
10 minute read
Mycotoxin-Induced Plant Stress and cpUPR in Chlamydomonas reinhardtii

from PCR - Fall 2022

by Ethan Boroditsky (V), Dr. John McLaughlin1
1Rutgers University, New Brunswick, NJ
Abstract
Every year, tens of billions of dollars are lost in harvest from the Fusarium graminearum fungus attacking and killing critical agricultural plants such as corn, wheat, barley. As it turns out, this fungus uses mycotoxins to kill these plants. Our goal is to better understand the mycotoxin’s mechanism of action. In this study, we used a small unicellular algae called Chlamydomonas reinhardtii as our model organism, because it allows us to better understand and experiment with this toxin. Our study took advantage of a comprehensive genome-wide mutant library for Chlamydomonas. Such a library allows for the performance of screens that simulate identical conditions for an organism with every single knockout mutant simultaneously. We looked at differences in growth as a phenotype representative of fitness. During this project, we wrote a Python program utilizing various libraries to automate the process of searching the internet for hundreds of unique accession codes that correspond to homologous mutants of interest in Arabidopsis thaliana. This allowed us to learn more about the molecular mechanism and cell components the genes were primarily involved in, while also beginning to make the jump to a higher organism.
Chlamydomonas reinhardtii is the optimal model organism for studying the plant stress response in the chloroplast for many reasons. Chlamydomonas is a eukaryotic single-cell organism that performs photosynthesis. Most importantly, Chlamydomonas can be transformed and mutated, and as of 2020, there is a genome-wide mutant library that is publicly available. This allows for the screening of the entire Chlamydomonas genome. In this study, mutants were screened with a mycotoxin believed to induce plant stress. Sensitivity was measured by assessing mutants’ growth and assigning a representative “fitness score.” It was hypothesized that the key genes in the plant stress response mechanism would be the ones with the lowest fitness scores in the respective mutants. Finally, we were able to utilize a gene ontology (GO) analysis to identify which molecular mechanism and cell components the genes were primarily involved in. Throughout the study, a few specific genes and proteins of interest were investigated individually, including D1, VIPP2, and MARS1. These three proteins are involved in the removal and repair of degradation products and aggregates that form as a result of translation inhibition, which is induced by plant stress.

Figure 1. Within each subset, each row was printed as a quad, as shown in (a), while we need to populate rows horizontally as shown in (b) Figure 2. The graph shows the fold change of VIPP2 and MARS1 expression after treatment with DON and Tcin after 1 and 24 hours. This data was gathered through RT-qPCR
This study was designed to elucidate the mechanisms through which mycotoxin-induced stress is dealt with in plants. The GO analysis shows what cellular components and molecular pathways the mutants are involved in [1]. The mutant library that we used was printed in quad format on 9 48x32 plates. This library also had a comprehensive spreadsheet in a 36x384 (columns x rows) format containing unique identification codes that corresponded to each mutant. To convert from the first format to the second, Python code was written using the Pandas library. The mutants were printed to each plate from subsets of 4x384 sheets. Within each subset, each row was printed as a quad, filling the 48x32 plates horizontally, two rows at a time. To reverse engineer this process, I took the 4x384 groups and broke them into 48x32 plates. As shown in Figure 1, each of the 9 plates can be broken down into smaller groups of two rows. Each of these two rows is filled by going down the 4x384 sheet, putting the first two mutants into the top row, and the remaining two mutants in the bottom row. Repeating this process for each 4x384 group generates the 9 separate mutant plates. The core of the python code for

this part of the project is shown in Appendix 1. After organizing the 9 plates, we were finally able to work with our physical library. The library was screened twice, once with Tcin and once with Lincomycin. After the screening, 156 mutants were identified as having the lowest fitness scores. These scores were assigned solely based off phenotypic growth. The phenotyping was done using the Balony software. Before performing the GO analysis, each of the 156 mutants had to be converted from LMJ format into another format which the Panther classification software could interpret. Through trial and error, we found that TAIR accession codes would be best for this task, especially since they represent the orthologs of a higher organism, bringing us closer to experimenting with larger plants. Scouring the web for TAIR codes would be a long and menial task without automation. To automate the process, we wrote a Python program. As shown in Appendix 2, the program follows a path of two URLs, the first converting from LMJ to Cre format, and the second from Cre to TAIR format. While the mutant library is known as “genome-wide,” it only encompassed about 25% of the Chlamydomonas genome. This is because the entire genome is still being mapped out to this day. Not every gene in the Chlamydomonas genome that is mapped has a defined function, so only about 62 of 156 mutants were found to have an Arabidopsis ortholog. In related studies, we identified Vipp2, Mars1, and D1 as genes of interest in the mycotoxin stress response [3][4]. We performed a RT-qPCR on the mRNA, the results of which are shown in Figure 2, measuring the expression of Vipp2 and Mars1 to confirm whether the two genes were involved in the stress response. We used wildtype Chlamydomonas and treated the samples with DON and Tcin, two types of mycotoxins, comparing treatment times of 1 and 24 hours. For our study of D1, we first treated samples of Chlamydomonas with DON. Following treatment, the samples were grown for two days, after which the protein was harvested. As shown in Figure 3, going from left to right, the concentration of DON treatment increased. The first and last lanes are ladder lanes. To quantify the immunoblot, we used an Odyssey machine. This tool measures the total amount of fluorescence emitted in a given area. In each lane of the blot, we then normalized the quantification to better understand the change in D1 quantity.
III. RESULTS
This study yielded findings that have brought us closer to achieving the goal of identifying genes in higher organisms that are involved in plant stress response. We found significant common-
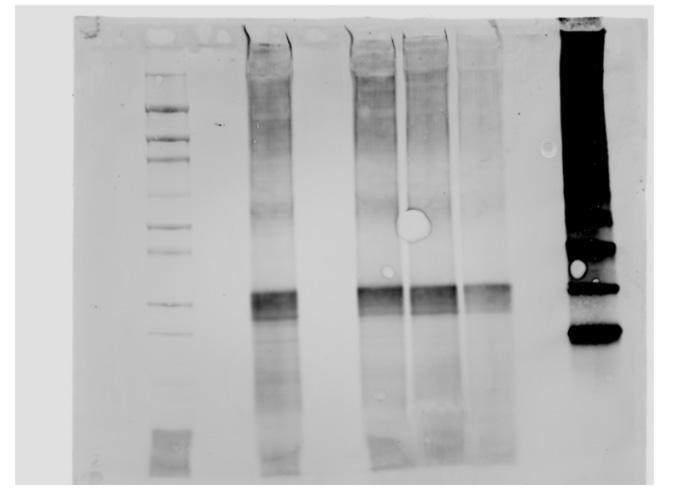
Figure 3. Western immunoblot with increasing DON concentration from left to right. The quantity of DON respectively decreases, corroborating the idea that toxic stress degrades DON. The first and last lanes are Magic Marker ladders. Contrary to expectation, no degredation products of D1 are visible in the blot.
alities in some of the mutants, suggesting some of the potential pathways involved. The results of the GO analysis, which are displayed in Figure 5, reveal that of the 28.6% of mutants implicated in binding, two thirds were involved in cyclic compound binding. A cyclic compound is a molecule whose atomic structure is organized in the shape of a ring. Lincomycin and Tcin are both cyclic compounds, whose structures are shown in Figure 4. This suggests that the mutants we identified in this category are likely responsible for directly binding to and detoxifying the mycotoxins. In the next stages of our research, we will look more into the genes that we inhibited in the sensitive mutants. Understanding exact function of their respective proteins may hold the key to understanding how to improve plant resistance to DON and Tcin. One of the more surprising findings was that only 3 out of the 156 sensitive mutants showed sensitivity to both DON and lincomycin. Lincomycin is known to bind in chloroplast-to-nucleus signaling. Therefore, our results are consistent with what was expected. D1 is a well-known Photosystem II protein that is highly sensitive to stressors such as high light [2]. In this study, we are propagating D1’s known sensitivity to phototoxic stress to include mycotoxic stress. D1 is known to be degraded as a result of light stress [2]. As shown in Figure 3, our blot is consistent with this, because the bands growth fainter as the DON concentration is increased. This is indicative of the degradation of D1. In this blot, we also were hoping to see degradation products of D1 below the band. However, no degradation products appear to be present below the band. The explanation for this may be that the treatment lasted too long. In future studies, we plan to sample the protein at earlier points after toxin treatment to look for aggregates and for D1 protein changes because it is likely given the concentrations we used, the cells can adapt and overcome damage by 2
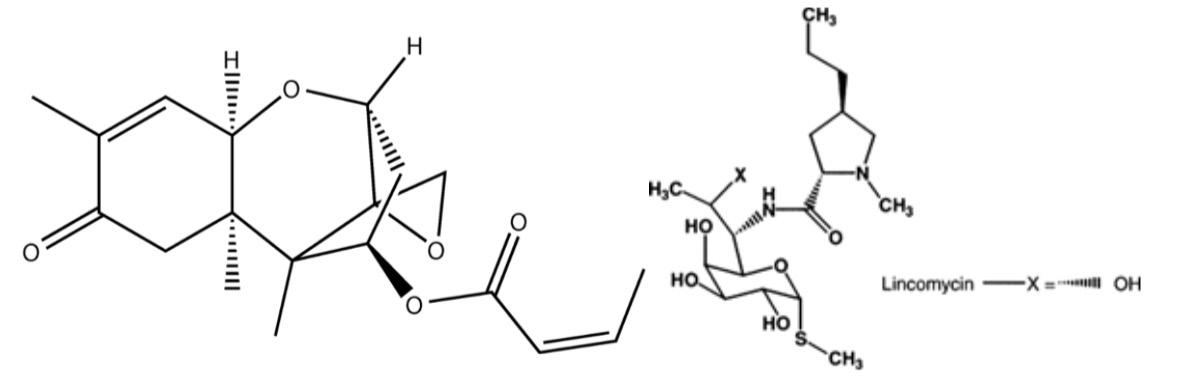
Figure 4. The molecular structures above represent Tcin and Lincomycin respectively. Both are cyclic molecules. to the ribosome and inhibit or alter translation. The lack of overlap indicates that mycotoxins act differently on the cell than lincomycin, meaning that mycotoxins act elsewhere in the cell. According to Figure 2, 1 hour after treatment with DON and Tcin, Vipp2 and Mars1 are both expressed significantly more than after 24 hours. After 24 hours, the cell is likely closer to returning to homeostasis. Increase in expression is a clear indicator of Vipp2’s and Mars1’s involvement in handling mycotoxin-induced stress. Although the fold increase is thousands of times lower in Mars1 and Vipp2, that does not mean that Mars1 is any less significant in stress response. Since Vipp2 is a membrane protein, it must be present in greater quantity to uniformly and densely cover the membrane. Mars1, however, is a kinase involved Figure 5. This pie chart represents the results of the GO analysis. It displays the individual molecular functions of the Arabidopsis ortholog mutants that were identified. 47.6% of mutants are involved in catalyic activity, while 28.6 % are involved in binding.

days in. The degradation products we had hoped to see may appear at shorter periods of time. IV. CONCLUSIONS
Here we report the early stage of the mycotoxin pathway study. Our final goal is to understand the genes that are involved in more significant agricultural organisms that contribute to the human diet. After we identify key genes in organisms such as Chlamydomonas, the research can move on to higher organisms. In the future, we plan to focus on the Arabidopsis orthologs involved in cyclic binding that we identified through the GO analysis. We will study their functions and discover which cellular component they are involved in and identify whether they interact with proteins of interest such as D1, VIPP2, AND MARS1. We plan to run shorter time intervals for harvesting D1 protein after treatment, to confirm the degradation of D1 from mycotoxic stress. As the Chlamydomonas library is updated, we will continue performing GO analyses to potentially identify new Arabidopsis orthologs as well. With the 156 mutants that were identified, we plan to create double knockouts, so that we can begin to understand the protein-protein interactions going on in the cells.
Works Cited
[1] Gene Ontology, C., The Gene Ontology resource: enriching Gold mine. Nucleic Acids Res, 2021. 49(D1): p. D325-D334. [2] Llamas, E. and P. Pulido, A proteostasis network safeguards the chloroplast proteome. Essays in Biochemistry, 2022. [3] Perlaza, K., et al., The Mars1 kinase confers photoprotection through signaling in the chloroplast unfolded protein response. Elife, 2019. 8. [4] Theis, J., et al., VIPP2 interacts with VIPP1 and HSP22E/F at chloroplast membranes and modulates a retrograde signal for HSP22E/F gene expression. Plant, Cell & Environment, 2020. 43(5): p. 1212-1229.
Appendix 1 APPENDICES
Appendix 2








