haematologica

VOL. 108 APRIL 2023 Journal of the Ferrata Storti Foundation ISSN 0390 - 6078 haematologica.org
J/"#,-#%3,0%&'()*#5*-K%*-%%
4'+5%+*$/1%6,'0-")%
78&"+$%!"+$,0%9:9;<%;;=:>%%% %?*$/@+,0/%9:9;<%;;=A%%%
!"#$%0/B*/C%&0,+/##%
@'(8*##*,-%→%;#$%1/+*#*,-<%;D%1"E#%

!"#$%&'()*+"$*,-%

."&/0#%*##'/1%2'#$%"3$/0%"++/&$"-+/
F,C%&'()*+"$*,-%+,#$%

G5/%&'()*#5/0%*#%"%-,-H&0,3*$% %!,'-1"$*,-%$5"$%I//&#%$5/%% %+,#$%3,0%"'$5,0#%"#%),C%"#%&,##*()/%
6,'0-")%,3%$5/%!/00"$"H%@$,0$*%!,'-1"$*,-%
h aematologica
haematologica
Editor-in-Chief
Jacob M. Rowe (Jerusalem)
Deputy Editors
Carlo Balduini (Pavia), Jerry Radich (Seattle)
Associate Editors
Shai Izraeli (Tel Aviv), Steve Lane (Brisbane), Pier Mannuccio Mannucci (Milan), Pavan Reddy (Houston), David C. Rees (London), Paul G. Richardson (Boston), Francesco Rodeghiero (Vicenza), Gilles Salles (New York), Kerry Savage (Vancouver), Aaron Schimmer (Toronto), Richard F. Schlenk (Heidelberg), Sonali Smith (Chicago)
Statistical Consultant
Catherine Klersy (Pavia)
Editorial Board
Walter Ageno (Varese), Sarit Assouline (Montreal), Andrea Bacigalupo (Roma), Taman Bakchoul (Tübingen), Pablo Bartolucci (Créteil), Katherine Borden (Montreal), Marco Cattaneo (Milan), Corey Cutler (Boston), Kate Cwynarski (London), Mary Eapen (Milwaukee), Francesca Gay (Torino), Ajay Gopal (Seattle), Alex Herrera (Duarte), Martin Kaiser (London), Marina Konopleva (Houston), Johanna A. Kremer Hovinga (Bern), Nicolaus Kröger (Hamburg), Austin Kulasekararaj (London), Shaji Kumar (Rochester), Ann LaCasce (Boston), Anthony R. Mato (New York), Matthew J. Mauer (Rochester), Neha Mehta-Shah (St. Louis), Moshe Mittelman (Tel Aviv), Alison Moskowitz (New York), Yishai Ofran (Haifa), Farhad Ravandi (Houston), John W. Semple (Lund), Liran Shlush (Toronto), Sara Tasian (Philadelphia), Pieter van Vlieberghe (Ghent), Ofir Wolach (Haifa), Loic Ysebaert (Toulouse)
Managing Director
Antonio Majocchi (Pavia)
Editorial Office
Lorella Ripari (Office & Peer Review Manager), Simona Giri (Production & Marketing Manager), Paola Cariati (Graphic Designer), Giulia Carlini (Graphic Designer), Debora Moscatelli (Graphic Designer), Igor Poletti (Graphic Designer), Marta Fossati (Peer Review), Diana Serena Ravera (Peer Review), Laura Sterza (Account Administrator)
Assistant Editors
Britta Dost (English Editor), Rachel Stenner (English Editor), Anne Freckleton (English Editor), Rosangela Invernizzi (Scientific Consultant), Marianna Rossi (Scientific Consultant), Massimo Senna (Information Technology), Luk Cox (Graphic Artist)
Haematologica | 108 - April 2023
Brief information on Haematologica
Haematologica (print edition, pISSN 0390-6078, eISSN 1592-8721) publishes peer-reviewed papers on all areas of experimental and clinical hematology. The journal is owned by a non-profit organization, the Ferrata Storti Foundation, and serves the scientific community following the recommendations of the World Association of Medical Editors (www.wame.org) and the International Committee of Medical Journal Editors (www.icmje.org).

Haematologica publishes Editorials, Original articles, Review articles, Perspective articles, Editorials, Guideline articles, Letters to the Editor, Case reports & Case series and Comments. Manuscripts should be prepared according to our guidelines (www.haematologica.org/information-for-authors), and the Uniform Requirements for Manuscripts Submitted to Biomedical Journals, prepared by the International Committee of Medical Journal Editors (www.icmje.org).
Manuscripts should be submitted online at http://www.haematologica.org/.
Conflict of interests. According to the International Committee of Medical Journal Editors (http://www.icmje.org/#conflicts), “Public trust in the peer review process and the credibility of published articles depend in part on how well conflict of interest is handled during writing, peer review, and editorial decision making”. The ad hoc journal’s policy is reported in detail at www.haematologica.org/content/policies.
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articles to the Ferrata Storti Foundation. No formal permission will be required to reproduce parts (tables or illustrations) of published papers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with commercial intent will require written permission and payment of royalties.
Subscription. Detailed information about subscriptions is available at www.haematologica.org. Haematologica is an open access journal and access to the online journal is free. For subscriptions to the printed issue of the journal, please contact: Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, E-mail: info@haematologica.org).
Rates of the printed edition for the year 2022 are as following:
Institutional: Euro 700
Personal: Euro 170
Advertisements. Contact the Advertising Manager, Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, e-mail: marketing@haematologica.org).
Disclaimer. Whilst every effort is made by the publishers and the editorial board to see that no inaccurate or misleading data, opinion or statement appears in this journal, they wish to make it clear that the data and opinions appearing in the articles or advertisements herein are the responsibility of the contributor or advisor concerned. Accordingly, the publisher, the editorial board and their respective employees, officers and agents accept no liability whatsoever for the consequences of any inaccurate or misleading data, opinion or statement. Whilst all due care is taken to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this journal, should only be followed in conjunction with the drug manufacturer’s own published literature.
Direttore responsabile: Prof. Carlo Balduini; Autorizzazione del Tribunale di Pavia n. 63 del 5 marzo 1955. Printing: Press Up, zona Via Cassia Km 36, 300 Zona Ind.le Settevene - 01036 Nepi (VT)
Associated with USPI, Unione Stampa Periodica Italiana. Premiato per l’alto valore culturale dal Ministero dei Beni Culturali ed Ambientali
Haematologica | 108 - April 2023
Table of Contents Volume 108, Issue 4: April 2023
About the Cover
Image taken from the review article by Xiang Zhou in this issue.
Landmark Paper in Hematology
937 The GPIIb-IIIa defect of platelets in Glanzmann thrombasthenia
Alan T. Nurden
https://doi.org/10.3324/haematol.2023.282836
Editorials
939
A double punch for plasma cell leukemia
Martin Kaiser
https://doi.org/10.3324/haematol.2022.281353
941 Plasma cell leukemia: another piece of the puzzle
Pellegrino Musto and Ralph Wäsch
https://doi.org/10.3324/haematol.2022.281432
945 CD4 T cells: the complicated key to unlocking the immune environment of classical Hodgkin lymphoma
Maher K. Gandhi and Colm Keane
https://doi.org/10.3324/haematol.2022.281440
947 The effects of chronic glucorticoid stimulation on erythropoiesis in Cushing syndrome
John Strouboulis and Sara El Hoss
https://doi.org/10.3324/haematol.2022.281355
949
Low-intensity induction in acute myeloid leukemia. Always in the patients' best interest?
Ehab Atallah
https://doi.org/10.3324/haematol.2022.281506
951 Getting (T cells) ENGaged
Susanne H.C. Baumeister
https://doi.org/10.3324/haematol.2022.281229
954 Splenectomy in sickle cell disease: do benefits outweigh risks?
Amina Nardo-Marino and Valentine Brousse
https://doi.org/10.3324/haematol.2022.281587
956 Lessons learned from therapy-related acute myeloid leukemia
Sabine Kayser
https://doi.org/10.3324/haematol.2022.281742
Review Article
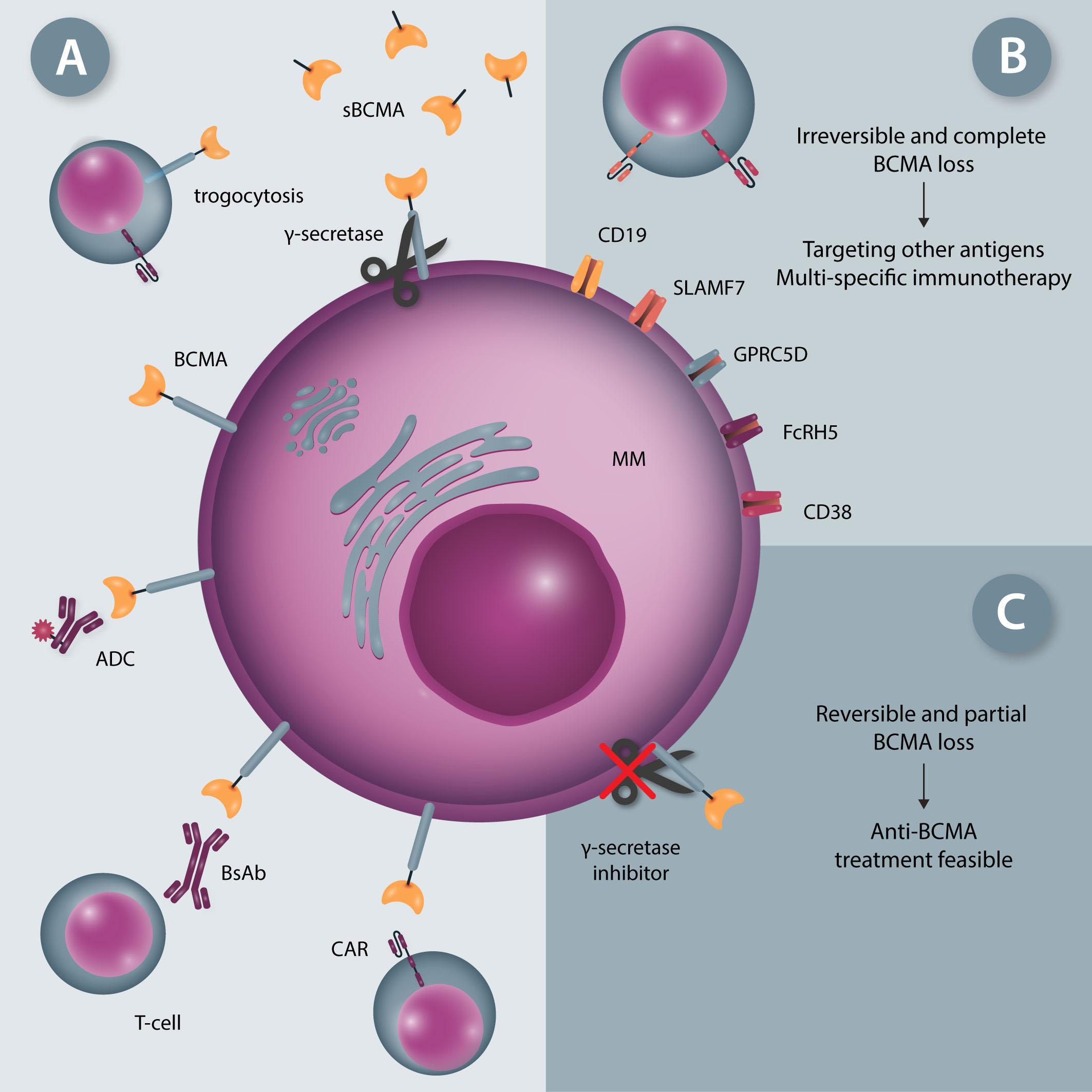
958 BCMA loss in the epoch of novel immunotherapy for multiple myeloma: from biology to clinical practice
Xiang Zhou et al.
https://doi.org/10.3324/haematol.2020.266841
Haematologica | 108 - April 2023 I
Articles
969 Acute Lymphoblastic Leukemia
Genomics improves risk stratifi cation of adults with T-cell acute lymphoblastic leukemia enrolled in measurable residual disease-oriented trials
Celia González-Gil et al.
https://doi.org/10.3324/haematol.2022.281196
981 Acute Lymphoblastic Leukemia
Hyperactive CREB subpopulations increase during therapy in pediatric B-lineage acute lymphoblastic leukemia
Dino Masic et al.
https://doi.org/10.3324/haematol.2022.281177
993 Acute Lymphoblastic Leukemia
Oncogenic TYK2 P760L kinase is effectively targeted by combinatorial TYK2, mTOR and CDK4/6 kinase blockade
Katharina Woess et al.
https://doi.org/10.3324/haematol.2021.279848
1006 Acute Myeloid Leukemia
Time spent at home among older adults with acute myeloid leukemia receiving azacitidine- or venetoclax-based regimens
Christopher E. Jensen et al.
https://doi.org/10.3324/haematol.2022.280728
1015 Acute Myeloid Leukemia
Characterization of therapy-related acute myeloid leukemia: increasing incidence and prognostic implications
Christer Nilsson et al.
https://doi.org/10.3324/haematol.2022.281233
1026 Bone Marrow Transplantation
A phase II, prospective, randomized, open-label study of defibrotide added to standard-of-care prophylaxis for the prevention of acute graft-versus-host disease after allogeneic hematopoietic cell transplantation
Michelle Hudspeth et al.
https://doi.org/10.3324/haematol.2022.281471
1039 Cell Therapy & Immunotherapy
Improving the anti-acute myeloid leukemia activity of CD123-specific engager T cells by MyD88 and CD40 co-stimulation
Abishek Vaidya et al.
https://doi.org/10.3324/haematol.2021.279301
1053
Hematopoiesis
Patients with hypercortisolemic Cushing disease possess a distinct class of hematopoietic progenitor cells leading to erythrocytosis
Lilian Varricchio et al.
https://doi.org/10.3324/haematol.2021.280542
1068 Hodgkin Lymphoma
PDL1 shapes the classical Hodgkin lymphoma microenvironment without inducing T-cell exhaustion
Joseph G. Taylor et al.
https://doi.org/10.3324/haematol.2022.280014
1083 Non-Hodgkin Lymphoma
Diffuse large B-cell lymphoma in octogenarians aged 85 and older can benefit from treatment with curative intent: a report on 129 patients prospectively registered in the Elderly Project of the Fondazione Italiana Linfomi (FIL)
Alessandra Tucci et al.
https://doi.org/10.3324/haematol.2022.281407
Haematologica | 108 - April 2023 II
1092 Non-Hodgkin Lymphoma
Overexpression of the key metabolic protein CPT1A defines mantle cell lymphoma patients with poor response to standard high-dose chemotherapy independent of MIPI and complement established high-risk factors
Anna Sandström Gerdtsson et al.
https://doi.org/10.3324/haematol.2022.281420
1105 Plasma Cell Disorders
Comparison of autologous and allogeneic hematopoietic cell transplantation strategies in patients with primary plasma cell leukemia, with dynamic prediction modeling
Sarah Lawless et al.
https://doi.org/10.3324/haematol.2021.280568
1115 Plasma Cell Disorders
An objective assessment in newly diagnosed multiple myeloma to avoid treatment complications and strengthen therapy adherence
Maximilian Holler et al.
https://doi.org/10.3324/haematol.2022.281489
1127 Platelet Biology & its Disorders
Immune-mediated thrombotic thrombocytopenic purpura plasma induces calcium- and IgG-dependent endothelial activation: correlations with disease severity
Edwige Tellier et al.
https://doi.org/10.3324/haematol.2022.280651
1141 Platelet Biology & its Disorders
Reduced platelet glycoprotein Ib a shedding accelerates thrombopoiesis and COX-1 recovery: implications for aspirin dosing regimen
Paola Simeone et al.
https://doi.org/10.3324/haematol.2022.281006
Letters
1158
Morbidity and mortality of sickle cell disease patients is unaffected by splenectomy: evidence from three decades of follow-up in a high-income setting
Valeria Maria Pinto et al.
https://doi.org/10.3324/haematol.2022.280815
1163
Axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma patients in complete metabolic response
Andrew P. Jallouk et al.
https://doi.org/10.3324/haematol.2022.281954
1168
IDH mutations are enriched in myelodysplastic syndrome patients with severe neutropenia and can be a potential for targeted therapy
Rami Komrokji et al.
https://doi.org/10.3324/haematol.2022.281607
1173
Landscape of immunoglobulin heavy chain γ gene class switch recombination in patients with adult T-cell leukemia–lymphoma
Hiroaki Hiramatsu et al.
https://doi.org/10.3324/haematol.2022.281435
1179
Targeting cytokine-induced leukemic stem cell persistence in chronic myeloid leukemia by IKK2-inhibition
Marlena Bütow et al.
https://doi.org/10.3324/haematol.2022.280922
1186
Methotrexate, cytarabine, thiotepa and rituximab (MATRix) chemoimmunotherapy for primary central nervous system lymphoma: a Toronto experience
Adam Suleman et al.
https://doi.org/10.3324/haematol.2022.282014
Haematologica | 108 - April 2023
III
1190 Modern, real-world patterns of care and clinical outcomes among patients with newly diagnosed diffuse large B-cell lymphoma with or without double/triple-hit status in the United States
Gaurav Goyal et al.
https://doi.org/10.3324/haematol.2022.281461
1196 Time without transfusion reliance: a novel patient-centric metric for new therapies in myelodysplastic syndromes
Joshua F. Zeidner et al.
https://doi.org/10.3324/haematol.2022.281856
Haematologica | 108 - April 2023 IV
The GPIIb-IIIa defect of platelets in Glanzmann thrombasthenia
Alan T. Nurden
Institut Hospitalo-Universitaire LIRYC, Pessac, France
E-mail: nurdenat@gmail.com
https://doi.org/10.3324/haematol.2023.282836
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license

AUTHORS Nurden AT, Caen JP.
Eduard Glanzmann was a Swiss pediatrician who in 1918 reported an inherited platelet functional disorder associated with a defective clot retraction. The clinical phenotype of this autosomal recessive bleeding disorder, later known as Glanzmann thrombasthenia (GT), was largely defined in the 1960s with major contributions from Jacques Caen in Paris and Marjorie Zucker in New York. My involvement in platelet research began in Oxford in 1968. Our project at that time was to define the components of the platelet “glycocalyx”, a carbohydrate-rich layer first highlighted on platelets by an electron microscopist, Olaf Behnke, in Copenhagen. I applied cytochemical techniques to identify negatively charged elements digested from this surface layer and separated by polyacrylamide gel electrophoresis (PAGE). Use of the detergent sodium dodecyl sulfate (SDS) and SDS-PAGE soon enabled the separation of the major intrinsic membrane glycoproteins (GP). Teams led by Ralph Nachman (New York) and David Phillips (Memphis) highlighted three major bands termed GPI (a sialic acid rich GP), GPII and GPIII. I continued my research in London and identified these GP in a range of mammals. However, I quickly realized that inherited platelet disorders held the key to identifying their function.
Early in 1973, I visited Jacques Caen in Paris to apply electrophoretic procedures to the platelets of his patients. How well I remember looking long and hard at my first carbohydrate-stained SDS-PAGE gels. I realized that, for each patient investigated, while the acidic GPI was present, the GPII and GPIII bands were hardly to be seen. The results were published in the British Journal of Haematology in 19741 and confirmed in Nature in 1975.2 A single dimension tube gel from a patient with Glanzmann thrombasthenia is shown in Figure 1. Meanwhile, on the other side of Paris, David Phillips and his co-workers were

independently studying the surface topography of GT platelets using lactoperoxidase-catalyzed iodination (125I) and in 1975 they published similar results to ours, also in Nature.3
As the complexity of the platelet surface constituents evolved, so did the nomenclature, and the affected GP became known as GPIIb and GPIIIa. Studies in my group,
Figure 1. The discovery of the glycoprotein IIb and glycoprotein IIIa defects in platelets in a patient with Glanzmann thrombasthenia. Sodium dodecyl sulfate (SDS)soluble platelet proteins were separated by SDSpolyacrylamide gel electrophoresis on single dimension tube gels prior to carbohydrate staining. Whereas the major glycoprotein I (GPI) band was clearly seen, the GPII and GPIII bands were absent or barely visible. (Figure adapted with permission from Nurden et al. Br J Haematol 1974)

JOURNAL British Journal of Haematology. 1974;28(2):253-260. PMID: 4473996.
Haematologica | 108 - April 2023 937 LANDMARK PAPER IN HEMATOLOGY A.T. Nurden
TITLE An abnormal platelet membrane glycoprotein pattern in three cases of Glanzmann's thrombasthenia.
first with Inger Hagen from Oslo and then with Tom Kunicki from Milwaukee, showed that, in fact, GPIIb and GPIIIa formed a Ca2++-dependent complex in the normal platelet membrane; a complex soon identified as the αIIbb3 integrin. James George (Oklahoma) and Uri Seligsohn (Tel Aviv) were major contributors in promoting a greater understanding of the clinical aspects of GT. The role of αIIbb3 as a fibrinogen receptor responsible for aggregation and clot retraction was progressively defined, while the nature of the mutations within the genes ITGA2B (encoding αIIb) and ITGB3 (encoding b3) gave rise to the
References
1. Nurden AT, Caen JP. An abnormal platelet glycoprotein pattern in three cases of Glanzmann’s thrombasthenia. Br J Haematol. 1974;28(2):253-260.
2. Nurden AT, Caen JP. Specific roles for platelet surface glycoproteins in platelet function. Nature. 1975;255(5511):720-722.
classic and variant forms of GT that are now used around the world as part of the diagnostic procedure.4 Pioneers in the early studies included Peter Newman (Milwaukee), Mark Ginsberg (La Jolla), Gerard Marguerie (La Jolla and Paris), Edward Plow (La Jolla), Joel Bennett (Philadelphia), Sandford Shattil (La Jolla), and Paul Bray (Baltimore), while a special mention goes to Barry S. Coller (New York), among many others.
Disclosure No
3. Phillips DR, Jenkins CSP, Luscher EF, Larrieu M-J. Molecular differences of exposed surface proteins on thrombasthenic platelet plasma membranes. Nature. 1975;257(5527):599-600.
4. Coller BS, Shattil SJ. The GPIIb/IIIa (integrin alphaIIbbeta3) odyssey: a technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112(9):3011-3025.
conflicts of interest to disclose. Haematologica | 108 - April 2023 938 LANDMARK PAPER IN HEMATOLOGY A.T. Nurden
A double punch for plasma cell leukemia
Martin Kaiser
The Institute of Cancer Research, The Royal Marsden Hospital, London, UK
Correspondence: M. Kaiser martin.kaiser@icr.ac.uk
Received: May 31, 2022.
Accepted: June 21, 2022.
Early view: June 30, 2022.
https://doi.org/10.3324/haematol.2022.281353
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The recent approval of multiple novel therapies for multiple myeloma makes it easy to forget that primary plasma cell leukemia (pPCL) remains a heavily understudied cancer with limited evidence-based treatment options. Its rare occurrence and the clinical urgency to start therapy, among other factors, have been substantive barriers to randomized trials for pPCL patients.
In this data-deprived context, the work by Lawless, Iacobelli and colleagues published in this issue of Haematologica offers highly welcome retrospective evidence from the European Group for Blood and Marrow Transplantation registry, which has been providing invaluable insights into rare blood cancers such as pPCL, long before ‘real-world data’ came into the focus of a wider audience.1 This analysis of 751 pPCL patients treated with single (autologous [auto] or allogeneic [allo]) or tandem transplants (auto-auto or auto-allo) between 1998 and 2014 (the ‘pre-maintenance therapy era’) focused particularly on the impact of tandem over single transplants, but also compared the tandem approaches, which had not been studied at this detail in pPCL before. Adjusting for baseline characteristics, patients treated with auto-allo showed the greatest improvement in progressionfree survival over those treated with single auto, while patients who underwent auto-auto showed a nominal, but non-significant improvement over those who underwent single auto. Interestingly, modeling suggested that patients in complete response after induction had longer progression-free survival with auto-auto than with auto-allo and that both of these transplantation strategies performed during complete response produced longer progression-free survival than that after single auto or auto-allo in patients not having achieved complete response post-induction. Although overall survival was nominally improved by tandem approaches, the difference over that with single auto did not reach statistical significance. While the clinically non-acceptable high and early non-relapse mortality of single allo was markedly lower with auto-allo, the long-term overall survival of patients treated with the two strategies was similar, as were acute and chronic graft-versus-host disease (GvHD) rates.
Intensive therapy including transplant remains standard care for younger pPCL patients in many healthcare systems - in-
duction and maintenance therapy often being laterally adopted from myeloma practice. In this context, results from the current study are highly informative. The longer progression-free survival and, in particular, the reduced nonrelapse mortality for auto-allo recipients are encouraging, and are likely to still hold up with contemporary induction therapies. Nevertheless, the majority of pPCL patients in this study still relapsed within 2-3 years after auto-allo, highlighting the general need for post-transplant maintenance strategies. Two potentially interlinked questions emerge in the context of maintenance: (i) would the advantage for auto-allo persist with maintenance? (ii) would maintenance be feasible in the context of acute and chronic GvHD after auto-allo?
While it is impossible to answer the former question without further data, there is some myeloma-derived evidence regarding the latter. For lenalidomide, the only approved maintenance therapy for myeloma, there are studies of reduced dosing to mitigate the adverse effect on GvHD reported earlier; however, post-allo therapy with lenalidomide can remain challenging.2 In contrast, a recent phase II study exploring bortezomib maintenance after tandem auto-allo in young and/or high-risk myeloma patients reported encouraging progression-free and overall survival, and lower rates of GvHD than in historic controls.3 Whether a general improvement in GvHD management may potentially further improve the deliverability of post-allo therapy for pPCL is yet to be seen. Currently the uncertainty regarding GvHD and its putative impact on subsequent therapies, including emergent ones, is probably one of the main barriers to wider utilization of auto-allo transplants in pPCL and myeloma.4
Moving forward, the data from Lawless and colleagues will need to be viewed in the context of a rapidly evolving treatment landscape for myeloma and, potentially, pPCL. Therapy with the anti-CD38 monoclonal antibody daratumumab in an intensive combination demonstrated promising responses in pPCL in the OPTIMUM/MUKnine trial. 5 Chimeric antigen receptor T-cell therapies and drug conjugate monoclonal antibodies against B-cell maturation antigen have already received regulatory approval for myeloma,6 and approvals for T-cell engager
Haematologica | 108 - April 2023 939 EDITORIAL M. Kaiser
monoclonal antibodies against multiple targets are pending.7 However, the activity and safety of these approaches are still to be established in pPCL and, accordingly, more trials including pPCL patients are urgently needed. The SWOG-1211 and OPTIMUM/MUKnine trials, both of which enrolled pPCL and high-risk myeloma patients, are examples of the feasibility of such inclusive approaches.8-10
Until the necessary evidence emerges and the ability of novel immunotherapies to supersede transplantation in pPCL is
References
1. Lawless S, Iacobelli S, Knelange NS, et al. Comparison of autologous and allogeneic hematopoietic cell transplantation strategies in patients with primary plasma cell leukemia, with dynamic prediction modeling. Haematologica. 2023;108(4):1105-1114.
2. Cook M, Panchal A, Brock K, et al. Reduced intensity stem cell transplantation followed by adjunctive lenalidomide is tolerable and safe and improves complete response rate in multiple myeloma: a UK NCRI phase 2 feasibility study (LenaRIC). Blood. 2017;130(Suppl 1):3308.
3. LeBlanc R, Ahmad I, Terra R, et al. Outcomes in newly diagnosed young or high-risk myeloma patients receiving tandem autologous/allogeneic transplant followed by bortezomib maintenance: a phase II study. Bone Marrow Transplant. 2022;57(2):252-260.
4. Zeiser R, von Bubnoff N, Butler J, et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N Engl J Med. 2020;382(19):1800-1810.
5. Kaiser MF, Hall A, Walker K, et al. Depth of response and minimal residual disease status in ultra high-risk multiple myeloma and plasma cell leukemia treated with daratumumab, bortezomib, lenalidomide, cyclophosphamide and dexamethasone (DaraCVRd): results of the UK Optimum/MUKnine trial. J Clin Oncol. 2021;39(15 Suppl):8001.
proven, the work by Lawless et al. provides very useful information on the value of tandem auto-allo in pPCL, in particular for carefully selected, younger patients.
Disclosures
MFK has received consultancy fees and travel support from Janssen, BMS and Takeda; consultancy fees from AbbVie, Pfizer, Seattle Genetics, and GSK; consultancy fees and honoraria from Amgen; and consultancy fees and research funding from Karyopharm, Janssen and Celgene Corporation.
6. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314-324.
7. Shah UA, Mailankody S. Emerging immunotherapies in multiple myeloma. BMJ. 2020;370:m3176.
8. Usmani SZ, Hoering A, Ailawadhi S, et al. Bortezomib, lenalidomide, and dexamethasone with or without elotuzumab in patients with untreated, high-risk multiple myeloma (SWOG-1211): primary analysis of a randomised, phase 2 trial. Lancet Haematol. 2021;8(1):e45-e54.
9. Brown S, Sherratt D, Hinsley S, et al. MUKnine OPTIMUM protocol: a screening study to identify high-risk patients with multiple myeloma suitable for novel treatment approaches combined with a phase II study evaluating optimised combination of biological therapy in newly diagnosed high-risk multiple myeloma and plasma cell leukaemia. BMJ Open. 2021;11(3):e046225.
10. Van De Donk NWCJ, van der Holt B, Schjesvold FH, et al. Treatment of primary plasma cell leukemia with carfilzomib and lenalidomide-based therapy: results of the first interim analysis of the phase 2 EMN12/HOVON129 study. Blood. 2019;134(Suppl 1):693.
Haematologica | 108 - April 2023 940 EDITORIAL M. Kaiser
Plasma cell leukemia: another piece of the puzzle
Pellegrino Musto1 and Ralph Wäsch2
1Department of Emergency and Organ Transplantation, "Aldo Moro" University School of Medicine, Unit of Hematology and Stem Cell Transplantation, AOUC Policlinico, Bari, Italy and 2University Medical Center Freiburg, Department of Hematology, Oncology and Stem Cell Transplantation, Faculty of Medicine, University of Freiburg, Freiburg, Germany
Correspondence: P. Musto pellegrino.musto@uniba.it
Received: June 15, 2022.
Accepted: June 21 2022.
Early view: June 30, 2022.
https://doi.org/10.3324/haematol.2022.281432
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Primary plasma cell leukemia (pPCL) is the most aggressive plasma cell neoplastic disorder. It is characterized by intrinsic genomic instability, high proliferative activity, and co-existence of multiple, adverse clinical and laboratory features, which result in a poorer outcome when compared to that of multiple myeloma.1 The introduction of proteasome inhibitors and immunomodulatory drugs into the treatment of pPCL has produced significant increases in overall (54-90%) and complete response (12-47%) rates compared to those achieved with previous “conventional” chemotherapy, although inducing only a moderate improvement in median overall survival (approximately 1 year for older patients, and 3 years for those who receive transplants).1-5
In this issue of Haematologica, Lawless and co-workers report an updated, retrospective European Blood and Marrow Transplantation Group (EBMT) analysis of 751 pPCL patients transplanted between 1998 and 2014, comparing four frontline transplant strategies: single autologous stem cell transplantation (auto-SCT), single allogeneic stem cell transplantation (allo-SCT), or a combined transplant, either tandem auto-SCT/allo-SCT or double autoSCT (Table 1A, B).6 With a median follow-up of approximately 4 years, the median progression-free survival and overall survival of all patients, irrespective of transplant type, were 14 and 33 months, respectively (Table 1A, B).
Three former retrospective registry studies in transplanteligible patients, two from the EBMT and one from the Center for International Blood and Marrow Transplant Research (CBMTR), evaluated 780 pPCL patients undergoing auto-SCT between 1980 and 2009 (therefore with a limited use of novel agents)7-9 (Table 1A). These surveys showed higher complete response rates in pPCL than in multiple myeloma, but also that auto-SCT was less effective in the long term due to increased non-relapse-related mortality and short duration of post-transplantation response. In particular, in the EBMT studies, the median overall survival was 26 months, while in the CBMTR study, 3-year overall survival was 56% versus 84% after single and double auto-SCT, respectively. More recently and expectedly, a positive effect on overall survival has been reported for maintenance therapy,2,3 low-risk cytogenetics
and achievement of complete remission after auto-SCT.4 Two of the EBMT and CBMTR studies also compared alloSCT in 112 patients, transplanted between 1995 and 2009, with similar populations treated with auto-SCT (Table 1B). The cumulative incidence of relapse was lower after alloSCT than after auto-SCT, but the risk of non-relapse mortality was much higher, without any evidence of survival benefit. In the EBMT study overall survival at 5 years was 19% after reduced-intensity conditioning and 27% after myelo-ablative conditioning.9 In the CBMTR study, 5-year overall survival following allo-SCT was 39% (32% for those undergoing myelo-ablative conditioning, 56% for those given reduced intensity conditioning).8 A plateau phase at 20% was observed.
More recently, on behalf of the CBMTR, Dhakal et al. retrospectively reviewed 348 patients with pPCL receiving auto-SCT (n=277) or allo-SCT (n=71) between 2008 and 2015, thus after the introduction of novel drugs (Table 1A, B).5 Four years after allo-SCT or auto-SCT the progressionfree survival (19% vs. 17%), non-relapse mortality (12% vs. 7%), relapse rate (69% vs. 76 %) and overall survival (31% vs. 28%) were similar in the two groups, confirming no differences in outcome.
Notably, only two prospective trials have been published regarding transplant-eligible pPCL patients, one by the Gruppo Italiano Malattie Ematologiche dell'Adulto (GIMEMA)10 and one by the Intergroupe Francophone du Myelome (IFM).11 Lenalidomide + demamethasone or alternating bortezomib, doxorubicin, dexamethasone (PAD)/bortezomib, cyclophosphamide, dexamethasone (VCD) induction, followed by low-dose lenalidomide or alternate bortezomib, lenalidomide, dexamethasone (VRD)/lenalidomide maintenance after auto-SCT were used, respectively. With a median follow-up of 34 and 28 months, the median overall survival was not reached after single or double auto-SCT in either study (Table 1A), while it was 36 months in the IFM trial patients with a suitable donor, who were planned to undergo auto-SCT followed by reduced intensity conditioning allo-SCT (Table 1B). What, therefore, does the new EBMT study add to our knowledge (Table 1A, B)? Albeit with several important limitations that the authors correctly report, the study by Lawless et al. sheds some light on an important, still
Haematologica | 108 - April 2023 941 EDITORIAL P. Musto and R. Wäsch
Frontline allo-SCT mPFS =11.7 months
Frontline allo-SCT 5-year PFS=19.9%
Frontline allo-SCT mOS= 17.5 months
Frontline allo-SCT 5-year OS= 34.6%
Mahindra et al.8
50 (48) MAC 34 NMA/RIC 16
CIBMTR (1995-2006) 3-year NRM MAC=41% NMA/RIC= 42%
EBMT (1998-2009)
Unspecified, but reported higher than in 411 similar pPCL patients undergoing auto-SCT
3-year PFS=20% MAC=21%
1-year PFS
MAC=39% RIC =43%
5-year PFS
MAC=19% RIC=11%
1-year OS
3-year OS=39% MAC=32% NMA/RIC=56% Morris et al.9 Total 66 MAC 49 (45.9) RIC 17 (52.9)
MAC=46% RIC=59% 5-year OS
MAC=27% RIC =19%
Plateau phase seen at 20%
Auto-SCT N of patients (median age in years) Study group (period of analysis) NRM PFS OS Lawless et al.6° (current study) Total 559 Single 442 (58.8) Double 117 (58.7) EBMT (1998-2014) Frontline auto-SCT NRM=7.3% Frontline auto-SCT mPFS=14.3 months Frontline auto-SCT 5-year PFS=14.3% Frontline auto-SCT mOS= 33.5 months Frontline auto-SCT 5-year OS=31.3% Drake et al.7* 272 (55) EBMT (1980-2006) Unspecified, but reported as increased with respect to registered myeloma patients mPFS=14.3 months mOS=25.7 months 1-year OS=69.3% 3-year OS=39.5% 5-year OS=27.2% Morris et al.9* 411 (55.9) EBMT (1984-2009) Unspecified, but reported lower than in 62 similar pPCL patients undergoing allo-SCT 1-year PFS= 51% 5-year PFS= 10% 1-year OS= 73% 5-year OS= 25% Mahindra et al.8** Total 97 (56) Single 68 Double 25 CIBMTR (1995-2006) 3-year NRM=5% 3-year PFS=34% Single=36% Double=37% 3-year OS=64% Single=56%, Double=84% Dakhal et al.5 Total 277 (60) Single 249 Double 28 CIBMTR (2008-2015) Cumulative 4-year NRM =7% Cumulative 4-year PFS=17% Cumulative 4-year OS=28% Musto et al.10§ Total 8 (58) Single 4 Double 4 GIMEMA (2009-2011) Cumulative NRM=0% Cumulative mPFS=27 months Cumulative OS=NR Royer et al.11^ (2010-2013) 7 (57) IFM (2010-2013) NA mPFS=NR mOS=NR A B Allo-SCT N of patients (median age in years) Study group (period of analysis) NRM PFS OS Lawless et al.6° (current study) Single allo-SCT 70 (47.2) Tandem autoSCT/allo-SCT 122 (51.6) EBMT (1998-2014) Frontline allo-SCT NRM=27%
Total
NMA/RIC=18%
Royer et al.11°° 16 (57) IFM (2010-2013) NRM=12% mPFS=17.9 months mOS=36.3 months Dakhal et al.5 Total. 71 (56) Single allo-SCT 43 Tandem autoSCT/allo-SCT 28 CIBMTR (2008-2015) Cumulative NRM=12% Cumulative 4-year PFS=19% Cumulative 4-year OS=31% Continued on following page.
Haematologica | 108 - April 2023 942 EDITORIAL P. Musto and R. Wäsch
Table
1. Selected studies evaluating the role of autologous (A) and allogeneic (B) stem cell transplantation in primary plasma cell leukemia. Excluding the GIMEMA and IFM prospective trials, all other reports are retropective, registry studies.
*Single auto-SCT. **Four patients underwent tandem auto-SCT/allo-SCT. §Prospective study with a median follow-up of 34 months. ^Prospective study with a median follow-up of 28.7 months; all patients received double auto-SCT. °Frontline auto-SCT included single auto-SCT, double auto-SCT and tandem auto-SCT/allo-SCT. Comparing single and double/tandem transplant procedures, patients undergoing frontline allo-SCT had the greatest risk of death in the first 100 days (but not later). Being transplanted in complete remission conferred significant benefit for both progression-free survival and overall survival after double auto-SCT, with respect to after single auto-SCT. Tandem autoSCT/allo-SCT positively influenced progression-free survival (but not overall survival) in patients not achieving complete remission after induction therapy. °°Prospective study with a median follow-up of 28.7 months; all patients received tandem auto-SCT/allo-SCT. Allo-SCT: allogeneic stem cell transplantation; auto-SCT: autologous stem cell transplantation; CIBMTR: Center for International Blood and Marrow Transplant Research; EBMT: European Group for Blood and Marrow Transplant; GIMEMA: Gruppo Italiano Malattie Ematologiche dell'Adulto; IFM: Intergroupe Francophone du Myélome; MAC: myeloablative conditioning; mOS: median overall survival; mPFS: median progression-free survival; NA: not available; NMA/RIC: non-myeloablative/reduced intensity conditioning; NR: not reached; NRM: non-relapse mortality; OS: overall survival; PFS: progression-free survival; pPCL: primary plasma cell leukemia; RIC reduced intensity conditioning.
unmet clinical need. Given that all previous studies clearly demonstrate the need for transplant(s) in pPCL patients who are eligible for such a procedure, which is the best option to use? Based on the data presented, the answer seems to be quite (and perhaps unexpectedly) clear, helping to guide clinical decisions on transplant strategy. First, the allo-SCT group had a lower relapse rate, but also a remarkable non-relapse mortality (particularly during the first 100 days) that, overall, negatively affected both progression-free and overall survival, at least for the first 2-3 years after transplantation. Interestingly, a plateau phase involving approximately a quarter of patients seemed to be present after 5 years. Although still based on a limited number of patients, this last observation constitutes a not negligible result in terms of a possible “cure” of the disease, which would warrant being discussed very thoroughly with eligible patients. Regarding tandem transplant strategies, double auto-SCT represented an effective option for patients achieving complete remission prior to their first transplant, while, on the other hand, tandem auto/allo-SCT reduced the short-term risk of non-relapse mortality following firstline single allo-SCT and showed a significant overall survival benefit when compared to single auto-SCT and double auto-SCT in patients without a complete response prior to the transplant. Thus, in these patients, disease status at the time of transplant may influence the outcome significantly. This is another important message for clinical practice, suggesting that the results achieved with transplant strategies may depend upon the efficacy of induction treatment. As a consequence, highly active initial therapies should be pursued before proceeding with transplant procedures.
According to currently available recommendations,12 firstline therapy for younger, transplant-eligible pPCL patients, should be oriented toward a short (2-3 cycles) induction phase with proteasome inhibitor and immunomodulatory drug-based triplet, considering the addition of chemotherapy (i.e. hyperfractionated cyclophosphamide, vincristine, doxorubicin, and prednisolone [hyper-CVAD] or cisplatin, doxorubicin, cyclophosphamide, and etoposide [PACE]) if rapid cytoreduction is required. The treatment should include double auto-SCT, consolidation, and maintenance therapy. The pros and cons of possible frontline
allo-SCT should be carefully discussed with eligible patients, who are younger individuals with poor prognostic characteristics at baseline and have achieved a good response to first-line induction.
However, the paradigm of first-line treatment in multiple myeloma is changing rapidly and, as a consequence, pPCL therapy will probably change as well, including new drugs (particularly monoclonal antibodies) in induction and in maintenance therapies after auto-SCT. In this setting, mature results of the recently concluded phase II, EMN12/HOVON129 study for newly-diagnosed pPCL, exploring carfilzomib and lenalidomide-based induction/maintenance therapy, integrated with double auto-SCT or tandem auto/allo-SCT in eligible patients, are eagerly awaited. Venetoclax, an oral inhibitor of BCL-2, may also represent an attractive option, either as a single agent or in combination with novel drugs, for patients with pPCL, given the high prevalence (30-50%) of the t(11;14) in the background of complex genomic characteristics in this population. Highly active new immunotherapies, currently employed in advanced multiple myeloma, such as chimeric antigen receptor T cells, and immuno-conjugated or bispecific antibodies, also warrant investigation for a desirable early use in the setting of pPCL. Further treatments, possibly based on recently recognized genomic characteristics of pPCL, could also be identified. Finally, the emerging role of measurable residual disease in multiple myeloma could be similarly useful in pPCL, i.e., for directing patients toward autologous and/or allogeneic procedures, in the near future.
On this basis, pPCL patients should always be considered for a clinical trial. As a rare disease, pPCL is, however, often excluded from studies performed in multiple myeloma. The new International Myeloma Working Group’s definition of pPCL, lowering the circulating plasma cells from 20% to 5%,13 seems to be one correct move to meet the goal of broader clinical trial availability for pPCL patients in great need of better therapies. It is therefore hoped that these patients will be enrolled in future multiple myeloma trials, with devoted endpoints, predefined plans to extrapolate specific data, and ad hoc analyses for pPCL populations. Such approaches could provide novel biological and clinical information in a short time, which would be useful to speed up the journey along the “long and winding road” of pPCL management.
Haematologica | 108 - April 2023 943 EDITORIAL P. Musto and R. Wäsch
Disclosures
References
1. Musto P, Statuto T, Valvano L, et al. An update on biology, diagnosis and treatment of primary plasma cell leukemia. Expert Rev Hematol. 2019;12(4):245-253.
2. Mina R, Joseph NS, Kaufman JL, et al. Survival outcomes of patients with primary plasma cell leukemia (pPCL) treated with novel agents. Cancer. 2019;125(3):416-423.
3. Gowda L, Shah M, Badar I, et al. Primary plasma cell leukemia: autologous stem cell transplant in an era of novel induction drugs. Bone Marrow Transplant. 2019;54(7):1089-1093.
4. Nandakumar B, Kumar SK, Dispenzieri A, et al. Clinical characteristics and outcomes of patients with primary plasma cell leukemia in the era of novel agent therapy. Mayo Clin Proc. 2021;96(3):677-687.
5. Dhakal B, Patel S, Girnius S, et al. Hematopoietic cell transplantation utilization and outcomes for primary plasma cell leukemia in the current era. Leukemia. 2020;34(12):3338-3347.
6. Lawless S, Iacobelli S, Knelange NS, et al. Comparison of autologous and allogeneic hematopoietic cell transplantation strategies in patients with primary plasma cell leukemia, with dynamic prediction modeling. Haematologica. 2023;108(4):1105-1114.
7. Drake MB, Iacobelli S, van Biezen A, et al. Primary plasma cell leukemia and autologous stem cell transplantation. Haematologica. 2010;95(5):804-809.
8. Mahindra A, Kalaycio ME, Vela-Ojeda J, et al. Hematopoietic cell
transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia. 2012;26(5):1091-1097.
9. Morris C, Iacobelli S, Gahrton G, et al. Has allogeneic transplantation a role in the management of plasma cell leukemia? A study on behalf of the Myeloma Subcommittee of the Chronic Leukemia Working Party of the EBMT. Blood. 2011;118(21): 2008.
10. Musto P, Simeon V, Martorelli MC, et al. Lenalidomide and lowdose dexamethasone for newly diagnosed primary plasma cell leukemia. Leukemia. 2014;28(1):222-225.
11. Royer B, Minvielle S, Diouf M, et al. Bortezomib, doxorubicin, cyclophosphamide, dexamethasone induction followed by stem cell transplantation for primary plasma cell leukemia: a prospective phase II study of the Intergroupe Francophone du Myelome. J Clin Oncol. 2016;34(18):2125-2132.
12. Gavriatopoulou M, Musto P, Caers J, et al. European Myeloma Network recommendations on diagnosis and management of patients with rare plasma cell dyscrasias. Leukemia. 2018;32(9):1883-1898.
13. Fernández de Larrea C, Kyle R, Rosiñol L, et al. Primary plasma cell leukemia: consensus definition by the International Myeloma Working Group according to peripheral blood plasma cell percentage. Blood Cancer J. 2021;11(12):192.
and RW wrote the manuscript.
No conflicts of interest to disclose. Contributions PM
Haematologica | 108 - April 2023 944 EDITORIAL P. Musto and R. Wäsch
CD4 T cells: the complicated key to unlocking the immune environment of classical Hodgkin lymphoma
Maher K. Gandhi1,2 and Colm Keane2,3
Immune checkpoint inhibition has revolutionized the treatment landscape of relapsed classical Hodgkin lymphoma (cHL). Remarkably, 60-70% of patients with relapsed disease will have a response to therapy with antiprogrammed cell death 1 (PD1) therapy.1 Despite its effectiveness, it is unclear why cHL is so responsive to this treatment approach. Clues may lie in the relatively small number of malignant Hodgkin and Reed-Sternberg (HRS) cells (approximately 5%) that sit within a large inflammatory immune cell-rich tumor milieu. In most cases these HRS cells have copy number gains in the locus of ligand 1 or ligand 2 for PD1 (PD-L1/-L2).2 Epstein-Barr virus expression is also found in 30-40% of cases, and this acts as a further driver of PD-L1 and PD-L2 expression within the malignant cell.
Despite this, the malignant cell still only contributes a small proportion of total PD-L1 within the tumor, with the majority emanating from the macrophages in the supporting tumor microenvironment (TME).3 This unique TME makes deciphering the role of the PD-1 axis in cHL extremely complex and challenging.
PD-1 blockade is an effective therapy in solid tumors that have a high mutational burden such as melanoma, particularly if accompanied by CD8 T-cell intratumoral infiltration. This indicates that the mechanism of action is in part related to reinvigoration of neoantigen-specific T cells. However, despite the clear genetic amplification and expression of PD1 axis molecules in the TME in cHL, there is a relative paucity of data to explain the excellent responses to anti-PD1 therapy. Indeed, understanding the excellent responses to anti-PD1 blockade is further complicated by the loss of major histocompatibility complex (MHC) class I and II expression on the malignant HRS cells in many cases (particularly Epstein-Barr virusnegative cHL), challenging the conventionally accepted understanding of immune checkpoint response.
In the current issue of Haematologica, Taylor et al. provide an insight into the cHL TME and an alternative explanation for anti-PD1 response in cHL which is quite distinct from that postulated in solid cancers.4 In support
Correspondence: C Keane
c.keane@uq.edu.au
Received: June 28, 2022.
Accepted: July 11, 2022.
Early view: July 14, 2022.
https://doi.org/10.3324/haematol.2022.281440
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
of previous work, they confirm that HRS cells do possess high levels of PD-L1 and are associated with high PD-L1 expression on surrounding macrophages. They do, however, demonstrate that T-cell exhaustion in the cHL microenvironment is not a predominant feature. They use various techniques to assess the level of T-cell exhaustion within the cHL microenvironment, and consistently show lower expression of PD1 and other markers of exhaustion on T cells from cHL samples, compared to those obtained from reactive lymph nodes. It appears that most of these T cells are functionally active rather than exhausted. In addition, high levels of PD-L1 in the cHL TME did not correlate with increasing Tcell exhaustion and in fact PD-L1 expression in the TME was not associated with PD-1 expression. It appears that the influence of PD-L1 expression in cHL may have its greatest impact on specific subsets of helper and regulatory T cells rather than effector T cells. TH1Reg cells were more common in PD-L1-rich environments and appeared to contribute to the exclusion of CD8 effector T cells. Thus, the authors speculate that a key mechanism for anti-PD1 response in cHL is the manipulation of the immune response away from a TReg/immunosuppressive environment to one in which the resident effector T cells can break through this immunosuppressive barrier and contribute to immune removal of tumor cells. Further functional work is required to test this hypothesis in models or cohorts of patients treated with anti-PD1 therapy.
The work by Taylor et al. is consistent with previous work in which CD4 T cells appear to play critical roles in the TME of cHL.5,6 Aoki et al. showed that TReg cells expressing LAG3 contributed significantly to an immunosuppressive TME in cHL, particularly where MHC class II molecules were lost. This work showed the likely benefit of targeting multiple immune checkpoints in cHL given the different mechanisms that the HRS employ under different genetic and TME conditions.
CD4 TReg were directly targeted in a recent phase I study that included a large number of cHL patients and utilized
1Mater Research, University of Queensland; 2Haematology Department, Princess Alexandra Hospital and 3Diamantina Institute, University of Queensland, Brisbane, Queensland, Australia
Haematologica | 108 - April 2023 945 EDITORIAL M.K. Gandhi and C. Keane
an anti-CD25 antibody that effectively depletes TReg cells. This study showed remarkable efficacy in patients with relapsed cHL, seemingly confirming the importance of TReg manipulation in the treatment of cHL.7
Emerging data from clinical cohorts treated with anti-PD1 agents appear to show possibly differing mechanisms of response at diagnosis and relapse. Updated results from the correlative work on the NIVAHL study seems to show that markers of peripheral T-cell exhaustion are predictors of response to anti-PD1 therapy and that specific tumor-associated antigen immune responses could be detected in a large number of treatment-naïve patients receiving frontline anti-PD1 therapy.8 Responses at relapse appear to be related to more diverse CD4 T-cell populations and innate immune cell expansions.9
In addition, it should be remembered that there is evidence of direct HRS cell survival with binding of PD-1 to its ligands on the tumor cells, and that other immune cells such as natural killer cells may also play critical roles in cHL, illustrating that the TME in cHL is unique and complex with likely multiple PD-1 pathways involved in disease propagation.10,11
References
1. Armand P, Engert A, Younes A, et al. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: extended follow-up of the multicohort single-arm phase II CheckMate 205 trial. J Clin Oncol. 2018;36(14):1428-1439.
2. Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34(23):2690-2697.
3. Carey CD, Gusenleitner D, Lipschitz M, et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood. 2017;130(22):2420-2430.
4. Taylor JG, Truelove E, Clear A, Calaminici M, Gribben JG. PDL1 shapes the classical Hodgkin lymphoma microenvironment without inducing T-cell exhaustion. Haematologica. 2023;108(4):1068-1082.
5. Aoki T, Chong LC, Takata K, et al. Single-cell transcriptome analysis reveals disease-defining T-cell subsets in the tumor microenvironment of classic Hodgkin lymphoma. Cancer Discov. 2020;10(3):406-421.
6. Nagasaki J, Togashi Y, Sugawara T, et al. The critical role of
In conclusion, despite the widespread adoption of antiPD1 therapy in cHL, important correlative work has lagged and this shortcoming needs to be addressed in future studies aimed at teasing out the complexity of the TME in cHL. It would appear, as suggested by Taylor et al., that dynamic changes of CD4 T cells in cHL are critical in the response to immune-based therapies.
Clinical studies that combine immune checkpoint therapies using anti-PD1 therapy as a backbone are emerging and will provide insights into how targeting TReg may be critical in cHL. The paper by Taylor et al. provides important findings that could influence the design of the next phase of immune-based therapies in cHL.
Disclosures
CK has participated in advisory boards for Karyopharm, Roche, Beigene and BMS and has received research funding from Beigene and MSD. MG received research funding from Beigene and Janssen.
Contributions
The authors contributed equally.
CD4+ T cells in PD-1 blockade against MHC-II-expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 2020;4(17):4069-4082.
7. Hamadani M, Collins GP, Caimi PF, et al. Camidanlumab tesirine in patients with relapsed or refractory lymphoma: a phase 1, open-label, multicentre, dose-escalation, dose-expansion study. Lancet Haematol. 2021;8(6):e433-e445.
8. Garcia-Marquez MA, Thelen M, Reinke S, et al. Reverted exhaustion phenotype of circulating lymphocytes as immune correlate of anti-PD1 first-line treatment in Hodgkin lymphoma. Leukemia. 2022;36(3):760-771.
9. Cader FZ, Hu X, Goh WL, et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat Med. 2020;26(9):1468-1479.
10. Jalali S, Price-Troska T, Bothun C, et al. Reverse signaling via PD-L1 supports malignant cell growth and survival in classical Hodgkin lymphoma. Blood Cancer J. 2019;9(3):22.
11. Vari F, Arpon D, Keane C, et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood. 2018;131(16):1809-1819.
Haematologica | 108 - April 2023 946 EDITORIAL M.K. Gandhi and C. Keane
The effects of chronic glucorticoid stimulation on erythropoiesis in Cushing syndrome
John Strouboulis and Sara El Hoss Red Cell Haematology, Comprehensive Cancer Centre, School of Cancer and Pharmaceutical Sciences, King’s College London, London, UK
Correspondence: J. Strouboulis
john.strouboulis@kcl.ac.uk
Received: June 30, 2022.
Accepted: July 12, 2022.
Early view: July 21, 2022.
https://doi.org/10.3324/haematol.2022.281355
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Varricchio et al. report on their use of Cushing syndrome to study stress erythropoiesis under conditions of chronic exposure to glucocorticoids.1 By studying patients with active Cushing syndrome and in remission, they describe a distinct population of glucocorticoid-responsive hematopoietic progenitors and provide intriguing new insights into the molecular basis of the loss of responsiveness to glucocorticoids in the treatment of anemias.
Glucocorticoids and stem cell factor (SCF) play key roles in stress erythropoiesis.2 Glucocorticoids bind to the glucocorticoid receptor (GR α in erythroid cells), which translocates to the nucleus to activate stress response genes. However, it is not clear precisely how the glucocorticoid/GR α pathway regulates stress erythropoiesis. This is of clinical significance as glucocorticoids are used to treat hyperproliferative anemias such as DiamondBlackfan anemia.3 Glucocorticoid therapy increases red cell mass, thereby alleviating anemia; however, patients become refractory for reasons that are not fully understood. Current models for studying glucocorticoids in erythropoiesis include animal models and ex vivo erythroid differentiation of human CD34 + hematopoietic stem and progenitor cells (HSPC), although these have limitations.4 These include species-specific differences in how murine and human proerythroblasts respond to glucocorticoids 5 and the near-ubiquitous use of dexamethasone and SCF in expanding proerythroblasts in vitro, giving rise to confounding effects.4 Endocrine disorders offer opportunities to study the effects of glucocorticoids on in vivo erythropoiesis under conditions of constitutively active (Cushing syndrome) or altogether absent (Addison disease) glucocorticoid/GR α activation.6 Hence it is surprising that only one publication has previously reported erythrocytosis (an increase in red blood cell mass) in one Cushing patient with an adrenocorticotropic hormone-secreting pituitary adenoma.7 This is redressed in the study by Varricchio et al., who recruited a relatively large (n=13) cohort of Cushing syndrome patients with active hypercortisolemia (active-phase pa-
tients) and an equal number of eucortisolemic patients in remission following surgical removal of the pituitary adenoma (remission-phase patients). Characterization of erythropoiesis showed that active-phase patients had erythrocytosis with normal HbF levels, the latter suggesting that chronic stress conditions do not induce fetal globin expression. Interestingly, CD14+ monocytes in active-phase patients had a distinct phenotype skewed towards a greater proportion of cells expressing CD163, presumably as a result of constitutive GR activation. 8 This was also maintained in remission-phase patients, suggesting a cellular memory of monocyte glucocorticoid activation after remission.
Varricchio et al. next assessed the immunophenotypic profile of circulating CD34+ cells in active- and remissionphase patients (and healthy controls) using a panel of antibodies that: (i) define a stress-progenitor cell population (CD110+ and CD36+); (ii) detect proteins that regulate (CALR) or respond to (CXCR4) GR activation; and (iii) monitor the response to SCF and interleukin-3 (IL-3) (CD117 and CD123, respectively) as growth factors used in culture to stimulate erythroid cells. CD133 (prominin), expressed in hematopoietic stem cells, was also included. This analysis showed that CD34+ HSPC from patients with active Cushing syndrome had a unique profile characterized by a higher proportion of cells expressing CD36, CD110, CXCR4 and CD133 and a lower proportion expressing CD117 and CD123, compared to cells from healthy controls. These observations are consistent with a stress-like phenotype, activated GR signaling and a greater responsiveness to SCF and IL-3, the receptors of which are down-modulated in response to stimulation.9 By contrast, CD34+ HSPC in remission-phase patients displayed a greater proportion of CXCR4-expressing cells, but no difference in the fraction of cells expressing stress-like features. These observations suggest that GR activation is sustained even after remission in Cushing disease patients who achieve this state. As expected, expansion of immature erythroid cells from the HSPC of active-phase patients was similar independently of the absence or
Haematologica | 108 - April 2023 947 EDITORIAL J. Strouboulis and S. El Hoss
presence of dexamethasone in culture. Interestingly, a similar effect was also seen with HSPC from remissionphase patients. Thus, the immunophenotypic profiles and erythroid expansion characteristics of CD34+ HSPC from remission-phase patients are consistent with GR signaling retaining some activity following surgical removal of the pituitary adenoma.
GR activation was also investigated at a molecular level by assessing the profile of GRα protein in erythroid cells from active- and remission-phase patients and healthy controls, differentiated ex vivo with and without dexamethasone. GILZ was analyzed as a gene target activated by GRα Using antibodies that detect total GRα protein, or the differentially phosphorylated GRα fractions that were marked either for cytoplasmic retention or for translocation to the nucleus, it was shown that, whereas total GRα levels were equivalent in cells from active-phase and remission-phase patients and healthy controls stimulated with dexamethasone, cytoplasmic GRα was lower in cells from patients with active-phase Cushing disease. In addition, GILZ levels were higher regardless of the presence of dexamethasone, indicating constitutive GRα activation in active-phase cells that does not respond further to glucocorticoid stimulation. Interestingly, cytoplasmic GR levels were higher in remission-phase patients than in active-phase patients, yet patients in remission are also insensitive to de-novo glucocorticoid stimulation. Taken together, these observations suggest that: (i) the lack of response to glucocorticoid treatment as a result of
References
1. Varricchio L GE, Martelli F, et al. Patients with hypercortisolemic Cushing disease possess a distinct class of hematopoietic progenitor cells leading to erythrocytosis Haematologica. 2023;108(4):1053-1067.
2. Varricchio L, Tirelli V, Masselli E, et al. The expression of the glucocorticoid receptor in human erythroblasts is uniquely regulated by KIT ligand: implications for stress erythropoiesis. Stem Cells Dev. 2012;21(15):2852-2865.
3. Da Costa L, Narla A, Mohandas N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Res. 2018;7:F1000 Faculty Rev-1350.
4. Migliaccio AR, Varricchio L. Concise review: advanced cell culture models for Diamond Blackfan anemia and other erythroid disorders. Stem Cells. 2018;36(2):172-179.
5. Ashley RJ, Yan H, Wang N, et al. Steroid resistance in Diamond Blackfan anemia associates with p57Kip2 dysregulation in erythroid progenitors. J Clin Invest. 2020;130(4):2097-2110.
chronic glucocorticoid exposure is most likely due to deregulation of the nuclear/cytoplasmic transport of GR α , rather than changes in GRα protein levels; and (ii) when in remission, the lack of response to dexamethasone, the higher levels of GRα retained in the cytoplasm and the increased fraction of CD163+ monocytes in the circulation may indicate a cellular memory mechanism that reflects the previous hypercorticosolemic state.
Overall, the study by Varricchio et al. adds to our understanding of glucocorticoid stimulation in erythropoiesis by refining our view of stress-like HSPC in conditions of chronic glucocorticoid exposure and in relation to previously characterized dexamethasone-responsive stress progenitors.5 This study also suggests an intriguing explanation for patients becoming refractory to glucocorticoid therapy, in that prolonged exposure to glucocorticoids leads to GRα retention in the cytoplasm, potentially as a moderating response to constitutive glucocorticoid exposure. Importantly, as the authors point out, this can be tested using inhibitors of cytoplasmic GRα phosphorylation. Lastly, a detailed investigation of the molecular (epi)genetic basis of constitutive GRα activation in stress progenitors and of the potential cellular memory mechanism described here is of great interest, also in relation to pathways and molecules that have been shown to be deregulated in anemias or following glucocorticoid treatment.5,10
Disclosures
No conflicts of interest to disclose.
6. Harris C. Clinical perspective: what do Addison and Cushing tell us about glucocorticoid action? Adv Exp Med Biol. 2015;872:83-96.
7. Gursoy A, Dogruk Unal A, Ayturk S, et al. Polycythemia as the first manifestation of Cushing's disease. J Endocrinol Invest. 2006;29(8):742-744.
8. Tippett E, Cheng WJ, Westhorpe C, et al. Differential expression of CD163 on monocyte subsets in healthy and HIV-1 infected individuals. PLoS One. 2011;6(5):e19968.
9. Federici G, Varricchio L, Martelli F, et al. Phosphoproteomic landscaping identifies non-canonical cKIT signaling in polycythemia vera erythroid progenitors. Front Oncol. 2019;9:1245.
10. Iskander D, Wang G, Heuston EF, et al. Single-cell profiling of human bone marrow progenitors reveals mechanisms of failing erythropoiesis in Diamond-Blackfan anemia. Sci Transl Med. 2021;13(610):eabf0113.
Haematologica | 108 - April 2023 948 EDITORIAL J. Strouboulis and S. El Hoss
Low-intensity induction in acute myeloid leukemia. Always in the patients' best interest?
Ehab Atallah Division of Hematology & Oncology, Medical College of Wisconsin, Milwaukee, WI, USA
Correspondence: E. Atallah
eatallah@mcw.edu
Received: July 6, 2022.
Accepted: July 12, 2022.
Early view: July 21, 2022.
https://doi.org/10.3324/haematol.2022.281506
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The amount of time patients with acute leukemia spend in a healthcare setting is an aspect that is frequently ignored. There are several ways to measure patients’ healthcare engagement. These range from simply calculating the percentage of days patients interact with healthcare, to calculating the percentage of hours spent in a healthcare setting to even adding commuting time. When evaluating low-intensity therapies for acute myeloid leukemia (AML), emphasis is usually placed on inpatient versus outpatient management of patients. In this issue of Haematologica, Jensen and colleagues address the issue of the amount of time patients with AML treated with low-intensity therapy spend in a healthcare setting.1
The investigators calculated the proportion of days spent at home (PDH) for patients with AML treated with azacitidine ± venetoclax across the University of North Carolina Health System using electronic health records. PDH was calculated as the number of days patients were
not engaged in cancer care divided by the total number of days. Days engaged in cancer care included admission to a hospital for any reason, emergency department visits for any reason, and oncology/infusion clinic appointments. The study included 113 patients with 44 and 54 patients receiving azacitidine and azacitidine + venetoclax, respectively. The overall median PDH was 0.63 with an increase in PDH over time. The PDH was 51%, 64% and 77% for patients surviving at month 1, 3 and 6. To put this in perspective, the PDH is 97% for patients with stage I-III breast cancer2 and 88% for patients with stage I lung cancer3 (Figure 1). Jensen et al. found no difference between patients who did or did not receive venetoclax, between patients who did or did not achieve remission or between those who had good versus poor prognostic factors. However, these comparisons may not be accurate due to the small sample size and different characteristics of the patients. In addition they are less clinically relevant given that azacitidine + venetoclax is now the standard of
Figure 1. Percentage of days at home for patients with solid cancers or acute myeloid leukemia during various stages of low- or high-intensity treatment. The percentage for breast cancer patients is the median over 18 months. The percentage for lung cancer patients is the median in the first 60 days. The percentage for intensive induction is an estimate.
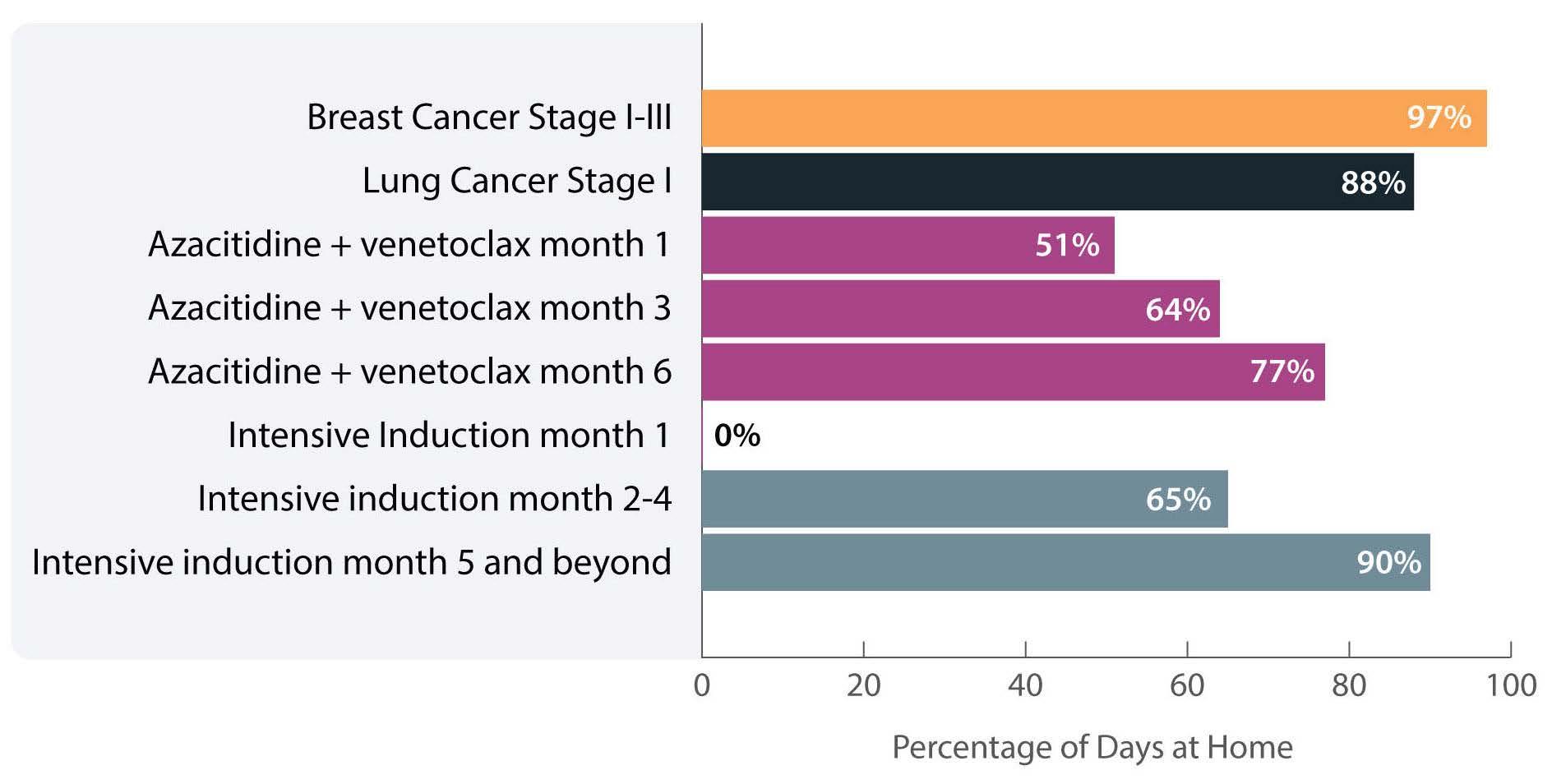
Haematologica | 108 - April 2023 949 EDITORIAL E. Atallah
care for all patients with AML. In fact, the study provides reassurance that adding venetoclax to the treatment regimen does not decrease PDH.
Inpatient hospital stay increased with increasing distance to the hospital. This is not an unexpected finding as we are more likely to admit patients who live farther away and do not have adequate supportive care close to their homes.
The more important comparison which is not reported here is the difference between high-intensity (e.g., 7+3) and low-intensity (e.g., azacitidine + venetoclax) therapy. In most institutions,4 all patients receiving intensive induction chemotherapy are admitted to the hospital for approximately 4 weeks. This is followed by three cycles of consolidation chemotherapy with supportive care (approximately 3-5 days of chemotherapy, with visits for laboratory checks and possible transfusions 3 times per week). If patients proceed to oral maintenance therapy after that the frequency of laboratory checks and office visits would range from 2-4/month. Based on this scenario, the estimated PDH for patients receiving intensive induction chemotherapy not proceeding to allogeneic hematopoietic stem cell transplantation is 0%, 65% and 90% for month 1, months 2-4 and month 5 thereafter, respectively (Figure 1).
So, azacitidine + venetoclax is clearly a step in the right direction to increase PDH, especially during induction. Following induction and for patients who are in remission we need to think of ways to decrease the amount of time patients spend in the healthcare setting. Patients with
References
1. Jensen CE, Heiling HM, Beke MH, et al. Time spent at home among older adults with acute myeloid leukemia receiving azacitidine- or venetoclax-based regimens. Haematologica. 2023;108(4):1006-1014.
2. Cheng AC, Levy MA. Data driven approach to burden of treatment measurement: a study of patients with breast cancer. AMIA Annu Symp Proc. 2017;2016:1756-1763.
3. Presley CJ, Soulos PR, Tinetti M, Montori VM, Yu JB, Gross CP. Treatment burden of Medicare beneficiaries with stage I nonsmall-cell lung cancer. J Oncol Pract. 2017;13(2):e98-e107.
4. Halpern AB, Walter RB, Estey EH. Outpatient induction and consolidation care strategies in acute myeloid leukemia. Curr Opin Hematol. 2019;26(2):65-70.
AML engage in cancer care for multiple reasons: inpatient hospital stays, outpatient treatment, outpatient clinic visits, and outpatient supportive care appointments
To reduce inpatient hospital stays, intensive induction chemotherapy can be administered in the outpatient setting. This approach is feasible in a select group of patients and in select institutions that can provide intensive outpatient monitoring and support. In such a setting, less than half of patients are eligible for outpatient intensive therapy. In addition, patients still spend a considerable number of days evaluated in a healthcare setting for laboratory checks and possible transfusions.5 For low-intensity therapy, home administration of chemotherapy, such as subcutaneous cytarabine or possibly subcutaneous azacitidine, would increase PDH. The development of oral substitutes would also increase PDH.
To reduce outpatient supportive care visits, home monitoring of laboratory parameters6 and home transfusions are possible. Logistically these are difficult as they require significant resources including home nursing, early and systematic sample collection and close outpatient monitoring.7 However, technology for remote visits, remote blood collection and vital sign monitoring8 is an important under-implemented tool that has the potential to significantly increase PDH, reduce costs and improve patients’ quality of life.
Disclosures
EA has acted as a consultant for Abbvie.
5. Halpern AB, Howard NP, Othus M, et al. Early hospital discharge after intensive induction chemotherapy for adults with acute myeloid leukemia or other high-grade myeloid neoplasm. Leukemia. 2020;34(2):635-639.
6. Kristian Kur D, Thøgersen D, Kjeldsen L, Friis-Hansen L. The HemoScreen hematology point-of-care device is suitable for rapid evaluation of acute leukemia patients. Int J Lab Hematol. 2021;43(1):52-60.
7. Benson K. Blood transfusions in the home sweet home: how to avoid a sour outcome. Cancer Control. 1997;4(4):364-367.
8. Nishikawa A, Fujimori Y, Sakano N, et al. Remote vital signs data monitoring during home blood transfusion: a pilot study. Health Sci Rep. 2021;4(3):e380.
Haematologica | 108 - April 2023 950 EDITORIAL E. Atallah
Getting (T cells) ENGaged
Susanne H.C. Baumeister
Department of Pediatric Oncology, Dana-Farber Cancer Institute; Boston Children’s Hospital, Division of Pediatric Hematology-Oncology and Harvard Medical School, Boston, MA, USA
Correspondence: S.H.C.
Baumeister
susanne_baumeister@dfci.harvard.edu
Received: June 16, 2022.
Accepted: July 21, 2022.
Early view: July 28, 2022.
https://doi.org/10.3324/haematol.2022.281229
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Vaidya and colleagues report on “Improving the anti-acute myeloid leukemia activity of CD123-specific engager T cells by MyD88 and CD40 co-stimulation”.1 This work focuses on engager T cells (ENG T cells), an interesting adoptive T-cell modality, aimed at combining the benefits of bispecific monoclonal antibodies (BsAb) to engage bystander T cells, regardless of their T-cell receptor (TCR)-specificity, with the longevity and trafficking capabilities of adoptively transferred T cells.
BsAb engaging both a tumor target and an immune effector cell have emerged as important therapeutic tools. This approach works by bridging T cells and target cells with bispecific monoclonal antibodies and prompts Tcell activation that is no longer major histocompatibility complex-restricted and independent of the specificity of the native TCR. BsAb can be generated in different formats and may include or lack an IgG backbone with a Fc domain. Configurations that do not include an Fc linker include bispecific T-cell engagers (BiTE), dual affinity retargeting (DART) and Diabodies which are differentiated by the type of linker and configuration of how the single chain variable fragment (scFv) recognizing the tumor target is linked to the scFv binding the T cell.2 Blinatumomab is an example of a BiTE that has shown tremendous clinical efficacy in engaging T cells to eliminate CD19+ B-lymphoblastic leukemia.3-5 Similarly, BsAb approaches are being developed for targets such as CD20 in CD20+ non-Hodgkin lymphoma and CD33 and CD123 in acute myeloid leukemia (AML).6
However, while these molecules can redirect resident T cells to target tumor targets, they have a short half-life (blinatumomab is being administered by continuous infusion) and do not self-amplify or promote ongoing Tcell engagement. In contrast, adoptive T-cell immunotherapies using antigen-specific T cells such as chimeric antigen-receptor (CAR) T cells mediate cytotoxic effects against tumor cells in a target-specific fashion, and can persist and mediate tumor control for years. However, although epitope-spreading has been described, they do not activate bystander T cells to mediate antigen-specific tumor-killing in the tumor microenvironment.
ENG T cells are a T-cell platform that secretes bispecific engagers, after having been transduced with a vector encoding for a BsAb consisting of two scFv, one specific for the tumor target and the other specific for CD3 e (Figure 1A, C). The production and delivery of BsAb by ENG T cells in vivo conceptually allows the continuous local delivery of BsAb at the tumor site without the need for a continuous infusion. The secreted BsAb engage both untransduced T cells in the microenvironment and the ENG T cells themselves and facilitate antigenspecific tumor killing. Additionally, ENG T cells can be engineered to provide co-delivery of co-stimulatory molecules or cytokines to improve ENG T-cell function and overcome immunosuppressive factors in the tumor microenvironment.
Several groups have reported preclinical activity with this approach utilizing different bispecific engagers in models of both hematologic malignancies and solid tumors.7-9 In the current study, the authors focus on CD123-specific ENG T cells, which are genetically modified T cells secreting a bispecific antibody (CD123-ENG) consisting of two scFv binding CD123 and CD3e to target AML. Previous studies documented the secretion of the bispecific engager protein (CD123 ENG) by CD123 ENG T cells and binding of the CD123 ENG protein to both CD123-ENG T cells and non-transduced bystander T cells. CD123 ENG T cells were able to kill CD123+ primary AML blasts in an antigen-specific manner, and redirected bystander T cells to induce antigen-specific AML target killing in transwell assays while demonstrating activity in AML xenograft models.7 However, a decrease in effector function of ENG T cells upon chronic antigen stimulation remains a limitation of this and other T-cell based therapies. To overcome this, Vaidya et al. explore the inclusion of a drug-inducible composite MyD88/CD40 activation receptor to provide co-stimulation, and confer improved expansion capability and antitumor activity, via downstream signaling pathways involving NF-kB and PI3K/AKT. The inducible MyD88/CD40 switch comprising a myristoylation-targeting sequence (to increase protein-protein interactions leading to subcellular localization of myristoylated proteins with their signaling partners), MyD88 (lacking its TIR sequence), the CD40 cytoplasmic domain and two tandem FKBP12v36
Haematologica | 108 - April 2023 951 EDITORIAL S.H.C. Baumeister
chemical inducer of dimerization (CID)-binding domains, which dimerize and activate the receptor upon administration of a dimerizing drug (i.e., a CID), has previously been described in the context of enhancing the function of dendritic cells10 and CAR T cells11,12 (Figure 1B).
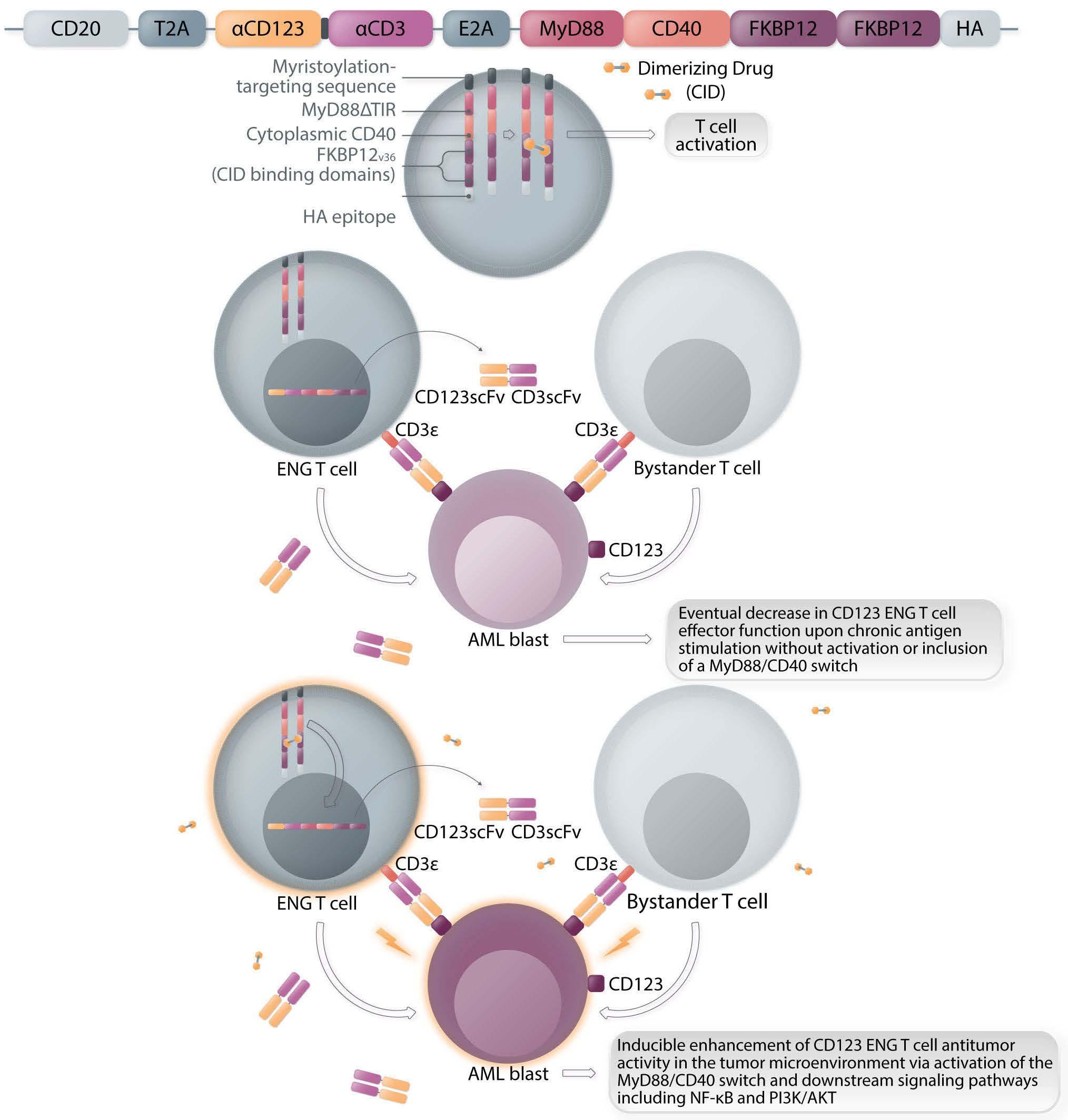
Here the authors report the effect of using an inducible co-stimulation system in CD123 ENG T cells (Figure 1C) and compare the effects of the inducible MyD88/CD40 molecule with inducible MyD88 and inducible CD40 alone. They demonstrate that upon activation of an inducible
encoding for the CD20 transduction marker, CD123 engager (CD123 ENG) T cells bispecific monoclonal antibody and MyD88/CD40 inducible switch. (B) Chemical inducer of dimerization (CID) activation of the inducible MyD88/CD40 co-stimulatory switch, containing a myristoylation-targeting sequence, MyD88 (lacking its TIR domain), the cytoplasmic CD40 domain and FKB12v36 CID-binding domains in tandem, leads to dimerization and activation of the inducible MyD88/CD40 switch activating downstream NF- k Btranscriptional activity. (C) Model of CD123 ENG T-cell action: transduced ENG T cells express the CD123 ENG bispecific diabody which binds to CD123 ENG T cells and bystander T cells, directing them to antigen-bearing AML cells. Upon antigen binding CD123 ENG and bystander T cells are activated and mediate antitumor activity. CD123 ENG T cells increase CD123 ENG diabody mRNA production. However, without activation or inclusion of a MyD88/CD40 switch, CD123 ENG T-cell function may decrease upon chronic antigen stimulation. (D) To overcome a decrease in effector function upon chronic antigen stimulation, CD123 ENG T cells can be engineered to express the MyD88/CD40 molecule which can be activated by administration of a CID as shown in (B). Inducible activation of the MyD88/CD40 switch results in superior CD123 ENG T-cell antitumor activity in the tumor microenvironment via downstream T-cell signaling pathways including NF- kB and PI3K/AKT.
Figure 1.
B C Haematologica | 108 - April 2023 952 EDITORIAL S.H.C. Baumeister A D
Features of the inducible co-stimulation system in CD123 engager T cells. (A) Transgene
MyD88/CD40 switch by a CID, CD123 ENG T cells maintained their antigen specificity, exhibited superior effector function under conditions of repeated stimulation and mediated enhanced anti-tumor activity in different AML xenograft models. Although the effect of the inducible MyD88/CD40 molecule on bystander T-cell
References
1. Vaidya A, Doherty E, Wu X, et al. Improving the anti-acute myeloid leukemia activity of CD123-specific engager T cells by MyD88 and CD40 co-stimulation. Haematologica. 2023;108(4):1039-1052.
2. Velasquez MP, Bonifant CL, Gottschalk S. Redirecting T cells to hematological malignancies with bispecific antibodies. Blood. 2018;131(1):30-38.
3. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
4. von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
5. Foa R, Bassan R, Vitale A, et al. Dasatinib-blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med. 2020;383(17):1613-1623.
6. Al-Hussaini M, Rettig MP, Ritchey JK, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood. 2016;127(1):122-131.
7. Bonifant CL, Szoor A, Torres D, et al. CD123-engager T cells as a
activation was not specifically evaluated, these data support further development of this promising engager Tcell platform.
Disclosures
No conflicts of interest to disclose.
novel immunotherapeutic for acute myeloid leukemia. Mol Ther. 2016;24(9):1615-1626.
8. Iwahori K, Kakarla S, Velasquez MP, et al. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther. 2015;23(1):171-178.
9. Liu X, Barrett DM, Jiang S, et al. Improved anti-leukemia activities of adoptively transferred T cells expressing bispecific T-cell engager in mice. Blood Cancer J. 2016;6(6):e430.
10. Narayanan P, Lapteva N, Seethammagari M, Levitt JM, Slawin KM, Spencer DM. A composite MyD88/CD40 switch synergistically activates mouse and human dendritic cells for enhanced antitumor efficacy. J Clin Invest. 2011;121(4):1524-1534.
11. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 2017;7(11):1306-1319.
12. Collinson-Pautz MR, Chang WC, Lu A, et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia. 2019;33(9):2195-2207.
Haematologica | 108 - April 2023 953 EDITORIAL S.H.C. Baumeister
Splenectomy in sickle cell disease: do benefits outweigh risks?
Amina Nardo-Marino1,2,3 and Valentine Brousse4,5
1Center for Hemoglobinopathies, Department of Hematology, Copenhagen University Hospital, Rigshospitalet, Copenhagen, Denmark; 2Department of Haematological Medicine, King's College Hospital, London, UK; 3Red Cell Haematology, Comprehensive Cancer Centre, School of Cancer and Pharmaceutical Sciences, King's College London, London, UK; 4Centre de Référence MCGRE, Service d'Hématologie-Immunologie, AP-HP, Hôpital Robert Debré, Paris, France and 5Université Paris Cité and Université des Antilles, Inserm, BIGR, France
The spleen is one of the first organs to be damaged in sickle cell disease (SCD). The unique environment of the spleen is particularly challenging for SCD erythrocytes, with constant intrasplenic sickling and trapping of erythrocytes in the red pulp causing repeated splenic injury from a very young age.1 This process is clinically silent in most children, manifesting mainly as progressive functional asplenia with a high risk of invasive bacterial infections,2-4 a complication that can be managed with daily prophylactic penicillin and appropriate immunizations. Nonetheless, some children with SCD experience other serious clinical complications related to the spleen, including acute splenic sequestration and chronic hypersplenism.1
Acute splenic sequestration is an unpredictable and lifethreatening complication particularly affecting young children with HbSS. Resulting from acute trapping of blood in the spleen caused by sudden enhanced intrasplenic sickling, symptoms include rapid splenic enlargement with acute anemia and potentially hypovolemia. Estimated rates of acute splenic sequestration vary according to age and sickle cell genotype. In HbSS, the first episode typically occurs between 6 months and 5 years of age, most within the first 2 years of life.5,6 After one episode of acute splenic sequestration, 67% of children with HbSS will experience a recurrent episode.5 In other SCD genotypes, such as HbSC disease and HbS/b+-thalassemia, acute splenic sequestration is more often reported in older children and young adults.7 Acute splenic sequestration is typically treated with blood transfusions and surgical splenectomy is often recommended for children with recurrent episodes.8,9 Another clinical indication for surgical splenectomy in SCD is hypersplenism. Persistent splenic enlargement is likely due to chronic intrasplenic erythrocyte sequestration and may be accompanied by low hemoglobin concentration, thrombocytopenia, and leukopenia, as well as abdominal pressure from the spleen and failure to thrive.
The benefit/risk ratio of splenectomy is particularly
Correspondence: A. Nardo-Marino amina.nielsen.nardo-marino@regionh.dk
Received: July 21, 2022.
Accepted: July 28, 2022.
Early view: August 4, 2022.
https://doi.org/10.3324/haematol.2022.281587
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
complex in SCD. On the one hand, surgical splenectomy provides an immediate clinical benefit in children with recurrent acute splenic sequestration or severe hypersplenism. On the other hand, splenectomy may be associated with both short- and long-term surgical, septic, and thromboembolic risks which are not fully understood nor evaluated. Additionally, most children with SCD become functionally asplenic at a very young age, adding to the complexity of a surgical risk evaluation as outcomes on morbidity and mortality related to the spleen are very difficult to interpret.
In a Letter to the Editor published in this issue of Haematologica, Pinto et al.10 compared survival and longterm complications in splenectomized and nonsplenectomized individuals with SCD in Italy. Presenting retrospective data from 534 individuals with SCD, 117 (32%) of whom had undergone surgical splenectomy, the study provides novel evidence on the absence of longterm increased mortality related to surgical splenectomy in SCD. With a median follow-up of 26 years, this study is the first to provide such reassuring long-term postsplenectomy follow-up data. However, some important study features must be considered when extrapolating results to other clinical settings, particularly if they are to be used as guidance for clinical decision-making.
The study included individuals with various genotypes reflecting the specific epidemiology of SCD in Italy: 32% had HbS/b+-thalassemia, 33% had HbS/b0-thalassemia, and only 35% had HbSS. The rate of surgical splenectomy varied significantly between these three genotypes, with as many as 53.4% of individuals with HbS/b0-thalassemia and 33.9% of individuals with HbS/b+-thalassemia having undergone surgical splenectomy, compared to just 9.6% of individuals with HbSS. Furthermore, the median age at splenectomy was 7 years in the HbSS group compared to 11 years in the HbS/b0-thalassemia group and 20 years in the HbS/b+-thalassemia group. Hypersplenism/recurrent splenic sequestration was the indication for splenectomy in 68.8% of patients, a percentage reflecting the high proportion of patients with HbS/b0-thalassemia and
Haematologica | 108 - April 2023 954 EDITORIAL A. Nardo-Marino and V. Brousse
HbS/b+-thalassemia. Findings may therefore not accurately capture the risks of surgical splenectomy when performed in young children with HbSS, which is likely the majority of children requiring surgical splenectomy in many settings.11 The study found no difference in the rate of fatal infectious complications between splenectomized and non-splenectomized individuals. This is not surprising for children with HbSS or HbS/b0-thalassemia who are known to become functionally asplenic in early childhood, thus rendering them highly susceptible to invasive bacterial infections regardless of whether they undergo surgical splenectomy or not.2-4 In contrast, splenic dysfunction in HbS/b+-thalassemia has not been widely evaluated, and for this group of patients the findings presented in this study are particularly valuable. A relationship that is not as well understood in SCD is the association between splenectomy (autosplenectomy or surgical splenectomy) and thromboembolic risk. This study did not demonstrate any specific risks of long-term thromboembolic complications.
There is a general paucity of research on the spleen in SCD.
References
1. Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol. 2014;166(2):165-176.
2. Pearson HA, Gallagher D, Chilcote R, et al. Developmental pattern of splenic dysfunction in sickle cell disorders. Pediatrics. 1985;76(3):392-397.
3. El Hoss S, Cochet S, Marin M, et al. Insights into determinants of spleen injury in sickle cell anemia. Blood Adv. 2019;3(15):2328-2336.
4. Nardo-Marino A, Braunstein TH, Petersen J, et al. Automating pitted red blood cell counts using deep neural network analysis: a new method for measuring splenic function in sickle cell anaemia. Front Physiol. 2022;13:859906.
5. Brousse V, Elie C, Benkerrou M, et al. Acute splenic sequestration crisis in sickle cell disease: cohort study of 190 paediatric patients. Br J Haematol. 2012;156(5):643-648.
6. Topley JM, Rogers DW, Stevens MC, Serjeant GR. Acute splenic sequestration and hypersplenism in the first five years in homozygous sickle cell disease. Arch Dis Child.
Although the study by Pinto et al.10 provides much needed novel evidence on the absence of long-term mortality related to surgical splenectomy, data on potential benefits of splenectomy are still sparse. Overall, international guidelines on the management of acute splenic sequestration and hypersplenism are lacking and recommendations for surgical splenectomy are based on weak evidence and pertain to all types of SCD despite genotypic variations in age and clinical presentation.8,9 Splenic complications in SCD are common and associated with high morbidity and mortality. Multicenter studies addressing parameters for and timing of surgical splenectomy, considering different genotypes, baseline splenic function and the variability in clinical settings, are necessary in order to provide adequate clinical guidelines and optimal care for all children and adults living with SCD.
Disclosures
No conflicts of interest to disclose.
Contributions
AN-M and VB co-wrote this editorial.
1981;56(10):765-769.
7. Aquino VM, Norvell JM, Buchanan GR. Acute splenic complications in children with sickle cell-hemoglobin C disease. J Pediatr. 1997;130(6):961-965.
8. Iolascon A, Andolfo I, Barcellini W, et al. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica. 2017;102(8):1304-1313.
9. Owusu-Ofori S, Remmington T. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database Syst Rev. 2017;11(11):CD003425.
10. Pinto VM, Gianesin B, Piel FB, et al. Morbidity and mortality of sickle cell disease patients is unaffected by splenectomy: evidence from three decades of follow-up in a high-income setting. Haematologica. 2023;108(4):1142-1146.
11. Wright JG, Hambleton IR, Thomas PW, Duncan ND, Venugopal S, Serjeant GR. Postsplenectomy course in homozygous sickle cell disease. J Pediatr. 1999;134(3):304-309.
Haematologica | 108 - April 2023 955 EDITORIAL A. Nardo-Marino and V. Brousse
Lessons learned from therapy-related acute myeloid leukemia
Sabine Kayser
NCT Trial Center, National Center of Tumor Diseases, German Cancer Research Center (DKFZ), Heidelberg and Institute of Transfusion Medicine and Immunology, Medical Faculty Mannheim, Heidelberg University, German Red Cross Blood Service Baden-Württemberg-Hessen, Mannheim, Germany
Correspondence: S. Kayser
s.kayser@dkfz-heidelberg.de
Received: August 10, 2022.
Accepted: August 19, 2022. Early view: August 25, 2022.
https://doi.org/10.3324/haematol.2022.281742
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Nilsson and colleagues present the results of a large retrospective analysis of realworld data regarding the incidence and prognostic implications of therapy-related acute myeloid leukemia (tAML, n=686) based on three Swedish nationwide population-based registries.1 This methodology allows an
unbiased view on prognostic implications and reliable observations of time trends in incidence. At the same time, this is one of the largest datasets investigating the clinical behavior of t-AML in comparison to de novo AML. The authors report that during the study period, ranging from 1997-2015, the incidence of t-AML almost doubled
1993-2008 Data not given
at 4 years:
at 4 years:
at 4 years after allo-HCT in 1st CR:
6779 (including 686 with t-AML)
70 (18-100) 1997-2015 47%**
in patients ≤ 60 years: t-AML: 22.9% de novo AML: 8.6%
t-AML: 6%* de novo AML: 10%*
at 3 years:
in patients ≤ 60 years: t-AML: 45.1% de novo AML: 46.3%
in patients > 60 years: t-AML: 85%* de novo AML: 75%*
at 3 years:
t-AML: 42.6% de novo AML: 58%
Kayser S, et al. Blood. 2011;117(7): 2137-2145 in patients > 60 years:
at 5 years:
in patients ≤ 60 years: t-AML: 49%** de novo AML: 28%**
in patients ≤ 60 years: t-AML: 21%** de novo AML: 22%**
t-AML: 48% de novo AML: 57% Nilsson C, et al. Haematologica. 2023;108(4): 1015-1025 in patients > 60 years: t-AML: 59%** de novo AML: 46%**
in patients > 60 years: t-AML: 28%** de novo AML: 32%**
*Estimated from cumulative incidence curves; **personal communication. allo-HCT: allogeneic hematopoietic cell transplantation; CIR: cumulative incidence of relapse; CR: complete remission; MAC: myeloablative conditioning; OS: overall survival; t-AML; therapy-related acute myeloid leukemia; t-MDS: therapy-related myelodysplastic syndrome; TRM: treatment-related mortality.
N of patients Age in years, median (range) Study period MAC TRM CIR OS Reference Allogeneic hematopoietic cell transplantation 868 (545 t-AML, 323 t-MDS) 40 (4-72) 1990-2004 77% 41% at 1 year 48% at 5 years 27% at 1 year 31% at 5 years 37% at 1 year 22% at 5 years Litzow MR, et al. Blood. 2010;115(9): 1850-1857 70 (39 t-AML, 31 t-MDS) 37 (16-55) 1980-1998 64% 49% at 2 years CIR not given 2-year relapse risk: 42% 30% at 2 years Yakoub-Agha I, et al. J Clin Oncol. 2000;18(5): 963-971 Intensively treated patients 2853 (200 t-AML, 2653 de novo AML) 54.5 (16.2-85)
Table 1. Outcomes of patients with therapy-related acute myeloid leukemia.
Haematologica | 108 - April 2023 956 EDITORIAL S. Kayser
with a yearly increase in t-AML of 4.5% (95% confidence interval: 2.8%-6.2%), most frequently due to t-AML after breast and prostate cancer. This is in part due to improvement of cancer treatments with decreased mortality during the same period, leading to better longterm survival after mutagenic cancer treatments.2,3 It also points to the increasing likelihood of encountering these patients in clinical practice. Thus, a better knowledge of, as well as better treatment approaches for, these patients is needed.
Secondly, the authors described the role of t-AML regarding prognosis within AML risk groups.4 Genetically, t-AML was underrepresented in patients with favorable risk, and overrepresented in patients with intermediate or adverse risk.5 Despite a good performance status (Eastern Cooperative Oncology Group score ≤2) patients with t-AML were less likely to receive intensive induction treatment (60% vs. 71%, P<0.001) and intensively treated patients were less likely to achieve complete remission (58% vs. 75%, P<0.001) as compared to those with de novo AML. The reason for the worse outcome of patients with intermediate- and poor-risk t-AML compared to their de novo counterparts, also when adjusting for other factors such as cytogenetics, age and performance status, is likely multifactorial. A higher frequency of unfavorable mutations and/or an enrichment of mutations originating from clonal hematopoiesis in patients with t-AML may contribute.6,7 Allogeneic hematopoietic cell transplantation (HCT), regardless of disease state, was performed in 9% of the patients with t-AML as compared to 16% in those with de novo AML (P<0.001). Corresponding rates of allogeneic HCT in first remission were 7% in t-AML and 12% in de novo AML (P=0.002), with no increase or decrease of transplantation rates over time. In multivariable analysis, t-AML was
References
1. Nilsson C, Linde F, Hulegardh E, et al. Characterization of therapy-related acute myeloid leukemia: increasing incidence and prognostic implications. Haematologica. 2023;108(4):1015-1025.
2. Arnold M, Rutherford MJ, Bardot A, et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995-2014 (ICBP SURVMARK-2): a population based study. Lancet Oncol. 2019;20(11):1493-1505.
3. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975-2014, featuring survival. J Natl Cancer Inst. 2017;109(9):djx030.
4. Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453-474.
5. Kayser S, Dohner K, Krauter J, et al. The impact of therapyrelated acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. 2011;117(7):2137-2145.
associated with poorer outcome in cytogenetically intermediate- and adverse-risk AML, but had no significant impact on outcome in favorable-risk AML, including core binding leukemias, acute promyelocytic leukemia and AML with mutated NPM1 without FLT3-ITD. This suggests that tAML patients with favorable-risk AML should be approached using the same treatment strategy as de novo favorable-risk patients and intensive chemotherapy including allogeneic HCT, if appropriate, should not be withheld from these patients. Biologically, this raises the question of whether t-AML with favorable risk (i.e., acute promyelocytic leukemia, NPM1 without FLT3-ITD and corebinding factor leukemias) are really therapy-related or more likely de novo AML. Comparative next-generation sequencing analysis may shed light on this issue.
Intermediate- and adverse-risk patients with t-AML have even poorer survival compared to their de novo counterparts. After allogeneic HCT, these patients suffer from high transplant-related mortality (Table 1), possibly reflecting cumulative toxicity of cancer treatment.5 However, the data should be interpreted with caution since relapses might be underreported, and thus the transplantrelated mortality might be overestimated. Nevertheless, novel treatment approaches for these patients are highly warranted.8 In addition, since intensifying the conditioning regimen was identified as a risk factor for worse outcome, the most appropriate conditioning regimen remains to be established.9
More recently, new therapeutic options, targeting FLT3, IDH1/2 and BCL2 have become available10 and may have the potential to improve outcome in certain subtypes of t-AML.
Disclosures
6. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367-1376.
7. Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552-555.
8. Lancet JE, Uy GL, Cortes JE, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36(26):2684-2692.
9. Yakoub-Agha I, de La Salmonière P, Ribaud P, et al. Allogeneic bone marrow transplantation for therapy-related myelodysplastic syndrome and acute myeloid leukemia: a long-term study of 70 patients-report of the French Society of Bone Marrow Transplantation. J Clin Oncol. 2000;18(5):963-971.
10. Kayser S, Levis MJ. The clinical impact of the molecular landscape of acute myeloid leukemia. Haematologica. 2023;108(4):956-957.
Haematologica | 108 - April 2023 957 EDITORIAL S. Kayser
No conflicts of interest to disclose.
BCMA loss in the epoch of novel immunotherapy for multiple myeloma: from biolog y to clinical practice
Xiang Zhou, Leo Rasche, K. Martin Kortüm, Julia Mersi and Hermann Einsele
Department of Internal Medicine II, University Hospital of Würzburg, Würzburg, Germany
Abstract
Correspondence: H.
Einsele einsele_h@ukw.de
Received: June 22, 2022.
Accepted: October 11, 2022.

Early view: October 20, 2022.
https://doi.org/10.3324/haematol.2020.266841
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The treatment of multiple myeloma (MM) is evolving rapidly. In the past few years, chimeric antigen receptor modified T cells and bispecific antibodies are bringing new treatment options to patients with relapsed/refractory MM. Currently, Bcell maturation antigen (BCMA) has emerged as the most commonly used target of T-cell-based immunotherapies for relapsed/refractory MM. Clinical data have demonstrated promising efficacy and manageable safety profiles of both chimeric antigen receptor T-cell and bispecific antibody therapies in heavily pretreated relapsed/refractory MM. However, most patients suffer from relapses at later time points, and the mechanism of resistance remains largely unknown. Theoretically, loss of antigen is a potential tumor-intrinsic resistance mechanism against BCMA-targeted immunotherapies. Strategies to overcome this kind of drug resistance are, therefore, needed. In this review, we discuss the loss of BCMA in the new epoch of immunotherapy for MM.
Introduction
Multiple myeloma (MM), the second most common hematologic malignancy, is characterized by uncontrolled plasma cell proliferation, which typically causes destructive osseous bone lesions, acute kidney injury, anemia, and hypercalcemia.1,2 In the past 20 years, integration of proteasome inhibitors and immunomodulatory drugs into the treatment of MM has significantly improved the survival outcomes of patients.3 Although MM is currently considered a largely incurable disease, the evolution of MM therapy is ongoing.4 In the mid-2010s, monoclonal antibodies targeting CD38 and signaling lymphocytic activation molecule F7 (SLAMF7), i.e. daratumumab and elotuzumab, were incorporated into the standard of care, bringing MM treatment into a new era of immunotherapy.5 Unlike conventional chemotherapies, these novel agents should recognize specific surface antigens in order to locate MM cells and, in turn, kill them selectively. In principle, the presence of a target antigen is an essential prerequisite for successful treatment.
The next revolution of immunotherapy for MM started recently with B-cell maturation antigen (BCMA)-directed treatments, including antibody-drug conjugates (ADC), bispecific antibodies (BsAb), and chimeric antigen recep-
tor (CAR) modified T-cell therapies.6 Although these novel immunotherapies are highly effective even in heavily pretreated relapsed/refractory (RR) MM patients, most patients suffer from relapses at later time points. A recent meta-analysis showed a median progression-free survival of merely 12.2 months in RRMM patients who were treated with BCMA-targeted CAR T cells.7 However, the underlying mechanism of resistance is currently not fully understood. To date, novel immunotherapies such as ADC, BsAb, and CAR T cells targeting other antigens have also been used in diverse hematologic malignancies including leukemia and lymphoma.8-10 Antigen loss has already been described as a tumor-intrinsic mechanism of resistance against BsAb and CAR T-cell therapies for leukemia and lymphoma. For instance, CD19 loss was detected in approximately 40% of patients with B-cell acute lymphoblastic leukemia treated with anti-CD19 CAR T cells, and point mutations in the CD19 gene were reported as a mechanism for CD19 loss in these patients.11 Likewise, in B-cell non-Hodgkin lymphoma, CD20-negative relapses were observed in patients who received REGN1979, a CD20/CD3-targeted BsAb.12,13 On the other hand, antigen loss following ADC treatments has been reported less frequently. Theoretically, antigen loss may also be a potential mechanism of resistance to anti-BCMA immunotherapies
Haematologica | 108 - April 2023 958 REVIEW ARTICLE
for MM. Indeed, in MM patients, biallelic BCMA loss has been reported in three cases relapsing from BCMA-targeted CAR T-cell therapies.14-16 In this review, we summarize the nature of BCMA loss based on the currently available data. Furthermore, strategies to overcome drug resistance caused by BCMA loss are discussed.
Biology of BCMA and anti-BCMA immunotherapies for multiple myeloma
The biology of BCMA as well as clinical data on BCMA-directed novel immunotherapies for MM have been summarized in previous review articles.17-20 Since the current review does not focus on these issues, at this point, we provide just a brief overview for completeness of the subject.
Biology of BCMA
BCMA, also referred to as tumor necrosis factor receptor superfamily 17 (TNFRSF17) or CD269, is a transmembrane glycoprotein highly expressed in plasma cells and almost absent in other human tissues. BCMA can be cleaved from the cell membrane by γ-secretase, releasing soluble BCMA (sBCMA) into the blood stream.21 The gene encoding BCMA is located on human chromosome band 16p13.1.22 In normal plasma cells, BCMA binds to B-cell activating factor (BAFF) and a proliferation inducing ligand (APRIL), regulating the maturation and differentiation of B cells into plasma cells and supporting survival of long-lived plasma cells.23-26 In MM, several survival and anti-apoptotic pathways could be activated by binding of BAFF or APRIL to BCMA, e.g. nuclear factor k light chain enhancer of activated B cells (NF-kB), mitogen activated protein kinase (MAPK), and protein kinase B (AKT), resulting in MM cell proliferation and immunosuppression in the bone marrow microenvironment.27 Importantly, the level of expression of BCMA is increased significantly on malignant cells compared to the level on healthy plasma cells.26,28 Based on these biological features of BCMA, it is considered a target of therapy for MM. To date, three classes of BCMA-directed immunotherapies have been investigated in humans, including ADC, BsAb, and CAR T cells (Figure 1A). As BCMA acts as an important factor contributing to survival of malignant plasma cells, loss of BCMA could be expected to place plasma cells at a selective growth disadvantage.
Recent advances in anti-BCMA immunotherapies for multiple myeloma
In August 2020, the US Food and Drug Administration and the European Medicines Agency approved the first BCMAtargeted ADC, belantamab mafodotin, for patients with RRMM.29 A few months later, the first anti-BCMA CAR Tcell therapy idecabtagene vicleucel (also referred to as
ide-cel or bb2121) was approved for RRMM patients who have received four or more prior lines of therapy, including an immunomodulatory drug, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.30 Most recently, the second anti-BCMA CAR T-cell therapy, ciltacabtagene autoleucel (cilta-cel), has been granted Food and Drug Administration approval for the same indication as ide-cel.31 Besides, multiple BCMA-directed T-cell engaging BsAb are under clinical investigation, and the early results have shown encouraging anti-MM efficacy.32,33 Based on the outstanding anti-tumor effect and acceptable toxicity, these novel anti-BCMA immunotherapies may become a part of standard MM treatment in the near future.
BCMA-targeted
antibody drug conjugates
An ADC is composed of a monoclonal antibody and a cytotoxic payload combined by a linker molecule. Belantamab mafodotin is the first-in-class ADC for MM patients. The pivotal randomized phase II DREAMM-2 trial demonstrated an overall response rate (ORR) of 31% and 34% in heavily pretreated RRMM patients receiving 2.5 mg/kg and 3.4 mg/kg of the drug every 3 weeks, respectively.34 Keratopathy is the most common toxicity of belantamab mafodotin with an incidence of up to 100%, which may lead to treatment discontinuation.35 Currently, belantamab mafodotin in combination with VRd (bortezomib, lenalidomide, and dexamethasone) is being evaluated in transplant-ineligible newly diagnosed MM.36 Other anti-BCMA ADC, e.g. AMG224 and MEDI2228, are under clinical investigation.37,38
BCMA-targeted bispecific antibodies
BsAb bind to MM and T cells via CD3 and a tumor-specific antigen, e.g. BCMA, to build an immune synapse, which subsequently leads to T-cell activation and cytotoxic effects. The first-in-class BCMA-directed BsAb AMG420 showed an ORR of 70% at a dose of 400 mg/day.33 However, further development of AMG420 has been stopped because of the product’s short half-life and the need for continuous infusion.17 For this reason, some other BsAb with extended half-lives have been developed, e.g. AMG701, teclistamab, REGN5458, TNB-383B, elranatamab, and CC-93269. Preliminary efficacy data for these novel agents demonstrated ORR of up to 90%, while some of the results were still immature.39-44 The most common adverse event of BsAb is cytokine release syndrome, which occurs with an incidence of up to 90%.45 Clinical trials evaluating BsAb in MM are ongoing.46
BCMA-targeted chimeric antigen receptor T cells
CAR T cells are another strategy to overcome tumor by utilizing the patient's own T cells. Genetically modified T cells with a CAR recognize tumor-specific antigens, e.g. BCMA, and activate T cells via the CD3ζ signaling domain. Additionally, some co-stimulatory domains such as CD28
Haematologica | 108 - April 2023 959 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
and 4-1BB are incorporated to enhance T-cell activation and proliferation. So far, there are more than 20 different BCMA-directed CAR T-cell products investigated within clinical trials, mainly in the USA and in China, producing an ORR of up to 100% in some studies.7,47-50 In the phase II KarMMa trial, the first-in-class BCMA-targeted CAR T-cell therapy ide-cel led to an ORR of 73%, and the median progression-free survival was only 8.8 months.51 Notably, the updated results of the phase Ib/II CARTITUDE-1 study
showed that cilta-cel not only produces a high ORR of 98% but also has encouraging long-term efficacy with a median duration of response of 21.8 months. Similarly to BsAb, BCMA-directed CAR T-cell therapy was associated with a very high rate of cytokine release syndrome of >80%, although this correlated with a good treatment response.20 At present, various clinical trials evaluating BCMA-targeted CAR T-cell products, including allogeneic CAR T cells, are enrolling patients.47
anti-BCMA chimeric antigen receptor modified (CAR) T cells, bispecific antibodies, and antibody-drug conjugates are available for the treatment of relapsed/refractory multiple myeloma (MM). γ-secretase can shed BCMA from the membrane of MM cells and can subsequently release soluble BCMA into the blood stream. An increase of soluble BCMA level can lead to a decline of BCMA-binding capacity on MM cells. BCMA could be transferred to CAR T cells via trogocytosis, resulting in reversible partial BCMA loss. (B) Irreversible complete BCMA loss. In patients with irreversible complete BCMA loss, which is caused by homozygous BCMA gene deletion, other immunotargets could be considered for further treatments, e.g. CD38, FcRH5, GPRC5D, CD19, and SLAMF7. Multi-specific immunotherapies targeting more than one antigen seem to be a promising strategy to prevent drug resistance due to the loss of a single antigen. (C) Reversible partial BCMA loss. γ-secretase inhibition is one option to increase BCMA density on MM cells. When the BCMA expression level recovers at later time-points, anti-BCMA retreatment could be considered. ADC: antibody drug conjugate; BCMA: B-cell maturation antigen; BsAb; bispecific antibody; CAR T-cell; chimeric antigen receptor modified T cell; FcRH5: Fc receptor-homolog 5; GPRC5D; G protein coupled receptor class C group 5 member D; MM: multiple myeloma; RR: relapsed/refractory; sBCMA; soluble BCMA; SLAMF7: signaling lymphocytic activation molecule F7.
Haematologica | 108 - April 2023 960
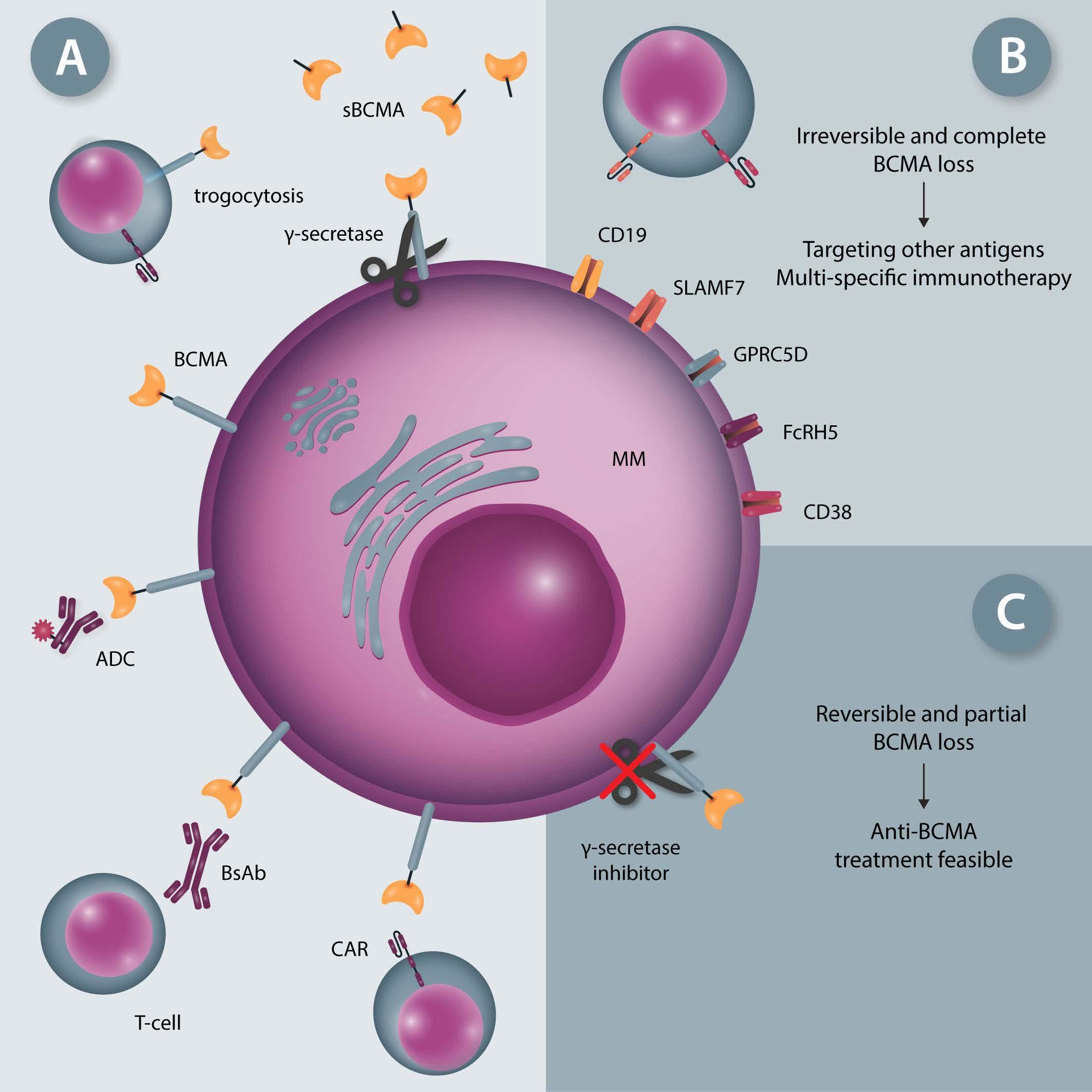 Figure 1. BCMA loss following targeted immunotherapies. (A) BCMA-directed immunotherapies. At present,
Figure 1. BCMA loss following targeted immunotherapies. (A) BCMA-directed immunotherapies. At present,
REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
BCMA loss: biology and clinical implication
The novel anti-BCMA immunotherapies, especially BsAb and CAR T cells, are highly effective in RRMM. However, the currently available data have demonstrated that the majority of patients relapse at later time-points. As these novel agents are being integrated into standard care, elucidating the mechanisms of resistance would be the next step in the development of these drugs to improve their anti-MM efficacy and to plan a precise treatment strategy for each given patient. Currently, as these novel anti-BCMA agents are still in their “infancy”, information on resistance mechanisms is still very limited. However, BCMA loss represents a potential tumor-intrinsic factor contributing to resistance against anti-BCMA immunotherapies. Here, we discuss the biology of BCMA loss and its clinical implications.
BCMA loss is not a common event
In general, the currently available data on anti-BCMA immunotherapies, mainly BsAb and CAR T cells, have demonstrated that BCMA loss after treatment is not a common event, with BCMA expression remaining positive in the majority of patients.22 BCMA loss was mostly detected when patients suffered an unexpected relapse after immunotherapies. In Table 1 we provide an overview of clinical cases with BCMA loss after immunotherapies in RRMM. The first case of BCMA loss was observed in a patient treated with BCMA-targeted CAR T cells. Ali et al. reported a patient who relapsed 2 months after CAR-BCMA treatment; flow cytometry of the patient’s bone marrow showed a population of BCMA-negative malignant plasma cells, whereas some other MM cells remained positive for BCMA.52 Similarly, Brudno et al. found a small number of MM cells that lacked BCMA expression, as determined by flow cytometry, in a patient 56 weeks after CAR-BCMA treatment. However, when resampling 8 weeks later, the MM cells of this patient presented mixed BCMA expression, suggesting a reversible BCMA loss.53 In addition, Green et al. described a patient with BCMA-negative MM cells 60 days after anti-BCMA CAR T-cell therapy. Although some BCMA-positive MM cells still existed in this patient, the BCMA expression level and the BCMA antigen-binding capacity were strongly reduced.54 Decreased BCMA expression levels were also observed by Cohen et al. in 12 out of 18 patients who received CAR-BCMA, including eight of nine responders and four of nine non-responders, while the BCMA expression “recovered” in later follow-up of these patients.55 Furthermore, BCMA loss was found in three of 71 patients (4%) at progression in the KarMMa study investigating ide-cel.51 With regard to BsAb treatment, Truger et al. reported a case of homozygous BCMA gene deletion after AMG420 treatment; this is the only pa-
tient with BCMA loss following BsAb therapies.56 BCMA loss after ADC has not yet been reported. Collectively, findings based on flow cytometry of bone marrow demonstrated that complete or partial BCMA loss could occur in a small proportion of RRMM patients following antiBCMA CAR T-cell therapy. However, at present, there is still limited experience with antigen loss following BCMA-targeted immunotherapies in MM patients, and determination of the level of BCMA expression is not part of routine tests at relapse. Therefore, the de facto incidence of BCMA loss is largely unknown. Theoretically, BCMA loss could appear at any time in the course of the disease.
Potential mechanisms of BCMA loss
Although BCMA loss has been reported in several studies, to date, the underlying mechanisms of this event are not fully understood. In this section, we summarize the potential mechanisms contributing to BCMA loss based on the currently available data.
With the rapid evolution of genomic diagnostics, such as whole-genome sequencing and single-cell RNA sequencing, the underlying biological mechanisms of BCMA loss after anti-BCMA immunotherapies began to be elucidated in the past few years. Da Vià et al. reported for the first time that homozygous BCMA gene deletion led to a complete and irreversible loss of BCMA expression in a RRMM patient who relapsed after ide-cel treatment. Furthermore, heterozygous BCMA gene deletion was present in 22% (37 out of 168) of MM patients (including a set of hyperhaploid MM cases) who had not received any antiBCMA therapy, indicating a higher risk of homozygous BCMA gene alteration after anti-BCMA immunotherapies.15 Likewise, Samur et al. and Leblay et al. found a similar biallelic BCMA loss (mutation + deletion or deletion + deletion) as a resistance mechanism in other RRMM patients treated with anti-BCMA CAR T cells.14,16 A homozygous BCMA gene deletion was also confirmed in a RRMM patient who received AMG420 BsAb therapy.56 The findings of these studies led to the hypothesis that the strong selection pressure exerted by these highly effective T-cellbased anti-BCMA immunotherapies might lead to selective expansion of a pre-existing minor population of BCMA-negative MM cells that could also appear in focal lesions and/or extramedullary manifestations due to spatial tumor heterogeneity. As a result of this, we observed permanent genetic and/or genomic changes after BCMAtargeted T-cell immunotherapies in these patients (Table 1). On the other hand, as anti-BCMA ADC are less effective than CAR T-cell or BsAb therapies in MM,17 the selection pressure of ADC should also be lower. Thus, the incidence of BCMA loss in patients treated with ADC might be lower than that in patients treated with CAR T cells or BsAb. Interestingly, most of the patients with BCMA gene deletion also had TP53 gene deletion, and TP53 mutations were
Haematologica | 108 - April 2023 961 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
Presence of BCMAnegative MM cells in one patient. On MM cells retaining BCMA
sion: 70% reduction of BCMA expression and 5-fold reduction in BCMA antigen binding capacity in this patient
Reduction of BCMA expression intensity in 67% (n=12) of the patients, including 8 of 9 responders and 4 of 9
Complete BCMA loss caused by homozygous BCMA gene deletion in one patient
Homozygous BCMA gene
Authors Year of publication Type of immunotherapy Product Clinical trial identi fi er Time point of BCMA loss Methods Frequency of BCMA loss in the study# Biological mechanism of BCMA loss Major fi ndings Reference Ali et al. 2016 CAR T-cell CAR-BCMA NCT02215967 2 months after treatment Flow cytometry 1/12 NR Partial loss of BCMA expression in MM cells at progression in one patient 52 Brudno et al. 2018 CAR T-cell CAR-BCMA NCT02215967 56 weeks after treatment Flow cytometry 1/16 NR Mixed BCMA ex
in one patient, with some MM cells negative for BCMA 53 Green et al. 2018 CAR T-cell NR NR 60 days after treatment Flow cytometry 1/7 NR
pression
54 Cohen et al. 2019 CAR T-cell CART-BCMA NCT02546167 1 month after treatment Flow cytometry 12/18 NR
expres
55 Truger et al. 2021 BsAb AMG420 NCT02514239 6 months after treatment IHC, WGS and RNA- seq Case report
56 Da Vià et al. 2021 CAR T-cell Idecabtagene- vicleucel NCT03361748 5 months after treatment IHC, WGS
Case report
15 Table 1. Selected
BCMA
targeted immunotherapies for relapsed/refractory
myeloma. Haematologica | 108 - April 2023 962 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al. Continued on following page.
non-responders
deletion
and RNA- seq
Homozygous BCMA gene deletion Complete BCMA loss caused by homozygous BCMA gene deletion in one patient
published cases on
loss following
multiple
BCMA loss caused by homozygous BCMA gene deletion in one patient
Loss of tumor BCMA expression was suspected in 3 of 71 patients (4%) at progression
One (5%) patient relapsed with BCMA-negative MM cells
BCMA: B-cell maturation antigen; BsAb: bispeci fi c antibody; CAR T-cell: chimeric antigen receptor modi fi ed T-cell; IHC: immunohistochemistry; MM: multiple myeloma; NR: not reported; RNA-seq: single-cell RNA sequencing; WGS: whole-genome sequencing; #Among the patients with evaluable BCMA expression a t baseline and relapse.
Samur et al. 2021 CAR T-cell Idecabtagene- vicleucel NCT02658929 8 months after treatment IHC,
Case report BCMA
tation Biallelic
one patient 14 Leblay et al. 2020 CAR T-cell NR NR NR Cellular indexing of
mes and epitopes by sequencing Case report Homozygous
16 Munshi et al. 2021 CAR T-cell Idecabtagene- vicleucel NCT03361748 NR NR 3/71 NR
WGS and RNA-seq
gene deletion + mu -
BCMA loss (mutation + deletion) in
transcripto
BCMA gene deletion Complete
51 Wang et al. 2022 CAR T-cell China ChiCTR-OIC- 17011272 NR Flow cytometry 1/21 NR
76 Authors Year of publication Type of immunotherapy Product Clinical trial identi fi er Time point of BCMA loss Methods
study#
loss Major fi ndings Reference
Frequency of BCMA loss in the
Biological mechanism of BCMA
Haematologica | 108 - April 2023 963 X. Zhou et al. REVIEW ARTICLE - BCMA loss in immunotherapy
also more frequent in patients with both del16p and del17p than in those who had only del16p or del17p. However, it is still unknown whether the co-occurrence of BCMA and TP53 loss is only an accidental event or whether there is some correlation between the two. This observation has also raised the question of whether patients with BCMA gene deletion have more genomic changes than do those without BCMA gene alterations. Moreover, due to the so-called branching evolution and spatial genomic heterogeneity, BCMA expression might be heterogeneous in various focal lesions and/or at different time points in the disease course.57-60 These issues have yet to be addressed in future studies.
As previously mentioned, γ-secretase can shed BCMA from the MM cell surface and release sBCMA into the blood.21 This might explain the reversible partial loss of BCMA expression reported in previous studies.53,55 Indeed, a preclinical study using cell lines and mouse models demonstrated that inhibition of γ-secretase activity could upregulate BCMA density on plasma cells and therefore increase the efficacy of anti-BCMA CAR T-cell therapy in vivo. 61 Thus, targeting γ-secretase might be a strategy to improve the anti-MM activity of BCMA-directed immunotherapies in patients without permanent BCMA loss. Another potential mechanism related to BCMA loss is interference by sBCMA. In a recent study by Chen et al., the authors showed that high levels of sBCMA in serum might lead to a consistent decrease in the binding of anti-BCMA antibody to tumor cells in patients with RRMM.62 Since the majority of RRMM patients display elevated sBCMA, the functionally available BCMA on the surface of MM cells may be significantly reduced by sBCMA, meaning a functional BCMA downregulation in these patients.
Theoretically BCMA loss could also be caused by so-called antigen masking. In B-cell acute lymphoid leukemia relapsing after anti-CD19 CAR T-cell therapy, it was reported that an unintentional introduction of a CAR gene into a CD19-positive blast cell could lead to expression of CAR on leukemic cells. Subsequently, these CAR on the leukemic blasts bound in cis to the CD19 epitope on the cell surface, masking the tumor cells from recognition by the “true” CAR T cells, causing a functional loss of antigen.63 However, this phenomenon has not yet been reported in CAR T-cell therapies for RRMM.
Further potential mechanisms that could be associated with BCMA loss include trogocytosis, and some epigenetic mechanisms, which have been described in other malignant hematologic diseases. Trogocytosis is a process in which the target antigen on tumor cells is transferred to CAR T cells. In mouse models of B-cell leukemia, trogocytosis could reduce the antigen (CD19) density on tumor cells, leading to fratricide killing, T-cell exhaustion, and a decreased anti-tumor effect by CAR T cells.64 Moreover, after rituximab-containing therapy, downregulation of
CD20 was observed in patients with diffuse large cell Bcell lymphoma. When the CD20-negative lymphoma cells were treated with 5-aza-2'-deoxycytidine in vitro, the expression of CD20 mRNA recovered within 3 days, suggesting that some epigenetic mechanisms might be involved in CD20 downregulation after rituximab.65 Theoretically, these mechanisms may also be related to BCMA loss in RRMM patients. However, the role of these mechanisms in BCMA has not yet been extensively evaluated.
Strategies to overcome BCMA loss
Although BCMA-directed immunotherapies may no longer be effective in some patients because of loss of the target antigen, several other treatment options could still restore a response in such patients. In principle, the strategies to overcome BCMA loss are dependent on the underlying mechanisms. The reversibility of BCMA loss is the most crucial determinant for planning further treatments. Here, we discuss some alternatives that could be considered in RRMM patients with BCMA loss.
Homozygous BCMA gene deletion may lead to irreversible and complete loss of BCMA expression on the MM cell surface.14,15 In these cases, BCMA-targeted therapies are irreversibly ineffective. One of the strategies to overcome this kind of BCMA loss is targeting other antigens such as CD38, G protein coupled receptor class C group 5 member D (GPRC5D), Fc receptor-homolog 5 (FcRH5), CD19, and SLAMF766 (Figure 1B). CAR T-cell and BsAb therapies targeting antigens other than BCMA have already been summarized in other review articles, which we would recommend for readers interested in this topic.18,20,46,67,68 For instance, in a recent phase I study, the GPRC5D-directed BsAb talquetamab showed an encouraging ORR of 70% in RRMM patients, 30% of whom had been previously treated with anti-BCMA agents.69 Moreover, cevostamab, an FcRH5-targeted BsAb, produced an ORR of 36.4% (8 out of 22) in RRMM patients previously exposed to anti-BCMA therapies.70 These findings suggest that targeting antigens other than BCMA might be feasible in RRMM patients previously treated with anti-BCMA therapies, and BCMA expression status was irrelevant for these agents. However, in a recent study, heterozygous deletions in GPRC5D and CD38 genes were found in, respectively, 15% and 10% of MM patients who were T-cell immunotherapy-naïve, suggesting an increased risk of antigen loss following highly effective immunotherapies. In contrast, gains of FCRH5 and SLAMF7 genes were significantly more frequent in RRMM than in newly diagnosed MM, indicating a low risk of antigen loss in the course of the disease. Importantly, heterozygous BCMA gene deletion was present in four out of 50 RRMM patients who were heavily pretreated with other drugs but had never received anti-BCMA immunotherapy.56 In conclusion, the expression of immunotherapy targets should be evaluated during the treatment decision-making process.
Haematologica | 108 - April 2023 964 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
Another option for patients with irreversible BCMA loss is so-called “multi-targeted” immunotherapy. For instance, a BCMA/CD200/CD16A trispecific antibody was developed to link NK cells and MM cells in vitro, and BCMA and CD200 double-positive MM cells were more effectively killed than cells positive for only one of the antigens.71 For CAR T cells, this could be achieved by co-administration of different mono-targeted CAR T cells or by constructing a CAR T cell that could simultaneously recognize more than one antigen.72 In preclinical settings with cell lines and mouse models, bispecific CAR T cells targeting BCMA and GPRC5D were able to enhance the interactions between MM and CAR T cells and were able to prevent relapse due to BCMA loss.73 Feng et al. reported on a BCMA/CD38-targeted bispecific CAR T cell that could trigger robust cytotoxicity against MM cells expressing either BCMA or CD38 in vitro and was able to achieve complete tumor clearance in mice.74 More recently, another bispecific CAR T cell targeting BCMA and CD24 has been developed by an US group, with a strong cytotoxic effect in xenograft mouse models. In a recently published phase I first-in-human trial, a bispecific CAR T-cell targeting BCMA and CD38 (BM38) produced an ORR of 87% and a median progression-free survival of 17.2 months. Interestingly, as demonstrated by flow cytometry, two patients had BCMA- or CD38-negative RRMM at baseline, and both patients responded to BM38 treatment. Unfortunately, the underlying mechanisms of CD38 and BCMA loss at baseline were not described in the report of the study.75 These results suggested the feasibility of bispecific CAR T cells even in RRMM patients with expression of only one target antigen. A combination of two different monotargeted CAR T cells is an alternative strategy to bispecific CAR T cells. Some clinical trials investigating combinations of anti-BCMA CAR T cells with CD19- or CD38-directed CAR T cells have shown similar antitumor efficacy in RRMM when compared with published data on mono-specific anti-BCMA CAR T-cell therapies.76,77 Loss of one of the two antigens has already been reported after co-administration of two different mono-specific CAR T cells in RRMM.76 Theoretically, because of the high immune selection pressure, loss of both antigens may be possible after multi-specific immunotherapies. However, studies addressing this issue are still lacking.
Compared with homozygous BCMA gene deletion, the more common type of BCMA loss is reversible downregulation of BCMA expression following anti-BCMA therapies.53,55 If the BCMA status remains positive at relapse, retreatment with an anti-BCMA therapy could be considered in RRMM patients previously exposed to BCMA-targeted therapies (Figure 1C). Indeed, effective belantamab mafodotin treatment after relapse from anti-BCMA CAR T-cell therapy has been described in case reports.78,79 The differences in the mechanisms of action between ADC and CAR T cells also support the rationale of anti-BCMA retreatment with belantamab
mafodotin in these cases, as ADC react with MM cells primarily via T-cell independent mechanisms.79 Another druggable target is γ-secretase, which can cleave BCMA from the MM cell membrane and lead to partial BCMA loss on the cell surface.21 In a phase I, first-in-human trial of antiBCMA CAR T cells in combination with JSMD194, a γ-secretase inhibitor, for RRMM patients, JSMD194 increased the level of BCMA expression and might augment the anti-MM activity of CAR T cells, with a comparable toxicity profile as that in other CAR T-cell trials. Moreover, teclistamab or belantamab mafodotin in combination with a γ-secretase inhibitor, i.e. LY-411575 or nirogacestat, is currently under clinical investigation in phase I trials.80,81
Conclusions
BCMA-targeted treatments such as CAR T cells, BsAb, and ADC have brought new hope for patients with RRMM, and will be administered in earlier lines of therapy. They help to improve tumor control and may cure MM. The persistence of target antigen on the MM cells is essential for these novel targeted immunotherapies. Irreversible complete BCMA loss, which is caused by homozygous BCMA gene deletion, seems to be relatively rare after BCMA-directed treatments, based on data from clinical trials investigating CAR T cells or BsAb. In contrast, reversible partial loss or downregulation of BCMA is a more common event following these targeted immunotherapies. As BCMA-directed treatments are becoming a part of standard care for RRMM, BCMA expression status should be considered in the selection of therapeutic agents for each patient. Monitoring of several biomarkers, such as sBCMA and tumor BCMA expression level, might be helpful when making therapeutic decisions. Analyses of BCMA status on different levels (whole-genome sequencing, single-cell RNA sequencing, flow cytometry and immunohistochemistry, etc.) will provide useful information to elucidate the underlying biological mechanisms of BCMA loss in each patient. These strategies are also aligned with the concepts of precision medicine. Data on BCMA loss are still very limited, as BCMA-directed immunotherapy is a young research field. In addition, resistance mechanisms are not fully understood, and BCMA loss is not the only cause of relapse after novel immunotherapies. Further studies are, therefore, needed.
Disclosures
No conflicts of interest to disclose.
Contributions
XZ, LR, and KMK performed the literature research, analyzed and interpreted the data, and drafted the work; JM and HE conceived the design of the work and substantially revised it. All the authors approved the submitted version.
Haematologica | 108 - April 2023 965 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
References
1. van de Donk N, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397(10272):410-427.
2. Cowan AJ, Green DJ, Kwok M, et al. Diagnosis and management of multiple myeloma: a review. JAMA. 2022;327(5):464-477.
3. Raza S, Safyan RA, Lentzsch S. Immunomodulatory drugs (IMiDs) in multiple myeloma. Curr Cancer Drug Targets. 2017;17(9):846-857.
4. Elnair RA, Holstein SA. Evolution of treatment paradigms in newly diagnosed multiple myeloma. Drugs. 2021;81(7):825-840.
5. Shah UA, Mailankody S. Emerging immunotherapies in multiple myeloma. BMJ. 2020;370:m3176.
6. Cho SF, Lin L, Xing L, et al. BCMA-targeting therapy: driving a new era of immunotherapy in multiple myeloma. Cancers (Basel). 2020;12(6):1473.
7. Roex G, Timmers M, Wouters K, et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J Hematol Oncol. 2020;13(1):164.
8. Haslauer T, Greil R, Zaborsky N, Geisberger R. CAR T-cell therapy in hematological malignancies. Int J Mol Sci. 2021;22(16):8996.
9. Ansell SM, Lin Y. Immunotherapy of lymphomas. J Clin Invest. 2020;130(4):1576-1585.
10. Barsan V, Ramakrishna S, Davis KL. Immunotherapy for the treatment of acute lymphoblastic leukemia. Curr Oncol Rep. 2020;22(2):11.
11. Orlando EJ, Han X, Tribouley C, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504-1506.
12. Bannerji R, Allan JN, Arnason JE, et al. Clinical activity of REGN1979, a bispecific human, anti-CD20 x anti-CD3 antibody, in patients with relapsed/refractory (R/R) B-cell non-Hodgkin lymphoma (B-NHL). Blood. 2019;134(Suppl 1):762.
13. Bannerji R, Arnason JE, Advani R, et al. Emerging clinical activity of REGN1979, an anti-CD20 x anti-CD3 bispecific antibody, in patients with relapsed/refractory follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), and other B-cell nonHodgkin lymphoma (B-NHL) subtypes. Blood. 2018;132(Suppl 1):1690.
14. Samur MK, Fulciniti M, Aktas Samur A, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. 2021;12(1):868.
15. Da Via MC, Dietrich O, Truger M, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. 2021;27(4):616-619.
16. Leblay N, Maity R, Barakat E, et al. Cite-seq profiling of T cells in multiple myeloma patients undergoing BCMA targeting CAR-T or Bites immunotherapy. Blood. 2020;136(Suppl 1):11-12.
17. Rasche L, Wasch R, Munder M, Goldschmidt H, Raab MS. Novel immunotherapies in multiple myeloma - chances and challenges. Haematologica. 2021;106(10):2555-2565.
18. Yu B, Jiang T, Liu D. BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol. 2020;13(1):125.
19. Sanchez L, Dardac A, Madduri D, Richard S, Richter J. B-cell maturation antigen (BCMA) in multiple myeloma: the new frontier of targeted therapies. Ther Adv Hematol. 2021;12:2040620721989585.
20. Zhou X, Rasche L, Kortum KM, Danhof S, Hudecek M, Einsele H. Toxicities of chimeric antigen receptor T cell therapy in multiple myeloma: an overview of experience from clinical trials, pathophysiology, and management strategies. Front Immunol. 2020;11:620312.
21. Laurent SA, Hoffmann FS, Kuhn PH, et al. Gamma-secretase
directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6:7333.
22. Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34(4):985-1005.
23. Tai YT, Acharya C, An G, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127(25):3225-3236.
24. Moreaux J, Legouffe E, Jourdan E, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103(8):3148-3157.
25. Tai YT, Li XF, Breitkreutz I, et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006;66(13):6675-6682.
26. Cho SF, Anderson KC, Tai YT. Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol. 2018;9:1821.
27. Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010;115(17):3541-3552.
28. Lee L, Bounds D, Paterson J, et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br J Haematol. 2016;174(6):911-922.
29. Wang B, Wu C, Zhong Q, et al. Belantamab mafodotin for the treatment of multiple myeloma. Drugs Today (Barc). 2021;57(11):653-663.
30. Mullard A. FDA approves first BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discov. 2021;20(5):332.
31. Mullard A. FDA approves second BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discov. 2022;21(4):249.
32. Usmani SZ, Garfall AL, van de Donk N, et al. Teclistamab, a Bcell maturation antigen x CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): a multicentre, open-label, single-arm, phase 1 study. Lancet. 2021;398(10301):665-674.
33. Topp MS, Duell J, Zugmaier G, et al. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J Clin Oncol. 2020;38(8):775-783.
34. Lonial S, Lee HC, Badros A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a twoarm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21(2):207-221.
35. Popat R, Nooka A, Stockerl-Goldstein K, et al. DREAMM-6: safety, tolerability and clinical activity of belantamab mafodotin (Belamaf) in combination with bortezomib/dexamethasone (BorDex) in relapsed/refractory multiple myeloma (RRMM). Blood. 2020;136(Suppl 1):19-20.
36. Usmani SZ, Alonso AA, Quach H, et al. DREAMM-9: phase I study of belantamab mafodotin plus standard of care in patients with transplant-ineligible newly diagnosed multiple myeloma. Blood. 2021;138(Suppl 1):2738.
37. Lee HC, Raje NS, Landgren O, et al. Phase 1 study of the antiBCMA antibody-drug conjugate AMG 224 in patients with relapsed/refractory multiple myeloma. Leukemia. 2021;35(1):255-258.
38. Kumar SK, Migkou M, Bhutani M, et al. Phase 1, first-in-human study of MEDI2228, a BCMA-targeted ADC in patients with relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):26-27.
Haematologica | 108 - April 2023 966 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
39. Harrison SJ, Minnema MC, Lee HC, et al. A phase 1 first in human (FIH) study of AMG 701, an anti-B-cell maturation antigen (BCMA) half-life extended (HLE) BiTE® (bispecific T-cell engager) molecule, in relapsed/refractory (RR) multiple myeloma (MM). Blood. 2020;136(Suppl 1):28-29.
40. Moreau P, Usmani SZ, Garfall AL, et al. Updated results from MajesTEC-1: phase 1/2 study of teclistamab, a B-cell maturation antigen x CD3 bispecific antibody, in relapsed/refractory multiple myeloma. Blood. 2021;138(Suppl 1):896.
41. Zonder JA, Richter J, Bumma N, et al. Early, deep, and durable responses, and low rates of cytokine release syndrome with REGN5458, a BCMAxCD3 bispecific monoclonal antibody, in a phase 1/2 first-in-human study in patients with relapsed/refractory multiple myeloma (RRMM). Blood.
2021;138(Suppl 1):160.
42. Kumar S, D'Souza A, Shah N, et al. A phase 1 first-in-human study of Tnb-383B, a BCMA x CD3 bispecific T-cell redirecting antibody, in patients with relapsed/refractory multiple myeloma. Blood. 2021;138(Suppl 1):900.
43. Sebag M, Raje NS, Bahlis NJ, et al. Elranatamab (PF-06863135), a B-cell maturation antigen (BCMA) targeted CD3-engaging bispecific molecule, for patients with relapsed or refractory multiple myeloma: results from MagnetisMM-1. Blood. 2021;138(Suppl 1):895.
44. Costa LJ, Wong SW, Bermúdez A, et al. First clinical study of the B-cell maturation antigen (BCMA) 2+1 T cell engager (TCE) CC93269 in patients (Pts) with relapsed/refractory multiple myeloma (RRMM): interim results of a phase 1 multicenter trial. Blood. 2019;134(Suppl_1):143.
45. Lancman G, Sastow DL, Cho HJ, et al. Bispecific antibodies in multiple myeloma: present and future. Blood Cancer Discov. 2021;2(5):423-433.
46. Zhou X, Einsele H, Danhof S. Bispecific antibodies: a new era of treatment for multiple myeloma. J Clin Med. 2020;9(7):2166.
47. Teoh PJ, Chng WJ. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J. 2021;11(4):84.
48. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726-1737.
49. Mailankody S, Htut M, Lee KP, et al. JCARH125, Anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: initial proof of concept results from a phase 1/2 multicenter study (EVOLVE). Blood. 2018;132(Suppl 1):957.
50. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 openlabel study. Lancet. 2021;398(10297):314-324.
51. Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705-716.
52. Ali SA, Shi V, Maric I, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128(13):1688-1700.
53. Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267-2280.
54. Green DJ, Pont M, Sather BD, et al. Fully human BCMA targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood. 2018;132(Suppl 1):1011.
55. Cohen AD, Garfall AL, Stadtmauer EA, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple
myeloma. J Clin Invest. 2019;129(6):2210-2221.
56. Truger MS, Duell J, Zhou X, et al. Single- and double-hit events in genes encoding immune targets before and after T cell–engaging antibody therapy in MM. Blood Adv. 2021;5(19):3794-3798.
57. Farswan A, Jena L, Kaur G, et al. Branching clonal evolution patterns predominate mutational landscape in multiple myeloma. Am J Cancer Res. 2021;11(11):5659-5679.
58. Rasche L, Chavan SS, Stephens OW, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun. 2017;8(1):268.
59. Terragna C, Martello M, Santacroce B, et al. A branching evolution model at relapse characterizes multiple myeloma patients who responded to up-front combination therapy including new drugs. Blood. 2016;128(22):2080.
60. Rasche L, Schinke C, Maura F, et al. The spatio-temporal evolution of multiple myeloma from baseline to relapserefractory states. Nat Commun. 2022;13(1):4517.
61. Pont MJ, Hill T, Cole GO, et al. Gamma-secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood. 2019;134(19):1585-1597.
62. Chen H, Li M, Xu N, et al. Serum B-cell maturation antigen (BCMA) reduces binding of anti-BCMA antibody to multiple myeloma cells. Leuk Res. 2019;81:62-66.
63. Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499-1503.
64. Hamieh M, Dobrin A, Cabriolu A, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112-116.
65. Hiraga J, Tomita A, Sugimoto T, et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood. 2009;113(20):4885-4893.
66. Bruno B, Wasch R, Engelhardt M, et al. European Myeloma Network perspective on CAR T-cell therapies for multiple myeloma. Haematologica. 2021;106(8):2054-2065.
67. Zhou X, Einsele H, Danhof S. [CAR T-cell therapy for multiple myeloma]. Internist (Berl). 2021;62(6):605-610.
68. Teoh PJ, Chng WJ. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J. 2021;11(4):84.
69. Krishnan AY, Minnema MC, Berdeja JG, et al. Updated phase 1 results from MonumenTAL-1: first-in-human study of talquetamab, a G protein-coupled receptor family C group 5 member D x CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma. Blood. 2021;138(Suppl 1):158.
70. Trudel S, Cohen AD, Krishnan AY, et al. Cevostamab monotherapy continues to show clinically meaningful activity and manageable safety in patients with heavily pre-treated relapsed/refractory multiple myeloma (RRMM): updated results from an ongoing phase I study. Blood. 2021;138(Suppl 1):157.
71. Gantke T, Weichel M, Herbrecht C, et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng Des Sel. 2017;30(9):673-684.
72. Simon S, Riddell SR. Dual targeting with CAR T cells to limit antigen escape in multiple myeloma. Blood Cancer Discov. 2020;1(2):130-133.
73. Fernandez de Larrea C, Staehr M, Lopez AV, et al. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discov. 2020;1(2):146-154.
Haematologica | 108 - April 2023 967 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
74. Feng Y, Liu X, Li X, et al. Novel BCMA-OR-CD38 tandem-dual chimeric antigen receptor T cells robustly control multiple myeloma. Oncoimmunology. 2021;10(1):1959102.
75. Mei H, Li C, Jiang H, et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J Hematol Oncol. 2021;14(1):161.
76. Wang Y, Cao J, Gu W, et al. Long-term follow-up of combination of B-cell maturation antigen and CD19 chimeric antigen receptor T cells in multiple myeloma. J Clin Oncol. 2022;40(20):2246-2256.
77. Zhang H, Liu M, Xiao X, et al. A combination of humanized antiBCMA and murine anti-CD38 CAR-T cell therapy in patients with relapsed or refractory multiple myeloma. Leuk Lymphoma. 2022;63(6):1418-1427.
78. Gazeau N, Beauvais D, Yakoub-Agha I, et al. Effective anti-BCMA retreatment in multiple myeloma. Blood Adv. 2021;5(15):3016-3020.
79. Cohen AD, Garfall AL, Dogan A, et al. Serial treatment of relapsed/refractory multiple myeloma with different BCMAtargeting therapies. Blood Adv. 2019;3(16):2487-2490.
80. Pillarisetti K, Powers G, Luistro L, et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020;4(18):4538-4549.
81. Nooka AK, Weisel K, van de Donk NW, et al. Belantamab mafodotin in combination with novel agents in relapsed/refractory multiple myeloma: DREAMM-5 study design. Future Oncol. 2021;17(16):1987-2003.
Haematologica | 108 - April 2023 968 REVIEW ARTICLE - BCMA loss in immunotherapy X. Zhou et al.
Genomics improves risk stratifi cation of adults with T-cell acute lymphoblastic leukemia enrolled in measurable residual disease-oriented trials
Celia González-Gil,1 Mireia Morgades,2+ Thaysa Lopes,1+ Francisco Fuster-Tormo,1 Jesús GarcíaChica,1 Ran Zhao,3 Pau Montesinos,4 Anna Torrent,2 Marina Diaz-Beya,5 Rosa Coll,6 Lourdes Hermosín,7 Santiago Mercadal,8 José González-Campos,9 Lurdes Zamora,2 Teresa Artola,10 Ferran Vall-Llovera,11 Mar Tormo,12 Cristina Gil-Cortés,13 Pere Barba,14 Andrés Novo,15 Jordi Ribera,1 Teresa Bernal,16 Paula López de Ugarriza,16 María-Paz Queipo,17 Pilar Martínez-Sánchez,18 Alicia Giménez,18 Teresa González-Martínez,19 Antonia Cladera,20 José Cervera,4 Rosa FernándezMartín,21 María Ángeles Ardaiz,22 María Jesús Vidal,23 Ángela Baena,24 Nuria López-Bigas,25 Anna Bigas,1,26 Jaroslaw Maciejewski,27 Alberto Orfao,28 Josep Maria Ribera1,2 and Eulalia Genescà1
1Institut d’Investigació contra la Leucemia Josep Carreras (IJC), Campus ICO-Germans Trias i Pujol, Universitat Autònoma de Barcelona, Barcelona, Spain; 2Departament d’Hematologia Clínica, ICO-Hospital Germans Trias i Pujol, Universitat Autònoma de Barcelona, Barcelona, Spain; 3Department of Quantitative Health Sciences and Leukemia Program, Department of Hematology and Medical Oncology, Cleveland Clinic, Cleveland, OH, USA; 4Hospital Universitari i Politècnic La Fe, Valencia, Spain; 5Servei d’Hematologia Clínica, Hospital Clínic de Barcelona, Barcelona, Spain; 6Institut Català d’Oncologia (ICO), Hospital Josep Trueta, Girona, Spain; 7Servicio Hematología Clínica, Hospital de Jerez, Jerez de la Frontera, Spain; 8Servei d’Hematologia Clínica, Hospital Duran i Reynals-ICO, Hospitalet del Llobregat, Spain; 9Servicio Hematología Clínica, Hospital Vírgen del Rocío, Sevilla, Spain; 10Servicio Hematología Clínica, Hospital Universitario de Donostia, Donostia, Spain; 11Servicio Hematología Clínica, Hospital Mútua de Terrassa, Terrassa, Spain; 12Hospital Clínico Universitario, Instituto de Investigación INCLIVA, Valencia, Spain; 13Servicio Hematología Clínica, Hospital General de Alicante, Alicante, Spain; 14Servicio Hematología Clínica, Hospital Universitari de la Vall d’Hebron, Barcelona, Spain; 15Servicio Hematología Clínica, Hospital Son Espases, Palma de Mallorca, Spain; 16Servicio Hematología Clínica, Hospital Central de Asturias, Instituto de Investigación Sanitario del Principado de Asturias (ISPA), Instituto Oncológico Universitario del Principado de Asturias (IUOPA), Oviedo, Spain; 17Servicio Hematología Clínica, Hospital Virgen de la Victoria, Málaga, Spain; 18Servicio Hematología Clínica, Hospital 12 de Octubre, Madrid, Spain; 19Servicio Hematología Clínica, Hospital Universitario de Salamanca, Salamanca, Spain; 20Servicio Hematología Clínica, Hospital Son LLátzer, Palma de Mallorca, Spain; 21Servicio Hematología Clínica, Hospital Insular de Gran Canarias, Las Palmas de Gran Canaria, Spain; 22Servicio Hematología Clínica, Complejo Hospitalario de Navarra, Pamplona, Spain; 23Servicio Hematología Clínica, Complejo Hospitalario de León, León, Spain; 24Servicio Hematología Clínica, Complejo Hospitalario de Jaén, Jaén, Spain; 25Institute for Research in Biomedicine (IRB Barcelona), Barcelona Institute of Science and Technology, Barcelona, Spain; 26Program in Cancer Research, Institut-Hospital del Mar d’Investigacions Mèdiques, CIBERONC, Barcelona, Spain; 27Department of Hematology and Medical Oncology, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH, USA and 28Centro de Investigación del Cáncer (IBMCC-CSIC/USAL), Departamento de Medicina, Universidad de Salamanca, Instituto Biosanitario de Salamanca, CIBERONC, Salamanca, Spain
+MM and TL contributed equally.
Abstract
Correspondence: E.G. Ferrer egenesca@carrerasresearch.org
Received: April 4, 2022.
Accepted: October 14, 2022.
Early view: November 3, 2022.
https://doi.org/10.3324/haematol.2022.281196
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Genetic information has been crucial to understand the pathogenesis of T-cell acute lymphoblastic leukemia (T-ALL) at diagnosis and at relapse, but still nowadays has a limited value in a clinical context. Few genetic markers are associated with the outcome of T-ALL patients, independently of measurable residual disease (MRD) status after therapy. In addition, the prognostic relevance of genetic features may be modulated by the specific treatment used. We analyzed the genetic profile of 145 T-ALL patients by targeted deep sequencing. Genomic information was integrated with the clinicalbiological and survival data of a subset of 116 adult patients enrolled in two consecutive MRD-oriented trials of the
Haematologica | 108 - April 2023 969 ARTICLE - Acute Lymphoblastic Leukemia
Spanish PETHEMA (Programa Español de Tratamientos en Hematología) group. Genetic analysis revealed a mutational profile defined by DNMT3A/ N/KRAS/ MSH2/ U2AF1 gene mutations that identified refractory/resistant patients. Mutations in the DMNT3A gene were also found in the non-leukemic cell fraction of patients with T-ALL, revealing a possible mutational-driven clonal hematopoiesis event to prime T-ALL in elderly. The prognostic impact of this adverse genetic profile was independent of MRD status on day +35 of induction therapy. The combined worse-outcome genetic signature and MRD on day +35 allowed risk stratification of T-ALL into standard or high-risk groups with significantly different 5year overall survival (OS) of 52% (95% confidence interval: 37-67) and 17% (95% confidence interval: 1-33), respectively. These results confirm the relevance of the tumor genetic profile in predicting patient outcome in adult T-ALL and highlight the need for novel gene-targeted chemotherapeutic schedules to improve the OS of poor-prognosis T-ALL patients.
Introduction
Acute lymphoblastic leukemia (ALL) is an infrequent aggressive cancer being more common in children.1 ALL includes B- and T-cell lineage subtypes. T-cell acute lymphoblastic leukemia (T-ALL) accounts for 10–15% of pediatric and 20–25% of adult cases.2,3 B-cell precursor ALL and T-ALL share some genetic features4 and are considered together for treatment in many trials. The classical prognostic factors such as age, white blood cell (WBC) count, measurable residual disease (MRD) and some genetic features have been used to predict outcome and stratify the therapy in both types of ALL.2,5-7 Although treatment response via MRD monitoring represents a milestone in virtually all ALL trials, MRD is not available at diagnosis and might not finely predict relapse in adult ALL patients,6-8 emphasizing the need for additional prognostic markers that may be readily available for these patients at diagnosis. Studies by the cooperative Group for Research in Adult Acute Lymphoblastic Leukemia (GRAALL) demonstrated the prognostic relevance of some genetic markers in adult T-ALL.7,9 The combination of mutations in the NOTCH1/FBXW7 signaling pathway, in the absence of N/KRAS gene mutations and alterations in PTEN, together with adequate MRD clearance, identified a group of adult T-ALL patients with a good prognosis, who might not benefit from further intensification treatment by allogeneic stem cell transplantation (allo-SCT).7 MRD and the oncogenetic pattern were independent prognostic factors used for patient stratification in the current protocols of the GRAALL Group.
It is well established that genetics plays a key role in the development and progression of T-ALL. The disease is sustained by genetic abnormalities that often determine the stage of maturation arrest (e.g., MLL-r, CALM::AFA10, HOXAr, TLX1-r, TLX3-r, SIL::TAL), and/or the proliferation and/or survival rate of leukemic cells (e.g., CDKN2A/B, K/NRAS, NOTCH1/FBXW7, NUP214::ABL1, JAK/STAT).10,11 However, the prognostic relevance of a genetic profile identified in a group of T-ALL patients may be influenced by the treatment protocol used, since several gene mutations generate resistance to treatment.12–17 Therefore, assessment of the
genetic profiles associated with patient outcome within specific clinical trials is essential for improved risk stratification and to move towards more personalized medicine. In the study reported here, we screened DNA from 145 TALL patients, based on a custom-built T-ALL-oriented next-generation sequencing panel (NGSp). Of these, 116 were adult patients treated as part of two consecutive high-risk MRD-oriented trials by the Spanish PETHEMA (Programa Español para el Tratamiento de Hemopatías Malignas) group. Our goal was to identify gene-mutational profiles (point mutations, small insertions and deletions and indels) at diagnosis that could help to predict response to therapy and outcome.
Methods
Targeted deep sequencing
DNA samples or cryopreserved leukemic cells from T-ALL patients (n=145) were collected from different national biobanks (see the Online Supplementary Appendix for information). The mutational profile was obtained employing a custom gene panel (SureSelectXT HS Target Enrichment System for Illumina Multiplexed Sequencing Platforms, Agilent Technologies, Santa Clara, CA, USA) and sequencing in a MiSeq instrument (Illumina, San Diego, CA, USA). Mutations were retrieved applying a home-made standard gold pipeline analysis (see detailed methods section in the Online Supplementary Appendix). Final selected variants were classified as pathogenic, benign or of uncertain significance if the majority (≥6/10), final version of the in silico predictors identified the variant as being in one of the categories. Information about predictors was extracted from dbNSFP18 via ANNOVAR annotation. Benign variants were excluded from further analyses. Information of genes and regions included in the panel has been previously described19 and it is shown in an Excel file in the Online Supplementary Appendix
Patients and treatment protocols
For clinical and outcome correlations, 29 patients from our initial cohort were excluded (see the Online Supplementary
Haematologica | 108 - April 2023 970 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
Appendix). Thus, a representative cohort of 116 adult T-ALL patients were studied. The diagnosis of T-ALL was made according to the World Health Organization criteria20 and the cytogenetic classification was based on the Genesca et al. 19 study, instead of the classical cut-off of five genetic alteration21 (see the Online Supplementary Appendix). Patients were treated with two consecutive MRD-oriented high-risk adult ALL protocols. Detailed information of both trials is shown in the Online Supplementary Appendix and the Online Supplementary Figure S1. In order to homogenize patient allocation according MRD levels at the end of induction treatment, we established a MRD cut-off of 0.1 to define a patient as good responder (≤ 0.1%) in the ALLAR [Ph-]-03 trial (clinicaltrials gov. Identifier: NCT00853008), similarly as it was established for the ALLHR [Ph-]-11 trial (clinicaltrials gov. Identifier: NCT01540812).
Informed consent was obtained from all patients. Samples and clinical data were stored in accordance with the declaration of Helsinki. The study was approved by the Institutional Review Board of the Hospital Germans Trias i Pujol.
Statistical analyses
Genes mutated in at least five patients were included in the initial screening for impact on OS and cumulative incidence of relapse (CIR), as individual genes, except for N/KRAs and JAK1/JAK3 mutations that were assessed together since are isoforms, respectively. OS curves were plotted using the Kaplan–Meier method and compared by the log-rank test. CIR was estimated by competing risks analysis using cumulative incidence functions. Gray’s test was used to compare CIR curves. A Cox proportional hazard regression model was used to identify predictive factors for OS. Statistical significance was concluded for two-sided values of P<0.05. All statistical analyses were performed using SPSS version 24 (IBM Corp. Armonk, NY, USA), GraphPad Prism® version 8 (GraphPad Software Inc., La Jolla, CA, USA) and R version 4.1.0.
Combinations of mutations (affecting ≥12 patients) and biological traits at the time of diagnosis were pairwise assessed using Fisher’s exact test. Multiple associations were corrected using a Benjamini-Hochberg q test, with significance concluded for co-existence for values of q<0.05.
Results
Somatic mutational landscape of T-cell acute lymphoblastic leukemia
The presence of point mutations and insertions and deletions (indels) was investigated in 145 T-ALL patients by TDS using a customized NGSp. Overall, genetic variants were detected in 136 of 145 patients (94%) for a median of four genetic variants and three mutated genes per patient. No
variants were observed in nine (6%) patients (Figure 1A). Of patients with variants, 92% (125/136) carried missense mutations and 32% (44/136) had nonsense mutations. Genes with predominate missense variants were JAK3 (96.3%), FBXW7 (92.3%), FAT1 (100%), JAK1 (100%) and NRAS (100%). In turn, insertions, deletions, and indels of up to 50 nucleotides, were identified in 67% (91/136) of the cases. Of these, 81% (74/91) presented short insertions (52 frameshift and 38 non-frameshift); 42% (38/91) had short deletions (28 frameshift and 11 non-frameshift); and 12% (11/91) had frameshift indels (small insertions or deletions). PTEN (71·4%) and IL7R (80%) were the two genes with the highest percentage of indels. Globally, combinations of point mutations and indels were observed in 60% (81/136) of patients, whereas isolated indels mutations were detected in 4% (6/136) of cases. According to the functional impact assigned to each variant, 89% (121/136) of patients presented pathogenic variants and 83% (113/136) had variants of uncertain significance, and only 15 of them exclusively presented variants of uncertain significance. Almost all pathogenic variants (98%) were missense mutations, whereas most variants of uncertain significance were nonsense variants (16%) or indels (53%) (Online Supplementary Figure S2).
Recurrently mutated genes found in at least five patients are listed in Figure 1B and all had been previously reported in T-ALL. Mutations were found in: i) transcription factor tumor suppressor genes (PTEN, BCL11B, RUNX1, GATA3, ETV6)23–27; ii) epigenetic regulators (PHF6, DNMT3A, EP300, KMT2C)27–29; iii) DNA mismatch repair genes (MSH2)30; iv) genes expressing ribosomal protein (RPL5)31; v) genes involved in RNA splicing (U2AF1)32; and vi) signaling pathways that regulate T-cell development, such as the RAS/MAPK signaling (NRAS)33 WNT (FAT1, FAT3),34,35 IL7R-JAK-STAT (JAK3, JAK1, IL7R, DNM2)36-39 and the NOTCH1/FBXW7 signalling.40,41 The latter was found in 71% (97/136) of patients (NOTCH1 and FBXW7), confirming the importance of this signaling pathway in the development of T-ALL.
Genetic-biological associations
Major biological features at diagnosis were available for 143/145 patients and their data were used to classify them by: i) age group: <18, 18-35, 36-60 and >60 years; ii) immunophenotype: ETP-ALL, pre-T, cortical, mature T-ALL and not determined immunophenotype (NDI); and iii) cytogenetic groups: 0-2 abnormalities, a complex karyotype with at least three abnormalities (CK ≥3) and non-evaluable (NE) cases.19 Previously reported correlations between genetic variants occurring in genes mutated in ≥12 patients and diagnostic traits included a direct association between the presence of variants in DNMT3A gene and both ETPALL (OR=8.56; q= 0.02)42,43 and advanced age.43 In our analysis, however, DNMT3A mutations were restricted to the oldest patients (>60 years [y]) (OR=9.6; q= 0.08) and
Haematologica | 108 - April 2023 971 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
Figure 1. Genetic profile of T-cell acute lymphoblastic patients at diagnosis. (A) Frequency of variants per patient in the cohort (n=145). (B) Frequency of patients showing recurrently mutated genes (cut-off of ≥5 mutations/gene). (C) Pairwise associations observed between the recurrently mutated genes and the biological characteristics of the disease at presentation. Positive and negative correlations are depicted as magenta and green circles, respectively. Circle diameters indicate the degree of significance. Y: years; ETP-ALL: early T-cell precursor acute lymphoblastic leukemia; NDI: not determined immunophenotype; abn: abnormalities; CK: complex karyotype; NE: non-evaluable karyotype.
were not associated to adolescents and young adults (AYA, 18-35 y) (OR=0.09; q=0.07). Other new significant associations included a lower incidence of FBXW7 variants in ETP-ALL (OR=0; q=0.07), higher frequency of FAT1 gene variants in patients with an NE karyotype (OR=6.36; q=0.07) at the expense of a negative association between this gene and patients with 0-2 abnormalities (OR=0.09; q=0.02), and association of NRAS mutations with and ETPALL immunophenotype (OR=6.13; q=0.07) that contrasts with the negative association of these mutations with the cortical subtype (OR=0; q=0.07) (Figure 1C).
Prognostic significance of gene mutations and mutational profiles
The analysis of the clinical impact of the genetic variants
and mutational profiles identified by TDS on patient outcome was restricted to 116 patients homogeneously treated in two high-risk consecutive MRD-oriented PETHEMA trials whose clinical, biological and outcome data were complete. Comparison of the outcome of the sequenced (n=116) versus non-sequenced patients (n=116) did not reach significant differences in terms of OS. Thus, our cohort was representative of the patients included in both trials. Like other adult T-ALL cohorts, this was mainly composed of males of median age (37 y). Overall, CR rate (Induction-1 + Induction-2) for the whole series was 88%, being 83% for cases who achieved MRD levels <0.1% at the end of Induction-1. Most patients (72%) were treated according to the chemotherapy schedules proposed in the trials. The remaining patients were assigned to allo-SCT.
A B C
Haematologica | 108 - April 2023 972 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
*results expressed as 2-year overall survival (OS) probability. CI: confidence interval; CIR: cumulative incidence of relapse.
The 5 year CIR and OS probabilities for the whole series was 54% (95% confidence interval [CI]: 43%-64%) and 36% (95% CI: 26-46), respectively.
In order to identify mutations in genes affecting patient outcome, we first assessed the impact on OS and CIR of each individual gene mutated in at least five patients. Strikingly, we could not confirm the previously reported association between NOTCH1 mutations and prolonged OS (Online Supplementary Figure S3A), or the lower CIR rate (data not shown).9,44 A more in-depth analysis of the distinct NOTCH1 gene variants identified with respect to their functional impact (pathogenic vs. uncertain significance) (Online Supplementary Figure S3B) and according to the variant allele frequency (VAF) with a cut-off of 25% to define a variant as clonal (Online Supplementary Figure S3C), confirmed the lack of prognostic impact of NOTCH1 variants on OS in our T-ALL patients. In contrast, patients carrying FBXW7 mutations, were associated with a better OS (Table 1). Sixty-five percent of patients carrying mutations in FBXW7 (13/20) also presented a mutation in the NOTCH1 gene, with no differences in OS between both groups (FBXW7 only vs. FBXW7 and NOTCH1) (data not shown), suggesting that the good outcome observed in patients carrying FBXW7 mutations could be attributed to a specific dysfunction of the FBXW7 protein (Online Supplementary Figure S3D). In addition, patients showing DNMT3A, N/KRAS, MSH2 and/or U2AF1 mutations had a lower OS than patients with no mutations in these genes (Table 1). It is of note that only patients with mutations in N/KRAS genes showed a high probability of CIR (Table 1). Based on these results, we grouped mutations in genes associated with a worse outcome according to the same homeostatic processes affected and basal biologic characteristics. Thus, patients with mutations in DNMT3A and U2AF1 were older (median age 54 y, P<0.001) and more frequently showed an ETP-ALL immunophenotype (P<0.001). Mutations in these two genes were consistent with clonal
hematopoiesis of indeterminate potential (CHIP).45–47 We grouped mutations in these two genes into the aginggenes cluster. In turn, N/KRAS, or MSH2 gene mutations have previously been described as being involved in generating resistance to ALL treatment.13,14,16 We named this group of gene mutations as treatment-resistance-gene cluster. Both clusters identified patients with a similarly poor response to treatment (Online Supplementary Table S1). Overall, 25% (29/116) of patients harbored mutations in genes conferring a worse outcome with more frequent slow response to initial treatment, high toxicity and early death (Figure 2). Of note, OS of patients with co-occurrence of more than one worse prognosis mutation was similar to that of patients carrying on only one worse outcome mutation (P=0.750). Therefore, we subsequently assessed the global prognostic impact of all adverse gene mutations collectively as a variable named worse-outcome genetics (WOG) by univariable and multivariable analyses. Similarly, patients carrying mutations in FBXW7 were also included as a variable named good-outcome genetics (GOG). Basal characteristics at the time of diagnosis and treatment response of patients belonging to the WOG- and WOG+ groups are shown in Table 2 (Online Supplementary Table S2 for patients GOG+ and GOG-). Importantly, only one patient presented mutations in FBXW7 (GOG) and U2AF1 (WOG), whose death was associated to transplantrelated mortality. This patient was included in the WOG group, to maintain both variables independent. Age, WBC count, central nervous system (CNS) infiltration, ETP-ALL phenotype, CK ≥3 alterations, MRD level at the 0.1% cutoff, WOG and GOG were included in the univariable analysis (Table 3). The two treatment protocols were also included in the analysis to exclude any possible protocoldependent bias. By multivariable analysis, MRD level ≥0.1% after induction-1 (day +35) and the WOG cluster were independently associated with shorter OS rates with hazard ratios (HR) of 2.187 (range, 1.087-4.400) and 3.040
Gene Number of patients (N=116) OS 5 years, % (95% CI) CIR 5 years, % (95% CI) Patients with mutations Patients without mutations Patients with mutations Patients without mutations P Patients with mutations Patients without mutations FBXW7 20 96 62 (36-88) 32 (21-43) 0.032 -DNMT3A 10 106 13 (0-37) 38 (27-49) 0.001 -N/KRAS 13 103 21 (0-45) 39 (28-50) 0.023 85 (37-97) 50 (39-61) MSH2 5 111 20 (0-55) 37 (26-48) 0.036 -U2AF1* 5 111 20 (0-55) 50 (40-60) 0.003 - -
(range,
Table 1. Prognostic impact of genes found to be recurrently mutated in the PETHEMA T-cell acute lymphoblastic leukemia
Haematologica | 108 - April 2023 973 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
Figure 2. Scheme of the genetic profile of each T-cell acute lymphoblastic patient, its response and evolution during treatment. Only genes recurrently mutated in ≥5 patients are shown. Each mutation is indicated by a square: brown squares correspond to genes contained in the worse-outcome genetics (WOG) signature; dark green squares correspond to mutations in the FBXW7 gene (good-outcome genetics [GOG] signature) and pink squares correspond to other mutated genes. Treatment response and patient evolution data are shown at the bottom. Induction + 14d indicates the percentage of blast cells in bone marrow 14 days after starting induction therapy; induction + 35d corresponds to measurable residual disease (MRD) values at the end of the first induction blocks (induction 1); induction-2 indicates patients that received or not an Induction-2 treatment block. Consolidation corresponds to MRD values at the end of consolidation chemo-block. Post-consolidation indicates treatment choice (allogeneic stem cell transplantation or chemotherapy) based on MRD values at the end of the consolidation treatment. On the right, the percentage of cases mutated in the different genes are indicated. NA: not available.
1.531-6.035) respectively (Table 3). The multivariable analysis for CIR, did not validate the predictive role of N/KRAS mutations observed in the univariable analysis (Online Supplementary Table S3). In fact, we could not identify any variable to predict CIR risk in our adult T-ALL cohort. Based on these results, we re-stratified our patients according to their MRD level on day +35 after induction-1 and the presence or absence of WOG mutations. Two groups of patients were identified: standard-risk patients with MRDlow plus WOG- (5-y OS of 52 %, 95% CI: 37-67, n=65); and highrisk patients that included MRDhigh and WOG- (n=11), MRDlow plus WOG+(n=9) and MRDhigh plus WOG+ (n=4) patients respectively, with a 5-y OS of 17% (95% CI: 1-33) (Figure 3).
Tracking aging cluster mutations in non-leukemic cells
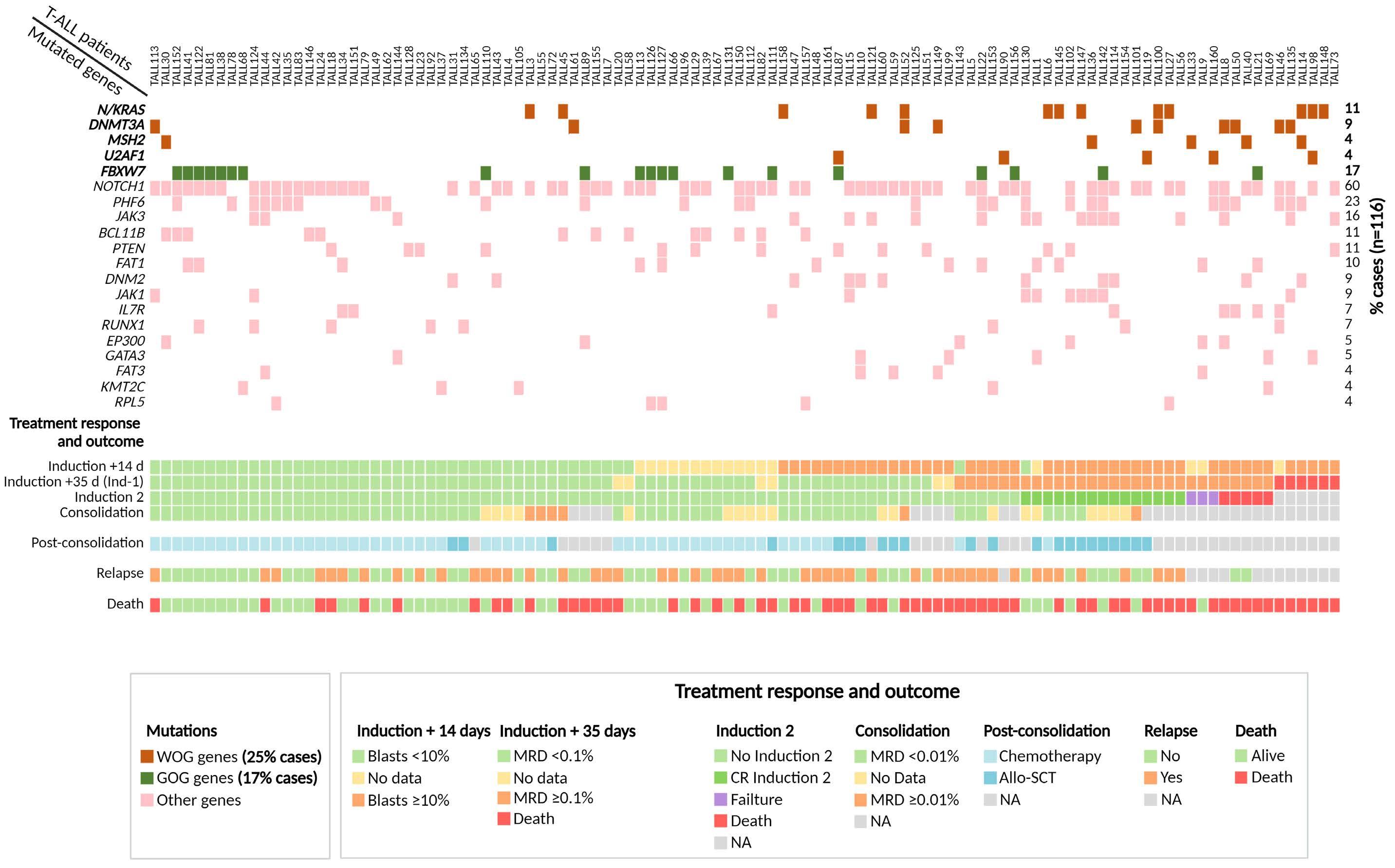
In order to determine whether the DNMT3A and U2AF1 genetic variants associated with the aging-genes cluster identified in our adult T-ALL patient cohort were related to
clonal hematopoiesis of indeterminate potential (CHIP), we investigated those variants in the non-leukemic fraction of peripheral blood cells of the same patients. For this purpose, genomic DNA from purified polymorphonuclear cells of nine patients carrying mutations in these two genes was sequenced. However, no genetic variants of the U2AF1 gene were identified in the polymorphonuclear cell fraction of the two patients with mutations in this gene in their leukemic cells. By contrast, identical DNMT3A mutations were found at lower or similar (VAF) in the polymorphonuclear fraction of seven of seven cases whose leukemic cells had mutations in this gene (Table 4), suggesting that these mutations were already present in a common lympho-myeloid early stem cell precursor and appear before the leukemia arises. Moreover, in two patients, copy number alterations and mutations in the DNMT3 gene were apparent in the blast cells, highlighting the importance of this gene in the development of their leukemia. Further analysis of non-
Haematologica | 108 - April 2023 974 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
hematopoietic cells would be needed to rule out a germinal basis for these alterations, although the in silico predictors indicated that those variants had an actual or potential impact on the function of the DNMT3A protein, which argues in favor of them being of non-germinal origin (Table 4).
Discussion
This study describes a gene mutational signature, named WOG, that identifies high-risk T-ALL patients with a slow
response to initial chemotherapy treatment and low CR rates. These patients more often needed two induction cycles to achieve a good MRD response which could also account for the higher rate of early deaths, due to toxicity, observed at the end of induction treatment, and thereby their shorter OS. However, our WOG signature did not have an impact on the CIR, since did not identify patients at high versus low risk of relapse. This may be due to the very poor clinical outcome of the patients with a WOG mutational profile, yet observed at early stages of the treatment, that would eliminate the possibility of a higher rate of sub-
Results expressed as number of cases/total cases (percentage). WOG: worse-outcome genetics; measurable residual disease (MRD) values were considered for those patients that reach complete remission (CR); M: male; F: female; WBC: white blood cells; ECOG: Eastern Cooperative Oncology Group; CNS: central nervous system; ETP-ALL: early T-cell precursor acute lymphoblastic leukemia; abn: abnormalities; CK: complex karyotype; day +14: 14 days after induction treatment; day +35: 35 days after induction treatment; allo-SCT: allogeneic stem cell transplantation. Ind: induction.
Genetic group P Total (N=116) WOG- (N=87) WOG+ (N=29) Patient-related features Median age, years (range) Sex, M/F 34 (16-61) 71/16 44 (19-60) 15/14 0.068 0.001 37 (16-61) 86/30 Disease-related features Median WBC, x109/L (range) 66.3 (0.5-525.4) 23.4 (0.6-495) ECOG, N (%) 0 1 2 ≥3 31/83 (37) 42/83 (51) 9/83 (11) 1/83 (1) 12/29 (41) 11/29 (38) 5/29(17) 1/29 (4) 0.546 43/112 (38) 53/112 (47) 14/112 (13) 2/112 (2) Adenopathies, N (%) 37/72 (51) 18/26 (69) 0.116 55/98 (56) Splenomegaly, N (%) 30/83 (36) 9/28 (32) 0.701 39/111 (35) Hepatomegaly, N (%) 20/82 (24) 5/28 (18) 0.476 25/110 (23) Mediastinal mass, N (%) 41/84 (49) 8/29 (28) 0.047 49/113 (43) CNS involvement, N (%) 10/83 (12) 4/27 (15) 0.743 14/110 (13) Immunophenotype, N (%) ETP-ALL Pre-T Cortical Mature 8/81 (10) 15/81 (18) 42/81 (52) 16/81 (20) 14/29 (48) 4/29 (14) 5/29 (17) 6/29 (21) <0.001 22/110 (19) 19/110 (16) 47/110 (41) 22/110 (19 Cytogenetics, N (%) 0-2 abn CK ≥3 NE 49/87 (56) 8/87 (9) 30/87 (35) 17/29 (59) 2/29 (7) 10/29 (34) 0.926 66 (57) 10 (9) 40 (34) Response-related features Slow response at day +14, N (%) 26/72 (36) 22/27 (82) <0.001 48/99 (48) Induction cycles to CR, N (%) 1 2 77/87 (89) 10/87 (11) 16/29 (55) 13/29 (45) <0.001 93 (80) 23 (20) CR post Ind-1, N (%) 81/87 (93) 14/29 (48) <0.001 95 (82) CR (Ind-1 + Ind-2), N (%) 82/87 (94) 20/29 (69) <0.001 102 (88) MRD <0.1% at day +35, N (%) 65/76 (86) 9/13 (69) 0.221 74/89 (83) Treatment Chemotherapy, N (%) Allo-SCT, N (%) 54/70 (77) 16/70 (23) 5/12 (42) 7/12 (58) 0.031 59/82 (72) 23/82 (28)
Table 2. Clinical-biological characteristics and response to treatment of T-cell acute lymphoblastic leukemia patients grouped according to their gene mutational profile.
Haematologica | 108 - April 2023 975 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
sequent relapses. Thus, the WOG mutational signature allows to identify at diagnosis those patients who will be resistant and refractory to conventional frontline treatment. The WOG signature was an independent risk factor for OS, together with the end-induction MRD levels (0.1% cut-off) indicating that the two parameters cooperate to confer a poor outcome to T-ALL patients. Thus, we demonstrated that the WOG signature contributes to the improved riskstratification of adult T-ALL patients, confirming the relevance of genetic data in this clinical setting. The lack of co-occurrence observed in the genes included in the WOG signature (Online Supplementary Figure S4) highlights the importance of the contribution of each individual gene in the outcome significance of the WOG signature, although we could not demonstrate its individual contribution in the multivariable analysis except for patients with DNMT3A mutations (data not showed), probably due to the limited number of patients included in each individual gene-group. Here also, the impact of WOG variable was probably underestimated since the MRD threshold employed in the two retrospective trials to discriminate patients with good versus worse response was not sensitive enough. With a more sensitive MRD cut-off, the number of patients with MRDhigh would be higher and therefore the WOG signature could contribute to discriminate an inter-
mediate-risk group (those with MRDhighplus WOG- and MRDlow plus WOG+) within the high-risk group. In addition, other genes that did not reach statistical significance when analyzed individually for OS impact, due to the limited number of mutated patients for the specific gene assessed (i.e., patients with JAK3 and JAK1 mutations), could also impair the selection of a very low-risk group of patients. The assessment of the WOG genetics in the current ALL-PETHEMA trial (ALL19), in which a more sensitive MRD cut-off is employed, will help to corroborate the utility of these genetic marker to stratify adult T-ALL patients within PETHEMA trials.
The WOG signature identified here is defined by the mutational status of genes known to be involved in ALL treatment resistance.13-16 These included activating mutations in the N/KRAS genes (n=13). The two G-proteins (N/KRAS) are involved in the RAF/MEK/ERK signaling pathway48 and have been identified as being responsible for steroid resistance.12,13 Importantly, these mutations are associated with poor prognosis in both pediatric and adult T-ALL.9,49 The WOG signature described here also included MSH2 gene mutations (n=5) that affected the interaction with MSH3 and MSH6 proteins, respectively, and the formation of MSH3-MSH2 and MSH6-MSH2 heterodimers, two components of the post-replicative DNA mismatch repair sys-
*Age and white blood cell (WBC) count were considered as continuous variables. N: number of cases; HR: hazard ratio; CI: confidence interval; OS: overall survival; ETP-ALL: early T-cell precursor acute lymphoblastic leukemia; abn: abnormalities; CK: complex karyotype; CNS: central nervous system; GOG: good-outcome genetics; WOG: worse-outcome genetics; MRD: measurable residual disease.
Disease/patient feature Univariable analyses Multivariable analyses N HR (95% CI) P N HR (95% CI) P Age* 95 1.007 (0.984–1.031) 0.540 - -WBC count* 93 1.001 (0.999–1.003) 0.440 - -CNS involvement No Yes 82 8 Reference 0.598 (0.185–1.937) 0.391 - -ETP-ALL No ETP-ALL ETP-ALL 77 11 Reference 1.687 (0.803–3.544) 0.168 - -Karyotype 0-2 abn. CK ≥3 59 6 Reference 2.623 (0.993–6.928) 0.052 - -PETHEMA treatment protocol ALL-HR-11 ALL-AR-03 69 26 Reference 1.162 (0.628–2.148) 0.633 - -GOG Non-mutated Mutated 77 18 Reference 0.343 (0.120–0.981) 0.046 - -WOG Non-mutated Mutated 81 14 Reference 2.636 (1.363 – 5.095) 0.004 76 13 Reference 2.187 (1.087 –4.400) 0.028 MRD at day +35 <0.1% ≥0.1% 74 15 Reference 3.339 (1.691 – 6.592) 0.001 74 15 Reference 3.040 (1.531 –6.035) 0.001
Haematologica | 108 - April 2023 976 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
Table 3. Prognostic factors for overall survival identified in the univariable and multivariable analyses in the PETHEMA adult Tcell acute lymphoblastic cohort.
tem (MMR).50 Under defective MMR function conditions, blasts may lack the capacity to recognize mismatched DNA pairs, leading to the generation of chemoresistance.14,15 Despite the pathogenic role of mutations in this gene, limited information is available about its clinical relevance, probably due to the low frequency of these mutations in adult T-ALL. Nevertheless, leukemic cells from relapsed pediatric cases are enriched in MSH2 gene mutations, suggesting that relapse in these patients may be due to a drug resistance mechanism driven by MSH2 mutations.17,51 The two other genetic components of WOG are the DNMT3A (n=10) and U2AF1 (n=5) genes. U2AF1 gene mutations have been previously identifi ed in adult and childhood T-ALL32,52 and predict poorer prognosis in de novo acute myeloid leukemia (AML) patients.53 However, as far as we know, no data are available concerning the consequences of this mutation in T-ALL. DMNT3A mutations have previously been identified in older patients and in
immature leukemic subtypes42,54-56 and like other epigenetic regulators, they are thought to be an early event in leukemogenesis, conferring self-renewal properties on uncommitted hematopoietic progenitors, facilitating the subsequent acquisition of secondary mutations.57,58 Importantly, most DNMT3A mutations observed in the blast cells were also present in the non-leukemic cell fraction of polymorphonucleated cells of the same patients at lower or similar VAF. It is of particular interest that the genetic signature that defines CHIP includes mutations in these two genes at very low VAF.45,46 These genes have also been described in AML at higher VAF.59,60 Together, these observations favor an explanation of a DNMT3A mutation-driven clonal hematopoiesis event in a common lymphoid-myeloid early progenitor that, together with other alterations, could determine the transformation into AML or T-ALL. In contrast, we did not find U2AF1 mutations in non-leukemic cells, although we were able to investigate only a few T-ALL cases.
Figure 3. Prognostic stratification of adult T-cell acute lymphoblastic patients according to overall survival defined by the presence of worse-outcome genetic mutations and measurable residual disease status 35 days after starting therapy. (A) Overall survival (OS) according to measurable residual disease (MRD) levels at 4 years (y) showed rates of (95% confidence interval [CI]: 35-63) in patients with MRDlow (<0.1%) and 8% (95% CI: 0-23) for those with MRDhigh (≥0.1%). (B) OS according to worse-outcome genetics (WOG) mutational status at 5 y showed rates of 13% (95% CI: 0-26) in the WOG-mutated patients (WOG+) and 45% (95% CI: 32-58) in non-mutated patients (WOG-). (C) OS according to WOG mutational status and MRD values (d+35) at 5 y was 52% (95% CI: 37-67) for patients with MRDlow (<0.1%) and WOG- (standard-risk-patients), compared with 17% (95% CI: 1-33) for high-risk patients including MRDhigh plus WOG-, MRDlow plus WOG+ and MRDhigh plus WOG+
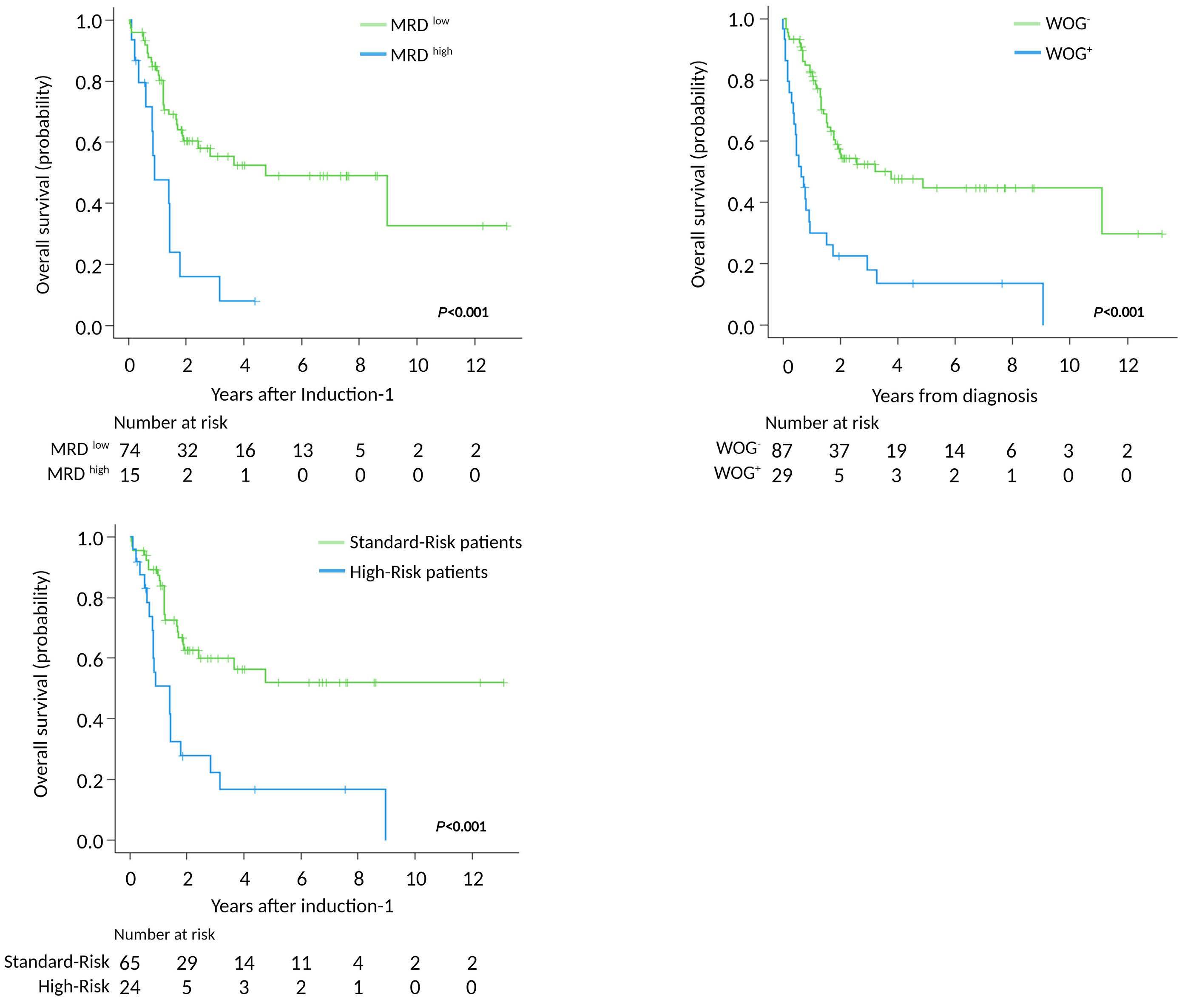
A C B
Haematologica | 108 - April 2023 977 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
ID: identifier; aa: amino acid; VAF: variant allelic frequency; CNA: copy number alteration; ETP-ALL: early T-cell precursor acute lymphoblastic leukaemia. RefSeq ID DNMT3A: NM_022552.
The incidence of NOTCH1 and FBXW7 mutations in our patients was similar to that reported in other adult and pediatric cohorts,40,44,61-63 but we found no association between mutations in NOTCH1 gene and patient outcome. In fact, the clinical impact of NOTCH1/FBXW7 mutations in adult T-ALL is still a matter of debate. Here we have shown that patients with mutations in FBXW7, including or not mutations in NOTCH1, have a better outcome comparing with patients carrying on only mutations in NOTCH1 gene, but this association do not reach statistical significance in the multivariable analysis. The GRAALL group showed that mutations in NOTCH1, together with FBXW7, identified patients with better event-free survival and OS, with an independent predictive value.44,61 They also showed that the combination of low-risk genetics (defined by the presence of mutations in the NOTCH1 signaling pathway [NOTCH1 and FBXW7] and wild-type N/KRAS or PTEN) and MRD negativity, allowed identification of a fraction of adult T-ALL patients with a very good outcome7. However, the same cooperative group also showed that the favorable prognostic impact of NOTCH1/FBXW7 mutations was influenced by the treatment protocol used.61 Other studies in adult T-ALL cohorts have not been able to confirm this benefit,62,64 whereas the large differences in the prevalence of NOTCH1/FBXW7 mutations reported in other series may compromise the clinical impact of these mutations in the studied cohort.65 Compared with the GRAALL trials,7,9 patients from the PETHEMA trials6,22 included in this study are older, and were treated slightly different (i.e., they did not receive cyclophosphamide dur-
ing the induction or late intensification stages of the treatment).
In conclusion, we describe a WOG mutational signature, which identifies older, refractory/resistant high-risk T-ALL patients with a poorer OS due to the suboptimal response to induction therapy. Patients in this group emerge as candidates for novel, personalized frontline therapeutic schedules.
Disclosures
No conflicts of interest to disclose.
Contributions
CG-G performed the experiments and analyzed the data, produced the figures and contributed to the writing process. MM did the statistical analyses. TL performed library preparations; FF-T analyzed the sequencing data. JG-C developed the initial NGSp design. PM, AT, MD-B, RC, JR, LH, SM, JG-C, LZ, TA, FV-L, MT, CG-C, PB, AN, TB, PLdU, M-PQ, PM-S, AG, TG, AC, JC, RF, MAA, MJV and ÁB provided clinical data. N-LB and AB provided support for the project through the AECC T-ALL consortium. RZ and JM created Figure 1C. AO and JMR contributed to the study conceptualization, data analysis and reviewed the manuscript. EG designed the study, reviewed the data, and wrote and reviewed the manuscript. All authors have read and approved the manuscript.
Acknowledgements
We would like to thank Carmen Benet, Maria Paz Garrastazul, Irene García-Cadenas, Gemma Azaceta, Beatriz Soria,
Patient ID Age, years Immunophenotype DNMT3A aa change VAF leukemic cells VAF non-leukemic cells CNA status Functional impact of DNMT3A mutations In silico predictors COSMIC TALL91 64 ETP-ALL p.F827fs 28% 34% 2 Uncertain significance p.R771X 59% 3.6% 3 Uncertain significance Pathogenic TALL95 61 ETP-ALL p.F732V 88% 38% 1 Pathogenic TALL93 78 ETP-ALL p.R882H 43% 3.5% .. Pathogenic Pathogenic TALL50 60 ETP-ALL p.R882H 48% 46% Pathogenic Pathogenic TALL101 48 Cortical p.R882C 50% 21% Pathogenic Pathogenic TALL46 57 ETP-ALL p.R882H 51% 40% Pathogenic Pathogenic TALL8 51 ETP-ALL p.D876E 41% 30% 2 Pathogenic p.I715fs 45% 31% 2 Uncertain significance
Table 4. DNMT3A genetic variants identified in patients associated with the aging cluster in the non-leukemic and leukemic cell.
Haematologica | 108 - April 2023 978 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
Silvia Monsalvo, María Lourdes Amador, Xavier Ortín, Jesús Feliu, Carlos Rodríguez, Irene Romero, María-Paz Martínez, Mª Jesús Peñarrubia and Alberto Gimenez for providing retrospective clinical data for this study. We would also like to thank to Dr. Dr. Yasunobu Nagata and Dr. Brian P. Hobbs for help with the design of the morphological-genetic associations analysis pipeline.
Funding
This project was supported by the AECC (GC16173697BIGA);
References
1. Yi M, Zhou L, Li A, Luo S, Wu K. Global burden and trend of acute lymphoblastic leukemia from 1990 to 2017. Aging. 2020;12(22):22869-22891.
2. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541-1552.
3. Teachey DT, Pui C-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019;20(3):e142-e154.
4. Kimura S, Mullighan CG. Molecular markers in ALL: clinical implications. Best Pract Res Clin Haematol. 2020;33(3):101193.
5. Thomas X, Le Q-H. Prognostic factors in adult acute lymphoblastic leukemia. Hematol Amst Neth. 2003;8(4):233-242.
6. Ribera J-M, Morgades M, Ciudad J, et al. Chemotherapy or allogeneic transplantation in high-risk Philadelphia chromosomenegative adult lymphoblastic leukemia. Blood. 2021;137(14):1879-1894.
7. Beldjord K, Chevret S, Asnafi V, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. 2014;123(24):3739-3749.
8. Lussana F, Intermesoli T, Gianni F, et al. Achieving molecular remission before allogeneic stem cell transplantation in adult patients with Philadelphia chromosome–positive acute lymphoblastic leukemia: impact on relapse and long-term outcome. Biol Blood Marrow Transplant. 2016;22(11):1983-1987.
9. Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al. Toward a NOTCH1/FBXW7/RAS/PTEN–based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a group for research in adult acute lymphoblastic leukemia study. J Clin Oncol. 2013;31(34):4333-4342.
10. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16(8):494-507.
11. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017;129(9):1113-1123.
12. Li Y, Buijs-Gladdines JGCAM, Canté-Barrett K, et al. IL-7 receptor mutations and steroid resistance in pediatric T cell acute lymphoblastic leukemia: a genome sequencing study. PLoS Med. 2016;13(12):e1002200.
13. Ariës IM, van den Dungen RE, Koudijs MJ, et al. Towards personalized therapy in pediatric acute lymphoblastic leukemia: RAS mutations and prednisolone resistance. Haematologica 2015;100(4):e132–e136.
14. Fordham SE, Matheson EC, Scott K, Irving JAE, Allan JM. DNA mismatch repair status affects cellular response to Ara-C and other anti-leukemic nucleoside analogs. Leukemia. 2011;25(6):1046-1049.
15. Diouf B, Cheng Q, Krynetskaia NF, et al. Somatic deletions of genes
ISCIII (PI19/01828 and PI19/01183), co-funded by ERDF/ESF, "A way to make Europe"/"Investing in your future", CERCA/Generalitat de Catalunya SGR 2017 288 (GRC)/ “La Caixa”. C González-Gil was supported by AGAUR grant (ref: 2020 FI_B2 00210).
Data-sharing statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17(10):1298-1303.
16. Fedier A, Schwarz VA, Walt H, Carpini RD, Haller U, Fink D. Resistance to topoisomerase poisons due to loss of DNA mismatch repair. Int J Cancer. 2001;93(4):571-576.
17. Li B, Brady SW, Ma X, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood. 2020;135(1):41-55.
18. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice site SNVs. Hum Mutat. 2016;37(3):235-241.
19. Genescà E, Morgades M, González-Gil C, et al. Adverse prognostic impact of complex karyotype (≥3 cytogenetic alterations) in adult T-cell acute lymphoblastic leukemia (T-ALL). Leuk Res. 2021;109:106612.
20. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
21. Moorman AV, Harrison CJ, Buck GAN, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109(8):3189-3197.
22. Ribera J-M, Oriol A, Morgades M, et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: final results of the PETHEMA ALL-AR-03 trial. J Clin Oncol. 2014;32(15):1595-1604.
23. Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13(10):1203-1210.
24. Gutierrez A, Kentsis A, Sanda T, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. 2011;118(15):4169-4173.
25. Della Gatta G, Palomero T, Perez-Garcia A, et al. Reverse engineering of TLX oncogenic transcriptional networks identifies RUNX1 as tumor suppressor in T-ALL. Nat Med. 2012;18(3):436-440.
26. Ting CN, Olson MC, Barton KP, Leiden JM. Transcription factor GATA-3 is required for development of the T-cell lineage. Nature. 1996;384(6608):474-478.
27. Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208(13):2571-2579.
Haematologica | 108 - April 2023 979
28. Van Vlierberghe P, Palomero T, Khiabanian H, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42(4):338-342.
29. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157-163.
30. Matheson EC, Hall AG. Expression of DNA mismatch repair proteins in acute lymphoblastic leukaemia and normal bone marrow. Adv Exp Med Biol. 1999;457:579-583.
31. De Keersmaecker K, Atak ZK, Li N, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2013;45(2):186-190.
32. Spinella J-F, Cassart P, Richer C, et al. Genomic characterization of pediatric T-cell acute lymphoblastic leukemia reveals novel recurrent driver mutations. Oncotarget. 2016;7(40):65485-65503.
33. von Lintig FC, Huvar I, Law P, Diccianni MB, Yu AL, Boss GR. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin Cancer Res. 2000;6(5):1804-1810.
34. Neumann M, Seehawer M, Schlee C, et al. FAT1 expression and mutations in adult acute lymphoblastic leukemia. Blood Cancer J. 2014;4(6):e224.
35. Morris LGT, Kaufman AM, Gong Y, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. 2013;45(3):253-261.
36. Shochat C, Tal N, Bandapalli OR, et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208(5):901-908.
37. Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43(10):932-939.
38. Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759-1767.
39. Tremblay CS, Brown FC, Collett M, et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia. 2016;30(10):1993-2001.
40. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269-271.
41. Thompson BJ, Buonamici S, Sulis ML, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204(8):1825-1835.
42. Neumann M, Heesch S, Schlee C, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121(23):4749-4752.
43. Bond J, Touzart A, Leprêtre S, et al. DNMT3A mutation is associated with increased age and adverse outcome in adult T-cell acute lymphoblastic leukemia. Haematologica. 2019;104(8):1617-1625.
44. Asnafi V, Buzyn A, Le Noir S, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113(17):3918-3924.
45. Kurosawa S, Iwama A. Aging and leukemic evolution of hematopoietic stem cells under various stress conditions. Inflamm Regen. 2020;40(1):1-10.
46. Buscarlet M, Provost S, Zada YF, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130(6):753-762.
47. Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med.
2018;24(7):1015-1023.
48. Knight T, Irving JAE. Ras/Raf/MEK/ERK pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front Oncol. 2014;4:160.
49. Oshima K, Khiabanian H, Silva-Almeida AC da, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2016;113(40):11306-11311.
50. Edelbrock MA, Kaliyaperumal S, Williams KJ. Structural, molecular and cellular functions of MSH2 and MSH6 during DNA mismatch repair, damage signaling and other noncanonical activities. Mutat Res. 2013;743–744:53-66.
51. Irving JAE. Towards an understanding of the biology and targeted treatment of paediatric relapsed acute lymphoblastic leukaemia. Br J Haematol. 2016;172(5):655-666.
52. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211-1218.
53. Hou H-A, Liu C-Y, Kuo Y-Y, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016;7(8):9084-9101.
54. Roller A, Grossmann V, Bacher U, et al. Landmark analysis of DNMT3A mutations in hematological malignancies. Leukemia. 2013;27(7):1573-1578.
55. Grossmann V, Haferlach C, Weissmann S, et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer. 2013;52(4):410-422.
56. Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, et al. Prognostic relevance of integrated genetic profiling in adult Tcell acute lymphoblastic leukemia. Blood. 2013;122(1):74-82.
57. Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284-299.
58. Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44(1):23-31.
59. Sasaki K, Kanagal-Shamanna R, Montalban-Bravo G, et al. Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the outcomes of patients with newly diagnosed acute myeloid leukemia. Cancer. 2020;126(4):765-774.
60. Saygin C, Hirsch C, Przychodzen B, et al. Mutations in DNMT3A, U2AF1, and EZH2 identify intermediate-risk acute myeloid leukemia patients with poor outcome after CR1. Blood Cancer J. 2018;8(1):1-12.
61. Ben Abdelali R, Asnafi V, Leguay T, et al. Pediatric-inspired intensified therapy of adult T-ALL reveals the favorable outcome of NOTCH1/FBXW7 mutations, but not of low ERG/BAALC expression: a GRAALL study. Blood. 2011;118(19):5099-5107.
62. Baldus CD, Thibaut J, Goekbuget N, et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adult acute T-lymphoblastic leukemia. Haematologica. 2009;94(10):1383-1390.
63. Breit S, Stanulla M, Flohr T, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108(4):1151-1157.
64. Mansour MR, Sulis ML, Duke V, et al. Prognostic Implications of NOTCH1 and FBXW7 Mutations in Adults With T-Cell Acute Lymphoblastic Leukemia Treated on the MRC UKALLXII/ECOG E2993 Protocol. J Clin Oncol. 2009;27(26):4352-4356.
65. Feng J, Li Y, Jia Y, et al. Spectrum of somatic mutations detected by targeted next-generation sequencing and their prognostic significance in adult patients with acute lymphoblastic leukemia. J Hematol Oncol. 2017;10(1):61.
Haematologica | 108 - April 2023 980 ARTICLE - Genomics improves risk stratification in T-ALL C. González-Gil et al.
Hyperactive CREB subpopulations increase during therapy in pediatric B-lineage acute lymphoblastic leukemia
Dino Masic,1 Kayleigh
1Wolfson Childhood Cancer Research Centre, Newcastle University Centre for Cancer, 2Haematology Department, Flow Cytometry Laboratory, Royal Victoria Infirmary, 3Newcastle University Flow Cytometry Core Facility, Newcastle University and 4Innovation, Methodology and Application Research Theme, Newcastle University, Newcastle upon Tyne, UK
Abstract
Correspondence: J. Irving julie.irving@newcastle.ac.uk
Received: June 21, 2022.
Accepted: November 10, 2022.
Early view: November 24, 2022.
https://doi.org/10.3324/haematol.2022.281177
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Persistence of residual disease in acute lymphoblastic leukemia (ALL) during the initial stages of chemotherapy is associated with inferior survival. To better understand clonal evolution and mechanisms of chemoresistance, we used multiparameter mass cytometry, at single-cell resolution, to functionally characterize pediatric B-ALL cells at disease presentation and those persisting during induction therapy. Analysis of ALL cells from presentation samples (n=42) showed that the most abundant phosphosignals were pCREB, pH2AX and pHH3 and we identified JAK-STAT and RAS pathway activation in five of six patients with JAK or RAS genetic aberrations. The clonal composition of ALL was heterogeneous and dynamic during treatment but all viable cell clusters showed pCREB activation. Levels of pCREB in ALL cells were increased or maintained during therapy and high dimensional analysis revealed a subpopulation of ALL cells at presentation that was positive for pCREB/pHH3/pS6 which increased during treatment in some patients, implicating this signaling node in conferring a survival advantage to multi-agent induction therapy. The small molecule CREB inhibitor, 666-15, was shown to reduce CREB transcriptional activity and induce apoptosis in ALL patient-derived xenograft cells of varying cytogenetic subtypes in vitro, both in the presence and absence of stromal support. Together, these data suggest that the cAMP signaling pathway may provide an opportunity for minimal residual disease-directed therapy for many patients at high risk of relapse.
Introduction
Childhood acute lymphoblastic leukemia (ALL) is the most common childhood malignancy and while outcome has improved dramatically over the last 50 years, relapsed ALL remains a major cause of cancer death in children.1,2 There are a number of well-recognized prognostic biomarkers at presentation of ALL including age, peripheral white blood cell count, morphology and key cytogenetic abnormalities.3 However, the most powerful, independent prognostic factor is the response of the leukemia to initial chemotherapy.4 Thus, levels of persisting leukemia cells assessed at 8, 15 or 28 days after the start of induction chemotherapy are highly prognostic. These are monitored initially by morphology, and subsequently by more sensitive methods to evaluate submicroscopic disease, known as minimal residual disease (MRD). Incorporation of residual disease assessment into contemporary trials has enabled risk-directed therapy
and has been fundamental in children receiving personalized, optimal therapy.5,6
Genetic analyses of paired ALL samples at presentation and relapse have revealed a number of recurrent pathways implicated in relapse, including RAS, JAK-STAT, cell cycle and B-cell development, as well as genes involved in epigenetic modification.7 Our own data, together with data from others, also implicate cell maturation as a resistance mechanism.8,9 Genomic analyses have revealed extensive clonal diversity and, in most cases, leukemic cells at relapse are related to a major or minor clone of cells found at presentation that have survived therapy and acquired additional mutations to give rise to the relapse.1016 This selection of mutated clones has been noted in the early stages of treatment, when their proportion relative to the total leukemic burden increases during the selective pressure of multi-agent induction therapy.14,17,18 Thus, the genotype, phenotype and therapeutic vulnerabilities of the leukemic clone persisting after induction chemo-
Fee,2 Hayden L. Bell,1 Marian Case,1 Gabby Witherington,1 Sophie Lansbury,1 Juan Ojeda-Garcia,3,4 David McDonald,3,4 Claire Schwab,1 Frederik W. van Delft,1 Andrew Filby3,4 and Julie Anne Elizabeth Irving1
Haematologica | 108 - April 2023 981 ARTICLE - Acute Lymphoblastic Leukemia
therapy may be very different from those at presentation. Drugs targeting MRD and mechanisms of chemoresistance may avert relapse.
MRD is routinely quantified by two different methodologies: molecular analyses of antigen receptor gene rearrangements and flow cytometry of aberrant immunophenotypes.19 Flow MRD relies fundamentally on the characterization of a leukemia-associated immunophenotype (LAIP) at presentation, an antibody combination that discriminates leukemic cells from normal lymphocyte progenitors and can thus be used for ‘on treatment’ samples to discriminate and quantify ALL cells. An advantage of cytometric methods is that, as cell-based assays, they can provide information beyond that of just MRD quantitation, including the mechanism behind the evasion of chemotherapy-induced killing and the presence of therapeutic targets. In this study, we used single-cell, high-dimensional mass cytometry to functionally characterize pediatric B-ALL cells both at presentation and persisting during therapy. We demonstrated the presence of activated cAMP response element-binding protein (CREB) across a broad spectrum of cytogenetic groups at presentation and found minor subpopulations with hyperactive CREB at presentation that appeared to have a selective advantage during induction therapy. We also validated CREB as a therapeutic target in ALL cells using a specific inhibitor of CREB transcriptional function. CREB and its signaling pathway may provide an opportunity for MRD-directed therapy.
Methods
Clinical samples
Bone marrow samples from children with B-lineage ALL were accessed through the Newcastle Haematology Biobank, after appropriate consent (reference numbers 2002/111 and 07/H0906). All patients were registered on the ALL2003 or the UKALL2011 trials which used an induction regimen consisting of three or four drugs, depending on National Cancer Institute risk factors. MRD was assessed using a standardized flow cytometry method that was adapted from four to seven or eight colors.20 The clinical details of the patients included in the study are listed in Online Supplementary Tables S1 and S2 and a CONSORT diagram is shown in Online Supplementary Figure S1
Cell lines and patient-derived xenograft cells
ALL cell lines were obtained from the European Collection of Authenticated Cell Cultures (ECACC), maintained in RPMI-1640 (Sigma-Aldrich, Dorset, UK) supplemented with 10% fetal bovine serum (Gibco, Rugby, UK), and incubated at 37°C in a 5% CO2 atmosphere. Patient-derived xenograft (PDX) cells were originally created by intrafemoral injection of presentation primary bone marrow samples into
NOD SCID γ null mice, as described previously.17 Clinical details of these grafts are also included in Online Supplementary Table S1.
Western blotting
Cells were washed in phosphate-buffered saline and proteins were extracted using PhosphoSafe extraction reagent (Merck, Nottingham, UK) supplemented with protease inhibitors (Roche, Hertfordshire, UK). Western blotting was carried out using a standard methodology with antibodies for pCREB (pS133), CREB, ERK2 (Santa Cruz, Dallas, TX, USA), p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), pSTAT5 (pT694), STAT5 (Cell Signaling Technology, Danvers, MA, USA) and α-tubulin (Sigma-Aldrich, St. Louis, MO, USA) which served as a loading control. Densitometry was carried out using AIDA image analysis software (Raytest, Straubenhardt, Germany).
Pharmacodynamic assays
Externalization of annexin V (Abcam, Cambridge, UK) was assessed by flow cytometry on a FACSCalibur (BD Biosciences, New Jersey, NJ, USA), fitted with a 488 nm laser. The CREB pathway was stimulated by dosing ALL cell lines with 50 mM forskolin and 100 mM IBMX diluted in dimethylsulfoxide for 30 minutes prior to treatment with 666-15. The mRNA levels of CREB gene targets that we had previously identified in ALL cells21 were assessed by quantitative real-time polymerase chain reaction (RQ-PCR) analysis. We included primer probe sets (Invitrogen, Carlsbad, CA, USA) for CXCR4 and MKNK2, with TBP used as a housekeeping control, as described previously.21
Statistical analyses
All statistical analyses were performed using Graphpad Prism. A P value of less than 0.05 was considered statistically significant. Additional methods are provided in the Online Supplementary Material.
Results
Mass cytometry analyses of presentation B-acute lymphoblastic leukemia samples revealed prominent pCREB/ATF1 signaling and identified JAK-STAT and RAS pathway activated leukemia
Mass cytometric analyses of B-lineage ALL at presentation (n=42) was performed; live, singlet, non-apoptotic ALL cells were gated by their specific LAIP and mature B cells were identified by the immunophenotype CD34–/CD10–/ CD22+/CD45+. The normalized mean mass intensity (MMI) of all phospho-antibodies in the ALL cells, relative to mature B cells, is shown in Figure 1A. The most prominent phospho-signals were pCREB/pATF1 [S133] (median, 23.95;
Haematologica | 108 - April 2023 982 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
range, -37.52 to 141.2), pH2AX [S139] (median, 7.53; range, -10.81 to 79.24) which is a marker of double-strand breaks, and pHH3 [Ser28] (median, -2.49; range, -107.1 to 71.5) which is activated in cells undergoing mitosis. For ALL cases at presentation for which there was sufficient stored material, we performed western blot analyses to validate our mass cytometry findings. Western blotting showed high expression of CREB in all ALL cells and confirmed the variable activation of both CREB and its close family member, ATF1 (activating transcription fac-
tor 1) (Figure 1B). Western blot analyses for pSTAT5 [Y694] showed that three of seven samples were positive and these had the highest MMI values (Figure 2A, Online Supplementary Figure S2). All three positive samples were Bother ALL, one with a PAX5-JAK2 and another with an IGH-CRLF2 translocation. With regard to pERK, three of seven samples were positive by western blot analyses and two of the positive samples had the highest pERK MMI in the group. These two samples were also B-other ALL, one of which had a known KRAS mutation.
Figure 1. pCREB/pATF1 [S133] is heterogeneously expressed in acute lymphoblastic leukemia at presentation. (A) Box and whisker plot of normalized phospho-signals showing median, upper and lower quartiles and range in acute lymphoblastic leukemia (ALL) samples at presentation normalized to the signals of mature B cells, as detected by mass cytometry. (B) Western blot analysis of ALL lysates for pCREB/ATF1 [S133], CREB and α-tubulin in a representative set of presentation ALL samples that were typical of the cohort.
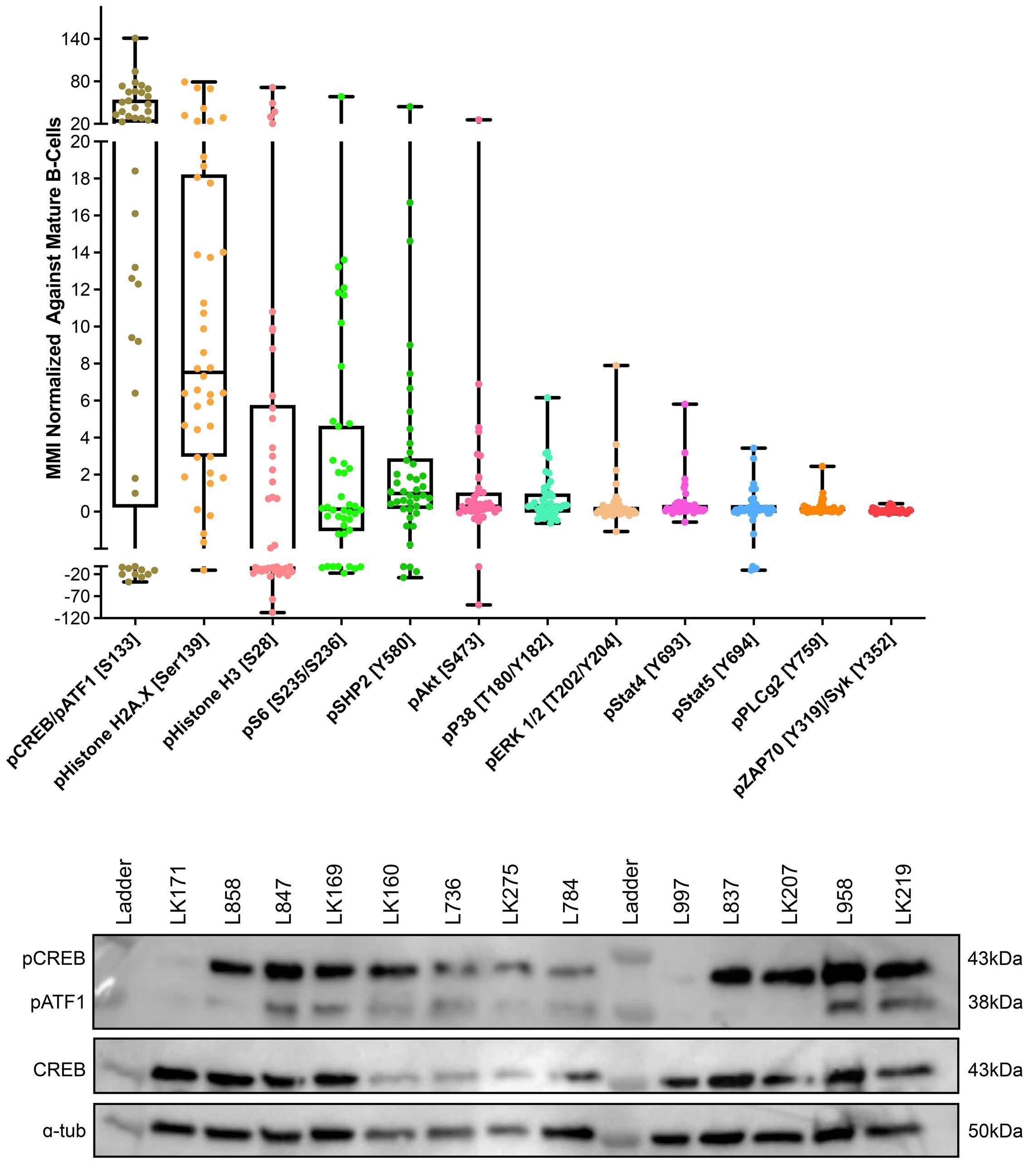
A B Haematologica | 108 - April 2023 983 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
When the cases of ALL were grouped by cytogenetic risk, levels of pSTAT5 MMI were lower in the good-risk cytogenetic group compared to the intermediate- and poorrisk groups, with means of -2.24 versus 0.48 versus 0.36, respectively (P<0.005) (Figures 2B). There was also a trend for higher pHH3 [Ser28] and pCREB/pATF1 [S133] levels with increasing cytogenetic risk, but the differences did not achieve statistical significance (P=0.067 and P=0.29, respectively) (Figure 2C, D).
Residual disease cells show maturation and increased phospho-signaling in pHH3 and pCREB MMI for antigens were again normalized to mature B cells within each sample and values compared between presentation samples (n=42) and MRD samples (n=15 in total; n=5 at day 8, n=8 at day 28 and n=2 at later time points), identified by sequential gating (Table 1A, Online Supple-
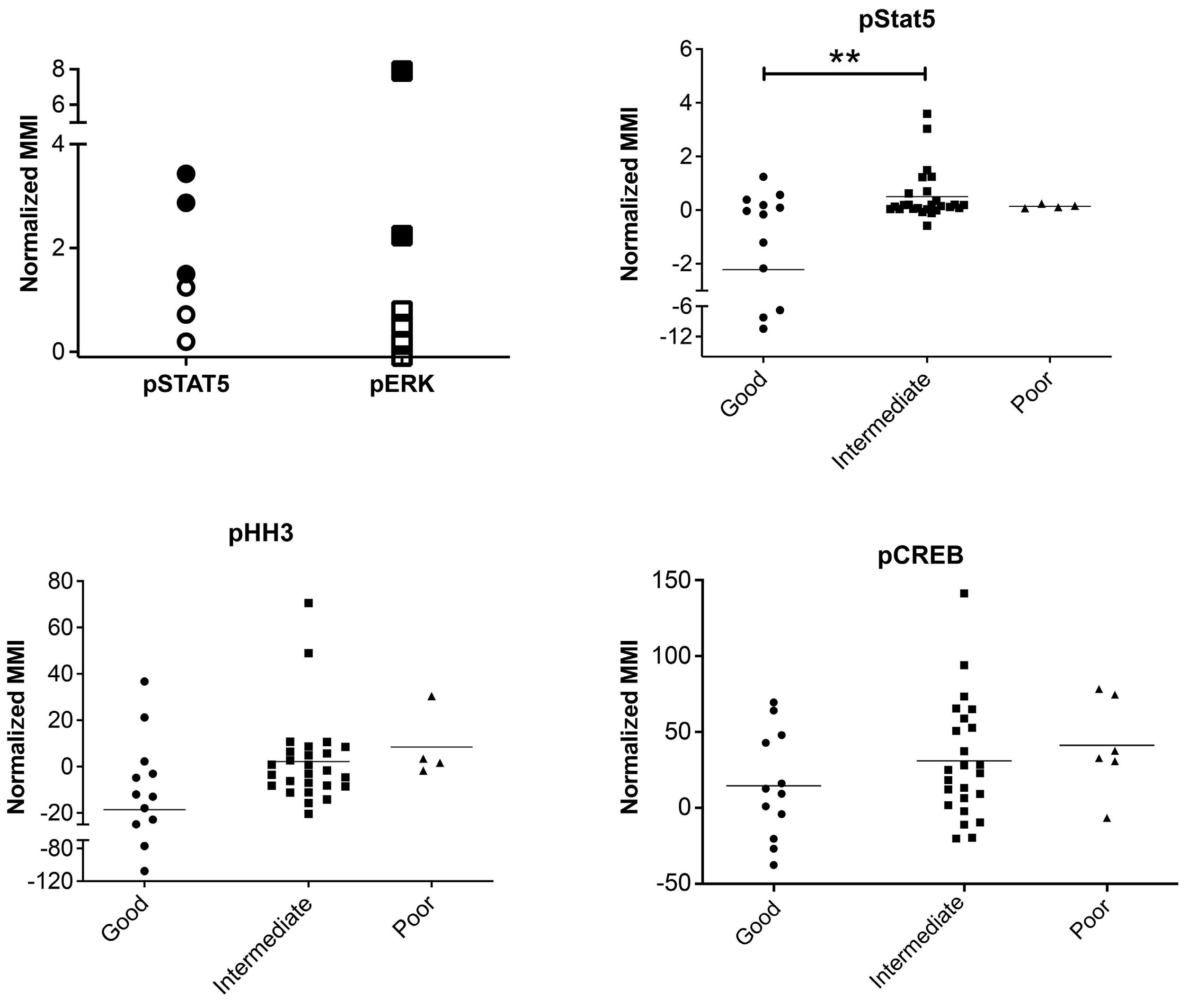
mentary Figure S3). MRD levels determined by flow or mass cytometry were highly concordant in this cohort and a pilot cohort (Online Supplementary Figure S4). Compared to presentation ALL cells, MRD showed a significant increase in the expression of the cell surface antigens, CD45 (P<0.01) and CD22 (P<0.05), and a trend to an increase in CD19 (P=0.078), consistent with maturation as previously reported.8 There was also a highly significant increase in the level of pHH3 (P<0.001), from a MMI of3.14 in presentation ALL cells to 62.48 in MRD cells. pCREB levels increased from 26.70 to 69.85 (P<0.01). There were also more modest increases in pp38, pSTAT5 and pZAP70 and a decrease in pSHP2 (P<0.05). In paired presentation and MRD samples (n=10), the same trends in antigen expression were observed but only the increase in pHH3 remained statistically significant (P <0.05) (Table 1B). A decrease of CD10 levels in MRD cells also gained signifi-
Figure 2. Phospho-signals determined by mass cytometry identify JAK-STAT and RAS pathway activation and a correlation of pSTAT5 activation with cytogenetic risk. (A) Normalized mean mass intensity (MMI) for pSTAT5 and pERK in presentation samples of acute lymphoblastic leukemia (ALL) with pathway activation assessed by both mass cytometry and western blot analysis. Solid black shapes denote ALL samples that are positive by western blot analysis. (B-D) Normalized MMI in good-, intermediate- and poor-risk cytogenetic groups for pSTAT5 (B), pHH3 (C) and pCREB (D). **P<0.01.
A C B D Haematologica | 108 - April 2023 984 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
cance (P<0.05) There was no difference in MMI values in mature B cells in samples taken at presentation or while on treatment, validating their reliability as an internal control (Online Supplementary Table S3).
The clonal composition of acute lymphoblastic leukemia is dynamic during therapy but all clusters have activated pCREB and high pCREB subpopulations in minimal residual disease are often enriched during induction therapy
To investigate the clonal composition of ALL cells during therapy, we used FlowSOM as part of the R Studio CyTOF workflow package to perform cluster analyses of ALL cells in paired presentation and MRD samples using only phospho-signals and identified 20 unique ALL cell clusters. Most ALL were complex and heterogeneous, showing multiple different clusters at both presentation and in MRD and clonal composition was dynamic during therapy (Figure 3). Despite the varying cytogenetics of the cohort, eight clusters made up more than 94% of the total cluster composition (Figure 4A). All clusters showed expression of pCREB, except clusters 6 and 7 which had very low ex-
pression of pHH3 and high levels of pH2AX and were, therefore, likely to be damaged cells that had not yet expressed the apoptotic markers that would have ensured their being gated out from the analyses. One cluster, cluster 10, was significantly more prevalent in MRD cells than in presentation, drug-naïve ALL cells (P<0.05) and was characterized by high levels of pCREB, pHH3 and pS6 (Figure 4B, C). We used the dimension reduction algorithm, Uniform Manifold Approximation and Projection (UMAP) to compare ALL cell clusters at presentation and at MRD. UMAP plots displayed one dominant island comprising the majority of generated clusters and showed an area populated only by presentation ALL at the top left of the plot, housing cluster 11, and two areas at the bottom populated predominantly by MRD (clusters 10, 15 and 16) but most areas having both (both clusters 3, 4 and 14) (Figure 5A, B). Two additional minor islands were generated: one was characterized by pSHP2 expression and comprised clusters 1, 2 and 8, the other expressed pH2AX and comprised clusters 6, 7, 9, and 17 (data not shown). Visualization of pCREB confirmed its ubiquitous ex-
The mean mass intensity (MMI), its range and the statistical significance of differences between values for pre-treatment (presentation) samples of acute lymphoblastic leukemia (ALL) cells and those during treatment (with minimal residual disease, MRD). (A) An unpaired, twotailed, equal variance t test was performed on presentation versus MRD ALL MMI signals. (B) A paired, two-tailed, equal variance t test was performed on presentation versus MRD ALL MMI signals. Statistically significant differences are shown in bold. *P<0.05, **P<0.01 and ***P<0.001.
A Antigen Presentation MRD P value MMI Range MMI Range pp38 0.74 6.78 2.73 25.90 0.034* pAKT -0.77 115.35 1.20 21.66 0.61 pCREB 26.70 178.72 69.85 281.80 0.004** pERK 0.45 8.15 0.38 2.33 0.85 pHH3 -3.14 178.60 62.48 246.49 0.00*** pPLCg2 0.22 2.50 0.21 0.81 0.89 pS6 2.71 76.02 4.93 40.27 0.49 pSHP2 2.69 58.00 -2.94 30.44 0.022* pSTAT4 0.49 5.91 1.02 8.23 0.21 pSTAT5 -0.32 14.10 1.65 12.60 0.019* pZAP70 0.07 0.54 0.38 3.12 0.015* pH2AX 14.66 90.05 6.46 60.87 0.16 CD10 49.47 183.61 33.47 119.83 0.21 CD45 -237.34 520.70 -101.02 406.49 0.003** CD38 74.48 295.45 47.89 277.39 0.24 CD22 -110.62 370.10 -55.33 199.50 0.013* CD123 21.02 144.11 35.43 158.89 0.20 CD34 25.19 141.89 24.95 78.19 0.98 CD19 -73.96 446.00 -5.08 614.66 0.078 CD58 16.63 98.51 21.30 66.51 0.43 B Antigen Presentation MRD P value MMI Range MMI Range pp38 0.90 6.78 1.93 25.90 0.57 pAKT 1.03 4.83 1.30 21.66 0.87 pCREB 42.40 114.30 67.11 253.70 0.30 pERK 1.39 8.10 0.30 2.33 0.14 pHH3 -2.70 54.80 56.38 243.10 0.014* pPLCg2 0.22 0.77 0.16 0.49 0.40 pS6 1.44 10.21 5.01 40.27 0.33 pSHP2 1.53 28.26 -2.58 35.22 0.22 pSTAT4 1.15 5.91 1.00 8.43 0.86 pSTAT5 -0.01 14.10 1.50 12.60 0.32 pZAP70 0.13 0.53 0.37 3.15 0.38 pH2AX 5.85 30.87 2.08 23.27 0.21 CD10 54.12 110.14 28.22 61.40 0.042* CD45 -171.36 236.40 -87.60 406.49 0.08 CD38 52.91 135.83 56.27 277.39 0.90 CD22 -83.23 135.86 -46.85 199.50 0.09 CD123 54.27 135.26 40.64 155.05 0.50 CD34 25.17 50.88 22.11 47.89 0.65 CD19 -21.20 196.20 20.63 609.95 0.41 CD58 16.15 45.39 22.11 66.51 0.42
Table 1. Mean mass intensity of acute lymphoblastic leukemia cells, normalized to mature B cells, in (A) unpaired and (B) paired samples taken at presentation and during treatment.
Haematologica | 108 - April 2023 985 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
pression across the UMAP in both presentation and MRD ALL cells, while pHH3 was more defined and concentrated in the MRD samples (Figure 5C, D). The MRD-enriched areas housed clusters 10, 15 and 16, which are characterized by high pCREB and pHH3. In samples for which there were sufficient MRD cells, individual UMAP showed that in five of seven patients, ALL cells with the highest pCREB
levels were enriched during therapy, with pCREB levels often increasing further (Figure 6).
To investigate the effect of drugs on pCREB activation, we treated PreB 697 cells with the half maximal inhibitory concentration (IC50) of the induction drugs, dexamethasone (67 nM) and vincristine (8.9 nM) on the CREB gene targets, CCRX4 and MKNK2 at various time-points. We
Figure 3. The clonal composition of acute lymphoblastic leukemia cells is complex and dynamic during therapy. Stacked bar plots show the percentages of clusters within acute lymphoblastic leukemia cells at presentation and at various time-points during treatment.
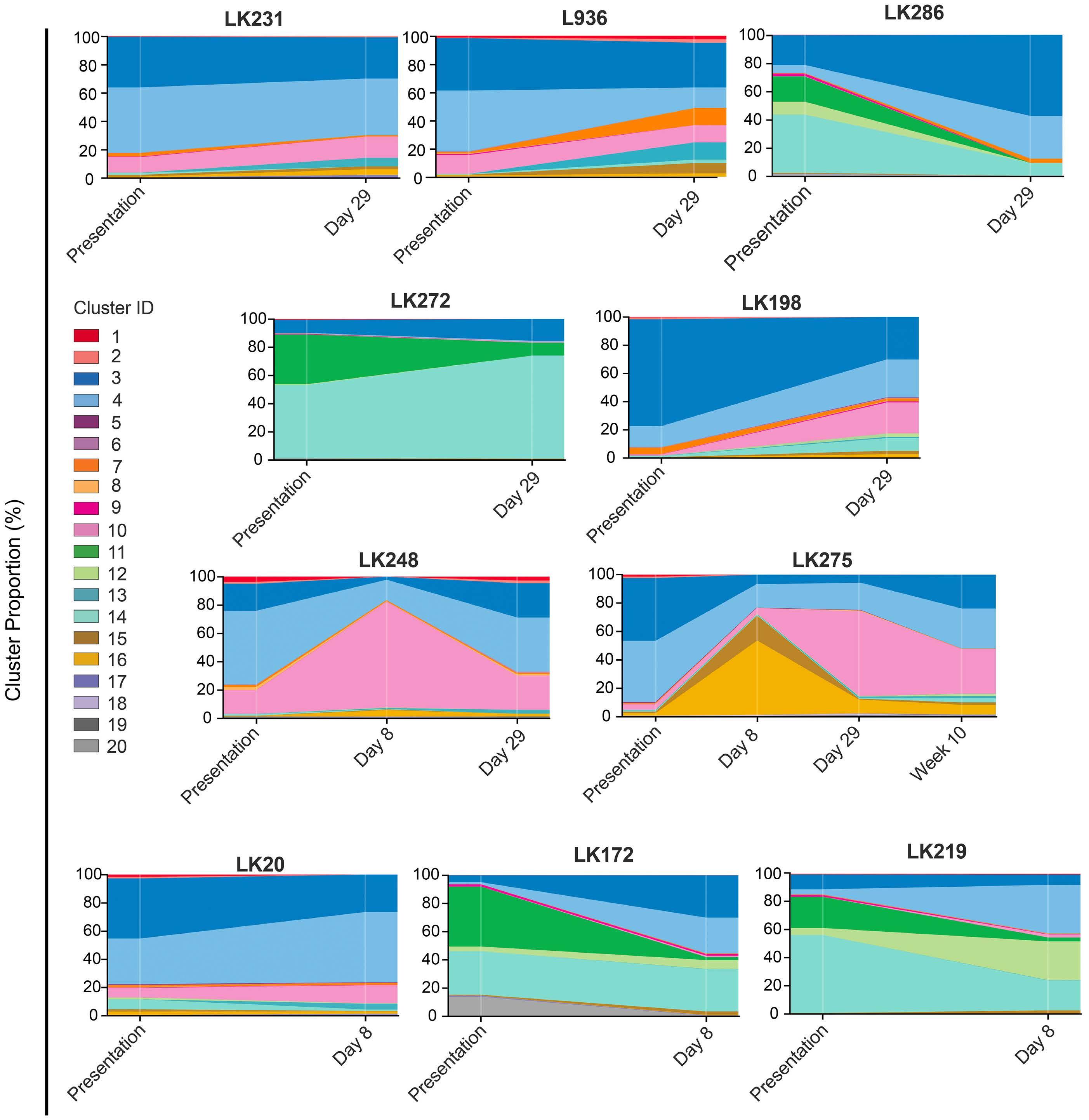
Haematologica | 108 - April 2023 986 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
documented a modest induction of CCRX4 mRNA levels at the 3-hour time-point after dexamethasone dosing (P<0.01) (Online Supplementary Figure S5).
CREB inhibitors are cytotoxic in acute lymphoblastic leukemia
Since cluster analyses showed pCREB in all clusters of ALL cells, and identi fi ed a signaling node involving pCREB/pHH3/pS6 that is enriched in MRD, we sought to evaluate pCREB as a therapeutic target. We therefore dosed ALL cells with varying concentrations of 666-15, a potent, selective CREB inhibitor, and assessed cell viability relative to that of cells treated with the vehicle control. ALL cell lines (n=3) and PDX cells (n=3) grown in suspension culture were assessed using a metabolic readout and primary (n=10) and PDX cells (n=3), supported on a layer of mesenchymal stromal cells, were analyzed using high throughput imaging microscopy
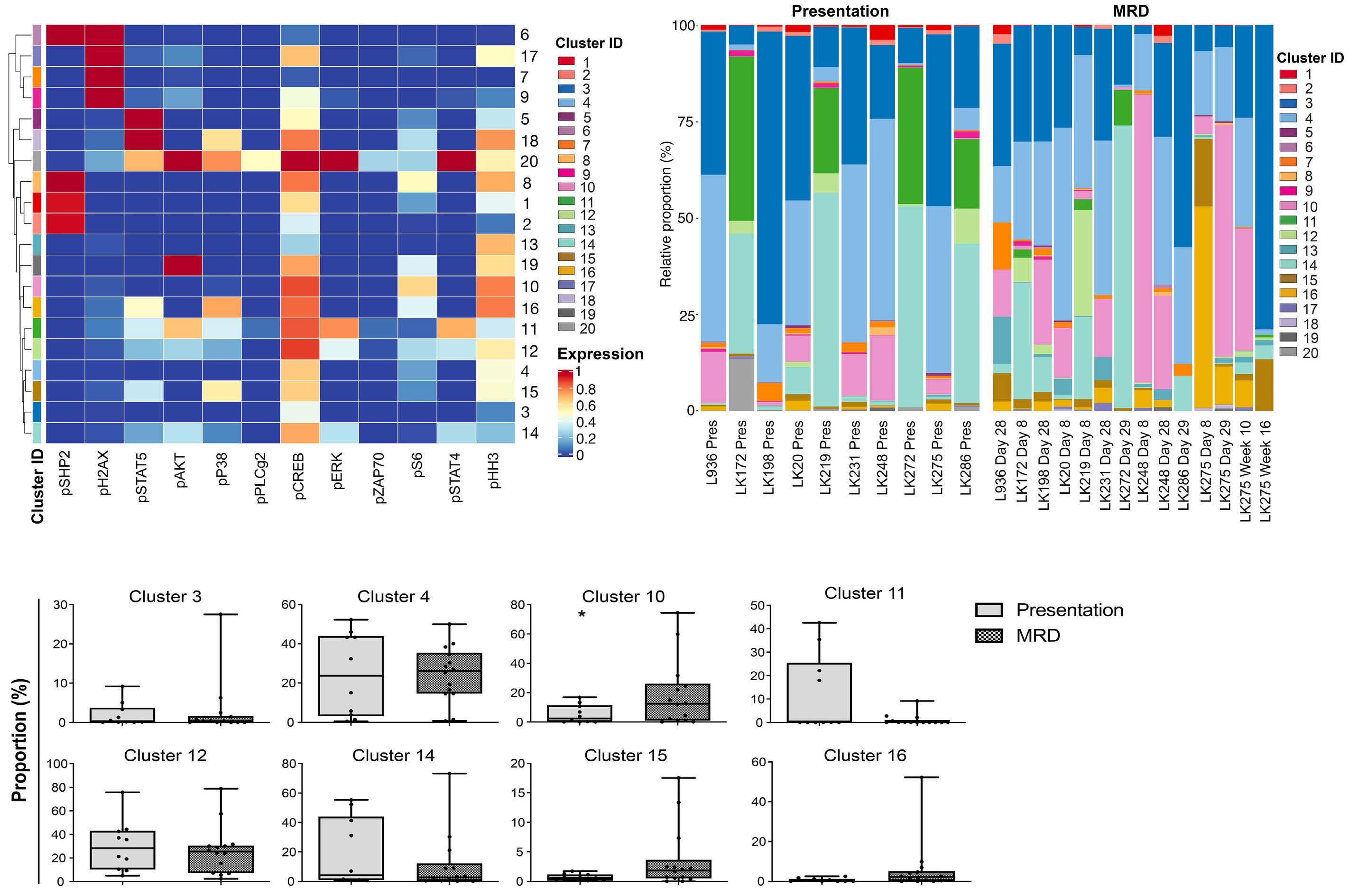
which yielded absolute cell numbers of both ALL blasts and mesenchymal stromal cells. IC50 values for cell lines ranged from 1.7 to 2.7 mM (mean, 2.4 mM), while those for primary/PDX cells ranged from 0.45 to 7 m M (mean, 2.6 mM) (Figure 7A, B). There was no difference in IC50 values of the three PDX samples grown in both suspension and on mesenchymal stromal support (P>0.75). There was no apparent correlation between levels of pCREB as assessed by western blot analysis and IC50 values (n=8, data not shown). Treating ALL cells with IC50 values of 666-15 was associated with induction of apoptosis as determined by phosphatidylserine externalization, with this effect being particularly marked in the PDX cells (P<0.05, paired t test) (Figure 7C). Pharmacodynamic experiments showed significant inhibition of CXCR4 (n=4) (P=0.013) and MKNK2 (n=4) ( P =0.033) gene expression (Figure 7D, E), confirming on- target inhibition of CREB activity at the IC50 values for 666-15.
Figure 4. Cluster analysis revealed that phosphorylation of acute lymphoblastic leukemia cells at presentation (drug-naïve) and during treatment (with minimal residual disease) is complex and heterogeneous with all clusters having activated CREB and a cluster characterized by high levels of pCREB, pHH3 and pS6 having an apparent selective advantage during induction therapy. (A) Heatmap showing expression of target markers within generated clusters, with marker expression intensity displayed from low (blue) to high (red). (B) Bar chart showing whole population cluster composition within patients, separated into presentation and minimal residual disease (MRD) samples. (C) Box plots comparing the incidence of the top eight expressed clusters in the cohort of patients, comparing proportions of presentation and MRD samples of acute lymphoblastic leukemia (ALL) cells in the various clusters. *P<0.05.
A C B Haematologica | 108 - April 2023 987 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
Discussion
The extensive panel of antibodies used in this mass cytometric analysis adds a new diagnostic, functional dimension for ALL. Normal B cells and ALL cells can be identified prior to and during therapy. Functional parameters such as proliferation, apoptosis, pathway activation and quantification of key antigens were assessed in ALL cells and compared to those of normal B cells within the same sample. Thus in one assay, therapeutically relevant antibody targets, including CD19, CD22 and CD38 and signaling pathways, such as the JAK-STAT and the RAS pathways,
can be assessed at the single-cell level. Importantly, this data-rich assay can be performed in samples with high or low leukemic burden. Thus for children with a poor response to induction or reinduction chemotherapy and at high risk of relapse, mass cytometry analysis could evaluate a range of predictive biomarkers to select the optimal targeted therapy directed at residual disease.
In our panel of presentation samples, levels of phosphorylated STAT5 were higher in the intermediate cytogenetic risk group than in the good-risk group, which is likely due to the high incidence of JAK-STAT genetic aberrations reported in Philadelphia chromosome-like and B-other sub-
Figure 5. Uniform manifold approximation and projection analyses showing enrichment of clusters high in pCREB and pHH3 in minimal residual disease samples. Uniform manifold approximation and projection (UMAP) visualization of all analyzed cells shows variation between presentation and minimal residual disease (MRD) samples. (A) The distribution of presentation and MRD blasts shows a focus of MRD blasts to the south of the figure. (B) UMAP analysis showing the distribution of generated clusters in presentation and MRD blasts, with enrichment of clusters 10, 15, and 16 observed in the MRD blasts. (C) UMAP analysis showing pCREB levels in presentation and MRD blasts, with greater expression observed in MRD-enriched areas. (D) UMAP analysis showing the distribution of pHH3 in presentation and MRD blasts with a focus of pHH3 expression observed in MRD cells.
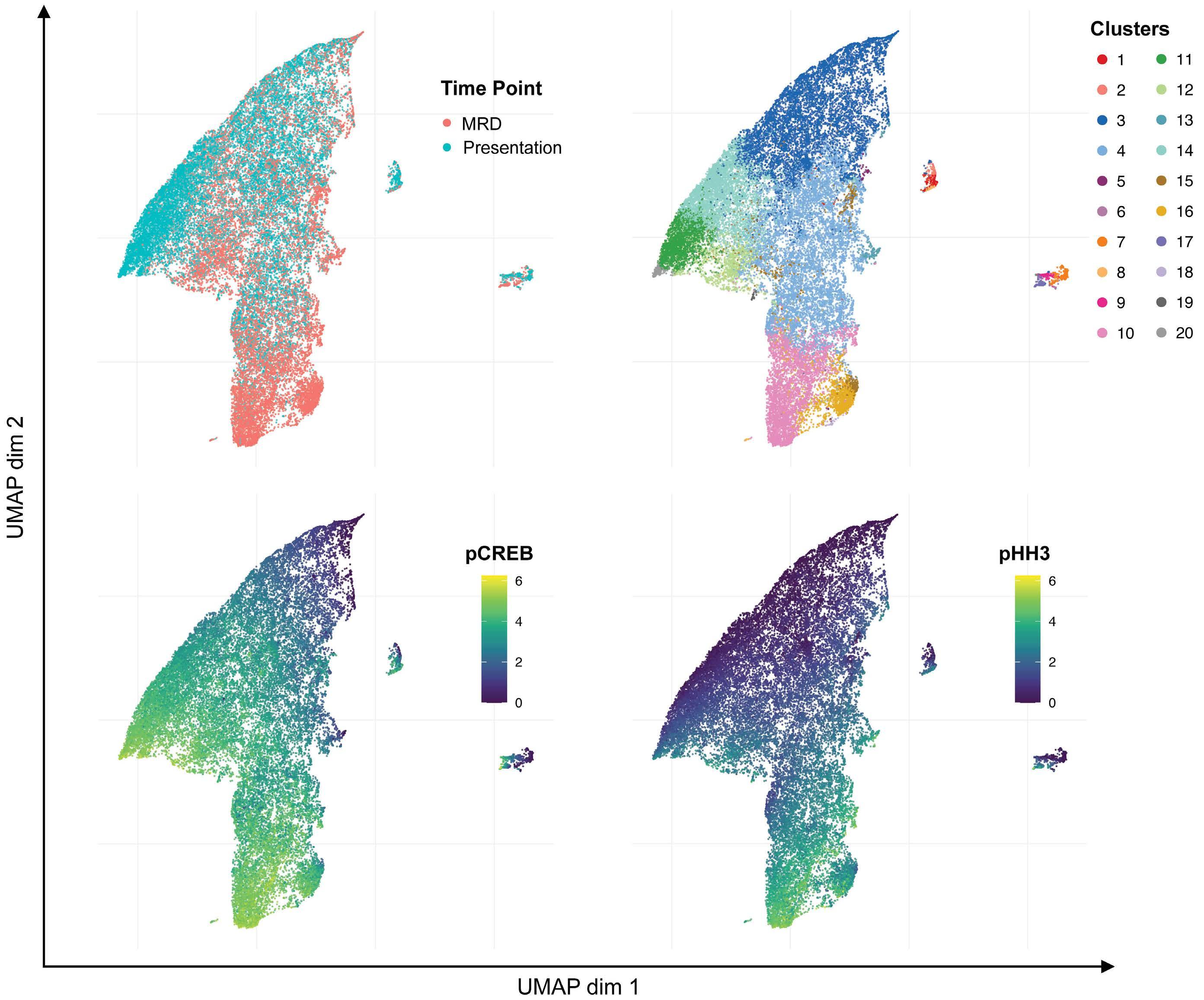
A B C D Haematologica | 108 - April 2023 988 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
Figure 6. Uniform manifold approximation and projection analysis of individual patients show selection of high pCREB cells during induction therapy. The same uniform manifold approximation and projection (UMAP) as in Figure 5 but with visualization of pCREB levels in presentation and minimal residual disease (MRD) blasts in individual patients.
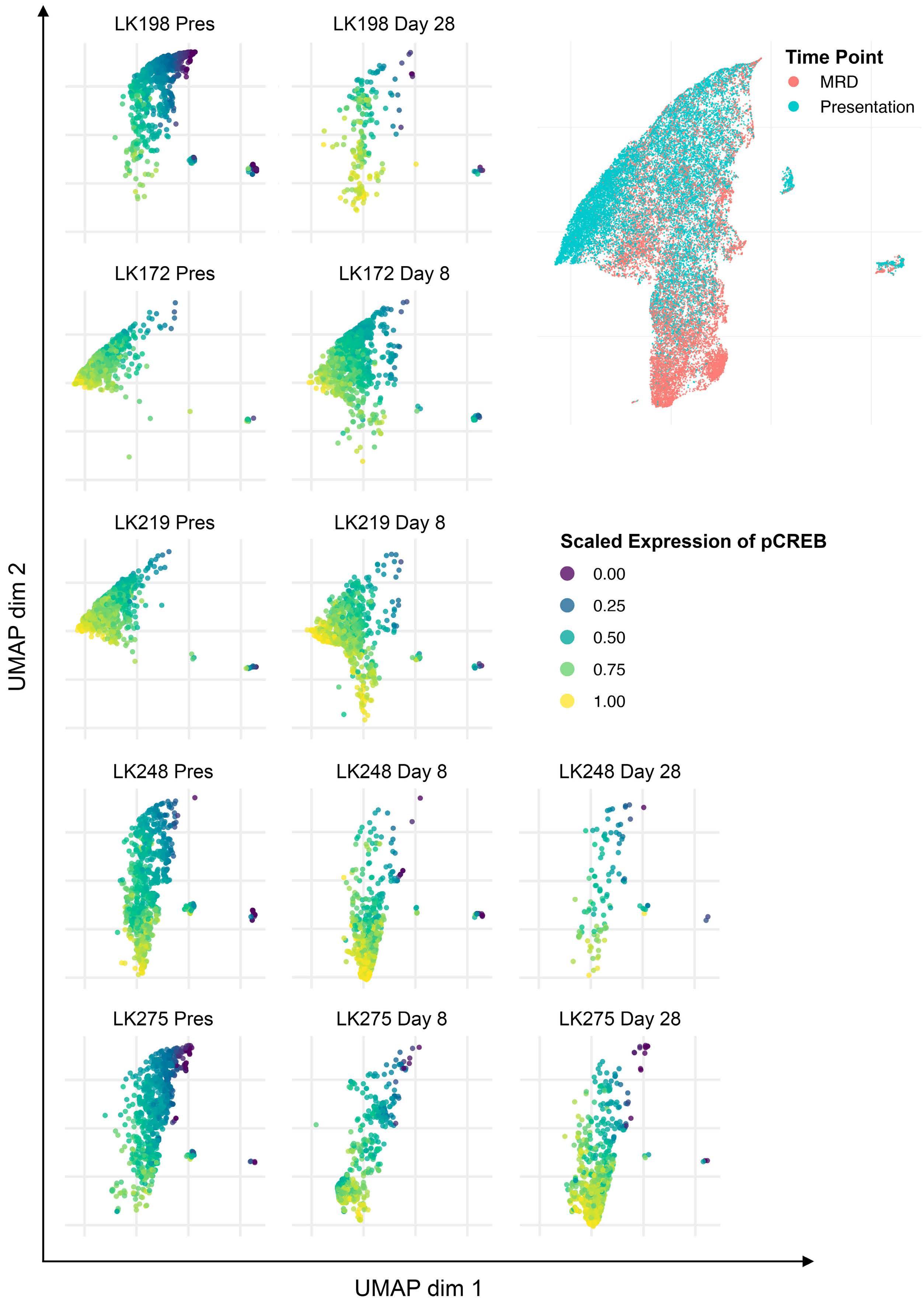
Haematologica | 108 - April 2023 989 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
groups of ALL.22 The high pSTAT5 and pERK levels were confirmed by western blotting and were often attributable to genetic aberrations known to activate these pathways and may serve as predictive biomarkers for sensitivity to JAK and MEK inhibitors.17,23 Our data also confirmed the more mature immunophenotype of MRD cells compared to that of cells at presentation,8,9 which suggests that anti-CD22 and anti-CD19 therapies such as inotuzumab and chimeric antigen receptor-modified T cells may be optimal at the end of induction when expression of antigens associated with B-cell maturation may be higher. We also identified a tendency for the levels of phosphorylated histone H3 (pHH3), a typical marker of mitotic cells, to be lower in good-risk cytogenetic ALL than in intermediate- and poor-risk groups. Phosphorylation of histone H3 at either Ser28 or Ser10 is a well-recognized prognostic biomarker in many cancer types but has not
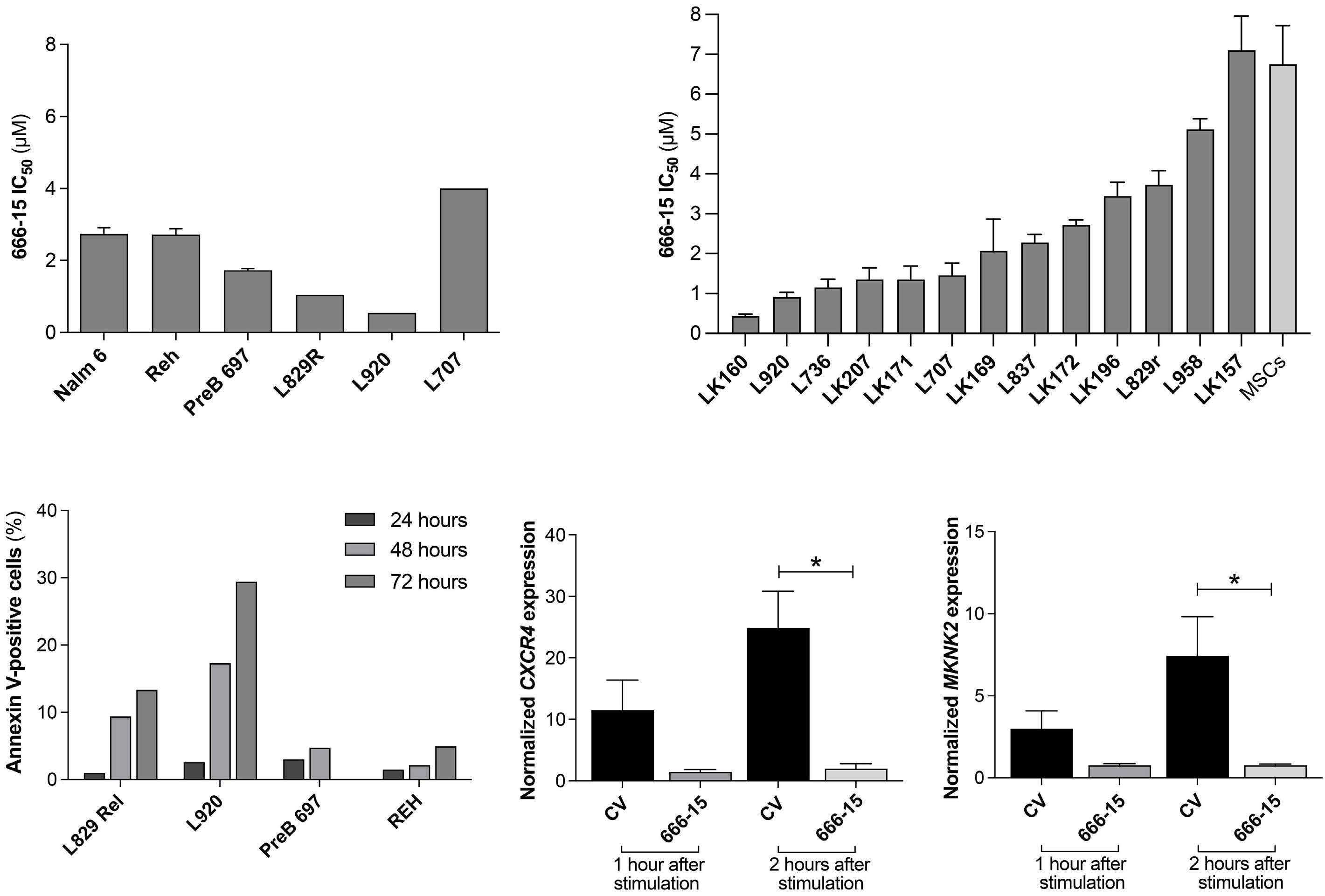
been explored in ALL.24 Our data suggest that pHH3 may have a role in prognostic classification in ALL.
A major novel finding stemming from our data is the demonstration of high expression of activated CREB as a common feature of ALL, its increase in MRD cells and the demonstration that this increase is commonly due to hyperactive pCREB/pHH3 subpopulations increasing during induction treatment and persisting in MRD. This demonstrates a possible selective advantage of ALL clones that have high pCREB/pHH3/pS6 signaling under chemotherapeutic pressure. Further investigations will be needed to decipher whether this signaling node represents cells in G2M or cells in interphase that have a small fraction of nucleosomes phosphorylated at Ser28 of HH3, related to transcriptional activities.25 Other studies have suggested that MRD cells are in fact dormant.26 Interestingly, our CREB data are supported by a small focused study in
Figure 7. The CREB inhibitor, 666-15, is cytotoxic in acute lymphoblastic leukemia cells. (A) The half maximal inhibitory concentration (IC50) of 666-15 for acute lymphoblastic leukemia (ALL) cell lines and patient-derived xenograft (PDX) ALL cells determined using an Alamar blue assay. The mean and standard error of mean (SEM) of three independent replicate experiments are shown for the cells lines. PDX are technical triplicates. (B) IC50 of 666-15 for primary and PDX ALL cells using mesenchymal stromal cell support. (C) Histogram showing the percentages of annexin V-positive cells after exposure to the specific IC50 of 666-15 for two PDX and two ALL cell lines after 24, 48 and 72 hours of incubation, normalized to that for the vehicle control. (D, E) Histogram of CXCR4 (n=4) (D) and MKNK2 (n=4) (E) gene expression in PreB697 cells after stimulation with forskolin and IBMX and subsequent dosing with the IC50 (1.7 mM) of 666-15 or control vehicle (CV) for 1 and 2 hours. The mean and SEM are shown. *P<0.05.
A B C D E Haematologica | 108 - April 2023 990 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
CRLF2-positive ALL which also showed activation of pCREB at presentation, increased activation in MRD cells and a strong connection between pCREB and pS6.27 Our study is the first to demonstrate this finding in a range of high-risk subtypes of ALL.
CREB and ATF1, along with cAMP response element modulator (CREM), are transcription factors of the basic leucine zipper superfamily which regulate gene expression through the activation of cAMP-dependent or -independent signal transduction. They can homo- or heterodimerize to bind cAMP response elements in target gene promoters. They are phosphorylated and activated by upstream serine-threonine kinases which increase their affinity to a number of transcriptional co-activators, including CREB-binding protein (CBP), p300 and transducers of regulated CREB (TORC). Phosphorylation of CREB at Ser133 is essential for CREBmediated transcription and target gene function in cell proliferation, differentiation and survival. Overexpression and/or overactivation of CREB has been described in many types of cancer and phosphorylation of CREB at Ser133 can be catalyzed by a variety of kinases, including calcium/calmodulin-dependent (Cam) kinases that are activated by calcium fluxes, Akt or p90Rsk which are downstream of ERK, as well as protein kinase A, which is activated by cAMP. Functional analyses suggest that overexpression of CREB contributes to ALL cell proliferation and survival through transcriptional activation of gene targets involved in glycolysis and anti-apoptosis, including Bcl-2, Bcl-xL, Mcl-1 and survivin.28-30 This array of anti-apoptotic CREB targets may explain the observed resistance to multi-agent induction chemotherapy. The importance of CREB in influencing clinical response has also been demonstrated in a study of adult ALL in which high levels of CREB and pCREB were associated with a shorter median overall survival; a similar trend was observed in pediatric disease.30 Furthermore, a recent study identified expanded leukemic cell populations that, if present at the diagnosis of ALL, were associated with relapse.31 One of the features of these expanded populations was high pCREB, again consistent with this pathway contributing to a chemoresistant phenotype.
Given their deregulation in many cancer types, CREB and CREB-specific signaling pathways have been proposed as targets for therapeutic intervention in cancer and inhibitors are being developed and have begun to enter early phase clinical trials.32 Despite the universal role of CREB signaling in cells, pharmacological inhibition ap-
References
1. Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185-3193.
2. Parker C, Waters R, Leighton C, et al. Effect of mitoxantrone on
pears well-tolerated, both in preclinical models and according to data emerging from the clinic. This tolerance may be due to cancer cells being differently dependent on CREB activity as compared to their normal counterparts.33 Thus, we preclinically evaluated a novel, specific small molecule CREB inhibitor in pCREB-positive ALL. 666-15 is a potent, selective inhibitor of CREB-mediated gene transcription, with an IC50 of 0.08 mM in cell-free assays. In a xenograft mouse model of breast cancer, it potently inhibited tumor growth, without overt toxicity and blood counts, blood chemistry, and tissue histology from liver, kidney and heart appeared similar to those of controls.34 Our data from cell lines and PDX ALL cells showed that these were sensitive to concentrations of 666-15 that were achievable in mice and that the CREB inhibitor induced robust apoptosis in PDX ALL cells, consistent with a cytotoxic action. Inhibition of CREB transcriptional activity was clearly demonstrated at these cytotoxic concentrations.
In summary, we have established and validated a onestop single-cell assay that can identify antigenic and signaling pathway targets in B-lineage ALL cells at both presentation of the disease and in ‘on treatment’ samples with high MRD. We show a role of hyperactive CREB, associated with an increase in subpopulations found in presentation samples, which appear preferentially selected for during induction therapy, suggesting that this pathway is involved in conferring chemoresistance to multidrug induction therapy. We propose that novel drugs affecting CREB activity or key downstream targets may be promising MRD-directed therapies for the majority of ALL patients at high risk of relapse.
Disclosures
JAEI has been awarded grant funding from Hoffmann LaRoche on an unrelated project. The other authors declare that they have no conflicts of interest to disclose.
Contributions
DM, HLB, KF, SL, GW, MC, DM, and CS performed research. CS, FWvD, HB, DM, JO-G, AF, and JAEI analyzed data. DM and JAEI wrote the manuscript. JAEI gained funding and supervised the study.
Data-sharing statement
Data are available on request.
outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet. 2010;376(9757):2009-2017.
3. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an
Haematologica | 108 - April 2023 991 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
update. J Clin Oncol. 2011;29(5):551-565.
4. Campana D. Minimal residual disease monitoring in childhood acute lymphoblastic leukemia. Curr Opin Hematol. 2012;19(4):313-318.
5. Vora A, Goulden N, Mitchell C, et al. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standardrisk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2014;15(8):809-818.
6. Vora A, Goulden N, Wade R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14(3):199-209.
7. Irving JA. Towards an understanding of the biology and targeted treatment of paediatric relapsed acute lymphoblastic leukaemia. Br J Haematol. 2016;172(5):655-666.
8. Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112(10):3982-3988.
9. Nicholson L, Evans CA, Matheson E, et al. Quantitative proteomic analysis reveals maturation as a mechanism underlying glucocorticoid resistance in B lineage ALL and resensitization by JNK inhibition. Br J Haematol. 2015;171(4):595-605.
10. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-361.
11. Davidsson J, Paulsson K, Lindgren D, et al. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia. 2010;24(5):924-931.
12. Dobson SM, Garcia-Prat L, Vanner RJ, et al. Relapse-fated latent diagnosis subclones in acute B lineage leukemia are drug tolerant and possess distinct metabolic programs. Cancer Discov. 2020;10(4):568-587.
13. Kuster L, Grausenburger R, Fuka G, et al. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood. 2011;117(9):2658-2667.
14. Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604.
15. van Delft FW, Horsley S, Colman S, et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood. 2011;117(23):6247-6254.
16. Yang JJ, Bhojwani D, Yang W, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112(10):4178-4183.
17. Irving J, Matheson E, Minto L, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124(23):3420-3430.
18. Irving JA, Minto L, Bailey S, Hall AG. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease
course. Cancer Res. 2005;65(21):9712-9718.
19. Campana D, Leung W. Clinical significance of minimal residual disease in patients with acute leukaemia undergoing haematopoietic stem cell transplantation. Br J Haematol. 2013;162(2):147-161.
20. Irving J, Jesson J, Virgo P, et al. Establishment and validation of a standard protocol for the detection of minimal residual disease in B lineage childhood acute lymphoblastic leukemia by flow cytometry in a multi-center setting. Haematologica. 2009;94(6):870-874.
21. Dixon ZA, Nicholson L, Zeppetzauer M, et al. CREBBP knockdown enhances RAS/RAF/MEK/ERK signaling in Ras pathway mutated acute lymphoblastic leukemia but does not modulate chemotherapeutic response. Haematologica. 2017;102(4):736-745.
22. Steeghs EMP, Jerchel IS, de Goffau-Nobel W, et al. JAK2 aberrations in childhood B-cell precursor acute lymphoblastic leukemia. Oncotarget. 2017;8(52):89923-89938.
23. Tasian SK, Teachey DT, Li Y, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017;129(2):177-187.
24. Hao Q, Dai C, Deng Y, et al. Pooling analysis on prognostic value of PHH3 expression in cancer patients. Cancer Manag Res. 2018;10:2279-2288.
25. Perez-Cadahia B, Drobic B, Davie JR. H3 phosphorylation: dual role in mitosis and interphase. Biochem Cell Biol. 2009;87(5):695-709.
26. Ebinger S, Ozdemir EZ, Ziegenhain C, et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30(6):849-862.
27. Sarno J, Savino AM, Buracchi C, et al. SRC/ABL inhibition disrupts CRLF2-driven signaling to induce cell death in B-cell acute lymphoblastic leukemia. Oncotarget. 2018;9(33):22872-22885.
28. Pigazzi M, Ricotti E, Germano G, Faggian D, Arico M, Basso G. cAMP response element binding protein (CREB) overexpression CREB has been described as critical for leukemia progression. Haematologica. 2007;92(10):1435-1437.
29. Shabestari RM, Safa M, Alikarami F, Banan M, Kazemi A. CREB knockdown inhibits growth and induces apoptosis in human pre-B acute lymphoblastic leukemia cells through inhibition of prosurvival signals. Biomed Pharmacother. 2017;87:274-279.
30. van der Sligte NE, Kampen KR, ter Elst A, et al. Essential role for cyclic-AMP responsive element binding protein 1 (CREB) in the survival of acute lymphoblastic leukemia. Oncotarget. 2015;6(17):14970-14981.
31. Good Z, Sarno J, Jager A, et al. Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nat Med. 2018;24(4):474-483.
32. Kimura K, Ikoma A, Shibakawa M, et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/beta-catenin inhibitor, in patients with hepatitis C virusrelated cirrhosis: a single-center, open-label, dose escalation phase 1 trial. EBioMedicine. 2017;23:79-87.
33. Li BX, Gardner R, Xue C, et al. Systemic inhibition of CREB is well-tolerated in vivo. Sci Rep. 2016;6:34513.
34. Xie F, Li BX, Kassenbrock A, et al. Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J Med Chem. 2015;58(12):5075-5087.
Haematologica | 108 - April 2023 992 ARTICLE - Hyperactive CREB and chemoresistance D. Masic et al.
Oncogenic TYK2 P760L kinase is effectively targeted by combinatorial TYK2, mTOR and CDK4/6 kinase blockade
Katharina Woess,1 Sabine Macho-Maschler,2 Dorette S. van Ingen Schenau,3 Miriam Butler,3 Caroline Lassnig,1,4 Daniel Valcanover,1 Andrea Poelzl,4 Katrin Meissl,1 Barbara Maurer,5 Tania Brandstoetter,5 Claus Vogl,1 Anna Koren,6 Stefan Kubicek,6 Anna Orlova,1 Richard Moriggl,1 Birgit Strobl,1 Veronika Sexl,5 Frank N. van Leeuwen,3 Roland P. Kuiper3,7 and Mathias Mueller1,4
1Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna, Vienna, Austria; 2Unit of Physiology, Pathophysiology and Experimental Endocrinology, University of Veterinary Medicine Vienna, Vienna, Austria; 3Princess Maxima Center for Pediatric Oncology, Utrecht, the Netherlands; 4University Center Biomodels Austria, University of Veterinary Medicine Vienna, Vienna, Austria; 5Institute of Pharmacology and Toxicology, University of Veterinary Medicine Vienna, Vienna, Austria; 6CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria and 7Department of Genetics, University Medical Center Utrecht, Utrecht, the Netherlands
Abstract
Correspondence: M. Mueller mathias.mueller@vetmeduni.ac.at
Received: September 7, 2021.
Accepted: January 5, 2022.
Early view: January 13, 2022.
https://doi.org/10.3324/haematol.2021.279848
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Tyrosine kinase 2 (TYK2) is a member of the Janus kinase/signal transducer and activator of transcription pathway, which is central in cytokine signaling. Previously, germline TYK2 mutations have been described in two patients developing de novo T-cell acute lymphoblastic leukemias (T-ALL) or precursor B-ALL. The mutations (P760L and G761V) are located within the regulatory pseudokinase domain and lead to constitutive activation of TYK2. We demonstrate the transformation capacity of TYK2 P760L in hematopoietic cell systems including primary bone marrow cells. In vivo engraftment of TYK2 P760L-expressing cell lines led to development of leukemia. A kinase inhibitor screen uncovered that oncogenic TYK2 acts synergistically with the PI3K/AKT/mTOR and CDK4/6 pathways. Accordingly, the TYK2-specific inhibitor deucravacitinib (BMS986165) reduces cell viability of TYK2 P760L-transformed cell models and ex vivo cultured TYK2 P760L-mutated patient-derived xenograft cells most efficiently when combined with mTOR or CDK4/6 inhibitors. Our study thereby pioneers novel treatment options for patients suffering from TYK2-driven acute leukemia.
Introduction
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway links extracellular cytokines with transcriptional regulation and reprogramming. As a core cancer pathway1 it connects to other cancer-driving signaling cascades such as the PI3K/AKT/mTOR or cell cycle progression pathways.2-4 Alterations in JAK/STAT signaling are associated with cancer and immune system disorders.5 Activating JAK1/2/3 mutations are frequent in various hematologic malignancies, whereas the role of the JAK family member tyrosine kinase 2 (TYK2) has only recently emerged.6-8 More evident are TYK2 mutations causative for immune system disorders and inflammatory diseases.9
TYK2 is associated with the cytokine family receptors for type I interferon (including interferon [IFN]-α and -b), interleukin (IL)-12 (including IL-23) and IL-10 (including type III IFN, IL-22 and IL-26). Under physiological conditions TYK2 activity may provoke tyrosine phosphorylation of all
STAT (STAT1-6).10 The sparse reports on aberrant TYK2 activity in cancers include TYK2 locus mutations, fusion proteins and crosstalk to oncogenic pathways.8 The first somatic activating TYK2 mutations have been described in T-cell acute lymphoblastic leukemia (T-ALL) cell lines.11 TYK2 fusion proteins have been found in various hematologic malignancies12-14 and TYK2 cross-talks to the nucleophosmin-anaplastic lymphoma kinase pathway in anaplastic large cell lymphoma.15
Next-generation sequencing is revealing an increasing number of germline gene alterations predisposing to leukemia.16 The germline TYK2 mutations (P760L, G761V) were identified in pediatric patients who developed multiple de novo ALL: the TYK2 P760L carrying patient showed two precursor B-ALLs and the TYK2 G761V patient two T-ALL. Both mutations affect the pseudokinase domain (JAK homology [JH]2 domain) and cause constitutive activation of TYK2 and of downstream STAT1/3/5.17
The first JAK inhibitors developed acted by competing ATP binding at the tyrosine kinase (JH1) domain. The high homology of the kinase domain within the JAK family or
Haematologica | 108 - April 2023 993 ARTICLE
- Acute Lymphoblastic Leukemia
other tyrosine kinases limits specificity of such inhibitors.18 The highly specific TYK2 inhibitor deucravacitinib acts differently and stabilizes the negative regulatory pseudokinase JH2 domain.19-21 The efficacy of deucravacitinib is currently evaluated in several advanced clinical trials for the treatment of autoimmune/inflammatory diseases.20,21 Combining inhibitors is beneficial, as combinatorial treatments allow for reduced drug concentrations to avoid side effects and may overcome potential resistances.22 Notably, JAK1/2/3 inhibitors in single or combined drug treatments are already applied for hematological malignancies.23 We here characterized TYK2-activating germline mutations (P760L, G761V) that have been described in childhood leukemia patients with respect to their transformation potential in vitro and in vivo. The TYK2 P760L mutation efficiently transforms hematopoietic cells and provokes cancer upon transplantation in mice. Viability of TYK2transformed cells is efficiently reduced by the highly specific TYK2 inhibitor deucravacitinib. A comprehensive inhibitor-based screen identified pathways co-operating with oncogenic TYK2. We show synergistic action of deucravacitinib with inhibitors of the top hit pathways
PI3K/AKT/mTOR or CDK4/6 in TYK2 P760L-transformed cell lines and in the TYK2 P760L-mutated patient-derived xenograft (PDX) cells. This reveals novel treatment options for acute leukemia in patients harboring gain-of-function (GOF) TYK2.
Methods
Details on plasmids, generation and cultivation of cell lines and primary cells, cell viability assays, flow cytometry, immunoblotting and histochemistry are described in the Online Supplementary Appendix
Ethics statement
Mice were housed under specific pathogen-free conditions according to FELASA guidelines. Animal experiments were approved by the Institutional Ethics and Animal Welfare Committee of the University of Veterinary Medicine Vienna, the Austrian authority according to §§ 26ff. of Animal Experiments Act, Tierversuchsgesetz 2012: TVG 2012 (BMBWF-68.205/0112-WF/V/3b/2016, BMBWF68.205/0174-V/3b/2018) and the Animal Experimental Committee of the Radboud University Medical Center (AVD1030020209324). PDX cells were generated by the Dutch Childhood Oncology Group. Informed consent for the use of spare specimens for research was obtained from study individuals, parents or legal guardians.
Drug screen
CellTiter-Glo Luminescent Cell Viability Assays (Promega, Madison, WI, US) in 384-well plates were performed in an
automated high-throughput approach (CeMM Molecular Discovery Platform - Chemical Screening). Six hundred and eighty drugs of kinase inhibitor libraries (L1600, Targetmol, Boston, MA, US and 10505, Cayman Chemicals, Ann Arbor, MI, US) were screened at 10 mM in duplicates on 2x103 Ba/F3 TYK2 P760L 1 cells/well for 3 days. Hits were selected having <30 percentage of control (POC) viability by setting the positive control to 0% (Bortezomib, 10 mM) and the negative control (0.1% dimethyl sulfoxide [DMSO]) to 100%. Two hundred and forty-six drugs from the first round were screened in triplicates with four different 10-fold dilutions on Ba/F3 TYK2 P760L 1 and parental Ba/F3 cells (cultivated with 1 ng/mL IL-3) for 3 days. For hit calling the mean difference of the area under the curve between Ba/F3 TYK2 P760L 1 and parental Ba/F3 cell POC curves was calculated.
In vivo experiments
Male and female age-matched (8-22 weeks) NOD.CgPrkdcscidIl2rgtm1Wjl/Sz (NSG) mice24 were used for xenografts. Bone marrow (BM)-derived patient cells were washed with phosphate-buffered saline (PBS) and digested with DNAse I (130 m g/mL, Roche, Basel, Switzerland) for 10 minutes (min) at room temperature. 5x105 cells were injected intrafemorally (Radboud University Medical Center, Nijmegen, the Netherlands). PDX cells in the blood were assessed bi-weekly and upon detection of human cells weekly. Mice were sacrificed once human cells reached >50% or at the humane endpoint (defined by scoring appearance, behavior, posture and mobility).
Ba/F3 and 32D cell lines were washed with PBS. 1x106 cells were injected intravenously, 5x106 cells subcutaneously. For survival studies mice were sacrificed at the humane endpoint (see above and for subcutaneous injection a tumor volume of 1,500 mm3). Tumor volume (=length*width2/2) was measured with a caliper. Organs were weighed and histologically examined. Blood parameters were measured by VetABC (scil, Viernheim, Germany). Blood was incubated with an ammoniumchloride-potassium lysis buffer for 5 min. Solid tissues were mashed through a 100 mm nylon cell strainer and red blood cell lysis was performed for spleen and liver. All isolated cells were analyzed by flow cytometry or cryopreserved.
Statistical analysis
One-way or two-way ANOVA with Tukey post hoc test and unpaired two-sided t-tests were performed on log or arcsine square root transformed data using GraphPad Prism version 7.0 for Mac (GraphPad Software, San Diego, CA, US). Mean ± standard deviation and statistical significance are shown (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
Haematologica | 108 - April 2023 994 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al.
Results
TYK2 P760L induces colony formation of primary bone marrow cells
We transduced primary murine BM cells with a retrovirus encoding human TYK2 constructs (Figure 1A) to study the effect of TYK2 P670L on colony formation. In order to test for TYK2-intrinsic kinase activity we introduced M978F as a kinase-inactivation mutation11 in the expression constructs. Co-expression of green fluorescent protein (GFP) allowed for the assessment of the transduction efficiency. On average 15% of cells were retrovirally transduced (Online Supplementary Figure S1) and an equal number of GFP+ cells was seeded into growth factor-free methylcellulose. TYK2 P760L-transduced BM cells formed colonies at largest size (Figure 1B) and highest number compared to cells expressing wild-type (WT) TYK2, TYK2 P760L, M978F or empty GFP vector (EV) (Figure 1C). TYK2 P760L-
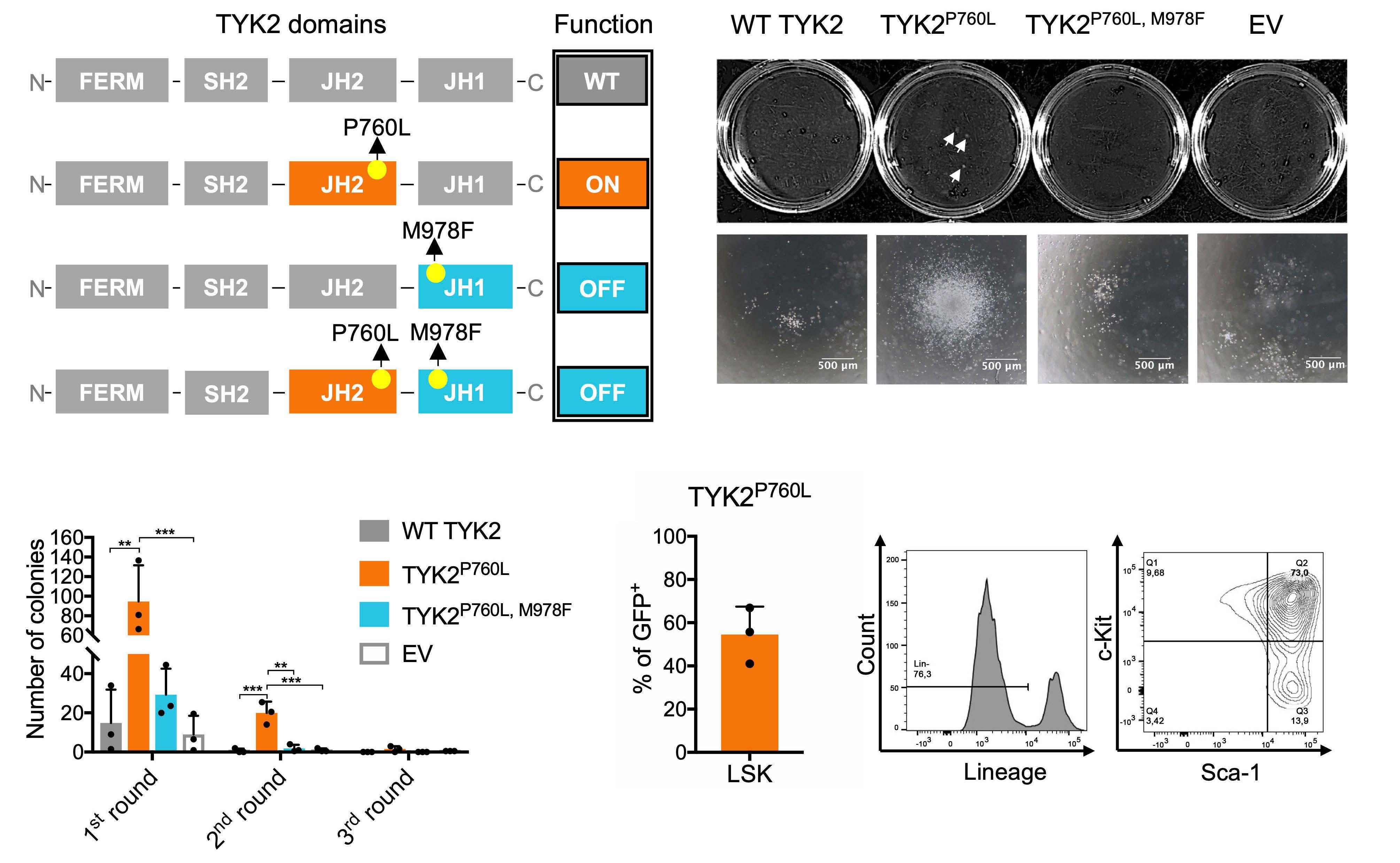
expressing BM cells were capable of replating for two rounds indicative for an enhanced self-renewal capacity (Figure 1C). The majority of the GFP+ TYK2 P760L-transduced BM cells showed characteristics of hematopoietic stem/progenitor cells (Lin- Sca-1+ c-Kit+)25 (Figure 1D). These data led us to conclude that TYK2 P760L enables colony formation of primary BM cells in growth factor-free conditions.
TYK2 P760L allows factor independence of hematopoietic cell lines and leukemogenesis in vivo
In order to further assess the ability of TYK2 mutants to provide growth factor independence, we used various hematopoietic cell lines including the stem cell factor (SCF)-dependent hematopoietic progenitor cell line HPC7, the IL-3-dependent pro-B cell line Ba/F3 and the BMderived progenitor cell line 32D.
In this experimental setting we employed the TYK2 P760L
Figure 1. TYK2 P760L-transduced primary bone marrow cells and factor-free colony formation. (A) Schematic illustration of combinations of kinase-activating germline and kinase-inactivating mutations: TYK2 consists of a four-point-one, ezrin, radixin, moesin (FERM), an atypical Src-homology 2 (SH2), a JAK homology (JH)2 and a JH1 domain. In wild-type (WT) TYK2 the JH2 pseudokinase domain inhibits the JH1 kinase domain at basal state; the JH1 domain is activated upon cytokine receptor engagement (WT, grey). P760L alters the structure of a conserved motif in the JH2 domain which is predicted to attenuate the kinase inhibitory JH2-JH1 interaction (ON, orange). M978F inactivates the JH1 domain (= OFF, blue). Combination of M978F with P760L abolishes the TYK2 kinase activity. (B) Representative colony pictures of transduced bone marrow (BM) cells, taken with 4-fold magnification objective for single colonies. Scale bars show 500 mm. White arrows indicate visible colonies. (C) Replating experiment of transduced BM cells in factor-free methylcellulose (n=3, duplicates, two-way ANOVA with log transformed data). (D) Percentage of Lin-, Sca-1+, c-Kit+ (LSK) cells among GFP+ TYK2 P760L-transduced BM cells after the first round in methylcellulose (n=3). Cells were gated on single cells, living, GFP+, Lin-, c-Kit+ and Sca-1+. Representative plots are shown. **P<0.01, ***P<0.001.
Haematologica | 108 - April 2023 995 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al. A B C D
and the TYK2 G761V mutant, another activating germline TYK2 mutant identified in pediatric ALL. In order to study the activation status of the TYK2-STAT axis, retrovirally transduced and GFP+-sorted HPC-7 cells (Figure 2A) were starved for 6 hours (h) as SCF may activate components of the JAK-STAT signaling pathway.26 As previously shown17 TYK2 P760L and TYK2 G761V enhanced TYK2 phosphorylation and increased phospho-STAT1 and -STAT3 levels. The effects on phospho-STAT5A/B were less consistent. The ectopic overexpression of WT TYK2 also induced activation of STAT1 and STAT3. Introduction of the TYK2 M978F mutation in the expression cassettes showed that the phosphorylation events were a direct consequence of TYK2 kinase activity (Figure 2B). Both TYK2-activating mutants induced the outgrowth of colonies of the immortalized progenitor cell line HPC-7 supplemented with a reduced SCF concentration. Colony numbers raised upon replating to a greater extent in TYK2 P760L cells (Figure 2C; Online Supplementary Figure S2A). This effect did not translate into a growth advantage in suspension culture upon titrated reduction of SCF (Online Supplementary Figure S2B).
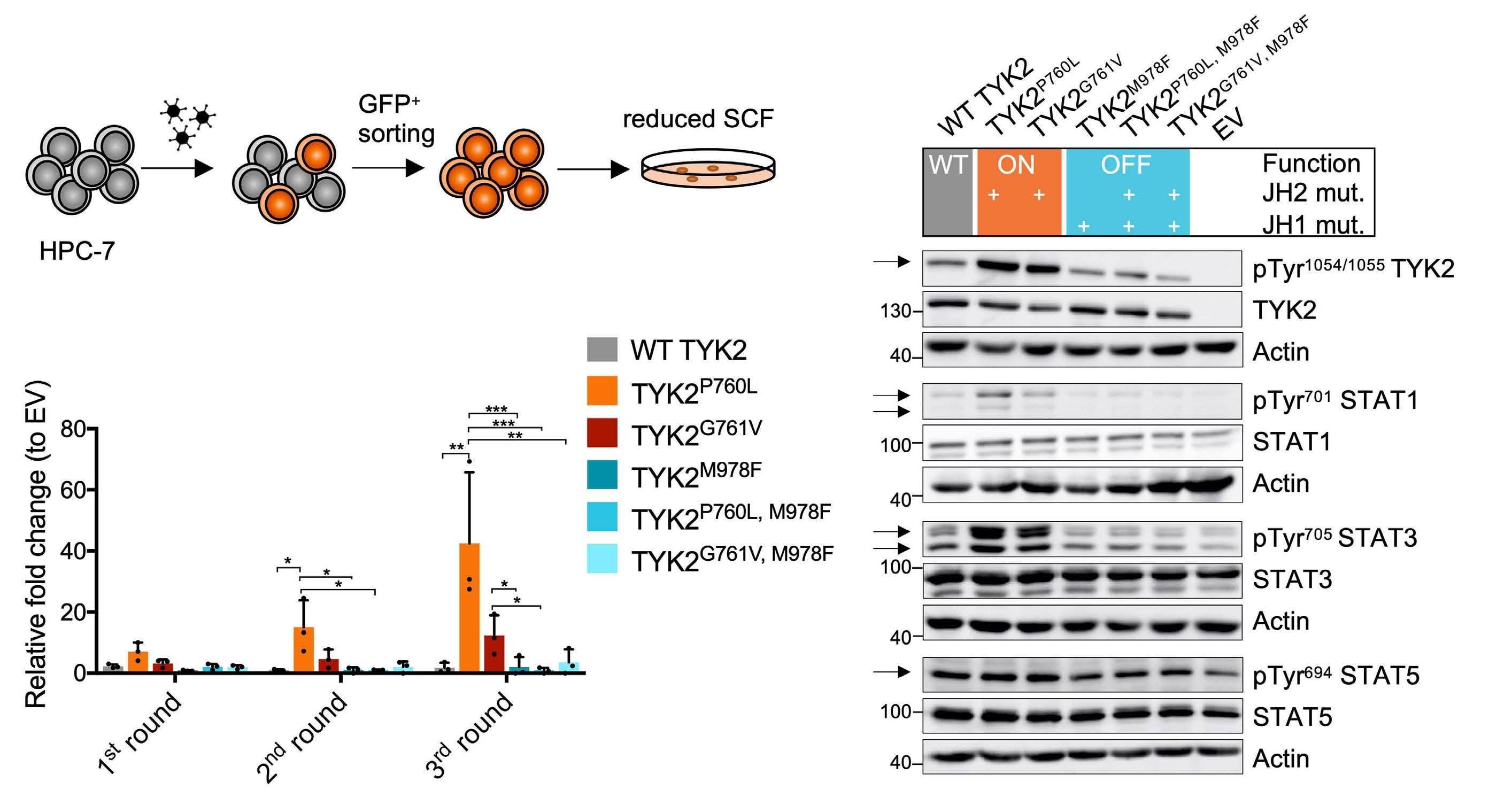
Next, we electroporated the TYK2 expression cassettes into Ba/F3 and 32D cells (Figure 3A). In both cell lines
TYK2 P760L expression enabled complete IL-3-independent growth while TYK2 G761V was only capable to transform 32D cells (Figure 3B; Online Supplementary Figure S3A). As in HPC-7 cells the TYK2 P760L-transformed cells showed constitutively activated TYK2-STAT1/3 (Online Supplementary Figure S3B). Taken together, both TYK2 germline mutations enhance STAT signaling in a TYK2 kinase-dependent manner and TYK2 P760L has a stronger transforming potential than TYK2 G761V in hematopoietic cell lines. The reasons for this remain to be elucidated. Notably, the activation state level mediated by TYK2 G761V seems to be lower than in TYK2 P760L-expressing cells as indicated by constitutive TYK2 or downstream STAT1 phosphorylation (Figure 2B and Waanders et al.17).
In order to assess the in vivo oncogenic potential, we injected Ba/F3 or 32D TYK2 P760L cells into NSG mice (Figure 3A) and used parental cells as controls. Upon systemic transplantation both cell lines harboring mutated TYK2 induced splenomegaly (Figure 3C; Online Supplementary Figure S3H) and led to high white blood cell counts (Figure 3D). Ba/F3 TYK2 P760L cells additionally caused enlarged livers (Figure 3E; Online Supplementary Figure S3H), thrombocytopenia (Figure 3F) and anemia (Figure 3G). As a consequence, the average survival of mice that had been
Figure 2. In vitro transformation capacity of TYK2 mutants. (A) Schematic outline of the HPC-7 cell experiment: cells were retrovirally transduced with the different TYK2 constructs (Figure 1A). HPC-7 cells were GFP+-sorted and seeded into methylcellulose containing reduced stem cell factor (SCF). (B) Western blot of transduced and GFP+-sorted HPC-7 cells starved for SCF for 6 hours (h) subjected to (phospho-) TYK2 (human) and STAT1/3/5 (murine) analysis. Wild-type (WT) TYK2 activity can be regulated (grey), JH2 mutations activate TYK2 signaling (ON, orange), JH1 mutation inhibits TYK2 signaling (OFF, blue). Actin was used as loading control. Numbers indicate molecular weight markers in kDa. (C) Replating experiment of transduced and GFP+-sorted HPC-7 cells expressing WT TYK2 (grey), TYK2 P760L (orange), TYK2 G761V (red) or kinase inactive TYK2 (blue) in methylcellulose with reduced SCF (n=3, two-way ANOVA with log transformed data, log colony number of empty vector (EV) transduced cells was subtracted from log number of TYK2-expressing colonies). *P<0.05, **P<0.01, ***P<0.001.
Haematologica | 108 - April 2023 996 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al. A B C
Figure 3. In vitro and in vivo transformation capacity of TYK2 mutants. (A) Schematic outline of the Ba/F3 and 32D cell experiment: cells were electroporated with the different TYK2 constructs (Figure 1A), IL-3 was withdrawn, and outgrowth was monitored. Transformed and parental cells were injected intravenously (i.v.) or subcutaneously (s.c.) into NOD.Cg-PrkdcscidIl2rgtm1Wjl/Sz (NSG) mice. (B) Representative growth curve of Ba/F3 (orange) and 32D cells (red) gaining IL-3 independence by expression of TYK2 P760L (n=3). (C to G) Spleen weight with representative spleen pictures, white blood cell count, liver weight, platelet count and hematocrit (HCT) of i.v. injected mice (Ba/F3 and 32D cells: n=5, Ba/F3 TYK2 P760L cells: n=4 (as humane endpoint was set for 2 mice), 32D TYK2 P760L cells: n=7, unpaired two-tailed t-test with log and arcsine square root [HCT] transformed data, from 2 experiments). (H) Kaplan Meier plot of i.v. injected mice (Ba/F3 and 32D cells [grey]: n=5, Ba/F3 TYK2 P760L cells: n=6, 32D TYK2 P760L cells: n=7, log-rank test: P-value =0.0002 between Ba/F3 TYK2 P760L cells and 32D TYK2 P760L cells, from 2 experiments). (I) Tumor growth of s.c. injected cells into right and left flank (Ba/F3 TYK2 P760L cells: n=5 [individual tumors, one site without tumor formation], 32D TYK2 P760L cells: n=6 [individual tumors]) and individual tumor weight (Ba/F3 TYK2 P760L cells: n=8 [2 sites without tumor formation], 32D TYK2 P760L cells: n=10, from 2 experiments). **P<0.01, ***P<0.001, ****P<0.0001.
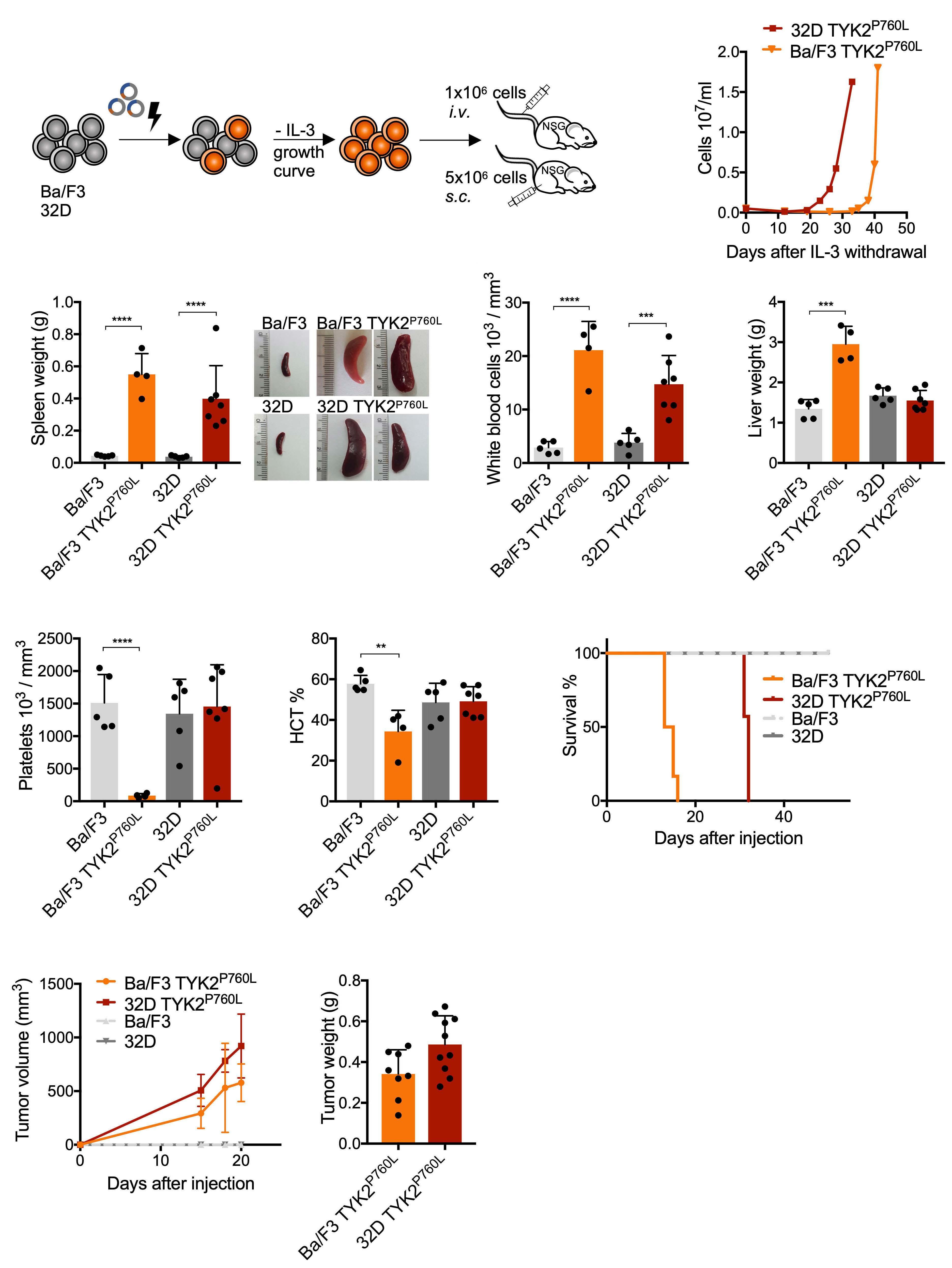
Haematologica | 108 - April 2023 997 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al. A B F G H I C D E
Figure 4. Viability of TYK2 P760L-transformed hematopoietic cell lines upon TYK2 inhibition and effect on signaling to STAT. (A and B) Dose response curves and half maximal inhibitory concentration (IC50) values of parental (black) and transformed Ba/F3 and 32D cells (shades of red) treated with deucravacitinib (TYK2inib) for 72 hours (h) supplemented with (dashed line/white filling) and without IL-3 (n≥3, in duplicates or triplicates, not all IC50 could be determined, one-way ANOVA with log transformed data). (C) Analysis of early apoptosis (Annexin V+ and 7-AAD-) and late apoptosis (Annexin V+ and 7-AAD+) of parental and transformed Ba/F3 cells treated with 1 mM deucravacitinib for 24 h, 48 h and 72 h (n=3 in duplicates). (D) Western blot of TYK2 P760L-transformed Ba/F3 and 32D cells and parental cells (with IL-3) subjected to (phospho-) TYK2-STAT1/3 analysis. Cells were treated with 1 mM deucravacitinib (TYK2i) for 6 h. Actin was used as loading control. Numbers indicate molecular weight markers in kDa. ****P<0.0001.
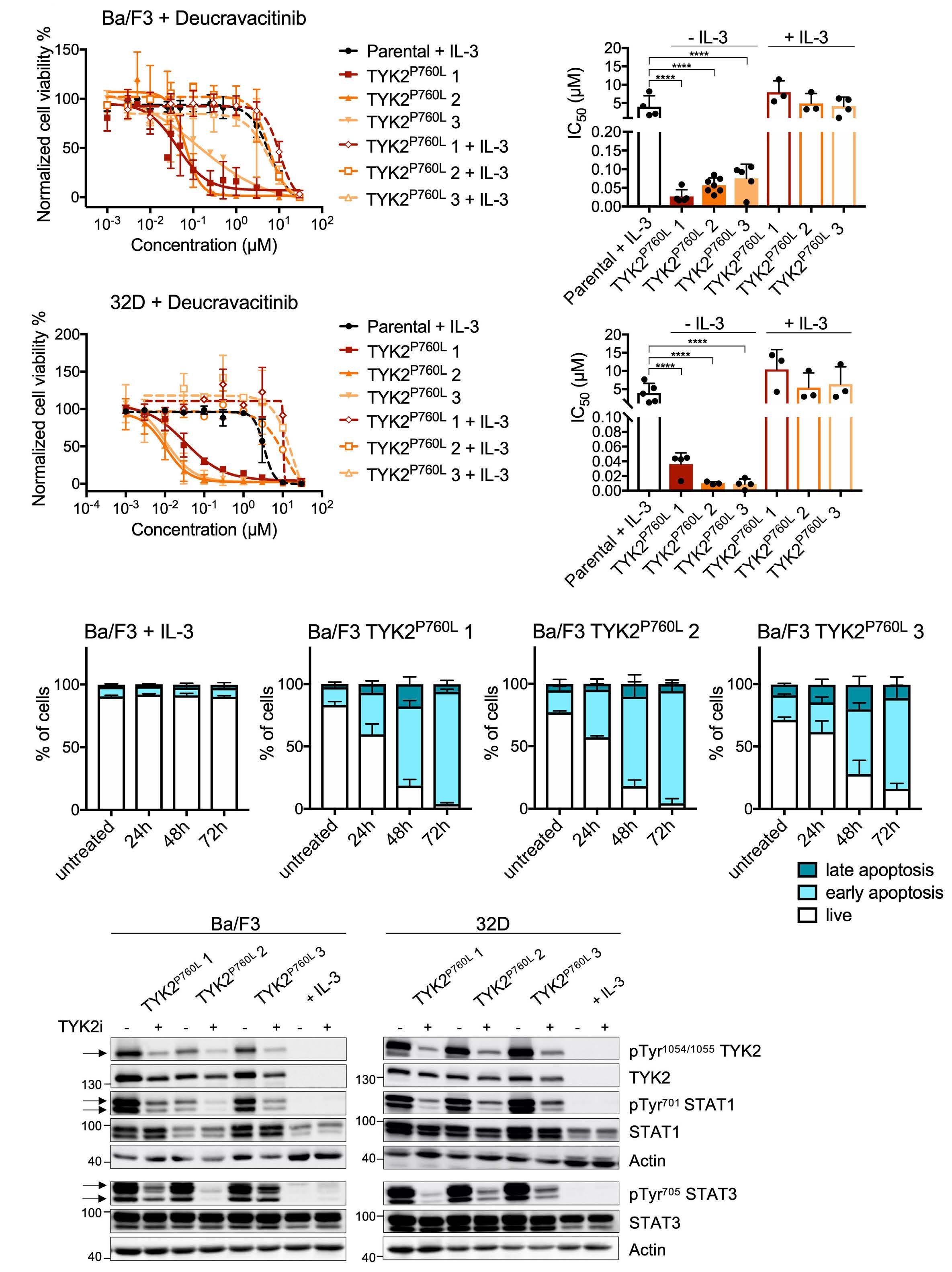
Haematologica | 108 - April 2023 998 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al. A B C D
injected with Ba/F3 TYK2 P760L cells was 14 days and with 32D TYK2 P760L cells 32 days (Figure 3H) whereas mice injected with parental Ba/F3 or 32D cells did not develop leukemia. In line we found that the subcutaneous injection of Ba/F3 or 32D TYK2 P760L cells induced local tumor growth (Figure 3I), while no tumors or signs of disease were detected upon injection of the parental cell lines. Ba/F3 TYK2 P760L cells engraftment was accompanied by hepatosplenomegaly caused by tumor cell infiltration (Online Supplementary Figure S3C to H) confirming the previous notion of oncogene carrying Ba/F3 cells as migratory cells.27
Collectively, these data show that TYK2 P760L-transformed murine cells cause cancer in systemic and local transplantation setups.
Pharmacological TYK2 inhibition abrogates oncogenic TYK2 P760L-driven signaling
We next investigated how TYK2 P760L-transformed cells react to JAK inhibitors (JAKinib). Deucravacitinib (BMS986165) is a highly selective and potent allosteric TYK2inib with excellent pharmacokinetic properties across cell types and species.20,21,28,29 Drug testing was performed on Ba/F3 and 32D TYK2 P760L cells from three outgrowth experiments (1-3; Online Supplementary Figure S3A) and on parental cells by assessing the metabolic activity. All transformed cell lines were highly sensitive to deucravacitinib with a half maximal inhibitory concentration (IC50) ranging from 10 to 90 nM (Figure 4A and B) which is around 100 times lower than IC50 values of control cell lines. Culture of Ba/F3 and 32D TYK2 P760L cells in the presence of IL-3 reversed the effect of TYK2 inhibition, indicating that factor-independent growth was governed by mutated TYK2 (Figure 4A and B). Treatment of Ba/F3 TYK2 P760L cells with deucravacitinib induced apoptosis (Figure 4C; Online Supplementary Figure 4A) and G0 cell cycle arrest (Online Supplementary Figure 4B). The efficacy of deucravacitinib was confirmed by western blotting that showed the decreased phosphorylation state of TYK2 and downstream STAT1/3 (Figure 4D).
In order to demonstrate the specificity of TYK2 inhibition in oncogenic transformed Ba/F3 cells and to exclude potential off-target effects of deucravacitinib, we tested three additional Ba/F3 cell lines driven by known oncogenes, i.e., TEL-JAK2, STAT5B N642H and BCR-ABL1 p210. In these settings IC50 values were comparable to parental Ba/F3 cells cultured with IL-3 (Online Supplementary Figure S5A). These experiments prove that deucravacitinib is selective for TYK2 and specifically interferes with TYK2dependent transformation. Further support for the unique function of TYK2 stems from experiments with the JAK1/3 inhibitor tofacitinib,30 the JAK1/2 inhibitor ruxolitinib31 and the JAK1 inhibitor filgotinib.32 None of these JAKinibwhen employed at specific concentrations - significantly
decreased the metabolic activity of TYK2 P760L-transformed cells (except Ba/F3 TYK2 P760L 1 with ruxolitinib) compared to parental Ba/F3 and 32D cells (Online Supplementary Figure S5B to G).
In summary we confirmed that only the TYK2-selective drug deucravacitinib efficiently reduces the cell viability of TYK2 P760L-transformed cells. This underscores the key role of TYK2 in maintaining the transformed state and excludes a major contribution of other JAK family members.
TYK2 P760L signaling co-operates with the PI3K/AKT/mTOR and CDK4/6 pathways
In order to understand whether and how oncogenic TYK2 P760L co-operates with other signaling pathways we designed a drug screen with commercial libraries of 680 approved or investigational kinase inhibitors (Figure 5A).
A first screen in Ba/F3 TYK2 P760L cells identified 246 drugs that reduce metabolically active cells to less than 30% compared to the control. In a second round, we included the Ba/F3 parental cell line as control and counter screen and looked for drugs that specifically block the TYK2 P760L-expressing cells. This narrowed the output down to 34 drugs, of which several target the PI3K/AKT/mTOR (8/34 drugs) and CDK4/6 (3/34 drugs) pathways (Figure 5A; Online Supplementary Table S4). Involvement of these pathways is also supported by increased phosphorylation of AKT in both TYK2 P760L-transformed cell lines and increased levels of CDK6 in transformed 32D cells. No changes in the mTOR target 4EBP1 were observed (Figure 5B).
The compound olverembatinib (GZD824) scored as top hit of the screen (Online Supplementary Table S4). Olverembatinib was initially described as inhibitor of the BCR-ABL1 fusion kinase but also interferes with PI3K/AKT and SRC kinase signaling.33 In order to validate our screening results, we determined dose response curves with inhibitors for the PI3K/AKT/mTOR or CDK4/6 pathway and used everolimus, vistusertib, olverembatinib, LY294002, abemaciclib and palbociclib (Figure 5A). We included the TYK2 P760Ltransformed Ba/F3 and 32D cells, and BCR-ABL1-, TELJAK2- and STAT5B N642H-transformed Ba/F3 cells as controls. TYK2 P760L-transformed Ba/F3 and 32D cells were more sensitive to all tested drugs compared to controls (Figure 5C to E; Online Supplementary Figures S6A to C and S7A to L). mTOR inhibitors everolimus and vistusertib were the most efficacious drugs with highest difference between transformed and parental Ba/F3 cell lines (Figure 5C and Online Supplementary Figure S6A). At the effective concentrations determined in the dose-response curves all tested drugs prevented IL-3-independent growth (Online Supplementary Figure S8A and B).
Cancer therapy regimens rely on drug combinatorial treatments.22 We thus combined the TYK2 inhibitor (deucravacitinib) with all tested co-operating inhibitors and
Haematologica | 108 - April 2023 999 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al.
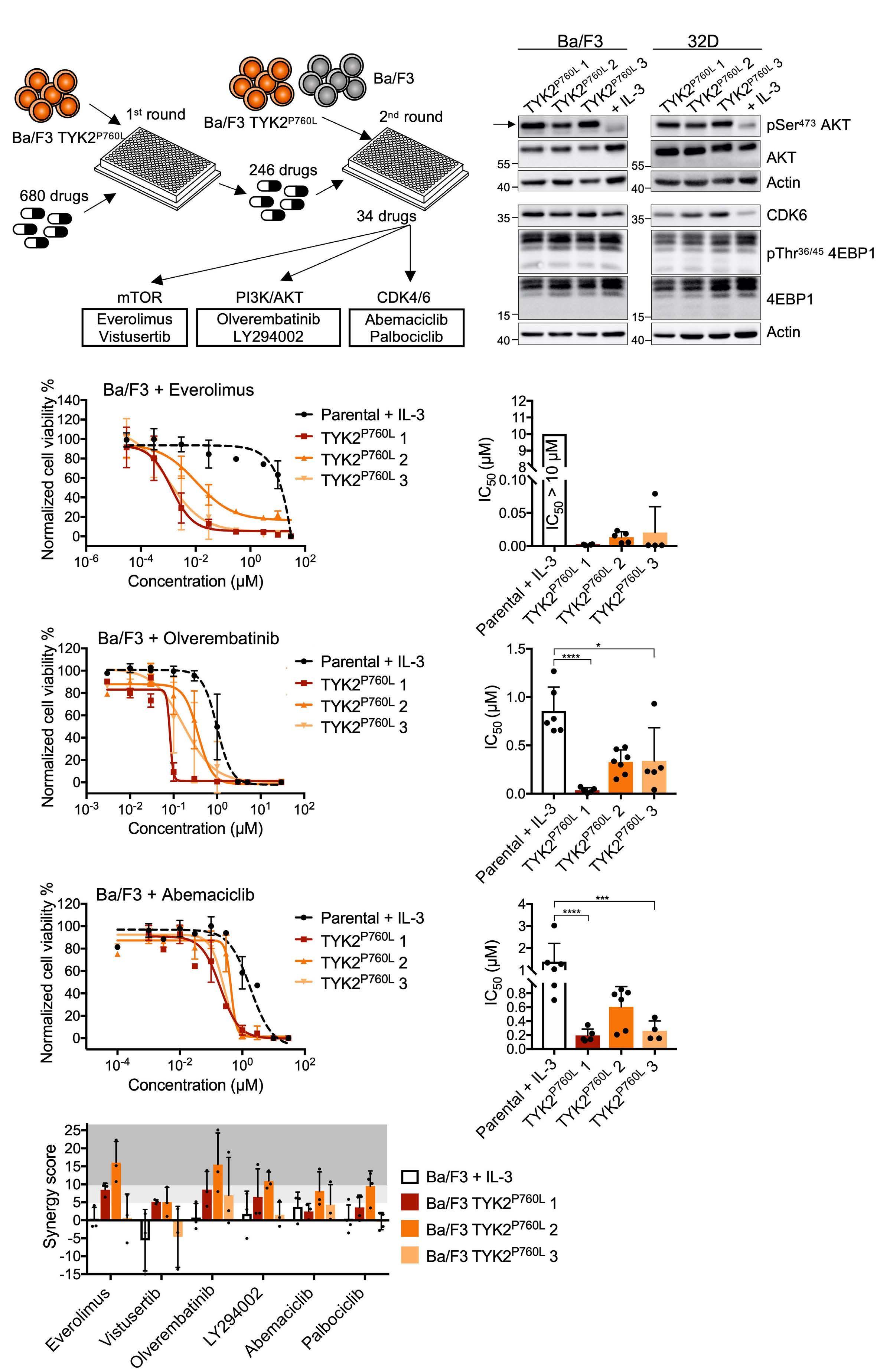
Continued on following page. Haematologica | 108 - April 2023 1000 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al. A B C D E F
Figure 5. Drug screen of TYK2 P760L-expressing cells and cross-talk with other kinase-dependent pathways. (A) Schematic outline of drug screen: Ba/F3 TYK2 P760L cells of the first outgrowth experiment were treated with 680 kinase inhibitors in a high throughput screen. In the second round 246 drugs identified (<30% viability of control) were used for comparison of Ba/F3 TYK2 P760L and parental cells. Among the top hits were mainly drugs targeting the mTOR, PI3K/AKT and CDK4/6 pathway. These targets were validated with the indicated drugs. (B) Western blot of TYK2 P760L-transformed Ba/F3 and 32D cells and parental cells (with IL-3) subjected to (phospho-) AKT, 4EBP1 and CDK6 analysis. Actin was used as loading control. Numbers indicate molecular weight markers in kDa. (C to E) Validation of screen result with dose-response curves and half maximal inhibitory concentration (IC50) values of parental (black dashed line/white filling, with IL-3) and transformed Ba/F3 cells (shades of red) treated for 72 hours (n≥3, in duplicates, not all IC50 could be determined, one-way ANOVA with log transformed data). (F) Synergy scores (n≥3, in duplicates) calculated with the zero interaction potency (ZIP) model. Light grey shows additivity, dark grey synergy. *P<0.05, ***P<0.001, ****P<0.0001.
calculated the synergy score by the zero interaction potency (ZIP) method.34,35 For Ba/F3 TYK2 P760L cells 11/18 and for 32D TYK2 P760L cells 16/18 combinations reached a synergy score higher than five indicating additive effects. Synergy scores ≥10 were found in Ba/F3 TYK2 P760L for the combination with PI3K/AKT/mTOR inhibitors (everolimus, LY294002 and olverembatinib), in 32D TYK2 P760L cells for the combination with the PI3K/AKT inhibitor LY294002 and the CDK4/6 inhibitor abemaciclib (Figure 5F; Online Supplementary Figure S8C to H)
Taken together, oncogenic TYK2 and co-operating pathways were successfully blocked by combinatorial treatment in murine cellular models.
Combined treatment with TYK2 and mTOR or CDK4/6 inhibitors decreases viability of TYK2 P760L xenograft cells
In order to translate these results in a patient-related setting, we analyzed the combinatorial treatments with deucravacitinib in human PDX cells harboring TYK2 P760L (Online Supplementary Figure S9A) co-cultured with human hTERT mesenchymal stem cells. The patient carrying the TYK2 P760L mutation first developed a leukemia classified as a chromosomal rearranged pre-B-ALL (leukemia 1) and subsequently a hyperdiploid common B-ALL (leukemia 2).17 PDX cells were generated from both leukemias and were co-treated with increasing concentrations of deucravacitinib and inhibitors of the identified co-operating pathways. Adding deucravacitinib to mTOR and CDK4/6 inhibitors resulted in a left-shift of the dose response curves (except for palbociclib in PDX cells leukemia 1) (Figure 6A to D; Online Supplementary Figure S9B to E). For both leukemias the synergy score was higher than 5 for the combination with abemaciclib (Figure 6C and D) and for the leukemia 2 the combination with vistusertib and palbociclib (Online Supplementary Figure S9C and E). The most synergistic area scores, which indicate the peak of the synergy matrix, were ≥5 for both leukemias with all treatments except leukemia 1 with palbociclib and ≥10 for both leukemias with abemaciclib, leukemia 1 with everolimus and leukemia 2 with vistusertib (Figure 6A to D; Online Supplementary Figure S9B to E). Combinations with PI3K/AKT inhibitors (olverembatinib and LY294002) failed to display synergism (data not shown).
These data show that the TYK2 inhibitor improves the efficacy of mTOR and CDK4/6 inhibitors in patient cells.
Discussion
We investigated the TYK2 P760L germline GOF mutation that was found in childhood B-ALL17 with regard to its oncogenic and druggable properties. Using in vitro and in vivo models we demonstrate that constitutively active human TYK2 P760L conferred growth/proliferation advantages on primary and immortalized hematopoietic cells. TYK2 P760L-harboring murine cells led to leukemic disease upon transplantation. A selective TYK2inib, but not JAKinib targeting JAK1-3, was highly efficacious in decreasing the metabolic activity of TYK2 P760L-transformed cells. A kinase inhibitor screen identified pathways co-operating with oncogenic TYK2 and established combinatorial TYK2 inhibition with mTOR or CDK4/6 pathway blockade as therapeutic option to eradicate TYK2-driven leukemia. We established the oncogenic potential of the TYK2 P760L GOF mutation in different cell systems ranging from primary BM cells to hematopoietic cell lines of various differentiation stages as exemplified by enhanced colony formation or growth factor-independent proliferation in vitro and migratory properties in vivo. Thus, the germline TYK2 mutation qualifies for inclusion in the expanding list of cancer driver gene mutations.36 This mutation is predicted to change the structure of the conserved DPG motif of JAK pseudokinases and thereby attenuates the inhibitory function.17 The DPG motif replaces the DFG motif of kinases and is involved in several non-covalent interactions within the JH2 domain.37 DPG mutations of other JAK listed in cancer databases are not further characterized. An activating JAK3 V674A mutation (adjacent to the DPG motif) found in T-ALL patients has been shown to cause leukemia in mice.38 Under the conditions analyzed, constitutively active TYK2 did not suffice to immortalize primary cells or allow for factor-independent growth of all cell lines tested. This is in line with the properties of germline-transmitted leukemia-predisposing mutations that frequently block the differentiation of lymphoid cells but need co-operating factors that enhance indefinite survival for malignant transformation.39 It is also in line with the reported weaker malignant
Haematologica | 108 - April 2023 1001
K. Woess et al.
ARTICLE - Co-operative targeting of oncogenic TYK2
transformation capacity of a given activating mutation of TYK2 compared to other JAK.40,41
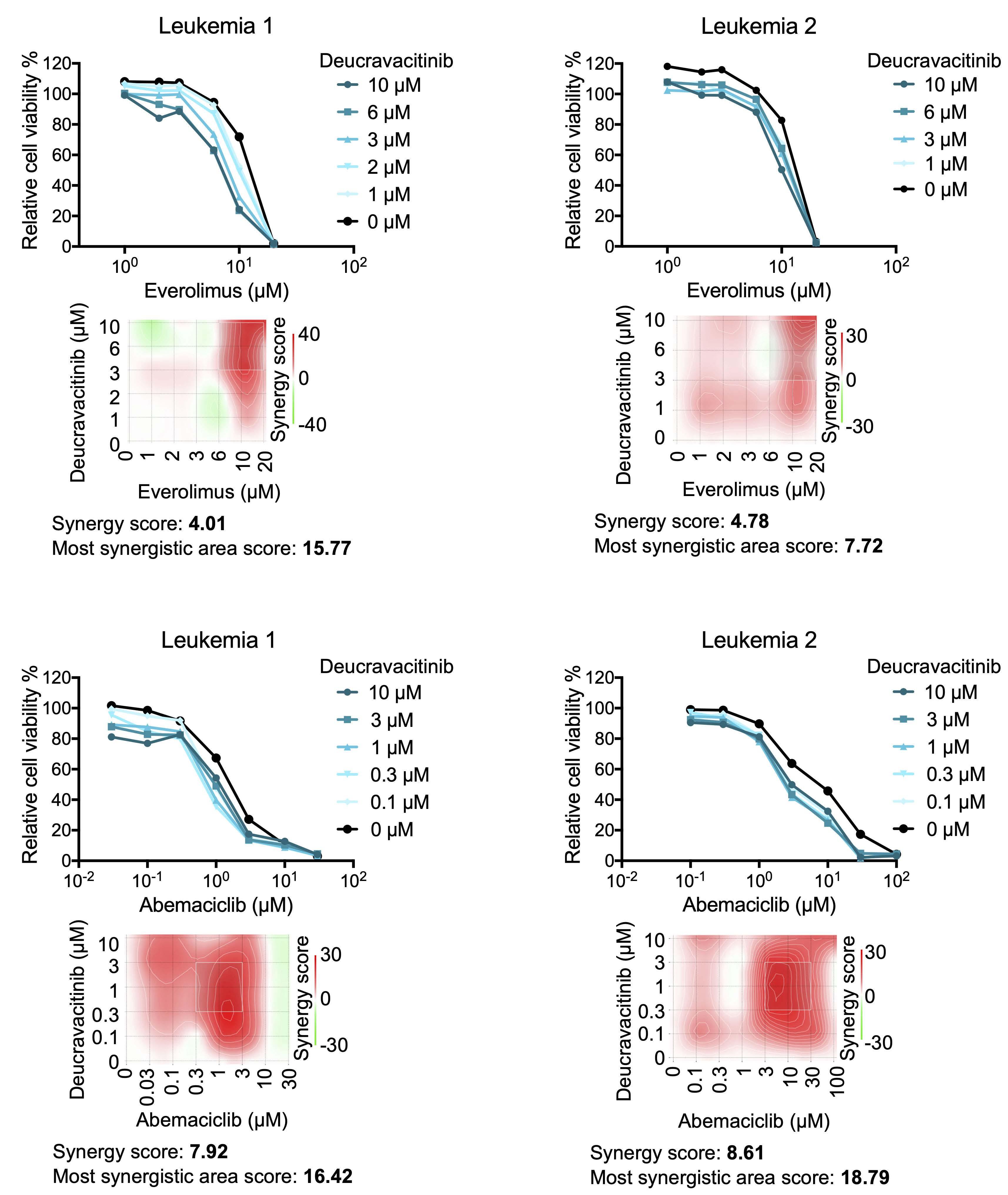
As previously shown TYK2 P760L expression leads to constitutive STAT1/3 signaling which is described to shape the oncogenic transcriptome.8,17 We provide strong evidence
that these signals stem directly from TYK2 activity as introduction of a kinase-inactivating mutation reverts STAT activation and cellular transformation. In addition, our studies with JAKinib revealed that only the TYK2-specific inhibitor deucravacitinib and not inhibitors with higher sel-
D) Cell viability curves of hTERT mesenchymal stem cells co-cultured patient-derived xenograft cells of leukemia 1 and leukemia 2 treated with different concentrations of the indicated drug and different concentrations of deucravacitinib (shades of blue) for 72 hours. A synergy map is shown for each drug combination. Synergy scores and most synergistic area score were calculated with the zero interaction potency (ZIP) model (n=1). Red shows synergism and green antagonism.
Figure 6.
Haematologica | 108 - April 2023 1002 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al. A B C D
TYK2 P760L patient-derived xenograft cells under combinatorial drug treatment. (A to
ectivity for JAK1-3 showed efficacy in cell viability assays with TYK2 P760L-transformed cells.
Deucravacitinib belongs to a novel class of JAKinib as it does not block the enzymatic activity in the JH1 domain but rather stabilizes the regulatory pseudokinase domain JH2.19-21 The inhibitor was shown to bind also the JH2 domain of JAK1, albeit with less affinity and selectivity.20,21
Importantly, the biochemical data translate in a weak activity against JAK1-dependent signaling.20,21,28,29 The efficiency of JH1-targeting TYK2 inhibitors in leukemia treatment has been shown for T-ALL xenografts.42 We are the first to prove the successful application of a JH2-targeting drug on a hyperactive TYK2 carrying the mutation in the JH2 domain. This might be important for future development of JAKinib.
Kinases represent one of the largest groups of druggable targets as they drive key signaling pathways and aberrant kinase activity leads or contributes to cancer onset and progression.43 In order to identify co-operation of oncogenic TYK2 with other kinase-driven pathways, we have screened inhibitor libraries using TYK2 P760L-transformed cells and identified the most potent hits in the PI3K/AKT/mTOR and CDK4/6 signaling axes. TYK2-dependent activation of PI3K and a crosstalk between JAK/STAT and mTOR signaling is well established.3,44,45 Aberrant TYK2-PI3K activity has been also reported for prostate cancer46 and T-ALL.11 The PI3K/AKT/mTOR pathway is frequently activated in ALL and specific inhibitors alone or in combination with JAKinib have been successfully applied in Ph-like ALL.47 At the molecular level CDK4/6 is reported to connect cell cycle progression with cell growth via mTOR activation, which may explain the efficacy of combined blocking of CDK4/6 and of PI3K/AKT/mTOR in solid tumors.48 A direct connection between CDK4/6 and TYK2 has not been described. However, STAT3, downstream of TYK2 P760L, is known to activate CDK4/6 during the cell cycle progression.49 CDK6 was shown to interact with STAT3 in tumorigenesis50 and CDK6 hyperactivation has been reported in ALL.51 Importantly, our combinatorial treatment of the TYK2 inhibitor with mTOR or CDK4/6 inhibitors was successfully applied in the TYK2 GOF PDX cells. Ex vivo assays tend to underestimate the effects of cell cycle drugs as even stromal co-cultures do not fully
References
1. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
2. Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173(2):321-337.
3. Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends Immunol. 2015;36(1):21-29.
4. Steelman LS, Pohnert SC, Shelton JG, et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression
support proliferation of ALL xenografts.52 In this context it is important to note that CDK6 also exhibits kinase- and cell cycle-independent functions in tumorigenesis.53,54
In summary, we provide proof of TYK2 acting as an oncogene in hematologic malignancies by firmly establishing the oncogenic potential of TYK2 germline mutations, revealing that TYK2 P760L is involved in leukemia progression. Moreover, our results indicate that combinatorial kinase inhibition in acute leukemia could be a valid strategy to combat hyperactivated TYK2-mutated leukemia.
Disclosures
No conficts of interest to disclose.
Contributions
KW performed and analyzed most experiments; SM-M, BS and MM supervised the project; SM-M, MB, KM, BM, TB and AO provided technical support; AK and SK performed and analyzed the kinase inhibitor screen; SM-M, DSvIS, CL and AP helped with in vivo mouse studies; CV helped with statistical analysis; DV performed some experiments; RM and VS provided crucial material and reagents; SM-M, RM, BS, VS, FNvL, RPK and MM were involved in study design and provided crucial scientific input; KW and MM wrote the manuscript with input from all authors. All authors approved the manuscript.
Acknowledgments
The authors thank Marion Bokor for histological sample preparations and H&E staining, Philipp Jodl for technical support, the mouse facility personnel for their help with in vivo experiments, Thomas Kolbe for provision of some NSG mice and Michael Dworzak for provision of a cell line.
Funding
This work was supported by the Austrian Science Fund (FWF) funded DK W1212 PhD program “Inflammation and Immunity” and the Special Research Program SFB F6101 and F6106 (BS and MM), F6105 (RM) and F6107 (VS).
Data-sharing statement
All novel reagents mentioned are available upon justified request.
and leukemogenesis. Leukemia. 2004;18(2):189-218.
5. O'Shea JJ, Schwartz DM, Villarino AV, et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311-328.
6. Hammaren HM, Virtanen AT, Raivola J, et al. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine. 2019;118:48-63.
7. Vainchenker W, Constantinescu SN. JAK/STAT signaling in
Haematologica | 108 - April 2023 1003 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al.
hematological malignancies. Oncogene. 2013;32(21):2601-2613.
8. Wöss K, Simonovic N, Strobl B, et al. TYK2: an upstream kinase of STATs in cancer. Cancers (Basel). 2019;11(11):1728.
9. Pellenz FM, Dieter C, Lemos NE, et al. Association of TYK2 polymorphisms with autoimmune diseases: A comprehensive and updated systematic review with meta-analysis. Genet Mol Biol. 2021;44(2):e20200425.
10. Strobl B, Stoiber D, Sexl V, et al. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci (Landmark Ed). 2011;16:3214-3232.
11. Sanda T, Tyner JW, Gutierrez A, et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013;3(5):564-577.
12. Velusamy T, Kiel MJ, Sahasrabuddhe AA, et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood. 2014;124(25):3768-3771.
13. Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27(4):516-532.
14. Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinaseactivating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005-1015.
15. Prutsch N, Gurnhofer E, Suske T, et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia. 2019;33(3):696-709.
16. Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol. 2019;16(4):227-240.
17. Waanders E, Scheijen B, Jongmans MC, et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia. 2017;31(4):821-828.
18. Bryan MC, Rajapaksa NS. Kinase inhibitors for the treatment of immunological disorders: recent advances. J Med Chem. 2018;61(20):9030-9058.
19. Tokarski JS, Zupa-Fernandez A, Tredup JA, et al. Tyrosine kinase 2-mediated signal transduction in T lymphocytes is blocked by pharmacological stabilization of its pseudokinase domain. J Biol Chem. 2015;290(17):11061-11074.
20. Burke JR, Cheng L, Gillooly KM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med. 2019;11(502):eaaw1736
21. Wrobleski ST, Moslin R, Lin S, et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J Med Chem. 2019;62(20):8973-8995.
22. Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30(7):679-692.
23. Klein K, Stoiber D, Sexl V, et al. Untwining anti-tumor and immunosuppressive effects of JAK inhibitors-A strategy for hematological malignancies? Cancers (Basel). 2021;13(11):2611.
24. Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477-6489.
25. Cheng H, Zheng Z, Cheng T. New paradigms on hematopoietic stem cell differentiation. Protein Cell. 2020;11(1):34-44.
26. Linnekin D, Mou S, Deberry CS, et al. Stem cell factor, the JAKSTAT pathway and signal transduction. Leuk Lymphoma. 1997;27(5-6):439-444.
27. Funakoshi-Tago M, Tago K, Sumi K, et al. The acute lymphoblastic leukemia-associated JAK2 L611S mutant induces
tumorigenesis in nude mice. J Biol Chem. 2009;284(19):12680-12690.
28. Chimalakonda A, Burke J, Cheng L, et al. Selectivity profile of the tyrosine kinase 2 inhibitor deucravacitinib compared with janus kinase 1/2/3 inhibitors. Dermatol Ther (Heidelb). 2021;11(5):1763-1776.
29. Catlett IM, Hu Y, Gao L, et al. Molecular and clinical effects of selective TYK2 inhibition with deucravacitinib in psoriasis. J Allergy Clin Immunol. 2021;S0091-6749(21)01690-0.
30. Flanagan ME, Blumenkopf TA, Brissette WH, et al. Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem. 2010;53(24):8468-8484.
31. Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109-3117.
32. Van Rompaey L, Galien R, van der Aar EM, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191(7):3568-3577.
33. Ye W, Jiang Z, Lu X, et al. GZD824 suppresses the growth of human B cell precursor acute lymphoblastic leukemia cells by inhibiting the SRC kinase and PI3K/AKT pathways. Oncotarget. 2017;8(50):87002-87015.
34. Yadav B, Wennerberg K, Aittokallio T, et al. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput Struct Biotechnol J. 2015;13:504-513.
35. Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020;48(W1):W488-W493.
36. Martinez-Jimenez F, Muinos F, Sentis I, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020;20(10):555-572.
37. Min X, Ungureanu D, Maxwell S, et al. Structural and functional characterization of the JH2 pseudokinase domain of JAK family tyrosine kinase 2 (TYK2). J Biol Chem. 2015;290(45):27261-27270.
38. Degryse S, de Bock CE, Cox L, et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood. 2014;124(20):3092-3100.
39. Tijchon E, Havinga J, van Leeuwen FN, Scheijen B. B-lineage transcription factors and cooperating gene lesions required for leukemia development. Leukemia. 2013;27(3):541-552.
40. Staerk J, Kallin A, Demoulin JB, et al. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005;280(51):41893-41899.
41. Shide K, Shimoda K, Kamezaki K, et al. Tyk2 mutation homologous to V617F Jak2 is not found in essential thrombocythaemia, although it induces constitutive signaling and growth factor independence. Leuk Res. 2007;31(8):1077-1084.
42. Akahane K, Li Z, Etchin J, et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br J Haematol. 2017;177(2):271-282.
43. Klaeger S, Heinzlmeir S, Wilhelm M, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367):eaan4368.
44. Rani MR, Leaman DW, Han Y, et al. Catalytically active TYK2 is essential for interferon-beta-mediated phosphorylation of STAT3 and interferon-alpha receptor-1 (IFNAR-1) but not for activation of phosphoinositol 3-kinase. J Biol Chem. 1999;274(45):32507-32511.
Haematologica | 108 - April 2023 1004 ARTICLE - Co-operative targeting of oncogenic TYK2 K. Woess et al.
45. Kusch A, Tkachuk S, Haller H, et al. Urokinase stimulates human vascular smooth muscle cell migration via a phosphatidylinositol 3-kinase-Tyk2 interaction. J Biol Chem. 2000;275(50):39466-39473.
46. Ide H, Nakagawa T, Terado Y, et al. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem Biophys Res Commun. 2008;369(2):292-296.
47. Tasian SK, Teachey DT, Li Y, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017;129(2):177-187.
48. Romero-Pozuelo J, Figlia G, Kaya O, et al. Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 2020;31(2):107504.
49. Fukada T, Ohtani T, Yoshida Y, et al. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle
transition. EMBO J. 1998;17(22):6670-6677.
50. Kollmann K, Heller G, Schneckenleithner C, et al. A kinaseindependent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013;24(2):167-181.
51. Nebenfuehr S, Kollmann K, Sexl V. The role of CDK6 in cancer. Int J Cancer. 2020;147(11):2988-2995.
52. Frismantas V, Dobay MP, Rinaldi A, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017;129(11):e26-e37.
53. Bellutti F, Tigan AS, Nebenfuehr S, et al. CDK6 antagonizes p53-induced responses during tumorigenesis. Cancer Discov. 2018;8(7):884-897.
54. Uras IZ, Maurer B, Nivarthi H, et al. CDK6 coordinates JAK2 (V617F) mutant MPN via NF-kappaB and apoptotic networks. Blood. 2019;133(15):1677-1690.
Haematologica | 108 - April 2023 1005
K. Woess et al.
ARTICLE - Co-operative targeting of oncogenic TYK2
Time spent at home among older adults with acute myeloid leukemia receiving azacitidine- or venetoclax-based regimens
Correspondence: D.R. Richardson daniel_richardson@med.unc.edu
Received: January 21, 2022.
Accepted: June 10, 2022.
Early view: July 21, 2022.
https://doi.org/10.3324/haematol.2022.280728
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
Time at home is a critically important outcome to adults with acute myeloid leukemia (AML) when selecting treatment; however, no study to date has adequately described the amount of time older adults spend at home following initiation of chemotherapy. We queried records from a multi-institution health system to identify adults aged ≥60 years newly diagnosed with AML who were treated with azacitidine or venetoclax and evaluated the proportion of days at home (PDH) following diagnosis. Days were considered “at home” if patients were not admitted or seen in the emergency department or oncology/infusion clinic. Assessed covariates included demographics and disease risk. Associations between PDH and baseline characteristics were evaluated via linear regression, adjusted for log length of follow-up. From 2015-2020, 113 older adults were identified. Most received azacitidine plus venetoclax (51.3%) followed by azacitidine monotherapy (38.9%). The mean PDH for all patients was 0.58 (95% confidence interval: 0.54-0.63, median 0.63). PDH increased among survivors over time. PDH did not differ between therapy groups (adjusted mean, azacitidine plus venetoclax: 0.68; azacitidine monotherapy: 0.66; P=0.64) or between disease risk categories (P=0.34). Compared to patients receiving azacitidine monotherapy, patients receiving azacitidine plus venetoclax had longer clinic visits (median minutes: 127.9 vs. 112.9, P<0.001) and infusion visits (median minutes: 194.3 vs. 132.5, P<0.001). The burden of care for older adults with AML treated with “less intense” chemotherapy is high. The addition of venetoclax to azacitidine did not translate into increased time at home. Future prospective studies should evaluate patient-centered outcomes, including time at home, to inform shared decision-making and drug development.
Introduction
The prognosis for older adults diagnosed with acute myeloid leukemia (AML) is poor. Historically, less than 40% of older adults (aged ≥60 years) survived 1 year from the time of diagnosis.1,2 Recently, however, treatment decisionmaking paradigms have shifted with the addition of venetoclax to azacitidine, a prior standard therapy. This combination has been associated with superior remission rates and overall survival compared to azacitidine alone.3 Prior research has demonstrated that patients with AML prefer treatments that allow increased time at home.4 In a national survey of patients with AML, most were willing to accept a reduction in remission rates in exchange for an increase in the amount of time spent at home, al-
though preferences for treatment outcomes varied.5 An accurate understanding of the amount of time patients can anticipate spending at home is therefore critical to inform patient-centered treatment decisions for older adults with AML.
Descriptions of patients’ time at home and treatment burden in the setting of advanced solid tumors have recently been published.6,7 However, no study has adequately described the amount of time older patients spend at home while receiving therapy for AML. To address this knowledge gap, we aimed to quantify the amount of time older patients with AML spend at home and the amount they spend engaged in AML-related care with initiation of first-line azacitidine- or venetoclax-containing regimens.
Christopher E. Jensen,1,2 Hilary M. Heiling,2,3 Konan E. Beke,1 Allison M. Deal,3 Ashley L. Bryant,3,4 Lorinda A. Coombs,3,4 Matthew C. Foster1,3 and Daniel R. Richardson1,3
1University of North Carolina School of Medicine; 2University of North Carolina Gillings School of Global Public Health; 3Lineberger Comprehensive Cancer Center, University of North Carolina and 4University of North Carolina School of Nursing, Chapel Hill, NC, USA
Haematologica | 108 - April 2023 1006 ARTICLE - Acute Myeloid Leukemia
Methods
Setting and patients
We queried records from University of North Carolina (UNC) Health to identify adults aged ≥60 years diagnosed with AML from 2015-2020. Those receiving first-line azacitidine and/or venetoclax were included. Records from 12 other health systems were available via electronic health data exchanges. Individuals receiving oncology care not reflected in available records were excluded. The UNC institutional review board approved the protocol.
Demographic and clinical variables
Demographic variables included age, race, sex, marital status, employment, and area-level measures (rurality8 and median household income9). Driving times/distances to clinical encounters from patients’ addresses were calculated via the Google Maps Application Program Interface (Google, LLC, Mountain View, CA, USA).10 Clinical variables included disease risk according to European LeukemiaNet (ELN) 2017 criteria11 and dates of diagnosis, death/last follow-up, and all clinic/emergency department/inpatient encounters.
Outcomes
The primary outcome was proportion of days at home (PDH),6,7,12 defined as the number of days subjects were not engaged in cancer-related care divided by total follow-up days. Individuals were deemed “engaged in care” if hospitalized (for any cause), seen in an emergency department (for any cause), or seen in an oncology/infusion clinic. Outpatient visits in non-oncology settings were not counted as engaged in cancer-related care. Counting began at diagnosis, with censoring at the end of 2020. Time commitment was also quantified via visit durations, calculated from check-in/check-out timestamps. Only appointments at UNC Health were included, as timestamps were not available for other health systems. Overall survival was calculated via the Kaplan-Meier method. Treatment response was assessed, with individuals achieving a best response of complete remission, complete remission with incomplete hematologic recovery, or a morphological leukemia-free state being considered to have achieved remission.11
Analysis
Descriptive statistics are provided as medians and interquartile ranges (IQR) or frequencies and percentages. Associations between patients’ characteristics and treatment regimens were assessed via modified Poisson models and characterized using relative risks. Cox proportional hazards models were used to examine associations between overall survival and demographic, disease, and treatment variables. Remission was used as a time-varying covariate in a Cox
proportional hazards model to assess association between remission and survival. Competing risks analysis was used to analyze the association between baseline characteristics and cumulative incidence of remission while adjusting for deaths. Paired t-tests were used to evaluate the impact of remission on pre- and post-remission time at home among those who entered remission. Median visit durations were stratified by treatment group with hypothesis testing via Wilcoxon rank-sum.
PDH was evaluated in terms of total PDH over the entire study period and on a monthly (30 days) basis. For monthly analyses, only patients surviving the entirety of a particular month were included in that month’s total. PDH was described via summary statistics for the overall cohort and stratified by categorical variables. Associations between PDH and baseline characteristics were evaluated via linear regression models. Regression models were adjusted for the log of duration of follow-up, and adjusted means are presented. Adjusted mean PDH values presented in this analysis were evaluated at the average follow-up time of 8.6 months. A multivariable linear regression model with distance from UNC Medical Center, age, ELN risk, and adjustment for duration of follow-up was performed to further characterize these associations.
Results
Demographics
We identified 372 individuals aged ≥60 years with a new diagnosis of AML treated within the UNC Health system from 2015 to 2020. Of these, 137 received azacitidine and/or venetoclax as first-line therapy. Twenty-four individuals were excluded because of incomplete records, yielding a final cohort of 113 patients.
The median age of these 113 individuals was 73 years (range, 61-95 years), with the majority (51.3%) of patients being between 70 and 79 years old (Table 1). The majority of the cohort was white (80.4%), and 56.6% were male. Most were retired or otherwise not currently employed (89.6%). Most were living with a spouse or long-term partner (66.4%) and had health insurance (89.4%). The median distance from home address to the UNC Medical Center was 42.3 miles (IQR, 27-93 miles).
Disease and treatment
ELN risk category was favorable for 8.1%, intermediate for 30.6%, and adverse for 61.3% of the cohort (Table 1). Patients received azacitidine plus venetoclax combination therapy (51.3%), azacitidine monotherapy (38.9%), and other venetoclax-containing regimens (9.7%) as first-line treatment. Baseline demographic covariates and ELN risk were similar across therapy groups (P for all associations >0.05) except that there were more individuals aged 80-
Haematologica | 108 - April 2023 1007 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
89 years in the azacitidine plus venetoclax group compared to the azacitidine monotherapy group (27.6% vs. 9.1%, respectively; P=0.036). Treatment was initiated in an
inpatient setting for 10.9% of individuals in the azacitidine plus venetoclax group and for 0% of the azacitidine monotherapy group.
AML: acute myeloid leukemia; AZA: azacitidine; VEN: venetoclax; ELN: European LeukemiaNet. Missing data not available in records: race (n=1 subject), employment status (n=17), marital status (n=3), rurality (n=2), household income (n=3), ELN risk (n=2). *Comparison between categorical variables assessed via the c2 test. Statistically significant result shown in bold.
Variable All patients (N=113) AZA monotherapy (N=44) AZA+VEN (N=58) P* Demographics Age group, N (%) 60-69 years 26 (23.0) 12 (27.3) 11 (19.0) (reference) 70-79 years 58 (51.3) 24 (54.5) 30 (51.7) 0.55 80-89 years 24 (21.2) 4 (9.1) 16 (27.6) 0.036 ≥90 years 5 (4.4) 4 (9.1) 1 (1.7) 0.34 Race, N (%) White 90 (80.4) 34 (79.1) 47 (81.0) (reference) Black or African-American 19 (17.0) 8 (18.6) 10 (17.2) 0.85 Other 3 (2.7) 1 (2.3) 1 (1.7) 0.83 Sex, N (%) Female 49 (43.4) 20 (45.5) 25 (43.1) 0.81 Male 64 (56.6) 24 (54.5) 33 (56.9) (reference) Employment status, N (%) Not employed or retired 86 (89.6) 33 (91.7) 46 (88.5) (reference) Employed or self-employed 10 (10.4) 3 (8.3) 6 (11.5) 0.59 Marital status, N (%) Not partnered 37 (33.6) 16 (38.1) 21 (30.9) 0.38 Partnered 73 (66.4) 26 (61.9) 47 (69.1) (reference) Insurance status, N (%) Uninsured 12 (10.6) 6 (13.6) 5 (8.6) 0.47 Insured 101 (89.4) 38 (86.4) 53 (91.4) (reference) Rurality, N (%) Rural 5 (4.5) 1 (2.3) 4 (6.9) 0.17 Urbanized area 9 (8.1) 5 (11.6) 4 (6.9) 0.36 Urbanized cluster 97 (87.4) 37 (86.0) 50 (86.2) (reference) Household income quartile, N (%) First (lowest income) 28 (25.5) 10 (23.3) 16 (28.1) (reference) Second 28 (25.5) 14 (32.6) 10 (17.5) 0.17 Third 29 (26.4) 8 (18.6) 16 (28.1) 0.71 Fourth (highest income) 25 (22.7) 11 (25.6) 15 (26.3) 0.78 Disease and treatment First-line treatment, N (%) Azacitidine 44 (38.9) 44 (100) Azacitidine + venetoclax 58 (51.3) 58 (100) Venetoclax + other medication 11 (9.7) ELN 2017 risk, N (%) Favorable 9 (8.1) 3 (7.1) 6 (10.3) 0.50 Intermediate 34 (30.6) 13 (31.0) 19 (32.8) 0.75 Adverse 68 (61.3) 26 (61.9) 33 (56.9) (reference)
Table 1. Demographic and disease characteristics of older adults with acute myeloid leukemia receiving azacitidine and/or venetoclax.
Haematologica | 108 - April 2023 1008 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
Response to treatment and survival
Among the full cohort, 34.5% of patients achieved remission (complete remission, complete remission with incomplete hematologic recovery, or a morphological leukemia-free state). The 6-month competing risk-adjusted event rate of remission was 0.31. Higher remission rates were observed among those receiving azacitidine plus venetoclax (6-month competing risk-adjusted event rate 0.50, unadjusted 58.9%) than among those receiving azacitidine alone (6-month competing risk-adjusted event rate 0.10, unadjusted 9.5%) (hazard ratio [HR]=7.50, 95% confidence interval [95% CI]: 2.76-20.40, P<0.001) (Online Supplementary Table S1). Increased distance from the UNC Medical Center was associated with lower remission rates in these competing risk-adjusted analyses (HR=0.93 for every 10 additional miles away, 95% CI: 0.870.997, P=0.042). No other factors, including demographic variables and ELN risk category, were associated with having achieved remission (P for all associations >0.05). The median overall survival for the full cohort was 7.7 months (95% CI: 3.8-10.9) and was similar across therapy groups (Online Supplementary Table S2). There were also no significant associations observed between overall survival and ELN risk, race, sex, or other demographic variables (P for all associations >0.05). When evaluated as a time-varying covariate, having achieved remission was associated with superior survival, with a hazard ratio for death of 0.56 (P=0.043), indicating achieving remission was associated with longer survival.
Proportion of days at home
Over the full follow-up period, the mean PDH was 0.58 (95% CI: 0.54-0.63) with a median of 0.63. PDH was positively associated with length of follow-up, with a statistically significant association between PDH and log length of follow-up time (regression coefficient 0.15, 95% CI: 0.120.17, P<0.0001). After adjusting for log follow-up time, the adjusted mean PDH was 0.67 (95% CI: 0.63-0.71). PDH was similar among those with adverse-risk (adjusted mean 0.65), intermediate-risk (0.69, P=0.34), and favorable-risk AML (0.64, P=0.81). PDH did not differ significantly among therapy groups: azacitidine plus venetoclax (adjusted mean 0.68), azacitidine alone (0.66, P=0.64 when compared to azacitidine plus venetoclax), and other venetoclax-containing regimens (0.65, P=0.65). There was also no significant difference in the proportion of days spent as inpatients or in outpatient clinics between patients in the azacitidine and azacitidine plus venetoclax groups (Online Supplementary Table S3).
Patients who had longer driving distances to UNC spent more time engaged in oncologic care, and thus less time at home (regression coefficient for PDH -0.0008 per mile, 95% CI: -0.0013 to -0.0002, P=0.011). Patients who lived farther than 50 miles from UNC spent approximately 7%
more days (amounting to 2.1 more days per month) engaged in oncology care than those who lived closer (adjusted mean PDH 0.63 vs. 0.70 for those <50 miles, P= 0.04). Patients who lived farther from UNC spent more days in hospital (26% of all follow-up days spent as inpatients for those living ≥50 miles vs. 20% for those living <50 miles from UNC). After adjusting for age, ELN risk, and median household income, the association between increased distance from the medical center and decreased PDH remained but was no longer statistically significant (P=0.08). There was no association seen between distance from UNC and chemotherapy regimen received. Other than driving distance, no individual covariate (including patient’s age) had a significant association with PDH when adjusted for length of follow-up (all P>0.05).
When evaluated on a month-by-month basis, the PDH rose over time among survivors (Table 2). For example, among patients who survived the full month, the mean PDH was 0.51 (95% CI: 0.47-54, n=105) in month 1 following diagnosis, 0.64 (95% CI: 0.58-0.70, n=75) in month 3, and 0.77 (95% CI: 0.72-0.82, n=57) in month 6. This pattern is demonstrated in Figures 1 and 2, which summarize person-days at home versus person-days engaged in care for 12 months following diagnosis, including contributions of person-days from those who did not survive the full month. Early in follow-up, most care days consisted of inpatient hospitalization (28% of all person-days in the first month), with a decline in hospitalization burden over the ensuing 12 months among survivors (4% of all person-days in month 12). The proportion of days seen in a clinic remained relatively constant over the year following diagnosis (in a range of 16-21% of all person-days). Patterns in monthly PDH were similar between those receiving azacitidine monotherapy and those receiving azacitidine plus venetoclax (Figure 3, Table 2). There was no significant difference in types of visits between those receiving azacitidine monotherapy and those receiving azacitidine plus venetoclax. The rate of early hospitalizations (days hospitalized during the first 30 days following diagnosis) was also similar between recipients of azacitidine monotherapy and azacitidine plus venetoclax, despite the higher percentage of patients within the azacitidine plus venetoclax group initiating therapy while admitted. Among the 38 patients who entered remission, more days were spent at home following remission than prior to remission. For this group, 53% (95% CI: 46-60%) of days prior to remission were spent at home compared to 72% (95% CI: 66-78%, P<0.0001) of days after remission.
Time spent in a clinic
For visits occurring within the UNC Health system, the median duration of oncology provider visits was 123.3 minutes (IQR, 80.1-185.2 minutes) (Table 3). The median duration of an infusion encounter was 169.3 minutes (IQR,
Haematologica | 108 - April 2023 1009 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
Table 2. Proportion of person-days spent at home by month from diagnosis among older adults with acute myeloid leukemia, stratified by first-line therapy.
AZA: azacitidine, VEN: venetoclax, PDH: proportion of days at home, 95% CI: 95% confidence interval. Number of subjects (N) reflects number of individuals in each group surviving at the end of the respective month. PDH for each month was calculated as the proportion of persondays not engaged in oncologic care among these survivors.
93.8-307.7) and the median duration of a laboratory encounter was 43.3 minutes (IQR, 25.7-83.3). Receipt of azacitidine plus venetoclax was associated with longer clinic visits (median 127.9 vs. 112.9 minutes, P<0.001) and infusion visits (median 194.3 vs. 132.5 minutes, P<0.001) compared to receipt of azacitidine monotherapy. Patients spent a median of 66 minutes (IQR, 30-110 minutes) in the two-way drive time to/from clinic for each day with an appointment. The median drive time was shorter among those receiving azacitidine plus venetoclax monotherapy than among those receiving azacitidine monotherapy (median 50 vs. 66 minutes, P<0.0001).
Discussion
The recent development of multiple novel effective chemotherapeutic agents has expanded treatment options for older adults with AML. Increasingly, patients are empowered to participate in a shared decision-making process that considers their values and preferences with respect to the anticipated outcomes of treatments. Time spent at home represents a critical but under-reported outcome of cancer therapies.7,13,14 We have previously shown that patients with AML are willing to sacrifice remission rates to achieve time at home.5 Currently, clinical trials in AML do not routinely capture, nor report, the time
at home experienced by patients following chemotherapy. Reliably capturing this outcome in routine care and clinical trials will enable rational, data-driven shared decisionmaking regarding chemotherapy. Here, we demonstrate the feasibility of quantifying the time at home older patients achieve following first-line therapy and report, for the first time, a comparison between current treatments. Older AML patients receiving azacitidine, azacitidine plus venetoclax, or other venetoclax-containing regimens spent an average of 42% of their days engaged in oncology care. This time commitment was observed for AML patients despite patients receiving “less intensive” chemotherapy compared to intensive induction chemotherapy that often necessitates a lengthy period as an inpatient. Even among survivors 1 year from diagnosis, 24% of days were devoted to their cancer treatment, reflecting the need for ongoing close monitoring and potential transfusion support among those with AML. These data are similar to those from a smaller group of patients receiving hypomethylating agents in an Australian study published prior to the approval of venetoclax.12
The time commitment faced by patients with AML is striking when compared to that by patients with advanced solid cancers. Rocque and colleagues reported that women with newly diagnosed metastatic breast cancer spent 7 to 10% of days in the 3 months following diagnosis engaged in oncology care.6 Among adults with newly di-
Month Full cohort (N=113) AZA monotherapy (N=44) AZA+VEN (N=58) Other VENcontaining regimen (N=11) N Mean PDH (95% CI) N Mean PDH (95% CI) N Mean PDH (95% CI) N Mean PDH (95% CI) 1 105 0.51 (0.47-0.55) 38 0.54 (0.48-0.61) 56 0.47 (0.41-0.53) 11 0.62 (0.47-0.76) 2 89 0.57 (0.52-0.62) 32 0.60 (0.52-0.68) 47 0.57 (0.51-0.63) 10 0.47 (0.28-0.65) 3 75 0.64 (0.58-0.70) 28 0.63 (0.54-0.72) 39 0.67 (0.58-0.75) 8 0.56 (0.30-0.83) 4 66 0.70 (0.65-0.76) 26 0.71 (0.62-0.79) 34 0.69 (0.60-0.78) 6 0.79 (0.56-1.02) 5 59 0.76 (0.71-0.81) 23 0.77 (0.71-0.84) 32 0.73 (0.65-0.81) 4 0.86 (0.59-1.12) 6 57 0.77 (0.72-0.82) 23 0.76 (0.70-0.82) 31 0.78 (0.70-0.86) 3 0.73 (0.45-1.02) 7 54 0.76 (0.71-0.82) 21 0.80 (0.73-0.87) 30 0.75 (0.66-0.84) 3 0.66 (0.00-1.32) 8 48 0.75 (0.68-0.82) 17 0.70 (0.61-0.79) 28 0.78 (0.68-0.88) 3 0.73 (0.30-1.17) 9 41 0.76 (0.69-0.82) 16 0.78 (0.71-0.86) 23 0.76 (0.66-0.86) 2 0.55 (0.34-0.76) 10 38 0.78 (0.70-0.85) 16 0.81 (0.72-0.90) 21 0.77 (0.65-0.89) 1 0.53 (---) 11 35 0.76 (0.68-0.84) 16 0.77 (0.66-0.89) 18 0.75 (0.62-0.87) 1 0.63 (---) 12 32 0.77 (0.69-0.85) 16 0.76 (0.65-0.88) 15 0.78 (0.64-0.93) 1 0.70 (---)
Haematologica | 108 - April 2023 1010 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
Figure 1. Proportion of person-days spent at home and engaged in cancer care among older adults with acute myeloid leukemia treated with azacitidine and/or venetoclax. The figure displays the person-days either at home (not engaged in care) or seen in each venue (clinic, emergency department, or hospital) among survivors. The proportion of days spent at home rose in the year following diagnosis, with a decline in the proportion of care-days consisting of inpatient hospitalization over the same period Values of the proportion of days at home in this figure differ slightly from monthly proportion values (PDH) in Table 2, as the PDH values in Table 2 were calculated only among survivors for a full month, whereas this figure reflects person-day contributions from individuals who did not survive the entirety of the respective month. ED: emergency department.
agnosed metastatic pancreatic cancer, who face a similarly poor prognosis as that of older adults with AML, Bange and colleagues described a cohort median of 10% of days devoted to cancer care following diagnosis.7 Older patients with AML spend over four times as many days engaged in oncology care as these patients. Time at home for patients with other hematologic malignancies has not been routinely reported outside of the context of end-oflife or hospice care.15
In a phase III trial of adults deemed ineligible for intensive chemotherapy, the addition of venetoclax to azacitidine increased remission rates and improved median overall survival from 9.6 to 14.7 months.16 In the current study, patients receiving azacitidine plus venetoclax had superior remission rates; however, this did not translate into a greater proportion of time at home for this group in aggregate. Patients receiving venetoclax also had longer clinic visits and longer visits for infusion, potentially related to a greater need for transfusion support. These data suggest that the clear benefit seen from the addition of venetoclax in clinical trials may not result in a substantial
improvement in time spent at home for patients. This finding was surprising as we anticipated that remission would translate into prolonged survival, fewer clinic visits, fewer visits to the infusion center, and fewer admissions. We therefore performed a post hoc analysis looking at time at home before and after remission. Among the minority of individuals who entered remission, more days were spent at home after remission than prior to achieving remission. However, in aggregate, remission status did not correlate with the overall PDH throughout follow-up. Assessment of this relationship is complicated, as multiple factors may contribute to increasing time at home further from diagnosis, including achieving remission, better management of an individual’s symptoms further into treatment, and early deaths of the sickest individuals who may account for the greatest time engaged in care. Survival in our azacitidine plus venetoclax cohort was also shorter than that described in some other realworld-data analyses17,18 which likely resulted in decreased time at home achieved by these patients. However, similar survival results have been described in other retrospective
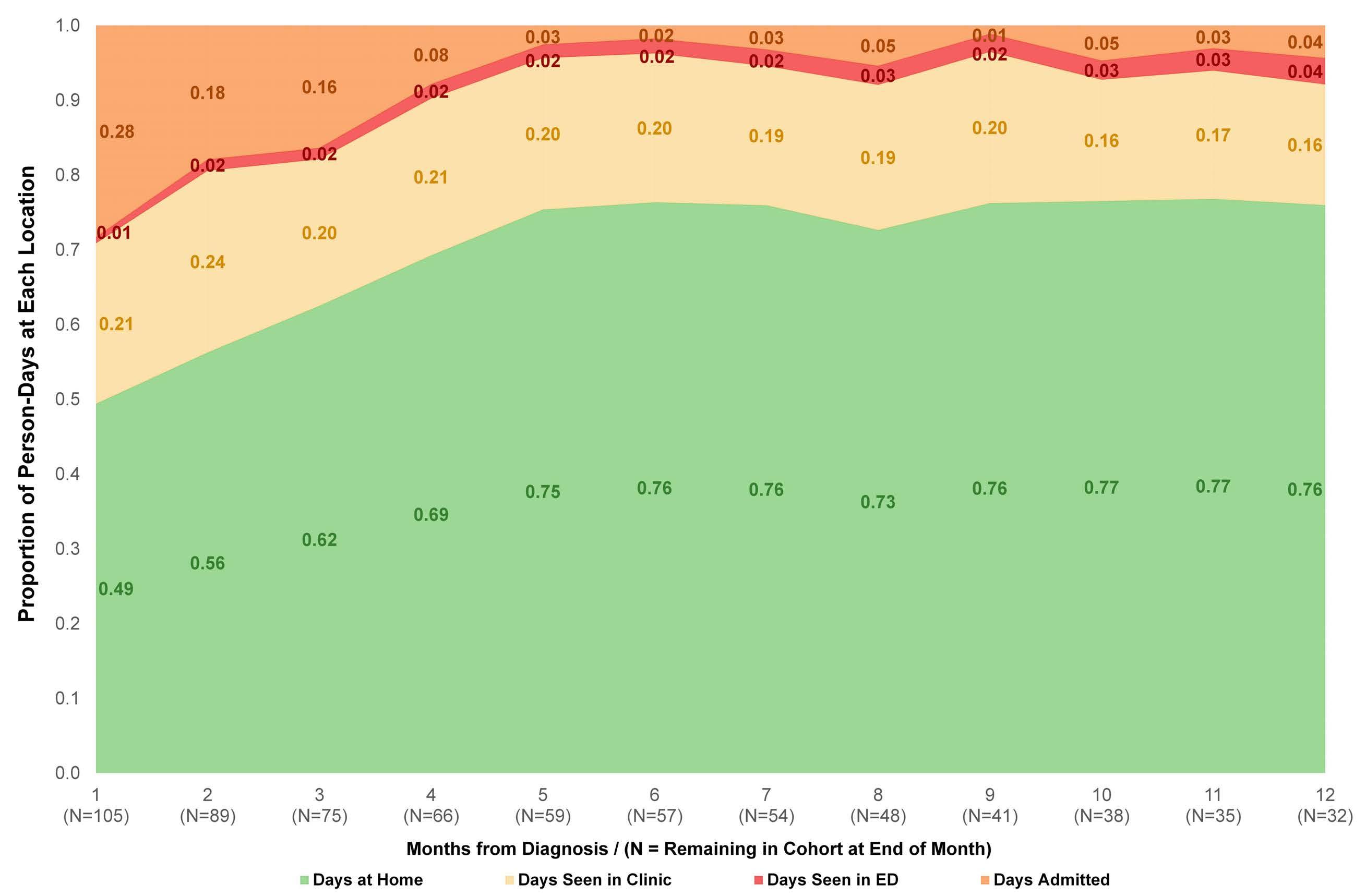
Haematologica | 108 - April 2023 1011 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
cohorts.19 Furthermore, although we examined multiple variables when comparing therapies, confounding by other factors may be present. In particular, we did not quantify comorbidity or frailty other than by age alone. Adjusting for these factors may have altered our findings, especially for patients between the age of 80 and 89 years, who were over-represented in the azacitidine plus venetoclax cohort. Additionally, developing expertise in optimizing delivery of a complicated treatment regimen such as azacitidine plus venetoclax requires time and experience. As oncologists and cancer centers become more familiar with delivering venetoclax-based regimens, time at home may improve.
No significant differences were noted in the PDH following AML diagnosis based on age, race, sex, or disease risk according to ELN criteria in our cohort. We did observe a significantly lower PDH among those living farther from our primary referral hospital and comprehensive cancer center. This difference is partially attributable to more inpatient days experienced by patients living farther from the cancer center, a care-delivery pattern that has been described in the setting of other malignancies.10 The as-
sociation between increased distance from the medical center and decreased PDH was no longer significant when adjusted for age, ELN risk, and median household income, possibly related to a degree of collinearity between driving distance and patients’ age.
This study has several additional limitations. Although records were captured from several health systems, this study includes only data from patients who were seen at least once at a single academic referral center. Consequently, the results may not be generalizable to adults treated entirely in community settings in which the burden of care may be different. Furthermore, the need for complete follow-up records to evaluate PDH may have biased our sample toward individuals with shorter survival and less time at home, as older adults with more stable disease may have experienced greater co-management with a local oncologist outside of the captured records. Simi-
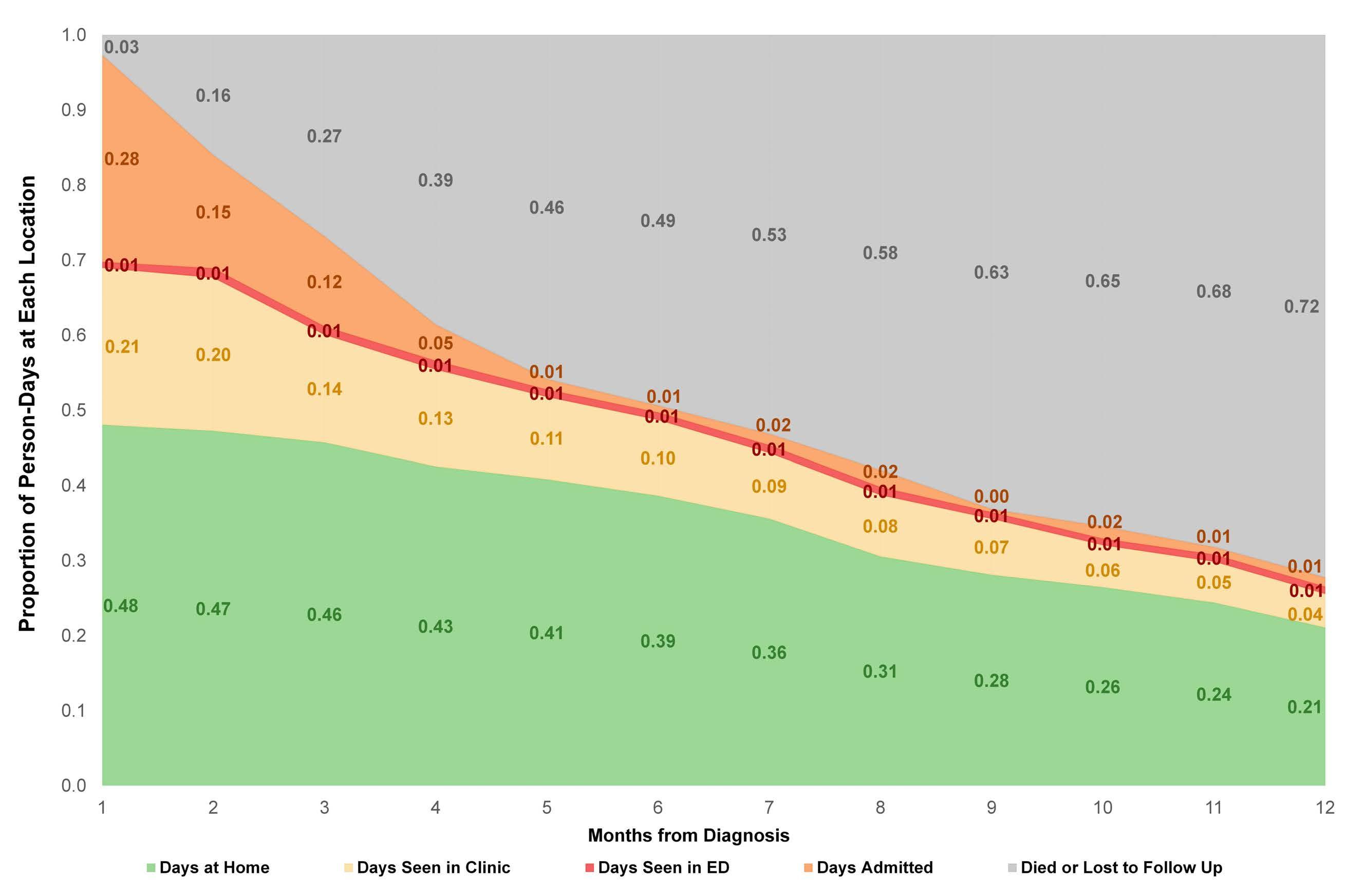 larly, we could not readily quantify days spent in inpatient rehabilitation facilities or nursing facilities, as these stays were not fully reflected in the medical records. A claimsbased approach may allow this component of care burden to be assessed more fully. No data have been published
larly, we could not readily quantify days spent in inpatient rehabilitation facilities or nursing facilities, as these stays were not fully reflected in the medical records. A claimsbased approach may allow this component of care burden to be assessed more fully. No data have been published
Haematologica | 108 - April 2023 1012 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
Figure 2. Proportion of person-days spent at home and engaged in cancer care among older adults with acute myeloid leukemia including decedents and those lost to follow-up. This figure displays the same data as in Figure 1, with the addition of individuals who died or were lost to follow-up. Values of the proportion of days at home in this figure differ slightly from monthly proportion values (PDH) in Table 2, as the PDH values in Table 2 were calculated only among survivors for a full month, whereas this figure reflects person-day contributions from individuals who did not survive the entirety of the respective month. ED: emergency department.
Figure 3. Proportion of person-days spent at home among older adults with acute myeloid leukemia treated with azacitidine with or without venetoclax in the year following diagnosis. Among survivors in each group, the proportion of days spent at home (and not engaged in oncologic care) rose over time, with generally similar proportions observed in each treatment group at each time point. Adjacent values for the two treatment groups are contemporaneous and have been offset for legibility. Error bars reflect 95% confidence intervals.
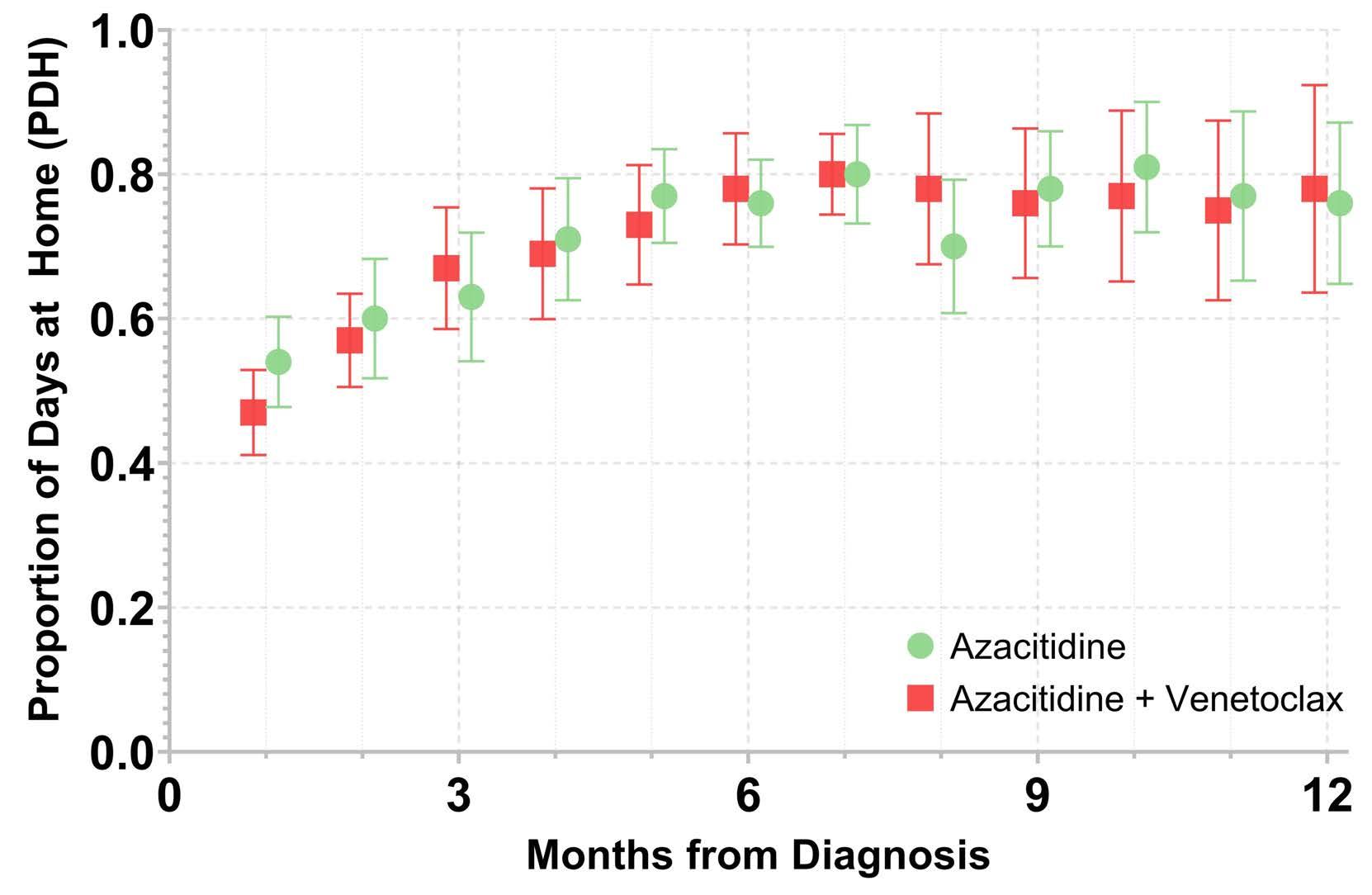
on the relative value patients with AML place on the specific health states in this study (e.g., clinic visits, infusion visits, hospitalization). We were, therefore, unable to adjust the reported time at home for perceived quality of this time to patients. Studies identifying the relative value of these health states would be informative to allow for quality-adjustment of the reported findings. This study represents an important step in understanding the experience of older adults with AML. A patient’s time is a finite resource that is increasingly consumed by complex cancer care. When compared to those with other advanced cancers, older adults with AML face a markedly increased burden on their time due to oncology care. The recent therapeutic paradigm shift in AML is an opportunity
to leverage these remarkable advancements into meaningful improvements in patients’ lives. Future prospective studies should evaluate time at home as an endpoint as part of a broader strategy to incorporate patient-centered outcomes into drug development and shared decisionmaking strategies. Additionally, interventions directed at increasing time at home, such as utilization of virtual visits/telehealth or clustering of care, are critically needed to maximize patients’ time at home. Early studies of increased utilization of telehealth in the context of the COVID-19 pandemic suggest patients often find this caredelivery approach satisfactory.14 More intensive outpatient care models that support patients’ time at home have also been described,20 including all-outpatient “intensive” in-
AZA: azacitidine; VEN: venetoclax. Median duration based on data from the University of North Carolina (UNC) Health system only. Numbers for total visits include data from UNC and other health systems (for provider visits and infusion visits) or UNC Health system only (for laboratory visits). *Oncology provider visits include encounters with physicians, nurse practitioners, physician assistants, or clinical pharmacist practitioners. †Drive times are calculated to the nearest minute with typical traffic and reflect time to/from the location of the clinical encounter, starting from the patients’ home addresses. One patient from the azacitdine group was excluded from the drive-time calculations because of a documented home address outside of North Carolina/bordering states. ‡P values reflect comparisons of duration of visits or drive time between therapy groups via the Wilcoxon rank-sum test.
Full
(N=113) AZA monotherapy
AZA+VEN
Median duration, minutes (IQR) Visits (total number) Visits per patient (mean) Median duration, minutes (IQR) Visits (total number) Visits per patient (mean) Median duration, minutes (IQR) Visits (total number) Visits per patient (mean) P‡ Oncology provider* 123.3 (80.1-185.2) 2,110 18.7 112.9 (73.2-165.2) 714 16.2 127.9 (85.7-192.2) 1,006 17.3 <0.0001 Infusion 169.3 (93.8-307.7) 5,539 49.0 132.5 (77.1-237.1) 2,083 47.3 194.3 (104.8-343.1) 2,606 44.9 <0.0001 Laboratory 43.3 (25.7-83.3) 1,878 16.6 58.9 (29-113.8) 573 13.0 39.3 (24.6-71.8) 1,216 21.0 <0.0001 Driving† (two-way) 66 (30-110) 5,639 50.3 66 (24-110) 2,392 55.6 50 (36-152) 2,717 46.8 <0.0001
cohort
(N=44)
(N=58)
Table 3. Time spent on oncology clinic appointments among older adults with acute myeloid leukemia treated with azacitidine and/or venetoclax.
Haematologica | 108 - April 2023 1013 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
duction strategies for AML.21 Whenever possible, such approaches to increase patients’ time at home should be implemented as overall survival remains tragically short for older patients with AML.
Disclosures
None of the authors has a relevant conflict of interest. CEJ reports research funding (Conquer Cancer) outside the submitted work. ALB reports research funding (Carevive, Jazz Pharmaceuticals) and honoraria (Servier Pharmaceuticals) outside the submitted work. LAC reports research funding (Alexion Pharmaceuticals , Machaon Diagnostics) outside the submitted work. MCF reports research funding (Bellicum Pharmaceuticals , Macrogenics, Rafael Pharmaceuticals) and consulting (Macrogenics, Daiichi Sankyo, Agios) outside the submitted work. DRR reports research funding (Conquer Cancer, Palliative Care Research Cooperative Group) outside the submitted work.
Contributions
CEJ and DRR conceptualized the study and identified the study cohort; CEJ and KEB extracted data from charts; CEJ,
References
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136-1152.
2. Acute myeloid leukemia: SEER stat fact sheets: 2019. Surveillance Epidemiology and End Results (SEER). Vol. 2019: National Cancer Institute. 2019.
3. DiNardo CD, Wei AH. How I treat acute myeloid leukemia in the era of new drugs. Blood. 2020;135(2):85-96.
4. Richardson DR, Oakes AH, Crossnohere NL, et al. Prioritizing the worries of AML patients: quantifying patient experience using best-worst scaling. Psychooncology. 2021;30(7):1104-1111.
5. Richardson DR, Crossnohere NL, Seo J, et al. Age at diagnosis and patient preferences for treatment outcomes in AML: a discrete choice experiment to explore meaningful benefits. Cancer Epidemiol Biomarkers Prev. 2020;29(5):942-948.
6. Rocque GB, Williams CP, Ingram SA, et al. Health care‐related time costs in patients with metastatic breast cancer. Cancer Med. 2020;9(22):8423-8431.
7. Bange EM, Doucette A, Gabriel PE, et al. Opportunity costs of receiving palliative chemotherapy for metastatic pancreatic ductal adenocarcinoma. JCO Oncol Pract. 2020;16(8):e678-e687.
8. United States Census Bureau. 2010 Census Urban and Rural Classification and Urban Area Criteria. Vol. 2021: United States Census Bureau. 2019.
9. United States Census Bureau. American Community Survey (ACS). Vol. 2021: United States Census Bureau. 2020.
10. Rocque GB, Williams CP, Miller HD, et al. Impact of travel time on health care costs and resource use by phase of care for older patients with cancer. J Clin Oncol. 2019;37(22):1935-1945.
11. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
12. Sharplin K, Wee LYA, Singhal D, et al. Outcomes and health care utilization of older patients with acute myeloid leukemia. J
HMH, AMD, and DRR designed the data analysis; HMH and AMD performed the data analysis; CEJ, KEB, ALB, LAC, MCF, and DRR contributed to interpretation of the results; CEJ drafted the manuscript. All authors reviewed the final manuscript.
Funding
The project described was supported by the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) through Grant Award Number UL1TR002489 and by a National Research Service Award Post-Doctoral Traineeship from the Agency for Healthcare Research and Quality sponsored by The Cecil G. Sheps Center for Health Services Research at The University of North Carolina at Chapel Hill through Grant Award Number T32-HS000032. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Data-sharing statement
Data can be requested via e-mail to the corresponding author.
Geriatr Oncol. 2021;12(2):243-249.
13. Fundytus A, Prasad V, Booth CM. Has the current oncology value paradigm forgotten patients’ time? JAMA Oncol. 2021;7(12):1757-1758.
14. Banerjee R, George M, Gupta A. Maximizing home time for persons with cancer. JCO Oncol Pract. 2021;17(9):513-516.
15. Cheung MC, Croxford R, Earle CC, Singh S. Days spent at home in the last 6 months of life: a quality indicator of end of life care in patients with hematologic malignancies. Leuk Lymphoma. 2020;61(1):146-155.
16. Dinardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629.
17. Garcia JS, Wolach O, Vachhani P, et al. Comparative effectiveness of venetoclax combinations vs other therapies among patients with newly diagnosed acute myeloid leukemia: results from the AML real world evidence (ARC) initiative. Blood. 2021;138(Suppl 1):2328.
18. Bouligny IM, Maher KR. Outcomes of induction with venetoclax in combination with decitabine, azacitidine, or low-dose cytarabine for treatment of AML: a real-world retrospective analysis. Blood. 2021;138(Suppl 1):2335.
19. Vachhani P, Abbas JA, Flahavan EM, et al. Real world treatment patterns and outcomes of venetoclax (Ven) and hypomethylating agents (HMA) in patients with newly diagnosed acute myeloid leukemia (AML) in the United States. Blood. 2021;138(Suppl 1):2290.
20. Handley NR, Bekelman JE. The oncology hospital at home. J Clin Oncol. 2019;37(6):448-452.
21. Mabrey FL, Gardner KM, Shannon Dorcy K, et al. Outpatient intensive induction chemotherapy for acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Adv. 2020;4(4):611-616.
Haematologica | 108 - April 2023 1014 ARTICLE - Time at home among older adults with AML C.E. Jensen et al.
Characterization of therapy-related acute myeloid leukemia: increasing incidence and prognostic implications
Christer
11Department of Medicine, Huddinge, Division of Hematology, Karolinska Institutet, Stockholm; 2Department of Medical Sciences, Uppsala University, Uppsala; 3Department of Hematology, Sahlgrenska University Hospital, Gothenburg; 4Department of Hematology, Skåne University Hospital, Lund, and Department of Hematology, Stem Cell Center, Department of Laboratory Medicine, Lund University, Lund; 5Department of Hematology, Linköping University Hospital, Linköping; 6Department of Hematology, Norrland University Hospital, Umeå and 7Department of Hematology, Sundsvall Hospital, Sundsvall, Sweden
Abstract
Correspondence: C. Nilsson
christer.nilsson@ki.se
Received: April 18, 2022.
Accepted: July 21, 2022.
Early view: August 25, 2022.
https://doi.org/10.3324/haematol.2022.281233
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Studies of therapy-related AML (t-AML) are usually performed in selected cohorts and reliable incidence rates are lacking. In this study, we characterized, defined the incidence over time and studied prognostic implications in all t-AML patients diagnosed in Sweden between 1997 and 2015. Data were retrieved from nationwide population-based registries. In total, 6,779 AML patients were included in the study, of whom 686 (10%) had t-AML. The median age for t-AML was 71 years and 392 (57%) patients were females. During the study period, the incidence of t-AML almost doubled with a yearly increase in t-AML of 4.5% (95% confidence interval: 2.8%-6.2%), which contributed significantly to the general increase in AML incidence over the study period. t-AML solidly constituted over 10% of all AML cases during the later period of the study. Primary diagnoses with the largest increase in incidence and decrease in mortality rate during the study period (i.e., breast and prostate cancer) contributed significantly to the increased incidence of t-AML. In multivariable analysis, t-AML was associated with poorer outcome in cytogenetically intermediate- and adverse-risk cases but t-AML had no significant impact on outcome in favorable-risk AML, including core binding leukemias, acute promyelocytic leukemia and AML with mutated NPM1 without FLT3-ITD. We conclude that there is a strong increase in incidence in t-AML over time and that t-AML constitutes a successively larger proportion of the AML cases. Furthermore, we conclude that t-AML confers a poor prognosis in cytogenetically intermediate- and adverse-risk, but not in favorable-risk AML.
Introduction
Therapy-related AML (t-AML) is a feared complication of treatment with chemotherapy and/or radiation. The World Health Organization (WHO) defines the disease entity therapy-related myeloid neoplasms as myeloid neoplasms secondary to cytotoxic treatment, in which t-AML is categorized along with therapy-related myelodysplastic syndrome (MDS) and therapy-related myeloproliferative neoplasms (MPN).1 Between 5-10% of all AML cases have been reported to be therapy-related,2,3 and treatment for a solid cancer increases the risk of AML up to 10-fold with chemotherapy4 and 2.5-fold after treatment with radiotherapy.5 While solid and hematologic tumors dominate as the primary disease, t-AML may also occur after treatment of non-malignant diseases, especially inflammatory dis-
orders.3,6-8 The mechanism behind the increased risk of AML after chemo- and/or radiation therapy are not fully understood, but it is currently regarded as a combination of mutagenic effects of chemotherapy and/or radiation and a selective pressure by this treatment, favoring existing premalignant clones in the blood and bone marrow.9-13 This may lead to clonal expansion of the premalignant clone as well as clonal evolution with accumulation of additional leukemogenic genetic aberrations that evolve to t-AML. t-AML is associated with a worse prognosis compared to that of de novo AML, and the disease presents with a higher rate of adverse genetic aberrations compared to de novo AML.3 Alkylating agents and radiation therapy have typically been associated with aberrations in chromosomes 5 and 7, while treatment with topoisomerase II inhibitors has been linked to rearrangements of
Nilsson,1 Fredrika Linde,2 Erik Hulegårdh,3 Hege Garelius,3 Vladimir Lazarevic,4 Petar Antunovic,5 Jörg Cammenga,5 Stefan Deneberg,1 Anna Eriksson,2 Martin Jädersten,1 Cecilia Kämpe Björkvall,6 Lars Möllgård,3 Lovisa Wennström,3 Emma Ölander,7 Martin Höglund,2 Gunnar Juliusson4 and Sören Lehmann1,2
Haematologica | 108 - April 2023 1015 ARTICLE - Acute Myeloid Leukemia
chromosome 11q23, involving the KMT2A gene.14 However, as chemotherapy treatment usually consists of a combination of agents, this subdivision of t-AML is difficult to identify clinically, and it is no longer part of the WHO classification.15 Complex and monosomal karyotypes as well as TP53 mutations are also overrepresented in t-AML.7,16,17 The spectrum of tumors that precedes t-AML depends on incidence rates, type of treatment and the likelihood of long-term survival for the particular cancer type.4,18 Thus, tumors treated with high intensity chemotherapy followed by high cure rates, are overrepresented as pre-t-AML diagnoses.3,19 Consequently, malignancies that are less frequently treated with intensive chemotherapy, such as prostate cancer, or malignancies with short survival are generally underrepresented as primary diseases in tAML.3,20
Most studies of t-AML are either small or performed in selected cohorts and within clinical trials. Larger populationbased studies in which t-AML is defined from a real-life perspective, and from which true incidence rates can be calculated, are exceedingly rare. Here, we use populationbased quality registries in Sweden to investigate changes in incidence and survival in t-AML over time, as well as to characterize t-AML in a large real-life AML cohort. Furthermore, we study prognostic factors for t-AML patients, as well as t-AML in itself as a prognostic factor within genetic subtypes of AML.
Methods
Data collection
Data were collected from three nationwide registries: the Swedish AML Registry (SAMLR), the Swedish Cancer Registry (SCR) and the Swedish Rheumatology Quality Register (SRQ). SAMLR has been validated against the SCR defining the coverage of SAMLR to 98% of all AML patients nationwide.21 Each registry, its data and the methods to collect data are described in more detail in the Online Supplementary Methods. All registries use national unique personal identification numbers enabling identification of individuals across the registries.
Patients
The study included all AML patients ≥8 years reported to SAMLR between January 1, 1997 and December 31, 2015.
Two t-AML patients with a prior AML diagnosis reported as the antecedent disease were excluded. The median follow-up time was 101 months during which 4,406 deaths were registered; 10 patients were lost to follow-up and 1,076 patients were still alive at the end of follow-up. Intensively treated patients received induction and consolidation therapy according to Swedish guidelines,22 with a chemotherapy intensity comparable to that of the classical
“3 + 7” AML induction. The ethical review board in Gothenburg approved the study (Dnr 781/13).
Definitions
t-AML was defined as AML with a prior diagnosis of a malignant or non-malignant disease treated with chemotherapy and/or radiation therapy. All chemotherapy regimens were considered, including methotrexate and cyclophosphamide for autoimmune diseases, but excluding immunosuppressive treatment. Patients treated with chemotherapy for MDS or MPN and progressing to AML were not defined as therapy-related cases, whereas patients developing MDS or MPN during the period between the treatment for the primary disease and the diagnosis of AML were considered to have t-AML. The 2010 European LeukemiaNet (ELN) criteria23 were used to stratify patients into cytogenetic risk groups. For risk classification of patients with available data on NPM1 and FLT3-ITD mutational status (starting from 2007) the ELN2010 criteria for NPM1 and FLT3-ITD were used. Complete remission was defined as <5% blasts in the bone marrow and recovery of peripheral blood counts.
Statistical analyses
Between-group comparisons of categorical and continuous variables were performed using the Pearson c2 test and the Mann-Whitney U test, respectively. The unequal variances t test was used to compare mean incidence rates between time periods. Overall survival was estimated using the Kaplan-Meier method and compared with the log-rank test or univariable Cox regression. Multivariable Cox regression was used for survival analyses with covariates specified where used. The proportional hazards assumption was tested for each covariate by graphical inspection of the scaled Schoenfeld residuals. Two-tailed tests with a 0.05 level of significance were used throughout the analyses. No imputation of missing data was performed. All data preparation and statistical analyses were conducted in R, version 3.6.0.24 Additional information about the statistical methods can be found in the Online Supplementary Material.
Results
Patients’ characteristics
A total of 5,492 patients were diagnosed with either t-AML (n=686) or de novo AML (n=4,806) in Sweden between 1997 and 2015, with an additional 1,287 patients being diagnosed with AML with an antecedent hematologic disease (AHD-AML). The aim of this study was to characterize t-AML patients specifically, which is why details on AHDAML patients are not further described. Of all AML cases in the study cohort, the proportion of t-AML was 10%, de novo AML 71% and AHD-AML 19%.
Haematologica | 108 - April 2023 1016 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al.
The clinical characteristics of patients with t-AML and de novo AML are shown in Table 1. Age at diagnosis was similar between t-AML and de novo AML (median 71 vs. 70 years, respectively) while t-AML had a female predominance (57%, compared to 49% in de novo AML). Performance status was similar with the majority of patients having Eastern Cooperative Oncology Group grade 0 or 1 (61% vs 63%, t-AML and de novo AML, respectively). Adverse cytogenetic risk was more common (46% vs. 28%, P<0.001) in t-AML, while favorable risk was somewhat less common
(12% vs. 16%, P=0.030). The proportion of APL was similar in t-AML compared to de novo AML (4% vs. 5%). Minor differences were observed in standard clinical parameters, with patients with t-AML having lower median bone marrow blast percentages (46% vs. 50%), lower white blood cell counts (6.1 vs. 8.7x109/L) and lower platelet counts (50 vs. 65 x109/L) compared to those with de novo AML. Inflammatory diseases and unspecified chronic diseases were more prevalent in t-AML than in de novo AML (13% vs. 3% and 23% vs. 9%, respectively), while the prevalence
Note: Data on FLT3-ITD, NPM1, CEBPA, comorbidities and blood chemistry are only reported since 2007. AML: acute myeloid leukemia; t-AML: therapy-related AML; ECOG: Eastern Cooperative Oncology Group; APL: acute promyelocytic leukemia; CR: complete remission;
allogeneic
stem cell transplantation; CR1: first
Overall De novo AML t-AML P N of patients 5,492 4,806 686 Age at diagnosis in years, median (range) 70 (18-100) 70 (18-98) 71 (18-98) 0.519 Female, N (%) 2,743 (50) 2,351 (49) 392 (57) <0.001 Year of diagnosis, N (%) <0.001 1997-2002 1,621 (30) 1,455 (30) 166 (24) 2003-2008 1,672 (30) 1,487 (31) 185 (27) 2009-2015 2,199 (40) 1,864 (39) 335 (49) ECOG performance status, N (%) 0.079 0 1,086 (20) 980 (21) 106 (16) 1 2,308 (42) 2,005 (42) 303 (45) 2 953 (17) 825 (17) 128 (19) 3 607 (11) 524 (11) 83 (12) 4 361 (7) 317 (7) 44 (6) Missing 134 (2) 118 (2) 16 (2) Cytogenetic risk, N (%) (data reported on 73% of cases) <0.001 Adverse 1,201 (30) 964 (28) 237 (46) Intermediate 2,187 (55) 1,974 (56) 213 (42) Favorable 619 (15) 559 (16) 60 (12) FLT3-ITD, N (%) (data reported on 23% of cases) 0.053 Present 316 (25) 295 (26) 21 (18) Absent 927 (75) 829 (74) 98 (82) NPM1, N (%) (data reported on 21% of cases) 0.172 Present 348 (30) 321 (31) 27 (24) Absent 805 (70) 720 (69) 85 (76) CEBPA, N (%) (data reported on 7% of cases) 0.709 Present 27 (7) 25 (8) 2 (5) Absent 348 (93) 307 (92) 41 (95) APL, N (%) 250 (5) 224 (5) 26 (4) 0.355 Treatment, N (%) <0.001 Hypomethylating agent 89 (2) 61 (1) 28 (4) Intensive 3,590 (70) 3,205 (71) 385 (60) None/palliative 1,485 (29) 1,251 (28) 234 (36) CR, intensively treated, N (%) 2,617 (73) 2,393 (75) 224 (58) <0.001 Allogeneic HCT, N (%) 0.001 In CR1 606 (11) 555 (12) 51 (7) In > CR1, relapsed or refractory state 208 (4) 191 (4) 17 (2) No 4,678 (85) 4,060 (84) 618 (90)
Table 1. Clinical characteristics of patients with therapy-related acute myeloid leukemia and de novo acute myeloid leukemia.
HCT:
hematopoietic
complete remission. Haematologica | 108 - April 2023 1017 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al.
of other comorbidities was similar (Online Supplementary Table S1).
Patients with t-AML were less likely to receive intensive induction treatment (60% vs. 71%, P<0.001) and intensively treated patients were less likely to reach complete remission (58% vs. 75%; P<0.001) compared to those with de novo AML. Allogeneic hematopoietic cell transplantation (HCT), regardless of disease state, was performed in 9% of the patients with t-AML compared to 16% of those with de novo AML (P<0.001). Corresponding rates of HCT in first remission were 7% in t-AML compared to 12% in de novo AML (P=0.002). There was no increase or decrease of transplantation rates over time (data not shown).
The incidence of therapy-related acute myeloid leukemia is increasing over time
The age-standardized incidence rate of t-AML increased during the study period from a mean of 0.39 cases per 100,000 adult inhabitants between 1997 and 2006 to 0.63 between 2007 and 2015 (P=0.004) (Figure 1A). During the last 2 years of the study period (2014-2015), the average age-adjusted incidence rate of t-AML was 0.95. There was also a slight increase in the incidence of de novo AML from 3.42 to 3.66 between the two study periods (P=0.02). For all AML (including de novo AML, t-AML and AHD-AML), the incidence rate increased from 4.69 (1997-2006) to 5.32

(2007-2015) (P=0.0006). The estimated annual percentage change was 4.5 % in t-AML (95% confidence interval [95% CI]: 2.8%-6.2%) compared to 0.7% in de novo AML (95% CI: 0.2%-1.2%). The relative increase in t-AML was substantial and contributed to a large proportion of the increased incidence of AML as a whole. As a result, the proportion of tAML of all AML cases increased from 8.3% in 1997-2006 to 11.8% in 2007-2015 (P=0.004, t test) (Figure 1B). For the last 2 years of the study period (2014-2015), the average proportion of t-AML was 16%.
To define how different primary tumors contributed to the increase in t-AML incidence, we compared the average number of t-AML cases per year per primary tumor disease between 1997-2002 and 2009-2015. Non-malignant diseases, breast cancer and prostate cancer contributed most to the rise in t-AML incidence with an absolute increase of 8.2, 5.6 and 5.2 cases per year per 10 million inhabitants, representing 40%, 27% and 25% of the increase in t-AML, respectively. A slightly increased contribution was also seen for lymphoma and gastrointestinal cancers. In contrast, gynecological cancers and multiple myeloma displayed a relatively stable or decreased contribution to tAML over time (Online Supplementary Table S2 and Online Supplementary Figure S1). To understand the contribution of the different primary diagnoses, we used official data from the Swedish Cancer Registry to compare the change
Haematologica | 108 - April 2023 1018 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al. A B
Figure 1. Incidence rates and proportion of therapy-related acute myeloid leukemias. (A) Age-standardized incidence rates of therapy-related acute myeloid leukemia (AML) and de novo AML. (B) Percentage of therapy-related AML cases of all AML cases. Figures show linear regression lines with 95% confidence intervals for visual aid.
in t-AML incidence to the change in incidence and mortality rate for the malignant diagnoses during the same period. As shown in Online Supplementary Figure S2A-L, the primary diagnoses that contributed most to the increase in t-AML, i.e. breast and prostate cancer, also displayed the largest increase in incidence and most prominent decrease in mortality rates. For gynecological cancers, incidence and mortality rates remained relatively unchanged for cervix and uterine corpus cancer while they decreased for ovarian carcinoma. Lymphoma displayed a small increase in incidence and a slightly improved survival. Thus, the increase in t-AML incidence seems to be at least partly driven by increased incidence and improved survival of some major tumors.
Characterization of diagnoses prior to therapy-related acute myeloid leukemia
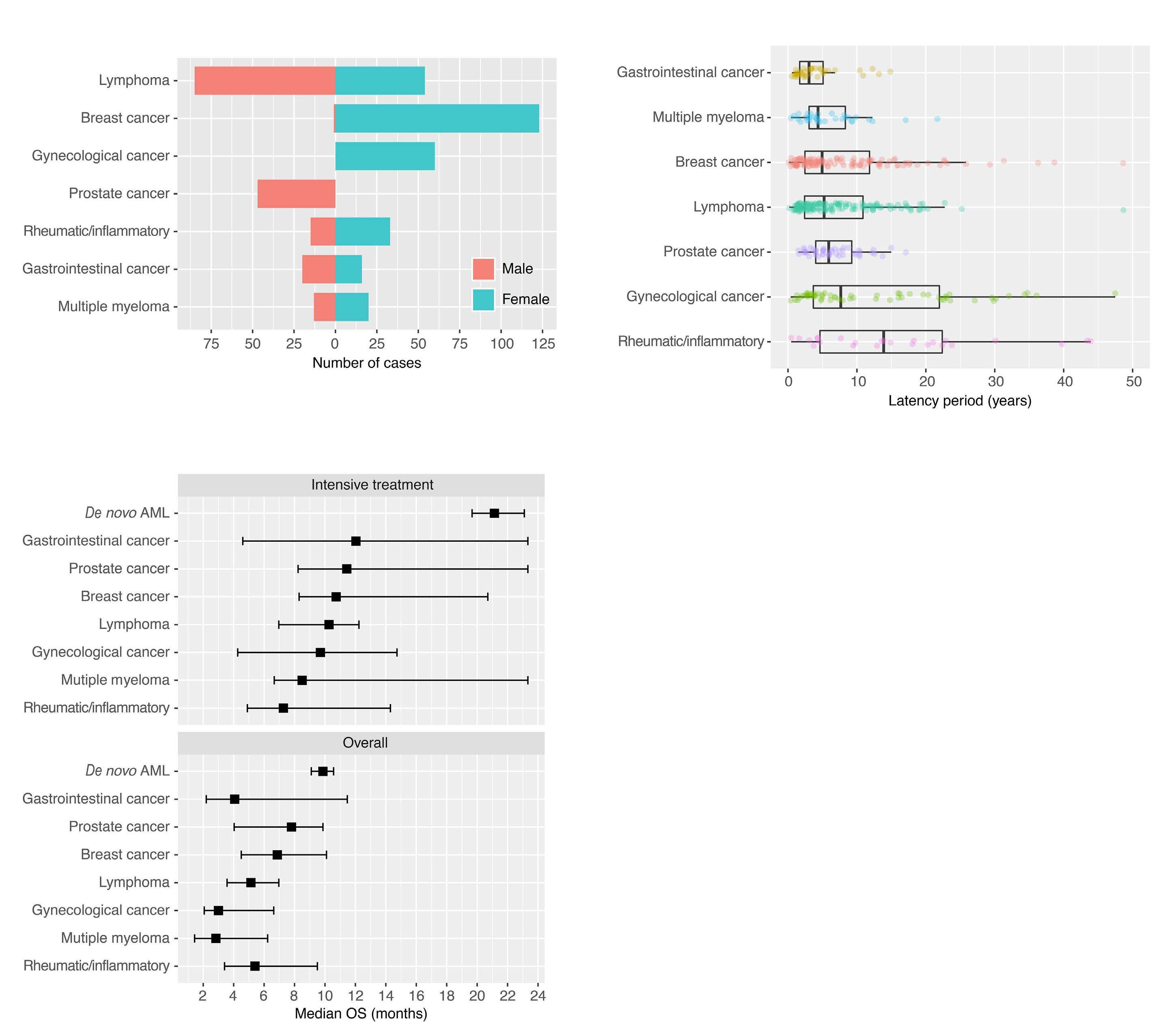
Of primary disorders, for which chemotherapy and/or radiation was given, 55% were non-hematologic solid cancers, 25% hematologic cancers and 18% non-malignant diseases. Figure 2A shows the number of cases and the gender distribution for each primary diagnosis and more details are provided in Online Supplementary Table S3. The most common primary diagnoses were lymphoma (n=139), followed by breast cancer (n=124), rheumatic and inflammatory disease (n=122), gynecological malignancies (n=60), prostate cancer (n=47), gastrointestinal cancer (n=36), and myeloma (n=33). Treatment for the primary disease was chemotherapy alone in 49%, radiation therapy alone in 25%, and chemotherapy and radiation therapy combined in 26% of the patients (Online Supplementary Table S3).
Figure 2. Primary diagnosis, gender, latency times and survival in therapy-related acute myeloid leukemia. (A) Number of cases and gender based on primary diagnoses. (B) Latencies between diagnosis of the primary disease and acute myeloid leukemia related to different primary diagnoses. (C) Median overall survival in intensively treated patients and overall in therapy-related acute myeloid leukemia grouped by primary disease. AML: acute myeloid leukemia; t-AML: therapy-related acute myeloid leukemia;
A Haematologica | 108 - April 2023 1019 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al. C B
Nineteen percent (n=133) of t-AML patients had more than one tumor diagnosis before the onset of t-AML, either occurring before, or more often after the malignancy for which the first chemotherapy and/or radiation therapy was given. Figure 3 displays in detail the complexity of sequential relations between the different diagnoses. Seventeen percent (n=118) of the patients had a diagnosis of MDS between the primary disease and t-AML.
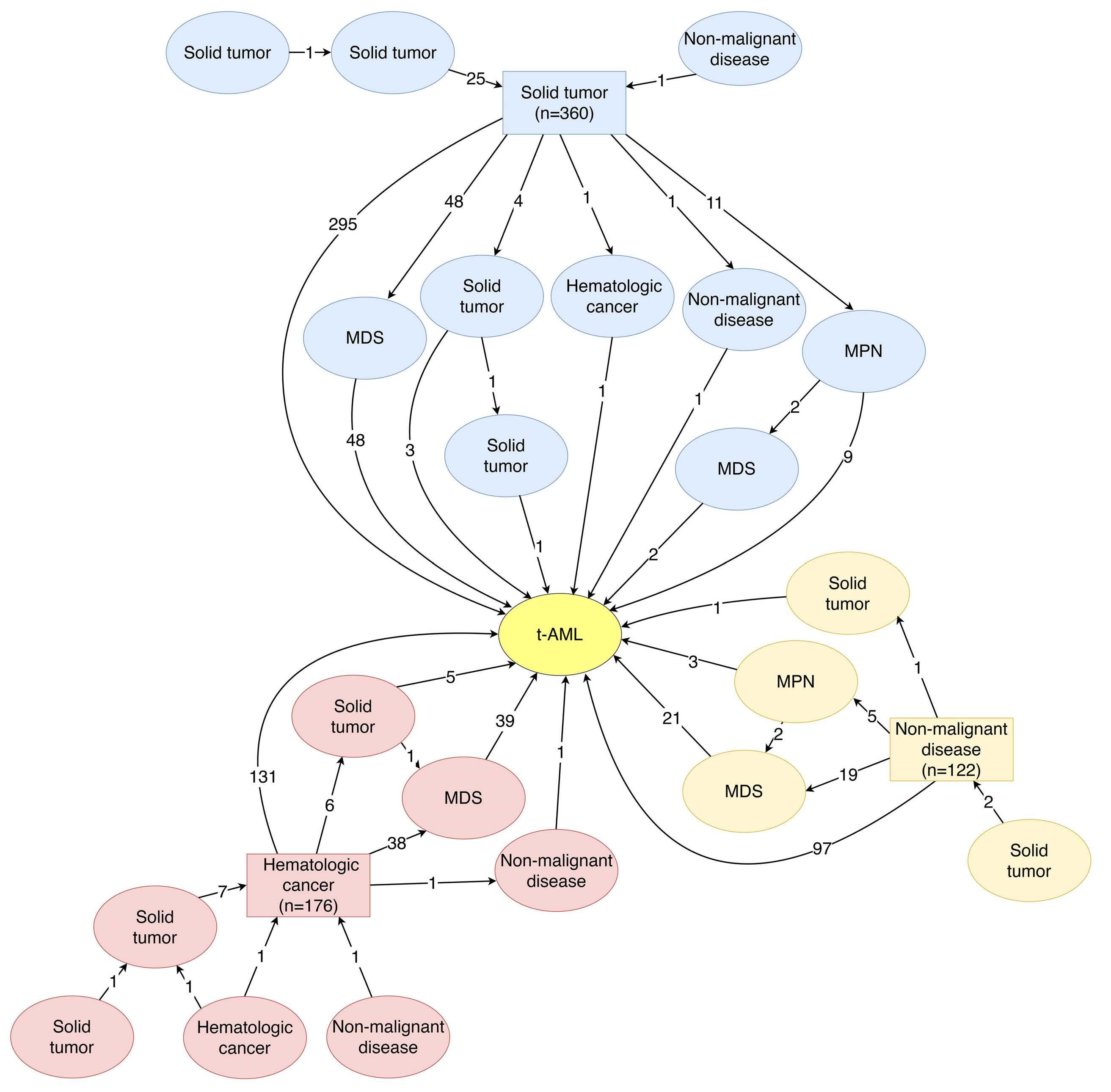
Latency periods
The latency period after a primary hematologic malignancy or a solid cancer to the diagnosis of t-AML was similar (median 57 vs. 61 months, respectively; P=0.287) while the latency period after diagnosis of a primary non-malignant disease (i.e., inflammatory or rheumatic disease) was longer (173 months; P<0.001), but also more variable (Online Supplementary Figure S3A). Figure 2B displays latency periods related to type of primary disease. The latency also varied with the type of primary treatment with similar la-
Figure 3. Paths to therapy-related acute myeloid leukemia. The figure shows the sequence of diagnoses of all malignant and/or hematologic diseases preceding the diagnosis of therapy-related acute myeloid leukemia (t-AML). Squares denote the primary disease for which cytotoxic treatment was given and ovals denote additional diseases, either prior to the primary disease or between the primary disease and t-AML. Numbers between squares and ovals represent number of cases. Twenty-eight cases for which the order of the disease could not be determined were excluded from the figure. MDS: myelodysplastic syndromes; MPN: myeloproliferative neoplasms.
Haematologica | 108 - April 2023 1020 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al.
tency after chemotherapy alone and combination therapy (chemotherapy + radiation) (median 54 vs. 55 months, respectively; P=0.848), but longer after radiation alone (median 97 months; P<0.001) (Online Supplementary Figure S3B). Both a primary non-malignant disease and radiation alone were associated with a longer latency period in multivariable analysis adjusting for age at primary diagnosis, type of primary treatment, type of primary disease and cytogenetic risk at diagnosis (Online Supplementary Table S4). The median latency periods of patients with -5/-7, 11q23/MLL and t(15;17) were 101 months (95% CI: 74-126 months), 41 months (95% CI: 29-75 months) and 84 months (95% CI: 41-190 months), respectively, compared to 62 months in t-AML overall (95% CI: 56-71 months). There was no association between the latency period and favorable, intermediate or adverse risk at large.
Improved survival for therapy-related acute myeloid leukemia patients over time
The estimated 5-year survival of patients with t-AML was 10% overall, 17% in intensively treated patients (n=385), and 48% in patients who underwent HCT (n=68). As a comparison, the 5-year survival rates in patients with de novo AML were 23% overall, 34% in intensively treated patients, and 57% in transplanted patients. Similarly, the median overall survival was 5.0 months for all t-AML, 9.5 months in intensively treated patients, and 48 months in patients who underwent HCT (Online Supplementary Figure S4A-C). Corresponding median overall survival times patients with de novo AML were 9.7 months overall, 20.7 months in intensively treated patients, and 113 months in transplanted patients. Overall survival among patients transplanted in first complete remission was worse in t-AML patients than in de novo AML patients in both univariable analysis (hazard ratio [HR]=1.56, 95% CI: 1.06-2.29) and after adjusting for age and cytogenetic risk (HR=1.50, 95% CI: 1.02-2.22). Outcome of patients with t-AML improved over time with a 5-year overall survival in intensively treated patients of 11%, 16% and 23% for 1997-2002, 2003-2008 and 20092015, respectively (Online Supplementary Figure S5A, B). Patients with an antecedent diagnosis of MDS had a worse outcome in univariable analysis compared to patients without prior MDS (HR=1.52, 95% CI: 1.09-2.12, P=0.012) but when adjusting for age and cytogenetic risk, no significant survival difference was observed (Online Supplementary Table S5). Patients with t-AML did worse compared to those with de novo AML regardless of primary disease (Figure 2C, details in Online Supplementary Table S6) and type of treatment for the primary disease (chemotherapy, radiotherapy or chemo-radiotherapy; Online Supplementary Figure S6A, B). No correlation between the type of cytotoxic treatment and cytogenetic risk was found (distribution of risk in patients treated with radiotherapy only vs. patients treated with chemotherapy or combination therapy,
P=0.06). The cumulative incidence of death and relapse was higher in patients older than 60 years, although the relapse difference was not statistically significant (Online Supplementary Figure S7).
Therapy-related acute myeloid leukemia had a strong negative impact on survival in cytogenetically intermediate- and adverse- but not favorable-risk acute myeloid leukemia
How t-AML contributes to prognosis within different AML risk groups remains unclear. Therefore, we analyzed the outcomes of patients with t-AML compared to those with de novo AML in each cytogenetic risk group. In crude analyses, t-AML did not have a statistically significant impact on outcome in favorable-risk AML, regardless of whether acute promyelocytic leukemia (APL) was included in the favorable-risk group or not. In contrast, there was a significant and strong negative impact of t-AML in intermediate- and adverse-risk AML (Online Supplementary Figure S8A-E). After adjusting for age and performance status, survival was still similar in favorable-risk t-AML (with or without APL) and in APL alone compared to de novo AML: HR=0.99 (P=0.95); HR=1.11 (P=0.73), and HR=0.89 (P=0.78), respectively. In contrast, after the same adjustments in the intermediate- and adverse-risk groups, t-AML had a strong negative impact on overall survival: HR=1.53 (P<0001) and HR=1.59 (P<0.001), respectively (Table 2).
Therapy-related acute myeloid leukemia does not confer poor prognosis in acute myeloid leukemia with mutated NPM1 and absence of FLT3-ITD
We further analyzed the impact of NPM1 mutations and FLT3-ITD (data available from 2007). There were 112 t-AML patients for whom information on NPM1 status was available and 119 for whom FLT3-ITD data were available; among de novo AML patients, the corresponding numbers were 1,041 and 1,124 patients, respectively. In t-AML patients, FLT3-ITD alone did not add prognostic information (Figure 4A), while t-AML patients with NPM1mut had significantly better overall survival compared to NPM1wt t-AML patients (Figure 4B). t-AML patients with NPM1mut in combination with FLT3wt had better survival compared to t-AML patients with any other combination of NPM1 and FLT3 status (Figure 4C). t-AML patients with NPM1mut/FLT3wt had a similar overall survival compared to that of NPM1mut/FLT3wt de novo AML patients (HR=0.82, 95% CI: 0.40-1.68, P=0.584 for t-AML vs. de novo AML) (Figure 4D). The comparison showed similar results when adjusting for age, performance status and cytogenetic risk (t-AML vs. de novo AML: HR=0.75, 95% CI: 0.36-1.58, P=0.454).
The role of t-AML in relation to the ELN2017 classification including a more comprehensive mutational characterization was outside the scope of the study as the study was based on retrospective registry data from a period before
Haematologica | 108 - April 2023 1021 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al.
comprehensive mutational analyses was performed routinely. Nevertheless, full mutational characterization was available for 58 of the patients and these data are provided in Online Supplementary Figure S9.
Discussion
In this study, we identified several features of t-AML that point to important current trends regarding t-AML and that provide real-world information on the role of t-AML in the prognostic outcome of AML. Firstly, we identified a significant increase in the incidence of t-AML during the last two to three decades. This increase is substantial and approaches a doubling during the study period. Consequently, it is much more likely that a newly diagnosed AML is a t-AML today compared to 20 to 30 years ago. This increase in t-AML was seen for primary tumors that have shown an increased incidence and decreased mortality during the same period, such as breast and prostate cancers. This is in part due to improvement of cancer therapy, leading to better long-term survival after mutagenic cancer treatments.25,26 With continuous improvements in survival of patients with malignant diseases, the number of individuals at risk of t-AML will continue to rise. However, the use of less mutagenic non-chemotherapeutic agents in cancer treatment may in part counteract this development.
Over the whole study period, the incidence of t-AML in our study was 0.51 cases per 100,000 inhabitants, which is substantially higher than that found in a recent American study using data from the Surveillance, Epidemiology, and End Results (SEER) registry in which the incidence rate of therapy-related myeloid neoplasms was 0.13 cases per 100,000 during 2001–2014.19 The reason for the discrepancy is unclear. Although it may be related to differences concerning treatment and/or survival of malignancies in general, it more likely reflects a difference in reporting and coverage of the registries as well as their sensitivity and specificity for recording previous treatment with chemotherapy and radiation. The advantage of the Swedish reporting system is the 98% coverage of the Swedish AML Registry, the compulsory reporting of cancer diagnoses to the Swedish Cancer Registry and the possibility of crosslinking the registries for each individual using personal identification numbers.
Regarding the basic characterization of the t-AML patients in this study, it is in line with most previous studies2,7,8 while we here can provide data from the largest population-based cohort of AML so far. The good performance status of t-AML patients is somewhat surprising, however, it is confirmed by several other studies.27,28 The good performance status contrasts with the fact that t-AML patients were less likely to receive intensive chemotherapy
Table 2. Multivariable Cox regression analysis of overall survival in therapy-related acute myeloid leukemia compared to de novo AML in intensively treated patients according to cytogenetic risk.
AML: acute myeloid leukemia; t-AML: therapy-related AML; APL: acute promyelocytic leukemia; OS: overall survival; HR: hazard ratio; CI: confidence interval; ECOG: Eastern Cooperative Oncology Group; PS: performance status.
and allogeneic transplantation. This could be due to fact that other factors rather than performance status can affect the treating physician’s assessment of the patient, such as sequelae from previous exposure to chemotherapy or radiation as well as the presence of a still existing primary tumor. Regarding the latency periods, they were associated with factors such as type of treatment (chemotherapy or radiation) and type of primary disease (malignant vs. non-malignant). In line with previous studies, patients with 11q23/MLL-rearranged AML had shorter latency periods, while those with -5/-7 had longer latency periods.7,29
Secondly, we were able to examine and describe the role of t-AML in the prognosis within AML risk groups. It is well established that karyotype is an independent prognostic factor among intensively treated patients with t-AML and that poor-risk karyotypes are overrepresented in tAML.7,29,30 However, data on the impact of t-AML within each cytogenetic risk group are conflicting. Our data showed a significantly worse survival in t-AML patients within the intermediate and adverse AML risk groups. In contrast, t-AML patients with favorable-risk AML had the same survival as that of patients with de novo AML, also when correcting for other prognostic factors. Schoch et al. previously reported data contrasting with ours, with no
HR (95% CI) P Favorable risk APL excluded t-AML vs. de novo 1.11 (0.62-1.97) 0.732 Age 1.04 (1.03-1.06) <0.001 ECOG PS 2-4 vs. 0-1 1.28 (0.84-1.96) 0.255 APL included t-AML vs. de novo 0.99 (0.62-1.57) 0.950 Age 1.05 (1.04-1.06) <0.001 ECOG PS 2-4 vs. 0-1 1.40 (1.00-1.94) 0.048 APL only t-AML vs. de novo 0.89 (0.40-2.00) 0.777 Age 1.06 (1.03-1.08) <0.001 ECOG PS 2-4 vs. 0-1 2.24 (1.27-3.98) 0.006 Intermediate risk t-AML vs. de novo 1.53 (1.25-1.87) <0.001 Age 1.04 (1.03-1.05) <0.001 ECOG PS 2-4 vs. 0-1 1.55 (1.34-1.79) <0.001 Adverse risk t-AML vs. de novo 1.59 (1.31-1.93) <0.001 Age 1.03 (1.02-1.04) <0.001 ECOG PS 2-4 vs. 0-1 1.56 (1.31-1.86) <0.001
Haematologica | 108 - April 2023 1022 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al.
survival differences in intermediate- and adverse-risk patients but a significant difference in the favorable-risk group.29 However, their study was considerably smaller with 93 t-AML patients in total, making the numbers of patients in each cytogenetic risk group very small. Aldoss and Pullarkat have reviewed whether the outcome of patients with favorable-risk t-AML is comparable to that of those with favorable risk de novo AML.31 The concluded that a therapy-related disease does not affect survival in APL but
that it negatively affects outcome in core-binding factor AML, although not to a degree that should change transplantation indications in therapy-related core-binding factor leukemia. Our data support the approach that indications for allogeneic transplantation should remain the same in t-AML as de novo AML in patients with favorable-risk cytogenetics.
Mutational screening of NPM1 and FLT3-ITD is part of clinical routine as well as ELN recommendations for prognostic
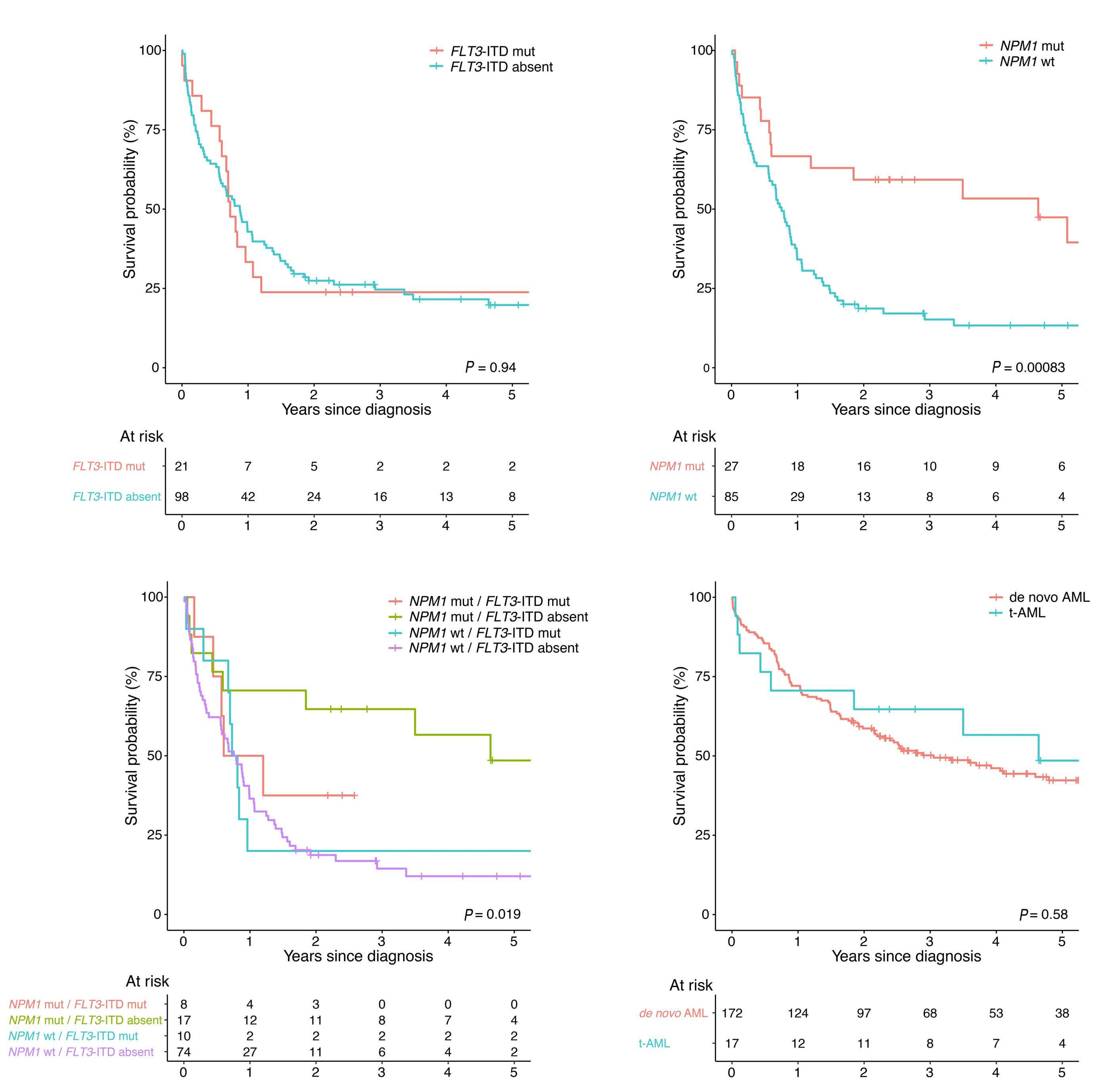
A B C Haematologica | 108 - April 2023 1023 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al. D
Figure 4. Survival in patients with therapy-related acute myeloid leukemia based on NPM1 and FLT3-ITD status. Overall survival by (A) FLT3-ITD status, (B) NPM1 mutational status and (C) FLT3-ITD/NPM1 status in therapy-related acute myeloid leukemia (tAML). (D) Comparison of overall survival between t-AML and de novo acute myeloid leukemia (AML) in patients with NPM1mut/FLT3-ITDabsent
classification32 and for the first time, we could study the role of t-AML in these mutational subcategories of AML. In our study, the presence of FLT3-ITD did not have a negative impact on survival in t-AML patients, while patients with NPM1mut had a significantly better survival compared to those with NPM1wt t-AML. The lack of impact of FLT3-ITD on survival may be due to the different cytogenetic pattern in t-AML, in which the prognostic role of FLT3-ITD can be dependent on the concomitant cytogenetic aberrations.33 Also, all FLT3-ITD mutations, regardless of allelic ratio were included, which may impaired the prognostic power of our analysis. As in de novo AML, the survival of patients with NPM1mut/FLT3wt was better than that of patients with any other combination of these mutations.34,35 Importantly, survival of t-AML patients with NPM1mut/FLT3wt, defined as favorable risk in ELN, did not differ significantly from that of NPM1mut/FLT3wt de novo patients in a multivariable analysis. This suggests that, similarly to cytogenetically favorablerisk AML, t-AML patients with NPM1mut/FLT3wt could be approached with the same treatment strategy as used for de novo NPM1mut/FLT3wt AML patients.
The reason for the worse outcome of intermediate- and poor-risk patients with t-AML, compared to their de novo counterparts, also when adjusting for other factors such as cytogenetics, age and performance status, is likely multifactorial. A higher frequency of unfavorable mutations and/or an enrichment of mutations originating from clonal hematopoiesis in patients with t-AML may contribute.9,13,36
As data in this study emerge from the clinical routine, which did not include mutational screening until very late during the study period, mutational data are lacking in this study. Therefore, there is a need for future studies that elucidate the role of t-AML in the setting of full mutation information. Standard induction treatment for AML remained mainly unchanged during the study period. More recently, new therapeutic options have become available, and these could potentially change the course of the disease in the future. For instance, prolonged survival in AML, including t-AML, has been seen among patients treated with CPX351.37 A broader use of hypomethylating agents alone or in combination with antibodies or novel inhibitors targeting FLT3, IDH1/2 and BCL2 also has the potential to im-
References
1. Swerdlow S, Campo E, Harris N, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). Lyon: IARC, 2017.
2. Granfeldt Ostgard LS, Medeiros BC, Sengelov H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national populationbased cohort study. J Clin Oncol. 2015;33(31):3641-3649.
3. Hulegardh E, Nilsson C, Lazarevic V, et al. Characterization and prognostic features of secondary acute myeloid leukemia in a
prove outcome in certain subtypes of t-AML.
In summary, this large population-based study reveals a significant increase in the incidence of t-AML over time, which points to the reality of an increasing likelihood of encounter patients with this malignancy in the clinical practice. Thus, we need more knowledge of, as well as better treatment approaches for, these patients. Given the poor survival in t-AML it is especially important to develop non-chemotherapeutic treatment approaches to malignant and inflammatory diseases, which should result in a decreased risk of t-AML. Our data support an approach in which the indication for HCT should be the same in patients with favorable-risk cytogenetics or NPM1mut/FLT3wt with respect to previous chemotherapy or radiation treatment. On the contrary, intermediate- and adverse-risk patients with t-AML have even poorer survival compared to their de novo counterparts and novel treatment approaches for these patients are highly warranted.
Disclosures
No conflicts of interest to disclose.
Contributions
CN and SL designed the study and gathered, assembled and classified all data. CN and SL performed all analyses and interpreted the data with contributions from FL, EH, HG and LM. CN and SL wrote the manuscript. All other authors contributed to the Swedish AML registry. All authors critically reviewed and approved the manuscript.
Funding
The study was supported by the Swedish Cancer Foundation, Swedish Research Council and Stockholm County Council.
Data-sharing statement
The datasets generated during and/or analyzed during the current study are not publicly available due to privacy concerns and limitations from the ethical review board but are available from the corresponding author upon reasonable request.
population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol. 2015;90(3):208-214.
4. Morton LM, Dores GM, Schonfeld SJ, et al. Association of chemotherapy for solid tumors with development of therapyrelated myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019;5(3):318-325.
5. Teepen JC, Curtis RE, Dores GM, et al. Risk of subsequent myeloid neoplasms after radiotherapy treatment for a solid cancer among adults in the United States, 2000-2014.
Haematologica | 108 - April 2023 1024 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al.
Leukemia. 2018;32(12):2580-2589.
6. Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102(1):43-52.
7. Kayser S, Dohner K, Krauter J, et al. The impact of therapyrelated acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. 2011;117(7):2137-2145.
8. Fianchi L, Pagano L, Piciocchi A, et al. Characteristics and outcome of therapy-related myeloid neoplasms: report from the Italian network on secondary leukemias. Am J Hematol. 2015;90(5):E80-85.
9. Bolton KL, Ptashkin RN, Gao T, et al. Oncologic therapy shapes the fitness landscape of clonal hematopoiesis. bioRxiv. 2019 Nov 20. doi: org/10.1101/848739 [preprint; not peer reviewed].
10. Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18(1):112-121.
11. Joannides M, Grimwade D. Molecular biology of therapy-related leukaemias. Clin Transl Oncol. 2010;12(1):8-14.
12. Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 2017;18(1):100-111.
13. Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552-555.
14. Andersen MK, Johansson B, Larsen SO, Pedersen-Bjergaard J. Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica. 1998;83(6):483-488.
15. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
16. Ok CY, Patel KP, Garcia-Manero G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39(3):348-354.
17. Shih AH, Chung SS, Dolezal EK, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98(6):908-912.
18. Morton LM, Dores GM, Tucker MA, et al. Evolving risk of therapyrelated acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975-2008. Blood. 2013;121(15):2996-3004.
19. Guru Murthy GS, Hamadani M, Dhakal B, Hari P, Atallah E. Incidence and survival of therapy related myeloid neoplasm in United States. Leuk Res. 2018;71:95-99.
20. Howlader N, Noone A, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2016. 2019 Available from: https://seer.cancer.gov/csr/1975_2016/
21. Juliusson G, Lazarevic V, Horstedt AS, Hagberg O, Hoglund M, Swedish Acute Leukemia Registry Group. Acute myeloid leukemia in the real world: why population-based registries are needed. Blood. 2012;119(17):3890-3899.
22. The Swedish AML group. Swedish national AML care program for adult patients. Akut myeloisk leukemi (AML) - Nationellt
vårdprogram 2019.
23. Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453-474.
24. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, 2019.
25. Arnold M, Rutherford MJ, Bardot A, et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995-2014 (ICBP SURVMARK-2): a population-based study. Lancet Oncol. 2019;20(11):1493-1505.
26. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975-2014, featuring survival. J Natl Cancer Inst. 2017;109(9):djx030.
27. Nagel G, Weber D, Fromm E, et al. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann Hematol. 2017;96(12):1993-2003.
28. Østgård LS, Nørgaard JM, Sengeløv H, et al. Comorbidity and performance status in acute myeloid leukemia patients: a nation-wide population-based cohort study. Leukemia. 2015;29(3):548-555.
29. Schoch C, Kern W, Schnittger S, Hiddemann W, Haferlach T. Karyotype is an independent prognostic parameter in therapyrelated acute myeloid leukemia (t-AML): an analysis of 93 patients with t-AML in comparison to 1091 patients with de novo AML. Leukemia. 2004;18(1):120-125.
30. Kern W, Haferlach T, Schnittger S, Hiddemann W, Schoch C. Prognosis in therapy-related acute myeloid leukemia and impact of karyotype. J Clin Oncol. 2004;22(12):2510-2511.
31. Aldoss I, Pullarkat V. Therapy-related acute myeloid leukemia with favorable cytogenetics: still favorable? Leuk Res. 2012;36(12):1547-1551.
32. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
33. Tao S, Wang C, Chen Y, et al. Prognosis and outcome of patients with acute myeloid leukemia based on FLT3-ITD mutation with or without additional abnormal cytogenetics. Oncol Lett. 2019;18(6):6766-6774.
34. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752-1759.
35. Thiede C, Koch S, Creutzig E, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107(10):4011-4020.
36. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367-1376.
37. Kolitz JE, Strickland SA, Cortes JE, et al. Consolidation outcomes in CPX-351 versus cytarabine/daunorubicin-treated older patients with high-risk/secondary acute myeloid leukemia. Leuk Lymphoma. 2020;61(3):631-640.
Haematologica | 108 - April 2023 1025 ARTICLE - Comprehensive characterization of t-AML C. Nilsson et al.
cell transplantation
Correspondence: M. Hudspeth hudspeth@musc.edu
Received: May 30, 2022.
1Medical University of South Carolina Children’s Hospital/Hollings Cancer Center, Charleston, SC, USA; 2Sarah Cannon Transplant and Cellular Therapy Program, Mountain View Hospital, Las Vegas, NV, USA; 3University Hospital for Internal Medicine V, Hematology & Oncology, Medical University, Innsbruck, Austria; 4Hospital Universitario Virgen del Rocío, Instituto de Biomedicina de Sevilla (IBIS/CISC), Universidad de Sevilla, Sevilla, Spain; 5Medizinische Klinik und Poliklinik 1, Universitätsklinikum Carl Gustav Carus an der TU Dresden, Dresden, Germany; 6University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 7Jazz Pharmaceuticals, Palo Alto, CA, USA; and 8Jazz Pharmaceuticals, Philadelphia, PA, USA and 9CHU de Lille, INSERM U1286, Infinite, Lille, France
Abstract
Accepted: December 2, 2022.
Early view: December 15, 2022.
https://doi.org/10.3324/haematol.2022.281471
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Acute graft-versus-host disease (aGvHD) is a life-threatening complication typically occurring within 100 days after allogeneic hematopoietic cell transplantation (allo-HCT). This hypothesis-generating, phase II, prospective, open-label, randomized study (clinicaltrials gov. Identifier: NCT03339297) compared defibrotide added to standard-of-care (SOC) GvHD prophylaxis (defibrotide prophylaxis arm) versus SOC alone (SOC arm) to prevent aGvHD post-transplant. This study estimated incidences of aGvHD and was not statistically powered to assess differences among treatment arms. Patients were randomized 1:1 to defibrotide prophylaxis arm (n=79; median age 57 years; range, 2-69 years) or SOC arm (n=73; median age 56 years; range, 2-72 years). Patient demographics in the two arms were similar except for conditioning regimen type (myeloablative: defibrotide, 76% vs. SOC, 61%) and stem cell source for allo-HCT (bone marrow: defibrotide, 34% vs. SOC, 26%). In the intent-to-treat primary endpoint analysis, the cumulative incidence of grade B-D aGvHD at day 100 post-transplant was 38.4% in the defibrotide prophylaxis arm versus 47.1% in the SOC arm (difference: –8.8%, 90% confidence interval [CI]: –22.5 to 4.9). The difference noted at day 100 became more pronounced in a subgroup analysis of patients who received antithymocyte globulin (defibrotide: 30.4%, SOC: 47.6%; difference: –17.2%; 90% CI: –41.8 to 7.5). Overall survival rates at day 180 post-transplant were similar between arms, as were the rates of serious treatment-emergent adverse events (defibrotide: 42%, SOC: 44%). While the observed differences in endpoints between the two arms were not substantial, these results suggest defibrotide prophylaxis may add a benefit to currently available SOC to prevent aGvHD following allo-HCT without adding significant toxicities.
Introduction
Graft-versus-host disease (GvHD), the most important life-threatening complication after allogeneic hematopoietic cell transplantation (allo-HCT), occurs when donor T cells are activated in response to major or minor histocompatibility mismatch or gene polymorphisms from the recipient, causing a cytotoxic effect in healthy tissues and organs.1,2 Acute GvHD (aGvHD) typically occurs within the first 100 days after allo-HCT.3,4 The pathophysiology of aGvHD broadly follows a three-stage process whereby tissue damage from the conditioning regimen activates the host antigen-presenting cells, which in turn activate donor
T cells to initiate GvHD. Subsequently, T-cell–induced cellular and inflammatory factors cause damage to organs,5 namely the skin, gastrointestinal tract, and liver.6 Patients who develop aGvHD exhibit a greater degree of endothelial damage and dysfunction compared to patients without this complication,7,8 as well as elevated biomarkers associated with endothelial cell damage (endothelial microparticles, E-selectin, intercellular adhesion molecule–1 [ICAM-1], and von Willebrand factor).9-12 Furthermore, factors in serum from patients with aGvHD have been shown to promote endothelial cell activation.7
Prophylactic regimens used to prevent aGvHD usually include a calcineurin inhibitor (e.g., cyclosporine A [CSA], ta-
Michelle Hudspeth,1 Shahram Mori,2 David Nachbaur,3 José Antonio Perez-Simon,4 Friedrich Stölzel,5 Marcie Riches,6 Wendy Wu,7 Peixin Zhang,8 Shirali Agarwal7 and Ibrahim Yakoub-Agha9
A phase II, prospective, randomized, open-label study of defibrotide added to standard-of-care prophylaxis for the prevention of acute graft-versus-host disease after allogeneic hematopoietic
Haematologica | 108 - April 2023 1026 ARTICLE - Bone Marrow Transplantation
crolimus) and methotrexate or mycophenolate mofetil.2,14 Despite the use of these immunosuppressive regimens, approximately 39% to 59% of patients receiving allo-HCT develop grade B-D aGvHD.15 Antithymocyte globulin (ATG) has been shown to lower the incidence of aGvHD after allo-HCT from an unrelated or sibling donor.16-18 Furthermore, cyclophosphamide taken post-HCT (PTCy) is widely used in both haploidentical and matched unrelated donor transplants.19,20 Although PTCy is on the path to becoming the new standard-of-care (SOC) for post-HCT aGvHD prophylaxis, it has been associated with graft dysfunction and infection.21 The mechanism of action of ATG and presumably of PTCy in the prevention of aGvHD involves T-cell depletion in blood and peripheral lymphatic tissues.20,22-24 Additionally, the selective T-cell co-stimulation modulator abatacept was recently approved by the US Food and Drug Administration for aGvHD prophylaxis when in combination with a calcineurin inhibitor and methotrexate.25 However, as abatacept is also a cytotoxic Tcell immunoglobulin, its primary mechanism of action involves immune suppression.25 The effectiveness of current aGvHD prophylactic regimens remains unsatisfactory, resulting in a need for safe and more effective therapies for the prevention of aGvHD.26
Defibrotide is a polydisperse mixture of predominantly single-stranded polydeoxyribonucleotide sodium salts.27 It reduces endothelial cell activation and enhances protection and stabilization of endothelial cells through antiinflammatory and anti-adhesive mechanisms and has been shown to protect the endothelium from toxic, inflammatory, and ischemic damage.27-29 In vitro evidence suggests that defibrotide protects endothelial cells, restores the thrombotic-fibrinolytic balance, and has immunosuppressive effects by inducing synthesis of prostaglandins that inhibit T-cell proliferation.30,31 Defibrotide has also been shown to suppress heparanase gene expression, and high levels of heparanase have been postulated as a risk factor for aGvHD development.32,33 Defibrotide has been approved for the treatment of hepatic veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS),34,35 a disease characterized by endothelial cell dysfunction.
The current SOC for prophylaxis of aGvHD works to suppress the immune system through either inhibition or depletion of T-cell lymphocytes or induction of tolerance to overcome the immune response from donor T-cell recognition that induces aGvHD; however, this can attenuate the beneficial graft-versus-tumor effect and increase the risk of opportunistic infection and disease relapse.2,36,37 Due to high morbidity and mortality associated with GvHD and the limitations of current therapies, prevention of aGvHD remains an area with significant unmet need. New treatment strategies for aGvHD prophylaxis are needed to improve clinical outcomes. Defibrotide is postulated to reduce the
incidence of aGvHD without an increase in opportunistic infections and relapse by (i) protecting endothelial cells in GvHD target organs from donor alloreactive T-cell infiltration and damage, (ii) not directly depleting T cells involved in the graft-versus-tumor effect, and (iii) ameliorating the inflammatory response and tissue damage associated with this immunopathological disease, reducing its incidence and severity.26
Several clinical studies have examined whether defibrotide can reduce the incidence of aGvHD.39-42 We designed the present hypothesis-generating, phase II, prospective, open-label, randomized study (clinicaltrials gov. Identifier: NCT03339297) to evaluate defibrotide added to SOC GvHD prophylaxis compared with SOC GvHD prophylaxis alone for the prevention of aGvHD following allo-HCT.
Methods
Study design and patients
Overall, 150 patients were planned for enrollment. Patients were stratified by age at screening (<17 years vs. ≥17 years), geographic region (North America vs. Europe), and use of ATG and were randomized 1:1 to defibrotide prophylaxis plus SOC (defibrotide prophylaxis arm) or SOC alone (SOC arm; Online Supplementary Figure S1). ATG type and dose were per the site SOC and varied by region. ATG use was limited to 30% of patients. Eligible patients (age ≥1 year) had to have a diagnosis of acute leukemia (in morphologic complete remission) or myelodysplastic syndrome and, after myeloablative or reduced-intensity conditioning, were scheduled for CD3+ T-cell replete peripheral blood stem cell or non-manipulated bone marrow graft transplantation from a human leukocyte antigen-matched or single-allele mismatched, unrelated donor. Key exclusion criteria were prior autologous or alloHCT and clinically significant acute bleeding within 24 hours before study treatment initiation.
Institutional Review Boards at participating centers approved the study, which was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference on Harmonization. All patients, parents, or legal guardians provided written informed consent.
Treatment
Patients in the defibrotide prophylaxis arm received defibrotide 25 mg/kg/day administered as 2-hour intravenous infusions of 6.25 mg/kg/dose every 6 hours prior to the start of conditioning therapy (1-4 doses) and continued for ≥21 days (ending no later than day 30 post-transplant). SOC GvHD immunoprophylaxis consisted of methotrexate or mycophenolate mofetil plus CSA or tacrolimus with or without ATG starting on the day of the conditioning
Haematologica | 108 - April 2023 1027 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
regimen. During the study, patients who developed VOD/SOS in either treatment arm could be treated with defibrotide. Per protocol, the use of medications that increased the risk of bleeding was closely monitored, and patients on defibrotide would be discontinued from defibrotide when bleeding developed or when taking medications that increased the risk of bleeding during the study. Thromboprophylaxis with heparin was allowed throughout the study for patients in both arms (maximum of 100 U/kg/day). Patients were followed for up to 180 days after transplantation.
Endpoints and assessments
The primary endpoint was cumulative incidence of grade B-D aGvHD by day 100 post-transplant. Key secondary endpoints included cumulative incidence of grade B-D aGvHD by day 180 post-transplant, grade C-D aGvHD by days 100 and 180 post-transplant, and safety. Grading of aGvHD for assessment of the primary and applicable secondary efficacy endpoints was based on the International Bone Marrow Transplant Registry Severity Index.43 An exploratory endpoint was overall survival (OS) by day 180 post-transplant. Safety assessments included monitoring treatment-emergent adverse events, serious treatmentemergent adverse events, and treatment-related treatment-emergent adverse events. Investigators classi fied adverse events using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
Statistical analysis
This study was designed to obtain estimates of the treatment difference of the cumulative incidence rates of aGvHD between the two treatment arms to compare the efficacy of defibrotide added to SOC (defibrotide prophylaxis arm) versus SOC alone (SOC arm). The study was for hypothesis generating and was not powered to detect minimal clinically meaningful differences between treatment arms at a significant level of 5%. A sample size of 75 patients per arm was estimated to provide a 90% confidence interval (CI): –0.28 to –0.03 for the treatment difference of the primary endpoint, assuming a cumulative incidence of 28.6% for the defibrotide prophylaxis arm and 44% for the SOC arm.41
Data were summarized by treatment arms using descriptive statistics for continuous variables, and numbers and percentages of patients for categorical variables. Computations were performed using SAS 9.4 (SAS Institute Inc., Cary, NC, USA). The primary analysis was performed on the intent-to-treat (ITT) population of all randomized patients. The safety population included all patients randomized to the defibrotide prophylaxis arm who received ≥1 dose of defibrotide and all patients randomized to the SOC arm. Analysis of the primary endpoint is detailed in the Online Supplementary Appendix.
Results
Patients
From February 21, 2018 to May 12, 2020, 152 patients participated in this study at 43 sites across 11 countries. The ITT population comprised 79 patients randomized to the defibrotide prophylaxis arm and 73 patients randomized to the SOC arm (Online Supplementary Figure S2). Five patients randomized to defibrotide prophylaxis did not undergo alloHCT and did not receive defibrotide; three patients randomized to the SOC did not undergo allo-HCT. The safety population included 74 patients in the defibrotide prophylaxis arm and 70 patients in the SOC arm. Fifty-six patients (71%) in the defibrotide prophylaxis arm and 59 patients (81%) in the SOC arm completed the study (Online Supplementary Figure S2).
The two treatment arms had similar baseline demographic characteristics (Table 1); however, the proportion of patients who received myeloablative conditioning prior to allo-HCT was higher in the defibrotide prophylaxis arm than in the SOC arm (56/74 patients [76%] vs. 43/70 patients [61%], respectively). Similarly, patients in the defibrotide prophylaxis arm more frequently received bone marrow as the source of stem cells compared to patients in the SOC arm (25/74 [34%] vs. 18/70 [26%], respectively). Among patients stratified to ATG use, baseline characteristics between the defibrotide prophylaxis (n=24) and SOC (n=21) arms followed a similar pattern as the total population (Table 2).
Treatment
The same percentage of patients (74%) in both the defibrotide prophylaxis and SOC arms received GvHD prophylaxis with methotrexate-based regimens (Online Supplementary Table S1), and 30% of patients received ATG in both study arms, per protocol. The percentage of patients who received CSA (47% vs. 47%) or tacrolimus (51% vs. 53%) was similar in the defibrotide prophylaxis and SOC arms, respectively. Median duration of exposure to defibrotide and duration of defibrotide treatment were both 25 days (range, 11-40 days) among the 74 patients who received defibrotide prophylaxis. A total of four (5%) patients in the defibrotide prophylaxis arm discontinued defibrotide treatment; three (4%) were due to adverse events and one (1%) was due to patient withdrawal. One patient (1%) in the defibrotide prophylaxis arm and four patients (6%) in the SOC arm received defibrotide for the treatment of VOD/SOS.
Cumulative incidence of acute graft-versus-host disease
Per the primary endpoint ITT analysis, where death without experiencing grade B-D aGvHD was treated as a competing risk, the cumulative incidence of grade B-D aGvHD by day 100 post-transplant in the defibrotide prophylaxis arm versus the SOC arm was 38.4% versus 47.1%, respectively (difference: –8.8%; 90% CI: –22.5 to 4.9; Figure 1A). In a planned
Haematologica | 108 - April 2023 1028 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
Table 1. Baseline demographic and clinical characteristics (intent-to-treat population).
aPercentages may not total 100 due to rounding. bIn either treatment arm. cIncludes MDS with single-lineage dysplasia, ringed sideroblasts, multilineage dysplasia, or isolated del5q and unclassifiable MDS. dIncludes AML with recurrent genetic abnormality, myelodysplasia-related changes, or not otherwise specified AML. eFive patients in the defibrotide prophylaxis arm and 3 patients in the SOC arm did not receive HCT. AML: acute myeloid leukemia; HCT: hematopoietic cell transplantation; HLA: human leukocyte antigen; MDS: myelodysplastic syndrome; SOC: standard-of-care.
sensitivity analysis for the primary endpoint using disease relapse as a competing risk in addition to death, the cumulative incidence of grade B-D aGvHD by day 100 post-transplant in the defibrotide prophylaxis arm compared to the SOC arm was 37.0% versus 45.7%, respectively (difference: –8.7; 90% CI: –22.4 to 4.9; Figure 1B). Both treatment arms had similar cumulative incidences of grade B-D aGvHD by day 180 post-transplant (49.0% vs. 50.2%, respectively).
In patients who received ATG, the cumulative incidence of grade B-D aGvHD by day 100 was consistent with the ITT population; however, the incidence was numerically lower in the defibrotide prophylaxis arm compared with the SOC arm. The cumulative incidence of grade B-D aGvHD by day 100 post-transplant in the defibrotide prophylaxis arm compared to the SOC arm was 30.4% versus 47.6%, respectively (difference: –17.2%; 90% CI: –41.8 to 7.5; Figure 2A). In patients who did not receive ATG, the cumulative incidence rates of grade B-D aGvHD by day 100 were similar between the defibrotide prophylaxis and SOC arms (42.0% vs. 46.9%, respectively; difference: –4.9%; 90% CI: –21.6 to 11.7; Figure 2B). Results of the cumulative incidence of grade C-D aGvHD
in ATG subgroups (Figure 3; Table 3) followed a similar pattern as the grade B-D aGvHD ATG subgroup analysis but with a more pronounced lowering of the cumulative incidence of grade C-D aGvHD by day 100 post-transplant with defibrotide prophylaxis versus SOC (4.3% vs. 28.9%, respectively; difference: –24.5%; 90% CI: –42.9 to –6.2). As shown in Table 2, in the ATG use subgroup, there was a slightly higher proportion of mismatched donors in the SOC arm (24% [5/21]) vs. the defibrotide prophylaxis arm (13% [3/24]); these proportions are somewhat higher than those seen in the non-ATG groups (DP: 7%; SOC 6%).
As in the subgroup of patients who received ATG, decreases in the cumulative incidence of grade B-D aGvHD at day 100 post-transplant were noted with defibrotide prophylaxis (n=56) compared with SOC alone (n=43) in patients who received myeloablative conditioning (41.8% vs. 55.8%, respectively; difference: –14.0%; 90% CI: –30.8 to 2.9; Table 3). In patients who did not receive myeloablative conditioning, the cumulative incidence of grade B-D aGvHD by day 100 was 27.8% in the defibrotide prophylaxis arm (n=18) versus 33.3% in the SOC arm (n=27;
–5.6%;
CI: –29.1
Characteristicsa Defibrotide prophylaxis (N=79) SOC (N=73) Sex, N (%) Male 41 (52) 36 (49) Female 38 (48) 37 (51) Race, N (%) Asian 1 (1) 4 (5) Black or African American 0 1 (1) White 66 (84) 63 (86) Not reported 12 (15) 5 (7) Age in years, median (range) 57 (2-69) 56 (2-72) Age group, N (%) <17 years 4 (5) 3 (4) ≥17 years 75 (95) 70 (96) Primary disease in >5% of patients, N (%)b MDSc 12 (15) 8 (11) AMLd 43 (54) 38 (52) B-lymphoblastic leukemia 8 (10) 10 (14) T-lymphoblastic leukemia 4 (5) 2 (3) Other 4 (5) 11 (15) Conditioning regimen, N (%) 74 70 Myeloablative conditioning 56 (76) 43 (61) Reduced-intensity conditioning 18 (24) 27 (39) Source of stem cells, N (%)e 74 70 Bone marrow 25 (34) 18 (26) Peripheral blood 49 (66) 52 (74) Degree of HLA matching, N (%) Full match of A, B, C, and DRB 67 (85) 62 (85) One mismatch of A, B, C, and DRB 7 (9) 8 (11)
difference:
90%
to
Haematologica | 108 - April 2023 1029 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
aTwo patients in the defibrotide prophylaxis arm were randomized and stratified as receiving ATG per the interactive response technology but were verified as not receiving ATG. bPercentages may not total 100 due to rounding. cIn any treatment subgroup. dIncludes MDS with single-lineage dysplasia, ringed sideroblasts, multilineage dysplasia, or isolated del5q and unclassifiable MDS. eIncludes AML with recurrent genetic abnormality, myelodysplasia-related changes, or not otherwise specified AML. fAmong patients stratified to ATG use, 1 patient in the defibrotide prophylaxis arm did not receive HCT. Among patients stratified to no ATG use, 4 patients in the defibrotide prophylaxis arm and 3 patients in the SOC arm did not receive HCT. AML: acute myeloid leukemia; ATG: antithymocyte globulin; HCT: hematopoietic cell transplantation; HLA: human leukocyte antigen; MDS: myelodysplastic syndrome; SOC: standard-of-care.
18.0; Table 3). Similarly, in patients who received bone marrow as the source of stem cells, decreases in the cumulative incidence of grade B-D and grade C-D aGvHD at day 100 post-transplant were noted with defibrotide prophylaxis (n=25) compared with SOC alone (n=18; Table 3).
Overall survival
OS rates were similar in the defibrotide prophylaxis and SOC arms by day 180 post-transplant (86.0% vs. 86.9%, respectively; Figure 4).
Safety
All patients experienced ≥1 treatment-emergent adverse event (Table 4). Fewer patients in the defibrotide prophylaxis arm experienced bleeding events compared to patients in
the SOC arm (34% vs. 41%, respectively). Nausea (78% vs 70%), diarrhea (65% vs. 76%), stomatitis (57% vs. 51%), and vomiting (53% vs. 54%) were the most common treatmentemergent adverse events reported in patients in both the defibrotide prophylaxis and SOC arms, respectively. In the defibrotide prophylaxis arm, 12 patients (16%) had treatment-related adverse events (Online Supplementary Table S2). No patients discontinued defibrotide due to treatmentrelated treatment-emergent adverse events.
Serious treatment-emergent adverse events were reported in 42% of patients in the defibrotide prophylaxis arm and 44% of patients in the SOC arm; none of the serious events were deemed to be related to defibrotide. Five patients in the defibrotide prophylaxis arm and three patients in the SOC arm had treatment-emergent adverse events leading
Table 2. Baseline demographic and clinical characteristics by antithymocyte globulin subgroup.a
Characteristicb ATG use No ATG use Defibrotide prophylaxis (N=24) SOC (N=21) Defibrotide prophylaxis (N=55) SOC (N=52) Sex, N (%) Male 10 (42) 11 (52) 31 (56) 25 (48) Female 14 (58) 10 (48) 24 (44) 27 (52) Race, N (%) Asian 0 2 (10) 1 (2) 2 (4) Black or African American 0 1 (5) 0 0 White 19 (79) 15 (71) 47 (85) 48 (92) Not reported 5 (21) 3 (14) 7 (13) 2 (4) Age in years, median (range) 54.5 (1.6-68.0) 56.0 (1.5-67.0) 58.0 (14-69) 58.5 (20-72) Age group, N (%) <17 years 3 (13) 3 (14) 1 (2) 0 ≥17 years 21 (88) 18 (86) 54 (98) 52 (100) Primary disease in >5% of patients, N (%)c MDSd 3 (13) 5 (24) 9 (16) 3 (6) AMLe 15 (63) 9 (43) 28 (51) 29 (56) B-lymphoblastic leukemia 5 (21) 5 (24) 3 (5) 5 (10) T-lymphoblastic leukemia 0 0 4 (7) 2 (4) Other 0 2 (10) 4 (7) 9 (17) Conditioning regimen, N (%) Myeloablative conditioning 20 (83) 13 (62) 36 (65) 30 (58) Reduced-intensity conditioning 3 (13) 8 (38) 15 (27) 19 (37) Source of stem cells, N (%)f Bone marrow 7 (29) 3 (14) 18 (33) 15 (29) Peripheral blood 16 (67) 18 (86) 33 (60) 34 (65) Degree of HLA matching, N (%) Full match of A, B, C, and DRB 20 (83) 16 (76) 47 (85) 46 (88) One mismatch of A, B, C, and DRB 3 (13) 5 (24) 4 (7) 3 (6) Haematologica | 108 - April 2023 1030 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
to death. The events leading to death were aGvHD (n=2), bacterial sepsis (n=1), respiratory failure (n=1), and VOD/SOS (n=1) in the defibrotide prophylaxis arm and multiple organ failure (n=1), lung disorder (n=1), and shock (n=1) in the SOC arm. None of these events were related to defibrotide.
Discussion
This hypothesis-generating, phase II, prospective, openlabel, randomized study is the first multi-national prevention study to use a novel approach directed towards endothelial injury. Here, defibrotide plus SOC GvHD pro-
phylaxis versus SOC alone was evaluated for the prevention of aGvHD after allo-HCT in 152 patients with acute leukemia or myelodysplastic syndrome. Patient baseline demographic and clinical characteristics were mostly similar between the two arms of the study and were consistent with those of a population at risk for aGvHD. There were differences between treatment arms in the type of conditioning regimen (myeloablative: defibrotide, 76% vs SOC, 61%) and source of stem cells (bone marrow: defibrotide, 34% vs. SOC, 26%). While the data from this study many only suggest a modest treatment effect of defibrotide, the findings add to the relatively small body of literature of randomized assessments for GvHD prevention.
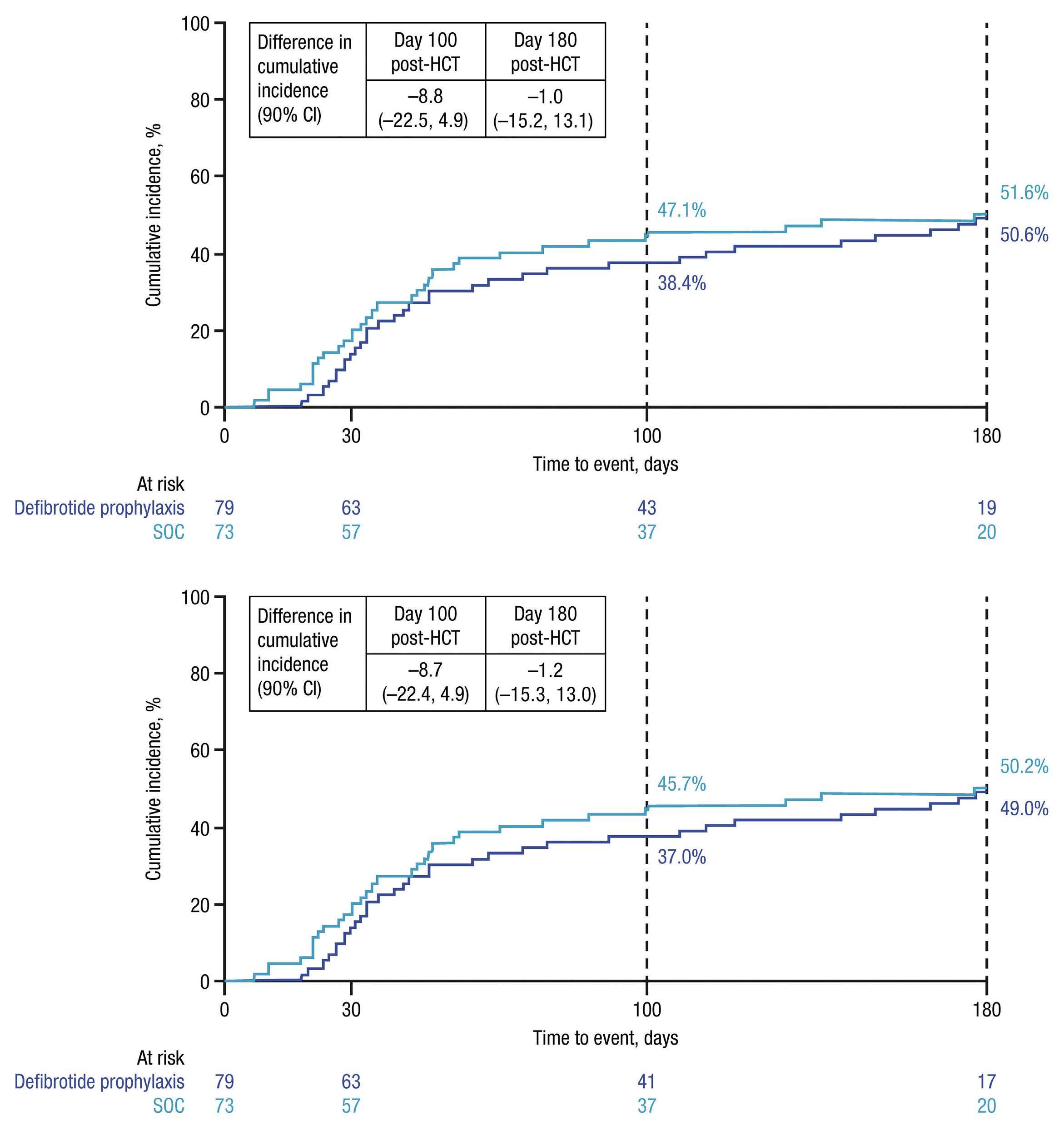
A B Haematologica | 108 - April 2023 1031 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
Figure 1. Cumulative incidence of grade B-D acute graft-versus-host disease by (A) day 100 and day 180 after allogeneic hematopoietic cell transplantation and (B) with disease relapse as a competing risk. The International Bone Marrow Transplant Registry (IBMTR) Severity Index was used to grade acute graft-versus-host disease. HCT: hematopoietic cell transplantation; CI: confidence interval; SOC: standard-of-care.
ITT analysis of the primary endpoint revealed that the cumulative incidence of grade B-D aGvHD by day 100 posttransplant was numerically lower in the defibrotide prophylaxis arm (38.4%) compared with the SOC arm (47.1%). By day 180 post-transplant, patients in the two treatment arms had similar cumulative incidences of grade B-D aGvHD. Similar results were reported in a study by Corbacioglu et al.41,42 in which patients who received defibrotide prophylaxis for VOD/SOS had a lower incidence of aGvHD by day 100 post-transplant versus control (no defibrotide; 47% vs. 65%, respectively; P=0.0046), and the inci-
dence of chronic GvHD by day 180 did not differ between study arms (defibrotide prophylaxis, 9%; control, 10%; P=0.8022). The absence of a noted effect of defibrotide on the incidence of chronic GvHD by day 180 may be explained by the different pathophysiologies of acute and chronic forms of this disease. Another study by Strouse et al.44 found notable differences in the cumulative incidence of grade B-D acute GvHD at day 100 post-HCT in patients who received defibrotide versus those who did not (23.1% vs. 37.7%; difference, –14.6; 95% CI: –33.1 to 3.9]). Furthermore, a study by
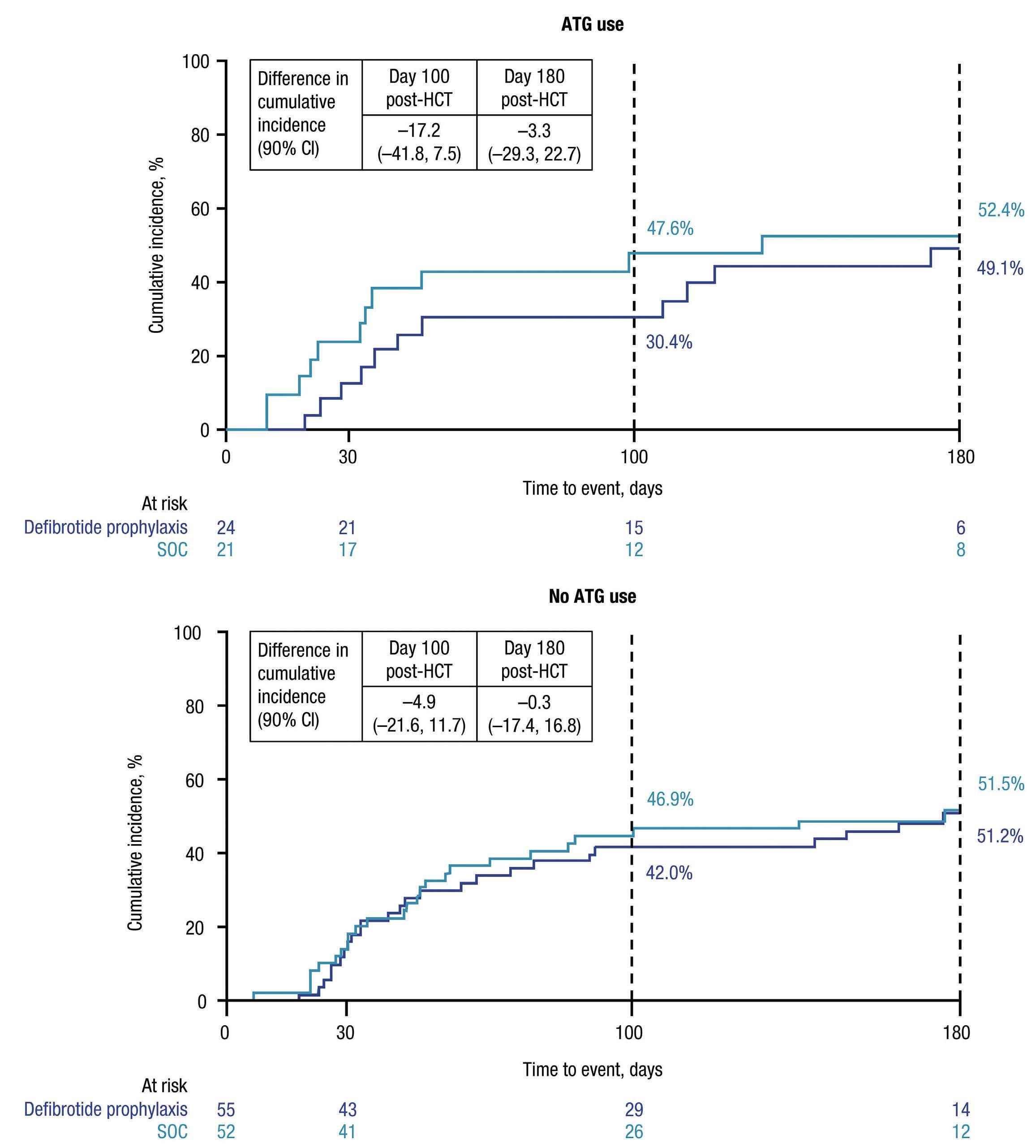 Tekgündüz et al.39 demonstrated that
Tekgündüz et al.39 demonstrated that
A B Haematologica | 108 - April 2023 1032 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
Figure 2. Cumulative incidence of grade B-D acute graft-versus-host disease by day 100 and day 180 after allogeneic hematopoietic cell transplantation by (A) antithymocyte globulin use and (B) no antithymocyte globulin use. The International Bone Marrow Transplant Registry (IBMTR) Severity Index was used to grade acute graft-versus-host disease. HCT: hematopoietic cell transplantation; ATG: antithymocyte globulin; CI: confidence interval; SOC: standard-of-care.
use of defibrotide prior to transplantation and concurrently with the conditioning regimen may decrease the incidence of aGvHD, and a separate study by Chalandon et al.40 indicated that defibrotide prophylaxis significantly reduced 1-year cumulative incidence of aGvHD. Interestingly, a retrospective study by Tilmont et al.45 showed no protective effect of defibrotide on the development or severity of aGvHD versus control (no defibrotide) in patients undergoing allo-HCT. However, in contrast to the current study, this was a retrospective observational study that
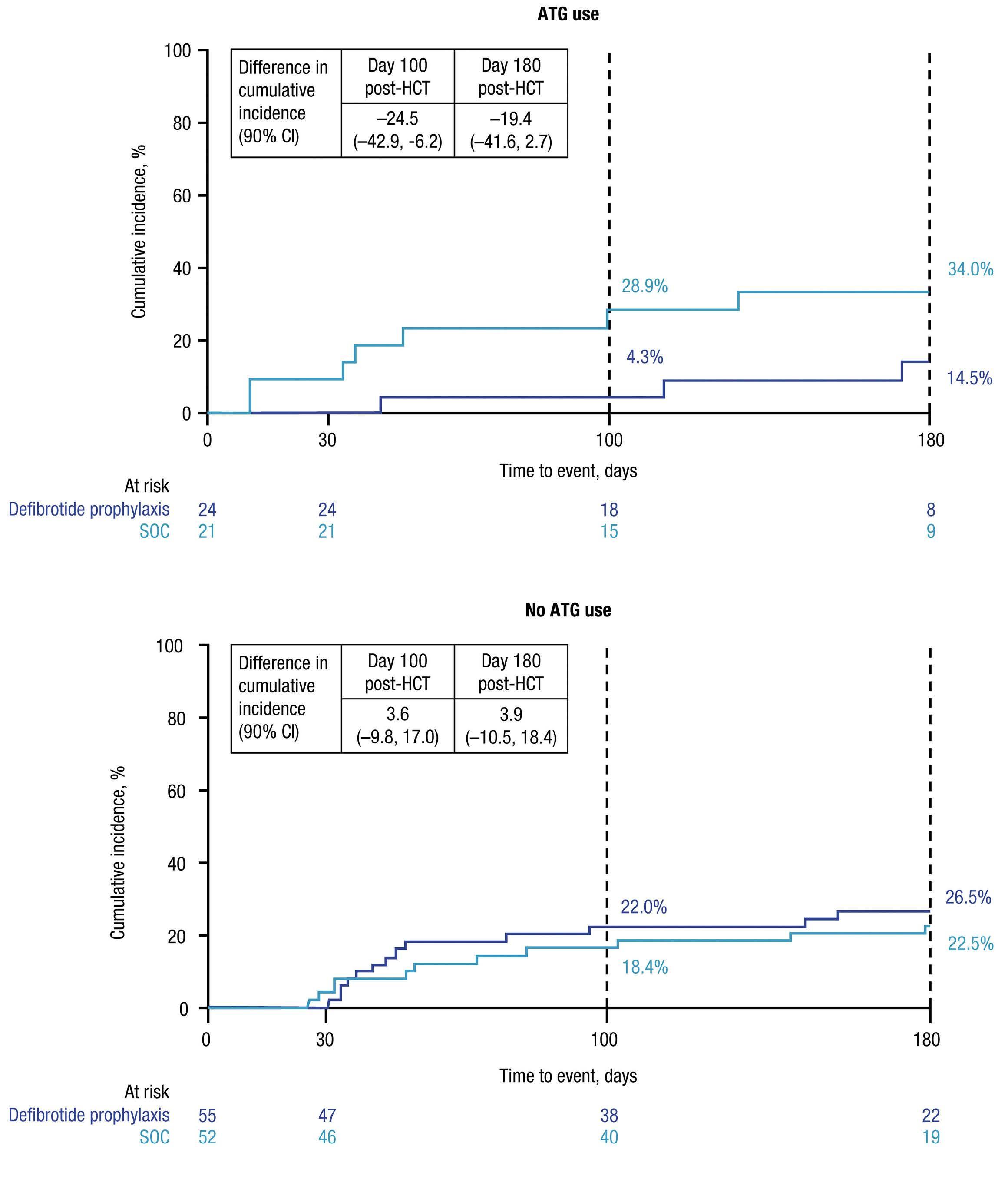
included a small number of patients who received defibrotide, the majority of whom received defibrotide for the treatment of VOD/SOS, and only a small number who received defibrotide as prophylaxis. Furthermore, more patients in the defibrotide group had progressive disease, which may have contributed to poorer outcomes.
In our study, the numerical difference noted by day 100 post-transplant in the ITT population became more pronounced in subgroup analyses of patients stratified by ATG use. T-cell depletion with ATG in addition to standard
A B Haematologica | 108 - April 2023 1033 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
Figure 3. Cumulative incidence of grade C-D acute graft-versus-host disease by day 100 and day 180 after allogeneic hematopoietic cell transplantation by (A) antithymocyte globulin use and (B) no antithymocyte globulin use. The International Bone Marrow Transplant Registry (IBMTR) Severity Index was used to grade acute graft-versus-host disease. HCT: hematopoietic cell transplantation; ATG: antithymocyte globulin; CI: confidence interval; SOC: standard-of-care.
GvHD prophylaxis has been shown to significantly reduce the occurrence and severity of GvHD in patients undergoing allo-HCT; ATG is also associated with impaired immune reconstitution and increased risk of infections.46,47 In patients receiving ATG in the current study, the cumulative incidence of the more severe grade C-D aGvHD by day 100 was lower with defibrotide prophylaxis compared to SOC alone; this difference was maintained through day 180 post-transplant. There was a slightly higher proportion of mismatched donors with grade C-D aGvHD in the SOC arm (24%) versus the defibrotide prophylaxis arm (13%). Although the small patient numbers in each group precludes the ability to draw solid conclusions, this could have led to the somewhat higher incidence of grade C-D aGvHD in the ATG SOC group. These results are consistent with those of Corbacioglu et al., 41 in which prophylaxis with defibrotide significantly reduced the occurrence and severity of aGvHD versus control; adjusting for ATG as a covariate confirmed the significant effects of defibrotide (adjusted risk difference for aGvHD grade B-D: –0.1470; 95% CI: –0.2618 to –0.0322; P=0.0121).42 Furthermore, the authors noted that defibrotide did not seem to interfere with the graft- versus-leukemia effect.42 In the current study, the two treatment arms had similar OS by day 180 post-transplant.
The effect of defibrotide on the cumulative incidence of aGvHD also appeared more pronounced in patients who had received bone marrow as the source of the stem cells. How-
ever, other studies have shown no difference in the incidence of aGvHD with these sources of progenitor cells.48,49 Similarly, in patients who received myeloablative conditioning, the cumulative incidence of aGvHD was also lower in the defibrotide prophylaxis arm versus the SOC arm. HCT recipients are exposed to insults that can stem from stressors such as conditioning regimen, engraftment, and infections that can cause endothelial cell activation and direct endothelial damage.50 Endothelial cells may be an important target for prophylaxis and therapeutic intervention for complications like GvHD, especially due to the role of endothelium in the pathophysiology of the condition. Despite the potential higher risk of developing aGvHD associated with myeloablative conditioning,15 defibrotide prophylaxis showed some benefit, which we hypothesize could be due to the known protective effect of defibrotide on the acute endothelial damage inflicted on these patients during conditioning.51 We speculate that the more intense the conditioning, the higher the endothelial damage. Preclinical studies suggest that defibrotide downregulates expression of key endothelial adhesion molecules (e.g., E-selectin, vascular cell adhesion molecule–1) involved in trafficking alloreactive immune cells to aGvHD target tissues.26 In a mouse model of allo-HCT, defibrotide prophylaxis prevented T cell and neutrophil tissue infiltration and aGvHD-associated tissue damage, resulting in reduced incidence of aGvHD and significantly improved survival versus untreated controls.26 Additionally, defibrotide may be acting synergis-
Subgroup Grade B-D Grade C-D Defibrotide prophylaxis SOC Defibrotide prophylaxis SOC ATG use N=24 N=21 N=24 N=21 Cumulative incidence rate (%) 30.4 47.6 4.3 28.9 Difference (90% CI) -17.2 (-41.8 to 7.5) -24.5 (-42.9 to -6.2) No ATG use N=55 N=52 N=55 N=52 Cumulative incidence rate grade (%) 42.0 46.9 22.0 18.4 Difference (90% CI) -4.9 (-21.6 to 11.7) 3.6 (-9.8 to 17.0) Myeloablative conditioning N=56 N=43 N=56 N=43 Cumulative incidence rate (%) 41.8 55.8 16.4 21.1 Difference (90% CI) -14.0 (-30.8 to 2.9) -4.7 (-18.0 to 8.7) No myeloablative conditioning N=18 N=27 N=18 N=27 Cumulative incidence rate (%) 27.8 33.3 17.0 22.2 Difference (90% CI) -5.6 (-29.1 to 18.0) -5.2 (-25.5 to 15.1) Peripheral blood transplant N=49 N=52 N=49 N=52 Cumulative incidence rate (%) 39.6 42.3 18.8 19.3 Difference (90% CI) -2.7 (-19.1 to 13.7) -0.6 (-13.7 to 12.5) Bone marrow transplant N=25 N=18 N=25 N=18 Cumulative incidence rate (%) 36.0 61.1 12.2 27.8 Difference (90% CI) -25.1 (-50.8 to 0.6) -15.6 (-36.7 to 5.5)
aGvHD: acute graft-versus-host disease; ATG: antithymocyte globulin; CI: confidence interval; SOC: standard-of-care. Haematologica | 108 - April 2023 1034 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
Table 3. Cumulative incidence of acute graft-versus-host disease by day 100 by subgroups.
aGvHD: acute graft-versus-host disease; SOC: standard-of-care. aIncidence was based on the number of patients, not the number of events. Percentages were calculated using the number of patients in each arm from the safety population as the denominator. bCoding was based on MedDRA version 21.1. cIn either treatment arm.
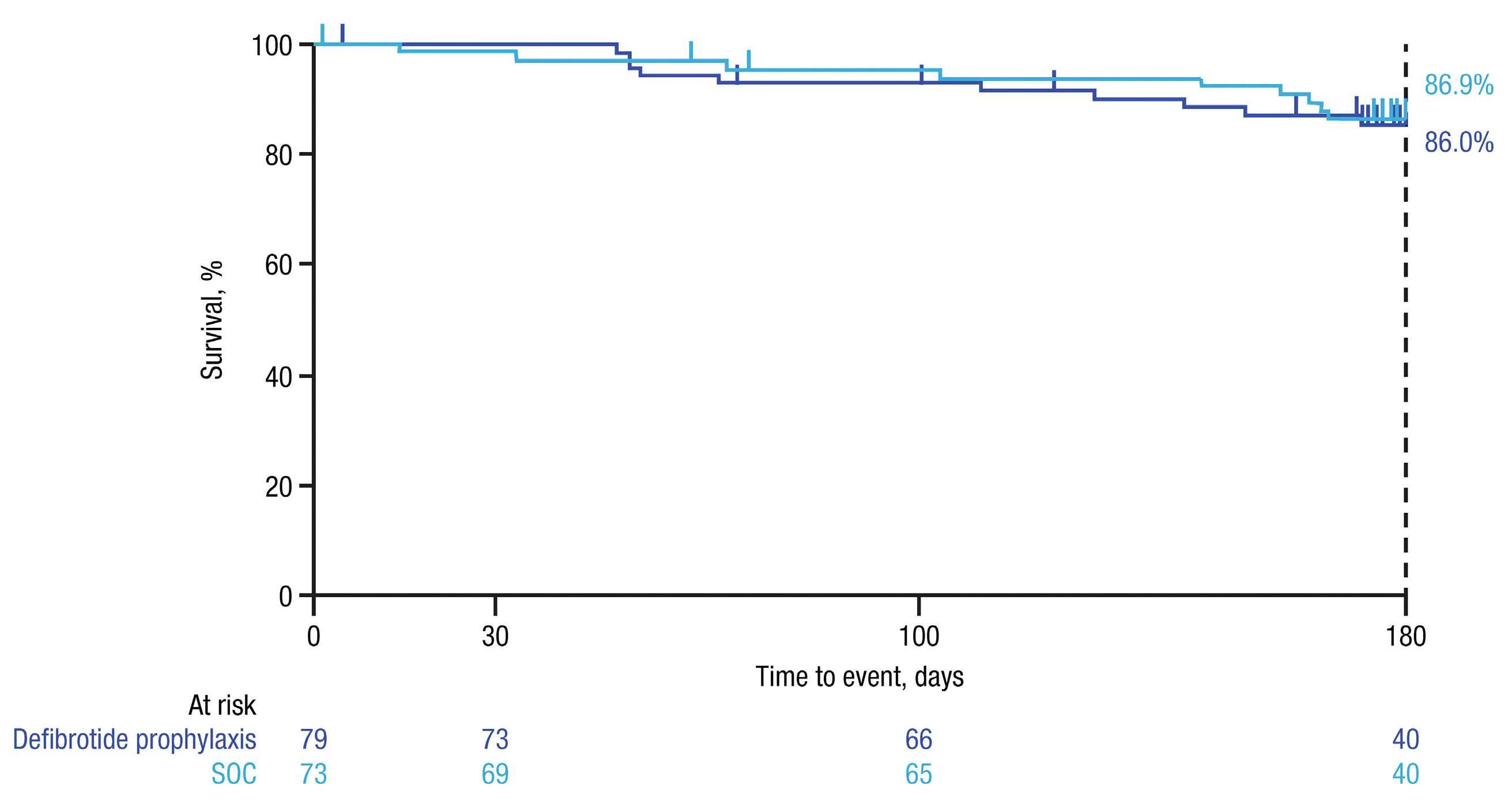
Number of patients, N (%)a Defibrotide prophylaxis (N=74) SOC (N=70) Treatment-emergent adverse eventsb ≥1 74 (100) 70 (100) Related to defibrotide 12 (16) ‒Treatment-emergent adverse events of special interest: bleeding 25 (34) 29 (41) Treatment-emergent adverse events occurring in >40% of patientsc Nausea 58 (78) 49 (70) Diarrhea 48 (65) 53 (76) Stomatitis 42 (57) 36 (51) Vomiting 39 (53) 38 (54) Febrile neutropenia 31 (42) 20 (29) Headache 31 (42) 23 (33) Decreased appetite 30 (41) 30 (43) Hypomagnesemia 30 (41) 35 (50) Constipation 28 (38) 29 (41) Anemia 27 (36) 30 (43) Hypertension 25 (34) 29 (41) Hypokalemia 24 (32) 28 (40) Serious treatment-emergent adverse eventsb ≥1 31 (42) 31 (44) Related to defibrotide 0 ‒Serious treatment-emergent adverse events in >2 patientsc aGvHD 4 (5) 1 (1) aGvHD in skin 1 (1) 4 (6) Acute kidney injury 3 (4) 2 (3) Diarrhea 3 (4) 2 (3) Dyspnea 3 (4) 0
Figure 4. Kaplan-Meier–estimated overall survival by day 180 after allogeneic hematopoietic cell transplantation. SOC: standard-of-care.
Table 4. Treatment-emergent adverse events and serious treatment-emergent adverse events.
Haematologica | 108 - April 2023 1035 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
tically with ATG’s polyclonal nature to produce a better response in patients who had received ATG. In support of a synergistic effect of defibrotide with other immunosuppressive agents, results from a preliminary study suggest that defibrotide prophylaxis combined with ATG, post-transplant cyclophosphamide, and CSA may be an effective strategy for preventing aGvHD.52 Furthermore, the role of cell subsets other than T cells, such as endothelial cells, in the pathophysiology of GvHD might be more pronounced in the absence of T lymphocytes, as it occurs with natural killer cell and killer Ig–like receptor disparities in the haploidentical transplant setting.53
The potential benefits of defibrotide in lowering the incidence of aGvHD, most commonly occurring in the first 100 days following HCT, may reflect defibrotide’s mechanism of action, especially its anti-inflammatory and endothelial protective properties, along with the suppression of heparanase gene expression. Additional studies are needed to further evaluate the effect of defibrotide in preventing aGvHD. Safety results in this study were consistent with the safety profile of defibrotide reported in other randomized studies.41,54 Importantly, there was no increased incidence of bleeding events with defibrotide prophylaxis compared to SOC, and there were no defibrotide-related serious treatment-emergent adverse events or deaths.
The main limitation of this study is the small sample size, which offered a low statistical power to detect differences between arms, particularly for the ATG and myeloablative conditioning subgroups. The study was intended to be hypothesis generating, with a goal of providing estimates of the cumulative incidence of aGvHD for defibrotide prophylaxis compared with SOC. The greater reductions in the incidence and severity of aGvHD with defibrotide prophylaxis versus SOC reported by Corbacioglu et al.41 in the phase III VOD/SOS prevention study may have been the result of a larger number of patients. Furthermore, the study by Corbacioglu et al.41 was performed in pediatric patients while this study included only a few pediatric patients. Another limitation of our study is the potential variability in SOC among patients, given that it was primarily defined by institutional guidelines that may vary among sites and regions. ATG type and dose were not specified in the protocol to be collected in the trial but rather were administered per the site SOC. In addition, although this study included many global HCT centers to include as many diverse populations as possible, the trial ended up with limited enrollment of minority populations and pediatric patients by nature. In order to extend the relevance of our findings to a broader patient population, future studies should have more stringent management of enrollment to ensure patient diversity. While not conclusive, the results of our study suggest that there may be a benefit to addition of defibrotide prophylaxis to SOC for the prevention of aGvHD after allo-HCT; however, further work is needed in the context of recently
adopted therapeutic approaches. Future studies are needed to determine which subgroups of patients might derive the most clinical benefit from defibrotide prophylaxis added to standard GvHD prophylaxis.
Disclosures
MH has participated on a Data Safety Monitoring Board or Advisory Board for Jazz Pharmaceuticals , Mesoblast, and Novartis. SM has received payment for speaker bureaus from Incyte Pharmaceuticals. JAP-S has received the following from Novartis, Jazz Pharmaceuticals, Janssen, Gilead, BMS, Amgen, Takeda, MSD, Alexion, and Abvvie: grants or contracts; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events; payment for expert testimony; and support for attending meetings and/or travel. FS has served on advisory boards and received honoraria from Jazz Pharmaceuticals, and has received support for attending meetings and/or travel from Jazz Pharmaceuticals . MR is a member of a Data Safety Monitoring Committee for Gamida Cell, is chair of the IT committee for the American Society of Transplantation and Cellular Therapy, is an employee of IQVIA Biotech, and her institution has receiv ed research funding and grants/contracts from Atara Biotherapeutics and Jazz Pharmaceuticals. WW, PZ, and SA are employees of Jazz Pharmaceuticals and hold stock and/or stock options in Jazz Pharmaceuticals. IY-A has received grants or contracts and consulting fees from Jazz Pharmaceuticals. DN has no other conflicts of interest to disclose. All authors received medical writing and editorial assistance for the development of this manuscript that was funded by Jazz Pharmaceuticals.
Contributions
MH, SM, MR, WW, SA, and IY-A performed the research, participated in the acquistion, analysis, or interpretation of the data, and critically revised the manuscript; DN, JAP-S, FS, and PZ participated in the acquisition, analysis, or interpretation of the data and critically revised the manuscript.
Acknowledgments
The authors would like to thank all the study participants and their families and caregiv ers. Medical writing and editorial assistance were provided by Monica Nicosia, PhD, and Heather Nyce, PhD, CMPP™, of Lumanity Scientific Inc ., and were financially supported by Jazz Pharmaceuticals.
Funding
The study was funded by Jazz Pharmaceuticals , Inc. (Palo Alto, CA, USA).
Data-sharing statement
All relevant data are provided within the manuscript and supporting files.
Haematologica | 108 - April 2023 1036 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
References
1. Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007;7(5):340-352.
2. Zeiser R, Blazar BR. Acute graft-versus-host disease - biologic process, prevention, and therapy. N Engl J Med. 2017;377(22):2167-2179.
3. Nassereddine S, Rafei H, Elbahesh E, Tabbara I. Acute graft versus host disease: a comprehensive review. Anticancer Res. 2017;37(4):1547-1555.
4. Ferrara JLM, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550-1561.
5. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT-related major complications. Front Immunol. 2017;8:79.
6. Harris AC, Ferrara JL, Levine JE. Advances in predicting acute GVHD. Br J Haematol. 2013;160(3):288-302.
7. Mir E, Palomo M, Rovira M, et al. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant. 2017;52(9):1317-1325.
8. Cordes S, Mokhtari Z, Bartosova M, et al. Endothelial damage and dysfunction in acute graft-versus-host disease. Haematologica. 2021;106(8):2147-2160.
9. Palomo M, Diaz-Ricart M, Carbo C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transplant. 2010;16(7):985-993.
10. Pihusch V, Rank A, Steber R, et al. Endothelial cell-derived microparticles in allogeneic hematopoietic stem cell recipients. Transplantation. 2006;81(10):1405-1409.
11. Matsuda Y, Hara J, Osugi Y, et al. Serum levels of soluble adhesion molecules in stem cell transplantation-related complications. Bone Marrow Transplant. 2001;27(9):977-982.
12. Biedermann BC, Tsakiris DA, Gregor M, Pober JS, Gratwohl A. Combining altered levels of effector transcripts in circulating T cells with a marker of endothelial injury is specific for active graft-versus-host disease. Bone Marrow Transplant. 2003;32(11):1077-1084.
13. Dietrich S, Falk CS, Benner A, et al. Endothelial vulnerability and endothelial damage are associated with risk of graft-versushost disease and response to steroid treatment. Biol Blood Marrow Transplant. 2013;19(1):22-27.
14. Gooptu M, Antin JH. GVHD prophylaxis 2020. Front Immunol. 2021;12:605726.
15. Jagasia M, Arora M, Flowers ME, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119(1):296-307.
16. Finke J, Bethge WA, Schmoor C, et al. Standard graft-versushost disease prophylaxis with or without anti-T-cell globulin in haematopoietic cell transplantation from matched unrelated donors: a randomised, open-label, multicentre phase 3 trial. Lancet Oncol. 2009;10(9):855-864.
17. Brissot E, Labopin M, Moiseev I, et al. Post-transplant cyclophosphamide versus antithymocyte globulin in patients with acute myeloid leukemia in first complete remission undergoing allogeneic stem cell transplantation from 10/10 HLAmatched unrelated donors. J Hematol Oncol. 2020;13(1):87.
18. Bonifazi F, Rubio MT, Bacigalupo A, et al. Rabbit ATG/ATLG in preventing graft-versus-host disease after allogeneic stem cell transplantation: consensus-based recommendations by an international expert panel. Bone Marrow Transplant. 2020;55(6):1093-1102.
19. Ciurea SO, Zhang MJ, Bacigalupo AA, et al. Haploidentical
transplant with posttransplant cyclophosphamide vs matched unrelated donor transplant for acute myeloid leukemia. Blood. 2015;126(8):1033-1040.
20. Bolaños-Meade J, Reshef R, Fraser R, et al. Three prophylaxis regimens (tacrolimus, mycophenolate mofetil, and cyclophosphamide; tacrolimus, methotrexate, and bortezomib; or tacrolimus, methotrexate, and maraviroc) versus tacrolimus and methotrexate for prevention of graft-versus-host disease with haemopoietic cell transplantation with reduced-intensity conditioning: a randomised phase 2 trial with a non-randomised contemporaneous control group (BMT CTN 1203). Lancet Haematol. 2019;6(3):e132-e143.
21. Irene GC, Albert E, Anna BV, et al. Patterns of infection and infectious-related mortality in patients receiving posttransplant high dose cyclophosphamide as graft-versus-host-disease prophylaxis: impact of HLA donor matching. Bone Marrow Transplant. 2021;56(4):818-827.
22. Martin PJ, Hansen JA, Buckner CD, et al. Effects of in vitro depletion of T cells in HLA-identical allogeneic marrow grafts. Blood. 1985;66(3):664-672.
23. Choi SW, Reddy P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat Rev Clin Oncol. 2014;11(9):536-547.
24. Mohty M. Mechanisms of action of antithymocyte globulin: Tcell depletion and beyond. Leukemia. 2007;21(7):1387-1394.
25. Orencia® (abatacept) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company; 2021. https://packageinserts.bms.com/pi/pi_orencia.pdf. Accessed 1 December 2022.
26. Garcia-Bernal D, Palomo M, Martinez CM, et al. Defibrotide inhibits donor leucocyte-endothelial interactions and protects against acute graft-versus-host disease. J Cell Mol Med. 2020;24(14):8031-8044.
27. Richardson PG, Palomo M, Kernan NA, Hildebrandt GC, Chao N, Carreras E. The importance of endothelial protection: the emerging role of defibrotide in reversing endothelial injury and its sequelae. Bone Marrow Transplant. 2021;56(12):2889-2896.
28. Bonifazi F, Barbato F, Ravaioli F, et al. Diagnosis and treatment of VOD/SOS after allogeneic hematopoietic stem cell transplantation. Front Immunol. 2020;11:489.
29. Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood. 2016;127(13):1719-1727.
30. Richardson PG, Corbacioglu S, Ho VT, et al. Drug safety evaluation of defibrotide. Expert Opin Drug Saf. 2013;12(1):123-136.
31. Ferraresso M, Rigotti P, Stepkowski SM, Chou TC, Kahan BD. Immunosuppressive effects of defibrotide. Transplantation. 1993;56(4):928-933.
32. Ostrovsky O, Shimoni A, Rand A, Vlodavsky I, Nagler A. Genetic variations in the heparanase gene (HPSE) associate with increased risk of GVHD following allogeneic stem cell transplantation: effect of discrepancy between recipients and donors. Blood. 2010;115(11):2319-2328.
33. Mitsiades CS, Rouleau C, Echart C, et al. Preclinical studies in support of defibrotide for the treatment of multiple myeloma and other neoplasias. Clin Cancer Res. 2009;15(4):1210-1221.
34. Defitelio® (defibrotide sodium) [prescribing information]. Palo Alto, CA: Jazz Pharmaceuticals, Inc.; 2016. https://pp.jazzpharma.com/pi/defitelio.en.USPI.pdf. Accessed 1
Haematologica | 108 - April 2023 1037 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
December 2022.
35. Defitelio® (defibrotide sodium) [summary of product characteristics]. Villa Guardia, Italy: Gentium SpA; 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/defitelio Accessed 1 December 2022.
36. Naserian S, Leclerc M, Shamdani S, Uzan G. Current preventions and treatments of aGVHD: from pharmacological prophylaxis to innovative therapies. Front Immunol. 2020;11:607030.
37. Jiang H, Fu D, Bidgoli A, Paczesny S. T cell subsets in graft versus host disease and graft versus tumor. Front Immunol. 2021;12:761448.
38. Martinez-Sanchez J, Hamelmann H, Palomo M, et al. Acute graft-vs.-host disease-associated endothelial activation in vitro is prevented by defibrotide. Front Immunol. 2019;10:2339.
39. Tekgunduz E, Kaya AH, Bozdag SC, et al. Does defibrotide prophylaxis decrease the risk of acute graft versus host disease following allogeneic hematopoietic cell transplantation? Transfus Apher Sci. 2016;54(1):30-34.
40. Chalandon Y, Simonetta F, Dantin C, et al. Efficient prophylaxis with defibrotide for sinusoidal obstruction syndrome (SOS) after allogeneic hematopoietic ttem cell transplantation (HSCT). Blood. 2016;128(22):2204-2204.
41. Corbacioglu S, Cesaro S, Faraci M, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379(9823):1301-1309.
42. Corbacioglu S, Cesaro S, Faraci M, et al. Impact of prophylaxis with defibrotide on the occurrence of acute GvHD in allogeneic HSCT. Blood. 2013;122(21):4591-4591.
43. Rowlings PA, Przepiorka D, Klein JP, et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol. 1997;97(4):855-864.
44. Strouse C, Richardson P, Prentice G, et al. Defibrotide for treatment of severe veno-occlusive disease in pediatrics and adults: an exploratory analysis using data from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2016;22(7):1306-1312.
45. Tilmont R, Yakoub-Agha I, Ramdane N, et al. Impact of
defibrotide in the prevention of acute graft-versus-host disease following allogeneic hematopoietic cell transplantation. Ann Pharmacother. 2022;56(9):1007-1015.
46. Yang X, Li D, Xie Y. Anti-thymocyte globulin prophylaxis in patients with hematological malignancies undergoing allogeneic hematopoietic stem cell transplantation: an updated metaanalysis. Front Oncol. 2021;11:717678.
47. Kekre N, Antin JH. ATG in allogeneic stem cell transplantation: standard of care in 2017? Counterpoint. Blood Adv. 2017;1(9):573-576.
48. Stem Cell Trialists' Collaborative Group. Allogeneic peripheral blood stem-cell compared with bone marrow transplantation in the management of hematologic malignancies: an individual patient data meta-analysis of nine randomized trials. J Clin Oncol. 2005;23(22):5074-5087.
49. Anasetti C, Logan BR, Lee SJ, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med. 2012;367(16):1487-1496.
50. Hildebrandt GC, Chao N. Endothelial cell function and endothelial-related disorders following haematopoietic cell transplantation. Br J Haematol. 2020;190(4):508-519.
51. Luft T, Dreger P, Radujkovic A. Endothelial cell dysfunction: a key determinant for the outcome of allogeneic stem cell transplantation. Bone Marrow Transplant. 2021;56(10):2326-2335.
52. Akpinar S, Kayikci O, tekgunduz E. Defibrotide combined with triple therapy including posttransplant cyclophosphamide, low dose rabbit anti-t-lymphocyte globulin and cyclosporine is effective in prevention of graft versus host disease after allogeneic peripheral blood stem cell transplantation for hematologic malignancies. Transfus Apher Sci. 2022;61(1):103367.
53. Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097-2100.
54. Richardson PG, Soiffer RJ, Antin JH, et al. Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol Blood Marrow Transplant. 2010;16(7):1005-1017.
Haematologica | 108 - April 2023 1038 ARTICLE - Defibrotide for preventing aGvHD post-transplant M. Hudspeth et al.
Improving the anti-acute myeloid leukemia activity of CD123-specific engager T cells by MyD88 and CD40 costimulation
Correspondence: P. Velasquez Paulina.velasquez@stjude.org
1Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN; 2Center for Cell and Gene Therapy, Texas Children’s Hospital, Baylor College of Medicine, Houston, TX; 3Department of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN and 4Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Abstract
Received: May 24, 2021.
Accepted: April 21, 2022.
Early view: July 28, 2022.
https://doi.org/10.3324/haematol.2021.279301
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The outcome of patients with acute myeloid leukemia remains poor, and immunotherapy has the potential to improve this. T cells expressing chimeric antigen receptors or bispecific T-cell engagers targeting CD123 are actively being explored in preclinical and/or early phase clinical studies. We have shown that T cells expressing CD123-specific bispecific T-cell engagers (CD123.ENG T cells) have anti-acute myeloid leukemia activity. However, like chimeric antigen receptor T cells, their effector function diminishes rapidly once they are repeatedly exposed to antigen-positive target cells. Here we sought to improve the effector function of CD123.ENG T cells by expressing inducible co-stimulatory molecules consisting of MyD88 and CD40 (iMC), MyD88 (iM), or CD40 (iC), which are activated by a chemical inducer of dimerization. CD123.ENG T cells expressing iMC, iM, or iC maintained their antigen specificity in the presence of a chemical inducer of dimerization, as judged by cytokine production (interferon-γ, interleukin-2) and their cytolytic activity. In repeat stimulation assays, activating iMC and iM, in contrast to iC, enabled CD123.ENG T cells to secrete cytokines, expand, and kill CD123-positive target cells repeatedly. Activating iMC in CD123.ENG T cells consistently improved antitumor activity in an acute myeloid leukemia xenograft model. This translated into a significant survival advantage in comparison to that of mice that received CD123.ENG or CD123.ENG.iC T cells. In contrast, activation of only iM in CD123.ENG T cells resulted in donor-dependent antitumor activity. Our work highlights the need for both toll-like receptor pathway activation via MyD88 and provision of co-stimulation via CD40 to consistently enhance the antitumor activity of CD123.ENG T cells.
Introduction
Acute myeloid leukemia (AML) is a disease with poor prognosis due to its high relapse rate and treatment-related mortality.1-4 Adoptive immunotherapy has the potential to improve outcomes in patients with AML, but overlapping antigen expression between tumor cells and healthy tissues as well as T-cell persistence in a hostile tumor microenvironment are problematic.5,6 CD123 is a promising immunotherapy target for AML because of its high expression on leukemia stem cells and lower expression on normal hematopoietic cells.7-9 Several T-cell-based immunotherapy approaches are currently being developed to target CD123, including T cells expressing chimeric antigen receptors (CAR) or strategies involving bispecific anti-
bodies (bispecific T-cell engagers, BiTE®; dual affinity retargeting antibodies, DART; bispecific engagers, ENG).8,10,11 We and others have previously reported on a T-cell platform that secretes bispecific engagers (ENG T cells) against solid tumors and hematologic malignancies.10,12-14 CD123-specific ENG T cells (CD123.ENG) secrete a bispecific antibody consisting of two single chain variable fragments, one able to bind CD123 and the other specific for CD3e. 10 We have shown that CD123.ENG T cells have antiAML activity in preclinical models.10 However, the effector function of ENG T cells, like that of CAR T cells, decreases rapidly upon repeated tumor exposure.15-17
Several approaches are being pursued to increase the ability of CAR and ENG T cells to sequentially kill tumor cells.18-21 These include transgenic expression of molecules
Abishek Vaidya,1 Erin Doherty,2 Xiya Wu,1 Sujuan Huang,3 Nikhil Hebbar,1 Unmesha Thanekar,1 Challice L. Bonifant,4 Cheng Cheng,3 Stephen Gottschalk1 and M. Paulina Velasquez1
Haematologica | 108 - April 2023 1039 ARTICLE - Cell Therapy & Immunotherapy
such as cytokines or co-stimulatory molecules,22,23 or knocking out negative regulators.24 We and others have shown that activating an inducible co-stimulatory molecule, consisting of a myristoylation-targeting sequence, MyD88 lacking its TIR domain, the cytoplasmic domain of CD40, and two tandem FKBP12v36 domains (iMC), significantly improves the effector function of CAR T cells, including their ability to repeatedly kill tumor cells.18,21,25 Here we explored whether an inducible co-stimulation system can be utilized to enhance the effector function of CD123.ENG T cells and determined the individual contribution of MyD88 and CD40. To achieve this, we generated retroviral vectors encoding CD123.ENG and inducible MyD88 (iM), inducible CD40 (iC), or iMC. We demonstrated that activation of iM and iMC improves the effector function of CD123.ENG T cells in vitro. However, for consistent benefit in vivo, activation of both MyD88 and CD40 was required in CD123.ENG T cells.
Methods
Cell lines and culture conditions
The MOLM-13 cell line was purchased from the Leibniz Institute (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany). MV-411, Kg1a, THP-1, K562 and HEK 293T were purchased from the American Type Culture Collection (Manassas, VA, USA). MOLM-13, MV-411, Kg1a, THP-1 and K562 cells expressing an enhanced green fluorescence protein firefly luciferase fusion gene (MOLM-13.GFP.ffluc and K562.GFP.ffluc) were generated by transducing cells with a retroviral vector encoding GFP.ffluc.26-28
Generation of retroviral vectors
The generation of SFG retroviral vectors encoding: (i) CD20 and CD123.ENG (CD20.T2A.CD123.ENG), (ii) CD20 and CD19.ENG (CD20.T2A.CD19ENG), and (iii) inducible costimulatory molecules encoding a myristoylation sequence, two FKBP dimerizer domains, and iM, iC or iMC with an HA-tag have been previously reported.10,13,18,25 Additional details are described in the Online Supplementary Appendix
Generation of bispecific engager T cells
All procedures involving human subjects were carried out in accordance with the Declaration of Helsinki. Human peripheral blood mononuclear cells from healthy donors were obtained, after acquiring informed consent, under a St. Jude Children’s Research Hospital protocol approved by the hospital’s institutional review board. Peripheral blood mononuclear cells were stimulated on CD3 (1 µg/mL, Miltenyi Biotec, Bergisch Gladbach, Germany) and αCD28 (1 µg/mL, Miltenyi Biotec) antibody-coated non-tis-
sue culture treated 24-well plates (144530, Thermo Fisher Scientific, Waltham, WA, USA). Human interleukin (IL)-7 and IL-15 (10 ng/mL and 5 ng/mL, respectively) (Biological Research Branch, National Cancer Institute, Frederick, MD, USA) were added to cultures on day 2. On day 3, T cells were transduced with retroviral particles on plates coated with retronectin (T100A, Takara Clontech, Mountain View, CA, USA) in the presence of IL-7 and IL-15. On day 5 transduced T cells were harvested and were subsequently expanded with IL-7 and IL-15. Non-transduced T cells were activated with CD3/CD28 and expanded in parallel with IL-7 and IL-15. Cells were cultured for 7-10 days prior to being used for in vitro or in vivo experiments.
Xenograft acute myeloid leukemia model
All animal experiments were performed on a protocol approved by the St. Jude Children’s Research Hospital’s Institutional Animal Care and Use Committee in accordance with the American Association for Laboratory Animal Science. Additional details are provided in the Online Supplementary Appendix.
Statistical analysis
Data were summarized using descriptive statistics. Measurement data are presented as mean ± standard deviation (SD). To examine overall differences in outcomes between constructs, an analysis of variance (ANOVA) test was used. This overall test was followed by pairwise comparisons using the t-test and when appropriate ANOVA was performed.
A generalized estimating equation was used to determine the overall difference in outcomes with repeated measurements over time, to account for the intra-subject correlation. A two-sided significance level of P<0.05 was used for all statistical tests. Adjustment for multiple testing was not performed because of the small sample size and the exploratory nature of the analysis. For the mouse experiments, survival, determined from the time of tumor cell injection, was analyzed by the Kaplan–Meier method and by the log-rank test. Statistical analyses were conducted with SAS 9.4 and GraphPad Prism 8 (GraphPad software). Additional experimental procedures are described in the Online Supplementary Appendix
Results
Generation of CD123.ENG T cells expressing inducible co-stimulatory molecules CD123.ENG, CD123.ENG.iMC, CD123.ENG.iM, CD123.ENG.iC, and CD19.ENG.iMC T cells were generated by transduction with retroviral vectors depicted in Figure 1A. Seven to 10 days after transduction, transduction efficiency was evaluated by determining CD20 expression via flow cyto-
Haematologica | 108 - April 2023 1040 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
metry analysis. Mean transduction efficiency was 57.4% (range, 52-63.4%) with no significant differences between constructs (n=12; Figure 1B, Online Supplementary Figure S2A). Expression of iM, iC, and iMC was confirmed by western blot for the HA-tag (Figure 1C). Following transduction, all T-cell populations expanded and there were no statistically significant differences (n=6; Online Supplementary Figure S2B). To confirm that CD123.ENG T cells secrete CD123.ENG protein, effector T cells were plated on a plate coated with recombinant CD123 protein with or without 0.5 nM chemical inducer of dimerization (CID). The concentration of secreted CD123.ENG protein was determined by enzyme-linked immunosorbent assay (ELISA) after 24 h. Unlike CD19.ENG.iMC T cells, effector T cells
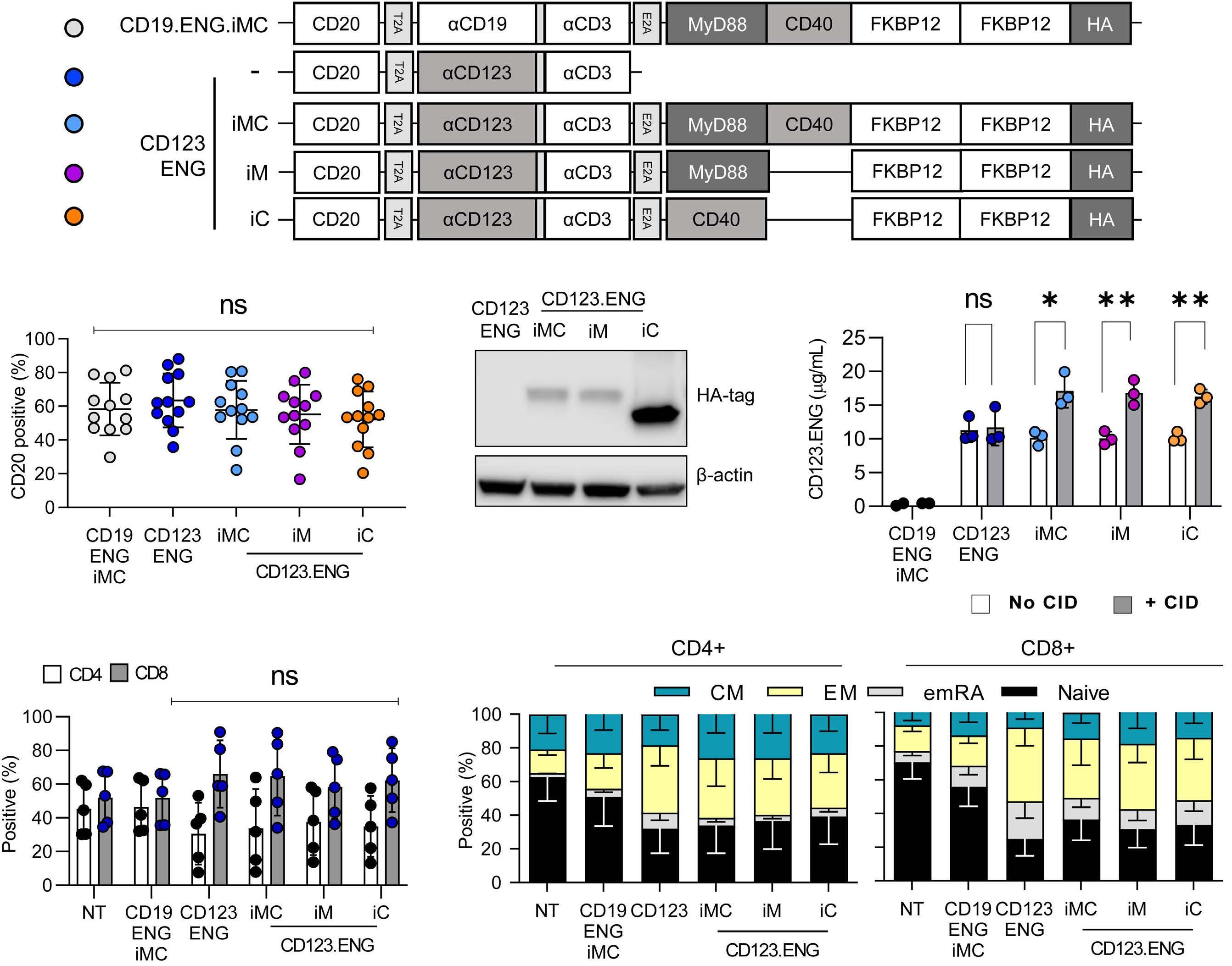
encoding CD123.ENG produced significant amounts of CD123.ENG protein (P<0.01) (Figure 1D). In addition, exposure to CID significantly increased CD123.ENG protein secretion of CD123.ENG T cells endowed with an inducible co-stimulation domain (iMC, iM, or iC) (P<0.05). T-cell subset analysis revealed that while CD123.ENG, CD123.ENG.iMC, CD123.ENG.iM, and CD123.ENG.iC T cells had a higher percentage of CD8+ T cells than non-transduced and CD19.ENG.iMC T cells, this difference did not reach statistical significance (Figure 1E). CD45RO and CCR7 cell surface markers were used to differentiate between naïve-like (N), central memory (CM), effector memory (EM) and terminally differentiated effector memory (EMRA) T cells. All CD123.ENG-expressing CD8+ T cells ex-
Figure 1. Generation of CD123.ENG T cells expressing iMC, iM, and iC. (A) Schema of vectors. Data throughout the manuscript are represented by the color code of the circles to the left of each construct. (B) Transduction efficiency was determined by CD20 expression (mean% ± standard deviation%: 57.4%±4.1%, n=12, P=ns). (C) Expression of iMC, iM, and iC was determined by western blot for the HA-tag. (D) CD123.ENG protein production by engager T cells plated in the presence of CD123 protein with or without 0.5 nM chemical inducer of dimerization was determined by enzyme-linked immunosorbent assay (n=3, *P<0.05, **P<0.01, Ttest). (E, F) T cells were stained 7 days after transduction for CD4, CD8, CD45RO and CCR7 to determine their immunophenotype (n=5). (E) CD4+ and CD8+ T-cell populations. (F) T-cell subsets: naïve-like, CCR7+CD45RO–; central memory (CM), CCR7+CD45RO+; terminally differentiated (emRA), CCR7–CD45RO–; and effector memory (EM), CCR7–CD45RO+). CID: chemical inducer of dimerization; iMC: inducible MyD88 and CD40; iM: inducible MyD88; iC: inducible CD40; ENG: engager; NT: not transduced.
A B C D E F Haematologica | 108 - April 2023 1041 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
cept CD123.ENG.iMC CD8+ T cells had a significantly (P<0.05) lower percentage of TN cells in comparison to non-transduced T cells. There were no statistically significant differences in the different CD4+ T-cell populations. (Figure 1F).
CD123.ENG T cells expressing inducible co-stimulatory molecules maintain antigen specificity
To evaluate whether constructs expressing inducible costimulation (CD123.ENG.iMC, CD123.ENG.iM, and CD123.ENG.iC T cells) maintained antigen specificity like CD123.ENG T cells, we performed co-culture assays in the presence or absence of 0.5 nM CID with CD123+ (MOLM13.ffLUC, MV-4-11.ffLUC, Kg1a.ffLUC) AML cells (Online Supplementary Figure S2C). Non-transduced and CD19.ENG.iMC T cells served as control T effector cells, while media and CD123– K562.ffLUC served as controls for target cells. Only CD123.ENG, CD123.ENG.iM, CD123.ENG.iC and CD123.ENG.iMC T cells produced significant amounts (n=3, P<0.05) of interferon (IFN)-γ and IL-2 in the presence of MOLM-13,, MV-4-11, or Kg1a in comparison to nontransduced and CD19.ENG.iMC T cells, confirming antigen specificity (Figure 2A, B, Online Supplementary Figure S3A). CD123.ENG.iM, CD123.ENG.iC and CD123.ENG.iMC T cells secreted higher amounts of IFN- γ and IL-2 in the presence of CID; however, the difference did not reach statistical significance (Figure 2A, B, Online Supplementary Figure S3A). Next, we examined the cytolytic activity of each T-cell population using a luciferase-based cytotoxicity assay. Effector T cells were plated in the presence of CD123+ (MOLM13.ffLUC, MV-4-11.ffLUC and Kg1a.ffLUC) or CD123– targets (K562.ffLUC) at a 1:1 effector to target (E:T) ratio. All constructs secreting CD123 ENG (CD123.ENG, CD123.ENG.iMC, CD123.ENG.iM and CD123.ENG.iC) recognized and killed CD123+ targets but did not have antitumor activity against K562.ffluc (CD123–). The addition of CID did not confer additional antitumor activity in these conditions. Effector T-cell controls (CD19.ENG.iMC and nontransduced T cells) showed no cytotoxicity towards CD123+ or CD123– targets (Figure 2C, Online Supplementary Figure S3B).
To further differentiate functional differences between CD123.ENG T-cell constructs, we performed a CD107a degranulation assay and analyzed TCF1 expression after exposing effector cells to CD123.ENG protein with or without CID. Our analysis showed increased degranulation in the groups secreting CD123.ENG in the presence of CD123+ targets in comparison to non-transduced cells, without any statistically significant differences between CD123.ENG constructs (Online Supplementary Figure S4A). In addition, TCF-1 expression was consistent across constructs and was not influenced by the addition of CID (Online Supplementary Figure S4B,C) Finally, we evaluated fold expression of genes associated
with T-cell function, activation and cell migration using quantitative polymerase chain reaction analysis. In the presence of CID and after overnight incubation, CD123.ENG.iMC T cells maintained a similar gene expression profile to that of CD123.ENG T cells (n=4, P=ns). However, CD123.ENG.iM and CD123.ENG.iC T cells showed significant upregulation of TIGIT, TIM3 and CCR5 in the presence of CID in comparison to iMC T cells (n=4, P<0.05). In addition, CD123.ENG.iC T cells expressed higher levels of MyD88, Blimp1, TCF7, Traf6 and EOMES, consistent with a more differentiated phenotype (Figure 2D, E).
iMC and iM enhance the effector function of CD123.ENG T cells in the setting of chronic antigen exposure Having observed limited benefit of activating iMC, iM, or iC with CID after a single exposure to tumor cells, we evaluated the effector function of CD123.ENG, CD123.ENG.iMC, CD123.ENG.iM, and CD123.ENG.iC in a sequential re-stimulation assay to mimic chronic antigen exposure (Online Supplementary Figure S4D). MOLM13.GFP.ffluc cells or Kg1a.GFP.ffluc cells were co-cultured with effector T cells at an E:T ratio of 1:1 with or without CID. Every 3-4 days, the presence of MOLM13.GFP.ffluc or Kg1a.GFP.ffluc cells was determined by a luciferase assay, and fresh tumor cells with or without CID were added if tumor cells had been killed (n=9 and n=3, respectively). T cells were also enumerated. A subset of donor cells that were co-cultured with MOLM13.GFP.ffluc underwent flow cytometric immunophenotype analysis (n=3) and media were collected for cytokine analysis 24 h after co-culture (n=3). In the absence of CID, all CD123.ENG T-cell populations were able to kill target cells for two to five stimulations, depending on the target cells used (Figure 3A, B left panels). T-cell expansion followed a similar pattern and cells failed to expand after two to five stimulations (Figure 3C, D left panels). The presence of CID did not affect antitumor activity or T-cell expansion for the CD123.ENG and CD123.ENG.iC groups. In comparison, CD123.ENG.iM and CD123.ENG.iMC T cells were able to kill target cells for five to 11 stimulations, depending on whether they were exposed to MOLM13.GFP.ffluc or Kg1a.GFP.ffluc (Figure 3A, B right panel). In the presence of CID, CD123.ENG.iM and CD123.ENG.iMC T cells continued to expand for six to ten stimulations whereas CD123.ENG and CD123.ENG.iC T cells stopped expanding after two to four stimulations (Figure 3C, D). Sequential immunophenotypic analysis in a subset of donors revealed that the expansion in CD123.ENG.iM and iMC groups was driven by an expansion of CD8+ T cells (Online Supplementary Figure S5A, B). CD19.ENG.iMC T cells had no cytolytic activity and did not expand in the presence of CID, confirming antigen specificity (Figure 3A, C).
To determine the cytokine expression profile in the setting of serial stimulation, we measured a Th1/Th2 panel of
Haematologica | 108 - April 2023 1042 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
Figure 2. Functional characterization of CD123.ENG.iM, CD123.ENG.iC and CD123.ENG.iMC T cells. (A, B) Effector cells were cocultured with media, K562 (CD123–), MOLM-13 (CD123+) or Kg1a (CD123+) cells at a 1:1 effector:target (E:T) cell ratio in the presence or absence of 0.5 nM chemical inducer of dimerization (CID) for 24 h. Supernatants were collected and evaluated for (A) interferon-γ (IFN-γ) and (B) interleukin-2 (IL-2) by enzyme-linked immunosorbent assay (ELISA). All T cells expressing CD123.ENG had a statistically significant increase in IFN-γ and IL-2 secretion in comparison to the secretion of T cells expressing CD19.ENG.iMC, when exposed to CD123+ target cells (n=3, *P<0.05,**P<0.01 paired t-test). (C) To determine antitumor activity, effector cells were co-cultured with target cells expressing firefly luciferase (MOLM-13.ffluc and Kg1a.ffluc) at an E:T ratio of 1:1. Tumor cell lysis was determined using a luciferase assay (n=3, P=ns for all CD123.ENG-expressing constructs). (D) Heatmap depicting fold expression increase in a panel of 14 genes by quantitative polymerase chain reaction in the presence or absence of CID. Gene expression was normalized to 18S RNA, and GAPDH. (E) Fold-expression increase for PD1 and CCL4. iMC: inducible MyD88 and CD40; iM: inducible MyD88; iC: inducible CD40; ENG: engager; NT: not transduced.
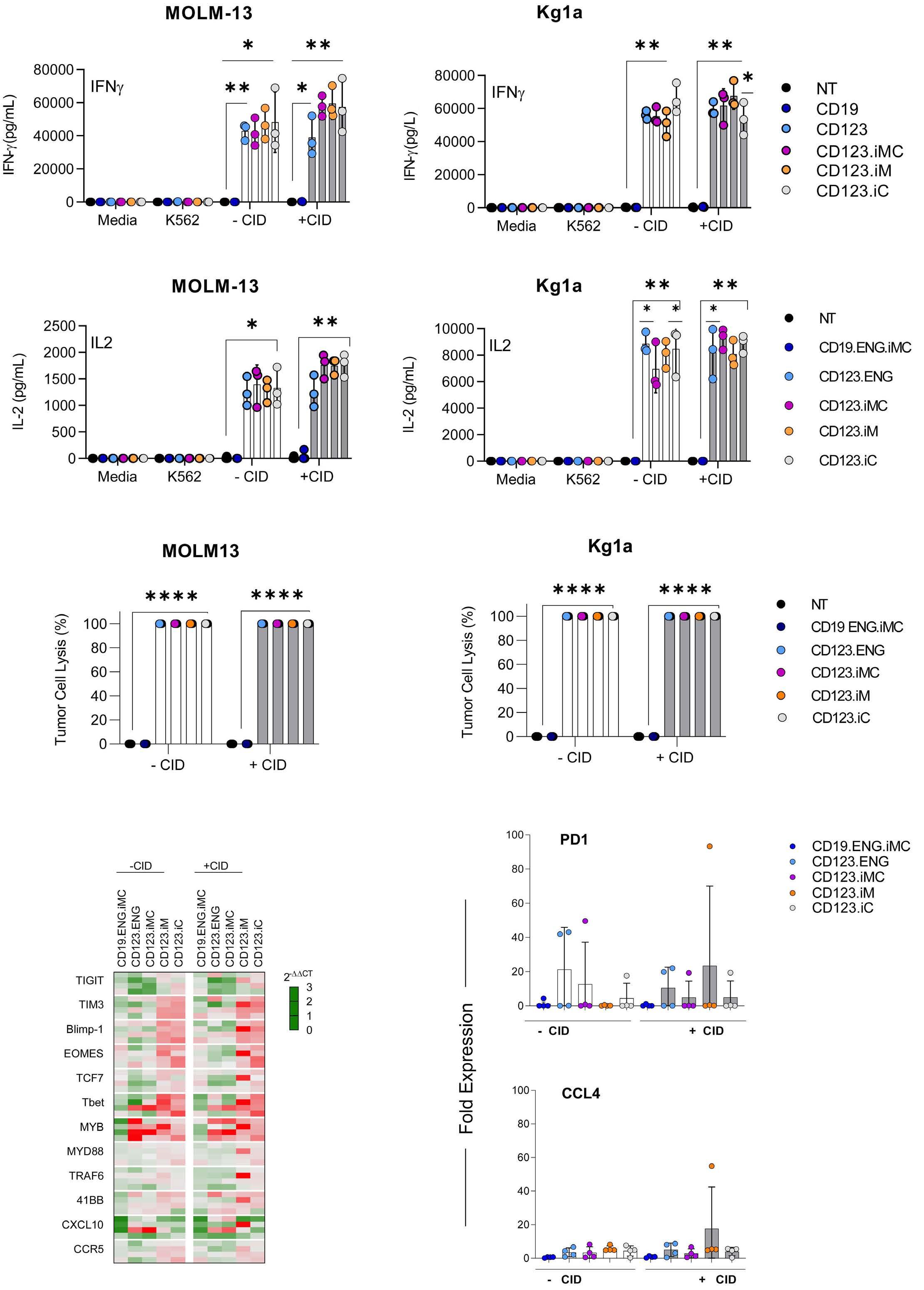
A B C D E Haematologica | 108 - April 2023 1043 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
cytokines including IFN-γ, TNFα, IL-2, and GM-CSF (Th1) and IL-4, IL-5, IL-6, IL-10 and IL-13 (Th2) 24 h after each stimulation (Figure 4A). CD123.ENG.iMC T cells secreted significantly higher amounts of IFN-γ than did CD123.ENG, CD123.ENG.iM and CD123.ENG.iC T cells. In addition, iMC cells secreted significantly higher levels of TNFα and GM-CSF compared to CD123. ENG and CD123.ENG.iM T cells after the first stimulation. CD123.ENG.iMC T cells
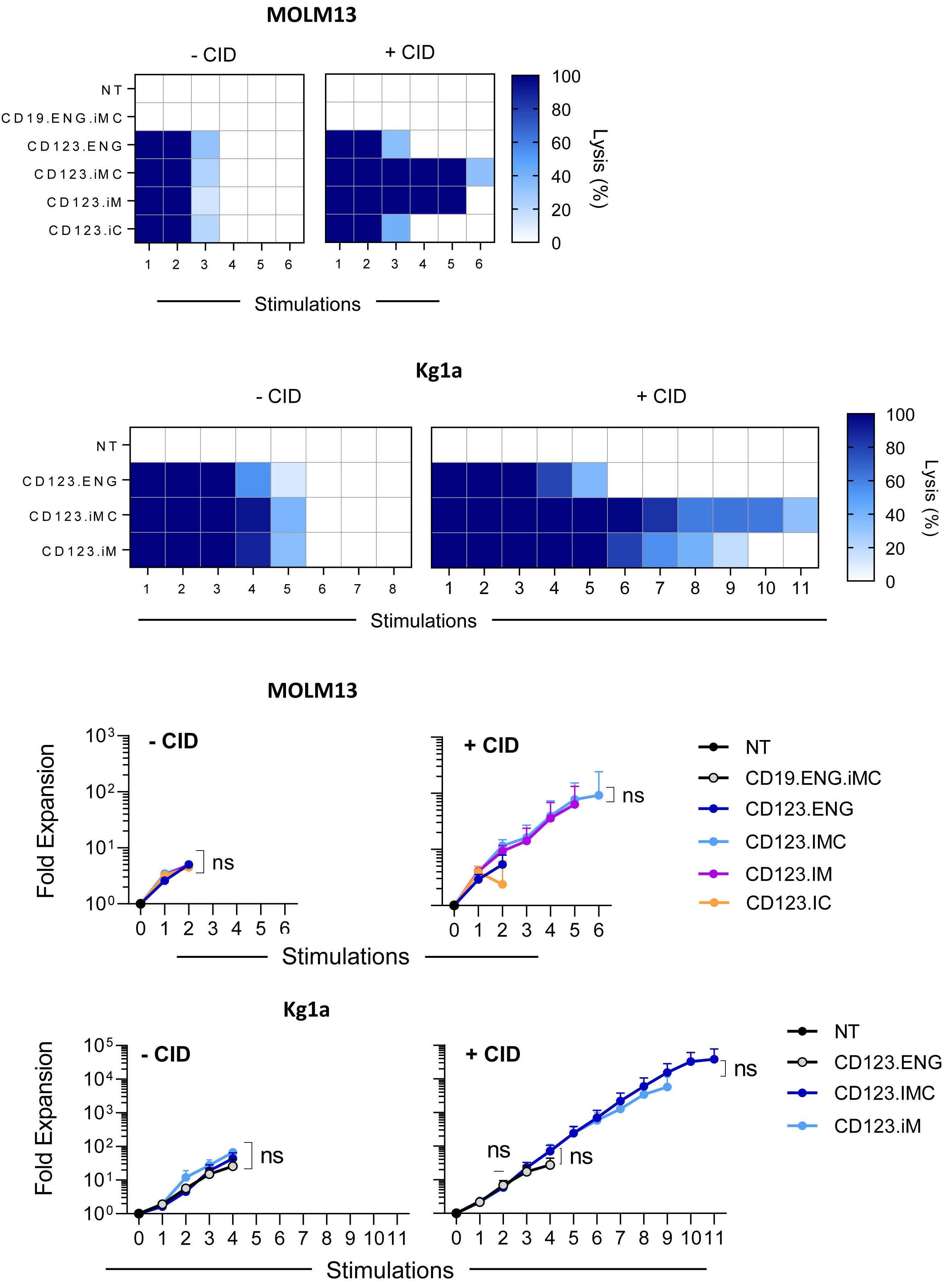
consistently maintained this increased secretion through stimulation 5. In addition, CD123.ENG.iMC consistently secreted higher levels of IL-13 with every stimulation.
CD123.ENG.iMC and CD123.ENG.iM T cells become predominantly CD8+ central memory T cells and transiently overexpress TIM3 after multiple stimulations We determined the CD4:CD8 ratio as well as TN, TCM, TEM,
Figure 3. CD123.ENG.iMC and CD123.ENG.iM T cells retain their effector function in the presence of a chemical inducer of dimerization in repeat stimulation assays. Effector T-cell populations were co-cultured with either MOLM-13.ffluc or Kg1a.ffluc cells at an effector:target (E:T) cell ratio of 1:1 with or without 0.5 nM chemical inducer of dimerization. Antitumor activity was assessed using a luciferase assay. Fresh tumor cells were added every 5 days resetting the E:T ratio to 1:1 until T cells no longer killed tumor cells. (A, B) Heat maps showing antitumor activity of effector cells in the presence of (A) MOLM13.ffluc (n=9) and (B) Kg1a.ffluc (n=3) (dark blue=100% killing, white=0% killing). (C, D) Fold expansion of effector T cells after exposure to (C) MOLM13.ffluc (n=6) or (D) Kg1a.ffluc (n=3). NT: not transduced; iMC: inducible MyD88 and CD40; iM: inducible MyD88; iC: inducible CD40; ENG: engager; ns: not statistically significant.
A B C D Haematologica | 108 - April 2023 1044 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
and TEMRA distribution after stimulation. All populations were predominantly CD8+, with CD123.ENG.iMC and CD123.ENG.iM T cells becoming predominantly TCM subsets, and CD123.ENG and CD123.ENG.iC T cells having a higher percentage of TEM and TEMRA (Figure 5A, Online Supplementary Figure S5). To further characterize cells and determine the exhaustion phenotype of T cells, we examined cell surface expression of PD1, TIM3 and LAG3 on CD123.ENG, CD123.ENG.iMC, CD123.ENG.iM, and CD123.ENG.iC T cells before each stimulation. While iMC and iM T cells expressed increased TIM3 between the second and fourth stimulation (Online Supplementary Figure S6), concomitant increased expression of two markers of exhaustion was not observed (Figure 5B).
CD123.ENG.iMC T cells consistently improve the antitumor activity of CD123.ENG T cells in vivo
To evaluate the antitumor activity of CD123.ENG T cells expressing iMC, iM, and iC we used our established MOLM13 NSG xenograft model that does not require prior sublethal irradiation. Mice were injected with 5x104 MOLM-
13.GFP.ffluc cells intravenously, and on day 7 received a single intravenous dose of 1x107 CD123.ENG, CD123.ENG.iMC, CD123.ENG.iM, CD123.ENG.iC, or CD19ENG.iMC T cells (n=10 mice per group with 2 T-cell donors, except for n=5 mice for CD19-ENG.iMC T cells; 1 T-cell donor). CID was given intraperitoneally every 3 to 4 days starting on the day of the T-cell injection (Figure 6A). Tumor burden was tracked by serial bioluminescence imaging. CD123.ENG.iMC T cells had significant antitumor activity in both donors, whereas CD123.ENG.iM T cells only controlled the tumor in one out of two donors (Figures 6 and 7). This resulted in a significant survival advantage over that of mice that had received CD123.ENG T cells for both donors for CD123.ENG.iMC T cells and for one donor for CD123.iM T cells (CD123.ENG vs. CD123.ENG.iMC T cells: P=0.0031, donor 1; P=0.0019, donor 2; CD123.ENG vs CD123.ENG.iM T cells; P=0.51, donor 1; P=0.0019, donor 2) (Figure 6B). In contrast, mice treated with CD123.ENG.iC T cells had no survival advantage in comparison to that of mice treated with CD123.ENG T cells. CD19.ENG.iMC T cells had no antitumor activity, demonstrating that the benefit
Figure 4. CD123.ENG.iMC T cells secrete increased Th1 cytokines upon stimulation. Heatmap showing cytokine secretion by effector T cells after stimulation as measured by multiplex assay (n=3; 2-way ANOVA). ENG: engager; iC: inducible CD40; iM: inducible MyD88; iMC: inducible MyD88 and CD40; IFN: interferon; TNF: tumor necrosis factor; IL: interleukin; GM-CSF: granulocyte-macrophage colony stimulating factor.
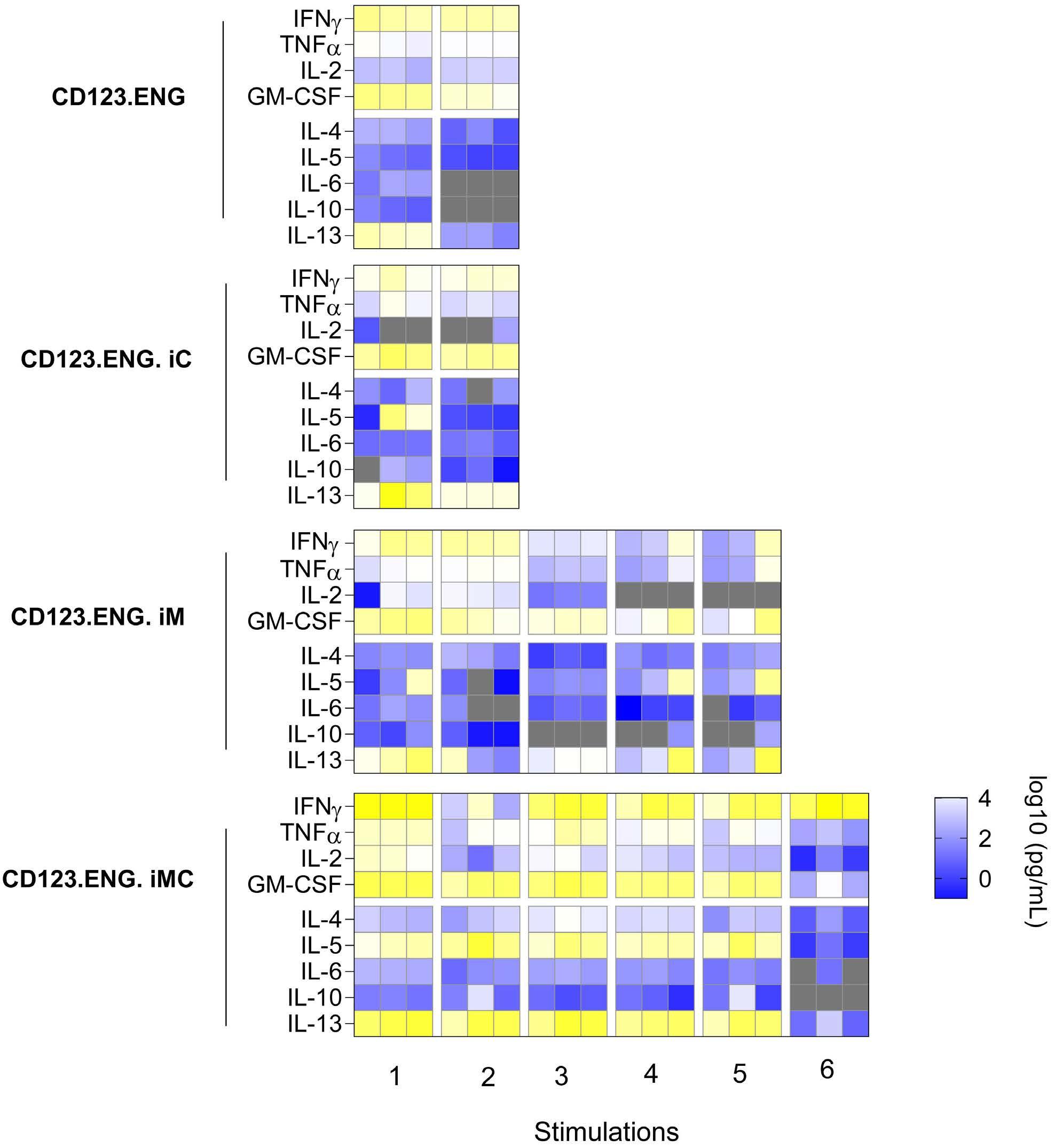
Haematologica | 108 - April 2023 1045 ARTICLE - Inducible co-stimulation
T-cell
A. Vaidya et al.
to enhance engager
function
Figure
iM, and iC after repeat stimulation. (A) Pie-chart representation of changes in immunophenotype in CD8+ populations of CD123.ENG.iM and CD123.ENG.iMC T cells after stimulations in the presence of chemical inducer of dimerization (CID) (n=3). The pie-charts were plotted using SPICE software38 (National Institute of Allergy and Infectious Diseases). (B) Summary of percentage of effector cells that stained double-positive for an inhibitory receptor (TIM3+LAG3+, PD1+LAG3+ or TIM3+PD1+) after repeated stimulations in the presence of CID. iMC: inducible MyD88 and CD40; iM: inducible MyD88; iC: inducible CD40; ENG: engager; CM: central memory; EM: effector memory; emRA: terminally differentiated effector memory.
of CID in vivo is strictly antigen-dependent, as observed in vitro. The weight of mice without tumors remained stable overall for long-term survivors (Online Supplementary Figure S7C). However, two of the ten mice in the CD123.ENG.iM T-cell group had to be euthanized due to non-tumor related morbidities on day 46 (paraphimosis) and day 68 (weight loss, presumptive graft-versus-host disease), and two of the ten mice in the CD123.ENG.iMC T-cell group on days 68 and 97 (weight loss, presumptive graft-versus-host disease). We confirmed these findings in a THP-1 model, in which disease was controlled in the
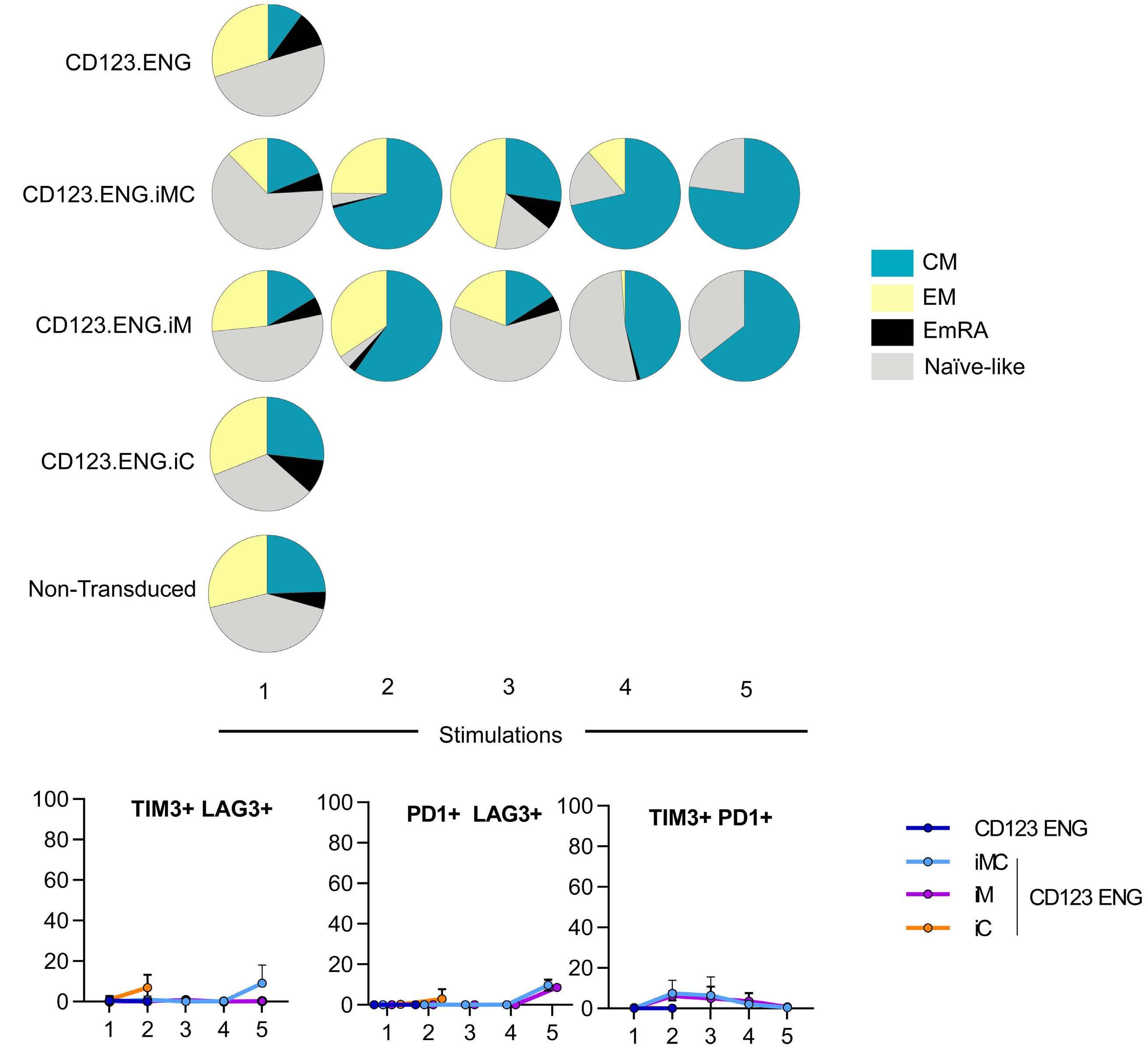
groups receiving either CD123.ENG.iM or CD123.ENG.iMC and CID, while it progresses in control groups (CD123.ENG vs. CD123.ENG.iMC or CD123.ENG.iM; P<0.0001) (Online
Supplementary Figure S8).
To evaluate the effect of iMC and iM activation in CD123.ENG T cells on in vivo T-cell expansion and persistence, we performed the same experiment as described above with T cells genetically modified to express GFP.ffluc instead of the tumor cells. Within the first 10 days after infusion, we observed a significantly greater expansion and persistence of CD123.ENG.iMC T cells, as
A B Haematologica | 108 - April 2023 1046 ARTICLE - Inducible co-stimulation to
engager T-cell
A. Vaidya et al.
5. Immunophenotype of CD123.ENG T cells expressing iMC,
enhance
function
Figure 6. CD123.ENG.iMC and CD123.ENG.iM T cells have potent antitumor activity in vivo. MOLM-13.GFP.ffluc-bearing mice received a single intravenous dose of 1x107 T cells on day 7 (n=10 animals per group, 2 T-cell donors for CD123.ENG, CD123.ENG iMC, CD123.ENG iM, or CD123.ENG iC T cells; n=5, 1 T-cell donor for CD19.ENG.iMC). Four doses of a chemical inducer of dimerization were given intraperitoneally every 3-4 days. (A) Experimental scheme and quantitative bioluminescence data for donor 1 and donor 2. (B) Kaplan-Meier survival curve (**P<0.01, log rank test). D0: day 0; D7: day 7; CID: chemical inducer of dimerization; ENG: engager; iMC: inducible MyD88 and CD40; iM: inducible MyD88; iC: inducible CD40;
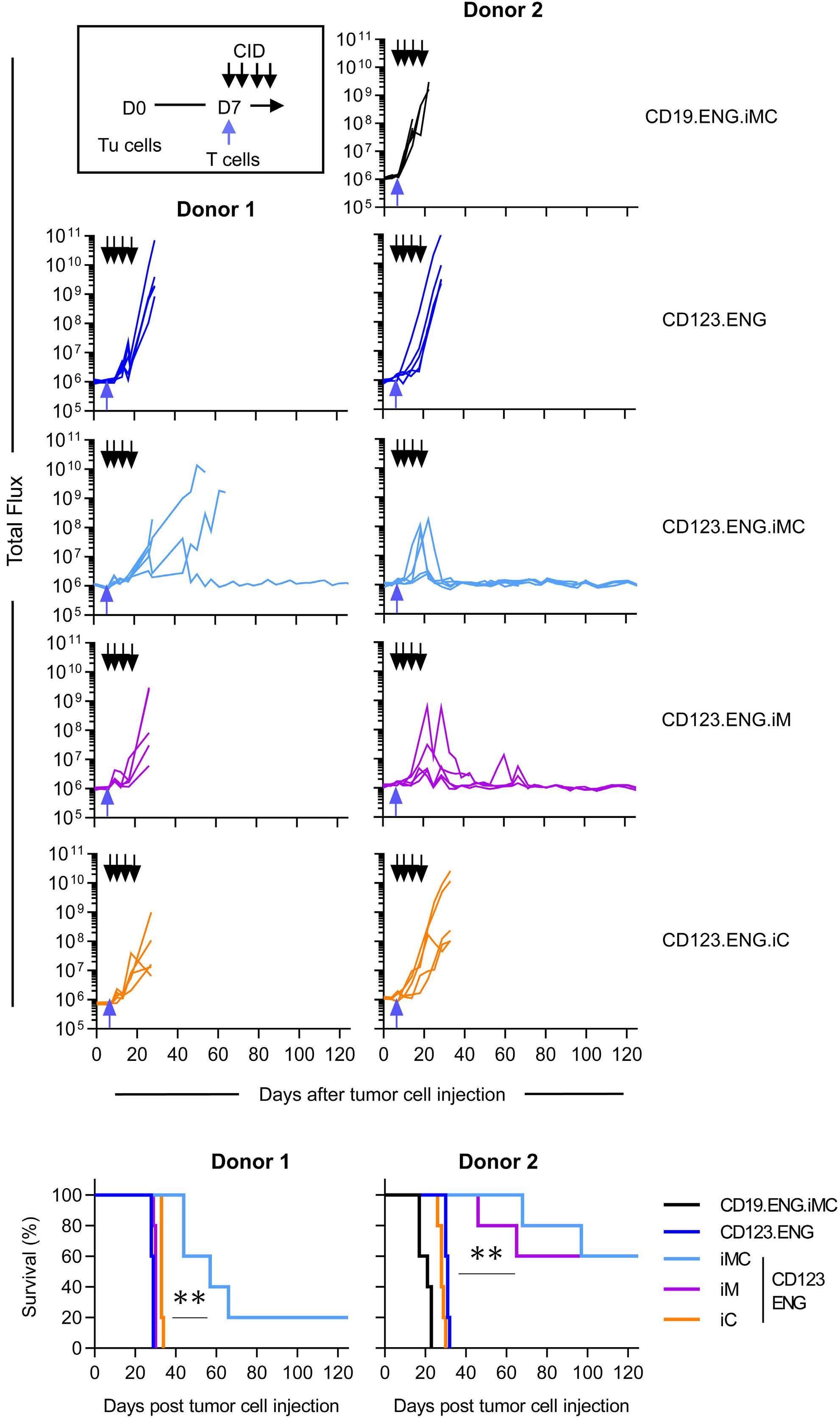
A B Haematologica | 108 - April 2023 1047 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
judged by area under the curve analysis (P<0.001), in comparison to that of CD123.ENG or CD123.ENG.iM T cells (Figure 8A, B, Online Supplementary Figure S9).
CD123.ENG.iMC T cells do not induce increased killing of normal CD123-positive hematopoietic progenitors and recognize primary acute myeloid leukemia blasts
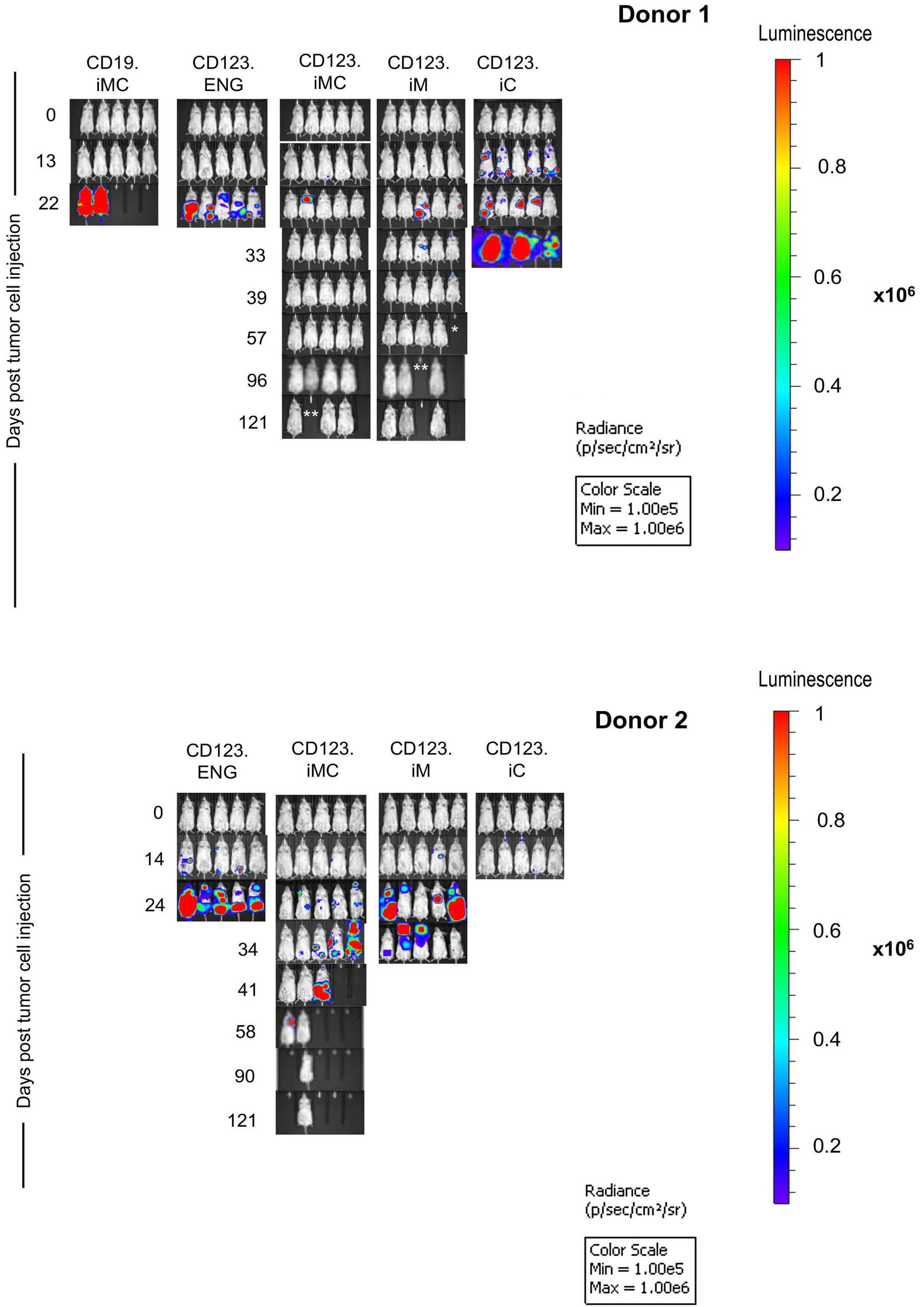
To determine whether expressing iMC, iM or iC could increase potential on-target/off-tumor toxicity of CD123.ENG T cells against bone marrow CD123+/CD34+ hematopoietic cells (Online Supplementary Figure S10) we performed standard colony-forming unit (CFU) assays at E:T ratios of 1:1 and 5:1 in the presence of CID. At both E:T
ratios, CD123.ENG.iMC cells did not induce greater myelotoxicity than non-transduced T cells in assays for granulocyte-erythroid-monocyte-megakaryocyte, erythroid, and granulocyte macrophage CFU (n=3 technical replicates, P=ns) (Figure 8C), but did show greater toxicity on burst-forming units at a 1:1 ratio (P=0.0038). CD123.ENG.iC T cells had some degree of myelotoxicity at both ratios and in all groups.
Lastly, we set out to determine whether we could generate genetically modified T cells from three diagnostic bone marrow samples of pediatric patients with AML and to evaluate whether they recognized CD123+ autologous AML blasts. We thawed primary bone marrow samples
Figure 7. Representative
images
the acute myeloid leukemia xenograft model. (A) Donor 1: representative IVIS images. Asterisks represent animals requiring euthanasia due to non-tumor related morbidities (*paraphimosis, **graft-versus-host disease). (B) Donor 2: representative IVIS images. Stars represent animals requiring euthanasia due to non-tumor related morbidities. iMC: inducible MyD88 and CD40; iM: inducible MyD88; iC: inducible CD40; ENG: engager.
A B Haematologica | 108 - April 2023 1048 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
IVIS
of
and evaluated surface expression of CD123 on AML blasts by flow cytometry (Figure 8D). We isolated CD3+ T cells using magnetic bead separation. The CD3–, AML blastcontaining cell fraction was frozen. We then generated CD123.ENG, CD123.ENG.iMC, CD123.ENG.iM, CD123.ENG.iC, and CD19-ENG.iMC T cells, and co-cultured these effector T cells with the primary, thawed AML blasts in the presence of 0.5 nM CID. After 24 h, media were collected and a limited cytokine evaluation by ELISA was performed by determining IFN- γ secretion. Only CD123.ENG and CD123.ENG.iMC T cells produced consistently more IFN-γ compared to non-transduced and CD19-ENG.iMC T cells (n=3, P<0.01 for CD123.ENG.iMC) (Figure 8E).
Discussion
Here we show that inducible activation of MyD88 and CD40 signaling in CD123.ENG T cells enhances their persistence and sequential killing capabilities without affecting antigen specificity. In the presence of CID, CD123.ENG.iMC had superior and sustained effector function in vitro and in vivo compared to CD123.ENG, CD123.ENG.iM and CD123.ENG.iC T cells.
Adoptive immunotherapy strategies to target AML are being actively explored, focusing on antigens such as CD123, CD33, CLL-1, LeY, among others.29 CD123 is an attractive target because of its high expression on leukemia
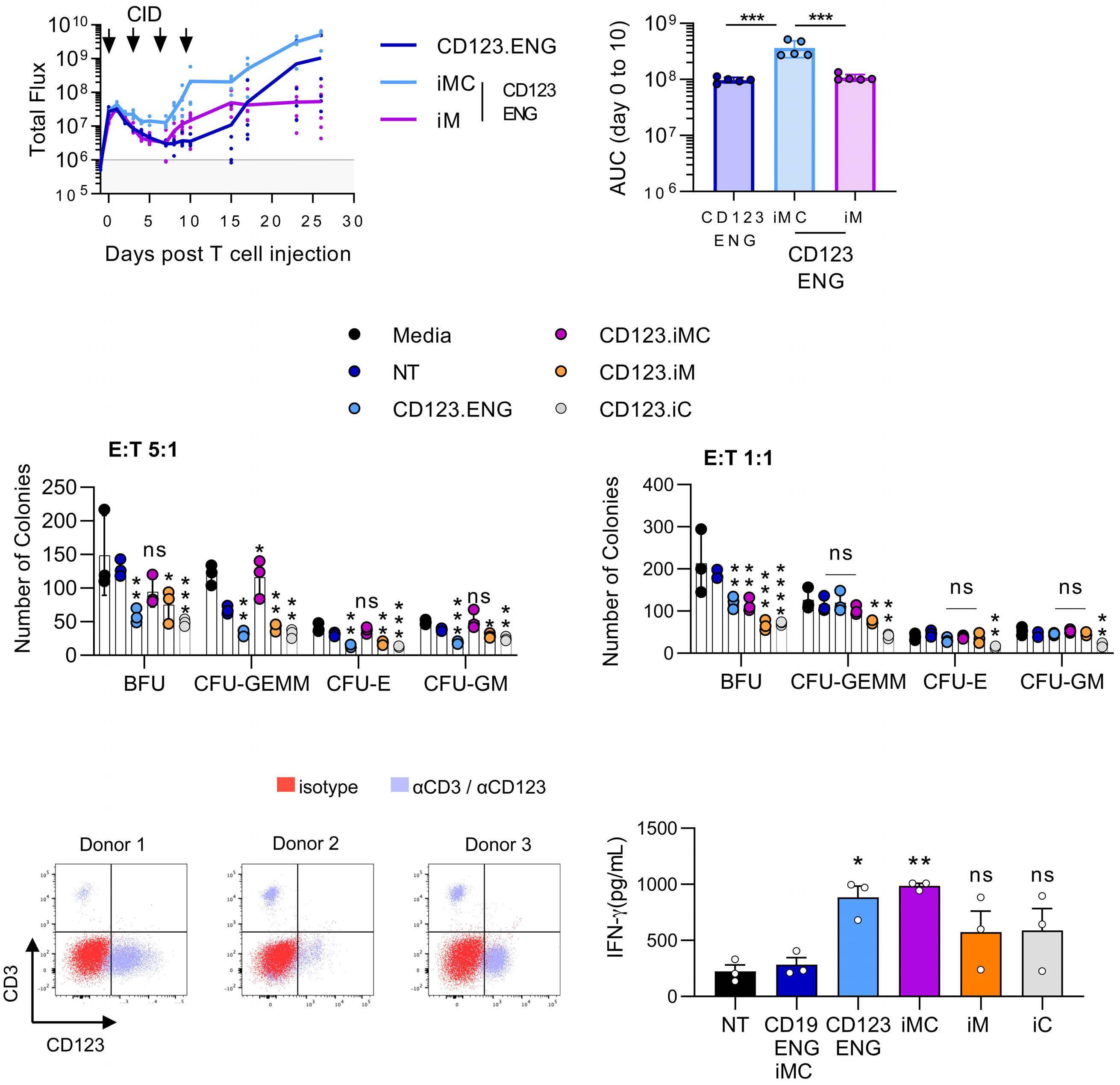
A B C D E Continued on following page. Haematologica | 108 - April 2023 1049 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
Figure 8. CD123.ENG.iMC T cells expand and persist in an in vivo acute myeloid leukemia model, do not have increased myelotoxicity, and recognize primary autologous acute myeloid leukemia blasts. (A,B) Analysis of T-cell persistence: MOLM-13 bearing mice (n=5 per group) received a single intravenous dose of 1x107 CD123.ENG, CD123.ENG iMC, or CD123.ENG iM T cells expressing GFP.ffluc on day 7; four doses of a chemical inducer of dimerization (CID) were given intraperitoneally every 3-4 days. (A) Quantitative bioluminescence data. (B) Area under the curve analysis for day 0 to day 10 after T-cell infusion (***P<0.001, 2-way analysis of variance [ANOVA]). (C) Colony-forming unit (CFU) assays: as indicated effector T cells were incubated with hematopoietic cells for 4 h at effector:target (E:T) ratios of 5:1 and 1:1, plated on semisolid media, and burst-forming units, CFU-granulocyte-erythroid-monocyte-megakaryocyte, CFU-erythroid and CFU-granulocyte-macrophage were enumerated after 12–14 days. (*P<0.05, **P<0.01, 2-way ANOVA). (D,E) Primary autologous T cells recognize primary acute myeloid leukemia (AML) blasts. (D) Flow cytometry analysis for CD123 and CD3 of bone marrow samples of pediatric patients with AML. (E) Co-culture assay in the presence of 0.5 nM CID at an E:T ratio of 1:1 with indicated effector T cells and autologous AML blasts. After 24 h media were obtained for enzyme-linked immunosorbent assay for interferon-γ (*P<0.05, **P<0.01, generalized linear model). CID: chemical inducer of dimerization; iMC: inducible MyD88 and CD40; iM: inducible MyD88; iC: inducible CD40; ENG: engager; AUC: area under the curve; NT: not transduced; E:T: effector-to-target; ns: not statistically significant; BFU: burst-forming unit; CFU: colony-forming unit; GEMM: granulocyte-erythroid-monocyte-megakaryocyte; E: erythroid; GM: granulocyte-macrophage; IFN: interferon.
stem cells and relatively low expression on normal hematopoietic cells. CD123-ENG T cells have been described as one of the approaches to target AML, but application of this strategy has been limited by modest T-cell persistence, likely due to the absence of co-stimulation.10 Optimal T-cell activation requires signal 1 delivered by Tcell receptor recognition of antigen-speci fi c peptide through major histocompatibility complex, signal 2 provided by co-stimulatory or inhibitory molecules expressed on the surface of antigen-presenting cells followed by a cytokine signal (signal 3) critical for T-cell expansion and response.30-33 Augmenting T-cell effector function by transgenic expression of co-stimulatory molecules has been explored by several groups.18,22,25 We have previously shown that constitutively expressing CD80 and 41BBL on the surface of CD19.ENG T cells enhances their effector function, resulting in increased Th1 cytokine secretion, T-cell expansion and superior antitumor activity.15 In addition, several groups have explored the use of inducible co-stimulatory systems, such as the MyD88.CD40 receptor controlled by CID.18,21,25 MyD88 is an adaptor for pathways downstream of TLR and IL-1, leading to functional outputs such as the activation of NFkB and MAP.34 CD40 is a co-stimulatory molecule member of the tumor necrosis factor receptor (TNFR) family that acts through TRAF-mediated pathways that can have overlap with MyD88.35,36 Together, they have been shown to be effective in increasing CAR T-cell activity and function. Mata et al. and Foster et al. showed that, in the presence of CID, two different types of fi rst-generation CAR T cells (HER2 ζ or PSCA ζ ), which also expressed an inducible MyD88 and CD40 receptor, had enhanced antitumor activity in vitro and in vivo . 18,21 Collison-Pautz et al . and Prinzing et al. further demonstrated that constitutive provision of MyD88/CD40 co-stimulation in CAR T cells resulted in increased expansion and antitumor activity of the T cells.35,36 In this study, we explored whether provision of inducible MyD88 and/or CD40 co-stimulation could improve the effector function of CD123.ENG T cells and determined which co-stimulatory domain combina-
tion is the most effective in maintaining T-cell persistence and sustained antitumor activity, without additional myelotoxicity as evidenced by CFU assays.
We demonstrated that expressing inducible MyD88 and/or CD40 switches do not change the antigen specificity or the immunophenotype of CD123.ENG T cells. All CD123.ENG T cells acquired a predominantly CD8+ effector memory phenotype, both in healthy donor T cells cocultured with tumor cell lines and using an autologous system of primary T cells and blasts.
To further characterize these cells, we determined LAG3, TIM3 and PD1 expression, 5 days after stimulation with fresh tumor cells. Inhibitory receptors such as LAG3, PD1 and TIM3 have been shown to dampen T-cell responses and have been associated with reduced Th1 cytokine secretion.37 These markers remained largely at baseline levels for CD123.ENG.iMC until the fifth stimulation, when there was a slight increase in TIM3 + LAG3 + as well as PD1 + LAG3 + expression on CD8 + T cells, which did not reach statistical significance. Consistent with these findings, CD123.iMC T cells secreted increased amounts of Th1 cytokines, specifically IFN-γ and TNF-α and GM-CSF. This increased cytokine secretion persisted throughout repeated stimulations and led to increased persistence. Expression of MyD88 in combination with CD40, was critical for consistent expansion and antitumor activity by CD123.ENG T cells through multiple stimulations in vitro and in two xenograft AML models. This was only evident in the presence of CID, highlighting the importance of TLR pathway activation to achieve an effective T-cell response.18,21 Response to CD123.ENG.iM in the presence of CID, on the other hand, proved to be dependent on both the donor and tumor model in vivo.
In summary, we demonstrated that CD123.ENG T cells expressing inducible co-stimulation maintain antigen specificity and that expression of MyD88 or MyD88 and CD40 confers improved expansion capability and potent antitumor activity in vitro and in vivo. In addition, the ability to remotely control the amount of T-cell engager secretion and T-cell persistence while maintaining antigen
Haematologica | 108 - April 2023 1050 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
speci fi city using this inducible system endows CD123.ENG.iMC cells with a desirable safety feature.
Disclosures
AV, CLB, SG and MPV have patent applications in the field of T-cell and gene-modified T-cell therapy for cancer. CLB has received research funding from Merck, Sharpe, and Dohme, Inc., Bristol Myers Squibb, and Kiadis Pharma. SG is a consultant for Catamaran Bio, Nektar Therapeutics, and TESSA Therapeutics, sits on the Scienti fi c Advisory Board of Tidal, and is a member of a Data Safety Monitoring Board for Immatics.
Contributions
AV, ED, SG and MPV conceived the study; AV, ED, XW, SH, NH, UT, SG and MPV analyzed data; AV, ED, XW, NH and UT performed investigations; CB and CC obtained resources; AV, ED, XW, SH, NH, UT and MPV conducted formal analyses; AV, SG and MPV wrote the original draft of the manuscript; AV, ED, XW, SH, NH, UT, CB, CC, SG and MPV contributed to writing, reviewing and editing the paper; SG and MPV acquired funds and supervised the study.
Acknowledgments
Preclinical procedures were performed in collaboration
References
1. Raetz EA, Bhatla T. Where do we stand in the treatment of relapsed acute lymphoblastic leukemia? Hematology Am Soc Hematol Educ Program. 2012;2012:129-136.
2. Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121(7):1077-1082.
3. Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14(6):e205-217.
4. Gokbuget N, Stanze D, Beck J, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120(10):2032-2041.
5. Epperly R, Gottschalk S, Velasquez MP. A bump in the road: how the hostile AML microenvironment affects CAR T cell therapy. Front Oncol. 2020;10:262.
6. Lamble AJ, Lind EF. Targeting the immune microenvironment in acute myeloid leukemia: a focus on T cell immunity. Front Oncol. 2018;8:213.
7. Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014;2(1):4.
8. Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138-3148.
9. Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389-401.
10. Bonifant CL, Szoor A, Torres D, et al. CD123-engager T cells as a
with St. Jude Children’s Animal Resource Center. Light microscopy was performed in collaboration with the Cell and Tissue Imaging Center. We would also like to thank other members of the Velasquez group and Diane Woods for their kind assistance with supporting this research work.
Funding
Preclinical imaging was performed by the Center for In vivo Imaging and Therapeutics, which is supported in part by National Institutes of Health (NIH) grants P01CA096832 and R50CA211481. Sequencing was performed by the Hartwell Center, which is supported in part by the National Cancer Institute (NCI)/NIH grant P30CA021765. The work was supported by the Leukemia Lymphoma Society (648316), the Cancer Prevention Research Institute of Texas (RP160693), St. Baldrick’s Foundation, the Assisi Foundation of Memphis, and the American Lebanese Syrian Associated Charities.
Data-sharing statement
Most data are available within the article or its supplementary materials. Any additional data are available upon reasonable request from the authors.
novel immunotherapeutic for acute myeloid leukemia. Mol Ther. 2016;24(9):1615-1626.
11. Cummins KD, Gill S. Chimeric antigen receptor T-cell therapy for acute myeloid leukemia: how close to reality? Haematologica. 2019;104(7):1302-1308.
12. Iwahori K, Kakarla S, Velasquez MP, et al. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther. 2015;23(1):171-178.
13. Velasquez MP, Torres D, Iwahori K, et al. T cells expressing CD19-specific engager molecules for the immunotherapy of CD19-positive malignancies. Sci Rep. 2016;6:27130.
14. Liu X, Barrett DM, Jiang S, et al. Improved anti-leukemia activities of adoptively transferred T cells expressing bispecific T-cell engager in mice. Blood Cancer J. 2016;6(6):e430.
15. Velasquez MP, Szoor A, Vaidya A, et al. CD28 and 41BB costimulation enhances the effector function of CD19-specific engager T cells. Cancer Immunol Res. 2017;5(10):860-870.
16. Krenciute G, Prinzing BL, Yi Z, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5(7):571-581.
17. Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumormediated inhibition. J Clin Invest. 2016;126(8):3130-3144.
18. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 2017;7(11):1306-1319.
Haematologica | 108 - April 2023 1051 ARTICLE - Inducible co-stimulation to enhance engager T-cell function A. Vaidya et al.
19. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. 2017;7(1):10541.
20. Alizadeh D, Wong RA, Yang X, et al. IL15 Enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res. 2019;7(5):759-772.
21. Foster AE, Mahendravada A, Shinners NP, et al. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol Ther. 2017;25(9):2176-2188.
22. Zhao Z, Condomines M, van der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415-428.
23. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423-431.
24. Choi BD, Yu X, Castano AP, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunolther Cancer. 2019;7(1):304.
25. Narayanan P, Lapteva N, Seethammagari M, Levitt JM, Slawin KM, Spencer DM. A composite MyD88/CD40 switch synergistically activates mouse and human dendritic cells for enhanced antitumor efficacy. J Clin Invest. 2011;121(4):1524-1534.
26. Chow KK, Naik S, Kakarla S, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629-637.
27. Kakarla S, Chow KK, Mata M, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21(8):1611-1620.
28. Braun DA, Wu CJ. Antigen discovery and therapeutic targeting in
hematologic malignancies. Cancer J. 2017;23(2):115-124.
29. Epperly R, Gottschalk S, Velasquez MP. Harnessing T cells to target pediatric acute myeloid leukemia: CARs, BiTEs, and beyond. Children (Basel). 2020;7(2):14.
30. Lafferty KJ, Misko IS, Cooley MA. Allogeneic stimulation modulates the in vitro response of T cells to transplantation antigen. Nature. 1974;249(454):275-276.
31. Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165(2):302-319.
32. Sharpe AH. Mechanisms of costimulation. Immunol Rev. 2009;229(1):5-11.
33. Chen L, Flies DB. Molecular mechanisms of T cell costimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227-242.
34. Warner N, Nunez G. MyD88: a critical adaptor protein in innate immunity signal transduction. J Immunol. 2013;190(1):3-4.
35. Collinson-Pautz MR, Chang WC, Lu A, et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia. 2019;33(9):2195-2207.
36. Prinzing B, Schreiner P, Bell M, Fan Y, Krenciute G, Gottschalk S. MyD88/CD40 signaling retains CAR T cells in a less differentiated state. JCI Insight. 2020;5(21):e136093.
37. Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, Wherry EJ. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog. 2011;7(7):e1002098.
38. Roederer M, Nozzi JL, Nason MC. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A. 2011;79A(2):167-174.
Haematologica | 108 - April 2023 1052
A. Vaidya et al.
ARTICLE - Inducible co-stimulation to enhance engager T-cell function
Patients with hypercortisolemic Cushing disease possess a distinct class of hematopoietic progenitor cells leading to erythrocytosis
Correspondence: A.R. Migliaccio a.migliaccio@unicampus.it
1Division of Hematology and Oncology, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 2Multidisciplinary Pituitary and Skull Base Tumor Center, Departments of Medicine and Neurosurgery, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 3National Center for Drug Research and Evaluation, Istituto Superiore di Sanità, Rome, Italy; 4Department of Biomedical and Neuromotorial Sciences, Alma Mater Studiorum University, Bologna, Italy; 5Altius Institute for Biomedical Sciences, Seattle, WA, USA; 6Department of Cell, Developmental, and Regenerative Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 7Division of Hematology, Department of Medicine, University of Washington, Seattle, WA, USA and 8Center for Integrated Biomedical Research, Campus Bio-medico, Rome, Italy.
*LV and EBG contributed equally as co-first authors. #TP and ARM contributed equally as co-senior authors.
Abstract
T. Papayannapoulou thalp@uw.edu
Received: January 3, 2022.
Accepted: May 11, 2022.
Early view: July 21, 2022.
https://doi.org/10.3324/haematol.2021.280542
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Although human cell cultures stimulated with dexamethasone suggest that the glucocorticoid receptor (GR) activates stress erythropoiesis, the effects of GR activation on erythropoiesis in vivo remain poorly understood. We characterized the phenotype of a large cohort of patients with Cushing disease, a rare condition associated with elevated cortisol levels. Results from hypercortisolemic patients with active Cushing disease were compared with those obtained from eucortisolemic patients after remission and from volunteers without the disease. Patients with active Cushing disease exhibited erythrocytosis associated with normal hemoglobin F levels. In addition, their blood contained elevated numbers of GR-induced CD163+ monocytes and a unique class of CD34+ cells expressing CD110, CD36, CD133 and the GR-target gene CXCR4. When cultured, these CD34+ cells generated similarly large numbers of immature erythroid cells in the presence and absence of dexamethasone, with raised expression of the GR-target gene GILZ. Of interest, blood from patients with Cushing disease in remission maintained high numbers of CD163+ monocytes and, although their CD34+ cells had a normal phenotype, these cells were unresponsive to added dexamethasone. Collectively, these results indicate that chronic exposure to excess glucocorticoids in vivo leads to erythrocytosis by generating erythroid progenitor cells with a constitutively active GR. Although remission rescues the erythrocytosis and the phenotype of the circulating CD34+ cells, a memory of other prior changes is maintained in remission.
Introduction
Genetic studies have well established that activation of the glucocorticoid receptor (GR),1 in cooperation with soluble stem cell factor (SCF),2 is required to recover from anemia induced by exogenous insults in mice. Mice carrying GR encoding a protein unable to respond to glucocorticoids (GRdim mice)1 have a normal hematocrit but do not readily recover from the anemia induced by hemolytic agents.2 In addition, stress inducers such as radiation, prompt the proteolytic cleavage of the membrane-bound form of SCF, increasing the concentration of soluble SCF in the circulation.2
As in the GRdim mice, mice that harbor a SCF gene encoding
a protein lacking this proteolytic site also exhibit a normal hematocrit and are unable to recover from anemia induced by radiation. Mechanistically, the link between GR activation and soluble SCF is provided by the observation that soluble human SCF is uniquely able to activate the ERK signal that stabilizes the GR protein which in turn enables the cells to respond to glucocorticoids.3
By contrast, in humans most of the information available on the functions of GR has been obtained using surrogate in vitro models, represented by ex-vivo culture of CD34+ cells derived from peripheral blood4 and stimulated by cocktails of growth factors containing the GR agonist dexamethasone plus soluble SCF.5 As a result of this, the picture of the ef-
Lilian Varricchio,1* Eliza B. Geer,2* Fabrizio Martelli,3 Maria Mazzarini,4,5 Alister Funnell,5 James J. Bieker,6 Thalia Papayannopoulou7# and Anna Rita Migliaccio5,8#
Haematologica | 108 - April 2023 1053 ARTICLE - Hematopoiesis
fects of GR on human erythropoiesis is confounded by the fact that in many of these in vitro culture systems, human hematopoietic cells are stimulated with soluble murine SCF, which indeed sustains their differentiation, but is unable to activate ERK and to stabilize GR, thus making the cells partially unresponsive to glucorticoids.3,6 Given that glucocorticoids are widely used for treatment of various disorders, including the anemia of patients with Diamond-Blackfan anemia, knowledge on the in vivo effects of chronic GR activation in vivo is of great clinical relevance. Unfortunately, the only information available on the effects of chronic GR activation in vivo in humans is provided by the erythrocytosis reported for one patient with Cushing disease,7 a rare endocrine disorder (incidence of 1.2-2.4 cases/million persons/year8) characterized by chronic excessive glucocorticoid production due to an adrenocorticotropic hormonesecreting pituitary adenoma.9 Cushing disease is characterized not only by excess levels of circulating glucocorticoids, but also by flattened cortisol diurnal rhythms and resistance to cortisol suppression by exogenous glucocorticoids. The latter is defined by the lack of appropriate serum cortisol suppression after administration of dexamethasone, due to autonomous production of adrenocorticotropic hormone by the tumor, resulting in increased systemic cortisol levels with lack of normal feedback inhibition. Eucortisolemia, normal circadian rhythms, and normal cortisol feedback inhibition are restored over time after surgical removal of the adrenocorticotropic hormone-secreting tumor, which is the first-line treatment for patients with Cushing disease.10 Untreated Cushing disease results in increased mortality and multiple morbidities due to chronic hypercortisolemia (visceral obesity, diabetes, hypertension, cardiovascular disease, myopathy, immune suppression, cognitive deficits and mood disorders).
We hypothesized that studies on hypercortisolemic and eucortisolemic Cushing patients would greatly increase our knowledge on the effects of long-term treatments with glucocorticoids in humans. To test this hypothesis, we characterized here the blood counts and the response to glucocorticoids in vitro of circulating CD34+ cells in what is a relatively large cohort of patients with Cushing disease, given the rarity of the disease: 13 patients with active disease and 13 in a remission phase). Given the prior observations that glucocorticoids stimulate increases in body mass and that fat cells in the bone marrow exert an important regulatory role on hematopoiesis,11 results were compared with those obtained in parallel from subjects of similar body mass and age without Cushing disease (matched controls, MC).
Methods
Human subjects
Blood from patients with Cushing disease in an active or re-
mission phase, i.e., before and after surgically induced remission (mean time since surgery, 14.5 months) and from MC was collected at the Mount Sinai Hospital for a prospective cohort study, the clinical registry of which contained 28 patients with active Cushing disease, 11 paired remission cases and 13 MC. Of these patients, this study analyzes blood from 13 with active disease, 13 (unpaired) cases in remission and eight MC (Table 1) provided as de-identified material according to the Mount Sinai Hospital Institutional Review Boardapproved protocol dedicated to studies on Cushing disease (Principal Investigator, Eliza Geer, at the time Medical Director, Mount Sinai Pituitary Care and Research Center).
Blood parameters and cortisol levels
Blood values from patients with Cushing disease and from MC were determined by the Mount Sinai Hospital clinical laboratory and were retrieved from the patients’ charts. Twenty-four hour urinary free cortisol and serum cortisol levels were quantified by gas chromatography-mass spectroscopy as described elsewhere.12 The limit of sensitivity of this assay is 2 ng with interassay coefficients of variation <10%.
High performance liquid chromatography
High performance liquid chromatography analysis of globin chains was performed using a Bio-Rad (Hercules, CA) CDM System (CDM 5.1 VII Minion) as described previously.13 Individual globin chain levels were quantified on a Shimadzu Prominence instrument with an SPD-10AV diode array detector and an LC-10AT binary pump (Shimadzu, Kyoto, Japan).
Erythroid cultures
Mononuclear blood cells (MNC) were separated by centrifugation at 400 g for 30 min over Ficoll-Hypaque (AmershamPharmacia Biotec, Uppsala, Sweden). Human erythroblasts were obtained by culturing 106 MNC/mL for 10 days in Iscove modified Dulbecco medium (Mascia Brunelli, Milan, Italy) containing 20% fetal bovine serum (Hyclone, Logan, UT, USA), SCF (10 ng/mL; Amgen, Thousand Oaks, CA, USA), erythropoietin (1 U/mL; Amgen), interleukin-3 (1 ng/mL; Bouty, Milan, Italy), dexamethasone (10 6 M) and estradiol (10 6 M) (both from Sigma), as described elsewhere.5 Selected experiments were conducted in parallel in cultures supplemented with growth factors and with or without dexamethasone.
Colony-forming cell assays
Colony-forming cells were assayed by culturing MNC (3x105 cells/mL) for 12 days in methylcellulose cultures (MethoCult, Stem Cell Techonologies, Inc., Vancouver, British Columbia, Canada) stimulated with human SCF (10 ng/mL; Amgen), interleukin-3 (10 ng/mL, Biosource, San Jose, CA, USA), and erythropoietin (Amgen) as described previously.14
Haematologica | 108 - April 2023 1054 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
Table 1. Clinical data of the subset of patients and matched controls from the prospective cohort study included in the present investigation.
Four patients analyzed both in the active and in the remission phase are indicated in italics. ID: identity; M: male; F: female; na: information not available.
Active phase Remission phase Controls Patients' number/ID Ethnicity Sex Weight Kg Serum cortisol m g/dL Patients' number/ID Ethnicity Sex Weight Kg Serum cortisol m g/dL Patients' number/ID Ethnicity Sex Weight Kg P1/02-030 Caucasian M 78.2 27.5 P25/02-030 Caucasian M 75.6 1 P16/C18 n.a. F na P6/02-034 Caucasian F 69.3 19.2 P36/02-034 Caucasian F na 19.6 P23/C23 Caucasian M 77.9 P8/02-035 Caucasian M 75.3 32.2 P26/02-035 Caucasian M 67.7 18.9 P24/C24 Asian M 107.3 P12/02-037 Hispanic F 71.7 15.2 P27/02-037 Hispanic F 67.4 3.5 P29/C26 Asian M 79.5 P2/02-031 Caucasian F 86.7 15 P14/02-029 Caucasian F 101.6 3.1 P33/C27 Caucasian F 86.9 P10/02-036 African American F 130.5 24.2 P5/02-023 Caucasian F 80.4 2.5 P34/C28 Caucasian F 62 P15/02-038 Caucasian M n.a. 5.2 P13/02-017 Caucasian F 64 7.1 P37/C29 na F 96 P19/02-040 Caucasian M 88.1 16.7 P21/02-018 Caucasian M 103.9 11.1 P38/C30 na F na P20/02-042 Caucasian F 98 17 P7/02-025 African American F 88.1 15.9 P28/02-044 Caucasian F 68.4 18.9 P32/02-014 Caucasian F 68.6 24.3 P30/02-045 Caucasian F 110.6 24.7 P3/02-032 African American F na 13.7 P31/02-046 Caucasian F 78.4 32.7 P4/02-033 Caucasian F na 2.3 P35/02-047 Caucasian M n.a. 35.2 P17/02-039 Caucasian F 59.7 9.7
Haematologica | 108 - April 2023 1055 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
Flow cytometry
CD34+ cell determinations and phenotyping were conducted by incubating MNC with primary antibodies for 30 min at 4°C and fluorescent signals were measured with a FACS Canto II (Becton and Dickinson, Franklin Lakes, NJ, USA) and analyzed using FlowJo v7.6.4/v10 (Ashland, OR, USA). Cells were exposed to allophycocyanin (APC)-CD34 with either fluorescein isothiocyanate (FITC)-CD36, phycoerythrin-cyanine7 (PE-Cy7)-CD117, Pacific blue-CD123, phycoerythrin (PE)CXCR4, PE-CD110, PE-CALR or FITC-CD133. Dead cells were excluded by Sytox Blue AlexaFluor 430 staining. Cultured erythroblasts were profiled with FITC-CD36 and APCCD235a.15 The frequency and phenotype of blood monocytes were assessed by analyzing the percentage of MNC that bound PE-Cy7-CD14, APC-CD169 and PE-CD163 (all from BD Biosciences, San Jose, CA, USA).
Western blot analysis
Whole cell extracts (30 mg protein/lane) were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes which were probed with primary antibodies against either GRα (sc8992H300), glucocorticoid-induced leucine zipper (GILZ, sc33780) (all from Santa Cruz Biotechnology, Dallas, TX, USA), pGRS211 (4161, Cell Signaling, Danvers, MA, USA), pGRS203 (Ab195703, Abcam, Cambridge, MA, USA) and GAPDH, (CB1001, Calbiochem, San Diego, CA, USA) and appropriate horseradish peroxidase-coupled secondary antibodies (Calbiochem).
Statistical analyses
Data are presented as box charts (plus minimum and maximum values) or as the mean (± standard error of mean), as indicated. Values from patients with active disease or disease in remission and from MC were compared using the Tukey multiple comparisons test while those obtained with and without dexamethasone within the same groups were compared by a paired t-test using GraphPad 9.2 (SAS Institute, Cary, NC, USA). Differences were considered statistically significant with a P<0.05.
Results
Patients with active Cushing disease exhibit erythrocytosis with nearly normal levels of fetal hemoglobin
To clarify the effects of dexamethasone on stress erythropoiesis in vivo, we conducted a thorough characterization of erythropoiesis in a relatively large cohort of hypercortisolemic Cushing patients in the active phase and after achieving eucortisolemia due to surgically induced remission (mean time since surgery, 14.5 months), along with non-diseased controls matched for weight (body mass index 33.3 kg/m2 in
active-phase patients and 30.7 kg/m2 in MC; range, 26.7-34.7 kg/m2, P=0.073) and age (mean age 41 years in both groups).
In the prospective Cushing disease cohort, as expected the mean serum cortisol levels in the active-phase patients were significantly higher than in MC and remission-phase patients (Table 2). The differences between patients in the active phase and those in remission remained statistically significant also in the cohorts of patients included in this study (Tables 1 and 2).
In the subset of 13 active-phase patients included in this study, hematocrit levels were higher than those of MC while hemoglobin and platelet counts were similar in the two groups (Figure 1A, C). Retrospective analyses of the data present in the clinical database indicated that, overall, active-phase patients had significantly higher hematocrit levels than remission-phase patients and that in the analyses conducted on a subset of 11 patients with paired samples, hemoglobin and platelet levels were significantly higher during the active phase than during remission (Figure 1B). Unexpectedly, hematocrit and cortisol (serum and urine) levels were not correlated either in active-phase or remissionphase patients (Online Supplementary Figure S1). This lack of correlation may in part be due to the wide intra-patient variability in cortisol values.16 Other biophysical properties of red blood cells (red blood cell distribution width, mean corpuscular volume, mean cell hemoglobin, and mean corpuscular hemoglobin concentration) were also similar among the two groups and MC (Online Supplementary Figure S2). White blood cell counts were higher in active-phase patients than in MC and remission-phase patients (Figure 1D). Analysis of white blood cell subpopulations indicated that the increases were due to greater numbers of neutrophils, while, as expected due to the anti-inflammatory effect of GR activation, frequencies of lymphocytes and eosinophils were lower in patients with active Cushing disease than in patients
Table 2. Mean cortisol levels in the serum of patients with active Cushing disease and patients in remission included in the whole prospective cohort study with respect to the levels observed in donors without Cushing disease matched for weight, age and sex and in those included in the study (see Table 1 for details).
na: not available; MC: matched controls; A: patients with active Cushing disease.
Mean cortisol levels, mg/dL (prospective cohort study) Mean cortisol levels, mg/dL (sub-set of patients in Table I) Matched controls 8.5±8 (N=13) na Active 22.2±6.3 (N=11) 21.3±8.8 (N=13) (P=3x10-7 vs MC) Remission 8.7±3.9 (N=11) 10.2±7.7 (N=14) (P=0.006 vs. A) (P=0.002 vs. A)
Haematologica | 108 - April 2023 1056 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
Figure 1. Patients with active Cushing disease have higher hematocrit, hemoglobin level, and platelet and white blood cell counts than patients in remission. (A) Photograph of blood tubes from one representative patient with active Cushing disease and one matched control (MC). (B) Hematocrit, hemoglobin levels and platelet counts for the whole cohort of patients with Cushing disease with active disease or in remission included in the clinical database (unpaired, left panel, and paired, right panel). (C) Hematocrit, hemoglobin levels and platelet counts of the MC and patients with active Cushing disease included in the study. (D) White blood cell counts for patients with active-phase or remission-phase Cushing disease and the MC included in the study. The number of patients included in each analysis is indicated by n. P values were calculated in (B) and (C) by a t test and in (D) by the Tukey multiple comparisons test. A: patients with active Cushing disease; R: patients with Cushing disease in remission; MC: matched controls (matched for age, weight and sex) without Cushing disease; Hct: hematocrit (%); Hb: hemoglobin (g/dL); Plt: platelet count (x109/L); WBC: white blood cell count.
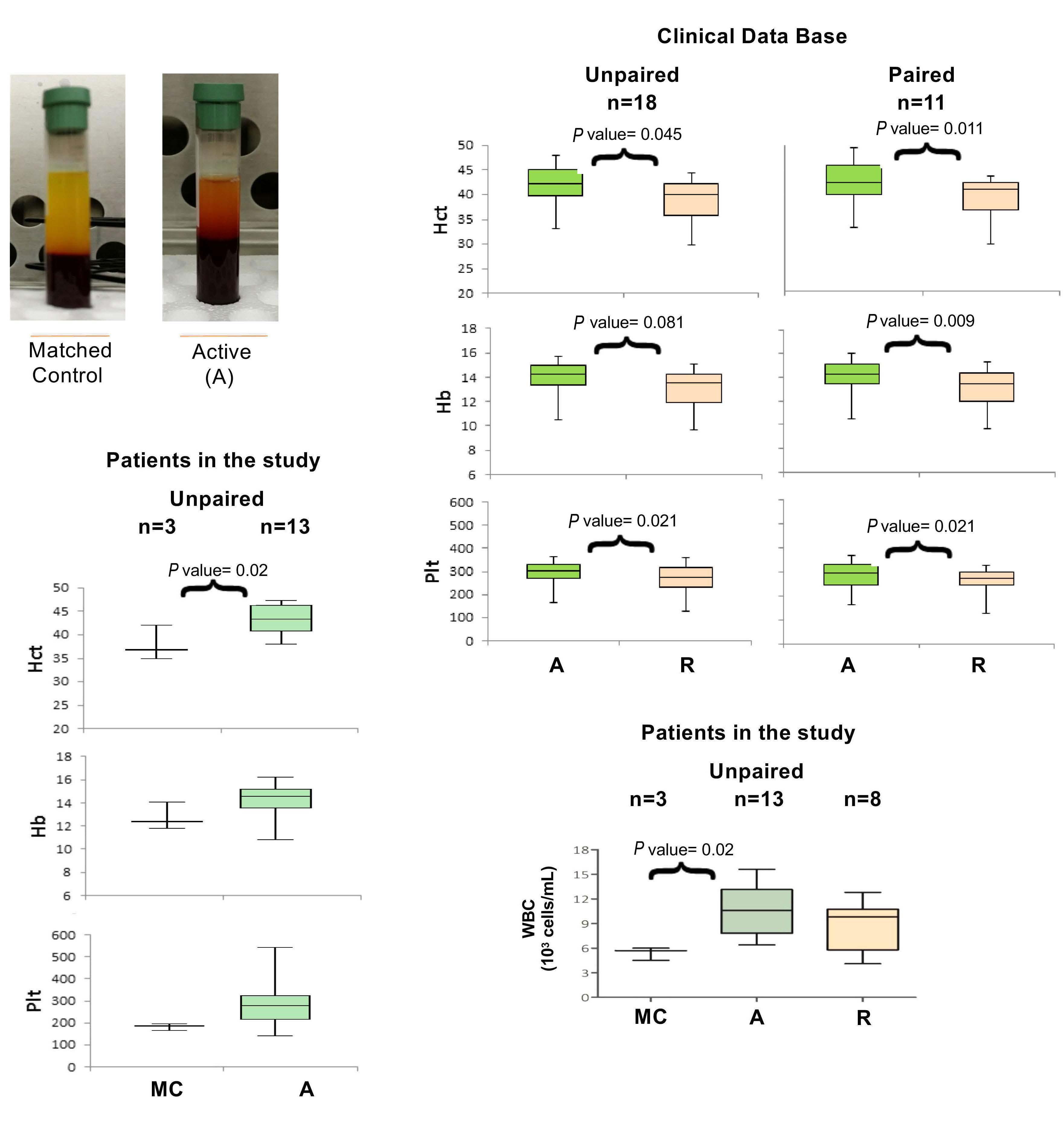
A B
Haematologica | 108 - April 2023 1057 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al. C D
Figure 2. Although the frequency of total monocytes in blood from Cushing patients (with active disease or in remission) and from matched contr ols is similar, the frequency of monocytes expressing CD163, which is induced by glucocorticoids in patients with Cushing disease, is greater than in ma tched controls. (A) Neutrophil, eosinophil, basophil, monocyte and lymphocyte counts in the blood from patients with Cushing disease (active and in remission) and ma tched controls (MC) included in the study. (B, C) Percentages of CD14 + cells and CD14 + cells expressing CD163 or CD169 in the blood from Cushing patients (active and in remission) and MC. Representative fl o w cytometry data are presented in (B) and the mean (± standard deviation) of multiple determinations are presented in (C). The number of patients included in each analysis is indicated by n. P values were calculated by the Tukey multiple comparisons test and those statistically signi fi can t are indicated in the panel. WBC: white blood cells; A: patients with active Cushing disease; R: patients with Cushing disease in remission; MC: matched contr ols (matched for age, weight and sex) without Cushing disease.
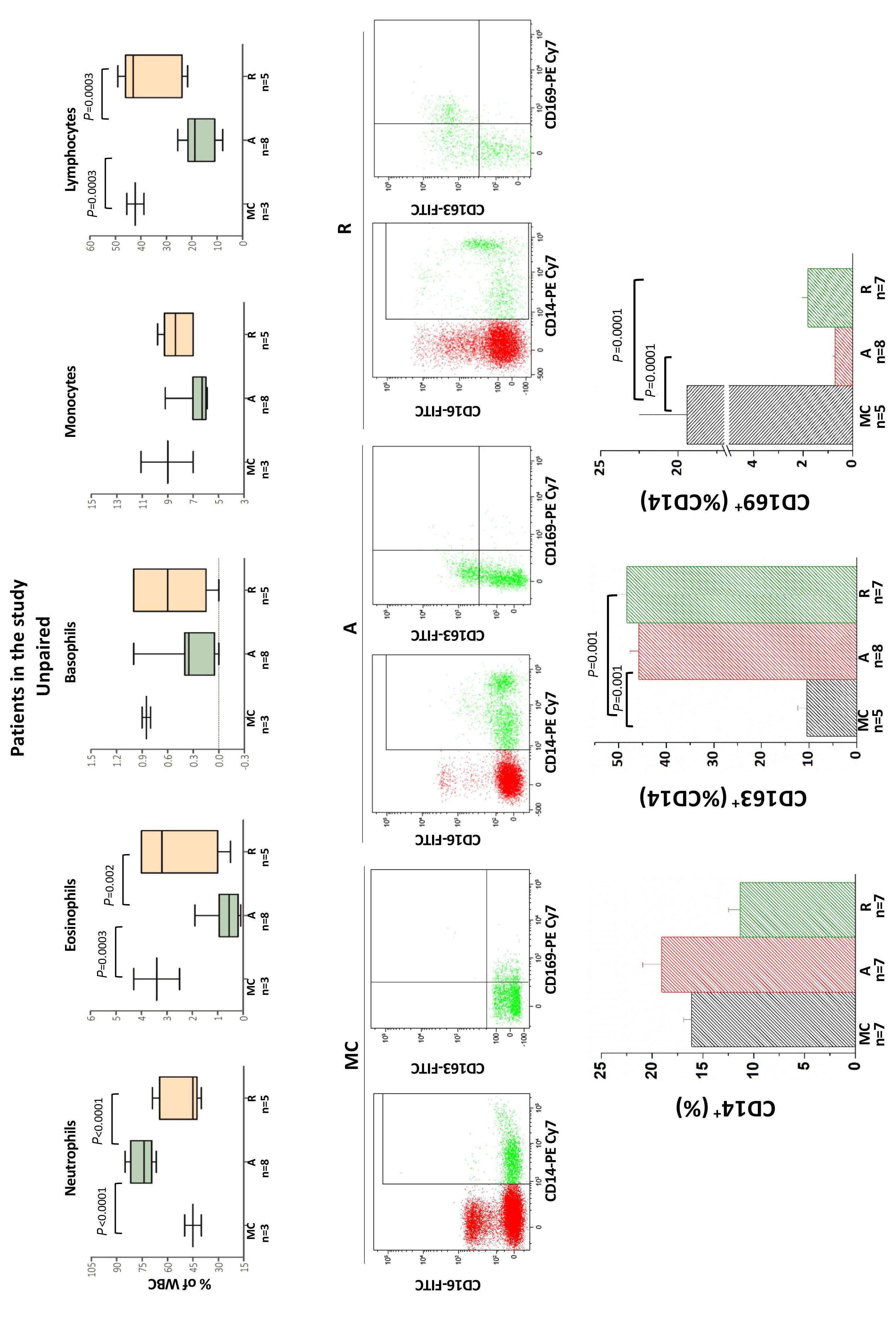
A B C
Haematologica | 108 - April 2023 1058 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
Figure 3. Patients with Cushing disease express overall normal levels of fetal hemoglobin. (A) High performance liquid chromatography determination of globin chain levels in red blood cells from representative patients with active Cushing disease and Cushing disease in remissions and matched controls (each graph represents a separate subject, as indicated; patients are identified with the same alpha-numerical code indicated in Table 1). (B, C) Average ratios of α/b-like and γ/β-like globins in red blood cells from active-phase and remission-phase patients and matched controls. P values were calculated by the Tukey multiple comparisons test. A: patients with active Cushing disease; R: patients with Cushing disease in remission; MC: matched controls (matched for age, weight and sex) without Cushing disease.
in remission or in MC (Figure 2A). Although the frequency of monocytes was comparable among groups, the phenotype of CD14+ cells in active-phase patients was skewed toward the CD163+ phenotype while the frequency of CD14+ cells expressing CD169 was reduced in these patients (Figure 2B, C). Since the haptoglobin-hemoglobin scavenger receptor CD163 is induced in monocytes by GR activation17,18 and CD169 is an interferon-α-induced inflammatory response probably suppressed by GR,19 the CD163+CD169– phenotype of the CD14+ cells from patients with active-phase Cushing disease provides an internal control showing that their hypercortisolemia is affecting their hematopoietic cells. In addition, the fact that the CD14+ cells from patients in remission remain skewed toward the CD163+/CD169– phenotype suggests that the monocytes maintain a memory of their activation state upon clinical remission. Whether the blood monocytes in Cushing disease, contribute to the erythroid-specific macrophage niche in the bone marrow is not known.
High performance liquid chromatography analyses of red blood cell lysates indicated that patients with active disease expressed slightly higher levels of α- and fetal γ-globin chains than those of patients in remission, although overall, these levels were within the range found in MC (Figure 3). These results indicate that patients with active Cushing disease experience erythrocytosis with normal hemoglobin F levels.
The blood from patients with active Cushing disease contains CD34+ cells with a stress-like phenotype
Recent studies of cultures stimulated with BMP4 or dexamethasone have identified a novel class of erythroid progenitor cells defined as stress-progenitor cells, which express CD110 (MPL, the thrombopoietin receptor20) and CD36 (the thrombospondin receptor21). In addition, murine erythroblasts generated with dexamethasone express higher levels of CXCR4 (the SDF1/CXCL12 receptor)22,23 and in human erythroblasts, GR is chaperoned to the nucleus
A B C
Haematologica | 108 - April 2023 1059 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
by the Ca2+-protein calreticulin (CARL) in response to dexamethasone.15 Therefore, to investigate whether constitutive GR activation in Cushing patients alters the phenotype of the erythroid progenitor cells, circulating CD34+ cells from Cushing patients with active disease and disease in remission, as well as from MC, were profiled with a panel of antibodies that identify stress-progenitor cells (CD110 and
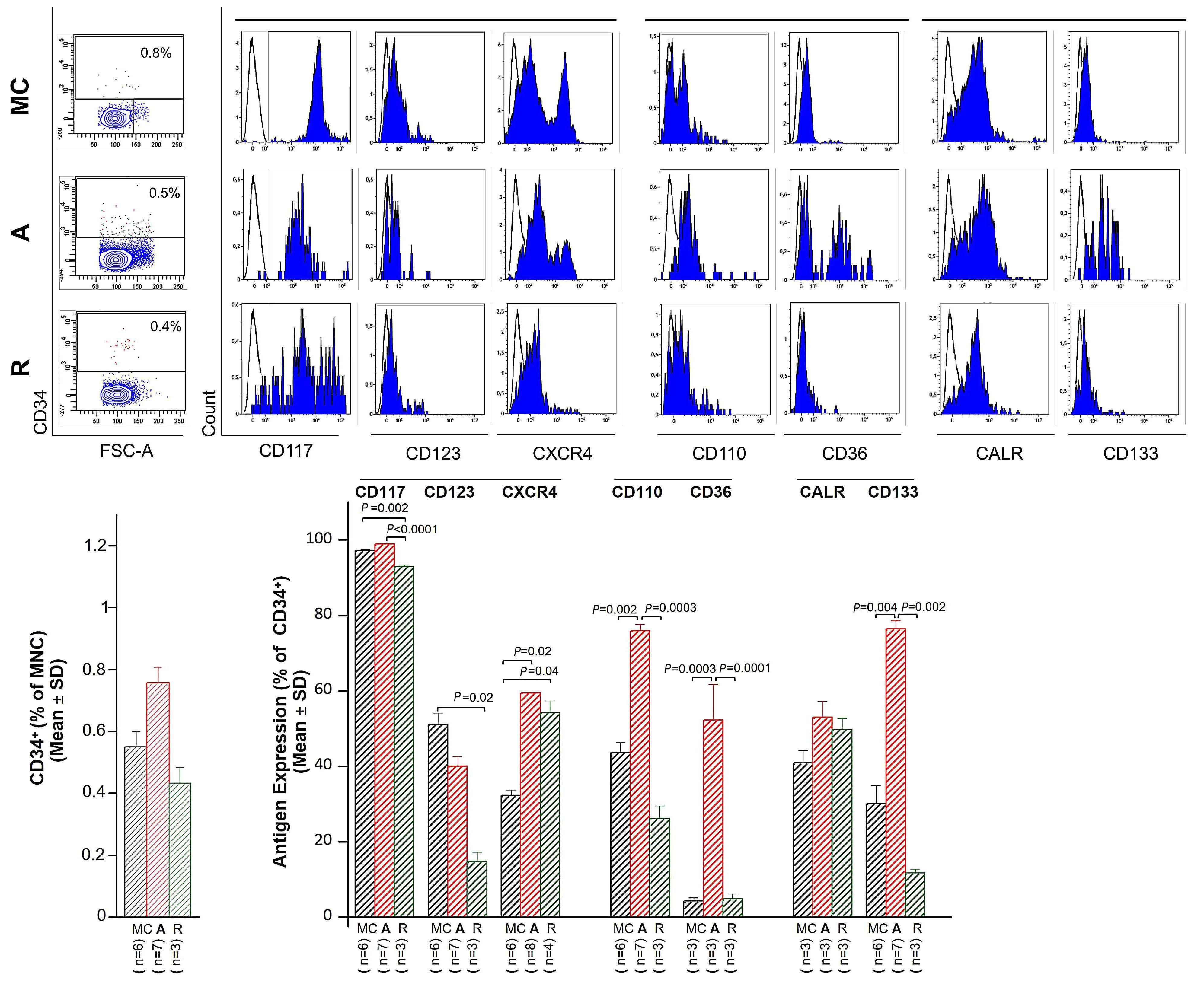
CD36)21,24 or proteins regulated by (CXCR4)23 or that regulate (CALR)15 the response to glucocorticoids. The panel also included CD117 and CD123, antibodies against receptors for SCF (cKIT25) and interleukin-3,26 as indicators of cell responsiveness to the growth factors used to stimulate the erythroid cultures (Figure 4).
The frequency of CD34+ cells in the blood of the three ex-
Figure 4. The blood from patients with active Cushing disease contains normal numbers of CD34+ cells that express a stresslike phenotype characterized by high cell surface levels of the glucocorticoid receptor target gene CXCR4, of CD110, CD36 and of the stem cell marker CD133. (A) Representative forward side scatter and CD34 staining of mononuclear cells from the blood of one representative Cushing disease patient with active disease and one in remission as well as the cells from a matched control (MC) are presented on the left. Cells in the CD34+ gate were then analyzed for expression of CD117, CD123, CXCR4, CD110, CD36, CALR and CD133, and the results presented as histograms in gray on the right. The numbers within the plots indicate the frequency of the cell populations within the gate. The blue histograms indicate the signal from irrelevant isotype-matched controls analyzed in parallel. (B) Frequency of CD34+ cells in the mononuclear blood cells from multiple active-phase and remission-phase patients and MC. (C) Frequency of CD34+ cells expressing CD117, CD123, CXCR4, CD110, CD36, CALR and CD133 from active-phase and remission-phase patients and MC. The number of different individuals included in the various groups is indicated by n. P values were calculated with the Tukey multiple comparisons test and those statistically significant are indicated in the panels. A: patients with active Cushing disease; R: patients with Cushing disease in remission; MC: matched controls (matched for age, weight and sex) without Cushing disease); FSC: forward scatter; MNC: mononuclear cells; SD: standard deviation.
A B C
Haematologica | 108 - April 2023 1060 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
perimental groups was similar (0.4-0.6% of the MNC) (Figure 4A, B). These frequencies among the three groups remained similar when the cells were functionally evaluated based on their ability to form colonies in semisolid cultures (30.2±27.2, 23.7±13.2 and 16.5±11.5 CFC/105 MNC in activephase patients, remission-phase patients and MC, respectively).
The frequency of CD34+ cells from active phase patients which bind CD117, CD123 and CALR was similar to that from MC (Figure 4A, C). Since response to SCF and interleukin-3 is associated with down-modulation of their receptors,27 the normal frequency of CD34+ cells expressing these receptors in active-phase patients suggests that they respond readily to SCF and interleukin-3 in vivo. In addition, the proportion of CD34+ cells that expressed CXCR4, CD36, CD110 and CD133 was greater in patients with active Cushing disease than in MC. A greater proportion of CD34+ cells from remission-phase patients expressed CXCR4 than did MC cells while a lower proportion of them expressed CD117/CD123, suggesting that CD34+ cells from Cushing patients in remission respond more and less readily than normal to CXCL12 and SCF/IL3, respectively. However, the proportions of CD34+ cells from remission-phase patients expressing all the other antigens were similar to those of the cells from MC (Figure 4A, C). A comparison of the antigen mean fluorescent intensity among groups indicates that, with the exception of CXCR4 which was expressed at levels lower than normal by remission-phase CD34+ cells, and of CD36 and CARL, which were respectively expressed at levels higher and lower than normal by active-phase CD34+ cells, the level of expression per cell of all the other antigens analyzed was comparable among groups (Online Supplementary Figure S3).
These results suggest that the circulating progenitor cells from patients with active Cushing disease constitute a unique population, likely generated from hematopoietic stem cells in response to the high cortisol levels found in these patients.
CD34+ cells from the blood of patients with active Cushing disease generate similarly high numbers of erythroid cells when cultured with or without glucocorticoids
MNC from patients with active Cushing disease generated similar numbers of erythroblasts in 10-day cultures with or without dexamethasone, a large proportion of which were immature under both conditions. In contrast, and as expected, cells from MC generated higher numbers of erythroblasts, a good proportion of which were immature, in culture with dexamethasone than without dexamethasone (Figure 5A, B). Surprisingly, MNC from patients in remission generated similarly low numbers of erythroblasts, with a similar maturation profile in cultures with or without dexamethasone (Figure 5C), suggesting that the apparently nor-
mal progenitor cells observed in these patients retain some memory of altered response to GR activation.
Erythroid cells generated in culture by CD34+ cells from patients with active Cushing disease display an activated glucocorticoid receptor signal when generated in culture stimulated or not with glucocorticoids
To provide mechanistic insights into the altered response to dexamethasone of erythroblasts from active-phase and remission-phase patients, biochemical studies of cells generated with and without dexamethasone from MC and Cushing patients with active disease or in remission were conducted (Figure 6). These studies compared the content of total GRα, the glucocorticoid binding isoform of the receptor, as well as of GRαS211, which is translocated to the nucleus, and of GRαS203, which is retained in the cytoplasm.28 Additionally, we analyzed levels of GILZ, a downstream target commonly used as a gold standard to measure the transcriptional activity of GR.29 As expected given the frequent polymorphism of GRα in the human population,29 great variability was observed in the GR content of erythroblasts obtained from different individuals (Figure 6A, B, Online Supplementary Figure S4). Erythroblasts generated with dexamethasone from active-phase and remission-phase patients as well as MC expressed equivalent levels of total GRα and GRαS211, but those from patients with active disease contained lower levels of GRαS203 than those from remission-phase patients or MC, although the low number (n=2) of active-phase patients investigated does not allow assessment of whether the difference is statistically significant (Figure 6B). The difference in GRαS203 content between the active and remission phases was conserved at the stoichiometry level (i.e., when normalized with respect to the corresponding GR content) (Figure 6C). Finally, erythroblasts from patients with active disease contained significantly greater amounts of GILZ than those from remission-phase patients or MC (Figure 6B) and erythroblasts from active-phase patients generated with or without dexamethasone expressed similar stoichiometric levels of GRαS203 and GRαS211 and similar levels of GILZ when compared to the levels of GRαS211 (Figure 6D).
These results suggest that the lack of response to exogenous glucocorticoids induced by chronic glucocorticoid exposure is mediated by mechanisms that alter the efficiency of the nuclear/cytoplasmic transport of GRα, rather than by those that interfere with the transcription/translation of its mRNA.
Discussion
By studying the erythroid compartments of a relatively large cohort of patients with Cushing disease in active and remission phases we confirm that chronic exposure to excess glucocorticoids in vivo leads to erythrocytosis and provide
Haematologica | 108 - April 2023 1061 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
Figure 5. Erythroid progenitor cells from patients with active Cushing disease generate similarly high numbers of erythroblasts in cultures stimulated or not with dexamethasone. (A) Total numbers of erythroblasts generated by day 10 by erythoid progenitor cells from matched controls (MC), patients with active Cushing disease and patients with Cushing disease in remission cultured with or without dexamethasone, as indicated. The total number of erythroblasts was calculated by multiplying the total number of cells in each culture by the corresponding percentage of cells with the CD235a/CD36 phenotype determined by fluorescence activated cell sorting. (B) Flow cytometry analyses for CD235a/CD36 expression of cells generated at day 10 by CD34+ cells from representative active- and remission-phase Cushing patients and MC. CD235a/CD36 staining divides erythroid cells in to CD235aneg/CD36pos (gate 1, proerythroblasts, purple), CD235alow/CD36pos (gate 2, basophilic erythroblasts, light green), and CD235amedium/CD36pos (gate 3, polychromatophilic erythroblasts, dark green) and CD235apos/CD36pos (gate 4, orthochromatic erythroblasts, light blue), as reported.17 Non-erythroid cells are CD235negCD36neg (dark blue) and are mainly lymphocytes. Results are presented as counterplots which allow for a better distinction of the clusters of the individual cell populations. (C) Frequency of cells in gates 1, 2, 3 and 4 generated by day 10 by CD34+ cells from MC and active-phase and remission-phase Cushing patients cultured with or without dexamethasone (the same color code as in panel B). Note that erythroblasts from MC are already mostly mature at day 10 in cultures without dexamethasone. By contrast, at day 10 similar numbers of immature erythroblasts are observed in cultures with or without dexamethasone from activephase and remission-phase patients. The number of different individuals included in the various groups is indicated by n. Statistical analyses between cultures with or without dexamethasone in the same group were performed by a paired t test and statistically significant values are indicated in the panel. Statistical analyses among groups was performed by the Tukey multiple comparisons test as appropriate. Results of the statistical analyses are as follows. (A) Paired t test: MC without dexamethasone vs. MC with dexamethasone, P=0.001; active- or remission-phase Cushing patient with or without dexamethasone, not statistically different. Tukey multiple comparisons test: MC with dexamethasone vs. remission-phase patient with dexamethasone, P=0.0003 and vs. remissionphase patient without dexamethasone, P=0.0002; MC without dexamethasone vs. remission-phase patient with dexamethasone, P=0.03 and vs. remission-phase patient without dexamethasone, P=0.02; active-phase patient with dexamethasone vs. remissionphase patient with dexamethasone, P=0.03 and vs. remission-phase patient without dexamethasone, P=0.02; active phase patient without dexamethasone vs. remission-phase patient without dexamethasone, P=0.04. All the other comparisons were not statistically different. (C) Paired t-test: gates 1 and 3 for MC with or without dexamethasone, P=0.02 for both; gates for active-phase and remissionphase patients with and without dexamethasone are not statistically different. Tukey multiple comparisons test: no statistically significant differences among gates for MC vs. active-phase patients or vs. remission-phase patients with or without dexamethasone. Erys: erythroblasts; SEM: standard error of mean; Dex: dexamethasone; A: patients with active Cushing disease; R: patients with Cushing disease in remission; MC: matched controls (matched for age, weight and sex) without Cushing disease; FITC; fluorescein isothiocyanate; APC: allophycocyanin.
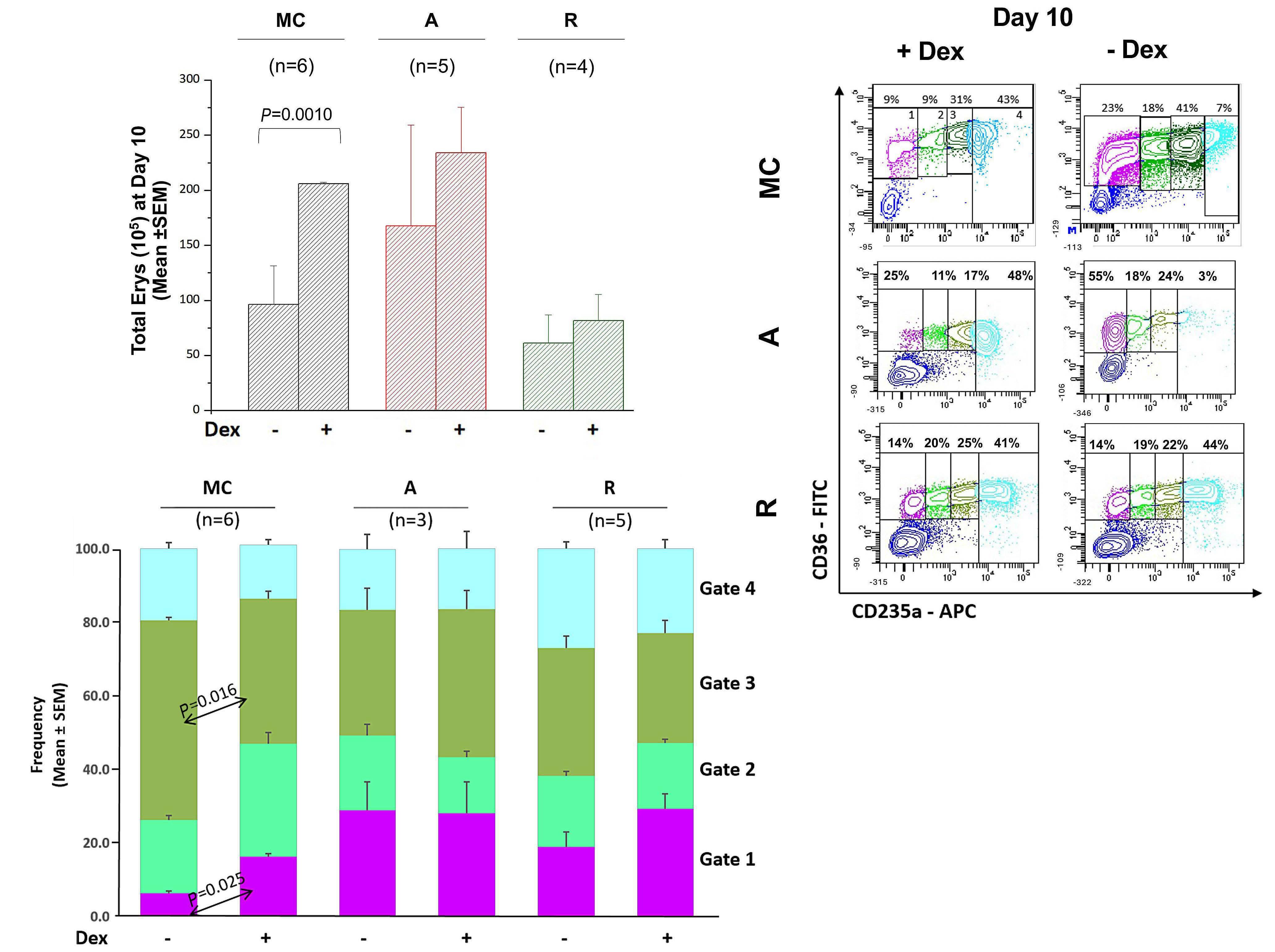
A B C
Haematologica | 108 - April 2023 1062 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
new information on the cells targeted by this receptor in vivo and on the biochemical/molecular consequences of its activation.
Cellular targets
There is considerable confusion in the literature on the
identity of the erythroid cells targeted by dexamethasone. Time-response and ex-vivo amplification studies indicate that the primary human cells that respond to dexamethasone are the burst-forming unit-erythroid (BFU-E) while colony-forming unit-erythroid (CFU-E) cells are insensitive to GR activation.30 Using cells prospectively isolated from
Continued on following page.
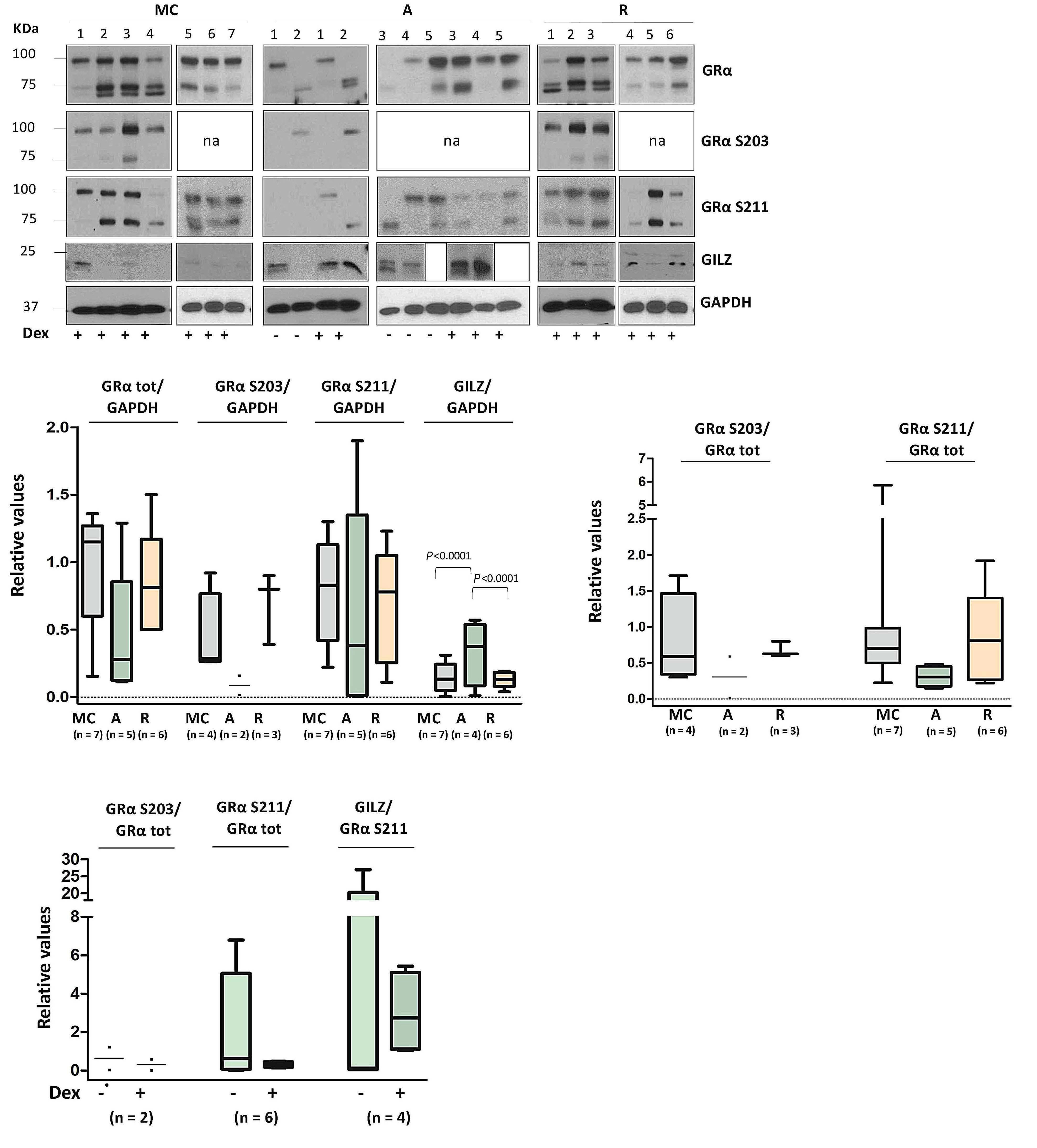
A B C D
Haematologica | 108 - April 2023 1063 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
Figure 6. Erythroblasts from patients with active Cushing disease contain normal levels of total glucocorticoid receptor (GR), which is poorly phosphorylated in the inactive cytoplasmic-retained S203 form, and express high levels of the S211 form and of the GR-target gene GILZ in cultures with and without dexamethasone. (A) Western blot analyses for total GRα and its nuclear (GRαS211) and cytoplasmic (GRαS203) forms, as well of the GR target gene GILZ of erythroblasts generated at day 10 by representative patients with active Cushing disease or disease in remission and matched controls (MC) (each number a different donor) in culture with and without (active-phase patients only) dexamethasone. The position of the molecular weight markers is indicated on the left. (B) Content of total GRα, GRαS203, GRαS211 and GRαS203 with respect to that of GAPDH in erythroblasts generated from cells from active- and remission-phase patients and from MC cultured with dexamethasone. (C) Stoichiometry levels of GRαS203 and GRαS211 in cells from active- and remission-phase patients and from MC cultured with dexamethasone. (D) Stoichiometry levels of GRαS203 and GRαS211 and levels of GILZ with respect to those of GRαS211 in cultures of cells from active-phase patients cultured with or without dexamethasone. In (B-D), results are expressed as box charts with minimum and maximum values observed in multiple individuals in each group. P values among groups were calculated by the Tukey multiple comparisons test in (B) and (C) and by a paired t test in (D). The number of individuals included in each group is indicated by n. A: patients with active Cushing disease; R: patients with Cushing disease in remission; MC: matched controls (matched for age, weight and sex), without Cushing disease; na: not available; Dex: dexamethasone.
mouse fetal liver, the team headed by Lodish confirmed these results by identifying steady-state BFU-E as the target of GR activation.31 By contrast, the sequence of differentiated cells that are progressively generated by human steady-state CD34+ cells ex-vivo in response to dexamethasone are all capable of responding to dexamethasone.21,24 Thus, whereas erythroblasts generated in vivo lose the ability to respond to dexamethasone (and SCF)14 between the BFU-E and the CFU-E stage, those generated in vitro with dexamethasone remain dexamethasone- (and SCF-)27 responsive up to the pro-erythroblast stage. The fact that the GR-sensitive BFUE and CFU-E express, respectively, CD110 and CD105, an antibody that recognizes endoglin,32,33 has suggested that in cultures stimulated with dexamethasone, multipotent CD34+ cells activate an alternative differentiation pathway that generates stress-specific erythroid progenitors. Whether these stress-specific cell populations are also generated in response to chronic erythroid stress in vivo in humans has not yet been investigated.
By profiling the CD34+ cells from the blood of a large cohort of patients with active (hypercortisolemic) Cushing disease, we identified for the first time a population of primary CD34+ cells which express CD36 and CD110, a phenotype that is similar to that of the stress progenitor cells observed in cultures of human CD34+ cells stimulated with dexamethasone.21,24 How representative these stress-specific cells are of the hematopoietic progenitors in the bone marrow of Cushing patients remains to be established. In addition to the CD36 and CD110 phenotype, another distinct feature in patients with Cushing disease was the increased frequency of CD34+CD133+ cells. Expression of CD133 has been previously noted among CD34+ cells in bone marrow34 and it was later documented that its presence was marking very long-term repopulating cells in transplantation experiments35 and early hematopoietic progenitors which, if they co-expressed VEGFR-2,36 represented endothelial progenitors. Whether the above quantitative changes in distinct phenotypes seen in Cushing patients reflect changes seen in vivo in other stress-like conditions is at present unclear and further studies are needed. Of further relevance, pre-
vious studies suggested that in vitro cultures of CD34+ cells from either peripheral blood or bone marrow from normal subjects gave rise to increased fetal globin levels. Examination of in vivo responses during acute erythropoietic demands also documented transient increases in fetal globin.37 However, such increases were not maintained under chronic stress conditions.38 Therefore, being under chronic steroid stress, it may not be surprising that Cushing patients did not show increases in fetal globin. Furthermore, it should be added that there is no evidence that addition of steroids in vitro specifically contributes to increases in fetal globin. In fact, the opposite is true since addition of steroids to cells at a developmental transition level accelerated the fetal to adult switch.39
Molecular mechanisms of glucocorticoid receptor resistance in Cushing disease and implications for chronic glucocorticoid therapy of hematopoietic disorders
The important role played by GR in the stress response inspired the development of GR agonists to treat several stress-related disorders (inflammation, autoimmune diseases, Diamond Blackfan anemia, etc.). A general feature of these therapies is that eventually patients become unresponsive to treatment. Given the clinical relevance of this unresponsiveness, the mechanisms leading patients to lose responsiveness to glucocorticoids have been the subject of extensive investigation. Human GR (GR/NR3C1) is highly polymorphic, displaying numerous single nucleotide polymorphisms in the coding region and in regions associated with alternative splicing and mRNA stabilization, resulting in more than 260 combinations of alternative GR isoforms with a wide range of affinities for glucocorticoids expressed in humans.29 Microenvironmental cues, such as its ligand and/or soluble SCF, may contribute negatively and/or positively to the variability of the response to glucocorticoids in the human population.3,40 In addition, prolonged GR activation may induce epigenetic modifications of the gene promoter, suppressing its expression,41 and/or post-transcriptional protein modifications, affecting its nuclear trafficking. In this regard, it has been demonstrated that
Haematologica | 108 - April 2023 1064 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
glucocorticoid stimulation may induce phosphorylation of GR not only at S211, resulting in nuclear translocation and activation of transcriptional activity,42 but also at S203 which instead retains the receptor in the cytoplasm, inhibiting its transcriptional activity.43 The data presented here indicate that CD34+ cells from all patients with Cushing disease (including those with active disease and those in remission) are resistant or insensitive to exogenous glucocorticoid stimulation. In the case of active Cushing disease, the observation that cells generated with or without dexamethasone in vitro express similarly high levels of GRαS211 and GILZ suggests constitutive activation of GR signaling. By contrast, the CD34+ cells from patients in remission, which are also not responsive to dexamethasone, do generate low numbers of mature erythroblasts in cultures with and without dexamethasone and these express low levels of GILZ, indicating that they are unable to activate GR signaling. Of interest, the large number of patients (and MC) included in the analyses allowed us to exclude, despite the great variability of GR content among cells obtained from different individuals, the hypothesis that variegation in response is due to differences in protein content, making it unlikely that lack of response in Cushing disease is mediated by the polymorphism of the gene and/or its epigenetic regulation. Instead, we observed an association with the levels of phosphorylation of GRS203. In fact, while erythroblasts from both active- and remission-phase patients contained GR phosphorylated at S211, which is required for nuclear translocation, the levels of GR phosphorylated at S203 in patients upon remission was much greater than those observed in two patients in the active phase. These data indicate that nuclear/cytoplasmic trafficking plays a significant role in restraining the cellular response to glucocorticoids also in patients with Cushing disease. They suggest, in fact, that activated GR is constitutively retained in the nucleus of cells from active patients, making them insensitive to de-novo stimulation. By contrast, patients in remission are insensitive to de-novo stimulation because, due to high levels of S203 phosphorylation, their GR is retained in the cytoplasm, as a memory of the mechanism which attempted to limit their response in the hypercortisolemic state. This hypothesis is consistent with the clinical observation that long-term comorbidities are seen in patients with Cushing disease despite remission, including persistent obesity, increased cardiovascular risk, evidence of abnormal systemic inflammation44–46 (and high numbers of CD163+ monocytes in blood) (Figure 2), as a memory of their previously hypercortisolemic features. This
hypothesis is testable and is consistent with studies that are investigating whether chemical inhibition of S203 may restore response to glucocorticoids in patients with other disorders who became resistant to dexamethasone.47,48 In addition, this hypothesis may be relevant to understand glucocorticoid resistance in patients with Diamond Blackfan anemia, 60% of whom became transfusion dependent upon treatment.49 The mechanism that induces loss of glucocorticoid response in Diamond Blackfan anemia is a subject of intensive investigation. Recently, the Blanc laboratory has proposed that patients with Diamond Blackfan anemia become glucocorticoid-unresponsive because their stressCFU-E fail to self-replicate in response to glucocorticoids since, through a mechanism still to be identified, they are unable to activate p57kip2 24 Based on our results in Cushing patients, we speculate that the stress-CFU-E from unresponsive Diamond Blackfan anemia patients fail to activate p57kip2 because they express high levels of GRαS203 which inhibits the transcriptional activity of the receptor. Thanks to the availability of inhibitors of GRS203, this hypothesis is testable and will be analyzed in separate studies.
In conclusion, blood from patients with active Cushing disease contains a unique CD34+ cell population in which GR is constitutively active and does not respond to exogenous dexamethasone. Surprisingly, CD34+ cells in the blood from patients in remission are phenotypically normal but retain an abnormal response to exogenous dexamethasone.
Disclosures
No conflicts of interest to disclose.
Contributions
LV and FM performed experiments and analyzed the data. MM performed statistical analyses. EG provided patients’ information, samples and clinical insights on Cushing disease. AF and JB analyzed the data and wrote the manuscript. TP and ARM designed the study, interpreted the data and wrote the manuscript. All the authors read the manuscript and concur with its content.
Funding
This study was supported by grants from the National Cancer Institute (P01-CA108671, to ARM), National Heart, Lung and Blood Institute (1R01-HL134684, to ARM and JB), and the Associazione Italiana Ricerca Cancro (AIRC IG23525, to ARM).
Data-sharing statement
Primary data will be disclosed upon request.
1999;13(22):2996-3002.
1. Bauer A, Tronche F, Wessely O, et al. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev.
References Haematologica | 108 - April 2023 1065 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
2. Tajima Y, Moore MAS, Soares V, Ono M, Kissel H, Besmer P.
Consequences of exclusive expression in vivo of kit-ligand lacking the major proteolytic cleavage site. Proc Natl Acad Sci U S A. 1998;95(20):11903-11908.
3. Varricchio L, Tirelli V, Masselli E, et al. The expression of the glucocorticoid receptor in human erythroblasts is uniquely regulated by KIT ligand: implications for stress erythropoiesis. Stem Cells Dev. 2012;21(15):2852-2865.
4. Ogawa M, Grush OC, O’Dell RF, Hara H, MacEachern MD. Circulating erythropoietic precursors assessed in culture: characterization in normal men and patients with hemoglobinopathies. Blood. 1977;50(6):1081-1092.
5. Migliaccio G, Di Pietro R, di Giacomo V, et al. In vitro mass production of human erythroid cells from the blood of normal donors and of thalassemic patients. Blood Cells Mol Dis. 2002;28(2):169-180.
6. Migliaccio AR, Varricchio L. Concise review: advanced cell culture models for Diamond Blackfan anemia and other erythroid disorders. Stem Cells. 2018;36(2):172-179.
7. Gursoy A, Dogruk Unal A, Ayturk S, et al. Polycythemia as the first manifestation of Cushing’s disease. J Endocrinol Invest. 2006;29(8):742-744.
8. Lindholm J, Juul S, Jørgensen JOL, et al. Incidence and late prognosis of Cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86(1):117-123.
9. Nieman LK, Biller BMK, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2008;93(5):1526-1540.
10. Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(8):2807-2831.
11. Zhong L, Yao L, Tower RJ, et al. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. Elife. 2020;9:e54695.
12. Moeller SJ, Couto L, Cohen V, et al. Glucocorticoid regulation of food-choice behavior in humans: evidence from Cushing’s syndrome. Front Neurosci. 2016;10:21.
13. Funnell APW, Prontera P, Ottaviani V, et al. 2p15-p16.1 microdeletions encompassing and proximal to BCL11A are associated with elevated HbF in addition to neurologic impairment. Blood. 2015;126(1):89-93.
14. Migliaccio G, Migliaccio AR, Druzin ML, Giardina P-JV, Zsebo KM, Adamson JW. Effects of recombinant human stem cell factor (SCF) on the growth of human progenitor cells in vitro. J Cell Physiol. 1991;148(3):503-509.
15. Falchi M, Varricchio L, Martelli F, et al. Dexamethasone targeted directly to macrophages induces macrophage niches that promote erythroid expansion. Haematologica. 2015;100(2):178-187.
16. Petersenn S, Newell-Price J, Findling JW, et al. High variability in baseline urinary free cortisol values in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2014;80(2):261-269.
17. Sulahian TH, Högger P, Wahner AE, et al. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. 2000;12(9):1312-1321.
18. Tippett E, Cheng W-J, Westhorpe C, et al. Differential expression of CD163 on monocyte subsets in healthy and HIV-1 infected individuals. PLoS One. 2011;6(5):e19968.
19. Affandi AJ, Olesek K, Grabowska J, et al. CD169 defines activated CD14+ monocytes with enhanced CD8+ T cell activation capacity. Front Immunol. 2021;12:697840.
20. Alexander WS. Thrombopoietin and the c-Mpl receptor: insights from gene targeting. Int J Biochem Cell Biol. 1999;31(10):1027-1035.
21. Heideveld E, Masiello F, Marra M, et al. CD14+ cells from
peripheral blood positively regulate hematopoietic stem and progenitor cell survival resulting in increased erythroid yield. Haematologica. 2015;100(11):1396-1406.
22. Broxmeyer HE. Chemokines in hematopoiesis. Curr Opin Hematol. 2008;15(1):49-58.
23. Kolbus A, Blazquez-Domingo M, Carotta S, et al. Cooperative signaling between cytokine receptors and the glucocorticoid receptor in the expansion of erythroid progenitors: molecular analysis by expression profiling. Blood. 2003;102(9):3136-3146.
24. Ashley RJ, Yan H, Wang N, et al. Steroid resistance in Diamond Blackfan anemia associates with p57Kip2 dysregulation in erythroid progenitors. J Clin Invest. 2020;130(4):2097-2110.
25. Broxmeyer HE, Maze R, Miyazawa K, et al. The kit receptor and its ligand, steel factor, as regulators of hemopoiesis. Cancer Cells. 1991;3(12):480-487.
26. Shi M, Su RJ, Parmar K-P, et al. CD123: a novel biomarker for diagnosis and treatment of leukemia. Cardiovasc Hematol Disord Drug Targets. 2019;19(3):195-204.
27. Federici G, Varricchio L, Martelli F, et al. Phosphoproteomic landscaping identifies non-canonical cKIT signaling in polycythemia vera erythroid progenitors. Front Oncol. 2019;9:1245.
28. Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277(29):26573-26580.
29. Zhou J, Cidlowski J. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70(5-7):407-417.
30. Narla A, Dutt S, McAuley JR, et al. Dexamethasone and lenalidomide have distinct functional effects on erythropoiesis. Blood. 2011;118(8):2296-2304.
31. Zhang L, Prak L, Rayon-Estrada V, et al. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature. 2013;499(7456):92-96.
32. Xiang J, Wu DC, Chen Y, Paulson RF. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood. 2015;125(11):1803-1812.
33. Mori Y, Chen JY, Pluvinage J V., Seita J, Weissman IL. Prospective isolation of human erythroid lineage-committed progenitors. Proc Natl Acad Sci U S A. 2015;112(31):9638-9643.
34. Yin AH, Miraglia S, Zanjani ED, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90(12):5002-5012.
35. Pasino M, Lanza T, Marotta F, et al. Flow cytometric and functional characterization of AC133 + cells from human umbilical cord blood. Br J Haematol. 2000;108(4):793-800.
36. Peichev M, Naiyer AJ, Pereira D, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95(3):952-958.
37. Papayannopoulou T, Vichinsky E, Stamatoyannopoulos G. Fetal Hb production during acute erythroid expansion. I. Observations in patients with transient erythroblastopenia and postphlebotomy. Br J Haematol. 1980;44(4):535-546.
38. Blau CA, Constantoulakis P, Al-Khatti A, et al. Fetal hemoglobin in acute and chronic states of erythroid expansion. Blood. 1993;81(1):227-233.
39. Zitnik G, Peterson K, Stamatoyannopoulos G, Papayannopoulou T. Effects of butyrate and glucocorticoids on gamma- to betaglobin gene switching in somatic cell hybrids. Mol Cell Biol. 1995;15(2):790-795.
40. Pujols L, Mullol J, Pérez M, et al. Expression of the human glucocorticoid receptor α and b isoforms in human respiratory
Haematologica | 108 - April 2023 1066 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
epithelial cells and their regulation by dexamethasone. Am J Respir Cell Mol Biol. 2001;24(1):49-57.
41. McGowan PO, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12(3):342-348.
42. Miller AL, Webb MS, Copik AJ, et al. p38 mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoidinduced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at Serine 211. Mol Endocrinol. 2005;19(6):1569-1583.
43. Matthews L, Johnson J, Berry A, et al. Cell cycle phase regulates glucocorticoid receptor function. PLoS One. 2011;6(7):e22289.
44. Webb SM, Valassi E. Morbidity of Cushing’s syndrome and impact of treatment. Endocrinol Metab Clin North Am. 2018;47(2):299-311.
45. Barahona M-J, Sucunza N, Resmini E, et al. Persistent body fat
mass and inflammatory marker increases after long-term cure of Cushing’s syndrome. J Clin Endocrinol Metab. 2009;94(9):3365-3371.
46. Shah N, Ruiz HH, Zafar U, Post KD, Buettner C, Geer EB. Proinflammatory cytokines remain elevated despite long-term remission in Cushing’s disease: a prospective study. Clin Endocrinol (Oxf). 2017;86(1):68-74.
47. Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22(8):1754-1766.
48. Anacker C, Cattaneo A, Musaelyan K, et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2013;110(21):8708-8713.
49. Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859-876.
Haematologica | 108 - April 2023 1067 ARTICLE - Erythrocytosis in Cushing disease L. Varricchio et al.
PDL1 shapes the classical Hodgkin lymphoma microenvironment without inducing T-cell exhaustion
Joseph G. Taylor,1,2 Edward Truelove,1,2 Andrew Clear,1 Maria Calaminici1,2 and John G. Gribben1,2

Abstract
Correspondence: J.G. Gribben j.gribben@qmul.ac.uk
Classical Hodgkin lymphoma (CHL) is unusually sensitive to PD1 inhibition and PDL1 is highly expressed on CHL cells and in the tumor microenvironment. This could be interpreted as evidence of exhaustion, but paradoxically, PD1+ lymphocyte infiltration does not predict response to PD1 inhibitors and no increase in cytotoxic markers is seen after PD1 therapy as might be expected with reversal of exhaustion. In contrast to PD1, elevated PDL1 does predict response to PD1 inhibitors and recent data associate both retained CHL MHC-II expression and increased T helper (TH) T-cell receptor diversity with response, suggesting a connection to the TH compartment. We performed a phenotypic, spatial and functional assessment of T-cell exhaustion in CHL and found co-expression of an exhaustion marker and lower PD1 expression in CHL than in reactive nodes whereas the proliferative and cytokine production capacity were similar in CHL and the reactive nodes. We found no correlation between PDL1 expression and exhaustion signatures. Instead, we identified a strong association between PDL1 expression and CHL MHC-II expression, TH recruitment, and enrichment of TH1 regulatory cells. These data suggest that a dominant effect of PDL1 expression in CHL may be TH engagement and promotion of a regulatory microenvironment rather than maintenance of exhaustion.
Introduction
Classical Hodgkin lymphoma (CHL) is the only licensed indication for CD279 (PD1, programmed cell death protein 1)inhibitors in hematologic malignancies and is unusually sensitive to this therapy.1,2 PD1 inhibitors are classically thought to act by reversing T-cell exhaustion, a state which limits the effectiveness of anti-tumor immune responses.3 Effector T cells become exhausted when they are chronically stimulated by low levels of antigen.4 Exhausted cells have sustained high expression of PD1, alongside other exhaustion markers, including CD223 (LAG3), TIM3, TBC21 (TBET) and EOMES, and progressively lose their effector functions.4,5 Exhaustion is partially reversible and PD1 inhibition can reinvigorate the T-cell response leading to improved tumor clearance.
CHL cells express high levels of ligand 1 for PD1 (PDL1) and polysomy, copy gains and amplifications of the PDL1 locus are seen in upwards of 95% of cases.6,7 PDL1 expression is also prominent within other cells in the tumor microenvironment. 8 However, functional data demonstrating exhaustion in CHL are limited and most
studies, including studies from our own laboratory, report only low PD1 expression in the CHL microenvironment. 9 Furthermore, PD1 + cell infiltration does not predict response to PD1 inhibitors in CHL. 9,10 Recent studies demonstrate that during PD1 inhibitor therapy the CHL microenvironment is characterized by a rapid reduction in PDL1 + macrophages and type 1 regulatory cells (T R 1) rather than cytotoxic T-cell expansion that might be expected with reversal of T-cell exhaustion.11 Another study found that expansion of singleton (putatively newly immigrant) T helper (T H) clones is associated with PD1 inhibitor response.12 This is in line with data from solid tumors and suggests that during PD1 therapy activated tumor-specific T cells are in fact newly immigrant and not derived from exhausted populations that were present before therapy.13 These studies run counter to the traditional model of PD1 inhibitors acting by reversing exhaustion and highlight a need to better understand the function of PDL1 within the CHL microenvironment.
In this study we phenotypically and functionally assessed exhaustion in CHL and its relationship to PDL1 ex-
1Centre for Haemato-Oncology, Barts Cancer Institute, Queen Mary University of London, and 2Barts NHS Trust, St Bartholomew’s Hospital, London, UK
Haematologica | 108 - April 2023 1068 ARTICLE - Hodgkin Lymphoma
September 25, 2021. Accepted: June 9, 2022. Early view: July 14, 2022. https://doi.org/10.3324/haematol.2022.280014 Published under a CC BY license
Received:
pression. Given the evidence of a close connection between PD1 inhibition and TH cells, we further assessed the relationship between PDL1 and TH effector subsets in CHL. We found low levels of exhaustion and no evidence of a connection between an exhausted phenotype and PDL1 expression. However, we identified a strong connection between PDL1 expression and enrichment for the T H 1 Reg regulatory phenotype, a recently characterized subset of T regulatory cells expressing both FOXP3 and TBET which may specialize in suppression of T H 1 cells that are numerous in the CHL microenvironment. These data support and expand upon recent findings that PDL1 expression in CHL may have a closer relationship to the maintenance of a protective tumor microenvironment than to the mediation of exhaustion.
Methods
Human tissue was accessed as part of study 05/Q0605/140, approved by the Regional Ethics Committee, with consent through Barts Tissue Bank and National Health Service Blood and Transplant. Cell lines were obtained from DSMZ. Samples were identified by a system-
atic search of adult cases at St Bartholomew’s Hospital between 1999 and 2016. The diagnosis was confirmed in all cases by an expert hematopathologist (MC) and Epstein-Barr virus (EBV) status was determined by EBV-encoded small RNA in situ hybridization (sample characteristics in Table 1). Reactive lymph nodes (RLN) from adult patients were selected as controls. Cases positive for human immunodeficiency virus or associated with malignancy were excluded. RLN histology was follicular hyperplasia (64%), progressive transformation of germinal centers (8%) or not otherwise specified (28%). RLN provide a comparative non-malignant immune response within lymphoid tissue and have been used in previous studies. 9 Non-reactive nodal tissue is scarce, usually collected during staging of known malignancy and generally small with bland infiltrate and so was considered a less robust control.
Immunohistochemistry
Immunohistochemistry (IHC) and multiplex IHC studies were performed on tissue microarrays. Forty-seven CHL cases were randomly selected for the multiplex experiments and grouped on a single array. Further details are provided in the Online Supplementary Methods
Characteristics of subjects
CHL: classical Hodgkin lymphoma; IHC: immunohistochemistry; mIHC: multiplex immunohistochemistry; IMC: imaging mass cytometry; ABVD: adriamycin, bleomycin; vincristine; doxorubicin; Unk: unknown; EBER-ISH: Epstein-Barr virus-encoded small RNA in situ hybridization; RLN: reactive lymph nodes.
Characteristics of the CHL patients (N=150) IHC cohort (N=150) mIHC cohort (N=47) IMC cohort (N=24) Functional cohort Male, % (N) 53 (80) 51 (24) 50 (12) 73 (11) Age >45 years, % (N) 24 (36) 27 (13) 25 (6) 0 (0) Advanced (IIB-IV), % (N) 69 (82), 32 Unk 74 (35) 71 (17) 42 (6), 1 Unk ABVD-treated, % (N) 92 (138) 91 (43) 100 (24) 100 (15) Radiotherapy only, % (N) 1 (2) 0 (0) 0 (0) 0 (0) Histological subtype, % (N) Nodular sclerosing 77 (116) 78 (37) 80 (20) 66 (10) Mixed cellularity 20 (30) 19 (9) 20 (5) 33 (5) Lymphocyte-rich 2 (3) 2 (1) 0 (0) 0 (0) Lymphocyte-depleted 0 (0) 0 (0) 0 (0) 0 (0) Unclassifiable 0.6 (1) 0 (0) 0 (0) 0 (0) EBER-ISH+, % (N) 31 (47) 34 (16) 25 (6) 46 (7) MMC-II, % (N) Positive 31 (47) 21 (10) 42 (10) 33 (5) Weak 15 (23) 10 (5) 12.5 (3) 27 (4) Negative 37 (55) 38 (18) 29 (7) 40 (6) Unclassifiable 17 (25) 30 (14) 12.5 (3) 0 (0)
with RLN (N=28) Male, % (N) 50 (14) 50 (14) Unk (0) Age >45 years, % (N) 25 (7) 25 (7) 50 (14)
Haematologica | 108 - April 2023 1069 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
Table 1. Characteristics of the cohort of patients.
Imaging mass cytometry
Imaging mass cytometry was performed on tissue microarrays on a subset of 24 cases from the multiplex IHC array. Further details are provided in the Online Supplementary Methods.
Cell culture and flow cytometry
For assessment of macrophage PDL1, KMH2-conditioned medium was harvested from high-viability, low-passage cells after 24 hours at 1x106/mL in RPMI medium. Monocytes from healthy donor peripheral blood mononuclear cells were positively selected using anti-CD14 microbeads according to the manufacturer’s instructions before being seeded in 2 mL RPMI medium at 5x105/mL in six-well plates with macrophage colony-stimulating factor 50 ng/mL and granulocyte-macrophage colony-stimulating factor 1 ng/mL. On day 7, macrophages were treated with RPMI or 50% RPMI/conditioned medium. Cells were harvested at 24 hours for flow cytometry analysis. For exhaustion assays, single cell suspensions were stimulated in 200 mL RPMI medium at 1x106/mL in 96-well plates. For cytokine and Ki67 assays, cells were stimulated with phorbol myristate acetate/ionomycin with protein transport inhibitors for 4 hours at 37°C. For proliferation assays the cells were stimulated with 10 mg/mL anti-CD3 and 1 mg/mL anti-CD28 and incubated for 4 days at 37°C after staining with CFSE. For co-cultures, healthy donor naïve CD4+ T cells were separated from peripheral blood mononuclear cells with the Stemcell EasySep kit according to the manufacturer’s instructions before co-culture at 1x106/mL with 4x104/mL KMH2 for 8 days before analysis by flow cytometry. Data were acquired using a BD LSRFortessa with compensation and fluorescence minus one controls at every run. The data were then analyzed using FlowJo software. Further details are available in the Online Supplementary Methods
Analyses
Images were analyzed in Visiopharm and R. Package details are provided in the Online Supplementary Methods. Spatial distributions were compared to the parent cell distribution (e.g., CD8+ to CD3+ cells) to avoid confounding data due to distribution of parent cell-type or complete spatial randomness where no biologically appropriate parent cell comparator was available. Tukey style boxplot marking was used to show medians and interquartile ranges.
Results
Classical Hodgkin lymphoma cells are PDL1hi and induce a PDL1+ myeloid microenvironment
We quantified PDL1 expression by IHC in 150 patients with
CHL and in 28 age-matched patients with RLN. The characteristics of the cohorts are summarized in Table 1. PDL1 expression on CHL cells (CD30+) and macrophages (CD68+) was further assessed by multiplex IHC in 47 CHL cases on a single tissue microarray. PDL1 expression was elevated in CHL (with expression seen on both CHL cells and within the microenvironment) (P<0.0001) (Figure 1A). This effect was seen irrespective of EBV status (data not shown). Mean PDL1 intensity was higher in CHL cells than in macrophages (P<0.0001) (Figure 1A). CD68+ macrophage frequency was increased in regions of high CHL density (P<0.0001). PDL1 intensity increased with macrophage proximity and density to CHL cells (P<0.0001) (Figure 1B). Consistent with this, media conditioned by the KMH2 CHL cell line induced upregulation of PDL1 on monocyte-derived macrophages in vitro (P<0.01) (Figure 1C). PDL1 upregulation in myeloid cells co-cultured with CHL cell lines is consistent with published data.14 Media conditioned with the non-Hodgkin lymphoma cell lines DHL4 and DHL6 also induced PDL1 expression on monocyte-derived macrophages, but at significantly lower levels than the CHL cell line (data not shown). Phenotyping of microenvironmental antigen-presenting cells (CD68+ macrophages, CD20+ B cells and CD14+ monocytes) by imaging mass cytometry and comparison to RLN demonstrated elevated CD206 intensity but lower B7H4 intensity (both M2 markers) in CHL than CD68+ in RLN. PDL1 expression was highest in CHL cells and CD68+ macrophages. CD14+ monocytes had higher CD16 and CD163 expression but lower mean CD14 expression than their RLN counterparts whereas CD20+ B cells were more likely to express HLADR, CX3CR1 and CD45RA (Figure 1D). Together, these data confirm, as previously reported, that CHL cells both express high levels of PDL1 and exist in a macrophage-enriched, PDL1+ microenvironment.8,15 Further to this, our data demonstrate that microenvironmental PDL1 expression is mainly seen within the CD68+ macrophage compartment, and that there is a correlation between macrophage number and the intensity of macrophage PDL1 expression (R=0.6, P<0.0001) (Figure 1E), suggesting that tumors promoting macrophage recruitment were also those promoting microenvironmental PDL1 expression.
PD1+ and phenotypically exhausted cells are infrequent in classical Hodgkin lymphoma, despite high PDL1 expression
After confirming that CHL cells and macrophages in the tumor microenvironment express high levels of PDL1 we sought evidence of a corresponding exhausted PD1+ T-cell population. Exhausted T cells are characterized by sustained PD1 expression and models suggest that cells with intermediate PD1 expression (PD1int) are most amenable to reversal by PD1 inhibition.3,16 We therefore quantified PD1 expression by IHC and stratified the results by PD1 ex-
Haematologica | 108 - April 2023 1070 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
pression intensity. Lymph node germinal centers (and rare residual germinal centers in 5/150 CHL cases) were excluded from analysis because of the high PD1 expression on T follicular helper cells (TFH). The analysis was performed using the most sensitive of three anti-PD1 monoclonal antibody clones tested and results were confirmed with a second clone (Online Supplementary Figure S4). PD1 expression was heterogeneous among cases, with patterns ranging from rare or no PD1 positivity to diffuse infiltration of PD1int, rosetting PD1hi cells and rare cases of
marked PD1hi infiltration (Figure 2A). Overall, despite prominent CD3, CD4 and CD8 infiltration, PD1 expression was significantly less frequent in CHL than in RLN (Figure 2B). PD1+ cells were seen at lower frequency in CHL than in RLN irrespective of PD1 expression intensity (Figure 2C). No difference in PD1 expression was seen according to EBV status, but enrichment was seen in the lymphocyte-rich histological subtype, consistent with previous reports.17 Isolated PD1 expression is not specific for exhaustion.18 We therefore assessed co-expression with other markers of
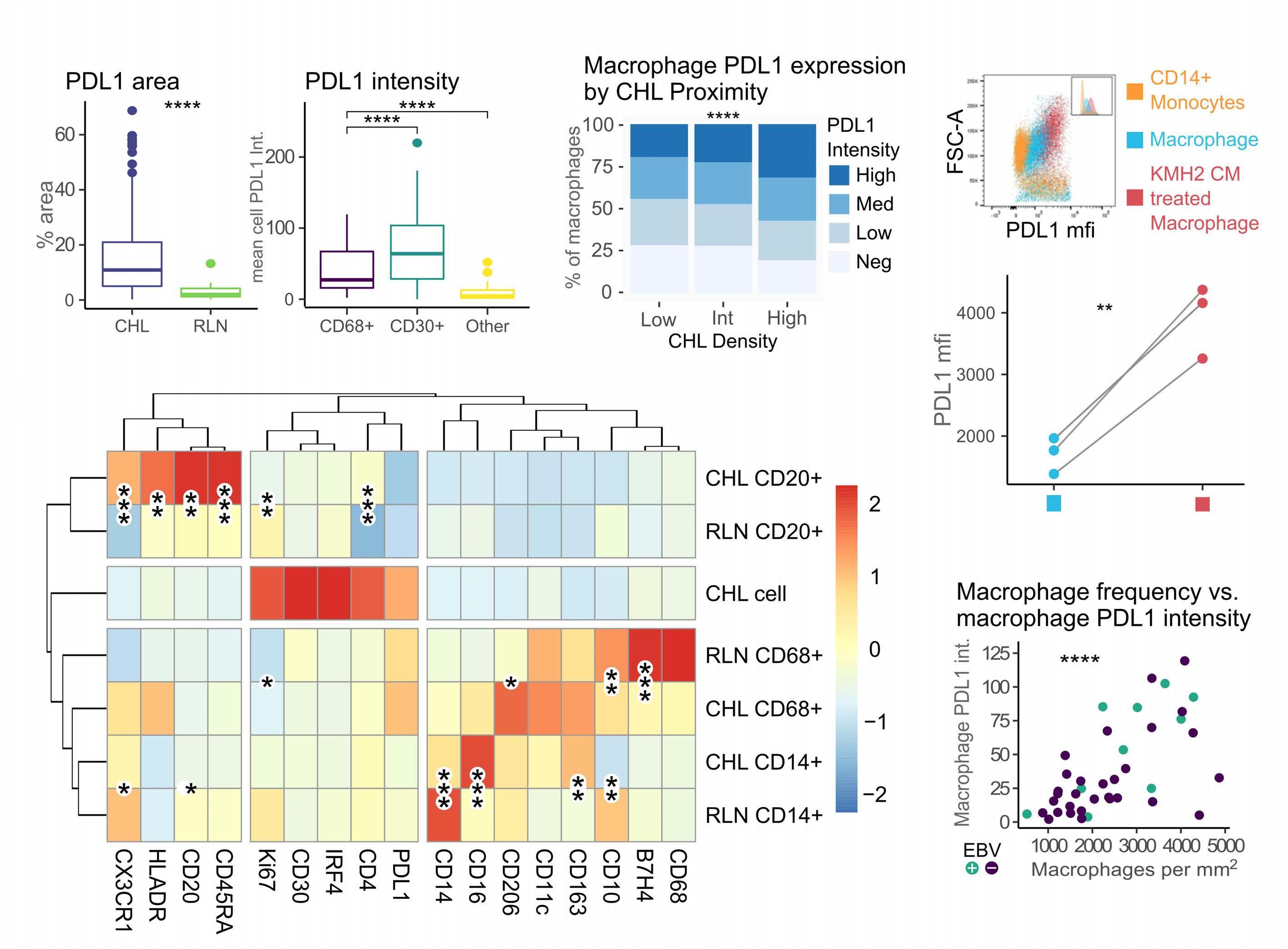
B C
Figure 1. Classical Hodgkin lymphoma cells express high levels of PDL1 and exist within a PDL1-positive microenvironment. (A) Classical Hodgkin lymphoma (CHL) is associated with increased PDL1 expression with highest expression intensity on CHL cells. Left: total PDL1 area by immunohistochemistry of 149 cases of CHL (EBV+ 46, EBV- 103) and 15 reactive lymph node (RLN). Right: mean cell PDL1 intensity by multiplex immunohistochemistry (IHC) in CD30+ (CHL cells), CD68+ (macrophages) and CD30–CD68–(other) cells in cases of CHL (n=46). (B) Macrophage PDL1 intensity increases with CHL density. Relative frequency of PDL1High, PDL1Med, PDL1Low and PDL1Neg macrophages within tessellation areas of CHL tumor. Tessellation areas were calculated by CHL cell density. The c2 test was used for statistical comparisons, P<0.0001, mean across 47 cases. (C) Monocyte-derived macrophages upregulate PDL1 in the presence of media conditioned by the KMH2 CHL cell line. Healthy donor CD14+ monocyte-derived macrophages cultured with (red) or without (blue) KMH2 conditioned media for 24 h. Flow cytometry analysis was performed on three biological replicates. mfi: mean fluorescence intensity. (D) Phenotyping of microenvironmental cells in CHL in comparison to RLN. The mean cell expression averaged by case of CD20+ (B cells), CD68+ (macrophages), CD14+ (monocytes) and CHL cells in CHL or RLN by imaging mass cytometry. Asterisks denote statistical significance of the comparison of mean cell intensity by case (20 CHL, 10 RLN). (E) Macrophage PDL1 intensity correlates with macrophage frequency. Mean PDL1 intensity per CD68+ cell compared to CD68+ cell frequency was assessed by multiplex immunohistochemistry in 47 CHL cases (EBV+ 9, EBV- 38), R=0.61, (EBV+ R= 0.72, EBV- R=0.55). Comparisons were performed with Mann-Whitney and Wilcoxon rank (three-way) tests. Correlations were performed by Pearson rank. ns: non-significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
D
Haematologica | 108 - April 2023 1071 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al. A E
exhaustion by multiplex IHC whereby tissue was stained with a panel of IHC antibodies and then the IHC antibody was serially stripped off before re-staining with a new one. Stained images were virtually aligned allowing assessment of cell-marker co-expression. Multiplex IHC was chosen over imaging mass cytometry due to the larger cohort size and analysis of larger tissue areas (three tissue microarray
cores versus 1x1 mm) making it preferable for rarer cell populations. PD1 expression was restricted to small lymphocytes and was expressed primarily on CD4+ cells, with some expression on CD8+ cells and minimal non-T-cell expression (Figure 2D). Since CD4 staining produced background when combined with TIM3 and LAG3 in stripping panels, after confirming PD1 T-cell restriction, CD4 was
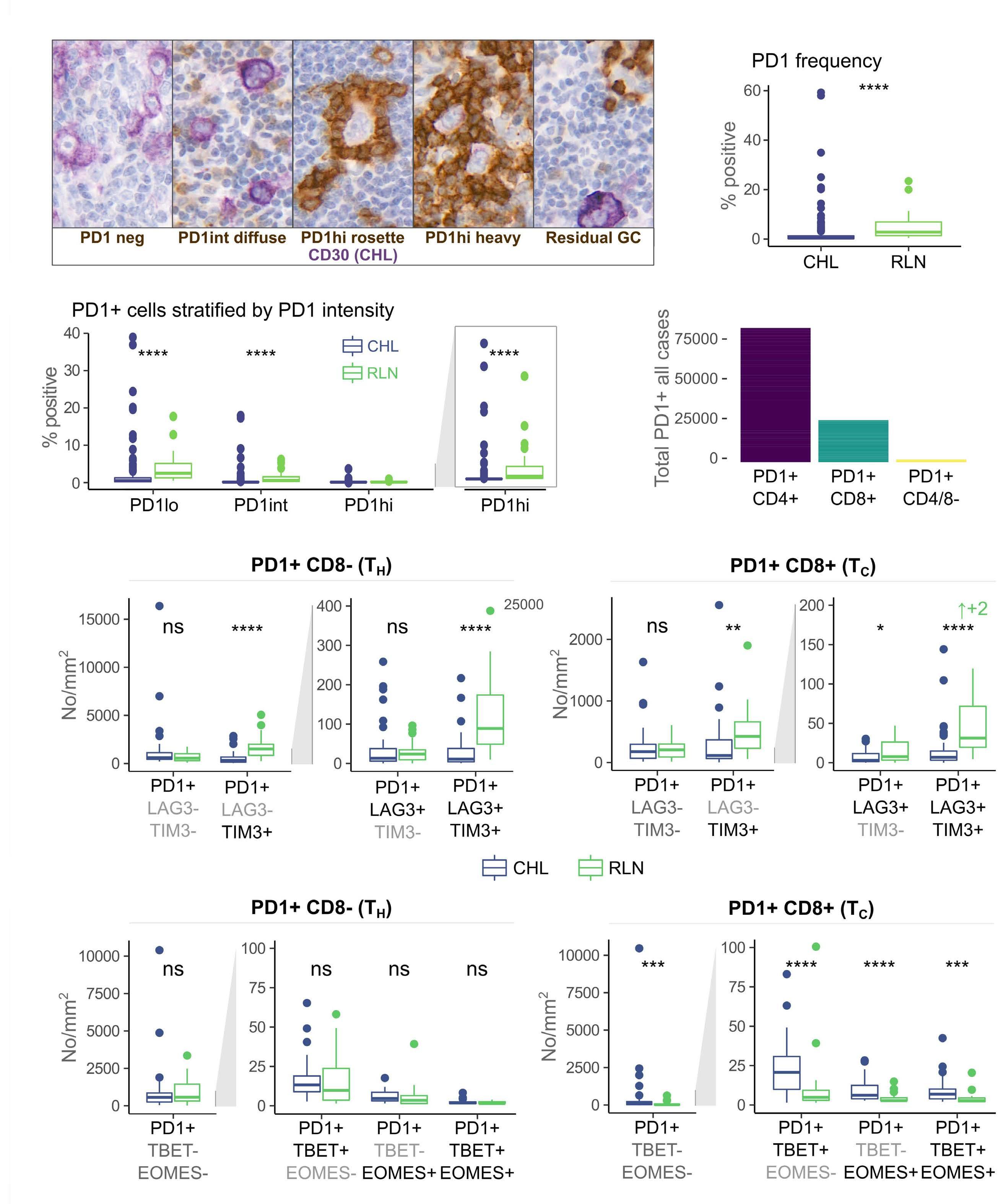
Continued on following page. A C E F B Haematologica | 108 - April 2023 1072 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al. D
Figure 2. PD1-positive lymphocytes are less frequent in classical Hodgkin lymphoma than in reactive lymph nodes. (A) Multiple PD1 infiltration patterns were observed. Most cases were characterized by rare/low or diffuse PD1weak/int infiltration. Rarely, PD1hi cells rosetted around classical Hodgkin lymphoma (CHL) cells or heavy infiltration with PD1hi cells was observed. Infrequent residual germinal centers were observed. Germinal centers were excluded from the analysis. In the images, PD1 is represented by brown, CD30 CHL by purple. (B) PD1+ infiltrating lymphocytes are reduced in CHL relative to reactive lymph node (RLN). Comparison by immunohistochemistry (IHC), germinal centers excluded (n=27/150). (C) PD1+ infiltrating lymphocytes are reduced in CHL irrespective of PD1 expression intensity. PD1+ area stratified by PD1 intensity by IHC. Germinal centers excluded. (right: PD1hi cells on expanded axis) (n=27/150). (D) PD1+ expression in CHL is T-cell restricted with most expression in CD4+ cells. Left: PD1, CD4 and CD8 co-expression as determined by multiplex IHC. Given this, CD8 positivity was used to differentiate cytotoxic T cells (TC) from T helper cells (TH) in multiplex panels for identifying exhaustion. (E) PD1+ cells in CHL co-express LAG3 and TIM3 at lower frequency than PD1+ cells in RLN. PD1 co-expression with TIM3 and LAG3 was assessed in TH and TC by multiplex IHC. Validation demonstrated T-cell-restricted PD1 positivity. PD1+ TH cells are defined as PD1+CD8- and PD1+ TC as PD1+CD8+. No significant difference was detected in PD1+ LAG3–TIM3– TH or TC frequency between CHL and RLN. Cells co-expressing PD1 with LAG3, TIM3 or both were significantly less frequent in CHL than in RLN. PD1+LAG3–TIM3– was the predominant TH phenotype in CHL whereas PD1+TIM3+LAG3–was predominant in CHL TC and RLN TH. (F) PD1+ TH cells co-express TBET and EOMES at similar frequency in CHL and RLN, while PD1+ TC co-express these markers at a higher frequency. PD1, TBET and EOMES co-expression was assessed in the same cohort by multiplex IHC. No significant difference in PD1 co-expression with TBET or EOMES was identified within the TH compartment. In contrast, increased co-expression was seen in the TC compartment in CHL as compared to RLN. The predominant TH and TC phenotype was that of isolated PD1+ expression with co-expression rarely identified. Comparisons were made by the Wilcoxon Mann-Whitney test. ↑+2 indicates that two outliers were excluded from the graph. All outliers were included in the statistical analysis. ns: non-significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
omitted from subsequent stripping panels. PD1+CD8+ and PD1+CD8– lymphocytes were significantly less frequent in CHL than in RLN controls (Figure 2E). PD1+CD8– lymphocytes co-expressed TBET or EOMES at similar levels to RLN controls while PD1+CD8+ lymphocytes co-expressed TBET and EOMES above levels in RLN (Figure 2F). Isolated PD1 expression was the dominant phenotype with very low levels of PD1/TBET/EOMES co-expression.
In summary, despite a more prominent T-cell infiltrate, PD1+ lymphocytes were less frequent in CHL than in RLN, irrespective of the intensity of PD1 expression and there was no enrichment of PD1+CD8– lymphocytes co-expressing exhaustion markers in CHL. The dominant phenotype of both PD1+CD8– and PD1+CD8+ lymphocytes in CHL was expression of PD1 in the absence of co-expression of any other exhaustion marker.
Classical Hodgkin lymphoma T cells are proliferative, cytokine-competent and contain fewer functionally exhausted cells than do reactive lymph node T cells
Proliferation, interleukin 2 (IL2) and interferon gamma (IFNγ) production in T cells from single cell suspensions from diagnostic CHL lymph nodes compared to five RLN single cell suspensions were assessed to determine functional features of TH and cytotoxic T (TC) exhaustion.3–5 Paired formalin-fixed, paraffin-embedded samples were evaluated to ensure that the single cell suspension samples were representative of the IHC cohort. No significant difference in PDL1, CD68, PD1, TBET, GATA3 or RORγT expression was seen, but FOXP3 expression was marginally higher in the single cell suspension group of samples than in the wider IHC cohort (Online Supplementary Figure S5). The characteristics of the cohorts are described in Table 1. CHL suspensions had higher baseline Ki67+ TH frequencies compared to the RLN controls. No significant differences in proliferation or IL2 and IFNγ production in response to
stimulation were seen between CHL and RLN (Figure 3A). Ki67–PD1+ (non-proliferative PD1+) CD3+CD4+ cells were significantly less frequent in CHL than in RLN, while Ki67–PD1+ CD3+CD4– cells were present at similar levels (Figure 3B). Non-cytokine-productive Ki67–PD1+ CD3+CD4+ and CD3+CD4– cells were also significantly less frequent in CHL (Figure 3B). By comparison, no significant difference was seen in numbers of PD1+Ki67+ cells (Figure 3C). On subgroup analysis, CD3+CD4– proliferation and CD3+CD4+ cytokine production capacity were significantly higher in EBV+ CHL than in EBV– CHL (Online Supplementary Figure S6). Further phenotyping by imaging mass cytometry demonstrated elevated expression of TIM3 and modest elevation of TBET in the PD1+Ki67–(CD4+CD3+) TH population, consistent with identification of an exhausted phenotype. In contrast, the proliferative Ki67+PD1–(CD8+CD3+) TC expressed higher levels of granzyme B, while the Ki67+PD1–(CD4+CD3+) TH fraction expressed increased FOXP3 and TBET. Interestingly LAG3 expression was higher in the TH proliferative fraction, perhaps reflecting the presence of TR1 cells which are known to be enriched in CHL (Figure 3D).19 Analysis of mean marker expression in immediate neighbors of phenotypically exhausted TH cells revealed higher expression of CD20, CD45RA (expressed on B cells in addition to naïve T cells) and HLADR with no relationship to CD30, PDL1 or IRF4 (CHL cell markers). In contrast, neighboring cells of phenotypically proliferative TH cells expressed higher levels of IRF4, CD30, PDL1 and B7H4 (Figure 3E).
PDL1 expression is not associated with PD1 expression in classical Hodgkin lymphoma
In the light of our findings of low PD1 expression and little functional evidence of exhaustion but high PDL1 expression we assessed the relationship between PD1 and PDL1 expression across CHL cases. Cases varied by both PDL1 and PD1 expression (Figure 4A), but counterintuitively, no associ-
Haematologica | 108 - April 2023 1073 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
ation was detected between PD1 and total PDL1 area or PDL1 intensity in CHL, irrespective of stratification for PD1 intensity or analysis of exhaustion marker co-expression (Figure 4B). Unexpectedly, in EBV+ CHL an inverse relationship was observed between PD1 expression and PDL1 intensity (Figure 4B). Analysis of exhaustion marker co-expression demonstrated that isolated PD1 expression rather than co-expression of exhaustion markers (LAG3/TIM3 or EOMES/TBET) was responsible for this effect (Figure 4B). Therefore, compared to RLN controls, we detected signifi-
cantly fewer phenotypic or functionally exhausted (noncytokine productive, non-proliferative, PD1+) CD3+CD4+ (TH) and CD3+CD4– (TC) cells and significantly more Ki67+ CD3+CD4+ (TH) cells in the CHL group, with no overall loss in proliferative or cytokine production capacity.
PDL1 is associated with classical Hodgkin lymphoma MHC-II expression, TH cell recruitment and local TC exclusion
In the light of the above data, we considered alternative
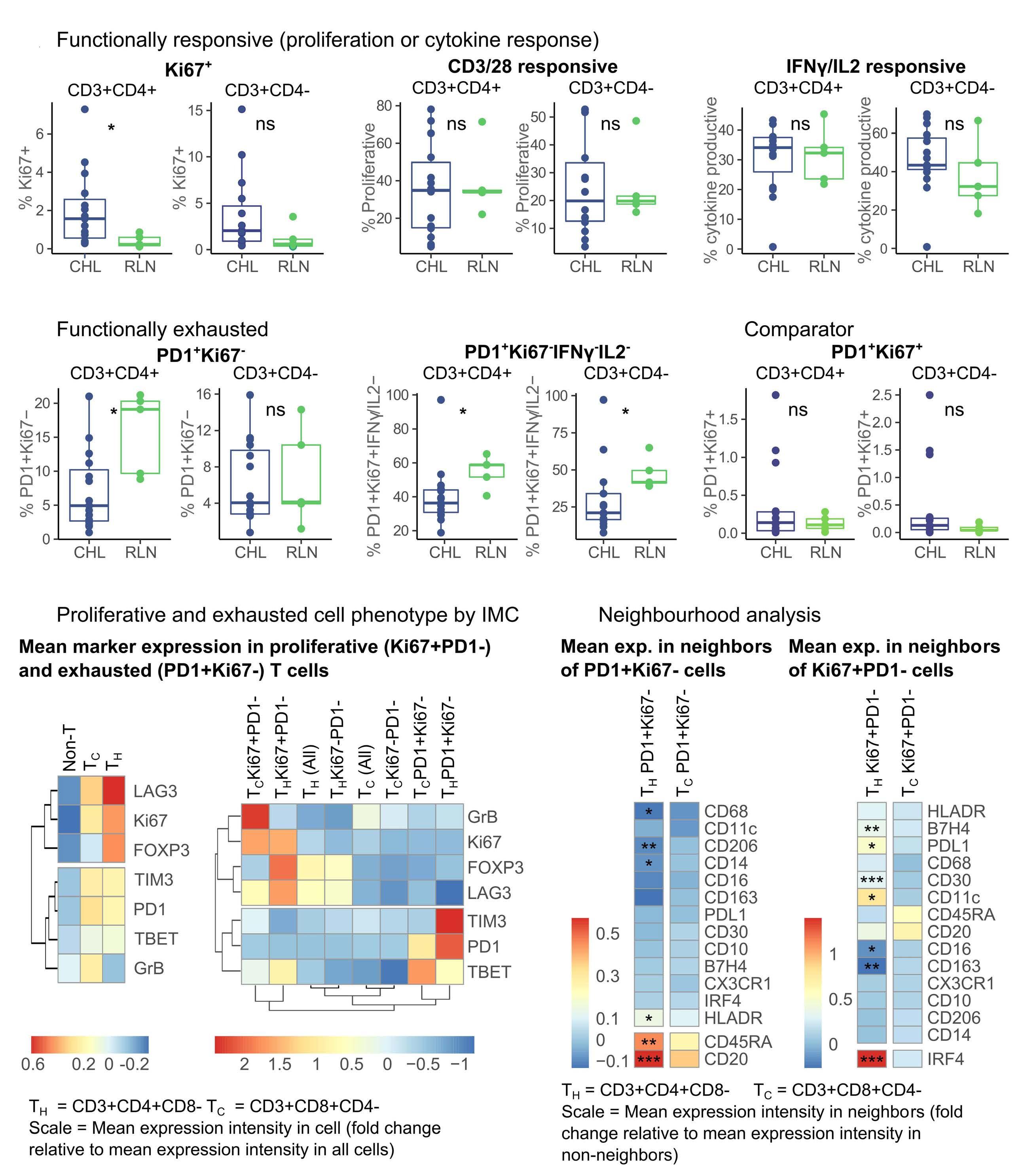
A B D E Continued on following page. Haematologica | 108 - April 2023 1074 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al. C
Figure 3. Classical Hodgkin lymphoma T cells are proliferative, cytokine-competent and contain fewer functionally exhausted cells than T cells from reactive lymph nodes. (A) Classical Hodgkin lymphoma (CHL) T cells are proliferative and cytokinecompetent. Left: unstimulated TH cells from CHL single cell suspensions expressed higher percentages of Ki67 than TH from reactive lymph node (RLN) single cell suspensions (P=0.017). Middle: CHL T cells were no less proliferative than RLN T cells to CD3/CD28 stimulation, as measured by CFSE assay after 3 days of culture in vitro. Right: CHL T cells produced no less cytokines than RLN T cells in response to phorbol myristate acetate (PMA)/ionomycin stimulation, as measured by intracellular flow cytometry after 4 h of PMA/ionomycin stimulation in vitro. (B) Functionally exhausted cells are less frequent in CHL. Left: nonproliferative (Ki67–) TH cells were less frequent in CHL (unstimulated, P=0.019). Right: non-proliferative non-cytokine productive (Ki67–IFNγ/IL2–) TC and TH cells were less frequent in CHL, as measured by intracellular flow cytometry after 4 h of PMA/ionomycin stimulation in vitro (P=0.025 and P=0.015, respectively). CHL, n=15; RLN, n=5. (C) Comparator cells. Indeterminate (both Ki67+ and PD1+) cells provided as a comparison group to proliferative and functionally exhausted groups (unstimulated, P=ns). CHL, n=15; RLN, n=5. Comparisons performed using the Mann-Whitney test. (D) Proliferative and exhausted cell phenotypes as determined by imaging mass cytometry. Mean per-cell marker expression intensity in the cell of interest was expressed as fold change from mean value across all cells. Results were calculated by sample and the overall mean results are displayed. Left: expression intensities in non-T (all CD3–), TC (CD3+CD8+CD4–) or TH (CD3+CD4+CD8–) cells. Right: expression intensities in proliferative and exhausted T-cell subgroups. (E) Neighborhood analysis of mean marker expression in cells adjacent to proliferative or exhausted T-cell subsets. Mean per-cell marker expression intensity in neighboring cells expressed as fold change from mean value across all cells. Results were calculated by sample and overall mean results are displayed. Comparisons were made by paired Mann-Whitney tests.
mechanisms by which PDL1 might influence the CHL microenvironment. We focused on its relationship to TH populations given that both CHL cell MHC-II expression and CD4+ T-cell receptor diversity predict PD1 inhibitor response.6,12 This is in contrast to MHC-I, whose expression does not predict PD1 inhibitor response.20
A positive association was observed between PDL1 expression and CHL MHC-II (Figure 5A), suggesting higher PDL1 expression in CHL cells retaining TH antigen-presenting capacity or deriving other survival advantage from interaction with TH cells in the tumor microenvironment. Of note, no association was identified between PD1 or exhaustion marker expression and CHL MHC II (Online Supplementary Figure S7). CD3 infiltration was increased in CHL and was positively associated with both PDL1 staining and CHL cell MHC II expression (Figure 5B). TH cells were responsible for this pattern: CD4 infiltration correlated positively with PDL1 expression, CHL cell PDL1 intensity, CD68+ macrophage PDL1 intensity and CHL MHC-II expression, while a negative relationship was seen between these markers and CD8 infiltration (Figure 5C, D). Spatial data supported these observations. CD4+ and CD8+ cells were spatially segregated within the tumor, with CD4+ cells proximal to CHL cells while CD8+ cells were frequently excluded from the tumor microenvironment (Figure 5E). The image shows CD8 versus overall CD3 distribution with PDL1 identifying tumor. FOXP3 demonstrates local TReg enrichment). Consistent with this, (CD3+CD8+) TC cells were located significantly further from CHL cells than both CD4+CD3+ TH and CD3+ T cells overall in the imaging mass cytometry analysis (Figure 5F). We therefore identified an association between PDL1, CHL MHC-II expression and CD4+ enrichment but CD8+ cell exclusion. Given that CHL cells secrete cytokines and actively recruit TH cells this suggests that CHL cases with high PDL1 expression may be those that actively recruit and interact with TH cells.21
PDL1 and classical Hodgkin lymphoma MHC-II are associated with differentiation of TH cells to TH1Reg but not to TH1 or non-polarized TReg cells
We next assessed the relationship between PDL1 and CHL MHC-II expression and TH cell differentiation in CHL. Naïve TH cells differentiate to effector subtypes with different functional roles based upon signals during activation. TH1 and TH17 cells are considered to have anti-tumor roles while TH2 and TReg are tumor protective. TReg also differentiate into subtypes mirroring effector types and may specialize in suppression of specific effector responses (TH1Reg being more effective at suppressing a TH1 response).22 PD1 is upregulated on activation and PDL1-PD1 signaling modulates differentiation, driving development away from TH1 towards TReg subtypes.23,24,24–27 PDL1 also regulates the conversion of differentiated TH1 to TReg and stabilizes inducible TReg conversion to TH1Reg.27–30 PDL1 expression in MHC-II-positive CHL may therefore drive TH1Reg differentiation to combat the anti-tumor TH1 response. To assess this, we first quantified the master transcription factors TBET, FOXP3, RORγT and GATA3 (for TH1, TReg, TH17 and TH2, respectively) by conventional IHC. TBET+ and FOXP3+ cells were enriched in CHL while RORγT+ cells were reduced and no difference was detected in GATA3+ (data not shown). We then used a multiplex IHC panel including CD4, TBET, FOXP3, ROR γ T and CD8 to define TH1 (TBET+CD4+, FOXP3–RORγT–CD8–), TH1Reg (TBET+FOXP3+CD4+, RORγT-CD8-), non-polarized TReg (FOXP3+CD4+, TBET–RORγT–CD8–), TH17 (ROR γ T+CD4+, TBET–FOXP3–CD8–) and TH17Reg (RORγT+FOXP3+CD4+, TBET–CD8–). TH1 and TH1Reg were enriched in CHL, whereas TH17, TH17Reg and, surprisingly, nonpolarized TReg were reduced (Figure 6A).
Next, we analyzed the spatial location of these T-cell subsets relative to CHL by imaging mass cytometry. TH1Reg were located significantly closer to (CD30+IRF4+) CHL cells than both TH1 and non-polarized (TBET–) TReg (Figure 6B). CD30 and IRF4 were selected as both are immunohisto-
Haematologica | 108 - April 2023 1075 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
Figure 4. PDL1 is not associated with markers of exhaustion. (A) Cases vary by PD1 and PDL1 infiltration. Cases dominated by PD1hi infiltration were consistently PDL1–, while PD1weak and PDL1 cases varied so cases were observed with infiltration of one or both. (B) No correlation was seen between PD1 and PDL1 in classical Hodgkin lymphoma, whereas negative correlations to PDL1 area and intensity were seen in the Epstein-Barr virus (EBV)+ CHL subgroup. Negative associations were observed between PDL1 area, CHL PDL1 intensity and macrophage PDL1 intensity and PD1 in EBV+ CHL, with no significant correlation detected in the EBV– group (EBV+ n=47, PD1+ P=0.03, PD1wk P=0.02, EBV– n=105. Pearson rank). Multiplex immunohistochemistry demonstrated that this effect was present in PD1+ cells not co-expressing exhaustion markers (LAG3/TIM3 or EOMES/TBET). PD1 expression in this context may represent an alternate state to exhaustion. No consistent association was seen in the EBV–group (EBV+ n=10, EBV– n=31, Pearson rank). The graph denotes strength of correlation and the dark color denotes statistical significance (P<0.05).
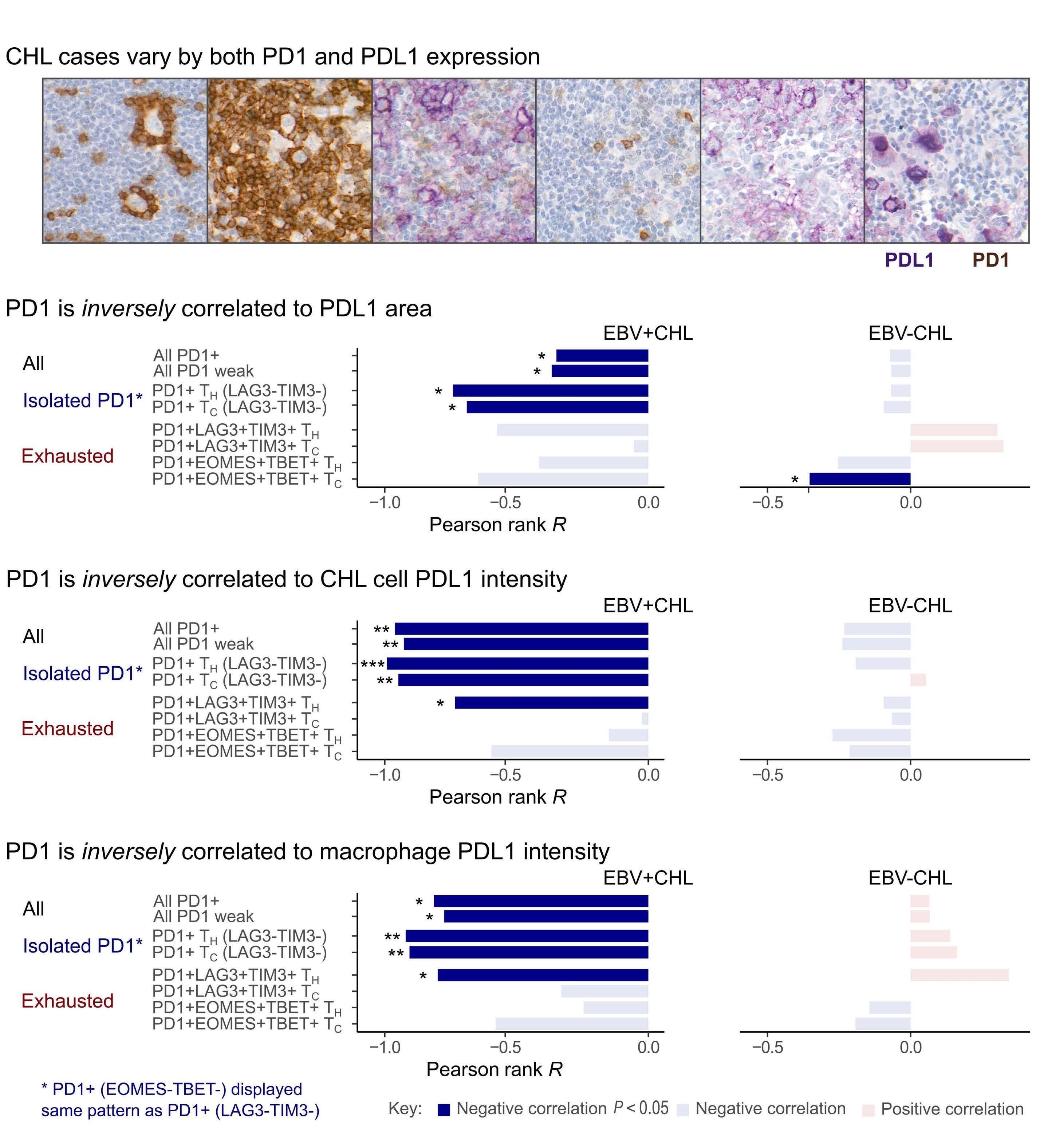
A B
Haematologica | 108 - April 2023 1076 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
Figure 5. PDL1 is associated with classical Hodgkin lymphoma MHC-II expression, TH cell recruitment and local TC cell exclusion. (A) PDL1 area, mean classical Hodgkin lymphoma (CHL) PDL1 intensity and macrophage PDL1 intensity are positively associated with CHL major histocompatibility complex II (MHC-II) expression, as determined by multiplex immunohistochemistry (IHC). PDL1 area to MHC-II positive versus negative status, compared by a Mann-Whitney test. MHC-II manual scoring (n=47/23/55), CHL cell PDL1 intensity to MHC-II (n=11/6/17), macrophage PDL1 intensity to MHC-II (n=11/6/17). (B) CD3+ T cells are enriched in CHL and are associated with both PDL1 and CHL MHC-II expression, as determined by IHC. Left: n=145/13. Middle: R=0.2 (n=144). Right: MHC-II positive versus negative status (n=48/23/54). (C) CD4+ TH but not CD8+ TC infiltration is positively associated with CHL MHC-II expression. MHC-II positive versus negative status (n=32), P=0.022 and P=0.19, respectively. (D) CD4+ TH infiltration in CHL is correlated with CHL cell and macrophage PDL1 intensity. Top: correlation by Pearson rank of CD4 and CD8 infiltration with CD30+ CHL cell PDL1 intensity, as determined by multiplex IHC. The graph denotes the strength of the correlation (R). Color intensity and stars denote statistical significance (n=32), (CD4 R=0.36). Bottom: CD4/CD8 to CD68+ macrophage PDL1 intensity (CD4 R=0.45, CD8 R= -0.39). (E) The image shows T-cell enrichment around PDL1+ tumor with local TReg enrichment and TC exclusion. Left: CD3 (T) cells are stained brown and PDL1 (PDL1+ tumor) cells purple. Right: FOXP3 (TReg) cells are stained brown and CD8 (TC) purple. (F) TC are locally depleted and TH enriched proximal to CHL cells. Mean nearest neighbor distance to CD30+IRF4+ CHL cells was assessed by imaging mass cytometry. TH cells are CD3+CD4+CD8–, TC cells are CD3+CD8+CD4–, T cells are CD3+, and non-T cells are CD3– (CHL cases, n=20, Wilcoxon paired rank). MHC-II expression –: negative; wk: weak, +: positive; TH: T helper cells; TC: cytotoxic T cells. ns: non-significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
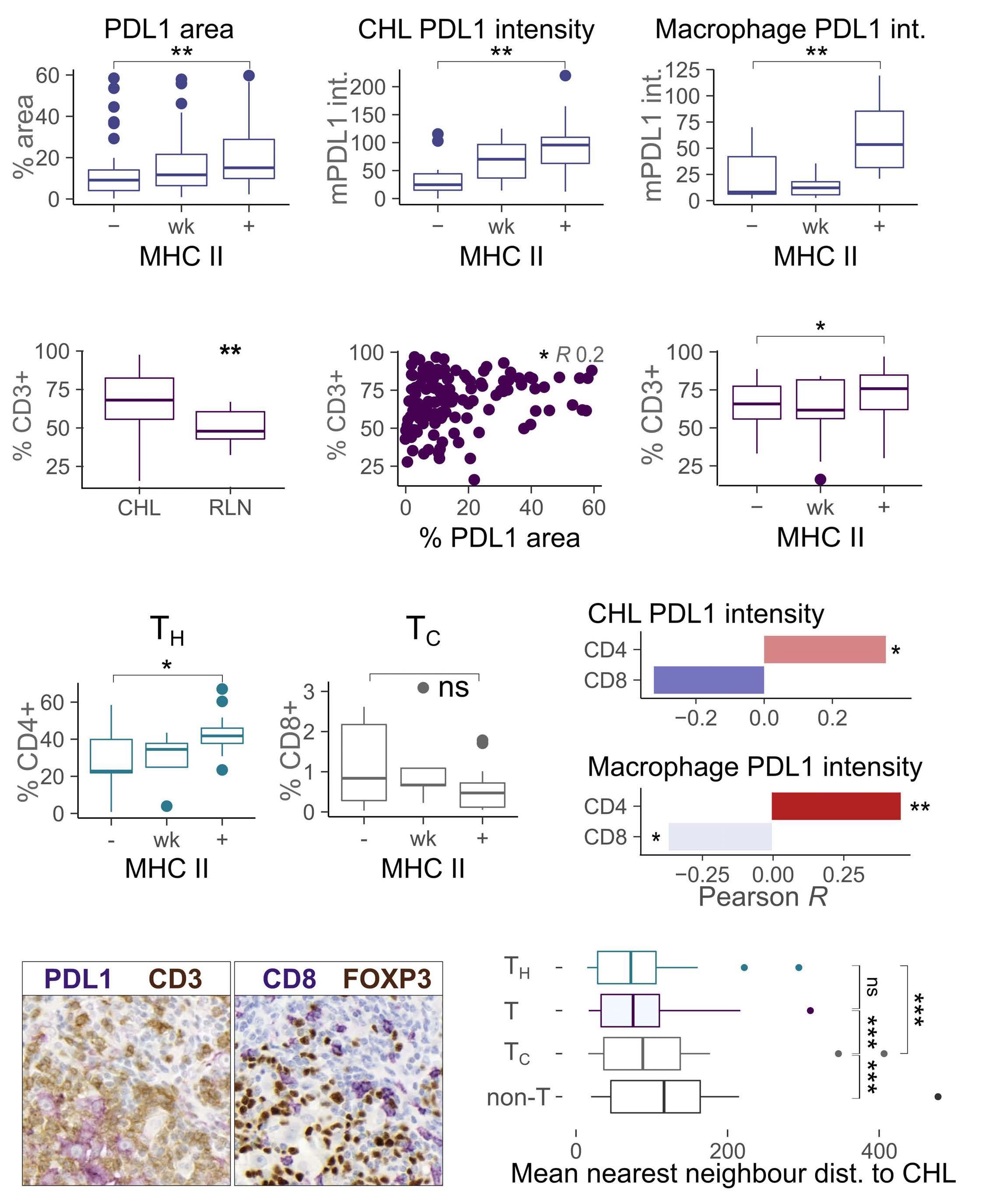
A B C E F D
Haematologica | 108 - April 2023 1077 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
chemical diagnostic markers of CHL. Given these findings we assessed the relationship between TH polarization and both PDL1 and CHL MHC-II expression. Retained expression of MHC-II on CHL was associated with increased TH1Reg, but no association was seen with TH1 or non-polarized TReg. Significant correlations were observed between TH1Reg and PDL1 area, CHL PDL1 intensity and macrophage PDL1 intensity, but again no association was seen with TH1 or non-polarized TReg (Figure 6C, D). On subgroup analysis TH1Reg associations with PDL1 retained significance in both
EBV+ CHL and EBV– CHL and in nodular sclerosing and mixed cellularity histological subtypes (data not shown). Associations with MHC-II expression were retained in EBV– CHL and nodular sclerosing subgroups. MHC-II negative cases were too infrequent in EBV+ and mixed cellularity groups for statistical comparison. Finally, co-culture of naïve TH from healthy donors with the KMH2 CHL cell line promoted marked TH1Reg differentiation (CD3+CD4+CD25hiCD127loFOXP3+TBET+) relative to media-activated controls. (Figure 6E) This is in line with previous publications.31 These data reveal that TH1Reg (but not
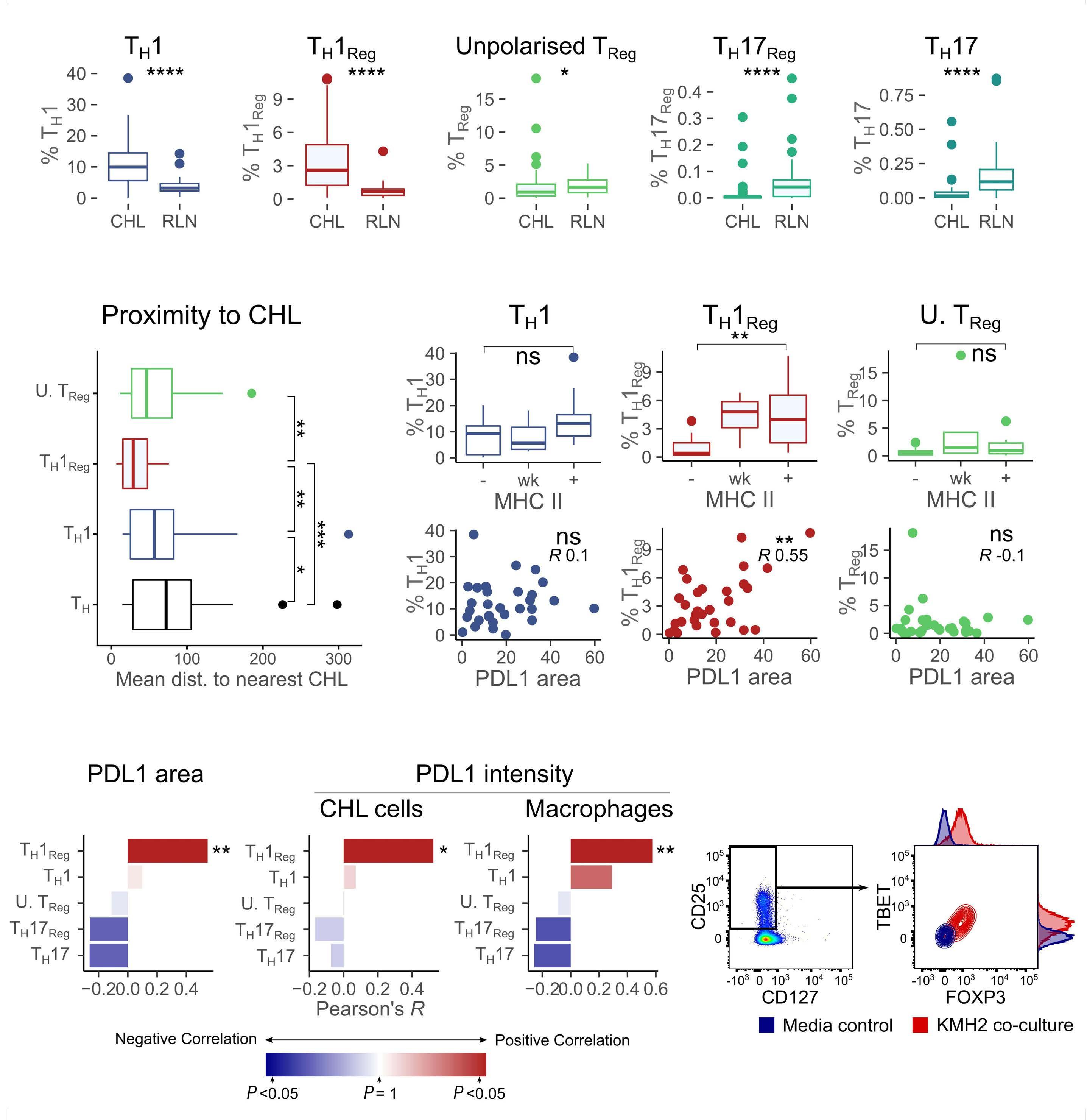
E D B A C Continued on following page. Haematologica | 108 - April 2023 1078 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
Figure 6. PDL1 and classical Hodgkin lymphoma MHC-II expression are associated with skewed TH differentiation towards TH1Reg but not TH1 or non-polarized TReg. (A) TH1 and TH1Reg are enriched in classical Hodgkin lymphoma (CHL) compared to reactive lymph nodes (RLN) while TH17, TH17Reg and unpolarized TReg are depleted. TBET+FOXP3+CD4+ TH1Reg (CD8–RORγT–) and TBET+CD4+ TH1 (FOXP3–RORγT–) were increased but FOXP3+CD4+ TReg (TBET–RORγT–), RORγT+CD4+TH17 (TBET–FOXP3–) and RORγT+FOXP3+CD4+ TH17Reg (CD8–TBET–) were reduced, as determined by multiplex immunohistochemistry (IHC) (n=49/31). (B) TH1Reg are located closer to CHL cells than either TH1 or non-polarized TReg. Mean nearest neighbor distance to CD30+IRF4+ CHL cells for TBET+FOXP3+CD4+CD3+ TH1Reg, TBET+CD4+CD3+ TH1 (FOXP3–) and non-polarized FOXP3+CD4+CD3+ TReg (TBET–) measured relative to TH (CD3+CD4+), as evaluated by imaging mass cytometry (n=20, CHL cases). (C) TH1Reg but not TH1 or unpolarized (U) TReg are associated with both PDL1 and CHL MHC-II expression. There was increased TBET+FOXP3+CD4+ TH1Reg (RORγT–) but not FOXP3+CD4+ TReg (TBET–RORγT–) or TBET+CD4+ TH1 (FOXP3–RORγT–) in CHL, as determined by multiplex IHC (MHC-II - to +; n=9/5/18). PDL1 area (n=32, TH1: R=0.1, TH1Reg: R=0.55, TReg: R= -0.1). (D) TH1Reg but not TH1 or non-polarized TReg correlate to PDL1 area and both macrophage and CHL cell mean PDL1 intensity. N=32, CD30+ CHL PDL1 intensity or CD68+ macrophage PDL1 intensity. (E) Co-culture of naïve TH cells with the KMH2 CHL cell line promotes TH1Reg differentiation. Naïve healthy donor TH cells were cocultured for 14 days with KMH2 cells or CD3/CD28 stimulation. KMH2 induced differentiation to CD3+CD4+CD25hiCD127loFOXP3+TBET+ TH1Reg relative to media-activated controls. TH1Reg were PD1hi. Comparisons were made by the Mann-Whitney test, with spatial comparison by Wilcoxon paired rank and correlation by Pearson rank analysis. MHC-II expression –: negative; wk: weak, +: positive; TH: T helper cells. ns: non-significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
TH1 or non-polarized TReg) enrichment is associated with high PDL1 and retained expression of MHC-II in CHL. Taken together these data show a lack of enrichment of PD1+ or exhausted cells in CHL and a negative or absent association between PD1 infiltration and PDL1 expression. Instead there is enrichment of Ki67+ cells and a positive association between PDL1 expression in CHL, retained MHC-II expression and both TH and TH1Reg cells (Figure 7).
Discussion
We present data suggesting a role for PDL1 in shaping the CHL tumor microenvironment. We found similar levels of T-cell exhaustion in RLN controls and CHL and no relationship between exhaustion signatures and PDL1 expression. Instead, we identified a consistent association between PDL1 and TH recruitment, TC exclusion and TH1Reg enrichment. These data support and add to recent evidence suggesting that the dominant role for PDL1 in CHL is to modulate the tumor-protective immune tumor microenvironment rather than maintaining an exhausted T-cell infiltrate.11,12
Reversal of exhaustion is the commonly accepted mechanism of action of PD1 inhibitors in both solid and hematologic malignancies. However, recent research on CHL has shown that expansion of novel T-cell clones, increased baseline TH receptor diversity and retained CHL MHC II expression but not increased TC signatures or reinvigoration of existing (putatively exhausted) clones are associated with PD1 inhibitor response.11,12 This raises questions as to whether reversal of exhaustion is the dominant mechanism of action of PD1 inhibitors in CHL. Exhaustion is assumed to be present because of high PDL1 expression and sensitivity to PD1 inhibition; however, this is largely extrapolated from solid tumors and chronic viral infection in which TC exhaustion is well characterized.3 In contrast to solid tumors, CHL cells are derived from professional
antigen-presenting cells within a TH-dominated immune environment and frequently lose MHC-I expression. It is therefore plausible that the interactions with immune cells would be different from the classical tumor-exhausted effector relationship. Evidence for exhaustion in CHL is limited. PD1 expression has been noted to be lower in CHL than in reactive tissue and similar to that in diffuse large B-cell lymphoma (which is much less responsive to PD1 inhibitors).9,32 Deep phenotyping has detected a terminally differentiated TH effector subset with TH1-like characteristics.33 No study has demonstrated the presence of functional exhaustion in CHL.
Our data inform and expand upon those in the literature and on previous work from our group in a new cohort, providing a more detailed functional assessment than any other to date but again finding no evidence of exhaustion above that seen in reactive tissue. Importantly, we also demonstrated that the effects of PDL1 both between CHL cases and spatially within individual tumors are not related to exhaustion signatures. This lack of relationship highlights that PDL1 is not a reliable surrogate for exhaustion, and the conclusion that PDL1 may therefore play roles beyond maintaining exhaustion perhaps explains why PDL1 has consistently been reported to predict PD1 inhibitor response in CHL while PD1 never has.6 We went on to identify a consistent link between PDL1 expression and TH1Reg enrichment, supporting our hypothesis that a dominant role of PDL1 within the tumor microenvironment is to sculpt the immune infiltrate by maintaining a regulatory TH environment.
The link between PDL1-PD1 signaling and the development of TH1Reg is supported by a body of literature. Activated T cells upregulate PD1 and become highly sensitive to PDL1PD1 signaling at even low levels of PD1 expression.23 Engagement of PD1 influences T-cell phenotype at multiple stages of differentiation. During activation PD1 engagement by PDL1 promotes differentiation of naïve TH cells towards TReg and limits the development of effectors such as
Haematologica | 108 - April 2023 1079 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
Figure 7. Summary of
associations. Comparison of classical Hodgkin lymphoma (CHL)
to reactive lymph node cells shows a significant increase in PDL1 expression with enrichment of macrophages, TH, TH1Reg and Ki67+ cells. PD1+/exhausted cells are seen at significantly lower frequency. Within CHL, there are positive associations between PDL1 and CHL cell MHC-II expression, macrophage PDL1 expression, and TH and TH1Reg infiltration whereas negative or absent associations are seen between PDL1 and both TC and PD1+ or exhausted cell infiltration. CHL: classical Hodgkin lymphoma; RLN: reactive lymph nodes; PDL1: programmed death ligand 1; MHC-II major histocompatibility cell type II; Mφ: macrophages; TH: T helper cells; TH1Reg: TH1 regulatory cells; PD1/Ex: programmed death protein-1-positive/exhausted cells; TC: cytotoxic T cells.
TH1.24,25,34–36 PDL1 promotes plasticity of fully differentiated TH1 towards TReg.27,28 PDL1 also stabilizes the conversion of inducible TReg to TH1Reg.29,30,37,38 These mechanisms point to a common concept – that of PDL1 expression shifting T-cell development away from anti-tumor effectors including TH1 and promoting development of tumor-protective regulatory cells including TH1Reg. This pattern is borne out by our data. The combined association we observed between PDL1 expression, retained CHL MHC-II expression and TH1Reg enrichment suggests this may at least in part be related to CHL antigen presentation and differentiation of naïve TH to TH1Reg and it is notable that CHL MHC-II expression is also predictive of response to PD1 inhibitors.6 Future work will examine whether the link between PDL1, CHL MHC-II and TH1Reg is via a direct or indirect effect and make comparisons to other PD1-responsive tumor types. The data cited support the possibility of a direct mechanistic link but further evidence is required to demonstrate this. It is noteworthy that while a strong correlation is seen, PD1 inhibitor response is not perfectly predicted by PDL1 or MHC-II expression.6,20 It is likely that PD1 inhibition acts via multiple pathways, beyond both exhaustion and the TH compartment, and other mechanisms including invigoration of innate immunity or B-cell compartments may play important roles.11,12,15 Our findings also provide a perspective on combination therapies such as PD1 and LAG3 co-inhibition, suggesting that rather than both focusing on exhausted cells these approaches may influence two prominent elements of the protective regulatory microenvironment (FOXP3+TBET+ TH1Reg and FOXP3–LAG3+ TR1). This may be particularly appealing given that while both are present irrespective of CHL MHC-II status, TH1Reg are enriched in
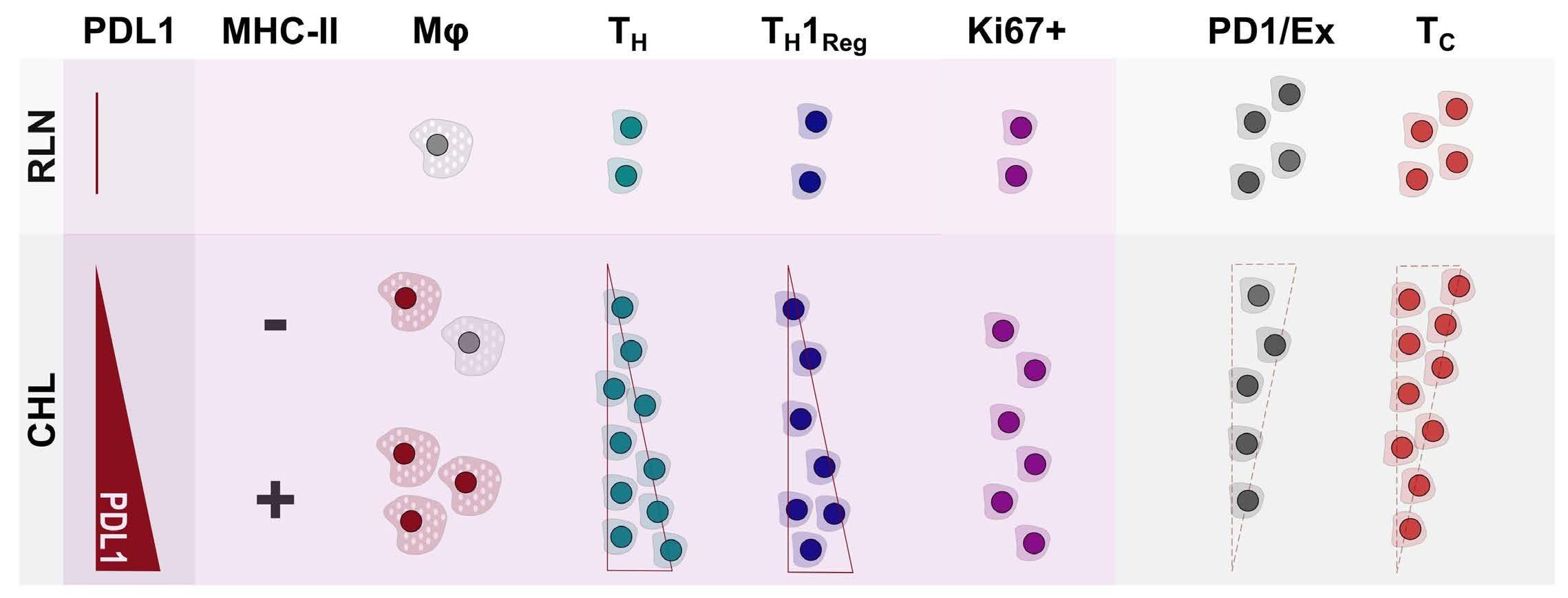
MHC-II-positive CHL, while TR1 are enriched in and mechanistically linked to CHL MHC-II loss.19
In conclusion, our data build on and enhance the current understanding of the roles of PDL1 within the CHL microenvironment. We provide phenotyping and functional evidence suggesting a limited role for exhaustion in the CHL microenvironment and identify a broader role for PDL1 within the microenvironment with links to MHC-II expression, TH recruitment and the regulatory cell infiltrate. These findings are informative for our understanding of CHL cell survival within the immune microenvironment and together with recent publications reinforce the concept that the reversal of exhaustion may be too narrow a lens with which to view the activity of the PD1-PDL1 axis in CHL.
Disclosures
No conflicts of interest to disclose.
Contributions
JGT and JG designed the experiments, JGT, ET, AC and MC performed the analyses. All authors contributed to writing the manuscript.
Funding
This investigation was supported by grant n. 200144/Z/15/Z to JT from the Wellcome Trust and grant funding to JGG from Barts and the London Charity, from Tom and Leona Baker and from The London Clinic Charity.
Data-sharing statement
Data and further methodological details are available on request.
Haematologica | 108 - April 2023 1080 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
key
cells
References
1. Bröckelmann PJ, Goergen H, Keller U, et al. Efficacy of nivolumab and AVD in early-stage unfavorable classic Hodgkin lymphoma: the randomized phase 2 German Hodgkin Study Group NIVAHL trial. JAMA Oncol. 2020;6(6):872-880.
2. Ramchandren R, Domingo-Domènech E, Rueda A, et al. Nivolumab for newly diagnosed advanced-stage classic Hodgkin lymphoma: safety and efficacy in the phase II CheckMate 205 study. J Clin Oncol. 2019;37(23):1997-2007.
3. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019;37457-495.
4. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486-499.
5. Crawford A, Angelosanto JM, Kao C, et al. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity. 2014;40(2):289-302.
6. Roemer MGM, Redd RA, Cader FZ, et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J Clin Oncol. 2018;36(10):942-950.
7. Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268-3277.
8. Carey CD, Gusenleitner D, Lipschitz M, et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for ReedSternberg cells in Hodgkin lymphoma. Blood. 2017;130(22):2420-2430.
9. Greaves P, Clear A, Owen A, et al. Defining characteristics of classical Hodgkin lymphoma microenvironment T-helper cells. Blood. 2013;122(16):2856-2863.
10. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311-319.
11. Reinke S, Bröckelmann PJ, Iaccarino I, et al. Tumor and microenvironment response but no cytotoxic T-cell activation in classic Hodgkin lymphoma treated with anti-PD1. Blood. 2020;136(25):2851-2863.
12. Cader FZ, Hu X, Goh WL, et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat Med. 2020;26(9):1468-1479.
13. Callahan MK, Wolchok JD. Recruit or reboot? How does anti-PD1 therapy change tumor-infiltrating lymphocytes? Cancer Cell. 2019;36(3):215-217.
14. Kawashima M, Carreras J, Higuchi H, et al. PD-L1/L2 protein levels rapidly increase on monocytes via trogocytosis from tumor cells in classical Hodgkin lymphoma. Leukemia. 2020;34(9):2405-2417.
15. Vari F, Arpon D, Keane C, et al. Immune evasion via PD-1/PD-L1 on NK-cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood. 2018;131(16):1809-1819.
16. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008;105(39):15016-15021.
17. Taylor JG, Clear A, Calaminici M, Gribben JG. Programmed cell death protein-1 (PD1) expression in the microenvironment of classical Hodgkin lymphoma is similar between favorable and adverse outcome and does not enrich over serial relapses with conventional chemotherapy. Haematologica. 2019;104(1):e42-e44.
18. Nam-Cha SH, Roncador G, Sanchez-Verde L, et al. PD-1, a
follicular T-cell marker useful for recognizing nodular lymphocyte-predominant Hodgkin lymphoma. Am J Surg Pathol. 2008;32(8):1252-1257.
19. Aoki T, Chong LC, Takata K, et al. Single cell transcriptome analysis reveals disease-defining T cell subsets in the tumor microenvironment of classic Hodgkin lymphoma. Cancer Discov. 2020;10(3):406-421.
20. Roemer MGM, Redd RA, Cader FZ, et al. Expression of major histocompatibility complex (MHC) class II, but not MHC class I, predicts outcome in patients with classical Hodgkin lymphoma (cHL) treated with nivolumab (programmed death-1 [PD-1] blockade). Blood. 2017;130(Suppl 1):1450.
21. Machado L, Jarrett R, Morgan S, et al. Expression and function of T cell homing molecules in Hodgkin’s lymphoma. Cancer Immunol Immunother. 2009;58(1):85-94.
22. Littringer K, Moresi C, Rakebrandt N, et al. Common features of regulatory T cell specialization during Th1 responses. Front Immunol. 2018;9:1344.
23. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229(1):114-125.
24. Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015-3029.
25. Chen X, Fosco D, Kline DE, et al. PD-1 regulates extrathymic regulatory T-cell differentiation. Eur J Immunol. 2014;44(9):2603-2616.
26. Zuazo M, Gato-Cañas M, Llorente N, et al. Molecular mechanisms of programmed cell death-1 dependent T cell suppression: relevance for immunotherapy. Ann Transl Med. 2017;5(19):385.
27. Amarnath S, Mangus CW, Wang JCM, et al. The PDL1-PD1 axis converts human Th1 cells Into regulatory T cells. Sci Transl Med. 2011;3(111):111ra120.
28. Kanamori M, Nakatsukasa H, Ito M, Chikuma S, Yoshimura A. Reprogramming of Th1 cells into regulatory T cells through rewiring of the metabolic status. Int Immunol. 2018;30(8):357-373.
29. Stathopoulou C, Gangaplara A, Mallett G, et al. Programmed death-1 receptor signaling downregulates asparaginyl endopeptidase to maintain Foxp3 stability in induced regulatory T cells. Immunity. 2018;49(2):247-263.e7.
30. Tahara M, Kondo Y, Yokosawa M, et al. T-bet regulates differentiation of forkhead box protein 3+ regulatory T cells in programmed cell death-1-deficient mice. Clin Exp Immunol. 2015;179(2):197-209.
31. Tanijiri T, Shimizu T, Uehira K, et al. Hodgkin’s Reed-Sternberg cell line (KM-H2) promotes a bidirectional differentiation of CD4+CD25+Foxp3+ T cells and CD4+ cytotoxic T lymphocytes from CD4+ naive T cells. J Leukoc Biol. 2007;82(3):576-584.
32. Péricart S, Tosolini M, Gravelle P, et al. Profiling immune escape in Hodgkin’s and diffuse large B-cell lymphomas using the transcriptome and immunostaining. Cancers (Basel). 2018;10(11):415.
33. Cader FZ, Schackmann RCJ, Hu X, et al. Mass cytometry of Hodgkin lymphoma reveals a CD4+ regulatory T-cell-rich and exhausted T-effector microenvironment. Blood. 2018;132(8):825-836.
34. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, HashimotoTane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med.
Haematologica | 108 - April 2023 1081 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
2012;209(6):1201-1217.
35. Hui E, Cheung J, Zhu J, et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science. 2017;355(6332):1428-1433.
36. Mizuno R, Sugiura D, Shimizu K, et al. PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front Immunol. 2019;10:630.
37. Cai J, Wang D, Zhang G, Guo X. The role of PD-1/PD-L1 axis In Treg development and function: implications for cancer immunotherapy. Onco Targets Ther. 2019;128437-8445.
38. Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: the role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol. 2018;9:2374.
Haematologica | 108 - April 2023 1082 ARTICLE - PDL1 shapes CHL without inducing exhaustion J.G. Taylor et al.
Diffuse large B-cell lymphoma in octogenarians aged 85 and older can benefit from treatment with curative intent: a report on 129 patients prospectively registered in the Elderly Project of the Fondazione Italiana Linfomi (FIL)
Alessandra Tucci,1 Francesco Merli,2 Alberto Fabbri,3 Luigi Marcheselli,4 Chiara Pagani,1
Benedetta Puccini,5 Dario Marino,6 Manuela Zanni,7 Elsa Pennese,8 Leonardo Flenghi,9
Annalisa Arcari,10 Barbara Botto,11 Melania Celli,12 Caterina Mammi,13 Alessandro Re,1 Giulia Campostrini,1 Agostino Tafuri,14 Vittorio R. Zilioli,15 Emanuele Cencini,3 Roberto Sartori,16
Chiara Bottelli,1 Michele Merli,17 Luigi Petrucci,18 Guido Gini,19 Monica Balzarotti,20 Federica Cavallo,21 Gerardo Musuraca,22 Stefano Luminari,2,23 Giuseppe Rossi1 and Michele Spina24
1Hematology Division, ASST Spedali Civili of Brescia, Brescia; 2Hematology Unit, Azienda USLIRCCS Reggio Emilia, Reggio Emilia; 3Hematology Unit, Azienda Ospedaliera Universitaria
Senese and University of Siena, Siena; 4Fondazione Italiana Linfomi Onlus, Modena; 5Hematology Unit, Careggi University Hospital, Firenze; 6Department of Clinical and Experimental Oncology, Medical Oncology 1, Veneto Institute of Oncology IOV-IRCCS, Padova;
7Hematology Unit, Antonio e Biagio e Cesare Arrigo Hospital Alessandria, Alessandria;
8Lymphoma Unit, Department of Hematology, Spirito Santo Hospital, Lymphoma Diagnosis and Therapy Center, Pescara; 9Hematology Unit, Santa Maria della Misericordia Hospital, Perugia; 10Hematology Unit, Ospedale Guglielmo da Saliceto, Piacenza; 11Hematology Division, Città della Salute e della Scienza Hospital and University of Torino, Torino; 12Hematology Unit, Ospedale degli Infermi, Rimini; 13Gruppo Amici dell’Ematologia GRADE-Onlus Foundation, Reggio Emilia; 14Hematology Division, Sant’Andrea Hospital, Rome; 15Hematology Division, ASST Grande Ospedale Metropolitano Niguarda, Milano; 16Department of Clinical and Experimental Oncology, Onco-hematology Unit, Veneto Institute of Oncology, IOV-IRCCS, Castelfranco Veneto; 17Hematology Division, Ospedale di Circolo e Fondazione Macchi—ASST Sette Laghi, University of Insubria, Varese; 18Hematology Institute, Department of Translational and Precision Medicine “Sapienza”, University of Roma, Roma; 19Hematology Division, Ospedali Riuniti Hospital and University of Ancona, Ancona; 20Medical Oncology and Hematology Department, Humanitas Clinical Research Hospital-IRCCS, Rozzano; 21Division of Hematology, Department of Molecular Biotechnologies and Health Sciences, Citta della Salute e della `Scienza di Torino” Hospital and University of Torino, Torino; 22Hematology Unit, IRCCS— Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) SRL, Meldola; 23Surgical, Medical and Dental Department of Morphological Sciences Related to Transplant, Oncology and Regenerative Medicine, University of Modena and Reggio Emilia, Modena and 24Division of Medical Oncology and Immune-related Tumors, Centro di Riferimento Oncologico di Aviano (CRO) IRCCS, Aviano, Italy
Abstract
Correspondence: A. Tucci alessandra.tucci@asst-spedalicivili.it
Received: May 26, 2022.
Accepted: November 10, 2022.
Early view: November 17, 2022.
https://doi.org/10.3324/haematol.2022.281407
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Octogenarian patients with diffuse large B-cell lymphoma are managed mainly with palliation, but recent improvement in their overall condition makes potentially curative treatment a possibility. Studies have shown that half of selected octogenarians may be cured using reduced-dose anthracycline chemoimmunotherapy. However, patients aged >85 (late octogenarians [LO]) were underrepresented, and selection criteria were poorly defined. We analyzed the clinical characteristics and outcomes of LO enrolled in the FIL Elderly Project in terms of the treatment received (palliative vs curative) and of their simplified geriatric assessment (sGA), then compared them with early octogenarians (EO) aged 8084 and with those aged 65-79 classified as UNFIT or FRAIL according to sGA enrolled in the same study. Of the 1,163 patients, 370 were >80 and 129 LO. Clinical characteristics were similar between LO and EO, but LO more frequently received palliation (50% vs. 23%; P=0.001) and had worse 2-year overall survival (OS) (48% vs. 63%; P=0.001) and 2-year progression-free survival (PFS) (43% vs. 56%; P=0.01). Patients receiving anthracycline did better than patients receiving palliation (P<0.001), without any difference between full or reduced doses. Rituximab within palliation improved outcome (2-yr OS with or without rituximab 42% vs. 22%; P=0.008). Elderly Prognostic Index (EPI) performed better than sGA in
Haematologica | 108 - April 2023 1083
ARTICLE - Non-Hodgkin Lymphoma
identifying different risk categories, and high-risk EPI retained an independent unfavorable effect on OS and PFS, together with treatment without anthracycline. In conclusion, late octogenarians can benefit from a curative approach with reduced-dose anthracycline and from rituximab within palliation. EPI may help in patient selection more than sGA can.
Introduction
Diffuse large B-cell lymphoma (DLCBL) is the most common lymphoid malignancy in Western countries, with a median age of 70 years at presentation.1,2 Given the progressive improvement in life expectancy,3,4 octogenarians will increasingly become more represented in this patient age group.5 Thanks to general improvement in socioeconomic conditions and in supportive care, the overall condition of octogenarian patients has become progressively better, making many of them suitable for treatment approaches with curative intent. Nevertheless, comorbidities and different limitations in daily living activities reduce the capacity of many of them to tolerate full-dose therapy. Thus, the need to tailor treatment choices for this category of patients affected by an aggressive but potentially curable disease is a growing clinical challenge for hematologists. Several studies have recently reported on the characteristics and prognosis of non-Hodgkin lymphoma (NHL) in octogenarian patients.6-9 The use of reduced doses of anthracycline in the rituximab-mini-CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) regimen has been proposed for patients aged >80 years by the French Lymphoma Study Association (LYSA); their favorable results represent a good compromise between efficacy and safety.10 However, in all the above-mentioned studies, the median age was <84 years, and most of the patients were under the age of 85 years. More recently, interest in the cohort of late octogenarians aged >85 has also been growing. Using the Swedish and Danish population-based lymphoma registers, the Nordic Lymphoma Group has recently analyzed the clinical characteristics and outcomes of 2,347 patients aged >85 years diagnosed with lymphoma, 924 of whom (39%) with DLBCL. Administration of active treatment was associated with a 2-year relative survival (RS) of 49% and a 5-year RS of 44% in patients with aggressive histology, but the lack of data regarding toxicity, dose reductions, and frailty assessment precluded more detailed analyses.11 In a retrospective study by the LYSA, active treatment improved the survival of 234 patients over the age of 90 years (113 with DLBCL - 48%). However, the geriatric characteristics of these very old patients were not formally assessed.12
Comprehensive geriatric assessment (CGA) has proven useful in guiding treatment decisions in older patients with DLBCL13 and is now recommended by international scientific societies.14-16 In particular, the identification of patients fit for curative intent has reduced the risk of
undertreating many older patients. However, the choice of therapies in octogenarians, particularly in those aged >85 years, still represents a problem in daily clinical practice. Recently, the Italian Lymphoma Foundation (FIL) conducted the Elderly Project (clinicaltrails gov. Identifier: NCT02364050), a large prospective study on 1,163 older patients with DLBCL. The aim of the study was to evaluate their outcome according to the treatment received and to the geriatric category defined by a simplified geriatric assessment (sGA).17
With the aim of potentially identifying the most appropriate match between patient characteristics and treatment intensity, the present study analyzed all patients enrolled in the Elderly Project study aged >85 years, defined as late octogenarians (LO), describing their clinical and geriatric characteristics as well as their outcome according to their fitness category and to the treatment actually received. It also compared LO with the cohorts of early octogenarians (EO) aged 80-84 years and of patients aged 65-79 years categorized as UNFIT or FRAIL according to sGA also enrolled in the Elderly Project.
Methods
The cohort of patients age >85 years consecutively enrolled in the Elderly Project was considered for this analysis. These patients were defined as LO. Patients aged 80–84 years, defined as EO, were also considered and compared with LO. All patients underwent an sGA (Online Supplementary Table S1), as previously reported.17 Accordingly, age >80 years precluded the classification of patients as FIT. Patients were classified as UNFIT if they had no limitations in activities of daily living (ADL) and instrumental ADL (IADL) and if they had fewer than five grade 2 comorbidities and no grade 3 according to the cumulative illness rating scale-geriatric (CIRS-G); in all the other conditions, patients were classified as FRAIL.17 Clinical characteristics were recorded in a database, and the elderly prognostic index (EPI) score was calculated as previously described.17 In particular, this score divides patients into three risk categories according to the International Prognostic Index, sGA, and hemoglobin levels (Online Supplementary Table S2). Treatment was delivered according to clinician’s decision and was classified as full-dose treatment (FDT) if it contained anthracycline (either liposomal or standard formulation) at a relative dose intensity greater than 70% of the standard dose, reduced-dose
Haematologica | 108 - April 2023 1084 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
treatment (RDT) if it contained between 50% and 70% of standard dose of anthracycline, or palliative treatment (PVT), in the absence of anthracycline in the treatment program. Palliative therapies included rituximab plus either bendamustine (R-B) or cyclophosphamide, vincristine, and prednisone (R-CVP), or other chemotherapy regimens without rituximab, including metronomic regimens, corticosteroids, or radiotherapy alone. UNFIT and FRAIL patients, aged 65–79 years, were also retrieved from the same Elderly Project database and acted as the control group.
Continuous variables are summarized as median with range; categorical variables are summarized as absolute and percentage frequencies. Comparisons between categorical variables and LO and EO groups were performed by Fisher’s exact probability or Chi-square test. All statistical tests were two-sided, and a significant P value was determined as <0.05.
Progression-free survival (PFS) was defined as time from diagnosis to relapse or progression or death from any cause or date of the last clinical visit when the patient was known to be alive. Overall survival (OS) was defined as time from diagnosis to the last visit when the patient was known to be alive or death from any cause. PFS and OS were calculated using the Kaplan-Meier method, and Cox proportional hazards regression models were used to evaluate characteristics associated with PFS and OS. The effect was expressed as hazard ratio (HR) with 95% confidence interval (CI). Association with the PVT approach was estimated by means of logistic regression, and the effect is expressed as odds ratio (OR), with 95% CI. In order to partially remove the selection bias from the estimation, we performed an inverse probability weight (IPW) analysis on Cox proportional hazards regression conducted on OS (see the Online Supplementary Appendix). Discrimination power was evaluated using the Harrell’s c-index, with 95% CI obtained from jackknife estimation. The median follow-up of observation was estimated by means of the reverse Kaplan-Meier method on OS.
Results
Of the 1,163 consecutive and fully evaluable patients enrolled in the Elderly Project from December 2013 to December 2017, 370 (32%) were older than 79 years: 129 (35%) were aged >85 years (LO) and 241 (65%) were aged 80-84 years (EO). All came from 36 Fondazione Italiana Linfomi (FIL) centers throughout Italy.
Clinical characteristics
The characteristics of LO are summarized in Table 1, which also shows the characteristics of EO for comparison. The median age of the 129 LO patients was 87 years (range,
LO: late octogenarians; EO: early octogenarians; sGA: simplified geriatric assessment; IPI: International Prognostic Index; Hb: hemoglobin; EPI: elderly prognostic index; FDT: full-dose treatment; RDT: reduceddose treatment; PVT: palliative treatment; w/o: without; R: rituximab. *28 (22%) >90 years (yr) old; **referred to patients receiving PVT. Percentages are given in brackets and refer to the proportions of evaluable patients for each variable. P value: Fisher’s exact probability of Chi-square test.
85–93 years), 22% were over 90, 40% were male, 62% had Ann Arbor Stage III-IV lymphoma, 16% had B symptoms, 59% belonged to the categories of intermediate-high or high-risk according to the IPI, and 46% were anemic. According to the sGA, 50 patients (39%) were classified as UNFIT and 79 (61%) as FRAIL, and 65% had a high EPI score. Among EO, there was a higher frequency of males (49%), stage III-IV (72%), B symptoms (29%), anemia (48%), sGA UNFIT (48%), and high EPI score (66%). However, differences between EO and LO were not statistically significant, with the exception of B symptoms. The frequency of EO patients with intermediate-high or high-risk IPI score was the same as that of LO patients (59%). Concerning treatment approach, the proportion of LO and EO patients who, based on clinical judgment, received FDT or PVT was significantly different. In LO, only 17 patients (13%) received FDT with curative intent, 47 (36%) received RDT, and 65 (50%) received PVT. In EO, FDT was given to
Factor Age groups LO N=129* EO N=241 P Age in years, median 87 82 sGA, N (%) UNFIT FRAIL 50 (39) 79 (61) 116 (48) 125 (52) 0.100 Sex, N (%) M F 52 (40) 77 (60) 118 (49) 123 (51) 0.126 Stage, N (%) I-II III-IV 48 (38) 79 (62) 67 (28) 174 (72) 0.058 B symptoms, N (%) 21 (16) 71 (29) 0.008 Bulky, N (%) 36 (28) 62 (26) 0.711 IPI score, N (%) 1 2 3-5 18 (16) 28 (25) 65 (59) 30 (14) 59 (27) 131 (59) 0.820 Hb g/dL, N (%) ≥12 <12 67 (54) 57 (46) 120 (52) 112 (48) 0.738 EPI score, N (%) Intermediate High 38 (35) 72 (65) 74 (34) 141 (66) 0.982 Treatment approach, N (%) FDT RDT PVT PVT w/o R 17 (13) 47 (36) 65 (50) 30 (46 ) 79 (33) 107 (44) 55 (23) 15 (27 ) <0.001 0.038
Table 1. Clinical characteristics of late and early octogenarians.
Haematologica | 108 - April 2023 1085 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
Figure
33%, RDT to 44%, and PVT to 23% of patients (P<0.001). The proportion of patients receiving rituximab in the context of PVT was higher in EO (73%) than in LO (54%) (P=0.0387). Among all octogenarians, prediction of palliative treatment by means of logistic regression showed age, female sex, limitation in IADL, and some comorbidities (hypertension, heart failure, and psychiatric illness) as the most relevant variables. In the palliative group, the variables correlated with the probability of being treated without rituximab by means of logistic regression were age, limitation in ADL, and impairment of kidney and muscle function on CIRS-G evaluation (Online Supplementary Tables S3 and S4).
Survival according to age
With a median follow-up of 30 months (range, 1–59 months), 2-year PFS and 2-year OS of LO were 43% and 48%, respectively, significantly lower than PFS (56%; P=0.01) and OS (63%; P=0.001) of EO (Figure 1). The most common cause of death was progressive disease in both age subgroups (67% and 76%, respectively; P=0.628), with no difference according to the treatment received. The cumulative incidence function (CIF) for specific cause of death is reported in the Online Supplementary Figures S1 and S2 Figure 1 also reports the estimated survival of the 157 consecutive patients aged 65-79 years enrolled in the Elderly Project and classified as UNFIT or FRAIL whose 2year PFS (52%) and OS (61%) were not significantly different from those of EO patients (P=0.764 and P=0.563,
age
Panel (A) shows overall survival (OS) and panel (B) shows progression-free survival (PFS) of different age categories: late octogenarians (LO) aged > 85 (85+), early octogenarians (EO) aged 80-84, and patients <80 years old (65-79) resulted UNFIT (UN) or FRAIL (FR) according to the simplified geriatric assessment.
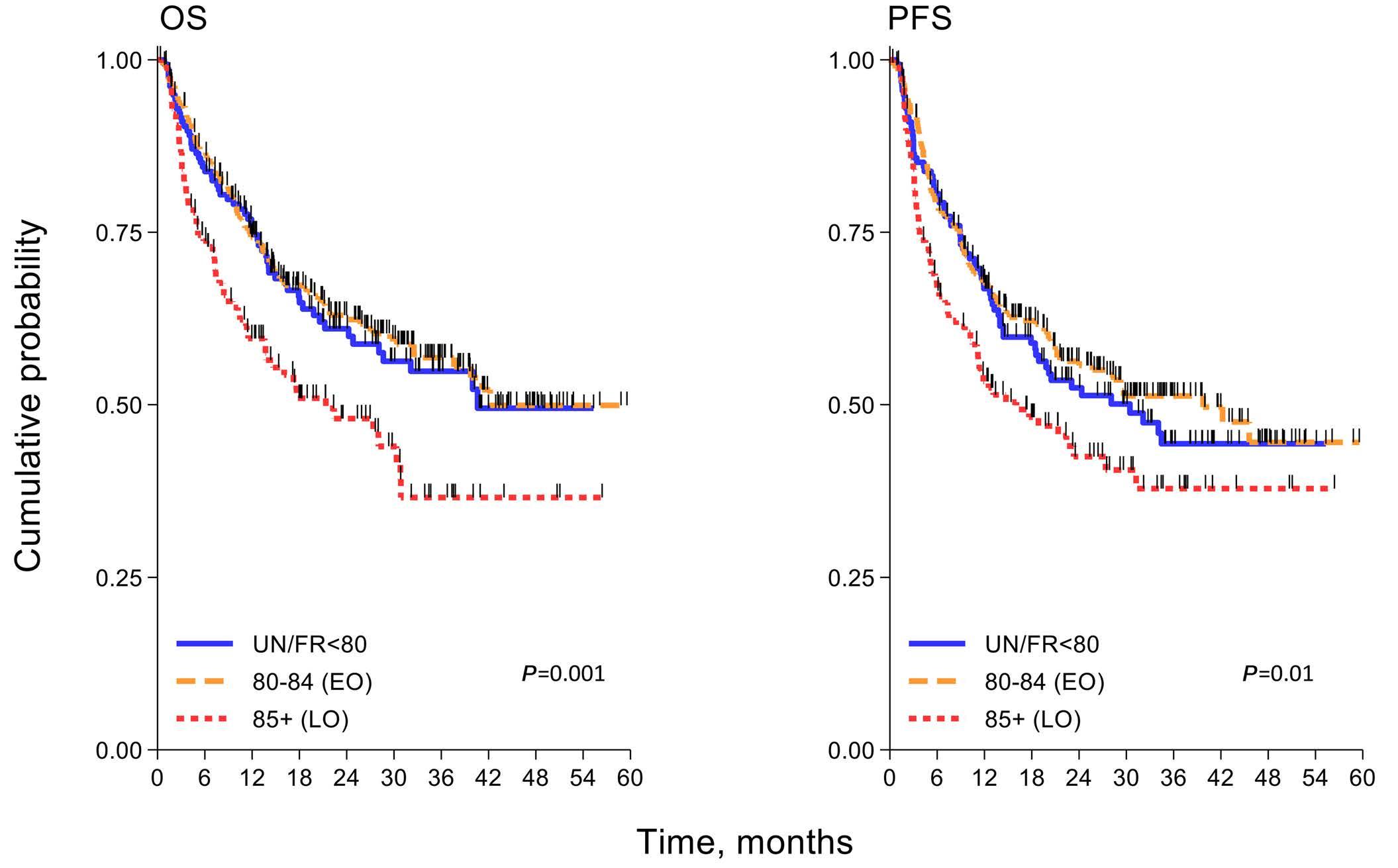
respectively). These two groups of patients had similar clinical characteristics, but they differed in terms of the treatment received (P<0.001): 59% of the patients aged 65–79 years received FDT, 24% received RDT, and 17% received PVT.
Survival according to treatment
Considering the effect of treatment intensity on patient outcome, no difference was observed between full and reduced doses of anthracycline in LO (2-year OS 64% with FDT vs. 67% with RDT) or in EO (2-year OS 70% with FDT vs. 68% with RDT). A palliative approach was associated with significantly worse 2-year survival both in LO (2-year OS 27% with PVT vs. 64% with FDT; P=0.001) and in EO (2year OS 42% with PVT vs. 70% with FDT; P=0.003) (Figure 2). Considering the whole population of octogenarians, HR associated with PVT was 2.42 (95% CI: 1.71-3.42); after weighted Cox regression by IPW, the HR associated with PVT was 1.66 (95% CI: 1.12-2.44; P=0.011). Among palliative approaches, the addition of rituximab improved the outcome in all octogenarians (2-year OS with rituximab 42% vs. 22% without rituximab; P=0.008) (Figure 3). Conversely, only small differences were observed when comparing the different chemotherapy regimens employed in association with rituximab (CVP vs. bendamustine vs. other combinations or rituximab alone) (Table 2).
Survival according to geriatric parameters
The outcome of patients was significantly predicted by their
A B Haematologica | 108 - April 2023 1086 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
1. Kaplan-Meier curves of survival in older patients according to different
groups.
geriatric category: 2-year OS was better in UNFIT versus FRAIL LO patients (66% vs. 37%; P=0.006) and in UNFIT versus FRAIL EO patients (70% vs. 56%; P=0.024). The same difference was observed concerning 2-year PFS (58% vs. 33%, P=0.012 in LO and 59% vs. 53%; P=0.07 in EO). According to the EPI score, 2-year OS of LO was 63% in intermediate versus 41% in high EPI score patients (P=0.025), and 2-year PFS was 57% versus 38% (P=0.015). Even in EO, EPI score maintained its prognostic significance: 2-year OS was 84% versus 52%; P=0.001, in intermediate versus high, respectively, and 2-year PFS was 77% versus 45%; P=0.001 (Figure 4). In a multivariable analysis (MVA) of variables influencing the survival of octogenarians, age >85 years had a modest independent adverse effect on OS but not on PFS. Bulky disease and B symptoms retained a prognostic role in MVA. On the other hand, anthracycline-containing treatment, either at full or reduced doses, had a strong independent effect
on both parameters compared to palliative treatment approaches. Among patients not receiving anthracycline, the inclusion of rituximab in the palliative treatment had an independent favorable effect on OS and PFS (Tables 3 and 4).
Focusing on geriatric parameters, EPI score performed better than sGA in identifying different risk categories having different OS and PFS, and high EPI score retained an independent unfavorable effect on OS and PFS, together with treatment without anthracycline. We performed an internal validation, whose results are reported in the Online Supplementary Table S5.
Discussion
Management of octogenarian DLBCL patients is becoming an increasingly relevant problem in clinical practice. Most
Figure 2.
overall survival in octogenarian patients according to treatment received. Panel (A) shows overall survival (OS) of late octogenarians (LO) and panel (B) shows OS of early octogenarians (EO), both according to different treatment received: full-dose treatment (FDT), reduced-dose treatment (RDT), or palliative treatment (PVT).
OS: overall survival; HR: hazard ratio; CI: confidence interval; R-CVP: rituximab, cyclophosphamide/vincristine/prednisone; RB: rituximab/bendamustine; R: rituximab. *5 R-monotherapy.
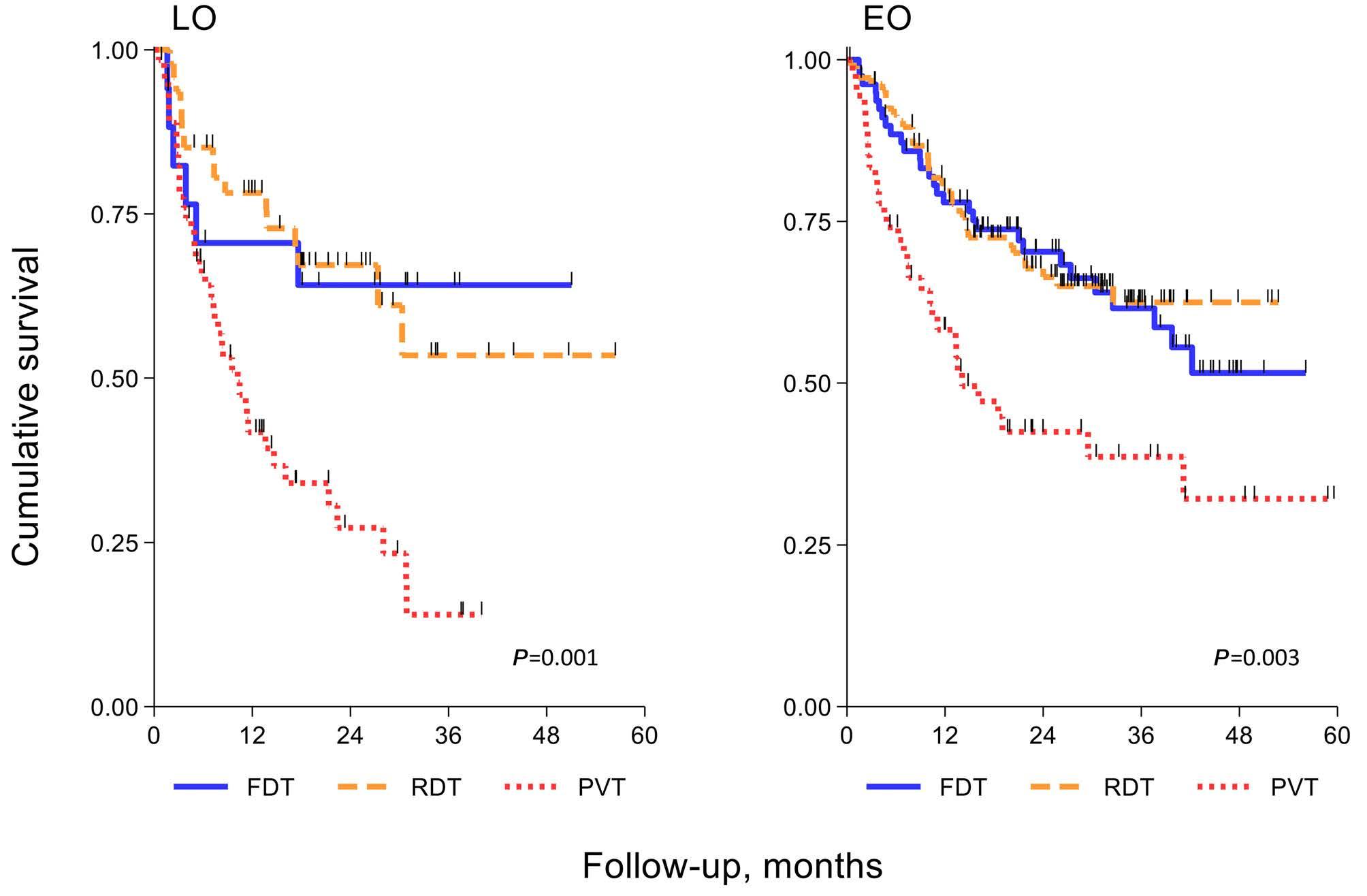
Treatment N (%) 2-year OS % (95% CI) HR (95% CI) P R-CVP 34 (28) 41 (22-59) 1.00 RB 12 (10) 50 (21-74) 0.77 (0.32-1.87) 0.562 R-other* 29 (24) 38 (18-38) 1.14 (0.58-2.25) 0.700 No R 45 (37) 22(9-38) 1.88 (1.05-3.35) 0.034 Total 120 35 (25-44) - -
Kaplan-Meier curves of
Table 2. Different palliative treatments with or without rituximab in patients aged ≥80 years.
A B Haematologica | 108 - April 2023 1087 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
of the studies available have analyzed early octogenarians (age <85 years), whereas LO aged >85 years have largely been underrepresented. The growing interest in these patients derives from the progressive improvement in their general condition, which makes considering treating at least some of them with curative intent a possibility. In a recent study from the Danish and Swedish lymphoma registers, the 2-year survival of patients who received active treatment was significantly better than that in the untreated group (49% vs. 12%, respectively). However, the geriatric characteristics of the two patient groups were not reported.11 The prospective Elderly Project study included all patients with DLBCL aged over 64 years and consecutively seen in 36 FIL centers. In the present study, we performed a detailed analysis of the LO enrolled in the Elderly Project, who accounted for more than 10% of the patients registered and for one-third of the patients over age 80. Their estimated 2-year OS was 48%, and the study confirmed a significantly better prognosis of those patients treated with curative intent with anthracyclinecontaining regimens (64%) than that of those receiving other regimens with palliative purpose (27%). Furthermore, in this latter group, the 2-year OS was significantly improved (37% vs. 15%) by including rituximab in the palliative treatment. The further improvement seen in our patients over the already remarkable survival reported in the Nordic Registry study may be explained by the fact that our patients were treated in experienced centers and cannot therefore be considered fully representative of the entire population of LO. The fact that all the participating centers used the FIL criteria to evaluate patient fitness using sGA may have also contributed.
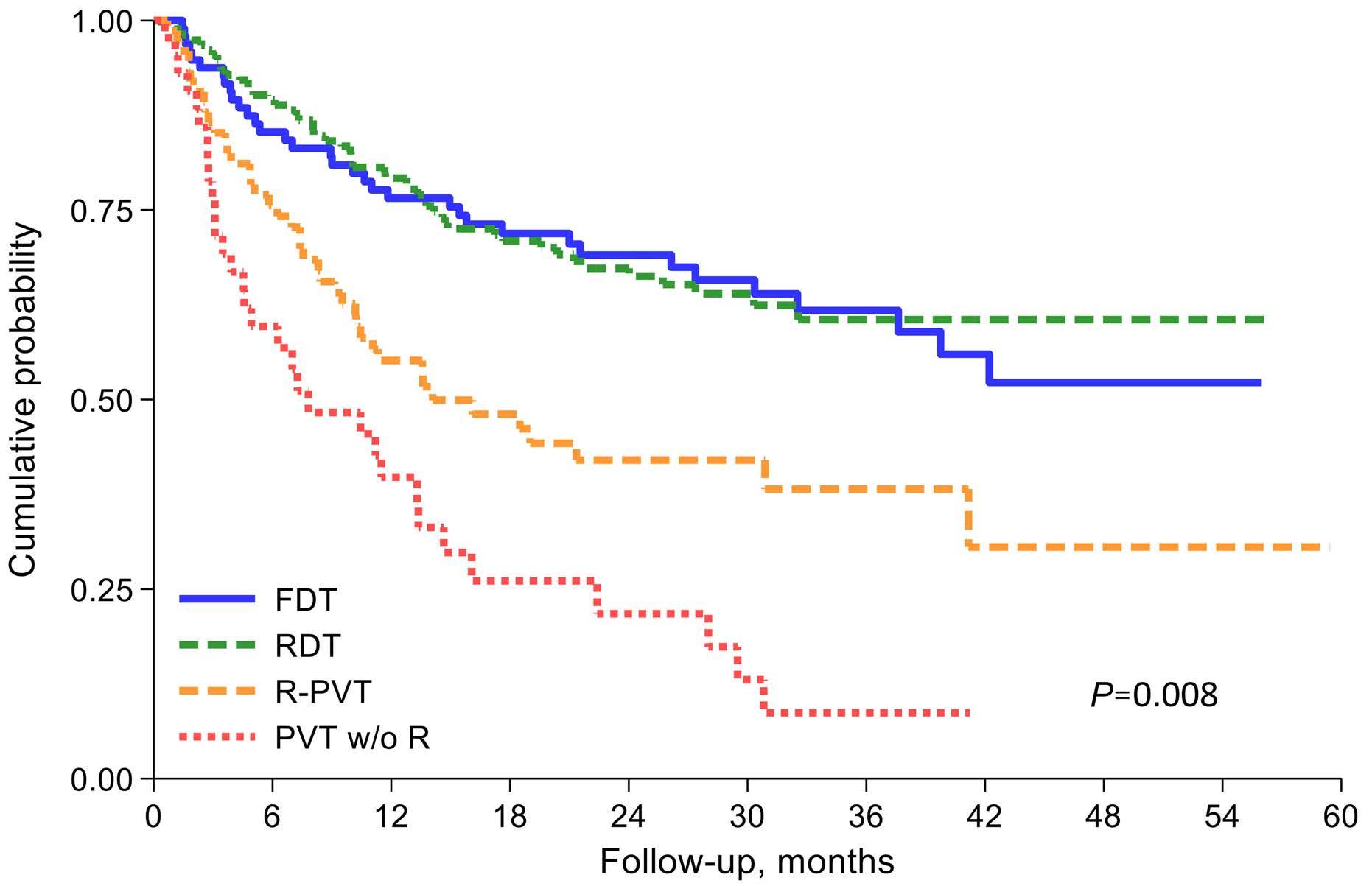
The OS of LO (2-year OS: 48%) was significantly shorter than of that of EO (2-year OS: 63%), despite the fact that the two cohorts had very similar clinical and geriatric characteristics. In addition to age and to a higher frequency of B symptoms among EO, the only difference between EO and LO was the proportion of patients treated either with full or reduced dose of anthracycline, which was 49% in LO and 77% in EO. On the other hand, the dose of anthracycline did not affect outcome, since survival was similar in patients receiving more or less than 70% of the full anthracycline dose. These data strongly support the advisability of using a reduced dose of anthracycline in all octogenarian patients and confirm the results reported by the LYSA group using a R-mini-CHOP regimen.10 The same group has recently reported a randomized study showing that the addition of lenalidomide to R-mini-CHOP did not improve survival of octogenarians, which was 66% at 2 years in both groups.18 By showing a 2-year OS of 67% in LO treated with reduced anthracycline doses, our study confirms this figure and extends it to LO.
In our study, sGA made it possible to subdivide octogenarian patients into one of two groups: those without any limitations in ADL and IADL and with limited comorbidities (UNFIT), or those with limitations and major comorbidities (FRAIL). These two groups had significantly different survival among both LO and EO, confirming the validity of this geriatric tool. In addition, the recently developed EPI score, which refines the sGA by adding two important clinical parameters, the International Prognostic Index and anemia, also proved able to predict both 2-year OS and 2-year PFS, confirming that the EPI score maintains its validity in these groups of very old patients as well.
Haematologica | 108 - April 2023 1088 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
Figure 3. Overall survival of the whole octogenarian population according to different treatment received. Full-dose treatment (FDT), reduced-dose treatment (RDT), palliative treatment with rituximab (R-PVT) or without rituximab (PVT w/o R).
Indeed, on multivariable analysis, the EPI score and the use of an anthracycline-containing treatment were the only two variables retaining an independent effect for predicting both OS and PFS in octogenarians, while age lost its significance, and the sGA performed less efficiently. In particular, EPI score identified a high-risk group whose needs are clearly unmet and who should be the subject of future investigations based on target molecules rather than standard chemotherapy regimens. Furthermore, these data suggest that in very advancedage patients such as LO, the geriatric criteria commonly used to define patient fitness in older DLBCL patients may not be completely adequate and may need further refinements by exploring different geriatric domains. The most frequent comorbidities observed in patients undergoing palliative treatments may suggest carefully evaluating kidney impairment, sarcopenia, and/or more subtle cognitive functions.
Rosko et al. recently underlined the need to carefully evaluate disease and patient-specific characteristics to define specific guidelines that incorporate GA measures.
This would guide treatment choices better among the increasingly wide range of options and supportive care available for older patients.19
Furthermore, as fitness and frailty are dynamic factors that can improve or deteriorate during the course of a disease and its treatment, repeated geriatric assessments are encouraged, especially in this very old population.20
An important observation is that the inclusion of rituximab in palliative regimens or its use as a single agent significantly improved survival compared with rituximabfree palliative regimens, regardless of age and patient fitness. The important gain in life span and the relatively good tolerability of this drug makes its use suitable when anthracyclines are not indicated, while the choice of the anthracycline-free chemotherapy regimen seems less important. Immunotherapy is an effective and well-tolerated approach that allows dose-intensity modulation of chemotherapy regimens. A recent study showed promising results obtained with a bispecific antibody used as a monotherapy in unfit patients with previously
Figure 4. Kaplan-Meier curves of overall survival and progression-free survival in octogenarian patients according to simplified geriatric assessment and Elderly Prognostic Index. Panel (A) shows (OS) and panel (B) shows progression-free survival (PFS) in late octogenarians (LO) and early octogenarians (EO) according to geriatric category (UNFIT and FRAIL); panel (C) shows OS and panel (D) PFS in LO and EO according to EPI (Elderly Prognostic Index) score (Intermediate and High).
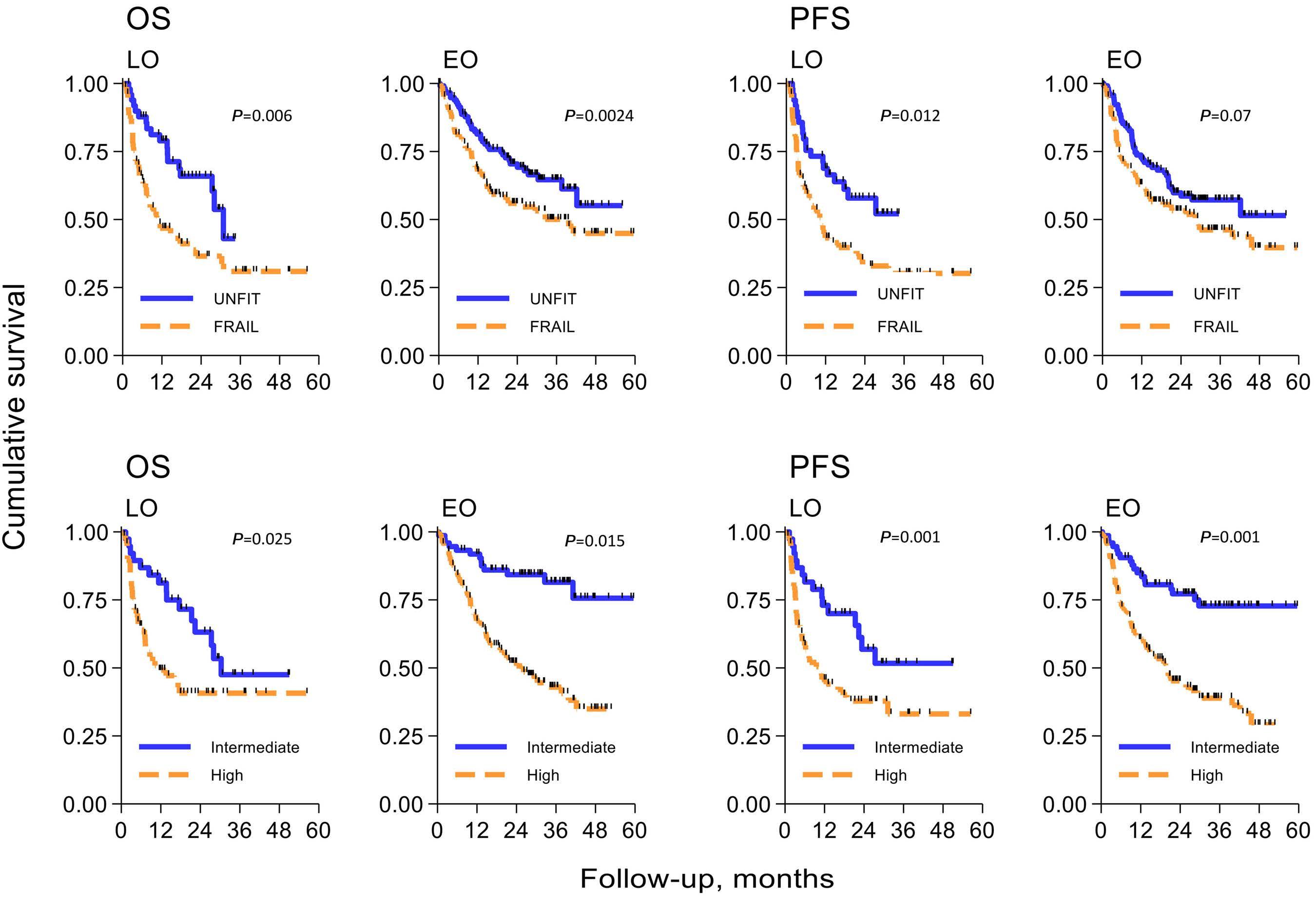
A B C D Haematologica | 108 - April 2023 1089 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
untreated aggressive B-cell lymphoma,21 opening the way to a chemotherapy-free approach in this very old population. A careful use of supportive care is another important way to improve outcome and quality of life in this group of patients.16,19,20
Although the reported results come from a large number of octogenarians consecutively treated in specialized centers, this study has some limitations. Patients in very compromised clinical conditions or with very advanced disease may be lost as they are often not referred to secondlevel centers. Furthermore, details on patient follow-up, including reports on exact treatment modalities, treatment-related toxicities, and/or the appearance of new comorbidities may have been incomplete. Moreover, the predictive capability of sGA and of EPI need to be formally
tested using an external validation dataset. Overall, the impact of the type of treatment on survival in the presence of many confounding factors is difficult to determine; only a randomized study can answer this question. In conclusion, while chronological age still has a negative impact on older patients’ survival, this study highlights the risk of undertreating patients solely because of their very advanced age. We demonstrate that late octogenarians can achieve long-term survival; a curative intent approach, with reduced anthracycline dose, is the best choice in this group of patients, and the choice may be effectively guided by considering the EPI score. The inclusion of rituximab in palliative treatment programs should always be considered, even in frail and very old patients.
EPI: Elderly Prognostic Index; HR: hazard ratio; CI: confidence interval; R-CHOP/COMP: rituximab-cyclophosphamide, doxorubicin hydrochloride, vincristine, prednisone/ cyclophosphamide, liposomal doxorubicin, vincristine, prednisone; sGA: simplified geriatric assessment; R: rituximab. Comparison c-Harrel: model with EPI – model with sGA =0.031 (95% CI: 0.001-0.060); P=0.041. The discriminant index Harrell's C refers to Cox model.
Table
proportional hazards regression on progression-free survival with Elderly Prognostic Index or simplified geriatric assessment in patients aged ≥80 years.
EPI: Elderly Prognostic Index; HR: hazard ratio; CI: confidence interval; R-CHOP/COMP: rituximab-cyclophosphamide, doxorubicin hydrochloride, vincristine, prednisone/ cyclophosphamide, liposomal doxorubicin, vincristine, prednisone; w/o: without; R: rituximab; sGI: simplified geriatric assessment. Comparison c-Harrel: model with EPI – model with sGA =0.031 (95% CI: 0.004-0.059), P=0.024. The discriminant index Harrell's C refers to Cox model.
Overall survival With EPI N=322 Variable HR (95% CI) P Age 85+ years 1.59 (1.11-2.30) 0.012 EPI Intermediate High 1.00 2.43 (1.59-3.74) <0.001 B symptoms 1.58 (1.09-2.29) 0.015 Bulky 1.59 (1.10-2.29) 0.013 Treatment R-CHOP/COMP/R-mini-CHOP Others with R Others w/o R Others w/o R vs. with R 1.00 1.99 (1.37-2.88) 3.92 (2.36-6.52) 1.97 (1.18-3.29) <0.001 <0.001 0.009 c-Harrell 0.717 (0.673-0.760) Overall survival With sGA N=322 Variable HR (95% CI) P Age 85+ years 1.52 (1.06-2.19) 0.023 sGA UNFIT FRAIL 1.00 1.16 (0.81-1.67) 0.416 B symptoms 1.83 (1.27-2.64) 0.001 Bulky 1.79 (1.24-2.58) 0.002 Treatment R-CHOP/COMP/R-miniCHOP Others with R Others w/o R Others w/o R vs. with R 1.00 2.06 (1.41-3.02) 3.99 (2.36-6.74) 1.94 (1.16-3.24) <0.001 <0.001 0.012 c-Harrel 0.686 (0.638-0.734) Progression-free survival With EPI N=322 Variable HR (95% CI) P Age 85+ years 1.36 (0.96-1.93) 0.084 EPI Intermediate High 1.00 2.21 (1.49-3.29) <0.001 B symptoms 1.62 (1.14-2.30) 0.008 Bulky 1.67 (1.19-2.35) 0.003 Treatment R-CHOP/COMP/R-mini-CHOP Others with R Others w/o R Others w/o R vs. with R 1.00 1.81 (1.27-2.57) 3.57 (2.17-5.89) 1.97 (1.19-3.27) 0.001 <0.001 0.007 c-Harrell 0.696 (0.653-0.739) Progression-free survival With sGA N=322 Variable HR (95% CI) P Age 85+ years 1.32 (0.93-1.87) 0.115 sGA UNFIT FRAIL 1.00 1.05 (0.75-1.48) 0.761 B symptoms 1.83 (1.29-2.60) 0.001 Bulky 1.87 (1.33-2.63) <0.001 Treatment R-CHOP/COMP/R-mini-CHOP Others with R Others w/o R Others w/o R vs. with R 1.00 1.91 (1.33-2.74) 3.84 (2.29-6.44) 2.01 (1.21-3.34) <0.001 <0.001 0.007
0.664 (0.618-0.711)
c-Harrel
Table 3. Multivariable Cox proportional hazards regression on overall survival with Elderly Prognostic Index or simplified geriatric assessment in patients aged ≥80 years.
4. Multivariable Cox
Haematologica | 108 - April 2023 1090 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
Disclosures
AT has a consultancy or advisory role at Janssen, Gentili, MSD, Takeda, Kiowa kyrin and Sanophi. FM has a consultancy or advisory role at Janssen, Gilead Sciences, MSD, Takeda and Roche; he has received travel and accommodation expenses from Janssen, Gilead Sciences, EUSA Pharma, Celgene, Roche and Takeda. LF has received travel and accommodation expenses from Roche and Janssen. AA has a consultancy or advisory role at Janssen-Cilag and has received travel and accommodation expenses from JanssenCilag and Takeda; AF has a consultancy and advisory role at Roche, Takeda, Incyte, Servier and Kyowa Kirin. FC is on the advisory board of Roche, he has received speaker fees from Servier and Gilead; he consults for Incyte and has an advisory role at Roche, Incyte and Janssen. GM has a consultancy and advisory role at Janssen Oncology and Servier. SL has a consultancy and advisory role at Roche, Gilead Sciences and Celegne; he has received travel and accommodation expenses
References
1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasm. Blood. 2016;127(20):2375-2390.
2. Smith SD, Chen A, Spurgeon S, et al. Diffuse large B-cell lymphoma in adults aged 75 years and older: a single institution analysis of cause-specific survival and prognostic factors. Ther Adv Hematol. 2013;4(6):349-353.
3. World Population Ageing 2019: Highlights (ST/ESA/SER.A/430)
4. Boccardi V. Population ageing: the need for a care revolution in a World 2.0. Geriatrics. 2019,4(3):47.
5. Bron D, Ades L, Fulop T, Goede V, Stauder R. Aging and blood disorders: new perspectives, new challenges. Haematologica. 2015;100(4):415-417.
6. Chihara D, Westin JR, Oki Y, et al. Management strategies and outcomes for very elderly patients with diffuse large B-cell lymphoma. Cancer. 2016;122(20):3145-3151.
7. Lee S, Fujita K, Negoro E, et al. Impact of relative dose intensity of standard regimens on survival in elderly patients aged 80 years and older with diffuse large B-cell lymphoma. Haematologica 2020;105(8):e415-e418.
8. Nabhan C, Smith SM, Helenowski I, et al. Analysis of very elderly (>/=80 years) non-hodgkin lymphoma: impact of functional status and co-morbidities on outcome. Br J Haematol. 2012;156(2):196204.
9. Thieblemont C, Grossoeuvre A, Houot R, et al. Non-Hodgkin’s lymphoma in very elderly patients over 80 years. A descriptive analysis of clinical presentation and outcome. Ann Oncol. 2008;19(4):774-779.
10. Peyrade F, Jardin F, Thieblemont C, et al. Groupe d'Etude des Lymphomes de l'Adulte (GELA) investigators. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2011;12(5):460-468.
11. Wasterlid T, Gradel KO, Eloranta S, et al. Clinical characteristics and outcomes among 2347 patients aged ≥85 years with major lymphoma subtypes: a Nordic Lymphoma Group study. Br J Haematol. 2021;192(3):551-559.
12. Trebouet A, Marchand T, Lemal R, et al. Lymphoma occurring in
from Janssen and Celgene. MS is employed by Bristol-Myers Squibb/Medarex and Sanofi; he has a consultancy or advisory role at Gilead Sciences and Incyte. All other authors have no conflicts of interest to disclose.
Contributions
AT, FM, LM, SL, GR, and MS conceived and designed the study; AT, FM, AF, CP, BP, DM, MZ, EP, LF, AA, BB, MC, AR, AT, VRZ, EC, RS, CB, MM, LP, GG, MB, FC, GM, SL, GR, and MS provided study materials and patients; AT, FM, LM, SL, GR, and MS performed data analysis and interpretation; AT and GR wrote the manuscript. All authors performed data collection and assembly and approved the final version of the manuscript.
Data-sharing statement
The authors will make their original data available to future researchers upon request directed to the corresponding author.
patients over 90 years of age: characteristics, outcomes, and prognostic factors. A retrospective analysis of 234 cases from the LYSA. Ann Oncol. 2013;24(10):2612-2618.
13. Wildiers H, Heeren P, Puts M, et al. International Society of Geriatric Oncology consensus on geriatric assessment in older patients with cancer. J Clin Oncol. 2014;32(24):2595-2603.
14. Morrison VA, Hamlin P, Soubeyran P, et al. Diffuse large B-cell lymphoma in the elderly: Impact of prognosis, comorbidities, geriatric assessment, and supportive care on clinical practice. An International Society of Geriatric Oncology (SIOG) expert position paper. J Geriatr Oncol. 2014;6(2):141-152.
15. Buske C, Hutchings M, Ladetto M, et al. ESMO consensus conference on malignant lymphoma: general perspectives and recommendations for the clinical management of the elderly patient with malignant lymphoma. Ann Oncol. 2018;29(3):544-562.
16. Akhtar OS, Huang LW, Tsang M, et al. Geriatric assessment in older adults with non-Hodgkin lymphoma: a Young International Society of Geriatric Oncology (YSIOG) review paper. J Geriatr Oncol. 2022;13(5):572-581.
17. Merli F, Luminari S, Tucci A, et al. Simplified geriatric assessment in older patients with diffuse large B-cell lymphoma: the prospective elderly project of the Fondazione Italiana Linfomi. J Clin Oncol. 2021;39(11):1214-1222.
18. Oberic L, Peyrade F, Puyade M, et al. Subcutaneous rituximabminiCHOP compared with subcutaneous rituximab-miniCHOP plus lenalidomide in diffuse large B-cell lymphoma for patients age 80 years or older. J Clin Oncol. 2021;39(11):1203-1213.
19. Rosko AE, Cordoba R, Abel G, et al. Advances in management for older adults with hematologic malignancies. J Clin Oncol. 2021;39(19):2102-2114.
20. Cordoba R, Eyre TA, Klepin HD, Wildes TM, Goede V. A comprehensive approach to therapy of haematological malignancies in older patients. Lancet Haematol. 2021;8(11):e840-e852.
21. Olszewski AJ, Avigdor A, Babu S, et al. Single-agent mosunetuzumab is a promising safe and efficacious chemotherapy-free regimen for elderly/unfit patients with previously untreated diffuse large B cell lymphoma. Blood. 2020;136(Suppl 1):S43-45.
Haematologica | 108 - April 2023 1091 ARTICLE - Late octogenarian DLBCL and curative approach A. Tucci et al.
Overexpression of the key metabolic protein CPT1A defines mantle cell lymphoma patients with poor response to standard high-dose chemotherapy independent
of MIPI and complement established highrisk factors
Correspondence: S. EK
sara.ek@immun.lth.se
Received: May 24, 2022.
1Department of Immunotechnology, Lund University, Lund, Sweden; 2Department of Hematology, Rigshospitalet, Copenhagen, Denmark; 3Department of Hematology, Helsinki University Hospital, Helsinki, Finland; 4Department of Oncology, Oslo University Hospital, Oslo, Norway; 5Department of Clinical Sciences, Oncology and Pathology, Lund University, Lund, Sweden and 6Department of Oncology, Lund University Hospital, Lund, Sweden
Abstract
Accepted: November 28, 2022.
Early view: December 15, 2022.
https://doi.org/10.3324/haematol.2022.281420
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The variable outcome to standard immunochemotherapy for mantle cell lymphoma (MCL) patients is a clinical challenge. Established risk factors, including high MCL International Prognostic Index (MIPI), high proliferation (Ki-67), non-classic (blastoid/pleomorphic) morphology, and mutated TP53, only partly identify patients in need of alternative treatment. Deepened understanding of biological factors that influence time to progression and relapse would allow for an improved stratification, and identification of novel targets for high-risk patients. We performed gene expression analyses to identify pathways and genes associated with outcome in a cohort of homogeneously treated patients. In addition to deregulated proliferation, we show that thermogenesis, fatty acid degradation and oxidative phosphorylation are altered in patients with poor survival, and that high expression of carnitine palmitoyltransferase 1A (CPT1A), an enzyme involved in fatty acid degradation, can specifically identify high-risk patients independent of the established high-risk factors. We suggest that complementary investigations of metabolism may increase the accuracy of patient stratification and that immunohistochemistry-based assessment of CPT1A can contribute to defining high-risk MCL.
Introduction
Mantle cell lymphoma (MCL) is a mature B-cell lymphoma with heterogeneous presentation and aggressive evolution upon progression.1 The primary pathogenic event is marked by the genetic translocation t(11;14)(q13,q32), which results in upregulation of CCND1 with constitutive overexpression of Cyclin D1 and deregulation of the cell cycle at the S1-G transition.2 Albeit essential for MCL development, Cyclin D1 has a limited oncogenic effect and secondary mechanisms are required to drive full malignant transformation. Genomic investigations have identified several genetic alterations, including ATM, TP53, BIRC3, NOTCH1, CCND1, KMT2D 3,4 The genomic landscape
of MCL is heterogeneous, which complicates the design of novel treatment strategies. In a recent study, we showed that only 21% of tumors harbored actionable mutations.4
The age and fitness of the patient are still the main factors for selecting frontline therapy, with young (≤65 years) and fit patients receiving chemoimmunotherapy, including high-dose cytarabine, rituximab and autologous stem cell transplantation (ASCT). However, response to treatment is variable, and more tailored regimens and companion biomarkers are required for improved survival of high-risk patients. Current clinical trials focus on the use of combinations of BTK inhibitors,5 CD20 antibodies, venetoclax and/or chimeric antigen receptor T-cell (CAR T) therapy
Anna Sandström Gerdtsson,1 Joana de Matos Rodrigues,1 Christian Winther Eskelund,2 Simon Husby,2 Kirsten Grønbæk,2 Riikka Räty,3 Arne Kolstad,4 Christian Geisler,2 Anna Porwit,5 Mats Jerkeman6 and Sara Ek1
Haematologica | 108 - April 2023 1092 ARTICLE - Non-Hodgkin Lymphoma
for high-risk or refractory patients.6 Therapeutic strategies complementary to these would enhance the possibilities to further adapt treatment both at diagnosis and in the relapsed setting based on the biological activity of the tumor.
The MCL International Prognostic Index (MIPI), is useful for prognostication but is currently not in clinical use to stratify patients. The known biological risk factors of MCL are frequently overlapping and include proliferation index assessed by Ki-67 staining, blastoid/pleomorphic (from here referred to as non-classic) morphology and TP53 mutations. It has been proposed that a combined strategy of assessing MIPI, proliferation and TP53 mutational status7 would allow clinicians to keep low-risk patients on standard treatment while identifying high-risk patients in need of alternative, possibly chemotherapy-free regimens. However, among low-risk patients, the response to standard high-dose chemotherapy is variable, indicating that additionally unknown biological factors contribute to a short time to progression (TTP).
The main aim of this study was to identify complementary strategies to find patients with low probability to respond to intensive chemoimmunotherapy, and to determine potential novel targets that are relevant for those patients. We found that thermogenesis, fatty acid degradation and oxidative phosphorylation are deregulated in patients with poor response to the Nordic MCL 2 and 3 (N-MCL2/3) clinical trials protocol which includes high-dose cytarabine. In particular, the overexpression of carnitine palmitoyltransferase 1A (CPT1A), a key factor for lipid metabolism, was validated on the protein level as negatively associated with TTP and overall survival (OS), both as a continuous and dichotomized variable. CPT1A overexpression was shown to be independent of established risk factors, such as proliferation and morphology. The association between CPT1A and OS was validated in an independent population-based patient cohort. We suggest that an improved risk stratification of MCL patients can be achieved through assessment of CPT1A at diagnosis.
Methods
Patient samples
Samples for gene expression (GEX) (n=70) and immunohistochemistry (IHC) (n=45) analysis were selected from the N-MCL2/3 clinical trial cohort (clinicaltrials gov. Identifiers: NCT00514475 and ISRCTN87866680).8,9 Samples had been previously collected from 2000 to 2006 (N-MCL2) and from 2005 to 2009 (N-MCL3), and patients had been treated with first-line R-CHOP (rituximab, cyclophosphamide, doxorubicin vincristine, and prednisone), high-dose cytarabine cycles and ASCT. Mutational status of ATM and
TP53 had been previously collected using custom design multiplex Ion Ampliseq.10 A population-based MCL cohort (n=135) from the biobank of lymphomas in Southern Sweden (BLISS) including patients diagnosed from 2000 to 2014, was used to validate the expression of individual proteins. The studies were approved by the Ethical Review Boards in Lund (Dnr 2011/593, BLISS; Dnr 2006/242, NMCL2/3) and Uppsala (Dnr 2009/428, N-MCL2/3). An overview of the cohort used is shown in Figure 1.
Transcriptome analysis
RNA was isolated using the RecoverAll total nucleic acid isolation kit (Ambion, Carlsbad, CA). RNA quantity (optical density [OD] 260 nm) and quality (260 nm/280 nm) were assessed using ND-1000 spectrophotometry (Thermo Scientific, Wilmington, DE). GEX was measured using GeneChip Human Gene ST 1.0 whole transcript arrays (Applied Biosystems, MA, USA), measuring >25 probes per transcript. R (version 4.1.1) and R studio (version 2021.09.0) were used for data analysis. The maEndtoEnd workflow (version 3.13)11 was used to identify differentially expressed genes (DEG) for TP53-mutated versus wild-type (wt), Ki67 high versus low, non-classic versus classic morphology, ATM-mutated versus wt, excluding TP53-mutated samples (n=10) from all but the first comparison. Survival-associated genes were identified by fitting a Cox regression model independently for each gene. PathfindR (version 1.6.2)12 was used for pathway analysis based on KEGG gene lists, Biogrid protein-protein interaction networks, and the number of iterations set to 5, with the following gene lists input criteria: log rank<0.01 (survival); P<0.05 (TP53 status); P<0.001 (morphology); P<0.0001 (proliferation).
Protein analysis
Tissue microarray blocks were assembled as previously described.13 For IHC, slides were pretreated using the DAKO PT link system (DAKO; Glostrup, Copenhagen, Denmark) and stained in an Autostainer Plus (DAKO; Glostrup, Copenhagen, Denmark) with the following antibodies: anti-FEN1 (1:1,500, ab109132 Abcam, Cambridge, UK), antiCPT1A (1:1,800, ab128568, Abcam, Cambridge, UK) and anti-WEE1 (1:200, sc-5285, Santa Cruz, Texas, USA). Slides were scanned at X20 magnification using a NanoZoomer 60 (Hamamatsu Photonics, Shizuoka, Japan) and evaluated with HALO® (Indica Labs, New Mexico, USA). Cox proportional hazard (PH) models were used to estimate the hazard ratio (HR) for CPT1A, FEN and WEE1, using date of diagnosis/treatment start as starting point, and date of death of any cause (BLISS and N-MCL2/3) and date of documented relapse or progression (N-MCL2/3) as endpoints for OS and TTP, respectively. The PH assumption was tested using the Therneau and Grambsch test of the Schoenfeld residuals. Maxstat14 was used to define cutoffs, log-rank statistics to evaluate differences between
Haematologica | 108 - April 2023 1093 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
survival curves, and Wilcoxon signed-rank test to compare differences between groups.
Results
Cohorts used for exploration and validation
Diagnostic biopsies (n=70) from the N-MCL2/3 clinical trials were used for whole-transcript expression analysis to identify genes and pathways associated with poor outcome (TTP and OS, respectively) and established risk factors (Table 1). Biopsies available as TMA from N-MCL2/3 (n=45) and BLISS (n=135) were used to validate expression of individual proteins, using IHC followed by digital scoring. The N-MCL2/3 inclusion criteria were age ≤65 years, Ann-Arbor stage II-IV and no previous cancer treatment. Patients were homogeneously treated, including immunochemotherapy. In contrast, the population-based BLISS cohort included MCL patients treated with different therapies, the most frequent being R-Bendamustine. Information on Ki-67, morphology, TP53 mutation status,10 p53 immunoreactivity15 was available for most samples. For Ki-67 and p53, a 30% cut-off was used to define high and
Figure 1. A flow diagram summarizing the cohorts used. RNA samples from the Nordic MCL2 and MCL3 trials were used for gene expression analyses. Partly overlapping samples from the same clinical trial material was available for validation of corresponding proteins. An independent population-based cohort, biobank of lymphomas in Southern Sweden (BLISS), was used to validate CPT1A, WEE1 and FEN1 protein expression.
low proliferation and/or p53. MIPI was available for most patients included in the N-MCL2/3 but only for a minority of patients in the population-based BLISS cohort. Patients were divided into MIPI low, intermediate, and high, according to the established scoring system.16 The majority (67-77%) of patients were male, reflecting the male dominance of MCL prevalence. Due to differences in inclusion criteria and treatments, N-MCL2/3 patients had lower risk (62% MIPI low) than BLISS (44% MIPI high). The median OS in the BLISS cohort was only 4.2 years, compared to 11 and 12.4 years in the N-MCL2/3 GEX and IHC cohorts, respectively. The discovery cohort had been selected from the N-MCL2/3 material to represent a wide survival range and to reflect variable MCL risk factors. The samples used for GEX analysis thus had higher frequencies of non-classic morphology and high proliferation cases, compared to the full N-MCL2/3 cohort (n=319)8-10. While OS was similar to the full N-MCL2/3 cohort (11 years vs. 12.5 years), TTP was shorter in the selected cohort (5.3 years vs. 8.2 years).
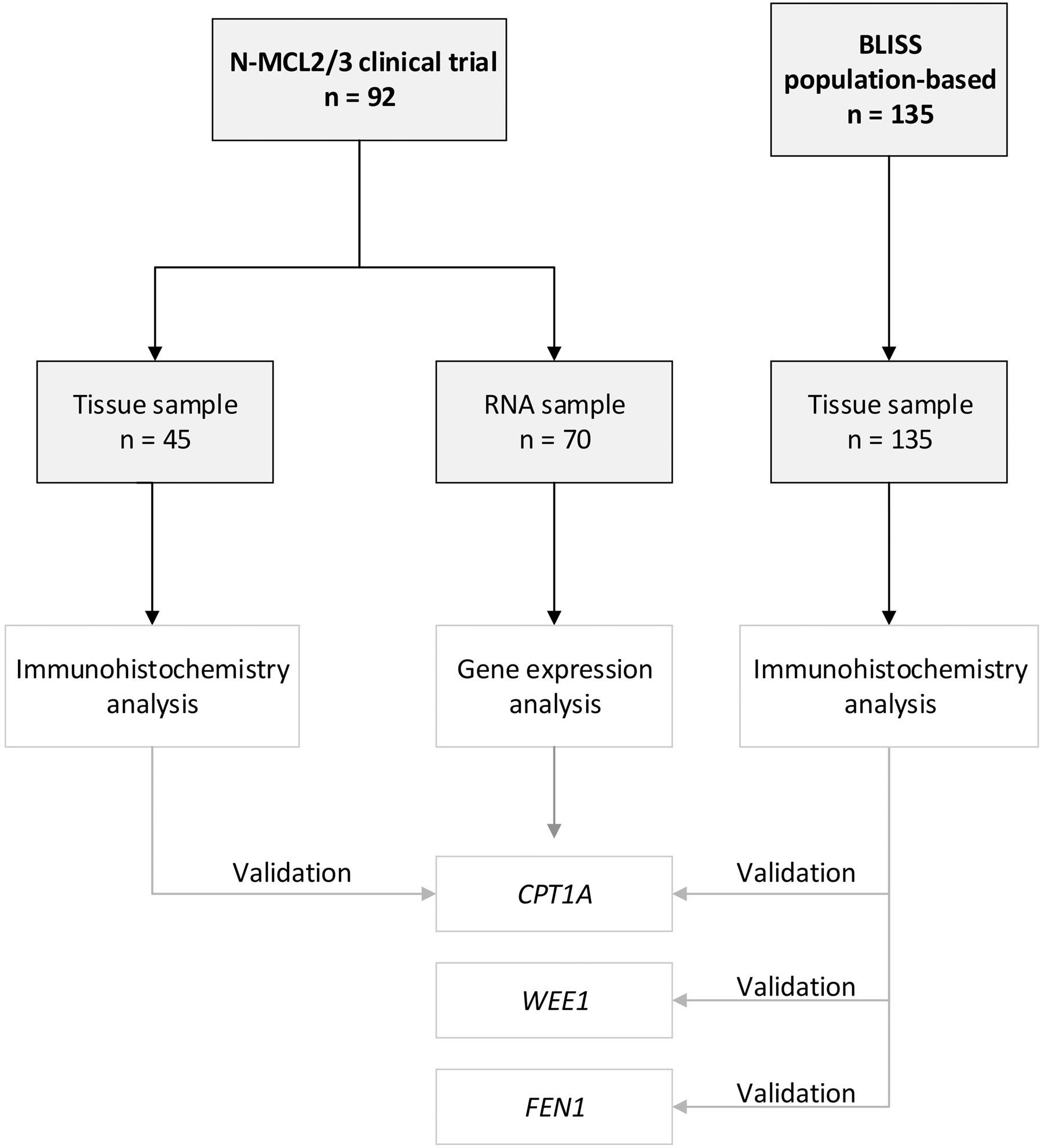
Pathways of poor prognosis in mantle cell lymphoma
Genes associated with inferior TTP and OS were identified using Cox regression. Upregulated cell cycle (P=6.6x10-11)
Haematologica | 108 - April 2023 1094 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Table 1. Clinicopathological characteristics of the different cohorts used in this study.
Percentages might not add to 100% due to rounding. IHC: immunohistochemistry; MIPI: mantle cell lymphoma International Prognostic Index; N-MCL2/3: Nordic MCL group clinical trials 2 and 3. Corresponding values for the full (n=319) N-MCL2/3 cohort have been reported previously.8-10
and related pathways were dominating among genes associated with short TTP, followed by DNA repair pathways, thermogenesis and fatty acid degradation (Figure 2A-C). Inferior OS was primarily associated with oxidative phosphorylation (P=6.8x10-08) and related pathways (Figure 3AC). Proliferation has repeatedly been demonstrated to be associated to poor MCL prognosis and adding proliferation to the MIPI score has been shown to improve the prognostic value.17-19 Concordantly, we identified strong upregulation of cell cycle-regulating genes including CCNE1, CDC45, CDC25C, WEE1, TTK, MAD2L2, CDC25A, SKP, CHEK2, and MCM4 in shorter TTP and/or OS. However, targeting proliferation by cytostatic drugs has proven insufficient to cure MCL, indicating that other mechanisms are in place and should be identified.
Mitochondrial fatty acid transportation and metabolism are associated with inferior survival
Investigations beyond cell cycle and DNA repair mechanisms identified upregulation of metabolic pathways of thermogenesis, oxidative phosphorylation and fatty acid degradation as significantly associated with shorter TTP
and OS. Upregulated genes were involved in fatty acid transportation into mitochondria, fatty acid degradation, and mitochondrial respiration, including CPT1A, SLC25A20, ACAA1, ACAA2, NADH dehydrogenases, Cytochrome c oxidases, and PPA1. Of interest, CPT1A and SLC25A20 (CACT) are involved in the carnitine shuttle and import of longchain fatty acids across the outer and inner mitochondrial membrane, respectively, while ACAA2 catalyses the last step of the mitochondrial fatty acid b -oxidation (Figure 4A). Upregulation of transcripts involved in the downstream mitochondrial respiratory chain, including NDUFB9, NDUFS4, COX10, COX6B1, COX6A1, COX5A, COA6, and PPA1, were also significantly correlated with poorer outcome.
CPT1A protein expression is a marker of poor prognosis
The correlation of CPT1A expression and poor prognosis was verified by IHC staining (Figure 4B). The N-MCL2/3 samples used for GEX (n=70) and IHC (n=45) analyses were overlapping by 23 patients, for which CPT1A gene and protein expression correlated (R=0.76; P=2.5x10-5; data not shown). Univariate Cox regression using CPT1A protein expression as a continuous value showed signifi-
N-MCL2/3 (mRNA) N-MCL2/3 (IHC) BLISS (IHC) Overall, N 70 45 135 Sex, N (%) Male Female 51 (73) 19 (27) 30 (67) 15 (33) 104 (77) 31 (23) Age in years at diagnosis, median (min-max) 55 (37-65) MIPI, N (%) Low risk Medium risk High risk Missing 34 (49) 17 (24) 19 (27) 28 (62) 10 (22) 7 (16) 11 (18) 24 (38) 27 (44) 73 Morphology, N (%) Classic Blastoid/pleomorphic Missing 51 (73) 19 (27) 39 (87) 6 (13) 116 (91) 12 (9) 7 KI-67, N (%) <30% >30% Missing 42 (60) 28 (48) 28 (68) 13 (32) 4 106 (80) 27 (20) 2 TP53, N (%) Wild-type Mutated Missing 35 (78) 10 (22) 25 22 (82) 5 (19) 18 48 (77)) 14 (23) 73 Overall survival time in years, median 11 12.4 4.2 Time to progression in years, median 5.3 10
Haematologica | 108 - April 2023 1095 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Figure 2. Pathways and associated genes related to inferior time to progression in mantle cell lymphoma. Top 10 enriched pathways associated with time to progression (TTP). Analysis was based on input of genes with log rank test P<0.01 as determined by cox regression using TTP as continuous variables. Bubble chart (A) x-axis displays enrichment based on cox regression bvalues and bubble size corresponds to the number of input genes in the given pathway. UpSet plot (B) displaying genes within the top 10 enriched pathways with red denoting positive b-values (increased hazard) and green negative b-values (decreased hazard) in relation to survival. Pathway clustering based on overlapping genes (C). Circle size correspond to number of significant genes, color correspond to pathway cluster and distance between circles represent pathway relatedness based on overlapping genes.
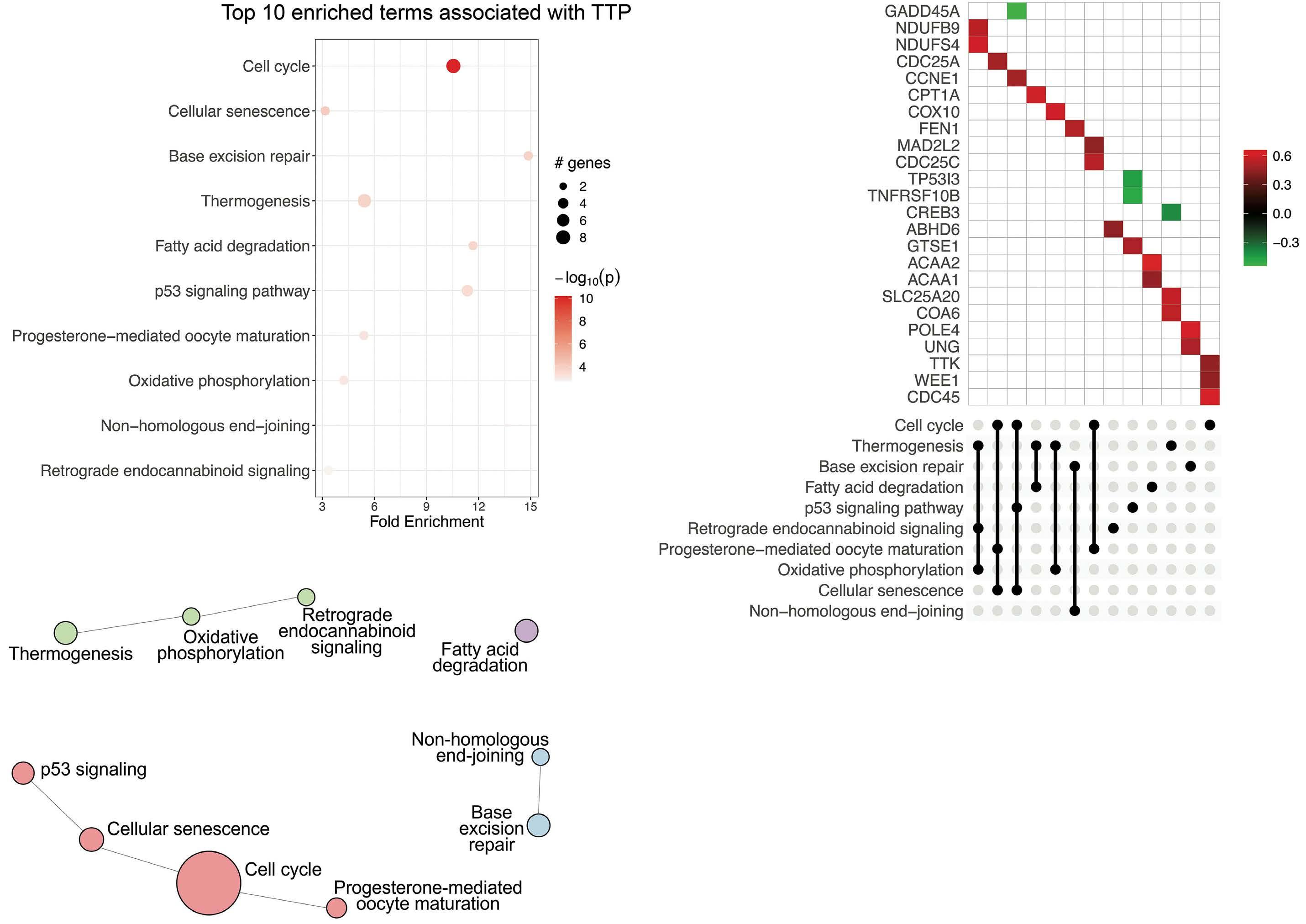
cant association to inferior survival (hazard ratio [HR]=1.02; 95% confidence interval [CI]: 1.01-1.04) at 1% increase of CPT1A for both TTP and OS in N-MCL2/3 (Table 2). The association of CPT1A and inferior OS was validated in the BLISS cohort (n=129) (HR=1.01; 95% CI: 1.004–1.02). Data on TTP was lacking in the BLISS cohort, and thus no validation of response to treatment could be performed. At the univariate gene level, HR for CPT1A was 1.81 (95% CI: 1.29-2.54) and 1.64 (95% CI: 1.13-2.37) for TTP and OS, respectively. In order to define thresholds of CPT1A expression for patient stratification into low- and high-risk, optimal cut-offs of 15% for TTP and 69% for OS, were identified in N-MCL2/3. (Figure 5A, B, respectively). Applying the 15% cut-off defined for TTP to OS data separated the patients into groups with different outcome, but the log-rank test was not statistically significant (Online Sup-
plementary Figure S1). The HR for patients with high CPT1A was 3.36 (95% CI: 1.47–7.68) for TTP and 8.12 (95% CI: 2.75–23.9) for OS. The 69% cut-off for OS was validated in the BLISS cohort, where a significant association to survival was confirmed (HR=2.16; 95% CI: 1.33-3.51) (Figure 5C), while data for TTP was not available for BLISS. The higher HR for patients treated with the N-MCL2/3 protocol was expected, as the BLISS cohort had significantly higher median age and frequency of established risk factors, and heterogeneously treated patients. The association between CPT1A and inferior survival was independent of MIPI, morphology, proliferation (Table 2), and TP53 (Online Supplementary Table S1) for TTP in N-MCL2/3. For OS, it was independent of morphology in BLISS, but not in NMCL2/3. High CPT1A expression was significantly associated with high Ki-67 and non-classic morphology in both
A B C Haematologica | 108 - April 2023 1096 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Figure 3. Pathways and associated genes related to inferior overall survival in mantle cell lymphoma. Top 10 enriched pathways associated with overall survival (OS). Analysis was based on input of genes with log-rank test P<0.01 as determined by cox regression using OS as continuous variables. Bubble chart (A) x-axis displays enrichment based on cox regression b-values and bubble size corresponds to the number of input genes in the given pathway. UpSet plot (B) displaying genes within the top 10 enriched pathways with red denoting positive b-values (increased hazard) and green negative b-values (decreased hazard) in relation to survival. Pathway clustering based on overlapping genes (C). Circle size correspond to number of significant genes, color correspond to pathway cluster and distance between circles represent pathway relatedness based on overlapping genes.
cohorts, but not to SOX11 (Online Supplementary Figure S2). We suggest that the prognostic value of CPT1A in relation to TTP is the most clinically relevant, and that future studies should use the defined 15% cut-off to assess high and low risk of short TTP.
Cell cycle is the dominating pathway associated with established high-risk mantle cell lymphoma factors
In order to investigate mechanisms underpinning known clinicopathological risk factors for MCL, differential GEX analysis was performed for Ki-67 high (>30% Ki-67-positive cells) versus Ki-67 low, non-classic versus classic morphology, and TP53-mutated versus wt in N-MCL2/3 (Figure 6A). A marked discrimination of high- versus lowproliferation cases was observed (21% DEG), with a significant enrichment peak of low P values and strong bias
of upregulated genes in the Ki-67-high group (Online Supplementary Figure S3A), concordant with the notion that proliferation heavily impacts transcriptomic analyses. The top DEG (P<1.0x10-4) were associated with 38 enriched pathways, and genes included in the top 10 pathways were exclusively upregulated in Ki-67-high (Figure 6B). The most significant pathway clusters were related to cell cycle, homologous recombination, and DNA replication (Figure 6C), with a significant part of key cell cycle genes being highly upregulated in Ki-67-high cases (Online Supplementary Figure S4).
With partly overlapping sample groups, pathways activated in non-classic versus classic morphology were also largely similar to those identified for high proliferation (Figure 6D, E). Compared to differential Ki-67, analysis based on morphology yielded weaker discrimination be-
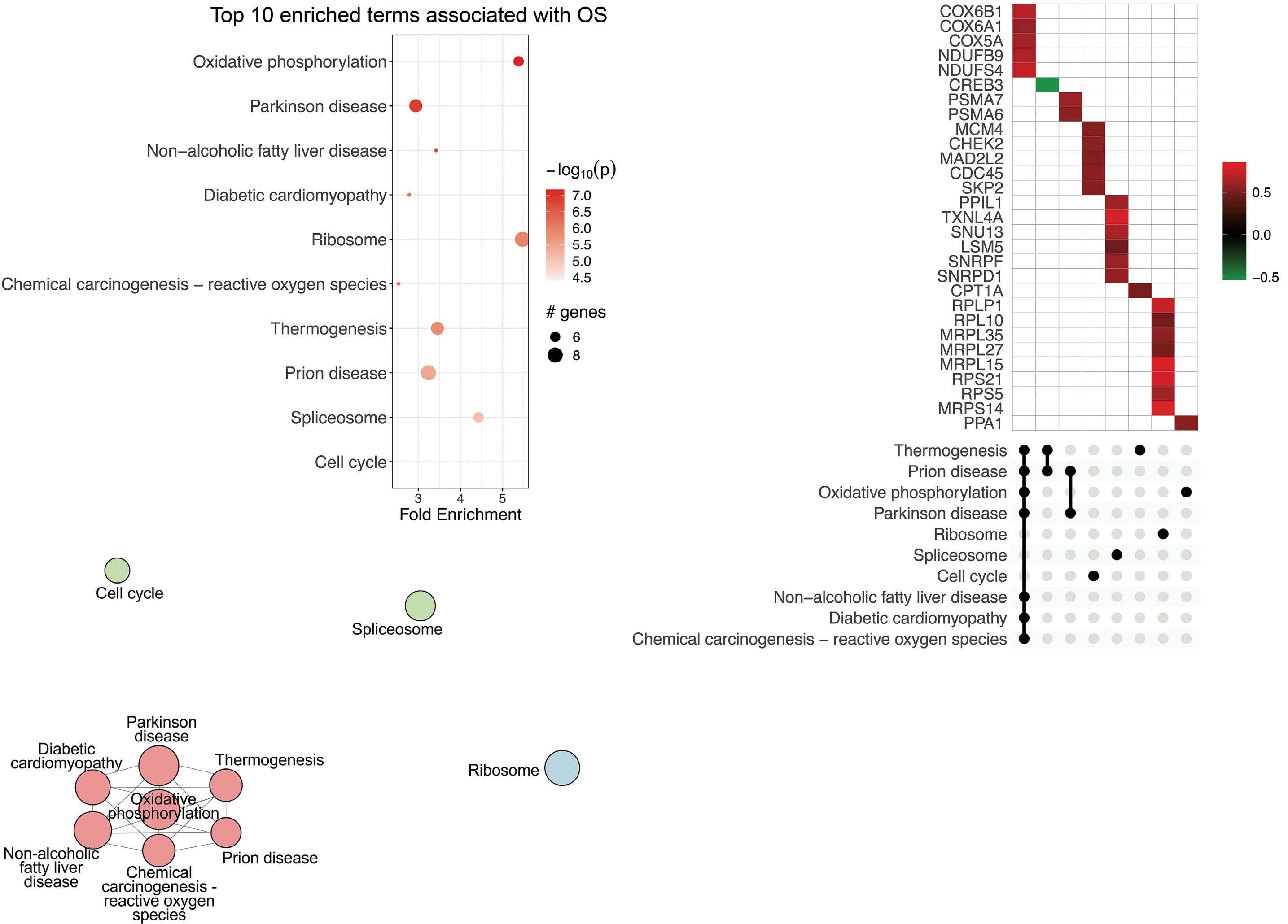
A B C Haematologica | 108 - April 2023 1097 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
tween groups, with 4.5% DEG ( Online Supplementary Figure S3B ). The top DEG ( P <0.01) corresponded to 25 enriched pathways, again with dominating clusters of cell cycle, DNA repair and replication (Figure 6E). Genes that were specifically deregulated in non-classic versus classic morphology, and not significantly associated with the other risk factors included MAP3K8 , MAP2K6 , and CXCL5 , involved in TNF signaling and related pathways.
FEN1 and WEE1 protein expression is associated with high-risk mantle cell lymphoma
Potentially targetable biomarkers were identified among upregulated genes in the high-risk MCL groups. FEN1 and WEE1 were found within the top deregulated pathways of Ki-67 high (Figure 5B) and non-classic morphology (Figure 6D) and were significantly associated with shorter TTP (Figure 2B). FEN1 encodes a key enzyme involved in DNA repair 20,21 and WEE1 , a kinase and key regulator of cell cycle through inhibition of Cdk1.22 IHC validated the association to Ki-67 high and non-classic morphology for both WEE1 (Online Supplementary Figure S5A, B ) and FEN1 ( Online Supplementary Figure S5D, E). While dichotomized FEN1 expression was significantly correlated to OS ( P<0.001) using a cut-off of 39% ( Online Supplementary Figure S5F ), no association to OS was seen for WEE1 on either gene (Figure 3B) or protein ( Online Supplementary Figure S5C ) level.
Transcriptional differences related to genetic alterations in MCL
Most genetic alterations in MCL are not associated with outcome, as for example ATM 4,23 . However, TP53 mutations are present in 15% of tumors,10,15 and define a highly aggressive sub-group of patients with poor response to currently available treatment regimens.10 As ATM and TP53 mutations were mutually exclusive in the patients included in the current study, we took the opportunity to investigate whether the transcriptional programs were distinct. The results are summarized in the Online Supplementary Figures S6 and S3C . Despite the strong impact on outcome, a low proportion (2.1%) of DEG was found in TP53 -mutated versus wt samples ( Online Supplementary Figure S3C ) while ATM aberrations were associated with deregulation of 10.7% of transcripts. Of interest, while TP53 expression was indicated to be lower in TP53 -mutated versus wt ( P =0.18) samples, it was significantly higher in ATM -mutated versu s wt ( P =0.002) samples. When comparing TP53 -mutated versus wt samples, the individual cell cycle pathway only appeared as the 16 th most significant pathway ( P =0.04), indicating that proliferation is not the sole dominating driver of TP53 -mutated cases. In ATM -mutated cases, the most significantly enriched pathway was steroid biosynthesis ( P =8x10 -10), but a network of pathways partly related to TP53 was also found to be enriched ( Online Supplementary Figure S6D ) together with genes
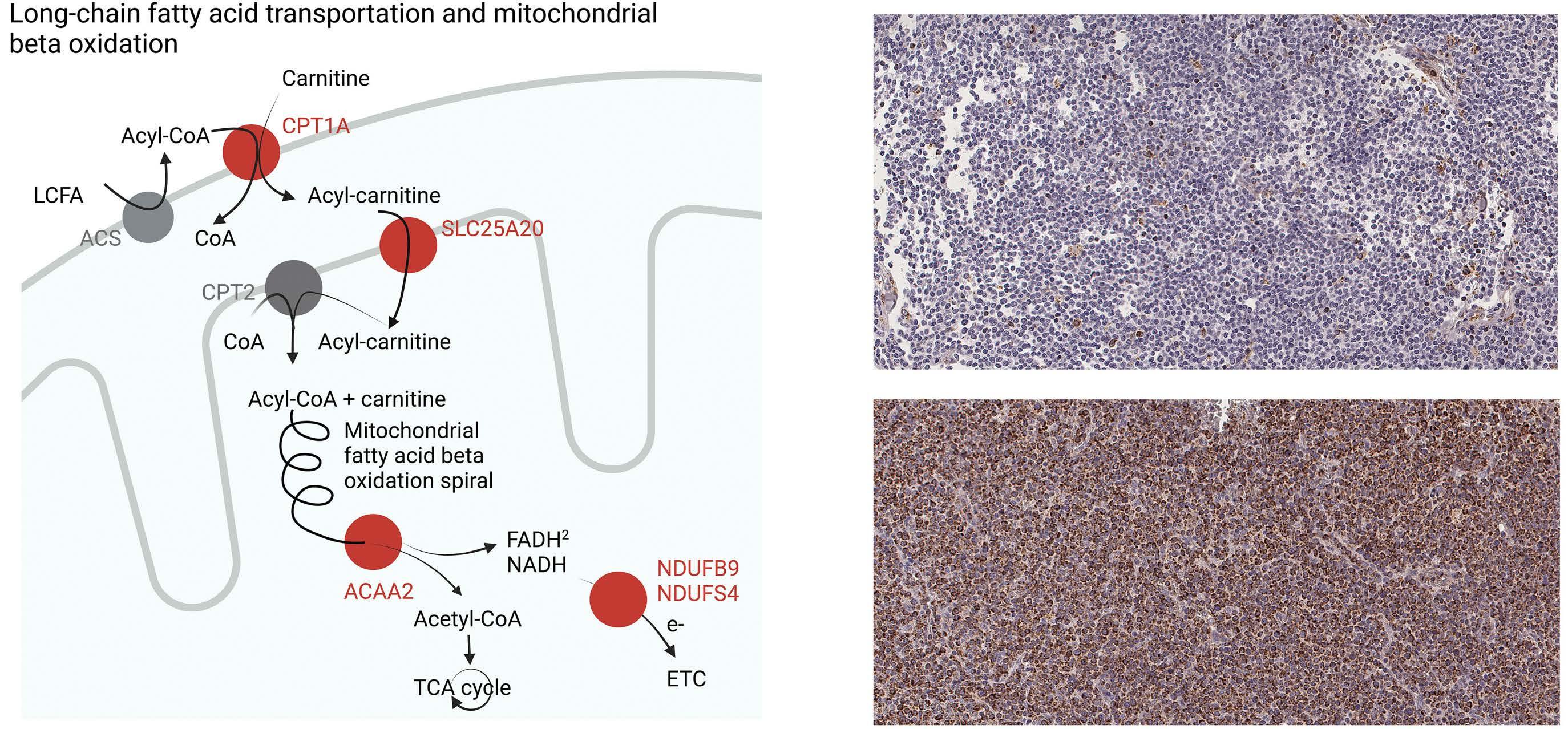
A B
Haematologica | 108 - April 2023 1098 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Figure 4. CPT1A expression, fatty acid transportation and mitochondrial fatty acid oxidation was associated with poor mantle cell lymphoma prognosis. (A) Active mitochondrial genes and pathways associated to shorter survival in the current cohort. Proteins marked in red represent some of the transcripts found to be upregulated in patients with poor outcome. LCFA: long-chain fatty acid; ACS: acetyl-CoA synthetase; TCA: tricarboxylic acid; ETC: electron transportation chain. (B) Representative high (94.5% positive cells) and low CPT1A (1.3% positive cells) immunohistochemistry staining.
Table 2. Univariate and multivariate Cox regression for CPT1A protein expression in both cohorts.
1Maximally selected rank statistics cut-off values. 2Mantle cell lymphoma International Prognostic Index (MIPI) low risk was used as reference category. *P<0.05; **P<0.01; ***P<0.001. CI: confidence interval; HR: hazard ratio; N: number of patients in the specific analyses; OS: overall survival; TTP: time to progression; N-MCL2/3: Nordic MCL group clinical trials 2 and 3; BLISS: biobank of lymphomas in Southern Sweden.
involved in apoptosis (Online Supplementary Figure S6E). The association of ATM mutations with tumor suppression and apoptosis provides a plausible explanation as to why ATM mutations do not confer a worse prognosis in MCL.
Discussion
More versatile treatment in combination with companion diagnostic strategies is needed to improve outcome for MCL patients. It has been proposed that MIPI,7 which is nowadays used for prognostication but not stratification, could be used as a treatment selection tool if combined with assessment of proliferation and TP53-mutational status. This would improve the possibility to identify patients in need of alternative non-chemotherapy-based treatment. However, such a combined index considers only a narrow range of risk factors, not including intrinsic transcriptional differences in, for example, BCL224 or extrinsic factors such as variation in angiogenesis25 or im-
mune composition.26,27 Deepened biological understanding of factors that influence early progression and relapse in patients treated with the current gold standard high-dose chemotherapy, would enable a more accurate treatment stratification and identification of novel targets for highrisk patients. To our knowledge, no/few studies have used outcome as a starting point to identify transcriptional profiles of MCL. Thus, to broaden the scope beyond established risk factors, we performed GEX analyses to decipher molecular mechanisms associated with outcome using a homogenously treated cohort of patients. For comparison and improved biological understanding of established risk factors, pathways and genes associated with TP53 and ATM mutations, proliferation and non-classic morphology were also identified and described. It is well known that the outcome of combinatorial treatment with immunochemotherapy, including rituximab, high-dose cytarabine and ASCT is related to several factors, including resistance to rituximab and/or cytarabine. Resistance to cytarabine has been extensively studied and involves the key rate-limiting step of the conversion of
Type of analysis Variable Sub-group HR (95% CI) Outcome N N-MCL2/3 Univariate CPT1A continuous 1.02 (1.01-1.03)** TTP 45 Univariate CPT1A continuous 1.02 (1.01-1.04)*** OS 45 Univariate CPT1A ≥5%1 3.36 (1.47-7.68)** TTP 45 Univariate CPT1A ≥69%1 8.12 (2.75-23.93)*** OS 45 Multivariate CTP1A ≥15%1 3.11 (1.15-8.42)* TTP 41 MIPI Intermediate2 1.25 (0.41-3.85) High2 6.72 (2.08-21.76)** Morphology Blastoid/pleomorphic 2.13 (0.52-8.74) Ki-67 ≥30% 1.08 (0.33-3.48) Multivariate CTP1A ≥69%1 6.18 (0.78-48.65) OS 41 MIPI Intermediate2 2.01 (0.52-7.69) High2 4.68 (0.98-22.39) Morphology Blastoid/pleomorphic 8.22 (1.59-42.36)* Ki-67 ≥30% 0.47 (0.08-2.74) BLISS Univariate CPT1A continuous 1.01 (1.004-1.02)** OS 129 Univariate CPT1A ≥ 69%1 2.16 (1.33-3.51)** OS 129 Multivariate CTP1A ≥ 69%1 1.79 (1.07-2.99)* OS 122 Morphology Blastoid/pleomorphic 0.66 (0.28-1.55) Ki-67 ≥ 30% 2.58 (1.3-5.13)
Haematologica | 108 - April 2023 1099 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Figure 5. CPT1A expression was associated with poor mantle cell lymphoma prognosis. Kaplan Meier curves representing the difference in (A) time to progression (TTP), log-rank test P=0.0024, for Nordic mantle cell lymphoma group clinical trials 2 and 3 (N-MCL2/3) patients, (B) Overall survival (OS) log-rank test P<0.0001, for N-MCL2/3 patients, (C) OS, log-rank test P=0.0015, for biobank of lymphomas in Southern Sweden (BLISS) patients.
Ara-C to the active substance Ara-CTP.28 We have previously shown that downregulation of deoxycytidine kinase (dCK) is a fundamental step during development of resistance, but no predictive markers are available. In line with previous observations, poor outcome was associated with key pathways associated with p53 regulation and proliferation. However, less-described pathways in MCL, including metabolic pathways such as thermogenesis, fatty acid degradation and oxidative phosphorylation, were also activated in patients with poor outcome. Of major interest, CPT1A and SLC25A20/CACT are both involved in the carnitine cycle,29 with CPT1A being the rate-limiting step for mitochondrial oxidation of long-chain fatty acids and thus a key protein for fatty acid metabolism. In order to explore the applicability of CPT1A as a biomarker, we validated the overexpression and association to outcome using IHC and digital scoring. Also on the protein level, CPT1A as a continuous variable was a marker of poor prognosis, as measured by both TTP and OS. Importantly, CPT1A showed prognostic significance for TTP independent of MIPI and established risk factors, including TP53mutational status, in multivariate analyses. Among the outcome variables, TTP is the most relevant prognostic
measurement for treatment stratification, and the cut-off identified at 15% CPT1A-positive cells, should be the focus for future validation studies. This cut-off was also applied to OS data and was close to significant in the N-MCL2/3 cohort. No validation cohort with TTP data was available; however, the optimal cut-off value for OS at 69% CPT1Apositive cells was validated in the independent population-based BLISS cohort and was shown to be independent of proliferation and morphology. The CPT1 gene is situated on the q-arm of chromosome 11 and CPT1A is the most expressed isoform. The gene is regulated through a plethora of mechanisms including hormones, NF-Y and Sp proteins,30 and microRNA.31 CPT1A is well known to be expressed in cancer and associated to treatment resistance and aggressiveness of disease. In breast cancer, CPT1A is regulated by c-MYC or AMPK, and promotes metastasis or therapeutic resistance through several oncogenic signaling pathways.32 Furthermore, it has been shown that BCL2 and CPT1A can interact on the surface of the mitochondria and regulate apoptosis in leukemia, and may thus antagonize apoptosis of BCL2targeting agents.33 When treating MCL patients with Bruton´s tyrosine kinase inhibitor Ibrutinib, enhanced
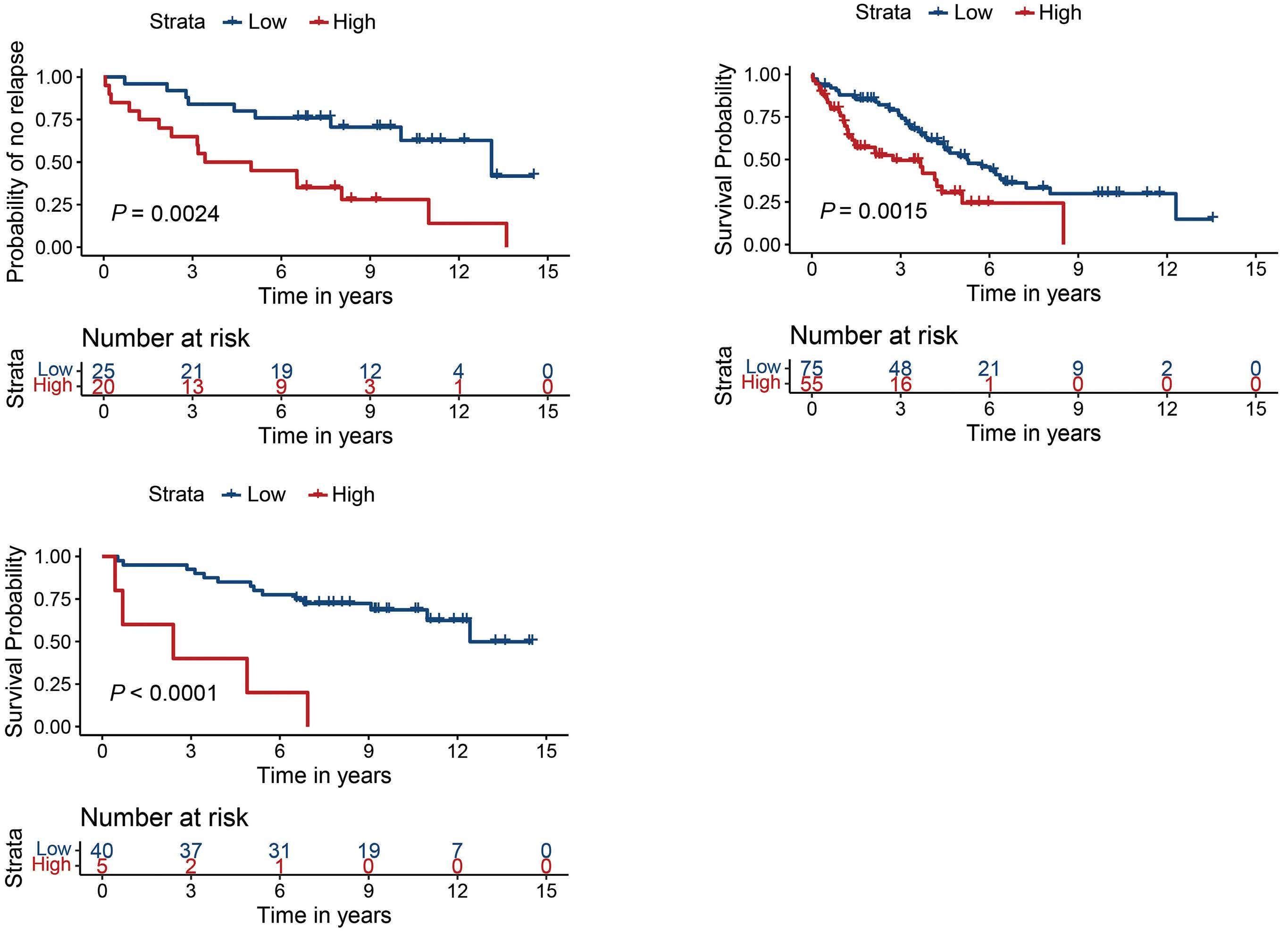
A B C Haematologica | 108 - April 2023 1100 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Continued on following page. A B C D E Haematologica | 108 - April 2023 1101 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Figure 6. Genes associated with clinicopathological risk factors of mantle cell lymphoma. (A) Distribution of clinicopathological risk factors assessed, with 1 patient per column. Grey cells denote unknown TP53-mutational status. (B) Top 10 significantly (P<1.4x10-4) enriched pathways associated with Ki-67 high mantle cell lymphoma (MCL) clustered by involvement of differentially expressed genes in an UpSet plot, colored by relative expression (all upregulated in Ki-67-high). Log fold-change (FC) values for Ki-67-high vs. -low, with non-classic vs. classic morphology (Morph.), and TP53-mutated vs. wild-type (wt) is shown for comparison. (C) Top 8 clusters of enriched pathways for Ki-67 high MCL. (D) Top 10 significantly (P<2.1x10-3) enriched pathways associated with non-classic MCL clustered by involvement of differentially expressed genes in an UpSet plot, colored by relative expression with red and green denoting up- and downregulation in non-classic vs. classic MCL, respectively. Log FC values for non-classic vs. classic, with Ki-67-high vs. -low, and TP53-mutated vs. wt added for comparison. (E) Top 8 clusters of enriched pathways for non-classic MCL.
oxidative phosphorylation is associated with poor clinical outcome.34 The activity of CPT1A inhibition in lymphomas is unknown, but in vitro evaluation of CPT1A inhibitors has shown high efficacy in several cancers such as high-grade serous ovarian cancer35 and hepatocellular carcinoma,36 and ability to sensitize cells to radiation in nasopharyngeal carcinoma.37 In addition, we found eight other markers of active oxidative phosphorylation upregulated including PPA1, supporting previous observations that enhanced oxidative phosphorylation is associated with treatment resistance in MCL34 and may be considered a potential target. Thus, the prognostic role associated to various treatments, including radiation in other types of cancers, indicates that CPT1A might be applicable for treatment stratification beyond the MCL2/3 regimen, although this remains to be determined. Multiple efforts have been made to define the MCL transcriptome associated to key features such as proliferation,38 the constitutive activation of NF k B and overexpression of CCND1. 39 As transcription-wide analyses are less suitable for clinical implementation, proliferationassociated signatures have been further developed into protein-based panels40 and a 35-gene nanostring-based mRNA assay (MCL35).41 It is evident, also from the current study, that proliferation is the factor associated with the largest transcriptional changes. Patients with non-classic morphology were highly overlapping with high proliferative cases, thus showing large commonality in pathways and genes that were deregulated. As expected, these pathways were mainly related to cell cycle and DNA repair. We identified FEN1 and WEE1 as associated to inferior TTP, as part of DNA repair and cell cycle pathways, respectively.
FEN1 protein was signifi cantly higher in Ki-67 high and non-classic morphology samples, and significantly associated with OS. FEN1/RAD27, encodes Flap endonuclease 1, which has been suggested as a promising target for patients with defects in homologous recombination, such as BRCA1/BRCA2-mutated tumors.42 WEE1 protein was also confirmed to be associated with high proliferation and non-classic morphology, but no association to OS was seen on gene or protein level. WEE1 is a key cell cycle regulator and the in vitro activity and synergistic effect of WEE1 and Chk1 inhibition has been demonstrated in MCL.43 However, targeting single proliferation-associated
genes has not been successful due to the many compensatory mechanisms.44
For long it was assumed that the overexpression of CCND1 mainly mediated increased proliferation of MCL cells. Today, it is more clear that it may also affect DNA repair, as shown in many cancers45 including MCL.46 Of interest, ATM, the most frequent (>40%) genetic aberration in MCL, affects DNA repair,3 but does not confer a survival disadvantage. While ATM mutations are common, TP53 mutations are the dominating molecular indicator of poor prognosis in MCL.3 As it has been shown by us4 and others23 that co-occurrence of TP53 and ATM mutations are infrequent in MCL, we set out to investigate if they share common molecular pathways. Pathways associated with ATM mutations were distinct from TP53-mutated MCL and dominated by the steroid biosynthesis pathway. Of interest, individuals with germline mutations in the ATM gene are known to have increased plasma cholesterol and triglyceride levels, and ATM has been demonstrated to facilitate clearance of apolipoproteins in plasma.49 Thus, ATM mutations may contribute to altered lipid metabolism and steroid biosynthesis. Today, proposed strategies for prediction of response to treatment in MCL are focused on MIPI in combination with assessment of proliferation and TP53 mutational status. In this comprehensive overview of the transcriptional landscape in MCL, we have explored high-risk features of MCL beyond these established factors. We show that fatty acid metabolism is deregulated in MCL and, thus propose that complementary investigations of metabolism may contribute to defining high-risk MCL through routine assessment of CPT1A.
Disclosures
CWE is currently employed by Genmab. MJ has received research support from Abbvie, AstraZeneca, Janssen, Gilead, BMS and Roche; and honoraria from Abbvie, AstraZeneca, BMS, Genmab, Janssen, Novartis, Incyte, EUSApharma, Gilead, and Roche. All other authors have no conflicts of interest to disclose.
Contributions
ASG performed data and statistical analysis and wrote the manuscript; JMR took part in the study plan, performed
Haematologica | 108 - April 2023 1102 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
data analysis and took part in the writing of the manuscript; CWE, SH, KG, RR, AK, CG and MJ were involved in the collection of the material and/or clinical data; AP took part in the pathology review; SE planned the study, interpreted results, and assisted in writing the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
The authors would like to thank the Nordic lymphoma group, and specifically the Nordic MCL network; Lina Olsson and May Hassan at the SpatialOmics@LU core facility at the Department of Immunotechnology, Lund University for assisting with expression analysis; Björn Nodin at the Department of Clinical Studies, Lund University for assisting with tissue cuts and IHC.
References
1. Grimm KE, O'Malley DP. Aggressive B cell lymphomas in the 2017 revised WHO classification of tumors of hematopoietic and lymphoid tissues. Ann Diagn Pathol. 2019;38:6-10.
2. Jain P, Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am J Hematol. 2019;94(6):710-725.
3. Hill HA, Qi X, Jain P, et al. Genetic mutations and features of mantle cell lymphoma: a systematic review and meta-analysis. Blood Adv. 2020;4(13):2927-2938.
4. Rodrigues JM, Porwit A, Hassan M, Ek S, Jerkeman M. Targeted genomic investigations in a population-based cohort of mantle cell lymphoma reveal novel clinically relevant targets. Leuk Lymphoma. 2021;62(11):2637-2647.
5. Rule S, Dreyling M, Goy A, et al. Ibrutinib for the treatment of relapsed/refractory mantle cell lymphoma: extended 3.5-year follow up from a pooled analysis. Haematologica. 2019;104(5):e211-e214.
6. Pu JJ, Savani M, Huang N, Epner EM. Mantle cell lymphoma management trends and novel agents: where are we going? Ther Adv Hematol. 2022;13:20406207221080743.
7. Silkenstedt E, Linton K, Dreyling M. Mantle cell lymphomaadvances in molecular biology, prognostication and treatment approaches. Br J Haematol. 2021;195(2):162-173.
8. Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood. 2014;123(19):2953-2959.
9. Geisler CH, Kolstad A, Laurell A, et al. Long-term progressionfree survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood. 2008;112(7):2687-2693.
10. Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903-1910.
11. Klaus B, Reisenauer S. An end to end workflow for differential gene expression using Affymetrix microarrays. F1000Res. 2016;5:1384.
12. Ulgen E, Ozisik O, Sezerman OU. pathfindR: an R package for comprehensive identification of enriched pathways in Omics
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement N 754299, Cancerfonden (2016/465, 19 0309Pj and 21 1561 Pj), Mats Paulssons Stiftelse för forskning, innovation och samhällsbyggande, Stiftelsen Stefan Paulssons cancerfond, and CREATE Health. All financial support was granted to SE.
Data-sharing statement
Original data and protocols are av ailable upon reasonable request.
data through active subnetworks. Front Genet. 2019;10:858.
13. Kononen J, Bubendorf L, Kallioniemi A, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4(7):844-847.
14. Hothorn T, Lausen B. On the exact distribution of maximally selected rank statistics. Computational Statistics & Data Analysis. 2003;43(2):121-137.
15. Rodrigues JM, Hassan M, Freiburghaus C, et al. p53 is associated with high-risk and pinpoints TP53 missense mutations in mantle cell lymphoma. Br J Haematol. 2020;191(5):796-805.
16. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111(2):558-565.
17. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol. 2016;34(12):1386-1394.
18. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol. 2014;32(13):1338-1346.
19. Hoster E. Prognostic relevance of clinical risk factors in mantle cell lymphoma. Semin Hematol. 2011;48(3):185-188.
20. Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997;16(11):3341-3348.
21. Querol-Audi J, Yan C, Xu X, et al. Repair complexes of FEN1 endonuclease, DNA, and Rad9-Hus1-Rad1 are distinguished from their PCNA counterparts by functionally important stability. Proc Natl Acad Sci U S A. 2012;109(22):8528-8533.
22. Harvey SL, Charlet A, Haas W, Gygi SP, Kellogg DR. Cdk1dependent regulation of the mitotic inhibitor Wee1. Cell. 2005;122(3):407-420.
23. Mareckova A, Malcikova J, Tom N, et al. ATM and TP53 mutations show mutual exclusivity but distinct clinical impact in mantle cell lymphoma patients. Leuk Lymphoma. 2019;60(6):1420-1428.
24. Montraveta A, Xargay-Torrent S, Rosich L, et al. Bcl-2 high mantle cell lymphoma cells are sensitized to acadesine with
Haematologica | 108 - April 2023 1103 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
ABT-199. Oncotarget. 2015;6(25):21159-21172.
25. Annese T, Ingravallo G, Tamma R, et al. Inflammatory infiltrate and angiogenesis in mantle cell lymphoma. Transl Oncol. 2020;13(3):100744.
26 Balsas P, Veloza L, Clot G, et al. SOX11, CD70, and Treg cells configure the tumor-immune microenvironment of aggressive mantle cell lymphoma. Blood. 2021;138(22):2202-2215.
27. Rodrigues JM, Nikkarinen A, Hollander P, et al. Infiltration of CD163-, PD-L1- and FoxP3-positive cells adversely affects outcome in patients with mantle cell lymphoma independent of established risk factors. Br J Haematol. 2021;193(3):520-531.
28. Di Francia R, Crisci S, De Monaco A, et al. Response and toxicity to cytarabine therapy in leukemia and lymphoma: from dose puzzle to pharmacogenomic biomarkers. Cancers (Basel). 2021;13(5):966.
29. Ramsay RR, Gandour RD, van der Leij FR. Molecular enzymology of carnitine transfer and transport. Biochim Biophys Acta. 2001;1546(1):21-43.
30. Steffen ML, Harrison WR, Elder FF, Cook GA, Park EA. Expression of the rat liver carnitine palmitoyltransferase I (CPTIalpha) gene is regulated by Sp1 and nuclear factor Y: chromosomal localization and promoter characterization. Biochem J. 1999;340( Pt 2)( Pt 2):425-432.
31. Schlaepfer IR, Joshi M. CPT1A-mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology. 2020;161(2):bqz046.
32. Tan Z, Zou Y, Zhu M, et al. Carnitine palmitoyl transferase 1A is a novel diagnostic and predictive biomarker for breast cancer. BMC Cancer. 2021;21(1):409.
33. Paumen MB, Ishida Y, Han H, et al. Direct interaction of the mitochondrial membrane protein carnitine palmitoyltransferase I with Bcl-2. Biochem Biophys Res Commun. 1997;231(3):523-525.
34. Zhang L, Yao Y, Zhang S, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. 2019;11(491):eaau1167.
35. Huang D, Chowdhury S, Wang H, et al. Multiomic analysis identifies CPT1A as a potential therapeutic target in platinumrefractory, high-grade serous ovarian cancer. Cell Rep Med. 2021;2(12):100471.
36. Ren M, Xu H, Xia H, Tang Q, Bi F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discov. 2021;7(1):125.
37. Tan Z, Xiao L, Tang M, et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics. 2018;8(9):2329-2347.
38. Demajo S, Albero R, Clot G, et al. A Cyclin D1-dependent transcriptional program predicts clinical outcome in mantle cell lymphoma. Clin Cancer Res. 2021;27(1):213-225.
39. Balaji S, Ahmed M, Lorence E, Yan F, Nomie K, Wang M. NFkappaB signaling and its relevance to the treatment of mantle cell lymphoma. J Hematol Oncol. 2018;11(1):83.
40. Hartmann E, Fernandez V, Moreno V, et al. Five-gene model to predict survival in mantle-cell lymphoma using frozen or formalin-fixed, paraffin-embedded tissue. J Clin Oncol. 2008;26(30):4966-4972.
41. Scott DW, Abrisqueta P, Wright GW, et al. New molecular assay for the proliferation signature in mantle cell lymphoma applicable to formalin-fixed paraffin-embedded biopsies. J Clin Oncol. 2017;35(15):1668-1677.
42. Guo E, Ishii Y, Mueller J, et al. FEN1 endonuclease as a therapeutic target for human cancers with defects in homologous recombination. Proc Natl Acad Sci U S A. 2020;117(32):19415-19424.
43. Chila R, Basana A, Lupi M, et al. Combined inhibition of Chk1 and Wee1 as a new therapeutic strategy for mantle cell lymphoma. Oncotarget. 2015;6(5):3394-3408.
44. Tchakarska G, Le Lan-Leguen A, Roth L, Sola B. The targeting of the sole cyclin D1 is not adequate for mantle cell lymphoma and myeloma therapies. Haematologica. 2009;94(12):1781-1782.
45. Jirawatnotai S, Hu Y, Michowski W, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011;474(7350):230-234.
46. Mohanty S, Mohanty A, Sandoval N, et al. Cyclin D1 depletion induces DNA damage in mantle cell lymphoma lines. Leuk Lymphoma. 2017;58(3):676-688.
47. Bellanger S, de Gramont A, Sobczak-Thepot J. Cyclin B2 suppresses mitotic failure and DNA re-replication in human somatic cells knocked down for both cyclins B1 and B2. Oncogene. 2007;26(51):7175-7184.
48. Liu Q, Li A, Tian Y, et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016;31:61-71.
49. Wu J, Xiao Y, Liu J, et al. Potential role of ATM in hepatocyte endocytosis of ApoE-deficient, ApoB48-containing lipoprotein in ApoE-deficient mice. Int J Mol Med. 2014;33(2):462-468.
Haematologica | 108 - April 2023 1104 ARTICLE - CPT1A expression defines MCL with poor prognosis A. Sandström Gerdtsson et al.
Comparison of autologous and allogeneic hematopoietic cell transplantation strategies in patients with primary plasma cell leukemia, with dynamic prediction modeling
Sarah Lawless,1* Simona Iacobelli,2* Nina Simone Knelange,3 Patrice Chevallier,4 Didier Blaise,5 Noel Milpied,6 Roberto Foà,7 Jan J. Cornelissen,8 Bruno Lioure,9 Ruben Benjamin,10 Xavier Poiré,11 Monique C. Minnema,12 Matthew Collin,13 Stig Lenhoff,14 John A. Snowden,15 Stella Santarone,16 Keith M. O. Wilson,17 Fernanda Trigo,18 Peter Dreger,19 Lara H. Böhmer,20 Hein Putter,21 Laurent Garderet,22 Nicolaus Kröger,23 Ibrahim Yaukoub-Agha,24 Stefan Schönland25 and Curly Morris26
1Belfast City Hospital, Belfast, Northern Ireland, 2Tor Vergata University, Rome, Italy, 3EBMT Data Office Leiden, Leiden, the Netherlands, 4CHU Nantes, Nantes, France, 5ICRCM, INSERM, CNRS, AMU and Institut Paoli Calmettes, Marseille, France, 6CHU Bordeaux, Pessac, France, 7Università La Sapienza`, Rome, Italy, 8Erasmus MC Cancer Institute, Rotterdam, the Netherlands, 9Nouvel Hopital Civil, Strasbourg, France, 10King’s College Hospital, London, UK, 11Cliniques Universitaires St. Luc, Brussels, Belgium, 12University Medical Center, Utrecht, Utrecht, the Netherlands, 13Freeman Hospital, Newcastle, UK, 14Skanes University Hospital, Lund, Sweden, 15Sheffield Teaching Hospitals Foundation Trust, Sheffield, UK, 16Ospedale Civile, Pescara, Italy, 17University Hospital of Wales, Cardiff, UK, 18Hospital Sao Joao, Porto, Portugal, 19University of Heidelberg, Heidelberg, Germany, 20Haga Teaching Hospital The Hague, the Netherlands, 21Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden, the Netherlands, 22Hospital Saint Antoine, Paris, France, 23University Hospital Eppendorf, Hamburg, Germany, 24CHU de Lille, University of Lille, INSERM U1286, Lille, France, 25University of Heidelberg, Heidelberg, Germany and 26Queen’s University of Belfast, Belfast, Northern Ireland
*SL and SI contributed equally as co-first authors.
Abstract
Correspondence: S. Schönland
Stefan.Schoenland@med.uni-heidelberg.de
Received: December 21, 2021.
Accepted: May 27, 2022.
Early view: June 30, 2022.
https://doi.org/10.3324/haematol.2021.280568
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Primary plasma cell leukemia (pPCL) is a rare and challenging malignancy. There are limited data regarding optimum transplant approaches. We therefore undertook a retrospective analysis from 1998-2014 of 751 patients with pPCL undergoing one of four transplant strategies; single autologous transplant (single auto), single allogeneic transplant (allofirst) or a combined tandem approach with an allogeneic transplant following an autologous transplant (auto-allo) or a tandem autologous transplant (auto-auto). To avoid time bias, multiple analytic approaches were employed including Cox models with time-dependent covariates and dynamic prediction by landmarking. Initial comparisons were made between patients undergoing allo-first (n=70) versus auto-first (n=681), regardless of a subsequent second transplant. The allo-first group had a lower relapse rate (45.9%, 95% confidence interval [95% CI]: 33.2-58.6 vs. 68.4%, 64.4-72.4) but higher nonrelapse mortality (27%, 95% CI: 15.9-38.1 vs. 7.3%, 5.2-9.4) at 36 months. Patients who underwent allo-first had a remarkably higher risk in the first 100 days for both overall survival and progression-free survival. Patients undergoing auto-allo (n=122) had no increased risk in the short term and a significant benefit in progression-free survival after 100 days compared to those undergoing single auto (hazard ratio [HR]=0.69, 95% CI: 0.52- 0.92; P=0.012). Auto-auto (n=117) was an effective option for patients achieving complete remission prior to their first transplant, whereas in patients who did not achieve complete remission prior to transplantation our modeling predicted that auto-allo was superior. This is the largest retrospective study reporting on transplantation in pPCL to date. We confirm a significant mortality risk within the first 100 days for allo-first and suggest that tandem transplant strategies are superior. Disease status at time of transplant influences outcome. This knowledge may help to guide clinical decisions on transplant strategy.
Introduction
Primary plasma cell leukemia (pPCL) is a rare plasma cell disorder. It follows an aggressive clinical course
with the median survival of affected patients being 1-3 years.1 Compared with multiple myeloma, pPCL is more likely to present with extramedullary involvement, thrombocytopenia, hypercalcemia, elevated serum b 2 -
Haematologica | 108 - April 2023 1105 ARTICLE - Plasma Cell Disorders
microglobulin and lactate dehydrogenase levels. 2 Due to the infrequent incidence and fulminant course of pPCL, there is a paucity of prospective data to guide clinicians managing this challenging disorder. 3-5
Analysis of the Surveillance, Epidemiology, and End Results (SEER) database of 445 pPCL patients between 1973 and 2009 shows an improvement in survival in recent years. 5 Novel agents, such as bortezomib 6-10 and lenalidomide,11 have been shown to be effective in pPCL used either alone or in combination 12-15 and may account for some of the improvements seen in recent years. Many of the reports also confirm the benefit of consolidation with hematopoietic stem cell transplantation, although all modalities of transplantation including autologous, allogeneic and tandem approaches have generally been considered together. Nevertheless, survival outcomes of pPCL patients in the SEER study are still inferior to those of multiple myeloma patients diagnosed during the same period when the analysis is adjusted for gender and age of the patients. 5
The European Society for Blood and Marrow Transplantation (EBMT) reported on the outcomes of 272 patients with pPCL undergoing autologous hematopoietic stem cell transplantation (auto).16 This study confirmed that auto can improve outcome, but results were markedly inferior to those achieved in patients with multiple myeloma. The Center for International Blood and Marrow Transplant Research (CIBMTR) has also demonstrated improvements in progression-free survival (PFS) and overall survival (OS) in pPCL following auto.17
However, the role of allogeneic hematopoietic stem cell transplantation (allo) remains uncertain. In 2012 the CIBMTR compared outcomes of 147 patients undergoing auto or allo between 1995-2006 and demonstrated that while allo patients had significantly lower relapse rates, their non-relapse mortality (NRM) was significantly higher with no OS benefit at 3 years.17
In 2020 the CIBMTR reported a further analysis of 348 patients with pPCL transplanted between 2008-2015. An increase in hematopoietic cell transplant utilization was noted from 12% in 1995 to 46% in 2009 but outcomes remained poor with no increase in OS in the allo group when compared with that in the previous study.18 The present study utilized the largest cohort of patients with pPCL (n=751) undergoing hematopoietic stem cell transplantation to examine various transplantation strategies and determine how these may be of most benefit. The study includes auto, allo and tandem transplants. To make statistically valid comparisons in this retrospective comparison of transplant strategies, non-standard statistical methods were employed, including dynamic prediction modeling.
Methods
A retrospective analysis was undertaken of the EBMT experience of patients with pPCL undergoing transplantation between 1998 and 2014. Only patients who had achieved a complete response, partial response, very good partial response or stable disease prior to transplantation were included. The objective was to compare patients undergoing a single autologous transplant (auto), a single allogeneic transplant (allo-first) or a combined tandem approach with an allogeneic transplant following an autologous transplant (auto-allo) or a tandem autologous transplant (auto-auto) as consolidation in first-line treatment. Tandem transplants were defined as given within 9 months in the absence of disease progression. The main endpoints of interest were OS and PFS. We also analyzed the cumulative incidence of relapse, NRM, and acute and chronic graft-versus-host disease. The problem and approaches used to compare transplant strategies are illustrated in the statistical methods section and in the Online Supplementary Material.
This study was conducted on behalf of the Chronic Malignancies Working Party of the EBMT. The EBMT represents more than 500 transplantation centers in and beyond Europe, which report minimum essential data on all transplants into a central database. EBMT centers are committed to obtain informed consent according to the local regulations applicable at the time of transplantation in order to report pseudonymized data to the EBMT. The study was planned and approved by the Chronic Malignancies Working Party of the EBMT. In addition, the study protocol was approved by the institutional review board at each site and complied with country-specific regulatory requirements. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Statistical methods
Events for OS and PFS were defined as death from any cause and the first between death and progression, respectively. The occurrence of relapse or progression and of death was analyzed as mutually competing (generating cumulative incidence of relapse and NRM cumulative incidence curves). For acute and chronic graft-versus-host disease, traditionally defined as occurring respectively within and after 100 days from allogeneic transplantation, relapse or progression and death were considered competing events. The standard methods indicated in the EBMT statistical guidelines19 were applied for the comparisons of groups according to the type of first transplant. Different approaches were applied for the comparison of single and tandem transplant strategies to avoid the risk of a time bias of retrospective analyses (Online Supplementary Material S1). A traditional landmark analysis is presented as a sec-
Haematologica | 108 - April 2023 1106 ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al.
ondary analysis (Online Supplementary Material S3) as it provides a partial view with some important limitations. An alternative landmark propensity score matched comparison (not shown) returned the same conclusions. The main analysis was done by Cox models including time-dependent covariates for the administration of the second transplant. Additionally, it was necessary to correct for the time-varying effect of an allogeneic transplant (when given as a first transplant or in a tandem transplant strategy) due to the higher early mortality. For simplicity, this time-dependent effect was modeled as being a stepwise constant in two periods measured from the time of allo: from day 0 to day 100 (“recent allo”) and after 100 days (“past allo”). The effects of the transplant strategies are thus measured as hazard ratios (HR) with respect to single auto as the baseline group. Candidate adjustment factors for the models were patients’ sex, age, disease status and performance status at first transplant, time from diagnosis to first transplant, and calendar year. Only age and disease
status were retained in the final models. A further insight into the effects on the probabilities of OS and PFS was obtained by applying a method of dynamic prediction (Online Supplementary Material S4), illustrating the evolution during the first 36 months of follow-up of the conditional 3-year OS and 1-year PFS. We applied the method of dynamic prediction by landmarking described by van Houwelingen and Putter20 based on Cox models with the same structure for the effects of the transplant strategies and the same adjustment factors as the main analysis. A second set of dynamic prediction curves was based on Cox models including interactions between patients’ characteristics and type of transplant strategy.
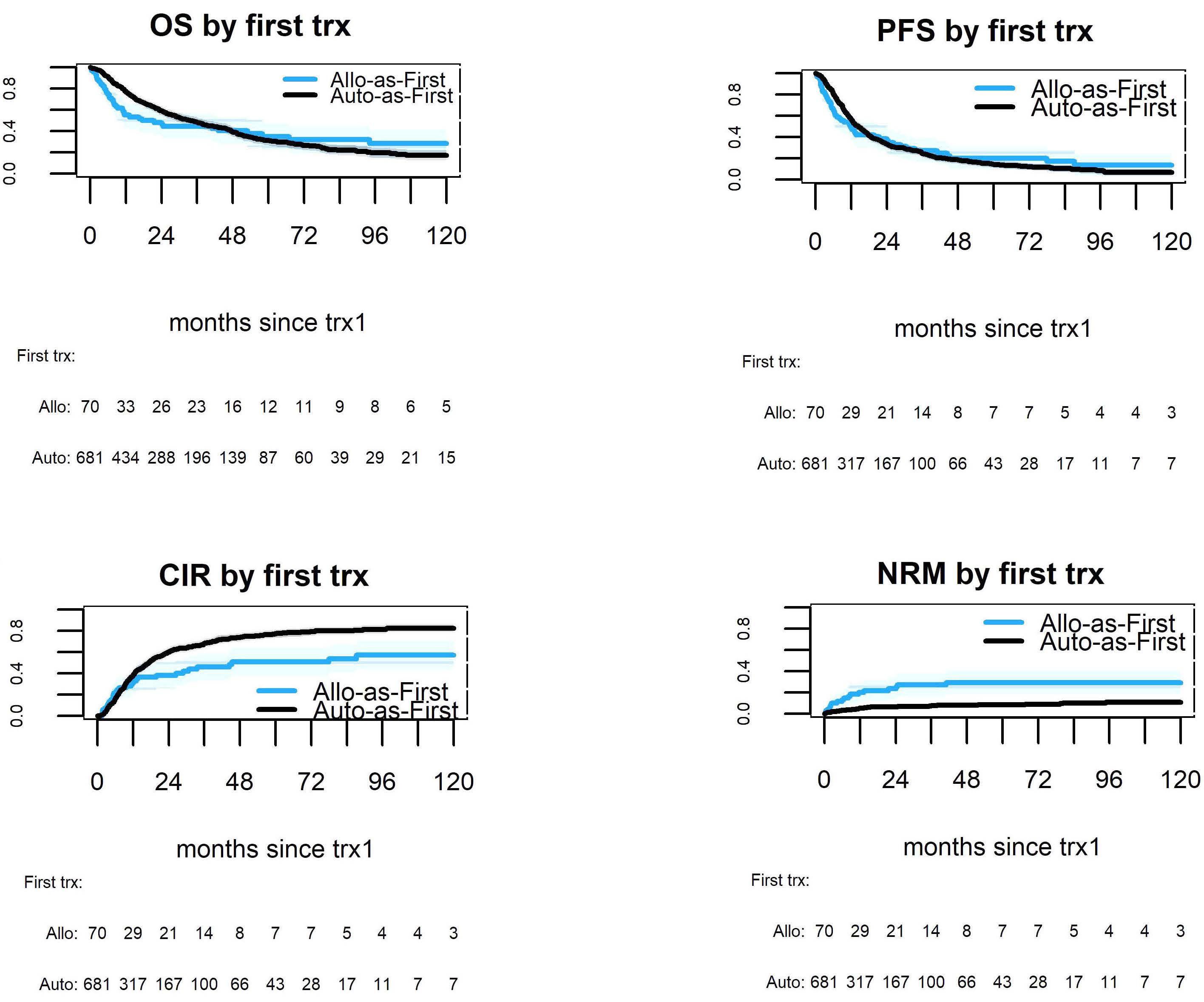
Results
A total of 751 patients were included in our analysis. The median OS of all patients irrespective of transplant type
Figure 1. Comparison of outcomes by type of first transplant (autologous or allogeneic). (A) Overall survival. (B) Progression-free survival. (C) Cumulative incidence of relapse. (D) Non-relapse mortality. trx: transplant; trx1: first transplant; allo: allogeneic hematopoietic stem cell transplant; auto: autologous hematopoietic stem cell transplantation; OS: overall survival; PFS: progression-free survival; CIR: cumulative incidence of relapse; NRM: non-relapse mortality.
Haematologica | 108 - April 2023 1107
A B
ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al. C D
Figure 2. Conditional probabilities of overall and progression-free survival estimated by dynamic prediction models. (A) Three-year overall survival. For each prediction time during the interval 0-36 months from the first transplant (x axis) the 3-year overall survival probability (on the y axis) is re-estimated taking into account the previous transplants received. For example, a patient in the tandem autologous-allogeneic group has the same probability of surviving for at least the next 3 years as a patient who underwent a single autologous transplant until the day of allogeneic transplantation, at 2 months, when the curves separate. Vertical changes of the curves for the allogeneic transplant first and the tandem autologous-allogeneic transplant recipients are due to the end of the first 100-day high-risk period after allogeneic transplantation. (B) One-year progression-free survival. As for overall survival, but with a horizon time of 1 year. In both (A) and (B) the baseline characteristics were age 55 and not in complete remission at first transplant. OS: overall survival; PFS: progression-free survival; yr: years; yo: years old; noCR: not in complete remission; allo: allogeneic hematopoietic stem cell transplant; auto: autologous hematopoietic stem cell transplantation; trx1: time since first transplant.
*Percentages computed among non-missing cases. °Test for linear trends in time. First auto: an autologous transplant first (regardless of whether a second transplant was subsequently performed); allo-first: single allogeneic transplant.
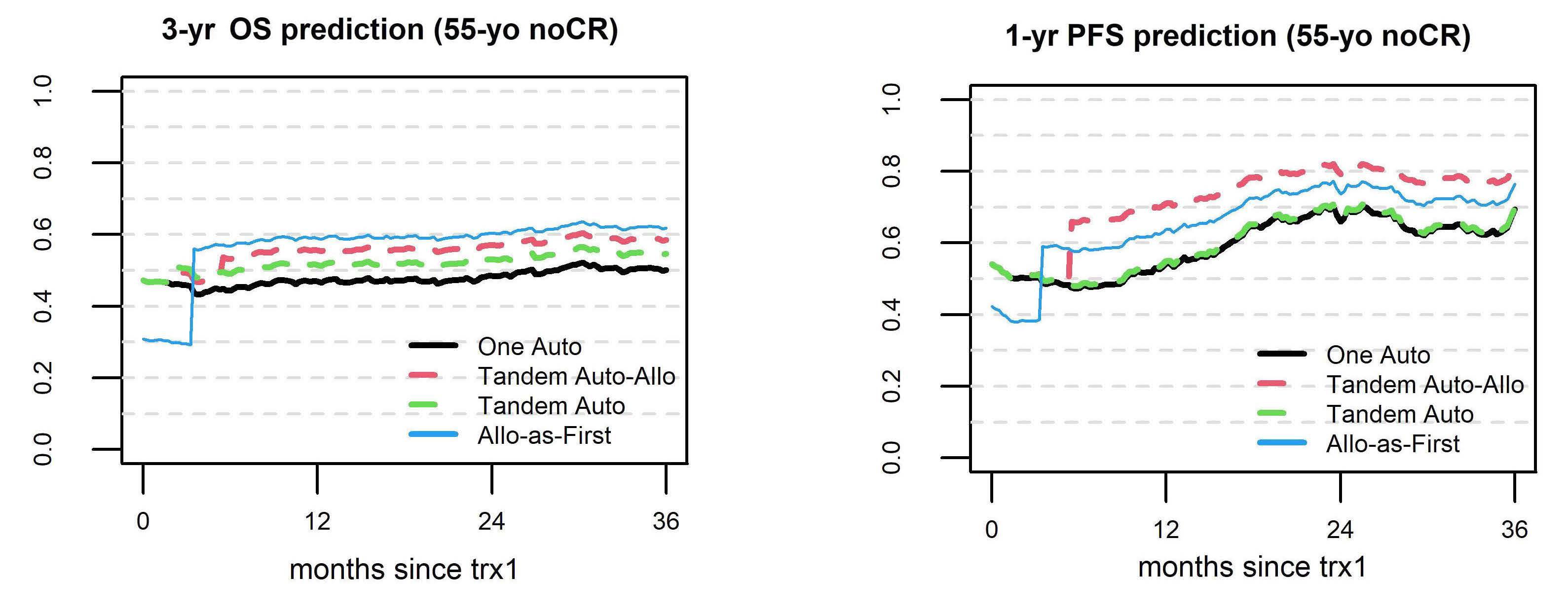
A B All cases First auto Allo-first P value N of patients 751 681 70 Age at 1st transplant, years Median 56.7 57.7 47.2 <0.001 Min-max 20-79 25-79 20-68 Sex, N (%) Male 378 (50.3) 334 (49.0) 44 (62.9) 0.028 Female 373 (49.7) 347 (51.0) 26 (37.1) Time from diagnosis to 1st transplant, N (%) ≤12 months 696 (92.7) 637 (93.5) 59 (84.3) 0.005 >12 months 55 (7.3) 44 (6.5.) 11 (15.7) Disease status at 1st transplant, N (%) Complete response 247 (32.9) 221 (32.5) 26 (37.1) <0.001 Partial response 460 (61.3) 427 (62.7) 33 (47.1) Stable disease 44 (5.9) 33 (4.8) 11 (15.7) Karnofsky performance status at 1st transplant*, N (%) ≥70 632 (96.3) 571 (96.3) 61 (96.8) 0.046 <70 24 (3.7) 22 (3.7) 2 (3.2.) Missing 95 (13) 88 (13) 7 (10) Calendar period of 1st transplant°, N (%) 1998-2003 153 (20.4) 132 (19.4) 21 (30.0) 0.132 2004-2007 143 (19.0) 131 (19.2) 12 (17.1) 2008-2010 144 (19.2) 133 (19.5) 11 (15.7) 2011-2012 149 (19.8) 136 (20.0) 13 (18.6) 2013-2014 162 (21.6) 149 (21.9) 13 (18.6)
Table 1. Characteristics of patients for all cases and split by the type of first transplant (autologous or allogeneic).
Haematologica | 108 - April 2023 1108 ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al.
was 33 months and the median PFS was 14 months. The median follow-up was 48.8 months.
Transplant strategies
Seventy patients received an allo-first and 681 patients received an auto as their first transplant. With respect to tandem strategies 122 patients proceeded to a tandem auto-allo and 117 underwent tandem auto-auto leaving 442 patients who underwent single auto only.
Comparison of autologous versus allogeneic as first transplant
Initial comparisons were made between patients undergoing allo-first versus first auto (regardless of subsequent administration of a second transplant). The characteristics of the patients are reported in Table 1. Allo-first patients were predominantly male, were significantly younger (median age 47.2 years vs . 57.7 years for first auto; P<0.001), had a longer time from diagnosis to transplant (P=0.005) and had a significantly higher proportion of patients both in complete remission and with stable disease (P<0.001). The median OS was 17.5 months for allo-first versus 33.5 months for first auto, while the median PFS was 11.7 months for allo-first and 14.3 months for first auto (Figure 1). The curves show a clear crossing so that at 60 months the OS and PFS probabilities were roughly similar (OS: allo 34.6% [95% CI: 21.6-47.6], auto 31.3% [95% CI: 26.8-35.9]; PFS: allo 19.9% [95% CI: 8.9-30.9], auto 14.3% [95% CI: 10.9 - 17.6]). Notably the NRM (Figure 1D) was 27% (95% CI: 15.9-38.1) at 36 months for allo versus 7.3% (95% CI: 5.2-9.4) for first auto while the cumulative incidence of relapse at 36 months was lower in the allofirst group (45.9%, 95% CI: 33.2-58.6) than in the auto group (68.4%, 95% CI: 64.4-72.4).
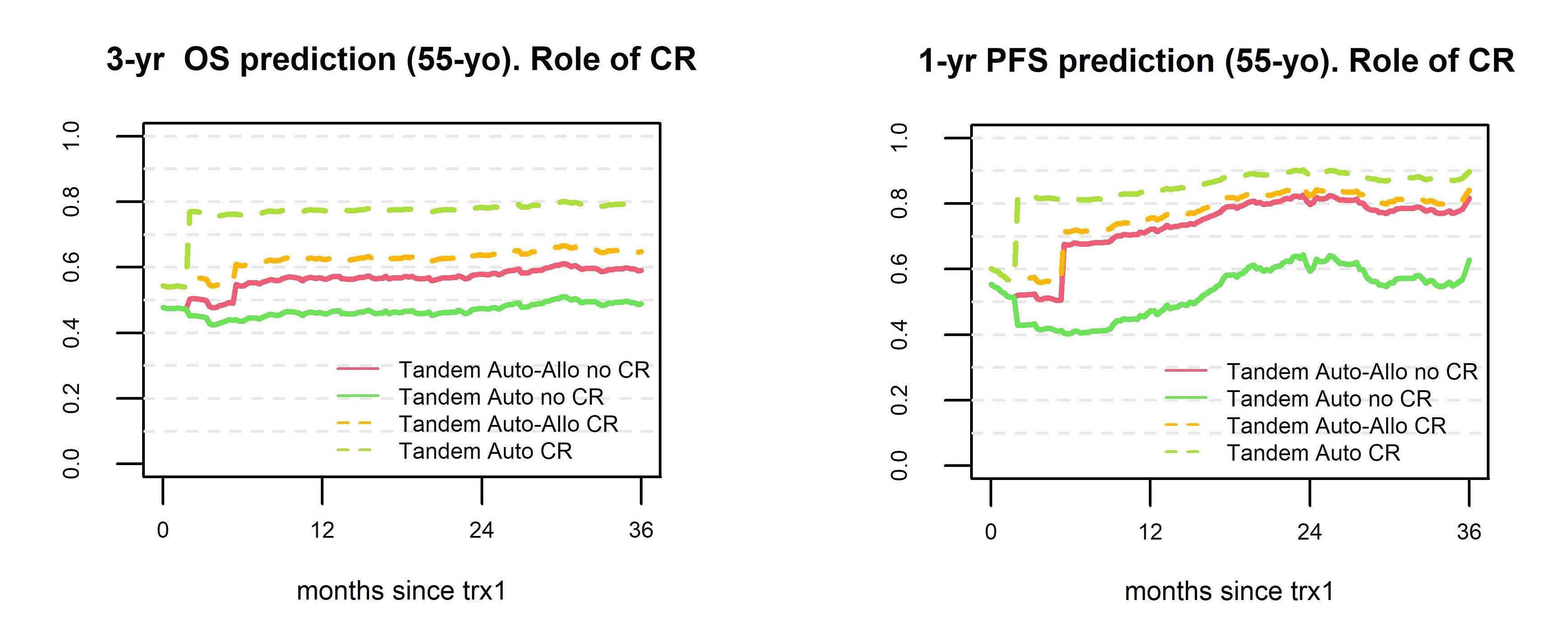
Comparison of single and tandem transplant strategies
The characteristics of patients grouped according to the actual transplantations received are illustrated in Tables 2 and 3. Patients receiving a tandem auto-allo were slightly older, had a shorter time from diagnosis to transplant, and had higher proportions of matched unrelated donors and reduced intensity conditioning than those who had allo-first (Table 3). They were also predominantly females and younger than the other patients undergoing first auto.
The characteristics of allogeneic transplants given as the first transplant or as part of a tandem auto-allo strategy are shown in Table 3. As previously noted, the administration of allo as a first or second transplant differed in several characteristics. Total body irradiation was administered more frequently to allo-first patients than to auto-allo patients if standard conditioning was used (47.1% allo-first vs 29.2% auto-allo), whereas it was given more frequently in auto-allo than allo if reduced intensity conditioning was used (42.3% allo-first vs. 11.1% auto-allo).
Any differences in conditioning did not translate into meaningful differences in graft-versus-host disease. Online Supplementary Table S1 shows the incidences of acute and chronic graft-versus-host disease, which appear similar to those seen in patients receiving allo first or auto-allo. Only 13 patients who underwent an allo received donor lymphocyte infusion and all were in the allo-first group. The median time to donor lymphocyte infusion was 5.7 months (range, 2.7-46.1) with six patients receiving the treatment before relapse and seven patients receiving it after relapse. In a preliminary approach, landmark analyses at 4 months were undertaken for OS, PFS, NRM and cumulative incidence of relapse (Online Supplementary Material S3; Online Supplementary Figure S1) which showed no significant A B
Haematologica | 108 - April 2023 1109 ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al.
Figure 3. Conditional probabilities estimated by dynamic prediction models with interaction terms. Role of status at first transplant. (A) Three-year overall survival. (B) one-year progression-free survival. See the legend to Figure 2 for a general explanation of the graphs. OS: overall survival; PFS: progression-free survival; yr: years; yo: years old; CR: complete response; allo: allogeneic hematopoietic stem cell transplant; auto: autologous hematopoietic stem cell transplantation; trx1: time since first transplant.
discrimination between transplant strategies, except for a remarkably higher NRM for allo-first. Due to the limitations of landmark analysis the main analysis was done using Cox models for OS and PFS. With single-auto as the baseline, comparisons were made with first-allo, tandem auto-auto and tandem auto-allo, adjusting for age and disease status (Table 4). It can be seen that allo-first patients had the greatest risk in the first 100 days (OS: HR=5.74, 95% CI: 2.66-12.40, P<0.001; PFS: HR=2.84, 95% CI: 1.57-5.15, P=0.001). Being transplanted in complete remission conferred a significant benefit and the effect of being younger at transplantation may also confer
a benefit. With consideration of the time-dependent effect, after 100 days the outcomes of allo-first became comparable to those of other strategies. Tandem auto-allo had a significant benefit on PFS after 100 days when compared to single-auto with a reduction of risk by 70% (HR=0.69, 95% CI: 0.52-0.92, P=0.012). Although some protective effect was also seen on OS (HR=0.80, 95% CI: 0.591.08, P=0.148), this did not reach statistical significance. For auto-auto the hazard ratios for PFS and OS were also reduced (models without interactions: HR=0.81 95% CI: 0.61-1.08, P=0.114 and HR=0.86, 95% CI: 0.67-1.11, P=0.254, respectively).
*Percentages computed among non-missing cases. Single auto; single autologous hematopoietic stem cell transplant; Tandem auto-auto; tandem autologous hematopoietic stem cell transplants; Tandem auto-allo; autologous hematopoietic stem cell transplant followed by an allogeneic transplant; Allo-first: single allogeneic transplant; na: not applicable.
Single auto Tandem auto-auto Tandem auto-allo Allo-first N of patients 442 117 122 70 Age at 1st transplant, years Median 58.7 58.7 51,6 47.2 Min-max (25,79) (37,75) (33,70) (20-68) Sex, N (%) Male 224 (50.7) 64 (54.7) 46 (37.7) 44 (62.9) Female 218 (49.3%) 53 (45.3) 76 (62.3) 26 (37.1) Time from diagnosis to 1st transplant, N (%) ≤12 months 403 (91.2) 114 (97.4) 120 (98.4) 59 (84.3) >12 months 39 (8.8) 3 (2.6) 2 (1.6) 11 (15.7) Disease status at 1st transplant, N (%) Complete response 155 (35.1) 28 (23.9) 38 (31.1) 26 (37.1) Partial response 268 (60.6) 79 (67.5) 80 (65.6) 33 (47.1) Stable disease 19 (4.3) 10 (8.5) 4 (3.3) 11 (15.7) Karnofsky performance status at 1st transplant*, N (%) ≥70 366 (95.1) 99 (98.0) 106 (99.1) 61 (96.8) <70 19 (4.9) 2 (2.0) 1 (0.9) 2 (3.2) Missing 57 (13) 16 (14) 15 (12) 7 (10) Calendar period of 1st transplant, N (%) 1998-2003 92 (20.8) 27 (23.1) 13 (10.7) 21 (30.0) 2004-2007 77 (17.4) 32 (27.4) 22 (18.0) 12 (17.1) 2008-2010 85 (19.2) 14 (12.0) 34 (27.9) 11 (15.7) 2011-2012 96 (21.7) 19 (16.2) 21 (17.2) 13 (18.6) 2013-2014 92 (20.8) 25 (21.4) 32 (26.2) 13 (18.6) Disease
transplant, N
Complete/partial response na 116 (99.1) 119 (97.5) na Stable disease/minimal response na 1 (0.9) 3 (2.5) na
status at 2nd
(%)
Table 2. Characteristics of patients according to transplant strategy.
Autologous
S. Lawless et al.
Haematologica | 108 - April 2023 1110 ARTICLE -
and allogeneic transplantation in PCL
Conditional overall and progression-free survival probabilities
The difference of outcomes of the four transplant strategies was further illustrated by dynamic prediction curves (Online Supplementary Material S4). Figure 2A and 2B show respectively the projected 3-year OS and 1-year PFS starting from any time during the first 36 months for a 55 yearold patient not in complete remission at the time of the first transplant (these being the median and the mode, respectively, of the two characteristics), according to the transplant strategy given. While it is clear that the OS outlook for the allo-first patients surviving the first 100 days
is at least as good as (or better than) any other strategy the high initial NRM is of concern. It can be seen that for 3-year OS there is no marked difference with respect to the transplant strategy used. A single auto is the least attractive option and is marginally improved by a second transplant, although the 1-year PFS is improved to a greater extent by an auto-allo than an auto-auto approach.
Effect of complete response
Further modeling detected an interaction of the disease status with auto-auto transplant strategy for both OS and PFS (Table 4, last two lines; Figure 3A, B). It can be seen that being in complete remission at the time of the first transplant provided a marginal benefit when combined with an auto-allo strategy (orange curves) whereas complete remission at first transplant was of great benefit if employing an auto-auto strategy (green curves).
Discussion
Despite the improvements brought about by the use of novel agents pPCL remains a challenging disorder for clinicians to manage. This retrospective study provides evidence to help guide transplant physicians in their decision-making process and offer patients an approach most suited to their circumstances following effective induction therapy.
*Percentages computed among non-missing cases. Information on total body irradiation is missing for one case in each group. T-cell depletion data are missing for nine (7.4%) and 11 (15.7%) cases, respectively. °For conditioning, the percentages for total body irradiation not given/given are computed within the subgroups of the standard and reduced intensity regimens. Tandem auto-allo; allogeneic hematopoietic stem cell transplant following an autologous transplant; Allo-first: single allogeneic transplant; allo: allogeneic hematopoietic stem cell transplant; CR: complete response; PR: partial response; SD: stable disease; MR: minimal response; HLA: human leukocyte antigen; TBI: total body irradiation.
Tandem transplants, both auto-auto and auto-allo, have been used in multiple myeloma for the past two decades but without great clarity on their place in the treatment paradigm. Two major prospective studies of patients with newly diagnosed multiple myeloma responding to therapy compared auto-auto to auto-allo.21,22 Although there was a dramatic improvement in NRM with the auto-allo approach compared with allo-first, the NRM remained significantly higher than with auto-auto and it was only after 5 years of follow-up that an advantage for the auto-allo approach became evident.22,23 Our study indicates that there may be a similar benefit from the auto-allo approach for patients with newly diagnosed pPCL in the longer term, particularly those not in complete remission at the time of the first transplant. We provided curves of the expected conditional probabilities of OS and PFS (using a dynamic prediction approach), to better quantify the differences, in addition to the hazard ratios provided by the Cox models. The predictions from this dynamic model suggest that if patients achieve a complete response prior to their first transplant then auto-auto is an effective option with outcomes similar to those of autoallo. This is an attractive option as it avoids the high NRM seen after allo and the potential morbidity and mortality of long-term graft- versus-host disease. However, if the
Tandem auto-allo Allo-first N of patients 122 70 Age at allo, years Median (min-max) 52.0 (33-71) 47.2 (20-68) Disease status at allo, N (%) CR/PR 119 (97.5) 59 (84.3) SD/MR 3 (2.5) 11 (15.7) Donor type, N (%) HLA matched sibling 58 (47.5) 46 (65.7) Matched unrelated donor 61 (50.0) 20 (28.6) Other donor 3 (2.5) 4 (5.7) Source of stem cells, N (%) Bone marrow 14 (11.5) 14 (20) Peripheral blood 108 (88.5) 56 (80) Conditioning*°, N (%) Standard 24 (19.8) 51 (73.9) - No TBI 17 (70.8) 27 (52.9) -TBI given 7 (29.2) 24 (47.1) Reduced intensity 97 (80.2) 18 (26.1) - No TBI 56 (57.7) 16 (88.9) - TBI given 41 (42.3) 2 (11.1) T cell depletion*, N (%) Not given 50 (44.2) 32 (54.2) Given 63 (55.8) 27 (45.8)
Table 3. Characteristics of allogeneic transplants (given as first transplant or in a tandem autologous-allogeneic strategy).
Haematologica | 108 - April 2023 1111 ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al.
patient does not have a complete response to induction therapy our model predicts that auto-allo is a superior approach with regard to survival.
In one of the few prospective studies in pPCL, the Intergroupe Francophone du Myélome (IFM) published results on 40 patients examining tandem auto-allo or tandem auto and maintenance therapy.23 The PFS and OS were better in the tandem auto and maintenance group than in the auto-allo group. The median PFS was 18.5 months for the tandem auto-allo patients and 50 months for the tandem auto and maintenance group, while the median OS was 39.3 months for the tandem auto-allo group and not reached in the tandem auto and maintenance group. Although we cannot draw direct comparisons between the IFM study and our findings it can be seen from the OS curves (Online Supplementary Material S1A) that the OS for the auto-allo group is comparable to the median OS in the IFM study. The median PFS reported by the IFM is higher than that observed in our study (Online Supplementary Material S1B) but the lack of maintenance in our cohort likely accounts for this.
There is growing evidence to indicate that consolidation and maintenance treatment improve PFS and OS in myeloma.24 Maintenance therapy is now standard of care for patients with myeloma following an autologous transplant. Although the findings in the IFM study appear encouraging, the number of patients who received maintenance is too small to draw firm conclusions on the role of maintenance therapy after transplantation in pPCL. This is an important area for future studies to consider and is currently being examined in the phase II
EMN12/HOVON129 study, one of the few prospective clinical trials underway in patients with pPCL. This trial is exploring the use of carfilzomib and lenalidomide induction (KRd), consolidation and maintenance in both young and elderly pPCL patients. The results of the first interim analysis included 33 patients under 65 years and 12 patients over 65 years old. It reported that KRd induced deep hematologic responses after four cycles of therapy (very good partial response or better in 80% and complete response in 33%) without early deaths.25
Our findings have focused on younger, transplant-eligible patients; older and less fit patients not eligible for transplantation treatment should be scheduled for personalized, continuous treatments, aiming to keep the patients on therapy for as long as possible.8
The initial results from EMN12/HOVON 129 are encouraging regarding efficient and rapid disease control with KRd induction. The importance of bringing pPCL under control early is vital to avoid early mortality in this aggressive plasma cell disorder. Due to the high incidence of t(11;14) translocation in pPCL bcl-2 inhibitors may play a role in pPCL in the near future.26,27 Monoclonal antibodies such as daratumumab and elotuzumab, directed against CD38 and SLAMF7, respectively, are currently widely used in multiple myeloma and may have a role in improving complete response rates in pPCL as has been shown in multiple myeloma.8 It is important to improve the outcomes of pPCL by combining highly effective (targeted) induction therapy to increase the chances of achieving CR prior to first transplant, followed by the selection of the most appropriate transplant modality in accordance with
In a model with interactions°:
°Models with interaction terms: only the hazard ratios for Tandem autologous transplants combined with Disease status are shown. The P value for the interaction was 0.060 for overall survival and 0.003 for progression-free survival. HR: hazard ratio; 95% CI: 95% confidence interval; CR: complete response; Allo-first: single allogeneic transplant; Tandem auto-allo; autologous hematopoietic stem cell transplant followed by an allogeneic transplant; Tandem auto-auto; tandem autologous hematopoietic stem cell transplants.
OS PFS HR 95% CI P value HR 95% CI P value Age: effect of +1 year 1.01 1.00-1.02 0.064 1.01 1.00-1.02 0.146 Disease status: no CR vs. CR 1.31 1.06-1.62 0.014 1.31 1.08-1.58 0.005 Allo-first, effect within 100 days 5.74 2.66-12.4 <0.001 2.84 1.57-5.15 0.001 Allo-first, effect after 100 days 0.92 0.61-1.38 0.677 0.83 0.57-1.20 0.317 Tandem auto-allo, effect within 100 days 0.89 0.45-1.79 0.751 1.01 0.62-1.64 0.967 Tandem auto-allo, effect after 100 days 0.80 0.59-1.08 0.148 0.69 0.52-0.92 0.012 Tandem auto-auto 0.81 0.60-1.08 0.144 0.86 0.67-1.11 0.254
Tandem auto-auto, no CR 0.94 0.68-1.28 0.678 1.08 0.82-1.42 0.602 Tandem auto-auto, CR 0.44 0.21-0.91 0.026 0.39 0.21-0.73 0.003
Table 4. Cox models for comparison of transplant strategies.
Haematologica | 108 - April 2023 1112 ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al.
the findings of the current analysis. Further international trials will be needed to determine the way forward, combining these agents with transplant strategies as outlined above.
As with all registry studies there are drawbacks in this work. The comparison of different transplant strategies could not be done based on information on intent-totreat, thus although the analyses were adjusted for the main baseline characteristics related to the administration of an elective second transplant (by use of Cox models or, not shown, propensity score matching) we cannot exclude a residual indication bias. The single auto group is, by construction, likely to include all patients who experienced an early relapse, and this could in part account for the worse outcome of this group compared to the groups of patients undergoing tandem transplants; however, the prevalence of relapse or progression as post-transplant response is limited (3.6%) (Online Supplementary Table S3 ). There was also a wide heterogeneity in treatments, for example for allogeneic transplantation we have described differences in modalities including the use of total body irradiation and donor lymphocyte infusion. While all of these factors may have relevance the potential number of subgroups generated would render statistical analysis meaningless. On the other hand it is unlikely that a series of interventional prospective studies could be set up for this rare disease such as to achieve strong evidence in favor of one of the multiple possible strategies. Our study is therefore an important source of background information for future
References
1. Gavriatopoulou M, Musto P, Caers J, et al. European myeloma network recommendations on diagnosis and management of patients with rare plasma cell dyscrasias. Leukemia. 2018;32(9):1883-1898.
2. van de Donk, Lokhorst HM, Anderson KC, Richardson PG. How I treat plasma cell leukemia. Blood. 2012;120(12):2376-2389.
3. Suska A, Vesole DH, Castillo JJ, et al. Plasma cell leukemia –facts and controversies: more questions than answers? Clin Haematol Int. 2020;2(4);133-142.
4. Iriuchishima H, Ozaki S, Konishi J, et al. Primary plasma cell leukemia in the era of novel agents: a multicenter study of the Japanese Society of Myeloma. Acta Haematol. 2016;135(2):113-121.
5. Gonsalves WI, Rajkumar SV, Go RS, et al. Trends in survival of patients with primary plasma cell leukemia: a population based analysis. Blood. 2014;124(6):907-912.
6. D'Arena G, Valentini CG, Pietrantuono G, et al. Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukemia: a retrospective study from GIMEMA Multiple Myeloma Working Party. Ann Oncol. 2012;23(6):1499-1502.
7. Musto P, Rossini F, Gay F, et al. Efficacy and safety of bortezomib in patients with plasma cell leukemia. Cancer.
studies. The use of proper statistical methodology to deal with the delayed definition of treatment groups was essential to avoid the time bias that typically affects retrospective comparisons. Additionally, our study did not assess the role of induction or maintenance therapy. However, most patients were unlikely to have received maintenance treatment after their auto, since their first transplant was performed in 2014 or earlier.
Thus, in conclusion, this study reinforces that significant NRM occurs in patients undergoing allo as a first transplant. Patients require careful selection and individual risk assessment when considering an allo. Our study supports a tandem transplant approach of upfront auto followed by either tandem allo or auto and our data suggest that remission status and especially a complete response prior to first transplant is an important determinant in selecting the optimal form of treatment for patients with pPCL.
Disclosures
No conflicts of interest to disclose.
Contributions
All authors submitted data and approved the manuscript. SI was responsible for the statistics . SL, SI, SS and CM designed the study and wrote the manuscript.
Data-sharing statement
The final analysis dataset will be available upon specific request to the Working Party, which can be sent to the corresponding author.
2007;109(11):2285-2290.
8. Musto P, Statuto T, Valvano L, et al. An update on biology, diagnosis and treatment of primary plasma cell leukemia. Expert Rev Hematol. 2019;12(4):245-253.
9. Brink M, Visser O, Zweegman S, et al. First-line treatment and survival of newly diagnosed primary plasma cell leukemia patients in the Netherlands: a population based study, 19892018. Blood Cancer J. 2021;11(2):22.
10. Katodritou E, Terpos E, Delimpasi S, et al. Real-world data on prognosis and outcome of primary plasma cell leukemia in the era of novel agents: a multicenter national study by the Greek Myeloma Study Group. Blood Cancer J. 2018;8(3):31.
11. Musto P, Simeon V, Martorelli MC, et al. Lenalidomide and low dose dexamethasone for newly diagnosed primary plasma cell leukemia. Leukemia. 2014;28(1):222-225.
12. Mina R, Joseph NS, Kaufman J, et al. Survival outcomes of patients with primary plasma cell leukemia (pPCL) treated with novel agents. Cancer. 2019;125(3):416-423.
13. Nandakumar B, Kumar S, Dispenzieri, et al. Clinical characteristics and outcomes of patients with primary plasma cell leukemia in the era of novel agent therapy. Mayo Clin Proc. 2021;96(3):677-687.
14. Fernandez de Larrea C, Kyle RA, Durie B, et al. Plasma cell
Haematologica | 108 - April 2023 1113 ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al.
leukemia. Consensus statement on diagnostic requirements, response criteria, and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013;27(4):780-791.
15. Levovic D, Zhang L, Alsina M, et al. Clinical outcomes of patients with primary plasma cell leukemia in the era or novel therapies and haematopoietic stem cell transplantation strategies: a single-institution experience. Clin Lymphoma Myeloma Leukemia. 2011;11(6):507-511.
16. Drake MB, Iacobelli S, Morris C, et al. European Group for Blood and Marrow Transplantation and the European Leukemia Net. Primary plasma cell leukemia and autologous stem cell transplantation. Haematologica. 2010;95(5):804-809.
17. Mahindra A, Kalaycio ME, Vela-Ojeda J, et al. Hematopoietic cell transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia. 2012;26(5):1091-109.
18. Dhakal, B, Patel S, Girnus S, et al. Hematopoietic cell transplantation utilization and outcomes for primary plasma cell leukemia in the current era. Leukemia. 2020;34(12):3338-3347.
19. Iacobelli S. Suggestions on the use of statistical methodologies in studies of the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2013;48(Suppl 1):S1-37.
20. van Houwelingen HC, Putter H., Dynamic predicting by landmarking as an alternative for multi-state modeling: an application to acute lymphoid leukemia data. Lifetime Data
Anal. 2008;14(4):447-463.
21. Gahrton G, Iacobelli S Bjorkstrand, et al. Autologous/reducedintensity allogeneic stem cell transplantation vs autologous transplantation in multiple myeloma: long-term results of the EBMT-NMAM2000 study. Blood. 2013;121(25):5055-5063.
22. Bruno B, Rotta M, Patriarca F, et al. A comparison of allografting with autografting in newly diagnosed myeloma. N Engl J Med. 2007;356(11):1110-1120.
23. Royer B, Diouf M, Roussel M, et al. Long term follow-up of hematopoietic stem cell transplantation (HSCT) for primary plasma cell leukemia (pPCL): final results of a prospective study of IFM group. Blood. 2016;128(22):4612.
24. Mohty M, Richardson PG, McCarthy PL, Attal M. Consolidation and maintenance therapy for multiple myeloma after autologous transplantation: where do we stand? Bone Marrow Transplant. 2015;50(8):1024-1029.
25. Van De Donk N, Van der Holt B, Schjesvold FH, et al. Treatment of primary plasma cell leukemia with carfilzomib and lenalidomide-based therapy: results of the first interim analysis of the phase 2 EMN12/HOVON129 study. Blood. 2019;134(Suppl 1):653.
26. Naighranyan S, Singh AP, Schinke C. The combination of venetoclax, bortezomib and dexamethasone for the treatment of refractory primary plasma cell leukaemia. Am J Haematol. 2020;95(2):E34-E35.
27. Touzeau C, Maciag P, Amiot M, Moreau P. Targeting Bcl-2 for the treatment of multiple myeloma. Leukemia. 2018;32(9):1899-1907.
Haematologica | 108 - April 2023 1114 ARTICLE - Autologous and allogeneic transplantation in PCL S. Lawless et al.
An objective assessment in newly diagnosed multiple myeloma to avoid treatment complications and strengthen therapy adherence
Correspondence: M. Engelhardt monika.engelhardt@uniklinik-freiburg.de
Received: May 30, 2022.
1Department of Medicine I Hematology and Oncology; 2Comprehensive Cancer Center Freiburg (CCCF), and 3Clinical Trials Unit, Medical Center - University of Freiburg, Faculty of Medicine, Freiburg, Germany
Abstract
Accepted: October 27, 2022.
Early view: November 3, 2022.
https://doi.org/10.3324/haematol.2022.281489
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
In heterogeneous multiple myeloma (MM) patients treatment decisions are challenging. The hypothesis was that adaptation of treatment intensity (dose reduction [DR] vs. none) according to an objective risk score (revised-myeloma comorbidity index [R-MCI]) rather than physician judgement alone may improve therapy efficacy and avoid toxicities. We performed this study in 250 consecutive MM patients who underwent a prospective fitness assessment at our center, after having received induction protocols based on physicians’ judgement. DR, serious adverse events (SAE), response, progression-free survival (PFS) and overall survival (OS) were compared in fitness (fit, intermediate-fit, frail), age (<60, ≥70 years [y]) and therapy intensity subgroups at baseline and follow-up. Fit and <60 y patients were mostly treated with full intensity, whereas frail and ≥70 y patients usually received DR. Hematological and non-hematological SAE were more frequently seen in frail versus ≥70 y patients. Dose adaptations were mainly necessary in frail patients. OS and PFS were similar in fit and intermediate-fit but significantly worse in frail patients (P=0.0245/P<0.0001), whereas in age-based subgroups, OS and PFS differences did not reach significance (P=0.1362/P=0.0569). Non-hematological SAE were another negative predictor for impaired OS and PFS (P=0.0054/P=0.0021). In the follow-up performed at a median of 11 months after the first fitness assessment, the R-MCI improved or remained stable in 90% versus deteriorated in only 10% of patients. In conclusion, separation by R-MCI/frailty-defined subgroups was superior to age-based subgroups and can be used to improve tailored treatment. Fitter patients benefit from intensive therapies, whereas frail patients bear a need for initial DR.
Introduction
Multiple myeloma (MM) is a hematological disease, which typically affects elderly patients.1 In the past decade, treatment options have substantially evolved and with inclusion of proteasome inhibitors (PI), immunomodulatory drugs (IMiDs) and antibodies (Ab)/immunotherapies into induction and relapse protocols, response rates, progression-free survival (PFS) and overall survival (OS) have improved impressively.2 Standard treatment in newly diagnosed MM (NDMM) includes triplets or quadruplets, plus - if patients are deemed fit enough - autologous stem cell transplantation (ASCT), followed by maintenance therapy.3,4 Even though patient assessment, via age,
Karnofsky Performance Status (KPS), comorbidities, patient history and examination, is performed, MM patients’ actual constitution and fitness can be over- or underestimated.5-8 Additionally, inclusion in clinical trials is especially rare in elderly patients over the age of 70 years.5,6 This suggests that more objective tools may assist physicians to find most suitable treatment regimens and to adapt dose intensity for elderly and/or MM-stricken patients.
In line, the long continuing COVID-19 pandemic imposes the need to prevent any unnecessary serious adverse events (SAE), hospitalization, time-consuming dose adjustments and therapy cessations.9-13 However, therapy decisions are often made without an objective fitness as-
Maximilian Holler,1,2 Gabriele Ihorst,3 Heike Reinhardt,1,2 Amelie Rösner,1,2 Magdalena Braun,1,2 Mandy-Deborah Möller,1,2 Esther Dreyling,1,2 Katja Schoeller,1,2 Sophia Scheubeck,1,2 Ralph Wäsch1,2 and Monika Engelhardt1,2
Haematologica | 108 - April 2023 1115 ARTICLE - Plasma Cell Disorders
sessment.7,14,15 In recent years, MM-specific risk scores (e.g., International Myeloma Working Group (IMWG)-frailty score, revised-myeloma comorbidity index (R-MCI), Mayorisk score, UK Myeloma Research Alliance Risk Profile) were developed to assist in this unsolved matter.16-19
The hypothesis of this study was that adaptation of therapy intensity according to an objective risk score (via RMCI; Online Supplementary Figure S1), rather than via physician judgement alone, may improve therapy efficacy and avoid therapy toxicities and discontinuation. More studies are now testing the feasibility of MM-specific riskscores for treatment assistance in MM patients, albeit additional studies should further be performed.14,20-22 We used the R-MCI, because this constitutes a repeatedly validated MM-specific risk tool, used routinely for MM patients at our institution, that has been integrated into our electronic tumor board (TB) online system.17,23 The R-MCI contains of five weighted risk factors, namely renal and lung function, KPS, frailty and age, plus allows to include cytogenetics.17,23 The R-MCI web tool allows the immediate calculation of the weighted R-MCI, which can be likewise performed by physicians or research assistants (www.myelomacomorbidityindex.org). We here investigated the applicability of the R-MCI for future therapy decision support by performing an analysis of patients receiving first-line treatment. Main aspects of this study were to track patients’ induction treatment, comparing RMCI- versus age subgroups in terms of therapy adaptations, SAE, response, OS and PFS. Since patients’ constitution and disease burden may change during treatment, we also re-evaluated constitution, fitness and R-MCI changes in a follow-up analysis (Online Supplementary Figure S2).
Methods
Data sources and study design
We performed this exploratory study in 250 consecutive MM patients, who received induction treatment ideally continuing until intolerance or progression at our Comprehensive Cancer Center Freiburg (CCCF) and catchment area of the Black Forest from 2000 to 2018. All patients were fully documented at our CCCF. The cohort with prospectively assessed R-MCI was pooled from two prior conducted analyses,8,17 retrieving patient- and therapy-relevant data through our electronic documentation system ‘Medoc’. Of the initial 359 patients, 109 had to be excluded either due to ongoing induction at the time of assessment (n=50) or incomplete data (n=59; Online Supplementary Figure S2 ). Patients’ characteristics included the International Staging System (ISS), R-MCI and IMWG-frailty scores at baseline. Via R-MCI and IMWGfrailty scores, all patients were grouped as fit, intermedi-
ate-fit or frail (Table 1; Online Supplementary Figure S1). Decisions of induction regimen, treatment intensity and dose reductions (DR) were based on physicians’ choice. Comparisons were performed for frailty- (R-MCI-fit, -intermediate-fit, vs. -frail), age- (<60, 60-69 vs. ≥70 years) and therapy-intensity (with vs . without initial DR) subgroups.
The study was performed according to the guidelines of the Declaration of Helsinki and Good Clinical Practice. All patients gave their written informed consent for institutionally initiated research studies and analyses of clinical outcome studies conforming to the institutional review board guidelines. The trial protocol was approved by the ethics committee of the University of Freiburg (EV 81/10).
Induction, dose reductions, serious adverse events, response and follow-up analysis
For each induction regimen, one lead agent was determined. Albeit any DR of any drug in combination first-line treatment could have been counted as a dose modification, this would not have allowed a less complex intensity calculation. Thus, any decrease in the lead agents’ standard dose intensity or change from triplets to doublets was defined as DR. Lead agents were defined, with regard to the severity of adverse events (AE) in the following order of priority: alkylating agents, subsequently IMiDs and PI/Ab, last anthracyclines or glucocorticoids. The standard dose was consistent with NCCN/EMN-guidelines and CCCF-chemotherapy manual.24
We used the Common Terminology Criteria (CTC) for AE version 5.0 to assess grade 3-4 hematological SAE (anemia, leukocytopenia, thrombocytopenia) and non-hematological SAE grades 3-5 (infections, renal, pulmonary, cardiac impairment).
Quality of response was assessed via IMWG-remission criteria until the end of induction.25,26
Six to 24 months after the first fitness assessment, we analyzed, if changes in remission status and patients’ constitution via a follow-up R-MCI analysis had occurred.
Statistical analysis
OS was defined as the time from start of induction to death from any cause and PFS as the time from start of induction to cancer recurrence or death from any cause. Data for patients alive at the time of the analysis were censored at the last follow-up. Probabilities of PFS and OS were estimated using Kaplan-Meier method and compared with log-rank tests. In order to avoid an immortal time bias, plots of OS and PFS of patients showing no or at least one SAE, included only those who survived at least 6 months after the start of induction treatment. Chi-squareand Mantel-Haenszel tests were utilized as appropriate in the comparisons of therapy protocols, DR, SAE, response rates in R-MCI and age groups. A P value of <0.05 was con-
Haematologica | 108 - April 2023 1116 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
sidered as statistically significant. Data were analyzed with SAS 9.2 (SAS Institute, Inc, Cary, North Carolina).
Results
Patient characteristics’ and induction regimen
Among the 250 analyzed patients the median age at baseline was 62 years and 61% were males. For a cohort, where first-line treatment was mainly applied at a tertiary and referral center, patient characteristics were typical (Table 1), likewise myeloma subtypes, ISS, median bone marrow infiltration (45%) underlying AL-amyloidosis rate (6%), and cytogenetics. ISS stage 2 and 3 were most common with 66% (Table 1).
Median IMWG-frailty score and R-MCI were 1 and 4, respectively, in line with prior test and validation analyses.15,17,23,27,28 Impaired constitution via KPS ≤70% was present in 42% and moderate or severe frailty was reported in 36%.8,17,23 Renal impairment with eGFR <60 mL/min/1.73m2 was present in 40% and moderate or se-
vere lung dysfunction as defined via R-MCI website in 12%, in line with previous analyses.8,17,23,29,30
VCD, VRd/RAD or other induction (Online Supplementary Figure S3) was predominantly performed with VCD and whenever possible in German study group (DSMM)-protocols (DSMM XI, XII, XIV).31,32 Induction was often initiated at the CCCF, but also at regional hospitals and private practices. Stem cell transplantation (SCT) was performed in 72% (Table 1).
Comparisons of patient- and therapy-relevant parameters in entire cohort, and in revised myeloma comorbidity index, age and dose intensity subgroups Patient and therapy parameters are summarized in Table 2 in entire cohort and in fit versus frail, younger versus older and full-dosed (no DR) and dose-reduced (initial DR) subgroups. No initial DR versus DR were performed in 59% and 41%, respectively, reflecting the complexity to treat an even fairly young MM cohort, and that initial DR was frequently performed. The number of hematological and non-hematological SAE were frequent with 157 and 123,
VCD/VRD/RAD/other*4
Place
Performed SCT/non-SCT
176/35/39 (70/14/16)
198/52 (79/21)
179/71 (72/28)
IMWG: International Myeloma Working Group; interm.: intermediate-fit; R-MCI: revised myeloma comorbidity score; KPS: Karnofsky performance status; eGFR: estimated glomerular filtration rate; VCD: bortezomib, cyclophosphamide, dexamethasone; VRd: bortezomib, lenalidomide, dexamethasone; RAD: lenalidomide, adriamycin, dexamethasone; CCCF: Comprehensive Cancer Center Freiburg; SCT: stem cell transplant. *1Unfavorable: del17p13, t(4;14), t(14;16), t(14;20), chromosome 1 abnormalities, c-myc, del(13q14), hypodiploidy. *2Frailty8,17,23: Karnofsky Index ≤70%; Time Up/Go >10 seconds; IADL ≤4 points; subjective fitness grade E or F. *3Lung dysfunction: mild: FEV1 <80%; moderate: FEV1 ≤60% or diffusion capacity ≤61%, severe: FEV1 <50%; mild: 0/1 parameter is correct; moderate: 2 parameters are correct; severe: >2 parameters are correct. *4Others: proteasome inhibitors, antibodies, anthracyclines, glucocorticoids. *5Others: regional hospitals/private practices.
N (%) Median (range) Age, years 62 (27-92) Sex, male/female 153/97 (61/39) Myeloma subtype IgG/IgA IgM Light chain only/biclonal/asecretory Κ/λ/biclonal/asecretory 131/47/2 (52/19/1) 64/2/4 (25/1/2) 160/88/1/1 (64/35/0.5/0.5) International Staging System, I/II/III 84/71/95 (34/28/38) Bone marrow infiltration, cytology/histopathology (%) 35/45 (0-90/0-100) AL-Amyloidosis, yes/no 16/234 (6/94) Cytogenetics, favorable/unfavorable*1/ missing 109/112/29 (43.6/44.8/11.6) Comorbidity indices IMWG-frailty score: fit/interm./frail R-MCI: fit/interm./frail 75/86/89 (30/34/36) 73/145/32 (29/58/13) 1 (0-4) 4 (0-9) KPS, 100/80-90/≤70% 15/130/105 (6/52/42) Frailty,*2 no/mild:moderate/severe
(64:36)
function, EGFR ≥90/89-60/<60 mL/min 50/101/99 (20/40/40)
dysfunction,*3 no/mild:moderate/severe 221:29 (88:12)
160:90
Renal
Lung
Therapy induction
of induction: CCCF/others*5
Table 1. Baseline multiple myeloma patient characteristics (n=250).
Haematologica | 108 - April 2023 1117 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
which accounted for SAE per patient in 0.63 and 0.49, respectively. The median induction duration was 62 days. Best IMWG response (≥ partial response [PR]) after induction was observed in 75% in line with prior data (Table 2).31 Results of R-MCI fit and younger (<60 years) patients were similar and merely differed in used protocols and SAE (Table 2). Intermediate-fit and frail patients (combined: 71%) showed increased median ages of 66 and 74 years, respectively. VRd/RAD first-line treatment in fit, intermediate-fit and frail patients were performed in 37%, 15% and 6%, thus decreased with frailty, whereas DR substantially increased (19%, 45% and 72%, respectively; Table 2). In line, SAE per patient increased from 0.23 in fit, to 0.72 in intermediate-fit and 1.1 in frail patients for hematological SAE, and 0.23, 0.48 and 1.13 for non-hematological SAE. Median induction duration in intermediate-fit patients was similar to fit patients, basically because SCT in intermediate-fit patients remained considerable with 72%. As expected, in frail patients longer induction (6-9 cycles plus maintenance) was performed and less SCT (Table 2). Divided into fit, intermediate-fit and frail patients, the response rates were 73%, 77% and 69%, respectively, thus higher in both former than latter subgroup (Table 2).
According to age subgroups, DR increased in <60, 60-69 and ≥70-year-old patients from 21% to 43% and 66%, respectively. SAE per patients increased less and seemed less predictable than in R-MCI subgroups (Table 2). Transplant-eligible patients aged 60-69 years did hardly show any differences in non-hematological SAE compared to patients ≥70 years (0.58/patient vs. 0.62/patient). SCT frequencies were typical in young in 93%, 81% in 60-69 and 32% in ≥70-year-old patients. Transplant-ineligible patients (≥70 years) were at treatment initiation in 29 of 73 (40%) >75 years old. Best responses (≥PR) in age cohorts ranged from 66-79% (Table 2).
In dose intensity subgroups, for patients without versus with DR, median age differences and R-MCI-differences were notable with 58 versus 69 years and 4 versus 5, respectively. This was in line with lesser performed SCT in the latter group (Table 2). Whereas hematological SAE/patient were similar in patients without versus with DR, these almost doubled for non-hematological SAE (0.36 versus 0.69/patient, respectively). Median induction duration was similar in both groups, while as expected, response rates (≥PR) were increased in patients without DR (Table 2).
DR: dose reduction; R-MCI: revised-myeloma comorbidity index; VCD: bortezomib, cyclophosphamide, dexamethasone; IMiDs: immunomodulatory drug; RAD: lenalidomide, adriamycin, dexamethasone; VRd: bortezomib, lenalidomide, dexamethasone; SAE: serious adverse event; SCT: stem cell transplant; CR: complete remission; vgPR: very good partial remission; PR: partial remission; SD: stable disease; PD: progressive disease. *1Lead agent prioritization was based on expected severity of adverse events; lead agents were rated: alkylating agents (mainly VCD) first, subsequently IMiDs (mainly RAD/VRd) and PI/Ab, and last anthracyclines or glucocorticoids; i.e., in VCD: cyclophosphamide was the lead agent. *2Others: proteasome inhibitors, antibodies, anthracyclines, glucocorticoids.
Entire group Fit Intermediate Frail <60 years 60-69 years ≥70 years No initial DR Initial DR Patients, N (%) 250 (100) 73 (29) 145 (58) 32 (13) 101 (40.4) 76 (30.4) 73 (29.2) 148 (59) 102 (41) Median age, years (range) 62 (27-92) 55 (29-77) 66 (27-85) 74 (61-92) 53 (27-59) 65 (60-69) 75 (70-92) 58 (29-80) 69 (27-92) Median R-MCI (range) 4 (0-9) 3 (0-3) 5 (4-6) 7 (7-9) 3 (0-6) 4 (2-8) 6 (2-9) 4 (0-9) 5 (2-9) Lead agents,*1 N (%) Alkylating agents IMiDs Others*2 190 (76) 51 (20) 9 (4) 42 (57.5) 27 (37) 4 (5.5) 119 (82) 22 (15) 4 (3) 29 (91) 2 (6) 1 (3) 71 (70) 24 (24) 6 (6) 56 (73.7) 18 (23.7) 2 (2.6) 63 (86.3) 9 (12.3) 1 (1.4) 109 (74) 37 (25) 2 (1) 79 (77) 12 (12) 11 (11) Initial DR, N (%) No Yes 148 (59) 102 (41) 59 (81) 14 (19) 80 (55) 65 (45) 9 (28) 23 (72) 80 (79) 21 (21) 43 (57) 33 (43) 25 (34) 48 (66) 148 (100) 0 (0) 0 (0) 102 (100) Hematological SAE, N (per patient) 157 (0.63) 17 (0.23) 105 (0.72) 35 (1.1) 54 (0.53) 44 (0.58) 59 (0.81) 92 (0.62) 65 (0.63) Non-hematological SAE, N (per patient) 123 (0.49) 17 (0.23) 70 (0.48) 36 (1.13) 34 (0.34) 44 (0.58) 45 (0.62) 53 (0.36) 70 (0.69) Median induction duration, days (range) 62 (2-365) 61 (14-365) 61 (2-299) 80 (5-271) 59 (10-217) 62 (2-365) 78 (5-299) 61 (5-299) 65 (2-365) Performed SCT, N (%) 179 (72) 68 (93) 104 (72) 7 (22) 94 (93) 62 (81) 23 (32) 126 (85) 53 (52) Obtained best response, N (%) CR/vgPR/PR SD/PD 187 (75) 63 (25) 53 (73) 20 (27) 112 (77) 33 (23) 22 (69) 10 (31) 79 (78) 22 (22) 50 (66) 26 (34) 58 (79) 15 (21) 118 (80) 30 (20) 69 (68) 33 (32)
Table 2. Patient and therapy parameters in entire cohort, in revised myeloma comorbidity score, age and dose intensity subgroups.
Haematologica | 108 - April 2023 1118 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
Serious adverse events: subgroup distribution for hematological and non-hematological serious adverse events
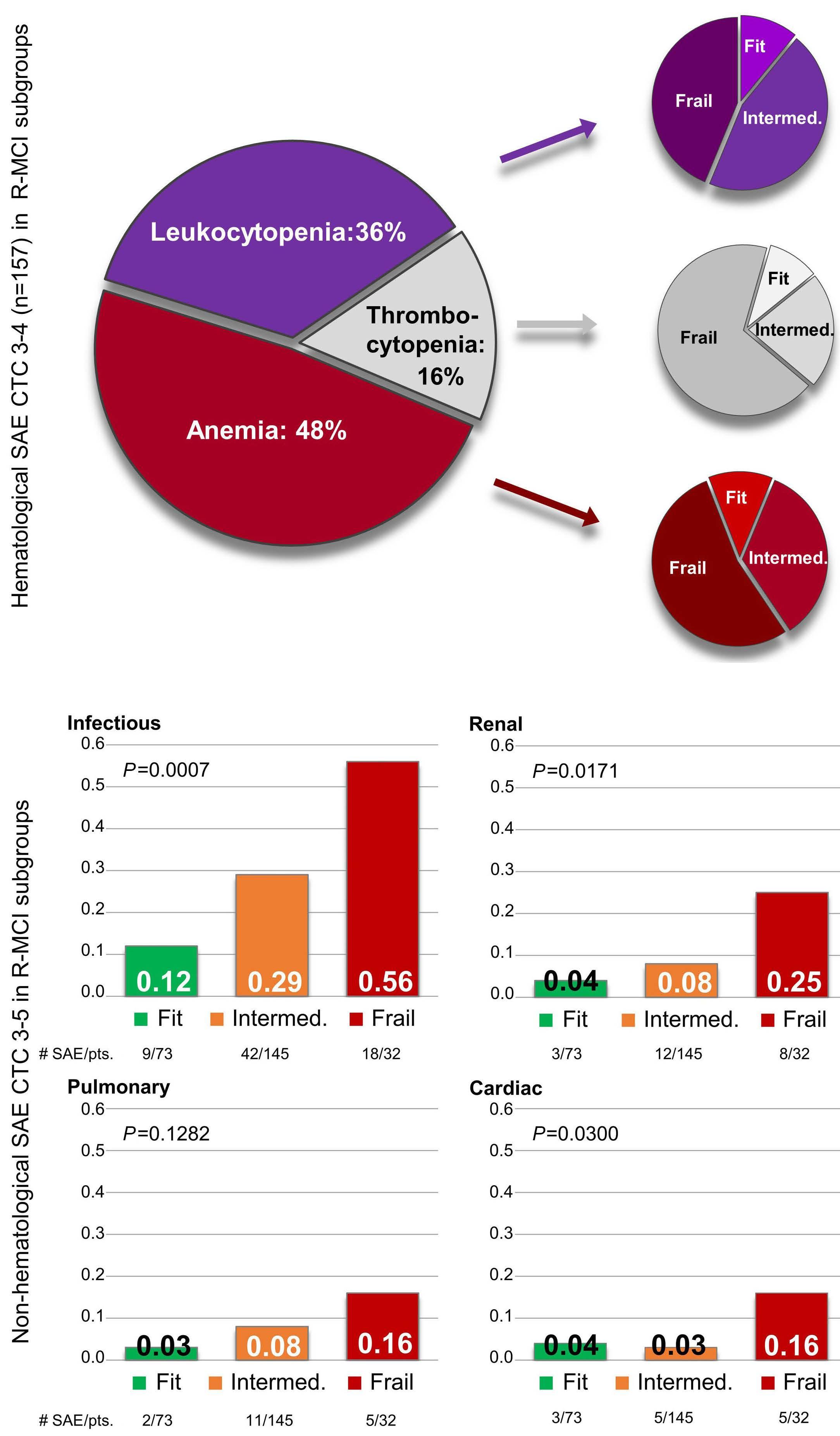
Distinct differences in types of hematological and nonhematological SAE are shown in Figure 1A and B, being more prevalent in frail than intermediate-fit or fit patients.
Anemia, leukocytopenia and thrombocytopenia SAE (CTC grade 3-4) occurred in 76, 56 and 25 patients, respectively. Leukocytopenia was equally prevalent in frail and intermediate-fit patients (44% and 45%, respectively), while anemia and thrombocytopenia were predominantly observed in frail patients (59% and 68%, respectively; Figure 1A). Hematological SAE appeared in patients
Figure 1. Serious adverse events in revised-myeloma comorbidity index subgroups. (A) Hematological serious adverse events (SAE) common toxicity criteria (CTC) 3-4: distribution in entire cohort and revised-myeloma comorbidity index (R-MCI) subgroups in percentage. Leukocytopenia: P=0.0008; thrombocytopenia: P=0.0007; anemia: P<0.0001. (B) Non-hematological SAE CTC grades 3-5 in R-MCI subgroups per patient. pt.: patient. Intermed: intermediate.
A B
1119 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
Haematologica | 108 - April 2023
treated with alkylating agents (VCD) in 47% and with IMiD-based protocols (VRd/RAD) in 20% (‘others’ were too few for meaningful conclusions).
Of 123 registered non-hematological SAE, 69 were attributed to infectious, 23 to renal, 18 to pulmonary and 13 to cardiac causes (Figure 1B). Comparison of different R-MCI subgroups with infectious, renal, pulmonary and cardiac SAE showed significant increases in frail as compared to fit or intermediate-fit patients, reaching significance in all except pulmonary SAE (Figure 1B).
Therapy adaptations in frailty and age subgroups
Our assessment of SAE in patients without or with initial DR is depicted in the Online Supplementary Table S1A and B. The occurrence of hematological SAE without or with DR did not show distinct differences in frailty subgroups. Therapy adaptations (DR, therapy pauses or discontinuation) after hematological SAE occurred in intermediatefit or frail patients only, both without and with performed initial DR (Online Supplementary Table S1A).
Non-hematological SAE occurred in R-MCI subgroups likewise more often in patients without DR than if performed, whereas this was less strikingly found for age subgroups (Online Supplementary Table S1B). Therapy adjustments or therapy discontinuations after non-hematological SAE increased both with frailty and age, and were more prevalent with full doses than if DR had been performed (Online Supplementary Table S1B).
Therapy discontinuation occurred in only eight of 250 (3%) patients (Online Supplementary Table S2), showing mostly advanced age and impaired R-MCI scores. Initial dose reductions had been performed in seven of eight patients. Patient constitution complications, myelomaand/or therapy-induced complications occurred in already the 1st (n=3) to 4th (n=3) induction cycle (range, 14). Retrospectively assessed therapy intensity by us (MH, ME) suggested in seven of eight patients, that these were overdosed. Thus, if on top of clinical judgement, R-MCItailored therapy had been performed - SAE and therapy cessation might have been avoided in seven of eight patients.
Serious adverse events leading to early death (<60 days after induction) or concomitant serious adverse events
SAE during induction which led to death were seen in three patients (3/250 i.e., in 1.2%). These showed cardiac complications, two of three associated with underlying AL-amyloidosis. Sixteen patients with concomitant AL-amyloidosis contributed to 28% of cardiac (n=13) and renal (n=23) SAE (CTC 3-5) of the entire cohort, thus showed a 5.6 higher risk for these complications. As expected, patients with CTC grade 3-4 leukocytopenia were more likely to acquire severe infections (Online Supplementary Table S3).
Overall survival and progression-free survival of entire cohort and in various subgroups
The median follow-up from start of induction was 65 months (range, 1-246). Detailed response in fit, intermediate-fit and frail patients are summarized in the Online Supplementary Table S4. The Kaplan-Meier curves for OS and PFS analyses are displayed in Figures 2 and 3. The estimated 3-year OS and PFS in the entire cohort were 85% (95% confidence interval [CI]: 79-89) and 53% (95% CI: 4760), respectively (Figure 2A and B). The three R-MCI subgroups in Figure 2C and D showed similar results for both fit and intermediate-fit patients. The 3-year OS in R-MCI fit, intermediate-fit and frail was 89% (95% CI: 79-94), 85% (95% CI: 77-90) and 70% (95% CI: 47-85), respectively (P=0.0688; Figure 2C). The 3-year PFS was 60% (95% CI: 47-70), 55% (95% CI: 47-63) and 21% (95% CI: 6-41), respectively (P<0.0001; Figure 2D).
Since results of fit and intermediate-fit patients were comparable and very different to frail patients, these were combined as displayed in Figure 2E and F. Of note, 3-year OS and PFS in fit/intermediate versus frail patients showed highly significant differences of 86% (95% CI: 81-90) versus 70% (95% CI: 47-85) and 57% (95% CI: 50-63) versus 21% (95% CI: 6-41), respectively (P=0.0245/P<0.0001).
Age subgroups revealed differences with best OS/PFS for <60-year-old patients, whereas 60-69 and ≥70-year subgroups revealed similar Kaplan Meier curves. Notably, 3year OS/PFS results via age displayed lesser and insignificant group distinctions (P=0.1362/P=0.0569; Figure 2G and H).
Serious adverse events and dose reduction impact on overall survival and progression-free survival
Any SAE led to impaired OS and PFS (Figure 3A and B), both for hematological (Figure 3C and D) and non-hematological SAE (Figure 3E and F), with more striking OS and PFS differences for the latter SAE. In line, DR versus no DR led to both OS and PFS differences in favor of patients with no DR, reflecting standard doses to be easier applied in fitter and younger patients and therefore accounting for these differences (Figure 3G and H).
Revised myeloma comorbidity index follow-up analysis
The follow-up analysis (T0 → T1) was possible in 180 patients (72%; Figure 4; Online Supplementary Figure S2). The median follow-up was 11 months (range, 6-24), in line with our previous study.8 Median R-MCI scores at T0 and T1 were both 4, reflecting intermediate-fit patients. Assessing also mean T0 versus T1 differences, the R-MCI improved from 4.3 to 3.7, respectively and accounted for a mean improvement of 0.6 points over an 11 month period (P<0.0001). Among all follow-up patients, 77 (43%) achieved a better, 84 (47%) a stable and only 19 (10%) a worse R-MCI (Figure 4). Maximum R-MCI changes were
Haematologica | 108 - April 2023 1120 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
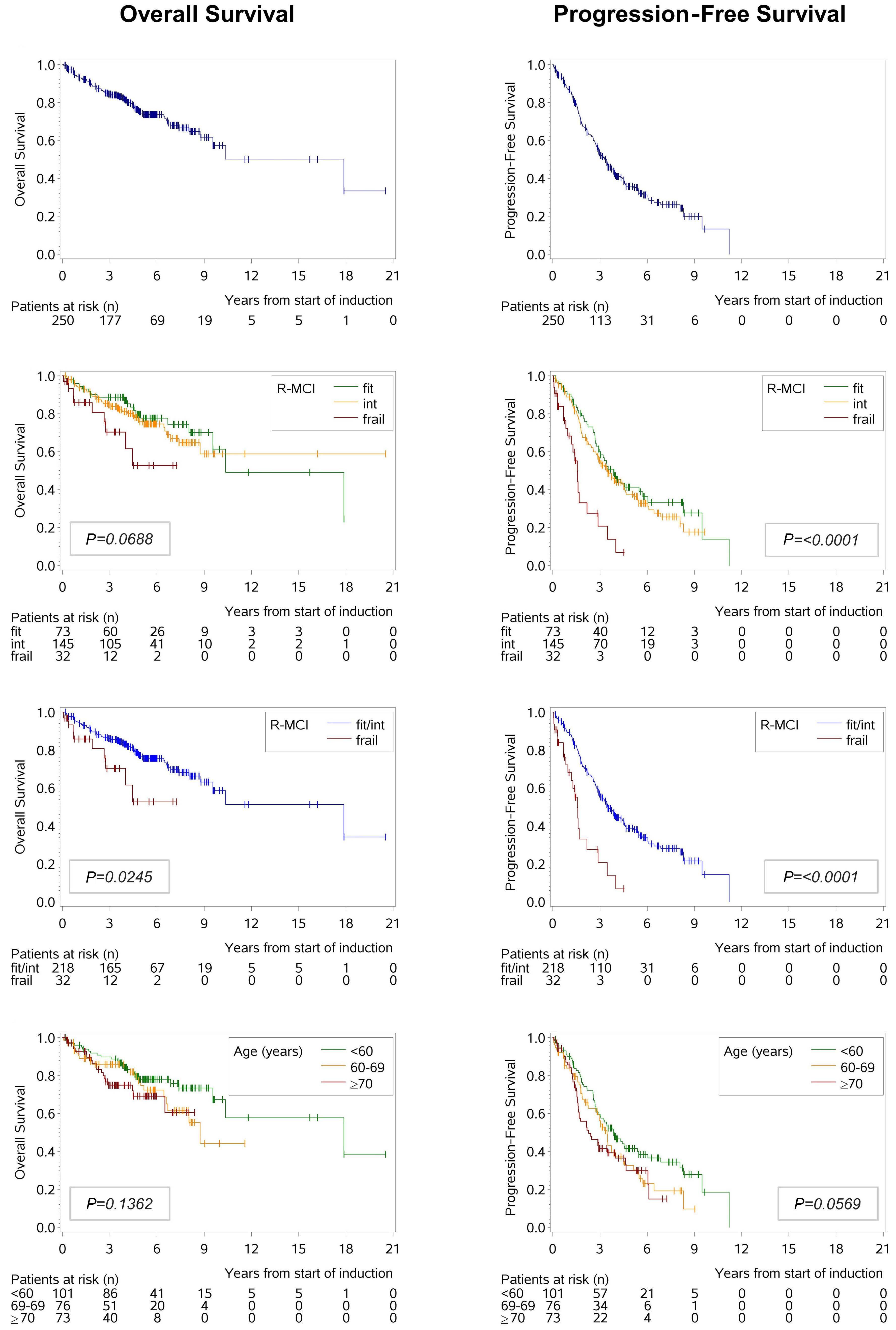
A B E F C D G H
Haematologica | 108 - April 2023 1121 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
Figure 2. Kaplan-Meier plots for entire cohort and different risk group distributions. (A) Overall survival (OS) and (B) progression-free survival (PFS) in entire cohort. (C) OS and (D) PFS in revised-myeloma comorbidity index (R-MCI) subgroups. (E) OS and (F) PFS in R-MCI subgroups fit and intermediate-fit (Int) vs. frail. (G) OS and (H) PFS in age groups.
Figure 3. Kaplan-Meier plots for patients without or with serious adverse events and patients without or with initial dose reduction. (A) Overall survival (OS) and (B) progression-free survival (PFS) in patients without or with any serious adverse events (SAE). (C) OS and (D) PFS in patients without or with any hematological SAE. (E) OS and (F) PFS in patients with or without any non-hematological SAE. (G) OS and (H) PFS in patients with or without DR. R-MCI: revised-myeloma comorbidity index; SAE: serious adverse events dose reduction; DR: dose reduction.
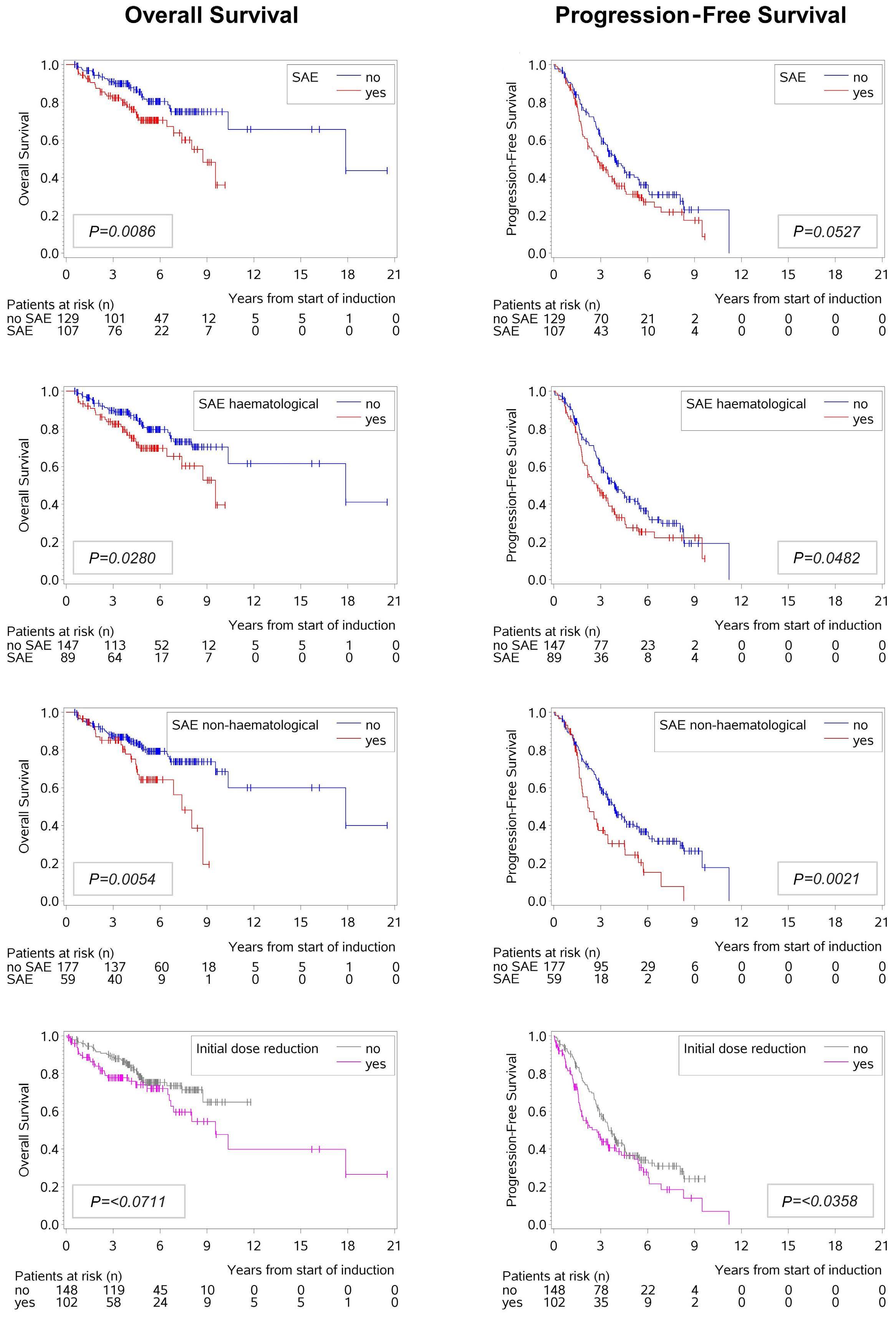
A B C D E F G H Haematologica | 108 - April 2023 1122 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
improvements by four points (R-MCI 6 → 2) and deteriorations by three points (R-MCI 4 → 7), impressively illustrating that R-MCI changes within a ~1 year follow-up can be more drastic than age shifts within this period.
Discussion
One crucial aspect for MM patients and physicians is the individual selection of the initial (and subsequent) treatment and its intensity.7,33 Our message is that we apply evidence generated from clinical trials that rarely include old or frail patients to treat such patients without knowledge of the need for modifications of the drugs used, the dosing or schedule. Although our study population received firstline MM treatment with dose modifications according to best clinical judgment and a prospective frailty assessment was performed, the frailty assessment was not used for decision making. Thus, clinicians were blinded to this information. This allowed to assign patients into those with versus without DR as compared to comorbidity and age subgroups (Table 2). Hence, we investigated physicians’ therapy decisions in a NDMM population during induction by analyzing the course of therapy and patient outcome. Our data demonstrates that the use of the R-MCI can assist to anticipate the likelihood of SAE. Those predictions can subsequently be used to optimize induction dose intensity (Table 2; Figure 1A and B) and identify patients at risk of treatment discontinuation with the associated risk of a more unfavorable outcome (Figure 2 and 3). We found compelling differences between R-MCI subgroups regarding protocols and doses prescribed. Overall, alkylating agents were the most used leading agent, in line with VCD being a commonly applied DSMM/GMMG and EMN-study regimen.31,34 With a median start of induction in 2016, most frail patients in our cohort were treated in compliance with
guidelines for dose-adjusted VCD or Vd as first-line treatment.34 The EHA-ESMO guideline from 2021 propagates nowadays to add a CD38-antibody (e.g., Dara-VMP, DaraRd) in NDMM patients ineligible for ASCT, reiterating that R-MCI- or other risk-tool-adapted triplets and quadruplets may profit from our approach described here to avoid SAE and therapy cessations.33
We were also able to show that patients at risk were mostly correctly identified and reasonable dose adaptations were made, but potential improvements remain. Moreover, we determined that fit patients had few initial DR (19%) and a low incidence of hematological or nonhematological SAE (both 0.14 per patient). In contrary, fulldosed frail patients (28%) were nine times more likely to suffer from hematological SAE and had a four times higher rate of non-hematological SAE (Online Supplementary Table S1A and B). The conjecture that some frail patients were possibly overtreated and dose-reduced fit patients undertreated is therefore plausible and has been described previously.6,17,23,34 Besides, the 3-year OS was superior in frail patients with initial DR versus in frail patients without initial DR (73% vs. 63%). Although numbers of this subgroup comparison were limited, other studies confirm this observation.11,22,35,36 Moreover, we observed that two of three events of death were associated with subclinically underlying AL-amyloidosis. These findings confirmed the need to critically evaluate induction protocols and protocol doses to avoid therapy complications and to reliably detect AL-amyloidosis.37,38 Indeed, the occurrence of any SAE was associated with a worse outcome (Figure 3A), validating prior studies.11,36,37
Since various treatment pathways continue to suggest age cut-offs (i.e., >60 or >70-years), we divided our cohort into patients aged <60, 60-69 and ≥70 years and compared them to R-MCI and therapy intensity (DR vs. no DR performed) subgroups (Table 2). In line with our and other
Figure 4. Changes in revised-myeloma comorbidity index scoring from start of induction (T0) to 6-24 months after first revised-myeloma comorbidity index assessment (T1) in 180 follow-up patients. R-MCI: revised-myeloma comorbidity index; Interm.-fit: intermediate-fit.
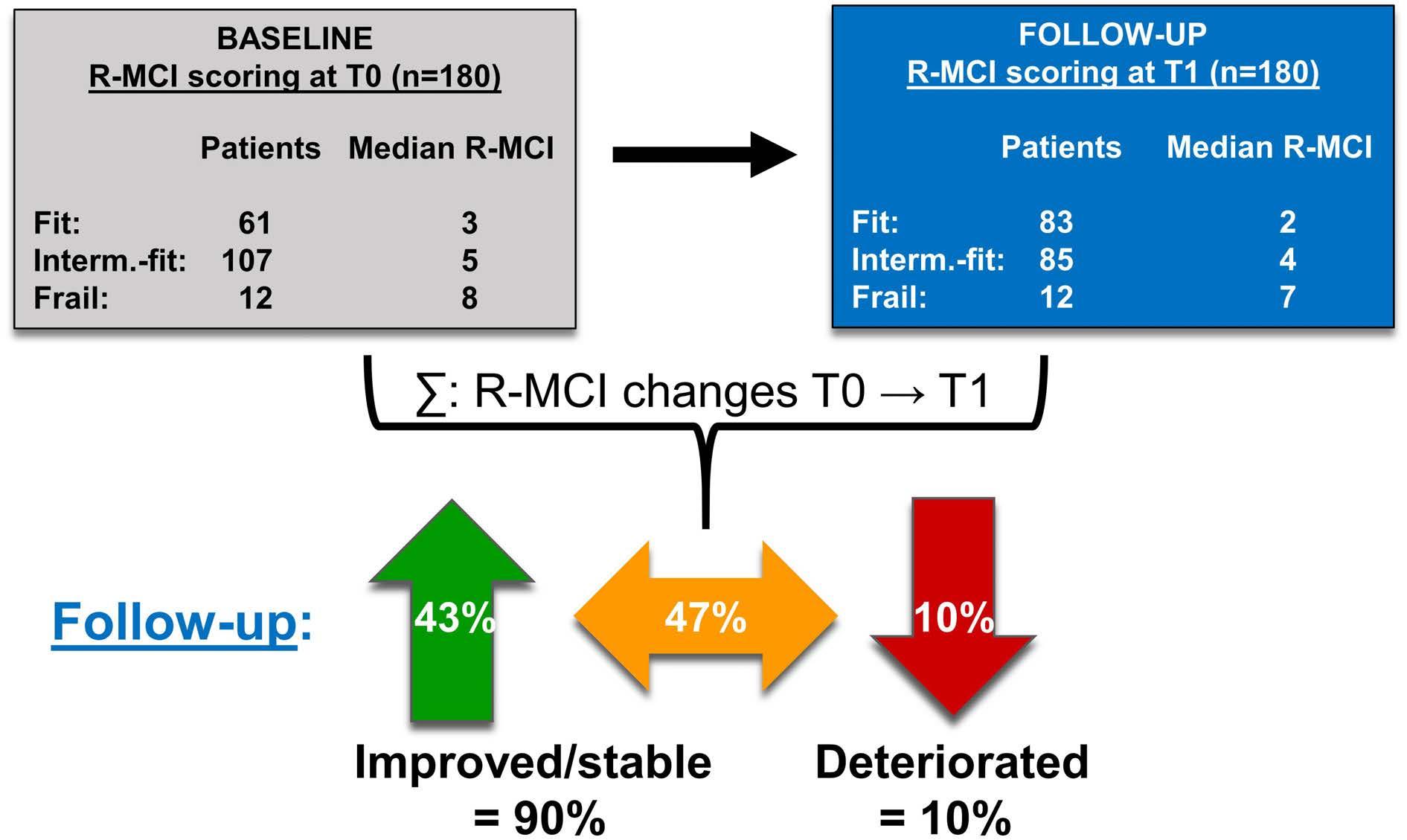
Haematologica | 108 - April 2023 1123 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
prior analyses, R-MCI subgroups differed much more than age or dose intensity subgroups, supporting the paradigm of an objective, functional GA and risk score.5,8,39-42 Notably, ≥70 and 60-69-year-old (transplant-eligible) patients showed similar results regarding SAE, OS and PFS and thereby differed from <60-year-old patients, whereas for fitness groups, fit and intermediate-fit patients were comparable and very distinct from frail patients. Since patients ≥70 years are often excluded from clinical trials and more intensive therapies, we assessed this age group more thoroughly.6,34,42,43 Of interest, 32% of our ≥70-year-old patients received a SCT after physicians’ appraisal (Table 2). Nevertheless, 56% (n=28) of patients ≥70 years, who did not receive a SCT, were classified as either intermediatefit or fit. Thus, those patients could have received intensive treatment. Larocca et al. elegantly demonstrated the benefit of a therapy-decision approach (dose-adjusted Rd-R vs continuous Rd) via IMWG-frailty index in intermediate-fit patients in a prospective study.20 An ongoing, equally important Medical Research Councils study randomizes unaltered to adjusted treatment according to fitness results (FiTNEss study, clinicaltrails gov. Identifier: NCT03720041, PI: G.Cook), both supporting our findings. Concerning patients’ outcome, we observed a substantial OS and PFS advantage in fit and intermediate-fit versus frail patients (Figure 2A and B). In line, Facon et al. propagated a simpler approach of the IMWG-frailty score, dividing patients likewise into non-frail versus frail patients, which seems straightforward for clinical routine and clinical trials.21 An understandable request is that these riskassessments should not be time-consuming and prospectively performed.7 Both, the R-MCI and IMWGfrailty index offer online tools for their automatic calculation.
Strengths of our analysis were the meticulous examination of a NDMM patient cohort, of performed treatment, doses, SAE, PFS and OS in R-MCI, age and therapy intensity subgroups. Moreover, 72% of the initially assessed patients could be included in our follow-up analysis and their constitution and fitness, as measured via R-MCI, revealed improvement or stabilization in 90% and deterioration in only 10%. We have previously described in an even more detailed functional analysis using 12 different comorbidity scores and functional tests: the assessments more frequently and significantly changed in younger patients (<70 years) and those with good response (≥PR), suggesting a better functional reconstitution in younger and responsive than in older and less responsive myeloma patients.8 These 10% of patients, whose R-MCI deteriorated in our follow-up analysis, should be reliably identified, because fittingly chosen therapies are relevant to perform and will ideally improve patients’ quality of life (QoL).8 This was also reflected in frequencies of nonhematological SAE, which were much lower in patients
with improved or stable R-MCI (both 0.39/per patient) than in those with deteriorated R-MCI (0.53/per patient). No difference in infections was observed, most likely, due to the use of detailed chemotherapy treatment plans, including strict antibiotic prophylaxis schedules therein.24 Although OS differences between these subgroups were hampered by limited patient numbers with deteriorated R-MCI, our follow-up analysis confirmed that MM treatment may indeed improve patients’ constitution. Renal impairment and/or frailty (including KPS) may recover under MM therapies, whereas in those not improving and deteriorating with QoL domains, therapeutic adjustments are important to consider.8 Additionally, our median observation period of 5.4 years was substantial, therefore our Kaplan-Meier results in all patients, in R-MCI, age, SAE-experiencing and dose-reduced patients were robust and mature. So far, there are few studies on prospective MM-specific risk tools for therapy-decision-support for NDMM and even lesser for relapsed/refractory (RR)MM patients, albeit these data and currently ongoing GIMEMA, MRC, HOVON and other studies support these endeavors.44-46 Lastly, these data impressively confirm that the R-MCI seems superior to age-based treatment pathways. Limitations of our study were the single institution approach, yet due to strict inclusion criteria regarding patients’ and therapy data, all patients included provided infinitely detailed information. Another criticism could be the heterogeneity in patients (age range, 27-92 years), with a considerable number of patients <70 years (71%), as is typical for tertiary centers in Germany (vs. more centralized institutions and countries). Since our university and catchment area-treated patient population was relatively young and the majority received ASCT, we refrained from non-ASCT versus ASCT-based subgroup analyses, but considered all patients as one group. Besides, one could criticize the use of other than VCD-induction protocols in rare subgroups. Underlying AL-amyloidosis could also been argued to be possibly excluded, which we decided against, because all patients were initially diagnosed with MM only, but determined with AL amyloidosis by us, thus initially remaining undetected.38 Lastly, we did not analyze the event-free survival as shown in prior studies,20 as we focused on OS/PFS. The former will be part of another upcoming study at our institution.
In conclusion, our results demonstrate the higher frequency of SAE, higher discontinuation rates and early mortality in frail patients, supporting MM patients' need for individualized induction and relapse protocols.17,23,46,47 Full-dose intensity for fit and reduced doses for frail patients appears pertinent, whereas intermediate-fi t patients need continuing consideration. The precise fitness assessment in MM, similar to other hematological malignancies, seems relevant to achieve favorable treatment results, less DR, SAE, as few unscheduled re-hospitaliza-
Haematologica | 108 - April 2023 1124 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
tions and preserved QoL.7,12,42,48,49 The latter demonstrated itself to be possible even after intensive regimens, after allogeneic transplantation or quadruplet RRMM treatment.8,17,23,27,28 The implementation of functional assessments in myeloma TB may also support physicians in treatment decisions, since this adds an objective assessment of patients’ individual constitution and possible treatment endurance. Future studies are needed to evaluate the benefits of a functionally adapted treatment approach versus ´treatment as usual´. Prospective studies using the R-MCI in TB for therapeutic decision support are in process at our CCCF.
Disclosures
No conflicts of interest to disclose.
Contributions
MH, ME, and all other authors performed the analysis. MH,
References
1. Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, riskstratification and management. Am J Hematol. 2020;95(5):548-567.
2. Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primer. 2017;3:17046.
3. Mateos M-V, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528.
4. Dimopoulos MA, Jakubowiak AJ, McCarthy PL, et al. Developments in continuous therapy and maintenance treatment approaches for patients with newly diagnosed multiple myeloma. Blood Cancer J. 2020;10(2):17.
5. Soto-Perez-de-Celis E, Li D, Yuan Y, Lau YM, Hurria A. Functional versus chronological age: geriatric assessments to guide decision making in older patients with cancer. Lancet Oncol. 2018;19(6):e305-e316.
6. Fakhri B, Fiala MA, Tuchman SA, Wildes TM. Undertreatment of older patients with newly diagnosed multiple myeloma in the era of novel therapies. Clin Lymphoma Myeloma Leuk. 2018;18(3):219-224.
7. Goede V, Neuendorff NR, Schulz R-J, Hormigo A-I, MartinezPeromingo FJ, Cordoba R. Frailty assessment in the care of older people with haematological malignancies. Lancet Healthy Longev. 2021;2(11):e736-e745.
8. Scheubeck S, Ihorst G, Schoeller K, et al. Comparison of the prognostic significance of 5 comorbidity scores and 12 functional tests in a prospective multiple myeloma patient cohort. Cancer. 2021;127(18):3422-3436.
9. Voorhees PM, Kaufman JL, Laubach J, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplanteligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136(8):936-945.
10. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115.
11. Bringhen S, Mateos MV, Zweegman S, et al. Age and organ damage correlate with poor survival in myeloma patients: meta-analysis of 1435 individual patient data from 4
GI, HR and ME analyzed results. MH, GI and ME prepared tables and figures. ME, RW, GI designed the research and MH and ME wrote the paper. All authors approved and carefully revised the paper.
Acknowledgments
The authors thank DSMM, GMMG, EMN and IMWG experts for their support and prior recommendations on this study. We thank all MM patients who participated in this study.
Funding
This work was supported by the Deutsche Krebshilfe (grants 1095969 and 111424 to ME and RW).
Data-sharing statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
randomized trials. Haematologica. 2013;98(6):980-987.
12. Klepin HD, Sun C-L, Smith DD, et al. Predictors of unplanned hospitalizations among older adults receiving cancer chemotherapy. JCO Oncol Pract. 2021;17(6):e740-e752.
13. Terpos E, Engelhardt M, Cook G, et al. Management of patients with multiple myeloma in the era of COVID-19 pandemic: a consensus paper from the European Myeloma Network (EMN). Leukemia. 2020;34(8):2000-2011.
14. Isaacs A, Fiala M, Tuchman S, Wildes TM. A comparison of three different approaches to defining frailty in older patients with multiple myeloma. J Geriatr Oncol. 2020;11(2):311-315.
15. Engelhardt M, Ihorst G, Duque-Afonso J, et al. Structured assessment of frailty in multiple myeloma as a paradigm of individualized treatment algorithms in cancer patients at advanced age. Haematologica. 2020;105(5):1183-1188.
16. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood. 2015;125(13):2068-2074.
17. Engelhardt M, Domm A-S, Dold SM, et al. A concise revised Myeloma Comorbidity Index as a valid prognostic instrument in a large cohort of 801 multiple myeloma patients. Haematologica. 2017;102(5):910-921.
18. Milani P, Vincent Rajkumar S, Merlini G, et al. N-terminal fragment of the type-B natriuretic peptide (NT-proBNP) contributes to a simple new frailty score in patients with newly diagnosed multiple myeloma. Am J Hematol. 2016;91(11):1129-1134.
19. Cook G, Royle K-L, Pawlyn C, et al. A clinical prediction model for outcome and therapy delivery in transplant-ineligible patients with myeloma (UK Myeloma Research Alliance Risk Profile): a development and validation study. Lancet Haematol. 2019;6(3):e154-e166.
20. Larocca A, Bonello F, Gaidano G, et al. Dose/schedule-adjusted Rd-R vs continuous Rd for elderly, intermediate-fit patients with newly diagnosed multiple myeloma. Blood. 2021;137(22):3027-3036.
21. Facon T, Dimopoulos MA, Meuleman N, et al. A simplified frailty
Haematologica | 108 - April 2023 1125 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
scale predicts outcomes in transplant-ineligible patients with newly diagnosed multiple myeloma treated in the FIRST (MM020) trial. Leukemia. 2020;34(1):224-233.
22. Brioli A, Manz K, Pfirrmann M, et al. Frailty impairs the feasibility of induction therapy but not of maintenance therapy in elderly myeloma patients: final results of the German Maintenance Study (GERMAIN). J Cancer Res Clin Oncol. 2020;146(3):749-759.
23. Engelhardt M, Dold SM, Ihorst G, et al. Geriatric assessment in multiple myeloma patients: validation of the International Myeloma Working Group (IMWG) score and comparison with other common comorbidity scores. Haematologica. 2016;101(9):1110-1119.
24. Engelhardt M, Mertelsmann R, Duyster J. Das Blaue Buch Chemotherapie-Manual Hämatologie und Onkologie. 7. Auflage. Springer Verlag. 2020.
25. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346.
26. Durie BGM, Harousseau J-L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20(9):1467-1473.
27. Greil C, Engelhardt M, Ihorst G, et al. Allogeneic transplantation of multiple myeloma patients may allow longterm survival in carefully selected patients with acceptable toxicity and preserved quality of life. Haematologica. 2019;104(2):370-379.
28. Waldschmidt JM, Keller A, Ihorst G, et al. Safety and efficacy of vorinostat, bortezomib, doxorubicin and dexamethasone in a phase I/II study for relapsed or refractory multiple myeloma (VERUMM study: vorinostat in elderly, relapsed and unfit multiple myeloma). Haematologica. 2018;103(10):e473-e479.
29. Dold SM, Möller M-D, Ihorst G, et al. Validation of the revised myeloma comorbidity index and other comorbidity scores in a multicenter German study group multiple myeloma trial. Haematologica. 2021;106(3):875-880.
30. Möller M-D, Ihorst G, Pahl A, et al. Physical activity is associated with less comorbidity, better treatment tolerance and improved response in patients with multiple myeloma undergoing stem cell transplantation. J Geriatr Oncol. 2021;12(4):521-530.
31. Einsele H, Engelhardt M, Tapprich C, et al. Phase II study of bortezomib, cyclophosphamide and dexamethasone as induction therapy in multiple myeloma: DSMM XI trial. Br J Haematol. 2017;179(4):586-597.
32. Bachmann F, Schreder M, Engelhardt M, et al. Kinetics of renal function during induction in newly diagnosed multiple myeloma: results of two prospective studies by the German Myeloma Study Group DSMM. Cancers. 2021;13(6):1322.
33. Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(3):309-322.
34. Tuchman SA, Moore JO, DeCastro CD, et al. Phase II study of dose-attenuated bortezomib, cyclophosphamide and dexamethasone (“VCD-Lite”) in very old or otherwise toxicityvulnerable adults with newly diagnosed multiple myeloma. J
Geriatr Oncol. 2017;8(3):165-169.
35. Mai EK, Miah K, Bertsch U, et al. Bortezomib-based induction, highdose melphalan and lenalidomide maintenance in myeloma up to 70 years of age. Leukemia. 2021;35(3):809-822.
36. Ludwig H, Delforge M, Facon T, et al. Prevention and management of adverse events of novel agents in multiple myeloma: a consensus of the European Myeloma Network. Leukemia. 2018;32(7):1542-1560.
37. Bringhen S, Milan A, Ferri C, et al. Cardiovascular adverse events in modern myeloma therapy - incidence and risks. A review from the European Myeloma Network (EMN) and Italian Society of Arterial Hypertension (SIIA). Haematologica. 2018;103(9):1422-1432.
38. Ihne S, Morbach C, Sommer C, Geier A, Knop S, Störk S. Amyloidosis - the diagnosis and treatment of an underdiagnosed disease. Dtsch Arzteblatt Int. 2020;117(10):159-166.
39. Möller M-D, Gengenbach L, Graziani G, Greil C, Wäsch R, Engelhardt M. Geriatric assessments and frailty scores in multiple myeloma patients: a needed tool for individualized treatment? Curr Opin Oncol. 2021;33(6):648-657.
40. Antoine-Pepeljugoski C, Braunstein MJ. Management of newly diagnosed elderly multiple myeloma patients. Curr Oncol Rep. 2019;21(7):64.
41. Cordoba R, Eyre TA, Klepin HD, Wildes TM, Goede V. A comprehensive approach to therapy of haematological malignancies in older patients. Lancet Haematol. 2021;8(11):e840-e852.
42. Rosko AE, Huang Y, Benson DM, et al. Use of a comprehensive frailty assessment to predict morbidity in patients with multiple myeloma undergoing transplant. J Geriatr Oncol. 2019;10(3):479.485.
43. Soekojo CY, Kumar SK. Stem-cell transplantation in multiple myeloma: how far have we come? Ther Adv Hematol. 2019;10:2040620719888111.
44. D’Agostino M, De Paoli L, Conticello C, et al. Continuous therapy in standard- and high-risk newly-diagnosed multiple myeloma: a pooled analysis of 2 phase III trials. Crit Rev Oncol Hematol. 2018;132:9-16.
45. Stege CAM, Nasserinejad K, Klein SK, et al. Improving the identification of frail elderly newly diagnosed multiple myeloma patients. Leukemia. 2021;35(9):2715-2719.
46. Cook G, Larocca A, Facon T, Zweegman S, Engelhardt M. Defining the vulnerable patient with myeloma-a frailty position paper of the European Myeloma Network. Leukemia. 2020;34(9):2285-2294.
47. Larocca A, Dold SM, Zweegman S, et al. Patient-centered practice in elderly myeloma patients: an overview and consensus from the European Myeloma Network (EMN). Leukemia. 2018;32(8):1697-1712.
48. Stauder R, Eichhorst B, Hamaker ME, et al. Management of chronic lymphocytic leukemia (CLL) in the elderly: a position paper from an international Society of Geriatric Oncology (SIOG) Task Force. Ann Oncol. 2017;28(2):218-227.
49. Abel GA, Klepin HD. Frailty and the management of hematologic malignancies. Blood. 2018;131(5):515-524.
Haematologica | 108 - April 2023 1126 ARTICLE - Minimizing treatment complications in NDMM M. Holler et al.
Immune-mediated thrombotic thrombocytopenic purpura plasma induces calcium- and IgG-dependent endothelial activation: correlations with disease severity
Edwige Tellier,1,2 Agnès Widemann,1 Raphaël Cauchois,2,3 Julien Faccini,1,2 Marie Lagarde,1,2 Marion Brun,3 Philippe Robert,4 Stéphane Robert,1 Richard Bachelier,1 Pascale Poullin,2,5 Elien Roose,6 Karen Vanhoorelbeke,6 Paul Coppo,2,7 Françoise Dignat-George1,8 and Gilles Kaplanski2,3 on behalf of the ENDO-13 study group
1Aix-Marseille Université, INSERM, INRAE, C2VN, Marseille, France; 2French Reference Center for Thrombotic Microangiopathies, Hôpital Saint-Antoine, Paris, France; 3Aix-Marseille Université, APHM, INSERM, INRAE, C2VN, CHU Conception, Service de Médecine Interne et Immunologie Clinique, Marseille, France; 4Aix-Marseille Université, APHM, CNRS, INSERM, CHU Conception, Laboratoire Immunologie, Marseille, France; 5APHM, Service d’Hémaphérèse, CHU Conception, Marseille, France; 6Laboratory for Thrombosis Research, IRF Life Sciences, KU Leuven Campus Kulak Kortrijk, Kortrijk, Belgium; 7Service d’Hématologie, Hôpital Saint-Antoine, APHP.6-SU, INSERM, UMRS 1138, Centre de Recherche des Cordeliers, Paris, France and 8Aix-Marseille Université, APHM, INSERM, INRAE, C2VN, CHU Conception, Laboratoire d’Hématologie, Marseille, France
Abstract
Correspondence: E. Tellier
edwige.tellier@univ-amu.fr
Received: January 12, 2022.
Accepted: November 21, 2022.
Early view: December 1, 2022.
https://doi.org/10.3324/haematol.2022.280651
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is characterized by a severe ADAMTS13 deficiency due to the presence of anti-ADAMTS13 auto-antibodies, with subsequent accumulation of circulating ultra-large von Willebrand factor (VWF) multimers. The role of endothelial cell activation as a trigger of the disease has been suggested in animal models but remains to be demonstrated in humans. We prospectively obtained plasma from the first plasma exchange of 25 patients during iTTP acute phase. iTTP but not control plasma, induced a rapid VWF release and P-selectin exposure on the surface of dermal human micro-vascular endothelial cell (HMVEC-d), associated with angiopoietin-2 and endothelin-1 secretion, consistent with Weibel-Palade bodies exocytosis. Calcium (Ca2+) blockade significantly decreased VWF release, whereas iTTP plasma induced a rapid and sustained Ca2+ flux in HMVEC-d which correlated in retrospect, with disease severity and survival in 62 iTTP patients. F(ab)’2 fragments purified from the immunoglobulin G fraction of iTTP plasma mainly induced endothelial cell activation with additional minor roles for circulating free heme and nucleosomes, but not for complement. Furthermore, two anti-ADAMTS13 monoclonal antibodies purified from iTTP patients’ B cells, but not serum from hereditary TTP, induced endothelial Ca2+ flux associated with Weibel-Palade bodies exocytosis in vitro, whereas inhibition of endothelial ADAMTS13 expression using small intering RNA, significantly decreased the stimulating effects of iTTP immunoglobulin G. In conclusion, Ca2+-mediated endothelial cell activation constitutes a “second hit” of iTTP, is correlated with the severity of the disease and may constitute a possible therapeutic target.
Introduction
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is a rare and life-threatening thrombotic microangiopathy (TMA) characterized by a severe thrombocytopenia (<30.109/L) and a mechanical hemolytic anemia. Consequently, ischemic events of variable severity occur, mainly affecting the brain, the heart, and the mesenteric tract. The diagnosis of iTTP relies on the demonstration of ADAMTS13 protease functional deficiency (<10%), due to the presence of anti-ADAMTS13 auto-antibodies.1–3 ADAMTS13 is responsible for the cleavage of ultra-large von Willebrand factor (UL-VWF) into smaller and less
thrombotic multimers.4,5 Deficiency of ADAMTS13 activity leads to the accumulation of highly prothrombotic UL-VWF in the patient plasma inducing the formation of multiple platelet-rich thrombi into the microcirculation, consumptive thrombocytopenia, mechanical hemolysis and clinical symptoms.1–3 Despite treatments based on therapeutic plasma exchange (PEX) and immunosuppressive drugs, the mortality rate remains as high as 5-10%.
Animal models demonstrate that in addition to ADAMTS13 deficiency, endothelial UL-VWF exocytosis is necessary to reproduce the disease, suggesting that endothelial cells (EC) may participate in a “second hit” of the disease.6 ULVWF is the main constituent of endothelial Weibel-Palade
Haematologica | 108 - April 2023 1127 ARTICLE - Platelet Biology & its Disorders
bodies (WPB), from which it can rapidly be released upon EC activation.6 In order to obtain a TTP-like disease, ADAMTS13 knockout (KO) mice need to be crossed with mice expressing high intracellular concentrations of VWF, then to be injected with shigatoxin to induce WPB degranulation leading to UL-VWF release, showing that inactivation of the adamts13 gene is not sufficient to induce TTP-like manifestations.7,8,9 In agreement, injections of large concentrations of recombinant VWF can induce TTP in ADAMTS13 KO mice. Moreover, vwf gene deletion results in complete protection in the shigatoxin-induced TTP murine model, demonstrating the absolute requirement of VWF to develop TTP.10,11 Another TTP model consisting in injection of murine anti-ADAMTS13 inhibitory monoclonal antibodies (mAb) into wild-type mice, led to plasma ADAMTS13 deficiency and UL-VWF accumulation without TTP-like symptoms.12 The additional injection of recombinant VWF in this model induces an iTTP-like disease, further demonstrating the essential role of large concentrations of circulating VWF. In a primate model of TTP, injection of a murine anti-human ADAMTS13 inhibitory antibody induced TTP, as demonstrated by the appearance of severe thrombocytopenia, hemolytic anemia, elevated LDH, schistocytes and the occurrence of microthrombi in kidney, heart, brain and spleen.13 However, the primates did not develop end-stage disease, suggesting once again that inhibition of ADAMTS13 alone, may not be sufficient to reproduce a full-spectrum human TTP.
Thus, these experimental models suggest that induction of TTP in animals is a “two-hit” process requiring first, ADAMTS13 protease inactivation and second, an increased VWF release by activated EC. Similar mechanisms however, remain to be demonstrated in humans. In this context, we asked whether iTTP-patient plasma was able to induce WPB exocytosis and tried to identify possible endothelial activators in iTTP-patient plasma. Using plasma prospectively collected from patients during the acute phase of iTTP, we observed induction of UL-VWF release from EC via WPB exocytosis in a Ca2+-dependent pathway. We identified IgG from iTTP patient plasma as the main inducer of endothelial activation and observed that Ca2+-dependent endothelial activation intensity correlated with disease severity.
Methods
Patient characteristics
We conducted a prospective study between 2008 and 2011 consisting in a National Clinical Research Project (#2007/23) approved by the Ethical Committee of the “Assistance Publique-Hôpitaux de Marseille”. Informed consent was provided according to the Declaration of Helsinki. Detailed information about the 62 patients can be found in the Online Supplementary Appendix and Table 1.
Immunofluorescence studies
Confluent human microvascular dermal EC (HMVEC-d) were grown in EC growth basal medium-2 (EBM2) containing 1% fetal bovine serum for 16 hours (h) and activated for 1 hour (h) with either control or iTTP plasma (1/100 in EBM2), washed twice in phosphate buffer saline (PBS), fixed in 1% paraformaldehyde for 10 minutes (min) and labeled with anti-VWF, anti-P-selectin antibodies or with rabbit non-immune serum like described in supplementary methods.
In order to visualize ADAMTS13, HMVEC-d were activated for 20 min with control or iTTP immunoglobulin G (IgG) (30 m g/mL), washed in PBS and labeled as described in the Online Supplementary Appendix. In order to exclude the influence of possible endotoxin contamination of biological samples, all experiments were performed in the presence of 10 m g/mL polymyxin B (Sigma-Aldrich, Saint Louis, USA).
iTPP: immune-mediated thrombotic thrombocytopenic purpura; IQR: interquartile range; LDH: lactate dehydrogenase.
Table 1. Demographics, clinical and biological data of the 62 immune-mediated thrombotic thrombocytopenic purpura patients included from Marseille and Paris.
iTTP N=25, Marseille Demographics Age in years, median (IQR) 39.5 (26.5-52.8) Male, N (%) 7 (28) Clinical features Neurological involvement, N (%) 14 (56) Mortality, N (%) 3 (12) Laboratory features, median (IQR) Platelets, x109/L 13 (11-21) Hemoglobin, g/L 81 (69-99) LDH, IU/L 1,228 (1,027-1577) Presence of schistocytes, N (%) 25 (100) ADAMTS13 activity, % 5 (0.5-6.5) Anti-ADAMTS13 IgG, IU/mL 87 (49.5-113.5) iTTP N=62, Marseille (25) + Paris (37) Demographics Age in years, median (IQR) 43 (28.8-54) Male, N (%) 18 (30) Clinical features Neurological involvement, N (%) 29 (46.8) Mortality, N (%) 21 (32.3) Laboratory features, median (IQR) Platelets, x109/L 13 (10-21) Hemoglobin, g/L 84 (68-101.8) LDH, IU/L 1,535 (1,115-2,461) Presence of schistocytes, N (%) 58 (93.5) ADAMTS13 activity, % 0 (0-5) Anti-ADAMTS13 IgG, IU/mL 94 (50-120) Haematologica | 108 - April 2023 1128
E. Tellier et al.
ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP
Soluble von Willebrand factor, endothelin-1 and angiopoietin-2 measurements
Specific enzyme-linked immunosorbent assays (ELISA) were performed to determine the concentrations of soluble VWF (American Diagnostica, Stanford, USA), endothelin-1 or angiopoietin-2 (R&D system, Minneapolis, USA), according to the manufacturer’s instructions.
Intracellular Ca2+ flux
HMVEC-d were incubated for 1 h with 5 mM fluo-4 AM fluorescent dye (ThermoFisher Scientific, USA) in PBS containing 1% bovine serum albumin (BSA), washed and incubated in PBS 1% BSA for 30 min at 37°C. Cells were stimulated with A23187 (10 µM in EBM2), thrombin (4 IU/mL or 0.5 mM), control or iTTP plasma samples (1/100 in EBM2) for 20 to 1,800 seconds, or with IgG (30 mg/mL in EBM2). Fluorescence analysis used a Cytofluor®, fluorescence multi-well plate reader (PerSeptive-Biosystems, Framingham, USA). Total fluorescence intensity was expressed in arbitrary unit (AU). Intracellular Ca2+ flux was also measured by microscopy (see the Online Supplementary Appendix).
Preparation of IgG-depleted plasma and purification of the IgG fraction
After washing with binding buffer (PBS containing 100 mM Na2HPO4 at pH 7.4), 100 mL of protein A magnetic sepharose beads (GEHealthcare) were incubated for 30 min at 4°C with 300 mL of 1:5 diluted plasma from control or iTTP patients. Beads were retrieved using a magnetic bench and plasma was used for Ca2+ flux induction and IF studies. IgG were eluted using 100 mL of elution buffer (100 mM glycine at pH 12.8) and 5 mL of 1 M TRIS hydrochloride at pH 9.
Human anti-ADAMTS13 monoclonal antibodies
The two anti-human ADAMTS13 mAb TTP73-1 and ELH2-1 were isolated and cloned from B cells of two different iTTP-patients. As previously reported, these two antibodies recognize cryptic epitopes in ADAMTS1314 and were used at 30 mg/mL in experiments.
ADAMTS13 gene silencing
Downregulation of ADAMTS13 mRNA level was achieved using small interfering RNA (siRNA) directed against ADAMTS13 (Ambion, ThermoFisher-Scientific, USA) and Jet Prime kit (Polyplus-transfection, USA) according to the manufacturer’s instructions. As the decrease in ADAMTS13 mRNA level was achieved 24 h after transfection by quantitative real-time polymerase chain reaction (RT-qPCR) and remained stable for 96 h, Ca2+ flux experiments were performed 48 h after transfection.
Statistical analysis
Graphpad Prism v.9.2.0 software (GraphPad-Software Inc.,
San Diego, USA) was used for statistical analysis. Further details of the methods are available in the Online Supplementary Appendix.
Results
Plasmas from immune-mediated thrombotic thrombocytopenic purpura patients induce von Willebrand factor string formation on the surface of HMVEC-d via Weibel-Palade bodies exocytosis
First, we examined VWF string formation on HMVEC-d by confocal microscopy using anti-VWF labeling on permeabilized HMVEC-d. After incubation with control plasma samples, anti-VWF labeling showed punctuated fluorescent spots, whereas incubation with iTTP plasma samples induced the appearance of typical UL-VWF strings (Figure 1A). Quantification of the VWF string fluorescence was performed after 30 min of incubation and demonstrated higher VWF fluorescence intensity (FI) when HMVEC-d were stimulated with iTTP than with control plasma samples (FI=6.77 AU/cellx104; interquartile range [IQR], 4.8-8.79 vs. FI=0.53 AU/cellx104; IQR, 0.36-0.87, respectively; P<0.0001; Figure 1B). Kinetic studies showed that VWF strings on the endothelial surface in response to iTTP plasma was observed after 5 min incubation and increased for up to 60 min (Figure 1C) (FI=6.75 AU/cellx104; IQR, 4.81-7.38; FI=8.62 AU/cellx104, IQR, 7.03-14.82; FI=14.5 AU/cellx104; IQR, 10.1-21.68 vs. FI=0.76 AU/cellx104, IQR, 0.712.99; P=0.0159; P=0.0079; P=0.0079 respectively at 5, 15 and 60 min vs. 0 min; Figure 1D)
Similar data were obtained in various conditions such as dynamic using a flow chamber device (Online Supplementary Figure S1A), macrovascular HUVEC (Online Supplementary Figure S1B), or when using either plasma obtained on citrated tubes or serum, instead of plasma from PEX in order to eliminate a possible Ca2+ overload due to the PEX process (Online Supplementary Figure S1C). In order to exclude possible passive absorption of soluble VWF contained in iTTP plasma on HMVEC-d surface, we used a VWF-affinity column to completely eliminate soluble VWF contained in plasma. Using ELISA, we measured soluble VWF in the supernatant of HMVEC-d stimulated with either VWF-free iTTP or VWF-free control plasma samples for 1 h and observed significantly higher concentrations of endothelial soluble VWF in the former condition (2.13 ng/mL, IQR, 1.76-2.50 vs. 1.05 ng/mL, IQR, 0.98-1.61; P=0.0159; Online Supplementary Figure S1D).
In order to determine whether VWF string formation on HMVEC-d was effectively due to WPB exocytosis, we measured the concentrations of endothelin-1 and of angiopoietin-2, two molecules stored in WPB. Significantly higher concentrations of endothelin-1 (19.02 pg/mL; IQR, 16.88-20.21 vs. 11.55 pg/mL; IQR, 5.79-17.9, respectively;
Haematologica | 108 - April 2023 1129 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
Figure 1. Induction of Weibel-Palade bodies exocytosis on endothelial cells after stimulation with immune-mediated thrombotic thrombocytopenic purpura plasma. (A) Confocal microscopy analysis of permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubated with either control plasma (A) or immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples (B) using rabbit anti-von Willebrand Factor (VWF) antibodies and Alexa Fluor 594-conjugated donkey anti-rabbit immunoglobulin G (IgG) (original magnification X63, scale bar=10 mm). (B) Quantification of VWF fluorescence intensity (FI) after HMVEC-d stimulation with either control plasma (Control: n=9) or iTTP plasma samples during the acute phase (iTTP: n=14, ***P<0.001). (C) Kinetics of VWF HMVEC-d exocytosis visualized by fluorescence microscopy after incubation with iTTP plasma samples (original magnification X40, scale bar=10 mm). (D) Quantification of VWF FI over time on HMVEC-d stimulated with iTTP plasma samples (iTTP: n=5, *P<0.05, **P<0.01). (E) Endothelin-1 and (F) angiopoietin-2 concentrations in HMVEC-d supernatants stimulated with either control plasma (n=6) or iTTP plasma samples for 1h (n=25, *P<0.05). (G) P-selectin expression detected by confocal microscopy analysis using anti-rabbit P-selectin antibodies and Alexa Fluor 488-conjugated donkey anti-rabbit IgG (green) in HMVEC-d incubated with control (A) or iTTP plasma samples (B) (original magnification X63, scale bar=10 mm). (H) Percentage of P-selectin FI per cell on HMVEC-d stimulated with control plasma (n=10) (representing 100%) or iTTP plasma samples (n=23, ***P<0.001).
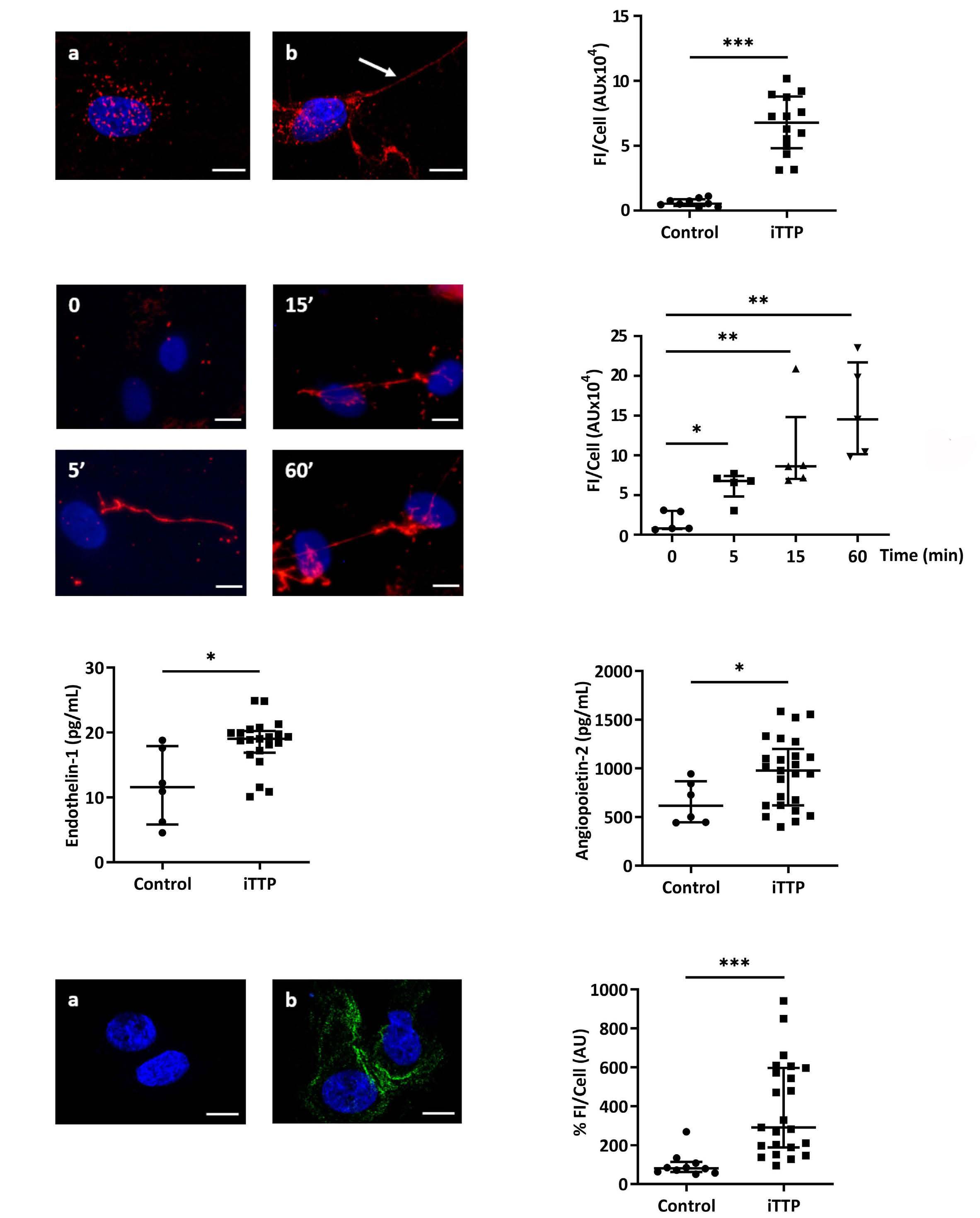
A C E G B D F H Haematologica | 108 - April 2023 1130 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
P=0.0102; Figure 1E) and angiopoietin-2 (978 pg/mL; IQR, 619.5-1,200 vs. 614.5 pg/mL; IQR, 445.8-868.3, respectively; P=0.0355; Figure 1F), were present in the supernatants of HMVEC-d when cultured with iTTP compared to control plasma samples. In addition, membrane P-selectin expression on HMVEC-d was observed after stimulation for 10 min with iTTP, but not with control plasma samples (Figure 1G), associated with a 3-time-increased P-selectin FI (FI=290.3 AU/cellx102; IQR, 188.2-596.6 vs. FI=81.23 AU/cellx102; IQR: 61.55-114.3; P<0.0001; Figure 1H). The same data concerning endothelin-1 secretion were observed when HUVEC were used instead of HMVEC-d (40.58 pg/mL; IQR, 25.66-43.52 vs. 19.28 pg/mL; IQR, 16.45-21.56; P=0.0070; Online Supplementary Figure S1E). The endothelin-1 secretion was equivalent when citrated plasma or serum of iTTP-patients were using instead of plasma from PEX (24.65 pg/mL; IQR, 20.39-28.41 and 25.33 pg/mL; IQR, 20.84-32.92 vs. 24.10 pg/mL; IQR, 19.9-24.9; Online Supplementary Figure S1F). Altogether, these data are consistent with the fact that iTTP plasmas obtained after PEX were able to induce WPB exocytosis.
Weibel-Palade bodies exocytosis induced by immune-mediated thrombotic thrombocytopenic purpura plasma is Ca2+-mediated
Two pathways of WPB exocytosis are known: a Ca2+-dependent pathway and a cAMP-dependent pathway. In order to determine whether the Ca2+ signaling cascade was involved in WPB exocytosis induced by iTTP plasma, we used the pharmacological inhibitor MAPTAM. Compared with iTTP plasma samples alone, we observed a significant decrease of VWF FI in the presence of MAPTAM (FI=1.09 AU/Cellx104; IQR, 0.78-1.79 vs. FI= 7.59 AU/Cellx104; IQR, 7.52-8.85; P=0.0079; Figure 2A, B). iTTP plasma-induced endothelin-1 secretion was also reduced by 92% in the presence of MAPTAM (82.1%; IQR, 73-134.8 vs. 8.5%; IQR, 6.9-9.6; P<0.0001; Figure 2C).
We then measured the ability of iTTP plasma to induce Ca 2+ -flux in EC, using a single-wavelength Ca 2+ fluorescent dye. After a 20-second incubation, iTTP plasma samples induced a significantly higher FI level than control plasma samples in HMVEC-d (94 AU; IQR, 78-125.5 vs. 45 AU; IQR, 35.5-76 respectively; P<0.0001; Figure 2D) or in HUVEC (68 AU; IQR, 55.38-114.9 vs . 41.75 AU; IQR, 37.75-54; P =0.0063; Online Supplementary Figure S1G). We also calculated the AUC for Ca2+ flux after 30 min of stimulation and observed that iTTP plasma induced significantly higher Ca 2+ mobilization than control plasma samples (1,011 AU; IQR, 472-1,334 vs. 242 AU, IQR, 43.5388.5; P=0.0002; Figure 2E). The endothelial Ca2+ mobilization was equivalent when using citrated plasma or serum instead of PEX plasma (74.5 AU; IQR, 59.5-94.75 and 66 AU; IQR, 41.75-85 vs. 70 AU; IQR, 48.75-87.5; Online Supplementary Figure S1H).
Immune-mediated thrombotic thrombocytopenic purpura plasma-induced endothelial Ca2+ flux correlates with immune-mediated thrombotic thrombocytopenic purpura severity
In order to establish whether iTTP-induced Ca2+ flux correlated with disease severity, we studied sera from 62 iTTP patients (25 from Marseille added with 37 from Paris) collected during the acute phase, and classified in two different groups depending on survival (Table 1). At 20 seconds, Ca2+ flux appeared significantly higher in cells treated with plasma from non-survivor patients compared with those of survivors (157 AU; IQR, 119.7-262.3 vs. 107 AU; IQR, 70-181, respectively; P=0.0031; Figure 3A). Similarly, after 30 min, area under the curve (AUC) from non-survivors was much higher than those in survivors (6,316 cm2; IQR, 3,066-8,503 vs. 1391 cm2; IQR: 833.5-4,235, respectively; P=0.0002; Figure 3B). In addition, plasma from iTTP patients in remission induced significantly less Ca2+ flux than plasma from the same patients during the acute phase (52.5 AU; IQR: 45-69 vs. 131.5 AU; IQR, 66-177; P=0.0013; Figure 3C) and levels of Ca2+ flux were not different from those induced by control plasma. In agreement, plasma obtained from patients in complete remission did not induce VWF release by HMVEC-d (Figure 3D) which was confirmed by fluorescence quantification (1.19 AUx104; IQR, 0.89-1.99) during remission versus 10.85 AUx104; IQR, 7.59-14.04 during the acute phase; P=0.0286; Figure 3E).
The purified IgG fraction from immune-mediated thrombotic thrombocytopenic purpura plasma induced Ca2+-dependent Weibel-Palade bodies exocytosis
In order to identify the soluble mediators contained in iTTP plasma involved in WPB exocytosis, we studied several candidates. As iTTP is an autoimmune disease, we first investigated the role of circulating IgG. After depletion of the IgG fraction from iTTP plasma, we observed a significant decrease of VWF FI release by HMVEC-d (Figure 4A). In agreement, the Ca2+ flux was reduced by 47% in HMVEC-d cells treated with IgG-depleted plasma compared with complete plasma (127.5 AU; IQR, 83.5-188 vs. 68 AU; IQR, 45.5-113, respectively; P=0.0003; Figure 4B). Conversely, we tested the ability of the purified IgG fraction from iTTP or control plasma to activate HMVEC-d. VWF exocytosis was observed in HMVEC-d cultured with iTTP IgG, but not with control IgG (Figure 4C). Using ELISA, we measured soluble VWF released in the supernatants of HMVEC-d and observed significantly increased VWF concentrations in HMVEC-d stimulated by iTTP IgG compared with control IgG (3.42 ng/mL; IQR, 2.68-4.18 vs. 2.72 ng/mL; IQR, 2.083.03, respectively; P=0.0273; Figure 4D). In agreement, we observed a significantly higher Ca2+ flux in HMVEC-d stimulated with iTTP IgG than with control IgG fractions (77.25 AU; IQR, 54.88-109.8 vs. 46.75 AU; IQR, 41-54, respectively;
Haematologica | 108 - April 2023 1131 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
Figure 2. Weibel-Palade bodies exocytosis induced by immune-mediated thrombotic thrombocytopenic purpura plasma is Ca2+mediated. (A) Von Willebrand Factor (VWF) secretion detected by fluorescence microscopy, using rabbit anti-VWF antibodies and Alexa Fluor 594-conjugated donkey anti-rabbit immunoglobulin G (IgG) after incubation of permeabilized human microvascular endothelial cells from derm (HMVEC-d) with immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples in the absence (A, B) or in the presence of the Ca2+ chelator MAPTAM (C, D) (original magnification X40, scale bar=30 mm). (B) VWF fluorescence intensity (FI) quantification by cell comparing HMVEC-d stimulated with iTTP plasma samples in absence or presence of MAPTAM (iTTP: n=5, **P<0.01). (C) Endothelin-1 concentration measured in the supernatants of HMVEC-d incubated with iTTP plasma samples (n=10) in the absence (representing 100%) or in the presence of MAPTAM (***P<0.001). (D) Fluorescence intensity (AU) of the Ca2+ flux in HMVEC-d incubated for 20 seconds with either control plasma samples (n=13), iTTP plasma samples (n=25, ***P<0.001) or A23187 Ca2+ ionophore (n=3). (E) Area under the curve (AUC) (cm2) from Ca2+ flux curves after 30 minutes of HMVEC-d incubation with either control plasma (n=13), iTTP plasma samples (n=25, ***P<0.001) or A23187 Ca2+ ionophore (n=3).
P=0.0011; Figure 4E). These results were confirmed by directly measuring Ca2+ flux in living cells stimulated with either iTTP or control IgG, using microscopy (Online Supplementary Figure S2A, B). In order to determine which fraction of the immunoglobulin was involved in EC activation, we purified the F(ab’)2 fragments from the IgG fraction, and observed that F(ab’)2 fragments induced both VWF string formation (Figure 4F) and a twice higher endothelial Ca2+ flux compared to the complete iTTP IgG fraction (130.8; IQR, 86.11-197.2 vs. 265.3; IQR, 206.6-315.3; P=0.002; Figure 4G).
Immune-mediated thrombotic thrombocytopenic purpura IgG induced Weibel-Palade bodies degranulation partly via ADAMTS13 recognition on endothelial cells
In order to identify the endothelial antigen target of iTTPIgG, we concentrated on ADAMTS13, the main protein involved in iTTP pathogenesis. Since it was impossible to create an ADAMTS13 affinity column in order to eliminate
anti-ADAMTS13 auto-antibodies contained in iTTP-purified IgG, we used different procedures. First, we compared the ability of hereditary TTP (hTTP) serum samples which do not contain anti-ADAMTS13 auto-antibodies to induce Weibel-Palade bodies degranulation, with iTTP serum samples. Compared to that induced by iTTP serum samples, we observed that VWF release induced by hTTP serum was 65% lower (6.61 ng/mL; IQR, 4.45-12.84 vs. 17.27 ng/mL; IQR, 11.09-24.88; P=0.0317; Figure 5A) and that the Ca2+ flux induced by hTTP serum samples appeared significantly reduced (113.5 AU; IQR, 112.7-118.4 vs. 126.7 AU; IQR, 123.3-131.7; P=0.0079; Figure 5B). After we verified that extracellular expression of endothelial ADAMTS13 was not modified when EC were stimulated with IgG from iTTPpatients or control plasma samples (Online Supplementary Figure S3A) (90.4%; IQR, 62.78-116.2 vs. 116.3 %; IQR, 73.37144.7; P=0.3357; Online Supplementary Figure S3B), we inhibited endothelial ADAMTS13 expression using specific siRNA (70% of inhibition, 1; IQR, 1-1 vs. 0.29; IQR, 0.16-0.41;
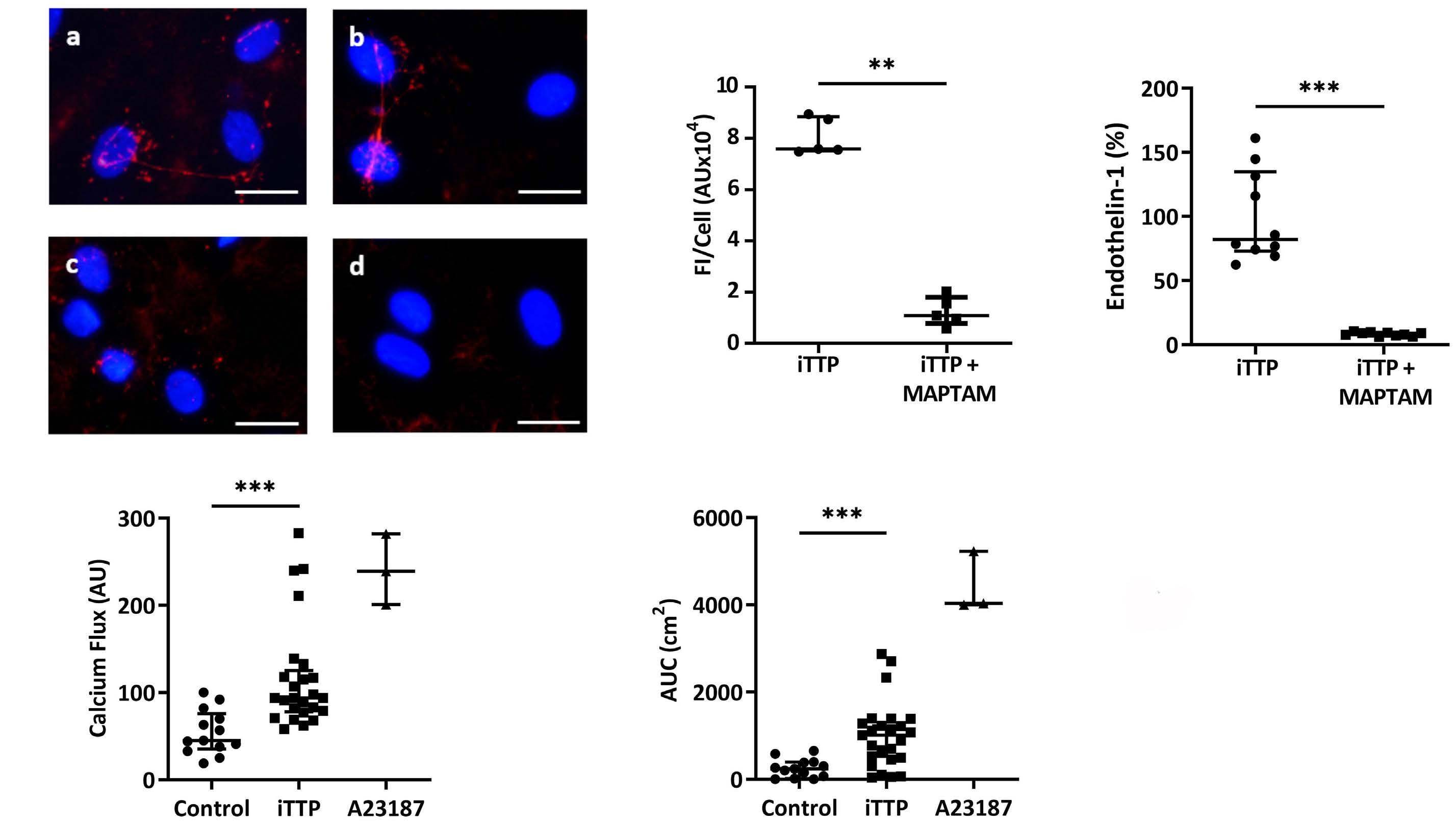
A C E B D Haematologica | 108 - April 2023 1132 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
Figure 3. In vitro endothelial Ca2+ flux intensity induced by immune-mediated thrombotic thrombocytopenic purpura plasma is associated with clinical severity and prognosis. (A) Fluorescence intensity (AU) of the Ca2+ flux measured at 20 seconds in permeabilized human microvascular endothelial cells from derm (HMVEC-d) stimulated with immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples of survivors (n=41) or non-survivors patients (n=21, **P<0.01). (B) Area under the curve (AUC) from Ca2+ flux curves after 30 minutes of HMVEC-d stimulated with iTTP plasma samples of survivors (n=41) or non-survivors (n=21, ***P<0.001). (C) Fluorescence intensity (AU) of the Ca2+ flux in HMVEC-d incubated for 20 seconds with plasma samples from iTTP patients in acute phase (iTTP-AP) or from same patients in remission (iTTP-R) (n=11, **P<0.01). Horizontal dotted line represents median Ca2+ mobilization induced in HMVEC-d by control plasma. (D) Von Willebrand Factor (VWF) secretion and tethering detected by fluorescence microscopy using rabbit anti-VWF antibodies and Alexa Fluor 594conjugated donkey anti-rabbit immunoglobulin (red) after 1-hour-incubation of HMVEC-d with iTTP plasma samples collected in acute phase (A) or during remission (B). Cell nuclei are labeled in blue using DAPI (original magnification X40, scale bar=10 mm). (E) VWF fluorescence quantification after the HMVEC-d stimulation with iTTP plasma from patients in acute phase (iTTP-AP: n=4), or in remission (iTTP-R: n=4, *P<0.05).
P=0.0313; Figure 5C,) and observed that inhibition of ADAMTS13 expression was associated with a significant reduction of the iTTP IgG-induced Ca2+ flux (33.44 AU; IQR, 25.88-35.22 vs. 47.15 AU; IQR, 31.42-73.9; P=0.0084; Figure 5D). We further tested the ability of two anti-ADAMTS13 antibodies, TTP73-1 or ELH2-1, previously isolated and cloned from the B cells of two iTTP-patients,14 to induce WPB exocytosis in HMVEC-d. Both TTP73-1 and ELH2-1 weakly induced VWF tethering on HMVEC-d membranes (Figure 5E). When cultured in the presence of TTP73-1 but not of or ELH2-1, higher soluble VWF concentrations were measured in HMVEC-d supernatants compared to stimulation with control IgG (3.7 ng/mL; IQR, 3.3-4.3 for TTP731 and 1.7 ng/mL; IQR, 1.6-2 for ELH2-1 vs. 0.8 ng/mL; IQR, 0.7-0.99; P=0.0357 and P=0.0571 respectively; Figure 5F), as well as higher endothelin-1 secretion (14.1 pg/mL; IQR, 11.8-15.4 for TTP73-1 and 11.5 pg/mL; IQR, 10.8-14.4 for
ELH2-1 vs. 6.4 pg/mL; IQR, 4.2-11.5; P=0.0317 and P=0.0952 respectively; Figure 5G). In addition, an increased but nonsignificant Ca2+ flux was also induced by TTP73-1 and ELH2-1, compared to control IgG (15.01 AU, IQR, 11.55-19.31 and 14.91 AU; IQR, 6.26-18.39, respectively, vs. 3.2 AU, IQR, 0.59-5.83; P=0.10; Figure 5H).
Free heme and nucleosomes, but not complement, play minor roles in immune-mediated thrombotic thrombocytopenic purpura plasma-induced
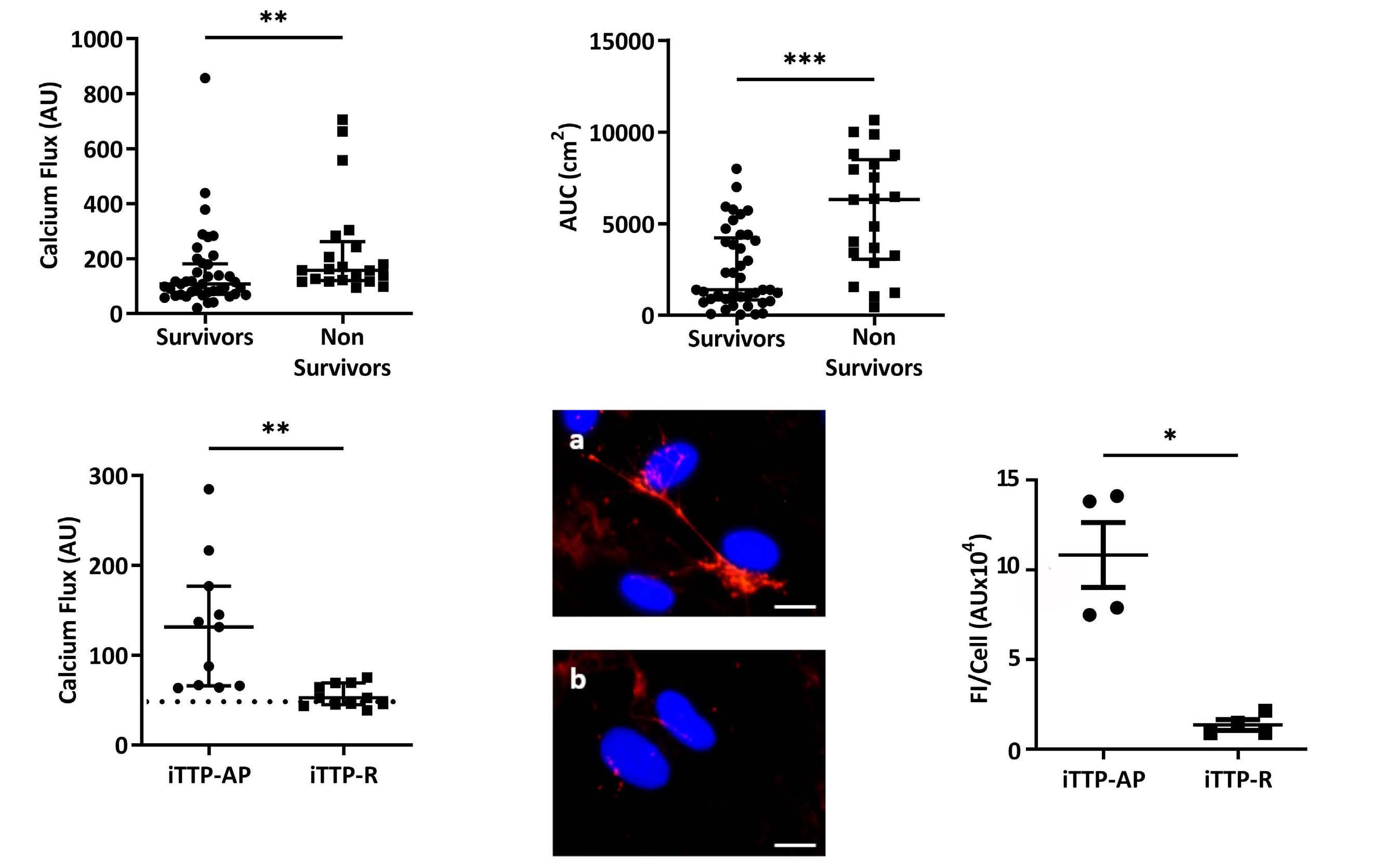
Weibel-Palade bodies exocytosis
The concentrations of free heme were significantly increased in iTTP compared to control plasma samples (11.2 mM; IQR, 6.98-20.35 vs. 9 mM; IQR, 8.2-9.6; P=0.0369; Figure 6A), but returned to control levels after remission (0.63 mM; IQR, 0.44-1.03 vs. 11.2 mM; IQR, 6.98-20.35; P<0.0001; Figure 6A). Free heme concentrations however, were
A C E B D Haematologica | 108 - April 2023 1133 ARTICLE -
E. Tellier et al.
Ca2+-mediated Weibel-Palade exocytosis in iTTP
Figure 4. Immunoglobulin G contained in immune-mediated thrombotic thrombocytopenic purpura plasma are mainly involved in permeabilized human microvascular endothelial cells from derm activation. (A) Von Willebrand Factor (VWF) secretion detected by fluorescence microscopy after permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubation with complete immune-mediated thrombotic thrombocytopenic purpura (iTTP) (A) or immunoglobulin (IgG)-depleted (B) plasma samples (original magnification X20, scale bar=30 mm). (B) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20-second incubation with complete iTTP or IgG-depleted plasma samples (n=15, ***P<0.001). (C) VWF secretion detected by fluorescence microscopy after HMVEC-d incubation with IgG purified from control (A) or iTTP plasma samples (b) (original magnification X40, scale bar=30 mm). (D) VWF quantification by enzyme-linked immunosorbant assay in the supernatants of cells treated with IgG purified from control (n=7) or iTTP plasma samples (n=16, *P<0.05). (E) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d after 20 seconds incubation with IgG from control (n=6) or iTTP plasma samples (n=16, **P<0.01). (F) VWF secretion detected by fluorescence microscopy after HMVEC-d incubation with IgG (a) from iTTP plasma samples or F(ab)’2 fragments (b) purified from these IgG (original magnification X63, scale bar=20 mm). (G) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d after a 20-second of incubation with medium alone (EBM2), A23187 calcium (Ca2+) ionophore, IgG (IgG) from iTTP plasma samples, or F(ab)’2 fragments purified from these IgG (n=10, **P<0.01).
equivalent between survivor and non-survivor patients (10.96 m M; IQR, 8.2-14.07 vs. 12.96 m M; IQR, 2.55-43.56; P=0.6368; Online Supplementary Figure S4A). Inhibition of heme by addition of large concentrations of hemopexin, weakly reduced iTTP plasma-induced VWF exocytosis (Figure 6B), endothelin-1 release (69.86%; IQR, 27.59-136.1 vs. 79.86%; IQR, 54.79-162.6; P<0.0494; Figure 6C) and iTTPinduced Ca2+ flux (30% inhibition, 133.3 AU; IQR, 89.25-
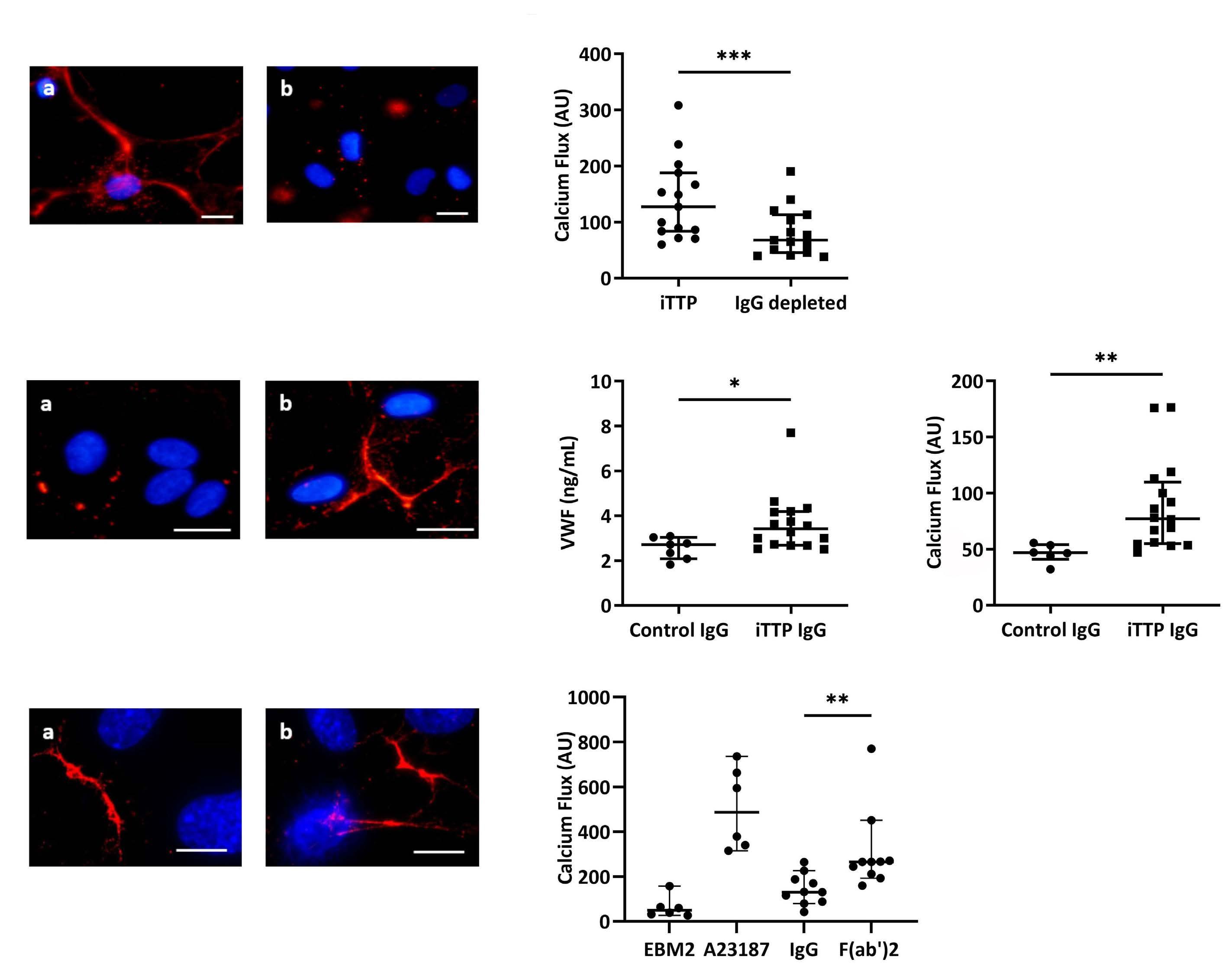
242.8 vs. 107.5 AU; IQR, 61.88-208.3; P=0.0063; Figure 6D).
iTTP plasma also contained significant enrichment in nucleosomes concentrations compared to control plasma samples (6.34-fold increase; IQR, 3.37-12.14 vs. 1.15-fold increase; IQR; 0.12-1.78; P<0.0001; Figure 6E) which was no longer observed after remission (0.81-fold increase; IQR, 0.53-1.52 vs. 6.34-fold increase; IQR, 3.37-12.14 in acute phase; P<0.0001; Figure 6E), in accordance with previous
A C E B D F G Haematologica | 108 - April 2023 1134 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
Figure 5. ADAMTS13 is involved in immune-mediated thrombotic thrombocytopenic purpura IgG-induced Weibel-Palade bodies degranulation. (A) Von Willebrand Factor (VWF) quantification by enzyme-linked immunosorbant assay in the supernatants of cells treated with immune-mediated thrombotic thrombocytopenic purpura (iTTP) serum (n=5) or hereditary TTP serum samples (hTTP, n=5, *P<0.05). (B) Fluorescence intensity (AU) of Ca2+ flux induction in permeabilized human microvascular endothelial cells from derm (HMVEC-d) after a 20-second incubation with serum from iTTP patients (n=5) or from hTTP patients (n=5, **P<0.01). (C) ADAMTS-13 mRNA relative quantification in HMVEC-d cultured in the presence of control small interfering RNA (siRNA) (Ct si) or siRNA targeting ADAMTS13 (ADAMTS13 Si) (n=6, *P<0.05). (D) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d transfected with control or anti-ADAMTS13 siRNA, then stimulated for 20 seconds with IgG from iTTP plasma samples (n=15, **P<0.01). (E) VWF expression detected by fluorescence microscopy in HMVEC-d incubated with IgG from control patient samples (a) TTP73-1 monoclonal antibody (mAb) (B) or ELH2-1 mAb (C) (original magnification X40, scale bar=30 mm). (F) VWF or (G) endothelin-1 concentrations in supernatants of HMVEC-d incubated with IgG from control patient samples (Control) (n=3), TTP73-1 or ELH2-1 mAb (n=5, *P<0.05). (H) Fluorescence intensity (AU) of Ca2+ flux measured at 20 seconds in HMVEC-d stimulated in a similar fashion as in (G) (n=3, ns: not significant).
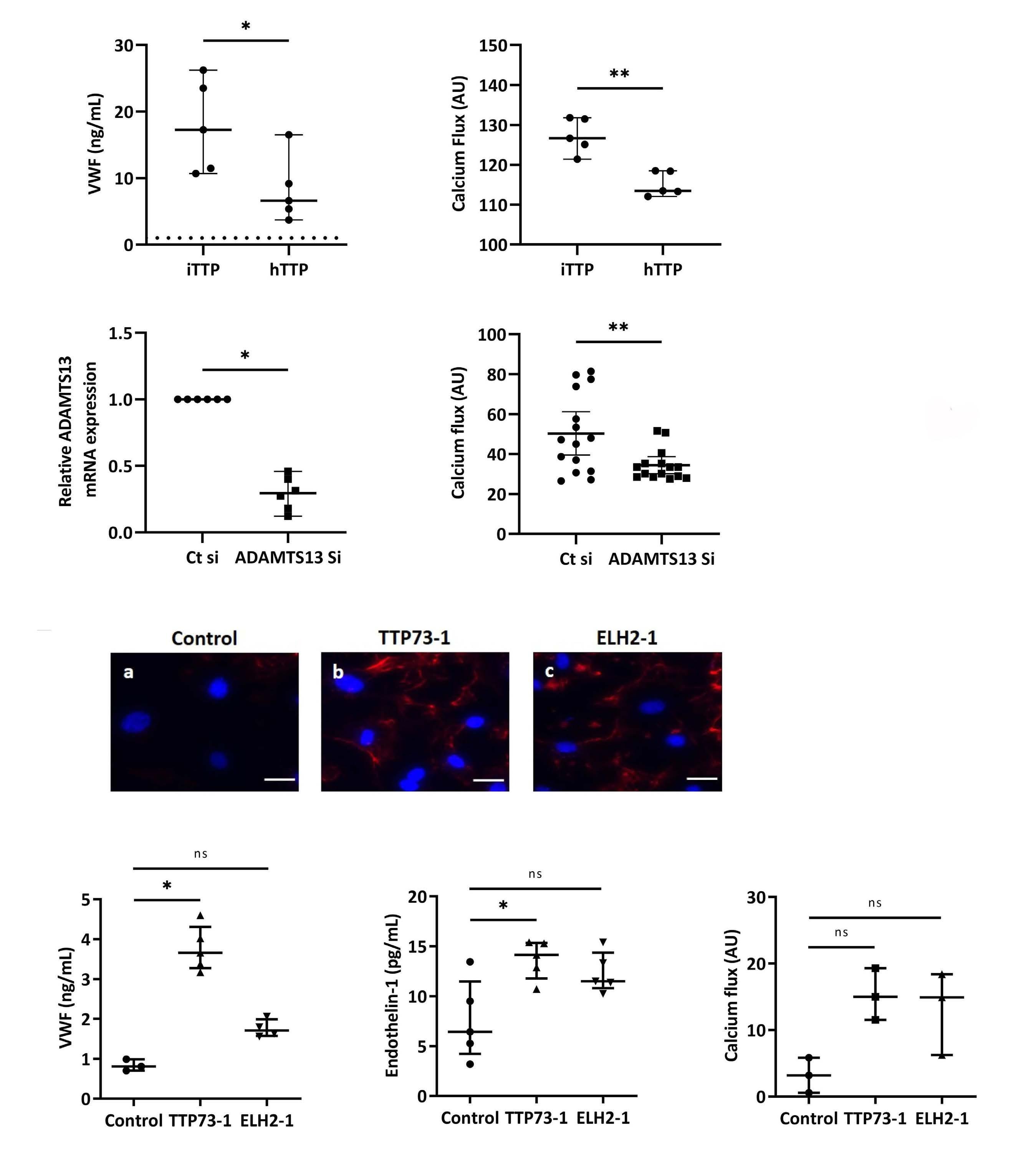
A C E B D F G H Haematologica | 108 - April 2023 1135 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
Figure 6. Heme and nucleosomes participate of the immune-mediated thrombotic thrombocytopenic purpura patient’s plasma induced Ca2+-mediated endothelial activation. (A) Free heme concentration (mM) measured in control (n=13), immune-mediated thrombotic thrombocytopenic purpura (iTTP) patients in acute phase (iTTP-AP, n=62) or iTTP patients in remission (iTTP-R, n=14) plasma samples (*P<0.05, ***P<0.001). (B) Von Willebrand Factor (VWF) expression detected by fluorescence microscopy after permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubation with heme or iTTP plasma samples in the absence (A, B) or in the presence (C, D) of hemopexin (+Hx) (original magnification X40, scale bar=30 mm). (C) Endothelin-1 concentrations in the supernatants of HMVEC-d incubated with iTTP plasma samples in the absence (representing 100%) or in the presence of hemopexin (n=14, *P<0.05). (D) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20second incubation with iTTP plasma samples alone or after preincubation with hemopexin (n=18, **P<0.01). (E) Total nucleosomes in control (n=13), iTTP patients in acute phase (iTTP-AP, n=62) or iTTP patients in remission (iTTP-R, n=14) plasma samples (***P<0.001). (F) VWF secretion detected by fluorescence microscopy after incubation of HMVEC-d with iTTP plasma samples in the absence (A) or in the presence of Annexin V (B) (original magnification X40, scale bar=30 mm). (G) Endothelin-1 concentrations in the supernatants of HMVEC-d incubated with iTTP plasma samples in the absence (representing 100%) or in the presence of Annexin V (n=16, **P<0.01). (H) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20second incubation with either iTTP plasma samples alone, or in the presence of Annexin V (n=17, **P<0.01).
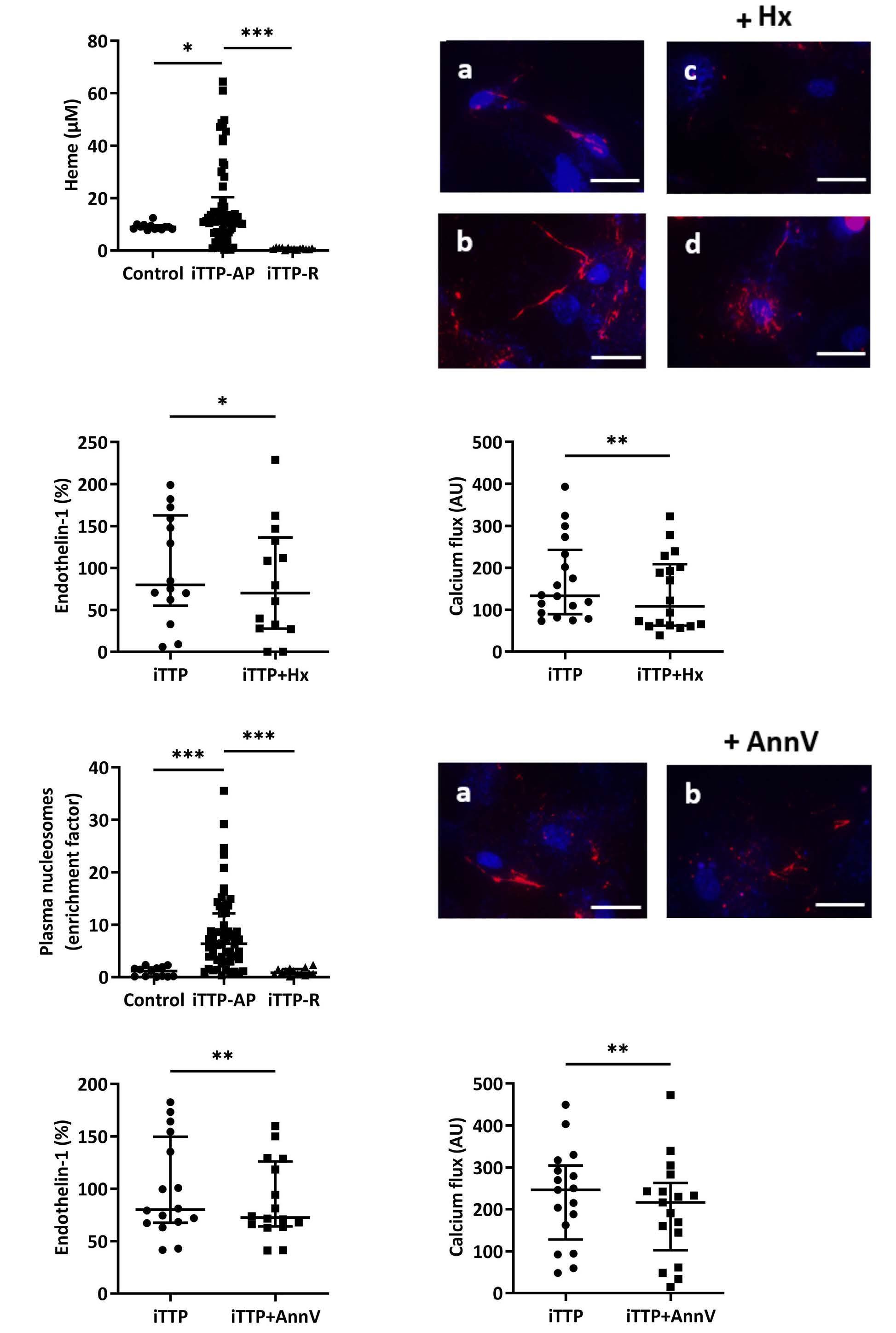
A C E B D F G H Haematologica | 108 - April 2023 1136 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
studies.15 Nucleosomes enrichment however, was not different between survivors and non-survivors (3.57-fold increase; IQR, 1.76-5.24 vs. 3.51-fold increase; IQR, 1.24-7.49; P=0.9295; Online Supplementary Figure S4B). Nucleosomes signalization is dependent of the phosphatidylserine activation and can be inhibited by Annexin V. Addition of Annexin V to iTTP plasma weakly but significantly reduced VWF exocytosis (Figure 6F) and endothelin-1 release by HMVEC-d (10% inhibition, 72.62%; IQR, 64.16-126.1 vs 80.11%; IQR; 67.52-149.6; P=0.0052; Figure 6G), as well as Ca2+ flux induced by iTTP plasma (13%-reduction, 216 AU; IQR, 102.8-262.8 vs. 246.5 AU; IQR, 128-304.5; P=0.0011; Figure 6H).
In order to investigate the role of complement on iTTP plasma-induced endothelial activation, we performed the same experiments with heated plasma or in the presence of eculizumab. None of these treatments significantly influenced endothelial VWF (Online Supplementary Figure S5A) or endothelin-1 secretion after 1 hour of stimulation (29.45 pg/mL; IQR, 23.61-41.76 and 20.85 pg/mL; IQR, 16.0127.10 vs. 23.27 pg/mL; IQR, 19.29-30.39, not significant; Online Supplementary Figure S5B), as well as endothelial Ca2+ flux intensity after 20 seconds (111.8 AU; IQR, 74.85-148.8 and 100.2 AU; IQR, 79.43-287.4 vs. 99.41 AU; IQR, 72.49210.7, not significant; Online Supplementary Figure S5C).
Since iTTP plasma is likely to contain thrombin, we reproduced the same experiments in the presence of hirudin and observed no significant modification (187.5 AU; IQR, 82.75-240 vs. 180 AU; IQR, 89.75-262.5, not significant; Online Supplementary Figure S5D). We also used the general serine protease inhibitor PPACK on the iTTP plasma induced VWF release and Ca2+ flux increase and observed no significant difference (6.298 ng/mL; IQR, 5.859-6.924 vs 6.345 ng/mL; IQR, 6.095-8.615, not significant; Online Supplementary Figure S5E, and 149.3 AU; IQR, 134.2-154.8 vs 144.4 AU; IQR, 138.5-155.9, not significant; Online Supplementary Figure S5F, respectively).
Discussion
In the present study, we observed for the first time that plasma or serum from iTTP patients in acute phase, but not in remission, were able to induce VWF secretion via WPB exocytosis by a mechanism involving a Ca2+ flux. We ruled out a possible passive adhesion of soluble VWF contained in iTTP plasma on the EC surface, since VWF tethering remained present on the EC membrane even after complete depletion of VWF contained in iTTP plasma and concluded that VWF secretion was an active mechanism likely due to WPB exocytosis. WPB exocytosis was indeed confirmed by the fact that iTTP plasma not only induced VWF and P-selectin membrane exposure, but also endothelin-1 and angiopoietin-2 secretion by EC, all com-
ponents of the WPB. WPB exocytosis is known to involve two different signaling pathways. One is rapid and Ca2+mediated, whereas the other is linked to adenylate cyclase activation.6 Both the WPB exocytosis and the rapid and sustained intracellular Ca2+ flux induced by iTTP plasma were inhibited by the Ca2+ chelator MAPTAM, demonstrating a Ca2+-mediated signaling pathway. The possible Ca2+ overload of iTTP plasma due to the PEX process was excluded by the observation that the serum of the iTTP-patients had similar effects as plasma and that plasma of non-TTP patients treated with PEX, was not able to activate EC.
Our objective was then to identify the main endothelial activators contained in iTTP plasma/serum and we showed that the IgG fraction purified from iTTP plasma was able to induce a Ca2+ flux and subsequent WPB exocytosis associated with VWF secretion, whereas IgG depletion resulted in an almost 60% reduction of EC activation by iTTP plasma. Noteworthy, was the fact that IgG stimulating effects were reproduced by the F(ab’)2 fractions and were thus linked to a putative antigen recognition on EC. Since iTTP is known to be caused by acquired antibodies directed against ADAMTS13, we investigated the role of these antibodies in EC activation. We did not succeed in directly purifying anti-ADAMTS13 Ab from the iTTP IgG fraction, thus we used different ways to determine whether ADAMTS13 played a role in endothelial activation by purified IgG. First, we compared the ability of serum from iTTP with those of hTTP to induce endothelial activation. Hereditary TTP are due to ADAMTS13 genetic deficiency and do not contain anti-ADAMTS13 auto antibodies. We observed a significant reduction (almost 60%) of endothelial activation using hTTP serum samples. Second, we observed that efficient inhibition of ADAMTS13 expression in HMVEC-d using siRNA was associated with a significant reduction of EC degranulation induced by iTTP IgG fraction. Third, we used two previously described human antiADAMTS13 mAb, cloned from iTTP isolated B cells,14 and observed that both of them weakly induced Ca2+ flux in EC as well as VWF and endothelin-1 secretion, confirming that anti-ADAMTS13 IgG were at least in part, responsible for iTTP-induced Ca2+-mediated EC activation. In most of the patients, anti-ADAMTS13 antibodies behave as inhibitors of the ADAMTS13 protease, but in 10-20% of iTTP-patients, these antibodies demonstrate other mechanisms of interaction with ADAMTS13, such as increased clearance,16,17 conformational changes,14,18 or increased metalloprotease activity19 without modifying ADAMTS13 quantification in ELISA.20,21 Our data suggest that in addition to these various interactions, anti-ADAMTS13 antibodies may also induce endothelial activation and VWF exocytosis, the well-known second hit of the disease.8 This observation may appear contradictory with animal models suggesting that antiADAMTS13 antibodies do not induce VWF release. Indeed,
Haematologica | 108 - April 2023 1137 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
injection of an inhibiting anti-human ADAMTS13 mAb into baboons did not increase circulating VWF concentrations, but induced histological lesions of TTP, therefore suggesting that at least local VWF occured.13 Also in humans the sole presence of anti-ADAMT13 auto antibodies seems insufficient to induce the disease, which appears frequently related to precipitating events such as pregnancy, surgery or infections.22 These apparently contradictory results may have several explanations. First that our observations in vitro may not have an important significance in vivo. However, despite some heterogeneity, our results show a significant association of Ca2+-induced endothelial activation in vitro with TTP prognosis and survival. Second, that not all anti-ADAMTS13 auto-antibodies have the capacity to induce WPB degranulation or that iTTP IgG may recognize additional endothelial targets to induce this effect. This is consistent with the fact that in our experiments, inhibition of ADAMTS13 expression did not completely suppress the effects of iTTP IgG and that anti-ADAMTS13 mAb weakly activated EC in vitro. Other anti-EC antibodies, as suggested by previous reports in iTTP and other autoimmune diseases, may participate in endothelial activation.23 Moreover, in support of the role of anti-ADAMTS13 in endothelial VWF release, it may be noticeable that during hTTP, even very low circulating concentrations of ADAMTS13 may not be associated with disease bouts, and that serum of hTTP which do not contain anti-ADAMTS13 auto-antibodies did not induce a strong endothelial activation in our experiments.
We also observed high free heme concentrations in iTTP plasma, in the order of magnitude of those observed in sickle cell disease and HUS patients.24,25 Heme-induced VWF exocytosis and participated in iTTP-induced Ca2+-mediated EC activation since VWF exocytosis was decreased by 30% by hemopexin. Free heme binds to TLR4 on EC and has been shown to induce WPB exocytosis.26,27 Interestingly, TLR4, also the receptor for LPS, may mediate Ca2+ flux and endothelial permeability, as shown in lung EC.28 Moreover, hemoglobin resulting from intravascular hemolysis has been shown to inactivate ADAMTS13.29 Furthermore, in accordance with previous reports showing that histones could induced WPB exocytosis,30 we observed elevated concentrations of nucleosomes in iTTP plasma samples which weakly participated in iTTP-induced Ca2+-mediated EC. DNA/histone complexes known to contribute to thrombosis have been reported elevated in the plasma of TMA patients15 and have been shown to induce TTP in a zebrafish model, underscoring their potential importance in iTTP pathogenesis.31 Despite previous evidence showing the amplifying role of complement in endothelial activation,32 our experiments did not show a role for complement in this very initial endothelial activation during iTTP. To date, except for markers of tissue ischemic injury, no real prognostic factor has been reported in iTTP, although
anti-ADAMTS13 antibodies of various isotypes, including IgA, may be associated with more severe forms.33,34 In our study, the amount of in vitro endothelial Ca2+ flux induced by 62 iTTP plasma or serum samples appeared to be associated with disease severity and death, whereas neither free heme, nor nucleosome concentrations correlated with iTTP severity. This result may indicate that several plasmatic components including anti-ADAMTS13 IgG, free heme, nucleosomes and possibly others converge to induce Ca2+-mediated EC activation. We have previously observed in a prospective study that circulating EC counts correlated with disease severity.35 Thus, iTTP early prognosis and death may be related to the intensity of endothelial activation/injury. Moreover, the identification of Ca2+ as the main cellular messenger of iTTP-induced WPB degranulation may possibly help to design a new therapeutic strategy targeting endothelial activation.
Disclosures
PC is a consultant for Sanofi, Alexion, Takeda and Roche. KV is a member of the scientific advisory boards of Ablynx-Sanofi and Shire-Takeda. PP is a member of the scientific advisory boards of Ablynx-Sanofi. All other authors have no conflicts of interest to disclose.
Contributions
ET, AW, MB, JF, RC and ML conducted the experiments and interpreted the data; MB, PP, PC, ENDO-13 study group members, ER, KV and GK contributed to sample collection and analysis of clinical data; ET, SR and JF contributed to image capture; PR and ET contributed to flow chamber experiments and fluorescence microscopy analysis; ER and KV provided TTP73 and ELH2-1 for the study and critically reviewed the manuscript; FDG handled funding and made a critical revision of the manuscript; RB critically reviewed the manuscript; GK took care of patients, handled funding and supervision of the project, helped with the interpretation of the experiments, and wrote the manuscript together with ET.
Acknowledgments
Members of the french ENDO-13 study group include D. Bourdessoule, CHU de Limoges; K. Clabault, CHU de Rouen; C. Daubin, CHU de Caen and G. Choukroun, CHU Amiens, France.
Funding
This study was supported by research funding from the French ministry of Health (Projet National de Recherche Clinique 2007, #2007/23) to GK.
Data-sharing statement
The original data and protocols are available to others investigators without unreasonable restrictions.
Haematologica | 108 - April 2023 1138 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
References
1. Kremer Hovinga JA, Coppo P, Lämmle B, Moake JL, Miyata T, Vanhoorelbeke K. Thrombotic thrombocytopenic purpura. Nat Rev Div Primers. 2017;3:17020.
2. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846.
3. Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312-322.
4. Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585-1594.
5. Furlan M, Robles R, Galbusera M, et al. von Willebrand factorcleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339(22):1578-1584.
6. Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26(5):1002-1007.
7. Banno F, Kokame K, Okuda T, et al. Complete deficiency in ADAMTS13 is prothrombotic, but it alone is not sufficient to cause thrombotic thrombocytopenic purpura. Blood. 2006;107(8):3161-3166.
8. Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115(10):2752-2761.
9. Nolasco LH, Turner NA, Bernardo A, et al. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106(13):4199-4209.
10. Schiviz A, Wuersch K, Piskernik C, et al. A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood. 2012;119(25):6128-6135.
11. Chauhan AK, Walsh MT, Zhu G, Ginsburg D, Wagner DD, Motto DG. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood 2008;111(7):3452-3457.
12. Deforche L, Tersteeg C, Roose E, et al. Generation of antimurine ADAMTS13 antibodies and their application in a mouse model for acquired thrombotic thrombocytopenic purpura. PLoS One. 2016;11(8):e0160388.
13. Feys HB, Roodt J, Vandeputte N, et al. Thrombotic thrombocytopenic purpura directly linked with ADAMTS13 inhibition in the baboon (Papio ursinus). Blood. 2010;116(12):2005-2010.
14. Roose E, Vidarsson G, Kangro K, et al. Anti-ADAMTS13 Autoantibodies against cryptic epitopes in immune-mediated thrombotic thrombocytopenic purpura. Thromb Haemost. 2018;118(10):1729-1742.
15. Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lämmle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120(6):1157-1164.
16. Shelat SG, Smith P, Ai J, Zheng XL. Inhibitory autoantibodies against ADAMTS-13 in patients with thrombotic thrombocytopenic purpura bind ADAMTS-13 protease and may accelerate its clearance in vivo. J Thromb Haemost. 2006;4(8):1707-1717.
17. Thomas MR, de Groot R, Scully MA, Crawley JTB. Pathogenicity
of anti-ADAMTS13 autoantibodies in acquired thrombotic thrombocytopenic purpura. EBioMedicine. 2015;2(8):942-952.
18. Roose E, Schelpe A-S, Tellier E, et al. Open ADAMTS13, induced by antibodies, is a biomarker for subclinical immune-mediated thrombotic thrombocytopenic purpura. Blood. 2020;136(3):353-361.
19. Schelpe A-S, Petri A, Roose E, et al. Antibodies that conformationally activate ADAMTS13 allosterically enhance metalloprotease domain function. Blood Adv. 2020;4(6):1072-1080.
20. Dekimpe C, Roose E, Tersteeg C, et al. Anti-ADAMTS13 autoantibodies in immune-mediated thrombotic thrombocytopenic purpura do not hamper ELISA-based quantification of ADAMTS13 antigen. J Thromb Haemost. 2020;18(4):985-990.
21. Dekimpe C, Roose E, Kangro K, et al. Determination of antiADAMTS-13 autoantibody titers in ELISA: influence of ADAMTS-13 presentation and autoantibody detection. J Thromb Haemost. 2021;19(9):2248-2255.
22. Morgand M, Buffet M, Busson M, et al. High prevalence of infectious events in thrombotic thrombocytopenic purpura and genetic relationship with toll-like receptor 9 polymorphisms: experience of the French Thrombotic Microangiopathies Reference Center. Transfusion. 2014;54(2):389-397.
23. Praprotnik S, Blank M, Levy Y, et al. Anti-endothelial cell antibodies from patients with thrombotic thrombocytopenic purpura specifically activate small vessel endothelial cells. Int Immunol. 2001;13(2):203-210.
24. Frimat M, Tabarin F, Dimitrov JD, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122(2):282-292.
25. Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383-1389.
26. Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vasoocclusion in murine sickle cell disease. Blood. 2014;123(3):377-390.
27. Figueiredo RT, Fernandez PL, Mourao-Sa DS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282(28):20221-20229.
28. Tauseef M, Knezevic N, Chava KR, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxininduced lung vascular permeability and inflammation. J Exp Med. 2012;209(11):1953-1968.
29. Studt J-D, Kremer Hovinga JA, Antoine G, et al. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood. 2005;105(2):542-544.
30. Michels A, Albánez S, Mewburn J, et al. Histones link inflammation and thrombosis through the induction of WeibelPalade body exocytosis. J Thromb Haemost. 2016;14(11):2274-2286.
31. Zheng L, Abdelgawwad MS, Zhang D, et al. Histone-induced thrombotic thrombocytopenic purpura in adamts13-/- zebrafish depends on von Willebrand factor. Haematologica. 2020;105(4):1107-1119.
32. Ruiz-Torres MP, Casiraghi F, Galbusera M, et al. Complement activation: the missing link between ADAMTS-13 deficiency and microvascular thrombosis of thrombotic microangiopathies. Thromb Haemost. 2005;93(3):443-452.
Haematologica | 108 - April 2023 1139 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
33. Ferrari S, Scheiflinger F, Rieger M, et al. Prognostic value of anti-ADAMTS 13 antibody features (Ig isotype, titer, and inhibitory effect) in a cohort of 35 adult French patients undergoing a first episode of thrombotic microangiopathy with undetectable ADAMTS 13 activity. Blood. 2007;109(7):2815-2822.
34. Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13
antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7(10):1703-1710.
35. Widemann A, Pasero C, Arnaud L, et al. Circulating endothelial cells and progenitors as prognostic factors during autoimmune thrombotic thrombocytopenic purpura: results of a prospective multicenter French study. J Thromb Haemost. 2014;12(10):1601-1609.
Haematologica | 108 - April 2023 1140 ARTICLE - Ca2+-mediated Weibel-Palade exocytosis in iTTP E. Tellier et al.
Reduced platelet glycoprotein Ib α shedding accelerates thrombopoiesis and COX-1 recovery: implications for aspirin
dosing regimen
Paola Simeone,1* Rossella Liani,1* Romina Tripaldi,1 Sonia Ciotti,1 Antonio Recchiuti,2 Vittorio Abbonante,3,4 Benedetta Porro,5 Piero Del Boccio,6 Augusto di Castelnuovo,7 Paola Lanuti,1 Marina Camera,5,8 Damiana Pieragostino,9 Melissa Lee-Sundlov,10 Myriam Luongo,11 Raffaella Auciello,12 Giuseppina Bologna,1 Maria Concetta Cufaro,9 Elena Tremoli,13 Karin M. Hoffmeister,10,14 Francesco Cipollone,1 Alessandra Balduini3 and Francesca Santilli1
1Department of Medicine and Aging Sciences, Center for Advanced Studies and Technology (CAST), University of Chieti, Chieti, Italy; 2Department of Medical, Oral, and Biotechnological Science, Center for Advanced Studies and Technology (CAST), Chieti, Italy; 3Department of Molecular Medicine, University of Pavia, Pavia, Italy; 4Department of Health Sciences, Magna Graecia University of Catanzaro, Catanzaro, Italy; 5Centro Cardiologico Monzino IRCCS, Milan, Italy; 6Department of Pharmacology, Center for Advanced Studies and Technology (CAST), Chieti, Italy; 7Mediterranea Cardiocentro, Napoli, Italy; 8Department of Pharmaceutical Sciences, Università degli Studi di Milano, Milan, Italy; 9Department of Innovative Technologies in Medicine and Dentistry, Center for Advanced Studies and Technology (CAST), Chieti, Italy; 10Versiti Translational Glycomics Center and Versiti Blood Research Institute, Milwaukee, WI, USA; 11Immunotransfusion Service, Clinical Hematology of Chieti University Hospital, Chieti, Italy; 12Clinical Pathology of Chieti University Hospital, Chieti, Italy; 13Maria Cecilia Hospital, Cotignola, Italy and 14Departments of Biochemistry and Medicine, Medical College of Wisconsin, Milwaukee, WI, USA
*PS and RL contributed equally as co-first authors.
Abstract
Correspondence: F. Santilli francesca.santilli@unich.it
Received: March 10, 2022.
Accepted: December 5, 2022.
Early view: December 22, 2022.
https://doi.org/10.3324/haematol.2022.281006
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Cardiovascular (CV) disease prevention with low-dose aspirin can be less effective in patients with a faster recovery of platelet (PLT) cyclooxygenase (COX)-1 activity during the 24-hour dosing interval. We previously showed that incomplete suppression of TXA2 over 24 hours can be rescued by a twice daily aspirin regimen. Here we show that reduced PLT glycoprotein (GP)Ibα shedding characterizes patients with accelerated COX-1 recovery and may contribute to higher thrombopoietin (TPO) production and higher rates of newly formed PLT, escaping aspirin inhibition over 24 hours. Two hundred aspirin-treated patients with high CV risk (100 with type 2 diabetes mellitus) were stratified according to the kinetics of PLT COX-1 activity recovery during the 10- to 24-hour dosing interval. Whole proteome analysis showed that PLT from patients with accelerated COX-1 recovery were enriched in proteins involved in cell survival, inhibition of apoptosis and cellular protrusion formation. In agreement, we documented increased plasma TPO, megakaryocyte maturation and proplatelet formation, and conversely increased PLT galactose and reduced caspase 3, phosphatidylserine exposure and ADAM17 activation, translating into diminished GPIbα cleavage and glycocalicin (GC) release. Treatment of HepG2 cells with recombinant GC led to a dose-dependent reduction of TPO mRNA in the liver, suggesting that reduced GPIbα ectodomain shedding may unleash thrombopoiesis. A cluster of clinical markers, including younger age, non-alcoholic fatty liver disease, visceral obesity and higher TPO/GC ratio, predicted with significant accuracy the likelihood of faster COX-1 recovery and suboptimal aspirin response. Circulating TPO/GC ratio, reflecting a dysregulation of PLT lifespan and production, may provide a simple tool to identify patients amenable to more frequent aspirin daily dosing.
Introduction
and in thrombus growth and vascular occlusion.1 Thromboxane (TX)-dependent platelet (PLT) activation, as reflected by 11-dehydro-TXB2 urinary excretion, has been associated with several conditions, including cardio-cerebrovascular diseases and cardiovascular (CV) risk factors, ARTICLE - Platelet Biology & its Disorders
Platelets play key roles in atherothrombosis, by acting as inflammatory mediators, and participating in the processes of forming and extending atherosclerotic plaques, Haematologica | 108 April 2023 1141
such as diabetes mellitus.2,3
The anti-PLT and cardioprotective properties of low-dose (81-100 mg daily) aspirin rely on its capacity to irreversibly inactivate cyclooxygenase (COX)-1 in anucleate PLT and TXA2 biosynthesis over 24 hours.3 Over the last decades, the concept of suboptimal response to aspirin, firstly inadequately referred to as aspirin resistance, has been affirmed, based on the evidence of lower-than-expected inhibition of PLT function assays, which, however, poorly reflect the mechanism of action of aspirin, and display scarce reproducibility over repeated measurements and poor correlation with COX-1 inhibition, as reflected by serum TXB2 (sTXB2).4–6
The variable turnover rate of the aspirin target, PLT COX1, is the most convincing determinant of the interindividual variability in the aspirin response. Under several pathological conditions, such as essential thrombocythemia, polycythemia vera, on-pump coronary artery by-pass surgery, as well as in patients at high CV risk, with or without type 2 diabetes mellitus (T2DM),7–10 a proportion of PLT with uninhibited COX-1 may determine faster recovery of COX-1 activity and TXA2 generation and limit the extent and duration of aspirin effect during the 12- to 24-hour dosing interval7,8 leading to substantial PLT activation and recovery of PLT function. As full suppression of TXA2-dependent PLT function requires >97% inhibition of COX-1 activity,6 even a modest recovery of this activity can sustain a substantial PLT activation responsible for the excess CV events in aspirin-treated subjects.
Shorter duration of TXA2 suppression during the 24-hour dosing interval can be rescued by a twice daily low-dose aspirin regimen,7,9,10 leading to persistently inhibited sTXB2 over 24 hours. An accelerated PLT turnover has been historically advocated as the underlying mechanism, although no direct evidence has been provided to substantiate enhanced megakaryopoiesis and accelerated destruction in subjects with shorter duration of the effect of aspirin. Patients’ classification based on clinical setting, including diabetes or acute coronary syndrome, failed to accurately identify patients escaping low-dose aspirin inhibition for whom different strategies or aspirin-dosing regimens may be required. Understanding the mechanisms driving faster COX-1 recovery in the usual dosing interval may help to identify novel biomarkers of suboptimal drug response.
Thrombopoietin (TPO) is the primary regulator of PLT production from megakaryocytes (Mk)11 and is produced by the liver due to a number of stimuli including inflammation.11 Conversely, PLT lifespan, glycan degradation and apoptosis mediate PLT clearance.11 Senescent PLT are cleared upon exposure of galactose to hepatic AshwellMorrell receptors (AMR), in turn stimulating TPO production.11,13 Glycoprotein I α (GPIb α ) shedding by metalloproteinases such as a disintegrin and metallopro-
tease (ADAM) 17 is another mechanism of PLT clearance,12 and plasma glycocalicin (GC), an extra cellular domain of GPIbα, released during PLT clearance, is an index of destruction and PLT turnover.14 PLT extracellular GPIbα domain is required for PLT-mediated TPO generation, as underscored in GPIbα / mice and patients with BernardSoulier syndrome.15
By employing an integrated approach including biochemistry, proteomics, fl ow-cytometry, cell biology, and a mechanism-based endpoint to monitor aspirin pharmacodynamics, we analyzed the PLT proteome, platelet turnover, as reflected by TPO and GC circulating levels, galactose exposure and GPIbα ectodomain shedding, Mk maturation and proplatelet formation (PPF) in patients at high CV risk with or without T2DM stratified according to the kinetics of COX-1 recovery. We show that reduced platelet GPIbα shedding characterizes patients with accelerated COX-1 recovery and may contribute to higher TPO production and higher rates of newly formed PLT, escaping aspirin inhibition over 24 hours. The TPO/GC ratio, a relatively simple, mechanism-based biochemical tool, may identify with significant diagnostic accuracy aspirinpoor responders due to accelerated renewal of the drug target.
Methods
Study design and participants
One hundred T2DM patients diagnosed according to the European Society of Cardiology (ESC) guidelines,16 with or without prior vascular disease, on 100 mg aspirin (entericcoated, Cardioaspirin, the anti-PLT dosage and formulation employed in Italy) once daily for at least 1 year and 100 patients without T2DM with comparable characteristics (Online Supplementary Table S1, study participants) were enrolled at the Diabetes and CV Prevention Clinics, Chieti SS Annunziata Hospital in Italy. Five healthy subjects were enrolled as control. All patients underwent a liver ultrasound (see the Online Supplementary Appendix). The study was preceded by a 7-day run-in during which patients were instructed to take aspirin at 8 am. Blood was collected before witnessed aspirin intake (8 am, day 1), and repeated at 10 and 24 hours after the last intake, to assess the kinetics of COX-1 activity recovery, as reflected by the slope of sTXB2 levels throughout the 24hour dosing interval between two witnessed aspirin administrations (Figure 1A). sTXB2 is indeed a marker of COX-1 activity ex vivo. 17 We previously reported that the absolute increase in sTXB2 levels between 12 and 24 hours after dosing predicts ~90% slope variability, as assessed by measuring sTXB2 every 3 hours in the 12- to 24-hour dosing interval.10
Based on the sTXB2 slope (sTXB2 T24 – sTXB2 T10)/14, we
Haematologica | 108 April 2023 1142 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
stratified patients in tertiles (n=~33 each) and carried out a cross-sectional comparison of PLT-related study variables between first (good aspirin responders) and third tertile (poor responders) within patients with and without T2DM.
Platelet isolation
PLT rich plasma (PRP) was separated (centrifugation 100xg, 15 minutes) from ACD-A anticoagulated blood, mixed with prostaglandin E1 (PGE1, 4 uM) and EDTA (10 mM), and filtered on Pall Purecell PL (Pall Medical, New York, USA) to remove leukocyte contaminants. Filtered PRP was analyzed by flow cytometery to exclude red blood cell (CD235+) contamination and platelet activation (CD62P+). PLT were lysed with DIGE buffer for western blot (WB) and proteomics analysis.
Statistics
We estimated that at least 30 patients would be required in each group to detect a mean difference in any of the investigated parameters with ≥1 standard deviation between the first and the third sTXB2 slope tertiles with α=0.01 and power=90%. All the statistical analyses were carried out separately in individuals with and without T2DM. Univariable comparisons between groups were performed by c2 tests or Mann-Whitney U or Spearman rank correlation test. Multivariable logistic regression models were constructed to identify factors associated with the likelihood of being in the third versus the first tertile for the sTXB2 recovery slope. A parsimonious backward-stepwise elimination of variables with P<0.20 was deemed appropriate in our setting. ROC curves were constructed for the predicted probabilities derived from the logistic regression models. The data analysis was generated using SAS software.
Study approval
The protocol was approved by the Institutional Ethics Committee (Prot 1129/2015, GR-2011-02350450). Participants provided written informed consent, and were identified by number, not by name. See the Online Supplementary Appendix (pages 2-8) for more details on study participants and an extended description of experimental procedures.
Results
Clinical characteristics
Clinical characteristics of patients are listed in the Online Supplementary Table S1 and detailed in the Online Supplementary Appendix. Patients’ characteristics according to sTXB2 recovery slope tertiles are reported in Table 1 and detailed in the Online Supplementary Table S2 and the Online Supplementary Appendix
COX-1 activity recovery
A fraction of patients showed faster recovery of COX-1 activity over the 10– to 24-hour aspirin dosing interval, higher COX-1 mRNA expression and TX-dependent platelet activation. In the first tertile of the recovery slope, sTXB2 was steadily suppressed over the 10– to 24-hour dosing interval (Figure 1B, C), as observed in healthy subjects,6 while patients in the third tertile (slope >=0.17 ngmL-1h-1 and slope >=0.18 ngmL-1h-1 in patients with and without diabetes, respectively) showed a significantly faster recovery of COX-1 activity, as reflected by the sTXB2 increase between 10 and 24 hours post-aspirin (Figure 1B, C). This indicates that at least a fraction of aspirin-treated patients at risk for CV events has an accelerated recovery of the drug target, and that the “poor aspirin responder”
Continued on following page.
A Haematologica | 108 April 2023 1143 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
Figure 1. Subjects with faster kinetics of recovery of serum thromboxane B2 during the 24-hour aspirin dosing interval, display increased platelet COX-1 expression and urinary 11-dehydro-thromboxane B2 excretion as compared to those with normal serum thromboxane B2 recovery. (A) Study design. Linear fitting of serum thromboxane B2 (sTXB2) measured 10 and 24 hours post-aspirin intake in patients without (B) (n=100), and with type 2 diabetes mellitus (T2DM) (C) (n=100) stratified in tertiles according to sTXB2 (ex vivo index of COX-1-dependent TXA2 production) slope (n=33/tertile). Patients in the third sTXB2 slope tertile display significantly faster recovery of sTXB2 vs. first tertile (P<0.001), during the 24 hours between 2 witnessed aspirin administrations. sTXB2 values are as median and interquartile range. Comparison of subjects in the third vs. first sTXB2 slope tertile for platelet transcript levels of COX-1 mRNA (D) (n=20 vs. n=24) and urinary 11-dehydro-TXB2 (E) (n=66 vs. n=66). Significance was calculated by Mann-Whitney U test.
phenotype is not specific for diabetes. In addition, the TXB2 recovery slope was not related to the duration of the aspirin treatment (Online Supplementary Table S2).
PLT COX-1 mRNA was significantly higher in the third versus the first tertile (P=0.014; Figure 1D), indicating a faster renewal of the drug target in the patients with suboptimal response to aspirin. In order to assess whether an accelerated recovery of sTXB2 ex vivo translated into enhanced in vivo TX-dependent PLT activation, we measured urinary 11-dehydro-TXB2 levels, that were higher in patients of the third versus the first tertile in the whole group (P=0.022; Figure 1E) and in T2DM (P=0.049; data not shown).
Characterization of the proteomic platelet profile PLT proteome indicated increased cell survival and inhibition of apoptosis in patients with faster COX-1 activity recovery. We characterized the functional proteomic profile of PLT from the third versus the first tertile within patients with or without T2DM, using pooled samples as described in the method section. The quantified proteins
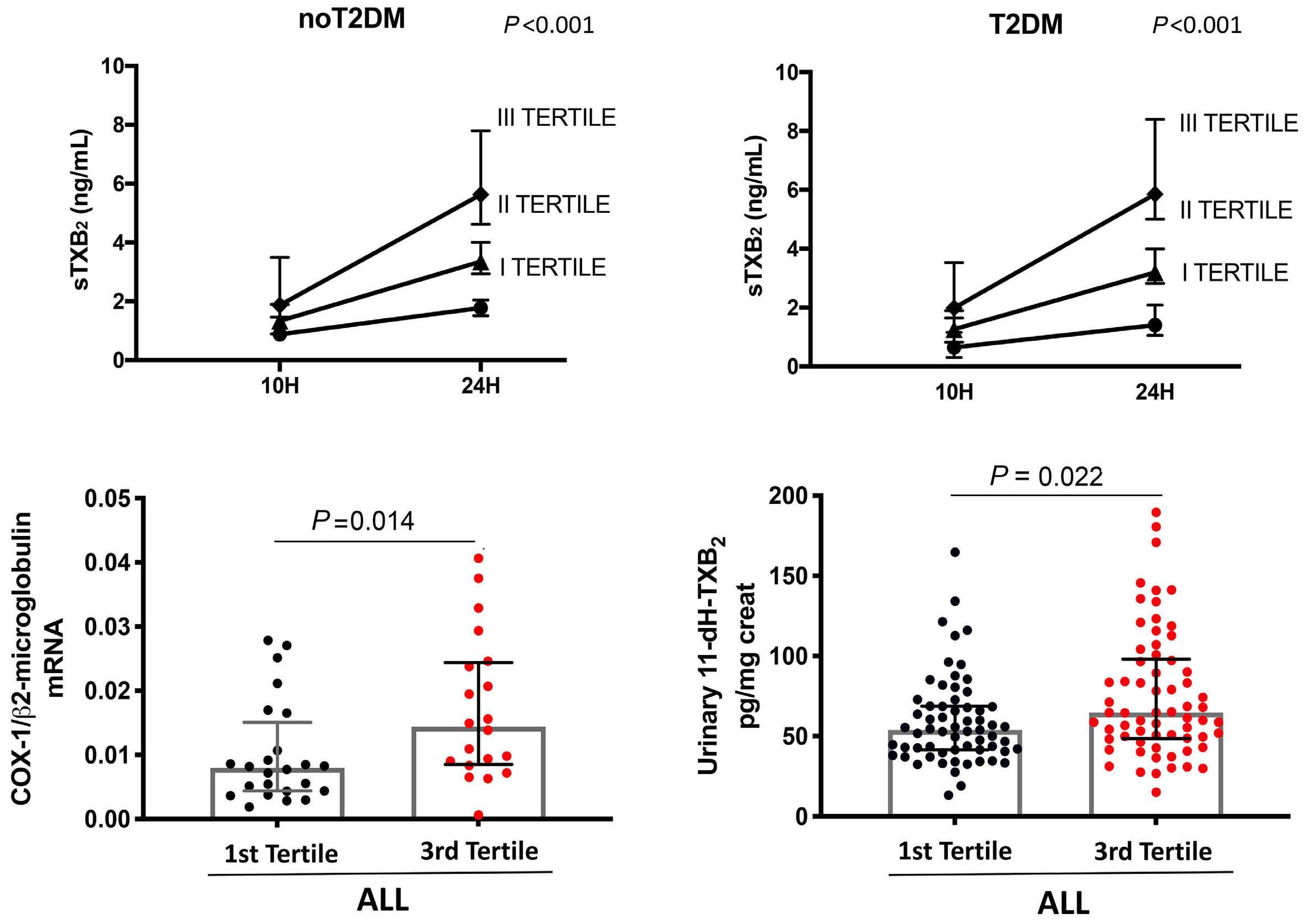
identified are reported in the Online Supplementary Tables S4, S5. In order to ensure the quality of our proteomics dataset we compared it to the reference PLT proteomic repository, obtaining more than 95% correlation (data not shown). Volcano plots show proteins differentially identified between the first versus the third tertile patients without and with T2DM (Online Supplementary Figure S3A, B).
The protein ratio was used for “core analysis” through the ingenuity pathway analysis (IPA software). We found a significant increase in the pathways “cell survival” (zscore=1.54 for noT2DM, z-score=2.01 for T2DM) “formation of cellular protrusions” (z-score=2.43 for noT2DM, zscore=1.92 for T2DM), and “production of reactive oxygen species” (z-score=2.06 for noT2DM, z-score=2.37 for T2DM), and a simultaneous inhibition of “apoptosis” (zscore=-0.58 for noT2DM, z-score=-1.86 for T2DM) in both clinical conditions in platelets from third tertile subjects in comparison to those from the first tertile (Figure 2A, B). We validated the proteomic data by WB analysis and
B C D E Haematologica | 108 April 2023 1144 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
Table 1. Characteristics of patients with and without type 2 diabetes mellitus in relation to tertiles of serum thromboxane B2 recovery slope.
Reference range of “normal values”: AST, 5-34 U/L; ALT, 0-55 U/L; hs-C-reactive protein, 0-47.62 nmol/L; platelet count, 150-450 x109/L; total cholesterol, 0.0-5.17 mmol/L; HDL cholesterol, 1.03-1.55 mmol/L; triglycerides, 0-1.69 mmol/L; fasting plasma glucose, 4.61-6.11 mmol/L; serum creatinine, 50.2-97.7 umol/L; fasting plasma glucose, 3.9-5.5 mmol/L; eGFR, >60 mL/min. BMI: body mass index; WC: waist circumference; WHR: waist-to-hip ratio; SAP: systolic arterial pressure; DAP: diastolic arterial pressure; NAFLD: non-alcoholic fatty liver disease; HbA1c: glycated hemoglobin; HDL: high-density lipoproteins; AST: aspartate aminotransferase; ALT: alanine amino transferase; hs-PCR: hs-C-reactive protein; FPG: fasting plasma glucose: eGFR: estimated glomerular filtration rate. Data are median (25th – 75th percentile). †Determined by Kruskal-Wallis or x2 test, as appropriate.
Variable T2DM noT2DM 1st tertile 2nd tertile 3rd tertile P value 1st tertile 2nd tertile 3rd tertile P value Age in years 69.0 (66.0-74.5) 69.0 (63.7-73.5) 67.0 (61.0-69.5) 0.047 69.0 (62.5-76.5) 67.0 (61.8-71.0) 69.0 (65.5-73.0) 0.468 Male sex, N (%) 20 (60.6) 22 (64.7) 22 (66.7) 0.872 17 (51.5) 19 (55.9) 21 (63.6) 0.602 Smokers, N (%) 4 (12.1) 5 (14.7) 4 (12.1) 0.936 4 (12.1) 2 (5.9) 4 (12.1) 0.616 Weight, kg 80.0 (70.5-90.0) 80.0 (71.3-95.5) 85.0 (73.3-92.5) 0.505 80.0 (70.5-90.0) 80.0 (71.3-95.5) 85.0 (73.3-92.5) 0.505 BMI, kg/m2 29.3 (25.5-31.5) 29.5 (25.7-33.7) 31.0 (28.1-34.7) 0.148 26.3 (25.4-30.0) 29.4 (24.4-32.5) 28.0 (25.9-32.7) 0.248 WC, cm 100.0 (100.0-110.0) 100.0 (90.0-110.0) 110.0 (100.0-110.0) 0.120 100.0 (90.0-110.0) 100.0 (90.0-110.0) 110.0 (100.0-120.0) 0.026 WHR 1.0 (0.9-1.0) 1.0 (0.9-1.0) 1.0 (0.9-1.0) 0.747 0.9 (0.9-1.0) 1.0 (0.9-1.0) 1.0 (0.9-1.0) 0.003 Obesity, N (%) 13 (39.4) 15 (44.1) 21 (63.6) 0.112 10 (30.3) 16 (47.1) 14 (42.4) 0.354 SAP, mmHg 145.0 (134.0-150.0) 146.0 (134.0-150.0) 148.0 (134.0-160.0) 0.675 140.0 (128.0-151.5) 138.5 (125.0-152.5) 142.0 (130.0-159.0) 0.413 DAP, mmHg 75.0 (70.0-80.0) 74.0 (69.0-80.0) 78.0 (70.0-83.0) 0.593 74.0 (67.0-80.0) 78.5 (70.0-81.5) 80.0 (70.5-86.0) 0.341 Hypertension, N (%) 29 (87.9) 30 (88.2) 31 (93.9) 0.653 24 (72.7) 28 (82.4) 28 (84.8) 0.429 Dyslipidemia, N (%) 17 (51.5) 22 (64.7) 21 (63.6) 0.476 24 (72.7) 13 (38.2) 17 (51.5) 0.017 Diabetes duration in years 4.0 (2.0-13.0) 5.0 (3.0-10.0) 6.5 (1.0-11.0) 0.983 - - -NAFLD, N (%) 19 (70.4) 30 (90.9) 27 (87.1) 0.040 21 (63.6) 13 (38.2) 18 (54.5) 0.322 HbA1c, mmol/mol 50.8 (42.1-56.3) 50.8 (45.4-56.3) 53.0 (45.4-61.7) 0.312 38.8 (34.4-41.0) 38.8 (34.4-42.1) 38.8 (36.6-41.0) 0.831 HbA1c, % 6.8 (6.0-7.3) 6.8 (6.3-7.3) 7.0 (6.3-7.8) 5.7 (5.3-5.9) 5.7 (5.3-6.0) 5.7 (5.5-5.9) Total cholesterol, mmol/L 4.5 (3.7-5.0) 4.2 (3.7-4.7) 4.6 (4.1-5.3) 0.223 4.9 (4.1-5.4) 5.3 (4.5-5.6) 4.4 (3.9-5.3) 0.130 HDL cholesterol, mmol/L 1.2 (1.0-1.5) 1.3 (1.0-1.4) 1.3 (1.1-1.5) 0.562 1.4 (1.1-1.5) 1.4 (1.2-1.6) 1.2 (1.1-1.4) 0.088 Triglycerides, mmol/L 1.4 (1.0-2.0) 1.4 (1.0-1.8) 1.5 (1.0-2.1) 0.914 1.2 (1.0-1.7) 1.2 (0.9- 1.6) 1.3 (1.0-1.9) 0.494 AST, U/L 21.0 (19.0-33.0) 24.5 (20.0-28.2) 26.0 (20.0-32.5) 0.513 25.0 (22.0-30.0) 22.5 (21.0-26.3) 24.0 (19.5-29.5) 0.593 ALT, U/L 28.0 (24.0-41.0) 31.0 (24.0-39.2) 34.0 (27.0-43.0) 0.270 31.0 (25.0-36.0) 25.5 (22.0-31.0) 30.0 (25.5-36.0) 0.005 Total bilirubin, umol/L 10.0 (7.0-14.0) 10.0 (9.0-14.0) 12.0 (9.0-1.0) 0.257 12.0 (10.0-15.0) 14.0 (0.6-15.0) 12.0 (10.0-17.0) 0.957 hs-PCR, nmol/L 19.0 (9.5-38.1) 28.6 (9.5-38.1) 19.0 (9.5-57.1) 0.592 19.0 (9.5-28.6) 19.0 (9.5-38.1) 28.6 (9.5-47.6) 0.546 Platelet count, x109/L 222.0 (179.0-241.0) 221.0 (193.0-251.0) 234.0 (206.0-284.0) 0.080 214.5 (178.2-240.5) 243.0 (193.8-288.8) 223.0 (198.5-278.8) 0.196 Mean platelet volume, fL 11.3 (10.6-11.9) 11.2 (10.5-11.7) 11.3 (10.7-11.8) 0.673 11.4 (11.1-12.0) 11.1 (10.2-11.6) 11.0 (10.2-11.5) 0.029 FPG, mmol/L 7.0 (5.7-7.6) 6.6 (5.7-7.2) 6.7 (5.6-7.8) 0.469 5.3 (4.7-5.5) 5.2 (4.8-5.5) 5.3 (4.9-5.7) 0.533 Serum creatinine, umol/L 79.2 (61.6-88.0) 70.4 (61.6-79.2) 70.4 (61.6-79.2) 0.689 70.4 (61.6-79.2) 70.4 (61.6-88.0) 70.4 (70.4-96.8) 0.396 eGFR, mL/min 85.0 (67.4-97.0) 90.1 (75.5-95.2) 91.0 (86.5-109.6) 0.034 88.6 (65.3-106.0) 88.3 (75.8-96.6) 82.0 (70.7-93.5) 0.570
Haematologica | 108 April 2023 1145 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
confirmed a significant upregulation of the endoplasmic reticulum stress marker, 78 kDa glucose-regulated protein (GRP78, P=0.042; Figure 2C), and COX-1 (P=0.051; Figure 2D). Patients of the third tertile showed consistently higher levels of GRP78 (P=0.042, P=0.017; Figure 2C) and COX-1 (P=0.051, P=0.004; Figure 2D) compared to first tertile patients and healthy subjects. In order to further explore the potential hypothesis of the formation of protrusions in PLT, we used the molecule activity predictor (MAP) function of IPA by selecting “formation of proplatelets” as downstream of interest. Our proteomic dataset was able to simulate directional consequences on this function by inferring its activation in PLT from third tertile subjects in both clinical conditions analyzed as shown by subnetworks in Figure 2E, with a significant upregulation of Rab27B, a protein actively involved in platelet biogenesis and proplatelet formation,18 in PLT of the third versus the first tertile (P=0.038; Figure 2F) and versus healthy subjects (P<0.001; Figure 2F).
Megakaryocyte maturation and proplatelet formation in patients
with faster COX-1 activity recovery
Based on the proteomic findings, we investigated whether the different recovery time of COX-1 activity reflects differences in megakaryopoiesis and proplatelet production by differentiating Mk in vitro from the hematopoietic progenitors derived from 18 peripheral blood samples of patients with and without T2DM belonging to the first and third sTXB2 tertile and from four healthy subjects (Figure 3A, B). The clinical characteristics of the patients’ groups
analyzed were comparable (Online Supplementary Table S6).
Mk differentiated from third tertile patients appeared more mature with a significantly higher staining of CD41 and CD42b (GPIb α ) Mk19 compared to first tertile and healthy subjects (Figure 3C, D), while the percentages of CD41+ and GPIb α + Mk on the total cultured cells were comparable (Figure 3C, D). Mk from third tertile patients extended more proplatelets compared to the first tertile or healthy subjects (Figure 3E). These data demonstrate that terminal Mk maturation and proplatelet formation were increased in patients presenting a faster recovery of platelet COX-1 activity.
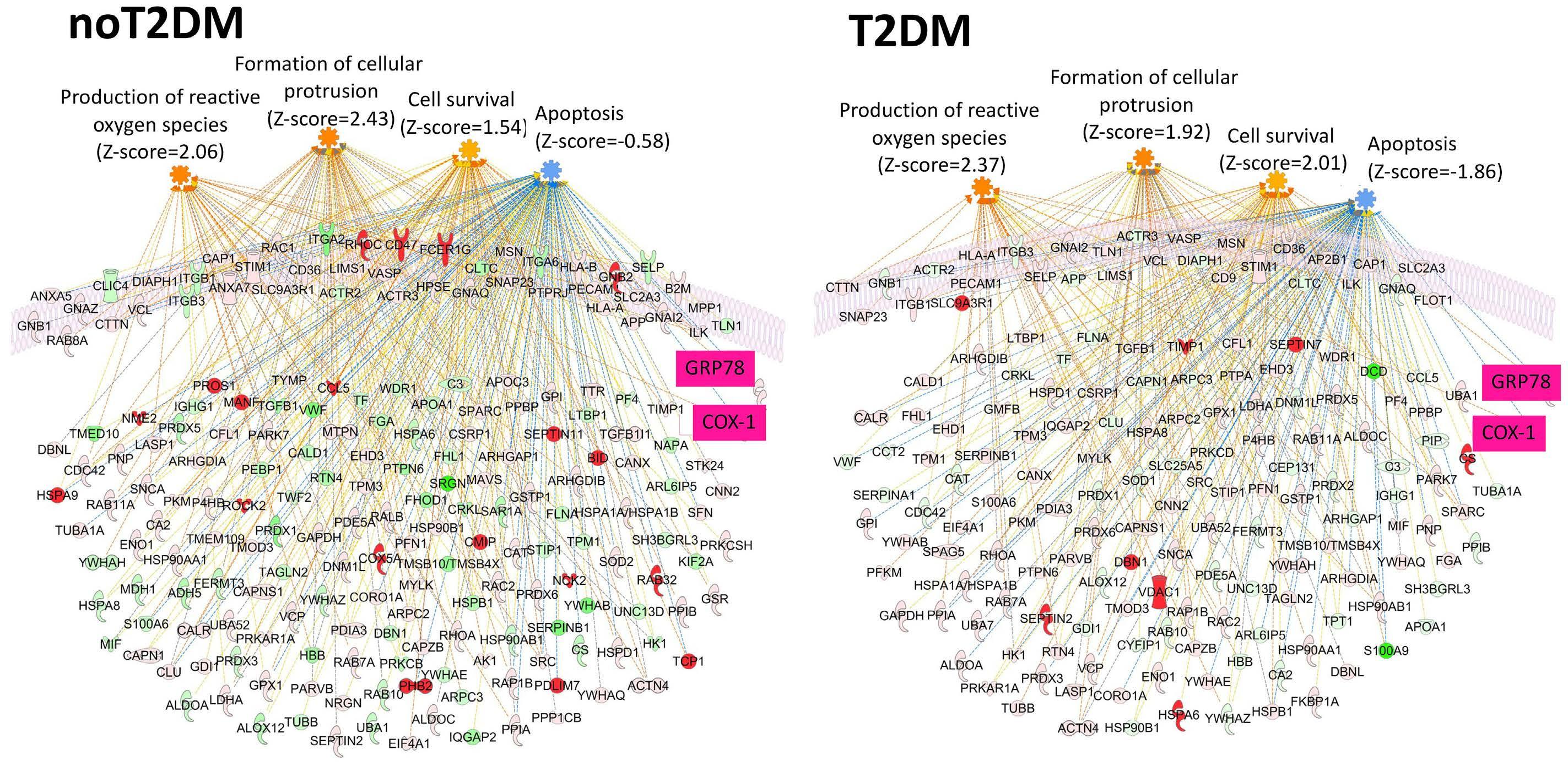
Association between COX-1 activity recovery and platelet production and destruction
We next evaluated the balance between PLT production and destruction by measuring TPO, GC and GC index (GCI), an index normalizing GC for PLT count (GC concentration µg/mLx250x109 PLT/L/individual platelet count x109/L), 24 hours after witnessed aspirin intake. Consistent with the proteomic findings, plasma TPO levels were higher (P=0.004, P=0.023; Figure 4A; Online Supplementary Figure 4SA), while plasma GC (P<0.001 both; Figure 4B; Online Supplementary Figure 4SB) and GCI (P<0.001 both; Figure 4D; Online Supplementary Figure 4SC) were lower in patients of the third versus the first tertile, in the whole group and in subjects with T2DM, with an inverse correlation between TPO, GC (rho=-0.216, P=0.013; data not shown) and GCI (whole group: rho=-0.245, P=0.006; T2DM: Continued on following page.
A B Haematologica | 108 April 2023 1146 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
Figure 2. Proteomic analysis shows activation of pathways “formation of cellular protrusions”, “formation of proplatelets”, “cell survival”, “production of reactive oxygen species”, and inhibition of “apoptosis” in platelets from patients with faster COX-1 recovery. Proteomic analysis using ingenuity pathway analysis (IPA) revealed activation of “formation of cellular protrusions”, “formation of proplatelets”, “cell survival”, “production of reactive oxygen species”, and inhibition of “apoptosis” pathways in platelets of third vs. first serum thromboxane B2 (sTXB2) tertile in patients without (A, E), and with type 2 diabetes mellitus (T2DM) (B, E). Further details are reported in the Online Supplementary Figures S1, S2 and S3. Validation of proteomic data by western blot, assessing 78 kDa glucose-regulated protein (GRP78) (C) (n=4/tertile), COX-1 (D) (n=4/tertile) and Rab27B (F) (n=4/tertile) in patients from third tertile vs. first tertile and healthy subjects (HS) (n=4), using b-Actin as loading control. Significance was calculated by Student’s t-test.
rho=-0.358, P=0.004; Figure 4E; Online Supplementary Figure 4SD). Similarly, among patients without T2DM, GC (P<0.001; Online Supplementary Figure 4SB) and GCI (P<0.001; Online Supplementary Figure 4SC) were significantly reduced in the third versus the first sTXB2 tertile, with a non-significant trend for increased TPO (Online Supplementary Figure 4SA). Levels of TPO were lower (P=0.001, P<0.001, P=0.009; Figure 4A; Online Supplementary Figure 4SA) and levels of GC were higher (P<0.001, P<0.001, P=0.005; Figure 4B; Online Supplementary Figure 4SB) in healthy subjects versus third tertile patients, in the whole group and in subjects without and with T2DM. Consistently, the platelet count was higher in the third versus first tertile in all patients (P=0.024; Figure 4C), and in T2DM patients (P=0.047; Online Supplementary Figure 4SE). In the whole group, the TXB2 recovery slope correlated directly with TPO (rho=0.252, P=0.003), and inversely with GC (rho=-0.432, P<0.001) and GCI (rho=-0.495, P<0.001; data not shown). Together, these results indicate
that patients in the third TXB2 slope tertile presented increased platelet production and reduced destruction.
Mechanisms underlying low circulating glycocalicin levels
In order to determine the mechanisms underlying lower GC in the third sTXB2 slope tertile patients, we measured GPIbα N-terminal fragment and ADAM17 expression in platelets.20 In all patients, we observed enhanced levels of GPIb α (P=0.016, P<0.001; Figure 5A) and lower levels of ADAM1721 (P=0.015, P=ns; Figure 5B) in the third versus first tertile and versus healthy subjects, respectively, which may explain the lower levels of circulating GC in the third slope tertile (Figure 4B; Online Supplementary Figure 4SB). In order to further understand the mechanisms underlying reduced GPIbα shedding, we analyzed the percentage of Annexin V+ platelets exposing phosphatidylserine (PS), since PS exposure is a signal for clearance of apoptotic platelets22 and is required for ADAM17 activation.20 The
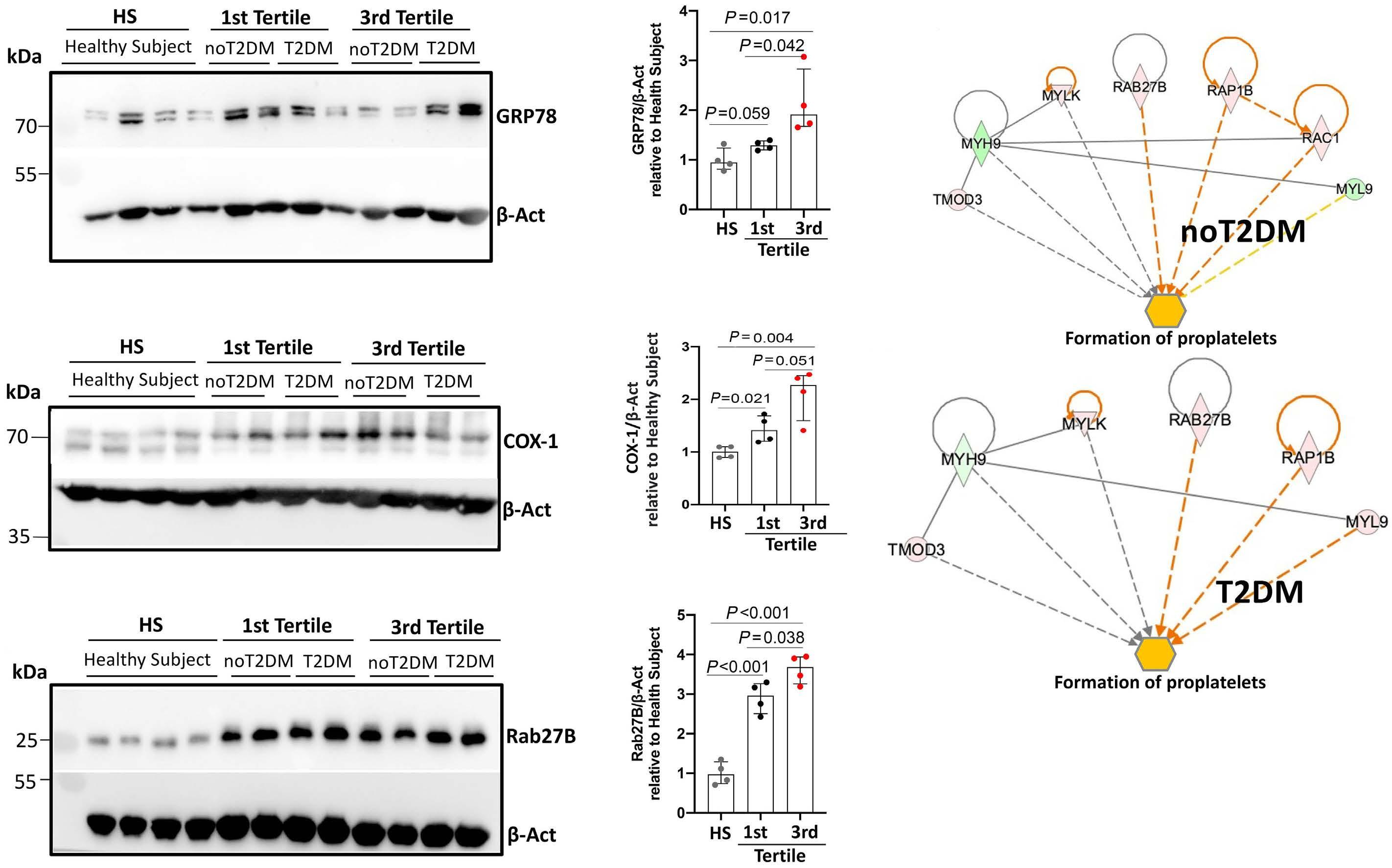
C D E F Haematologica | 108 April 2023 1147 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
Figure 3. Enhanced megakaryocyte maturation and proplatelet formation in patients with faster COX-1 recovery. Representative immunofluorescence of megakaryocytes (Mk) (A) and proplatelet formation (PPF) (B). The proportion of mature Mk was measured by flow cytometry, as the percentage and mean fluorescence intensity (MFI) of CD41-positive cells (healthy subjects n=5; first tertile: all n=8, type 2 diabetes mellitus (T2DM) n=4, no T2DM n=4; third tertile: all n=10, T2DM n=4, no T2DM n=6) (C, D); and as the percentage and MFI of GPIbα (CD42b)-positive cells (healthy subjects n=5; first tertile: all n=8, T2DM n=4, no T2DM n=4; third tertile: all n=11, T2DM n=5, no T2DM n=6) (C, D). PPF was quantified as the proportion of Mk displaying at least one proplatelet with respect to the total number of adhered Mk (healthy subjects n=5; first tertile: all n=7, T2DM n=4, no T2DM n=3; third tertile: all n=8, T2DM n=4, no T2DM n=4) (E).
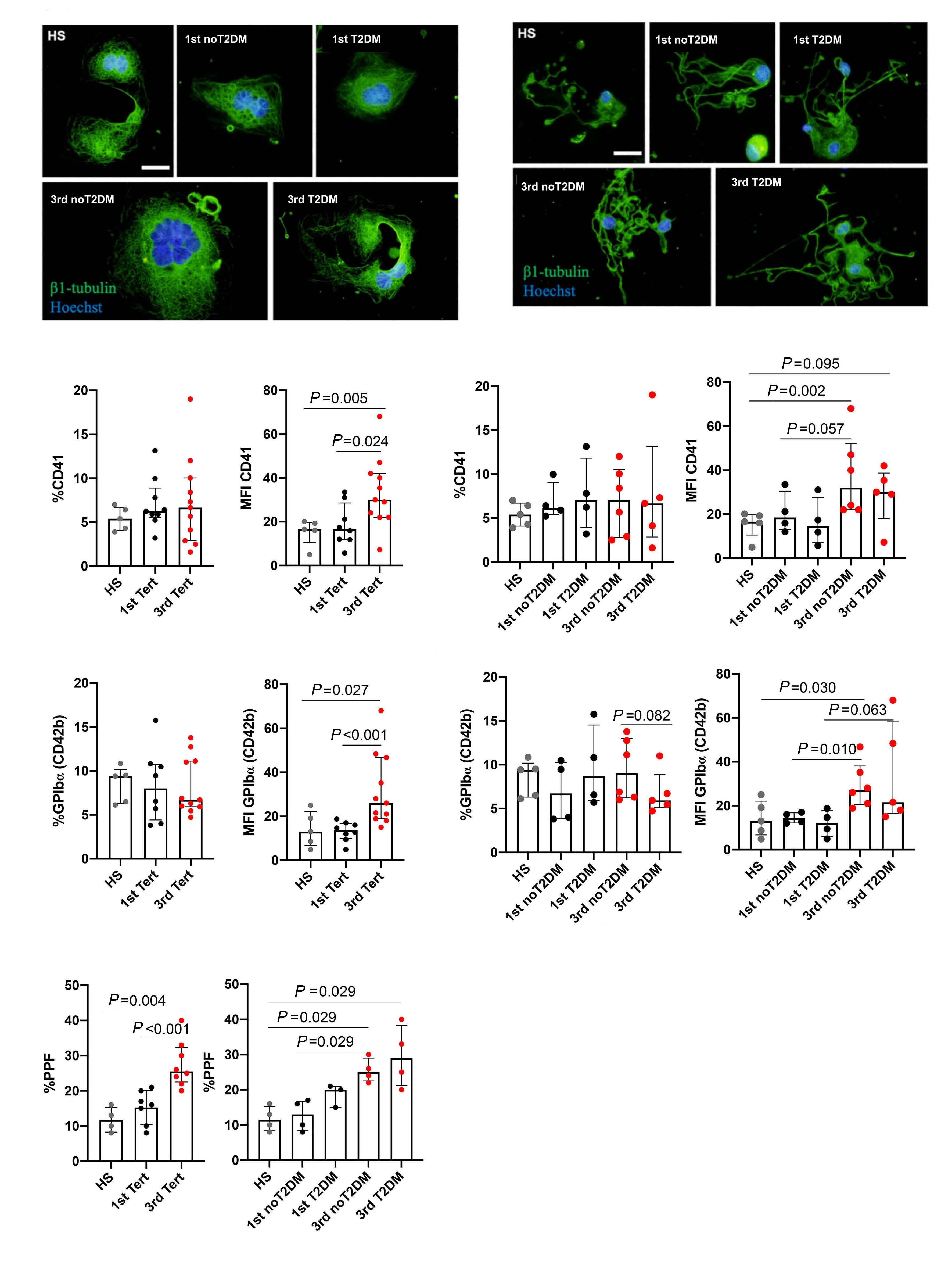
A B C D E Haematologica | 108 April 2023 1148 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
percentage of Annexin V+ platelets was significantly lower in the third versus first tertile in all patients (P=0.007; Figure 5C) and in patients with T2DM (P <0.001; data not shown) with a non-significant trend for patients without T2DM (data not shown).
In healthy volunteers, aspirin administration was associated with an increase of PS exposure (P=0.002; Figure 5D), and activation of ADAM1723 after 10 and 24 hours (Figure 5E). Thus, reduced PS exposure and ADAM17 activation, translating into diminished GPIb α cleavage and GC release, characterizes patients with accelerated recovery of platelet COX-1, and may be related to a lower aspirin effect.
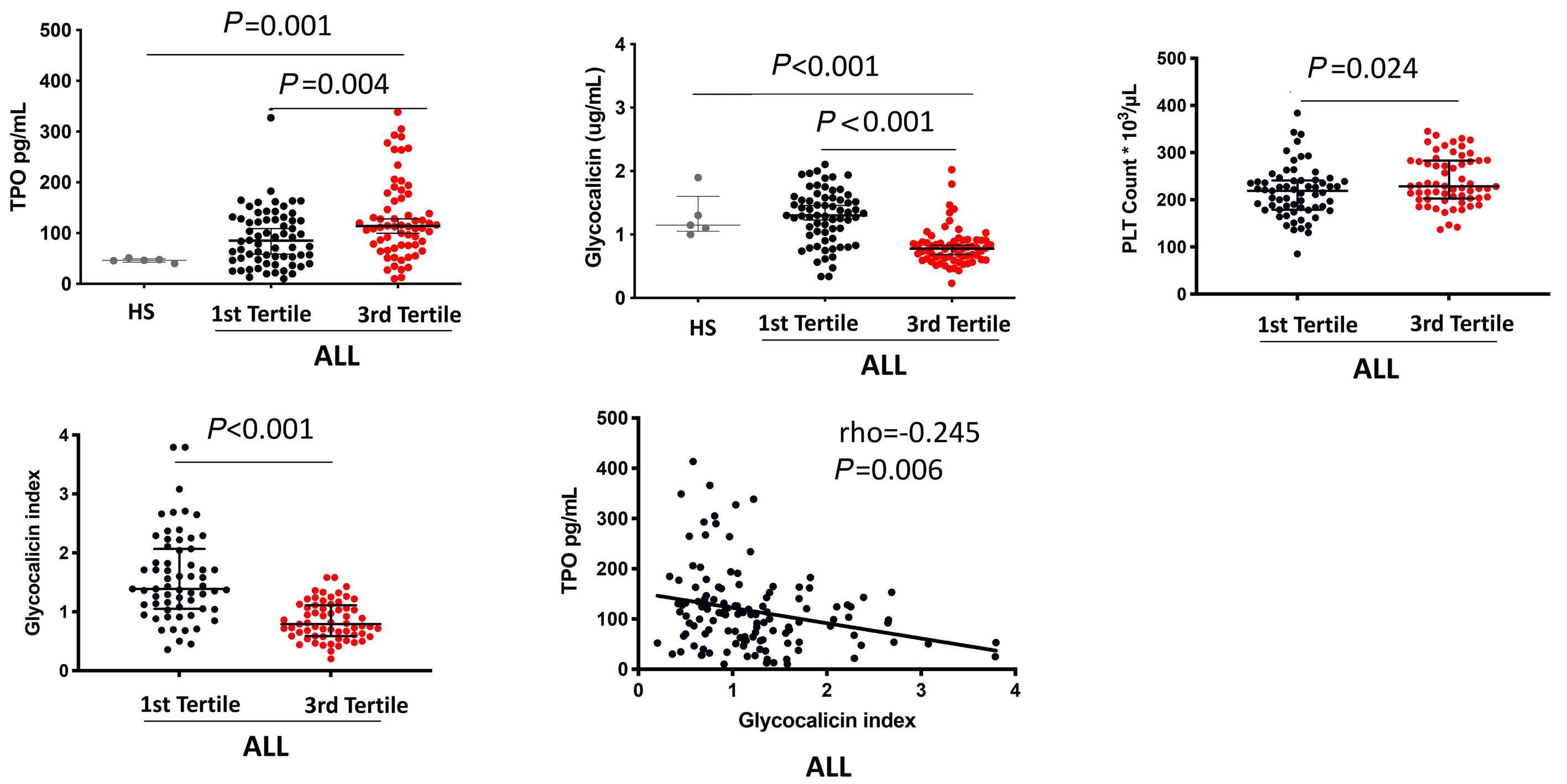
Regulation of thrombopoietin expression
We next hypothesized a role for the GPIbα ectodomain in the regulation of TPO expression, since its uncleaved form on the surface of platelets is a trigger of TPO synthesis in hepatocytes.15 We asked whether the fragment of GC per se was able to modulate TPO expression. Treatment of HepG2 cells with an increasing concentration of human recombinant GC was associated, after 1 hour of incubation, with a significant dose-dependent reduction in TPO mRNA (Figure 5F). Thus, lower GC levels, as observed in the third TXB2 slope tertile, may explain the higher liver expression and circulating levels of TPO.
Platelet galactose exposure in patients with faster COX1 activity recovery
Since TPO production is regulated by the binding of galactose exposed on aged platelets to the AMR in hepatocytes, we analyzed galactose-recognizing lectins, RCA-I and ECL, to assess platelet expression of the terminal b4N-acetyllactosamine (LacNAc).24 PLT of the third tertile were characterized by higher ECL (P=0.034, P=0.064) and RCA-I levels (P=0.002, P=0.004) versus first tertile and healthy subjects (Figure 6A, B), indicating an increase of PLT galactose exposure in aspirin poor responders. More pronounced PLT galactose exposure in the third versus first tertile was accompanied by higher expression of neuraminidase (Neu)1 (P=0.061; Figure 6C), the sialidase that removes sialic acid from GPIbα 25 These data suggest that PLT from patients with accelerated COX-1 recovery are characterized by a higher degree of terminal galactose.
Determinants of the accelerated recovery of COX-1 and predictive value of the thrombopoietin/ glycocalicin ratio Finally, we carried out a multivariable analysis to identify clinical and biochemical determinants of accelerated recovery of COX-1. In T2DM patients, starting from a panel of potential determinants including age, sex, body mass index (BMI), HbA1c, non-alcoholic fatty liver disease
Figure 4. Higher circulating levels of thrombopoietin and platelet count and lower glycocalicin and glycocalicin index at 24 hours after witnessed aspirin intake in patients with faster COX-1 recovery. Comparison of thrombopoietin (TPO) (A), glycocalicin (GC) (B), platelet (PLT) count (C) and glycocalicin index (GCI) (D) between first vs. third serum thromboxane B2 (sTXB2) slope tertile in all patients (n=132). Comparison of TPO (A) and GC (B) between healthy subjects (HS) (n=5) vs. first and vs. third sTXB2 slope tertile in all patients (n=132). Significance was calculated by Mann-Whitney U test. Correlation between GCI and TPO in all investigated patients (E). Spearman correlation coefficient and P value are reported. Significance was calculated by Mann-Whitney U test.
A B C D E Haematologica | 108 April 2023 1149 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
(NAFLD), established atherosclerotic CV disease (ASCVD), glomerular filtration rate, PLT count, mean platelet volume (MPV), PDW and tertiles of TPO/GC ratio, stepwise multivariable logistic regression analysis identified younger age, presence of NAFLD, higher platelet count and higher TPO/GC ratio as independent predictors of the likelihood of being in the third sTXB2 slope tertile (Figure 7A). In patients without T2DM, starting from a panel of potential determinants including age, sex, BMI, waist-to-hip ratio (WHR), PLT count, MPV, PDW, hemoglobin, statin treatment, and tertiles of TPO/GC ratio, stepwise multivariable
Figure 5. Lower glycocalicin circulating levels in platelets from patients with faster COX-1 recovery depend on higher GPIbα expression, lower phosphatidylserine expression and lower ADAM17 activation, and enhance thrombopoietin mRNA transcription in liver cells. GPIbα protein levels in platelets of healthy subjects (HS) (n=4) vs. first (n=4) vs. third (n=4) tertile in all patients (A). ADAM17 levels in platelets of HS (n=3) vs. first (n=4) vs. third serum thromboxane B2 (sTXB2) slope tertile (n=4) in all patients (B). Phosphatidylserine (PS)-positive platelets (%CD41a+/AnV+) in the first (n=34) vs. third tertile (n=20) in all patients (C). PS-positive platelets (%CD41a+/AnV+) (D) and activeADAM17 cleaved form (E) in 4 healthy subjects treated with low-dose aspirin, at 10 and 24 hours post aspirin. Treatment of HepG2 cells (n=4) with an increasing concentrations of human recombinant glycocalicin (rGC, 0.5, 1 and 2 µg/mL) is associated, after 1 hour of incubation, with a significant dose-dependent reduction in thrombopoietin (TPO) mRNA (F). Significance was calculated by Mann-Whitney U test or by Student’s t test.
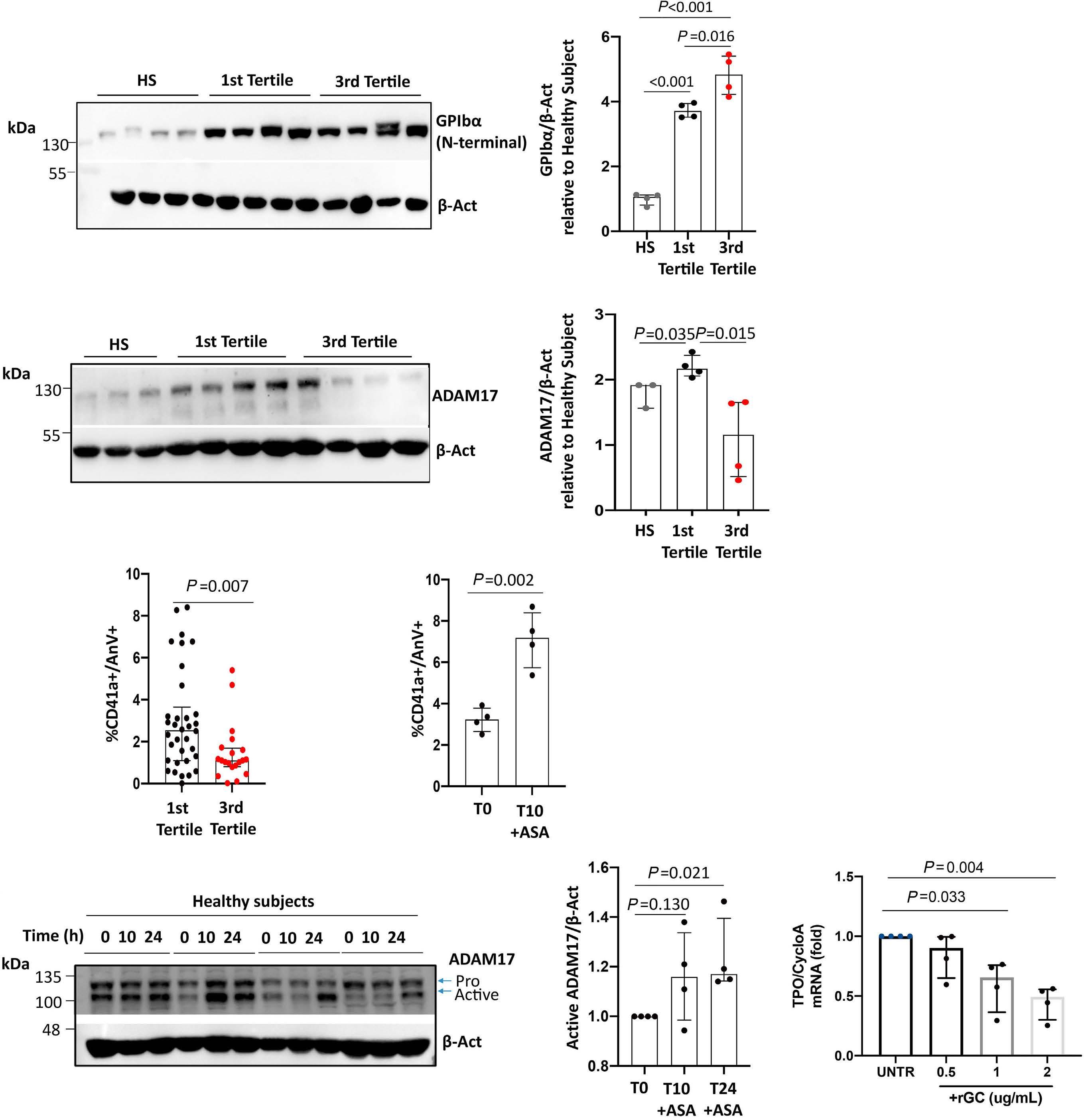
logistic regression analysis identified higher WHR, lower MPV and higher TPO/GC ratio as independent predictors of the likelihood of being in the third sTXB2 slope tertile (Figure 7B). Analysis of the ROC curves revealed an outstanding diagnostic accuracy (AUC ≥0.88) for the two models in the prediction of the third tertile status versus first tertile (Figure 7 C, D).
In order to translate our mechanistic findings of increased PLT production and reduced clearance characterizing accelerated COX-1 recovery into a clinically useful tool, we challenged the predictive value of TPO/GC ratio. Multivari-
A B C D E F Haematologica | 108 April 2023 1150 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
able logistic regression analysis revealed that among subjects with T2DM, those in the third tertile of the TPO/GC ratio (threshold=138 pg/ug) were 33 times more likely (Figure 7A) to be in the third sTXB2 slope tertile, while those with TPO/GC between 60 and 137 were 11 times more likely (Figure 7A) to be in the third tertile. The addition of tertiles of TPO/GC ratio to the model including only clinical or hemocromocytometric variables yielded a significant increase in area under the curve (AUC) (from 0.754 to 0.883; P for difference 0.015). Among patients without T2DM,
those with TPO/GC above 147 were ten times more likely (Figure 7B) to be in the third tertile, while those with TPO/GC between 76 and 146 were nine times more likely (Figure 7B) to be in the third tertile. Thus, a clinical and biochemical signature may unravel patients with shorter duration of sTXA2 inhibition for whom more frequent dosing regimens may prevent the steep recovery of platelet COX-1 activity. See the Online Supplementary Appendix (pages 9-10) for more details on clinical characteristics and study findings.
Figure 6. Higher platelet desialylation rate in patients with faster COX-1 recovery. Expression levels of galactoserecognizing lectins, Erythrina cristagalli agglutinin (ECL) (A), and Ricinus communis agglutinin I (RCA-I) (B), in platelets of healthy subjects (HS) (n=4) vs. first (n=4) vs. third (n=4) tertile in all patients. Expression levels of the sialidase Neu1 in the same subset (C). b -Actin was used as loading control. Significance was calculated by Student’s t test.
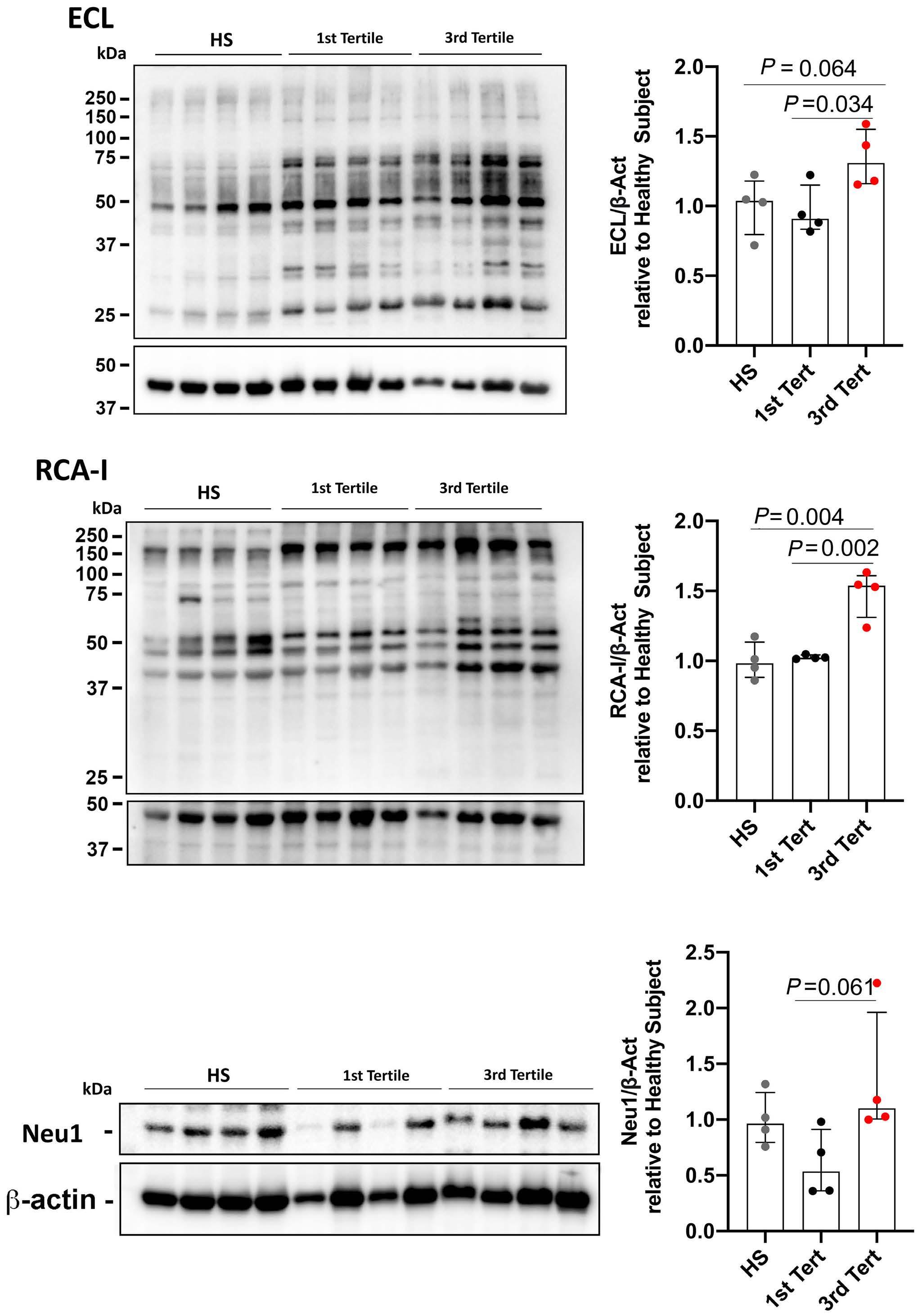
A B C Haematologica | 108 April 2023 1151 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
Discussion
In this work we demonstrate that the shorter duration of TXB2 suppression by aspirin over the 10- to 24-hour dosing interval, in a fraction of high-risk patients on chronic aspirin treatment, may be explained by i) accelerated recovery of COX-1 through increased functionally active COX-1 and COX-1 mRNA, higher TX-dependent PLT activa-
tion, enhanced TPO production, Mk maturation and proplatelet formation, leading to increased PLT number; ii) reduced PLT PS exposure, GPIb α ectodomain shedding and higher galactose exposure, fostering thrombopoiesis through liver TPO synthesis; iii) a clinical and molecular signature, including younger age, NAFLD, visceral obesity and high TPO/GCI, identifying with high accuracy aspirinpoor responders.
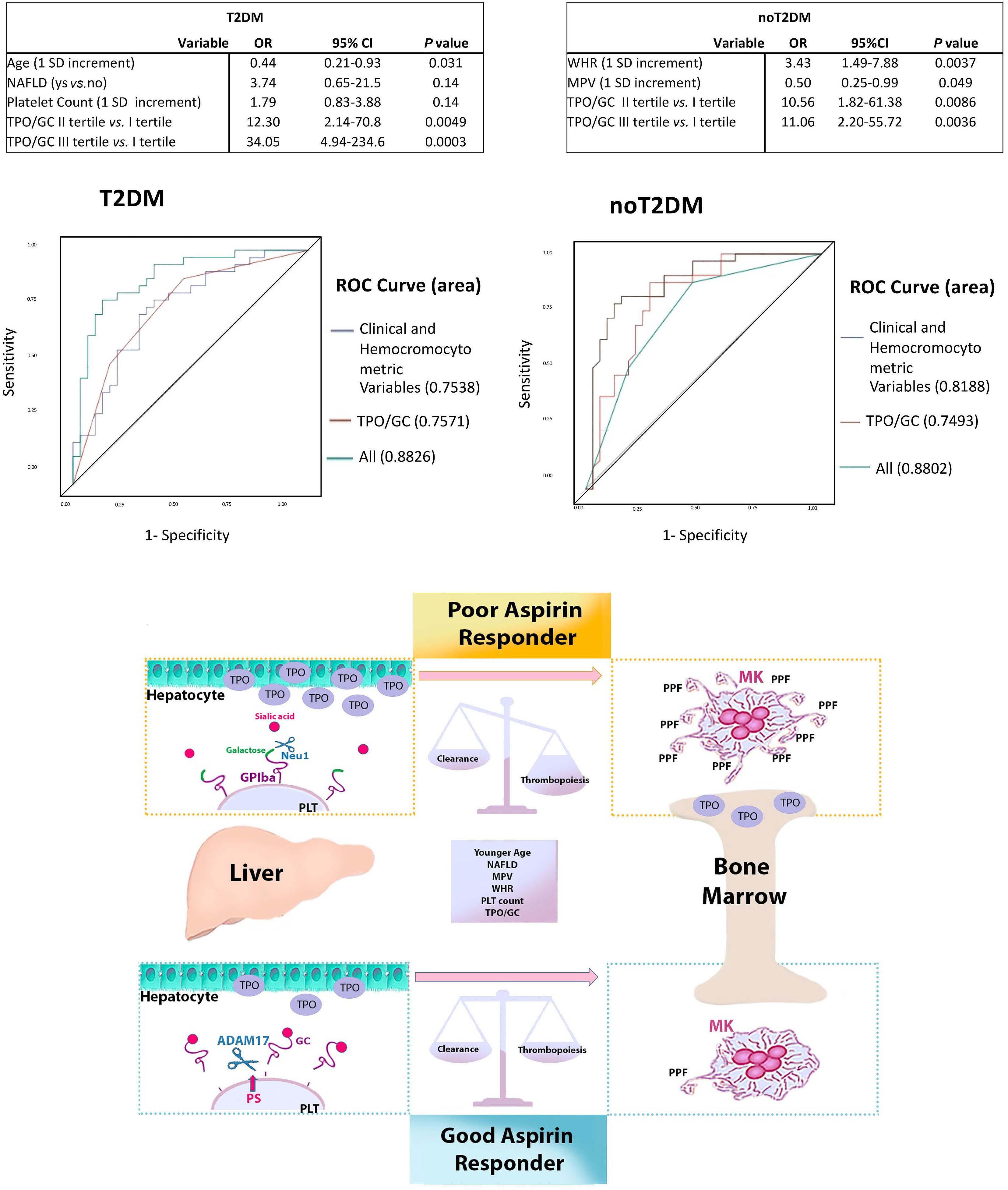
on following page. A B C D E Haematologica | 108 April 2023 1152 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
Continued
Figure 7. Multivariable logistic regression analyses, receiver operating characteristic curve for the prediction of poor aspirin response and proposed model depicting the mechanisms involving platelet lifespan that may limit the extent and duration of aspirin effect over 24 hours. Determinants of the accelerated recovery of COX-1 activity in patients with (A) and without type 2 diabetes mellitus (T2DM) (B) assessed by multivariable logistic regression analysis. Receiver operating characteristic (ROC) curve for the prediction of poor aspirin response (C, D). ROC and the relative area under the curve (AUC) showing the ability of the model in discriminating between the third vs. first serum thromboxane B2 (sTXB2) slope tertile. Among patients with T2DM, the combination of younger age (standard deviation [SD]=6.38 years), presence of non-alcoholic fatty liver disease (NAFLD), higher platelet (PLT) count (SD=55.66 mL) and higher thrombopoietin/glycocalicin (TPO/GC) ratio (1st tertile: <60; 2nd tertile: from 60 to 138; third tertile >138) yielded an AUC value of 0.883 (95% confidence interval [CI]: 0.799-0.966) in distinguishing patients in third sTXB2 slope tertile from first tertile patients (C). In comparison with a model including only clinical/hemocromocytometric variables (age, NAFLD and PLT count) the addition of TPO/GC ratio yielded a significant increase in AUC (from 0.754 to 0.883; P for difference 0.015). Among patients without T2DM, higher waist-to-hip ratio (WHR) (SD=0.066), lower mean platelet volume (SD=0.93 mL) and higher TPO/GC ratio (first tertile: <76; second tertile: from 76 to 147; third tertile >147) yielded an AUC value of 0.880 (95% CI: 0.794-0.966) in distinguishing patients in third sTXB2 slope tertile from first tertile patients (D). Aspirin-treated patients were stratified according to the kinetics of COX-1 recovery over the 10- to 24-hour dosing interval. In poor aspirin responders we showed: i) increased plasma thrombopoietin, megakaryocyte (Mk) maturation and proplatelet formation (PPF) reflecting enhanced PLT production; ii) increased PLT desialylation, lower phosphatidylserine exposure, lower PLT sheddase ADAM17 and plasma glycocalicin and increased glycoprotein (GP)Ibα expression, altogether reflecting defective PLT GPIbα shedding; iii) a proteomic signature characterized by activation of cell survival and inhibition of apoptosis. Younger age, NAFLD and visceral obesity, higher PLT count together with higher thrombopoietin-to-glycocalicin ratio, predict suboptimal aspirin response (E).
Over the last decades, the concept of suboptimal response to aspirin has been affirmed, but the prevalence of this phenomenon is unclear. This is due to the heterogeneity of methods used to quantity the anti-PLT effect of aspirin in these studies, which poorly reflect the biochemical pathway affected by aspirin, i.e., PLT COX-1 activity6 and variably reflect the aspirin-sensitive TX-dependent component of PLT aggregation.1 Even when using sTXB2, a mechanism-based endpoint with the highest specificity and sensitivity to monitor aspirin pharmacodynamics, we and others have previously characterized an interindividual variability in PLT COX-1 recovery during the 12- to 24-hour dosing interval in patients at high CV risk, or undergoing coronary artery by-pass surgery or with essential thrombocythemia.7,9,10 This phenomenon was reverted by shortening the dosing interval, suggesting an accelerated COX-1 renewal within the dosing interval. While previous evidence was largely indirect and based on increased MPV or higher levels of reticulated PLT,10,26 here we demonstrated increased circulating TPO, enhanced in vitro Mk maturation and proplatelet formation in patients with accelerated kinetics of platelet COX-1. Our data substantiate the hypothesis that, during the 24-hour dosing interval, newly generated PLT entering circulation, after aspirin effect waning, contain unacetylated COX-1 and synthesize new TXA2
Accelerated MK maturation and proplatelet formation may be driven, at least in part, by higher concentrations of circulating TPO. We hypothesize that the higher TPO in vivo biases the commitment of hematopoietic progenitors toward the Mk lineages that we observe in vitro cultures.27
We next sought to analyze circulating TPO and GC, markers of PLT production and destruction, respectively. We unraveled that reduced PLT destruction, as reflected by lower circulating GC, lower levels of caspase 328 (Online Supplementary Figure S5) and platelet PS exposure, identify pla-
telets from patients with a shorter response durability to aspirin. Indeed, proteomics profiling indicated activation of cell survival and inhibition of apoptosis pathways in third sTXB2 tertile patients, which may characterize younger platelets.29
The observation that PLT from patients with accelerated recovery of COX-1 are less prone to apoptosis was corroborated by lower caspase 3, lower Annexin V staining, reduced ADAM17 activation, increased expression of uncleaved GPIbα, mirroring reduced destruction30–33 leading to lower circulating GC and GCI and higher PLT count. Even if PS alone is regarded as a common marker for both procoagulant or apoptotic platelets, our results on caspase 3, along with proteomics analysis, corroborate our conclusion that third slope tertile patients have less apoptotic platelets.34
Of interest, PS exposure is required for ADAM17-mediated cleavage of GPIbα20 whose constitutive proteolysis is considered as a signature event of platelet aging. Indeed, treatment with artificial agents mimicking platelet aging induces GC release30,31 and accelerated removal of transfused platelets occurs following GPIbα proteolysis from stored platelets.32,33 Inhibition of GPIbα shedding by kinase inhibitors33 or antibodies32 can mitigate PLT clearance and prolong the lifespan of transfused PLT in mice.
Our present results do not allow to draw final conclusions regarding the lifespan of PLT. Although circulating GC has been consistently regarded as an index of PLT destruction, and several lines of evidence converge to support inhibited apoptosis, no direct demonstration of reduced PLT clearance in the third COX-1 recovery tertile has been obtained in our cohort. The reduced percentage of PS-exposing platelets and reduced apoptosis may alternatively be regarded as a feature of young, newly released PLT, or may be the result of earlier PLT clearance over the 24-hour time interval. However, MPV was not higher in third tertile of either group, despite the higher prevalence of larger, newly formed PLT,
Haematologica | 108 April 2023 1153 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
and lower, rather than higher, MPV was a significant predictor of belonging to the upper sTXB2 slope tertile among nondiabetic patients, raising the hypothesis of a longer PLT lifespan in poor aspirin responders, with coexistence of larger and smaller size PLT. Whether increased PLT survival/reduced apoptosis is a feature of the “poor-responder” PLT or a consequence of poor aspirin response, is not unraveled. Aspirin induces PLT apoptosis35 and shedding of GPIbα and GPV through activation of ADAM17.23 Consistently, in a small number of our healthy volunteers, aspirin treatment was associated with enhanced Annexin V+ PLT and increased expression of active ADAM17. Vice versa, patients with accelerated COX-1 recovery displayed lower Annexin V+ platelets and lower PLT ADAM17 expression, concomitant with higher expression of platelet GPIbα N-terminal domain and lower GC and GCI, versus normal COX-1 recovery patients, suggesting lack of apoptosis induction by aspirin. In order to establish a link between the extent of COX-1 acetylation or inhibition and GPIbα clustering, on the one hand, and PS exposure, on the other hand, we measured complexes of the adapter protein 14-3-3ξ with GPIbα and COX-1 in platelets from first and third tertile patients. It was previously shown that arachidonic acid (AA) accumulation due to COX-1 inactivation in cold-stored PLT induce 14-3-3 ζ -GPIb α association, 14-3-3 ζ release from phospho-Bad, Bad activation, PS exposure, and apoptosis.36 GPIbα clustering is also linked to galactose exposure.37 In our setting, 14-3-3 ξ :GPIb α complexes were significantly less in third tertile patients, while 14-3-3 ξ :COX-1 complexes were significantly higher (Online Supplementary Figure S5). Therefore, it is possible that in first tertile patients, treatment with aspirin, which leads to arachidonic acid accumulation due to inhibition of its biochemical utilization by platelet COX-1, results in displacement of 14-3-3 ξ from the proapoptotic protein Bad in favor of GPIb α and subsequent activation of platelet death. In contrast, in third tertile patients, which have a faster recovery of COX-1 activity, conversion of AA into TXA2 may determine a lower degree of interaction of 14-3-3ξ with GPIbα and a reduced activation of apoptosis. In keeping with this, we found that apoptosis was inactivated in PLT from third tertile patients.
Increased GPIbα and reduced GC characterize and predict poor aspirin response and may play a role in promoting PLT activation and escape from aspirin. While it is assumed that ADAM17 restrains continuous GPIbα-mediated PLT activation,38 the phenotype observed in third tertile patients may indicate hyperreactive PLT since GPIbα clustering triggers TXA2.39 Along this line, poor aspirin responders with high GPIbα have higher platelet COX-1 and persistent TXA2 biosynthesis. The unexpected inverse relationship between PLT destruction and production, as reflected by GC or GCI and
TPO, respectively, prompted us to hypothesize that defective GPIb α ectodomain shedding may contribute to sustain enhanced thrombopoiesis in patients with shorter duration of COX-1 inhibition. Indeed, the extracellular domain of GPIbα per se, independently of platelet clearance, is required for liver TPO production.12 Thus, we challenged the effect of a commercially available recombinant human soluble GC expressed in murine myeloma cells on liver cells in vitro, showing a dose-dependent inhibition in TPO mRNA expression. Together our findings suggest that GP1bα ectodomain shedding by ADAM17 leads to soluble GC binding to hepatocytes, thus reducing liver TPO release. Conversely, low levels of GC shedding in subjects with accelerated COX-1 recovery, may unleash TPO mRNA transcription. We recognize that sugar additions on rGC synthesized in murine myelomas may differ from GC found in circulating human GC, including non-human Nglycolylneuraminic acids, which could affect the binding and recognition of rGC by hepatocytes. Further studies, which will require a careful glycoproteomics approach, are necessary to understand the role of the protein backbone and sugar additions in the binding of GC to hepatocytes. However, glycoproteomics of rGC and human GC is out of scope of this report.
PLT galactose exposure also triggers thrombopoiesis through the interaction with AMR.9 In our study, patients with a shorter duration of the aspirin response showed increased lectin binding and Neu-1 expression, suggesting a possible further mechanism activating TPO production in these subjects. Thus, both PLT galactose exposure and GPIb α expression may contribute, with a feed-forward mechanism, to accelerated thrombopoiesis escaping aspirin inhibition at the usual dosing interval. More terminal galactose moieties would be expected to lead to increased PLT clearance and decreased circulating platelet count. Other evidence shows that PLT isolated from myeloproliferative diseases, often associated with change in circulating platelet count, have a significant increase in terminal galactose expression that correlated with the high allele burden regardless of the underlying identified mutation. Mk derived in vitro from these patients showed an increased expression of the B4GALT1 gene encoding b -1,4- galactosyltransferase 1 ( b 4 GalT1) and terminal galactose expression relative to healthy controls. Altered expression of B4GALT1 in mutant Mk can lead to the production of platelets with aberrant galactosylation, which in turn promote hepatic TPO synthesis regardless of platelet mass.27 These data suggest a more complex role for B4GALT1-dependent galactose decorations to balance platelet clearance and production. A pathologic increase in galactose could result in both increased PLT production and PLT clearance to perpetuate TPO production.
Finally, we identified a cluster of clinical and biochemical
Haematologica | 108 April 2023 1154 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
markers predicting the likelihood of suboptimal aspirin response, with particular reference to the TPO/GC ratio, mirroring our mechanistic findings.
This may help identifying those patients for whom a more frequent aspirin dosing regimen (bis in die) may be required. The twice daily regimen has already been suggested for the management of myeloproliferative neoplasms40 and a phase II trial is ongoing to assess the safety of this approach in this setting. Moreover, the ongoing ANDAMAN trial is testing the efficacy and safety of aspirin twice a day in patients with acute coronary syndrome and diabetes, obesity, or aspirin failure (clinicaltrials gov. Identifier: NCT02520921). However, no clinical setting has been shown to accurately identify those with faster recovery of COX-1 activity. Obesity is known to impair aspirin responsiveness by affecting systemic drug availability, i.e., absorption and biotransformation, leading to reduced, albeit steady, 24-hour inhibition of COX-1–dependent TX production.4 Here we show an additional role of obesity in shortening the effect of aspirin to less than 24 hours. Indeed, in our study, visceral obesity and NAFLD, in patients without and with T2DM, respectively, were independent clinical predictors of shorter duration of aspirin effect, suggesting a role for insulin resistance as the pathophysiological hallmark of both conditions. Indeed, plasma TPO was directly related to waist circumference, hs-CRP, insulinemia, and HOMA-IR.
Diabetes per se is also a recognized setting of platelet hyperreactivity,41 persistent TX-dependent platelet activation42,43 and enhanced PLT turnover, with suboptimal PLT responsiveness.44 Hyperglycemia is a trigger of IL-6 mediated liver TPO production.45 Not surprisingly, the predictive power of the TPO/GC ratio is substantially higher in patients with T2DM versus non-diabetic subjects, regardless of underlying CV risk: indeed, patients in the upper tertile for the TPO/GC ratio, have a 33-fold higher risk to be poor aspirin responders, versus 11-fold higher risk in subjects without T2DM.
However, the diagnostic accuracy of single clinical features, such as obesity, or diabetes, in discriminating subjects with faster COX-1 recovery is poor and does not allow a personalized, disease-based approach. On the other hand, assessment of aspirin response in the individual patient based on the kinetics of COX-1 recovery is complex and requires repeated measurements. At variance, the TPO/GC ratio identified here is calculated with one blood sampling and provides alone good diagnostic accuracy in detecting subjects with faster COX-1 recovery and for whom the efficacy and safety of more frequent anti-PLT dosing regimens should be tested.
Limitations of the study include its observational nature and the lack of reticulated PLT data, although previously shown by our group and others and overcome by a direct evaluation of Mk maturation and proplatelet formation.
Also information about COX-1 acetylation or salicylate measurement is lacking. However, in addition, we performed lectin blots using galactose binding lectins, showing all proteins with terminal galactose, instead of flow cytometry, which would have revealed most of surface-exposed terminal galactose moieties. It is noteworthy that most intracellular proteins are not glycosylated, exception being the Golgi apparatus and OGlcNAcylated proteins, which seem to be relatively low expressed in PLT (data not shown) and PLT contain only few Golgi-like granules.46 Hence, we speculate that most proteins with exposed galactose would reside on the platelet surface. The healthy subject group is very small, although differences in results pre versus post aspirin administration are quite evident. Strengths are accurate clinical and biochemical characterization and CV risk stratification; ascertainment of compliance to low-dose aspirin; accurate timing of blood sampling; use of a mechanism-based biochemical endpoint to monitor aspirin pharmacodynamics and renewal of the drug target; a combined approach including biochemistry, proteomics, flow cytometry, cell biology.
In conclusion, an imbalance between platelet production and clearance, with accelerated megakaryopoiesis/ PLT production and reduced clearance/prolonged survival, characterizes patients with poor aspirin response, as reflected by the accelerated recovery of platelet COX-1 activity, with or without diabetes (Figure 7E). This imbalance translated into increased PLT count (especially in patients with T2DM) and enhanced TX-dependent PLT activation. Integration of clinical data with TPO to GC ratio may provide a relatively simple tool to identify patients amenable to more frequent aspirin daily dosing, and should be tested in larger, independent cohorts.
Disclosures
FS has received research grant support and consulting fees from Bayer, all unrelated to this manuscript. The remaining authors have no conflicts of interest to disclose.
Contributions
FS conceived and designed the research studies and obtained funds for the project; PS enrolled patients and acquired data; RL, RT, SC, AR, VA, BP, MLS, ML, RA, GB, PDB, DP, and MCC conducted experiments; AdC, RL, RT, SC and PS analyzed data; FS, PS, RL, AR and AB wrote the manuscript with input from the other authors; FS, AB, PL, MC, DP, ET, KMH, PDB and FC revised and provided critical interpretation for important intellectual content. All authors approved the final version.
Acknowledgments
The authors thank Prof Carlo Patrono for his invaluable suggestions and for critical reading of the manuscript. We
Haematologica | 108 April 2023 1155 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
also thank Dr Laura Creati, Silvio Basile, Mariapia Blasetti, Luciano Giacci, Diego Ferrara, Rosalba Silvestri, Moreno D’Emilio, who provided assistance with patients, and Dr Valeria Creato, Damiano D’Ardes , Andrea Boccatonda, Raffaele Pepe for help in the patients’ recruitment and Dr Pasquale Simeone who contributed to perform flow-cytometry analysis.
References
1. Davì G, Patrono C. Mechanisms of disease: platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482-2494.
2. Davì G, Catalano I, Averna M, et al. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N Engl J Med. 1990;322(25):1769-1774.
3. Santilli F, Simeone P, Liani R, Davì G. Platelets and diabetes mellitus. Prostaglandins Other Lipid Mediat. 2015;120:28-39.
4. Patrono C, Rodríguez LAG, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353(22):2373-2383.
5. Roth GJ, Stanford N, Majerus PW. Acetylation of prostaglandin synthase by aspirin. Proc Natl Acad Sci. 1975;72(8):3073-3076.
6. Santilli F, Rocca B, Cristofaro RD, et al. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays. Implications for aspirin “resistance.” J Am Coll Cardiol. 2009;53(8):667-677.
7. Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. 2012;119(15):3595-3603.
8. Santilli F, Romano M, Recchiuti A, et al. Circulating endothelial progenitor cells and residual in vivo thromboxane biosynthesis in low-dose aspirin-treated polycythemia vera patients. Blood. 2008;112(4):1085-1090.
9. Cavalca V, Rocca B, Veglia F, et al. On-pump cardiac surgery enhances platelet renewal and impairs aspirin pharmacodynamics: effects of improved dosing regimens. Clin Pharmacol Ther. 2017;102(5):849-858.
10. Rocca B, Santilli F, Pitocco D, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost. 2012;10(7):1220-1230.
11. Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Novel mechanisms of platelet clearance and thrombopoietin regulation. Curr Opin Hematol. 2015;22(5):445-451.
12. Xu M, Li J, Neves MAD, et al. GPIba is required for plateletmediated hepatic thrombopoietin generation. Blood. 2018;132(6):622-634.
13. Kile BT. Aging platelets stimulate TPO production. Nat Med. 2015;21(1):11-12.
14. Barsam SJ, Psaila B, Forestier M, et al. Platelet production and platelet destruction: assessing mechanisms of treatment effect in immune thrombocytopenia. Blood. 2011;117(21):5723-5732.
15. Karakas D, Xu M, Ni H. GPIbα is the driving force of hepatic thrombopoietin generation. Res Pract Thromb Haemost. 2021;5(4):e12506.
16. Cosentino F, Grant PJ, Aboyans V, et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J. 2020;41(2):255-323.
17. Patrono C, Rocca B. Measurement of thromboxane biosynthesis in health and disease. Front Pharmacol. 2019;10:1244.
Funding
The research was supported by grants from the Italian Ministry of Health (COD WF GR 2011-02350450 to FS) and PRIN 2017Z5LR5Z to AB.
Data-sharing statement
For original data, please contact the corresponding author.
18. Tiwari S, Italiano JE, Barral DC, et al. A role for Rab27b in NFE2-dependent pathways of platelet formation. Blood. 2003;102(12):3970-3979.
19. Liu ZJ, Italiano J, Ferrer-Marin F, et al. Developmental differences in megakaryocytopoiesis are associated with upregulated TPO signaling through mTOR and elevated GATA-1 levels in neonatal megakaryocytes. Blood. 2011;117(15):4106-4117.
20. Sommer A, Kordowski F, Büch J, et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat Commun. 2016;7(7):11523.
21. Schlöndorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem J. 2000;347(1):131-138.
22. Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Ann Rev Biophys. 2010;39:407-427.
23. Aktas B, Pozgajova M, Bergmeier W, et al. Aspirin induces platelet shedding via ADAM17 (TACE). J Biol Chem. 2005;280(48):39716-39722.
24. Grozovsky R, Begonja AJ, Liu K, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2STAT3 signaling. Nat Med. 2015;21(1):47-54.
25. Jansen AJG, Josefsson EC, Rumjantseva V, et al. Desialylation accelerates platelet clearance after refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice. Blood. 2012;119(5):1263-1273.
26. Spectre G, Arnetz L, Östenson CG, Brismar K, Li N, Hjemdahl P. Twice daily dosing of aspirin improves platelet inhibition in whole blood in patients with type 2 diabetes mellitus and micro-or macrovascular complications. Thromb Haemost. 2011;106(3):491-499.
27. Di Buduo CA, Giannini S, Abbonante V, Rosti V, Hoffmeister KM, Balduini A. Increased B4GALT1 expression is associated with platelet surface galactosylation and thrombopoietin plasma levels in MPNs. Blood. 2021;137(15):2085-2089.
28. Dasgupta SK, Argaiz ER, Mercado JEC, et al. Platelet senescence and phosphatidylserine exposure. Transfusion. 2010;50(10):2167-2175.
29. Allan HE, Hayman MA, Marcone S, et al. Proteome and functional decline as platelets age in the circulation. J Thromb Haemost. 2021;19(12):3095-3112.
30. Schoenwaelder SM, Jarman KE, Gardiner EE, et al. Bcl-xLinhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118(6):1663-1674.
31. Bergmeier W, Burger PC, Piffath CL, et al. Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro-aged or -injured mouse platelets. Blood. 2003;102(12):4229-4235.
32. Chen W, Liang X, Syed AK, et al. Inhibiting GPIbα shedding preserves post-Transfusion recovery and hemostatic function of
Haematologica | 108 April 2023 1156 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
platelets after prolonged storage. Arterioscler Thromb Vasc Biol. 2016;36(9):1821-1828.
33. Canault M, Duerschmied D, Brill A, et al. p38 mitogen-activated protein kinase activation during platelet storage: consequences for platelet recovery and hemostatic function in vivo. Blood. 2010;115(9):1835-1842.
34. Lebois M, Josefsson EC. Regulation of platelet lifespan by apoptosis. Platelets. 2016;27(6):497-504.
35. Zhao L, Zhang W, Chen M, Zhang J, Zhang M, Dai K. Aspirin Induces platelet apoptosis. Platelets. 2013;24(8):637-642.
36. van der Wal DE, Gitz E, Du VX, et al. Arachidonic acid depletion extends survival of cold-stored platelets by interfering with the glycoprotein Ib - 14-3-3 association. Haematologica. 2012;97(10):1514-1522.
37. Rumjantseva V, Grewal PK, Wandall HH, et al. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat Med. 2009;15(11):1273-1280.
38. Berndt MC, Karunakaran D, Gardiner EE, Andrews RK. Programmed autologous cleavage of platelet receptors. J Thromb Haemost. 2007;5(SUPPL. 1):212-219.
39. Van Der Wal DE, Verhoef S, Schutgens REG, Peters M, Wu Y, Akkerman JWN. Role of glycoprotein Ibα mobility in platelet function. Thromb Haemost. 2010;103(5):1033-1043.
40. Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk‐stratification and management. Am J Hematol. 2020;95(12):1599-1613.
41. Rodriguez BAT, Johnson AD. Platelet measurements and type 2 diabetes: investigations in two population-based cohorts. Front Cardiovasc Med. 2020;7:118.
42. Santilli F, Davì G, Consoli A, et al. Thromboxane-dependent CD40 ligand release in type 2 diabetes mellitus. J Am Coll Cardiol. 2006;47(2):391-397.
43. Santilli F, Zaccardi F, Liani R, et al. In vivo thromboxanedependent platelet activation is persistently enhanced in subjects with impaired glucose tolerance. Diab Metab Res Rev. 2020;36(2):e3232.
44. Santilli F, Simeone P, Liani R. The role of platelets in diabetes mellitus. In: Michelson A, Cattaneo M, Frelinger A, Newman P, editors. Platelets. Elsevier. 2019:469-503.
45. Kraakman MJ, Lee MKS, Al-Sharea A, et al. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J Clin Invest. 2017;127(6):2133-2147.
46. Wandall HH, Rumjantseva V, Sørensen ALT, et al. The origin and function of platelet glycosyltransferases. Blood. 2012;120(3):626-635.
Haematologica | 108 April 2023 1157 ARTICLE - Platelet lifespan imbalance and aspirin response P. Simeone et al.
Morbidity and mortality of sickle cell disease patients is unaffected by splenectomy: evidence from three decades of follow-up in a high-income setting
Sickle cell disease (SCD) is a globally widespread hereditary red cell disorder characterized by the production of pathological hemoglobin S (HbS).1 Patients with SCD include homozygous subjects for HbS (SS) and compound heterozygotes with HbS/HbC (SC) or HbS/b+/0-thalassemia (Sb0/b+). In Italy, SCD is endemic with HbS/b+/0-thalassemia being prevalent in areas of southern Italy. In the last two decades, the number of SCD patients across Italy has increased due to migration from sub-Saharan Africa and the Middle East.2,3 Italian expert centers for hemoglobinopathies, in which the vast majority of patients are managed, have registered about 2,300 patients with SCD, distributed over the whole territory with the highest prevalence in Sicily (10 patients/100,000 inhabitants) and in regions of the north of Italy (~5 patients/100,000 inhabitants) (Online Supplementary Figure S1A). In line with European Hematology Association guidelines, the main indications for splenectomy in SCD in Italy are splenic sequestration and hypersplenism.4 Although studies on short-term post-splenectomy follow-up (e.g., 2-10 years) are available, results of long-term follow-on mortality are lacking.
Here we report on 11,195 patient-years of follow-up using a large cohort of SCD patients. We designed a retrospective observational cohort study, which was supported by the Italian Society of Thalassemia and Hemoglobinopathies (SITE; www.site-italia.org). We identified six reference centers of the Italian Hemoglobinopathy Comprehensive Care Network (Online Supplementary Figure S1A) with SCD patients followed from the 1990s with continuous follow-up data covering 30 years. The aim of the study was to compare survival, causes of death and complications in splenectomized versus not-splenectomized SCD patients. Data were collected between 20162018, curated and analyzed since then. Centers involved in the present study are geographically located in areas of high SCD prevalence and collectively follow up more than a third (n=801) (Online Supplementary Figure S1B) of registered SCD patients in Italy. Inclusion criteria were continuous long-term follow-up considered from the creation of the centers if the year of birth was before 1990 or from the first contact with the center before the age of 10 years.
For each patient, we collected data on gender, age at last follow-up, year of last follow-up, age of first access to the center, ethnicity, genotype (Sb+, Sb0 and SS, confirmed by molecular analysis), splenectomy, age and year of sple-
nectomy, type of common therapy (chronic transfusion regimen, hydroxyurea or iron chelation treatment), age at first therapy, death, age and year of death, and cause of death. The definition of ethnicity was based on self-reported ancestry. No data on the method of splenectomy were available. The study was approved by the Ethics Committee of the Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy.
After exclusions, data from 534 patients (272 males, 51%) with genotypes Sb+ (n=171, 32%), Sb0 (n=176, 33%) and SS (n=187, 35%) were analyzed (Table 1, Online Supplementary Figure S1B). Gender was balanced overall and within the three genotypes considered. Altogether, in the period 1990-2018, 50 patients (10%) died, and 17 patients (3%) underwent their last visit before the start of survey in 2016 (lost to follow-up). The median follow-up was 26 years (interquartile range, 25th - 75th percentile, [IQR], 15-27 years; minimum – maximum, 1-28 years). Patients with the SS genotype - predominantly migrants from African countries and the Middle East - were younger than subjects with other genotypes (Table 1). A subset of 170 patients (32%), equally spread between males and females, underwent splenectomy. The age of splenectomy was similar between genders. The indications for splenectomy were acute splenic sequestration in 30/170 (17.6%), hypersplenism/recurrent splenic sequestration in 117/170 (68.8%), and unknown/other in 23/170 (13.5%). We found that SCD patients with the SS genotype were splenectomized earlier (7 years; IQR, 5-10 years; P<0.001) than those with either the S b 0 (11 years; IQR, 7.5-18.5 years; P=0.0024) or Sb+ genotype (20 years; IQR, 11-27 years). This is in line with previous reports in other cohorts of SS or Sb0 patients, for whom splenic sequestration is the main indication for splenectomy.5-7 For the Sb+ genotype, the indication for splenectomy is hypersplenism more than splenic sequestration, which is consistent with the older ages observed. The probability of being splenectomized was greater in patients with Sb0 and Sb+ than in SS patients (P<0.001). Pairwise comparisons of proportions with the Bonferroni correction showed that the percentage of patients who underwent splenectomy was greater in the group with the Sb0 genotype (53%) than in the Sb+ group (34%; P=0.0012) and the SS group (9.6%; P<0.001) (Sb0 > Sb+ > SS) (Table 1). In our cohort, the rate of splenectomy in SS patients was close to that reported in other studies with a similar SS population (Online Supplementary Figure S2).8-14 It is noteworthy that the rate of splenectomy in our
Haematologica | 108 - April 2023 1158 LETTER TO THE EDITOR
Sb patients was higher than that described by Belhani et al. and Diagne et al., who carried out their studies on small Sb patient populations in African countries.9,12,14
The long-term follow-up of our cohort of patients allowed us to analyze whether changes in the management of SCD (e.g. hydroxyurea or chronic transfusion regimen) affected the indication for splenectomy in SCD patients over time. To achieve this, we considered four different cohorts based on quartiles of the year of birth of patients (before 1966, 1967-1979, 1980-2000 and after 2001), each one including about 130 patients. The analysis (Figure 1A) suggested that indications for splenectomy did not change over time, being similar in different birth cohorts. Using the Kaplan-Meier method, the 10-year survival probabilities were estimated to be 87% (95% confidence interval [95% CI]: 81-93%), 86% (95% CI: 81-92), 83% (95% CI: 7790%) and 88% (95% CI: 80-96%), respectively, for each of the four periods (P=0.71). This was confirmed when we analyzed the age-adjusted incidence rate of splenectomy over time considering different birth cohorts (Figure 1B). We then analyzed the survival rate and the causes of death within our SCD cohort. No statistically significant differences were observed in the survival or age of death between splenectomized and non-splenectomized patients with SCD (P=0.7 and P=0.9, respectively) (Figure 1C). The survival curves were similar for the three genotypes (P=0.29) with an overall median survival time of 72 years
(Sb0: 73 years; Sb+: 68 years; SS: 68 years) (Figure 2A). As expected, the survival rate was significantly reduced in children splenectomized before 5 years of age, whereas no major differences were observed for the other age groups (Figure 2B). When we considered the impact of different treatments (chronic transfusion regimen, hydroxyurea or iron chelation treatment) versus no therapy on the mortality of patients with SCD, the mortality rate was worse in treated patients than in untreated ones (Online Supplementary Figure S3A). This might be related to the milder phenotype of untreated SCD patients compared to treated SCD subjects. Indeed, the percentage of sickle cell-related events was higher in treated SCD patients than in untreated ones (Online Supplementary Figure S3B). We registered 50 deaths, which occurred at a median age of 49.5 years (IQR, 39.1-57.5 years; minimum - maximum, 31-73 years), which was similar among genotypes (P=0.9). The four main causes of deaths were acute chest syndrome (n=15), liver failure (n=12), stroke (n=7) and solid cancer (n=7: 3 liver, 2 lung, 1 breast, 1 colon) (Figure 1D).
Among the deaths due to liver failure, ten were related to chronic hepatitis C virus infection. In addition, two out of three patients with hepatocellular carcinoma had chronic hepatitis C virus infection. Splenectomy was reported in 20 out of 50 (40%) of the patients who died. Moreover, considering the subgroup of patients whose death was due to either acute chest syndrome, stroke or pulmonary
Table 1. Characteristics of the studied cohort of patients with sickle cell disease.
Total S b+ S b° SS P-value Demographics N of patients (N of males) 534 (272) 171 (93) 176 (89) 187 (90) 0.7 Age in years at last follow-up, median (IQR) 38 (17-49) 39 (27-52) 42 (32-53) 18 (7-43) <0.001 N of patients in age groups Age ≤19 years 151 32 19 100 <0.001 Age 20-39 years 138 55 50 33 0.005 Age 40-59 years 206 67 89 50 <0.001 Age ≥60 years 39 17 18 4 0.003 Follow-up in years, median (IQR) 26 (15-27) 26 (22-28) 26 (26-27) 17 (7-26) <0.001 Splenectomy, N/total N in group 170/534 58/171 94/176 18/187 <0.001 Age at splenectomy in years, median (IQR) 13 (7-22) 20 (11-27) 11 (7.5-18.5) 7 (5-10) <0.001 Ethnicity Caucasian, N 432 168 174 90 <0.001 African, N 99 3 2 94 <0.001 African-American, N 3 - - 3Deaths N of deaths 50 17 19 14 0.6 Splenectomy, N/N of deaths 20/50 6/17 11/19 2/14 0.04 Age at death in years, median (IQR) 49.5 (39-58) 49 (38-59) 48 (39-58) 50 (46-54) 0.9 Haematologica | 108 - April 2023 1159 LETTER TO THE EDITOR
IQR: interquartile range (25th - 75th percentile); ACS: acute chest syndrome.
Figure 1. Splenectomy does not affect survival rate and the incidence of fatal infectious events in patients with sickle cell disease. (A) Probability of splenectomy in patients with sickle cell disease (SCD) analyzed for different birth cohorts (~130 patients/cohort). (B) Age-adjusted incidence rates of splenectomy in 5-year periods (mean value, 95% confidence interval). The age distribution of the SCD population in the period 2014-2018 was used to adjust rates. (C) Survival probability of patients with SCD according to splenectomy. (D) Causes of death in splenectomized (YES) or non-splenectomized (NO) patients with SCD. Pts-yrs: patient-years; ACS: acute chest syndrome.
hypertension, we did not observe a predominance of splenectomized patients versus non-splenectomized individuals (10 out 25, P=0.6). When we considered genotypes and causes of death in splenectomized SCD patients, we found that S b 0 patients had an increased risk of acute chest syndrome, liver failure and solid cancer compared to splenectomized SCD patients with either the SS or Sb+ genotype (Online Supplementary Figure S3C). Concerning the risk of death from sepsis, we did not find any difference between splenectomized and non-splenectomized patients with SCD. Our cohort of SCD patients received anti-pneumococcal, anti-meningococcal, anti-Haemophilus influenzae and anti-influenza virus vaccines. Based on our records, antibiotic prophylaxis was generally discontinued either after the age of 14 years or at 1 year after splenectomy, associated with education for the patients and caregivers. Our findings are concordant with results from four different studies which analyzed smaller SCD populations and for a shorter period of time compared to our study.6,7,15,16 Similar results were also reported for two
different studies from low-income countries with a follow-up of 18 months and 3 years after splenectomy.10,14,17 In our cohort, the absence of a significant difference in fatal infectious events between splenectomized and nonsplenectomized patients with SCD might be related to a combination of vaccination, patient education and the intensive follow-up program conducted in comprehensive care centers for hemoglobinopathies by expert medical staff. Although 3.4% of analyzed patients were born before 1980 and splenectomized before the age of 5 years, our results on fatal infectious events in splenectomized versus non-splenectomized patients were unaffected by including or excluding this population from our analysis. This was expected given that sickle cell patients are characterized by asplenia, which might expose them to an increased risk of infection compared to that of the healthy population. Overall, our data support the observation that patient education, vaccination programs and early identification and treatment of severe infections by expert medical staff help to prevent mortality due to sepsis.18 The
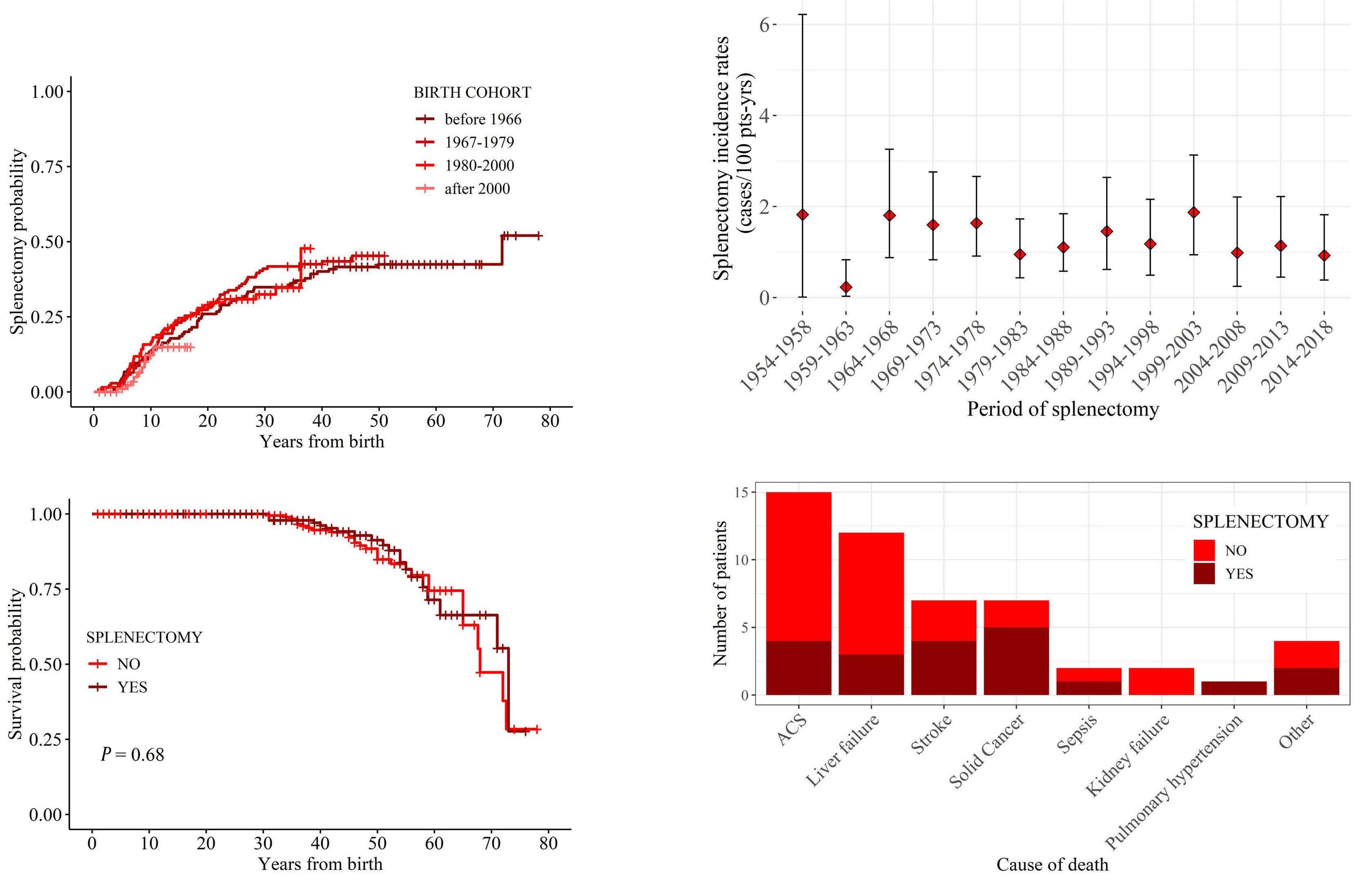
B Haematologica | 108 - April 2023 1160 LETTER TO THE EDITOR A D C
present study has some limitations due to its retrospective design (e.g., lack of details on surgical approaches and acute post-splenectomy complications) and a possible selection bias, our cohort being composed of well-treated patients followed from birth in comprehensive care centers for hemoglobinopathies. In conclusion, this 26-year long-term follow-up cohort study of SCD patients highlights that Sb patients require surgical splenectomy more frequently than SS patients, who in turn may undergo auto-splenectomy. The study provides crucial new evidence of the absence of negative impacts of splenectomy on fatal outcomes, supporting splenectomy as a recommended therapeutic approach in the treatment of patients with SCD.
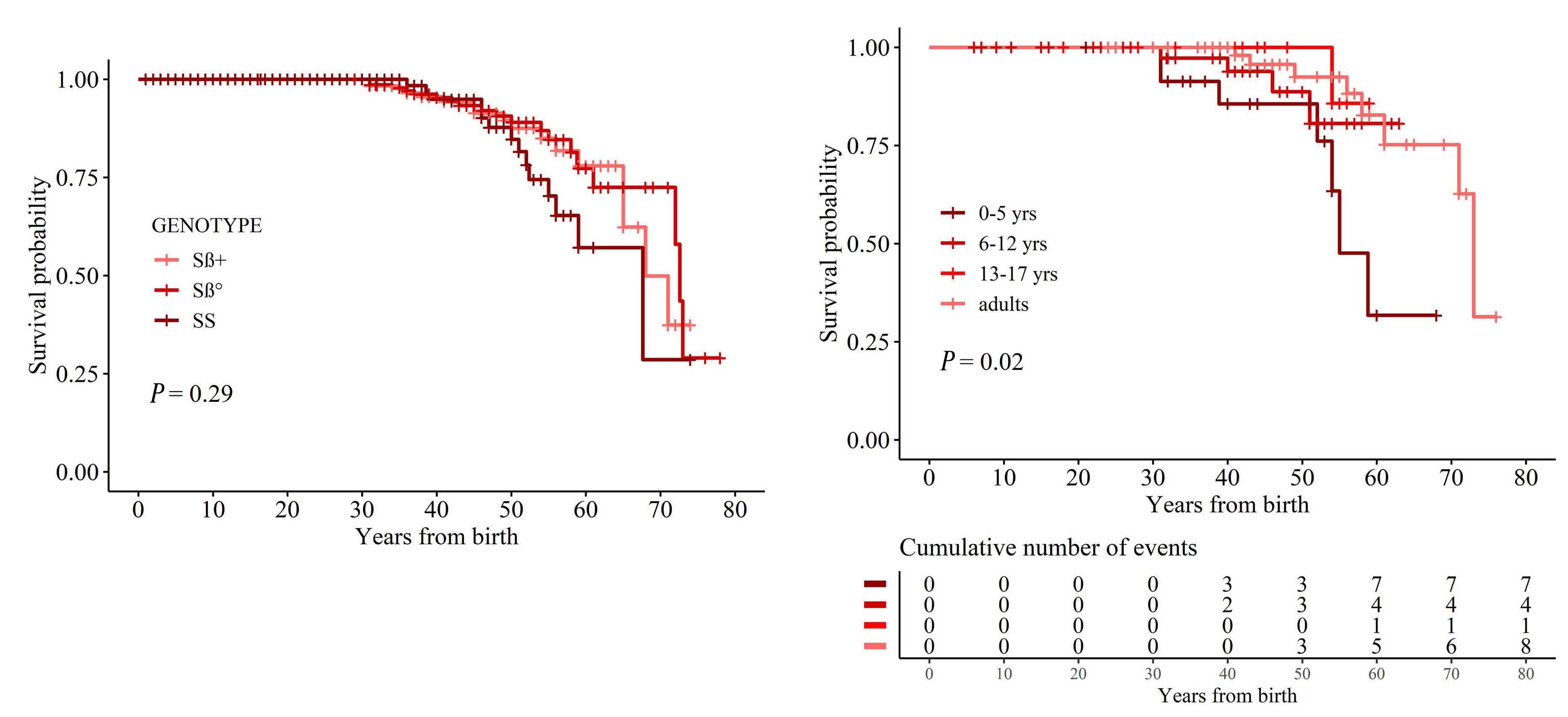
Authors
Valeria Maria Pinto,1* Barbara Gianesin,2* Frédéric B. Piel,3 Filomena Longo,4 Paolo Rigano,5 Alessandra Quota,6 Vincenzo Spadola,7 Giovanna Graziadei,8 Filippo Mazzi,9 Maria Domenica Cappellini,8 Aurelio Maggio,5 Antonio Piga,10 Lucia De Franceschi9# and Gian Luca Forni1#
1Center for Microcythemia, Congenital Anemia and Iron Dysmetabolism, Galliera Hospital, Genoa, Italy; 2ForAnemia Foundation, Genoa, Italy; 3Department of Epidemiology and Biostatistics, School of Public Health, Imperial College London, London, UK; 4Reference Center for Hemoglobinopathies, AOU San Luigi Gonzaga Hospital, Orbassano, Italy; 5Campus of Hematology
Franco and Piera Cutino, AOOR Villa Sofia-V. Cervello, Palermo, Italy; 6Thalassemia Unit, P.O. Vittorio Emanuele III, Gela, Caltanissetta, Italy; 7Thalassemia Center, P.O. Giovanni Paolo II, Ragusa, Italy; 8Department of Medicine and Medical Specialities, IRCCS Ca' Granda Foundation, Maggiore Policlinico Hospital, Milan, Italy; 9Department of Medicine, University of Verona & AOUI Verona, Policlinico GB Rossi, Verona, Italy and 10Department of Clinical and Biological Sciences, University of Turin, Turin, Italy.
*VMP and BG contributed equally as co-first authors. #LDF and GLF contributed equally as co-senior authors.
Correspondence:
GIAN LUCA FORNI - gianluca.forni@galliera.it
https://doi.org/10.3324/haematol.2022.280815
Received: February 10, 2022.
Accepted: June 15, 2022.
Early view: August 4, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
VMP, LDF and GLF contributed to the conceptualization and design of the study; acquisition, curation, analysis and interpretation of the data; and writing, critically appraising, commenting on, reviewing and
B Haematologica | 108 - April 2023 1161 LETTER TO THE EDITOR A
Figure 2. Probability of survival of patients with sickle cell disease. (A) Survival probability of patients according to genotypes. (B) Survival probability of patients with sickle cell disease who underwent splenectomy by age of splenectomy.
editing the manuscript. FP and BG contributed to data analysis and interpretation; and writing, critically appraising, commenting on, reviewing and editing the manuscript. PR, AQ, CF, GG, FM, MDC, AU and AP contributed to the acquisition and curation of the data, and critical appraisal of and comments on the manuscript. All authors have read and agreed to the published version of the manuscript.
References
1. Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311-323.
2. De Franceschi L, Lux C, Piel FB, et al. Access to emergency department for acute events and identification of sickle cell disease in refugees. Blood. 2019;133(19):2100-2103.
3. Rigano P, De Franceschi L, Sainati L, et al. Real-life experience with hydroxyurea in sickle cell disease: a multicenter study in a cohort of patients with heterogeneous descent. Blood Cells Mol Dis. 2018;69:82-89.
4. Iolascon A, Andolfo I, Barcellini W, et al. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica. 2017;102(8):1304-1313.
5. George A, Conneely SE, Mangum R, Lupo PJ, Scheurer ME. Splenic complications in sickle cell disease: a retrospective cohort review. Blood. 2021;138(Suppl 1):766.
6. Sakran W, Levin C, Kenes Y, Colodner R, Koren A. Clinical spectrum of serious bacterial infections among splenectomized patients with hemoglobinopathies in Israel: a 37-year follow-up study. Infection. 2012;40(1):35-39.
7. Machado NO, Grant CS, Alkindi S, et al. Splenectomy for haematological disorders: a single center study in 150 patients from Oman. Int J Surg. 2009;7(5):476-481.
8. Durosinmi MA, Salawu L, Ova YA, Lawal OO, Fadiran OA. Haematological parameters in sickle cell anaemia patients with and without splenomegaly. Niger Postgrad Med J. 2005;12(4):271-274.
9. Diagne I, Diagne-Guèye NR, Fall AL, et al. Aspects épidémiologiques et évolutifs de la splénomégalie chez les enfants et adolescents porteurs de syndromes drépanocytaires majeurs au Sénégal. Arch Pediatr.
Funding
This study was partially supported by the ForAnemia Foundation.
Data-sharing statement
Data are available on request to the authors.
2010;17(7):1017-1025.
10. Okoro BA, Kaine WN, Okeahialam TC. Splenectomy in Nigerian children with sickle cell anaemia. Trop Geogr Med. 1989;41(2):123-127.
11. Gale HI, Bobbitt CA, Setty BN, et al. Expected sonographic appearance of the spleen in children and young adults with sickle cell disease. J Ultrasound Med. 2016;35(8):1735-1745.
12. Belhani M, Morle L, Godet J, et al. Sickle cell b-thalassaemia compared with sickle cell anaemia in Algeria. Scand J Haematol. 2009;32(4):346-350.
13. Tolo-Diebkilé A, Koffi KG, Nanho DC, et al. Drépanocytose homozygote chez l’adulte ivoirien de plus de 21 ans. Sante. 2010;20(2):63-67.
14. Ladu AI, Aiyenigba AO, Adekile A, Bates I. The spectrum of splenic complications in patients with sickle cell disease in Africa: a systematic review. Br J Haematol. 2021;193(1):26-42.
15. Hall BJ, Reiter AJ, Englum BR, et al. Long-term hematologic and clinical outcomes of splenectomy in children with hereditary spherocytosis and sickle cell disease. Pediatr Blood Cancer. 2020;67(8):e28290.
16. Wright JG, Hambleton IR, Thomas PW, Duncan ND, Venugopal S, Serjeant GR. Postsplenectomy course in homozygous sickle cell disease. J Pediatr. 1999;134(3):304-309.
17. Gnassingbe K, Akakpo-Numado GK, Attipou K, Gbadoe A, Tekou H. [Prophylactic splenectomy to prevent complications of splenomegaly in children with sickle cell anemia?]. Sante. 17(4):207-211.
18. De Montalembert M, Lenoir G. Antibiotic prevention of pneumococcal infections in asplenic hosts: admission of insufficiency. Ann Hematol. 2004;83(1):18-21.
Haematologica | 108 - April 2023 1162 LETTER TO THE EDITOR
Axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma patients in complete metabolic response
Axicabtagene ciloleucel (axi-cel) is an anti-CD19 autologous chimeric antigen receptor (CAR) T-cell product approved by the Food and Drug Administration for patients with large B-cell lymphoma (LBCL) who progress or relapse within 12 months of frontline chemoimmunotherapy (CIT) or after two lines of systemic therapy. These approvals are based on results of the ZUMA-7 and ZUMA-1 studies, respectively.1,2 However, both trials required active lymphoma for eligibility and prohibited bridging chemotherapy, so there are limited data to support the use of axi-cel in patients with complete metabolic response (CMR) on positron emission tomography (PET) scan before CAR T-cell infusion. In real-world practice, the management of patients who achieve CMR before axi-cel infusion remains controversial, with some providers recommending observation or consolidation with autologous stem cell transplant (SCT).3-7 Concerns regarding axi-cel use in patients achieving CMR also include potentially decreased efficacy, based on the theoretical possibility of suboptimal CAR T-cell expansion in the absence of an adequate number of CD19+ tumor cells. Here, we present a retrospective analysis of 13 patients with relapsed or refractory LBCL who achieved a CMR prior to axi-cel infusion and compare their clinical outcomes and CAR T-cell expansion levels to matched cohorts of patients with active LBCL. We find that patients in CMR at the time of axi-cel infusion had similar rates of cytokine release syndrome (CRS)
and immune cell-associated neurotoxicity syndrome (ICANS), as well as similar progression-free survival (PFS) and overall survival (OS) as patients with active disease. CAR T-cell peak levels were comparable to higher in patients with CMR at the time of axi-cel infusion compared to those with active disease. These findings support the feasibility of axi-cel in patients with relapsed or refractory LBCL who achieve a pre-infusion CMR.
We conducted a retrospective cohort analysis of 240 consecutive patients with relapsed or refractory LBCL treated with standard-of-care axi-cel at our institution between Januray 2018 and December 2021. Of these, 196 had a PET-computerized tomography scan performed after their most recent therapy and before lymphodepleting chemotherapy (LDC). Thirteen of these patients were in CMR at the time of axi-cel infusion. The immediate treatment before achieving CMR consisted of CIT in seven patients (platinum-based in 5 patients, methotrexate-based in 2 patients), radiation therapy in three patients, and targeted therapy in three patients (lenalidomide and rituximab in 2 patients, and ibrutinib in 1 patient). Baseline characteristics are shown in Table 1. Bridging therapy was defined as therapy received after apheresis and before LDC. C-reactive protein (CRP) and ferritin levels were measured at the initiation of LDC.
Of the 183 patients with metabolically active disease, 41 achieved a partial response (PR) prior to axi-cel infusion
SD: stable disease; PD: progressive disease; PR: partial response; CMR: complete metabolic response; DLBCL/HGBCL: diffuse large B-cell lymphoma/high grade B-cell lymphoma (as compared to transformed follicular lymphoma and primary mediastinal lymphoma); ECOG: European Cooperative Oncology Group; CRP: C-reactive protein; SCT: stem cell transplant; na: not available.
Total (N=196) SD/PD (N=142) PR (N=41) CMR (N=13) P value CMR vs. SD/PD P value CMR vs. PR DLBCL/HGBCL, N (%) 105 (74) 29 (71) 10 (77) 0.814 0.664 Age in years, median (range) 59 (21-85) 61 (18-78) 53 (39-72) 0.446 0.485 Male, N (%) 92 (65) 29 (71) 7 (54) 0.432 0.260 ECOG 2-4, N (%) 22 (16) 7 (17) 1 (8) 0.449 0.407 CRP mg/L, median (range) 21.9 (0.3-283.7) 7.1 (0.3-246.2) 7.7 (1.8-29.1) 0.023 0.879 Ferritin mg/L, median (range) 810.5 (13-38,964) 679 (69-3,834) 602 (101-3,190) 0.640 0.671 Previous therapies >2, N (%) 115 (81) 35 (85) 8 (62) 0.097 0.063 Bridging therapy use, N (%) 63 (44) 28 (68) 6 (46) 0.901 0.150 Bridging chemotherapy, N (%) 43 (30) 22 (54) 4 (31) 0.971 0.150 Disease refractory to prior therapy, N (%) 113 (80) 31 (76) 6 (46) 0.006 0.046 Previous autologous SCT, N (%) 34 (24) 5 (12) 4 (31) 0.584 0.117 Previous allogeneic SCT, N (%) 4 (3) 0 (0) 0 (0) 0.540 na
Table 1. Baseline characteristics of overall population.
Haematologica | 108 - April 2023 1163 LETTER TO THE EDITOR
while 142 had stable disease (SD) or progressive disease (PD). When compared to the 13 patients in CMR, patients with SD or PD had higher CRP levels at initiation of LDC (21.9 mg/L vs. 7.7 mg/L; P=0.023) and more frequently had disease refractory to prior therapy (80% vs. 46%; P=0.006). Patients with PR also more frequently had disease refractory to prior therapy (76% vs. 46%; P=0.046) compared to patients in CMR. Therefore, we performed propensity score matching to match cohorts of patients in SD/PD and in PR to patients in CMR based on these covariates (CRP and prior refractoriness for SD/PD; prior refractoriness for PR). A propensity score was calculated using logistic regression and patients in SD/PD were matched 5:1 with patients in CMR while patients in PR were matched 2:1 with patients in CMR. Baseline characteristics of these cohorts are shown in Table 2. No significant differences between the matched cohorts were identified.
When comparing patients in CMR to matched cohorts of patients with SD/PD and PR, there were no significant differences in the rates of CRS of any grade (100% vs. 91% vs. 91%) and grade 2-4 CRS (62% vs. 35% vs. 50%). Grade 3-4 CRS was observed in two (15%) patients in CMR, six (9.2%) patients with SD/PD and one (4.5%) patient in PR. There were also no significant differences in the rates of ICANS of any grade (85% vs . 59% vs. 46%), grade 3-4 ICANS (23% vs. 39% vs. 36%) and grade 3-4 cytopenias at day 30 (62% vs. 57% vs. 46%) across groups (Figure 1A). After a median follow-up of 26 months (95% confidence interval [CI]: 17-35), when comparing patients in CMR to matched cohorts of patients with SD/PD and PR, significant differences for 2-year PFS (44% vs . 35% vs . 60%) and 2-year OS rates (71% vs. 51% vs. 63%) were not found (Figure 1B).
CAR T-cell amplification in peripheral blood at day 7 (peak expansion) was measured in seven patients from the cohort in CMR and in 19 patients from the matched cohorts with active disease (17 patients with SD/PD, 2 patients in PR). Significantly higher levels of expansion were observed in patients who achieved CMR (geometric mean [GM] 47.7 cells/mL, coefficient of variation [CV] 124%) compared to patients who did not (GM 5.0 cells/mL, CV 307%; P=0.035) (Figure 1C). No CD19+ B cells were detected in peripheral blood at day 0 or day 7 for any of the seven patients in CMR (Figure 1D). All three patients who relapsed and were assessed for CD19 expression were CD19+ at the time of relapse.
In this study, we show for the first time that patients with relapsed or refractory LBCL who achieve pre-infusional CMR and are treated with axi-cel have improved CAR Tcell expansion and comparable safety and efficacy profiles to those with pre-infusional active disease. Our results are consistent with those of several prior studies. For instance, adequate CAR T-cell expansion has been reported in the JULIET study among seven patients with relapsed or refractory LBCL who achieved CMR before tisagenlecleucel infusion.8 However, due to significant differences in construct and kinetics, similar findings could not be inferred for axi-cel. Additionally, a recent retrospective study showed significantly prolonged survival in patients with LBCL who had a low total metabolic tumor volume before axi-cel infusion.9 Our data also suggest that decreasing tumor burden before CAR T-cell infusion does not diminish cell expansion or worsen clinical outcomes. Larger numbers of patients in CMR at time of infusion are needed to better determine the clinical efficacy of axi-cel in this setting.
SD: stable disease; PD: progressive disease; PR: partial response; CMR: complete metabolic response; DLBCL/HGBCL: diffuse large B-cell lymphoma/high grade B-cell lymphoma (as compared to transformed follicular lymphoma and primary mediastinal lymphoma); ECOG: European Cooperative Oncology Group; CRP: C-reactive protein; SCT: stem cell transplant; na: not available.
Total (N=100) SD/PD (N=65) PR (N=22) CMR (N=13) P value CMR vs. SD/PD P value CMR vs. PR DLBCL/HGBCL, N (%) 47 (72) 17 (77) 10 (77) 0.732 0.981 Age in years, median (range) 59 (21-81) 59 (18-72) 53 (39-72) 0.668 0.625 Male, N (%) 41 (63) 17 (77) 7 (54) 0.532 0.149 ECOG 2-4, N (%) 8 (12.3) 5 (23) 1 (8) 0.634 0.254 CRP mg/L, median (range) 7.7 (0.3-283.7) 8.3 (0.3-142.6) 7.7 (1.8-29.1) 0.639 0.987 Ferritin mg/L, median (range) 606.5 (13-14,361) 637 (69-3,834) 602 (101-3,190) 0.856 0.749 Previous therapies >2, N (%) 53 (82) 18 (82) 8 (62) 0.111 0.185 Bridging therapy use, N (%) 28 (43) 15 (69) 6 (46) 0.838 0.199 Bridging chemotherapy, N (%) 18 (28) 13 (59) 4 (31) 0.822 0.105 Disease refractory to prior therapy, N (%) 46 (71) 12 (55) 6 (46) 0.086 0.631 Previous autologous SCT, N (%) 17 (26) 2 (9) 4 (31) 0.732 0.100 Previous allogeneic SCT, N (%) 3 (5) 0 (0) 0 (0) 0.430 na
Table 2. Baseline characteristics of matched cohorts.
Haematologica | 108 - April 2023 1164 LETTER TO THE EDITOR
One mechanism by which high tumor burden may suppress CAR T-cell efficacy is through an increased presence of protumoral macrophages that contribute to an immunosuppressive microenvironment.10-12 However, it is difficult to determine what role this mechanism plays when residual disease is too small to be detected by PET. The continued presence of an immunosuppressive microenvironment in subclinical residual disease may in part explain why outcomes from patients in CMR are not clearly superior to those in PR or SD/PD. Nevertheless, our data suggest that LBCL patients who achieve a CMR after salvage CIT may still benefit from CAR T-cell therapy, although prospective validation and comparison with autologous SCT in this setting is needed.13
One theoretical concern with the use of axi-cel in patients achieving CMR is that the limited presence of tumor cells expressing CD19 would negatively impact CAR T-cell expansion. However, our data suggest that patients in CMR at the time of axi-cel infusion have comparable to high peak CAR T-cell levels than those with active disease. The degree of CAR T-cell expansion is closely related to development of CRS and ICANS.14 Thus, our finding of high peak CAR T-cell levels in patients in CMR is consistent with our finding that these patients experience similar rates of CRS and ICANS as those with active disease. The robust CAR T-cell expansion noted in patients in CMR may be due in part to a decrease in the immunosuppressive components of the tumor micro -
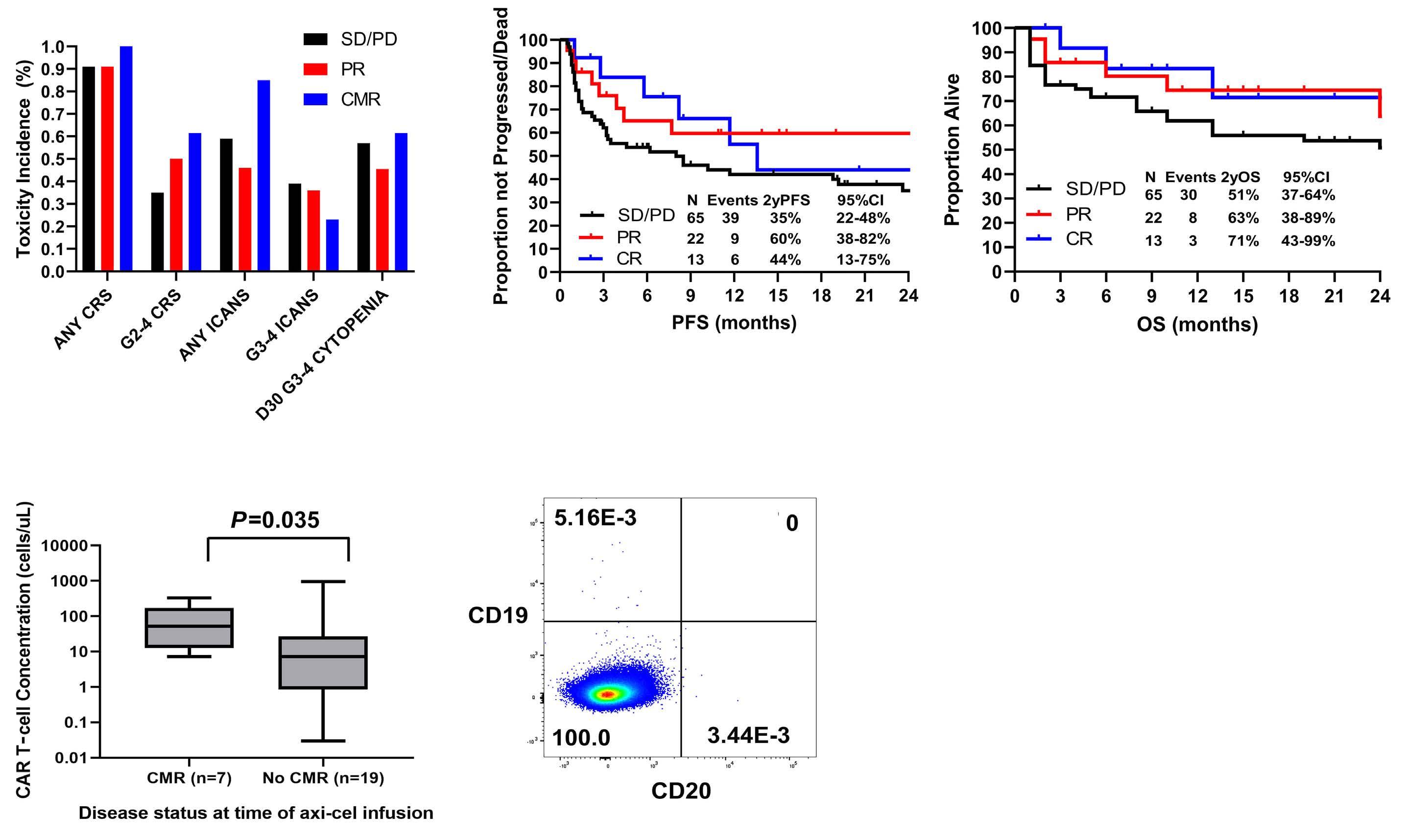
Figure 1. Safety, efficacy, CAR T-cell expansion and CD19 expression in peripheral blood according to response before infusion. (A) Cytokine release syndrome (CRS), immune cell-associated neurotoxicity syndrome (ICANS) and day 30 grade 3-4 cytopenia according to pre-infusion response in matched cohorts. (B) Progression-free survival (PFS) and overall survival (OS) according to pre-infusion (PFS) response in matched cohorts. (C) Day 7 (peak) chimeric antigen receptor CAR T-cell levels among patients with and without pre-infusion complete metabolic response (CMR). (D) CD19 expression on day 0 peripheral blood mononuclear cells from patients with pre-infusion CMR (representative image). SD/PD: stable disease/progressive disease; PR: partial response; CR: complete response; CI: confidence interval.
environment as discussed above. Potential sources of antigen driving CAR T-cell expansion in patients achieving CMR include subclinical residual disease and B cells circulating in peripheral blood or residing in lymph nodes or other tissues. Of note, no circulating CD19+ B cells were detected in the peripheral blood of any patient on day 0 or day 7, making these an unlikely source of antigen stimulation. Furthermore, in three patients who relapsed and were assessed for tumoral CD19 expression, all were CD19+ at the time of relapse. This finding suggests that mechanisms other than CD19 antigen loss are responsible for relapse in this patient population. Further investigation is warranted to better understand the kinetics of CAR T-cell expansion and persistence as well as the mechanisms of relapse in these patients. We acknowledge multiple limitations of this study, including its single-center and retrospective nature, its relatively small sample size and the lack of data regarding measurable residual disease or long-term CAR T-cell persistence in these patients.
In conclusion, our data support the use of axi-cel in patients with relapsed or refractory LBCL who achieve a CMR before axi-cel infusion and warrant further investigation of its activity as a consolidative strategy in future trials. Identification of effective and biologically rational bridging therapies aimed at decreasing disease burden and improving outcomes in patients treated with axi-cel is needed.
A B D C
Haematologica | 108 - April 2023 1165 LETTER TO THE EDITOR
Authors
Andrew P. Jallouk,1* Sushanth Gouni,1* Jason Westin,1 Lei Feng,2 Haleigh Mistry,1 Raphael E. Steiner,1 Jinsu James,1 Mansoor Noorani,1 Sandra Horowitz,1 Nahum Puebla-Osorio,3 Luis E. Fayad,1
Swaminathan P. Iyer,1 Misha Hawkins,1 Christopher R. Flowers,1 Sairah Ahmed,1 Loretta J Nastoupil,1 Partow Kebriaei,4 Elizabeth J. Shpall,4 Sattva S. Neelapu,1 Yago Nieto4# and Paolo Strati1#
1Department of Lymphoma and Myeloma, 2Department of Biostatistics, 3Department of Radiation Oncology, 4Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
*APJ and SG contributed equally as co-first authors. #YN and PS contributed equally as co-senior authors.
Correspondence:
P.S. STRATI - pstrati@mdanderson.org
https://doi.org/10.3324/haematol.2022.281954
Received: August 17, 2022.
Accepted: November 10, 2022.
Early view: November 17, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
PS is a consultant for Kite-Gilead, Roche-Genentech, Hutchinson MediPharma, ADC Therapeutics, Incyte Morphosis and TG Therapeutics; and received research funds from Sobi Pharmaceuticals, Astrazeneca-Acerta, ALX Oncology and ADC Therapeutics. RES has received research funding from Seagen, BMS,
References
1. Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. 2022;386(7):640-654.
2. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531-2544.
3. Johnston PB, Wiseman GA, Micallef IN. Positron emission tomography using F-18 fluorodeoxyglucose pre- and postautologous stem cell transplant in non-Hodgkin's lymphoma. Bone Marrow Transplant. 2008;41(11):919-925.
4. Filmont JE, Gisselbrecht C, Cuenca X, et al. The impact of preand post-transplantation positron emission tomography using 18-fluorodeoxyglucose on poor-prognosis lymphoma patients undergoing autologous stem cell transplantation. Cancer. 2007;110(6):1361-1369.
5. Svoboda J, Andreadis C, Elstrom R, et al. Prognostic value of
Rafael Pharmaceuticals and GSK. SA received research funding from Seattle Genetics, Merck, Xencor, and Tessa Therapeutics and has membership on Tessa Therapeutic’s advisory committee. JN reports honoraria from Celgene, Genentech, Gilead, Janssen, Juno, Novartis, Spectrum, TG Therapeutics and research support from Celgene, Genentech, Janssen, Karus Therapeutics, and Merck. SSN served as consultant to Kite, a Gilead Company, Merck, Bristol-Myers Squibb, Novartis, Celgene, Pfizer, Allogene Therapeutics, Cell Medica/Kuur, Incyte, Precision Biosciences, Legend Biotech, Adicet Bio, Calibr, and Unum Therapeutics; received research support from Kite, a Gilead Company, Bristol-Myers Squibb, Merck, Poseida, Cellectis, Celgene, Karus Therapeutics, Unum Therapeutics, Allogene Therapeutics, Precision Biosciences, and Acerta; received royalties from Takeda Pharmaceuticals, and has intellectual property related to cell therapy.
Contributions
APJ and SG analyzed data, and wrote the paper; JW, RES, LEF, SPI, CRF, SA, LJN, PK, EJS and SSN provided clinical care to patients and co-authored the paper; HM, JJ, MN and SH collected clinical data and co-authored the paper; APJ, NPO and SSN performed correlative studies and co-authored the paper; LF provided statistical support and co-authored the paper; PS and YN designed the study, analyzed the data, provided clinical care to patients, and wrote the paper.
Funding
This research is supported in part by The University of Texas MD Anderson Cancer Center Support Grant from the National Institutes of Health (P30 CA016672). PS salary is supported by the Lymphoma Research Foundation Career Development Award, the Leukemia Lymphoma Society Scholar in Clinical Research Career Development Program, the Sabin Fellowship Award, and by an R21 NIH grant.
Data-sharing statement
De-identified data are available upon request from the corresponding author.
FDG-PET scan imaging in lymphoma patients undergoing autologous stem cell transplantation. Bone Marrow Transplant. 2006;38(3):211-216.
6. Spaepen K, Stroobants S, Dupont P, et al. Prognostic value of pretransplantation positron emission tomography using fluorine 18-fluorodeoxyglucose in patients with aggressive lymphoma treated with high-dose chemotherapy and stem cell transplantation. Blood. 2003;102(1):53-59.
7. Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38(27):3119-3128.
8. Bishop MR, Maziarz RT, Waller EK, et al. Tisagenlecleucel in relapsed/refractory diffuse large B-cell lymphoma patients without measurable disease at infusion. Blood Adv. 2019;3(14):2230-2236.
Haematologica | 108 - April 2023 1166 LETTER TO THE EDITOR
9. Dean EA, Mhaskar RS, Lu H, et al. High metabolic tumor volume is associated with decreased efficacy of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020;4(14):3268-3276.
10. Poorebrahim M, Melief J, Pico De Coaña Y, Wickström SL, CidArregui A, Kiessling R. Counteracting CAR T cell dysfunction. Oncogene. 2021;40(2):421-435.
11. Jain MD, Zhao H, Wang X, et al. Tumor interferon signaling and suppressive myeloid cells are associated with CAR T-cell failure in large B-cell lymphoma. Blood. 2021;137(19):2621-2633.
12. Sakemura R, Cox MJ, Hefazi M, Siegler EL, Kenderian SS. Resistance to CART cell therapy: lessons learned from the treatment of hematological malignancies. Leuk Lymphoma. 2021;62(9):2052-2063.
13. Westin J, Sehn LH. CAR T cells as a second-line therapy for large B-cell lymphoma: a paradigm shift? Blood. 2022;139(18):2737-2746.
14. Neelapu SS. Managing the toxicities of CAR T cell therapy. Hematol Oncol. 2019;37(S1):48-52.
Haematologica | 108 - April 2023 1167 LETTER TO THE EDITOR
IDH mutations are enriched in myelodysplastic syndrome patients with severe neutropenia and can be a potential for targeted therapy
Myelodysplastic syndromes (MDS) are a heterogenous group of neoplastic bone marrow failure diseases.1 The Revised International Prognostic Scoring System (IPSS-R) is the most widely used prognostic scoring system to tailor therapy for MDS patients. The IPSS-R incorporated severe neutropenia (SN) defined as absolute neutrophil count (ANC) <0.8x109/L as a prognostic variable. Among MDS patients (pts), 18% had ANC <0.8x109/L.2 Current treatment guidelines recommend considering hypomethylating agents or immunosuppressive therapy for treating MDS pts with neutropenia with low neutrophil response reported in clinical studies (<10-20%).3 Recurrent infections remain a major cause of morbidity and mortality in MDS pts.1 Identification of the genomic landscape of MDS pts with SN is crucial given the large unmet clinical need in this patient population which may assist identifying potential targeted therapy.
IDH somatic mutations (MT) are described in 8-12% of acute myeloid leukemia (AML) cases and MDS.4,5 These recurrent MT in key metabolic enzymes lead to the production of the oncometabolite 2-hydroxyglutarate (2-HG), which promotes leukemogenesis through a block in normal myeloid differentiation. Selective oral inhibitors of mutant IDH1 and IDH2 have subsequently been developed and are now approved for AML4 and are under investigation for MDS.6,7
We analyzed all MDS pts treated at Moffitt Cancer Center with known ANC values around time of diagnosis and who had next-generation sequencing (NGS) as part of routine clinical care using standard Illumina platform as previously described.8 We defined SN around time of diagnosis for the purpose of this study according to the IPSS-R cut-off (ANC 0.8x109/L) and stratified pts into two groups based on this definition.
We identified 1,972 MDS pts among whom 466 pts (24%) had SN. Table 1 summarizes baseline characteristics comparing SN and non-SN pts. Neutropenic pts were slightly younger, had higher myeloblasts percentage, lower platelet counts, higher risk disease and were more likely to be classified as MDS-EB subtypes. Ninety-three pts had isolated SN (hemoglobin [Hgb] >10 g/dL and platelets >100x109/L).
IDH MT (IDH-1/IDH-2) were the only MT observed at higher rate among neutropenic pts. Figure 1A summarizes landscape of common MT observed comparing SN and nonSN pts in the whole group and stratified by IPSS-R (lower
risk defined as very low to intermediate and higher risk as high and very high groups). Among the whole cohort, 13% of MDS pts (61/462) with SN harbored IDH MT compared to 6% in non-SN pts (85/1,489) (P<0.005). Both IDH-1 and IDH-2 MT were more common in SN pts and among both lower and higher risk IPSS-R groups. The most common observed hot spot in IDH-2 was R140, although the R172 hotspot was observed more in SN pts. Among pts with isolated SN, 18% harbored IDH MT compared to 12% in non-isolated SN (P=0.1). IDH-1 MT were more common in pts with isolated SN (11% vs. 4%; P=0.01) but no difference in IDH-2 MT (8% in both isolated SN and non-isolated SN groups; P=0.8). TP53 was observed in 26% compared to 19% respectively for SN and non-SN pts, P<0.005 but no statistical difference was observed when examined among IPSS-R risk groups.
Figure 1B illustrates the presence of SN among MT and wild-type (WT) commonly observed somatic MT in MDS pts. Among pts with IDH1/2 MT 42% of pts had SN compared to 22% among WT, 40% IDH-1 MT MDS pts had SN compared to 23% of IDH-1 WT, and 44% of IDH-2 MT had SN compared to 23% of IDH-2 WT. SN was present in 30% of TP53 MT MDS pts compared to 23% among those with WT. SF3B1 MT MDS pts were less likely to have SN. The median overall survival (mOS) was shorter (25 months [mo] vs. 42 mo; P<0.005) and the rate of AML transformation higher (49% vs. 26%; P<0.005) in SN versus non-SN pts respectively. SN was not associated with worse outcome when adjusted for myeloblast percentage, (hazard ratio [HR]: 1.0; 95% confidence interval [CI]: 0.83-1.2; P<0.98). The mOS was worse for SN IDH WT compared to non-SN IDH-WT, (24 mo vs. 43.5 mo; P<0.005). This observation reflects enrichment of TP53 MT among SN IDHWT (29%) compared to non-SN IDH WT (8%) (P=0.001). There was no difference in mOS comparing SN IDH-MT compared to non-SN IDH MT (mOS 33 mo vs . 30 mo; P=0.3). Among SN pts, there was no difference in mOS among IDH MT compared to WT (mOS 33 mo vs. 24 mo; P=0.1). Among non-SN pts IDH-MT was associated with worse OS with a mOS 31 mo compared to 42 mo for nonSN IDH-WT (P=0.04) in univariable analysis. In multivariable analysis adjusting for IPSS-R, IDH MT in non-SN pts was not statistically significantly associated with worse outcome (HR: 1.3; P=0.08)
The complete response rate (CR) to azacitidine was 20% among SN pts. There was no difference in response to
Haematologica | 108 - April 2023 1168 LETTER TO THE EDITOR
t-MDS: therapy-related myelodysplastic syndrome; WHO: World Health Organization; SLD: single lineage dysplasia; MLD: multilineage dysplasia; RS: ring sideroblasts; EB: excess blasts; AML: acute myeloid leukemia; MDS-U: MDS unclassifiable; MPN: myeloproliferative neoplasm; MDS-RS-T: MDS-RS with thrombocytosis; R-IPSS: Revised International Prognostic Scoring System; Hgb: hemoglobin; ANC: absolute neutrophile count; WBC: white blood cell.
azacitidine among SN pts based on IDH MT status (CR rates 20% [n=51/254] for IDH-WT and 15% [n=5/33] for IDH-MT; P=0.9).
Given the lack of effective treatment options for neutropenia in general, two symptomatic IDH1 SN lower risk MDS pts have been treated with ivosidenib. The first pt had IDH1 R123 C (variant allele frequency [VAF] 44%) and SRSF2 P95R (VAF 43%). Hemoglobin improved from 9.4 g/dL to 14 g/dL, platelets were normal at baseline. There were 1-2% circulating peripheral blood blasts which resolved on therapy. The pt has been in remission for 31 months. The second pt had DNMT3A and IDH1 R132 C mutations at baseline (VAF at 7% for both). Platelets improved from 111x109/L to 180x109/L. Hgb also improved from 11.8 g/dL to 14.1 g/dL. The pt has been in durable remission for 11 months now. Both pts achieved a complete hematologic response within 2 weeks of initiation of therapy (ANC 0.3 to 2.8 and ANC 0.21 to 2.4), which has been durable, with therapy ongoing.
Severe neutropenia is present in almost one fourth of MDS pts and it is associated with worse outcome.2 SN is
more commonly observed with higher-risk disease, complex karyotype and excess myeloblasts. SN is less encountered in lower-risk MDS which may dictate choice of therapy and isolated neutropenia as sole indication for treatment in lower-risk MDS is even more rare.9 There are limited options for treating neutropenia.9 Granulocyte colony stimulating factors have not been shown to improve outcomes.10 Anti-thymocyte globulin/cyclosporine may yield trilineage response including neutrophil response in selected subset of young or hypoplastic lower-risk MDS but is rarely utilized.11 Hypomethylating agents, widely used to treat patients with bi/pancytopenia, only yield up to 20% neutrophil response compared to 19% with conventional care regimens.12
We observed that IDH MT are enriched among SN MDS pts regardless of IPSS-R risk group. Notably, in two of two IDH-1 MT SN pts, treatment with ivosidenib resulted in ongoing, durable complete hematologic responses.
The IDH MT genotype and the neutropenia phenotype association have been observed in patients with AML. IDH mutations were also commonly observed among patients
Severe neutropenia (ANC <0.8x109/L) N=466 No severe neutropenia (ANC ≥0.8x109/L) N=1,506 P value Age in years, mean 67.6 69 0.001 Sex (male), N (%) 292 (63) 950 (63) 0.9 Race (white), N (%) 415 (89) 1,371 (92) 0.15 t-MDS, N (%) 92 (20) 275 (18) 0.47 WHO classification MDS SLD/MLD, % MDS SLD/MLD RS, % MDS-EB, % AML <30% blasts, % del 5q, % MDS-U, % MDS/MPN, % MDS-RS-T, % 21 4 52 21 1 1 0 0 33 16 29 6 4 2 6 4 <0.005 R-IPSS Very low, % Low, % Intermediate, % High, % Very high, % 4 11 17 23 45 16 36 18 14 16 <0.005 Myeloblasts, % Hgb g/dL, mean Platelets x109/L, mean ANC x109/L, mean WBC x109/L, mean 12 9.4 98 0.46 2.2 6 10 147 3.2 6 <0.005 0.1 <0.005 <0.005 <0.005
Table 1. Baseline characteristics comparing neutropenic and non-neutropenic myelodysplastic syndromes patients.
Haematologica | 108 - April 2023 1169 LETTER TO THE EDITOR
Figure 1. Correlation of somatic mutations and severe neutropenia among myelodysplastic syndromes patients. (A) Somatic mutations among patients with severe neutropenia compared to non-severe neutropenia and (B) frequency of severe neutropenia among commonly observed somatic mutations in myelodysplastic syndromes patients. LR: low risk; HR: high risk.
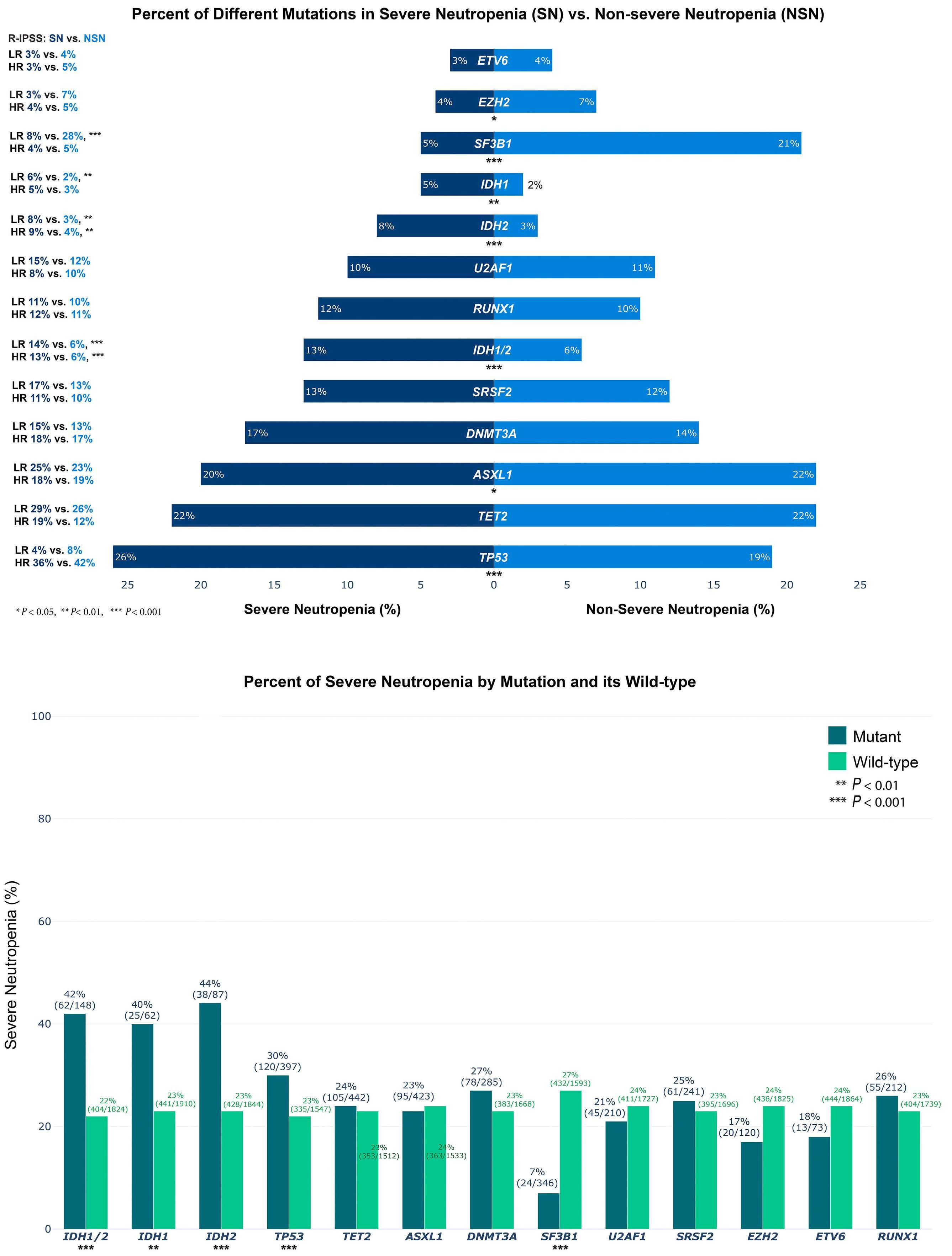
A B Haematologica | 108 - April 2023 1170 LETTER TO THE EDITOR
with chronic idiopathic neutropenia and evidence of clonal hematopoiesis.13 Potentially, treatment early on in disease course may lead to higher response rates, particularly in the absence of other driver co-mutations.
Patnaik et al . reported IDH MT in 12% of MDS patients There was no difference in ANC based on IDH MT status.
Patients with IDH-1 MT had a lower white blood cell count and were all red blood cell transfusion dependent. IDH-1 but not IDH-2 mutation in multivariable analysis was associated with inferior OS and LFS.5
The molecular IPSS was recently proposed to refine the IPSS-M prognostic utility and incorporate molecular data. Notably, the new molecular model excluded neutropenia as a clinical variable.14 A new personalized precision model using artificial intelligence retained neutrophil count as a clinical variable but did not include IDH MT.15
Early promising data using IDH inhibitors in MDS were reported in different setting including post hypomethylating agent failure higher-risk disease, first-line higher-risk MDS as single agent and in lower risk after erythroid stimulating agents’ failure.6,7 Responses were reported in 50% of lower-risk MDS patients treated with IDH inhibitors after erythroid stimulating agents failure.
Our study limitation includes its retrospective nature, not fully examining the co-occurrence of somatic mutations and the interplay with other clinical variables. The underlying biology of this observation (likely differentiation block or inhibition of dioxygenase enzymes) for MDS pts with neutropenia should be further explored. IDH inhibitors through reduction of 5-HG and promotion of differentiation may improve granulopoiesis. Our data demonstrating enrichment of IDH MT among MDS pts with SN and the anecdotal durable responses observed in two cases of lower risk MDS with SN merit further exploring this targeted therapy in the context of clinical trials.
Authors
Rami Komrokji, Najla al Ali, Onyee Chan, Kendra Sweet, Andrew Kuykendall, Jeffrey Lancet, Eric Padron and David A. Sallman
Department of Malignant Hematology, H Lee Moffitt Cancer Center, Tampa, Fl, USA
Correspondence:
R. KOMROKJI - Rami.komrokji@moffitt.org
https://doi.org/10.3324/haematol.2022.281607
Received: July 4, 2022.
Accepted: November 11, 2022.
Early view: November 24, 2022.
©2023
Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
RM received honoraria from BMS, Novartis, Geron, Abbvie, JAZZ, Servier and PharmaEssentia; he is part of the Speaker's Bureau of Acceleron, JAZZ, Servier and PharmaEssentia. AK received honoraria from BluePrint Medicines, Novartis, PharmaEssentia, Incyte, Prelude and Celgene/BMS; he is part of the Speaker's Bureau of Novartis and Celgene/BMS; he received research funding from Abbvie and PharmaEssentia; he consults for PharmaEssentia and CTI Biopharma; he is a member of PharmaEssentiaity's Board of Directors or advisory committees. KS is a member of Gilead, Novartis, Astellas, AROG and Bristol Meyers Squibb Board of Directors or advisory committees; he received honoraria from Novartis and Bristol Meyers Squibb. JL consults for BerGenBio, AbbVie, ElevateBio Management, Celgene/BMS, Astellas, Daiichi Sankyo, Millenium Pharma/Takeda, Servier and Jazz.EP consults for Taiho; he received honoraria from Blueprint, Kura and Stemline; he received research funding from Incyte and BMS. DAS is part of the Speaker's Bureau of Incyte, AbbVie; he is a member of Shattuck Labs, Syndax, Bristol-Myer, Magenta, Aprea, Kite and Servier Board of Directors or advisory committees; he is part of the Speaker's Bureau of Intellia; he consults for Takeda, Novartis and Aprea; he received research funding from Kite. All other authors have no conflicts of interest to disclose.
Contributions
RK designed the study, analyzed data and wrote the manuscript; NA collected and analyzed data; OC, KS, AK, JL and EP reviewed, edited and approved the final version of the manuscript and contributed patients; DS designed the study, reviewed the manuscript and contributed patients.
Data-sharing statement
No data will be shared.
1. Volpe VO, Garcia-Manero G, Komrokji RS. Myelodysplastic syndromes: a new decade. Clin Lymphoma Myeloma Leuk. 2022;22(1):1-16.
2. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes.
Blood. 2012;120(12):2454-2465.
3. Greenberg PL, Stone RM, Al-Kali A, et al. Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Canc Netw. 2017;15(1):60-87.
4. Issa GC, DiNardo CD. Acute myeloid leukemia with IDH1 and
Haematologica | 108 - April 2023 1171 LETTER TO THE EDITOR
References
IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 2021;11(6):107-107.
5. Patnaik MM, Hanson CA, Hodnefield JM, et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic Study of 277 patients. Leukemia. 2012;26(1):101-105.
6. Ades L, Dimicoli-Salazar S, Sebert M, et al. Enasidenib (ENA) is effective in patients with IDH2 mutated myelodysplastic syndrome (MDS) : the ideal phase 2 study by the GFM group. Blood. 2021;138(Suppl 1):S63.
7. Sebert M, Cluzeau T, Beyne Rauzy O, et al. Ivosidenib monotherapy is effective in patients with IDH1 mutated myelodysplastic syndrome (MDS): the Idiome phase 2 study by the GFM group. Blood. 2021;138(Suppl 1):S62.
8. Sallman DA, Komrokji R, Vaupel C, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia. 2016;30(3):666-673.
9. Volpe VO, Komrokji RS. Treatment options for lower-risk myelodysplastic syndromes. Where are we now? Ther Adv Hematol. 2021;12:2040620720986641.
10. Hutzschenreuter F, Monsef I, Kreuzer KA, Engert A, Skoetz N.
Granulocyte and granulocyte-macrophage colony stimulating factors for newly diagnosed patients with myelodysplastic syndromes. Cochrane Database Syst Rev. 2016;2:CD009310.
11. Komrokji RS, Mailloux AW, Chen DT, et al. A phase II multicenter rabbit anti-thymocyte globulin trial in patients with myelodysplastic syndromes identifying a novel model for response prediction. Haematologica. 2014;99(7):1176-1183.
12. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223-232.
13. Tsaknakis G, Galli A, Papadakis S, et al. Incidence and prognosis of clonal hematopoiesis in patients with chronic idiopathic neutropenia. Blood. 2021;138(14):1249-1257.
14. Bernard E, Tuechler H, Greenberg PL, et al. Molecular international prognosis scoring system for myelodysplastic syndromes. NEJM Evid. 2022;1(7).
15. Nazha A, Komrokji R, Meggendorfer M, et al. Personalized prediction model to risk stratify patients with myelodysplastic syndromes. J Clin Oncol. 2021;39(33):3737-3746.
Haematologica | 108 - April 2023 1172 LETTER TO THE EDITOR
Landscape of immunoglobulin heavy chain γ gene class switch recombination in patients with adult T-cell leukemia–lymphoma
We are currently conducting the multicenter prospective study “Monitoring of immune responses following mogamulizumab-containing treatment in patients with adult Tcell leukemia–lymphoma (ATL)” (MIMOGA), (Trial registration number: UMIN000008696).1-4 We previously reported the importance of humoral immune measures, such as the proportion of CD2-CD19+ B cells in peripheral blood mononuclear cells (PBMC), for the outcome of ATL patients.5 Subsequently, we demonstrated that lower immunoglobulin G (IgG) B-cell diversity in PBMC was a significant unfavorable prognostic factor for overall survival (OS) in patients.6 In that study, we had focused on the diversity-generating mechanism for the IgG variable regions. Therefore, here we focused on the diversity-generating mechanism in the IgG constant region, namely class switch recombination (CSR),7-9 and explored its detailed status in ATL patients. The present investigation was affiliated with the MIMOGA study, and 81 ATL patients were enrolled according to the criteria used in that previous study.5,6 Unbiased amplification and high-throughput sequencing of the immunoglobulin heavy chain γ (IGHG) genes was conducted using PBMC at enrollment in the MIMOGA study.6,10 The sequence reads whose immunoglobulin heavy chain constant (IGHC) region was determined to be assigned to immunoglobulin heavy constant γ P were excluded from the analysis. IGHG unique reads, with identical IGHV (variable), IGHD (diversity) and IGHJ (joining) gene usage and identical deduced amino acid sequences of the CDR3 complementarity determining region 3 (CDR3), were designated “VDJ/CDR3 identical unique reads”. Among IGHG unique reads in PBMC, the percentage of VDJ/CDR3 identical reads which were shared between two or more different subclass genes was designated “the percentage of CSR of IGHG unique reads in PBMC”, abbreviated to “%CSR of IGHG”. In practice, the %CSR of IGHG was calculated as follows: the total number of VDJ/CDR3 identical unique reads which were shared between two subclass genes (IGHG3-IGHG1, IGHG3-IGHG2, IGHG3-IGHG4, IGHG1IGHG2, IGHG1-IGHG4, or IGHG2-IGHG4), or three subclass genes (IGHG3-IGHG1-IGHG2, IGHG3-IGHG1-IGHG4, IGHG3IGHG2-IGHG4, or IGHG1-IGHG2-IGHG4), or four subclass genes (IGHG3-IGHG1-IGHG2-IGHG4) was divided by the total number of IGHG unique reads.
We first analyzed VDJ/CDR3 identical unique reads according to the type of subclass genes in PBMC of ATL patients, compared to those of 12 healthy individuals, who
had also participated in our previous study.6,10 The percentage of IGHG unique reads which had a single subclass gene of IGHG1 tended to be higher in ATL patients than in healthy individuals. Among IGHG unique reads, the percentages of VDJ/CDR3 identical reads which were shared between two different functional subclass genes, namely, IGHG3-IGHG1, IGHG3-IGHG2, IGHG3-IGHG4, or IGHG1-IGHG2 were significantly lower in ATL patients. The remaining two pairs consisting of two different subclasses, such as IGHG1-IGHG4 and IGHG2-IGHG4, tended to be lower in ATL patients. Additionally, those shared between three different functional subclass genes, in all four available pairs, i.e., IGHG3-IGHG1-IGHG2, IGHG3-IGHG1-IGHG4, IGHG3IGHG2-IGHG4, or IGHG1-IGHG2-IGHG4, were significantly lower in ATL patients. Accordingly, the %CSR of IGHG was significantly lower in ATL patients than in healthy controls (Table 1). These data indicate that the CSR of IGHG unique reads occurs significantly less frequently in ATL patients. These differences in the frequency of CSR among IGHG are likely to reflect the degree of impairment of the humoral immune system.
Second, we sought correlations between the %CSR of IGHG, and the Shannon–Weaver diversity index (SWDI) for the IGHG repertoire in PBMC.6,10 We found a significant positive correlation of the %CSR of IGHG with the SWDI (Spearman rank correlation coefficients [Rs]=0.860; P<0.001) in ATL patients. This was also the case in healthy controls (Rs=0.846; P=0.001), likely because both CSR and somatic hypermutation (SHM) leading to the diversification of antibody variable regions are mediated by activation-induced cytidine deaminase (AID).12-14
Third, we analyzed the clinical characteristics of ATL patients according to the %CSR of IGHG. Clinically meaningful cut-off values of the %CSR of IGHG had not been determined. Hence, we divided the patients into two groups according to the %CSR. Subsequently, univariate analysis for survival was performed using a Cox proportional hazards regression model at each of the six cut-off points. In the present study, the cut-off point yielding the minimum P value was selected as the most meaningful cut-off value, as previously described.5,11 As a result, the cut-off value of the %CSR of IGHG was set at 9.363% (Online Supplementary Table S1). In this context, the median age was 67 years (range, 41–86) and 70 years (range, 58–83) in patients with a lower and higher %CSR, respectively (not significantly different; P=0.119). There were also no
Haematologica | 108 - April 2023 1173 LETTER TO THE EDITOR
Table 1. VDJ/CDR3 identical unique reads according to the types of shared subclass genes in adult T-cell leukemia–lymphoma patients and healthy individuals.
The IGHG unique reads which have a single subclass gene (IGHG3, IGHG1, IGHG2, or IGHG4), and the VDJ/CDR3 identical unique reads according to the types of shared subclass genes in peripheral blood mononuclear cells (PBMC) of adult T-cell leukemia–lymphoma (ATL) patients and healthy individuals are shown. Subclass genes employed by the VDJ/CDR3 identical unique reads are represented by the filled gray cells. The mean and median values (percentages) of each type of subclass gene are indicated before and after the / symbol, respectively. Significant differences in the proportions between ATL patients and healthy individuals are indicated as P values. P<0.050/15 was considered statistically significant after Bonferroni correction. Significantly higher values are indicated in bold. The total proportions of all types of subclass genes employed by the VDJ/CDR3 identical unique reads are 100% in each healthy individual or ATL patient. IGHG unique reads, with identical IGHV (immunoglobulin heavy chain variable), IGHD (diversity), and IGHJ (joining), gene usage and identical deduced amino acid sequences of the CDR3 (complementarity determining region 3), were designated as “VDJ/CDR3 identical unique reads”. Among IGHG unique reads, the percentage of VDJ/CDR3 identical reads which were shared between two or more different subclass genes (IGHG3, IGHG1, IGHG2, or IGHG4) was designated as “the percentage of CSR of IGHG unique reads”, abbreviated to “%CSR of IGHG”. CSR: class switch recombination; IGHG: immunoglobulin heavy chain γ gene; IGHG3: immunoglobulin heavy constant γ3 gene.
significant differences between patients with a lower or higher %CSR regarding previous systemic chemotherapy (yes or no; P=0.579), sex (P=1.000), or Eastern Cooperative Oncology Group Performance Status (ECOG PS) (0, 1 vs. 2, 3, 4; P=0.207). In addition, there were also no significant differences between patients with a lower or higher %CSR for clinical subtype (chronic, smoldering vs. acute, lymphoma; P=1.000), or in serum soluble interleukin-2 receptor (sIL-2R) (mean and median, 17,971, and 8,600, vs. 13,879 and 4,590; P= 0.127). Accordingly, this makes it hard to estimate the status of IGHG CSR based on clinical characteristics.
Fourth, we analyzed the immunological characteristics of ATL patients according to the %CSR of IGHG. The percentages of CD2-CD19+ B cells were significantly higher in the patients with a higher %CSR of IGHG compared to those with a lower %CSR. This suggests that as more B cells, in-
cluding IgG B cells, are present in PBMC, more CSR of IGHG can occur. On the other hand, there was no significant difference in the percentage of CD3+CD8+ or CD3+CD4+ T cells in the patients with a higher or lower %CSR. With respect to CD16+CD56+ natural killer (NK) cells, or CD11c+ monocytes, the former were significantly higher, and the latter tended to be higher in the patients with a higher %CSR. These NK cells or monocytes might assist CSR for IgG, but no responsible mechanisms have been clearly identified. Serum IgG and IgA titers were significantly higher in patients with a higher %CSR than in those with a lower %CSR, but the difference in serum IgM was not significant (Table 2). These results likely reflect the gene order in the IGHC locus on chromosome 14q32, such as IGHM (immunoglobulin heavy constant m)-IGHD (immunoglobulin heavy constant δ )-IGHG3-IGHG1-IGHA1 (immunoglobulin heavy constant α1)-IGHG2-IGHG4-IGHE
Subclass gene Healthy (N=12) mean/median (%) ATL (N=81) mean/median (%) P IGHG3 IGHG1 IGHG2 IGHG4 3.66/3.25 7.94/3.51 0.766 27.52/26.61 41.25/37.99 0.009 31.72/31.60 39.05/40.15 0.242 1.06/0.93 2.81/0.47 0.449 1.29/0.97 0.58/0.44 0.003 1.56/1.49 0.65/0.52 < 0.001 0.10/0.09 0.05/0 0.003 19.00/19.15 4.06/3.46 < 0.001 0.22/0.23 0.18/0.01 0.005 0.17/0.15 0.12/0.01 0.023 10.58/10.55 1.94/1.58 < 0.001 0.15/0.16 0.072/0 < 0.001 0.09/0.06 0.0306/0 0.001 0.99/0.78 0.20/0.03 < 0.001 1.88/1.42 1.06/0.72 0.059 %CSR of IGHG 36.04/37.38 8.95/8.38 < 0.001
Haematologica | 108 - April 2023 1174 LETTER TO THE EDITOR
ATL: adult T-cell leukemia-lymphoma; CSR: class switch recombination; IGHG: immunoglobulin heavy
(immunoglobulin heavy constant e)-IGHA2 (Online Supplementary Figure S1).8 That is to say, the IGHG gene region overlaps IGHA on chromosome 14q32. Therefore, the frequent CSR of IGHG would be likely associated with frequent CSR of IGHA, thus leading to higher serum IgG and IgA, but not IgM, titers. The significantly higher SWDI for the IGHG repertoire in patients with a higher %CSR of IGHG would be expected, due to AID,12-14 and SHM and CSR might both reflect the degree of impairment of the hu-
moral immune system.
Finally, we analyzed the prognostic impact of %CSR of IGHG. The OS of patients with a higher %CSR of IGHG was significantly longer than of those with a lower %CSR (median OS, 30.9 vs. 13.2 months; P=0.043) (Online Supplementary Figure S2A). Multivariate analysis of OS in the 81 ATL patients was performed using the following six variables: sex, age, clinical subtype, ECOG PS, sIL-2R, and %CSR of IGHG. Of these, two variables significantly af-
Characteristics %CSR of IGHG in PBMC P lower (<9.363%) higher (>9.363%) N (%) 50 (62) 31 (38) CD2-CD19+ cells (%)* mean median range 1.16 0.39 0.00-8.17 4.89 1.25 0.03-32.91 0.009 CD3+CD8+ cells (%)* mean median range 14.07 7.45 0.10-71.73 13.41 9.86 0.74-52.33 0.620 CD16+CD56+ cells (%)* mean median range 6.74 3.62 0.07-31.57 11.97 8.22 0.43-39.42 0.016 CD11c+ monocytes (%)** mean median range 50.50 53.05 0.50-97.18 68.88 78.60 0.34-95.76 0.059 CD3+CD4+ cells (%)* mean median range 65.64 69.34 11.16-98.14 57.31 61.62 14.60-97.48 0.108 Serum IgG (mg/dL) mean median range 908 941 303-1,663 1,142 1,177 512-2,310 0.002 Serum IgA (mg/dL) mean median range 168 139 5-522 274 216 93-1,076 0.002 Serum IgM (mg/dL) mean median range 41 38 5-112 48 35 15-99 0.270 SWDI for IgG heavy-chain repertoire in PBMC mean median range 3.643 3.487 1.004-6.516 6.385 6.355 3.233-10.096 <0.001
Table 2. Immunological characteristics of T-cell leukemia–lymphoma patients according to the percentage of class switch recombination of IGHG unique reads of IGHG in peripheral blood mononuclear cells.
chain γ gene; PBMC: peripheral blood mononuclear cells; SWDI: Shannon-Weaver diversity index. *Percentage among whole lymphocytes in PBMC. **Percentage among whole monocytes in PBMC.
Haematologica | 108 - April 2023 1175 LETTER TO THE EDITOR
Table 3. Multivariate analysis including the percentage of class switch recombination of IGHG unique reads in peripheral blood mononuclear cells for overall survival of patients with T-cell leukemia–lymphoma.
CSR: class switch recombination; IGHG: immunoglobulin heavy chain γ gene; PBMC: peripheral blood mononuclear cells; CI: confidence interval; ECOG PS: Eastern Cooperative Oncology Group Performance Status; sIL-2R: soluble interleukin-2 receptor.
fected OS, namely a higher serum sIL-2R, and a higher %CSR of IGHG (HR, 0.439; 95% confidence interval [CI]: 0.229–0.839) (Table 3). CSR is a process resulting in improved ability of antibodies to eliminate pathogens.8,9,14 Thus, a higher frequency of efficient CSR possibly implies better humoral immune status, leading to a more favorable prognosis. Considering the previously identified prognostic factor, the frequency of CD2-CD19+ B cells within lymphocytes,5 the OS of patients with both a lower %CSR and a lower CD2-CD19+ B cells was significantly worse than of the other patients (median OS 7.2 vs. 18.8 months; P=0.004) (Online Supplementary Figure S2B). Based on the present observations of host immune status of the patients, together with an assessment of somatic alterations in the tumor cells,15 the establishment of precision medicine for patients with ATL is imminent. The present investigation offers significant observations regarding associations of IGHG CSR with clinical outcomes in ATL patients. However, a limitation of the study should be recognized, namely, the possibility that the %CSR of IGHG was affected by the absolute B-cell count in the patients’ blood used for the IGHG sequencing, which cannot be completely excluded. Nonetheless, mitigating against this in the present patient cohort, there was no significant correlation between these two factors (Rs=0.214; P=0.056). In conclusion, the present study demonstrated that IGHG CSR occurs less frequently in ATL patients than in healthy individuals. Additionally, the lower frequency of CSR of
IGHG was a significant independent unfavorable prognostic factor in patients with ATL receiving mogamulizumab-containing treatment. These observations provide novel insights into the mechanism of impaired humoral immunity in ATL patients, the degree of dysfunction of which may be reflected in the status of IGHG CSR, which is associated with the clinical outcome. Further investigation of strategies to enhance the quality of humoral immunity is warranted.
Authors
Hiroaki Hiramatsu,1 Kisato Nosaka,2 Shigeru Kusumoto,3 Nobuaki
Nakano,4 Ilseung Choi,5 Makoto Yoshimitsu,6 Yoshitaka Imaizumi,7 Michihiro Hidaka,8 Hidenori Sasaki,9 Junya Makiyama,10 Eiichi
Ohtsuka,11 Tatsuro Jo,12 Masao Ogata,13 Asahi Ito,3 Kentaro Yonekura,14
Hiro Tatetsu,15 Takeharu Kato,7 Toshiro Kawakita,8 Youko Suehiro,5,16
Kenji Ishitsuka,6 Shinsuke Iida,3 Takaji Matsutani,17 Hiroyoshi
Nishikawa,1 Atae Utsunomiya,4 Ryuzo Ueda1 and Takashi Ishida1,3
1Department of Immunology, Nagoya University Graduate School of Medicine, Nagoya; 2Cancer Center, Kumamoto University Hospital, Kumamoto; 3Department of Hematology and Oncology, Nagoya City University Graduate School of Medical Sciences, Nagoya;
4Department of Hematology, Imamura General Hospital, Imamura; 5Department of Hematology, National Hospital Organization Kyushu
Variables N Hazard Ratio 95% CI P Sex Male Female 50 31 1.000 1.0570.560-1.993 Reference 0.865 Age in years ≤70 >70 52 29 1.000 1.2720.659-2.456 Reference 0.474 Clinical subtype chronic, smoldering acute, lymphoma 13 68 1.000 2.2870.755-6.925 Reference 0.143 ECOG PS 0,1 2,3,4 58 23 1.000 1.2140.611-2.413 Reference 0.580 sIL-2R (U/mL) <20,000 >20,000 64 17 1.000 4.9242.303-10.527 Reference <0.001 % CSR of IGHG unique reads in PBMC <9.363 >9.363 50 31 1.000 0.4390.229-0.839 Reference 0.013
Haematologica | 108 - April 2023 1176 LETTER TO THE EDITOR
Cancer Center Hospital, Kyushu; 6Department of Hematology and Rheumatology, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima; 7Department of Hematology, Nagasaki University Hospital, Nagasaki; 8Department of Hematology, National Hospital Organization Kumamoto Medical Center, Kumamoto; 9Division of Medical Oncology, Hematology, and Infectious Diseases, Department of Medicine, Fukuoka University Hospital, Fukuoka; 10Department of Hematology, Sasebo City General Hospital, Sasebo; 11Department of Hematology, Oita Prefectural Hospital, Oita; 12Department of Hematology, Japanese Red Cross Nagasaki Genbaku Hospital, Nagasaki; 13Department of Hematology, Oita University Hospital, Oita; 14Department of Dermatology, Imamura General Hospital, Imamura; 15Department of Hematology, Kumamoto University Hospital, Kumamoto; 16Department of Cell Therapy National Hospital Organization Kyushu Cancer Center Hospital, Kyushu and 17Osaka Laboratory, Repertoire Genesis Incorporation, Osaka, Japan
Correspondence:
T. ISHIDA - itakashi@med.nagoya-u.ac.jp
https://doi.org/10.3324/haematol.2022.281435
Received: May 21, 2022.
Accepted: November 15, 2022.
Early view: November 24, 2022.
©2023
Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
KN has received consultancy fees, research funding and honoraria from Kyowa Kirin, research funding from Chugai Pharmaceutical, and honoraria from Celgene, Eisai, Meiji Seika Pharma, Janssen Pharmaceutical and Bristol Myers Squibb. SK has received consultancy fees, research funding and honoraria from Chugai Pharmaceutical, research funding and honoraria from Kyowa Kirin, Daiichi Sankyo, Takeda Pharmaceutical, Janssen Pharmaceutical and honoraria from Otsuka Pharmaceutical and Eisai. NN has received consultancy fees from JIMRO and honoraria from Novartis, Takeda pharmaceutical, Chugai Pharmaceutical, Celgene, Otsuka Pharmaceutical, Nippon Shinyaku, Kyowa Kirin and Asahi Kasei Pharma. MY has received honoraria from Novartis International, Takeda Pharmaceutical and Sanofi YI has received honoraria from Kyowa Kirin, Celgene, Eisai, Bristol Myers Squibb and Sanofi, Meiji Seika Pharma Co., Ltd., SymBio Pharmaceuticals Limited, Sumitomo Dainippon Pharma, and Nippon Shinyaku Co., Ltd. MH has received research funding from Chugai Pharmaceutical and honoraria from JCI Pharma. KY has received honoraria from Janssen, and Taiho Pharmaceutical. HT has received honoraria from Bristol Myers Squibb, Chugai Pharmaceutical, Eisai, Ono Pharmaceutical, SymBio Pharmaceuticals Limited and patents and royalties from Mesoblast. YS has received research funding and honoraria from Kyowa Kirin and honoraria from Celgene and Bristol Myers Squibb. KI has
received honoraria from Abbvie, Astellas Pharma, Bristol Myers Squibb, Celgene, Chugai Pharmaceutical, CSL Behring, Daiichi Sankyo, Eizai, Janssen, Kyowa Kirin, Meiji Seika, Ono Pharmaceutical, Otsuka, Pfizer, Sanofi, Takeda, Yakult, and consultancy from Daiichi Sankyo, Meiji Seika, and Yakult, and research funding from Kyowa Kirin, and Ono Pharmaceutical. SI has received honoraria from Janssen, Sanofi, Bristol-Myers Squibb, Pfizer, Takeda, Ono, and Celgene, and research funding from Janssen, Sanofi, Bristol-Myers Squibb, Takeda, Ono, Pfizer, Amgen, Abbvie, Glaxo SmithKlein, and Daichi Sankyo, and donation of commercial property from Chugai, and Sanofi. TM is an employee and a stockholder and an employee of Repertoire Genesis Incorporation. HN has received research funding and honoraria from Ono, Bristol-Myers, MSD, Chugai, and research funding from Taiho, Daiichi-Sankyo, Kyowa Kirin, Zenyaku, Oncolys BioPharma, Debiopharma, Asahi-Kasei, Sysmex, Fujifilm, SRL, Astellas, Sumitomo Dainippon and BD Japan. AU has received honoraria from Bristol-Myers, and Meiji Seika Pharma, and has received consulting fees from JIMRO and Otsuka Medical Devices. RU has received research funding from Kyowa Kirin, Chugai Pharmaceutical and Ono Pharmaceutical. All other authors have no conflicts of interest to disclose.
Contributions
HH, RU and TI developed the concept and design of the study; KN, SK, NN, IC, MY, YI, MH, HS, JM, EO, TJ, MO, AI, KY, HT, TK, TK, YS, KI, SI, AU and TI aquired and analyzed data; HH, TM, HN, RU and TI interpreted data. All authors wrote and approved the final version of the manuscript.
Acknowledgments
We thank all nurses and clinical research coordinators who were involved in this study, for their patient care and schedule management. We also thank the Japan Institute of Statistical Technology (Saitama, Japan) for their critical review of the statistical analyses, and for providing a certificate attesting the validity of the statistical methods used for the data analyses in the present manuscript. We are grateful to Mr. Hiroshi Iwata (SRL Medisearch Inc., Tokyo, Japan) for his support in scheduling samples from patients, and for sample preservation. We are also grateful to Mr. Satoshi Shinohara (Repertoire Genesis Inc.) for his fruitful discussions with us.
Funding
This work was supported by a Grant-in-Aid for Early-Career Scientists (N 21K15567 to HH), a Grant-in-Aid for Challenging Research (Exploratory) (N 21K19900 to TI), a Grant-in-Aid for Scientific Research (B) (N 22H02918 to TI), and by Grants-in-Aid from the Japan Agency for Medical Research and Development (N 21ae0101074h0001, and 22ae0101074h0001 to RU).
Data-sharing statement
Original data can be made available in response to a reasonable, written request to the corresponding author
Haematologica | 108 - April 2023 1177 LETTER TO THE EDITOR
References
1. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult Tcell leu-kemia: clinical and hematologic features of 16 cases. Blood. 1977;50(3):481-492.
2. Ishii T, Ishida T, Utsunomiya A, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res. 2010;16(5):1520-1531.
3. Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30(8):837-842.
4. Ishida T, Jo T, Takemoto S, et al. Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemialymphoma: a randomized phase II study. Br J Haematol. 2015;169(5):672-682.
5. Yonekura K, Kusumoto S, Choi I, et al. Mogamulizumab for adult T-cell leukemia-lymphoma: a multicenter prospective observational study. Blood Adv. 2020;4(20):5133-5145.
6. Nosaka K, Kusumoto S, Nakano N, et al. Clinical significance of the immunoglobulin G heavy-chain repertoire in peripheral blood mononuclear cells of adult T-cell leukaemia-lymphoma patients receiving mogamulizumab. Br J Haematol. 2022;196(3):629-638.
7. Honjo T, Kataoka T. Organization of immunoglobulin heavy chain genes and allelic deletion model. Proc Natl Acad Sci U S A. 1978;75(5):2140-2144.
8. Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-
switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol. 2012;12(7):517-531.
9. Westra ER , Sünderhauf D, Landsberger M, Buckling A. Mechanisms and consequences of diversity-generating immune strategies. Nat Rev Immunol. 2017;17(11):719-728.
10. Kitaura K, Yamashita H, Ayabe H, Shini T, Matsutani T, Suzuki R. Different somatic hypermutation levels among antibody subclasses disclosed by a new next-generation sequencingbased antibody repertoire. Analysis. Front Immunol. 2017;8:389.
11. Mazumdar M, Glassman JR. Categorizing a prognostic variable: review of methods, code for easy implementation and applications to decision-making about cancer treatments. Stat Med. 2000;19(1):113-132.
12. Muramatsu M, Sankaranand VS, Anant S, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA editing deaminase family in germinal center B cells. J Biol Chem. 1999;274(26):18470-18476.
13. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation- induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553-563.
14. Feng Y, Seija N, Di Noia JM, Martin A. AID in antibody diversification: there and back again. Trends Immunol. 2021;42(1):89.
15. Tanaka N, Mori S, Kiyotani K, et al. Genomic determinants impacting clinical outcome of mogamulizumab treatment for adult T-cell leukemia/lymphoma. Haematologica. 2022;107(10):2418-2431.
Haematologica | 108 - April 2023 1178 LETTER TO THE EDITOR
Targeting cytokine-induced leukemic stem cell persistence in chronic myeloid leukemia by IKK2-inhibition
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder, arising from a hematopoietic stem cell (HSC) that acquires the chromosomal translocation t(9;22), resulting in the BCR-ABL oncoprotein. While BCRABL-targeting tyrosine kinase inhibitors (TKI) eliminate the majority of CML cells, the most primitive disease-initiating leukemic stem cells (LSC) are frequently spared. CML development is accompanied by increasing levels of inflammatory cytokines, such as IL-1 α , IL-1 b , IL-6, or TNF. Correspondingly, LSC are characterized by a TKI-persisting, inflammatory cytokine response pattern that was observed even during prolonged therapy.1 Our preliminary work confirmed that TNF signaling is active in murine CML stem cells and TNF-targeted antibody treatment enhanced therapeutic efficiency of TKI treatment.2 Moreover IKK-dependent activation of NF- k B has been shown to contribute to BCR-ABL-induced myeloid and lymphoid leukemogenesis.3,4 In CML stem and progenitor cells, an autocrine TNF loop and NF- k B signaling were found to persist during TKI treatment in vitro 5 NF-kB serves as a central inflammatory hub as several cytokines induce IKKdependent phosphorylation at serine 32/36 and subsequent degradation of IkBα, which releases the NF-kB complex, allowing its activation by phosphorylation at serine 536 and transport to the nucleus.6 Thereby, NF-kB induces transcription of target genes such as several cytokines or the NF-kB signaling molecules themselves.7 Here we confirmed TKI-persistent TNF-induced NF- k B signaling in CML mice and human cells. As TNF can induce pro-proliferative signaling via NF-kB but also pro-apoptotic cascades via RIPK1-induced CASP8 activation, we here aimed to specifically inhibit the pro-proliferative activity by targeting IKK2. Therefore, we performed pharmacologic IKK2 inhibition, using the small molecule IKK2 inhibitor LY2409881 (LY), previously shown to be well tolerable in pre-clinical application.8 Combined BCRABL/IKK2-targeting blocked malignant NF- k B signaling and enhanced apoptosis in TKI-sensitive but also TKI-resistant cells. In vivo, NF- k B-mediated TNF activity was elevated despite nilotinib (NIL) treatment. IKK2 targeting severely reduced LSC, which was further reflected by preventing disease onset upon secondary transplantation. As TNF-induced NF- k B-signaling was evident in chronicphase (CP) but likewise blast crisis (BC) CML, IKK2 targeting affected the clonogenic potential in both disease stages. Finally, in CP CML CD34+ cells, combined BCRABL/IKK2 inhibition significantly induced apoptosis, also in quiescent LSC showing that this approach enables the
eradication of the TKI-persisting malignant stem cell population in CML.
Aiming to get insight into the role of malignant TNF signaling in CML, we started to compare BCR-ABL-positive with ABL-negative cell lines. We observed significant upregulation of TNF signaling in 32D BCR-ABL versus empty vector control cells, as represented by increased expression of relevant TNF targets in BCR-ABL positive cells (IkBα, fold change (fc): 1.9; Nf-kb, fc: 2.2, A20, fc: 6.5; Online Supplementary Figure S1A). In order to mimic the malignant inflammatory niche, TNF was subsequently added to both cell types at a physiological concentration and this still resulted in increased TNF target gene expression in BCR-ABL versus control cells (I k B α , fc: 1.7; Nf- kb, fc: 3, A20, fc: 3.4). Notably, this upregulation of TNF signaling persisted in the presence of NIL (Online Supplementary Figure S1A). TKI-persistent NF-kB activation was likewise evident in vivo, as shown by bone marrow (BM) immunohistochemical p65 staining of Scl-tTa-BCR-ABL or Scl-tTa control mice (Figure 1A). Although NIL slightly reduced p65 protein expression, reversion to healthy control levels was not achieved.
Next, we studied the effects induced by pharmacologic inhibition of the Ik B-kinase-subunit, IKK2 using LY. The small molecule inhibitor prevents IKK2 phosphorylation and subsequent degradation of IkBα, which thus remains bound to the NF-kB complex to block its phosphorylation. This prevents nuclear translocation of NF-kB and thereby expression of target genes. Analysis of A20 expression in human KCL22 CML cells suggested a NIL-induced upregulation (Figure 1B; fc: 3.37), that was reduced by LY treatment (A20, fc: 2.02). We observed similar effects when adding imatinib (IM) or dasatinib (DAS) (Online Supplementary Figure S1B). We studied signaling activity via western blot analyses in KCL-22 cells (Figure 1C). As expected, phosphorylation of NF-kB subunit p65 was not reduced by NIL treatment alone. Moreover, pIkBα at serine 32/36 was significantly inhibited by LY single treatment (fc: 0.27) and this was still achieved in the presence of NIL (fc: 0.17), as shown by significant downregulation as compared to the corresponding DMSO control or NIL single treatment, in the presence of TNF. This further resulted in reduced phosphorylation of p65 at serine 536 in the presence of LY (fc: 0.42) or both drugs (fc: 0.29), as compared to the corresponding DMSO control.
Annexin V staining of treated KCL-22 cells showed the advantage of IkB kinase inhibition as this induced a significant increase in apoptosis by IKK2 inhibition alone (fc: 2.4)
Haematologica | 108 April 2023 1179 LETTER TO THE EDITOR
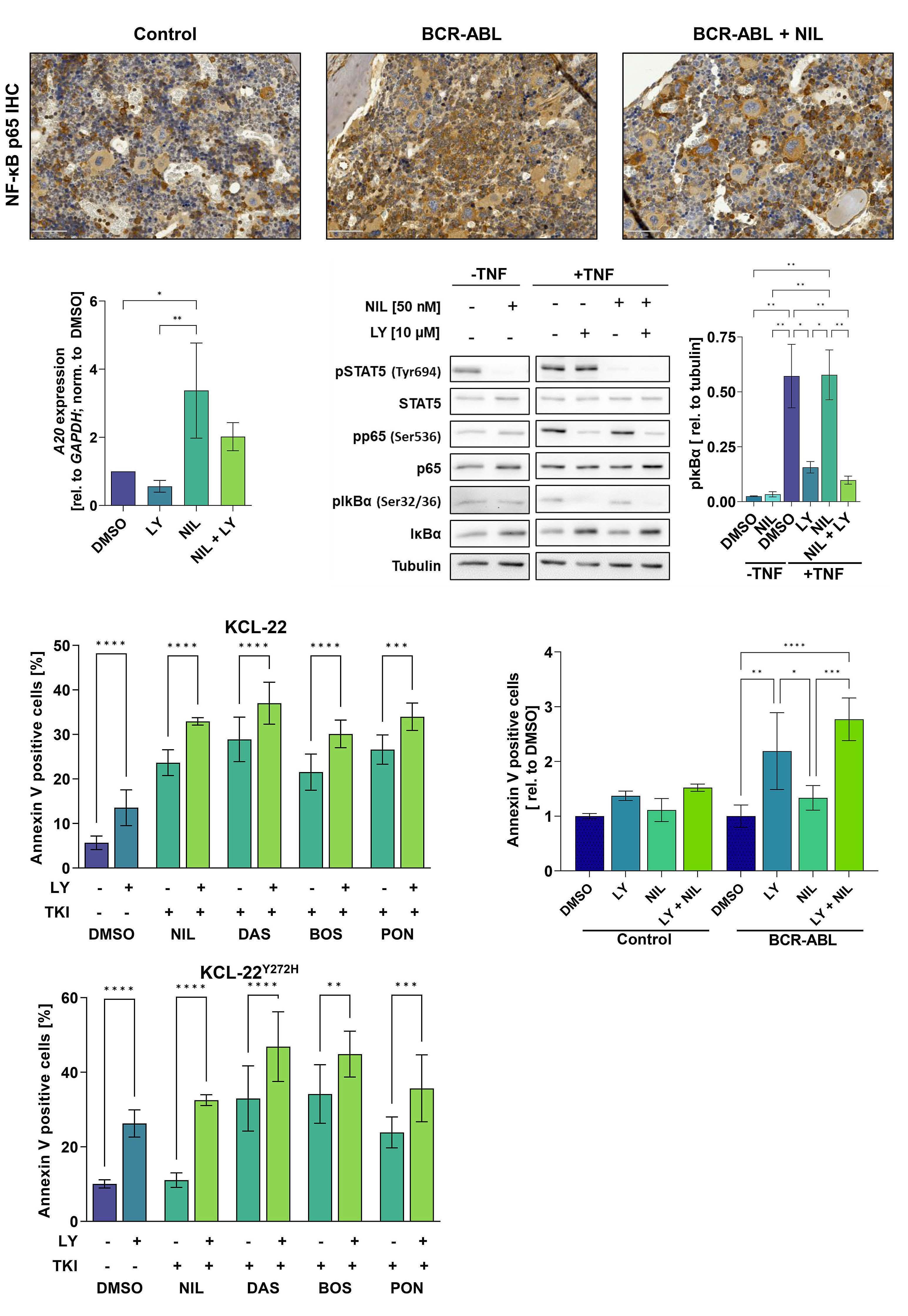
Continued on following page. A B C D F E Haematologica | 108 - April 2023 1180 LETTER TO THE EDITOR
Figure 1. Targeting tyrosine kinase inhibitor-persistent TNF-signaling by pharmacological IKK2 inhibition. (A) Elevated and tyrosine kinase inhibitor (TKI)-persisting NF-kB activation in chronic myeloid leukemia (CML) mice is shown by a representative immunohistochemical bone marrow (BM) staining of NF-kB p65 (brown) and hematoxylin (blue) in BM sections from mice, which were either transplanted with Scl-tTa-BCR-ABL or Scl-tTa control BM and subsequently treated using nilotinib (NIL) or vehicle control for 3 weeks. White scale bar represents 50 mm. (B) CML KCL-22 cells were treated using Ly2409881 (LY) (10 mM), NIL (50 nM), or LY + NIL, in the presence of 1 ng/mL TNF for 16 hours (h). Expression of A20 was analyzed via quantitative real time polymerase chain reaction (qRT-PCR) and is expressed as % of GAPDH. Shown are mean values of n=3, normalized to DMSO control. (C) Western blot analysis of KCL-22 cells using the indicated antibodies, pretreated with 1 ng/mL TNF for 1 h and subsequently treated using NIL (50 nM) and/or LY (5 mM) for again 1 h. Shown is 1 representative result of n=3 (left panel). Phosphorylation was quantified using densitometry (n=3, right panel). Shown are means ± standard error of the mean. (D) Flow cytometry analysis of Annexin V-positive KCL-22 cells in the presence of 1 ng/mL TNF after 48 h of treatment using LY (5 mM), NIL (50 nM), LY + NIL, dasatinib (DAS) (10 nM), LY + DAS, bosutinib (BOS) (5 nM), LY + BOS, ponatinib (PON) (5 nM), LY + PON. (E) Annexin V flow cytometry analysis of BM lin- MACS-isolated cells from Scl-tTa or Scl-tTa-BCR-ABL mice that were induced to express BCR-ABL for 10 days prior to BM-isolation. In vitro treatment was performed using LY (10 mM), NIL (50 nM), LY + NIL or DMSO control in the presence of 1 ng/mL TNF for 16 h (n=3). (F) Flow cytometry analysis of Annexin V-positive KCL-22Y272H cells after 48 h of treatment using LY (5 mM), NIL (50 nM), LY + NIL, DAS (10 nM), LY + DAS, BOS (5 nM), LY + BOS, PON (5 nM), LY + PON in the presence of TNF (1 ng/mL; n=3). Significance was calculated using one or two-way ANOVA; Mean ± standard deviation; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
that further enhanced NIL-induced cell death by 1.4-fold (Figure 1D). The therapeutic effect of IKK2 inhibition was also evaluated in the context of other clinically relevant TKI. For all TKI tested, we observed increased apoptosis levels upon combined treatment (DAS fc: 1.3, BOS fc: 1.4, PON fc: 1.3) (Figure 1D). We next treated isolated lin- BM cells from Scl-tTa-BCR-ABL or Scl-tTa control mice (Figure 1E), showing that control cells remained largely unaffected whereas apoptosis was induced upon LY monotherapy (fc: 2.2) in murine primary CML cells. Moreover, combined LY and NIL treatment enhanced NIL-induced apoptosis by 2.1-fold.
In order to study the role of TNF signaling in BCR-ABL kinase-mutated TKI resistance, we utilized the KCL-22Y272H9 and KCL-22T315I-cell lines.10 In KCL-22Y272H cells, LY therapy induced apoptosis, while NIL alone had no effect (Figure 1F). As DAS, bosutinib (BOS), and ponatinib (PON) could bypass the P-loop mutation, these TKI indeed induced KCL-22Y272H cell death that was further elevated with IKK2 inhibition, as reflected by a further rise in Annexin V positivity (DAS fc: 1.4, BOS fc: 1.3, PON fc: 1.5). In KCL-22T315I cells, effects were very modest with LY application only slightly inducing apoptosis and this was unaffected by TKI treatment (Online Supplementary Figure S1C).
In order to investigate whether IKK2 targeting could affect TKI-persistent LSC, we implemented the Scl-tTa-BCR-ABL mouse model (Figure 2A). As expected, splenomegaly and white blood cell count were normalized by NIL treatment (Online Supplementary Figure S2A, B). Comprehensive immunophenotyping revealed a predominant expansion of malignant B cells (B220low) that responded to BCR-ABL inhibition (Online Supplementary Figure S2C, D). Moreover, we confirmed our previous findings on reduced BM-derived long-term (LT)-HSC (Lin-, Sca1+, c-kit+, CD48low, CD150+) upon disease development (Figure 2B).11 In line with the concept that TKI treatment selects for primitive LSC, we observed increased LT-HSC in NIL- or NIL/LYtreated mice. Interestingly, LY monotherapy decreased LT-
HSC by 2.3-fold. While Tnf expression was decreased upon treatment in BM, NIL monotherapy, as well as combined NIL/LY therapy apparently increased Tnf transcript levels in the spleen (Figure 2C). As TNF acts both, autocrine and paracrine, this likely resulted in elevated A20 expression in both organs (Figure 2C). In light of this, A20 is not only described to be increased in response to TNF, but also known to protect cells from TNF–induced apoptosis.12
Treatment efficacy was further shown by a reduction of BCR-ABL expression, with the strongest reduction upon combined BCR-ABL- and IKK2 inhibtion (Figure 2D). We assessed the malignant stem cell potential via serial transplantation of BM cells into irradiated wild-type recipients (Figure 2A). In line with our finding that BM-derived LT-HSC are spared by TKI or TKI + LY treatment, secondary recipients showed disease recurrence, accompanied by slightly increased spleen weights (Online Supplementary Figure S2E), as well as elevated BCR-ABL transcript levels (Figure 2E). Interestingly, LY monotherapy prevented spleen expansion and severely impaired re-expansion of malignant cells as shown by only residual BCRABL transcript levels, in secondary recipients.
In order to determine whether TNF-signaling is specifically active in LSC, we re-analyzed microarray data (GSE40721) revealing upregulation of TNF signaling via NF- k B in CD34+CD38- stem cells versus CD34+CD38+ progenitor cells (Figure 3A). We proceeded to evaluate the clonogenic potential in CML CD34+ cells upon treatment and this was strongly reduced by both single treatments, as compared to DMSO control (LY: 12.1-fold, NIL: 3.8-fold) and almost completely abrogated upon combined IKK2 and BCR-ABL targeting (77.5-fold; Figure 3B). In order to assess the effect on most primitive cells, replating was performed without any further treatment and this showed that previous NIL treatment had spared malignant stem cells while LY-treated cells were severely impaired in the ability to form colonies (Figure 3C), suggesting that LY monotherapy targets the most primitive malignant stem cells. In
Haematologica | 108 - April 2023 1181 LETTER TO THE EDITOR
healthy donor-derived CD34+ cells, IKK2 inhibition showed an effect in two of six samples (Online Supplementary Figure S3A). As these cells were partially provided by osteoarthritis patients, we assume that IKK2 inhibition could affect these samples, as pro-inflammatory cytokines could be likewise involved.13 In order to dissect if IKK2 tar-
geting induces apoptosis in the most primitive CML cells we performed Annexin V staining in CD34+/CFSEMAX quiescent LSC. Indeed, in vitro apoptosis was significantly increased by IKK2 inhibition alone (fc: 7.4) or in combination with NIL (fc: 8.5; Figure 3D), suggesting that LY treatment exerts its anti-leukemic effects observed upon secondary
Figure 2. IKK2 inhibition reduces long-term hematopoietic stem cells and inhibits disease re-initiation in vivo. (A) Schematic overview of the experimental setup: 2x106 bone marrow (BM) cells of either FVB/N Scl-tTa or Scl-tTa-BCR-ABL mice were transplanted into irradiated wild-type FVB/N recipients. Expression of BCR-ABL was induced 1 week after transplantation and treatment was started at 2 weeks after transplant and continued for 3 weeks. During that time, LY2409881 (LY) was administered via intraperitoneal injection, 3 times per week (50 mg/kg body weight, dissolved in 5% dextrose) and/or nilotinib (NIL) treatment was performed via oral gavage, daily (50 mg/kg body weight, dissolved in 10% N-methyl-2-pyrrolidon and 90% polyethylene glycol) and/or the corresponding vehicle control for 3 weeks daily. Following treatment groups were evaluated: ScltTa (vehicle, designated as control) or Scl-tTa-BCR-ABL (vehicle, LY, NIL, and LY + NIL). After sacrifice, 2x106 BM cells of treated animals were transplanted into secondary irradiated recipients. These mice were sacrificed for analyses 6 weeks after transplantation. (B) BM-derived long-term hematopoietic stem cells (LT-HSC) (lin-,Sca1+,ckit+,CD48low,CD150+) of treated recipients, shown as % of living cells. (C) Quantitative real time polymerase chain reaction (qRT-PCR) analyses of Tnf and A20 mRNA expression in BM and spleen of treated primary recipients is shown relative to Gapdh (%). (D) BCR-ABL mRNA expression in BM of primary and secondary (E) recipients. Significance was calculated using one or two-way ANOVA; mean ± standard deviation; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
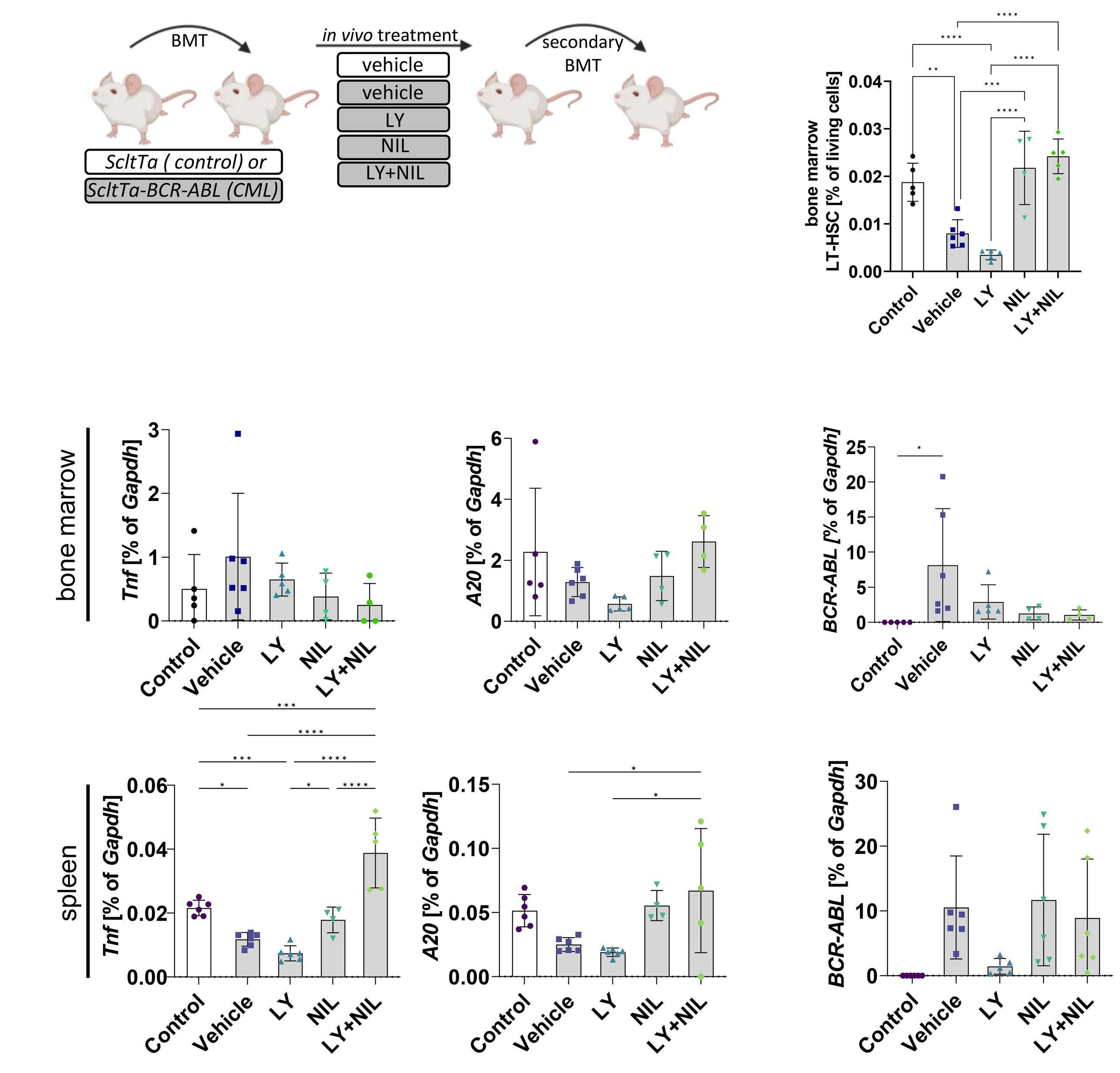
A B C D E Haematologica | 108 - April 2023 1182 LETTER TO THE EDITOR
Figure 3. Targeting elevated NF- kB signaling substantially reduces chronic phase and blast crisis chronic myeloid leukemia stem cells. (A) Gene set enrichment analysis (GSEA) of published expression data set GSE40721 comparing CML CD34+;CD38+ progenitor cells with CML CD34+;CD38- stem cells for the ‘HALLMARK_TNFA_SIGNALING_VIA_NFKB’ gene set. (B) Colony-forming unit (CFU) assays using primary patient-derived CD34+ bone marrow (BM) cells after being treated for 72 hours (h) using LY2409881 (LY) (10 mM), nilotinib (NIL) (50 nM), LY + NIL or DMSO control, in the presence of TNF (1 ng/mL). (C) CFU counts upon serial-plating using cells obtained in (B), without any further treatment. (D) Analysis of apoptotic and CFSEMax/Annexin V CD34+ chronic phase chronic myeloid leukemia (CML-CP) patient-derived BM cells after being treated for 72 h using LY (10 mM), NIL (50 nM), LY + NIL, or DMSO control, in the presence of TNF (1 ng/mL). Shown is 1 of 3 representative results. (E) GSEA of published expression data set GSE47927 comparing CML-CP hematopoietic stem cells (HSC) with blast crisis CML (CML-BC) HSC analyzed for the ‘HALLMARK_TNFA_SIGNALING_VIA_NFKB’-gene set. (F) CFU assays of BC-CML-derived mononucleated (MNC) BM cells being treated for 72 h using LY (10 mM), NIL (50 nM), LY + NIL, or DMSO control in the presence of TNF (1 ng/mL). Significance was calculated using one or two-way ANOVA; mean ± standard deviation; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
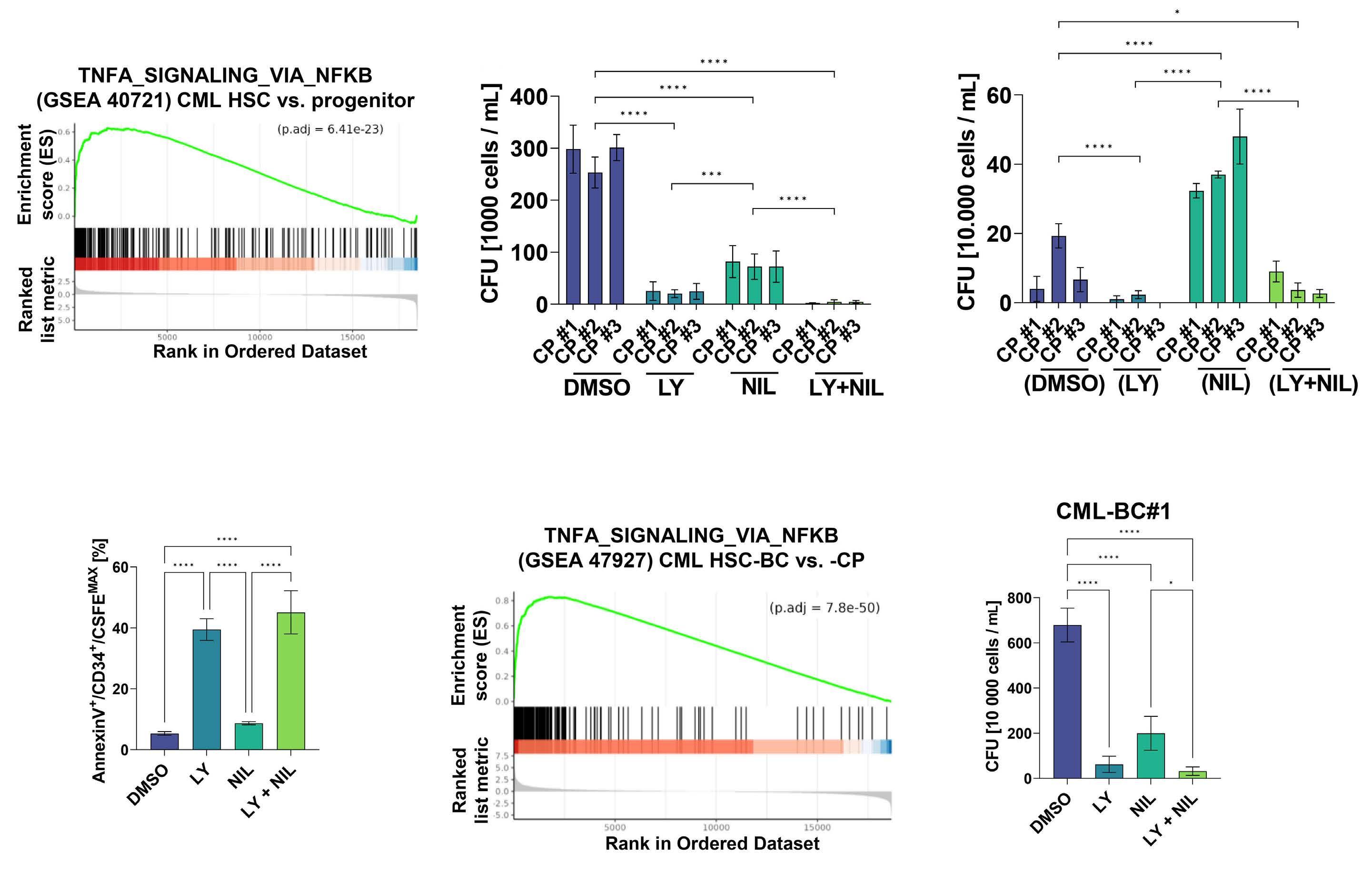
transplantation at least partially by induction of apoptosis within the LSC compartment.
As further LSC persistence-mediating cytokines activate the NF-kB pathway, we next tested the treatment effect in the presence of physiological IL-1α or IL-1b levels (Online Supplementary Figure S3B). Again, colony formation was significantly reduced by inhibition of IKK2 alone (IL-1α: 1.5fold, IL-1b: 1.7-fold) as well as by combined IKK2- and BCRABL-targeting (IL-1α: 3.4-fold, IL-1b: 5.2-fold), suggesting that IKK2 targeting could exerts its effects by inhibiting, not only TNF-mediated NF-kB activation but also due to targeting further pro-leukemic inflammatory signaling.
In the context of progressed disease, IKK2 was described as a resistance-associated gene.14
In line with this, TNF signaling increased in BM CD34+ cells upon CML progression (Figure 3E). Therefore, we studied
IKK2 targeting also in primary BC-CML samples, showing that impaired colony formation was evident upon LY exposure in all patient samples tested (CML-BC #1: 10.9-fold; #2: 7.4-fold; #3: 13.9-fold; Figure 3F; Online Supplementary Figure S3C). Again, in combination this was further reduced by NIL (CML-BC #1: 21.4-fold; #2: 42-fold; #3: 93.7-fold). In conclusion, our results underline the potential of NF-kB targeting, via IKK2-inhibition, to eliminate TKI-persisting LSC. In vivo treatment revealed the complexity. While NIL alone and in combination was able to reduce leukemic cells, LSC persisted and re-initiated the disease upon secondary transplant. Treatment with NIL expanded CD3+ T cells, which was still observed when combined with LY but not in LY monotherapy-treated mice (Online Supplementary Figure S2C). As T cells in CML display a TNF-dominated cytokine secretion profile,15 we assume this could be a source of enhanced
A B C D E F Haematologica | 108 - April 2023 1183 LETTER TO THE EDITOR
splenic Tnf expression, probably exerting paracrine effects on the BM, thereby potentially protecting BM-residing LSC.
Authors
Marlena Bütow,1,2 Fabio J. Testaquadra,1,2 Julian Baumeister,1,2 Tiago Maié,3 Nicolas Chatain,1,2 Timo Jaquet,1,2 Stefan Tillmann,1,2 Martina Crysandt,1,2 Ivan G. Costa,3 Tim H. Brümmendorf1,2 and Mirle Schemionek1,2
1Department of Hematology, Oncology, Hemostaseology, and Stem Cell Transplantation, Faculty of Medicine, RWTH Aachen University; 2Center for Integrated Oncology Aachen Bonn Cologne Düsseldorf (CIO ABCD) and 3Institute for Computational Genomics, Joint Research Center for Computational Biomedicine, RWTH Aachen University, Aachen, Germany
Correspondence:
M. SCHEMIONEK-REINDERS - mschemionek@ukaachen.de
https://doi.org/10.3324/haematol.2022.280922
Received: March 10, 2022.
Accepted: November 23, 2022. Early view: December 1, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
MB designed research, performed experiments, analyzed the data
and wrote the manuscript; FJT and JB performed experiments and revised the manuscript; NC, TJ and ST assisted experimental work and revised the manuscript; IGC and TM reanalyzed publicly available expression data sets; MC contributed patient samples and clinical data; THB contributed research material and revised the manuscript; MS designed research, analyzed the data and revised the manuscript. Final approval of the manuscript was provided by all authors.
Acknowledgments
The authors thank Dr. Vignir Helgason and Dr. Eric Kalkman for providing KCL22 T315I cells, Julia Plum and Kristina Pannen for excellent technical assistance, and Dr. Jörg Eschweiler for providing healthy donor-derived cells. This work was supported by the IHC (immunohistochemistry) facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen, within the Faculty of Medicine at RWTH Aachen University. Figures were created with BioRender.com.
Funding
This work was supported by the Clinical Research Group entitled “Untangling and Targeting Mechanisms of Myelofibrosis in Myeloproliferative Neoplasms (MPN)” (CRU344) funded by the German Research Foundation (DFG) (SCHE 1938/3-1 [AOBJ 659838]), in the framework of the Research Training Group “Tumor-targeted Drug Delivery" grant 331065168 as well as by the START-Program of the Faculty of Medicine, RWTH Aachen (grant 691706).
Data-sharing statement
All technical information pertaining to the experimentation performed is available on request.
See also: [GSE40721]
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse40721 [GSE47927]
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse47927
1. Giustacchini A, Thongjuea S, Barkas N, et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat Med. 2017;23(6):692-702.
2. Herrmann O, Kuepper MKMK, Bütow M, et al. Infliximab therapy together with tyrosine kinase inhibition targets leukemic stem cells in chronic myeloid leukemia. BMC Cancer. 2019;19(658):1-14.
3. Hsieh MY, Van Etten RA. IKK-dependent activation of NF-kB contributes to myeloid and lymphoid leukemogenesis by BCRABL1. Blood. 2014;123(15):2401-2411.
4. Yamashita M, Passegué E. TNF-α coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25(3):357-372.
5. Gallipoli P, Pellicano F, Morrison H, et al. Autocrine TNF-α production supports CML stem and progenitor cell survival and enhances their proliferation. Blood. 2013;122(19):3335-3339.
6. Sakurai H, Suzuki S, Kawasaki N, et al. Tumor necrosis factor-αinduced IKK phosphorylation of NF-kB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278(38):36916-36923.
7. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15(6):362-374.
8. Deng C, Lipstein M, Rodriguez R, et al. The novel IKK2 inhibitor LY2409881 potently synergizes with histone deacetylase inhibitors in preclinical models of lymphoma through the downregulation of NF-kB. Clin Cancer Res. 2015;21(1):134-145.
9. Kuepper MKMK, Bütow M, Herrmann O, et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia. 2019;33(8):1964-1977.
10. Yuan H, Wang Z, Gao C, et al. BCR-ABL Gene expression is required for its mutations in a novel KCL-22 cell culture model for acquired resistance of chronic myelogenous leukemia. J Biol Chem.
References Haematologica | 108 - April 2023 1184 LETTER TO THE EDITOR
2010;285(7):5085-5096.
11. Schemionek M, Elling C, Steidl U, et al. BCR-ABL enhances differentiation of long-term repopulating hematopoietic stem cells. Blood. 2010;115(16):3185-3195.
12. Priem D, Devos M, Druwé S, et al. A20 protects cells from TNFinduced apoptosis through linear ubiquitin-dependent and -independent mechanisms. Cell Death Dis. 2019;10(10):692.
13. Zhao Y, Li Y, Qu R, et al. Cortistatin binds to TNF-α receptors and
protects against osteoarthritis. EBioMedicine. 2019;41:556-570.
14. Villuendas R, Steegmann JL, Pollán M, et al. Identification of genes involved in imatinib resistance in CML: a gene-expression profiling approach. Leukemia. 2006;20(6):1047-1054.
15. Westermann J, Van Lessen A, Schlimper C, et al. Simultaneous cytokine analysis by cytometric bead array for the detection of leukaemia-reactive T cells in patients with chronic myeloid leukaemia. Br J Haematol. 2006;132(1):32-35.
Haematologica | 108 - April 2023 1185 LETTER TO THE EDITOR
Methotrexate, cytarabine, thiotepa and rituximab (MATRix) chemoimmunotherapy for primary central nervous system lymphoma: a Toronto experience
The MATRix (methotrexate, cytarabine, thiotepa, rituximab) regimen has significantly improved outcomes of patients with primary central nervous system lymphoma (PCNSL). The original trial population, however, was young and fit with few comorbidities. We show that, in a realworld setting, the majority of patients treated with MATRix experienced hematologic toxicity, and 65% of patients developed grade 3 or 4 infectious complications. Dose reductions were made in 68% of patients and dose delays occurred in 49% of patients. Survival was comparable to what has been reported in the literature, but the rate of toxicity was higher than previously described. The randomized International Extranodal Lymphoma Study Group trial IELSG32 examined outcomes of MATRix followed by consolidation therapy with either autologous stem cell transplant (ASCT) or whole brain radiation in patients with PCNSL.1 The addition of thiotepa to MATRix had prolonged both overall survival (OS) and progression-free survival (PFS), producing sustained improved outcomes with a 7-year PFS of 50% and an OS of 70%.2 The IELSG32 trial population was a young and fit population, with a median age of 57 years and the majority of patients (67%) had Eastern Cooperative Oncology Group (ECOG) performance status 0-1. One retrospective multicenter analysis documented poorer outcomes in a population with poorer performance status.3 We therefore set out to examine the use of MATRix as an induction regimen in a real-world clinical setting in Toronto, Canada. We performed a retrospective review of patients with a diagnosis of B-cell PCNSL treated at Sunnybrook Health Sciences Centre, Princess Margaret Cancer Centre and St. Michael’s Hospital from October 2017 to March 2020. Exclusion criteria included positive serology for human immunodeficiency virus, or systemic lymphoma. A review of electronic health records was performed to retrieve the patients’ baseline characteristics, medical history, and treatment details including dose intensity, timing, and response to therapy. For patients deemed eligible for ASCT, induction with MATRix was given at any of the three hospitals, but ASCT was only performed at one center. As such, patients were often seen for transplant later in their treatment course. The local practice evolved to omit thiotepa from the third cycle to allow for improved stem cell collection off the third or fourth cycle. All other dose adjustments were made at the treating physician’s discretion. The primary outcome was assessment of adverse events
related to treatment. Secondary outcomes included OS and PFS at 2 years, and response to treatment. Response was assessed by magnetic resonance imaging or computed tomography of the brain using published criteria.4 Descriptive statistics were used to summarize patients’ demographics, treatments and outcomes. The Kaplan-Meier estimator was used to determine the cumulative incidence of OS and PFS at 2 years. Cox regression models were used to investigate the impact of baseline characteristics on outcomes.
A total of 37 patients with a diagnosis of diffuse large Bcell PCNSL were included in this study. The median age of the patients was 58 years (range, 30-69 years). Patients were followed for an average of 16.9 months (range, 3-48 months). The majority of patients received MATRix as firstline therapy (89%). A high number of patients had an ECOG performance status of 2 or more (41%) and 18 patients (49%) had a Charlson Comorbidity Index score of 2 or more. Twenty-four patients (65%) received Pneumocystis jiroveci pneumonia prophylaxis throughout treatment with sulfamethoxazole-trimethoprim or atovaquone. Granulocyte colony-stimulating factor was used in all patients for primary prophylaxis.
Dose reductions of any degree were performed in 25 (68%) of the 37 patients during any cycle of MATRix, as shown in Table 1. Treatment was delayed in 18 patients (49%), most often because of infectious complications. Thirty patients (81%) completed all four cycles of MATRix, and 22 patients went on to ASCT. Two (29%) of the seven patients not completing all cycles died before all cycles could be delivered, and the remaining five patients did not complete all four cycles because of toxicity or disease progression while on treatment.
The majority of patients experienced grade 3 or 4 hematologic toxicity during the first cycle of treatment (24.3% grade 3 or 4 anemia, 73.0% grade 3 or 4 thrombocytopenia, 70.3% grade 3 or 4 neutropenia), which often persisted through each cycle of MATRix. Table 2 lists the non-hematologic toxicities experienced by patients. Notably, 24 patients (65%) developed infections on treatment, for a total of 33 individual infectious episodes. Eleven of these episodes were from bacteremia, three from viral infections requiring admission, and twelve were other infections including cellulitis, urinary tract infections, or pneumonia requiring treatment. Seven patients developed severe opportunistic infections, including blastomycosis pneumonia, herpes simplex virus encephalitis, disseminated varicella
Haematologica | 108 - April 2023 1186 LETTER TO THE EDITOR
of patients
*Routine omission prior to peripheral blood stem cell collection based on local institutional protocols.
zoster virus, Pneumocystis jiroveci pneumonia (2 patients), cytomegalovirus pneumonia and pulmonary aspergillosis. Both episodes of Pneumocystis jiroveci pneumonia occurred in patients who were not on prophylaxis, and one occurred in a patient receiving MATRix as second-line therapy. Seven patients (19%) developed episodes of febrile neutropenia without any organism being identified. Eight patients (22%) required admission to an intensive care unit during treatment, with a mean duration of stay of 7.5 days. Six patients (16%) developed a venous thromboembolic event including a catheter-associated thrombosis after the third cycle. Two patients developed deep vein thrombosis during the first cycle of treatment, with one episode occurring in a patient receiving MATRix as third-line therapy.
Thirty-five patients (95%) were alive at the end of treatment, and 30 patients had magnetic resonance imaging scans available for analysis. A complete response was seen in seven patients (23%) and a partial response in 18 patients (60%). One patient had stable disease and four patients had progressive disease. Of the 25 patients who responded to treatment, 21 went on to have consolidation therapy with ASCT with thiotepa and carmustine conditioning, and four had consolidation with whole brain radiation. Consolidation was not administered to ten patients because of disease progression, worsening performance status or ineligibility as determined by the treating physician.
The 2-year OS for all patients was 74%, and the median OS was not reached (Figure 1). The 2-year OS was 100% for patients who received ASCT and 49% for patients who did not (P<0.01). The 2-year PFS was 54% and the median PFS was
not reached. The 2-year PFS was 78% for patients who received ASCT and 21% for patients who did not (P<0.01). The median PFS was not reached for the 21 patients who received ASCT, and was 10 months (95% confidence interval: 2.2-17.9 months) for patients who did not. Cox regression analyses found that patients with an ECOG performance status greater than 1 showed a trend toward increased mortality, although this was not statistically significant (odds ratio=5.08, 95% confidence interval: 0.80-32.37). MATRix delivered as second- or third-line therapy was not associated with decreased OS (P=0.99). Six patients (16.2%) died during the study period, four from progression of their PCNSL, and two from infectious complications of treatment.
Number
100% dose 75% dose 50% dose or less Omission Cycle 1 (37 patients) Methotrexate Cytarabine Thiotepa Rituximab 33 30 31 34 0 3 1 0 0 2 0 0 4 2 5 3 Cycle 2 (36 patients) Methotrexate Cytarabine Thiotepa Rituximab 31 30 30 36 2 5 2 0 1 1 1 0 2 0 3 0 Cycle 3 (33 patients) Methotrexate Cytarabine Thiotepa Rituximab 28 25 7 32 3 5 2 0 1 2 2 0 1 1 22* 1 Cycle 4 (30 patients) Methotrexate Cytarabine Thiotepa Rituximab 27 22 23 29 2 1 0 0 1 6 1 0 0 1 6 1
Table 1. Doses delivered of the components of the MATRix protocol.
Table 2. Non-hematologic toxicities experienced during the delivery of MATRix.
Frequency (37 patients) Percentage Infection 24 64.9 Febrile neutropenia (no organism identified) 7 18.9 Venous thromboembolism 6 16.2 ICU admission 8 21.6 Acute kidney injury 9 24.3 Gastrointestinal injury/upset 7 18.9 Transaminitis 12 32.4 Mucositis 8 21.6 Neurotoxicity 10 27.0 Haematologica | 108 - April 2023 1187 LETTER TO THE EDITOR
ICU: Intensive Care Unit.
Figure 1. Survival outcomes of patients who received MATRix chemotherapy for the treatment of primary central nervous system lymphoma. (A) Two-year overall survival (74%) for all patients. (B) Two-year overall survival for patients who underwent autologous stem-cell transplantation (100%) and for patients who did not (49%) (P<0.01). (C) Two-year progression-free survival (54%) for all patients. (D). Two-year progression-free survival for patients who underwent autologous stem-cell transplantation (78%) and for patients who did not (21%) (P<0.01). ASCT: autologous stem-cell transplantation.
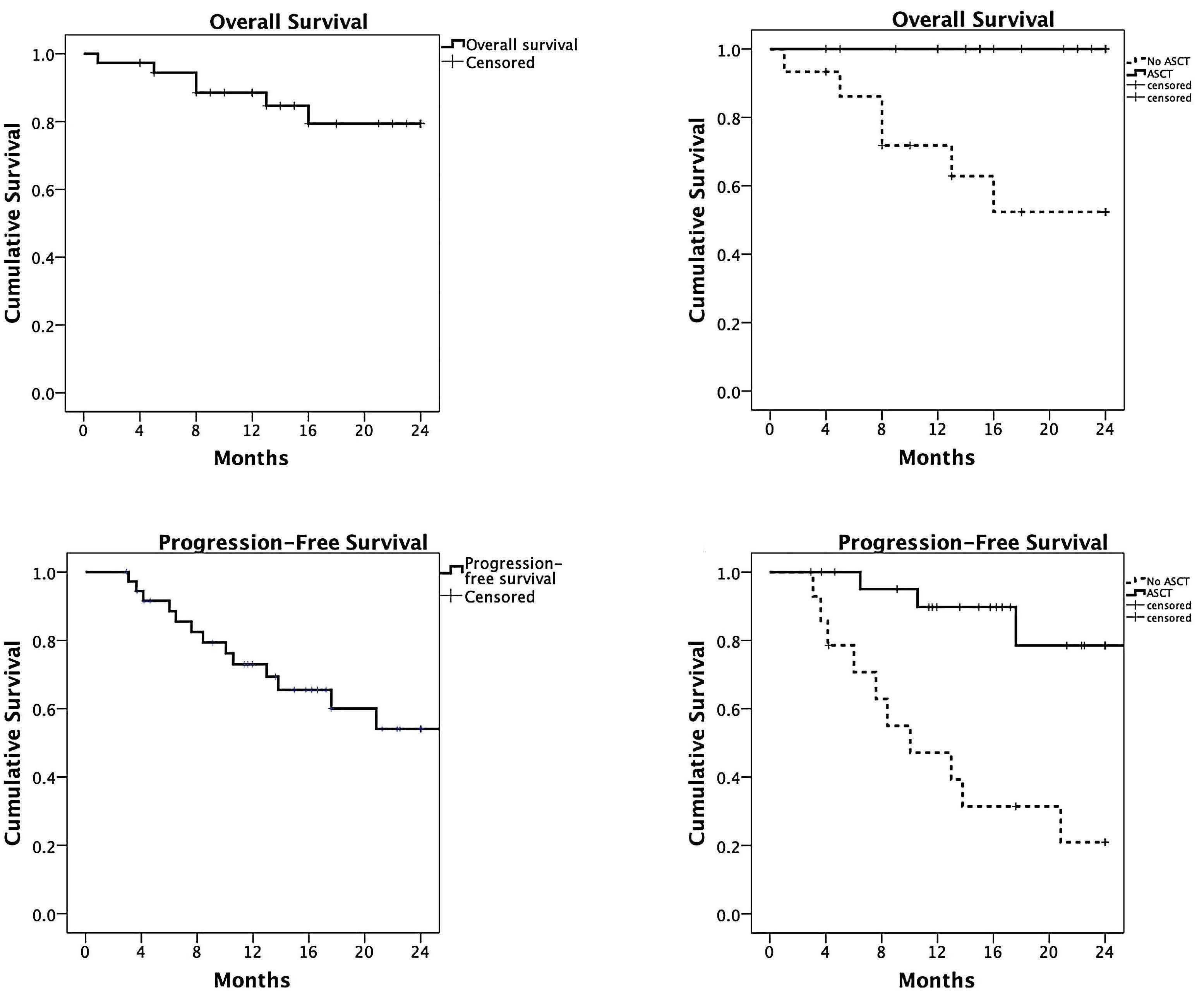
The rate of toxicity in our study was much higher than that in the IELSG32 study. Only 23% of patients in the original trial developed infections, whereas in our non-selected, real-world population, the infection rate was 65%. Although only two patients died directly from an infectious complication, four out of 25 patients with severe infectious complications were excluded from consolidation therapy because of their severe deconditioning. The infectious complications were more severe than those previously described3 and included invasive opportunistic infections such as blastomycosis and aspergillosis. In addition, 22% of patients in our cohort required admission to an intensive care unit, for a mean of 7.5 days at some point during treatment; this is a higher percentage than previously reported.1,3 This high likelihood of admission to an intensive care unit raises questions about the tolerability of MATRix, particularly in patients who are less fit.
There were a significant number of prophylactic or reactive dose reductions, and yet the rate of infections was still high. Although fewer patients achieved a complete response, ef-
ficacy was not impacted compared to the IELSG32 trial in this early analysis. High efficacy despite dose reductions was also seen in the study by Schorb et al., in which dose reductions during the first cycle also did not have an impact on PFS or OS.3 Dose reductions in the IELSG32 trial were not reported. Further studies exploring alternative dosing that could maintain efficacy with less toxicity would be of value. In their real-world experience, Schorb et al. found that 2year OS and PFS were significantly affected by both age and ECOG performance status.3 The demographics of the populations in our study and that by Schorb et al. were similar with a median age of 58 years and 62 years, respectively, and a performance status of >2 in 40% of patients in our study versus 51% in that by Schorb et al. 3 OS was not significantly affected in either trial, but newer second-line treatments such as ibrutinib and lenalidomide may enhance survival compared to that in the original IELSG32 trial.5-7
In conclusion, this is one of the first studies to look at the use of MATRix in a real-world setting with a varied pa-
A B C D
Haematologica | 108 - April 2023 1188 LETTER TO THE EDITOR
tient population. There were no restrictions to inclusion based on performance status or age, allowing for a diverse cohort. We highlight infectious complications with a focus on opportunistic infections. Efficacy was maintained even with a significant number of dose reductions. Reducing infections by carefully selecting patients and providing prophylaxis against infections should be considered.
Authors
1Faculty of Medicine, University of Toronto, Toronto, Ontario, Canada;
2Division of Medical Oncology and Hematology, St. Michael’s Hospital, Toronto, Ontario, Canada; 3AbbVie, Inc., North Chicago, IL, USA;
4Division of Medical Oncology and Hematology, Princess Margaret Cancer Centre, Toronto, Ontario, Canada and 5Sunnybrook Health Sciences Centre, Odette Cancer Centre, Toronto, Ontario, Canada
#AP and NB contributed equally as co-senior authors. °AbbVie had no role in this study and solely reflects this author’s current affiliation.
References
1. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol. 2016;3(5):e217-e227.
2. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Long-term efficacy, safety and neurotolerability of MATRix regimen followed by autologous transplant in primary CNS lymphoma: 7-year results of the IELSG32 randomized trial. Leukemia. 2022;36(7):1870-1878.
3. Schorb E, Fox CP, Kasenda B, et al. Induction therapy with the MATRix regimen in patients with newly diagnosed primary diffuse large B-cell lymphoma of the central nervous system –an international study of feasibility and efficacy in routine clinical practice. Br J Haematol. 2020;189(5):879-887.
4. Abrey LE, Batchelor TT, Ferreri AJM, et al. Report of an
Correspondence:
A. SULEMAN - asule095@uottawa.ca
https://doi.org/10.3324/haematol.2022.282014
Received: August 29, 2022.
Accepted: November 23, 2022.
Early view: December 1, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose. AKD is currently affiliated with AbbVie, but this company had no role in the study.
Contributions
AS and AKD collected data for the manuscript. JL, AP and NB conceptualized the manuscript. AS, JL, LKH, AKD, MC, RK, AP and NB all contributed to writing the manuscript.
Data-sharing statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
international workshop to standardize baseline evaluation and response criteria for primary CNS lymphoma. J Clin Oncol. 2005;23(22):5034-5043.
5. Ghesquieres H, Chevrier M, Laadhari M, et al. Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective “proof of concept” phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA). Ann Oncol. 2019;30(4):621-628.
6. Choquet S, Houillier C, Bijou F, et al. Ibrutinib monotherapy in relapse or refractory primary CNS lymphoma (PCNSL) and primary vitreo-retinal lymphoma (PVRL). Result of the interim analysis of the iLOC phase II study from the LYSA and the French LOC Network. Blood. 2016;128(22):784.
7. Grommes C, Nayak L, Tun HW, Batchelor TT. Introduction of novel agents in the treatment of primary CNS lymphoma. Neuro Oncol. 2019;21(3):306-313.
Adam Suleman,1 Jiajia Liu,1 Lisa K. Hicks,1,2 Adi Klil Drori,3° Michael Crump,1,4 Robert Kridel,1,4 Anca Prica1,4# and Neil Berinstein1,5#
Haematologica | 108 - April 2023 1189 LETTER TO THE EDITOR
Modern, real-world patterns of care and clinical outcomes among patients with newly diagnosed diffuse large B-cell lymphoma with or without double/triple-hit status
in the United States
Patients with diffuse large B-cell lymphoma (DLBCL) and double-hit or triple-hit cytogenetics (DHL/THL) historically had a poor prognosis when treated with the standard front-line regimen consisting of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).1,2 Patterns of care and subsequent clinical outcomes of patients with DHL/THL had been largely determined from multiple retrospective studies, most without any direct comparisons to patients without DHL/THL. A single-arm, phase II study utilizing dose-adjusted rituximab, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin (R-EPOCH) in 24 patients with DHL/THL showed a 4-year event-free survival of 73%, which was significantly higher than that of historical controls.3 These data led to the adoption of dose-adjusted R-EPOCH as the preferred treatment for DHL/THL. A recent phase III study comparing R-CHOP versus dose-adjusted R-EPOCH included a small cohort of patients with DHL/THL, which precluded a comparison of efficacy.10
In this study, we aimed to evaluate the patterns of care and clinical outcomes of DLBCL from 2011 to the present day among a contemporary cohort of patients with newly diagnosed DLBCL. We also compared the outcomes of patients with DHL/THL DLBCL morphology treated with RCHOP and R-EPOCH regimens in this population. This retrospective, observational study utilized the nationwide Flatiron Health electronic health record-derived database. This is a longitudinal database containing de-identified patient-level data, curated via a combination of tech-enabled search engines and abstractors.4 During the study period, data originated from approximately 280 US cancer clinics (~800 sites of care). We included adult patients (>18 years at diagnosis) with a confirmed diagnosis of the following histological subtypes of lymphoma: DLBCL not otherwise specified, DHL, Epstein-Barr viruspositive DLBCL, and T-cell/histiocyte-rich large B-cell lymphoma. We included patients diagnosed on or after January 1, 2011 who had at least two electronic health record-documented visits on or after that date, with at least 6 months of potential follow-up (diagnosis prior to January 31, 2021).
Cytogenetic testing groups were defined by the documentation of cytogenetic test results based on fluorescence in situ hybridization (FISH) studies and/or karyotyping be-
fore or up to 6 weeks after initiation of first-line treatment. Patients were categorized into three groups: documented DHL/THL, documented testing but negative for DHL/THL (not DHL/THL), and no documented FISH/karyotype testing results (no documented testing for MYC).
The primary outcome measure was real-world overall survival (OS), defined as time from initiation of first-line treatment to date of death or censoring at last confirmed activity.5 The secondary outcome was time to next treatment (TTNT) as a proxy for progression-free survival, defined as time from initiation of first-line treatment to second-line treatment, or date of death (whichever came first), or censoring at last confirmed activity. Adjusted covariates included age, sex, race/ethnicity, Eastern Cooperative Oncology Group (ECOG) performance status at first-line treatment initiation, elevated lactate dehydrogenase and more than one site of extranodal disease. ECOG performance status was based on the value reported within 30 days prior to and 7 days after initiation of first-line treatment.
We used the Kaplan-Meier method and log-rank tests to estimate real-world OS and TTNT by cytogenetic testing group among all eligible patients with DLBCL. Among patients with DHL/THL who received R-CHOP or R-EPOCH as first-line treatment, the Kaplan-Meier method was used to estimate median real-world OS and TTNT. Multivariable Cox proportional hazards models were used to assess the impact of first-line treatment group (R-CHOP vs. R-EPOCH) on real-world OS and TTNT, adjusting for age, sex, race, ECOG performance status, stage, year of diagnosis, extranodal disease status and elevated lactate dehydrogenase levels. Statistical analyses were conducted using RStudio 3.6.1 software.
We included 6,412 patients with DLBCL in our total cohort (Table 1, Online Supplementary Figure S1A), with a mix of patients being cared for in the community (87%) and academic settings (13%). Among them, 2,604 (40.6%) patients did not have any documented cytogenetic testing before or up to 6 weeks after initiation of first-line treatment. Among patients with documented cytogenetic testing, 304 (8%) had DHL/THL and 3,504 (92%) patients did not (Table 1). Among the cases of DHL/THL, there were 176 (57.9%) patients with MYC/BCL2, 57 (18.8%) patients with MYC/BCL6, and 71 (23.4%) patients with MYC/BCL2/BCL6
Haematologica | 108 April 2023 1190 LETTER TO THE EDITOR
ECOG PS at initiation of first-line therapy, N
N (%)
DHL: double-hit lymphoma; THL: triple-hit lymphoma; ECOG PS: Eastern Cooperative Oncology Group performance status; LDH: lactate dehydrogenase; ULN: upper limit of normal; R-CHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; R-EPOCH: rituximab, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin.
Baseline characteristics All N=6,412 No documented tests N=2,604 Not DHL/THL N=3,504 DHL/THL N=304 P overall Age at diagnosis, N (%) ≤40 years 41-60 years 61-75 years >75 years 279 (4.4) 1,490 (23.2) 2,870 (44.8) 1,773 (27.7) 86 (3.3) 573 (22.0) 1,145 (44) 800 (30.7) 181 (5.2) 834 (23.8) 1,594 (45.5) 895 (25.5) 12 (3.9) 83 (27.3) 131 (43.1) 78 (25.7) <0.001 Sex, N (%) Female Male 2,796 (43.6) 3,615 (56.4) 1,200 (46.1) 1,403 (53.9) 1,466 (41.8) 2,038 (58.2) 130 (42.8) 174 (57.2) 0.004 Race/ethnicity, N (%) Non-Hispanic white Non-Hispanic black Hispanic or Latino Asian Other/unknown 4,307 (67.2) 378 (5.9) 402 (6.3) 140 (2.2) 1,185 (18.5) 1,791 (68.8) 150 (5.8) 167 (6.4) 42 (1.6) 454 (17.4) 2,313 (66.0) 215 (6.1) 216 (6.2) 94 (2.7) 666 (19.0) 203 (66.8) 13 (4.3) 19 (6.2) 4 (1.3) 65 (21.4) 0.042
0 1 ≥2 Unknown 1,528 (23.8) 1,451 (22.6) 681 (10.6) 2,752 (42.9) 496 (19.0) 515 (19.8) 282 (10.8) 1,311 (50.3) 945 (27.0) 870 (24.8) 360 (10.3) 1,329 (37.9) 87 (28.6) 66 (21.7) 39 (12.8) 112 (36.8) <0.001 Cell of origin, N (%) Germinal B cell Non-germinal B cell/activated B cell Unknown/not documented 2,096 (32.7) 1,433 (22.3) 2,883 (45.0) 660 (25.3) 455 (17.5) 1,489 (57.2) 1,267 (36.2) 949 (27.1) 1,288 (36.8) 169 (55.6) 29 (9.5) 106 (34.9) <0.001 Stage at diagnosis, N (%) I-II III-IV Unknown 1,762 (27.5) 3,283 (51.2) 1,367 (21.3) 717 (27.5) 1,247 (47.9) 640 (24.6) 996 (28.4) 1,854 (52.9) 654 (18.7) 49 (16.1) 182 (59.9) 73 (24.0) <0.001 B symptoms, N (%) Other/unknown B symptoms With B symptoms Without B symptoms 834 (13.0) 2,325 (36.3) 3,253 (50.7) 417 (16.0) 894 (34.3) 1,293 (49.7) 384 (11.0) 1,317 (37.6) 1,803 (51.5) 33 (10.9) 114 (37.5) 157 (51.6) <0.001 ≥1 Extra nodal site, N (%) Yes No/unknown 3,527 (55.0) 2,885 (45.0) 1,405 (54.0) 1,199 (46.0) 1,947 (55.6) 1,557 (44.4) 175 (57.6) 129 (42.4) 0.300 Transformation, N (%) Yes No/unknown 1,104 (17.2) 5,308 (82.8) 450 (17.3) 2,154 (82.7) 578 (16.5) 2,926 (83.5) 76 (25.0) 228 (75.0) 0.001 LDH ratio at initiation of first-line treatment, N (%) Normal Elevated, up to 3xULN >3x ULN Unknown 1,846 (28.8) 1,500 (23.4) 226 (3.5) 2,840 (44.3) 678 (26.0) 505 (19.4) 82 (3.1) 1,339 (51.4) 1,106 (31.6) 902 (25.7) 114 (3.3) 1,382 (39.4) 62 (20.4) 93 (30.6) 30 (9.9) 119 (39.1) <0.001 Duration from diagnosis to initiation of first-line treatment, N (%) ≤14 days >14 days 2,210 (34.5) 4,202 (65.5) 958 (36.8) 1,646 (63.2) 1,126 (32.1) 2,378 (67.9) 126 (41.4) 178 (58.6) <0.001
treatment,
R-CHOP R-EPOCH Other 4,643 (72.4) 456 (7.1) 1,313 (20.5) 1,917 (73.6) 92 (3.5) 595 (22.8) 2,629 (75) 233 (6.6) 642 (18.3) 97 (31.9) 131 (43.1) 76 (25.0) <0.001
(%)
First-line
Table 1. Baseline characteristics by cytogenetic testing groups.
Haematologica | 108 April 2023 1191 LETTER TO THE EDITOR
(THL) rearrangements. The annual rates of documented cytogenetic testing increased consistently over the last decade, from 37% in 2011 to 82% in 2021 (Online Supplementary Figure S1B).
Among patients with documented cytogenetic test results, patients with DHL/THL were more likely to be younger, have a germinal center B-cell subtype, stage III-IV disease, transformed lymphoma, elevated lactate dehydrogenase, and to have started first-line treatment within 14 days of diagnosis, when compared to patients without DHL/THL. Among patients with DHL/THL, those who received R-CHOP as firstline treatment were more likely to be older and have THL, compared with patients who received R-EPOCH. Other baseline characteristics were similar between the two treatment groups.
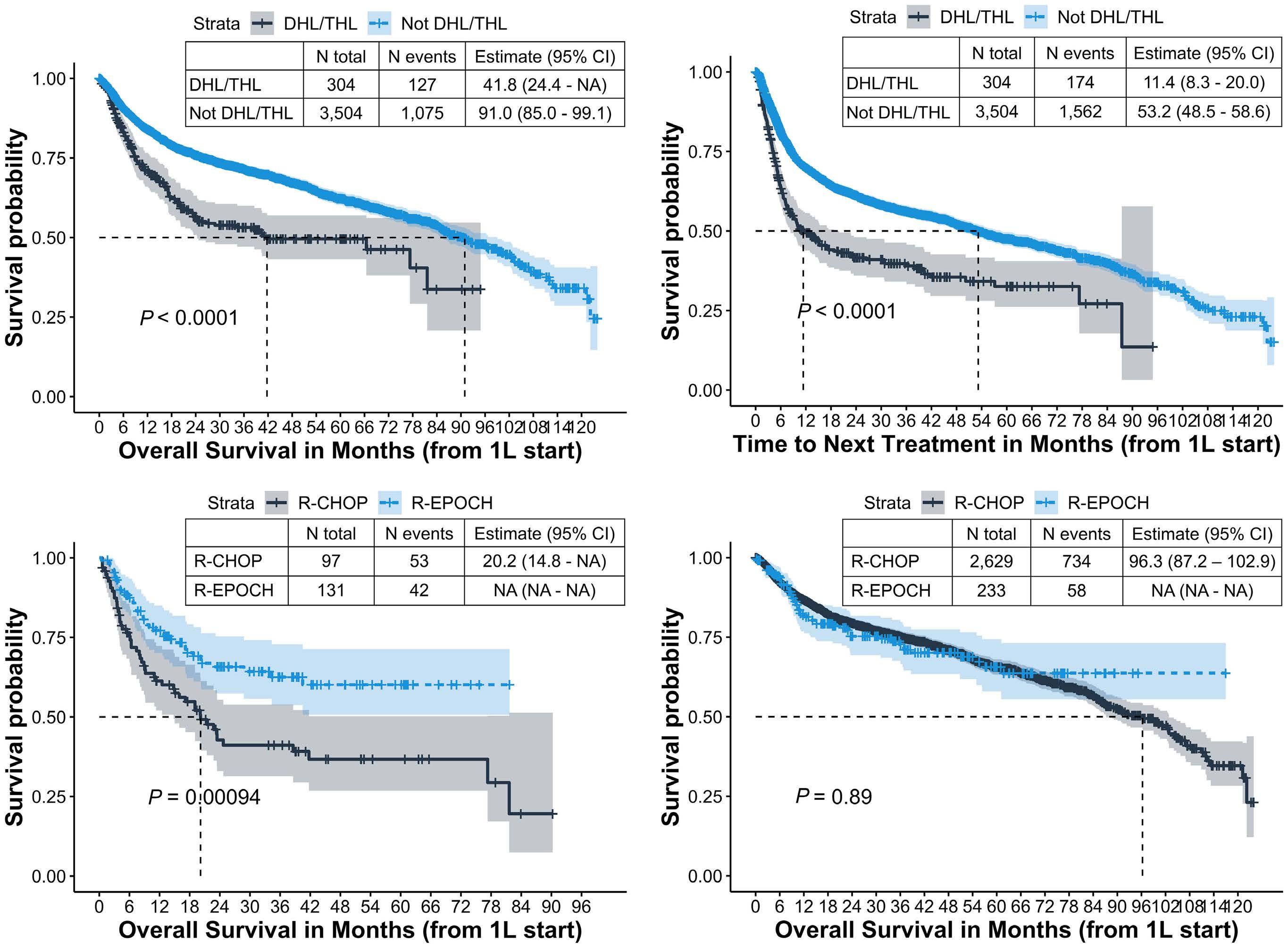
R-CHOP was the most common first-line treatment overall,
but the proportion of patients who received first-line RCHOP was much lower in the DHL/THL cohort (29%) than in the group of patients who did not have DHL/THL (69%) or did not have documented test results (70%) (Online Supplementary Figure S2). Patients with DHL/THL were more likely to receive R-EPOCH as first-line treatment (43%) than were patients who did not have DHL/THL (4.7%). Choices for second- and third-line treatments varied widely between all groups; second-line therapy was received by 38% of patients with DHL/THL, by 27% without DHL/THL, and by 28% of patients without documentation of cytogenetic testing (Online Supplementary Figure S2). The proportion of patients who received second-line therapy or underwent transplantation was higher among patients with DHL/THL than among those without DHL/THL.
The median duration of follow-up for the entire cohort was
THL: triple-hit lymphoma; 95% CI: 95% confidence interval; 1L; first-line treatment; NA: not available; R-CHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; R-EPOCH: rituximab, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin.
B A C D
Figure 1. Kaplan-Meier curves of overall survival. (A, B) Real-world overall survival (A) and time to next treatment (B) stratified by gene-rearrangement status among patients who had undergone cytogenetic testing. (C, D) Real-world overall survival stratified by first-line treatment (R-CHOP vs. R-EPOCH) among patients with diffuse large B-cell lymphoma divided by cytogenetic group into those with double/triple hit lymphoma (C) and those without double/triple hit lymphoma (D). DHL: double-hit lymphoma;
Haematologica | 108 April 2023 1192 LETTER TO THE EDITOR
Figure 2. Multivariable analysis among cytogenetic testing groups.
(A, B) Adjusted hazard ratios for real-world overall survival by firstline treatment among patients with diffuse large B-cell lymphoma documented as having double/triple-hit lymphoma (A) or not having double/triple hit lymphoma (B). 1L: first-line treatment; R-CHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; R-EPOCH: rituximab, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin; ECOG PS: Eastern Cooperative Oncology Group performance status; LDH: lactate dehydrogenase; AIC: Akaike information criterion.
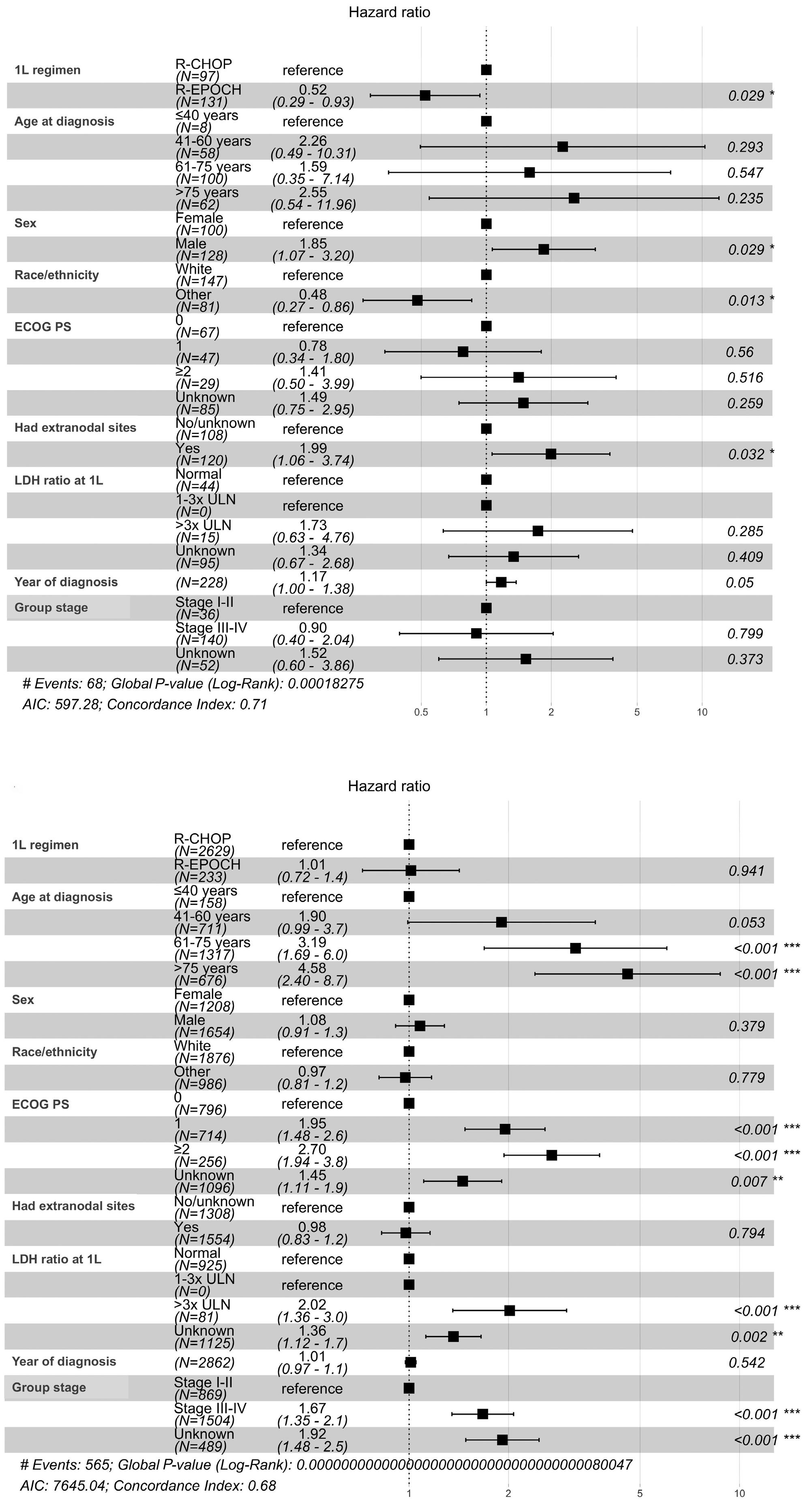
A B Haematologica | 108 April 2023 1193 LETTER TO THE EDITOR
27.5 months (interquartile range, 10.2-56.3). Among all the patients, those with DHL/THL had worse real-world OS from initiation of first-line treatment (median: 41.7 months; 95% confidence interval [95% CI]: 24.4-not available [NA]), compared with patients who did not have DHL/THL (median: 91.0 months; 95% CI: 85.0-99.1) and patients without documented cytogenetic tests (median: 88.1 months; 95% CI: 83.3-95.0) (P<0.001) (Figure 1A and data not shown). Similar patterns were observed for TTNT; patients with DHL/THL had a shorter median TTNT from first-line treatment initiation (11.4 months; 95% CI: 8.3-20.0), compared with patients who did not have DHL/THL (53.2 months; 95% CI: 48.5-58.6) and patients without documented cytogenetic tests (50.7 months; 95% CI: 45.7-57.3) (P<0.001) (Figure 1B).
Among patients with DHL/THL who received R-CHOP (n=97) or R-EPOCH (n=131) as first-line treatment, patients receiving R-EPOCH had significantly longer real-world OS (median: not reached) compared with patients receiving RCHOP (median: 20 months; 95% CI: 14.8-NA; P=0.001) (Figure 1C). Among patients who did not have DHL/THL, there was no difference in real-world OS between patients treated with R-CHOP or R-EPOCH (Figure 1D).
After adjusting for demographic and clinical characteristics among patients with DHL/THL, those who received REPOCH had a 50% lower risk of death (adjusted hazard ratio: 0.50; 95% CI: 0.33-0.77) than that of patients receiving RCHOP as first-line treatment (Figure 2). Patients with an ECOG performance status greater than 2 (n=29) and male patients (n=128) also had higher hazard ratios for death. Additional sensitivity analysis adjusting for time to treatment initiation, or excluding patients who received methotrexate, patients undergoing transplant in first-line, those aged ≥80 years, those with transformed DLBCL, and patients who switched therapies within 18 weeks of starting first-line treatment did not change the results materially (Online Supplementary Table S1). We also conducted multiple imputation analysis when data were missing in >10% cases as well as inverse probability of treatment weighting analysis; however, the association of improved OS with R-EPOCH among DHL/THL persisted.
To the best of our knowledge, this is the largest direct comparison of the outcomes of DLBCL with and without DHL/THL among more than 6,000 patients in the real-world setting. With rapidly evolving standards of care in DLBCL in terms of cytogenetic testing and treatments, this study gives us the most recent assessment of survival outcomes. Although we found that 40% of newly diagnosed DLBCL cases in the entire period of our study did not have documented cytogenetic test results, we noted that testing rates increased steadily over time, with almost 80% of cases having documented testing in 2021.
Our study was able to compare outcomes between the two commonly used regimens, R-CHOP and R-EPOCH, in newly
diagnosed DHL/THL with DLBCL morphology. Prior studies showed an improved progression-free survival, but not OS, with the use of R-EPOCH compared with R-CHOP in DHL/THL.2,6 In a real-world setting we found that patients treated with R-EPOCH had a longer median OS compared with those treated with R-CHOP, even after adjusting for demographic and clinical factors. Furthermore, the longer duration of OS in our study compared with older studies for DHL/THL suggests an improvement in therapies over the last decade.
The biggest strength of our study is the large number of diverse USA oncology practices and patients involved, which allowed a pragmatic assessment of outcomes in the realworld setting.4 Furthermore, our source data provided granularity of clinical variables at the patient level, enabling more robust outcome analyses across several sub-cohorts of interest than possible from prior single-center or pooled analyses, and we used a real-world composite mortality endpoint with high reliability.5
However, our study also has limitations and potential for biases. We mitigated for potential immortal time bias in testing by limiting our inclusion criteria to a testing window at diagnosis. Missing data was another limitation, but we conducted sensitivity analysis using multiple imputation which did not change results significantly. Although we controlled for several known prognostic variables in a multivariate approach, we were unable to account for additional factors that may contribute to the choice of treatments such as patient/physician preference, comorbidities, and overall health status. Additionally, we did not have data on the specific MYC translocation partner (IgH or non-IgH), which precluded assessment of its impact on prognosis, as shown before.7
In summary, our data showed, in a real-world setting, that R-EPOCH was associated with better OS compared with RCHOP among patients with DHL/THL, and no difference among patients without DHL/THL. Our study also suggests that cytogenetic assessment may be an imperfect prognostic tool, with 5-year survival rates among DHL/THL cases of over 60% in the R-EPOCH group and 30% in the R-CHOP cohort. In the last 5 years, novel prognostic categories beyond cell of origin and DHL/THL status have been defined using next-generation sequencing,27-29 and may offer better opportunities for designing future prospective trials as well as real-world studies.
Authors Gaurav Goyal,1 Tylan Magnusson,2 Xiaoliang Wang,3 James Roose,3 Mayur Narkhede1
Erlene Seymour3 1Division
Haematologica | 108 April 2023 1194 LETTER TO THE EDITOR
and
of Hematology-Oncology, University of Alabama at
Birmingham, Birmingham, AL; 2Department of Internal Medicine, University of Alabama at Birmingham, Birmingham, AL and 3Flatiron Health, New York, NY, USA
Correspondence:
G. GOYAL - ggoyal@uabmc.edu
https://doi.org/10.3324/haematol.2022.281461
Received: May 24, 2022.
Accepted: November 24, 2022.
Early view: December 1, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
MN has received institutional research funding from Gilead/Forty seven, TG Therapeutics, Roche/Genentech and EUSA
References
1. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol. 2017;35(20):2260-2267.
2. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in doublehit lymphoma: a multicenter retrospective analysis. Blood. 2014;124(15):2354-2361.
3. Dunleavy K, Fanale MA, Abramson JS, et al. Dose-adjusted EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) in untreated aggressive diffuse large B-cell lymphoma with MYC rearrangement: a prospective, multicentre, single-arm phase 2 study. Lancet Haematol. 2018;5(12):e609-e617.
4. Ma X, Long L, Moon S, Adamson BJS, Baxi SS. Comparison of population characteristics in real-world clinical oncology databases in the US: Flatiron Health, SEER, and NPCR. medRxiv.
pharmaceuticals.
Contributions
GG and ES conceived and designed the study; GG, TM, WW, JR and ES analyzed and interpreted the data; all authors were involved in writing the manuscript, gave their final approval of the submitted version and are accountable for all aspects of the work.
Funding
This work was supported by the Walter B. Frommeyer, Jr., Fellowship Award in Investigative Medicine, University of Alabama at Birmingham (to GG).
Data-sharing statement
The data that support the findings of this study originated from Flatiron Health, Inc. The de-identified data may be made available upon request, and are subject to a license agreement with Flatiron Health; interested researchers should contact DataAccess@flatiron.com to determine licensing terms.
May 30. doi: 10.1101/2020.03.16.20037143 [preprint, not peerreviewed].
5. Zhang Q, Gossai A, Monroe S, Nussbaum NC, Parrinello CM. Validation analysis of a composite real-world mortality endpoint for patients with cancer in the United States. Health Serv Res. 2021;56(6):1281-1287.
6. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, doseescalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol. 2015;170(4):504-514.
7. Rosenwald A, Bens S, Advani R, et al. Prognostic significance of MYC rearrangement and translocation partner in diffuse large B-cell lymphoma: a study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol. 2019;37(35):3359-3368.
Haematologica | 108 April 2023 1195 LETTER TO THE EDITOR
Time without transfusion reliance: a novel patient-centric metric for new therapies in myelodysplastic syndromes
Myelodysplastic syndromes (MDS) are clonal myeloid malignancies associated with ineffective hematopoiesis, consequent cytopenias, and for many progression to acute myeloid leukemia (AML), which is associated with shortened survival and worse health-related quality of life (HRQoL).1,2 Most patients with MDS are affected by anemia and its attendant symptoms, which can lead to blood transfusion dependency. Blood transfusions can be burdensome, costly, and are major contributors to poor HRQoL in patients with MDS.2,3
The efficacy of new therapies in MDS should be evaluated using both standardized measures of response rates and overall survival (OS), along with outcomes reflecting patient experience, including disease symptoms and HRQoL.4 However, the demonstration of treatment benefit through patient-reported outcomes (PRO) has been challenging in the evaluation of novel therapies in hematologic malignancies. Some of the limitations of PRO in hematologic malignancy trials to date include lack of good quality HRQoL data, suboptimal timing of assessment of HRQoL methods, use of weak HRQoL instruments, low compliance over time of patients enrolled on prospective clinical trials, and inadequate statistical analyses.1,2 Further, HRQoL measures do not distinguish between different causes of anemia in MDS, so similar HRQoL may be observed due to improvement of anemia from treatment versus continued red blood cell (RBC) transfusions. To date, analyses pertaining to RBC transfusions largely include descriptions of the number of transfused units received or aggregated assessments estimating the duration of transfusion-independence for each patient. Such approaches do not account for possible differential followup resulting from unbalanced efficacy between trial treatment arms, such as disease progression or OS, thus confounding treatment arm comparisons. We propose a new aggregated measure combining clinical outcomes (OS, transformation to AML) and transfusiondependency: the time without transfusion reliance (TWiTR) approach, inspired by the time without symptoms and toxicity (TWiST) analysis.5,6 In TWiST analyses,6-8 periods of treatment toxicity and disease progression assumed to reduce HRQoL are subtracted from the OS time for each patient. Similar to the TWiST approach, the TWiTR analysis subtracts periods of time from the OS of each patient when HRQoL is assumed to have deteriorated: i) the time period when patients experience disease progression and ii) the time period when patients are transfusion-dependent. Hence, three health states are defined
in the TWiTR analysis: a transfusion dependence (TD) state that is defined as the sum of all TD periods experienced by the patient (replacing the toxicity state in the TWiST approach); a relapse (REL) state that is defined as the time between disease progression and death; and the TWiTR state that is defined as the time without TD or REL. Health state durations (OS or event-free survival [EFS] minus TD and REL) are then calculated using KaplanMeier estimates. The mean duration in each state can be estimated by the area under each survival curve obtained with Kaplan-Meier estimates9 and can be weighted using a utility value, which can be derived from any utility-based instrument, attached to each state to obtain a quality-adjusted TWiTR (Q-TWiTR) that reflects both the duration of each state, and the corresponding HRQoL experienced by patients in this state.5,7,9 A bootstrap approach10 can be applied to estimate the 95% confidence interval (CI) for the mean Q-TWiTR for each arm and mean Q-TWiTR difference between arms.
The TWiTR approach was implemented in the context of a randomized phase II study of pevonedistat plus azacitidine (PEVO+AZA) versus azacitidine (AZA) alone in higher-risk MDS (HR-MDS), chronic myelomonocytic leukemia (CMML), and low-blast AML naïve to hypomethylating agents (P2001; clinicaltrials.gov Identifier: NCT02610777 11). The primary endpoint of the study was EFS, defined as transformation to AML or death for HR MDS/CMML, or death for low-blast AML, which was used to define the health states in the TWiTR analysis. The P-2001 study included two HRQoL measures: the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire-Core 30 items (QLQ-C30), an instrument designed for use in a wide range of cancer patient populations;12 and the EQ-5D-5L, a self-administered instrument developed for use as a generic, preferencebased measure of health outcomes, from which utilities can be derived.13
The P-2001 study enrolled 120 patients in a 1:1 randomization (PEVO+AZA: n=58, AZA: n=62). Of the total enrolled patients, 63 patients with HR-MDS had an HRQoL assessment at baseline, and at least one post-baseline HRQoL assessment. HRQoL measures were associated with different clinical outcomes independently of treatment arms (e.g., improved HRQoL in patients experiencing a complete remission compared to baseline; worse HRQoL in patients whose MDS transformed to AML); however, the HRQoL measures were similar between both treatment arms (PEVO+AZA vs. AZA) (Online Supplementary Figure S2).1, 11
Haematologica | 108 April 2023 1196 LETTER TO THE EDITOR
In this study, the TD state was defined according to the 2006 International Working Group response criteria in MDS,14 which defines TD as any transfusion (blood or platelets) within an 8-week period. In order to calculate the duration of the TD state while incorporating multiple episodes of TD during the follow-up period, the number of days of all TD periods after the start of treatment and before death or disease progression were summed. Thus, a TD period started at the first transfusion of a series of transfusions and ended after a period of 8 weeks without any transfusion. An ongoing TD period at the time of death or disease progression ended at the date of the event (Online Supplementary Figure S1).
The TWiTR analysis was conducted in the subpopulation of HR-MDS patients. The TD state was defi ned as the total of TD periods from the start of treatment period and before transformation to AML or death (EFS). The REL state was defi ned as the time between EFS and death (OS). The TWiTR state was defined as the time without TD or REL (i.e., EFS minus TD). For each arms, KaplanMeier curves were calculated for each health state to partition the OS time. The mean time spent by patients in each state was estimated using the area under the Kaplan-Meier curve. The observed utility values for each health state (TD, REL, and TWiTR) were calculated in HRMDS patients. The TWiTR analysis revealed that HR-MDS
Figure 1. Partitioned survival plot of the transfusion dependence, time without transfusion reliance and relapse states in highrisk myelodysplatic syndromes subpopulation from the P-2001 study who had a patient-reported outcome assessment at baseline and at least one post-baseline assessment (N=63). TD: transfusion dependence; TWiTR: time without transfusion reliance; REL: relapse.
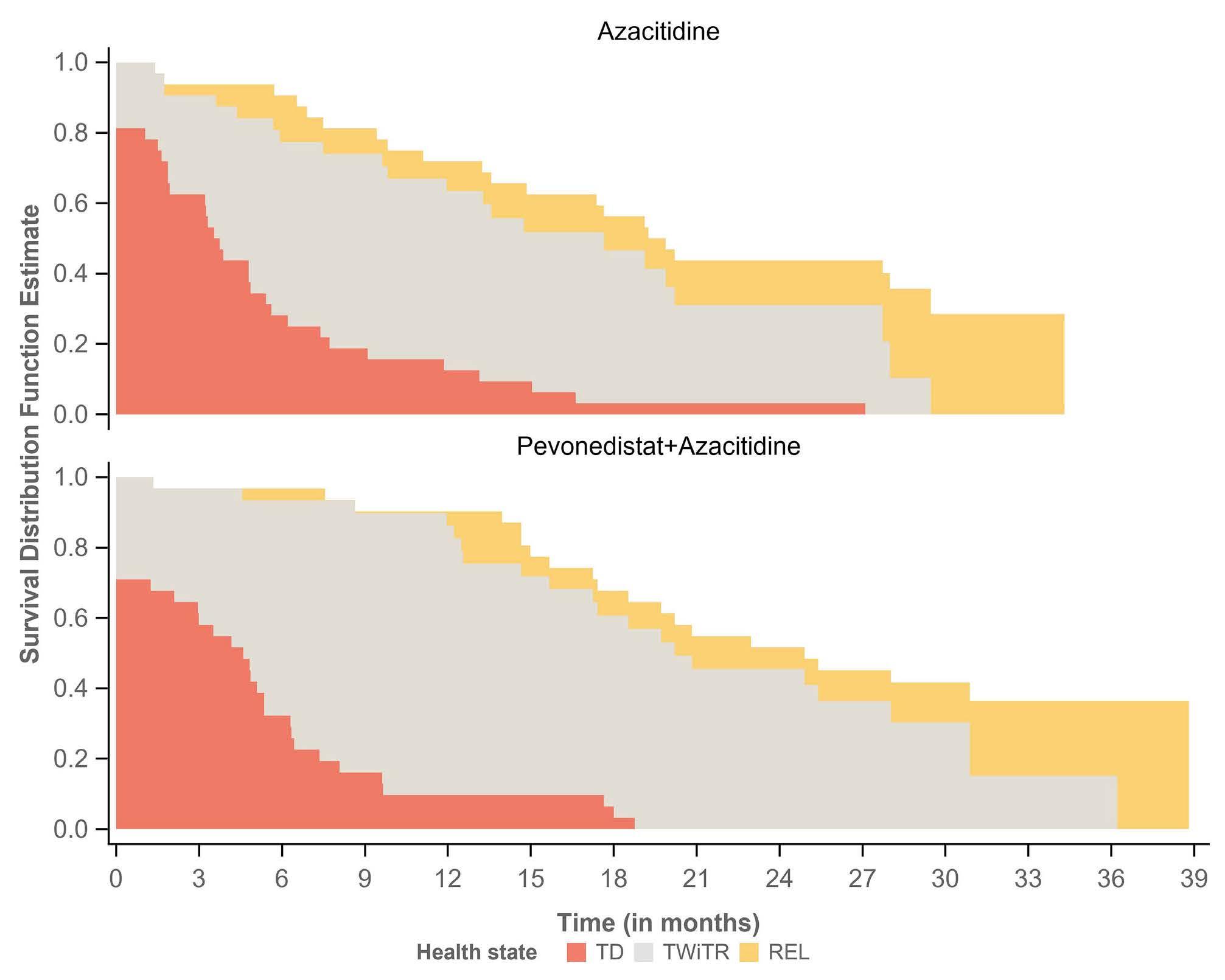
Table 1. Mean duration (in months) of transfusion dependence, time without transfusion reliance and relapse health states of the time without transfusion reliance analysis in the high-risk myelodysplastic syndromes subpopulation from the P-2001 study (n=63).
*95% CI: 95% confidence interval estimated using a bootstrap approach; AZA: azacitidine arm; PEVO+AZA: pevonedistat+azacitidine arm; TD: transfusion dependence; TWiTR: time without transfusion reliance; REL: relapse. Difference PEVO+AZA vs. AZA: difference in mean duration in months between pevonedistat+azacitidine arm and azacitidine alone arm for each health state of the TWiTR analysis.
Health state PEVO+AZA (N=31) AZA (N=32) Difference PEVO+AZA vs. AZA Duration in each state in months, mean (95% CI ) TD TWiTR REL 5.0 (3.1-7.0) 16.0 (13.2-18.8) 1.8 (-1.5 to 5.1) 5.3 (3.2-7.5) 11.2 (8.2-14.3) 2.9 (-0.8 to 6.6) -0.3 (-3.2 to 2.5) 4.8 (0.7-8.9) -1.1 (-6.0 to 3.8)
Haematologica | 108 April 2023 1197 LETTER TO THE EDITOR
patients in the PEVO+AZA arm had a significantly longer duration of TWiTR compared with the AZA arm (Table 1: mean TWiTR duration 16.0 months vs. 11.2 months, respectively). As the mean TD duration was similar in both arms, for this study the TWiTR benefit was largely driven by a longer EFS in the PEVO+AZA arm (Table 1). Additionally, the mean EQ-5D-5L utility value was higher for the TWiTR state than for the TD and REL states (mean EQ-5D-5L utility value of 0.82 in TWiTR state vs. 0.77 in TD and REL states), suggesting that transfusion dependence may have negatively impacted HRQoL in MDS regardless of treatment. The partitioned survival plot from the TWiTR approach provides an overall picture of the duration in months in each health state in the HR-MDS subpopulation (Figure 1). In conclusion, we have demonstrated a “proof of concept” application of the novel TWiTR analysis, a promising, innovative, patient-centered metric for the evaluation of new therapies for MDS in which transfusion dependence is clinically burdensome. TWiTR analysis involves a rigorous methodology that can be used as an assessment for the evaluation and comparison of novel treatment strategies in MDS and may serve as an effective complement to well-designed HRQoL analyses. Transfusion independence is a critical endpoint for improving the HRQoL of patients with MDS, even though hematologic improvement/transfusion independence may not always be included as an endpoint in HR-MDS clinical trials. Further application and investigation of TWiTR is warranted to determine its utility in MDS. Additionally, the TWiTR analysis can be potentially applied to prospective studies in lower-risk MDS in which transfusion independence and HRQoL may be the ultimate goal.15
Authors
Joshua F. Zeidner,1 Flora Mazerolle,2 Jonathan Norton,3 Antoine Regnault,2 Fjoralba Kristo,3 Heather Romero,3 Robert J. Fram,3 Douglas V. Faller,3 Mehul Dalal,3 Lionel Ades4,5 and Mikkael A. Sekeres6
1University of North Carolina, Lineberger Comprehensive Cancer Center, Chapel Hill, NC, USA; 2Modus Outcomes, a division of THREAD, Lyon, France; 3Millennium Pharmaceuticals, Inc. a wholly owned subsidiary of Takeda Pharmaceutical Ltd. Company, Cambridge, MA, USA; 4AP-HP, Hôpital Saint Louis, Paris, France; 5University of Paris, INSERM U944, Paris, France and 6Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL, USA
Correspondence:
J.F. ZEIDNER - joshua_zeidner@med.unc.edu
https://doi.org/10.3324/haematol.2022.281856
Received: July 28, 2022.
Accepted: November 28, 2022.
Early view: December 7, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
JFZ has received consulting fees from AbbVie, Gilead, and Servier; honoraria from advisory boards from AbbVie, Bristol Myers Squibb, Daiichi Sankyo, Genentech, Gilead, Immunogen, Servier, Shattuck Labs; research funding from AbbVie, Arog, ASTEX, Gilead, Merck, Stemline, Sumitomo Dainippon Pharma, and Takeda. AR and FM are employees of Modus Outcomes, a division of THREAD, which received payment from Millennium Pharmaceuticals, Inc., Cambridge, MA, a wholly-owned subsidiary of Takeda Pharmaceutical Ltd. Company. JN, FK, HR, RJF, DVF, and MD are all employees of Millennium Pharmaceuticals, Inc., a wholly-owned subsidiary of Takeda Pharmaceutical Ltd. Company. RJF is an equity holder of Takeda, BMS, Baxter, Pfizer, Gilead, Teva, Viatris, Medtronics, Zimmer Biomet Hldgs Inc, and Zimvie Inc. DVF discloses stock options in Viracta Therapeutics, Inc., Briacell, Inc.; former employment, and membership on an entity's board of directors or advisory committees at Viracta Therapeutics, Inc. LA discloses research funding from Celgene, Jazz; honoraria from Jazz, Takeda, Celgene/BMS, Novartis, AbbVie. MAS discloses membership on an entity's board of directors or advisory committees for BMS, Takeda/Millenium and Novartis.
Contributions
All authors had substantial contribution in the drafting of the manuscript content and its critical revision; JFZ, JN, AR, LA and MAS conceived the analysis; AR designed the analysis, contributed to the conduct of the analysis, and interpreted the analysis; FM, JN, FK, HR and MD contributed to the design and the interpretation of the analysis; FM conducted the analysis.
Acknowledgments
The authors wish to acknowledge and thank all the patients for participating in P-2001 study, as well as the investigators and the staff at all clinical sites. Lori Bacarella (Modus Outcomes) provided editorial support, which was funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Ltd. Company.
Funding
This work and the P-2001 study were funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Ltd. Company.
Data-sharing statement
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participants data supporting the results reported in this article, will be made available within 3 months from initial request, to researchers who provide a
Haematologica | 108 April 2023 1198 LETTER TO THE EDITOR
methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.
References
1. Zeidner JF, Mazerolle F, Bell JA, et al. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine in higher-risk myelodysplastic syndromes/chronic myelomonocytic leukemia or low-blast acute myeloid leukemia: exploratory analysis of patient-reported outcomes. Blood. 2020;136(Suppl 1):S39-40.
2. Oliva EN, Platzbecker U, Fenaux P, et al. Targeting health-related quality of life in patients with myelodysplastic syndromes –current knowledge and lessons to be learned. Blood Rev. 2021;50:100851.
3. Bell JA, Galaznik A, Blazer M, et al. Transfusion-free interval is associated with improved survival in patients with higher-risk myelodysplastic syndromes engaged in routine care. Leuk Lymphoma. 2019;60(1):49-59.
4. Cannella L, Caocci G, Jacobs M, Vignetti M, Mandelli F, Efficace F. Health-related quality of life and symptom assessment in randomized controlled trials of patients with leukemia and myelodysplastic syndromes: what have we learned? Crit Rev Oncol Hematol. 2015;96(3):542-554.
5. Gelber RD, Cole BF, Gelber S, Goldhirsch A. Comparing treatments using quality-adjusted survival: the Q-TWiST method. Am Stat. 1995;49(2):161-169.
6. Gelber RD, Goldhirsch A, Cole BF, Wieand HS, Schroeder G, Krook JE. A quality-adjusted time without symptoms or toxicity (Q-TWiST) analysis of adjuvant radiation therapy and chemotherapy for resectable rectal cancer. J Natl Cancer Inst. 1996;88(15):1039-1045.
7. Oza AM, Lorusso D, Aghajanian C, et al. Patient-centered outcomes in ARIEL3, a phase III, randomized, placebocontrolled trial of rucaparib maintenance treatment in patients
Data requests should follow the process described in the datasharing section on https://clinicaltrials.takeda.com/ and https://vivli.org/ourmember/takeda/.
with recurrent ovarian carcinoma. J Clin Oncol. 2020;38(30):3494-3505.
8. Patil S, Figlin RA, Hutson TE, et al. Q-TWiST analysis to estimate overall benefit for patients with metastatic renal cell carcinoma treated in a phase III trial of sunitinib vs interferon-α Br J Cancer. 2012;106(10):1587-1590.
9. Billingham LJ, Abrams KR, Jones DR. Methods for the analysis of quality-of-life and survival data in health technology assessment. Health Technol Assess. 1999;3(10):1-152.
10. Efron B, Tibshirani RJ. An introduction to the bootstrap. New York (NY): CRC press; 1994.
11. Sekeres MA, Watts J, Radinoff A, et al. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine for higherrisk MDS/CMML or low-blast AML. Leukemia. 2021;35(7):2119-2124.
12. Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376.
13. Group TE. EuroQol-a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208.
14. Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108(2):419-425.
15. Zeidan AM, Platzbecker U, Garcia-Manero G, et al. Longer-term benefit of luspatercept in transfusion-dependent lower-risk myelodysplastic syndromes with ring sideroblasts. Blood. 2022;140(20):2170-2174.
Haematologica | 108 April 2023 1199 LETTER TO THE EDITOR

haematologica
Journal of the Ferrata Storti Foundation
haematologica.org