
Τόµος 86 | Τεύχος 2 Μάιος | Ιούνιος | Ιούλιος | Αύγουστος 2023 Τ ετ ρ αµ η ν ι α ί ο επ ι στ η µο νικό πε ρ ι ο δ ι κό της Ελλ ηνι κ ή ς Παι δ ιατρ ι κ ή ς Ετα ι ρε ί α ς Volume 86 | Number 2 May | June | July | August 2023 Fo ur mo n t hl y scientific jour n al of th e Gr eek Pae d iat r ic Socie t y
Προσοχή για πρώιμα προειδοποιητικά συμπτώματα της νωτιαίας μυϊκής ατροφίας (SMA)1–3
Η πρώιμη διάγνωση της νωτιαίας μυϊκής ατροφίας είναι ζωτικής σημασίας επειδή η βλάβη που προκαλείται πριν από τη θεραπεία είναι μη αναστρέψιμη4,5. Ελέγξτε για τα παρακάτω συμπτώματα σε παιδιά ηλικίας έως και 3 μηνών1–3 ,6,7
4,8 Βιβλιογραφία: 1. Kolb SJ and Kissel JT. Neurol Clin. 2015;33(4):831–46. 2. Prior TW, et al. NCBI Bookshelf 2019. Available at https://www.ncbi.nlm. nih.gov/books/NBK1352. Date accessed: Δεκέμβριος 2022. 3. Wang CH, et al. J Child Neurol. 2007;22(8):1027–49. 4. Govoni A, et al. Mol Neurobiol. 2018;55(8):6307–18. 5. Stifani N. Front Cell Neurosci. 2014;8:293. doi: 10.3389/fncel.2014.00293. 6. Pera MC, et al. PLoS One. 2020;15(3):e0230677. 7. Lin CW, et al. Pediatr Neurol. 2015;53(4):293–300. 8. Mercuri E, et al. Neuromuscul Disord. 2018;28(2):103–115.


ΔΥΣΚΟΛΊΑ ΣΤΉΝ ΑΝΑΠΝΟΉ/ ΔΥΣΚΑΤΑΠΟΣΊΑ (ΔΥΣΚΟΛΊΑ ΣΤΉΝ ΚΑΤΑΠΟΣΉ)

ΣΥΜΠΤΩΜΑΤΑ ΤΗΣ
Απευθυνθείτε επειγόντως
GR2305307266 Αυτές
του κοινού
καμία περίπτωση δεν μπορούν να αντικαταστήσουν τη συμβουλή γιατρού ή άλλου αρμοδίου επαγγελματία υγείας. ΥΠΟΤΟΝΊΑ ΚΕΦΑΛΉΣ ΊΝΊΔΊΣΜΟΣ ΤΉΣ ΓΛΏΣΣΑΣ ΥΠΟΤΟΝΊΑ ΑΠΏΛΕΊΑ ΑΝΤΑΝΑΚΛΑΣΤΊΚΏΝ
σε παιδονευρολόγο εάν παρατηρήσετε τα συμπτώματα
οι πληροφορίες προορίζονται για γενική πληροφόρηση και ενημέρωση
και σε
071
Τόμος 86 | Τεύχος 2 | Μάιος - Ιούνιος - Ιούλιος - Αύγουστος 2023
Τετραμηνιαία έκδοση της Eλληνικής Παιδιατρικής Eταιρείας
Περιεχόμενα
Στέλιος Αντωνιάδης
60ο Πανελλήνιο Παιδιατρικό Συνέδριο
Στρογγυλό Τραπέζι με θέμα «ΠΑΛΙΑ ΚΑΙ ΝΕΑ ΑΥΤΟΦΛΕΓΜΟΝΩΔΗ ΝΟΣΗΜΑΤΑ, ΠΟΥ ΠΡΕΠΕΙ ΝΑ ΓΝΩΡΙΖΕΙ ΚΑΙ ΝΑ ΑΝΑΓΝΩΡΙΖΕΙ Ο ΚΛΙΝΙΚΟΣ ΠΑΙΔΙΑΤΡΟΣ »
Συντονισμός-Προεδρείο: Κανακούδη-Τσακαλίδου Φλωρεντία, Πρατσίδου-Γκέρτση Πολυξένη
Ομιλητές εισηγήσεων: Σγουροπούλου Βασιλική, Καρανάνου Παναγιώτα, Πρατσίδου-Γκέρτση
Πολυξένη, Κουτσονικολή Άρτεμις
110
Κανακούδη-Τσακαλίδου Φλωρεντία
Εισαγωγή στα Αυτοφλεγμονώδη νοσήματα
120
Σγουροπούλου Βασιλική
Οικογενής μεσογειακός πυρετός, σύγχρονη αντιμετώπιση / Σύνδρομο PFAPA με μερική
επικάλυψη Οικογενούς Μεσογειακού Πυρετού: αλλάζει η θεραπεία;
130
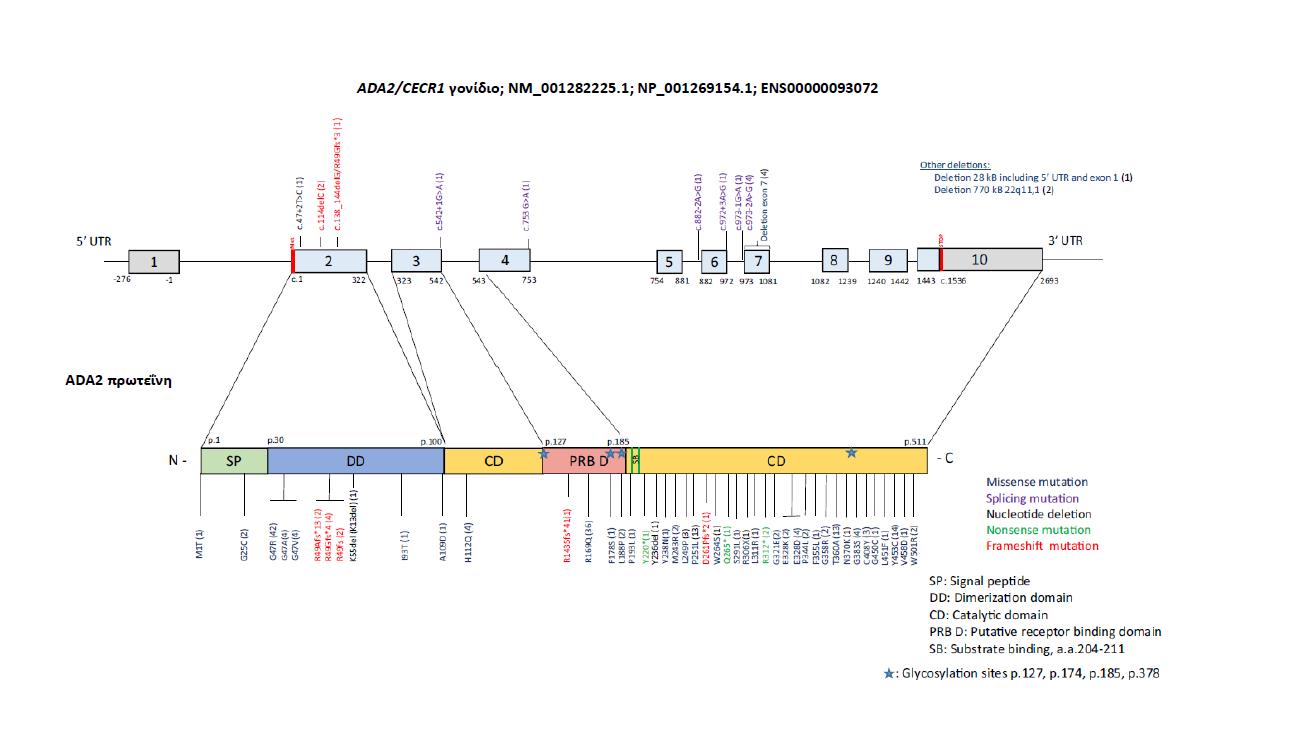
Καρανάνου Παναγιώτα Απλοανεπάρκεια της πρωτεΐνης Α20 και Ανεπάρκεια της απαμινάσης της αδενοσίνης 2 (DADA2): Ανοσοανεπάρκειες ή Αυτοφλεγμονώδη νοσήματα;
144
Πρατσίδου-Γκέρτση Πολυξένη
Εισαγωγή στις Ιντερφερονοπάθειες. Σύνδρομα AICARDI-GOUTIERES και CANDLE
Πρόεδρος Α. Κωνσταντόπουλος
Συντακτική επιτροπή Διευθυντής Σ. Αντωνιάδης
Mέλη Σ. Ανδρονίκου Ε. Γαλανάκης Α. Ευαγγελίου Λ. Θωμαΐδου Μ. Κανάριου Α. Καπόγιαννης Σ. Kίτσιου-Τζέλη Ε. Μανταδάκης Π. Παναγιωτοπούλου-Γαρταγάνη Α. Παπαδοπούλου Β. Παπαευαγγέλου Α. Παπαθανασίου Α. Σιαμοπούλου-Μαυρίδου Α. Συρίγου-Παπαβασιλείου
072
108 ΕΠΙΣΤΟΛΗ ΑΠΟ ΤΟN ΔΙΕΥΘΥΝΤΗ ΣΥΝΤΑΞΗΣ
Υποβολή εργασιών e-mail: grammateia@e-child.gr
Οδηγίες προς τους συγγραφείς: http://e-child.gr/publications/ instructions-to-authors
Iδιοκτήτης
Eλληνική Παιδιατρική Eταιρεία
Μπακοπούλου 15,15451, Ν.
Ψυχικό
Tηλ.: 2107771140 e-mail: grammateia@e-child.gr
Eτήσια συνδρομή: €40
Eιδικευόμενοι, φοιτητές: €20
152
Κουτσονικολή Άρτεμις Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονο-ΣΕΛ) και Σύνδρομο SAVI (STINGAssociated Vasculopathy with onset in Infancy)
160
Κανακούδη-Τσακαλίδου Φλωρεντία
Επίλογος
174
ΣΥΝΑΔΕΛΦΙΚΑ
Στέλιος Αντωνιάδης
176
ΚΡΙΤΙΚΗ ΒΙΒΛΙΟΥ
Στέλιος Αντωνιάδης
180
ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ
ΣΥΓΓΡΑΦΕΙΣ
Volume 86 | Number 2 | May - June - July - August 2023
Four monthly publication of the Greek Paediatric Society
Contents
108 EDITORIAL
Stelios Antoniadis
60th Panhellenic Pediatric Congress
Round Table: OLD AND NEW AUTOINFLAMMATORY DISEASES WHICH A GENERAL PEDIATRICIAN SHOULD BE AWARE OF AND RECOGNISE
Moderators/ Chairpersons: Kanakoudi-Tsakalidou Florence, Pratsidou-Gertsi Polyxeni
Speakers: Sgouropoulou Vasiliki, Karananou Panagiota, Pratsidou-Gertsi Polyxeni, Koutsonikoli Artemis
110
Kanakoudi-Tsakalidou Florence Introduction to Autoinflammatory diseases
120
Sgouropoulou Vasiliki
Familial Mediterranean Fever and PFAPA Syndrome: Contemporary Disease management and treatment when partial overlap is present
130
Karananou Panagiota
Haploinsufficiency of A20 protein (HA20) and Deficiency of adenosine deaminase 2 (ADA2) enzyme (DADA2): Immunodeficiencies or Autoinflammatory diseases?
144
Pratsidou-Gertsi Polyxeni
Introduction to Interferonopathies. Aicardi Goutières and CANDLE syndromes: new members in the genetically determined Autoinflammatory diseases family
President
A. Constantopoulos
Editorial board
Editor- in- Chief
S. Antoniadis
Members
S. Andronikou
E. Galanakis
A. Evangeliou
L. Thomaidou
M. Kanariou
A. Kapogiannis
S. Kitsiou-Tzeli
E. Mantadakis
P. Panagiotopoulou-Gartagani
A. Papadopoulou
V. Papaevagelou
A. Papathanassiou
A. Siamopoulou-Mavridou
A. Syrigou-Papavasiliou
074
Manuscript submission
e-mail: grammateia@e-child.gr
Instructions to authors: http://e-child.gr/publications/ instructions-to-authors
Owner Greek Paediatric Society
15, Mpakopoulou st. GR - 15451, Ν. Psychiko Tel.: +302107771140
e-mail: grammateia@e-child.gr
Annual subscription
All foreign countries: US$50
152
Koutsonikoli Artemis Monogenic Systemic Lupus Erythematosus (mono-SLE) and SAVI syndrome (STING-Associated Vasculopathy with onset in Infancy)
160
Kanakoudi-Tsakalidou Florence Conclusions
174 BETWEEN COLLEAGUES Stelios Antoniadis
176 BOOK PRESENTATION Stelios Antoniadis
180 INSTRUCTIONS TO AUTHORS
ΕΠΙΣΤΟΛΗ ΑΠΟ ΤΟΝ ΔΙΕΥΘΥΝΤΗ
ΣΥΝΤΑΞΗΣ Α
γαπητοί συνάδελφοι εκλεκτοί φίλοι
Από ότι φαίνεται αργά και σταθερά η πανδημία βαδίζει προς το ιστορικό της τέλος το οποίο θα
την κατατάξει δίπλα σε αυτές που ταλαιπώρησαν την ανθρωπότητα στη διάρκεια του εικοστού
αιώνα όπως η Ισπανική Γρίπη (1918), η Ασιατική Γρίπη (1957), η Γρίπη του Hong-Kong (1968).
Οι νεκροί από την πρώτη υπολογίζονται σε περίπου 40 εκατομμύρια, από τη δεύτερη σε 2
και από την τρίτη σε 1. Και δεν πρέπει να ξεχνάμε πως ο πληθυσμός της γης ήταν τότε πολύ
μικρότερος του σημερινού.
Ένα άλλο ευχάριστο νέο αποτελεί η μεγάλη επιτυχία του τελευταίου Πανελλήνιου Παιδιατρικού
Συνεδρίου με πολύ αυξημένο αριθμό συνέδρων οι οποίοι το παρακολούθησαν συμμετέχοντας
στις περισσότερες συνεδρίες. Στην επιτυχία των Συνεδρίων όπως είναι πολύ καλά γνωστό
πάντα συμβάλλουν παράγοντες όπως η άρτια οργάνωση, η προσεκτική επιλογή των θεμάτων
που θα παρουσιαστούν καθώς και αυτοί που θα τα παρουσιάσουν. Ευχόμαστε και του χρόνου
η επιτυχία να είναι μεγαλύτερη.
Στο περασμένο τεύχος είχα επισημάνει αυτό που με απασχολεί και σίγουρα απασχολεί
όλους μας, το θέμα της βίας στα παιδιά εφηβικής αλλά και προεφηβικής ηλικίας. Βία, παραβατικότητα και αξιόποινες πράξεις σε συχνότητα που δεν είχαμε δει μέχρι τώρα.
Αναρωτιέμαι συνεχώς για τα αίτια. Να είναι ο εγκλεισμός που επέβαλε η πανδημία; Να είναι η καθημερινή απίστευτη βία που παρουσιάζουν οι ταινίες που βλέπουμε στην τηλεόραση; Να
υπάρχουν άλλα αίτια; Όπως και να είναι αδιαμφισβήτητα πιστεύω στον πολύ σημαντικό ρόλο
076
που μπορεί να παίξει ο παιδίατρος για αυτό και τονίζω ξανά ότι κάτι πρέπει να κάνουμε μια
και κανείς δεν γνωρίζει το παιδί όσο εμείς.
Το τεύχος αυτό είναι, για πρώτη φορά, μονοθεματικό, ελπίζω να σας αρέσει μια και παρουσιάζεται ένα πολύ ενδιαφέρον θέμα.
Σας εύχομαι ένα καλό, ευχάριστο, ξεκούραστο και χαρούμενο καλοκαίρι
Με μεγάλη όπως πάντα εκτίμηση και συναδελφική αγάπη
Στέλιος Αντωνιάδης
077
Παιδίατρος – Παιδοκαρδιολόγος Καθηγητής – Διευθυντής
Εισαγωγή στα Αυτοφλεγμονώδη Νοσήματα
Φλωρεντία Κανακούδη-Τσακαλίδου
ΕΙΣΑΓΩΓΗ ΣΤΑ ΑΥΤΟΦΛΕΓΜΟΝΩΔΗ ΝΟΣΗΜΑΤΑ
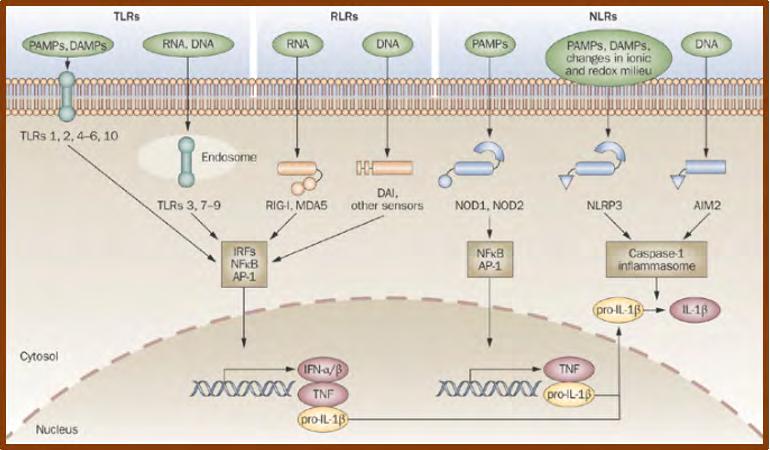
Βασική ιδιότητα του Ανοσιακού Συστήματος είναι η αναγνώριση του «εαυτού» (συστατικού)
από το «ξένο». Το «ξένο» μπορεί να είναι ενδογενέςυλικό, αλλοιωμένο από διάφορες αιτίες
(υπολείμματα καταστραφέντων κυττάρων, ιστών) ή να είναι εξωγενές υλικό, όπως διάφοροι
παθογόνοι μικροοργανισμοί (ιοί, βακτήρια, μύκητες, παράσιτα...). Αυτοί οι ενδογενείς και εξωγενείς παράγοντες που δεν αναγνωρίζονται από το Ανοσιακό Σύστημα ως «εαυτά
συστατικά-αντιγόνα» αποτελούν για τον οργανισμό «σήματα κινδύνου», γνωστά στη διεθνή
βιβλιογραφία ως DAMPs (Danger Associated Molecular Patterns ή Μοριακές δομές που
σχετίζονται με κίνδυνο). Τα σήματα κινδύνου αναγνωρίζονται από τους αντίστοιχους υποδοχείς
/αισθητήρες (sensors) των κυττάρων της φυσικής ανοσίας. Ακολουθεί η ενεργοποίηση
των μηχανισμών εξουδετέρωσης του «ξένου» από το Ανοσιακό Σύστημα (ενδοκυττάρωση,
φαγοκυττάρωση, φλεγμονώδης αντίδραση και επί αποτυχίας, επεξεργασία και παρουσίαση του «ξένου» στα Τ και Β κύτταρα της ειδικής ανοσίας με αποτέλεσμα την πρόκληση ειδικής ανοσιακής απάντησης/απόκρισης). (1)
Οι μηχανισμοί αναγνώρισης και εξουδετέρωσης των «σημάτων κινδύνου» ελέγχονται από
άλλους ρυθμιστικούς μηχανισμούς ώστε να σταματούν έγκαιρα και πριν αποβούν επιβλαβείς
αντί για ωφέλιμοι στον οργανισμό. Όταν υπάρχουν γενετικές ή επιγενετικές διαταραχές στη
λειτουργία αυτών των ρυθμιστικών μηχανισμών, όπως για παράδειγμα στη λειτουργία της
φλεγμονώδους αντίδρασης, το αποτέλεσμα της προσπάθειας να εξουδετερωθεί το «ξένο»
είναι να δημιουργηθεί παρατεταμένη ή υποτροπιάζουσα φλεγμονή με συστηματικές ή/και εντοπισμένες εκδηλώσεις από διάφορα όργανα/συστήματα, κατάσταση που χαρακτηρίζει μια ετερογενή ομάδα νοσημάτων, τα οποία αποκαλούνται «ΑυτοφλεγμονώδηΝοσήματα» (ΑΦΝ). (2,3,4).
Τα ΑΦΝ αναγνωρίστηκαν ως ξεχωριστές κλινικές οντότητες μόλις 20 χρόνια πριν. Πρόκειται για νοσήματα, τα οποία εντάσσονται τόσο στη Ρευματολογία (εξαιτίας του κλινικού τους φαινότυπου) όσο και στην Ανοσολογία (εξαιτίας της αιτιο-παθογένειάς τους). Στα νοσήματα αυτά αυτοπυροδοτείται μια άσηπτη, υποτροπιάζουσα ή παρατεταμένη φλεγμονή, που προσβάλλει σχεδόν όλα τα συστήματα και πολύ συχνά το δέρμα, το οποίο εμφανίζει χαρακτηριστικές αλλοιώσεις. Oι φλεγμονώδεις εκδηλώσεις μπορεί να συνοδεύονται από
υποτροπιάζοντα επεισόδια πυρετού (με ή χωρίς περιοδικότητα), μπορεί όμως και όχι. Εκτός των κλινικών, έχουν και ορισμένα άλλα κοινά χαρακτηριστικά, όπως:
• Η παθογένειά τους συνδέεται με πρωτοπαθείς λειτουργικές διαταραχές του συστήματος της
φυσικής ανοσίας, οι οποίες αποδίδονται κυρίως σε μεταλλάξεις διαφόρων πρωτεϊνών που παίρνουν μέρος στην ενεργοποίηση της φλεγμονώδους αντίδρασης.
• Για τα επεισόδια της φλεγμονής, μπορεί να υπάρχουν ορισμένοι προδιαθεσικοί παράγοντες, που συνεργούν στην εκδήλωσή τους (π.χ. σωματική ή ψυχική καταπόνηση, ιογενείς λοιμώξεις, έμμηνος ρύση στα κορίτσια κ.ά.) κατά κανόνα όμως, δενυπάρχειεμφανήςαιτία. • Για την αυτοπυροδότηση των φλεγμονωδών επεισοδίων ευθύνονται μοριακές διαταραχές στο σύστημα της φυσικής (innate immunity) και όχι της ειδικής ανοσίας (adaptive immunity), όπως συμβαίνει στα αυτοάνοσα νοσήματα. Ως εκ τούτου, τα ΑΦΝ διακρίνονται από τα
αυτοάνοσα διότι απουσιάζουν τα αυτοαντισώματα (χυμική ανοσία) και τα ειδικά αυτοδραστικά
Τ κύτταρα (κυτταρική ανοσία). Ωστόσο, τα όρια δεν είναι πάντα τόσο στεγανά με αποτέλεσμα
ορισμένα νοσήματα να θεωρείται ότι ανήκουν και στις δύο κατηγορίες, όπως π.χ. η ν. του
Αλληλογραφία
Φλωρεντία ΚανακούδηΤσακαλίδη Μ. Μπότσαρη 2, 54643 Θεσσαλονίκη Τ. 2310839291
K. 6972127338
e-mail: flkan@auth.gr
Φλωρεντία ΚανακούδηΤσακαλίδου Α΄Παιδιατρική Κλινική Α.Π.Θ. - Γενικό Νοσοκομείο Θεσσαλονίκης «Ιπποκράτειο»
078
Crohn, το σ. Αδαμαντιάδη-Bechet, η νεανική ιδιοπαθής αρθρίτιδα συστηματικού τύπου έναρξης, η αγκυλοποιητική σπονδυλαρθρίτιδα κ.ά.
Στην πλειονότητα των ΑΦΝ νοσημάτων έχει ταυτοποιηθεί το γενετικό υπόστρωμά τους, που αφορά μεταλλάξεις σε ένα μόνο γονίδιο γι’ αυτό και τα περισσότερα είναι γνωστά
ως μονογονιδιακά ΑΦΝ. Οι γονιδιακές μεταλλάξεις συνεπάγονται: απώλεια /ελαττωμένη
λειτουργία (Loss of function mutations) ή αύξηση λειτουργίας/ών (Gain of function mutations) των φλεγμονογόνων/φλεγμονωδών πρωτεϊνών που κωδικοποιούνται από τα
αντίστοιχα γονίδια. Για τους λόγους αυτούς τα ΑΦΝ μπορεί να προέλθουν από διαφορετικούς
παθογενετικούς μηχανισμούς σε οποιοδήποτε επίπεδο, (5) όπως από:
• Διαταραχές στην παραγωγή κυτταροκινών δια μέσου του φλεγμονοσώματος ή της αυξημένης σηματοδότησης και ενεργοποίησης του πυρηνικού παράγοντα NF-κΒ
• Ενδοκυτταρικό stress από συσσώρευση «ξένου» υλικού
• Διαταραχές στους ρυθμιστικούς παράγοντες μονοπατιών της φλεγμονής
• Διαταραχές στην αναδίπλωση των πεπτιδικών αλυσίδων των πρωτεϊνών καθώς και στη
λειτουργία των ουβικουϊτινών
• Αυξημένη έκφραση γονιδίων IFN/υπερπαραγωγή IFN
• Αυξημένη ενεργοποίηση του συμπληρώματος
Τέλος, υπάρχουν και ΑΦΝ που μπορεί να προέλθουν από πολυγονιδιακές μεταλλάξεις ή
πολυπαραγοντικές διαταραχές. Τα πιο σημαντικά ΑΦΝ, σύμφωνα με τον προέχοντα μηχανισμό που εμπλέκεται στη φλεγμονή, διακρίνονται σε:
• Φλεγμονοσωμοπάθειες ή Σύνδρομα που σχετίζονται με την IL-1β
• Νοσήματα που σχετίζονται με την ενεργοποίηση του NF-κΒ
• Νοσήματα που σχετίζονται με τη σηματοδότηση των φλεγμονογόνων/φλεγμονωδών κυτταροκινών
• Ιντερφερονοπάθειες ή σύνδρομα που σχετίζονται με αυξημένη έκφραση γονιδίων IFN/ υπερπαραγωγή IFN (Εικ. 1)
Εικόνα 1. Ταξινόμηση των πιο γνωστών παιδιατρικών ΑΦΝ σύμφωνα με τον κυρίαρχο
μηχανισμό που επηρεάζει τη φλεγμονώδη αντίδραση. di Donato, et al., Int J Mol Sci 2021; 22: 6360.
079
Από τις κατηγορίες αυτές θα περιγραφούν συνοπτικά παρακάτω μόνο τα ΑΦΝ, που είναι μέχρι τώρα πιο γνωστά στα παιδιά και μπορεί να τα υποψιαστεί ο παιδίατρος επειδή έχουν χαρακτηριστικό φαινότυπο, όπως δείχνουν οι φωτογραφίες που εμπλουτίζουν τα κείμενα των συγγραφέων που συμμετείχαν στο στρογγυλό τραπέζι. Τα πιο συχνά από αυτά είναι οι φλεγμονοσωμοπάθειες ή σύνδρομα που σχετίζονται με την ιντερλευκίνη 1 (IL-1), άμεσα ή έμμεσα. Οι ιντερφερονοπάθειες είναι πιο σπάνιες στην παιδική ηλικία, αλλά ολοένα και περισσότερα νοσήματα της κατηγορίας αυτής αναδύονται συνεχώς.(4) Αντίθετα με τις
φλεγμονοσωμοπάθειες, στα υπόλοιπα ΑΦΝ και ειδικά σε αυτά που προβάλλουν ως γενετικά
μεταβιβαζόμενες αγγειίτιδες, οι κλινικοί φαινότυποι δεν σχετίζονται πάντα με συγκεκριμένες
μεταλλάξεις γι’ αυτό και μπορεί να αλληλο-καλύ¬πτονται, δηλαδή ένας φαινότυπος να μην
είναι χαρακτηριστικός μόνο ενός συγκεκριμένου ΑΦΝ.
Στη διάρκεια της τελευταίας 20ετίας η εντόπιση των μεταλλαγμένων γονιδίων που
συνδέονται με τα ΑΦΝ νοσήματα έχει επιτρέψει, αφενός την καλύτερη κατανόηση του
γενετικού υποστρώματος και των παθογενετικών μηχανισμών και αφετέρου το να ανοίξουν
νέες προοπτικές στις στοχευμένες θεραπείες. Επιπλέον, η ανάπτυξη των τεχνικών της
αλληλούχισης επόμενης γενιάς (NGS) έχει αυξήσει την έρευνα στο πεδίο αυτό, το οποίο
αποκτά όλο και μεγαλύτερο ενδιαφέρον. (6)
Για τους λόγους αυτούς η di Donato και oι συν, σ’ ένα ανασκοπικό άρθρο που δημοσίευσαν
τον Ιούνιο του 2021 (3), αναφέρουν:
«Ο Γενικός Παιδίατρος πρέπει να γνωρίζει τη σπουδαιότητα αυτών των νοσημάτων και να τα αναγνωρίζει σε ασθενείς με χαρακτηριστικά ευρήματα ώστε να συστήνει την περαιτέρω διερεύνησήτουςαπότουςειδικούςρευματολόγουςστανοσήματααυτά».
«General pediatricians need to be aware of the importance of this group of diseases and they should consider autoinflammatory diseases in patients with clinical hallmarks, in ordertoguidefurtherexaminationsandreferthepatientstoaspecialistrheumatologist».
ΒΙΒΛΙΟΓΡΑΦΙΑ
1.Κανακούδη-Τσακαλίδου, Φ, Παπαχρήστου, Φ, Δρόσου-Αγακίδου Β, Ζαφειρίου, Δ (2023) Βασική Παιδιατρική, 4η έκδ., University Studio Press.
2. Rood JE, Behrens EM. Inherited Autoinflammatory Syndromes. Annu Rev Pathol 2022 Jan 24;17:227-249
3. di Donato G, d’Angelo DM, Breda L, Chiarelli F. Monogenic Autoinflammatory Diseases: State of the Art and Future Perspectives. Int J Mol Sci 2021, 22,6360. https://doi.org/10.3390/ ijms22126360.
4.Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol 2022;22(8):471-483.
5. Savic S, Caseley EA, Michael F. McDermott MF. Moving towards a systems-based classification of innate immune mediated diseases. Nature Reviews Rheumatology, 2020; 16: 222-237
6. The expanding pathways of autoinflammation: a lesson from the first 100 genes related to autoinflammatory manifestations. Adv Protein Chem Struct Biol 2020;120:1-44. doi: 10.1016/bs.apcsb.2019.11.001. Epub 2019 Dec 12.
080
Εισαγωγή στα Αυτοφλεγμονώδη Νοσήματα
081 Εισαγωγή στα Αυτοφλεγμονώδη Νοσήματα
ΕΡΓΑΣΙΑ
Οικογενής Μεσογειακός Πυρετός
και Σύνδρομο PFΑPA: Σύγχρονη
αντιμετώπιση και θεραπεία όταν
συνυπάρχει μερική επικάλυψη
Βασιλική Σγουροπούλου
Περίληψη
Ο Οικογενής Μεσογειακός Πυρετός (ΟΜΠ) και το σύνδρομο PFAPA αποτελούν τα πιο
κοινά αυτοφλεγμονώδη νοσήματα της παιδικής ηλικίας. O ΟΜΠ αποτελεί μία γενετική
διαταραχή στο σύστημα της φυσικής ανοσίας που οφείλεται σε μετάλλαξη στο γονίδιο MEFV.
Σήμερα, εκτός από τα παλαιότερα κλινικά κριτήρια, διαθέτουμε νέα διαγνωστικά κριτήρια
που συμπεριλαμβάνουν και το γονότυπο, ενώ για την παρακολούθηση του νοσήματος
χρησιμοποιούνται σύγχρονα εργαλεία. Παράλληλα στην θεραπευτική μας φαρέτρα έχουν προστεθεί, εκτός από την κλασική θεραπεία με την κολχικίνη, σύγχρονες βιολογικές
θεραπείες που στοχεύουν στην πλήρη ύφεση του νοσήματος.
Το σύνδρομο PFAPA αποτελεί ένα πολυπαραγοντικό νόσημα με μη αναγνωρισμένο μέχρι
σήμερα γονιδιακό υπόστρωμα. Η έναρξη της νόσου στην πλειονότητα των περιπτώσεων
σημειώνεται στην πρώτη παιδική ηλικία μέχρι τα 6 έτη, ενώ καταγραφές υπάρχουν με έναρξη ακόμη και στην ενήλικη ζωή. Ιδιαίτερα συχνή είναι η συνύπαρξη του νοσήματος
με τις μεταλλάξεις στο γονίδιο MEFV, η οποία έχει συσχετιστεί με όψιμη έναρξη της νόσου
και με επεισόδια βραχύτερης διάρκειας και μικρότερης συχνότητας. Οι παραδοσιακές
μέθοδοι αντιμετώπισης της νόσου με κορτικοστεροειδή και αμυγδαλεκτομή έχουν πλέον
αντικατασταθεί από τη χορήγηση κολχικίνης. Η πρόγνωση του συνδρόμου είναι άριστη, με υποχώρηση των επεισοδίων συνήθως στα 3-6 έτη μετά την έναρξη, ενώ μόνο 20% των ασθενών θα συνεχίσει να εμφανίζει επεισόδια και στην ενήλικη ζωή.
Λέξεις-κλειδιά: Οικογενής Μεσογειακός Πυρετός, Σύνδρομο PFAPA
Αλληλογραφία
Βασιλική Σγουροπούλου Ολυμπιάδος 3Β, 57010 Πεύκα Θεσσαλονίκης Τ. 2310676202
K. 6972232877
e-mail: vsgouro@hotmail. com
Βασιλική Σγουροπούλου Α΄Παιδιατρική Κλινική Α.Π.Θ. - Γενικό Νοσοκομείο Θεσσαλονίκης "Ιπποκράτειο"
082
ΒΡΑΒΕΥΜΕΝΗ
Correspondence
Vasiliki Sgouropoulou Olimpiados 3Β, 57010 Pefka Thessaloniki
Τ. +30 2310676202, M. +30 6972232877 e-mail: vsgouro@hotmail. com
Familial Mediterranean Fever and PFAPA Syndrome: Contemporary Disease management and treatment when partial overlap is present
Vasiliki Sgouropoulou
Abstract
Familial Mediterranean Fever (FMF) and PFAPA syndrome are the most common autoinflammatory diseases in childhood. FMF is a genetic disorder of the innate immune system, caused by mutations in the MEFV gene. Today, in addition to the former clinical criteria, we have new diagnostic criteria that include genotype, while modern tools are used for disease monitoring. At the same time, in addition to the standard treatment with colchicine, innovative biological agents are used, aiming at complete remission of the disease.
PFAPA syndrome is a multifactorial disease with an unknown genetic basis. The onset of the disease in the majority of the cases is observed in early childhood up to 6 years of life, while there are records of onset even in adulthood. The coexistence of the disease with mutations in the MEFV gene is particularly common and has been associated with late onset of the disease and with episodes of shorter duration and lower frequency. The conventional treatment methods with corticosteroids and tonsillectomy have now been replaced by colchicine. The prognosis of the syndrome is excellent, with episodes resolving 3-6 years after onset, while only 20% of patients will continue to have episodes in adulthood.
Keywords: Familial Mediterranean Fever, PFAPA syndrome
Α. ΟΙΚΟΓΕΝΗΣ ΜΕΣΟΓΕΙΑΚΟΣ ΠΥΡΕΤΟΣ
Ο Οικογενής Μεσογειακός Πυρετός αποτελεί το πιο συχνό μονογονιδιακό αυτοφλεγμονώδες νόσημα και προσβάλλει κυρίως τους λαούς γύρω από τη λεκάνη της Μεσογείου.
Ο επιπολασμός του εμφανίζει μεγάλη ετερογένεια παγκοσμίως, ακόμη και μέσα στις ίδιες τις χώρες, οι οποίες παραδοσιακά χαρακτηρίζονται από συχνή εμφάνιση του νοσήματος.
Μελέτες που προέρχονται από τέτοιες χώρες, όπως η Τουρκία αναφέρουν επιπολασμό που ανέρχεται στο 1/1.000, ενώ σε άλλες περιοχές τις ίδιας χώρας καταγράφεται επιπολασμός ανά
Vasiliki Sgouropoulou
1st Department of Paediatrics, Paediatric Immunology and Rheumatology Referral Center, Hippokration General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
0.6/1000 (1) Άλλοι λαοί που εμφανίζουν υψηλό επιπολασμό είναι οι Αρμένιοι, οι μη Εσκενάζυ Εβραίοι και οι Άραβες, στους οποίους αναφέρονται ποσοστά φορείας που φτάνουν στο 25%. Αναφορές του νοσήματος καταγράφονται πλέον και σε άλλες περιοχές, όπως η Βόρεια Μεσόγειος, η Αμερική και η Ιαπωνία. (2,3)
Το νόσημα ανήκει στις φλεγμονοσωμοπάθειες, στις οποίες παρατηρείται διαταραχή στο σύστημα της φυσικής ανοσίας και συγκεκριμένα στην πρωτεΐνη που κωδικοποιεί το γονίδιο
MEFV (MΕditerranean FeVer) (Εικ.1)(4).
083
AWARDED PAPER
084
Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA:
Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
Εικόνα 1. Παθοφυσιολογία του Οικογενούς Μεσογειακού Πυρετού.
Η μεταλλαγμένη πρωτεΐνη καλείται πυρίνη ή μαρενοστρίνη. Το όνομα πυρίνη προέρχεται
από την ελληνική ονομασία του πυρετού, ενώ το μαρενοστρίνη από τη λατινική ονομασία της θάλασσας (mare nostrum).
Τυπικά, το νόσημα μεταβιβάζεται σύμφωνα με το πρότυπο της αυτοσωματικής υπολειπόμενης κληρονομικότητας. Έχουν όμως αναφερθεί και περιπτώσεις στις οποίες δεν διαπιστώνεται
η ύπαρξη δεύτερης μετάλλαξης στους πάσχοντες, οδηγώντας στο συμπέρασμα ότι η νόσος μπορεί να κληρονομείται κατά τον αυτοσωματικό επικρατούντα χαρακτήρα. Μέχρι σήμερα
έχουν καταγραφεί 389 μεταλλάξεις του γονιδίου, σύμφωνα με τη διεθνή βάση δεδομένων Infevers, με συχνότερες αυτές που φαίνονται στον Πίνακα 1.
Πίνακας 1. Οι συχνότερες MEFV μεταλλάξεις
Συχνότερες μεταλλάξεις παγκοσμίως
Στο 25-60% των ασθενών η έναρξη της νόσου παρατηρείται στην ηλικία κάτω των 10 ετών, ενώ στο 64-90% κάτω των 20 ετών (5). Τα επεισόδια είναι αυτοτροφοδοτούμενα, αλλά μπορεί
να υπάρχουν και προδιαθεσικοί πυροδοτικοί μηχανισμοί, όπως η σωματική καταπόνηση, το συναισθηματικό stress, η έμμηνος ρύση, η διατροφή και οι λοιμώξεις. Η διάγνωση της
νόσου παραμένει κλινική, ενώ ο γονιδιακός έλεγχος μπορεί να επιβεβαιώσει, αλλά όχι να
αποκλείσει τη διάγνωση.
Κλινικές εκδηλώσεις: Οι συχνότερες κλινικές εκδηλώσεις του νοσήματος είναι τα
υποτροπιάζοντα εμπύρετα επεισόδια που συνοδεύονται από κοιλιακό άλγος, ρευματικές
εκδηλώσεις (αρθρίτιδα, αρθραλγίες) συχνότερα στα κάτω άκρα, θωρακικό άλγος (πλευρίτιδα,
M694V M680I V726A A744S T267I E148Q P369S E167D F479L K695R M694I I692del R761H I591T
Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA:
Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
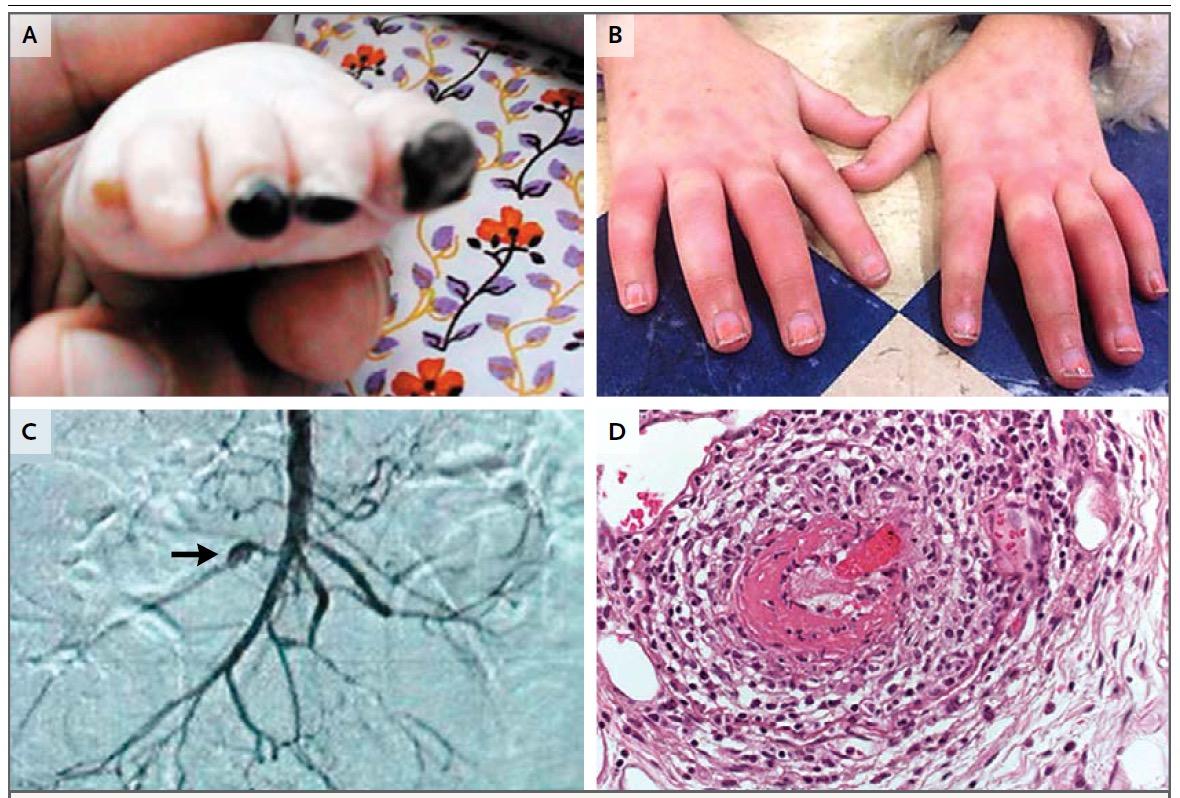
περικαρδίτιδα), ερυσιπελατοειδές εξάνθημα (συνήθως στα άκρα), έμετοι, άφθες, διάρροιες, λεμφαδενοπάθεια, μυαλγίες, ηπατική συμμετοχή, μεγαλοσπληνία και ορχίτιδα. (Εικ.2,3)
Εικόνα 2. Μεταβολισμός κολχικίνης.
Εικόνα 3. Bonnetblanc m et al, Am J Clin Dermatol 2003; 4(3)
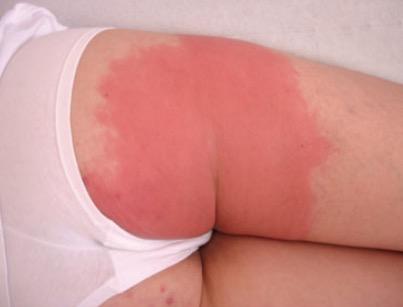
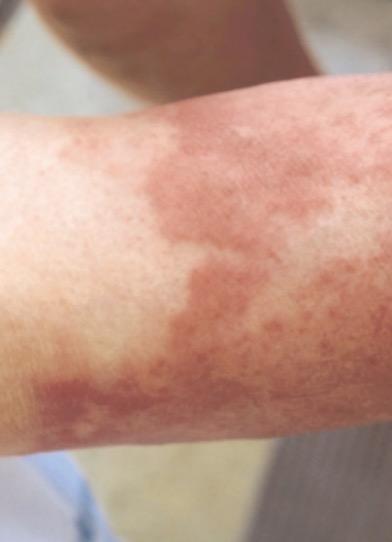
Δερματική βλάβη ερυσιπελατοειδούς εξανθήματος.
Διακρίνονται 3 φαινότυποι της νόσου.
Ο φαινότυπος Ι χαρακτηρίζεται από τυπικές κλινικές εκδηλώσεις, όπως περιγράφηκαν
παραπάνω.
ΟφαινότυποςΙΙ χαρακτηρίζεται από την ύπαρξη
πρωτεϊνουρίας ή νεφρικής ανεπάρκειας, ως
εκδήλωση της αμυλοείδωσης Α, πριν την εμφάνιση κλινικών εκδηλώσεων ή ως μεμονωμένο
εύρημα σε ένα μέλος οικογένειας, στην οποία υπάρχουν άτομα που πάσχουν από ΟΜΠ.
085
Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA:
Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
Το αναφερόμενο ποσοστό επίπτωσης κυμαίνεται από 7-25% σύμφωνα με μεγάλες μελέτες
που έγιναν στη δεκαετία του 1990 (6). Στους ασθενείς αυτούς, οι τυπικές κρίσεις μπορεί
να εμφανιστούν αρκετά χρόνια μετά την εκδήλωση της αμυλοείδωσης. Σε ορισμένες
περιπτώσεις, μπορεί να μην αναπτύξουν συμπτώματα καθ’ όλη τη διάρκεια της ζωής τους,
να έχουν όμως επίμονα αυξημένα επίπεδα SAA πρωτεΐνης παρά την απουσία κρίσεων. Στους
ασθενείς αυτούς επιβάλλεται η διενέργεια μοριακού ελέγχου, για την ανίχνευση πιθανών
MEFV μεταλλάξεων.
Ο φαινότυπος ΙΙΙ (σιωπηλή/silent μορφή) χαρακτηρίζεται από απουσία κλινικών
συμπτωμάτων ή αμυλοείδωσης, αλλά ο γονιδιακός έλεγχος αποκαλύπτει την ύπαρξη δύο
μεταλλάξεων (ομοζυγωτία ή διπλή ετεροζυγωτία). Η επίπτωση της νόσου υπολογίζεται σε
1:300 στους Εσκενάζυ Εβραίους και 1:25 στους Ιρακινούς Εβραίους. Η φορεία δύο “σιωπηλών
μεταλλάξεων”, παρά την απουσία κλινικών εκδηλώσεων, προδιαθέτει στην ανάπτυξη ΑΑ
αμυλοείδωσης, σκιαγραφώντας κατά τον τρόπο αυτόν τον φαινότυπο II. Για τον λόγο αυτόν, προτείνεται να ελέγχονται γονιδιακά τα μέλη των οικογενειών, στις οποίες υπάρχουν ασθενείς
με ΟΜΠ. Παράλληλα συστήνεται η παρακολούθηση των ατόμων με φαινότυπο ΙΙΙ για πιθανή
ανάπτυξη ΟΜΠ συμπτωμάτων, ασυμπτωματικής πρωτεϊνουρίας ή αυξημένων δεικτών οξείας
φάσης, προκειμένου να αναγνωριστεί έγκαιρα η αλλαγή σε φαινότυπο Ι ή ΙΙ.
Τα πρώτα κλινικά κριτήρια θεσπίστηκαν το 1997 και ονομάστηκαν κριτήρια Tel Hashomer.
Από το 2019 έχουμε στην διάθεσή μας τα κριτήρια ταξινόμησης Eurofever της PRINTO που
εξυπηρετούν κυρίως για τη διαφοροδιάγνωση μεταξυ των αυτοφλεγμονωδών νοσημάτων και
είναι τα πρώτα που συμπεριέλαβαν τον γονότυπο (6).
Τα κριτήρια περιλαμβάνουν:
Α. Την παρουσία επιβεβαιωτικού MEFV γονοτύπου (παθογόνες ή πιθανόν παθογόνες
μεταλλάξεις σε ομοζυγωτία ή διπλή ετεροζυγωτία) και τουλάχιστον 1 από τα ακόλουθα:
• Διάρκεια επεισοδίων 1- 3 ημέρες
• Αρθρίτιδα
• Θωρακικό άλγος
• Κοιλιακό άλγος
Β. Την παρουσία μη επιβεβαιωτικού MEFV γονότυπου (διπλή ετεροζυγωτία με μία παθογόνο
μετάλλαξη και μία άγνωστης σημασίας μετάλλαξη ή δύο άγνωστης σημασίας μεταλλάξεις
ή ετεροζυγωτία με μία παθογόνο μετάλλαξη) και τουλάχιστον 2 από τα προηγουμένως
αναφερθέντα κριτήρια.
Εκτός από τους κλασικούς δείκτες υποκλινικής φλεγμονής, που είναι λευκοκυττάρωση,
αυξημένες CRP και ΤΚΕ, στα μεσοδιαστήματα της νόσου είναι σημαντική η αξιολόγηση των επιπέδων του αμυλοειδούς Α (SAA) (serum amyloid alpha) και του NLR (neutrophil/lymphocyte ratio) λόγος πολυμορφοπυρήνων/ λεμφοκύτταρα) (7). Η παρουσία επίσης μυαλγίας
και ερυσιπελατοειδούς εξανθήματος στις κρίσεις έχει συσχετιστεί με αυξημένο κίνδυνο
υποκλινικής φλεγμονής (8).
Από τις μεταλλάξεις που έχουν καταγραφεί, η ομοζυγωτία της M694V έχει συσχετιστεί με
βαρύτερο κλινικό φαινότυπο, αυξημένο κίνδυνο για πρώιμη έναρξη της νόσου, καθώς και με
αυξημένα ποσοστά επιπλοκών και κυρίως νεφρική αμυλοείδωση (9).
Από τα διεθνή δεδομένα, ποσοστό ασθενών 40-70% αναφέρονται ως ομοζυγώτες, στο 2025% απομονώνεται μόνο μία μετάλλαξη, ενώ στο 5-10% δεν εντοπίζεται καμία μετάλλαξη (10),(11),(12).
Θεραπεία εκλογής από τη δεκαετία του ’70 μέχρι και σήμερα παραμένει η κολχικίνη (13). Η
κολχικίνη μειώνει τη συχνότητα και βαρύτητα των κρίσεων, προφυλάσσει από υποκλινική
φλεγμονή, προστατεύει από αμυλοείδωση, συμβάλλει στην αντιμετώπισή της και βελτιώνει
την ποιότητα ζωής των ασθενών. Στους μηχανισμούς δράσης της περιλαμβάνεται η δέσμευση
της β-τουμπουλίνης, η αναστολή του πολυμερισμού των μικροσωληνίσκων, καθώς και η
086
μείωση του πολλαπλασιασμού και της χημειοταξίας των λευκοκυττάρων, καταστέλλοντας με
τον τρόπο αυτόν τη φλεγμονώδη διαδικασία (14),(15).
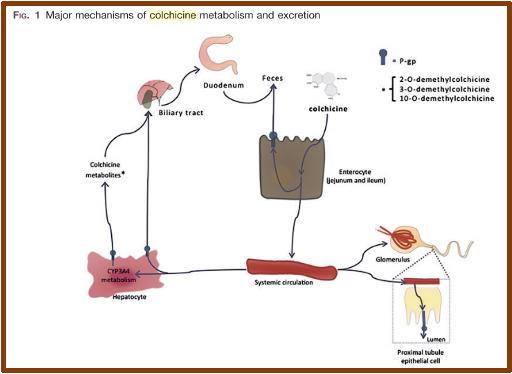
Η κολχικίνη μεταβολίζεται κατά κύριο λόγο στο ήπαρ μέσω του κυτοχρώματος P450 και σε μικρότερο βαθμό στους νεφρούς (Εικ.4) (13).
Εικόνα 4. Kolivras A et al, J Cutan Pathol 2013: 40: 585–590 Δερματική βλάβη ερυσιπελατοειδούς εξανθήματος.
Αποτελεί επίσης υπόστρωμα της γλυκοπρωτεΐνης P, που είναι υπεύθυνη για την απέκκρισή της. Με τον τρόπο αυτό, φάρμακα και ουσίες που αναστέλλουν το κυτόχρωμα P450 (CYP3A4) και την γλυκοπρωτεΐνη P μπορεί να προκαλέσουν αύξηση της κολχικίνης σε τοξικά επίπεδα
και πρέπει να αποφεύγεται η λήψη τους (Πίνακας 2) (13,16).
Πίνακας 2. Αλληλεπιδράσεις της κολχικίνης με φάρμακα και ουσίες
Ισχυροί αναστολείς του CYP3A4 Μέτριοι αναστολείς του CYP3A4
Clarithromycin
Cobicistat
Αναστολείς Pγλυκοπρωτεΐνης
Στατίνες που μεταβολίζονται στo CYP3A4
Cimetidine Amiodarone Atorvastatin
Ciprofloxacine Carvedilol Simvastatin
Diltiazem Cyclosporin Clarithromycin Lovastatin
Intraconazole Erythromycin Intraconazol Fluvastatin
Ketoconazol Fluconazol Quinidine Pravastatin
Ritonavir Fluvoxamine Ranolazine
Telithromycin Imatinib Ritonavir
Voriconazole Verapamil Verapamil
087
Grape fruit Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA: Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA:
Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
Η δοσολογία της κολχικίνης καθορίζεται από την ηλικία του παιδιατρικού ασθενούς και τη
βαρύτητα της νόσου. Συνήθως, με μέγιστη δόση 2 mg στα παιδιά και 3 mg στους ενήλικες
οι κρίσεις ελέγχονται. Υψηλότερη δόση μπορεί να χρειαστεί σε αυξημένη δραστηριότητα της
νόσου, σε ομοζυγωτία της M694V και όταν προϋπάρχουν επιπλοκές, όπως η αμυλοείδωση (17). Επίσης, αναπροσαρμογή της δόσης διενεργείται σε ύπαρξη υποκλινικής φλεγμονής ή σε
μη ικανοποιητικό έλεγχο των επεισοδίων της νόσου (18).
Σε γενικές γραμμές, η κολχικίνη είναι φάρμακο ασφαλές και καλά ανεκτό από τους ασθενείς.
Ανεπιθύμητες ενέργειες (κοιλιακό άλγος, διάρροιες, έμετοι) παρατηρούνται σε ένα ποσοστό
γύρω στο 20%. Στις περιπτώσεις αυτές, ο περιορισμός των γαλακτοκομικών προϊόντων και η
διαίρεση του φαρμάκου σε περισσότερες της μίας δόσης μπορεί να μειώσουν την ένταση και
τη συχνότητα των ανεπιθύμητων ενεργειών, ανάλογα με την ανοχή και τη συμμόρφωση των
ασθενών (13).
Ως ανθεκτικότητα στη θεραπεία με κολχικίνη χαρακτηρίζεται «η κατάσταση νόσου» με ≥ 1
κρίση/μήνα για 6 μήνες ή η παρουσία υποκλινικής φλεγμονής παρά το γεγονός ότι υπάρχει
καλή συμμόρφωση του ασθενούς στην αγωγή ή το ότι παίρνει τη μέγιστη ανεκτή δόση των
φαρμάκων (17). Υπάρχει, όμως ένα ποσοστό ασθενών 5% που δεν απαντούν στην αγωγή, ενώ 30-40% είναι μερικοί απαντητές (19). Οι ασθενείς αυτοί είναι υποψήφιοι για βιολογική
θεραπεία (φάρμακα 2ης γραμμής) που αποτελούν οι αναστολείς της ιντερλευκίνης-1 (IL-1)
και που πρέπει να λαμβάνονται παράλληλα με την κολχικίνη(20).
Ο βιολογικός παράγοντας Anakinra αποτελεί έναν ανταγωνιστή του υποδοχέα της
ανθρώπινης ιντερλευκίνης-1 (r-metHuIL-1ra), εγκεκριμένο από τους FDA και ΕΜΑ, για τον
οποίο οι μελέτες έχουν δείξει ικανοποιητική αποτελεσματικότητα. Το δοσολογικό του σχήμα περιλαμβάνει τη χορήγηση καθημερινών ενέσεων υποδορίως σε δόση: ΒΣ<50 kg: 1-2 mg/ kg/ημέρα υποδορίως και ΒΣ >50 kg: 100 mg/ημέρα υποδορίως. Σε ανεπαρκή ανταπόκριση, η δοσολογία μπορεί να ανέλθει στα 4 mg/kg/ημέρα. Μειονέκτημα της θεραπευτικής αυτής προσέγγισης αποτελεί το δοσολογικό του σχήμα, που απαιτεί την καθημερινή χορήγηση
ενέσεων.
Άλλος βιολογικός παράγοντας εγκεκριμένος από το 2016, για τη θεραπεία του ανθεκτικού
Οικογενούς Μεσογειακού Πυρετού για ηλικίες 2 ετών και άνω, αποτελεί το Canakinumab.
Πρόκειται για ένα πλήρως εξανθρωποποιημένο μονοκλωνικό αντίσωμα της ανθρώπινης
ιντερλευκίνης-1β του ισοτύπου IgG1/κ, που δεσμεύεται με μεγάλη συγγένεια ειδικά στην
ανθρώπινη IL-1β και εξουδετερώνει τη βιολογική δράση της. Σε ασθενείς με βάρος σώματος
≥ 7.5 kg και ≤ 40 kg η δοσολογία είναι 2 – 4 mg/kg, ενώ σε βάρος > 40 kg η δόση είναι 150 mg κάθε 4 εβδομάδες υποδορίως με μέγιστη δόση τα 300 mg. Σε πρόσφατη μετα-ανάλυση
που συμπεριέλαβε όλες τις μελέτες μέχρι το 2019 με συνολικά 221 ασθενείς παρατηρήθηκε
πλήρης απάντηση στην κολχικίνη στο 79.9%, μερική απάντηση στο 19.3% και καμία απάντηση
στο 1.1% των ασθενών (21).
Β. ΣΥΝΔΡΟΜΟ PFΑPA
Το σύνδρομο PFAPA περιγράφτηκε για 1η φορά το 1987 από τον Marshall, από τον οποίον έλαβε και το αρχικό του όνομα σύνδρομο Marshall. Αποτελεί το πιο κοινό αυτοφλεγμονώδες
νόσημα της παιδικής ηλικίας και είναι ένα πολυγονιδιακό ή πολυπαραγοντικό νόσημα με μη
αναγνωρισμένη μέχρι σήμερα τη γονιδιακή βλάβη. Η επίπτωση του νοσήματος παραμένει
άγνωστη, με μία μόνο μελέτη από τη Νορβηγία, η οποία την υπολογίζει στα 2.3/10.000 παιδιά έως τα 5 έτη (22). Στην πλειονότητα των περιπτώσεων η έναρξη της νόσου παρατηρείται στην
πρώτη παιδική ηλικία μέχρι τα 6 έτη, ενώ καταγραφές υπάρχουν με έναρξη ακόμη και στην
ενήλικη ζωή (23, 24). Θετικό οικογενειακό ιστορικό καταγράφεται με διάφορα ποσοστά που
κυμαίνονται από 13.8%-78% σε μια πρόσφατη μελέτη (25).
Στην αιτιοπαθογένεια της νόσου φαίνεται να εμπλέκονται μηχανισμοί τόσο της φυσικής, όσο
και της επίκτητης ανοσίας. Ειδικότερα, η σχέση του συνδρόμου με μεταλλάξεις γονιδίων, όπως
MEFV, NLRP3, CARD8, που οδηγούν σε διαταραχή της λειτουργίας του φλεγμονοσώματος με
088
Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA: Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
αποτέλεσμα την υπερέκκριση της IL-1, υποδηλώνει τη συμμετοχή της φυσικής ανοσίας.
Παράλληλα όμως, η ενεργοποίηση των Τ-λεμφοκυττάρων που επάγουν την Τh1 ανοσιακή
απάντηση, υποδεικνύει τη συμμετοχή και της ειδικής ανοσίας (26, 27). Ιδιαίτερα συχνή είναι η
συνύπαρξη του νοσήματος με τις μεταλλάξεις στο γονίδιο MEFV του Οικογενούς Μεσογειακού
Πυρετού M694V, E148Q και V726A. Η συνύπαρξη αυτή έχει συσχετιστεί με όψιμη έναρξη της
νόσου και με επεισόδια βραχύτερης διάρκειας και μικρότερης συχνότητας (28, 29, 30).
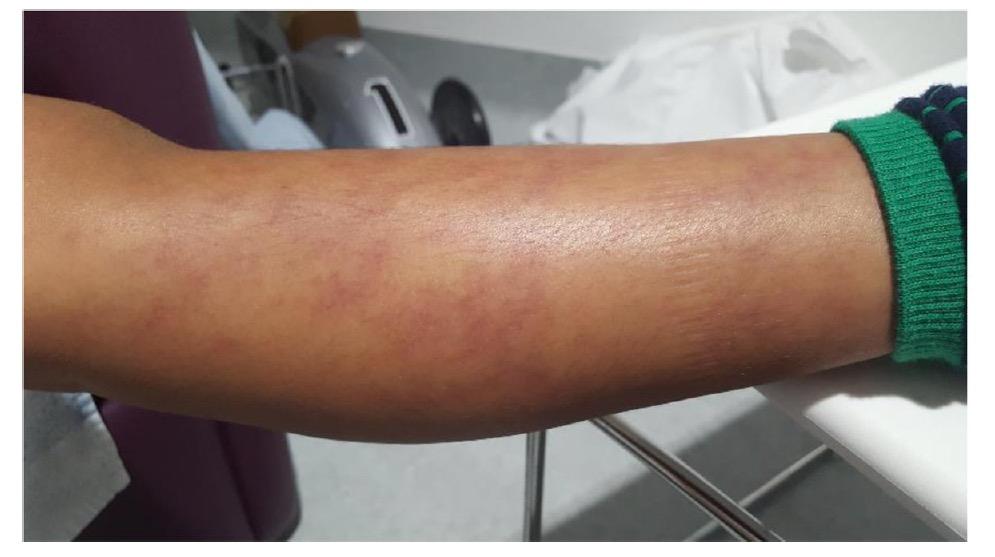
Κλινική εικόνα: Το σύνδρομο χαρακτηρίζεται από περιοδικά εμπύρετα επεισόδια με
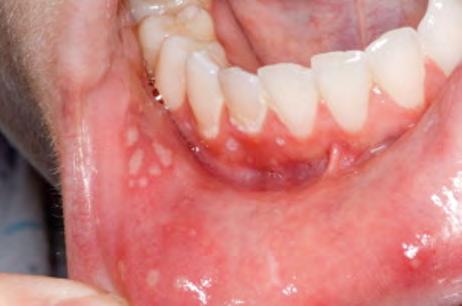
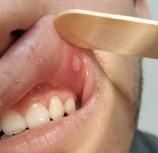
εικόνα φαρυγγοαμυγδαλίτιδας, έκθυση αφθών στον στοματικό βλεννογόνο και τραχηλική
λεμφαδενίτιδα. (Εικ.5.) Τα επεισόδια διαρκούν 3-7 ημέρες και εμφανίζουν περιοδικότητα
που κυμαίνεται από 2-8 εβδομάδες. Πρόδρομα συμπτώματα εμφανίζονται στο 60% των ασθενών και σε αυτά περιλαμβάνονται η καταβολή, η κεφαλαλγία, το κοιλιακό άλγος και η ευερεθιστότητα (27). Το 2019 θεσπίστηκαν τα κλινικά κριτήρια ταξινόμησης EUROFEVER της PRINTO (6), για τα οποία απαιτούνται 7 από τα 8 κριτήρια: Παρουσία:
• Φαρυγγοαμυγδαλίτιδας
• Διάρκεια επεισοδίων 3-6 ημέρες
• Τραχηλικής λεμφαδενίτιδας
• Περιοδικότητας
Απουσία:
• Διάρροιας
• Θωρακικού άλγους
• Εξανθήματος
• Αρθρίτιδας
Εικόνα 5. Αφθώδεις βλάβες σε στοματικό βλεννογόνο.
Στα εργαστηριακά ευρήματα της νόσου περιλαμβάνονται οι αυξημένοι δείκτες οξείας φάσης (λευκοκυττάρωση, αυξημένες CRP, ΤΚΕ), φυσιολογικά ή αυξημένα ουδετερόφιλα και φυσιολογικοί δείκτες οξείας φάσης στα μεσοδιαστήματα των κρίσεων (31).
Στα φάρμακα πρώτης γραμμής ανήκουν τα κορτικοστεροειδή. Χορηγείται πρεδνιζολόνη
σε δοσολογία 0.5 - 2 mg/kg/ημέρα ή βηταμεθαζόνη σε δοσολογία 0.1 – 0.2 mg/kg/ημέρα.
Τα κορτικοστεροειδή επιφέρουν καταστολή του επεισοδίου μέσα σε 24 ώρες στο 80-95%
των περιπτώσεων, σε ορισμένες όμως περιπτώσεις μπορεί να χρειαστεί και 2η δόση στο 2ο 24ωρο(32). Η δοκιμασία χορήγησης της κορτιζόνης χρησιμοποιείται συχνά και για επιβεβαίωση της διάγνωσης(33). Η δράση όμως του φαρμάκου είναι παροδική και δεν αποτρέπει την εμφάνιση νέου επεισοδίου. Αντίθετα, μπορεί να αυξήσει την συχνότητά τους στο 25-50% των ασθενών(34). Επιπλέον, η μακροχρόνια λήψη τους έχει συσχετιστεί με ποικίλες ανεπιθύμητες ενέργειες.
Η θεραπεία που τα τελευταία χρόνια έχει αποδειχθεί αποτελεσματική και πλέον υποστηρίζεται
και συστήνεται είναι η κολχικίνη. Συστήνεται έναρξη της χορήγησης στην ακόλουθη δοσολογία
με δυνατότητα αύξησης αυτής: ηλικία < 5 ετών: 0.5 mg/Η, 5–10 ετών : 1.5 mg/Η, > 10 ετών : 1 mg/ημέρα.
089
Οι μελέτες δείχνουν παράταση των μεσοδιαστημάτων στο 85% των ασθενών, ενώ σε συνύπαρξη με μεταλλάξεις η αποτελεσματικότητα της θεραπείας με κολχικίνη αυξάνει σημαντικά σε
σχέση με τους ασθενείς που δεν φέρουν καμία μετάλλαξη (96% vs 80%) (35, 28).
Στη διεθνή βιβλιογραφία μόνο 2 διπλές τυφλές τυχαιοποιημένες μελέτες με μικρό αριθμό
ασθενών έχουν καταγραφεί, που δείχνουν μείωση της συχνότητας και της διάρκειας των
επεισοδίων στο 60-90% (36,37). Ακολούθησε όμως μία συστηματική ανασκόπηση που
κατέληξε ότι η απόδειξη αυτή είναι μέτριας ποιότητας (38). Παράλληλα, δεν είναι λίγες
οι καταγραφές που περιγράφουν υποτροπή μετά από αμυγδαλεκτομή, αδενοειδεκτομή.
Δεδομένων λοιπόν, των σημαντικά αυξημένων ποσοστών βελτίωσης με φαρμακευτική
αγωγή, της ύπαρξης χειρουργικών επιπλοκών που εγκυμονεί η αμυγδαλεκτομή και της
αυτοπεριοριζόμενης φύσης του νοσήματος, η χειρουργική επέμβαση δεν περιλαμβάνεται
σήμερα στους ενδεδειγμένους τρόπους διαχείρισης του συνδρόμου PFAPA (39). Η πρόγνωση
του συνδρόμου είναι άριστη. Τα επεισόδια συνήθως υποχωρούν αυτόματα πριν την εφηβεία, 3-6 έτη μετά την έναρξη, ενώ 20% των ασθενών θα συνεχίσει να εμφανίζει επεισόδια και
στην ενήλικη ζωή (32). Τα παιδιά έχουν φυσιολογική σωματική αύξηση και δεν εμφανίζουν
μακροπρόθεσμες επιπτώσεις (40).
Συμπερασματικά, διαπιστώνεται ότι ο παιδίατρος κατέχει ρόλο κλειδί στην έγκαιρη διάγνωση
και αντιμετώπιση των παραπάνω αυτοφλεγμονωδών νοσημάτων, που είναι εύκολο να τα
υποπτευθεί διότι: εκδηλώνονται κατά κανόνα στην παιδική ηλικία, η συχνότητα των ιατρικών
επισκέψεων είναι αυξημένη στην ηλικία αυτή και τα συμπτώματά τους είναι χαρακτηριστικά
ώστε να θέσει την κλινική διάγνωση, να συντονίσει τη συνεργασία με τις άλλες εξειδικεύσεις
προκειμένου να την επιβεβαιώσει και να ξεκινήσει άμεσα η θεραπευτική αγωγή.
Βιβλιογραφία
1. Georgin-Lavialle S, Hentgen V, Stankovic Stojanovic K, Bachmeyer C, Rodrigues F, Savey L, et al. Familial Mediterranean fever Vol. 39, Revue de Medecine Interne. 2018 p. 240–55.
2. Burhan Fatih Kocyigit, Ahmet Akyol. Bibliometric analysis of publication activity in the field of familial Mediterranean fever in 2010-2019: a Scopus-based study. Rheumatol Int. 2021 Nov;41(11):2015-2023.
3. Koga T, Sato S, Mishima H, Migita K, Endo Y, Umeda Y et al. Next-generation sequencing of the whole MEFV gene in Japanese patients with familial Mediterranean fever: a casecontrol association study. Clin Exp Rheumatol. 2020 Sep-Oct;38 Suppl 127(5):35-41.
4. Theofilopoulos AN, Gonzalez-Quintial R, Lawson BR, Koh YT, Stern ME, Kono DH, et al. Sensors of the innate immune system: their link to rheumatic diseases. Nat Rev Rheumatol [Internet]. 2010 Mar;6(3):146–56.
5. Georgin-Lavialle S, Hentgen V, Stankovic Stojanovic K, Bachmeyer C, Rodrigues F, Savey L, et al. Familial Mediterranean fever Vol. 39, Revue de Medecine Interne. Rev Med Interne; 2018. p. 240–55.
6. Saatçi Ü, Ozen S, Özdemir S, et al. Familial Mediterranean fever in children: Report of a large series and discussion of the risk and prognostic factors of amyloidosis. Eur J Pediatr. 1997. doi:10.1007/s004310050677
7. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis 2019 Aug 1;78(8):1025–32.
8. Ahsen A, Ulu MS, Yuksel S, Demir K, Uysal M, Erdogan M, et al. As a new inflammatory marker for familial mediterranean fever: Neutrophil-to-lymphocyte ratio. Inflammation. 2013 Dec;36(6):1357–62.
9. Özer S, Yılmaz R, Sönmezgöz E, Karaaslan E, Taskin S, Bütün I, et al. Simple markers for subclinical inflammation in patients with familial mediterranean fever. Med Sci Monit. 2015 Jan 23;21:298–303.
10. Giancane G, Haar NMT, Wulffraat N, Vastert SJ, Barron K, Hentgen V, et al. Evidencebased recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis. 2015 Apr 1;74(4):635–41.
11. Soriano A, Pras E. Familial mediterranean fever: Genetic update. Isr Med Assoc J. 2014;16(5):274–6.
12. Jéru I, Hentgen V, Cochet E, Duquesnoy P, Le Borgne G, Grimprel E, et al. The Risk of
090
Familial Mediterranean Fever in MEFV Heterozygotes: A Statistical Approach. PLoS One. 2013 Jul 3;8(7).
13. Ben-Chetrit E, Ozdogan H. Can we make a diagnosis of autoinflammatory diseases based upon clinical features only?. Vol. 35, Clinical and Experimental Rheumatology. Clin Exp Rheumatol; 2017. p. S16–8.
14. Slobodnick A, Shah B, Krasnokutsky S, Pillinger MH. Update on colchicine, 2017. Vol. 57, Rheumatology (Oxford, England). Rheumatology (Oxford); 2018. p. i4–11.
15. Manukyan G, Aminov R. Update on pyrin functions and mechanisms of familial mediterranean fever. Vol. 7, Frontiers in Microbiology. Front Microbiol; 2016.
16. Sahakyan H, Abelyan N, Arakelov V, Arakelov G, Nazaryan K. In silico study of colchicine resistance molecular mechanisms caused by tubulin structural polymorphism. PLoS One. 2019 Aug 1;14(8).
17. Finkelstein Y, Aks SE, Hutson JR, Juurlink DN, Nguyen P, Dubnov-Raz G, et al. Colchicine poisoning: The dark side of an ancient drug. Vol. 48, Clinical Toxicology. Clin Toxicol (Phila); 2010. p. 407–14.
18. Ozen S, Demirkaya E, Erer B, Livneh A, Ben-Chetrit E, Giancane G, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis. 2016 Apr 1;75(4):644–51.
19. Hentgen V, Grateau G, Kone-Paut I, Livneh A, Padeh S, Rozenbaum M, et al. Evidencebased recommendations for the practical management of Familial Mediterranean Fever. Semin Arthritis Rheum. 2013 Dec;43(3):387–91.
20. Ozen S, Kone-Paut I, Gül A. Colchicine resistance and intolerance in familial mediterranean fever: Definition, causes, and alternative treatments. Vol. 47, Seminars in Arthritis and Rheumatism. Semin Arthritis Rheum; 2017. p. 115–20.
21. Cetin P, Sari I, Sozeri B, Cam O, Birlik M, Akkoc N, et al. Efficacy of Interleukin-1 Targeting Treatments in Patients with Familial Mediterranean Fever. Inflammation. 2015 Feb 1;38(1):27–31.
22. Kacar M, Savic S, van der Hilst JCH. The efficacy, safety and tolerability of canakinumab in the treatment of familial mediterranean fever: A systematic review of the literature. Vol.
13, Journal of Inflammation Research. J Inflamm Res; 2020. p. 141–9.
23. Førsvoll J, Kristoffersen EK, Øymar K. Incidence, clinical characteristics and outcome in Norwegian children with periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome; A population-based study. Acta Paediatr Int J Paediatr. 2013 Feb];102(2):187–92.
24. Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis phayngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J. 2008;10(5):358–60.
25. Cantarini L, Vitale A, Bartolomei B, Galeazzi M, Rigante D. Diagnosis of PFAPA syndrome applied to a cohort of 17 adults with unexplained recurrent fevers. Clin Exp Rheumatol. 2012;30(2):269–71.
26. Kraszewska-Głomba B, Matkowska-Kocjan A, Szenborn L. The Pathogenesis of Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Cervical Adenitis Syndrome: A Review of Current Research. Vol. 2015, Mediators of Inflammation. Mediators Inflamm; 2015.
27. Perko D, Debeljak M, Toplak N, Avčin T. Clinical features and genetic background of the periodic fever syndrome with aphthous stomatitis, pharyngitis, and adenitis: A single center longitudinal study of 81 patients. Mediators Inflamm 2015;2015.
28. Asna Ashari K, Rezaei N. PFAPA (periodic fever, aphthous stomatitis, pharyngitis, and adenitis) syndrome: an overview of genetic background Vol. 40, Clinical Rheumatology. Clin Rheumatol; 2021 p. 4437–44.
29. Welzel T, Ellinghaus M, Wildermuth AL, Deschner N, Benseler SM, Kuemmerle-Deschner JB. Colchicine Effectiveness and Safety in Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis. Front Pediatr 2021 Nov 25;9.
30. Yildiz M, Adrovic A, Ulkersoy I, Gucuyener N, Koker O, Sahin S, et al. The role of Mediterranean fever gene variants in patients with periodic fever, aphthous stomatitis, pharyngitis, and adenitis syndrome. Eur J Pediatr. 2021 Apr 1;180(4):1051–8.
31. Taniuchi S, Nishikomori R, Iharada A, Tuji S, Heike T, Kaneko K. MEFV Variants in Pa-
091
tients with PFAPA Syndrome in Japan. Open Rheumatol J. 2013 Apr 27;7(1):22–5.
32. Feder HM, Salazar JC. A clinical review of 105 patients with PFAPA (a periodic fever syndrome) Vol. 99, Acta Paediatrica, International Journal of Paediatrics. Acta Paediatr; 2010 p. 178–84.
33. Gaggiano C, Rigante D, Sota J, Grosso S, Cantarini L. Treatment options for periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome in children and adults: a narrative review. Vol. 38, Clinical Rheumatology. Clin Rheumatol; 2019. p. 11–7.
34. Soriano A, Soriano M, Espinosa G, Manna R, Emmi G, Cantarini L, et al. Current Therapeutic Options for the Main Monogenic Autoinflammatory Diseases and PFAPA Syndrome: Evidence-Based Approach and Proposal of a Practical Guide Vol. 11, Frontiers in Immunology. Front Immunol; 2020
35. Rigante D, Gentileschi S, Vitale A, Tarantino G, Cantarini L. Evolving frontiers in the treatment of periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis (PFAPA) syndrome Vol. 19, Israel Medical Association Journal. 2017 p. 444–7.
36. Gunes M, Cekic S, Kilic SS. Is colchicine more effective to prevent periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis episodes in Mediterranean fever gene variants? Pediatr Int 2017 Jun 1;59(6):655–60.
37. Renko M, Salo E, Putto-Laurila A, Saxen H, Mattila PS, Luotonen J, et al. A Randomized, Controlled Trial of Tonsillectomy in Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis Syndrome. J Pediatr 2007 Sep;151(3):289–92.
38. Garavello W, Romagnoli M, Gaini RM. Effectiveness of Adenotonsillectomy in PFAPA Syndrome: A Randomized Study. J Pediatr 2009 Aug;155(2):250–3.
39. Burton MJ, Pollard AJ, Ramsden JD, Chong LY, Venekamp RP. Tonsillectomy for periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA) Vol. 2014, Cochrane Database of Systematic Reviews. Cochrane Database Syst Rev; 2014
40. Vigo G, Martini G, Zoppi S, Vittadello F, Zulian F. Tonsillectomy efficacy in children with PFAPA syndrome is comparable to the standard medical treatment: A long-term observational study. Clin Exp Rheumatol 2014;32:S156–9.
41. Wang A, Manthiram K, Dedeoglu F, Licameli GR. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome: A review [Internet]. Vol. 7, World Journal of Otorhinolaryngology - Head and Neck Surgery. World J Otorhinolaryngol Head Neck Surg; 2021 p. 166–73.
092
Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA: Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
093 Οικογενής Μεσογειακός Πυρετός και Σύνδρομο PFΑPA: Σύγχρονη αντιμετώπιση και θεραπεία όταν συνυπάρχει μερική επικάλυψη
Απλοανεπάρκεια Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της
αδενοσίνης 2: Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
Παναγιώτα Καρανάνου
Περίληψη
Η απλοανεπάρκεια
της Α20 πρωτεΐνης (ΗΑ20) αποτελεί ένα αυτοφλεγμονώδες νόσημα, που
οφείλεται σε μεταλλάξεις τύπου «απώλειας λειτουργικότητας» στο γονίδιο TNFAIP3 (Τumor
Νecrosis Factor α-Induced Protein 3). Οι κλινικές εκδηλώσεις της είναι παρόμοιες με
αυτές του συνδρόμου Αδαμαντιάδη-Bechet με βασική διαφορά την πρωϊμότερη εμφάνισή τους (σε μικρότερη ηλικία). O ασθενής μπορεί να εκδηλώσει πυρετό και λιποδυστροφία.
Το κυριότερο όμως χαρακτηριστικό της νόσου είναι τα υποτροπιάζοντα επώδυνα έλκη στον
βλεννογόνο του γαστρεντερικού σωλήνα, του στόματος και των γεννητικών οργάνων. Άλλα
συχνά συμπτώματα είναι οι εκδηλώσεις γαστρεντερίτιδας, αρθραλγιών, πολυαρθρίτιδας
και δερματικών εξανθημάτων ενώ λιγότερο συχνά αναφέρεται οφθαλμοπάθεια, καρδιακή
συμμετοχή και νεφρικές εκδηλώσεις.
H ανεπάρκεια της ADA2 (Adenosine Deamine 2 [ADA2] Deficiency, DADA2) είναι ένα μονογονιδιακό αυτοφλεγμονώδες νόσημα, που προσομοιάζει στην οζώδη πολυαρτηρίτιδα
και οφείλεται σε μεταλλάξεις του γονιδίου της ADA2 πρωτεΐνης. Η έναρξη των συμπτωμάτων
της νόσου τοποθετείται στην πρώτη δεκαετία της ζωής. H κλινική της εικόνα χαρακτηρίζεται
από διαλείποντα πυρετό με εκδηλώσεις αγγειίτιδας που περιγράφoνται στην ομάδα των ρευματικών νοσημάτων της μεγαλύτερης παιδικής και εφηβικής ηλικίας (κυρίως δερματικές, νευρολογικές και γαστρεντερικές με ή χωρίς συμμετοχή του καρδιαγγειακού συστήματος
και των νεφρών). Οι εκδηλώσεις αυτές μπορεί να συνδυάζονται και με κλινική εικόνα ανοσοανεπάρκειας.
Μέχρι σήμερα δεν υπάρχει ειδική θεραπεία για την ΗΑ20 και την DADA2. Η αντιμετώπιση
είναι ανάλογη με τις κλινικές εκδηλώσεις και τη βαρύτητα της νόσου. Συμπερασματικά, σε παιδιατρικούς ασθενείς με εκδηλώσεις, όπως αυτές του συνδρόμου Αδαμαντιάδη-Bechet
σε πολύ μικρή ηλικία, καθώς και αυτές με συνύπαρξη οζώδους πολυαρτηριίτιδας και ανοσοανεπάρκειας στην 1η 10ετία της ζωής, πρέπει να τίθεται η υποψία αυτοφλεγμονώδους
νοσήματος.
Λέξεις-κλειδιά: Απλοανεπάρκεια της Α20 πρωτεΐνης (ΗΑ20), ανεπάρκεια της ADA2 (Adenosine Deamine 2 [ADA2] Deficiency, DADA2), αυτοφλεγμονώδη νοσήματα, ανοσοανεπάρκειες
Αλληλογραφία
Παναγιώτα Καρανάνου Αιγαίου 41, 55133 Καλαμαριά, Θεσσαλονίκη
Τ. 2310249878
K. 6974842552
e-mail: panagiotakarananou@gmail.com
Παναγιώτα Καρανάνου Δ΄Παιδιατρική Κλινική Α.Π.Θ, Γενικό Νοσοκομείο Παπαγεωργίου, Θεσσαλονίκη
094
Correspondence
Panagiota Karananou
Aigaiou 41, 55133
Kalamaria, Thessaloniki
Τ. +30 2310249878
M. +30 6974842552
e-mail: panagiotakarananou@gmail.com
Haploinsufficiency of A20 protein (HA20) and Deficiency of adenosine deaminase 2 (ADA2) enzyme (DADA2): Immunodeficiencies or Autoinflammatory diseases?
Panagiota Karananou
Abstract
Haploinsufficiency of A20 protein (HA20) is an autoinflammatory disease caused by highpenetrance loss-of-function germline mutations in TNFAIP3(Τumor Νecrosis Factor α-Induced Protein 3). Patients may present with symptoms of Adamantiades-Behcet-like disease with the main difference of the earlier onset (childhood). Fever is also reported as well as lipodystrophy. However, the hallmark feature of the disease is the recurrent painful oral, genital and/or gastrointestinal ulcers. Other common symptoms that occur at various time points during disease course include gastrointestinal complaints, polyarthritis and/ or arthralgia, skin involvement and less frequently, ocular and cardiovascular and renal involvement.
Deficiency of adenosine deaminase 2 (ADA2) enzyme (DADA2) is a monogenic autoinflammatory disease, with polyarteritis nodosa (PAN)–like features, associated with mutations in ADA2 protein. The first symptoms of the disease occur early, before the age of 10 years. DADA2 can manifest with intermittent fevers, and vasculitis/vasculopathy that are described in the group of rheumatic diseases in older children and adolescents (mainly skin, neurological, gastrointestinal with or without cardiovascular and renal involvement). These manifestations may also be combined with a clinical picture of immunodeficiency. To date there is no specific treatment for HA20 and DADA2. The treatment depends on the clinical manifestations and severity of the disease. In conclusion, in pediatric patients with manifestations, such as those of Adamantiades-Bechet syndrome at a very young age, as well as the coexistence of polyarteritis nodosa and immunodeficiency in the first decade of life, an autoinflammatory disease should be considered and suspected.
Keywords: Haploinsufficiency of A20 protein (HA20), Deficiency of adenosine deaminase 2 (ADA2) enzyme (DADA2), autoinflammatory diseases, immunodeficiencies.
Panagiota Karananou 4th Department of Pediatrics, Aristotle University of Thessaloniki, School of Medicine, Papageorgiou General Hospital, Ring Road Nea Efkarpia 56403, Thessaloniki, Greece
Εισαγωγή
Τα αυτοφλεγμονώδη νοσήματα, σύμφωνα με την πιο πρόσφατη κατάταξη της Διεθνούς
Ένωσης για τις Πρωτοπαθείς Ανοσοανεπάρκειες (IUIS) το 2022, αποτελούν μία από τις 10 (δέκα) ομάδες των Πρωτοπαθών Ανοσοανεπαρκειών (1).
Απλοανεπάρκεια της Α20 πρωτεΐνης (ΗΑ20)
Η Απλοανεπάρκεια της Α20 πρωτεΐνης (ΗΑ20) περιγράφηκε για πρώτη φορά το 2016, από
τον Zhou και συν (2) ως ένα πρώιμης έναρξης αυτοφλεγμονώδες νόσημα που οφείλεται σε
μεταλλάξεις του γονιδίου TNFAIP3.
095
096
Απλοανεπάρκεια Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της αδενοσίνης 2: Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
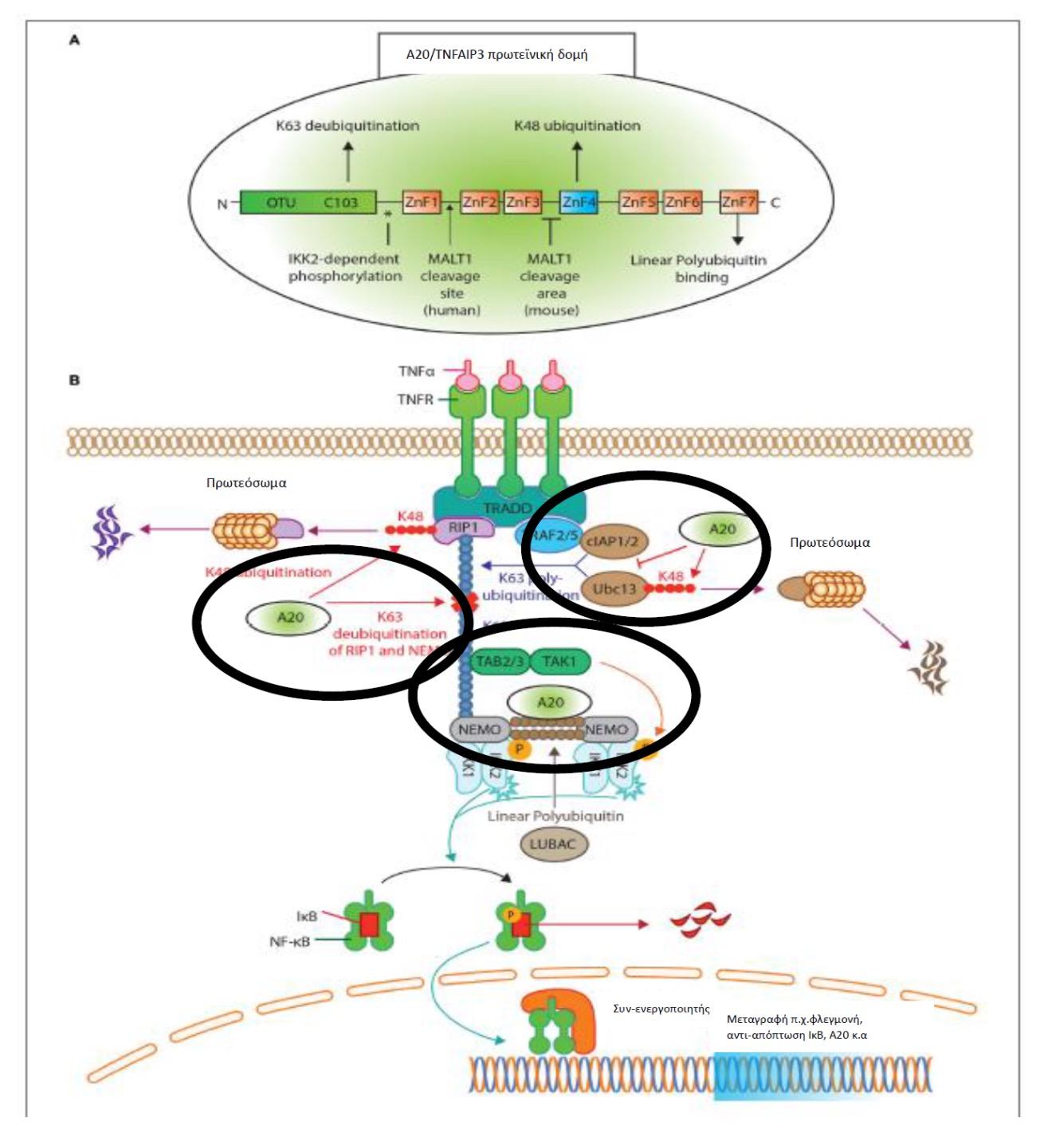
Η πρωτεΐνη A20 που κωδικοποιείται από το γονίδιο TNFAIP3 διαδραματίζει σημαντικό
ρόλο στην αρνητική ανοσορρύθμιση και καταστολή της φλεγμονής δρώντας ως ένζυμο των
ουβικουιτινών, είναι δηλαδή μία αποουβικουιτινάση. [Οι ουβικουιτίνες είναι ρυθμιστικές
πρωτεΐνες που βρίσκονται στους περισσότερους ιστούς για αυτό και χαρακτηρίζονται ως «πανταχού παρούσες» (από το ubiquitous). Επηρεάζουν τις πρωτεΐνες με πολλούς τρόπους (ουβικουιτυλίωση), όπως: επισημαίνοντας και προσδένοντας τις πρωτεΐνες του υποστρώματος
που προορίζονται για αποδόμηση μέσω του πρωτεασώματος, αλλάζοντας τη θέση τους ενδοκυτταρικά και επάγοντας ή αναστέλλοντας τη δραστηριότητά τους με αλληλεπιδράσεις
μεταξύ τους (3)]. Η μετάλλαξη της πρωτεΐνης TNFAIP3/ Α20 οδηγεί στην απώλεια της ενζυμικής
λειτουργίας ως αποουβικουιτινάση και συνεπώς στην αναποτελεσματική κατασταλτική δράση
των ουβικουιτινών στα φλεγμονογόνα μόρια «ενεργοποιητής του πυρηνικού παράγοντα NFκB(NF-κB essential modifier – NEMO)», και «παράγοντας 6» που σχετίζεται με τον υποδοχέα
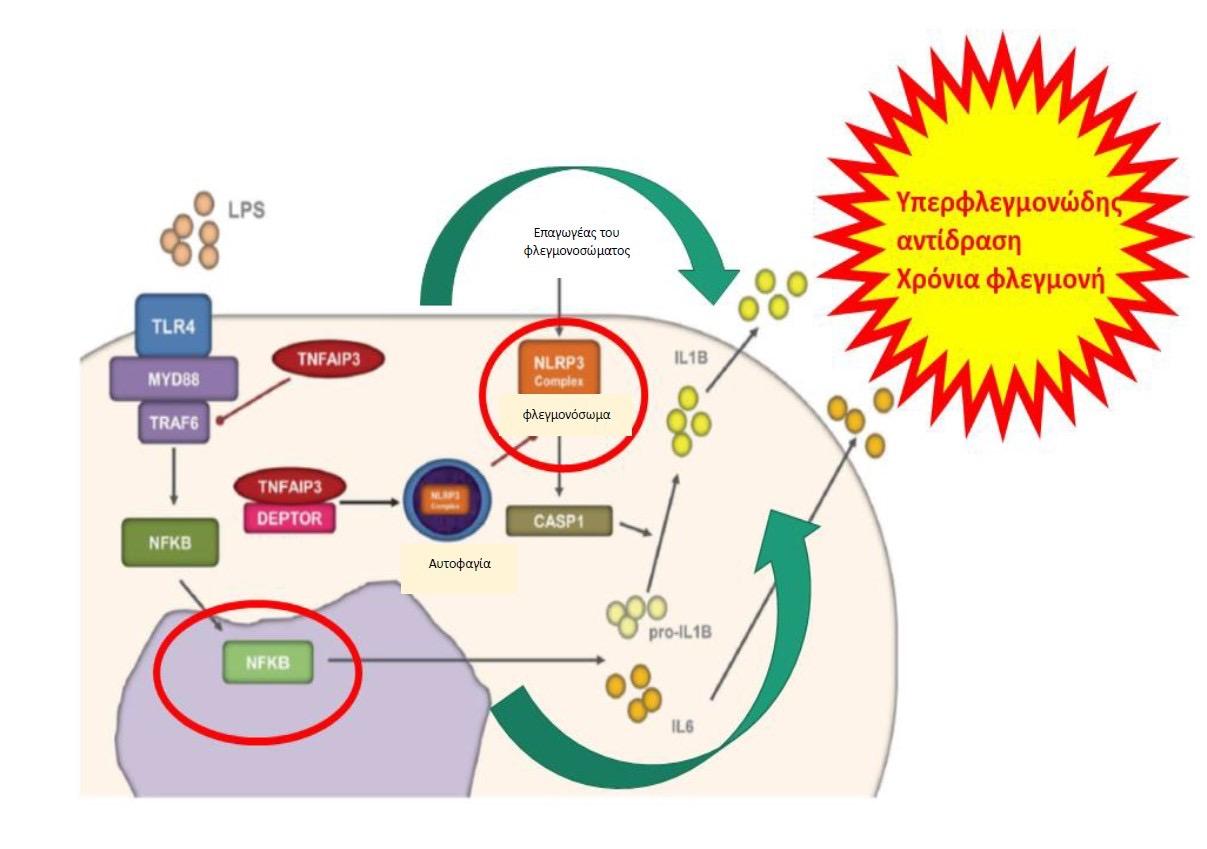
του TNF (TNF receptor-associated factor 6 - TRAF6). (4), (Εικ. 1). Αυτό έχει ως αποτέλεσμα
να απουσιάζει η καταστολή του φλεγμονογόνου μονοπατιού ενεργοποίησης του NFκ-B ή
να ενισχύεται η φλεγμονώδης δραστηριότητα της πρωτεΐνης NLRP3 του φλεγμονοσώματος,
με κατάληξη την υπέρμετρη έκκριση φλεγμονογόνων κυτταροκινών (IL-1β,IL-6, IL-18 και
TNF-α) (5), (Εικ. 2). Μέχρι σήμερα, έχουν αναφερθεί στη βιβλιογραφία 24 διαφορετικές
μεταλλάξεις που προκαλούν νόσο, 14 κληρονομούμενες με τον αυτοσωματικό επικρατούντα
χαρακτήρα και 10 με de novo μετάλλαξη (6).
Εικόνα 1. A,Β Δομή και λειτουργία της πρωτεϊνης TNFAIP3 (TNF-a Induced Protein 3)/Α20
μέσω του TNFR (Tumor Necrosis Factor Receptor). Πηγή: Τροποποιημένο από (4)
Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της αδενοσίνης 2: Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
Κατάλογος
συντομογραφιών
ADA2 enzyme: Adenosine deaminase 2 enzyme, ένζυμο της απαμινάσης της αδενοσίνης
ADA2: Deficiency of adenosine deaminase 2 (ADA2) enzyme
ΗΑ20: Haploinsufficiency of A20 protein, Aπλοανεπάρκεια της Α20 πρωτεΐνης
JAK: janus kinase, janus κινάση
IL: Interleukin, ιντερλευκίνη
NF-κB: Nuclear factor of activated B cells
NEMO: NF-κB essential modifier, NF-κB απαραίτητος ρυθμιστικός παράγοντας
NLRP3: NOD-like receptor protein 3
TNF: Tumor necrosis factor, παράγοντας νέκρωσης των όγκων
TNFAIP3: TNF Alpha Induced Protein 3
TRAF6: Tumor necrosis factor receptor-associated factor 6
Εικόνα 2. Σχηματική αναπαράσταση της αναποτελεσματικής καταστολής του NFκ-B σηματοδοτικού μονοπατιού και/ή της ενίσχυσης της φλεγμονώδους δραστηριότητας της
NLRP3 πρωτεΐνης του φλεγμονοσώματος που έχει ως αποτέλεσμα την υπερφλεγμονώδη
αντίδραση ή την χρόνια φλεγμονή που προκαλείται από τη μετάλλαξη της πρωτεΐνης
TNFAIP3. Πηγή: Τροποποιημένο από (5)
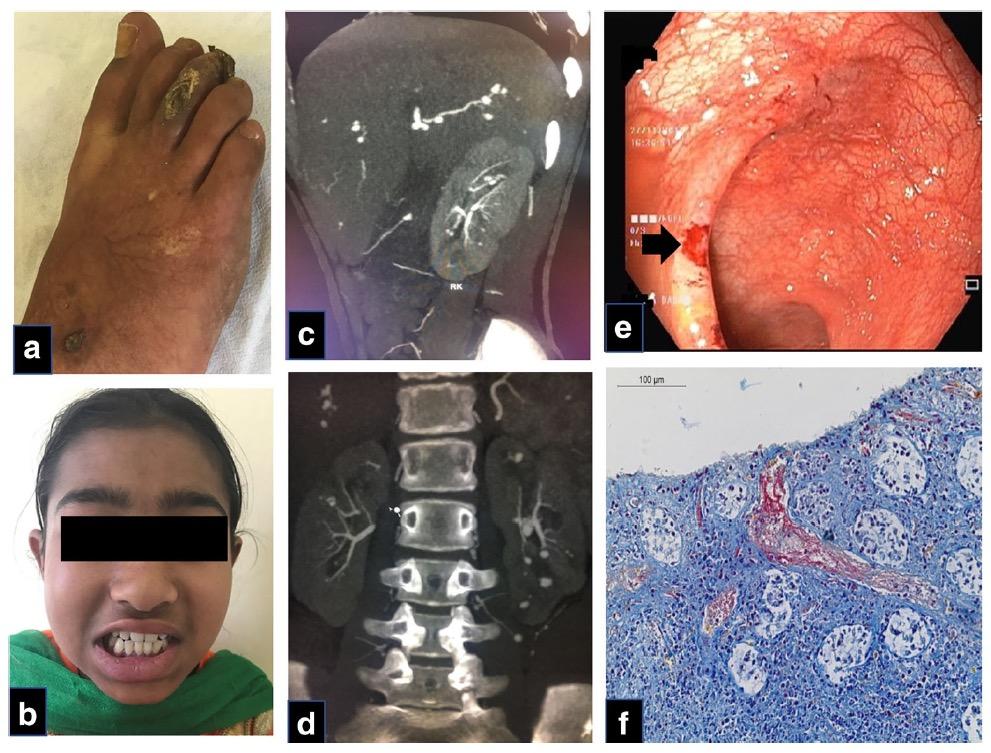
Κλινική εικόνα. Οι κλινικές εκδηλώσεις της ΗΑ20 είναι παρόμοιες με αυτές του συνδρόμου Αδαμαντιάδη-Bechet με βασική διαφορά την πρωϊμότερη εμφάνιση των συμπτωμάτων και ειδικότερα σε ηλικία μικρότερη των 10 ετών με ένα μέσο όρο έναρξης την ηλικία των 6,3 ετών (7). Η αναλογία άρρενα:θήλεα είναι 1:2. Πυρετός και λιποδυστροφία μπορεί να εκδηλωθούν σε κάποιους ασθενείς αλλά το κυριότερο χαρακτηριστικό της νόσου, όπως και στη νόσο Αδαμαντιάδη-Beçhet, είναι τα υποτροπιάζοντα, επώδυνα έλκη κατά μήκος του γαστρεντερικού σωλήνα, στο βλεννογόνο του στόματος καθώς και στα γεννητικά όργανα. Άλλα συχνά συμπτώματα, που εκδηλώνονται σε διάφορες φάσεις της νόσου, είναι οι γαστρεντερικές εκδηλώσεις (κοιλιακό άλγος, διάρροια), οι αρθραλγίες, η πολυαρθρίτιδα και οι δερματικές εκδηλώσεις (μη ειδικό εξάνθημα, ερυθηματώδες έως φολιδωτό, δισκοειδές, ψωρίαση, θυλακίτιδα, φλύκταινες, αποστήματα). Λιγότερα συχνά αναφέρεται οφθαλμοπάθεια, καρδιακή συμμετοχή και νεφρικές εκδηλώσεις (πρωτεϊνουρία, αιματουρία) (6,7,8,9), (Εικ. 3, Εικ. 4).
Μέχρι σήμερα δεν υπάρχει ειδική θεραπεία για την ΗΑ20. Η αντιμετώπιση βασίζεται στον κύριο κλινικό φαινότυπο του ασθενούς. Για την καταστολή της φλεγμονής, έχουν χορηγηθεί μέχρι σήμερα, κορτικοστεροειδή, διάφορα συνήθη ανοσοκατασταλτικά (κυκλοσπορίνη, μεθοτρεξάτη, αζαθειοπρίνη, μυκοφαινολικό μοφετίλ) καθώς και κολχικίνη με σχετικά καλά έως τώρα αποτελέσματα. Επίσης, έχουν δοκιμαστεί οι βιολογικοί παράγοντες (κατά των φλεγμονογόνων κυτταροκινών), anti-TNF-a, anti-IL-1 και anti-IL-6. Επίσης, τα AntiCD20 μονοκλωνικά αντισώματα (rituximab) και οι JAK-1, JAK-3 αναστολείς. Σε έναν μικρό αριθμό ασθενών έχει γίνει μεταμόσχευση αρχέγονων αιμοποιητικών κυττάρων (HSCT) με ενθαρρυντικά έως τώρα αποτελέσματα (7). Μελλοντικά, η ταυτοποίηση περισσότερων
ασθενών με ΗΑ20 θα διευρύνει το φαινοτυπικό φάσμα της νόσου και ίσως επιτρέψει τη χρήση πιο εξειδικευμένης θεραπείας.
097 Απλοανεπάρκεια
098
Απλοανεπάρκεια Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της αδενοσίνης 2: Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
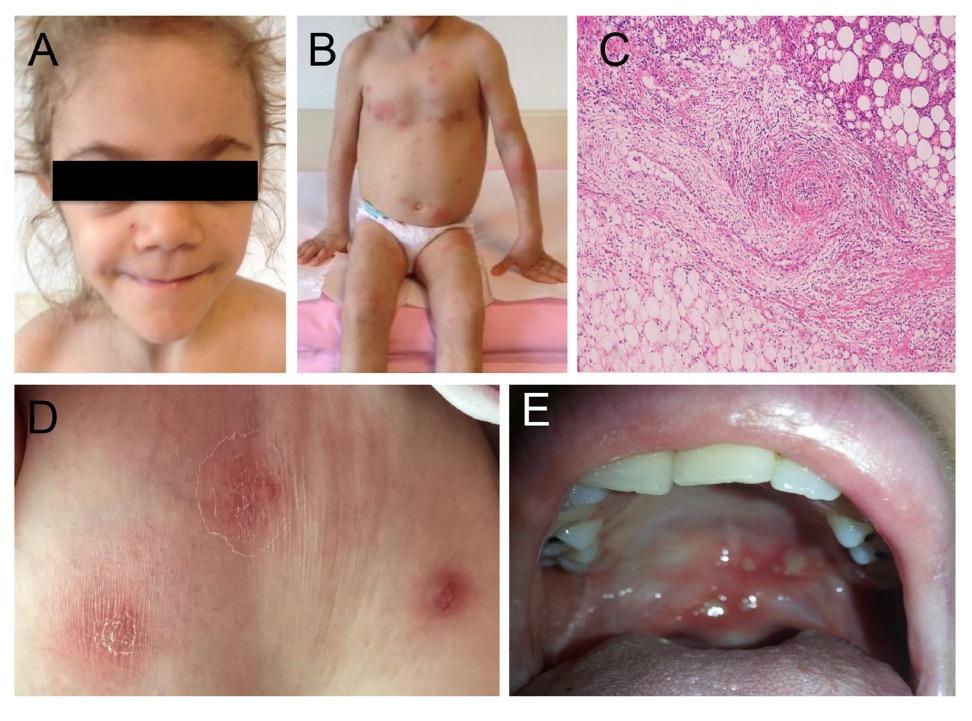
Εικόνα 3. Κλινικές και απεικονιστικές εκδηλώσεις της Απλοανεπάρκειας της Α20 πρωτεΐνης A. Λιποδυστροφία B. Ερυθηματώδεις δερματικές πλάκες και υποδόρια οζίδια C. Βιοψία του
δέρματος: πυκνή φλεγμονώδης διήθηση σε υποδόριους ιστούς, υποδόρια λοβιακή ατροφία ή λιποδυστροφία, αγγειίτιδα σε μεσαίου μεγέθους αρτηρία D. Δερματικά αποστήματα E. Αφθώδη έλκη στην υπερώα. . Πηγή: Τροποποιημένο από (8)
Εικόνα 4. Κλινικές και απεικονιστικές εκδηλώσεις της Απλοανεπάρκειας της Α20 πρωτεΐνης
A. α) Έλκη και στένωση στο ανιόν κόλον b) Έλκος στην εντερική αναστόμωση και στένωση
στο εγκάρσιο κόλον Β. Βιοψία: διατοιχωματική φλεγμονή του εντέρου με απώλεια
των κρυπτών και υπερπλασία του ινώδους ιστού C. Αξονική εντερογραφία: πάχυνση
του τοιχώματος και στένωση στην καμπυλότητα της ηπατικής καμπής D. Mαγνητική
αγγειογραφία (MRA): πολλαπλές αρτηριακές στενώσεις στα άνω και κάτω άκρα. Πηγή:
Τροποποιημένο από (9)
Ανεπάρκεια της πρωτεΐνης «απαμινάση της αδενοσίνης 2» (Adenosine Deaminase 2 deficiency, DADA2)
Η ανεπάρκεια της ADA2 (Adenosine Deaminase 2) περιγράφηκε για πρώτη φορά από
δυο ανεξάρτητες ερευνητικές ομάδες το 2014 από τον Zhou και συν. (10) και την NavonElkan και συν. (11) ως ένα μονογονιδιακό νόσημα, παρόμοιο με την οζώδη πολυαρτηρίτιδα.
Το γονίδιο της ADA2 (CECR1, Cat Eye syndrome Chromosome Region 1 gene) εδράζεται
στο χρωμόσωμα 22q11.1 και κωδικοποιεί την πρωτεΐνη ADA2. Η ADA1 εκφράζεται σε όλα
τα κύτταρα και είναι η κύρια ενδοκυττάρια ADA περισσότερο γνωστή γιατί η ανεπάρκειά
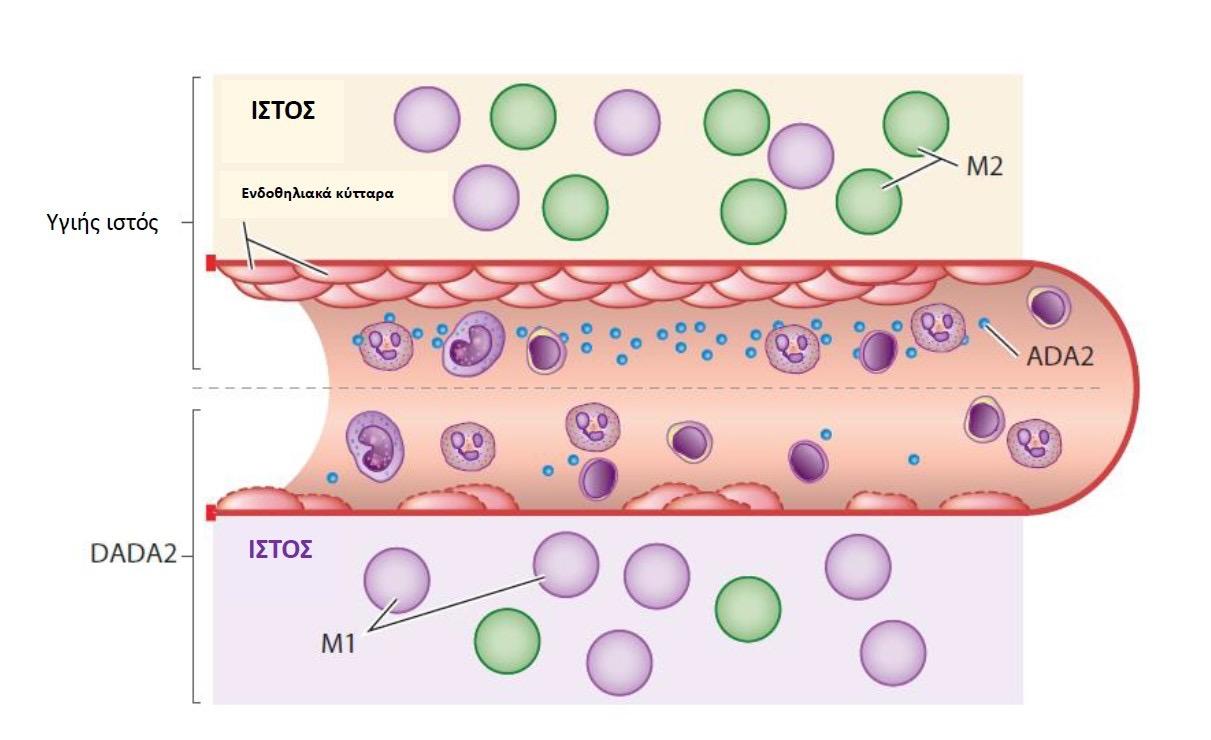
της οδηγεί στη βαριά συνδυασμένη ανοσοανεπάρκεια (Severe Combined Immunodeficiency-SCID). Αντιθέτως, η ADA2 εκφράζεται κυρίως στα κύτταρα της μυελικής σειράς και έχει καθοριστικό ρόλο στον πολλαπλασιασμό των μονοκυττάρων, στη διαφοροποίηση των μακροφάγων προς Μ1 υποπληθυσμούς με φλεγμονώδη δράση και στον πολλαπλασιασμό των CD4+ T λεμφοκυττάρων (12). Επίσης, η ADA2 λειτουργεί ως αυξητικός παράγοντας για τα ενδοθηλιακά κύτταρα και τα Μ2 μακροφάγα. Η παθοφυσιολογία της νόσου βασίζεται στο ότι, τα μειωμένα επίπεδα ADA2 στο αίμα οδηγούν σε μειωμένη ακεραιότητα του ενδοθηλίου και διαταραχή στην ισορροπία των Μ1/Μ2 μακροφάγων με υπερίσχυση των Μ1, που είναι φλεγμονογόνα. Έτσι προκαλείται ένας φαύλος κύκλος αγγειίτιδας και φλεγμονής (15), (Εικ. 6).
Εικόνα 6. Παθοφυσιολογία της DADA2. Πηγή: Τροποποιημένο από (15)
Η DADA2 κληρονομείται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα και μέχρι σήμερα
έχουν αναφερθεί 140 παθογόνες μεταλλάξεις. Η πιο συχνή μετάλλαξη στη Μέση Ανατολή
είναι η p.Gly47Arg, η οποία έχει ανιχνευθεί σε ποσοστό 10% φορέων σε γεωργιανόεβραϊκό πληθυσμό. Ακολουθεί η μετάλλαξη p.Arg169Gln με αυξημένη συχνότητα φορείας (1:500) σε πληθυσμούς της Βόρειας και Δυτικής Ευρώπης (Ολλανδία, Φιλανδία, Βέλγιο). Η
συχνότητα ανίχνευσης μεταλλάξεων σε αφρικανικούς, ασιατικούς και λατίνο/ισπανόφωνους
πληθυσμούς είναι συγκριτικά μικρότερη. Οι μέχρι τώρα δημοσιευμένες σειρές ασθενών με DADA2 αποδεικνύουν ότι δεν μπορεί να υπάρξει συσχέτιση μεταξύ γονοτύπου και φαινοτύπου καθώς υπάρχει μεγάλη φαινοτυπική ετερογένεια ακόμη και σε ασθενείς με την ίδια μετάλλαξη (και σε συγγενείς) κάτι που υποδηλώνει την επίδραση περιβαλλοντικών και επιγενετικών παραγόντων στον φαινότυπο και στη σοβαρότητα της νόσου (12,13), (Εικ. 5).
Σύμφωνα με μαθηματικά υπολογιστικά μοντέλα, η εκτιμώμενη φορεία της DADA2 είναι 1/236
άτομα και ο αναμενόμενος επιπολασμός της νόσου ~1 στα 222.000 άτομα (13).
099
Απλοανεπάρκεια Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της αδενοσίνης 2: Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
Απλοανεπάρκεια Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της αδενοσίνης 2:
Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
Εικόνα 5. Σχηματική απεικόνιση των μεταλλάξεων που σχετίζονται με νόσο στο ADA2
γονίδιο/πρωτεΐνη. Πηγή: Τροποποιημένο από (12)
Κλινική εικόνα. Η έναρξη των συμπτωμάτων της νόσου τοποθετείται στην πρώτη δεκαετία
της ζωής μπορεί όμως να συμβεί ακόμη και μέσα στον 1ο χρόνο της ζωής. H κλινική της
εικόνα χαρακτηρίζεται από διαλείποντα πυρετό με εκδηλώσεις αγγειίτιδας μικρού και
μεσαίου μεγέθους αγγείων από διάφορα συστήματα όπως συμβαίνει και στην οζώδη πολυαρτηρίτιδα, η οποία εμφανίζεται σε μεγαλύτερα παιδιά και εφήβους. Συνοδά ευρήματα
μπορεί να υπάρχουν από το δέρμα (δικτυωτή πελίωση, αιμορραγικό νεκρωτικό εξάνθημα, έλκη, νέκρωση, γάγγραινα). Οι νευρολογικές εκδηλώσεις με λανθάνοντα ισχαιμικά και /ή
αιμορραγικά εγκεφαλικά επεισόδια μπορεί να είναι προέχουσες. Λόγω της αγγειίτιδας, μπορεί ακόμη να συνυπάρχει διόγκωση του ήπατος και του σπλήνα, συμμετοχή του γαστρεντερικού (κοιλιακό άλγος, μεσεντέρια ισχαιμία, παγκρεατίτιδα), των νεφρών (αρτηριακή υπέρταση) και
του καρδιαγγειακού συστήματος (ανευρύσματα στεφανιαίων, διατατική μυοκαρδιοπάθεια)
(11,16,17), (Εικ. 7, 8, 9). Οι ασθενείς μπορεί επίσης να παρουσιάζουν αιματολογικές διαταραχές
(λεμφοπενία) και ανοσοανεπάρκεια (υπογαμμασφαιριναιμία και λεμφοϋπερπλασία).
Σε κάποιους ασθενείς, συνυπάρχει και καθυστέρηση της σωματικής αύξησης (18). Η
εκδήλωση φαινοτύπου οζώδους πολυαρτηρίτιδας σε μικρή ηλικία θέτει την υποψία της
νόσου. Η διάγνωση στηρίζεται στη μέτρηση της δραστηριότητας του ενζύμου στο πλάσμα
και επιβεβαιώνεται με τη γονοτύπηση (NGS). Η γενετική διάγνωση πρέπει να ακολουθείται
από screening τόσο προγεννητικό σε επόμενη κύηση, όσο και των γονέων και των αδερφών
της οικογένειας. Η θεραπεία είναι ανάλογη με τις κλινικές εκδηλώσεις και τη βαρύτητα της νόσου. Έχουν χορηγηθεί κορτικοστεροειδή μαζί με συμβατικά ανοσοτροποποιητικά (μεθοτρεξάτη, μυκοφαινολικό μοφετίλ), βιολογικοί παράγοντες (κυρίως anti-TNF και antiIL6) και σε ανθεκτικές περιπτώσεις, διενεργήθηκε μεταμόσχευση αρχέγονων αιμοποιητικών
κυττάρων (17).
Συμπερασματικά, σε παιδιατρικούς ασθενείς που εκδηλώνουν νoσήματα με χαρακτηριστικά
αυτοφλεγμονής ή και αυτοανοσίας σε πολύ μικρή ηλικία πρέπει να τίθεται η υποψία
αυτοφλεγμονώδους νοσήματος και να διενεργείται αλληλούχιση DNA νέας γενιάς (NGS= Next Generation Sequencing). Με τη βοήθειά της έχουν αναγνωριστεί πολλά νέα μονογονιδιακά
αυτοφλεγμονώδη νοσήματα καθώς και οι παθογενετικοί μηχανισμοί τους. H έγκαιρη διάγνωση είναι πολύ σημαντική γιατί οδηγεί στην κατάλληλη και στοχευμένη θεραπεία.
100
Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της αδενοσίνης 2: Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
Εικόνα 7. Κλινικές και απεικονιστικές εκδηλώσεις της DADA2 Α. Νέκρωση άκρων δακτύλων Β. Φαινόμενο Raynaud και δικτυωτή πελίωση C. Ανεύρυσμα σε κλάδο της κοιλιακής αορτής (αγγειογραφία) D. Βιοψία άνω μεσεντέριας αρτηρίας: περιαρτηρίτιδα, ινώδης νέκρωση, καταστροφή του μέσου και έσω ελαστικού πετάλου των αγγειακού τοιχώματος. Πηγή: Τροποποιημένο από (11)
Εικόνα 8. Δικτυωτή πελίωση (livedo racemosa) στο κάτω άκρο ασθενούς με DADA2. Πηγή: Τροποποιημένο από (16)
101 Απλοανεπάρκεια
Απλοανεπάρκεια Α20 πρωτεΐνης & Ανεπάρκεια
Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
Εικόνα 9. Κλινικές και απεικονιστικές εκδηλώσεις της DADA2 a. Γαγγραινώδης νέκρωση
3ου δακτύλου άκρου ποδός b. Παράλυση προσωπικού νεύρου c. Αξονική αγγειογραφία:
ηπατικά και νεφρικά ανευρύσματα d. Αξονική αγγειογραφία: νεφρικά ανευρύσματα και
έμφρακτα e. Έλκη πρωκτού σε ασθενή με μεσεντέρια ισχαιμία (σιγμοειδοσκόπηση) f.
Βιοψία από το σιγμοειδές κόλον: ινώδης νέκρωση με θρόμβωση. Πηγή: Τροποποιημένο από (17)
Βιβλιογραφία
1. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022 Jun 24.
2. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2016 Jan;48(1):67-73.
3. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem (1998) 67:425–79.
4. Das T, Chen Z, Hendriks RW, Kool M. A20/Tumor Necrosis Factor α-Induced Protein 3 in Immune Cells Controls Development of Autoinflammation and Autoimmunity: Lessons from Mouse Models. Front Immunol. 2018 Feb 21;9:104.
5. Zhai Y, Lin P, Feng Z, Lu H, Han Q, Chen J, et al. TNFAIP3-DEPTOR complex regulates inflammasome secretion through autophagy in ankylosing spondylitis monocytes. Autophagy. 2018;14(9):1629-1643.
6. Yu MP, Xu XS, Zhou Q, Deuitch N, Lu MP. Haploinsufficiency of A20 (HA20): updates on the genetics, phenotype, pathogenesis and treatment. World J Pediatr. 2020 Dec;16(6):575-584.
7. Zhang D, Su G, Zhou Z, Lai J. Clinical characteristics and genetic analysis of A20 haploinsufficiency. Pediatr Rheumatol Online J. 2021;19(1):75.
8. Aksentijevich I, Zhou Q. NF-κB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front Immunol. 2017 Apr 19;8:399.
9. Chen Y, Huang H, He Y, Chen M, Seidler U, Tian D, et al. A20 Haploinsufficiency in a Chinese Patient With Intestinal Behcet's Disease-Like Symptoms: A Case Report. Front Immunol. 2020 Jul 3;11:1414.
10. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke
102
της απαμινάσης της αδενοσίνης 2:
and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014 Mar 6;370(10):91120.
11. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014 Mar 6;370(10):921-
31.
12. Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J Clin Immunol. 2018 Jul;38(5):569-578.
13. Aksentijevich I, Sampaio Moura N, Barron K. Adenosine Deaminase 2 Deficiency. 2019 Aug 8. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022.
14. Jee H, Huang Z, Baxter S, Huang Y, Taylor ML, Henderson LA, et al. Comprehensive analysis of ADA2 genetic variants and estimation of carrier frequency driven by a functionbased approach. J Allergy Clin Immunol. 2022 Jan;149(1):379-387.
15. Stoffels M, Kastner DL. Old Dogs, New Tricks: Monogenic Autoinflammatory Disease Unleashed. Annu Rev Genomics Hum Genet. 2016 Aug 31;17:245-72.
16. Clarke K, Campbell C, Omoyinmi E, Hong Y, Al Obaidi M, Sebire N, et al. Testicular ischemia in deficiency of adenosine deaminase 2 (DADA2). Pediatr Rheumatol Online J. 2019 Jul 10;17(1):39.
17. Pinto B, Deo P, Sharma S, Syal A, Sharma A. Expanding spectrum of DADA2: a review of phenotypes, genetics, pathogenesis and treatment. Clin Rheumatol. 2021 Oct;40(10):38833896.
18. Moens L, Hershfield M, Arts K, Aksentijevich I, Meyts I. Human adenosine deaminase 2 deficiency: A multi-faceted inborn error of immunity. Immunol Rev. 2019 Jan;287(1):62-72.
103
Απλοανεπάρκεια Α20 πρωτεΐνης & Ανεπάρκεια της απαμινάσης της αδενοσίνης 2: Aνοσοανεπάρκειες ή αυτοφλεγμονώδη νοσήματα;
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE: Νέα
μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών
νοσημάτων
Πολυξένη Πρατσίδου-Γκέρτση
Περίληψη
Οι Ιντερφερονοπάθειες αποτελούν ετερογενή ομάδα αυτοφλεγμονωδών νοσημάτων που
οφείλονται σε μεταλλάξεις (απορρυθμισμένες δηλαδή γονιδιακές εκφράσεις) της παραγωγής
ή/ και της λειτουργίας των ιντερφερονών τύπου Ι (IFN-Ι). Είναι πολυσυστηματικά νοσήματα
με εκδηλώσεις πρώιμης ηλικιακής έναρξης, ενδεικτικές συστηματικής αγγειίτιδας και έχουν
αυξημένη νοσηρότητα και θνητότητα. Η έγκαιρη αναγνώρισή τους στηρίζεται στην αποτύπωση
των μεταλλαγμένων γονιδίων της IFN-Ι ενώ η εκτίμηση του “score ενεργότητας” των
εμπλεκόμενων γονιδίων βοηθάει στην αντιμετώπιση των ασθενών με τη χορήγηση βιολογικών παραγόντων που βελτιώνουν την έκβασή τους. Το πρότυπο των ιντερφερονοπαθειών είναι το σύνδρομο Aicardi Goutières (AGS) και από τις πρωτεασωμοπάθειες (μία υποομάδα των ιντερφερονοπαθειών), το σύνδρομο CANDLE (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature). Στο AGS εμπλέκονται μεταλλαγμένα γονίδια του μονοπατιού αναγνώρισης και επεξεργασίας νουκλεϊνικών οξέων. Το σύνδρομο εισβάλλει πρώιμα, ακόμη και από τη νεογνική περίοδο με σοβαρή εικόνα νευρο-αυτοφλεγμονής, παρόμοιας των συγγενών λοιμώξεων. Έχει εξελικτική πορεία με αυξημένη θνητότητα και
όσοι επιβιώσουν έχουν σοβαρές αναπηρίες. Συνοδές κλινικές εκδηλώσεις είναι τα χείμετλα άκρων και ώτων, ενώ τα εργαστηριακά ευρήματα θυμίζουν Συστηματικό Ερυθηματώδη Λύκο. Ανάλογες εκδηλώσεις αναγνωρίζονται και στις πρωτεασωμοπάθειες που οφείλoνται σε μεταλλάξεις στο πρωτεάσωμα και οδηγούν σε ενδοπλασματική συγκέντρωση αποδομημένων
πρωτεϊνών και ενδοκυττάριο stress. Το σύνδρομο CANDLE, εκδηλώνεται με πυρετούς, δερματώσεις, συγκάμψεις αρθρώσεων, υποδορίτιδες, προσβολή του ΚΝΣ, οφθαλμών και αργότερα μεταβολικό σύνδρομο και ηπατική στεάτωση. Η νόσος ελέγχεται μερικώς με τη χορήγηση anti-TNF παραγόντων και πρόσφατα υποσχόμενες θεραπείες είναι οι αναστολείς των κινασών JAK.
Λέξεις-κλειδιά: αυτοφλεγμονώδη νοσήματα, ιντερφερόνη, ιντερφερονοπάθειες, σύνδρομο
Aicardi Goutières, σύνδρομο CANDLE
Αλληλογραφία
Πολυξένη ΠρατσίδουΓκέρτση
Βασιλικού 13, 54636 Θεσσαλονίκη
Τ. 2310204872
K. 6944598159
e-mail: jennypratsidou. gertsi@gmail.com
Πολυξένη ΠρατσίδουΓκέρτση
Α΄ Παιδιατρική
Κλινική Α.Π.Θ. - Γενικό Νοσοκομείο Θεσσαλονίκης «Ιπποκράτειο»
104
Correspondence
Jenny Pratsidou-Gertsi
Vasilikou 13, 54636
Thessaloniki
Τ. +30 2310204872
M. +30 6944598159
e-mail: jennypratsidou. gertsi@gmail.com
Interferonopathies, Aicardi Goutières and CANDLE syndromes: new members in the genetically determined autoinflammatory diseases Family
Jenny Pratsidou-Gertsi
Abstract
Interferonopathies are heterogeneous diseases resulting from an immune dysregulation of the interferon type I (IFN-I) expression. They are attributed to genetic disorders concerning the production and/or function of IFN-I. Interferonopathies are multisystem overlapping diseases with an early onset, recurrent or persistent inflammatory phenotype indicative of vasculitis, with an increased morbidity and mortality, and no cure. An early recognition is based on gene expression profiling with Next-Generation Sequencing and IFN gene signature, and the genetic monitoring is based on the IFN activity score. Targeted therapy with contemporary biologic agents has improved their outcome. The prototype of interferonopathies is Aicardi Goutières syndrome (AGS) and of proteasomopathies- a subgroup of interferonopathies-, CANDLE syndrome (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature). AGS is attributed to mutated genes of the nucleic acid recognition and processing pathway. It has an early onset- even from the neonatal period- with a severe phenotype of neuro-autoinflammation similar to congenital infections. AGS has a progressive course and an increased mortality, with severe disabilities evident in the survivors. The escorting clinical manifestations are limp and ear pernio-like lesions and the laboratory findings reminiscent of Systemic Lupus Erythematosus. Similar manifestations are also recognized in proteasomopathies, entities due to proteasome mutations that lead to an endoplasmic concentration of degraded proteins and intracellular stress. CANDLE syndrome is characterized by fevers, dermatoses, joint contractures, panniculitis, CNS and eye involvement and later, development of metabolic syndrome and hepatic steatosis. It partially responds to biologic therapy with anti-TNF agents; recently, JAK inhibitors are promising.
Keywords: Autoinflammatory disease, interferon, interferonopathies, Aicardi Goutières syndrome, CANDLE syndrome
Jenny Pratsidou-Gertsi
1st Department of Paediatrics, Paediatric Immunology and Rheumatology Referral Center, Hippokration General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
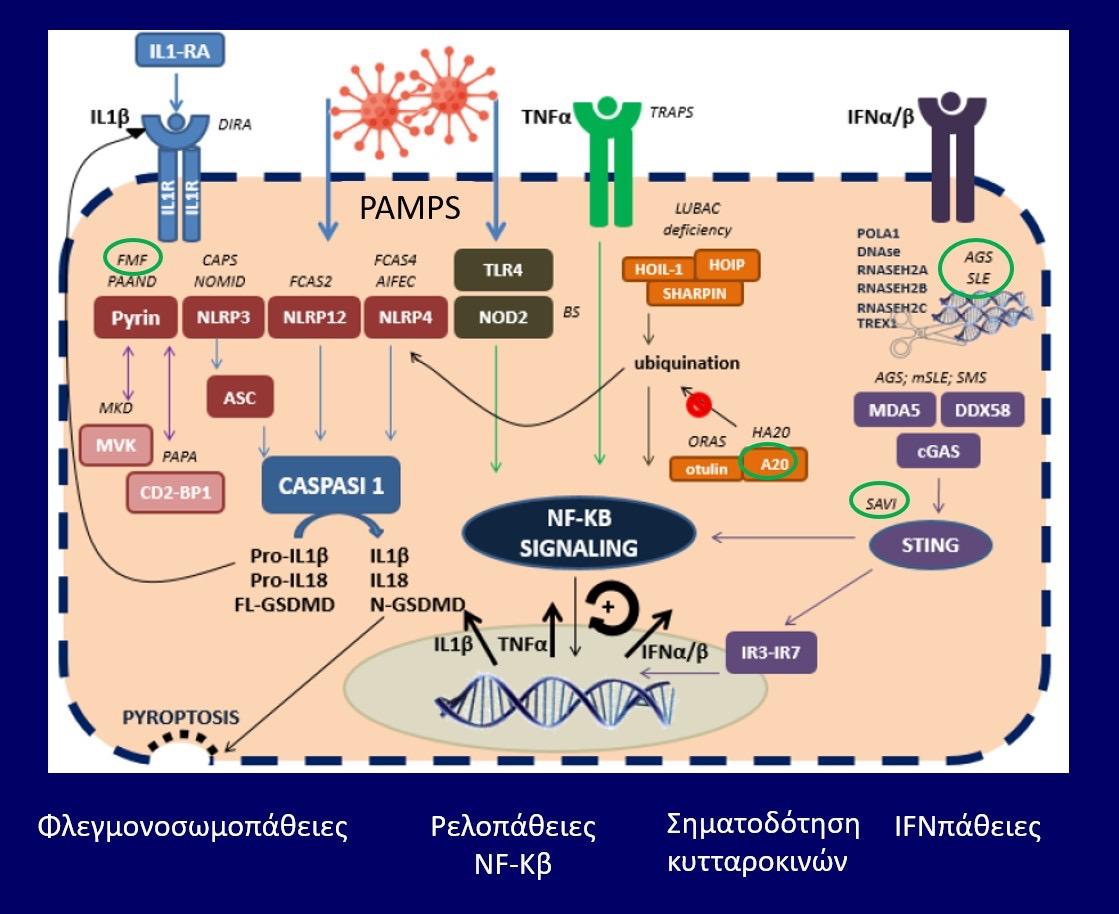
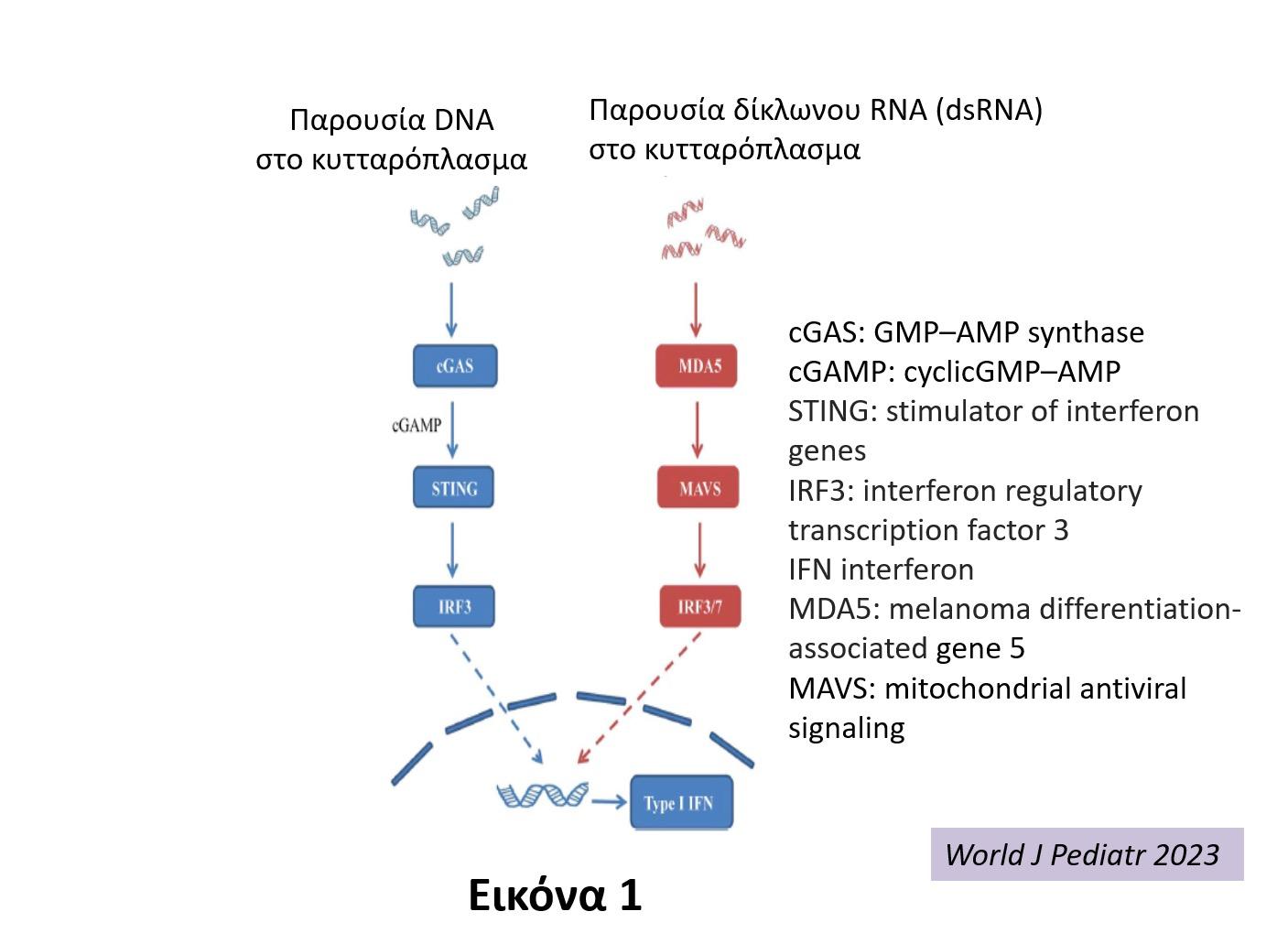
οφείλονται σε μεταλλάξεις απορρυθμισμένες δηλαδή γονιδιακές εκφράσεις της παραγωγής ή/ και της λειτουργίας των ιντερφερονών τύπου Ι (IFN-Ι). Οι IFN-Ι, εκτός των άλλων λειτουργιών τους, έχουν και δράση φλεγμονογόνων κυτταροκινών. Στο πολύπλοκο μονοπάτι της φλεγμονώδους αντίδρασης που πυροδοτείται από τις φλεγμονογόνες IFN-Ι, εμπλέκονται διαδοχικά πολλές ρυθμιστικές
πρωτεΐνες και ένζυμα: αρχικά ενεργοποιούνται οι DNA-RNA νουκλεάσες, μετά το σύμπλεγμα STING (Stimulation of Interferon Genes complex) και στη συνέχεια ρυθμιστικοί μηχανισμοί
της παραγωγής IFN, όπως οι παράγοντες IR3-IR7 (Εικ 1). (1-9). Η παρουσία μεταλλάξεων σε διάφορα γονίδια που φυσιολογικά κωδικοποιούν τις προαναφερθείσες
105
ΕΙΣΑΓΩΓΗ Οι Ιντερφερονοπάθειες είναι αυτοφλεγμονώδη νοσήματα που
πρωτεΐνες/
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE:
Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
ένζυμα που ενεργοποιούν ή/και περιορίζουν τη φλεγμονώδη αντίδραση, οδηγεί, χωρίς
εμφανές ερέθισμα, σε υπερενεργοποίηση ή /και παρατεταμένη ενεργοποίηση των γονιδίων
αυτών με αποτέλεσμα την υπερέκκριση ή/και υπερλειτουργία των IFN-I. Αυτή οδηγεί στην
εκδήλωση ετερογενών μονογονιδιακών ή διγονιδιακών αυτοφλεγμονωδών νοσημάτων, που
ονομάστηκαν το 2011 από τον ερευνητή Yanick Crow, ιντερφερονοπάθειες (IFNπάθειες).
(1,3,4,9) Αντίθετα από τις φλεγμονοσωμοπάθειες, οι IFNπάθειες μπορεί να συνοδεύονται
από την παρουσία διαφόρων αυτοαντισωμάτων, ως αποτέλεσμα της υπερπαραγωγής IFN-
I, η οποία επάγει την αυτοανοσία. (2,7,9) Αρχικά αναγνωρίστηκαν IFNπάθειες με πρώιμη
έναρξη του κλινικού φαινότυπου, ακόμη και από την ενδομήτρια ή από την πρώτη εβδομάδα
της ζωής. Γενικά, είναι πολυσυστηματικά νοσήματα, με κλινική ετερογένεια, συνήθως όμως
προβάλλουν με εικόνα πρώιμης αγγειίτιδας (Πίν.1). (3,10,11) Οι φαινότυποι μπορεί να είναι αλληλο-επικαλυπτόμενοι μεταξύ των διαφόρων συνδρόμων, όπως του μονογονιδιακού
Συστηματικού Ερυθηματώδους Λύκου, του συνδρόμου Aicardi-Goutières (Aicardi-Goutières syndrome/ AGS), του συνδρόμου SAVI (STING Associated Vasculopathy with onset in Infancy), των συνδρόμων που σχετίζονται με το πρωτεάσωμα (Proteasome-Associated Autoinflammatory Syndromes /PRAAS) και συνεχώς η ομάδα αυτή εμπλουτίζεται με νέα νοσήματα.
Οι IFNπάθειες έχουν αυξημένη νοσηρότητα και θνητότητα από την πρώιμη ακόμη ηλικία. Για
τον λόγο αυτόν, η αναγνώρισή τους είναι απαραίτητη, ώστε να αντιμετωπίζονται έγκαιρα με
νέες, στοχευμένες θεραπείες. (1-3,7,12)
Εικόνα 1. Πρωτεΐνες και ένζυμα που ενεργοποιούν το μονοπάτι παραγωγής IFN-1
Πίνακας 1. Η ετερογένεια των κλινικών εκδηλώσεων στις ιντερφερονοπάθειες
Συχνότερες μεταλλάξεις παγκοσμίως
Συστηματική φλεγμονή Κλινικές εκδηλώσεις
CANDLE/PRAAS, SAVI, AGS Υποτροπιάζων πυρετός, διόγκωση ήπατος/ σπλήνα Εργαστηριακά ευρήματα: Αυξημένη ΤΚΕ και CRP, γονιδιακή «υπογραφή» της IFN
106
Κατάλογος συντομογραφιών
ADAR1: Adenosine Deaminase Acting on RNA 1, Απαμινάση αδενοσίνης με
δράση στο RNA1
AGS: Aicardi-Goutières syndrome, σύνδρομο Aicardi-Goutières
ANA: ANtinuclear Antibodies, αντιπυρηνικά
αντισώματα
c-ANCA: AntiNeutrophil Cytoplasmic Antibodies, Κυτταροπλασματικά
αντισώματα έναντι των
ουδετεροφίλων
CANDLE: Chronic
Atypical Neutrophilic Dermatosis with Lipodystrophy and ELevated temperature/ Χρόνια άτυπη
ουδετεροφιλική δερμάτωση με λιποδυστροφία και
αυξημένη θερμοκρασία
DAMPS: Damage-Associated Molecular Patterns, Μοριακά πρότυπα που
σχετίζονται με βλάβη/
καταστροφή ή «σήματα κινδύνου»
DNAase: Deoxyribonuclease, δεοξυριβονουκλεάση
ENA: Extractable Nuclear Antibodies, αντισώματα
έναντι αντιγόνων πυρηνικού εκχυλίσματος
JAKI: Janus Kinase Inhibitors, αναστολείς των Janus κινασών
IFN: interferon, ιντερφερόνη
IFN-Ι: IFN τύπου Ι
IFNπάθειες: Ιντερφερονοπάθειες
Δερματικές εκδηλώσεις
CANDLE/PRAAS
SAVI
AGS
Εκδηλώσεις ΚΝΣ
CANDLE/PRAAS
Ουδετεροφιλική υποδορίτιδα, οζώδες εξάνθημα, ερυθροιώδες δακτυλιοειδές εξάνθημα, λιποδυστροφία
Αγγειοπάθεια (πχ Χείμετλα του Λύκου, ισχαιμία άκρων φαινόμενο Raynaud έως γάγγραινα, απώλεια δακτύλων)
Χείμετλα, αλλοιώσεις άκρων (όπως φαινόμενο Raynaud), υποδορίτιδα
Κλινικές εκδηλώσεις: κεφαλαλγία, γνωστική δυσλειτουργία
ΕΝΥ: Άσηπτη πλειοκυττάρωση Νευροαπεικόνιση: Επασβεστώσεις βασικών γαγγλίων
SAVI Νευροαπεικόνιση: Επασβεστώσεις βασικών γαγγλίων (σπάνια)
AGS
Κλινικές εκδηλώσεις: Υποξεία ή οξεία έναρξη νευρολογικών συμπτωμάτων όπως αναπτυξιακή καθυστέρηση, ευερεθιστότητα, νευρολογική επιδείνωση ή υποστροφή, δυστονία και σπαστικότητα, εστιακά κινητικά ευρήματα, εξελικτική μικροκεφαλία, σπασμοί
ΕΝΥ: Άσηπτη πλειοκυττάρωση, αυξημένα επίπεδα στο ΚΝΣ βιοδεικτών ανοσιακής ενεργοποίησης (νεοπτερίνης και τετραυδροβιοπτερίνης), αυξημένη IFNα Νευροαπεικόνιση: Λευκοεγκεφαλοπάθεια, εγκεφαλικές επασβεστώσεις, πρώιμη και ταχέως εξελισσόμενη εγκεφαλική ατροφία με ή χωρίς επασβεστώσεις,
Πνευμονικές εκδηλώσεις
CANDLE/PRAAS Πνευμονική υπέρταση χωρίς ίνωση
SAVI Διάμεση πνευμονοπάθεια με/χωρίς δευτεροπαθή πνευμονική υπέρταση
AGS
Ηπατικές εκδηλώσεις
Πνευμονική υπέρταση
CANDLE/PRAAS Αυξημένες τρανσαμινάσες, ηπατική στεάτωση
AGS
Μεταβολικές και ενδοκρινικές εκδηλώσεις
CANDLE/PRAAS
AGS
Μυοσκελετικές εκδηλώσεις
Αυξημένες τρανσαμινάσες, αυτοάνοση ηπατίτιδα
Υπέρταση, υπερλιπιδαιμία, δυσανοχή στη γλυκόζη (=μεταβολικό σύνδρομο)
Υποθυρεοειδισμός, άποιος διαβήτης, διαβήτης
CANDLE/PRAAS, SAVI, AGS Μυοσίτιδα,αρθρίτιδα, συγκάμψεις αρθρώσεων
107 Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE:
νοσημάτων
Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών
Συχνότερες μεταλλάξεις παγκοσμίως
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE:
Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων Συχνότερες μεταλλάξεις παγκοσμίως
Αύξηση και ανάπτυξη
CANDLE/PRAAS, SAVI, AGS Καθυστέρηση αύξησης, οστεόπόρωση, καθυστέρηση οστικής ανάπτυξης, καθυστέρηση ήβης
Αιματολογικές εκδηλώσεις
CANDLE/PRAAS, SAVI, AGS Αναιμία, λευκοπενίε, λεμφοπενία, και/ή θρομβοπενία
Οφθαλμολογικές εκδηλώσεις
CANDLE/PRAAS Επισκληρίτιδα και κερατίτιδα
Εκδηλώσεις από την καρδιά Καρδιομυοπάθεια, επασβεστώσεις βαλβίδων
CANDLE/PRAAS: Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature)/ Proteasome-associated autoinflammatory syndrome
SAVI: Stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy
AGS: Aicardi-Goutières syndrome
Διάγνωση. Η διαγνωστική προσέγγιση και η σφαιρική εκτίμηση «της κατάστασης νόσου»
μιας IFNπάθειας, όπως και όλων των ΑΦΝ, απαιτεί τη συνεργασία ομάδας ειδικών, αφενός
για την κλινική διάγνωση και αφετέρου για την εργαστηριακή επιβεβαίωση που γίνεται με
μοριακές δοκιμασίες (αλληλούχιση επόμενης γενιάς, Next Generation Sequencinq/NGS) και άλλες εξετάσεις σε εξειδικευμένα κέντρα, (6-9) όπως: α) το γονιδιακό αποτύπωμα με τη δυνατότητα χρωματικής αποτύπωσης της υπερέκφρασης
γονιδίων, όπως π.χ. της IFN-I που ονομάστηκε γονιδιακή «υπογραφή» ή γονιδιακό αποτύπωμα
(Interferon Gene Response Signature/IGS, Εικόνα 3). Στο αποτύπωμα αυτό, η υπερέκφραση
των γονιδίων της IFN-I αντιστοιχεί στο κόκκινο χρώμα που υπερισχύει. (9,11)
β) Πολύ πρόσφατα προστέθηκε και η μελέτη του μεταγραφώματος (αλληλούχιση του RNA στο περιφερικό αίμα) σε συνδυασμό με λογισμικά της τεχνητής νοημοσύνης (transcriptomic profiling), ως διαγνωστικό εργαλείο της πρώιμης διαφορικής διάγνωσης IFNπαθειών από άλλα περιοδικά εμπύρετα (όπως λοιμώξεις) και ρευματικά νοσήματα. (13)
γ) Το«ScoreενεργότηταςτηςIFNπάθειας», μετράται με ειδική ποσοτική PCR (reverse transcription quantitative polymerase chain reaction, RT-qPCR). Αυτό υπολογίζεται με βάση τον βαθμό ενεργοποίησης των εμπλεκόμενων γονιδίων και προκύπτει η αντίστοιχη βαθμολογία, που καλείται «Interferon Score (IS)». Ελάττωση του IS υπό αγωγή, υποδηλώνει θεραπευτική ανταπόκριση. (3)
Τα καινοτόμα αυτά εργαλεία συνέβαλαν καθοριστικά σε εξατομικευμένες θεραπευτικές
αποφάσεις, ιδίως σε περιπτώσεις αδιαφοροποίητων αυτοφλεγμονωδών νοσημάτων. (3,9-12, 14-15)
Είναι επομένως προφανές ότι για την παρακολούθηση της πορείας της νόσου, όπως και για τα περισσότερα ΑΦΝ απαιτείται συνεχής εκτίμηση από την ίδια πολυεπιστημονική ομάδα
ειδικών και, κατά περίπτωση, η συνεργασία και με άλλους επαγγελματίες υγείας. (3,15)
Αντιμετώπιση. Οι IFNπάθειες μέχρι στιγμής δεν έχουν ίαση και η αντιμετώπιση για τα
σύνδρομα Aicardi-Goutières (AGS), SAVI και πρωτεασωμοπάθειες (CANDLE/PRAAS),
στοχεύει στον έλεγχο της δραστηριότητας της νόσου ή έστω σε επίτευξη χαμηλής
δραστηριότητας ώστε να αποτραπεί η εξέλιξη και η εγκατάσταση σοβαρών βλαβών. (3,12, 16-20) Προς το παρόν, δοκιμάζονται 3 θεραπευτικά μονοπάτια: α) περιορισμός παραγωγής
εαυτών νουκλεϊνικών οξέων, β)ευόδωση της αποδόμησής τους, γ)αναστολή του καταρράκτη
108
Κατάλογος συντομογραφιών
IFIH1/MDA5: Inteferon Induced With Helicase
C Domain 1 / melanoma differentiationassociated protein 5, Ιντερφερόνη επαγόμενη
από τον τομέα 1 της
ελικάσης/διαφοροποίηση
μελανώματος που
σχετίζεται με την πρωτεΐνη
5
NGS: Next Generation Sequencing, αλληλούχηση
επόμενης γενιάς
NOMID: Neonatal-onset multisystem inflammatory disease / Νεογνικής
έναρξης πολυσυστηματικό
φλεγμονώδες σύνδρομο
PRAAS: ProteasomeAssociated Autoinflammatory Syndromes, σύνδρομα
που σχετίζονται με το
πρωτεάσωμα
RNASEH2B: Ribonuclease H2 subunit B, Υπομονάδα Β
της Ριβονουκλεάσης H2
RT-qPCR: Reverse
Transcription quantitative polymerase chain reaction Ανάστροφη
μεταγραφή με την ποσοτική αλυσιδωτή αντίδραση της
πολυμεράσης
SAMHD1: SAM-domainand HD-domain containing protein 1, SAM τομέας και
HD τομέας που περιέχουν
την πρωτεΐνη 1
SAVI: STING-Associated Vasculopathy with onset in Infancy, Αγγειοπάθεια
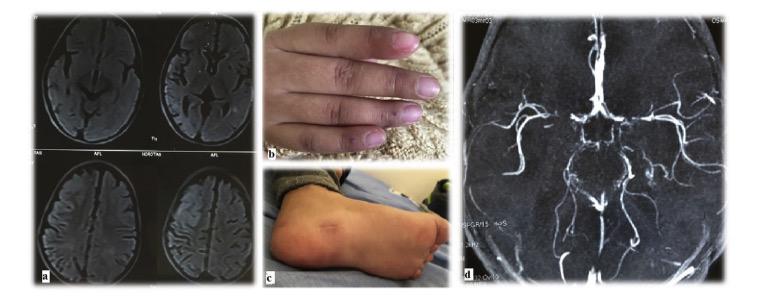
σχετιζόμενη με το STING με
έναρξη στη βρεφική ηλικία
STING: STimulator of INterferon Genes, σύμπλεγμα
που διεγείρει γονίδια
ιντερφερόνης
TORCH: Τoxoplasmosis, Οthers (Syphilis, Hepatitis B), Rubella, Cytomegalovirus (CMV), and Herpes simplex
Εικόνα 2. Αποτύπωση της αυξημένης λειτουργικής έκφρασης μεταλλαγμένων ρυθμιστικών
γονιδίων των ιντερφερονών (Gain of function mutations), σε ασθενείς με SAVI συγκριτικά

με άλλα AΦΝ (CANDLE, NOMID) και με υγιείς μάρτυρες. To έντονο κόκκινο χρώμα δηλώνει
αυξημένη έκφραση IFN.

109 Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE: Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE:
Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
σηματοδότησης της IFN. Δοκιμάστηκαν ήδη αναστολείς νουκλεοσιδών (nucleoside reversetranscriptase inhibitors) σε συνδυασμό με JAKI, που αναστέλλουν τη σηματοδότηση της IFN
με ποικίλη ανταπόκριση. Ωστόσο, συνυπάρχει ο κίνδυνος σοβαρών λοιμώξεων λόγω αυτής
της ανοσοκαταστολής, επομένως πρέπει να υπάρχει αυξημένη επαγρύπνηση και χορήγηση χημειοπροφύλαξης (κοτριμοξαζόλης). (3,7,8,16,17)
Γενικά, οι θεραπείες με βιολογικούς παράγοντες anti-TNF, anti-IL6 και πρόσφατα αναστολείς
των Janus κινασών (Janus Kinase Inhibitors, JAKI ) βελτιώνουν τα συμπτώματα των CANDLE/
PRAAS, SAVI, και AGS αλλά χρειάζεται στενή φαρμακοεπαγρύπνιση για έγκαιρη εντόπιση
ανεπιθύμητων ενεργειών. (3,7,16-18,20-22) Υπάρχουν επίσης μεμονωμένες αναφορές για αλλογενή μεταμόσχευση αιμοποιητικών βλαστοκυττάρων σε ανθεκτικές περιπτώσεις. (23)
Οι ασθενείς δεν έχουν μεγαλύτερο κίνδυνο για λοίμωξη COVID, και για τους εμβολιασμούς, συστήνεται ωστόσο να εμβολιάζονται σύμφωνα με το Εθνικό Πρόγραμμα Εμβολιασμών. (17,18)
Από την συνεχώς διευρυνόμενη ομάδα των IFNπαθειών θα περιγραφούν δύο από τα συχνότερα και πιο χαρακτηριστικά νοσήματα αυτής της οικογένειας: Το σύνδρομο Aicardi Goutières (AGS), που είναι το πρότυπο σύνδρομο IFNπαθειών και το σύνδρομο CANDLE (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature/
Χρόνια άτυπη ουδετεροφιλική δερμάτωση με λιποδυστροφία και αυξημένη θερμοκρασία), που ανήκει στις πρωτεασωμοπάθειες, μία υποομάδα των IFNπαθειών.
ΣΥΝΔΡΟΜΟ AICARDI-GOUTIERES (AGS)
Το σύνδρομο Aicardi-Goutières (AGS) προκαλείται από διάφορες μεταλλάξεις, συνήθως
απώλειας της λειτουργικής έκφρασης γονιδίων που εμπλέκονται στην αναγνώριση και επεξεργασία νουκλεϊνικών οξέων (Πίν. 2). Η πρώτη περιγραφή του δημοσιεύθηκε το 1984. Κληρονομείται συνήθως με τον αυτοσωματικό υπολειπόμενο χαρακτήρα. (3-6,19,20-22)
Χαρακτηριστική εκδήλωση του AGS είναι η νευρο-αυτοφλεγμονή. Κλινικά μιμείται σύνδρομο συγγενών λοιμώξεων (όπως το TORCH), με εξελικτική προσβολή του ΚΝΣ, χρόνια λεμφοκυτταρική μηνιγγίτιδα, αμφοτερόπλευρη σπαστικότητα, δυστονία, σπασμούς, ως και τύφλωση, μικροκεφαλία, διαταραγμένη επικοινωνία και πρόωρο θάνατο (Εικ.3).
Στην οφθαλμική συμμετοχή αναφέρεται συχνότερα καταρράκτης, γλαύκωμα, θηλίτιδα και αμφιβληστροειδική αγγειοπάθεια. (3,8, 20,22)
Ηλικία έναρξης πορεία και έκβαση του AGS
Η νόσος εισβάλλει πρώιμα με βαριά προσβολή του ΚΝΣ. Στο 20% έχει ενδομήτρια έναρξη
ή στη νεογνική ή πρώτη βρεφική περίοδο με εμφάνιση του προαναφερόμενου φαινότυπου
στο 70% των περιπτώσεων μέσα στον 1ο χρόνο ζωής. Μετά την αρχική προσβολή συνήθως
δεν ακολουθεί περαιτέρω νευρολογική επιδείνωση. Οι συνοδές κλινικές εκδηλώσεις είναι
χείμετλα άκρων και ώτων (30%) και τα συχνότερα εργαστηριακά ευρήματα στην ενεργό
νόσο, είναι πέραν των δεικτών φλεγμονής, ανοσολογικά ευρήματα που θυμίζουν νεανικό
Συστηματικό Ερυθηματώδη Λύκο (ΣΕΛ). Στη μεγαλύτερη σε αριθμό ασθενών δημοσίευση
μέχρι σήμερα, 19% αυτών κατέληξαν και 74% από τους επιζήσαντες είχαν πολύ σοβαρή
αναπηρία, ευρήματα που αναδεικνύουν την ανάγκη πρώιμης διάγνωσης και στοχευμένης
θεραπείας. (8,20) Οι βλάβες του ΚΝΣ που συχνά ξεκινούν στην ενδομήτρια περίοδο δεν είναι
αναστρέψιμες παρά τη θεραπεία. (3-8,20)
110
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE:
Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
Πίνακας 2. Συσχετίσεις φαινοτύπων στο σ. Aicardi Goutieres με τα πιο συχνά γονίδια
Κατάλογος συντομογραφιών
TREX1 3': Three Prime Repair Exonuclease 1
ΣΕΛ: Συστηματικός
Ερυθηματώδης Λύκος
Χαρακτηριστικό φαινότυπου
Νεογνική έναρξη νόσου
Αμφοτερόπλευρη νέκρωση του ραβδωτού
σώματος, σοβαρή δυστονία
Προσβολή Αγγειακό εγκεφαλικών αγγείων
Σχετιζόμενα γονίδια
TREX1
ADAR1
SAMHD1 Κληρονομούμενη σπαστική παραπληγία ADAR1, IFIH1, SAMHD1, RNASEH2B
(ενδοκράνιες στενώσεις και ανευρύσματα), έλκη στόματος, αρθοπάθεια, γλαύκωμα
Γονίδια
TREX1 3': Three Prime Repair Exonuclease 1
ADAR1: Αdenosine Deaminase Αcting on RNA 1
SAMHD1: SAM-domain- and HD-domain containing protein 1
RNASEH2B: ribonuclease H2 subunit B
SAMHD1: SAM-domain- and HD-domaincontaining protein 1, RNASEH2B: ribonuclease H2 subunit B
ΣΥΝΔΡΟΜΟ CANDLE (CHRONIC ATYPICAL NEUTROPHILIC DERMATOSIS WITH LIPODYSTROPHY AND ELEVATED TEMPERATURE SYNDROME)

Ανάλογες με τις προαναφερθείσες εκδηλώσεις μπορεί να προκαλούνται και από άλλες ιντερφερονοπάθειες, που οφείλoνται σε μεταλλάξεις στο πρωτεάσωμα, με κυριότερο το σ. CANDLE (Χρόνια άτυπη ουδετεροφιλική δερμάτωση με λιποδυστροφία και αυξημένη θερμοκρασία). Φυσιολογικά, το πρωτεάσωμα εμπλέκεται στην αποδόμηση άχρηστων, δυσλειτουργικών ή κατεστραμμένων πρωτεϊνών. Αντίθετα, το δυσλειτουργικό λόγω μεταλλάξεων πρωτεάσωμα οδηγεί σε ενδοπλασματική συγκέντρωση των προϊόντων αποδόμησης αυτών των πρωτεϊνών και σε ενδοκυττάριο stress. (15,16,24-26)
111
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE:
Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
Κληρονομείται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα και δεν έχει φυλετική ή εθνική
προδιάθεση. Εισβάλλει συνήθως με υποτροπιάζοντα πρωινό πυρετό, με/ή ετερόχρονες
χρόνιες άτυπες ουδετεροφιλικές δερματώσεις (χρόνια φλεγμονή με μικροοζώδεις
χειμετλώδεις αλλοιώσεις), μυική ατροφία, αρθρίτιδες, συγκάμψεις και υποδορίτιδα (Εικ. 4).
Μπορεί επίσης να συνυπάρχει μικροκυτταρική αναιμία, δυστροφία και από το μυοσκελετικό
αρθραλγίες, χονδρίτιδα, μυοσίτιδα που καταλήγει σε μυοπάθεια με απεικονιστικά ευρήματα
παρόμοια Δερματομυοσίτιδας. Από το ΚΝΣ περιγράφονται άσηπτη μηνιγγίτιδα, ενδοκράνιες
επασβεστώσεις και από τους οφθαλμούς οιδηματώδη βλέφαρα, ραγοειδίτιδα, και αγγειίτιδα αμφιβληστροειδούς. Στην 1η δεκαετία της ζωής, οι μισοί ασθενείς αναπτύσσουν υπέρταση, μεταβολικό σύνδρομο και ηπατική στεάτωση. (2,7, 9, 15,24-26)
Διάγνωση-Αντιμετώπιση
Όπως σε κάθε υποψία IFNπάθειας, η διάγνωση στηρίζεται στην ανεύρεση της μοριακής βλάβης.
Η τρέχουσα αντιμετώπιση περιλαμβάνει ώσεις ή συστηματική χορήγηση κορτικοστεροειδών, βιολογικών παραγόντων (anti-TNF, anti-IL6) και πρόσφατα δοκιμάστηκαν με επιτυχία οι JAKαναστολείς (Εικ. 5). (7,10,15,16, 19,24-26)
ΒΙΒΛΙΟΓΡΑΦΙΑ

1. di Donato G, d’Angelo DM, Breda L, Chiarelli F. Monogenic Autoinflammatory Diseases: State of the Art and Future Perspectives. Int J Mol Sci 2021, 22,6360. https://doi.org/10.3390/ ijms22126360.
2. Rood JE, Behrens EM. Inherited Autoinflammatory Syndromes. Annu Rev Pathol 2022 Jan 24;17:227-249
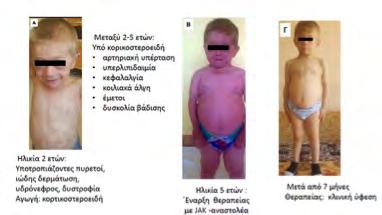
3. Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol 2022;22(8):471-483.
4. Negishi H, Taniguchi T, Yanai H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb Perspect Biol 2018;10:a028423
5. Gao KM, Marshak-Rothstein A, Fitzgerald KA Type-1 interferon-dependent and -independent mechanisms in cyclic GMP-AMP synthase-stimulator of interferon genes-driven auto-inflammation. Curr Opin Immunol 2023 Feb;80:102280. doi: 10.1016/j.coi.2022.102280.
112
Epub 2023 Jan 11.
6. Moghaddas F, Masters SL. The classification, genetic diagnosis and modelling of monogenic autoinflammatory disorders. Clin Sci 2018;132:1901-1924.
7. Eleftheriou D, Brogan PA. Genetic interferonopathies: An overview. Best Practice 2017; 31(4): 441-459
8. D'Angelo DM , Di Filippo P, Breda L , Chiarelli F. Type I Interferonopathies in Children: An Overview. Front Pediatr 2021 Mar 31;9:631329. doi: 10.3389/fped.2021.631329. eCollection 2021.
9. Papa R, Picco P, Gattorno M. The expanding pathways of autoinflammation: a lesson from the first 100 genes related to autoinflammatory manifestations. Adv Protein Chem Struct Biol 2020;120:1-44. doi: 10.1016/bs.apcsb.2019.11.001. Epub 2019 Dec 12.
10. de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Yan Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases: J Clin Invest 2020;130(4):1669–1682. https://doi.org/10.1172/JCI129301.
11. Liu Υ, Jesus ΑΑ, Marrero Β, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med 2014;371:507-18.
12. Melki I, Fremond ML. Type I Interferonopathies: From a novel concept to targeted therapies. Curr Rheum Rep 2020; 22:32 doi:10.1007/s1926-020-00909-4
13. Ha MK, Bartholomeus E, Van Os L, Dandelooy J, Leysen J, Aerts O, et al. Blood transcriptomics to facilitate diagnosis and stratification in pediatric rheumatic diseases – a proof of concept study. Pediatric Rheumatology 2022; 20:91
14. Savic S, Caseley EA, Michael F. McDermott MF. Moving towards a systems-based classification of innate immune mediated diseases. Nat Rev Rheumatol 2020; 16: 222-237
15. Miyamoto T, Honda Y, Izawa K, Kanazawa N, Kadowaki S, Ohnishi H, et al. Assessment of type I interferon signatures in undifferentiated inflammatory diseases: A Japanese multicenter experience. Front Immunol 2022 Sep 23;13:905960. doi: 10.3389/fimmu.2022.905960. eCollection 2022.
16. Rice GI, Park S, Gavazzi F, Adang LA, Ayuk LA, Van Eyck L, et al. Genetic and phenotypic spectrum associated with IFIH1 gain-of-function Hum Mutat 2020;41:837-849. doi: 10.1002/humu.23975.
17. Gedik KC, Lamot L, Romano M, Demirkaya E, Piskin D, Torreggiani S, et al. The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology. Points to Consider for Diagnosis and Management of Autoinflammatory Type I Interferonopathies: CANDLE/PRAAS, SAVI, and AGS. Arthritis & Rheumatol 2022;74:735-751. doi:10,1002/art.42087
18. Hanson EP, Lee-Kirsch MA, Montealegre Sanchez GA, Neven B, Orcesi S, Ozen S, et al. The 2021 EULAR and ACR points to consider for diagnosis and management of autoinflammatory type I interferonopathies: CANDLE/PRAAS, SAVI and AGS. Ann Rheum Dis 2022;81:601-613. doi: 10.1136/annrheumdis-2021-221814
19. Volpi S, Picco P, Caorsi R, Candotti F, Gattorno M. Type I interferonopathies in pediatric rheumatology. Pediatr Rheumatol Online J 2016 Jun 4;14(1):35
20. Dell'Isola GB, Dini G, Culpepper KL, Portwood KE, Ferrara P, Di Cara G, et al. Clinical spectrum and currently available treatment of type I interferonopathy Aicardi-Goutières syndrome. World J Pediatr 2023 Jan 17. doi: 10.1007/s12519-022-00679-2. Online ahead of print
21. Du Y, Liu M, Nigrovic PA, Dedeoglu F, Pui Y, Lee PY. Biologics and JAK inhibitors for the treatment of monogenic systemic autoinflammatory diseases in children. J Allergy Clin Immunol 2023 Jan 25;S0091-6749(22)02589-1.
22. Krusche M, Kallinich T. Autoinflammation – Unterschiede bei Kindern und Erwachsenen. [Article in German] Z Rheumatol 2021 https://doi.org/10.1007/s00393-021-01115-y Published Online: 11 Nov 2021
23. Signa S, Dell'Orso G, Gattorno M, Faraci M. Hematopoietic stem cell transplantation in systemic autoinflammatory diseases - the first one hundred transplanted patients. Expert Rev Clin Immunol 2022;18:667-689. doi: 10.1080/1744666X.2022.2078704.
24. Papendorf JJ, Krüger E, Ebstein F. Proteostasis Perturbations and Their Roles in Causing Sterile Inflammation and Autoinflammatory Diseases. Cells 2022 Apr 22;11(9):1422. doi: 10.3390/cells11091422.
113
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE: Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE: Νέα μέλη στην οικογένεια των γενετικά
25. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-offunction proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production J Clin Invest 2015;125:4196-211. doi: 10.1172/JCI81260. Epub 2015 Oct 20.
26. Boyadzhiev M, Marinov L, Boyadzhiev V, Iotova V, Aksentijevich I, Hambleton S. Disease course and treatment effects of a JAK inhibitor in a patient with CANDLE syndrome. Pediatr Rheumatol Online J. 2019 May 2;17(1):19. doi: 10.1186/s12969-019-0322-9.
114
καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
115 Ιντερφερονοπάθειες, σύνδρομα Aicardi-Goutières και CANDLE: Νέα μέλη στην οικογένεια των γενετικά καθοριζόμενων αυτοφλεγμονωδών νοσημάτων
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και
Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Άρτεμις Κουτσονικολή
Περίληψη
Ο Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) αποτελεί αυτοφλεγμο-
νώδες νόσημα, που οφείλεται σε μεταλλάξεις ενός μόνο γονιδίου. Τα περίπου 30 υπεύθυνα
γονίδια, που έχουν αναγνωριστεί μέχρι σήμερα, εμπλέκονται στη φλεγμονή και συμμετέχουν:
α. στο μονοπάτι ενεργοποίησης συμπληρώματος ή β. στο μονοπάτι σηματοδότησης
ιντερφερονών (IFN) ή γ. στη λειτουργία εξωκυττάριων DNAσών ή δ. στους μηχανισμούς
ανοσιακής ανοχής. Χαρακτηριστικά του μονοΣΕΛ είναι η πρώιμη εκδήλωση σε παιδιά
ηλικίας <5 ετών και η συχνά οικογενής εμφάνιση. Ανάλογα με το ανοσιακό μονοπάτι που
διαταράσσεται λόγω της εκάστοτε μετάλλαξης, η παθοφυσολογία, η κληρονομικότητα και
η κλινική εικόνα του νοσήματος διαφέρουν. Οι ασθενείς γενικά παρουσιάζουν συμπτώματα
από διάφορα συστήματα και ενίοτε ο κλινικός φαινότυπος θυμίζει κάποιο αυτοάνοσο νόσημα, όπως τον νεανικό ΣΕΛ. Από τις πιο χαρακτηριστικές δερματικές εκδηλώσεις είναι τα χείμετλα (chilblain lupus). Η θεραπεία αποτελεί πρόκληση, καθώς συχνά δεν υπάρχει ανταπόκριση
στην καθιερωμένη αγωγή που χρησιμοποιείται στον νΣΕΛ. Το σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Infancy) είναι σπάνιο μονογονιδιακό αυτοφλεγμονώδες
σύνδρομο. Οφείλεται σε μετάλλαξη του γονιδίου της πρωτεΐνης STING (STimulator of INterferon Genes), που οδηγεί σε αυξημένη ενεργοποίησή της, υπερπαραγωγή IFN και επαγωγή συστηματικής φλεγμονής. Συχνά εκδηλώνεται από τη νεογνική ηλικία. Μπορεί
να κληρονομείται με αυτοσωματικό επικρατούντα χαρακτήρα αλλά συνηθέστερα οφείλεται σε de novo μετάλλαξη. Χαρακτηρίζεται από δερματικές εκδηλώσεις βαριάς αγγειοπάθειας
και διάμεση πνευμονική νόσο (με δευτεροπαθή πνευμονική ίνωση). Συμπτώματα από
άλλα συστήματα ενδέχεται να συνυπάρχουν. Η πρόγνωση είναι δυσμενής λόγω κυρίως της πνευμονικής προσβολής. Η ανταπόκριση σε καθιερωμένους ανοσοτροποποιητικούς
παράγοντες είναι ανεπαρκής. Ενθαρρυντικά αποτέλεσματα προκύπτουν από τη χρήση των αναστολέων κινασών janus (JAK).
Λέξεις-κλειδιά: μονοΣΕΛ, σύνδρομο SAVI, αυτοφλεγμονώδη
Αλληλογραφία
Άρτεμις Κουτσονικολή Πανταζίδου 38, Πυλαία Τ. 2310892498
K. 6944590243
e-mail: artemis_kou@ yahoo.com
Άρτεμις Κουτσονικολή Α΄Παιδιατρική Κλινική Α.Π.Θ. - Γενικό Νοσοκομείο Θεσσαλονίκης «Ιπποκράτειο»
116
Correspondence
Artemis Koutsonikoli
Pantazidou 38, Pylaia
Τ. +30 2310892498
M. +30 6944590243
e-mail: artemis_kou@ yahoo.com
Monogenic Systemic Lupus Erythematosus (monoSLE) and SAVI syndrome
(STING-Associated Vasculopathy with onset in Infancy)
Artemis Koutsonikoli
Abstract
Monogenic Systemic Lupus Erythematosus (monoSLE) is an autoinflammatory disease, caused by a single gene’s mutation. The approximately 30 described genes are involved in inflammation and participate in the: a. complement’s activation pathway or b. interferon (IFN) signaling pathway or c. extracellular DNAses’ function or d. mechanisms of immune tolerance. Characteristics of monoSLE are the early onset in children <5 years old and the frequent familial presentation. Depending on the immune pathway, disrupted by each mutation, the pathophysiology, inheritance and clinical picture differ. Patients generally present with symptoms from multiple systems and sometimes the clinical phenotype is reminiscent of an autoimmune disease, like juvenile SLE. Among the most characteristic skin manifestations are chilblains (chilblain lupus). Treatment is challenging, as there is often no response to the standard regimens used in jSLE. SAVI syndrome (STING-Associated Vasculopathy with onset in Infancy) is a rare monogenic autoinflammatory syndrome. It is caused by a mutation in the gene encoding the protein STING (STimulator of INterferon Genes), which leads to its increased activation, overproduction of IFN and induction of systemic inflammation. The syndrome’s onset is often in the neonatal period. The manner of inheritance is autosomal dominant but de novo mutations are more common. SAVI typically manifests with cutaneous signs of severe vasculopathy and interstitial lung disease (with secondary pulmonary fibrosis). Symptoms from other systems may coexist. The prognosis is poor mainly due to the pulmonary involvement. Response to established immunomodulatory agents is inadequate. Encouraging results are emerging from the use of janus kinase (JAK) inhibitors.
Keywords: monoSLE, SAVI syndrome, autoinflammatory
Artemis Koutsonikoli
1st Department of Pediatrics, Pediatric Immunology and Rheumatology Referral Center, Hippokration Hospital, Aristotle University, Thessaloniki, Greece
Η αιτιοπαθογένεια του νεανικού Συστηματικού Ερυθηματώδους Λύκου (νΣΕΛ), όπως
και του ΣΕΛ των ενηλίκων, δεν έχει ακόμη πλήρως αποσαφηνιστεί. Γνωρίζουμε όμως ότι, όπως φαίνεται στην εικόνα 1, μια πλειάδα παραγόντων αλληλεπιδρούν μεταξύ τους, προκαλώντας δυσλειτουργία του ανοσιακού συστήματος, που με τη σειρά της οδηγεί στην ανάπτυξη φλεγμονής σε διάφορα όργανα. Ανάμεσα στους παράγοντες που ενοχοποιούνται περιλαμβάνονται και οι γονιδιακοί. Έχουν αναγνωριστεί γενετικοί πολυμορφισμοί σε αρκετά γονίδια, που επιφέρουν γενετική επιβάρυνση. Ο νΣΕΛ, λοιπόν, είναι μια πολυπαραγοντική και πολυγονιδιακή νόσος.(1)
117
ΜΟΝΟΓΟΝΙΔΙΑΚΟΣ ΣΥΣΤΗΜΑΤΙΚΟΣ ΕΡΥΘΗΜΑΤΩΔΗΣ ΛΥΚΟΣ
(μονοΣΕΛ)
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Εικόνα 1. Συνοπτική απεικόνιση της παθογένειας του νεανικού Συστηματικού Ερυθηματώδους Λύκου [Προσαρμοσμένη αναπαραγωγή από (1)]
Σε αντίθεση, ο μονογονιδιακός ΣΕΛ (μονοΣΕΛ) οφείλεται σε μεταλλάξεις σε ένα μόνο γονίδιο.
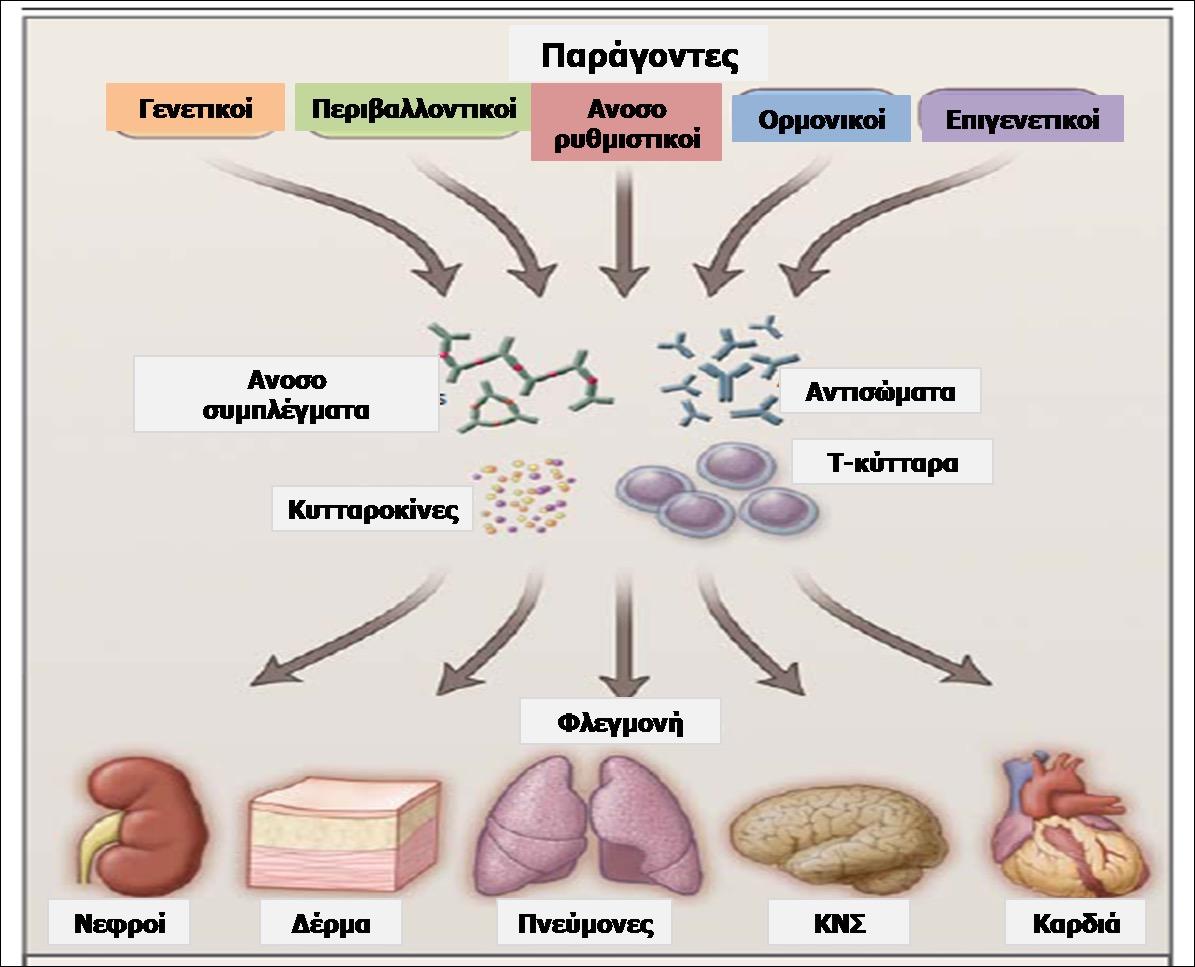
Μέχρι σήμερα έχουν αναγνωρισθεί περίπου 30 γονίδια, μεταλλάξεις των οποίων οδηγούν στην εμφάνιση της νόσου. Κάθε ασθενής φέρει μετάλλαξη σε ένα μόνο από τα γονίδια αυτά.
Τα προϊόντα των εν λόγω γονιδίων εμπλέκονται στη διεργασία της φλεγμονής και μπορεί να κατηγοριοποιηθούν στις εξής ομάδες. 1. Γονίδια που σχετίζονται με το μονοπάτι ενεργοποίησης
του συμπληρώματος. 2. Γονίδια που σχετίζονται με το μονοπάτι σηματοδότησης ιντερφερονών (IFN). 3. Γονίδια που σχετίζονται με τη λειτουργία των εξωκυττάριων DNAσών και 4. Γονίδια που σχετίζονται με τους μηχανισμούς ανοσιακής ανοχής. Η κάθε ομάδα μεταλλαγμένων
γονιδίων χαρακτηρίζεται και από διαφορές στον κλινικό φαινότυπο της νόσου.(2-5)
Διαφορές μεταξύ μονοΣΕΛ και νΣΕΛ.
Ενώ ο νΣΕΛ εμφανίζεται σε παιδιά προεφηβικής και κυρίως εφηβικής ηλικίας και προσβάλει
με μεγαλύτερη συχνότητα τα κορίτσια, ο μονοΣΕΛ εμφανίζεται σε μικρά παιδιά, συνήθως
ηλικίας μικρότερης των 5 ετών και προσβάλει ισότιμα τα 2 φύλα. Οι ασθενείς με μονοΣΕΛ
συχνά έχουν συγγενείς με παρόμοιο φαινότυπο, ενώ στο νΣΕΛ συνηθέστερα παρατηρείται
θετικό οικογενειακό ιστορικό αυτοάνοσων νόσων γενικά. Στις περιπτώσεις μονοΣΕΛ επίσης
αναφέρεται κάποιες φορές συγγένεια μεταξύ των γονέων.(2-5)
Παθοφυσιολογία.
Στην εικόνα 2 παρουσιάζεται σχηματικά η παθοφυσιολογία του μονοΣΕΛ ανάλογα με το μονοπάτι που διαταράσσεται από την εκάστοτε υπεύθυνη μετάλλαξη.(6)
118
Κατάλογος συντομογραφιών
αντι-CCP: anti-Cyclic Citrullinated Peptide, αντι-κυκλικού
κιτρουλλινιωμένου
πεπτιδίου
μονοΣΕΛ: Μονογονιδιακός
Συστηματικός
Ερυθηματώδης Λύκος
νΣΕΛ: νεανικός
Συστηματικός
Ερυθηματώδης Λύκος
ACP5/TRAP: Acid Phosphatase 5, Tartrate-resistant acid phosphatase
ADAR1: Adenosine deaminase acting on RNA 1
ANA: ANtinuclear Antibodies, αντιπυρηνικά
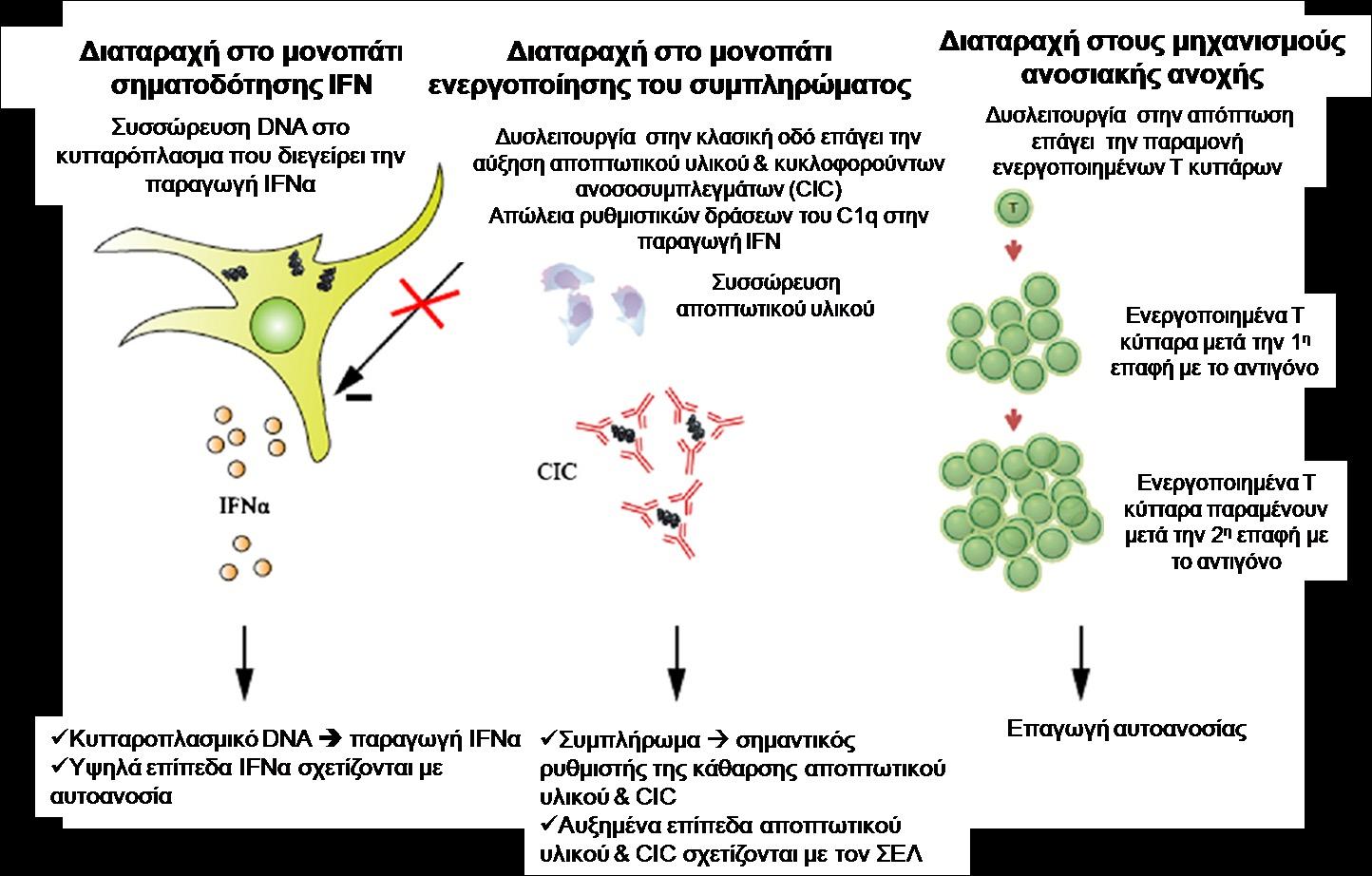
αντισώματα
CD95: Cluster of differentiation 95
CH50: hemolytic complement 50, ολική αιμολυτική δραστηριότητα
συμπληρώματος
CIC: Circulating Immune Complexes, κυκλοφορούντα ανοσοσυμπλέγματα
DNAάση: deoxyribonuclease, δεοξυριβονουκλεάση
dsDNA: double stranded deoxyribonucleic acid, δίκλωνο δεοξυριβονουκλεϊκό οξύ
IFIH1/MDA5: Interferon
Induced With Helicase
C Domain 1 / melanoma
differentiation-associated protein 5
IFN: interferon, ιντερφερόνη
JAK: janus kinase, janus κινάση
KRAS: Kirsten rat sarcoma viral oncogene homolog
PRKCD/PKC-δ: Protein kinase C delta type
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Εικόνα 2. Σχηματική απεικόνιση της παθοφυσιολογίας του μονογονιδιακού Συστηματικού Ερυθηματώδους Λύκου (μονοΣΕΛ) ανάλογα με το μονοπάτι που διαταράσσεται από την εκάστοτε υπεύθυνη μετάλλαξη. Ειδικότερα, μετάλλαξη σε γονίδιο που σχετίζεται με το μονοπάτι σηματοδότησης IFN (αριστερό μονοπάτι) μπορεί να οδηγήσει σε συσσώρευση DNA στο κυτταρόπλασμα, που με τη σειρά της διεγείρει την παραγωγή IFNα. Τα υψηλά επίπεδα IFNα σχετίζονται με την αυτοανοσία. Μετάλλαξη σε γονίδιο σχετιζόμενο με το μονοπάτι ενεργοποίησης του συμπληρώματος (κεντρικό μονοπάτι) οδηγεί σε δυσλειτουργία της κλασικής οδού του συμπληρώματος. Το συμπλήρωμα αποτελεί σημαντικό ρυθμιστή της κάθαρσης αποπτωτικού υλικού και των κυκλοφορούντων ανοσοσυμπλεγμάτων (Circulating Immune Complexes, CIC). Λόγω της δυσλειτουργίας του συσσωρεύονται αποπτωτικά υλικά και CIC, χαρακτηριστική κατάσταση στον ΣΕΛ. Μετάλλαξη σε γονίδιο που σχετίζεται με τους μηχανισμούς ανοσιακής ανοχής (δεξί μονοπάτι) μπορεί να επιφέρει δυσλειτουργία στην απόπτωση αυτοδραστικών Τ κυττάρων, τα οποία μετά την επαφή τους με το αντιγόνο, πολλαπλασιάζονται και επάγουν την αυτοανοσία. [Προσαρμοσμένη αναπαραγωγή από (6)]
Μεταλλάξεις στο μονοπάτι του συμπληρώματος στον μονοΣΕΛ.
Τα κυριότερα γονίδια, μεταλλάξεις των οποίων σχετίζονται με την εμφάνιση μονοΣΕΛ, είναι αυτά που κωδικοποιούν τα κλάσματα C1q, C1r, C1s, C2 και C4 του συμπληρώματος.
Η επίδραση της μετάλλαξης των γονιδίων αυτών είναι η μειωμένη κάθαρση αποπτωτικού υλικού και κυκλοφορούντων ανοσοσυμπλεγμάτων (Circulating Immune Complexes, CIC). Κληρονομούνται με τον αυσωματικό υπολειπόμενο χαρακτήρα. Η κλινική εικόνα είναι παρόμοια με εκείνη του νΣΕΛ και χαρακτηρίζεται από υψηλή συχνότητα δερματικών εκδηλώσεων και σοβαρή «κατάσταση νόσου» (disease status), που επιβαρύνεται ακόμη περισσότερο από τις υποτροπιάζουσες σοβαρές λοιμώξεις που συνυπάρχουν. Τα αντιπυρηνικά αντισώματα (ANtinuclear Antibodies, ANA) είναι συχνά θετικά, ενώ θετικά μπορεί να είναι και άλλα αυτοαντισώματα, χαρακτηριστικά του νΣΕΛ, όπως τα αντι-dsDNA (έναντι του δίκλωνου DNA) και τα αντι-Ro. Η ολική αιμολυτική δραστηριότητα του συμπληρώματος (CH50) είναι ελαττωμένη, ενώ τα κλάσματα C3 και C4 μπορεί, ανάλογα με τη μετάλλαξη, να είναι φυσιολογικά.(2-5)
Μεταλλάξεις στο μονοπάτι σηματοδότησης IFN.
Τα κυριότερα μεταλλαγμένα γονίδια που σχετίζονται με την εμφάνιση μονοΣΕΛ και τα προϊόντα κωδικοποίησής τους είναι τα: ACP5/TRAP, TREX1, IFIH1/MDA5, ADAR1, RNASEH2A, B, C.
Η τελική επίδραση των μεταλλάξεών τους είναι η υπερπαραγωγή IFN. Άλλες μεταλλάξεις
119
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
κληρονομούνται με τον υπολειπόμενο και άλλες με τον επικρατούντα αυτοσωματικό
χαρακτήρα. Η κλινική εικόνα χαρακτηρίζεται από επικάλυψη με φαινότυπους άλλων
συνδρόμων τόσο ως προς τις δερματικές βλάβες που είναι χαρακτηριστικές (χείμετλα), όσο
και ως προς τις εκδηλώσεις από τα άλλα συστήματα. Ο μονοΣΕΛ με χείμετλα (chilblain lupus) χαρακτηρίζεται από την εμφάνιση συχνά επώδυνων ή κνησμωδών ερυθρών ή σκούρων
ιωδών πλακών (χιονίστρες), βλατίδων ή οζιδίων μετά από έκθεση στο κρύο. Οι βλάβες
αφορούν κυρίως τα δάκτυλα, σπανιότερα τη μύτη και τα αυτιά, και μπορεί να επιπλακούν
από υπερκεράτωση και έλκη (Εικ. 3). Μια άλλη δερματική οντότητα, που μπορεί πιο σπάνια
να εμφανιστεί σε μεταλλάξεις αυτού του μονοπατιού, είναι η κληρονομική συμμετρική
δυσχρωμάτωση (Dyschromatosus symmetrica hereditaria), που χαρακτηρίζεται από υπέρ-
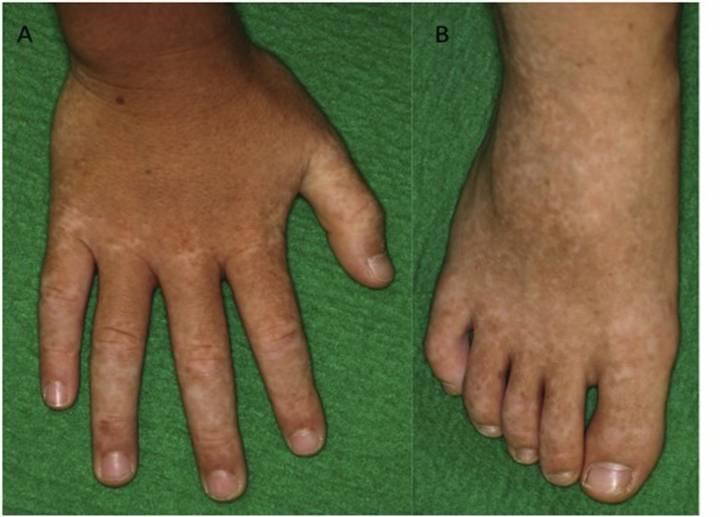
και υπό- χρωμες κηλίδες στη ραχιαία επιφάνεια χεριών και ποδιών (Εικ. 4). Σε αυτήν την κατηγορία μεταλλάξεων μπορεί να βρεθούν μερικές φορές θετικά τα αυτοαντισώματα που χαρακτηρίζουν τον νΣΕΛ. (2-5, 7-9)
Εικόνα 3. Λύκος με χείμετλα (chilblain lupus) [Αναπαραγωγή από (2,7,8)]
Εικόνα 4. Κληρονομική συμμετρική δυσχρωμάτωση [Αναπαραγωγή από (9)]
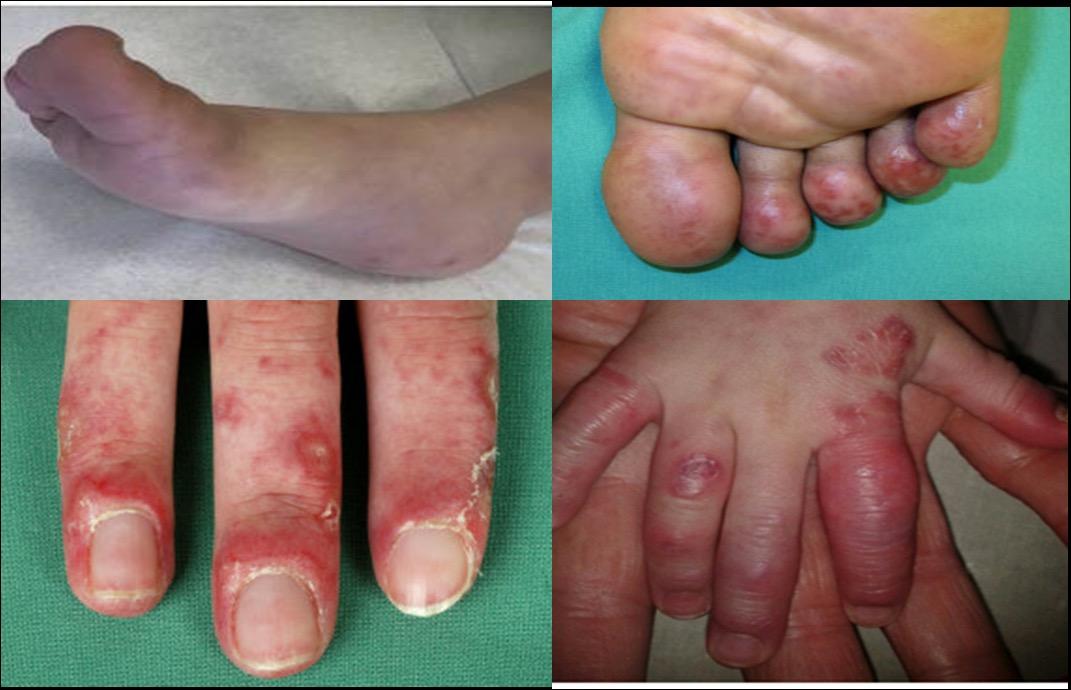
120
Κατάλογος συντομογραφιών
PTPN11: Protein Tyrosine Phosphatase Non-Receptor Type 11
RNASEH2A: Ribonuclease
H2 subunit A
SAVI: STING-Associated Vasculopathy with onset in Infancy, αγγειοπάθεια σχετιζόμενη με το STING με
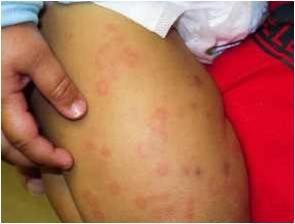
έναρξη στη βρεφική ηλικία
SHOC2: Soc-2 suppressor of clear homolog
STING: STimulator of INterferon Genes, διεγείρων τα γονίδια ιντερφερόνης
TNFRSF6: tumor necrosis factor receptor superfamily
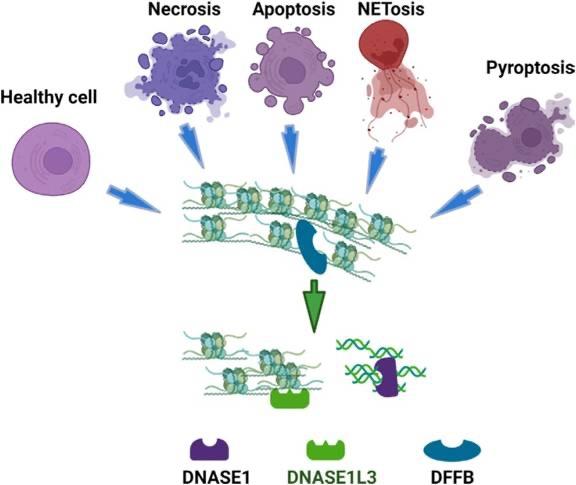
TMEM173: transmembrane protein 173
TREX1: Three Prime Repair Exonuclease 1
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Ανεπάρκεια εξωκυττάριων DNAσών.
Τα κυριότερα γονίδια των οποίων οι μεταλλάξεις σχετίζονται με την εμφάνιση μονοΣΕΛ της
κατηγορίας αυτής, είναι τα της DNASE1 και της DNASE1L3. Στην εικόνα 5 παρουσιάζεται
η δράση των κυριοτέρων εξωκυττάριων DNAσών, δηλαδή η αποδόμηση των εξωκυττάριων
νουκλεϊκών οξέων, που προκύπτουν από τις διάφορες διαδικασίες καταστροφής των κυττάρων. Οι ενοχοποιούμενες μεταλλάξεις οδηγούν σε μειωμένη αποδόμηση των εξωκυττάριων νουκλεϊκών οξέων. Κληρονομούνται είτε με τον υπολειπόμενο είτε με τον επικρατούντα αυτοσωματικό χαρακτήρα. Στην κλινική εικόνα μπορεί να συνυπάρχουν συμπτώματα κνιδωτικής αγγειίτιδας με χαμηλό συμπλήρωμα (Εικ. 6) ή συνδρόμου Sjögren. Ανευρίσκονται συχνά θετικά ANA και αντι-dsDNA και χαμηλά τα κλάσματα C3 και C4 του συμπληρώματος.(2-5,10,11)
[Αναπαραγωγή από (10)] Εικόνα 6. Εξάνθημα κνιδωτικής αγγειίτιδας με χαμηλό συμπλήρωμα [Αναπαραγωγή από (11)]
Μεταλλάξεις σχετιζόμενες με την ανοσιακή ανοχή. Τα κυριότερα μεταλλαγμένα γονίδια και τα προϊόντα κωδικοποίησής τους που σχετίζονται με την εμφάνιση μονοΣΕΛ της κατηγορίας αυτής είναι τα: TNFRSF6/CD95, PRKCD/PKC-δ, PTPN11, KRAS, SHOC2. Η επίδραση των μεταλλάξεων είναι η ελαττωμένη απόπτωση αυτοδραστικών Β και Τ κυττάρων. Κληρονομούνται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα. Εκτός των εκδηλώσεων του νΣΕΛ παρατηρείται λεμφαδενοπάθεια, διόγκωση ήπατος και σπλήνα και μερικές φορές το αυτοάνοσο λεμφοϋπερπλαστικό σύνδρομο ενώ οι δερματικές εκδηλώσεις και η νεφρίτιδα είναι συχνές.(2-5)
121
Εικόνα 5. Σχηματική απεικόνιση της δράσης των εξωκυττάριων DNAσών
Μονογονιδιακός Συστηματικός Ερυθηματώδης
Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Θεραπευτική αντιμετώπιση.
Η θεραπεία του μονοΣΕΛ αποτελεί πρόκληση καθώς συχνά η νόσος είναι ανθεκτική στην
καθιερωμένη αγωγή που χρησιμοποιείται στον νΣΕΛ. Στις περιπτώσεις ανεπάρκειας του
συμπληρώματος μπορεί να χρειαστεί μετάγγιση νωπού πλάσματος ή ακόμη και μεταμόσχευση
μυελού των οστών. Στις μεταλλάξεις που αφορούν το μονοπάτι σηματοδότησης IFN μπορεί
να χορηγηθούν οι αναστολείς κινασών janus (JAK inhibitors), καθώς και παράγοντες έναντι
της IFNα ή των υποδοχέων της. Στις περιπτώσεις διαταραχών των μηχανισμών ανοσιακής
ανοχής έχουν θέση οι βιολογικοί παράγοντες κατά των Β κυττάρων (Rituximab), αλλά και εδώ
περιγράφονται περιπτώσεις που χρειάστηκαν τελικά μεταμόσχευση μυελού των οστών.(2-5)
ΣYΝΔΡΟΜΟ SAVI (STING-ASSOCIATED VASCULOPATHY WITH ONSET IN INFANCY)
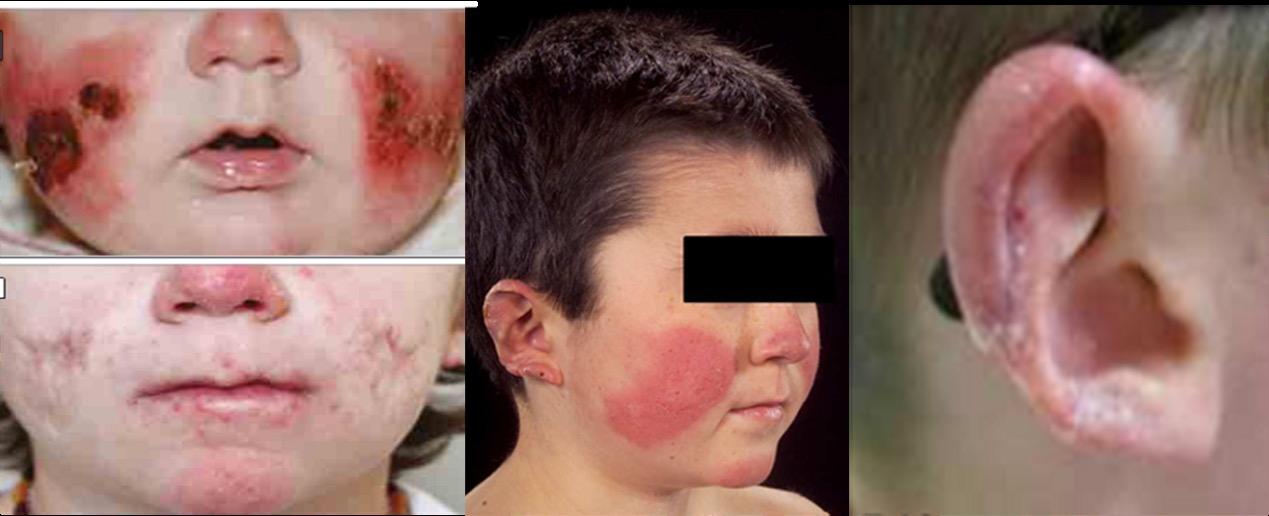
Πρόκειται για σπάνιο μονογονιδιακό αυτοφλεγμονώδες σύνδρομο που περιγράφηκε για
πρώτη φορά το 2014. Οφείλεται σε μετάλλαξη στο γονίδιο TMEM173, το οποίο κωδικοποιεί
την πρωτεΐνη STING (STimulator of INterferon Genes). Η μετάλλαξη οδηγεί σε αυξημένη
ενεργοποίηση της STING, με αποτέλεσμα την υπερπαραγωγή IFN και την επακόλουθη
επαγωγή συστηματικής φλεγμονής.(12)
Επιδημιολογία.
Στις περισσότερες περιπτώσεις το σύνδρομο εκδηλώνεται στη νεογνική ηλικία, αλλά
συνήθως η διάγνωση τίθεται σε μεγαλύτερη ηλικία. Θήλεα και άρρενα προσβάλλονται με την ίδια συχνότητα. Η μετάλλαξη κληρονομείται με τον αυτοσωματικό επικρατούντα χαρακτήρα.
Συχνότερες όμως είναι οι επίκτητες (de novo) μεταλλάξεις.(12)
Κλινική εικόνα.
Το σύνδρομο SAVI χαρακτηρίζεται από τον συνδυασμό δερματικών εκδηλώσεων βαριάς αγγειοπάθειας και διάμεσης πνευμονικής νόσου με δευτεροπαθή πνευμονική ίνωση. Μπορεί να συνυπάρχει πυρετός. (12)
Οι δερματικές εκδηλώσεις περιλαμβάνουν διάφορα εξανθήματα αγγειίτιδας, κυρίως στο πρόσωπο, στα χέρια και στα πόδια. Τα εξανθήματα είναι συνήθως τηλαγγειεκτασίες, χείμετλα, ερυθηματώδεις ή ιώδεις διηθητικές βλάβες με απολέπιση, που μπορεί να επιπλακούν με έλκη, εσχαροποίηση, γάγγραινα, ουλοποίηση και απώλεια ιστών. Παρατηρείται επίσης δυστροφία και απώλεια νυχιών. (Εικ. 7) Χαρακτηριστική της αγγειίτιδας του δέρματος
είναι επίσης η δικτυωτή πελίωση (livedo reticularis ή racemosa). (Εικ. 8). Στα πλαίσια της
αγγειίτιδας μπορεί να συμβεί διάτρηση του ρινικού διαφράγματος. Ενδέχεται να συνυπάρχει
εξάνθημα πεταλούδας και φωτοευαισθησία, συμπτώματα που θυμίζουν ΣΕΛ. Ωστόσο, στην
τυπική του μορφή το εξάνθημα του συνδρόμου SAVI προσβάλλει τα πιο προέχοντα σημεία
της μύτης (κορυφή) και των παρειών και είναι κρυοεπαγώμενο, ενώ στον νΣΕΛ το εξάνθημα
εκτείνεται από τη ράχη της μύτης προς τις παρειές με την κατανομή πεταλούδας και είναι
φωτοευαίσθητο. (Εικ. 9) (12-24)
122
Εικόνα 7α.
Εικόνα 7β.
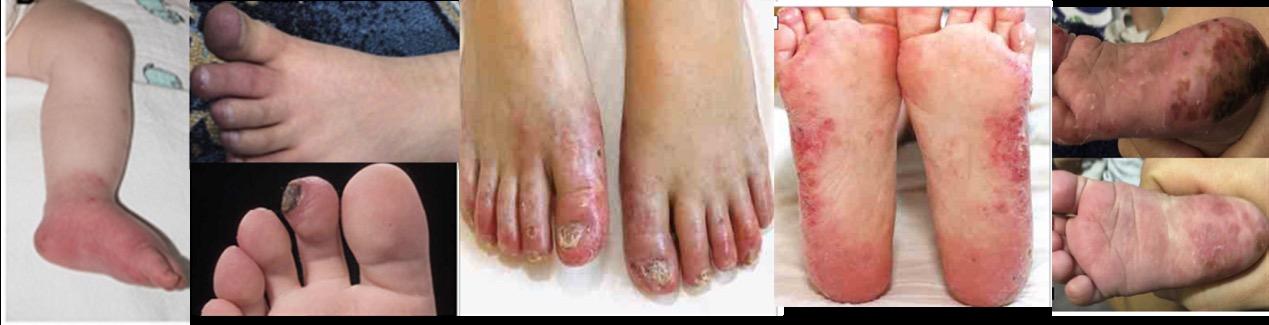
Εικόνα 7γ.
Εικόνα 7. Δερματικές εκδηλώσεις στο πρόσωπο και αυτιά (α) και στα άκρα (β,γ) ασθενών με σύνδρομο SAVI. Εξάνθημα αγγειίτιδας, ερυθηματώδεις διηθητικές πλάκες με τηλαγγειεκτασικές βλάβες, απολέπιση, εξελκώσεις, εσχάρες και ουλοποίηση. Όνυχες με δυστροφία [Αναπαραγωγή από (13-16,21-23)]
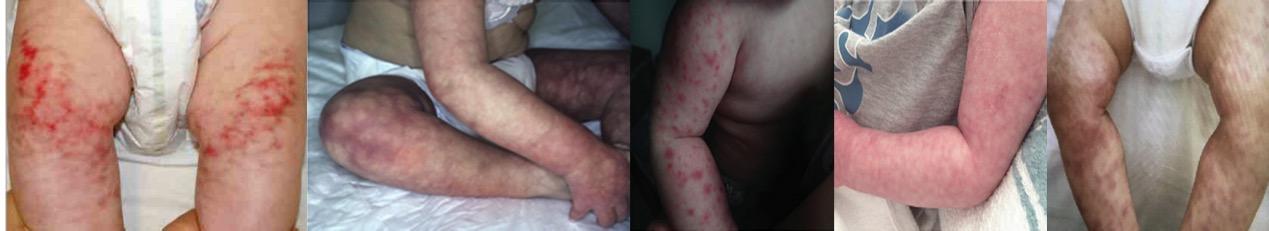
Εικόνα 8. Δικτυωτή και φλεγμονώδης πελίωση (livedo reticularis και racemosa) σε ασθενείς με σύνδρομο SAVI [Αναπαραγωγή από (15,18,19,21)]
123
Μονογονιδιακός Συστηματικός
Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Εικόνα 9. Σύγκριση εξανθήματος προσώπου (μύτη και παρειές) σε ασθενείς με σύνδρομο SAVI (πάνω) και νεανικό Συστηματικό Ερυθηματώδη Λύκο (κάτω) [Αναπαραγωγή από (24)]
Οι πνευμονικές εκδηλώσεις χαρακτηρίζονται από την ανάπτυξη διάμεσης πνευμονικής νόσου, η οποία σχετικά γρήγορα μπορεί να οδηγήσει σε πνευμονική ίνωση. Μπορεί να εμφανιστεί παρατραχειακή λεμφαδενοπάθεια, κυψελιδική αιμορραγία και πνευμονική υπέρταση. Η
πνευμονική προσβολή μπορεί αρχικά να εκδηλώνεται ως αποφρακτική βρογχιολίτιδα, υποτροπιάζουσα πνευμονία ή ως χρόνιος βήχας ανεξήγητης αιτίας. Στις εικόνες 10-12
παρουσιάζονται απεικονιστικά και κλινικά ευρήματα ασθενών με πνευμονική προσβολή
συνδρόμου SAVI.(12,19,23)
Συνυπάρχουσες εκδηλώσεις του σ.SAVI.
Αναφέρονται εκδηλώσεις σχεδόν από όλα τα συστήματα, οι οποίες όμως δεν είναι ειδικές του
συνδρόμου (πίνακας 1).(12)
Εργαστηριακά ευρήματα.
Δεν υπάρχουν ειδικά του συνδρόμου εργαστηριακά ευρήματα. Τις περισσότερες φορές
παρατηρείται αύξηση των δεικτών φλεγμονής. Επιπλέον, μπορεί να ανιχνεύονται διάφορα
αντιπυρηνικά και άλλα αυτοαντισώματα, όπως τα αντι-CCP (anti-Cyclic Citrullinated Peptide)
και ο ρευματοειδής παράγοντας). (12)
124
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Εικόνα 10. Απεικονιστικά ευρήματα ασθενών με σύνδρομο SAVI και πνευμονική προσβολή. Αριστερά: αμφοτερόπλευρη διάμεση πνευμονική νόσος (εικόνα «θολής υάλου»). Δεξιά: σχεδόν πλήρης κατάληψη του δεξιού πνεύμονα και του αριστερού κατώτερου λοβού από περιοχές με εικόνα «μακροκυστικής μελισσοκηρύθρας», ενδεικτική πνευμονικής ίνωσης.
Διάχυτη θολερότητα τύπου «θολής υάλου» στις περιοχές του αριστερού πνεύμονα που δεν έχουν υποστεί ίνωση. [Αναπαραγωγή από (19,23)]
Εικόνα 11. Απεικονιστικά ευρήματα ασθενών με σύνδρομο SAVI και πνευμονική προσβολή.
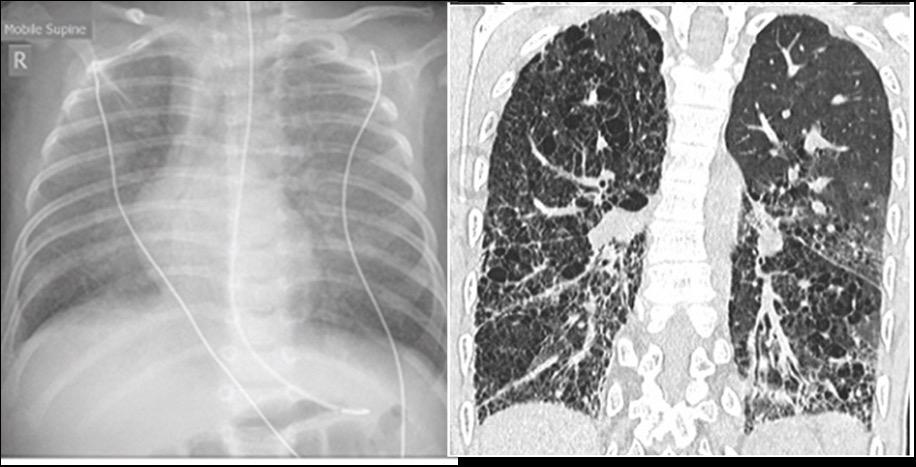
Πάνω: Ασύμμετρη πνευμονική προσβολή κυρίως στο δεξιό πνεύμονα και στον κατώτερο
αριστερό πνευμονικό λοβό. Θολερότητα τύπου «θολής υάλου», πάχυνση των ενδολοβιακών
γραμμών σχετιζόμενη με «μικροκυστική μελισσοκηρύθρα». Λοβία με εμφυσηματικές
περιοχές. (αριστερά) Διάχυτη θολερότητα τύπου «θολής υάλου» σχετιζόμενη με πάχυνση
των ενδολοβιακών γραμμών και υποπλευρικές μικροκυστικές βλάβες. Διάσπαρτες
μακροκυστκές βλάβες, κάποιες περιέχουσες κεντρικό αρτηριόλιο, ενδεικτικό βλαβών με κεντρολοβιακό εμφύσημα παρά την παρουσία παχέων τοιχωμάτων (δεξιά). [Αναπαραγωγή
από (23)] Κάτω: Διάχυτη θολερότητα τύπου «θολής υάλου» και τοπικός υπεραερισμός των πνευμόνων [Αναπαραγωγή από (12,23)]
125
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
Εικόνα 12. Πληκτροδακτυλία σε ασθενή με σύνδρομο SAVI [Αναπαραγωγή από (12)]
Συστηματικές εκδηλώσεις
Ανεπάρκεια αύξησης, κακουχία, αναιμία χρόνιας νόσου
Δέρμα Αλωπεκία
Ενδοκρινείς Αυτοάνοση θυρεοειδοπάθεια-Υποθυρεοειδισμός
Δόντια-στοματική κοιλότητα Περιοδοντίτιδα
Νεφροί Λευκωματουρία, μη ειδική νεφροπάθεια
Μυοσκελετικό Αρθρίτιδα, μυοσίτιδα
Αιματολογικά Κυτταροπενία
Κεντρικό Νευρικό Επασβεστώσεις βασικών γαγγλίων
Οφθαλμοί Αμφιβληστροειδοπάθεια, γλαύκωμα
Ευπάθεια σε λοιμώξεις Υποτροπιάζουσες βακτηριακές λοιμώξεις
Θεραπευτική αντιμετώπιση.
Η απάντηση σε ανοσοτροποποιητικούς συμβατικούς και βιολογικούς παράγοντες είναι μη
ικανοποιητική. Ενθαρρυντικά αποτελέσματα αναφέρονται από τη χρήση JAK αναστολέων.(12)
Πρόγνωση.
Η πρόγνωση είναι δυσμενής. Η θνησιμότητα είναι υψηλή στις 2 πρώτες δεκαετίες της ζωής
κυρίως από πνευμονικές επιπλοκές.
Συμπερασματικά, η υποψία για την παρουσία μονοΣΕΛ θα πρέπει να τίθεται σε:
• Κλινικές εκδηλώσεις παρόμοιες του νΣΕΛ αλλά με εμφάνιση σε ασθενείς μικρής ηλικίας.
• Ιστορικό συγγένειας γονέων ή παρόμοιων εκδηλώσεων και σε άλλα μέλη της οικογένειας.
• Ανθεκτικότητα στις καθιερωμένες θεραπείες του νΣΕΛ.
Η υποψία για την παρουσία συνδρόμου SAVI θα πρέπει να τίθεται σε: Πρώιμη έναρξη
126
Πίνακας 1. Συνυπάρχουσες εκδηλώσεις του συνδρόμου SAVI
πολυσυστηματικών εκδηλώσεων με προέχουσες τις χαρακτηριστικές αλλοιώσεις/βλάβες του δέρματος (χείμετλα, εξανθήματα και δερματικές αλλοιώσεις ΣΕΛ, ισχαιμικές νεκρωτικές βλάβες βαριάς αγγειίτιδας) και την προσβολή των πνευμόνων.
ΒΙΒΛΙΟΓΡΑΦΙΑ
1. Tsokos G. N Engl J Med. Systemic lupus erythematosus. N Engl J Med 2011;365:2110-21.
2. Omarjee O, Picard C, Frachette C, Moreews M, Rieux-Laucat F, Soulas-Sprauel P et al. Monogenic lupus: Dissecting heterogeneity. Autoimmun Rev 2019;18:102361.
3. Alperin J, Ortiz-Fernández L, Sawalha A. Monogenic Lupus: A Developing Paradigm of Disease. Front Immunol 2018;9: 2496.
4. Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond. J Clin Med 2020;9:712.
5. Costa-Reis P, Sullivan K. Monogenic lupus: it's all new! Curr Opin Immunol 2017;49:87-95.
6. Belot A, Cimaz R. Monogenic forms of systemic lupus erythematosus: new insights into SLE pathogenesis. Pediatr Rheumatol Online J 2012;10:21.
7. Melki I, Frémond ML. Type I Interferonopathies: from a Novel Concept to Targeted Therapeutics. Curr Rheumatol Rep 2020;22:32.
8. https://dermnetnz.org/topics/chilblain-lupus-erythematosus
9. Kono M, Akiyama M. Dyschromatosis symmetrica hereditaria and reticulate acropigmentation of Kitamura: An update. J Dermatol Sci 2019;93:75-81.
10. Mathapathi S, Chu CQ. Contribution of impaired DNASE1L3 activity to anti-DNA autoantibody production in systemic lupus erythematosus. Rheumatol Immunol Res 2022;3:17-22.
11. https://dermnetnz.org/topics/urticarial-vasculitis
12. Wang Y, Wang F, Zhang X. STING-associated vasculopathy with onset in infancy: a familial case series report and literature review. Ann Transl Med 2021;9(2):176.
13. Omoyinmi E, Melo Gomes S, Nanthapisal S, Woo P, Standing A, Eleftheriou D et al. Stimulator of interferon genes-associated vasculitis of infancy. Arthritis Rheumatol 2015;67:808.
14. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014;371:507-518.
15. Munoz J, Rodière M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI et al. JAMA Dermatol 2015;151:872-7.
16. Picard C, Thouvenin G, Kannengiesser C, Dubus JC, Jeremiah N, Rieux-Laucat F et al. Severe Pulmonary Fibrosis as the First Manifestation of Interferonopathy (TMEM173 Mutation). Chest 2016;150:e65-71.
17. Frémond ML, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol 2016;138:1752-1755.
18. Melki I, Rose Y, Uggenti C, Van Eyck L, Frémond ML, Kitabayashi N et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J Allergy Clin Immunol 2017;140:543-552.
19. Saldanha RG, Balka KR, Davidson S, Wainstein BK, Wong M, Macintosh R et al. A Mutation Outside the Dimerization Domain Causing Atypical STING-Associated Vasculopathy With Onset in Infancy. Front Immunol 2018;9:1535.
20. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018;128:3041-3052.
21. Balci S, Ekinci RMK, de Jesus AA, Goldbach-Mansky R, Yilmaz M. Baricitinib experience on STING-associated vasculopathy with onset in infancy: A representative case from Turkey. Clin Immunol 2020;212:108273.
22. Lin B, Berard R, Al Rasheed A, Aladba B, Kranzusch PJ, Henderlight M et al. A novel STING1 variant causes a recessive form of STING-associated vasculopathy with onset in infancy (SAVI). J Allergy Clin Immunol. 2020;146:1204-1208.e6.
23. Frémond ML, Hadchouel A, Berteloot L, Melki I, Bresson V, Barnabei L et al. Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) Among 21 Patients. J Allergy Clin Immunol Pract 2021;9:803-818.e11.
127
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI
Vasculopathy with onset in Ιnfancy)
(STING-Associated
(STING-Associated Vasculopathy with onset in Ιnfancy)
24. Kim H, Sanchez GA, Goldbach-Mansky R. Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus. J Mol Med (Berl) 2016;94:11111127.
128
Μονογονιδιακός Συστηματικός Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI
Ερυθηματώδης Λύκος (μονοΣΕΛ) και Σύνδρομο SAVI (STING-Associated Vasculopathy with onset in Ιnfancy)
129 Μονογονιδιακός
Συστηματικός
Επίλογος
Φλωρεντία Κανακούδη-Τσακαλίδου
Κλείνουμε αυτό το τραπέζι παραθέτοντας ως «επίλογο» ορισμένες χαρακτηριστικές
εκδηλώσεις που θα θέσουν την υποψία ενός μονογονιδιακού ΑΦΝ:
ΠΟΤΕ Ο ΠΑΙΔΙΑΤΡΟΣ ΘΑ ΥΠΟΠΤΕΥΘΕΙ ΜΟΝΟΓΟΝΙΔΙΑΚΟ ΑΦΝ;
Όταν υπάρχουν:
Υποτροπιάζονταεμπύρεταμεήχωρίςπεριοδικότηταή Πρώτοεμπύρετο(παρατεταμένο) σε συνδυασμό με ένα ή περισσότερα από τα παρακάτω:
1. Θετικό οικογενειακό ιστορικό (παρόμοιες ή υποτροπιάζουσες, αδιευκρίνιστες εκδηλώσεις
και σε άλλα μέλη ή/ και αδιάγνωστοι θάνατοι)
2. Ανθεκτικές περιπτώσεις σε ανοσοτροποποιητική θεραπεία
3. Διάμεση πνευμονοπάθεια, υποδορίτιδα με ή χωρίς λιποδυστροφία
4. Εμφάνιση σε μικρή ηλικία εκδηλώσεων από το ΚΝΣ (επασβεστώσεις βασικών γαγγλίων, λευκοεγκεφαλοπάθεια, λεμφοκυτταρικό ΕΝΥ) ή το δέρμα (οζώδες ερύθημα, χείμετλα στα άκρα)
www.nomidalliance.org/compchart.php
Αλληλογραφία
Φλωρεντία ΚανακούδηΤσακαλίδη
Μ. Μπότσαρη 2, 54643 Θεσσαλονίκη
Τ. 2310839291
K. 6972127338
e-mail: flkan@auth.gr
Φλωρεντία ΚανακούδηΤσακαλίδου Α΄Παιδιατρική Κλινική Α.Π.Θ. - Γενικό Νοσοκομείο Θεσσαλονίκης «Ιπποκράτειο»
130
131
ΣΥΝΑΔΕΛΦΙΚΑ
Βρίσκομαι στο τέλος του δεύτερου χρόνου στην Παιδοκαρδιολογική Κλινική του
Great Ormond Street. Οι περισσότερες μέρες της εβδομάδας περνάνε στο αιμοδυναμικό τμήμα
όπου κάνω καθετηριασμούς σε μικρής ηλικίας και χαμηλού βάρους παιδιά με σύμπλοκες και
βαριές καρδιοπάθειες. Δύσκολη, επικίνδυνη και απαιτητική δουλειά που γίνεται ακόμα πιο
απαιτητική με το πρωτόκολλο του τμήματος που θέλει την κάθε αιμοδυναμική μελέτη να
γίνεται σε όλη της τη λεπτομέρεια. Πρέπει να μπούμε σε όλους τους χώρους της καρδιάς, σε όλα τα αγγεία για να πάρουμε πιέσεις και οξυγονώσεις και να κάνουμε αγγειογραφίες.
Πρωτόκολλο διαφορετικό από αυτό που είδα μετά όταν πήγα στις ΗΠΑ στο Παιδιατρικό
Νοσοκομείο του Πανεπιστημίου Harvard.
Από την αρχή της εργασίας μου στο νοσοκομείο ακούγαμε για την νέα πτέρυγα και το νέο Παιδοκαρδιολογικό τμήμα στο οποίο θα μετακομίζαμε. Στους τελευταίους έξι μήνες, ενάμισι
χρόνο από την αρχή είχαμε ξεκινήσει τις πρόβες. Από που θα μπαίνουμε και θα βγαίνουμε, που θα στέκεται ο γιατρός, που οι νοσηλεύτριες και που οι τεχνολόγοι, πρόβες με κούκλα
για το πως θα κάνουμε τις μελέτες, από ποια μεριά θα στεκόμαστε και άλλες πάρα πολλές λεπτομέρειες. Τελικά ήρθε η στιγμή που μεταφερθήκαμε και αρχίσαμε να δουλεύουμε στο
νέο τμήμα. Όλα καλά, κανένα πρόβλημα και πως να υπάρχει, αφού τα είχαμε προβάρει όλα
επί σχεδόν μισό χρόνο.
Ένα πρωινό καθετηριάζω μια δύσκολη περίπτωση ενός μικρού παιδιού. Στο μέσο περίπου της
μελέτης βλέπω από το τζάμι που μας χωρίζει έναν από τους διευθυντές να μου κάνει κάποια
περίεργα σήματα και να μου λέει τελείωνε γρήγορα. Ήταν ένας από τους πιο απαιτητικούς
και αυτά που έλεγε δεν τα είχα ξανακούσει έτσι που μου φάνηκαν περίεργα. Φώναξα λοιπόν
την προϊσταμένη του αιμοδυναμικού που ήταν φίλη μου για να τη ρωτήσω αν καταλαβαίνω
καλά και τι ακριβώς συμβαίνει για να πάρω την απάντηση κάνε όσο πιο γρήγορα μπορείς
και τελείωνε γιατί: “The building is collapsing” με άλλα λόγια το κτήριο γκρεμίζεται. Έμεινα
άναυδος και στην επανάληψη της ερώτησης πήρα την ίδια απάντηση. Τελικά έκανα ότι
καλύτερο μπορούσα χωρίς να βάζω σε κίνδυνο τη ζωή του ασθενούς και τελείωσα.
Στη συνέχεια έμαθα ότι στους επαναληπτικούς ελέγχους που είχαν γίνει, βρήκαν κάποιες
παραλείψεις στην κατασκευή του κτηρίου οι οποίες θα μπορούσαν, έστω και με πολύ μικρές
πιθανότητες, να δημιουργήσουν στατικά προβλήματα!
132
ΣΥΝΑΔΕΛΦΙΚΑ
Τελικά επιστρέψαμε στο παλιό κτήριο, στον γνωστό και αγαπημένο μας οικείο χώρο όπου
συνεχίσαμε τη δουλειά μας.
Ούτε έως ότου να φύγω, ούτε και στα πολλά επόμενα χρόνια που πέρασαν δεν έγινε τίποτα
και φυσικά το κτήριο μένει όρθιο. Όπως και να είναι όμως όλη η ιστορία δείχνει πόσο μεγάλη
σημασία δίνουν οι Άγγλοι στη λεπτομέρεια και ίσως όχι άδικα.
133
BETWEEN COLLEAGUES
ΦΛΟΓΟΖΑΛΗ ΛΟΥΛΟΥΔΙΩΝ
ΜΝΗΜΗΣ ΛΑΜΠΥΡΙΣΜΑΤΑ
Του ΓΙΑΝΝΗ ΜΕΛΕΤΗ
Ένα βιβλίο φορτωμένο με δεκάδες αναμνήσεις. Αναμνήσεις ενός γιατρού που αφιέρωσε τη
ζωή του στην ιατρική επιστήμη και στους ασθενείς του. Ενός ανθρώπου με Α κεφαλαίο που
τιμά το προνόμιο του να είσαι άνθρωπος όπως έλεγε και ο μεγάλος φιλόσοφός μας Κώστας
Δεσποτόπουλος.
Ένας ιατρός που έταξε τη ζωή του στην ανακούφιση του πόνου, στην αντιμετώπιση της
αρρώστιας, στη θεραπεία των ασθενών του χωρίς να έχει ποτέ του σκοπό το κέρδος και τον
πλουτισμό αλλά μόνο τη βοήθεια στον άνθρωπο που πάσχει, στον απελπισμένο άνθρωπο
που κινδυνεύει να χάσει ό,τι πολύτιμο έχει, τη ζωή του. Με σκληρή εργασία, ατέλειωτες

νοσοκομειακές ώρες, αφιερωμένος στην έρευνα, στη διδασκαλία αλλά και στην κλινική
ιατρική πράξη με αγάπη και καλοσύνη, αλτρουισμό και ανιδιοτέλεια έκανε πάντα ό,τι καλύτερο
μπορούσε να γίνει. Χαρούμενος με την ικανοποίηση του να βλέπει τους μαθητές του να
διαπρέπουν εφαρμόζοντας αυτά που τους δίδαξε ο Δάσκαλός τους χωρίς ποτέ να τον ξεχνούν χαρίζοντάς του για πάντα τον πολύτιμο τίτλο του Δασκάλου. Του δασκάλου ιατρού πρότυπου
που αντιμετωπίζει με αγάπη και καλοσύνη, δοτικότητα και ανιδιοτέλεια τον συνάνθρωπό του.
Μέσα στο βαρύ νοσοκομειακό περιβάλλον με την αίσθηση του υψηλού χιούμορ που τον
χαρακτηρίζει, και τις αθώες ζαβολιές, συνέβαλε σημαντικά στην ελάφρυνση της καταθλιπτικής
ατμόσφαιρας. Πάντα με το χαμόγελο και τη γλυκύτητα του ύφους του δημιουργούσε το
καλύτερο περιβάλλον για ιατρούς και αρρώστους. Εκτός από το νοσοκομείο στο οποίο
αφιέρωσε όλη του τη ζωή τις ίδιες καλές υπηρεσίες πρόσφερε και ως αγροτικός ιατρός.
Ένας ευλογημένος ιατρός από αυτούς που χρειάζεται η ανθρωπότητα για να γλυκάνει τους
πόνους και τις δυστυχίες της.
Το βιβλίο του αυτό, είναι ένα ευχάριστο ανάγνωσμα όχι μόνο για ιατρούς αλλά και για
οποιονδήποτε αγαπάει το διάβασμα. Γραμμένο σε απλή ρέουσα καθομιλουμένη γλώσσα
την οποία συνηθίζει να χρησιμοποιεί ο συγγραφέας, δείχνει ότι εκτός από την ποίηση με
τα θαυμάσια ποιήματα του μπορεί να διαπρέπει και στον πεζό λόγο. Την ποίησή του την
απολαμβάνουμε σχεδόν καθημερινά στη Σελίδα του Συλλόγου μας: Σύλλογος Ιατρών φίλων
του Λόγου, της Τέχνης, της Μουσικής στο Facebook. Ελπίζουμε στο μέλλον να απολαμβάνουμε
και αποσπάσματα από τον πεζό του λόγο.
134
ΚΡΙΤΙΚΗ ΒΙΒΛΙΟΥ
135 Μια καλαίσθητη έκδοση με εξώφυλλο στο οποίο υπάρχει ζωγραφικό έργο του πραγματικά ταλαντούχου εγγονού του Γιαννάκη τον οποίο λατρεύει ο συγγραφέας-ποιητής παππούς Γιάννης Μελέτης. Εύχομαι το βιβλίο να αγαπηθεί από πολλούς. ΣΤΕΛΙΟΣ ΑΝΤΩΝΙΑΔΗΣ BOOK PRESENTATION
A. Γενικές πληροφορίες
Το περιοδικό “Παιδιατρική” είναι η επιστημονική έκδοση και ιδιοκτησία της Ελληνικής
Παιδιατρικής Εταιρείας, που διανέμεται στα μέλη της. Έχει ως βασικούς στόχους την
αποτύπωση του παιδιατρικού επιστημονικού έργου και τη συνεχή ενημέρωση των Παιδιάτρων.
Για το σκοπό αυτό δέχεται για δημοσίευση ποικιλία άρθρων και συγκεκριμένα:
1. Άρθρα σύνταξης (μετά από πρόσκληση της Συντακτικής Επιτροπής)
2. Ανασκοπήσεις
3. Βραβευμένες εργασίες
4. Ερευνητικές μελέτες
5. Κλινικά κουίζ
6. Επιλεγμένες συζητήσεις στρογγυλών τραπεζών
7. Επίκαιρα θέματα
8. Θέματα εκπαίδευσης και οργάνωσης υγείας
9. Ενδιαφέρουσες περιπτώσεις
10. Σύντομα νέα
11. Βραχείες δημοσιεύσεις
12. Επιστολές προς τη σύνταξη
13. Περιλήψεις της βιβλιογραφίας
14. Ανακοινώσεις προσεχών συνεδρίων και επιστημονικών εκδηλώσεων
15. Αναφορές και κριτική νέων εκδόσεων, ελληνικών και ξένων, παιδιατρικού ενδιαφέροντος
Η Συντακτική Επιτροπή διατηρεί το δικαίωμα να δημοσιεύει άρθρα με ιδιαίτερο επιστημονικό
ενδιαφέρον, καθώς και άρθρα με επίκαιρα θέματα χωρίς να τηρείται η σειρά υποβολής.
Επίσης, μπορεί να αποφασίζει για τη δημοσίευση εργασιών που έχουν παρουσιαστεί στο ετήσιο Παιδιατρικό Συνέδριο, την ολική ή μερική δημοσίευση παρουσιάσεων ιδιαίτερου ενδιαφέροντος και την ολική ή μερική δημοσίευση επιστολών που αναφέρονται σε δημοσιευμένα επιστημονικά άρθρα του περιοδικού.
Στις εργασίες με επίκαιρα θέματα πρέπει να αναγράφεται στην πρώτη σελίδα η πρόθεση των συγγραφέων για την αναγκαιότητα της ταχείας δημοσίευσης. Η Επιτροπή Σύνταξης διατηρεί το
δικαίωμα της αποδοχής της ταχείας δημοσίευσης.
Όλες οι εργασίες πρέπει να μην έχουν εν μέρει ή εξ ολοκλήρου δημοσιευθεί ή υποβληθεί
για κρίση σε άλλο περιοδικό. Επιπλέον, στην εργασία θα πρέπει να αναφέρονται τυχόν
επιδοτήσεις-χορηγίες ή άλλες πηγές υποστήριξης. Όλες οι κλινικές έρευνες πρέπει να έχουν
γίνει μετά από πληροφορημένη συναίνεση των μετεχόντων ή των νομίμων εκπροσώπων τους, σύμφωνα με τις διακηρύξεις του Ελσίνκι και του Τόκιο. Επίσης, πρέπει να έχουν τηρηθεί οι οδηγίες για τη φροντίδα πειραματόζωων του Εθνικού Ινστιτούτου Υγείας των ΗΠΑ (DHEW Publication, ΝΙΗ, 80-23). Οι κλινικές μελέτες πρέπει να έχουν εγκριθεί από την επιτροπή Ηθικής και Δεοντολογίας του Νοσοκομείου.
Οι απόψεις και τα συμπεράσματα των συγγραφέων που διατυπώνονται στις δημοσιευμένες εργασίες δεν αντικατοπτρίζουν απαραίτητα αυτές του περιοδικού. Η καταχώριση διαφημίσεων
στο περιοδικό δεν υποδηλώνει οπωσδήποτε έγκριση των περιεχομένων τους από την Ελληνική
Παιδιατρική Εταιρεία, τη Συντακτική Επιτροπή ή από τον εκδότη του περιοδικού.
Όλες οι δημοσιευμένες εργασίες θεωρούνται ιδιοκτησία του περιοδικού “Παιδιατρική” και η
ολική ή μερική αναδημοσίευσή τους επιτρέπεται μόνο μετά από έγγραφη συγκατάθεση του
περιοδικού.
Β. Σύνταξη των εργασιών
Όλες οι εργασίες πρέπει να ακολουθούν “Κοινές Προδιαγραφές για Χειρόγραφα που
υποβάλλονται σε Βιοϊατρικά Περιοδικά” (Uniform Requirements for Manuscripts Submit-
136 ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ ΣΥΓΓΡΑΦΕΙΣ
ted to Biomedical Journals), οι οποίες τροποποιήθηκαν πρόσφατα και δημοσιεύονται στις διευθύνσεις: www.icmje.org και www.icmje.org/icmje.pdf
Το κείμενο πρέπει να έχει την ακόλουθη διάταξη: σελίδα τίτλου, βραχύς τίτλος, περίληψη στα
ελληνικά και τα αγγλικά, κατάλογος συντομογραφιών, κείμενο, ευχαριστίες καθώς και αναφορά
σε επιδοτήσεις-χορηγίες ή άλλες πηγές υποστήριξης, βιβλιογραφία, πίνακες, εικόνες, τίτλοι εικόνων.
Η έκταση προσδιορίζεται:
• Για τις ανασκοπήσεις σε 2000-3000 λέξεις.
• Για τις ερευνητικές εργασίες σε 1500-2500 λέξεις και τις ενδιαφέρουσες περιπτώσεις σε 1000-1500 λέξεις.
• Για τις βραχείες δημοσιεύσεις σε 1000-1500 λέξεις.
• Για τις επιστολές σε 250-500 λέξεις.
Σελίδα τίτλου
Πρέπει να περιλαμβάνει:
• Τον τίτλο (<14 λέξεις) του άρθρου. Δεν επιτρέπονται συντμήσεις.
• Το όνομα και το επώνυμο κάθε συγγραφέα στην ονομαστική.
• Το επιστημονικό κέντρο (ίδρυμα, κλινική, εργαστήριο) από όπου προέρχεται η εργασία. Σε
περίπτωση έλλειψης συνεργασίας με συγκεκριμένα κέντρα, πρέπει να αναφέρεται η ιδιότητα του/των συγγραφέα/ων (π.χ. ιδιώτης παιδίατρος) και ο τόπος διαμονής του/τους.
• Την πλήρη διεύθυνση, e-mail και το τηλέφωνο του συγγραφέα με τον οποίο γίνεται η αλληλογραφία.
Περιλήψεις
Η περίληψη πρέπει να ανακεφαλαιώνει τους στόχους της εργασίας, τη μεθοδολογία, τα
κυριότερα αποτελέσματα και τα συμπεράσματα της μελέτης.
• Θα πρέπει να περιλαμβάνει τουλάχιστον 200 λέξεις, όμως δεν θα πρέπει να υπερβαίνει τις 250 λέξεις.
• Η περίληψη των εργασιών πρέπει να είναι δομημένη στις εξής παραγράφους: εισαγωγή, μέθοδοι, αποτελέσματα και συμπεράσματα.
Στην περίληψη στα αγγλικά πρέπει να αναγράφονται στην αρχή του κειμένου ο τίτλος της
εργασίας και τα ονόματα των συγγραφέων στα αγγλικά. Το περιεχόμενο του αγγλικού κειμένου πρέπει να είναι δομημένο σε: εισαγωγή (background), μέθοδοι (methods), αποτελέσματα (results) και συμπεράσματα (conclusions). Η περίληψη στα αγγλικά δεν πρέπει να διαφέρει από την αντίστοιχη στα ελληνικά.
Κάτω από την ελληνική και την αγγλική περίληψη σημειώνονται τρεις έως πέντε λέξεις κλειδιά που θα χρησιμοποιηθούν για το θεματικό ευρετήριο.
Κείμενο Οι ερευνητικές εργασίες περιλαμβάνουν: εισαγωγή, μεθόδους, αποτελέσματα και συζήτηση.
Η εισαγωγή περιλαμβάνει τα τελευταία δεδομένα της έρευνας στο συγκεκριμένο θέμα με τις κυριότερες βιβλιογραφικές παραπομπές και το σκοπό της εργασίας. Η περιγραφή των μεθόδων
πρέπει να είναι ακριβής και αρκετά λεπτομερής, ώστε να επιτρέπει την αναπαραγωγή τους από άλλους ερευνητές. Επίσης, πρέπει να αναφέρονται με λεπτομέρειες οι στατιστικές μέθοδοι ανάλυσης και αξιολόγησης των αποτελεσμάτων. Τα αποτελέσματα πρέπει να παρουσιάζονται
με σαφήνεια και να συνοδεύονται από την απαραίτητη στατιστική ανάλυση. Η συζήτηση πρέπει
να περιλαμβάνει τα συμπεράσματα που προκύπτουν από την εργασία, τη σημασία που μπορεί
να έχουν και την πιθανή συσχέτισή τους με παρατηρήσεις άλλων ερευνητών.
Οι ενδιαφέρουσες περιπτώσεις περιλαμβάνουν σύντομη εισαγωγή, περιγραφή της περίπτωσης
και βραχεία συζήτηση, με έμφαση στη διαφορική διάγνωση.
137 ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ ΣΥΓΓΡΑΦΕΙΣ
Τα υπόλοιπα θέματα έχουν ελεύθερη δομή κατά την κρίση των συγγραφέων τους.
Ευχαριστίες ή αναγνωρίσεις (αναφορά σε επιδοτήσεις-χορηγίες ή άλλες πηγές υποστήριξης)
θα πρέπει να αναφέρονται στο τέλος του κειμένου και πριν από τις βιβλιογραφικές
παραπομπές.
Τιμές εργαστηριακών εξετάσεων
Οι τιμές των εργαστηριακών εξετάσεων πρέπει να εκφράζονται στο Διεθνές Σύστημα Μονάδων
(SI Units) και στο Μετρικό (Conventional-Συμβατικό) Σύστημα μέσα σε παρένθεση. Πίνακες
μετατροπής περιλαμβάνονται στις διευθύνσεις: www.icmje.org και www.icmje.org/icmje.pdf.
Συντομογραφίες
Οι συντομογραφίες που έχουν επιβληθεί διεθνώς, δημοσιεύονται σε κάθε τεύχος του
περιοδικού. Σύνθετοι και μακροσκελείς όροι που επαναλαμβάνονται συχνά στο κείμενο
μπορούν να αντικαθίστανται από συντομογραφίες που επεξηγούνται από τους συγγραφείς σε
ειδικό κατάλογο, ο οποίος υποβάλλεται μαζί με την εργασία. Οι συντομογραφίες αναφέρονται
σε παρένθεση μόνο στις περιλήψεις.
Βιβλιογραφικές παραπομπές
Στο τμήμα της βιβλιογραφίας καταχωρούνται όλες οι βιβλιογραφικές παραπομπές με τη σειρά
που αναφέρονται στο κείμενο. Στο κείμενο, οι βιβλιογραφικές παραπομπές αναφέρονται
με αραβικούς αριθμούς σε παρένθεση. Οι βιβλιογραφικές παραπομπές δεν πρέπει να υπερβαίνουν:
• τις 70 στις ανασκοπήσεις
• τις 40 στις ερευνητικές εργασίες
• τις 20 στα επίκαιρα θέματα και τις ενδιαφέρουσες περιπτώσεις
• τις 10 στις βραχείες δημοσιεύσεις και τις επιστολές
Η σύνταξη των βιβλιογραφικών παραπομπών γίνεται σύμφωνα με τις πρόσφατα τροποποιημένες προδιαγραφές της International Committee of Medical Journal Editors/Uniform Requirements for Manuscripts Submitted to Biomedical Journals, (www.icmje.org και www.icmje.org/icmje.pdf). Οι συντμήσεις των τίτλων των περιοδικών γίνονται με βάση το Cumulated Index Medicus [List of Journals Indexed in Index Medicus (www.nlm.nih.goν/bsd/ uniform_requirements.html)].
Παραδείγματα βιβλιογραφικών παραπομπών
Ι. ΠΕΡΙΟΔΙΚA
Αν οι συγγραφείς είναι έως έξι, αναγράφονται όλοι, ενώ αν είναι επτά ή περισσότεροι, αναγράφονται οι πρώτοι έξι και προστίθεται “et al” ή “και συν”.
Τακτική έκδοση περιοδικού: Αντωνίου Δ, Μπάρα Χ, Γιαννάτου Χ. Εμβόλια: επίδραση στην επιδημιολογία των λοιμωδών
νοσημάτων και την παιδιατρική πράξη. Παιδιατρική 1999;59:272-279.
Proesmans W. Bartter syndrome and its neonatal νariant. Eur J Pediatr 1997;156:669-679.
Συμπληρωματικό τεύχος περιοδικού:
Flyvbjerg Α. Role of growth hormone, insulin-like growth factors (IGFs) and IGF-binding proteins in the renal complications of diabetes. Kidney Ιnt 1997;52 (60 Suppl):S12-S19.
Χωρίς συγγραφέα:
National Institutes of Health Consensus Deνelopment Conference. Neurofibromatosis conference statement. Arch Neurol1988;45:575-578.
138 ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ ΣΥΓΓΡΑΦΕΙΣ
ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ ΣΥΓΓΡΑΦΕΙΣ
Προσδιορισμός τύπου άρθρου: Schreiner GF, Lange L. Ethanol modulation of macrophage influx in glomerulonephritis [Abstract]. J Am Soc Nephrol 1991;2:562.
Should antileukotriene therapies be used instead of inhaled corticosteroids in asthma? [Editorial]. Am J Respir Crit Care Med 1998;158:1697-1701.
Laux-End R, Inaebnit D, Gerber ΗΑ, Bianchetti MG. Vasculitis associated with leνamisole and circulating autoantibodies [Let ter]. Arch Dis Child 1996;75:355-356.
II. ΒΙΒΛΙΑ Κεφάλαιο σε βιβλίο: Clark AG, Barratt ΤΜ. Steroid-responsiνe nephrotic syndrome. Ιn: Barratt ΤΜ, Arner ED, Harmon WE, editors. Pediatric Nephrology. 4th ed. Baltimore: Lippincott William Wilkins; 1999. p. 742.
Σύγγραμμα ή μονογραφία: Gorlin RJ, Cohen ΜΜ, Leνin LS. Syndromes of the head and neck. 3rd ed. New York: Oxford Uniνersity Press; 1990.
Δημοσίευση σε τόμο πρακτικών: Bauer ΑW. The two definitions of bacterial resistance. In: Smith AJ, Rogers CA, eds. Proceedings of the Third International Congress of Chemotherapy; 1962 May 29-31; New York: International Society of Chemotherapy; 1963. p. 484-500.
Διδακτορική διατριβή: Παπαδόπουλος Χ. Η θεραπεία του στραβισμού
Αθηνών; 1979.
Kaplan SJ. Post hospital home health care: the elderly’s access and utilization [dissertation]. St. Louis (Μο): Washington Univ.;1995.
III. CD-ROM
Andersoη SC, Poulsen ΚΒ. Anderson’s electronic atlas of hematology [CD-ROM]. Philadelphia: Lippincott Williams & Wilkins; 2002.
IV. ΣΤΟ ΔΙΑΔΙΚΤΥΟ
Άρθρο σε περιοδικό:
Abood S. Quality improνement initiatiνe in nursiηg homes: the ΑΝΑ acts in an adνisory role. Am J Nurs [Internet]. 2002 Jun:
Webpage: http://www.nursingworld.org/AJN/2002/june/Wawatch.htm
Μονογραφία: Foley ΚΜ, Gelbaηd Η, editors. Improving palliative care for cancer [Monograph, Internet]. Washington: National Academy Press; 2001.
Webpage: http://www.nap.edu/books/0309074029/html
Ιστοσελίδες: Cancer-Pain.org [Webpage, Internet]. New York: Association of Cancer Online Resources, Ιnc.; 2002: http://www.cancer-pain.org/
Πίνακες και Εικόνες
Οι πίνακες αριθμούνται με αραβικούς αριθμούς με τη σειρά που εμφανίζονται στο κείμενο. Περιλαμβάνουν βραχύ τίτλο και επεξήγηση των συντμήσεων στο κάτω μέρος.
139
[διδακτορική διατριβή]. Αθήνα: Πανεπιστήμιο
ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ ΣΥΓΓΡΑΦΕΙΣ
Όλο το απεικονιστικό υλικό (σχήματα, διαγράμματα, φωτογραφίες, κ.λπ.) χαρακτηρίζεται ως
εικόνες και θα πρέπει να αποστέλλεται σε ηλεκτρονική μορφή και σε αρχεία υψηλής ανάλυσης.
Σε περίπτωση που αυτό δεν είναι δυνατό, το υλικό θα αποστέλλεται ταχυδρομικώς και στο
πίσω μέρος των εικόνων θα σημειώνεται με μολύβι ο αριθμός της εικόνας και το όνομα του
πρώτου συγγραφέα, καθώς και ένα βέλος το οποίο να δείχνει το πάνω μέρος της εικόνας. Στις
φωτογραφίες ασθενών δεν πρέπει να αναγνωρίζεται η ταυτότητά τους. Οι ασθενείς δεν πρέπει
να αναφέρονται ονομαστικά.
Γ. Υποβολή και Δημοσίευση των Εργασιών
Όλες οι εργασίες πρέπει να αποστέλλονται στην ηλεκτρονική διεύθυνση της εταιρείας ( grammateia@e-child.gr). Σε ξεχωριστό μήνυμα θα δηλώνεται ότι η εργασία δεν έχει
εν μέρει ή εξ ολοκλήρου δημοσιευθεί ή υποβληθεί για κρίση σε άλλο περιοδικό και ότι οι
συγγραφείς εγκρίνουν τη δημοσίευσή της στο περιοδικό “Παιδιατρική”. Επιπλέον, θα πρέπει
να αναφέρονται τυχόν επιδοτήσεις-χορηγίες ή άλλες πηγές υποστήριξης.
Εφόσον η εργασία γίνει αποδεκτή, το διορθωμένο σύμφωνα με τις υποδείξεις των κριτών
κείμενο πρέπει να υποβάλεται στη Συντακτική Επιτροπή μέσω e-mail που να περιλαμβάνει
την εργασία σε πρόγραμμα Word. Σε ξεχωριστό αρχείο θα περιλαμβάνεται επιστολή όπου θα
αναφέρονται αναλυτικά οι τροποποιήσεις ή οι αντιρρήσεις στις προτάσεις των κριτών.
Η καθυστέρηση της επανυποβολής του τροποποιημένου κειμένου πέραν των 30 ημερών
συνεπάγεται νέα υποβολή.
Τα κείμενα των εργασιών που δε γίνονται αποδεκτές για δημοσίευση, δεν επιστρέφονται.
Μπορεί να επιστραφούν, εφόσον ζητηθεί εντός εξαμήνου, τα σχήματα και οι φωτογραφίες που τα συνοδεύουν.
Πριν αποστείλετε ηλεκτρονικά την εργασία σας, βεβαιωθείτε ότι το αρχείο περιλαμβάνει:
1. Σελίδα τίτλου που περιέχει:
• τίτλο και βραχύ τίτλο εργασίας
• όνομα και επώνυμο συγγραφέων (ολογράφως)
• επιστημονικό/ά κέντρο/α όπου έγινε η εργασία
• όνομα, διεύθυνση, ηλεκτρονική διεύθυνση και τηλέφωνο συγγραφέα για αλληλογραφία
2. Περίληψη ελληνική-αγγλική (200-250 λέξεις), δομημένη στις εξής παραγράφους: εισαγωγή, μέθοδοι, αποτελέσματα και συμπεράσματα
3. Λέξεις κλειδιά
4. Κατάλογο συντομογραφιών
5. Κείμενο
6. Ευχαριστίες ή αναγνωρίσεις (αναφορά σε επιδοτήσεις-χορηγίες ή άλλες πηγές υποστήριξης)
7. Βιβλιογραφία
8. Πίνακες (ο καθένας σε χωριστή σελίδα)
9. Εικόνες (η καθεμία σε χωριστή σελίδα)
10. Τίτλους εικόνων
140
ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ ΣΥΓΓΡΑΦΕΙΣ
141 ΟΔΗΓΙΕΣ ΠΡΟΣ ΤΟΥΣ ΣΥΓΓΡΑΦΕΙΣ
crea v e o m m u n ca o n serv ce s crea v e o m m u n ca o n serv ce s
