VARIANTE ESCLEROSANTE DIFUSA INCIDENTAL PAPILAR MIOMATOSIS UTERINA MANEJO CARDIOVASCULAR EN EL PACIENTE CON CÁNCER: NUEVAS ESTRATEGIAS ECOANSIEDAD: ENFERMOS POR EL PLANETA MANEJO DECÁNCER DE PRÓSTATA: PROSTATECTOMÍA RADICAL LAPAROSCÓPICA 2D FRENTE A LA PROSTATECTOMÍA RADICAL LAPAROSCÓPICA 3DHD
ACR20 at week 2 was not controlled for type 1 error; therefore, statistical conclusions cannot be made.
Consistent joint symptom results regardless of TNFi
SPIRIT-P1 (BIOLOGIC-NAIVE): rates at week NRI2,4 SPIRIT-P2 (TNFi-EXPERIENCED): rates week

NRI of
SPIRIT-P1 and -P2 Trial Design3-6SPIRIT-P1 (N=417) and -P2 (N=363) were phase 3, randomized, doubleblind, placebo-controlled trials to evaluate the efficacy and safety of Taltz compared with placebo in patients with active psoriatic arthritis. Patients in SPIRIT-P1 were biologic-naive. Patients in SPIRIT-P2 were tumor necrosis factor inhibitor (TNFi)- experienced, having had an inadequate response and/or intolerance to 1 or 2 prior TNFis. In both trials, the primary efficacy endpoint was the proportion of patients achieving ACR20 response at week 24. All patients were ≥18 years of age and had ≥3 swollen and ≥3 tender joints. Patients were randomized to placebo or Taltz 80 mg every 2 or 4 weeks following a 160 mg starting dose. In SPIRIT-P1, an active reference arm of adalimumab 40 mg every 2 weeks was included. Patients in all study
SPIRIT-P1 (N=417) and -P2 (N=363) were phase 3, randomized, doubleblind, placebo-controlled trials to evaluate the efficacy and safety of Taltz compared with placebo in patients with active psoriatic arthritis. Patients in SPIRIT-P1 were biologic-naive. Patients in SPIRIT-P2 were tumor necrosis
FOR PATIENTS WITH ACTIVE PSORIATIC ARTHRITIS ACR20 response seen as early as week in some patients Instructions included
arms were allowed to continue taking stable background medications during the trial. Inadequate responders (as defined by blinded criteria of <20% improvement in tender and in swollen joint counts) at week 16 received rescue therapy and were analyzed as nonresponders after week 16 until the
arms were allowed to continue taking stable background medications during the trial. Inadequate responders (as defined by blinded criteria of <20% improvement in tender and in swollen joint counts) at week 16 received rescue therapy and were analyzed as nonresponders after week 16 until the primary endpoint. After receiving rescue therapy, inadequate responders in the placebo and adalimumab arms were re-randomized to Taltz 80 mg every 2 or 4 weeks. NRI methods were used for categorical efficacy analyses during the double-blind treatment period.
ACR=American College of Radiology; TNFi=tumor necrosis factor inhibitor; NRI=nonresponder imputation.
Rapid
2
1-3 Please see Brief Summary of Prescribing Information on the following pages. Please see
for Use
with the device.
ACR response
24,
ACR response
at
24, NRI3,4 P A TIENTS A CHIEVING RESPONSE (% ) ACR20 5 8%* 30 % 100 80 60 40 20 0 *P≤.001 vs placebo at week 24 for ACR20 2 Taltz 80 mg every 4 weeks (n=107) Placebo (n=106) P A TIENTS A CHIEVING RESPONSE (% ) ACR20 5 3%† 20 % 100 80 60 40 20 0 † P≤.0001 vs placebo at week 24 for ACR20 3 Taltz 80 mg every 4 weeks (n=122) Placebo (n=118) FDA Approved4SPIRIT-P1 ACR20 AT WEEK 2: TALTZ 39% VS PLACEBO=13% SPIRIT-P2 ACR20 AT WEEK 2: TALTZ 38% VS PLACEBO=12%
experience2-4
intent-to-treat population through week 24. Inadequate responders (<20% improvement in tender and in swollen joint counts) at week 16 were analyzed as nonresponders after week 16 until the primary endpoint 1 Primary endpoint=ACR20 response at week 24.
FOR PATIENTS WITH ACTIVE PSORIATIC ARTHRITIS Rapid ACR20 response seen as early as week 2 in some patients 1-3 Please see Important Safety Information on adjacent page. Please see Brief Summary of Prescribing Information on the following pages. Please see Instructions for Use included with the device. ACR20 at week 2 was not controlled for type 1 error; therefore, statistical conclusions cannot be made. SPIRIT-P1 (BIOLOGIC-NAIVE): ACR response rates at week 24, NRI2,4 SPIRIT-P2 (TNFi-EXPERIENCED): ACR response rates at week 24, NRI3,4 P A TIENTS A CHIEVING RESPONSE (% ACR20 5 8%* 30 % 100 80 60 40 20 0 *P≤.001 vs placebo at week 24 for ACR20 2 Taltz 80 mg every 4 weeks (n=107) Placebo (n=106) P A TIENTS A CHIEVING RESPONSE (% ACR20 5 3%† 20 % 100 80 60 40 20 0 † P≤.0001 vs placebo at week 24 for ACR20 3 Taltz 80 mg every 4 weeks (n=122) Placebo (n=118) FDA Approved4SPIRIT-P1 ACR20 AT WEEK 2: TALTZ 39% VS PLACEBO=13% SPIRIT-P2 ACR20 AT WEEK 2: TALTZ 38% VS PLACEBO=12% Consistent joint symptom results regardless of TNFi experience2-4 NRI of intent-to-treat population through week 24. Inadequate responders (<20% improvement in tender and in swollen joint counts) at week 16 were analyzed as nonresponders after week 16 until the primary endpoint 1 Primary endpoint=ACR20 response at week 24. SPIRIT-P1 and -P2 Trial Design3-6
No new National Drug Codes (NDCs)
No new Rx needed for existing Taltz patients
No new PAs to transition existing Taltz patients
No gaps in therapy
Once inventory of Taltz
Taltz Citrate-Free Bioequivalence Study Design7
The Citrate-Free Bioequivalence Study (N=245) was a 2-arm, subjectblind, parallel-design study in healthy subjects age 18-75 years to evaluate bioequivalence of Taltz citrate-free (CF) formulation compared to the original formulation of Taltz. Subjects were stratified into 1 of 3 weight categories (low: <70.0 kg; medium: 70.0-80.0 kg; high: >80.0 kg). Participants were then randomized within the 3 weight categories 1:1 to a single subcutaneous dose of either 80 mg Taltz original formulation (n=126) or 80 mg Taltz CF formulation (n=119). Subjects in each group were sub-randomized 1:1:1 to injection site (arm, thigh, or abdomen). Injections were administered by a medical professional using an autoinjector. The primary endpoint was bioequivalence as measured by maximum concentration (Cmax) of serum ixekizumab and area under the concentration versus time curve (AUC) of ixekizumab from time of injection through day 85 and time of injection through infinity.
Taltz Citrate-Free Injection Pain Study Design7 Citrate-Free Injection Pain Study (N=70) was a subject-blind, randomized, crossover study in healthy subjects age 18-75 years to determine injection site pain differences between Taltz citrate-free formulation compared to the original formulation of Taltz. The primary endpoint was pain intensity on injection, as measured by VAS Pain 0-100. Subjects were randomized 1:1:1 to receive a single 1 mL subcutaneous injection of 80 mg Taltz original formulation, 80 mg Taltz citrate-free formulation 1 (CF1), or 80 mg Taltz citrate-free formulation 2 (CF2) in 1 of 3 possible treatment sequences on Days 1, 8, and 15 in a 3-period cross-over design. Injections were administered in the abdomen by a medical professional using a prefilled syringe. CF2 is not an approved formulation. Only data on the commercially available CF1 will be presented.
INDICATIONS AND IMPORTANT SAFETY
INFORMATION
Taltz is indicated for adult patients with active ankylosing spondylitis, for adult patients with active psoriatic arthritis (PsA), and for adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation. Taltz is also indicated for adult patients and pediatric patients aged 6 years or older with moderate-to-severe plaque psoriasis (PsO) who are candidates for systemic therapy or phototherapy.
CONTRAINDICATIONS
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients.
WARNINGS AND PRECAUTIONS
Infections
Taltz may increase the risk of infection. In clinical trials of adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). A similar increase in risk of infection was seen in placebocontrolled trials of adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis. Serious infections have occurred. Instruct patients to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur.
If a serious infection develops, discontinue Taltz until the infection resolves.
Pre-Treatment Evaluation for Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Closely monitor patients receiving Taltz for signs and symptoms of active TB during and after treatment.
Hypersensitivity
Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz. If a serious hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy.
Inflammatory Bowel Disease
Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than the placebo group. During Taltz treatment, monitor patients for onset or exacerbations of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management.
Immunizations
Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz.
ADVERSE REACTIONS
Most common adverse reactions (≥1%) associated with Taltz treatment are injection site reactions, upper respiratory tract infections, nausea, and tinea infections. Overall, the safety profiles observed in adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis were consistent with the safety profile in adult patients with plaque psoriasis, with the exception of influenza and conjunctivitis in psoriatic arthritis and conjunctivitis, influenza, and urticaria in pediatric psoriasis.
Please see Brief Summary of Prescribing Information on the following pages. Please see Instructions for Use included with the device.
IX HCP ISI 07MAY2020
References: 1. Data on file. Lilly USA, LLC. DOF-IX-US-0304. 2. Mease PJ, van der Heijde D, Ritchlin CT, et al; on behalf of SPIRIT-P1 Study Group. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017;76:79-87. 3. Nash P, Kirkham B, Okada M, et al; on behalf of SPIRIT-P2 Study Group. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317-2327. 4. Taltz. Prescribing information. Lilly, USA. LLC. 5. Mease PJ, van der Heijde D, Ritchlin CT, et al; on behalf of SPIRIT-P1 Study Group. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebocontrolled and active (adalimumab)-controlled period of the phase 3 trial SPIRIT-P1. Ann Rheum Dis. 2017;76(suppl):1-30. 6. Nash P, Kirkham B, Okada M, et al; on behalf of SPIRIT-P2 Study Group. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317-2327. Supplementary appendix. 7. Chabra S, Gill BJ, Gallo G, et al. Ixekizumab citrate-free formulation: results from two clinical trials. Adv Ther. 2022;Epub (Incl Suppl Inf):1-11, 1-4. https://doi.org/10.1007/s12325-022-02126-0.
Taltz® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-IX-US-5471 05/2022 ©Lilly USA, LLC 2022. ALL RIGHTS RESERVED.
VAS=Visual Analogue Scale; LSM=least squares mean; PA=prior authorization.
original formulation is depleted, Only citrate-free formulation will be available
Simple transition to Taltz Citrate-Free4 Taltz is FDA approved in a citrate-free formulation 4 ¶P<.0001; based on VAS 0-100 ‡Same active ingredient §Vs original formulation; immediately after injection; based on VAS 0-100 Same Taltz,‡ less injection site pain§ VAS Injection Site Pain Score Immediately Following Injection7 LSM VAS SCORE 0 20 40 60 Taltz 80 mg original formulation (n=61) Taltz 80 mg Citrate-Free formulation (n=63) 3.5¶ 25.2
Taltz® (ixekizumab) injection
Brief Summary: Consult the package insert for complete prescribing information. INDICATIONS AND USAGE
Plaque Psoriasis—Taltz is indicated for the treatment of patients aged 6 years and older with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
Psoriatic Arthritis—Taltz is indicated for the treatment of adult patients with active psoriatic arthritis. Ankylosing Spondylitis—Taltz is indicated for the treatment of adult patients with active ankylosing spondylitis.
Non-radiographic Axial Spondyloarthritis—Taltz is indicated for the treatment of adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation. CONTRAINDICATIONS
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients (Warnings and Precautions) WARNINGS AND PRECAUTIONS
Infections—Taltz may increase the risk of infection. In clinical trials in adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). Upper respiratory tract infections, oral candidiasis, conjunctivitis and tinea infections occurred more frequently in the Taltz group than in the placebo group. A similar increase in risk of infection was seen in placebo-controlled trials in patients with pediatric psoriasis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis (Adverse Reactions)
Instruct patients treated with Taltz to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a serious infection or is not responding to standard therapy, monitor the patient closely and discontinue Taltz until the infection resolves. Pre-treatment Evaluation for Tuberculosis—Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Consider anti-TB therapy prior to initiating Taltz in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Patients receiving Taltz should be monitored closely for signs and symptoms of active TB during and after treatment.
Hypersensitivity—Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz (Adverse Reactions). If a serious hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy. Inflammatory Bowel Disease—Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than in the control group (Adverse Reactions). During Taltz treatment, monitor for onset or exacerbation of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management. Immunizations—Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz. No data are available on the response to live vaccines.
ADVERSE REACTIONS
The following adverse drug reactions are discussed in greater detail in other sections of the label:
• Infections (Warnings and Precautions)
• Hypersensitivity Reactions (Contraindications and Warnings and Precautions)
• Inflammatory Bowel Disease (Warnings and Precautions)
Clinical Trials Experience—Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Plaque Psoriasis
Weeks 0 to 12: Three placebo-controlled trials in subjects with plaque psoriasis were integrated to evaluate the safety of Taltz compared to placebo for up to 12 weeks. A total of 1167 subjects (mean age 45 years; 66% men; 94% White) with plaque psoriasis received Taltz (160 mg at Week 0, 80 mg every 2 weeks [Q2W] for 12 weeks) subcutaneously. In two of the trials, the safety of Taltz (use up to 12 weeks) was also compared with an active comparator, U.S. approved etanercept.
In the 12-week, placebo-controlled period, adverse events occurred in 58% of the Taltz Q2W group (2.5 per subject-year of follow-up) compared with 47% of the placebo group (2.1 per subject-year of follow-up). Serious adverse events occurred in 2% of the Taltz group (0.07 per subject-year of follow-up), and in 2% of the placebo group (0.07 per subject-year of follow-up). Table 1 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the Taltz group than the placebo group during the 12-week placebo-controlled period of the pooled clinical trials.
Table 1: Adverse Reactions Occurring in ≥1% of the Taltz Group and More Frequently than in the Placebo Group in the Plaque Psoriasis Clinical Trials through Week 12 Adverse Reactions Taltz 80 mg Q2W (N=1167) (n%) Etanerceptb (N=287) (n%) Placebo (N=791) (n%)
Injection site reactions 196 (17) 32 (11) 26 (3)
Upper respiratory tract infectionsa 163 (14) 23 (8) 101 (13)
Nausea 23 (2) 1 (<1) 5 (1)
Tinea infections 17 (2) 0 1 (<1)
a Upper respiratory tract infections cluster includes nasopharyngitis and rhinovirus infection. b U.S. approved etanercept.
Adverse reactions that occurred at rates less than 1% in the Taltz group and more frequently than in the placebo group during the 12-week induction period included rhinitis, oral candidiasis, urticaria, influenza, conjunctivitis, inflammatory bowel disease, and angioedema.
Weeks 13 to 60: A total of 332 subjects received the recommended maintenance regimen of Taltz 80 mg dosed every 4 weeks. During the maintenance period (Weeks 13 to 60), adverse events occurred in 80% of subjects treated with Taltz (1.0 per subject-year of follow-up) compared to 58% of subjects treated with placebo (1.1 per subject-year of follow-up). Serious adverse events were reported in 4% of subjects treated with Taltz (0.05 per subject-year of follow-up) and none in the subjects treated with placebo.
Weeks 0 to 60: Over the entire treatment period (Weeks 0 to 60), adverse events were reported in 67% of subjects treated with Taltz (1.4 per subject-year of follow-up) compared to 48% of subjects treated with placebo (2.0 per subject-year of follow-up). Serious adverse events were reported in 3% of subjects treated with Taltz (0.06 per subject-year of follow-up), and in 2% of subjects treated with placebo (0.06 per subject-year of follow-up).
Specific Adverse Drug Reactions: Injection Site Reactions: The most frequent injection site reactions were erythema and pain. Most injection site reactions were mild-to-moderate in severity and did not lead to discontinuation of Taltz.
Infections: In the 12-week, placebo-controlled period of the clinical trials in plaque psoriasis, infections occurred in 27% of subjects treated with Taltz (1.2 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.4% of subjects treated with Taltz (0.02 per subject-year of follow-up) and in 0.4% of subjects treated with placebo (0.02 per subject-year of follow-up) (Warnings and Precautions)
During the maintenance treatment period (Weeks 13 to 60), infections occurred in 57% of subjects treated with Taltz (0.70 per subject-year of follow-up) compared to 32% of subjects treated with placebo (0.61 per subject-year of follow-up). Serious infections occurred in 0.9% of subjects treated with Taltz (0.01 per subject-year of follow-up) and none in the subjects treated with placebo.
Over the entire treatment period (Weeks 0 to 60), infections were reported in 38% of subjects treated with Taltz (0.83 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.7% of subjects treated with Taltz (0.02 per subject-year of follow-up), and in 0.4% of subject treated with placebo (0.02 per subject-year of follow-up).
Inflammatory Bowel Disease: In adult subjects with plaque psoriasis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the TALTZ 80 mg Q2W group (Crohn’s disease 0.1%, ulcerative colitis 0.2%) than the placebo group (0%) during the 12-week, placebo-controlled period in clinical trials (Warnings and Precautions).
Laboratory Assessment of Cytopenia:
Neutropenia—Over the entire treatment period (Weeks 0 to 60), neutropenia occurred in 11% of subjects treated with Taltz (0.24 per subject-year of follow-up) compared to 3% of subjects treated with placebo (0.14 per subject-year of follow-up). In subjects treated with Taltz, the incidence rate of neutropenia during Weeks 13 to 60 was lower than the incidence rate during Weeks 0 to 12.
In the 12-week, placebo-controlled period, neutropenia ≥ Grade 3 (<1,000 cells/mm3) occurred in 0.2% of the Taltz group (0.007 per subject-year of follow-up) compared to 0.1% of the placebo group (0.006 per subject-year of follow-up). The majority of cases of neutropenia were either Grade 2 (2% for Taltz 80 mg Q2W versus 0.3% for placebo; ≥1,000 to <1,500 cells/mm3) or Grade 1 (7% for Taltz 80 mg Q2W versus 3% for placebo; ≥1,500 cells/mm3 to <2,000 cells/mm3). Neutropenia in the Taltz group was not associated with an increased rate of infection compared to the placebo group.
Thrombocytopenia—Ninety eight percent of cases of thrombocytopenia were Grade 1 (3% for Taltz 80 mg Q2W versus 1% for placebo; ≥75,000 cells/mm3 to <150,000 cells/mm3). Thrombocytopenia in subjects treated with Taltz was not associated with an increased rate of bleeding compared to subjects treated with placebo.
Active Comparator Trials: In the two clinical trials that included an active comparator, the rate of serious adverse events during weeks zero to twelve was 0.7% for U.S.-approved etanercept and 2% for Taltz 80 mg Q2W, and the rate of discontinuation from adverse events was 0.7% for U.S. approved etanercept and 2% for Taltz 80 mg Q2W. The incidence of infections was 18% for U.S. approved etanercept and 26% for Taltz 80 mg Q2W. The rate of serious infections was 0.3% for both Taltz 80 mg Q2W and U.S. approved etanercept.
Pediatric Plaque Psoriasis
Taltz was evaluated in a placebo-controlled trial in pediatric subjects with moderate-to-severe psoriasis 6 to less than 18 years of age. A total of 171 subjects were studied (115 subjects on Taltz and 56 subjects on placebo). Overall, the safety profile observed in pediatric subjects with plaque psoriasis treated with Taltz every 4 weeks is consistent with the safety profile in adult subjects with plaque psoriasis with the exception of the frequencies of conjunctivitis (2.6%), influenza (1.7%), and urticaria (1.7%).
In this clinical trial, Crohn’s disease occurred at a greater frequency in the Taltz group (0.9%) than the placebo group (0%) during the 12-week, placebo-controlled period. Crohn’s disease occurred in a total of 4 Taltz treated subjects (2.0%) in the clinical trial (Warnings and Precautions).
Psoriatic Arthritis
Taltz was studied in two placebo-controlled trials in patients with psoriatic arthritis. A total of 678 patients were studied (454 patients on Taltz and 224 on placebo). A total of 229 patients in these trials received Taltz 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with psoriatic arthritis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis with the exception of the frequencies of influenza (1.3%) and conjunctivitis (1.3%).
Taltz® (ixekizumab) injection IX HCP BS 06JAN2022 Taltz® (ixekizumab) injection IX HCP BS 06JAN2022
Ankylosing Spondylitis
Taltz was studied in two placebo-controlled trials in patients with ankylosing spondylitis. A total of 566 patients were studied (376 patients on Taltz and 190 on placebo). A total of 195 patients in these trials received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with ankylosing spondylitis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis.
In adult patients with ankylosing spondylitis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred in 2 patients (1.0%) and 1 patient (0.5%), respectively, in the Taltz 80 mg Q4W group and 1 patient (0.5%) and 0%, respectively, in the placebo group during the 16-week, placebo-controlled period in clinical trials. Of these patients, serious events occurred in 1 patient in the Taltz 80 mg Q4W group and 1 patient in the placebo group (Warnings and Precautions).
Non-radiographic Axial Spondyloarthritis
Taltz was studied in a placebo-controlled trial in patients with non-radiographic axial spondyloarthritis. A total of 303 patients were studied (198 patients on Taltz and 105 on placebo). A total of 96 patients in this trial received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with non-radiographic axial spondyloarthritis treated with Taltz 80 mg Q4W up to Week 16 is consistent with the previous experience of Taltz in other indications. Immunogenicity—As with all therapeutic proteins, there is the potential for immunogenicity with Taltz. The assay to test for neutralizing antibodies has limitations detecting neutralizing antibodies in the presence of ixekizumab; therefore, the incidence of neutralizing antibodies development could be underestimated.
Plaque Psoriasis Population
By Week 12, approximately 9% of adult subjects treated with Taltz every 2 weeks developed antibodies to ixekizumab. Approximately 22% of subjects treated with Taltz at the recommended dosing regimen developed antibodies to ixekizumab during the 60-week treatment period. The clinical effects of antibodies to ixekizumab are dependent on the antibody titer; higher antibody titers were associated with decreasing drug concentration and clinical response.
Of the adult subjects who developed antibodies to ixekizumab during the 60-week treatment period, approximately 10%, which equates to 2% of subjects treated with Taltz at the recommended dosing regimen, had antibodies that were classified as neutralizing. Neutralizing antibodies were associated with reduced drug concentrations and loss of efficacy.
In pediatric psoriasis subjects treated with ixekizumab at the recommended dosing regimen up to 12 weeks, 21 subjects (18%) developed anti-drug antibodies, 5 subjects (4%) had confirmed neutralizing antibodies associated with low drug concentrations. No conclusive evidence could be obtained on the potential association of neutralizing antibodies and clinical response and/or adverse events due to small number of pediatric subjects in the study.
Psoriatic Arthritis Population
For subjects treated with Taltz 80 mg every 4 weeks for up to 52 weeks (PsA1), 11% developed anti-drug antibodies, and 8% had confirmed neutralizing antibodies.
Ankylosing Spondylitis Population
For patients treated with Taltz 80 mg every 4 weeks for up to 16 weeks (AS1, AS2), 5.2% developed anti-drug antibodies, and 1.5% had neutralizing antibodies.
Non-radiographic Axial Spondyloarthritis Population
Of patients treated with Taltz 80 mg every 4 weeks for up to 52 weeks (nr-axSpA1), 8.9% developed anti-drug antibodies, all of which were low titer. No patient had neutralizing antibodies. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to Taltz across indications or with the incidences of antibodies to other products may be misleading.
Postmarketing Experience—The following adverse reactions have been identified during postapproval use of Taltz. Because the reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to Taltz exposure.
Immune system disorders: anaphylaxis (Contraindications and Warnings and Precautions)
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Pregnant women should be encouraged to enroll themselves in the registry by calling 1-800-284-1695.
Risk Summary—There are no available data on Taltz use in pregnant women to inform any drug associated risks. Human IgG is known to cross the placental barrier; therefore, Taltz may be transmitted from the mother to the developing fetus. An embryofetal development study conducted in pregnant monkeys at doses up to 19 times the maximum recommended human dose (MRHD) revealed no evidence of harm to the developing fetus. When dosing was continued until parturition, neonatal deaths were observed at 1.9 times the MRHD [see Data]. The clinical significance of these nonclinical findings is unknown.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data Animal Data—An embryofetal development study was conducted in cynomolgus monkeys administered ixekizumab. No malformations or embryofetal toxicity were observed in fetuses
from pregnant monkeys administered ixekizumab weekly by subcutaneous injection during organogenesis to near parturition at doses up to 19 times the MRHD (on a mg/kg basis of 50 mg/ kg/week). Ixekizumab crossed the placenta in monkeys.
In a pre- and post-natal development toxicity study, pregnant cynomolgus monkeys were administered weekly subcutaneous doses of ixekizumab up to 19 times the MRHD from the beginning of organogenesis to parturition. Neonatal deaths occurred in the offspring of two monkeys administered ixekizumab at 1.9 times the MRHD (on a mg/kg basis of 5 mg/kg/week) and two monkeys administered ixekizumab at 19 times the MRHD (on a mg/kg basis of 50 mg/kg/ week). These neonatal deaths were attributed to early delivery, trauma, or congenital defect. The clinical significance of these findings is unknown. No ixekizumab-related effects on functional or immunological development were observed in the infants from birth through 6 months of age.
Lactation
Risk Summary—There are no data on the presence of ixekizumab in human milk, the effects on the breastfed infant, or the effects on milk production. Ixekizumab was detected in the milk of lactating cynomolgus monkeys. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Taltz and any potential adverse effects on the breastfed infant from Taltz or from the underlying maternal condition.
Pediatric Use—The safety and effectiveness of Taltz have been established in pediatric subjects aged 6 years to less than 18 years with moderate-to-severe plaque psoriasis. The safety and effectiveness of Taltz in other pediatric indications and for pediatric subjects less than 6 years of age have not been established.
Geriatric Use—Of the 4204 psoriasis subjects exposed to Taltz, a total of 301 were 65 years or older, and 36 subjects were 75 years or older. Although no differences in safety or efficacy were observed between older and younger subjects, the number of subjects aged 65 and over is not sufficient to determine whether they respond differently from younger subjects.
PATIENT COUNSELING INFORMATION—Advise the patient and/or caregiver to read the FDAapproved patient labeling (Medication Guide and Instructions for Use) before the patient starts using Taltz and each time the prescription is renewed, as there may be new information they need to know.
Instructions on Self-Administration: Provide guidance to patients and caregivers on proper subcutaneous injection technique, including aseptic technique, and how to use the autoinjector or prefilled syringe correctly (Instructions for Use)
Infection: Inform patients that Taltz may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider, and contacting their healthcare provider if they develop any symptoms of infection (Warnings and Precautions).
Allergic Reactions: Advise patients to seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions (Warnings and Precautions).
Pregnancy: Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Advise patients to contact the registry at 1-800-284-1695 to enroll (Use in Specific Populations)
Additional information can be found at www.Taltz.com
See
Taltz® (ixekizumab) HCP BS 06JAN2022 Taltz® (ixekizumab)
injection IX
injection IX HCP BS 06JAN2022
Instructions for Use accompanying the product device. Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA Copyright © 2016, 2017, 2019, 2020, 2021, 2022 Eli Lilly and Company. All rights reserved. IX HCP BS 06JAN2022 PP-IX-US-5359




EARLY BIRD REGISTRATION: $795.00 BY AUGUST 31, 2022 FOR MORE INFORMAION: 7 8 7 4 0 8 0 0 2 4 i n f o @ a d v a n c e d a n t i a g i n g s u m m i t . c o m 1 A D V A N C E D A N T I - A G I N G S U M M I T D A T E P L A C E : F A I R M O N T E L H O T E L S A N J U A N , I S L A V E R D E , P U E R T O R I C O S T S A V E T H E N O V E M B E R 5 , 2 0 2 2
EDITOR FUNDADOR Juan Carlos Orengo Valverde, MD, MPH, PhD EDITOR Alberto Santiago Cornier, MD, PhD CONSEJO ASESOR Oscar Soto Raíces, MD, Ahmed Morales, MD, FACP, FACG, FASGE, AGAF, Lcda. Wanda González PRINCIPAL OFICIAL EJECUTIVO Pedro Carlos Lugo Hernández III, P.A.C. PRESIDENTA Y FUNDADORA Glorybelle Hernández Figueroa, MBA VICEPRESIDENTA Y FUNDADORA Laila Paloma Lugo, MBA CONTABILIDAD Julio Soto ADMINISTRACIÓN Marta Ivelisse Vélez Ramos, MBA, MARKETING Y SERVICIOS 360 Alexelena Cayere, Yasmin Morell PERIODISTAS Belinda Burgos, Grenda Rivera, Mayra Acevedo, Luis Penchi, Limarys Suárez DIRECCIÓN GRÁFICA Natalia Zoé Rivera Torres DIRECTORA AUDIOVISUAL Fabiola Plaza REALIZADORA AUDIOVISIAL Salomé Mateus FOTOS Revista Medicina y Salud Pública DIRECCIÓN GENERAL / FUNDADOR Carlos Alexis Lugo Marrero DISTRIBUCIÓN OFICINAS Y TORRES MÉDICAS Editorial Mundo ENVÍO DE REVISTAS Y DISTRIBUCIÓN A GRUPOS MÉDICOS Servicio de correo postal/Comunicación Inteligente Para ventas y otros servicios pueden comunicarse al 787.848.3333, msp@editorialmundo.com o www.medicinaysaludpublica.com Revista Puertorriqueña de Medicina y Salud Pública ISSN 1937-8521
COMITÉ EDITORIAL CIENTÍFICO

COMITÉ EDITORIAL Olga Rodríguez, MD - Decana Escuela de Medicina de Ponce (Puerto Rico), Vivian Green, LND, MS, PhD, Sub editora y fundadora (Puerto Rico), José Cordero, MD, MPH - Exdecano Escuela Graduada Salud Pública Recinto de Ciencias Médicas UPR (Puerto Rico), Ángeles Rodríguez, MD, MPH (Puerto Rico), Simón Carlo, MD (Puerto Rico), Bárbara Rosado, MD (Puerto Rico), Idhaliz Flores PhD (Puerto Rico), Jesús Cruz-Correa, MD, FACOG (Puerto Rico), Rafael Bredy, MD, LicMTo, MBE, MS (Puerto Rico), David Caseida, MD, FACOG, (Puerto Rico), José Capriles, MD, MHSA (Puerto Rico) Joaquín Laboy, MD, FACOG (Puerto Rico), Luis Adrian Rivera Pomales, MD, PEMBA, MPH, CMQ (Puerto Rico), Juan Fernández, MS, PhD (Puerto Rico), Nuria Sebate, MD (Puerto Rico), Pedro Amador, MD, MPH (Puerto Rico), Nydia Cappas, PsyD (Puerto Rico), Luis Franco, MD (Puerto Rico), Federico Montealegre, DVM, PhD, Msc (Puerto Rico), Nydia Ortiz, PsyD (Puerto Rico), José Pons, PhD, FPPR (Puerto Rico), Esdrás Vélez, JD, MPH (Puerto Rico), Diego Zavala, MSc, PhD, (Puerto Rico), Ana Torres-Martín, MD (Puerto Rico), Julio Cádiz, MD, MPH (Puerto Rico), Rafael Gómez-Cuevas (Colombia), José Javier Orengo, PhD(c) (España), Cesar A. Del Rey, MD (Panamá), Pedro Serrano, MD, PhD (España), Luis Serra-Majem, MD, PhD (España), José Ramón Calvo, MD, PhD (España).
Síguenos en www.medicinaysaludpublica.com, www.facebook.com/revistamsp, en Twitter @revistamsp, en LinkedIn como Revista Puertorriqueña de Medicina y Salud Pública. Las normas editoriales de la Revista Puertorriqueña de Medicina y Salud Pública para la publicación de artículos originales y cartas al editor pueden ser accesadas en la página web: www.medicinaysaludpublica.com, y solicitadas a través de msp@editorialmundo.com.
Medicina y Salud Pública es propiedad de publicaciones mundo. Medicina es una publicación de la REVISTA PUERTORRIQUEÑA DE MEDICINA Y SALUD PÚBLICA. Medicina y Salud Pública tiene como política corregir y aclarar cualquier información incorrecta que pueda ser publicada en su revista. Medicina y Salud Pública no asume responsabilidad alguna por los anuncios, artículos y otros servicios anunciados en nuestra publicación.
Revista Puertorriqueña de Medicina y Salud Pública 2D FRENTE 3DHD EL CORAZÓN
7 CONTENIDO 08 22 34 62 58 ECOANSIEDAD: ENFERMOS POR EL PLANETA MANEJO DE CÁNCER DE PRÓSTATA: PROSTATECTOMÍA RADICAL LAPAROSCÓPICA
A LA PROSTATECTOMÍA RADICAL LAPAROSCÓPICA
MIOMATOSIS UTERINA LA QUIMIOTERAPIA Y
CUIDADO DEL PACIENTE CON DIABETES EN TEMPORADA DE HURACANES


Revista Puertorriqueña de Medicina y Salud Pública8 MANEJO DECÁNCER DE PRÓSTATA: Prostatectomía Radical Laparoscópica 2D frente a la Prostatectomía Radical Laparoscópica 3DHD
 Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública
9
ABSTRACTO
Propósito: Nuestro estudio compara los resultados perioperatorios entre la prostatectomía radical laparos cópica (LRP) bidimensional (2D) y la LRP tridimensional / de alta definición (3DHD) de 4ª generación. Métodos: Se evaluaron los datos adquiridos retrospectiva mente de pacientes que se sometieron a LRP 2D (n = 75) y LRP 3DHD (n = 75) desde marzo de 2013 hasta octubre de 2015. Los procedimientos fueron realizados por un solo cirujano. Se utilizó el abordaje extraperi toneal con 5 trócares. Los resultados perioperatorios, la potencia y la continencia se compararon entre los grupos. Resultados: Las características de los pa cientes fueron similares entre los dos grupos en cuanto a edad (p=0,44), niveles de antígeno prostático es pecífico (PSA) (p=0,34) y puntuaciones de Gleason (p=0,14). El índice de masa corporal (IMC) fue signi ficativamente mayor en el grupo 3DHD (p=0,0036). Después de la operación, no se observaron diferen cias significativas en la pérdida de Hgb (p = 0,50), los márgenes quirúrgicos positivos (p = 1,00) y las puntuaciones de Gleason postoperatorias (p = 0,30). Se observaron diferencias significativas en cuanto a la duración de la estancia hospitalaria (p<0,001) y el drenaje de Jackson-Pratt (JP) (p<0,001). En cuanto a la potencia, el 73,7% y el 51,6% de los pacientes en los grupos 3DHD y 2D recuperaron la potencia a los 6 meses, respectivamente (p=0,0025). Casi el 43% de los pacientes en el grupo 3DHD recuperaron la conti nencia a 1 mes, mientras que para los grupos 2D fue solo el 17,3% (p = 0,0008). Conclusión: 3DHD y 2D LRP han resultado en buenos resultados en los pe ríodos perioperatorios. Nuestros resultados muestran una disminución del drenaje de JP, una menor duración de la estancia hospitalaria, un retorno más temprano del control urinario y un retorno más temprano de la función sexual en el grupo de LRP 3DHD. En centros de menor volumen donde el equipo de robótica no es factible debido a las barreras económicas, 3DHD se puede realizar de manera segura como una alternati va mínimamente invasiva.
PALABRAS CLAVE
Continencia; Prostatectomía laparoscópica; Prostatectomía laparoscópica tridimensional de alta definición, prostatectomía abierta; Cáncer de próstata; Prostatectomía; Prostatectomía asistida por robot.
KEY WORDS
Continence; laparoscopic prostatectomy; High-definition three-dimensional laparoscopic prostatectomy, open prostatectomy; Prostate cancer; Prostatectomy; Robotic-assisted prostatectomy.
ABSTRACT
Purpose: Our study compares periope rative outcomes between two-dimensional (2D) laparoscopic radical prostatectomy (LRP) and 4th generation three-dimen sional/high-definition (3DHD) LRP. Methods: Retrospectively acquired data from patients who underwent 2D LRP (n = 75) and 3DHD LRP (n = 75) from March 2013 to October 2015 were evaluated. Procedures were performed by a single surgeon. The extraperitoneal approach with 5 trocars was used. Perioperative outcomes, potency, and continence were compared between groups. Results: Pa
tient characteristics were similar between the two groups in terms of age (p=0.44), prostate-specific antigen (PSA) levels (p=0.34), and Gleason scores (p=0.14). ). Body mass index (BMI) was significantly higher in the 3DHD group (p=0.0036). Postoperatively, no significant differences were observed in Hgb loss (p = 0.50), positive surgical margins (p = 1.00), and postoperative Gleason scores (p = 0.30).
Significant differences were observed for length of hospital stay (p<0.001) and Jackson-Pratt (JP) drainage (p<0.001). Regarding potency, 73.7% and 51.6% of patients in the 3DHD and 2D groups re
gained potency at 6 months, respectively (p=0.0025). Almost 43% of the patients in the 3DHD group regained continence at 1 month, while for the 2D groups it was only 17.3% (p = 0.0008). Conclusion: 3DHD and 2D LRP have resulted in good results in the perioperative periods. Our results show decreased JP drainage, shorter len gth of hospital stay, earlier return of uri nary control, and earlier return of sexual function in the LRP 3DHD group. In lower volume centers where robotics equipment is not feasible due to economic barriers, 3DHD can be performed safely as a mini mally invasive alternative.
Revista Puertorriqueña de Medicina y Salud Pública10
INTRODUCCIÓN
El cáncer de próstata es el cáncer más común diagnosticado en los hombres y la segunda causa principal de muertes re lacionadas con el cáncer en los Estados Unidos. Se estima que 268,490 hombres serán diagnosticados y 34,500 hombres morirán de cáncer de próstata en 2022 [1]. En 2020, casi 1,4 millones de hom bres fueron diagnosticados con cáncer de próstata, lo que representa el 15% de los cánceres diagnosticados en hombres de todo el mundo. Los países menos de sarrollados tienen una menor incidencia pero mayores tasas de mortalidad por cáncer de próstata que los países más desarrollados (Australia/Nueva Zelanda, América del Norte, Europa). Tangen et al. (2012) afirmaron que el uso de herra mientas de detección como las pruebas de antígeno prostático específico (PSA) y los tratamientos mejorados en los países más desarrollados permite una detección más temprana del cáncer de próstata y una disminución de las tasas de morta lidad [2].
Puerto Rico posee una población es timada de 3.200.000 habitantes. Los puertorriqueños son particularmente vul nerables a las disparidades del cáncer debido a las cualidades socioeconómi cas. En 2020, aproximadamente el 44% de la población en Puerto Rico vivía en la pobreza, en comparación con el 17% de los hispanos estadounidenses y el 9% de los blancos no hispanos (NHW) que viven en los Estados Unidos continenta les. Chinea et al. (2017) [3] informaron que los subgrupos hispanos/latinos (H/L) tienen diferentes tasas de mortalidad es pecífica por cáncer de próstata (PCSM) en comparación con los hombres negros no hispanos (NHB) de NHW, utilizan do datos de 2000-2013 que incluyeron 486,865 hombres. Las tasas de inciden cia y mortalidad por cáncer de próstata (PCa) en hombres H/L fueron similares a las de NHW; sin embargo, los hombres puertorriqueños hispanos/latinos (PR H/L) tenían un PCSM significativamente más alto que el NHW y tenían la mayor mortalidad entre los subgrupos hispanos. En 2018, la tasa general de incidencia de PCa en Puerto Rico fue de 145.2 ca sos por 100,000 habitantes y una tasa de mortalidad ajustada por edad de 18.2 muertes por cada 100,000 habitan tes (Figura 1).
Tenemos como objetivo avanzar signifi cativamente en la investigación de la bio logía del cáncer de próstata letal (PCa) y además reducir la carga de las dispa ridades de salud de PCa en los hombres puertorriqueños hispanos / latinos (PR H / L). El PCa es el cáncer más prevalente, tanto en términos de incidencia como de mortalidad en Puerto Rico. En este escri to exponemos una técnica costo efectiva para tratar a pacientes con cáncer de próstata. Esto tomando en consideración la importancia de proveer la mejor tecno logía de tratamiento posible a población con desventajas económicas.
Aunque la primera prostatectomía radi cal laparoscópica 2D (PRL) se realizó en 1992 [4], se informaron modificaciones adicionales en 1999 a la técnica original que resultaron en una disminución de la pérdida de sangre y la duración de la es tancia hospitalaria [5]. Esta técnica no lo gró un uso generalizado a pesar de ofre cer ventajas sobre el estándar de criterio de prostatectomía radical abierta (ORP) debido a su empinada curva de apren dizaje de 40-60 casos [6]. En compa ración con los sistemas visuales 2D, la 1ª y 2ª generación de laparoscopia 3D se vieron limitadas por la reducción de la resolución, la iluminación más tenue y la visualización periférica borrosa que dio lugar a síntomas de fatiga ocular, dolor de cabeza y náuseas [7]. Los sistemas
LRP 3D de tercera generación causaron fatiga al cirujano ya que los cirujanos te nían que usar un casco pesado que aco modaba la visión 3D [8]
La primera prostatectomía radical asisti da por robot utilizando el sistema da Vin ci se realizó en 2001 [9]. Con una curva de aprendizaje de 10-20 casos [6] y una visión 3D cómoda, se está realizando un número creciente de prostatectomías radicales con el enfoque asistido por ro bot, suplantando a cirugía convencional abierta (CCA) como la alternativa de prostatectomía radical más común [10]. En comparación con la CCA, los estudios informan que los enfoques mínimamente invasivos (es decir, LRP y prostatectomía radical asistida por robótica [RARP]) re ducen la pérdida de sangre, la duración de la estancia hospitalaria y las tasas de transfusión, al tiempo que mantienen los mismos resultados funcionales y oncoló gicos [11]. Para obtener las ventajas del enfoque laparoscópico mínimamente in vasivo sin las limitaciones vinculadas a la LRP 2D o a generaciones anteriores de LRP 3D, los cirujanos han preferido elegir el enfoque asistido por robótica a pesar de los costos más altos [12].
Casi 20 años después del primer LRP, se inventó el laparoscopio 3D de alta defini ción de 4ª generación caracterizado por la tecnología “Patterned Retarder tech nology” de tipo Película. En 2013, Buchs
Revista Puertorriqueña de Medicina y Salud Pública 11
Tabla 1. Características preoperatorias de los grupos de estudio.
Variables 2D (n=75) 3D (n=75) p-valor
Edad ± SD ± 6,1 ± 6,7 0.44
PSA ± SD (ng/ml) ± 3.3 ± 3.0 0.34
Puntuación de Gleason para biopsia preoperatoria 0.14
≤5 (0%) (2%)
6(3+3) (77%) (64%) (16%) (22%) (3%) (8%) (4%) (4%)
IMC ± SD ± 3,4 ± 4,2 0.0036*
2D-LRP: prostatectomía radical laparoscópica de 2 dimensiones, 3D-LRP: prostatectomía radical laparoscópica de 3 dimensiones, PSA: antígeno prostático específico, SD: desviación estándar
et al. informó que con el sistema visual 3DHD, se obtuvo percepción de profundi dad sin fatiga visual. Las gafas son lige ras, cómodas y no perjudican la visión al apartar la vista de la pantalla [13]. Con una visión aumentada que facilita cómo damente la navegación espacial, el nue vo sistema visual 3DHD parece abordar las limitaciones de los sistemas visuales 2D y los sistemas visuales 3D de genera ciones anteriores. Dado que los estudios comparativos con respecto a los resul tados perioperatorios, de continencia y potencia entre el LRP 2D y el LRP 3DHD son escasos, tenemos la intención de comparar ambas técnicas para evaluar la seguridad del LRP 3DHD y discutirlo como una posible alternativa al costoso RARP en centros de bajo volumen. Con el aumento de la popularidad de RARP, nuestros hallazgos son relevantes no solo para los urólogos en centros de volumen medio o bajo, sino también para los pro veedores de atención médica en general, al considerar factores económicos.
MÉTODOS
Población del estudio: Se evaluaron los datos adquiridos retrospectivamen te de 150 pacientes que se sometieron a cirugía de prostatectomía desde mar zo de 2013 hasta octubre de 2015. La Junta de Revisión Institucional de PHSU/ PRI revisó y aprobó el estudio (IRB no. 1909021277R001). Los pacientes (n = 150) incluidos en este estudio se estrati
ficaron en dos grupos: un grupo se so metió a LRP 2D (n = 75) y otro grupo se sometió a 4ª generación de LRP 3DHD (n = 75). Todos los procedimientos fueron realizados por un solo cirujano utilizan do el abordaje extraperitoneal con 5 tró cares en ambos grupos. El cirujano tiene mucha experiencia, habiendo realizado más de 500 LRP. Se intentó preservar los nervios en todos los pacientes. Se extra jeron los datos epidemiológicos y clíni cos para cada paciente.
Evaluación de los resultados clínicos: Los resultados perioperatorios se com pararon entre los grupos de estudio. La continencia se evaluó a los 1, 3, 6 y 12 meses. Los pacientes se definieron como “secos” cuando usaban 0-1 almohadilla al día. La potencia se evaluó a los 6 y 12 meses. La potencia se definió como la capacidad de lograr una erección su ficiente para las relaciones sexuales al menos el 50% de las veces.
Análisis estadístico: Los datos epide miológicos y clínicos fueron analizados utilizando tablas de contingencia. La sig nificación estadística se evaluó mediante pruebas de Chi-cuadrado o exactas de Fisher, según sea necesario. P<0,05 se consideró estadísticamente significativo.
RESULTADOS
Características del paciente preoperatorio
Las características de los pacientes se resumen en la Tabla 1. No se detectaron
diferencias significativas entre los grupos de estudio en cuanto a la edad, el nivel de PSA preoperatorio y las puntuaciones de Gleason de biopsia preoperatoria. El índice de masa corporal (IMC) fue signi ficativamente mayor en el grupo de LRP 3D (p = 0,0036).
Resultados postoperatorios
Los resultados postoperatorios se resu men en la Tabla 2. Todas las cirugías fueron exitosas. La conversión a cirugía abierta no fue necesaria y no se produje ron complicaciones que requirieran una reintervención temprana. Hubo esteno sis anastomótica en el cuello de vejiga en menos del 3% de todos los pacien tes; no se informó perforación rectal ni transfusiones de sangre para ninguno de los grupos. La pérdida media de HgB es comparable entre los grupos, aunque fue ligeramente menor en el gru po de LRP 2D (p = 0,50). Los pacientes operados por 3DHD LRP, en promedio, fueron dados de alta 10 horas antes del hospital (p<0.001). Además, los pacien tes con LRP 3D drenaron, en promedio, 90 ml menos en las primeras 24 horas (p<0,001). Aunque no es estadísticamen te significativo, se observó un porcentaje ligeramente menor de márgenes quirúr gicos positivos en el grupo de LRP 3DHD (10% vs 13%). Las puntuaciones de Glea son postoperatorias fueron similares en ambos grupos.
Revista Puertorriqueña de Medicina y Salud Pública12
LRP
LRP
62,5
62,7
5.0
6.0
0
1
58
48
7(3+4) 12
17
7(4+3) 2
6
≥8 3
3
27,4
9,1
Erección
Los datos de potencia se resumen en la Tabla 3. Sólo los pacientes considera dos potentes antes de la cirugía fueron incluidos en este análisis. Un total de 62 pacientes en el grupo de LRP 2D y 61 pacientes en el LRP 3D se consideraron potentes antes del procedimiento. Los pacientes fueron considerados potentes antes y después del procedimiento si po dían lograr erecciones suficientes para las relaciones sexuales el 50% de las veces. Seis meses después del procedi miento, casi el 74% de los pacientes en el LRP 3D informaron ser potentes, en com paración con casi el 52% en los grupos de LRP 2D (p = 0,0025). A los 12 meses, 16 pacientes adicionales en el grupo de LRP 2D y 4 pacientes en el LRP 3D infor maron recuperar la potencia.
Continencia
Los datos de continencia urinaria pre y postoperatoria se resumen en la Tabla 3. Antes de la cirugía, ninguno de los 150 pacientes reportó incontinencia. Después de la cirugía, los pacientes se conside raron “secos” cuando usaron 0-1 almo hadilla por día. A 1 mes, alrededor del 43% de los pacientes en el grupo de LRP 3D recuperaron la continencia, mientras que para el grupo de LRP 2D fue solo el 17% de los pacientes. La mayoría de los pacientes en el grupo de LRP 2D recupe raron la continencia 3 meses después de la cirugía (p = 0,0008).
DISCUSIÓN
Nuestro estudio compara los resultados clínicos de los pacientes que se sometie ron a LRP 2D y 3D. Sorprendentemente, el drenaje JP y la duración de la estancia hospitalaria se redujeron significativa mente en el grupo de LRP 3D. La reduc ción de las fugas podría deberse a una anastomosis uretral-vesicular superior como resultado del aumento de los cam pos visuales y la mayor precisión de la manipulación quirúrgica utilizando lapa roscopios 3DHD reticulantes. La duración de la estancia hospitalaria en nuestro estudio es comparable con la literatura previa que informa duraciones de 1-2 días [14,15]. La estenosis anastomótica en menos del 3% de los casos incluidos en este estudio es comparable a informes anteriores con tasas de estenosis anasto mótica del 1% al 17%. La aposición cui dadosa de la mucosa, la eversión de la mucosa del cuello de la vejiga y el ma
nejo adecuado del tejido que se puede realizar fácilmente con la ayuda de una excelente visualización del área pueden explicar las tasas de complicaciones en nuestro estudio.
Curiosamente, los pacientes con 3DHD LRP informaron un aumento en las tasas de potencia temprana en comparación con el grupo clásico de LRP 2D. Los estu dios sugieren que la función eréctil con tinúa mejorando, en algunos hombres, años después de la prostatectomía radi cal [16]. Al limitar el seguimiento en nues tros pacientes a 12 meses, el porcentaje de pacientes que eventualmente recupe ran la función sexual podría ser mayor que lo que se informa en nuestro estudio. Elpreservamiento del nervio exitoso re quiere una disección precisa, una visuali zación clara, el control de los vasos indi viduales en la superficie de las vesículas seminales y evitar la electro-cauteriza ción lateralmente [17]. Una reconstruc ción tridimensional de la región objetivo facilita en gran medida la precisión de la manipulación quirúrgica y la estima ción de la profundidad anatómica [18], lo que resultaría útil durante los pasos de preservación del nervio. Encontramos que realizar la disección con el endosco pio reticulador y las tijeras articuladoras nos permitió llegar al haz neurovascular desde diferentes ángulos, incluyendo un
fácil acceso a la superficie prostática posterior que no se puede lograr con un endoscopio rígido.
En cuanto a la continencia, los resulta dos de nuestro estudio son sobresalien tes. Los pacientes en el grupo de LRP 3D informaron una recuperación más tem prana del control urinario que el grupo de LRP 2D. La recuperación temprana de la continencia parece estar relacionada con un mayor ahorro de esfínteres y una reducción del trauma [19]. La angulación del endoscopio facilita la visualización del ápice prostático en relación con el esfínter permitiéndonos preservar el es fínter sin riesgo de entrar en la próstata.
A medida que la atención administrada se vuelve más frecuente, los urólogos de ben evaluar críticamente el aspecto eco nómico y la satisfacción del paciente con los patrones de práctica urológica. Los datos de la muestra de pacientes hospi talizados a nivel nacional informaron en 2009 que el 61% de las prostatectomías eran RARP [20]. En los últimos años, el RARP ha reemplazado a la CCA como el enfoque quirúrgico más común para el cáncer de próstata a pesar de la falta de evidencia que demuestre resultados on cológicos o funcionales superiores [21]. A pesar de producir resultados quirúrgi cos similares a costos significativamente más bajos, LRP ha sido constantemente
Tabla 2. Resultados postoperatorios de los grupos de estudio Variables 2D (n=75) LRP 3D (n=75) p-valor
Diferencia de HgB ( media ± DE) ± 0,9 1.3 ± 0.9 0.50
Horas de hospitalización ( media ± DE) 46 ± 0,69 36 ± 12 <0.001*
Drenaje JP de 24 horas ( promedio ± DE) 208 ± 125,7 118 ± 89 <0.001*
Márgenes quirúrgicos positivos 10 (13%) 8 (10%) 1.00
Estenosis anastomótica 2 (2%) 1 (1%) 1.00
Puntuación de Gleason postoperatoria 0.30
≤5 2 (3%) 3 (4%) 6(3+3) 54 (72%) 45 (61%) 7(3+4) 13 (17%) 16 (21%) 7(4+3) 1 (2%) 6 (8%)
≥8 5 (6%) 5 (6%)
2D-LRP: prostatectomía radical laparoscópica de 2 dimensiones, 3D-LRP: prostatectomía radical laparoscópica de 3 dimensiones, SD: desviación estándar, HgB: hemoglobina, JP: Jackson-Pratt
Revista Puertorriqueña de Medicina y Salud Pública 13
LRP
1,2
TABLA 3
Tabla 3. Asociación entre la potencia y el tiempo de recuperación de la continencia y el tipo de cirugía.
Tiempo de recuperación LRP 2D (n=62) LRP 3D (n=61) p-valor
Potencia
6 meses 32 (51.6%) 45 (73.7%) 0.0025
12 meses 16 (25.8%) 4 (6.6%)
Continencia LRP 2D (n=75) LRP 3D (n=75) p-valor
1 mes 13 (17.3%) 32 (42.6%) 0.0008
3 meses 37 (49.3%) 34 (45.3%)
6 meses 15 (20.0%) 3 (4.0%)
12 meses 5 (6.7%) 3 (4.0%)
2D-LRP: prostatectomía radical laparoscópica 2 dimensiones, 3D-LRP: prostatectomía radical laparoscópica tridimensional.
eclipsado por RARP. Estar limitado a una pan talla 2D y una curva de aprendizaje empina da de 40-60 casos ha hecho que los ciruja nos prefieran el enfoque asistido por robótica a pesar de los costos más altos [12]. Queda por documentar si la visualización 3D acorta la curva de aprendizaje de LRP en cirujanos no capacitados. Estudios anteriores han su gerido que la visión 3D es beneficiosa tanto para cirujanos no entrenados como experimen tados [22,23]. Un estudio de Votanopoulos et al. (2008) presenta una ventaja significativa en el uso de imágenes 3D para enseñar habi lidades laparoscópicas a cirujanos inexpertos [24]. Cuando se trata de cirujanos expertos, la transición de imágenes 2D a imágenes 3D se produjo rápidamente sin comprometer la seguridad del paciente [21] .3D la laparosco pia puede reducir los errores de rendimiento al tiempo que aumenta la velocidad en compara ción con la laparoscopia 2D [15]. Se necesitan estudios dirigidos específicamente a estudiar las curvas de aprendizaje de 3DHD LRP.
CONCLUSIÓN
Nuestra serie resultó en una disminución del drenaje de JP, una menor duración de la estan cia hospitalaria, un retorno más temprano de la continencia y un retorno más temprano de la potencia en el grupo de LRP 3D. LRP se pue de realizar de forma segura con la ayuda de las imágenes 3DHD y laparoscopios de reticu lación con excelentes resultados a costos más bajos que RARP. Con el aumento de la popula ridad de RARP, nuestros hallazgos son impor tantes no solo para los urólogos sino también
para los proveedores de atención médica al considerar los problemas económicos en cen tros que no tienen el volumen para hacer que el uso de la robótica sea sostenible. Los exper tos sugieren que la visión 3D parece facilitar el aprendizaje en cirujanos novatos. Sin em bargo, se necesitan estudios dirigidos específi camente a estudiar las curvas de aprendizaje de 3DHD LRP. Entendemos la limitante de la experiencia del cirujano en esta serie de ca sos, pero como mencionamos, esta tecnología podría ser replicada mas facil que laparosco pia convencional 2D.
CONTRIBUCIONES
AER: Desarrollo de protocolos/proyectos, Recopilación o gestión de datos, Redacción/ edición de manuscritos. IRI: Recopilación o gestión de datos, redacción/edición de ma nuscritos. NER: Recopilación o gestión de da tos, redacción/edición de manuscritos. COS: Análisis de datos, Redacción/edición de ma nuscritos. JEM: Análisis de datos, redacción/ edición de manuscritos. GRD: Desarrollo de protocolos/proyectos, Redacción/edición de manuscritos.
CONFLICTO DE INTERESES
Todos los autores declaran que no hay divul gaciones financieras o intereses concurrentes no financieros.
REFERENCIAS:
1.ACS. Datos y cifras sobre el cáncer 2022.
; Sociedad Americana del Cáncer: Atlanta, 2022.
Revista Puertorriqueña de Medicina y Salud Pública14
2.Tangen, C.M.; Hussain, M.H.; Higa no, C.S.; Eisenberger, M.A.; Pequeño, E.J.; Wilding, G.; Donnelly, B.J.; Schel hammer, P.F.; Crawford, E.D.; Vogelzang, N.J., et al. Mejora de las tendencias de supervivencia general de los hombres con cáncer de próstata M1 recién diag nosticado: una experiencia de ensayo de fase III SWOG (S8494, S8894 y S9346). J Urol 2012, 188, 1164-1169.
3. Chinea, F.M.; Patel, V.N.; Kwon, D.; Lamichhane, N.; Lopez, C.; Punnen, S.; Kobetz, E.N.; Abramowitz, M.C.; Pollack, A. Ethnic heterogeneity and prostate can cer mortality in Hispanic/Latino men: a population-based study. Oncotarget 2017, 8,69709-69721, doi:10.18632/on cotarget.19068.
4.Schuessler, W.W.; Schulam, P.G.; Clayman, R.V.; Kavoussi, L.R. Prostatecto mía radical laparoscópica: experiencia inicial a corto plazo. Urología 1997, 50, 854-857.
5.Guillonneau, B.; Cathelineau, X.; Ba rret, E.; Rozet, F.; Vallancien, G. Prostatec tomía radical laparoscópica: evaluación técnica y oncológica temprana de 40 operaciones. Eur Urol 1999, 36, 14-20.
6.Ahlering, T.E. Prostatectomía radical robótica versus laparoscópica. Nature Clinical Practice Urology 2004, 1, 58-59, doi:10.1038/ncpuro0040.
7.Cheng, J.; Gao, J.; Shuai, X.; Wang, G.; Tao, K. Laparoscopia bidimensional versus tridimensional en la eficacia qui rúrgica: una revisión sistemática y un metanálisis. Oncotarget 2016, 7, 7097970990.
8.Bhayani, S.B.; Andriole, G.L. Visión tridimensional (3D): ¿mejora las habili dades laparoscópicas? Una evaluación de un sistema de visualización montado en la cabeza 3D. Rev Urol 2005, 7, 211214.
9.Abbou, C.C.; Hoznek, A.; Salomón, L.; Olsson, L.E.; Lobontiu, A.; Santo, F.; Cicco, A.; Antífona, P.; Chopin, D. Pros tatectomía radical laparoscópica con un robot a control remoto. J Urol 2001, 165, 1964-1966.
10.Sood, A.; Jeong, W.; Peabody, J.O.; Hemal, A.K.; Menon, M. Prostatectomía radical asistida por robot: avanzando hacia el estándar de oro. Urol Clin North Am 2014, 41, 473-484.
11.Healy, K.A.; Gomella, L.G. Prostatec tomía radical retropúbica, laparoscópica o robótica: ¿hay alguna diferencia real? Semin Oncol 2013, 40, 286-296.
12.Bolenz, C.; Gupta, A.; Hotze, T.; Ho, R.; Cadeddu, J.A.; Roehrborn, C.G.; Lotan, Y. Comparación de costos de la prostatectomía radical robótica, laparos cópica y abierta para el cáncer de prós tata. Eur Urol 2010, 57, 453-458.
13.Buchs, Carolina del Norte; Volonte, F.; Pugin, F.; Toso, C.; Morel, P. Laparos copia tridimensional: un paso hacia la navegación quirúrgica avanzada; Surg Endosc. 2013 Febrero;27(2):692-3. doi: 10.1007/s00464-012-2481-3. Epub 2012 Jul 18.
14.Brown, J.A.; Rodin, D.; Lee, B.; Dahl, D.M. Enfoque transperitoneal versus ex traperitoneal para la prostatectomía ra dical laparoscópica: una evaluación de 156 casos. Urología 2005, 65, 320-324.
15.Smith, J.A. Prostatectomía laparoscó pica asistida por robot: una evaluación de su papel contemporáneo en el trata miento quirúrgico del cáncer de próstata localizado. The American Journal of Sur gery 2004, 188, 63-67, doi:https://doi. org/10.1016/j.amjsurg.2004.08.006.
16.Miller, D.C.; Sanda, M.G.; Dunn, R.L.; Montie, J.E.; Pimentel, H.; Sandler, H.M.; McLaughlin, W.P.; Wei, J.T. Resul tados a largo plazo entre sobrevivientes localizados de cáncer de próstata: cam bios en la calidad de vida relacionados con la salud después de la prostatecto mía radical, la radiación externa y la braquiterapia. J Clin Oncol 2005, 23, 2772-2780.
17.Tewari, A.; Peabody, J.O.; Fischer, M.; Sarle, R.; Vallancien, G.; Delmas, V.; Hassan, M.; Bansal, A.; Hemal, A.K.; Guillonneau, B., et al. Un estudio qui rúrgico y anatómico para ayudar en el preservamiento de los nervios durante la prostatectomía radical laparoscópica y robótica. Eur Urol 2003, 43, 444-454.
18.Zhao, D.; Huang, Z.; Zou, Z. [Progre so de la investigación del sistema de la paroscopio tridimensional]. Nan Fang Yi Ke Da Xue Xue Bao 2014, 34, 594-596.
19.Schlomm, T.; Heinzer, H.; Steuber, T.; Salomón, G.; Engel, O.; Michl, Estados Unidos; Haese, A.; Graefen, M.; Huland, H. Preservación completa del esfínter
uretral de longitud funcional durante la prostatectomía radical. Eur Urol 2011, 60, 320-329.
20.Trinh, Q.D.; Sammón, J.; Sol, M.; Ravi, P.; Ghani, K.R.; Bianchi, M.; Jeong, W.; Shariat, S.F.; Hansen, J.; Schmitges, J., et al. Resultados perioperatorios de la prostatectomía radical asistida por robot en comparación con la prostatectomía radical abierta: resultados de la muestra de pacientes hospitalizados a nivel na cional. Eur Urol 2012, 61, 679-685.
21.Eifler, J.B.; Cookson, M.S. Mejor evi dencia con respecto a la superioridad o inferioridad de la prostatectomía radical asistida por robot. Urol Clin North Am 2014, 41, 493-502.
22.Kyriazis, I.; Özsoy, M.; Kallidonis, P.; Vasilas, M.; Panagopoulos, V.; Liatsikos, E. Integración de la visión tridimensional en la laparoscopia: la curva de aprendi zaje de un experto. J Endourol 2015, 29, 657-660.
23.Özsoy, M.; Kallidonis, P.; Kyriazis, I.; Panagopoulos, V.; Vasilas, M.; Sakellaro poulos, G.C.; Liatsikos, E. Cirujanos no vatos: ¿se benefician de la laparoscopia 3D? Láseres Med Sci 2015, 30, 13251333.
24.Votanopoulos, K.; Brunicardi, F.C.; Thornby, J.; Fuelles, C.F. Impacto de la visión tridimensional en el entrenamiento laparoscópico. World J Surg 2008, 32, 110-118.
25.Sørensen, S.M.; Savran, M.M.; Kon ge, L.; Bjerrum, F. Visión tridimensional versus bidimensional en laparoscopia: una revisión sistemática. Surg Endosc 2016, 30, 11-23.
Revista Puertorriqueña de Medicina y Salud Pública 15
Brief Summary of Prescribing Information for ERLEADA® (apalutamide)
ERLEADA® (apalutamide) tablets, for oral use
See package insert for Full Prescribing Information
INDICATIONS AND USAGE
ERLEADA is indicated for the treatment of patients with
• Metastatic castration-sensitive prostate cancer (mCSPC)
• Non-metastatic castration-resistant prostate cancer (nmCRPC) CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Cerebrovascular and Ischemic Cardiovascular Events
Cerebrovascular and ischemic cardiovascular events, including events leading to death, occurred in patients receiving ERLEADA. Monitor for signs and symptoms of ischemic heart disease and cerebrovascular disorders. Optimize management of cardiovascular risk factors, such as hypertension, diabetes, or dyslipidemia. Consider discontinuation of ERLEADA for Grade 3 and 4 events.
In a randomized study (SPARTAN) of patients with nmCRPC, ischemic cardiovascular events occurred in 3.7% of patients treated with ERLEADA and 2% of patients treated with placebo. In a randomized study (TITAN) in patients with mCSPC, ischemic cardiovascular events occurred in 4.4% of patients treated with ERLEADA and 1.5% of patients treated with placebo. Across the SPARTAN and TITAN studies, 4 patients (0.3%) treated with ERLEADA, and 2 patients (0.2%) treated with placebo died from an ischemic cardiovascular event.
In the SPARTAN study, cerebrovascular events occurred in 2.5% of patients treated with ERLEADA and 1% of patients treated with placebo [see Adverse Reactions]. In the TITAN study, cerebrovascular events occurred in 1.9% of patients treated with ERLEADA and 2.1% of patients treated with placebo. Across the SPARTAN and TITAN studies, 3 patients (0.2%) treated with ERLEADA, and 2 patients (0.2%) treated with placebo died from a cerebrovascular event.
Patients with history of unstable angina, myocardial infarction, congestive heart failure, stroke, or transient ischemic attack within six months of randomization were excluded from the SPARTAN and TITAN studies.
Fractures
Fractures occurred in patients receiving ERLEADA. Evaluate patients for fracture risk. Monitor and manage patients at risk for fractures according to established treatment guidelines and consider use of bone-targeted agents.
In a randomized study (SPARTAN) of patients with non-metastatic castrationresistant prostate cancer, fractures occurred in 12% of patients treated with ERLEADA and in 7% of patients treated with placebo. Grade 3-4 fractures occurred in 2.7% of patients treated with ERLEADA and in 0.8% of patients treated with placebo. The median time to onset of fracture was 314 days (range: 20 to 953 days) for patients treated with ERLEADA. Routine bone density assessment and treatment of osteoporosis with bone-targeted agents were not performed in the SPARTAN study.
In a randomized study (TITAN) of patients with metastatic castrationsensitive prostate cancer, fractures occurred in 9% of patients treated with ERLEADA and in 6% of patients treated with placebo. Grade 3-4 fractures were similar in both arms at 1.5%. The median time to onset of fracture was 56 days (range: 2 to 111 days) for patients treated with ERLEADA. Routine bone density assessment and treatment of osteoporosis with bone-targeted agents were not performed in the TITAN study.
Falls
Falls occurred in patients receiving ERLEADA with increased frequency in the elderly [see Use in Specific Populations]. Evaluate patients for fall risk.
In a randomized study (SPARTAN), falls occurred in 16% of patients treated with ERLEADA compared to 9% of patients treated with placebo. Falls were not associated with loss of consciousness or seizure.
Seizure
Seizure occurred in patients receiving ERLEADA. Permanently discontinue ERLEADA in patients who develop a seizure during treatment. It is unknown whether anti-epileptic medications will prevent seizures with ERLEADA. Advise patients of the risk of developing a seizure while receiving ERLEADA and of engaging in any activity where sudden loss of consciousness could cause harm to themselves or others.
In two randomized studies (SPARTAN and TITAN), five patients (0.4%) treated with ERLEADA and one patient treated with placebo (0.1%) experienced a seizure. Seizure occurred from 159 to 650 days after initiation of ERLEADA. Patients with a history of seizure, predisposing factors for seizure, or receiving drugs known to decrease the seizure threshold or to induce seizure were excluded. There is no clinical experience in re-administering ERLEADA to patients who experienced a seizure.
Embryo-Fetal Toxicity
The safety and efficacy of ERLEADA have not been established in females. Based on findings from animals and its mechanism of action, ERLEADA can cause fetal harm and loss of pregnancy when administered to a pregnant female. In an animal reproduction study, oral administration of apalutamide to pregnant rats during and after organogenesis resulted in fetal abnormalities and embryo-fetal lethality at maternal exposures ≥ 2 times the human clinical exposure (AUC) at the recommended dose. Advise males with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ERLEADA [see Use in Specific Populations and Clinical Pharmacology (12.1) in Full Prescribing Information].
ERLEADA® (apalutamide) tablets
ADVERSE REACTIONS
The following are discussed in more detail in other sections of the labeling:
• Cerebrovascular and Ischemic Cardiovascular Events [see Warnings and Precautions]
• Fractures [see Warnings and Precautions].
• Falls [see Warnings and Precautions]
• Seizure [see Warnings and Precautions]
Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions (≥ 10%) that occurred more frequently in the ERLEADA-treated patients (≥ 2% over placebo) from the randomized placebo-controlled clinical trials (TITAN and SPARTAN) were fatigue, arthralgia, rash, decreased appetite, fall, weight decreased, hypertension, hot flush, diarrhea, and fracture.
Metastatic Castration-sensitive Prostate Cancer (mCSPC)
TITAN, a randomized (1:1), double-blind, placebo-controlled, multi-center clinical study, enrolled patients who had mCSPC. In this study, patients received either ERLEADA at a dose of 240 mg daily or placebo. All patients in the TITAN study received a concomitant gonadotropin-releasing hormone (GnRH) analog or had prior bilateral orchiectomy. The median duration of exposure was 20 months (range: 0 to 34 months) in patients who received ERLEADA and 18 months (range: 0.1 to 34 months) in patients who received placebo.
Ten patients (1.9%) who were treated with ERLEADA died from adverse reactions. The reasons for death were ischemic cardiovascular events (n=3), acute kidney injury (n=2), cardio-respiratory arrest (n=1), sudden cardiac death (n=1), respiratory failure (n=1), cerebrovascular accident (n=1), and large intestinal ulcer perforation (n=1). ERLEADA was discontinued due to adverse reactions in 8% of patients, most commonly from rash (2.3%). Adverse reactions leading to dose interruption or reduction of ERLEADA occurred in 23% of patients; the most frequent (>1%) were rash, fatigue, and hypertension. Serious adverse reactions occurred in 20% of ERLEADAtreated patients and 20% in patients receiving placebo.
Table 1 shows adverse reactions occurring in ≥10% on the ERLEADA arm in TITAN that occurred with a ≥2% absolute increase in frequency compared to placebo. Table 2 shows laboratory abnormalities that occurred in ≥15% of patients, and more frequently (>5%) in the ERLEADA arm compared to placebo.
Table 1: Adverse Reactions in TITAN (mCSPC)
ERLEADA N=524 Placebo N=527
System/Organ Class
Adverse reaction
Musculoskeletal and connective tissue disorders
All Grades % Grade 3-4 % All Grades % Grade 3-4 %
Arthralgiaa 17 0.4 15 0.9
Skin and subcutaneous tissue disorders
Rashb 28 6 9 0.6
Pruritus 11 0.2 4.6 0.2
Vascular disorders
Hot flush 23 0 16 0
Hypertension 18 8 16 9
a Per the Common Terminology Criteria for Adverse Reactions (CTCAE), the highest severity for these events is Grade 3 b Includes rash, rash maculo-papular, rash generalized, urticaria, rash pruritic, rash macular, conjunctivitis, erythema multiforme, rash papular, skin exfoliation, genital rash, rash erythematous, stomatitis, drug eruption, mouth ulceration, rash pustular, blister, papule, pemphigoid, skin erosion, dermatitis, and rash vesicular
Additional adverse reactions of interest occurring in 2%, but less than 10% of patients treated with ERLEADA included diarrhea (9% versus 6% on placebo), muscle spasm (3.1% versus 1.9% on placebo), dysgeusia (3.2% versus 0.6% on placebo), and hypothyroidism (3.6% versus 0.6% on placebo).
Table 2: Laboratory Abnormalities Occurring in ≥ 15% of ERLEADA-Treated Patients and at a Higher Incidence than Placebo (Between Arm Difference > 5% All Grades) in TITAN (mCSPC)
ERLEADA N=524 Placebo N=527
Laboratory Abnormality
Hematology
All Grades % Grade 3-4 % All Grades % Grade 3-4 %
White blood cell decreased 27 0.4 19 0.6
Chemistry
Hypertriglyceridemiaa 17 2.5 12 2.3 a Does not reflect fasting values
Non-metastatic Castration-resistant Prostate Cancer (nmCRPC) SPARTAN, a randomized (2:1), double-blind, placebo-controlled, multi-center clinical study, enrolled patients who had nmCRPC. In this study, patients received either ERLEADA at a dose of 240 mg daily or a placebo. All patients in the SPARTAN study received a concomitant gonadotropin-releasing hormone (GnRH) analog or had a bilateral orchiectomy. The median duration of exposure was 33 months (range: 0.1 to 75 months) in patients who received ERLEADA and 11 months (range: 0.1 to 37 months) in patients who received placebo. Twenty-four patients (3%) who were treated with ERLEADA died from adverse reactions. The reasons for death with ≥ 2 patients included infection (n=7), myocardial infarction (n=3), cerebrovascular event (n=2), and unknown reason (n=3). ERLEADA was discontinued due to adverse reactions in 11% of patients, most commonly from rash (3.2%). Adverse reactions leading to dose interruption or reduction of ERLEADA occurred in 33% of patients; the most common (>1%) were rash, diarrhea, fatigue, nausea, vomiting, hypertension, and hematuria. Serious adverse reactions occurred in 25% of ERLEADAtreated patients and 23% in patients receiving placebo. The most frequent serious adverse reactions (>2%) were fracture (3.4%) in the ERLEADA arm and urinary retention (3.8%) in the placebo arm.
Table 3 shows adverse reactions occurring in ≥10% on the ERLEADA arm in SPARTAN that occurred with a ≥2% absolute increase in frequency compared to placebo. Table 4 shows laboratory abnormalities that occurred in ≥15% of patients, and more frequently (>5%) in the ERLEADA arm compared to placebo.
Table 3: Adverse Reactions in SPARTAN (nmCRPC)
ERLEADA N=803 Placebo N=398
System/Organ Class
Adverse reaction
General disorders and administration site conditions
All Grades % Grade 3-4 % All Grades % Grade 3-4 %
Fatiguea,b 39 1.4 28 0.3
Musculoskeletal and connective tissue disorders
Arthralgiab 16 0 8 0
Skin and subcutaneous tissue disorders
Rashc 25 5.2 6 0.3
Metabolism and nutrition disorders
Decreased appetited 12 0.1 9 0
Peripheral edemae 11 0 9 0
Injury, poisoning and procedural complications
Fallb 16 1.7 9 0.8
Fracturef 12 2.7 7 0.8
Investigations
Weight decreasedb 16 1.1 6 0.3
Vascular disorders
Hypertension 25 14 20 12 Hot flush 14 0 9 0
Gastrointestinal disorders
Diarrhea 20 1.1 15 0.5
Nausea 18 0 16 0
a Includes fatigue and asthenia
b Per the Common Terminology Criteria for Adverse Reactions (CTCAE), the highest severity for these events is Grade 3
c Includes rash, rash maculo-papular, rash generalized, urticaria, rash pruritic, rash macular, conjunctivitis, erythema multiforme, rash papular, skin exfoliation, genital rash, rash erythematous, stomatitis, drug eruption, mouth ulceration, rash pustular, blister, papule, pemphigoid, skin erosion, dermatitis, and rash vesicular
d Includes appetite disorder, decreased appetite, early satiety, and hypophagia
e Includes peripheral edema, generalized edema, edema, edema genital, penile edema, peripheral swelling, scrotal edema, lymphedema, swelling, and localized edema
f Includes rib fracture, lumbar vertebral fracture, spinal compression fracture, spinal fracture, foot fracture, hip fracture, humerus fracture, thoracic vertebral fracture, upper limb fracture, fractured sacrum, hand fracture, pubis fracture, acetabulum fracture, ankle fracture, compression fracture, costal cartilage fracture, facial bones fracture, lower limb fracture, osteoporotic fracture, wrist fracture, avulsion fracture, fibula fracture, fractured coccyx, pelvic fracture, radius fracture, sternal fracture, stress fracture, traumatic fracture, cervical vertebral fracture, femoral neck fracture, and tibia fracture
Additional clinically significant adverse reactions occurring in 2% or more of patients treated with ERLEADA included hypothyroidism (8% versus 2% on placebo), pruritus (6% versus 1.5% on placebo), and heart failure (2.2% versus 1% on placebo).
Table 4: Laboratory Abnormalities Occurring in ≥ 15% of ERLEADA-Treated Patients and at a Higher Incidence than Placebo (Between Arm Difference > 5% All Grades) in SPARTAN (nmCRPC)
ERLEADA N=803 Placebo N=398
Laboratory Abnormality
Hematology
All Grades % Grade 3-4 % All Grades % Grade 3-4 %
Anemia 70 0.4 64 0.5
Leukopenia 47 0.3 29 0
Lymphopenia 41 1.8 21 1.6
Chemistry
Hypercholesterolemiaa 76 0.1 46 0 Hyperglycemiaa 70 2 59 1.0
Hypertriglyceridemiaa 67 1.6 49 0.8
Hyperkalemia 32 1.9 22 0.5
a Does not reflect fasting values
Rash
In the combined data of two randomized, placebo-controlled clinical studies, SPARTAN and TITAN, rash associated with ERLEADA was most commonly described as macular or maculo-papular. Adverse reactions of rash were reported for 26% of patients treated with ERLEADA versus 8% of patients treated with placebo. Grade 3 rashes (defined as covering > 30% body surface area [BSA]) were reported with ERLEADA treatment (6%) versus placebo (0.5%).
The onset of rash occurred at a median of 83 days of ERLEADA treatment. Rash resolved in 78% of patients within a median of 78 days from onset of rash. Rash was commonly managed with oral antihistamines, topical corticosteroids, and 19% of patients received systemic corticosteroids. Dose reduction or dose interruption occurred in 14% and 28% of patients, respectively. Of the patients who had dose interruption, 59% experienced recurrence of rash upon reintroduction of ERLEADA.
Hypothyroidism
In the combined data of two randomized, placebo-controlled clinical studies, SPARTAN and TITAN, hypothyroidism was reported for 8% of patients treated with ERLEADA and 1.5% of patients treated with placebo based on assessments of thyroid-stimulating hormone (TSH) every 4 months. Elevated TSH occurred in 25% of patients treated with ERLEADA and 7% of patients treated with placebo. The median onset was at the first scheduled assessment. There were no Grade 3 or 4 adverse reactions. Thyroid replacement therapy was initiated in 4.9% of patients treated with ERLEADA. Thyroid replacement therapy, when clinically indicated, should be initiated or dose-adjusted [see Drug Interactions] Post-Marketing Experience
The following additional adverse reactions have been identified during postapproval use of ERLEADA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Respiratory, Thoracic and Mediastinal Disorders: interstitial lung disease Skin and Subcutaneous Tissue Disorders: Stevens-Johnson syndrome/toxic epidermal necrolysis
DRUG INTERACTIONS
Effect of Other Drugs on ERLEADA Strong CYP2C8 or CYP3A4 Inhibitors
Co-administration of a strong CYP2C8 or CYP3A4 inhibitor is predicted to increase the steady-state exposure of the active moieties (sum of unbound apalutamide plus the potency-adjusted unbound N-desmethyl-apalutamide). No initial dose adjustment is necessary however, reduce the ERLEADA dose based on tolerability [see Dosage and Administration (2.2) in Full Prescribing Information]. Mild or moderate inhibitors of CYP2C8 or CYP3A4 are not expected to affect the exposure of apalutamide.
Effect of ERLEADA on Other Drugs CYP3A4, CYP2C9, CYP2C19 and UGT Substrates
ERLEADA is a strong inducer of CYP3A4 and CYP2C19, and a weak inducer of CYP2C9 in humans. Concomitant use of ERLEADA with medications that are primarily metabolized by CYP3A4, CYP2C19, or CYP2C9 can result in lower exposure to these medications. Substitution for these medications is recommended when possible or evaluate for loss of activity if medication is continued. Concomitant administration of ERLEADA with medications that are substrates of UDP-glucuronosyl transferase (UGT) can result in decreased exposure. Use caution if substrates of UGT must be co-administered with ERLEADA and evaluate for loss of activity [see Clinical Pharmacology (12.3) in Full Prescribing Information].
ERLEADA® (apalutamide) tablets
ERLEADA® (apalutamide) tablets
ERLEADA® (apalutamide) tablets
P-gp, BCRP or OATP1B1 Substrates
Apalutamide was shown to be a weak inducer of P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and organic anion transporting polypeptide 1B1 (OATP1B1) clinically. At steady-state, apalutamide reduced the plasma exposure to fexofenadine (a P-gp substrate) and rosuvastatin (a BCRP/ OATP1B1 substrate). Concomitant use of ERLEADA with medications that are substrates of P-gp, BCRP, or OATP1B1 can result in lower exposure of these medications. Use caution if substrates of P-gp, BCRP or OATP1B1 must be co-administered with ERLEADA and evaluate for loss of activity if medication is continued [see Clinical Pharmacology (12.3) in Full Prescribing Information].
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
The safety and efficacy of ERLEADA have not been established in females. Based on findings from animals and its mechanism of action, ERLEADA can cause fetal harm and loss of pregnancy when administered to a pregnant female [see Clinical Pharmacology (12.1) in Full Prescribing Information]
There are no available data on ERLEADA use in pregnant women to inform a drug-associated risk. In an animal reproduction study, oral administration of apalutamide to pregnant rats during and after organogenesis resulted in fetal abnormalities and embryo-fetal lethality at maternal exposures ≥ 2 times the human clinical exposure (AUC) at the recommended dose (see Data)
Data
Animal Data
In a pilot embryo-fetal developmental toxicity study in rats, apalutamide caused developmental toxicity when administered at oral doses of 25, 50 or 100 mg/kg/day throughout and after the period of organogenesis (gestational days 6-20). Findings included embryo-fetal lethality (resorptions) at doses ≥50 mg/kg/day, decreased fetal anogenital distance, misshapen pituitary gland, and skeletal variations (unossified phalanges, supernumerary short thoracolumbar rib(s), and small, incomplete ossification, and/or misshapen hyoid bone) at ≥25 mg/kg/day. A dose of 100 mg/kg/day caused maternal toxicity. The doses tested in rats resulted in systemic exposures (AUC) approximately 2, 4 and 6 times, respectively, the AUC in patients.
Lactation
Risk Summary
The safety and efficacy of ERLEADA have not been established in females. There are no data on the presence of apalutamide or its metabolites in human milk, the effect on the breastfed child, or the effect on milk production. Females and Males of Reproductive Potential
Contraception
Males
Based on the mechanism of action and findings in an animal reproduction study, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ERLEADA. [see Use in Specific Populations].
Infertility
Males
Based on animal studies, ERLEADA may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1) in Full Prescribing Information].
Pediatric Use
Safety and effectiveness of ERLEADA in pediatric patients have not been established.
Geriatric Use
Of the 1327 patients who received ERLEADA in clinical studies, 19% of patients were less than 65 years, 41% of patients were 65 years to 74 years, and 40% were 75 years and over.
No overall differences in effectiveness were observed between older and younger patients.
Of patients treated with ERLEADA (n=1073), Grade 3-4 adverse reactions occurred in 39% of patients younger than 65 years, 41% of patients 65-74 years, and 49% of patients 75 years or older. Falls in patients receiving ERLEADA with androgen deprivation therapy was elevated in the elderly, occurring in 8% of patients younger than 65 years, 10% of patients 65-74 years, and 19% of patients 75 years or older.
OVERDOSAGE
There is no known specific antidote for apalutamide overdose. In the event of an overdose, stop ERLEADA, undertake general supportive measures until clinical toxicity has been diminished or resolved.
ERLEADA® (apalutamide) tablets
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information). Cerebrovascular and Ischemic Cardiovascular Events
• Inform patients that ERLEADA has been associated with cerebrovascular and ischemic cardiovascular events. Advise patients to seek immediate medical attention if any symptoms suggestive of a cardiovascular or a cerebrovascular event occur [see Warnings and Precautions]
Falls and Fractures
• Inform patients that ERLEADA is associated with an increased incidence of falls and fractures [see Warnings and Precautions].
Seizures
• Inform patients that ERLEADA has been associated with an increased risk of seizure. Discuss conditions that may predispose to seizures and medications that may lower the seizure threshold. Advise patients of the risk of engaging in any activity where sudden loss of consciousness could cause serious harm to themselves or others. Inform patients to contact their healthcare provider right away if they experience a seizure [see Warnings and Precautions].
Rash
• Inform patients that ERLEADA is associated with rashes and to inform their healthcare provider if they develop a rash [see Adverse Reactions].
Dosage and Administration
• Inform patients receiving concomitant gonadotropin-releasing hormone (GnRH) analog therapy that they need to maintain this treatment during the course of treatment with ERLEADA.
• Instruct patients to take their dose at the same time each day (once daily). ERLEADA can be taken with or without food. Each tablet should be swallowed whole.
• Inform patients that in the event of a missed daily dose of ERLEADA, they should take their normal dose as soon as possible on the same day with a return to the normal schedule on the following day. The patient should not take extra tablets to make up the missed dose [see Dosage and Administration (2.1) in Full Prescribing Information].
• Instruct patients who have difficulty swallowing tablets whole to mix the recommended dose of ERLEADA tablets with applesauce. Do not crush tablets [see Dosage and Administration (2.3) in Full Prescribing Information].
Embryo-Fetal Toxicity
• Inform patients that ERLEADA can be harmful to a developing fetus. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ERLEADA. Advise male patients to use a condom if having sex with a pregnant woman [see Warnings and Precautions]
Infertility
• Advise male patients that ERLEADA may impair fertility and not to donate sperm during therapy and for 3 months following the last dose of ERLEADA [see Use in Specific Populations].
Manufactured for: Janssen Products, LP Horsham, PA 19044, USA © 2019 Janssen Pharmaceutical Companies cp-50509v8
WE ARE NOT INVISIBLE. NO SOMOS INVISIBLES. WE ARE NOT “CRAZY.” NO SOMOS “LOCAS.” WE ARE RESILIENT. SOMOS RESILIENTES. AND WE HAVE RIGHTS. Y TENEMOS DERECHOS. no somos invisibles JÓVENES LATINAS, LA SALUD MENTAL Y LAS ESCUELAS DE FILADELFIA Por servicios de SALUD MENTAL para todos en cualquier situación, en cualquier parte de Puerto Rico.
MIOMATOSIS UTERINA
Por: Kevin Santisteban1,3, Glenda Ramos2,4, Flori Martínez1,5

1Médico y Cirujano, Facultad de Ciencias Médicas, Universidad de San Carlos de Guatemala.
2Médica y Cirujana, Facultad de Ciencias de la Salud, Universidad Mariano Gálvez de Guatemala.


PALABRAS CLAVE
mioma, cirugía, histerectomía, ginecología.
KEY WORDS myoma, surgery, hysterectomy, gynecology.
ABSTRACTO
Los miomas son los tumores benignos uteri nos más frecuentes, sin embargo, el diagnós tico puede pasar inadvertido ya que solo el 30% presenta sintomatología; cuya resolución será quirúrgica en su mayoría con previo tra tamiento médico y preparación preoperato ria, considerando la opción quirúrgica perti nente según el objetivo de la paciente.
ABSTRACT
Myomas are the most frequent benign uterine tumors, however, the diagnosis can go unnoti ced since only 30% present symptoms; whose resolution will be mostly surgical with previous medical treatment and preoperative prepara tion, considering the pertinent surgical option according to the objective of the patient.
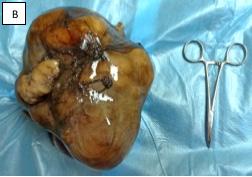

Revista Puertorriqueña de Medicina y Salud Pública22
INTRODUCCIÓN
Los miomas uterinos, son tumores benig nos mesenquimatosos monoclonales que aparecen en el miometrio, con una preva lencia del 20 al 50% en las mujeres en edad fértil, y hasta el 70% en mujeres en la menopausia. Los miomas pueden ser únicos o múltiples, en su mayoría asinto máticos, sin embargo, aproximadamente el 30% presentan principalmente menome trorragia y dolor pélvico. Además, se pue de presentar anemia, dismenorrea, dolor abdominal y dorsal, subfertilidad, poliuria y abortos espontáneos. Siendo una de las indicaciones más frecuentes de histerecto mía, 40 al 60%. [1][2][3]
Los factores de riesgo son, predisposición genética (en familiares de primer grado que poseen fibromas, aumenta un 40%), nuliparidad, mujeres afroamericanas (au menta la probabilidad en 3 a 9 veces), menarquia temprana (<10 años), trata mientos hormonales, consumo de café y alcohol, diabetes mellitus, obesidad, die tas ricas en vitamina A y consumo elevado de carnes rojas.[1][4][5]
Los miomas se pueden desarrollar en cualquier parte del miometrio, clasificán dose según la Federación Internacional de Ginecología y Obstetricia: submucosos (fibromas 0, 1 y 2), intramurales o intesti ciales (fibromas 3, 4 y 5), subserosos (6 y 7), e independientes del útero (fibromas 8). [1][2][3]
PRESENTACIÓN DE CASO
Mujer de 36 años de edad, nulípara, con antecedente de quistectomía ovárica iz quierda y prediabetes sin tratamiento, sin otros antecedentes de importancia, quien consulta por menometrorragia de 2 meses de evolución.
Refiere que el 11 de mayo del 2022 ini cia con menometrorragia abundante por lo que auto receta Vitamina K ampollas en tres dosis, sin embargo, no presenta mejo ría decide consultar con médico particular, quien realiza ultrasonido pélvico repor tando, útero aumentado de tamaño, de contornos lobulados donde se observan al menos 10 miomas de distintos tamaños, mediciones de 3.8-7.5cm existiendo intra murales y subserosos. Medidas del útero 12.5x9.2x10cm con volumen 612cc, con cluyendo miomatosis uterina (Fig. 1A).
Se realiza laboratorios, presentando leucocitosis (10,190 mm3), hemoglobina 9.8g/dl, hematocrito 30.6%, recuento de plaquetas 498,000 mm3, TSH 0.5 μUI/ml, hemoglobina glicosilada 6.34%, nitróge
no de urea 8.7mg/dl, creatinina 0.9 mg/dl, glucosa 103 mg/dl; orina: ligeramente tur bia, pH 5, bacterias +++, leucocitos 8 por campo/400, epitelios +, moco ++, eritroci tos 2eri/uL, por lo que indican que debe compensar valor de hemoglobina para in gresar a sala de operaciones, iniciando tra tamiento con anticonceptivos orales y hie rro por 4 semanas. Dos semanas después continúa con menometrorragia por lo que consulta con médico particular quien rece ta Vitamina K en dos dosis y Eritropoyetina 4,000 UI cada 72 horas por 3 dosis. Consultando el día 8 de julio del 2022, en examen físico presenta leve dolor a la palpación en hipogastrio y sangrado vagi nal, resto normal. Laboratorios control, gló bulos blancos 8,900mm3, hemoglobina 10.8 g/dl, hematocrito 35%, recuento de plaquetas 481,000mm3, tiempo de pro trombina 10s, tiempo parcial de trombo plastina 28s, índice internacional norma lizado 1, hemoglobina glicosilada 5.7%, siendo llevada a sala de operaciones para realización de Histerectomía abdominal total, con hallazgos, miomatosis uterina de grandes elementos con abundantes adhe rencias de omento (Fig. 1B); posoperatorio con adecuada evolución clínica, durante el cual es transfundida con una unidad de células empacadas.
La pieza es llevada a patología, quienes reportan, miometrio grisáceo de hasta 10 cm de espesor, con múltiples nódulos gri sáceos de hasta 7 cm de diámetro mayor; concluyendo: cuello uterino con cervicitis y endocervicitis crónica, quistes de Naboth. Endometrio: basal. Miometrio: Leiomiomas.
DISCUSIÓN:
La miomatosis uterina es el tumor benigno más frecuente en las mujeres con edad fér til, los cuales pueden ser diagnosticados por medio del uso de la ecografía. Las pa cientes que poseen indicación quirúrgica se debe indicar previamente tratamiento médico para minimizar el sangrado, dis minuir el tamaño de los miomas, el cual consistirá en tratamiento hormonal con anticonceptivos orales, y no hormonales, con procoagulantes (ácido tranexámico) y antiinflamatorios no esteroideos.[1][2]
El 30% de todos los miomas son sintomá ticos, requiriendo en su mayoría tratamien to quirúrgico para su resolución, existiendo tres opciones de intervención, tratamiento definitivo (histerectomía), preservar el úte ro (miomectomía, criomiólisis y termocoa gulación laparoscópica, y oclusión lapa roscópica de arterias uterinas) y preservar
el útero sin función obstétrica (ablación endometrial, embolización de las arterias uterinas, cirugía de ultrasonido focalizado guiado por resonancia magnética y oclu sión vaginal de las arterias uterinas). Por lo que el tratamiento dependerá en oca siones del tamaño y presentación de la sin tomatología de los miomas, y a su vez del deseo de fertilidad de la paciente.[2][3]
MANIFESTACIÓN DE CONFLICTO DE INTERÉS:
Los autores declaran que no existe conflic to de interés.
REFERENCIAS:
Viroga S. Miomatosis uterina, enfoque del tratamiento médico. Tendencias en Me dicina [Internet]. 2018 [Consultado 11 Jul 2022]; 13: 132-40. Disponible en: http:// www.tendenciasenmedicina.com/Image nes/imagenes13p/art_23.pdf
Debras E, Neveu M, Capmas P, Fernán dez H. Mioma e infertilidad. EMC-Gineco logía-Obstetricia [Internet]. 2022 [Consul tado 11 Jul 2022]; 50(1): 1-12. Disponible en: https://reader.elsevier.com/reader/ sd/pii/S1283081X22460516?token= 39D30353BC583362C6A43CCEE24EA 1DE2705EF45D3F3C877904F2DFACE0 64DBD309080ADE2794A49C7CE6F3B 05DE9207&originRegion=us-east-1&ori ginCreation=20220713045006
Toncel C, Gallego L. Miomatosis ute rina: enfoque terapéutico. Memorias Curso de Actualización en Ginecología y Obstetricia [Internet]. 2022 [Consul tado 12 Jul 2022]; 25-34. Disponible en: https://revistas.udea.edu.co/index. php/ginecologia_y_obstetricia/article/ view/347149/20808546
Méndez A, Moya C, Moré A, Día Y, Ro dríguez V, Rodríguez O. Miomatosis uteri na complicada con aborto de un mioma submucoso. Medicent Electrón [Internet]. 2018 [Consultado 12 Jul 2022]; 22(3): 289-96. Disponible en: http://scielo.sld.cu/ pdf/mdc/v22n3/mdc15318.pdf
Monleón J, Cañete M, Caballero V, Lo sada M, Del Campo M, Doménech A, Calaf J. Epidemiology of uterine myomas and clinical practice in Spain: An obser vational study. Eur J Obstet Gynecol Re prod Biol [Internet]. 2018 [Consultado 12 Jul 2022]; 226: 59-65. Disponible en: https://www.ejog.org/action/showPdf?pi i=S0301-2115%2818%2930249-5
Revista Puertorriqueña de Medicina y Salud
Pública 23
Important Safety Information (continued)
Risk of Thyroid C-cell Tumors: Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro. Such monitoring may increase the risk of unnecessary procedures, due to the low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin values may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
Pancreatitis: Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists. Pancreatitis has been reported in Mounjaro clinical trials. Mounjaro has not been studied in patients with a prior history of pancreatitis. It is unknown if patients with a history of pancreatitis are at higher risk for development of pancreatitis on Mounjaro. Observe patients for signs and symptoms, including persistent severe abdominal pain sometimes radiating to the back, which may or may not be accompanied by vomiting. If pancreatitis is suspected, discontinue Mounjaro and initiate appropriate management. Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin: Concomitant use with an insulin secretagogue (e.g., sulfonylurea) or insulin may increase the risk of hypoglycemia, including severe hypoglycemia. The risk of hypoglycemia may be lowered by reducing the dose of sulfonylurea (or other concomitantly administered insulin secretagogue) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
Hypersensitivity Reactions: Hypersensitivity reactions, sometimes severe, have been reported with Mounjaro in clinical trials. If hypersensitivity reactions occur, discontinue use of Mounjaro; treat promptly per standard of care, and monitor until signs and symptoms resolve. Do not use in patients with a previous serious hypersensitivity to Mounjaro. Use caution in patients with a history of angioedema or anaphylaxis with a GLP-1 receptor agonist because it is unknown if such patients will be predisposed to these reactions with Mounjaro.
Acute Kidney Injury: Mounjaro has been associated with gastrointestinal adverse reactions, which include nausea, vomiting, and diarrhea. These events may lead to dehydration, which if severe could cause acute kidney injury. In patients treated with GLP-1 receptor agonists, there have been postmarketing reports of acute kidney injury and worsening of chronic renal failure, sometimes requiring hemodialysis. Some of these events have been reported in patients without known underlying renal disease. A majority of reported events occurred in patients who had experienced nausea, vomiting,
diarrhea, or dehydration. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe adverse gastrointestinal reactions.
Severe Gastrointestinal Disease: Use of Mounjaro has been associated with gastrointestinal adverse reactions, sometimes severe. Mounjaro has not been studied in patients with severe gastrointestinal disease, including severe gastroparesis, and is therefore not recommended in these patients.
Diabetic Retinopathy Complications in Patients with a History of Diabetic Retinopathy: Rapid improvement in glucose control has been associated with a temporary worsening of diabetic retinopathy. Mounjaro has not been studied in patients with non-proliferative diabetic retinopathy requiring acute therapy, proliferative diabetic retinopathy, or diabetic macular edema. Patients with a history of diabetic retinopathy should be monitored for progression of diabetic retinopathy.
Acute Gallbladder Disease: In clinical trials, acute gallbladder disease was reported by 0.6% of Mounjarotreated patients and 0% of placebo-treated patients. If cholelithiasis is suspected, gallbladder diagnostic studies and appropriate clinical follow-up are indicated.
The most common adverse reactions reported in ≥5% of Mounjaro-treated patients in placebo-controlled trials were nausea, diarrhea, decreased appetite, vomiting, constipation, dyspepsia, and abdominal pain.
Drug Interactions: When initiating Mounjaro, consider reducing the dose of concomitantly administered insulin secretagogues (such as sulfonylureas) or insulin to reduce the risk of hypoglycemia. Mounjaro delays gastric emptying, and thereby has the potential to impact the absorption of concomitantly administered oral medications, so caution should be exercised.
Pregnancy: Limited data on Mounjaro use in pregnant women are available to inform on drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Based on animal reproduction studies, there may be risks to the fetus from exposure to tirzepatide. Use only if potential benefit justifies the potential risk to the fetus.
Lactation: There are no data on the presence of tirzepatide in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Mounjaro and any potential adverse effects on the breastfed infant from Mounjaro or from the underlying maternal condition.
Females of Reproductive Potential: Advise females using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation.
Pediatric Use: Safety and effectiveness of Mounjaro have not been established and use is not recommended in patients less than 18 years of age.
Please see the Brief Summary on the following page, including the Boxed Warning about possible thyroid tumors, including thyroid cancer, and Medication Guide. Please see Instructions for Use included with the pen.
Reference: 1. Mounjaro. Prescribing Information. Lilly USA, LLC.
Mounjaro™ and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-TR-US-0193 05/2022 ©Lilly USA, LLC 2022. All rights reserved.
TR HCP ISI MAY2022
Mounjaro™ (tirzepatide) Brief Summary: Consult the package insert for complete prescribing information.
INDICATIONS AND USAGE Mounjaro™ is a glucose-dependent insulinotropic polypeptide (GIP) receptor and glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use: Mounjaro has not been studied in patients with a history of pancreatitis. Mounjaro is not indicated for use in patients with type 1 diabetes mellitus.
WARNING: RISK OF THYROID C-CELL TUMORS
In both male and female rats, tirzepatide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined.
Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro.
CONTRAINDICATIONS: Mounjaro is contraindicated in patients with: A personal or family history of medullary thyroid carcinoma (MTC), in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2) and in patients with a known serious hypersensitivity to tirzepatide or any of the excipients in Mounjaro.
WARNINGS AND PRECAUTIONS:
Risk of Thyroid C-Cell Tumors: In both sexes of rats, tirzepatide caused a dose-dependent and treatment-duration-dependent increase in the incidence of thyroid C-cell tumors (adenomas and carcinomas) in a 2-year study at clinically relevant plasma exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined. Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2. Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro. Such monitoring may increase the risk of unnecessary procedures, due to the low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin values may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
Pancreatitis: Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists. In clinical studies, 14 events of acute pancreatitis were confirmed by adjudication in 13 Mounjarotreated patients (0.23 patients per 100 years of exposure) versus 3 events in 3 comparator-treated patients (0.11 patients per 100 years of exposure). Mounjaro has not been studied in patients with a prior history of pancreatitis. It is unknown if patients with a history of pancreatitis are at higher risk for development of pancreatitis on Mounjaro. After initiation of Mounjaro, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back and which may or may not be accompanied by vomiting). If pancreatitis is suspected, discontinue Mounjaro and initiate appropriate management. Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin: Patients receiving Mounjaro in combination with an insulin secretagogue (e.g., sulfonylurea) or insulin may have an increased risk of hypoglycemia, including severe hypoglycemia. The risk of hypoglycemia may be lowered by a reduction in the dose of sulfonylurea (or other concomitantly administered insulin secretagogue) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
Hypersensitivity Reactions: Hypersensitivity reactions have been reported with Mounjaro in clinical trials (e.g., urticaria and eczema) and were sometimes severe. If hypersensitivity reactions occur, discontinue use of Mounjaro; treat promptly per standard of care, and monitor until signs and symptoms resolve. Do not use in patients with a previous serious hypersensitivity reaction to tirzepatide or any of the excipients in Mounjaro. Anaphylaxis and angioedema have been reported with GLP-1 receptor agonists. Use caution in patients with a history of angioedema or anaphylaxis with a GLP-1 receptor agonist because it is unknown whether such patients will be predisposed to these reactions with Mounjaro.
Acute Kidney Injury: Mounjaro has been associated with gastrointestinal adverse reactions, which include nausea, vomiting, and diarrhea. These events may lead to dehydration, which if severe could cause acute kidney injury.
In patients treated with GLP-1 receptor agonists, there have been postmarketing reports of acute kidney injury and worsening of chronic renal failure, which may sometimes require hemodialysis. Some of these events have been reported in patients without known underlying renal disease. A majority of the reported events occurred in patients who had experienced nausea, vomiting, diarrhea, or dehydration. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe gastrointestinal adverse reactions.
Severe Gastrointestinal Disease: Use of Mounjaro has been associated with gastrointestinal adverse reactions, sometimes severe. Mounjaro has not been studied in patients with severe gastrointestinal disease, including severe gastroparesis, and is therefore not recommended in these patients.
Diabetic Retinopathy Complications in Patients with a History of Diabetic Retinopathy: Rapid improvement in glucose control has been associated with a temporary worsening of diabetic retinopathy. Mounjaro has not been studied in patients with non-proliferative diabetic retinopathy requiring acute therapy, proliferative diabetic retinopathy, or diabetic macular edema. Patients with a history of diabetic retinopathy should be monitored for progression of diabetic retinopathy.
Acute Gallbladder Disease: Acute events of gallbladder disease such as cholelithiasis or cholecystitis have been reported in GLP-1 receptor agonist trials and post marketing. In Mounjaro placebo-controlled clinical trials, acute gallbladder disease (cholelithiasis, biliary colic, and cholecystectomy) was reported by 0.6% of Mounjaro-treated patients and 0% of placebo-treated patients. If cholelithiasis is suspected, gallbladder diagnostic studies and appropriate clinical follow-up are indicated.
ADVERSE REACTIONS
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Pool of Two Placebo-Controlled Clinical Trials: The data in Table 1 are derived from 2 placebo-controlled trials (1 monotherapy trial [SURPASS-1] and 1 trial in combination with basal insulin with or without metformin [SURPASS-5]) in adult patients with type 2 diabetes mellitus. These data reflect exposure of 718 patients to Mounjaro and a mean duration of exposure to Mounjaro of 36.6 weeks. The mean age of patients was 58 years, 4% were 75 years or older and 54% were male. The population was 57% White, 27% Asian, 13% American Indian or Alaska Native, and 3% Black or African American; 25% identified as Hispanic or Latino ethnicity. At baseline, patients had type 2 diabetes mellitus for an average of 9.1 years with a mean HbA1c of 8.1%. As assessed by baseline fundoscopic examination, 13% of the population had retinopathy. At baseline, eGFR was ≥90 mL/min/1.73 m2 in 53%, 60 to 90 mL/min/1.73 m2 in 39%, 45 to 60 mL/min/1.73 m2 in 7%, and 30 to 45 mL/min/1.73 m2 in 1% of patients.
MOUNJAROTM (tirzepatide) Injection, for subcutaneous use
Pool of Seven Controlled Clinical Trials: Adverse reactions were also evaluated in a larger pool of adult patients with type 2 diabetes mellitus participating in seven controlled clinical trials which included two placebo-controlled trials (SURPASS-1 and -5), three trials of Mounjaro in combination with metformin, sulfonylureas, and/or SGLT2 inhibitors (SURPASS-2, -3, -4), and two additional trials conducted in Japan. In this pool, a total of 5119 adult patients with type 2 diabetes mellitus were treated with Mounjaro for a mean duration of 48.1 weeks. The mean age of patients was 58 years, 4% were 75 years or older and 58% were male. The population was 65% White, 24% Asian, 7% American Indian or Alaska Native, and 3% Black or African American; 38% identified as Hispanic or Latino ethnicity. At baseline, patients had type 2 diabetes mellitus for an average of 9.1 years with a mean HbA1c of 8.3%. As assessed by baseline fundoscopic examination, 15% of the population had retinopathy. At baseline, eGFR was ≥90 mL/min/1.73 m2 in 52%, 60 to 90 mL/min/1.73 m2 in 40%, 45 to 60 mL/min/1.73 m2 in 6%, and 30 to 45 mL/min/ 1.73 m2 in 1% of patients.
Common Adverse Reactions: In the pool of placebo-controlled trials, the following adverse reactions were reported in ≥5% of Mounjaro-treated adult patients with type 2 diabetes mellitus (excluding hypoglycemia): (frequencies listed, respectively, as: placebo [N=235]; 5 mg [N=273]; 10 mg [N=240]; 15 mg [N=241]): nausea (4%, 12%, 15%, 18%), diarrhea (9%, 12%, 13%, 17%), decreased appetite (1%, 5%, 10%, 11%), vomiting (2%, 5%, 5%, 9%), constipation (1%, 6%, 6%, 7%), dyspepsia (3%, 8%, 8%, 5%), and abdominal pain (4%, 6%, 5%, 5%). Percentages reflect the number of patients who reported at least 1 occurrence of the adverse reaction. In the pool of seven clinical trials, the types and frequency of common adverse reactions, not including hypoglycemia, were similar.
Gastrointestinal Adverse Reactions: In the pool of placebo-controlled trials, gastrointestinal adverse reactions occurred more frequently among patients receiving Mounjaro than placebo (placebo 20.4%, Mounjaro 5 mg 37.1%, Mounjaro 10 mg 39.6%, Mounjaro 15 mg 43.6%). More patients receiving Mounjaro 5 mg (3.0%), Mounjaro 10 mg (5.4%), and Mounjaro 15 mg (6.6%) discontinued treatment due to gastrointestinal adverse reactions than patients receiving placebo (0.4%). The majority of reports of nausea, vomiting, and/or diarrhea occurred during dose escalation and decreased over time.
The following gastrointestinal adverse reactions were reported more frequently in Mounjaro-treated patients than placebo-treated patients (frequencies listed, respectively, as: placebo; 5 mg; 10 mg; 15 mg): eructation (0.4%, 3.0%, 2.5%, 3.3%), flatulence (0%, 1.3%, 2.5%, 2.9%), gastroesophageal reflux disease (0.4%, 1.7%, 2.5%, 1.7%), abdominal distension (0.4%, 0.4%, 2.9%, 0.8%).
Other Adverse Reactions: Hypoglycemia: Hypoglycemia adverse reactions in placebo-controlled trials in adults with type 2 diabetes mellitus were as follows. For the 40-week monotherapy study (frequencies listed, respectively, as: placebo [N=115]; 5 mg [N=121]; 10 mg [N=119]; 15 mg [N=120]): blood glucose <54 mg/dL (1%, 0%, 0%, 0%), severe hypoglycemia (0%, 0%, 0%, 0%). For the 40week add-on to basal insulin with or without metformin study (placebo [N=120], 5 mg [N=116], 10 mg [N=119], 15 mg [N-120]): blood glucose <54 mg/dL (13%, 16%, 19%, 14%), severe hypoglycemia (0%, 0%, 2%, 1%). Data include events occurring during 4 weeks of treatment-free safety follow up. Events after introduction of a new glucose-lowering treatment are excluded. Severe hypoglycemia was defined as episodes requiring the assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions.
Hypoglycemia was more frequent when Mounjaro was used in combination with a sulfonylurea. In a clinical trial up to 104 weeks of treatment, when administered with a sulfonylurea, hypoglycemia (glucose level <54 mg/dL) occurred in 13.8%, 9.9%, and 12.8%, and severe hypoglycemia occurred in 0.5%, 0%, and 0.6% of patients treated with Mounjaro 5 mg, 10 mg, and 15 mg, respectively.
Heart Rate Increase: In the pool of placebo-controlled trials, treatment with Mounjaro resulted in a mean increase in heart rate of 2 to 4 beats per minute compared to a mean increase of 1 beat per minute in placebo-treated patients. Episodes of sinus tachycardia, associated with a concomitant increase from baseline in heart rate of ≥15 beats per minute, also were reported in 4.3%, 4.6%, 5.9% and 10% of subjects treated with placebo, Mounjaro 5 mg, 10 mg, and 15 mg, respectively. For patients enrolled in Japan, these episodes were reported in 7% (3/43), 7.1% (3/42), 9.3% (4/43), and 23% (10/43) of patients treated with placebo, Mounjaro 5 mg, 10 mg, and 15 mg, respectively. The clinical relevance of heart rate increases is uncertain.
Hypersensitivity Reactions: Hypersensitivity reactions have been reported with Mounjaro in the pool of placebo-controlled trials, sometimes severe (e.g., urticaria and eczema); hypersensitivity reactions were reported in 3.2% of Mounjaro-treated patients compared to 1.7% of placebo-treated patients.
In the pool of seven clinical trials, hypersensitivity reactions occurred in 106/2,570 (4.1%) of Mounjaro-treated patients with antitirzepatide antibodies and in 73/2,455 (3.0%) of Mounjaro-treated patients who did not develop anti-tirzepatide antibodies.
Injection Site Reaction: In the pool of placebo-controlled trials, injection site reactions were reported in 3.2% of Mounjarotreated patients compared to 0.4% of placebo-treated patients.
In the pool of seven clinical trials, injection site reactions occurred in 119/2,570 (4.6%) of Mounjaro-treated patients with antitirzepatide antibodies and in 18/2,455 (0.7%) of Mounjaro-treated patients who did not develop anti-tirzepatide antibodies.
Acute Gallbladder Disease: In the pool of placebo-controlled clinical trials, acute gallbladder disease (cholelithiasis, biliary colic and cholecystectomy) was reported by 0.6% of Mounjaro-treated patients and 0% of placebo-treated patients.
Laboratory Abnormalities: Amylase and Lipase Increase: In the pool of placebo-controlled clinical trials, treatment with Mounjaro resulted in mean increases from baseline in serum pancreatic amylase concentrations of 33% to 38% and serum lipase concentrations of 31% to 42%. Placebo-treated patients had a mean increase from baseline in pancreatic amylase of 4% and no changes were observed in lipase. The clinical significance of elevations in lipase or amylase with Mounjaro is unknown in the absence of other signs and symptoms of pancreatitis.
DRUG INTERACTIONS
Concomitant Use with an Insulin Secretagogue (e.g., Sulfonylurea) or with Insulin When initiating Mounjaro, consider reducing the dose of concomitantly administered insulin secretagogues (e.g., sulfonylureas) or insulin to reduce the risk of hypoglycemia.
Oral Medications:
Mounjaro delays gastric emptying, and thereby has the potential to impact the absorption of concomitantly administered oral medications. Caution should be exercised when oral medications are concomitantly administered with Mounjaro. Monitor patients on oral medications dependent on threshold concentrations for efficacy and those with a narrow therapeutic index (e.g., warfarin) when concomitantly administered with Mounjaro. Advise patients using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation with Mounjaro. Hormonal contraceptives that are not administered orally should not be affected.
USE IN SPECIFIC POPULATIONS
Pregnancy: Risk Summary: Available data with Mounjaro use in pregnant women are insufficient to evaluate for a drug-related risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy. Based on animal reproduction studies, there may be risks to the fetus from exposure to tirzepatide during pregnancy. Mounjaro should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. In pregnant rats administered tirzepatide during organogenesis, fetal growth reductions and fetal abnormalities occurred at clinical exposure in maternal rats based on AUC. In rabbits administered tirzepatide during organogenesis, fetal growth reductions were observed at clinically relevant exposures based on AUC. These adverse embryo/fetal effects in animals coincided with pharmacological effects on maternal weight and food consumption. The estimated background risk of major birth defects is 6–10% in women with pre-gestational diabetes with an HbA1c >7% and has been reported to be as high as 20–25% in women with an HbA1c >10%. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively. Clinical Considerations: Disease-Associated Maternal and/or
TR HCP BS MAY2022 MOUNJAROTM (tirzepatide) Injection, for subcutaneous use
TR HCP BS MAY2022
Embryo/Fetal Risk: Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia-related morbidity. Data: Animal Data: In pregnant rats given twice weekly subcutaneous doses of 0.02, 0.1, and 0.5 mg/kg tirzepatide (0.03-, 0.07-, and 0.5-fold the MRHD of 15 mg once weekly based on AUC) during organogenesis, increased incidences of external, visceral, and skeletal malformations, increased incidences of visceral and skeletal developmental variations, and decreased fetal weights coincided with pharmacologically-mediated reductions in maternal body weights and food consumption at 0.5 mg/kg. In pregnant rabbits given once weekly subcutaneous doses of 0.01, 0.03, or 0.1 mg/kg tirzepatide (0.01-, 0.06, and 0.2-fold the MRHD) during organogenesis, pharmacologically-mediated effects on the gastrointestinal system resulting in maternal mortality or abortion in a few rabbits occurred at all dose levels. Reduced fetal weights associated with decreased maternal food consumption and body weights were observed at 0.1 mg/kg. In a pre- and post-natal study in rats administered subcutaneous doses of 0.02, 0.10, or 0.25 mg/kg tirzepatide twice weekly from implantation through lactation, F1 pups from F0 maternal rats given 0.25 mg/kg tirzepatide had statistically significant lower mean body weight when compared to controls from post-natal day 7 through post-natal day 126 for males and post-natal day 56 for females.
Lactation: Risk Summary: There are no data on the presence of tirzepatide in animal or human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Mounjaro and any potential adverse effects on the breastfed infant from Mounjaro or from the underlying maternal condition.
Females and Males of Reproductive Potential: Contraception: Use of Mounjaro may reduce the efficacy of oral hormonal contraceptives due to delayed gastric emptying. This delay is largest after the first dose and diminishes over time. Advise patients using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation with Mounjaro.
Pediatric Use: Safety and effectiveness of Mounjaro have not been established in pediatric patients (younger than 18 years of age).
Geriatric Use: In the pool of seven clinical trials, 1539 (30.1%) Mounjaro-treated patients were 65 years of age or older, and 212 (4.1%) Mounjaro-treated patients were 75 years of age or older at baseline. No overall differences in safety or efficacy were detected between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Renal Impairment: No dosage adjustment of Mounjaro is recommended for patients with renal impairment. In subjects with renal impairment including end-stage renal disease (ESRD), no change in tirzepatide pharmacokinetics (PK) was observed. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe adverse gastrointestinal reactions.
Hepatic Impairment: No dosage adjustment of Mounjaro is recommended for patients with hepatic impairment. In a clinical pharmacology study in subjects with varying degrees of hepatic impairment, no change in tirzepatide PK was observed.
OVERDOSE: In the event of an overdosage, contact Poison Control for latest recommendations. Appropriate supportive treatment should be initiated according to the patient’s clinical signs and symptoms. A period of observation and treatment for these symptoms may be necessary, taking into account the half-life of tirzepatide of approximately 5 days.
PATIENT COUNSELING INFORMATION: Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use)
Missed Doses: Inform patients if a dose is missed, it should be administered as soon as possible within 4 days after the missed dose. If more than 4 days have passed, the missed dose should be skipped and the next dose should be administered on the regularly scheduled day. In each case, inform patients to resume their regular once weekly dosing schedule.
Additional information can be found <at www.Mounjaro.com/at lillymedical.com/by calling The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979)/ on Twitter @MounjaroHCP/at Click to Chat/etc.>.
See Brief Summary of Prescribing Information on adjacent page. See User Guide accompanying the product device.
MOUNJAROTM (tirzepatide) Injection, subcutaneous
for
use TR HCP BS MAY2022
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA Copyright © 2022, Eli Lilly and Company. All rights reserved. TR HCP BS MAY2022 PP-TR-US-0344
CASO CLÍNICO
The diffuse sclerosing variant papillary thyroid carcinoma (DSPTC) is an uncommon form of this neoplasm. Some studies describe its high propensity for tumor invasion, me-tastasis, and mortality compared with classic papillary thy-roid carcinoma. Histologic features of DSPTC may resemble diffuse inflam mation as seen with Grave’s or Hashimoto’s thyroiditis, which makes initial diagnosis challen ging.
A 27-year-old female with Noonan’s Syndrome was evaluated on an outpatient basis after developing atrial fi-brillation de novo. Thyroid function tests were consistent with hyperthyroidism with TSH: <0.005 (n: 0.300-3.000 uIU/ mL), FT4: 3.59 (0.71-1.85 ng/ mL) and FT3: 16.77 (n: 2.0-7.0 pmol/L). Diffuse goiter was noted on physical exam, but no ophthalmopathy or dermopathy was present. TRAB and TSI

Revista Puertorriqueña de Medicina y Salud Pública28
VARIANTE ESCLEROSANTE DIFUSA INCIDENTAL PAPILAR CÁNCER DE TIROIDES
EN LA ENFERMEDAD DE GRAVES
Por: Nicolle Canales, MD1, Yadiel Rivera Nieves, MD1, Nydia Ivette Burgos Ortega, MD1, Janet Marie Colon Castellano, MD1, Nicole Hernández Cordero, MD1, Alberto Javier Grana Santini, MD1, Melba Feliciano-Emmanuelli, MD1, Loida Alejandra Gonzalez-Rodriguez, MD1, Milliette Alvarado, MD1, Margarita Ramirez, MD1, Victor J. Carlo, MD2, Wilma Rodriguez Mojica, MD3, Marcel Mesa Pabón, MD4
1University of Puerto Rico Endocrine Fellowship Program, San Juan, PR, United States, San Juan, PR, USA, 2Puerto Rico Pathology, San Juan, PR, USA, 3University of Puerto Rico Department of Radiology, San Juan, PR, USA, 4University of Puerto Rico, Department of Medicine, Cardiology Division, San Juan, PR, USA.
were elevated at 38.4 (n: <16%) and 423% (n: <140%) respectively.
A twenty-four-hour radioiodine uptake was 48% (n:10-35%) and described as essentially homoge-nous with two foci of decreased radiotracer concen tration suggestive of cold nodules. Thyroid ultrasound showed diffuse nodularity bilaterally with associated clusters of calcifications and no discrete nodules. No abnormal appearing lymph nodes were identi fied. Fine-needle aspi-ration of both nodular areas was positive for DSPTC.
Total thyroidectomy with central neck dissection was performed.
Gross and microscopic post-surgical pathology confirmed the presence of diffuse sclerosing papillary thyroid cancer, along with local metastasis to one central lymph node. Patient was scheduled for radioactive iodine therapy. Diffuse sclerosing variant is considered an aggres-sive histotype of papillary thyroid cancer. Ultrasound features include diffuse scattered microcalcifications with or without discrete nodules that may be
confused with chronic inflammatory changes. Despite the limited number of cases, DSPTC is recognized to have specific characteris-tics, a high female to male ratio, and a young patient age. DSPTC has a high potential for aggressive biologic beha-vior if not treated promptly at the time of diagnosis. When suspected, total thyroidectomy with lymph node excision followed by radioiodine therapy has been proposed as the correct management to decrease the risk of persistent or recurrent disease.
Revista Puertorriqueña de Medicina y Salud Pública 29

LA QUIMIOTERAPIA Y EL CORAZÓN
Por: Luis A. Rosado Carrillo MD, FACC, FISC Cardiólogo Expresidente de la Sociedad Puertorriqueña de Cardiología

RESUMEN
La quimioterapia es una de las formas más utili zadas para tratar el cáncer y algunas condiciones asociadas. Sin embargo, los fármacos utilizados en este procedimiento pueden afectar el funcionamien to del corazón. A este daño provocado por la qui mioterapia se le llama toxicidad cardíaca.
ABSTRACT
Chemotherapy is one of the most used ways to treat cancer and some associated conditions. Howe ver, the drugs used in this procedure can affect the functioning of the heart. This damage caused by chemotherapy is called cardiac toxicity.
RESUMEN
Por décadas, la quimioterapia ha sido el tratamiento más utiliza do para tratar un sinnúmero de condiciones relacionadas con el cáncer. Por ello, es importante entender que el beneficio del uso de estos fármacos en la inmensa mayoría de los casos va por encima de los riesgos de toxicidad que estos tratamientos pueden causar en el organismo. En este artículo vamos a hablar especí ficamente del daño que estos medicamentos pueden causar al corazón y como prevenirlos.

Toxicidad cardíaca, es el daño que ocurre al corazón por qui mioterapia. Primero, cabe destacar que no todas las quimiotera pias son tóxicas al corazón. Sin embargo, el fármaco más co mún relacionado a toxicidad cardíaca es la doxorrubicina. La doxorrubicina es un tipo de fármaco de quimioterapia llamado antraciclina. Las antraciclinas se pueden utilizar para tratar leu cemia, linfoma, mieloma múltiple y con frecuencia, también en el tratamiento del cáncer de mama.
PALABRAS CLAVE
Quimioterapia, toxicidad cardíaca, corazón, problemas cardiovasculares.
KEY WORDS
Chemotherapy, cardiac toxicity, heart, cardiovascular problems.
Revista Puertorriqueña de Medicina y Salud Pública34
SIGNOS Y SÍNTOMAS
En los pacientes donde ocurre daño por el uso de estos fármacos, lo más frecuente es el desarrollo de fallo cardíaco, lo que significa que el fármaco promue ve debilidad al músculo cardíaco. Como consecuencia, esto hace al corazón incapaz de bombear la cantidad suficiente de sangre con oxígeno para suministrarle al cuerpo. Estos pacientes presentan principalmente fatiga al mínimo esfuerzo, debilidad, retención de lí quidos, molestia o dolor en el pecho, palpitaciones, arritmias cardíacas, alta presión y en algunos casos in farto cardíaco. También, puede presentar desórdenes de coagulación.
TRATAMIENTO
¿Cómo se puede prevenir la toxicidad cardíaca? Los problemas cardíacos se pueden prevenir modificando la dosis del fármaco administrado, el método de administra ción y el tipo de antraciclina. Los predictores de cardio toxicidad incluyen también los factores de riesgo cardio vasculares previos al tratamiento y la edad del paciente. Es extremadamente importante que antes de ser sometido al tratamiento se sepa qué tan bien o mal se encuentra el corazón del paciente en términos de su función.
Por tal razón, es que frecuentemente el oncólogo envía al paciente a realizarse una evaluación con el cardiólogo para conocer su riesgo antes de comen zar el tratamiento y ofrecerle (en los casos meritorios) terapias que pueden reducir su riesgo de desarrollo de complicaciones, principalmente de fallo cardíaco. Las terapias pueden incluir, pero no limitarse, a inhi bidores de la ECA, betabloqueadores y diuréticos. Es también muy común que el cardiólogo continúe el mo nitoreo cardíaco durante el tratamiento oncológico, dependiendo del tipo de quimioterapia que reciba el paciente. El monitoreo también puede continuar des pués del tratamiento.
CONCLUSIÓN
Cabe destacar que las terapias oncológicas en la inmensa mayoría de los casos prolongan y/o salvan vidas. La toxicidad cardíaca es algo que es prevenible y muy fácil de cuantificar si se da el seguimiento apro piado. Siguiendo las recomendaciones del oncólogo y el cardiólogo trabajando como equipo, podremos reducir significativamente los riesgos.

Revista Puertorriqueña de Medicina y Salud Pública 35
Patients with PAD are complex.1
Helping protect them against thrombosis doesn’t have to be.
LER = lower extremity revascularization. PAD = peripheral artery disease.
INDICATIONS
XARELTO® (rivaroxaban) is indicated to reduce the risk of stroke and systemic embolism in adult patients with nonvalvular atrial fibrillation (AF).
There are limited data on the relative effectiveness of XARELTO® and warfarin in reducing the risk of stroke and systemic embolism when warfarin therapy is well controlled.
XARELTO® is indicated for the treatment of deep vein thrombosis (DVT). XARELTO® is indicated for the treatment of pulmonary embolism (PE). XARELTO® is indicated for the reduction in the risk of recurrence of DVT and/or PE in adult patients at continued risk for recurrent DVT and/or PE after completion of initial treatment lasting at least 6 months.
XARELTO® is indicated for the prophylaxis of DVT, which may lead to PE in adult patients undergoing knee or hip replacement surgery.








XARELTO® is indicated for the prophylaxis of venous thromboembolism (VTE) and VTE-related death during hospitalization and post hospital discharge in adult patients admitted for an acute medical illness who are at risk for thromboembolic
IMPORTANT SAFETY INFORMATION (cont’d) DRUG INTERACTIONS (cont'd)


• Coadministration of enoxaparin, warfarin, aspirin, clopidogrel, and chronic NSAID use may increase risk of bleeding.
• Avoid concurrent use of XARELTO® with other anticoagulants due to increased bleeding risk, unless benefit outweighs risk. Promptly evaluate signs or symptoms of blood loss if patients are treated concomitantly with aspirin, other platelet aggregation inhibitors, or NSAIDs.
USE IN SPECIFIC POPULATIONS
• Pregnancy: The limited available data on XARELTO® in pregnant women are insufficient to inform a drug-associated risk of adverse developmental outcomes. Use XARELTO® with caution in pregnant patients because of the potential for pregnancy-related hemorrhage and/or emergent delivery. The anticoagulant effect of XARELTO® cannot be reliably monitored with standard laboratory testing. Consider the benefits and risks of XARELTO® for the mother and possible risks to the fetus when prescribing to a pregnant woman.
Fetal/Neonatal adverse reactions: Based on the pharmacologic activity of Factor Xa inhibitors and the potential to cross the placenta, bleeding may occur at any site in the fetus and/or neonate.
Labor or delivery: The risk of bleeding should be balanced with the risk of thrombotic events when considering use in this setting.
There are no adequate or well-controlled studies of XARELTO® in pregnant women, and dosing for pregnant women has not been established. Postmarketing experience is currently insufficient to determine a rivaroxabanassociated risk for major birth defects or miscarriage.
• Lactation: Rivaroxaban has been detected in human milk. There are insufficient data to determine the effects of rivaroxaban on the breastfed child or on milk production. Consider the developmental and health benefits of breastfeeding along with the mother’s clinical need for XARELTO® and any potential adverse effects on the breastfed infant from XARELTO® or from the underlying maternal condition.
• Females and Males of Reproductive Potential: Females of reproductive potential requiring anticoagulation should discuss pregnancy planning with their physician.




The risk of clinically significant uterine bleeding, potentially requiring gynecological surgical interventions, identified with oral anticoagulants, including XARELTO®, should be assessed in females of reproductive potential and those with abnormal uterine bleeding.

• Pediatric Use: XARELTO® was not studied and therefore dosing cannot be reliably determined or recommended in children less than 6 months who
XARELTO® is licensed from Bayer HealthCare AG, 51368 Leverkusen, Germany.







© Janssen Pharmaceuticals, Inc. 2022 02/22 cp-237997v2





















NVAF Reduce stroke risk
CAD
Reduce the risk of major CV events

PAD









Reduce the risk of major thrombotic vascular events


VTE Prophylaxis
Acutely ill medical patients
DVT/PE
Initial treatment
DVT/PE Extended treatment
DVT Prophylaxis After hip/knee replacement surgery
The XARELTO® vascular dose* is the FIRST and ONLY treatment approach indicated to help reduce the risks of major thrombotic vascular events† in patients with PAD, including patients after recent LER due to symptomatic PAD.2,3
* XARELTO® 2.5 mg twice daily plus aspirin 75 mg to 100 mg once daily.
† Reduction of a composite of acute limb ischemia, major amputation of vascular etiology, myocardial infarction, ischemic stroke, or CV death.
complications due to moderate or severe restricted mobility and other risk factors for VTE, and not at high risk of bleeding.
XARELTO®, in combination with aspirin, is indicated to reduce the risk of major cardiovascular events (cardiovascular death, myocardial infarction, and stroke) in adult patients with coronary artery disease (CAD).
XARELTO®, in combination with aspirin, is indicated to reduce the risk of major thrombotic vascular events (myocardial infarction, ischemic stroke, acute limb ischemia, and major amputation of a vascular etiology) in adult patients with peripheral artery disease (PAD), including patients who have recently undergone a lower extremity revascularization procedure due to symptomatic PAD
XARELTO® is indicated for the treatment of venous thromboembolism (VTE) and reduction in the risk of recurrent VTE in pediatric patients from birth to less than 18 years after at least 5 days of initial parenteral anticoagulant treatment.




XARELTO® is indicated for thromboprophylaxis in pediatric patients aged 2 years and older with congenital heart disease who have undergone the Fontan procedure.
were less than 37 weeks of gestation at birth, had less than 10 days of oral feeding, or had a body weight of less than 2.6 kg.
Clinical studies that evaluated safety, efficacy, and pharmacokinetic/ pharmacodynamic data support the use of XARELTO® 10-mg, 15-mg, and 20-mg tablets in pediatric patients. For the XARELTO® 2.5-mg tablets, there are no safety, efficacy, and pharmacokinetic/pharmacodynamic data to support the use in pediatric patients. Therefore, XARELTO® 2.5-mg tablets are not recommended for use in pediatric patients.
Although not all adverse reactions identified in the adult population have been observed in clinical trials of children and adolescent patients, the same warnings and precautions for adults should be considered for children and adolescents.
• Geriatric Use: In clinical trials the efficacy of XARELTO® in the elderly (65 years or older) was similar to that seen in patients younger than 65 years. Both thrombotic and bleeding event rates were higher in these older patients.
OVERDOSAGE
• Overdose of XARELTO® may lead to hemorrhage. Discontinue XARELTO® and initiate appropriate therapy if bleeding complications associated with overdosage occur. An agent to reverse the anti-factor Xa activity of rivaroxaban is available.
ADVERSE REACTIONS
• Most common adverse reactions in adult patients with XARELTO® were bleeding complications.
• Most common adverse reactions in pediatric patients were bleeding, cough, vomiting, and gastroenteritis.
Please read accompanying Brief Summary of full Prescribing Information, including Boxed WARNINGS for XARELTO®.




REFERENCES:
1. Gerhard-Herman MD, Gornik HL, Barret C, et al. 2016 AHA/ACC Guideline on the management of patients with lower extremity peripheral artery disease: Executive summary. Circulation. 2016;135:e686-e725. 2. XARELTO® [prescribing information]. Titusville, NJ: Janssen Pharmaceuticals, Inc. 3. Bonaca MP, Bauersachs RM, Anand SS, et al. Rivaroxaban in peripheral artery disease after revascularization [article and supplementary appendix]. N Engl J Med. 2020;382(21):1994-2004.
cp-62551v9
WARNING: (A) PREMATURE DISCONTINUATION OF XARELTO® INCREASES THE RISK OF THROMBOTIC EVENTS, (B) SPINAL/EPIDURAL HEMATOMA
A. Premature discontinuation of XARELTO® increases the risk of thrombotic events
Premature discontinuation of any oral anticoagulant, including XARELTO®, increases the risk of thrombotic events. If anticoagulation with XARELTO® is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant.
B. Spinal/epidural hematoma
Epidural or spinal hematomas have occurred in patients treated with XARELTO® who are receiving neuraxial anesthesia or undergoing spinal puncture. These hematomas may result in long-term or permanent paralysis. Consider these risks
CONTRAINDICATIONS
• Active pathological bleeding
• Severe hypersensitivity reaction to XARELTO® (eg, anaphylactic reactions)
WARNINGS AND PRECAUTIONS
• Increased Risk of Thrombotic Events after Premature Discontinuation: Premature discontinuation of any oral anticoagulant, including XARELTO®, in the absence of adequate alternative anticoagulation increases the risk of thrombotic events. An increased rate of stroke was observed during the transition from XARELTO® to warfarin in clinical trials in atrial fibrillation patients. If XARELTO® is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant.
• Risk of Bleeding: XARELTO® increases the risk of bleeding and can cause serious or fatal bleeding. Promptly evaluate any signs or symptoms of blood loss and consider the need for blood replacement. Discontinue in patients with active pathological hemorrhage.
– An agent to reverse the anti-factor Xa activity of rivaroxaban is available. Because of high plasma protein binding, rivaroxaban is not dialyzable.
– Concomitant use of other drugs that impair hemostasis increases risk of bleeding. These include aspirin, P2Y12 platelet inhibitors, dual antiplatelet therapy, other antithrombotic agents, fibrinolytic therapy, NSAIDs, selective serotonin reuptake inhibitors (SSRIs), and serotonin norepinephrine reuptake inhibitors (SNRIs).
– Risk of Hemorrhage in Acutely Ill Medical Patients at High Risk of Bleeding: Acutely ill medical patients with the following conditions are at increased risk of bleeding with the use of XARELTO® for primary VTE prophylaxis: history of bronchiectasis, pulmonary cavitation, or pulmonary hemorrhage; active cancer (ie, undergoing acute, in-hospital cancer treatment); active gastroduodenal ulcer or history of bleeding in the three months prior to treatment; or dual antiplatelet therapy. XARELTO® is not for use for primary VTE prophylaxis in these hospitalized, acutely ill medical patients at high risk of bleeding.
• Spinal/Epidural Anesthesia or Puncture: When neuraxial anesthesia (spinal/epidural anesthesia) or spinal puncture is employed, patients treated with anticoagulant agents for prevention of thromboembolic complications are at risk of developing an epidural or spinal hematoma, which can result in longterm or permanent paralysis. To reduce the potential risk of bleeding associated with concurrent use of XARELTO® and epidural or spinal anesthesia/analgesia or spinal puncture, consider the pharmacokinetic profile of XARELTO®. Placement or removal of an epidural catheter or lumbar puncture is best performed when the anticoagulant effect of XARELTO® is low; however, the exact timing to reach a sufficiently low anticoagulant effect in each patient is not known. An indwelling epidural or intrathecal catheter should not be removed before at least 2 half-lives have elapsed (ie, 18 hours in young patients aged 20 to 45 years and 26 hours in elderly patients aged 60 to 76 years), after the last administration of XARELTO®. The next dose should not be administered earlier than 6 hours after the removal of the catheter. If traumatic puncture occurs, delay the administration of XARELTO® for 24 hours. Monitor frequently to detect signs or symptoms of neurological impairment, such as midline back pain, sensory and motor deficits (numbness, tingling, or weakness in lower limbs), or bowel and/or bladder dysfunction. Instruct patients to immediately report any of the above signs or symptoms. If signs or symptoms of spinal hematoma are suspected, initiate urgent diagnosis and treatment including consideration for spinal cord decompression even though such treatment may not prevent or reverse neurological sequelae
• Use in Patients with Renal Impairment:
– Nonvalvular Atrial Fibrillation: Periodically assess renal function as clinically indicated (ie, more frequently in situations in which renal function may decline) and adjust therapy accordingly. Consider dose adjustment or discontinuation in patients who develop acute renal failure while on XARELTO®. Clinical efficacy and safety studies with XARELTO® did not enroll patients with CrCl <30 mL/min or end-stage renal disease (ESRD) on dialysis
– Treatment of Deep Vein Thrombosis (DVT), Pulmonary Embolism (PE), and Reduction in the Risk of Recurrence of DVT and of PE: In patients with CrCl <30 mL/min, rivaroxaban exposure and pharmacodynamic effects are increased compared to patients with normal renal function. There are limited clinical data in patients with CrCl 15 to <30 mL/min; therefore, observe closely and promptly evaluate any signs or symptoms of blood loss in these patients. There are no clinical data in patients with CrCl <15 mL/min (including patients on dialysis); therefore, avoid the use of XARELTO® in these patients. Discontinue XARELTO® in patientswho develop acute renal failure while on treatment.
– Prophylaxis of Deep Vein Thrombosis Following Hip or Knee Replacement Surgery: In patients with CrCl <30 mL/min, rivaroxaban exposure and pharmacodynamic effects are increased compared to patients with normal renal function. There are limited clinical data in patients with CrCl 15 to <30 mL/min; therefore, observe closely and promptly evaluate signs or symptoms of blood loss in
when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
• Use of indwelling epidural catheters
• Concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, other anticoagulants, see Drug Interactions
• A history of traumatic or repeated epidural or spinal punctures
• A history of spinal deformity or spinal surgery
• Optimal timing between the administration of XARELTO® and neuraxial procedures is not known
Monitor patients frequently for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary.
Consider the benefits and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis.
these patients. There are no clinical data in patients with CrCl <15 mL/min (including patients on dialysis); therefore, avoid the use of XARELTO® in these patients. Discontinue XARELTO® in patients who develop acute renal failure while on treatment.
– Prophylaxis of Venous Thromboembolism in Acutely Ill Medical Patients at Risk for Thromboembolic Complications Not at High Risk of Bleeding: In patients with CrCl <30 mL/min, rivaroxaban exposure and pharmacodynamic effects are increased compared to patients with normal renal function. There are limited clinical data in patients with CrCl 15 to <30 mL/min; therefore, observe closely and promptly evaluate any signs or symptoms of blood loss in these patients. There are no clinical data in patients with CrCl <15 mL/min (including patients on dialysis); therefore, avoid the use of XARELTO® in these patients. Discontinue XARELTO® in patients who develop acute renal failure while on treatment.
– Reduction of Risk of Major Cardiovascular Events in Patients with CAD and Reduction of Risk of Major Thrombotic Vascular Events in Patients with PAD, Including Patients after Recent Lower Extremity Revascularization Due to Symptomatic PAD: For patients with CrCl <15 mL/min, no data are available, and limited data are available for patients with a CrCl of 15 to 30 mL/min. In patients with CrCl <30 mL/min, a dose of 2.5 mg XARELTO® twice daily is expected to give an exposure similar to that in patients with moderate renal impairment (CrCl 30 to <50 mL/min), whose efficacy and safety outcomes were similar to those with preserved renal function. Clinical efficacy and safety studies with XARELTO® did not enroll patients with end-stage renal disease (ESRD) on dialysis.
– Pediatric Patients: There are limited clinical data in pediatric patients 1 year or older with moderate or severe renal impairment (eGFR <50 mL/min/1.73 m²); therefore, avoid use of XARELTO® in these patients.
There are no clinical data in pediatric patients younger than 1 year with serum creatinine results above 97.5th percentile; therefore, avoid the use of XARELTO® in these patients.
• Use in Patients with Hepatic Impairment: No clinical data are available for adult patients with severe hepatic impairment. Avoid use in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment or with any hepatic disease associated with coagulopathy, since drug exposure and bleeding risk may be increased. No clinical data are available in pediatric patients with hepatic impairment.
• Use with P-gp and Strong CYP3A Inhibitors or Inducers: Avoid concomitant use of XARELTO® with known combined P-gp and strong CYP3A inhibitors or inducers
• Risk of Pregnancy-Related Hemorrhage: In pregnant women, XARELTO® should be used only if the potential benefit justifies the potential risk to the mother and fetus. XARELTO® dosing in pregnancy has not been studied. The anticoagulant effect of XARELTO® cannot be monitored with standard laboratory testing. Promptly evaluate signs or symptoms suggesting blood loss (eg, a drop in hemoglobin and/or hematocrit, hypotension, or fetal distress)
• Patients with Prosthetic Heart Valves: Use of XARELTO® is not recommended in patients who have had transcatheter aortic valve replacement (TAVR), based on the results of the GALILEO study, which reported higher rates of death and bleeding in patients randomized to XARELTO® compared to those randomized to an antiplatelet regimen. Safety and efficacy of XARELTO® have not been studied in patients with other prosthetic heart valves or other valve procedures. Use of XARELTO® is not recommended in patients with prosthetic heart valves.
• Acute PE in Hemodynamically Unstable Patients/Patients Who Require Thrombolysis or Pulmonary Embolectomy: Initiation of XARELTO® is not recommended acutely as an alternative to unfractionated heparin in patients with pulmonary embolism who present with hemodynamic instability or who may receive thrombolysis or pulmonary embolectomy
• Increased Risk of Thrombosis in Patients with Antiphospholipid Syndrome: Direct-acting oral anticoagulants (DOACs), including XARELTO®, are not recommended for use in patients with triple-positive antiphospholipid syndrome (APS). For patients with APS (especially those who are triple positive [positive for lupus anticoagulant, anticardiolipin, and anti-beta 2-glycoprotein I antibodies]), treatment with DOACs has been associated with increased rates of recurrent thrombotic events compared with vitamin K antagonist therapy.
DRUG INTERACTIONS
• Combined P-gp and strong CYP3A inhibitors increase exposure to rivaroxaban and may increase risk of bleeding.
• Combined P-gp and strong CYP3A inducers decrease exposure to rivaroxaban and may increase risk of thromboembolic events.
• XARELTO® should not be used in patients with CrCl 15 to <80 mL/min who are receiving concomitant combined P-gp and moderate CYP3A inhibitors (eg, erythromycin) unless the potential benefit justifies the potential risk.
IMPORTANT SAFETY INFORMATION
Please read accompanying Brief Summary of full Prescribing Information, including Boxed WARNINGS for XARELTO® (continued on next page)
Brief Summary of Prescribing Information for XARELTO® (rivaroxaban)
XARELTO (rivaroxaban) tablets, for oral use XARELTO (rivaroxaban) for oral suspension
See package insert for Full Prescribing Information
WARNING: (A) PREMATURE DISCONTINUATION OF XARELTO INCREASES THE RISK OF THROMBOTIC EVENTS, (B) SPINAL/EPIDURAL HEMATOMA
A. Premature discontinuation of XARELTO increases the risk of thrombotic events
Premature discontinuation of any oral anticoagulant, including XARELTO, increases the risk of thrombotic events. If anticoagulation with XARELTO is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant [see Dosage and Administration (2.3, 2.4) in Full Prescribing Information, Warnings and Precautions, and Clinical Studies (14.1) in Full Prescribing Information].
B. Spinal/epidural hematoma
Epidural or spinal hematomas have occurred in patients treated with XARELTO who are receiving neuraxial anesthesia or undergoing spinal puncture. These hematomas may result in long-term or permanent paralysis. Consider these risks when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
• use of indwelling epidural catheters
• concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, other anticoagulants
• a history of traumatic or repeated epidural or spinal punctures
• a history of spinal deformity or spinal surgery
• optimal timing between the administration of XARELTO and neuraxial procedures is not known [see Warnings and Precautions and Adverse Reactions].
Monitor patients frequently for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary [see Warnings and Precautions]
Consider the benefits and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis [see Warnings and Precautions]
INDICATIONS AND USAGE
Reduction of Risk of Stroke and Systemic Embolism in Nonvalvular Atrial Fibrillation
XARELTO is indicated to reduce the risk of stroke and systemic embolism in adult patients with nonvalvular atrial fibrillation.
There are limited data on the relative effectiveness of XARELTO and warfarin in reducing the risk of stroke and systemic embolism when warfarin therapy is well-controlled [see Clinical Studies (14.1) in Full Prescribing Information].
Treatment of Deep Vein Thrombosis
XARELTO is indicated for the treatment of deep vein thrombosis (DVT). Treatment of Pulmonary Embolism
XARELTO is indicated for the treatment of pulmonary embolism (PE). Reduction in the Risk of Recurrence of Deep Vein Thrombosis and/or Pulmonary Embolism
XARELTO is indicated for the reduction in the risk of recurrence of DVT and/or PE in adult patients at continued risk for recurrent DVT and/or PE after completion of initial treatment lasting at least 6 months.
Prophylaxis of Deep Vein Thrombosis Following Hip or Knee Replacement Surgery
XARELTO is indicated for the prophylaxis of DVT, which may lead to PE in adult patients undergoing knee or hip replacement surgery.
Prophylaxis of Venous Thromboembolism in Acutely Ill Medical Patients at Risk for Thromboembolic Complications Not at High Risk of Bleeding
XARELTO is indicated for the prophylaxis of venous thromboembolism (VTE) and VTE related death during hospitalization and post hospital discharge in adult patients admitted for an acute medical illness who are at risk for thromboembolic complications due to moderate or severe restricted mobility and other risk factors for VTE and not at high risk of bleeding [see Warnings and Precautions and Clinical Studies (14.5) in Full Prescribing Information].
Reduction of Risk of Major Cardiovascular Events in Patients with Coronary Artery Disease (CAD)
XARELTO, in combination with aspirin, is indicated to reduce the risk of major cardiovascular events (cardiovascular death, myocardial infarction, and stroke) in adult patients with coronary artery disease.
Reduction of Risk of Major Thrombotic Vascular Events in Patients with Peripheral Artery Disease (PAD), Including Patients after Lower Extremity Revascularization due to Symptomatic PAD
XARELTO, in combination with aspirin, is indicated to reduce the risk of major thrombotic vascular events (myocardial infarction, ischemic stroke, acute limb ischemia, and major amputation of a vascular etiology) in adult patients with PAD, including patients who have recently undergone a lower extremity revascularization procedure due to symptomatic PAD.
Treatment of Venous Thromboembolism and Reduction in Risk of Recurrent Venous Thromboembolism in Pediatric Patients
XARELTO is indicated for the treatment of venous thromboembolism (VTE) and the reduction in the risk of recurrent VTE in pediatric patients from birth to less than 18 years after at least 5 days of initial parenteral anticoagulant treatment.
Thromboprophylaxis in Pediatric Patients with Congenital Heart Disease after the Fontan Procedure
XARELTO is indicated for thromboprophylaxis in pediatric patients aged 2 years and older with congenital heart disease who have undergone the Fontan procedure.
CONTRAINDICATIONS
XARELTO is contraindicated in patients with:
• active pathological bleeding [see Warnings and Precautions]
• severe hypersensitivity reaction to XARELTO (e.g., anaphylactic reactions) [see Adverse Reactions]
WARNINGS AND PRECAUTIONS
Increased Risk of Thrombotic Events after Premature Discontinuation
Premature discontinuation of any oral anticoagulant, including XARELTO, in the absence of adequate alternative anticoagulation increases the risk of thrombotic events. An increased rate of stroke was observed during the transition from XARELTO to warfarin in clinical trials in atrial
fibrillation patients. If XARELTO is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant [see Dosage and Administration (2.3, 2.4) and Clinical Studies (14.1) in Full Prescribing Information].
Risk of Bleeding
XARELTO increases the risk of bleeding and can cause serious or fatal bleeding. In deciding whether to prescribe XARELTO to patients at increased risk of bleeding, the risk of thrombotic events should be weighed against the risk of bleeding.
Promptly evaluate any signs or symptoms of blood loss and consider the need for blood replacement. Discontinue XARELTO in patients with active pathological hemorrhage. The terminal elimination half-life of rivaroxaban is 5 to 9 hours in healthy subjects aged 20 to 45 years.
Concomitant use of other drugs that impair hemostasis increases the risk of bleeding. These include aspirin, P2Y12 platelet inhibitors, dual antiplatelet therapy, other antithrombotic agents, fibrinolytic therapy, non-steroidal anti-inflammatory drugs (NSAIDs) [see Drug Interactions], selective serotonin reuptake inhibitors, and serotonin norepinephrine reuptake inhibitors.
Concomitant use of drugs that are known combined P-gp and strong CYP3A inhibitors increases rivaroxaban exposure and may increase bleeding risk [see Drug Interactions].
Risk of Hemorrhage in Acutely Ill Medical Patients at High Risk of Bleeding Acutely ill medical patients with the following conditions are at increased risk of bleeding with the use of XARELTO for primary VTE prophylaxis: history of bronchiectasis, pulmonary cavitation, or pulmonary hemorrhage, active cancer (i.e., undergoing acute, in-hospital cancer treatment), active gastroduodenal ulcer in the three months prior to treatment, history of bleeding in the three months prior to treatment, or dual antiplatelet therapy. XARELTO is not for use for primary VTE prophylaxis in these hospitalized, acutely ill medical patients at high risk of bleeding.
Reversal of Anticoagulant Effect
An agent to reverse the anti-factor Xa activity of rivaroxaban is available. Because of high plasma protein binding, rivaroxaban is not dialyzable [see Clinical Pharmacology (12.3) in Full Prescribing Information]. Protamine sulfate and vitamin K are not expected to affect the anticoagulant activity of rivaroxaban. Use of procoagulant reversal agents, such as prothrombin complex concentrate (PCC), activated prothrombin complex concentrate or recombinant factor VIIa, may be considered but has not been evaluated in clinical efficacy and safety studies. Monitoring for the anticoagulation effect of rivaroxaban using a clotting test (PT, INR or aPTT) or anti-factor Xa (FXa) activity is not recommended.
Spinal/Epidural Anesthesia or Puncture
When neuraxial anesthesia (spinal/epidural anesthesia) or spinal puncture is employed, patients treated with anticoagulant agents for prevention of thromboembolic complications are at risk of developing an epidural or spinal hematoma which can result in long-term or permanent paralysis [see Boxed Warning]
To reduce the potential risk of bleeding associated with the concurrent use of XARELTO and epidural or spinal anesthesia/analgesia or spinal puncture, consider the pharmacokinetic profile of XARELTO [see Clinical Pharmacology (12.3) in Full Prescribing Information]. Placement or removal of an epidural catheter or lumbar puncture is best performed when the anticoagulant effect of XARELTO is low; however, the exact timing to reach a sufficiently low anticoagulant effect in each patient is not known.
An indwelling epidural or intrathecal catheter should not be removed before at least 2 half-lives have elapsed (i.e., 18 hours in young patients aged 20 to 45 years and 26 hours in elderly patients aged 60 to 76 years), after the last administration of XARELTO [see Clinical Pharmacology (12.3) in Full Prescribing Information]. The next XARELTO dose should not be administered earlier than 6 hours after the removal of the catheter. If traumatic puncture occurs, delay the administration of XARELTO for 24 hours.
Should the physician decide to administer anticoagulation in the context of epidural or spinal anesthesia/analgesia or lumbar puncture, monitor frequently to detect any signs or symptoms of neurological impairment, such as midline back pain, sensory and motor deficits (numbness, tingling, or weakness in lower limbs), bowel and/or bladder dysfunction. Instruct patients to immediately report if they experience any of the above signs or symptoms. If signs or symptoms of spinal hematoma are suspected, initiate urgent diagnosis and treatment including consideration for spinal cord decompression even though such treatment may not prevent or reverse neurological sequelae.
Use in Patients with Renal Impairment Nonvalvular Atrial Fibrillation
Periodically assess renal function as clinically indicated (i.e., more frequently in situations in which renal function may decline) and adjust therapy accordingly [see Dosage and Administration (2.1) in Full Prescribing Information]. Consider dose adjustment or discontinuation of XARELTO in patients who develop acute renal failure while on XARELTO [see Use in Specific Populations]
Treatment of Deep Vein Thrombosis (DVT), Pulmonary Embolism (PE), and Reduction in the Risk of Recurrence of DVT and of PE
In patients with CrCl <30 mL/min, rivaroxaban exposure and pharmacodynamic effects are increased compared to patients with normal renal function. There are limited clinical data in patients with CrCl 15 to <30 mL/min; therefore, observe closely and promptly evaluate any signs or symptoms of blood loss in these patients. There are no clinical data in patients with CrCl <15 mL/min (including patients on dialysis); therefore, avoid the use of XARELTO in these patients.
Discontinue XARELTO in patients who develop acute renal failure while on treatment [see Use in Specific Populations]
Prophylaxis of Deep Vein Thrombosis Following Hip or Knee Replacement Surgery
In patients with CrCl <30 mL/min, rivaroxaban exposure and pharmacodynamic effects are increased compared to patients with normal renal function. There are limited clinical data in patients with CrCl 15 to <30 mL/min; therefore, observe closely and promptly evaluate any signs or symptoms of blood loss in these patients. There are no clinical data in patients with CrCl <15 mL/min (including patients on dialysis); therefore, avoid the use of XARELTO in these patients.
Discontinue XARELTO in patients who develop acute renal failure while on treatment [see Use in Specific Populations]
Prophylaxis of Venous Thromboembolism in Acutely Ill Medical Patients at Risk for Thromboembolic Complications Not at High Risk of Bleeding
In patients with CrCl <30 mL/min, rivaroxaban exposure and pharmacodynamic effects are increased compared to patients with normal
renal function. There are limited clinical data in patients with CrCl 15 to <30 mL/min; therefore, observe closely and promptly evaluate any signs or symptoms of blood loss in these patients. There are no clinical data in patients with CrCl <15 mL/min (including patients on dialysis); therefore, avoid the use of XARELTO in these patients.
Discontinue XARELTO in patients who develop acute renal failure while on treatment [see Use in Specific Populations]
Pediatric Patients
There are limited clinical data in pediatric patients 1 year or older with moderate or severe renal impairment (eGFR <50 mL/min/1.73 m2); therefore, avoid the use of XARELTO in these patients.
There are no clinical data in pediatric patients younger than 1 year with serum creatinine results above 97.5th percentile; therefore, avoid the use of XARELTO in these patients [see Dosage and Administration (2.2) in Full Prescribing Information and Use in Specific Populations].
Use in Patients with Hepatic Impairment
No clinical data are available for adult patients with severe hepatic impairment.
Avoid use of XARELTO in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment or with any hepatic disease associated with coagulopathy since drug exposure and bleeding risk may be increased [see Use in Specific Populations]
No clinical data are available in pediatric patients with hepatic impairment.
Use with P-gp and Strong CYP3A Inhibitors or Inducers
Avoid concomitant use of XARELTO with known combined P-gp and strong CYP3A inhibitors [see Drug Interactions].
Avoid concomitant use of XARELTO with drugs that are known combined P-gp and strong CYP3A inducers [see Drug Interactions].
Risk of Pregnancy-Related Hemorrhage
In pregnant women, XARELTO should be used only if the potential benefit justifies the potential risk to the mother and fetus. XARELTO dosing in pregnancy has not been studied. The anticoagulant effect of XARELTO cannot be monitored with standard laboratory testing. Promptly evaluate any signs or symptoms suggesting blood loss (e.g., a drop in hemoglobin and/or hematocrit, hypotension, or fetal distress) [see Warnings and Precautions and Use in Specific Populations].
Patients with Prosthetic Heart Valves
On the basis of the GALILEO study, use of XARELTO is not recommended in patients who have had transcatheter aortic valve replacement (TAVR) because patients randomized to XARELTO experienced higher rates of death and bleeding compared to those randomized to an anti-platelet regimen. The safety and efficacy of XARELTO have not been studied in patients with other prosthetic heart valves or other valve procedures. Use of XARELTO is not recommended in patients with prosthetic heart valves.
Acute PE in Hemodynamically Unstable Patients or Patients Who Require Thrombolysis or Pulmonary Embolectomy
Initiation of XARELTO is not recommended acutely as an alternative to unfractionated heparin in patients with pulmonary embolism who present with hemodynamic instability or who may receive thrombolysis or pulmonary embolectomy.
Increased Risk of Thrombosis in Patients with Triple Positive Antiphospholipid Syndrome
Direct-acting oral anticoagulants (DOACs), including XARELTO, are not recommended for use in patients with triple-positive antiphospholipid syndrome (APS). For patients with APS (especially those who are triple positive [positive for lupus anticoagulant, anticardiolipin, and anti-beta 2-glycoprotein I antibodies]), treatment with DOACs has been associated with increased rates of recurrent thrombotic events compared with vitamin K antagonist therapy.
ADVERSE REACTIONS
The following clinically significant adverse reactions are also discussed in other sections of the labeling:
• Increased Risk of Stroke After Discontinuation in Nonvalvular Atrial Fibrillation [see Boxed Warning and Warnings and Precautions]
• Bleeding Risk [see Warnings and Precautions]
• Spinal/Epidural Hematoma [see Boxed Warning and Warnings and Precautions]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
During clinical development for the approved indications, 34,947 adult patients were exposed to XARELTO.
Hemorrhage
The most common adverse reactions with XARELTO were bleeding complications [see Warnings and Precautions]
Nonvalvular Atrial Fibrillation
In the ROCKET AF trial, the most frequent adverse reactions associated with permanent drug discontinuation were bleeding events, with incidence rates of 4.3% for XARELTO vs. 3.1% for warfarin. The incidence of discontinuations for non-bleeding adverse events was similar in both treatment groups.
Table 1 shows the number of patients experiencing various types of bleeding events in the ROCKET AF trial.
Table 1: Bleeding Events in ROCKET AF*- On Treatment Plus 2 Days Parameter XARELTO N=7111 n (%/year) Warfarin N=7125 n (%/year) XARELTO vs. Warfarin HR (95% CI)
Major Bleeding† 395 (3.6) 386 (3.5) 1.04 (0.90, 1.20)
Intracranial Hemorrhage (ICH)‡ 55 (0.5) 84 (0.7) 0.67 (0.47, 0.93)
Hemorrhagic Stroke§ 36 (0.3) 58 (0.5) 0.63 (0.42, 0.96)
Other ICH 19 (0.2) 26 (0.2) 0.74 (0.41, 1.34)
Gastrointestinal (GI)¶ 221 (2.0) 140 (1.2) 1.61 (1.30, 1.99)
Fatal Bleeding# 27 (0.2) 55 (0.5) 0.50 (0.31, 0.79)
ICH 24 (0.2) 42 (0.4) 0.58 (0.35, 0.96)
Non-intracranial 3 (0.0) 13 (0.1) 0.23 (0.07, 0.82)
XARELTO® (rivaroxaban) XARELTO® (rivaroxaban)
Abbreviations: HR = Hazard Ratio, CI = Confidence interval, CRNM = Clinically Relevant Non-Major.
* Major bleeding events within each subcategory were counted once per patient, but patients may have contributed events to multiple subcategories. These events occurred during treatment or within 2 days of stopping treatment.
† Defined as clinically overt bleeding associated with a decrease in hemoglobin of ≥2 g/dL, a transfusion of ≥2 units of packed red blood cells or whole blood, bleeding at a critical site, or with a fatal outcome.
‡ Intracranial bleeding events included intraparenchymal, intraventricular, subdural, subarachnoid and/or epidural hematoma.
§ Hemorrhagic stroke in this table specifically refers to non-traumatic intraparenchymal and/or intraventricular hematoma in patients on treatment plus 2 days.
¶ Gastrointestinal bleeding events included upper GI, lower GI, and rectal bleeding.
# Fatal bleeding is adjudicated death with the primary cause of death from bleeding.
Figure 1 shows the risk of major bleeding events across major subgroups.
Figure 1: Risk of Major Bleeding Events by Baseline Characteristics in ROCKET AF – On Treatment Plus 2 Days
Table 3: Bleeding Events* in EINSTEIN CHOICE
Parameter
XARELTO† 10 mg N=1127 n (%)
Table 5: Bleeding Events in MAGELLAN* Study–Safety Analysis Set - On Treatment Plus 2 Days
Acetylsalicylic Acid (aspirin)† 100 mg N=1131 n (%)
Major bleeding event 5 (0.4) 3 (0.3)
Fatal bleeding 0 1 (<0.1)
Non-fatal critical organ bleeding 2 (0.2) 1 (<0.1)
Non-fatal non-critical organ bleeding‡ 3 (0.3) 1 (<0.1)
Clinically relevant non-major (CRNM) bleeding§ 22 (2.0) 20 (1.8)
Any bleeding 151 (13.4) 138 (12.2)
* Bleeding event occurred after the first dose and up to 2 days after the last dose of study drug. Although a patient may have had 2 or more events, the patient is counted only once in a category.
† Treatment schedule: XARELTO 10 mg once daily or aspirin 100 mg once daily.
‡ Major bleeding which is not fatal or in a critical organ, but resulting in a decrease in Hb ≥ 2 g/dL and/or transfusion of ≥ 2 units of whole blood or packed red blood cells.
§ Bleeding which was clinically overt, did not meet the criteria for major bleeding, but was associated with medical intervention, unscheduled contact with a physician, temporary cessation of treatment, discomfort for the patient, or impairment of activities of daily life.
In the EINSTEIN CHOICE study, there was an increased incidence of bleeding, including major and CRNM bleeding in the XARELTO 20 mg group compared to the XARELTO 10 mg or aspirin 100 mg groups.
Prophylaxis of Deep Vein Thrombosis Following Hip or Knee Replacement Surgery
In the RECORD clinical trials, the overall incidence rate of adverse reactions leading to permanent treatment discontinuation was 3.7% with XARELTO.
The rates of major bleeding events and any bleeding events observed in patients in the RECORD clinical trials are shown in Table 4.
Table 4: Bleeding Events* in Patients Undergoing Hip or Knee Replacement Surgeries (RECORD 1 3)
XARELTO 10 mg Enoxaparin†
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and all of which were pre-specified (diabetic status was not pre-specified in the subgroup but was a criterion for the CHADS2 score). The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
Treatment of Deep Vein Thrombosis (DVT) and/or Pulmonary Embolism (PE) EINSTEIN DVT and EINSTEIN PE Studies
In the pooled analysis of the EINSTEIN DVT and EINSTEIN PE clinical studies, the most frequent adverse reactions leading to permanent drug discontinuation were bleeding events, with XARELTO vs. enoxaparin/ Vitamin K antagonist (VKA) incidence rates of 1.7% vs. 1.5%, respectively. The mean duration of treatment was 208 days for XARELTO-treated patients and 204 days for enoxaparin/VKA-treated patients.
Table 2 shows the number of patients experiencing major bleeding events in the pooled analysis of the EINSTEIN DVT and EINSTEIN PE studies.
Table 2: Bleeding Events* in the Pooled Analysis of EINSTEIN DVT and EINSTEIN PE Studies
Parameter
XARELTO† N=4130 n (%) Enoxaparin/ VKA† N=4116 n (%)
Major bleeding event 40 (1.0) 72 (1.7)
Fatal bleeding 3 (<0.1) 8 (0.2)
Intracranial 2 (<0.1) 4 (<0.1)
Non-fatal critical organ bleeding 10 (0.2) 29 (0.7)
Intracranial‡ 3 (<0.1) 10 (0.2)
Retroperitoneal‡ 1 (<0.1) 8 (0.2)
Intraocular‡ 3 (<0.1) 2 (<0.1)
Intra-articular‡ 0 4 (<0.1)
Non-fatal non-critical organ bleeding§ 27 (0.7) 37 (0.9)
Decrease in Hb ≥ 2 g/dL 28 (0.7) 42 (1.0)
Transfusion of ≥2 units of whole blood or packed red blood cells 18 (0.4) 25 (0.6)
Clinically relevant non-major bleeding 357 (8.6) 357 (8.7) Any bleeding 1169 (28.3) 1153 (28.0)
* Bleeding event occurred after randomization and up to 2 days after the last dose of study drug. Although a patient may have had 2 or more events, the patient is counted only once in a category.
† Treatment schedule in EINSTEIN DVT and EINSTEIN PE studies: XARELTO 15 mg twice daily for 3 weeks followed by 20 mg once daily; enoxaparin/VKA [enoxaparin: 1 mg/kg twice daily, VKA: individually titrated doses to achieve a target INR of 2.5 (range: 2.0-3.0)]
‡ Treatment-emergent major bleeding events with at least >2 subjects in any pooled treatment group
§ Major bleeding which is not fatal or in a critical organ, but resulting in a decrease in Hb ≥ 2 g/dL and/or transfusion of ≥2 units of whole blood or packed red blood cells
Reduction in the Risk of Recurrence of DVT and/or PE EINSTEIN CHOICE Study
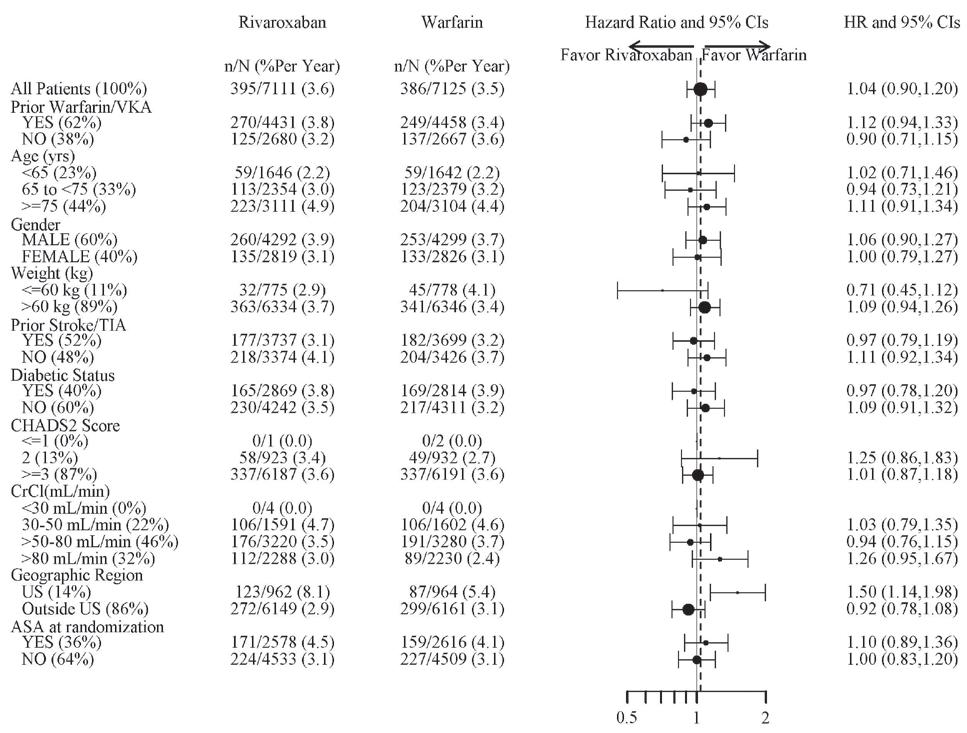
In the EINSTEIN CHOICE clinical study, the most frequent adverse reactions associated with permanent drug discontinuation were bleeding events, with incidence rates of 1% for XARELTO 10 mg, 2% for XARELTO 20 mg, and 1% for acetylsalicylic acid (aspirin) 100 mg. The mean duration of treatment was 293 days for XARELTO 10 mg-treated patients and 286 days for aspirin 100 mg-treated patients.
Table 3 shows the number of patients experiencing bleeding events in the EINSTEIN CHOICE study.
Total treated patients N=4487 n (%) N=4524 n (%)
Major bleeding event 14 (0.3) 9 (0.2)
Fatal bleeding 1 (<0.1) 0
Bleeding into a critical organ 2 (<0.1) 3 (0.1)
Bleeding that required re-operation 7 (0.2) 5 (0.1)
Extra-surgical site bleeding requiring transfusion of >2 units of whole blood or packed cells 4 (0.1) 1 (<0.1)
Any bleeding event‡ 261 (5.8) 251 (5.6)
Hip Surgery Studies N=3281 n (%) N=3298 n (%)
Major bleeding event 7 (0.2) 3 (0.1)
Fatal bleeding 1 (<0.1) 0
Bleeding into a critical organ 1 (<0.1) 1 (<0.1)
Bleeding that required re-operation 2 (0.1) 1 (<0.1)
Extra-surgical site bleeding requiring transfusion of >2 units of whole blood or packed cells 3 (0.1) 1 (<0.1)
Any bleeding event‡ 201 (6.1) 191 (5.8)
Knee Surgery Study N=1206 n (%) N=1226 n (%)
Major bleeding event 7 (0.6) 6 (0.5)
Fatal bleeding 0 0
Bleeding into a critical organ 1 (0.1) 2 (0.2)
Bleeding that required re-operation 5 (0.4) 4 (0.3)
Extra-surgical site bleeding requiring transfusion of >2 units of whole blood or packed cells 1 (0.1) 0
Any bleeding event‡ 60 (5.0) 60 (4.9)
* Bleeding events occurring any time following the first dose of doubleblind study medication (which may have been prior to administration of active drug) until two days after the last dose of double-blind study medication. Patients may have more than one event.
† Includes the placebo-controlled period for RECORD 2, enoxaparin dosing was 40 mg once daily (RECORD 1-3)
‡ Includes major bleeding events
Following XARELTO treatment, the majority of major bleeding complications (≥60%) occurred during the first week after surgery.
Prophylaxis of Venous Thromboembolism in Acutely Ill Medical Patients at Risk for Thromboembolic Complications Not at High Risk of Bleeding
In the MAGELLAN study, the most frequent adverse reactions associated with permanent drug discontinuation were bleeding events. Cases of pulmonary hemorrhage and pulmonary hemorrhage with bronchiectasis were observed. Patients with bronchiectasis/pulmonary cavitation, active cancer (i.e., undergoing acute, in-hospital cancer treatment), dual antiplatelet therapy or active gastroduodenal ulcer or any bleeding in the previous three months all had an excess of bleeding with XARELTO compared with enoxaparin/placebo and are excluded from all MAGELLAN data presented in Table 5. The incidence of bleeding leading to drug discontinuation was 2.5% for XARELTO vs. 1.4% for enoxaparin/placebo.
Table 5 shows the number of patients experiencing various types of bleeding events in the MAGELLAN study.
MAGELLAN Study¶ XARELTO 10 mg N=3218 n (%) Enoxaparin 40 mg / placebo N=3229 n (%)
Major bleeding‡† 22 (0.7) 15 (0.5)
Critical site bleeding 7 (0.2) 4 (0.1)
Fatal bleeding§ 3 (<0.1) 1 (<0.1)
Clinically relevant non-major bleeding events (CRNM) 93 (2.9) 34 (1.1)
* Patients at high risk of bleeding (i.e. bronchiectasis/pulmonary cavitation, active cancer, dual antiplatelet therapy or active gastroduodenal ulcer or any bleeding in the previous three months) were excluded.
† Major bleeding events within each subcategory were counted once per patient, but patients may have contributed events to multiple subcategories. These events occurred during treatment or within 2 days of stopping treatment.
‡ Defined as clinically overt bleeding associated with a drop in hemoglobin of ≥2 g/dL, a transfusion of ≥2 units of packed red blood cells or whole blood, bleeding at a critical site, or with a fatal outcome.
§ Fatal bleeding is adjudicated death with the primary cause of death from bleeding.
¶ Patients received either XARELTO or placebo once daily for 35 ±4 days starting in hospital and continuing post hospital discharge or received enoxaparin or placebo once daily for 10 ±4 days in the hospital.
Reduction of Risk of Major Cardiovascular Events in Patients with CAD In the COMPASS trial overall, the most frequent adverse reactions associated with permanent drug discontinuation were bleeding events, with incidence rates of 2.7% for XARELTO 2.5 mg twice daily vs. 1.2% for placebo on background therapy for all patients with aspirin 100 mg once daily. The incidences of important bleeding events in the CAD and PAD populations in COMPASS were similar.
Table 6 shows the number of patients experiencing various types of major bleeding events in the COMPASS trial.
Table 6: Major Bleeding Events in COMPASS - On Treatment Plus 2 Days*
Parameter XARELTO† N=9134 n (%/year) Placebo† N=9107 n (%/year) XARELTO vs. Placebo HR (95 % CI)
Modified ISTH Major Bleeding‡ 263 (1.6) 144 (0.9) 1.8 (1.5, 2.3)
- Fatal bleeding event Intracranial hemorrhage (ICH) Non-intracranial 12 (<0.1) 6 (<0.1) 6 (<0.1) 8 (<0.1) 3 (<0.1) 5 (<0.1) 1.5 (0.6, 3.7) 2.0 (0.5, 8.0) 1.2 (0.4, 4.0)
- Symptomatic bleeding in critical organ (non-fatal)
- ICH (fatal and non-fatal) Hemorrhagic Stroke Other ICH 58 (0.3) 23 (0.1) 18 (0.1) 6 (<0.1) 43 (0.3) 21 (0.1) 13 (<0.1) 9 (<0.1) 1.4 (0.9, 2.0) 1.1 (0.6, 2.0) 1.4 (0.7, 2.8) 0.7 (0.2, 1.9)
- Bleeding into the surgical site requiring reoperation (non-fatal, not in critical organ)
7 (<0.1) 6 (<0.1) 1.2 (0.4, 3.5)
- Bleeding leading to hospitalization (non-fatal, not in critical organ, not requiring reoperation) 188 (1.1) 91 (0.5) 2.1 (1.6, 2.7)
Major GI bleeding 117 (0.7) 49 (0.3) 2.4 (1.7, 3.4)
* Major bleeding events within each subcategory were counted once per patient, but patients may have contributed events to multiple subcategories. These events occurred during treatment or within 2 days of stopping treatment in the safety analysis set in COMPASS patients.
† Treatment schedule: XARELTO 2.5 mg twice daily or placebo. All patients received background therapy with aspirin 100 mg once daily.
‡ Defined as i) fatal bleeding, or ii) symptomatic bleeding in a critical area or organ, such as intraarticular, intramuscular with compartment syndrome, intraspinal, intracranial, intraocular, respiratory, pericardial, liver, pancreas, retroperitoneal, adrenal gland or kidney; or iii) bleeding into the surgical site requiring reoperation, or iv) bleeding leading to hospitalization.
CI: confidence interval; HR: hazard ratio; ISTH: International Society on Thrombosis and Hemostasis
Reduction of Risk of Major Thrombotic Vascular Events in Patients with Peripheral Artery Disease (PAD), Including Patients after Lower Extremity Revascularization due to Symptomatic PAD
The incidence of premature permanent discontinuation due to bleeding events for XARELTO 2.5 mg twice daily vs. placebo on background therapy with aspirin 100 mg once daily in VOYAGER was 4.1% vs. 1.6% and in COMPASS PAD was 2.7% vs. 1.3%, respectively.
Table 7 shows the number of patients experiencing various types of TIMI (Thrombolysis in Myocardial Infarction) major bleeding events in the VOYAGER trial. The most common site of bleeding was gastrointestinal.
Table 7: Major Bleeding Events* in VOYAGER- On Treatment Plus 2 Days XARELTO† N=3256 Placebo† N=3248 XARELTO vs. Placebo HR (95 % CI)
Parameter n (%) Event rate %/year n (%) Event rate %/year
TIMI Major Bleeding (CABG/non-CABG) 62 (1.9) 0.96 44 (1.4) 0.67 1.4 (1.0, 2.1)
Fatal bleeding 6 (0.2) 0.09 6 (0.2) 0.09 1.0 (0.3, 3.2)
Intracranial bleeding 13 (0.4) 0.20 17 (0.5) 0.26 0.8 (0.4, 1.6)
Clinically overt signs of hemorrhage associated with a drop in hemoglobin of ≥5 g/dL or drop in hematocrit of ≥15% 46 (1.4) 0.71 24 (0.7) 0.36 1.9 (1.2, 3.2)
XARELTO® (rivaroxaban) XARELTO® (rivaroxaban) XARELTO® (rivaroxaban)
* Major bleeding events within each subcategory were counted once per patient, but patients may have contributed events to multiple subcategories.
† Treatment schedule: XARELTO 2.5 mg twice daily or placebo. All patients received background therapy with aspirin 100 mg once daily.
CABG: Coronary artery bypass graft; CI: confidence interval; HR: hazard ratio; TIMI: Thrombolysis in Myocardial Infarction Bleeding Criteria
Other Adverse Reactions
Non-hemorrhagic adverse reactions reported in ≥1% of XARELTO-treated patients in the EINSTEIN DVT and EINSTEIN PE studies are shown in Table 8.
Table 8: Other Adverse Reactions* Reported by ≥1% of XARELTO-Treated Patients in EINSTEIN DVT and EINSTEIN PE Studies
Body System
Adverse Reaction
EINSTEIN DVT Study
Gastrointestinal disorders
XARELTO 20 mg N=1718 n (%)
† Treatment schedule: body weight-adjusted doses of XARELTO; randomized 2:1 (XARELTO: Comparator).
‡ Unfractionated heparin (UFH), low molecular weight heparin (LMWH), fondaparinux or VKA.
§ Defined as clinically overt bleeding associated with a decrease in hemoglobin of ≥2 g/dL, a transfusion of ≥2 units of packed red blood cells or whole blood, bleeding at a critical site, or with a fatal outcome.
¶ Defined as clinically overt bleeding, which did not meet the criteria for major bleeding, but was associated with medical intervention, unscheduled contact with a physician, temporary cessation of treatment, discomfort for the patient, or impairment of activities of daily life.
Non-bleeding adverse reactions reported in ≥5% of XARELTO-treated patients are shown in Table 11.
DRUG INTERACTIONS
General Inhibition and Induction Properties
Rivaroxaban is a substrate of CYP3A4/5, CYP2J2, and the P-gp and ATPbinding cassette G2 (ABCG2) transporters. Combined P-gp and strong CYP3A inhibitors increase exposure to rivaroxaban and may increase the risk of bleeding. Combined P-gp and strong CYP3A inducers decrease exposure to rivaroxaban and may increase the risk of thromboembolic events.
Drugs that Inhibit Cytochrome P450 3A Enzymes and Drug Transport Systems
Interaction with Combined P-gp and Strong CYP3A Inhibitors
Avoid concomitant administration of XARELTO with known combined P-gp and strong CYP3A inhibitors (e.g., ketoconazole and ritonavir) [see Warnings and Precautions and Clinical Pharmacology (12.3) in Full Prescribing Information]
Enoxaparin/VKA N=1711 n (%)
Abdominal pain 46 (2.7) 25 (1.5)
General disorders and administration site conditions
Fatigue 24 (1.4) 15 (0.9)
Musculoskeletal and connective tissue disorders
Back pain 50 (2.9) 31 (1.8)
Muscle spasm 23 (1.3) 13 (0.8)
Nervous system disorders
Dizziness 38 (2.2) 22 (1.3)
Psychiatric disorders
Anxiety 24 (1.4) 11 (0.6)
Depression 20 (1.2) 10 (0.6)
Insomnia 28 (1.6) 18 (1.1)
EINSTEIN PE Study
Skin and subcutaneous tissue disorders
XARELTO 20 mg N=2412 n (%)
Table 11: Other Adverse Reactions* Reported in XARELTO-Treated Patients by ≥5% in EINSTEIN Junior Study
Adverse Reaction
XARELTO N=329 n (%) Comparator Group N=162 n (%)
Pain in extremity 23 (7) 7 (4.3)
Fatigue† 23 (7) 7 (4.3)
* Adverse reaction with Relative Risk >1.5 for XARELTO versus comparator.
† The following terms were combined: fatigue, asthenia.
A clinically relevant adverse reaction in XARELTO-treated patients was vomiting (10.6% in the XARELTO group vs 8% in the comparator group).
Thromboprophylaxis in Pediatric Patients with Congenital Heart Disease (CHD) after the Fontan Procedure
Although clarithromycin is a combined P-gp and strong CYP3A inhibitor, pharmacokinetic data suggests that no precautions are necessary with concomitant administration with XARELTO as the change in exposure is unlikely to affect the bleeding risk [see Clinical Pharmacology (12.3) in Full Prescribing Information]
Interaction with Combined P-gp and Moderate CYP3A Inhibitors in Patients with Renal Impairment
XARELTO should not be used in patients with CrCl 15 to <80 mL/min who are receiving concomitant combined P-gp and moderate CYP3A inhibitors (e.g., erythromycin) unless the potential benefit justifies the potential risk [see Warnings and Precautions and Clinical Pharmacology (12.3) in Full Prescribing Information]
Drugs that Induce Cytochrome P450 3A Enzymes and Drug Transport Systems
Enoxaparin/VKA N=2405 n (%)
Pruritus 53 (2.2) 27 (1.1)
* Adverse reaction with Relative Risk >1.5 for XARELTO versus comparator
Non-hemorrhagic adverse reactions reported in ≥1% of XARELTOtreated patients in RECORD 1-3 studies are shown in Table 9.
Table 9: Other Adverse Drug Reactions* Reported by ≥1% of XARELTOTreated Patients in RECORD 1-3 Studies
Body System Adverse Reaction
Injury, poisoning and procedural complications
XARELTO 10 mg N=4487 n (%) Enoxaparin† N=4524 n (%)
Wound secretion 125 (2.8) 89 (2.0)
Musculoskeletal and connective tissue disorders
Pain in extremity 74 (1.7) 55 (1.2)
Muscle spasm 52 (1.2) 32 (0.7)
Nervous system disorders
Syncope 55 (1.2) 32 (0.7)
Skin and subcutaneous tissue disorders
Pruritus 96 (2.1) 79 (1.8)
Blister 63 (1.4) 40 (0.9)
* Adverse reaction occurring any time following the first dose of doubleblind medication, which may have been prior to administration of active drug, until two days after the last dose of double-blind study medication
† Includes the placebo-controlled period of RECORD 2, enoxaparin dosing was 40 mg once daily (RECORD 1-3)
Pediatric Patients
Treatment of Venous Thromboembolism and Reduction in Risk of Recurrent Venous Thromboembolism in Pediatric Patients
The safety assessment is based on data from the EINSTEIN Junior Phase 3 study in 491 patients from birth to less than 18 years of age. Patients were randomized 2:1 to receive body weight-adjusted doses of XARELTO or comparator (unfractionated heparin, low molecular weight heparin, fondaparinux or VKA).
Discontinuation due to bleeding events occurred in 6 (1.8%) patients in the XARELTO group and 3 (1.9%) patients in the comparator group.
Table 10 shows the number of patients experiencing bleeding events in the EINSTEIN Junior study. In female patients who had experienced menarche, ages 12 to <18 years of age, menorrhagia occurred in 23 (27%) female patients in the XARELTO group and 5 (10%) female patients in the comparator group.
Table 10: Bleeding Events in EINSTEIN Junior Study – Safety Analysis Set - Main Treatment Period*
Parameter
XARELTO† N=329 n (%) Group‡ N=162 n (%)
Major bleeding§ 0 2 (1.2)
Clinically relevant non-major bleeding¶ 10 (3.0) 1 (0.6)
Trivial bleeding 113 (34.3) 44 (27.2)
Any bleeding 119 (36.2) 45 (27.8)
* These events occurred after randomization until 3 months of treatment (1 month for patients <2 years with central venous catheter-related VTE (CVC-VTE). Patients may have more than one event.
The data below are based on Part B of the UNIVERSE study which was designed to evaluate the safety and efficacy of XARELTO for thromboprophylaxis in 98 children with CHD after the Fontan procedure who took at least one dose of study drug. Patients in Part B were randomized 2:1 to receive either body weight-adjusted doses of XARELTO or aspirin (approximately 5 mg/kg).
Discontinuation due to bleeding events occurred in 1 (1.6%) patient in the XARELTO group and no patients in the aspirin group.
Table 12 shows the number of patients experiencing bleeding events in the UNIVERSE study.
Table 12: Bleeding Events in UNIVERSE Study - Safety Analysis SetOn Treatment Plus 2 Days
Parameter
XARELTO* N=64 n (%) Aspirin* N=34 n (%)
Major Bleeding† 1 (1.6) 0
Epistaxis leading to transfusion 1 (1.6) 0
Clinically relevant non-major (CRNM) bleeding§ 4 (6.3) 3 (8.8)
Trivial bleeding 21 (32.8) 12 (35.3)
Any bleeding 23 (35.9) 14 (41.2)
* Treatment schedule: body weight-adjusted doses of XARELTO or aspirin (approximately 5 mg/kg); randomized 2:1 (XARELTO: Aspirin).
† Defined as clinically overt bleeding associated with a decrease in hemoglobin of ≥2 g/dL, a transfusion of the equivalent of ≥2 units of packed red blood cells or whole blood, bleeding at a critical site, or with a fatal outcome.
§ Defined as clinically overt bleeding, which did not meet the criteria for major bleeding, but was associated with medical intervention, unscheduled contact with a physician, temporary cessation of treatment, discomfort for the patient, or impairment of activities of daily life.
Non-bleeding adverse reactions reported in ≥5% of XARELTO-treated patients are shown in Table 13.
Table 13: Other Adverse Reactions* Reported by ≥5% of XARELTOTreated Patients in UNIVERSE Study (Part B)
Adverse Reaction
XARELTO N=64 n (%) Aspirin N=34 n (%)
Cough 10 (15.6) 3 (8.8)
Vomiting 9 (14.1) 3 (8.8)
Gastroenteritis† 8 (12.5) 1 (2.9)
Rash† 6 (9.4) 2 (5.9)
* Adverse reaction with Relative Risk >1.5 for XARELTO versus aspirin.
† The following terms were combined: Gastroenteritis: gastroenteritis, gastroenteritis viral Rash: rash, rash maculo-papular, viral rash
Postmarketing Experience
The following adverse reactions have been identified during postapproval use of XARELTO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: agranulocytosis, thrombocytopenia Hepatobiliary disorders: jaundice, cholestasis, hepatitis (including hepatocellular injury)
Immune system disorders: hypersensitivity, anaphylactic reaction, anaphylactic shock, angioedema
Nervous system disorders: hemiparesis
Skin and subcutaneous tissue disorders: Stevens-Johnson syndrome, drug reaction with eosinophilia and systemic symptoms (DRESS)
Avoid concomitant use of XARELTO with drugs that are combined P-gp and strong CYP3A inducers (e.g., carbamazepine, phenytoin, rifampin, St. John’s wort) [see Warnings and Precautions and Clinical Pharmacology (12.3) in Full Prescribing Information]
Anticoagulants and NSAIDs/Aspirin
Coadministration of enoxaparin, warfarin, aspirin, clopidogrel and chronic NSAID use may increase the risk of bleeding [see Clinical Pharmacology (12.3) in Full Prescribing Information]
Avoid concurrent use of XARELTO with other anticoagulants due to increased bleeding risk unless benefit outweighs risk. Promptly evaluate any signs or symptoms of blood loss if patients are treated concomitantly with aspirin, other platelet aggregation inhibitors, or NSAIDs [see Warnings and Precautions]
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
The limited available data on XARELTO in pregnant women are insufficient to inform a drug-associated risk of adverse developmental outcomes. Use XARELTO with caution in pregnant patients because of the potential for pregnancy related hemorrhage and/or emergent delivery. The anticoagulant effect of XARELTO cannot be reliably monitored with standard laboratory testing. Consider the benefits and risks of XARELTO for the mother and possible risks to the fetus when prescribing XARELTO to a pregnant woman [see Warnings and Precautions].
Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnancy is a risk factor for venous thromboembolism and that risk is increased in women with inherited or acquired thrombophilias. Pregnant women with thromboembolic disease have an increased risk of maternal complications including pre-eclampsia. Maternal thromboembolic disease increases the risk for intrauterine growth restriction, placental abruption and early and late pregnancy loss.
Fetal/Neonatal Adverse Reactions
Based on the pharmacologic activity of Factor Xa inhibitors and the potential to cross the placenta, bleeding may occur at any site in the fetus and/or neonate.
Labor or Delivery
All patients receiving anticoagulants, including pregnant women, are at risk for bleeding and this risk may be increased during labor or delivery [see Warnings and Precautions]. The risk of bleeding should be balanced with the risk of thrombotic events when considering the use of XARELTO in this setting.
Data Human Data
There are no adequate or well-controlled studies of XARELTO in pregnant women, and dosing for pregnant women has not been established. Post-marketing experience is currently insufficient to determine a rivaroxaban-associated risk for major birth defects or miscarriage. In an in vitro placenta perfusion model, unbound rivaroxaban was rapidly transferred across the human placenta.
Animal Data
Rivaroxaban crosses the placenta in animals. Rivaroxaban increased fetal toxicity (increased resorptions, decreased number of live fetuses, and decreased fetal body weight) when pregnant rabbits were given oral doses of ≥10 mg/kg rivaroxaban during the period of organogenesis. This dose corresponds to about 4 times the human exposure of unbound drug, based on AUC comparisons at the highest recommended human dose of 20 mg/day. Fetal body weights decreased when pregnant rats were given oral doses of 120 mg/kg during the period of organogenesis. This dose corresponds to about 14 times the human exposure of unbound drug. In rats, peripartal maternal bleeding and maternal and fetal death occurred at the rivaroxaban dose of 40 mg/kg (about 6 times maximum human exposure of the unbound drug at the human dose of 20 mg/day).
Comparator
XARELTO® (rivaroxaban) XARELTO® (rivaroxaban) XARELTO® (rivaroxaban)
Lactation
Risk Summary
Rivaroxaban has been detected in human milk. There are insufficient data to determine the effects of rivaroxaban on the breastfed child or on milk production. Rivaroxaban and/or its metabolites were present in the milk of rats. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for XARELTO and any potential adverse effects on the breastfed infant from XARELTO or from the underlying maternal condition (see Data)
Data
Animal Data
Following a single oral administration of 3 mg/kg of radioactive [14C]-rivaroxaban to lactating rats between Day 8 to 10 postpartum, the concentration of total radioactivity was determined in milk samples collected up to 32 hours post-dose. The estimated amount of radioactivity excreted with milk within 32 hours after administration was 2.1% of the maternal dose.
Females and Males of Reproductive Potential
Females of reproductive potential requiring anticoagulation should discuss pregnancy planning with their physician.
The risk of clinically significant uterine bleeding, potentially requiring gynecological surgical interventions, identified with oral anticoagulants including XARELTO should be assessed in females of reproductive potential and those with abnormal uterine bleeding.
Pediatric Use
The safety and effectiveness of XARELTO have been established in pediatric patients from birth to less than 18 years for the treatment of VTE and the reduction in risk of recurrent VTE. Use of XARELTO is supported in these age groups by evidence from adequate and wellcontrolled studies of XARELTO in adults with additional pharmacokinetic, safety and efficacy data from a multicenter, prospective, open-label, active-controlled randomized study in 500 pediatric patients from birth to less than 18 years of age. XARELTO was not studied and therefore dosing cannot be reliably determined or recommended in children less than 6 months who were less than 37 weeks of gestation at birth; had less than 10 days of oral feeding, or had a body weight of less than 2.6 kg [see Dosage and Administration (2.2) in Full Prescribing Information, Adverse Reactions, Clinical Pharmacology (12.3) and Clinical Studies (14.8) in Full Prescribing Information].
The safety and effectiveness of XARELTO have been established for use in pediatric patients aged 2 years and older with congenital heart disease who have undergone the Fontan procedure. Use of XARELTO is supported in these age groups by evidence from adequate and wellcontrolled studies of XARELTO in adults with additional data from a multicenter, prospective, open-label, active controlled study in 112 pediatric patients to evaluate the single- and multiple-dose pharmacokinetic properties of XARELTO and the safety and efficacy of XARELTO when used for thromboprophylaxis for 12 months in children with single ventricle physiology who had the Fontan procedure [see Dosage and Administration (2.2) in Full Prescribing Information, Adverse Reactions, Clinical Pharmacology (12.3) and Clinical Studies (14.9) in Full Prescribing Information].
Clinical studies that evaluated safety, efficacy, pharmacokinetic and pharmacodynamic data support the use of XARELTO 10 mg, 15 mg, and 20 mg tablets in pediatric patients. For the XARELTO 2.5 mg tablets, there are no safety, efficacy, pharmacokinetic and pharmacodynamic data to support the use in pediatric patients. Therefore, XARELTO 2.5 mg tablets are not recommended for use in pediatric patients.
Although not all adverse reactions identified in the adult population have been observed in clinical trials of children and adolescent patients, the same warnings and precautions for adults should be considered for children and adolescents.
Geriatric Use
Of the total number of adult patients in clinical trials for the approved indications of XARELTO (N=64,943 patients), 64 percent were 65 years and over, with 27 percent 75 years and over. In clinical trials the efficacy of XARELTO in the elderly (65 years or older) was similar to that seen in patients younger than 65 years. Both thrombotic and bleeding event rates were higher in these older patients [see Clinical Pharmacology (12.3) and Clinical Studies (14) in Full Prescribing Information]
Renal Impairment
In pharmacokinetic studies, compared to healthy adult subjects with normal creatinine clearance, rivaroxaban exposure increased by approximately 44 to 64% in adult subjects with renal impairment. Increases in pharmacodynamic effects were also observed [see Clinical Pharmacology (12.3) in Full Prescribing Information]
Nonvalvular Atrial Fibrillation
Patients with Chronic Kidney Disease not on Dialysis
In the ROCKET AF trial, patients with CrCl 30 to 50 mL/min were administered XARELTO 15 mg once daily resulting in serum concentrations of rivaroxaban and clinical outcomes similar to those in patients with better renal function administered XARELTO 20 mg once daily. Patients with CrCl <30 mL/min were not studied, but administration of XARELTO 15 mg once daily is expected to result in serum concentrations of rivaroxaban similar to those in patients with moderate renal impairment [see Clinical Pharmacology (12.3) in Full Prescribing Information].
Patients with End-Stage Renal Disease on Dialysis
Clinical efficacy and safety studies with XARELTO did not enroll patients with end-stage renal disease (ESRD) on dialysis. In patients with ESRD maintained on intermittent hemodialysis, administration of XARELTO 15 mg once daily will result in concentrations of rivaroxaban and pharmacodynamic activity similar to those observed in the ROCKET AF study [see Clinical Pharmacology (12.2, 12.3) in Full Prescribing Information]. It is not known whether these concentrations will lead to similar stroke reduction and bleeding risk in patients with ESRD on dialysis as was seen in ROCKET AF.
Treatment of DVT and/or PE and Reduction in the Risk of Recurrence of DVT and/or PE
In the EINSTEIN trials, patients with CrCl values <30 mL/min at screening were excluded from the studies, but administration of XARELTO is expected to result in serum concentrations of rivaroxaban similar to
those in patients with moderate renal impairment (CrCl 30 to <50 mL/min) [see Clinical Pharmacology (12.3) in Full Prescribing Information] Observe closely and promptly evaluate any signs or symptoms of blood loss in patients with CrCl 15 to <30 mL/min. Avoid the use of XARELTO in patients with CrCl <15 mL/min.
Prophylaxis of DVT Following Hip or Knee Replacement Surgery
The combined analysis of the RECORD 1-3 clinical efficacy studies did not show an increase in bleeding risk for patients with CrCl 30 to 50 mL/min and reported a possible increase in total venous thromboemboli in this population. In the RECORD 1-3 trials, patients with CrCl values <30 mL/min at screening were excluded from the studies, but administration of XARELTO 10 mg once daily is expected to result in serum concentrations of rivaroxaban similar to those in patients with moderate renal impairment (CrCl 30 to <50 mL/min) [see Clinical Pharmacology (12.3) in Full Prescribing Information]. Observe closely and promptly evaluate any signs or symptoms of blood loss in patients with CrCl 15 to <30 mL/min. Avoid the use of XARELTO in patients with CrCl <15 mL/min.
Prophylaxis of Venous Thromboembolism in Acutely Ill Medical Patients at Risk for Thromboembolic Complications Not at High Risk of Bleeding Patients with CrCl values <30 mL/min at screening were excluded from the MAGELLAN study. In patients with CrCl <30 mL/min a dose of XARELTO 10 mg once daily is expected to result in serum concentrations of rivaroxaban similar to those in patients with moderate renal impairment (CrCl 30 to <50 mL/min) [see Clinical Pharmacology (12.3) in Full Prescribing Information]. Observe closely and promptly evaluate any signs or symptoms of blood loss in patients with CrCl 15 to <30 mL/min. Avoid use of XARELTO in patients with CrCl <15 mL/min.
Reduction of Risk of Major Cardiovascular Events in Patients with CAD and Reduction of Risk of Major Thrombotic Vascular Events in Patients with PAD, Including Patients After Recent Lower Extremity Revascularization due to Symptomatic PAD
Patients with Chronic Kidney Disease not on Dialysis Patients with a CrCl <15 mL/min at screening were excluded from COMPASS and VOYAGER, and limited data are available for patients with a CrCl of 15 to 30 mL/min. In patients with CrCl <30 mL/min, a dose of 2.5 mg XARELTO twice daily is expected to give an exposure similar to that in patients with moderate renal impairment (CrCl 30 to <50 mL/min) [see Clinical Pharmacology (12.3) in Full Prescribing Information], whose efficacy and safety outcomes were similar to those with preserved renal function.
Patients with End-Stage Renal Disease on Dialysis
No clinical outcome data is available for the use of XARELTO with aspirin in patients with ESRD on dialysis since these patients were not enrolled in COMPASS or VOYAGER. In patients with ESRD maintained on intermittent hemodialysis, administration of XARELTO 2.5 mg twice daily will result in concentrations of rivaroxaban and pharmacodynamic activity similar to those observed in moderate renal impaired patients in the COMPASS study [see Clinical Pharmacology (12.2, 12.3) in Full Prescribing Information]. It is not known whether these concentrations will lead to similar CV risk reduction and bleeding risk in patients with ESRD on dialysis as was seen in COMPASS.
Pediatric Use
No dosage adjustment is required in patients 1 year of age or older with mild renal impairment (eGFR 50 to ≤ 80 mL/min/1.73 m2). There are limited clinical data in pediatric patients 1 year or older with moderate or severe renal impairment (eGFR <50 mL/min/1.73 m2); therefore, avoid the use of XARELTO in these patients.
There are no clinical data in pediatric patients younger than 1 year with serum creatinine results above 97.5th percentile; therefore, avoid the use of XARELTO in these patients [see Dosage and Administration (2.2) in Full Prescribing Information]
Hepatic Impairment
In a pharmacokinetic study, compared to healthy adult subjects with normal liver function, AUC increases of 127% were observed in adult subjects with moderate hepatic impairment (Child-Pugh B).
The safety or PK of XARELTO in patients with severe hepatic impairment (Child-Pugh C) has not been evaluated [see Clinical Pharmacology (12.3) in Full Prescribing Information]
Avoid the use of XARELTO in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment or with any hepatic disease associated with coagulopathy.
No clinical data are available in pediatric patients with hepatic impairment.
OVERDOSAGE
Overdose of XARELTO may lead to hemorrhage. Discontinue XARELTO and initiate appropriate therapy if bleeding complications associated with overdosage occur. Rivaroxaban systemic exposure is not further increased at single doses >50 mg due to limited absorption. The use of activated charcoal to reduce absorption in case of XARELTO overdose may be considered. Due to the high plasma protein binding, rivaroxaban is not dialyzable [see Warnings and Precautions and Clinical Pharmacology (12.3) in Full Prescribing Information]. Partial reversal of laboratory anticoagulation parameters may be achieved with use of plasma products. An agent to reverse the anti-factor Xa activity of rivaroxaban is available.
Active Ingredient Made in Germany
Manufactured for: Janssen Pharmaceuticals, Inc. Titusville, NJ 08560
Licensed from: Bayer HealthCare AG 51368 Leverkusen, Germany
© 2011-2021 Janssen Pharmaceutical Companies cp-62545v11
XARELTO® (rivaroxaban) XARELTO® (rivaroxaban)
MANEJO CARDIOVASCULAR EN EL PACIENTE CON CÁNCER: NUEVAS ESTRATEGIAS
Por: Gilberto Rivera Gautier, MD, FACC, FSIAC, RPVI Board Certified por la American Board of Internal Medicine en Medicina Interna y Enfermedades Cardiovasculares Pasado Presidente de la Sociedad Puertorriqueña de Cardiología Director del Laboratorio Cardiovascular Invasivo y Laboratorio Cardiovascular No-Invasivo del Hospital Auxilio Mutuo

RESUMEN
























Los daños cardiovasculares en pacientes con cáncer de ben ser analizados exhaustivamente con base en la ma lignidad que se trata el individuo, la primera opción del tratamiento definida por el oncólogo y el historial clínico. De estos y otros factores dependerá el riesgo que presente el paciente con cáncer de desarrollar enfermedad cardio vascular y de la cardiotoxicidad inducida por los fármacos y agentes químicos a los que los pacientes deben ser some tidos. Por ende, en este artículo se exponen nuevos métodos que minimizan los riesgos y destacan la importancia del tra bajo multidisciplinar.
ABSTRACT













































Cardiovascular damage in cancer patients should be analyzed exhaustively based on the malignancy presented by the indivi dual, the first choice of treatment defined by the oncologist and the clinical record. Of these and other factors will de pend the risk that the cancer patient develops cardiovascular disease and of the cardio-toxicity induced by the drugs and chemical agents to which the patients should be subjected. This article sets out new methods that minimize risks and emphasize the importance of multidisciplinary work.
PALABRAS CLAVE
Enfermedad cardiovascular, cáncer, cardiotoxicidad, tratamiento













KEY WORDS
































Cardiovascular disease, cancer, cardiac toxicity, treatment

















Revista Puertorriqueña de Medicina y Salud Pública42
La evaluación y manejo de pacientes con cáncer y enfermedad cardiovascular debe tomar en consideración la etiología de la enfermedad cardiovascular y el tipo de terapia recibida. Podemos encontrarnos con pacientes con cáncer y enfermedad cardiovascular primaria concurrente, así como pacientes con cáncer que desarrollan enfermedad cardiovascular como complicación del tratamiento para el propio cáncer.
Hemos definido la presencia de cardiotoxicidad inducida por quimioterapia como una disminución en la fracción de expulsión del ventrículo izquierdo igual o mayor del 5 % a una función menor de 55% en un paciente con una presentación clínica de fallo cardíaco, o, una disminución igual o mayor a un 10% a una función menor de 55% en un paciente asintomático.
Tomando en consideración los posibles efectos cardiovasculares de agentes far macológicos, considerados cardiotóxicos, podemos dividirlos en diferentes grupos: los que afectan primordialmente la fun ción cardíaca (ej. antraciclinas y trastuzu mab), los que afectan la función vascular (ej. 5-fluorouracil y capecitabine) y los que afectan ambos (ej. bevacizumab y sunitinib). La terapia de radiación puede ser causante de daño cardiovascular en diferentes niveles, incluyendo daño al mio cardio, pericardio, aparatos valvulares y vasos sanguíneos (ej. arterias coronarias). La cardiotoxicidad inducida por quimiote rapia ha sido clasificada en 2 grupos: la tipo 1, caracterizada por daño estructural irreversible al músculo cardíaco -típica mente encontrada con el tratamiento con antraciclinas- y la tipo 2, caracterizada por ausencia de anormalidades estructu rales y reversibilidad posible -típicamente encontrada con el tratamiento con tras tuzumab-. Se ha observado el desarrollo de enfermedad cardiovascular más agre siva en pacientes que reciben la combi nación de farmacoterapia con potencial cardiotóxico y radioterapia.
El manejo cardiovascular del paciente con cáncer debe comenzar con un aná lisis de riesgo del paciente, combinando los factores de riesgo cardiovascular pre existentes con el riesgo de cardiotoxicidad del medicamento a utilizarse. La combina ción de estos factores nos permiten esta blecer una puntuación de riesgo para cardiotoxicidad, utilizado para dictar las recomendaciones de monitoreo y manejo previo, durante y después del tratamiento. El electrocardiograma y ecocardiograma son frecuentemente utilizados como herra mienta para la estratificación de los pa cientes. Una vez iniciado el tratamiento del paciente, se debe decidir si se requie re un seguimiento cardiovascular tomando en cuenta el perfil de riesgo cardiovas cular de base, el régimen de tratamiento para cáncer utilizado y el desarrollo de síntomas cardiovasculares o eventos car diovasculares mayores.
Los primeros protocolos validados para monitoreo cardiovascular de los pacien tes con cáncer tratados con antraciclinas fueron desarrollados para las décadas
de los 70’s-80’s. Éstos se sustentaron con estudios de ventriculografía por radionu cleótidos (MUGA) que permitían identi ficar cambios en la función ventricular previo el desarrollo de fallo cardíaco clí nico. Estas pruebas nucleares han sido paulatinamente sustituidas por la ecocar diografía bidimensional. Recientemente, la ecocardiografía bidimensional con con traste y la ecocardiografía tridimensional han sido considerados como herramien tas comparables al MUGA (Multigated Analysis) en su capacidad de evaluación de función ventricular, añadiendo también la posibilidad de evaluación de otra área del corazón como el pericardio, válvulas y otros datos hemodinámicos. La modali dad en ecocardiograma del “strain rate imaging” ha sido más introducida en la evaluación cardíaca y permite identificar ciertas deformidades en el músculo del co razón, que tienden a preceder el proceso de disminución en contracción cardíaca. Esta modalidad permite una identificación aún más temprana del paciente a riesgo de desarrollo de cambios en función car díaca por toxicidad. El biomarcador que mejor ha demostrado capacidad de pre dicción de cardiotoxicidad ha sido la Tro ponina, que al aumentar ofrece indicios de cardiotoxicidad, aunque su utilidad no ha superado la evaluación con ecocardio grafía.
La primera línea de intervención para prevención cardiovascular en pacientes con cancer es precisamente el control de los factores de riesgo tradicionales para el desarrollo de enfermedad cardiovascular.. Control adecuado de diabetes, hiperten sión y lípidos, así como una alimentación saludable y ejercicio rutinario son de gran importancia en esta población de pacien tes. El uso de dexrazoxane, un agente quelante, puede ser utilizado en pacientes con cancer de seno que han recibido al tas dosis de antraciclinas para prevención de cardio toxicidad.
El tratamiento de pacientes que han de sarrollado cardiomiopatía por cardiotoxi cidad asociada a terapia de cáncer debe realizarse con base en las recomendacio nes establecidas por la Asociación Ame ricana del Corazón/Colegio Americano de Cardiología (AHA/ACC). El uso de
Revista Puertorriqueña de Medicina Salud
y
Pública 43
los agentes inhibidores de la enzima con vertidora de angiotensina (ECA) son consi derados como primera línea de tratamiento en pacientes con reducción de contracción cardíaca. El uso de los bloqueadores de re ceptores de angiotensina (ARB) pueden ser utilizados en pacientes con intolerancia a los inhibidores de ECA. El uso de inhibido res de aldosterona puede ser considerado en pacientes con síntomas de fallo cardíaco y una función menor o igual al 35%. Los bloqueadores beta, particularmente carve dilol y nebivolol son considerados el segun do grupo mayor de medicamentos a ser utilizados en el tratamiento de cardiomio patía y, en combinación con los inhibidores de ECA, han demostrado un efecto signifi cativo en mejoría de función cardíaca. El uso de sacubitril (un inhibidor de neprilisin) con valsartan (un bloqueador de receptor de angiotensina) ha demostrado una reduc ción significativa en mortalidad y hospita lización de pacientes con fallo cardíaco y reducción de la fracción de expulsión así como pacientes con función preservada. Esta terapia ha comenzado a ser evaluada en pacientes con cancer y cardiomiopa tia por cardiotoxicidad; los efectos bene ficiosos de esta terapia aparentan estar también presentes en esta población de pacientes. De la misma manera, el uso de fármacos SGLT2 inhibidores, los cuales fueron inicialmente aprobados para el tra
tamiento de diabetes, han logrado un gran impacto en el tratamiento de fallo cardia co y, de la misma forma, parecen tener un efecto beneficioso en pacientes con cancer y fallo cardiaco por toxicidad.
Los pacientes que fallan a la terapia mé dica podrían beneficiarse de dispositivos para apoyo hemodinámico como terapia aguda temporera o como puente para tras plante cardíaco. El uso de desfibrilador au tomático implantable puede también consi derarse en pacientes con cardiomiopatía severa y/o arritmias malignas cuya sobre vida esperada es mayor a un año y toman do en consideración otros aspectos clínicos asociados al pronóstico general.
El pobre pronóstico asociado a la cardio miopatía inducida por terapia de cáncer, ha promovido el desarrollo de protocolos de tratamiento preventivo utilizando los agentes cardiovasculares disponibles y el desarrollo de nuevos agentes. Dexrazoxa ne, un agente quelante intracelular, ha de mostrado una reducción significativa de fallo cardíaco, pero su utilidad ha sido li mitada por su reducción en el efecto antitu moral de los tratamientos. Los betabloquea dores que han demostrado mayor beneficio en estos pacientes han sido el carvedilol y nebivolol. La evidencia del uso de estatinas como prevención para cardiomiopatía es limitada, pero se ha recomendado su consi deración en pacientes de alto riesgo. El uso
rutinario de terapia preventiva para cardio toxicidad debe ser considerado en pacien tes con un índice de riesgo alto según se establezca al evaluar sus factores preexis tentes y el tipo de terapia a recibirse.
En conclusión, los pacientes de cáncer se encuentran en riesgo de desarrollar proble mas cardiovasculares significativos asocia do al tratamiento recibido. Una evaluación temprana y manejo por un equipo multidis ciplinario cardio-oncológico es una gran herramienta para la detección temprana y manejo apropiado para contribuir a una mejor sobrevida y calidad de vida de los afectados.

REFERENCES
Khouri M, Greene S, et al. Sodium-Glu cose Co-Transporter-2 Inhibitor Therapy During Anthracycline Treatment. J Am Coll Cardiol HF. 2022 Aug, 10 (8) 568–570. https://doi.org/10.1016/j.jchf.2022.04.014
Khouri MG, Douglas PS, Mackey JR, Martin M, Scott JM, Scherrer-Crosbie M, Jones LW. Cancer therapy-induced car diac toxicity in early breast cancer: ad dressing the unresolved issues. Circulation. 2012;126:2749–3276. doi: 10.1161/CIRCU LATIONAHA.112.100560.

Plana JC, Galderisi M, Barac A, et al: Ex pert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: A report from the Ameri can Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging 15:1063-1093, 2014 Crossref, Medline, Google Scholar
Ky B, Putt M, Sawaya H, et al: Early in creases in multiple biomarkers predict subsequent cardiotoxicity in patients with breast cancer treated with doxorubicin, ta xanes, and trastuzumab. J Am Coll Cardiol 63:809-816, 2014 Crossref, Medline, Goo gle Scholar
Koene RJ (2016) Shared risk factors in car diovascular disease and cancer. Circulation 133: 1104-1114.
Mac MJ, Packer M, et al. Angioten sin- Neprilysin inhibition versus Ena lapril in heart failure. N Engl J Med. 2014;371:993–1004.
Christina E. Sheppard, Maria Anwar. The use of sacubitril/valsartan in Anthracycline-in duced cardiomyopathy: a mini case series. J Oncol Pharm Pract. 2019;25(5):1231–4.
Revista Puertorriqueña de Medicina y Salud Pública44
What can

look like for your patients living with Crohn’s disease?
SKYRIZI®
150 mg/mL single-dose
600 mg/10 mL single-dose vial
360 mg/2.4 mL single-dose prefilled cartridge on-body
INDICATIONS AND USAGE
Plaque Psoriasis
SKYRIZI® is indicated for the treatment of moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
Psoriatic Arthritis
SKYRIZI is indicated for the treatment of active psoriatic arthritis in adults.
Crohn’s Disease
SKYRIZI is indicated for the treatment of moderately to severely active Crohn’s disease in adults.
CONTRAINDICATIONS
SKYRIZI is contraindicated in patients with a history of serious hypersensitivity reaction to risankizumab-rzaa or any of the excipients [see Warnings and Precautions]
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been reported with use of SKYRIZI. If a serious hypersensitivity reaction occurs, discontinue SKYRIZI and initiate appropriate therapy immediately [see Adverse Reactions]
Infections
SKYRIZI may increase the risk of infections [see Adverse Reactions]
Treatment with SKYRIZI should not be initiated in patients with any clinically important active infection until the infection resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing SKYRIZI. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If a patient develops such an infection or is not responding to standard therapy, monitor the patient closely and do not administer SKYRIZI until the infection resolves.
Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with SKYRIZI. Across the Phase 3 psoriasis clinical studies, of the 72 subjects with latent TB who were concurrently treated with SKYRIZI and appropriate TB prophylaxis during the studies, none developed active TB during the mean follow-up of 61 weeks on SKYRIZI. Two subjects taking isoniazid for treatment of latent TB discontinued treatment due to liver injury. Of the 31 subjects from the PsO-3 study with latent TB who did not receive prophylaxis during the study, none developed active TB during the mean follow-up of 55 weeks on SKYRIZI. Consider anti-TB therapy prior to initiating SKYRIZI in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after SKYRIZI treatment. Do not administer SKYRIZI to patients with active TB.
Hepatotoxicity in Treatment of Crohn’s Disease
A serious adverse reaction of drug-induced liver injury was reported in a patient with Crohn’s disease (ALT 54x ULN, AST 30x ULN, and total bilirubin 2.2x ULN) following two intravenous doses of SKYRIZI 600 mg in conjunction with a rash that required hospitalization. The liver test abnormalities resolved following administration of steroids. SKYRIZI was subsequently discontinued. For the treatment of Crohn’s disease, evaluate liver enzymes and bilirubin at baseline, and during induction at least up to 12 weeks of treatment. Monitor thereafter according to routine patient management. Consider other treatment options in patients with evidence of liver cirrhosis. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. Interrupt treatment if drug-induced liver injury is suspected, until this diagnosis is excluded. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of hepatic dysfunction.
Administration of Vaccines
Avoid use of live vaccines in patients treated with SKYRIZI. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating therapy with SKYRIZI, complete all age-appropriate vaccinations according to current immunization guidelines. No data are available on the response to live or inactive vaccines.
ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of labeling:
• Hypersensitivity Reactions [see Warnings and Precautions]
• Infections [see Warnings and Precautions]
• Tuberculosis [see Warnings and Precautions]
• Hepatotoxicity in Treatment of Crohn’s disease [see Warnings and Precautions] Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse drug reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Plaque Psoriasis
A total of 2234 subjects were treated with SKYRIZI in clinical development trials in plaque psoriasis. Of these, 1208 subjects with psoriasis were exposed to SKYRIZI for at least one year. Data from placebo- and active-controlled trials were pooled to evaluate the safety of SKYRIZI for up to 16 weeks. In total, 1306 subjects were evaluated in the SKYRIZI 150 mg group.
Table 1 summarizes the adverse drug reactions that occurred at a rate of at least 1% and at a higher rate in the SKYRIZI group than the placebo group during the 16-week controlled period of pooled clinical trials.
Table 1. Adverse Drug Reactions Occurring in ≥ 1% of Subjects on SKYRIZI through Week 16
Adverse Drug Reactions
SKYRIZI N = 1306 n (%) Placebo N = 300 n (%)
Upper respiratory infectionsa 170 (13.0) 29 (9.7)
Headacheb 46 (3.5) 6 (2.0)
Fatiguec 33 (2.5) 3 (1.0)
Injection site reactionsd 19 (1.5) 3 (1.0)
Tinea infectionse 15 (1.1) 1 (0.3)
a Includes: respiratory tract infection (viral, bacterial or unspecified), sinusitis (including acute), rhinitis, nasopharyngitis, pharyngitis (including viral), tonsillitis
b Includes: headache, tension headache, sinus headache, cervicogenic headache
c Includes: fatigue, asthenia
d Includes: injection site bruising, erythema, extravasation, hematoma, hemorrhage, infection, inflammation, irritation, pain, pruritus, reaction, swelling, warmth
e Includes: tinea pedis, tinea cruris, body tinea, tinea versicolor, tinea manuum, tinea infection, onychomycosis
Adverse drug reactions that occurred in < 1% but > 0.1% of subjects in the SKYRIZI group and at a higher rate than in the placebo group through Week 16 were folliculitis and urticaria.
Specific Adverse Drug Reactions
Infections
In the first 16 weeks, infections occurred in 22.1% of the SKYRIZI group (90.8 events per 100 subject-years) compared with 14.7% of the placebo group (56.5 events per 100 subject-years) and did not lead to discontinuation of SKYRIZI. The rates of serious infections for the SKYRIZI group and the placebo group were ≤0.4%. Serious infections in the SKYRIZI group included cellulitis, osteomyelitis, sepsis, and herpes zoster. In Studies PsO-1 and PsO-2, through Week 52, the rate of infections (73.9 events per 100 subject-years) was similar to the rate observed during the first 16 weeks of treatment.
Safety Through Week 52
Through Week 52, no new adverse reactions were identified, and the rates of the adverse reactions were similar to those observed during the first 16 weeks of treatment. During this period, serious infections that led to study discontinuation included pneumonia.
Psoriatic Arthritis
The overall safety profile observed in subjects with psoriatic arthritis treated with SKYRIZI is generally consistent with the safety profile in subjects with plaque psoriasis. Additionally, in the Phase 3 placebocontrolled trials the incidence of hepatic events was higher in the SKYRIZI group (5.4%, 16.7 events per 100 patient years) compared to the placebo group (3.9%, 12.6 events per 100 patient years). Of these, the most common events that were reported more frequently in both the placebo group and the SKYRIZI group were ALT increased (placebo: n=12 (1.7%); SKYRIZI: n=16 (2.3%)), AST increased (placebo: n=9 (1.3%); SKYRIZI: n=13 (1.8%)), and GGT increased (placebo: n=5 (0.7%); SKYRIZI: n=8 (1.1%)). There were no serious hepatic events reported. The incidence of hypersensitivity reactions was higher in the SKYRIZI group (n=16, 2.3%) compared to the placebo group (n=9, 1.3%). In the Phase 3 placebo-controlled trials, hypersensitivity reactions reported at a higher rate in the SKYRIZI group included rash (placebo: n=4 (0.6%); SKYRIZI: n=5 (0.7%), allergic rhinitis (placebo: n=1 (0.1%); SKYRIZI: n=2 (0.3%), and facial swelling (placebo: n=0 (0.0%); SKYRIZI n=1 (0.1%). One case of anaphylaxis was reported in a subject who received SKYRIZI in the Phase 2 clinical trial.
Crohn’s Disease SKYRIZI was studied up to 12 weeks in subjects with moderately to severely active Crohn’s disease in two randomized, double-blind, placebo-controlled induction studies (CD-1, CD-2) and a randomized, double-
blind, placebo-controlled, dose-finding study (CD-4; NCT02031276). Long-term safety up to 52 weeks was evaluated in subjects who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (CD-3).
In the two induction studies (CD-1, CD-2) and the dose finding study (CD-4), 620 subjects received the SKYRIZI intravenous induction regimen. In the maintenance study (CD-3), 142 subjects who achieved clinical response defined as a reduction in CDAI of at least 100 points from baseline after 12 weeks of induction treatment with intravenous SKYRIZI in studies CD-1 and CD-2, received SKYRIZI subcutaneously as a maintenance regimen. Adverse reactions reported in > 3% of subjects in induction studies and at a higher rate than placebo are shown in Table 2.
Table 2. Adverse Drug Reactions Reported in > 3% of Subjects with Crohn’s Disease Treated with SKYRIZI in Placebo-Controlled 12-Week Induction Studies
Adverse Drug Reactions
SKYRIZI 600 mg Intravenous Infusiona N = 620 n (%)
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and embryo/fetal risk
Published data suggest that the risk of adverse pregnancy outcomes in women with inflammatory bowel disease is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Fetal/Neonatal adverse reactions
Placebo N = 432 n (%)
Upper respiratory infectionsb 66 (10.6) 40 (9.3)
Headachec 41 (6.6) 24 (5.6)
Arthralgia 31 (5.0) 19 (4.4)
a SKYRIZI 600 mg as an intravenous infusion at Week 0, Week 4, and Week 8.
b Includes: influenza like illness, nasopharyngitis, influenza, pharyngitis, upper respiratory tract infection, viral upper respiratory tract infection, COVID-19, nasal congestion, respiratory tract infection viral, viral pharyngitis, tonsillitis, upper respiratory tract inflammation
c Includes: headache, tension headache
Adverse reactions reported in >3% of subjects in the maintenance study and at a higher rate than placebo are shown in Table 3.
Table 3. Adverse Reactions Reported in >3% of Subjects with Crohn’s Disease Treated with SKYRIZI in Placebo-Controlled 52-Week Maintenance Study (CD-3)
Adverse Drug Reactions
SKYRIZI 360 mg Subcutaneous Injectiona N = 142 n (%) Placebo N = 143 n (%)
Arthralgia 13 (9.2) 12 (8.4)
Injection site reactionsb,c 8 (5.6) 4 (2.8)
Abdominal paind 12 (8.5) 6 (4.2)
Anemia 7 (4.9) 6 (4.2)
Pyrexia 7 (4.9) 4 (2.8)
Back pain 6 (4.2) 3 (2.1)
Arthropathy 5 (3.5) 2 (1.4)
Urinary tract infection 5 (3.5) 4 (2.8)
a SKYRIZI 360 mg at Week 12 and every 8 weeks thereafter for up to an additional 52 weeks b Includes: injection site rash, injection site erythema, injection site swelling, injection site urticaria, injection site warmth, injection site pain, injection site hypersensitivity, injection site reaction c Some subjects had multiple occurrences of injection site reactions. The adverse reaction is included only once per subject.
dIncludes: abdominal pain, abdominal pain upper, abdominal pain lower Specific Adverse Drug Reactions
Infections
In the maintenance study (CD-3) through Week 52, the rate of infections was 36.6% (60.8 events per 100 subject-years) in subjects who received SKYRIZI compared to 36.4% (60.3 events per 100 subject-years) in subjects who received placebo after SKYRIZI induction. The rate of serious infections was 5.6% (7.4 events per 100 subject-years) in subjects who received SKYRIZI compared to 2.1% (2.4 events per 100 subject-years) in subjects who received placebo after SKYRIZI induction.
Lipid Elevations
Elevations in lipid parameters (total cholesterol and low-density lipoprotein cholesterol [LDL-C]) were first assessed at 4 weeks following initiation of SKYRIZI in the induction trials (CD-1, CD-2). Increases from baseline and increases relative to placebo were observed at Week 4 and remained stable to Week 12. Following SKYRIZI induction, mean total cholesterol increased by 9.4 mg/dL from baseline to a mean absolute value of 175.1 mg/dL at Week 12. Similarly, mean LDL-C increased by 6.6 mg/dL from baseline to a mean absolute value of 92.6 mg/dL Week 12. Following maintenance treatment with SKYRIZI, mean LDL-C increased by 2.3 mg/dL from baseline to Week 52, to an absolute value of 102.2 mg/dL.
Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products, including other risankizumab products, may be misleading.
Plaque Psoriasis
By Week 52, approximately 24% (263/1079) of subjects treated with SKYRIZI at the recommended dose developed antibodies to risankizumab-rzaa. Of the subjects who developed antibodies to risankizumabrzaa, approximately 57% (14% of all subjects treated with SKYRIZI) had antibodies that were classified as neutralizing. Higher antibody titers in approximately 1% of subjects treated with SKYRIZI were associated with lower risankizumab-rzaa concentrations and reduced clinical response.
Psoriatic Arthritis
By Week 28, approximately 12.1% (79/652) of subjects treated with SKYRIZI at the recommended dose developed antibodies to risankizumab-rzaa. None of the subjects who developed antibodies to risankizumabrzaa had antibodies that were classified as neutralizing. Antibodies to risankizumab-rzaa were not associated with changes in clinical response for psoriatic arthritis. A higher proportion of subjects with anti-drug antibodies experienced hypersensitivity reactions (6.3% (5/79)) and injection site reactions (2.5% (2/79)) compared to subjects without anti-drug antibodies (3.8% (22/574) with hypersensitivity reactions and 0.7% (4/574) with injection site reactions). None of these hypersensitivity and injection site reactions led to discontinuation of risankizumab-rzaa.
Crohn’s Disease
By Week 64, approximately 3.4% (2/58) of subjects treated with SKYRIZI at the recommended induction and maintenance dosages developed antibodies to risankizumab-rzaa. None of the subjects who developed antibodies to risankizumab-rzaa had antibodies that were classified as neutralizing.
Postmarketing Experience
The following adverse reactions have been reported during post-approval of SKYRIZI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to SKYRIZI exposure: • Skin and subcutaneous tissue disorders eczema and rash
USE IN SPECIFIC POPULATIONS
Pregnancy Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors outcomes in women who become pregnant while treated with SKYRIZI. Patients should be encouraged to enroll by calling 1-877-302-2161 or visiting http://glowpregnancyregistry.com.
Risk Summary
Available pharmacovigilance and clinical trial data with risankizumab use in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Although there are no data on risankizumab-rzaa, monoclonal antibodies can be actively transported across the placenta, and SKYRIZI may cause immunosuppression in the in utero-exposed infant There are adverse pregnancy outcomes in women with inflammatory bowel disease (see Clinical Considerations)
In an enhanced pre- and post-natal developmental toxicity study, pregnant cynomolgus monkeys were administered subcutaneous doses of 5 or 50 mg/kg risankizumab-rzaa once weekly during the period of organogenesis up to parturition. Increased fetal/infant loss was noted in pregnant monkeys at the 50 mg/kg dose (see Data) The 50 mg/kg dose in pregnant monkeys resulted in approximately 10 times the exposure (AUC) in humans administered the 600 mg induction regimen and 39 times the exposure (AUC) to the 360 mg maintenance doses, respectively. No risankizumab-rzaa-related effects on functional or immunological development were observed in infant monkeys from birth through 6 months of age. The clinical significance of these findings for humans is unknown.
Transport of endogenous IgG antibodies across the placenta increases as pregnancy progresses, and peaks during the third trimester. Because risankizumab may interfere with immune response to infections, risks and benefits should be considered prior to administering live vaccines to infants exposed to SKYRIZI in utero. There are insufficient data regarding infant serum levels of risankizumab at birth and the duration of persistence of risankizumab in infant serum after birth. Although a specific timeframe to delay live virus immunizations in infants exposed in utero is unknown, a minimum of 5 months after birth should be considered because of the half-life of the product.
Data Animal Data
An enhanced pre- and post-natal developmental toxicity study was conducted in cynomolgus monkeys. Pregnant cynomolgus monkeys were administered weekly subcutaneous doses of risankizumab-rzaa of 5 or 50 mg/kg from gestation day 20 to parturition, and the cynomolgus monkeys (mother and infants) were monitored for 6 months after delivery. No maternal toxicity was noted in this study. There were no treatment-related effects on growth and development, malformations, developmental immunotoxicology, or neurobehavioral development. However, a dose-dependent increase in fetal/infant loss was noted in the risankizumab-rzaa-treated groups (32% and 43% in the 5 mg/kg and 50 mg/kg groups, respectively) compared with the vehicle control group (19%). The increased fetal/infant loss in the 50 mg/kg group was considered to be related to risankizumab-rzaa treatment. The no observed adverse effect level (NOAEL) for maternal toxicity was identified as 50 mg/kg and the NOAEL for developmental toxicity was identified as 5 mg/kg. On an exposure (AUC) basis, the 5 mg/kg dose in pregnant monkeys resulted in approximately 1.24 times the exposure in humans administered the 600 mg induction regimen and 5 times the exposure in humans administered the 360 mg maintenance doses, respectively. In the infants, mean serum concentrations increased in a dose-dependent manner and were approximately 17%-86% of the respective maternal concentrations. Following delivery, most adult female cynomolgus monkeys and all infants from the risankizumab-rzaa-treated groups had measurable serum concentrations of risankizumab-rzaa up to 91 days postpartum. Serum concentrations were below detectable levels at 180 days postpartum.
Lactation
Risk Summary
There are no data on the presence of risankizumab-rzaa in human milk, the effects on the breastfed infant, or the effects on milk production. Endogenous maternal IgG and monoclonal antibodies are transferred in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to risankizumab-rzaa are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SKYRIZI and any potential adverse effects on the breastfed infant from SKYRIZI or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of SKYRIZI have not been established in pediatric patients.
Geriatric Use
Of the 2234 subjects with plaque psoriasis exposed to SKYRIZI, 243 subjects were 65 years or older and 24 subjects were 75 years or older. No overall differences in SKYRIZI exposure, safety, or effectiveness were observed between older and younger subjects who received SKYRIZI. However, the number of subjects aged 65 years and older was not sufficient to determine whether they respond differently from younger subjects. Clinical studies of SKYRIZI for the treatment of Crohn’s disease did not include sufficient numbers of subjects 65 years of age and older to determine whether they respond differently from younger adult subjects. No clinically meaningful differences in the pharmacokinetics of risankizumab-rzaa were observed in geriatric subjects compared to younger adult subjects with Crohn’s disease.
PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Hypersensitivity Reactions
Advise patients to discontinue SKYRIZI and seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions].
Infections
Inform patients that SKYRIZI may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider and contacting their healthcare provider if they develop any symptoms of an infection [see Warnings and Precautions]
Hepatotoxicity in Treatment of Crohn’s Disease
Inform patients that SKYRIZI may cause liver injury, especially during the initial 12 weeks of treatment. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of liver dysfunction. (e.g., unexplained rash, nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine) [see Warnings and Precautions].
Administration of Vaccines
Advise patients that vaccination with live vaccines is not recommended during SKYRIZI treatment and immediately prior to or after SKYRIZI treatment. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Instruct patients to inform the healthcare practitioner that they are taking SKYRIZI prior to a potential vaccination [see Warnings and Precautions] Administration Instruction
Instruct patients or caregivers to perform the first self-injected dose under the supervision and guidance of a qualified healthcare professional for training in preparation and administration of SKYRIZI, including choosing anatomical sites for administration, and proper subcutaneous injection technique.
If using SKYRIZI 75 mg/0.83 mL, instruct patients or caregivers to administer two 75 mg single-dose syringes to achieve the full 150 mg dose of SKYRIZI.
Instruct patients or caregivers in the technique of pen or syringe disposal.
Pregnancy
Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to SKYRIZI during pregnancy and patients can call 1-877-302-2161 [see Use in Specific Populations]
Manufactured by: AbbVie Inc.
North Chicago, IL 60064, USA
US License Number 1889
SKYRIZI® is a registered trademark of AbbVie Biotechnology Ltd. © 2019-2022 AbbVie Inc.
Ref: 20070464 Revised: June, 2022 LAB-7524 MASTER
(sky-RIZZ-ee) (risankizumab-rzaa) injection, for subcutaneous use
pen and prefilled syringe
with
injector PROFESSIONAL BRIEF SUMMARY CONSULT PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION
20070464 Skyrizi PB-10.5x13(1).indd 1 /21/Jun2022 2:50 PM US-SKZG-220333

LA MEDICINA DE LONGEVIDAD: EL MOMENTO ES AHORA

Afortunadamente, los avances tecnológicos, la demanda por estos servicios y la evolución de estas especialidades nos permite ahora ofrecer estos entrenamientos y congresos en Puerto Rico y el Caribe. El tiempo nos ha llegado.
 Dr. Luis Martínez
Dr. Luis Martínez
Revista Puertorriqueña de Medicina y Salud Pública50
Es una realidad. Los seres humanos no queremos envejecer. Y los avances médicos nos acercan cada vez más a la posibilidad de una vida mucho más larga y saludable de lo que hoy día tenemos. A nivel mundial, la tasa de vida promedio sigue, generalmente, en aumento. Pero existen retos aso
ciados a este aumento de vida promedio. Prime ro, no solo es vivir más, sino mantener la salud durante esos años adicionales de vida. Segundo, ¿cuánto más podemos seguir extendiendo la vida? ¿Existe un límite máximo, o lograremos alcanzar la inmortalidad?
Por estas preguntas y necesidades es que ha ido evolucionando la medici na de longevidad, también conocida como la medicina antiaging. La misma busca, de manera proactiva, maximizar las intervenciones que pue dan brindar salud y mayor longevidad a los seres humanos.
En sus comienzos, la medicina antiaging se de sarrolló con un enfoque particular de reemplazo hormonal y nutricional en las poblaciones de enve jecientes, notando una mejor calidad de vida con dichas intervenciones. Sin embargo, la evolución de esta disciplina ahora incorpora muchos otros trata mientos como lo son las terapias celulares y regene rativas, los senolíticos, las terapias con péptidos y la medicina personalizada, entre otras.
El interés por estas intervenciones no solo ha sido una iniciativa de los profesionales de la sa lud. Hoy día, muchas de las grandes compañías, como lo son Google y Apple, entre otras, están invirtiendo recursos y dinero significativo en tratamientos que puedan combatir y revertir el proceso
del envejecimiento. Incluso, hasta naciones y gobier nos se están uniendo a la iniciativa de reconocer el envejecimiento como una enfermedad que se debe atender y revertir.
Uno de los retos previos para el desarrollo y ofre cimiento de estas terapias en Puerto Rico era el acceso a educación y a los entrenamientos espe cializados de estas disciplinas. Antes, los médicos puertorriqueños que buscaban entrenarse en estas disciplinas, se veían obligados a viajar a Estados Unidos o a Europa para asistir a cursos especiali zados. Esto significaba una inversión substancial de tiempo y recursos. Y por eso en parte, el campo de la longevidad ha crecido más lentamente en Puerto Rico que en otras partes del mundo.
Por eso estamos tan entusiasmados con el primer Antiaging Sum mit de Puerto Rico, el cual se celebrará en el mes de noviembre, en San Juan. El mismo cuenta con presentadores internaciona les de renombre en el campo del antiaging y longevidad. Se cu brirán temas de alto interés como lo son la genómica personalizada, los últimos avances en terapias hormonales y de péptidos, la medicina estética regenerativa, los senolíticos y muchos más. El evento además contará con múltiples exhibidores ofreciendo lo último en productos y tecnología para clínicas antiaging. Y el contenido científico es apto tanto para principiantes del tema como para profe sionales establecidos con años de experiencia.
Para la medicina de longe vidad, el momento es ahora. Y con el primer Antiaging Summit, se busca posicionar a Puerto Rico como centro (hub) de longevidad y antia ging en el Caribe y las Amé ricas.
Los esperamos, Dr. Luis Martínez
Revista Puertorriqueña de Medicina y Salud Pública 51
IMPORTANT SAFETY INFORMATION1
SERIOUS INFECTIONS
Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. If a serious infection develops, interrupt RINVOQ until the infection is controlled.
Reported infections include:
• Active tuberculosis (TB), which may present with pulmonary or extrapulmonary disease. Test patients for latent TB before RINVOQ use and during therapy. Consider treatment for latent TB infection prior to RINVOQ use.
• Invasive fungal infections, including cryptococcosis and pneumocystosis.
• Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
Carefully consider the risks and benefits of treatment with RINVOQ prior to initiating therapy in patients with chronic or recurrent infection. Monitor patients closely for the development of signs and symptoms of infection during and after treatment with RINVOQ, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy.
MORTALITY
In a large, randomized, postmarketing safety study comparing another Janus kinase (JAK) inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years old with at least one cardiovascular (CV) risk factor, a higher rate of all-cause mortality, including sudden CV death, was observed with the JAK inhibitor. Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ.
MALIGNANCIES
Lymphoma and other malignancies have been observed in patients treated with RINVOQ.
In a large, randomized, postmarketing safety study comparing another JAK inhibitor with TNF blockers in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with the JAK inhibitor. Patients who are current or past smokers are at additional increased risk.
With RINVOQ, consider the benefits and risks for the individual patient prior to initiating or continuing therapy, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers. NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer. Advise patients to limit sunlight exposure by wearing protective clothing and using sunscreen.
MAJOR ADVERSE CARDIOVASCULAR EVENTS
In a large, randomized, postmarketing study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years old with at least one CV risk factor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke) was observed with the JAK inhibitor. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and patients with other CV risk factors. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
INDICATION1
THROMBOSIS
Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. Many of these adverse events were serious and some resulted in death.
GASTROINTESTINAL PERFORATIONS
HYPERSENSITIVITY
LABORATORY ABNORMALITIES
Neutropenia
Lymphopenia
Anemia
SAFETY CONSIDERATIONS1
In a large, randomized, postmarketing study comparing another JAK inhibitor to TNF blockers in RA patients ≥50 years old with at least one CV risk factor, a higher rate of thrombosis was observed with the JAK inhibitor. Avoid RINVOQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ and be promptly evaluated.
RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients. Serious hypersensitivity reactions, such as anaphylaxis and angioedema, were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ and institute appropriate therapy.
Lipids
Malignancies: Lymphoma and other malignancies have been observed in RINVOQ-treated patients. A higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with another JAK inhibitor when compared with TNF blockers in RA patients. Patients who are current or past smokers are at additional increased risk.
Gastrointestinal (GI) perforations have been reported in clinical trials with RINVOQ. Monitor RINVOQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs). Promptly evaluate patients presenting with new onset abdominal pain for early identification of GI perforation.
Treatment with RINVOQ was associated with an increased incidence of neutropenia (absolute neutrophil count [ANC] <1000 cells/mm3). Treatment with RINVOQ is not recommended in patients with an ANC <1000 cells/mm3. Evaluate neutrophil counts at baseline and thereafter according to routine patient management.
Major Adverse Cardiovascular Events: A higher rate of CV death, myocardial infarction, and stroke was observed with a JAK inhibitor in a study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years of age with at least one CV risk factor. Current or past smokers are at additional increased risk.
Thrombosis: Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. A higher rate of thrombosis was observed with another JAK inhibitor when compared with TNF blockers in RA patients.
Absolute lymphocyte counts (ALC) <500 cells/mm3 were reported in RINVOQtreated patients. Treatment with RINVOQ is not recommended in patients with an ALC <500 cells/mm3. Evaluate at baseline and thereafter according to routine patient management.
Decreases in hemoglobin levels to <8 g/dL were reported in RINVOQ-treated patients. Treatment should not be initiated or should be interrupted in patients with hemoglobin levels <8 g/dL. Evaluate at baseline and thereafter according to routine patient management.
Hypersensitivity: RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients.
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol. Manage patients according to clinical guidelines for the management of hyperlipidemia. Evaluate patients 12 weeks after initiation of treatment and thereafter according to the clinical guidelines for hyperlipidemia.
Liver enzyme elevations
Other Serious Adverse Reactions: Hypersensitivity Reactions (anaphylaxis and angioedema), Gastrointestinal Perforations, Laboratory Abnormalities (neutropenia, lymphopenia, anemia, lipid elevations, liver enzyme elevations), and Embryo-Fetal Toxicity.
EMBRYO-FETAL TOXICITY
VACCINATION
LACTATION
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevation compared to placebo. Evaluate at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury.
If increases in aspartate aminotransferase (AST) or alanine aminotransferase (ALT) are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ should be interrupted until this diagnosis is excluded.
a SELECT-EARLY (RA-I; MTX-naïve) [primary endpoint at Week 12: ACR50 response vs MTX, select ranked secondary endpoint at Week 24: Δ mTSS vs MTX]; SELECT-MONOTHERAPY (RA-II; MTX-IR) [primary endpoint at Week 14: ACR20 response vs MTX, select ranked secondary endpoints at Week 14: DAS28-CRP<2.6 vs MTX, DAS28-CRP≤3.2 vs MTX]; SELECT-NEXT (RA-III; csDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD]; SELECT-COMPARE (RA-IV; MTX-IR) [RINVOQ + MTX; primary endpoint at Week 12: ACR20 response vs placebo + MTX, select ranked secondary endpoints at Week 26: Δ mTSS vs placebo + MTX]; SELECT-BEYOND (RA-V; bDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD, select ranked secondary endpoints at Week 12: DAS28-CRP≤3.2 vs placebo + csDMARD.] 1,2 SELECT-CHOICE (bDMARD-IR) [RINVOQ + csDMARDs; primary endpoint at Week 12: Δ DAS28-CRP (noninferiority)
HEPATIC IMPAIRMENT
RINVOQ is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
INDICATION 1 RINVOQ is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
Based on findings in animal studies, RINVOQ may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with RINVOQ and for 4 weeks after the final dose. Verify pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ.
Limitation of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
SAFETY CONSIDERATIONS 1
Avoid use of live vaccines during, or immediately prior to, RINVOQ therapy. Prior to initiating RINVOQ, patients should be brought up to date on all immunizations, including varicella zoster or prophylactic herpes zoster vaccinations, in agreement with current immunization guidelines.
There are no data on the presence of RINVOQ in human milk, the effects on the breastfed infant, or the effects on milk production. Available data in animals have shown the excretion of RINVOQ in milk. Advise patients that breastfeeding is not recommended during treatment with RINVOQ and for 6 days after the last dose.
Serious Infections: Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. These infections include tuberculosis (TB), invasive fungal, bacterial, viral, and other infections due to opportunistic pathogens. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids.
Mortality: A higher rate of all-cause mortality, including sudden cardiovascular (CV) death, was observed with a Janus kinase (JAK) inhibitor in a study comparing another JAK inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years of age with at least one CV risk factor.
RINVOQ is not recommended for use in patients with severe hepatic impairment.
ADVERSE REACTIONS
Limitation of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
Malignancies: Lymphoma and other malignancies have been observed in RINVOQ-treated patients. A higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with another JAK inhibitor when compared with TNF blockers in RA patients. Patients who are current or past smokers are at additional increased risk.
The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenzalike illness, elevated liver enzymes, and rash.
Major Adverse Cardiovascular Events: A higher rate of CV death, myocardial infarction, and stroke was observed with a JAK inhibitor in a study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years of age with at least one CV risk factor. Current or past smokers are at additional increased risk.
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.
Serious Infections: Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. These infections include tuberculosis (TB), invasive fungal, bacterial, viral, and other infections due to opportunistic pathogens. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids.
Dosage Forms and Strengths: RINVOQ is available in 15 mg, 30 mg, and 45 mg extended-release tablets.
Mortality: A higher rate of all-cause mortality, including sudden cardiovascular (CV) death, was observed with a Janus kinase (JAK) inhibitor in a study comparing another JAK inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years of age with at least one CV risk factor.
Thrombosis: Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. A higher rate of thrombosis was observed with another JAK inhibitor when compared with TNF blockers in RA patients.
Hypersensitivity: RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients.
Other Serious Adverse Reactions: Hypersensitivity Reactions (anaphylaxis and angioedema), Gastrointestinal Perforations, June 2-5, 2021; E-Congress. Rubbert-Roth A, Enejosa J, Pangan AL, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med. 2020;383(16):1511-1521.
RINVOQ® and its design are registered trademarks of AbbVie Biotechnology Ltd. ©2022 AbbVie Inc. North Chicago, IL 60064 US-RNQR-220149 April 2022 Printed in Puerto Rico
WITH INHIBITOR EXPECTATIONS or 14, SELECT-BEYOND and absence of and patient7,a,b
Laboratory Abnormalities (neutropenia, lymphopenia, anemia, lipid elevations, liver enzyme elevations), and Embryo-Fetal Toxicity. Please see additional Important Safety Information, including BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis, on the previous page of this advertisement. Please see Brief Summary of full Prescribing Information on previous pages of this advertisement. a SELECT-EARLY (RA-I; MTX-naïve) [primary endpoint at Week 12: ACR50 response vs MTX, select ranked secondary endpoint at Week 24: Δ mTSS vs MTX]; SELECT-MONOTHERAPY (RA-II; MTX-IR) [primary endpoint at Week 14: ACR20 response vs MTX, select ranked secondary endpoints at Week 14: DAS28-CRP<2.6 vs MTX, DAS28-CRP≤3.2 vs MTX]; SELECT-NEXT (RA-III; csDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD]; SELECT-COMPARE (RA-IV; MTX-IR) [RINVOQ + MTX; primary endpoint at Week 12: ACR20 response vs placebo + MTX, select ranked secondary endpoints at Week 26: Δ mTSS vs placebo + MTX]; SELECT-BEYOND (RA-V; bDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD, select ranked secondary endpoints at Week 12: DAS28-CRP≤3.2 vs placebo + csDMARD.] 1,2 bSELECT-CHOICE (bDMARD-IR) [RINVOQ + csDMARDs; primary endpoint at Week 12: Δ DAS28-CRP (noninferiority) vs active comparator + csDMARDs]. 8 c RINVOQ 15 mg; upadacitinib 30 mg; RINVOQ 15 mg is the approved dose.1,7 ACR=American College of Rheumatology; AEs=adverse events; bDMARD=biologic DMARD; csDMARD=conventional synthetic DMARD; DAS28-CRP=Disease Activity Score 28 joints, C-reactive protein; DMARD=disease-modifying antirheumatic drug; HAQ-DI=Health Assessment Questionnaire Disability Index; IR=intolerance or inadequate response; JAK=Janus kinase; mTSS=modified total Sharp score; MTX=methotrexate; TNFi=tumor necrosis factor inhibitor. CHALLENGE TREATMENT GOALS IN RA WITH A ONCE-DAILY ORAL JAK INHIBITOR EXPECTATIONS For moderate to severe rheumatoid arthritis (RA) in adult TNFi-IR patients1 Discover our commitment to exceptional access and patient support at RinvoqHCPPR.com LONG-TERM REMISSION AND LOW DISEASE ACTIVITY DATA observed up to 84 weeks with or without MTX1,3-6 • DAS28-CRP<2.6* and DAS28-CRP≤3.2 evaluated at Week 12 or 14, with response rates from 60 to 84 weeks (in SELECT-BEYOND and SELECT-MONOTHERAPY, respectively) *Clinical remission does not mean drug-free remission or complete absence of disease activity. LONG-TERM SAFETY DATA AEs observed in long-term analysis with ~4.5 years maximum and ~2.6 years median exposure to RINVOQ 15 mg as of 6/30/207,a,b • >4400 patients evaluated on upadacitinib,c with >7000 patientyears of long-term exposure to RINVOQ 15 mg as of 6/30/207,a,b RINVOQ met primary (ACR20 or ACR50 at Week 12 or 14) and ranked secondary endpoints in clinical trials, with some patients achieving ACR20 as early as Week 1 in SELECT-BEYOND1-3,a,b Please see Brief Summary of full Prescribing Information on previous pages of this advertisement. References: 1. RINVOQ [package insert]. North Chicago, IL: AbbVie Inc; 2022. 2. Data on file, AbbVie Inc. ABVRRTI68885. 3. Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139): 2513-2524. 4. Smolen JS, Emery P, Rigby W, et al. Upadacitinib as monotherapy in patients with rheumatoid arthritis and prior inadequate response to methotrexate: results at 84 weeks from the SELECTMONOTHERAPY study. Poster presented at: The European Congress of Rheumatology; June 3-6, 2020; E-Congress. 5. Smolen JS, Pangan AL, Emery P, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393(10188):2303-2311. Erratum in: Lancet. 2019;393(10191):2590. 6. Genovese MC, Combe B, Hall S, et al. Upadacitinib in patients with rheumatoid arthritis and inadequate response or intolerance to biological DMARDs: Results at 60 weeks from the SELECT-BEYOND study. Poster presented at: The American College of Rheumatology; November 8-13, 2019. 7. Cohen SB, van Vollenhoven R, Curtis JR, et al. Integrated safety profile of upadacitinib with up to 4.5 years of exposure in patients with rheumatoid arthritis. Poster presented at: The European Congress of Rheumatology;
8.
b
vs active comparator + csDMARDs]. 8 c RINVOQ 15 mg; upadacitinib 30 mg; RINVOQ 15 mg is the approved dose. 1,7 CHALLENGE TREATMENT GOALS IN RA WITH A ONCE-DAILY ORAL JAK INHIBITOREXPECTATIONS Discover our commitment to exceptional access and patient support at RinvoqHCPPR.com LONG-TERM REMISSION AND LOW DISEASE ACTIVITY DATA observed up to 84 weeks with or without MTX 1,3-6 • DAS28-CRP<2.6* and DAS28-CRP≤3.2 evaluated at Week 12 or 14, with response rates from 60 to 84 weeks (in SELECT-BEYOND and SELECT-MONOTHERAPY, respectively) * Clinical remission does not mean drug-free remission or complete absence of disease activity. LONG-TERM SAFETY DATA AEs observed in long-term analysis with ~4.5 years maximum and ~2.6 years median exposure to RINVOQ 15 mg as of 6/30/20 7,a,b • >4400 patients evaluated on upadacitinib, c with >7000 patientyears of long-term exposure to RINVOQ 15 mg as of 6/30/20 7,a,b RINVOQ met primary (ACR20 or ACR50 at Week 12 or 14) and ranked secondary endpoints in clinical trials, with some patients achieving ACR20 as early as Week 1 in SELECT-BEYOND 1-3,a,b
CONTRAINDICATIONS
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, and THROMBOSIS SERIOUS INFECTIONS
Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions, Adverse Reactions]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt RINVOQ until the infection is controlled.
Reported infections include:
• Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before RINVOQ use and during therapy. Treatment for latent infection should be considered prior to RINVOQ use.
• Invasive fungal infections, including cryptococcosis and pneumocystosis.
• Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with RINVOQ should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with RINVOQ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions]
MORTALITY
In a large, randomized, postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another Janus kinase (JAK) inhibitor to tumor necrosis factor (TNF) blockers, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed with the JAK inhibitor [see Warnings and Precautions].
MALIGNANCIES
Lymphoma and other malignancies have been observed in patients treated with RINVOQ. In RA patients treated with another JAK inhibitor, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk [see Warnings and Precautions]
MAJOR ADVERSE CARDIOVASCULAR EVENTS
In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke [see Warnings and Precautions].
THROMBOSIS
Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. Many of these adverse events were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of thrombosis was observed when compared with TNF blockers. Avoid RINVOQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ and be promptly evaluated [see Warnings and Precautions]
INDICATIONS AND USAGE
Rheumatoid Arthritis
RINVOQ® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic disease-modifying antirheumatic drugs (DMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
Psoriatic Arthritis
RINVOQ is indicated for the treatment of adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
Atopic Dermatitis
RINVOQ is indicated for the treatment of adults and pediatric patients 12 years of age and older with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies are inadvisable.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
Ulcerative Colitis
RINVOQ is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for ulcerative colitis, or with potent immunosuppressants such as azathioprine and cyclosporine.
Ankylosing Spondylitis
RINVOQ is indicated for the treatment of adults with active ankylosing spondylitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
PROFESSIONAL BRIEF SUMMARY CONSULT PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION
RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients [see Warnings and Precautions].
WARNINGS AND PRECAUTIONS
Serious Infections
Serious and sometimes fatal infections have been reported in patients receiving RINVOQ. The most frequent serious infections reported with RINVOQ included pneumonia and cellulitis [see Adverse Reactions]. Among opportunistic infections, tuberculosis, multidermatomal herpes zoster, oral/esophageal candidiasis, and cryptococcosis, were reported with RINVOQ.
Avoid use of RINVOQ in patients with an active, serious infection, including localized infections. Consider the risks and benefits of treatment prior to initiating RINVOQ in patients:
• with chronic or recurrent infection
• who have been exposed to tuberculosis
• with a history of a serious or an opportunistic infection
• who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
• with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with RINVOQ. Interrupt RINVOQ if a patient develops a serious or opportunistic infection.
A patient who develops a new infection during treatment with RINVOQ should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, the patient should be closely monitored, and RINVOQ should be interrupted if the patient is not responding to antimicrobial therapy. RINVOQ may be resumed once the infection is controlled.
Tuberculosis
Evaluate and test patients for latent and active tuberculosis (TB) infection prior to administration of RINVOQ. Patients with latent TB should be treated with standard antimycobacterial therapy before initiating RINVOQ. RINVOQ should not be given to patients with active TB. Consider anti-TB therapy prior to initiation of RINVOQ in patients with previously untreated latent TB or active TB in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent TB but who have risk factors for TB infection.
Consultation with a physician with expertise in the treatment of TB is recommended to aid in the decision about whether initiating anti-TB therapy is appropriate for an individual patient.
During RINVOQ use, monitor patients for the development of signs and symptoms of TB, including patients who tested negative for latent TB infection prior to initiating therapy.
Viral Reactivation
Viral reactivation, including cases of herpes virus reactivation (e.g., herpes zoster) and hepatitis B virus reactivation, were reported in clinical trials with RINVOQ [see Adverse Reactions]. The risk of herpes zoster appears to be higher in patients treated with RINVOQ in Japan. If a patient develops herpes zoster, consider temporarily interrupting RINVOQ until the episode resolves.
Screening for viral hepatitis and monitoring for reactivation should be performed in accordance with clinical guidelines before starting and during therapy with RINVOQ. Patients who were positive for hepatitis C antibody and hepatitis C virus RNA, were excluded from clinical trials. Patients who were positive for hepatitis B surface antigen or hepatitis B virus DNA were excluded from clinical trials. However, cases of hepatitis B reactivation were still reported in patients enrolled in the Phase 3 trials of RINVOQ. If hepatitis B virus DNA is detected while receiving RINVOQ, a liver specialist should be consulted.
Mortality
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed in patients treated with the JAK inhibitor compared with TNF blockers.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ.
Malignancy and Lymphoproliferative Disorders
Malignancies, including lymphomas, were observed in clinical trials of RINVOQ [see Adverse Reactions]
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. In this study, current or past smokers had an additional increased risk of overall malignancies.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Non-Melanoma Skin Cancer
NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
Exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen.
Major Adverse Cardiovascular Events
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of major adverse cardiovascular events (MACE) defined as cardiovascular death, non-fatal myocardial infarction (MI), and non-fatal stroke was observed with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients
should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke.
Thrombosis
Thrombosis, including deep venous thrombosis (DVT), pulmonary embolism (PE), and arterial thrombosis, have occurred in patients treated for inflammatory conditions with JAK inhibitors, including RINVOQ. Many of these adverse events were serious and some resulted in death.
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, higher rates of overall thrombosis, DVT, and PE were observed compared to those treated with TNF blockers.
If symptoms of thrombosis occur, patients should discontinue RINVOQ and be evaluated promptly and treated appropriately. Avoid RINVOQ in patients that may be at increased risk of thrombosis.
Hypersensitivity Reactions
Serious hypersensitivity reactions such as anaphylaxis and angioedema were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ and institute appropriate therapy [see Adverse Reactions].
Gastrointestinal Perforations
Gastrointestinal perforations have been reported in clinical trials with RINVOQ.
Monitor RINVOQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs).
Evaluate promptly patients presenting with new onset abdominal pain for early identification of gastrointestinal perforation.
Laboratory Abnormalities
Neutropenia
Treatment with RINVOQ was associated with an increased incidence of neutropenia (ANC less than 1000 cells/mm3).
Evaluate neutrophil counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation and interrupt RINVOQ treatment in patients with a low neutrophil count (i.e., ANC less than 1000 cells/mm3).
Lymphopenia
ALC less than 500 cells/mm3 were reported in RINVOQ-treated patients in clinical trials.
Evaluate lymphocyte counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low lymphocyte count (i.e., less than 500 cells/mm3).
Anemia
Decreases in hemoglobin levels to less than 8 g/dL were reported in RINVOQ-treated patients in clinical trials.
Evaluate hemoglobin at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low hemoglobin level (i.e., less than 8 g/dL).
Lipids
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol [see Adverse Reactions] Elevations in LDL cholesterol decreased to pre-treatment levels in response to statin therapy. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Assess lipid parameters approximately 12 weeks after initiation of treatment, and thereafter according to the clinical guidelines for hyperlipidemia. Manage patients according to clinical guidelines for the management of hyperlipidemia.
Liver Enzyme Elevations
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevations compared to treatment with placebo.
Evaluate liver enzymes at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury.
If increases in ALT or AST are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ should be interrupted until this diagnosis is excluded.
Embryo-Fetal Toxicity
Based on findings in animal studies, RINVOQ may cause fetal harm when administered to a pregnant woman. Administration of upadacitinib to rats and rabbits during organogenesis caused increases in fetal malformations. Verify the pregnancy status of patients of reproductive potential prior to starting treatment. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception during treatment with RINVOQ and for 4 weeks following completion of therapy [see Use in Specific Populations]
Vaccinations
Avoid use of live vaccines during, or immediately prior to, RINVOQ therapy. Prior to initiating RINVOQ, it is recommended that patients be brought up to date with all immunizations, including varicella zoster or prophylactic herpes zoster vaccinations, in agreement with current immunization guidelines.
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
• Serious Infections [see Warnings and Precautions]
• Mortality [see Warnings and Precautions]
• Malignancy and Lymphoproliferative Disorders [see Warnings and Precautions]
• Major Adverse Cardiovascular Events [see Warnings and Precautions]
• Thrombosis [see Warnings and Precautions]
• Hypersensitivity Reactions [see Warnings and Precautions]
• Gastrointestinal Perforations [see Warnings and Precautions]
• Laboratory Abnormalities [see Warnings and Precautions]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
DO NOT RE-SIZE RINVOQ® (RIN-VOKE) (upadacitinib) extended-release tablets, for oral use
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 1 04 May 2022 9:00 AM US-RNQR-220149
Adverse Reactions in Patients with Rheumatoid Arthritis
A total of 3833 patients with rheumatoid arthritis were treated with upadacitinib in the Phase 3 clinical trials of whom 2806 were exposed for at least one year.
Patients could advance or switch to RINVOQ 15 mg from placebo, or be rescued to RINVOQ from active comparator or placebo from as early as Week 12 depending on the trial design.
A total of 2630 patients received at least 1 dose of RINVOQ 15 mg, of whom 1860 were exposed for at least one year. In trials RA-I, RA-II, RA-III and RA-V, 1213 patients received at least 1 dose of RINVOQ 15 mg, of which 986 patients were exposed for at least one year, and 1203 patients received at least 1 dose of upadacitinib 30 mg, of which 946 were exposed for at least one year.
Table 1: Adverse Reactions Reported in ≥ 1% of Rheumatoid Arthritis Patients Treated with RINVOQ 15 mg in Placebo-controlled Trials
Adverse Reaction Placebo RINVOQ 15 mg n=1042 (%) n=1035 (%)
Upper respiratory tract infection (URTI)* 9.5 13.5
Nausea 2.2 3.5
Cough 1.0 2.2
Pyrexia 0 1.2
*URTI includes: acute sinusitis, laryngitis, nasopharyngitis, oropharyngeal pain, pharyngitis, pharyngotonsillitis, rhinitis, sinusitis, tonsillitis, viral upper respiratory tract infection
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg group and at a higher rate than in the placebo group through Week 12 included pneumonia, herpes zoster, herpes simplex (includes oral herpes), and oral candidiasis.
Four integrated datasets are presented in the Specific Adverse Reaction section:
Placebo-controlled Trials: Trials RA-III, RA-IV, and RA-V were integrated to represent safety through 12/14 weeks for placebo (n=1042) and RINVOQ 15 mg (n=1035). Trials RA-III and RA-V were integrated to represent safety through 12 weeks for placebo (n=390), RINVOQ 15 mg (n=385), and upadacitinib 30 mg (n=384). Trial RA-IV did not include the 30 mg dose and, therefore, safety data for upadacitinib 30 mg can only be compared with placebo and RINVOQ 15 mg rates from pooling trials RA-III and RA-V.
MTX-controlled Trials: Trials RA-I and RA-II were integrated to represent safety through 12/14 weeks for MTX (n=530), RINVOQ 15 mg (n=534), and upadacitinib 30 mg (n=529).
12-Month Exposure Dataset: Trials RA-I, II, III, and V were integrated to represent the long-term safety of RINVOQ 15 mg (n=1213) and upadacitinib 30 mg (n=1203).
Exposure adjusted incidence rates were adjusted by trial for all the adverse events reported in this section.
Specific Adverse Reactions
Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, infections were reported in 218 patients (95.7 per 100 patient-years) treated with placebo and 284 patients (127.8 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, infections were reported in 99 patients (136.5 per 100 patient-years) treated with placebo, 118 patients (164.5 per 100 patient-years) treated with RINVOQ 15 mg, and 126 patients (180.3 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Infections were reported in 127 patients (119.5 per 100 patient-years) treated with MTX monotherapy, 104 patients (91.8 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 128 patients (115.1 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Infections were reported in 615 patients (83.8 per 100 patient-years) treated with RINVOQ 15 mg and 674 patients (99.7 per 100 patient-years) treated with upadacitinib 30 mg.
Serious Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, serious infections were reported in 6 patients (2.3 per 100 patient-years) treated with placebo, and 12 patients (4.6 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, serious infections were reported in 1 patient (1.2 per 100 patientyears) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 7 patients (8.2 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Serious infections were reported in 2 patients (1.6 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 8 patients (6.4 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Serious infections were reported in 38 patients (3.5 per 100 patient-years) treated with RINVOQ 15 mg and 59 patients (5.6 per 100 patient-years) treated with upadacitinib 30 mg. The most frequently reported serious infections were pneumonia and cellulitis.
Tuberculosis
Placebo-controlled Trials and MTX-controlled Trials: In the placebo-controlled period, there were no active cases of tuberculosis reported in the placebo, RINVOQ 15 mg, and upadacitinib 30 mg groups. In the MTX-controlled period, there were no active cases of tuberculosis reported in the MTX monotherapy, RINVOQ 15 mg monotherapy, and upadacitinib 30 mg monotherapy groups.
12-Month Exposure Dataset: Active tuberculosis was reported for 2 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg. Cases of extra-pulmonary tuberculosis were reported.
Opportunistic Infections (excluding tuberculosis)
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, opportunistic infections were reported in 3 patients (1.2 per 100 patient-years) treated with placebo, and 5 patients (1.9 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, opportunistic infections were reported in 1 patient (1.2 per 100 patient-years) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (7.1 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Opportunistic infections were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 0 patients
treated with RINVOQ 15 mg monotherapy, and 4 patients (3.2 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Opportunistic infections were reported in 7 patients (0.6 per 100 patient-years) treated with RINVOQ 15 mg and 15 patients (1.4 per 100 patient-years) treated with upadacitinib 30 mg.
Malignancies
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, malignancies excluding NMSC were reported in 1 patient (0.4 per 100 patient-years) treated with placebo, and 1 patient (0.4 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, malignancies excluding NMSC were reported in 0 patients treated with placebo, 1 patient (1.1 per 100 patient-years) treated with RINVOQ 15 mg, and 3 patients (3.5 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Malignancies excluding NMSC were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 0 patients treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Malignancies excluding NMSC were reported in 13 patients (1.2 per 100 patient-years) treated with RINVOQ 15 mg and 14 patients (1.3 per 100 patient-years) treated with upadacitinib 30 mg.
Gastrointestinal Perforations
Placebo-controlled Trials: There were no gastrointestinal perforations (based on medical review) reported in patients treated with placebo, RINVOQ 15 mg, and upadacitinib 30 mg.
MTX-controlled Trials: There were no cases of gastrointestinal perforations reported in the MTX and RINVOQ 15 mg group through 12/14 weeks. Two cases of gastrointestinal perforations were observed in the upadacitinib 30 mg group.
12-Month Exposure Dataset: Gastrointestinal perforations were reported in 1 patient treated with RINVOQ 15 mg and 4 patients treated with upadacitinib 30 mg.
Thrombosis
Placebo-controlled Trials: In RA-IV, venous thrombosis (pulmonary embolism or deep vein thrombosis) was observed in 1 patient treated with placebo and 1 patient treated with RINVOQ 15 mg. In RA-V, venous thrombosis was observed in 1 patient treated with RINVOQ 15 mg. There were no observed cases of venous thrombosis reported in RA-III. No cases of arterial thrombosis were observed through 12/14 weeks.
MTX-controlled Trials: In RA-II, venous thrombosis was observed in 0 patients treated with MTX monotherapy, 1 patient treated with RINVOQ 15 mg monotherapy and 0 patients treated with upadacitinib 30 mg monotherapy through Week 14. In RA-II, no cases of arterial thrombosis were observed through 12/14 weeks. In RA-I, venous thrombosis was observed in 1 patient treated with MTX, 0 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg through Week 24. In RA-I, arterial thrombosis was observed in 1 patient treated with upadacitinib 30 mg through Week 24.
12-Month Exposure Dataset: Venous thrombosis events were reported in 5 patients (0.5 per 100 patient-years) treated with RINVOQ 15 mg and 4 patients (0.4 per 100 patient-years) treated with upadacitinib 30 mg. Arterial thrombosis events were reported in 0 patients treated with RINVOQ 15 mg and 2 patients (0.2 per 100 patient-years) treated with upadacitinib 30 mg.
Laboratory Abnormalities
Hepatic Transaminase Elevations
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, alanine transaminase (ALT) and aspartate transaminase (AST) elevations ≥ 3 x upper limit of normal (ULN) in at least one measurement were observed in 2.1% and 1.5% of patients treated with RINVOQ 15 mg, and in 1.5% and 0.7% of patients treated with placebo, respectively. In RA-III and RA-V, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 1.0% of patients treated with RINVOQ 15 mg, 1.0% and 0% of patients treated with upadacitinib 30 mg and in 1.3% and 1.0% of patients treated with placebo, respectively.
In MTX-controlled trials, for up to 12/14 weeks, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 0.4% of patients treated with RINVOQ 15 mg, 1.7% and 1.3% of patients treated with upadacitinib 30 mg and in 1.9% and 0.9% of patients treated with MTX, respectively.
Lipid Elevations
Upadacitinib treatment was associated with dose-related increases in total cholesterol, triglycerides and LDL cholesterol. Upadacitinib was also associated with increases in HDL cholesterol. Elevations in LDL and HDL cholesterol peaked by Week 8 and remained stable thereafter. In controlled trials, for up to 12/14 weeks, changes from baseline in lipid parameters in patients treated with RINVOQ 15 mg and upadacitinib 30 mg, respectively, are summarized below:
• Mean LDL cholesterol increased by 14.81 mg/dL and 17.17 mg/dL.
• Mean HDL cholesterol increased by 8.16 mg/dL and 9.01 mg/dL.
• The mean LDL/HDL ratio remained stable.
• Mean triglycerides increased by 13.55 mg/dL and 14.44 mg/dL.
Creatine Phosphokinase Elevations
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related increases in creatine phosphokinase (CPK) values were observed. CPK elevations > 5 x ULN were reported in 1.0%, and 0.3% of patients over 12/14 weeks in the RINVOQ 15 mg and placebo groups, respectively. Most elevations >5 x ULN were transient and did not require treatment discontinuation. In RA-III and RA-V, CPK elevations > 5 x ULN were observed in 0.3% of patients treated with placebo, 1.6% of patients treated with RINVOQ 15 mg, and none in patients treated with upadacitinib 30 mg.
Neutropenia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in neutrophil counts, below 1000 cells/mm3 in at least one measurement occurred in 1.1% and <0.1% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in neutrophil counts below 1000 cells/mm3 in at least one measurement occurred in 0.3% of patients treated with placebo, 1.3% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg. In clinical trials, treatment was interrupted in response to ANC less than 1000 cells/mm3
Lymphopenia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.9%
and 0.7% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.5% of patients treated with placebo, 0.5% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg.
Anemia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, hemoglobin decreases below 8 g/dL in at least one measurement occurred in <0.1% of patients in both the RINVOQ 15 mg and placebo groups. In RA-III and RA-V, hemoglobin decreases below 8 g/dL in at least one measurement were observed in 0.3% of patients treated with placebo, and none in patients treated with RINVOQ 15 mg and upadacitinib 30 mg.
Adverse Reactions in Patients with Psoriatic Arthritis
A total of 1827 patients with psoriatic arthritis were treated with upadacitinib in clinical trials representing 1639.2 patient-years of exposure, of whom 722 were exposed to upadacitinib for at least one year. In the two Phase 3 trials, 907 patients received at least 1 dose of RINVOQ 15 mg, of whom 359 were exposed for at least one year.
Two placebo-controlled trials were integrated (640 patients on RINVOQ 15 mg once daily and 635 patients on placebo) to evaluate the safety of RINVOQ 15 mg in comparison to placebo for up to 24 weeks after treatment initiation.
Overall, the safety profile observed in patients with active psoriatic arthritis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis. During the 24-week placebo-controlled period, the frequencies of herpes zoster and herpes simplex were ≥1% (1.1% and 1.4%, respectively) with RINVOQ 15 mg and 0.8% and 1.3%, respectively with placebo. A higher incidence of acne and bronchitis was also observed in patients treated with RINVOQ 15 mg (1.3% and 3.9%, respectively) compared to placebo (0.3% and 2.7%, respectively).
Adverse Reactions in Patients with Atopic Dermatitis
Three Phase 3 (AD-1, AD-2, and AD-3) and one Phase 2b (AD-4) randomized, double-blind, placebo-controlled, multicenter trials evaluated the safety of RINVOQ in patients with moderate-to-severe atopic dermatitis. The majority of patients were White (68%) and male (57%). The mean age was 34 years (ranged from 12 to 75 years) and 13% of the patients were 12 to less than 18 years. In these 4 trials, 2612 patients were treated with RINVOQ 15 mg or 30 mg orally once daily, with or without concomitant topical corticosteroids (TCS).
In the Phase 3 clinical trials (AD-1, AD-2, and AD-3), a total of 1239 patients received RINVOQ 15 mg, of whom 791 were exposed for at least one year and 1246 patients received RINVOQ 30 mg, of whom 826 were exposed for at least one year.
Trials AD-1, AD-2, and AD-4 compared the safety of RINVOQ monotherapy to placebo through Week 16. Trial AD-3 compared the safety of RINVOQ + TCS to placebo + TCS through Week 16. Weeks 0 to 16 (Trials AD-1 to AD-4)
In RINVOQ trials with and without TCS (Trials AD-1, 2, 3 and 4) through Week 16, the proportion of patients who discontinued treatment because of adverse reactions in the RINVOQ 15 mg, 30 mg and placebo groups were 2.3%, 2.9% and 3.8%, respectively. Table 2 summarizes the adverse reactions that occurred at a rate of at least 1% in the RINVOQ 15 mg or 30 mg groups during the first 16 weeks of treatment.
Table 2: Adverse Reactions Reported in ≥ 1% of Patients with Atopic Dermatitis Treated with RINVOQ 15 mg or 30 mg
Adverse Reaction
Placebo RINVOQ 15 mg RINVOQ 30 mg n=902 (%) n=899 (%) n=906 (%)
Upper respiratory tract infection (URTI)* 17 23 25 Acne** 2 10 16
Herpes simplex*** 2 4 8 Headache 4 6 6
Increased blood creatine phosphokinase 2 5 6 Cough 1 3 3 Hypersensitivity**** 2 2 3 Folliculitis 1 2 3 Nausea 1 3 3
Abdominal pain***** 1 3 2 Pyrexia 1 2 2
Increased Weight 1 2 2 Herpes zoster****** 1 2 2 Influenza <1 2 2 Fatigue 1 1 2 Neutropenia <1 1 2 Myalgia 1 1 2 Influenza like illness 1 1 2
* Includes: laryngitis, laryngitis viral, nasopharyngitis, oropharyngeal pain, pharyngeal abscess, pharyngitis, pharyngitis streptococcal, pharyngotonsillitis, respiratory tract infection, respiratory tract infection viral, rhinitis, rhinolaryngitis, sinusitis, tonsillitis, tonsillitis bacterial, upper respiratory tract infection, viral pharyngitis, viral upper respiratory tract infection
** Includes: acne and dermatitis acneiform
*** Includes: genital herpes, genital herpes simplex, herpes dermatitis, herpes ophthalmic, herpes simplex, nasal herpes, ophthalmic herpes simplex, herpes virus infection, oral herpes
**** Includes anaphylactic reaction, anaphylactic shock, angioedema, dermatitis exfoliative generalized, drug hypersensitivity, eyelid oedema, face oedema, hypersensitivity, periorbital swelling, pharyngeal swelling, swelling face, toxic skin eruption, type hypersensitivity, urticaria
***** Includes abdominal pain and abdominal pain upper ****** Includes herpes zoster and varicella
DO NOT RE-SIZE
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 2 04 May 2022 9:00 AM US-RNQR-220149
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg and/or 30 mg group and at a higher rate than in the placebo group through Week 16 included anemia, oral candidiasis, pneumonia, and the adverse event of retinal detachment.
The safety profile of RINVOQ through Week 52 was generally consistent with the safety profile observed at Week 16.
Overall, the safety profile observed in patients with AD treated with RINVOQ was similar to the safety profile in patients with RA. Other specific adverse reactions that were reported in patients with AD included eczema herpeticum/Kaposi’s varicelliform eruption.
Eczema Herpeticum/Kaposi’s Varicelliform Eruption
Placebo-controlled Period (16 weeks): Eczema herpeticum was reported in 4 patients (1.6 per 100 patient-years) treated with placebo, 6 patients (2.2 per 100 patient-years) treated with RINVOQ 15 mg and 7 patients (2.6 per 100 patient-years) treated with RINVOQ 30 mg.
12-Month Exposure (Weeks 0 to 52): Eczema herpeticum was reported in 18 patients (1.6 per 100 patient-years) treated with RINVOQ 15 mg and 17 patients (1.5 per 100 patient-years) treated with RINVOQ 30 mg.
Adverse Reactions in Patients with Ulcerative Colitis
RINVOQ was studied up to 8 weeks in patients with moderately to severely active ulcerative colitis in two randomized, double-blind, placebo-controlled induction studies (UC-1, UC-2) and a randomized, double-blind, placebo controlled, dose-finding study (UC-4; NCT02819635). Long term safety up to 52-weeks was evaluated in patients who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (UC-3) and a long-term extension study.
In the two induction studies (UC-1, UC-2) and a dose finding study (UC-4), 1097 patients were enrolled of whom 719 patients received RINVOQ 45 mg once daily.
In the maintenance study (UC-3), 746 patients were enrolled of whom 250 patients received RINVOQ 15 mg once daily and 251 patients received RINVOQ 30 mg once daily.
Adverse reactions reported in ≥2% of patients in any treatment arm in the induction and maintenance studies are shown in Tables 3 and 4, respectively.
Table 3. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 45 mg in Placebo-Controlled Induction Studies (UC-1, UC-2 and UC-4)
Adverse Reaction Placebo RINVOQ 45 mg Once Daily N= 378 (%) N = 719 (%)
Upper respiratory tract infection* 7 9
Acne* 1 6
Increased blood creatine phosphokinase 1 5
Neutropenia* <1 5 Rash* 1 4
Elevated liver enzymes** 2 3 Lymphopenia* 1 3 Folliculitis 1 2 Herpes simplex* <1 2
* Composed of several similar terms ** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury and cholestasis.
Other adverse reactions reported in less than 2% of patients in the RINVOQ 45 mg group and at a higher rate than in the placebo group through Week 8 included herpes zoster and pneumonia.
Table 4. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 15 mg or 30 mg in the Placebo-Controlled Maintenance Study (UC-3)1
Adverse Reaction Placebo RINVOQ 15 mg Once Daily RINVOQ 30 mg Once Daily n = 245 (%) n = 250 (%) n = 251 (%)
Upper respiratory tract infection* 18 16 20
Increased blood creatine phosphokinase 2 6 8
Neutropenia* 2 3 6
Elevated liver enzymes** 1 6 4 4
Herpes zoster 0 4 Folliculitis 2 2
Hypercholesterolemia* 1 2 Influenza 1 3
Herpes simplex* 1 2
Lymphopenia* 2 3 2
Hyperlipidemia* 0 2 2
1 Patients who were responders to 8 weeks induction once of several similar Elevated liver enzymes composed of elevated ALT, AST, ALP, liver transaminases, hepatic enzymes, and cholestasis.
The safety profile of RINVOQ in the long-term extension study was similar to the safety profile observed in the placebo-controlled induction and maintenance periods.
Overall, the safety profile observed in patients with ulcerative colitis treated with RINVOQ was generally similar to the safety profile in patients with RA and AD.
Specific Adverse Reactions
Serious Infections
Induction Studies: In UC-1, UC-2, and UC-4, serious infections were reported in 5 patients (8.4 per 100 patient-years) treated with placebo and 9 patients (8.4 per 100 patient-years) treated with RINVOQ 45 mg through 8 weeks.
Placebo-controlled Maintenance Study: In UC-3, serious infections were reported in 8 patients (6.3 per 100 patient-years) treated with placebo, 8 patients (4.5 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (3.1 per 100 patient-years) treated with RINVOQ 30 mg through 52 weeks.
Laboratory Abnormalities
Hepatic Transaminase Elevations
In studies UC-1, UC-2, and UC-4, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 1.5% of patients treated with RINVOQ 45 mg, and 0% of patients treated with placebo for 8 weeks. AST elevations to ≥ 3 x ULN occurred in 1.5% of patients treated with RINVOQ 45 mg, and 0.3% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN occurred in 0.4% of patients treated with RINVOQ 45 mg and 0% of patients treated with placebo.
In UC-3, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 4% of patients treated with RINVOQ 30 mg, 2% of patients treated with RINVOQ 15 mg, and 0.8% of patients treated with placebo for 52 weeks. Elevations of AST to ≥ 3 x ULN in at least one measurement were observed in 2% of patients treated with RINVOQ 30 mg, 1.6% of patients treated with RINVOQ 15 mg and 0.4% of patients treated with placebo.
Elevations of ALT to ≥ 5 x ULN were observed in 0.8% of patients treated with 30 mg, 0.4% of patients treated with 15 mg, and 0.4% of patients treated with placebo.
Overall, laboratory abnormalities observed in patients with ulcerative colitis treated with RINVOQ were similar to those described in patients with RA.
Adverse Reactions in Patients with Ankylosing Spondylitis
A total of 596 patients with ankylosing spondylitis were treated with RINVOQ 15 mg in the two clinical trials representing 577.3 patient-years of exposure, of whom 228 were exposed to RINVOQ 15 mg for at least one year.
Overall, the safety profile observed in patients with active ankylosing spondylitis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis and psoriatic arthritis.
During the 14-week placebo-controlled period in Trial AS-I, the frequency of headache was 5.4% with RINVOQ 15 mg and 2.1% with placebo. During the 14-week placebo-controlled period in Trial AS-II, the frequency of headache was 3.3% with RINVOQ 15 mg and 1.4% with placebo.
DRUG INTERACTIONS
Strong CYP3A4 Inhibitors
Upadacitinib exposure is increased when RINVOQ is co-administered with a strong CYP3A4 inhibitor (such as ketoconazole and clarithromycin), which may increase the risk of RINVOQ adverse reactions. Monitor patients closely for adverse reactions when co-administering RINVOQ 15 mg once daily with strong CYP3A4 inhibitors.
For patients with atopic dermatitis, coadministration of RINVOQ 30 mg once daily with strong CYP3A4 inhibitors is not recommended.
For patients with ulcerative colitis taking strong CYP3A4 inhibitors, reduce the RINVOQ induction dosage to 30 mg once daily. The recommended maintenance dosage is 15 mg once daily.
Strong CYP3A4 Inducers
Upadacitinib exposure is decreased when RINVOQ is co-administered with strong CYP3A4 inducers (such as rifampin), which may lead to reduced therapeutic effect of RINVOQ. Coadministration of RINVOQ with strong CYP3A4 inducers is not recommended.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Available data from the pharmacovigilance safety database and postmarketing case reports on use of RINVOQ in pregnant women are not sufficient to evaluate a drug-associated risk for major birth defects or miscarriage. Based on animal studies, RINVOQ has the potential to adversely affect a developing fetus. Advise patients of reproductive potential and pregnant patients of the potential risk to the fetus.
In animal embryo-fetal development studies, oral upadacitinib administration to pregnant rats and rabbits at exposures equal to or greater than approximately 1.6 and 15 times the 15 mg dose, 0.8 and 7.6 times the 30 mg dose, and 0.6 and 5.6 times the maximum recommended human dose (MRHD) of 45 mg (on an AUC basis) resulted in dose-related increases in skeletal malformations (rats only), an increased incidence of cardiovascular malformations (rabbits only), increased post-implantation loss (rabbits only), and decreased fetal body weights in both rats and rabbits. No developmental toxicity was observed in pregnant rats and rabbits treated with oral upadacitinib during organogenesis at exposures approximately 0.29 and 2.2 times the 15 mg dose, 0.15 times and 1.1 times the 30 mg dose, and at 0.11 and 0.82 times the MHRD (on an AUC basis).
In a pre- and post-natal development study in pregnant female rats, oral upadacitinib administration at exposures approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and the same as the MRHD (on an AUC basis) resulted in no maternal or developmental toxicity (see Data)
The background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages are 2-4% and 15-20%, respectively.
Report pregnancies to the AbbVie Inc.’s Adverse Event reporting line at 1-888-633-9110, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or ulcerative colitis. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Data
Animal Data
In an oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 5, 25, and 75 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that consisted of misshapen humerus and bent scapula) at exposures equal to or greater than approximately 1.7 times the
15 mg dose, 0.9 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 5 mg/kg/day and higher). Additional skeletal malformations (bent forelimbs/hindlimbs and rib/vertebral defects) and decreased fetal body weights were observed in the absence of maternal toxicity at an exposure approximately 84 times the 15 mg dose, 43 times the 30 mg dose, and 31 times the MRHD (on an AUC basis at a maternal oral dose of 75 mg/kg/day).
In a second oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 1.5 and 4 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that included bent humerus and scapula) at exposures approximately 1.6 times the 15 mg dose, 0.8 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 4 mg/kg/day). No developmental toxicity was observed in rats at an exposure approximately 0.29 times the 15 mg dose, 0.15 times the 30 mg dose, and 0.11 times the MRHD (on an AUC basis at a maternal oral dose of 1.5 mg/kg/day).
In an oral embryo-fetal developmental study, pregnant rabbits received upadacitinib at doses of 2.5, 10, and 25 mg/kg/day during the period of organogenesis from gestation day 7 to 19. Embryolethality, decreased fetal body weights, and cardiovascular malformations were observed in the presence of maternal toxicity at an exposure approximately 15 times the 15 mg dose, 7.6 times the 30 mg dose, and 5.6 times the MRHD (on an AUC basis at a maternal oral dose of 25 mg/kg/day). Embryolethality consisted of increased post-implantation loss that was due to elevated incidences of both total and early resorptions. No developmental toxicity was observed in rabbits at an exposure approximately 2.2 times the 15 mg dose, 1.1 times the 30 mg dose, and 0.82 times the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
In an oral pre- and post-natal development study, pregnant female rats received upadacitinib at doses of 2.5, 5, and 10 mg/kg/day from gestation day 6 through lactation day 20. No maternal or developmental toxicity was observed in either mothers or offspring, respectively, at an exposure approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and at approximately the same exposure as the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
Lactation Risk Summary
There are no data on the presence of upadacitinib in human milk, the effects on the breastfed infant, or the effects on milk production. Available pharmacodynamic/toxicological data in animals have shown excretion of upadacitinib in milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions in the breastfed infant, advise patients that breastfeeding is not recommended during treatment with RINVOQ, and for 6 days (approximately 10 half-lives) after the last dose.
Data
A single oral dose of 10 mg/kg radiolabeled upadacitinib was administered to lactating female Sprague-Dawley rats on post-partum days 7-8. Drug exposure was approximately 30-fold greater in milk than in maternal plasma based on AUC0-t values. Approximately 97% of drug-related material in milk was parent drug.
Females and Males of Reproductive Potential Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ [see Use in Specific Populations]
Contraception Females
Based on animal studies, upadacitinib may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations]. Advise female patients of reproductive potential to use effective contraception during treatment with RINVOQ and for 4 weeks after the final dose.
Pediatric Use
Juvenile Idiopathic Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The safety and effectiveness of RINVOQ in pediatric patients with juvenile idiopathic arthritis, psoriatic arthritis, and ankylosing spondylitis have not been established.
Atopic Dermatitis
The safety and effectiveness of RINVOQ in pediatric patients 12 years of age and older weighing at least 40 kg with atopic dermatitis have been established. A total of 344 pediatric patients aged 12 to 17 years with moderate to severe atopic dermatitis were randomized across three trials (AD-1, AD-2 and AD-3) to receive either RINVOQ 15 mg (N=114) or 30 mg (N=114) or matching placebo (N=116) in monotherapy or combination with topical corticosteroids. Efficacy was consistent between the pediatric patients and adults. The adverse reaction profile in the pediatric patients was similar to the adults [see Adverse Reactions].
The safety and effectiveness of RINVOQ in pediatric patients less than 12 years of age with atopic dermatitis have not been established.
Ulcerative Colitis
The safety and effectiveness of RINVOQ in pediatric patients with ulcerative colitis have not been established.
Geriatric Use
Rheumatoid Arthritis and Psoriatic Arthritis
Of the 4381 patients treated in the five clinical trials, a total of 906 rheumatoid arthritis patients were 65 years of age or older, including 146 patients 75 years and older. Of the 1827 patients treated in the two psoriatic arthritis Phase 3 clinical trials, a total of 274 patients were 65 years of age or older, including 34 patients 75 years and older. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of overall adverse events, including serious infections, in patients 65 years of age and older.
Atopic Dermatitis
Of the 2583 patients treated in the three Phase 3 clinical trials, a total of 120 patients with atopic dermatitis were 65 years of age or older, including 6 patients 75 years of age. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of serious infections and malignancies in those patients 65 years of age or older in the 30 mg dosing group in the long-term trials.
Ulcerative Colitis
Of the 1097 patients treated in the controlled clinical trials, a total of 95 patients with ulcerative colitis were 65 years and older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with ulcerative colitis to determine whether they respond differently from younger adult patients.
DO NOT RE-SIZE
Rash*
5 5
4
4
4
3
3
therapy with RINVOQ 45 mg
daily * Composed
terms **
GGT,
bilirubin, drug-induced liver injury,
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 3 04 May 2022 9:00 AM US-RNQR-220149
Renal Impairment
For patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (eGFR 60 to < 90 mL/min/1.73 m2), moderate (eGFR 30 to < 60 mL/min/1.73 m2), or severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2).
For patients with atopic dermatitis, the maximum recommended dosage is 15 mg once daily for patients with severe renal impairment. No dosage adjustment is needed in patients with mild or moderate renal impairment.
For patients with ulcerative colitis, the recommended dosage for severe renal impairment is 30 mg once daily for induction and 15 mg once daily for maintenance. No dosage adjustment is needed in patients with mild or moderate renal impairment.
RINVOQ has not been studied in patients with end stage renal disease (eGFR <15 mL/min/1.73m2). Use in patients with atopic dermatitis or ulcerative colitis with end stage renal disease is not recommended.
Hepatic Impairment
The use of RINVOQ has not been studied in patients with severe hepatic impairment (Child Pugh C), and therefore not recommended for use in patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, ulcerative colitis, or ankylosing spondylitis.
For patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment.
For patients with ulcerative colitis, the recommended dosage for mild to moderate hepatic impairment is 30 mg once daily for induction and 15 mg once daily for maintenance
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Serious Infections
Inform patients that they may be more likely to develop infections when taking RINVOQ. Instruct patients to contact their healthcare provider immediately during treatment if they develop any signs or symptoms of an infection [see Warnings and Precautions]
Advise patients that the risk of herpes zoster is increased in patients taking RINVOQ and in some cases can be serious [see Warnings and Precautions]
Malignancies
Inform patients that RINVOQ may increase their risk of certain cancers and that periodic skin examinations should be performed while using RINVOQ. Advise patients that exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen [see Warnings and Precautions]
Major Adverse Cardiovascular Events
Inform patients that RINVOQ may increase their risk of major adverse cardiovascular events (MACE) including myocardial infarction, stroke,
and cardiovascular death. Instruct all patients, especially current or past smokers or patients with other cardiovascular risk factors, to be alert for the development of signs and symptoms of cardiovascular events [see Warnings and Precautions]
Thrombosis
Inform patients that events of deep venous thrombosis and pulmonary embolism have been reported in clinical trials with RINVOQ. Instruct patients to seek immediate medical attention if they develop any signs or symptoms of a DVT or PE [see Warnings and Precautions]
Hypersensitivity Reactions
Advise patients to discontinue RINVOQ and seek immediate medical attention if they develop any signs and symptoms of allergic reactions [see Warnings and Precautions].
Gastrointestinal Perforations
Inform patients that gastrointestinal perforations have been reported in clinical trials with RINVOQ and that risk factors include the use of NSAIDS or history of diverticulitis. Instruct patients to seek medical care immediately if they experience new onset of abdominal pain, fever, chills, nausea, or vomiting [see Warnings and Precautions].
Retinal Detachment
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ [see Adverse Reactions]
Laboratory Abnormalities
Inform patients that RINVOQ may affect certain lab tests, and that blood tests are required before and during RINVOQ treatment [see Warnings and Precautions]
Vaccinations
Advise patients to avoid use of live vaccines with RINVOQ. Instruct patients to inform their healthcare practitioner that they are taking RINVOQ prior to a potential vaccination [see Warnings and Precautions]
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that exposure to RINVOQ during pregnancy may result in fetal harm. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions and Use in Specific Populations]
Advise females of reproductive potential that effective contraception should be used during treatment and for 4 weeks following the final dose of upadacitinib [see Use in Specific Populations]
Advise females patients who are exposed to RINVOQ during pregnancy to contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Lactation
Advise women not to breastfeed during treatment with RINVOQ and for 6 days after the last dose [see Use in Specific Populations]
Administration
Advise patients not to chew, crush, or split RINVOQ tablets. Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® is a registered trademark of AbbVie Biotechnology Ltd. ©2019-2022 AbbVie Inc.
Ref: 20071734 Revised: April 2022 LAB-7082 MASTER
US-RNQR-220149
DO NOT RE-SIZE
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 4 04 May 2022 9:00 AM US-RNQR-220149
ECOANSIEDAD: ENFERMOS POR EL PLANETA


TRASTORNOS
Cambio climático, desaparición de biodiversidad, catástrofes naturales, migraciones, conflictos, penurias: para muchas personas, los retos medioambientales se han convertido en la preocupación principal. Y para una parte de ellas, en un nuevo motivo de ansiedad
Por: Charline Schmerber Psicoterapeuta
Revista Puertorriqueña de Medicina y Salud Pública58
Tras el verano de 2019 y sus récords de calor, recibí en mi consulta de psicoterapia en Montpellier a pa cientes que presentaban trastornos de ansiedad de un nuevo tipo. ¿El motivo de su visita? El miedo al futuro; en particu lar, a un futuro sombrío e incierto anuncia do con regularidad en los informes cien tíficos sobre el calentamiento climático y la desaparición de la biodiversidad. Para estas personas, proyectarse en un porve nir así resultaba, simple y llanamente, te rrible, ansiógeno y desesperante. El término ecoansiedad ha aparecido re cientemente en el discurso público y los medios de comunicación. Una encuesta actual revela que el medioambiente ya es la primera preocupación de los fran ceses, por delante del poder adquisiti
vo y los asuntos sociales. ¿Qué implica exactamente esa noción? ¿Cómo debe interpretarla un terapeuta que se enfren ta a menudo a los trastornos de ansiedad «clásicos»? Y ¿qué nos revela este nuevo fenómeno sobre el estado de la sociedad y del planeta?
Los pacientes que acudían a mi consul ta por estas razones me convencieron de que debía de haber un fe nómeno de gran alcance ligado a estas nuevas inquietu des. Por esa razón, decidí rea lizar una encuesta en línea. Para ello publiqué un anuncio en Internet. Iba diri gido a todas aquellas personas que qui sieran responder a cuestionarios sobre su ecoansiedad, tal y como la estuvieran viviendo. En unas semanas, estaba des bordado. A mí, que no tengo el poder
de convocatoria de una institución sani taria, me «llovían» las respuestas al rit mo de 40 al día, hasta alcanzar un total de 1200 entre el 10 de septiembre y el 10 de octubre de 2019. Algunos de los participantes que respondieron al cues tionario hacían declaraciones que invi taban a la reflexión: «A menudo estoy postrado. No consigo proyectarme en el futuro. Soy muy cínico con la gente de mi edad que tiene hijos, porque vivirán en un mundo apocalíptico». Este tipo de posturas deberían hacernos reflexionar. ¿Estas personas se encuentran enfermas como los pacientes que sufren un tras torno de ansiedad «clásicos» o, simple mente, demuestran lucidez ante un futu ro que perciben con más clarividencia que los demás?
¿Quiénes son los ecoansiosos?
Mi encuesta permaneció en línea durante un mes y se difundió, sobre todo, en las redes sociales. En total engloba las declaraciones de 1264 voluntarios, de los cuales 1066 respondieron al cuestionario completo (el 84,3 por ciento de los participan tes). No se trata de una muestra representativa de la población, ya que únicamente las personas muy preocupadas por el tema accedieron a colaborar. Sin embargo, una vez que se comprometieron a dar el paso, el elevado índice de participación demuestra una necesidad real de expresión.
De la muestra se desprenden cuatro tendencias. En primer lugar, se trata de un público femenino en su mayoría (más del 64 por ciento), que proviene de categorías socioprofesionales superiores y reside en ciudades. Más del 60 por ciento de los encuestados pertenecen a la franja de edad comprendida entre los 26 y los 45 años.
¿A las mujeres les concierne en mayor medida esta encuesta por la dimensión emo cional vinculada a ella (sabemos que las mujeres tienen más facilidad que los hom bres para expresar sus sentimientos)? ¿La mayoría de las personas que proceden de categorías socioprofesionales favorecidas y constituían mi muestra era también un indicador del tipo de público sensible a las cuestiones medioambientales? Una pun tualización al respecto: el desarrollo sostenible es el tercer sector de actividad más representado entre mis encuestados, por detrás de las profesiones sanitarias y las re lacionadas con la educación. Por consiguiente, se trata de un sector de la población en contacto directo con la problemática ecológica.
Entre los factores desencadenantes destaca la degradación del ambiente en más del 90 por ciento de los participantes. Esta preocupación genera un alto nivel de ansiedad: una media del 3,78 en una escala del 1 al 5, frente al 3,08 para el estrés cotidiano. Para más del 68 por ciento de los encuestados, la ansiedad relacionada con la situación económica se considera importante o grave. Junto con la pérdida de la biodiversidad, los recursos hidrológicos y el calentamiento climático son los facto res citados con mayor frecuencia.
EN SÍNTESIS : ANSIEDAD POR EL FUTURO
-1-
Ante el futuro amenazante que depara el calentamien to climático, ha surgido un nuevo tipo de angustia: la llamada «ecoansiedad».
-2-
Los ecoansiosos experimen tan una forma de traumatismo por anticipación: el choque pretraumático. Mientras algu nos sufren tetania (espasmos musculares), otros cambian de vida radicalmente.
-3-
La ecoansiedad no es una enfermedad. Antes bien se trata de una conciencia profunda sobre los problemas futuros que desempeña la función de centinela de la sociedad.
Revista Puertorriqueña de Medicina y Salud Pública 59
Incertidumbre globalizada
El origen de las inquietudes que se reflejan en esta muestra va más allá de los meros problemas medioambientales. Se trata de una ansiedad sistémica, anclada tanto en la esfera de la intimidad como en las cues tiones geopolíticas y mundiales. Así pues, para más del 33 por ciento de los partici pantes, los riesgos ansiógenos son múlti ples. «Guerras, hambrunas, enfermedades, medicina/sanidad (disponibilidad de medi camentos), muertes masivas, conflictos, ley del más fuerte, individualismo, riesgo de accidentes nucleares...» Este comentario de uno de los participante resume bien el tono general. La «nebulosa» de los temores pare ce dividirse en seis grandes temáticas, que abarcan desde los miedos individuales a las preocupaciones políticas, pasando por las amenazas sistémicas, medioambienta les, sociales y económicas.
Pero la ansiedad no es la única emoción que sentían los participantes de esta en cuesta. Podemos enumerar 175 términos que utilizaron para definir sus sentimien tos. Entre dichos términos, 114 pueden ca lificarse como «negativos» (miedo, tristeza, inquietud, preocupación, desánimo, entre otros) y 61, como «positivos» (motivación, fascinación, por ejemplo). Entre las emocio nes más representadas se encuentran la ira, la tristeza, la impotencia o el miedo, pero también la esperanza. «Tristeza por perder los tesoros naturales; alegría por poder con templar todavía algunos de ellos; ira hacia los destructores; culpabilidad de formar par te de ellos; miedo por las consecuencias de nuestras sociedades.»
¿Cómo superar la angustia?
Enfrentados a estas emociones y a esa carga de angustia, los ecoansiosos no se quedan de brazos cruzados. Según los re sultados de la encuesta, la mayoría de ellos reacciona con actos, más en concreto, con la palabra. Hablar de sus sentimientos y com partirlos es la estrategia más utilizada. Así, para más del 60 por ciento, conversar con personas de su entorno les permite no que darse «enfrascados» en su angustia. Se trata de evocar las preocupaciones, transmitir las amenazas que se perciben e intentar sensibi lizar a los demás sobre uno mismo, ya sean amigos, miembros de la familia, compañeros o conocidos de las redes sociales.
En general, se observan tres tipos de re acciones en esta muestra de personas: la parálisis, una ambivalencia entre las ganas de actuar y el sentimiento de impotencia o comenzar a moverse y pasar a la acción. La primera reacción concierne al 30 por ciento de los participantes, a quienes sen tir ecoansiedad les hace experimentar una forma de tetania (espasmos musculares en las extremidades) que les impide actuar. Pre domina la duda, así como la dificultad de tomar decisiones para el futuro. En estas per sonas aparece una inseguridad existencial y de identidad que suele afectar la vida profe sional, la cual es cuestionada (la persona ya no se identifica con su trabajo). La problemá tica de los niños y el futuro es habitual, pues les resulta complicado proyectarse en el fu turo. Algunos participantes ven afectado su bienestar cotidiano, pues sufren obsesiones, insomnio o mecanismos compensatorios, como el juego o las compras compulsivas, el consumo de alcohol excesivo, así como difi cultad de concentración y dudas bloquean tes acerca de elecciones fundamentales, como comprar una vivienda, tener un bebé o la orientación profesional.
El segundo tipo de reacción es la ambi valencia. Para el 9 por ciento de los parti cipantes se trata de una oscilación entre las ganas de actuar y la impotencia. Los encuestados mencionan fluctuaciones im portantes entre estos dos extremos y, a me nudo, la incapacidad de encontrar un punto medio. Estas personas recuerdan a aquellas que acaban de atravesar un duelo: tras una fase descendente que empieza por un shock y una incredulidad, experimentan una fase ascendente en la que pasan a la acción.
En resumen, al 61 por ciento de los parti cipantes la ecoansiedad les activa. Para un 87,43 por ciento, desencadena el deseo de realizar acciones concretas y precisas en el
plano individual y colectivo. Aplican o re fuerzan una transición ecológica dentro del hogar (residuos cero, productos locales, me dios de transporte más responsables, entre otras medidas). A veces se trata de un nue vo lugar donde habitar, más autónomo, de un cambio de trayectoria profesional para vivir con más coherencia, de adquirir nue vas habilidades y de no tener otro hijo. La naturaleza empieza a ocupar un lugar pre ponderante (limpieza de espacios naturales, creación de huertos de barrio, entre otras acciones). Algunos se dedican al arte para sublimar su angustia o comienzan un traba jo psicológico de introspección personal. En la mayoría de los casos, se orientan ha cia la colectividad, desarrollan o refuerzan su participación en el mundo asociativo e inten tan sensibilizar a su entorno de los problemas medioambientales. La militancia es una de las acciones más mencionadas (en un partido político o movimiento social, como Rebelión contra la Extinción). Ya sea la acción indivi dual o colectiva, ante todo es importante que cada uno pueda aceptar sus emociones, que las acepten otras personas, y que esto tenga lugar en un contexto específico para ello o de manera informal. No hay que rechazar los pequeños gestos cotidianos, pues generan un sentimiento de utilidad muy valioso frente a la impotencia. Reflexionar sobre lo que tiene sen tido para uno mismo a medio y largo plazo ayuda a recuperar la coherencia en el camino vital. Sin duda, de los cuestionarios se des prende que un mundo dominado por el indivi dualismo no nos permitirá afrontar las distintas luchas que nos esperan. La noción de ayuda mutua parece una estrategia imprescindible para construir un futuro duradero. Otro con cepto consiste en un proceso de reconexión con la naturaleza, considerado esencial por el 96 por ciento de los encuestados. Estos mencionan una correlación entre medioam biente y salud física y psíquica. Cuando se les pregunta por las emociones asociadas a los momentos vividos en un medio natural, descri ben una gama de sentimientos positivos, con cinco términos que se repiten constantemente: calma, serenidad, paz, bienestar y alegría.
Cuidado con el desgaste ecológico
Para un terapeuta, la ecoansiedad repre senta un desafío: ¿cómo debe abordarse esta nueva forma de sufrimiento? En mi con sulta intento identificar el malestar de los pa cientes y las causas relacionadas con él. No niego la realidad de lo que les trae hasta mi consulta. No busco poner pomada en su dolor. Me doy cuenta de que para ellos ya
Revista Puertorriqueña de Medicina y Salud Pública60
Las fuentes de ansiedad Los Grandes Motivos de Preocupación Riesgos sanitarios (epidemias) Violencia Crisis económica Escasez de alimentos y agua Guerra Otras causas 6 % 7 % 7 % 8 % 12 % 60 %
supone un alivio encontrarse cara a cara fren te a una persona que reconoce lo que están viviendo y los acepta como son.
En la actualidad, observo numerosos perfiles de pacientes. Algunos llegan con un «desgas te ecológico». Su impresión de estar ascen diendo una montaña infranqueable en las ac ciones cotidianas que a menudo emprenden en su vida profesional o asociativa provoca el agotamiento de sus capacidades cognitivas. Ello implica, en ocasiones, la incapacidad de elegir, aunque solo se trate de un destino vacacional. Por esa razón trabajo con ellos para restaurar el sentimiento de identidad. ¿Qué desean? ¿Qué necesitan? ¿Qué tiene sentido para ellos? Por lo general, se trata de personas que denominamos «límite», que presentan dificultades para percibir sus pro pias limitaciones, para definir su campo de acción. También eso es necesario trabajarlo.
Otra categoría de pacientes acude a mí a través de la colapsología, una corriente de análisis del cambio climático y de la socie dad que plantea la posibilidad de que se desplome el sistema de producción industrial, de las finanzas y de las estructuras sociales, lo cual provocaría a la vez el agotamiento de los recursos naturales y cambios drásticos del ecosistema debido al desajuste climático y la degradación de la biodiversidad. Son perso nas generalmente inquietas, con una urgen cia y una necesidad de «prepararse», con una especie de bulimia de información que también les lleva a sobrepasar los límites de su poder de asimilación de datos y reacción específica a esos problemas. Suelo orientar las hacia el camino de la «desintoxicación mediática». No se trata de caer en la nega ción de la realidad, sino de protegerse ante el exceso de información, que acaba por ser nefasto para el equilibrio emocional de la persona. Sobre todo les propongo soltar lastre momentáneamente respecto a las cues tiones del futuro a largo plazo y centrarse en los actos a corto y medio plazo. El objetivo consiste en vivir mejor la cotidianeidad, salir de la lógica de la urgencia y efectuar peque ños gestos en el presente, lo cual reinstaura la sensación de que se recuperaran las riendas de los acontecimientos.
Me parece importante tener en cuenta toda la dimensión emocional de los pacientes, que va más allá de la ansiedad, ya que enmarca los factores afectivos en un cuadro racional, mediante la palabra y la escucha atenta, lo cual implica seguir el ritmo de las palabras del paciente, sin imponer nada. A veces ne cesitan permanecer en la oscuridad del céle bre «nada tiene arreglo» durante un tiempo.
Para que aparezca la luz, hay que aceptar que se necesita atravesar zonas de sombra.
Constato con regularidad que un pará metro muy relevante es localizar dónde se sitúa el deseo del paciente, qué constituye su energía e impulso vital, pues estos pue den activarlo y aportarle recursos. Es lo que generará nuevos proyectos. Por tanto, la fun ción del terapeuta, en la medida de lo posi ble, es acompañar estas iniciativas haciendo que la persona visualice la forma concreta y real que podrían tener.
Para concluir, animo a mis pacientes a incor porar en su día a día momentos para una me ditación que incluya técnicas de respiración (como la sofrología), así como ratos dedica dos al ser y no al hacer. No hay que buscar siempre la acción o llenar la inacción, sino hallar instantes para estar con los tuyos, reen contrar el sentido del tiempo que pasa, de la belleza y la contemplación.
Los centinelas de lo vivo
Queda pendiente una cuestión de fondo para los profesionales de la psique. Frente a la ecoansiedad nos preguntamos: ¿dónde se encuentra el límite entre un trastorno que indicaría una disfunción psíquica (una per sona con ansiedad que tiene palpitaciones cardíacas sin motivo aparente o la impresión de ahogarse o asfixiarse) y una angustia pro vocada por la proyección dolorosa en un futuro hecho de incertidumbre y amenazas? Tenemos poca perspectiva ante el fenómeno y una cierta dificultad para identificarlo. Sin duda, las personas ecoansiosas tienen una mayor sensibilidad para el medioambien te que sus congéneres. Desde ese punto de vista, la ecoansiedad podría considerarse la consecuencia de una mirada lúcida hacia el mundo actual, más que un problema que con vendría tratar desde una perspectiva médica. De cualquier modo, este discurso merece ser revisado cuando la ansiedad es tan grave que inmoviliza al afectado o este cae en una depresión grave con ideas suicidas.
Podría plantearse un debate similar sobre las enfermedades respiratorias vinculadas a la contaminación. En el conjunto de la pobla ción existen personas más sensibles que otras a las impurezas del aire y a los gases tóxicos. En una gran aglomeración, estas son las per sonas que acuden más a menudo a urgen cias o a consulta por asma o insuficiencias respiratorias. Sin embargo, no son en un principio individuos con trastornos, pero su umbral de sensibilidad a los agentes conta minantes es más bajo y nos demuestran que la composición del aire ya no responde a las
necesidades del organismo.
La idea de centinela sanitario se convier te en un concepto esclarecedor dentro de un mundo cuyos dife rentes parámetros medioambientales (químicos, sociales, atmosféricos, alimentarios, etcétera) tras pasan con frecuencia nuevos límites. La imagen más ilustrativa, que obtuve del psi quiatra y psicoterapeuta Christophe André, es la del canario que los mineros se lleva ban a las minas de carbón para detectar las emanaciones de monóxido de carbono, un gas muy tóxico, incoloro e inodoro. Los canarios, más sensibles que los humanos a este gas, sucumbían antes, una señal que todo el mundo interpretaba como la obliga ción de evacuar urgentemente el yacimien to. Si los ecoansiosos son los canarios de nuestros desajustes medioambientales, qui zá nos convendría escucharlos.
Referencia:
«Solastalgia», a new concept in health and identity. Glenn Albrecht en PAN: Phi losophy, Activism, Nature, vol. 3, pág. 45, 2005.
UNA ANGUSTIA CON VARIAS FACETAS
Los riesgos que citan los afectados de ecoansiedad se encuentran relacionados con seis grandes temáticas que engloban numerosos problemas referentes al funcio namiento del mundo.
Contexto social
Miedo a que se constituya una población «a dos velocidades» (una despierta; la otra poco informada, inactiva, sumida en la negación) y al riesgo de tensiones e injusticias sociales.
Situación personal
Riesgos asociados a la naturaleza humana, a las cuestiones existenciales (soledad, muerte, extinción de la especie humana), al futuro, a los hijos y a las condiciones de vida.
Esfera medioambiental
Riesgo de catástrofes naturales e industriales (entre ellos, las nucleares), contaminación del agua y suelo, problema de los residuos, impacto en el bienestar animal.
Coyuntura económica
Miedo a que se desplome el sistema financiero.
Situación política
Situación política nacional, internacional; miedo al éxito del extremismo, a perder libertades individuales y a la gestión de las migraciones climáticas.
Amenazas sistémicas
Conjunto de fenómenos que pondrían en peligro las necesidades primarias: alimentación, salud y seguridad.
Revista Puertorriqueña de Medicina y Salud
Pública 61
CUIDADO DEL PACIENTE CON DIABETES EN TEMPORADA DE HURACANES
-Estudios revelan la importancia de tener un plan de acción adecuado-
En plena temporada de huracanes -que para Puerto Rico es la mitad del año- y luego de haber sufrido dos huracanes de proporciones mayores como Irma y María en el 2017, una de las principales preocu paciones cada año son los pacientes con condiciones crónicas.
Según datos del Departamento de Salud de Puerto Rico, se estima que para el 2021 unos 537,000 millones de adultos entre las edades de 20 a 79 años tenían un diagnós tico de diabetes. En la Isla casi medio millón de adultos, o dos de cada 13, padecen esta condición crónica según cifras hasta el 2020.
Ante este panorama, y en el pico de la temporada de huracanes, es inevitable preguntarse: ¿saben las personas con diabetes cómo prepararse para enfrentar una tormenta o huracán? ¿Existe un plan para que este 15.8% de la población pueda seguir con su condición bajo control? ¿Estamos preparados para proteger a estos
pacientes en el caso de enfrentarnos a un evento atmosférico de grandes propor ciones?
Las personas con diabetes no deben interrumpir sus medicamentos, ni siquiera durante una emergencia. Por lo tanto, es un tema que se debe tocar entre familiares y médicos, para tener un plan de acción que contribuya a manejar esta condición adecuadamente en lo que la emergencia culmina.
Como hemos visto en otras situaciones, la población de 65 años o más tiene el impacto mayoren este tipo de situaciones. Para el 2020 se observó en la Isla que, de las personas con diabetes, un 38% pertenecía a ese grupo de edad. Además, dichas estadísticas revelan que estos pacientes tienen comorbilidades como hipertensión, asma, colesterol elevado y enfermedad renal, entre otras, lo cual puede complicar su salud.

Revista Puertorriqueña de Medicina y Salud Pública62
1. Guardar copia de su plan médico e identificaciones.
2. Tener disponible suficiente medicamento de mantenimiento en el hogar. Los suplidos a 90 días son la alternativa ideal en estos casos. Puede preguntar en la farmacia si tiene remanente en su receta para este despacho o solicitar a su médico una receta para 90 días.
3. Preparar un botiquín con artículos básicos que le puedan ayudar en el caso de una cortadura o lesión, como: vendajes adhesivos (curas), gasas estériles, toallitas antisépticas, alcohol y crema triple antibiótico, entre otros.
4. Mantener los medicamentos lejos de la exposición directa del sol y calor. En el caso de un evento atmosférico, mantenlos en un envase o bolsa con cierre hermético, para protegerlos del agua.
5. La insulina puede usarse aún sin estar refrigerada, por al menos 28 días, siempre y cuando se mantenga en una temperatura entre 59°F y 86°F. Es importante que no esté expuesta a tempera turas extremas, no se congele y mantenerla lejos del calor excesivo y exposición directa del sol. . Al conseguir insulina nueva y restablecerse el servicio eléctrico, debe desechar la insulina que no estuvo en refrigeración. Si la insulina muestra cambios en su apariencia, ya sea en color, textura o forma, debe consultar con su médico o farmacéutico de inmediato.
6. Tener un glucómetro con suficientes tiras para monitorear los niveles de glucosa en la sangre.
7. Tener alimentos no perecederos que puedan comerse a temperatura ambiente sin tener que cocinarlos. Prefiera aquellas alternativas bajas en azúcar, sal y carbohidratos.

8. Tener agua potable almacenada para evitar deshidratación. No tomar suficiente líquido puede elevar los niveles de azúcar en sangre.
9. Evitar bebidas con cafeína, energizantes y el alcohol, ya que pueden elevar el azúcar en sangre.
Además, es importante prestar atención a la salud emocional que es parte esencial de una salud completa. Altos niveles de estrés y ansiedad pueden contribuir a un aumento de azúcar en la sangre. De hecho, el Departamento de Salud revela que en la Isla un 22% de los pacientes con diabetes mayores de 65 años presenta depresión, siendo una de las comorbilidades de mayor prevalencia.
¿QUÉ SE DEBE HACER?
• Mantenerse informado con medios confiables.
• Tener a la mano los números de teléfono de familiares y vecinos, así como de médicos, líneas de servicio y apoyo del plan médico, hospitales, farmacias, servicio de ambulancia, y entidades locales que atiendan emergencias.
• Tener una lista de las condiciones de salud de todos en el hogar, y sus medicamentos, y tenerlos a la mano.
• Tratar de descansar para evitar la fatiga.
• Mientras sea posible, mantener comunicación con familiares y amigos. Y de ser necesario, pedir ayuda y compañía.
Ante cualquier emergencia, la tranquilidad de estar preparado es lo que contribuye a disfrutar de salud completa.
En MCS buscamos contribuir a que toda situación de salud inesperada, sobre todo durante períodos de emergencia pueda ser atendida.
REFERENCIAS: Diabetes en PR Boletín 2020 https://www.salud.gov.pr/CMS/ DOWNLOAD/5675#:~:text=2%20 de%20cada%2013%20adultos,viven%20 con%20Diabetes%20(2020).&text=Du rante%20el%202020%2C%20se%20 observa,a%C3%B1os%20o%20m% C3%A1s%20(38.0%25).
Departamento de Salud, Programa para la Prevención y el Control de la Diabetes https://www.salud.gov.pr/CMS/430 Centros para el Control y la Preven ción de Enfermedades (CDC) https://www.cdc.gov/es/disasters/pdf/ cdc_hurricane_preparednesssafetymes saging_july2018_spanish_508.pdf
ANTE ESTE PANORAMA, MCS RECOMIENDA:
Revista Puertorriqueña de Medicina y Salud Pública 63
20 de agosto de
20 de agosto de 2022 Creando Fuerzas 2022 los desafíos del VIH”
20 de agosto de 2022
24 al 27 de agosto de 2022
Caribe Hilton Hotel San Juan, PR
Ponce Hilton Hotel & Casino Ponce, PR
Hotel San Juan Marriott Resort & Stellaris Casino, San Juan, PR
Anfiteatro de la Escuela de Enfermería del Recinto de Ciencias Médicas
IC Planners ivettecolon@icplannerspr.com / 787-504-3655
AMEC 787-289-8989 amec@amec-pr.com

Ariadne Comualda, KAZ Event 787-960-5875 kaz@kaz-events.com // Lazos@prconcra.net
Conferencia
Conferencia
Simposio
Royal Sonesta SJ Hotel, Carolina, PR Planners- Merna Morales 787-645-9914 bplanner21@gmail.com Hilton Hotel San Juan, PR Planners ivettecolon@icplannerspr.com / 787-504-3655
Revista Puertorriqueña de Medicina y Salud Pública64 www.medicinaysaludpublica.com EVENTOS Y CONVENCIONES AGENDA MÉDICA 2022 Puerto Rico Fecha Actividad Lugar Contacto TIPO DE EVENTO 11 al 14 de agosto Convención Sunshine Seminar 2022 Wyndham Grand Rio Mar Río Grande, PR PRO GYN (787) 763-0838 Convención 13 de agosto 1st. Allergy and Immunology Symposium in WESTAsociación Puertorriqueña Médicos Alergistas Rincón Beach Resort IC Planners ivettecolon@icplannerspr.com / 787-504-3655 Simposio 13 al 14 de agosto Convención Sociedad de Cirujanos Vasculares y Endovasculares de PR Hotel Sheraton Convention Center, San Juan, PR AMEC 787-289-8989 amec@amec-pr.com Simposio 18 al 21 de agosto de 2022 Convención anual del Colegio de Farmacéuticos de PR Wyndham Grand Rio Mar Río Grande, PR Colegio de Farmacéuticos de PR 787-753-7157 Convención 20 de agosto de 2022 Convención anual de la Asociación de Gastroenterología y Hepatología Pediátrica de PR
Convención
2022 The Arrhythmia Group: 3rd ElectroCardiovascular Conference 2022
Conferencia: Lazos
“Superando
2nd Annual Palliative Care Symposium: Selected Issues in Clinical Ethics
Convención Sociedad Puertorriqueña de Nefrología
Business
/
Convención Agosto 26 al 28 de 2022 10mo Congreso anual Coalición de Asma y otras condiciones Respiraciones Crónicas de PR Caribe
IC
Congreso Agosto 26 al 28 de 2022 Convención Anual de la Academia Puertorriqueña de Neurología: “Jornadas Neurológicas” Hotel Hyatt Regency Reserve, Rio Grande, PR AMEC 787-289-8989 amec@amec-pr.com Convención 3-5 de septiembre 2022 Convención Anual de la Asociación de Médicos Pediatras de la Región Oeste (AMPRO) Mayagüez Resort & Casino AMPRO ampropediatras@gmail.com Convención 24 de septiembre 2022 Conferencia Epilepsia del Caribe Embassy Suite Isla Verde Sociedad Puertorriqueña de Epilepsia info@sociedadepilepsiapr.org / (787) 782-6200 Conferencia 2-3 de Septiembre 2022 Convención Sociedad Puertorriqueña de Cardiología Intervencional Por confirmar Sociedad Puertorriqueña de Cardiología Intervencional Enid Rivera / (804) 774-6326 Convención Septiembre 2022 Pain Management Conference-Pain Management Academy Por confirmar AMEC 787-289-8989 amec@amec-pr.com Conferencia ACTIVIDADES ESTÁN SUJETAS A CAMBIO
EVENTOS Y CONVENCIONES AGENDA MÉDICA 2021
www.medicinaysaludpublica.com
Puerto Rico
Fecha Actividad Lugar
9 al 10 de septiembre
Octubre 2022
17 al 20 de octubre de 2022
17 al 20 de octubre de 2022
Octubre 2022
Octubre 2022
26 al 29 de octubre de 2022
15 de octubre
11 -13 noviembre
18 al 20 de noviembre de 2022
2do Congreso y Elecciones Sociedad Puertorriqueña de Neumología
Convención anual de la Asociación de Hematología y Oncología Médica de PR (AHOMPR)
Convención annual de la Asociación de Hospitales de PR
Ponce Hilton Hotel & Casino Ponce, PR
Hyatt Regency Grand Reserve PR Río Grande, PR
Sheraton Centro de Convenciones
Convención annual de la Asociación de Hospitales de PR Sheraton Centro de Convenciones
Cardiovascular Innovation Forum- Asociación de Cardiólogos del Noroeste
Convención Psiquiátrica anual - Asociación de Psiquiatras de Bayamón (APREBA)
Convención anualPuerto Rico Urological Association
Convención anual de la Asociación Puertorriqueña de Médicos Alergistas
Convención Anual de la Asociación de Médicos Pediatras de la Región Este (AMPRE)
Convención Anual Sociedad Puertorriqueña de Neumología
Contacto
Business Planners- Merna Morales 787-645-9914 / bplanner21@gmail.com
Germaine Quiñones ahomprgq@gmail.com / 787-608-1477
Business Planners- Merna Morales 787-645-9914 / bplanner21@gmail.com
Business Planners- Merna Morales 787-645-9914 / bplanner21@gmail.com
Por confirmar AMEC 787-289-8989 amec@amec-pr.com
Por confirmar AMEC 787-289-8989 amec@amec-pr.com
Caribe Hilton Hotel San Juan, PR
Sheraton Convention Center Hotel San Juan, PR
Embassy Suites Hotel Isla Verde
Wyndham Grand Rio Mar Río Grande, PR
Aixa Vélez genteinc@gmail.com / 787-649-7681
IC Planners ivettecolon@icplannerspr.com / 787-504-3655
Business Planners- Merna Morales 787-645-9914 / bplanner21@gmail.com
Business Planners- Merna Morales 787-645-9914 bplanner21@gmail.com
Noviembre Pro Events- Rafy Nieto (787) (866)
TIPO DE EVENTO
Educación Continua
Convención
Convención
Convención
Educación Continua
Convención
Convención
Convención
Convención
Centro de Convenciones de Médicos de
Diciembre
Revista Puertorriqueña de Medicina y Salud Pública 65
/
Convención
ElectroCardio Workshops Conference Arrhytmia Group Por confirmar AMEC 787-289-8989 amec@amec-pr.com Conferencia Noviembre Convención anual de la Academia Puertorriqueña de Neurología Por confirmar AMEC 787-289-8989 amec@amec-pr.com Convención 2 y 3 de diciembre Mini Convención de la Sociedad Dermatológica de PR Por confirmar RN
368-7939 /
232-2068 rafinieto@rnproevents.com Convención Diciembre 2022 Convención Anual SPED Por confirmar Educational Partners & Coaching 787-646-0780 / perez.vilma@gmail.com Convención
2022 Convención anual Colegio de Médicos Cirujanos de PR
de PR San Juan, PR Colegio
Cirujanos
PR 787-751-5979 / info@colegiomedicopr.org Convención ACTIVIDADES ESTÁN SUJETAS A CAMBIO
MSP SERVICIO PÚBLICO
El suplemento de género del estudio “Beyond Intervention” analiza por qué es urgente mejorar la equidad en salud para entender la salud cardiovascular de las mujeres. Las enfermedades cardio vasculares (ECV) son la principal causa de muerte en el mundo, con alrededor de 18 millones de muertes por año. Al respecto, la OMS indica que más de las tres cuartas partes de las muertes por estas enfermedades ocurren en
países de ingreso bajo y medio.
La Asociación Estadounidense del Corazón indica que, en Puerto Rico, las enfermedades cardíacas son la principal causa de muerte y tienen varias condiciones de riesgo asociadas, como presión arterial alta, colesterol alto y estilos de vida nocivos, que se pueden prevenir o controlar. La concien tización y la implementación de tecnología son necesarias para combatir estas enfermedades.
Con el llamado de ¡Vacuna ción al día! se inauguró el nuevo centro de vacunación de VOCES Coalición de Inmunización y Promoción de la Salud de Puerto Rico junto al Departamento de Salud (DS), en el segundo nivel de Plaza Las Américas, para continuar recibiendo a la población desde los 3 años en adelante con el fin de que todos obtengan protección contra el COVID-19 y tengan su vacunación al día.
“Vacunación al día significa tener toda la serie primaria
recomendadas para cada edad , así como los refuerzos pertinentes. La vacunación sigue siendo la mejor opción de protección y evitar complicaciones. Por fin hemos regresado a nuestros trabajos y esta semana regresan nuestros niños y jóvenes a sus centros de estudios y es gracias a los avances de la vacunación. Sigamos acercando la normalidad. Tengamos nuestra vacunación al día. No regresemos al pasado”, expresó Lilliam Rodríguez Capó, CEO y fundadora de VOCES.
Como parte del compromiso de brindar servicios de salud de excelencia y calidad el Sistema de Salud Episcopal San Lucas adquirirá una institución hospitalaria y un centro de cuidado prolongado en San Juan con una inversión millonaria.


“Hoy es un día histórico para el Sistema de Salud Episcopal (SSE), con este proyecto empren demos un nuevo camino para extender nuestros servicios a los residentes de la zona metropoli tana. Hoy inicia la última etapa del proceso de lo que será el Hospital Episcopal San Lucas Metro y el Centro de Cuidado Prolongado San Lucas. Con la adquisición alcanzamos un sueño y una meta dentro de nuestro plan de desarrollo estratégico”, expresó el obispo Rafael Morales Maldonado, presidente de las Juntas de Directores.
El acuerdo de adquisición incluye la compra de todos los activos inmuebles y muebles que componen el hospital secundario con licencia operacional para 60 camas, una sala de emergencia, laboratorio clínico y las principales modalidades de radiología. Además, cuenta con servicios de farmacia institucional y los servicios de nutrición, entre otros.
“Nos sentimos honrados de servir a nuestros hermanos. Con estos nuevos servicios continuamos afianzando nuestro compromiso con la industria de la salud en la Isla y procurar el bienestar holístico de los puertorriqueños que tanto lo necesitamos”, destacó Juan Salazar Trogolo, principal oficial ejecutivo del SSE.
En esta ocasión, la marca San Lucas continuará impactando con excelentes servicios hospitalarios, por los cuales se ha distinguido por los pasados 115 años, y expande su alcance con la incursión de los servicios del centro de cuidado prolongado donde se brinda atención especializada a pacientes internados que por su condición no necesitan supervisión médica directa que provee un hospital.

“Con la llegada de San Lucas al área metro reafirmamos que continuaremos brindando servicios de salud con la calidad
y seguridad que el paciente necesita. Sobre todo procuraremos que el cuidado se brinde con la compasión, eficiencia, integridad y respeto que distingue a la Facultad Médica y personal que forman parte de la familia de San Lucas”, manifestó el Lcdo. Elyonel Pontón Cruz, Director Ejecutivo Operacional del Centro Médico Episcopal San Lucas, quien será la entidad administradora de los nuevos servicios adquiridos.
El Hospital Episcopal San Lucas Metro y el Centro de Cuidado Prolongado San Lucas comenzará operaciones el próximo mes de septiembre.
Para información sobre el Hospital Episcopal San Lucas Metro y otros servicios puede visitar su página web www.sanlucaspr.org, seguirles a través de Facebook e Instagram/Hospita lEpiscopalSanLucasMetro o comunicarse al 787-844-2080.
EL PAPEL DE LA PERSPECTIVA DE GÉNERO EN EL TRATAMIENTO DE ENFERMEDADES CARDIOVASCULARES
SISTEMA DE SALUD EPISCOPAL SAN LUCAS ADQUIRIRÁ INSTITUCIÓN HOSPITALARIA Y DE CUIDADO PROLONGADO EN SAN JUAN
¡VACUNACIÓN AL DÍA,AHORA MÁS QUE NUNCA!
Revista Puertorriqueña de Medicina y Salud Pública66
TRANSPARA: ÚLTIMO EN INTELIGENCIA PARA LA TEMPRANA CANCER DE SENO
Grupo Hospitalario anunció la adquisi ción de TRANSPARA; un sistema de inteligencia artificial que tiene la capacidad de pre-analizar e identificar las caracte rísticas de las imágenes radiográficas (sospechosas) de mamografía y sonoma mografía del seno. TRANSPARA organiza el orden de lectura del radiólogo para que éste pueda leer primero aquellas imágenes que tienen mayor riesgo de cáncer de seno y cáncer oculto. El Dr. Samuel Padua, radiólogo especialista en imágenes del seno en el Women ‘s Imaging Institute de Manatí Medical Center asegura que esta tecnología ayuda a salvar vidas pues facilita la detección temprana de cáncer de seno. “Cuanto antes se detecte el cáncer de mama, más fácil es tratarlo. Por tanto, es de vital importancia que todas las mujeres tengan acceso a las últimas innovaciones capaces de detectar el cáncer en sus estadios más tempranos. Un cáncer diagnos ticado a tiempo tiene una alta probabilidad de supervivencia versus uno detectado tarde”. Dijo el Dr. Padua.
“TRANSPARA toma un rol protagónico
en la manera en que yo le doy lectura a los estudios; la computadora ayuda a encontrar los casos más complejos, y se atienden de inmediato, porque esta tecnología me organiza los casos según la prioridad que tienen. La tecnología va avanzando, y debemos usarla en nuestro beneficio”, explicó.
Es importante resaltar que, los expertos coinciden en que el cáncer de mama es actualmente el tipo de cáncer más frecuente en las mujeres a nivel mundial, y su incidencia va en aumento, por ello el especialista enfatizó en que este mecanismo de inteligencia artificial, “definitivamente, va a agilizar el proceso a través de detección temprana para salvar vidas. Cabe resaltar ese miedo que le tenemos a la inteligencia artificial, pero siempre la decisión final va a recaer sobre el doctor, y la computadora no va a tomar decisiones por mí, pero sí va a ser mi complemento; me va a ayudar a organizar, a identificar y a levantar esa bandera en donde debo poner más atención” concluyó.
El Dr. Samuel Padua destacó que el CT
Radiology Complex Imaging Center del Bayamón Medical Center, al igual que el Women’s Imaging Institute del Manatí Medical Center del Grupo Hospitalario son los primeros centros de imágenes en Puerto Rico en implementar la innovadora tecnología de TRANSPARA, y que cada paciente que se realice su mamografía o sonomamografía en estos centros, contará con el beneficio de tener a su disposición la eficiencia y prioridad del este mecanismo para un diagnóstico rápido y certero.
Admitió que dar el diagnóstico no es sencillo, pero “pero siempre le digo a las pacientes que tenemos las armas para tratarlo, contamos con oncólogos y cirujanos excelentes en Puerto Rico para operar, tratar y curar el cáncer de seno. Una mamografía al año puede salvar la vida de una mujer. Es importante reconocer los riesgos, saber si tienen historial familiar de cáncer de seno, o si hay algún defecto genético que pueda disponer a la paciente a cáncer de seno para hacer las pruebas pertinentes y poner manos a la obra.” Concluyó el especialista.
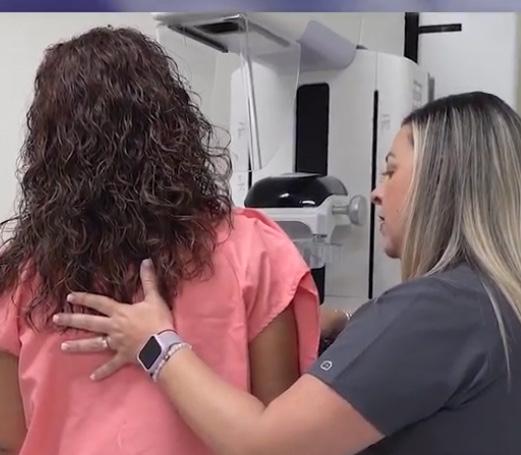
Revista Puertorriqueña de Medicina y Salud Pública 67
LO
ARTIFICIAL
DETECCIÓN
DE

Dr.
Dr.
Dr. Eric Adler
Cirujano Plástico
Dr. Eric Adler



Cirujano Plástico
Dr.


Dr. Álvarez
Dr. Álvarez

Dr. Álvarez Romagosa Ginecólogo/Obstetra
Dr. Álvarez Romagosa Ginecólogo/Obstetra









Salud completa es un equilibrio entre tu salud física, mental y emocional.
Salud completa es un equilibrio entre tu salud física, mental y emocional.
Salud completa es un equilibrio entre tu salud física, mental y emocional.
Salud completa es un equilibrio entre tu salud física, mental y emocional.
Salud completa es un equilibrio entre tu salud física, mental y emocional.
Salud completa es un equilibrio entre tu salud física, mental y emocional.
Con MCS cuentas con una amplia red de médicos y hospitales para atender tus necesidades de salud y con beneficios innovadores que transforman tu calidad de vida.
Con MCS cuentas con una amplia red de médicos y hospitales para atender tus necesidades de salud y con beneficios innovadores que transforman tu calidad de vida.
Con MCS cuentas con una amplia red de médicos y hospitales para atender tus necesidades de salud y con beneficios innovadores que transforman tu calidad de vida.
Con MCS cuentas con una amplia red de médicos y hospitales para atender tus necesidades de salud y con beneficios innovadores que transforman tu calidad de vida.
Con MCS cuentas con una amplia red de médicos y hospitales para atender tus necesidades de salud y con beneficios innovadores que transforman tu calidad de vida.


Con MCS cuentas con una amplia red de médicos y hospitales para atender tus necesidades de salud y con beneficios innovadores que transforman tu calidad de vida. TECUIDA
 Dr.
Dr.
787.758.2500 | mcs.com.pr @MCSPuertoRico
SALUD QUE TECUIDA
Eric Adler Cirujano Plástico Facial
Romagosa Ginecólogo/Obstetra 787.758.2500 | mcs.com.pr @MCSPuertoRico
SALUD QUE TECUIDA Dr. Eric Adler Cirujano Plástico Facial
Romagosa Ginecólogo/Obstetra 787.758.2500 | mcs.com.pr @MCSPuertoRico
SALUD QUE TECUIDA
Eric Adler Cirujano Plástico Facial Dr. Álvarez Romagosa Ginecólogo/Obstetra 787.758.2500 | mcs.com.pr @MCSPuertoRico
SALUD QUE
Eric Adler Cirujano Plástico Facial
Álvarez Romagosa Ginecólogo/Obstetra 787.758.2500 | mcs.com.pr @MCSPuertoRico
SALUD QUE TECUIDA
Facial
787.758.2500 | mcs.com.pr @MCSPuertoRico
SALUD QUE TECUIDA
Facial
C A R R E R A P OR L A SALU D DE L ÓRGA NO M Á S I M P ORTA N T E: LA PIEL
Humanismo, medicina, y tres décadas en la carrera dermatoló gica en beneficio de los pacientes del País.
La oración no le hace justicia, pero aspira a compilar de la manera más clara la vida de la Dra. Damaris Torres Paoli, quien con valentía asumió la presidencia de la Sociedad Dermatológica de Puerto Rico, en medio de un trastoque salubrista debido a una pandemia mundial y el compromiso que acarreó la posición frente a uno de los campos más importantes para la salud de los puertorriqueños, como lo es la dermatología.

comunicando que estos pacientes con psoriasis además sufren de depresión, debido a la falta de educación de tratamientos disponi bles, y de otra parte, reconoce que en el País hacen falta más especia listas en dermatología, debido al gran cúmulo de pacientes que cada día más necesita de este especia lista. Igualmente, ha sido vocal enfatizando que la condición tiene alternativas de tratamiento y lograr su control.
DRA. DAMARIS TORRES PAOLI
Graduada del Recinto de Ciencias Médicas, hace unos 30 años Torres Paoli emplea su misión entre tantas vidas trastocadas por condiciones como la psoriasis, melanoma, dermatitis atópica, acné severo, infecciones en piel, entre otras condiciones, que provocan que en ocasiones esta población de pacientes reciba rechazo o sean víctimas de la estigmatización.
Ha sido clara en su posición
Su pasión por educar ha hecho de su práctica una cautiva y de devoción por cada paciente que atiende. Su sensibilidad la ha destacado entre todos sus colegas, y precisamente, sus pacientes. Precisamente fue el legado que dejó cuando culminó su presidencia este año en la sociedad que acoge a los dermatólogos del País.
Precisamente ha sido el eco de vidas que han decido hablar de su condición para educar a otros que lo han necesitado. Por tal razón, hoy es merecedora de nuestro A Ciencia Cierta.
Pasada presidenta Sociedad Dermatológica de Puerto Rico
Revista Puertorriqueña de Medicina y Salud Pública70
¿PODRÍA ESTAR EXPERIMENTANDO MIGRAÑA?
¿Experimenta ataques de dolor de cabeza que duran entre 4 y 72 horas?
¿Su dolor de cabeza se concentra en un lado de la cabeza, de manera pulsante y se agrava con la actividad?
¿Experimenta sensibilidad a la luz o al sonido cuando sufre un dolor de cabeza?
Cuando sufre un dolor de cabeza, ¿experimenta náuseas o vómitos?1
ES POSIBLE QUE SUFRA DE MIGRAÑA
La migraña es la segunda causa principal de discapacidad en todo el mundo a partir del 2016.2 Según el estudio American Migraine Prevalence and Prevention (Prevalencia y prevención de la migraña en Estados Unidos), más del 35 % de las personas con migraña eran candidatos para un tratamiento preventivo, pero solo el 10 % de esas personas estaban en un tratamiento preventivo.3 13 % de los puertorriqueños experimentan migraña.4

La migraña afecta la capacidad de las personas para funcionar y realizar las actividades diarias de su vida personal y profesional. Para las personas con migraña, un día de dolor de cabeza por semana podría significar una posibilidad de más de 50 días al año afectados por la migraña.3
Converse con su médico sobre sus opciones
Referencias: 2007;68:343-349. 2003;43:774-778.
PP-LU-US-0514 07/2020 ©Lilly USA, LLC 2020. Todos los derechos reservados.
1. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38(1):1-211. 2. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1211-1259. 3. Lipton RB, Bigal ME, Diamond M, et al; on behalf of the AMPP Advisory Group. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology.
4. Miranda H, Ortiz G, Figueroa S., et al. Prevalence of headache in Puerto Rico. Headache.