
PERFUSION
Gesellschaft für Arterioskleroseforschung Current Contents/


ISSN 0935-0020
Clinical Medicine Mitteilungen, Kongressberichte REDAKTIONELLER TEIL Mehr Zeit im Zielbereich nach Umstellung auf Lyumjev® auch Neue Studiendaten zu Dapagliflozin zeigen signifikante Verbesserungen bei Patienten mit Herzinsuffizienz oder Amplatzer Piccolo™ verschließt persistierenden Ductus arteriosus sicher und effektiv Vorteile der dualen antithrombotischen Therapie mit Rivaroxaban plus ASS bei KHK und pAVK Kein erhöhtes Schlaganfallrisiko durch die Impfung Langzeitdaten zu Inclisiran zeigen anhaltende Wirksamkeit Defibrillatorweste – ein wirksamer Schutz Homöopathie unter dem Radarstrahl der Wissenschaft inkl. Video zur Entstehung der HCM Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015;65(12):1249-1254. 1 HCM. Gehen wir es an. www.HCM-angehen.de © 2022 MyoKardia, Inc., a Bristol-Myers Squibb Company CV-DE-2200372
uns näher
gedacht .
Offizielles Organ der Deutschen
HCM-Patienten sind
als
5/6 2022
Kreislauf- und Stoffwechselerkrankungen in Klinik und Praxis
Jahrgang 35, Heft 5/6 Dezember 2022
VERL AG PERFUSION
Offizielles Organ der Deutschen Gesellschaft für Arterioskleroseforschung
Current Contents/ Clinical Medicine
ORIGINALARBEIT
Homöopathie unter dem Radarstrahl der Wissenschaft
ÜBERSICHTSARBEIT
Defibrillatorweste – ein wirksamer Schutz vor dem plötzlichen Herztod
FOREN
Forum diabeticum: Mehr Zeit im Zielbereich nach Umstellung auf Lyumjev® auch bei Typ-2-Diabetes
Forum cardiologicum:
• Neue Studiendaten zu Dapagliflozin zeigen signifikante Verbesserungen bei Patienten mit Herzinsuffizienz oder chronischer Nierenerkrankung
• Amplatzer Piccolo™ verschließt persistierenden Ductus arteriosus sicher und effektiv
Forum antihromboticum: Vorteile der dualen antithrombotischen Therapie mit Rivaroxaban plus ASS bei KHK und pAVK
Forum Ernährung: Wie sinnvoll ist eine Zuckersteuer?
Forum Schlaganfall: Kein erhöhtes Schlaganfallrisiko durch die Impfung gegen SARS-CoV-2
Forum Lipidsenker: Langzeitdaten zu Inclisiran zeigen anhaltende Wirksamkeit und Sicherheit über vier Jahre
REDAKTIONELLER TEIL
Mitteilungen, Kongressberichte
ISSN 0935-0020
Gemeinsam bringen wir die Therapie Ihrer Patienten auf eine neue Ebene

VESOXX® (1 mg/ml Oxybutynin-HCl) wirkt durch seinen multimodalen Wirkmechanismus2–5 und schützt dadurch langfristig die Nieren.6, 7 Die Dosierung von VESOXX® wird entsprechend urodynamischer Parameter patientenindividuell festgelegt.1 Die direkten Wirkungen von VESOXX® in der Blase und die geringere Verstoffwechselung lassen eine hohe Effektivität und eine bessere Verträglichkeit im Vergleich zur oralen Therapie erwarten.1, 7–11
VESOXX (1 mg/ml) wird angewendet zur Unterdrückung einer neurogenen Detrusorüberaktivität bei Kindern ab 6 Jahren u. bei Erwachsenen, d. ihre Blase mittels sauberer intermittierender Katheterisierung entleeren und nicht adäquat mit oralen Anticholinergika eingestellt werden können.1
1. VESOXX® Fachinformation. 2. Murakami S. et al. Urol Int. 2003; 71(3):290–298; (präklinische Studie an isoliertem humanen Blasengewebe mit Antimuskarinika). 3. Chapple C.R. et al. Urology. 2002; 60(5 Suppl 1):82–88; discussion 88–89; (Übersichtsartikel). 4. Kim Y. et al. Urology. 2005; 65(2):238–242; (präklinische Studie an Ratten mit intravesikalen Antimuskarinika). 5. De Wachter S. and Wyndaele J.J. J Urol. 2003; 169(5):1892–1895; (präklinische Studie an Ratten mit intravesikalem Oxybutynin). 6 Pannek J. et al. Urology. 2000; 55(3):358–362; (prospektive Open-Label-Studie mit intravesikalem Oxybutynin in Kombination mit oralem Oxybutynin, n = 25). 7. Humblet M. et al. Neurourol Urodyn. 2015; 34(4):336–342; (retrospective Kohortenstudie mit intravesikalem Oxybutynin, n = 10 bei Re-Evaluation). 8. Krause P. et al. J Urol. 2013; 190(5):1791–1797; (prospektive, randomisierte Cross-Over Open-Label-Studie (Periode I und II: orales oder intravesikales Oxybutynin, Periode III: intravesikales Oxybutynin), n = 20). 9. Buyse G. et al. 1998; 160(3 Pt 2):1084–1087; discussion 1092; (prospektive Open-Label-Studie mit intravesikalem Oxybutynin, n = 15). 10. Oki T. et al. J Urol. 2004; 172(5 Pt 1): 2059–2064; (präklinische Studie an Ratten mit oralem und intravesikalem Oxybutynin). 11. Schröder A. et al. Neurourol Urodyn. 2016; 35(5):582–588; (randomisierte, prospektive, aktiv kontrollierte, multizentrische Open-Label-Studie mit intravesikalem Oxybutynin (n = 18) und oralem Oxybutynin (n = 17)).
VESOXX 1 mg/ml, Lösung zur intravesikalen Anwendung. Wirkst.: Oxybutyninhydrochlorid. Zus.: 1 ml Lös. enth. 1 mg Oxybutyninhydrochlorid; 1 skalierte Fertigspritze m. 10 ml Lös. enth. 10 mg Oxybutyninhydrochlorid. Sonst. Bestandt.: Salzsäure, Natriumchlorid, Wasser f. Inj.-zwecke. Anw.: Zur Unterdrück. einer neurogenen Detrusorüberaktivität (Neurogenic Detrusor Overactivity; NDO) b. Kdrn. ab 6 J. u. b. Erw., die ihre Blase mittels sauberer intermittierender Katheterisier. (CIC) entleeren, wenn sie durch eine Beh. m. oralen Anticholinergika aufgrund mangelnder Wirksamkeit und/oder unerträglicher Nebenwirk. nicht adäquat eingestellt werden können. Gegenanz.: Überempfindlichk. gg. d. Wirkst. od. sonst. Bestandt.; schwere gastrointest. Erkrank. (z. B. schwere Colitis ulcerosa u. tox. Megakolon); Myasthenia gravis; Engwinkelglaukom u. Pat. m. einem Risiko dafür; begleit. Sauerstoffther. Nebenwirk.: Harnwegsinfekt.; asymptomat. Bakteriurie; Hyperprolaktinämie; Prolaktin erhöht; Teilnahmslosigk.; Halluzinat.; kognitive Stör.; Hyperaktivität; Schlaflosigk.; Schlafstör.; Agoraphobie; Orientierungsstör.; Aufmerksamkeitsstör.; Schwindelgefühl; Kopfschmerz; Somnolenz; Erschöpf.; Dysgeusie; getrübter Bewusstseinszustand; Bewusstlosigk.; anticholinerges Syndr.; Krampfanfall; Vertigo; Trockenes Auge; anomale Sinnesempfind. d. Auges; Akkommodationsstör.; supraventrik. Tachykardie; Hypotonie; Gesichtsröt.; Obstipat.; Mundtrockenh.; abdominale Beschwerden; Schmerzen im Unter- od. Oberbauch; Übelk.; Dyspepsie; Diarrhö; Hypohidrose; Ausschlag; nächtl. Schwitzen; (verstärkter) Harndrang; Proteinurie; Hämaturie; Stör. b. d. Entleer. d. Harnblase; Schmerzen an d. Instill.-stelle; Durst; Brustkorbbeschwerden; Kältegefühl. Verring. Sauerstoffsätt. im Rahmen einer Sauerstoffther. Bek. NW einer anticholinergen Ther. (bisher b. intravesikaler Anw. v. Oxybutynin nicht beob.): Erbrechen; Anorexie; vermind. Appetit; Dysphagie; gastroösophag. Refluxkrankh.; Pseudoobstrukt. b. Risikopat. (ältere Personen od. Pat. m. Obstipat. u. b. Behandl. m. and., die intest. Motilität verring. AM); Verwirrth.-zustand; Agitiert.; Angst; Alpträume; Paranoia; Sympt. einer Depress.; Abhängigk. v. Oxybutynin (b. Pat. m. einer Vorgeschichte v. Drogen- od. Substanzmissbrauch); Arrhythmie; Hitzschlag; (Engwinkel-) Glaukom; Augeninnendruck erhöht; trockene Haut; Angioödem; Urtikaria; Photosensitivität; Überempfindlichk. Kdr. könnten empfindlicher f. d. Wirk. d. Produktes sein, insbes. in Hinblick auf psychiat. u. d. ZNS betreff. NW. Warnhinw.: Enth. d. sonst. Bestandteil m. bek. Wirk. Natrium (3,56 mg/ml). Weit. Angaben: s. Fach- u. Gebrauchsinfo. Verschreibungspflichtig.
FARCO-PHARMA GmbH, Gereonsmühlengasse 1-11, D 50670 Köln. Stand: 08/2022.
VESOXX® (1
Oxybutynin-HCl) – die intravesikale Lösung zur Behandlung der neurogenen Detrusorüberaktivität1 Alle Informationen zu Verordnung und Erstattung finden Sie unter www.vesoxx.de NEU: VESOXX® für NDO Patient*innen – unabhängig ihrer Grunderkrankung z. B. Spina bifida, Querschnittlähmung, Multiple Sklerose, Morbus Parkinson
mg/ml
Warum ich wieder Deutscher wurde
Der Prozess begann vor über 2 Jahren, als ich zur deutschen Botschaft in London ging, um meinen lange abgelaufenen Pass erneuern zu lassen. Dort reichte ich der Beamtin meinen alten Reisepass und bat darum, ihn zu verlängern. Sie schaute ihn an, dann mich, dann wieder den Pass. Sodann konfiszierte sie meinen alten Pass und meinte unmissverständlich: Sie sind kein Deutscher mehr! Wenn ich wieder Deutscher werden wolle, müsse ich ganz von vorne anfangen und mich neu einbürgern lassen. Als ich 1999 die britische Staatsbürgerschaft annahm, hätte ich die doppelte Staatsbürgerschaft beantragen müssen. Da ich dies unterlassen hatte, verlor ich meine deutsche Staatsangehörigkeit. Um ehrlich zu sein, damals wäre mir das alles sowieso egal gewesen –ich hatte einen Europäischen Pass, und das war mir genug. Aber das war 1999, und jetzt hatte ich meine Meinung geändert. Nun folgte ein zweijähriger Antragsprozess. Das bedeutete, Dokumente über Dokumente zu finden, zu beschaffen und einzureichen. Ich hätte mich nicht gewundert, wenn man mich nach den Fingerabdrücken meiner Großmutter gefragt hätte. Aber warum wollte ich überhaupt wieder ein Deutscher werden? Wer mich gut kennt, weiß, dass ich nie wirklich stolz darauf war, Deutscher zu sein. Als ich heranwuchs, habe ich mich oft sogar dafür geschämt. Eines meiner Forschungsthemen war lange Zeit die Medizin im Dritten Reich, und es war dieses Thema, das mich dann gänzlich
Prof. Dr. med. E. Ernst, Exeter, U.K. von Deutschland entzaubert hat. Nachdem ich 1993 einen Ruf an die Uni Exeter angenommen hatte, schien es mir völlig logisch, Brite zu werden. Ich war sogar stolz darauf; das Vereinigte Königreich war meine Wahlheimat, und diese Entscheidung sollte fürs Leben sein. Nicht in meinen kühnsten Träumen hätte ich damals gedacht, dass Großbritannien eines Tages dem kollektiven Todeswunsch erliegen würde, die EU zu verlassen. Nach dem zutiefst unehrlichen Referendum im Jahr 2016 war ich immer noch überzeugt, dass dieser Akt außergewöhnlicher Selbstschädigung verhindert werden würde. Aber dann geschah es doch, nicht zuletzt aufgrund der Lügen des rechten Flügels der Tory-Partei und der doppelzüngigen Inkompetenz von Jeremy Corbyn und seinen Anhängern. Ich erinnere mich an den Morgen, an dem ich das Ergebnis des Referendums im Radio hörte – ich war den Tränen nahe. Der Brexit an sich wäre schon schlimm genug gewesen, aber die Art und Weise, wie er die Atmosphäre in Großbritannien veränderte, war meiner Meinung nach noch schlimmer. Mit den Nationalisten an der Macht schien sich jeder Schwachkopf berechtigt zu fühlen, seine fremdenfeindlichen Gedanken nach Belieben kundzutun. Lügen und Korruption in der Politik wurden zur neuen Norm.
Die Presse schwieg weitgehend oder unterstützte sogar diejenigen, die behaupteten, der Brexit würde nur Vorteile haben und das große britische Empire zurückbringen. Die wenigen, die den Mut hatten zu widersprechen, wurden zu „Volksfeinden“ erklärt. Ich und viele Gleichgesinnte hatten das Gefühl, dass Großbritannien schnell zu einer Bananenrepublik wurde. Das meiste von dem, was mich einst dazu gebracht hatte, ein Brite zu sein, war innerhalb weniger Monate verschwunden. Es war also an der Zeit, meinen Standpunkt zu überdenken. Der erste Schritt, den meine Frau und ich unternahmen, war, nach Cambridge zu ziehen. Wir empfanden diesen Ort als eine Enklave der Vernunft in einem Land, das sich selbst zu zerstören schien. Der zweite Schritt bestand darin, mehr Zeit in der Bretagne zu verbringen, dem Geburtsort meiner Frau. Unsere französischen Freunde waren ausnahmslos der Meinung, dass das Vereinigte Königreich den Verstand verloren habe und zur Lachnummer Europas geworden war. Der dritte Schritt bestand darin, wieder die deutsche Staatsbürgerschaft zu beantragen. Jetzt, da dies alles erledigt ist, bin ich froh und traurig zugleich. Glücklich, wieder Mitglied der Europäischen Union zu sein, und traurig, zu sehen, was aus dem Land geworden ist, das ich so geliebt habe.
Edzard Ernst, Emeritus Professor, University of Exeter
Perfusion 5-6/2022 35.
© Verlag PERFUSION GmbH 5-6/2022 133
Jahrgang
EDITORIAL
148 Forum diabeticum 150, 157 Forum cardiologicum 152 Forum Ernährung 155 Forum Schlaganfall 158 Forum Lipidsenker 143, 147, Mitteilungen 149, 159 161 Kongressberichte
Offizielles Organ der Deutschen Gesellschaft für Arterioskleroseforschung Current Contents/Clinical Medicine
148 Forum diabeticum 150, 157 Forum cardiologicum 152 Forum nutrition 155 Forum stroke 158 Forum lipid lowering drugs 143, 147, Informations 149, 159 161 Congress reports
INHALT
EDITORIAL
133 Warum ich wieder Deutscher wurde E. Ernst
ORIGINALARBEIT
136 Homöopathie unter dem Radarstrahl der Wissenschaft Norbert Aust, Viktor Weisshäupl
144
ÜBERSICHTSARBEIT
Defibrillatorweste – ein wirksamer Schutz vor dem plötzlichen Herztod Brigitte Söllner
CONTENTS
EDITORIAL
133 Why I became a German again E. Ernst
ORIGINAL PAPER
136 Homeopathy under the science radar Norbert Aust, Viktor Weisshäupl
144 REVIEW
Defibrillator vest –- effective protection before sudden cardiac death Brigitte Söllner
Heft 5/6
Dezember 2022

Raisa Pavlova flieht vor den Kämpfen
unsere Mitarbeiterin
KRIEGEN SETZEN WIR HOFFNUNG ENTGEGEN Mit Ihrer Spende rettet ÄRZTE OHNE GRENZEN Leben: Mit 52 Euro können wir zum Beispiel 40 Menschen auf der Flucht drei Monate lang mit den wichtigsten Medikamenten versorgen. Private Spender*innen ermöglichen unsere weltweite Hilfe –jede Spende macht uns stark Spendenkonto: Bank für Sozialwirtschaft IBAN: DE72 3702 0500 0009 7097 00 BIC: BFSWDE33XXX www.aerzte-ohne-grenzen.de/spenden
REPUBLIK MOLDAU:
in der Ukraine,
Svetlana Bujac bietet ihr Hilfe an. © Peter Bräunig
ORIGINALARBEIT
Homöopathie unter dem Radarstrahl der Wissenschaft
Norbert Aust1, Viktor Weisshäupl2 1 Informationsnetzwerk Homöopathie, Deutschland 2 Initiative für Wissenschaftliche Medizin, Österreich PERFUSION 2022; 35: 136 – 142
Im November 2020 wurde eine Studie zur homöopathischen Zusatzbehandlung von Patienten mit nicht-kleinzelligem Lungenkrebs veröffentlicht [1]. Hauptautor ist Professor Michael Frass, der die Medizinische Universität Wien (MedUni Wien) als Institutszugehörigkeit angibt, allerdings zum Zeitpunkt der Veröffentlichung pensionsbedingt dort nicht mehr tätig war. Diese Studie ragt in vielerlei Hinsicht aus den üblichen Homöopathiestudien heraus:
• Sie wurde nicht in einem von den in großer Zahl am Markt befindlichen Journalen der Komplementär- und Alternativmedizin veröffentlicht, sondern in der angesehenen Fachzeitschrift „The Oncologist“ (Impact Factor >5) unter Peer Review [2].
• Das untersuchte Krankheitsbild, der nicht-kleinzellige Lungenkrebs, stellt ohne Zweifel eine sehr schwere Erkrankung dar, an der 4 von 5 Patienten innerhalb von 5 Jahren versterben [3].
• Die Studie wurde als randomisierte, doppelt verblindete, placebokontrollierte Vergleichsstudie ausgeführt, dazu an mehreren Standorten in Österreich. Damit wird der „Goldstandard“ noch deutlich übertroffen. Die beschriebenen Verfahren recht-
Zusammenfassung
Im November 2020 wurde eine Studie zur Homöopathie als Zusatzbehandlung bei Patienten mit fortgeschrittenem nicht-kleinzelligem Lungenkrebs veröffentlicht. Dabei soll es den HomöopathiePatienten wesentlich besser ergangen sein und sie die Diagnose deutlich länger überlebt haben als die Patienten, die stattdessen nur Placebo erhielten. Eine eingehende Analyse und der Vergleich mit Daten aus dem Studienregister ergaben jedoch, dass die Autoren die Daten sehr wahrscheinlich manipuliert oder gefälscht haben. Das Versuchsprotokoll wurde erst zu einem Zeitpunkt aufgestellt, als die Ergebnisse der Versuchspersonen bekannt waren. Dabei wurden die Beobachtungszeit von 104 auf 18 Wochen erheblich gekürzt und die Ausschlusskriterien von 1 auf 11, später auf 22 beträchtlich erweitert. Diese Festlegungen können somit zur Beeinflussung der Studienergebnisse genutzt worden sein. Bestimmte Merkmale der veröffentlichten Überlebenskurven deuten darauf hin, dass dies auch geschehen ist. Die Österreichische Agentur für Wissenschaftliche Integrität hat diesen Befund inzwischen bestätigt und die Retraktion der Studie empfohlen. Dieser Fall macht auch deutlich, dass Autoren bisweilen darin erfolgreich sind, Studien zur Homöopathie in renommierten Journalen zu veröffentlichen. Daher wird an alle am Veröffentlichungsprozess Beteiligte appelliert, solche Arbeiten sehr genau zu prüfen, ob positive Ergebnisse der Homöopathie oder anderer unplausibler Behandlungsverfahren wirklich auf solider Forschung beruhen und entsprechend Stellung zu nehmen.
Schlagwörter: Homöopathie, Lungenkrebs, Datenmanipulation, Wissenschaftliche Veröffentlichungen
Summary
A study on the adjunct homeopathic treatment of patients in advanced stages of non small-cell lung cancer was published in November 2020. Apparently patients who received this treatment fared much better and had a much prolonged survival compared
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
N. Aust, V. Weisshäupl: Homöopathie unter dem Radarstrahl der Wissenschaft
136
to patients that received placebo instead. However, a thorough analysis and comparison with data from the study register showed, that very likely the authors might have modified or falsified their results. The protocol was compiled only after the study was completed and the patient data known to the authors. Follow-up time was reduced drastically from 104 weeks to 18 weeks only. The number of exclusion criteria was inflated from originally 1 to 11, later to 22 items. These settings could have been deployed to modify the results. The survival functions as published show characteristics that suggest, this really has happened. The Austrian Agency for Scientific integrity confirmed these findings and recommended the study to be withdrawn. This case is an example that authors try to publish studies on homeopathy in respectable journals, sometimes successfully. All the parties involved in the publication of scientific papers should double-check manuscripts about homeopathy and other implausible treatments very thoroughly if they really were the outcome of solid scientific research and should take appropriate action if not.
Keywords: homeopathy, lung cancer, data manipulation, scientific publications
fertigen es ohne weiteres, der Studie nur ein geringes Risiko eines Bias zuzusprechen.
• Die Untersuchung wurde lange vor dem Start bei ClinicalTrials. gov registriert [4], das Studienprotokoll wurde als begleitendes Dokument hochgeladen [5].
• Die berichteten Resultate sind sensationell: Die mediane Überlebenszeit war unter der homöopathischen Zusatzbehandlung um 70 % höher als unter Placebo. Lebensqualität und subjektives Wohlbefinden verbesserten sich mit der Homöopathie kontinuierlich und deutlich in allen Aspekten, während es den Patienten nur mit Placebo immer schlechter ging [1]. Ohne Zweifel handelt es sich hier um signifikante und klinisch relevante Effekte, was in den bisher vorliegenden hochwertigen Studien zur Homöopathie noch nie zu verzeichnen war.
Damit könnte die Studie als valider Beleg für eine durchgreifende Wirksamkeit der Homöopathie gelten. Die Vermutung, dass die Homöopathie auch in anderen, weniger tiefgreifenden Indikationen eine Wirksamkeit über Placebo hinaus entfalten könnte, wäre zumindest naheliegend.
Die zutage getretene starke Wirksamkeit weckt indes erhebliche Zweifel. Bisher ist in noch keiner Studie ein derartig durchschlagender Effekt aufgetreten [6], obwohl weitgehend dieselben Mittel eingesetzt wurden wie in vielen anderen Studien auch. Warum haben sie nicht in anderen weniger schweren Krankheitsbildern deutlichere Effekte gezeigt als die eher geringen Effektstärken, wie sie in allen systematischen Übersichtsarbeiten zitiert werden? Präparate, die praktisch keine Bestandteile aus dem Ausgangsstoff mehr enthalten und nur aus geschütteltem Lösungsmit-
tel bzw. dem Arzneiträger bestehen, sollen in der Lage sein, prinzipiell zum Tode führende pathologische Prozesse deutlich zu verzögern? Aufgrund dieser Zweifel haben das Informationsnetzwerk Homöopathie aus Deutschland und die Initiative für Wissenschaftliche Medizin aus Österreich eine mehrköpfige Arbeitsgruppe gebildet, um die Studie genauer zu untersuchen.
Methode
Gegenstand der Untersuchungen waren alle Unterlagen, die über das Internet frei verfügbar sind. Dies sind:
• Der Text der Studie, in der Folge als „Veröffentlichung“ bezeichnet [1]
• Die Registerdaten bei Clinical Trials.gov in den verschiedenen Datenständen, sowohl die aktuellen Einträge [4] als auch die älteren Versionen, die über die „History of Changes“ zugänglich sind [7]. Zum Zeitpunkt der Untersuchung war der Eintrag vom 6. November 2020 aktuell.
• Das zu den Registerdaten hochgeladene Studienprotokoll, datiert auf den 10. Januar 2011 [5] sowie im weiteren Verlauf die aktualisierte Fassung, datiert auf den 6. Februar 2014 [8].
Die einzelnen Angaben wurden in eine zeitliche Reihenfolge gebracht und miteinander verglichen. Dabei wurde versucht, den Ablauf zu verstehen, um die Vorgehensweise mit den Anforderungen einer validen Forschungsarbeit zu vergleichen. Nachdem die Ergebnisse vorlagen, wurden die Studienautoren im Juni 2021 angeschrieben und über den Befund informiert. Dem Haupt-
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
N. Aust, V. Weisshäupl: Homöopathie unter dem Radarstrahl der Wissenschaft
137
Registrierung Januar 2012 [10] Veröffentlichung November 2020
Zahl der Teilnehmer 600 150
Zahl der Studienarme 2 3
Zahl der Ausschlusskriterien 1 22
Beobachtungszeit Lebensqualität 104 Wochen* 18 Wochen
Zahl der Krebsarten 3 1
* Abgeleitet aus der Angabe „Time Frame: 7 Years“ abzüglich der Rekrutierungszeit von 5 Jahren
Tabelle 1
[1]
Register 5. August 2018 [12] Protokoll 18. September 2019** [5]
Zahl der Teilnehmer 150 300
Zahl der Studienarme 3 3
Zahl der Ausschlusskriterien 1 11
Beobachtungszeit Lebensqualität 104 Wochen* 18 Wochen
Zahl der Krebsarten 1 1
* Abgeleitet aus der Angabe „Time Frame: 7 Years“ abzüglich der Rekrutierungszeit von 5 Jahren
** Datum des Hochladens zu ClinicalTrials.gov, das Dokument trägt das Datum vom 10. Januar 2011.
Tabelle 2
autor wurde expressis verbis die Möglichkeit zur Stellungnahme gegeben. Dies ist jedoch nicht erfolgt. Neben den Autoren wurden auch der Vorsitzende der Ethikkommission sowie die Forschungsrektorin der MedUni Wien über die Ergebnisse in Kenntnis gesetzt, die ihrerseits bei der Österreichischen Agentur für Wissenschaftliche Integrität eine Untersuchung der Arbeit anregte. Dort wurden zusätzlich Originaldaten und die Protokolle ausgewertet, wie sie der Ethikkommission vorlagen. Die Ergebnisse sind nur insoweit publik, wie sie im Nachrichtenmagazin „Profil“ veröffentlicht wurden [9], der Originalbericht ist leider nicht öffentlich zugänglich.
parameter vorgenommen worden, zu denen aber in der Veröffentlichung nichts ausgesagt wird (Tab. 1). Erkennbar sind die beträchtlich angestiegene Zahl der Ausschlusskriterien von 1 auf 22 und die drastische Abkürzung der Beobachtungszeit für die Lebensqualität von 104 auf 18 Wochen, die auch für das subjektive Wohlbefinden gilt. Beides sind die Hauptzielkriterien dieser Studie.
Ergebnisse
Während der Studie sind erhebliche Änderungen wesentlicher Studien-
35. Jahrgang
Die Angaben zu diesen Studienparametern in der ersten hochgeladenen Version des auf den Januar 2011 datierten Protokolls stimmen jedoch besser mit der Veröffentlichung überein als die Angaben in der späteren Erstregistrierung (Tab. 2). Allerdings wurde das Protokoll nicht vor Beginn der Studie publiziert, wie es das angegebene Datum suggeriert, sondern es wurde erst sehr viel später, am 18. September 2019, zu den Registerdaten hochgeladen [11]. Dies ist ein Zeitpunkt
ca. 2 Monate nach Abschluss der Datenerfassung im Juli 2019. Es ist anzunehmen, dass zu diesem Datum bereits erste Auswertungen vorlagen. Angaben im Protokoll deuten ebenfalls auf einen wesentlich späteren Entstehungszeitpunkt hin als das angegebene Datum vom Januar 2011: So wird eine Softwareversion beschrieben (SPSS –25.0), die erst 2017 verfügbar war, und es wird eine Literaturangabe offenbar durch Copy und Paste übernommen, die mit dem Literaturverzeichnis der Veröffentlichung übereinstimmt, während es im Protokoll kein Quellenverzeichnis gibt.
Daraus ist zu schließen, dass die Angaben im Protokoll, sofern sie vom vorher aktuellen Datenstand des Registers abweichen, erst post hoc festgelegt wurden, während mit der Datumsangabe des Protokolls ein Zeitpunkt vor Studienbeginn vorgespiegelt wird.
Perfusion 5-6/2022
©
PERFUSION GmbH
Verlag
N. Aust, V. Weisshäupl: Homöopathie unter dem Radarstrahl der Wissenschaft
138
Tabelle 2 zeigt den Vergleich der ersten Protokollfassung mit den vor dem Upload aktuellen Registereintrag. Man erkennt die hier erstmals eingeführte Abkürzung der Beobachtungszeit und den starken Anstieg der Ausschlusskriterien, die bis zur Veröffentlichung, also zu einem noch späteren Zeitpunkt, auf 22 anwuchs. Beide Änderungen sind demnach post hoc erfolgt. Etwa 2 Wochen nachdem die Autoren über unsere Ergebnisse informiert worden waren, wurde am 14. Juni 2021 eine zweite Protokollfassung hochgeladen, die hinsichtlich der Studienparameter vollkommen mit der Veröffentlichung übereinstimmt [8]. Der Text wurde überarbeitet, sodass die Anhaltspunkte für einen späteren Entstehungszeitpunkt nicht mehr enthalten sind. Diese Protokollversion wurde auf den 6. Februar 2014 vordatiert. Zu diesem Zeitpunkt lief die Studie bereits 2 Jahre und die ersten Patienten könnten ihre Nachbeobachtungszeit gerade abgeschlossen haben. Mithin wäre dies immer noch ein angemessener Punkt für genauere Festlegungen der Studienparameter. Die ältere Protokollversion wird nicht erwähnt. Die zweite Protokollversion kann nur als Versuch verstanden werden, den wahren Sachverhalt erneut zu verschleiern und die Ergebnisse der Arbeitsgruppe als gegenstandslos erscheinen zu lassen.
Die Eintragungen und das Protokoll vermitteln den unzutreffenden Eindruck, die Studienparameter wären zu einem Zeitpunkt festgelegt worden, als die Resultate der einzelnen Patienten noch weitestgehend unbekannt waren, und dies sei während der gesamten weiteren Studiendauer konsequent eingehalten worden.
Das alles legt die Vermutung nahe, dass die Studienergebnisse auf manipulierten oder verfälschten Daten beruhen und nicht auf soliden Forschungsergebnissen.
Post-hoc-Festlegungen
Die in der Veröffentlichung nicht dokumentierte post hoc eingeführte drastische Verkürzung der Beobachtungszeit für die Hauptzielkriterien „Lebensqualität“ und „subjektives Wohlbefinden“ von 104 auf 18 Wochen führen zu einem selektiven Berichten der Studienergebnisse. Man darf unterstellen, dass der Bericht dann abgebrochen wurde, als die Ergebnisse für die Homöopathie den Optimalpunkt erreicht hatten. Damit wird zwangsläufig die wahre Wirkung ins Positive überzeichnet dargestellt. Die berichteten Zahlenwerte sind nicht repräsentativ für das Gesamtgeschehen und daher ungeeignet für eine Bewertung. Dies ist ein schwerwiegender Studienmangel, weil eine solche Vorgehensweise eine nachträgliche Manipulation der Daten darstellt [13].
Die zweite starke post hoc erfolgte Änderung ist der beträchtliche Zuwachs an Ausschlusskriterien, was ebenfalls nicht in der Veröffentlichung beschrieben wird. Die auf diese Weise ausgeschlossenen Patienten erscheinen auch nicht im Consort-Flussdiagramm, das die Entwicklung der Teilnehmerzahlen in den verschiedenen Stufen darstellen soll [1].
Bei den Ausschlusskriterien ist kein Schema zu erkennen, nach dem es selbsterklärend wäre, warum Patienten etwa mit einer koronaren Herzerkrankungen, mit Nieren- oder Lebererkrankungen nicht teilnehmen
durften, Diabetes, Bluthochdruck oder Magenerkrankungen hingegen keine Ausschlusskriterien darstellten. Angesichts der vergleichsweise geringen Teilnehmerzahl in Homöopathie- und Placebogruppe, in Summe nur 98 Personen, ist es denkbar, dass ein Ausschlusskriterium nur jeweils 1 oder 2 Teilnehmer betroffen hatte. Es könnte also sein, dass mit den post hoc erfolgten Festlegungen Daten einzelner Teilnehmer gezielt aus der Auswertung entfernt wurden.
Ein an anderer Stelle veröffentlichtes Zahlenexperiment zeigt, dass man durch gezielte Ausschlüsse einzelner Patienten das Resultat in eine bestimmte Richtung verschieben kann [14]. Streicht man in der Homöopathiegruppe Patienten, die früh verstorben sind, und in der Placebogruppe solche, die lange überlebt haben, dann entsteht aus ansonsten gleichwertigen Kurvenzügen ein charakteristischer Verlauf, der auch in den veröffentlichten Überlebenskurven erkennbar ist:
• Die Unterschiede zwischen beiden Gruppen resultieren nur aus einer kurzen Zeitspanne zu Anfang, danach verlaufen die Kurven mit mehr oder weniger großen Streuungen parallel. So auch in der Studie: In den ersten 9 Wochen sterben in der Homöopathiegruppe nur 2 von 51 Patienten (4 %), in der Placebogruppe aber 11 von 47 (23 %). Während der restlichen Beobachtungszeit von 95 Wochen versterben in beiden Gruppen dann etwa gleich viele Patienten: 26 unter der Homöopathie (51 % der ursprünglichen Anzahl), 25 unter Placebo (53 %).
• Die mediane Überlebenszeit der Placebo-Patienten verringert sich,
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
N. Aust, V. Weisshäupl: Homöopathie unter dem Radarstrahl der Wissenschaft
139
da Patienten mit langen Überlebenszeiten fehlen. In der Studie erreichen die Patienten unter Placebo bei Weitem nicht die 303 Tage als mediane Überlebenszeit, wie sie in der Veröffentlichung als Durchschnittswert für eine rein konventionelle Behandlung zitiert werden. Vielmehr lag sie mit nur 257 Tagen deutlich darunter.
• Die mediane Überlebenszeit in der Homöopathiegruppe steigt hingegen an, wenn früh verstorbene Patienten ausgeschlossen werden. So auch in der Studie: Bei den Homöopathie-Patienten lag die mediane Überlebenszeit mit 435 Tagen deutlich über dem Erwartungswert.
Es bleibt also festzuhalten, dass die nachträgliche Festlegung der zahlreichen Ausschlusskriterien es ermöglicht, die Daten dadurch zu manipulieren, dass Patienten mit unpassenden Resultaten ausgeschlossen werden. Die veröffentlichten Lebensdauerkurven zeigen genau die Merkmale, wie sie bei einer solchen Manipulation entstehen. Aus den vorhandenen Unterlagen folgt zumindest, dass die in der Veröffentlichung präsentierten Resultate wahrscheinlich nicht durch solide Forschungsarbeit zustande gekommen sind. Vielmehr ist der Schluss naheliegend, dass die erstaunlichen und einzigartigen Effekte nicht real sind, sondern bewusst in eine gewünschte Richtung verändert wurden. Alleine die eindeutige Rückdatierung der Protokolle lässt kaum eine andere Erklärung zu.
Die Österreichische Agentur für Wissenschaftliche Integrität (ÖAWI) kommt mit anderen Me-
thoden zu sehr ähnlichen Ergebnissen. Auch dort werden die Festlegungen des Protokolls als post hoc erfolgt angesehen. Die Kommission kommt zu der Schlussfolgerung [9]: “The Committee concludes that there are numerous breaches in scientific integrity in the study, as reported in the publication. Several of the results can only be explained by data manipulation or falsification. The publication is not a fair representation of the study.”
Das Journal, in dem die Studie veröffentlicht wurde, hat inzwischen eine „Expression of Concern“ veröffentlicht [15], die Retraktion der Studie steht noch aus (Stand 19.11.2022).
Schlussfolgerungen
Im Bestreben, die Homöopathie als eine wissenschaftsbasierte Lehre zu positionieren, ist in der Vergangenheit eine Reihe von klinischen Studien veröffentlicht worden. Dabei sind solche Untersuchungen für die homöopathische Lehre nutzlos: Die therapeutischen Eigenschaften der einzelnen Mittel werden in sogenannten homöopathischen Arzneimittelprüfungen ermittelt, bei denen gesunde Testpersonen die Mittel einnehmen. Die daraufhin erlebten Symptome bilden gesammelt in Repertorien die Grundlagen der Medikamentierung, die nach dem von Hahnemann (1755 – 1843) postulierten Ähnlichkeitsgesetz verordnet werden. Demnach ist eine Substanz in der Lage, bei einem Kranken die Symptome erfolgreich zu bekämpfen, die sie bei einem Gesunden hervorrufen kann. Die bisherige Evidenzlage für die
Homöopathie ist eher dünn: Sämtliche systematischen Reviews seit 1991 kommen zu dem Schluss, dass es zwar, wenn man alle Studien indikationsübergreifend zusammenfasst, kleine Effekte zugunsten der Homöopathie gibt, diese Ergebnisse aber wegen der unzureichenden Qualität der einzelnen zugrunde liegenden Studien nicht belastbar sind [16]. Bislang ist es noch nicht gelungen, einen über Placebo hinausgehenden, therapeutisch relevanten Effekt in einer hochwertigen klinischen Studie nachzuweisen. Dennoch werden auch die kleinsten Erfolge, und seien sie in noch so zweifelhaften Studien zustande gekommen, viele sogar nur in Pilotstudien, von interessierter Stelle werbewirksam als Beleg dafür präsentiert, dass die Homöopathie per se doch eine sinnvolle Therapieoption darstelle. So wird diese Studie auf einer Reihe einschlägiger Webseiten präsentiert, beispielsweise im Auftritt der Clinica Santa Croce, die sich auf die homöopathische Behandlung von Krebserkrankungen spezialisiert hat [17].
Der Hauptautor erhielt 2021 den Peithner-Forschungspreis, der von der Muttergesellschaft des größten österreichischen Herstellers homöopathischer Präparate vergeben wurde [18].
In diesem Zusammenhang ist der Schluss recht einfach zu ziehen: Es gibt nach wie vor keine hochwertige Studie, in der signifikante und klinisch relevante Effekte homöopathischer Präparate zweifelsfrei aufgetreten sind. Diese Aussage ist durch die Studie zum nicht-kleinzelligen Lungenkrebs nicht widerlegt worden.
Allerdings ist die erfolgte kritische Auseinandersetzung der ÖAWI
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
N. Aust, V. Weisshäupl: Homöopathie unter dem Radarstrahl der Wissenschaft
140
keine Folge eines aus dem Wissenschaftsbetrieb heraus initiierten Prozesses. Vielmehr wäre die ganze Affäre nicht ins Rollen gekommen, wenn nicht eine Gruppe von Kritikern der Homöopathie die Initiative ergriffen hätte, die Studie zu analysieren und die Ergebnisse publik zu machen. Dies deutet auf blinde Flecken in der medizinischen Forschung hin, die von Anhängern dubioser Heilslehren wie der Homöopathie ausgenutzt werden können. Denn:
• Wie war es möglich, dass eine solche fragwürdige Arbeit in einer renommierten Fachzeitschrift veröffentlicht werden konnte?
• Wie war es möglich, dass die kritische Auseinandersetzung mit der Studie nicht durch das Fachpublikum aus der Leserschaft erfolgte?
• Was wäre passiert, wenn der Hauptautor nicht die MedUni Wien als seine Institutszugehörigkeit angegeben hätte? Dann hätte diese nicht die ÖAWI eingeschaltet, die als Ergebnis eine Retraktion der Studie empfohlen hat.
Im Prinzip haben alle Beteiligten des Prozesses, der am Ende eigentlich zu validen Erkenntnissen und bestmöglichem Wissen führen sollte, dies nicht mit der erforderlichen Intensität verfolgt:
Die Ethikkommission: Angesichts der theoretischen Unmöglichkeit, dass homöopathische Präparate über Placebo hinaus wirksam sind, widerspricht es ethischen Grundsätzen, klinische Untersuchungen der Phase III, die die Wirksamkeit der Präparate nach-
weisen sollen, freizugeben. Patienten wird unnötig Hoffnung auf eine positive Wirkung gemacht. Prinzipiell begrenzte Etatmittel werden für unnütze Pseudoforschung aufgewendet.
Die Co-Autoren: Unter den CoAutoren findet sich eine Reihe bekannter Wissenschaftler auf dem Gebiet der Onkologie, die zwar die Patienten konventionell behandelt haben, aber mit der Homöopathie nur sehr wenige Berührungspunkte haben. Deren Namen sind in Fachkreisen sicher aus einer Reihe seriöser Veröffentlichungen bekannt – und scheinen für eine Seriosität der Arbeit zu bürgen. Es erscheint daher notwendig, dass sich CoAutoren mit der Forschungsfrage auseinandersetzen – und bei unsinnigen Vorhaben eine Co-Autorschaft ablehnen.
Der Verlag (1): Sofern der Verlag eine Arbeit zur Homöopathie überhaupt annimmt, sollte dafür gesorgt werden, dass ein wirklich kritisches Peer Review stattfindet. Das führt natürlich in einen Zwiespalt: Da die Homöopathie als Ganzes nach einer wissenschaftlichen Validierung sucht, werden Anhänger dieser Lehre einer Studie mit positiven Resultaten eher wenig kritisch gegenüber stehen. Seriöse Forscher aus der wissenschaftlich orientierten Medizin werden hingegen kaum bereit sein, hierfür Arbeitszeit aufzuwenden. Vielleicht sollten die Verlage ohnehin dazu übergehen, die Aufwendungen für die Peer Reviews, die ja eine essentielle Lebensgrundlage eines wissenschaftlichen Journals sind, auch zu honorieren.
Das Peer Review: Ein Peer Review sollte sich nicht nur auf den Text der Veröffentlichung konzentrieren und diesen nach mehr oder weniger formalen Punkten überprüfen. Hier hätte ein Blick in die Daten der Erstregistrierung die eklatanten Abweichungen zur Veröffentlichung offenbart, die im Manuskript nicht beschrieben worden sind. Die unerklärte Abkürzung der Beobachtungszeit und die Erweiterung der Ausschlusskriterien hätte sofort auffallen müssen und die Arbeit hätte mit der Forderung nach Klärung der offenen Punkte zurückgewiesen werden müssen.
Das Studienregister: Glücklicherweise hat das US-Amerikanische Studienregister ClinicalTrials.gov alle Versionen der Registerdaten konserviert, allerdings fällt es beim Aufruf der Webseite nicht auf, dass wesentliche Studienparameter modifiziert wurden. Hier ist eine deutlichere Kennung auf der ersten Seite erforderlich, die sofort ins Auge fällt. Es gibt jedoch auch viele nationale Studienregister, in denen alte Datenstände nicht konserviert werden und die Autoren die Möglichkeit haben, während der Laufzeit der Studie Parameter anzupassen, ohne dass dies irgendwo erscheint. Dies nimmt dem Review die Möglichkeit, eben solche Veränderungen zweifelsfrei zu erkennen und verfehlt damit den Zweck, der mit einer Registrierung erreicht werden soll.
Das Fachpublikum: Bis dato, Stand November 2022, ist kein einziger Leserbrief zur Studie veröffentlicht worden. Das heißt, das Fachpublikum hat diese Arbeit bestenfalls mit einem Schulterzu-
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
N. Aust, V. Weisshäupl: Homöopathie unter dem Radarstrahl der Wissenschaft
141
cken ignoriert. Dies ist zwar verständlich, lässt aber die Studie als unkommentiert und unwidersprochen erscheinen. Das wiederum sieht so aus, als wäre die Studie in den einschlägigen Fachkreisen akzeptiert, was zur Werbung für homöopathische Therapien genutzt werden kann. Es sei daher an die fachkundigen Leser von Journalen appelliert, zu dubiosen Veröffentlichungen in Leserbriefen Stellung zu nehmen.
Die Verlage/Herausgeber (2): Die Verlage bzw. die Herausgeber müssen sich verpflichten, Leserbriefen zu ihren Artikeln auch dann zu prüfen, wenn sie nicht direkt vom angesprochenen Fachpublikum stammen. In diesem Fall ist Folgendes geschehen: Bereits im Juni 2021 hatte die Arbeitsgruppe einen „Letter to the Editor“ verfasst und darin die wesentlichen Kritikpunkte mit den zugehörigen Belegen aufgeführt. Dieser ist bislang nicht veröffentlicht worden, es hat auch offenbar keine inhaltliche Prüfung stattgefunden. Erst nach dem Abschlussbericht der ÖAWI ist man tätig geworden und hat eine „Expression of Concern“ veröffentlicht [15]. Eine Retraktion steht noch aus. Man stelle sich vor, der Hauptautor hätte nicht die MedUni Wien als Institutszugehörigkeit genannt, dann hätte man dort auch nicht die ÖAWI eingeschaltet, es gäbe keinen Abschlussbericht und die Arbeit stünde immer noch als unangefochtene Evidenz im Raum.
Fazit: Wenn sich in der Zukunft solche Fälle nicht wiederholen, dann hätte diese Studie die evidenzbasierte Medizin tatsächlich vorangebracht.
Literatur*
1 Frass M, Lechleitner P, Gründling C. et al. Homeopathic treatment as an add-on therapy may improve quality of life and prolong survival in patients with nonsmall cell lung cancer: a prospective, randomized, placebo-controlled doubleblind, three-arm multicenter study. The Oncologist 2020;25:1-26 (Open Access). https://doi.org/10.1002/onco. 13548
2 Oxford Academic “The Oncologist”. https://academic.oup.com/oncolo
3 Robert Koch Institut: Krebsregister, Daten für Lungenkrebs. https://www. krebsdaten.de/Krebs/DE/Content/ Krebsarten/Lungenkrebs/lungenkrebs_ node.html
4 ClinicalTrails.gov: Registerdaten zur Studie Frass_2020: https://clinicaltrials. gov/ct2/show/NCT01509612
5 Frass M. Homeopathy in cancer (HINC) – Study protocol; Version date: January 10, 2011. https://clinicaltrials. gov/ProvidedDocs/12/NCT01509612/ Prot_SAP_000.pdf
6 Beispielhaft: Mathie RT, Lloyd SM, Legg LA et al.: Randomised placebocontrolled trials of individualised homeopathic treatment: systematic review and meta-analysis. Systematic Reviews 2014;3:142. https://systematicreviewsjournal.biomedcentral.com/articles/10.1186/2046-4053-3-142
NHMRC Information Paper: Evidence on the effectiveness of homeopathy for treating health conditions, Canberra: NHMRC;2015. https://www.nhmrc. gov.au/_files_nhmrc/publications/attachments/cam02a_information_paper. pdf
7 ClinicalTrails.gov: History of changes zur Studie Frass_2020. https://clinicaltrials.gov/ct2/history/NCT01509612
8 Frass M. Homeopathy in Cancer (HINC) –- Study protocol; Version date: February 6, 2014. https://clinicaltrials.gov/ProvidedDocs/12/ NCT01509612/Prot_SAP_001.pdf
9 Schönberger A. Homöopathie bei Krebspatienten: Fast zu schön, um wahr zu sein. Profil, 28.10.2022 https://www. profil.at/wissenschaft/homoeopathiebei-krebspatienten-fast-zu-schoen-umwahr-zu-sein/402198219
10 ClinicalTrials.gov: Registerdaten zur Studie Frass_2020: Datenstand January 13, 2012. https://clinicaltrials.gov/ct2/ history/NCT01509612?V_2=View#Stu dyPageTop
11 ClinicalTrials.gov: Registerdaten zur Studie Frass_2020: Datenstand October 29, 2020. https://clinicaltrials.gov/ct2/ history/NCT01509612?V_8=View#Stu dyPageTop
12 ClinicalTrials.gov: Registerdaten zur Studie Frass_2020: Datenstand August 15, 2018. https://clinicaltrials.gov/ct2/ history/NCT01509612?V_7=View#Stu dyPageTop
13 Higgins J, Thomas J. Cochrane Handbook for Systematic Reviews of Interventions; Version 6.3, 2022, Chapter 8.14.1: Rationale for concern about bias [of selective outcome reporting]. https://handbook-5-1.cochrane.org/ chapter_8/8_14_1_rationale_for_concern_about_bias.htm
14 Aust N, Weisshäupl V. Verbesserung beim Überleben von Lungenkrebspatienten mit Homöopathischer Komplementärbehandlung? (Frass et al. 2020) – Eine Studienkritik; Webseite des Informationsnetzwerks Homöopathie: https://netzwerk-homoeopathie.info/ studienkritik-zu-lungenkrebsstudiefrass-et-al-2020/
15 NN. Expression of Concern: Homeopathic treatment as an add-on therapy may improve quality of life and prolong survival in patients with non-small cell lung cancer: a prospective, randomized, placebo-controlled double-blind, threearm multicenter study. https://doi. org/10.1093/oncolo/oyac221
16 NN. Systematische Reviews zur Homöopathie – Überblick; Webseite „Homöopedia“ des Informationsdnetzwerks Homöopathie: https://www.homöopedia.eu/index.php?title=Artikel:Systematische_Reviews_zur_Homöopathie_-_Übersicht
17 NN. Neue Studie zur Homöopathie bestätigt die klinische Erfahrung der Clinica Dr. Spinedi; Eintrag vom 04.02.2021. https://clinicaspinedi.ch/de/ neue-studie-zur-homoopathie-bestatigtdie-klinische-erfahrung-der-clinica-drspinedi/
18 NN. Wissenschaftspreis für Homöopathie-Studien aus Österreich; Beitrag auf MedMEdia.at vom 13.05.2021. https:// www.medmedia.at/relatus-pharm/wissenschaftspreis-fuer-homoeopathie-studien-aus-oesterreich/
Anschriften der Verfasser: Dr.-Ing. Norbert Aust Fritz-Heeg-Erasmus-Straße 25 79650 Schopfheim Deutschland
Dr. phil. Dr. med. Viktor Weisshäupl Graf-Starhemberg-Gasse 32-1-11 1040 Wien Österreich
* Alle Links abgerufen am 17.11.2022.
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
N. Aust, V. Weisshäupl: Homöopathie unter dem Radarstrahl der Wissenschaft
142
MITTEILUNGEN
Stellungnahme der Deutschen Hochdruckliga zum Einnahmezeitpunkt von Antihypertensiva
Morgens oder abends? Eine kontrollierte randomisierte Studie aus Großbritannien kam zu dem Ergebnis: Wann man die blutdrucksenkenden Medikamente einnimmt, ist egal. Hier eine zusammenfassende Stellungnahme der Deutschen Hochdruckliga zu diesem Thema.
Es ist unklar, ob eine abendliche Antihypertensivagabe Vorteile hinsichtlich der Blutdruckeinstellung und der Verhinderung kardiovaskulärer Endpunkte gegenüber einer morgendlichen Dosierung hat. Zwei große spanische Studien (MAPEC, HYGIA) wurden u.a. wegen implausibel hoher Effektstärke bei nur sehr geringen Blutdruckunterschieden und implausibel niedriger Dropout-Raten (0,4 – 2 %) sowie nicht festgelegter antihypertensiver Medikation in den Behandlungsarmen stark kritisiert [1]. Deshalb wurden weitere große Studien initiiert, um Klarheit in dieser wichtigen Frage zu bringen: TIME („Treatment In Morning vs Evening“ [2]) sowie die noch laufenden Studien BedMed und BedMed-Frail [1].
TIME-Studie bringt Klarheit
Die TIME-Studie [2] wurde online am 11. Oktober 2022 im Lancet publiziert und kam zu dem klaren Ergebnis, dass eine Randomisie-
rung des Einnahmezeitpunktes aller Antihypertensiva am Morgen oder am Abend bei 21.104 Patienten mit medikamentös behandelter Hypertonie keinen Effekt auf den primären Endpunkt aus vaskulärem Tod und Hospitalisierung wegen Herzinfarkt oder Schlaganfall nach einem Follow-up-Zeitraum von im Median 5,2 Jahren hatte (HR: 0,95; 95%-KI: 0,83–1,10]. Für relevante sekundäre Endpunkte wie die vaskuläre Mortalität und Gesamtsterblichkeit erbrachte die abendliche Gabe mit 1,1 % bzw. 4,2 % im Vergleich zur morgendlichen Gabe mit 1,0 % bzw. 4,1 % ebenfalls keinen Vorteil.
Bei abendlicher Einnahme der Antihypertensiva lag der morgendliche systolische/diastolische Blutdruck um 1,8/0,4 mmHg niedriger und der abendliche systolische/diastolische Blutdruck um 1,1/0,9 mmHg höher als bei morgendlicher Antihypertensivagabe.
Das untersuchte Patientenkollektiv war zu Studienbeginn 65 Jahre alt, mit einem mittleren Blutdruck von 135/79 mmHg unter der Einnahme von durchschnittlich 1,5 Antihypertensiva, hatte in 13 % eine kardiovaskuläre Vorerkrankung und bestand zu 57 % aus Männern und zu 90 % aus Kaukasiern.
Fazit
Die TIME-Studie [2] unterstützt den Konsens verschiedener Hypertoniefachgesellschaften [1], dass entsprechend der wissenschaftlichen Datenlage die abendliche Antihypertensivagabe keinen Vorteil gegenüber der morgendlichen Antihypertensivagabe aufweist. Somit kann die Antihypertensivagabe in
gleicher Weise am Morgen oder am Abend erfolgen.
Entscheidend ist, wie sich die Antihypertensivaeinnahme am besten in den Tagesrhythmus des Patienten einfügt und welcher Einnahmezeitpunkt mit der höheren Adhärenz assoziiert ist (im Allgemeinen höher bei morgendlicher Einnahme).
Natürlich spielen auch mögliche Nebenwirkungen eine Rolle, die z.B. die Gabe eines Diuretikums am Morgen als günstiger erscheinen lassen.
Bernhard K. Krämer, Martin Hausberg, Markus van der Giet, Ulrich Wenzel
Literatur
1 Stergiou G, Brunström M, MacDonald T et al. Bedtime dosing of antihypertensive mediactions: Systematic review and consensus statement. International Society pf Hypertension position paper endorsed by World Hypertension League and European Society of Hypertension. J Hypertens 2022;40:18471858
2 Mackenzie IS, Rogers A, Poulter NR et al. Cardiovascular outcomes in adults with hypertension with evening versus morning dosing of usual antihypertensives in the UK (TIME study): a prospective, randomized, open-label, blindedendpoint clinical trial. Lancet 2022; 400:1417-1425
143 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
Mitteilungen
2022; 35: 144 – 147
Der plötzliche Herztod (PHT) ist trotz stetigen medizinischen Fortschritts und verbesserter medikamentöser Therapie noch immer die Ursache von 50 % der kardiovaskulären Todesfälle und zählt zu den häufigsten Todesursachen in der westlichen Welt. Allein in Deutschland versterben jährlich über 100.000 Menschen daran [1]. Häufigster Auslöser sind schnelle Herzrhythmusstörungen wie anhaltende Kammertachykardien und Kammerflimmern. Die einzige Behandlung, die diese lebensgefährlichen Herzrhythmusstörungen beenden kann, ist eine Defibrillation, d.h. die Abgabe eines elektrischen Behandlungsschocks. Das Herz soll damit wieder einen normalen, koordinierten Rhythmus aufnehmen. Je schneller die Defibrillation durchgeführt werden kann, desto höher ist die Wahrscheinlichkeit des Überlebens – mit jeder Minute Verzögerung einer Defibrillation sinkt die Überlebenschance um etwa 10 % [2].
Im Krankenhaus ist die zeitnahe Defibrillation häufig erfolgreich, da der Patient durchgängig überwacht wird und geschultes Krankenhauspersonal schnell verfügbar ist, um den Schock zu verabreichen. Außerhalb des Krankenhauses ist eine
ÜBERSICHTSARBEIT
Defibrillatorweste – ein wirksamer Schutz vor dem plötzlichen Herztod
Brigitte Söllner, Erlangenerfolgreiche zeitnahe Defibrillation davon abhängig, ob der Betroffene überwacht wird und ob der Rettungsdienst rechtzeitig eintrifft. Daher werden Patienten mit einem bekannten dauerhaft hohen PHTRisiko oft mit einem implantierbaren Cardioverter-Defibrillator (ICD) geschützt. Bevor der Eingriff erfolgt, empfehlen die Leitlinien der nationalen und internationalen Fachgesellschaften eine Wartezeit: nach Myokardinfarkt 40 Tage, bei nicht ischämischer Kardiomyopathie mindestens 3 Monate optimale medikamentöse Therapie. Denn in vielen Fällen verbessert sich in diesem Wartezeitraum die Pumpleistung des Herzens und das PHT-Risiko sinkt, sodass kein ICD mehr implantiert werden muss [3]. Gerade in der Zeit früh nach einem kardialen Ereignis besteht jedoch das höchste Risiko für einen PHT [4, 5]. Um die Patienten in dieser vulnerablen Phase vor dem PHT zu schützen, gibt es 2 Möglichkeiten: die kostenintensive Überwachung im Krankenhaus oder die Versorgung der Patienten mit einem tragbaren Cardioverter-Defibrillator (WCD, LifeVest®). Geschützt durch den WCD können die Patienten das Krankenhaus verlassen und
ihr alltägliches Leben wie gewohnt weiterführen. Darüber hinaus ermöglicht die Monitoringfunktion des WCD eine individuellere Risikostratifizierung. Ziel ist es ja, die Behandlung langfristig zu optimieren und zu entscheiden, ob ein anhaltender Schutz mittels eines ICD nötig ist.
Indikationen
Die LifeVest® Defibrillatorweste wird bei einer Vielzahl von Indikationen eingesetzt, die Patienten einem erhöhten PHT-Risiko aussetzen [6]:
• bei Herzinsuffizienz mit einer LVEF ≤35 %, wenn noch kein permanent erhöhtes Risiko für eine PHT festgestellt wurde
• nach kurz zurückliegendem (<40 Tage) Myokardinfarkt mit einer LVEF ≤35 % (für 40 – 90 Tage)
• nach einer koronaren Revaskularisierung (PCI, aortokoronarer venöser Bypass; für 3 – 4 Monate)
• bei neu diagnostizierter Kardiomyopathie, wenn eine Besserung der linksventrikulären Funktion zu erwarten ist (für 3 – 6 Monate)
144 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
Brigitte Söllner: Defibrillatorweste – ein wirksamer Schutz vor dem plötzlichen Herztod
PERFUSION
• bei akuter Myokarditis, wenn eine Besserung der linksventrikulären Funktion zu erwarten ist (für 3 – 6 Monate)
• bei fortgeschrittener koronarer Herzkrankheit während der Wartezeit auf eine geplante Herzoperation (für 3 – 4 Monate)
• bei erhöhtem Risiko unmittelbar nach der Explantation eines ICD, wenn eine sofortige Reimplantation nicht möglich ist (für 1 – 2 Monate)
• bei indizierter ICD-Implantation, die z.B. aufgrund von Nebenerkrankungen noch nicht möglich ist Über die unmittelbare Schutzfunktion hinaus dient der WCD auch der Risikostratifizierung von Patienten. Außerdem kann der WCD tagesaktuelle telemedizinische Informationen bereitstellen, sodass Änderungen des Gesundheitszustands des Patienten frühzeitig erkannt und bei Bedarf behandelt werden können. Somit trägt der WCD zu einer optimierten und patientenindividuellen Therapie bei.
Breite Evidenz für die Schutzfunktion und sichere Risikostratifizierung
Umfangreiche Daten aus retrospektiven und prospektiven Registern mit mehr als 20.000 Patienten belegen die klinische Wirksamkeit des WCD in der Terminierung ventrikulärer Tachyarrhythmien bei gleichzeitig sehr niedrigem Risiko inadäquater Therapien [7, 8, 9, 10]. Nach den Ergebnissen des WEARIT II-Registers benötigen nur 42 % der Patienten mit ischämischer, nicht ischämischer oder
So funktioniert die LifeVest® Defibrillatorweste
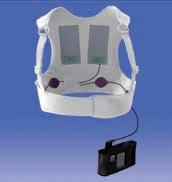
Die LifeVest® besteht aus einer Stoffweste, in der sich ein Gürtel mit Mess- und Therapieelektroden befindet, und einem Monitor. Die Stoffweste wird unter der Kleidung direkt am Körper, der Monitor an der Hüfte oder an einem Schulterriemen getragen, und zwar rund um die Uhr, also auch während des Schlafens. Die Defibrillatorweste sollte vom Patienten nur zum Waschen oder Duschen abgelegt werden. Das Gerät überwacht das Herz des Patienten kontinuierlich. Wird ein lebensgefährlicher Herzrhythmus erkannt, benachrichtigt das integrierte Alarmmodul den Patienten mit hör-, sicht- und fühlbaren Warnsignalen. Ist der Patient bei Bewusstsein und toleriert die Arrhythmie, kann er einen unnötigen Schock verhindern, indem er gleichzeitig die beiden Reaktionstasten am Monitor drückt. Ist der Patient bewusstlos, gibt das Gerät erst ein leitendes BlueTM Gel über die Therapieelektroden und dann einen elektrischen Behandlungsschock ab, um den normalen Herzrhythmus wieder herzustellen. Für die LifeVest® ist – anders als beim automatischen externen Defibrillator (AED) – kein Eingreifen durch andere Personen notwendig. Die Behandlungssequenz von der Erkennung der lebensbedrohlichen Arrhythmie bis zur Abgabe des Behandlungsschocks dauert in der Regel weniger als 1 Minute.
kongenitaler Kardiomyopathie und schwer eingeschränkter linksventrikulärer Funktion nach einer mittleren Tragedauer des WCD von 90 Tagen (22,5 Stunden/Tag) noch einen ICD [6]. In einer deutschen Registerstudie mit 6.000 Patienten betrug die mediane Tragezeit 23,1 Stunden pro Tag [9].
Dass der WCD bei konsequenter Anwendung einen klaren Überlebensvorteil bietet, verdeutlichen die Ergebnisse der randomisierten, kontrollierten Studie „Vest Prevention of Early Sudden Death Trial“ (VEST), die 2.302 Patienten mit stark eingeschränkter Pumpfunktion nach Myokardinfarkt einschloss [11].
In der Intention-to-treat-Analyse konnte zwar keine signifikante Senkung des primären Endpunkts arrhythmische Mortalität gezeigt werden (relatives Risiko: 0,67; p = 0,18); die Gesamtmortalität hingegen – der wichtigste sekundäre Endpunkt – wurde signifikant reduziert (relatives Risiko: 0,64; p = 0,04). Dieses Ergebnis wurde in einer im Jahr 2020 veröffentlichten Per-Protocol-Analyse der VESTStudie nochmals näher beleuchtet [12]. In diese Analyse wurden alle Patientendaten vom Zeitpunkt der Randomisierung bis zum letzten protokollgemäßen Tragetag des WCD aufgenommen, nicht aber diejenigen Tage nach einem vor-
145 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
Brigitte Söllner: Defibrillatorweste – ein wirksamer Schutz vor dem plötzlichen Herztod
zeitigen Abbruch, z.B. wegen der ICD-Implantation. Die Per-Protocol-Analyse ergab, dass bei Patienten unter konsequenter WCD-Therapie nicht nur die Gesamtmortalität um 75 % (p < 0,001), sondern auch der primäre Endpunkt eines arrhythmischen Todes um 62 % (p = 0,02) signifikant reduziert wurde. Die tägliche mediane Tragezeit in der Per-Protocol-Analyse lag bei ≥22 Stunden [12].
Dass eine effiziente Risikostratifizierung möglich ist, bestätigten die Daten der PROLONG-II-Studie, die 353 Patienten mit Herzinsuffizienz (ICM oder NICM; LVEF: 25 ± 8 %) einschloss, die zwischen 2012 und 2017 mit einem WCD versorgt wurden [13]. Hier konnte über einen langen Nachbeobachtungszeitraum (2,8 ± 1,5 Jahre) gezeigt werden, dass bei 53 % der Patienten aufgrund der Verbesserung der LVEF keine Indikation mehr für einen ICD bestand. Die mittlere Tragzeit pro Patient betrug 104 ± 76 Tage. Bei 4 % der Patienten wurden lebensbedrohliche Arrhythmien durch adäquate Schocks erfolgreich behandelt, sodass die Patienten im Anschluss kein erhöhtes Risiko für einen PHT mehr hatten [13].
Von Leitlinien empfohlen
Seit 2000 ist die LifeVest® Defibrillatorweste in Europa CE-zertifiziert, und seit 2001 besitzt sie die Zulassung der US-amerikanischen Food and Drug Administration (FDA). Im Jahr 2015 wurde die LifeVest® in die Leitlinien der Deutschen Gesellschaft für Kardiologie (DGK) [10] sowie der European Society of Cardiology (ESC)
[14] für das Management ventrikulärer Arrhythmien und die Prävention des plötzlichen Herztods aufgenommen. Im Zuge der letzten Aktualisierungen der ESC Heartfailure Guidelines 2021 [15] sowie der ESC VT/VF Guidelines 2022 [16] wurde der Leitlinienansatz der ESC weiterentwickelt, indem Empfehlungen für bestimmte Patientenindikationen in verschiedene Leitlinien aufgenommen wurden. Die Defibrillatorweste hat nun für ischämische und nicht ischämische Patienten in den ESC HF Guidelines eine IIb-Empfehlung mit Evidenzgrad B [15] und für Post-MyokardPatienten eine IIb-Empfehlung mit Evidenzgrad B [16]. Auch die Leitlinien der American Heart Association (AHA) und der Heart Rhythm Society (HRS) beinhalten die Empfehlung, die Anwendung des WCD bei ischämischen und nicht ischämischen Patienten in Betracht zu ziehen [17].
Leitlinienempfehlungen zum Einsatz der LifeVest®
• NICM- und ICM-Patienten mit Herzinsuffizienz sind in den ESC HF-Leitlinien (2021) mit einer Empfehlung der Klasse IIb (Level B) eingestuft [15]
• Post-MI-Patienten sind in den ESC-Leitlinien für VT/VF (2022) aufgeführt und bewahren die Klasse IIb (Level B), vorher Evidenzlevel C [16]
• Für die Sekundärprophylaxe bleibt die Empfehlung Klasse IIa (Level C) [16]
• Für Patienten, die auf eine Herztransplantation warten, lautet die Empfehlung weiterhin Klasse IIb (Level C) [16]
Kostenerstattung
Die LifeVest® Defibrillatorweste ist seit 2005 im Hilfsmittelverzeichnis des GKV-Spitzenverbandes gelistet [6] und wird als Hilfsmittel verordnet. Die Kosten werden in der Regel von den Krankenkassen erstattet. Im Juli 2019 wurde das Hilfsmittelverzeichnis aktualisiert und es wurden zusätzlich alle primärprophylaktischen Indikationen mit einer hochgradig eingeschränkten Pumpfunktion (LVEF ≤35 %) aufgenommen [6].
Hierzu zählen Patienten mit einem temporär erhöhten PHT-Risiko nach akutem Myokardinfarkt (<40 Tage zurückliegend), einer erwarteten LVEF-Verbesserung bei akuter Myokarditis oder bei Erstdiagnose einer dilatativen Kardiomyopathie. Weitere Einsatzbereiche sind die peripartale Kardiomyopathie, die fortgeschrittene koronare Herzkrankheit während der Wartezeit auf eine geplante Herzoperation sowie post PTCA und post ACVB. Weiterhin wird die Defibrillatorweste für eine prolongierte Risikostratifizierung aufgeführt.
Die Ergänzung des Hilfsmittelverzeichnisses unterstreicht die breite wissenschaftliche Datenlage und bestätigt die seit Jahren gängige Verordnungspraxis.
Literatur
1 Kauferstein S et al. Plötzlicher Herztod bei jungen Menschen durch kardiale Gendefekte. Dtsch Arztebl Int 2009; 106:41-47
2 Perkins GD et al. Basismaßnahmen zur Wiederbelebung Erwachsener und Verwendung automatisierter externer Defibrillatoren. Kap. 2 der Leitlinien zur Reanimation 2015 des European Resuscitation Council. Notfall Rettungsmed 2015;18:748-769
146 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
Brigitte Söllner: Defibrillatorweste – ein wirksamer Schutz vor dem plötzlichen Herztod
Brigitte Söllner: Defibrillatorweste – ein wirksamer Schutz vor dem plötzlichen Herztod
3 Deutsche Gesellschaft für Kardiologie – Herz- und Kreislaufforschung e.V. (Hrsg.). Ventrikuläre Arrhythmien und Prävention des plötzlichen Herztodes, ESC Pocket Guidelines, Version 2015
4 Adabag AS et al. Sudden death after myocardial infarction. JAMA 2008; 300:2022-2029
5 Solomon SD et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. NEJM 2005;352:25812588
6 GKV-Spitzenverband. Nachtrag zum Hilfsmittelverzeichnis vom 10. Juli 2019. Bundesanzeiger vom 23. Juli 2019
7 Epstein AE et al. Wearable cardioverter-defibrillator use in patients perceived to be at high risk early post-myocardial infarction. J Am Coll Cardiol 2013; 62:2000-2007
8 Nguyen E et al. Wearable cardioverter-defibrillators for the prevention of sudden cardiac death: A meta-analysis. J Innov Card Rhythm Manag 2018;9:3151-3162
9 Wäßnig N et al. Experience with the wearable cardioverter-defibrillator in patients at high risk for sudden cardiac death. Circulation 2016;134:635-643
10 Deneke T et al. Kommentar zu den ESC-Leitlinien 2015 „Ventrikuläre Arrhythmien und Prävention des plötzlichen Herztodes“. Kardiologe 2017;11: 27-43
11 Olgin JE et al. Wearable cardioverter-defibrillator after myocardial infarction. N Engl J Med 2018;379:12051215
12 Olgin JE et al. Impact of wearable cardioverter-defibrillator compliance on outcomes in the VEST trial: As-treated and per-protocol analyses. J Cardiovasc Electrophysiol 2020;31:1009-1018
13 Mueller-Leisse J et al. Extended follow-up after wearable cardioverter-defibrillator period: the PROLONG-II study. ESC Heart Fail 2021;8:51425148
14 Priori SG et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the preven-
MITTEILUNGEN
Neue KfH-Patientenbroschüre „Diabetes verstehen. Nierenfunktion erhalten.“

Einer von 10 Erwachsenen weltweit lebt derzeit mit Diabetes. In Deutschland sind zirka 8,5 Millionen an einem Typ-2-Diabetes erkrankt, Tendenz steigend. Während ein festgestellter Diabetes heutzutage sehr gut behandelbar ist, können dauerhaft zu hohe Blutzuckerwerte die Blutgefäße der Organe schädigen und so zu schwerwiegenden Komplikationen für Nieren, Augen, Nerven und das Herz-KreislaufSystem führen. Diabetes ist eine der häufigsten Ursachen für ein chronisches Nierenversagen. „Des-
Perfusion 5-6/2022 35. Jahrgang
tion of sudden cardiac death. Eur Heart J 2015;36:2793-2867
15 McDonagh TA et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021;42:3599-3726
16 Zeppenfeld K et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 2022;43:3997-4126
17 Al-Khatib SM et al. 2017 AHA/ACC/ HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Circulation 2017; CIR.0000000000000 549
Anschrift der Verfasserin: Brigitte Söllner
Medizinjournalistin und Wissenschaftliche Lektorin Lärchenweg 10 91058 Erlangen E-Mail: brigitte.soellner@online.de
halb sind eine frühe Diagnose sowie eine frühzeitige, konsequente Behandlung auch für die Nierengesundheit von großer Bedeutung“,
betont Professor Dr. med. Dieter Bach, Vorstandsvorsitzender des KfH Kuratorium für Dialyse und Nierentransplantation e.V. Mit seiner seiner neuen Patientenbroschüre „Diabetes verstehen. Nierenfunktion erhalten“ informiert das KfH über mögliche Folgeerkrankungen, insbesondere für die Nieren, und wie man diesen vorbeugen kann.
Die Broschüre wurde mit medizinischer Beratung von Professor Carsten Böger, leitender Arzt des KfHNierenzentrums Traunstein, erstellt und steht zum Download auf www. kfh.de/infomaterial/patientenratgeber/ zur Verfügung oder kann unter info@kfh-dialyse.de bestellt werden.
147
© Verlag PERFUSION GmbH
S.
M.
Das weiterentwickelte Insulin lispro Lyumjev® imitiert mit seinem besonders schnellen Wirkeintritt die physiologische Insulinwirkung noch genauer als herkömmliches Insulin lispro (Humalog®) [1]. Was das für Patienten mit Typ-2-Diabetes bedeutet, die von ihrer bisherigen Bolusinsulintherapie auf Lyumjev® umgestellt werden, und wie sich wichtige Zielparameter der Glukosekontrolle verändern, untersuchte die Studie PRONTO Time in Range [2].
An der prospektiven Open-LabelStudie mit einem Behandlungsarm nahmen 176 Patienten teil, die seit über einem Jahr Typ-2-Diabetes hatten und neben weiteren Medikamenten seit mindestens 3 Monaten eine intensivierte Insulintherapie (ICT) bekamen. Die Studienteilnehmer wurden von ihrer gewohnten ICT auf eine Behandlung mit
FORUM DIABETICUM
Mehr Zeit im Zielbereich nach Umstellung auf Lyumjev® auch bei Typ-2-Diabetes
dem Mahlzeiteninsulin Lyumjev® in der Formulierung U100 umgestellt. Als Basalinsulin verwendeten sie Insulin glargin.
Während zu Studienbeginn nur 22 % ein System zur kontinuierlichen Glukosekontrolle (CGM) nutzten, wurden während der Beobachtungszeit von 12 Wochen bei allen Teilnehmern die Glukosewerte per CGM erhoben. Diese Studie ist die erste, bei der CGMSysteme Wirksamkeitsdaten zu Lyumjev® bei Typ-2-Diabetikern lieferten.
Primärer Endpunkt der Studie war die Zeit im Zielbereich von 70–180
mg/dl tagsüber (zwischen 6 und 24 Uhr). Als sekundäre Endpunkte wurden die gesamte Zeit im Zielbereich und der HbA1c definiert [2].
Verbesserte Glukosekontrolle nach 12 Wochen
Nach 12 Wochen zeigte sich eine deutliche Verbesserung mehrerer Parameter der Glukosekontrolle gegenüber den Ausgangswerten. So hatte sich die Zeit im Zielbereich tagsüber signifikant um durchschnittlich 45 Minuten erhöht, ebenso wie die gesamte Zeit im
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 148
Abbildung 1: Durch die Umstellung auf das Mahlzeiteninsulin Lyumjev® erhöhte sich bei Typ-2-Diabetikern die Zeit im Zielbereich (TIR) um 45 Minuten täglich [2]. 63 % Woche 12 n=133 Ausgangswert n=153 Veränderung der Zeit im Zielbereich 12 Wochen nach Umstellung auf LYUMJEV® 59 % 680 635 65 % 60 % 55 % 50 % 45 % 40 % 35 % 30 % + 45 Minuten Daten sind Kleinste-Quadrate-Mittelwerte +/- Standardabweichungen ** p<0,1 (Δ 4 %**) Zeit im Zielbereich (in Min.) TIR 70-180 mg/dL, %
FORUM DIABETICUM
Zielbereich (Abb. 1). Dieser Anstieg war das Resultat der Abnahme hyperglykämischer Werte, insbesondere nach den Mahlzeiten. Die Zeit im hypoglykämischen Bereich blieb dagegen unverändert. Gleichzeitig kam es zu einer durchschnittlichen signifikanten Reduktion des HbA1c-Werts von 8,2 % auf 7,8 %. Bereits die Zulassungsstudie PRONTO T2D hatte gezeigt, dass bei Menschen mit Typ-2-Diabetes unter Lyumjev® weniger Glukosespitzen und niedrigere postprandiale Glukosewerte auftreten als unter herkömmlichem Insulin lispro [3]. Eine längere Zeit im Zielbereich konnte bisher jedoch nur bei Typ1-Diabetikern nachgewiesen werden [4].
Die Ergebnisse dieser ersten Studie mit Anwendung von CGM-Systemen bei Patienten mit Typ-2-Diabetes zeigen nun, dass auch diese Patientengruppe deutlich von einer Umstellung auf Lyumjev® profitieren und dadurch eine verbesserte Glukosekontrolle, auch im Sinne von mehr Zeit im Zielbereich und niedrigerem HbA1c, erreichen kann.
Fabian Sandner, NürnbergLiteratur
1 Fachinformation Lyumjev®, aktueller Stand
2 Wang Q et al. Association of Diabetes Care & Education Specialists – 2022 Annual Conference; Baltimore, USA; 12.–15. August 2022, Poster IP 11
3 Blevins T et al. Diabetes Care 2020; 43:2991-2998
4 Klaff L et al. Diabetes Obes Metab 2020;22:1799-1807
MITTEILUNGEN
Tempo
Smart Button™ erleichtert das digitale DiabetesManagement mit
Fertigpens
Für Menschen mit Diabetes, die Insulinpens verwenden, ist es wichtig, die verabreichte Insulinmenge und den Zeitpunkt der Dosisgabe genau nachzuverfolgen. Denn in Verbindung mit den Glukosedaten kann auf dieser Basis über notwendige Anpassungen der Therapie entschieden werden. Eine deutliche Erleichterung wäre es, wenn die Datenerfassung automatisiert erfolgen würde.
Mit dem Tempo Smart Button™ hat Lilly nun ein Modul entwickelt, das als Bestandteil eines neuen personalisierten Diabetes-Management-Systems in Echtzeit verlässliche Daten zu den verabreichten Insulindosen liefert.
Automatisierte Protokollierung der verabreichten Insulindosen
Der Tempo Smart Button™ ist ein kleines Modul, das am Tempo Pen™ angebracht wird, einer modifizierten Version des bestehenden vorgefüllten Einweg-Insulinpens KwikPen™ von Lilly. Es kann den Zeitpunkt und die Menge der mit dem Pen injizierten Insulindosen sowie das verwendete Insulin registrieren und diese Daten automatisiert an eine mit ihm über Bluetooth verbundene App senden.

Fließen dort auch die Werte aus der kontinuierlichen Glukosemessung ein, lässt sich die Qualität der Diabetestherapie zeitnah bewerten und verbessern. Da alle relevanten Daten an das digitale Tagebuch übermittelt werden, braucht der Patient kein Diabetestagebuchs mehr führen.
Das Modul ist kompatibel mit der Software und den medizinischen Geräten mehrerer Anbieter von Diabetes-Management-Plattformen, darunter z.B. Glooko®, mySugr® und DexCom Inc.
Der Tempo Smart Button™ hat die Zertifizierung abgeschlossen und die CE-Kennzeichnung erhalten. Er kann mit allen Tempo Pens™ verwendet werden und wird damit allen erwachsenen Patienten zur Verfügung stehen, die mit Lyumjev®, Abasaglar® und/oder Humalog® (jeweils in der Formulierung U100) behandelt werden.
Lilly plant, das Tempo® System 2023 in Deutschland auf den Markt zu bringen.
B. S.Perfusion
© Verlag PERFUSION GmbH 149
5-6/2022 35. Jahrgang
AstraZeneca präsentierte auf den diesjährigen Kongressen der American Society of Nephrology (ASN, Kidney Week) und der American Heart Association (AHA, Scientific Sessions) aktuelle Daten zum Natrium-Glucose-Cotransporter2-Inhibitor (SGLT2i) Dapagliflozin (Forxiga®). Diese neuen Erkenntnisse betreffen sowohl das kardiovaskuläre als auch das renale Wirkspektrum des SGLT2i und haben wichtige Implikationen für Patienten und das Gesundheitssystem [1, 2]. Wie eine neue präspezifizierte Analyse der Phase-III-Studie DELIVER zeigt, kann Dapagliflozin die Symptombelastung und die gesundheitsbezogene Lebensqualität von Patienten mit Herzinsuffizienz verbessern – und dies sowohl bei leicht reduzierter als auch erhaltener Ejektionsfraktion. Außerdem ergaben Auswertungen der DAPACKD-Studie, dass die Dapagliflozin-Therapie die Rate der Krankenhauseinweisungen bei Patienten mit chronischer Nierenerkrankung (CKD) aller Ursachen reduzieren kann.
FORUM CARDIOLOGICUM
HF-Patienten profitieren durch eine Verbesserung ihres gesamten Gesundheitszustands
Patienten mit Herzinsuffizienz (HF) mit leicht reduzierter oder erhaltener Ejektionsfraktion (EF) haben – neben einem höheren Risiko für Krankenhauseinweisungen und Mortalität – weitere physiologische Einschränkungen und belastende Symptome sowie eine reduzierte Lebensqualität [2]. Die
Verbesserung des gesamten Gesundheitszustands stellt daher ein wesentliches Therapieziel dar. In einer präspezifizierten Analyse der Phase-III-Studie DELIVER wurde der Kansas City Cardiomyopathy Questionnaire (KCCQ) angewendet, um die gesundheitlichen Auswirkungen von Dapagliflozin mit Hinblick auf Symptombelastung, körperliche Einschränkungen und Lebensqualität zu untersuchen [2]. Bei allen mittels KCCQ erhobenen Parametern führte Dapagliflozin –zusätzlich zur Standardbehandlung und im Vergleich zu Placebo – zu einer signifikanten Verbesserung: bei der körperlichen Einschränkung (1,9 Punkte), der klinischen Krankheitsmanifestation (2,3 Punkte), beim Gesamtzustand (2,1 Punkte) und beim Gesamtsymptomwert (2,4 Punkte; alle p < 0,001). Diese Vorteile wurden bereits nach 1 Monat registriert und über die Dauer von 8 Monaten hinweg aufrechterhalten. Nach 8 Monaten erzielten außerdem mehr Patienten unter Dapagliflozin eine kleine, moderate oder große (um 5, 10 bzw. 15 Punkte) Verbesserung des Gesundheitszustands in den bewerteten KCCQ-Bereichen. Der Nutzen von
Dapagliflozin in Bezug auf kardiovaskuläre Todesfälle und eine Verschlechterung der HF bei Patienten mit leicht reduzierter oder erhaltener EF schien bei Betroffenen, die bei Studienbeginn einen höheren Grad an symptomatischer Beeinträchtigung aufwiesen, besonders deutlich ausgeprägt zu sein. Das Verträglichkeitsprofil von Dapagliflozin entsprach dabei dem bewährten Sicherheitsprofil des Medikaments [2].
Diese Ergebnisse unterstützen die gemeinsamen HF-Leitlinien 2022 des American College of Cardiology, der American Heart Association und der Heart Failure Society of America, in denen ein breiterer Einsatz von SGLT2i in der klinischen Praxis und ein früherer Beginn einer leitliniengerechten medikamentösen Therapie empfohlen wird [3]. Darüber hinaus stehen sie im Einklang mit einer aktuellen Veröffentlichung, laut der eine frühzeitige und anhaltende Verringerung klinischer Ereignisse bei HF-Patienten mit leicht reduzierter oder erhaltener EF einen statistisch signifikanten Nutzen zeigt – und dies bereits binnen 4 Wochen nach Behandlungsbeginn [4].
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 150
Neue Studiendaten zu Dapagliflozin zeigen signifikante Verbesserungen bei Patienten mit Herzinsuffizienz oder chronischer Nierenerkrankung
Renaler Benefit bei CKDPatienten
Zusätzlich zum klinischen Nutzen von Dapagliflozin im Bereich kardiovaskulärer Erkrankungen belegt eine aktuelle, auf der Kidney Week der ASN präsentierte Analyse der DAPA-CKD-Studie auch den renalen Benefit des SGLT2i bei Patienten mit chronischer Nierenerkrankung (CKD) [5]. Die Auswertung ergab, dass die Dapagliflozin-Therapie die Rate der Krankenhauseinweisungen aller Ursachen reduzierte – und dies unabhängig vom Vorliegen eines Typ-2-Diabetes. Diese Ergebnisse haben nicht nur Auswirkungen auf die Lebensqualität der Betroffenen, sondern auch auf das Gesundheitswesen. Denn neben dem Einfluss, den die CKD auf Patientenebene nimmt, verursacht sie auch für die Gesundheitssysteme weltweit erhebliche Kosten – insbesondere wenn die CKD in eine terminale Niereninsuffizienz mündet oder die Entwicklung kardiorenaler Ereignisse begünstigt [1]. Die Studie INSIDECKD schätzte daher das Einsparpotenzial, das durch die Behandlung mit Dapagliflozin erreicht werden könnte [1]. Die Analyse von 23 Ländern mit über 100.000 Patienten ergab, dass sich durch die Addon-Therapie mit Dapagliflozin versus alleiniger Standardbehandlung über einen Zeitraum von 3 Jahren Kosten in Höhe von 205 Mio. USD einsparen ließen. Dies entspricht einer Kostensenkung in Höhe von 33 % [1].
FORUM CARDIOLOGICUM
Dieses Einsparpotenzial verwundert nicht, denn weltweit sind mehr als 850 Millionen Menschen von CKD betroffen – mit steigender Tendenz [6]. Allerdings bleibt die Erkrankung häufig unerkannt [7]. Daher untersuchte die internationale Studie REVEAL-CKD die Rate an CKD-Diagnosen in USA, Italien, Deutschland, Japan und Frankreich. Dabei ergaben sich beachtlich hohe Unterdiagnoseraten von 61,6 % bis 95,5 % für Patienten mit manifester CKD im 3. Stadium (entsprechend einer geschätzten glomerulären Filtrationsrate (eGFR) von ≥30 und <60 ml/min/1,73 m2) [8]. Erfreulich war jedoch, dass – sobald die Diagnose gestellt wurde – die Patienten von den entsprechenden Monitoring- sowie Managementstrategien profitieren [8]. Der positive Einfluss der frühzeitigen Diagnosestellung auf den Erhalt der Nierenfunktion ließ sich dabei an der jährlichen Abnahme der eGFR ablesen, die anhand der US-Datenbank TriNetX erhoben wurde [9]. Bei den berücksichtigten 27.000 Patienten bezifferte sich der Abfall der eGFR vor CKD-Diagnose auf –4,12 (95%-KI: –4,23 bis –4,02). In den 2 Jahren nach der Diagnose war er mit nur –0,30 (95%-KI: –0,44 bis –0,14) wesentlich geringer [9].
Fabian Sandner, NürnbergLiteratur
1 McEwan P et al. Translating the findings of DAPA-CKD to reductions in healthcare resource utilization from a
global perspective. American Society of Nephrology (ASN) Kidney Week 2022, 3.–6. November 2022 in Orlando
2 Kosiborod MN, et al. The effects of dapagliflozin on symptoms, function and quality of life in patients with heart failure and mildly reduced or preserved ejection fraction: results from the DELIVER Trial. American Heart Association (AHA) Scientific Sessions 2022, 5.–7. November 2022 in Chicago
3 Heidenreich PA et al. 2022 AHA/ACC/ HFSA Guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022;145:e895-e1032
4 Vaduganathan M et al. Time to clinical benefit of dapagliflozin in patients with heart failure with mildly reduced or preserved ejection fraction: a prespecified secondary analysis of the DELIVER randomized clinical trial. JAMA Cardiol 2022;7:2380-6583
5 Schechter M et al. Dapagliflozin effect on hospital admissions in patients with chronic kidney disease: a post hoc analysis of the DAPA-CKD trial. American Society of Nephrology (ASN) Kidney Week 2022, 3.–6. November 2022 in Orlando
6 Jager KJ et al. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Nephrol Dial Transplant 2019;34:1803-1805
7 Bikbov B et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020;395:709-733
8 Tangri N et al. REVEAL-CKD: Management and monitoring of patients with CKD stage 3 in France, Germany, Italy, Japan, and the USA. American Society of Nephrology (ASN) Kidney Week 2022, 3.–6. November 2022 in Orlando
9 Tangri N et al. REVEAL CKD: Estimated glomerular filtration Rate (eGFR) decline before and after a CKD diagnosis among patients with CKD stage 3. American Society of Nephrology (ASN) Kidney Week 2022, 3.–6. November 2022 in Orlando
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 151
FORUM ERNÄHRUNG
Wie sinnvoll ist eine Zuckersteuer?
Franz-Werner Dippel, Hohen NeuendorfAus zahlreichen Studien wissen wir, dass Zucker eines der größten Gesundheitsrisiken in unserer Ernährung darstellt [1]. Deshalb empfiehlt die WHO, höchstens 5 Energieprozent des täglichen Kalorienbedarfs in Form von zugesetztem Zucker zu sich zu nehmen [2]. Bei einem normalgewichtigen Erwachsenen entspricht das etwa 8 Stück Würfelzucker pro Tag (ca. 25 g). Der durchschnittliche Zuckerverzehr der Deutschen liegt jedoch aktuell bei dem Vierfachen [3]. Der tatsächliche Verbrauch dürfte aber noch deutlich höher sein, da es für zahlreiche Zuckerarten keine belastbaren Verbrauchszahlen gibt.
Zucker hat viele Gesichter Unter Zucker im engeren Sinne versteht man nur den weißen, kristallinen Haushaltszucker aus Zuckerrüben oder Zuckerrohr. Chemisch handelt es sich dabei um einen Doppelzucker (Disaccharid), der zu je einem Teil aus den beiden Einfachzuckern (Monosacchariden) Glukose (Traubenzucker) und Fruktose (Fruchtzucker) besteht, die chemisch fest miteinander zu einem Doppelmolekül (Saccharose) verbunden sind.

Darüber hinaus wird der Begriff „Zucker“ aber auch als Sammelbegriff für zahlreiche weitere süß schme-
ckende Kohlenhydrate aus anderen pflanzlichen Quellen verwendet. So lassen sich Glukose und Fruktose mittlerweile enzymatisch aus stärkehaltigen Pflanzen herstellen [4]. Vorzugsweise werden dafür Mais, Weizen, Reis und Hirse sowie Kartoffeln verwendet. Daraus gewinnt man einerseits ein Zuckergemisch aus kurz-, mittel- und langkettigen Stärkeabbauprodukten (Maltose, Maltodextrin), andererseits eine Mischung aus den beiden Einfachzuckern Glukose und Fruktose (Glukose-Fruktose-Sirup, High-Fructose-Corn-Sirup bzw. Isoglukose). Aus lebensmittelrechtlichen Gründen darf der so gewonnene „Stärkezucker“ jedoch nicht als Zucker bezeichnet werden, da es sich nicht um Saccharose handelt.
Verschiedene exotische Zuckerpflanzen dienen ebenfalls der Zuckergewinnung. Die daraus hergestellten Pflanzensirupe (z.B. Agaven-, Ahorn- und Dattelsirup) bzw. Pflanzenzucker (z.B. Kokosblütenzucker, Palmzucker) bestehen aus den bei-
den Monosacchariden Glukose und Fruktose sowie aus dem Disaccharid Saccharose in unterschiedlicher Zusammensetzung.
Schließlich lassen sich durch Eindicken von Fruchtsäften aller Art zuckerhaltige Konzentrate herstellen (Traubensüße, Birnendicksaft, Bananenmark etc.). Auch sie enthalten die Zuckerarten Saccharose, Glukose und Fruktose in unterschiedlichen Mischungsverhältnissen.
Bleiben zu guter Letzt noch die beiden tierischen Zuckerquellen Milch und Honig. Beim Milchzucker (Laktose) handelt es sich um ein Doppelmolekül aus Traubenzucker (Glukose) und Schleimzucker (Galaktose). Der natürliche Laktosegehalt von Vollmilch(produkten) liegt bei ca. 4,7 %. Damit fällt Kuhmilch in die Klasse der Lebensmittel, die offiziell als zuckerreduziert gelten. Honig, das zuckerreiche Ausscheidungsprodukt der Bienen, besteht zu etwa gleichen Teilen aus den beiden Einfachzuckern Glukose und Fruktose sowie einer geringen Menge des Zweifachzuckers Saccharose.
Zucker als Lebensmittelzutat
Zur Süßung von Fertiggerichten und Getränken werden von der Lebensmittelindustrie bevorzugt Haushaltszucker (Saccharose), Stärkezucker (Glukose-Fruktose-Sirup, Maltodex-
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 152
FORUM ERNÄHRUNG
Bezeichnung des Zuckers
Haushaltszucker, Kristallzucker
Maissirup, Glukose-Fruktose-Sirup, Isoglukose, Stärkezucker, High-Fructose-Corn-Sirup (HFCS) etc.
Malzzucker, Bierzucker
Ausgangsprodukt
Zuckerrübe Zuckerrohr
Enthaltene Zuckerarten
Saccharose = Disaccharid aus Glukose und Fruktose
Getreide (Mais, Weizen, Hirse, Reis), Kartoffeln Gemisch aus Glukose und Fruktose
Maltose = Disaccharid aus Glukose und Glukose
Vor allem Gerste
Maltodextrin, Gerstenmalz(extrakt), Malzsirup Gemisch aus Oligo- und Polymeren der Glukose
Ahorn-, Agaven- und Dattelsirup etc. Kokosblüten- und Palmzucker
Exotische Zuckerpflanzen
Gemisch aus Glukose, Fruktose und Saccharose Frucht(saft)konzentrat, Dicksaft etc. Dörrobst, Trockenfrüchte Obst, Früchte und Beeren
Bienenhonig Pflanzennektar
Molken- bzw. Süßmolkenpulver, Vollmilch- bzw. Magermilchpulver Vorwiegend Kuhmilch
Tabelle 1: In industriell hergestellten Fertigprodukten verwendete Zuckerarten.
trin) sowie diverse Fruchtkonzentrate (Glukose, Fruktose, Saccharose) und Milchzucker (Laktose) eingesetzt. Neben seiner geschmacks- und strukturgebenden Funktion hat Zucker auch konservierende und farbgebende Eigenschaften. Exotische Sirupe und Honig kommen wegen ihrer Konsistenz sowie aus Kostengründen seltener zum Einsatz. In geringem Umfang werden auch natürlich süßende Zutaten wie Frischobst oder Trockenfrüchte verwendet. Es gilt also festzuhalten, dass unser Nahrungszucker vorwiegend aus 3 Zweifachzuckern (Saccharose, Maltose, Laktose) sowie deren Komponenten Glukose, Fruktose und Galaktose besteht (Tab. 1). Glukose kommt als Baustein in allen 3 Zweifachzuckern vor und ist damit der häufigste Einfachzucker in unserer Nahrung. Rechnet man die Glukose aus den stärkehaltigen Lebensmitteln (z.B. Brot, Nudeln, Reis, Kartoffeln) noch hinzu, so wird schnell deutlich,
dass Glukose heute die zentrale Rolle im menschlichen Kohlenhydratstoffwechsel spielt. Das lässt sich auch daran erkennen, dass Glukose von allen Körperzellen als Energiequelle genutzt werden kann. Fruktose und Galaktose hingegen werden vom Körper erst je nach Bedarf in Glukose, Glykogen oder Fett umgewandelt und tragen auf diese Weise ebenfalls zu Übergewicht sowie zur Entwicklung von Zivilisationskrankheiten bei.
Laktose = Disaccharid aus Glukose und Galaktose
Kennzeichnung von Zucker auf der Verpackung
Wegen ihrer unterschiedlichen Herkunft bzw. Herstellung müssen alle zugesetzten Zuckerarten in der Zutatenliste von Fertigprodukten separat ausgewiesen werden. Das gilt sowohl für Haushalts-, Stärke- und Milchzucker, Pflanzensirupe, Fruchtkonzentrate und Honig als auch für natürlich
süßende Zutaten (z.B. Rosinen, Cranberries etc.). Zahlreiche Bezeichnungen erschweren dabei den Überblick. Bei den meisten Zuckerarten taucht der Begriff Zucker nämlich gar nicht im Namen auf (Glukose, Saccharose, Dextrose, Maissirup, Isoglukose, Fruktose-Sirup, Gerstenmalz, Rübenkraut, Bananenmark, Apfelpüree, Laktose, Magermilchpulver, Molkenpulver etc.). Um dennoch den Gesamtzuckergehalt eines Fertigprodukts zu ermitteln, lohnt ein Blick auf die Nährwerttabelle. Hier werden, ungeachtet ihrer Herkunft, alle unterschiedlichen Ein- und Zweifachzucker addiert und mengenmäßig als Untergruppe der Gesamtkohlenhydrate in der Rubrik „davon Zucker“ ausgewiesen.
Zuckersteuer ohne Ausnahmen
Eine Zuckersteuer auf Fertigprodukte sollte alle zugesetzten Zuckerarten
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 153
FORUM ERNÄHRUNG
berücksichtigen, unabhängig davon, aus welcher Quelle sie stammen. Als Bemessungsgrundlage für den Steuersatz könnte der in der Nährwerttabelle ausgewiesene Gesamtzuckergehalt dienen.
In Anlehnung an die Empfehlungen des Verbraucherzentrale Bundesverbands (VZBV) wären 3 Steuerklassen denkbar:
• Zuckergehalt bis 5,0 %: Zuckersteuer 5 %
• Zuckergehalt bis 12,5 %: Zuckersteuer 10 %
• Zuckergehalt >12,5 %: Zuckersteuer 20 %
Für Getränke gilt der halbe Zuckergehalt als Bemessungsgrenze.
In die höchste Steuerklasse von 20 % würden neben Softdrinks, Limonaden, Fruchtsäften und Smoothies auch zahlreiche Fertigprodukte (z.B. Cerealien, süße Brotaufstriche, Süßwaren) sowie alle extrahierten und angereicherten Zuckerarten fallen, die als süßende Zutaten in der Industrie sowie im Privathaushalt eingesetzt werden (Haushaltszucker, Pflanzenzucker bzw. -sirupe, Honig, Dicksäfte Trockenfrüchte etc.).
Lediglich frisches und tiefgekühltes Obst wird von der Zuckersteuer ausgenommen, da es neben Glukose, Fruktose und Saccharose in physiologisch verträglichen Konzentrationen auch Ballaststoffe, Vitamine, Mineralien, Spurenelemente und sekundäre Pflanzenstoffe enthält.
tische Konsistenz ihrer Produkte zu erzielen, werden die Hersteller die traditionellen Zuckerarten gegen Zuckerersatzstoffe austauschen. Diese Entwicklung zeigt sich zumindest in solchen Ländern, in denen es bereits eine Zuckersteuer gibt [5]. Unter den Oberbegriff Zuckerersatzstoffe fallen Substanzen, die den süßen Geschmack des Zuckers mitbringen, jedoch keine echten Kohlenhydrate sind. Dazu gehören die Zuckeralkohole (z.B. Sorbit, Erythrit, Xylit) sowie die synthetischen Süßstoffe (z.B. Aspartam, Cyclamat, Sucralose). In beiden Fällen handelt es sich – wie auch bei den herkömmlichen Zuckerarten – um stark verarbeitete Substanzen, deren Langzeitwirkung auf die menschliche Gesundheit noch nicht abschließend beurteilt werden kann [6, 7].
Entwicklung von Übergewicht, Typ2-Diabetes und zahlreichen Krebserkrankungen bereits seit Jahrzehnten bekannt ist [9, 10], steht der Nachweis einer relevanten gesundheitlichen Verbesserung durch die Einführung einer Zuckersteuer jedoch noch aus. In einer randomisiert kontrollierten Studie konnte mittlerweile auch der kausale Zusammenhang zwischen dem Konsum stark verarbeiteter Fertigprodukte und der Entwicklung von Übergewicht belegt werden [11].
Am Geldbeutel ansetzen
Zuckeralternativen sind keine Lösung
Um einen vergleichbaren Geschmack sowie eine annähernd iden-
Ernährungspolitische Gesamtstrategie notwendig
Die skizzierte Datenlage legt nahe, dass die Einführung einer singulären Zuckersteuer vermutlich zu kurz greift. Da industriell hergestellte Fertigprodukte neben Zucker in der Regel gleichzeitig auch noch weitere stark verarbeitete Zutaten wie z.B. modifizierte Stärke, gehärtete und umgeesterte Fette, Proteinextrakte, Salz und zahlreiche Zusatz- bzw. EStoffe enthalten, könnte eine Besteuerung von Lebensmitteln nach dem Grad der Verarbeitung möglicherweise eine stärkere gesundheitliche Wirkung in der Bevölkerung entfalten [8]. Während der Zusammenhang zwischen dem Konsum hochverarbeiteter Fertigprodukte und der
Nach Ansicht zahlreicher Wissenschaftler hat die Besteuerung ungesunder Lebensmittel ein hohe Lenkungswirkung. So konnte eine Modellrechnung zeigen, dass durch eine Änderung der Steuerstruktur für Lebensmittel, bei der adipogene Lebensmittel verteuert und Obst und Gemüse verbilligt werden, eine Reduktion der Adipositasprävalenz sowie eine Senkung der Krankheitskosten im deutschen Gesundheitswesen erreicht werden kann [12]. Fiskalpolitische Maßnahmen sind also durchaus geeignet, durch ihre verhältnispräventive Wirkung eine Ernährungswende zu unterstützen, wobei ihre Effektivität durch ordnungspolitische Maßnahmen wie z.B. verbindliche Werbebeschränkungen und verbesserte Kennzeichnungspflichten für stark verarbeitete Fertigprodukte noch gesteigert werden könnte [13]. Eine derart gestaltete Ernährungsumgebung wäre die Basis dafür, dem Ziel einer gesundheitsförderlichen Lebensweise endlich näher zu kommen.
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 154
Literatur
1 Lustig RH. Die bittere Wahrheit über Zucker. Wie Übergewicht, Diabetes und andere chronische Krankheiten entstehen und wie wir sie besiegen können. München: Riva-Verlag; 2016
2 WHO. Guideline: Sugars intake for adults and children. Geneva; 2015
3 Heuer T. Zuckerkonsum in Deutschland. Akt Ernährungsmed 2018; 43 (Suppl. 1):S8-S11
4 Takasaki Y. Studies on sugar-isomerizing enzyme. Agricultural and Biological Chemistry 1966;30:1247-1253
5 Bandy LK, Scarborough P, Harrington RA et al. Reductions in sugar sales from soft drinks in the UK from 2015 to 2018. BMC Medicine 2020;18:20
6 Pollmer U. Zusatzstoffe von A bis Z. Was Etiketten verschweigen. Deutsches Zusatzstoffmuseum (Hrsg.), Hamburg; 2017
7 Debras C, Chazelas E, Srour B et al. Artificial sweeteners and cancer risk: results from the NutriNet-Santé population-based cohort study. PLoS Med 2022;19:e1003950
8 Monteiro CA, Cannon G, Levy RB et al. NOVA. The star shines bright. World Nutrition 2016;7:28-38
9 Pagliai G, Dinu M, Madarena MP et al. Consumption of ultra-processed foods and health status: a systematic review and meta-analysis. Br J Nutr 2021;125: 308-318
10 Lane MM, Davis JA, Beattie S et al. Ultraprocessed food and chronic noncommunicable diseases: a systematic review and meta-analysis of 43 observational studies. Obesity Rev 2020;1-19
11 Hall KD, Ayuketah A, Brychta R et al. Ultra-processed diets cause excess calorie intake and weight gain: an inpatient randomized controlled trial of ad libitum food intake. Cell Metab 2019;30:67-77
12 Effertz T. Die Auswirkungen der Besteuerung von Lebensmitteln auf Ernährungsverhalten, Körpergewicht und Gesundheitskosten in Deutschland. Universität Hamburg 2017
13 Garlichs D. Nationale Strategien der Reduktion von Zucker, Fett und Salz in Lebensmitteln. Akt Ernährungsmed 2018;43 (Suppl. 1):S37-S41
Anschrift des Verfassers: Dr. rer. med. Franz-Werner Dippel 16540 Hohen Neuendorf
E-Mail: franz-werner.dippel@t-online.de
Ende März 2021 wurde eine schwere, wenn auch seltene Nebenwirkung nach COVID-19-Impfung mit Vektor-basierten Vakzinen beobachtet: Impfassoziiert traten vor allem bei jüngeren Frauen Sinus- und Hirnvenenthrombosen auf und es kam zu Todesfällen. Bei Impfung mit mRNA-Vakzinen wurde diese unerwünschte Nebenwirkung nicht beobachtet, zumindest nicht in einer Häufigkeit, die einen Zusammenhang vermuten ließ. Der Vektor-basierte Impfstoff ChAdOx1 (AstraZeneca) wurde daraufhin nicht mehr jungen Frauen verabreicht, außerdem wurden Geimpfte für das Leitsymptom Kopfschmerzen nach Impfung sensibilisiert und Ärztinnen und Ärzte auf das Phänomen der Bildung von antiPF4-Antikörpern hingewiesen. Der Nachweis dieser Antikörper kann Betroffene identifizieren, bevor klinische Symptome von Sinus- und Hirnvenenthrombosen auftreten, und erlaubt somit eine frühzeitige Therapie und Prävention dieser seltenen Komplikation. Es wurde aber auch ein leicht erhöhtes Risiko für hämorrhagische Schlaganfälle (sog. Hirnblutungen) nach Impfung mit einem mRNA-Vakzin beschrieben.
Eine im Oktober 2021 in „Nature Medicine“ publizierte Auswertung [1] zeigte diesbezüglich ein erhöhtes Risiko an den Tagen 1–7 und den Tagen 15–-21 nach Impfung mit BNT162b2 (IRR: 1,27 und 1.38). Seitdem haftet allen Impfstoffen gegen SARS-CoV-2 das Stigma an, sie könnten Schlaganfälle auslösen – eine Sorge, die verständlicherweise zu Ängsten führt und zur Impfskepsis beiträgt. Doch 2 aktuelle Studien zeigen nun, dass die Impfung gegen SARS-CoV-2 nicht mit einem erhöhten Schlaganfallrisiko einhergeht.
Schlaganfallrate nach Impfung ist mit der in der Allgemeinbevölkerung vergleichbar
In einem in „Neurology“ publizierten, systematischen Review [2] wurden 2 randomisierte Studien, 3 Kohortenstudien und 11 Register-basierte Studien ausgewertet. Insgesamt wurden 17.481 Fälle ischämischer Schlaganfälle erfasst – bei einer Gesamtzahl von 782.989.363 Impfungen. Die Schlaganfallrate betrug insgesamt 4,7 Fälle pro 100.000 Impfungen.
© Verlag PERFUSION GmbH 155
Perfusion 5-6/2022 35. Jahrgang
FORUM SCHLAGANFALL
Kein erhöhtes Schlaganfallrisiko durch die Impfung gegen SARS-CoV-2
Nur bei 3,1 % der Schlaganfälle infolge einer SARS-CoV-2-Impfung lag eine thrombotisch-thrombozytopenische Purpura (TTP) zugrunde. Wie die Autoren schlussfolgern, ist damit die Schlaganfallrate nach Impfung mit der in der Allgemeinbevölkerung vergleichbar – und die TTP, die zu Sinus- und Hirnvenenthrombosen führte, zumindest nach den Vorkehrungen, die getroffen wurden, eine sehr seltene Komplikation. Des Weiteren betonen sie, dass die Schlaganfallrate bei SARS-CoV-2-infizierten Menschen hingegen deutlich höher liegt [2].
Keine Assoziation zwischen mRNA-Impfstoffen und dem Auftreten schwerer kardiovaskulärer Komplikationen
Bei der zweiten Studie handelt es sich um eine aktuelle Auswertung des „French National Health Data System“ (Système National des Données de Santé [SNDS]) [3]. Untersucht wurde, wie häufig nach erster und zweiter Gabe von Vakzinen gegen SARS-CoV-2 bei Menschen im Alter von 18–75 Jahren kardiovaskuläre Ereignisse (Myokardinfarkte, Lungenembolien oder Schlaganfälle) auftraten. Insgesamt waren 73.325 Ereignisse dokumentiert worden, bei 37 Millionen geimpften Personen.
Im Ergebnis zeigte die Studie, dass es keine Assoziation zwischen mRNA-Impfstoffen und dem Auftreten dieser schweren kardiovaskulären Komplikationen gab. Die erste Dosis des Vektor-basierten Impfstoffs ChAdOx1 war in
FORUM SCHLAGANFALL
Woche 2 nach der Impfung mit einer erhöhten Rate an Myokardinfarkten und Lungenembolien vergesellschaftet (RI: 1,29 und 1,41), auch beim Impfstoff von JanssenCilag konnte eine Assoziation mit dem Auftreten von Myokardinfarkten in Woche 2 nach Vakzinierung nicht ausgeschlossen werden. In Bezug auf die Schlaganfallrate ergab die Auswertung aber für keinen der Impfstoffe ein höheres Risiko.
Impfung halbiert das Schlaganfallrisiko während einer COVID-19-Erkrankung
Die vorliegenden Daten zeigen zumindest für die mRNA-Impfstoffe keinerlei Sicherheitssignale in Bezug auf ein erhöhtes Schlaganfallrisiko. Die Tatsache, dass beide Erhebungen sehr große Kohorten ausgewertet haben und beide zum gleichen Ergebnis kommen, gibt zusätzliche Sicherheit: mRNAVakzine gegen SARS-CoV-2 erhöhen nicht das Schlaganfallrisiko, die Sorge davor sollte also Menschen nicht davon abhalten, sich impfen zu lassen. Ganz im Gegenteil: Die SARSCoV-2-Infektion geht mit einer höheren Schlaganfallrate einher und die Impfung schützt somit vor Schlaganfällen. Das zeigte jüngst eine koreanische Studie [4]: Von 592.719 SARS-CoV-2-positiven Patientinnen und Patienten im Studienzeitraum von Juli 2020 bis Dezember 2021) wurden 231.037 in die Studie eingeschlossen. 62.727 waren ungeimpft, 168.310 vollständig geimpft (2 Dosen eines
mRNA- oder Vektorimpfstoffs), sie hatten sich aber trotzdem mit Corona infiziert. Die geimpften Studienteilnehmer waren älter und wiesen mehr Komorbiditäten auf. Dennoch waren schwere oder gar kritische COVID-19-Verläufe in dieser Gruppe seltener ebenso wie die Rate an Folgeerkrankungen. Das adjustierte Risiko betrug für den ischämischen Schlaganfall 0,40 bei den geimpften Teilnehmern, was bedeutet, dass die Impfung das Schlaganfallrisiko im Vergleich zur Gruppe der ungeimpften Studienteilnehmer mehr als halbierte.
Pressemeldung der DGNLiteratur
1 Patone M et al. Neurological complications after first dose of COVID-19 vaccines and SARS-CoV-2 infection. Nat Med 2021;27: 2144-2153
2 Stefanou MI et al. Acute arterial ischemic stroke following COVID-19 vaccination: a systematic review and meta-analysis. Neurology 2022 Aug 24:10.1212/WNL.0000000000200996. doi: 10.1212/WNL.0000000000200996. Epub ahead of print. PMID: 36002319
3 Botton J et al. Risk for myocardial infarction, stroke, and pulmonary embolism following COVID-19 vaccines in adults younger than 75 years in France. Ann Intern Med 2022 Aug 23. doi: 10.7326/M22-0988. Epub ahead of print. PMID: 35994748
4 Kim YE et al. Association between vaccination and acute myocardial infarction and ischemic stroke after COVID-19 infection. JAMA 2022; 328:887-889. doi:10.1001/jama.2022. 12992
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 156
Beim Fötus verbindet der Ductus arteriosus die Aorta mit dem Truncus pulmonalis, sodass das sauerstoffreiche Blut von der Mutter die noch nicht funktionierende Lunge des Kindes umgehen kann und direkt in den Körperkreislauf fließt. Wenn nach der Geburt die Atmung des Kindes einsetzt, verschließt sich der Ductus arteriosus normalerweise sukzessive von selbst. Ist das nach 4–8 Wochen nicht der Fall, führt die verbleibende Öffnung, der persistierende Ductus arteriosus (PDA), zu einer Volumenzunahme im Lungenkreislauf und zu einer linksventrikulären Volumenbelastung. Denn das Blut strömt dann aus der Aorta über den Truncus pulmonalis und den Lungenkreislauf zurück in das linke Herz. Während das Blut im Lungenkreislauf eine hohe Sauerstoffsättigung aufweist, erhält der Körper zu wenig sauerstoffreiches Blut. Infolge dieser Mangelversorgung entwickeln sich die Neugeborenen schlecht. Daher sollte ein PDA stets verschlossen werden. Dies erfolgt heute bei größeren Säuglingen, Kindern und Erwachsenen standardmäßig minimalinvasiv durch das Einsetzen eines Coils oder Okkluders per Katheter.
Bei Frühgeborenen, bei denen ein PDA sehr häufig auftritt (bei bis zu 50 % der Frühgeborenen und bei
FORUM CARDIOLOGICUM
Amplatzer Piccolo™ verschließt persistierenden Ductus arteriosus sicher und effektiv
mehr als 80 % der Frühgeborenen mit extrem niedrigem Geburtsgewicht <1.000 g) [1] und ein hohes Mortalitätsrisiko birgt, wird der Transkatheterverschluss dagegen bislang noch nicht routinemäßig durchgeführt. Mit Einführung des Amplatzer Piccolo™ wird sich das mit Sicherheit ändern. Denn der kleinste Okkluder der Welt hat sich in der Behandlung des PDA bei Frühgeborenen bislang sehr gut bewährt, wie die auf dem Symposium der Pediatric and Congenital Interventional Cardiovascular Society in Chicago vorgestellten aktuellen Studienergebnisse zeigen.
Überzeugende 3-JahresErgebnisse
Sicherheit und Wirksamkeit des Amplatzer Piccolo™ Okkluders wurden in der ADO-II-AS-Studie bei 200 Frühgeborenen (Alter >3 Tage, Gewicht ≥700 g) mit einem PDA ≤4 mm im Durchmesser bzw. einem PDA ≥3 mm Länge untersucht [2]. 100 Patienten wogen ≤2 kg. Allen in die Studie aufgenommenen Frühgeborenen wurde der Okkluder perkutan mittels 4-FKatheter über die Femoralvene unter echokardiografischer Kontrolle in den Ductus arteriosus eingesetzt. Primärer Wirksamkeitsendpunkt war die Rate des PDA-Verschlusses nach 6 Monaten, als primärer Sicherheitsendpunkt wurde die Rate schwerer Komplikationen über 6 Monate definiert. Sekundärer Endpunkt war die Rate signifikanter Lungen- oder Aortenobstruktionen während der 6-monatigen Nachbeobachtung. Mittlerweile liegen die Daten für einen Zeitraum von 3 Jahren vor. Sie zeigen:
Abbildung 1: Der Okkluder Amplatzer Piccolo™ ist kleiner als eine Erbse und ist als weltweit erste und einzige minimalinvasive Transkatheterbehandlung für den Verschluss eines PDAs bei Frühgeborenen zugelassen. Der selbstexpandierende Schirm besteht aus einem dichtgewebten Drahtgeflecht, das den Rest-Shunt nach der Platzierung im Ductus arteriosus minimiert (© Abbott).
• eine hohe Implantationserfolgsrate von 95,5 % (99,0 % für Patienten ≤ 2kg, 92,0 % für Patienten >2 kg)
• einen vollständigen PDA-Verschluss nach 6 Monaten bei 99,4 % (100 % für Patienten ≤ 2kg, 98,8 % für Patienten >2 kg)

Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH 157
• eine hohe Überlebensrate von 95,5 % (keine eingriffsbedingten Todesfälle)
• eine geringe Rate systembedingter, schwerwiegender unerwünschter Ereignisse (2,1 %) und keine späten (>1 Jahr) systembedingten Ereignisse Damit erweist sich der Amplatzer Piccolo™ Okkluder als lebensrettendes System für Frühgeborene mit PDA, die dringend eine Behandlung brauchen, um überleben zu können, für die ein chirurgischer Eingriff aber zu risikoreich ist. Da es sich um ein minimalinvasives Verfahren handelt, können viele der schwerkranken Frühgeborenen auf der Neugeborenen-Intensivstation schon bald nach der Implantation von der künstlichen Beatmung genommen werden. Die US-Arzneimittelbehörde FDA hat den Amplatzer Piccolo™ Okkluder im Jahr 2019 zugelassen, seit 2019 hat er das CE-Zeichen.
Brigitte Söllner, ErlangenLiteratur
1 Hamrick SE, Hansmann G. Pädiatrie 2010;125:1020-1030
2 Sathanandam SK, Gutfinger D, O‘Brien L et al. Catheter Cardiovasc Interv 2020; 96:1266-1276
FORUM LIPIDSENKER
Langzeitdaten zu Inclisiran zeigen
Wirksamkeit und Sicherheit über vier Jahre
Das Low-Density-LipoproteinCholesterin (LDL-C) ist ein kausaler, aber gut adressierbarer Risikofaktor für atherosklerotische Herz-Kreislauf-Erkrankungen (ASCVD). Trotz des weit verbreiteten Einsatzes von Statinen erreichen jedoch 4 von 5 Patienten nicht die in den Leitlinien empfohlenen LDLC-Ziele, sodass sie weitere verlässliche Therapieoptionen benötigen, um ihr kardiovaskuläres Risiko zu senken. Eine vielversprechende Option ist das seit dem 9. Dezember 2020 in der Europäischen Union zugelassene Inclisiran (Leqvio®), das in den zulassungsrelevanten Studien – zusätzlich zu einer maximal tolerierten Statin-Therapie –eine signifikante (p < 0,0001) und über 6 Monate anhaltende LDL-CSenkung um 50,7 % im Vergleich zum Ausgangswert bewirkte [1]. Aktuelle Daten zeigen, dass Inclisiran, eine small interfering RNA (siRNA), auch langfristig zur LDLC-Senkung beitragen kann [2, 3].
Wegweisende Ergebnisse der ORION-3-Studie
Auf dem Kongress der American Heart Association (AHA) im November 2022 wurden die Ergebnisse der ORION-3-Studie, einer offenen, nicht randomisierten
Verlängerung der Phase-II-Studie ORION-1 bekannt gegeben. Eingeschlossen in ORION-1 wurden 233 Patienten mit ASCVD (~1.210 Patientenjahre) oder ASCVD-Risikoäquivalenten und jeweils erhöhtem LDL-C trotz maximal verträglicher lipidsenkender Therapie. Primäre Studienendpunkt war die prozentuale LDL-C-Veränderung vom Ausgangswert (Tag 1 von ORION-1) bis Tag 210. Die LDL-C-Werte wurden bis zu Tag 1.440 (4 Jahre) gemessen. Die Patienten erhielten in dieser Studie die siRNA gemäß Zulassung*.
In der Verlängerungsstudie ORION-3 wurde die Senkung der LDL-C-Spiegel über den gesamten Studienzeitraum von 4 Jahren aufrechterhalten: Bei den mit Inclisiran behandelten Patienten kam es zu einer durchschnittlichen Senkung des LDL-C um 47,5 % im Vergleich zum Ausgangswert (Tag 1 von ORION-1) bis zu Tag 210 (95%-KI: –50,69 bis –44,27) und zu einer zeitlich gemittelten Senkung des LDL-C-Wertes um 44,2 % über 4 Jahre bei zweimal jährlicher Gabe (ab der zweiten Injektion*) [1, 2]. Unter der Therapie mit der der siRNA erreichten
* Inclisiran (Leqvio®) wird nach einer einer initialen Dosis und einer weiteren nach 3 Monaten in der Dauertherapie alle 6 Monate subkutan injiziert [1].
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
158
anhaltende
FORUM LIPIDSENKER
Inclisiran
Bei Inclisiran (Leqvio®) handelt es sich um eine small interfering RNA (siRNA). Diese kurzen Ribonukleinsäure-Moleküle codieren keine Proteine, sondern verbinden sich im Zellkern gezielt mit komplementären einzelsträngigen mRNA-Molekülen eines Gens und blockieren so die Übersetzung der Erbinformation für den Bau eines Proteins. Inclisiran nutzt diesen körpereigenen Prozess der RNA-Interferenz (sog. Gen-Stilllegung), um durch den gezielten Abbau der mRNA die Translation der vom entsprechenden Gen ausgelesenen Information in das Protein PCSK9 in der Leber zu hemmen. PCSK9, das Enzym Proprotein Convertase Subtilisin/Kexin type 9, ist eine wichtige Stellschraube für die Regulierung der Anzahl der LDL-Rezeptoren auf der Oberfläche der Hepatozyten, denn es bindet an den Komplex aus LDL-Cholesterin und LDL-Rezeptor und führt dadurch zum Abbau dieser Rezeptoren. Wird die PCSK9-Synthese durch die RNA-Interferenz gehemmt, stehen mehr LDLRezeptoren in der Leber zur Verfügung, sodass die LDL-C-Aufnahme durch die Hepatozyten erhöht wird und LDL-C-Serumwerte deutlich sinken. Inclisiran ist zugelassen zur Behandlung Erwachsener mit primärer Hypercholesterinämie (heterozygot familiär und nicht-familiär) oder gemischter Dyslipidämie zusätzlich zu einer diätetischen Therapie:
• in Kombination mit einem Statin oder einem Statin mit anderen lipidsenkenden Therapien bei Patienten, die mit der maximal tolerierten StatinDosis die LDL-C-Ziele nicht erreichen, oder
• allein oder in Kombination mit anderen lipidsenkenden Therapien bei Patienten mit Statin-Intoleranz oder für die ein Statin kontraindiziert ist [1].
ca. 80 % der Studienteilnehmer zu jedem Zeitpunkt der Studie einen LDL-C-Wert <70 mg/dl [2, 3].
In dieser bisher längsten Followup-Studie zur Sicherheit von Inclisiran stimmte das Nutzen-RisikoProfil mit den Ergebnissen früherer Phase-III-Studien mit Laufzeiten von 18 Monaten überein. Die häufigsten behandlungsbedingten unerwünschten Ereignisse waren Reaktionen an der Injektionsstelle, die zumeist leicht bis mäßig ausgeprägt waren [2–4].
Brigitte Söllner, ErlangenLiteratur
1 Fachinformation Leqvio®, aktueller Stand
2 Ray KK et al. Efficacy and safety of twice yearly subcutaneous inclisiran in patients with high cardiovascular risk and elevated low-density lipoprotein cholesterol up to 4 years: the ORION3 trial. AHA Scientific Sessions 2022. November 2022, Chicago
3 Ray KK et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL-C (ORION 3): results from the 4-year open-label extension of the ORION-1 trial. AHA Scientific Sessions 2022. November 2022, Chicago
4 Ray KK et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N Engl J Med 2020; 382:1507-1519
DOAK-Therapie
Direkte orale Antikoagulanzien (DOAK) gehören zum heutigen Therapiestandard in der Prävention und Behandlung von thromboembolischen Ereignissen wie z.B. Schlaganfällen bei erwachsenen Patienten mit nicht valvulärem Vorhofflimmern (nvVHF). Derzeit sind 4 DOAK in der EU zugelassen. Herausforderungen bei der Versorgung mit DOAK sind mögliche Interaktionen mit anderen Wirkstoffen und Substanzen bei häufig multimorbiden Patienten, aber auch die Auswahl und Dosisanpassung für jeweils unterschiedliche Indikationen und mit verschiedenen Kriterien für die Dosisreduktion. Somit gehören DOAK zu den Medikamenten, die relativ häufig Krankenhauseinweisungen verursachten – allerdings nur aufgrund von Fehldosierungen wie Off-label-, Über- oder Unterdosierungen. Diese potenziell vermeidbaren Faktoren erhöhen die Gesamtmortalität und Hospitalisierungsrate – ein Hinweis darauf, wie hoch der Unterstützungsbedarf bei der Medikation mit DOAK ist.
Kostenlose digitale Entscheidungshilfe
Vor dem Hintergrund dieser Versorgungsrealität entwickelte das Universitätsklinikum Heidelberg (UKHD) in enger Kooperation mit Daiichi Sankyo Deutschland und der BAYOOCARE GmbH die App
Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
159
MITTEILUNGEN
easyDOAC: eine neue App zur Unterstützung bei der
Auswahl und Dosierung von DOAK
(Hauptmodul)
Wenn noch keine Einträge gemacht wurden, weist das Hauptmodul darauf hin, dass Indikation und Nierenfunktion (Kreatinin-Clearance) eingegeben werden müssen (Abbildung 13). Die Indikationen sind zur Auswahl automatisch aufgeklappt.
easyDOAC. Im Fokus dieser innovativen Anwendung stehen die leichte Handhabung und der direkte Nutzeffekt – mit dem Ziel, ärztliche Leistungsträger bei der Auswahl und Dosisanpassung mit DOAK zu unterstützen. Das zertifizierte Medizinprodukt der Risikoklasse I ist als Progressive Web-App (PWA) online sowie offline über Smartphone, Tablet, Laptop oder Desktop nutzbar. easyDOAC steht kostenlos zur Verfügung und kann über das Internet aufgerufen (www.easydoac.de) oder im Play Store und im iOS App Store heruntergeladen werden. Die App kann in bestehende Arztsoftwaresysteme eingebettet oder als Favorit auf allen Devices abgelegt werden. Ein hinterlegtes Benutzerhandbuch gibt detaillierte Anwendungshinweise und Hintergrundinformationen. Datengrundlage der App sind kontinuierlich aktualisierte Fachinformationen, Forschungsdaten des UKHD und peer-reviewte Quellen wie internationale Studien und Guidelines.
easyDOAC in der klinischpraktischen Anwendung
Die Anwendung lässt sich leicht in klinische Abläufe integrieren: Über ein benutzerfreundliches Dashboard werden patientenspezifische Parameter eingegeben, die zur Therapieeinstellung nötig sind (Nierenfunktion, Alter, Gewicht, Kreatinin; Abb.1). Auf Basis validierter Fachinformationen erhalten Ärzte und Apotheker konkrete Dosierungsempfehlungen zu allen vier zugelassenen DOAK. Zudem können das Schlaganfallrisiko bei VHF-Patienten und das Blutungsrisiko unter DOAK-Therapie über
Validierter Mehrwert bei Auswahl und Dosierung von DOAK
Abbildung 1: Eingabeoberfläche des Hauptmoduls von easyDOAC mit automatisch aufgeklappten Indikationen und den anderen Filtermöglichkeiten.
Abbildung 13: Eingabeoberfläche des Hauptmoduls mit automatisch aufgeklappten Indikationen und den anderen Filtermöglichkeiten.
Nach der Auswahl der interessierenden Indikation werden die anderen Indikationen geschlossen, ein Löschsymbol (Eimer) zum Löschen der Indikation dargestellt und automatisch die Eingabefläche für die Nierenfunktion aufgeklappt (Abbildung 14).

den eingebetteten CHA2DS2-VAScScore- bzw. HAS-BLED-ScoreRechner bestimmt werden. Über ein separates Nierenmodul lässt sich die individuelle Nierenfunktion einschätzen. Durch Angabe der Begleitmedikation kann man mit der App auch relevante Arzneimittelinteraktionen und Kontraindikationen der jeweiligen Produkte prüfen sowie Informationen zu wirtschaftlichen Rahmenbedingungen (Tagestherapiekosten, Nutzenbewertung) aufrufen.
Was sagen Anwender zum konkreten Nutzen und Mehrwert der App? Um diese Frage zu beantworten, führte die figus GmbH eine Studie zur Evaluierung von easyDOAC durch. Die teilnehmenden Ärzte wurden vor und nach der 8-wöchigen Erprobung der App über Fokusgruppeninterviews befragt, welche Mehrwerte easyDOAC ihnen hinsichtlich einer adäquaten Therapieentscheidung bietet. Das Ergebnis: Unterstützungsbedarf zur Medikationseinstellung besteht vor allem bei komplexen Krankheitssituationen mit Begleitmedikation und eingeschränkter Nierenfunktion. Auch die gesonderte Prüfung der Begleitmedikation ist ein häufiger Grund für die Nutzung der App. Ein hoher Mehrwert besteht dabei in der Zeit- und Arbeitsersparnis, da keine zusätzliche Informationsbeschaffung über unterschiedliche Quellen nötig ist. Hinsichtlich der Nutzungshäufigkeit ergab sich, dass vor allem jüngere und noch nicht so erfahrene Ärzte von der App profitierten. Ein deutlicher Mehrwert ist demnach auch der Lerneffekt und damit eine höhere Sicherheit in der Therapieentscheidung. Letztlich profitieren auch – und vor allem – die Patienten von der ärztlich genutzten Entscheidungshilfe, weil Über- und Unterdosierungen vermieden und Kontraindikationen sicher identifiziert werden können. Das wirkt sich positiv auf das Patienten-Outcome aus und hat damit auch eine hohe gesundheitspolitische Relevanz.
Version 1.3 Datum 5.4.2022
160 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH Mitteilungen
Autor: WEH
B. S.
KONGRESSE
Behandlung der Hypertonie –Quo vadis?
Vom 1. bis 3. Dezember fand in Berlin der 46. Deutsche HypertonieKongress statt. Unter dem Motto „Hypertonie in allen Lebenslagen“ wurden die neuesten Erkenntnisse aus Wissenschaft, Forschung und Praxisalltag in Vorträgen, Symposien und Workshops sowohl theoretisch als auch praxisnah präsentiert und diskutiert. Im Rahmen des Kongresses lud Servier unter der Leitung von Professor Peter Trenkwalder, Gauting, zum Industriesymposium „Behandlung der Hypertonie – Quo vadis?“ ein. Hier erläuterte Professor Florian Limbourg, Hannover, die Bedeutung der Diuretika: Sie werden oft „gescholten“, sind aber unersetzliche Medikamente für die Hypertoniebehandlung. Im weiteren Verlauf präsentierte Trenkwalder die Ergebnisse einer deutschen Analyse, wonach die Verschreibung von antihypertensiven Wirkstoffen als Fixkombination entgegen den Leitlinienempfehlungen sowie den Vorlieben der Patienten rückläufig ist. Dies unterstreicht die Bedeutung der Patientenbeteiligung für den Therapieerfolg, was auch Professor Roland Schmieder, Erlangen, in seinem Vortrag weiter beleuchtete.
Welchen Stellenwert haben Diuretika in der BluthochdruckTherapie?
Wie Limbourg betonte, sind Diuretika eine für die Bluthochdruck-
Therapie unersetzliche Wirkstoffklasse. Unterschieden werden die Thiaziddiuretika (z.B. Hydrochlorothiazid) und die thiazidartigen Diuretika (z.B. Indapamid, Chlortalidon), wobei letztere durch eine längere Wirkdauer von bis zu 24 Stunden und somit auch eine länger anhaltende Blutdrucksenkung überzeugen. Außerdem ist unter thiazidartigen Diuretika das Risiko für Herzinsuffizienzen (HFrEF und HFpEF) geringer als bei anderen antihypertensiven Wirkstoffklassen und ihr Einsatz hat sich auch in besonderen Risikoindikationen wie z.B. höherem Alter oder eingeschränkter Nierenfunktion bewährt. Allerdings ist zu beachten, dass sich unter der Diuretika-Therapie eine Hyponatriämie oder Hypokaliämie entwickeln kann. „Alles in allem ist das Nutzen-Risiko-Profil der Diuretika dennoch günstig“, konstatierte Limbourg. Diuretika sollten den europäischen Leitlinien entsprechend in allen Stufen des Therapiealgorithmus als Kombinationstherapie bei Hypertonie eingesetzt werden. Die Evidenzen für diese Empfehlungen stammen aus zahlreichen randomisierten kontrollierten Studien, in denen u.a. die Wirksamkeit von Indapamid in Kombination mit anderen Wirkstoffklassen untersucht wurde und verschiedenste Patientenkollektive (z.B. Typ-2-Diabetiker in ADVANCE, Schlaganfallpatienten in PROGRESS und über 80-Jährige in HYVET) abgebildet wurden. Auch im Langzeitverlauf konnte die Wirksamkeit von Diuretika bestätigt werden: Im Rahmen der Brisighella-Heart-Studie wurden Patienten, die verschiedene antihypertensive Dreifachkombinationen erhielten, bis zu 12 Jahre
lang beobachtet. Die Kombination aus Perindopril, Amlodipin und Indapamid (in Deutschland als Fixkombination Viacorind® verfügbar) bewirkte dabei eine stabilere Blutdrucksenkung als andere eingesetzte Kombinationstherapien bei gleichzeitig protektivem Stoffwechselprofil.
Diuretika sind demnach bei allen Bluthochdruckpatienten eine essenzielle Wirkstoffklasse und aus der modernen antihypertensiven Therapie nicht mehr wegzudenken, wobei Indapamid laut Limbourg das günstigste Wirkungs- und Nebenwirkungsprofil besitzt.
Moderne Hypertoniebehandlung – the patient’s view
Die suboptimale Adhärenz bei der antihypertensiven Therapie ist eine der Hauptursachen für eine schlechte Blutdruckkontrolle und mit einem erhöhten Risiko für kardiovaskuläre Ereignisse, Komorbiditäten und Mortalität assoziiert. Laut Schmieder liegt es daher in der Verantwortung des Behandelnden, die Bedürfnisse der Patienten zu berücksichtigen und gemeinsam mit ihm die optimale Therapiestrategie festzulegen. Wichtigster Aspekt ist dabei, die Anzahl der Tabletten möglichst gering zu halten bzw. idealerweise verschiedene Wirkstoffkombinationen in einer einzigen Tablette als Single Pill Combination (SPC) zu verabreichen. Zwar wird dies seit 2018 in den europäischen Leitlinien bereits als Initialtherapie bei unkompliziertem Bluthochdruck empfohlen, aber immer noch zu selten praktiziert. Dabei wurden die Vorteile der Kombinationspräparate in zahlrei-
161 Perfusion 5-6/2022 35.
© Verlag PERFUSION GmbH Kongresse
Jahrgang
chen Studien nachgewiesen. Höhere Therapietreue, weniger Komplikationen und geringere Kosten bei Verordnung einer SPC – dies sind die Ergebnisse der START-Studie, einer groß angelegten Analyse von Real-World-Versorgungsdaten mit fast 200.000 Fällen der AOK PLUS. Auch in einer italienischen Beobachtungsstudie war eine SPC bestehend aus Perindopril, Amlodipin und Indapamid (in Deutschland als Fixkombination Viacorind® verfügbar) einer freien Kombination derselben Wirkstoffe hinsichtlich der Adhärenz signifikant überlegen: Fast 60 % der Patienten in der SPC-Gruppe zeigten eine hohe Therapietreue, während es in der Gruppe mit der freien Kombination nur 27 % waren (p < 0,001). Der Anteil an nicht adhärenten Patienten war unter der freien Kombination mit 54 % gegenüber 21 % in der SPC-Gruppe mehr als doppelt so hoch (p < 0,001). Dies spiegelte sich auch in verbesserten Outcomes wider: So waren die Mortalitätsrate (139,9 vs. 105,8 pro 1000 Personenjahre; p < 0,001) sowie die Inzidenz kardiovaskulärer Ereignisse (33,7 vs. 29,9 pro 1000 Personenjahre; p < 0,05) unter der freien Kombination signifikant höher als unter der SPC-Behandlung. „Die Vereinfachung des Therapieregimes ist somit eine simple, aber wirkungsvolle Maßnahme – SPC kann Leben retten“, resümierte Schmieder.
Was sollten die neuen Leitlinien berücksichtigen?
Die derzeit aktuellen europäischen Leitlinien für die Hypertoniebehandlung stammen aus dem Jahr
2018 und enthalten Empfehlungen, die auf den Daten aus jüngeren klinischen Studien und MetaAnalysen basieren. So wurden die Interventionsgrenzen angepasst, Blutdruckzielbereiche neu definiert, das Behandlungsschema durch die initiale Therapie mit einer Zweifachkombination vereinfacht sowie ein intensivierter Einsatz von SPC zur Besserung der Therapieadhärenz gefordert.
In naher Zukunft dürfte wieder mit einer neuen europäischen Leitlinie zu rechnen sein – welche Veränderungen sind dann zu erwarten oder wären notwendig?
Den Expertenmeinungen zufolge bildet der aktuelle empfohlene Therapiealgorithmus das optimale Vorgehen bei der Hypertoniebehandlung ab. Lediglich 3 Wünsche hätten die Experten für eine Aktualisierung: Erstens ist das Kollektiv der Älteren nicht optimal abgebildet; der Fokus sollte statt auf das kalendarische Alter ab 65 Jahren eher auf das biologische Alter gerichtet sein. Zweitens sollte die Heimblutdruckmessung häufiger angewendet werden und drittens rutscht im klinischen Praxisalltag eine besondere Population „durchs Raster“: die jüngeren Männer zwischen der letzten Untersuchung im Jugendalter und dem ersten Auftreten von Komplikationen mit Anfang 50. Trotz der guten Grundlagen besteht bei der Umsetzung im klinischen Alltag noch Luft nach oben – ein Ziel, das durchaus erreichbar ist.
ElisabethKonsistente
Blutdrucksenkung
nach renaler Denervierung mit Ultraschall
Auf der Jahrestagung 2022 der American Heart Association (AHA) präsentierte ReCor Medical die Ergebnisse der Analyse der gepoolten Daten von 3 prospektiven, randomisierten und placebokontrollierten klinischen Studien, in denen die Wirksamkeit und Sicherheit des endovaskulären Paradise™-Systems zur renalen Denervierung mit Ultraschall (uRDN) bei Patienten mit unkontrolliertem Bluthochdruck untersucht wurden. Im Einzelnen handelte es sich um die Studien RADIANCE-HTN TRIO bei Patienten mit resistenter Hypertonie sowie RADIANCE-HTN SOLO und RADIANCE II, in denen Patienten mit leichter bis mittelschwerer Hypertonie untersucht wurden. Alle 3 Studien waren individuell auf den primären Endpunkt des ambulanten systolischen Tagesblutdrucks nach 2 Monaten ausgerichtet, und alle drei erreichten diesen primären Wirksamkeitsendpunkt mit statistischer Signifikanz nach 2 Monaten. Insgesamt wurden bei der RADIANCE-Pool-Analyse die Daten von mehr als 500 Patienten ausgewertet.
Wirksam bei verschiedenen Hypertonieformen und -schweregraden
Wilhelmi, München
Wie Professor Ajay Kirtane vom NewYork-Presbyterian/Columbia University Irving Medical Center betonte, führte die uRDN bei den eingeschlossenen Patienten mit
162 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
Kongresse
Abbildung 1: Das Paradise™ System zur renalen Denervierung mit Ultraschall (© ReCor Medical).
unkontrolliertem Bluthochdruck zu einer konsistenten und signifikante Blutdrucksenkung, und das unabhängig von Alter, Geschlecht, Ausgangsblutdruck, Medikamentenspiegel und ethnischer Zugehörigkeit.
Der ambulante systolische Tagesblutdrucks sank in der uRDN-Gruppe um –8,5 mmHg (p < 0,0001) mit einem Unterschied zwischen Therapie und Scheinbehandlung nach 2 Monaten von –5,9 mmHg (p < 0,0001), was für die uRDN spricht. Die Blutdruckergebnisse waren bei den 24-Stunden-, Nacht-, Heim- und Praxis-Messungen ähnlich positiv.
„Die gemeinsame Auswertung der Daten aus dem RADIANCE-Programm zeigt, dass die Behandlung mit dem Paradise™ uRDN-System zu einer konsistenten Senkung des Blutdrucks bei unterschiedlichen Schweregraden der Hypertonie führt. Diese durchgängige und klinisch bedeutsame Senkung des Blutdrucks über mehrere Patientengruppen hinweg verstärkt unser Interesse an der Verwendung von uRDN als potenzielle therapeuti-
sche Option, wenn sie mit einer Änderung des Lebensstils und Medikamenten für unsere Patienten mit unkontrolliertem Blutdruck kombiniert wird“, resümierte Kirtane, einer der Hauptprüfer der Studie. Und Studienleiter Professor Michel Azizi, Paris, ergänzte: „Es ist sehr wichtig, dass die gepoolte RADIANCE-Analyse eine konsistente Blutdrucksenkung bei Patienten mit verschiedenen Hypertonieformen und sowohl mit als auch ohne blutdrucksenkende Medikamente gezeigt hat, wodurch die potenzielle Anwendbarkeit von uRDN erweitert wird. Ebenso wichtig ist, dass mehr als 50 % der mit uRDN behandelten Patienten in der gepoolten Analyse entweder eine Kontrolle des ambulanten Blutdrucks am Tag erreichten oder eine Senkung des ambulanten systolischen Blutdrucks am Tag um mehr als 10 mmHg nach 2 Monaten aufwiesen. Das belegt den potenziellen Nutzen von uRDN als Bestandteil eines Behandlungsschemas für Patienten mit unkontrolliertem Bluthochdruck.“
Brigitte Söllner, Erlangen
Versorgungslücken im AdipositasManagement schließen
Laut WHO ist die chronische Erkrankung Adipositas nicht zuletzt in Europa weiterhin ungebremst auf dem Vormarsch und hat PandemieCharakter. Vor diesem Hintergrund diskutierten Experten auf dem Kongress der Deutschen Adipositas Gesellschaft (DAG) in München im Rahmen eines Symposiums von Novo Nordisk Versorgungslücken sowie das zeitgemäße Management von Adipositas und Komorbiditäten in Deutschland.
Hier ist fast jeder vierte Erwachsene von Adipositas betroffen, bei Kindern und Jugendlichen zwischen 5 und 19 Jahren sind es etwa 10 % (11,4 % und 7,8 %). Dass es in der Regelversorgung von Menschen mit dieser chronischen Erkrankung größerer Anstrengung bedarf, zeigten unter anderem auch Daten der ACTION IOStudie, die die Wahrnehmungen, Einstellungen, Verhaltensweisen und Hindernisse für eine wirksame Behandlung bei Menschen mit Adipositas untersuchte. 77 % der Befragten fanden es gut, dass ihr Hausarzt Gespräche über das Gewichtsmanagement initiierte, und 65 % derjenigen, die zuvor noch nicht über ihr Gewicht gesprochen hatten, wünschten sich, dass ihr Hausarzt ein entsprechendes Gespräch suchen würde. Von den Personen, die ihr Gewicht besprochen hatten, hielten 44 % die Gespräche jedoch für wenig oder gar nicht hilfreich. Auf Seiten der Ärztinnen und Ärzte waren 71 % bzw. 68 % davon überzeugt, dass die Patienten wenig Interesse oder Motivation zum Abnehmen haben, und un-
163 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH Kongresse
terließen deshalb Gespräche über das Gewichtsmanagement.
Chronische Erkrankung mit vielen Komorbiditäten
„Für die Diagnose Adipositas braucht es keine Bluttests. Es reicht die Kenntnis von Größe und Gewicht“, konstatierte Professor Jens Aberle, Präsident der DAG. Mit Adipositas gehen bis zu 200 Komorbiditäten einher, die von Typ2-Diabetes und kardiovaskulären Erkrankungen bis zum obstruktiven Schlafapnoe-Syndrom und bestimmten Krebsarten reichen. Außerdem sinken Lebenserwartung und Lebensqualität deutlich mit steigendem Body-Mass-Index (BMI). Umgekehrt senkt eine Abnahme des BMI um 5 kg/m2 oberhalb von 25 kg/m2 die Gesamtmortalität um 30 %. Und bereits eine Gewichtsreduktion im Bereich von 5 – 15 % des Ausgangsgewichts verbessert diverse klinisch relevante Risikofaktoren und kann sich
vorteilig auf die genannten Komorbiditäten auswirken. Diese Zusammenhänge im Gespräch deutlich zu machen, kann helfen, der Entstehung von Folgeerkrankungen vorzubeugen: „Eine frühe Behandlung verhindert Komorbiditäten“, bekräftigte Aberle. „Und selbst wenn bereits etwa ein Typ-2-Diabetes oder eine kardiovaskuläre Erkrankung besteht, kann man therapeutisch noch viel erreichen, wie man aus Studien weiß“. Schließlich umfasst die Adipositastherapie, die stets multimodal sein sollte, gemäß Leitlinie zusätzlich zu Lebensstilinterventionen und Verhaltenstherapie auch bariatrische und medikamentöse Behandlungen.
Therapielücke schließen: Stellenwert medikamentöser Therapie
„Inkretin-basierte Therapien können wir bei einer sehr guten Nutzen-Risiko-Bewertung über einen längeren Zeitraum bei Menschen
mit Adipositas einsetzen“, erklärte Dr. med. Markus Menzen, Bonn. Zugelassen ist in Deutschland aus dieser Substanzklasse z.B. der Glucagon-like peptide-1 (GLP-1) Rezeptoragonist Liraglutid (Saxenda®). Early Responder* erreichten unter Liraglutid (3 mg) nach einem Jahr Therapiedauer etwa einen Gewichtsverlust von 11,2 %. Eine noch stärkere Gewichtsreduktion lässt sich mit der verwandten Substanz Semaglutid (2,4 mg; Wegovy®) erzielen. In STEP 3, einer randomisierten klinischen Studie, an der 611 Erwachsene mit Übergewicht oder Adipositas teilnahmen, führte eine 68-wöchige Behandlung mit einmal wöchentlich subkutan verabreichtem Semaglutid im Vergleich zu Placebo (in Kombination mit einer intensiven Verhaltenstherapie
einer
* Early Responder: Patienten, die in den ersten 12 Wochen mit der 3,0-mg-Erhaltungsdosis eine Gewichtsreduktion ≥5 % erzielten; gepoolte Analyse der SCALEStudien zu Adipositas und Prädiabetes und SCALE Diabetes.
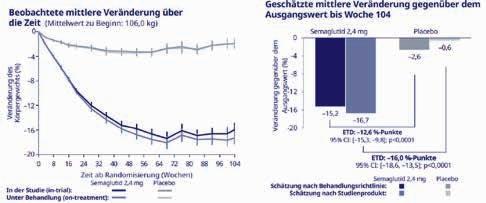
164 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH Kongresse
und
Abbildung 1: Veränderung des Körpergewichts über 104 Wochen Therapie mit Semaglutid (Wegovy®) in der Studie STEP 5 (nach Garvey WT et al., 2022).
kalorienarmen Ernährung) in den ersten 8 Wochen zu einer Verringerung des Körpergewichts um 16 % versus 5,7 % (Differenz –10,3 %; 95%-KI: –12 bis –8,6; p < 0,001).
Eine Langzeitbehandlung über 104 Wochen führte zu einer mittleren Veränderung des Körpergewichts von –15,2 % in der SemaglutidGruppe gegenüber –2,6 % unter Placebo (Abb. 1), was einem geschätzten Behandlungsunterschied von –12,6 % entspricht (95%-KI –15,3 bis –9,8; p < 0,0001).
Neben der Gewichtsreduktion beeinflusste die Behandlung mit Semaglutid auch metabolische und kardiovaskuläre Risikofaktoren positiv, wie etwa HbA1c-Wert, Triglyzeride, systolischer und diastolischer Blutdruck oder C-reaktives Protein.
„Nicht zu vernachlässigen ist auch die Tatsache, dass Semaglutid einen Einfluss auf den glykämischen Status ausübt und eine Rückkehr vom Prädiabetes zu Normoglykämie fördert“, betonte der Experte. Keiner der Patienten aus der prädiabetischen Subpopulation entwickelte unter dem Prüfpräparat einen Typ-2-Diabetes (Placebo 3,7 %). „Diese Daten belegen, dass wir die Lücke zwischen Verhaltenstherapie und bariatrischer Therapie heute mithilfe von medikamentösen Therapien effektiv schließen können“, resümierte Menzen.
Fabian Sandner, NürnbergIMPRESSUM
Überzeugende Daten auch zu MitraClip™ und Amplatzer™ Amulet™
Herausgeber: Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School,Salmon Pool Lane, Exeter EX2 4SG, UK
Prof. Dr. med. Wolfgang Koenig Deutsches Herzzentrum München Technische Universität München Lazarettstraße 36 80636 München
Auf dem EuroPCR präsentierte Abbott außerdem die positiven Ergebnisse der EXPAND-Studie. Diese belegte, dass die Therapie mit dem MitraClip™-G4-System (Abb. 3) bei HerzinsuffizienzPatienten mit Mitralinsuffizienz zu einer Verbesserung der Symptome und der Lebensqualität führt. Bei Patienten mit Vorhofflimmern kann zum Schutz vor kardioembolischen Schlaganfällen das linke Vorhofohr (left atrial appendage, LAA) minimalinvasiv mit dem LAA-Okkluder Amplatzer™ Amulet™ (Abb. 4) verschlossen werden. Obwohl bei Frauen häufiger Frühkomplikationen nach dem LAA-Verschluss auftreten als bei Männern, belegen die Ergebnisse der Amulet-IDE-Studie nun, dass Frauen und Männer, denen der Amplatzer™ Amulet™ LAAOkkluder implantiert wurde, langfristig ähnliche Vorteile aus dem LAA-Verschluss ziehen. Brigitte Söllner, Erlangen
Wissenschaftlicher Beirat: Prof. Dr. med. T. von Arnim (Kardiologie), München Prof. Dr. med. G. V. R. Born (Arterioskleroseforschung), London Prof. Dr. med. C. Diehm (Angiologie), Karlsbad Priv.-Doz. Dr. med. Dr. phil. C. Drosde (Kardiologie), Freiburg Dr. med. J. Dyerberg MD, Ph. D. (Klin. Chemie), Aalborg Sygehus, Dänemark Univ.-Prof. Dr. med. H. W. Eichstädt, (Kardiologie), Berlin Doz. Dr. rer. nat. F.-D. Ernst (Hämorheologie), Dresden Dr. med. J. Gehring (Kardiologie, Rehabilitation), München Prof. Dr. med. J. D. Gruß (Gefäßchirurgie), Kassel Prof. Dr. J. Harenberg (Hämostaseologie), Mannheim Prof. Dr. med. L. Heilmann (Gynäkologie), Rüsselsheim Prof. Dr. med. H. M. Hoffmeister (Kardiologie), Solingen Prof. Dr. med. H. U. Janka (Diabetologie), München Dr. med. J. Janzen MPhil (Pathologie), Bern, Schweiz Prof. Dr. med. L. Kollár M.D., PhD (Gefäßchirurgie), Universität Pécs, Ungarn Prof. Dr. med. M. Marshall (Phlebologie), Rottach Egern Prof Dr. med. J. Matsubara (Chirurgie), Ishikawa, Japan Prof. Dr. med. G. Mchedlishvilli (Mikrozirkulation), Tbilisi, Georgien Prof. Dr. med. V. Mitrovic (Kardiologie, Klinische Pharmakologie), Bad Nauheim Prof. Dr. med. H. Mörl (Angiologie), Mannheim Prof. Dr. med. F. J. Neumann (Kardiologie), Bad Krozingen Prof. Dr. med. K. L. Resch (Medizin-Statistik), Bad Elster Prof. Dr. med. G. Rettig (Kardiologie), Homburg PD Dr. med. Rainer Röttgen (Radiologie), Berlin Prof. Dr. med. G. Schmid-Schönbein (Biomechanik), La Jolla, USA Prof. Dr. med. H. Schmid-Schönbein (Physiologie), Aachen
Prof. Dr. med. A. Schrey (Pharmakologie), Düsseldorf
Prof. Dr. med. H. Sinzinger (Nuklearmedizin), Wien, Österreich
Prof. Dr. med. T. Störk (Kardiologie, Angiologie), Göppingen
Prof. Dr. med. I. Szirmai M.D. (Neurologie), Universität Budapest, Ungarn
Prof. Dr. med. G. Trübestein (Angiologie), Bonn Prof. Dr. med. B. Tsinamdzvrishvili (Kardiologie, Hypertonie), Tbilisi, Georgien
Prof. Dr. med. W. Vanscheidt (Dermatologie), Freiburg
Prof. Dr. med. H. Weidemann (Kardiologie, Sozialmedizin), Bad Krozingen
Schriftleitung: Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School, Salmon Pool Lane, Exeter EX2 4SG, UK E-Mail: Edzard.Ernst@pms.ac.uk Tel: +44 (0) 1392 726029 Fax: +44 (0) 1392 421009
Die Zeitschrift erscheint 6-mal im Jahr; Jahresabonnement 27,–; Einzelheft 5,50, inklusive MwSt., zuzüglich Versandspesen. Der Abonnementpreis ist im voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich.
Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 – Rolf Wolle (verantwortlich) Schwabacherstr. 117, 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna (verantwortlich) Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Satz: Rolf Wolle, Schwabacherstr. 117, 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM GmbH Austraße 7 96114 Hirschaid
PERFUSION is listed in Current Contents/Clinical Medicine (CC/CM) and listed in The Genuine Article.
VERL AG PERFUSION
Verlag PERFUSION GmbH Storchenweg 20 90617 Puschendorf Telefon: 09101/990 11 10 Fax: 09101/990 11 19 www.Verlag-Perfusion.de E-Mail: perfusion@t-online.de
165 Perfusion 5-6/2022 35. Jahrgang © Verlag PERFUSION GmbH
Kongresse
OFFIZIELLES ORGAN DER DEUTSCHEN GESELLSCHAFT FÜR ARTERIOSKLEROSEFORSCHUNG
PERFUSION
Journal
SARCLISA® + Kd
HERAUSRAGENDES MEDIANES PFS VON 3 JAHREN!
IKEMA-Studie zeigt das längste mediane PFS eines Anti-CD38-Antikörpers in Kombination mit einem Proteasominhibitor beim RRMM.1
Tieferes Ansprechen
CR-Rate 44 % vs. 29 % (SARCLISA® + Kd vs. Kd)1
S3-Leitlinie
Längeres PFSa,1
mPFS 35,7 vs. 19,2 Mon. mit Kd alleina, 1

Verbesserte MRDNegativitätsrate 34 % MRD-Negativitätb vs. 15 % mit Kd allein1
SARCLISA® ist in Kombination mit Kd in der 2. Linie für Patient*innen mit MM empfohlen. 2
Genau
hinschauen
lohnt sich!
Update zur IKEMA-Studie
Die Ergebnisse zum Nachlesen finden Sie hier.
a Bei medianem Follow-up von 44 Monaten. b Intention-To-Treat-Population, Next-Generation-Sequenzierung, Sensitivität 10–5 CD38 = Cluster of Differentiation 38; CR = komplette Remission; Kd = Carfilzomib und Dexamethason; Mon. = Monate; mPFS = medianes PFS; MRD = minimale Resterkrankung; PFS = progressionsfreies Überleben; RRMM = rezidiviertes, refraktäres Multiples Myelom.
1. Moreau P, Dimopoulos MA, Mikhael J, et al. Updated progression-free survival (PFS) and depth of response in IKEMA, a randomized Phase 3 trial of isatuximab, carfilzomib and dexamethasone (Isa-Kd) vs Kd in relapsed multiple myeloma (MM). Präsentiert beim Controversies in Multiple Myeloma (COMy) World Congress, 12.–15. Mai 2022. https://comylive.cme-congresses.com/wp-content/uploads/2022/05/Moreau.pdf. (Zugriff am 08.08.2022) 2. Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): Diagnostik, Therapie und Nachsorge für Patienten mit monoklonaler Gammopathie unklarer Signifikanz (MGUS) oder Multiplem Myelom, Langversion 1.0, 2022, AWMFRegisternummer: 018/035OL, https://www.leitlinienprogrammonkologie.de/leitlinien/multiples-myelom/. (Zugriff am 21.03.2022)
Sarclisa 20 mg/ml Konzentrat zur Herstellung einer Infusionslösung. Wirkstoffe: Isatuximab. Zusammens.: Arzneil. wirks. Bestandt.: 1 Durchstechfl. m. 5/25 ml Konzentrat enth. 100/500 mg Isatuximab, entspr. 20 mg/ml. Sonst.Bestandt.:Sucrose, Histidinhydrochlorid-Monohydrat, Histidin, Polysorbat 80, Wasser f. Injektionszwecke. Anw.-geb.: In Kombination m. Pomalidomid u. Dexamethason z. Behandl. d. rezidivierten u. refraktären Multiplen Myeloms b. Erwachsenen, d. mind. 2 vorausgegangene Ther., darunter Lenalidomid u. e. Proteasom-Inhibitor, erhalten haben u. unter d. letzten Ther. e. Krankheitsprogression zeigten. In Kombination m. Carfilzomib u. Dexamethason z. Behandl. des Multiplen Myeloms b. Erwachsenen, d. mind. 1 vorausgegangene Ther. erhalten haben. Gegenanz.:. Überempfindlichk. ggü. d. Wirkstoff od. e. d. sonst. Bestandt. Warnhinw. u. Vorsichtsm.: Nicht schütteln. Nebenw. Isatuximab m. Pomalidomid: Infekt.u.parasit.Erkr.: Sehr häufig: Pneumonie, Infekt. d. ob. Atemw., Bronchitis, Häufig: Herpes zoster Gutart., bösart. u. unspez. Neubild.: Häufig: Plattenepithel-Ca d. Haut. Blut u. Lymphsyst.: Sehr häufig: Neutropenie, febrile Neutropenie. Immunsystem: Gelegentl.: anaphyl. Reaktionen. Stoffw. u. Ernähr.-stör.: Häufig: vermind. Appetit. Herz: Häufig: Vorhofflimmern. Atemw.,Brustr.,Mediast.: Sehrhäufig: Dyspnoe.GIT: Sehrhäufig: Diarrhö,Übelk.,Erbrechen.Untersuchungen: Häufig: Gewichtsabnahme.Verletz.,Vergift.u. durch Eingriffe bedingte Komplikat.: Sehr häufig: infusionsbedingte Reaktion. Nebenw. Isatuximab m. Carfilzomib: Infekt. u. parasit. Erkr.: Sehr häufig: Pneumonie, Infekt. d. ob. Atemw., Bronchitis, Häufig: Herpes zoster Gefäßerkr.: Sehr häufig: Hypertonie. Gutart., bösart. u. unspez. Neubild.: Häufig: Hautkrebs, solide Tumore außer Hautkrebs. Blut u. Lymphsyst.: Häufig: Neutropenie. Immunsystem: Gelegentl.: anaphyl. Reaktionen. Atemw., Brustr., Mediast.: Sehr häufig: Dyspnoe, Husten. GIT: Sehr häufig: Diarrhö, Erbrechen. Allg. Erkr. u. Beschw. am Verabreichungsort: Sehr häufig: Fatigue. Verletz., Vergift. u. durch EingriffebedingteKomplikat.: Sehr häufig: infusionsbedingte Reaktion. Verschreibungspflichtig Sanofi-aventis groupe, 54 rue La Boétie, 75008 Paris, Frankreich Stand der Information: Juni 2022 Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
Sanofi-Aventis Deutschland GmbH Lützowstraße 107 | 10785 Berlin | Telefon 0800 0436996 | www.sanofi.de

Monoklonaler Anti-CD38-Antikörper mit multimodaler Wirkung
MAT-DE-2201827 – 2.0 – 08/2022