8 minute read
The Sonochemical Degradation of PFOA and PFOS

from PCR - Fall 2022

by Katia Krishtopa (V), Jitendra A. Kewalramani1, Jay N. Meegoda1
1Department of Civil and Environmental Engineering, New Jersey Institute of Technology, Newark, New Jersey
Abstract
There is a serious public concern about the environmental contamination of soil and water with per- and polyfluoroalkyl substances (PFAS). These compounds are manmade chemicals and are considered to be toxic and persistent in the environment [1]. PFOA (Perfluorooctanoic Acid) and PFOS (Perfluorooctanesulfonic Acid) are part of the vast PFAS group of chemicals and most commonly occurring PFAS pollutants. In this study the sonochemical degradation technology was used to remove PFOA and PFOS from water. The destruction of PFOA and PFOS mixtures in ultrasonic reactor was studied at ambient temperature and in the air atmosphere at various sonication treatment times. The destruction efficiencies of PFOS and PFOA were demonstrated by direct measurement of PFOS and PFOA as well as the defluorination of PFOA and PFOS by the detection of fluoride ions in the water under treatment.
PFOA and PFOS are perfluorooctanoic acid and perfluorooctanesulfonic acid respectively (Figure 1) and are part of the group called PFAS (polyfluoroalkyl substances). PFAS do not exist in nature, they are man-made chemicals. The sources of PFAS in the environment, particularly in ground and surface water, include the firefighting foams, discharge from the manufacturing industries, and consumer goods such as coated fabrics and food packaging. Due to their unique chemistry (C-F bond is the shortest and strongest bond in nature), PFAS are environmentally persistent and bioaccumulative meaning that they are very stable and cannot degrade naturally. Because of its potential carcinogenic effect, PFAS are considered to be potential hazards to human health. The United States EPA (Environmental Protection Agency) recently announced that the PFAS health advisory limit is 0.004 nanograms/ liter in contrast to the previous limit of 70 nanograms/liter which calls for new more advanced remediation technologies which are able to reduce PFAS concentrations in the environment. Sonication or, in other words, the use of sound energy (ultrasound), in environmental engineering is found to be very promising remediation technique for treatment of contaminated soils and waste waters. Since no toxic chemicals are used or produced during this type of treatment it is considered to be environmentally friendly. Among other benefits are high efficiency and a low cost. In this study the sonochemical degradation technology was used to remove PFAS from water. II. METHODS
PFOA (>98.0%) was purchased from TCI America. Potassium perfluorooctanesulfonate (K PFOS) (≥98.0%) was purchased from Sigma-Aldrich. Standards of PFOA (>98%), K-PFOS (>98%) and EPA 533 PFAS standards, were obtained from Wellington Laboratories. Milli-Q water (18.2 MΩ·cm) was used in all experiments and analyses. A high-frequency ultrasonic bath reactor equipped with a 700 kHz plate transducer provided by PCT systems was used for experimental work. The detailed reactor set up is described elsewhere [2,3]. The total maximum power applied was 450 W. Experiments were conducted at ambient temperature (25±2oC) in the air atmosphere in a closed system to avoid any evaporation. The experiments were performed as follows: The desired concentration of PFOA and PFOS mixtures were prepared in Milli-Q water. For testing, 10 Liters of the above prepared solution were placed in the bath reactor for the following ultrasonic treatment. The typical ultrasonic treatment run was 5 hours. For further analyses, duplicate five mL samples were collected before the experiment and every thirty minutes over the treatment time (300 minutes). These samples were further analyzed separately for PFAS and fluoride concentrations. The only known method which allows quantifying individual PFAS destruction is LC/MS/ MS. Therefore, the collected samples were analyzed for PFOA and PFOS concentrations according to the EPA 8327 [4] and EPA 533 methods [5] using liquid chromatography with tandem mass spectrometry (Agilent 6470 Triple Quadrupole LC/MS System [6, 7]). Prior to the LC/MS analysis, all collected water samples were diluted with methanol to have a concentration below 100 ppb and then filtered. A fluoride-ion selective electrode (Thermo-Scientific, F-ISE) was used to follow the formation of inorganic fluoride ions in solution. Collected water samples were diluted with TISAB II (1:1 ratio) for the analysis.
The destruction of PFOA and PFOS mixture in water in ultrasonic reactors was studied at various sonication treatment times. The normalized concentrations of PFOS and PFOA as function of the treatment time for a mixture containing the initial concentrations of PFOS and PFOA of 3.7µM and 6µM, respectively, were determined. The destruction efficiencies of PFOS and PFOA in percentages are demonstrated in Figure 2. As seen in Fig 2, the decomposition is sonication time dependable and at the end of the five hours of continuous treatment, 99% decomposition of PFOA and almost 96% decomposition of PFOS were observed.
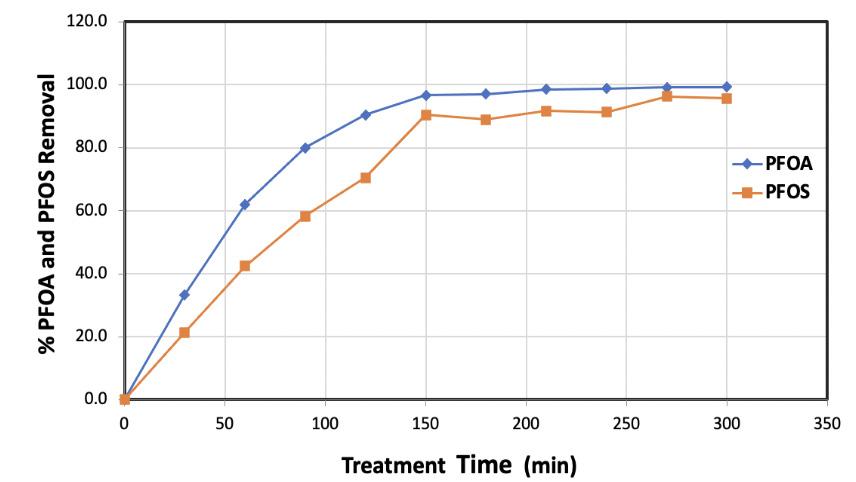
Figure 2. PFOA and PFOS Removal as a function of treatment time
According to the previous studies on the decomposition of PFAS by different technologies [8-5] there is a possibility of a complete destruction (or so called complete mineralization) when the perfluoroalkyl chain of the PFAS molecule is completely defluorinated. The anticipated byproducts formed in this case are exclusively carbon dioxide, carbon oxide or inorganic carbon, and hydrogen fluoride. Depending on the PFAS treatment/destructive techniques, another alternative is a partial defluorination of PFAS molecules leading to the potential formation of by products such as “short chain” hydrocarbons in addition to CO2 and CO. In order to follow the defluorination path of the decomposition, the released fluoride ions were monitored in collected water samples. The concentrations of released fluoride ions into solution after treatment of the described above mixture of PFOA and PFOS were determined. The fluoride concentration in the reactor increases with the time of treatment and maximum achieved concentration after treatment was 112 µM. If we assume that the complete defluorination of both PFOA and PFOS occurs and all fluorine atoms are released into a solution, the anticipated concentration yield of fluoride ions in 10 L of treated solution can be calculated. Knowing that PFOS (C8HF17O3S, Molar Mass=500.13 g/ mol) molecule contained 64.6% fluorine atoms and PFOA (C8HF15O2, Molar Mass=414.07 g/ mol) molecule contained 68.8% fluorine atoms, for the given initial concentrations ([PFOS] o=3.7µM and [PFOA]o=6µM) the resulting total concentration of fluoride ions should be approximately 152 µM. Figure 3 shows the percentage of defluorination achieved in the experiment. There is about 26 % difference (152 µM versus 112 µM) between theoretical fluoride yield and observed in the current experiment. This difference can be attributed to the insufficient sonication time and based on the trend line (Figure 3) could be potentially achieved. However, the untreated PFAS (combined PFOA and PFOS) left after five hours of treatment can be accountable for only a tiny amount of “missing” fluorine atoms (about 9%). This reasonably leads to the possibility of existence of other fluorine containing by-products.
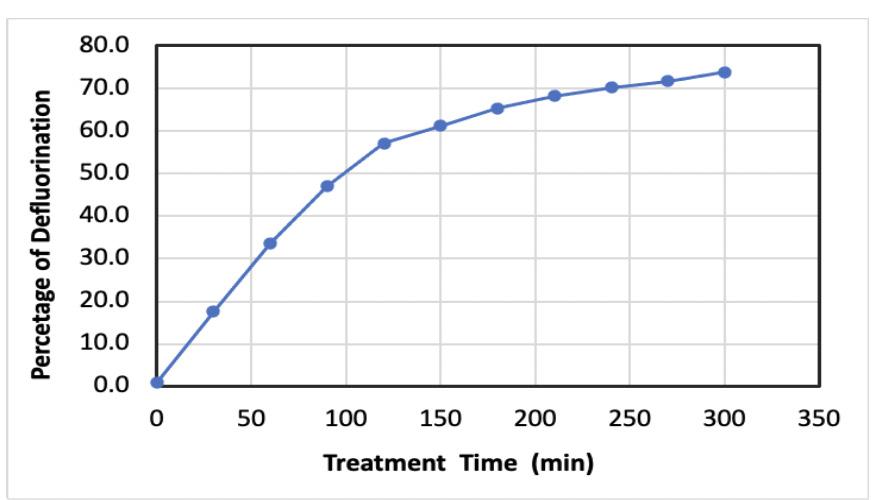
Figure 3. Percentage of defluorination as a function of treatment time
This study was focused on the sonochemical decomposition of the mixture of PFOA and PFOS and its relation to the treatment time. The experiments were conducted in the ultrasonic batch reactor at ambient temperature and in the air atmosphere. After five hours of treatment under experimental conditions, 99% decomposition of PFOA and almost 96% decomposition of PFOS were achieved. The formation of fluoride ions in treated water was detected and 74 % of defluorination of PFAS (PFOS + PFOA) was observed. However, incomplete defluorination leads us to believe in the existence of other fluorine-containing by-products which will be the subject of future study. V. ACKNOWLEDGMENTS
I would like to thank New Jersey Institute of Technology and its High School Summer Research Internship Program (HSSRI) for the opportunity to perform research over the summer. I would like to express my sincere gratitude to my advisor Professor Jay Meegoda and my laboratory mentor, doctoral candidate Jitendra Kewalramani, for providing me the unique opportunity to join and work with their wonderful research group. I would also like to thank the whole research team and my fellow HSSRI students for creating a friendly lab atmosphere and for all the help.
kyl Substances in Drinking Water by Isotope Dilution Anion Exchange Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry, United States Environmental Protection Agency, Office of Ground Water and Drinking Water, https://www.epa.gov/sites/default/files/2019-12/documents/method-533-815b19020.pdf [6] Analysis of Per/Polyfluoroalkyl substances in Water Using Agilent 6470 Triple Quadrupole LC/MS, Agilent Technologies, Application Note 5991-7851EN, 2017, https://www.agilent.com/cs/library/applications/5991-7951EN.pdf [7] EPA Method 533 for Analysis of Per/Polyfluoroalkyl substances in Drinking Water Using Agilent 6470 Triple Quadrupole LC/MS, Agilent Technologies, Application Note 5991-7863EN, 2020, https://www.agilent.com/ cs/library/applications/5991-7863EN.pdf [8] R. K. Singh, S.Fernando, S. F. Baygi, N. Multari, S. M. Thagard, T. M. Holsen, “Breakdown products from perfluorinated alkyl substances (PFAS) degradation in a plasma based water treatment process”, Environ. Sci. Technol. 53 (2019) 2731-2738 [9] H. Moriwaki, Y. Takagi, M. Tanaka, K. Tsuruho, K. Okitsu, Y. Maeda, “Sonochemical Decomposition of perfluorooctane sulfonate and perfluorooctanoic acid”, Environ. Sci. Technol. 39 (2005) 3388-3392. [10] J. Horst, J. McDonough, I. Ross, E. Houtz, “Understanding and managing the potential by-products of PFAS destruction,” Groundwater Monitoring & Remediation. 2020, National Ground Water Association [11] S. Stockenhuber, N. Weber, L. Dixon, J. Lucas, C. Grimison, M. Bennett, M. Stockenhuber, J. Mackie, E. Kennedy, “Thermal degradation of perfluorooctanoic acid (PFOA)”, 16th International Conference on Environmental Science and Technology, 2019.
Works Cited
[1] R.C. Buck, J.Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S.A. Mabury, S. PJ. Van Leeuwen, “Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and Origin, Integrated Environmental Assessment and Management”, 7 (2011) number 4, 513541. [2] J. A. Kewalramani, B. Wang, R.W. Marsh, J. N. Meegoda, L. Rodriguez, “Coupled high and low-frequency ultrasound remediation of PFAS-contaminated soils”, Ultrasonic Sonochemistry 88 2022, 106063. [3] J. A. Kewalramani, R.W. Marsh, D. Prajapati, J. N. Meegoda, “Sonochemical degradation of PFAS and PFOA in Concentrated Waste: Impact of Power Density and Initial Concentration,” submitted to Ultrasonic Sonochemistry, 2022 [4] SW-846 Test Method 8327: Per- and Polyfluorinated Substances (PFAS) by Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS), Hazardous Waste Test Methods, United States Environmental Protection Agency, https://epa.gov/system/files/documents/2021-07/8327.pdf [5] Method 533: Determination of Per- and Polyfluoroal-







