zugelassene
Gentherapie bei Hämophilie B
Inhibitoren in der Vorgeschichte erhielt [3].
24-Monats-Daten der HOPE-BStudie zeigen langfristig stabile FIX-Aktivität
Zulassungsrelevant waren die Ergebnisse der offenen einarmigen Studie HOPE-B, an der 54 Erwachsene mit mittelschwerer und schwerer Hämophilie B (FIX-Aktivität ≤ 2 %) mit Faktor-IX-(FIX) Prophylaxe teilnahmen [4, 5]. In einer sechsmonatigen Lead-in-Phase, in der die Patienten weiterhin ihre Standardtherapie verwendeten, wurde die jährliche Blutungsrate (annualized bleeding rate, ABR) als
Referenzwert ermittelt. Danach erhielten die Studienteilnehmer eine einmalige Infusion Etranacogen dezaparvovec in einer Dosis von 2 1013 gc/kg Körpergewicht (gc: genome copies, Vektorgenom).
Primärer Endpunkt war die 52-wöchige ABR nach Erreichen einer stabilen FIX-Transgenexpression, gemessen von Monat 7 bis 18 nach Infusion [4].
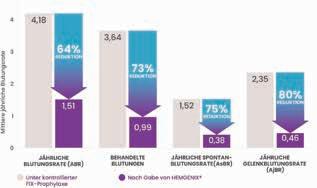
Von der laufenden Studie liegen bereits 24-Monats-Daten vor: Im Vergleich zur Lead-in-Phase verringerte sich die bereinigte ABR signifikant um 64 % (mean ABR 1,51; p = 0,0002), die Rate aller mit FIX behandelten Blutungen um 73 % (p = 0,0001). Die bereinigte annualisierte spontane Blutungsrate
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 44 FORUM HÄMOPHILIE
Abbildung 1: Häufigkeit und Charakteristika von Einblutungen bei Hämophilie (© CSL Behring).
(AsBR) ging um 75 % (p = 0,0005) und die bereinigte annualisierte Gelenkblutungsrate (AjBR) um 80 % (p = 0,0001) zurück – alle Differenzen waren signifikant (Abb. 2) [4]. 96 % der Patienten konnten die Faktor-IX-Prophylaxe beenden. Die mittlere FIX-Aktivität stieg stabil und dauerhaft auf 36,66 IE/dl (±19,0; 4,7–99,2; Abb. 3) [4].
Ausgewogenes Sicherheitsprofil: Stabile FIX-Aktivität trotz AntiAAV5-Antikörper
Etranacogen dezaparvovec verfügt über ein ausgewogenes Sicherheitsprofil, schwerwiegende unerwünschte Wirkungen wurden nicht festgestellt. Zu den häufigen Nebenwirkungen gehören u.a. Kopfschmerzen, grippeähnliche Symptome sowie infusionsbedingte Reaktionen. In der HOPE-B-Studie entwickelten 9 von 54 Teilnehmern (16,7 %) eine behandlungsbedürftige Erhöhung der AlaninAminotransferase (ALT). Der Anstieg der Leberenzymwerte ist eine immunvermittelte Reaktion auf die intravenöse Verabreichung des Adeno-assoziierten viralen Vektors vom Serotyp 5 (AAV5) und trat meist in den ersten 3 Monaten auf. Um die Transgenexpression nicht zu gefährden, erfolgte in diesen Fällen vorübergehend eine Behandlung mit einem Kortikosteroid (Prednisolon). Wie erwartet, wurde bei allen Patienten eine anhaltende humorale Immunantwort auf den infundierten AAV5-Vektor beobachtet, FIX-Hemmkörper entwickelte keiner der Studienteil-
Überlegener Blutungsschutz im Vergleich zur FIX-Prophylaxe
Abbildung 2: Ergebnisse der HOPE-B-Studie nach einem Follow-up von 24 Monaten. Die einmalige Verabreichung von Etranacogen dezaparvovec (Hemgenix®) führte zu einer stabilen, signifikanten Reduktion der Blutungen im Vergleich zur 6-monatigen Lead-in-Phase unter bestehender Prophylaxe. Die ABR für alle Arten von Blutungen nach stabiler FIX-Expression sank von einem Mittelwert von 4,18 in der Lead-inPhase (alle Patienten befanden sich hier unter einer optimierten und kontrollierten durchgeführten Prophylaxe) auf einen Mittelwert von 1,51 (p = 0,0002) in den Monaten 7 – 24 nach der Infusion von Hemgenix® [4].
Langfristig gesteigerte Faktor-IX-Spiegel
Abbildung 3: Die mittlere FIX-Aktivität (gemessen mit einem Einstufen-FIX-Assay, OSA; n = 50) lag 18 Monate nach der Verabreichung bei 36,90 % (SD 21,40) und blieb auch 24 Monate nach der Verabreichung stabil (36,66 %; SD: 18,96; Median 33,85 %).
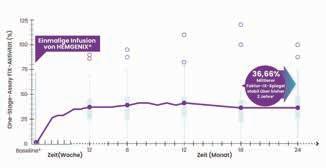
* Mittlere Baseline-Fix-Aktivität 1 % [4].
nehmer. Patienten mit und ohne vorbestehende neutralisierende Anti-AAV5-Antikörper zeigten bis
zu einem Titer von 1:687 keine klinisch relevanten Unterschiede im Ansprechen [4].
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 45 FORUM HÄMOPHILIE
Wirkmechanismus von Etranacogen dezaparvovec
Hämophilie B wird verursacht durch vererbte oder spontane Mutationen im F9-Gen (d.h. monogenetisch), das für den Gerinnungsfaktor IX (FIX) kodiert. Bei der Gentherapie wird mittels eines vermehrungsunfähigen Adeno-assoziierten viralen (AAV) Vektors ein funktionierendes Gen in die Leberzellen eingeführt, das die FIX-Produktion kodiert, sodass dem Körper wieder Faktor-IX-Protein zur Verfügung steht.
Etranacogen dezaparvovec (Hemgenix®) besteht aus einem rekombinanten AAV5-Kapsid und der natürlich vorkommende Padua-Genvariante des Faktor-IX mit einem Leber-spezifischen Promotor. Die Padua-Variante ist 6- bis 8-mal aktiver als der in der Allgemeinbevölkerung normalerweise vorkommende Wildtyp. In den Hepatozyten bildet das Transgen ein stabiles Episom [5]. Obwohl der AAV5-Vektor nicht-integrierend ist, besteht ein theoretisches Risiko für eine Insertionsmutagenese – jedoch ist ein solches Ereignis in der Vergangenheit nicht beobachtet worden. Erkrankungen, verursacht durch natürlich vorkommende AAVs, sind bislang nicht bekannt und der Vektor weist eine vergleichsweise geringe Immunogenität auf.
Nach erfolgreicher Transduktion in der Zielzelle kommt es zu einer effektiven endogenen FIX-Produktion, was für einen kontinuierlichen und anhaltenden Schutz vor Blutungen sorgen kann [3].
Wirksamkeitsdauer: Modelle gehen von über 25 Jahren FIX-Aktivität aus
Eine wichtige Frage im Zusammenhang mit der Gentherapie ist, wie lange eine ausreichend hohe FIX-Aktivität nach der einmaligen Behandlung bestehen bleibt. Um eine Vorhersage für Etranacogen dezaparvovec treffen zu können,
wurden zwei statistische Analyseverfahren kombiniert. Die eingeflossenen Daten stammen aus der Phase-IIb- und der HOPE-B-Studie. Das Ergebnis der Modellierung*: Über 80 % der künftigen Patienten mit Hämophilie B, die einmalig mit Etranacogen dezaparvovec behandelt werden, benötigen bis zu 25,5 Jahre keine routinemäßige FIX-Ersatztherapie mehr (Aktivi-
tätsgrenze im Modell ≥2 IE/dl). Ein weiteres Modell mit Grenzziehung bei ≥5 IE/dl (milder Phänotyp) sagt für über die Hälfte der Patienten ein prophylaxefreies Leben für eine Dauer von mehr als 25 Jahren nach Infusion voraus [6].*
Fabian Sandner, Nürnberg
Literatur
1 Gualtierotti R et al. J Thromb Haemost 2021;19:2112-2121
2 Zanon E et al. Blood Transfus 2019; 17:378-384
3 Fachinformation Hemgenix®; Stand: Februar 2023
4 Pipe SW et al. Poster 2141, 64. ASH; https://ash.confex.com/ash/2022/webprogram/Paper166135.html
5 Pipe SW et al. N Engl J Med 2023; 388:706-718
6 Srivastava A et al. WFH Guidelines for the Management of Hemophilia, 3rd edition, Haemophilia 2020;Suppl 6:1-158. Erratum in: Haemophilia 2021;27:699
* Die Modellrechnungen beruhen auf einer geringen Datenbasis und sind nicht geeignet, tatsächlich belastbare Voraussagen für den Einzelfall zu treffen.
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 46 FORUM HÄMOPHILIE
Rund 40 % der Patienten mit Typ2-Diabetes (T2D) entwickeln im Verlauf eine chronische Nierenerkrankung (CKD) [1]. Bei dieser Konstellation besteht trotz der Standardtherapie (z.B. Behandlung mit SGLT2- und RAS-Inhibitoren), die vorrangig auf hämodynamische und metabolische Faktoren abzielt (Abb. 1) [2, 3], ein hohes Risiko für eine Progression der CKD und für kardiovaskuläre Ereignisse. Die kardiovaskuläre Mortalität steigt mit zunehmenden Einbußen der Nierenfunktion rapide an [4]. Bei T2D mit Albuminurie und eingeschränkter geschätzter glomerulärer Filtrationsrate (eGFR) ist die Mortalität um bis zu mehr als das Fünffache höher als bei T2D mit intakter Nierenfunktion (Abb. 2). Das Zusammentreffen von Diabetes und CKD geht demnach mit drastischen Einbußen der Lebenserwartung einher [5].

Prof. Dr. Christoph Wanner, Würzburg
Finerenon ist zur Behandlung von chronischer Nierenerkrankung (mit Albuminurie) in Verbindung mit Typ-2-Diabetes bei Erwachsenen zugelassen.
Innovativer Nieren- und Herzschutz:
Finerenon jetzt auch für frühe Stadien der chronischen Nierenerkrankung bei Typ-2Diabetes zugelassen
Treibende Faktoren der CKD-Progression bei T2D ACE-Hemmer und ARBs Thiazid-artige Diuretika und Dihydropyridin-Ca2+-KanalBlocker

Hämodynamische (erhöhter BD und/oder intraglomerulärer Druck)
SGLT2-Inhibitoren
Keycharts Prof. Dr. Thomas Ebert, Leipzig
SGLT2-Inhibitoren
GLP-1-RAs
Metformin Sonstige Antidiabetika

Eine neue MRA-Generation zum Schutz von Nieren und Herz
Mit Finerenon (Kerendia®), einem innovativen nichtsteroidalen Mineralokortikoidrezeptor-Antagonisten (nsMRA), eröffnet sich nun die Möglichkeit, einen bislang nicht adressierten treibenden Faktor der CKD-Progression bei T2D anzugehen. Als erster und bislang einziger nsMRA, der zur Therapie der CKD bei Erwachsenen mit T2D zugelassen ist, greift sein innovativer Wirkmechanismus am bei T2D überaktivierten Mineralokortikoidrezeptor an und entfaltet auf diese Weise antientzündliche und antifi-
PP-KER-DE-0176-01
Entzündung und Fibrose
???
Metabolische (schlechte glykämische Kontrolle)
Abbildung 1: Die Standardtherapien der CKD bei T2D zielen vorrangig auf hämodynamische und metabolische Faktoren ab [2, 3].
Zusätzliches Risiko
Abbildung 2: Standardisierte kumulative 10-Jahres-Inzidenz der Mortalität bei Typ2-Diabetes nach CKD-Status (mod. nach [2]). Verglichen mit T2D allein erhöht eine gleichzeitige CKD die Mortalität um bis zu mehr als das Fünffache und kann die Lebenserwartung von Patienten mit T2D im Vergleich zur Allgemeinbevölkerung um bis zu 16 Jahre verkürzen [3]. CKD ist definiert als erhöhte Albuminurie (UACR ≥30 mg/g), eingeschränkte eGFR (≤60 ml/min/1,73 m2) oder beides.
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 47 FORUM DIABETICUM
0 10 20 30 40 50 60 70 T2D & keine Niereninsuffizienz T2D & Albuminurie T2D & eingeschränkte GFR T2D,
10-Jahres-Inzidenz der Mortalität (95-%-KI) ×2,5 ×
>
Albuminurie und eingeschränkte GFR
3
×5
von Nieren und Herz
Finerenon reduziertenach4 Monatendie UACR um 32
imVergleichzuPlacebo
%
Über den gesamten Studienzeitraum wurde unter Finerenoneine niedrigere UACR als unter Placebo beibehalten.
brotische Effekte [6]. Auf pharmakologischer Ebene unterscheidet sich der Wirkstoff also deutlich von herkömmlichen MR-Antagonisten. Aufgrund der überzeugenden Ergebnisse der Studie FIDELIO-DKD [7], die die Wirkung von Finerenon im Vergleich zu Placebo jeweils zusätzlich zur leitliniengerechten Standardtherapie hinsichtlich renaler und kardiovaskulärer Ergebnisse untersuchte, erhielt Finerenon die Zulassung zur Behandlung der CKD Stadien 3 und 4 mit Albuminurie bei Erwachsenen mit TD2. Im Februar 2023 hat die Europäische Kommission die Zulassung erweitert. Finerenon kann nun auch in frühen Stadien (Stadien 1 – 2, mit einer eGFR von ≥60 ml/min/1,73 m2 gemäß KDIGO-Klassifizierung) der mit T2D assoziierten CKD eingesetzt werden. Finerenon ist jetzt für die Behandlung der CKD (mit Albuminurie) im Zusammenhang mit T2D bei Erwachsenen indiziert [6].
Signifikante Risikosenkung bei einem breiten Patientenspektrum
Ausschlaggebend für die Zulassungserweiterung waren die Ergebnisse der Phase-III-Studie
FIGARO-DKD [8], die – wie die von FIDELIO-DKD [7] – bestätigten, dass Finerenon im Vergleich zu Placebo das Risiko von kardiovaskulären Ereignissen bei einem breiten Spektrum von erwachsenen Patienten mit CKD (mit Albuminurie) und T2D signifikant senken kann. Gemeinsam mit den Daten der Pha-

Die statistischen Analysen sind explorativ. Es wurden keine konfirmatorischen Hypothesentests durchgeführt. Falls statistische Tests durchgeführt wurden, sind die p-Werte explorativ. Daten in KlammernsindmittlereÄnderungengegenüberBaseline.
Abbildung 3: Ergebnisse der FIDELITY-Analyse für den kombinierten Nierenendpunkt (Zeit bis zum Nierenversagen, anhaltender eGFR-Rückgang um ≥57 % vs. Baseline oder Tod durch Nierenversagen): Zusätzlich zur maximal verträglichen RASTherapie senkte Finerenon (Kerendia®) den kombinierten Nierenendpunkt signifikant um 23 % [9].
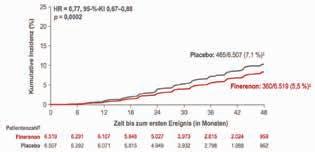
‡ Kumulative Inzidenz berechnet mittels Aalen-Johansen-Schätzer unter Verwendung von Todesfällen aufgrund anderer Ursachen als konkurrierendem Risiko. ¶ Anzahl Patienten mit einem Ereignis über die mediane Beobachtungszeit von 3,0 Jahren.
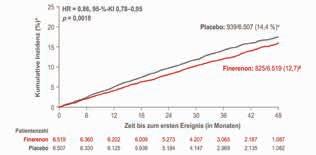
Abbildung 4: Ergebnisse der FIDELITY-Analyse für den kombinierten kardiovaskulären Endpunkt (Zeit bis zum CV-Tod, zum nicht tödlichen MI, nicht tödlichen Schlaganfall oder bis zur Hospitalisierung aufgrund von HF): Zusätzlich zur maximal verträglichen RAS-Therapie senkte Finerenon (Kerendia®) den kombinierten CV-Endpunkt signifikant um 14 % [9].
* Kumulative Inzidenz berechnet mittels Aalen-Johansen-Schätzer unter Verwendung von Todesfällen aufgrund anderer Ursachen als konkurrierendem Risiko, # Anzahl Patienten mit einem Ereignis über einen medianen Nachbeobachtungszeitraum von 3,0 Jahren.
se-III-Studie FIDELIO-DKD liegen nunmehr Ergebnisse von mehr als 13.000 Patienten mit CKD und T2D vor, die in der präspezifizierten
explorativen gepoolten Analyse FIDELITY [9] ausgewertet wurden. Diese Analyse deckt ein breites Patientenspektrum ab: T2D-Patienten
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 48 FORUM DIABETICUM
UACR:Urin-Albumin-Kreatinin-Quotient Agarwal R, et al. EurHeart J 2022;43:474–484 0,2 0,4 0,6 0,8 1,0 1,2 0 12 24 36 48 GeometrischesMittel(gSD) UACR zuBaseline: Placebo: 454,3 (3,5) Finerenon: 445,4(3,5) 6.519 6.507 Patientenanzahl Finerenon Placebo 6.273 6.239 4.867 4.829 2.745 2.706 (–36,3%) (–6,4%) (–43,1%) (–5,5%) (–41,1%) (–4,8%) (–35,8%) (–1,1%) UACR LS mittlereRatio vs. Baseline Placebo Finerenon MonateseitRandomisierung Ratio LS Mittel 0,68; 95% KI 0,66–0,70 (+1,8%) 899 872 (–27,0%) 4 5.988 5.973 p<0,0001
mit den CKD-Stadien 1 – 4 plus Albuminurie, die eine optimierte Renin-Angiotensin-Inhibitor-Therapie erhielten und gut eingestellte Blutdruck- und HbA1c-Werte hatten. Kombinierter renaler Endpunkt war die Zeit bis zum Nierenversagen, ein anhaltender Rückgang der eGFR um mindestens 57 % gegenüber dem Ausgangswert oder Tod durch Nierenversagen. Für den kombinierten kardiovaskulären Endpunkt wurde die Zeit bis zu einem kardiovaskulär bedingten Tod, einem nicht tödlichen Myokardinfarkt oder Schlaganfall und einer Herzinsuffizienz-bedingten Hospitalisierung (HHF) erfasst. Zusätzlich zu einer optimierten RAS-Blockade verabreicht, senkte Finerenon signifikant das Risiko für den kombinierten Nierenendpunkt um 23 % (HR: 0,77; 95%-KI: 0,67 – 0,88; p = 0,0002; Abb. 3). Das Risiko für terminale Niereninsuffizienz nahm signifikant um 20 % ab. Bereits nach 4 Monaten Therapie zeigte sich eine Verbesserung der UACR (Albumin-Kreatinin-Quotient im Urin) um 32 % im Vergleich zu Placebo. Auch das Risiko für den kombinierten kardiovaskulären Endpunkt verringerte sich unter Finerenon signifikant um 14 %, wobei der Nutzen von Finerenon hier hauptsächlich auf die Abnahme der HHF und der kardiovaskulären Mortalität zurückzuführen war [9].
Nebenwirkungen weitgehend auf Placeboniveau
Finerenon erwies sich in der FIDELITY-Kohorte als gut verträglich.
Die Inzidenz unerwünschter Ereignisse lag weitgehend auf Placeboniveau. Es gab kaum geschlechtshormonelle Nebenwirkungen. Der Blutdruck verminderte sich im Durchschnitt um 3 – 4 mmHg. Es kann zwar zu einer leichten Zunahme des Serumkaliums kommen, die Hyperkaliämien lassen sich aber durch routinemäßige Kontrollen des Kaliumspiegels und mit kurzen Therapiepausen gut beherrschen. Empfehlenswert ist, vor Therapiebeginn die eGFR sowie das Serumkalium zu bestimmen und bei Werten <5 mmol/l mit der Anfangsdosis von 10 mg Finerenon 1 × täglich zu starten*. Nach 4 Wochen sollten das Serumkalium und die eGFR erneut kontrolliert werden, um dann die Dosis entsprechend anzupassen. Die empfohlene Ziel- und Höchstdosis von Finerenon beträgt 20 mg 1 × täglich.
Empfehlungen für Finerenon in den Leitlinien auf höchstem Niveau
Auf Basis der umfangreichen Studiendaten haben internationale Fachgesellschaften den klinischen Nutzen von Finerenon erkannt und den Wirkstoff bereits als neues, einzigartiges Wirkprinzip konsistent mit dem höchsten Evidenz-Level
* Bei einem Serumkalium-Wert von ≤4,8 mmol/l kann die Behandlung mit Finerenon begonnen werden, bei >4,8 bis 5,0 mmol/l kann der Behandlungsbeginn unter zusätzlicher Überwachung des Serumkaliums während der ersten 4 Wochen auf Basis der Patientencharakteristika und des Serumkalium-Spiegels erwogen werden.
A in relevante internationale Leitlinien und Praxisempfehlungen zum Diabetes-Management bei chronischen Nierenerkrankungen aufgenommen.
Laut den Empfehlungen des ADA/ KDIGO-Konsensusberichts ist die Gabe von Finerenon bei Patienten mit T2D, einer eGFR ≥25 ml/ min/1,73m2, normalem Serumkaliumspiegel und Albuminurie (≥30 mg/g) trotz maximal verträglicher Renin-Angiotensin-InhibitorDosis zu empfehlen [10], da die Patienten deutlich von dem multifaktoriellen Ansatz profitieren können.
Brigitte Söllner, Erlangen
Literatur
1 Doshi SM et al. Clin J Am Soc Nephrol 2017;12:1366-1373
2 Alicic RZ et al. Clin J Am Soc Nephrol 2017;12:2032-2045
3 Mora-Fernández C et al. J Physiol 2014;18:3997
4 Afkarian M et al. J Am Soc Nephrol 2013;24:302-308
5 Wen CP et al. Kidney Int 2017;92:388396
6 Fachinformation Kerendia®; Stand: Februar 2023
7 Bakris GB et al. N Engl J Med 2020; 383:2219-2229
8 Pitt B et al. N Eng J Med 2021; 385:2252-2263
9 Agarwal R et al. Eur Heart J 2021;00:112
10 de Boer IH et al. Diabetes Care 2022; 45:3075-3090
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 49 FORUM DIABETICUM
Die Diagnose chronische Herzinsuffizienz (HF) bringt für die Betroffenen ein erhebliches Risiko für Morbidität und Mortalität mit sich: Die Sterblichkeitsrate innerhalb von 5 Jahren nach der Diagnose liegt bei 42 % [1]. Die aktuellen Leitlinien zur effektiven Therapie der Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF) empfehlen die Kombination von HF-Medikamenten verschiedener Wirkstoffklassen, die gleichzeitig kombiniert werden sollen. Dennoch bleibt für die Patienten unter dieser Basistherapie ein hohes Restrisiko für eine Verschlechterung: So zeigen klinische Studien, dass trotzdem immer noch ein 15%iges Risiko für eine Hospitalisierung besteht.
Kommt es trotz der Basistherapie zu einem Dekompensationsereignis, wirkt dieses als Trigger, sodass sich die 1-Jahres-Mortalität um den Faktor 4 erhöht. In dieser Hochrisiko-Situation benötigen die Patienten besondere Aufmerksamkeit und müssen intensiv sowie schnellst möglich behandelt werden. Einen zusätzlichen Schutz, von dem die Patienten neben der Basistherapie profitieren können, bietet der sGCStimulator Vericiguat (Verquvo®*) [2, 3], der in der aktuellen ESCLeitlinie in dieser Indikation empfohlen wird [4].
Herzinsuffizienz nach Dekompensation: Vericiguat bietet zusätzlichen Schutz zur Basistherapie
GB-A-Nutzenbewertung: Die EU Label Population profitiert von Vericiguat
bedingten Hospitalisierung um 6,3 pro 100 Patientenjahre (HR: 0,86; p = 0,001; NNT = 16).
* Vericiguat (Verquvo®) ist zugelassen zur Behandlung der symptomatischen, chronischen Herzinsuffizienz bei Erwachsenen mit reduzierter Ejektionsfraktion, die nach einem kürzlich aufgetretenen Dekompensationsereignis, das eine i.v. Therapie erforderte, stabilisiert wurden [3].
Vericiguat überzeugt in der Therapie der chronischen Herzinsuffizienz nach Dekompensation. Laut der Nutzenbewertung des GB-A zeigt Vericiguat in der Praxis robuste Effekte in der für den praktischen Einsatz relevanten EU Label Population mit einer HF bei einer Ejektionsfraktion von weniger als 40 % [5]. Die Gabe von Vericiguat erfolgte laut Nutzenbewertung zusätzlich zur Standardtherapie, das heißt bei Patienten, die eine leitliniengerechte patientenindividuelle Behandlung der Herzinsuffizienz sowie der Grund- und Begleiterkrankungen erhalten hatten. In dieser für den praktischen Einsatz relevanten EU Label Population führte Vericiguat im Vergleich zu Placebo, jeweils zusätzlich zur Standardtherapie, zu einer statistisch signifikanten absoluten Reduktion des primär kombinierten Endpunkts aus kardiovaskulärem Tod oder Herzinsuffizienz-bedingter Hospitalisierung um 5,3 % pro 100 Patientenjahre (Hazard Ratio: 0,88; p = 0,008; Number Needed to Treat: 19) sowie zu einer absoluten Risikoreduktion der gesamten Herzinsuffizienz-
Die Nutzenbewertung umfasste auch Daten zur Lebensqualität, die mit dem Kansas City Cardiomyopathy Questionnaire (KCCQ) –ein Messinstrument zur Erfassung der Lebensqualität bei chronischer Herzinsuffizienz – erhoben wurden. Demnach zeigten mehr Patienten mit Vericiguat eine Verbesserung der Lebensqualität und weniger eine Verschlechterung [5].
Nebenwirkungen auf Placeboniveau
Das günstige Sicherheitsprofil von Vericiguat wurde bereits in der Zulassungsstudie VICTORIA [2] dokumentiert. Die Rate unerwünschter Ereignisse lag unter Vericiguat plus Basistherapie auf Placeboniveau (Basistherapie allein, Abb. 1) und die Adhärenz für die Zieldosis (10 mg Vericiguat einmal täglich) betrug ca. 90 %. Vericiguat zeigte in der VICTORIA-Studie keinen signifikanten Einfluss auf den Blutdruck im Vergleich zu Placebo und ist bei Patienten bis zu einer eGFR ≥15 ml/min/1,73 m² anwendbar. Es ist keine Elektrolyt-Überwachung
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 50 FORUM CARDIOLOGICUM
Abbildung 1: In der VICTORIA-Studie waren die Raten unerwünschter Ereignisse (UE) und schwerer unerwünschter Ereignisse (SUE) unter Vericiguat numerisch niedriger als unter Placebo [2].
Vericiguat
Der Wirkstoff Vericiguat (Verquvo®) ist ein einmal täglich oral einzunehmender, direkter Stimulator der löslichen Guanylatzyklase (sGC). Dieses Enzym stellt einen essenziellen Bestandteil des NO-sGC-cGMP-Signalweges im Körper dar, denn es unterstützt die Funktion des Herzens und der Blutgefäße. Bei Patienten mit Herzinsuffizienz wird die sGC jedoch nur unzureichend stimuliert, sodass es zu einer Fehlfunktion des Herzmuskels und zur Progression der Herzinsuffizienz kommt. Vericiguat stellt die beeinträchtigte Funktion dieses Signalweges wieder her. Die lösliche Guanylatzyklase, das Zielenzym von Vericiguat, wird bisher von keiner verfügbaren Herzinsuffizienztherapie adressiert. Damit wirkt Vericiguat komplementär und additiv zu anderen HF-Therapien [3].
notwendig. Die Rate symptomatischer Hypotonien unter Vericiguat war vergleichbar mit der unter Placebo (9,1 % versus 7,9 %, Differenz nicht signifikant) [6].
Fazit für die Praxis
Aufgrund seiner sehr guten Verträglichkeit eignet sich Vericiguat
auch bei vulnerablen Patienten und ist mit bestehenden Basistherapien gut kombinierbar. Bei dieser progredienten Erkrankung bietet Vericiguat mit seinem in der Herzinsuffizienz-Therapie bisher nicht verwendeten Wirkansatz eine wirksame, sehr gut verträgliche und einfach zu kombinierende Therapieoption für Patienten mit einem hohen Risiko für eine weitere Ver-
schlechterung der Herzinsuffizienz. In Frage kommen Patienten, die sich trotz optimaler Basistherapie wieder verschlechtern oder hospitalisiert werden müssen, bei denen die Basistherapien aufgrund von Kontraindikationen nicht angewendet werden können oder diese aufgrund von aktuellen klinischen Parametern nicht eingesetzt oder vollständig aufdosiert werden können.
Der Einsatz des sGC-Stimulators ist schon frühzeitig nach der ersten Dekompensation parallel zur Optimierung der Basistherapie möglich, nachdem die Patienten stabilisiert wurden. Die Aufdosierung bis zur maximalen Zieldosis von 10 mg erfolgt initial mit 2,5 mg einmal täglich für 14 Tage, anschließend 5 mg einmal täglich für 14 Tage und schließlich 10 mg/d.
Brigitte Söllner, Erlangen
Literatur
1 Loehr LR et al. Am J Cardiol 2008;101: 1016-1022
2 Armstrong PW, et al. N Engl J Med 2020;382:1883-1893
3 Fachinformation Verquvo®; Stand: Juli 2021
4 McDonagh TA et al. Eur Heart J 2021; 36:3599-3726
5 1. Nutzenbewertung nach § 35a SGB V zum Wirkstoff Vericiguat V (Vorgangsnummer 2021-09-15-D-724), Anhaltspunkt für einen geringen Zusatznutzen
6 Armstrong PW et al. JACC Heart Fail 2018,6:96-104
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 51 FORUM CARDIOLOGICUM
81,0 100 90 80 70 60 50 40 30 20 10 0 Patienten (%) Placebo Vericiguat 80,5 UE SUE 34,8 32,8
Obwohl sich bei Patienten mit symptomatischer Herzinsuffizienz und eingeschränkter Pumpfunktion (HFrEF, Ejektionsfraktion ≤35 %) mit der leitliniengerechten medikamentösen Therapie (guideline-directed medical therapy, GDMT [1]) die immer noch hohe Morbidität und Mortalität reduzieren lassen, ist die Verbesserung der körperlichen Leistungsfähigkeit und Lebensqualität der Patienten nicht immer ausreichend. Die Zahl an Hospitalisierungen ist noch immer hoch, und jede Einweisung bedeutet eine Verschlechterung des Allgemeinzustands sowie einen Verlust an Lebensqualität. Die GDMT verbessert zwar die Herzfunktion, aber die Patienten leiden häufig unter chronischer Müdigkeit, Dyspnoe und peripheren Ödemen. Das zeigt den hohen ungedeckten medizinischen Bedarf, kardiovaskuläre Erkrankungen wie Herzinsuffizienz und Hypertonie neben der medikamentösen Therapie auch mit innovativen Technologien zu behandeln.
Symptomatische Herzinsuffizienz:
Länger besser leben mit der Baroreflex-Aktivierungstherapie
Halsschlagader sendet (Abb. 1). Diese Mechanorezeptoren werden normalerweise durch die Dehnung der Gefäßwand aktiviert und halten den arteriellen Blutdruck auf einem konstanten Niveau, indem sie entsprechende Signale an die Medulla oblongata senden, die den Blutdruck nach dem Prinzip der negativen Rückkopplung reguliert: Steigt der Blutdruck, erhöht sich die Aktivität der Barorezeptoren. Als Folge wird die Aktivität des Parasympathikus am Herzen und an anderen
Organen gesteigert und gleichzeitig die Aktivität des Sympathikus am Herzen, an den Blutgefäßen, Nebennieren, Nieren, der Lunge und anderen Organen verringert. Das autonome Nervensystem wird wieder ins Gleichgewicht gebracht, sodass Blutdruck und Herzfrequenz sinken und sich die linksventrikuläre Auswurffraktion verbessert. Ein entscheidender Vorteil dieser Therapie ist, dass keine Sonde ins Herz oder in Gefäße implantiert wird. Sie kommt daher auch für
Das Barostim™-System –die erste neuromodulative Device-Therapie

Eine vielversprechende Option ist die Baroreflex-Aktivierungstherapie mit dem Barostim™-System [2]. Diese von CVRx entwickelte innovative Technologie nutzt die Neuromodulation, um die Symptome von HFrEF-Patienten zu verbessern. Dazu wird dem Patienten ein Impulsgenerator implantiert, der elektrische Impulse an die Barorezeptoren in der Wand der
Abbildung 1: Das Barostim™-System. Das Gerät zur Barorezeptorstimulation wird dem Patienten in Vollnarkose implantiert. Dabei wird eine Elektrode auf die Barorezeptoren im Bereich der Halsschlagader platziert und mit einem Impulsgeber verbunden. Dieser wird ähnlich wie ein Herzschrittmacher unter die Haut oder zwischen den Brustmuskel eingesetzt (© CVRx).
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 52 FORUM CARDIOLOGICUM
Abbildung 2: Ergebnisse der BeAT-HF-Studie (Prä-Market-Phase) für die primären Endpunkte [3].
Patienten infrage, die beispielsweise bereits einen Herzschrittmacher bzw. eine kardiale Resynchronisationstherapie (CRT) oder einen implantierbaren Kardioverter-Defibrillator (ICD) erhalten haben oder aller Voraussicht nach benötigen werden.
Das Barostim™-System verfügt über die CE-Kennzeichnung und ist in der EU zur Behandlung von herzinsuffizienten Patienten (NYHAKlasse III und einer LVEF ≤35 %
trotz Behandlung mit der entsprechenden leitliniengerechten Herzinsuffizienz-Therapie) sowie von Hypertoniepatienten zugelassen.
Signifikante klinisch relevante Verbesserungen
Dass die neuromodulative DeviceTherapie mit Barostim™ direkt die autonome Dysfunktion bei HFrEF adressieren kann, sodass sich die
Herzinsuffizienssymptome und die körperliche Belastbarkeit signifikant verbessern, belegen die Ergebnisse der Zulassungsstudie BeAT-HF [3]. Diese Studie besteht aus einer Prä-Market-Phase mit 264 Studienteilnehmern, deren Ergebnisse für die Zulassung relevant waren, und einer Post-Market-Phase mit weiteren 59 Patienten, in der Langzeitdaten für die Wirksamkeit und Sicherheit der Therapie mit Barostim™ erhoben wurden. Die eingeschlossenen HFrEF-Patienten erhielten entweder eine leitliniengerechte Herzinsuffizienztherapie (GDMT) plus eine Baroreflex-Aktivierungstherapie (BAT-Gruppe) oder nur eine GDMT (KontrollGruppe).
Nach Abschluss der 6 Monate dauernden Prä-Market-Phase zeigte sich, dass die kombinierte Therapie aus GDMT plus BAT hinsichtlich aller primären Endpunkte der alleinigen GDMT überlegen war: In der BAT-Gruppe (n = 130) kam es zu einer signifikanten Verbesserung der körperlichen Leistungsfähigkeit (erhoben mit dem 6-MinutenGehtest), der Lebensqualität und der NYHA-Klasse, außerdem sank der NT-proBNP-Spiegel signifikant im Vergleich zur Kontrollgruppe (n = 134; Abb. 2).
Überzeugende Wirksamkeit und Sicherheit auch nach 24 Monaten
Auf der diesjährigen Jahrestagung der Deutschen Gesellschaft für Kardiologie (DGK) wurden die Daten der Post-Market-Phase der
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 53 FORUM CARDIOLOGICUM
-14 -21 0 -5 -10 -15 -20 -25 Barostim Kontrolle Diff. -14 Punkte Klinisch aussagekräftig -5 Punkte p < 0,001 -6 60 49 70 60 50 40 30 20 10 0 -10 -20 Barostim Kontrolle Diff. +60 Meter Klinisch aussagekräftig 25 Meter -8 p < 0,001 2 % 29 % 13 % 52 % 70 % 60 % 50 % 40 % 30 % 20 % 10 % 0 % Barostim Kontrolle 34 % Verbessert Verbessert um 2 NYHAKlassen Verbessert um 1 NYHAKlasse p < 0,001 -25 % -21 % 10 % 5 % 0 % -5 % -10 % -15 % -20 % -25 % -30 % Barostim Kontrolle Diff. -25 % NT-proBNPSpiegel Klinisch aussagekräftig -10 % Relative Senkung p < 0,004 3 % -14 -21 0 -5 -10 -15 -20 -25 Barostim Kontrolle Diff. -14 Punkte Klinisch aussagekräftig -5 Punkte p < 0,001 -6 60 49 70 60 50 40 30 20 10 0 -10 -20 Barostim Kontrolle Diff. +60 Meter Klinisch aussagekräftig 25 Meter -8 p < 0,001 2 % 29 % 13 % 52 % 70 % 60 % 50 % 40 % 30 % 20 % 10 % 0 % Barostim Kontrolle 34 % Verbessert Verbessert um 2 NYHAKlassen Verbessert um 1 NYHAKlasse p < 0,001 -25 % -21 % 10 % 5 % 0 % -5 % -10 % -15 % -20 % -25 % -30 % Barostim Kontrolle Diff. -25 % NT-proBNPSpiegel Klinisch aussagekräftig -10 % Relative Senkung p < 0,004 3 % -14 Diff. -14 Punkte Klinisch aussagekräftig -5 Punkte < 0,001 2 % 29 % 13 % 52 % 70 % 60 % 50 % 40 % 30 % 20 % 10 % 0 % Barostim Kontrolle 34 % Verbessert Verbessert um 2 NYHAKlassen Verbessert um 1 NYHAKlasse p < 0,001 -14 -21 0 -5 -10 -15 -20 -25 Barostim Kontrolle Diff. -14 Punkte Klinisch aussagekräftig -5 Punkte p < 0,001 -6 60 49 70 60 50 40 30 20 10 0 -10 -20 Barostim Kontrolle Diff. +60 Meter Klinisch aussagekräftig 25 Meter -8 p < 0,001 2 % 29 % 13 % 52 % 70 % 60 % 50 % 40 % 30 % 20 % 10 % 0 % Barostim Kontrolle p < 0,001 -25 % -21 % 10 % 5 % 0 % -5 % -10 % -15 % -20 % -25 % -30 % Barostim Kontrolle Diff. -25 % NT-proBNPSpiegel Klinisch aussagekräftig -10 % Relative Senkung p < 0,004 3 % Körperliche Leistungsfähigkeit (6MHW) Lebensqualität (MLWHF) NYHA-Klasse NT-proBNP-Spiegel Veränderung im 6MHW –nach 6 Monaten Veränderung der NYHA-Klasse –nach 6 Monaten Veränderung des MLWHF-Scores –nach 6 Monaten Veränderung des NT-proBNP –nach 6 Monaten
Tabelle 1: Die Langzeitdaten der BeAT-HF-Studie zeigen, dass die durch die Baroreflex-Aktivierungstherapie erzielten Verbesserungen der Herzinsuffizienzsymptome auch nach 24 Monaten noch anhielten [4].
BeAT-HF-Studie präsentiert [4]. In dieser Phase wurden weitere 59 Patienten mit symptomatischer Herzinsuffizienz und eingeschränkter Pumpfunktion (Ejektionsfraktion ≤35 %) eingeschlossen (BATGruppe: n = 33; Kontrollgruppe: n = 26). Primärer Endpunkt war in dieser zweiten Studienphase die Kombination aus kardiovaskulärer Mortalität (kardiovaskulärer Tod, left ventricular assist device [LVAD] oder Herztransplantation) und Herzinsuffizienz-Morbidität (Herzinsuffizienz-bedingte Hospitalisierung oder Herzinsuffizienzbedingte Aufnahme in die Notaufnahme).
Obwohl der primäre Endpunkt keine signifikanten Unterschiede zwischen BAT- und Kontroll-Gruppe zeigte (Rate Ratio: 0,94; p = 0,82), zeigte sich ein Effekt auf die Gesamtmortalität: Während in der BAT-Gruppe die Ereignisrate für Tod jeglicher Ursache, LVAD oder Herztransplantation bei 7,0 Ereignissen pro 100 Patientenjahre lag, betrug sie in der Kontrollgruppe 10,4 Ereignisse pro 100 Patientenjahre. Dies entspricht einer rela-
tiven Risikoreduktion von 34 % (Hazard Ratio: 0,662; p = 0,054). Außerdem bestätigen die BeATHF-Langzeitdaten die Resultate der 6-Monats-Analyse. Die bereits nach 6 Monaten beobachtete Verbesserung der HerzinsuffizienzSymptome blieb auch über 12 bzw. 24 Monate erhalten (Tab. 1). Auch die Sicherheit der Prozedur hielt über die 24 Monate unverändert an: 97 % der Studienteilnehmer waren frei von schwerwiegenden neurologischen, kardiovaskulären oder prozedurbezogenen Ereignissen (p < 0,001).
Fazit
Die aktuell veröffentlichten klinischen Studiendaten zeigen, dass die Baroreflex-Aktivierungstherapie mit Barostim™ bei Herzinsuffizienz-Patienten mit reduzierter linksventrikulärer Ejektionsfraktion die Symptome der Herzinsuffizienz deutlich reduziert, die Belastungstoleranz erhöht sowie den funktionellen Status (NYHA) und insbesondere die Lebensqualität
(nach MLWHF, Minnesota Living With Heart Failure) verbessert. Dabei profitieren die Patienten auch langfristig von der hohen Therapiesicherheit, sind belastbarer und erhalten so ein großes Stück Lebensqualität zurück.
Brigitte Söllner. Erlangen
Literatur
1 ESC-Leitlinien. Eur Heart J 2021;42, 3427-3520. doi:10.1093/eurheartj/ehab 364
2 Barostim Neo™ Systemreferenzhandbuch, Stand: 07. Mai 2020
3 Zile MR et al. J Am Coll Cardiol 2020; 76:1-13
4 Zile MR et al. Baroreflex activation therapy (BAT) in patients with heart failure and a reduced ejection fraction (BeAT-HF Trial): long-term outcomes. Präsentation auf dem Jahreskongress
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 54 FORUM CARDIOLOGICUM
der Deutschen Gesellschaft für Kardiologie 2023 in Mannheim Endpunkt Nach 12 Monaten Nach 24 Monaten BAT Kontrolle BAT Kontrolle 6-Minuten-Gehstrecke +40,6 Meter –3 Meter Daten nicht erhoben Differenz: 43,5 Meter; p < 0,001 Lebensqualität 17 Punkte Verbesserung 8,6 Punkte Verbesserung 18 Punkte Verbesserung 8 Punkte Verbesserung Differenz: 8,4 Punkte Verbesserung p < 0,001 Differenz: 10 Punkte Verbesserung p < 0,001 NYHA-Klasse: Anteil mit Verbesserung um ≥1 Klasse 72,7 % 40,8 % 68,0 % 41,1 % Differenz: 31,9 Prozentpunkte p < 0,001 Differenz: 26,9 Prozentpunkte p < 0,001
Die European Society of Cardiology (ESC) empfiehlt in ihrer Leitlinie von 2021 für die medikamentöse Erstlinientherapie der chronischen, symptomatischen Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF) einen vereinfachten 4-Säulen-Therapiealgorithmus [1]. Wie diese Empfehlungen im Versorgungsalltag in Deutschland wahrgenommen und umgesetzt werden, zeigen die Ergebnisse einer aktuellen repräsentativen bundesweiten Befragung von niedergelassenen und in der Klinik tätigen Kardiologen (jeweils n = 100) sowie Allgemeinmedizinern, Praktikern und Internisten (APIs, n = 150), die durch das Marktforschungsinstitut Ipsos im Auftrag der Novartis Pharma GmbH vom 14.11. bis 21.12.2022 durchgeführt wurde.
Die wichtigsten Ergebnisse
Die Kerninhalte der aktuellen ESCLeitlinie sind den Ärzten weitestgehend bekannt. So nennen nahezu alle (99 %) den neuen schnellen Behandlungsstart mit allen empfohlenen Wirkstoffklassen der 4-SäulenTherapie. Außerdem ist fast allen Befragten (92 %) bekannt, dass der Angiotensin-Rezeptor-Neprilysin-Inhibitor (ARNI) Sacubitril/ Valsartan (Entresto®) jetzt auch für Neueinstellungen alternativ zum ACE-Hemmer empfohlen wird (Klasse-IIb-Empfehlung).
Dass ein Angiotensin-1-Rezeptorblocker (ARB) nicht mehr empfohlen und bei symptomatischen Patienten durch ARNI ersetzt werden
ESC-Leitlinienempfehlungen sind im HFrEF-Behandlungsalltag angekommen
sollte, wissen jedoch etwa 30 % der APIs und circa 20 % der niedergelassenen Kardiologen bislang noch nicht.
Unter den Befragten zeigte sich eine Präferenz für den ARNI (Sacubitril/Valsartan) gegenüber einem ACE-Hemmer: Zwei Drittel aller Ärzte geben an, zukünftig initial bevorzugt den ARNI anstelle eines ACE-Hemmers einzusetzen. Die Präferenz für den ARNI ist bei allen Fachgruppen vorhanden, wenn auch unterschiedlich stark ausgeprägt, und reicht von 55 % bei den APIs bis 94 % bei den klinisch tätigen Kardiologen. Als Gründe für den präferierten Einsatz des ARNI nennen die Befragten unter anderem die Datenlage.
So zeigen die Ergebnisse der PARADIGM-HF-Studie mit 8.442 Patienten im Vergleich zu Enalapril, dass Sacubritril/Valsartan
• das Risiko für kardiovaskulären Tod um 20 % (p < 0,00004) reduzierte,
• das Risiko für Herzinsuffizienz-bedingte stationäre Einweisungen um 21 % (p < 0,00004) senkte und
• das Gesamtsterblichkeitsrisiko um 16 % (p < 0,0005) verringerte.
Hinsichtlich des kombinierten primären Endpunkts aus kardio-
vaskulärem Tod oder Herzinsuffizienz-bedingter stationärer Einweisung wurde bei den mit Sacubritril/Valsartan behandelten Patienten das Risiko um 20 % reduziert (p < 0,0000004) [2].
Gefragt nach dem Therapieziel Nummer 1 nennen die Ärzte die Verbesserung der Lebensqualität (44 %), dicht gefolgt von der Verbesserung der Symptomatik (38 %). Bei einer leitliniengerechten Behandlung beobachten über 90 % bei ihren Patienten eine Symptomverbesserung, 87 % eine Verbesserung der Lebensqualität, die sich u.a. in einer Steigerung der physischen und sozialen Aktivität äußert. Leitliniengemäß ist für die Befragten vor allem die Symptomatik ausschlaggebend dafür, ob die 4-Säulen-Therapie eingesetzt wird: Wird sie nicht umgesetzt, nennen 62 % als Grund, dass die Patienten stabil bzw. nicht symptomatisch sind. Wichtig ist der Kontakt auch zu diesen Patienten: Um bei ihnen ein asymptomatisches Fortschreiten der Herzinsuffizienz rechtzeitig erkennen und behandeln zu können, empfiehlt die Leitlinie ein regelmäßiges Follow-up.
Literatur
B. S.
1 McDonagh TA et al. Eur Heart J 2021; 42:3599-3726
2 McMurray J et al. N Engl J Med 2014; 371:993-1004
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 55 FORUM CARDIOLOGICUM
Auf der „Technology and Heart Failure Therapeutics“-Konferenz (THT) in Boston, Massachusetts, stellte Abbott aktuelle Studienergebnisse vor, wonach die Fernüberwachung des Pulmonalarteriendrucks mit einem hämodynamischen Drucksensor wie dem CardioMEMS™ HF-System das Überleben von HerzinsuffizienzPatienten mit reduzierter Ejektionsfraktion (HFrEF; EF ≤40 %) signifikant verbessern kann. Denn anders als die traditionell zur Verlaufskontrolle in der Praxis erhobenen physiologischen Parameter und kardialen Marker BNP und NTproBNP wird der PA-Druck vom CardioMEMS™-Sensor kontinuierlich erfasst und gemeldet, sodass auf Verschlechterungen sofort reagiert und damit einer Progression vorgebeugt werden kann.
Senkung des Mortalitätsrisikos um 25 Prozent
In einer Metaanalyse von drei randomisierten, kontrollierten Studien zur Wirksamkeit und Sicherheit des CardioMEMS™ HF-Systems (CHAMPION, GUIDE-HF und LAPTOP-HF*) wurden die Effekte der hämodynamischen Überwachung auf die Hospitalisierung wegen Herzinsuffizienz und das Überleben untersucht [1]. Eingeschlossen in die Metaanalyse wurden 1.350 HFrEF-Patienten (Ausgangs-EF ≤40 %). 666 Pati-
* Der in der LAPTOP-HF-Studie untersuchte Sensor ist nicht für den klinischen Einsatz zugelassen oder verfügbar.

Fernüberwachung mit CardioMEMS™ verbessert die Überlebensrate von Herzinsuffizienz-Patienten
enten wurden einer hämodynamischen Überwachung unterzogen, 684 Patienten befanden sich in der Kontrollgruppe. Die Hospitalisierungen aufgrund von Herzinsuffizienz wurden über einen Nachbeobachtungszeitraum von 12 Monaten analysiert, die Gesamtmortalität wurde über 24 Monate ausgewertet [1].
Die Ergebnisse der Metaanalyse zeigen erstmals, dass ein Überwachungssystem wie Cardio-
Das CardioMEMS™ HF-System
MEMS™, das die Veränderung des pulmonalarteriellen Drucks kontinuierlich misst, rechtzeitig vor einer Verschlechterung der Herzinsuffizienz warnen und dadurch sowohl die Hospitalisierungsrate nach 12 Monaten um 36 % (HR: 0,64; 95%KI: 0,55 – 0,76) als auch das Sterberisiko bei HFrEF-Patienten nach 2 Jahren um 25 % (HR: 0,75; 95%KI: 0,57 – 0,99; p = 0,04) senken kann [1]. Denn die behandelnden Ärzte können rechtzeitig proaktive
Der CardioMEMS™-Sensor ist ein büroklammergroßes Implantat, das nach der minimalinvasiven Platzierung in der Lungenarterie Druckveränderungen anzeigt, die auf eine sich verschlechternde Herzinsuffizienz hinweisen. Er überträgt die täglichen Messwerte drahtlos an das betreuende klinische Team und ermöglicht es, den Zustand des Patienten von praktisch überall aus kontinuierlich zu überwachen.
Das CardioMEMS™ HF-System ist zugelassen für Patienten mit Herzinsuffizienz der NYHA-Klasse II oder III, die entweder im vorangegangenen Jahr wegen Herzinsuffizienz hospitalisiert wurden oder bei denen ein Bluttest erhöhte Werte der natriuretischen Peptide ergab, die auf eine Verschlechterung der Herzinsuffizienz hinweisen.
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 56 FORUM CARDIOLOGICUM
Änderungen am Behandlungsplan der Patienten vornehmen, bevor die Krankheit fortschreitet. Dadurch lassen sich nicht nur wiederholte Krankenhausaufenthalte vermeiden, sondern auch das Sterberisiko senken, das sich mit jeder herzinsuffizienzbedingten Hospitalisierung deutlich erhöht – fast die Hälfte der HFrEF-Patienten, die aufgrund einer Herzinsuffizienz ins Krankenhaus eingeliefert werden, sterben innerhalb eines Jahres nach ihrer ersten Einweisung [2]. Mit dem CardioMEMS™ HF-System zur kontinuierlichen Überwachung des Pulmonalarteriendrucks steht für diese Patienten nun eine klinisch erprobte Option zur Verfügung, die ihre Lebensqualität verbessern und das Sterberisiko signifikant senken kann.
Brigitte Söllner, Erlangen
Studie bestätigt Nutzen von Rivaroxaban zur Rezidivprophylaxe venöser Thromboembolien bei Krebspatienten
Eine venöse Thromboembolie (VTE) ist die häufigste Ursache für Morbidität und Mortalität bei Krebspatienten, gleich nach der Krebserkrankung selbst. Die Betroffenen haben ein höheres Risiko, an einer VTE zu erkranken, als Patienten ohne Krebs – und auch ein höheres Risiko für wiederkehrende VTE und Blutungen.
Neue Daten aus der klinischen Praxis von 2013 bis 2020 zeigen, dass der orale Faktor-Xa-Inhibitor Rivaroxaban (Xarelto®) bei der Behandlung von krebsbedingten Thromboembolien (CAT) bei einer Gruppe von Patienten mit verschiedenen Krebsarten, für die die Gabe direkter oraler Antikoagulanzien (DOAC) indiziert ist, eine vergleichbare Wirksamkeit und Sicherheit aufweist wie Apixaban. Die Daten wurden auf der 64. Jahrestagung der American Society of Hematology (ASH) in New Orleans vorgestellt.
der Einsatz von DOAC als Alternative zu niedermolekularem Heparin empfohlen wird. Eingeschlossen wurden Patienten mit einer aktiven Krebserkrankung und einer VTE (Indexereignis), die am Tag 7 nach der Diagnose der VTE eine therapeutische Dosis Rivaroxaban oder Apixaban erhalten hatten. Primärer kombinierter Studienendpunkt war die Zeitspanne bis zum Auftreten einer rezidivierenden VTE oder einer Blutung, die zu einem Krankenhausaufenthalt führte. Die Nachbeobachtungszeit betrug mindestens 3 Monate. Weitere Endpunkte waren die Kombination aus rezidivierender VTE oder einer kritischen Organblutung, rezidivierender VTE, einer Blutung, die zu einem Krankenhausaufenthalt führte, und einer kritischen Organblutung nach 3 und 6 Monaten nach dem Indexereignis.
Kein signifikanter Unterschied hinsichtlich Wirksamkeit und Sicherheit
Literatur
1 Lindenfeld J, on behalf of the GUIDEHF, CHAMPION, and LAPTOP-HF investigators. Longer term effects of hemodynamic monitoring on outcomes: a combined data analysis of patients with HFrEF in CHAMPION, GUIDE-HF, and LAPTOP-HF. Presented at THT Conference, Boston, MA. March 2023
2 Setoguchi S et al. Repeated hospitalizations predict mortality in the community population with heart failure. Am Heart J 2007;154:260-266
Vergleich von Rivaroxaban und Apixaban
Für die retrospektive Studie wurden elektronische Gesundheitsdaten von Januar 2013 bis Dezember 2020 analysiert. In die Studie aufgenommen wurden die Daten von 2.437 Patienten mit einer tumorassoziierten VTE, für die in den Leitlinien
Nach jeweils 3 und 6 Monaten war Rivaroxaban bei dem zusammengesetzten Endpunkt aus rezidivierender VTE oder blutungsbedingter Krankenhauseinweisung ebenso wirksam und sicher wie Apixaban. Nach 3 Monaten hatten die mit Rivaroxaban behandelten Patienten eine numerisch niedrigere Rate des primären Endpunkts (5,3 % gegenüber 6,0 %; HR = 0,87; 95%-KI: 0,60 – 1,27). Auch nach 6 Monaten wurden keine signifikanten Unterschiede zwischen den Gruppen für diesen Endpunkt sowie für die anderen Endpunkte beobachtet.
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH
MITTEILUNGEN 57
B. S.
Die small interfering RNA (siRNA) Inclisiran (Leqvio®) ist zur Behandlung von Erwachsenen mit primärer Hypercholesterinämie (heterozygot familiär [HeFH] und nicht familiär) oder gemischter Dyslipidämie zusätzlich zu einer Statin-Basistherapie und/oder anderen lipidsenkenden Therapien zugelassen [1]. In den Phase-IIIStudien ORION-9, -10 und -11 ließ sich mit dem Wirkstoff eine signifikante und über 6 Monate anhaltende LDL-Cholesterin-Senkung erreichen [2, 3]. Da es noch keine Daten aus der klinischen Routine gibt, wurde im Januar 2022 die multizentrische nicht interventionelle Studie VICTORION-Implement gestartet, die die Wirksamkeit und Sicherheit der siRNA Inclisiran und anderer oraler lipid-
Erste Verlaufsdaten aus dem klinischen Alltag zeigen
vergleichbare LDL-Senkung unter Inclisiran wie in den klinischen Studien
senkender Medikamente (LLT) im klinischen Alltag in Deutschland untersucht [4].
Zielsetzung der VICTORIONImplement-Studie
Eingeschlossen in VICTORIONImplement wurden Patienten, für die laut Arzneimittel-Richtlinie (AM-RL) Anlage III in Deutschland eine Behandlung mit Inclisiran erstattet werden kann [5].
Primäres Studienziel ist die Untersuchung und Charakterisierung von Patientengruppen, die neu auf die siRNA oder andere orale lipidsenkende Therapien eingestellt sind. Untersucht wird, wie viele Patienten ihre empfohlenen LDLC-Zielwerte gemäß der aktuellen
PCSK9: Ein Target zur LDL-C-Senkung
ESC- und EAS-Leitlinie im klinischen Alltag in Deutschland erreichen [6]. Patienten mit hohem und sehr hohem kardiovaskulärem Gesamtrisiko werden eine LDL-CSenkung um >50 % gegenüber dem Ausgangswert sowie ein LDL-CZiel von <70 mg/dl bei hohem und < 55 mg/dl bei sehr hohem Risiko empfohlen [6].
Als weiteres Studienziel soll untersucht werden, ob sich durch die Inclisiran-Therapie die Apheresefrequenz reduzieren lässt [4]. Die siRNA hemmt die Synthese des Enzyms Proproteinkonvertase
Subtilisin Kexin Typ 9 (PCSK9) [1]. Monoklonale Antikörper gegen PCSK9 konnten in Studien die Apherese komplett ersetzen und bei HeFH-Patienten die Frequenz verlängern [7].
Das Enzym Proproteinkonvertase Subtilisin/Kexin Typ 9 (PCSK9) ist ein zentraler Regulator im Stoffwechsel des LDLCholesterins. Bindet PCSK9 zusätzlich zum LDL-C an den für die Leberzellen spezifischen LDL-Rezeptor (LDL-R), wird dieser nach der Endozytose zusammen mit dem LDL-C abgebaut. Somit wird das „Recycling“ des LDL-R verhindert und die Konzentration von LDL-C im Blut steigt. Mutationen im Gen für PCSK9 können zu einer Überaktivität führen, die eine Ursache für erbliche Hypercholesterinämien sein kann. Für moderne Wirkstoffe dient PCSK9 daher als Target zur Cholesterinsenkung. Monoklonale Antikörper gegen PCSK9 binden dieses Enzym außerhalb der Hepatozyten. Die small interfering Ribonukleinsäure (siRNA) Inclisiran hemmt über den natürlichen Mechanismus der RNA-Interferenz die PCSK9-Synthese in den Leberzellen, sodass das Enzym erst gar nicht gebildet wird. Über beide Wege wird die LDLR-Zahl auf der Zelloberfläche erhöht. Dies führt in der Folge zur Senkung der LDL-C-Plasmakonzentration [10].
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 58 FORUM LIPIDSENKER
eine
Hemmung der PCSK9-Synthese durch Inclisiran in den Leberzellen
Bei Inclisiran(Leqvio®) handelt es sich um eine small interfering RNA (siRNA). Diese kurzen Ribonukleinsäure-Moleküle codieren keine Proteine, sondern verbinden sich im Zellkern gezielt mit komplementären einzelsträngigen mRNAMolekülen eines Gens und blockieren so die Übersetzung der Erbinformation für den Bau eines Proteins. Inclisiran nutzt diesen körpereigenen Prozess der RNA-Interferenz (sog. Gen-Stilllegung), um durch den gezielten Abbau der mRNA die Translation der vom entsprechenden Gen ausgelesenen Information in das Protein PCSK9 in der Leber zu hemmen [4].
Die siRNA besteht aus einem Antisense-Strang und einem Sense-Strang, an den ein N-Acetylgalactosamin (GalNAc)Rest gekoppelt ist . Letzterer ermöglicht durch Bindung an den für Leberzellen spezifischen AsialoglykoproteinRezeptor (ASGPR) die Aufnahme und selektive Wirkung der siRNA in den Hepatozyten. Der über GalNAc an den ASGPR gebundene Wirkstoff gelangt per Endozytose in die Zelle , wird aus dem Endosom ins Zytoplasma freigesetzt und an den RNA-induzierten Silencing-Komplex (RISC) gebunden , der den Sense-Strang abspaltet . Der AntisenseStrang im RISC erkennt die PCSK9-mRNA, welche gebunden wird . Daraufhin wird die PCSK9-mRNA abgebaut . Der RISC bleibt erhalten und kann im Anschluss erneut seine Target-mRNA binden . Das Enzym wird gar nicht erst synthetisiert und kann daher auch nicht an den LDL-R binden und dessen Abbau induzieren . Dessen Recycling ist wieder möglich und zirkulierendes LDL-C kann vermehrt in die Leberzellen aufgenommen und abgebaut werden , wodurch seine Plasmakonzentration sinkt [4].
Letztlich sollen alle gewonnenen Daten dazu dienen, die Belastung durch die Erkrankung im Hinblick auf die Lebensqualität sowie den Einfluss von Behandlungsstrategien und Sicherheitsprofilen in der klinischen Routine besser zu verstehen. Hervorzuheben sind noch die Be-
sonderheit des Studien-Set-Ups: Die VICTORION-Implement-Studie liefert Prüfärzten Echtzeitdaten von LDL-C-Werten im Zeitverlauf, die über ein digitales Dashboard dargestellt werden. Dabei können je nach Bedarf eigene Patienten mit allen Studienteilnehmern, mit
den einzelnen Behandlungsarmen oder mit verschiedenen Regionen verglichen werden. Darüber hinaus wird auf der Grundlage der eingegebenen Daten der SMART-Risiko-Score berechnet und den Ärzten werden die Veränderungen im Zeitverlauf angezeigt [4].
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 59 FORUM LIPIDSENKER
2 1 3 4 5 6 7 8 Zellkern DNA ASGPR PSCK9-mRNA abgebaute PSCK9-mRNA Endosom Endozytose Freisetzung ins Zytoplasma RISC-Bindung Sense-StrangAbspaltung Bindung von
RISC PCSK9-mRNADegradation RISC-Erhalt RISC inhibitierte PSCK9-Synthese Sense-Strang doppelsträngige siRNA Inclisiran GaINAc Antisense-Strang Hepatozyt LDL-CPartikel 11 10 LDL-R-Synthese und -Verankerung GolgiApparat LDL-CDegradation LDL-RRecycling 9 (© Novartis)
PCSK9-mRNA und
Studiendesign
Nach dem Screening wurden die in ca. 300 Zentren in Deutschland rekrutierten 2030 teilnehmenden Patienten (>18 Jahre) gemäß ihrer in der klinischen Routine angewendeten Therapie in 3 Kohorten aufgeteilt [4]:
1. Kohorte A: 1.000 Patienten mit diagnostizierter atherosklerotischer Herz-Kreislauf-Erkrankung (ASCVD), die zusätzlich zu einem Statin neu auf eine orale lipidsenkende Therapie eingestellt werden.
2. Kohorte B: 1.000 Patienten, die neu auf Inclisiran eingestellt sind, bei denen Inclisiran gemäß AM-RL Anlage III indiziert ist [5] und die gemäß individueller Zugangsvoraussetzungen nicht das in den EAS/ESC-Leitlinien 2019 [6] empfohlene LDL-CZiel gemäß ihrem kardiovaskulären Risiko erreicht haben. Mindestens 60 % von ihnen sind nicht mit einem Inhibitor der PCSK9 vorbehandelt.
3. Kohorte C: 30 Patienten, die zusätzlich zu regelmäßiger Apherese (einmal wöchentlich, oder einmal alle 2 Wochen) neu auf Inclisiran eingestellt sind.
Überzeugende Ergebnisse
nach 3 Monaten
Erste Verlaufsdaten der VICTORION-Implement-Studie wurden nach 3 Monaten ausgewertet. Die Interimsdaten von 317 Patienten zeigen, dass die Behandlung mit Inclisiran bei neu eingestellten Pa-
tienten (n = 108) zu einer LDL-CSenkung von im Median 45,8 % (42,0 mg/dl) führte. Die mediane LDL-C-Reduktion mit neu begonnener oraler LLT (n = 209) betrug 36,1 % (28,6 mg/dl) [4]. Die LDL-C-Senkung mit Inclisiran im Real-World-Einsatz zeigt damit vergleichbare Ergebnisse wie im klinischen Studiensetting: So lag die LDL-C-Reduktion vom Ausgangswert in der klinischen Studie ORION-1 nach 3 Monaten Behandlung und damit der ersten Injektion nach der Initialdosis im Mittel bei 44,2 % [8].
Laut der Zwischenauswertung erhalten 58,3 % der mit Inclisiran behandelten Patienten als Begleitmedikation eine Statintherapie, davon 51,8 % eine hochintensive und 6,5 % eine niedrigintensive. Von diesen Patienten wird der Großteil (67,2 %) mit Inclisiran + Statin + Ezetimib behandelt. Ein geringerer Anteil erhält eine Therapie mit Inclisiran + Statin (19,7 %) bzw. mit Inclisiran + Statin + Ezetimib + Bempedoinsäure (13,1 %). 41,7 % der mit der siRNA behandelten Patienten werden nicht mit einem Statin behandelt, davon erhalten 70,7 % eine Inclisiran-Monotherapie [4].
Betrachtet man die Baseline-Charakteristika, waren beim Start der Studie neu auf Inclisiran im Vergleich zu neu auf eine orale LLT eingestellte Patienten jünger, häufiger weiblich, das erste kardiovaskuläre Ereignis lag länger zurück und es bestand häufiger eine periphere arterielle Verschlusskrankheit. Außerdem waren die Ausgangswerte von LDL-C, HDL-C, Gesamtcho-
lesterin und Lipoprotein (a) in dieser Gruppe höher.
Fazit
Die ersten Verlaufsdaten der VICTORION-Implement-Studie unterstreichen die Bedeutung der LDL-C-Laborwerte für die Therapieentscheidung. Bei den Charakteristika der Inclisiran-Patienten fällt auf, dass sie deutlich höhere LDL-C-Baseline-Werte aufweisen als Patienten mit einer reinen oralen lipidsenkenden Therapie. Sind die LDL-C-Werte im klinischen Alltag also sehr hoch, ist es empfehlenswert, über eine schnellere Therapieeskalation nachzudenken, um den individuellen LDL-C-Zielwert zu erreichen.
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Leqvio®, aktueller Stand
2 Raal F et al. N Engl J Med 2020;382: 1520-1530
3 Ray KK al. N Engl J Med 2020;382: 1507-1519
4 Studienprotokoll, Novartis Data on File
5 Gemeinsamer Bundesausschuss (GBA). Beschluss über Änderungen der Arzneimittel-Richtlinie Anlage III Abrufbar unter: https://www.g-ba.de/ downloads/39-261-5072/2021-10-21_ AM-RL-III_Nr35c-Inclisiran.pdf
6 Mach F et al. Eur Heart J 2020;41:111188
7 Moriarty PM et al. Eur Heart J 2016;37: 3588-3595
8 Ray KK et al. Lancet Diabetes Endocrinol 2023;11:109-119
9 Lambert G et al. J Lipid Res 2012; 53(12):2515-2524
10 Khvorova A. N Engl J Med 2017;376: 4-7
Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH 60 FORUM LIPIDSENKER
Antikoagulation bei Hochrisikopatienten: Studiendaten und Praxiserfahrungen unterstreichen den klinischen Stellenwert von Edoxaban
In den Indikationen nicht valvuläres Vorhofflimmern (nvVHF) und venöse Thromboembolien (VTE) sind Nicht-Vitamin-K-abhängige orale Antikoagulanzien (NOAKs) wie Edoxaban seit einigen Jahren etablierter Behandlungsstandard. Herausfordernd ist dabei nach wie vor die Antikoagulation von Hochrisikopatienten mit Begleiterkrankungen wie etwa Krebs, Atherosklerose und Nierenerkrankungen sowie bei perkutanen Interventionen. Diese Themen diskutierten Experten während eines Symposiums von Daiichi Sankyo im Rahmen der 89. Jahrestagung der Deutschen Gesellschaft für Kardiologie –Herz- und Kreislaufforschung e. V. (DGK) 2023 in Mannheim.
Peri- und postprozedurale Therapie mit Edoxaban auch im Alltag effektiv und sicher
Wie Professor Andreas Götte, Paderborn, darlegte, konnte die ENTRUST-AF-PCI-Studie für das klinische Setting zeigen, dass ein Regime aus Edoxaban und einem P2Y12Inhibitor einer Triple-Therapie aus VKA und 2 Plättchenhemmern im Hinblick auf schwere Blutungen bei Patienten mit nvVHF nach einer perkutanen Koronarintervention (PCI) nicht unterlegen ist. Die
Edoxaban
Edoxaban (Lixiana®) ist in der Europäischen Union seit Juni 2015 für die Prophylaxe von Schlaganfällen und systemischen embolischen Ereignissen (SEE) bei erwachsenen Patienten mit nicht valvulärem Vorhofflimmern und mindestens einem Risikofaktor zugelassen. Zu den Risikofaktoren zählen kongestive Herzinsuffizienz, Hypertonie, Alter von ≥75 Jahren, Diabetes mellitus, Schlaganfall oder transitorische ischämische Attacke in der Anamnese. Darüber hinaus ist Edoxaban für die Behandlung venöser Thromboembolien (VTE; tiefe Venenthrombosen und Lungenembolien) sowie zur Prophylaxe rezidivierender VTE bei erwachsenen Patienten indiziert. Die Standarddosierung von Edoxaban beträgt einmal täglich 60 mg für alle zugelassenen Indikationen (bei VTE nach initialer Anwendung eines parenteralen Antikoagulans über mindestens 5 Tage). Patienten, die gleichzeitig mit den P-Glykoprotein-Inhibitoren Ciclosporin, Dronedaron, Erythromycin oder Ketoconazol behandelt werden, sowie Patienten mit mäßig oder stark eingeschränkter Nierenfunktion (Kreatinin-Clearance: 15 – 50 ml/min) oder einem Körpergewicht ≤60 kg erhalten eine reduzierte Edoxaban-Dosis von 30 mg einmal täglich.
aktuellen ESC-Leitlinien empfehlen für diese Indikation eine risikoadaptierte, möglichst kurze Dauer der Tripeltherapie. Die nicht interventionelle Registerstudie ENCOURAGE-AF zur peri- und postoperativen Anwendung von Antikoagulanzien und Thrombozytenaggregationshemmern bei Patienten mit nvVHF, die nach einer erfolgreichen PCI mit Edoxaban behandelt werden, lieferte für Deutschland erste Ergebnisse zur Effektivität und Sicherheit aus der klinischen Praxis. Somit ermöglicht sie Rückschlüsse auf die Beachtung der Leitlinie.
Laut Götte wurden die Leitlinienvorgaben der europäischen kardiologischen Gesellschaft in der klinischen Praxis demnach gut umgesetzt: So war die Acetylsalicylsäure-Gabe zum Datenerhebungspunkt der Snapshot-Analyse bei 91 % der Patienten spätestens an Tag 30 beendet.
Tödliche Blutungsereignisse traten unter Edoxaban innerhalb von 30
Tagen nicht auf. Die Raten schwerer und klinisch relevanter, nicht schwerer Blutungen waren in den ersten 30 Tagen mit 3,5 % niedrig. Kardiovaskuläre Ereignisse waren selten; nach 30 Tagen wurden 0,3 % Schlaganfälle, 0,2 % ungeplante perkutane Interventionen und keine Myokardinfarkte registriert.
„Es ist phantastisch, dass diese wichtige, prospektive Registerstudie auch unter den schwierigen Corona-Bedingungen durchgeführt wurde und sogar die Rate unerwünschter kardiovaskulärer Ereignisse der ENTRUST-AF-PCI-Studie in der klinischen Routine noch unterboten werden konnte“, fasste Götte zusammen.
Krebspatienten haben ein erhöhtes Risiko für kardiovaskuläre Erkrankungen
Eine große, im klinischen Alltag relevante Gruppe sind Patienten mit
61 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse KONGRESSE
Tumorerkrankungen, die gegenüber Menschen ohne Malignome per se ein deutlich erhöhtes Risiko für VTE und Schlaganfälle haben. Eine wirksame Antikoagulation wird für diese Patienten aber auch deshalb immer wichtiger, weil sie dank der Entwicklung zahlreicher neuer, potenter Tumortherapeutika in den letzten Jahren immer länger überleben.
„Mit der höheren Lebenserwartung steigt aber zugleich ihr Risiko für kardiovaskuläre Erkrankungen – und zwar unabhängig von der Schwere der Tumorerkrankung“, berichtete Professor Matthias Totzeck, Essen. Da für verschiedene Tumortherapeutika zudem beträchtliche Arzneimittelwechselwirkungen mit Antikoagulanzien bekannt sind, kann die gerinnungshemmende Therapie bei Krebspatienten herausfordernd sein.
Der Nutzen von Edoxaban in dieser Indikation wurde in der Hokusai VTE Cancer-Studie untersucht. Ein zusätzlicher Vorteil des NOAK: Tumorpatienten neigen aufgrund der Chemotherapie laut Totzeck häufig zu behandlungsbedürftigen Pilzinfektionen. Dabei kann Edoxaban in reduzierter Dosis (30 mg einmal täglich) gleichzeitig mit dem P-Glykoprotein (P-gp)-Inhibitor Ketoconazol, der zur Behandlung der Mykose eingesetzt wird, verabreicht werden.
gleich der wichtigste Risikofaktor für die Entstehung kardiogener Schlaganfälle und negativ mit der Atherosklerose assoziiert. Das Gleiche gilt für Menschen mit VTE – auch bei ihnen ist die Inzidenz der durch Atherosklerose bedingten kardiovaskulären Ereignisse deutlich erhöht.
„Die pathophysiologische Verbindung zwischen nvVHF und VTE lässt sich über die Inflammation in den Gefäßen ziehen. Wir wissen, dass diese Entzündungsprozesse – und damit auch das Risiko für VTE-Rezidive – durch lipidsenkende Therapien wie Statine oder Proprotein-Convertase-SubtilisinKexin-Typ 9 (PCSK9)-Inhibitoren reduziert werden können, wofür möglicherweise pleiotrope Effekte der Wirkstoffe eine Rolle spielen“, erläuterte die Kardiologin.
Die Registerstudie ETNA-AF-Europe hat für Patienten mit nvVHF, die mit Edoxaban antikoaguliert wurden, gezeigt, dass das Risiko für ischämische Schlaganfälle unter einer zusätzlichen lipidsenkenden Therapie im Vergleich zu keiner Therapie signifikant niedriger ausfiel (Hazard Ratio: 0,54; 95%-Konfidenzintervall: 0,35 – 0,85).
48-Studie wurden mittlerweile über 70 weitere Analysen publiziert. Zu den untersuchten Subpopulationen zählen auch die Hochrisikopatienten mit chronischer Nierenerkrankung (CKD). Die relative Wirksamkeit und Sicherheit von Edoxaban im Vergleich zu Warfarin entsprach für diese Gruppe laut Professor Gunnar Heine, Frankfurt/M., den Gesamtstudienergebnissen: „In den nach der Nierenfunktion präspezifizierten Subgruppen mit einer Kreatinin-Clearance (CrCl) von 30 – 50 ml/min und CrCl >50 ml/min zeigten sich unter der Gabe von Edoxaban 60/30 mg einmal täglich im Vergleich zu gut eingestelltem Warfarin konsistent eine ähnliche Wirksamkeit (Schlaganfall/systemische embolische Ereignisse) und ein positives Sicherheitsprofil (schwere Blutungen)“, berichtete der Nephrologe.
Vorhofflimmern kommt selten allein
Wie Professor Christine Espinola-Klein, Mainz, hervorhob, ist nvVHF im klinischen Alltag die häufigste relevante, anhaltende Herzrhythmusstörung. Sie ist zu-
Dass auch Edoxaban pleiotrope Effekte aufweist, legen die Ergebnisse tierexperimenteller Untersuchungen nahe, die Götte zusammenfassend vorstellte. Demnach bestehen zahlreiche positive Effekte auf zellulärer Ebene in verschiedenen Geweben.
Sicher und wirksam bei chronischer Nierenerkrankung
Basierend auf der Primärpublikation zur ENGAGE-AF-TIMI-
Heine stellte außerdem die Ergebnisse einer Metaanalyse vor, die bei Patienten mit moderat eingeschränkter Nierenfunktion und VTE ein überlegenes Nutzen-Risiko-Profil für NOAKs gegenüber VKA belegen. „Aussagen zum Nutzen von NOAKs bei Patienten mit terminaler Niereninsuffizienz und unter Dialyse sind aufgrund fehlender Daten jedoch nicht möglich“, betonte Heine.
Elisabeth Wilhelmi, München
62 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH Kongresse
Jetzt profitieren auch Patienten mit mittelschwerer Hämophilie A von Emicizumab
Der Fokus der Hämophilie-ABehandlung wurde bisher auf eine effektive und langfristige Blutungskontrolle bei Patienten mit schwerer Erkrankung gelegt. Aber auch Patienten mit mittelschwerer Erkrankung sind größtenteils von häufigen Blutungsereignissen und den damit einhergehenden Einschränkungen betroffen, wie Experten auf der Jahrestagung der Gesellschaft für Thrombose- und Hämostaseforschung (GTH) darlegten. Da diese Patienten ein erhöhtes Risiko für Folgeerkrankungen aufweisen, ist eine frühe und effektive Prophylaxe indiziert. Die Zulassungserweiterung von Emicizumab (Hemlibra®) ermöglicht nun auch eine Intervention bei Patienten mittelschwerer Hämophilie A ohne Faktor VIII (FVIII)-Hemmkörper mit schwerem Blutungsphänotyp.
Auch Blutungsphänotyp und Gelenkstatus bei der Therapieentscheidung einbeziehen!
Ziel der Therapie bei Hämophilie A ist die Vorbeugung von Blutungen sowie deren Konsequenzen, um so den Patienten ein möglichst normales Leben zu ermöglichen und eine hohe Lebensqualität zu erhalten. „In den letzten Jahren hat sich dabei immer mehr herauskristallisiert, dass man sich nicht allein auf die schweren Fälle der Erkrankungen fokussieren darf“, berichtete Dr. Carmen Escuriola-Ettingshausen,
Mörfelden. Vielmehr sollten auch der Blutungsphänotyp, der Gelenkstatus, die individuelle Pharmakokinetik sowie der Patientenwunsch in die Therapieentscheidung mit einfließen. Viele Patienten mit mittelschwerer Hämophilie A und einem schweren Blutungsphänotyp erkennen Blutungen erst sehr spät, sodass es zu einer Unterdiagnostik und einer Verschleppung der Therapieinitiierung kommen kann.
Wegweisende Ergebnisse der HAVEN-6-Studie
Die Studie HAVEN 6 untersuchte die Prophylaxe mit Emicizumab bei Patienten mit leichter und mittelschwerer Hämophilie A ohne Faktor VIII-Hemmkörper. Wie Professor Johannes Oldenburg, Bonn, erläuterte, standen dabei die jährliche Blutungsrate (ABR) sowie Sicherheitsaspekte im Fokus. Die Patienten erhielten nach einer vierwöchigen Initialdosis mit wöchentlich 3 mg/kg Emicizumab subkutan dann in der Erhaltungsphase entweder 1,5 mg/kg einmal wöchentlich, 3 mg/kg alle 2 Wochen oder 6 mg/ kg alle 4 Wochen. Nach einem medianen Follow-up von 55,6 Wochen betrug die ABR 0,9 für behandlungsbedürftige Blutungen, 0,2 für behandlungsbedürftige Gelenkblutungen ebenso wie für behandlungsbedürftige Spontanblutungen. Es wurden keine neuen Sicherheitshinweise detektiert.
Auf Basis dieser Ergebnisse wurde Hemlibra nun auch für Patienten ohne Faktor-VIII-Hemmkörper mit mittelschwerer Hämophilie A mit schwerem Blutungsphänotyp zugelassen. „Durch subkutane Injek-
tionen und Applikationsintervallen von bis zu 4 Wochen steht diesen Patienten jetzt eine effektive Prophylaxe mit minimaler Therapielast zur Verfügung“, kommentierte Oldenburg.
Vorteile der frühen Prophylaxe
Um die Gelenke dauerhaft gesund zu halten, ist die Vermeidung von Blutungen essenziell. Etwa 85 % aller Betroffenen erleiden im Laufe ihres Lebens Blutungen, die langfristig zu Gelenkproblemen führen können. Daher ist eine frühe Prophylaxe indiziert. Doch gerade bei sehr jungen Kindern stellt vor allem der problematische venöse Zugang Behandelnde und Angehörige immer wieder vor große Herausforderungen. „Hier kann die subkutane Verabreichung von Hemlibra® eine effektive Alternative darstellen“, sagte Escuriola-Ettingshausen und untermauerte dies mit den Interimsergebnissen der Studie HAVEN 7, die die Wirksamkeit und Sicherheit einer Prophylaxe mit Hemlibra bei sehr jungen Patienten unter 12 Monaten mit schwerer Hämophilie A ohne FVIII-Hemmkörper untersucht. „Die Interimsanalyse zeigt eine durchgängige Wirksamkeit über alle Blutungsendpunkte hinweg. Bei keinem Betroffenen traten behandlungsbedürftige Spontanblutungen auf. Die Rate der Patienten ohne behandlungsbedürftige Muskelblutungen lag bei 98,1 % und die ohne behandlungsbedürftige Blutungen bei 77,8 %. Insgesamt blieben 42,6 % der Patienten blutungsfrei“, so die Expertin. Neue Sicherheitssignale traten nicht auf und die Behandlung wurde gut vertragen.
63 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
Auch im Praxisalltag überzeugend
Die Ergebnisse der klinischen Studien lassen sich laut sich laut Dr. Swee Wenning, Heidelberg, auch im Praxisalltag bestätigen. Dies zeigen auch die bisherigen Ergebnisse der nicht interventionellen Studie EMIIL, die die Wirksamkeit von Emicizumab unter RealWorld-Bedingungen in allen Altersgruppen beleuchtet. Die zweite Interimsanalyse ergab eine ABR von 0,6, wobei eine große Anzahl von Patienten ohne Blutungen blieb und sich das bekannte Sicherheitsprofil bestätigte.
PD Dr. Sylvia von Mackensen, Hamburg, brachte die Relevanz einer effektiven Blutungsprophylaxe abschließend noch einmal auf den Punkt, indem sie aufzeigte, dass in Deutschland viele erwachsene Hämophilie-A-Patienten immer noch unter chronischen Gelenkschäden leiden, die zu chronischen Schmerzen führen und die Lebensqualität erheblich einschränken. Ihr Fazit: „Gelenkblutungen und damit Gelenkschäden zu vermeiden, kann die Lebensqualität deutlich steigern und den Betroffenen zu einem möglichst normalen Alltag verhelfen.“
Elisabeth Wilhelmi, München
Mit Suliqua®
Kontrolle
„Die Titration des Basalinsulins im Rahmen einer basalunterstützten oralen Therapie (BOT) bei Typ-2-Diabetes hat Grenzen – mit einer wei-
teren Dosiserhöhung wird dann keine Verbesserung der glykämischen Kontrolle mehr erreicht“, erklärte Professor Matthias Blüher, Leipzig, im Rahmen einer von der SanofiAventis Deutschland GmbH veranstalteten virtuellen Fachpressekonferenz und ergänzte: „Die Betroffenen können vom Einsatz einer Fixkombination aus Basalinsulin und einem kurzwirksamen GLP-1-RA (Glukagon-ähnliches Peptid-1-Rezeptoragonist) wie iGlarLixi (Suliqua®) profitieren.“ Der Konsensreport 2022 von ADA (American Diabetes Association) und EASD (European Association for the Study of Diabetes) betont, dass Fixkombinationen aus Basalinsulin und GLP-1-RA wie iGlarLixi eine sehr hohe blutzuckersenkende Effektivität aufweisen und dabei gleichzeitig die Komplexität und Belastung der Behandlung reduzieren.
iGlarLixi ist die einzige in Deutschland verfügbare Fixkombination aus Basalinsulin und GLP-1-RA und besteht aus Insulin glargin 100 Einheiten (E)/ml und Lixisenatid. Sie wird bei Erwachsenen mit unzureichend kontrolliertem Typ2-Diabetes zur Verbesserung der Blutzuckerkontrolle, ergänzend zu Diät und Bewegung sowie zusätzlich zu Metformin ± SGLT2-(Natrium-Glukose Cotransporter 2)-Inhibitoren mit einmal täglicher Applikation eingesetzt.
Auch deutsche Therapieempfehlungen beurteilen es positiv, ein Basalinsulin und einen GLP-1-RA zu kombinieren, wenn das Potenzial einer BOT ausgeschöpft ist: Die Kombination hat laut der Deutschen Diabetes Gesellschaft e.V. (DDG)
im Vergleich zur intensivierten Insulintherapie Vorteile in Bezug auf Therapieadhärenz, Unterzuckerungen und die Gewichtsentwicklung.
Mit täglichen Dosisanpassungen schneller ans Ziel
Warum zu Beginn der Therapie mit iGlarLixi eine tägliche Dosistitration erfolgen sollte, machte Blüher anhand der Phase-III-Studie LixiLan ONE CAN deutlich: Die randomisierte Studie über 26 Wochen untersuchte unterschiedliche Titrationsalgorithmen bei der Therapieinitiierung. „Die Auswertung der Daten zeigte, dass ein einmal täglicher Titrationsalgorithmus mit iGlarLixi der einmal wöchentlichen Titration überlegen ist. Wurde die Dosis täglich um einen Dosisschritt angepasst, war es Menschen mit Typ-2-Diabetes schneller möglich, ihre Erhaltungsdosis und ihr HbA1cZiel zu erreichen“, berichtete Blüher. Hinsichtlich Hypoglykämien und gastrointestinaler Ereignisse gab es keinen Unterschied zwischen den beiden Algorithmen.
In der Versorgungsrealität vergleichbar effektiv wie BasalBolus-Therapien, aber mit zusätzlichen Vorteilen
Aktuelle Real-World-Daten zeigten, dass die Fixkombination im klinischen Alltag effektiv war. In der retrospektiven Beobachtungsstudie SoliSimplify wurden 2 Kohorten – Erwachsene unter iGlarLixi oder unter Basal-Bolus-Therapieformen (BBT; BOTplus bzw. intensivierte Insulintherapie [ICT]) – mittels Propensity Score Matching vergli-
64 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH Kongresse
einfacher hin zu einer guten glykämischen
chen. Eingeschlossen wurden 1.628 Patienten mit Typ-2-Diabetes, die von einer BOT auf iGlarLixi oder auf eine BBT umgestellt wurden. Nach 6 Monaten kam es bei beiden Kohorten zu einer vergleichbaren Verbesserung der glykämischen
Kontrolle: Der mittlere HbA1c-Wert war von 9,2 % (beide Gruppen) auf 8,5 % (iGlarLixi) bzw. 8,4 % (BBT) gesunken (p = 0,0032 für Nichtunterlegenheit von iGlarLixi).
Einen klaren Vorteil zeigte iGlarLixi bei der Gewichtsentwicklung: Die Studienteilnehmer nahmen unter der Fixkombination im Mittel leicht um 0,1 kg ab, unter der BBT dagegen um 0,7 kg zu (p = 0,0069; zweiseitiger p-Wert für Überlegenheit von iGlarLixi). Entsprechend positiv kommentierte Blüher die Studiendaten: „Die Daten aus der Versorgungsrealität zeigen, dass iGlarLixi nach 6 Monaten zu ähnlichen Verbesserungen der Blutzuckerkontrolle führte wie eine BBT, mit den zusätzlichen Vorteilen der Gewichtsneutralität und eines einfacheren Behandlungsregimes.“
Fazit: iGlarLixi – wenn die BOT ausgereizt ist
„iGlarLixi, die einzige in Deutschland erhältliche Fixkombination aus Basalinsulinanalogon und kurzwirksamem GLP-1-RA in einer Injektion, ist bei Erwachsenen mit Typ-2-Diabetes, die unter einer BOT nicht ausreichend kontrolliert sind, eine vergleichbar effektive, aber deutlich weniger komplexe Intensivierungsoption im Vergleich zur Zugabe von Mahlzeiteninsulin,“ fasste Blüher zusammen.
Fabian Sandner, Nürnberg
MITTEILUNGEN
Hypertrophe obstruktive Kardiomyopathie: CHMP empfiehlt Zulassung des selektiven kardialen Myosin-Inhibitors
Mavacamten
Die hypertrophe obstruktive Kardiomyopathie (HOCM) ist eine familiär gehäuft auftretende, progressiv verlaufende Erkrankung, bei der es aufgrund der übermäßigen Kontraktilität des Herzmuskels und der verringerten Füllkapazität des linken Ventrikels zu Problemen in der Blutzirkulation und infolgedessen zu beeinträchtigenden Symptomen und Herzfunktionsstörungen kommt. Die häufigste Ursache der HOCM sind Mutationen der Herzmuskelproteine des Sarkomers. Bislang gibt es für HOCM-Patienten in der EU aber noch keine zugelassene Therapie, die auf die dieser Erkrankung zugrunde liegenden Pathomechanismen abzielt. Dies
Mavacamten
könnte sich bald ändern, denn der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Arzneimittelagentur (EMA) hat am 26. April die Zulassung des kardialen Myosin-Inhibitors Mavacamten (Camzyos®) zur Behandlung erwachsener Patienten mit symptomatischer HOCM empfohlen. Die endgültige Entscheidung der Europäischen Kommission wird innerhalb von 67 Tagen nach Erhalt der Zulassungsempfehlung des CHMP erwartet.
Überzeugende Studienergebnisse
Grundlage für die positive Empfehlung des CHMP sind die Ergebnisse der beiden randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien EXPLORERHCM und VALOR-HCM.
In EXPLORER-HCM wurden die Wirksamkeit und Sicherheit von Mavacamten versus Placebo untersucht. Alle primären und sekundären Endpunkte wurden mit statistischer Signifikanz erreicht:
Mavacamten (Camzyos®) ist ein first-in-class oraler allosterischer Inhibitor des kardialen Myosins und zielt damit selektiv auf die der obstruktiven hyprtrophen Kardiomyopathie (HOCM) zugrunde liegende Pathophysiologie ab. Kennzeichnend für die HOCM sind eine übermäßige Bildung von Myosin-Aktin-Querbrücken und die Dysregulation des entspannten Zustands. Mavacamten verschiebt die gesamte Myosin-Population in Richtung eines energiesparenden, rekrutierbaren, entspannten Zustands. Durch die Myosin-Inhibition verringert sich die dynamische LVOT-Obstruktion und der Füllungsdruck des Herzens verbessert sich.
Mavacamten ist bereits in den USA für die Behandlung von Erwachsenen mit symptomatischer (NYHA-Klasse II–III) HOCM zur Verbesserung der funktionellen Leistungsfähigkeit und der Symptome zugelassen, ebenso in Australien, Kanada, Brasilien und der Schweiz.
65 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse/Mitteilungen
Es zeigten sich ein deutlicher Behandlungseffekt mit klinisch bedeutsamen Verbesserungen der körperlichen Leistungsfähigkeit, der Symptome und des Gesundheitsstatus sowie eine klinisch relevante Verringerung der Obstruktion im linksventrikulären Ausflusstrakt. Die VALOR-HCM-Studie untersuchte die Wirksamkeit von Mavacamten bei Patienten mit symptomatischer HOCM versus Placebo, die für eine Septumreduktionstherapie (SRT) infrage kamen. Auch in dieser Studie wurden alle primären und sekundären Endpunkte mit statistischer Signifikanz erreicht. Bei den behandelten HOCM-Patienten kam es zu einer Verbesserung der wichtigsten kardialen Messgrößen, wodurch sich die Notwendigkeit einer invasiven SRT signifikant verringerte.
mit chronischem Koronarsyndrom und/oder chronischer Herzinsuffizienz mit eingeschränkter systolischer linksventrikulärer Funktion, die bereits mit den Monopräparaten in derselben Dosierung eingestellt sind.
Die neue Fixkombination vereint zwei bewährte und häufig verordnete Wirkstoffe zur Blutdrucksenkung in einer Kapsel und bietet so die Möglichkeit einer effektiven Hypertoniebehandlung bei gleichzeitig verbesserter Compliance.
Leitlinien-Empfehlung:
Initial mit Zweifachkombination behandeln
S. Ramiprolol® –ACE-Hemmer und Betablocker jetzt
B.
in einer Kapsel
Seit März 2023 ist eine neue Fixkombination von Aristo Pharma zur Hypertonie-Behandlung in Deutschland verfügbar: Ramiprolol®, die Fixkombination aus dem breit eingesetzten ACE-Hemmer Ramipril und dem selektiven Betablocker Bisoprolol, ist zugelassen zur Behandlung von Hypertonie, Hypertonie mit gleichzeitig bestehendem chronischem Koronarsyndrom und/oder chronischer Herzinsuffizienz mit eingeschränkter systolischer linksventrikulärer Funktion bzw. zur Verringerung des kardialen Risikos bei Patienten
Für die medikamentöse Behandlung der arteriellen Hypertonie stehen verschiedene Wirkstoffklassen (ACE-Hemmer, Diuretika, Betablocker, Kalziumantagonisten, Angiotensin-II-Rezeptorblocker [ARB; Sartane]) zur Verfügung. Die Wahl des geeigneten Präparats richtet sich dabei u.a. nach Alter, konkreter Diagnose und Komorbiditäten. Häufig wird durch ein einzelnes Präparat allerdings keine ausreichende Blutdrucksenkung erzielt, sodass die einschlägige Leitlinie empfiehlt, bereits initial mit einer Zweifachkombination zu behandeln. Eine weitere Empfehlung findet sich in der Leitlinie zur Behandlung des chronischen Koronarsyndroms von 2019: Diese empfiehlt zur Hochdruckbehandlung auch eine Kombinationstherapie mit einem Betablocker – allerdings nicht pauschal, sondern bei vorliegenden Indikationen, wie z.B. der systolischen Herzinsuffizienz, Rhythmusstörungen oder Angina pectoris.
Dr. Wolfgang Derer, Kardiologe und Oberarzt am Helios Klinikum in Berlin-Buch, hält diese Empfehlung für „differenzierter“ – räumt aber ein: „Trotzdem wird bei einer Vielzahl von Patienten mit chronischem Koronarsyndrom eine initiale antihypertensive Therapie mit einem RAS-Blocker (z.B. ACE-Hemmer) und einem Betablocker indiziert sein, da das chronische Koronarsyndrom häufig durch die eben genannten Sachverhalte verkompliziert wird.“ Diese Empfehlungen führen, insbesondere beim Vorliegen weiterer Erkrankungen, mitunter zu einer sehr hohen Tablettenlast, was sich ungünstig auf die Compliance auswirken kann. Um die BlutdruckZielwerte (<130/80 mmHg bzw. <140/80 mmHg im Alter von >65 Jahren) zu erreichen und gleichzeitig die Tablettenlast zu reduzieren, bieten sich daher Fixdosiskombinationen aus zwei oder mehreren Wirkstoffen in einer Tablette bzw. Kapsel an. Solche Fixkombinationen gibt es in der Kardiologie z.B. bereits aus ACE-Hemmer und Diuretikum oder Betablocker und Diuretikum. Nun ist in Deutschland mit Ramiprolol® erstmals eine Fixkombination aus dem ACE-Hemmer Ramipril und dem Betablocker Bisoprolol auf dem Markt.
Als Inhibitor des Angiotensin Converting Enzyme (ACE) hemmt Ramipril die Umwandlung von Angiotensin I zu Angiotensin II. Somit kann dieses seine gefäßverengende Wirkung nicht entfalten. Daneben hemmt Ramipril über die
66 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH Mitteilungen
Ramipril: Allrounder des kardiovaskulären Kontinuums
Inhibition des ACE auch die Freisetzung des Hormons Aldosteron, was zusätzlich eine schwache diuretische Wirkung bedingt. Zudem wird durch den ACE-Hemmer die Inaktivierung des vasodilatatorischen Gewebshormons Bradykinin verhindert. Eine Behandlung mit Ramipril hat daher einen kombinierten vasodilatatorischen und somit blutdrucksenkenden Effekt. Der ACE-Hemmer Ramipril kommt primär bei der Therapie von arterieller Hypertonie und systolischer Herzinsuffizienz zum Einsatz, aber die Indikationen schließen auch weitere kardiovaskuläre Erkrankungen ein, wie beispielsweise die zur Sekundärprävention nach einem akuten Myokardinfarkt. Aufgrund seiner günstigen Wirkung auf die Nierenfunktion ist Ramipril auch Mittel der Wahl in der antihypertensiven Therapie, etwa bei diabetischer Nephropathie und Proteinurie. Zudem gelten ACE-Hemmer allgemein als gut mit anderen Wirkstoffen kombinierbar.
rung der Schlagfrequenz) Wirkung auf das Herz erklären. Deshalb werden Betablocker nicht nur bei Hypertonie, sondern auch bei KHK oder Herzinsuffizienz eingesetzt. Speziell der Betablocker Bisoprolol verbessert bei Herzinsuffizienz nachweislich die Prognose. Der Wirkstoff besitzt keine intrinsische Aktivität und ist β1-selektiv, entfaltet seine Wirkung also vor allem am Herzen, was für ein günstigeres Nebenwirkungsprofil sorgt. Darüber hinaus ist Bisoprolol im Vergleich etwa zu Metoprolol inerter in Bezug auf Wechselwirkungen mit neurologischen/ psychiatrischen Medikamenten (z.B. Antidepressiva). Dank seiner Halbwertszeit von 10 – 12 Stunden reicht eine einmal tägliche Gabe in der Regel aus.
Meistverordnete Substanzen fix kombiniert
die Wirkstoffe bei erwachsenen Patienten, deren Blutdruck bereits mit diesen beiden Wirkstoffen als Monopräparate in gleichbleibender Dosis hinreichend kontrolliert ist, in Form der neuen Fixkombination Ramiprolol® verordnet werden. Da die Hartkapsel zwei Mechanismen zur Vasodilatation vereint, lässt sich der Blutdruck – und dadurch das kardiovaskuläre Risiko – effektiv senken.
Patienten mit kardiologischen Problemen wie Herzschwäche, Herzinfarkt, koronarer Herzkrankheit Vorhofflimmern, die auch unter manifester Hypertonie leiden, sind Dechend zufolge, „besonders prädestiniert“ für die „Kombination aus Gefäßprotektion (Ramipril) und Herzprotektion (Bisoprolol)“.
Gleichzeitig vereinfacht die Gabe der Fixkombination, die nur 1 × täglich (morgens) eingenommen wird, das Therapieregime und reduziert die Tablettenlast.
Bisoprolol: ein bewährter Betablocker
Betablocker blockieren β-Adrenozeptoren (u.a. an den Herzkranzgefäßen) für Noradrenalin und Adrenalin und sorgen somit für eine Verringerung der Herzfrequenz und der Kontraktilität. Zusätzlich kommt es in der Niere zu einer verminderten Ausschüttung des Hormons Renin. In der Folge werden auch geringere Mengen Angiotensin und Aldosteron freigesetzt, woraus sich der vasodilatatorische Effekt und die negativ inotrope (Verringerung der Kontraktilität) und negativ chronotrope (Verringe-
Für Professor Ralf Dechend, Oberarzt in der Kardiologie am Helios Klinikum und der Charité in Berlin-Buch, gehören beide Wirkstoffe in der Kardiologie heute zu einer „State-of-the-art-Therapie“, da sie sich sowohl in klinischen Studien als auch im Praxisalltag bewährt haben. Sowohl Ramipril als auch Bisoprolol sind die meistverordneten Substanzen ihrer jeweiligen Wirkstoffklasse. Dazu kommt: „Beide sind bei vielen Patienten mit kardiovaskulären Erkrankungen ohnehin schon als Einzelsubstanzen Teil des medikamentösen Therapieregimes – und unter Kardiologen, Allgemeinmedizinern und auch Nephrologen entsprechend bekannt“, weiß auch Derer aus Erfahrung. Nun können
Beides bedeutet für die Patienten häufig nicht nur eine Verbesserung der Lebensqualität: Studienergebnisse zur Anwendung von Fixkombinationen zeigen, dass so die Compliance im Vergleich zur Einnahme als freie Kombination um 29 % gesteigert werden kann.
Die 6 Wirkstärken von Ramiprolol® (Ramipril/Bisoprolol 2,5 mg/ 1,25 mg; 2,5 mg/2,5 mg; 5 mg/2,5 mg; 5 mg/5 mg;10 mg/5 mg;10 mg/10 mg) ermöglichen dabei eine individuelle Dosisanpassung.
B. S.
67 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH
Mitteilungen
Luspatercept jetzt auch für Patienten mit nicht-transfusionsabhängiger Beta-Thalassämie zugelassen
Luspatercept (Reblozyl®) erhielt am 28. Februar 2023 von der Europäischen Kommission die Zulassung zur Behandlung erwachsener Patienten mit Anämie, die mit einer nicht-transfusionsabhängigen BetaThalassämie verbunden ist. Dies ist die dritte zugelassene Indikation für Luspatercept, dem ersten und einzigen Erythrozyten-ReifungsAktivator zur Behandlung von Patienten mit Erkrankungen, die mit einer Anämie einhergehen (vgl. Insert).
Signifikante Hb-Anstiege in der BEYOND-Studie
Die Zulassungserweiterung durch die Europäische Kommission beruht auf den Ergebnissen der doppelblinden, randomisierte, placebokontrollierte Phase-II-Studie BEYOND. Diese untersuchte die Wirksamkeit und Sicherheit von Luspatercept im Vergleich zu Placebo bei 145 erwachsenen Patienten mit nicht-transfusionsabhängiger Beta-Thalassämie. Die Studienteilnehmer wurden im Verhältnis 2:1 entweder auf Luspatercept (n = 96) oder Placebo (n = 49) randomisiert und konnten bedarfsweise zusätzlich Best Supportive Care, einschließlich Erythrozytenkonzentrat (EK)-Transfusionen, Eisenchelatoren, einer antibiotischen, antiviralen und antimykotischen Therapie und/oder einer Ernährungstherapie, erhal-
Beta-Thalassämie
Beta-Thalassämie ist eine Blutkrankheit, die durch einen genetischen Defekt im Beta-Globin-Gen verursacht wird. Es ist eine der häufigsten autosomalrezessiv vererbten Erkrankungen. Die Gesamtinzidenz symptomatischer Personen wird pro Jahr auf 1:100.000 Einwohner weltweit geschätzt. Die Krankheit ist mit einer ineffektiven Erythropoese verbunden, die zu einem Mangel an gesunden Erythrozyten führt. In der Folge kommt es häufig zu einer Anämie, die für die Patienten oftmals eine hohe Belastung darstellt und mit schweren Komplikationen einhergehen kann.
Die Behandlungsmöglichkeiten für chronische Anämien im Zusammenhang mit Beta-Thalassämie sind begrenzt. Sie bestehen hauptsächlich aus häufigen EK-Transfusionen, die das Risiko einer sekundären Eisenüberladung bergen und schwerwiegende Komplikationen wie zum Beispiel Organschäden verursachen können.
Der Begriff nicht-transfusionsabhängige Beta-Thalassämie wird für Patienten verwendet, die nicht ihr Leben lang auf regelmäßige EK-Transfusionen angewiesen sind, um zu überleben. Allerdings können bei ihnen verschiedene klinische Komplikationen auftreten und gelegentliche oder sogar häufiger wiederkehrende Transfusionen erforderlich sein.
Luspatercept
Der Erythrozyten-Reifungs-Aktivator Luspatercept (Reblozyl®) ist der erste und einzige Vertreter dieser Wirkstoffklasse. Das Medikament kann die Ausreifung der Erythrozyten im Spätstadium der Erythropoese fördern. In der EU ist Luspatercept zugelassen für
• die Behandlung erwachsener Patienten mit Anämie, die mit transfusionsabhängiger und nicht-transfusionsabhängiger Beta-Thalassämie verbunden ist, sowie für
• die Behandlung erwachsener Patienten mit transfusionsabhängiger Anämie aufgrund von myelodysplastischen Syndromen (MDS) mit Ringsideroblasten, mit sehr niedrigem, niedrigem oder intermediärem Risiko, die auf eine Erythropoetin-basierte Therapie nicht zufriedenstellend angesprochen haben oder dafür nicht geeignet sind.
Cave: Luspatercept ist nicht indiziert als Ersatz für EK-Transfusionen bei Patienten, die eine sofortige Behandlung ihrer Anämie benötigen.
ten. Primärer Studienendpunkt war der Anteil an Patienten mit einem mittleren Anstieg des Hämoglobin (Hb)-Wertes um ≥1,0 g/dl ohne EK-Transfusionen ab Studienbeginn über einen kontinuierlichen Zeitraum von 12 Wochen (Woche 13
24).
Wichtige sekundäre Endpunkte waren die mittleren Veränderungen der Patient-Reported-OutcomesScores für die Domänen Müdigkeit und Schwäche bei nicht-transfusionsabhängiger Beta-Thalassämie und des Hb-Wertes seit Studienbeginn.
68 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH
Mitteilungen
–
Mitteilungen
Einen mittleren Anstieg des HbWertes um ≥1,0 g/dl ab Studienbeginn und damit den primären Endpunkt der Studie erreichten 77,1 % der Patienten unter Luspatercept gegenüber 0 % der Patienten im Placebo-Arm (p < 0,0001).
Der wichtige sekundäre Endpunkt, ein mittlerer Anstiegs des Hb-Wertes um ≥1,5 g/dl ohne EK-Transfusionen im Vergleich zu Studienbeginn wurde von 49,0 % der mit Luspatercept behandelten Patienten in Woche 37 – 48 gegenüber 0 % im Placebo-Arm erreicht.
In Woche 1 – 24 blieben 89,6 % der Patienten im Luspatercept-Arm transfusionsfrei gegenüber 67,3 % der Patienten im Placebo-Arm. Bei den Verbesserungen der Patient-Reported-Outcomes zu Aspekten der Lebensqualität (Müdigkeit und Schwäche) wurde ebenfalls eine Korrelation mit dem Anstieg des Hb-Wertes beobachtet.
Schwerwiegende Nebenwirkungen traten bei 11,5 % der Patienten unter Luspatercept sowie bei 25 % der Patienten im Placebo-Arm auf. Die häufigsten Nebenwirkungen, die bei ≥10 % der mit Luspatercept behandelten Patienten registriert wurden, waren Knochenschmerzen (36 %), Kopfschmerzen (30 %), Arthralgie (29 %), Rückenschmerzen (28 %), Prähypertonie (23 %), Hypertonie (20 %), Husten (18 %), Diarrhö (17 %), grippeartige Erkrankung (17 %), Asthenie (13 %), Grippe (13 %), Schlaflosigkeit (11 %) und Übelkeit (10 %).
B. S.
IMPRESSUM
OFFIZIELLES ORGAN DER DEUTSCHEN GESELLSCHAFT FÜR ARTERIOSKLEROSEFORSCHUNG
Herausgeber:
Überzeugende Daten auch zu MitraClip™ und Amplatzer™
Amulet™
Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School,Salmon Pool Lane, Exeter EX2 4SG, UK
Prof. Dr. med. Wolfgang Koenig Deutsches Herzzentrum München Technische Universität München
Lazarettstraße 36 80636 München
Wissenschaftlicher Beirat:
Prof. Dr. med. T. von Arnim (Kardiologie), München
Prof. Dr. med. G. V. R. Born (Arterioskleroseforschung), London
Schriftleitung:
Auf dem EuroPCR präsentierte Abbott außerdem die positiven Ergebnisse der EXPAND-Studie. Diese belegte, dass die Therapie mit dem MitraClip™-G4-System (Abb. 3) bei HerzinsuffizienzPatienten mit Mitralinsuffizienz zu einer Verbesserung der Symptome und der Lebensqualität führt.
Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School, Salmon Pool Lane, Exeter EX2 4SG, UK
E-Mail: Edzard.Ernst@pms.ac.uk
Tel: +44 (0) 1392 726029
Fax: +44 (0) 1392 421009
Die Zeitschrift erscheint 6-mal im Jahr; Jahresabonnement 27,–; Einzelheft 5,50, inklusive MwSt., zuzüglich Versandspesen. Der Abonnementpreis ist im voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Prof. Dr. med. C. Diehm (Angiologie), Karlsbad
Priv.-Doz. Dr. med. Dr. phil. C. Drosde (Kardiologie), Freiburg
Dr. med. J. Dyerberg MD, Ph. D. (Klin. Chemie), Aalborg Sygehus, Dänemark Univ.-Prof. Dr. med. H. W. Eichstädt, (Kardiologie), Berlin
Doz. Dr. rer. nat. F.-D. Ernst (Hämorheologie), Dresden
Dr. med. J. Gehring (Kardiologie, Rehabilitation), München
Prof. Dr. med. J. D. Gruß (Gefäßchirurgie), Kassel
Prof. Dr. J. Harenberg (Hämostaseologie), Mannheim
Prof. Dr. med. L. Heilmann (Gynäkologie), Rüsselsheim
Prof. Dr. med. H. M. Hoffmeister (Kardiologie), Solingen
Prof. Dr. med. H. U. Janka (Diabetologie), München
Dr. med. J. Janzen MPhil (Pathologie), Bern, Schweiz
Prof. Dr. med. L. Kollár M.D., PhD (Gefäßchirurgie), Universität Pécs, Ungarn Prof. Dr. med. M. Marshall (Phlebologie), Rottach Egern Prof Dr. med. J. Matsubara (Chirurgie), Ishikawa, Japan
Prof. Dr. med. G. Mchedlishvilli (Mikrozirkulation), Tbilisi, Georgien Prof. Dr. med. V. Mitrovic (Kardiologie, Klinische Pharmakologie), Bad Nauheim
Bei Patienten mit Vorhofflimmern kann zum Schutz vor kardioembolischen Schlaganfällen das linke Vorhofohr (left atrial appendage, LAA) minimalinvasiv mit dem LAA-Okkluder Amplatzer™ Amulet™ (Abb. 4) verschlossen werden. Obwohl bei Frauen häufiger Frühkomplikationen nach dem LAA-Verschluss auftreten als bei Männern, belegen die Ergebnisse der Amulet-IDE-Studie nun, dass Frauen und Männer, denen der Amplatzer™ Amulet™ LAAOkkluder implantiert wurde, langfristig ähnliche Vorteile aus dem LAA-Verschluss ziehen. Brigitte Söllner, Erlangen
Prof. Dr. med. H. Mörl (Angiologie), Mannheim
Prof. Dr. med. F. J. Neumann (Kardiologie), Bad Krozingen
Prof. Dr. med. K. L. Resch (Medizin-Statistik), Bad Elster
Prof. Dr. med. G. Rettig (Kardiologie), Homburg
PD Dr. med. Rainer Röttgen (Radiologie), Berlin
Prof. Dr. med. G. Schmid-Schönbein (Biomechanik), La Jolla, USA
Prof. Dr. med. H. Schmid-Schönbein (Physiologie), Aachen
Prof. Dr. med. A. Schrey (Pharmakologie), Düsseldorf
Prof. Dr. med. H. Sinzinger (Nuklearmedizin), Wien, Österreich
Prof. Dr. med. T. Störk (Kardiologie, Angiologie), Göppingen
Prof. Dr. med. I. Szirmai M.D. (Neurologie), Universität Budapest, Ungarn
Prof. Dr. med. G. Trübestein (Angiologie), Bonn
Prof. Dr. med. B. Tsinamdzvrishvili (Kardiologie, Hypertonie), Tbilisi, Georgien Prof. Dr. med. W. Vanscheidt (Dermatologie), Freiburg
Prof. Dr. med. H. Weidemann (Kardiologie, Sozialmedizin), Bad Krozingen
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 – Rolf Wolle (verantwortlich) Schwabacherstr. 117, 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna (verantwortlich) Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf
Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Satz:
Rolf Wolle, Schwabacherstr. 117, 90763 Fürth
Druck und Verarbeitung:
DRUCK_INFORM_W.R.
Roland Welker
Austraße 7, 96114 Hirschaid
PERFUSION is listed in Current Contents/Clinical Medicine (CC/CM) and listed in The Genuine Article.
Verlag PERFUSION GmbH
69 Perfusion 2/2023 36. Jahrgang © Verlag PERFUSION GmbH
Storchenweg 20 90617 Puschendorf Telefon: 09101/990 11 10 Fax: 09101/990 11 19 www.Verlag-Perfusion.de E-Mail: perfusion@t-online.de
PERFUSION VERL AG PERFUSION Journal





