
200 minute read
TABLAS Y FIGURAS


TABLAS Y FIGURAS
Tabla 1.3.1. Ejemplo de búsqueda sobre acreditación de formación continuada
Formación Continuada AND Acreditación
OR
OR
education continuing accreditation*
education, medical, continuing
education, nursing, continuing
Figura 1.3.1. Figura 1.3.2.

Figura 1.3.3.

Figura 1.3.4. Figura 1.3.5.
Figura 1.3.6.
Figura 1.3.7.
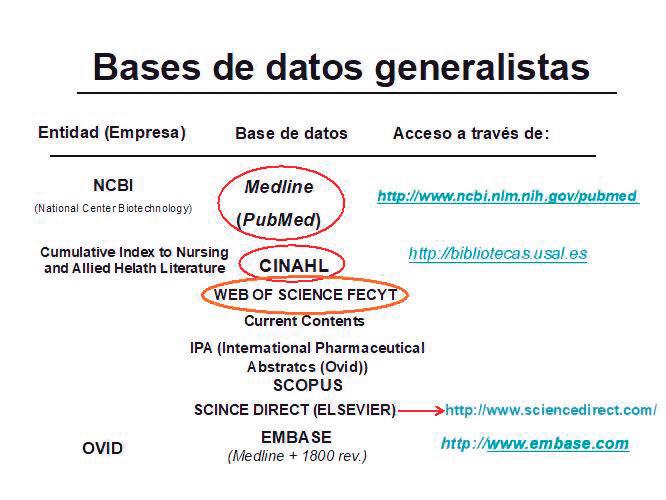

Figura 1.3.8.

Figura 1.3.9.

Figura 1.3.10.
Figura 1.3.11.
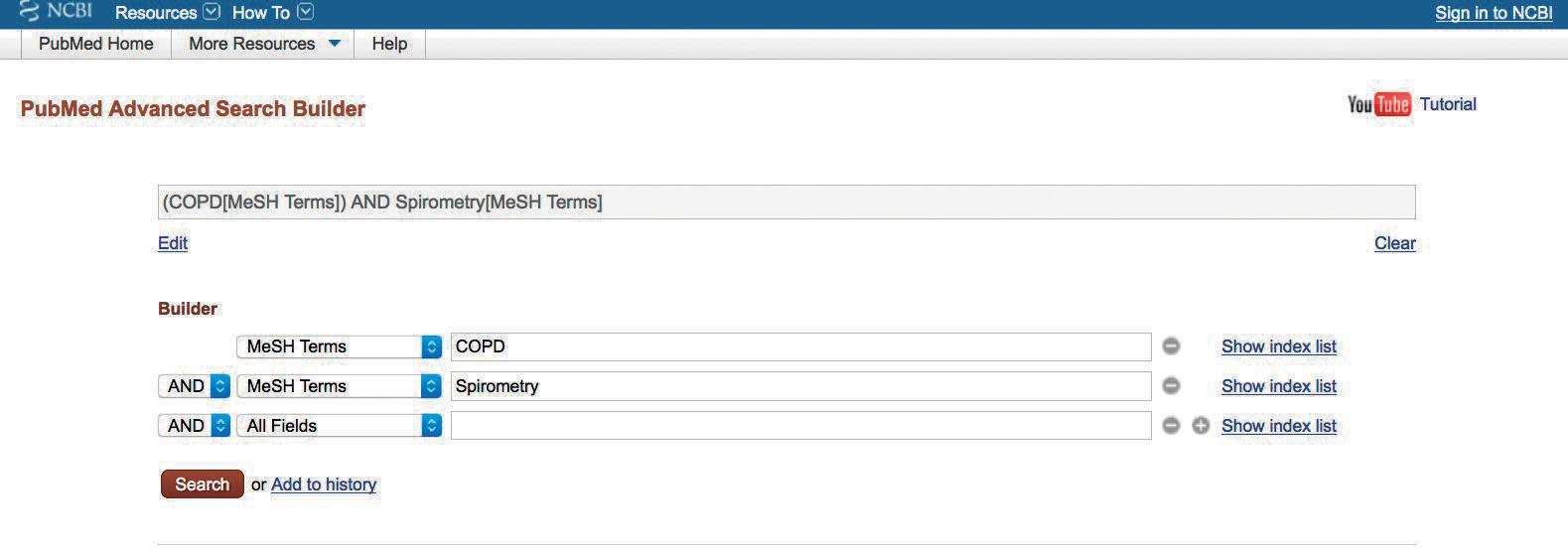
Figura 1.3.12.

Figura 1.3.13.
Figura 1.3.14.
Figura 1.3.15.
Figura 1.3.16.
Figura 1.3.17.

Figura 1.3.18.

Figura 1.3.19.
Figura 1.3.20.
Modulo 2:
METODOLOGÍA CIENTÍFICA
Responsable: Dr. Joan Soriano2.
ÍNDICE
2.1. TIPOS DE DISEÑOS: GENERALIDADES ........................................................................................ 61 INTRODUCCIÓN .................................................................................................................................. 61 DISEÑO DE UN ESTUDIO ................................................................................................................. 61 Ejemplos PUNTOS CLAVE ................................................................................................................................... 63 BIBLIOGRAFÍA ..................................................................................................................................... 64 TABLAS Y FIGURAS ........................................................................................................................... 65
2.2. ESTUDIOS DESCRIPTIVOS ............................................................................................................. 69 INTRODUCCIÓN .................................................................................................................................. 69 CARACTERÍSTICAS GENERALES ................................................................................................... 69 ESTUDIOS ECOLÓGICOS ................................................................................................................. 71 Estudios descriptivos de mapas Estudios de series temporales ESTUDIOS DE BASE INDIVIDUAL ................................................................................................. 72 Series de casos Estudios transversales PUNTOS CLAVE ................................................................................................................................... 74 BIBLIOGRAFÍA ..................................................................................................................................... 75 TABLAS Y FIGURAS ........................................................................................................................... 76
2.3. ESTUDIOS ANALÍTICOS: COHORTES Y CASO-CONTROL ...................................................... 77 INTRODUCCIÓN .................................................................................................................................. 77 ESTUDIOS DE COHORTE .................................................................................................................. 77 Proceso de un estudio de cohorte Ventajas de un estudio de cohorte Inconvenientes de un estudio de cohorte Análisis ESTUDIOS DE CASOS Y CONTROLES .......................................................................................... 80 Tipos de estudio de casos y controles Proceso de un estudio de casos y controles Ventajas de los estudios de casos y controles Inconvenientes de los estudios de casos y controles Análisis INTERPRETACIÓN ............................................................................................................................... 82 PUNTOS CLAVE ................................................................................................................................... 83
BIBLIOGRAFÍA ..................................................................................................................................... 84 TABLAS Y FIGURAS ........................................................................................................................... 86
2.4. ESTUDIOS EXPERIMENTALES Y CUASI EXPERIMENTALES ................................................. 91 INTRODUCCIÓN .................................................................................................................................. 91 ESTUDIOS EXPERIMENTALES ........................................................................................................ 91 Características Ventajas y limitaciones ESTUDIOS CUASI EXPERIMENTALES .......................................................................................... 94 Características Ventajas y limitaciones PRINCIPALES SESGOS EN LOS ESTUDIOS EXPERIMENTALES Y CUASI EXPERIMENTALES ................................................................................................................ 95 ENSAYO CLÍNICO CON MEDICAMENTOS ................................................................................... 96 CONSIDERACIONES ÉTICAS DE LOS ESTUDIOS EXPERIMENTALES ................................ 96 PUNTOS CLAVE ................................................................................................................................... 97 BIBLIOGRAFÍA ..................................................................................................................................... 98 TABLAS Y FIGURAS ........................................................................................................................... 99
2.5. TIPOS DE ERROR. VALIDEZ, SESGOS Y CONFUSIÓN ............................................................. 101 EL ERROR EN INVESTIGACIÓN ....................................................................................................... 101 ERROR ALEATORIO. PRECISIÓN .................................................................................................... 101 Defi nición, causas y efectos del error aleatorio Evaluación del error aleatorio ERROR SISTEMÁTICO. VALIDEZ .................................................................................................... 102 Sesgo de selección. Defi nición, causas y efectos Sesgo de información. Defi nición, causas y efectos Confusión. Defi nición, causas y efectos Evaluación del error sistemático (sesgo de selección, sesgo de información y confusión). Análisis de sensibilidad VALIDEZ EXTERNA. GENERALIZACIÓN ........................................................................................ 105 REDUCIR LOS ERRORES ALEATORIOS Y SISTEMÁTICOS .................................................... 105 Evitar o reducir el error aleatorio Evitar o reducir el sesgo de selección Evitar o reducir el sesgo de información Evitar o reducir la confusión EQUÍVOCOS Y DUDAS FRECUENTES EN TORNO A LOS ERRORES EN INVESTIGACIÓN .................................................................................................................................. 108 CONSIDERACIONES FINALES ......................................................................................................... 109 PUNTOS CLAVE ................................................................................................................................... 109 BIBLIOGRAFÍA ..................................................................................................................................... 110 TABLAS Y FIGURAS ........................................................................................................................... 111
2.6. REVISIONES SISTEMÁTICAS Y METAANÁLISIS ...................................................................... 119 INTRODUCCIÓN .................................................................................................................................. 119 PROTOCOLO DE LA REVISIÓN SISTEMÁTICA ........................................................................... 119 DEFINICIÓN DE LA PREGUNTA DE REVISIÓN ............................................................................ 120 CRITERIOS DE INCLUSIÓN Y EXCLUSIÓN ................................................................................. 120 BÚSQUEDA BIBLIOGRÁFICA ......................................................................................................... 121 PROCESO DE REVISIÓN ................................................................................................................... 122 Equipo de revisión Estudios excluidos PROCESO DE EXTRACCIÓN DE DATOS ....................................................................................... 123 EVALUACIÓN DEL RIESGO DE SESGOS ...................................................................................... 123 ANÁLISIS DE RESULTADOS ............................................................................................................ 124 Metaánalisis Metasíntesis EVALUACIÓN DE LA CALIDAD DE LA EVIDENCIA ...................................................................... 125 REDACCIÓN DE RESULTADOS ....................................................................................................... 125 LIMITACIONES DE LOS RESULTADOS ......................................................................................... 125 REVISANDO UNA REVISIÓN .......................................................................................................... 125 PUNTOS CLAVE ................................................................................................................................... 126 BIBLIOGRAFÍA ..................................................................................................................................... 127 TABLAS Y FIGURAS ........................................................................................................................... 128
2.7. VALIDACIÓN DE PRUEBAS DIAGNÓSTICAS ............................................................................. 131 INTRODUCCIÓN .................................................................................................................................. 131 FASES DE UN ESTUDIO DE EVALUACIÓN DE PRUEBAS DIAGNÓSTICAS ....................... 131 ELEMENTOS CLAVE PARA AUMENTAR LA CALIDAD DE LOS ESTUDIOS DE VALIDACIÓN DIAGNÓSTICA .................................................................................................... 132 Estructura básica Pasos a seguir en los estudios VALIDEZ, FIABILIDAD Y PRECISIÓN DE LOS ESTUDIOS DE PRUEBAS DIAGNÓSTICAS .................................................................................................................................. 133 Validez Fiabilidad Precisión CONTROL DE CALIDAD DE LOS ESTUDIOS DE PRUEBAS DIAGNÓSTICAS .................... 136 PUNTOS CLAVE ................................................................................................................................... 136 BIBLIOGRAFÍA ..................................................................................................................................... 137 TABLAS Y FIGURAS ........................................................................................................................... 138
2.8. VALIDACIÓN DE CUESTIONARIOS ............................................................................................... 145 INTRODUCCIÓN .................................................................................................................................. 145 ELABORACIÓN DE UN CUESTIONARIO ....................................................................................... 146 VALIDACIÓN DEL CUESTIONARIO ................................................................................................ 147 Fiabilidad Validez RESPUESTA AL CAMBIO .................................................................................................................. 150 Cambio homogéneo Cambio heterogéneo CUESTIONES A CONSIDERAR ........................................................................................................ 151 PUNTOS CLAVE ................................................................................................................................... 152 BIBLIOGRAFÍA ..................................................................................................................................... 153 TABLAS Y FIGURAS ........................................................................................................................... 154
2.1. TIPOS DE DISEÑOS: GENERALIDADES
Dra. Elena Gimeno
INTRODUCCIÓN
En este capítulo se presentan los tipos de diseño en investigación y sus generalidades. En capítulos posteriores se describirán las características de cada uno de ellos de manera más detallada. El diseño de un proyecto de investigación se defi ne como el conjunto de procedimientos, métodos y técnicas para la selección de sujetos, evaluación de las variables del estudio, recogida de datos y análisis para la interpretación de resultados.
DISEÑO DE UN ESTUDIO
En investigación existen diferentes tipos de diseños de estudios. La elección del diseño más adecuado va a depender de la pregunta de investigación que se quiera responder (objetivo principal), de la frecuencia de la condición clínica o enfermedad a estudiar, de la consideración de los aspectos éticos, así como de los recursos económicos, de infraestructura y de tiempo del equipo investigador. Los factores a tener en cuenta para el diseño de un estudio se pueden clasifi car según diferentes criterios:
• Naturaleza de los datos: cuantitativo o cualitativo. • Propósito del estudio: descriptivo o analítico. • Asignación de la exposición: experimental u observacional. • Temporalidad del seguimiento: transversal o longitudinal. • Relación cronológica del inicio del estudio y aparición del fenómeno de estudio: prospectivo o retrospectivo. • Unidad de estudio: individuo o grupo de individuos.
En la tabla 2.1.1 se describen de manera general las características de cada uno de los criterios de clasifi cación para el diseño de un estudio.
La tigura 2.1.1 presenta un algoritmo de clasifi cación de los estudios teniendo en cuenta algunos de los criterios anteriormente descritos. Este algoritmo no presenta una clasifi cación universalmente aceptada (ya que puede variar en función de los criterios que se usen), pero puede ayudar a encuadrar la pregunta de investigación y decidir el diseño de estudio más adecuado.
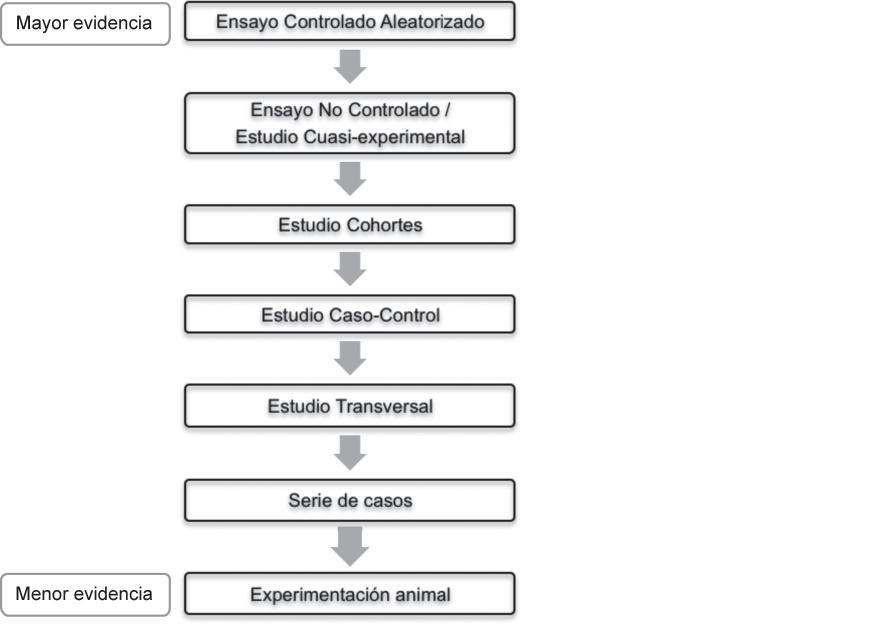
Otra de las características a tener en cuenta y que está relacionada con un tipo de diseño u otro es la evidencia científi ca que los resultados de un estudio proporcionarán. La Figura 2.1.2 esquematiza de manera general la jerarquía de evidencia científi ca que cada tipo de estudio aporta.
Como se mencionó anteriormente, la estructura del diseño de nuestro estudio combinará las diferentes características o criterios que aparecen en la tabla y en el algoritmo. A continuación, se presentan algunos ejemplos de estudios, ya publicados, con el objetivo de analizar los criterios de clasifi cación vistos en este capítulo.
Ejemplos
Ejemplo 1. Garcia-Aymerich J, Gómez FP, Benet M, et al. PAC-COPD Study Group. Identifi cation and prospective validation of clinically relevant chronic obstructive pulmonary disease (COPD) subtypes. Th orax. 2011;66:430-437.
Este estudio recoge datos demográfi cos, clínicos, funcionales, etc. (estudio cuantitativo) de 342 pacientes con enfermedad pulmonar obstructiva crónica (EPOC) durante su primer ingreso por agudización en 9 hospitales terciarios de 3 comunidades autónomas españolas, sin hacer ningún tipo de intervención (estudio observacional), con el objetivo de: 1) identifi car los subtipos de los pacientes que ingresan por primera vez por agudización de la EPOC (analítico transversal) y 2) validar los subtipos identifi cados durante un seguimiento de 4 años (analítico longitudinal prospectivo) con variables clínicamente relevantes, como hospitalización y mortalidad (relación causa-efecto: estudio de cohorte).
Ejemplo 2. McKeever T, Harrison TW, Hubbard R, Shaw D. Inhaled corticosteroids and the risk of pneumonia in people with asthma: a case-control study. Chest. 2013;144:1788-1794.
En una cohorte de pacientes con diagnóstico de asma se identifi có a los pacientes que hubieran sido diagnosticados de neumonía (casos) y a pacientes de la misma edad y sexo que no hubieran tenido diagnóstico de neumonía (controles) (estudio cuantitativo) con el objetivo de estudiar la asociación (estudio analítico) de neumonía y el uso de corticoides inhalados en pacientes con asma (estudio observacional, longitudinal, relación efecto-causa: estudio de casos-controles).
Ejemplo 3. Miravitlles M, Soriano JB, García-Río F, et al. Prevalence of COPD in Spain: impact of undiagnosed COPD on quality of life and daily life activities. Th orax. 2009;64:863-868.
En este estudio se recogen datos demográfi cos y clínico-funcionales (cuantitativo) mediante envío de un cuestionario (transversal) a una muestra poblacional de más de 4.200 sujetos de edades comprendidas entre 40 y 80 años de 10 ciudades de España (descriptivo) para estimar la prevalencia de EPOC (transversal: prevalencia).
Ejemplo 4. Ehlken N, Lichtblau M, Klose H, et al. Exercise training improves peak oxygen consumption and haemodynamics in patients with severe pulmonary arterial hypertension and inoperable chronic thrombo-embolic pulmonary hypertension: a prospective, randomized, controlled trial. Eur Heart J. 2016;37:3544.
En este estudio se selecciona una muestra de 87 pacientes con hipertensión arterial pulmonar con el objetivo de estudiar los efectos de 15 semanas de entrenamiento físico en parámetros clínicos como el pico en el consumo de oxígeno y en la hemodinámica (estudio cuantitativo analítico). Los pacientes son asignados aleatoriamente a grupo intervención (entrenamiento físico) o grupo control (tratamiento habitual), y son
evaluados antes y después de la intervención (estudio prospectivo, experimental, aleatorizado y con grupo control: ensayo clínico controlado aleatorizado – ECA).
Ejemplo 5. Dobbels F, De Jong C, Drost E, et al. Th e PROactive innovative conceptual framework on physical activity. Eur Respir J. 2014;44:1223-1233.
Este estudio es un ejemplo de diseño cualitativo, ya que aunque los autores aportan información numérica sobre las características de los sujetos de estudio (edad, función pulmonar, prueba de la marcha, etc.), los resultados principales son básicamente narrativos: un marco conceptual y anotaciones extraídas de entrevistas y grupos focales con pacientes con EPOC para conocer su experiencia de actividad física.
PUNTOS CLAVE
• La pregunta de investigación y, por lo tanto, el objetivo principal del estudio constituye los factores que determinarán en mayor medida la selección del diseño más adecuado. • Los diferentes tipos de estudios se distribuyen de manera jerárquica en función de la evidencia científi ca que generan. En humanos, la mayor evidencia la proporcionarán los ensayos controlados aleatorizados (ECA), mientras que las series de casos son los que menor evidencia producen. • Los estudios cualitativos recogen información narrativa (no numérica) basada en percepciones, opiniones y explicaciones, mientras que los estudios cuantitativos recogen información que puede ser medida o registrada numéricamente. • Los estudios descriptivos se centran en el estudio de características generales, distribución de la población y descripción de la historia natural de una enfermedad. Los estudios analíticos permiten estudiar la relación causal entre la exposición (o intervención) y el efecto (o resultado). • En los estudios observacionales no existe manipulación por parte del investigador, quien se limita a observar lo que ocurre. Sin embargo, en los estudios experimentales se manipula el factor de estudio para evaluar el efecto. • Los estudios transversales recogen la información en un momento temporal determinado (ideales para estudiar prevalencia). Los estudios longitudinales recogen los datos después de un periodo de seguimiento (ideales para estudiar causalidad). • En los estudios prospectivos, el evento de interés se producirá después del inicio del estudio, mientras que en los estudios retrospectivos el evento de interés ya se produjo (los datos se pueden recoger mediante revisión de archivos históricos).
BIBLIOGRAFÍA
Argimón Pallás JM, Jiménez Villa J. Métodos de investigación clínica y epidemiológica. Madrid, Elsevier, 4ª ed., 2013. Garcia-Aymerich J, Soriano JB. “Aspectos epidemiológicos en neumología”. En: Casán P, García Río F, Gea J (editores). Fisiología y biología respiratorias. Madrid, ERGON, 2007, p. 689-701. Chatburn RL. Handbook for respiratory care research. Cleveland, Mandu Press, 1ª ed. 2002.
TABLAS Y FIGURAS
Tabla 2.1.1. Criterios de clasifi cación para el diseño de un estudio
Naturaleza de los datos Cualitativo Cuantitativo
Recoge información narrativa extraída de entrevista no estructurada, observación del participante, análisis documental, grupos focales, etc. Estudia las percepciones, opiniones y explicaciones en un grupo de sujetos de interés en situaciones o contextos dinámicos. Análisis inductivo (desarrollo de teorías). Recoge información que puede ser medida numéricamente. Estudia la asociación o relación entre variables cuantifi cables a través de una muestra representativa en contextos más controlados. Análisis deductivo (análisis de la teoría).
Propósito del estudio Descriptivo Analítico
Permite estudiar características generales y distribución poblacional, así como describir la historia natural de las enfermedades. Útil para generar hipótesis etiológicas que deberían contrastarse con estudios analíticos. Muchas veces clasifi cados como observacionales por su naturaleza implícita de no manipulación del factor de estudio. Permite evaluar una relación causal entre un factor, exposición o intervención y el efecto o resultado. Útil para confi rmar hipótesis etiológicas.
Asignación del factor de estudio Observacional Experimental
No existe manipulación del factor de estudio por parte del investigador, este simplemente se limita a observar, medir y analizar las variables de estudio en los sujetos de interés. Permite probar hipótesis y estudiar relaciones causales. Manipulación (asignación) del factor de estudio por parte del investigador, mediante aleatorización y según un protocolo previamente establecido. Si la asignación no es aleatorizada será un diseño
cuasi-experimental.
En general, se usa para evaluar el efecto de una intervención.
Temporalidad del seguimiento Transversal Longitudinal
Los datos se obtienen en un momento temporal determinado (cross-sectional). Se mide a la vez la frecuencia de la exposición y la enfermedad de interés, y por ello no permiten establecer relación causa-efecto. Ideal para estudiar (describir) la prevalencia de una enfermedad o una característica clínica, y para generar hipótesis causales que deberían contrastarse con estudios longitudinales. Los datos se obtienen después de un periodo de seguimiento (follow-up). Se establece una secuencia temporal entre las variables medidas lo que permite estudiar causalidad. Si la dirección temporal va de la causa hacia el desenlace (efecto) será un estudio experimental o uno de cohortes. Si la dirección temporal va desde el desenlace (efecto) hacia la causa será un estudio de casos y controles.
Relación cronológica del inicio del estudio y la aparición del fenómeno Prospectivo Retrospectivo
El desenlace o evento de interés se produce después del inicio del estudio. Los datos de las variables de interés se recogen a medida que van sucediendo. El desenlace o evento de interés se ha producido antes del inicio del estudio. Los datos se recogen mediante revisión de archivos o de historia clínica, o a través de entrevista sobre los eventos sucedidos.
Unidad de estudio
Individuo Grupo de individuos (ecológicos)
La información se recoge de manera individual. La información se recoge de manera agregada según criterios previamente establecidos, por ejemplo, geográfi cos o temporales.
Figura 2.1.1. Algoritmo de clasifi cación de los tipos de diseño de estudio
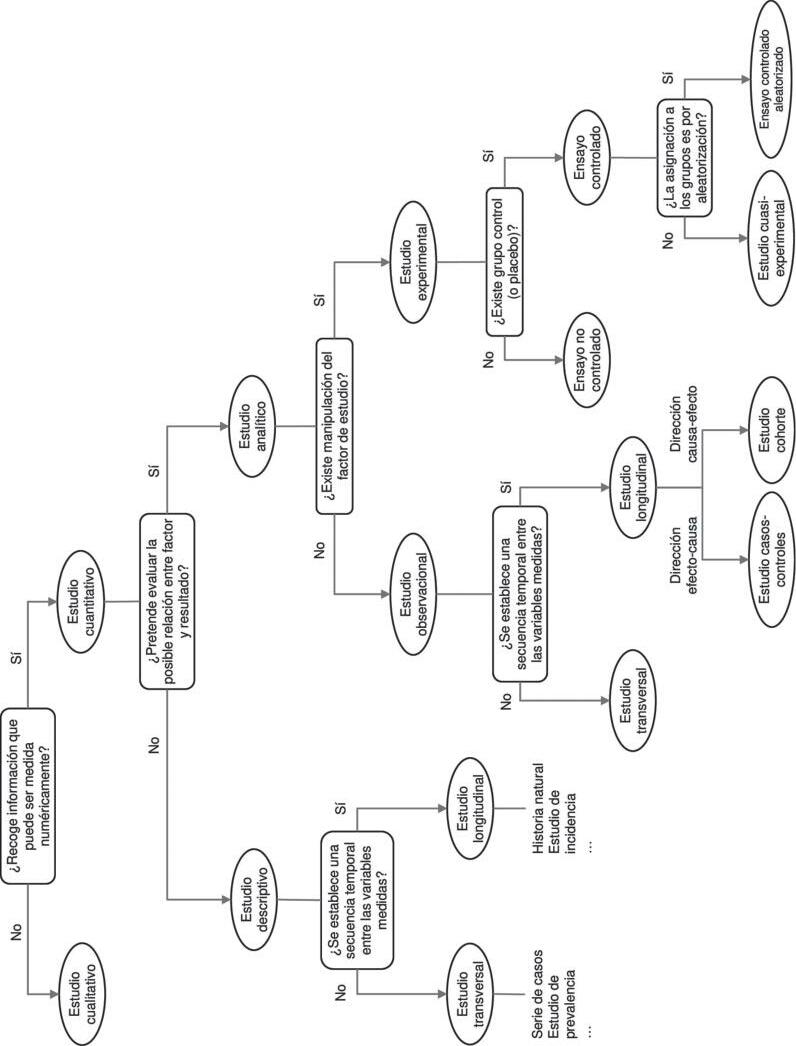
Figura 2.1.2. Jerarquía de evidencia científi ca según el tipo de estudio
2.2. ESTUDIOS DESCRIPTIVOS
Dr. José Luis López-Campos Bodineau
INTRODUCCIÓN
Las principales fuentes de orientación ética para la realización de investigaciones clínicas son tres: la DeclarA pesar de la extendida creencia de que los estudios descriptivos pueden tener un menor interés, esta opinión no se corresponde con la realidad. Los estudios descriptivos permiten tener una primera aproximación a un problema de salud, detectar de manera rápida un nuevo evento clínico, comparar situaciones entre áreas geográfi cas o momentos temporales y, además, son la principal fuente de generación de hipótesis para ulteriores estudios. Se trata, por tanto, de diseños sencillos, pero que pueden tener un gran poder de información. En concreto, la epidemiología descriptiva permite responder preguntas relevantes sobre la distribución de las enfermedades en el espacio y el tiempo y determinar los factores asociados a esta distribución. De esta manera, los estudios descriptivos aportan información sobre las poblaciones que desarrollan una determinada enfermedad o un efecto clínico, su frecuencia en comparación con otras poblaciones, cómo varía esta frecuencia en el tiempo o entre localizaciones geográfi cas y los factores de riesgo asociados. Por este motivo, es necesario conocer bien los estudios descriptivos y saber diseñarlos, ejecutarlos y analizarlos adecuadamente. Sus dos grandes ventajas son posibilitar la síntesis de información compleja y extensa y la generación de hipótesis. Se trata, por tanto, de diseños denominados observacionales o no experimentales, puesto que su objetivo es describir una situación clínica concreta en un contexto temporal y geográfi co sin intervenir de ninguna manera en la población observada.
CARACTERÍSTICAS GENERALES
Para llevar a cabo un estudio descriptivo, es necesario realizar una serie de pasos que nos ayudarán a defi nir el objetivo de nuestro trabajo y que deben tenerse en cuenta en la metodología. En su diseño deberemos defi nir: la enfermedad a estudiar, la población diana, las variables que vamos a registrar y la elección de la fuente de información.
Problema de salud a estudiar. En el diseño del estudio descriptivo es necesario identifi car y defi nir bien la enfermedad que va a ser objeto de estudio. Este paso es crucial para interpretar los resultados y compararlos con otros estudios previos. En este punto, además de identifi car la enfermedad en concreto, es necesario defi nir bien los criterios diagnósticos. Un ejemplo cercano está representado por los estudios epidemiológicos sobre enfermedad pulmonar obstructiva crónica (EPOC). Aunque el concepto de la enfermedad está claro,
los criterios diagnósticos para defi nir un caso en un estudio epidemiológico pueden variar, ya que pueden utilizarse el cociente fi jo FEV1/FVC de la espirometría o bien su límite inferior de la normalidad, y puede hacerse con la espirometría, la pre o la post-broncodilatación. Según se elija unos u otros, la prevalencia de la enfermedad cambiará.1
Población diana. Aunque siempre es interesante conocer cómo se comporta la población respecto a una determinada enfermedad o factor de riesgo, la población a estudio no tiene por qué ser la población general. En ocasiones interesará estudiar una población concreta. Por ejemplo, el cáncer de pulmón en mujeres, o en población no fumadora, o en un determinado rango de edad. Esta población debe quedar perfectamente identifi cada en el diseño del trabajo.
Variables a registrar. Es importante defi nir previamente qué información se va a registrar de cada caso. Para ello, no sólo es importante seleccionar las variables, sino que también hay que concretar dos aspectos relevantes:
• Cómo se van a defi nir estas variables. Por ejemplo, si queremos estudiar la distribución de pacientes con frecuentes agudizaciones, no sólo tendremos que elegir el número de agudizaciones como variable, también tendremos que defi nir qué vamos a considerar una agudización y qué vamos a considerar como agudizador frecuente. Otro ejemplo podría ser el nivel socioeconómico, cuya defi nición y estratifi cación deberemos defi nir en la metodología. Situaciones parecidas se pueden dar en la categorización de la gravedad de una enfermedad, la intensidad de la exposición a un factor de riesgo o los valores de una determinación analítica. • Cómo se van a registrar. En este sentido, deberemos defi nir si se va a registrar como una variable continua o categórica. Para las variables cualitativas está claro que se deben registrar como categóricas, pero deberemos defi nir las categorías que vamos a emplear. En ocasiones será sencillo (por ejemplo, hombre o mujer), pero en otras será más complicado (por ejemplo, nivel de estudios o situación socioeconómica). En cuanto a las variables cuantitativas, podremos registrarlas como números o categorizarlas; en este último caso también deberemos defi nir las categorías. Esta decisión es de especial relevancia, ya que dependiendo de si usamos la variable cuantitativa cruda o categorizada realizaremos después el análisis estadístico de los datos en la misma línea.
Elección de la fuente de información. En nuestro caso, la fuente de información será por lo general la historia clínica del paciente o bien datos almacenados en bases de datos sanitarias. Aquí es importante tener presente dos aspectos:
• La fuente de la información. En función de la variable que queramos medir, deberemos recurrir al historial médico, al de enfermería o a otros registros. Por ejemplo, si vamos a registrar la temperatura corporal de los pacientes durante una hospitalización, no tendría mucha validez si extrajéramos este dato de la historia médica, puesto que allí se recoge de manera inconstante. Sin embargo, el historial de enfermería la recoge sistemáticamente. • Si la información que vamos a usar se puede extraer directamente como dato primario del origen o si, por el contrario, la información tiene que ser procesada antes de su análisis. Por ejemplo, podemos analizar los niveles de potasio en suero, que es un dato primario de la historia clínica; sin embargo si queremos estudiar el uso de un índice compuesto, tendremos que calcularlo a partir de los datos de la historia. En este último caso, será necesario defi nir muy bien en la metodología cómo tratan los datos y de qué fuente provienen.
Clasifi cación. Los estudios descriptivos se han clasifi cado de distintas formas. En este capítulo hablaremos de dos grandes tipos de estudio en función de la unidad de análisis. Si la unidad de análisis son personas,
hablaremos de estudio de base individual. Si, por el contrario, se analizan datos de grupos de poblaciones, también llamados datos agregados, entonces hablaremos de estudios ecológicos.
ESTUDIOS ECOLÓGICOS
Los estudios ecológicos usan datos agregados de grupos poblacionales. Su función es describir la distribución de las enfermedades en una población, durante un período de tiempo concreto y en un ámbito geográfi co defi nido. De esta manera, pueden dar información sobre la frecuencia de la enfermedad o del fenómeno a estudiar y cuáles son los factores asociados. Este tipo de estudio permite, además, identifi car aumentos en el tiempo o el espacio de una agregación de casos o eventos que son de una ayuda considerable al planifi car políticas de salud.
Hasta hace unas décadas, estos estudios eran difíciles de llevar a cabo, puesto que no existían muchas fuentes de información de donde extraer los datos. Con la informatización de los datos y la disponibilidad a través de internet, se ha conseguido que estos datos estén disponibles para los investigadores interesados en responder preguntas de investigación. Por lo general, las bases de datos de salud suelen estar abiertas siempre que se cumplan los requisitos éticos, incluyendo la confi dencialidad de los datos, y que se utilicen para el fi n de investigación defi nido.
Entre sus ventajas destaca que son relativamente rápidos de realizar, siempre que se tenga la información adecuada. Además, son bastante económicos, ya que, al tener acceso a la información, realmente el principal recurso necesario para llevarlos a cabo es tiempo para hacer un análisis correcto. Su principal limitación es que sirven para plantear hipótesis, pero generalmente no para verifi carlas. Otra limitación metodológica se deriva de la comparación entre estudios. Debido a que son estudios de datos agregados, puede ser erróneo comparar datos de áreas geográfi cas o momentos temporales distintos, puesto que la estructura de la población que se estudia puede infl uir en los resultados. Por ejemplo, podríamos detectar una mayor mortalidad por EPOC en un país en comparación con otro, pero si el primer país tuviera una población más envejecida, entonces las diferencias podrían deberse a esta diferencia y no a un porcentaje de mortalidad por EPOC distinto. Para corregir esto, los estudios ecológicos ajustan sus valores a la población de estudio. De esta manera, las denominadas tasas crudas se transforman en tasas ajustadas a la población de esa zona y en ese momento. Es lo que se llama estandarización o ajuste de tasas. Las formas de estandarización de tasas quedan fuera de los objetivos de este capítulo, pero básicamente existen dos métodos, denominados método directo e indirecto. Una consideración metodológica que limita estos estudios es que asumen que toda la población está expuesta en la misma medida a los factores de riesgo en estudio, por lo que sus resultados no deben aplicarse a un caso concreto, puesto que incurriríamos en la denominada falacia ecológica.
Para entender la falacia ecológica, es importante recordar que los estudios ecológicos nos dan información agregada de la población en estudio, pero no de los individuos concretos. La falacia ecológica se produce cuando queremos extrapolar los resultados de un grupo a un individuo concreto o bien cuando queremos extrapolar los resultados de un individuo o grupo de individuos al grupo completo. Los estudios ecológicos se pueden clasifi car en dos grandes grupos: estudios descriptivos de mapas y estudios de series temporales.
Estudios descriptivos de mapas
Los estudios de mapas estudian un determinado parámetro epidemiológico, como puede ser la incidencia, la prevalencia o la mortalidad, buscando patrones según su distribución entre diversas áreas geográfi cas. Estas áreas pueden ser de cualquier tamaño: localidades, provincias, países o continentes. Generalmente se hace un mapa de calor en el que se colorea cada ámbito geográfi co en mayor o menor intensidad de color
según el mayor o menor valor del parámetro estudiado. La generación de estos mapas siempre ha necesitado una gran cantidad de tiempo, puesto que tradicionalmente se han hecho de manera manual con soft ware no específi co. En los últimos años han comenzado a aparecer aplicaciones específi camente diseñadas para el tratamiento de los datos y su distribución geográfi ca. Estas aplicaciones generan los mapas necesarios una vez se han introducido los datos. Entre ellas fi guran aplicaciones independientes, aplicaciones que se integran en los paquetes de ofi mática más comunes del mercado y aplicaciones online a través de páginas web específi cas. La fi gura 2.2.1 es un ejemplo de este tipo de mapas realizado por una aplicación online de acceso gratuito.
Estudios de series temporales
Los estudios de series o tendencias temporales describen la evolución de un problema de salud en el tiempo. En este tipo de estudio, es el tiempo y no el espacio lo que se analiza para apreciar los cambios del problema de salud. La principal limitación es que si la información es considerable y la variabilidad de los datos también, entonces puede ser difícil ver un patrón. Para esto existen métodos estadísticos que simplifi can la información y muestran una tendencia única. El método más conocido es la denominada regresión de joinpoint.2 Este análisis de regresión identifi ca puntos de infl exión (denominados joinpoints) en la curva de evolución temporal, con el objeto de identifi car los años en que se produjeron cambios signifi cativos en la pendiente lineal de la tendencia temporal. Este enfoque tiene dos ventajas principales: identifi ca el momento en que hay cambios en la tendencia y estima la magnitud del aumento o de la disminución observada en cada intervalo mediante el cálculo del denominado porcentaje de cambio anual entre dos joinpoints. Actualmente existe soft ware gratuito que hace este tipo de análisis. En la fi gura 2.2.2 se presenta un ejemplo de estudio de serie temporal con análisis de regresión de joinpoint.
ESTUDIOS DE BASE INDIVIDUAL
Series de casos
Los estudios de series de casos, o incluso de un caso concreto, son los estudios más simples. La descripción de uno o varios casos clínicos con características clínicas relevantes constituye la base de la investigación biomédica, sus orígenes, y tiene una gran relevancia clínica. A pesar de que su valor epidemiológico es escaso por no tener una muestra representativa ni contar con un grupo control, al compartir con colegas una presentación clínica peculiar, junto con la actitud diagnóstica o terapéutica llevada a cabo su valor clínico y docente es indudable. Además, este tipo de estudios también cumple la función de establecer hipótesis clínicamente relevantes que más adelante deberán confi rmarse con otro tipo de estudio. Por ejemplo, en 1935 dos médicos de Baltimore, Louis Hamman y Arnold Rich, publicaron una serie de 4 casos clínicos, la mayoría de pacientes de raza negra, entre los 20 y los 30 años de edad, que desarrollaban un cuadro clínico de un fracaso respiratorio agudo acompañado de infi ltrados pulmonares y con un pronóstico ominoso.3 Esta descripción inicial dio lugar al estudio de la patología intersticial pulmonar con un cuadro que en su día se denominó como pulmón de Hamman-Rich y que actualmente se ha asociado con la neumonía intersticial aguda. Entre sus ventajas fi guran que son fáciles, económicos y rápidos de realizar, tienen sentido clínico, permiten llamar la atención sobre un hecho clínico poco habitual y sirven para generar hipótesis.
Estudios transversales
Los estudios transversales, también llamados estudios de prevalencia, tienen por objetivo evaluar la presencia de una enfermedad y de los factores de riesgo asociados en un mismo momento temporal y en una población bien defi nida. Como resultado obtenemos la prevalencia de una determinada enfermedad o factor de riesgo en esa población defi nida.
Ventajas
Entre sus ventajas fi guran que proporciona información fi able y estática, una fotografía, para la planifi cación sanitaria, ya que permite identifi car grupos más vulnerables a un determinado problema de salud y además describe las enfermedades y las relaciona con la distribución de sus factores de riesgo en la población. Además, proporciona información basal para estudios de seguimiento y puede dar información sobre el impacto de una determinada intervención terapéutica en la población al repetir el estudio tras esta intervención. Finalmente, otra ventaja es su coste relativamente bajo en comparación con los estudios de seguimiento.
Limitaciones
El diseño de los estudios transversales, por otro lado, tiene algunos retos metodológicos. En primer lugar, por lo general resulta difícil estudiar toda la población diana, por lo que suelen tener una primera fase de selección de una muestra sobre la que se hace el estudio. El reto está en asegurar que esta muestra seleccionada es representativa de la población completa. Para conseguirlo, es necesario hacer algún tipo de muestreo bien defi nido y defi nir bien los criterios de inclusión y exclusión en el estudio. Los tipos de muestreos, sus ventajas e inconvenientes quedan fuera de los objetivos de este capítulo, pero este aspecto es importante para darle validez a los resultados. Es vital que las personas estudiadas sigan este proceso de muestreo para evitar sesgos en la selección de la muestra que la haga no representativa de la población a estudio. En este sentido, se han descrito diversos tipos de sesgos de selección que deben ser tenidos en cuenta, como el del voluntario sano, el de los no respondedores o el de supervivencia. En segundo lugar, es importante recordar que la información es estática en el tiempo, es una fotografía de la situación. Por tanto, al medir simultáneamente la presencia o no de enfermedad y sus factores de riesgo no tenemos noción de la dirección temporal de la asociación. En otras palabras, no sabemos si fue antes la enfermedad o el factor de riesgo. En tercer lugar, generalmente se asume que la enfermedad a estudio está representada por una variable dicotómica (tiene o no tiene la enfermedad), pero esto no siempre es así.
Análisis
El análisis de los estudios transversales nos aporta un resultado más inmediato, la prevalencia, entendida como número de sujetos que tienen la enfermedad entre el número de sujetos estudiados. Para estudiar su relación con un factor de riesgo determinado, podemos calcular la prevalencia entre los expuestos a ese factor de riesgo y entre los no expuestos. Esto nos permite estimar cómo de frecuente es la enfermedad entre expuesto y no expuesto mediante el cálculo de la razón de prevalencia, que es el cociente de ambas. Como alternativa a esta aproximación, un estudio de prevalencia se puede analizar como un estudio de casos-control y calcular la odds ratio de prevalencia, como se verá en el capítulo de los estudios de casos-control.
PUNTOS CLAVE
• Los estudios descriptivos permiten tener una primera aproximación a un problema de salud, comparar situaciones entre áreas geográfi cas o momentos temporales y disponer de una fuente de generación de hipótesis. • Los estudios ecológicos usan datos agregados de grupos poblacionales con objeto de describir la distribución de las enfermedades en una población, durante un período de tiempo concreto y en un ámbito geográfi co defi nido. • A pesar de que su valor epidemiológico es escaso por no tener una muestra representativa ni contar con un grupo control, los estudios de series de casos constituyen la base de la investigación biomédica y sus orígenes y tienen una gran relevancia clínica. • Los estudios transversales, también llamados estudios de prevalencia, tienen por objetivo evaluar la presencia de una enfermedad y de los factores de riesgo asociados en un mismo momento temporal.
BIBLIOGRAFÍA
1. Tilert T, Dillon C, Paulose-Ram R, Hnizdo E, Doney B. Estimating the U.S. prevalence of chronic obstructive pulmonary disease using pre- and post-bronchodilator spirometry: the National Health and
Nutrition Examination Survey (NHANES) 2007-2010. Respir Res. 2013;14:103. 2. Kim HJ, Fay MP, Feuer EJ, Midthune DN. Permutation tests for joinpoint regression with applications to cancer rates. Stat Med. 2000;19(3):335-351. 3. Hamman L, Rich AR. Fulminating Diff use Interstitial Fibrosis of the Lungs. Trans Am Clin Climatol
Assoc. 1935;51:154-163. 4. López-Campos JL, Tan W, Soriano JB. Global burden of COPD. Respirology. 2015. 5. López-Campos JL, Ruiz-Ramos M, Soriano JB. COPD mortality rates in Andalusia, Spain, 1975-2010: a joinpoint regression analysis. Int J Tuberc Lung Dis. 2013;17(1):131-136.
TABLAS Y FIGURAS
Figura 2.2.1. Ejemplo de estudio ecológico de mapas. La fi gura muestra la distribución de la mortalidad por EPOC en el mundo

Reproducido de López-Campos JL, et al. Respirology. 2015(4).
Figura 2.2.2. Ejemplo de estudio de serie temporal. La gráfi ca representa un análisis de regresión de joinpoint sobre la mortalidad por EPOC en Andalucía en hombres (A) y mujeres (B)

SMR: tasa de mortalidad estandarizada. TMR: tasa de mortalidad estandarizada y truncada para las edades entre 40 y 70 años. Reproducido de López-Campos JL, et al. Int J Tuberc Lung Dis. 2012(5).
2.3. ESTUDIOS ANALÍTICOS: COHORTES Y CASO-CONTROL
Dr. Joan B. Soriano Ortiz Dra. Patricia Pérez
INTRODUCCIÓN
Gran parte de la investigación biomédica es de tipo observacional. Al contrario de los ensayos clínicos, en que la calidad se da por supuesta, en la investigación observacional la calidad es a menudo insufi ciente, lo que difi culta la evaluación de sus puntos fuertes y débiles para la generalización de un estudio y su aplicación traslacional a los pacientes y a la salud general. Dentro de la sistemática de los diseños de estudio en investigación, los estudios observacionales no descriptivos y analíticos, que permiten probar hipótesis y estudiar causalidades, se clasifi can en:
• Estudios de cohorte: Siguen a lo largo del tiempo a un grupo de sujetos (cohorte) inicialmente libres de enfermedad midiendo en el momento de su exposición al factor de interés y estudiando la aparición posterior de la enfermedad. • Estudios de casos y controles, también llamados de caso-control: Comparan determinadas características (exposiciones) entre personas con la enfermedad (casos) y personas libres de la enfermedad (controles).
A continuación se repasan sucintamente las características y usos de cada uno.
ESTUDIOS DE COHORTE
También denominados estudios de seguimiento, de proyección o de incidencia. La palabra cohorte deriva del latín cohors, que era una unidad táctica del ejército romano formada por 480 hombres o legionarios, que marchaban juntos a la batalla. Diez cohortes, numeradas del I al X, constituían una legión romana. Durante el gobierno del emperador Augusto, el ejército romano totalizó 28 legiones. Es de interés repasar las distintas unidades: contubernio, 8 hombres; centuria, 10 contubernios (80 hombres); manípulo, 2 centurias (160 hombres); cohorte, 6 centurias (480 hombres); legión, 10 cohortes más 120 jinetes (5.240 hombres). Una vez entraban en una cohorte, liderada por seis centuriones, los soldados romanos podían cambiar de posición según su jerarquía y experiencia dentro de su cohorte, hasta que se licenciaban o morían.
Este mismo concepto se aplica a la cohorte en investigación clínica: un individuo sano o sin enfermedad/ evento de interés es reclutado en una cohorte y aporta información hasta que, voluntariamente, involuntariamente o por fallecimiento, abandona la cohorte.
En investigación epidemiológica y clínica, un estudio de cohortes es un estudio epidemiológico, observacional, analítico y longitudinal, prospectivo en el que compara la frecuencia de una enfermedad (o de un determinado desenlace) entre muestras de una población o entre dos o más poblaciones, una de las cuales está sometida a un determinado factor de exposición o factor de riesgo al que no está expuesta la otra. Los individuos que componen los grupos de estudio se seleccionan en función de la presencia de una determinada característica o exposición.
Estos individuos no tienen la enfermedad de interés y son seguidos durante un cierto periodo de tiempo para observar la frecuencia con que la enfermedad aparece en cada uno de los grupos. Los estudios de cohorte tienen por objetivo medir causalidad entre los factores de riesgo y la enfermedad a estudiar.
Proceso de un estudio de cohorte
El proceso para realizar un estudio de cohorte incluye las siguientes etapas:
• Seleccionar una muestra de estudio de individuos sanos de una población. El concepto sano se entiende en su acepción más amplia, y signifi ca que antes de iniciar el periodo de seguimiento los participantes no han experimentado la enfermedad o evento de interés. • Medir variables de exposición en la muestra, si el factor de riesgo está ausente o presente. • Seguir la cohorte. • Medir las variables de resultado, es decir, la presencia o ausencia de enfermedad. • Analizar mediante la estadística adecuada.
Ventajas de un estudio de cohorte
Un diseño de estudio de cohorte permite:
1. Estudiar factores de exposición extraños. 2. Visualizar los múltiples efectos (riesgos y benefi cios) que una exposición pueda tener. 3. Observar simultáneamente los efectos de varias exposiciones (siempre y cuando esta posibilidad sea planteada desde el principio del estudio). 4. Posibilitar la muestra de la secuencia temporal entre exposición y desenlace. 5. Permitir la estimación de incidencia y riesgo relativo. 6. Establecer claramente la secuencia de sucesos de interés, como es la de exposición-enfermedad. 7. Evitar el sesgo de supervivencia. 8. Tener mejor control sobre la selección de sujetos. 9. Tener mayor control de las medidas. 10. Tener movimiento.
Inconvenientes de un estudio de cohorte
Sin embargo, también deben considerarse sus potenciales limitaciones:
1. A menudo requiere grandes tamaños muestrales. 2. No es efi ciente en eventos raros o con largos períodos de latencia (tiempo transcurrido desde la exposición al evento).
3. Su ejecución puede requerir mucho tiempo y dinero. 4. No es útil para enfermedades raras (pues no hay sufi cientes sujetos). 5. Son estudios caros por la cantidad de personas y el tiempo de seguimiento necesarios, así como por los esfuerzos que han de llevarse a cabo para que la calidad del seguimiento minimice las pérdidas entre las poblaciones que se siguen, manteniéndolas en un nivel aceptable. 6. No se dispone de resultados hasta después de transcurrido mucho tiempo. 7. Sólo evalúan la relación entre el evento del estudio y la exposición a ciertos factores defi nidos y/o cuantifi cados al inicio del estudio.
Los estudios de seguimiento son los estudios prospectivos clásicos, caracterizados por el hecho de que el planteamiento del estudio y la medición de la exposición (o no exposición) se producen con anterioridad al desarrollo de la enfermedad. A pesar de que se trata de estudios muy costosos, pues requieren grandes recursos económicos y de tiempo, y de gran solidez, ya que la probabilidad de que estén sesgados es menor, hay que cuidar extremadamente los aspectos del diseño y evitar sesgos, especialmente de clasifi cación y el de los trabajadores sanos, entre otros.
Como alternativa, existe otro tipo de estudios de cohortes, el estudio de las cohortes retrospectivas, en que la investigación se plantea después de haberse producido la enfermedad, para lo cual tanto la presencia de exposición como el desenlace deben proceder de registros históricos o clínicos de alta fi abilidad en los que se hayan registrado con precisión en el momento de producirse.
En función de la existencia de medidas repetidas, se distinguen dos tipos de estudios de cohorte: estudios longitudinales y estudios sin medidas repetidas.
El estudio de cohorte perfecto no existe, y quizás sea literalmente imposible de conseguir.1 En una enfermedad crónica, asociada al envejecimiento y al tabaco, como la EPOC,2 o con aspectos ambientales, como el asma, sería quizás una versión ampliada al ámbito respiratorio del Estudio de Framingham3 lo que nos permitiera hacer lo que los cardiólogos ya han hecho. Además, se debe tener en cuenta el conocimiento (o la falta de conocimiento) de la genética y el origen precoz de las enfermedades de aparición tardía. Desde este punto de vista, un tanto ingenuo, el estudio de cohortes para explorar la historia natural de una enfermedad debe tener las siguientes características: ser grande; comenzar antes del nacimiento; incluir el seguimiento de por vida; incluir unos pocos cientos de páginas de formularios de encuestas, pruebas de función pulmonar estandarizadas y muchas otras, mediciones de biomarcadores (incluyendo imágenes) y medidas de exposición directa y, por supuesto, la genética y todas las “ómicas” (por ejemplo, la genómica, la proteómica, la metabolómica). Obviamente, no es una tarea fácil. Una alternativa podría ser la puesta en común de los resultados de estudios de historia natural individuales. Utilizando un enfoque metaanalítico, podría ser una solución parcial, positiva, como se ha propuesto en cohortes de nacimiento que estudian asma y atopia, y se está explorando actualmente para la función pulmonar y la EPOC. Como consecuencia, el estudio de cohortes ideal para investigar la historia natural de las enfermedades respiratorias crónicas podría consistir en múltiples estudios, cada uno de los cuales se dirigiría a responder una pregunta científi ca específi ca.
Existen cohortes de muy diversos orígenes y modelos a seguir. Una cohorte puede representar desde una muestra de la población general en un lugar determinado geográfi camente (p. ej., la cohorte de Framingham mencionada anteriormente) hasta un grupo de trabajadores pertenecientes a una industria o un grupo de pacientes con una exposición (p. ej., hipertensos tratados). Algunas cohortes seleccionan a individuos de los que se espera un alto grado de colaboración para minimizar las pérdidas y aumentar la calidad de los datos recogidos. Es el caso de la cohorte de enfermeras de la Universidad de Harvard o la cohorte de voluntarios de la Sociedad Americana del Cáncer. Existen cohortes que fueron establecidas hace años y cuyos miembros son objeto de un seguimiento regular. Aunque en su día fueron diseñadas para un estudio concreto, normal-
mente con el paso del tiempo sus usos se van ampliando y llegan a ser una valiosa fuente de datos para múltiples enfermedades. Otras cohortes que se han utilizado son las llamadas históricas (p. ej., supervivientes a la bomba atómica de Hiroshima). En SEPAR, las cohortes ya establecidas y de gran utilidad incluyen, entre otros, los trabajadores de tareas de rescate del Prestige4 y la cohorte de pacientes CHAIN.5
Análisis
La medida de asociación usada normalmente en los estudios de cohorte es el riesgo relativo (RR). Tal como se aprecia en la tabla 2.3.1, el análisis para un estudio de cohorte permite calcular la incidencia en cada grupo y el riesgo relativo en los expuestos en comparación con los no expuestos, de modo que riesgo relativo = (a/a+b)/(c/c+d).6
Si los tiempos de seguimiento son desiguales, sea por abandono, fallecimiento por una causa diferente al evento de estudio, fi nalización del periodo de estudio establecido en el protocolo u otros, una forma de compensarlo es realizar análisis basados en tiempo-persona o en el promedio de tiempo durante el que se ha seguido a los sujetos de la cohorte. Esto posibilita calcular la tasa de incidencia por una unidad de tiempo determinado, de modo que riesgo relativo = (a/tpe) / (c/tpne), donde tpe es el tiempo-persona de seguimiento de los sujetos expuestos y tpne es el tiempo-persona de seguimiento de los sujetos no expuestos.7 (Tabla 2.3.2)
ESTUDIO DE CASOS Y CONTROLES
Un estudio de casos y controles es un estudio epidemiológico, observacional, analítico, con seguimiento retrospectivo, en el cual los sujetos se seleccionan en función de que tengan (casos) o no tengan (controles) una determinada enfermedad o, en general, un determinado evento. Una vez seleccionados los individuos de cada grupo, se investiga si estuvieron expuestos o no a una característica de interés y se compara la proporción de expuestos en el grupo de casos con la del grupo de control.
Tipos de estudio de casos y controles
Estudio de casos y controles retrospectivos: Todos los casos han sido diagnosticados antes del inicio del estudio.
Estudio de casos y controles prospectivos: Los casos son diagnosticados con posterioridad al inicio del estudio, y así pueden incluirse los nuevos casos detectados durante un cierto tiempo establecido previamente.
Estudio de casos y controles de base poblacional: Combina elementos del estudio de cohorte y de casos y controles. Se sigue a un grupo de individuos hasta que aparezca la enfermedad de interés, de igual forma que en el estudio de cohortes. Estos casos se comparan con un grupo control, cuyos componentes pertenecen a la misma población. Una vez obtenidos todos los casos y controles, se analiza el tipo de exposición previa o actual, como en un estudio de casos y controles. Este tipo de estudio es recomendable para enfermedades raras, puesto que un sujeto inicialmente identifi cado como control podría desarrollar el evento de interés durante el tiempo de seguimiento y ser seleccionado como caso, lo que pone en peligro el valor estadístico del estudio.
Estudio de casos y controles anidados: Es un estudio caso-control en el cual tanto los casos como los controles son tomados de la población que participa en un estudio de cohortes. Como los datos de esta población se obtienen a lo largo del tiempo, se reduce la posibilidad de sesgos de selección e información, que son comunes en los diseños caso-control. Es decir, no se genera sesgo porque el sujeto puede pasar de control a caso si desarrolla el evento de estudio.
Proceso de un estudio de casos y controles
El proceso empieza seleccionando una muestra de población con la enfermedad o problema de estudio. A estos individuos se les llama casos. A continuación se selecciona una muestra de la población de riesgo que no presenta enfermedad, que será el grupo control. Finalmente, se miden las variables predictoras, que son los factores de riesgo.
Ventajas de los estudios de casos y controles
Los diseños de estudio de casos y controles:
1. Estudian eventos raros o enfermedades y también enfermedades con un largo periodo de latencia; además, pueden ser un estudio preliminar para conocer la asociación entre un factor de riesgo y una determinada enfermedad. 2. Permiten estudiar muestras relativamente pequeñas. 3. Requieren tiempos cortos de ejecución en comparación con los estudios de cohortes prospectivos. 4. Son relativamente baratos comparados con los estudios de cohortes (aunque los de diseño de base poblacional suelen ser más caros). 5. Determinan la odds ratio. 6. Evalúan muchos factores de riesgo para una enfermedad o suceso.
Inconvenientes de los estudios de casos y controles
De nuevo, también deben considerarse sus potenciales limitaciones:
1. 1Son inefi cientes para el estudio de efectos de exposición raros, para los que es preferible considerar un estudio de cohorte. 2. No establecen la secuencia de eventos de interés, lo que difi culta la recogida de información fi able sobre el estado de la exposición del sujeto al evento de estudio en el tiempo. 3. Pueden incurrir en posibles errores de selección de casos y controles por la difi cultad para conseguir un grupo de comparación adecuado que cumpla los criterios de Hill.8-9 Este sesgo es menor en el diseño de base poblacional. 4. Pueden dar posibles sesgos de supervivencia. 5. Sólo pueden estudiar una variable de resultado (enfermedad). 6. No proporcionan estimadores de prevalencia, incidencia o riesgo atribuible. (En el diseño de base poblacional sí se puede estimar la incidencia.) 7. Son inapropiados cuando el resultado de interés no se conoce al comienzo del estudio o cuando el resultado es una variable continua.
Algunos ejemplos de estudios de casos y controles son: el estudio de las causas de las epidemias por aceite de colza y síndrome del aceite tóxico,10 el asma de soja en Barcelona,11 las neumonías y el síndrome Ardistyl.12
Análisis
La medida de asociación usada normalmente en los estudios de caso-control es la razón de probabilidades, habitualmente denominada odds ratio (OR) (Tabla 2.3.1). Un estudio de caso-control no permite calcular ni la incidencia ni el RR, pero sí permite calcular la odds ratio como medida de asociación, de la siguiente manera: 1º odds entre los expuestos = a/b; 2º odds ratio entre los no expuestos = c/d; y 3º odds ratio = (a/b)/ (c/d)= ad/bc. Esta medida, en el caso concreto de los estudios “anidados”, es siempre un estimador válido del riesgo relativo que se habría obtenido en un estudio de cohortes. Mas allá de la deducción matemática de esta propiedad de las OR, varios estudios han demostrado esto empíricamente. De lo cual se deduce que
los resultados en un estudio de casos y controles “anidados” son virtualmente los mismos que los obtenidos en el correspondiente estudio de cohortes.
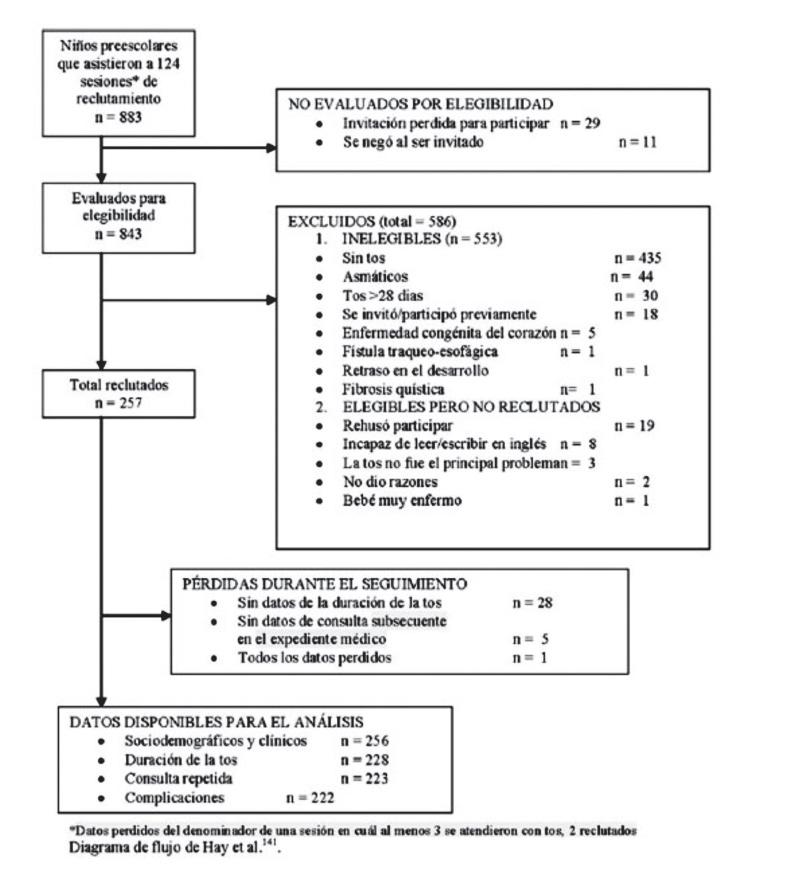
Finalmente, debe mencionarse la iniciativa STROBE13 (siglas de Strengthening the Reporting of Observational Studies in Epidemiology, en español Fortalecimiento de la Presentación de estudios observacionales en epidemiología), que tanto para los estudios de cohorte o de casos y controles, como para el resto de estudios observacionales pretende incrementar la calidad de su realización, informe e interpretación. STROBE ha desarrollado recomendaciones sobre lo que debería incluirse en un informe preciso y completo de un estudio observacional en las tres grandes modalidades de estudio: de cohortes, de casos y controles y transversales. Se desarrolló un listado de 22 elementos (la declaración STROBE) que se relacionan con el título, resumen, introducción, métodos, resultados y discusión de los artículos. Por ejemplo, la obtención de muestra de estudio fi nal debe poder reconstruirse con un diagrama de fl ujo de participantes (Figura 2.3.1), semejante al estándar CONSORT de un ensayo clínico. En particular, 18 puntos son comunes a los tres diseños de estudio y cuatro son específi cos para la cohorte, casos y controles, o estudios transversales.14 Se estima que la declaración STROBE contribuye a mejorar la calidad de los informes de los estudios observacionales. STROBE también está disponible en castellano.15
INTERPRETACIÓN
Los investigadores clínicos que tengan capacidad para integrar información de diferentes disciplinas y puedan realizar o interpretar los resultados de estudios epidemiológicos con diseño de cohortes o caso-control, entre otros, y que también puedan interactuar con otros investigadores básicos y clínicos, junto con matemáticos y bioinformáticos, tendrán ventajas añadidas al aplicar su investigación a todos los niveles. Sólo se necesita una receta de éxito para superar con creces estos nuevos desafíos: monitorizar las tendencias de las enfermedades y sus determinantes mediante la epidemiología llamada de “suela de zapato”,16-17 es decir, la epidemiología clásica, quizás complementada por Big Data y otras nuevas tecnologías,18 como la inteligencia artifi cial. La implementación de herramientas como las descritas recientemente será de gran ayuda.19-21
Esta nueva frontera en la investigación médica será probablemente más efi ciente y potencialmente más precisa. El uso de información de las historias clínicas, en papel o en versión digital, combinadas con el texto de los informes de radiología, bioquímica, función pulmonar u otras variables fi siológicas, no es ni sencillo ni directo.20-21 Superar estos desafíos conjugando algoritmos de inteligencia artifi cial y aprendizaje de máquinas (machine learning), con el reconocimiento del texto libre con la tecnología de procesamiento de lenguaje natural (NLP por sus siglas en inglés) dará una verdadera “nueva vida” a los datos recogidos en las historias clínicas, que el sistema de atención médica recopila por defecto, y las oportunidades que surgen de su reutilización pueden ser más que atractivas.22-24
Para concluir, la tabla 2.3.3 resume las ventajas e inconvenientes de los estudios de casos y controles frente a los estudios de cohortes.
PUNTOS CLAVE
• Los estudios de cohorte y los de casos y controles constituyen dos tipos de estudios observacionales, no descriptivos y analíticos. • Ambos diseños de estudio son similares a pesar de usar métodos y estrategias analíticas diferentes. • La diferencia fundamental entre estos dos tipos de estudios radica en el método de selección de los sujetos de estudio, que en los primeros se basa en el grado de exposición, y en los segundos, en el evento de interés. • Estos estudios representan una alternativa válida a los estudios experimentales. No obstante, debido a su mayor complejidad y a su mayor vulnerabilidad a los diferentes sesgos, se requiere un conocimiento avanzado en el campo de la metodología y el análisis epidemiológico para poder utilizarlos sin que lleven a resultados erróneos o de difícil interpretación. • Los estudios observacionales, de cohorte y de casos y controles son fundamentales para generar hipótesis de causalidad, que idealmente deben confi rmarse en ensayos clínicos controlados.
BIBLIOGRAFÍA
1. Kohansal R, Soriano JB, Agusti A. Investigating the natural history of lung function: facts, pitfalls, and opportunities. Chest. 2009;135:1330-1341. 2. Mannino DM, Watt G, Hole D, et al. Th e natural history of chronic obstructive pulmonary disease. Eur
Respir J. 2006;27:627-643. 3. Kannel WB, Feinleib M, McNamara PM, et al. An investigation of coronary heart disease in families: the
Framingham off spring study. Am J Epidemiol. 1979;110:281-290. 4. Zock JP, Rodríguez-Trigo G, Pozo-Rodríguez F, Barberà JA. Health eff ects of oil spills: lessons from the
Prestige. Am J Respir Crit Care Med. 2011;184:1094-1096. 5. López-Campos JL, Peces-Barba G, Soler-Cataluña JJ, Soriano JB, De Lucas Ramos P, De-Torres JP, Marín JM, Casanova C (en nombre del grupo de estudio CHAIN). Chronic obstructive pulmonary disease history assessment in Spain: a multidimensional chronic obstructive pulmonary disease evaluation.
Study methods and organization. Arch Bronconeumol. 2012;48:453-459. 6. De Stavola BL, Nitsch D, Silva ID, et al. Statistical issues in life course epidemiology. Am J Epidemiol. 2006;163:84-96. 7. LAZCANO-PONCE E, FERNANDEZ E, SALAZAR-MARTINEZ E, HERNANDEZ-AVILA M. Estudios de cohorte. Metodología, sesgos y aplicación. Salud pública Méx [online]. 2000, vol. 42, n.3, pp. 230-241. ISSN 0036-3634. http://dx.doi.org/10.1590/S0036-36342000000300010. 8. Hill AB. “Th e Environment and Disease: Association or Causation?”, en Proceedings of the Royal Society of Medicine, 1965;58:295-300. 9. Delgado M, Llorca J, Doménech JM. Investigación científi ca: Fundamentos metodológicos y estadísticos. Barcelona, Signo; 5ª ed., 2012. 10. Posada de la Paz M, Philen RM, Borda AI. Toxic oil syndrome: the perspective aft er 20 years. Epidemiol
Rev. 2001;23:231-247. 11. Sunyer J, Antó JM, Rodrigo MJ, Morell F. Case-control study of serum immunoglobulin-E antibodies reactive with soybean in epidemic asthma. Lancet. 1989;1:179-182. 12. Solé A, Cordero PJ, Morales P, Martínez ME, Vera F, Moya C. Epidemic outbreak of interstitial lung disease in aerographics textile workers -the “Ardystil syndrome”: a fi rst year follow up. Th orax. 1996;51:9495. 13. Vandenbroucke JP, Von Elm E, Altman DG, Gøtzsche PC, Mulrow CD, Pocock SJ, Poole C, Schlesselman JJ, Egger M; STROBE initiative Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): explanation and elaboration. Ann Intern Med. 2007;147:163-194. 14. http://www.strobe-statement.org/index.php?id=available-checklists [acceso el 21 de dic. de 15]. 15. Von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP; Iniciativa STROBE.
Declaración de la Iniciativa STROBE (Strengthening the Reporting of Observational studies in Epidemiology): directrices para la comunicación de estudios observacionales. Gac Sanit. 2008;22:144-150. 16. Koo D, Th acker SB. In Snow’s Footsteps: Commentary on Shoe-Leather and Applied Epidemiology. Am
J Epidemiol. 2010;172:737-739. 17. Tong SY. Genomic polish for shoe-leather epidemiology. Nat Rev Microbiol. 2013;11(1):8. 18. Gené Badia J, Gallo de Puelles P, De Lecuona I. Big data y seguridad de la información. Aten Primaria. 2018;50(1):3-5. 19. Johnston IG, Hoff mann T, Greenbury SF, Cominetti O, Jallow M, Kwiatkowski D, Barahona M, Jones
NS, Casals-Pascual C. Precision identifi cation of high-risk phenotypes and progression pathways in severe malaria without requiring longitudinal data. NPJ Digit Med. 2019;10:2:63. 20. Divita G, Carter M, Redd A, Zeng Q, Gupta K, Trautner B, Samore M, Gundlapalli A. Scaling-up NLP
Pipelines to Process Large Corpora of Clinical Notes. Methods Inf Med. 2015;54(6):548-552.
21. MacRae J, Darlow B, McBain L, Jones O, Stubbe M, Turner N, Dowell A. Accessing primary care Big
Data: the development of a soft ware algorithm to explore the rich content of consultation records. BMJ
Open. 2015;5(8):e008160. 22. Hernández-Medrano I, Carrasco G. El profesional de la salud ante el mundo del Big Data. Rev Calid
Asist. 2016;31(5):250-253.
TABLAS Y FIGURAS
Tabla 2.3.1. Tabla de 2x2 y su análisis
Expuestos
No expuestos Enfermos No enfermos
a b a+b
c d Prueb
a+b b+d N
Análisis para un estudio de cohorte:
Permite calcular la incidencia en cada grupo y el riesgo relativo en los expuestos respecto a los no expuestos Riesgo relativo = (a/a+b)/(c/c+d) Incidencia en el grupo de expuestos: a/(a+b) Incidencia en el grupo de no expuestos: c/(c+d) Diferencia de incidencia acumulada: a/(a+b) – c/(c+d) (Si la exposición es protectora, se calcula al revés: c/(c+d) - a/(a+b) )
Análisis para un estudio caso-control:
No permite calcular la incidencia ni el RR, pero sí permite calcular la odds ratio como medida de asociación. Odds entre los expuestos = a/b Odds entre los no expuestos = c/d Odds ratio = (a/b)/(c/d) = ad/bc
Tabla 2.3.2. Principales diseños de estudios en Epidemiología
Usos
Unidad de análisis
Temporalidad
Ventajas
Inconvenientes
Ejemplo Ensayos clínicos Estudios transversales Estudios de casos y controles Estudios de cohorte
Comparar tratamientos (preventivos o terapéuticos) Investigar la prevalencia de enfermedades Estudiar la relación de las enfermedades con exposiciones “fi jas” de los individuos (sexo, raza…) Generar hipótesis Estudiar los factores de riesgo de una enfermedad Evaluar las intervenciones terapéuticas Identifi car las causas de epidemias Estudiar factores de riesgo de las enfermedades Establecer tasas de incidencia Estudiar la historia natural de las enfermedades Establecer pronóstico
Individuo Individuo/grupo Individuo Individuo
Prospectivos Retrospectivos Retrospectivos Prospectivos o retrospectivos
“Libres” de sesgos mayormente Económicos, rápidos y sencillos Permiten estudiar diferentes exposiciones y enfermedades al mismo tiempo Habitualmente representan a la población general Económicos, rápidos y sencillos Permiten estudiar diversas exposiciones de una enfermedad Útiles para enfermedades raras Conceptualmente sencillos Permiten estudiar todos los efectos asociados a una exposición
No permiten probar hipótesis etiológicas No permiten hacer inferencias causales (¿un factor es causa o efecto?) Sólo identifi can casos prevalentes (no incidentes) de enfermedad Sólo permiten estudiar un efecto asociado a una o más exposiciones Susceptibles de error de medida (evaluación retrospectiva de los factores de exposición) y de sesgo de selección Costosos y lentos (requieren largos períodos) Poco útiles para enfermedades raras o con largos períodos de latencia
Medical Research Council Working Party. Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Lancet 1981;1):681-6.
Objetivo: Estudiar la efectividad de la oxigenoterapia crónica do miciliaria en pacientes respiratorios crónicos. Diseño y sujetos: Ensayo clínico. Pacientes (n=87) con bronquitis crónica o enfi sema con obstrucción irreversible del fl ujo aéreo, insufi ciencia respiratoria e insufi ciencia cardíaca congestiva, aleatorizados a grupo tratamiento (oxígeno domicilario mínimo 15h/d) o control (no oxígeno). Resultados: Tras 5 años de seguimiento, murieron el 45% de los sujetos en el grupo tratamiento y el 67% en el grupo control. No se observaron diferencias en los días de estancia hospitalaria o de ausencia laboral.
Sobradillo V, Miravitlles M, Jimenez CA, Gabriel R, Viejo JL, Masa JF, Fernandez-Fau L, Villasante C. [Epidemiological study of chronic obstructive pulmonary disease in Spain (IBERPOC): prevalence of chronic respiratory symptoms and airfl ow limitation] Arch Bronconeumol 1999;35:159-66.
Objetivo: Estudiar la prevalencia de síntomas respiratorios crónicos y limitación crónica al fl ujo aéreo en España. Diseño y sujetos: Estudio transversal en 7 áreas españolas. Selección de una muestra aleatoria poblacional de más de 4000 sujetos entre 40 y 69 años a partir del censo. Resultados: Se encontraron las siguientes prevalencias: síntomas respiratorios crónicos 48% (IC 95%:46.4-49.5), bronquitis crónica 4.8% (4.15.4), asma 4.9% (4.2-5.5), limitación crónica al fl ujo aéreo 10.6 (9.6-11.5). Excepto el asma, todos los trastornos fueron más frecuentes en hombres que en mujeres y en fumadores o exfumadores que en no-fumadores. Se encontraron diferencias geográfi cas importantes entre áreas.
Teran-Santos J, Jimenez-Gomez A, Cordero-Guevara J. Th e association between sleep apnea and the risk of traffi c accidents. Cooperative Group Burgos-Santander. N Engl J Med 1999;340:847-51.
Objetivo: Estudiar la relación entre apnea del sueño y accidentes de tráfi co. Diseño y sujetos: Casos y controles. Casos: 102 conductores visitados en urgencias tras accidentes de tráfi co. Controles: 152 sujetos seleccionados al azar a partir de atención primaria de la misma zona geográfi ca, apareados por sexo y edad. Resultados: Los sujetos con índice de apnea-hipopnea igual o mayor a 10 mostraron una razón de odds de 6.3 (IC 95%: 2.4-16.2) de tener un accidente de tráfi co, independiente de confusores como ingesta de alcohol, consumo de fármacos u otros.
Doll R, Peto R. Mortality in relation to smoking: 20 years’ observations on male British doctors. Br Med J 1976;2:1525-36.
Objetivo: Estudiar la relación entre el hábito tabáquico y la mortalidad. Diseño y sujetos: Cohorte de más de 34000 hombres que respondieron en 1951 un cuestionario sobre tabaco y fueron seguidos durante 20 años. Resultados: La razón de riesgo de morir en los fumadores respecto a los no fumadores de la misma edad fue de 2. Las causas de muerte asociadas al tabaco fuer on enfermedad cardíaca, cáncer de pulmón, enfermedad pulmonar obstructiva crónica y enfermedades vasculares.
Tabla 2.3.3. Pros y contras de los estudios de casos y controles frente a los de cohortes
Características Casos y controles Cohortes
Conocer historia natural enfermedad Mal Bien Planifi cación Mejor en prospectivos Bien Incidencia No (sí en híbridos) Sí Cálculo de riesgos Parcial Todos Valoración de exposición Tras efecto Mejor, sobre todo si hay cambios en el tiempo Multiefectividad No Sí Sesgos Muchos Pocos Infl uencia de la mortalidad Difícil de estudiar Más fácil Control de calidad de la información Más difícil Más fácil
Exposiciones raras No Sí Enfermedades raras Sí No Coste Variable (menor que las cohortes prospectivas) Variable (en general más caros)
Tiempo Menor que las cohortes prospectivas
Mayor en las cohortes prospectivas Reproducibilidad Alta Escasa (prospectivos) Intervención No Sí, aunque discreta Cantidad de personal Depende Depende Pérdidas No Sí Riesgo de obsoleto Bajo Mayor Como exploratorios Sí No Asociaciones débiles entre exposición y respuesta No Sí Comprensibilidad Baja Alta
Figura 2.3.1. Ejemplo de diagrama de fl ujo STROBE (adaptado de referencia 12)
Dra. Carmen Fuentelsaz Gallego
INTRODUCCIÓN
Los estudios experimentales, también llamados estudios de intervención, son estudios de tipo analítico que permiten conocer la relación entre la variable independiente (la intervención que se realiza) y la o las dependientes (las principales variables de resultado, que son las que se verán afectadas por la intervención). La principal diferencia entre ambos tipos es que en los estudios experimentales se realiza una intervención, mientras que en los estudios observacionales sólo se observan determinadas características de los sujetos de estudio (Figura 2.4.1).
En los estudios experimentales hay, al menos, una intervención (o manipulación), puede haber un grupo control y uno experimental, y una asignación aleatoria a ambos grupos.
Son realmente experimentales cuando hay una intervención, grupo control y experimental y asignación aleatoria a ambos grupos. Se consideran cuasi experimentales cuando no hay grupo control y experimental o, en caso de que los haya, cuando la asignación a ambos grupos no se realiza de forma aleatoria.
ESTUDIOS EXPERIMENTALES
Características
Los estudios experimentales (también llamados ensayos clínicos) son de tipo prospectivo, es decir, se hace un seguimiento de los sujetos de estudio para ver el efecto de la intervención.1-3 Por tanto, el objetivo de estos estudios es conocer si una intervención es más efi caz o efectiva que otra. Esto es, cuando se plantea una hipótesis de superioridad, pero también se pueden plantear hipótesis de no inferioridad o de equivalencia.4
El esquema de un estudio experimental se presenta en la fi gura 2.4.2. Como cualquier otro tipo de estudio, se inicia defi niendo las características de la población de referencia mediante los criterios de selección, es decir, los criterios de inclusión y los de exclusión, que deben estar claramente determinados. En estos estudios, los criterios de selección suelen ser muy estrictos, sobre todo en los ensayos clínicos con medicamentos, lo que puede perjudicar su validez externa.
Si la población de referencia es demasiado grande, por lo que no es pertinente estudiarla toda, se seleccionará una muestra de sujetos, que serán los que realmente se estudiarán. Para ello, se debe estimar el tamaño de muestra necesario para poder valorar el efecto de la intervención en la variable principal de resultado y se debe determinar la técnica de muestreo a utilizar.
Cabe señalar la diferencia entre la técnica de muestreo aleatoria y la asignación aleatoria a los grupos control y experimental, ya que a veces son términos que se confunden.
La técnica de muestreo es la que se utiliza para seleccionar la muestra de la población de referencia. En los estudios experimentales no se suele utilizar una técnica de muestreo aleatoria (aunque sería lo ideal), y generalmente se utiliza un muestreo consecutivo, incluyendo a los sujetos a medida que van ingresando en el hospital o acuden a la consulta.
La asignación es la forma en que se decide si los sujetos de estudio se incluyen en el grupo control o en el grupo experimental. Esta asignación debe ser aleatoria, es decir, al azar, de forma que cualquier sujeto tenga la misma probabilidad de formar parte del grupo control que del grupo experimental. Lo más habitual es utilizar programas estadísticos para hacer la asignación, mediante la generación de números aleatorios.
Con la asignación aleatoria se consigue que los grupos sean comparables, lo que signifi ca que sus características al inicio del estudio (llamadas características basales) son muy similares (aunque esta similitud no se puede garantizar al 100%) y que las variables que pueden infl uir en la relación entre la intervención realizada y las variables de resultado se distribuyen de igual forma en ambos grupos. Por lo tanto, si se encuentran diferencias entre el grupo control y el experimental al fi nal del estudio, se podría afi rmar que son debidas al efecto de la intervención, no a que los grupos fuesen diferentes cuando se inició el estudio. Las características basales suelen ser variables sociodemográfi cas, clínicas u otras de interés.
Esta asignación a los grupos experimental y control debe ser oculta, lo que signifi ca que las personas implicadas en el estudio no deben conocer ni tener acceso a la secuencia de inclusión de los participantes a cada grupo.
Cuando se va asignando cada participante al grupo control o al experimental, se le realizará la intervención que le corresponda. En general, al grupo experimental se le realiza el tratamiento nuevo a experimentar, y al grupo control, el tratamiento habitual o un placebo (aunque éste último no siempre es posible utilizarlo). En el caso de que se realice este tratamiento habitual o de que se impartan los cuidados enfermeros habituales, habrá que defi nirlos muy bien, puesto que pueden ser diferentes según el centro donde se desarrolle el estudio. Ambos grupos deben ser tratados de la forma más similar posible a lo largo del estudio, a excepción de la intervención asignada.
Lo habitual es que los sujetos del grupo control y del grupo experimental reciban la intervención al mismo tiempo, por lo que el estudio se denomina “ensayo clínico paralelo”. En caso contrario, será no paralelo.
Se deben indicar y defi nir claramente las variables principales del estudio: la independiente, que será la intervención a realizar, y la variable principal de resultado. Deben indicarse y describirse las intervenciones que se harán al grupo control y al grupo experimental. También hay que describir claramente cuál o cuáles son las variables principales de resultado, que preferiblemente deben poder medirse de forma objetiva, para no incluir sesgos en el estudio.
Para que el estudio tenga mayor calidad y rigor metodológico, se pueden utilizar las técnicas de cegamiento o ciego:
1. Ensayo clínico abierto o no ciego. Tanto el investigador como el sujeto de estudio conocen el grupo al que ha sido asignado dicho sujeto. 2. Simple ciego. Los participantes desconocen si han sido asignados al grupo control o al experimental. 3. Doble ciego. Tanto el investigador como el sujeto de estudio desconocen el grupo al que ha sido asignado dicho sujeto. 4. Triple ciego. Además de los participantes y el investigador, tampoco la persona que analiza los resultados del ensayo conoce el grupo al que ha sido asignado el sujeto. 5. Evaluación ciega por terceros. Cuando no se puede utilizar alguna de las técnicas de ciego anteriores, la opción es realizar una evaluación ciega por terceros, en la que una persona, generalmente ajena al equipo investigador y que desconoce el grupo al que ha sido asignado cada sujeto, evalúa la variable principal de resultado.
El equipo investigador determinará el tiempo de seguimiento que se realizará a los participantes en el estudio. Al fi nal de este periodo se medirá la variable principal de resultado, para ver si ha habido cambios tras la intervención en cada uno de los dos grupos. En esta fase de seguimiento es muy importante controlar las pérdidas de participantes en cada grupo, las causas de estas pérdidas y las características de dichos sujetos, así como los efectos adversos que se puedan producir, para saber si pueden estar relacionados con la intervención realizada a cada sujeto. También se deberán tener en cuenta los incumplidores del tratamiento asignado, tanto en el grupo control como en el experimental, para evaluar si el incumplimiento tuviese relación con dicho tratamiento.
Además del diseño clásico del ensayo clínico que se ha mostrado en la fi gura 2.4.3, existen otros tipos, como el “ensayo clínico cruzado”, en el que cada sujeto es su propio control, por lo que todos los participantes reciben los dos tratamientos o intervenciones. A la mitad de los sujetos de la muestra se les asigna de forma aleatoria al tratamiento A o al B, y después de un periodo (llamado de blanqueo o de lavado, para que los efectos del tratamiento recibido desaparezcan), se les cambia el tratamiento, a los que recibieron el A se les asigna el B, y viceversa.
Cuando en un estudio experimental la intervención no se realiza sobre cada uno de los individuos sino que se realiza sobre un grupo o sobre una comunidad de la que forman parte dichos individuos, estos estudios se denominan “ensayos comunitarios”. Generalmente se evalúan intervenciones relacionadas con hábitos de salud. Los resultados obtenidos en el estudio son del grupo, no se pueden extraer de cada individuo en particular.
Un grupo de expertos ha elaborado unas recomendaciones para la publicación de estudios experimentales en revistas científi cas, pero que pueden ser útiles para investigadores noveles que deseen desarrollar un estudio de este tipo. Estas directrices se recogen en la lista de comprobación CONSORT (Consolidated Standards of Reporting Trials).5 En la página web del grupo (http://www.consort-statement.org/) se pueden consultar diferentes instrumentos según el tipo de estudio (ensayos grupales, no inferioridad y equivalencia y ensayos pragmáticos),6-8 según el tipo de intervención (hierbas medicinales, tratamientos no farmacológicos y acupuntura)9-11 u otros instrumentos (efectos adversos y elaboración de resúmenes).12-13
Ventajas y limitaciones
A continuación se señalan algunas de las principales ventajas de los estudios experimentales.
• Una de las mayores ventajas de los estudios experimentales es el control que los investigadores tienen sobre el diseño del estudio. Se puede controlar tanto la intervención que se realiza como todo el desarrollo del estudio y, por tanto, las condiciones en que se realiza el ensayo.
• Cuando se trata de evaluar efectividad o efi cacia, son los que aportan un mayor nivel de evidencia científi ca y, en consecuencia, una mayor calidad de la evidencia. • El riesgo de introducir sesgos en los ensayos clínicos es menor que en otro tipo de estudios epidemiológicos, debido principalmente a la asignación aleatoria a los grupos control y experimental, que permite que las variables confusoras se distribuyan de igual forma en ambos grupos. • Son estudios fácilmente reproducibles. Y si las intervenciones evaluadas y las variables principales de resultado son iguales que las de otros estudios, los resultados obtenidos serán fácilmente comparables. • Se pueden utilizar técnicas de ciego, lo que reduce el riesgo de introducir en el ensayo sesgos de información.
Estos estudios también presentan una serie de limitaciones, que se detallan a continuación.
• Una de las principales limitaciones es la difi cultad para generalizar los resultados. Con el control estricto del diseño del ensayo se garantiza la validez interna, pero a veces la validez externa se ve perjudicada debido a los estrictos criterios de selección de los participantes y también a la propia intervención, que puede alejarse de la práctica habitual e imposibilitar que se trasladen los resultados a la práctica. • Muchas veces son estudios de elevado coste, dependiendo de las intervenciones que se realicen y del tiempo de seguimiento de los participantes. • También pueden presentar limitaciones de tipo ético, debido a la asignación aleatoria de las intervenciones a evaluar, sobre todo cuando la intervención del grupo control es menos efectiva que la del grupo experimental. • En cada estudio sólo se podrá evaluar una intervención experimental, ya que, si se realiza más de una, no se sabrá a qué intervención se debe el efecto producido. • Con los estudios experimentales sólo se pueden responder las preguntas de investigación relacionadas con efectividad o efi cacia, por lo que muchas otras preguntas no se pueden responder con este tipo de estudios.
ESTUDIOS CUASI EXPERIMENTALES
Características
Los diseños cuasi experimentales son también muy utilizados en investigación clínica, ya que a veces no es posible tener un grupo control y otro experimental o asignar a los participantes de forma aleatoria a dichos grupos.14 Seguidamente, se exponen algunos diseños de este tipo.
a. Cuasi experimentales pretest-postest con grupo control. La intervención sólo se realiza en el grupo experimental, mientras que al grupo control se le realizan las mismas mediciones de la variable de resultado pero no se le hace ninguna intervención. Las mediciones se efectúan antes y después de la intervención.
A veces, en este tipo de estudios también se realiza una intervención en el grupo control.
b. Cuasi experimentales postest con grupo control. En este tipo de estudios, aunque hay grupo experimental y control, sólo se hace una medición de la variable de resultado tras la realización de la intervención, no se realiza la medición previa a la intervención.
c. Cuasi experimentales pretest-postest de un solo grupo. Después de incluir a los sujetos en el estudio, se les realiza una primera medición, a continuación se les hace la intervención y fi nalmente se les hace otra medición para ver el efecto de la intervención.
Ventajas y limitaciones
Loa estudios cuasi experimentales son más fáciles de desarrollar que los experimentales y, por tanto, más económicos en cuanto a recursos materiales, personales y de tiempo. Además, a veces son la única alternativa para responder a la pregunta de investigación. En ocasiones, la validez externa es más factible que en los experimentales.
La principal limitación de estos estudios es que no hay asignación aleatoria a los grupos control y experimental, por lo que puede darse el caso de que los grupos no sean homogéneos y, por tanto, no se puedan comparar. Además, la inclusión de sesgos es más fácil en estos estudios que en los experimentales.
Como con cualquier otro tipo de diseño, en los estudios experimentales y cuasi experimentales se deben evitar o minimizar los sesgos para no perjudicar la validez interna del estudio. Los principales sesgos de estos diseños están relacionados con la selección de los pacientes, la asignación aleatoria a los grupos control y experimental, la realización de la intervención y el seguimiento de los participantes.
Se producirá un sesgo de selección cuando haya diferencias en los grupos control y experimental al inicio del estudio. Este sesgo se evitará o minimizará defi niendo claramente los criterios de selección de los participantes (criterios de inclusión y exclusión), con la estimación del tamaño muestral y con la asignación aleatoria oculta a ambos grupos. Cabe señalar, que la primera fase del análisis estadístico en estos estudios será comprobar que los dos grupos, experimental y control, sean similares en las variables defi nidas como basales y, por tanto, sean comparables, ya que la asignación aleatoria no garantiza totalmente que los grupos sean homogéneos.
Cuando el estudio ya se ha iniciado, se puede producir un sesgo de realización cuando se hace la intervención en los grupos control y experimental, si se dan diferencias en la atención habitual proporcionada a ambos grupos, a parte de la intervención que se evalúe. Este sesgo se puede evitar utilizando las técnicas de ciego indicadas anteriormente.15
Otro sesgo que se puede introducir en estos estudios es el de seguimiento (llamado también de desgaste o pérdida), cuando los participantes que fi nalizan el estudio tienen características diferentes a los que lo han abandonado. Para evitar este sesgo, debemos hacer un buen control del seguimiento de los participantes y registrar con todo detalle la información sobre las pérdidas o abandonos, sus causas y las características de todos los sujetos perdidos. Otra forma de evitarlo es con un análisis estadístico de los datos al fi nalizar el estudio, realizando el llamado análisis por intención de tratar. Se trata de hacer el análisis con los sujetos dentro del grupo al que han sido asignados inicialmente, independientemente del grupo en el que fi nalicen el estudio y de la intervención recibida.16
En los estudios cuasi experimentales, como no hay asignación aleatoria o no hay grupo control, es mucho más fácil y frecuente que se produzcan sesgos.
ENSAYO CLÍNICO CON MEDICAMENTOS
Como se ha comentado anteriormente, un estudio experimental se denomina también ensayo clínico, aunque algunos ensayos tienen unas implicaciones legales y administrativas que otros estudios experimentales no tienen. Los estudios que se realizan con medicamentos o con algunos productos sanitarios deben cumplir una serie de requisitos marcados por la legislación española17 y por la europea.18
En el Real Decreto 1090/201517 se indican los diferentes conceptos relacionados con este tipo de estudios: ensayo clínico, medicamento de investigación, comités de ética de la investigación con medicamentos, etc.
Ensayo clínico. “Un estudio clínico que cumpla cualquiera de las siguientes condiciones: 1) Se asigna de antemano al sujeto de ensayo a una estrategia terapéutica determinada, que no forma parte de la práctica clínica habitual del Estado miembro implicado; 2) La decisión de prescribir los medicamentos en investigación se toma junto con la de incluir al sujeto en el estudio clínico; 3) Se aplican procedimientos de diagnóstico o seguimiento a los sujetos de ensayo que van más allá de la práctica clínica habitual.”
Medicamento de uso humano: “Toda sustancia o combinación de sustancias que se presente como poseedora de propiedades para tratamiento o prevención de enfermedades en seres humanos o que pueda usarse en seres humanos o administrarse a seres humanos con el fi n de restaurar, corregir o modifi car las funciones fi siológicas ejerciendo una acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico médico”.
Reacción adversa. “Cualquier respuesta nociva y no intencionada a un medicamento”.
Producto sanitario. “Cualquier instrumento, dispositivo, equipo, programa informático, material u otro artículo, utilizado solo o en combinación, incluidos los programas informáticos destinados por su fabricante a fi nalidades específi cas de diagnóstico y/o terapia y que intervengan en su buen funcionamiento, destinado por el fabricante a ser utilizado en seres humanos.”
Todos los estudios clínicos deben ser aprobados por un Comité de Ética de la Investigación con Medicamentos (CEIM). El CEIM revisará que el estudio cumpla todos los requisitos legales y administrativos, así como los aspectos metodológicos, que en ocasiones tienen repercusión en la seguridad de los participantes. Entre los aspectos importantes a revisar, cabe destacar: evaluar el balance riesgo/benefi cio para los participantes; evaluar las modifi caciones de la práctica habitual y las intervenciones invasivas; tener en consideración las poblaciones vulnerables (niños, ancianos, mujeres embarazadas, etc.); y respetar en todo momento la ley de protección de datos de carácter personal.
Antes del inicio del estudio, todos los participantes deben tener información del estudio y deben fi rmar un consentimiento para su participación. En el capítulo II del Real Decreto 1090/2015,17 se indican todos los aspectos relacionados con la protección de los sujetos del ensayo y del consentimiento informado.
PUNTOS CLAVE
• En los estudios observacionales los investigadores no realizan ninguna intervención, en cambio en los experimentales sí. Esta es la principal diferencia entre ambos diseños. • Las tres características fundamentales de los estudios experimentales son: manipulación (intervención) por parte del investigador, grupo control y grupo experimental y asignación aleatoria a ambos grupos. • Los aspectos más importantes en el diseño de los estudios experimentales son: los criterios de selección, la asignación aleatoria a los grupos control y experimental y el control en el seguimiento de pérdidas, abandonos y efectos adversos. • Para evaluar efectividad o efi cacia, los estudios experimentales aportan el nivel de evidencia más elevado. • En los estudios cuasi experimentales se pueden producir más sesgos que en los experimentales. • Los estudios con medicamentos o productos sanitarios deben seguir las pautas indicadas en la legislación española y europea.
BIBLIOGRAFÍA
1. Bermejo B. Estudios experimentales. Matronas Prof. 2008;9(1):15-20. 2. Manterola C, Otzen T. Estudios experimentales. 1ª Parte. El ensayo clínico. Int J Morphol. 2015;33(1):342349. 3. Laporte JR. Principios básicos de investigación clínica.. Barcelona: AstraZeneca, 2ª ed., 2001. Disponible en: http://www.icf.uab.cat/llibre/llibre.htm. 4. Delgado O, Puigventós F, Pinteño M, Ventayol P. Equivalencia terapéutica: concepto y niveles de evidencia. Med Clin (Barc). 2007;129(19):736-745. 5. Cobos A. Ensayos clínicos aleatorizados (CONSORT). Med Clin (Barc). 2005;125(Supl. 1):21-27. 6. Campbell MK, Piaggio G, Elbourne DR, Altman DG; CONSORT Group. Consort 2010 statement: extension to cluster randomised trials. BMJ. 2012;345:e5661. Doi: 10.1136/bmj.e5661. 7. Piaggio G, Elbourne DR, Altman DG, Pocock SJ, Evans SJ; CONSORT Group. Reporting of noninferiority and equivalence randomized trials: an extension of the CONSORT statement. JAMA. 2006;295(10):1152-1160. 8. Zwarenstein M, Treweek S, Gagnier JJ, Altman DG, Tunis S, Haynes B, et al; CONSORT group; Pragmatic Trials in Healthcare (Practihc) group. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ. 2008; 337:a2390. doi: 10.1136/bmj.a2390. 9. Boutron I, Moher D, Altman DG, Schulz KF, Ravaud P; CONSORT Group. Methods and processes of the CONSORT Group: example of an extension for trials assessing nonpharmacologic treatments. Ann
Intern Med. 2008;148(4):60-66. 10. Gagnier JJ, Boon H, Rochon P, Moher D, Barnes J, Bombardier C; CONSORT Group. Reporting randomized, controlled trials of herbal interventions: an elaborated CONSORT statement. Ann Intern Med. 2006;144(5):364-367. 11. MacPherson H, Altman DG, Hammerschlag R, Youping L, Taixiang W, White A, Moher D; STRIC-
TA Revision Group. Revised Standards for Reporting Interventions in Clinical Trials of Acupuncture (STRICTA): extending the CONSORT statement. PLoS Med. 2010;7(6):e1000261. 12. Hopewell S, Clarke M, Moher D, Wager E, Middleton P, Altman DG, Schulz KF and the CONSORT
Group (2008) CONSORT for reporting randomised trials in journal and conference abstracts. Lancet. 371:281-283. 13. Ioannidis JP, Evans SJ, Gøtzsche PC, O’Neill RT, Altman DG, Schulz K, et al; CONSORT Group. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med. 2004;141(10):781-788. 14. Manterola C, Otzen T. Estudios experimentales 2ª Parte. Estudios Cuasi-Experimentales. Int J Morphol. 2015;33(1):382-387. 15. Rodríguez JL, Casado A. Doble ciego. El control de los sesgos en la realización de ensayos clínicos. Contradicciones, insufi ciencias e implicaciones. Med Clin (Barc). 2002;118(5):192-195. 16. Abraira V. ¿Qué es el análisis por intención de tratar?. SEMERGEN. 2000;26:393-394. 17. Boletín Ofi cial del Estado. Nº 307, jueves 24 de diciembre de 2015. Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos. 18. Diario Ofi cial de la Unión Europea del 27/05/2014. Reglamento (UE) Nº 536/2014 del Parlamento Europeo y del Consejo, sobre los ensayos clínicos de medicamentos de uso humano.
TABLAS Y FIGURAS
Figura 2.4.1. Estudios experimentales y cuasi experimentales

Figura 2.4.2. Esquema de los estudios experimentales

E = enfermedad o característica en estudio
Figura 2.4.3. Esquema de los ensayos cruzados

2.5. TIPOS DE ERROR. VALIDEZ, SESGOS Y CONFUSIÓN
Dra. Judith García Aymerich
EL ERROR EN INVESTIGACIÓN
Cualquier trabajo de investigación puede ser considerado un ejercicio de medida y, como tal, es propenso a tener errores. Tales errores pueden resultar en infra o sobreestimaciones de la prevalencia de exposiciones o enfermedades así como provocar asociaciones falsas u ocultar asociaciones verdaderas entre exposiciones y enfermedades. A menudo, estas situaciones llevan a contradicciones entre estudios y suscitan críticas externas a la investigación como disciplina. Por ello, es esencial que los investigadores tengan formación sobre los diferentes tipos de errores y sus causas.
Existen múltiples fuentes de error, que además pueden coexistir. En general, el error se clasifi ca en aleatorio y sistemático. Este capítulo revisa, en primer lugar, las defi niciones, causas, efectos y métodos para evaluar los errores aleatorios y sistemáticos. A continuación se presenta y discute el concepto de validez externa, tradicionalmente explicado junto con el error sistemático. Posteriormente se presentan los métodos para evitar o reducir los errores aleatorio y sistemático. Finalmente, se revisan algunos equívocos frecuentes en torno a los errores, para concluir con unas consideraciones fi nales. El capítulo se acompaña de ejemplos reales e hipotéticos que, por simplifi cación para fi nes pedagógicos, se limitan a preguntas de investigación sobre prevalencia (de exposición o enfermedad) o sobre asociación entre exposición y enfermedad.
ERROR ALEATORIO. PRECISIÓN
Defi nición, causas y efectos del error aleatorio
El error aleatorio se puede defi nir como “aquella parte de nuestra experiencia que no podemos predecir”,1 y puede atribuirse al fenómeno biológico que se está midiendo o al propio proceso de medición.2 La ausencia de error aleatorio se denomina precisión (Figura 2.5.1).
La principal fuente de error aleatorio en investigación es el uso de muestras para estudiar fenómenos de poblaciones. Ante la imposibilidad de estudiar toda una población, las mediciones se realizan en una muestra de sujetos, que puede representar erróneamente el conjunto de dicha población. Por este motivo, el error aleatorio se denomina a menudo error muestral. Otra fuente de error aleatorio relacionada con el error muestral es la inefi ciencia muestral. La efi ciencia muestral se defi ne como la capacidad de maximizar la
información obtenida y requiere disponer de sufi cientes sujetos en todas las categorías de interés. Si no se contempla la efi ciencia muestral, puede suceder que, incluso con un tamaño muestral elevado, el tamaño de ciertos subgrupos de interés (por ejemplo, “sujetos de 30-45 años no fumadores con hipertensión”) sea reducido y se convierta en fuente de error aleatorio. Finalmente, la variabilidad en un mismo individuo, los instrumentos de medida, los entrevistadores o incluso el proceso de entrada de datos pueden también ser origen de error aleatorio.
El error aleatorio tiene tantas probabilidades de provocar observaciones individuales con valores superiores a los reales como inferiores a éstos. Como consecuencia, la media de las observaciones individuales tiende a acercarse al valor real.
Evaluación del error aleatorio
Para tener en cuenta la presencia de error aleatorio, los resultados de las investigaciones no se presentan a través de un único valor (estimación puntual) sino que se acompañan de medidas de precisión (intervalo de confi anza, desviación estándar u otras). Estas medidas informan del intervalo de valores que es probable que incluyan el valor real.
Para evaluar el error aleatorio se utilizan las pruebas de contraste de hipótesis, que estiman la probabilidad de que el resultado de una investigación se haya obtenido por azar. Estas pruebas, detalladas en los capítulos 26-32, son las que proporcionan los familiares “valores de p”.
ERROR SISTEMÁTICO. VALIDEZ
El error sistemático o sesgo se defi ne como la diferencia sistemática entre un valor observado y el valor real.3 La ausencia de error sistemático se denomina validez. El término sesgo también signifi ca prejuicio y, aunque no se discute en este capítulo, es importante reconocer que los prejuicios (opiniones preconcebidas y preferencias) de los investigadores también pueden producir sesgo al interpretar los resultados de una investigación.4
En general, el error sistemático en las investigaciones se debe a factores implicados en el proceso de selección de sujetos y a factores que intervienen en la defi nición y medición de las variables del estudio. Así, aunque se han descrito múltiples tipos de sesgos, se distinguen tres grandes grupos: el sesgo de selección, el sesgo de información y la confusión (o sesgo de confusión).
Sesgo de selección. Defi nición, causas y efectos
El sesgo de selección se defi ne como la distorsión en los resultados de una investigación debida a errores sistemáticos en el proceso de identifi cación, selección o seguimiento de los sujetos a estudiar. Este sesgo se relaciona inversamente con la representatividad, defi nida como el grado de similitud entre las características de los sujetos participantes y las de todos los sujetos elegibles (participantes y no participantes). En general, la relación entre la exposición y la enfermedad de los sujetos participantes es distinta a la de los sujetos elegibles. Como resultado, la asociación observada en los participantes es una combinación entre la asociación real (la que se habría observado en todos los sujetos elegibles) y los efectos de los factores que determinan la participación. La tabla 2.5.1 describe diversos ejemplos de sesgo de selección.5
El sesgo de selección puede alterar los resultados en cualquier sentido.
Sesgo de información. Defi nición, causas y efectos
El sesgo de información se defi ne como la distorsión en los resultados de la investigación debida a errores sistemáticos en la medida de la exposición, la enfermedad u otras variables relevantes para un estudio. Si este error se produce en la medida de la exposición, da lugar a la denominada “mala clasifi cación de la exposición”, es decir, algunos sujetos clasifi cados como expuestos en realidad no lo están, y viceversa. Si el error se produce en la medida de la enfermedad, da lugar a la llamada “mala clasifi cación de la enfermedad”, es decir, algunos sujetos clasifi cados como enfermos en realidad no lo son, y viceversa. La tabla 2.5.2 muestra diversos ejemplos de sesgo de información.
Las consecuencias del sesgo de información dependen de si la mala clasifi cación se presenta de forma diferencial o de forma no diferencial entre los grupos de interés. La mala clasifi cación diferencial es aquella que depende de otras variables. Por ejemplo, en un estudio sobre el riesgo de malformaciones congénitas potencialmente atribuibles a cierto medicamento, las madres cuyos hijos sufren alguna malformación son más propensas a recordar todos los medicamentos que tomaron durante el embarazo, lo que resulta en una estimación errónea del efecto del medicamento (Tabla 2.5.3). Este tipo concreto de sesgo de información se denomina sesgo de memoria, y puede aparecer en cualquier estudio que utiliza la anamnesis para asignar exposiciones, ya que la experiencia de enfermedad afecta la memoria. En segundo lugar, la mala clasifi cación no diferencial es aquella que no depende de los valores de otras variables. La tabla 2.5.4 ilustra un hipotético estudio de casos y controles en el que el instrumento para medir la exposición, tanto en casos como en controles, tiene una sensibilidad del 80% y una especifi cidad del 100%. Como resultado, la asociación observada es diferente a la real.
El sesgo de información de tipo mala clasifi cación diferencial puede tanto exagerar como infraestimar la magnitud de una asociación. En el caso de mala clasifi cación no diferencial, la dirección del sesgo es, en general, hacia la hipótesis nula. Sin embargo, en situaciones de extrema mala clasifi cación, de medidas continuas de la exposición o la enfermedad, o cuando coinciden mala clasifi cación no diferencial de la exposición y de la enfermedad, es posible que el sesgo, aun siendo no diferencial, se aleje de la hipótesis nula (es decir, sobreestime la asociación).1
Confusión. Defi nición, causas y efectos
La confusión es la situación en la que la asociación observada entre una determinada exposición y una determinada enfermedad se debe a la infl uencia de una tercera variable que confunde o se combina con la asociación real entre exposición y enfermedad. Por ejemplo, en el estudio del efecto del consumo de alcohol en la enfermedad coronaria, la asociación entre estas variables puede estar confundida por el papel del consumo de tabaco; este es más frecuente en los consumidores de alcohol y causa de enfermedad coronaria (Figura 2.5.2). Habitualmente se denomina variable confusora (confounder o confounding variable en inglés) a la variable responsable de la confusión. Ésta se utiliza como variable de ajuste para eliminar el sesgo producido por la confusión.
En general, una variable confusora debe ser un factor de riesgo para la enfermedad, debe estar asociada (causalmente o no) con la exposición, y no puede ser un paso intermedio en la cadena causal entre la exposición y la enfermedad1 (Figura 2.5.2). Es importante resaltar que la evaluación de la confusión requiere combinar el conocimiento experto (clínico y/o fi siopatológico) con las estrategias estadísticas. Así, una evaluación (y eventual manejo) de la confusión basada solamente en criterios estadísticos o solamente en conocimiento experto puede conducir a errores.
La confusión puede llevar a sobreestimar o infraestimar el efecto real de una exposición en la enfermedad, e incluso puede cambiar el sentido de la asociación.
Evaluación del error sistemático (sesgo de selección, sesgo de información y confusión). Análisis de sensibilidad
Es muy importante resaltar que el sesgo es un fenómeno cuantitativo, no cualitativo. Por ello, es la cantidad de sesgo la que se debe evaluar (y eventualmente corregir), no su mera presencia o ausencia. La tabla 2.5.5 muestra cómo se puede evaluar cuantitativamente el error sistemático utilizando como ejemplo un caso real de confusión por indicación. En la confusión por indicación, los sujetos expuestos a un tratamiento teóricamente benefi cioso muestran peor pronóstico que los sujetos no expuestos, debido a que la indicación de tratamiento es motivada por la gravedad de la enfermedad, que también es la responsable de la evolución. En este ejemplo, en el que los pacientes con EPOC que son controlados por el neumólogo toman anticolinérgicos inhalados, participan en programas de rehabilitación pulmonar o reciben oxigenoterapia domiciliaria tienen mayor riesgo de ingresar por agudización de la EPOC que los correspondientes grupos de referencia, el ajuste por las variables disponibles de gravedad (parámetros de función pulmonar, intercambio de gases e ingresos previos) conseguía explicar hasta un 72% de la asociación.6 Sin embargo, el modelo ajustado todavía mostraba una asociación entre ser controlado por el neumólogo y tomar anticolinérgicos y peor evolución, lo que podría atribuirse a confusión residual (debida a variables de gravedad no medidas en el estudio), a efectos adversos de los tratamientos, o a una combinación de ambos.7
El análisis de sensibilidad es un análisis habitualmente planifi cado a priori por los investigadores para evaluar el efecto de los errores, es decir, para medir la cantidad de sesgo. El análisis de sensibilidad consiste en repetir de manera sistemática el análisis estadístico utilizando cada vez diferentes asunciones sobre la población de estudio, la medida de la exposición y la enfermedad, y las variables confusoras, para ver hasta qué punto los estimadores son sensibles a los cambios en las asunciones.1 Si los estimadores cambian poco en respuesta a los cambios en las asunciones, los resultados se califi can de razonablemente aceptables y se considera altamente improbable que el sesgo en estudio sea responsable de la asociación observada. Si los estimadores cambian mucho, es necesario aceptar que el sesgo podría ser responsable de los resultados y, por tanto, que los resultados obtenidos no se corresponden con los valores reales. A continuación se presentan diversos métodos para analizar el efecto de los sesgos de información, confusión y selección. El análisis de sensibilidad para sesgo de selección se presenta en último lugar porque combina estrategias del análisis de sensibilidad del sesgo de información y del de confusión.
Respecto al sesgo de información, la asunción (habitualmente no declarada) de todos los estudios es que la información sobre la exposición y la enfermedad es correcta, es decir, que todos los sujetos clasifi cados como expuestos o como enfermos en realidad lo están. Una estrategia frecuente para medir el sesgo producido por la mala clasifi cación en la exposición o la enfermedad consiste en repetir los análisis ponderando los valores de la variable exposición o enfermedad potencialmente “errónea” a partir de datos sobre sensibilidad y especifi cidad (obtenidos en el propio estudio o de la literatura) del instrumento de medida. Por ejemplo, la broncoscopia puede tener una sensibilidad inferior al 50% para diagnosticar cáncer de pulmón cuando la lesión no es visible endoscópicamente; si en un estudio de casos y controles se ha utilizado la broncoscopia para defi nir los casos de cáncer, se puede repetir el análisis del estudio reclasifi cando a ciertos sujetos del grupo control como casos de cáncer (ver más detalles en la referencia 1). En estas situaciones, se puede asumir tanto mala clasifi cación diferencial como no diferencial y se pueden estimar los resultados bajo diferentes supuestos. Otro análisis habitual consiste en modifi car la defi nición de la exposición o la enfermedad haciéndola más específi ca para reducir la posible mala clasifi cación. Por ejemplo, en un estudio poblacional sobre EPOC se pueden excluir los sujetos que, aun cumpliendo con la defi nición espirométrica de EPOC, declaran tener síntomas de asma.
Para eliminar el sesgo por confusión, habitualmente se ofrecen los resultados tras ajustar por las variables confusoras, pero es frecuente que alguna posible confusora no se haya recogido en un estudio concreto. Por ejemplo, la inactividad física aumenta el riesgo de múltiples enfermedades, pero raramente se mide de
manera rutinaria, como sí se mide el consumo de tabaco. En este caso, se debe descartar si la asociación observada en el análisis multivariable puede ser debida a confusión residual por las confusoras no medidas. Si la variable en cuestión se ha medido en una submuestra de la población, aunque sea pequeña, se pueden comparar los estimadores entre el análisis con toda la muestra sin la mencionada variable y el análisis sólo con la submuestra adicionalmente ajustado por dicha variable. Aunque en el último caso los intervalos de confi anza aumentarán debido a la reducción de tamaño de la muestra (y quizás los valores de p sean > 0,05), los estimadores de la asociación se pueden comparar, como en la tabla 2.5.5, lo que permitirá evaluar la cantidad de sesgo debida a la confusión residual. Otra opción es estimar el valor de una variable no medida a partir de datos en la literatura sobre su prevalencia y su relación con la enfermedad.1 Este método se denomina ajuste externo o ajuste indirecto (más detalles en la referencia 1).
Finalmente, para evaluar el sesgo de selección, el mínimo análisis consiste en comparar las características individuales de los sujetos participantes con los no participantes. Cuando se conocen los factores que afectan a la selección (por ejemplo, la no participación o el no seguimiento), este sesgo se puede controlar cuantitativamente. Se pueden usar estrategias similares al control de la confusión si se dispone de la información sobre estos factores en el propio estudio. Si se dispone de la distribución de estos factores en la población, se pueden utilizar estrategias similares al control del sesgo de información.1
VALIDEZ EXTERNA. GENERALIZACIÓN
Tradicionalmente, en cuanto a la validez (falta de error sistemático) se han distinguido dos componentes: la validez interna, que hace referencia al “grado en que los resultados del estudio pueden aplicarse a la población de estudio” (aquella en la que se obtuvo la muestra) y la validez externa (o generalización), que se defi ne como “el grado en que los resultados de un estudio pueden aplicarse a poblaciones que no participaron en dicho estudio”.8 Hoy en día, se considera que la validez externa va más allá de las discusiones sobre muestras y poblaciones. Utilizando un argumento extremo, si sólo fuera posible generalizar a las poblaciones en las que se han reclutado las muestras, los resultados de la investigación en animales jamás se aplicarían a humanos y cada población necesitaría repetir todas las investigaciones adaptadas a su realidad física, biológica y social. En realidad, si aceptamos que el objetivo fi nal de la investigación es contribuir al conocimiento científi co, cabe reconocer que el proceso de sintetizar la evidencia disponible sobre un fenómeno de interés requiere una gran capacidad de abstracción y que va más allá de aplicar a una muestra más amplia lo que se ha observado.
REDUCIR LOS ERRORES ALEATORIOS Y SISTEMÁTICOS
Evitar o reducir el error aleatorio
Aunque el error aleatorio, a diferencia del sistemático, no se puede predecir, es más fácil de resolver. El principal instrumento para reducir el error aleatorio es aumentar el tamaño de la muestra. El tamaño de la muestra es inversamente proporcional al error aleatorio. Intuitivamente, todos estamos familiarizados con la idea de que, al lanzar una moneda al aire 1.000 veces, aproximadamente el 50% de las ocasiones caerá de cara, mientras que si la lanzamos solamente 10 veces, esta proporción puede alejarse del 50% simplemente por azar. De la misma manera, a mayor tamaño de la muestra, mayor precisión en las estimaciones. El capítulo 18 describe cómo calcular el tamaño muestral más adecuado según la pregunta de investigación, el diseño del estudio y las características de las variables clave.
Escoger el diseño más adecuado para una pregunta de investigación permite también aumentar la precisión. Por ejemplo, en una enfermedad poco frecuente, un diseño de casos y controles es más efi ciente estadísti-
camente que un estudio de cohortes. Además, si el estudio prevé análisis estratifi cados y/o ajustados, es necesario que el cálculo de tamaño muestral asegure sufi cientes sujetos en cada uno de los estratos de interés.
El error aleatorio se puede reducir también intentando minimizar el papel del azar en la recogida e incluso en la entrada de datos. Por ejemplo, la estandarización de las condiciones del instrumental, del paciente y del entorno para medir la tensión arterial permitirá reducir la variabilidad aleatoria en las estimaciones. De manera similar, la doble entrada de los datos permitirá identifi car errores tipográfi cos, reduciendo así la variabilidad aleatoria.
Evitar o reducir el sesgo de selección
Para reducir el sesgo de selección, lo más importante es defi nir bien la población de estudio y utilizar técnicas de muestreo que permitan que la muestra sea lo más representativa posible de la población de estudio. En general, una muestra de pacientes seleccionados al azar en centros de atención primaria será más representativa de la población general que una muestra de conveniencia de pacientes hospitalarios. Sin embargo, si la población de interés son pacientes con enfermedad avanzada, la muestra hospitalaria puede ser, no sólo más factible, sino también más adecuada.
Otro aspecto esencial para reducir el sesgo de selección es garantizar que la población de referencia (o población control) sea lo más parecida posible a la población expuesta, excepto en la exposición. Algunos diseños (ensayos cruzados o estudios de casos cruzados) permiten estudiar a los mismos sujetos bajo los dos supuestos (exposición y no exposición), pero por desgracia no son aplicables para la mayoría de preguntas de investigación. En ausencia de esta posibilidad, los investigadores deben recopilar el máximo de información sobre las variables que difi eren entre ambas poblaciones con el fi n de tenerlas en cuenta en el análisis (por ejemplo, como variables de ajuste). De hecho, algunos tipos de sesgo de selección se pueden interpretar como un problema de confusión.
Ante el sesgo de selección llamado de no respuesta, los investigadores deberían intentar obtener alguna información sobre los no participantes (sexo, edad, hábito tabáquico) que permitiera estimar las diferencias en variables clave entre los participantes y la población de estudio. También en los estudios de seguimiento donde se producen pérdidas que pueden dar lugar al sesgo de selección denominado sesgo de supervivencia, disponer de las características que se asocian al seguimiento (por ejemplo, factores que aumentan el riesgo de morir) permitirá ajustar por estos factores en el análisis.
Recientemente se han utilizado técnicas de imputación para controlar el sesgo de selección, especialmente, el sesgo debido a las pérdidas de seguimiento.9 Aunque el concepto de imputar valores a los fallecidos ha sido discutido,10 los estudios existentes muestran que tales técnicas acostumbran a reducir el sesgo.9
Evitar o reducir el sesgo de información
Para reducir el sesgo de información, es necesario utilizar los mejores métodos para defi nir la enfermedad, la exposición y las otras variables relevantes para la pregunta de investigación.
Los datos sobre las enfermedades se pueden obtener, entre otros métodos, a través de cuestionarios, registros o pruebas diagnósticas. En general, se considera buena práctica verifi car los diagnósticos siempre que sea posible. Por ejemplo, para asignar un sujeto a las categorías EPOC sí o EPOC no, utilizar una espirometría forzada es preferible a utilizar un cuestionario, y en el caso del cáncer es mejor disponer de una confi rmación anatomopatológica que de un cuestionario.
En ocasiones no es factible realizar pruebas diagnósticas a todos los participantes. En estos casos se recomienda realizar estudios de validación en una submuestra. Por ejemplo, el Nurses Health Study dispone de una simple pregunta autodeclarada por las enfermeras sobre diagnóstico de asma. Para acompañar esta medida de algún dato de validación, se revisó el diagnóstico a partir de registros médicos en una submuestra, siendo éste confi rmado en un 96% de los casos.11
Respecto a la información sobre exposiciones, existen situaciones especialmente complicadas, como la investigación sobre factores ambientales, en la que resulta difícil recoger mediciones individuales, o la investigación sobre exposiciones laborales, en la que se debe recopilar la historia ocupacional completa. En general, la exposición más reciente es más fácil de recordar que la exposición lejana. También para reducir el error en la medida de la exposición, se recomienda realizar estudios de validación. Por ejemplo, para estudiar los efectos de la actividad física en una muestra muy grande de población general se podría utilizar un cuestionario para todos los sujetos y un acelerómetro en una submuestra. Estas dos medidas permitirían validar la medida de actividad física del cuestionario e incluso ponderarla a partir de los datos del acelerómetro. Otra posibilidad es recoger la información de diferentes fuentes (por ejemplo, tabaco a partir de entrevista al paciente, entrevista al familiar y análisis de cotinina en orina).
En el caso de utilizar cuestionarios, el entrenamiento de los entrevistadores, las condiciones de la entrevista e incluso el orden de las preguntas son esenciales para reducir los errores sistemáticos en la declaración, que acostumbran a ir en el sentido de lo que se percibe como socialmente deseado (Tabla 2.5.2).
Finalmente, el enmascaramiento (o ciego) permite evitar algunos errores sistemáticos debidos al participante o al observador y es una condición requerida en los estudios experimentales. Las técnicas de enmascaramiento más comunes son el simple ciego (sólo el paciente desconoce el grupo al que ha sido asignado, es decir, ignora cuál de los posibles tratamientos recibe), el doble ciego (tanto el paciente como el investigador/ médico desconocen el grupo de asignación) y triple ciego (cuando también el análisis y evaluación de los datos se hace sin conocer la identidad de los grupos).
Evitar o reducir la confusión
El control de la confusión se debe llevar a cabo en la fase de diseño del estudio y en el análisis estadístico. Existen tres técnicas que permiten evitar la confusión: la aleatorización, el apareamiento y la restricción.
La aleatorización para asignar sujetos a un tratamiento u otro en estudios experimentales permite crear subgrupos similares en todas sus características excepto en la exposición de interés, especialmente si las muestras son sufi cientemente grandes. En el caso de muestras pequeñas, sin embargo, es probable que ciertas características, poco frecuentes pero que afectan el riesgo de la enfermedad, por azar se distribuyan de manera diferente entre los subgrupos, evitando el control de la confusión.
El apareamiento se usa sobre todo en estudios de casos y controles. Consiste en seleccionar a los sujetos de tal manera que la distribución de los posibles factores confusores sea igual (o muy similar) entre los grupos a comparar. En general, los sujetos se aparean por sexo, edad, hábito tabáquico o clase social, intentando que ambos elementos de la pareja sólo se distingan por la exposición. Existen diferentes métodos de apareamiento (por ejemplo, individual o de frecuencia), y se pueden utilizar en diferentes diseños, pero es necesario disponer de conocimiento extenso sobre el fenómeno de interés y sobre metodología epidemiológica y estadística, ya que el apareamiento puede también provocar sesgos.
La restricción es la manera más efectiva de evitar la confusión. Si la variable confusora se mide en categorías, como en el sexo o la etnia, la restricción se consigue aceptando en el estudio solamente alguna de las categorías. Si la variable confusora se mide de forma continua, como la edad, se puede defi nir un rango
relativamente estrecho como para que la muestra sea homogénea. Un ejemplo reciente es el reanálisis del efecto del asma en la enfermedad cardiovascular: durante décadas, los estudios observacionales han sugerido un riesgo excesivo de morbilidad y mortalidad cardiovascular en los pacientes asmáticos, incluso tras ajustar por las variables disponibles de tabaco. Sin embargo, un estudio reciente no encontró diferencias en enfermedades cardiovasculares entre asmáticos y no asmáticos tras restringir el análisis a los sujetos que no habían fumado nunca.12 Estos resultados sugieren que la asociación previamente descrita se debía a confusión residual por tabaco.13 El problema de la restricción es que puede difi cultar la generalización de los resultados, especialmente si el efecto de las exposiciones puede ser diferente entre substratos de las variables confusoras.
Desgraciadamente en la mayoría de investigaciones no es posible aleatorizar ni restringir, y el apareamiento es difícil y/o no recomendado, motivo por el que la aproximación más habitual (y acertada) para prevenir la confusión no es precisamente intentar evitarla, sino intentar controlarla. Para ello es necesario recopilar información detallada sobre las posibles variables confusoras y utilizarlas adecuadamente en el análisis estadístico. La identifi cación de las variables que provocan confusión es específi ca de cada pregunta de investigación y requiere conocimiento experto clínico y/o fi siopatológico. La medición de las variables de confusión requiere también experiencia en metodología de la investigación. Finalmente, es necesario disponer de conocimiento estadístico para utilizar las variables de confusión como covariables en los modelos multivariables y/o para estratifi car los resultados (ver capítulos módulo 5).
EQUÍVOCOS Y DUDAS FRECUENTES EN TORNO A LOS ERRORES EN INVESTIGACIÓN
Azar versus leyes de la física. Algunas personas creen en el azar como origen de los fenómenos físicos y biológicos. Otras personas, deterministas, consideran que todos los fenómenos son predecibles. En la práctica, todos aquellos fenómenos impredecibles con el conocimiento actual son indistinguibles de los producidos por azar, por lo que la distinción entre error aleatorio y sistemático descrita en este capítulo es válida bajo todas las creencias.
Valor de la p, tamaño muestral y sesgo. A menudo se interpreta el resultado de las pruebas de contraste de hipótesis (por ejemplo, el valor de p) como una medida de evaluación del sesgo. No es correcto. El sesgo es debido a la presencia de error sistemático y, por tanto, no se puede analizar con las técnicas que miden error aleatorio. De manera similar, el aumento del tamaño de la muestra no puede corregir ningún sesgo.
¿Existe el sesgo? ¿Sí o no? El sesgo es un fenómeno cuantitativo, no cualitativo. Como se ha mencionado previamente, es la cantidad de sesgo la que se debe evaluar (y eventualmente corregir), no su presencia o ausencia.
¿Confusión o sesgo de confusión? ¿Confusión por indicación o sesgo por indicación? Diversos de los términos mencionados en este capítulo (error sistemático, sesgo, validez…) son utilizados por diversas disciplinas y en diversos contextos, con signifi cado similar, pero no siempre idéntico. Por ejemplo, en estadística sesgo se refi ere a la diferencia entre el valor medio de un estimador y el valor real del parámetro que se pretende estimar; en psicometría, la validez se refi ere al grado en que un instrumento de medición mide el constructo que se supone que debe medir; y aunque la distinción entre errores aleatorios y sistemáticos se encuentra en muchas disciplinas, no siempre estos dos tipos de error están completamente separados. En cualquier caso, los investigadores deberían preocuparse por entender el fenómeno, sus causas y sus efectos, más que discutir sobre los nombres.
Tras ajustar, desaparece la asociación, ergo, no existe asociación. Cualquier asociación puede ser debida a confusión, pero continúa siendo asociación. Si tras ajustar la asociación por una variable confusora la asociación desaparece, la interpretación correcta es que la asociación observada sin ajustar (que es real) no se debe a un efecto causal, sino al efecto de una tercera variable.
La variable X es confusora. Ninguna variable X es solo confusora. Cualquier variable X puede ser confusora en el estudio del efecto de una variable Y en otra variable Z. La confusión es un problema de relaciones; en ausencia de con quién relacionarse, no existe confusión.
CONSIDERACIONES FINALES
Todo investigador debe, en diversas fases de la investigación (planifi cación del estudio, análisis estadístico, interpretación de resultados, lectura de artículos, etc.), revisar sistemática y críticamente las cuatro principales fuentes de error más relevantes mencionadas en este capítulo: error aleatorio (tamaño de la muestra), sesgo de selección, sesgo de información y confusión. La tabla 2.5.6 propone una serie de recomendaciones a tener en cuenta. Cabe insistir en la necesidad de disponer de conocimiento experto clínico y/o fi siopatológico, metodológico y estadístico para poder abordar correctamente los errores en investigación. Finalmente, es necesario recordar que sólo se puede realizar una interpretación causal de los resultados de la investigación (tanto observacional como experimental) tras descartar las cuatro fuentes de error revisadas en este capítulo.
PUNTOS CLAVE
• Tipos de error en investigación: error aleatorio y error sistemático (o sesgo). • Precisión: ausencia de error aleatorio. • Validez: ausencia de error sistemático. • Tipos de sesgo: sesgo de selección, sesgo de indicación (o mala clasifi cación) y confusión. • El sesgo es un fenómeno cuantitativo, no cualitativo. • Cuándo considerar los errores: ¡siempre! (diseño del estudio, recogida de datos, análisis estadístico, interpretación de resultados…). • Cómo reducir el error aleatorio: aumentar el tamaño de la muestra. • Cómo reducir el sesgo de selección: defi nir bien la población de estudio y las técnicas de muestreo. • Cómo reducir el sesgo de información: utilizar los mejores métodos para defi nir la enfermedad, la exposición y las demás variables relevantes. • Cómo reducir la confusión: aleatorizar (en estudios experimentales), aparear (en algunos estudios observacionales) o restringir (si la pregunta de investigación lo permite) y, sobre todo, recopilar información detallada sobre las posibles variables confusoras y utilizarla adecuadamente en el análisis estadístico. •- Para prevenir e interpretar correctamente los errores en investigación, es necesario disponer de conocimiento experto clínico y/o fi siopatológico, metodológico y estadístico.
BIBLIOGRAFÍA
1. Rothman KJ, Greenland S. Modern epidemiology. Philadelphia, Lippincott-Raven, 2ª ed. 1998. 2. Fletcher R, Fletcher S, Wagner E. Epidemiología Clínica: aspectos fundamentales. Barcelona, MAS-
SON-Williams & Wilkins España, 2ª ed. 1998. 3. Porta M. A Dictionary of Epidemiology. Nueva York, Oxford University Press, 6ª ed. 2014. 4. Abramson JH. Survey Methods in Community Medicine. Edinburgh, Churchill Livingstone, 2ª ed. 1979. 5. Sackett DL. Bias in analytic research. J Chronic Dis. 1979;32:51-63. 6. Szklo M, Nieto J. Epidemiology Beyond the Basics. Geithersburg, Aspen Publishers, 2000. 7. Garcia-Aymerich J, Marrades RM, Monsó E, Barreiro E, Farrero E, Antó JM; EFRAM Investigators.
Paradoxical results in the study of risk factors of chronic obstructive pulmonary disease (COPD) re-admission. Respir Med. 2004;98:851-857. 8. Porta M. A Dictionary of Epidemiology. Nueva York, Oxford University Press, 6ª ed. 2014. 9. Weuve J, Tchetgen Tchetgen EJ, Glymour MM, Beck TL, Aggarwal NT, Wilson RS, Evans DA, Mendes de Leon CF. Accounting for bias due to selective attrition: the example of smoking and cognitive decline.
Epidemiology. 2012;23:119-128. 10. Chaix B, Evans D, Merlo J, Suzuki E. Commentary: Weighing up the dead and missing: refl ections on inverse-probability weighting and principal stratifi cation to address truncation by death. Epidemiology. 2012;23:129-131. 11. Camargo CA, Weiss ST, Zhang S, Willett WC, Speizer FE. Prospective study of body mass index, weight change, and risk of adult-onset asthma in women. Arch Intern Med. 1999;159:2582-2588. 12. Çolak Y, Afzal S, Nordestgaard BG, Lange P. Characteristics and Prognosis of Never-Smokers and Smokers with Asthma in the Copenhagen General Population Study. A Prospective Cohort Study. Am J
Respir Crit Care Med. 2015;192:172-181. 13. Garcia-Aymerich J. Th e Role of Smoking in the Association between Asthma and Cardiovascular Disease. An Example of Poorly Controlled Confounding. Am J Respir Crit Care Med. 2015;192:123.
TABLAS Y FIGURAS
Tabla 2.5.1. Ejemplos de sesgos de selección
Nombre del sesgo de selección Ejemplo
Autoselección / sesgo del voluntario / sesgo de no respuesta
Falacia de Berkson / selección en el hospital En un estudio para estimar la prevalencia de síntomas respiratorios en población general, los sujetos que sufren algún síntoma (o que tienen algún familiar que sufre síntomas) son más propensos a participar que los que no tienen síntomas respiratorios. Como consecuencia, la muestra participante tendrá una mayor prevalencia de síntomas que la población real.
En un estudio de casos y controles para conocer si el consumo de tabaco aumenta el riesgo de fracturas, los casos se seleccionan en el hospital (donde es fácil identifi car un número elevado de sujetos con fracturas). Se observará una asociación entre tabaco y fracturas superior a la asociación real debido a que en el caso de sufrir una fractura es más probable ser ingresado si existen enfermedades concomitantes (cardiovasculares, respiratorias o cáncer) y al hecho de que estas enfermedades a menudo tienen relación con el consumo de tabaco.
Trabajador sano En un estudio para conocer los efectos respiratorios de cierta exposición laboral continuada, los sujetos que han desarrollado algún problema respiratorio han dejado de trabajar y no participan en el estudio, por lo que el efecto observado será inferior al efecto real.
Sesgo de supervivencia En un estudio de seguimiento para analizar la relación entre el tabaquismo activo y los cambios en la función pulmonar, algunos sujetos mueren tras ser reclutados y no pueden participar en las visitas de seguimiento. Dado que la muerte está probablemente relacionada con el consumo de tabaco, los sujetos que continúan en el estudio exhibirán características diferentes a los que han fallecido y la asociación observada entre tabaco y cambios en función pulmonar estará sesgada.
Tabla 2.5.2. Ejemplos de sesgos de información
Fuente del error de medida Ejemplo
Error de medida dependiente del sujeto Cambios fi siológicos Función pulmonar reducida en los asmáticos agudizados
Respuesta socialmente aceptable / Cambios por saber que está siendo investigado Declaración de no fumar en un cuestionario sobre síntomasrespiratorios
Factores que afectan la respuesta a las preguntas: despiste, motivación, memoria, etc. Olvido sobre haber consumido un fármaco en el pasado según el estado de salud en el presente
Error de medida dependiente del observador Errores sistemáticos al administrar preguntas “Usted no toma alcohol, ¿verdad?”
Error de medida dependiente de los instrumentos Factores relacionados con la experiencia
Propiedades del instrumento de medida
Errores en el instrumento de medida En un estudio de función pulmonar con múltiples observadores, un observador con más experiencia en la técnica de la espirometría conseguirá valores más altos que el resto
Baja sensibilidad en la broncoscopia para diagnosticar cáncer de pulmón no visible endoscópicamente
Pequeña fuga de aire en el espirómetro
Tabla 2.5.3. Sesgo de información por mala clasifi cación diferencial de la exposición debida a falta de memoria selectiva (sesgo de memoria, datos hipotéticos)
Exposición No exposición
Datos reales
Casos Controles
Datos declaradosa
Casos Controles 10 10 40 40 OR=1
10 2 40 48 OR=6
OR: Odds ratio
a Los sujetos enfermos (casos) recuerdan la exposición perfectamente y los datos declarados concuerdan con la realidad. Sin embargo, la mayoría de los sujetos sanos (controles) han olvidado haber estado expuestos; esta mala clasifi cación deriva en una estimación errónea de la asociación entre exposición y enfermedad (OR observada = 6 versus OR real = 1).
Tabla 2.5.4. Sesgo de información por mala clasifi cación no diferencial de la exposición debida al instrumento de medida (datos hipotéticos)
Exposición No exposición
Datos reales
Casos Controles
Datos observadosa Casos Controles 25 10 25 40 OR=4
20 8 30 42 OR=3,5
OR: Odds ratio
a Tanto en los sujetos enfermos (casos) como en los sanos (controles), el instrumento de medida tiene una sensibilidad del 80%, por lo que el 20% de los expuestos se clasifi can como no expuestos. Esta mala clasifi cación deriva en una estimación errónea de la asociación entre exposición y enfermedad (OR observada = 3,5 versus OR real = 4).
Tabla 2.5.5. Evaluación de la confusión por indicación. Asociación entre diversas intervenciones médicas y riesgo de ingreso por agudización de la EPOC
HR crudo HR ajustadoa % exceso de riesgob
Control por el neumólogo (referencia: médico de familia) 2.16c 1.73c 37%
Uso de anticolinérgicos inhalados 3.52c
Rehabilitación pulmonar 1.77c
Oxigenoterapia domiciliaria 2.36c 2.10c 56%
1.28 64%
1.38 72%
HR: hazard ratio (razón de riesgos) a Ajustado por FEV1, PaO2 e ingresos previos por agudización de la EPOC. b Porcentaje del exceso de riesgo explicado por FEV1, PaO2 e ingresos previos, calculado como [(HR crudo – HR ajustado)/HR crudo-1)]*100. c p < 0,05. Fuente: Garcia-Aymerich J, Marrades RM, Monsó E, Barreiro E, Farrero E, Antó JM; EFRAM Investigators. Paradoxical results in the study of risk factors of chronic obstructive pulmonary disease (COPD) re-admission. Respir Med. 2004;98:851-857.
Tabla 2.5.6. Recomendaciones sobre cómo actuar ante los posibles errores en diferentes fases de la investigación
Diseñar un estudio
Describir las limitaciones de un estudio Interpretar un resultado / leer un artículo
Tamaño muestral Calcular el tamaño de la muestra necesaria, incluyendo previsión de ps y análisis de subgrupos.
Sesgo de selección Identifi car la población de estudio, muestra y técnica de muestreo más adecuada a la pregunta de investigación y el diseño. Planifi car el análisis de sensibilidad.
Sesgo de información Utilizar los mejores instrumentos para defi nir las variables de exposición, enfermedad y otras relevantes. Considerar estudios de validación en una submuestra. Planifi car el análisis de sensibilidad.
Confusión Identifi car y medir las principales variables que pudieran provocar confusión. Planifi car el análisis multivariable. Planifi car el análisis de sensibilidad. Discutir las diferencias entre los modelos crudo y ajustado. Identifi car si existen factores de confusión no medidos y discutir el posible efecto de la confusión residual en los resultados.
Identifi car si se ha conseguido el tamaño muestral previsto, incluyendo los subgrupos.
Discutir si se ha conseguido una muestra que represente a toda la población de estudio o si las no respuestas, pérdidas o exclusiones pueden haber afectado la representatividad. Discutir el posible efecto del sesgo de selección en los resultados.
Discutir sobre validez y fi abilidad de los instrumentos utilizados para defi nir las variables de exposición, enfermedad y otras relevantes. Discutir el posible efecto del sesgo de información en los resultados. ¿Es la muestra sufi cientemente grande? ¿Se presentan los cálculos de tamaño muestral? ¿Se discute en limitaciones sobre el tamaño de la muestra, especialmente en los subgrupos para los que se presentan resultados?
¿Están los individuos bien seleccionados? ¿Se describe la población de estudio? ¿Se describe el sistema de muestreo? ¿Se informa de no respuestas, pérdidas, muertes o exclusiones y se comparan sus características con las de los participantes? ¿Se discuten los posibles sesgos y sus efectos? ¿Se incluye algún análisis de sensibilidad?
¿Están las variables de exposición, enfermedad y confusoras bien defi nidas? ¿Se describen los instrumentos utilizados para medir las variables y su validez y precisión? ¿Se incluyen análisis de sensibilidad? ¿Se discuten los posibles sesgos y sus efectos?
¿Están medidas y consideradas en el análisis todas las posibles v ariables confusoras? ¿Se describen las variables consideradas confusoras? ¿Se describe cómo se han construido los modelos multivariables? ¿Se incluye el análisis de sensibilidad?
Figura 2.5.1. Representación gráfi ca de los conceptos de precisión y validez
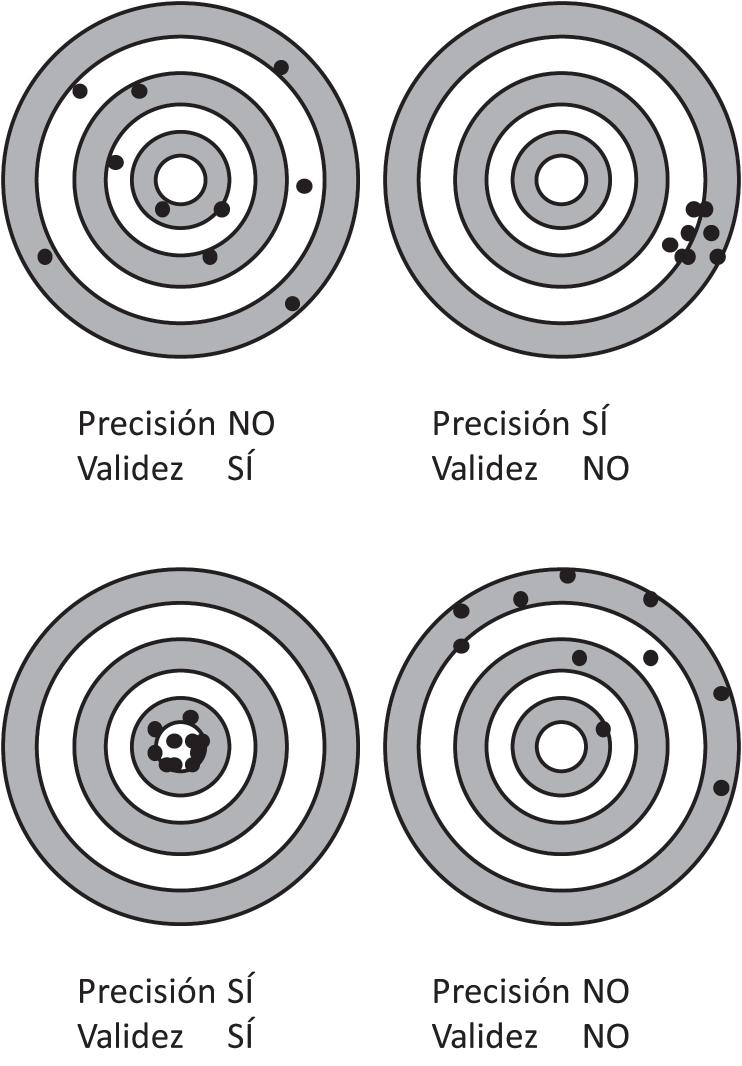
Figura 2.5.2. Representación gráfi ca del efecto del tabaco (variable confusora) en el estudio de la asociación entre el consumo de alcohol (variable exposición) y la enfermedad coronaria

2.6. REVISIONES SISTEMÁTICAS Y METAANÁLISIS
Dra. Elena Gimeno Santos
INTRODUCCIÓN
Una revisión sistemática (RS) tiene como objetivo recopilar toda la evidencia para responder una pregunta de investigación concreta, minimizando el sesgo gracias a la utilización de métodos explícitos, sistemáticos y rigurosos. Es por este rigor metodológico que las RS son el estándar de referencia para la síntesis de evidencias en salud y se utilizan para el desarrollo de guías de práctica clínica y apoyar la toma de decisiones clínicas.
Por lo tanto, una RS tiene unas características similares a cualquier estudio de investigación. Sin embargo no usa datos de sujetos individuales, sino que recoge datos de estudios ya realizados.
Aunque muchas RS contienen metaanálisis, estos dos términos no deberían usarse como sinónimos, aunque habitualmente ocurre. El término metaanálisis corresponde al método estadístico que combina resultados de estudios individuales para dar una estimación global más precisa de los efectos estudiados.
Asimismo, debe diferenciarse una RS de una revisión de la literatura realizada por uno o varios expertos. La primera, la RS, es una revisión protocolizada que identifi ca, selecciona, sintetiza y evalúa todas las evidencias de investigaciones primarias. La revisión de la literatura es un resumen cualitativo de las evidencias en un tema concreto que usa métodos informales o subjetivos para recolectar e interpretar los estudios elegidos.
PROTOCOLO DE LA REVISIÓN SISTEMÁTICA
Igual que cualquier proyecto de investigación, una RS debe seguir un protocolo que se habrá escrito y detallado antes de iniciar el proceso de revisión en sí, a fi n de que cualquier investigador pueda reproducir el proceso de revisión. El protocolo especifi cará los métodos usados durante las diferentes fases del proceso de RS, las cuales se resumen a continuación.
• Defi nición de la pregunta de investigación. Formulación de objetivos claros y concretos. • Concreción de criterios de selección de las referencias bibliográfi cas (criterios inclusión y exclusión) y de la estrategia de búsqueda. • Búsqueda sistemática de la literatura y aplicación de los criterios de elegibilidad de los estudios.
• Inclusión de estudios y extracción de datos. • Evaluación del riesgo de sesgos y de la calidad de la evidencia. • Síntesis de los datos mediante procedimientos estadísticos, como el metaanálisis (si es necesario) y la interpretación de datos. • Redacción de los resultados.
DEFINICIÓN DE LA PREGUNTA DE REVISIÓN
Una RS debería establecer preguntas claras. Se recomienda que la defi nición de la pregunta sea precisa para conseguir la mejor evidencia para responderla. La pregunta de revisión, especialmente en RI sobre intervenciones, debe estar enmarcada en términos de población, intervención, comparación, variables de resultado (Outcomes) y frecuentemente también el diseño del estudio (Study design). Estos elementos, que se resumen en el acrónimo PICO(S), facilitan la defi nición de los criterios de inclusión y exclusión que se usarán para la selección de los estudios a revisar. A continuación, un ejemplo de pregunta siguiendo la “fórmula” PICO(S).
• Objetivo de la revisión: Evaluar los efectos de la rehabilitación pulmonar tras una exacerbación de la
EPOC en hospitalizaciones, mortalidad, calidad de vida y capacidad de ejercicio. • Población: Pacientes con EPOC que hayan sufrido una exacerbación de la enfermedad (con o sin ingreso hospitalario). • Intervención: Cualquier programa de rehabilitación pulmonar que incluya al menos ejercicio físico, tanto en pacientes hospitalizados como ambulatorios, que hayan sufrido una exacerbación de la EPOC. El programa de rehabilitación debe comenzar inmediatamente después o hasta tres semanas después del inicio del tratamiento médico de la exacerbación. • Comparación: Rehabilitación pulmonar versus tratamiento convencional tras exacerbación de la EPOC. • Outcomes: Hospitalizaciones (como mínimo una durante el periodo de seguimiento). Outcomes secundarios: mortalidad, calidad de vida y capacidad de ejercicio. • Diseño ((S)tudy design): Ensayos clínicos aleatorizados y controlados.
CRITERIOS DE INCLUSIÓN Y EXCLUSIÓN
Los criterios de inclusión y exclusión (criterios de elegibilidad) de la revisión han de ser explícitos y claros y deben permitir guiar de manera correcta la inclusión de los estudios. Los criterios de inclusión y exclusión servirán también para preparar la estrategia de búsqueda bibliográfi ca y defi nir los fi ltros que se añadirán a esta.
Los criterios de elegibilidad son la combinación de los diferentes aspectos de la pregunta de revisión: tipos de participantes, tipos de intervenciones, tipos de variables de resultado (outcomes) y tipos de estudio. A continuación se detallará los factores a considerar en cada punto.
1. Tipos de participantes • Defi nir la condición clínica o enfermedad diagnosticada. • Detallar las características más importantes de los participantes. • Especifi car el entorno de los participantes (hospital, atención primaria, comunidad, etc.). • Defi nir si hay algún otro tipo de participante o condición clínica que debería ser excluido de la revisión. • Concretar cómo se gestionarán los estudios que incluyan sólo una submuestra de los participantes de interés.
2. Tipos de intervenciones • Defi nir la intervención experimental y control (o comparación) de interés. • Especifi car si la intervención puede tener variaciones en relación a dosis, modo, frecuencia, duración y tiempo de administración o personal que lo administra. • Concretar cómo se gestionarán los estudios que incluyan sólo una parte de la intervención.
3. Tipos de outcomes o variables de resultado • Generalmente se incluyen todas las variables de resultado que puedan tener una importancia para los clínicos, pacientes, responsables de políticas de salud, así como, población general. • Es importante decidir cómo se han medido los outcomes (medidas objetivas o subjetivas) y en qué momento temporal se midieron (una estrategia de ayuda es preespecifi car intervalos: corto, medio y largo plazo). • Asegurar que las variables de resultado incluyen tanto los efectos de la intervención como los efectos adversos de esta.
4. Tipos de estudios • En el momento de escribir el protocolo de la revisión sistemática, los autores deben considerar qué tipo/s de diseño/s son los más adecuados para responder los objetivos de su revisión. • Generalmente, las revisiones sistemáticas se centran en ensayos clínicos aleatorizados porque así se asegura la comparación entre grupos. Sin embargo, y según la pregunta de investigación planteada, se pueden utilizar otro tipo de estudios, como los observacionales. Hay que tener en cuenta que el uso de estudios que no sean ensayos clínicos difi cultará el proceso de análisis de la magnitud del efecto y la combinación de los datos.
BÚSQUEDA BIBLIOGRÁFICA
La búsqueda de las referencias bibliográfi cas debería ser lo más sensible posible, aunque esto implique baja precisión en un inicio. El objetivo de la búsqueda debe ser identifi car todos los estudios existentes sobre el tema en cuestión, esto implica detectar tanto los estudios publicados como los no publicados. La estrategia de búsqueda debe ser replicable y deberá documentarse adecuadamente en el momento de escribir el manuscrito de la RS (generalmente como anexo).
Es recomendable contar con la participación de un documentalista que ayude a realizar una correcta estrategia de búsqueda bibliográfi ca combinando los términos clave (basados en la pregunta PICO) con los operadores booleanos (AND, OR y NOT), y asegurando que esté libre de sesgos (sesgo de publicación, de idioma, etc.).
Reducir el sesgo de publicación implica que la búsqueda bibliográfi ca incluya búsqueda en bases de datos como Medline, Embase, Psycinfo, Biblioteca Cochrane, entre otras (generalmente se recomienda buscar en más de una). Pero también es necesario complementarla con la búsqueda manual a través de las referencias localizadas en la búsqueda electrónica, en libros o en otras revisiones, contactar con expertos en el tema, así como intentar localizar literatura no publicada y literatura “gris” (tesis doctorales, comunicaciones en congresos, etc.). Asimismo es importante no aplicar ninguna restricción de idioma y/o fecha a la estrategia de búsqueda para acceder al máximo de referencias bibliográfi cas.
Una vez realizada la búsqueda, es recomendable tratar las referencias mediante soft ware específi co para ello (EndNote, RefWorks, Reference Manager, Mendeley, etc.). La decisión de qué gestor bibliográfi co utilizar muchas veces está supeditado a los accesos que tenga la propia institución de trabajo.
PROCESO DE REVISIÓN
El proceso de revisión de las referencias bibliográfi cas encontradas debe estar detallado en el protocolo. Los pasos a seguir para seleccionar estudios e incluirlos en la revisión deberían ser los que se detallan a continuación.
1. Reunir en el gestor bibliográfi co todas las referencias encontradas y descartar referencias duplicadas.
Esto ocurre al utilizar varias bases de datos para la búsqueda. 2. Revisar los títulos y los resúmenes de las publicaciones para eliminar aquellas referencias que no sean relevantes para la RS. Este proceso se realiza en parejas de revisores, por lo que si el número de referencias es muy alto se recomienda realizar dos o más equipos de revisores. Cada revisor explorará de manera independiente las referencias asignadas a la pareja de revisores. Una vez terminado el proceso, se compararán las decisiones y se discutirán las discrepancias que puedan existir (si es necesario, un tercer revisor tomará la decisión fi nal). En esta fase se recomienda a los revisores ser muy conservadores (“más vale que sobren referencias que no que falten”), ya que si alguna referencia fi nalmente no debe incluirse en la revisión se podrá descartar en el proceso de revisión de texto completo. 3. Encontrar el texto completo de las referencias marcadas como incluidas durante el proceso de revisión de títulos y resúmenes. Este punto puede ser complejo por las restricciones de acceso a revistas (no subscripción) en cada institución. De nuevo será recomendable trabajar de manera coordinada con un documentalista. 4. Revisar el texto completo y aplicar los criterios de elegibilidad (previamente defi nidos en el protocolo) para decidir si el artículo o publicación se incluye o se excluye de la revisión. Este proceso vuelve a realizarse por parejas de revisores. Una vez completado, se comparan las decisiones que cada revisor ha tomado de manera independiente para comprobar si existe consenso en la decisión. En caso de desacuerdo entre revisores, de nuevo será un tercer revisor quien se encargará de tomar la decisión. 5. Complementar el proceso de revisión con una revisión manual de las referencias de cada publicación incluida. Y repetir los pasos anteriores en caso de añadir alguna referencia nueva al proceso de revisión. 6. Proceder a la extracción de datos de las publicaciones incluidas en la revisión.
Equipo de revisores
El equipo de revisores que llevará a cabo la RS debería tener conocimientos de metodología concreta sobre RS, así como experiencia en el área de estudio y en métodos estadísticos específi cos. Se recomienda que como mínimo dos investigadores/revisores estén implicados en la revisión, pues así se reduce al máximo el sesgo y la presencia de errores durante el proceso de revisión. Sin embargo, en algunos momentos del proceso de revisión se puede requerir la participación de un tercer investigador, por ejemplo ante la discrepancia de los dos revisores respecto a incluir o no un estudio en la RS.
Asimismo, en algunas ocasiones el equipo de revisores puede necesitar asesoramiento de expertos clínicos o metodológicos externos, por lo que se recomienda (aunque no es obligado) tener un grupo asesor asociado a la RS.
Estudios excluidos
Es recomendable listar los estudios o publicaciones que pasan a la fase de “revisión de texto completo” pero que fi nalmente quedan excluidas, así como recoger el motivo principal de exclusión.
PROCESO DE EXTRACCIÓN DE DATOS
Los datos que se extraerán de cada publicación/estudio y la estrategia para realizar la obtención de datos deben contemplarse en el protocolo de la RS. Los formularios de extracción de datos deben estar diseñados cuidadosamente para alcanzar los objetivos de la RS. Dichos formularios pueden ser en formato electrónico o en formato papel, a elección de los investigadores. También es importante hacer una prueba piloto con el formulario y redefi nirlo y perfeccionarlo adecuadamente.
La extracción de datos, al igual que el resto de procesos de la revisión, deben realizarlo como mínimo dos revisores independientes. Es importante valorar la concordancia entre revisores para garantizar que los datos extraídos sean adecuados.
La tabla 2.6.1 resume los principales datos que se extraen de cada artículo o publicación incluida en la RS.
En relación a las variables principales y al estimador que se va a utilizar para comparar todos los estudios (diferencia de medias, odds ratio, risk ratios, etc.), si la publicación en sí no proporciona datos sufi cientes será necesario contactar con los autores para intentar obtener dicho estimador o los datos originales y poder calcularlo.
EVALUACIÓN DEL RIESGO DE SESGOS
La evaluación de la validez de los estudios incluidos en una RS es muy importante, ya que puede alterar los análisis, la interpretación de los datos y, por lo tanto, las conclusiones de la RS.
Para la evaluación del riesgo de sesgo se pueden encontrar diferentes herramientas, la mayoría escalas y listas de comprobación que permiten cuantifi car el riesgo. A continuación, y por su frecuencia de uso en las RS, se resumen los diferentes tipos de sesgo que se deberían evaluar en ensayos clínicos.
• Sesgo de selección: diferencias sistemáticas en las características basales de los grupos de comparación.
Tiene relación con cómo se generó la secuencia de asignación de grupos y la ocultación de esta. • Sesgo de actuación: diferencias sistemáticas en la intervención o cuidado proporcionado a cada grupo.
Tiene relación con el proceso de enmascaramiento de los participantes y de los profesionales que administran la intervención. • Sesgo de detección: diferencias sistemáticas entre grupos por cómo se evalúan las variables de resultado (outcomes). Tiene relación con el proceso de enmascaramiento del evaluador. • Sesgo por pérdida de datos: diferencias sistemáticas entre grupos por exclusión de sujetos de los análisis o datos no disponibles. Tiene relación con datos incompletos de las variables de resultado. • Sesgo de descripción de datos: diferencias sistemáticas entre datos reportados y datos no reportados. Tiene relación con la presentación o descripción selectiva de los resultados.
Además de lo anterior, hay sesgos que pueden afectar a los resultados de la RS y que no tienen relación directa con el sesgo que pueda presentar cada estudio de manera individual, pero también se tendrán en cuenta en el momento de reportar las conclusiones que se extraigan de la RS. Estos sesgos se puede clasifi car en:
• Sesgo de publicación: cuando no se han podido detectar todos los estudios, por ejemplo porque presentan resultados negativos y son difíciles de publicar.
• Sesgo de idioma: cuando en la revisión sólo se incluyen estudios publicados en un solo idioma, por ejemplo en inglés. • Sesgo de localización de estudios en las bases de datos: suele ocurrir si sólo se utiliza una base de datos para la búsqueda de las referencias bibliográfi cas. • Sesgo de publicación múltiple: suele ocurrir cuando se incluyen datos de diferentes publicaciones pero que provienen del mismo estudio (típico de estudios multicéntricos).
ANÁLISIS DE LOS RESULTADOS
Los análisis de una RS pueden ser narrativos (resumen estructurado de los estudios encontrados) o cuantitativos (uso de análisis estadístico con los estimadores de los estudios individuales).
El protocolo de la RS también debería considerar el plan de análisis que se llevará a cabo. Este suele ser el apartado del protocolo que está más supeditado a cambios, ya que es imposible anticipar qué datos se van a encontrar y a extraer de los estudios.
Metaanálisis
El metaanálisis es una herramienta estadística que permite combinar los resultados de dos o más estudios individuales.
Un metaanálisis es una herramienta complementaria a todo el proceso de RS, y se debe tener en cuenta junto a las consideraciones realizadas para el análisis en general. En otras palabras, el efecto global resumido por el rombo en la fi gura del forest plot no es el único resultado de la RS.
Los objetivos del metaanálisis se centran en: 1) hacer una valoración cualitativa y cuantitativa de los resultados individuales de cada estudio, 2) mejorar la precisión del estimador, es decir, aumentar el poder estadístico, 3) resolver incongruencias entre los resultados de los estudios individuales, 4) responder a nuevas preguntas de investigación que no se habían planteado en los estudios originales y/o no tenían sufi ciente poder estadístico para hacerlo, y 5) generar nuevas ideas para futuros trabajos de investigación.
Para que el metaanálisis tenga sentido, se deben combinar los resultados si los estudios son lo sufi cientemente homogéneos, por lo que en un primer paso del análisis es importante analizar correctamente la heterogeneidad (I2) de los estudios. En caso de que exista mucha heterogeneidad, se debería tratar de identifi car qué factores están relacionados y qué puede explicarla. Una I2 mayor al 50% ya representa una heterogeneidad importante y, por lo tanto, debe considerarse en el momento de la interpretación de resultados y conclusión de la RS.
La fi gura 2.6.1 representa un ejemplo de la representación gráfi ca (forest plot) de un metaanálisis para datos dicotómicos.
Metasíntesis
Cuando realizar el metaanálisis no es posible o no es apropiado, la única opción para reportar los resultados es realizarlo de manera narrativa (mediante texto, tablas resumen, etc.).
Esto suele ocurrir cuando en la RS existe mucha heterogeneidad, como por ejemplo si se incluyen diferentes tipos de estudios, con diferentes estimadores, y por lo tanto, se intenta comparar “peras con manzanas”.
EVALUACIÓN DE LA CALIDAD DE LA EVIDENCIA
Los datos de una RS y su respectivo metaanálisis (si se ha realizado) no deberían mostrarse sin haber evaluado la calidad de la evidencia encontrada. Esto signifi ca poder responder a “cuán confi ados estamos de que el efecto global calculado para cada variable resultado está cercano a la verdad”.
Para responder esta pregunta existen diferentes enfoques. Uno de los más recomendados y utilizados es el sistema GRADE (Grading of Recommendations Assessment, Development and Evaluation). El método GRADE especifi ca 4 niveles de calidad (alta, moderada, baja y muy baja) que dependen de diversos factores que los autores deberán evaluar para cada variable resultado siguiendo unos criterios que permiten reducir o incrementar la puntuación del nivel de calidad de la evidencia.
REDACCIÓN DE LOS RESULTADOS
Toda RS debería reportarse siguiendo la guía específi ca para ello: la normativa PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses). Esta consiste en un listado de 27 ítems y un diagrama de fl ujo para representar los resultados de las diferentes fases de una RS. Es importante destacar que PRISMA tiene como objetivo ayudar a los investigadores a reportar los resultados de una RS de manera transparente y adecuada, pero no es una herramienta que evalúe o proporcione información sobre la calidad de la RS.
Como en todo manuscrito, la sección de resultados de una RS debe ser clara y lógica. Los autores pueden usar las tablas y fi guras que crean más convenientes. Es adecuado mostrar los resultados en relación a las características de los estudios incluidos (se recomienda el uso de tablas), los datos analizados (si se ha realizado metaanálisis, se suelen representar usando forest plot), las fi guras (diagrama de fl ujo, otros forest plots, gráfi cas y tablas del riesgo de sesgo, etc.) y otros resultados y tablas que se quiera mostrar.
LIMITACIONES DE LOS RESULTADOS
Aunque la RS y el metanálisis se consideran la mejor evidencia para obtener una respuesta defi nitiva a una pregunta de investigación, los defectos inherentes asociados y presentados durante el capítulo (ej.: selección de estudios, heterogeneidad, pérdida de información, etc.) pueden limitar la toma de decisiones clínicas o cambios en actuaciones de promoción de la salud. Así pues, se debe asumir que tras realizar una RS puede seguir habiendo resultados no concluyentes sobre la pregunta de investigación, que puede no haber sufi cientes estudios o que todos los estudios encontrados pueden tener una baja evidencia.
REVISANDO UNA REVISIÓN
Como revisores o lectores de un manuscrito de una RS, también se recomienda seguir un proceso sistemático para evaluarla de forma crítica. Las siguientes preguntas, basadas en el propio proceso de una RS, pueden ayudar:
• ¿La pregunta de la revisión está claramente defi nida en términos de población, intervención, comparación, resultados y diseño de los estudios (PICO(S))? • ¿La estrategia de búsqueda es adecuada y apropiada? ¿Hay restricciones de idioma, tipo de publicación o fecha de publicación?
• ¿Los criterios para evaluar la calidad de los estudios originales son adecuados? • ¿Se ha intentado minimizar el sesgo y los errores en el proceso de selección de los estudios? • ¿Se ha intentado minimizar el sesgo y los errores en el proceso de extracción de datos? • ¿Los detalles presentados para cada uno de los estudios primarios son adecuados y sufi cientes? • ¿Los métodos utilizados para la síntesis de los datos han sido adecuados? • ¿Las conclusiones de los autores refl ejan con precisión la evidencia revisada?
PUNTOS CLAVE
• Una revisión sistemática: búsqueda sistemática, evaluación crítica y síntesis de todos los estudios relevantes sobre un tema específi co. • El objetivo: recopilar toda la evidencia para responder una pregunta de investigación concreta minimizando el sesgo mediante la utilización de métodos explícitos, sistemáticos y rigurosos. • Es semejante a un diseño de investigación en sí misma en que las unidades a estudio son trabajos originales ya publicados. • El metaanálisis es una herramienta estadística que permite combinar los resultados de dos o más estudios individuales siempre y cuando los estudios sean lo sufi cientemente homogéneos.
BIBLIOGRAFÍA
Higgins JPT, Green S (ed.). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [actualización Marzo 2011]. Th e Cochrane Collaboration, 2011. Disponible en www.cochrane-handbook.org. Argimón Pallás JM, Jiménez Villa J. Métodos de investigación clínica y epidemiológica. Madrid, Elsevier, 4ª ed., 2013. Gisbert JP, Bonfi ll X. ¿Cómo realizar, evaluar y utilizar revisiones sistemáticas? Gastroenterol Hepatol. 2004;27(3):129-149. Cochrane Database of Systematic Reviews http://www.cochranelibrary.com/cochrane-database-of-systematic-reviews/. Formación online de Cochrane Collaboration sobre revisiones sistemáticas http://training.cochrane.org/. Normativa PRISMA (Preferred Reportin Items for Systematic Reviews and Meta-Analyses) http://www. prisma-statement.org/. Normativa GRADE (Grading of Recommendations Assessment, Development and Evaluation) http://www. gradeworkinggroup.org/.
TABLAS Y FIGURAS
Tabla 2.6.1. Datos que se extraerán de cada artículo o publicación incluida en una revisión sistemática
Datos identifi cativos • Identifi cador del estudio (creado por el revisor); • Identifi cador del revisor (generalmente las iniciales); • Referencia bibliográfi ca y detalles de contacto; Inclusión/Exclusión • Confi rmación de inclusión (sí/no); • Motivo de exclusión; Métodos • Diseño del estudio; • Duración del estudio; • Generación de la secuencia de asignación de grupos; • Ocultación de la secuencia de asignación; • Enmascaramiento; • Otras información relacionada con los sesgos; Participantes • Número total de sujetos; • Entorno (setting); • Diagnóstico y datos relevantes relacionados (ej.: FEV1); • Edad; • Sexo; • Otros datos demográfi cos relevantes para la revisión (ej.: raza); Intervenciones • Número total de grupos de intervención; Para cada intervención: • Intervención; • Detalles de la intervención;
Variables resultado (outcomes)
• Variable resultado y momento de recogida; Para cada variable resultado: • Defi nición de la variable resultado; • Unidad de medida (si es relevante); Resultados • Número de participantes asignados a cada grupo de intervención; Para cada variable resultado: • Tamaño de la muestra; • Participantes perdidos (missings); • Datos resumen para cada grupo intervención (ej.: Tabla 2x2 para datos dicotómicos, media y desviación estándar para datos continuos); Otra información que se considere oportuna para la revisión o por el revisor: fi nanciación del estudio, referencias relacionadas, comentarios del revisor, etc.
Figura 2.6.1. Ejemplo de la representación gráfi ca (forest plot) de metaanálisis para datos dicotómicos

2.7. VALIDACIÓN DE PRUEBAS DIAGNÓSTICAS
Dr. Alberto Fernández Villar
INTRODUCCIÓN
Las decisiones clínicas se basan en la información obtenida de los pacientes mediante diferentes procedimientos, como la historia clínica, la exploración física y las pruebas de laboratorio o de imagen. Éstas aportan datos que pueden modifi car la probabilidad de un diagnóstico al reducir la incertidumbre sobre si un paciente tiene una afección de interés.1-2 El diagnóstico consiste, pues, en decidir, en base a la información obtenida de las pruebas diagnósticas, si una persona está o no enferma, si bien en la mayoría de las veces existirá alguna incertidumbre. Aunque la evaluación de las pruebas diagnósticas desempeña un papel central en la actividad clínica, los profesionales sanitarios reciben un escaso entrenamiento formal para ella, cuya incorporación a la práctica clínica históricamente no ha tenido el mismo control que las intervenciones terapéuticas.1,3 En múltiples estudios se ha demostrado que los trabajos referidos a pruebas diagnósticas publicados, incluso en prestigiosas revistas, adolecen de importantes sesgos y problemas metodológicos que generalmente llevan a la sobreestimación de la validez de las mismas.3 En el presente capítulo se describen los aspectos más importantes que permitan a diseñar y llevar a cabo estudios de validación de pruebas diagnósticas de calidad.
FASES DE UN ESTUDIO DE EVALUACIÓN DE PRUEBAS DIAGNÓSTICAS
Al igual que el diseño de un ensayo clínico, estos estudios constan de diferentes fases, cada una de las cuales debe responder a una pregunta concreta. Solo se puede pasar a la siguiente fase en el caso de que la respuesta a la cuestión de la anterior sea afi rmativa.2
• Fase I: ¿Los resultados de la prueba diagnóstica en pacientes con la enfermedad difi eren de los de las personas normales (no enfermas)? • Fase II: ¿Son los individuos con determinados resultados de la prueba más propensos a tener la enfermedad que los pacientes con otros resultados de la prueba? • Fase III: ¿El resultado de la prueba diagnóstica discrimina a los individuos con y sin la enfermedad en los individuos en los que es clínicamente razonable sospechar que la enfermedad está presente? • Fase IV: ¿A los pacientes que se someten a esta prueba diagnóstica les va mejor (“consecuencias” en salud) que a pacientes similares que no han sido testados?
ELEMENTOS CLAVE PARA AUMENTAR LA CALIDAD DE LOS ESTUDIOS DE VALIDACIÓN DIAGNÓSTICA
La estructura básica de este tipo de estudio está constituida por las etapas óptimas, el patrón de referencia (gold standard), los posibles tipos de diseño y la descripción del método dirigido a describir de forma adecuada los resultados del estudio. La combinación óptima de estos ingredientes va a condicionar la calidad del estudio al disminuir los posibles sesgos que se producen con más frecuencia.1-2,4-8
Estructura básica
Estamos ante un problema clínico que es posible medir de forma fi able mediante una prueba de referencia o gold standard, pero existe otro test (tal vez más económico o invasivo) que también podría diagnosticar la enfermedad. El objetivo de nuestro estudio será averiguar si la nueva prueba podría ser sustituta de la habitual en todos o algunos casos.
Pasos a seguir en los estudios
Primera: Defi nir de forma clara las pruebas que se van a analizar en el estudio (prueba nueva y el patrón de referencia o gold standard). El patrón de referencia debe ser el mejor método accesible para determinar si un paciente tiene la enfermedad o estado de interés. Como este método muchas veces es invasivo y/o caro, motivo por el cual pretendemos validar otras pruebas, realizar estos patrones a individuos no enfermos suele generar problemas, o no es del todo ético, por lo que estaríamos ante el sesgo de referencia o verifi cación parcial. En algunos estudios se opta por realizar un seguimiento del paciente al no poder realizarse el patrón de referencia, lo que implicaría un posible sesgo de verifi cación diferencial. Ambas soluciones suelen conllevar una sobreestimación del rendimiento diagnóstico de la nueva prueba a validar. En relación con el patrón de referencia, resulta también importante considerar si es capaz de clasifi car el estado de enfermedad en todas las observaciones. En el caso de que existan observaciones con un diagnóstico indeterminado, si éstas son excluidas del análisis se producirán estimaciones sesgadas de las características operativas de la prueba diagnóstica. Este sesgo, conocido como sesgo por exclusión de indeterminados, ocasiona habitualmente sobrestimaciones de la sensibilidad y de la especifi cidad.
Segunda: Enmascarar la interpretación de las pruebas de modo que ambas sean evaluadas de forma independiente y ciegas entre sí. Esta exigencia es tanto más importante cuanto mayor componente de interpretación subjetiva tengan las pruebas. Este paso evita el sesgo de revisión, que puede llevar tanto a sobrestimación como a subestimación.
Tercera: Evitar que la prueba de referencia “dependa de” o “incorpore a” la prueba a analizar, con lo que así evitamos el sesgo de incorporación.
Cuarta: Elegir adecuadamente la población a estudiar. La prueba debe validarse en los individuos en los que se pretende evaluar en el futuro y, si es posible, en una muestra consecutiva de pacientes. También deben incluirse a sujetos con diferentes grados de gravedad de la enfermedad, es decir, no solo los muy enfermos, también los no enfermos y, entre estos, a pacientes en los que la enfermedad entre en el diagnóstico diferencial, y no solo exclusivamente sanos, con lo que disminuiremos el sesgo de espectro.
Quinta: Seleccionar correctamente el diseño de estudio.
Podemos hacerlo a través de una única muestra, mediante un diseño transversal en el que incluiremos a sujetos a los que se les realizaría la prueba. Aunque este diseño tiene la ventaja de que la estimación de la
validez y la exactitud diagnóstica puede hacerse forma directa, su inconveniente es que si la prevalencia es baja se precisan tamaños muestrales grandes.
Otro posible diseño sería seleccionar dos muestras bien a partir del gold standard (diagnóstico defi nitivo) o bien a partir de la prueba a validar. El primero sería un diseño caso-control, siendo los casos los sujetos enfermos (prueba de referencia positiva), y los controles, los sujetos no enfermos, ya que ésta fue negativa. La ventaja principal de este diseño es su mayor potencia, al necesitarse tamaños muestrales más bajos. Sin embargo, como desventajas deben destacar que solo puede estimarse parcialmente la validez diagnóstica –pues puede determinarse de forma directa los valores predictivos que dependen de la prevalencia–, que solo puede calcularse de forma indirecta y que tiende a sobreestimar el verdadero valor de la prueba a validar.
Si partimos del resultado de la nueva prueba, hablaríamos de un estudio de cohortes, una en la que el resultado fue positivo y otra en la que fue negativo. Sus ventajas e inconvenientes son contrarios a los de caso-control, pudiendo calcularse solamente la sensibilidad y la especifi cidad de forma directa y precisando tamaños muestrales amplios en el caso de una prevalencia de enfermedad baja. Al comprobar solo los casos positivos, incurriríamos en el sesgo de verifi cación o confi rmación.
Sexta: Describir con detalle los métodos realizados para permitir su reproducción en otro entorno y los términos de normalidad y anormalidad que se van a considerar y el análisis de la interpretación de los resultados.
Séptima: Tener en cuenta los posibles resultados del estudio, que serán la capacidad discriminatoria de la prueba entre enfermos y no enfermos (sensibilidad y especifi cidad), el rendimiento de la misma (valores predictivos) o evaluar la utilidad y posible satisfacción de la prueba diagnóstica. La utilidad de una prueba dependerá de la capacidad para producir los mismos resultados cada vez que se aplica en condiciones similares (fi abilidad) y de que sus mediciones refl ejen exactamente el fenómeno que se intenta medir (validez y exactitud), pero también de su rendimiento clínico y de su coste.
Por su importancia, sobre todo en la evaluación de los estudios de validación de pruebas diagnósticas, este aspecto se comenta de forma más detallada en el siguiente apartado.
En la fi gura 2.7.1 se representa un esquema de los pasos clave de un estudio de validación de una prueba diagnóstica.
VALIDEZ, FIABILIDAD Y PRECISIÓN DE LOS ESTUDIOS DE PRUEBAS DIAGNÓSTICAS
Validez
Para evaluar la validez de una prueba diagnóstica se requiere un patrón de referencia. El grado de acuerdo entre la prueba diagnóstica y el patrón de referencia puede ser representado en una tabla de contingencia (2x2), de donde se obtiene, entre otros, la sensibilidad (S), especifi cidad (E), valor predictivo positivo (VPP) y valor predictivo negativo (VPN). En la tabla 2.7.1 se expresan los indicadores de validez de una prueba diagnóstica a través de los resultados de un estudio hipotético sobre la utilidad de una prueba diagnóstica utilizando su patrón de referencia, tomado de la referencia 4.
La S considera la validez de la prueba entre los enfermos (corresponde a la probabilidad de que la prueba sea positiva en sujetos con la enfermedad) y la E la validez de la prueba entre los sanos (corresponde a la
probabilidad de que el resultado de la prueba sea negativo en sujetos sanos). Los valores complementarios de la S y la E corresponden a las probabilidades de los resultados falsos negativos (FN) y falsos positivos (FP), respectivamente. Cuando una prueba tiene una S muy alta, un resultado negativo sirve para descartar la enfermedad (la probabilidad de un FN es muy pequeña); si la prueba tiene una E muy alta, un resultado positivo es prácticamente diagnóstico (la probabilidad de un FP es muy pequeña).4-5
El VPP es la probabilidad de encontrar la enfermedad en individuos con un resultado positivo de la prueba. El VPN es la probabilidad de no encontrar la enfermedad en individuos con un resultado negativo de la prueba. S y E son propiedad intrínseca de las pruebas diagnósticas, que presentan una relación recíproca entre ambas y cuyos resultados son independientes de la prevalencia de la enfermedad, a diferencia de los valores predictivos. Así, cuanto mayor sea la prevalencia de la enfermedad, mayor será el VPP y menor el VPN; los valores predictivos tienen una utilidad postprueba.2,4-5
Las características operativas tradicionales de las pruebas diagnósticas –la S, la E, el VPP y el VPN– tienen algunas limitaciones para su uso en la práctica clínica diaria. La S y la E no brindan información sufi ciente para tomar decisiones junto al paciente, y consideradas aisladamente, es imposible concluir si un paciente tiene o no una determinada enfermedad. La interpretación del resultado de una prueba, en el contexto del diagnóstico, requiere el uso de los valores “predictivos”, y éstos, a su vez, pueden tener valores diferentes en cada escenario clínico de acuerdo con la prevalencia o probabilidad previa de la enfermedad.
Como alternativa para vencer esas limitaciones, en la literatura médica de los últimos años se ha propuesto otra herramienta con algunas ventajas prácticas para la evaluación y utilización de las pruebas diagnósticas: el cociente de probabilidades (CP, Likelihood Ratio en inglés). El CP compara la probabilidad de encontrar el resultado de la prueba (positivo, negativo, o cualquiera que éste sea) en personas enfermas, con la probabilidad de encontrar ese mismo resultado en personas sin la enfermedad. Cuando el resultado de la prueba en estudio es positivo, este resultado debe ser más frecuente en los enfermos que en los no enfermos; y cuando el resultado de la prueba es negativo, éste debe ser más frecuente en los no enfermos que en los enfermos. De este modo, el CP dice cuántas más –o menos– veces es frecuente el resultado de una prueba en personas enfermas comparado con personas sin la enfermedad.4-5
Para la situación más simple, tenemos dos tipos: cociente de probabilidades de un resultado positivo (CP+) y cociente de probabilidades de un resultado negativo (CP-) (Tabla 2.7.2).
1). Dado que los CP relacionan la S y la E, no varían con la prevalencia. Los CP adoptan valores entre 0 e infi nito, siendo el valor nulo el 1, y cuya interpretación se valora según los criterios expresados en la tabla 2.8.2. El CP es una medida de gran utilidad en la práctica clínica. Su principal utilidad es permitir calcular la probabilidad postprueba (Ppost = probabilidad de que un sujeto tenga la enfermedad si obtiene un resultado positivo en la prueba o VPP ajustado) a partir de cualquier probabilidad preprueba (Ppre = probabilidad de que un sujeto tenga la enfermedad antes de aplicarle la prueba) o prevalencia, y de esta forma evaluar cuánto puede ganarse si se realiza. El objetivo de la realización de la prueba diagnóstica es, una vez conocido el resultado, modifi car esta Ppre hasta obtener una Ppost; la magnitud y dirección de ese cambio va a depender de las características operativas de la prueba diagnóstica, pero en todo caso debemos tener en cuenta que el punto de partida, la Ppre, va a resultar muy importante en ese proceso.4,6
Para operar con los CP en el cálculo de probabilidades, éstas deben transformarse en ventajas (odds) (Tabla 2.7.1); los pasos a seguir son: 1) transformar la Ppre en odds preprueba = Ppre/1-Ppre; 2) obtener la odds postprueba = CP+ x odds preprueba; 3) obtener la Ppost= odds postprueba/1 + odds postprueba. La diferencia que exista entre la Ppre y Ppost informa de la utilidad de una determinada prueba diagnóstica; a mayor diferencia entre una y otra probabilidad, mayor contribución de la prueba al proceso diagnóstico.
Afortunadamente, existe un modo más simple de realizar estos cálculos, como es el Nomograma de Fagan6,8 (Figura 2.7.2). Existen muchas calculadoras web y aplicativos para dispositivos móviles que permiten realizar estos cálculos de forma directa.
Todos estos estimadores de validez pueden ser aplicados a pruebas con resultados discretos con más de dos categorías e incluso a resultados expresados en variables continuas. En este caso, podemos establecer puntos de corte para convertir el resultado en una variable discreta con dos o más categorías. Otra alternativa que permite explorar la capacidad diagnóstica de una prueba en sus distintos valores son las curvas ROC (iniciales del término inglés Receiver Operating Characteristics), con las que podemos conocer su validez global y seleccionar el punto o puntos de corte más adecuados. La curva ROC es un gráfi co en el que se observan todos los pares S/E resultantes de la variación continua de los puntos de corte en todo el rango de resultados observados. En el eje Y de coordenadas se sitúa la S o fracción de VP, defi nida como se expuso anteriormente y calculada en el grupo de enfermos (Figura 2.7.3). En el eje X se sitúa la fracción de FP o 1-especifi cidad, defi nida como FP/VN + FP y calculada en el subgrupo no afectado. Cada punto de la curva representa un par S/1-E correspondiente a un nivel de decisión determinado. Una prueba con discriminación perfecta, sin solapamiento de resultados en las dos poblaciones, tiene una curva ROC que pasa por la esquina superior izquierda, donde S y E toman valores máximos (S y E = 1). Una prueba sin discriminación, con igual distribución de resultados en los dos subgrupos, da lugar a una línea diagonal de 45º, desde la esquina inferior izquierda hasta la superior derecha. La mayoría de las curvas ROC caen entre estos dos extremos. Si cae por debajo de la diagonal de 45º, se corrige cambiando el criterio de positividad de “mayor que” a “menor que”, o viceversa. De lo anterior se desprende que el área bajo la curva ROC (ABC) se puede utilizar como un índice conveniente de la exactitud global de la PD, donde la validez máxima corresponde a un valor de ABC de1 y la mínima a uno de 0,5. El uso de estas gráfi cas aporta también la ventaja de permitir comparar visualmente de forma directa el poder discriminativo de dos pruebas diferentes4-5 (Figura 2.7.3).
Fiabilidad
La fi abilidad de una prueba viene determinada por la estabilidad de sus mediciones cuando se repite en condiciones similares. La variabilidad de las mediciones va a estar infl uida por múltiples factores que interesa conocer y controlar. Entre ellos, tiene especial importancia distinguir las variaciones de interpretación intraobservador e interobservador. La fi abilidad puede ser evaluada para resultados discretos nominales mediante el índice kappa (IK), para resultados discretos ordinales mediante el índice kappa ponderado y para resultados continuos mediante el coefi ciente de correlación intraclase y el método de Bland-Altman. El IK nos ofrece una estimación del grado de acuerdo no debido al azar a partir de la proporción de acuerdo observado (Po) y la proporción de acuerdo esperado (Pe) = Po-Pe/ 1-Pe. El IK puede adoptar valores entre -1 y + 1: es 1 si existe un acuerdo total, 0 si Po es igual a Pe, y menor de 0 si Po es inferior a Pe por azar.4,6 Para su interpretación, diferentes autores han propuesto varias clasifi caciones (Figura 2.7.4).
Precisión
Al igual que en otros tipos de estudios, la valoración de la validez de las pruebas diagnósticas se hace sobre muestras, por lo que los resultados obtenidos son sólo estimaciones puntuales, sujetas a variabilidad aleatoria, y por lo tanto deben expresarse con sus intervalos de confi anza (IC). Estos IC tendrán que ser aplicados en el cálculo de la Ppost para poder juzgar la utilidad de la prueba diagnóstica.
Como se comentaba en la introducción, con la investigación en pruebas diagnósticas, a pesar de ser un campo muy dinámico debido al desarrollo tecnológico, no ha ocurrido lo mismo que en los estudios sobre intervenciones terapéuticas que siguen un cuidado y detallado proceso marcado por las agencias reguladoras en el que se suceden estudios de complejidad creciente. Al no producirse este razonable control, han surgido iniciativas para corregir la situación. Algunas han ido dirigidas a que el proceso de validación de pruebas diagnósticas sea un proceso conceptual y paralelo al proceso de evaluación de fármacos con estudios preliminares o exploratorios, de validación retrospectiva y prospectiva, y, por último, de impacto y utilidad.1-2
Otras iniciativas van dirigidas a los editores de las revistas y los autores e incluyen unos listados de puntos que resultan esenciales para que se facilite la identifi cación de posibles sesgos del estudio (atentados a la validez interna) y se puedan valorar en su justa medida la aplicabilidad y la posibilidad de generalizar los resultados (validez externa). Una de ellas ha partido de un grupo de trabajo de la Colaboración Cochrane y tiene como fi n mejorar la calidad de las pruebas diagnósticas. Para ello, este grupo recomienda que los artículos que se publiquen reúnan unos STARD (Standars for Reporting of Diagnosis Accuracy), y proponen un listado de 25 elementos de información referidos a elementos de diseño del estudio, del test de evaluación y de sus resultados2,9 (Tabla 2.7.3).
Por último, existen iniciativas que, aunque en principio se dirigieron a los investigadores que realizan revisiones sistemáticas de estudios de validez diagnóstica, indirectamente también se dirigen a quienes realizan la investigación primaria, como la norma QUADAS1,10 (Tabla 2.7.4). Otra herramienta útil podría ser el Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD),11 publicado de forma simultánea en 11 revistas y a cuyas listas de comprobación es posible acceder a través del link https://www.equator-network.org/reporting-guidelines/tripod-statement/.
Estos listados proporcionan un método muy útil para medir la calidad de los distintos estudios y la detección de posibles errores y deben ser sistemáticamente utilizados por los investigadores que realizan estudios de validación diagnóstica.
PUNTOS CLAVE
• Un estudio de pruebas diagnósticas debe seguir una serie de fases, cada una de las cuales debe responder a una pregunta concreta sobre los resultados e individuos a analizar. Solo puede pasarse a la siguiente fase en el caso de que la respuesta a la cuestión de la anterior sea afi rmativa. • Los elementos clave para aumentar la calidad de los estudios de validación diagnóstica son la estructura básica de este tipo de estudios, las etapas óptimas, el patrón de referencia (gold standard), los posibles tipos de diseño y la descripción del método dirigido a describir de forma adecuada los resultados del estudio. La combinación óptima de estos ingredientes va a condicionar la calidad del estudio, pues disminuirá los sesgos que se producen con más frecuencia. • En este tipo de estudios debe analizarse su validez (sensibilidad, especifi cidad, valores predictivos y cocientes de probabilidad), fi abilidad (índice Kappa o coefi ciente de correlación intraclase) y precisión (intervalos de confi anza). • Existen una serie de listados y herramientas muy útiles para medir la calidad de los distintos estudios y la detección de posibles errores, y se recomienda su utilización sistemática en las investigaciones de validación diagnóstica.
BIBLIOGRAFÍA
1. Abraira V, Zamora J. Criterios de calidad de los estudios de pruebas diagnósticas. FMC. 2008;15:460461. 2. Rodríguez Álvarez MX. Diseñando un estudio de pruebas diagnósticas. Itinerario de Investigación e
Innovación Biosanitaria. FEGAS/SERGAS. En: http://fegasmultimedia.sergas.es/default.aspx?action=play&conferenceGUID=8b18ce66-1722-40a6-86b7-10e89b5d20cb. Consultado el 1 de diciembre de 2019. 3. Carnero-Pardo C. Evaluación de pruebas diagnósticas. Rev Neurol. 2005;40:641-643. 4. Ochoa Sangrador C, González de Dios J, Buñuel Álvarez JC. Evaluación de artículos científi cos sobre pruebas diagnósticas. Evid Pediatr. 2007;3:24. 5. Jaeschke R, Guyatt G, Sackett DL. Users’ guides to the medical literature. III. How to use an article about a diagnostic test. A. Are the results of the study valid? Evidence-Based Medicine Working Group.
JAMA. 1994;271:389-391. 6. Jaeschke R, Guyatt GH, Sackett DL. Users’ guides to the medical literature. III. How to use an article about a diagnostictest. B. What are the results and will they help me in caringfor my patients? Th e Evidence-Based Medicine Working Group. JAMA. 1994;271:703-707. 7. Grimes DA, Schulz Kf. Refi ning clinical diagnosis with likelihood ratios. Lancet. 2005;365:1500-1505. 8. Fagan TJ. Letter: Nomogram for Bayes theorem. N Engl J Med. 1975;293:257. 9. Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, et al. Towards complete and accurate reporting of studies of diagnostic accuracy: Th e STARD Initiative. Ann Intern Med. 2003;138: 40-44. 10. Whiting P, Rutjes AW, Reitsma JB, Bossuyt PM, Kleijnen J. Th e developmentof QUADAS: a tool for the quality assessment of studies ofdiagnostic accuracy included in systematic reviews. BMC Med Res Methodol. 2003;3:25. 11. Moons KG, Altman DG, Reitsma JB, Ioannidis JP, Macaskill P, Steyerberg EW, et al. Transparent Reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): Explanation and Elaboration. Ann Intern Med. 2015;162:W1-W73.
TABLAS Y FIGURAS
Tabla 2.7.1. Medidas de utilidad de una prueba diagnóstica. Resultados de un estudio hipotético de la utilidad de una prueba diagnóstica, utilizando su patrón de referencia
Prueba de referencia
Prueba diagnóstica Positiva Negativa
Positiva 225 VP (a) 25 FP (b) 250 (a+b)
Negativa 100 FN (c) 400 VN (d) 500 (c+d)
325 (a+c) 425 (b+d) 750 (a+b+c+d)
(a) Verdaderos positivos (VP): enfermos con la prueba positiva (b) Falsos positivos (FP): no enfermos con la prueba positiva (c) Falsos negativos (FN): enfermos con la prueba negativa (d) Verdaderos negativos (VN): no enfermos con la prueba negativa a+c: casos con patrón de referencia positivo (enfermos) b+d: casos con patrón de referencia negativo (no enfermos) a+b: casos con la prueba diagnóstica positiva c+d: casos con la prueba diagnóstica negativa Sensibilidad (S) = a/(a+c) = 225/325 = 69,2% Especifi cidad (E) = d/(b+d) = 400/425 = 94,1% Falsos negativos (FN) = 1-S = 30,8% Falsos positivos (FP) = 1-E = 5,9% Valor predictivo positivo (VPP) = a/(a+b) = 225/250 = 90,0% Valor predictivo negativo (VPN) = d/(c+d) = 400/500 = 80,0% Cociente de probabilidad positivo (CP+) = S/(1-E) = 69,2/5,9 = 11,7 Cociente de probabilidad negativo (CP-) = 1-S/E = 30,8/94,1 = 0,32 Prevalencia o probabilidad preprueba (Ppre) = (a+c)/(a+b+c+d) = 325/750 = 43,03% Odds preprueba = Ppre/(1-Ppre) = 43,3/56,7 = 0,76 Odds postprueba = CP+ x odds preprueba = 11,7 x 0,76 = 8,89 Probabilidad postprueba (Ppost) = odds postprueba/(odds postprueba + 1) = 8,89/9.89 = 89,8%
Tabla 2.7.2. Interpretación de los cocientes de probabilidad
Valor del CP + y CP- Interpretación CP+ >10 ó CP- <0,1 Generan cambios amplios y a menudo concluyentes desde una probabilidad preprueba hasta una probabilidad postprueba CP+ 5-10 y CP- 0,1-0,2 Generan cambios moderados desde la probabilidad preprueba hasta la probabilidad postprueba CP+ 2-5 y CP- 0,5 – 0,2 Generan cambios pequeños (pero en ocasiones pueden ser importantes) de la probabilidad CP+ 1-2 y CP- 0,5-1 Alteran la probabilidad en un grado insignifi cante (y rara vez importante)
CP+: cociente de probabilidad positivo; CP-: cociente de probabilidad negativo
Tabla 2.7.3. Lista STARD para el informe de los estudios de precisión diagnóstica
TÍTULO / RESUMEN 1 El artículo se identifi ca como un estudio de VD INTRODUCCIÓN 2 Consta que el objetivo es estimar la VD o compararla con la de otro test
MÉTODOS
Participantes 3 Describe la población estudiada y los criterios de selección y exclusión 4 Describe el criterio de reclutamiento: síntomas, otros test, hacer el test actual
5 Describe el muestreo: consecutivo, aleatorio, no especifi cado 6 Describe la recogida de datos: prospectivo o retrospectivo
Métodos de las pruebas
7 Describe la prueba de referencia y su justifi cación
8 Describe o referencia las especifi caciones técnicas y como tomar las medidas
9 Describe las unidades de medida, puntos de corte o categorías de los r esultados
10 Describe el número, entrenamiento y experiencia de los que realizan el test 11 Describe si la aplicación dela prueba y del test de referencia se realizó de forma ciega Métodos estadísticos 12 Describe los métodos para calcular o comparar la VD y el grado de incertidumbre
13 Describe cálculo de la fi abilidad y cómo se ha hecho
RESULTADOS
Participantes 14 Informa cuando se realizó el estudio (fecha de inicio y fi nalización del r eclutamientende
15 Informa de los caracteres demográfi cos y clínicos 16 Informa de las razones para la pérdida (diagrama de fl ujo)
Resultados de las pruebas
17 Informa del tiempo y circunstancias (tratamiento) entre el test y el estándar
18 Informa del grado de afectación de los enfermos y del diagnóstico de los no enfermos
19 En caso de valores discretos hay tabla 2x2 con los resultados. En caso de valores continuos se informa de la distribución. Se describe los resultados perdidos e indeterminados 20 Se describen los efectos adversos del test y del estándar Estimación 21 Se describe la VD y su incertidumbre estadística (p. ej. IC 95%) 22 Informa como se ha manejado los perdidos, indeterminados o extremos 23 Informa de la variabilidad de la VD entre evaluadores, centros y subgrupos 24 Informa sobre la fi abilidad en el caso de que se haya evaluado DISCUSIÓN 25 Discute la aplicabilidad clínica de los hallazgos del estudio
2.7. Validación de pruebas diagnósticas
Tabla 2.7.4. Ítems de la herramienta QUADAS
1. ¿Es representativa la muestra? 2. ¿Se describen bien los criterios de selección? 3. ¿Clasifi ca bien el patrón de referencia? 4. Si el patrón y la prueba no se aplican simultáneamente, ¿es razonable pensar que la enfermedad no haya evolucionado en ese tiempo 5. ¿Sesgo de verifi cación parcial? 6. ¿Sesgo de verifi cación diferencial? 7. ¿Es el patrón de referencia independiente de la prueba? (p. ej., la prueba no forma parte del patrón) 8. Detalle de la ejecución de la prueba 9. Detalle de la ejecución del estándar 10. ¿Se interpretó la prueba sin conocer el resultado del patrón? 11. ¿Se interpretó el patrón sin conocer el resultado de la prueba? 12. ¿La información clínica disponible en la interpretación de la prueba es la misma que habrá cuando se use en la práctica? 13. ¿Se informa de los resultados no interpretables? 14. ¿Se explican las pérdidas?
Figura 2.7.1. Esquema de los pasos claves de un estudio de evaluación de los estudios de pruebas diagnósticas

Figura 2.7.2. Nomograma de Fagan para la interpretación de los cocientes de probabilidad

Figura 2.7.3. Curvas ROC. Ejemplos de dos pruebas con diferente nivel de discriminación
Ejemplo 1: Sabemos que la probabilidad preprueba de una determinada prueba diagnóstica es del 10% y el cociente de probabilidad negativa es de 0,2. El resultado negativo de esta prueba haría que la probabilidad postprueba fuera del 2% (línea continua azul).
Ejemplo 2: Sabemos que la probabilidad preprueba de una determinada prueba diagnóstica es del 30% y el cociente de probabilidad negativa es de 0,05. El resultado negativo de esta prueba haría que la probabilidad postprueba fuera del 2% (línea discontinua roja).

Figura 2.7.4. Clasifi caciones propuestas para la interpretación del Índice de Kappa

2.8. VALIDACIÓN DE CUESTIONARIOS
Dr. Ignasi Serra Pons Dra. Judith García Aymerich
INTRODUCCIÓN
El cuestionario es una de las técnicas más utilizadas en el campo de la investigación para la recogida de datos. Es una herramienta económica que nos permite recoger información relevante del aspecto de salud que queremos estudiar. Esta información será más o menos útil en función de la calidad del cuestionario. Por esa razón, es de vital importancia su validación. Si se utiliza información extraída de un cuestionario de salud que no haya sido validado, las conclusiones obtenidas de esos datos carecerán de un valor y no las podremos creer. No será una buena herramienta.
La validación de cuestionarios es un tema complejo en el campo de la salud. Su principal complejidad viene dada por el objetivo que tienen este tipo de herramientas: medir de forma objetiva una característica (dimensión) o características de salud que provienen de una defi nición conceptual. Por ejemplo: la ansiedad, el bienestar físico, la calidad de vida, la cantidad de actividad física, el nivel nutricional… En todos los casos pretendemos cuantifi car un concepto no observable directamente, tenemos una idea subjetiva de lo que queremos. El cuestionario es el instrumento que consigue acercarnos a esta idea empíricamente.
Es difícil desarrollar una herramienta que nos proporcione una escala de valoración cuantitativa a partir de ideas, actitudes, sentimientos y percepciones personales. En consecuencia, valorar la calidad de estos instrumentos resulta complejo. Lo que está claro es que, cuanto más precisos y minuciosos seamos en todos los pasos de elaboración de un cuestionario, éste será de más calidad, y por lo tanto, será más válido.
En general, para desarrollar una herramienta válida y fi able es necesario tener en cuenta dos fases: la elaboración del cuestionario y su validación (Figura 2.8.1). Sin embargo, cabe mencionar que la validación es un aspecto a considerar en todo el proceso de desarrollo de la herramienta, desde la elaboración de cada uno de los ítems hasta conseguir una escala útil para medir el constructo del estado de salud en el cual estamos interesados. En este capítulo desarrollamos los conceptos y métodos más importantes para realizar la validación de cuestionarios durante todo el proceso.
ELABORACIÓN DE UN CUESTIONARIO
En la primera fase se defi ne el aspecto a medir o constructo1 y sus dimensiones (puede tener más de una dimensión) y se elaboran las preguntas o ítems del cuestionario. Esta fase suele ser costosa, pues requiere una investigación cualitativa, varias revisiones bibliográfi cas y el contacto con expertos en la materia de interés. Se recomienda formar un comité lo más multidisciplinar posible que reúna aspectos de salud, metodológicos y de redacción. A continuación se detallan los puntos a defi nir claramente.
(1) El objetivo, el aspecto a medir (constructo) y sus dimensiones. Hay que llegar a una idea clara de lo que se quiere medir y para qué. Un mismo constructo se puede defi nir de varias formas. Hay que consensuar una defi nición lo más precisa posible del fenómeno que queremos cuantifi car y si lo que medimos tiene una sola dimensión o varias. Por ejemplo, el estudio PROactive pretendía desarrollar un cuestionario para medir la experiencia de actividad física de los pacientes con enfermedad pulmonar obstructiva crónica (EPOC). Tras la investigación cualitativa (entrevistas individuales y grupos focales, ambos con pacientes y expertos), se identifi có que el constructo “experiencia de actividad física” era descrito por los pacientes en tres dimensiones: “cantidad de actividad física”, “síntomas experimentados durante la actividad física” y “adaptaciones realizadas para facilitar la actividad física”.13
(2) La población a quien va dirigida. Hay que delimitar la población objetivo en la cual administraremos la herramienta. Según la población objetivo, las preguntas del cuestionario y su enfoque serán distintos aunque pretendan medir el mismo constructo. Por ejemplo, no es lo mismo evaluar el nivel de actividad física en pacientes con EPOC mayores de 45 años que en adolescentes que van al instituto. Estas dos poblaciones son muy distintas y, por lo tanto, el enfoque del cuestionario también será diferente.
(3) La forma de administración. Según el constructo y la población objetivo, podemos administrar el cuestionario de varias formas: telefónicamente, autoadministrado, a modo de entrevista personal o a través de plataformas digitales. Por ejemplo, en una población mayor y con problemas de salud, probablemente sea más fácil recoger la información a través de una entrevista personal en el hospital que utilizando nuevas tecnologías que requieran cierto nivel de práctica y entrenamiento. Utilizando una forma u otra podemos obtener información de calidad distinta.
(4) Los ítems. Crear los ítems implica defi nir el formato, la redacción, el orden de los mismos, la codifi cación de las respuestas y la puntuación de cada una de ellas para generar una escala clara. El ítem es la unidad primaria del cuestionario. Consiste en una pregunta con una respuesta cerrada con diferentes opciones. Cada opción de respuesta tiene un valor que contribuye a la puntuación de la escala fi nal. El diseño, la redacción y el orden de los ítems dependerán del constructo a medir, de sus dimensiones y de la forma de administración.
(5) Prueba piloto. Antes de la siguiente fase, se realiza una primera valoración de la herramienta para, si es preciso, mejorarla. En este punto se valora la viabilidad (feasibility, en inglés) del cuestionario con una muestra de unos 30-40 participantes con los mismos criterios de inclusión que la población objetivo. Este número puede variar en función del tipo de cuestionario elaborado. El objetivo es hacer un informe con las principales difi cultades de comprensión del cuestionario entrevistando personalmente a los participantes. Se valorará la calidad de las preguntas, el tiempo utilizado en responderlas, la facilidad de entendimiento, el formato de administración y otros aspectos cualitativos del instrumento. La viabilidad es una validación cualitativa del cuestionario antes de su redacción fi nal.
En esta primera fase de elaboración del cuestionario se utilizan diversas técnicas estadísticas. Las más relevantes para los investigadores clínicos son el análisis factorial y los modelos de la teoría de respuesta de los ítems.
El primero, el análisis factorial, nos indica si hemos defi nido correctamente las dimensiones del constructo de interés. Es un análisis que nos muestra la variabilidad capturada por todos los ítems dentro de las diferentes dimensiones teóricas que hemos usado para defi nir el constructo. Nos crea unos factores latentes que deberían representar las dimensiones teóricas descritas en la primera fase. Por ejemplo, en el estudio PROactive se crearon los ítems correspondientes a las dimensiones etiquetadas por los pacientes como “cantidad de actividad física”, “síntomas” y “adaptaciones”. En el siguiente paso se realizó el análisis factorial con los resultados de los ítems medidos en 236 pacientes con EPOC. Se identifi có que los ítems del cuestionario se distribuían en dos dimensiones, “cantidad de actividad física” (correspondiente a los ítems desarrollados con los pacientes en la dimensión “cantidad”) y “difi cultad con la actividad física” (incluyendo los ítems de las dimensiones “síntomas” y “adaptaciones”).14 En este caso, el análisis factorial sugirió que la mejor manera de agrupar la información era en dos factores independientes (las dos dimensiones) y nos informó de los ítems asociados a cada una de ellas así como de la variabilidad explicada por estos ítems en la dimensión a la cual pertenecían.
El segundo, la teoría de respuesta de los ítems, nos permite estudiar las propiedades de cada uno de los ítems según la probabilidad de sus respuestas. En función de las probabilidades de respuesta en cada categoría de cada ítem podemos reducir el número de ítems del cuestionario fi nal e incluso agrupar categorías que se solapan dentro de un ítem determinado. Por ejemplo, en la dimensión “difi cultad de actividad física” del estudio PROactive se partió de 26 ítems iniciales, y después, mediante análisis de teoría de respuesta de los ítems, se mejoró gradualmente el cuestionario reduciendo esta dimensión a tan sólo 5 ítems. Se consiguió omitir ítems redundantes que explicaban la misma información que otros dentro de la propia dimensión “difi cultad”.
VALIDACIÓN DEL CUESTIONARIO
Una vez elaborado el instrumento que cuantifi ca el constructo defi nido, debemos analizar sus principales propiedades psicométricas: la fi abilidad, la validez y la respuesta al cambio.2 Los conceptos de validez y fi abilidad, así como el error aleatorio y sistemático, a los cuales también haremos referencia, se presentan en este mismo capítulo. La respuesta al cambio es una propiedad de los cuestionarios que algunos autores analizan separadamente de la validación. Nosotros la presentamos conjuntamente por cuestiones pedagógicas.
Es importante mencionar que la bibliografía está repleta de múltiples teorías e índices estadísticos para valorar las propiedades psicométricas de los cuestionarios. En este capítulo se muestran las principales y más utilizadas que todo investigador clínico debe conocer. A partir de aquí, se puede afi nar más o menos según las características propias de cada estudio.
Fiabilidad
La fi abilidad (reliability en inglés) es la capacidad del instrumento para proporcionar medidas con precisión, es decir, con el mínimo error posible. La fi abilidad está sometida solamente al error aleatorio y mide la proporción de la variabilidad en las mediciones debida únicamente a diferencias entre los individuos.1,3 La fi abilidad se valora a través de la coherencia interna, la fi abilidad intra-observador o test-retest y la fi abilidad inter-observador.
Coherencia interna
La coherencia interna de un cuestionario valora en qué medida los ítems que lo forman están relacionados entre sí y, por lo tanto, en qué medida presentan una homogeneidad común para medir el constructo de interés. Si el cuestionario está formado por ítems que miden diferentes dimensiones del mismo constructo,
los ítems relacionados con cada una de las dimensiones deben presentar una buena homogeneidad entre ellos. Esta homogeneidad se mide con el estadístico Alfa de Cronbach, un índice con valores entre 0 y 1. Se considera que el instrumento tiene una buena coherencia interna a partir del valor 0,7, siendo aconsejable tener valores comprendidos entre 0,7 y 0,9.4 Los valores mayores de 0,9 indican una coherencia muy buena, pero también pueden indicar la presencia de algún ítem redundante, es decir, la existencia de un ítem que mide exactamente lo mismo que otro. Por ejemplo, en la versión diaria del cuestionario PROactive de actividad física mencionado anteriormente5 los cuatro ítems de la dimensión “cantidad” muestran un Alfa de Cronbach de 0,77, y los cinco ítems de la dimensión “difi cultad” toman valores de Alfa de Cronbach de 0,85. Podemos considerar entonces que el cuestionario tiene una buena coherencia interna.
Fiabilidad intra-observador (test-retest)
El test-retest se refi ere a la concordancia obtenida en la puntuación de nuestro constructo de interés al repetirlo en dos o más momentos diferentes en el tiempo, es decir, analizamos la repetibilidad de la herramienta. Si el cuestionario tiene una escala cuantitativa, realizamos el análisis mediante el coefi ciente de correlación intraclase (CCI).6 Es difícil valorar la correcta aplicabilidad de esta técnica porque el tiempo que debe pasar entre las dos administraciones del cuestionario no puede ser ni muy largo ni muy breve (habitualmente se consideran dos semanas). Si el tiempo es demasiado breve, nos veremos infl uenciados por el efecto recordatorio, y si es demasiado largo, pueden haber cambiado algunas condiciones del fenómeno medido. El CCI es un estadístico que se basa en la ANOVA de medidas repetidas y se calcula mediante el cociente de la variación debida a las dos medidas en el tiempo entre la variación total de las medidas. Es de fácil interpretación, ya que se parece a un índice de correlación, y su valor oscila entre 0 y 1. Son deseables CCI’s superiores a 0,8.4 Una alternativa (o complemento) a este índice es el método gráfi co de Bland y Altman,7 en el que se representa la diferencia entre las dos medidas contra la media de ambas. Por ejemplo, la dimensión “difi cultad” del cuestionario PROactive5 tiene un CCI = 0,87 y el Bland-Altman muestra diferencias prácticamente inexistentes entre las dos administraciones para todos los valores del cuestionario.
Finalmente, si la escala medida es una escala cualitativa, lo cual sucede con mucha menos frecuencia, analizaremos la repetibilidad a través del índice de concordancia Kappa de Cohen.8
Fiabilidad inter-observador
La fi abilidad inter-observador analiza el grado de acuerdo entre dos o más evaluadores diferentes. Utilizamos también los métodos ICC, Bland Altman y el índice de Kappa de Cohen según sea la escala de medida, cuantitativa o cualitativa. Este tipo de fi abilidad no es evaluable en cuestionarios auto administrados, en los que no existe evaluador.
Validez
La validez es la capacidad del instrumento de medir el constructo para el que ha sido elaborado. Por ejemplo, un cuestionario elaborado para medir la ansiedad debe medir la ansiedad y no la depresión. La validez está sometida al error sistemático, es decir, cuanto menos error sistemático tenga el instrumento, más válido va a ser. Es la característica que nos permite hacer buenas inferencias de las medidas del cuestionario al constructo de interés. Para estudiar la correcta validación de los cuestionarios debemos analizar sus cuatro dimensiones: validez aparente o lógica, de contenido, de constructo y de criterio.
La validez aparente y la de contenido se evalúan de forma cualitativa, básicamente en la primera fase de elaboración del cuestionario, y se revisa después de la prueba piloto. Esta evaluación es subjetiva y requiere revisiones bibliográfi cas y expertos en el tema de interés. En este capítulo nos centramos en la validez de
constructo y de criterio, ya que son las más relevantes para el investigador clínico y se evalúan de forma objetiva mediante estadísticos conocidos.
Validez de constructo
La validez de constructo evalúa de forma empírica y objetiva si las mediciones obtenidas a partir de la escala generada del fenómeno de interés se pueden considerar válidas y útiles para cuantifi carlo. Una técnica utilizada para analizar este tipo de validez es el análisis factorial confi rmatorio. Este método parte de un modelo teórico en el que se defi nen los diferentes factores (dimensiones del fenómeno de interés) y verifi ca que cada factor se corresponda con las dimensiones defi nidas a priori. Cuantifi ca la relación que existe entre los ítems y las dimensiones del constructo.
Otra manera más fácil de analizar la validez de constructo es a través de la validez convergente y de la validez discriminante, ambas basadas en el cálculo de correlaciones.
La validez convergente estudia la relación entre la escala creada y otras mediciones que ya sabemos que son válidas y están relacionadas con el fenómeno que estudiamos.1-3 Por ejemplo, para analizar la validez de constructo del cuestionario PROactive de actividad física5 calculamos la correlación entre la escala (y subescalas) del cuestionario y las variables “distancia recorrida en la prueba de la marcha” y “escala de disnea mMRC”, ya que existe abundante literatura que demuestra que, en pacientes con EPOC, la actividad física se relaciona con estos parámetros.9 El grado de validez del cuestionario se determina al comparar estas correlaciones con las esperadas por conocimiento experto o por la literatura. Se esperan siempre correlaciones menores de 0,9, ya que si son mayores se considera que nuestra escala mide exactamente lo mismo que la medición externa.4
De la misma forma, la validez discriminante estudia la relación entre la escala creada y otras variables que ya sabemos que no tienen relación alguna con nuestro constructo. Se considera que las correlaciones deben ser < 0,3. Siguiendo con el ejemplo anterior, para medir la validez discriminante del cuestionario PROactive5 se calculó la correlación entre el mismo y las variables “altura” y “presión arterial sistólica” del paciente, ya que sabemos que no están relacionadas con la actividad física. Los resultados obtenidos fueron de correlaciones inferiores a 0,3.
Validez de criterio
La validez de criterio se analiza comparando el instrumento nuevo con un estándar de referencia ampliamente validado y reconocido. Se usará como criterio externo ya validado.10 Para evaluar la validez de criterio, se requiere administrar el cuestionario y también el estándar de referencia a los mismos sujetos, como si fueran medidas independientes. Dependiendo del tipo de escala y de las variables medidas, se usarán coefi cientes de correlación (variables cuantitativas) o los índices de sensibilidad y especifi cidad (variables cualitativas). Si las dos escalas están correlacionadas entre sí, podemos decir que nuestro cuestionario tiene validez de criterio.
Por ejemplo, para validar el cuestionario Yale de actividad física se compararon sus resultados con los resultados de un acelerómetro, utilizando tanto coefi cientes de correlación (entre otros, para la medida de tiempo en actividad física) como los índices de sensibilidad y especifi cidad (para evaluar la capacidad del cuestionario de detectar inactividad física).15
RESPUESTA AL CAMBIO
La respuesta al cambio (responsiveness) es la capacidad de un instrumento para medir si se ha producido un cambio verdadero en el estado de salud de interés. Existen muchas maneras de evaluar la respuesta al cambio, debido, entre otros motivos, a la falta de una defi nición estándar y controversia en los conceptos,11 ya que la sensibilidad al cambio depende de las características de la muestra, del cambio que se espera y del diseño del estudio.
El principal criterio para escoger entre unas u otras técnicas es la homogeneidad/heterogeneidad esperada en el cambio. Por ejemplo, en un grupo de pacientes la puntuación global de un cuestionario de síntomas respiratorios puede cambiar de forma parecida en todos los sujetos (cambio homogéneo en la muestra), mientras que en otro grupo de pacientes, por sus diferencias en la gravedad de la enfermedad o por cualquier otra razón, la puntuación puede cambiar de forma distinta entre pacientes (cambio heterogéneo en la muestra). En este capítulo exponemos los estadísticos principales que evalúan la sensibilidad al cambio según la homogeneidad/heterogeneidad del cambio en nuestra muestra.
Cambio homogéneo
Si suponemos que nuestra muestra está formada por un grupo de pacientes que pueden tener un cambio parecido entre dos administraciones de un cuestionario, por ejemplo, después de un tratamiento conocido,12 utilizaremos estadísticos que nos cuantifi quen la media del cambio. Estos estadísticos son los que se detallan a continuación.
(1) Estadístico t para datos apareados. Este estadístico evalúa la hipótesis de cambio nulo en la media de nuestra escala de interés entre dos momentos en el tiempo. Dado que se trata de los mismos sujetos, se utiliza la t para datos apareados:
t = media de la diferencia / (desviación estándar de la diferencia / √n).
Este estadístico se basa en las diferencias estadísticamente signifi cativas, y depende del tamaño de la muestra y de la variabilidad entre las dos administraciones. No es muy potente para la sensibilidad al cambio, ya que depende de n, es decir, puede detectar como estadísticamente signifi cativos cambios que no sean clínicamente relevantes para muestras grandes. Si t es grande, rechazamos la hipótesis nula “no hay cambio”.
(2) Tamaño del efecto estandarizado de Cohen. Es la media de las diferencias entre las dos administraciones dividida por la desviación estándar de la administración basal:
Cohen = media de la diferencia / desviación estándar basal.
Un valor de 0,25, por ejemplo, nos indica que el cambio es una cuarta parte de la desviación estándar basal y se considera pequeño. En general, se considera que los valores menores de 0,5 indican poca sensibilidad al cambio, entre 0,5 y 0,7 indican moderada sensibilidad, y mayores de 0,7 indican alta sensibilidad a los cambios.11-12
(3) Respuesta media estandarizada (SMR). Este estadístico tiene en cuenta la variabilidad del cambio entre las dos administraciones. Si observamos una gran variabilidad en el cambio obtendremos valores pequeños en el SMR.
El SMR es el estadístico más adecuado ya que además de no depender del tamaño de la muestra, también controla la variabilidad del cambio entre las dos medidas. El SMR se interpreta igual que Cohen.11-12
Cambio heterogéneo
En este caso, partimos de la premisa que nuestra muestra está formada por un grupo de pacientes que pueden cambiar de diferente manera. Se evalúa también un estándar de referencia externo y se comparan ambas medidas de cambio. Los métodos que se utilizan para evaluar la respuesta al cambio son el coefi ciente de correlación y los modelos de regresión.
(1) Coefi ciente de correlación. Se calcula el coefi ciente de correlación entre las dos medidas del cambio, la de nuestra herramienta de interés y la del estándar externo. Este estadístico tiene valores entre -1 y +1, donde un valor cercano a |1| nos indica que nuestra herramienta es sensible al cambio. La sensibilidad al cambio depende del estándar externo elegido, por lo que la selección de este estándar es muy importante. Una limitación de este estadístico es la presencia de valores extremos.
(2) Modelos de regresión. En este caso se evalúa si el cambio en el instrumento de interés puede predecir el cambio del estándar externo, o viceversa:
Cambio en la medida estándar = constante + coefi ciente* (Cambio en el instrumento) + error.
Unos valores del coefi ciente altos nos indican que los cambios producidos en el instrumento producen también cambios importantes en el estándar. La ventaja de este modelo es que se puede ajustar por niveles basales del estado de salud de interés y por otras variables que consideremos relevantes. Si el cambio en nuestro instrumento puede predecir el cambio del estándar externo, podemos considerar que nuestra herramienta es sensible al cambio.
CUESTIONES A CONSIDERAR
Los cuestionarios forman parte del día a día de los investigadores clínicos. En la mayoría de casos, estos investigadores son “simplemente usuarios”, otras veces necesitan validar un cuestionario ya elaborado para una nueva población o un nuevo idioma, y, en el más infrecuente pero no menos importante de los casos, deben desarrollar completamente un cuestionario desde cero. Todos ellos necesitan conocer los conceptos y métodos descritos en este capítulo para saber interpretar la literatura existente, tomar decisiones sobre instrumentos a escoger en las investigaciones y, eventualmente, llevar a cabo una validación completa.
Hay que ser rigurosos en los pasos descritos para construir herramientas de medición adecuadas, válidas y fi ables. Sólo de esta manera se pueden obtener resultados realmente útiles para la comunidad científi ca y la salud de nuestras poblaciones.
PUNTOS CLAVE
• Constructo: concepto no observable que se pretende cuantifi car. • Dimensión (o factor): característica de un aspecto del constructo. El constructo puede tener más de una dimensión. • Ítem: unidad primaria de que se compone un cuestionario o instrumento de medida. • Fases de desarrollo de un cuestionario: elaboración (defi nición del constructo y sus dimensiones, la población objetivo y el modo de administración, elaboración de ítems y prueba piloto) y validación (estudio de la fi abilidad, validez y respuesta al cambio). • Fiabilidad: capacidad de un instrumento de medir sin error, con precisión. La fi abilidad se mide a través de la coherencia interna, la fi abilidad intra-observador y la fi abilidad inter-observador. • Coherencia interna: grado de coherencia de todos los ítems de un instrumento. • Fiabilidad intra-observador o test-retest: fi abilidad que mide el acuerdo entre dos medidas de un mismo instrumento en los mismos sujetos en diferentes momentos del tiempo. • Fiabilidad inter-observador: fi abilidad que mide el acuerdo entre dos medidas de un mismo instrumento administrado por diferentes evaluadores en los mismos sujetos. • Validez: capacidad del instrumento de medir el constructo para el que ha sido diseñado. La medimos a través de la validez aparente y la validez de contenido, que son estudiadas cualitativamente según bibliografía y expertos en el tema, y a través de la validez de constructo y la validez de criterio, las cuales son estudiadas empíricamente según medidas externas.
Respuesta al cambio: es la capacidad del instrumento para medir si se producen cambios verdaderos en el aspecto de salud de interés.
BIBLIOGRAFÍA
1. Argimón Pallás JM, Jiménez Vila J. Métodos de investigación clínica y epidemiológica. Madrid, Ediciones Harcourt, 3ª ed., 2006. 2. Martín Arribas MC. Diseño y validación de cuestionarios. Matronas profesión. 2004; Vol. 5, núm. 17. 3. García de Yébenes Prous MJ, Rodríguez Salvanés F, Carmona Ortells L. Validación de cuestionarios.
Reumatología Clínica. 2009; 5(4):171-177. 4. Pesudovs K, Burr JM, Harley C, et al. Th e development, assessment, and selection of questionnaires.
Optom Vis Sci. 2007;84:663-674. 5. Gimeno-Santos E, Raste Y, Demeyer H, Louvaris L, De Jong C, Rabinovich RA, et al. Th e PROactive instruments to measure physical activity in patients with chronic obstructive pulmonary disease. Eur
Respir J. 2015;46(4):988-1000. 6. Müller R, Büttner P. A critical discussion of intraclass correlation coeffi cients. Stat Med. 1994;13:24652476. 7. Altman DG, Bland JM. Measurement in medicine: the analysis of method comparison studies. Statistician. 1983;32:307-317. 8. Ato García M, López García JJ. Análisis estadístico para datos categóricos. Madrid, Síntesis, 1ª ed., 1996. 9. Gimeno-Santos E, Frei A, Steurer-Stey C, De Batlle J, Rabinovich RA, Raste Y, et al. Determinants and outcomes of physical activity in patients with COPD: a systematic review. Th orax. 2014; 69(8):731-739. 10. Ramada-Rodilla JM, Serra-Pujadas C, Delclós-Clanchet GL. Adaptación cultural y validación de cuestionarios de salud: revisión y recomendaciones metodológicas. Salud Publica Mex. 2013;55:57-66. 11. García de Yébenes Prous MJ, Rodríguez Salvanés F, Carmona Ortells L. Sensibilidad al cambio de las medidas de desenlace. Reumatología Clínica- 2008;4:240-247. 12. Husted JA, Cook RJ, Farewell VT, Gladman DD. Methods for assessing responsiveness: a critical review and recommendations. J Clin Epidemiol. 2000;53:459-468. 13. Dobbels F, De Jong C, Drost E, Elberse J, Feridou C, Jacobs L, et al. Th e PROactive innovative conceptual framework on physical activity. Eur Respir J. 2014;44:1223-1233. 14. Gimeno-Santos E, Raste Y, Demeyer H, Louvaris Z, De Jong C, Rabinovich RA, et al. Th e PROactive instruments to measure physical activity in patients with chronic obstructive pulmonary disease. Eur
Respir J. 2015;46:988-1000. 15. Donaire-Gonzalez D, Gimeno-Santos E, Serra I, Roca J, Balcells E, Rodríguez E, et al. Validation of the
Yale Physical Activity Survey in chronic obstructive pulmonary disease patients. Arch Bronconeumol. 2011;47:552-560.
TABLAS Y FIGURAS
Figura 2.8.1. Fases de la elaboración y validación de cuestionarios

Modulo 3:
PROYECTO DE INVESTIGACIÓN
3Responsable: Dr. Francisco García Río .
ÍNDICE
3.1. PROTOCOLO DE INVESTIGACIÓN: GENERALIDADES ............................................................. 161 INTRODUCCIÓN Y CONCEPTO ....................................................................................................... 161 FINALIDADES ........................................................................................................................................ 161 CONSIDERACIONES GENERALES PARA LA ELABORACIÓN DE UN PROTOCOLO DE INVESTIGACIÓN ................................................................................................................................... 162 ESTRUCTURA DE UN PROTOCOLO DE INVESTIGACIÓN ........................................................ 163 Identifi cación Resumen Antecedentes y estado actual del tema. Justifi cació Bibliografía Objetivos Hipótesis Metodología Ejemplos OTROS ASPECTOS DEL PROTOCOLO DE INVESTIGACIÓN .................................................... 167 PUNTOS CLAVE .................................................................................................................................... 167 BIBLIOGRAFÍA ...................................................................................................................................... 168 TABLAS Y FIGURAS ........................................................................................................................... 169
3.2. HIPÓTESIS Y OBJETIVO DE ESTUDIO ........................................................................................... 175 CONTRASTE DE HIPÓTESIS ............................................................................................................. 175 Errores en el contraste de hipótesis: error tipo i y error tipo ii OBJETIVO DE ESTUDIO ...................................................................................................................... 176 Formulación de un objetivo PUNTOS CLAVE .................................................................................................................................... 177 BIBLIOGRAFÍA ...................................................................................................................................... 178 TABLAS Y FIGURAS ........................................................................................................................... 179
3.3. POBLACIÓN DIANA, POBLACIÓN DE ESTUDIO Y MUESTRA. TÉCNICAS DE MUESTREO ...................................................................................................................................... 181 INTRODUCCIÓN ................................................................................................................................... 181 POBLACIÓN, MUESTRA Y OTROS CONCEPTOS ASOCIADOS .............................................. 181 MUESTREO PROBABILÍSTICO ........................................................................................................ 182 Muestreo aleatorio simple Muestreo sistemático Muestreo aleatorio estratifi cado Muestreo aleatorio por conglomerados o en etapas múltiples MUESTREO NO PROBABILÍSTICO ................................................................................................. 184 Muestreo por cuotas Muestreo discrecional Muestreo por bola de nieve Muestreo de conveniencia ERROR ALEATORIO Y SESGOS ....................................................................................................... 185 PUNTOS CLAVE ................................................................................................................................... 185 BIBLIOGRAFÍA ..................................................................................................................................... 186 TABLAS Y FIGURAS ........................................................................................................................... 187
3.4. TAMAÑO DE LA MUESTRA ............................................................................................................... 193 INTRODUCCIÓN .................................................................................................................................. 193 ESTIMACIÓN DE PARÁMETROS...................................................................................................... 193 Estimación de una proporción Estimación de una medida PRUEBAS DE CONTRASTE DE HIPÓTESIS .................................................................................. 196 ESTIMACIÓN DE UNA ODDS RATIO .............................................................................................. 198 EQUIVALENCIA DE DOS INTERVENCIONES ................................................................................ 199 EL TAMAÑO MUESTRAL AJUSTADO A LAS PÉRDIDAS ........................................................... 200 Calculadoras de tamaño de muestra PUNTOS CLAVE ................................................................................................................................... 200 BIBLIOGRAFÍA ..................................................................................................................................... 201 TABLAS Y FIGURAS ........................................................................................................................... 202
3.5. CREACIÓN Y MEDICIÓN DE VARIABLES. FUENTES DE INFORMACIÓN ............................ 203 TIPOS DE VARIABLES ....................................................................................................................... 203 Variables cuantitativas Variables cualitativas Variables según su infl uencia MEDICIÓN DE LAS VARIABLES ....................................................................................................... 205 FUENTES DE INFORMACIÓN ............................................................................................................ 205 PUNTOS CLAVE ................................................................................................................................... 206 BIBLIOGRAFÍA ..................................................................................................................................... 207 TABLAS Y FIGURAS ........................................................................................................................... 208
3.6. TRABAJO DE CAMPO ......................................................................................................................... 209 CARACTERÍSTICAS DEL TRABAJO DE CAMPO .......................................................................... 209 TIPOS DE DISEÑO DEL TRABAJO DE CAMPO ............................................................................. 210 Diseño experimental Experimento post-facto Diseño encuesta Diseño panel Estudio de casos TÉCNICAS Y FASES DEL TRABAJO DE CAMPO ......................................................................... 210 Proceso en la investigación de campo PREPARACIÓN DEL INVESTIGADOR PARA INICIAR EL TRABAJO DE CAMPO ................ 213 RECOGIDA DE DATOS EN EL TRABAJO DE CAMPO ................................................................... 213 Encuestas Cuestionario Entrevista Grupos de enfoque Observación PUNTOS CLAVE ................................................................................................................................... 214 BIBLIOGRAFÍA ..................................................................................................................................... 215 TABLAS Y FIGURAS ........................................................................................................................... 216
3.7. PROCESAMIENTO Y GESTIÓN DE MUESTRAS BIOLÓGICAS. BIOBANCOS ................... 217 MUESTRAS BIOLÓGICAS PARA INVESTIGACIÓN BIOMÉDICA ........................................... 217 PROCESAMIENTO Y ALMACENAMIENTO DE MUESTRAS Y DATOS ASOCIADOS .......... 218 Procesamiento y almacenamiento de muestras DATOS ASOCIADOS A LAS MUESTRAS ...................................................................................... 219 Datos del biobanco/biorepositorio Datos del paciente o donante de muestras Datos de la donación Datos de la muestra Datos del análisis de las muestras APLICATIVOS DE GESTIÓN DE MUESTRAS ................................................................................ 220 GESTIÓN DE MUESTRAS BIOLÓGICAS ........................................................................................ 220 BIOBANCOS .......................................................................................................................................... 220 PUNTOS CLAVE ................................................................................................................................... 222 BIBLIOGRAFÍA ..................................................................................................................................... 223
3.8. FINANCIACIÓN DE UN PROYECTO. MEMORIA ECONÓMICA ............................................... 225 NORMAS GENERALES ........................................................................................................................ 225 FUENTES DE FINANCIACIÓN ............................................................................................................ 227 QUÉ SE PUEDE FINANCIAR .............................................................................................................. 228 MEMORIA ECONÓMICA ..................................................................................................................... 229 PUNTOS CLAVE ................................................................................................................................... 229 TABLAS Y FIGURAS ........................................................................................................................... 230
3.1. PROTOCOLO DE INVESTIGACIÓN: GENERALIDADES
Dr. Francisco García Río
INTRODUCCIÓN Y CONCEPTO
En el presente módulo se revisarán algunos de los aspectos más relevantes a tener en cuenta para plantear un proyecto de investigación. Aunque en los sucesivos capítulos se proporcionará una explicación más detallada sobre aspectos concretos, en este primer capítulo del módulo se plantea una visión global de qué es un protocolo de investigación, sus fi nalidades y su estructura. Además, se abordarán algunos aspectos generales que pueden resultar de utilidad a la hora de elaborar un protocolo.
El protocolo de un proyecto de investigación es un documento que refl eja una descripción ordenada y sistemática del estudio propuesto y proporciona una visión general de los aspectos del estudio. Debe ser redactado con sufi ciente claridad para permitir que otro investigador pueda realizar el estudio o para que pueda ser realizado en otro momento.1
Necesariamente, es un documento que antecede a la investigación y que se presentará ante las instituciones correspondientes para dar a conocer los aspectos del estudio que se pretende realizar, a fi n de que sean evaluadas su pertinencia y relevancia, así como su oportunidad, originalidad, practicidad y aplicabilidad.
Todo lo anterior justifi ca la importancia de elaborar un buen protocolo o proyecto de investigación. Así pues, la claridad con que se enuncien los objetivos y el método del estudio redundará tanto en la calidad de los resultados como en la evaluación de su pertinencia.2-3
FINALIDADES
Desde un punto de vista general, puede considerarse que un protocolo de investigación tiene tres fi nalidades: comunicación, planifi cación y compromiso.4 La primera de sus tareas es comunicar, ya que el proyecto de investigación sirve para dar a conocer los planes de investigación de su autor. La segunda función es planifi car, pues el protocolo es un plan de acción que se detalla paso por paso. Finalmente, la tercera función implica un compromiso entre el investigador, sus colaboradores y la institución para la cual se llevará a cabo el trabajo.
De una forma más concreta, resulta claro que el protocolo de investigación es el instrumento que permite una discusión concreta y precisa del proyecto e identifi ca los aspectos que resultan menos adecuados.5 En
este sentido, es muy recomendable que un grupo de investigación novel someta su protocolo a revisión, que llevarán a cabo investigadores más expertos de su propio centro, sociedad científi ca o instituciones afi nes. Cuando ya se dispone de una propuesta explicitada y detallada de lo que se pretende realizar, es el momento idóneo para detectar aspectos de mejora y plantear cambios que el equipo investigador pueda asumir. Desde luego, una vez iniciado el proyecto resulta mucho más difícil (y casi siempre no recomendable) introducir modifi caciones metodológicas. A su vez, sobre una propuesta concreta de investigación detallada en un protocolo resulta factible concretar posibles colaboraciones y defi nir con precisión el ámbito en que se va a llevar a cabo cada una de ellas.
También es necesario considerar que el protocolo de investigación deberá ser sometido a la aprobación o autorización institucional correspondiente.6 Dado que la actividad investigadora no es una tarea aislada, cada vez existen más requerimientos para que un grupo de investigación someta su propuesta al organismo establecido por su institución (comisión de investigación, dirección, etc.) con objeto de determinar si resulta pertinente y adecuada y si se corresponde con los intereses o líneas de investigación de la institución. Este suele ser un requisito previo para la autorización formal a la realización del proyecto. Además, para llevar a cabo el estudio también es necesario remitir el protocolo de investigación al comité de ética de investigación correspondiente para que de su aprobación.
Otra fi nalidad del protocolo de investigación es la captación de recursos. En un entorno cada vez más competitivo, en el que resulta necesario captar fondos de ayudas destinadas a la investigación por parte de agencias de fi nanciación externa, el protocolo de investigación constituye habitualmente un elemento crucial para determinar el interés en fi nanciar una propuesta.3 Incluso en convocatorias no competitivas y para la formalización de cualquier contrato de investigación, se requiere un protocolo de investigación.
Por último, tampoco debe dejar de considerarse que el protocolo de investigación supone el instrumento para comunicar al resto de la comunidad científi ca el proyecto que estamos realizando y que, como tal, en muchas ocasiones puede interesar su publicación o registro en algún repertorio internacional.7
El protocolo de investigación debe contener las distintas fases de la investigación (Figura 3.1.1). Obviamente, requiere una revisión del modelo teórico del conocimiento que permita formular la pregunta a investigar. Debe explicitar con el máximo detalle posible el diseño de la investigación y concretar cómo se pretende efectuar la recogida de los datos, su análisis y la interpretación de los resultados.8 Incluso puede resultar recomendable que el protocolo de investigación recoja brevemente cómo se van a comunicar los resultados y las estrategias de publicación previstas.
En la tabla 3.1.1 se enumeran los principales pasos a seguir en la elaboración de un protocolo de investigación. Aunque a continuación se analizarán los distintos elementos a considerar, cabe plantear como recomendación general que su extensión sea de unos 15-20 folios y que la redacción se realice preferiblemente de forma impersonal. Es decir, resulta aconsejable utilizar expresiones del tipo se analizará o se evaluará y evitar construcciones en primera persona. También se recomienda evitar el uso excesivo de abreviaturas y desarrollarlas la primera vez que aparezcan en el texto.
Por supuesto, es necesario tener presente la fi nalidad del protocolo y, en caso necesario, adaptarlo al formulario específi co de un comité evaluador o agencia de fi nanciación, tanto pública como privada o de sociedad científi ca (Figura 3.1.2).
No está de más recordar que para la elaboración de un buen protocolo de investigación suele ser necesario el contacto previo con personal cualifi cado, como estadísticos, epidemiólogos, informáticos, etc., que orienten, no sólo en su preparación, sino también en aspectos metodológicos, como técnicas estadísticas a aplicar, diseños de investigación más complejos, análisis de costes, etc.
ESTRUCTURA DE UN PROTOCOLO DE INVESTIGACIÓN
Aunque puede existir cierta variabilidad en función del destino del protocolo, existen unos elementos comunes que deben desarrollarse en todos los casos (Tabla 3.1.2).3,9-10 A continuación, se describen de forma breve.
Identifi cación
El título debe ser conciso y específi co, refl ejar la acción que se va a realizar (evaluación, comparación, determinación, variabilidad, etc.) y sobre qué tipo de pacientes, evitando expresiones superfl uas o circunstanciales. Se recomienda seleccionar en el título el mayor número posible de descriptores del proyecto de investigación evitando términos que no clarifi can nada, como estudio o informe. Su extensión no debería superar las quince palabras, y no suele ser conveniente incluir abreviaturas.
Además del título, puede tener interés generar un acrónimo que permita identifi car más fácilmente el proyecto de investigación. También es necesario consignar el número de versión del protocolo y su fecha. En este apartado se debe identifi car a los investigadores que desarrollarán el proyecto y sus fi liaciones.
Resumen
Es un esquema estructurado que proporciona una visión general del proyecto. Consiste en una descripción concreta y objetiva del contenido global de la propuesta de investigación, que debe contener: objetivo (principal), diseño, ámbito del estudio (centro de salud, hospital, área sanitaria, ciudad, comunidad, etc.), sujetos del estudio, instrumentación y determinaciones. Habitualmente, se recomienda que su extensión no exceda las 250 palabras. Debe ir acompañado de una selección de palabras clave que permitan la clasifi cación del protocolo.
Antecedentes y estado actual del tema. Justifi cación
Ante todo, es importante destacar que este apartado no debe ser una revisión bibliográfi ca, sino que debe centrarse en el proyecto de investigación. Los antecedentes deben orientar al trabajo que se va a realizar formulando el problema de investigación, siempre con adecuado soporte bibliográfi co. En la redacción del estado actual del tema es aconsejable mencionar cifras de prevalencia o incidencia de los estudios encontrados, siempre partiendo de un ámbito más general para ir luego hacia uno más local. El último apartado de esta sección corresponde a la justifi cación de por qué realizar el estudio (por ejemplo, “no hay estudios previos en nuestro medio”, “se aporta nueva información”, “se analizan otras variables”, etc.).
En su conjunto, esta sección debe contar con la estructura y la claridad adecuadas para que el lector intuya cuáles son los objetivos del proyecto. Un esquema orientativo podría ser el siguiente:
• Magnitud, frecuencia y distribución del problema a estudiar. Áreas geográfi cas involucradas y grupos de población afectados por el problema. • Causas probables del problema: ¿cuál es el conocimiento actual sobre el problema y sus causas?, ¿hay consenso?, ¿hay discrepancias?, ¿hay evidencias conclusivas?
• Soluciones posibles: ¿cuáles han sido las formas de resolver el problema?, ¿qué se ha propuesto?, ¿qué resultados se han obtenido? • Preguntas sin respuesta: ¿qué sigue siendo un interrogante?, ¿qué no se ha logrado conocer, determinar, verifi car, probar?
Bibliografía
Debe apoyar toda la argumentación del apartado anterior, por lo que hay que seleccionar las referencias más adecuadas y acordes con lo que se quiere investigar. Las citas deberían tener una antigüedad máxima de 5-10 años, siempre que no se trate de una referencia fundamental, en cuyo caso habrá que incluirla aunque sea más antigua. Resulta muy importante evitar la omisión de referencias fundamentales sobre el tema, sobre todo si se refi eren a aspectos parcialmente relacionados con el proyecto que se presenta. En general, las referencias se citan en el texto de forma consecutiva y se editan siguiendo la normativa de Vancouver.
Objetivos
Aunque un capítulo posterior de este módulo se dedica específi camente a la formulación de hipótesis y objetivos, es conveniente recordar que deben ser precisos, concisos, medibles y alcanzables.10 Ser precisos signifi ca que se deben expresar de forma clara, con lenguaje sencillo y sin ambigüedades. Ser concisos implica que se deben formular de la manera más resumida posible, sin rodeos y utilizando sólo las palabras necesarias, y ser medibles supone que deben expresarse de modo tal que permitan medir las cualidades o características del objeto de investigación. Finalmente, los objetivos deben ser alcanzables, es decir, deben existir posibilidades reales de lograr los objetivos planteados. En su redacción, se recomienda emplear verbos como determinar, comparar, analizar, diferenciar y evitar otros como investigar o estudiar, que resultan más imprecisos.
Se suele plantear un objetivo general, muy parecido al título del proyecto, aunque redactado en infi nitivo, que constituye el propósito central del proyecto. En esencia, abarca todo el problema e informa de manera global para qué se hace la investigación. Además, se suelen exponer de cuatro a seis objetivos específi cos, que contienen un desarrollo pormenorizado del objetivo general y comprenden acciones que no están descritas en éste, procurando evitar duplicidades.
Hipótesis
Guían el análisis de datos y deben ser la consecuencia de un marco teórico bien establecido. Suelen referirse al objetivo principal y se recomienda que se redacten en afi rmativo (“el tratamiento A es más efectivo que el tratamiento B”), antes que como expresiones condicionales.
Se suelen establecer distintos niveles de hipótesis. La hipótesis conceptual formula la idea general que se plantea, mientras que la operativa establece una mayor concreción de la misma. Por último, las hipótesis estadísticas serán las que se verifi quen o refuten en el análisis de datos. Las hipótesis estadísticas contienen la nula y la alternativa. La hipótesis nula o hipótesis de diferencias nulas, que niega la existencia de relación signifi cativa entre las variables de estudio, se formula habitualmente con la intención de ser rechazada. La hipótesis alternativa afi rma la existencia de relaciones signifi cativas entre las variables, diferencias entre los grupos a comparar, etc.
Metodología
Constituye la sección más importante del protocolo de investigación y deberá responder a la pregunta: ¿Cómo se procederá para alcanzar los objetivos planteados? Debe exponer cómo se realizará el estudio con
el mayor grado de detalle posible, pues ello garantiza que cualquier interesado pueda reproducirlo. De forma más o menos esquemática, contiene los siguientes subapartados.3,9
Ejemplos
Ámbito del estudio. Constituye la población de referencia o población total sobre la que se espera extrapolar los resultados y donde se enmarca el estudio.
Población del estudio o diana. Es el subgrupo de la población de referencia, que debe cumplir una serie de criterios de selección. Está formada por los casos candidatos a participar en el estudio.
Criterios de selección. Especifi can las características que los pacientes que formen parte del estudio deben cumplir para que su reclutamiento se haga de la manera más homogénea posible y se eviten sesgos. Consisten en los criterios de inclusión, de exclusión y de retirada del estudio. Los primeros corresponden a las características que necesariamente deberán tener los sujetos de estudio, mientras que los criterios de exclusión recopilan las características cuya existencia obliga a no incluir un sujeto como elemento de estudio. Los criterios de retirada defi nen las características que, al presentarse en los sujetos ya incluidos en el estudio, obliguen a prescindir de ellos.
Tamaño muestral y procedimiento de muestreo. Aunque también se desarrolla con mayor extensión en otro capítulo, este apartado del protocolo detalla el número de pacientes necesario para alcanzar los objetivos del estudio. Debe contener una breve descripción de las premisas aplicadas para su cálculo. También se debe precisar el procedimiento de muestreo que se utilizará para seleccionar a los pacientes del estudio (aleatorio, sistemático, consecutivo o de otro tipo). Si se trata de un ensayo clínico, se especifi cará la forma de asignar los sujetos a los distintos grupos del estudio.
Diseño. Debe adecuarse a la pregunta de investigación y describirse de forma clara y concisa, empleando la terminología epidemiológica al uso.11 Este es un aspecto crítico en cualquier protocolo de investigación, que ya ha sido tratado en más detalle en un capítulo previo. Como norma general, se recomienda no mezclar diseños en un mismo estudio.
Intervención. Obviamente, este apartado sólo se incluirá cuando el proyecto planteado contenga algún tipo de intervención. Podrá tratarse de estudios comparativos con diseños experimentales, cuasiexperimentales, de antes y después, o de ensayos clínicos. Se deberá describir la intervención tan detalladamente como sea posible explicando las actividades en el orden que van a ocurrir.
Variables. Se defi nirá con claridad y precisión qué variables se van a registrar, cómo se medirán y cómo se codifi carán. El proceso de defi nición de variables comienza en cuanto se determina el problema de estudio y se formulan sus objetivos, y es uno de los pasos más difíciles de la investigación. Hay variables que no son difíciles de describir, defi nir o medir, como la edad o el sexo, cuya compresión resulta muy intuitiva en la práctica de la vida diaria. Sin embargo, otras variables son más complejas, por su composición o variación de criterios entre regiones, países, especialidades y autores. La calidad de la atención y la accesibilidad a los servicios son ejemplos de variables más problemáticas. Por ello es importante conceptualizar y operacionalizar las variables, ya que cada investigador puede utilizar un criterio diferente y, por tanto, obtener datos muy distintos.10
El proceso de llevar una variable de un nivel abstracto a un plano concreto se denomina operacionalización, y la función básica de dicho proceso es precisar o concretar al máximo el signifi cado o alcance que se otorga a una variable en un determinado estudio. Dicha operacionalización se logra mediante la transformación de una variable en otras que tengan el mismo signifi cado y que sean susceptibles de medición empírica. Para
lograrlo, las variables principales se descomponen en otras más específi cas llamadas dimensiones, las cuales a su vez se deben traducir en indicadores para permitir la observación directa. Algunas veces, la variable puede ser operacionalizada mediante un solo indicador, mientras que en otros casos es necesario hacerlo por medio de un conjunto de indicadores.
En el protocolo se deben identifi car las variables independientes (factores de exposición, de confusión, etc.) y una o pocas dependientes (variables de respuesta, resultado o desenlace). En caso de variables categóricas, se señalarán todas las categorías de medida, mientras que si son numéricas se especifi carán las escalas y unidades de medida.
Recogida de datos. Se describirán los instrumentos de medida o fuentes de información que se utilizarán, cómo se van a llevar a cabo las mediciones sobre los pacientes y quién las va a realizar, dónde y cuándo. Puede resultar muy útil incluir en los anexos el cuaderno de recogida de datos.
Análisis de los datos. Debe estar claramente descrito, ya que se conoce de antemano las variables contenidas en el estudio y su escala de medición. Suele ser recomendable incluir una parte descriptiva, otra de análisis básico de los datos y una tercera con análisis más complejos. Se deben describir los métodos y modelos de análisis de los datos, según el tipo de variables. También se detallará brevemente los programas informáticos que se utilizarán.
Difi cultades y limitaciones del estudio. Es muy recomendable que el protocolo cuente con un apartado en el que se especifi quen los problemas que se prevé encontrar en la realización del estudio, las clases de sesgos, las variables de confusión, las interacciones, las pérdidas, etc. Los investigadores deberían esforzarse en justifi car que estos problemas no tienen por qué modifi car lo que se quiere medir y qué métodos de control se proponen implantar.
Plan de trabajo. El protocolo debe contener un cronograma que especifi que las tareas que se realizarán durante el proyecto, algunas de las cuales pueden solaparse, indicando el tiempo estimado para su cumplimiento así como las personas que las llevarán a cabo. (Figura 3.1.3). Este apartado debe completarse con la descripción de la distribución de tareas entre los miembros del equipo investigador.
Experiencia del equipo investigador sobre el tema. Proporciona elementos necesarios para valorar la factibilidad del proyecto y su adecuación a las líneas de investigación en que hayan trabajado los solicitantes del proyecto dentro del tema que se desea investigar.
Medios disponibles para la realización del proyecto. Contiene una breve relación de los recursos humanos y materiales de los que dispone el equipo investigador. Resulta imprescindible en los protocolos que se presenten a convocatorias de fi nanciación e importante en todos los tipos de protocolo para estimar las garantías sobre la ejecución del proyecto de investigación.
Justifi cación de la ayuda solicitada. En aquellos proyectos que se presenten a convocatorias de fi nanciación, este apartado contendrá la explicación de por qué se necesita la ayuda que se solicita para el estudio. En dichos casos, esta sección suele ir acompañada por una memoria económica en la que se detalla el coste de todas las partidas seleccionadas, que obviamente debe adecuarse a las características de la convocatoria a la que se concurre.
Anexos. Pueden incluir información muy diversa, desde datos previos del equipo investigador, todavía no publicados pero útiles para la planifi cación del proyecto de investigación, hasta el modelo de hoja informativa y consentimiento del paciente, o cuaderno de recogida de datos.
OTROS ASPECTOS DEL PROTOCOLO DE INVESTIGACIÓN
En el caso de proyectos de investigación en seres humanos, en los criterios de selección se establecerá la exigencia de recogida del consentimiento informado. No obstante, dependiendo de las características de la propuesta, puede ser conveniente completar este apartado con algunas consideraciones éticas. Puede contener un conjunto de refl exiones en torno a las posibles implicaciones de la realización del estudio y el compromiso del investigador frente a las personas participantes, los datos obtenidos, el grupo de control si la experiencia es benéfi ca y acerca del manejo de las fuentes de consulta, entre otros aspectos. Esta información adicional puede resultar de ayuda en la evaluación del comité de ética de la investigación correspondiente.
Como ya se ha mencionado, también se recomienda acompañar el protocolo del cuaderno de recogida de datos, lo que facilita una interpretación más concreta de muchos aspectos metodológicos. También resulta de gran utilidad elaborar un manual de procedimientos, en el que se describirán con el mayor detalle posible (tipo enumeración por pasos) todos los procedimientos que se van a llevar a cabo en el proyecto de investigación. Además de reforzar la calidad de las determinaciones, este documento resulta muy importante en proyectos multicéntricos o si se pretende incorporar al equipo nuevos investigadores.
En casi todas las convocatorias competitivas se solicita describir el impacto y la repercusión sociosanitaria del proyecto de investigación. En esta sección, habitualmente denominada aplicabilidad y utilidad práctica de los resultados, se explicará el interés que tendrán los resultados del proyecto y para qué servirán, con una indicación de aplicación futura. Se debe redactar en términos prácticos y realistas.
Aunque no resulta imprescindible en todos los protocolos, se recomienda considerar la inclusión de un plan de publicaciones y de explotación de resultados. Este apartado ayuda a estimar la relevancia del proyecto y puede ser muy útil para estratifi car el análisis de resultados así como para defi nir precozmente criterios de autoría.
Por último, se debe considerar que si el proyecto de investigación propuesto es un ensayo clínico, resulta obligado el registro del protocolo en repositorios en línea específi cos. El más habitualmente utilizado es clinicaltrials.gov, y exige una estructura del protocolo predeterminada (Figura 3.1.4).
En conclusión, el protocolo de investigación consiste en la descripción detallada de los fundamentos, objetivos y métodos del estudio. Supone un elemento crucial para mejorar el planteamiento de un proyecto de investigación y resulta un requisito imprescindible previo a su ejecución.
PUNTOS CLAVE
Cualquier protocolo de investigación debe responder afi rmativamente a estas diez preguntas:
1. ¿El título refl eja lo que se pretende investigar? 2. ¿El planteamiento del problema explica la situación general que origina la interrogante? 3. ¿La formulación del problema constituye una interrogante precisa, delimitada en espacio y tiempo? 4. ¿Los objetivos expresan los aspectos que se pretende conocer? 5. ¿La justifi cación explica por qué se realizará el estudio, así como sus posibles aportaciones? 6. ¿Los antecedentes son investigaciones previas relacionadas con el problema? 7. ¿Las bases teóricas constituyen posiciones de distintos autores que permiten sustentar la investigación? 8. ¿La defi nición de términos expresa el signifi cado de los vocablos inmersos en el problema? 9. ¿El nivel y diseño de la investigación son pertinentes para el logro de los objetivos? 10. ¿Las técnicas e instrumentos corresponden al diseño de investigación?
BIBLIOGRAFÍA
1. Villareal Ríos E. El protocolo de investigación en las ciencias de la salud: el proyecto de investigación, metodología y ejemplos de protocolos, aspectos éticos, administrativos y legales. México: Trillas, 2011. 2. García Roldán JL. Cómo elaborar un proyecto de investigación. Alicante: Universidad de Alicante, 1995. 3. Rodríguez del Águilar MM, Pérez Vicente S, Sordo del Castillo L, Fernández Sierra MA. Cómo elaborar un protocolo de investigación en salud. Med Clin (Barc) 2007;129:299-302. 4. Lawrence FL, Silverman S, Spirduso WW. Reading and understanding research. Los Angeles, SAGE Publications Inc., 3a ed., 2009. 5. Guyatt G. Preparing a research protocol to improve chances for success. J Clin Epidemiol. 2006;59:893899. 6. Roca JA. Cómo y para qué hacer un protocolo. Med Clin (Barc). 1996; 106:257-262. 7. Godlee F. Publishing study protocols: making them visible will improve registration, reporting and recruitment. BMC News and Views. 2001;2:4. 8. Organización Panamericana de la Salud. Guía para escribir un protocolo de investigación. Consultado el 20 de septiembre de 2010. Disponible en www.sld.cu/galerias/pdf/sitios/rehabilitacion.../ops_protocolo. pdf. 9. Suárez Pérez EL, Pérez Cardona CM. Basic components in the development of research proposals in health sciences. P R Health Sci J. 1999;18 Suppl A:i-iv,1-49. 10. González Labrador I. Partes componentes y elaboración del protocolo de investigación y del trabajo de terminación de la residencia. Rev Cub Med Gen Integr. 2010;26:387-406. 11. Hulley SB, Cummings SR. Diseño de la investigación clínica. Un enfoque epidemiológico. Barcelona:
Doyma; 1993.
TABLAS Y FIGURAS
Tabla 3.1.1. Pasos para la elaboración de un protocolo de investigación
1. Presentar el título completo del estudio. 2. Determinar y especifi car de manera clara el planteamiento del problema. 3. Elaborar una justifi cación del proyecto. 4. Recolectar la información que se incluirá en el apartado de antecedentes/marco teórico. 5. Formulación de una hipótesis válida y precisa. 6. Defi nición concreta de los objetivos. 7. Selección y explicación de la metodología. 8. Listado y descripción de los recursos. 9. Elaboración del cronograma (opcional). 10. Listado de las referencias bibliográfi cas consultadas. 11. Presentación de los anexos, sólo si es necesario.
Tabla 3.1.2. Epígrafes más importantes de un protocolo de investigación en salud
Título
Resumen Objetivo Diseño Ámbito del estudio Sujetos del estudio Instrumentación Determinaciones Palabras clave Antecedentes y estado actual del tema Justifi cación del estudio Bibliografía Objetivos Objetivos generales Objetivos específi cos Hipótesis Metodología Población de referencia y de estudio Criterios de inclusión y exclusión Tamaño muestral y procedimientos de muestreo Diseño del estudio Variables Recogida de datos y fuentes de información Análisis de datos Difi cultades y limitaciones del estudio Plan de trabajo Experiencia del equipo investigador sobre el tema Aplicabilidad y utilidad práctica de los resultados Medios disponibles para la realización del proyecto Justifi cación de la ayuda solicitada Presupuesto solicitado Anexos
Figura 3.1.1.
FASE CONCEPTUAL FASE METODOLÓGICA
Modelo teórico Formulación de la pregunta
Diseño de la investigación Recogida de los datos
Análisis de los datos
Publicación de los resultados
Redacción del informe de investigación
Interpretación de los resultados
FASE EMPÍRICA
Figura 3.1.2.



Figura 3.1.3.




Figura 3.1.4.

3.2. HIPÓTESIS Y OBJETIVO DE ESTUDIO
Máster Meritxell Peiró i Fàbregas
Existen dos tipos de hipótesis, la hipótesis conceptual y la operativa.
La hipótesis conceptual es la que se formula como resultado de las explicaciones teóricas aplicables a nuestro problema. Nos ayuda a explicar desde el punto de vista teórico el fenómeno que estamos investigando. En cambio, la hipótesis operativa es aquella que le sirve al investigador como base de su estudio. Este tipo de hipótesis trata de dar una explicación al fenómeno que se está investigando y presenta cuantitativamente (en términos medibles) la hipótesis conceptual.
CONTRASTE DE HIPÓTESIS
Para representar un resultado con un margen de error con límite superior e inferior se utilizan los intervalos de confi anza. Estos límites son los utilizados para estimar parámetros de una población. Aun así, existe otro enfoque más indirecto y al que se suele dar preferencia en biomedicina. Se trata del contraste de hipótesis.
Un contraste de hipótesis supone una comparación. Se establecen dos hipótesis de trabajo posibles y antagónicas y se elige una. El contraste comienza con la formulación de dos hipótesis sobre el valor de algún parámetro poblacional, cada una de ellas contrapuesta o antagónica respecto a la otra (una es cierta y la otra es falsa). Supondremos cierta una de ellas, a la cual llamaremos hipótesis nula (H0).
La hipótesis nula mantiene que el efecto de interés NO infl uye, es decir, vale 0 en la población de donde sale la muestra.
La hipótesis alternativa (H1) mantiene que existe algún efecto distinto de 0 en la población de la que procede la muestra.
Por tanto, se empieza planteando siempre una hipótesis nula, que indicará que el efecto sobre la población es cero, y a continuación utilizaremos los datos para decidir si podemos rechazar esta hipótesis y adoptar una hipótesis alternativa. El contraste de hipótesis consistirá en decidir entre una u otra. Si rechazamos la hipótesis nula (H0), diremos que el resultado es estadísticamente signifi cativo.
Un ejemplo podría ser un estudio para valorar la efi cacia en términos de abstinencia anual de una intervención para la deshabituación tabáquica en pacientes fumadores con EPOC durante un ingreso hospitalario.
Al azar, se asignarían en dos grupos. Uno sería el grupo control, en el que la intervención sería la práctica clínica habitual, y el otro sería el grupo intervención, en el que la intervención sería más intensiva con tratamiento farmacológico y seguimiento durante un año después del alta hospitalaria. Por lo tanto, la hipótesis H0 sería: La tasa de abstinencia anual será la misma para los fumadores con EPOC ingresados hayan recibido intervención o no.
Errores en el contraste de hipótesis: error tipo i y error tipo ii
En la tabla 3.2.1 podremos observar las cuatro posibles combinaciones dependiendo de la decisión tomada y de la certeza o no de la hipótesis nula.
Hablaremos de error de tipo I cuando hayamos descartado la hipótesis nula, siendo ésta cierta, es decir, hablaremos de una signifi cación estadística de los resultados cuando no es verdad. Este error se considera muy grave. Finalmente, acontecerá un error de tipo II cuando hayamos optado por no descartar la hipótesis nula y resulte que esta es falsa. O, dicho de otro modo, hablaremos de resultados no estadísticamente signifi cativos cuando en realidad sí lo son.
Las pruebas de signifi cación estadística nos proporcionarán un valor p. El valor p será la probabilidad de encontrar la diferencia observada en la muestra u otra más alejada a la hipótesis nula, condicionada a que esta fuera cierta. Cuanto menor sea el valor de la p de signifi cación estadística, mayores argumentos habrá para rechazar la hipótesis nula y apoyar la alternativa. Para considerar un valor p bajo, se suele escribir p < 0,05. En este caso, se rechaza la hipótesis nula y se dice que el resultado es estadísticamente signifi cativo. En cambio, cuando el valor de la p es p ≥ 0,10 no se rechaza la hipótesis nula y se dice que los resultados no son estadísticamente signifi cativos. Pero, ¿qué sucede cuando obtenemos resultados entre 0,05 y 0,10? Podremos decir que hay una tendencia o que se aproxima a la signifi cación estadística. Pero lo relevante no será la signifi cación estadística, sino la signifi cación clínica del resultado. La signifi cación clínica en la práctica diaria dependerá del tamaño muestral y de la magnitud del efecto. Podría ser sufi cientemente importante para no rechazar la hipótesis nula con una muestra grande pero no con una muestra de un número de sujetos muy reducido.
Para la elaboración de un manuscrito se deberá tener en cuenta la forma de transcripción, evitando los valores dados por programas estadísticos como p = ,000 o escribir “ns” (no signifi cativo), numerosos decimales, e incluso evitaremos la p < o > 0,005 al referirnos a si los resultados son o no signifi cativos estadísticamente. En la tabla 3.2.2 se muestran los modos correctos de presentar los resultados de un contraste de hipótesis según la p.
OBJETIVO DE ESTUDIO
La hipótesis es una respuesta tentativa a la pregunta que nos planteamos al formular un problema. Por tanto, la hipótesis tiene una relación directa con el problema que se está investigando. Pero cuando queremos resolver un problema, haremos el planteamiento en forma de preguntas. Y de esas preguntas elaboraremos los objetivos, es decir, lo que nos preguntamos, lo que nos proponemos conocer e investigar.
Los objetivos deben corresponderse con las preguntas realizadas. Los objetivos generales apuntan al tipo más general de conocimiento que se espera producir con la investigación. Los objetivos específi cos son aquellos que deben lograrse para alcanzar los objetivos generales y se enumerarán por orden de importancia, por orden temporal, etc.
Los objetivos del estudio deberán ser:
a) Claros y concretos, sin provocar errores de interpretación. b) Concisos. c) Medibles, es decir, los resultados se podrán medir o cuantifi car de forma objetiva.
Es necesario tener en cuenta que todos los aspectos del problema de investigación estén contenidos en el objetivo general, así como que todos los objetivos específi cos se desprendan del objetivo general, ya que si alguno de ellos no aporta conocimiento al mismo se llevará a cabo sin fundamento, pudiendo, incluso, ser objeto de otra investigación.
Los errores más comunes en la defi nición de los objetivos son:
• Ser demasiado amplios y generalizados. • Tener unos objetivos específi cos no contenidos en los generales. • Hacer un planteamiento de pasos como si fueran objetivos (confundir métodos, caminos, con objetivos). • Carecer de relación entre los objetivos, el marco teórico y la metodología: los objetivos son el destino de la tesis; el marco teórico, el terreno y la metodología son el camino a seguir.
Formulación de un objetivo
Para la correcta formulación de un objetivo, la composición de este deberá constar de 3 partes principales:
a) un verbo en infi nitivo b) un qué c) un cómo.
Un ejemplo podría ser: Evaluar (a) la efi cacia de un programa educativo en asma (b) en términos de prevención de exacerbaciones y mejora del control del asma (c).
PUNTOS CLAVE
• La hipótesis nula mantiene que no existe asociación entre los factores y que las diferencias observadas se deben al azar. • La hipótesis alternativa es la que contradice la hipótesis nula y que se aceptará en caso de rechazar la hipótesis nula. • La interpretación de una p es la probabilidad de observar diferencias mayores a las observadas en la muestra si la hipótesis nula fuera cierta. • Cuanto menor sea el valor de p, mayores argumentos tendremos para rechazar la hipótesis nula y aceptar la hipótesis alternativa.
BIBLIOGRAFÍA
Martínez-González MA, Sánchez Villegas A, Toledo Atucha E, Faulín Fajardo F. Bioestadística amigable. Madrid, ELSEVIER, 3ª ed., 2014. Hernández Sampieri R, Fernández Collado C, Baptista Lucio P. Fundamentos de metodología de la Investigación,. México, Mc. Graw - Hill, 2ª ed., 1998.
TABLAS Y FIGURAS
Tabla 3.2.1. Posibles resultados en un contraste de hipótesis
Decisión Hipótesis nula H0
Verdadera Falsa
No se descarta la H0 Decisión correcta tipo A (1-α) Error tipo II Probabilidad β
Se rechaza la H0 Error tipo I Probabilidad α Decisión correcta tipo B (1-β)
Tabla 3.2.2. Modo correcto de presentación de los resultados de un contraste de hipótesis
MODO INCORRECTO
MODO CORRECTO
p = ,000, p = 0,0000001 P = < 0,001
p = ns, ns, p > 0,05 Ejemplo: p = 0,16, p = 0,38
p < 0,05 p = 0,04
p = 0,036598700025 p = 0,04





