MEDICINA LABORATORIAL
Da Prescrição à Interpretação Clínica
HEMATO-ONCOLOGIA


Rui Abrantes Pimentel
Licenciado em Medicina pela Faculdade de Medicina da Universidade de Lisboa; Internato Complementar de Patologia Clínica no Serviço de Patologia Clínica do Hospital de São José; Grau de Assistente Hospitalar de Patologia Clínica pelo Hospital Curry Cabral; Especialista em Patologia Clínica pela Ordem dos Médicos; Consultor de Patologia Clínica da Carreira Médica Hospitalar no Centro Hospitalar Vila Nova de Gaia/Espinho, EPE – Unidade do Monte da Virgem; Responsável pela Coordenação do Controlo da Qualidade Analítica e pela Área Core Laboratorial no Dr. Joaquim Chaves, Laboratório de Análises Clínicas SA; Diretor Técnico Substituto no Dr. Joaquim Chaves, Laboratório de Análises Clínicas SA; esteve envolvido na atividade docente da cadeira de Farmacologia da Faculdade de Medicina da Universidade de Lisboa; foi convidado para fazer parte de vários grupos de trabalho.
A todos aqueles que foram, e muitos continuam a ser, os meus mestres, colegas e amigos, e que me têm acompanhado e acrescentado valor à minha formação pessoal, académica e profissional, em particular ao Dr. Joaquim Chaves pela confiança que sempre depositou em mim, pela sua amizade e por todas as oportunidades que me concedeu.
À minha grande amiga Arminda Maria Braz Vilares, que conheço e com quem tenho o maior orgulho de trabalhar há mais de 25 anos, pela generosidade com que se prontificou a partilhar a sua experiência profissional de mais de 10 anos no âmbito da imunofenotipagem, na Dr. Joaquim Chaves Laboratório de Análises Clínicas SA, e contribuiu, nesta área do conhecimento, para enriquecer e melhorar de forma significativa o conteúdo científico deste trabalho.
Técnicas, Lda.
Edições
Lidel –
Conheço o Doutor Rui Pimentel há mais de quarenta anos, desde que nos cruzámos no Internato de Patologia Clínica no Hospital de São José.
A sua dedicação à Medicina, consubstanciada na Patologia Clínica, foi sempre muito grande e proporcionou, ao longo dos anos, a necessidade de aprofundar conhecimentos e de os transmitir, como agora evidenciam estes volumes de Medicina Laboratorial. Trata-se da compilação de um exaustivo trabalho de pesquisa, de estudo e de prática laboratorial de grande utilidade para os cursos e aulas ministrados.
Os volumes de Medicina Laboratorial são, antes de mais, a seleção de matéria e a sistematização de conhecimentos que interessam à Medicina Laboratorial nesta sua vertente, aos quais não seria possível aceder senão pela consulta de extensa bibliografia, também aqui disponível.
Este volume, Hemato-Oncologia, da obra Medicina Laboratorial, encontra-se dividido em três partes:
• Citomorfologia hematológica;
• Mielograma;
• Hemato-Oncologia.
A citomorfologia relata detalhadamente as características morfológicas das células, permitindo, mediante a sua consulta, a caracterização de todos os elementos celulares passíveis de serem encontrados, classificados e avaliados nas preparações coradas tanto pelo May-Grünwald-Giemsa, como nas colorações citoquímicas de sangue periférico ou de medula óssea.
O mielograma, exame da medula óssea, permite a avaliação qualitativa e semiquantitativa da hematopoiese e ajuda no diagnóstico de doenças hereditárias e adquiridas tanto benignas, como malignas. Salienta-se ainda que os volumes de Medicina Laboratorial sugerem, para a sua execução, uma metodologia que me parece muito assertiva, permitindo avaliar sequencialmente as etapas necessárias à observação e as técnicas a utilizar, tendo presente a natureza pluridisciplinar do diagnóstico hemato-oncológico.
Reforço a importância dos cadernos para o estudo de uma matéria de grande complexidade, como é a Hemato-Oncologia, e que tem evoluído substancialmente nas últimas décadas com a obrigatoriedade de um diagnóstico pluridisciplinar, que engloba a morfologia (mielograma), a biópsia osteomedular, a imunofenotipagem, a citogenética e a biologia molecular clínica.
Pegando em dois dos múltiplos exemplos possíveis refiro-me às mais-valias evidenciadas em dois dos capítulos que se referem, a meu ver, às patologias mais difíceis de compreender e diagnosticar:
• Leucemias agudas, em que a descrição pormenorizada das diversas entidades com as diferentes alterações citogenéticas e mutações, e a correlação com os achados morfológicos observados em cada uma delas torna este trabalho uma avaliação integrada, atual e objetiva, permitindo o diagnóstico pluridisciplinar que possibilita ao clínico estabelecer a terapêutica e ter ferramentas para a monitorização, acompanhamento e avaliação de recaídas, da doença residual mínima e da eficácia terapêutica;
• Síndromas mielodisplásicas, em que está patente a necessidade da cooperação entre o laboratório e a clínica, o papel fundamental da Morfologia na avaliação da displasia, elemento-chave deste diagnóstico, assim como a cooperação com outras valências, nomeadamente a Citogenética e a Biologia molecular, que aqui sai reforçada.
Sendo a Hemato-Oncologia uma parte substancial da Hematologia, os volumes de Medicina Laboratorial dão-nos a possibilidade de a conhecer de a estudar e interpretar à luz dos conhecimentos atuais, realçando a necessidade da colaboração com a clínica para colmatar o distanciamento existente entre estas duas áreas.
Congratulo-me com publicação desta obra, que vai preencher uma grande lacuna no acesso ao conhecimento desta matéria, e que vai permitir à população-alvo o conhecimento detalhado e o estudo da Hemato-Oncologia.
Margarida Silveira
Ex-Diretora do Serviço de Patologia Clínica do Instituto Português de Oncologia de Lisboa Francisco Gentil, EPE
O presente trabalho é, essencialmente, uma compilação modulada de boas práticas no âmbito da prescrição médica, de todas as fases do processo laboratorial (pré-analítico, analítico e pós-analítico) e da interpretação dos resultados analíticos. Baseia-se na minha experiência clínica e laboratorial, que foi sendo adquirida e amadurecida ao longo de mais de quatro décadas, mais de metade das quais na Dr. Joaquim Chaves, Laboratório de Análises Clínicas, e nas evidências publicadas na literatura científica das diversas especialidades médicas.
Aceitei o desafio proposto pelas minhas colegas e amigas, Catarina Rombo, Marina Pires e Rute Pereira, e pela minha mulher, Margarida Ponte, de publicar Medicina Laboratorial: Da Prescrição à Interpretação Clínica por estar convencido de que a partilha, em todas as áreas do conhecimento, incluindo na Medicina Laboratorial, é uma janela de oportunidade imperdível, por permitir criar as condições necessárias para explicar a importância da relação entre a clínica e o laboratório, cujo diálogo entre pares deve ser permanente, assim como o significado de muitos sinais do âmbito da Patologia Clínica. Estou de convicto que esta obra constitui um importante contributo para melhorar a compreensão e o processo de interpretação dos resultados dos parâmetros laboratoriais (mesurandos) prescritos, uma vez que é neles que se baseiam entre 60 e 70% das decisões médicas, que condicionam a qualidade de vida das pessoas e, muitas vezes, o seu tempo de sobrevida.
Revisitada a literatura médica publicada, quer em livros de texto, quer em artigos científicos, nacionais e internacionais, é evidente a existência de uma enorme lacuna, que se vem traduzindo pela insuficiência de articulação, cooperação e coordenação de esforços interdisciplinares. É patente, também, um afastamento cada vez maior entre o que é pretendido e tomado como necessidade por cada uma das especialidades médicas e cirúrgicas, e as limitações e potencialidades do estado da arte da Medicina Laboratorial, que importa conhecer e colmatar.
Só há uma Medicina. A Medicina como a Arte Maior de ajudar as pessoas a terem a melhor qualidade de vida (bem-estar físico, psíquico e social) durante o máximo tempo possível.
No âmbito da Hemato-Oncologia e mielograma, Medicina Laboratorial abrange uma parte significativa da área do conhecimento da Hematologia. Neste contexto, são descritas as características citomorfológicas mais importantes dos elementos celulares, normais e patológicos, que podem ser observados quer no sangue periférico, quer nos aspirados de medula óssea. É evidenciada a importância do mielograma no estudo dos doentes e são identificados os critérios de diagnóstico das patologias hematológicas agudas e crónicas mais importantes, com repercussões clinicamente significativas no sangue periférico, na medula óssea ou em ambos. São ainda caracterizados os mesurandos com maior valor semiológico no âmbito do rastreio, do diagnóstico, da monitorização da evolução clínica e da resposta ao tratamento, e da estratificação do risco (prognóstico) das diferentes entidades nosológicas, assim como a sua sensibilidade e especificidade, tendo em consideração o estado da arte atual.
Edições Técnicas, Lda.
Lidel –
Os médicos recorrem às análises clínicas para efetuar diagnósticos, estratificar o risco de morbilidade e de mortalidade, identificar recaídas ou recorrências, e monitorizar a evolução clínica e a resposta ao tratamento de diversas entidades nosológicas, assim como para despistar apresença destas entidades em indivíduos aparentemente saudáveis.
A principal responsabilidade da Medicina Laboratorial é contribuir para melhorar de forma consistente a qualidade de vida das pessoas (estado de completo bem-estar físico, psíquico e social) durante o máximo tempo possível, devendo para isso garantir que, de forma sustentada, todos os resultados reportados se caracterizem pelos seguintes aspetos:
• Não estejam comprometidos por enviesamentos inaceitáveis em relação ao valor real (in vivo);
• Sejam clinicamente úteis para permitir a tomada de decisões médicas mais adequadas;
• Sejam reportados em tempo útil.
Tem sido evidenciado que até 25% dos erros de avaliação clínica relacionados com resultados laboratoriais afetam o processo de decisão médica, tendo como consequência atrasos na prestação dos cuidados de saúde mais adequados em tempo útil.
A caracterização da variabilidade fisiológica, fisiopatológica e iatrogénica, e a identificação e monitorização da variabilidade que decorre do processo analítico, assim como a garantia de que se estas mantêm dentro de limites clinicamente aceitáveis, são fundamentais para que as informações veiculadas pelos resultados reportados não induzam os clínicos prescritores a cometer erros de julgamento em relação a um diagnóstico, prognóstico (estratificação do risco) ou tratamento e, com isso, diminuir a qualidade de vida dos pacientes.
A variabilidade dos resultados laboratoriais está relacionada com diversos fatores e condições dos quais se destacam de acordo com a sua origem os seguintes:
• A variabilidade biológica intraindividual específica de cada mesurando (equilíbrio homeostático);
• As alterações do estado de saúde dos indivíduos (variabilidade fisiopatológica);
• As alterações decorrentes de atos ou de intervenções no âmbito da terapêutica ou do diagnóstico (variabilidades iatrogénicas);
• O processo pré -analítico, analítico e pós -analítico, designadamente a competência dos profissionais envolvidos (fatores humanos) e a qualidade do desempenho dos diferentes procedimentos de medida e ensaios instalados no laboratório (exemplos: precisão e exatidão próprias do método, intervalo de medição e linearidade, limites de deteção e especificidade analítica);
• A estabilidade dos mesurandos (analitos e parâmetros) nas amostras biológicas em função das suas características, da preparação pré-analítica efetuada, e das condições de colheita, de conservação e do seu processamento;
• As interferências analíticas positivas ou negativas que se traduzem pela diferença entre o resultado obtido e o seu valor real (in vivo).
Antes de ser tomada uma decisão perante resultados laboratoriais anormais ou discordantes, seja através da história clínica, epidemiológica ou ambas, seja com recurso a critérios interparamétricos, ou ainda através dos resultados obtidos em colheitas anteriores, é importante que o processo de interpretação, tanto a nível laboratorial como clínico, permita determinar se estão relacionados com o estado
de saúde dos pacientes ou se são resultantes de variáveis pré-analíticas, analíticas ou pós-analíticas, nas quais se incluem as interferências.
O impacto negativo que os resultados incorretos podem ter para os pacientes está relacionado com os seguintes fatores:
• Estado de saúde e características das patologias que o condicionam;
• Sensibilidade e especificidade diagnósticas (valor semiológico) dos mesurandos objeto de análise;
• Grandeza da diferença entre o resultado reportado e o seu valor real (in vivo);
• Competência e experiência quer dos profissionais do laboratório, quer dos médicos prescritores em identificar e lidar com este tipo de resultados, visando minimizar a gravidade do dano.
Técnicas, Lda.
Edições
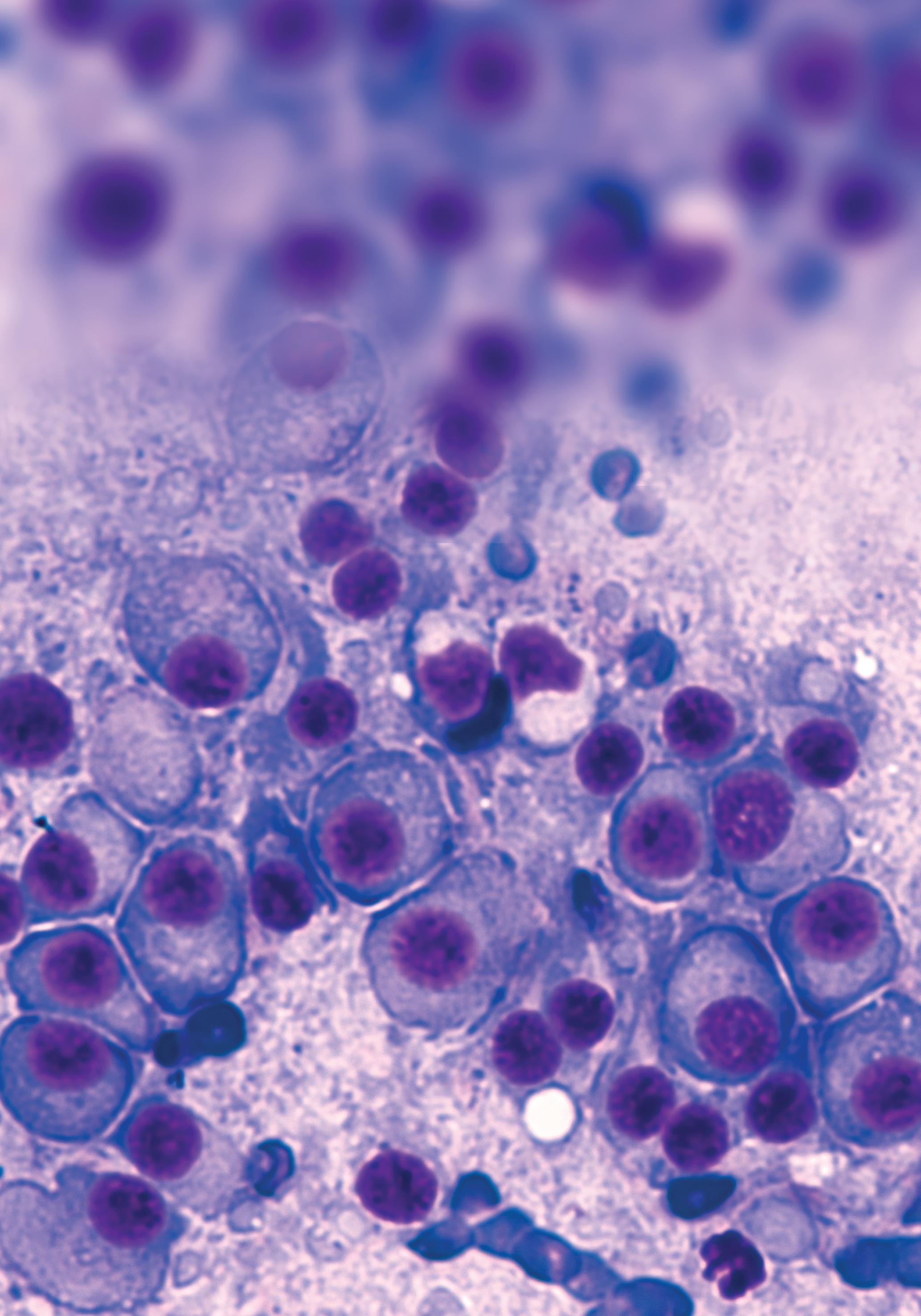
1.1. Macrófagos norMais
São células reticulares ou histiócitos que cons tituem um estádio de evolução dos monócitos (fase tecidular):
• Tamanho médio a grande (15 a 80 μm de diâmetro), com contorno irregular;
• Relação entre o tamanho do núcleo e o do citoplasma (N:C) diminuída (< 0,8);
• Núcleo ligeiramente excêntrico, arredondado, ovalado ou alongado, de contorno regular, ou indentado; membrana nuclear bem marcada;
• Cromatina pouco condensada reticulada ou esponjosa dispersa, de cor avermelhada, com membrana nuclear nítida; geralmente, 1 a 3 nucléolos basófilos bem visíveis;
• Citoplasma abundante, pálido ou azul‑claro, de aspeto esponjoso (vacuolizado) e de con tornos irregulares mal definidos (franjado ou pseudópodes filiformes), podendo apre sentar grânulos mais ou menos grosseiros ou células fagocitadas (exemplos: série eri troide, mieloide ou ambas) no seu interior (hemofagocitose).
1.2. Macrófagos anorMais
• Células de tamanho médio a grande (15 a 80 μm de diâmetro);
• Relação N:C diminuída (< 0,8);
• Núcleo grande redondo ou oval, por vezes con voluto, polilobado ou com núcleos múltiplos;
• Cromatina grosseira em faixas nitidamente separadas umas das outras; nucléolo grande com condensação ou reforço perinucleolar, por vezes com bordos angulados;
• Citoplasma abundante muito basófilo com numerosos vacúolos.
• Células grandes (20 a 90 μm de diâmetro);
• Relação N:C diminuída (< 0,8);
• Citoplasma abundante com múltiplos vacúo los;
• Observados em número significativo nos aspirados medulares, associados a diversas patologias, nomeadamente:
– Doença de Niemann‑Pick [os vacúolos são pequenos e uniformes e contêm caracteris ticamente esfingomielina; também estão presentes nos linfócitos e nos monócitos do sangue periférico (SP)];
– Doença de Gaucher, gangliosidoses e doen ça de Fabry;
– Hiperlipemias;
– Síndromas talassémicas, doença drepa nocítica, trombocitopenia imune crónica, mononucleose infeciosa e aplasia medular induzida pela quimioterapia;
– Artrite reumatoide (AR), hepatite viral e doença renal crónica (DRC).
Também designados por sea‑blue macrophage/ /histiocyte, são macrófagos com núcleo central ou periférico e citoplasma não vacuolizado com nu merosos grânulos que apresentam as seguintes ca racterísticas:
• São densos, arredondados, com aspeto homo géneo e podem cavalgar o núcleo;
• Coram de azul‑marinho ou azul esverdeado pelo May‑Grünwald‑Giemsa (MGG), ver melho pelo ácido periódico de Schiff (PAS) e negro pelo Sudão B, e não coram pelo Perls nem pela coloração álcool‑ácido resistente;
• Têm uma estrutura membranosa ou lamelar e são constituídos por glicolípidos, fosfolí pidos e/ou ceroide; este material acumula ‑se no citoplasma por incapacidade dos
8.1.5.1. Coloração de fundo
• “Não se observou”;
• “Presente” (exemplo: eosinofílica por edema da BM).
8.1.5.2. Material proteináceo intercelular
•“Não se observou”;
• “Presente”; material proteináceo amorfo eosinofílico com elementos celulares hema topoiéticos difíceis de caracterizar no seu interior (alterações morfológicas e tintoriais) por necrose da BM (áreas ou focos de maior ou menor dimensão do tecido hematopoié tico e do estroma resultante da falência da microcirculação com isquemia, na maior parte dos casos, por processos mecânicos, imunoinflamatórios e/ou tóxicos induzidos, por exemplo, por fármacos ou radiações ionizantes) – ver Caixa “Caracterização da necrose medular”:
– Etiologia:
■ Neoplásica (é a mais frequente, estando na origem de cerca de 90% dos casos): sobretudo as neoplasias hematológicas (agudas e crónicas) – nomeadamente as leucemias mieloides e linfoblásticas agu das, os linfomas (exemplos: células B e T maduras e LH) e as MPN e MDN/MPN –, e as metástases dos tumores sólidos [exemplos: adenocarcinoma da próstata, sarcoma de Ewing ou tumor neuroec todérmico primitivo (PNET), rabdo miossarcoma alveolar, neuroblastoma, carcinoma ductal da mama, carcinoma neuroendócrino, carcinoma do urotélio, adenocarcinoma do pulmão, carcinoma de pequenas células do pulmão e tumo res das células germinais];
■ Não neoplásica: infeções (exemplos: VIH, parvovírus humano B19, bacteria nas, fúngicas e a Mycobacterium spp), parasitoses (exemplos: Leishmania spp e Toxoplasma gondii), choque sético, anemia megaloblástica, síndromas talas sémicas e hemoglobinopatias – em par ticular a doença drepanocítica –, CID, síndroma hemolítica‑urémica, doenças
sistémicas (exemplos: diabetes mellitus, gota, hipertensão arterial, alcoolismo e cirrose hepática), cirurgia, fármacos [exemplos: fatores de crescimento gra nulocítico – G‑CSF ou GM‑CSF; fluda rabina, imatinib, mesilato e interferão‑α (IFN‑α); ciclofosfamida, fenobarbital, estrogénios e colchicina], radiações ioni zantes e exposição a agentes químicos tóxicos, anorexia, hiperparatiroidismo, doenças autoimunes [exemplos: lúpus eritematoso sistémico (LES) e síndro ma antifosfolipídica (APLS)], patologias trombóticas (estados de hipercoagulabi lidade), e lesões traumáticas (exemplos: stress biomecânico, contusões e fratu ras), degenerativas, isquémicas, metabó licas (exemplo: gota) e inflamatórias.
■ Idiopática (muito rara).
Caracterização da necrose medular
A necrose medular tem uma incidência inferior a 2%; é definida pela destruição do tecido hematopoiético e do estroma, sem envolvimento do osso cortical, que se manifesta clinicamente por desconforto ou por dores ósseas (80% casos), febre e, por vezes, icterícia ligeira. Laboratorialmente, caracteriza-se por anemia (90% dos casos), trombocitopenia (90% dos casos), reação leucoeritroblástica (50% dos casos) e aumento da atividade da lactato desidrogenase (LDH) (41% dos casos) e da ALP (51% dos casos).
8.1.5.3. Cristais
• “Não se observaram”;
• “Presentes” (exemplos):
– Cristais de Charcot‑L eyden (CLC): cris tais bipiramidais com aspeto de agulha de bússola formados por lisofosfolipase; é o constituinte proteico encontrado nos grâ nulos dos Eo e dos Ba, geralmente extra celulares; também podem ser obser vados no citoplasma dos macrófagos e dos neu trófilos; estão associados frequentemente a necrose medular:
■ Característicos de doença eosinofílica reativa ou neoplásica;
■ AML com mutações no gene NPM1 sem eosinofilia (associada a necrose medular);
Lidel –Edições Técnicas, Lda.
©
Tabela 2.1 • Critérios utilizados para determinar e reportar as reservas de Fe em amostras de aspirado medular –reação de Perls (avaliação semiquantitativa das reservas de Fe pelo método de Gale):
Scores Critérios
0 [ausentes]
1+ [vestigiais]
2 + [reduzidas]
3 + [normais]
4 + [aumentadas]
5 + [muito aumentadas]
6 + [marcadamente aumentadas]
8.2.3.2. Siderócitos
Não se observa Fe no citoplasma dos macrófagos (poalho ou grânulos) com ampliação de 1000x
Coloração citoplasmática difusa ou raros grânulos azulados intracelulares dispersos de pequeno tamanho (1000x)
Alguns grânulos azulados intracelulares dispersos de pequeno tamanho, observáveis com ampliação de 100x
Numerosos grânulos azulados intracelulares de pequeno e médio tamanho, observáveis nos grumos e nos macrófagos isolados com ampliação de 100x
Muitos grânulos azulados intracelulares de médio e grande tamanho com tendência para se agregarem (grânulos grosseiros), observáveis nos grumos e nos macrófagos isolados com ampliação de 100x
Grânulos azulados intracelulares grosseiros, observáveis nos grumos e nos macrófagos isolados com ampliação de 100x
Grânulos azulados de grande tamanho (grosseiros), intra e extracelulares, observáveis nos grumos com ampliação de 100x, obscurecendo os detalhes celulares
Os siderócitos são eritrócitos maduros que apresentam, predominantemente na sua perife ria, grânulos de hemossiderina ou siderossomas (designados por corpos de Pappenheimer: geral mente, 2 a 5 pequenas inclusões por eritrócito).
Intervalos de referência por 100 eritrócitos:
• Recém‑nascidos (RN): 0,3 a 1,7%;
• Crianças e adultos: até 0,3%.
Os seus valores estão aumentados, paralela mente aos dos sideroblastos, na eritropoiese side roblástica e na IE excessiva, designadamente, nas anemias sideroblásticas, nas hemoglobinopatias, nas talassemias major, nas AH, no hipoesplenis mo/asplenia e na cirrose hepática alcoólica.
8.2.3.3. Sideroblastos
Os sideroblastos são os eritroblastos policro matófilos e ortocromáticos (precursores eritroi des), que apresentam no seu citoplasma grânu los de hemossiderina ou siderossomas revelados pela reação de Perls:
• Normais: grânulos delicados, dispersos ale atoriamente pelo citoplasma, que corres pondem ao Fe da hemossiderina associado à membrana dos lisossomas;
• Patológicos:
– Perinucleares relacionados com a acumu lação anormal de Fe da hemossiderina nas mitocôndrias;
– Presença de numerosos grânulos grossei ros (grandes).
Embora a sua caracterização e contagem di ferencial seja efetuada a partir de amostras de aspirado de BM, em determinadas situações pa tológicas também é possível serem identificados no SP [exemplo: está descrita a presença transi tória de eritroblastos em anel ou tipo 3 associada à fase aguda de uma AH secundária a PV com mutação pontual no gene JAK2, que origina uma substituição da valina por fenilalanina na proteí na codificada (JAK2V617F)].
Os sideroblastos são classificados em 3 tipos, de acordo com o número e o padrão da distribui ção citoplasmática dos grânulos de hemossideri na (critérios):
6.1. fases Da evolução temporal Da et
• Progressão da ET e frequência com que ocorre:
– MF (disfunção da via de sinalização JAK ‑STAT) com metaplasia mieloide (5 a 15% dos casos após 15 anos);
– Fase blástica (< 5% dos casos ao fim de 20 anos): a progressão para esta fase está frequentemente associada à presença de mutações envolvendo os genes TP53, EZH2, SH2B3, SF3B1, U2AF1 e/ou IDH2.
• Causas de fibrose medular que devem ser excluídas no âmbito do diagnóstico diferen cial da fase de MF pós‑ET:
– Outras neoplasias mieloides (exemplos: PMF e PV);
– Doenças não hematológicas (fibrose medular secundária ou mieloftise): metás tases de neoplasias sólidas, MF autoimune, LES, leishmaníase (Kala‑Azar), tubercu lose, doença de Paget, infeção pelo VIH, raquitismo (deficiência de vitamina D), osteodistrofia renal, hiperparatiroidismo, síndroma das plaquetas cinzentas, MF infantil familiar e hipertensão pulmonar idiopática.
O diagnóstico da ET requer a presença dos 4 critérios major ou dos 3 primeiros major e o minor:
• Critérios major (pré‑requisitos):
– Trombocitose (≥ 450x109/L) persistente (≥ 3 meses), inexplicada por outras causas;
– BM: celularidade normal para a idade; aumento do número e do tamanho dos MK maduros (células grandes – 50 a 110 μm, núcleo hiperlobado, com aspeto em chifre de veado ou de largada de balões, citoplasma maduro abundante ligeira mente basófilo com múltiplos grânulos azurófilos) sem displasia, não se apresen tando em aglomerados densos (até 6 MK); pode observar‑se a presença de emperipo lese. Não se observa um aumento signifi cativo da eritropoiese, nem desvio esquer
do da granulopoiese neutrofílica (ausência de hiperplasia eritroide e granulocítica significativas), assim como de fibrose (até grau I);
– Sem evidência (critérios de diagnóstico) de PV, PMF, CML ou de outra neoplasia mieloide;
– Presença de mutação do gene JAK2 (envolvendo o exão 14, em 60 a 65% dos casos, e o exão 12, raramente; associado a um maior número de casos de trombose), do CALR (19p13.13; 20 a 25% dos casos; associado a trombocitoses mais elevadas e a uma maior taxa de evolução para MF) ou do MPL (1p34.2; 5% dos casos).
• Critério minor (critério de suporte):
– Presença de um marcador clonal/neo plásico (citogenética e/ou mutações) ou ausência de evidência de trombocitose de origem reativa (exemplos: hemorragia aguda, recuperação de trombocitopenia, infeção/inflamação aguda, cirurgia, esple nectomia, neoplasias, doenças linfoproli ferativas e exercício físico).
Presença de dois critérios requeridos e de, pelo menos, dois critérios adicionais:
• Critérios requeridos: diagnóstico prévio (confirmado) de ET e presença de mielofi brose grau 2 ou 3;
• Critérios adicionais: anemia e diminui ção do valor da Hb > 2g/dL em relação à mediana do valor basal; leucoeritroblastose ou eritromielemia (eritroblastos e mielémia associados frequentemente à presença de dacriócitos); esplenomegalia (de novo ou aumento > 5 cm em relação ao valor basal anterior); aumento da atividade no soro ou no plasma da LDH (lactato desidrogenase); presença de, pelo menos, 2 dos 3 sintomas constitucionais: febre > 37,5ºC inexplicada por outras causas; sudação noturna; perda de peso não intencional > 10% nos últimos 6 meses.
Critérios de diagnóstico da CMUS:
• Monocitose (≥ 0,5x109/L e ≥ 10%) persisten te (≥ 3 meses) no SP, inexplicada por outras causas (exemplos: processos inflamatórios sistémicos de qualquer natureza, necrose tecidular extensa, doenças de armazenamen to dos lípidos, hemólise, fase de regeneração medular ou neoplasias de órgãos sólidos);
• Citopenia(s) (critérios da OMS): a sua ausência caracteriza a CMUS e a sua presen ça a CCMUS;
• Presença de clonalidade: pelo menos uma mutação associada a neoplasia mieloide (VAF ≥ 2%);
• Ausência de displasia significativa, de blas tos aumentados e de achados morfológicos ao nível da BM compatíveis com CMML;
• Ausência de critérios de diagnóstico de neoplasia hematológica.
/MIELOPROLIFERATIVA (MDN/MPN)
COM
É uma doença rara (BCR:ABL1 negativa) que tem pior prognóstico do que a CML.
É mais frequente estes doentes apresentarem anemia, trombocitopenia e esplenomegalia do que na forma clássica. A anemia e a displasia também são mais acentuadas do que na CML.
3.1. critérios De DiagNóstico
• Leucocitose é sempre ≥ 13,0x109/L, estando associada às seguintes alterações no SP:
– Neutrofilia;
– Mielémia (promielócitos, mielócitos e metamielócitos) ≥ 10%;
– Disgranulopoiese acentuada;
– Ausência de eritroblastose;
– Basofilia absoluta mínima ou ausente < 2%;
– Monocitose absoluta mínima ou ausente (< 10%);
– Eosinofilia ausente ou mínima (< 10%).
• Blastos no SP e na BM < 20%;
• Citopenia(s) (critérios da OMS);
• BM:
– Hipercelular com proliferação predomi nantemente granulocítica;
– Disgranulopoiese acentuada, associada ou não a dismegacariopoiese e a diseritro poiese;
– Relação M:E muitas vezes > 10,0.
• Não satisfaz os critérios de diagnóstico de nenhuma MPN (exemplos: CML, PMF, PV, ET e CNL);
• Citogenética e genética molecular:
– Ausência do gene de fusão BCR:ABL1, de mutações dos JAK2, CALR e MPL, e de outras anomalias genéticas compatíveis com o diagnóstico de neoplasias mieloi des/linfoides com eosinofilia e rearran jos de genes tirosina quinase (exemplos: ETV6:ABL1, PDGFRA, PDGFRB, FGFR1, JAK2 e FLT3).
– MDN/MPN com neutrofilia ou leucemia mieloide crónica atípica: está associa da frequentemente a mutações isoladas ou associadas dos SETBP1, ASXL1 e/ou ETNK1. As mutações do CSF3R são raras (presente em < 10% dos casos obrigando a excluir o diagnóstico de CNL).
E TROMBOCITOSE (MDN/ /MPN ‑T ‑ SF3B1), E MDN/MPN COM SIDEROBLASTOS EM ANEL E TROMBOCITOSE (MDN/MPN ‑RS ‑T)
A idade média dos doentes em que é efetuado odiagnóstico situa‑se entre os 71 e os 75 anos. O tempo de sobrevida médio depois do diag nóstico é de 6 anos. São fatores de mau prognós tico, independentes da idade (≥ 70 anos), a Hb ≤ 10,0 g/dL e a presença de cariotipo anormal (exceto o que envolve o comossoma Y).
4.10. linFoma b da zona marginal nodal (nmzl) e variante Pediátrica (Pnmzl)
A população de linfócitos neoplásicos tem origem na MZ nodal das células B (morfologia monocitoide).
Cerca de 10 a 20% dos casos transformam‑se em DLBCL.
4.10.1. Características
Características observáveis no SP:
• Pequenos linfócitos (mais frequentes) com contorno nuclear irregular, cromatina com pequenos grumos e cordões finos, e citoplas ma escasso; ou
• Linfócitos monocitoides de tamanho médio a grande, núcleo reniforme (seme lhante ao dos monócitos), cromatina pouco condensada e presença ocasional de nuclé olo; citoplasma amplo e basófilo (difere dos monócitos porque estes têm uma tonalidade mais acinzentada).
4.10.2. Imunofenotipagem
• SmIgM/D fortemente positivo e L positivas;
• CD19, CD20, CD79a, CD79b e CD22 positivos; CD23, CD5 e CD10 negativos; FMC7dim/+; ciclina D1 negativo; Bcl2 posi tivo, Bcl6 negativo.
4.10.3. Anomalias genéticas
• Ganho 2p e 6p, e perda 1p e 6q;
• Trissomia dos cromossomas 3 e 18;
• Ausência de fusões e rearranjos genéticos característicos.
4.10.4. Diagnóstico diferencial
• FL [CD10 e t(14;18) habitualmente presen tes];
• Linfoma das células do manto [presença de CD5 com ciclina D1 ou t(11:14)];
• B‑SLL;
• Hiperplasia das células B monocitoides da MZ (BCL2 negativo).
4.11. linFoma Folicular (Fl) e outros lnH relacionados com o Fl
É o linfoma de pequenas células mais frequen te, representando cerca de 20 a 30% dos LNH (2.º mais frequente).
O FL tem origem nas células B do centro ger minal (centro do folículo).
É uma doença incurável de natureza hetero génea, cuja evolução é indolente (crónica), re cidivante, com um tempo de sobrevida longo, afetando predominantemente os adultos (muito rara antes dos 20 anos; idade mediana em que é efetuado o diagnóstico: entre os 55 e os 65 anos), e não tendo predomínio de sexo.
Em 10 anos, cerca de 30% dos casos transformam‑se em DLBCL de alto grau ou com características de BL e, mais raramente, em PTCL e MZL.
Na maioria dos doentes, o FL é assintomático na altura do diagnóstico. Nos casos sintomáticos, os doentes apresentam as seguintes manifesta ções clínicas:
• Sintomas B;
• Envolvimento dos gânglios linfáticos (linfa denopatia generalizada incluindo o anel de Waldeyer);
• Infiltração do baço (esplenomegalia; trom bocitopenia relacionada com o hiperesple nismo);
• Infiltração da BM (está presente em cerca de 50% dos doentes na altura do diagnóstico; anemia e trombocitopenia);
• Infiltração de outros tecidos extranodais;
• Leucemização com presença de células neo plásicas/células de LNH no SP (leucemiza ção em 5 a 10% dos casos).
4.11.1. Diagnóstico e prognóstico (abordagem)
• Biópsia ganglionar e da BM (histologia e IHC);
• Aspirado de BM;
• Imagiologia (funcional e anatómica): FDG ‑PET‑CT (fluorodeoxyglucosepositron emission tomography/computerised tomography);
• Outros exames laboratoriais recomendados:
Vol. 2
Esta coleção de 5 volumes apresenta as fases da prescrição médica, do processo laboratorial (pré-analítico, analítico e pós-analítico) e da interpretação dos resultados analíticos. É explicada e evidenciada a importância da relação entre a clínica e o laboratório, cujo diálogo entre pares deve ser permanente, e clarificado o significado de muitos sinais do âmbito da Patologia Clínica, que são representados e se traduzem nas diferentes formas como os resultados dos parâmetros laboratoriais prescritos podem ser reportados e devem ser interpretados.
Destina-se a internos e especialistas em Patologia Clínica; estudantes de Medicina; médicos de diversas especialidades que, na prática clínica diária, prescrevem análises clínicas e interpretam os seus resultados; estudantes e profissionais das áreas de Ciências Farmacêuticas, Ciências Biomédicas, Biologia e Bioquímica em formação e especialistas em Análises Clínicas; interessa também a técnicos de análises clínicas e saúde pública.
Hemato-Oncologia
O volume abrange uma parte significativa da área do conhecimento da Hematologia. Neste contexto, são descritas as características citomorfológicas mais importantes dos elementos celulares, normais e patológicos, que podem ser observados quer no sangue periférico, quer nos aspirados de medula óssea. É evidenciada a importância do mielograma no estudo dos doentes e são identificados os critérios de diagnóstico das neoplasias hematológicas agudas e crónicas mais importantes, com repercussões clinicamente significativas no sangue periférico, na medula óssea ou em ambos. São ainda caracterizados os parâmetros laboratoriais com maior valor semiológico no âmbito do rastreio, do diagnóstico, da monitorização, da evolução clínica e da resposta ao tratamento, e da estratificação do risco (prognóstico) das diferentes entidades nosológicas, assim como a sua sensibilidade e especificidade, tendo em consideração o estado da arte atual.

9 789897 528705
ISBN 978-989-752-870-5 www.lidel.pt
Edição apoiada por:
