26 minute read
Optimising the synthesis of the 4-iodo analogue of pyrimethamine


Maxine Wu
Barker College Theemergence and rapid spread of drug resistant P. falciparum, the most prevalent malaria pathogen, has threatened to curtail the efficacy and therapeutic lifetime of antifolate drug treatments like pyrimethamine, due to point mutations in the dihydrofolate reductase (DHFR) enzyme of the parasite. Currently, the burden of malaria is highest in developing counties, and the deteriorating efficacy of existing anti-malarial drugs have increased the cost and complexity of routine prophylaxis and treatment. The present work proposes to overcome such resistance, using a simple rational drug design strategy which focuses on changing substituents on the pyrimethamine ring to synthesise new pyrimethamine analogues as potential drug leads. This report discusses methods used to successfully optimise the synthesis of the 4-iodopyrimethamine analogue and its possible future applications in cross-coupling reactions.
Literature Review
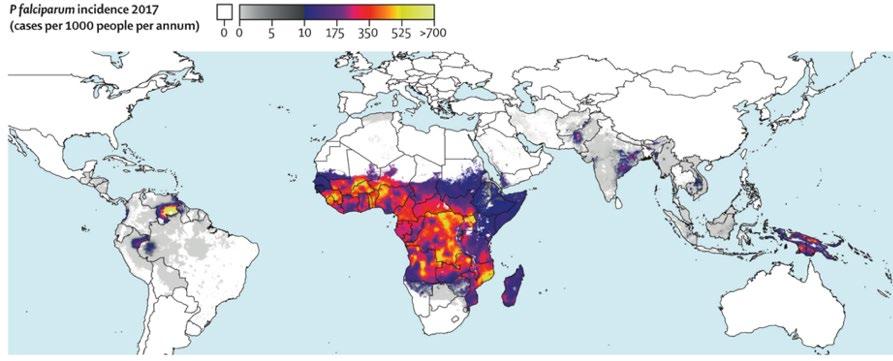
Malaria is a protozoan disease that continues to present major public health concerns for most endemic areas of the world (Weiss et al., 2019). Once infected, malaria attacks the body’s red blood cells, and if not treated within 24 hours, severe complications often lead to death (WHO, 2020). The burden of malaria is highest in developing countries, affecting vulnerable and marginalised populations – particularly children, pregnant women, migrants and refugees (Murphy, 2006). The optimisation of access to malaria intervention is essential for achieving universal health coverage and promoting well-being of all ages, having the potential to alleviate poverty, improve equity, hence contributing to overall socio-economic development in affected areas. Globally, there were 229 million cases of malaria in 2019, with an estimated death toll of 409,000 (WHO, 2020). Of the five Plasmodium parasite species, Plasmodium falciparum is the most virulent causative agent, and in 2018, accounted for 99.7% of cases in the African region, 71% in the Eastern Mediterranean, 65% in the Western Pacific and 50% in the South-East Asian region (Figure 1).
Since 2000, the scale-up of malaria control interventions has fuelled bold aims for disease eradication, having significantly reduced global morbidity and mortality (Weiss et al., 2019). This was driven by several factors, in particular, increased funding, effective vector control, and improved case reporting and surveillance (Cotter, Sturrock & Hsiang, 2013). Although remarkable
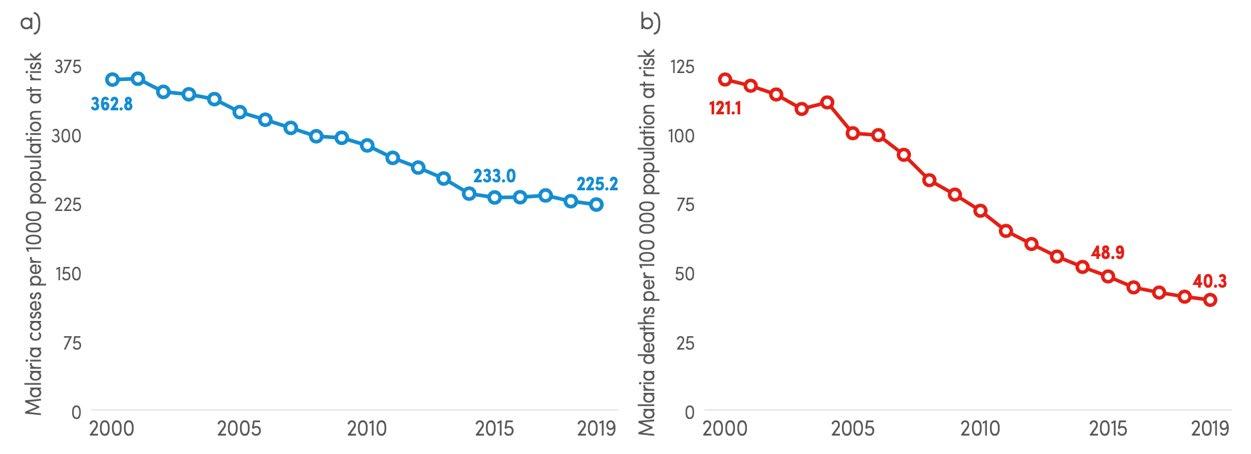
Figure 2: Graphs displaying the trends in a) malaria case incidence rate between 2000 and 2019 and b) mortality rate between 2000 and 2019 (Source: WHO, 2020)
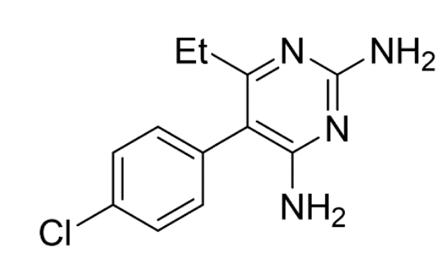
Figure 3: Structure of pyrimethamine (1)
Figure 4: Bio-synthetic pathway of DHFR catalysed hydrogenation of dihydrofolate (After: Sardarian et al., 2003)
progress has been made, the rate of reduction in both mortality and morbidity has slowed dramatically (Figure 2) (WHO, 2020).
Currently, malaria parasites have developed resistance to almost every anti-malarial drug available (Dhiman, 2019). Initially, these agents were highly effective, but due to drug pressure, the selection of resistant parasites has allowed for the continued proliferation of malaria (White, 2004). The subsequent propagation of resistant strains by local transmission and migration of parasite reservoirs are key factors contributing to the increased cost and complexity of achieving parasitological cure (Wernsdorfer, 1994). Accordingly, since present malaria control interventions are unlikely to address these epidemiological changes, the systematic exploration of novel strategies and methods are urgently needed.
The antimalarial drug pyrimethamine (1, Figure 3) is a specific derivative of 2,4- diaminopyrimidine and is currently sold under the trade name Daraprim (Tse, Korsik & Todd 2019). Since its development in 1952, pyrimethamine has been most widely used as an anti-malarial drug. However, the antiprotozoal agent has also been used in the treatment of other protozoan diseases such as toxoplasmosis and trypanosomiasis caused by Toxoplasma gondii and Trypanosoma brucei, respectively (Adane & Bharatam, 2008). In addition, pyrimethamine has also been used to treat Pneumocystis carinii pneumonia that often affects immune-compromised patients (Cirioni et al., 2000).
Drugs used in malaria treatment target specific biochemical processes vital to the growth of the disease causing parasite (Warhurst, 2002). The
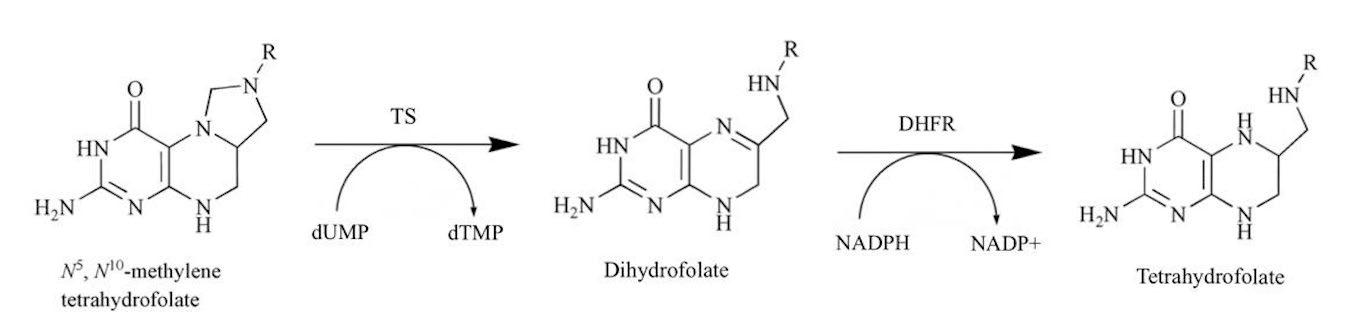
mechanistic function of pyrimethamine owes its effectiveness to its structural similarities with one of the domains of the bifunctional dihydrofolate reductase-thymidylate synthase (DHFR-TS) of P. falciparum, namely, the dihydrofolate reductase (DHFR) enzyme (Sirawaraporn et al., 1997). Due to its ability to inhibit the DHFR enzyme domain in the folate bio-synthetic pathway (Figure 4), pyrimethamine prevents the catalysation of the NADPH-dependent reduction of dihydrofolate to tetrahydrofolate. Consequently, this affects the biosynthesis of purines and several amino acids as well as cofactors imperative for DNA synthesis and cell multiplication, and hence, pyrimethamine prevents further proliferation of the plasmodia (Peterson, Walliker & Wellems, 1988).
Unfortunately, the efficacy of pyrimethamine over the years has been blunted by the rapid spread of resistance that has enervated scientific efforts (Bloland 2001). Knowledge of the molecular structure of the DHFR enzyme and its interactions with inhibitors and substrates, as well as how mutations can affect these functions are imperative for rational drug design.
Recently, solved crystal structures of the wild-type and mutant P. falciparum DHFR complexed with pyrimethamine have yielded insights into the mechanisms of resistance resulting from mutations (Figure 5A) (Yuthavong et al., 2005).
Although the connectivity of the P. falciparum DHFR-TS domains (Figure 5A, denoted in red and blue respectively) are still relatively unknown, the structure of this enzyme-inhibitor complex confirms that several amino acid residues at the active site are engaged in hydrogen bonding with dihydrofolate, and highlights other interactions which holds the substrate molecule in an orientation that allows for inhibition of the enzyme function (Figure 5B). However, mutations in the side chains as well as in the main chain configuration at the active site have created a steric constraint which is detrimental to the binding of the rigid parachlorophenyl group in pyrimethamine (Yuthavong et al., 2005). This has caused the subsequent dislocation of the phenyl ring, and significantly reduced the binding affinity for pyrimethamine (Lozovsky et al., 2009).
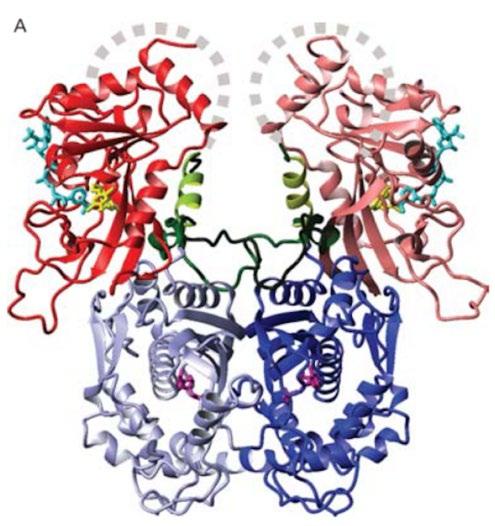
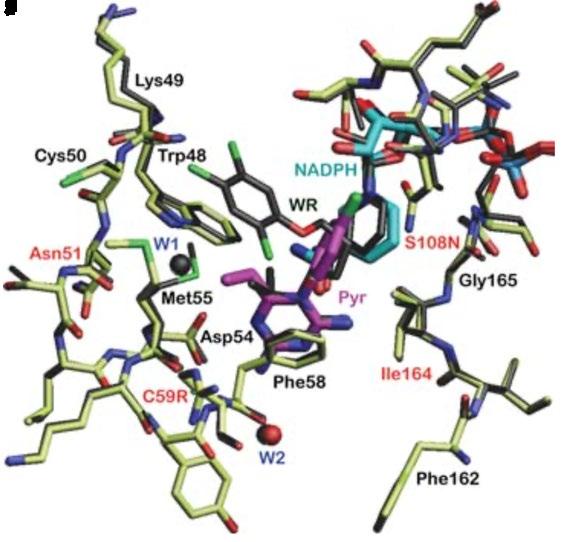
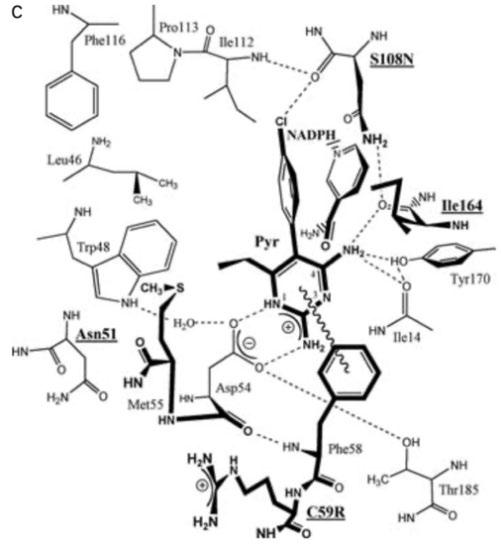
Figure 5: A) Crystal structure for P. falciparum DHFR-TS (Different shades denote individual sub-units. Grey dashed curves represent possible linkages based on intermolecular space in crystal packing. Junction between DHFR (blue) and TS (red) are drawn as dark green. Ligands bound in active sites are coloured as follows: dUMP in magenta and NADPH in cyan). B) Enzyme-inhibitor interactions at the active site of the wild-type DHFR. Four mutated amino acid residues are denoted with red labels. C) Diagram of interactions, four residues responsible for resistance are bolded and underlined. (Source: Yuthavong et al., 2005)
Furthermore, the Serine residue 108 (S108N) has also been found to be a crucial point mutation found in all pyrimethamine resistant strains (Sirawaraporn et al., 1997). Cumulative effects on inhibitor binding are seen with ancillary mutations at residue Asparagine 51 (N51L), Cysteine 59 (C59R) and Isoleucine 164 (1164L) which have caused a significant reduction in the binding affinity for pyrimethamine (Figure 5C) (Yuthavong et al., 2012). Such mutation-based resistance poses questions about research into designing new and potent antifolate drugs with improved affinity against wild-type and mutant DHFR as a means to minimise the burden of P. falciparum malaria (Adane & Bharatam, 2008).
One approach to overcoming drug resistance involves the development of new analogues where small structural changes to the chemical structure of the compound can significantly improve drug efficacy (Yuthavong, et al., 2005). Although crossresistance may be expected for inhibitors with similar structures, given that the DHFR enzyme needs to remain functional, it is likely that there are limitations to the number of mutational combinations that can occur in the DHFR domain (Tarnchompoo et al., 2018). Thus, this approach assumes that suitable structural modifications can be exploited.
This research aims to optimise the synthesis of a novel 4-iodopyrimethamine analogue (2) that was completed by a Barker College student in 2020 (Wong, 2020)(Figure 6B).The iodine analogue (2) is of interest due to the larger atomic radius and lower electronegativity compared to the chlorine atom in pyrimethamine (1). By replacing the chlorine atom with an iodine atom at the R4 position (Figure 6A), it is hypothesised that this could potentially enhance binding interactions at the DHFR enzyme site, or at least provide interesting structure-activity information. Additionally, given the significant effort into the synthesis of other various pyrimethamine analogues, the collection of inhibition constants (KI and IC50 values) acquired via biological testing against both mutant and wildtype P. falciparum from previous studies can be used quantitively to compare and assess the drug efficacy and inhibition potency of the novel 4iodopyrimethamine analogue (Nattee et al., 2017).
A
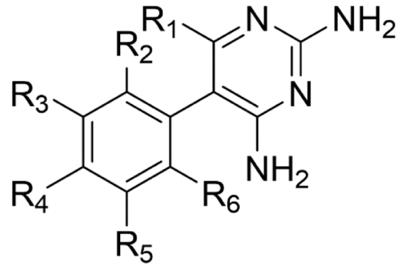
B
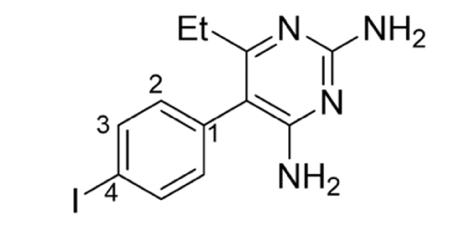
Figure 6: A) General structure of pyrimethamine analogues. B) Proposed 4-iodopyrimethamine analogue (2)
Furthermore, the carbon-iodine bond can be used as a “synthetic handle” for metal catalysed crosscoupling reactions, creating a pathway for the synthesis of more complex analogues with increased binding affinity (Mao et al., 2009). In the current literature, efforts have been made to exploit the Suzuki Miyaura cross-coupling procedure, and halogenated pyrimidines with boronic acids have been used as a common approach for the preparation of a diverse set of substituted pyrimidines (Richardson & Stevens, 2002). Since the carbon-iodine bond at the R4 position can be easily functionalised, the optimisation of the synthesis of the 4-iodopyrimethamine analogue can create a starting point for a library of potential antimalarial drugs in the future.
Previously, the successful synthesis of compound 2 was conducted following the 2016 synthetic pathway developed by Sydney Grammar School (SGS, 2016) for the synthesis of pyrimethamine (1) (Figure 7) (Wong, 2020). However, due to issues with synthetic yields and product purity, the final mass of 4-iodopyrimethamine was insufficient to conduct biological testing against P. falciparum. The focus of this report aims to resolve these issues and optimise yields such that biological data on the efficacy of 4-iodopyrimethamine in combating malarial pathogens can be conducted.
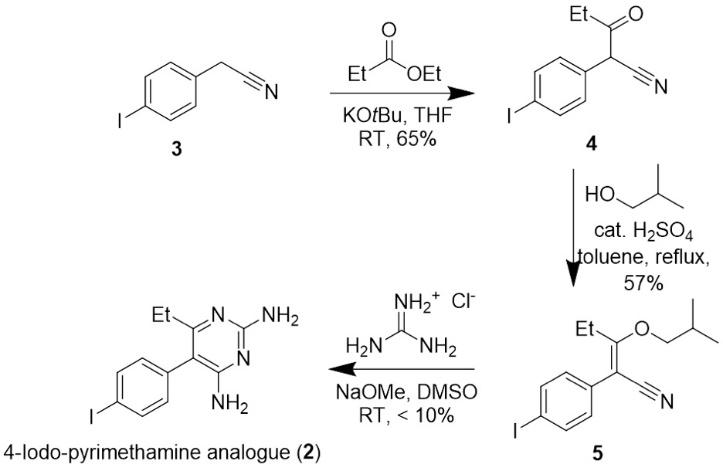
Figure 7: Pathway for the synthesis of 4-iodopyrimethamine analogue (2) with the yields from the previous synthesis
Scientific research question
How can the synthesis of 4-iodopyrimethamine (2) be optimized to improve yields and purity to allow for biological testing?
Scientific hypothesis
That an optimized synthesis of 4iodopyrimethamine (2) can be successfully undertaken to produce enough material for biological testing.
Methodology
General experiment details
1H spectra were recorded at 300 K using a Bruker Avance DRX400 NMR spectrometer in deuterated solvents. Residual acetone (δ 2.05) and chloroform (δ 7.26) were used as internal reference for 1H NMR spectra. The data is reported as chemical shift (δH ppm), relative integral, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet) and assignment. Atom labels on structures are to illustrate 1H NMR spectral assignments and do not necessarily correspond to the IUPAC names given. Analytical thin layer chromatography was performed with Merck Kieselgel 60 F254 (0.2 mm) pre-coated aluminium sheets, and visualisation was achieved by inspection under UV light. Throughout the reaction process Thin Layer Chromatography (TLC) was conducted to gauge the progress of the reaction and determine the point of completion. TLC analysis was conducted with either 50:50 Dichloromethane (DCM) : Hexane, or pure DCM.
Step 1: Synthesis of 2-(4-iodophenyl)-3oxopentanenitrile
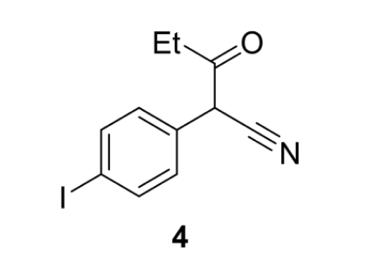
Figure 8: 2-(4-iodophenyl)-3-oxopentanenitrile (4)
4-iodophenylacetonitrile (10.00 g, 0.046 mol, 1 equiv.), ethyl propionate (5.00 g, 0.049 mol, 1.05 equiv.) and potassium tert-butoxide (10.47 g, 0.093 mol, 2 equiv.) were combined in THF (100 mL) at room temperature, with stirring in a round bottom flask. The reaction mixture turned to a dark red and heated up rapidly. The reaction was sealed and stirred for 2 hours.
The reaction mixture was worked up by the addition of 1.0 M HCl (100 mL) to the reaction vessel. The acidified reaction mixture was transferred to a separating funnel and the aqueous layer was extracted with DCM (3 x 65 mL). The combined organic layer was washed with brine (100mL), dried with anhydrous sodium sulfate, filtered, and concentrated in vacuo to afford 2-(4-iodophenyl)-3oxopentanenitrile (4) (12.699 g, 0.042 mol, 91%) as a reddish oil. TLC was conducted with 100% DCM as the eluent. The crude 2-(4-iodophenyl)-3oxopentanenitrile (4) was used without purification in the second step of the synthesis.
Step 2: Synthesis of 2-(4-iodophenyl)-3-(s2methylpropoxy)-pent-2-enenitrile
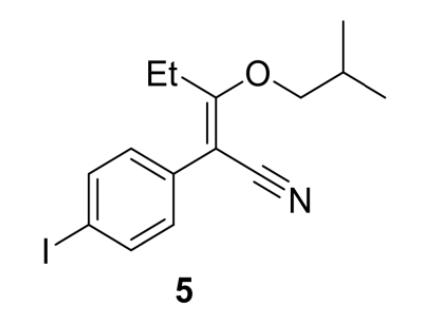
Figure 9: 2-(4-iodophenyl)-3-(s2-methylpropoxy)-pent-2enenitrile (5)
2-(4-iodophenyl)-3-oxopentanenitrile (12.699 g, 0.042 mol, 1 equiv.) was dissolved in a mixture of toluene (65.0 mL) and 2-methylpropan-1-ol (6.50 mL, 0.21 mol). 18M H2SO4 (1.00 mL) was added, and the mixture was refluxed for 10 hours in a Dean Stark apparatus. The reaction mixture was poured onto a saturated sodium hydrogen carbonate in a separating funnel and the aqueous phase was extracted with DCM (2 x 50 mL). The combined organic extracts were dried over anhydrous sodium sulfate.
Addition of 5.0 mL of triethylamine to the reaction mixture converted the unreacted starting material to its very polar triethylammonium enolate salt. Chromatography silica (50.0 g) was added to the organic phase, which was made up to 200mL with dichloromethane and stirred for 1.5 hours. The organic phase was filtered using vacuum filtration and rinsed with 1M HCL (2 x 50 mL) and deionised water (50 mL) in a separating funnel to remove all traces of triethylamine. The combined organic extracts were dried over anhydrous sodium sulfate and filtered. The solvent was removed in vacuo to yield 2-(4-iodophenyl)-3-(2-methylpropoxy)-pent2-enenitrile (5) (11.36 g, 0.032 mol, 75%) as a red oil. This product was used in the next step of synthesis.
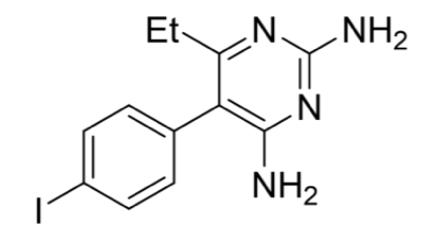
Figure 10: 4-iodopyrimethamine analogue (2)
Step 3: Synthesis of 4-iodopyrimethamine analogue
2-(iodophenyl)-3-(2-methylpropoxy)-pent-2enenitrile (11.36 g, 0.0320 mol, 1 equiv.) was dissolved in DMSO (90.0 mL). Guanidine hydrochloride (6.40 g, 0.0640 mol, 2 equiv.) was stirred into the solution followed by sodium methoxide powder (4.00 g, 0.0704 mol, 2.2 equiv.). The solution became dark red in colour on addition of the sodium methoxide, which dissolved into the solution within an hour. No precipitation of sodium chloride was observed. The solution was allowed to stand at room temperature for 48 hours. Crystals appeared in the reaction mixture. These were filtered and isolated for analysis. The remaining reaction mixture was poured onto water and extracted with DCM. 100% ethanol was added to the organic layerand left for 48 hours.No additional crystallisation occurred.
Results
Step 1: Synthesis of 2-(4-iodophenyl-3oxopentanetitrile (4)
.
Figure 11: 1H NMR spectra of crude product 4
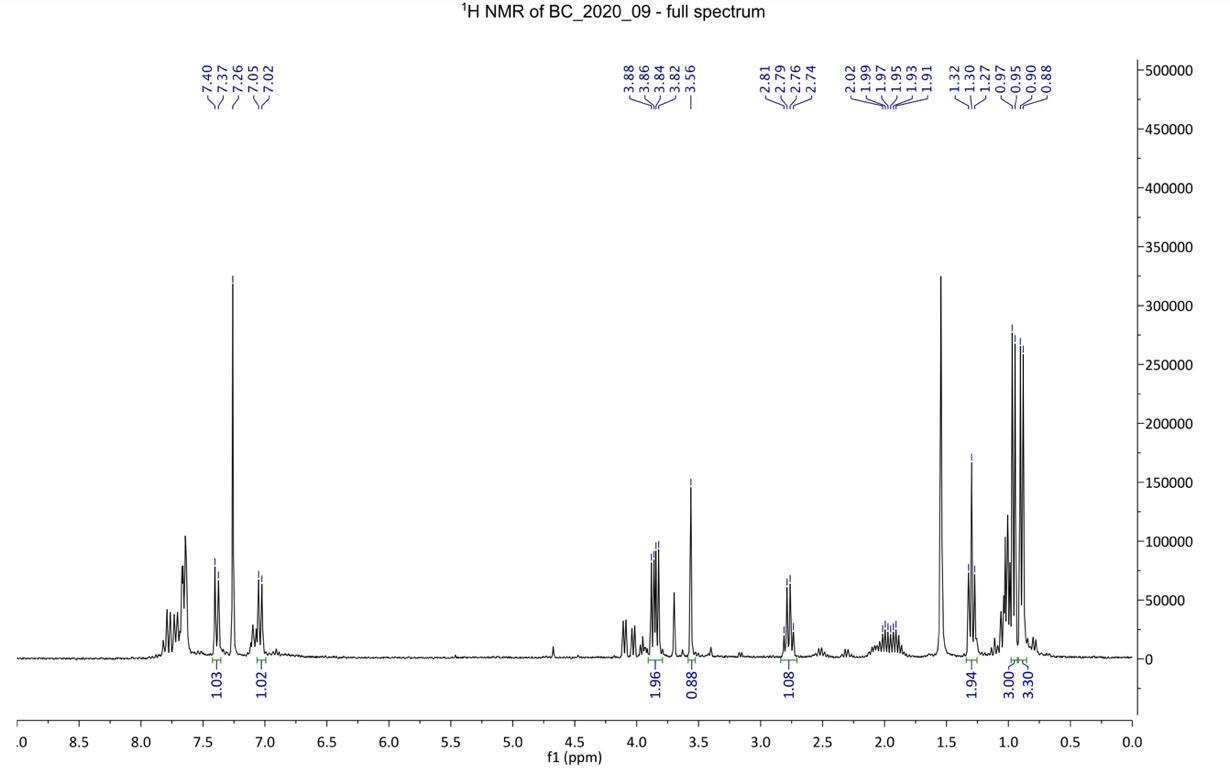
1H NMR (500 MHz, chloroform-d): δ 7.77-7.73 (2H, d, H1), 7.12-7.07 (2H, d, H2), 4.61 (1H, s, H3), 2.69-2.64 (2H, m, CH2), 1.06 (3H, t, CH3)

Figure 12: TLC of step 1 after reaction was stirred for 2hrs Figure 13: Previous report’s TLC of step 1

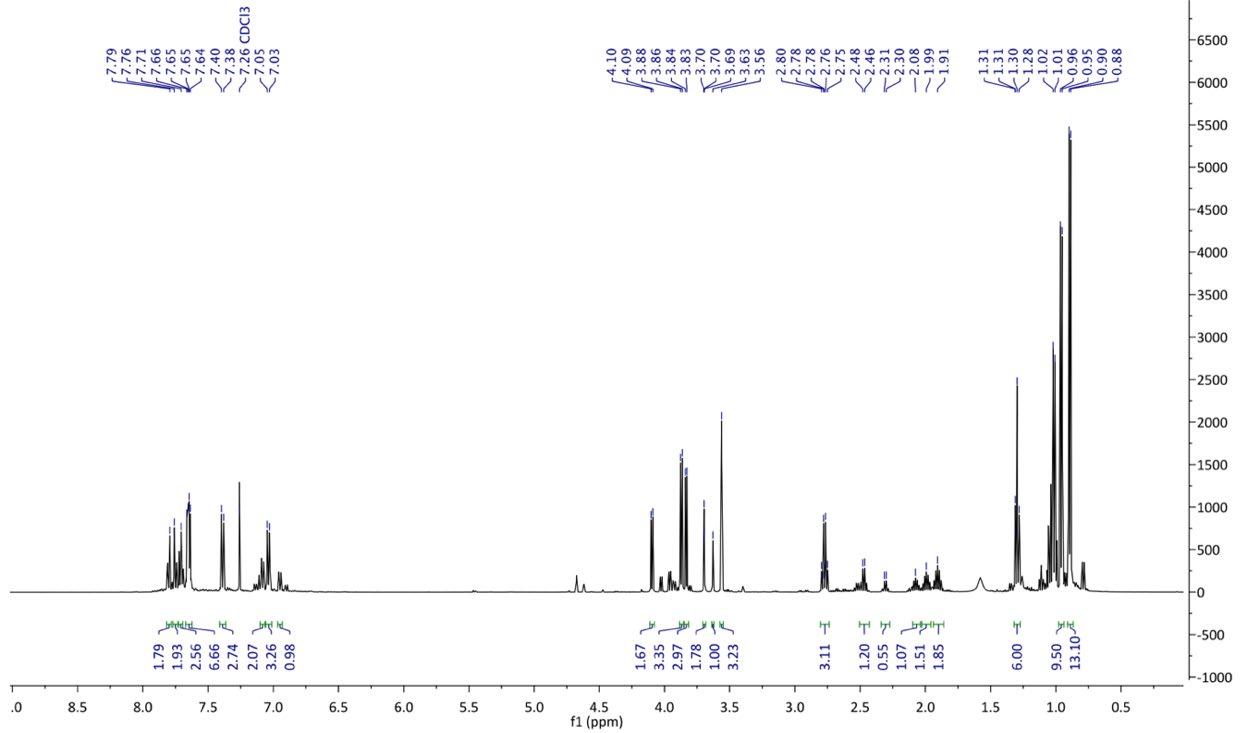
Figure 14: 1H NMR of crude product 5.
1H NMR (500 MHz, chloroform-d): δ 7.70 (2H, d, H1), 7.21 (2H, d, H2), 3.85 (2H, d, CH2), 2.78 (2H, q, CH2), 2.02-1.91 (1H, m, CH), 1.30 (3H, t, CH3), 0.93 (6H, d, CH3)
Step 2: Synthesis of 2-(4-iodophenyl)-3-(2-methylpropoxy)-pent-2-enenitrile (5)
Figure 13: Previous report’s TLC of step 1
Figure 15: Previous report’s 1H NMR experiment after step 2. Hydrogen environments cannot be assigned due to complexity

Figure 16: TLC after work up in step 2

Figure 17: Previous synthesis’ TLC after work up in step 2
Discussion
Step 1: Synthesis of 2-(4-iodophenyl)-3oxopentanenitrile (4)

Figure 18: Reaction of 4-iodophenylacetonitrile (3) to form 2(4-iodophenyl)-3-oxopentanenitrile (4)
Step 1 of the synthesis was performed to afford Compound 4 from a condensation reaction between 4-iodoacetonitrile (3) and ethyl propionate. More specifically, addition of the strong base potassium tert-butoxide caused deprotonation at the CH2 group of Compound 3, and reaction with ethyl propionate led to the elimination of ethanol (Figure 19). The yield obtained for this step was 91% which is significant increase from the previous report’s 65% yield and is likely a result of optimising the reaction (Wong, 2020). By performing a scaled-up synthesis with 11.33g of starting material, a greater amount than the 7g used previously, loss of the target compound 4 was minimised during extraction and separation from aqueous impurities.
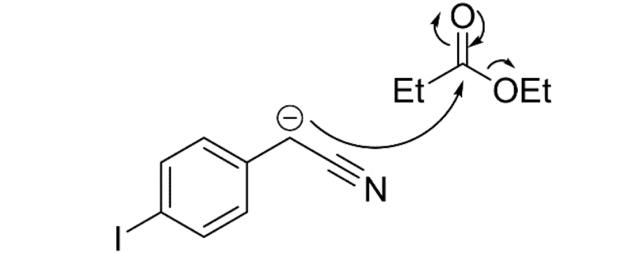
Figure 19: Mechanism of the reaction between 4iodophenylacetonitrile and ethyl propionate
Additionally, filtration of compound 4 from sodium sulfate after drying was optimised by using a sintered funnel rather than filter paper, as was used previously. These considerations attributed to a yield percentage increase of 40%. The appearance of polar baseline material on the TLC (Figure 12) corresponded to the formation of the more polar Compound 4. In comparison to the TLC plate from the previous report, this TLC is cleaner (Figure 12), indicating that the compound has a higher purity than the synthesis prior (Figure 13). The 1HNMR spectrum is highly complex, likely due to the combination of enol and keto tautomers in equilibrium (Figure 20). The doublet signal between 7.77-7.73 ppm and 7.12-7.07 ppm were assigned to the H1 and H2 aromatic protons, respectively. This was expected since hydrogen atoms on a benzene ring characteristically appear in the 7-8 ppm region. The H3 singlet was assigned to the signal further upfield at 4.61 ppm, due to the electron withdrawing nature of the nitrile group and carbon-oxygen double bond causing a deshielding of the hydrogen atom in this environment. The quartet signal between 2.69-2.64 ppm was assigned to the CH2 on the ethyl group. This is characteristically a quartet but due to possible leftover impurities or the appearance of enol tautomer, the spectra has become more complicated. The distinctive triplet splitting pattern
at 1.06 ppm indicated the presence of the CH3 on the ethyl group.

Figure 20: Mechanism of reaction between enol and keto tautomers of compound 4 in equilibrium
The information provided by the 1HNMR was able to confirm the formation of Compound 4. Although comparisons cannot be made with the 1HNMR analysis of the previous compound since it was conducted with a different solvent (acetone-d6, (CD3)2CO), there was enough evidence to conclude that the optimisation of step 1 was achieved. Future trials can focus on obtaining a cleaner sample by undertaking further purification of the crude product, such that complete characterisation of this compound can be undertaken.
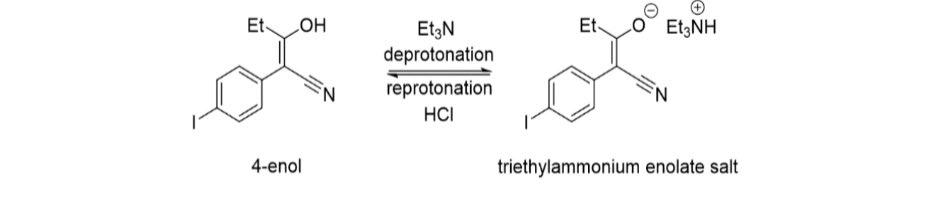
Figure 22: Mechanism of the reaction between 4iodophenylacetonitrile and ethyl propionate
Step 2: Synthesis of 2-(4-iodophenyl)-3-(2methylpropoxy)-pent-2-enenitrile (5)
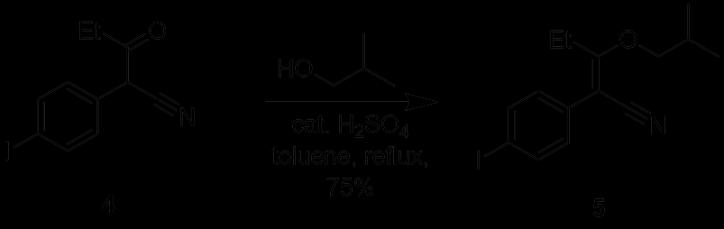
Figure 21: Reaction of 2-(4-iodophenyl)-3-oxopentanenitrile (4) to form 2-(4-iodophenyl)-3-(2-methylpropoxy)-pent-2enenitrile (5)
Step 2 of the synthesis was performed under reflux whereby a substitution reaction led to the formation of Compound 5 from Compound 4. A Dean Stark apparatus was used to remove water and drive the equilibrium reaction in the forward direction. To remove the remaining starting material, triethylamine was added to convert it into triethylammonium enolate salt. Similar to the previous synthesis, the addition of silica gel at this stage facilitated the removal of the salt from the reaction mixture. However, after the work up, the previous TLC (Figure 17) indicated that there were still remnants of the starting compound leftover, most likely due to inefficient separation of the silica gel via decanting, which reappeared in the separating funnel. The re-protonation of the triethylammonium enolate salt stuck to the silica gel occurred after addition of HCl, reforming the enol tautomer of Compound 4 (Figure 22). To optimise this step, the sintered funnel was again incorporated as an alternative to separate the silica gel from the crude product mixture, and the resultant purity was evident in the TLC which showed that the starting material reaction spot was less apparent than the previous attempt. (Figure 16). Furthermore, the 1HNMR spectra of this compound appeared to be of a higher purity than the previous synthesis. Thus, by optimising the reaction, a yield of 75% was obtained from this step, which is a 32% increase from the previous yield of 57%.
In the previous synthesis, impurities in the 1H NMR made the spectrum difficult to analyse and as a result, 13C NMR was used to verify formation of product. However, the 1H NMR for this optimised procedure was able to confirm the successful synthesis of Compound 5. The doublet signal at 7.70-7.21 ppm were assigned to the aromatic H1 and H2 hydrogen environments which were previously seen in step 1. The presence of two new doublet signals further downfield at 3.85 ppm were assigned to the H4 hydrogen protons due to their position next to the highly electronegative oxygen atom which can cause a deshielding of the hydrogen in that environment. Compound 5 can also be confirmed by the appearance of the new multiplet signal from 2.02-1.91 ppm which was assigned to the H5 hydrogen environment. This is because it has many neighbouring protons which can cause the signal to split multiple times. The characteristic quartet splitting pattern at 2.78 ppm corresponded to CH2 group from the ethyl group. Lastly, the distinctive triplet splitting pattern at 1.30 ppm and the doublet splitting pattern at 0.92 indicate the presence of three CH3 groups, thus confirming the successful synthesis of Compound 5.
Step 3: Synthesis of 4-iodopyrimethamine analogue (2)
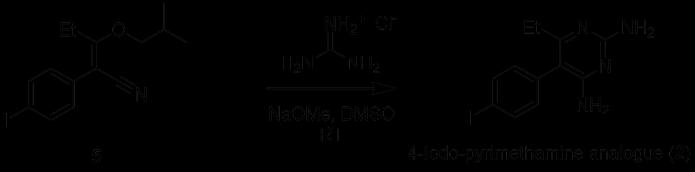
Figure 23: Reaction of 2-(4-iodophenyl)-3-(2-methylpropoxy)pent-2-enenitrile (5) to form 4-iodopyrimethamine (2)
Step 3 of synthesis was initiated by the deprotonation of the guanidine ion by sodium methoxide. Following this, guanidine reacted at the nitrile carbon of Compound 5, and subsequent electron rearrangement and the elimination of 2methylpropanol afforded the pyrimidine ring seen in Compound 2. Unfortunately, the optimisation of step 3 was not effective due to time constraints which only permitted one experimental attempt at isolation and purification. Although there was a higher amount of solid material obtained, 1H NMR results were not conclusive due to the problems encountered with dissolving the compound in chloroform-d. Given that the previous synthesis performed the 1H NMR experiment in this solvent, the compound crystallised in this step may not have been the desired 4-iodopyrimethamine analogue. Another possibility could be that the compound contained too many impurities, making it insoluble. Instead, MeOD solvent was used to run the experiment, but no signals were observed from the 1H NMR spectra. Thus, the identification of the solid is still awaiting structural confirmation. Although different solvents were used in combination to crystallise the product, like the previous synthesis, the same difficulties were encountered during the third stage of the synthesis which could potentially be a result of the 4-iodopyrimethamine analogue (2) having a higher degree of solubility compared to the original pyrimethamine compound (1). Due to these issues, the pyrimethamine analogue will not be able to undergo biological testing, but future efforts could focus on finding a suitable solvent to precipitate out the desired compound such that a sufficient amount can be produced. Overall, the optimisation of the first two steps of the synthetic pathway for 4-iodopyrimethamine (2) was successful. Future considerations such as using column chromatography for further purification at the end of each step could be carried out such that a complete characterisation and analysis of 4iodopyrimethamine can be conducted.
Future Research
Future possibilities of the 4-iodopyrimethamine analogue include applications in the preparation of a diverse set of other analogues via substitution at the R4 position. Previously, Suzuki coupling reactions have been able to successfully effect the cross-coupling of pyrimidines, allowing the construction of carbon-carbon bonds between functionalised substrates that were previously inaccessible or required multi-step procedures (Biajoli et al., 2014). In one pilot experiment, a successful reaction scheme utilising a Sandmeyer reaction afforded an iodo-pyrimethamine analogue that was then cross-coupled with 4methoxybenzeneboronic acid to furnish a substituted biphenyl at the R3 position of the analogue (Richardson & Stevens 2002) (Figure 24).
For the 4-iodopyrimethamine analogue, although the iodine is in the R4 rather than R3 position (Figure 24), the highly reactive electrophile can still act as an effective additive for accelerating these couplings with satisfactory results (Figure 25) (Dobrounig, Trobe & Breinbauer 2017). Thus, even if the potency and inhibitory activity of the 4iodopyrimethamine analogue isn’t particularly effective, given that the optimised synthetic pathway is much simpler than current analogues reported in the literature, it can function as a precursor for the future synthesis of more complex structures and molecules that can effectively inhibit the DHFR enzyme substrate.
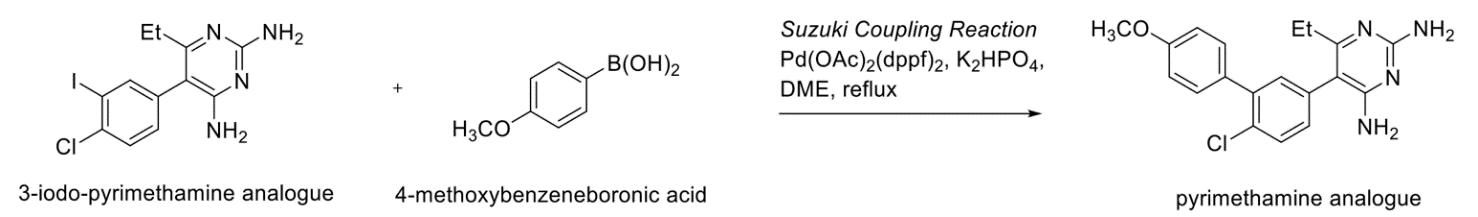
Figure 25: Proposed possible future applications of 4-iodopyrimethamine analogue via. Suzuki-Miyaura pathway
Conclusion
This report details the successful optimisation of the first two steps of a synthetic pathway developed previously for the synthesis of the 4iodopyrimethamine analogue (2). The results of the optimisation of step three cannot be confirmed due to difficulties encountered with dissolving the solid obtained at the conclusion of step 3, hence the product is still waiting for 1H NMR analysis so that structural confirmation can be made.
In this experiment, thin layer chromatography (TLC) was carried out to gauge the progress of each reaction, and structural conformation was undertaken by using 1H NMR data to confirm that the desired product was produced. Further comparisons with the previous synthesis’ TLC and 1H NMR spectra allowed for the efficacy of the optimisation to be confirmed. Significant quantities have been made, and thus, future research can focus on the purification of each reaction product to obtain full characterisation. Additionally, after successful optimisation of the final step of the synthesis, cross-coupling reactions can be investigated for their potential as novel synthetic pathways towards the synthesis of new pyrimethamine analogues in the future.
Acknowledgements
I wish to thank Dr Katie Terrett for her guidance throughout the entire project. In particular, her extensive knowledge and expert advice, supervision of the synthesis and for overseeing the completion of this report proved to be invaluable. I would also like to acknowledge the collaborators of Breaking Good who assisted with providing the 1H NMR experimental data which was greatly appreciated.
References
Adane, L. & Bharatam, P.V. 2008, ‘Modelling and informatics in the analysis of P. falciparum DHFR enzyme inhibitors.’, Current medicinal chemistry.
Biajoli, A.F.P., Schwalm, C.S., Limberger, J., Claudino, T.S. & Monteiro, A.L. 2014, ‘Recent Progress in the Use of PdCatalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds’, Journal of the Brazilian Chemical Society, viewed 20 February 2021, <http://www.gnresearch.org/doi/10.5935/01035053.20140255>.
Bloland, P.B. 2001, ‘Drug resistance in malaria’, World Health Organisation, p. 32.
Cirioni, O., Giacometti, A., Barchiesi, F. & Scalise, G. 2000, ‘Inhibition of growth of Pneumocystis carinii by lactoferrins alone and in combination with pyrimethamine, clarithromycin and minocycline’, Journal of Antimicrobial Chemotherapy, vol. 46, no. 4, pp. 577–82.
Cotter, C., Sturrock, H. & Hsiang, M. 2013, ‘The changing epidemiology of malaria elimination: new strategies for new challenges (vol 382, pg 900, 2013)’, The Lancet, vol. 382, pp. 858–858.
Dhiman, S. 2019, ‘Are malaria elimination efforts on right track? An analysis of gains achieved and challenges ahead’, Infectious Diseases of Poverty, vol. 8, no. 1, p. 14.
Dobrounig, P., Trobe, M. & Breinbauer, R. 2017, ‘Sequential and iterative Pd-catalyzed cross-coupling reactions in organic synthesis’, Monatshefte Fur Chemie, vol. 148, no. 1, pp. 3–35.
Lozovsky, E.R., Chookajorn, T., Brown, K.M., Imwong, M., Shaw, P.J., Kamchonwongpaisan, S., Neafsey, D.E., Weinreich, D.M. & Hartl, D.L. 2009, ‘Stepwise acquisition of pyrimethamine resistance in the malaria parasite’, Proceedings of the National Academy of Sciences, vol. 106, no. 29, pp. 12025–30.
Mao, J., Hua, Q., Xie, G., Guo, J., Yao, Z., Shi, D. & Ji, S. 2009, ‘Iodine-Catalyzed Suzuki-Miyaura Coupling Performed in Air’, Advanced Synthesis & Catalysis, vol. 351, pp. 635–41.
Murphy, S.C. 2006, ‘Malaria and Global Infectious Diseases: Why Should We Care?’, AMA Journal of Ethics, vol. 8, no. 4, pp. 245–50.
Nattee, C., Khamsemanan, N., Lawtrakul, L., Toochinda, P. & Hannongbua, S. 2017, ‘A novel prediction approach for antimalarial activities of Trimethoprim, Pyrimethamine, and Cycloguanil analogues using extremely randomized trees’, Journal of Molecular Graphics & Modelling, vol. 71, pp. 13–27.
Peterson, D.S., Walliker, D. & Wellems, T.E. 1988, ‘Evidence that a point mutation in dihydrofolate reductase-thymidylate
synthase confers resistance to pyrimethamine in falciparum malaria’, Proceedings of the National Academy of Sciences, vol. 85, no. 23, pp. 9114–8.
Richardson, M.L. & Stevens, M.F.G. 2002, ‘Structural Studies on Bioactive Compounds. Part 37.1 Suzuki Coupling of Diaminopyrimidines: A New Synthesis of the Antimalarial Drug Pyrimethamine’, Journal of Chemical Research, vol. 2002, no. 10, pp. 482–4.
Sardarian, A., Douglas, K.T., Read, M., Sims, P.F.G., Hyde, J.E., Chitnumsub, P., Sirawaraporn, R. & Sirawaraporn, W. 2003, ‘Pyrimethamine analogs as strong inhibitors of double and quadruple mutants of dihydrofolate reductase in human malaria parasites’, Organic & Biomolecular Chemistry, vol. 1, no. 6, pp. 960–4.
Sirawaraporn, W., Sathitkul, T., Sirawaraporn, R., Yuthavong, Y. & Santi, D.V. 1997, ‘Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase’, Proceedings of the National Academy of Sciences, vol. 94, no. 4, pp. 1124–9.
Sydney Grammar School Synthesis 2016, ‘Daraprim Synthesis’, Pyrimethamine Synthesis: Status at the end of 2016, viewed 20 February 2021, <https://malaria.ourexperiment.org/daraprim_synthesis>.
Tarnchompoo, B., Chitnumsub, P., Jaruwat, A., Shaw, P.J., Vanichtanankul, J., Poen, S., Rattanajak, R., Wongsombat, C., Tonsomboon, A., Decharuangsilp, S., Anukunwithaya, T., Arwon, U., Kamchonwongpaisan, S. & Yuthavong, Y. 2018, ‘Hybrid Inhibitors of Malarial Dihydrofolate Reductase with Dual Binding Modes That Can Forestall Resistance’, ACS Medicinal Chemistry Letters, vol. 9, no. 12, pp. 1235–40.
Tse, E.G., Korsik, M. & Todd, M.H. 2019, ‘The past, present and future of anti-malarial medicines’, Malaria Journal, vol. 18, no. 1, p. 93.
Warhurst, D.C. 2002, ‘Resistance to Antifolates in Plasmodium Falciparum, the Causative Agent of Tropical Malaria’, Science Progress, vol. 85, no. 1, pp. 89–111.
Weiss, D.J., Lucas, T.C.D., Nguyen, M., Nandi, A.K., Bisanzio, D., Battle, K.E., Cameron, E., Twohig, K.A., Pfeffer, D.A., Rozier, J.A., Gibson, H.S., Rao, P.C., Casey, D., Bertozzi-Villa, A., Collins, E.L., Dalrymple, U., Gray, N., Harris, J.R., Howes, R.E., Kang, S.Y., Keddie, S.H., May, D., Rumisha, S., Thorn, M.P., Barber, R., Fullman, N., Huynh, C.K., Kulikoff, X., Kutz, M.J., Lopez, A.D., Mokdad, A.H., Naghavi, M., Nguyen, G., Shackelford, K.A., Vos, T., Wang, H., Smith, D.L., Lim, S.S., Murray, C.J.L., Bhatt, S., Hay, S.I. & Gething, P.W. 2019, ‘Mapping the global prevalence, incidence, and mortality of Plasmodium falciparum, 2000–17: a spatial and temporal modelling study’, The Lancet, vol. 394, no. 10195, pp. 322–31.
Wernsdorfer, W.H. 1994, ‘Epidemiology of drug resistance in malaria’, Acta Tropica, vol. 56, no. 2, pp. 143–56.
White, N.J. 2004, ‘Antimalarial drug resistance’, Journal of Clinical Investigation, vol. 113, no. 8, pp. 1084–92.
WHO 2020, World Malaria Report 2020, viewed 20 February 2021, <https://www.who.int/publications/i/item/9789240015791>. Wong, K. 2020, ‘Synthesis of a pyrimethamine analogue’, Barker College Science Extension Journal, vol. 2, no. Number 1, pp. 47–60.
Yuthavong, Y., Tarnchompoo, B., Vilaivan, T., Chitnumsub, P., Kamchonwongpaisan, S., Charman, S.A., McLennan, D.N., White, K.L., Vivas, L., Bongard, E., Thongphanchang, C., Taweechai, S., Vanichtanankul, J., Rattanajak, R., Arwon, U., Fantauzzi, P., Yuvaniyama, J., Charman, W.N. & Matthews, D. 2012, ‘Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target’, Proceedings of the National Academy of Sciences, vol. 109, no. 42, pp. 16823–8.
Yuthavong, Y., Yuvaniyama, J., Chitnumsub, P., Vanichtanankul, J., Chusacultanachai, S., Tarnchompoo, B., Vilaivan, T. & Kamchonwongpaisan, S. 2005, ‘Malarial (Plasmodium falciparum) dihydrofolate reductase-thymidylate synthase: structural basis for antifolate resistance and development of effective inhibitors’, Parasitology, vol. 130, no. Pt 3, pp. 249–59.







