JAHRGANG 34
HEFT 1
Februar 2025

JAHRGANG 34
HEFT 1
Februar 2025
Behandlung der akuten Pharyngitis mit GeloTonsil® Gurgelgel
Plaque-Psoriasis: Daten aus dem deutschen Behandlungsalltag belegen Verbesserung der Erkrankungslast durch Deucravacitinib
Refraktäres diffus großzelliges B-Zell-Lymphom: CAR-T-Zelltherapie mit Axi-Cel überzeugt als neuer Zweitlinienstandard
Daratumumab-VRd + DR-Erhaltung: Neue Erstlinientherapie für transplantationsgeeignete Myelom-Patienten
Secukinumab: Real-World-Daten bestätigen die über 5 Jahre anhaltende Wirksamkeit bei PsA und r-axSpA
Apalutamid beim mHSPC: Real-World-Daten untermauern die Ergebnisse der Zulassungsstudie
Multiple Sklerose: Aktuelle Daten bestätigen Sicherheit von Ocrelizumab in Schwangerschaft und Stillzeit

WIRKSAM in RCT 9 – 10,16,# und RWE 3 – 7,§
EINFACH im Praxisalltag 11,$,‡ auch bei Vorerkrankungen 8,11 – 15 VERTRÄGLICH
Mehr Informationen finden Sie unter www.ibrance.de PARSIFAL-LONG1 ist eine Follow-up-Studie mit 80,5 % der Patientinnen der internationalen, multizentrischen, prospektiven Phase-2-Studie PARSIFAL 2 und mit einer medianen Nachbeobachtungszeit von 59,7 Monaten. * Im IBRANCE® + FUL-Arm wurden de novo metastasierte Patientinnen eingeschlossen.1, 2 Die Anwendung von IBRANCE® + FUL ist nur nach vorheriger endokriner Therapie zugelassen.11 Bitte beachten Sie die IBRANCE® Fachinformation.
Referenzen: 1. Llombart-Cussac A et al. SABCS 2023; oral presentation RF01-03.; 2. Llombart-Cussac A et al. JAMA Oncol 2021;7(12):1791-1799.; 3. Porte B et al. Breast. 2020;54:303–310.; 4. DeMichele A et al. Breast Cancer Res. 2021;23(1):37.; 5. Brufsky A et al. Target Oncol. 2021;16(5):601–611.; 6. Mycock K et al. Future Oncol. 2022;18(3):349–362.; 7. Rugo HS et al. NPJ Breast Cancer. 2022;8(1):114.; 8. Harbeck N et al. Future Oncol. 2021;17(16):2107–22.; 9. Rugo HS et al. Breast Cancer Res Treat. 2019;174(3):719–729.; 10. Turner NC et al. N Engl J Med. 2018;379(20):1926–36.; 11. IBRANCE® Fachinformation, aktueller Stand.; 12. Roncato R et al. Int J Mol Sci. 2020;21(17):6350.; 13. Rugo HS et al. Ann Oncol. 2018;29(4):888–894.; 14. Harbeck N et al. Ann Oncol. 2016;27(6):1047–54.; 15. Gelmon K et al. Breast. 2021;59:321-326.; 16. Cristofanilli M. et al. Lancet Oncol. 2016 Apr;17(4):425-439. AI = Aromatase-Inhibitor; CDK4/6i = CDK4/6-Inhibitor; FUL = Fulvestrant; HR+/HER2- = Hormonrezeptor-positiv, humaner epidermaler Wachstumsfaktorrezeptor 2-negativ; mBC = metastasierter Brustkrebs; LET = Letrozol; RCT = randomisierte kontrollierte Studien; RWE = Real-World-Evidence; (rw)PFS = (Real-World) progressionsfreies Überleben § Signifikanter OS- und rwPFS-Vorteil für IBRANCE® + AI in der klinischen Praxis (basierend auf Patientendaten aus der Flatiron Health Analytic Datenbank). 4,7; # Signifikanter PFS-Vorteil unter IBRANCE® + LET in der Erstlinie sowie unter IBRANCE® + FUL bei endokrin-vorbehandelten Patientinnen.9,10; $ 3 Wochen anwenden, 1 Woche pausieren, Therapieschema laut Fachinformation.; ‡ Obligate Blutbildkontrolle gemäß Fachinformation als einzige routinemäßige Monitoringanforderung.
IBRANCE ® 75 mg Filmtabletten; IBRANCE ® 100 mg Filmtabletten; IBRANCE ® 125 mg Filmtabletten; Wirkstoff: Palbociclib; Zusammensetzung: Wirkstoff: 1 Filmtabl. enth. 75 mg/ 100 mg/ 125 mg Palbociclib. Sonst. Bestandteile: Tablettenkern: mikrokristalline Cellulose, hochdisperses Siliciumdioxid, Crospovidon (Ph.Eur.), Magnesiumstearat, Bernsteinsäure. Filmüberzug: Hypromellose (E 464), Titandioxid (E 171), Triacetin, Indigocarmin-Aluminiumsalz (E 132), Eisen(III)-oxid (E 172) (nur 75 mg u. 125 mg Tabletten), Eisen(III)-hydroxid-oxid x H2O (E 172) (nur 100 mg Tabletten). Anwendungsgebiete: Zur Behandl. v. Hormonrezeptor (HR)-pos., humanem epidermalen Wachstumsfaktor-Rezeptor-2 (HER2)neg. lokal fortgeschr. od. metastasiertem Brustkrebs, i. Komb. m. e. Aromatasehemmer od. i. Komb. m. Fulvestrant b. Frauen, d. zuvor e. endokrine Ther. erhielten. B. prä- od. perimenopausalen Frauen sollte d. endokrine Ther. m. e. LHRH-Agonisten komb. werden. Gegenanzeigen: Überempfindlichk. gg. d. Wirkstoff od. e. d. sonst. Bestandt. Die Anw. v. Arzneim., d. Johanniskraut enthalten. Nebenwirkungen: Sehr häufig: Infektionen; Neutropenie (Neutrophilenzahl vermindert), Leukopenie (Leukozytenzahl vermindert), Anämie (Hämoglobin erniedrigt, Hämatokrit vermindert), Thrombozytopenie (Thrombozytenzahl vermindert); vermind. Appetit; Stomatitis (Stomatitis aphthosa, Cheilitis, Glossitis, Glossodynie, Mundulzeration, Schleimhautentzünd., Mundschmerzen, Beschwerden i. Oropharynx, Schmerzen i. Oropharynx), Übelk., Diarrhö, Erbrechen; Ausschlag (Ausschlag makulo-papulös, Ausschlag m. Juckreiz, Ausschlag erythematös, Ausschlag papulös, Dermatitis, Dermatitis acneiform, toxischer Hautausschlag), Alopezie, trockene Haut; Fatigue, Asthenie, Pyrexie; ALT erhöht, AST erhöht. Häufig: febrile Neutropenie; Dysgeusie; verschwommenes Sehen, verstärkte Tränensekretion, trockenes Auge; venöse Thromboembolie (Lungenembolie, Embolie, tiefe Venenthrombose, periphere Embolie, Thrombose); Epistaxis, ILD/ Pneumonitis; Palmar-plantares Erythrodysästhesie-Syndrom; erhöhtes Kreatinin im Blut. Gelegentlich: Kutaner Lupus erythematodes, Erythema multiforme. Weitere Informationen s. Fach- u. Gebrauchsinformation. Abgabestatus: Verschreibungspflichtig. Pharmazeutischer Unternehmer: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Brüssel, Belgien. Repräsentant in Deutschland: PFIZER PHARMA GmbH, Friedrichstr. 110, 10117 Berlin. Stand: Mai 2024.
Wer ein Arzneimittel neu zulassen möchte, muss Studien vorlegen, die die Wirksamkeit dieses Medikaments belegen, genauer gesagt, die Wirksamkeit des konkreten Inhaltsstoffes, der für die Wirksamkeit ursächlich ist. Folgerichtig wird die Wirksamkeit des gesamten pharmazeutischen Produkts mit einer Variante verglichen, die äußerlich völlig identisch ist, der aber der Wirkstoff fehlt – also mit einem Placebo. Der Unterschied zwischen den beiden Gruppen lässt sich dann quantitativ und kausal auf den Wirkstoff zurückführen. Das macht unmittelbar Sinn, denn ein wirksames Medikament ist in aller Regel deutlich teurer als die Herstellungskosten für ein Placebo, und dieser Preisunterschied muss zuverlässig mit der Wirkstoff-basierten Wirksamkeit begründet werden können und nicht etwa durch z.B. Form oder Farbe oder eine attraktive Verpackung.
Diese Art von „Beweisführung“ setzt zwingend voraus, dass weder die Studienteilnehmer noch die übrigen Beteiligten wissen, wer welche Variante bekommen hat, was als „doppelte Verblindung“ bezeichnet wird. Keine Frage, wenn es um den möglichst effizienten Einsatz von Mitteln (Mitgliedsbeiträgen) der Krankenkassen geht, ist diese Vorgehensweise plausibel, angemessen und richtig. Offensichtlich sind aber immer mehr Wissenschaftler so sehr auf diese Methodik fixiert, dass sie entweder nicht mehr willens oder nicht mehr in der Lage sind zu hinterfragen, ob und wann diese Methodik indiziert ist. Diese leidvolle Erfahrung habe ich zusammen mit meinen Mitautoren erst jüngst wieder machen müssen. Das Manuskript für eine zweiarmige Interventionsstudie [1] wurde mehrfach unmittelbar und ohne weitere Prüfung mit lapidaren Begründungen zurückgewiesen wie der, dass das Journal wegen des Risikos auf Verzerrung durch andere Einflussfaktoren grundsätzlich nur randomisierte Studien mit
doppelter Verblindung zur Prüfung annimmt. Haben wir bei der Studienplanung also einen wissenschaftlich unverzeihlichen Fehler eingebaut? Urteilen Sie selbst: Thema unserer Studie war die Behandlung von Säuglingen, die in den ersten Lebensmonaten durch exzessives Schreien auffallen. Leider gibt es dafür bis heute kein nebenwirkungsarmes, sicher und kausal wirksames Medikament, weshalb Pädiater, vor allem aber Hebammen, den Eltern zunehmend häufig raten, eine osteopathische Praxis aufzusuchen. Wer die beiden Begriffe Schreibaby und Osteopathie googelt, bekommt Tausende von Links und Testimonials. Vergleichsweise bescheiden ist demgegenüber die Evidenz aus hochwertigen klinischen Studien. Unsere Studie mit über 100 Teilnehmern sollte konkret die Frage untersuchen, was Eltern von Schreikindern erwarten können, wenn sie sich (bewusst!) entschließen, ihr Baby osteopathisch behandeln zu lassen. Die Eltern sollten vor, während und nach der zweiwöchigen Behandlungsphase auf einer Skala von 0 bis 10 einschätzen, wie sehr sie sich durch das exzessive Schreien beeinträchtigt/gestresst fühlten. In der Kontrollgruppe begann die Behandlung erst 2 Wochen später, zum Vergleich herangezogen wurde der Zeitraum vom Erstkontakt bis zum Beginn der Behandlungsphase. Die Studie untersuchte also die Perspektive verzweifelter Eltern, konkret die Frage, ob sich die Entscheidung für eine osteopathische Behandlung aus ihrer Sicht „rentiert“. Um die Studie so realitätsnah wie möglich zu gestalten, waren auch alle anderen therapeutischen

Prof. Dr. med. Karl-Ludwig Resch
Bemühungen („usual care“) explizit erlaubt.
Das Ergebnis unserer Studie: Die subjektiven Beeinträchtigungen gingen bei den Eltern in der Interventionsgruppe auf der 10-stufigen Ratingskala um 3,5 Stufen mehr zurück als im gleichen Zeitraum bei den Eltern in der Gruppe der unbehandelten Säuglinge.
Die Studie orientierte sich methodisch am Ansatz des „Comparative Effectiveness Research“ (CER), bei dem es um den Vergleich zweier therapeutischer Strategien – z.B. pharmakologischer versus nichtpharmakologischer Ansatz – geht bzw. um die Frage, ob sich eine Therapie „lohnt“ oder ob man es lieber bleiben lassen soll [2]. Letzteres ist besonders nahe an der Realität, wenn versorgungsmedizinisch keine Behandlungsnotwendigkeit oder -bedürftigkeit besteht oder kein erfolgversprechender Therapieansatz zur Verfügung steht. Dann stellt sich für Betroffene die Frage, ob sie
ihr eigenes Geld „investieren“ sollen oder nicht bzw. was sie dafür bekommen („value for money“).
Bei Zulassungsstudien für (rezeptpflichtige!) Arzneimittel wird eine möglichst hohe „interne Validität“ gefordert, also eine Minimierung des Risikos, dass irgendetwas anderes als der „spezifische Effekt“ einer Substanz als Ursache für den Unterschied zwischen den beiden Studiengruppen infrage kommen kann. Dies erfolgt oft sehr zu Lasten der „externen Validität“, also der Übertragbarkeit in die Alltagssituation.
Genau diese stand auch in der in diesem Heft veröffentlichten Originalarbeit [3] im Fokus, und das mit Recht. Wer von den Betroffenen erwartet, dass sie ein Medikament aus eigener Tasche bezahlen, muss Untersuchungen ernst nehmen bzw. durchführen, die die Sicht und Interessen der Kunden in den Mittelpunkt stellen. Deshalb hätte meiner Meinung nach eine randomisiert kontrollierte Studie mit doppelter Verblindung hier wohl für die Praxis eher wertlose Erkenntnisse gebracht. Die gewählte Methodik einer handwerklich guten Beobachtungsstudie mit praxisrelevanten Zielgrößen hat indes recht überzeugende Zahlen generiert, mit wie viel „gesundheitlichem Mehrwert“ im Falle einer Entscheidung für das Gurgelgel gerechnet werden kann.
Karl-Ludwig Resch, Nürnberg
Quellen
1 Schwerla F, Zimmer M, Göpfert J et al. Osteopathic treatment of infants with infantile colic/excessive crying: a prospective, multicentric, randomized controlled trial and nested observational trial. BMC Pediatr 2025; 25:Article 77. https://doi.org/10.1186/s12887025-05413-1
2 Institute of Medicine. Initial national priorities for comparative effectiveness research. National Academies Press 2009. doi:10.17226/12648
3 Behrbohm H. Behandlung der akuten Pharyngitis mit GeloTonsil® Gurgelgel. J Pharmakol Ther 2025;1:4-7
Behandlung der akuten Pharyngitis mit GeloTonsil® Gurgelgel 4 Hans Behrbohm
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Plaque-Psoriasis: Daten aus dem deutschen Behandlungsalltag belegen Verbesserung der Erkrankungslast durch Deucravacitinib 10
Refraktäres diffus großzelliges B-Zell-Lymphom: CAR-T-Zelltherapie mit Axi-Cel überzeugt als neuer Zweitlinienstandard 12
Filgotinib: Anhaltende Wirksamkeit und Sicherheit bei Colitis ulcerosa 14
Daratumumab-VRd + DR-Erhaltung: Neue Erstlinientherapie für transplantationsgeeignete Myelom-Patienten 18
Secukinumab: Real-World-Daten bestätigen die über 5 Jahre anhaltende Wirksamkeit bei PsA und r-axSpA 20
Apalutamid beim mHSPC: Real-World-Daten untermauern die Ergebnisse der Zulassungsstudie 22
Multiple Sklerose: Aktuelle Daten bestätigen Sicherheit von Ocrelizumab in Schwangerschaft und Stillzeit 26
Wissenswertes 7, 8, 15, 16, 21, 32 Kongresse 28
MIT ERLEADA® IM mHSPC
• Einfach 1 Tablette täglich
• Stärker wirksam für ein längeres Leben*
• Starten und Flexibilität für Folgetherapien erhalten


Mehr erfahren auf jmc.link/ stärker-starten

ERLEADA® ist u. a. indiziert zur Behandlung erwachsener Männer mit metastasiertem hormonsensitivem Prostatakarzinom (mHSPC) in Kombination mit Androgendeprivationstherapie (ADT). * vs. ADT
ERLEADA® 60 mg Filmtabletten, ERLEADA® 240 mg Filmtabletten. Wirkstoff: Apalutamid. Zusammensetz.: ERLEADA® 60 mg: Jede Filmtabl. enth. 60 mg Apalutamid. Sonst. Bestandt.: Tabl.kern: Hochdisp. Siliciumdioxid, Croscarmellose-Natrium, Hypromelloseacetatsuccinat, Magnesiumstearat, mikrokrist. Cellulose; mikrokrist. Cellulose, Siliciumdioxid-beschichtet. Filmüberzug: Eisen(II,III)oxid (E172), Eisen(III)-hydroxid-oxid x H2O (E172), Macrogol, Poly(vinylalkohol), Talkum, Titandioxid (E171). ERLEADA® 240 mg: Jede Filmtabl. enth. 240 mg Apalutamid. Sonst. Bestandt.: Tabl.kern: Hochdisp. Siliciumdioxid, Croscarmellose-Natrium, Hypromelloseacetatsuccinat, Magnesiumstearat, mikrokrist. Cellulose, Siliciumdioxid-beschichtet. Filmüberzug: Glycerolmonocaprylocaprat (Ph.Eur.) (Typ I), Eisen(II,III)-oxid (E172), Poly(vinylalkohol), Talkum, Titandioxid (E171), Macrogol-Poly(vinylalkohol)-Pfropfcopolymer. Anw.geb.: Bhdlg. erwachs. Männer m. nicht-metastasiertem kastrationsresistentem Prostatakarzinom (nm-CRPC), d. e. hohes Risiko f. d. Entwicklg. v. Metastasen aufweisen, Bhdlg. erwachs. Männer m. metastasiertem hormonsensitivem Prostatakarzinom (mHSPC) i. Komb. m. ADT (Androgendeprivationstherapie). Gegenanz.: Überempf. gg. d. Wirkst. od. e. d. sonst. Bestandt.; Schwangersch. od. Frauen, d. schwang. werden könnten, Stillzeit. Nebenwirk.: Vermind. Appetit, Hitzewallung, Hypertonie, Diarrhö, Hautausschl., Fraktur, Arthralg., Ermüdung, Gewichtsverlust, Sturz, Hypothyreose, Hypercholesterin., Hypertriglyzerid., Dysgeusie, ischäm. Herzerkr., ischäm. zerebrovask. Erkr., Pruritus, Alopezie, Muskelspasm., Krampfanf., Restless-Legs-Syndr., QT-Zeitverläng., Stevens-Johnson-Syndr./tox. epiderm. Nekrolyse, Arzneim. wirk. m. Eosinophilie u. system. Sympt., lichenoider Ausschlag, interstitielle Lungenerkr.. Warnhinw.: Arzneim. f. Kdr. unzugängl. aufbew.. Verschreibungspflichtig. Pharmazeut. Unternehmer: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgien. Örtl. Vertreter für Deutschland: Janssen-Cilag GmbH, Johnson & Johnson Platz 1, D-41470 Neuss. Stand d. Inform.: 11/24.
Janssen-Cilag GmbH
ZUSAMMENFASSUNG
Die akute Pharyngitis als Folge einer viralen sowie in seltenen Fällen bakteriellen Infektion ist eine schmerzhafte, jedoch selbstlimitierende Erkrankung des Rachens, ohne eine gleichzeitig vorliegende Entzündung der Tonsillen. Die Therapie zielt daher auf deren symptomatische Behandlung ab. In einer aktuellen Studie wurde die mögliche Linderung von häufigen Symptomen der akuten Pharyngitis wie Halsschmerzen, Schluckbeschwerden und ein trockener Hals durch die Anwendung von GeloTonsil® Gurgelgel über 4 Tage untersucht. Die Ergebnisse belegen die starke Verbesserung der Symptome unter der Behandlung mit GeloTonsil® Gurgelgel. 91,5 % der Patienten sprachen auf die Therapie an und ein Großteil hatte bei Abschluss der Behandlung keine oder nur noch geringe Beschwerden. Ebenso positiv wurde die Verträglichkeit bewertet. Zusammenfassend belegt die Studie die gute Wirkung und sichere Anwendung von GeloTonsil® Gurgelgel. Daher kann es zur Behandlung der akuten Pharyngitis empfohlen werden.
Schlüsselwörter: Pharyngitis, virale Infektion, Halsschmerzen, GeloTonsil® Gurgelgel, Symptomlinderung, Antibiotikaverordnung
Hans Behrbohm, Park-Klinik Weißensee, Berlin
Halsschmerzen als Hauptsymptom einer akuten Pharyngitis sind einer der häufigsten Beweggründe für den Hausarztbesuch. Eine akute Pharyngitis ist fast immer Folge einer viralen Infektion, wodurch es zu einer schmerzhaften Entzündung der Schleimhaut im Rachenbereich kommt, die häufig mit weiteren unspezifischen Erkältungsbeschwerden einhergeht. Eine Vielzahl von Erregern kann die Erkrankung auslösen, darunter fallen Rhinoviren, Adenoviren, das Eppstein-Barr-Virus, Herpesviren, Influenzaviren, Parainfluenzaviren und Coronaviren wie z.B. SARSCoV-2 [1]. Bei entsprechender Symptomatik mit akutem, heftigem Hals- und Schluckschmerz in Verbindung mit Gliederschmerzen und Abgeschlagenheit ist daher immer ein Coronatest zu empfehlen [2].
Aufgrund der viralen Ätiologie sollte bei unkompliziertem Verlauf eine Behandlung mit Antibiotika vermieden werden, zumal die Erkrankung selbstlimitierend ist und im Regelfall innerhalb von einer Woche von selbst abklingt. Nur in etwa 30 % der Pharyngitiden, jedoch vermehrt insbesondere bei kleineren Kindern, liegt eine bakterielle Infektion vor, dabei am
häufigsten induziert durch Gruppe A Streptokokken (GAS), was den Heilungsverlauf deutlich verzögern kann. Doch selbst in diesen Fällen ist der Einsatz von Antibiotika nur selten sinnvoll, da er häufig nur zu einer geringfügigen Verkürzung der Krankheitsdauer führt [3]. Unglücklicherweise sind die beiden am häufigsten verwendeten diagnostischen Scores McIsaac and Centor, die genutzt werden, um zwischen einer GAS und einer viralen Pharyngitis zu unterscheiden, häufig nicht spezifisch [4]. Daher kommt es häufig zu einem unsachgemäßen Einsatz von Antibiotika, der durch die Vorgaben in der aktuellen ärztlichen Behandlungsleitlinie unterbunden werden soll [5]. Da eine Pharyngitis jedoch mit schmerzhaften Symptomen verbunden sein kann, ist der Bedarf an wirkungsvollen Medikamenten zur Linderung der Beschwerden groß, und bedarfsgerecht eingesetzt, können diese einen Beitrag zu Senkung des Antibiotikaeinsatzes leisten. Es gibt eine Vielzahl von rezeptfreien Produkten. Gemäß Leitlinie wird die lokale Anwendung von Anästhetika oder nichtsteroidalen Antirheumatika zur Symptomlinderung empfohlen, jedoch wird ausdrücklich darauf hingewiesen, von Antiseptika und lokalen Antibiotika abzusehen, wenn diese
nicht expliziert indiziert sind [5]. Das neu entwickelte GeloTonsil® Gurgelgel mit seinem rein physikalischen Wirkprinzip stellt eine gut verträgliche Alternative dar. Durch das Gurgeln mit GeloTonsil® Gurgelgel werden Erreger physikalisch entfernt und es legt sich ein befeuchtender Schutzfilm über die entzündeten Areale des Rachens.
In einer ersten monozentrischen „Proof-of-concept“-Studie wurde die Wirksamkeit von GeloTonsil® Gurgelgel bereits untersucht und die lindernde Wirkung des Gurgelgels auf klassische PharyngitisSymptome belegt [6]. In der aktuellen Studie sollten die Ergebnisse nun in einer deutlich größer angelegten, multizentrischen Untersuchung bestätigt werden.
Ergebnisse der Studie mit GeloTonsil® Gurgelgel
An der aktuellen Studie nahmen insgesamt 521 volljährige Patientinnen und Patienten teil, die an einer akuten Pharyngitis litten. Ihr mittleres Alter betrug 41 Jahre, 59,2 % waren Frauen.
Vor Beginn der Behandlung litten
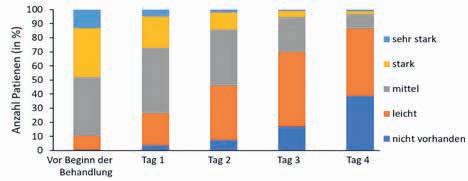
48 % der Studienteilnehmer an starken oder sehr starken Halsschmerzen. Bereits nach den ersten Anwendungen ging dieser Anteil auf 26,8 % zurück und zugleich stieg die Zahl der Patientinnen und Patienten mit lediglich leichten oder keinen Halsschmerzen von 10,2 auf 25,8 %. Bis zum Ende der Behandlung an Tag 4 verbesserten sich die Beschwerden weiter: 85,6 % gaben an, keine oder nur noch leichte Halsschmerzen zu haben (Abb. 1).
Auch die übrigen Symptome zeigten einen vergleichbaren Heilungsverlauf. So berichteten bei Abschluss der Behandlung über 80 % aller Studienteilnehmer von einem starken bis vollständigen Rückgang der Symptome Schluckbeschwerden, Halskratzen, Husten und einem trockenem Hals (Abb. 2).
Außerdem wurden die Patientinnen und Patienten vor Beginn und nach Abschluss der Behandlung auf Halsrötungen sowie -schwellungen untersucht. Vor Behandlungsbeginn wiesen 31,4 % starke oder sehr starke Rötungen und 14,6 % entsprechend schwere Halsschwellungen auf. Bei der Abschlussvisite waren diese Be-
SUMMARY
Acute pharyngitis as a result of a viral and, in rare cases, bacterial infection is a painful but self-limiting disease of the throat without concomitant inflammation of the tonsils. Therapy is therefore aimed at symptomatic treatment. In a recent study, the possible relief of common symptoms of acute pharyngitis such as sore throat, difficulty swallowing and a dry throat was investigated by using GeloTonsil® Gurgelgel for 4 days. The results confirm the strong improvement in symptoms under treatment with GeloTonsil® gargle. 91.5 % of patients responded to the therapy and the majority had no or only minor symptoms at the end of treatment. Tolerability was also rated positively. In summary, the study confirms the good effect and safe use of GeloTonsil® Gurgelgel. It can therefore be recommended for the treatment of acute pharyngitis.
Keywords: pharyngitis, viral infection, sore throat, GeloTonsil® Gurgelgel, symptom relief, antibiotic prescription
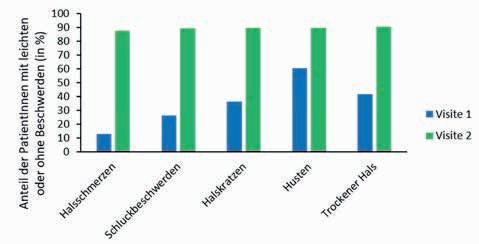
Abbildung 2: Anteil der Patientinnen und Patienten mit leichten oder ohne Symptome vor Beginn (blau) und nach Abschluss (grün) der Behandlung.
schwerden deutlich auf 1,4 % bzw. 0,8 % gesunken. Im Mittel verspürten die teilnehmenden Patientinnen und Patienten bereits innerhalb von 10 Minuten nach dem Gurgeln eine Linderung ihrer Beschwerden, die am ersten Tag durchschnittlich über 2, am vierten Tag sogar über 3 Stunden anhielt (Abb. 3).
Mit einer Symptomverbesserung und keiner Verschlechterung eines Symptoms sprachen 91,5 % der Patientinnen und Patienten auf die Therapie an. Dieser Wert deckt sich mit 78,3 % der Studienteilnehmer, die im Verlauf der Behandlung eine leichte oder deutliche Verbesserung ihrer Symptome bemerkten, während eine Verschlechterung nur bei 2,8 % der Patientinnen und Patienten auftrat. GeloTonsil® Gurgelgel weist zudem ein gutes Nutzen-RisikoProfil auf. Nur 3,9 % aller Studienteilnehmer berichteten von einer unerwünschten Nebenwirkung, deren Schwere größtenteils leicht und in lediglich 3 Fällen moderat
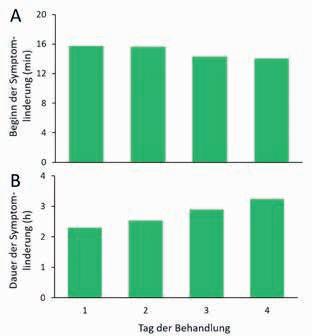
Abbildung 3: Mittlere Zeit bis zum Einsetzen (A) und Dauer der Symptomlinderung (B) nach Anwendung von GeloTonsil® Gurgelgel.
war. Die Verträglichkeit wurde von 90,2 % der Patientinnen und Patienten als sehr gut oder gut bewertet.
Aufgrund der guten Wirkung und Verträglichkeit des Produktes waren 41,9 % der Studienteilnehmer zufrieden oder sogar sehr zufrieden (27,3 %) mit der Therapie.
Die Ergebnisse dieser Studie bestätigen die gute Wirkung und Verträglichkeit von GeloTonsil® Gurgelgel bei der Behandlung der akuten Pharyngitis. Patientinnen und Patienten sowie Ärztinnen und Ärzte berichteten übereinstim-
mend von einer guten bis sehr guten und schnellen sowie langanhaltenden Linderung der Symptome. GeloTonsil® Gurgelgel stellt daher eine wirksame Therapieoption dar, die einen Beitrag zur Verringerung von Antibiotikaverordnungen leisten kann.
1 Avcı D, Muluk NB, Hao SP. Pharyngitis: causes, symptoms and treatment. In: Cingi C et al., Hrsg.: Airway Diseases. Cham: Springer Nature Switzerland AG; 2023:1-11
2 Behrbohm H, Thiele T. Prospektive Studie zu Schnelltests bei COVID-19-Patienten. ZWP Online, Wissenschaft und Forschung 2021
3 Krüger K, Oltrogge JH. Halsschmerzen –leitlinienbasierte Diagnostik und Therapie. Z Allg Med 2022;98:126-132
Endometriumkarzinom: Zulassungserweiterung von Dostarlimab für MMRp/MSS-Tumoren
Am 20. Januar 2025 hat die Europäische Kommission Dostarlimab (Jemperli) in Kombination mit Carboplatin-Paclitaxel (Chemotherapie) für die Erstlinientherapie bei erwachsenen Patientinnen mit primär fortgeschrittenem oder rezidivierendem Endometriumkarzinom zugelassen, für die eine systemische Therapie infrage kommt. Diese Zulassung erweitert die bisherige Indikation für Dostarlimab plus Carboplatin-Paclitaxel in der EU auf Patientinnen mit Mismatch-Reparatur-profizienten (MMRp)/mikrosatellitenstabilen (MSS) Tumoren, die bei etwa 75 % der Patientinnen mit Endometriumkarzinom vorliegen und für die ein hoher Bedarf an Behandlungsoptionen besteht. Das
Dostarlimab (Jemperli) ist ein humanisierter monoklonaler anti-programmed cell death protein-1 (PD-1)-Antikörper, der mit hoher Affinität an den PD-1-Rezeptor bindet und dessen Wechselwirkung mit den Liganden PD-L1 und PD-L2 blockiert.
fortgeschrittene oder rezidivierende Endometriumkarzinom hat eine sehr schlechte Prognose mit einer 5-Jahres-Überlebensrate von nur 17 %.
Verlängerung des medianen Gesamtüberlebens um 16,4 Monate
Die Zulassungserweiterung basiert auf den Ergebnissen von Teil 1 der randomisierten, doppelblinden, placebokontrollierten Phase-IIIStudie RUBY* mit 785 Patientin-
4 Kanagasabai A, Evans C, Jones HE et al. Systematic review and meta-analysis of the accuracy of McIsaac and Centor score in patients presenting to secondary care with pharyngitis. Clin Microbiol Infect 2024;30:445-452
5 Oltrogge J, Krüger K. DEGAM S3-Leitlinie Nr. 14 „Halsschmerzen“, AWMF-Register-Nr. 053-010. Deutsche Gesellschaft für Allgemeinmedizin und Familienmedizin 2020
6 Behrbohm H. Linderung von PharyngitisSymptomen durch die Behandlung mit GeloTonsil® Gurgelgel. J Pharmakol Ther 2023;32:4-7
Anschrift desVerfassers:
Prof. Dr. Hans Behrbohm Park-Klinik Weißensee Schönstraße 80 13086 Berlin E-Mail: behrbohm@park-klinik.com
nen mit primär fortgeschrittenem oder rezidivierendem Endometriumkarzinom. Nach einer mittleren Nachbeobachtungsdauer von mehr als 3 Jahren zeigte sich ein klinisch bedeutsamen und statistisch signifikanter Vorteil im medianen Gesamtüberleben: Unter der Therapie mit Dostarlimab plus CarboplatinPaclitaxel erreichten die Patientinnen im Median ein Gesamtüberleben von 44,6 Monaten im Vergleich zu 28,2 Monaten in der Kontrollgruppe, die mit Placebo plus Carboplatin-Paclitaxel behandelt wurde. Dies entspricht einem um 31 % reduzierten Sterberisiko (HR: 0,69; 95%-KI: 0,54 – 0,89). Das Sicherheitsprofil für Dostarlimab plus Carboplatin-Paclitaxel war mit den bekannten Sicherheitsprofilen der einzelnen Wirkstoffe vergleichbar.
B. S.
* Powell MA et al. Annals of Oncology 2024:35:728-738
Ribociclib erhält Zulassung für frühen HR+/HER2–Brustkrebs mit hohem Rezidivrisiko
Die Europäische Kommission hat Ribociclib (Kisqali®) nun auch in Kombination mit einem Aromatasehemmer für die adjuvante Behandlung von Patientinnen und Patienten mit Hormonrezeptorpositivem, humanem epidermalem Wachstumsfaktor-Rezeptor2-negativen (HR+/HER2– frühen Brustkrebs zugelassen, die ein hohes Rückfallrisiko haben.
Die Zulassung beruht auf den Ergebnissen der randomisierten, offenen Phase-III-Studie NATALEE [1], in die eine breite Population von Patientinnen und Patienten mit frühem HR+/HER2– Brustkrebs im Stadium II und III einschließlich solcher mit nodal-negativer Erkrankung mit Hochrisikomerkmalen eingeschlossen war. Ein hohes Rezidivrisiko bestand, wenn einer der folgenden Faktoren zutraf: ≥1 Lymphknoten betroffen (≥N1), Tumor >5 cm (N0) oder Tumor >2 – 5 cm (N0), wenn Grad 2 mit hohem genomischem Risiko oder Ki-67 ≥20 % oder Grad 3.
Signifikante Reduktion des Rezidivrisikos um bis zu 28,5 Prozent
NATALEE verglich die Wirksamkeit und Sicherheit von Ribociclib in Kombination mit einer endokrinen Therapie (ET) im Vergleich zur ET alleine als adjuvante Behandlung. Diese bestand in beiden Behandlungsarmen aus einem nichtsteroidalen Aromatasehemmer (NSAI, Anastrozol oder
Ribociclib (Kisqali®) ist ein selektiver Cyclin-abhängiger Kinaseinhibitor (CDKi). Er gehört zu einer Medikamentenklasse, die dazu beitragen kann, das Fortschreiten von Krebs durch Hemmung der Cyclin-abhängigen Kinasen 4 und 6 (CDK4/6) zu verlangsamen. Bei Überaktivität können diese Proteine Krebszellen zu einer schnellen Teilung veranlassen und so zum Tumorwachstum beitragen. In der EU ist Ribociclib zur Behandlung von Frauen mit einem HR+/ HER2– lokal fortgeschrittenen oder metastasierten Mammakarzinom in Kombination mit einem Aromatasehemmer oder Fulvestrant als initiale endokrinbasierte Therapie oder bei Frauen mit vorangegangener endokriner Therapie zugelassen sowie in Kombination mit einem Aromatasehemmer für die adjuvante Behandlung von Erwachsenen mit HR+/HER2– frühem Mammakarzinom mit hohem Rückfallrisiko.
Letrozol), der bei Männern und prämenopausalen Frauen durch die Hinzunahme von Goserelin ergänzt wurde. Primärer Endpunkt war das invasive krankheitsfreie Überleben (iDFS).
Die empfohlene Dosis lag bei 400 mg Ribociclib einmal täglich für 21 aufeinanderfolgende Tage, gefolgt von einer 7-tägigen Pause zur Komplettierung des 28-tägigen Zyklus für insgesamt 36 Monate. Die Ergebnisse zeigten für Ribociclib in Kombination mit ET eine signifikante Reduktion des Rezidivrisikos um 25,1 % im Vergleich zur alleinigen ET (HR: 0,749; 95%-KI: 0,628 – 0,892; zweiseitiger p-Wert = 0,0012). In allen vorab definierten Subgruppen wurde für Ribociclib in Kombination mit ET ein klinisch bedeutsamer, konsistenter Vorteil hinsichtlich des iDFS im Vergleich zu alleiniger ET beobachtet [1].
Eine zwischenzeitlich im Nachgang des Zulassungsantrags vorgestellte 4-Jahres-Landmarkanalyse der laufenden NATALEE-Studie zeigte darüber hinaus einen zunehmenden Nutzen von Ribociclib plus ET hinsichtlich des iDFS: ein um 28,5 % reduziertes Rezidivrisi-
ko gegenüber alleiniger ET (HR: 0,715; 95%-KI: 0,609 – 0,840; nominaler einseitiger p-Wert <0,0001) [2]. Zu diesem Zeitpunkt hatten alle Studienteilnehmer die Therapie mit Ribociclib abgeschlossen.
B. S.
Quellen
1 Hortobagyi GN et al. A phase III trial of adjuvant ribociclib plus endocrine therapy vs endocrine therapy alone in patients with HR+/HER2– early breast cancer: final invasive disease-free survival results from the NATALEE trial. Ann Oncology 2024, doi: https://doi.org/10.1016/j.annonc.2024.10.01
2 Fasching PA. Adjuvant ribociclib plus nonsteroidal aromatase inhibitor in patients with HR+/HER2− Early Breast Cancer: 4-year outcomes from the NATALEE trial. LBA13. Proffered Paper presented at the European Society for Medical Oncology Congress (ESMO); September 16, 2024; Barcelona

Kisqali in Kombination mit ET kann dazu beitragen, dass mehr Patientinnena mit frühem HR+/HER2— Brustkrebs krebsfrei bleiben.1, b Die Wirksamkeit ist nachgewiesen durch Reduktion des relativen Rezidivrisikos um 28,5 %.2 Damit Sie bei der Nachsorge noch öfter sagen können: „Alles unauffällig“.
a Bei frühem HR+/HER2– Brustkrebs ist Kisqali auch für Männer zugelassen.1 b Kisqali wird in Kombination mit einem Aromatasehemmer als adjuvante Behandlung bei Patientinnen und Patienten mit frühem HR+/HER2– Brustkrebs mit hohem Rezidivrisiko einschließlich Nodalstatus N0 mit Hochrisikomerkmalen (T3, T4 und T2 nur bei G3 oder G2 mit hohem genomischen Risiko oder Ki-67 ≥ 20 %) angewendet. Bei prä- oder perimenopausalen Frauen und bei Männern sollte der Aromatasehemmer mit einem LHRH-Agonisten kombiniert werden.1 ET Endokrine Therapie. G2 Mäßig differenzierter Tumor. G3 Schlecht differenzierter Tumor. HER2— Humaner epidermaler Wachstumsfaktor-Rezeptor-2-negativ. HR+ Hormonrezeptorpositiv. LHRH Luteinising Hormone-Releasing Hormone. N0 Keine Lymphknotenbeteiligung. T2 Tumor > 2 cm, aber ≤ 5 cm. T3 Tumor > 5 cm. T4 Tumor mit direkter Ausdehnung auf die Brustwand und/oder die Haut mit sichtbaren Veränderungen. Daten zu Wirksamkeit und Sicherheit von Ribociclib finden Sie in der Publikation zur Zulassungsstudie.
1. Fachinformation Kisqali. 2. Fasching PA, et al. ESMO 2024. Oral presentation LBA13.
Kisqali® 200 mg Filmtabletten Wirkstoff: Ribociclib. Zus.-setz.: 1 Tablette enth.: Arzneil. wirksamer Bestandt.: 200 mg Ribociclib. Sonst. Bestandt.: Mikrokristalline Cellulose, Crospovidon (Typ A), Hyprolose (5,0 - 16,0 % m/m Hydroxypropoxy-Gruppen), Magnesiumstearat, Hochdisperses Siliciumdioxid, Eisen(II,III)- oxid (E172, schwarz), Eisen(III)-oxid (E172, rot), Phospholipide aus Sojabohnen (E322), Poly(vinylalkohol), Talkum, Titandioxid (E171), Xanthangummi. Anwend.-gebiete: Adjuvante Behandlung bei Patientinnen und Patienten mit einem Hormonrezeptor(HR)-positiven, humanen epidermalen Wachstumsfaktor-Rezeptor-2(HER2)-negativen frühen Mammakarzinom mit hohem Rezidivrisiko. Bei prä- oder perimenopausalen Frauen und bei Männern sollte der Aromatasehemmer mit einem Luteinisierendes-Hormon-Release Hormon (LHRH)-Agonisten kombiniert werden. Behandlung von Frauen mit einem HR-positiven, HER2-negativen, lokal fortgeschrittenen od. metastasierten Mammakarzinom in Kombination mit einem Aromatasehemmer oder Fulvestrant als initiale endokrin-basierte Therapie oder bei Frauen mit vorangegangener endokriner Therapie. Bei prä- oder perimenopausalen Frauen sollte die endokrine Therapie mit einem LHRH-Agonisten kombiniert werden. Gegenanzeigen: Überempfindlichkeit gegen d. Wirkstoff od. gegen Erdnuss, Soja oder e. d. sonst. Bestandt. Nebenwirkungen: Patientinnen und Patienten mit frühem Mammakarzinom mit einer Anfangsdosis von 400 mg Ribociclib: Sehr häufig: Infektionen (wie Harnwegsinfektionen, Atemwegsinfektion). Neutropenie, Leukopenie. Kopfschmerzen. Husten. Übelkeit, Diarrhö, Obstipation, Abdominalschmerzen (Bauchschmerzen, Oberbauchschmerzen). Alopezie. Fatigue, Asthenie, Pyrexie. Abnormale Ergebnisse von Leberfunktionstests (ALT erhöht, AST erhöht, Bilirubin im Blut erhöht). Häufig: Anämie, Thrombozytopenie, Lymphopenie. Hypokalzämie, Hypokaliämie, verminderter Appetit. Benommenheit. Dyspnoe, interstitielle Lungenkrankheit (ILD)/Pneumonitis. Erbrechen, Stomatitis, Mukositis. Hepatotoxizität (hepatische Zytolyse, arzneimittelbedingter Leberschaden (< 1 %), Hepatotoxizität, autoimmune Hepatitis (Einzelfall)). Hautausschlag (einschließlich makulopapulöser Hautausschlag, juckender Hautausschlag), Pruritus. Peripheres Ödem, Oropharyngeale Schmerzen. Erhöhter Kreatininwert im Blut, verlängerte QT-Zeit im Elektrokardiogramm. Gelegentlich: Febrile Neutropenie. Patientinnen mit fortgeschrittenem oder metastasiertem Mammakarzinom mit einer Anfangsdosis von 600 mg Ribociclib: Sehr häufig: Infektionen (wie Harnwegsinfektionen, Atemwegsinfektion, Gastroenteritis, Sepsis (<1 %)). Neutropenie, Leukopenie, Anämie, Lymphopenie. Verminderter Appetit. Kopfschmerzen, Benommenheit. Dyspnoe, Husten. Übelkeit, Diarrhö, Erbrechen, Obstipation, Abdominalschmerzen (Bauchschmerzen, Oberbauchschmerzen), Stomatitis, Dyspepsie. Alopezie, Hautausschlag (einschließlich makulopapulöser Hautausschlag, juckender Hautausschlag), Pruritus. Rückenschmerzen. Fatigue, peripheres Ödem, Pyrexie, Asthenie. Abnormale Ergebnisse von Leberfunktionstests (ALT erhöht, AST erhöht, Bilirubin im Blut erhöht). Häufig: Thrombozytopenie, febrile Neutropenie. Hypokalzämie, Hypokaliämie, Hypophosphatämie. Schwindel. Erhöhter Tränenfluss, trockenes Auge. Synkope. Interstitielle Lungenkrankheit (ILD)/Pneumonitis. Dysgeusie. Hepatotoxizität (hepatische Zytolyse, hepatozelluläre Schädigung, arzneimittelbedingter Leberschaden (< 1 %), Hepatotoxizität, Leberversagen, autoimmune Hepatitis (Einzelfall)). Hauttrockenheit, Erythem, Vitiligo. Oropharyngeale Schmerzen, Mundtrockenheit. Erhöhter Kreatininwert im Blut, verlängerte QT-Zeit im Elektrokardiogramm. Selten: Erythema multiforme. Nicht bekannt: Toxische epidermale Nekrolyse (TEN). Warnhinweise: Enthält Phospholipide aus Sojabohnen. Verschreibungspflichtig. Weitere Hinweise: Siehe Fachinformation. Stand: November 2024 (MS 11/24.21). Novartis Pharma GmbH, Sophie-Germain-Str. 10, 90443 Nürnberg. Tel.: (0911) 273-0. www.novartis.de
Auf dem 33. Kongress der European Academy of Dermatology & Venereology hat Bristol Myers Squibb erste Interimsdaten aus der prospektiven, multizentrischen, nicht interventionellen Beobachtungsstudie DELPHIN vorgestellt [1]. Ziel der Studie ist es, Real-World-Daten aus dem deutschen Behandlungsalltag mit Deucravacitinib (Sotyktu®) zu gewinnen. Untersucht werden dabei die Wirksamkeit des oralen Tyrosinkinase 2 (TYK2)-Inhibitors sowie der Effekt der Therapie auf die gesundheitsbezogene Lebensqualität über einen Gesamtzeitraum von 5 Jahren bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis an ca. 100 Zentren in Deutschland [1, 2].
Primäres Studienziel der DELPHIN-Studie ist die Ermittlung des Anteils der Patienten, die unter der Therapie mit Deucravacitinib einen absoluten Psoriasis Area and Severity Index (aPASI-Score) ≤3 erreichen. Sekundäre Wirksamkeitsparameter schließen weitere klinische Daten ein, so z.B. den static Physician Global Assessment (sPGA) 0/1-Score, das PASI 75/90/100-Ansprechen, Veränderungen der betroffenen Body Surface Area (BSA) sowie die Veränderung der gesundheitsbezogenen Lebensqualität mittels PatientReported Outcomes wie etwa dem Dermatology Life Quality Index (DLQI) [2].
Baseline-Charakteristika der in die Interimsanalyse eingeschlossenen Patienten
Bei der Interimsanalyse in Woche 24 der DELPHIN-Studie (Cut-off: 1. Juli 2024) wurden die Daten der zu diesem Zeitpunkt eingeschlossenen 340 Patienten mit mittelschwe-
rer bis schwerer Plaque-Psoriasis ausgewertet. Bei Behandlungsbeginn mit Deucravacitinib lag das mittlere Alter der Patienten bei 48 Jahren; 42,9 % waren weiblich und ihr mittlerer Body-Mass-Index betrug 28,3 kg/m². Im Mittel waren seit der initialen Diagnosestellung 15,1 Jahre (SD ± 14,2 Jahre) vergangen. Die Patienten hatten einen durchschnittlichen PASI-Score von 13,3 (SD ± 8,8) und einen mittleren BSA-Wert von 19,6 % (SD ± 16,0 %). Bei 38,5 % der Patienten bestand eine genitale Beteiligung, bei 46,2 % lag eine intertriginöse und bei 30,0 % eine palmoplantare Psoriasis vor. Bei 78,2 % der Patienten waren die Kopfhaut und bei 33,8 % die Fingernägel betroffen. Der mittlere DLQI von 12,5 (SD ± 7) bei Baseline wies auf eine starke Beeinträchtigung der gesundheitsbezogenen Lebensqualität durch die Plaque-Psoriasis hin.
204 (60,0 %) der 340 in die Interimsanalyse eingeschlossenen Patienten hatten vor der Behandlung mit Deucravacitinib bereits mindestens eine systemische Therapie erhalten. Von diesen Patienten waren 131 (64,2 %) zuvor ausschließlich mit konventionellen systemischen Therapien behandelt worden. Insgesamt hatten vor
Studieneinschluss 170 Patienten (50,0 %) eine konventionelle systemische Therapie, 48 Patienten (14,0 %) eine Biologika-Therapie und 28 Patienten (8,2 %) eine Therapie mit Apremilast erhalten. 171 Patienten (50,3 %) hatten eine topische Vortherapie, 89 Patienten (26,2 %) hatten eine Phototherapie und 3 Patienten (0,9 %) hatten eine Klimatherapie bekommen [1].
Verbesserungen des aPASI und der gesundheitsbezogenen Lebensqualität nach 24 Wochen
Die Ergebnisse der Interimsanalyse zeigen, dass sich Deucravacitinib auch im Behandlungsalltag als wirksam erweist: Bei Studienbeginn besaßen die Patienten einen mittleren PASI von 13,3, nach 4 Wochen lag der Wert bei 8,9 (SD ± 7,5; n = 282) und nach 16 Wochen bei 4,3 (SD ± 5,1; n = 148).
In Woche 24 betrug der mittlere PASI 3,0 (SD ± 3,9; n = 76). Der Anteil der Patienten mit einem aPASI-Wert ≤3 lag bei 3,0 % zu Studienbeginn, bei 18,4 % nach 4 und 54,7 % nach 16 Wochen. In Woche 24 hatten 65,8 % der Patienten einen aPASI ≤3 [1].
Darüber hinaus verbesserte sich die gesundheitsbezogene Lebens-
Deucravacitinib (Sotyktu®) ist ein oraler, selektiver, allosterischer Tyrosinkinase 2 (TYK2)-Inhibitor und repräsentiert pharmakologisch eine neue Klasse innerhalb der niedermolekularen Wirkstoffe (Small Molecules). Als erster Vertreter seiner Wirkstoffklasse ist er seit März 2023 zur Behandlung von erwachsenen Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis zugelassen, die für eine systemische Therapie infrage kommen [3].
Grundlage der Zulassung sind die Ergebnisse der Phase-III-Studien POETYK PSO-1 und POETYK PSO-2. In beiden klinischen Studien erreichten im Vergleich zu Placebo und Apremilast signifikant mehr Patienten unter Deucravacitinib nach 16 bzw. 24 Wochen einen sPGA-Score von 0 oder 1 sowie ein PASI 75-Ansprechen und ein PASI 90-Ansprechen [4, 5]. In der Langzeit-Extensionsstudie POETYK PSO-LTE wurde nach 4 Jahren kontinuierlicher Behandlung mit Deucravacitinib das klinische Ansprechen, gemessen am PASI 75, weiterhin bei mehr als 70 % der Patienten aufrechterhalten. Das Sicherheitsprofil des TYK2-Inhibitors blieb dabei konsistent zu dem bekannten Sicherheitsprofil aus POETYK PSO-1 und POETYK PSO-2 [6].
Deucravacitinib ist selektiv gegen TYK2 gerichtet und hemmt damit die Signalkaskade von Interleukin (IL)-23, IL-12 und Typ-1-Interferon (IFN), die zu den zentralen Zytokinen der Pathogenese zahlreicher immunvermittelter Erkrankungen gehören. Einen hohen Grad an Selektivität erreicht Deucravacitinib infolge der Bindung des Wirkstoffs an die regulatorische Domäne von TYK2, was die allosterische Hemmung von TYK2 und deren nachgelagerten Funktionen zur Folge hat. Deucravacitinib hemmt in physiologisch relevanten Konzentrationen lediglich TYK2 selektiv. In zugelassener therapeutischer Dosis inhibiert Deucravacitinib JAK1, JAK2 bzw. JAK3 nicht [3].
qualität der Patienten unter der Behandlung mit Deucravacitinib. So lag der mittlere Score im DLQI in Woche 24 bei 4,3 (SD ± 5,6; n = 76) und war somit um 8,2 Punkte niedriger als der Ausgangswert [1].
Laut Dr. Ralph Michael von Kiedrowski, dem wissenschaftlichen Studienleiter der DELPHIN-Studie, sind diese anhand der DELPHIN-Studie erhobenen RealWorld-Daten ebenso relevant für
Dermatologen und die Patientenversorgung wie die aus den Zulassungsstudien resultierenden Daten (vgl. Insert). „Denn wir sehen in der Plaque-Psoriasis sehr unterschiedliche Patientenkollektive. Um diese bestmöglich behandeln zu können, ist es wichtig, dass neue Wirkstoffe nach ihrer Zulassung zügig in den dermatologischen Behandlungskanon integriert werden. Die mit dieser Studie gewonnenen Erkenntnisse deuten darauf hin, dass der orale TYK2-Inhibitor Deucravacitinib auch im deutschen Behandlungsalltag eine wirksame Therapieoption bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis darstellt.“ Brigitte Söllner, Erlangen
Literatur
1 Von Kiedrowski RM et al. Trial-in-progress – deucravacitinib in routine clinical practice: a 5-year, multicenter, prospective, noninterventional cohort study to evaluate effectiveness and quality of life in patients with moderate to severe plaque psoriasis in Germany (DELPHIN). EADV 2024, Poster: 3302
2 https://clinicaltrials.gov/study/NCT 06104644
3 Fachinformation Sotyktu®, aktueller Stand
4 Armstrong AW et al. J Am Acad Dermatol 2023;88:29-39
5 Strober B et al. J Am Acad Dermatol 2023;88:40-51
6 Armstrong AW et al. EADV Spring 2024; Präsentation FC02
Das diffus großzellige BZell-Lymphom (DLBCL) ist die häufigste Neoplasie des lymphatischen Systems und mit 30 % nicht nur das häufigste Non-Hodgkin-Lymphom, sondern auch ein besonders aggressiver Subtyp [1]. Die Erkrankung ist grundsätzlich heilbar, unbehandelt jedoch schnell fortschreitend mit ungünstigem Verlauf. In der Erstlinie basiert die Therapie auf dem Einsatz einer ImmunChemotherapie mit Rituximab, Cyclophosphamid, Doxorubicin, Vincristin und Prednison (R-CHOP-Protokoll) [2]. Jedoch sprechen einige Patienten nicht auf die Erstlinientherapie an oder erleiden einen Rückfall und sind daher auf wirksame Folgetherapien angewiesen [3]. Für die Gruppe der primär progredienten/früh rezidivierten Hochdosis-fähigen Patienten mit r/r DLBCL empfiehlt die S3-Leitlinie DLBCL ab der zweiten Therapielinie eine gegen CD19 gerichtete CAR-T-Zelltherapie wie Axicabtagen ciloleucel (Axi-Cel, Yescarta®) als potenziell kurative Behandlungsoption [1]. Diese Empfehlung deckt sich mit der Onkopedia-Leitlinie 2022, die zudem seit Januar 2024 bei Rezidiv bzw. Refraktärität nach der Eignung für eine CAR-T-Zelltherapie in „CAR-T-fähig“ bzw. „nicht CAR-T-fähig“ differenziert – entgegen der vorherigen Einteilung in „HDT-ASZT-fähig (Hochdosistherapie gefolgt von einer allogenen Stammzelltransplantation)“ versus „nicht HDT-ASZT-fähig“ [4]. Ausschlaggebend für die Empfehlung der CAR-TZelltherapie als neuen Zweitlinienstandard waren u.a. die positiven Daten der Studie ZUMA-7 zu Axi-Cel, die beim r/r DLBCL einen signifikanten Gesamtüberlebensvorteil gezeigt hat [5].
neuer
Langzeitdaten sprechen für ein kuratives Potenzial – auch bei älteren Patienten
Die offene, multizentrische, 1:1 randomisierte Phase-III-Studie ZUMA-7 untersuchte die Wirksamkeit einer Einzelinfusion AxiCel im Vergleich zur damaligen Standardbehandlung (SOC*) bei 359 Erwachsenen mit LBCL, deren Erkrankung innerhalb von 12 Monaten nach Abschluss einer Erstlinien-Chemoimmuntherapie mit Rituximab und Anthracyclin rezidiviert oder gegenüber dieser refraktär war. Die Studienteilnehmer erhielten randomisiert entweder Axi-Cel (n = 180) oder SOC (n = 179). Primärer Endpunkt war das ereignisfreie Überleben (EFS), sekundäre Endpunkte waren das progressionsfreie Überleben (PFS) und das Gesamtüberleben (OS).
Nach 24 Monaten war das EFS mit 40,5 % im Axi-Cel-Arm signifikant höher als im SOC-Arm mit 16,3 %. Zum Zeitpunkt der primären EFSAnalyse (medianes Follow-up 24,9 Monate) betrug das mediane progressionsfreie Überleben (mPFS)
* Definiert als 2–3 Zyklen einer StandardChemoimmuntherapie (R-ICE, R-DHAP oder R-DHAX, R-ESHAP oder R-GDP) gefolgt von einer Hochdosistherapie mit allogener Stammzelltransplantation (HDTASZT) bei Ansprechen der Erkrankung.
entsprechend 14,7 vs. 3,7 Monate. Das mediane Gesamtüberleben wurde in der Axi-Cel-Gruppe nicht erreicht und lag in der SOCGruppe bei 31,1 Monaten. Nach 4 Jahren betrug die geschätzte OSRate unter Axi-Cel 54,6 % gegenüber 46,0 % unter SOC (HR: 0,73; 95%-KI: 0,54 – 0,98; p = 0,03) [5].
Auch die primäre OS-Analyse 5 Jahre nach Einschluss des ersten Patienten (medianes Follow-up 47,2 Monate) ergab einen signifikanten Gesamtüberlebensvorteil zugunsten von Axi-Cel – entsprechend einer Reduktion des Sterberisikos um 27,4 % [5].
Der Überlebensvorteil zeigte sich über alle prädefinierten Gruppen hinweg. Wie eine Subgruppenanalyse von ZUMA-7 belegt, profitierten auch Patienten im Alter von ≥65 bzw. ≥70 Jahren von der CAR-T-Zelltherapie mit einer effektiveren Risikoreduktion im Vergleich zur SOC [6].
Gut dokumentiertes Sicherheitsprofil
Die in der Studie ZUMA-7 bedeutendsten, häufigsten unerwünschten Ereignisse unter der Therapie mit Axi-Cel waren das Zytokinfreisetzungssyndrom (92 %), Enzephalopathie (49 %) und Infektionen (45 %). Schwerwiegende
Die CAR-T-Zelltherapie ist eine für jeden Patienten individuell angefertigte Immuntherapie, die im Kampf gegen bestimmte rezidivierte/refraktäre Krebserkrankungen wie z.B. Non-Hodgkin-Lymphome und Leukämien eingesetzt wird [10, 11]. Dazu werden dem Erkrankten körpereigene T-Zellen entnommen und gentechnisch so verändert, dass sie auf ihrer Oberfläche chimäre Antigenrezeptoren (CAR), bilden, die gegen die krebsspezifischen Oberflächenproteine gerichtet sind. Die CAR-T-Zellen werden dem Patienten reinfundiert. Ihre Wirkung beruht darauf, dass sie auf den Tumorzellen die entsprechenden Antigene (z.B. CD19) erkennen und die CD19-exprimierenden Zellen durch ihre zytotoxische Aktivität zerstören.
Zu den akuten Nebenwirkungen, die im Nachgang einer Therapie mit CAR-T-Zellen auftreten können, zählen u.a. das Zytokinfreisetzungssyndrom (CRS) und das Immuneffektor-assoziierte Neurotoxizitätssyndrom (ICANS). Die Patienten müssen daher in den ersten 7 Tagen nach der Infusion täglich auf Anzeichen und Symptome eines potenziellen CRS, neurologischer Ereignisse und anderer Toxizitäten überwacht werden.
In der Europäischen Union sind aktuell 6 CAR-T-Zellprodukte zur Therapie von Patienten mit verschiedenen hämatologischen oder lymphatischen Malignitäten zugelassen, darunter Axicabtagen ciloleucel (Axi-Cel, Yescarta®).
Nebenwirkungen traten bei 54 % der Patienten auf. Um CRS-Symptome und neurologische Nebenwirkungen zu lindern, wurden spezielle Behandlungsalgorithmen entwickelt [7].
Zuordnung der geeigneten Patienten muss systematisch verbessert werden
„Ist mein r/r DLBCL-Patient CART-fähig?“ – das ist die entscheidende Frage zur Therapiesteuerung bei r/r DLBCL. Grundsätzlich kann diese Frage auch für Patienten mit höherem Alter, Komorbiditäten, schlechtem Allgemeinzustand oder einer Erkrankung mit dem humanen Immundefizienz-Virus HIV bejaht werden – unabhängig davon, ob die Patienten für eine Stammzelltransplantation geeignet
sind [1, 6]. Trotz der Empfehlung der Onkopedia-Leitlinie 2024, allen CAR-T-fähigen Patienten eine CAR-T-Zelltherapie anzubieten und trotz der positiven Ergebnisse dieser Immuntherapie scheint der Fokus beim r/r DLBCL aber noch immer nicht auf der CART-Zelltherapie zu liegen. Wie eine aktuelle Analyse der Daten von 126 Patienten von 50 Ärzten in Deutschland zeigte, gibt es hierzulande noch immer Unsicherheit bei der Einschätzung einer CAR-TEignung. Denn 21 % aller geeigneten Patienten wurden fälschlich als „nicht-CAR-T-fähig“ eingestuft [8]. Das entspräche hochgerechnet 460 Patienten in Deutschland, die statt einer CAR-T-Zelltherapie eine alternative Behandlung zugewiesen bekommen hätten – wobei für jeden dieser Patienten theoretisch eine durchschnittlich um
mehr als 8 Monate verringerte Lebenserwartung errechnet wurde [8].
Um beim r/r DLBCL CAR-T-fähige Patienten systematisch besser zu identifizieren, sollten laut Empfehlung der „Nationalen Strategie für gen- und zellbasierte Therapien (GCT)“ interdisziplinäre GCT-Behandlungseinrichtungen etabliert und Qualifizierungsprozesse effizienter gestaltet werden, um den Betroffenen den regelhaften Zugang zu dieser innovativen Therapie zu ermöglichen und die Chance auf eine längere Lebenszeit zu nutzen [9].
Brigitte Söllner, Erlangen
Literatur
1 Lenz G et al. Onkopedia-Leitlinie Diffuses großzelliges B-Zell-Lymphom. Stand: Januar 2024
2 S3-Leitlinie Diagnostik, Therapie und Nachsorge für erwachsene Patient*innen mit einem diffusen großzelligen B-ZellLymphom und verwandten Entitäten (Version 1.0, Langversion). AWMF-Registernummer: 018-038OL. Stand: Oktober 2022
3 Melchardt T et al. ESMO Open 2023;8: 100750
4 Lenz G et al. Onkopedia-Leitlinie Diffuses großzelliges B-Zell-Lymphom. Stand: Juli 2022
5 Westin JR et al. N Engl J Med 2023; 389:148-157
6 Kersten MJ et al. Blood 2023;142(Suppl. 1):1761
7 Fachinformation Yescarta®; Stand: Juli 2024
8 Buecklein VL et al. Blood 2024; 44(Suppl. 1):5037
9 Berlin Institute of Health at Charité (BIH). Translationsforschungsbereich der Charité (Hrsg). Im Auftrag des Bundesministeriums für Bildung und Forschung (BMBF). Nationale Strategie für genund zellbasierte Therapien (GCT), Stand: Juni 2024. https://www.bihealth.org/fileadmin/GZT/NationaleStrategie_GCT_ DE.pdf
10 Finck AV et al. Nat Med 2022;28:678689
11 Dagar et al. J Transl Med 2023;21:449
Colitis ulcerosa (CU) ist eine chronisch-entzündliche Darmerkrankung, die eine lebenslange Behandlung erfordert. Daher sind die 5-Jahres-Daten der Studie SELECTION LTE für die betroffenen Patienten besonders relevant, denn sie bestätigen die bisherigen anhaltend hohen Remissionsraten sowie die gute Verträglichkeit des Januskinase 1 (JAK1)-Inhibitors Filgotinib (Jyseleca®) [1].
85 Prozent Remission nach 5 Jahren
Die 5-Jahres-Daten der Studie SELECTION LTE, einer offenen Langzeitverlängerungsstudie der randomisierten kontrollierten Studie SELECTION [2], in der alle Patienten 200 mg Filgotinib 1 täglich erhielten, bestätigen die anhaltende Wirksamkeit von Filgotinib: 85,0 % der Completer der SELECTION-Studie (Induktions- und Erhaltungsphase unter Filgotinib 200 mg 1 täglich abgeschlossen) waren in Woche 192 der Verlängerung SELECTION LTE in Remission. Unter den initialen Non-Respondern der SELECTION-Studie (keine klinische Remission oder klinisches Ansprechen in Woche 10 unter Filgotinib 200 mg oder 100 mg 1täglich; Open-LabelTherapie mit Filgotinib 200 mg 1 täglich in SELECTION LTE) lag der Anteil in Woche 240 bei 65,4 % bzw. 66,7 % [1].
91,2 % der Completer und 85,7 % bzw. 100 % der Patienten der initialen Non-Responder-Kohorte, die von 200 mg bzw. 100 mg Filgotinib 1 täglich auf 200 mg Filgotinib 1 täglich umgestellt wurden, erreichten in Woche 192 bzw. 240 der Langzeitverlängerungsstudie eine steroidfreie Remission [1].
Anhaltende Verbesserung der Lebensqualität
Die Langzeittherapie mit Filgotinib ging außerdem mit einer anhaltenden Verbesserung der gesundheitsbezogenen Lebensqualität einher: Eine IBDQ-Remission (Inflammatory Bowel Disease Questionnaire) erzielten in SELECTION LTE 84,8 % der Completer in Woche 192 und 79,5 bzw. 77,8 % der initialen Non-Responder (Umstellung von 100 mg Filgotinib auf 200 mg 1 täglich bzw. Fortführung von Filgotinib 200 mg 1 täglich) in Woche 240 [1].
Filgotinib
RCT-Daten im Einklang mit Real-World-Studien
Da die in randomisierten kontrollierten Studien (RCTs) wie SELECTION eingeschlossenen Patienten aufgrund der rigiden Ein- und Ausschlusskriterien nicht immer dem realen Patientenkollektiv entsprechen, sind Real-World(RW)Studien eine wichtige Ergänzung. Die bisher größte voll publizierte RW-Studie zur Wirksamkeit und Sicherheit von Filgotinib bei CU – eine multizentrische, retrospektive, in Japan durchgeführte Erhebung – erfasst 238 Patienten, die eine Therapie mit Filgotinib jüngst begonnen hatten. Die Auswertung der Daten erfolgte nach Woche 10, 26 und 58 [3].
Ein klinisches Ansprecheng in Woche 58 erreichten unter Filgotinib 85,4 % der Teilnehmer, für die zu diesem Zeitpunkt Daten vorlagen (n = 48). 64,6 % erzielten eine klinische Remission (n = 48) und 60,9 % profitierten vom Erreichen
Der präferenzielle Januskinase 1 (JAK1)-Inhibitor Filgotinib (Jyseleca®) ist zugelassen zur Behandlung der mittelschweren bis schweren aktiven Colitis ulcerosa bei erwachsenen Patienten, die auf eine konventionelle Therapie oder ein Biologikum unzureichend angesprochen haben, nicht mehr darauf ansprechen oder diese nicht mehr vertragen haben.
Filgotinib kann als Monotherapie oder in Kombination mit Methotrexat (MTX) angewendet werden [5].
des Endpunkts einer Kortikosteroid-freien Remission (n = 46) [3].
Auf breiter Linie bestätigtes Sicherheitsprofil
Die Sicherheitsanalyse der RealWorld-Studie ergab ebenso wie die 5-Jahres-Daten von SELECTION LTE keine Hinweise auf neue, bisher nicht bekannte Signale [1, 3]. Insgesamt ist das Sicherheitsprofil von Filgotinib gut belegt: So erfasste eine integrierte Analyse der Studien SELECTION und SELECTION LTE 3.326 Patientenjahre unter Filgotinib [4]. Der präferenzielle JAK1-Inhibitor wurde dabei gut vertragen, das Sicherheitsprofil war mit früheren Studien vergleichbar.
Fabian Sandner,
Nürnberg
Neues Serviceangebot der Fach-Website www.muskelschmerzenbehandeln.de
Die Plattform www.muskelschmerzenbehandeln.de unterstützt Ärzte bereits seit einem Jahr dabei, muskulär bedingte Rückenschmerzen gezielt zu therapieren. Nun erweitert sie ihr Angebot und stellt Ärzten neue Ressourcen für eine optimierte Behandlung muskulär bedingter Schmerzen zur Verfügung. Neben wissenschaftlichen Informationen zu den Indikationen und zur Anwendung von Muskelrelaxanzien wie Pridinol bietet die Plattform zusätzliche Publikationen und eine Mediathek mit Videovorträgen und Diskussionsrunden, die dabei helfen, Patienten noch gezielter zu betreuen und umfassend aufzuklären. Rückenvideos zeigen alltagstaugliche Übungen, die Ärzte ihren Patienten empfehlen können, um muskuläre Verspannungen zu lindern und die Rückengesundheit zu fördern.
dien. Ergänzend können Mediziner den Fach-Newsletter PraxisKompakt – SchmerzRadar abonnieren, der regelmäßig über neue Entwicklungen in der Therapie von Myalgien informiert.
White Paper zur PROVIDENCE-Register-Studie
Literatur
1 Feagan BG et al. United European Gastroenterology Week 2024; Präsentation OP105
2 Feagan BG et al. Lancet 2021;397:23722384
3 Akiyama S et al. Aliment Pharmacol Ther 2024;59:1413-1424
4 Schreiber S et al. Aliment Pharmacol Ther 2023;58:874-887
5 Fachinformation Jyseleca®, aktueller Stand
Presseinformation
Ein neues White Paper, das direkt auf der Plattform abrufbar ist, fasst die wesentlichen wissenschaftlichen Erkenntnisse der Registerdaten-Studie PROVIDENCE zusammen: Die Ergebnisse weisen Pridinol (z.B. Myditin®), als überlegene Alternative zu NSAR bei akuten Kreuz-/Rückenschmerzen aus, mit einer signifikant höheren Responserate im Vergleich zu NSAR.
Fach-Website www.muskelschmerzen-behandeln.de Serviceangebot
Alsdorf – 15 Januar 2025 – Die Fachplattform www.muskelschmerzen behandeln.de erweitert ihr Angebot und stellt Ärzten* neue Ressourcen optimierte Behandlung muskulär bedingter Schmerzen zur Verfügung. wissenschaftlichen Informationen zu Indikationen und zur Anwendung Muskelrelaxantien wie Pridinol bietet die Plattform zusätzliche Inhalte, klinische Praxis unterstützen und den Zugang zu aktuellen medizinischen Erkenntnissen erleichtern. Die neuen Materialien helfen, Patienten betreuen und umfassend aufzuklären.
„Highlights“-Rubrik und Fach-Newsletter
Mit der regelmäßig aktualisierten Rubrik Highlights erhalten Ärzte einen schnellen Überblick über die neuesten Informationen zur Schmerztherapie, zu Fortbildungsmöglichkeiten und relevanten Stu-
Die Plattform www.muskelschmerzen-behandeln.de unterstützt Ärzte bereits dabei, muskulär bedingte Rückenschmerzen gezielt zu therapieren, indem wissenschaftliche Erkenntnisse und klinische Praxis verbindet. Sie bietet Publikationen und eine Mediathek mit Videovorträgen und Diskussionsrunden Wissen, das unmittelbar in der Patientenversorgung genutzt werden kann. Rubriken eingeführt, die das bestehende Angebot erweitern und die Versorgung Patienten mit muskulären Beschwerden weiter optimieren.
Neue Informationsangebote: „Highlights“-Rubrik und Fach-Newsletter
Pridinol, das für die Anwendung bei zentralen und peripheren Muskelspasmen, Lumbalgie, Torticollis sowie allgemeinen Muskelschmerzen bei Erwachsenen zugelassen ist blockiert den muskarinischen Acetylcholinrezeptor. Es wirkt höchstwahrscheinlich im Rückenmark an den α-Motoneuronen, die das Skelettmuskelgewebe innervieren. Durch die Reduktion polysynaptischer Reflexe, die den Muskeltonus erhöhen, wird die muskuläre Spannung gelöst, ohne die normale Muskelaktivität zu beeinträchtigen. Dies ermöglicht eine schnelle Mobilisation der Patienten und hilft ihnen, rasch wieder am Alltag teilzunehmen.
B. S.
Mit der regelmäßig aktualisierten Rubrik Highlights bleiben Ärzte stets Laufenden: Hier erhalten sie einen schnellen Überblick über die neuesten Schmerztherapie, Fortbildungsmöglichkeiten und relevante Studien. Ergänzend Mediziner den Fach-Newsletter PraxisKompakt – SchmerzRadar abonnieren, regelmäßig über neue Entwicklungen in der Therapie von Myalgien informiert. Ärzte können sich direkt auf der Plattform https://muskelschmerzenbehandeln.de/de/service-hub/newsletter/ für den Newsletter anmelden folgenden QR-Code:
Interessierte Ärzte konnen sich über den QR-Code direkt auf der Plattform https://muskelschmerzenbehandeln.de/de/service-hub/newsletter/ für den Newsletter anmelden.

Theorie und Praxis vereint: White Paper zur PROVIDENCE-Register Rückenvideos
Die Plattform verbindet theoretisches Wissen und praktische Anwendungen. White Paper fasst die wesentlichen wissenschaftlichen Erkenntnisse der Studie PROVIDENCE zusammen: Die Ergebnisse weisen Pridinol als zu NSAR bei akuten Kreuz /Rückenschmerzen aus, mit einer
AlPaCa – das neue Alexion Patient Care Program für Menschen mit seltenen Erkrankungen
Seltene Erkrankungen stellen besondere Herausforderungen an das Gesundheitswesen. Auch wenn einzelne Erkrankungen selten sind, ist die Gesamtzahl der Betroffenen aufgrund der Vielzahl an verschiedenen Erkrankungen hoch – allein in Deutschland leben etwa 4 Millionen Menschen mit seltenen Erkrankungen. Es gibt jedoch nur wenige Experten, die auf die jeweilige seltene Erkrankung spezialisiert sind. Auch gute Behandlungs- und Betreuungsmöglichkeiten sind nicht immer leicht zu finden. All dies kann dazu führen, dass Betroffene sich mit ihrer Erkrankung allein gelassen fühlen und es zu Verzögerungen bei der Behandlung kommt. Ein unterstützendes Umfeld kann für sie ähnlich wichtig sein wie der richtige Arzt und die adäquate medizinische Behandlung. AlPaCa bietet Betroffenen und ihren Angehörigen Informationen, Orientierung und individuelle Unterstützung bei einer Vielzahl an seltenen Erkrankungen (s. Infokasten) – als Zusatz zur medizinischen Behandlung.
Neben umfangreichen Informationsmaterialien, mit denen die Patienten ihr Wissen zur Erkrankung und zum Alltag erweitern und weitere Ansprechpartner finden können, ist auch eine kostenlose Telefon-Hotline rund um die Uhr erreichbar. Darüber hinaus steht den Patienten ein persönli-
AlPaCa bietet Unterstützung bei folgenden seltenen Erkrankungen:
• Atypisches hämolytisch-urämisches Syndrom (aHUS), gekennzeichnet durch die Trias mikroangiopathische hämolytische Anämie, Thrombozytopenie und akute Nierenschädigung, bedingt durch ein Versagen der Komplementregulation.
• Lysosomale saure Lipase-Defizienz (LAL-D), eine lebensbedrohliche genetische Stoffwechselerkrankung, bei der sich aufgrund des Mangels an dem Enzym lysosomale saure Lipase Fette und Cholesterinester übermäßig in verschiedenen Geweben anhäufen und zu Organschäden führen.
• Hypophosphatasie (HPP), bei der durch eine Fehlfunktion des Enzyms alkalische Phosphatase der Knochenaufbau gestört ist, und es infolgedessen zu Schmerzen, Zahnausfall, wiederholten Frakturen und anderen orthopädischen Problemen kommen kann.
• Myasthenia gravis (MG), eine chronische Autoimmunerkrankung, die zu einer belastungsabhängigen Muskelschwäche und -ermüdbarkeit führt. Dies zeigt sich u.a. in hängenden Augenlidern, Doppelbildern, Schwierigkeiten beim Sprechen, Schlucken oder Atmen.
• Neurofibromatose Typ 1 (NF1), eine genetische Erkrankung, bei der multiple kutane oder plexiforme (PN) Neurofibrome entstehen. Das Wachstum der PNs verursacht Schmerzen, Skoliose und Tumorprädisposition, was zu einer verkürzten Lebenserwartung führt.
• Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD), die aufgrund von antikörpervermittelten Entzündungen des Rückenmarks, des Sehnervs oder bestimmter Hirnareale zu Sehverlust, Rollstuhlabhängigkeit oder Tod führen können und eine wichtige Differenzialdiagnose zur Multiplen Sklerose sind.
• Paroxysmale Nächtliche Hämoglobinurie (PNH), die aus einer chronischen Zerstörung der roten Blutkörperchen durch das Komplementsystem resultiert. Die Folgen können u.a. schwere Nierenschäden und hepatische, zerebrale oder abdominale Thrombosen sein.
cher Coach zur Seite, der ihnen per Telefon oder Videocall bei Alltagsfragen, mit Informationen zur Erkrankung oder bei der Suche nach Unterstützung hilft. Um die Einnahme von Arzneimitteln oder wichtige Arzttermine nicht zu verpassen, können die Patienten die Erinnerungsfunktion in Anspruch nehmen, die sie pünktlich per SMS, E-Mail oder Anruf informiert. Die Nutzung von AlPaCa ist
dabei unabhängig von der eingesetzten Arzneimitteltherapie. Für Patienten, die eine Therapie von Alexion erhalten, gibt es zusätzliche Informationen und Angebote. Unter www.alpaca-program.de können sie selbst die verschiedenen Services von AlPaCa kennenlernen.
S. M.

Hier mehr über DARZALEX® erfahren. jmc.link/drd-dvrd1
DARZALEX®-Rd*
außergewöhnlich langes
7,5 Jahre mOS 84 % PFS nach 4 Jahren progressionsfrei ²,±,°
Gesamtüberleben¹,**,# transplant non-transplant
* DARZALEX® ist indiziert in Kombination mit Lenalidomid und Dexamethason (Rd) bei nicht-transplantationsgeeigneten erwachsenen Patienten mit neu diagnostiziertem Multiplen Myelom. ** Ergebnisse der internationalen, offenen, randomisierten Phase-III-Studie MAIA mit neu diagn., nicht-transplantationsgeeigneten MM-Patienten (DRd (n = 368), Rd (n = 369)). Prim. Endpunkt: PFS, sek. Endpunkte u. a.: OS, MRD, ORR, ≥ VGPR, Zeit bis zum u. Dauer des Ansprechens. Medianes Follow-up: 64,5 Monate. # mOS unter DRd 90,3 Monate vs. 64,1 Monate unter Rd. HR für Versterben 0,67 (95 % KI, 0,55–0,82; nominaler p < 0,0001; medianes Follow-up: 89,3 Monate). + Indikation: DARZALEX® ist indiziert in Kombination mit Bortezomib, Lenalidomid und Dexamethason für die Behandlung erwachsener Patienten mit neu diagnostiziertem Multiplen Myelom, die für eine autologe Stammzelltransplantation geeignet sind; Therapieablauf: Induktion u. Konsolidierung mit DVRd, Erhaltungstherapie mit DR. ± Ergebnisse der multizentrischen, offenen, randomisierten Phase-III-Studie PERSEUS mit neu diagn., transplantationsgeeigneten MM-Patienten (DVRd + DR (n = 355), VRd + R (n = 354)). Prim. Endpunkt: PFS, sek. Endpunkte u. a.: ≥ CR, MRD, OS. Medianes Follow-up: 47,5 Monate. ° Geschätztes 48-Monats-PFS unter DVRd + DR 84,3 % vs. 67,7 % unter VRd + R. HR für Progress oder Versterben 0,42 (95 % KI, 0,30–0,59; medianes Follow-up: 47,5 Monate).
CR: Komplettes Ansprechen; DR: DARZALEX® + Dexamethason; DRd: DARZALEX® + Lenalidomid + Dexamethason; DVRd: DARZALEX® + Bortezomib + Lenalidomid + Dexamethason; HR: Hazard Ratio; KI: Konfidenzintervall; MM: Multiples Myelom; (m)OS: (Medianes) Gesamtüberleben; MRD: Minimale Resterkrankung; ORR: Gesamtansprechrate; PFS: Progressionsfreies Überleben; R: Lenalidomid; Rd: Lenalidomid + Dexamethason; VGPR: Sehr gutes partielles Ansprechen; VRd: Bortezomib + Lenalidomid + Dexamethason
1. Facon T et al. Final Survival Analysis of Daratumumab Plus Lenalidomide and Dexamethasone in Transplant-Ineligible Patients With Newly Diagnosed Multiple Myeloma: MAIA Study. Poster P968 presented at EHA; 13–16 June, 2024; Madrid, Spain. 2. Sonneveld P et al. N Engl J Med. 2024;390(4):301–313.
DARZALEX® 20 mg/ml Konzentrat zur Herstellung einer Infusionslösung; DARZALEX® 1.800 mg Injektionslösung. Wirkstoff: Daratumumab. Zusammensetz.: Infusionslsg.: Durchstechfl. (5 ml) enth. 100 mg; Durchstechfl. (20 ml) enth. 400 mg Daratumumab. Hum. monokl. IgG1 kappa-Ak gg. CD38 Ag. Sonst. Bestandt.: Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 20, Sorbitol (E420), Wasser f. Injektionszw.; Inj.lsg.: Durchstechfl. (15 ml) enth. 1.800 mg Daratumumab. Hum. monokl. IgG1 kappa-Ak gg. CD38 Ag. Sonst. Bestandt.: rekomb. hum. Hyaluronidase (rHuPH20), Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 20, Sorbitol (E420), Wasser f. Injektionszw.. Anw.geb.: Infusionslsg.: Nur f. d. Bhdlg. erw. Pat.: In Komb. m. Lenalidomid u. Dexamethason od. m. Bortezomib, Melphalan u. Predn. b. neu diagn. multipl. Myelom (MM), wenn ungeeign. f. e. autologe Stammzelltransplantation (ASZT). In Komb. m. Bortezomib, Thalidomid u. Dexamethason b. neu diagn. MM wenn geeignet f. e. ASZT. In Komb. m. Lenalidomid u. Dexamethason oder Bortezomib und Dexamethason b. MM m. mind. e. Vorbhdlg.. Monother.: B. rezidiv. u. refrakt. MM, soweit vorbehand. m. e. Proteasom-Inh. u. Immunmodul. u. Krankh.-progr. währ. d. letzt. Bhdlg.. Inj.lsg: Nur f. d. Bhdlg. erw. Pat.: In Komb. m. Lenalidomid u. Dexamethason od. m. Bortezomib, Melphalan u. Predn. b. neu diagn. multipl. Myelom (MM), wenn ungeeign. f. e. autologe Stammzelltransplantation (ASZT). In Komb. m. Bortezomib, Lenalidomid u. Dexamethason b. neu diagn. MM wenn geeignet f. e. ASZT. In Komb. m. Bortezomib, Thalidomid u. Dexamethason b. neu diagn. MM wenn geeignet f. e. ASZT. In Komb. m. Lenalidomid u. Dexamethason od. Bortezomib u. Dexamethason b. MM m. mind. e. Vorbhdlg.. In Komb. m. Pomalidomid u. Dexamethason b. MM u. Vorbhdlg. m. e. Proteasom-Inh. u. Lenalidomid u. refrakt. gg. Lenalidomid od. b. mind. zwei Vorther. einschl. Lenalidomid u. e. Proteasom-Inh. u. Krank.-progr. währ. od. nach d. letzt. Ther.. Monother.: B. rezidiv. u. refrakt. MM, soweit vorbehand. m. e. Proteasom-Inh. u. Immunmodul. u. Krankh.-progr. währ. d. letzt. Bhdlg.. In Komb. m. Cyclophosphamid, Bortezomib u. Dexamethason f. d. Bhdlg. b. neu diagn. system. Leichtketten (AL-)Amyloidose. Gegenanz.: Überempf. gg. d. Wirkst. od. e. d. sonst. Bestandt.. Nebenwirk. i.v. und s.c.: Infekt. ober. Atemwege, COVID-19, Pneumonie, Bronchitis, Neutropenie, Thrombozytopenie, Anämie, Lymphopenie, Leukopenie, vermind. Appetit, Schlaflosigk., periph. sensorische Neuropathie, Kopfschm., Husten, Dyspnoe, Diarrhö, Obstipation, Übelk., Erbrechen, Ausschlag, Rückenschm., Arthralg., Muskelspasm., Ermüd./Fatigue, periph. Ödem, Pyrexie, Asthenie, infusionsbedingte Reakt., Harnwegsinfekt., Influenza, Sepsis, Hypo-gammaglobulinämie, Hyperglykämie, Hypokalzämie, Dehydratation, Schwindelgef., Parästhesie, Synkope, Vorhofflimmern, Hypertonie, Lungenödem, Pankreatitis, Pruritus, Schm. im Brustr., Schüttelfrost, Reaktionen a. d. Injektionsstelle, Zytomegalievirus-Infektion, HBV-Reaktivierung, anaphyl. Reakt.,Warnhinw.: Arzneim. f. Kdr. unzugängl. aufbew.. Nicht schütteln. Verschreibungspflichtig. Pharmazeut.
Unternehmer: Janssen-Cilag International NV, Turnhoutseweg 30, 2340 Beerse, Belgien. Örtl. Vertreter für Deutschland: Janssen-Cilag GmbH, Johnson & Johnson Platz 1, 41470 Neuss. Stand d. Inform.: 11/24.
Janssen-Cilag GmbH
Seit Oktober 2024 ist der antiCD38-Antikörper Daratumumab (Darzalex®) für Patienten mit neu diagnostiziertem Multiplen Myelom (NDMM), die für eine autologe Stammzelltransplantation (ASZT) infrage kommen, in Kombination mit Bortezomib plus Lenalidomid und Dexamethason (DVRd) sowie Lenalidomid (DR) in der Erhaltung zugelassen [1]. Grundlage für die Indikationserweiterung durch die Europäische Arzneimittel Agentur (EMA) waren die Ergebnisse der Phase-III-Studie PERSEUS [2].
Signifikanter PFS-Vorteil und langanhaltendes, tiefes Ansprechen
PERSEUS war eine offene, randomisierte Phase-III-Studie bei Patienten mit NDMM, die für eine autologe Stammzelltransplantation (ASZT) geeignet waren. Die Studienteilnehmer erhielten entweder unverblindet in wöchentlichen bis mehrwöchigen Abständen subkutan Daratumumab in Kombination mit einer VRd-Induktions- und Konsolidierungstherapie, gefolgt von Daratumumab + Lenalidomid (DR) in der Erhaltung (DVRd-Gruppe, n = 355) oder eine VRd-Induktions- und Konsolidierungstherapie sowie eine Lenalidomid-Erhaltungstherapie allein (VRd-Gruppe, n = 354). Primärer Endpunkt war das progressionsfreie Überleben (PFS).
Zu den sekundären Endpunkten gehörte u.a. ein komplettes Ansprechen oder besser (≥CR) sowie ein negativer Status für minimale Resterkrankung (MRD-Negativität) bei Patienten mit ≥CR [2].
Nach einer medianen Nachbeobachtungszeit von 47,5 Monaten
Daratumumab-VRd
geeignete
betrug das progressionsfreie Überleben 84,3 % in der DVRd-Gruppe und 67,7 % in der VRd-Gruppe (HR: 0,42; 95%-KI: 0,30 – 0,59; p <0,001; Abb. 1). Das Risiko für Krankheitsprogression oder Tod lag damit in der DVRd-Gruppe um 58 % unter dem der Standardbehandlung mit VRd [2].
Neben dem signifikant besseren PFS zeigte sich auch eine hohe, sich im Therapieverlauf vertiefende ≥CR-Rate [2]:
• ≥CR-Rate am Ende der Induktion: 22,5 % unter DVRd+DR vs. 21,2 % unter VRd+R
• ≥CR-Rate am Ende der ASZT: 27,9 % unter DVRd+DR vs. 23,4 % unter VRd+R
• ≥CR-Rate am Ende der Konsolidierung 44,5 % unter DVRd+DR vs. 34,7 % unter VRd+R
• Gesamt ≥CR-Rate (beliebiger Zeitpunkt während Studiendauer): 87,9 % unter DVRd+DR vs. 70,1 % unter VRd+R (p < 0,001)
Auch der Anteil der Patienten mit nicht nachweisbarer minimaler Resterkrankung (MRD-Negativität, Sensitivität 10–5) war mit 75,2 % in der DVRd-Gruppe höher als in der VRd-Gruppe mit 47,5 % (p < 0,001). Bei 65 % der Patienten unter DVRd+DR gegenüber 30 %
der Patienten unter Vrd+R ließ sich ein Anhalten der MRD-Negativität über ≥12 Monate beobachten [3].
Das Sicherheitsprofil erwies sich als handhabbar und zeigte sich konsistent mit den bekannten unerwünschten Ereignissen der Einzelwirkstoffe. Häufigste hämatologische unerwünschte Ereignisse (UEs) waren Neutropenien (DVRd-Gruppe: 69,2 % vs. VRdGruppe: 58,8 %) und Thrombozytopenien (48,4 % vs. 34,3 %). Als häufigste nicht-hämatologische UE wurden Diarrhö (61,0 % v. 54,2 %), periphere sensorische Neuropathie (53,6 % vs. 51,6 %), Obstipation (33,9 % vs. 34,0 %) oder Pyrexie (31,6 % vs. 31,4 %) gemeldet. Unter DVRd+DR brachen mit 8,8 % weniger Patienten die Behandlung aufgrund von unerwünschten Ereignissen ab als unter VRD+R mit 21,3 %. Im Beobachtungszeitraum von median 47,5 Monaten verstarben 34 Patienten in der DVRd-Gruppe und 44 in der Gruppe ohne Daratumumab [2].
Fazit
Patienten mit Multiplem Myelom haben in der Erstlinientherapie die größte Chance auf eine gute Re-
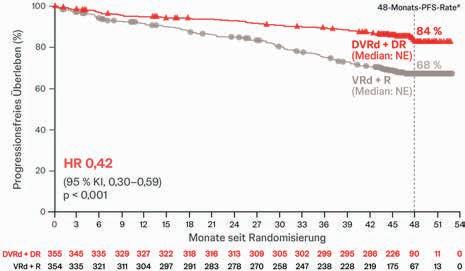
Abbildung 1: Ergebnis der PERSEUS-Studie für den primären Studienendpunkt, das progressionsfreie Überleben. Unter der Therapie mit Daratumumab (Darzalex®) in Kombination mit einer VRd-Induktions- und Konsolidierungstherapie, gefolgt von Daratumumab + Lenalidomid in der Erhaltung (DVRd+DR) verbesserte sich das PFS signifikant im Vergleich zur alleinigen VRd + Erhaltung mit Lenalidomid (VRd + R) (mod. nach [2]). # Kaplan-Meier-Schätzung.
mission und ein langes krankheitsfreies Überleben. Der frühe Einsatz effektiver Therapieoptionen ist daher entscheidend. Für Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine autologe Stammzelltransplantation (ASZT) infrage kommen, kann die neu zugelassene Erstlinie mit Daratumumab-VRd + DR das Überleben verbessern. Wie die Ergebnisse der PERSEUS-Studie belegen, kann das Hinzufügen von Daratumumab s.c. zu einer Standard-Erstlinien-
therapie die Raten des kompletten Ansprechens erhöhen und das Progressionsrisiko signifikant senken. Zudem ist die Behandlung sicher und interferiert nicht mit einer geplanten ASZT. Für Patienten, die aufgrund von Alter und/oder Komorbiditäten nicht für eine ASZT infrage kommen, steht Daratumumab in Kombination mit Lenalidomid und Dexamethason (DRd) als Standardtherapie zur Verfügung. Die zulassungsrelevante Phase-III-Studie MAIA
zeigte hier unter DRd ein medianes OS von 7,5 Jahren (HR: 0,67; 95% KI: 0,55 – 0,82; pnominal < 0,0001) [4].
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Darzalex®, aktueller Stand
2 Sonneveld P et al. N Engl J Med 2024; 390:301-313
3 Rodriguez-Otero P et al. presented ASCO Annual Meeting 2024
4 Facon T et al. Poster P968 presented at EHA 2024
Auf dem Kongress des American College of Rheumatology (ACR) wurden neue Daten zur Wirksamkeit des Interleukin (IL)-17A-Inhibitors Secukinumab (Cosentyx®) bei Psoriasis-Arthritis (PsA) und röntgenologischer axialer Spondyloarthritis (r-axSpA) präsentiert [1]. Die Ergebnisse einer Analyse der auf 5 Jahre ausgelegten, longitudinalen, nicht-interventionellen Real-World-Beobachtungsstudie SERENA [2] bestätigen die langfristige Wirksamkeit von Secukinumab bei PsA und r-axSpA. In die Analyse der SERENA-Daten wurden 522 Patienten mit PsA und 474 Betroffene mit r-axSpA einbezogen, die vor der Aufnahme in die Studie ≥16 Wochen (im Durchschnitt ein Jahr) lang eine Secukinumab-Behandlung erhalten hatten [1].
Über 70 % der Patienten waren auch nach 5 Jahren ohne geschwollene oder schmerzhafte Gelenke
Bei den PsA-Patienten kam es unter der Therapie mit Secukinumab
über den Beobachtungszeitraum von 5 Jahren zu einer deutlichen Reduktion der geschwollenen und schmerzhaften Gelenke, gemessen am Swollen Joint Count (SJC) ≤1 und Tender Joint Count (TJC) ≤1. Einen SJC ≤1 erreichten im ersten Jahr 86,5 % (n = 399) und im fünften Jahr 90,0 % (n = 221). Einen TJC ≤1 erzielten im ersten Jahr 75,7 % (n = 399) und im fünften Jahr 81,0 % (n = 221) der PsA-Patienten. Außerdem wiesen im ersten Jahr 61,3 % (n = 431) keinerlei geschwollene oder schmerzhafte Gelenke mehr auf, im fünften Jahr war dies bei 70,7 % (n = 232) der Fall (Tab. 1) [1].
Auch bei den r-axSpA-Patienten verbesserte sich die Krankheitsaktivität, gemessen am Bath Ankylo-
sing Spondylitis Disease Activity Index (BASDAI), unter der Therapie mit Secukinumab: Im ersten Jahr nahm der BASDAI um –0,09 Punkte ab (n = 340) und im fünften Jahr um –0,34 Punkte (n = 176) (Tab. 1). Somit lag der BASDAI im ersten Jahr bei 3,1 (n = 351) und im fünften Jahr bei 2,6 (n = 178) [1]. Ein BASDAI unter 4 definiert eine niedrige Krankheitsaktivität [3]. Die Krankheitsaktivität hatte sich somit über den Beobachtungszeitraum von 5 Jahren deutlich verbessert (Tab. 1) [1].
Ergänzend zur Krankheitsaktivität wurde die Therapietreue der PsAEndpunkte
Aktive Psoriasis-Arthritis (PsA, n = 522)
Keine schmerzhaften oder geschwollenen Gelenke
SJC ≤1
(239/512)
(333/441)
(264/431)
(345/399)
(225/364)
(293/331)
(204/321)
(263/296)
(190/291)
(238/270)
(164/232)
(199/221) TJC≤1 66,7% (294/441)
(302/399)
(259/331)
Radiologisch nachweisbare axiale Spondyloarthritis (r-axSpA, n = 474)
BASDAI-Score (0–10)
Mittelwert ± SD 3,2 ± 2,3 (n = 448) 3,1 ± 2,3 (n = 351) 2,9 ±
(n = 300)
(237/296)
(208/270)
(179/221)
(n =255)
BASDAI CFB Mittelwert ± SD – –0,09 ± 1,9 (n =340) –0,15 ± 2,1 (n = 290) –0,24 ± 2,1 (n= 246) –0,28 ± 1,9 (n = 213) –0,34 ± 2,0 (n = 176) PtGA NRS ≤2
(98/378)
Tabelle 1: Wirksamkeitsreaktionen unter der Therapie mit Secukinumab (Cosentyx®) bei Patienten mit PsA und r-axSpA vom ersten bis zum fünften Jahr [1]. SJC: Swollen Joint Count, TJC: Tender Joint Count, BASDAI: Bath AS Disease Activity Index, CFB: Change from Baseline, PtGA: Patient’s Global Assessment of Disease Activity. NRS: Numeric Rating Scale.
Secukinumab (Cosentyx®) ist ein vollhumaner, monoklonaler Antikörper, der direkt gegen Interleukin (IL)-17A gerichtet ist [4]. Das Zytokin IL-17A ist an Entzündungsprozessen und der Entstehung von Plaque-Psoriasis, Psoriasis-Arthritis (PsA) axialer Spondyloarthritis (axSpA) und Hidradenitis suppurativa (HS) beteiligt.
Secukinumab ist seit 8 Jahren für mittlerweile 8 Indikationen zugelassen und verfügt über umfangreiche klinische Evidenz in allen Indikationen: Dazu zählen die Plaque-Psoriasis, pädiatrische Plaque-Psoriasis, HS, PsA, nicht-röntgenologische axSpA (nr-axSpA), ankylosierende Spondylitis (AS, r-axSpA) sowie Enthesitis-assoziierte Arthritis (EAA) und juvenile Psoriasis-Arthritis (JPsA), zwei Unterformen der juvenilen idiopathischen Arthritis (JIA).
und r-axSpA-Patienten erfasst, die ebenfalls Aufschluss über die Wirksamkeit einer Therapie geben kann.
Der Therapieverbleib der PsABetroffenen lag im ersten Jahr bei 86,9 % (n = 437) und im fünften
Mit der von der Galderma Laboratorium GmbH ins Leben gerufenen Podcast-Reihe „Rosacea.fm“ erhalten interessierte Dermatologen relevante Informationen rund um die Therapie der Rosacea. In dem Audio-Format tauschen sich Experten aus dem Fachbereich Dermatologie zum Thema „Rosacea“ aus und geben wichtige Insights aus der Praxis.
Im Gespräch fühlt Gastgeber Professor Peter Arne Geber, Düsseldorf, dabei seinen Kollegen auf den sprichwörtlichen Zahn und orientiert sich mit seinen Gesprächspartnern an den aktuellen Bedürfnissen der Patienten und behandelnden Ärzte. Dabei stehen wissenschaftliche Inhalte, thera-
Jahr bei 55,7 % (n = 123). Von den r-axSpA-Patienten blieben im ersten Jahr 86,2 % (n = 391) und im fünften Jahr 61,1 % (n = 131) der Therapie mit Secukinumab treu (Schätzungen durch Kaplan-Meyer-Verfahren) [1] Trotz des langen
Abrufbar ist der Podcast „Rosacea.fm“ auf der Galderma-Website: www.rosacea-info.de/fachkreise/podcast

Beobachtungszeitraums von 5 Jahren und des damit einhergehenden zu erwartenden Abfalls der Therapietreue waren also noch über die Hälfte der im fünften Jahr verbliebenen PsA- und r-axSpA-Patienten auf Secukinumab eingestellt.
Brigitte Söllner, Erlangen
Literatur
1 Kiltz U et al. Arthritis Rheumatol 2024; 76 (Suppl 9)
2 Kiltz U et al. Adv Ther 2020;37:28652883
3 Kiltz U et al. S3-Leitlin Axiale Spondyloarthritis inkl. Morbs-Bechterew-Frühformen. AWMF Leitlinien Regist Nr. 060003, Version 2019
4 Fachinformation Cosentyx®, aktueller Stand
pie- und praxisrelevante Informationen sowie patientenorientierte Themen rund um die Rosacea im Fokus.
Über Galderma Wir sind ein führendes Dermatologie-Unternehmen und in etwa 90 Ländern vertreten. Wir bieten ein innovatives, wissenschaftlich fundiertes Portfolio von Premium-Marken und -Dienstleistungen an, das die gesamte Breite des schnell wachsenden Dermatologie-Marktes abdeckt – von ästhetischen Injektionsbehandlungen über Hautkosmetik bis hin zur therapeutischen Dermatologie. Seit unserer Gründung im Jahr 1981 richten wir unser Engagement und unseren Fokus auf das größte Organ des menschlichen Körpers – die Haut – und erfüllen die individuellen Verbraucher- und Patientenbedürfnisse in partnerschaftlicher Zusammenarbeit mit Angehörigen der Gesundheitsberufe und mit überragenden Ergebnissen. Wir wissen, dass unsere Haut die eigene Lebensgeschichte prägt. Darum ist unser Ziel die Weiterentwicklung der Dermatologie – für jede Hautgeschichte.
Weitere Informationen finden Sie unter: www.galderma.com
Galderma Pressekontakt
Antje Saßenberg
Teamlead Communication & Congress
Galderma Laboratorium GmbH
Toulouser Allee 23a, D-40211 Düsseldorf
Telefon +49 211 58601-4243 Fax +49 211 9367 8811
E-mail antje.sassenberg@galderma.com
In der aktuellen Folge dreht sich alles um die Haarbalgmilbe Demodex folliculorum, die in der Infektiologie eine wichtige Rolle spielt. Die meisten Dermatologen kennen sie im Zusammenhang mit der Er-
krankung Rosacea, bei der sie das Entzündungsgeschehen entscheidend beeinflussen kann. Über den aktuellen Stand der Wissenschaft zum Thema Demodex und die Therapeutika, die in diesem Zusammenhang eingesetzt werden können, diskutiert Professor Gerber mit Professor Martin Schaller, Tübingen. Als ausgewiesener Rosacea-Experte erläutert Professor Schaller neueste Studiendaten, die Auswirkung von Demodex auf den Krankheitsverlauf sowie die Therapieoptionen.
B. S.
Abrufbar ist der Podcast „Rosacea.fm“ auf der Galderma-Website: www.rosacea-info.de/fachkreise/podcast
Abrufbar ist der Podcast „Rosacea.fm“ auf der Galderma-Website: www.rosacea-info.de/ fachkreise/podcast.

Standard in der Therapie des metastasierten hormonsensitiven Prostatakarzinoms (mHSPC) ist die in den Leitlinien mit dem höchsten Empfehlungsgrad aufgeführte Kombination aus einem Androgenrezeptor-SignalwegInhibitor (ARSI) und einer Androgenentzugstherapie (ADT) [1, 2]. Der Androgenrezeptorantagonist Apalutamid (Erleada®) ist bereits seit etwa 5 Jahren für die Therapie des mHSPC zugelassen und wurde seither weiter untersucht, sodass es ergänzend zur Zulassungsstudie TITAN [3] mittlerweile auch Daten aus dem Behandlungsalltag gibt. Bislang stammten diese vor allem aus den USA. Im Herbst 2024 wurden entsprechende Beobachtungsstudien auch aus Europa und Deutschland veröffentlicht [2, 3, 4]. Diese Real-World-Daten stimmen gut mit den Post-hoc-Daten der TITAN-Studie überein und bestätigen den tiefen PSA-Abfall unter Erleada® im Praxisalltag [4, 5, 6].
ProgrammPresseinformation
KeyslidesReferenten
Baseline (PSA90-Ansprechen). Ein Abfall des PSA-Werts auf die Nachweisgrenze von <0,2 ng/ml war nach im Median 3,48 Monaten zu beobachten. Innerhalb von 12 Monaten erreichten 81,5 % der Patienten ein PSA90-Ansprechen und 60,7 % einen PSA-Wert von <0,2 ng/ml. Der tiefe PSA-Abfall im Behandlungsalltag war vergleichbar mit dem in der TITANStudie (Abb. 1).
Johnson & Johnson Oncology
Prof. Dr. med. Axel Hegele Wirksamkeit von ERLEADA® in der TITAN-Studie und im Behandlungsalltag
Europäische Real-World-Studie zum PSA-Ansprechen
ArtemisR ist die erste europäische, länderübergreifende Beob-
unter Apalutamid/ADT
achtungsstudie, die retrospektiv Daten aus den Krankenakten von mHSPC-Patienten sammelt. Ausgewertet wurden die Daten von 242 Patienten in einem medianen Alter von 71 Jahren, die von Februar 2020 bis August 2021 mit Apalutamid/ADT behandelt und im Median über 25,5 Monate nachverfolgt wurden. Die Ergebnisse wurden im November 2024 beim 16th European Multidisciplinary Congress on Urological Cancers (EMUC) in Lissabon vorgestellt [4]. Demnach dauerte es unter einer Apalutamid-Therapie im Alltag im Median 1,94 Monate bis zum Abfall des Werts des prostataspezifischen Antigens (PSA) um ≥90 % gegenüber
Zugleich lebten nach 12 Monaten noch 94,2 % der Patienten, bei 91,0 % hatte noch kein Progress zum metastasierten kastrationsresistenten Prostatakarzinom (mCRPC) stattgefunden und 80,8 % nahmen Apalutamid weiterhin ein [4].
Post-hoc-Analyse TITAN-Studie
PSA-Abfall unter Apalutamid/ADT
Abbildung 1: In der europäischen Beobachtungsstudie ArtemisR war der PSA-Abfall unter der Behandlung mit Apalutamid (Erleada®)/ADT vergleichbar mit dem PSA-Abfall in der Zulassungsstudie TITAN [3, 4].

AmPeL-Studie
PSA-Abfall unter Apalutamid/ADTnach 3 Monaten
Post-hoc-Analyse TITAN-Studie
PSA-Abfall unter Apalutamid/ADT nach 3 Monaten
Abbildung 2: In der AmPel-Studie erreichten die unselektierten Patienten dasselbe PSA-Ansprechen unter Apalutamid (Erleada®)/ADT wie die Patienten in der Zulassungsstudie TITAN [3, 5].
AmPel – eine deutsche Real-World-Studie zum PSAAnsprechen
Die deutsche retrospektive Beobachtungsstudie AmPel untersuchte das PSA-Ansprechen unter Apalutamid bei 117 mHSPC-Patienten (medianes Alter 73 Jahre) aus 9 urologischen Schwerpunktpraxen [5]. Die im September 2024 auf dem Kongress der Deutschen Gesellschaft für Urologie (DGU) in Leipzig vorgestellten Ergebnisse zeigen unter der Therapie mit Apalutamid ein rasches und anhaltendes Ansprechen (PSA-Abfall um 98,2 % nach 3 Monaten und um 99 % nach 9 Monaten). Der PSA-Wert war nach 3 Monaten Apalutamid-Therapie bei 51 % der Patienten auf <0,2 ng/ml gefallen –auch hier spiegeln die Ergebnisse aus dem Alltag die Post-hoc-Auswertungen der TITAN Studie wider, bei der das PSA-Ansprechen nach 3 Monaten ebenfalls 51 % betragen hatte [3] (Abb. 2). Zu ergänzen ist, dass das PSAAnsprechen in der AmPel-Studie vergleichbar war, obwohl die unselektierten Patienten in der Beobachtungsstudie zu Studienbeginn
ein höheres medianes Alter (73 vs. 69 Jahre) sowie häufiger einen ECOG-Performance-Status von ≥1 (74 % vs. 37,5 %) und somit einen schlechteren Allgemeinzustand als die Patienten aus dem Verum-Arm der TITAN-Studie [3] hatten.
AmPel1 (n=117)
Verum-Arm TITAN2 (n=525)
Alter, Median in Jahren (range) 73 (54-87) 69 (45-94)
Allgemeinzustand (ECOG)
Deutsche Real-World-Studie zum Ultra-Low-PSA-Ansprechen
vor Beginn Apalutamid (ng /ml) 11,7 (0,04-4124) 5,97 (0–2682)
ECOG, Eastern Cooperative Oncology Group; PSA, prostataspezifisches Antigen. Mod. nach 1. Hegele A et al. DGU-Kongress 2024: #V03 & Vortrag; 2. Chi KN et al. N Engl J Med. 2019;381:13-24.
In einer weiteren deutschen, unizentrischen, retrospektiven RealWorld-Studie, die Ende August 2024 publiziert wurde, wurde bei 107 mHSPC-Patienten unter Apalutamid-Therapie aus der Uniklinik Frankfurt/Main das Ultra-Low-(UL)-PSA-Ansprechen im Alltag ausgewertet [6]. Die untersuchten UL-PSA-Bereiche befinden sich noch unterhalb der Nachweisgrenze von ≤0,2 ng/ml: Der Ultra-Low-1-PSA-Bereich reicht von 0,2 bis >0,02 ng/ml, der Ultra-Low-2-PSA-Bereich liegt bei ≤0,02 ng/ml [6]. In der Beobachtungsstudie war während eines medianen Followup von 23 Monaten bei rund 2 Drittel der Patienten ein Ultra-LowPSA-Ansprechen zu beobachten.
Dabei betrug der Anteil der Patienten mit UL-1-PSA-Ansprechen 27,1 % und der Anteil mit UL2-PSA-Ansprechen 40,2 %. Damit wurde in der Beobachtungsstudie ein ähnliches UL-PSA-Anspre chen unter Apalutamid erreicht wie in in einer Post-hoc-Analyse der TITAN-Studie (Abb. 3) [6, 7]. Außerdem korrelierte auch hier ein tieferes PSA-Ansprechen mit einem längeren Gesamtüberleben und einer längeren Zeit bis zur Kastrationsresistenz. In beiden Studien kam es bei Patienten mit einem UL-2-Ansprechen zu einem günstigeren Verlauf als bei Patienten mit einem UL-1-Ansprechen; am ungünstigsten war der Verlauf bei Patienten mit PSA-Werten von >0,2 ng/ml [6, 7].


Wie in der AmPel-Studie stimmten auch in der Beobachtungsstudie aus Frankfurt die Ergebnisse mit den Post-hoc-Daten der TITANStudie überein, obwohl die Patienten aus dem Behandlungsalltag eine schlechtere Prognose hatten: So lag der mediane AusgangsPSA-Wert in der Real-World-Studie bei 29 ng/ml, im Verum-Arm der TITAN-Studie dagegen bei 6 ng/ml [3].

UL1-PSA
UL2-PSA
zur Wirksamkeit von Apalutamid in der mHSPC-Therapie gut auf den Behandlungsalltag übertragen werden können.
Brigitte Söllner, Erlangen
Abbildung 3: In der deutschen Real-World-Studie zum Ultra-Low-PSA-Ansprechen erreichten etwa 2 Drittel der Patienten unter Apalutamid (Erleada®)/ADT ein Ultra-Low-PSA-Ansprechen (≤0,2 ng/ml). Der Anteil mit UL-1- bzw. UL-2-PSA-Ansprechen war vergleichbar mit dem in der Zulassungsstudie TITAN erreichten Anteil [3, 6]. UL1: PSA = 0,2 – >0,02 ng/ml; UL2: PSA = ≤0,02 ng/ml.
In der Zulassungsstudie TITAN verbesserte Apalutamid (Erleada)®/ADT im Vergleich zu Placebo/ ADT das Gesamtüberleben (OS) sowie das radiologische progressionsfreie Überleben (rPFS) bei Patienten mit metastasiertem hormonsensitivem Prostatakarzinom (mHSPC). Die Analysen konnten außerdem zeigen, dass ein tieferes PSA-Ansprechen mit einem längeren OS und rPFS korreliert [3].
Die knapp 5 Jahre nach der Zulassung verfügbaren Real-WorldDaten aus 3 Beobachtungsstudien untermauern diese Ergebnisse: Unter Alltagsbedingungen kam es zu einem ähnlichen PSA-Ansprechen und zu einem ähnlich tiefen Abfall des PSA-Werts auf <0,2 ng/ ml nach 3 Monaten wie im Setting der TITAN-Studie, obwohl die Patienten im Alltag z.T. ungünstigere Baseline-Charakteristika hatten. Daraus lässt sich schließen, dass die Ergebnisse der TITAN-Studie
Literatur
1 S3-Leitlinie Prostatakarzinom 2024. www.leitlinienprogramm-onkologie.de/ leitlinien/prostatakarzinom/
2 EAU Guidelines on Prostate Cancer 2024: uroweb.org/guidelines/prostatecancer
3 Chi KN et al. J Clin Oncol 2021;39:22942303
4 Kramer G et al. ArtemisR: Final analysis of a European real-world retrospective study of mHSPC patients treated with apalutamide. 16th EMUC 2024: #P128 & Poster. https://urosource.uroweb.org/resource-centres/EMUC24/261070/abstract
5 Hegele A et al. PSA-Ansprechen unter Apalutamid beim metastasierten hormonsensitiven Prostatakarzinom (mHSPC) –„Real World“ Daten aus der AmPel-Studie. DGU 2024: #V03 & Vortrag. https:// dgu2024.abstractserver.com/program_ 76/#/details/presentations/854
6 Wenzel M et al. Prostate-specific antigen nadir and cancer-control outcomes in real-world apalutamide-treated metastatic hormone-sensitive prostate cancer patients: a single-center analysis. Eur Urol Oncol 2024. doi: 10.1016/j.euo.2024.08. 007
7 Merseburger AS et al. Targeted investigational treatment analysis of novel anti-androgen (TITAN) study: ultralow prostatespecific antigen decline with apalutamide plus androgen-deprivation therapy. BJU Int 2024. doi: 10.1111/bju.16449
bei mittelschwerer bis schwerer Plaque-Psoriasis §












# SO einfach* # SO langanhaltend wirksam** # SO verträglich***
* Oral, 1× täglich – keine Laborkontrollen unter Therapie §§,1
** Langanhaltende§§§ Wirksamkeit in einer Tablette1,2
*** Au ällig unau älliges Sicherheits- und Verträglichkeitsprofil2,3
LTE = long-term extension (Langzeitextension), PASI = Psoriasis Area and Severity Index
§ Zugelassen für Erwachsene mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Therapie infrage kommen1
§§ Vor Einleitung der Behandlung mit SOTYKTU sollten die Patient:innen auf Tuberkuloseinfektionen untersucht werden.1
§§§ Konsistente PASI-Ansprechraten über 4 Jahre (Ergebnisse der Studien POETYK PSO-1, PSO-2 und PSO-LTE)1,2
Referenzen: 1. Fachinformation SOTYKTU, aktueller Stand. 2. Armstrong AW et al. EADV Spring 2024;Präsentation FC02. 3. Lebwohl M et al. Br J Dermatol. 2024;190:668-679.
SOTYKTU 6 mg Filmtabletten. Wirksto : Deucravacitinib. Zusammensetzung: Wirksto : 6 mg Deucravacitinib. Sonst. Bestandteile: Hypromelloseacetatsuccinat, wasserfreie Lactose, mikrokristalline Cellulose, Croscarmellose-Natrium, Siliciumdioxid-Hydrat, Magnesiumstearat, Polyvinylalkohol, Titandioxid, Macrogol, Talkum, Eisen(III)-oxid, Eisen(III)-hydroxid-oxid × H2O. Anwendungsgebiete: Behandlung erw. Pat. mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine system. Therapie infrage kommen. Gegenanzeigen: Überempfindlichkeit gg. den Wirksto o.e.d. sonst. Bestandteile; Klinisch bedeutsame aktive Infektionen (z. B. aktive Tuberkulose). Nebenwirkungen: Sehr häufig: Infektionen d. oberen Atemwege. Häufig: Herpes simplex, orale Ulzerationen, akneiformer Ausschlag, Follikulitis, CPK im Blut erhöht. Gelegentlich: Herpes zoster. Weitere Hinweise: siehe Fachinformation. Verschreibungspflichtig. Pharmazeutischer Unternehmer: Bristol-Myers Squibb Pharma EEIG, Plaza 254, Blanchardstown Corporate Park 2, Dublin 15, D15 T867, Irland. Stand: v1
Frauen sind dreimal häufiger von Multipler Sklerose (MS) betroffen als Männer, und viele von ihnen erhalten die Diagnose mitten in der Familienplanungsphase. Kinderwunsch und Schwangerschaft werden so zu einer Herausforderung, die von großer Unsicherheit geprägt und ein häufiger Grund für den Abbruch einer immunmodulatorischen Therapie ist [1]. Die Sicherheit für Mutter und Kind hat bei der Behandlung der MS höchste Priorität, doch die Entscheidungen gleichen derzeit, insbesondere bei den hocheffektiven Therapien, einem Balanceakt. Aktuelle Daten zu Ocrelizumab (Ocrevus®), das seit 2018 zur Behandlung erwachsener Patienten mit schubförmiger MS (RMS) sowie mit primär progredienter MS (PPMS) zugelassen ist, machen Hoffnung, dass zukünftig eine hocheffektive MS-Therapie nicht mehr aufgrund eines Kinderwunsches depriorisiert werden muss. Denn die Phase-IV-Studien MINORE [2] und SOPRANINO [3] bestätigen die Sicherheit einer Therapie mit Ocrelizumab bei MS während der Schwangerschaft sowie nach der Geburt und Stillzeit. In beiden klinischen Studien lagen die B-Zell-Spiegel der Säuglinge im Normbereich und sowohl der plazentare Ocrelizumab-Transfer als auch die Übertragung von Ocre-
lizumab in die Muttermilch waren größtenteils nicht nachweisbar. Außerdem zeigten Real-World-Daten aus dem mit ca. 4.000 registrierten Schwangerschaften bisher umfangreichsten Datenregister zu einer Anti-CD20-Antikörper-Therapie keine neuen Sicherheitssignale [4].
Minimaler Plazentatransfer und normale B-Zell-Spiegel bei Säuglingen
Wie die Ergebnisse der Phase-IVStudie MINORE zeigen, verändert sich nach Einnahme von Ocrelizumab während der Schwangerschaft die B-Zell-Konzentration bei Säuglingen nicht [2]. Der Anti-
CD20-Antikörper war sowohl im Nabelschnur-Serum bei der Geburt als auch im Säuglings-Serum in der 6. Lebenswoche fast nicht nachweisbar (Abb. 1). Weiterhin entsprachen die beobachteten unerwünschten Ereignisse (UE) den typischerweise bei Schwangerschaft, Geburt, Wochenbett und im Säuglingsalter beobachteten UE [2].
B-Zell-Spiegel von gestillten Säuglingen im Normbereich
In der Phase-IV-Studie SOPRANINO wurde eine vernachlässigbare Übertragung von Ocrelizumab in die Muttermilch festgestellt, ohne Auswirkungen auf die normalen
Ocrelizumab (Ocrevus®) ist ein humanisierter monoklonaler Antikörper, der selektiv gegen CD20-positive B-Zellen gerichtet ist. Dies sind spezielle Immunzellen, die vermutlich wesentlich zur Schädigung der Myelinscheide, der Axone und der daraus resultierenden Behinderung bei Patienten mit MS beitragen. Wie präklinische Studien gezeigt haben, bindet Ocrelizumab an CD20-Oberflächenproteine, die auf bestimmten B-Zellen exprimiert werden, nicht jedoch auf Stammzellen oder Plasmazellen. Deshalb bleiben wichtige Funktionen des Immunsystems erhalten. In der EU ist Ocrelizumab seit Januar 2018 zur Behandlung der schubförmigen sowie der primär progredienten MS bei Erwachsenen zugelassen. Die Zulassungsänderung von Ocrelizumab für schwangere und stillende Frauen wird für das erste Quartal 2025 erwartet [5].
Abbildung 1: In der MINORE-Studie war Ocrelizumab (OCR) sowohl im Nabelschnur-Serum bei der Geburt als auch im Säuglings-Serum in der 6. Lebenswoche fast nicht nachweisbar. CCOD: Klinisches Cut-Off-Datum, GA: Gestationsalter, LLOQ: untere Nachweisgrenze, LMP: letzte Menstruation; a: Wo Ocrelizumab nachweisbar war, lag der Wert nahe der unteren Nachweisgrenze, b: n/N (mod. nach [2]).
B-Zell-Spiegel der gestillten Säuglinge [3]. Ähnlich wie bei der Studie MINORE wiesen auch die gestillten Säuglinge keine messbaren Ocrelizumab-Konzentrationen im Serum auf und ihr allgemeines Gesundheitsbild war mit dem anderer Kinder im 1. Lebensjahr vergleichbar [3].
zeigte keine erhöhte Risikoassoziation für UE während der Schwangerschaft und beim Säugling nach einer In-utero-Exposition mit Ocrelizumab [4].
Fazit für die Praxis
über die Familienplanungszeit mit Schwangerschaft und Stillzeit bis hin zur Menopause und der damit verbundenen Hormonumstellung. Elisabeth Wilhelmi, München
SOPRANINO: B-Zell-Spiegel von gestillten Säuglingen im Normbereich
Real-World-Datenregister bestätigt Sicherheit
Obwohl Ocrelizumab gemäß Label bisher nicht für die Schwangerschaft zugelassen ist, nimmt die Zahl der Patientinnen, die vor, während und nach einer Schwangerschaft Ocrelizumab erhalten haben, stetig zu* [4]. Eine Analyse des bisher umfangreichsten Schwangerschafts-Datenregister
In der Phase -IV-Studie SOPRANINO wurde eine vernachlässigbare Übertragung von Ocrelizumab in die Muttermilch festgestellt, ohne Auswirkungen auf die normalen B -Zell Spiegel der gestillten Säuglinge.2 Ähnlich wie bei der Studie MINORE wiesen auch die gestillten Säuglinge keine messbaren Ocrelizumab -Konzentrationen im Serum auf und allgemeines Gesundheitsbild war mit dem anderer Kinder im 1. Lebensjahr vergleichbar.
Ocrelizumab kann als erste MSTherapie ein umfangreiches PhaseIV-Programm vor, während und nach der Schwangerschaft vorweisen. Die dadurch gewonnene Evidenz gibt dem behandelnden Arzt mehr Sicherheit bei der Therapiebegleitung von MS-Patientinnen
Literatur
1 MS-Register, Kinderwunsch und Schwangerschaft, aktiv! 2023; Nr. 278 1/23: 26 f.
Umfangreiches Real-World-Datenregister bestätigt Sicherheit
* Während der Behandlung mit Ocrelizumab sowie für 6 Monate nach der letzten Infusionsgabe wird bislang zu einer Empfängnisverhütung geraten. Dennoch können während dieses Zeitraums Schwangerschaften auftreten.
2 Hellwig K et al. ECTRIMS 2024; P087
3 Bove R et al. ECTRIMS 2024; O039
4 Dobson R et al. ECTRIMS 2024; P085
5 Europäische Arzneimittel-Agentur (EMA), Periodic Benefit Risk Evaluation Report (PBRER); 30.08.2024
Obwohl OCREVUS gemäß Label bisher nicht für die Schwangerschaft zugelassen ist, nimmt die Zahl der Patientinnen, die vor, während und nach einer Schwangerschaft OCREVUS erhalten haben, stetig zu. 3,a Eine Analyse des bisher umfangreichsten Schwangerschafts Datenregister zeigte keine erhöhte Risikoassoziation für UE während der Schwangerschaft und beim Säugling nach einer in utero-Exposition mit OCREVUS. 3
Best first: zielgerichtete Therapien für die individualisierte Tumortherapie
Progressionfreies Überleben von Patienten unter Palbociclib+ET / Ribociclib+ET in der Erstlinie nach IPTW Palbociclib+ET
Ereignisse n (%) Median [95% KI]
Palbociclib 201 (52,1%) 26,7 [23,2, 30,7]
Ribociclib 124 (53,3%) 27,0 [21,1, 30,7]
Überleben von Patienten unter Palbociclib+ET / Ribociclib+ET in der Erstlinie nach IPTW
Die Einführung der Cyclin-abhängige Kinase 4/6 (CDK4/6)Inhibitoren hat die Therapielandschaft beim HR-positiven, HER2-negativen fortgeschrittenen oder metastasierten Brustkrebs (mBC) nachhaltig verändert. Mit umfangreichen Wirksamkeits- und Verträglichkeitsdaten aus klinischen Studien, ergänzt durch RealWorld-Evidenz, hat sich Palbociclib (Ibrance®) hier breit bewährt. Auf einem von Pfizer ausgerichteten Symposium anlässlich des Kongresses der Deutschen Gesellschaft für Hämatologie und Medizinische Onkologie (DGHO) am 11. Oktober 2024 erläuterte Professor Nadia Harbeck, Brustzentrum der Ludwig-Maximilians-Universität München, die Therapieoptionen in der metastasierten Situation am Beispiel einer 69-jährigen Frau mit Knochen- und Lebermetastasen. CDK4/6-Inhibitoren zeigen laut Harbeck alle auch bei älteren Patientinnen ein konsistentes Sicherheitsprofil. Bei typischen Komorbiditäten wie Herzinsuffizienz biete Palbociclib eine verträgliche Option: „Wir können Palbociclib geben, ohne uns um die KardioToxizität kümmern zu müssen.“ Die Lebensqualität im Alltag ist ein entscheidender Faktor. Auch im Vergleich mit anderen CDK4/6Inhibitoren sei der Benefit von Palbociclib in allen Subgruppen sichtbar. Harbeck fasste die Therapieentscheidung für ihre Patientin mit Komorbiditäten zusammen: „Sie tun der Patientin auf jeden Fall einen Gefallen, wenn Sie mit Palbociclib arbeiten.“
Ereignisse n (%) Median [95% KI]
Palbociclib 201 (52,1%) 26,7 [23,2, 30,7]
Ribociclib 124 (53,3%) 27,0 [21,1, 30,7]
Anzahl unter Risiko
Gesamtüberleben von Patienten unter Palbociclib+ET / Ribociclib+ET in der Erstlinie nach IPTW
Ereignisse n (%) Median [95% KI]
Palbociclib 131 (33,8%) 41,4 [38,8, 50,3]
Ribociclib 77 (32,8%) 49,3 [36,9, NA]
Anzahl unter Risiko
Abbildung 1: OPAL-Register: Wirksamkeit der Therapie mit Palbociclib (Ibrance®) versus Ribociclib in der Erstlinienbehandlung des HR+/HER2– aBC. IPTW: inverse Wahrscheinlichkeitsbehandlungsgewichtung (mod. nach Thill M et al. San Antonio Breast Cancer Symposium 2023, P01-04-21).
Anzahl unter Risiko
Deutsches OPAL-Register: Erstlinientherapie unter Palbociclib versus Ribociclib
Daten aus Real-World-Studien und Registern ergänzen die Evidenzlage für Palbociclib. Ein Beispiel aus der Versorgungsrealität sind die Registerdaten aus dem OPALRegister. Harbeck beschrieb die Ergebnisse einer vergleichenden Effektivitätsbewertung in der Praxis für Palbociclib und Ribociclib in der Erstlinie beim HR+/HER2–aBC (Abb. 1). Von 1049 Patientinnen der Kohorte erhielten 388 Palbociclib plus endokrine Therapie (ET) und 235 Ribociclib plus ET als Erstlinientherapie.
Das adjustierte mediane progressionsfreie Überleben (PFS) betrug 26,7 Monate (95%-KI: 23,2 – 30,7) für die Palbociclib-Gruppe und 27,0 Monate (95%-KI: 21,1 – 30,7) für die Ribociclib-Gruppe (HR: 1,01; 95%-KI: 0,80 – 1,26).
Das adjustierte mediane Gesamtüberleben (OS) lag bei 41,4 Monaten (95%-KI: 38,8 – 50,3) unter Palbociclib plus ET und bei 49,3 Monaten (95%-KI: 36,9 – NA) unter Ribociclib plus ET (HR: 0,99; 95%-KI: 0,72 – 1,29).
Die Analyse zeigt laut Autoren ein vergleichbares Ergebnis – auch wenn keine direkten Vergleiche zwischen den Präparaten gezogen werden können, da es sich nicht um eine prospektive Vergleichsstudie handelt. Wie immer die Entscheidung des Tumorboards aussehen mag: Harbeck betonte, der Grundsatz in der der Mamma-Onkologie sei „best first“. Sie appellierte, die wirklich wirksamen Substanzen möglichst bald zu geben und nicht für spätere Therapielinien aufzusparen.
Martina Freyer, München
Nicht schubförmige sekundär progrediente Multiple Sklerose: Weniger Behinderungsprogression unter Tolebrutinib
„Bisher haben wir die MS als sequenzielles Ereignis verstanden – mit einer präklinischen Phase, einer inflammatorischen Phase, in der die Schübe stattfinden, und schließlich dem Übergang in die progressiven neurodegenerativen Phasen, in der die Krankheitsprogression im Vordergrund steht. Heute gehen wir davon aus, dass die verschiedenen pathologischen Prozesse nebeneinander ablaufen und die neurodegenerative Pathologie von Beginn an eine wichtige Rolle spielt“, erklärte Professor Mathias Mäurer, Würzburg, auf einer Pressekonferenz von Sanofi. Denn trotz des frühzeitigen Einsatzes bisher verfügbarer Disease Modifying Therapies (DMT) zeigen viele Patienten eine schubunabhängige Behinderungsprogression, die sogenannte PIRA (Progression Independent of Relapse Activity). PIRA gilt als Treiber der Progression und tritt über das gesamte MS-Kontinuum hinweg auf. „Der kleinste Teil der Progression unter einer hocheffizienten antiinflammatorischen Therapie geht auf Schübe zurück, der Großteil, also 80 – 90 %, tritt unabhängig von Schüben auf“, erläuterte Mäurer. Professor Luisa Klotz, Münster, ergänzte: PIRA ist häufig und startet oft schon früh – so zeigt etwa ein Drittel der Patienten in den ersten 5 Jahren nach Symptombeginn bereits eine schubunabhängige Progression.“
Die Behinderungsakkumulation wird laut Klotz wahrscheinlich durch eine schwelende Neuroinflammation (Smoldering MS) im ZNS angetrieben, die ein wichtiger
pathophysiologischer Faktor von PIRA ist. „Neben der von peripher getriebenen fokalen Entzündung spielen intrinsische Prozesse im Hirngewebe eine wichtige Rolle. Diese pathologischen Prozesse, an denen Mikroglia und B-Zellen beteiligt sind, finden hinter der geschlossenen Blut-Hirn-Schranke statt und sind demnach für peripher wirksame Medikamente schwer zu erreichen“, verdeutlichte Mäurer. Es besteht daher ein großer ungedeckter medizinischer Bedarf an neuen Therapiemöglichkeiten, die das Fortschreiten der Behinderung adressieren und sowohl peripher als auch zentral im ZNS wirken.
Tolebrutinib kann die Blut-Hirn-Schranke überwinden
Der gehirngängige BTK-Inhibitor (BTKi) Tolebrutinib kann gezielt die biologischen Prozesse adressieren, die die Krankheitsprogression im Gehirn vorantreiben, denn er erreicht die erforderlichen Konzentrationen im ZNS, um auf B-Lymphozyten und krankheitsassoziierte Mikroglia einzuwirken. Tolebrutinib zeigt laut der bislang verfügbaren Daten eine bessere ZNS-Gängigkeit als Evobrutinib, zudem wurde das Studienprogramm zu Tolebrutinib wesentlich breiter angelegt und umfasst das gesamte MS-Spektrum.
HERCULES-Studie: Erstmalig weniger Behinderungsprogression bei nrSPMS unter Tolebrutinib
Für Menschen mit nicht schubförmiger sekundär progredienter MS (nrSPMS) gibt es derzeit noch keine zugelassenen Therapien, daher besteht für diese Gruppe ein hoher Bedarf an neuen Therapieansätzen. Die Phase-III-Studie HERCULES
untersuchte die Wirksamkeit und Sicherheit von Tolebrutinib gegenüber Placebo bei Patienten mit nrSPMS. Die Teilnehmer erhielten randomisiert (2:1) entweder eine tägliche orale Dosis Tolebrutinib oder Placebo über einen Zeitraum von bis zu 48 Monaten. „In der Studie ist es gelungen, eine Population mit geringer fokaler Entzündungsaktivität zu rekrutieren, die weiterhin eine messbare Krankheitsprogression zeigt“, unterstrich Klotz die Besonderheit des Studienkollektivs. Bei Studienbeginn hatte die Mehrheit der Teilnehmer (ca. 87 %) keine Gd- anreichernden Läsionen.
„Der primäre Endpunkt, die Verzögerung der Zeit bis zum Einsetzen einer nach 6 Monaten bestätigten Behinderungsprogression (Confirmed Disability Progression, CDP) konnte mit Tolebrutinib im Vergleich zu Placebo sehr überzeugend erreicht werden. Die Kurven trennen sich kontinuierlich immer weiter voneinander“, berichtete Klotz. „Mit Tolebrutinib konnte erstmalig die Behinderungsprogression bei Menschen mit nrSPMS signifikant reduziert werden. Die relative Risikoreduktion für den primären Endpunkt (Zeit bis zur 6-Monats-CDP) betrug 31 % nach 45 Monaten (p = 0,0026), außerdem konnte sogar eine Verbesserung der Behinderung gezeigt werden – das sind beeindruckende Daten“. Unter Tolebrutinib kam es bei proportional mehr Teilnehmern zu einer nach 6 Monaten bestätigten Verbesserung der Behinderung (Confirmed Disability Improvement; CDI; sekundärer Endpunkt): Im Vergleich zu Placebo wurde eine doppelt so hohe Quote nach 45 Monaten beobachtet (10 % vs. 5 %; p = 0,021).
Darüber hinaus war die jährliche Rate neuer/sich vergrößernder T2-
Läsionen unter Tolebrutinib im Vergleich zu Placebo signifikant reduziert (p = 0,011).
„Erhöhungen der Leberenzymwerte sind ein bekannter Substanzklasseneffekt bei BTKi“, erinnerte Klotz. Unter Tolebrutinib zeigten 4,1 % der Teilnehmer erhöhte Leberenzymwerte (>3ULN) verglichen mit 1,6 % in der Placebogruppe. Bei 0,5 % der Patienten in der Tolebrutinib-Gruppe kam es zu ALT-Spitzenwerten von >20×ULN, die alle innerhalb der ersten 90 Tage der Behandlung auftraten und mit Ausnahme eines Falles ohne Folgeerscheinungen abklangen. Vor Änderung des Studienprotokolls mit stringenterem Monitoring erhielt ein Patient in der Tolebrutinib-Gruppe (n = 754) eine Lebertransplantation und verstarb aufgrund postoperativer Komplikationen. Bis zum heutigen Zeitpunkt hat die häufigere Überwachung derart schwerwiegende Ereignisse im Hinblick auf die Leber minimiert.
GEMINI-Studien:
Tolebrutinib versus. Teriflunomid bei schubförmiger MS
Die Phase-III-Studien GEMINI I und GEMINI II bewerteten Wirksamkeit und Sicherheit von Tolebrutinib im Vergleich zu Teriflunomid bei Patienten mit schubförmiger MS (RMS). Beide Studien verfehlten den primären Endpunkt einer statistisch signifikanten Reduktion der jährlichen Schubrate (ARR) im Vergleich zu Teriflunomid (gepoolte Analyse GEMINI I und II: ARR 0,12 Tolebrutinib vs. 0,12 Teriflunomid).
„Insgesamt war die Krankheitsaktivität bei den Patienten der Studienpopulationen niedrig. Es wurde kein Unterschied im Hinblick auf eine weitere Senkung der ARR
zwischen Tolebrutinib und Teriflunomid beobachtet“, berichtete Mäurer.
„Interessanterweise sieht man aber einen signifikanten Unterschied zwischen Tolebrutinib und Teriflunomid bei schubförmiger MS, wenn man die Zeit bis zur bestätigten 6-monatigen Behinderungszunahme betrachtet“, hob er hervor. So zeigte die Auswertung eines wichtigen sekundären Endpunkts, die gepoolten 6-Monatsdaten zur Bestätigung der Behinderungszunahme (Confirmed disability worsening; CDW), eine bedeutende Verzögerung: Tolebrutinib reduzierte das Risiko in der Zeit bis zum Auftreten einer 6-MonatsCDW um 29 % (nominaler pWert = 0,023). „Dies weist darauf hin, dass Tolebrutinib die pathophysiologischen Prozesse, die zur Krankheitsprogression führen beeinflusst, ohne dass dies durch seine antiinflammatorische Wirkung alleine erklärt werden kann“, kommentierte Mäurer. Die Ergebnisse zur CDW in den GEMINI-Studien bei RMS stehen im Einklang mit der 31-prozentigen Risikoreduktion der Behinderungsprogression (CDP) bei den Patienten in der HERCULES-Studie mit nrSPMS. „Wenn man alle 3 Studien zusammen betrachtet, kann man ganz klar sagen, dass unabhängig von der formalen Eingruppierung der Patienten ein konsistenter Effekt von Tolebrutinib auf die Behinderungsprogression zu sehen ist. Und das ist für unsere Patienten einer der wichtigsten Endpunkte überhaupt“, fasste Klotz zusammen. Mit Spannung werden in 2025 die Ergebnisse der PERSEUS-Studie bei primär progredienter MS erwartet.
Fabian Sandner, Nürnberg
Bei CED immer die Leber mitdenken
Die Leber rückt immer weiter in das Blickfeld von Gastroenterologen, die sich mit chronisch entzündlichen Darmerkrankungen (CED) wie Colitis ulcerosa (CU) und Morbus Crohn (MC) beschäftigen. Wie auf auf einem von der Dr. Falk Pharma GmbH organisierten Satellitensymposium anlässlich des Kongresses „Viszeralmedizin 2024“ deutlich wurde, leidet jeder zweite Patient mit einer CED an extraintestinalen Manifestationen [1]. Häufig sind die Leber und die Gallenblase betroffen [2]. Bei etwa einem Drittel der CED-Patienten sind die Leberwerte erhöht [3], 5 % der Betroffenen entwickeln eine chronische Lebererkrankung [4] und bis zu 8 % der CU-Patienten leiden gleichzeitig an einer primär sklerosierenden Cholangitis (PSC) [5].
Bleiben Lebererkrankungen bei CED-Patienten unentdeckt, kann dies zu Komplikationen führen. Eine regelmäßige Überwachung der Leber ist deshalb bei CED besonders wichtig.
PSC als Muster-Ko-Morbidität bei CED
Einer primär sklerosierenden Cholangitis liegt offenbar eine komplexe Interaktion aus genetischen und Umweltfaktoren zugrunde, ihre genaue Ursache ist aber nicht hinreichend bekannt. Von der chronisch cholestatischen Lebererkrankung sind sowohl die intra- als auch die extrahepatischen Gallengänge betroffen. Etwa 70 % der Menschen mit einer PSC leiden gleichzeitig an einer CED, meist an einer CU [6]. Umgekehrt wird bei schätzungsweise 3 % der Patienten mit
MC und bei bis zu 8 % derer mit CU auch eine PSC festgestellt [5]. Die überwiegende Mehrzahl der Patienten mit einer PSC zeigt laborchemisch eine erhöhte alkalische Phosphatase (AP). Eine erhöhte γ-Glutamyl-Transferase (γ-GT) ergänzt oft das typische cholestatische Laborprofil, die Serumtransaminasen sind meist nur diskret erhöht. Das SerumBilirubin ist bei der Erstdiagnose der PSC überwiegend normal und erhöht sich erst bei zunehmendem Schweregrad der Erkrankung. In der klinischen Untersuchung zeigt sich vor allem eine Hepatomegalie bzw. Splenomegalie. Da die laborchemischen Parameter keine eindeutigen Hinweise auf die PSC liefern, sollte die Diagnose durch eine Magnetresonanz-Cholangiopankreatikographie (MRCP) erfolgen [5].
Im Vergleich zur isolierten CED ist beim Vorliegen einer PSC-CED das Risiko für ein kolorektales Karzinom erhöht. Aus diesem Grund hat die PSC-Diagnose einen hohen Wert. Liegen sowohl hepatobiliäre Erkrankungen als auch chronisch entzündliche Darmerkrankungen vor, sollte das KolonkarzinomScreening früher begonnen und jährlich durchgeführt werden [5].
Das Vorliegen einer PSC bei CED beeinflusst also die Prognose erheblich. Weitere Komplikationen können eine Cholangitis und Cholezystolithiasis, Cholangiokarzinome, Osteoporose, Vitaminmangel und Steatorrhö sein.
CED-Therapien und ihr Einfluss auf die Leber
Neben Ko-Morbiditäten wie der PSC können auch die medikamentösen Behandlungsoptionen der CED ein erhöhtes Risiko für eine
Hepatotoxizität tragen. Deshalb empfiehlt die European Crohn’s and Colitis Organization (ECCO) alle 1 – 3 Monate eine Leberwertbestimmung [1].
5-ASA (5-Aminosalicylsäure)Präparate gehen selten mit einer Drug-Induced Liver Injury (DILI) einher. Allerdings werden unter der Therapie mit Methotrexat bei rund 10 % der Patienten mit CED erhöhte Transaminasen-Werte beobachtet. Auch bei manchen Biologika wurden Fälle von Hepatotoxizität beschrieben [1]. In der klinischen Praxis sollte ein gezieltes Monitoring angestrebt werden, um DILI frühzeitig zu erkennen.
Derzeit existiert keine Standardtherapie der PSC. Die Off-label-Behandlung mit Ursodesoxycholsäure (UDCA) in mittlerer Dosierung kann zu einer Verbesserung der Laborparameter führen [5]. Abhängig vom Erkrankungsstadium können zusätzliche medikamentöse und/ oder endoskopische Verfahren zum Einsatz kommen. Angesichts der derzeit limitierten Therapiemöglichkeiten besteht daher ein hoher Forschungsbedarf an wirksamen medikamentösen Therapien.
Fabian Sandner, Nürnberg
Quellen
1 Harbord M et al. European Crohn’s and Colitis Organisation. J Crohns Colitis 2016;10:239-254
2 Mazza S et al. World J Hepatol 2021; 13:1828-1849
3 Gaspar R et al. World J Hepatol 2021; 13:1956–1967
4 Restellini S et al. Liver Int 2017;37:475489
5 Palmela C et al. Gut Liver 2018;12:17-29
6 Dyson JK et al. Lancet 2018;391:25472559
RSV-Impfung nun auch Kassenleistung bei älteren Erwachsenen
munisierung sollte zeitnah – idealerweise vor Beginn der Erkältungssaison im Herbst – begonnen werden.
Der gemeinsame Bundesausschuss (G-BA) ist der Empfehlung der STIKO [1] gefolgt und hat die Aufnahme der Schutzimpfung gegen Infektionen mit dem Respiratorischen Synzytial-Virus (RSV) für Erwachsene ab 75 Jahren sowie für Menschen von 60 – 74 Jahren, die an schweren Grunderkrankungen leiden oder in Pflegeeinrichtungen leben, in die SchutzimpfungsRichtlinie beschlossen. Damit ist die Impfung gegen RSV für diese Patientengruppe Pflichtleistung der gesetzlichen Krankenversicherungen (GKV) und die Kosten werden vollumfänglich erstattet. Zur Immunisierung soll eine einmalige Impfung mit einem proteinbasierten RSV-Impfstoff erfolgen. Diese wird für die Standardimpfung mit der Ziffer 89137 für die Indikationsimpfung mit der Ziffer 89138 dokumentiert. Mit der Im-
Das Respiratorische SynzytialVirus (RSV) ist ein weltweit vorkommender, ansteckender Erreger, der akute Atemwegserkrankungen auslösen kann.
Erhöhtes Risiko im Alter durch Immunoseneszenz und Grunderkrankungen
Saisonales Auftreten –in Europa von November bis April
Befall der oberen und unteren Atemwege
Symptome und Verlauf
Erkältungsähnlicher Verlauf
Niesen, Schnupfen, Husten, Fieber, verminderter Appetit. Ein symptomloser Verlauf ist möglich.
Infektionen mit dem RS-Virus stellen gerade für ältere Menschen und Personen mit bestimmten Grunderkrankungen eine Gefahr dar (Abb. 1). Denn bei ihnen ist das Risiko erhöht, dass es zu einem schweren Verlauf der Infektion kommt. Angaben des RKI zufolge liegt die Hospitalisierungsinzidenz aufgrund einer RSV-Erkrankung bei Menschen ab 60 Jahren bei 11,4/100.000 und für Personen ab 75 Jahren bei 21,2/100.000. Die Wahrscheinlichkeit, aufgrund einer RSV-Erkrankung im Kran kenhaus zu versterben, wird dabei für die 65–74-Jährigen mit 6,3 % und für 75–79-Jährigen mit 7,5 % angegeben [2]. Ursachen für das erhöhte Risiko von älteren Erwachsenen sind die altersbedingt
Übertragung durch Tröpfcheninfektion; Basisreproduktionszahl R0: ca. 3 ± 0,6: jede infizierte Person steckt im Durchschnitt drei weitere Personen an.1 RSV ist infektiöser als das Influenzavirus (R0 1,16–2,10)2
nachlassende Leistungsfähigkeit des Immunsystems sowie das häufigere Auftreten u.a. von kardiologischen und respiratorischen Grunderkrankungen, die sich im Falle einer RSV-Infektion negativ auf deren Verlauf auswirken können [3]. Umgekehrt können sich auch bestehende Grunderkrankungen durch eine Infektion mit dem RS-Virus verschlimmern [4, 5]. Mit der STIKO-Empfehlung zur Impfung für Risikogruppen und der Übernahme in die Schutzimpfungsrichtlinie können vulnerable ältere Menschen nun per Impfung vor den Folgen einer RSV-Infektion geschützt werden.

Adjuvantierter RSV-Impfstoff nachweislich hochwirksam

Schwerer Verlauf
Befall der unteren Atemwege: Pneumonie sowie Verschlechterung von Grunderkrankungen (COPD, Asthma, Herzerkrankungen). Hospitalisierung und Tod als mögliche Folgen.
RSV betrifft alle Altersgruppen
RSV- Erkrankungen






>> Erhöhtes Risiko für einen schweren Verlauf haben:
• Säuglinge und Kleinkinder
• Ältere Erwachsene (ab 60 Jahren)

Für die Impfung stehen inzwischen verschiedene Impfstoffe zur Verfügung. Der speziell für ältere Erwachsene entwickelter adjuvantierter RSV-Impfstoff Arexvy hat seine hohe Wirksamkeit bei Menschen ab 60 Jahren mit chronischen Grunderkrankungen in
>> Besonders hohes Risiko für schwere RSV-Infektionen und Komplikationen bei älteren Erwachsenen mit chronischen Grunderkrankungen:
• Asthma
• Diabetes
• COPD
• Herzerkrankungen
• Menschen mit chronischen Grunderkrankungen: pulmonale, endokrin-metabolische, kardiovaskuläre und renale Erkrankungen, Immunschwäche (durch Erkrankung oder Medikation)
Abbildung 1: RSV verursacht wiederholte Infektionen während des gesamten Lebens, nicht nur in der Kindheit. Innerhalb des 1. Lebensjahres haben 50 – 70 % und bis zum Ende des 2. Lebensjahres nahezu alle Kinder mindestens eine Infektion mit RSV durchgemacht. Eine langfristige Immunität besteht nicht. Vor allem bei älteren Menschen kann eine Re- oder Neuinfektion zu einem schweren Verlauf führen [2].
Aktive Immunisierung für Erwachsene ab 60 Jahre
Kausale Therapie bei akuter Infektion4 keine
der Zulassungsstudie gezeigt: Bei Vorliegen mindestens einer relevanten Komorbidität (z. B. COPD, Asthma, Diabetes Typ 1 oder 2, chronische Herzinsuffizienz, fortgeschrittene Leber- oder Nierenerkrankung) konnte der Impfstoff das Risiko für eine RSV-bedingte Erkrankung der unteren Atemwege (RSV-LRTD) während der ersten RSV-Saison um 94,6 % reduzie-
ren [6, 7]. Bei Erwachsenen in der Altersgruppe der 70- bis 79-Jährigen erreichte die Impfung einen Schutzeffekt von 93,8 % [7]. Die hohe Wirksamkeit hielt über 2 volle RSV-Saisons an [8].
B. S.
Quellen
1 RKI Epidemiologisches Bulletin 32/2024
2 Robert-Koch-Institut. RKI Ratgeber RSV-
Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Infektionen. Epidemiologisches Bulletin 01/2024
3 Branche AR et al. Clin Infect Dis 2022; 74:1004-011
4 Zwaans WA et al. J Clin Virol 2014;61: 181-188
5 Ivey KS et al. J Am Coll Cardiol 2018; 71:1574-1583
6 Fachinformation Arexvy, aktueller Stand
7 Papi A et al. N Engl J Med 2023;388:595608
8 Ison M et al. Clin Infect Dis, 2024, DOI 10.1093/cid/ciae010
Titelbild: Multiple Sklerose ist eine Autoimmunerkrankung, bei der die körpereigenen Abwehrzellen die Nervenzellen angreifen und Entzündungen auslösen (Quelle: iStock, peterschreiber.media).
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden im Journal für Pharmakologie und Therapie personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz: HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM_W.R. Roland Welker
Austraße 7, 96114 Hirschaid
Verlag PERFUSION GmbH
Storchenweg 20 90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de
E-Mail: perfusion@t-online.de
> 1,6 Millionen Patient*innen1
1 Jahrzehnt, das Vertrauen für die Zukunft schafft: a damals, heute und morgen
Evidenz aus über 200 Studien3
Erfahrung aus 8 Indikationen mit > 1,6 Millionen Patient*innen1,4
Vorreiter als 1. IL-17A-Inhibitor in der Immunologieb,5
a Im 10. Jahr in den Indikationen mittelschwere bis schwere Plaque-Psoriasis (seit Januar 2015), aktive ankylosierende Spondylitis (seit November 2015) sowie aktive Psoriasis-Arthritis (seit November 2015) zugelassen.2 b In den Indikationen Plaque-Psoriasis (bei Erwachsenen), Psoriasis-Arthritis, axiale Spondyloarthritis (AS und nr-axSpA) und Hidradenitis suppurativa.
1. Novartis financial report Q3/2024 - Supplementary Data, Novartis Pharma AG, Basel. https://www.novartis.com/sites/novartis_com/files/2024-10-interim-financial-report-en.pdf (zuletzt abgerufen am 18.11.2024). 2. Cosentyx®: EPAR – Procedural steps taken and scientific information after authorisation. https://www.ema.europa.eu/en/medicines/human/EPAR/cosentyx#authorisation-details (zuletzt abgerufen am 20.11.2024). 3. ClinicalTrials.gov. https://clinicaltrials.gov/search?intr=Secukinumab (zuletzt aufgerufen am 20.11.2024). 4. Fachinformation Cosentyx. 5. EPAR Assessment Report Variation; EMA/CHMP/665405/2015. https://www.ema.europa.eu/en/documents/variation-report/cosentyx-h-c-3729-ii-0002-epar-assessment-report-variation_en.pdf (zuletzt aufgerufen am 20.11.2024). Cosentyx® 75 mg Injektionslösung in einer Fertigspritze, Cosentyx® 150 mg Injektionslösung in einer Fertigspritze, Cosentyx® 150 mg Injektionslösung in einem Fertigpen, Cosentyx® 300 mg Injektionslösung in einer Fertigspritze, Cosentyx® 300 mg Injektionslösung in einem Fertigpen. Wirkstoff: Secukinumab (in Ovarialzellen d. chines. Hamsters [CHO-Zellen] produzierter, gg. Interleukin-17A gerichteter, rekombinanter, vollständig humaner monoklonaler Antikörper d. IgG1/κ-Klasse). Zus.-setz.: Arzneil. wirks. Bestandt.: 1 Fertigspritze enthält 75 mg Secukinumab in 0,5 ml bzw. 1 Fertigspritze/Fertigpen enthält 150 mg Secukinumab in 1 ml bzw. 300 mg Secukinumab in 2 ml. Sonst. Bestandt.: Trehalose-Dihydrat, Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 80, Wasser f. Inj.-zwecke. Anwend.: Behandl. v. Kindern u. Jugendl. ab 6 J. mit mittelschwerer bis schwerer Plaque-Psoriasis, d. für eine system. Therapie in Frage kommen. Behandl. v. Kindern u. Jugendl. ab 6 J. mit Enthesitis-assoziierter Arthritis od. juveniler Psoriasis-Arthritis, allein od. in Kombination mit Methotrexat (MTX), wenn Erkrankung unzureich. auf eine konventionelle Therapie angesprochen hat od. d. diese nicht vertragen. 150/300 mg Injektionslösung zusätzl.: Behandl. erw. Pat. mit mittelschwerer bis schwerer Plaque-Psoriasis, d. für eine system. Therapie in Frage kommen. Behandl. erw. Pat. mit mittelschwerer bis schwerer aktiver Hidradenitis suppurativa (Acne inversa), d. auf eine konventionelle system. HS-Therapie unzureichend angesprochen haben. Behandl. erw. Pat. mit aktiver Psoriasis-Arthritis, allein od. in Kombination mit MTX, wenn d. Ansprechen auf eine vorhergeh. Therapie mit krankheitsmodifizierenden Antirheumatika (DMARD) unzureich. gewesen ist. Behandl. erw. Pat. mit aktiver ankylosierender Spondylitis, d. auf eine konventionelle Therapie unzureich. angesprochen haben. Behandl. erw. Pat. mit aktiver nichtröntgenolog. axialer Spondyloarthritis mit objektiven Anzeichen d. Entzündung, angez. durch erhöhtes C-reaktives Protein (CRP) u./od. Nachweis durch Magnetresonanztomographie (MRT), d. unzureich. auf nichtsteroid. Antirheumatika (NSAR) angesprochen haben. Gegenanz.: Überempfindlichkeit gg. d. Wirkstoff od. einen d. sonst. Bestandt. Klinisch relevante, aktive Infekt. (z. B. aktive Tuberkulose). Nebenw.: Sehr häufig: Infekt. d. oberen Atemwege. Häufig: Oraler Herpes. Kopfschmerzen. Rhinorrhö. Diarrhö, Übelkeit. Ekzem. Ermüdung. Gelegentl.: Orale Candidose, Otitis externa, Infekt. d. unteren Atemwege, Tinea pedis. Neutropenie. Konjunktivitis. Entzündl. Darmerkrankungen. Dyshidrot. Ekzem. Urtikaria. Selten: Anaphylakt. Reakt., Angioödem. Exfoliative Dermatitis, Hypersensitivitätsvaskulitis. Häufigkeit nicht bekannt: Mukokutane Candidose (einschl. ösophageale Candidose). Pyoderma gangraenosum. Verschreibungspflichtig. Weit. Angaben: S. Fachinformationen. Stand: November 2024 (MS 12/24.24). Novartis Pharma GmbH, Sophie-Germain-Str. 10, 90443 Nürnberg. Tel.: (09 11) 273-0. www.novartis.de


12/2024