change (allgemeine Einschätzung der Veränderung durch den Patienten), EBA: Bewertung der Augenbrauen (eyebrow assessment), ELA: Bewertung der Wimpern (eyelash assessment), * Unterschied statistisch signifkant mit Adjustierung für Mehrfachtestung, ** Unterschied statistisch signifkant.
eine mindestens 80%ige Bedeckung der Kopfhaut mit Haaren (SALT-Score ≤20) gegenüber 1,6 % unter Placebo.
• 49,2 % der mit Ritlecitinib behandelten Studienteilnehmer berichteten von einer „moderaten“ bis „großen“ Verbesserung ihrer Alopecia areata, verglichen mit 9,2 %unter Placebo (erhoben mittels PGI-C).
• Auch hinsichtlich des Nachwachsens von Augenbrauen und Wimpern schnitt Ritlecitinib signifkant besser ab.
Akzeptables Sicherheitsprofil
Zu den häufgsten Nebenwirkungen, die im Zusammenhang mit Ritlecitinib berichtet wurden, gehörten Durchfall (9,2 %), Akne (6,2 %), Infektionen der oberen Atemwege (6,2 %), Urtikaria (4,6 %), Hautausschlag (3,8 %), Follikulitis (3,1 %) und Schwin-
del (2,3 %). Das bei Jugendlichen beobachtete Sicherheitsprofl war dem der erwachsenen Population ähnlich [10].
Anmerkung: Bei der Therapieentscheidung müssen die bei Anwendung von JAK-Inhibitoren generell möglichen erhöhten Risiken für schwere Infektionen, kardiovaskuläre Ereignisse, maligne Erkrankungen, thrombotische Ereignisse sowie die erhöhte Gesamtsterblichkeit beachtet werden. Aus diesem Grund sollte Ritlecitinib nur bei den schwersten Formen der Alopecia areata eingesetzt und das Nutzen-Risiko-Verhältnis der Behandlung in regelmäßigen Abständen neu bewertet werden.
Brigitte Söllner, Erlangen
Literatur
1 Pratt CH et al. Alopecia areata. Nat Rev Dis Primers 2017;3:17011
2 Islam N et al. The autoimmune basis of alopecia areata: a comprehensive review. Autoimmun Rev 2015;14:81-89
3 Stefanaki C et al. A retrospective study on alopecia areata in children: clinical characteristics and treatment choices. Skin Appen Dis 2021;7:454-459
4 Sterkens A et al. Alopecia areata: a review on diagnosis, immunological etiopathogenesis and treatment options. Clin Exp Med 2021;21:215-230
5 Macbeth AE et al. The associated burden of mental health conditions in alopecia areata: a population-based study in UK primary care. Br J Dermatol 2022;187: 73-83
6 Strazzulla LC et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol 2018;78:15-24
7 Phan K et al. Methotrexate for alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol 2019;80:120127.e2
8 van der Steen PH et al. Topical immunotherapy for alopecia areata: re-evaluation of 139 cases after an additional follow-up period of 19 months. Dermatology 1992; 184:198-201
9 Fachinformation Olumiant® 2 mg/4 mg Filmtabletten; Stand: Mai 2023
10 Fachinformation Litfulo® 50 mg Hartkapseln; Stand: September 2023
11 King B et al. Efficacy and safety of ritlecitinib in adults and adolescents with alopecia areata: a randomised, double-blind, multicentre, phase 2b-3 trial. Lancet 2023;401:1518-1529
Avelumab (Bavenico®) ist ein humaner monoklonaler Antikörper, der gegen den programmierten Zelltod-Liganden 1 (PD-L1) gerichtet ist. Er zählt zu den Checkpoint-Inhibitoren. Seit September 2017 ist Avelumab zur Therapie des metastasierten Merkelzellkarzinoms (mMCC) zugelassen. Patienten, die an diesem seltenen und aggressiven Hautkrebs leiden, haben eine sehr schlechte Prognose: Weniger als 20 % der Betroffenen überleben länger als 5 Jahre. Wie die Zulassungsstudie JAVELIN Merkel 200 damals zeigte, betrug die objektive Ansprechrate bei Patienten mit mMCC, deren Erkrankung nach mindestens einer vorausgegangenen Chemotherapie fortgeschritten war, 33 %, wobei 11 % der Patienten eine Vollremission (CR) und 22 % eine Teilremission erreichten. Die Ansprechdauer reichte von 2,8 Monaten bis über 24,9 Monate. In 93 % der Fälle betrug sie mindestens 6 Monate bei, bei 71 % mindestens 12 Monate (n = 13) [1].
Positives Wirksamkeits- und Sicherheitsprofil in allen Therapielinien
Heute gilt Avelumab als Standardtherapie des mMCC, und das mit
gutem Grund: Aus der JAVELIN Merkel 200 Studie liegen aktuell Follow-up-Daten über median 21,2 Monate in der Erstlinientherapie [2] und über median 65,1 Monate in allen anderen Therapielinien [3] vor. Unter der Erstlinientherapie wurde eine objektive Ansprechrate (ORR) von 39,7 % beobachtet, das mediane progressionsfreie Überleben (PFS) betrug 4,1 Monate, das mediane Gesamtüberleben (OS) 20,3 Monate. Hervorzuheben ist ferner die mit 18,1 % niedrige Inzidenzrate behandlungsassoziierter unerwünschter Ereignisse der Grade 3 und 4 [2].
Die Auswertung zu Avelumab in späteren Therapielinien ergab ein medianes Gesamtüberleben von 12,6 Monaten, behandlungsassoziierte unerwünschte Ereignisse mit Todesfolge waren nicht aufgetreten [3].
Hohe Ansprechraten und gute Verträglichkeit auch im Praxisalltag
Wie Real-World-Daten zeigen, lassen sich diese Resultate auch im klinischen Alltag erreichen. In einer retrospektiven Analyse von US-Patientenakten betrug die ORR 73 %, das mediane PFS 24,4 Monate und das mediane OS 30,7 Monate (mediane Beobachtungs-
dauer 20,8 Monate) [4]. Erfreuliche Resultate lieferte auch eine Analyse von Patientendaten aus Israel. Sie ergab eine ORR von 59,3 %, ein medianes PFS von 8,1 Monaten und ein medianes OS von 23,5 Monaten (mediane Beobachtungsdauer 12,8 Monate). Die Inzidenzrate für behandlungsassoziierte unerwünschte Ereignisse der Grade 3 und 4 betrug 14 % [5].
Klinischer Nutzen auch in speziellen Subgruppen
Mittlerweile liegen für Avelumab auch Daten zu besonderen klinischen Fragestellungen vor – beispielsweise, ob eine Reinduktion nach einer CheckpointInhibitor(CI)-Therapie sinnvoll ist. Eine Registerauswertung gibt dazu wichtige Hinweise. Darin erreichten 4 von 8 Patienten unter einer neuerlichen CI-Therapie ein partielles Ansprechen [6]. Eine weitere Fragestellung ist, inwieweit CI bei Patienten mit Immunsuppression und speziell nach Organtransplantation eingesetzt werden können. Denn es ist denkbar, dass Immunsuppressiva die Wirkung der Immunonkologika beeinträchtigen und CI nach einer Transplantation das Risiko für eine Transplantatabstoßung erhöhen. Auch hierzu gibt es Er-
Wirkstoffklasse
Avelumab ist ein humaner monoklonaler Antikörper, der gegen den programmierten ZelltodLiganden 1 (PD-L1) gerichtet ist.1 Er zählt zu den sogenannten Checkpoint-Inibitoren.1
Wirkprinzip
PD-L1 wird unter anderem auf Tumorzellen exprimiert, der entsprechende Rezeptor PD-1 auf T-Zellen 2 Durch die Bindung von PD-L1 an PD-1 werden die betroffenen T-Zellen in ihrer Aktivität unterdrückt.2 Damit kann sich der Krebs einer anti-tumoralen Immunantwort entziehen.
Neue Therapiekonzepte beim mMCC?
Avelumab bindet spezifisch an PD-L1 und blockiert so die Bindung zwischen PD-L1 und PD-1 1 Dadurch bleiben tumorspezifische T-Zellen aktiviert und können eine entsprechende Immunantwort einleiten (Abb. 1) 1 In präklinischen Studien zeigte sich außerdem, dass Avelumab mittels antikörperabhängiger zellulärer Zytotoxizität (Antibody Dependent Cell Cytotoxicity, ADCC) zu einer direkten Tumorzelllyse führt, die durch Natürliche Killerzellen (NK-Zellen) vermittelt wird.1
Avelumab (Bavencio®) ist ein humaner monoklonaler Antikörper, der gegen den programmierten Zelltod-Liganden 1 (PD-L1) gerichtet ist [9]. PD-L1 wird unter anderem auf Tumorzellen exprimiert, der entsprechende Rezeptor PD-1 auf T-Zellen. Durch die Bindung von PD-L1 an PD-1 werden die betroffenen T-Zellen in ihrer Aktivität unterdrückt. Damit kann sich der Krebs einer antitumoralen Immunantwort entziehen [10].
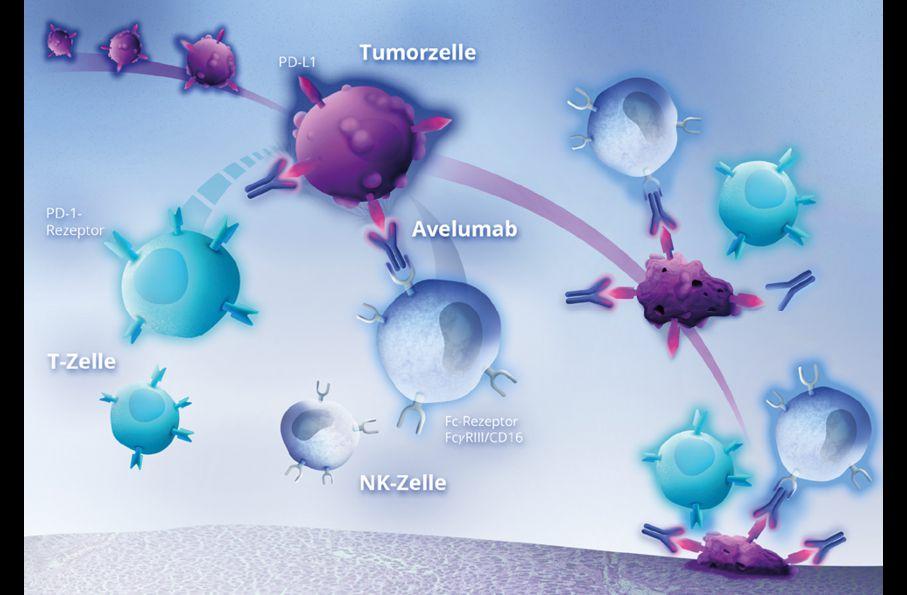
Abbildung 1: Wirkmechanismus Avelumab (modifiziert nach 1,2)
Avelumab bindet spezifisch an PD-L1 und blockiert so die Bindung zwischen PD-L1 und PD-1. Dadurch bleiben tumorspezifische TZellen aktiviert und können eine entsprechende Immunantwort einleiten. In präklinischen Studien zeigte sich außerdem, dass Avelumab mittels antikörperabhängiger zellulärer Zytotoxizität zu einer direkten Tumorzelllyse führt, die durch natürliche Killerzellen (NK-Zellen) vermittelt wird [9].
1
Das Potenzial der CI scheint mit dem Einsatz beim mMCC jedoch noch nicht ausgeschöpft. Derzeit werden mehrere Wirkstoffe in (neo-)adjuvanten Settings geprüft. Die Hoffnung ist, dass sich dadurch die hohen Rezidivraten selbst nach vollständiger Resektion eindämmen lassen. Erste Hinweise, dass solche Konzepte das krankheitsfreie Überleben verlängern könnten, gibt es bereits [8].
Fabian Sandner, Nürnberg
gebnisse aus der bereits zitierten Beobachtungsstudie aus Israel: Hier schnitten immunsupprimierte Patienten (n = 14) bezüglich Wirksamkeit und Sicherheit unter Avelumab ebenso gut ab wie immunkompetente [5]. In einer
Studie mit nierentransplantierten Patienten mit diversen malignen Erkrankungen erlitt keiner der mit einem PD-L1-Inhibitor (Avelumab, Atezolizumab) behandelten Studienteilnehmer (n = 6) eine Abstoßungsreaktion [7].
Literatur
1 Kaufman HL et al. Lancet Oncol 2016; 17:1374-1385
2 D’Angelo SP et al. J Immunother Cancer 2021;9:e002646
3 D’Angelo SP et al. ESMO Open 2021; 6:100290
4 Bhatia S et al. J Immunother Cancer 2022;10:e004904
5 Averbuch I et al. Cancer Med 2023; 12:12065-12070
6 Stege HM et al. SN Compr Clin Med 2020;2:2202-2207
7 Murakami N et al. Kidney Intern 2021; 100:196-205
8 Becker JC et al. Lancet 2023;402:798808
9 Fachinformation Bavencio®, aktueller Stand
10 Collins JM et al. Human Vaccin Immunother 2019;15:891-908
Die atopische Dermatitis (AD; Synonym: atopisches Ekzem, Neurodermitis) ist die häufgste chronisch entzündliche Hauterkrankung bei Kindern. In Deutschland leiden etwa 13 % aller Kinder und etwa 3,5 % aller Erwachsenen zumindest zeitweilig unter einer AD [1]. Starker Juckreiz und daraus resultierende Schlafstörungen, rezidivierende, teils großfächige Ekzeme und eine ausgeprägte Hauttrockenheit stellen eine erhebliche Krankheitslast dar [2]. Dementsprechend hoch ist der Versorgungsbedarf, wobei aus Sicht der Patienten die Schmerzlinderung, die Abheilung der Hautveränderungen sowie die langfristige Krankheitskontrolle im Vordergrund stehen [3]. Neben den körperlichen Beschwerden können psychosoziale Belastungen wie Stigmatisierung und Probleme an Schul- und Arbeitsplätzen die Lebensqualität beeinträchtigen.
Die Behandlung der AD erfordert eine Vielzahl von Maßnahmen, die individuell auf die Patienten abgestimmt werden sollten. Hierzu gehören die Reduktion und Vermeidung von Exazerbationsfaktoren, eine topische Basistherapie mit Emollienzien und Moisturizern sowie eine individuell angepasste symptom- und schweregradorientierte medikamentöse Behandlung [4]. Sprechen die Patienten nicht auf die antientzündliche topische Therapie mit Glukokortikosteroiden und Calcineurin-Inhibitoren an, ist eine sytemische Therapie indiziert [4].
Mit Lebrikizumab (Ebglyss®) hat die Europäische Kommission im November 2023 ein neues Medikament zugelassen, das aufgrund seines selektiven Wirkmechanismus eine vielversprechende Behandlungsoption für Patienten mit
mittelschwerer bis schwerer AD darstellt, die mit einer topischen Therapie nicht kontrolliert werden können. Lebrikizumab ist ein monoklonaler Antikörper, der mit hoher Affnität an IL-13 bindet und dessen nachgeschaltete Sig-
Lebrikizumab
nalübertragung selektiv hemmt [5]. Indiziert ist Lebrikizumab zur Behandlung von erwachsenen und jugendlichen Patienten (ab 12 Jahren mit einem Körpergewicht von mindestens 40 kg) mit mittelschwerer bis schwerer AD, die für
Lebrikizumab (Ebglyss®) ist ein monoklonaler Immunglobulin (IgG4) Antikörper. Er bindet selektiv an Interleukin (IL)13, ein proinflammatorisch wirkendes Zytokin, das eine wesentliche Rolle bei der Typ-2-Immunität spielt und in der Haut von Patienten mit atopischer Dermatitis überexprimiert ist. IL-13 treibt den Typ-2-Entzündungskreislauf an, der zu einer Dysfunktion der Hautbarriere sowie zu Juckreiz, einer Hautverdickung und Infektionen führt. Durch die Bindung an IL-13 verhindert Lebrikizumab dessen Interaktion mit seinen Rezeptoren, insbesondere dem heterodimeren Rezeptor-Signalkomplex, bestehend aus dem IL-4-Rezeptor alpha (IL4Rα) und dem Interleukin-13-Rezeptor-alpha-1-Protein (IL-13Rα1). Hierdurch werden die nachgelagerten IL-13-vermittelten Effekte blockiert [5] und damit die Entzündungsreaktionen reduziert [5]. Lebrikizumab inhibiert jedoch nicht den IL-4-Signalweg. IL-13 kann in Gegenwart von Lebrikizumab zudem immer noch an den DecoyRezeptor IL-13Rα2 binden, was eine anschließende Internalisierung des Zytokins mit nachfolgendem lysosomalem Abbau ermöglicht. Es wird angenommen, dass IL-13Rα2 ein Decoy-Rezeptor ist, der die endogene Regulierung von IL-13 vermittelt [5]. Die Elimination von IL13 über IL-13Rα2 bleibt somit unbeeinflusst [5, 7].
EBGLYSS® steht als vorgefüllte Fertigspritze und als Fertigpen mit jeweils 250 mg Lebrikizumab in 2 ml Lösung zur Verfügung und wird subkutan injiziert [6].
Bei Lebrikizumab handelt es sich um einen monoklonalen Immunglobulin selektiv an Interleukin (IL)13. IL-13 ist ein proinflammatorisch wirkendes II-Immunität spielt und in der Haut von Patienten mit atopischer als das Schlüsselzytokin bei atopischer Dermatitis in der Haut[1,2].
In der Haut bei atopischer Dermatitis wirkt IL-13 pro-inflammatorisch, störung und erhöhte kutane Infektanfälligkeit, begünstigt die Verdickung die Entstehung von Juckreiz [2]. Lebrikizumab verhindert die Bildung komplexes bestehend aus Interleukin-13-Rezeptor-alpha-1-Protein Hierdurch werden die nachgelagerten IL-13-vermittelten Effekte
Lebrikizumab inhibiert jedoch nicht den IL-4 Signalweg. IL-13 kann immer noch an den Decoy-Rezeptor IL-13Rα2 binden, was eine anschließende mit nachfolgendem lysosomalen Abbau ermöglicht. Es wird angenommen, zeptor ist, der die endogene Regulierung von IL-13 vermittelt [1]. Die bleibt somit unbeeinflusst [1,2].
1. Okragly AJ et al. Binding, Neutralization and Internalization of the Interleukin-13 2023;13(7):1535-1547.
2. Bieber T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis.
Woche 16
Pruritus Numerical Rating Scale (NRS, Verbesserung um ≥4 Punkte) c
Dermatology Life Quality Index (DLQI, Erwachsene; Verbesserung um ≥4 Punkte) d
Tabelle 1: Ergebnisse zur Wirksamkeit von Lebrikizumab (LEB, Ebglyss®) in den zulassungsrelevanten klinischen Studien ADvocate-1 und ADvocate-2 in Woche 16 [7].
* p < 0,001 vs. Placebo.
a Patienten mit einem IGA von 0 oder 1 („erscheinungsfrei“ oder „fast erscheinungsfrei“) mit Reduktion um ≥2 Punkte gegenüber Baseline auf einer IGA-Skala von 0–4.
b Patienten mit einer Reduktion des EASI um 75 % bzw. 90 % von Baseline bis Woche 16.
c Der Prozentsatz wird relativ zur Anzahl der Patienten mit einer Baseline-Pruritus-NRS-Wert ≥4 berechnet.
d Der Prozentsatz wird relativ zur Anzahl der Patienten mit einem Baseline-DLQI-Wert ≥4 berechnet.
eine systemische Therapie in Betracht kommen [6].
Frühe klinische Wirksamkeit und anhaltendes Ansprechen bei einer 4-wöchentlichen Erhaltungsdosis
Die Zulassung von Lebrikizumab stützt sich auf 3 Phase-III-Studien, in denen ein langfristiges Therapieansprechen mit anhaltender Juckreizlinderung nachgewiesen wurde. In ADvocate-1 und ADvocate-2 wurde Lebrikizumab als Monotherapie bei erwachsenen und jugendlichen Patienten mit mittelschwerer bis schwerer AD geprüft [8]. An diesen beiden internationalen Studien (unter deutscher Beteiligung) nahmen 851 Patienten teil. Sie erhielten über 16 Wochen alle 2 Wochen eine subkutane Injektion mit entweder 250 mg Lebrikizumab (nach initialer Aufsättigung mit 2 250-mgInjektionen in Woche 0 und 2) oder Placebo. Die coprimären Endpunkte waren ein Investigator Global Assessment Score (IGA)
von 0 oder 1 („erscheinungsfrei“ oder „fast erscheinungsfrei“) bei einer Score-Verbesserung um mindestens 2 Punkte und die Reduktion des Eczema Area and Severity Index (EASI) um 75 % verglichen mit Baseline in den ersten 16 Wochen.
Einen IGA-Score von 0 oder 1 erreichten in ADvocate-1 43,1 % der mit Lebrikizumab behandelten Patienten gegenüber 12,7 % in der Placebogruppe. In ADvocate-2 waren es 33,2 % der Patienten in der Lebrikizumab-Gruppe und 10,8 % in der Placebogruppe. Die Unterschiede waren signifkant (p < 0,001). Lebrikizumab zeigte in der Monotherapie in Woche 16 eine frühe klinische Wirksamkeit und reduzierte bei fast 60 % der Patienten das Ausmaß und den Schweregrad der Erkrankung um mindestens 75 % (EASI-75) (Tab. 1) [8]. In Kombination mit topischen Kortikosteroiden, die in der ADhere-Studie untersucht wurde, war dies bei fast 70 % der Fall [9]. Nahezu 80 % der Patienten, die in Woche 16 auf die Behandlung
mit Lebrikizumab ansprachen* und diese sowohl als Monotherapie als auch in Kombination mit topischen Kortikosteroiden über einen Zeitraum von bis zu 2 Jahren fortsetzten, zeigten unter der vierwöchentlichen Erhaltungsdosis ein anhaltendes klinisches Ansprechen der Hautveränderungen, des Pruritus und eine Verringerung des Schweregrads der Erkrankung (Tab. 2) [10, 11].
Konsistente gute Verträglichkeit
Im Rahmen des klinischen Studienprogramms der Phase III wurde auch das Sicherheitsprofl von Lebrikizumab untersucht. Die meisten unerwünschten Ereignisse in den Studien waren leicht oder mittelschwer und führten nicht
* Responder waren definiert als diejenigen, die in Woche 16 eine 75%ige Verringerung des EASI-75 oder einen IGA 0 oder 1 („erscheinungsfrei“ oder „fast erscheinungsfrei“) mit einer Verbesserung von mindestens 2 Punkten und ohne Einsatz von Notfallmedikamenten erreichten.
Woche 52
IGA 0 oder 1 a
EASI 75 b
Pruritus Numerical Rating Scale (NRS, Verbesserung um ≥4 Punkte) c
ADvocate-1 und ADvocate-2 (gepoolt)
Placebo d (LEB abgesetzt) n = 60 LEB 250 mg Q2W n = 118
DTabelle 2: Ergebnisse zum Erhalt des klinischen Ansprechens von Lebrikizumab (LEB, Ebglyss®) in den zulassungsrelevanten klinischen Studien ADvocate-1 und ADvocate-2 in Woche 16 [10, 11].
* p < 0,05, ** p < 0,01 versus Placebo.
a Patienten mit IGA 0/1 mit einer Verbesserung um ≥2 Punkte gegenüber Baseline in Woche 1, die in Woche 52 weiterhin IGA 0/1 mit einer Verbesserung um ≥2 Punkte aufwiesen.
b Patienten, die in Woche 16 EASI 75 erreichten und in Woche 52 weiterhin EASI 75 aufwiesen, oder Patienten, die in Woche 16 EASI 75 erreichten und in Woche 52 EASI 90 aufwiesen.
c Der Prozentsatz wird relativ zur Anzahl der Patienten mit einer Baseline-Pruritus-NRS-Wert ≥4 berechnet.
d Patienten, die in Woche 16 auf LEB 250 mg Q2W ansprachen (IGA 0/1 oder EASI 75) und dann auf Placebo randomisiert wurden.
zum Abbruch der Behandlung. Die häufgsten Nebenwirkungen waren Konjunktivitis (6,9 %), Reaktionen an der Injektionsstelle (2,6 %), allergische Konjunktivitis (1,8 %) und trockenes Auge (1,4 %).
Fazit für die Praxis
Die klinischen Daten zeigen, dass Lebrikizumab eine wirksame und gut verträgliche Therapieoption bei mittelschwerer bis schwerer AD und damit einen Fortschritt für Patienten darstellt, die an einer durch eine topischen Therapie nicht kontrollierbaren AD leiden. Denn die Therapie mit Lebrikizumab führt zu einer langfristigen Verbesserung der klinischen Zeichen und Symptome der Krankheit [1]. Aufgrund seiner nachgewiesenen kurz- und langfristigen Wirksamkeit, seiner vierwöchentlichen Erhaltungsdosis und seines konsis-
tenten Sicherheitsprofls hat Lebrikizumab durchaus das Potenzial, ein Biologikum für die Erstlinientherapie zu sein.
Brigitte Söllner, Erlangen
Literatur
1 Bergmann KC et al. Allergo J Int 2016; 25:6-10
2 Beikert FC et al. Arch Dermatol Res 2014;306:279-286
3 Augustin M et al. J Eur Acad Dermatol Venereol 2020;34:142-152
4 S3-Leitlinie „Atopische Dermatitis“, AWMF-Registernr. 013-027, 2023
5 Okragly A et al. Dermatol Ther (Heidelb) 2023;13:1535-1547
6 Fachinformation Ebglyss®; Stand: 11/ 2023
7 Bieber T. Allergy 2020;75:54-62
8 Silverberg JI et al. N Engl J Med 2023; 388:1080-1091
9 Simpson EL et al. JAMA Dermatol 2023; 159:182-191
10 Blauvelt A et al. Br J Dermatol 2023; 188:740-748
11 Guttman-Yassky E et al. Presented at the Fall Clinical Dermatology Conference, October 20, 2023
ie Behandlung des nicht kleinzelligen Lungenkarzinoms (NSCLC) hat sich in den vergangenen Jahren tiefgreifend gewandelt. Neben zielgerichteten Therapien, die Treiberaberrationen adressieren, spielen Immuntherapien mit Checkpoint-Inhibitoren wie Cemiplimab (Libtayo®) heute eine zentrale Rolle [1].
Seit März 2023 ergänzt eine Indikationserweiterung für Cemiplimab das therapeutische Spektrum. Der PD (programmed cell death)-1-Inhibitor ist indiziert in Kombination mit einer platinbasierten Chemotherapie für die Erstlinienbehandlung von erwachsenen Patienten mit einem NSCLC, das PD-L1 in ≥1 % der Tumorzellen exprimiert und keine EGFR-, ALK- oder ROS1-Aberrationen aufweist [2]. Die Zulassung umfasst Patienten mit lokal fortgeschrittenem NSCLC, die keine Kandidaten für eine defnitive Radiochemotherapie sind, und Patienten mit metastasiertem NSCLC [2].
Grundlage der Indikationserweiterung sind Daten der Zulassungsstudie EMPOWER-Lung 3 [3].
Die eingeschlossenen neu diagnostizierten Patienten mit fortgeschrittenem NSCLC erhielten 2:1 randomisiert entweder Cemiplimab plus Chemotherapie oder Chemotherapie allein [3]. Außerdem waren auch Patienten im Stadium IIIB/C eingeschlossen, die nicht für eine defnitive Radiochemotherapie geeignet waren (14 %) [2].
Auch bei Plattenepithelkarzinomen effektiv
In der Primäranalyse (Datenschnitt 14. Juni 2021) lag das mediane Gesamtüberleben (OS) unter Cemiplimab plus Chemotherapie für
Patienten mit PD-L1-Expression ≥1 % bei 21,9 Monaten gegenüber 12,6 Monaten bei alleiniger Chemotherapie (HR: 0,55; 95%KI: 0,39 – 0,78) [2]. Eine Posthoc-Analyse zeigt zudem, dass die Erstlinientherapie mit Cemiplimab und Chemotherapie auch in der prognostisch ungünstigen Subgruppe der Patienten mit Plattenepithel-Histologie (43 %) wirksam ist. Das mediane OS lag hier bei 23,2 vs. 12,6 Monaten (HR: 0,61; 95%-KI: 0,40 – 0,94; nominaler p-Wert = 0,0241) zugunsten der Kombination. Das mediane progressionsfreie Überleben (PFS) war mit 8,2 gegenüber 4,6 Monaten in der Kontrollgruppe signifkant überlegen (HR: 0,53; 95%-KI: 0,36 – 0,78; nominaler p-Wert = 0,001) [4].
Die Zulassungserweiterung von Cemiplimab ist gerade für NSCLC-Patienten im Stadium IIIB/C relevant, die bislang eine schlechte Prognose haben. Nach 5 Jahren beträgt die mediane Überlebenswahrscheinlichkeit für Patienten im Stadium IIIB 24 – 26 %, im Stadium IIIC nur 12 – 13 % [5, 6]. Und auch der Einsatz bei Patienten, die nicht für die eine defnitive Radiochemotherapie geeignet sind, schließt eine wichtige Lü-
cke. So zeigen Auswertungen des CRISP-Registers, dass nur 41 % der NSCLC-Patienten im Stadium IIIB/C eine Radiochemotherapie erhalten [7].
Negativer TTF-1-Status als Zeichen für ein reduziertes Ansprechen auf Pemetrexed
Die Effektivität von CheckpointInhibitoren beim NSCLC ist stark von der PD-L1-Expression abhängig. Gerade bei geringer Expression steigt der Stellenwert einer zusätzlichen Chemotherapie. Ein Biomarker für eine reduzierte PD-L1-Expression ist der thyreoidale Transkriptionsfaktor (TTF-1) [8]. Etwa 25 % aller Adenokarzinome sind TTF-1-negativ und mit einem ungünstigen Verlauf verbunden [9, 10]. Der TTF1-negative Phänotyp ist überdurchschnittlich häufg männlich, im reduzierten Allgemeinzustand sowie mit ausgeprägter Tumorlast und oft mit Nebennierenmetastasen assoziiert – Eigenschaften, die sonst eher bei Plattenepithelkarzinomen auftreten [11]. Das ist ein möglicher Hinweis darauf, dass es sich hierbei um eine Tumorentität handelt, die zwischen Adeno- und Plattenepithelkarzinom steht. Gleichzeitig zeigen Patienten mit TTF-1-negativem Status ein reduziertes Ansprechen auf Pemetrexed. Wie eine aktuelle retro-
spektive Analyse ergab, sind bei diesen Patienten Gemcitabin-, Taxan- oder Vinorelbin-basierte Chemotherapien gegenüber Pemetrexed-Regimen mit einem signifkant verbesserten PFS (HR: 0,42; p = 0,001) und OS (HR: 0,40; p < 0,001) verbunden [12]. Daher ist gerade für diese Patienten eine Flexibilität hinsichtlich der Chemotherapie entscheidend – eine Anforderung, der nun auch mit der Cemiplimab-Kombinationstherapie Rechnung getragen werden kann.
Fabian Sandner, Nürnberg
Literatur
1 Onkopedia Leitlinien: Lungenkarzinom, nicht-kleinzellig. www.onkopedia.com/ de/onkopedia/guidelines/lungenkarzinom-nicht-kleinzellig_nsclc/@@guideline/html/index.html
2 Fachinformation Libtayo®; Stand: Juni 2023
3 Gogishvili M et al. Nat Med 2022;28: 2374-2380
4 Baramidze A et al. ESMO 2023;1495P
5 Casal-Mourino A et al. Transl Lung Cancer Res 2021;10:506-518
6 Goldstraw P et al. J Thorac Oncol 2016; 11:39-51
7 Eberhardt WEE, DGHO Jahrestagung 2021;V288
8 Di Federico A et al. WCLC 2023; MA20.06
9 Yamaguchi T et al. Cancer Cell 2013; 23:718-723
10 Kim JH et al. J Cancer 2018;9:4279-4286
11 Park WY et al. Mod Pathol 2012;25: 1265-1274
12 Frost N et al. Clin Lung Cancer 2020; 21:607-621
Levodopa, die für die Bewegungskontrolle wichtige Dopaminvorstufe, ist nach wie vor Goldstandard in der symptomatischen Parkinson-Therapie [1]. Während die orale Pharmakotherapie in den frühen ParkinsonStadien noch gut anschlägt, erfordert der fortschreitende Verlust dopaminerger Zellen im Krankheitsverlauf immer komplexere Therapieregime. Versuche, die Dosis der Basistherapie zu optimieren, stoßen mit der Zeit an ihre Grenzen und auch unter optimierter Basistherapie verbringen die Patienten im fortgeschrittenen Stadium bis zu 4 Stunden ihres Tages im OFF [2] – also in einer Phase, in der die Wirkung der Basismedikation bereits vor der nächsten Medikamentengabe nachlässt („Wearing-OFF“) und erneut motorische oder nicht motorische Symptome der Erkrankung auftreten. Die Symptome während dieser Schwankungen belasten die Betroffenen erheblich und schränken ihren Lebensradius stark ein [1, 3]. Effektive, verlässliche und einfach anzuwendende Bedarfsmedikamente zur Unterbrechung der OFF-Episoden sind daher dringend erforderlich. Inbrija® ist das erste inhalative Levodopa-Medikament, das für die On-Demand-Therapie von OFFEpisoden bei erwachsenen Parkinson-Patienten zur Verfügung steht, die mit Levodopa und einem Dopa-
Decarboxylase-Hemmer (DDCI) behandelt werden [4]. Durch die Einnahme über die Lunge lassen sich mehrere Barrieren umgehen, die bei Parkinson-Patienten häufg auftreten [2, 3].
Verbesserung der Wirksamkeit durch Umgehung der Magen-Darm-Passage
Bevor Levodopa im Striatum in Dopamin umgewandelt werden kann, muss es verschiedene Hürden überwinden, die die Wirksamkeit der Therapie beeinträchtigen können [5]. Dazu gehören die im Krankheitsverlauf zunehmenden Schluckstörungen, aber auch andere gastrointestinale Barrieren, wie vor allem ein verändertes Mikrobiom mit Levodopa abbauenden Darmbakterien und die bei Parkinson-Patienten sehr häufg auftretende Gastroparese [6]. Beide sind mit einem Delayed- bzw. No-ON verbunden [5, 6]. Insbesondere eiweißreiche Nahrungsmittel werden mit einer verzögerten, schlechteren, kürzeren und auch fehlenden Wirkung von Levodopa assoziiert [5, 6]. Die Therapie sollte daher dahingehend optimiert werden, dass möglichst viel des Wirkstoffs möglichst schnell im Gehirn ankommt, um den Patienten eine rasche und langanhaltende Verbesserung der Krankheitssymptome zu ermöglichen.
Durch die Aufnahme über die Lunge vermeidet inhalatives Levodopa die Metabolisierung in der Leber (First-Pass-Effekt), kann sowohl Schluckstörungen (Dysphagien) als auch andere gastrointestinale Hürden effektiv umgehen und schnell, on-demand und ohne Berücksichtigung der Nahrungsaufnahme in den Blutkreislauf gelangen [4]. Dies belegen die Ergebnisse einer pharmakokinetischen Studie, in der 84 mg inhalatives Levodopa zusammen mit 25 mg Carbidopa schneller resorbiert wurden als orales Carbidopa/ Levodopa [7]. Postprandial konnte im Vergleich eine geringere Varianz des Levodopas im Plasma bei Einnahme des Pulverinhalats aufgezeigt werden [7].
Rasche und anhaltende Wirkung bei guter Verträglichkeit
Die pulmonale Verabreichung von inhaliertem Levodopa-Pulver sorgt für einen vorhersehbaren und schnellen Behandlungseffekt. In der zulassungsrelevanten PhaseIII-Studie SPAN-PD, in die 351 Patienten mit Parkinson und OFFEpisoden eingeschlossen waren, kam es bereits nach 10 Minuten nach der Levodopa-Inhalation im Vergleich zu Placebo zu einer klinisch relevanten Verbesserung der motorischen Symptome [8]. Primärer Wirksamkeitsend-
• Die nachfolgenden Grafiken finden Sie ebenfalls in der digitalen Pressemappe.
• Die Abbildungen können honorarfrei unter der Angabe „Esteve Pharmaceuticals GmbH“ bei Veröffentlichungen zum Thema INBRIJA® verwendet werden.
Abbildung 1: INBRIJA® 33 mg Packshot
Inbrija® ist das erste Levodopa-Pulverinhalat auf dem Markt, das direkt in die Lunge verabreicht wird. Dadurch wird die Magen-DarmPassage umgangen, deren Motilität durch die Erkrankung bereits eingeschränkt ist. Auch ein First-Pass-Effekt und ein dadurch bedingt verzögerter Wirkeintritt, wie er etwa bei oralem Levodopa auftritt, werden umgangen. Durch inhalatives Levodopa kann der Wirkstoff schneller freigesetzt werden und die Wirkung ohne Verzögerung einsetzen.
Inhalatives Levodopa ist zugelassen für die intermittierende Behandlung von episodischen motorischen Fluktuationen (OFF-Episoden) bei erwachsenen Patienten mit Parkinson-Krankheit, die Levodopa in Kombination mit einem Dopa-Decarboxylase-Inhibitor (DDCI) erhalten.
Abbildung 2: INBRIJA® Inhaler
Die Levodopa-Hartkapseln werden unmittelbar vor der Anwendung in den Inhalator eingelegt. Sobald Symptome einer OFF-Phase spürbar sind, inhalieren die Patienten den Inhalt von 2 Kapseln nacheinander. Eine Titration ist für die Anwendung nicht notwendig. Die empfohlene Höchstdosis liegt bei 10 Kapseln pro Tag, das entspricht 5 Anwendungen mit je 2 Kapseln. Jede der Kapseln enthält 42 mg Levodopa, davon werden 33 mg abgegeben. Die Maximaldosis beträgt 330 mg pro Tag [4].

punkt war die mittlere Änderung des Punktwertes auf der Unifed Parkinson’s Disease Rating Scale (UPDRS) Teil III* 30 Minuten nach der Dosis in Woche 12 gegenüber dem Ausgangswert. Der primäre Endpunkt wurde erreicht: 30 Minuten nach der Applikation des inhalativen Levodopas verbesserte sich der UPDRS-Punktwert
statistisch signifkant um 10 Punkte gegenüber 6 Punkten unter Placebo (p =0,0088). Der Effekt hielt auch nach 60 Minuten noch an (p = 0,0027) [8].
Das Sicherheitsprofl von inhaliertem Levodopa entsprach dem der Levodopa-DDCI-Therapie, allerdings traten zusätzlich pulmonal bedingte Nebenwirkungen
* Der UPDRS-Teil III bezieht sich auf die Beurteilung des Schweregrades der grundlegenden motorischen Befunde (z.B. Tremor, Rigidität, Bradykinesie, posturale Instabilität) bei Patienten mit Morbus Parkinson. Dieser Endpunkt wurde in der Studie in einem klinischen Setting untersucht: Die Patienten nahmen die übliche Morgendosis ihres oralen Levodopa-/Dopa-Decarboxylase-Hemmers ein und suchten 2 – 5 Stunden später die Klinik auf. Bei Eintreten einer OFF-Phase erhielten die Patienten Placebo oder inhalatives Levodopa. Jeweils vor und 30 Minuten nach der Anwendung wurde der UPDRS-III-Score bestimmt [4].
(z.B. Husten) auf. Die häufgsten Nebenwirkungen für die 84 mgDosierung waren Husten (15 %), Infektion der oberen Atemwege (6 %), Übelkeit (5 %), verfärbtes Sputum (5 %) und Dyskinesie (4 %) [8].
Inhalatives Levodopa ist ein einfach anzuwendendes, innovatives Medikament für die Überbrückung von OFF-Episoden, das fexibel als On-Demand-Therapie, d.h. zusätzlich zur Basistherapie, eingesetzt werden kann. Durch die Inhalation und die damit verbundene Umgehung der Magen-Darm-Passage kann das Medikament mahlzeitenunabhängig eingenommen werden und entfaltet schnell seine Wirkung. Wichtig ist die früh- und damit rechtzeitige Identifzierung der für die On-Demand-Medikation geeigneten Parkinson-Patienten, die – nach entsprechender Schulung der richtigen Inhalationstechnik – mit Inbrija® die Möglichkeit haben, ihr Leben wieder fexibler zu gestalten.
Brigitte Söllner, Erlangen
Literatur
1 Jost WH et al. J Neural Transm (Vienna) 2023;130:821-826
2 Ferreira JJ et al. Lancet Neurol 2016; 15:154-165
3 Thach A et al. BMC Neurol 2021;21:46
4 Fachinformation Inbrija® 33 mg Hartkapseln mit Pulver zur Inhalation; Stand Mai 2023
5 Leta V et al. Eur J Neurol 2023;30:14651480
6 Pfeiffer RF et al. Parkinsonism Relat Disord 2020;76:63-71
7 Safirstein BE et al. Clin Ther 2020; 42:1034-1046
8 LeWitt PA et al. Lancet Neurol 2019; 18:145-154
Das multiple Myelom (MM) ist eine aggressive und derzeit nicht heilbare Form von Blutkrebs, die die im Knochenmark gebildeten Plasmazellen angreift, und die zweithäufgste hämatologische Krebserkrankung in westlichen Industriestaaten mit jährlich rund 7.000 – 8.000 Neudiagnosen in Deutschland [1]. Kennzeichnend für das MM ist die Ansammlung von Mutationen, die zunehmend zu Therapieresistenzen und häufgen Rezidiven führten. Daher ist das Überleben der Patienten zwangsläufg dadurch limitiert, dass es ab einem gewissen Punkt keine wirksamen Therapieoptionen mehr gibt. Eine wichtige Erweiterung des Therapiespektrums für stark vorbehandelte Patienten mit rezidiviertem und refraktärem multiplem Myelom (RRMM) ist der subkutan zu verabreichende Antikörper Elranatamab* (Elrexfo®, 40 mg/ml Injektionslösung). Er bindet bispezifsch an das B-Zell-Reifungsantigen (BCMA) auf den Myelomzel-
* Elranatamab ist zugelassen als Monotherapie zur Behandlung erwachsener Patienten mit rezidiviertem und refraktärem multiplem Myelom, die zuvor bereits mindestens 3 Therapien erhalten haben, darunter einen immunmodulatorischen Wirkstoff, einen Proteasom-Inhibitor und einen Anti-CD38Antikörper, und die während der letzten Therapie eine Krankheitsprogression gezeigt haben [2].
len sowie an den CD3-Rezeptoren auf den T-Zellen, wodurch die T-Zellen aktiviert werden, proinfammatorische Zytokine freisetzen und so die Lyse der Myelomzellen zu bewirken [2].
Tiefes und dauerhaftes Ansprechen
Basis für die im Dezember 2023 erteilte Zulassung sind die Ergebnisse für die Kohorte A der PhaseII-Studie MagnetisMM-3, die ein deutliches Ansprechen bei stark vorbehandelten (mit mindestens 3 Therapien, darunter ein immunmodulatorischer Wirkstoff, ein Proteasom-Inhibitor und ein AntiCD38-Antikörper) Patienten zeigte [3]. Die 123 eingeschlossenen Patienten erhielten Elranatamab als erste BCMA-gerichtete Therapie. Die Analyse der Daten ergab eine objektive Ansprechrate von 61 % und eine Wahrscheinlichkeit von ca. 72 % für die Aufrechterhaltung des Ansprechens über einen Zeitraum von 14,7 Monaten. Neuere Daten (Data-Cut-off: September 2023) bestätigen die Ergebnisse und zeigen ebenfalls eine Ansprechrate von 61 % [4]. Aus dieser Analyse ergab sich außerdem ein medianes progressionsfreies Überleben von 17,2 Monaten [4].
Darüber hinaus lässt sich aus den Studienergebnissen ableiten, dass nach 24 Wochen mit wöchentlicher Elranatamab-Behandlung die Medikation bei Respondern auf eine zweiwöchentliche Gabe reduziert werden kann. Die Patienten müssen somit seltener in die Klinik bzw. Praxis kommen und können von einer möglicherweise verlängerten Langzeitverträglichkeit der Therapie proftieren. Bei 80 % der Patienten, die nach mindestens 6 Monaten vor Daten-Cut-Off auf eine zweiwöchentliche Gabe umgestellt wurden (n = 50), blieb das Ansprechen nach der Umstellung aufrechterhalten oder verbesserte sich sogar: 38 % erreichten nach der Umstellung eine Komplettremission oder ein noch besseres Ergebnis [4].
Die häufgste Nebenwirkung von Elranatamab war mit 58 %das Zytokinfreisetzungssyndrom (CRS), gefolgt von Anämie (54 %), Neutropenie (45 %), Fatigue (44 %), Infektion der oberen Atemwege (39%) und Pneumonie (37%).
Patienten und Ärzte profitieren von der einfachen Anwendung
Neben den überzeugenden Wirksamkeitsdaten ein zusätzlicher Vorteil ist die einfache Anwendung von Elranatamab, der einen Einsatz
einer breiten Patientenpopulation erlaubt. Denn der bispezifsche Antikörper wurde als gebrauchsfertige Immuntherapie konzipiert, die ohne Hospitalisierung auch ambulant erfolgen kann. Elranatamab wird subkutan in fxer gewichtsunabhängiger Dosis injiziert. Nach einer Step-up-Dosierung er (12 mg an Tag 1 und 32 mg an Tag 4) wird die vollständige Behandlungsdosis von 76 mg ab der 2. Behandlungswoche einmal wöchentlich appliziert. Bei Respondern kann die Dosierung nach Woche 24 auf eine zweiwöchentliche Gabe reduziert werden [2]. Dies bedeutet nicht nur eine verbesserte Lebensqualität für die Patienten, sondern auch eine deutliche Erleichterung der Therapie in der klinischen Praxis. Da in den höheren Therapielinien komplexe Therapie- und Kombinationsoptionen dominieren, kann die Monotherapie mit Elranatamab dazu beitragen, das Behandlungskonzept beim RRMM zu vereinfachen.
Brigitte Söllner, Erlangen
Budesonid-Zäpfchen vereinfacht die antientzündliche rektale Therapie bei Proctitis ulcerosa
Eine Colitis ulcerosa, bei der sich die Entzündung nur auf die letzten 10 – 15 cm des Enddarms beschränkt, wird als Proctitis ulcerosa bezeichnet. Die Patienten leiden außer an analen Blutungen und Schmerzen vor allem unter dem imperativem Stuhldrang, häufg begleitet von Dranginkontinenz. Da die Entzündung auf den zugänglichen Teil der Darmschleimhaut begrenzt ist, ist hier auch eine topische Therapie möglich. Allerdings haben die Patienten mit bisher verfügbaren topischen Behandlungsoptionen wie Klysmen und Rektalschäumen oft Anwendungsprobleme – ein Grund, warum in der täglichen Praxis rektale Therapien zu wenig genutzt werden.
Eine wirksame Alternative sind Budenofalk® 4 mg Zäpfchen, die ersten und einzigen Suppositorien mit dem topisch wirksamen Kortikosteroid Budesonid. Indiziert sind sie zur rektalen Anwendung bei leicht bis moderat ausgeprägter Proctitis ulcerosa.
Hinsichtlich Wirksamkeit und Sicherheit mit Rektalschaum vergleichbar
mit leichter bis moderater Proctitis ulcerosa. Sie erhielten morgens und abends jeweils ein Zäpfchen und den Rektalschaum, ohne zu wissen, in welchem der Präparate der Wirkstoff und in welchem Placebo enthalten war.
Nach 8 Wochen hatten 75,1 % der Patienten unter BUS 4 mg und 70,3 % der Patienten unter BUF 2 mg eine klinische Remission und 76,2 % bzw. 75,9 % der Patienten eine Mukosaheilung erlangt. Auch bei den sekundären Studienendpunkten, dem Erreichen einer tiefen klinischen Remission bzw. tiefen mukosalen Heilung, bestätigte sich die Nichtunterlegenheit. Außerdem führte die Behandlung mit BUS 4 mg bei 70,7 % der Patienten mit zuvor erfolgloser Mesalazin-Therapie zur klinischen Remission.
Auch die Sicherheitsprofle beider Präparate erwiesen sich als vergleichbar. Die morgendlichen Kortisolspiegel im Serum lagen trotz Verringerung zu Beginn der Studie stets im Normalbereich. Es wurde keine klinisch signifkante oder anhaltende Suppression der endogenen Kortisolproduktion nach Abschluss der Behandlung festgestellt.
Mehrzahl präferiert Zäpfchen
Literatur
1 Deutsches Krebsforschungszentrum DKFZ: Multiples Myleom (Knochenmarkkrebs). https://www.krebsinformationsdienst.de/tumorarten/mulitplesmyelom/ index.php
2 Fachinformation Elreexfio®, aktueller Stand
3 Lesokhin AM et al. Nat Med 2023;29: 2259-2267
4 Tomasson MH et al. 65th ASH 2023, Poster 3385
In einer randomisierten, aktiv kontrollierten Nichtunterlegenheitsstudie mit einem doppelblinden Double-Dummy-Design* wurden die Sicherheit und Wirksamkeit der Budenofalk® 4 mg Zäpfchen (BUS 4 mg) und des Budenofalk® 2 mg Rektalschaums (BUF 2 mg) miteinander verglichen. Eingeschlossen wurden 577 Patienten
* Kruis W et al. J Crohn’s Colitis 2022; 16:1714-1724
Nach der Präferenz gefragt, sprachen sich in der Studie 46,9 % der Studienteilnehmer für das Zäpfchen und 22,1 % für den Rektalschaum aus. Ihnen war außerdem wichtig, ab wann sie mit einer spürbaren Besserung rechnen können. Mit dem Verschwinden der Symptome ist im Mittel innerhalb von etwa 40 Tagen zu rechnen – eine wichtige Information mit Blick auf die erforderliche Therapieadhärenz.
F. S.
Daridorexant (Quviviq™) ist seit einem Jahr in Deutschland zur Behandlung der chronischen Insomnie von Erwachsenen mit Schlafstörungen (Insomnie) verfügbar, deren Symptome seit mindestens 3 Monaten anhalten und eine beträchtliche Auswirkung auf die Tagesaktivität haben [1]. Der erste in Europa zugelassene duale Orexin-RezeptorAntagonist kann als derzeit einziges Arzneimittel bei chronischer Insomnie ohne zeitliche Einschränkung zu Lasten der Gesetzlichen Krankenkassen verordnet werden und ist damit für die Langzeitanwendung geeignet. Diese sollte jedoch so kurz wie möglich sein und innerhalb der ersten 3 Monate sowie anschließend in regelmäßigen Abständen neu bewertet werden. Basis für diese Ausnahmeregelung ist die einstimmige Entscheidung des Gemeinsamen Bundesausschusses (G-BA), die Anlage III AM-RL* am 11. November 2023 für Daridorexant zu öffnen [2]. Maßgeblich dafür waren die fehlenden Abhängigkeitsanzeichen selbst bei längerer Einnahme und das fehlende Risiko einer Medikalisierung [2].
Chronische Insomnie und ihre langfristigen Folgen
Bei einer chronischen Insomnie leiden die Betroffenen unter Einund/oder Durchschlafstörungen, die 3 oder mehr Nächte in der Woche auftreten und mindestens 3 Monaten anhalten. Dies kann zu klinisch relevanten Problemen
* Die Anlage III der Arzneimittel-Richtlinie beinhaltet eine Übersicht aller bereits bestehenden Verordnungseinschränkungen und -ausschlüsse in der Arzneimittelversorgung von verschreibungspflichtigen Schlafmitteln.
oder Beeinträchtigungen der Tagesaktivität mit Symptomen wie Schläfrigkeit, Konzentrations-, Aufmerksamkeits- und Gedächtnisschwierigkeiten sowie zu Stimmungsschwankungen führen und im langfristigen Verlauf sogar ernsthafte Folgeerkrankungen nach sich ziehen. So erhöht sich beispielweise die Wahrscheinlichkeit, an einer kardiovaskulären Erkrankung wie Bluthochdruck, Herzinfarkt und Herzversagen zu erkranken. Zudem stellt die chronische Insomnie einen Risikofaktor für Diabetes mellitus Typ 2 dar und begünstigt das Auftreten späterer psychiatrischer sowie neurologischer Erkrankungen [3]. Insbesondere bei Patienten, die eine längere Therapie ihrer Schlafstörungen benötigen, bestand bislang ein hoher ungedeckter medizinischer Bedarf. Da die Einnahmedauer von Schlafmitteln wie z.B. Z-Substanzen und Benzodiazepinen aufgrund des potenziellen körperlichen Abhängigkeitsrisikos auf 28 Tage beschränkt war, wurden die Patienten oft mit der Verordnung von Antidepressiva und allgemeinen Tipps zur Schlafhygiene „vertröstet“. Mit Daridorexant steht nun ein innovativer dualer Orexin-Rezeptor-Antagonist zur Verfügung, der im Gegensatz zu anderen verschreibungspfichtigen Medikamenten auch zur Langzeittherapie der chronischen Insomnie eingesetzt werden kann.
Vorteile des dualen OrexinRezeptor-Antagonisten
Daridorexant verfügt über einen anderen Wirkmechanismus als die bisherigen verschreibungspfichtigen Arzneimittel zur Behandlung der chronischen Insomnie. Der duale Orexin-Rezeptor-Antagonist (DORA) blockiert die OrexinRezeptoren und verhindert so die Weiterleitung von Wachheitssignalen durch Orexin (vgl. Insert). Dies kann das Einschlafen erleichtern und das Durchschlafen ermöglichen, ohne das Verhältnis der natürlichen Schlafphasen zu verändern [4, 5, 6]. Daridorexant setzt damit bei der pathologisch erhöhten nächtlichen Wachheit (Hyperarousal) als mögliche Ursache der chronischen Insomnie an [4, 7, 8]. Der Orexin-Rezeptor-blockierende Mechanismus reduziert die nächtliche Übererregung, vermindert Weckreaktionen und unterstützt den Organismus darin, seinen eigenen, gesunden Schlaf zu generieren. Im Gegensatz zu Benzodiazepinen und Z-Substanzen fanden sich in klinischen Studien mit Daridorexant auch nach 12-monatiger Einnahme keine Hinweise auf die Entwicklung einer körperlichen Abhängigkeit. Die Patienten zeigten zudem keine Anzeichen von Entzugserscheinungen, Toleranzentwicklung gegenüber der eingenommenen Dosierung, Rebound oder morgendliche Residualeffekte [9].
Daridorexant (Quviviq™) besitzt einen grundlegend anderen Wirkmechanismus als die bislang verfügbaren verschreibungspflichtigen Medikamente, die bei Schlafstörungen eingesetzt werden: Es blockiert die Orexin-Rezeptoren und verhindert somit die Weiterleitung der Wachheitssignale durch Orexin. Anders als bei anderen Schlafmitteln tritt nach der Einnahme keine Sedierung ein. Die unterschiedlichen Schlafphasen bleiben somit erhalten. Dies ist besonders relevant, da das Durchlaufen der unterschiedlichen Schlafphasen essenziell für einen erholsamen Schlaf ist [10]. Damit setzt Daridorexant bei der pathologisch erhöhten Wachheit (Hyperarousal) als mögliche Ursache der chronischen Insomnie an [1]. Der duale Orexin-Rezeptor-Antagonist wird aufgrund seiner pharmakokinetischen Eigenschaften rasch resorbiert und hat eine optimierte Halbwertszeit von etwa 8 Stunden und damit eine Wirksamkeit für die Dauer eines regulären Schlaffensters. Die orale Einnahme erfolgt idealerweise 8 – 9 Stunden vor der gewünschten Weckzeit. Es wird empfohlen, nach der Einnahme von Daridorexant etwa 9 Stunden bis zum Führen von Fahrzeugen oder das Bedienen von Maschinen zu warten [1].
Wachheits- und Schlafsignale werden durch komplizierte neuronale Schaltkreise im Gehirn gesteuert. Eine Schlüsselkomponente dieses Prozesses ist das Orexin-System, das die Wachheit fördert. Dieses System besteht aus den beiden Orexin-Neuropeptiden Orexin A und Orexin B (kleine proteinähnliche Moleküle, über die Nervenzellen miteinander im Gehirn kommunizieren) sowie den dazugehörigen Rezeptoren OX1R und OX2R. Das Orexin-System stimuliert gezielt Neuronen im Wachsystem und führt zur Freisetzung verschiedener wachheitsfördernder Botenstoffe (Serotonin, Histamin, Acetylcholin, Norepinephrin). Unter normalen Umständen steigt der Orexinspiegel im Laufe des Tages an, um die Wachheit zu unterstützen, und fällt zur Nacht wieder ab, um Schlaf zu ermöglichen. Die Überaktivität des Wachheitssystems kann ein Auslöser der Insomnie sein [4, 11].
Besseres Ein- und Durchschlafen, längere Schlafdauer und geringere Tagesschläfrigkeit
Relevant für die Zulassung von Daridorexant waren die Ergebnisse zweier dreimonatiger placebokontrollierter Phase-III-Studien mit identischem Design [4] sowie einer doppelblinden 40-wöchigen Verlängerungsstudie [9]. In das Studienprogramm wurden insgesamt 1.854 Patienten mit chronischer Insomnie aufgenommen. Da Insomnie häufg erst im fortgeschrittenen Lebensalter auftritt und ältere Erwachsene anfälliger für fragmentierten Schlaf, frühmorgendliches Erwachen und Tagesmüdigkeit sind, waren rund 40 % der rekrutierten Population mindestens 65 Jahre alt [4]. Primäre Endpunkte waren die Einschlafatenz, gemessen mittels Polysomnographie in Monat 1 und 3, und das Durchschlafen, sekundäre Endpunkte waren die Schlafdauer sowie die Tagesaktivität, gemessen mittels der Schläfrigkeits-Symptomskala des Insomnia Daytime Symptoms and Impacts Questionnaire Fragebogens (IDSIQ©) [4]. Daridorexant zeigte in der Dosierung 50 mg die höchste Wirksamkeit mit einer Verbesserung sowohl der nächtlichen Symptome als auch der Tagesaktivität: Die Therapie mit Quviviq™ 50 mg verkürzte signifkant die Einschlafzeit (Abb. 1) und verbesserte signifkant das Durchschlafen (Abb. 2). Mit Quviviq™ 50 mg schliefen die Patienten im 3. Monat jede Nacht etwa 1 Stunde länger (insgesamt 6,5 Stunden Schlaf), was im Vergleich zum Ausgangswert rechnerisch einer zusätzlichen Nacht Schlaf pro Woche entspricht. Außerdem fühlten sich die Patienten im 3. Monat tagsüber weniger müde, weniger schläfrig und energiegeladener als zu Studienbeginn – die
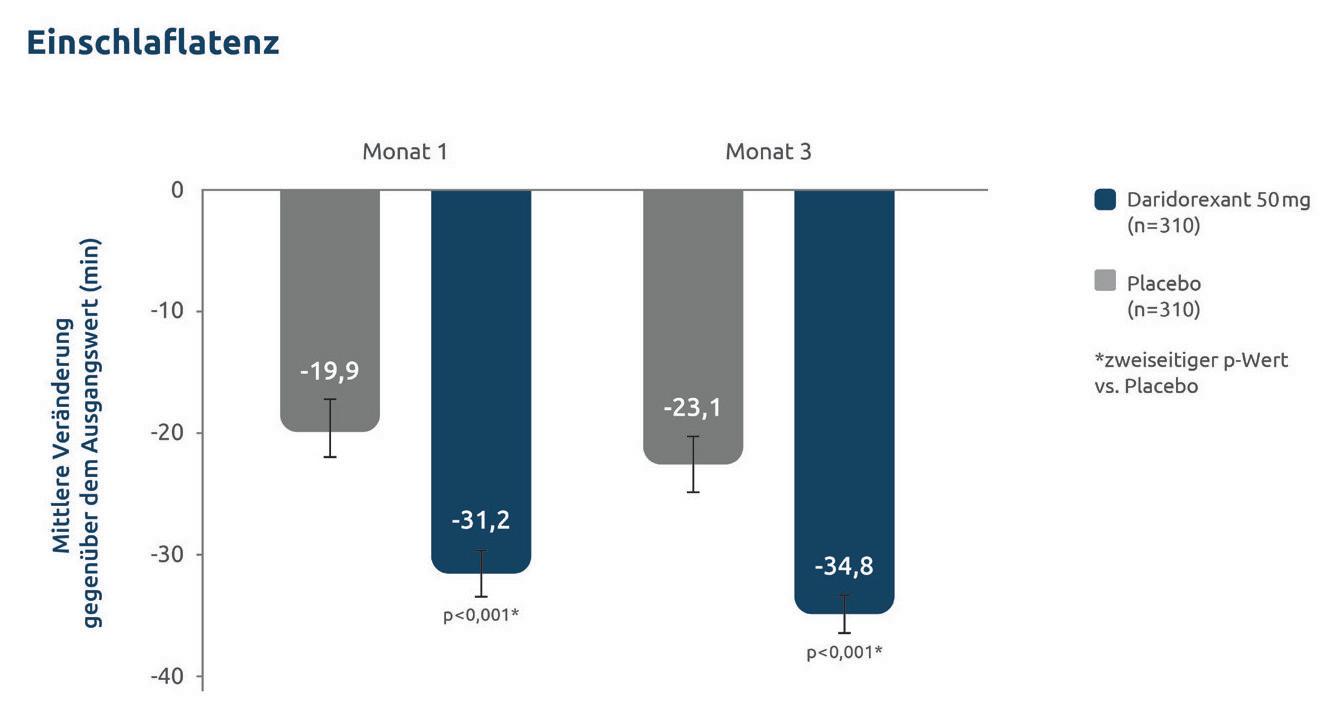
Abbildung 1: Schon nach einem Monat führte die Therapie mit Daridorexant (Quviviq™ 50 mg) zu einer signifkanten Verkürzung der Einschlafzeit um 30 Minuten im Vergleich zum Ausgangswert (mod. nach [4]).
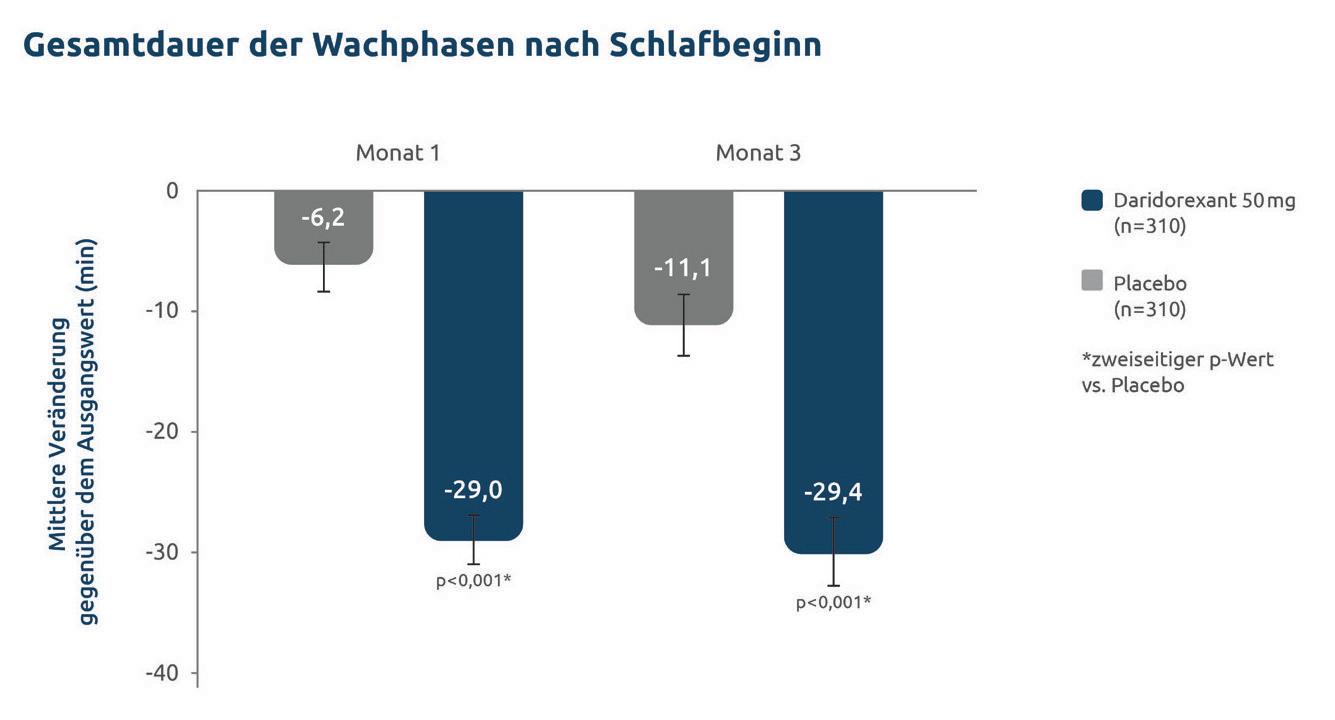
Abbildung 2: In Monat 1 und 3 kam es unter der Therapie mit Daridorexant (Quviviq™ 50 mg) zu einer signifkante Reduktion der Wachzeiten nach Schlafbeginn um 30 Minuten im Vergleich zum Ausgangswert (mod. nach [4]).
Auswertungen des IDSIQ zeigten über ein Jahr stabile Verbesserungen in allen Punkten [4].
Der durch Daridorexant verbesserte Schlaf blieb auch in der 40-wöchigen Verlängerungsstudie, an der 804 Patienten aus den beiden 3-monatigen Studien teilnahmen, stabil – es gab keine Adaption, keinen Wirkungsverlust und keine Rebound-Insomnie [9]. Insgesamt besticht Daridorexant durch ein hervorragendes Sicherheits- und Verträglichkeitsprofl.
Die Rate an unerwünschten Ereignissen lag in den beiden Studien auf Placeboniveau. Die häufgsten unerwünschten Ereignisse waren Kopfschmerzen und Nasopharyngitis [1].
Für die Praxis von Bedeutung ist vor allem, dass die Studienteilnehmer keine Anzeichen von Entzugserscheinungen, Toleranzentwicklung gegenüber der eingenommenen Dosierung, eines Rebounds oder morgendliche Residualeffekte zeigten [9].
In den Zulassungsstudien von Daridorexant wurden selbst bei täglicher Anwendung über 12 Monate keine Anzeichen für eine körperliche Abhängigkeit nach dem Absetzen der Behandlung beobachtet. Dies macht den dualen Orexin-Rezeptor-Antagonisten zu einem wertvollen, innovativen Arzneimittel zur Behandlung der Insomnie, da er im Gegensatz zu anderen verschreibungspfichtigen Medikamenten ohne Einschränkungen verordnet und auch zur Langzeittherapie der chronischen Insomnie eingesetzt werden kann.
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Quviviq™; Stand: Juni 2023
2 www.g-ba.de/downloads/39-261-6139/ 2023-08-17_AM-RL-III_Nr32- Daridorexant.pdf
3 Riemann D et al. S-3 Leitlinie Nicht erholsamer Schlaf/Schlafstörungen. AWMF Registriernummer 063/003, Update 2016, Version 2.0. Somnologie 2017 (Suppl s2):S97-S180
4 Mignot E et al. Lancet Neurol 2022; 21:125-139
5 Riemann D et al. Somnologie 2017;21:244
6 Roch C et al. Psychopharmacology 2021; 238:2693-2708
7 Janto K et al. J Clin Sleep Med 2018; 14:1399-1408
8 Clifford BS et al. Trends Neurosci 2001; 24:726-731
9 Kunz D et al. CNS Drugs 2023; 37:93106
10 Brinkman JE et al. Physiology of Sleep [Updated 2023 Apr 3]. In: StatsPearls. Treasure Island, FL: StatsPearls Publishing; Januar 2022. https://www.ncbi. nlm.nih.gov/books/NBK482512/2023
11 Levenson JC et al. Chest 2015;147:1179
RSV-Impfstoff Arexvy bietet älteren Menschen mit kardiorespiratorischen Grunderkrankungen besonders hohen Schutz
Das Respiratorische Synzytial-Virus (RSV) ist ein weit verbreitetes, ansteckendes Atemwegsvirus, das eine erhebliche Gefahr für ältere Erwachsene darstellt. Denn sie weisen bei einer RSV-Infektion aufgrund ihres nachlassenden Immunsystems und vermehrt auftretenden Grunderkrankungen wie COPD, Diabetes oder chronische Herz- und Lungenerkrankungen ein erhöhtes Risiko für einen schweren Verlauf mit Hospitalisierung und Tod auf. Ihr Risiko, bei einer stationären Behandlung aufgrund von RSV eine Exazerbation zu erleiden, ist hoch: Laut einer US-Studie beträgt es bei Patienten mit kongestiver Herzinsuffzienz 38 %, bei COPD 80 % und bei Asthma 50 % – entsprechend hoch ist auch die Komplikations- und Mortalitätsrate.
Wie Experten auf einem von GSK veranstalteten Symposium anlässlich des Kongresses der European Respiratory Society (ERS) in Mailand betonten, können gerade die älteren Patienten mit Vorerkrankungen besonders vom neuen RSV-Impfstoff Arexvy proftieren, der seit dem 1. August 2023 in Deutschland zur aktiven Immunisierung älterer Erwachsener (ab 60 Jahren) zur Prävention von RSV-bedingten Erkrankungen der unteren Atemwege zur Verfügung steht.
Hoch wirksame Prävention durch die aktive Immunisierung
Arexvy hat in der placebokontrollierten Zulassungsstudie AReSVi-006 eine hohe Wirksamkeit bewiesen: Der Impfstoff reduzierte die Rate an RSV-bedingten Erkrankungen der unteren Atemwege bei Erwachsenen im Alter von 60 Jahren oder älter in der ersten RSV-Saison statistisch signifkant um 82,6 % gegenüber einer Placebo-Impfung. Bei älteren Erwachsenen mit mindestens einer relevanten Grunderkrankung* lag die Wirksamkeit bei 94,6 % gegenüber Placebo.
Eine auf dem ERS-Kongress erstmals von Professor Alberto Papi, Ferrara/Italien, präsentierte Subgruppenanalyse der Zulassungsstudie AResVi-006 von Studienteilnehmern mit kardiorespiratorischen Grunderkrankungen zeigte die Wirksamkeit von Arexvy für diese Patientenpopulation mit einem höheren Risiko für schwere RSV-Erkrankungen (etwa 20 % der Studienteilnehmer): Der Impfstoff konnte mit einer Wirksamkeit von 92,1 % gegenüber Placebo durch RSV bedingte Erkrankungen der unteren Atemwege verhindern. Auch der Schutz vor einer durch RSV verursachten akuten respiratorischen Insuffzienz war mit 88,1 % vergleichsweise hoch. Diese besonders vulnerablen vorerkrankten Patienten proftierten somit besonders deutlich von einer Immunisierung mit dem RSVImpfstoff. Wie Papi hervorhob, erwies sich die Impfung mit Arexvy bei diesen Studienteilnehmern als gut verträglich.
Mit Therapie und Prävention COPD-Exazerbationen vermindern
Wie bereits der GOLD-Report 2023 betont auch der GOLD-Report 2024, wie wichtig die genaue Betrachtung des Exazerbationsrisikos bei COPD ist, da Exazerbationen einen großen Einfuss auf die Morbidität und Mortalität von COPD-Patienten haben. Gerade in dieser Population zeigt sich der Nutzen einer RSV-Prävention mit Arexvy: Denn für Patienten mit COPD kann die neue Impfung einen wichtigen Baustein zur Prävention bzw. Verminderung von Exazerbationen darstellen. Angesichts des erhöhten Hospitalisierungsrisikos bei einer RSV-Infektion ist es gerade für diese Gruppe bedeutsam, wenn Erkrankungen der unteren Atemwege vermieden werden können.
Die Risikoeinschätzung sollte sich in Zukunft auf objektive Kriterien stützen, wie das Ausmaß der Atemnot sowie die Atem- und Herzfrequenz. Das Thema Exazerbation stellt neben der Dyspnoe eines der neu defnierten „behandelbaren Charakteristika“ in der COPDTherapie dar, erläuterten die Referenten beim ERS-Symposium zum omnipräsenten Themenblock „Treatable Traits“. Sie zogen übereinstimmend folgendes Fazit: Zur Verminderung von COPD-Exazerbationen ist ein ganzheitlicher, multidisziplinärer ManagementAnsatz empfehlenswert: sowohl therapeutisch mit einer LAMA/ LABA/ICS-Triple-Therapie (z.B. Trelegy Ellipta) als auch präventiv mit einer geeigneten Impfstrategie – dazu gehören auch die GOLDImpfempfehlungen gegen Infuenza, Pneumokokken und COVID-19.
* COPD, Asthma, jede chronische Atemwegs-/Lungenerkrankung, Diabetes Typ 1 oder Typ 2, kongestive Herzinsuffizienz, fortgeschrittene Leber- oder Nierenerkrankungen
Brigitte Söllner, Erlangen
Eligard® EPSS und Tivozanib – zwei neue Therapieoptionen in der Urologie
Im Rahmen eines von Recordati veranstalteten Pressegesprächs auf dem Kongress der Deutschen Gesellschaft für Urologie e.V. (DGU) wurden zwei Innovationen vorgestellt, von denen Patienten proftieren können, die an einem Prostata- oder Nierenzellkarzinom leiden: das Eligard® Pre-connected Syringe System (EPSS) und der VEGFR-Tyrosinkinaseinhibitor Tivozanib.
Einfachere Handhabung mit dem neuen Eligard® Pre-connected Syringe System
Seit über 20 Jahren bietet Eligard® mit seiner einzigartigen Gel-Depotformulierung (Atrigel®) den Vorteil einer konstanten Freisetzung des Wirkstoffs Leuprorelinacetat über 1, 3 und 6 Monate, entsprechend den verfügbaren Dosierungen 7,5 mg, 22,5 mg und 45 mg und ermöglicht damit eine zuverlässig konstante Testosteronsenkung beim hormonabhängigen, fortgeschrittenen und metastasierten Prostatakarzinom. In den Zulassungsstudien erreichten bis zu 97,5 % der Patienten eine anhaltende wirksame Testosteronsenkung von ≤20 ng/dl – dem anzustrebenden Kastrationsgrenzwert nach der aktuellen Empfehlung der European Association of Urology. Die mittlere Testosteronkonzentration lag mit Eligard® am Ende der Untersuchung bei 6,1 (±0,4) ng/dl beim 1-Monatsdepot, bei 10,1 (±0,7) ng/dl beim 3-Monatsdepot und bei 12,6 (±2,1) ng/ dl beim 6-Monatsdepot. 91 % der mit Eligard® behandelten Patienten
erreichten im Schnitt sogar Testosteronwerte <5 ng/dl. Bei weniger als 1 % der Patienten traten dabei Testosterondurchbrüche auf. „Diese stabile und langanhaltend tiefe Testosteronsenkung ist bei der Behandlung des Prostatakarzinoms besonders wichtig“, betonte Professor Carsten Ohlmann, Bonn. „Schließlich ist sie mit einem niedrigeren Progressions- wie auch Sterberisiko sowie einer verbesserten Dauer des Therapieansprechens verbunden.“
Damit einher geht eine tiefe und langanhaltende Senkung des Prostata-spezifschen Antigens (PSA). Der PSA-Wert sank im Schnitt um 96 % und 95 % der Patienten erreichten bis Studienende einen PSA-Wert von <4 ng/ml.
„Die Effektivität von Eligard® ist dabei unabhängig vom Alter und Körpergewicht der Patienten, was die Anwendung zusätzlich erleichtert“, berichtete Ohlmann. Dies macht Eligard® zu einer wichtigen Option für diese Patientengruppen und bietet gleichermaßen Flexibilität durch die 3 unterschiedlichen Monatsdepots.
„Basierend auf diesen klinischen Erkenntnissen kann Eligard® als die bevorzugte ADT bei fortgeschrittenem Prostatakrebs angesehen werden“, fasste Ohlmann die Daten zusammen und ergänzte: „Mit dem innovativen Eligard® Pre-connected Syringe System (EPSS) ist die Vorbereitung der subkutanen Injektion nun noch einfacher, intuitiver und anwenderfreundlicher.“
Das EPSS besteht aus zwei miteinander vorverbundenen Spritzen. Darin befndet sich zum einen der Wirkstoff Leuprorelinacetat, zum anderen die Depotformulierung (Atrigel®). Durch eine Verriegelung sind die Inhaltsstoffe während der Aufbewahrung voneinander
getrennt, vor der Anwendung kann diese durch ein einfaches Klicken gelöst werden. Wiederholtes Drücken des Inhalts von der einen in die andere Spritze vermischt die Inhaltstoffe miteinander, sodass nach 60 Zyklen eine homogene, viskose Lösung zur Injektion entsteht.
„Das neue vorverbundene Spritzensystem wird den Praxisalltag deutlich erleichtern“, begrüßte Ohlmann die Neuerung. „So wurde Bewährtes noch besser gemacht, denn selbstverständlich bietet Eligard® weiterhin eine therapeutische Flexibilität mit 3 Dosierungsmöglichkeiten bei gleichzeitig hoher Zuverlässigkeit.“ Eligard® könne daher als die Androgendeprivationstherapie (ADT) der Wahl beim fortgeschrittenen Prostatakarzinom betrachtet werden.
Tivozanib: Überlegene Therapieoption bei mRCC „Good-Risk“-Patienten
Wie Professor Christian Doehn, Lübeck, ausführte, gibt die aktuelle S3-Leitlinie beim fortgeschrittenen und/oder metastasierten Nierenzellkarzinom (mRCC) eine starke Empfehlung für die Monotherapie mit einem VEGFRTyrosinkinaseinhibitor (TKI), wie z.B. Tivozanib (Fotivda®), bei Patienten mit einem günstigen Risikoprofl („Good-Risk“-Patienten; gemäß IMDC-Score), wenn eine Kombinationstherapie mit einem Immuncheckpoint-Inhibitor (ICI) ausgeschlossen ist. Studiendaten weisen zudem darauf hin, dass die Monotherapie mit einem VEGFRTKI der Kombinationstherapie bei dieser Patientenpopulation vorzuziehen ist.
Laut einer aktuellen Metaanalyse bietet die ICI-Kombinationsthera-
pie keinen Überlebensvorteil gegenüber der alleinigen Gabe eines VEGFR-TKI (HR: 1,24; 95%-KI: 0,86 – 1,78). „Im Vergleich zur ICIKombinationstherapie ist die Monotherapie mit einem VEGFR-TKI jedoch deutlich verträglicher, was im Hinblick auf das in der Regel häufg ältere und komorbide Patientenklientel ein wichtiger Aspekt ist“, erläuterte Doehn. Verglichen mit anderen VEGFRTKI hat die Therapie mit Tivozanib wesentliche Vorteile. Als VEGFRTKI der dritten Generation besitzt der Inhibitor besondere pharmakologische Eigenschaften. So zeigt Tivozanib unter allen VEGFR-TKI die höchste Bindungsselektivität zu den VEGFR-1 bis -3. Hierdurch werden unerwünschte Off-TargetEffekte mit anderen zellulären Kinasen oder Wechselwirkungen mit Begleitmedikationen minimiert. In Studien führte dies zu einer guten Wirksamkeit und Verträglichkeit der Behandlung.
Dass sich diese Ergebnisse auf die reale Situation im Therapiealltag übertragen lassen, verdeutlichte Doehn anhand von aktuellen Ergebnissen der prospektiven, nicht interventionellen Studie T-REX: „Die in Deutschland erhobenen Real-World-Daten ergaben konstant hohe Ansprechraten und ein sehr gutes Nebenwirkungsprofl der Therapie mit Tivozanib“, betonte der Experte. Dabei zeigte sich, dass etwa jeder vierte Patient im realen Setting ein „Good-Risk“-Patient ist. Speziell für diese Patientenpopulation stellt Tivozanib aufgrund seiner Wirksamkeits- und Verträglichkeitsvorteile laut Doehn eine wichtige Therapieoption in der Erstlinie dar.
Fabian Sandner, Nürnberg
Aktuelle Real-World-Daten unterstützen den Einsatz von Vedolizumab als Firstline-Biologikum bei Morbus Crohn
Anhand welcher Kriterien das „richtige“ Firstline-Biologikum zur Behandlung des Morbus Crohn (MC) ausgewählt werden kann, diskutierten Experten im Rahmen eines von Takeda unterstützten Symposiums anlässlich der „Viszeralmedizin 2023“ der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS).
Als „einen großen Strauß an Möglichkeiten“ bezeichnete Professor Torsten Kucharzik, Lüneburg, die modernen Therapieoptionen, die für die Behandlung des Morbus Crohn zur Verfügung stehen. Dazu zählen u.a. Anti-TNFα-Therapien (Adalimumab, Infiximab), der α4β7-Integrin-Antagonist Vedolizumab, der IL-12/23-Antagonist Ustekinumab, der IL-23-Antagonist Risankizumab sowie der JAKInhibitor Upadacitinib. Professor Carsten Büning, Berlin, ergänzte: „Die Vielfalt der Therapien macht es heute nicht einfach, sich für eine bestimmte Medikation zu entscheiden. Wir haben die sprichwörtliche Qual der Wahl.“ Zumal auch die aktuelle S3-Leitlinie zur MC-Therapie die zielgerichteten Optionen nicht wertet, sondern nur in alphabetischer Reihenfolge nennt.
VEDOIBD-Studie zeigt deutliche Vorteile für Vedolizumab gegenüber Anti-TNFα-Therapien
Bei der Auswahl einer bestimmten Therapieoption müssen laut Büning daher verschiedene Kriterien berücksichtigt werden, wie etwa die Erfahrungen der Behandler,
die Sicherheit der Therapie und die Präferenz der Patienten (z.B. i.v. oder s.c.). Wichtig für die Auswahl einer geeigneten Therapie sind natürlich auch Effektivität und Persistenz. Hier können RealWorld-Studien hilfreich sein, da sie die Behandlungsrealität abbilden. Aussagekräftige Daten zur Effektivität und Persistenz hat die groß angelegte prospektive RealWorld-Studie VEDOIBD erbracht. „In der VEDOIBD-Studie wurde über 2 Jahre der Einsatz von Vedolizumab (Entyvio®) und TNFαAntagonisten bei 1.200 Biologikanaiven und -erfahrenen Patienten mit MC oder Colitis ulcerosa untersucht“, berichtete Professor Timo Rath, Erlangen. Er stellte die 2-Jahres-Analyse der Biologikanaiven MC-Kohorte vor, die 327 Patienten umfasste, die neu mit Vedolizumab (n = 86) oder einer Anti-TNFα-Therapie (n = 241; 57,7 % Adalimumab, 42,3 % Infiximab) behandelt wurden. Primärer Endpunkt war die klinische Remission*. Außerdem wurden u.a. die steroidfreie Remission** und die Therapiepersistenz über 2 Jahre ermittelt. Um möglichst valide Ergebnisse zu generieren, erfolgte ein Propensity-Score-Matching. Analysiert wurde die mITT (modifzierte Intention-to-treat)-Population. Dabei wurden Patienten mit einem Wechsel der Behandlung als Therapieversager gewertet. Während nach 14 Wochen noch signifkant mehr Patienten eine klinische Remission unter TNFαAntagonisten im Vergleich zu
* Klinische Remission: Harvey-BradshawIndex (HBI) ≤4 am Ende der Induktionstherapie (Woche 14) sowie während der Erhaltungstherapie nach 1 und 2 Jahren.
** Steroidfreie Remission: HBI ≤4 und keine systemische Anwendung von Steroiden oder Budenosid oral bis zur vorherigen Visite.
Vedolizumab erreichten, drehte sich das Verhältnis nach 1 Jahr numerisch und nach 2 Jahren signifkant um: So waren nach 2 Jahren 64,2 % der Patienten unter Vedolizumab vs. 44,7 % unter TNFα-Therapien in klinischer Remission (p = 0,037). Auch bei der steroidfreien Remission schnitt nach 2 Jahren Vedolizumab signifkant besser ab (62,5 % vs. 41,6 %; p = 0,035). „Interessant ist auch, dass sich Vedolizumab bei MC als die Therapie mit der höheren Persistenz im Vergleich zu den Anti-TNFα-Therapien erwiesen hat, betonte Rath. 2 Jahre nach Behandlungsbeginn hatten nur 17 % in der Vedolizumab-Gruppe auf eine andere Therapie gewechselt, in der Anti-TNFα-Gruppe waren es 44 %. „Die deutlichen RealWorld-Ergebnisse aus VEDOIBD unterstützen somit den Einsatz von Vedolizumab als Firstline-Biologikum bei Morbus Crohn“, folgerte der Experte.
Firstline-Biologikum beeinflusst Folgebehandlungen
„Bei der Auswahl des ersten Biologikums trägt man eine besondere Verantwortung, denn man trifft eine Entscheidung, die den weiteren Verlauf beeinfussen kann“, konstatierte Büning. Und Rath ergänzte: „Wir wollen natürlich das effektivste Biologikum zuerst einsetzen. Vedolizumab hat als Firstline-Biologikum den Vorteil, dass es die Wirksamkeit einer Zweitlinien-Behandlung mit AntiTNFα-Therapien nicht negativ beeinfusst. Dies hat die retrospektive EVOLVE-Studie gezeigt. Daten dieser Studie belegen eine leichte numerisch bessere Effektivität von Vedolizumab als Firstline-Biologikum im Vergleich zu Firstline-
Anti-TNFα bei Biologika-naiven Patienten. Beim Einsatz als Zweitlinie – nach Versagen von AntiTNFα – war Vedolizumab dann jedoch nicht so wirksam wie in der Firstline. Bei Anti-TNFα war es tendenziell umgekehrt. Laut Bressler et al. könnte dies – auch aufgrund des guten Sicherheitsprofls – für den Einsatz von Vedolizumab als Firstline-Biologikum sprechen.
CDST können die Therapiewahl unterstützen
„Neben den genannten Kriterien könnten auch Clinical Decision Support Tools (CDST) bei der Therapieentscheidung hilfreich sein“, erklärte Kucharzik. Mit CDST-Algorithmen für Biologika bei der CED-Behandlung lässt sich die Wahrscheinlichkeit einer klinischen Remission unter einer bestimmten Therapieoption berechnen. Ein einfach anzuwendendes CDST für Vedolizumab ist als Medizinprodukt in Entwicklung. Kucharzik stellte die Entwicklung und Validierung der Prädiktoren für die Wahrscheinlichkeit eines Ansprechens auf die Vedolizumab-Therapie vor: Auf Basis der GEMINI-2-Daten (814 MCPatienten) wurden 5 Prädiktoren für das Erreichen einer steroidfreien, klinischen und dauerhaften Remission ermittelt und anhand des VICTORY-Konsortiums (336 MCPatienten) gewichtet und validiert: keine vorherige Darmoperation (+2 Punkte), keine vorherige AntiTNFα-Therapie (+3 Punkte), keine vorherige fstulierende Erkrankung (+2 Punkte), Albumin zu Baseline (+0,4 Punkte pro g/l) sowie C-reaktives Protein zu Baseline (–0,5 Punkte falls 3,0 – 10,0 mg/l; –3,0 Punkte falls >10 mg/l).
Patienten mit einem Wert von >19 Punkten haben eine hohe Wahrscheinlichkeit für ein Vedolizumab-Ansprechen. Sie zeigten nach 26 Wochen bessere klinische Ergebnisse, einen deutlich schnelleren Wirkeintritt und geringere Operationsraten nach 12 Monaten im Vergleich zu Patienten mit einer geringeren Punktzahl. Laut Kucharzik ist dieser CDST-Algorithmus spezifsch für Vedolizumab. In einer retrospektiven Studie mit 180 Anti-TNFα-erfahrenen MCPatienten wurde gezeigt, dass das Vedolizumab-CDST die klinische sowie die steroidfreie Remission nach 48 Wochen für Vedolizumab, aber nicht für Ustekinumab vorhersagte.
„CDST sind in erster Linie eine Hilfe für die Entscheidungsfndung, inwieweit eine Therapie für den Patient infrage kommt. Berücksichtigt man dann noch patientenindividuelle Präferenzen, z.B. für die Applikationsart, dann kann man das Profl einer Substanz schärfen“, so die abschließende Beurteilung von Rath.
Elisabeth Wilhelmi, München
STIKO empfiehlt bei Erwachsenen ausschließlich die Impfung mit dem 20-valenten PneumokokkenKonjugatimpfstoff Apexxnar®
Die Ständige Impfkommission (STIKO) hat ihre Empfehlung hinsichtlich der PneumokokkenImpfung aktualisiert und empfehlt ab sofort nur noch den 20-valenten Pneumokokken-Konjugatimpfstoff Apexxnar® (PCV20) für alle
Personen ab 60 Jahren sowie alle Erwachsenen ab 18 Jahren mit relevanten Grunderkrankungen (angeborene oder erworbene Immundefekte, chronische Krankheiten sowie anatomische und fremdkörperassoziierte Risiken für eine Pneumokokken-Meningitis) bzw. einem berufichen Risiko. Bislang wurde, abhängig von der Indikation, entweder die alleinige Impfung mit dem 23-valenten Polysaccharidimpfstoff (PPSV23) oder die sequenzielle Impfung mit PCV13, gefolgt von PPSV23 empfohlen. PCV20 ist nach der neusten Evaluation der STIKO diesen Impfstoffen überlegen – für die Pati-
Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
enten bedeutet das nicht nur einen verbesserten Schutz, sondern häufg auch einen geringerer Aufwand, da die sequenzielle Impfung entfällt.
Die STIKO begründet die Änderung der Empfehlung mit der breiten Serotypenabdeckung und der durch die Konjugattechnologie zu erwartenden besseren und länger anhaltenden Immunantwort. Bei Konjugatimpfstoffen ist das Kapselpolysaccharid an ein immunogenes Protein gebunden. Dadurch
wird, zusätzlich zu der durch das Polysaccharid ausgelösten B-ZellAntwort, eine T-Zell-abhängige Immunantwort ausgelöst. Durch diese wird u.a. die Freisetzung von B-Zell-aktivierenden Botenstoffen verstärkt, sodass die Bildung eines immunologischen Gedächtnisses ermöglicht wird.
Die bessere Immunogenität kann gerade bei älteren oder immunsupprimierten Menschen wichtig sein, da sowohl eine Immunschwäche als auch Veränderungen des Immunsystems im Alter zu einer abgeschwächten Impfantwort führen können.
Titelbild: Impfstoff gegen Krebs als onkologisches Behandlungskonzept mittels Immuntherapie mit Zellen aus dem menschlichen Körper als 3D-Illustration (Quelle: iStock).
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf
Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden im Journal für Pharmakologie und Therapie personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz:
HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM_W.R. Roland Welker Austraße 7, 96114 Hirschaid
Verlag PERFUSION GmbH
Storchenweg 20 90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de
E-Mail: perfusion@t-online.de
NEU:
Eligard® EPSS
Pre-connected Syringe System
Anwenderfreundlich – intuitv –einfach

Wirtschaflich – Partner ALLER Kassen mit öfentlichen Rabatverträgen für Leuprorelin*
Einzigartge Atrigel Galenik für eine kontrollierte und langanhaltende Freisetzung1,2
T-Senkung unter 20 ng/dL3-6 (= EAU-Empfehlung)
Referenzen: 1. Sartor O. Eur Urolw Suppl 2006; 5: 905-10. 2. Merseburger AS, Roesch MC. Expert Rev Antcancer Ther. 2022;22(7):703-715. doi:10.1080/14737140.2022.2082947. 3. Aktuelle Fachinformaton Eligard® 4. Perez-Marrero R, et al. Clin Ther 2002;24: 1902-14. 5. Chu FM, et al. J Urol 2002; 168: 1199-203. 6. Crawford ED, et al J Urol 2006; 175: 533-6. * Stand: Januar 2024.
Eligard® 7,5 mg / 22,5 mg / 45 mg Pulver und Lösungsmitel zur Herstellung einer Injektonslösung. Wirkstof: Leuprorelinacetat. Zusammensetzung: Eine vorgefüllte Spritze mit Pulver zur Herstellung einer Injektonslösung (Spritze B) enthält: Wirkstof: 7,5 mg Leuprorelinacetat (entsprechend 6,96 mg Leuprorelin) bzw. 22,5 mg Leuprorelinacetat (entsprechend 20,87 mg Leuprorelin) bzw. 45 mg Leuprorelinacetat (entsprechend 41,7 mg Leuprorelin). Eine vorgefüllte Spritze mit Lösungsmitel zur Herstellung einer Injektonslösung (Spritze A) enthält: Sonstge Bestandteile: Poly(glycolsäure-co-milchsäure), N-Methylpyrrolidon (Ph.Eur.). Anwendungsgebiete: Zur Behandlung des hormonabhängigen, fortgeschritenen Prostatakarzinoms und in Kombinaton mit Radiotherapie zur Behandlung von lokalisiertem Hochrisiko- und lokal fortgeschritenem hormonabhängigem Prostatakarzinom. Gegenanzeigen: Frauen und Kindern; bei Überempfndlichkeit gegen Leuprorelinacetat, andere GnRH-Agonisten oder einen der sonstgen Bestandteile; orchiektomierte Patenten als alleinige Behandlung bei Prostatakarzinom-Patenten mit Rückenmarkkompression oder Anzeichen von Metastasen im Rückenmark. Nebenwirkungen: Unerwünschte Reaktonen auf Eligard hängen hauptsächlich mit spezifschen pharmakologischen Wirkungen von Leuprorelinacetat zusammen (Ansteg und Absinken bestmmter Hormonspiegel). Am häufgsten wird über Hitzewallungen, Übelkeit, Unwohlsein und Müdigkeit, über vorübergehende lokale Reizung an der Injektonsstelle sowie über leichte bis mitelschwere Hitzewallungen berichtet. Sehr häufg: Hitzewallungen; Ekchymose, Erythem; Müdigkeit, Brennen an der Injektonsstelle, Parästhesie an der Injektonsstelle. Häufg: Nasopharyngits; Übelkeit, Diarrhö,Gastroenterits/Colits; Pruritus, Nachtschweiß; Arthralgie, Schmerzen in den Extremitäten, Myalgie, Rigor, Schwäche; seltenes Wasserlassen, Miktonsbeschwerden, Dysurie, Nykturie, Oligurie; Druckempfndlichkeit der Brust, Hodenatrophie, Hodenschmerzen, Unfruchtbarkeit, Brusthypertrophie, erektle Dysfunkton, reduzierte Penisgröße; Unwohlsein, Schmerzen an der Injektonsstelle, Bluterguss an der Injektonsstelle, Stechen an der Injektonsstelle; hämatologische Veränderungen, Anämie; Erhöhung der Kreatninphosphokinase im Blut, Verlängerung der Gerinnungszeit. Gelegentlich: Harnwegsinfekton, lokale Infekte der Haut; Verschlechterung eines Diabetes mellitus; abnorme Träume, Depression, Abnahme der Libido; Schwindel, Kopfschmerzen, Insomnie, Geschmacks- und Geruchsstörungen, verminderte Reizempfndung, Vertgo; Hypertonie, Hypotonie; Rhinorrhoe, Dyspnoe; Obstpaton, Mundtrockenheit, Dyspepsie, Erbrechen; feuchtkalte Haut, vermehrtes Schwitzen; Rückenschmerzen, Muskelkrämpfe; Spasmen der Harnblase, Hämaturie, erhöhte Harnfrequenz, Harnretenton; Gynäkomaste, Impotenz, Hodenerkrankung; Juckreiz an der Injektonsstelle, Verhärtung an der Injektonsstelle, Lethargie, Schmerzen, Fieber; Erhöhung der Alaninaminotransferase, Erhöhung der Blutriglyceride, Verlängerung der Prothrombinzeit, Gewichtszunahme. Selten: abnorme unwillkürliche Bewegungen; Synkope, Kollaps; Flatulenz, Aufstoßen; Alopezie, Hautausschlag; Schmerzen in der Brust; Ulzeraton an der Injektonsstelle. Sehr selten: Nekrose an der Injektonsstelle. Nicht bekannt: Idiopathische intrakranielle Hypertonie; QT-Verlängerung; Intersttelle Lungenerkrankung. Zu anderen unerwünschten Ereignissen, über die im Zusammenhang mit einer Leuprorelinacetat-Behandlung gewöhnlich berichtet wird, gehören periphere Ödeme, Lungenembolie, Palpitatonen, Myalgie, Muskelschwäche, veränderte Hautsensibilität, Schütelfrost, Hautausschlag, Amnesie und Sehstörungen. In dieser Präparategruppe wurde nach Langzeitanwendung eine Muskelatrophie beobachtet. In seltenen Fällen wurde nach Verabreichung von GnRH-Agonisten mit Kurz- oder Langzeitwirkung über einen Infarkt einer bereits bestehenden Hypophysenapoplexie berichtet. Über Thrombozytopenie und Leukopenie wurde in seltenen Fällen berichtet. Über Veränderungen der Glucosetoleranz liegen Berichte vor. Nach Verabreichung von GnRH-Analoga wurde über Konvulsionen berichtet. Die nach Injekton von Eligard berichteten unerwünschten Ereignisse am Verabreichungsort entsprechen den im Zusammenhang mit ähnlichen subkutan injizierten Präparaten beschriebenen unerwünschten Ereignissen. Im Allgemeinen werden diese lokal begrenzten unerwünschten Ereignisse nach s. c. Injektonen als leicht und kurzzeitg beschrieben. In seltenen Fällen wurde nach Verabreichung von GnRH-Analoga über anaphylaktsche/anaphylaktoide Reaktonen berichtet. Veränderungen der Knochendichte: Bei Männern wurde in der Fachliteratur über eine Verminderung der Knochendichte infolge einer Orchiektomie oder einer Behandlung mit GnRH-Analoga berichtet. Daher ist unter einer Langzeitherapie mit Leuprorelinacetat mit einer Verstärkung von Osteoporosesymptomen zu rechnen. Erhöhtes Frakturrisiko infolge einer Osteoporose. Verschlechterung der Zeichen und Symptome der Erkrankung: In den ersten Wochen einer Behandlung mit Leuprorelinacetat kann es zu einer Verschlechterung der Zeichen und Symptome der Erkrankung kommen. Wenn sich Erkrankungen wie Wirbelsäulenmetastasen und/oder Harnwegsobstrukton oder Hämaturie verschlechtern, können neurologische Probleme wie Schwäche und/oder Parästhesie der unteren Extremitäten oder Verschlechterung der Harnwegsymptome aufreten. Verschreibungspfichtg. Stand: September 2022. Pharmazeut. Unternehmer: Recordat Industria Chimica e Farmaceutca S.p.A., Via Mateo Civitali 1, 20148 Mailand (Italien). Mitvertrieb: Recordat Pharma GmbH, Eberhard-Finckh-Str. 55, 89075 Ulm (Deutschland).