SPRAWDŹ
Pacjenci z chorobą
Fabry’ego mogą żyć dłużej i lepiej s5
EKSPERT
HAE to obrzęk zagrażający życiu s6
WAŻNE
Potrafimy w Polsce dobrze leczyć i diagnozować SMA s9
INSPIRACJE
Choroba Stilla dziś nie zatrzyma mojej córki! s10


SPRAWDŹ
Pacjenci z chorobą
Fabry’ego mogą żyć dłużej i lepiej s5
EKSPERT
HAE to obrzęk zagrażający życiu s6
WAŻNE
Potrafimy w Polsce dobrze leczyć i diagnozować SMA s9
INSPIRACJE
Choroba Stilla dziś nie zatrzyma mojej córki! s10


Prof. dr hab. n. med. Katarzyna Kotulska-Jóźwiak W ataksji Friedreicha celem jest spowolnienie progresji

Prof. dr hab. n. med. Anna Liberek Polscy pacjenci z PFIC mogą być skutecznie leczeni

Prof. dr hab. n. med. Marek Hus Czym charakteryzuje się zaawansowana mastocytoza układowa?

Prof. dr hab. n. med.
Anna Latos-Bieleńska Genetyka kluczem do diagnostyki chorób rzadkich
Senior Business Developer: Magdalena Nędza (+48 530 746 777, magdalena.nedza@mediaplanet.com) Content and Production Manager: Izabela Krawczyk Managing Director: Krystyna Miłoszewska
Skład: Mediaplanet Web Editor & Designer: Tatiana Anusik Fotografie: stock.adobe.com, zasoby własne Kontakt e-mail: pl.info@mediaplanet.com Adres: MEDIAPLANET PUBLISHING HOUSE SP. Z O.O., ul. Zielna 37, 00-108 Warszawa
Rządowy Plan dla Chorób Rzadkich zakłada wprowadzenie platformy informacyjnej, rejestru pacjentów i chorób oraz cyfrowej Karty Pacjenta. Rząd zamierza też zwiększyć liczbę ośrodków wyspecjalizowanych w chorobach rzadkich.

Urszula
Podsekretarz Stanu w Ministerstwie Zdrowia; profesor nauk medycznych, ma specjalizacje w dziedzinie chorób wewnętrznych (drugiego stopnia), immunologii, alergologii i diagnostyki laboratoryjnej
Jak wygląda aktualnie proces diagnozy w kierunku chorób rzadkich w Polsce? Większość tych chorób ma na tyle nietypowe i niespecyficzne objawy, że samo postawienie szybkiej i właściwej diagnozy jest wyzwaniem. W związku z tym chorzy narażeni są na tzw. odyseję diagnostyczną – przechodzą dziesiątki badań, wizytują wielu specjalistów i lekarzy, narażeni są na nerwy i niepewność. Choroby rzadkie dotykają m.in. niemowląt, u których trzeba być szczególnie wyczulonym na wszelkie sygnały dotyczące nieprawidłowego rozwoju. Jeśli pacjent trafi do wyspecjalizowanego ośrodka, na ogół w dużym mieście, to może liczyć na szybką diagnozę, ale dla chorych z mniejszych miast ta odyseja diagnostyczna jest bardziej uciążliwa. Jako Ministerstwo Zdrowia przygotowaliśmy Plan dla Chorób Rzadkich, który ma na celu m.in. skrócenie tej ścieżki – od pierwszych objawów po finalną diagnozę i objęcie chorego opieką zespołu specjalistów.
Na jaką pomoc mogą liczyć chorzy? Obecnie w Polsce mamy 44 ośrodki eksperckie, które specjalizują się w konkretnych typach chorób rzadkich. Oceniamy, że w Polsce mamy ok. 3 mln osób z chorobami rzadkimi, co oznacza, że tych ośrodków jest za mało. Tym bardziej, że choroby dotyczą różnych narządów – od przypadków neurologicznych, przez choroby nerek, serca, skóry, aż po narządy endokrynne. Czasami mamy do czynienia z zespołami chorób kilku narządów. Plan dla Chorób Rzadkich zakłada rozszerzanie sieci tych ośrodków na każde województwo.
Jak przebiega wdrażanie Planu dla Chorób Rzadkich na lata 2024-2025 i jakie są jego kluczowe punkty?
mało znane, więc informacje o dostępnych lekach i terapiach, o ośrodkach, o ryzyku wystąpienia choroby u kolejnego dziecka są niezwykle cenne. I to nie tylko dla chorych, ale też dla lekarzy z Podstawowej Opieki Zdrowotnej. Plan zakłada także powstanie pełnego rejestru osób z chorobami rzadkimi oraz cyfrowej Karty Pacjenta. To pozwoli nam ocenić, ile mamy dokładnie przypadków każdej choroby, jakie są efekty leczenia, gdzie występują największe braki i potrzeby. Rejestr będzie też przydatnym narzędziem badawczym dla lekarzy i naukowców, by mogli poszerzać swoją wiedzę o tych chorobach.





Czytaj więcej : chorobyrzadkie.com


Poprzedni plan, na lata 2022-23, wygasł wraz z końcem 2023 r., więc musieliśmy przeprowadzić proces legislacyjny na nowo. Uchwała Rady Ministrów została podjęcia w sierpniu, dokument jest uchwalony i można go wdrażać. Plan zawiera wiele elementów ważnych dla pacjentów – np. ruszyła platforma informacyjna dotycząca chorób rzadkich, gdzie chorzy i ich bliscy mogą pozyskać wiele cennych informacji. Choroby te są








Obecnie
w Polsce
mamy
44 ośrodki eksperckie, które specjalizują się w konkretnych typach chorób rzadkich.
Jakie inne inicjatywy podejmuje ministerstwo, aby poprawić systemową opiekę i dostęp do leków dla pacjentów z chorobami rzadkimi?
Wprowadzamy nowe terapie, wdrażamy w Polsce nowoczesne metody leczenia, które pojawiają się w najlepiej rozwiniętych krajach świata. Chcemy, żeby jak najwięcej tych metod było refundowanych i dostępnych także dla polskich pacjentów. Barierą są przepisy prawne, bo nie zawsze producenci tych nowoczesnych leków są otwarci na negocjacje i wprowadzenie leku do polskiego systemu. Ogromnym sukcesem Polski w ostatnim czasie było włączenie do systemu refundacji leku na SMA, czyli rdzeniowy zanik mięśni. Nowa metoda jest krokiem milowym w walce z tą chorobą i cieszymy się, że jest już dostępna również dla polskich pacjentów.











To dokument, który wytycza zadania mające doprowadzić do tego, by choroby rzadkie były w Polsce leczone w sposób optymalny. To ważne, bo mówimy o około 10 tys. jednostek chorobowych i grupie około 3,5 mln pacjentów. Plan dla Chorób Rzadkich ma dać recepty na to, jak przyspieszyć diagnostykę i zwiększyć dostęp do nowoczesnych terapii.

Prof. dr hab. n. med.
Alicja Chybicka
Lekarka i polityk, profesor nauk medycznych, profesor
Uniwersytetu
Medycznego we Wrocławiu. Przez 21 lat kierowała obecną kliniką
Przylądek Nadziei we Wrocławiu. Była prezes
Polskiego Towarzystwa
Pediatrycznego, senator VIII i X kadencji, posłanka na Sejm VIII i X kadencji.
Jakie kluczowe punkty zawiera nowa wersja Planu dla Chorób Rzadkich na lata 2024-2025?
To zwieńczenie ośmioletniej pracy zespołu wybitnych ekspertów. Pierwotnie plan był przyjęty w 2022 roku i nic z niego nie zostało wdrożone. Okazało się jednak, że trzeba go tylko zmodyfikować. To był mój priorytet, kiedy objęłam prowadzenie parlamentarnego zespołu ds. chorób rzadkich.
Plan na lata 2024-2025 dotyczy sześciu obszarów. Pierwszy to powołanie ośrodków eksperckich chorób rzadkich, tzw. OCR-ów, oraz analiza produktów rozliczeniowych dla nich. Dziesięć ośrodków zostało już wybranych, a będzie ich łącznie 42. Obszar drugi to poprawa dostępu do badań diagnostycznych. To bardzo ważna, wręcz kluczowa sprawa, bo w tej chwili każdy pacjent przeżywa odyseję diagnostyczną, krążąc latami między lekarzami i nie słysząc poprawnej diagnozy. Trzeci punkt to poprawa dostępu do leków i środków specjalnego przeznaczenia żywieniowego. Droga do refundacji jest obecnie bardzo długa. Tracimy czas, a ceną tego niestety jest życie części pacjentów. Obszar czwarty to polski rejestr chorób rzadkich, w którym znajdzie się każda osoba cierpiąca na takie schorzenia. Kolejny dotyczy karty pacjenta z chorobą rzadką, która będzie tworzona w formie elektronicznej. Osoby starsze dostaną ją też w formie papierowej. Szósty obszar to prowadzenie
platformy informacyjnej. Chodzi o bazę rzetelnej i szczegółowej wiedzy o około 10 tys. chorób.
Jakie jest znaczenie międzynarodowej współpracy w leczeniu chorób rzadkich? To jest kluczowe i żeby to wyjaśnić posłużę się przykładem z innej dziedziny. Każde polskie dziecko, które zachoruje na chorobę nowotworową, jest już na wstępie wpięte w europejski program, oparty na światowych trendach, w którym Polska od początku brała udział. To samo marzy nam się w chorobach rzadkich i tak niestety nie jest, a przynajmniej nie we wszystkich dziedzinach. Europa powinna wytypować swoje najlepsze ośrodki, które zajmą się daną grupą chorób rzadkich.
Jakie jest znaczenie innowacji w diagnostyce i leczeniu chorób rzadkich? Badania diagnostyczne są podstawą tego, co się z pacjentem będzie działo dalej. Kluczowe jest rozszerzenie badań prenatalnych i noworodkowych, co pozwoli szybko postawić diagnozę i skierować dziecko do właściwego ośrodka. Dzięki temu w wielu przypadkach będzie mogło wyrosnąć na zdrowego człowieka. Co ważne, większość, bo aż 80 proc., tych chorób, ma podłoże genetyczne, więc jest potrzebna zaawansowana diagnostyka genetyczna.
Dziś już rzadkie choroby genetyczne często możemy leczyć przyczynowo.
Niestety, wszystkie terapie celowane w tej dziedzinie są opracowywane w Stanach Zjednoczonych, a nie ma żadnego ośrodka w Europie, który mógłby to opracowywać i sprzedawać, a to wydłuża drogę do dostępności dla polskich chorych.
Kluczowe jest rozszerzenie badań prenatalnych i noworodkowych, co pozwoli szybko postawić diagnozę i skierować dziecko do właściwego ośrodka.
Jakie zadania stoją teraz przed rządzącymi, jeśli chodzi o choroby rzadkie?
Teraz czeka nas wdrażanie założeń wynikających z Planu dla Chorób Rzadkich. Do tego potrzebne jest stworzenie odpowiednich ustaw i nowelizacji. Tym, co powinniśmy zrobić niewątpliwie jest opracowanie ustawy o chorobach rzadkich, w której się zawrze wszystko, co będzie do tego potrzebne.
Pacjenci cierpiący na tę chorobę przez wiele lat krążą między różnymi lekarzami zanim usłyszą prawidłową diagnozę. W tym czasie dochodzi do uszkodzenia różnych tkanek. Pacjent cierpi, a zmiany często zagrażają jego życiu. Jak pomóc osobom z chorobą Fabry’ego?

Prof. dr hab.
n. med. Anna
Kostera-Pruszczyk
Klinika Neurologii
Warszawskiego Uniwersytetu Medycznego
Czym jest choroba Fabry’ego i jakie są jej typowe objawy?
To choroba ultrarzadka, która dotyka ok. 1 osoby na 150-180 tys. Zdecydowanie częściej i wcześniej jej objawy mają chłopcy czy mężczyźni. Jej bezpośrednią przyczyną są mutacje genu, który koduje enzym o nazwie alfa-galaktozydaza A, odpowiedzialny za rozkładanie glikosfingolipidów. Dochodzi do ich gromadzenia w różnych tkankach, w tym w nerkach lub sercu. Objawy są zróżnicowane i niespecyficzne, dlatego pacjenci czasem trafiają najpierw do neurologa, a czasem do dermatologa, reumatologa, laryngologa czy okulisty. Częstymi objawami są silny ból kończyn, głównie stóp i dłoni, upośledzona tolerancja wysokich temperatur związana ze zmniejszonym wydzielaniem potu czy grudkowa wysypka, najczęściej na pośladkach i udach. Możliwe są bóle brzucha, nudności, wymioty, zawroty głowy czy omdlenia. Istnieje ryzyko wystąpienia postępującej niewydolności nerek, przebiegającej z białkomoczem, czy przerostu lewej komory
serca. Pokaźną grupę wśród ludzi, którzy w młodym wieku doznają udaru niedokrwiennego mózgu, stanowią właśnie pacjenci z chorobą Fabry’ego.
Jak wygląda proces diagnostyczny i dlaczego wczesne rozpoznanie jest tak ważne?
Niestety, niewielu lekarzy ma doświadczenie w rozpoznawaniu i leczeniu tej choroby, a przekłada się to również na zazwyczaj wieloletnią wędrówkę pacjenta pomiędzy specjalistami. Podstawą diagnostyki jest badanie enzymatyczne, możliwe do przeprowadzenia przy pobraniu krwi na bibułę. Pacjent nie musi być nawet na czczo. Powinno zostać wykonane u pacjenta z takimi właśnie wieloukładowymi objawami. Rozpoznanie choroby Fabry’ego jest możliwe, jeśli uwzględniamy ją w diagnostyce różnicowej. Warto podkreślić, że w przypadku tej choroby jest dostępne leczenie, również w programie lekowym NFZ. Pozwala ono łagodzić objawy, do których już doszło, ale też znacząco chroni przed postępem choroby.
Jak poprawić wykrywalność choroby?
Coraz więcej krajów rozważa możliwość rozpoznawania tej choroby w okresie przedobjawowym, np. poprzez włączenie jej do programu badań przesiewowych u noworodków.
Niewielu lekarzy ma doświadczenie w rozpoznawaniu i leczeniu tej choroby, a przekłada się to również na zazwyczaj wieloletnią wędrówkę pacjenta pomiędzy specjalistami.
Dzięki temu można aktywnie monitorować stan pacjenta i zapewnić mu leczenie wtedy, kiedy pojawią się pierwsze objawy. Potrzebne są też działania edukacyjne i czujność diagnostyczna.
WYZWANIA
Pacjenci cierpiący na chorobę Fabry’ego mierzą się nie tylko z jej poważnymi skutkami zdrowotnymi. Dla wielu z nich problemem są też częste i niekiedy dalekie podróże, które muszą odbywać, by otrzymać leczenie.

Anna Moskal
Prezes Stowarzyszenia
Rodzin z Chorobą Fabry’ego
Jakie są największe wyzwania, z którymi mierzą się pacjenci z chorobą Fabry’ego?
To chociażby problemy z uzyskaniem orzeczenia o niepełnosprawności i z dostępem do świadczeń socjalnych, takich jak renty. Orzecznicy często wydają decyzje odmowne, od których pacjenci muszą się odwoływać. Problemem jest także uwzględnienie harmonogramu leczenia w życiu codziennym, szczególnie jeśli mówimy o enzymatycznej terapii zastępczej, która wymaga wizyt w szpitalu co dwa tygodnie. Chorzy mierzą się też z tym, co wynika z samej choroby. To m.in. przewlekłe zmęczenie, problemy ze wzrokiem czy kłopoty natury neurologicznej. Często muszą przez to rezygnować z życia zawodowego.
Jak ważna jest rola pacjentów w procesie podejmowania decyzji dotyczących ich leczenia?
Zarówno w tej, jak i w każdej innej chorobie decyzja o rozpoczęciu leczenia i jego formie powinna być podejmowana w porozumieniu pomiędzy lekarzem i pacjentem. Nie powinniśmy się skupiać wyłącznie na aspektach medycznych.
Trzeba też uwzględnić potrzeby, plany i sytuację życiową oraz zawodową pacjenta.
Jakie są oczekiwania pacjentów z chorobą Fabry’ego i na jakie zmiany w systemie opieki zdrowotnej czekają?
Obecnie największą potrzebą są dla nas dożylne podania leku w warunkach domowych. Pacjenci z Fabrym rozsiani są po całym kraju i nie każdy ma możliwość szybkiego dotarcia do placówki realizującej program lekowy. Ważny jest w tym też aspekt ekonomiczny, bo czasem wiąże się to z koniecznością odbywania nawet kilkusetkilometrowych podróży przez wiele lat – i to co dwa tygodnie. Oczywiście dostępne jest również leczenie doustne, z którego niestety nie każdy pacjent może skorzystać. Potrzebny jest też lepszy dostęp do rehabilitacji i konsultacji specjalistycznych.
Gdzie pacjenci mogą szukać wsparcia i informacji o chorobie?
W pierwszej kolejności polecałabym kontakt z naszym stowarzyszeniem. Staramy się przekierowywać pytania od
pacjentów na adres mailowy lekarza, który z nami współpracuje. Co ważne, mamy merytoryczne wsparcie Rady Medycznej, dzięki czemu informacje, które trafiają od nas do pacjentów, są sprawdzone. Mamy też grupę na Facebooku. Tam chorzy wymieniają się swo-
Czytaj więcej : chorobyrzadkie.com
Pacjenci z Fabrym rozsiani są po całym kraju i nie każdy ma możliwość szybkiego dotarcia do placówki realizującej program lekowy.
imi doświadczeniami. Od niedawna na naszej stronie dostępny jest „Przewodnik w Chorobie Fabry’ego”, gdzie zebraliśmy najważniejsze informacje na temat choroby, diagnostyki oraz leczenia.

Polska jest ostatnim krajem w Europie, który zdecydował się na finansowanie terapii dla pacjentów w chorobie Fabry’ego. Tymczasem leczenie sprawia, że chorzy stopniowo wracają do aktywności społecznej i zawodowej. Jakość ich życia jest uwarunkowana również tym, w jakiej formie otrzymują leczenie.

Dr hab. n. med.
i n. o zdr. Longin
Niemczyk
Klinika Nefrologii, Dializoterapii i Chorób
Wewnętrznych, Warszawski
Uniwersytet Medyczny
Jakie terapie dostępne są dla polskich pacjentów z chorobą Fabry’ego? Pacjentom możemy zaproponować enzymatyczną terapię zastępczą, a dla pacjentów z obecnością wrażliwej mutacji genetycznej dostępne jest leczenie w formie doustnej w tabletkach.
W jaki sposób terapie poprawiają komfort życia pacjentów?
Wszystkie wymienione terapie są skuteczne i poprawiają jakość życia naszych pacjentów. W trakcie leczenia zmniejszają się dolegliwości bólowe i poprawia się też termoregulacja To ważne, bo u nieleczonych pacjentów z chorobą Fabry’ego obserwujemy zmniejszone wydzielanie potu, a przez to chorzy przegrzewają się i mają udary cieplne. Pod wpływem leczenia pojawia się pocenie, które dla zdrowych ludzi jest właśnie podstawowym mechanizmem termoregulacyjnym. Chorzy, zwłaszcza ci jeszcze niezdiagnozowani i nieleczeni, mają problemy z normalną aktywnością, bo ból nie pozwala im wstać z łóżka. Często muszą brać zwolnienia, przez co niejednokrotnie tracą pracę. Pacjenci, którzy mają mniej dolegliwości bólowych czy zawrotów głowy, na pewno lepiej będą funkcjonować w społeczeństwie. Należy mieć jednak na uwadze to, że każda wizyta w szpitalu wpływa na jakość życia pacjenta, a osoby leczone za pomocą wlewów enzymatycznych muszą zgłosić się na leczenie do placówki medycznej co drugi tydzień.
Dodatkowo wszyscy pacjenci dwa razy w roku przez kilka dni przechodzą diagnostykę konieczną do kwalifikacji do kontynuacji leczenia. Dlatego diagnostyka i leczenie powodują ograniczenie aktywności, też zawodowej, pacjentów na blisko miesiąc w ciągu roku. Pacjenci, którzy przyjmują leki doustne, mają trochę lepiej, bo widzimy się z nimi dwa razy w roku na badaniach kontrolnych i dostają od nas lek, a przyjmują go już w domu.
Jakie są kryteria kwalifikacji pacjenta do leczenia?
Podstawą kwalifikacji chorego do leczenia jest potwierdzenie choroby w badaniach genetycznych, a następnie wykonanie badań potwierdzających uszkodzenia narządowe. W przypadku terapii doustnej musi być genetycznie potwierdzona wrażliwa mutacja, która się poddaje takiemu leczeniu. Pozostali pacjenci są kwalifikowani do jednej z dostępnych terapii dożylnych. Ich skuteczność jest zbliżona, więc decyzja o tym, którą wdrożymy, w dużej mierze jest podejmowana przez chorego. Wlewy różnią się dawką i tym, jak długo trwa podanie leku. Terapię enzymatyczną można rozpocząć w wieku co najmniej 8 lat, a leczenie doustne nie wcześniej niż po ukończeniu 16. roku życia.
Czy pacjenci powinni mieć wpływ na swoje leczenie? Mają przecież swoje plany, potrzeby, preferencje.
Zawsze bierzemy pod uwagę te czynniki i staramy się dostosować do pacjenta. Nie zawsze jednak możemy spełnić jego życzenie, bo nie mamy wpływu np. na obecność mutacji pozwalającej na leczenie formą doustną. Przyjmuje się jednak, że z tej formy leczenia może korzystać 30-35 proc. chorych.
Chorzy, zwłaszcza ci jeszcze niezdiagnozowani i nieleczeni, mają problemy z normalną aktywnością, bo ból nie pozwala im wstać z łóżka.
Jakie szanse daje chorym dostęp do terapii i jak ten dostęp pozycjonuje nas na tle Europy?
Dostęp do terapii daje pacjentom szansę na dłuższe i lepsze życie. Polska jest natomiast ostatnim krajem w Europie, który rozpoczął aktywne leczenie choroby Fabry’ego, finansowane ze środków publicznych. W tym momencie już całkiem nieźle gonimy Europę. Mamy coraz więcej pacjentów i mamy już też narzędzia do tego, żeby ich diagnozować i leczyć.
Czytaj więcej : chorobyrzadkie.com



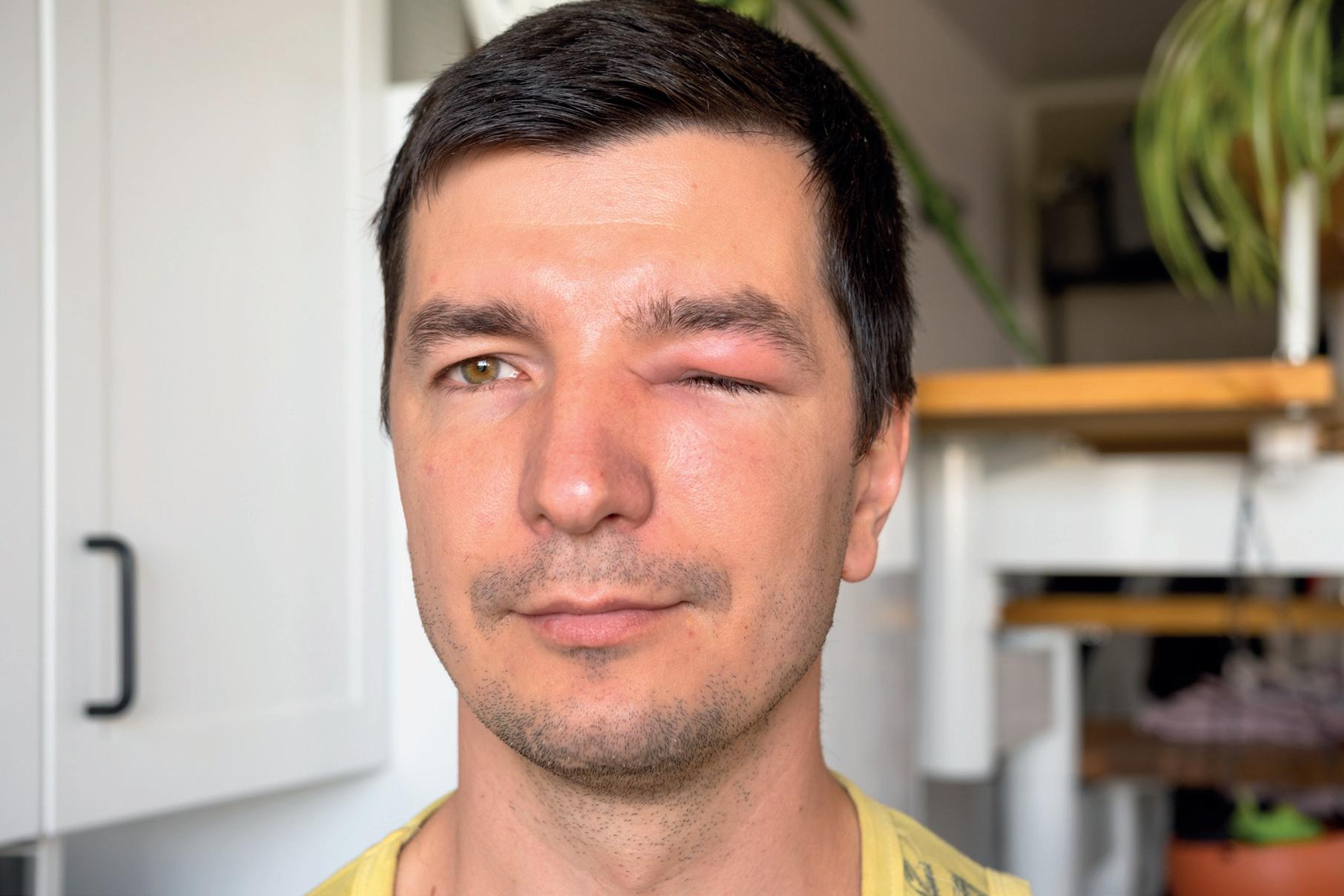
Wrodzony obrzęk naczynioruchowy (HAE) to schorzenie, które prowadzi do znacznego pogorszenia jakości życia, a w niektórych przypadkach grozi nawet śmiercią. W Polsce jest możliwe leczenie obrzęków ostrych HAE oraz, od niedawna, leczenie profilaktyczne. Brakuje refundacji dla profilaktyki długoterminowej dla kobiet w ciąży.
Czytaj więcej : chorobyrzadkie.com
Czym właściwie jest wrodzony obrzęk naczynioruchowy?
To choroba genetyczna, dziedziczona w sposób autosomalny dominujący, co oznacza, że 50 proc. potomstwa ma ryzyko odziedziczenia jej. Choroba dotyka zarówno kobiet, jak i mężczyzn, przy czym w 60-75 proc. chorują panie. W Polsce mamy obecnie rozpoznanych około 600 pacjentów.
Jak i kiedy objawia się ta choroba?
Objawia się występowaniem napadowych obrzęków, które dotyczą np. dłoni, stóp, goleni, pleców czy pośladków, ale też krtani, gardła
Obrzęki bradykininowe na pierwszy rzut oka są podobne do histaminowych, ale narastają wolniej i dłużej się utrzymują.
i szyi lub jamy brzusznej. Dwa ostatnie przypadki to stany zagrażające życiu. W pierwszym związane jest to z zamknięciem lub znacznym ograniczeniem pasażu powietrza przez
górne drogi oddechowe. W drugim może dojść do znacznego spadku ciśnienia tętniczego.
U podstaw choroby leży brak białka inhibitora C1-esterazy, przez co występuje nadmierna produkcja bradykininy. Konsekwencją tego jest rozszczelnienie naczyń krwionośnych i przesiąkanie płynu z łożyska naczyniowego do okolicznych tkanek. Czynnikami wywołującymi napady są najczęściej stres oraz równomierny ucisk w jednym miejscu. Bardzo często są one też wywoływane przez estrogeny, więc u kobiet do nasilenia objawów dochodzi podczas dojrzewania, w okresie okołomiesiączkowym i w ciąży.
Jak choroba jest rozpoznawana?
Do wstępnego rozpoznania wystarczy dobrze zebrany wywiad. Obrzęki bradykininowe na pierwszy rzut oka są podobne do histaminowych, ale narastają wolniej i dłużej się utrzymują. Nie reagują na sterydy, leki przeciwhistaminowe i adrenalinę. Rozpoznanie potwierdza się poprzez badania laboratoryjne – ocenę funkcji i poziomu inhibitora C1-esterazy.
Jakie konsekwencje dla zdrowia i komfortu życia niesie wrodzony obrzęk naczynioruchowy?
Najpoważniejszą konsekwencją choroby może być śmierć. Z kolei obrzęki obwodowe nie zagrażają życiu, ale znacznie pogarszają jego jakość. Proszę sobie wyobrazić osobę, u której dwa razy w miesiącu występuje trwający pięć dni obrzęk stopy i nie może założyć
butów. Mam dwie pacjentki, które są siostrami. Każda z nich ma po dwie szafy. W jednej trzyma ubrania w swoim normalnym rozmiarze, a w drugiej te na okres rzutu choroby.
Jak wygląda leczenie?
Pierwsza refundowana ścieżka to leczenie ostrych napadów, zwłaszcza tych zagrażających życiu. Drugą jest profilaktyka krótkoterminowa, którą stosujemy w przypadku konieczności przeprowadzenia zabiegów medycznych, laryngologicznych czy stomatologicznych. Trzecia, na której nam najbardziej zależy, to już nie leczenie napadów, ale zapobieganie im. To długoterminowe leczenie, refundowane u osób spełniających ścisłe kryteria włączenia.
Co jest jeszcze potrzebne, by polscy pacjenci mogli być leczeni optymalnie?
Te potrzeby dotyczą kobiet w ciąży. Z jednej strony nie ma przeciwwskazań do tego, żeby nasze pacjentki rodziły dzieci. Z drugiej – jednym z czynników wywołujących napady są estrogeny. Chore z reguły fatalnie znoszą ciążę. Doświadczają zaostrzenia i napadów, niestety najczęściej dotyczących jamy brzusznej, połączonych z wymiotami, silnymi bólami oraz biegunkami. Walczymy o to, by zapewnić im dostęp do stosowanej na całym świecie profilaktyki długoterminowej. Teoretycznie możemy w Polsce stosować leczenie zapobiegawcze u kobiet z wrodzonym obrzękiem naczynioruchowym w ciąży, ale niestety bez refundacji, a koszt prowadzenia takiej ciąży to wtedy ponad milion złotych.

Zeylandów
Codzienność
Codzienne życie pacjentów z dziedzicznym obrzękiem naczynioruchowym (HAE) pełne jest wyzwań, zarówno fizycznych, jak i psychicznych.
Nieprzewidywalność ataków, ich wpływ na życie zawodowe oraz rodzinne, a także ograniczenia systemowe w dostępności do leczenia znacząco utrudniają prowadzenie normalnej egzystencji.
Jakie są najczęstsze wyzwania pacjentów z HAE w życiu codziennym?
Pacjenci z HAE, zwłaszcza ci z częstymi atakami, doświadczają ograniczeń w codziennym życiu. Napady choroby są nieprzewidywalne, wywoływane m.in. przez stres, infekcje, obniżoną odporność, a w przypadku kobiet przez ciążę i menstruację. Aż 87 proc. pacjentów wskazuje stres jako czynnik wywołujący ataki. W przypadku częstych ataków, które pojawiają się kilka razy w miesiącu, choroba ogranicza aktywność zawodową, edukacyjną, społeczną i towarzyską. Pacjenci często boją się podróżować, zakładać rodziny czy nawet mieć dzieci. Wielu z nich żyje w chronicznym stresie, co dodatkowo wpływa na ich zdrowie psychiczne.
Jakiej pomocy potrzebują pacjenci z HAE w życiu zawodowym i rodzinnym?
Najważniejsze jest zagwarantowanie pacjentom stałego dostępu do leczenia – zarówno
doraźnego, jak i zapobiegawczego. Obecnie ograniczona dostępność terapii wpływa na poczucie bezpieczeństwa pacjentów, którzy nie mogą być pewni, czy w razie potrzeby będą mieli dostęp do odpowiednich leków. Nadal pacjentki w ciąży i kobiety karmiące nie mają zapewnionej profilaktyki napadów HAE. Konieczna jest reforma systemowa, która zagwarantuje nieograniczoną refundację terapii dla chorych z ultrarzadkimi chorobami, takimi jak HAE. Powinien to być dla decydentów priorytet, zwłaszcza w kontekście ostatnich wydarzeń, czyli przyjęcia przez Radę Ministrów Planu dla Chorób Rzadkich na lata 2024-2025.
Jak stowarzyszenie Pięknie Puchnę wspiera pacjentów z HAE?
Stowarzyszenie Pięknie Puchnę działa od 2005 roku, zwiększając świadomość choroby zarówno wśród pacjentów, jak i lekarzy. Dążymy do skrócenia czasu diagnozy oraz zapewnienia

Michał Rutkowski jest współzałożycielem i prezesem Zarządu Polskiego Stowarzyszenia Pomocy Chorym z Obrzękiem Naczynioruchowym Pięknie Puchnę. Jest również wiceprezydentem międzynarodowej organizacji parasolowej działającej na rzecz chorych z dziedzicznym obrzękiem naczynioruchowym – HAE International.
nieograniczonej dostępności do leczenia. Organizujemy konferencje, warsztaty, wspieramy badania kliniczne i dostarczamy pacjentom nowoczesne technologie monitorowania objawów. Jesteśmy także w stałym kontakcie z decydentami, informując o potrzebach chorych. Warto podkreślić, że jesteśmy organizacją całkowicie neutralną, dla której zawsze na pierwszym miejscu jest pacjent. Nasza oferta przeznaczona dla chorych jest bardzo rozwinięta i daje pacjentom wiele możliwości.
Czytaj więcej : chorobyrzadkie.com




INSPIRACJE
Rdzeniowy zanik mięśni (SMA) powstaje na skutek mutacji w genie SMN1. Jeśli terapia genowa zostanie zastosowana przed wystąpieniem pierwszych objawów, jest w stanie zapobiec pojawieniu się symptomów choroby.

Maria Mazurkiewicz-Bełdzińska Kierownik Kliniki
Neurologii Rozwojowej, Gdański Uniwersytet Medyczny, przewodnicząca
Zarządu Głównego Polskiego Towarzystwa Neurologów Dziecięcych
Czym charakteryzuje się rdzeniowy zanik mięśni i jaki jest profil chorego?
W wyniku wady genetycznej obumierają neurony w rdzeniu kręgowym, co w konsekwencji prowadzi do osłabienia i zanikania mięśni szkieletowych oraz ostatecznie – osłabienia siły mięśniowej do całkowitego porażenia. U większości przypadków pierwsze objawy pojawiają się w okresie niemowlęctwa lub wczesnego dzieciństwa, jednak mogą wystąpić też później – w zależności od momentu pojawienia określany jest typ SMA (wyróżniamy typy opisane liczbami od 1 do 4, gdzie 1 wskazuje na wystąpienie pierwszych objawów przed 6. miesiącem życia, a 4 – powyżej 30. roku życia). Na SMA w Polsce choruje około 1000 osób. Ponadto rocznie rodzi się tu około 50 dzieci, u których rozpoznawane jest SMA, a u około 35 z nich choroba ma ostry przebieg, czyli rozpoczyna się w okresie niemowlęcym.
Jak wygląda proces diagnostyczny SMA? Obecnie dzieci rodzące się w Polsce mają wykonywane badanie przesiewowe m.in. w kierunku SMA. Dzięki tak wcześnie
wdrożonej diagnostyce możliwe jest zastosowanie leczenia jeszcze przed wystąpieniem pierwszych objawów, co daje chorym szanse na normalne życie.
Czym jest program leczenia SMA? Na czym polega leczenie przyczynowe terapią genową?
Od 2019 r. chorzy mogą skorzystać z nowoczesnych terapii w ramach refundowanego programu lekowego. Obecnie leczenie obejmuje stosowanie trzech substancji, w tym także od 2022 r. terapii genowej, która jest najnowocześniejszym osiągnięciem medycyny – działa na przyczynę choroby, poprawiając działanie nieprawidłowych genów. Co ważne, wdrożenie leczenia przed wystąpieniem pierwszych objawów jest w stanie zapobiec pojawieniu się symptomów choroby. Terapie te są bezpłatne, a skorzystanie z nich możliwe po zakwalifikowaniu do programu lekowego.
Co ubiegły rok przyniósł polskim pacjentom z SMA?
Przede wszystkim możemy pochwalić się tym, że program leczenia SMA
w Polsce obejmuje wszystkie zarejestrowane na świecie nowoczesne terapie, które w połączeniu z wdrożonym w marcu 2022 r. programem badań przesie-
Dzięki wcześnie wdrożonej diagnostyce możliwe jest zastosowanie leczenia jeszcze przed wystąpieniem pierwszych objawów, co daje chorym szanse na normalne życie.
wowych noworodków zmieniają życie chorych i ich rodzin na lepsze. Trzeba podkreślić, że terapia genowa wymaga jedynie jednorazowego podania substancji, a dzięki niej dziecko może być w pełni sprawne i rozwijać się jak jego zdrowi rówieśnicy.
WYZWANIA
Obecny program lekowy obejmuje trzy terapie, m.in. terapię genową. Leczenie i badania przesiewowe sprawiły, że dzieci rozwijają się jak ich zdrowi rówieśnicy.

Dorota Raczek
Prezes Fundacji SMA od 2020 roku, żona i matka syna i dwóch córek – jedna z nich choruje na SMA; PR-owiec z wykształcenia, dyrektor zarządzający szczególnie domowym ogniskiem, kobieta ze Śląska
Jakie wyzwania na co dzień stawia przed pacjentami i ich opiekunami diagnoza rdzeniowego zaniku mięśni (SMA)?
Do tej pory SMA było chorobą postępującą, dlatego regularną fizjoterapią można było jedynie opóźnić jej skutki, a to sprawiało, że codzienność pacjentów była wymagająca. Jednak po wdrożeniu programu lekowego i zastosowaniu nowoczesnych terapii u dzieci przed wystąpieniem pierwszych objawów leczenie jest w stanie zapobiec pojawieniu się choroby, dzięki czemu chorzy po terapiach programu lekowego mogą rozwijać się jak ich zdrowi rówieśnicy. Czekamy jeszcze na terapie, które mogą wspomóc mięśnie. Trzeba pamiętać, że nadal jest grupa chorych, których codzienność wymaga m.in. rehabilitacji zwiększającej sprawność mięśni, stosowania wentylacji po utracie umiejętności oddychania, karmienia dojelitowego w przypadku utraty zdolności przełykania czy stosowania sprzętów ułatwiających poruszanie. Musimy pamiętać, że są to osoby
potrzebujące specjalistycznej, skoordynowanej opieki.
Gdzie pacjenci i ich opiekunowie mogą szukać wsparcia i informacji o chorobie?
Zachęcam do skontaktowania się z naszą fundacją, która działa już od ponad 10 lat, dlatego jesteśmy w stanie odpowiadać na różne potrzeby chorych, a także służyć jako rzetelne źródło wiedzy. Wiemy, że otrzymanie diagnozy SMA jest szokujące, dlatego wspieramy na każdym etapie choroby – zarówno rodziców najmłodszych dzieci, które zdiagnozowano dzięki programowi badań przesiewowych, jak również chorych, u których wykryto chorobę po pojawieniu się objawów.
Jakie są główne trudności i ograniczenia związane z chorobą? Co w tym kontekście zmienił przełom w dostępie do leczenia SMA w Polsce? Od końca marca 2022 r. funkcjonuje program badań przesiewowych m.in. w kierunku wykrywania SMA, dzięki czemu diagnoza stawiana jest szybko i tym
samym noworodek może być kwalifikowany do nowoczesnego leczenia, w tym np. terapii genowej, które pozwala zatrzymać chorobę. To niewątpliwie odpowiedź na potrzeby tej grupy chorych, bo do 2019 r. trudności i ograniczenia wynikały z braku odpowiedniej diagnostyki i dostępu do refundowanego, innowacyjnego leczenia.
Jak terapia genowa zmieniła życie pacjentów z SMA i ich rodzin?
Zmieniła je zdecydowanie na lepsze, bo po wdrożeniu leczenia widzimy, że te rodziny funkcjonują prawie normalnie. U małych pacjentów rozwój fizyczny jest prawidłowy, czyli potrafią m.in. siadać, raczkować i chodzić, zupełnie tak jak ich zdrowi rówieśnicy, z czego niewątpliwie wszyscy się cieszymy. Poza tym jednorazowe podanie leku w terapii genowej wydaje się najlepszym wyjściem, jeżeli chodzi o noworodki z SMA – pośrednim efektem leczenia jest też to, że zyskuje na nim cała rodzina, której życie toczy się po prostu normalnie.


Rdzeniowy zanik mięśni (SMA) jest uwarunkowaną genetycznie chorobą, charakteryzującą się postępującą utratą neuronów ruchowych. Prowadzi do osłabienia mięśni i niepełnosprawności fizycznej oraz niewydolności oddechowej. Na ciężkość przebiegu choroby wpływa liczba kopii genu SMN2. Im jest wyższa, tym późniejszy początek choroby i łagodniejszy jej przebieg. Zawsze jednak wiąże się on z postępującą niepełnosprawnością.
Czytaj więcej : chorobyrzadkie.com
Polski program leczenia rdzeniowego zaniku mięśni, który w obecnym kształcie funkcjonuje od 1 września 2022 roku, jest uznawany za modelowy. Obejmuje wszystkie trzy dopuszczone do stosowania opcje leczenia, w tym terapię genową, którą podaje się raz w życiu. Obecnie w Polsce może być ona stosowana u niemowląt w wieku do 6 miesięcy, mających nie więcej niż 3 kopie genu SMN2. Dzięki badaniom przesiewowym noworodków chorobę można zdiagnozować i leczyć jeszcze przed pojawieniem się objawów. Jak najszybsze rozpoczęcie terapii jest kluczowe zwłaszcza w przypadku



chorych z jedną lub dwoma kopiami genu SMN2. Jak podkreślają specjaliści w dziedzinie neurologii, ale też eksperci ochrony zdrowia i farmakoekonomiści, leczenie możliwie najszerszej populacji chorych na SMA przy pomocy wszystkich zarejestrowanych w tym wskazaniu leków jest opłacalne pomimo bardzo wysokich kosztów jednostkowych. Pozwala to pozytywnie modyfikować przebieg choroby, która nieleczona prowadzi do głębokiej niepełnosprawności lub śmierci. Co ważne, opcja ta jest leczeniem preferowanym przez rodziców pacjentów.
Klinicyści specjalizujący się w leczeniu SMA uważają ponadto, że należy rozważyć możliwość stosowania terapii genowej w Polsce również w populacji
Jak najszybsze rozpoczęcie terapii jest kluczowe zwłaszcza w przypadku chorych z jedną lub dwoma kopiami genu SMN2.
pacjentów z czterema kopiami genu SMN2. Światowe doświadczenia pokazują, że włączenie jej w tej grupie chorych pozwala na osiąganie kamieni milowych w rozwoju w czasie zbliżonym do zdrowych dzieci.
Prof. dr hab. n. med. Katarzyna Kotulska-Jóźwiak Przewodnicząca Zespołu Koordynacyjnego ds. leczenia chorych na rdzeniowy zanik mięśni, kierownik Kliniki Neurologii i Epileptologii Instytutu „Pomnik – Centrum Zdrowia Dziecka”
Co dla pacjentów z SMA i ich rodzin oznacza możliwość zastosowania terapii genowej? Podanie terapii ma na celu zahamowanie procesu chorobowego, czyli obumierania komórek ruchowych rdzenia kręgowego (motoneuronów). Oznacza to, że terapia daje możliwość uratowania tych komórek, które jeszcze nie zostały nieodwracalnie uszkodzone wskutek choroby. Działanie leku, zarówno terapii genowej, jak i nusinersenu oraz trzeciej zarejestrowanej w SMA cząsteczki – rysdyplamu – polega na zwiększaniu ilości brakującego
białka SMN. W przypadku terapii genowej odbywa się to dzięki dostarczeniu prawidłowej kopii genu produkującego to białko. Gen ten nie zastępuje zmutowanego genu, tylko funkcjonuje obok. Kluczową kwestią dla powodzenia terapii jest czas, rozumiany zarówno jako wiek pacjenta, jak i etap choroby. Dotyczy to wszystkich trzech leków stosowanych w SMA. Terapia genowa jest najbardziej skuteczna u pacjentów, którzy otrzymają ją w okresie przedobjawowym, czyli zanim choroba spowoduje spustoszenie. Wówczas istnieje szansa
na to, że objawy SMA w ogóle się nie rozwiną albo jeśli się rozwiną, to będą stosunkowo niewielkie i nie będą wpływały istotnie na jakość życia pacjenta. Wiele tak wcześnie leczonych dzieci rozwija się prawidłowo, tj. zgodnie z kalendarzem rozwojowym dziecka. Ponadto, z wyników badań klinicznych już wiemy na pewno, że dzięki wczesnemu leczeniu nie dochodzi do najcięższych powikłań choroby, jakimi są niewydolność oddechowa, głęboka niepełnosprawność czy nawet śmierć z powodu rdzeniowego zaniku mięśni.
Hania po czterech latach wróciła do szkoły. Może być aktywna fizycznie. Chodzi na basen, wybiera się na dłuższe spacery, wróciła też do jazdy na rowerze – tak o efektach leczenia inhibitorem IL-1beta mówi Justyna Hasiuk, mama 15-letniej Hani, zmagającej się z chorobą Stilla.

Justyna Hasiuk
Mama 15-letniej
Hani, zmagającej się z chorobą Stilla
Jak wyglądała droga Hani do usłyszenia diagnozy?
Pierwsze objawy nie były charakterystyczne dla choroby Stilla. Myślę, że to sprawiło, że czas, jaki upłynął do postawienia diagnozy, był długi. Wszystko zaczęło się w lutym 2018 roku, kiedy Hania miała 9 lat. Obudziła się i nie mogła stanąć. Mówiła, że ma sztywną nogę, boli ją i nie może chodzić. Lekarz powiedział, że może doznała jakiegoś urazu przy zabawie, i kazał podawać jej ibuprofen. Wszystko wróciło do normy, jednak po miesiącu sytuacja się powtórzyła. Lekarz dał skierowanie na badanie krwi, ale wszystko było w normie, więc zalecił suplementację magnezem i potasem oraz znów ibuprofen. Powiedział, że objawy mogą mieć związek z okresem wzrastania. W sierpniu stan Hani pogorszył się gwałtownie. Pojawiła się długotrwała, wysoka gorączka, nawet do 40 stopni. Leki zbijały ją na 4-6 godzin. Lekarze włączali kolejne antybiotyki, pomimo że nie było żadnych cech infekcji. Wskaźniki zapalne były wysokie. Postawiono wstępną diagnozę sepsy. W szpitalu Hania była leczona kolejnymi antybiotykami. Córka praktycznie przestała chodzić, spuchły jej wszystkie palce, skarżyła się na ból w całym ciele i ciągle gorączkowała. Wtedy lekarze zaczęli podejrzewać, że może to uogólniona postać zapalenia stawów. Na oddziale reumatologicznym potwierdzono diagnozę choroby Stilla.
Jak choroba wpłynęła na życie Hani i całej rodziny?
Kiedy pojawił się przełom?
To był moment, kiedy trafiliśmy na leczenie do oddziału reumatologicznego w Krakowie, bo pojawiły się nowe możliwości leczenia. We wcześniejszym ośrodku wyczerpaliśmy wszystkie dostępne opcje. W Krakowie zaproponowano nam leczenie lekiem biologicznym, podawanym podskórnie.

Czytaj więcej : chorobyrzadkie.com
Choroba zmieniła nasze życie. Jej przebieg był bardzo agresywny i córka słabo reagowała na leczenie. Problemy były już nie tylko z normalnym poruszaniem się, ale i nawet z jedzeniem. Musiałam ją karmić, ubierać, pomagać w umyciu się. Hania często i na długo trafiała do szpitala, więc opuszczała dużo lekcji, a ja musiałam brać wolne w pracy. Przeszliśmy na nauczanie indywidualne w domu. Córka musiała zrezygnować z aktywności, które bardzo lubiła, takich jak jazda na rowerze, i straciła kontakt z rówieśnikami. Ja ostatecznie zrezygnowałam z zatrudnienia, a mąż zmienił system pracy. Musieliśmy przeprowadzić w domu gruntowny remont, np. zlikwidować wannę.
Choroba zmieniła nasze życie. Jej przebieg był bardzo agresywny i córka słabo reagowała na leczenie.
Jak dobrane leczenie wpłynęło na życie Hani?
Bóle i sztywności stawów znacznie zmalały, pojawiają się tylko raz na jakiś czas. Sporadycznie zdarzają się stany podgorączkowe. Hania po czterech latach wróciła do szkoły. Bardzo poprawiła się też jej kondycja psychiczna. Ma koleżanki, utrzymuje kontakt z rówieśnikami. Jest weselsza i może być aktywna fizycznie. Chodzi na basen, wybiera się na dłuższe spacery, wróciła też do jazdy na rowerze. Lek jest podawany w zastrzykach podskórnych co cztery tygodnie, więc łatwiej nam cokolwiek zaplanować. Wcześniejszy lek był podawany w kroplówkach, a Hania ma bardzo słabo widoczne żyły. Zakładanie wenflonu zawsze wiązało się u niej ze stresem.
Jakie są teraz potrzeby pacjentów?
Uważam, że bardzo ważne jest to, by w przypadku każdego pacjenta to lekarz prowadzący miał możliwość wyboru odpowiedniej terapii i dostosowania częstotliwości jej podawania. Dostęp do różnych opcji leczenia nie powinien być utrudniony.



Dzięki nowatorskiej metodzie leczenia pacjenci z chorobą Stilla mogą powrócić do normalnego trybu życia. Niestety, wciąż pozostaje problem z właściwą diagnozą tego ciężkiego schorzenia i pełną dostępnością do leczenia.

Prof. KAAFM
dr hab. n. med.
Zbigniew Żuber
Katedra Pediatrii
KAAFM w Krakowie, Klinika Pediatrii
i Reumatologii, Szpital
św. Ludwika w Krakowie
Na czym polega terapia choroby Stilla?
Aby przejść do samej terapii, najpierw trzeba poprawnie zdiagnozować tę chorobę, co niestety jest bardzo trudne. Po pierwsze, jej objawy nie są charakterystyczne – zaczyna się od zwykłej gorączki, bóli stawów czy wysypki. Następnie pojawiają się liczne zmiany narządowe: zaburzenia pracy wątroby i jej powiększenie, wysięki do osierdzia, powiększenie węzłów chłonnych. Objawy mogą wskazywać na zakażenie uogólnione (sepsę) czy ostrą białaczkę, co utrudnia i opóźnia właściwą terapię w kierunku choroby Stilla. Wielu lekarzy nie spotyka się z chorobami układowymi, jedynie specjaliści, jak reumatolodzy i hematolodzy, mają większą świadomość występowania tej choroby. Jest to schorzenie bardzo poważne, które zagraża życiu. Jeśli chodzi o terapię, to podstawowym zadaniem lekarzy jest wyeliminowanie objawów, czyli zablokowanie tzw. burzy cytokinowej, która występuje w chorobie Stilla oraz z jeszcze większą gwałtownością w przebiegu zespołu aktywacji makrofagów, będącego powikłaniem choroby Stilla.
Jakie są najlepsze metody leczenia i czy wszystkie możliwe terapie są dostępne dla polskich pacjentów?
Mamy już w Polsce w różnym zakresie dostępne leczenie, które od wielu lat jest praktykowane w innych krajach. Chodzi o blokery interleukiny pierwszej i blokery interleukiny szóstej (IL-1 i IL-6).
Od dwóch lat w Polsce możemy leczyć blokerami interleukiny pierwszej o krótkim działaniu i o długim działaniu, z czego się bardzo cieszymy, bo mówimy tu o pewnym przełomie w leczeniu choroby Stilla, choć zmniejszenie ograniczeń do inhibitora IL-1 o długim działaniu byłoby pomocne. Te substancje znakomicie poprawiają sytuację polskich pacjentów. Obecnie jest to podstawowa linia leczenia, od której zaczynamy terapię.
Chorzy wracają do pełni zdrowia i normalnego funkcjonowania?
Choroba Stilla ma podłoże genetyczne, nie jesteśmy w stanie całkowicie jej
zlikwidować, ale możemy wyeliminować jej objawy. Trzeba mieć świadomość, że może ona w każdej chwili powrócić. Natomiast dzięki stosowaniu blokerów interleukiny pierwszej i szóstej pacjenci faktycznie mogą normalnie funkcjonować, nie mają żadnych ograniczeń jeśli chodzi o wysiłek fizyczny czy dietę. Chory wraca do normalnego trybu życia, do pracy czy szkoły. Te leki przywracają całkowicie jego sprawność. Pacjent musi jedynie stawiać się na kontrole, natomiast samo przyjmowanie leków – czy to w formie zastrzyków podskórnych, czy doustnie – może odbywać się w warunkach domowych. To jest właśnie ta główna korzyść z zastosowania innowacyjnej metody leczenia.
Jaka jest różnica w częstotliwości podawania leczenia oraz jaki wpływ ma to na jakość życia pacjentów podczas terapii? Długodziałający inhibitor IL-1 beta to lek biologiczny do wstrzykiwań, podawany raz na cztery tygodnie, co stanowi niewielkie obciążenie dla pacjenta z ciężką chorobą. Natomiast w Polsce dostępny jest inhibitor IL-1 w postaci zastrzyków, który podaje się codziennie, co jest dużym obciążeniem – zarówno fizycznym, jak i psychicznym. Jednorazowe wkłucie igły może nie stanowić problemu (choć nie u wszystkich, ze względu na igłofobię), natomiast częste iniekcje mogą prowadzić do wystąpienia odczynów miejscowych oraz skumulowania drobnych urazów. Oba leki są znakomite, jednak powinniśmy mieć możliwość wyboru, zwłaszcza w populacji dziecięcej. Choroba Stilla, będąca rzadką chorobą, częściej występuje u dzieci, a jej leczenie nie stanowi nadmiernego obciążenia finansowego.
Czy wszyscy pacjenci w Polsce kwalifikują się do tego innowacyjnego leczenia? Choroba Stilla to choroba heterogenna i może mieć różny przebieg, a od niego uzależnia się decyzje, jakiego leczenia wymagają konkretne przypadki.
Choroba Stilla może przyjąć formę jednorazowego rzutu, wielu rzutów z regularnymi nawrotami lub utrzymywać się stale. Jeśli przebieg choroby jest łagodny, to w ogóle nie musimy korzystać z tych substancji. Natomiast jeśli objawy są nasilone, to jest to wskazana forma leczenia i wtedy każdy potrzebujący pacjent może być poddany terapii przy zastosowaniu blokerów interleukiny pierwszej i szóstej.
Choroba Stilla ma podłoże genetyczne, nie jesteśmy w stanie całkowicie jej zlikwidować, ale możemy wyeliminować jej objawy.
Jakie zmiany powinny nastąpić, aby polskim pacjentom z tym schorzeniem żyło się lepiej?
Problemem w Polsce jest fakt, że pełna oferta terapeutyczna nie jest dostępna dla wszystkich potrzebujących pacjentów. Część leczenia (blokery interleukiny pierwszej o długim działaniu) jest dostępna tylko w ramach programu RDTL, czyli Ratunkowym Dostępie do Technologii Lekowych, który finansuje stosowanie tej terapii, a nie wszystkie ośrodki lecznicze mogą z niej korzystać. To spore utrudnienie dla pacjenta, który na etapie diagnozy, leczenia i późniejszej kontroli może korzystać tylko z wybranych kilku ośrodków w Polsce. Dostęp w ramach programu lekowego znacząco zmieniłby sytuację pacjentów i lekarzy reumatologów. Druga kwestia dotyczy samej diagnostyki – uważam, że potrzebna jest kampania edukacyjna wśród lekarzy. Przebieg choroby Stilla jest nietypowy, niecharakterystyczny, co utrudnia prawidłową diagnozę i szybkie rozpoczęcie terapii.
Ataksja Friedreicha to postępująca, rzadka choroba genetyczna, która prowadzi do niepełnosprawności ruchowej, a w większości przypadków również do uszkodzenia serca i przedwczesnego zgonu. O życiu w cieniu choroby i iskierce nadziei, jaką jest zarejestrowany niedawno lek, opowiada Teresa Nieczaj, mama Kacpra i Karola – bliźniaków zmagających się z tym schorzeniem.
Jak wyglądało rozpoznanie choroby u pani synów?
Chłopcy urodzili się jako wcześniaki, ale do 10. roku życia byli praktycznie zupełnie zdrowymi dziećmi. Wtedy zauważyłam, że zaczęli się garbić. Ortopeda stwierdził u nich skoliozę idiopatyczną. Mimo rehabilitacji problem się pogłębiał, więc lekarze zalecili stosowanie specjalnych gorsetów. Chłopcy buntowali się przeciwko temu, m.in. z uwagi na reakcje rówieśników. Wciąż jednak intensywnie ćwiczyli. Gdy byli w wieku około 12 lat, zauważyłam, że zmienił się ich chód – zaczęli się chwiać. Kolejni lekarze bezradnie rozkładali ręce. Kiedy chłopcy mieli prawie 15 lat, moja siostra poleciła mi neurologa. Powiedział, że
EKSPERT
choroba na pewno jest poważna, i wspomniał o ataksji Friedreicha, ale uznał takie rozpoznanie za mało prawdopodobne. Po badaniach genetycznych potwierdziła się jednak właśnie ta diagnoza. Plany i marzenia chłopców nagle runęły. Do dziś pamiętam ich wbity w podłogę wzrok, kiedy usłyszeli diagnozę.
Jak choroba wpływa na życie państwa rodziny?
Zmieniło się o 180 stopni. Ja zrezygnowałam z pracy, w opiekę nad chłopcami zaangażował się też nasz starszy syn. Chłopcy wciąż intensywnie się rehabilitują. Pojawiło się leczenie hamujące postęp tej choroby, ale nie jest refundowane.
Staramy się walczyć o nie i o świadomość społeczną na temat choroby, bo jest bardzo mała.
Jak choroba wpłynęła na relacje synów z rówieśnikami i otoczeniem?
Kiedyś nasz dom tętnił życiem. Jednak im bardziej choroba się pogłębiała, tym wizyty rówieśników stawały się coraz rzadsze. Kontakty zaczęły się urywać. Chłopcy są teraz na trzecim roku studiów, ale nie nawiązali tam żadnych bliższych znajomości.
Co aktualnie dla was jest najważniejsze w walce z chorobą?
To przede wszystkim ciągła rehabilitacja. Żyjemy też nadzieją na refundację leku.

Kacper i Karol Bliźniacy zmagający się z ataksją Friedreicha
Ataksja Friedreicha to postępująca choroba neurodegeneracyjna, prowadząca do niepełnosprawności i przedwczesnej śmierci. Jedynym, co można zaproponować pacjentom jeszcze do niedawna, była rehabilitacja. Pojawiła się jednak pierwsza terapia, dzięki której można spowolnić postęp choroby.

Prof. dr hab. n. med.
Katarzyna Kotulska-Jóźwiak
Przewodnicząca Zespołu Koordynacyjnego
ds. leczenia chorych na rdzeniowy zanik
mięśni, kierownik
Kliniki Neurologii i Epileptologii Instytutu
„Pomnik – Centrum
Zdrowia Dziecka”
Czym jest ataksja Friedreicha (FA) i jak liczna jest grupa polskich pacjentów?
To jedna z chorób neurodegeneracyjnych. Wskutek mutacji w genie frataksyny dochodzi do postępującego uszkodzenia układu nerwowego. Osiowym objawem choroby są zaburzenia równowagi i zborności ruchów. U pacjentów pojawiają się także inne dolegliwości, w tym kardiologiczne i ortopedyczne, a często także cukrzyca. Objawy zaczynają się najczęściej we wczesnym wieku nastoletnim. FA cechuje się zmiennością występowania w różnych krajach. Szacujemy, że w Polsce może być ok. 150 pacjentów z tą chorobą.
W jaki sposób diagnozuje się chorobę?
Podstawą diagnozowania jest badanie genetyczne wykonywane metodą PCR, które na pierwszym etapie polegającym na stwierdzeniu liczby powtórzeń tripletu GAA nie jest trudne. Jest kilka laboratoriów, które wykonują takie badanie, i bez problemu lekarz może zlecić taką
diagnostykę. Niestety, krążenie pacjenta między różnymi specjalistami, nim usłyszy diagnozę, trwa zwykle co najmniej kilka lat.
Czego może spodziewać się pacjent lub jego rodzic, słysząc taką diagnozę?
W swoim naturalnym przebiegu, czyli gdyby nie było żadnej możliwości interwencji, średnio po ok. 10 latach od rozpoznania pacjent byłby zmuszony do korzystania z wózka inwalidzkiego. Przeżycie to średnio ok. 40 lat, więc choroba zdecydowanie skraca życie. Ataksja Friedreicha jest postępująca, zatem objawy z biegiem czasu się nasilają. Pacjenci doświadczają też stygmatyzacji, bo często są przez otoczenie uznawani za osoby nietrzeźwe lub odurzone narkotykami.
Na czym polega leczenie FA?
Można poprawić jakość życia pacjenta, stosując odpowiednie sposoby rehabilitacji, nadzorując objawy kardiologiczne
i ortopedyczne i dobrze kontrolując cukrzycę, jeśli ona występuje. Od ubiegłego roku w USA i od początku tego roku w Europie jest zarejestrowany lek,
Krążenie pacjenta między różnymi specjalistami, nim usłyszy diagnozę, trwa zwykle co najmniej kilka lat.
który w istotny sposób spowalnia postęp ataksji, a nawet powoduje niewielką poprawę. W Polsce lek nie jest jeszcze refundowany, ale dostęp do niego jest możliwy w ramach ratunkowego dostępu do technologii lekowych (RDTL).
Czy wiesz, że...
Ataksja Friedreicha (FA) to rzadkie, postępujące schorzenie neurodegeneracyjne, które dotyka osoby na całym świecie, także w Polsce?
To broszura, która zawiera szczegółowe informacje na temat tej choroby!
1. Czym jest Ataksja Friedreicha?
• Rzadka choroba neurodegeneracyjna
• Dziedziczona genetycznie
• Powoduje stopniową utratę koordynacji ruchowej
• Dotyka również serca, wzroku, mowy
3. Kogo Dotyczy?

• Pierwsze objawy: 8-15 lat
• Średnia długość życia: 37 lat
• W Polsce: około 150 chorych
5. Leczenie
• Brak całkowitego wyleczenia
• Fizjoterapia, rehabilitacja
7. Poznaj Historie Pacjentów
• Życie z FA to codzienne wyzwania
• Inspirujące historie walki z chorobą
po broszurę!
Kliknij tutaj i poznaj pełen obraz życia z FA!
Bürk, K. (2017). Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias 4, 4. doi:10.1186/s40673-017-0062-x;
2. Główne Objawy
• Niezborność (ataksja)
• Trudności z mówieniem (dyzartria)
• Problemy z połykaniem (dysfagia)
• Kardiomiopatia
• Skolioza i deformacje stóp
4. Diagnoza
• Objawy kliniczne
• Badanie genetyczne (PCR)
• Wczesne wykrycie kluczowe dla leczenia
6. Wsparcie Społeczne
• Opieka wielospecjalistyczna
• Pomoc rodzin i organizacji charytatywnych
Dowiedz się, jakie są główne wyzwania, przed którymi stają pacjenci i ich opiekunowie oraz jakie wsparcie można uzyskać w walce z tym trudnym schorzeniem Zeskanuj kod QR, aby dowiedzieć się więcej o Ataksji Friedreicha.
5. Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007 Mar;6(3):245-57. doi: 10.1016/S1474-4422(07)70054-6. PMID: 17303531; 6. Parkinson MH, Boesch S, Nachbauer W, Mariotti C, Giunti P. Clinical features of Friedreich’s ataxia: classical and atypical phenotypes. J Neurochem. 2013 Aug;126 Suppl 1:103-17. doi: 10.1111/jnc.12317. PMID: 23859346.
7. Friedreich’s Ataxia Research Alliance. “What is FA?” Dostępne pod adresem: https://www.curefa.org/what-is-friedreichs-ataxia#. (dostęp: kwiecień 2024)
8. Indelicato E, Nachbauer W, Eigentler A, et al. Onset features and time to diagnosis in Friedreich’s ataxia. Orphanet J Rare Diseases. 2020;15:198
9. Corben LA, Collins V, Milne S, Farmer J, Musheno A, Lynch D, Subramony S, Pandolfo M, Schulz JB, Lin K, Delatycki MB; Clinical Management Guidelines Writing Group. Clinical management guidelines for Friedreich ataxia: best practice in rare diseases. Orphanet J Rare Dis. 2022 Nov 12;17(1):415. doi: 10.1186/s13023-022-02568-3. PMID:36371255; PMCID: PMC9652828;

5 października obchodzony jest Dzień Świadomości PFIC . Co wiemy o tej rzadkiej chorobie?

Prof. dr hab. n. med.
Anna Liberek
COPERNICUS PL, Szpital św. Wojciecha
w Gdańsku, Oddział Pediatryczny
Czym charakteryzuje się postępująca rodzinna cholestaza wewnątrzwątrobowa (PFIC) i jak często jest rozpoznawana?
W Polsce mamy obecnie zarejestrowanych kilkadziesiąt przypadków, natomiast jeśli badania genetyczne byłyby wykonywane powszechnie wśród chorych, u których przebieg kliniczny sugeruje PFIC, to jestem przekonana, że tych pacjentów byłoby znacznie więcej. PFIC to grupa rodzinnych cholestaz, składająca się z kilkunastu typów, które są szczegółowo diagnozowane za pomocą badań genetycznych. Poszczególne typy mają podobny przebieg, ale ich podłoże genetyczne jest różne. Najczęściej występujące typy PFIC to 1, 2 i 3.
Pierwsze objawy PFIC mogą wystąpić już we wczesnym dzieciństwie. Jakie symptomy powinny zaniepokoić rodziców?
W okresie początkowym przebieg tych schorzeń może być bezobjawowy. Dzieci są w dobrym stanie ogólnym, prawidłowo przybierają na wadze i zwykle pierwszym objawem PFIC jest żółtaczka. Jednak – jak powszechnie wiadomo – żółtaczka występuje u noworodków oraz najmłodszych niemowląt stosunkowo często, również u zdrowych dzieci (np. może być związana z karmieniem piersią), co utrudnia rozpoznanie żółtaczek patologicznych, w tym także PFIC. Objawem, który może być sygnałem alarmowym, jest świąd skóry. U noworodka czy niemowlęcia manifestuje się to zwykle nasilonym niepokojem. Kolejnym sygnałem mogącym sugerować PFIC są okresowo odbarwione stolce (u zdrowych dzieci powinny być żółte lub brązowe). W tych przypadkach bezwzględnie konieczna jest konsultacja lekarska w celu wykluczenia innych przyczyn odbarwionych stolców.
PFIC jest chorobą o podłożu genetycznym. Jak zatem wygląda proces diagnostyczny?
Jeżeli u noworodka przedłuża się żółtaczka (powyżej 2 tygodni) bądź pojawią się wyżej wspomniane objawy, takie jak niepokój czy odbarwione stolce, to lekarz rodzinny w POZ powinien bezwzględnie zlecić badania w celu określenia stężenia bilirubiny całkowitej oraz jej frakcji (bilirubina pośrednia i bezpośrednia) we krwi. Są wprawdzie dostępne narzędzia (bilirubinometr) do nieinwazyjnego przezskórnego pomiaru stężenia bilirubiny, jednak
ten pomiar nie różnicuje jej frakcji. Następnie należy ocenić funkcję wątroby oraz aktywność GGT – enzymu, którego pomiar ma istotne znaczenie w różnicowaniu przyczyn cholestaz, czyli stanów, w których poziom bilirubiny bezpośredniej przekracza 1 mg/dl. W sytuacji stwierdzenia cholestazy, dziecko powinno być skierowane do ośrodka specjalistycznego zajmującego się schorzeniami wątroby. Kolejnym bardzo ważnym parametrem jest ocena stężenia kwasów żółciowych we krwi, których wartość w cholestazach jest wysoka. W trakcie dalszego poszukiwania przyczyny cholestazy – już w ośrodku specjalistycznym – przeprowadza się szeroką diagnostykę różnicową. W przypadku cholestaz, gdy aktywność GGT jest niska, można podejrzewać PFIC (m.in. typ 1 lub 2, natomiast w typie 3 aktywność GGT jest wysoka), dlatego wskazane jest wykonanie badania genetycznego, które może pozwolić na ostateczne ustalenie rozpoznania.
jej transplantacji. Oczywiście transplantacja jest wielkim dokonaniem medycyny, ratującym zdrowie i życie, ale pacjenci po przeszczepieniu przez całe życie otrzymują leki immunosupresyjne, są podatni na infekcje, dodatkowo w przypadku części pacjentów z PFIC typu 1 może dochodzić do stłuszczenia przeszczepu, zaś u chorych na PFIC typu 2 – do nawrotu choroby w przeszczepionym narządzie. Wczesne rozpoznanie choroby, w tym potwierdzenie w badaniu genetycznym, stwarza możliwość szybkiego włączenia nowoczesnego i skutecznego leczenia.
Wczesne rozpoznanie choroby, w tym potwierdzenie w badaniu genetycznym, stwarza możliwość szybkiego włączenia nowoczesnego i skutecznego leczenia.
Dlaczego wczesne rozpoznanie PFIC jest tak ważne? PFIC, czyli postępująca rodzinna cholestaza wewnątrzwątrobowa, to grupa chorób – jak sama nazwa wskazuje – postępujących. Jeżeli szybko nie zostanie wdrożone odpowiednie leczenie, to konsekwencją choroby jest postępujące uszkodzenie wątroby, włóknienie, marskość czy niewydolność tego narządu, co prowadzi do konieczności
Jakie możliwości terapeutyczne mają polscy pacjenci chorujący na PFIC? W okresie początkowym leczenie dzieci z PFIC polega na włączeniu preparatu kwasu ursodeoksycholowego (UDCA), witamin rozpuszczalnych w tłuszczach, leków mających na celu zmniejszenie świądu skóry. Jeżeli ten etap leczenia okaże się nieskuteczny, do niedawna w kolejnym etapie stosowano metody operacyjne – takie jak m.in. częściowe zewnętrzne odprowadzenie żółci (pacjent był skazany na życie z przetoką i workiem stomijnym, co zasadniczo obniża jakość życia), zespolenie omijające krętniczo-kątnicze czy częściowe wewnętrzne odprowadzenie żółci. W przypadku braku pożądanego efektu wyżej wymienionych metod i postępu choroby, jedynym ratunkiem pozostawało przeszczepienie wątroby. Obecnie dla chorych z PFIC dostępne są leki, które zmniejszając wchłanianie kwasów żółciowych w przewodzie pokarmowym, redukują świąd skóry i – według doniesień z piśmiennictwa naukowego w obserwacji długofalowej – stwarzają możliwość wieloletniego przeżycia bez konieczności transplantacji wątroby. Są to selektywne inhibitory transportera kwasów żółciowych w jelicie krętym (ang. ileal bile acid transporter) – w skrócie IBAT. Takie leczenie jest obecnie dostępne dla polskich pacjentów. Może być zastosowane u dzieci powyżej 6. miesiąca życia, z masą ciała powyżej 5 kg, z potwierdzonym genetycznie PFIC typu 1 lub PFIC typu 2, u których dotychczasowe leczenie kwasem UDCA i/lub zastosowanie ww. metod operacyjnych okazało się nieskuteczne. Dla młodych pacjentów z PFIC jest to ogromna szansa na zminimalizowanie skutków choroby i uniknięcie okaleczających zabiegów chirurgicznych czy przeszczepienia wątroby.

Rzadkie choroby wątroby o podłożu genetycznym – takie jak postępująca rodzinna cholestaza wewnątrzwątrobowa (PFIC) i zespół Alagille’a – objawiają się żółtaczką oraz uporczywym świądem i dużym cierpieniem chorych. W ostatnich latach postęp medycyny przyniósł rozwiązania, na które czekali chorzy i ich bliscy.

Prof. dr hab. n. med.
Irena Jankowska
Klinika Gastroenterologii, Hepatologii, Zaburzeń Odżywiania i Pediatrii, Instytut „Pomnik – Centrum Zdrowia Dziecka”
Postępująca rodzinna cholestaza wewnątrzwątrobowa (PFIC) to grupa rzadkich, genetycznie uwarunkowanych chorób wątroby. Na czym one polegają?
Najczęściej objawiają się one u niemowląt i małych dzieci. Wczesna diagnoza jest bardzo istotna dla szybkiego zastosowania odpowiedniego leczenia. Na podstawie połączenia objawów, takich jak świąd skóry i żółtaczka, oraz wyników badań czynności wątroby (w większości typów prawidłowa aktywność enzymu GGT i wysokie stężenie kwasów żółciowych, stężenie bilirubiny bezpośredniej powyżej 1 mg/dl) można postawić wstępne rozpoznanie postępującej rodzinnej cholestazy wewnątrzwątrobowej – PFIC. Dopiero wynik badania genetycznego pozwala ostatecznie potwierdzić diagnozę i określić typ PFIC.
Jak rozpoznanie typu PFIC wpływa na rokowania i leczenie?
Aktualnie wyróżnia się minimum 12 typów genetycznych postępującej rodzinnej cholestazy wewnątrzwątrobowej. W Polsce najczęściej mamy do czynienia z PFIC 1, 2, 3, a także 4. Ich rokowanie jest zmienne i trudne do przewidzenia u poszczególnych chorych. Choroba nieleczona lub niewłaściwie leczona może prowadzić do postępującej niewydolności wątroby i konieczności jej transplantacji. W przypadku PFIC typu 1 u części pacjentów po przeszczepieniu dochodzi do stłuszczenia przeszczepionego narządu i zaostrzenia objawów pozawątrobowych – uporczywych, przewlekłych biegunek. Należy więc starać się wykorzystać wszystkie opcje terapeutyczne przed ewentualną decyzją o transplantacji wątroby. PFIC typu 2 może pojawić się w okresie niemowlęcym lub później, nawet w wieku nastoletnim. Pacjenci z tym typem mają zwiększone ryzyko rozwoju nowotworu wątroby. PFIC typu 3 jako jedyny występuje z podwyższoną aktywnością enzymu GGT, dlatego bez badania genetycznego nie potrafimy go rozpoznać. Zwykle objawia się w wieku starszym – u osób młodocianych i dorosłych. U pacjentów z PFIC typu 3 występuje zwiększone ryzyko rozwoju kamicy
pęcherzyka żółciowego i dróg żółciowych, a także podwyższone ryzyko rozwoju nowotworu wątroby czy dróg żółciowych. W PFIC typu 4 objawy pojawiają się u niemowląt, ale w wieku starszym dochodzą też inne – pozawątrobowe, takie jak np. utrata słuchu.
Jak obecnie wygląda dostęp do nowoczesnych metod terapii PFIC w Polsce? Jeszcze w latach 80. nasze możliwości obejmowały jedynie podawanie kwasu ursodeoksycholowego, leków przeciwświądowych (np. rifampicyny), a także uzupełnienie niedoborów witamin, zwłaszcza witaminy E. Większość dzieci z PFIC umierała wówczas z powodu marskości wątroby przed drugą dekadą życia. Pod koniec lat 80. opracowano zabieg częściowego zewnętrznego odprowadzenia żółci poprzez wyłonienie stomii, który pozwalał ocalić dzieciom życie. Jednak takie leczenie znacząco zmniejszało jakość życia pacjentów, ponieważ część z nich nie akceptowała posiadania na brzuchu worka, w którym gromadziła się żółć. U części dzieci zabieg był nieskuteczny i konieczne było też przeszczepienie wątroby, wiążące się m.in. z dożywotnim leczeniem immunosupresyjnym. Za największy przełom, który dokonał się w ostatnich latach, uważam opracowanie terapii selektywnymi inhibitorami transportera kwasów żółciowych (IBAT). Widzimy, że po podaniu leków o takim mechanizmie działania zmniejsza się lub nawet ustępuje świąd skóry, a stężenie kwasów żółciowych we krwi maleje. Takie leczenie jest już dostępne dla polskich pacjentów.
Jakie znaczenie dla pacjentów miało poszerzenie możliwości leczenia?
Jeśli wcześnie rozpoznamy chorobę i wdrożymy leczenie, to dziecko może normalnie funkcjonować, bez uporczywych objawów. W życiu codziennym pacjentom z PFIC najbardziej doskwiera silny świąd skóry, który zaburza życie nie tylko pacjenta, ale także jego rodziny.
Nowe możliwości leczenia pozwalają uniknąć inwazyjnych zabiegów chirurgicznych i ich konsekwencji. Co więcej, kilkuletnie obserwacje wskazują, że wczesna diagnostyka i włączenie terapii inhibitorami IBAT może opóźnić konieczność wykonania przeszczepienia wątroby lub nawet spowodować długoletnie przeżycie bez tego zabiegu.
Do rzadkich schorzeń wątroby o podłożu genetycznym zalicza się również zespół Alagille’a. Na czym polega ta choroba i jak ją rozpoznać? Zespół Alagille’a to rzadka choroba wielonarządowa, która w okresie noworodkowym może objawiać się przedłużającą się żółtaczką. U starszych dzieci i dorosłych przebieg może wiązać się ze zwiększoną aktywnością enzymu GGT, wysokimi stężeniami cholesterolu w surowicy, a także świądem skóry i charakterystycznymi guzkami podskórnymi – żółtakami. Aby postawić rozpoznanie, musimy stwierdzić 3 z 5 cech. Są to: 1. żółtaczka cholestatyczna (stężenie bilirubiny bezpośredniej powyżej 1 mg/dl); 2. charakterystyczne rysy twarzy (wydatne, wypukłe czoło, głęboko osadzone oczy, spiczasty podbródek); 3. charakterystyczna wada serca (np. zwężenie zastawki płucnej czy obwodowych tętnic płucnych); 4. wady kręgosłupa (m.in. widoczne w badaniu RTG tzw. kręgi motyle); 5. specyficzna wada narządu wzroku. Do tego wszystkiego dochodzi bardzo silny świąd skóry.
Co do zaoferowania chorym na zespół Alagille’a ma współczesna medycyna?
Ścieżka terapeutyczna jest podobna jak w przypadku PFIC – rozpoczynamy od podania pacjentowi kwasu ursodeoksycholowego, leczenia świądu skóry oraz uzupełnienia niedoborów witamin. Również tu bardzo dobrze sprawdzają się wspomniane selektywne inhibitory transportera kwasów żółciowych (IBAT). Leki z tej grupy są zarejestrowane także w leczeniu świądu u pacjentów z zespołem Alagille’a, ale niestety nie są jeszcze w Polsce refundowane.
Szacuje się, że ogólnie zaawansowana mastocytoza układowa występuje u około 5 przypadków na milion osób rocznie. Choroba prowadzi do postępującej niewydolności wielonarządowej, a przeżycie pomimo stosowania leczenia cytoredukcyjnego wynosi od kilku miesięcy do kilku lat.

Prof. dr hab. n. med.
Marek Hus
Kieruje Kliniką Hematoonkologii i Transplantacji Szpiku Samodzielnego Publicznego Szpitala Klinicznego nr 1 w Lublinie, profesor Uniwersytetu Medycznego w Lublinie
Czym charakteryzuje się zaawansowana mastocytoza układowa?
Zaawansowana mastocytoza układowa to najcięższa postać mastocytozy, w przebiegu której dochodzi do niekontrolowanego namnażania się komórek tucznych w narządach wewnętrznych, co zaburza lub nawet uniemożliwia ich prawidłowe funkcjonowanie. Obejmuje grupę najcięższych zaburzeń dotyczących komórek tucznych. Wszystkie postaci zaawansowanej mastocytozy układowej stanowią rozpoznania chorób nowotworowych, które wymagają intensywnego leczenia.
Istnieją trzy rodzaje zaawansowanej mastocytozy układowej: • agresywna mastocytoza układowa, • mastocytoza układowa z powiązanym nowotworem układu krwiotwórczego, • białaczka mastocytarna (MCL).
Każdy z tych rodzajów choroby występuje niezwykle rzadko. Szacuje się, że ogólnie zaawansowana mastocytoza układowa występuje u około 5 przypadków na milion osób rocznie.
Choroba prowadzi do postępującej niewydolności wielonarządowej, a przeżycie pomimo stosowania leczenia cytoredukcyjnego wynosi od kilku miesięcy do kilku lat.
Jakie opcje terapeutyczne są dostępne dla pacjentów chorych na zaawansowaną mastocytozę układową?
Podstawą leczenia zaawansowanej mastocytozy układowej jest terapia cytoredukcyjna, której celem jest ustąpienie objawów uszkodzenia narządowego oraz organomegalii, zmniejszenie nacieczenia szpiku kostnego przez komórki tuczne, z następczą ewentualną allogeniczną transplantacją szpiku, która jak dotychczas jest jedyną opcją wyleczenia.
Midostauryna jest pierwszym zarejestrowanym wielotorowym inhibitorem kinazy tyrozynowej. Wykazuje zdolność hamowania szlaków przekazywania sygnału i proliferacji komórkowych drogą receptora FLT3. Wykazuje zdolność hamowania szlaków sygnałowych KIT, proliferacji komórek i uwalniania histaminy oraz indukuje apoptozę w komórkach tucznych.
Awaprytynib to silny i selektywny inhibitor kinaz KIT i PDGFRA, wykazuje wysoką skuteczność w leczeniu zaawansowanej mastocytozy układowej, z odpowiedzią u 57 proc. pacjentów. W Polsce lek nie jest objęty refundacją
i jest dostępny wyłącznie w ramach Ratunkowego Dostępu do Technologii Lekowych (RDTL).
Interferon-α (IFN-α) stosowany przed erą inhibitorów kinazy tyrozynowej uznawany był za lek pierwszego wyboru u pacjentów z zaawansowaną mastocytozą układową, jakkolwiek obserwowano zmienną skuteczność leczenia, a dokładne dawkowanie i czas trwania leczenia pozostawały nieznane.
Imatynib jest skuteczny w rzadkich przypadkach mastocytozy z mutacjami KIT innymi niż D816V. U pacjentów z tą mutacją lek nie przynosi odpowiedzi klinicznej.
Kladrybina wykazuje aktywność przeciwnowotworowym mastocytom. Znajduje zastosowanie w leczeniu właściwie wszystkich postaci klinicznych SM. Znajduje zatem ograniczone zastosowanie w leczeniu rzadkich przypadków mastocytozy układowej, które wykazują wrażliwą na imatynib mutację KIT, bez mutacji KIT D816V lub o nieznanym statusie. Wskazanie to dotyczy zaledwie około 10 proc. chorych z SM. Odsetki uzyskiwanych odpowiedzi szacuje się na 20-30 proc.
Choć przeszczepienie szpiku kostnego pozostaje jedyną opcją potencjalnie prowadzącą do wyleczenia, jest obarczone dużym ryzykiem powikłań i zarezerwowane dla młodszych pacjentów bez ciężkich chorób współistniejących.
Dlaczego allogeniczny przeszczep szpiku kostnego nie jest zalecany wszystkim pacjentom?
Allogeniczna transplantacja komórek macierzystych jest jedynym sposobem wyleczenia zaawansowanej mastocytozy układowej. Wysokie ryzyko powikłań związane z procedurą wyraźnie jednak ogranicza liczbę potencjalnych kandydatów. Wyniki leczenia zależą od wariantu choroby i zastosowanego kondycjonowania. Z niekorzystnym rokowaniem wiązało się rozpoznanie MCL. Odnotowano korzyści ze stosowania leczenia mieloablacyjnego w porównaniu z kondycjonowaniem o zredukowanej intensywności. Śmiertelność związana z leczeniem wynosiła 20 proc. po jednym roku. Znaczenie terapii z użyciem inhibitorów KIT, tj. midostauryny po przeprowadzonym alloSCT na przeżycie długoterminowe (wolne od progresji), jest obecnie nadal nieznane. Wydaje się jednak celowe kontynuowanie leczenia midostauryną, aby stłumić resztkowe klony z mutacją KIT.
Dlaczego białaczka mastocytarna (MCL) jest tak trudna w leczeniu?
Białaczka mastocytarna to bardzo szybko rosnący, agresywny nowotwór układu krwiotwórczego, w przebiegu którego w szpiku kostnym oraz we krwi występuje nadmiar komórek tucznych. Białaczka mastocytarna może wystąpić u pacjentów, u których wcześniej rozpoznano zaburzenie dotyczące komórek tucznych (np. w przebiegu agresywnej mastocytozy układowej) lub u osób bez żadnych innych poprzedzających objawów mastocytozy.
Jest to niezwykle rzadka choroba. Wystąpi ona u zaledwie 1 proc. wszystkich osób cierpiących na zaawansowaną mastocytozę układową. Rozpoznanie MCL wiąże się z najgorszym rokowaniem – mediana czasu przeżycia nie przekracza 6 miesięcy.
Choć przeszczepienie szpiku kostnego pozostaje jedyną opcją potencjalnie prowadzącą do wyleczenia, jest obarczone dużym ryzykiem powikłań i zarezerwowane dla młodszych pacjentów bez ciężkich chorób współistniejących.
Jak przedstawia się dostępność leczenia zaawansowanej mastocytozy układowej dla pacjentów w Polsce?
Od 1 maja 2021 midostauryna jest objęta refundacją w ramach programu lekowego MZ. W przypadku awaprytynibu na chwilę obecną leczenie nie jest objęte refundacją przez NFZ, ale dostęp do terapii jest możliwy poprzez Ratunkowy Dostęp do Terapii Lekowych (RDTL). Niedostępne w Polsce jest leczenie Interferonem-α. Kladrybina jest dostępna w ramach katalogu chemioterapii, ale lek nie jest zarejestrowany w tym wskazaniu. Leczenie imatynibem jest dostępne w ramach katalogu chemioterapii, ale jego skuteczność ogranicza się w leczeniu bardzo rzadkich przypadków zaawansowanej mastocytozy układowej.

Dzięki metodom, takim jak sekwencjonowanie następnej generacji (NGS), szanse na wczesne wykrycie chorób rzadkich znacznie wzrosły. Jednak w Polsce nadal brakuje dostępu do kluczowych badań, a potrzeba inwestycji w specjalistów genetyki rośnie.

Prof. dr hab.
n. med. Anna Latos-Bieleńska
Konsultant Krajowa w dziedzinie genetyki klinicznej, Katedra i Zakład Genetyki Medycznej Uniwersytetu Medycznego im. K. Marcinkowskiego w Poznaniu
Jaka część chorób rzadkich jest uwarunkowana genetycznie? Dlaczego badania genetyczne w przypadku chorób rzadkich mają szczególne znaczenie?
Choroby rzadkie to choroby o różnorodnej etiologii, ale aż 80 proc. chorób rzadkich to choroby genetyczne. Do chorób rzadkich prowadzą różnego rodzaju zmiany materiału genetycznego, najczęściej są to mutacje pojedynczych genów. Diagnostyka molekularna w takich przypadkach nie jest prosta, ponieważ rzadko się zdarza, że przyczyną danej choroby jest jedna określona i łatwa do zbadania zmiana materiału genetycznego. Najczęściej przyczyną choroby rzadkiej może być nie tylko wiele (setki, tysiące) różnych zmian w danym genie, ale także jedna określona choroba rzadka może być spowodowana zmianami w wielu różnych genach. To właśnie skomplikowane podłoże molekularne spowodowało, że chorzy na choroby rzadkie pozostawali bez rozpoznania i musieli czekać na postęp w genetyce. Obecnie, dzięki sekwencjonowaniu następnej generacji (NGS), rozpoznawalność chorób rzadkich ogromnie się poprawiła. Chorzy i my – ich lekarze – z ogromną niecierpliwością czekamy na objęcie refundacją trzech nowych rodzajów badań genetycznych: porównawcza hybrydyzacja genomowa do mikromacierzy (aCGH), panele celowane NGS oraz sekwencjonowanie całego eksomu (WES). Badania są po wszystkich pozytywnych ocenach. Ustalenie rozpoznania choroby rzadkiej i poznanie jej podłoża molekularnego jest ważne dla objęcia pacjenta właściwą opieką medyczną.
Diagnostyka genetyczna jest także niezwykle efektywna ekonomicznie. Jako środowisko genetyków przygotowaliśmy „Białą księgę” badań genetycznych, wykazując, że odpowiednio wcześnie zastosowana diagnostyka genetyczna to zaledwie 20 proc. kosztów diagnostyki innymi metodami, w dodatku na ogół nieprowadzącymi do ustalenia rozpoznania. Potwierdzają to także dane z piśmiennictwa. Dzisiaj już nikt nie wątpi, że diagnostyka genetyczna to wielka oszczędność środków.
Z czym związane są te największe wyzwania w diagnostyce genetycznej chorób rzadkich?
Aktualnie w Polsce realizowane są dwa wielkie projekty systemowe: Plan dla Chorób Rzadkich oraz Narodowa Strategia Onkologiczna. W obu nowoczesna diagnostyka genetyczna jest jednym w ważnych obszarów, co powoduje gwałtowny wzrost zapotrzebowania na badania genetyczne. Trzeba pilnie inwestować w kadry genetyczne. Potrzeba dużej liczby wyszkolonych genetyków laboratoryjnych – specjalistów laboratoryjnej genetyki medycznej oraz medycznej genetyki molekularnej. Wynik badania genetycznego musi być zawsze interpretowany w kontekście danych rodowodowo-klinicznych, a chorzy na choroby genetyczne i ich rodziny muszą zostać objęci poradnictwem genetycznym. Potrzebni są lekarze specjaliści genetyki klinicznej, wspierani przez pielęgniarki genetyczne. W onkologii potrzebny jest nowy zawód medyczny – doradca genetyczny w onkologii. Konieczne jest przyjęcie Ustawy o Genetyce Medycznej.
Jakie działania podejmowane są aktualnie w Polsce, aby przyśpieszyć rozpoznanie choroby rzadkiej?
Plan dla Chorób Rzadkich, który wygasł po 19 miesiącach realizacji (20222023), został reaktywowany, a poprawa diagnostyki jest nadal jednym z najważniejszych obszarów Planu. Szybkie uzupełnienie koszyka świadczeń gwarantowanych o nowe badania genetyczne będzie momentem zwrotnym w ocenie Planu przez pacjentów, którzy od razu odczują, że Plan działa.
Ustalenie rozpoznania choroby rzadkiej i poznanie jej podłoża molekularnego jest ważne dla objęcia pacjenta właściwą opieką medyczną.
Ustalenie rozpoznania choroby rzadkiej i nadanie kodu ORPHA (w ośrodkach eksperckich klinicznych i genetycznych) spowoduje, że informacja ta znajdzie się w Polskim Rejestrze Chorób Rzadkich, a pacjent otrzyma Kartę Pacjenta z Chorobą Rzadką. Jest to nowoczesne rozwiązanie, możliwe w Polsce dzięki bardzo dobrej informatyzacji w ochronie zdrowia. Liczymy też na przyjęcie Ustawy o Chorobach Rzadkich.

Skrócenie tzw. odysei diagnostycznej w przypadku chorób rzadkich możliwe jest m.in. dzięki rosnącej świadomości i wiedzy lekarzy rodzinnych.

Dr hab. n. med.
Jolanta Sykut-Cegielska
Prof. IMiD, kierownik
Kliniki Wrodzonych Wad Metabolizmu i Pediatrii w Instytucie Matki i Dziecka, konsultant krajowa w dziedzinie pediatrii metabolicznej
Jak wygląda kwestia rozpoznawania chorób rzadkich na ich wczesnym etapie?
Od wielu lat zajmuję się chorobami rzadkimi, w szczególności wrodzonymi wadami metabolizmu, więc najłatwiej mi odpowiadać na tym przykładzie. Tym bardziej, że z 8 tys. zidentyfikowanych chorób rzadkich w aż 2 tys. z nich podłożem są właśnie genetycznie uwarunkowane wrodzone wady metabolizmu.
Ścieżki diagnostyczne są dwojakie: skrining populacyjny i selektywny. Niektóre choroby rozpoznaje się poprzez badania przesiewowe noworodków, co oznacza, że każde żywo urodzone dziecko jest badane obecnie pod kątem 26 różnych wad metabolizmu, które spełniają definicję choroby rzadkiej. Jeśli wynik badania przesiewowego jest nieprawidłowy, to noworodek kierowany jest na dodatkowe badania specjalistyczne. Druga metoda diagnostyki to skrining selektywny i dotyczy osób, u których pojawiają się objawy kliniczne. Wówczas prowadzona jest wieloetapowa diagnostyka, która dopiero niestety po długim czasie doprowadza do ostatecznego rozpoznania choroby rzadkiej. Diagnostyce również podlega rodzeństwo chorego, jeśli wykryta wrodzona wada metabolizmu dziedziczy się w sposób autosomalny recesywny.
Jak ważna w ścieżce diagnostycznej jest właściwa reakcja na podejrzane objawy lekarza podstawowej opieki zdrowotnej?
Rola lekarza pierwszego kontaktu jest absolutnie kluczowa, bo to osoba,
która najczęściej widzi danego pacjenta, może obserwować nasilenie, współwystępowanie czy zmiany objawów. Dlatego tak ważna jest czujność diagnostyczna lekarzy medycyny rodzinnej, a szczególnie pediatrów. W tym aspekcie bardzo ważna jest sama świadomość lekarzy, że choroby rzadkie faktycznie występują w populacji i że mając jakiekolwiek podejrzenia, należy pacjenta kierować na bardziej specjalistyczne badania. Szczęśliwie możemy powiedzieć, że sytuacja z roku na rok się poprawia. Lekarze wykazują coraz większą czujność, co znacznie skraca tzw. odyseję diagnostyczną, na jaką narażeni są chorzy. W skrajnych wypadkach od wystąpienia pierwszych objawów do prawidłowej diagnozy nadal może minąć nawet kilka czy kilkanaście lat, ale co do zasady rozpoznania są już szybsze niż jeszcze dekadę temu. Poprawia się także dostępność informacji o chorobach rzadkich oraz zakres opieki, jaką w specjalistycznych ośrodkach referencyjnych otaczani są chorzy i ich rodziny.
Jakie objawy wskazują na podwyższone ryzyko występowania choroby rzadkiej? W przypadku wrodzonych wad metabolizmu nie ma jednego charakterystycznego objawu. I w drugą stronę – brak danego objawu nie daje pewności wykluczenia choroby rzadkiej o podłożu metabolicznym. Objawy mogą pojawić się z czasem oraz dotyczyć każdego narządu i układu. Sygnałem alarmowym jest opóźniony rozwój
psychoruchowy dziecka lub regres w jego rozwoju, czyli sytuacja, w której dziecko rozwija się prawidłowo tylko do pewnego momentu. Niepokojąca może być też już słaba dynamika rozwoju, a także dysfunkcja wątroby, zaburzenia
Rola lekarza pierwszego kontaktu jest absolutnie kluczowa, bo to osoba, która najczęściej widzi danego pacjenta, może obserwować nasilenie, współwystępowanie czy zmiany objawów.
ze strony mięśnia sercowego, zmiany w ośrodkowym układzie nerwowym, jak np. leukodystrofia mózgu. Symptomatologia chorób rzadkich, w tym wrodzonych wad metabolizmu, jest bardzo złożona i szeroka. Polscy pacjenci mają jednak szeroki dostęp do ośrodków referencyjnych, gdzie można przeprowadzić kompleksową diagnostykę, która wymaga specjalistycznego sprzętu oraz kadry doświadczonych lekarzy.
Czytaj więcej : chorobyrzadkie.com
Krajowe Forum na Rzecz Terapii Chorób Rzadkich ORPHAN (KFO) jest pacjencką organizacją parasolową skupiającą stowarzyszenia pacjentów i fundacje działające na rzecz pacjentów z chorobami rzadkimi w Polsce.

Stanisław Maćkowiak
Prezes Krajowego Forum
ORPHAN i Federacji
Pacjentów Polskich
Wsierpniu 2024 r. KFO zrzeszało 75 organizacji pacjentów z chorobami rzadkimi. Działania KFO skupiają się wokół nadrzędnego celu, jakim jest szeroko rozumiana poprawa jakości życia ponad 3 milionów osób dotkniętych chorobami rzadkimi w Polsce. Jako organizacja parasolowa dbamy, by głos pacjentów z chorobami rzadkimi był słyszalny.
Już w 2008 r., kiedy zaczynaliśmy w Polsce prace nad Planem dla Chorób Rzadkich, to z naszej pacjenckiej strony padały propozycje, żeby ten dokument był wprowadzony aktem prawnym ustawowym, czyli ustawą. W tym momencie mamy więc naprawdę dobrą sytuację, bo – tak jak państwo słyszą – wszyscy decydenci mówią o tym, że musi to być akt prawny w formie ustawy. Pierwszy krok to jest ta uchwała, która jest przyjęta
jako dokument kierunkowy przez rząd. Kolejny krok to ustawa, a w międzyczasie możemy robić różne rzeczy, aby nie zostawiać pacjentów z chorobami rzadkimi samym sobie.
Postulujemy kilka rzeczy – takich, które można zrobić bez konieczności wprowadzania rozwiązań ustawowych, np. rozszerzenie przesiewu noworodków o nowe choroby, podania domowe czy wspólne zakupy leków na poziomie Unii Europejskiej. Przez wiele lat mówiliśmy o tym, żeby znowelizować ustawę refundacyjną w ten sposób, żeby Minister miał możliwość zainicjowania refundacji tanich leków dla pacjentów z chorobami rzadkimi. W tej chwili najważniejsze jest to, żeby chorzy na choroby rzadkie wreszcie dostali kompleksową opiekę. Powinna być zorganizowana zgodnie z Europlanem, bo Plan dla Chorób Rzadkich na lata 2024-2025 to dokument dotyczący tylko części medycznej opieki dla chorych na choroby rzadkie. Musimy pamiętać,
że oprócz części medycznej, tak jak jest w Europlanie, musi być również zintegrowana pomoc socjalna, nauka i edukacja oraz wszystkie inne elementy, które są w tym dokumencie opisane. Chorzy na choroby rzadkie nie oczekują specjalnego traktowania, pacjenci oczekują wyrównania szans do życia, jakie mają inni obywatele. O tych potrzebach mówimy na podstawie czteroletnich badań organizacji pacjentów z chorobami rzadkimi, które publikujemy w corocznych AUDYTACH KRAJOWEGO FORUM ORPHAN „Potrzeby pacjentów z chorobami rzadkimi w zakresie dostępu do technologii medycznych i optymalizacji opieki w Polsce”. Więcej szczegółów w wersjach elektronicznych audytów za lata 2021, 2022, 2023, 2024 na stronie www.rzadkiechoroby.org/audyt
To twarde dane pokazujące stan opieki oraz niezaspokojonych potrzeb pacjentów z chorobami rzadkimi w Polsce. Zapraszamy państwa do lektury.
INFOGRAFIKA





Około 3,5 mln osób w Polsce cierpi na choroby rzadkie Istnieje aż 10 tys. jednostek chorobowych



Tzw. „odyseja diagnostyczna" trwa często latami, opóźniając leczenie
Brakuje specjalistów genetyków i wyspecjalizowanych ośrodków



80% chorób rzadkich ma podłoże genetyczne
Skrining noworodków obejmuje obecnie 26 wad metabolicznych Nowoczesna diagnostyka genetyczna (NGS) umożliwia szybsze wykrywanie tych schorzeń



Rozszerzenie liczby ośrodków (OCR) Wprowadzenie rejestru chorób rzadkich oraz cyfrowej Karty Pacjenta
Szybsza diagnoza dzięki badaniom prenatalnym i noworodkowym
Swixx BioPharma jest firmą farmaceutyczną, reprezentującą międzynarodowe koncerny z obszaru biotechnologii, preparatów dostępnych bez recepty (OTC) oraz wyrobów medycznych, działając z ich pełnomocnictwa lub w zastępstwie na rynkach, na których nie zdecydowały się one zaangażować lub zdecydowały się z nich wyjść. Jesteśmy zaangażowani w poprawę zdrowia pacjentów, nasz Zespół niestrudzenie dokłada wszelkich starań, aby zapewnić pacjentom dostęp do nowoczesnych leków naszych Partnerów.
Swixx BioPharma koncentruje się na czterch strategicznych obszarach terapeutycznych, wprowadzając na rynek wysoce innowacyjne, nowe terapie w chorobach rzadkich, onkologii i hematologii, opiece specjalistycznej i szczepionkach.

SWIXX BIOPHARMA SP. Z O.O.
UL. PROSTA 51, 00-838 WARSZAWA
TELEFON: +48 22 460 07 20 E-MAIL: poland.info@swixxbiopharma.com
PM-PL-2023-12-6931 data zatw. materiału: 20.12.2023 r.