Criterios diagnósticos y manejo del mieloma múltiple
Evaluación de los nódulos de la tiroides
Cuidado preventivo para pacientes con enfermedad inflamatoria de intestino
La diabetes y su manejo cambiante
Sociedad
Puertorriqueña de Pediatría celebra su convención anual número 70 bajo el lema ‘Excelencia en Pediatría’
Asociación
Puertorriqueña de Gastroenterología celebra congreso enfocado en la endoscopia terapéutica
Dr. Norman
Maldonado, un legado de sabiduría, servicio e investigación
CRITERIOS DIAGNÓSTICOS ASOCIADOS A LA PSORIASIS Y SU TRATAMIENTO

Por: Hiram A. Ruiz Santiago, MD, FAAD Dermatólogo, expresidente de la Sociedad Dermatológica de Puerto Rico.
@RevistaMSP
34 42 50 56 78 80 79
Dr. Fernando Cabanillas y el Centro de Cáncer del Hospital Auxilio Mutuo marcan un legado en la oncología puertorriqueña
FOR ADULTS WITH MODERATE TO SEVERE PLAQUE PSORIASIS
Data on long-term complete clearance in challenging body areas
1-7
Scalp, nail, genital, and palmoplantar psoriasis
AVAILABLE
Taltz is indicated for patients aged 6 years or older with moderate-to-severe plaque psoriasis (PsO) who are candidates for systemic therapy or phototherapy. Taltz is also indicated for adult patients with active psoriatic arthritis (PsA), adult patients with active ankylosing spondylitis (AS), and adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation.
COMPLETE CLEARANCE IN SCALP PSORIASIS
87%
PSSI=0 by year 5, observed Nx=173
Mean baseline PSSI=20 mNRI analysis=69% (n=349)
COMPLETE CLEARANCE IN NAIL PSORIASIS
77%
NAPSI=0 by year 5, observed Nx=127

Mean baseline NAPSI=27 mNRI analysis=64% (n=229)
Scalp and nail psoriasis often are predictors of a patient developing PsA18
COMPLETE CLEARANCE IN GENITAL PSORIASIS
60%
sPGA-G 0 by year 1, NRI* n=75
Mean baseline sPGA-G=3.4
COMPLETE CLEARANCE IN PALMOPLANTAR PSORIASIS
90%
PPASI 100 by year 5, observed Nx=41
Nail, scalp, and palmoplantar data are from an open-label extension of UNCOVER-3 and are post hoc, subgroup analyses of patients who had nail, scalp, and palmoplantar psoriasis at baseline. sPGA-G 0 was a prespecified, exploratory endpoint from an open-label extension of IXORA-Q. The open-label phase of the studies has limitations (eg, no placebo comparison, patients remaining in the extension phase may be those with better results).
*At week 12, 73% of patients taking Taltz vs 8% of patients taking placebo achieved the primary endpoint in IXORA-Q of sPGA-G 0,1.7
#1 prescribed IL-17A antagonist in Dermatology*
*Based on new prescriptions and total prescriptions, inclusive of inhibitors targeting IL-17 A/F.
Co-primary endpoint results (NRI) in UNCOVER-1 (Taltz n=433; placebo n=431), UNCOVER-2 (Taltz n=351; placebo n=168), and UNCOVER-3 (Taltz n=385; placebo n=193): 89%, 90%, and 87% of Taltz patients, respectively, achieved PASI 75 at week 12 vs 4%, 2%, and 7% for placebo. Also, 82%, 83%, and 81% of Taltz patients, respectively, achieved sPGA 0,1 at week 12 vs 3%, 2%, and 7% for placebo.8
Additional week 12 results NRI: In UNCOVER-1, UNCOVER-2, and UNCOVER-3: 35%, 40%, and 38% of Taltz patients achieved PASI 100 vs 0%, 1%, and 0% for placebo, respectively.
In the maintenance period of UNCOVER-1 and -2, among patients who achieved sPGA 0,1 at week 12 with Taltz 80 mg every 2 weeks (NRI), 75% of patients who were re-randomized to Taltz 80 mg every 4 weeks (n=181† ) and 7% of patients who were rerandomized to placebo (n=203† ) achieved sPGA 0,1 at week 60.8,9
†Evaluable patients at week 60
PSSI=Psoriasis Scalp Severity Index; mNRI=modified nonresponder imputation; NAPSI=Nail Psoriasis Severity Index; sPGA-G=static Physician’s Global Assessment of Genitalia; NRI=nonresponder imputation; PPASI=Palmoplantar Psoriasis Area Severity Index.
2 Revista Puertorriqueña de Medicina y Salud Pública
See more Taltz efficacy Taltz.com/CBA
Mean baseline PPASI=10 mNRI analysis=75% (n=96) PP-IX-US-6026 Taltz Derm PPG and Upcoming Journal Ad Placement-PR.indd 1-2
Important Safety Information
CONTRAINDICATIONS
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients.
WARNINGS AND PRECAUTIONS
Infections
Taltz may increase the risk of infection. In clinical trials of adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). A similar increase in risk of infection was seen in placebo-controlled trials of adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis. Serious infections have occurred. Instruct patients to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a serious infection develops, discontinue Taltz until the infection resolves.
Pre-Treatment Evaluation for Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Closely monitor patients receiving Taltz for signs and symptoms of active TB during and after treatment
Hypersensitivity
Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz. If a serious
Trial Designs
UNCOVER-1, -2, and -38,11: The Taltz plaque psoriasis clinical trial program included 3 randomized, double-blind, placebo-controlled trials to evaluate the efficacy and safety of Taltz. All patients were ≥18 years of age and had plaque psoriasis with a body surface area involvement of ≥10%, a static Physician’s Global Assessment (sPGA) score ≥3, and a Psoriasis Area and Severity Index (PASI) score ≥12, and were candidates for phototherapy or systemic therapy. Participants were randomized to receive placebo or Taltz 80 mg every 2 weeks following a 160 mg starting dose. In UNCOVER-2 and -3, an additional arm of US-approved Enbrel® (etanercept) (50 mg twice weekly) was included. Co-primary efficacy endpoints were proportion of patients with an sPGA 0,1 and at least a 2-point improvement from baseline and proportion of patients achieving PASI 75 (at least a 75% reduction in the PASI composite score) at week 12. Nonresponder imputation (NRI) methods were used for categorical efficacy analyses.
Patients originally randomized to Taltz who were responders at week 12 (ie, sPGA 0,1) in UNCOVER-1 and -2 were rerandomized to either Taltz 80 mg every 4 weeks or placebo. In UNCOVER-3, after week 12 all patients received open label Taltz 80 mg every 4 weeks. In all UNCOVER trials, any patients who relapsed (sPGA ≥3) at any time during the maintenance period were classified as nonresponders.
hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy.
Inflammatory Bowel Disease
Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than the placebo group. During Taltz treatment, monitor patients for onset or exacerbations of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management.
Immunizations
Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz.
ADVERSE REACTIONS
Most common adverse reactions (≥1%) associated with Taltz treatment are injection site reactions, upper respiratory tract infections, nausea, and tinea infections. Overall, the safety profiles observed in adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis were consistent with the safety profile in adult patients with plaque psoriasis, with the exception of influenza and conjunctivitis in psoriatic arthritis and conjunctivitis, influenza, and urticaria in pediatric psoriasis. Please see Brief Summary of full Prescribing Information on the following pages. See Instructions for Use included with the device.
IX HCP ISI 07MAY2020
IXORA-Q8,10: IXORA-Q is a placebo-controlled, randomized, double-blind study comparing the efficacy and safety of Taltz vs placebo in patients with moderate to severe genital psoriasis, as measured by the proportion of patients achieving sPGA-G 0,1 at week 12 (primary endpoint).
The study enrolled 149 adult patients with plaque psoriasis who had minimum BSA of ≥1%, sPGA ≥3 (moderate psoriasis) and sPGA-G score of ≥3 (moderate genital psoriasis), who failed to respond to or were intolerant of at least one topical therapy used for treatment of psoriasis affecting the genital area (corticosteroids, calcineurin inhibitors, and/or vitamin D analogues), and who were candidates for phototherapy and/or systemic therapy. Patients were randomized to receive placebo or Taltz 80 mg every 2 weeks after a 160 mg starting dose. Nonresponder imputation (NRI) methods were used for categorical efficacy analyses.
References: 1. Data on file. Lilly USA, LLC. DOF-IX-US-0264. 2. Blauvelt A, Lebwohl MG, Mabuchi T, et al. Long-term efficacy and safety of ixekizumab: 5-year analysis of the UNCOVER-3 randomized controlled trial [published online ahead of print November 27, 2020]. J Am Acad Dermatol. doi:10.1016/j.aad.2020.11.022. 3. Data on file. Lilly USA, LLC. DOF-IX-US-0271. 4. Data on file. Lilly USA, LLC. DOF-IX-US-0259. 5. Data on file. Lilly USA, LLC. DOF-IX-US-0258. 6. Data on file. Lilly USA, LLC. DOFIX-US-0263. 7. Ryan C, Menter A, Guenther L, et al; on behalf of IXORA-Q Study Group. Efficacy and safety of ixekizumab in a randomized, double-blinded, placebo-controlled phase IIIb study of patients with moderate-to-severe genital psoriasis. Br J Dermatol. 2018;179:844-852. 8. Taltz [package insert]. Indianapolis, IN: Eli Lilly and Company; 2022. 9. Data on file. Lilly USA, LLC. DOF-IX-US-0274. 10. Data on file. Lilly USA, LLC. DOF-IX-US-0273. 11. Data on file. Lilly USA, LLC. DOF-IX-US-0183.
Taltz® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries or affiliates. PP-IX-US-5432 06/2022 ©Lilly USA, LLC 2022. All rights reserved.
Revista Puertorriqueña de Medicina y Salud Pública 3
1/27/23
Taltz® (ixekizumab) injection
Brief Summary: Consult the package insert for complete prescribing information.
INDICATIONS AND USAGE
Plaque Psoriasis—Taltz is indicated for the treatment of patients aged 6 years and older with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. Psoriatic Arthritis—Taltz is indicated for the treatment of adult patients with active psoriatic arthritis.
Ankylosing Spondylitis—Taltz is indicated for the treatment of adult patients with active ankylosing spondylitis.
Non-radiographic Axial Spondyloarthritis—Taltz is indicated for the treatment of adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation.
CONTRAINDICATIONS
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients (Warnings and Precautions)
WARNINGS AND PRECAUTIONS
Infections—Taltz may increase the risk of infection. In clinical trials in adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). Upper respiratory tract infections, oral candidiasis, conjunctivitis and tinea infections occurred more frequently in the Taltz group than in the placebo group. A similar increase in risk of infection was seen in placebo-controlled trials in patients with pediatric psoriasis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis (Adverse Reactions). Instruct patients treated with Taltz to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a serious infection or is not responding to standard therapy, monitor the patient closely and discontinue Taltz until the infection resolves.
Pre-treatment Evaluation for Tuberculosis—Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Consider anti-TB therapy prior to initiating Taltz in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Patients receiving Taltz should be monitored closely for signs and symptoms of active TB during and after treatment.
Hypersensitivity—Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz (Adverse Reactions). If a serious hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy.
Inflammatory Bowel Disease—Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than in the control group (Adverse Reactions). During Taltz treatment, monitor for onset or exacerbation of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management.
Immunizations—Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz. No data are available on the response to live vaccines.
ADVERSE REACTIONS
The following adverse drug reactions are discussed in greater detail in other sections of the label:
• Infections (Warnings and Precautions)
• Hypersensitivity Reactions (Contraindications and Warnings and Precautions)
• Inflammatory Bowel Disease (Warnings and Precautions)
Clinical Trials Experience—Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Plaque Psoriasis
Weeks 0 to 12: Three placebo-controlled trials in subjects with plaque psoriasis were integrated to evaluate the safety of Taltz compared to placebo for up to 12 weeks. A total of 1167 subjects (mean age 45 years; 66% men; 94% White) with plaque psoriasis received Taltz (160 mg at Week 0, 80 mg every 2 weeks [Q2W] for 12 weeks) subcutaneously. In two of the trials, the safety of Taltz (use up to 12 weeks) was also compared with an active comparator, U.S. approved etanercept.
In the 12-week, placebo-controlled period, adverse events occurred in 58% of the Taltz Q2W group (2.5 per subject-year of follow-up) compared with 47% of the placebo group (2.1 per subject-year of follow-up). Serious adverse events occurred in 2% of the Taltz group (0.07 per subject-year of follow-up), and in 2% of the placebo group (0.07 per subject-year of follow-up).
Table 1 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the Taltz group than the placebo group during the 12-week placebo-controlled period of the pooled clinical trials.
Table 1: Adverse Reactions Occurring in ≥1% of the Taltz Group and More Frequently than in the Placebo Group in the Plaque Psoriasis Clinical Trials through Week 12
Adverse reactions that occurred at rates less than 1% in the Taltz group and more frequently than in the placebo group during the 12-week induction period included rhinitis, oral candidiasis, urticaria, influenza, conjunctivitis, inflammatory bowel disease, and angioedema.
Weeks 13 to 60: A total of 332 subjects received the recommended maintenance regimen of Taltz 80 mg dosed every 4 weeks. During the maintenance period (Weeks 13 to 60), adverse events occurred in 80% of subjects treated with Taltz (1.0 per subject-year of follow-up) compared to 58% of subjects treated with placebo (1.1 per subject-year of follow-up). Serious adverse events were reported in 4% of subjects treated with Taltz (0.05 per subject-year of follow-up) and none in the subjects treated with placebo.
Weeks 0 to 60: Over the entire treatment period (Weeks 0 to 60), adverse events were reported in 67% of subjects treated with Taltz (1.4 per subject-year of follow-up) compared to 48% of subjects treated with placebo (2.0 per subject-year of follow-up). Serious adverse events were reported in 3% of subjects treated with Taltz (0.06 per subject-year of follow-up), and in 2% of subjects treated with placebo (0.06 per subject-year of follow-up).
Specific Adverse Drug Reactions:
Injection Site Reactions: The most frequent injection site reactions were erythema and pain. Most injection site reactions were mild-to-moderate in severity and did not lead to discontinuation of Taltz.
Infections: In the 12-week, placebo-controlled period of the clinical trials in plaque psoriasis, infections occurred in 27% of subjects treated with Taltz (1.2 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.4% of subjects treated with Taltz (0.02 per subject-year of follow-up) and in 0.4% of subjects treated with placebo (0.02 per subject-year of follow-up) (Warnings and Precautions)
During the maintenance treatment period (Weeks 13 to 60), infections occurred in 57% of subjects treated with Taltz (0.70 per subject-year of follow-up) compared to 32% of subjects treated with placebo (0.61 per subject-year of follow-up). Serious infections occurred in 0.9% of subjects treated with Taltz (0.01 per subject-year of follow-up) and none in the subjects treated with placebo.
Over the entire treatment period (Weeks 0 to 60), infections were reported in 38% of subjects treated with Taltz (0.83 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.7% of subjects treated with Taltz (0.02 per subject-year of follow-up), and in 0.4% of subject treated with placebo (0.02 per subject-year of follow-up).
Inflammatory Bowel Disease: In adult subjects with plaque psoriasis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the TALTZ 80 mg Q2W group (Crohn’s disease 0.1%, ulcerative colitis 0.2%) than the placebo group (0%) during the 12-week, placebo-controlled period in clinical trials (Warnings and Precautions).
Laboratory Assessment of Cytopenia:
Neutropenia—Over the entire treatment period (Weeks 0 to 60), neutropenia occurred in 11% of subjects treated with Taltz (0.24 per subject-year of follow-up) compared to 3% of subjects treated with placebo (0.14 per subject-year of follow-up). In subjects treated with Taltz, the incidence rate of neutropenia during Weeks 13 to 60 was lower than the incidence rate during Weeks 0 to 12.
In the 12-week, placebo-controlled period, neutropenia ≥ Grade 3 (<1,000 cells/mm3) occurred in 0.2% of the Taltz group (0.007 per subject-year of follow-up) compared to 0.1% of the placebo group (0.006 per subject-year of follow-up). The majority of cases of neutropenia were either Grade 2 (2% for Taltz 80 mg Q2W versus 0.3% for placebo; ≥1,000 to <1,500 cells/mm3) or Grade 1 (7% for Taltz 80 mg Q2W versus 3% for placebo; ≥1,500 cells/mm3 to <2,000 cells/mm3). Neutropenia in the Taltz group was not associated with an increased rate of infection compared to the placebo group.
Thrombocytopenia—Ninety eight percent of cases of thrombocytopenia were Grade 1 (3% for Taltz 80 mg Q2W versus 1% for placebo; ≥75,000 cells/mm3 to <150,000 cells/mm3). Thrombocytopenia in subjects treated with Taltz was not associated with an increased rate of bleeding compared to subjects treated with placebo.
Active Comparator Trials: In the two clinical trials that included an active comparator, the rate of serious adverse events during weeks zero to twelve was 0.7% for U.S.-approved etanercept and 2% for Taltz 80 mg Q2W, and the rate of discontinuation from adverse events was 0.7% for U.S. approved etanercept and 2% for Taltz 80 mg Q2W. The incidence of infections was 18% for U.S. approved etanercept and 26% for Taltz 80 mg Q2W. The rate of serious infections was 0.3% for both Taltz 80 mg Q2W and U.S. approved etanercept.
Pediatric Plaque Psoriasis
Taltz was evaluated in a placebo-controlled trial in pediatric subjects with moderate-to-severe psoriasis 6 to less than 18 years of age. A total of 171 subjects were studied (115 subjects on Taltz and 56 subjects on placebo). Overall, the safety profile observed in pediatric subjects with plaque psoriasis treated with Taltz every 4 weeks is consistent with the safety profile in adult subjects with plaque psoriasis with the exception of the frequencies of conjunctivitis (2.6%), influenza (1.7%), and urticaria (1.7%).
In this clinical trial, Crohn’s disease occurred at a greater frequency in the Taltz group (0.9%) than the placebo group (0%) during the 12-week, placebo-controlled period. Crohn’s disease occurred in a total of 4 Taltz treated subjects (2.0%) in the clinical trial (Warnings and Precautions).
Psoriatic Arthritis
Taltz was studied in two placebo-controlled trials in patients with psoriatic arthritis. A total of 678 patients were studied (454 patients on Taltz and 224 on placebo). A total of 229 patients in these trials received Taltz 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with psoriatic arthritis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis with the exception of the frequencies of influenza (1.3%) and conjunctivitis (1.3%).
Ankylosing Spondylitis
a Upper respiratory tract infections cluster includes nasopharyngitis and rhinovirus infection. b U.S. approved etanercept.
Taltz was studied in two placebo-controlled trials in patients with ankylosing spondylitis. A total of 566 patients were studied (376 patients on Taltz and 190 on placebo). A total of 195 patients in these trials received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with ankylosing spondylitis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis.
4 Revista Puertorriqueña de Medicina y Salud Pública
Adverse Reactions Taltz 80 mg Q2W (N=1167) (n%) Etanerceptb (N=287) (n%) Placebo (N=791) (n%) Injection site reactions 196 (17) 32 (11) 26 (3) Upper respiratory tract infectionsa 163 (14) 23 (8) 101 (13) Nausea 23 (2) 1 (<1) 5 (1) Tinea infections 17 (2) 0 1 (<1) IX HCP BS 27SEP2022 Taltz® (ixekizumab) injection PP-IX-US-6026 Taltz Derm PPG and Upcoming Journal Ad Placement-PR.indd 3-4
In adult patients with ankylosing spondylitis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred in 2 patients (1.0%) and 1 patient (0.5%), respectively, in the Taltz 80 mg Q4W group and 1 patient (0.5%) and 0%, respectively, in the placebo group during the 16-week, placebo-controlled period in clinical trials. Of these patients, serious events occurred in 1 patient in the Taltz 80 mg Q4W group and 1 patient in the placebo group (Warnings and Precautions).
Non-radiographic Axial Spondyloarthritis
Taltz was studied in a placebo-controlled trial in patients with non-radiographic axial spondyloarthritis. A total of 303 patients were studied (198 patients on Taltz and 105 on placebo).
A total of 96 patients in this trial received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with non-radiographic axial spondyloarthritis treated with Taltz 80 mg Q4W up to Week 16 is consistent with the previous experience of Taltz in other indications.
Immunogenicity—As with all therapeutic proteins, there is the potential for immunogenicity with Taltz. The assay to test for neutralizing antibodies has limitations detecting neutralizing antibodies in the presence of ixekizumab; therefore, the incidence of neutralizing antibodies development could be underestimated.
Plaque Psoriasis Population
By Week 12, approximately 9% of adult subjects treated with Taltz every 2 weeks developed antibodies to ixekizumab. Approximately 22% of subjects treated with Taltz at the recommended dosing regimen developed antibodies to ixekizumab during the 60-week treatment period. The clinical effects of antibodies to ixekizumab are dependent on the antibody titer; higher antibody titers were associated with decreasing drug concentration and clinical response.
Of the adult subjects who developed antibodies to ixekizumab during the 60-week treatment period, approximately 10%, which equates to 2% of subjects treated with Taltz at the recommended dosing regimen, had antibodies that were classified as neutralizing. Neutralizing antibodies were associated with reduced drug concentrations and loss of efficacy.
In pediatric psoriasis subjects treated with ixekizumab at the recommended dosing regimen up to 12 weeks, 21 subjects (18%) developed anti-drug antibodies, 5 subjects (4%) had confirmed neutralizing antibodies associated with low drug concentrations. No conclusive evidence could be obtained on the potential association of neutralizing antibodies and clinical response and/or adverse events due to small number of pediatric subjects in the study.
Psoriatic Arthritis Population
For subjects treated with Taltz 80 mg every 4 weeks for up to 52 weeks (PsA1), 11% developed anti-drug antibodies, and 8% had confirmed neutralizing antibodies.
Ankylosing Spondylitis Population
For patients treated with Taltz 80 mg every 4 weeks for up to 16 weeks (AS1, AS2), 5.2% developed anti-drug antibodies, and 1.5% had neutralizing antibodies.
Non-radiographic Axial Spondyloarthritis Population
Of patients treated with Taltz 80 mg every 4 weeks for up to 52 weeks (nr-axSpA1), 8.9% developed anti-drug antibodies, all of which were low titer. No patient had neutralizing antibodies. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to Taltz across indications or with the incidences of antibodies to other products may be misleading.
Postmarketing Experience—The following adverse reactions have been identified during post-approval use of Taltz. Because the reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to Taltz exposure.
Immune system disorders: anaphylaxis (Contraindications and Warnings and Precautions)
Infections: esophageal candidiasis.
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry—There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Pregnant women exposed to Taltz are encouraged to enroll in the Taltz Pregnancy Registry by calling 1-800-284-1695. Contact information for the registry is also available on https://www.taltz.com.
Risk Summary—Available data from the published literature and the pharmacovigilance database with Taltz use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes.
Human IgG is known to cross the placental barrier; therefore, Taltz may be transmitted from the mother to the developing fetus. An embryofetal development study conducted in pregnant monkeys during organogenesis at doses up to 19 times the maximum recommended human dose (MRHD) revealed no evidence of harm to the developing fetus. When dosing was continued until parturition, neonatal deaths were observed at 1.9 times the MRHD [see Data]. The clinical significance of these nonclinical findings is unknown.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. Data
Animal Data—An embryofetal development study was conducted in cynomolgus monkeys administered ixekizumab. No malformations or embryofetal toxicity were observed in fetuses from pregnant monkeys administered ixekizumab weekly by subcutaneous injection during organogenesis to near parturition at doses up to 19 times the MRHD (on a mg/kg basis of 50 mg/kg/week). Ixekizumab crossed the placenta in monkeys.
In a pre- and post-natal development toxicity study, pregnant cynomolgus monkeys
were administered weekly subcutaneous doses of ixekizumab up to 19 times the MRHD from the beginning of organogenesis to parturition. Neonatal deaths occurred in the offspring of two monkeys administered ixekizumab at 1.9 times the MRHD (on a mg/kg basis of 5 mg/kg/week) and two monkeys administered ixekizumab at 19 times the MRHD (on a mg/kg basis of 50 mg/kg/week). These neonatal deaths were attributed to early delivery, trauma, or congenital defect. The clinical significance of these findings is unknown. No ixekizumab-related effects on functional or immunological development were observed in the surviving infants from birth through 6 months of age.
Lactation
Risk Summary—There are no available data on the presence of ixekizumab in human milk, the effects on the breastfed infant, or the effects on milk production. Ixekizumab was detected in the milk of lactating cynomolgus monkeys. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Taltz and any potential adverse effects on the breastfed infant from Taltz or from the underlying maternal condition.
Pediatric Use—The safety and effectiveness of Taltz have been established in pediatric subjects aged 6 years to less than 18 years with moderate-to-severe plaque psoriasis. The safety and effectiveness of Taltz in other pediatric indications and for pediatric subjects less than 6 years of age have not been established.
Geriatric Use—Of the 4204 psoriasis subjects exposed to Taltz, a total of 301 were 65 years or older, and 36 subjects were 75 years or older. Although no differences in safety or efficacy were observed between older and younger subjects, the number of subjects aged 65 and over is not sufficient to determine whether they respond differently from younger subjects.
PATIENT COUNSELING INFORMATION—Advise the patient and/or caregiver to read the FDAapproved patient labeling (Medication Guide and Instructions for Use) before the patient starts using Taltz and each time the prescription is renewed, as there may be new information they need to know.
Instructions on Self-Administration: Provide guidance to patients and caregivers on proper subcutaneous injection technique, including aseptic technique, and how to use the autoinjector or prefilled syringe correctly (Instructions for Use)
Infection: Inform patients that Taltz may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider, and contacting their healthcare provider if they develop any symptoms of infection (Warnings and Precautions).
Allergic Reactions: Advise patients to seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions (Warnings and Precautions).
Pregnancy: Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Advise patients to contact the registry at 1-800-284-1695 to enroll (Use in Specific Populations)
Additional
See Instructions for Use accompanying the product device.
IX HCP BS 27SEP2022
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
Copyright © 2016, 2017, 2019, 2020, 2021, 2022, 2023 Eli Lilly and Company. All rights reserved. PP-IX-US-6026
Revista Puertorriqueña de Medicina y Salud Pública 5
information can be found at www.Taltz.com
IX HCP BS 27SEP2022
Taltz® (ixekizumab) injection
1/27/23 12:10 AM
CONTENIDO
CRITERIOS DIAGNÓSTICOS Y MANEJO DEL MIELOMA MÚLTIPLE
CRITERIOS DIAGNÓSTICOS ASOCIADOS A LA PSORIASIS Y SU TRATAMIENTO
DERMATITIS ATÓPICA CRITERIOS DIAGNÓSTICOS Y EXACERBACIONES ASOCIADAS A LA CONDICIÓN
EVALUACIÓN DE LOS NÓDULOS DE LA TIROIDES
DIABETES
SOCIEDAD PUERTORRIQUEÑA DE PEDIATRÍA CELEBRA SU CONVENCIÓN ANUAL NÚMERO 70 BAJO EL LEMA ‘EXCELENCIA EN PEDIATRÍA’
DR. NORMAN MALDONADO, UN LEGADO DE SABIDURÍA, SERVICIO E INVESTIGACIÓN
EDITOR FUNDADOR Juan Carlos Orengo Valverde, MD, MPH, PhD EDITOR Alberto Santiago Cornier, MD, PhD CONSEJO ASESOR Oscar Soto Raíces, MD, Ahmed Morales, MD, FACP, FACG, FASGE, AGAF, Lcda. Wanda González fisióloga del ejercicio PRINCIPAL OFICIAL EJECUTIVO Pedro Carlos Lugo Hernández III, P.A.C. PRESIDENTA Y FUNDADORA Glorybelle Hernández Figueroa, MBA
VICEPRESIDENTA Y FUNDADORA Laila Paloma Lugo, MBA CONTABILIDAD Julio Soto ADMINISTRACIÓN Marta Ivelisse Vélez Ramos, MBA, MARKETING Y SERVICIOS 360 Darlene Rodríguez, Yasmin Morell, Belinda Burgos PERIODISTAS Mayra Acevedo, Luis Penchi, Limarys Suárez DIRECCIÓN GRÁFICA Natalia Zoé Rivera Torres ARTISTA GRÁFICO Jhorman González
DIRECTOR AUDIOVISUAL Christopher Soto REALIZADORA AUDIOVISIAL Salomé Mateus, Duban Valencia FOTOS Revista Medicina y Salud Pública DIRECCIÓN GENERAL / FUNDADOR Carlos Alexis Lugo Marrero DISTRIBUCIÓN OFICINAS Y TORRES MÉDICAS Editorial Mundo ENVÍO DE REVISTAS Y DISTRIBUCIÓN A GRUPOS MÉDICOS Servicio de correo postal/Comunicación Inteligente
Para ventas y otros servicios pueden comunicarse al 787.848.3333, msp@editorialmundo.com o www.medicinaysaludpublica.com
Revista Puertorriqueña de Medicina y Salud Pública ISSN 1937-8521
COMITÉ EDITORIAL CIENTÍFICO
COMITÉ EDITORIAL Olga Rodríguez, MD - Decana Escuela de Medicina de Ponce (Puerto Rico), Vivian Green, LND, MS, PhD, Sub editora y fundadora (Puerto Rico), José Cordero, MD, MPH - Exdecano Escuela Graduada Salud Pública Recinto de Ciencias Médicas UPR (Puerto Rico), Ángeles Rodríguez, MD, MPH (Puerto Rico), Simón Carlo, MD (Puerto Rico), Bárbara Rosado, MD (Puerto Rico), Idhaliz Flores PhD (Puerto Rico), Jesús Cruz-Correa, MD, FACOG (Puerto Rico), Rafael Bredy, MD, LicMTo, MBE, MS (Puerto Rico), David Caseida, MD, FACOG, (Puerto Rico), José Capriles, MD, MHSA (Puerto Rico) Joaquín Laboy, MD, FACOG (Puerto Rico), Luis Adrian Rivera Pomales, MD, PEMBA, MPH, CMQ (Puerto Rico), Juan Fernández, MS, PhD (Puerto Rico), Nuria Sebate, MD (Puerto Rico), Pedro Amador, MD, MPH (Puerto Rico), Nydia Cappas, PsyD (Puerto Rico), Luis Franco, MD (Puerto Rico), Federico Montealegre, DVM, PhD, Msc (Puerto Rico), Nydia Ortiz, PsyD (Puerto Rico), José Pons, PhD, FPPR (Puerto Rico), Esdrás Vélez, JD, MPH (Puerto Rico), Diego Zavala, MSc, PhD, (Puerto Rico), Ana Torres-Martín, MD (Puerto Rico), Julio Cádiz, MD, MPH (Puerto Rico), Rafael Gómez-Cuevas (Colombia), José Javier Orengo, PhD(c) (España), Cesar A. Del Rey, MD (Panamá), Pedro Serrano, MD, PhD (España), Luis Serra-Majem, MD, PhD (España), José Ramón Calvo, MD, PhD (España).
Síguenos en www.medicinaysaludpublica.com, www.facebook.com/revistamsp, en Twitter @revistamsp, en LinkedIn como Revista Puertorriqueña de Medicina y Salud Pública. Las normas editoriales de la Revista Puertorriqueña de Medicina y Salud Pública para la publicación de artículos originales y cartas al editor pueden ser accesadas en la página web: www.medicinaysaludpublica.com, y solicitadas a través de msp@editorialmundo.com.
6 Revista Puertorriqueña de Medicina y Salud Pública
8 50 16 94 42 62 34 98
PACIENTES CON ENFERMEDAD INFLAMATORIA DE INTESTINO
CUIDADO PREVENTIVO PARA
CAMBIANTE
LA
Y SU MANEJO
MENSAJE ESPECIAL
La piel es el órgano más grande de nuestro cuerpo. Entre sus múltiples funciones se encuentra la de proteger los órganos internos de posibles traumas, así como controlar la temperatura corporal. La piel nos protege ante la exposición de sustancias tóxicas e infecciones que nos puedan causar daño y, selectivamente, controla las moléculas que entran y salen de nuestro cuerpo.
La piel posee su propio sistema inmunológico que sirve
Dr. José R. González Chávez

Dermatólogo, investigador y fundador de las clínicas de Psoriasis de Puerto Rico.
para neutralizar los organismos patogénicos que nos puedan enfermar. Adicionalmente, actúa como sistema endocrino capaz de secretar hormonas tales como la vitamina D, interleucinas y sustancias vasoactivas que ayudan a amplificar nuestra respuesta inmunológica.
Debido a la multiplicidad de funciones de la piel, y el estar expuesta a numerosos agentes agresivos como la irradiación solar, las infecciones , elementos carcinógenos, enfermedades genéticas y las de origen autoinmune, la piel actúa como una ventana que nos permite diagnosticar condiciones importantes de acuerdo a sus manifestaciones cutáneas. Es por esto que la dermatología cobra una relevancia enorme entre todas las ramas de la medicina.
Muchas enfermedades de la piel, tales como la psoriasis, la dermatitis atópica, el lupus eritematoso entre otras, han servido de modelo para los
científicos poder estudiar y descifrar la patogénesis de muchísimas otras enfermedade, tanto cutáneas como sistémicas.
A primera luz, la evaluación dermatológica puede parecer inconsecuente para aquel que no la ha estudiado, pero el dermatólogo debe dominar muchas habilidades, de diferentes disciplinas. Además de tener los conocimientos básicos sobre la estructura y función de la piel, el dermatólogo debe dominar temas como la inmunología, genética, enfermedades infecciosas, farmacología, terapéutica, alergias y cáncer. Además, debe poseer destrezas quirúrgicas. El dermatólogo puede ser un gran recurso para otros especialistas en el diagnóstico y manejo de casos complicados.
Las enfermedades de la piel abarcan un número significativo de las evaluaciones en las salas de emergencias, así como en las visitas a médicos internistas, pediatras, reumatólogos y muchos otros especialistas en su consulta.
Expuesto de esta manera, esta revista se compromete en sus próximas ediciones a generar artículos en los que se aborden temas de interés para la comunidad científica y que muestren los avances en materia de enfermedades de la piel, con el apoyo de dermatólogos de renombre y prestigio a nivel nacional e internacional.
Revista Puertorriqueña de Medicina y Salud Pública 7
CRITERIOS DIAGNÓSTICOS ASOCIADOS A LA PSORIASIS Y SU TRATAMIENTO

Fuentes: Notimex, Mayoclinic, Novartis
8 Revista Puertorriqueña de Medicina y Salud Pública
Hiram A. Ruiz Santiago, MD, FAAD

Dermatólogo, expresidente de la Sociedad Dermatológica de Puerto Rico.
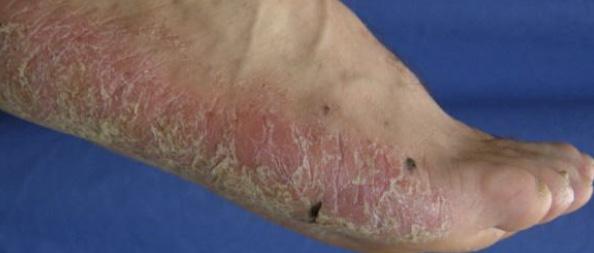
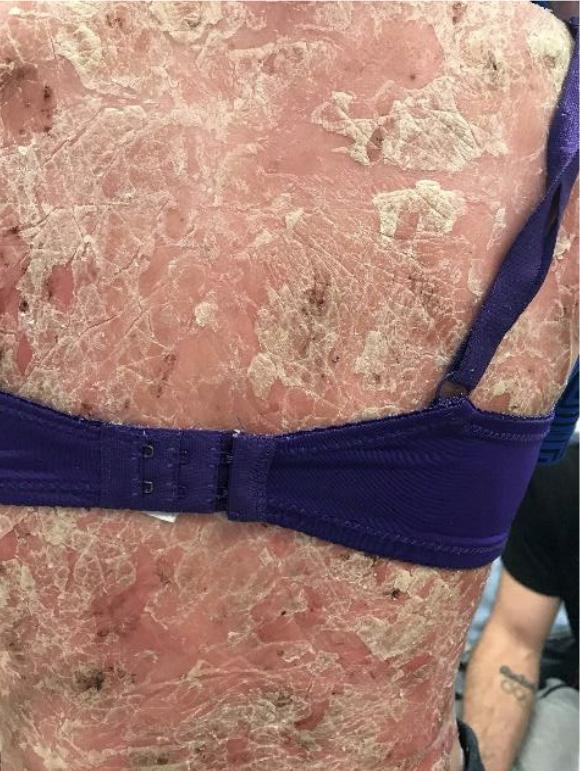
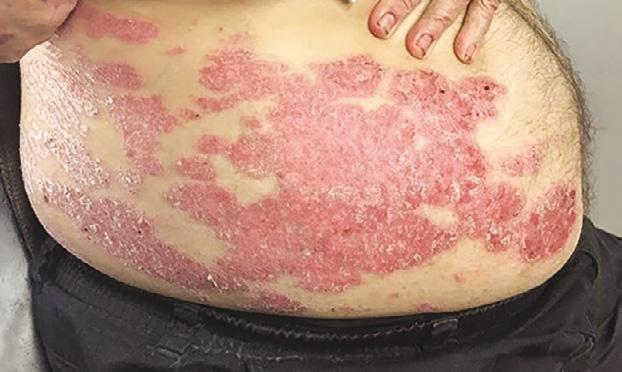
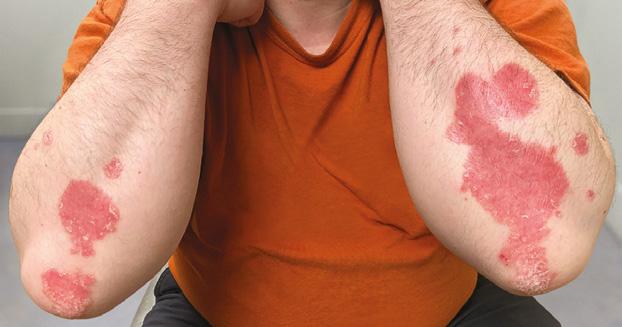
La Psoriasis es una enfermedad inflamatoria que envuelve principalmente la piel y en algunos casos las articulaciones. Es importante recalcar que es una condición crónica y que no es contagiosa. Aunque hay varios tipos clínicos como la psoriasis eritrodérmica, la psoriasis pustular y la psoriasis en gota, la presentación más común se conoce como psoriasis en placas Fig.1. Esto por su manifestación clínica de placas rojas bien delimitadas con escamas gruesas. Estas pueden ocasionar picor y/o ardor al paciente.
Además, los casos severos pueden estar asociados a obesidad, síndrome metabólico, hipertensión, diabetes y enfermedad aterosclerótica.
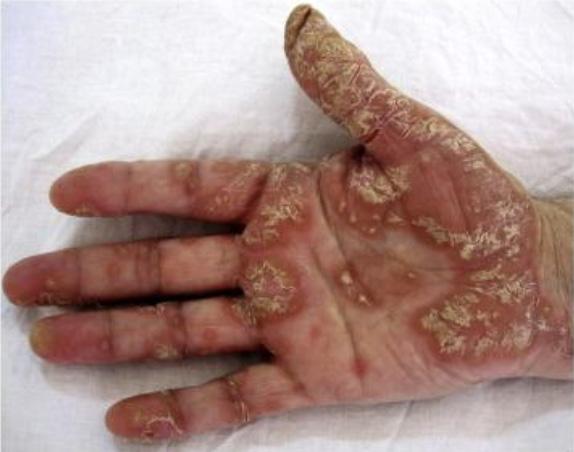
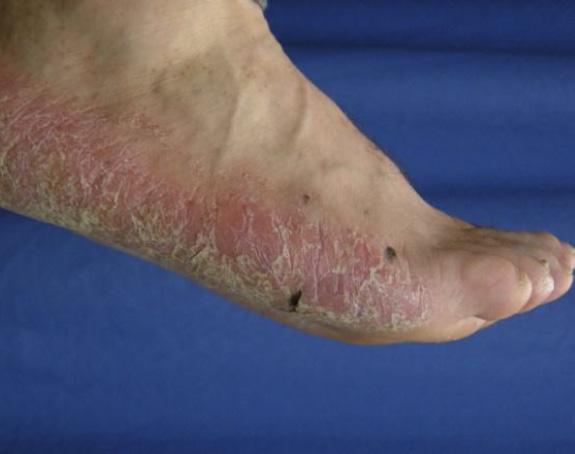
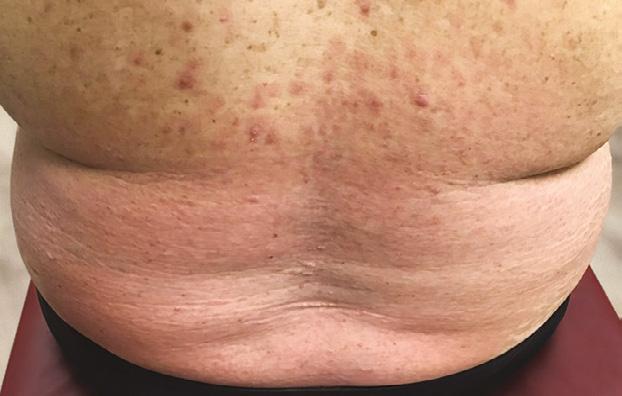
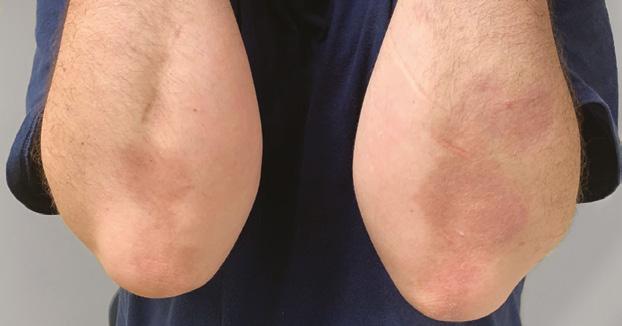
La Psoriasis en placa generalmente se manifiesta en cuero cabelludo, codos, rodillas y glúteos, pero puede envolver cualquier parte del cuerpo incluyendo las uñas, palmas y plantas, oídos y el área genital. Cuando la Psoriasis afecta las manos y los pies, puede producir dolor debido a fisuras en la piel. Además, la persona con Psoriasis palmar, frecuentemente se siente retraídas pues no se atreven estrecharle la mano a otra persona al saludarla. Fig 2.
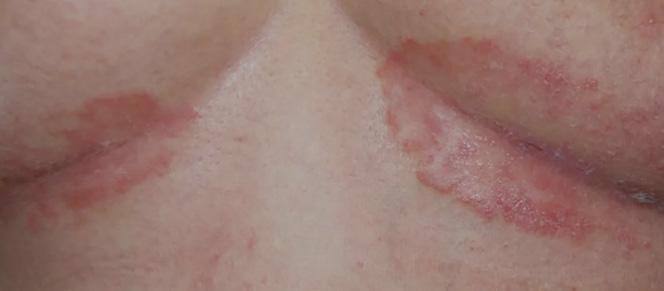
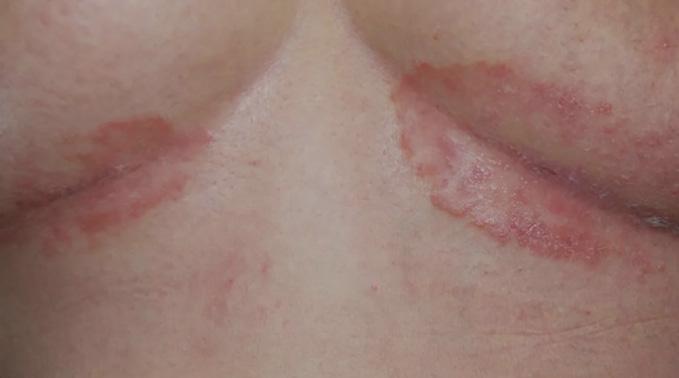
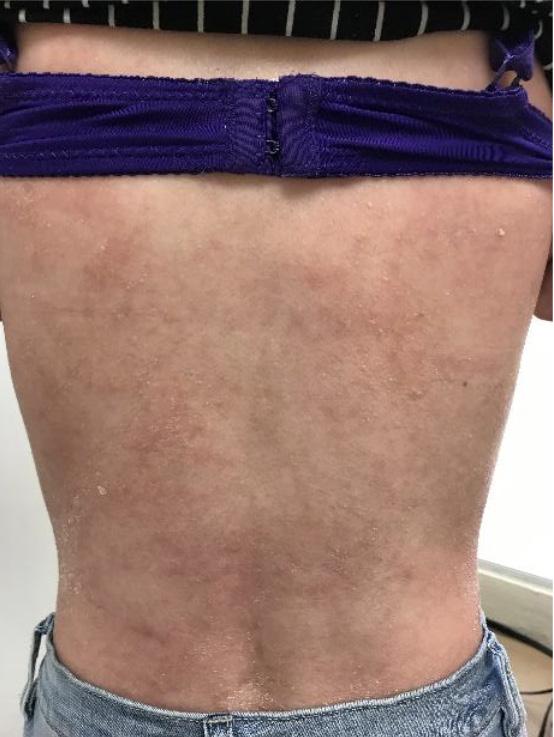
El diagnóstico de la psoriasis se hace mediante el examen físico. Ciertas áreas pueden presentar hallazgos diferentes a las placas escamosas. Por ejemplo, en las áreas intertriginosas (axila, ingle, debajo de los senos), las lesiones son rojas, pero sin escamas y

Revista Puertorriqueña de Medicina y Salud Pública 9
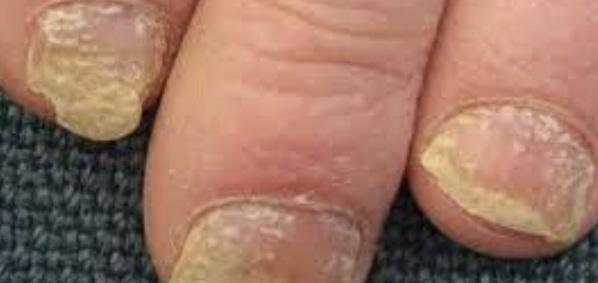
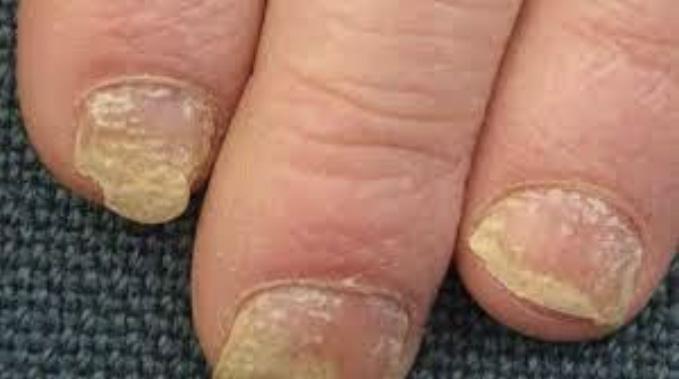
pueden confundirse con infección por hongos o levaduras Fig.3A. En las uñas la psoriasis puede dar cambios bien parecidos a la infección micótica Fig.3B. En estos casos, se puede descartar este diagnóstico mediante una prueba que se realiza en la oficina para identificar microscópicamente el hongo (KOH). En casos en donde el diagnóstico clínico sea equivoco, se puede confirmar mediante una biopsia de la piel.
El tratamiento de la psoriasis debe tomar en consideración la calidad de vida del paciente. Cuando la condición es leve, se puede controlar con tratamientos tópicos. Esto incluye los corticosteroides de alta potencia preferiblemente en ungüento. Una vez se logra una mejoría significativa, estos deben ser utilizados con menos frecuencia pues pueden afinar la piel o perder su efectividad. Otros tratamientos tópicos incluyen retinoides como el tazarotene, derivados de brea e inhibidores de calcineurina como pimecrolimus y tacrolimus. Estos últimos dos pueden ser efectivos en áreas donde no deben usarse esteroides tópicos potentes como en las áreas intertriginosas. Recientemente otros dos medicamentos tópicos, Tapinarof y Roflumilast, no relacionados a los corticosteroides, han sido aprobados por la FDA.
Casos severos o que afecten funciones cotidianas del paciente, deben ser tratados con medicamentos sistémicos. Con la llegada de los tratamientos biológicos, hace aproximadamente 20 años, comienza una nueva era donde en la mayoría de los casos se logra devolverle al paciente su calidad de vida, manteniendo un excelente perfil de seguridad. Los tratamientos biológicos se caracterizan por su mecanismo de acción. En vez de suprimir el sistema inmunológico por completo, lo hacen de manera más selectiva, inhibiendo moléculas especificas que se producen excesivamente en los casos de psoriasis. De esta forma logran una efectividad superior a otras terapias anteriores como el metotrexate y un mejor perfil de seguridad.
Los inhibidores del “tumor necrosis factor-alpha”(TNF-a) como el Etanercept, Infliximab, Adalimumab y Cetrolizumab pegol, fueron de los primeros biológicos aprobados para tratar la psoriasis y la artritis psoriásica. Adalimumab logró una mejoría de un 75% en 53% de los pacientes en 12 semanas. En otro estudio demostró ser más efectivo que el metotrexato4. Los inhibidores del TNF-a tienen el potencial de activar infecciones latentes como la tuberculosis y otras infecciones. Aunque se advierte de un posible aumento de riesgos de malignidad, estudios a largo plazo no
han podido confirmar esa asociación.
Otras citoquinas, como la Interleukina 17(IL-17) y la 23(IL-23) son aún más importantes en la patofisiología de la psoriasis. Entre los inhibidores de la IL-17 disponibles están: Secukinumab, Ixekizumab, y Brodalumab. Por lo general se consideran más efectivos que los inhibidores del TNF-a y tienen menos advertencias de seguridad. Están asociados a un leve aumento en infecciones por Cándida. En estudios clínicos 74% de los pacientes tratados con Ixekizumab lograron un aclaramiento total o con lesiones mínimas a las 12 semanas6. Los inhibidores de la IL-23 incluyen: Ustekinumab, Tildrakizumab, Guselkumab, y Risankizumab. Estos últimos 2 han demostrado ser igual o más efectivos que los inhibidores de IL-17 pero requiriendo menos dosis al año.
Entre las terapias no inyectables (orales) para tratar la Psoriasis está Apremilast, un inhibidor de la fosfodiesterasa. Aunque menos efectiva que los biológicos, no es considerado un inmunosupresor y aparte de problemas gastro-intestinales, generalmente es bien tolerado. EL año pasado el FDA aprobó el uso de Deucarvacitinib como tratamiento oral para la Psoriasis moderada a severa. La dosis estándar es de una pastilla (6mg) diaria. En estudios comparativos demostró ser más efectiva que el Apremilast, bien tolerada y con pocos efectos adversos a corto plazo (nasofaringitis e infecciones respiratorias).
En los próximos meses y años se espera que sigan saliendo al mercado otros medicamentos que bloquean receptores o proteínas especificas para tratar la Psoriasis sin afectar considerablemente otras funciones del sistema inmunológico.
ESTO TRATAMIENTOS BIOLÓGICOS HAN REVOLUCIONADO EL MANEJO DE LOS PACIENTES CON PSORIASIS MODERADA A SEVERA. LA GRAN MAYORÍA DE LOS PACIENTES LOGRAN UN ACLARAMIENTO EN LA PIEL SIGNIFICATIVO Y UNA MUCHO MEJOR CALIDAD DE VIDA.
10 Revista Puertorriqueña de Medicina y Salud Pública
Fig.2 Psoriasis palmoplantar
Palabras Clave: Psoriasis, tratamientos biológicos
Aunque se sigue demostrando su efectividad a largo plazo, hay veces que los medicamentos biológicos van perdiendo su efectividad debido a la formación de anticuerpos neutralizantes. En estos casos, por lo general, se cambia a otro medicamento que tenga un mecanismo de acción diferente.
El cuidado multidisciplinario del paciente con Psoriasis envuelve al Dermatólogo, al Internista y al Reumatólogo. El problema principal con estos medicamentos biológicos no son sus efectos secundarios sino el acceso a los pacientes. Son bien costosos y los planes médicos restringen el despacho de estos constantemente. Aun así, las farmacéuticas ofrecen programas de asistencia para cubrir los deducibles y existen fundaciones que en algunos casos proveen el medicamento, al menos por un tiempo, sin costo alguno para el paciente.
CONCLUSIÓN
La psoriasis es mucho más que una enfermedad de la piel, además de sus comorbilidades sistémicas, causa problemas emocionales y sociales severos. Aunque no es contagiosa en muchos casos los pacientes son discriminados por su apariencia, se retraen socialmente y la incidencia de depresión es mas alta. Con los avances médicos, en la gran mayoría de los casos, el paciente de psoriasis moderada a severa puede recobrar la calidad de vida que todo ser humano merece.
REFERENCIAS
Vtama (tapinarof) cream, for topical use. US FDA approved product information; Long Beach, CA: Dermavant Sciences, Inc; May 2022. https://www. accessdata.fda.gov/drugsatfda_docs/ label/2022/215272s000lbl.pdf (Accessed on June 02, 2022).
Zoryve (roflumilast) cream, for topical use. US FDA approved product information; Westlake Village, CA: Arcutis Biotherapeutics, Inc; July 2022. https:// www.accessdata.fda.gov/drugsatfda_ docs/label/2022/215985s000lbl.pdf (Accessed on August 03, 2022).
Clinical response to adalimumab treatment in patients with moderate to severe psoriasis: double-blind, randomized controlled trial and open-label extension study.Gordon KB, Langley RG, Leonardi C, Toth D, Menter MA, Kang S, Heffernan M, Miller B, Hamlin R, Lim L, Zhong J, Hoffman R, Okun MM. J Am Acad Dermatol. 2006;55(4):598. Epub 2006 Aug 10.
Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. methotrexate vs. placebo in patients with psoriasis (CHAMPION). Saurat JH, Stingl G, Dubertret L, Papp K, Langley RG, Ortonne JP, Unnebrink K, Kaul M, Camez A, CHAMPION Study Investigators
Br J Dermatol. 2008;158(3):558. Epub 2007 Nov 28.
Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies.
Gottlieb AB, Kalb RE, Langley RG, Krueger GG, de Jong EM, Guenther L, Goyal K, Fakharzadeh S, Chevrier M, Calabro S, Langholff W, Menter A
Revista Puertorriqueña de Medicina y Salud Pública 11
Fig. 3A Psoriasis inversa
Fig.4A Pre-tratamiento
Fig 4B semanas después de 1 dosis (Risankizumab)
Fig. 3B Psoriasis de la uña
An IL-23 inhibitor for adults with moderate to severe plaque psoriasis (Ps) 1

PICTURE WHAT IMPROVEMENT COULD LOOK LIKE
SKYRIZI EFFICACY: IN P s AT WEEK 16 IN TWO PIVOTAL PHASE 3 STUDIES (NRI)2


CO-PRIMARY ENDPOINTS ( P <0.0001)
MAINTENANCE OF RESPONSE 1
88 %
STUDY DESIGN 2
UltIMMa-1 (N=506) and UltIMMa-2 (N=491) were replicate phase 3, randomized, double-blind, placebo- and active-controlled studies to evaluate the efficacy and safety of SKYRIZI (150 mg) vs placebo over 16 weeks and biologic active control over 52 weeks in adult patients with moderate to severe plaque psoriasis. SKYRIZI (150 mg) was given as 2 subcutaneous injections at Weeks 0, 4, and 16, and every 12 weeks thereafter. Co-primary endpoints were PASI 90 and sPGA 0/1 at Week 16 vs placebo in each study (assessed by non-responder imputation).

proprietary method on diagnosis classification.1,3
SKYRIZI PLACEBO PASI 90 at Week 16 (220/294) (2/98) 75 % 5 % 75 % 2% (229/304) (5/102) ULTIMMA-1 ULTIMMA-2 sPGA 0/1 at Week 16 (246/294) (5/98) 88 % 8 % 84 % 5 % (267/304) (8/102) ULTIMMA-1 ULTIMMA-2 NRI=Non-Responder Imputation. As
10/2021.
BIOLOGIC
of
New patients defined as bio-naïve; switch patients defined as bio-experienced switching biologics. Source: Integrated Symphony Health (PatientSource) and IQVIA (NSP) through proprietary method on diagnosis classification.1,3 PRESCRIBED
IN NEW AND SWITCHING PLAQUE PSORIASIS PATIENTS
In the randomized psoriasis trials, among patients who achieved PASI 90 at Week 16 MAINTAINED PASI 90 AT WEEK 52 (n=398/450)
ACTUAL PSORIASIS PATIENTS TREATED WITH SKYRIZI 4

WEEK 0 (BASELINE)
WEEK 16 (AFTER 2 DOSES)
INDICATION1
SKYRIZI is indicated for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.

IMPORTANT SAFETY INFORMATION1
Hypersensitivity Reactions
SKYRIZI® (risankizumab-rzaa) is contraindicated in patients with a history of serious hypersensitivity reaction to risankizumab-rzaa or any of the excipients. Serious hypersensitivity reactions, including anaphylaxis, have been reported with the use of SKYRIZI. If a serious hypersensitivity reaction occurs, discontinue SKYRIZI and initiate appropriate therapy immediately.
Infection
SKYRIZI may increase the risk of infection. Do not initiate treatment with SKYRIZI in patients with a clinically important active infection until it resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing SKYRIZI. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If a patient develops such an infection or is not responding to standard therapy, closely monitor and discontinue SKYRIZI until the infection resolves.
Tuberculosis (TB)
Prior to initiating treatment with SKYRIZI, evaluate for TB infection and consider treatment in patients with latent or active TB for whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after SKYRIZI treatment. Do not administer SKYRIZI to patients with active TB.
Administration of Vaccines
Avoid use of live vaccines in patients treated with SKYRIZI. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating SKYRIZI, complete all age appropriate vaccinations according to current immunization guidelines.
Adverse Reactions
Most common (≥1%) adverse reactions associated with SKYRIZI include upper respiratory infections, headache, fatigue, injection site reactions, and tinea infections.
In psoriatic arthritis phase 3 trials, the incidence of hepatic events was higher with SKYRIZI compared to placebo.
SKYRIZI is available in a 150 mg/mL prefilled syringe and pen.
Please see the Brief Summary of the Full Prescribing Information on the following page.
AbbVie. All rights reserved. US- SKZD-220291 Printed in U.S.A. August 2022 References: 1. SKYRIZI [package insert]. North Chicago, IL: AbbVie Inc. 2. Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-tosevere plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 2018;392(10148):650-661. 3. Data on file, AbbVie Inc. In-play patient share. 2021. 4. Data on file, AbbVie Inc. Rep-fielded images. Presented: June 16, 2020.
© 2022 AbbVie. All rights reserved. US- SKZD-220291 Printed in U.S.A. August 2022
PATIENT FROM ILLINOIS PATIENT FROM TEXAS PATIENT FROM PENNSYLVANIA
PATIENT FROM ILLINOIS PATIENT FROM TEXAS PATIENT FROM PENNSYLVANIA
Find more real patient images at www.SkyriziHCP.com/dermatology
Photos represent results captured at baseline and Week 16 from non–trial participant patients. Co-primary endpoints for patients in the trial were PASI 90 and sPGA 0/1 at Week 16.2
Actual SKYRIZI-treated patients. PASI and/or sPGA achievement undefined. Left: Photos courtesy of Dr. Meyer Horn. Middle: Photos courtesy of Dr. Matthew Bruno, PA-C. Right: Photos courtesy of Dr. David Andrew Kasper.
The patients depicted here have moderate to severe plaque psoriasis with an affected body surface area ≥10%.
SKYRIZI® (sky-RIZZ-ee) (risankizumab-rzaa) injection, for subcutaneous use
150 mg/mL single-dose pen and prefilled syringe
600 mg/10 mL single-dose vial
360 mg/2.4 mL single-dose prefilled cartridge with on-body injector
INDICATIONS AND USAGE
Plaque Psoriasis
SKYRIZI® is indicated for the treatment of moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
Psoriatic Arthritis
SKYRIZI is indicated for the treatment of active psoriatic arthritis in adults.
Crohn’s Disease
SKYRIZI is indicated for the treatment of moderately to severely active Crohn’s disease in adults.
CONTRAINDICATIONS
SKYRIZI is contraindicated in patients with a history of serious hypersensitivity reaction to risankizumab-rzaa or any of the excipients [see Warnings and Precautions]
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been reported with use of SKYRIZI. If a serious hypersensitivity reaction occurs, discontinue SKYRIZI and initiate appropriate therapy immediately [see Adverse Reactions]
Infections
SKYRIZI may increase the risk of infections [see Adverse Reactions]
Treatment with SKYRIZI should not be initiated in patients with any clinically important active infection until the infection resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing SKYRIZI. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If a patient develops such an infection or is not responding to standard therapy, monitor the patient closely and do not administer SKYRIZI until the infection resolves.
Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with SKYRIZI. Across the Phase 3 psoriasis clinical studies, of the 72 subjects with latent TB who were concurrently treated with SKYRIZI and appropriate TB prophylaxis during the studies, none developed active TB during the mean follow-up of 61 weeks on SKYRIZI. Two subjects taking isoniazid for treatment of latent TB discontinued treatment due to liver injury. Of the 31 subjects from the PsO-3 study with latent TB who did not receive prophylaxis during the study, none developed active TB during the mean follow-up of 55 weeks on SKYRIZI. Consider anti-TB therapy prior to initiating SKYRIZI in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after SKYRIZI treatment. Do not administer SKYRIZI to patients with active TB.
Hepatotoxicity
in Treatment of Crohn’s Disease
A serious adverse reaction of drug-induced liver injury was reported in a patient with Crohn’s disease (ALT 54x ULN, AST 30x ULN, and total bilirubin 2.2x ULN) following two intravenous doses of SKYRIZI 600 mg in conjunction with a rash that required hospitalization. The liver test abnormalities resolved following administration of steroids. SKYRIZI was subsequently discontinued.
For the treatment of Crohn’s disease, evaluate liver enzymes and bilirubin at baseline, and during induction at least up to 12 weeks of treatment. Monitor thereafter according to routine patient management.
Consider other treatment options in patients with evidence of liver cirrhosis. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. Interrupt treatment if drug-induced liver injury is suspected, until this diagnosis is excluded. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of hepatic dysfunction.
Administration of Vaccines
Avoid use of live vaccines in patients treated with SKYRIZI. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating therapy with SKYRIZI, complete all age-appropriate vaccinations according to current immunization guidelines. No data are available on the response to live or inactive vaccines.
ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of labeling:
• Hypersensitivity Reactions [see Warnings and Precautions]
• Infections [see Warnings and Precautions]
• Tuberculosis [see Warnings and Precautions]
• Hepatotoxicity in Treatment of Crohn’s disease [see Warnings and Precautions]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse drug reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Plaque Psoriasis
A total of 2234 subjects were treated with SKYRIZI in clinical development trials in plaque psoriasis. Of these, 1208 subjects with psoriasis were exposed to SKYRIZI for at least one year.
Data from placebo- and active-controlled trials were pooled to evaluate the safety of SKYRIZI for up to 16 weeks. In total, 1306 subjects were evaluated in the SKYRIZI 150 mg group.
Table 1 summarizes the adverse drug reactions that occurred at a rate of at least 1% and at a higher rate in the SKYRIZI group than the placebo group during the 16-week controlled period of pooled clinical trials.
a Includes: respiratory tract infection (viral, bacterial or unspecified), sinusitis (including acute), rhinitis, nasopharyngitis, pharyngitis (including viral), tonsillitis
b Includes: headache, tension headache, sinus headache, cervicogenic headache
c Includes: fatigue, asthenia
d Includes: injection site bruising, erythema, extravasation, hematoma, hemorrhage, infection, inflammation, irritation, pain, pruritus, reaction, swelling, warmth
e Includes: tinea pedis, tinea cruris, body tinea, tinea versicolor, tinea manuum, tinea infection, onychomycosis
Adverse drug reactions that occurred in < 1% but > 0.1% of subjects in the SKYRIZI group and at a higher rate than in the placebo group through Week 16 were folliculitis and urticaria.
Specific Adverse Drug Reactions
Infections
In the first 16 weeks, infections occurred in 22.1% of the SKYRIZI group (90.8 events per 100 subject-years) compared with 14.7% of the placebo group (56.5 events per 100 subject-years) and did not lead to discontinuation of SKYRIZI. The rates of serious infections for the SKYRIZI group and the placebo group were ≤0.4%. Serious infections in the SKYRIZI group included cellulitis, osteomyelitis, sepsis, and herpes zoster. In Studies PsO-1 and PsO-2, through Week 52, the rate of infections (73.9 events per 100 subject-years) was similar to the rate observed during the first 16 weeks of treatment.
Safety Through Week 52
Through Week 52, no new adverse reactions were identified, and the rates of the adverse reactions were similar to those observed during the first 16 weeks of treatment. During this period, serious infections that led to study discontinuation included pneumonia.
Psoriatic Arthritis
The overall safety profile observed in subjects with psoriatic arthritis treated with SKYRIZI is generally consistent with the safety profile in subjects with plaque psoriasis. Additionally, in the Phase 3 placebo-controlled trials the incidence of hepatic events was higher in the SKYRIZI group (5.4%, 16.7 events per 100 patient years) compared to the placebo group (3.9%, 12.6 events per 100 patient years). Of these, the most common events that were reported more frequently in both the placebo group and the SKYRIZI group were ALT increased (placebo: n=12 (1.7%); SKYRIZI: n=16 (2.3%)), AST increased (placebo: n=9 (1.3%); SKYRIZI: n=13 (1.8%)), and GGT increased (placebo: n=5 (0.7%); SKYRIZI: n=8 (1.1%)). There were no serious hepatic events reported. The incidence of hypersensitivity reactions was higher in the SKYRIZI group (n=16, 2.3%) compared to the placebo group (n=9, 1.3%). In the Phase 3 placebo-controlled trials, hypersensitivity reactions reported at a higher rate in the SKYRIZI group included rash (placebo: n=4 (0.6%); SKYRIZI: n=5 (0.7%), allergic rhinitis (placebo: n=1 (0.1%); SKYRIZI: n=2 (0.3%), and facial swelling (placebo: n=0 (0.0%); SKYRIZI n=1 (0.1%). One case of anaphylaxis was reported in a subject who received SKYRIZI in the Phase 2 clinical trial.
Crohn’s Disease
SKYRIZI was studied up to 12 weeks in subjects with moderately to severely active Crohn’s disease in two randomized, double-blind, placebo-controlled induction studies (CD-1, CD-2) and a randomized, double-blind, placebocontrolled, dose-finding study (CD-4; NCT02031276). Long-term safety up to 52 weeks was evaluated in subjects who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (CD-3).
In the two induction studies (CD-1, CD-2) and the dose finding study (CD-4), 620 subjects received the SKYRIZI intravenous induction regimen. In the maintenance study (CD-3), 142 subjects who achieved clinical response defined as a reduction in CDAI of at least 100 points from baseline after 12 weeks of induction treatment with intravenous SKYRIZI in studies CD-1 and CD-2, received SKYRIZI subcutaneously as a maintenance regimen. Adverse reactions reported in > 3% of subjects in induction studies and at a higher rate than placebo are shown in Table 2.
PROFESSIONAL BRIEF SUMMARY CONSULT PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION
Adverse Drug Reactions Reported in > 3% of Subjects with Crohn’s Disease Treated with SKYRIZI in Placebo-Controlled 12-Week Induction Studies
Table
600 mg as an intravenous infusion at Week 0, Week 4, and Week 8.
b Includes: influenza like illness, nasopharyngitis, influenza, pharyngitis, upper respiratory tract infection, viral upper respiratory tract infection, COVID-19, nasal congestion, respiratory tract infection viral, viral pharyngitis, tonsillitis, upper respiratory tract inflammation c Includes: headache, tension headache
Adverse reactions reported in >3% of subjects in the maintenance study and at a higher rate than placebo are shown in Table 3.
Table 3. Adverse Reactions Reported in >3% of Subjects with Crohn’s Disease Treated with SKYRIZI in Placebo-Controlled 52-Week
SKYRIZI 360 mg at Week 12 and every 8 weeks thereafter for up to an additional 52 weeks
b Includes: injection site rash, injection site erythema, injection site swelling, injection site urticaria, injection site warmth, injection site pain, injection site hypersensitivity, injection site reaction c Some subjects had multiple occurrences of injection site reactions. The adverse reaction is included only once per subject. dIncludes: abdominal pain, abdominal pain upper, abdominal pain lower
Specific
Infections
In the maintenance study (CD-3) through Week 52, the rate of infections was 36.6% (60.8 events per 100 subject-years) in subjects who received SKYRIZI compared to 36.4% (60.3 events per 100 subject-years) in subjects who received placebo after SKYRIZI induction. The rate of serious infections was 5.6% (7.4 events per 100 subject-years) in subjects who received SKYRIZI compared to 2.1% (2.4 events per 100 subject-years) in subjects who received placebo after SKYRIZI induction.
Lipid Elevations
Elevations in lipid parameters (total cholesterol and low-density lipoprotein cholesterol [LDL-C]) were first assessed at 4 weeks following initiation of SKYRIZI in the induction trials (CD-1, CD-2). Increases from baseline and increases relative to placebo were observed at Week 4 and remained stable to Week 12. Following SKYRIZI induction, mean total cholesterol increased by 9.4 mg/dL from baseline to a mean absolute value of 175.1 mg/dL at Week 12. Similarly, mean LDL-C increased by 6.6 mg/dL from baseline to a mean absolute value of 92.6 mg/dL Week 12. Following maintenance treatment with SKYRIZI, mean LDL-C increased by 2.3 mg/dL from baseline to Week 52, to an absolute value of 102.2 mg/dL.
Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products, including other risankizumab products, may be misleading.
Plaque Psoriasis
By Week 52, approximately 24% (263/1079) of subjects treated with SKYRIZI at the recommended dose developed antibodies to risankizumabrzaa. Of the subjects who developed antibodies to risankizumab-rzaa, approximately 57% (14% of all subjects treated with SKYRIZI) had antibodies that were classified as neutralizing. Higher antibody titers in approximately 1% of subjects treated with SKYRIZI were associated with lower risankizumab-rzaa concentrations and reduced clinical response.
Psoriatic Arthritis
By Week 28, approximately 12.1% (79/652) of subjects treated with SKYRIZI at the recommended dose developed antibodies to risankizumab-rzaa. None of the subjects who developed antibodies to risankizumab-rzaa had antibodies that were classified as neutralizing. Antibodies to risankizumab-
14 Revista Puertorriqueña de Medicina y Salud Pública DO NOT RE-SIZE
Drug Reactions SKYRIZI N = 1306 n (%) Placebo N = 300 n (%) Upper respiratory infectionsa 170 (13.0) 29 (9.7) Headacheb 46 (3.5) 6 (2.0) Fatiguec 33 (2.5) 3 (1.0) Injection site reactionsd 19 (1.5) 3 (1.0) Tinea infectionse 15 (1.1) 1 (0.3)
Table 1. Adverse Drug Reactions Occurring in ≥ 1% of Subjects on SKYRIZI through Week 16
Adverse
Drug Reactions SKYRIZI 600 mg Intravenous Infusiona N = 620 n (%) Placebo N = 432 n (%) Upper respiratory infectionsb 66 (10.6) 40 (9.3) Headachec 41 (6.6) 24 (5.6) Arthralgia 31 (5.0) 19 (4.4) a
2.
Adverse
SKYRIZI
Maintenance Study (CD-3) Adverse Drug Reactions SKYRIZI 360 mg Subcutaneous Injectiona N = 142 n (%) Placebo N = 143 n (%) Arthralgia 13 (9.2) 12 (8.4) Injection site reactionsb,c 8 (5.6) 4 (2.8) Abdominal paind 12 (8.5) 6 (4.2) Anemia 7 (4.9) 6 (4.2) Pyrexia 7 (4.9) 4 (2.8) Back pain 6 (4.2) 3 (2.1) Arthropathy 5 (3.5) 2 (1.4) Urinary tract infection 5 (3.5) 4 (2.8)
a
Adverse Drug
Reactions
20070464 Skyrizi PB-7.625 x 10.5 (1.75).indd 1 /22/Jun2022 8:39 AM US-SKZD-220291
rzaa were not associated with changes in clinical response for psoriatic arthritis. A higher proportion of subjects with anti-drug antibodies experienced hypersensitivity reactions (6.3% (5/79)) and injection site reactions (2.5% (2/79)) compared to subjects without anti-drug antibodies (3.8% (22/574) with hypersensitivity reactions and 0.7% (4/574) with injection site reactions). None of these hypersensitivity and injection site reactions led to discontinuation of risankizumab-rzaa.
Crohn’s Disease
By Week 64, approximately 3.4% (2/58) of subjects treated with SKYRIZI at the recommended induction and maintenance dosages developed antibodies to risankizumab-rzaa. None of the subjects who developed antibodies to risankizumab-rzaa had antibodies that were classified as neutralizing.
Postmarketing Experience
The following adverse reactions have been reported during post-approval of SKYRIZI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to SKYRIZI exposure:
• Skin and subcutaneous tissue disorders: eczema and rash
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors outcomes in women who become pregnant while treated with SKYRIZI. Patients should be encouraged to enroll by calling 1-877-302-2161 or visiting http://glowpregnancyregistry.com.
Risk Summary
Available pharmacovigilance and clinical trial data with risankizumab use in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Although there are no data on risankizumab-rzaa, monoclonal antibodies can be actively transported across the placenta, and SKYRIZI may cause immunosuppression in the in utero-exposed infant There are adverse pregnancy outcomes in women with inflammatory bowel disease (see Clinical Considerations)
In an enhanced pre- and post-natal developmental toxicity study, pregnant cynomolgus monkeys were administered subcutaneous doses of 5 or 50 mg/kg risankizumab-rzaa once weekly during the period of organogenesis up to parturition. Increased fetal/infant loss was noted in pregnant monkeys at the 50 mg/kg dose (see Data). The 50 mg/kg dose in pregnant monkeys resulted in approximately 10 times the exposure (AUC) in humans administered the 600 mg induction regimen and 39 times the exposure (AUC) to the 360 mg maintenance doses, respectively. No risankizumab-rzaa-related effects on functional or immunological development were observed in infant monkeys from birth through 6 months of age. The clinical significance of these findings for humans is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and embryo/fetal risk
Published data suggest that the risk of adverse pregnancy outcomes in women with inflammatory bowel disease is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Fetal/Neonatal adverse reactions
Transport of endogenous IgG antibodies across the placenta increases as pregnancy progresses, and peaks during the third trimester. Because risankizumab may interfere with immune response to infections, risks and benefits should be considered prior to administering live vaccines to infants exposed to SKYRIZI in utero. There are insufficient data regarding infant serum levels of risankizumab at birth and the duration of persistence of risankizumab in infant serum after birth. Although a specific timeframe to delay live virus immunizations in infants exposed in utero is unknown, a minimum of 5 months after birth should be considered because of the half-life of the product.
Data
Animal Data
An enhanced pre- and post-natal developmental toxicity study was conducted in cynomolgus monkeys. Pregnant cynomolgus monkeys were administered weekly subcutaneous doses of risankizumab-rzaa of 5 or 50 mg/kg from gestation day 20 to parturition, and the cynomolgus monkeys (mother and infants) were monitored for 6 months after
delivery. No maternal toxicity was noted in this study. There were no treatment-related effects on growth and development, malformations, developmental immunotoxicology, or neurobehavioral development. However, a dose-dependent increase in fetal/infant loss was noted in the risankizumab-rzaa-treated groups (32% and 43% in the 5 mg/kg and 50 mg/kg groups, respectively) compared with the vehicle control group (19%). The increased fetal/infant loss in the 50 mg/kg group was considered to be related to risankizumab-rzaa treatment. The no observed adverse effect level (NOAEL) for maternal toxicity was identified as 50 mg/kg and the NOAEL for developmental toxicity was identified as 5 mg/kg. On an exposure (AUC) basis, the 5 mg/kg dose in pregnant monkeys resulted in approximately 1.24 times the exposure in humans administered the 600 mg induction regimen and 5 times the exposure in humans administered the 360 mg maintenance doses, respectively. In the infants, mean serum concentrations increased in a dose-dependent manner and were approximately 17%-86% of the respective maternal concentrations. Following delivery, most adult female cynomolgus monkeys and all infants from the risankizumab-rzaa-treated groups had measurable serum concentrations of risankizumab-rzaa up to 91 days postpartum. Serum concentrations were below detectable levels at 180 days postpartum.
Lactation
Risk Summary
There are no data on the presence of risankizumab-rzaa in human milk, the effects on the breastfed infant, or the effects on milk production. Endogenous maternal IgG and monoclonal antibodies are transferred in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to risankizumab-rzaa are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SKYRIZI and any potential adverse effects on the breastfed infant from SKYRIZI or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of SKYRIZI have not been established in pediatric patients.
Geriatric Use
Of the 2234 subjects with plaque psoriasis exposed to SKYRIZI, 243 subjects were 65 years or older and 24 subjects were 75 years or older. No overall differences in SKYRIZI exposure, safety, or effectiveness were observed between older and younger subjects who received SKYRIZI. However, the number of subjects aged 65 years and older was not sufficient to determine whether they respond differently from younger subjects.
Clinical studies of SKYRIZI for the treatment of Crohn’s disease did not include sufficient numbers of subjects 65 years of age and older to determine whether they respond differently from younger adult subjects. No clinically meaningful differences in the pharmacokinetics of risankizumab-rzaa were observed in geriatric subjects compared to younger adult subjects with Crohn’s disease.
PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Hypersensitivity Reactions
Advise patients to discontinue SKYRIZI and seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions].
Infections
Inform patients that SKYRIZI may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider and contacting their healthcare provider if they develop any symptoms of an infection [see Warnings and Precautions]
Hepatotoxicity in Treatment of Crohn’s Disease
Inform patients that SKYRIZI may cause liver injury, especially during the initial 12 weeks of treatment. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of liver dysfunction. (e.g., unexplained rash, nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine) [see Warnings and Precautions].
Administration of Vaccines
Advise patients that vaccination with live vaccines is not recommended during SKYRIZI treatment and immediately prior to or after SKYRIZI treatment. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Instruct patients to inform the healthcare practitioner that they are taking SKYRIZI prior to a potential vaccination [see Warnings and Precautions]
Administration Instruction
Instruct patients or caregivers to perform the first self-injected dose under the supervision and guidance of a qualified healthcare professional for training in preparation and administration of SKYRIZI, including choosing anatomical sites for administration, and proper subcutaneous injection technique.
If using SKYRIZI 75 mg/0.83 mL, instruct patients or caregivers to administer two 75 mg single-dose syringes to achieve the full 150 mg dose of SKYRIZI. Instruct patients or caregivers in the technique of pen or syringe disposal.
Pregnancy
Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to SKYRIZI during pregnancy and patients can call 1-877-302-2161 [see Use in Specific Populations]
Manufactured by: AbbVie Inc.
North Chicago, IL 60064, USA
US License Number 1889
SKYRIZI® is a registered trademark of AbbVie Biotechnology Ltd. © 2019-2022 AbbVie Inc.
Ref: 20070464 Revised: June, 2022
LAB-7523 MASTER
US-SKZD-220291
Revista Puertorriqueña de Medicina y Salud Pública 15 DO NOT RE-SIZE
20070464 Skyrizi PB-7.625 x 10.5 (1.75).indd 2 /22/Jun2022 8:39 AM
US-SKZD-220291
DERMATITIS ATÓPICA CRITERIOS
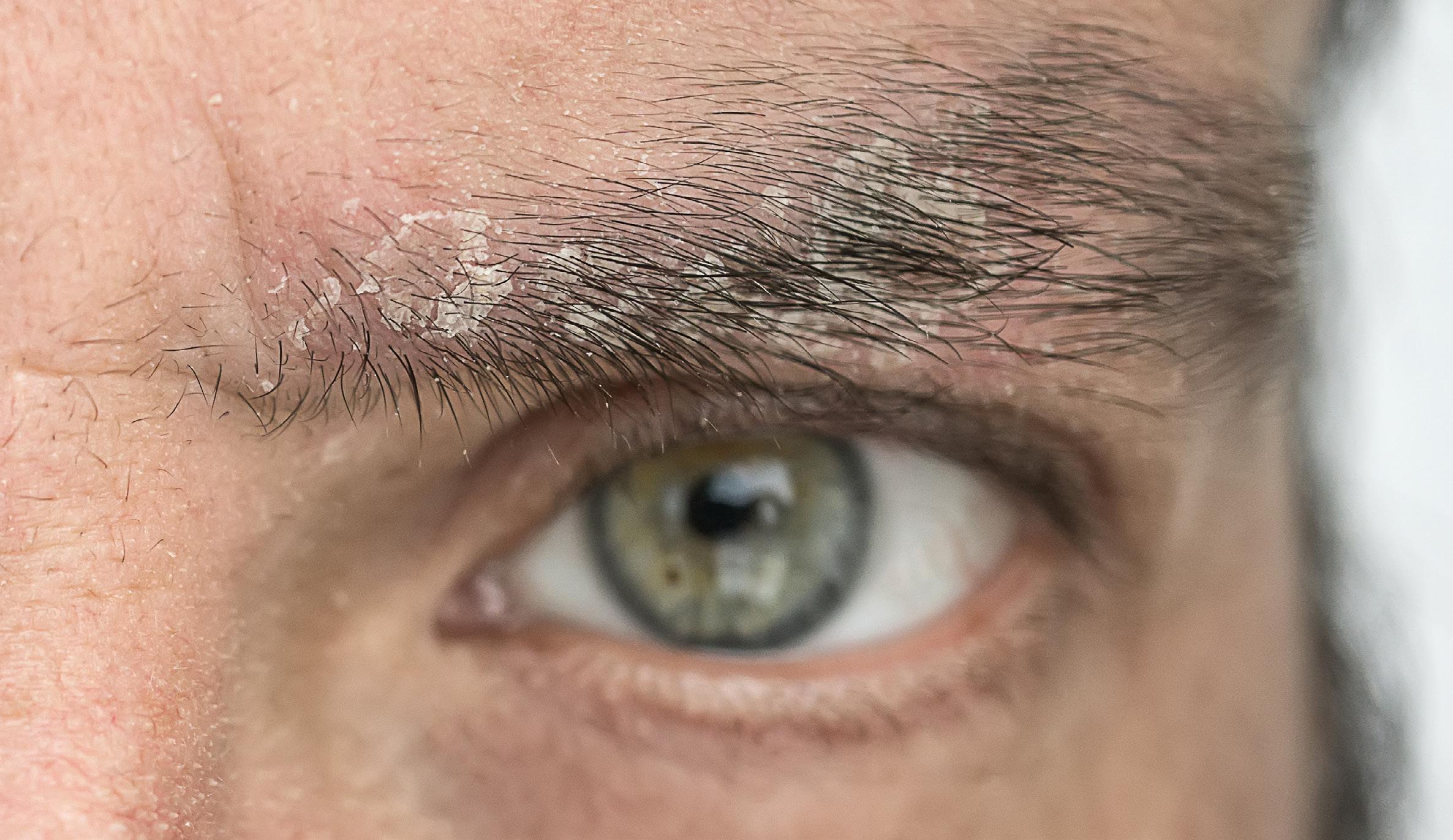
DIAGNÓSTICOS Y EXACERBACIONES ASOCIADAS A LA CONDICIÓN
Palabras clave
Dermatitis atópica, triada atópica, picor, eczema
RESUMEN
La dermatitis atópica (DA) es una condición crónica caracterizada por exacerbaciones y remisiones. Esta condición afecta a pacientes de todas las edades, muchos tienen un historial familiar o personal de condiciones atópicas. Los síntomas cardinales de la condición son picor, morfología típica y
16 Revista Puertorriqueña de Medicina y Salud Pública
MSP ARTÍCULO / DE REVISIÓN
Diana V. Rodríguez Rivera MD Residente de tercer año, Departamento

distribución de acuerdo con la edad del paciente. Los extremos de temperatura, alergias ambientales e infecciones pueden provocar una exacerbación. La condición afecta la calidad de vida de los pacientes y se ha visto asociada con susceptibilidad a infecciones, trastornos del sueño, enfermedades oculares, enfermedades mentales, entre otras. Existen diversos tratamientos comenzando con evitar alergenos e irritantes, corticoesteroides tópicos (ET), agentes tópicos no esteroidales, fototerapia, inmunosupresores, biológicos e inhibidores de JAK.
INTRODUCCIÓN
urbanas y países de altos ingresos. La prevalencia está aumentando y se entiende que el 10-30% de los niños y 2-10% de los adultos la padecen. En PR, la prevalencia en niños de 6-7 años es de 24.8%. La frecuencia de visitas médicas ha aumentado en los últimos años, principalmente a médicos primarios debido a problemas agudos en niños de 0-4 años. Estos pacientes presentan una alta utilización de los recursos del sistema de salud representando un alto costo para el mismo. A pesar de la alta prevalencia de la condición, estudios han encontrado un bajo diagnóstico ya que muchos de los pacientes con síntomas no han sido diagnosticados ni tratados apropiadamente. Esta condición presenta un problema de salud pública en nuestro país.
Revista Puertorriqueña de Medicina y Salud Pública 17 MSP ARTÍCULO / DE REVISIÓN
de Dermatología, Universidad de Puerto Rico
La DA es una condición crónica común que se ve principalmente en zonas
DISCUSIÓN
La mitad de los pacientes con DA debutan durante el primer año de vida y la mayoría de ellos antes de los 5 años. Existen tres subtipos-comienzo temprano, el cual presenta durante los primeros dos años de vida; comienzo tardío, luego de la pubertad y senil, luego de los 60 años. La DA es uno de los componentes de la triada atópica (TA) la cual está compuesta por asma, rinitis alérgica y DA. Muchos de los pacientes que padecen de DA tienen alguna otra condición de la TA. Estudios recientes encontraron una prevalencia de asma en pacientes con DA de 25.7%. En PR, la alta prevalencia de DA correlaciona con la alta prevalencia de asma observada en la población². La mayor parte de los pacientes con DA tienen familiares con condiciones atópicas, a mayor duración de su dermatitis, mayor el riesgo a padecer de alergias y se encuentra más frecuentemente un historial familiar de eczema, rinitis alérgica y asma.
Se entiende que múltiples factores contribuyen al desarrollo de la condición, incluyendo la alteración en la función de barrera de la piel, una flora microbiana alterada, alergenos e irritantes y desregulación inmunológica. En muchos de los pacientes se ve una mutación en el gen de filagrina el cual afecta la integridad de la piel y la hace más susceptible a factores externos. La comida no parece influenciar el riesgo a padecer DA. Los criterios diagnósticos incluyen picor, morfología típica y patrones de distribución específicos a la edad. La presentación varía por edad y usualmente mejora a medida que el paciente crece.
La presentación infantil ocurre en niños menores de dos años, principalmente en la cara (mejillas), cuero cabelludo y superficies extensoras con pápulas o pápulovesiculas edematosas, palidez o enrojecimiento facial y puede coexistir con la dermatitis seborreica.
La presentación de niñez ocurre entre los 2 y 12 años, es más crónica, favorece las superficies flexoras y la resequedad de la piel es difusa y evidente.
La presentación en adolescentes y adultos es similar y ocurre en pacientes mayores de 12 años. Esta se caracteriza por placas liquenificadas, ocurre en áreas flexoras, cara, cuello, brazos, espalda y superficies acrales. Si
está presente desde la niñez suele ser más severa y resistente a tratamiento. También tiene variantes de prurigo nodularis, dermatitis de los párpados o dermatitis en las manos.
LA PRESENTACIÓN SENIL OCURRE EN ADULTOS MAYORES DE 60 AÑOS Y SE CARACTERIZA POR RESEQUEDAD.
Las exacerbaciones agudas se caracterizan por enrojecimiento, hinchazón, vesículas, exudado y costra. La presentación subaguda y crónica se caracteriza por liquenificación, pápulas, nódulos y excoriaciones.
Existen múltiples factores que pueden provocar una exacerbación de la condición como los extremos en temperatura, alergias ambientales (ácaros del polvo, polen, alergenos de contacto), irritantes (telas ásperas, detergentes) e infecciones tanto cutáneas (Staphylococcus aureus, molusco contagioso) como sistémicas (infección de tracto respiratorio alto). La presencia de moho en los hogares puertorriqueños se ha asociado con la DA. Las alergias a alimentos son un factor provocante en una minoría de los pacientes. Los alergenos más comunes son el huevo, leche, maní, frutos secos, crustáceos, soya y/o trigo.
Existen varias características asociadas a la DA entre ellas los pliegues en el párpado inferior (líneas de Dennie Morgan), las ojeras alérgicas, palidez centrofacial, pitiriasis alba, pliegues en el cuello anterior, hipo o hiper pigmentación post inflamatoria, prominencia folicular, keratosis pilaris, excoriaciones, xerosis, ictiosis vulgar, hiperlinearidad de las palmas y/o plantas.
También existen variantes de la DA, entre ellas, la dermatitis de la cabeza y cuello, eczema de los oídos, eczema de los párpados, eczema del pezón, dishidrosis, dermatitis de las manos, dermatitis numular, entre otras.
Los pacientes son más susceptibles a
infecciones por virus (herpes, molusco contagioso, y papiloma humano), bacterias (Staphylococcus aureus) y dermatofitos (tricophyton rubrum). También presentan complicaciones oculares (keratoconjuntivitis, keratoconus, desprendimiento de la retina, entre otras). Las comorbilidades asociadas a la condición se han estudiado de manera extensa y se encontró relación a condiciones autoinmunes (vitiligo, alopecia areata), enfermedades mentales (ansiedad, trastornos del estado de ánimo, síndrome de déficit de atención e hiperactividad, abuso de sustancias controladas), trastorno de sueño, desórdenes hematológicos y linfoides, osteopenia y osteoporosis, y síndrome metabólico. También se encontró una alta incidencia de ausentismo al trabajo y gran afección a la calidad de vida de los pacientes y sus familiares. Se propone que el impacto en la calidad de vida en pacientes pediátricos se debe en parte a la baja utilización de terapias sistémicas, pocas opciones de tratamiento y preocupación acerca de los posibles efectos adversos relacionados al uso a largo plazo de tratamientos inmunosupresores.
Se deben evitar irritantes, utilizar detergente, jabones y humectantes (crema preferiblemente) sin fragancia ni olor y limitar la cantidad de baños ya que esto puede empeorar la resequedad y, por lo tanto, llevar a falta de control de la condición. Se recomienda evitar alergenos como los ácaros, polen, esporas y algún alimento que se haya visto que exacerbe la condición. En algunas ocasiones se recomiendan baños de cloro o antibióticos antiestafilococo en aquellos pacientes que estén colonizados. Los antihistamínicos sedantes pueden ayudar en la fase aguda para aliviar la alteración del sueño, pero los estudios más recientes encontraron que no ayudan con el picor a largo plazo ya que no es mediado por histamina.
En cuanto al manejo, se recomienda un acercamiento proactivo comenzando con la educación al paciente, para que entiendan su condición y sepan cómo manejarla.
18 Revista Puertorriqueña de Medicina y Salud Pública MSP ARTÍCULO / DE REVISIÓN
Los tratamientos para la condición son variados y van acorde a la severidad. Se comienza con ET, la potencia se elige basado en la localización y severidad de las lesiones. Es importante la educación al paciente acerca del uso apropiado, beneficios y posibles efectos secundarios de los ET. El miedo que existe al uso de ET lleva a pobre adherencia al tratamiento y falta de control de la condición. Los inhibidores de calcineurina tópicos (tacrolimus o pimecrolimus) y el crisaborole son antiinflamatorios no esteroidales, por lo tanto, no conllevan los mismos efectos secundarios de los ET. En casos de condición moderada a severa, se puede utilizar la fototerapia, cursos cortos de esteroides sistémicos, e inmunosupresores clásicos (metotrexato, micofenolato mofetil, azatioprina, o ciclosporina).
En los últimos años, gracias a la investigación científica, se ha estudiado mucho la patofisiología de la condición. Se entiende que los nervios sensoriales aferentes tienen un rol clave en la transmisión del picor en la DA utilizando citoquinas, neurotransmisores, lípidos, proteasas, péptidos antimicrobianos, agonistas de los canales iónicos y varios receptores asociados a proteínas G.
A medida que se entienden las causas de la condición, se van desarrollando medicamentos dirigidos a la misma. En el 2017, el FDA aprobó el primer medicamento biológico dirigido hacia el receptor de la interleukina 4/13, el dupilumab, para el tratamiento de la DA. En el último año (2022), el FDA aprobó varios medicamentos para la DA, el biológico tralokinumab (anti-IL13), los inhibidores de JAK, upadacitinib (JAK1) y abrocitinib (JAK1).
CONCLUSIÓN
La DA es muy común y representa un problema de salud pública en PR. Esta condición se ve en todas las edades y los pacientes van a buscar ayuda tanto de sus médicos primarios y emergenciólogos, como dermatólogos y/o alergistas. Es trabajo de todos conocer los síntomas cardinales de la condición para hacer un diagnóstico y principios de manejo para ayudar a nuestros pacientes. También es importante tener en cuenta las comorbilidades que los pueden
afectar para llevar a cabo las pruebas de cernimiento pertinentes y proveer un cuidado holístico a nuestros pacientes.
REFERENCIAS
Bolognia JL, Schaffer JV, Cerroni L. Dermatology Fourth Edition (2018) Volume 1 Chapter 12 Atopic Dermatitis McAleer MA, O’Reagan GM, Irvine AD.
Maymi MA, Somolinos AL, Nazario CM, Sanchez JL. The prevalence of atopic dermatitis in Puerto Rican school children PR Health Sci J. 2007 Jun; 26(2):127-33
Singh P, Silverberg JI. Outpatient utilization patterns for atopic dermatitis in the United States. J Am Acad Dermatol. 2023 Feb; 88(2): 357-363
Eckert L, Gupta S, Amand C, Gadkari A, Mahajan P, Gelfand JM. The burden of atopic dermatitis in US adults: Health care resource utilization data from the 2013 National Health and Wellness Survey. J Am Acad Dermatol 2018 Jan; 78 (1): 5461
Yang G, Han YY, Forno E, Acosta-Perez E, Colon-Semidey A, Alvarez M, Canino G, Chen W, Celedon JC. Under-diagnosis of atopic dermatitis in Puerto Rican children World Allergy Organ J. 2019 Jan 26; 12(1): 100003.
Ravnborg N, Ambikaibalan D, AgnihotriG, Price S, Rastogi S, Patel KR, Singam V, Andersen Y, Halling AS, Silverberg JI, EgebergA, Thyssen JP. Prevalence of asthma in patients with atopic dermatitis: a systematic review and meta-analysis. J Am Acad Dermatol. 2021 Feb; 84(2): 471-478
Del Pozo DV, Zhu Y, Mitra N, Hoffstad OJ, Margolis DJ. The risk of atopic comorbidities and atopic march progression among Black and White children with mild-to-moderate atopic dermatitis: a cross-sectional study. J Am Acad Dermatol. 2022 Nov; 87(5): 11451147
Shaheen MS, Silverberg JI. Atopic dermatitis is associated with osteoporosis and osteopenia in older adults. J Am Acad Dermatol. 2019 Feb; 80(2): 550-551
Eckert L, Gupta S, Amand C, Gadkari A, Mahajan P, Gelfand JM. Impact of atopic dermatitis on health-related quality of life and productivity in adults in the United States: an analysis using the National Health and Wellness Survey. J Am Acad
Dermatol. 2017 Aug; 77(2): 274-279
Paller AS, Guttman-Yassky E, Schuttelaar MLA, Irvine AD, Baselga E, Kataoka Y, Antila M, de Bruin-Weller MS, Marcoux D, Abramova A, Rizova E, Liu C, Zhang A. Disease characteristics, comorbidities, treatment patterns and quality of life impact in children <12 years old with atopic dermatitis: Interim results from the PEDISTAD Real-World Registry. J Am Acad Dermatol. 2022 Nov; 87(5): 1104-1108
Fan R, Leasure AC, Damsky W, Cohen JM. Association of atopic dermatitis with attention-deficit hyperactivity disorder among US adults in the “All of Us” research program: A case-control study. J Am Acad Dermatol. 2022 Sep; 87(3): 691-692
Huang AH, Roh YS, Sutaria N, Choi J, Williams KA, Canner JK, Grossberg AL, Kwatra SG. Real-world comorbidities of atopic dermatitis in the pediatric ambulatory population in the United States. J Am Acad Dermatol. 2021 Oct; 85(4): 893-900
Roh YS, Huang AH, Sutaria N, Choi U, Wongvibulsin S, Choi J, Bordeaux ZA, Parthasarathy V, Deng J, Patel DP, Canner JK, Grossberg AL, Kwatra SG. Real-world comorbidities of atopic dermatitis in the US adult ambulatory population. J Am Acad Dermatol. 2022 Apr; 86 (4) 835-845.
Davis DMR, Drucker AM, Alikhan A, Bercovitch L, Cohen DE, Darr JM, Eichenfield LF, Frazer-Green L, Paller AS, Silverberg JI, Singh AM, Sidbury R. American Academy of Dermatology Guidelines: Awareness of comorbidities associated with atopic dermatitis in adults. J Am Acad Dermatol. 2022 Jun; 86(6)1335-1336
Li AW, Yin ES, Antaya RJ. Topical Corticosteroid Phobia in Atopic Dermatitis: A Systematic Review. JAMA Dermatol. 2007 Oct 1; 153(10): 1036-1042
Kwatra SG, Misery L, Clibborn C, Steinhoff M. Molecular and cellular mechanisms of itch and pain in atopic dermatitis and implications for novel therapeutics. Clin Transl Immunology. 2022 May 9; 11(5): e1390
Revista Puertorriqueña de Medicina y Salud Pública 19 MSP ARTÍCULO / DE REVISIÓN

20 Revista Puertorriqueña de Medicina y Salud Pública

Revista Puertorriqueña de Medicina y Salud Pública 21

22 Revista Puertorriqueña de Medicina y Salud Pública

Revista Puertorriqueña de Medicina y Salud Pública 23

24 Revista Puertorriqueña de Medicina y Salud Pública

Revista Puertorriqueña de Medicina y Salud Pública 25

26 Revista Puertorriqueña de Medicina y Salud Pública

For moderate to severe rheumatoid arthritis (RA) in adult TNFi-IR patients1
For moderate to severe rheumatoid arthritis (RA) in adult TNFi-IR patients1
EXPECTATIONS
EXPECTATIONS
CHALLENGE TREATMENT GOALS IN RA WITH A ONCE-DAILY ORAL JAK INHIBITOR
CHALLENGE TREATMENT GOALS IN RA WITH A ONCE-DAILY ORAL JAK INHIBITOR
RINVOQ met primary (ACR20 or ACR50 at Week 12 or 14) and ranked secondary endpoints in clinical trials, with some patients achieving ACR20 as early as Week 1 in SELECT-BEYOND1-3,a,b
RINVOQ met primary (ACR20 or ACR50 at Week 12 or 14) and ranked secondary endpoints in clinical trials, with some patients achieving ACR20 as early as Week 1 in SELECT-BEYOND1-3,a,b
LONG-TERM REMISSION AND LOW DISEASE ACTIVITY DATA observed up to 84 weeks with or without MTX1,3-6
LONG-TERM REMISSION AND LOW DISEASE ACTIVITY DATA observed up to 84 weeks with or without MTX1,3-6
• DAS28-CRP<2.6* and DAS28-CRP≤3.2 evaluated at Week 12 or 14, with response rates from 60 to 84 weeks (in SELECT-BEYOND and SELECT-MONOTHERAPY, respectively)
• DAS28-CRP<2.6* and DAS28-CRP≤3.2 evaluated at Week 12 or 14, with response rates from 60 to 84 weeks (in SELECT-BEYOND and SELECT-MONOTHERAPY, respectively)
*Clinical remission does not mean drug-free remission or complete absence of disease activity.
Clinical remission does not mean drug-free remission or complete absence of disease activity.

LONG-TERM SAFETY DATA
AEs observed in long-term analysis with ~4.5 years maximum and ~2.6 years median exposure to RINVOQ 15 mg as of 6/30/207,a,b
LONG-TERM SAFETY DATA
AEs observed in long-term analysis with ~4.5 years maximum and ~2.6 years median exposure to RINVOQ 15 mg as of 6/30/207,a,b
• >4400 patients evaluated on upadacitinib,c with >7000 patientyears of long-term exposure to RINVOQ 15 mg as of 6/30/207,a,b
• >4400 patients evaluated on upadacitinib,c with >7000 patientyears of long-term exposure to RINVOQ 15 mg as of 6/30/207,a,b
Discover our commitment to exceptional access and patient support at RinvoqHCPPR.com

Discover
our commitment to exceptional access and patient support at RinvoqHCPPR.com
a SELECT-EARLY (RA-I; MTX-naïve) [primary endpoint at Week 12: ACR50 response vs MTX, select ranked secondary endpoint at Week 24: Δ mTSS vs MTX]; SELECT-MONOTHERAPY (RA-II; MTX-IR) [primary endpoint at Week 14: ACR20 response vs MTX, select ranked secondary endpoints at Week 14: DAS28-CRP<2.6 vs MTX, DAS28-CRP≤3.2 vs MTX]; SELECT-NEXT (RA-III; csDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD]; SELECT-COMPARE (RA-IV; MTX-IR) [RINVOQ + MTX; primary endpoint at Week 12: ACR20 response vs placebo
+ MTX, select ranked secondary endpoints at Week 26: Δ mTSS vs placebo + MTX]; SELECT-BEYOND (RA-V; bDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD, select ranked secondary endpoints at Week 12: DAS28-CRP≤3.2 vs placebo + csDMARD.] 1,2 bSELECT-CHOICE (bDMARD-IR) [RINVOQ + csDMARDs; primary endpoint at Week 12: Δ DAS28-CRP (noninferiority) vs active comparator + csDMARDs]. 8 c RINVOQ 15 mg; upadacitinib 30 mg; RINVOQ 15 mg is the approved dose.1,7
INDICATION1
a SELECT-EARLY (RA-I; MTX-naïve) [primary endpoint at Week 12: ACR50 response vs MTX, select ranked secondary endpoint at Week 24: Δ mTSS vs MTX]; SELECT-MONOTHERAPY (RA-II; MTX-IR) [primary endpoint at Week 14: ACR20 response vs MTX, select ranked secondary endpoints at Week 14: DAS28-CRP<2.6 vs MTX, DAS28-CRP≤3.2 vs MTX]; SELECT-NEXT (RA-III; csDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD]; SELECT-COMPARE (RA-IV; MTX-IR) [RINVOQ + MTX; primary endpoint at Week 12: ACR20 response vs placebo + MTX, select ranked secondary endpoints at Week 26: Δ mTSS vs placebo + MTX]; SELECT-BEYOND (RA-V; bDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD, select ranked secondary endpoints at Week 12: DAS28-CRP≤3.2 vs placebo + csDMARD.] 1,2 bSELECT-CHOICE (bDMARD-IR) [RINVOQ + csDMARDs; primary endpoint at Week 12: Δ DAS28-CRP (noninferiority) vs active comparator + csDMARDs]. 8 c RINVOQ 15 mg; upadacitinib 30 mg; RINVOQ 15 mg is the approved dose.1,7
RINVOQ is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
INDICATION1


RINVOQ is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
Limitation of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
Limitation of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
SAFETY CONSIDERATIONS1
Malignancies: Lymphoma and other malignancies have been observed in RINVOQ-treated patients. A higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with another JAK inhibitor when compared with TNF blockers in RA patients. Patients who are current or past smokers are at additional increased risk.
Malignancies: Lymphoma and other malignancies have been observed in RINVOQ-treated patients. A higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with another JAK inhibitor when compared with TNF blockers in RA patients. Patients who are current or past smokers are at additional increased risk.
Major Adverse Cardiovascular Events: A higher rate of CV death, myocardial infarction, and stroke was observed with a JAK inhibitor in a study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years of age with at least one CV risk factor. Current or past smokers are at additional increased risk.
SAFETY CONSIDERATIONS1
Serious Infections: Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. These infections include tuberculosis (TB), invasive fungal, bacterial, viral, and other infections due to opportunistic pathogens. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids.
Serious Infections: Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. These infections include tuberculosis (TB), invasive fungal, bacterial, viral, and other infections due to opportunistic pathogens. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids.
Mortality: A higher rate of all-cause mortality, including sudden cardiovascular (CV) death, was observed with a Janus kinase (JAK) inhibitor in a study comparing another JAK inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years of age with at least one CV risk factor.
Mortality: A higher rate of all-cause mortality, including sudden cardiovascular (CV) death, was observed with a Janus kinase (JAK) inhibitor in a study comparing another JAK inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years of age with at least one CV risk factor.
Major Adverse Cardiovascular Events: A higher rate of CV death, myocardial infarction, and stroke was observed with a JAK inhibitor in a study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years of age with at least one CV risk factor. Current or past smokers are at additional increased risk.
Thrombosis: Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. A higher rate of thrombosis was observed with another JAK inhibitor when compared with TNF blockers in RA patients.
Thrombosis: Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. A higher rate of thrombosis was observed with another JAK inhibitor when compared with TNF blockers in RA patients.
Hypersensitivity: RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients.
Hypersensitivity: RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients.
Other Serious Adverse Reactions: Hypersensitivity Reactions (anaphylaxis and angioedema), Gastrointestinal Perforations, Laboratory Abnormalities (neutropenia, lymphopenia, anemia, lipid elevations, liver enzyme elevations), and Embryo-Fetal Toxicity.
Other Serious Adverse Reactions: Hypersensitivity Reactions (anaphylaxis and angioedema), Gastrointestinal Perforations, Laboratory Abnormalities (neutropenia, lymphopenia, anemia, lipid elevations, liver enzyme elevations), and Embryo-Fetal Toxicity.
Please see additional Important Safety Information, including BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis, on the previous page of this advertisement. Please see Brief Summary of full Prescribing Information on previous pages of this advertisement.
Please see additional Important Safety Information, including BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis, on the previous page of this advertisement.
ACR=American College of Rheumatology; AEs=adverse events; bDMARD=biologic DMARD; csDMARD=conventional synthetic DMARD; DAS28-CRP=Disease Activity Score 28 joints, C-reactive protein; DMARD=disease-modifying antirheumatic drug; HAQ-DI=Health Assessment
Please see Brief Summary of full Prescribing Information on previous pages of this advertisement.
Questionnaire Disability Index; IR=intolerance or inadequate response; JAK=Janus kinase; mTSS=modified total Sharp score; MTX=methotrexate; TNFi=tumor necrosis factor inhibitor.
ACR=American College of Rheumatology; AEs=adverse events; bDMARD=biologic DMARD; csDMARD=conventional synthetic DMARD; DAS28-CRP=Disease Activity Score 28 joints, C-reactive protein; DMARD=disease-modifying antirheumatic drug; HAQ-DI=Health Assessment Questionnaire Disability Index; IR=intolerance or inadequate response; JAK=Janus kinase; mTSS=modified total Sharp score; MTX=methotrexate; TNFi=tumor necrosis factor inhibitor.
28 Revista Puertorriqueña de Medicina y Salud Pública
RINVOQ® (RIN-VOKE) (upadacitinib) extended-release tablets, for oral use
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, and THROMBOSIS
SERIOUS INFECTIONS
Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions, Adverse Reactions]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. If a serious infection develops, interrupt RINVOQ until the infection is controlled.
Reported infections include:
• Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before RINVOQ use and during therapy. Treatment for latent infection should be considered prior to RINVOQ use.
• Invasive fungal infections, including cryptococcosis and pneumocystosis.
• Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with RINVOQ should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with RINVOQ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions]
MORTALITY
In a large, randomized, postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another Janus kinase (JAK) inhibitor to tumor necrosis factor (TNF) blockers, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed with the JAK inhibitor [see Warnings and Precautions]
MALIGNANCIES
Lymphoma and other malignancies have been observed in patients treated with RINVOQ. In RA patients treated with another JAK inhibitor, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk [see Warnings and Precautions]
MAJOR ADVERSE CARDIOVASCULAR EVENTS
In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke [see Warnings and Precautions].
THROMBOSIS
Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. Many of these adverse events were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of thrombosis was observed when compared with TNF blockers. Avoid RINVOQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ and be promptly evaluated [see Warnings and Precautions]
INDICATIONS AND USAGE
Rheumatoid Arthritis
RINVOQ® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic disease-modifying antirheumatic drugs (DMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
Psoriatic Arthritis
RINVOQ is indicated for the treatment of adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
Atopic Dermatitis
RINVOQ is indicated for the treatment of adults and pediatric patients 12 years of age and older with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies are inadvisable.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
Ulcerative Colitis
RINVOQ is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for ulcerative colitis, or with potent immunosuppressants such as azathioprine and cyclosporine.
Ankylosing Spondylitis
RINVOQ is indicated for the treatment of adults with active ankylosing spondylitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
CONTRAINDICATIONS
PROFESSIONAL BRIEF SUMMARY
CONSULT PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION
RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients [see Warnings and Precautions].
WARNINGS AND PRECAUTIONS
Serious Infections
Serious and sometimes fatal infections have been reported in patients receiving RINVOQ. The most frequent serious infections reported with RINVOQ included pneumonia and cellulitis [see Adverse Reactions]. Among opportunistic infections, tuberculosis, multidermatomal herpes zoster, oral/esophageal candidiasis, and cryptococcosis, were reported with RINVOQ.
Avoid use of RINVOQ in patients with an active, serious infection, including localized infections. Consider the risks and benefits of treatment prior to initiating RINVOQ in patients:
• with chronic or recurrent infection
• who have been exposed to tuberculosis
• with a history of a serious or an opportunistic infection
• who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
• with underlying conditions that may predispose them to infection. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with RINVOQ. Interrupt RINVOQ if a patient develops a serious or opportunistic infection.
A patient who develops a new infection during treatment with RINVOQ should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, the patient should be closely monitored, and RINVOQ should be interrupted if the patient is not responding to antimicrobial therapy. RINVOQ may be resumed once the infection is controlled.
Tuberculosis
Evaluate and test patients for latent and active tuberculosis (TB) infection prior to administration of RINVOQ. Patients with latent TB should be treated with standard antimycobacterial therapy before initiating RINVOQ. RINVOQ should not be given to patients with active TB. Consider anti-TB therapy prior to initiation of RINVOQ in patients with previously untreated latent TB or active TB in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent TB but who have risk factors for TB infection. Consultation with a physician with expertise in the treatment of TB is recommended to aid in the decision about whether initiating anti-TB therapy is appropriate for an individual patient.
During RINVOQ use, monitor patients for the development of signs and symptoms of TB, including patients who tested negative for latent TB infection prior to initiating therapy.
Viral Reactivation
Viral reactivation, including cases of herpes virus reactivation (e.g., herpes zoster) and hepatitis B virus reactivation, were reported in clinical trials with RINVOQ [see Adverse Reactions]. The risk of herpes zoster appears to be higher in patients treated with RINVOQ in Japan. If a patient develops herpes zoster, consider temporarily interrupting RINVOQ until the episode resolves. Screening for viral hepatitis and monitoring for reactivation should be performed in accordance with clinical guidelines before starting and during therapy with RINVOQ. Patients who were positive for hepatitis C antibody and hepatitis C virus RNA, were excluded from clinical trials. Patients who were positive for hepatitis B surface antigen or hepatitis B virus DNA were excluded from clinical trials. However, cases of hepatitis B reactivation were still reported in patients enrolled in the Phase 3 trials of RINVOQ. If hepatitis B virus DNA is detected while receiving RINVOQ, a liver specialist should be consulted.
Mortality
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed in patients treated with the JAK inhibitor compared with TNF blockers.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ.
Malignancy and Lymphoproliferative Disorders
Malignancies, including lymphomas, were observed in clinical trials of RINVOQ [see Adverse Reactions]
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. In this study, current or past smokers had an additional increased risk of overall malignancies.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Non-Melanoma Skin Cancer
NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
Exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen.
Major Adverse Cardiovascular Events
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of major adverse cardiovascular events (MACE) defined as cardiovascular death, non-fatal myocardial infarction (MI), and non-fatal stroke was observed with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients
should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke.
Thrombosis
Thrombosis, including deep venous thrombosis (DVT), pulmonary embolism (PE), and arterial thrombosis, have occurred in patients treated for inflammatory conditions with JAK inhibitors, including RINVOQ. Many of these adverse events were serious and some resulted in death.
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, higher rates of overall thrombosis, DVT, and PE were observed compared to those treated with TNF blockers. If symptoms of thrombosis occur, patients should discontinue RINVOQ and be evaluated promptly and treated appropriately. Avoid RINVOQ in patients that may be at increased risk of thrombosis.
Hypersensitivity Reactions
Serious hypersensitivity reactions such as anaphylaxis and angioedema were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ and institute appropriate therapy [see Adverse Reactions].
Gastrointestinal Perforations
Gastrointestinal perforations have been reported in clinical trials with RINVOQ.
Monitor RINVOQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs). Evaluate promptly patients presenting with new onset abdominal pain for early identification of gastrointestinal perforation.
Laboratory Abnormalities
Neutropenia
Treatment with RINVOQ was associated with an increased incidence of neutropenia (ANC less than 1000 cells/mm3).
Evaluate neutrophil counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation and interrupt RINVOQ treatment in patients with a low neutrophil count (i.e., ANC less than 1000 cells/mm3).
Lymphopenia
ALC less than 500 cells/mm3 were reported in RINVOQ-treated patients in clinical trials.
Evaluate lymphocyte counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low lymphocyte count (i.e., less than 500 cells/mm3).
Anemia
Decreases in hemoglobin levels to less than 8 g/dL were reported in RINVOQ-treated patients in clinical trials.
Evaluate hemoglobin at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low hemoglobin level (i.e., less than 8 g/dL).
Lipids
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol [see Adverse Reactions]
Elevations in LDL cholesterol decreased to pre-treatment levels in response to statin therapy. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Assess lipid parameters approximately 12 weeks after initiation of treatment, and thereafter according to the clinical guidelines for hyperlipidemia. Manage patients according to clinical guidelines for the management of hyperlipidemia.
Liver Enzyme Elevations
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevations compared to treatment with placebo.
Evaluate liver enzymes at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury.
If increases in ALT or AST are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ should be interrupted until this diagnosis is excluded.
Embryo-Fetal Toxicity
Based on findings in animal studies, RINVOQ may cause fetal harm when administered to a pregnant woman. Administration of upadacitinib to rats and rabbits during organogenesis caused increases in fetal malformations. Verify the pregnancy status of patients of reproductive potential prior to starting treatment. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception during treatment with RINVOQ and for 4 weeks following completion of therapy [see Use in
Specific Populations]
Vaccinations
Avoid use of live vaccines during, or immediately prior to, RINVOQ therapy. Prior to initiating RINVOQ, it is recommended that patients be brought up to date with all immunizations, including varicella zoster or prophylactic herpes zoster vaccinations, in agreement with current immunization guidelines.
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
• Serious Infections [see Warnings and Precautions]
• Mortality [see Warnings and Precautions]
• Malignancy and Lymphoproliferative Disorders [see Warnings and Precautions]
• Major Adverse Cardiovascular Events [see Warnings and Precautions]
• Thrombosis [see Warnings and Precautions]
• Hypersensitivity Reactions [see Warnings and Precautions]
• Gastrointestinal Perforations [see Warnings and Precautions]
• Laboratory Abnormalities [see Warnings and Precautions]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
30 Revista Puertorriqueña de Medicina y Salud Pública DO NOT RE-SIZE
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 1 04 May 2022 9:00 AM US-RNQR-220149
Adverse Reactions in Patients with Rheumatoid Arthritis
A total of 3833 patients with rheumatoid arthritis were treated with upadacitinib in the Phase 3 clinical trials of whom 2806 were exposed for at least one year.
Patients could advance or switch to RINVOQ 15 mg from placebo, or be rescued to RINVOQ from active comparator or placebo from as early as Week 12 depending on the trial design.
A total of 2630 patients received at least 1 dose of RINVOQ 15 mg, of whom 1860 were exposed for at least one year. In trials RA-I, RA-II, RA-III and RA-V, 1213 patients received at least 1 dose of RINVOQ 15 mg, of which 986 patients were exposed for at least one year, and 1203 patients received at least 1 dose of upadacitinib 30 mg, of which 946 were exposed for at least one year.
Table 1: Adverse Reactions Reported in ≥ 1% of Rheumatoid Arthritis Patients Treated with RINVOQ 15 mg in Placebo-controlled Trials
treated with RINVOQ 15 mg monotherapy, and 4 patients (3.2 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Opportunistic infections were reported in 7 patients (0.6 per 100 patient-years) treated with RINVOQ 15 mg and 15 patients (1.4 per 100 patient-years) treated with upadacitinib 30 mg.
Malignancies
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, malignancies excluding NMSC were reported in 1 patient (0.4 per 100 patient-years) treated with placebo, and 1 patient (0.4 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, malignancies excluding NMSC were reported in 0 patients treated with placebo, 1 patient (1.1 per 100 patient-years) treated with RINVOQ 15 mg, and 3 patients (3.5 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Malignancies excluding NMSC were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 0 patients treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Malignancies excluding NMSC were reported in 13 patients (1.2 per 100 patient-years) treated with RINVOQ 15 mg and 14 patients (1.3 per 100 patient-years) treated with upadacitinib 30 mg.
Gastrointestinal Perforations
Placebo-controlled Trials: There were no gastrointestinal perforations (based on medical review) reported in patients treated with placebo, RINVOQ 15 mg, and upadacitinib 30 mg.
and 0.7% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.5% of patients treated with placebo, 0.5% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg.
Anemia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, hemoglobin decreases below 8 g/dL in at least one measurement occurred in <0.1% of patients in both the RINVOQ 15 mg and placebo groups. In RA-III and RA-V, hemoglobin decreases below 8 g/dL in at least one measurement were observed in 0.3% of patients treated with placebo, and none in patients treated with RINVOQ 15 mg and upadacitinib 30 mg.
Adverse Reactions in Patients with Psoriatic Arthritis
A total of 1827 patients with psoriatic arthritis were treated with upadacitinib in clinical trials representing 1639.2 patient-years of exposure, of whom 722 were exposed to upadacitinib for at least one year. In the two Phase 3 trials, 907 patients received at least 1 dose of RINVOQ 15 mg, of whom 359 were exposed for at least one year.
Two placebo-controlled trials were integrated (640 patients on RINVOQ 15 mg once daily and 635 patients on placebo) to evaluate the safety of RINVOQ 15 mg in comparison to placebo for up to 24 weeks after treatment initiation.
*URTI includes: acute sinusitis, laryngitis, nasopharyngitis, oropharyngeal pain, pharyngitis, pharyngotonsillitis, rhinitis, sinusitis, tonsillitis, viral upper respiratory tract infection
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg group and at a higher rate than in the placebo group through Week 12 included pneumonia, herpes zoster, herpes simplex (includes oral herpes), and oral candidiasis.
Four integrated datasets are presented in the Specific Adverse Reaction
section:
Placebo-controlled Trials: Trials RA-III, RA-IV, and RA-V were integrated to represent safety through 12/14 weeks for placebo (n=1042) and RINVOQ 15 mg (n=1035). Trials RA-III and RA-V were integrated to represent safety through 12 weeks for placebo (n=390), RINVOQ 15 mg (n=385), and upadacitinib 30 mg (n=384). Trial RA-IV did not include the 30 mg dose and, therefore, safety data for upadacitinib 30 mg can only be compared with placebo and RINVOQ 15 mg rates from pooling trials RA-III and RA-V.
MTX-controlled Trials: Trials RA-I and RA-II were integrated to represent safety through 12/14 weeks for MTX (n=530), RINVOQ 15 mg (n=534), and upadacitinib 30 mg (n=529).
12-Month Exposure Dataset: Trials RA-I, II, III, and V were integrated to represent the long-term safety of RINVOQ 15 mg (n=1213) and upadacitinib 30 mg (n=1203).
Exposure adjusted incidence rates were adjusted by trial for all the adverse events reported in this section.
Specific Adverse Reactions
Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, infections were reported in 218 patients (95.7 per 100 patient-years) treated with placebo and 284 patients (127.8 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, infections were reported in 99 patients (136.5 per 100 patient-years) treated with placebo, 118 patients (164.5 per 100 patient-years) treated with RINVOQ 15 mg, and 126 patients (180.3 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Infections were reported in 127 patients (119.5 per 100 patient-years) treated with MTX monotherapy, 104 patients (91.8 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 128 patients (115.1 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Infections were reported in 615 patients (83.8 per 100 patient-years) treated with RINVOQ 15 mg and 674 patients (99.7 per 100 patient-years) treated with upadacitinib 30 mg.
Serious Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, serious infections were reported in 6 patients (2.3 per 100 patient-years) treated with placebo, and 12 patients (4.6 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, serious infections were reported in 1 patient (1.2 per 100 patientyears) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 7 patients (8.2 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Serious infections were reported in 2 patients (1.6 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 8 patients (6.4 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Serious infections were reported in 38 patients (3.5 per 100 patient-years) treated with RINVOQ 15 mg and 59 patients (5.6 per 100 patient-years) treated with upadacitinib 30 mg.
The most frequently reported serious infections were pneumonia and cellulitis.
Tuberculosis
Placebo-controlled Trials and MTX-controlled Trials: In the placebo-controlled period, there were no active cases of tuberculosis reported in the placebo, RINVOQ 15 mg, and upadacitinib 30 mg groups. In the MTX-controlled period, there were no active cases of tuberculosis reported in the MTX monotherapy, RINVOQ 15 mg monotherapy, and upadacitinib 30 mg monotherapy groups.
12-Month Exposure Dataset: Active tuberculosis was reported for 2 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg.
Cases of extra-pulmonary tuberculosis were reported.
Opportunistic Infections (excluding tuberculosis)
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, opportunistic infections were reported in 3 patients (1.2 per 100 patient-years) treated with placebo, and 5 patients (1.9 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, opportunistic infections were reported in 1 patient (1.2 per 100 patient-years) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (7.1 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Opportunistic infections were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 0 patients
MTX-controlled Trials: There were no cases of gastrointestinal perforations reported in the MTX and RINVOQ 15 mg group through 12/14 weeks. Two cases of gastrointestinal perforations were observed in the upadacitinib 30 mg group.
12-Month Exposure Dataset: Gastrointestinal perforations were reported in 1 patient treated with RINVOQ 15 mg and 4 patients treated with upadacitinib 30 mg.
Thrombosis
Placebo-controlled Trials: In RA-IV, venous thrombosis (pulmonary embolism or deep vein thrombosis) was observed in 1 patient treated with placebo and 1 patient treated with RINVOQ 15 mg. In RA-V, venous thrombosis was observed in 1 patient treated with RINVOQ 15 mg. There were no observed cases of venous thrombosis reported in RA-III. No cases of arterial thrombosis were observed through 12/14 weeks.
MTX-controlled Trials: In RA-II, venous thrombosis was observed in 0 patients treated with MTX monotherapy, 1 patient treated with RINVOQ 15 mg monotherapy and 0 patients treated with upadacitinib 30 mg monotherapy through Week 14. In RA-II, no cases of arterial thrombosis were observed through 12/14 weeks. In RA-I, venous thrombosis was observed in 1 patient treated with MTX, 0 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg through Week 24. In RA-I, arterial thrombosis was observed in 1 patient treated with upadacitinib 30 mg through Week 24.
12-Month Exposure Dataset: Venous thrombosis events were reported in 5 patients (0.5 per 100 patient-years) treated with RINVOQ 15 mg and 4 patients (0.4 per 100 patient-years) treated with upadacitinib 30 mg. Arterial thrombosis events were reported in 0 patients treated with RINVOQ 15 mg and 2 patients (0.2 per 100 patient-years) treated with upadacitinib 30 mg.
Laboratory Abnormalities
Hepatic Transaminase Elevations
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, alanine transaminase (ALT) and aspartate transaminase (AST) elevations ≥ 3 x upper limit of normal (ULN) in at least one measurement were observed in 2.1% and 1.5% of patients treated with RINVOQ 15 mg, and in 1.5% and 0.7% of patients treated with placebo, respectively. In RA-III and RA-V, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 1.0% of patients treated with RINVOQ 15 mg, 1.0% and 0% of patients treated with upadacitinib 30 mg and in 1.3% and 1.0% of patients treated with placebo, respectively.
In MTX-controlled trials, for up to 12/14 weeks, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 0.4% of patients treated with RINVOQ 15 mg, 1.7% and 1.3% of patients treated with upadacitinib 30 mg and in 1.9% and 0.9% of patients treated with MTX, respectively.
Lipid Elevations
Upadacitinib treatment was associated with dose-related increases in total cholesterol, triglycerides and LDL cholesterol. Upadacitinib was also associated with increases in HDL cholesterol. Elevations in LDL and HDL cholesterol peaked by Week 8 and remained stable thereafter. In controlled trials, for up to 12/14 weeks, changes from baseline in lipid parameters in patients treated with RINVOQ 15 mg and upadacitinib 30 mg, respectively, are summarized below:
• Mean LDL cholesterol increased by 14.81 mg/dL and 17.17 mg/dL.
• Mean HDL cholesterol increased by 8.16 mg/dL and 9.01 mg/dL.
• The mean LDL/HDL ratio remained stable.
• Mean triglycerides increased by 13.55 mg/dL and 14.44 mg/dL.
Creatine Phosphokinase Elevations
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related increases in creatine phosphokinase (CPK) values were observed. CPK elevations > 5 x ULN were reported in 1.0%, and 0.3% of patients over 12/14 weeks in the RINVOQ 15 mg and placebo groups, respectively. Most elevations >5 x ULN were transient and did not require treatment discontinuation. In RA-III and RA-V, CPK elevations > 5 x ULN were observed in 0.3% of patients treated with placebo, 1.6% of patients treated with RINVOQ 15 mg, and none in patients treated with upadacitinib 30 mg.
Neutropenia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in neutrophil counts, below 1000 cells/mm3 in at least one measurement occurred in 1.1% and <0.1% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in neutrophil counts below 1000 cells/mm3 in at least one measurement occurred in 0.3% of patients treated with placebo, 1.3% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg. In clinical trials, treatment was interrupted in response to ANC less than 1000 cells/mm3
Lymphopenia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.9%
Overall, the safety profile observed in patients with active psoriatic arthritis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis. During the 24-week placebo-controlled period, the frequencies of herpes zoster and herpes simplex were ≥1% (1.1% and 1.4%, respectively) with RINVOQ 15 mg and 0.8% and 1.3%, respectively with placebo. A higher incidence of acne and bronchitis was also observed in patients treated with RINVOQ 15 mg (1.3% and 3.9%, respectively) compared to placebo (0.3% and 2.7%, respectively).
Adverse Reactions in Patients with Atopic Dermatitis
Three Phase 3 (AD-1, AD-2, and AD-3) and one Phase 2b (AD-4) randomized, double-blind, placebo-controlled, multicenter trials evaluated the safety of RINVOQ in patients with moderate-to-severe atopic dermatitis. The majority of patients were White (68%) and male (57%). The mean age was 34 years (ranged from 12 to 75 years) and 13% of the patients were 12 to less than 18 years. In these 4 trials, 2612 patients were treated with RINVOQ 15 mg or 30 mg orally once daily, with or without concomitant topical corticosteroids (TCS).
In the Phase 3 clinical trials (AD-1, AD-2, and AD-3), a total of 1239 patients received RINVOQ 15 mg, of whom 791 were exposed for at least one year and 1246 patients received RINVOQ 30 mg, of whom 826 were exposed for at least one year.
Trials AD-1, AD-2, and AD-4 compared the safety of RINVOQ monotherapy to placebo through Week 16. Trial AD-3 compared the safety of RINVOQ + TCS to placebo + TCS through Week 16.
Weeks 0 to 16 (Trials AD-1 to AD-4)
In RINVOQ trials with and without TCS (Trials AD-1, 2, 3 and 4) through Week 16, the proportion of patients who discontinued treatment because of adverse reactions in the RINVOQ 15 mg, 30 mg and placebo groups were 2.3%, 2.9% and 3.8%, respectively. Table 2 summarizes the adverse reactions that occurred at a rate of at least 1% in the RINVOQ 15 mg or 30 mg groups during the first 16 weeks of treatment.
Table 2: Adverse Reactions Reported in ≥ 1% of Patients with Atopic Dermatitis Treated with RINVOQ 15 mg or 30 mg
* Includes: laryngitis, laryngitis viral, nasopharyngitis, oropharyngeal pain, pharyngeal abscess, pharyngitis, pharyngitis streptococcal, pharyngotonsillitis, respiratory tract infection, respiratory tract infection viral, rhinitis, rhinolaryngitis, sinusitis, tonsillitis, tonsillitis bacterial, upper respiratory tract infection, viral pharyngitis, viral upper respiratory tract infection
** Includes: acne and dermatitis acneiform
*** Includes: genital herpes, genital herpes simplex, herpes dermatitis, herpes ophthalmic, herpes simplex, nasal herpes, ophthalmic herpes simplex, herpes virus infection, oral herpes
**** Includes anaphylactic reaction, anaphylactic shock, angioedema, dermatitis exfoliative generalized, drug hypersensitivity, eyelid oedema, face oedema, hypersensitivity, periorbital swelling, pharyngeal swelling, swelling face, toxic skin eruption, type I hypersensitivity, urticaria
***** Includes abdominal pain and abdominal pain upper
****** Includes herpes zoster and varicella
Revista Puertorriqueña de Medicina y Salud Pública 31 DO NOT RE-SIZE
Adverse Reaction Placebo RINVOQ 15 mg n=1042 (%) n=1035 (%) Upper respiratory tract infection (URTI)* 9.5 13.5 Nausea 2.2 3.5 Cough 1.0 2.2 Pyrexia 0 1.2
Adverse Reaction Placebo RINVOQ 15 mg RINVOQ 30 mg n=902 (%) n=899 (%) n=906 (%) Upper respiratory tract infection (URTI)* 17 23 25 Acne** 2 10 16 Herpes simplex*** 2 4 8 Headache 4 6 6 Increased blood creatine phosphokinase 2 5 6 Cough 1 3 3 Hypersensitivity**** 2 2 3 Folliculitis 1 2 3 Nausea 1 3 3 Abdominal pain***** 1 3 2 Pyrexia 1 2 2 Increased Weight 1 2 2 Herpes zoster****** 1 2 2 Influenza <1 2 2 Fatigue 1 1 2 Neutropenia <1 1 2 Myalgia 1 1 2 Influenza like illness 1 1 2
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 2 04 May 2022 9:00 AM US-RNQR-220149
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg and/or 30 mg group and at a higher rate than in the placebo group through Week 16 included anemia, oral candidiasis, pneumonia, and the adverse event of retinal detachment.
The safety profile of RINVOQ through Week 52 was generally consistent with the safety profile observed at Week 16.
Overall, the safety profile observed in patients with AD treated with RINVOQ was similar to the safety profile in patients with RA. Other specific adverse reactions that were reported in patients with AD included eczema herpeticum/Kaposi’s varicelliform eruption.
Eczema Herpeticum/Kaposi’s Varicelliform Eruption
Placebo-controlled Period (16 weeks): Eczema herpeticum was reported in
4 patients (1.6 per 100 patient-years) treated with placebo, 6 patients (2.2 per 100 patient-years) treated with RINVOQ 15 mg and 7 patients (2.6 per 100 patient-years) treated with RINVOQ 30 mg.
12-Month Exposure (Weeks 0 to 52): Eczema herpeticum was reported in
18 patients (1.6 per 100 patient-years) treated with RINVOQ 15 mg and
17 patients (1.5 per 100 patient-years) treated with RINVOQ 30 mg.
Adverse Reactions in Patients with Ulcerative Colitis
RINVOQ was studied up to 8 weeks in patients with moderately to severely active ulcerative colitis in two randomized, double-blind, placebo-controlled induction studies (UC-1, UC-2) and a randomized, double-blind, placebo controlled, dose-finding study (UC-4; NCT02819635). Long term safety up to 52-weeks was evaluated in patients who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (UC-3) and a long-term extension study.
In the two induction studies (UC-1, UC-2) and a dose finding study (UC-4), 1097 patients were enrolled of whom 719 patients received RINVOQ 45 mg once daily.
In the maintenance study (UC-3), 746 patients were enrolled of whom 250 patients received RINVOQ 15 mg once daily and 251 patients received RINVOQ 30 mg once daily.
Adverse reactions reported in ≥2% of patients in any treatment arm in the induction and maintenance studies are shown in Tables 3 and 4, respectively.
Table 3. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 45 mg in Placebo-Controlled Induction Studies (UC-1, UC-2 and UC-4)
Specific Adverse Reactions
Serious Infections
Induction Studies: In UC-1, UC-2, and UC-4, serious infections were reported in 5 patients (8.4 per 100 patient-years) treated with placebo and 9 patients (8.4 per 100 patient-years) treated with RINVOQ 45 mg through 8 weeks. Placebo-controlled Maintenance Study: In UC-3, serious infections were reported in 8 patients (6.3 per 100 patient-years) treated with placebo, 8 patients (4.5 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (3.1 per 100 patient-years) treated with RINVOQ 30 mg through 52 weeks.
Laboratory Abnormalities
Hepatic Transaminase Elevations
In studies UC-1, UC-2, and UC-4, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 1.5% of patients treated with RINVOQ 45 mg, and 0% of patients treated with placebo for 8 weeks. AST elevations to ≥ 3 x ULN occurred in 1.5% of patients treated with RINVOQ 45 mg, and 0.3% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN occurred in 0.4% of patients treated with RINVOQ 45 mg and 0% of patients treated with placebo.
In UC-3, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 4% of patients treated with RINVOQ 30 mg, 2% of patients treated with RINVOQ 15 mg, and 0.8% of patients treated with placebo for 52 weeks. Elevations of AST to ≥ 3 x ULN in at least one measurement were observed in 2% of patients treated with RINVOQ 30 mg, 1.6% of patients treated with RINVOQ 15 mg and 0.4% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN were observed in 0.8% of patients treated with 30 mg, 0.4% of patients treated with 15 mg, and 0.4% of patients treated with placebo.
Overall, laboratory abnormalities observed in patients with ulcerative colitis treated with RINVOQ were similar to those described in patients with RA. Adverse Reactions in Patients with Ankylosing Spondylitis
A total of 596 patients with ankylosing spondylitis were treated with RINVOQ 15 mg in the two clinical trials representing 577.3 patient-years of exposure, of whom 228 were exposed to RINVOQ 15 mg for at least one year.
Overall, the safety profile observed in patients with active ankylosing spondylitis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis and psoriatic arthritis.
During the 14-week placebo-controlled period in Trial AS-I, the frequency of headache was 5.4% with RINVOQ 15 mg and 2.1% with placebo. During the 14-week placebo-controlled period in Trial AS-II, the frequency of headache was 3.3% with RINVOQ 15 mg and 1.4% with placebo.
DRUG INTERACTIONS
Strong CYP3A4 Inhibitors
Upadacitinib exposure is increased when RINVOQ is co-administered with a strong CYP3A4 inhibitor (such as ketoconazole and clarithromycin), which may increase the risk of RINVOQ adverse reactions. Monitor patients closely for adverse reactions when co-administering RINVOQ 15 mg once daily with strong CYP3A4 inhibitors.
For patients with atopic dermatitis, coadministration of RINVOQ 30 mg once daily with strong CYP3A4 inhibitors is not recommended.
For patients with ulcerative colitis taking strong CYP3A4 inhibitors, reduce the RINVOQ induction dosage to 30 mg once daily. The recommended maintenance dosage is 15 mg once daily.
Strong CYP3A4 Inducers
15 mg dose, 0.9 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 5 mg/kg/day and higher). Additional skeletal malformations (bent forelimbs/hindlimbs and rib/vertebral defects) and decreased fetal body weights were observed in the absence of maternal toxicity at an exposure approximately 84 times the 15 mg dose, 43 times the 30 mg dose, and 31 times the MRHD (on an AUC basis at a maternal oral dose of 75 mg/kg/day).
In a second oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 1.5 and 4 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that included bent humerus and scapula) at exposures approximately 1.6 times the 15 mg dose, 0.8 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 4 mg/kg/day). No developmental toxicity was observed in rats at an exposure approximately 0.29 times the 15 mg dose, 0.15 times the 30 mg dose, and 0.11 times the MRHD (on an AUC basis at a maternal oral dose of 1.5 mg/kg/day).
In an oral embryo-fetal developmental study, pregnant rabbits received upadacitinib at doses of 2.5, 10, and 25 mg/kg/day during the period of organogenesis from gestation day 7 to 19. Embryolethality, decreased fetal body weights, and cardiovascular malformations were observed in the presence of maternal toxicity at an exposure approximately 15 times the 15 mg dose, 7.6 times the 30 mg dose, and 5.6 times the MRHD (on an AUC basis at a maternal oral dose of 25 mg/kg/day). Embryolethality consisted of increased post-implantation loss that was due to elevated incidences of both total and early resorptions. No developmental toxicity was observed in rabbits at an exposure approximately 2.2 times the 15 mg dose, 1.1 times the 30 mg dose, and 0.82 times the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
In an oral pre- and post-natal development study, pregnant female rats received upadacitinib at doses of 2.5, 5, and 10 mg/kg/day from gestation day 6 through lactation day 20. No maternal or developmental toxicity was observed in either mothers or offspring, respectively, at an exposure approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and at approximately the same exposure as the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
Lactation
Risk Summary
There are no data on the presence of upadacitinib in human milk, the effects on the breastfed infant, or the effects on milk production. Available pharmacodynamic/toxicological data in animals have shown excretion of upadacitinib in milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions in the breastfed infant, advise patients that breastfeeding is not recommended during treatment with RINVOQ, and for 6 days (approximately 10 half-lives) after the last dose.
Data
A single oral dose of 10 mg/kg radiolabeled upadacitinib was administered to lactating female Sprague-Dawley rats on post-partum days 7-8. Drug exposure was approximately 30-fold greater in milk than in maternal plasma based on AUC0-t values. Approximately 97% of drug-related material in milk was parent drug.
Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ [see Use in Specific Populations]
* Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury and cholestasis.
Other adverse reactions reported in less than 2% of patients in the RINVOQ 45 mg group and at a higher rate than in the placebo group through Week 8 included herpes zoster and pneumonia.
Table 4. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 15 mg or 30 mg in the Placebo-Controlled Maintenance Study (UC-3)1
Upadacitinib exposure is decreased when RINVOQ is co-administered with strong CYP3A4 inducers (such as rifampin), which may lead to reduced therapeutic effect of RINVOQ. Coadministration of RINVOQ with strong CYP3A4 inducers is not recommended.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Available data from the pharmacovigilance safety database and postmarketing case reports on use of RINVOQ in pregnant women are not sufficient to evaluate a drug-associated risk for major birth defects or miscarriage. Based on animal studies, RINVOQ has the potential to adversely affect a developing fetus. Advise patients of reproductive potential and pregnant patients of the potential risk to the fetus.
In animal embryo-fetal development studies, oral upadacitinib administration to pregnant rats and rabbits at exposures equal to or greater than approximately 1.6 and 15 times the 15 mg dose, 0.8 and 7.6 times the 30 mg dose, and 0.6 and 5.6 times the maximum recommended human dose (MRHD) of 45 mg (on an AUC basis) resulted in dose-related increases in skeletal malformations (rats only), an increased incidence of cardiovascular malformations (rabbits only), increased post-implantation loss (rabbits only), and decreased fetal body weights in both rats and rabbits. No developmental toxicity was observed in pregnant rats and rabbits treated with oral upadacitinib during organogenesis at exposures approximately 0.29 and 2.2 times the 15 mg dose, 0.15 times and 1.1 times the 30 mg dose, and at 0.11 and 0.82 times the MHRD (on an AUC basis).
In a pre- and post-natal development study in pregnant female rats, oral upadacitinib administration at exposures approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and the same as the MRHD (on an AUC basis) resulted in no maternal or developmental toxicity (see Data)
The background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages are 2-4% and 15-20%, respectively. Report pregnancies to the AbbVie Inc.’s Adverse Event reporting line at 1-888-633-9110, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Considerations
Contraception
Females
Based on animal studies, upadacitinib may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations]. Advise female patients of reproductive potential to use effective contraception during treatment with RINVOQ and for 4 weeks after the final dose.
Pediatric Use
Juvenile Idiopathic Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The safety and effectiveness of RINVOQ in pediatric patients with juvenile idiopathic arthritis, psoriatic arthritis, and ankylosing spondylitis have not been established.
Atopic Dermatitis
The safety and effectiveness of RINVOQ in pediatric patients 12 years of age and older weighing at least 40 kg with atopic dermatitis have been established. A total of 344 pediatric patients aged 12 to 17 years with moderate to severe atopic dermatitis were randomized across three trials (AD-1, AD-2 and AD-3) to receive either RINVOQ 15 mg (N=114) or 30 mg (N=114) or matching placebo (N=116) in monotherapy or combination with topical corticosteroids. Efficacy was consistent between the pediatric patients and adults. The adverse reaction profile in the pediatric patients was similar to the adults [see Adverse Reactions].
The safety and effectiveness of RINVOQ in pediatric patients less than 12 years of age with atopic dermatitis have not been established.
Ulcerative Colitis
The safety and effectiveness of RINVOQ in pediatric patients with ulcerative colitis have not been established.
Geriatric Use
Rheumatoid Arthritis and Psoriatic Arthritis
Of the 4381 patients treated in the five clinical trials, a total of 906 rheumatoid arthritis patients were 65 years of age or older, including 146 patients 75 years and older. Of the 1827 patients treated in the two psoriatic arthritis Phase 3 clinical trials, a total of 274 patients were 65 years of age or older, including 34 patients 75 years and older. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of overall adverse events, including serious infections, in patients 65 years of age and older.
1 Patients who were responders to 8 weeks induction therapy with RINVOQ 45 mg once daily
* Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury, and cholestasis.
The safety profile of RINVOQ in the long-term extension study was similar to the safety profile observed in the placebo-controlled induction and maintenance periods.
Overall, the safety profile observed in patients with ulcerative colitis treated with RINVOQ was generally similar to the safety profile in patients with RA and AD.
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or ulcerative colitis. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Data
Animal Data
In an oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 5, 25, and 75 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that consisted of misshapen humerus and bent scapula) at exposures equal to or greater than approximately 1.7 times the
Atopic Dermatitis
Of the 2583 patients treated in the three Phase 3 clinical trials, a total of 120 patients with atopic dermatitis were 65 years of age or older, including 6 patients 75 years of age. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of serious infections and malignancies in those patients 65 years of age or older in the 30 mg dosing group in the long-term trials.
Ulcerative Colitis
Of the 1097 patients treated in the controlled clinical trials, a total of 95 patients with ulcerative colitis were 65 years and older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with ulcerative colitis to determine whether they respond differently from younger adult patients.
32 Revista Puertorriqueña de Medicina y Salud Pública DO NOT RE-SIZE
Adverse Reaction Placebo RINVOQ 45 mg Once Daily N= 378 (%) N = 719 (%) Upper respiratory tract infection* 7 9 Acne* 1 6 Increased blood creatine phosphokinase 1 5 Neutropenia* <1 5 Rash* 1 4 Elevated liver enzymes** 2 3 Lymphopenia* 1 3 Folliculitis 1 2 Herpes simplex* <1 2
Adverse Reaction Placebo RINVOQ 15 mg Once Daily RINVOQ 30 mg Once Daily n = 245 (%) n = 250 (%) n = 251 (%) Upper respiratory tract infection* 18 16 20 Increased blood creatine phosphokinase 2 6 8 Neutropenia* 2 3 6 Elevated liver enzymes** 1 6 4 Rash* 4 5 5 Herpes zoster 0 4 4 Folliculitis 2 2 4 Hypercholesterolemia* 1 2 4 Influenza 1 3 3 Herpes simplex* 1 2 3 Lymphopenia* 2 3 2 Hyperlipidemia* 0 2 2
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 3 04 May 2022 9:00 AM US-RNQR-220149
Renal Impairment
For patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (eGFR 60 to < 90 mL/min/1.73 m2), moderate (eGFR 30 to < 60 mL/min/1.73 m2), or severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2). For patients with atopic dermatitis, the maximum recommended dosage is 15 mg once daily for patients with severe renal impairment. No dosage adjustment is needed in patients with mild or moderate renal impairment. For patients with ulcerative colitis, the recommended dosage for severe renal impairment is 30 mg once daily for induction and 15 mg once daily for maintenance. No dosage adjustment is needed in patients with mild or moderate renal impairment.
RINVOQ has not been studied in patients with end stage renal disease (eGFR <15 mL/min/1.73m2). Use in patients with atopic dermatitis or ulcerative colitis with end stage renal disease is not recommended.
Hepatic Impairment
The use of RINVOQ has not been studied in patients with severe hepatic impairment (Child Pugh C), and therefore not recommended for use in patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, ulcerative colitis, or ankylosing spondylitis.
For patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment. For patients with ulcerative colitis, the recommended dosage for mild to moderate hepatic impairment is 30 mg once daily for induction and 15 mg once daily for maintenance
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Serious Infections
Inform patients that they may be more likely to develop infections when taking RINVOQ. Instruct patients to contact their healthcare provider immediately during treatment if they develop any signs or symptoms of an infection [see Warnings and Precautions]
Advise patients that the risk of herpes zoster is increased in patients taking RINVOQ and in some cases can be serious [see Warnings and Precautions]
Malignancies
Inform patients that RINVOQ may increase their risk of certain cancers and that periodic skin examinations should be performed while using RINVOQ. Advise patients that exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen [see Warnings and Precautions]
Major Adverse Cardiovascular Events
Inform patients that RINVOQ may increase their risk of major adverse cardiovascular events (MACE) including myocardial infarction, stroke,
and cardiovascular death. Instruct all patients, especially current or past smokers or patients with other cardiovascular risk factors, to be alert for the development of signs and symptoms of cardiovascular events [see Warnings and Precautions]
Thrombosis
Inform patients that events of deep venous thrombosis and pulmonary embolism have been reported in clinical trials with RINVOQ. Instruct patients to seek immediate medical attention if they develop any signs or symptoms of a DVT or PE [see Warnings and Precautions]
Hypersensitivity Reactions
Advise patients to discontinue RINVOQ and seek immediate medical attention if they develop any signs and symptoms of allergic reactions [see Warnings and Precautions].
Gastrointestinal Perforations
Inform patients that gastrointestinal perforations have been reported in clinical trials with RINVOQ and that risk factors include the use of NSAIDS or history of diverticulitis. Instruct patients to seek medical care immediately if they experience new onset of abdominal pain, fever, chills, nausea, or vomiting [see Warnings and Precautions].
Retinal Detachment
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ [see Adverse Reactions]
Laboratory Abnormalities
Inform patients that RINVOQ may affect certain lab tests, and that blood tests are required before and during RINVOQ treatment [see Warnings and Precautions]
Vaccinations
Advise patients to avoid use of live vaccines with RINVOQ. Instruct patients to inform their healthcare practitioner that they are taking RINVOQ prior to a potential vaccination [see Warnings and Precautions]
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that exposure to RINVOQ during pregnancy may result in fetal harm. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions and Use in Specific Populations]
Advise females of reproductive potential that effective contraception should be used during treatment and for 4 weeks following the final dose of upadacitinib [see Use in Specific Populations]
Advise females patients who are exposed to RINVOQ during pregnancy to contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Lactation
Advise women not to breastfeed during treatment with RINVOQ and for 6 days after the last dose [see Use in Specific Populations]
Administration
Advise patients not to chew, crush, or split RINVOQ tablets. Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® is a registered trademark of AbbVie Biotechnology Ltd.
©2019-2022 AbbVie Inc.
Ref: 20071734 Revised: April 2022
LAB-7082 MASTER
US-RNQR-220149
Revista Puertorriqueña de Medicina y Salud Pública 33 DO NOT RE-SIZE
20071734 Rinvoq PB-7.625 x 10.5(3.5).indd 4 04 May 2022 9:00 AM US-RNQR-220149
CRITERIOS DIAGNÓSTICOS Y MANEJO DEL MIELOMA MÚLTIPLE
TRATAMIENTO
El tratamiento de MM se caracteriza por la terapia de inducción seguida por la consolidación con trasplante autólogo de células hematopoyéticos (TPH). Por lo tanto, al decidir el tratamiento inicial, es importante determinar si el paciente es candidato
a trasplante. Anteriormente, la terapia de inducción consistía en esteroides en altas dosis y una combinación de quimioterapia. Pero a través de los años se ha expandido el tratamiento para los pacientes de MM y contamos con terapias inmunológicas, biológicas
y moleculares que son igual de efectivas que la quimioterapia. Es importante destacar que, al escoger un tratamiento para el paciente, se debe escoger una combinación de tres drogas en vez de dos drogas, ya que sé que tiene más beneficio en la sobrevida.
34 Revista Puertorriqueña de Medicina y Salud Pública
Joel López Figueroa MD Hematólogo Oncólogo Especialista en Mieloma Múltiple Clínica de Mieloma Múltiple, Programa de Trasplante de Medula Ósea, Hospital Auxilio Mutuo
Mariola A. Vázquez Martínez, MD Fellow Hematología Oncología Hospital Municipal de San Juan y Hospital de Veteranos de San Juan Puerto Rico
 Rafael Mestres, MD Fellow Hematología Oncología
Rafael Mestres, MD Fellow Hematología Oncología

Hospital Municipal de San Juan y Hospital de Veteranos de San Juan Puerto Rico
INTRODUCCIÓN
Mieloma Múltiple (MM) es una malignidad de las células plasmáticas. El MM representa el 10% de las malignidades hematológicas y la edad promedio es alrededor de 65 años. Según la Sociedad Americana del Cáncer, se estima alrededor de 35,730 casos para el 2023 en los Estados Unidos.
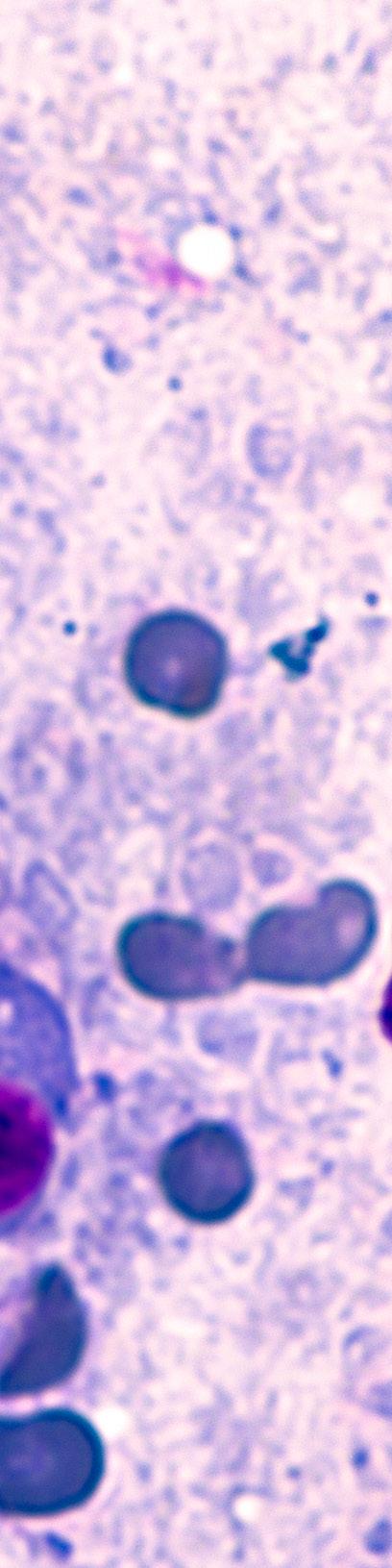
MM se caracteriza por la proliferación de las células plasmáticas, infiltrando la médula ósea. Esto produce una proteína anormal en la sangre y la orina, conocido como pico monoclonal o M-spike. Para diagnosticar MM se necesita múltiples laboratorios, una biopsia de médula ósea y de estudios radiológicos para detectar lesiones óseas o plasmacitoma. Los laboratorios más importantes son: electroforesis de proteína con inmunofluorescencia en suero y orina, niveles de cadena pesadas y ligeras en suero y colección de orina de 24 horas para proteínas. El MM se diagnostica al haber una proteína monoclonal en suero sobre 3 g/dL (pico monoclonal o M-spike) o una infiltración de células plasmáticas en la medula ósea mayor de 10%. El MM podría producir solamente cadenas ligeras de inmunoglobulina, que debido a su pequeño tamaño no se detectan como una proteína monoclonal en suero por la
Entre las drogas más comunes están los Inhibidores de proteasoma.
Estos bloquean la acción de las proteasomas, que están encargados en la descomposición de las proteínas. Esto puede evitar la degradación de factores pro-apoptoticos, que permite
electroforesis, pero si midiendo sus niveles en suero o en una colección de orina. Estas se conocen como proteínas de Bence Jones.
Las características clínicas de MM son: lesiones osteolíticas sintomáticas, anemia, insuficiencia renal e hipercalcemia. Estos síntomas se conocen con el acrónimo ingles CRAB (Calcium, Renal Insuficiency, Anemia, Bone Lesions). En los pacientes de MM, tenemos algunos factores pronósticos como la edad, el estado de salud general, anomalidades genéticas y respuestas al tratamiento inicial. Esto nos ayuda a estratificar al paciente como de riesgo estándar o alto riesgo y de la misma manera nos puede ayudar a decidir que tratamiento recibirá el paciente.
Originalmente solo se comenzaba tratamiento para el MM al tener síntomas (CRAB). Pero a través de los años se reconoció que hay algunos pacientes con MM asintomático que tienen un mayor riesgo a progresar y se benefician de tratamiento en el momento. Este grupo de pacientes se caracteriza por tener más de 60% de células plasmáticas en la medula ósea, cadenas ligeras de inmunoglobulina anormales>100 y presencia de alguna lesión ósea en MRI.
la activación de la muerte celular programada en células de cáncer. Esta clase de medicamento es de suma importancia para los pacientes con MM de alto riesgo. En esta clase están bortezomib, carfilzomib e ixazomib.
Los Agentes inmunomoduladores (IMiDs) se usan para mejorar la respuesta inmunitaria del cuerpo contra el cáncer. Estos son análogos a la talidomida y tienen múltiples mecanismos de acción como la apoptosis de las células tumorales malignas, la interferencia
Revista Puertorriqueña de Medicina y Salud Pública 35
con las interacciones del tumor con el microambiente celular en la medula ósea entre otras. Entre ellos está la, lenalidomida y la pomalidomida.
Los Anticuerpos monoclonales (MoAb) han revolucionado el tratamiento de MM. Pueden actuar induciendo una citotoxicidad directa en la célula tumoral atreves de receptores o vías de señales celular. También trabajan al inducir una respuesta inmune en contra de las células tumorales. Los MoAb trabajan de forma eficiente con menos toxicidad que otras drogas. El primer MoAb aprobado para MM fue elotuzumab, anticuerpo en contra de SLAMF7 (Signaling Lymphocytic Activation Molecule family member 7). Esta droga trabaja al unirse
a las células con la proteína SLAMF7 y las descompone. También actúa al activar el sistema inmune y mejora la capacidad de los linfocitos citolíticos (NK) naturales para atacar a las células cancerosa. Elotuzuma está aprobado en combinación con lenalidomida o pomalidomida junto a dexametasona para pacientes que han recibido 1 a 3 líneas de tratamiento previas.
Daratumuab e isatuximab son anticuerpos en contra de CD38. Estos tienen efectos antitumorales a través de citotoxicidad directa (ADCC, ADCP, CDC) y tiene efecto efectos directo sobre las células efectoras del sistema inmunológico. Es importante recalcar que estos dos MoAB se dirigen a
distintos epítopos de CD 38. Isatuximab puede inducir directamente apoptosis, mientras que daratumuamb no puede inducir apoptosis sin combinarse con agentes reticulantes (cross-linking).

Selinexor es un inhibidor de la exportación nuclear. Inhibe la exportación de ciertas proteínas supresoras de tumores y reguladoras del crecimiento de tumores al bloquear la exportación 1. Esto conduce a un aumento en la supresión del tumor, lo que conduce a la muerte de las células cancerosas. Esta droga está aprobada en combinación con dexametasona o bortezomib en pacientes con MM en recaída o relapso que han recibido al menos 4 terapias previas.
El TPH sigue siendo un componente clave de la terapia del MM. Este se puede incorporar como parte de la terapia inicial o posponerse hasta la primera recaída. Aunque el TPH no es curativo, puede retrasar la progresión de la enfermedad. El escoger si el TPH se hace luego de la primera recaída o como terapia inicial es una decisión individualizada y se debe considerar algunos factores como la preferencia del paciente, edad, el tipo de riesgo de MM, respuesta a la terapia inicial, entre otras.
El TPH autólogo temprano da como resultado respuestas más profundas y una mejor sobrevida libre de enfermedad (PFS), aunque la sobrevida general (OS) no cambia mucho en comparación con el TPH tardío. Hay dos investigaciones grandes sobre TPH temprano versus tardío DETERMINATION y el IFM 2009. En ambos estudios, el TCH temprano dio como resultado respuestas más profundas con más respuestas completas y más pacientes que lograron un estado de MRD negativo. La PFS era más duradera, pero OS no era diferente entre TPH temprano versus tardío.
36 Revista Puertorriqueña de Medicina y Salud Pública
Trasplante de Medula Ósea
Una manera de prolongar la sobrevida libre de enfermedad luego de un TPH, es utilizar alguno de los fármacos como terapia de mantenimiento post trasplante. El mantenimiento se dar por dos años. Esto puede retrasar la progresión de la enfermedad y mejorar OS. Usualmente si el paciente es de riesgo estándar el mantenimiento es con lenalidomida y si es MM de alto riesgo es una combinación de in inhibidor de proteosoma más lenalidomida.
En algunos casos donde el paciente sufre una recaída a un año y medio o más luego de un primer TPH autólogo y que responda un tratamiento de rescate pueden considerarse para un segundo TPH autólogo.
La terapia celular CAR-T es otra opción de tratamiento prometedora para el mieloma múltiple avanzado. El CAR-T para el MM es una forma de inmunoterapia que utiliza las células T genéticamente alteradas del propio paciente para reconocer y atacar las células cancerosas, específicamente dirigidas al receptor BCMA. Las células
T del paciente se extraen del cuerpo y se modifican en un entorno de laboratorio para producir receptores especializados conocidos como receptores de antígenos quiméricos (CAR) en su superficie, que reconocen y se unen a antígenos específicos asociados al cáncer. Las células T alteradas se reintroducen en el paciente, donde pueden identificar y destruir eficazmente las células cancerosas. Los estudios han demostrado que la terapia CAR-T para el MM puede ser muy eficiente para controlar la progresión de la enfermedad e inducir una remisión completa en pacientes, que previamente no existía otra línea de tratamiento. Actualmente existen dos CAR-T aprobados por la Administración de Alimentos y Medicamentos (FDA); Idecabtagene vicleucel(ide-cel) y ciltacabtagene autoleucel(cilta-cel). Esto ofrece una posible nueva opción de tratamiento para los pacientes con MM más avanzado luego de recibir varias líneas de tratamiento. Sin embargo, este tipo de terapia también puede conllevar un alto riesgo de efectos secundarios, entre ellos el CRS y toxicidad neurológica.
CONCLUSIÓN
Los recientes avances en el campo del MM han mejorado significativamente el pronóstico de los pacientes con dicha
Terapias dirigida y CART
Con cada tratamiento de MM, es menos probable que el cáncer responda, y asi la enfermedad vuelve más rápido. Es por eso que el campo de la oncología y hematología sigue creciente y ahora tenemos nuevos tratamientos más específicas para el MM.
Las células de MM expresan una proteína llamada antígeno de maduración de célula B (BCMA), y esto a sido una estrategia para crear terapias dirigidas. Los dos tipos de terapias hechas para BCMA son los anticuerpos biespecificos (BiTe) y la terapia de CAR-T. Teclistamab es un BiTe que se une a dos antígenos diferente
al mismo tiempo (BCMA y CD3). El CD3 es una proteína presente en las células T. Fue aprobado para pacientes que hayan usado al menos 4 terapias anteriormente incluyendo un anti-CD38, un inhibidor de proteosoma y IMiDs. Alguna de sus toxicidades son síndrome de liberación de citoquinas (CRS), toxicidad neurológica, hepatotoxicidad, infecciones y neutropenia.
enfermedad considerada como una condición incurable. Como resultado, ahora se utilizan tratamientos como la inmunoterapia y nuevos agentes para ralentizar e incluso detener la progresión de la enfermedad. Además, los investigadores activamente estudian nuevas formas de mejorar aún más la eficacia del tratamiento, la calidad de vida y los resultados generales de los pacientes. Se trata del uso de agentes novedosos, una mejor comprensión de las vías moleculares y el desarrollo de terapias dirigidas. La introducción de estos nuevos tratamientos ha aportado
una mayor sensación de esperanza a los pacientes con mieloma múltiple. Con la continuación de la investigación y de los ensayos clínicos, es probable que aún se produzcan más avances en este campo que conduzcan a nuevas mejoras en la atención a los pacientes. En definitiva, las perspectivas para los pacientes con mieloma múltiple son más halagadoras que nunca, con gran esperanza de mejores resultados en un futuro y potencial cura del MM. Con el tratamiento y los cuidados adecuados, los pacientes pueden vivir más tiempo y disfrutar de una mejor calidad de vida.
Revista Puertorriqueña de Medicina y Salud Pública 37
¿ Q u é es SARCL I SA ? SARCLISA es un medicamento recetado usado en combinación con:
• Los medicamentos pomalidomida y dexametasona para tratar a adultos que han recibido al menos 2 terapias previas, incluidos lenalidomida y un inhibidor de proteasoma, para tratar el mieloma múltiple.
mieloma múltiple que han recibido de 1 a 3 líneas de tratamiento y estos no funcionaron o han dejado de funcionar.
Está comprobado que SARCLISA ayuda a extender el tiempo que las personas viven sin empeoramiento del mieloma múltiple

En un estudio d e 307 p er sonas con mi elo ma múl t ipl e recividante y refrac t ar io, SARCLISA ad minist rado con Po malyst ® (po malido mida) y d exam et asona (Pd) ex tendió el ti e mp o que las p er sonas vivi eron sin e mp eorami ento d el mi elo ma múl t ipl e en co mparación con Pd sola (m ediana d e 11 5 m eses vs 6 5 m eses).

Información de Seguridad Importante
No reciba SARCLISA si tiene antecedentes de una reacción alérgica severa a isatuximab-irfc o a cualquiera de los ingredientes de SARCLISA (vea una lista de los ingredientes en la Información completa para la Prescripción)
Antes de recibir SARCLISA, infórmele a su profesional del cuidado de la salud acerca de todas sus condiciones médicas, incluido si usted:
• Tiene problemas cardiacos, si su profesional del cuidado de la salud le receta
• Ha tenido culebrilla (herpes zóster)
• Está embarazada o tiene planes de quedar embarazada. SARCLISA puede hacerle daño al feto No debe recibir SARCLISA durante el embarazo
Las mujeres que pueden quedar embarazadas deben usar un método de control del embarazo efectivo durante el tratamiento y por 5 meses después de la última dosis de SARCLISA Hable con su profesional del cuidado de la salud acerca de los métodos de control del embarazo que puede usar durante este tiempo
Déjele saber a su profesional del cuidado de la salud de inmediato si cree que está embarazada o si queda embarazada durante el tratamiento de SARCLISA.
• Está lactando o piensa lactar No se conoce si SARCLISA pasa a la leche materna. No debe lactar durante el tratamiento con SARCLISA

Déjele saber a su profesional del cuidado de la salud acerca de todos los medicamentos que usa, incluidos los recetados o los que no requieren receta, vitaminas y suplementos naturales
¿Cómo recibiré SARCLISA?
• SARCLISA lo administra su profesional del cuidado de la salud po (IV) en una vena
• SARCLISA se administra en ciclos de tratamientos de 28 días
En el ciclo 1, SARCLISA se administra, por lo general, semanalmente Comenzando en el ciclo 2, SARCLISA se administra, po semanas
• Si no asiste a alguna cita, llame a su profesional del cuidado de la salud lo ante posible para reprogramar su cita.
• Su profesional del cuidado de la salud le dará medicinas antes de cada dosis d SARCLISA, para ayudar a reducir el riesgo de reacciones a la i menos frecuentes y severas)
¿Cuáles son los efectos secundarios posibles de SARCLISA
SARCLISA puede causar efectos secundarios graves, entre ell
• Reacciones a la infusión. Las reacciones a la infusión son comunes co SARCLISA, y algunas veces son severas o mortales
Su profesional del cuidado de la salud le recetará medicinas antes de cad infusión de SARCLISA para ayudar a reducir el riesgo de las la infusión o para ayudar a que cualquier reacción a la infusi severa. Se le dará seguimiento por la posibilidad de reacciones a la infusió durante cada dosis de SARCLISA

Su profesional del cuidado de la salud puede reducir la velocidad o detene la infusión, o suspender el tratamiento de SARCLISA completamente si tien una reacción a la infusión

©
Cu a ndo e l m i e l o ma m ú l tiple r ec u r r e, mi r e h ac i a e l f ut u r o con SA RCLI SA
Conozca si SARCLISA puede ser adecuado para usted. Visite SARCLISA.com/considering-sarclisa.
Busque ayuda médica de inmediato si desarrolla alguno de los siguientes síntomas de reacción a la infusión durante o después de una infusión de
SARCLISA:
falta de aliento, sibilancia o para respirar hinchazón de la cara, la boca, la garganta o la lengua
apretamiento de la garganta palpitaciones mareo, aturdimiento o desmayo dolor de cabeza

tos erupción o picor náuseas goteo o congestión nasal escalofríos
• Disminución del recuento de glóbulos blancos La disminución del recuento de glóbulos blancos es común con SARCLISA y ciertos glóbulos blancos pueden disminuir de manera severa. Puede correr un mayor riesgo de adquirir ciertas infecciones, como infecciones del tracto respiratorio superior e inferior e infecciones del tracto urinario
sanguíneas durante el tratamiento con SARCLISA Su profesional del cuidado de la salud puede recetarle antibióticos o antivirales para ayudar a prevenir infección, o una medicina para ayudar a aumentar sus recuentos de glóbulos blancos durante el tratamiento con SARCLISA Déjele saber a su profesional del cuidado de la salud de inmediato si amiento con
SARCLISA
• Riesgo de cánceres nuevos durante el tratamiento con SARCLISA Su profesional del cuidado de la salud le
SARCLISA
• Cambios en las pruebas de sangre SARCLISA puede afectar los resultados de las pruebas de sangre para determinar su tipo de sangre. Su profesional del
antes de comenzar el tratamiento con SARCLISA Déjele saber a todos sus profesionales del cuidado de la salud que está recibiendo tratamiento con SARCLISA antes de recibir transfusiones de sangre pomalidomida y dexametasona incluyen:
• infección del tracto respiratorio superior
• infección pulmonar (neumonía)
• diarrea
• infección del tracto respiratorio superior
• cansancio y debilidad
• presión arterial alta
• problemas para dormir
• bronquitis
• disminución del recuento de glóbulos rojos (anemia)
• disminución del recuento de plaquetas (trombocitopenia)
• tos
• dolor de espalda
• diarrea
• infección pulmonar (neumonía) para respirar
• disminución del recuento de glóbulos rojos (anemia)
• disminución del recuento de plaquetas (trombocitopenia)
• para respirar
• tos
• hinchazón de tobillos, pies o piernas
Puede producirse un fallo cardiaco durante el tratamiento con SARCLISA en Déjele saber a su profesional del cuidado de la salud de inmediato si desarrolla alguno de los siguientes síntomas: Vea el resumen breve de SARCLISA en la próxima página.
¿Qué es SARCLISA?
SARCLISA es un medicamento recetado usado en combinación con:
• Pomalidomida y dexametasona para tratar a adultos que han recibido al menos 2 terapias previas, incluidos lenalidomida y un inhibidor de
• Los medicamentos carfilzomib y dexametasona, para tratar a adultos con funcionaron o han dejado de funcionar.
Información de Seguridad Importante
No reciba SARCLISA si isatuximab-irfc o a cualquiera de los ingredientes de SARCLISA (vea una lista de los ingredientes en la Información completa para la Prescripción)
Antes de recibir SARCLISA, infórmele a su profesional del cuidado de la salud acerca de todas sus condiciones médicas, incluido si usted:
• Tiene problemas cardiacos, si su profesional del cuidado de la salud le receta SARCLISA en combinación con carfilzomib y dexametasona.
• Ha tenido culebrilla (herpes zóster).
hacerle daño al feto. No debe recibir SARCLISA durante el embarazo. Las mujeres que pueden quedar embarazadas deben usar un método de cuidado de la salud acerca de los métodos de control del embarazo que Déjele saber a su profesional del cuidado de la salud de inmediato si cree que está embarazada o si queda embarazada durante el tratamiento de SARCLISA.
• Está lactando o piensa lactar. No se conoce si SARCLISA pasa a la leche materna. No debe lactar durante el tratamiento con SARCLISA.
Déjele saber a su profesional del cuidado de la salud acerca de todos los medicamentos que usa, incluidos los recetados o los que no requieren receta, vitaminas y suplementos naturales.
¿Cómo recibiré SARCLISA?
• SARCLISA lo administra su profesional del cuidado de la salud por vía intravenosa (IV) en una vena.
• SARCLISA se administra en ciclos de tratamientos de 28 días (4 semanas), junto con las medicinas pomalidomida y dexametasona, o carfilzomib y dexametasona.
En el ciclo 1, SARCLISA se administra, por lo general, semanalmente. Comenzando en el ciclo 2, SARCLISA se administra, por lo general, cada 2 semanas.
• Si no asiste a alguna cita, llame a su profesional del cuidado de la salud lo antes posible para reprogramar su cita.
• Su profesional del cuidado de la salud le dará medicinas antes de cada dosis de SARCLISA, para ayudar a reducir el riesgo de reacciones a la infusión (hacerlas menos frecuentes y severas).
¿Cuáles son los efectos secundarios posibles de SARCLISA?
SARCLISA puede causar efectos secundarios graves, entre ellos:
• Reacciones a la infusión. Las reacciones a la infusión son comunes con SARCLISA, y algunas veces son severas o mortales.
Su profesional del cuidado de la salud le recetará medicinas antes de cada infusión de SARCLISA para ayudar a reducir el riesgo de las reacciones a la infusión o para ayudar a que cualquier reacción a la infusión sea menos severa. Se le dará seguimiento por la posibilidad de reacciones a la infusión durante cada dosis de SARCLISA.
Su profesional del cuidado de la salud puede reducir la velocidad o detener la infusión, o suspender el tratamiento de SARCLISA
Busque ayuda médica de inmediato si desarrolla alguno de los siguientes síntomas de reacción a la infusión durante o después de una infusión de SARLCISA:
falta de aliento, sibilancia o dificultad para respirar — hinchazón de la cara, la boca, la garganta o la lengua
apretamiento de la garganta palpitaciones mareo, aturdimiento o desmayo dolor de cabeza
tos erupción o picor náuseas goteo o escalofríos
• Disminución del recuento de glóbulos blancos. La disminución del recuento de glóbulos blancos es común con SARCLISA y ciertos glóbulos blancos pueden disminuir de manera severa. Puede correr un mayor riesgo de adquirir ciertas infecciones, como infecciones del tracto respiratorio superior e inferior e infecciones del tracto urinario.
Su profesional del cuidado de la salud verificará sus recuentos de células sanguíneas durante el tratamiento con SARCLISA. Su profesional del cuidado infección, o una medicina para ayudar a aumentar sus recuentos de glóbulos blancos durante el tratamiento con SARCLISA.
Déjele saber a su profesional del cuidado de la salud de inmediato si desarrolla alguna fiebre o síntomas de infección durante el tratamiento con SARCLISA.
• Riesgo de cánceres nuevos. Han ocurrido cánceres nuevos en personas durante el tratamiento con SARCLISA. Su profesional del cuidado de la salud le dará seguimiento por la posibilidad de cánceres nuevos durante el tratamiento con SARCLISA.
• Cambios en las pruebas de sangre. SARCLISA puede afectar los resultados
sangre antes de comenzar el tratamiento con SARCLISA. Déjele saber a todos sus profesionales del cuidado de la salud que está recibiendo tratamiento con SARCLISA antes de recibir transfusiones de sangre.
Los efectos secundarios más comunes de SARCLISA en combinación con pomalidomida y dexametasona incluyen:
• infección del tracto respiratorio superior
• infección pulmonar (neumonía)
• diarrea
• disminución del recuento de glóbulos rojos (anemia)
• disminución del recuento de plaquetas (trombocitopenia)
Los efectos secundarios más comunes de SARCLISA en combinación con carfilzomib y dexametasona incluyen:
• infección del tracto respiratorio superior
• cansancio y debilidad
• presión arterial alta
• problemas para dormir
• tos
• dolor de espalda
• diarrea
• infección pulmonar (neumonía)
• dificultad para respirar
• disminución del recuento de glóbulos rojos (anemia)
• disminución del recuento de plaquetas (trombocitopenia)
Puede producirse un fallo cardiaco durante el tratamiento con SARCLISA en combinación con carfilzomib y dexametasona. Déjele saber a su profesional del cuidado de la salud de inmediato si desarrolla alguno de los siguientes síntomas:
• dificultad para respirar
• tos
• hinchazón de tobillos, pies o piernas
Estos no son todos los efectos secundarios posibles de SARCLISA. Para más información, pregúntele a su profesional del cuidado de la salud o al
40 Revista Puertorriqueña de Medicina y Salud Pública
C M Y CM MY CY CMY K C M Y CM MY CY CMY K C M Y CM MY CY CMY K







Revista Puertorriqueña de Medicina y Salud Pública 41 DAMAS - Cardiologia-Anuncio Medicina y Salud-FP-NEW-FINAL.pdf 1 2/10/23 5:58 PM DAMAS - Cardiologia-Anuncio Medicina y Salud-FP-NEW-FINAL.pdf 1 2/10/23 5:58 PM DAMAS - Cardiologia-Anuncio Medicina y Salud-FP-NEW-FINAL.pdf 1 2/10/23 5:58 PM
EVALUACIÓN DE LOS NÓDULOS DE LA TIROIDES
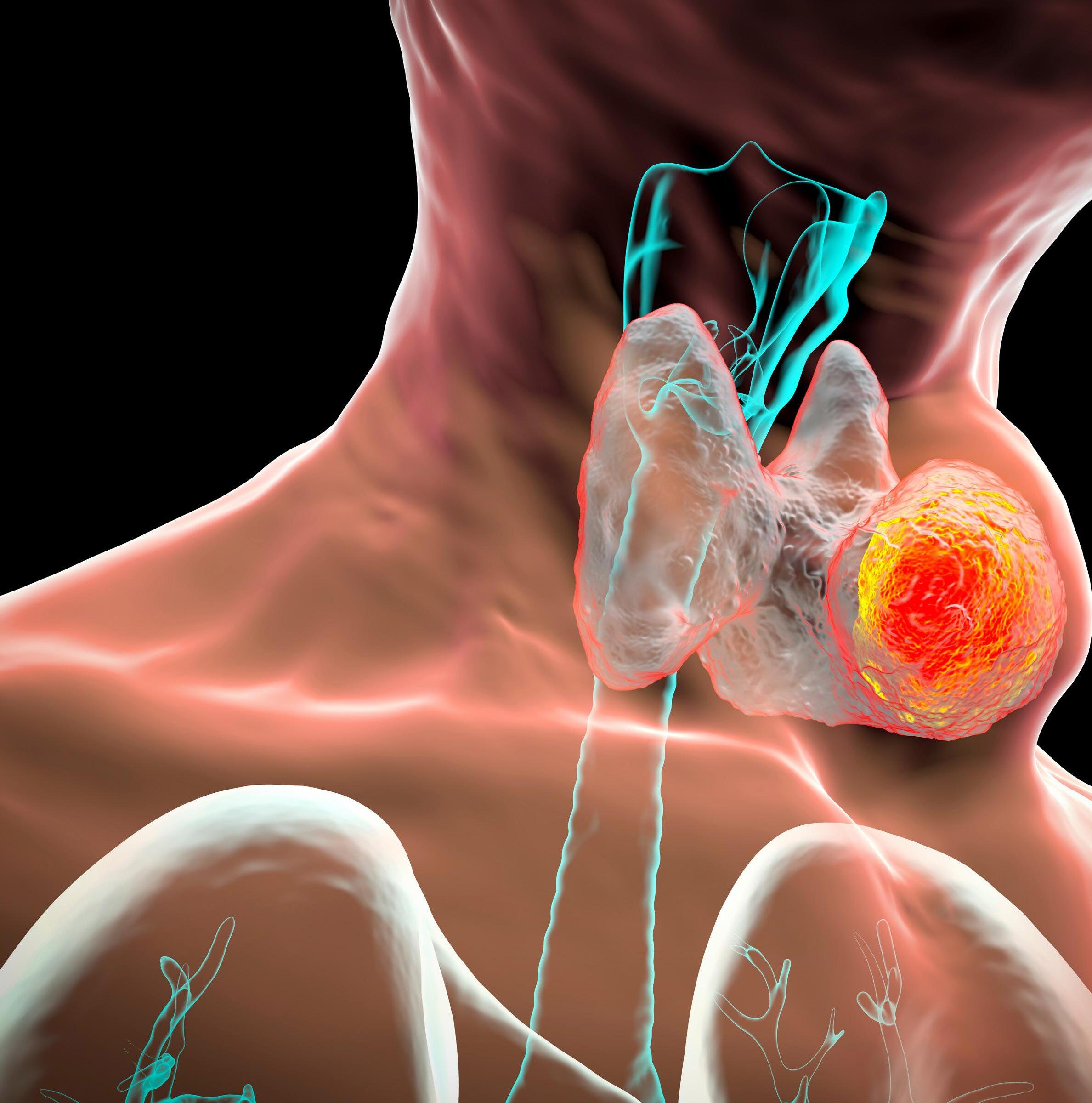
42 Revista Puertorriqueña de Medicina y Salud Pública
MSP ARTÍCULO / MÉDICO
Luis N. Madera Marin, MD

Sección de Endocrinología del Recinto de Ciencias Médicas

RESUMEN
Los nódulos tiroideos son masas o protuberancias sólidas o llenas de líquido que se forman dentro de la tiroides, una pequeña glándula ubicada en la base del cuello, justo encima del esternón (hueso). La mayoría de los nódulos tiroideos no son graves y no causan síntomas. Solo un pequeño porcentaje de los nódulos tiroideos son cancerosos. A menudo, no sabrá que tiene un nódulo tiroideo hasta que su médico lo descubra durante un examen médico de rutina, también su médico puede descubrirlo durante una exploración que se realizó
por otro motivo de salud, de manera incidental. Sin embargo, algunos nódulos tiroideos pueden volverse lo suficientemente grandes como para ser visibles o dificultar el tragado, el habla y/o la respiración. Las opciones de tratamiento dependen del tipo de nódulo tiroideo que tenga. Los nódulos tiroideos son frecuentes. Se presentan con más frecuencia en las mujeres que en los hombres. Y la probabilidad de que una persona desarrolle un nódulo tiroideo incrementa con la edad.
Revista Puertorriqueña de Medicina y Salud Pública 43
MSP ARTÍCULO / MÉDICO
INTRODUCCIÓN
Los nódulos tiroideos llaman la atención clínica cuando el paciente los nota; por un médico durante el examen físico de rutina; o durante un procedimiento radiológico, como un sonograma de cuello y/o tiroides, una ecografía carotídea, una tomografía computarizada (TC) de cuello o tórax. Su importancia clínica se relaciona principalmente con la necesidad de excluir el cáncer de tiroides.
DISCUSIÓN
La mayoría de los nódulos tiroideos no presentan signos ni síntomas. Pero ocasionalmente, algunos nódulos se vuelven tan grandes que pueden:
• Ser sentido(s) por la persona misma
• Ser visto o notado, a menudo como una hinchazón en la base de su cuello
• Presión sobre la tráquea o el esófago, lo que provoca falta de aire o dificultad para tragar
-En algunos casos, los nódulos tiroideos producen hormona tiroidea adicional por la glándula tiroides y pudiese causar síntomas de exceso de esta.
-Solo una pequeña cantidad de nódulos tiroideos son cancerosos.
Pero determinar qué nódulos son cancerosos, no se puede hacer solo evaluando sus síntomas sino un análisis conlleva otros estudios y/o procedimientos.
La mayoría de los nódulos tiroideos cancerosos crecen lentamente y pueden
CAUSAS
ser pequeños cuando su médico los descubre.
Datos Ante Los Que Se Debe Sospechar Que Un Nódulo Tiroideo Sea Canceroso Son:
HISTORIAL
• Tiene un nódulo duro y/o firme
• Tiene un nódulo adherido a estructuras cercanas (como, por ejemplo: nervios, esófago, vías aéreas)
• Tiene antecedentes familiares de cáncer de tiroides
• Ha notado un cambio en la voz(habiendo descartado otras causas como el reflujo)
• Es menor de 20 años o mayor de 70 (edades extremas de la vida)
• Es de género masculino
• Tiempo de evolución de este nódulo(s)- (rápido crecimiento)
• Historia de haber recibido o estar expuesto a radiación de cabeza, cuello o tórax
• Historia familiar de Neoplasia Endocrina Múltiple tipo II (MEN II)
HACER LABORATORIOS
Pruebas tiroideas para ver si es o no funcional el nódulo, es decir, ver si produce exceso de hormona tiroidea (T4 libre, TSH)- la mayoría no tienen alteración en las pruebas de sangre.
HACER SONOGRAMA DE CUELLO
Permite establecer tamaño y/o características: sólido, quístico o mixto, presencia de calcificaciones, además detecta nódulos no palpables, entre otras características…
HACER BIOPSIA O ASPIRACIÓN DEL NÓDULO(S) EN LA TIROIDES
Es una aspiración con aguja fina que no requiere de anestesia y se pudiera recomendar dependiendo del tamaño del nódulo y características encontradas en el sonograma que pudiesen preocupar y aumentar el riesgo de malignidad
Se reportaría como benigno, maligno, sospechoso, indeterminado que pudiesen realizársele pruebas genéticas para determinar riesgo de malignidad o que se reporte una muestra insuficiente que pudiese haber la necesidad de repetir el procedimiento.
EXAMEN FÍSICO
Verificando el tamaño del nódulo, si es duro y/o está fijo a las estructuras del cuello, si hay ganglios o nódulos linfáticos en otras áreas del cuello palpables.
Un nódulo que tenga una biopsia positiva o le cause síntomas de compresión pudiera requerir ser enviado a un cirujano que le remueva los nódulos.
Luego de cirugía pudiera requerir de reemplazo hormonal con hormona de tiroides.
Varias condiciones pueden hacer que se desarrollen nódulos en la glándula tiroides, que incluyen:
• Quistes tiroideos
• Inflamación crónica de la tiroides (como por ejemplo la Enfermedad de Hashimoto)
• Bocio (agrandamiento de la tiroides) multinodular
• Cáncer tiroideo
• Deficiencia de yodo- poco común en los Estados Unidos, donde el yodo se agrega rutinariamente a la sal de mesa y otros alimentos.
REFERENCIAS
Mayo Clinic, American Thyroid Association
44 Revista Puertorriqueña de Medicina y Salud Pública
¿QUÉ ESPERAR DE SER DIAGNOSTICADO CON NÓDULO(S) TIROIDEO?
Los nódulos tiroideos no cancerosos no son potencialmente mortales. Muchos de ellos no requieren tratamiento. Los exámenes de rutina son suficientes.
Todos los nódulos de tiroides que tienen cáncer o que son altamente sospechosos de cáncer deben ser removidos por un cirujano experto de tiroides, como mencionado anteriormente. La mayoría de los canceres de tiroides son curables y rara vez causan problemas que amenazan la vida.
Los nódulos de tiroides que son benignos o que son muy pequeños para hacerles biopsia deben ser seguidos con ultrasonido cada 6-12 meses, y un examen físico anual por su médico. También, aun cuando la biopsia sea benigna, se le podríá
recomendar cirugía para sacar el nódulo si este sigue creciendo, o si desarrolla características de alto riesgo en el ultrasonido.
El pronóstico para el cáncer de tiroides depende del tipo de cáncer. Para los tipos de cáncer de tiroides más comunes, el pronóstico es muy bueno después del tratamiento.
¿CUÁNDO VER A UN MÉDICO?
Aunque la mayoría de los nódulos tiroideos no son cancerosos y no causan problemas, pídale a su médico que evalúe o lo refiera al Endocrinólogo, es importante evaluar cualquier hinchazón inusual en su cuello, especialmente si tiene problemas para respirar o tragar. Es importante evaluar la posibilidad de cáncer.
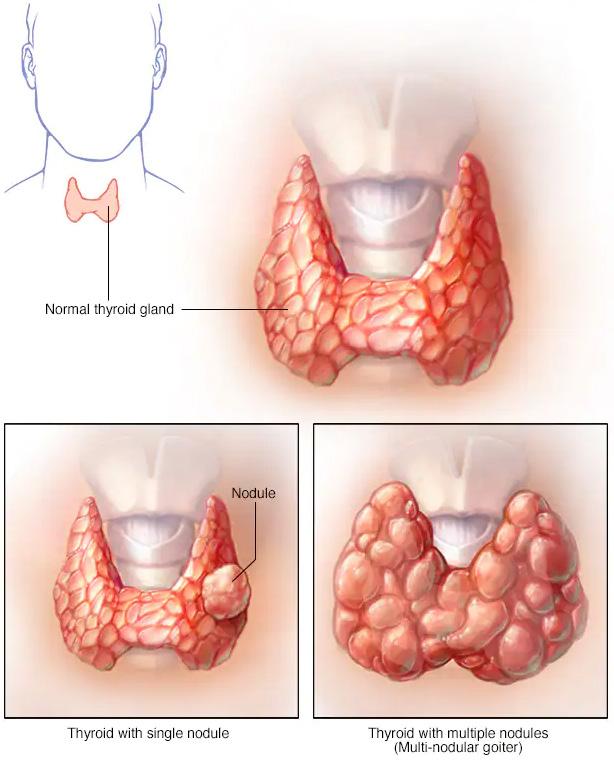
Busque atención médica si presenta signos y síntomas de hipertiroidismo. Los síntomas más habituales son:
LCOMPLICACIONES
Las complicaciones asociadas con algunos nódulos tiroideos incluyen:
• Problemas para tragar o respirar: Los nódulos grandes o un bocio multinodular pueden interferir con la deglución o la respiración.
• Hipertiroidismo: Pueden ocurrir problemas cuando un nódulo o bocio produce hormona tiroidea, lo que lleva a una cantidad excesiva de la hormona en el cuerpo. El hipertiroidismo puede provocar pérdida de peso, debilidad muscular, aumento de la sudoración, temblores, nerviosismo, latidos cardíacos rápidos o irregulares.
• Problemas relacionados con la cirugía del nódulo tiroideo: Si su médico recomienda una cirugía para extirpar un nódulo, es posible que deba tomar una terapia de reemplazo de hormona tiroidea por el resto de su vida, dependiendo de la extensión de la cirugía.
• Nerviosismo excesivo.
• Insomnio, palpitaciones.
• Cansancio inexplicable.
• Sudoración fácil, mala tolerancia al calor.
• Temblor de manos.
• Pérdida de peso y diarreas. También consulte a su médico si tiene signos y síntomas que pueden significar que su glándula tiroides no está produciendo suficiente hormona tiroidea (hipotiroidismo), que incluyen:
• Sensación de frio inexplicable
• Sentirse cansado con más facilidad
• Piel seca
• Problemas de memoria
• Depresión
• Estreñimiento
Revista Puertorriqueña de Medicina y Salud Pública 45

46 Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública 47
48 Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública 49
CUIDADO PREVENTIVO PARA PACIENTES CON ENFERMEDAD INFLAMATORIA DE INTESTINO
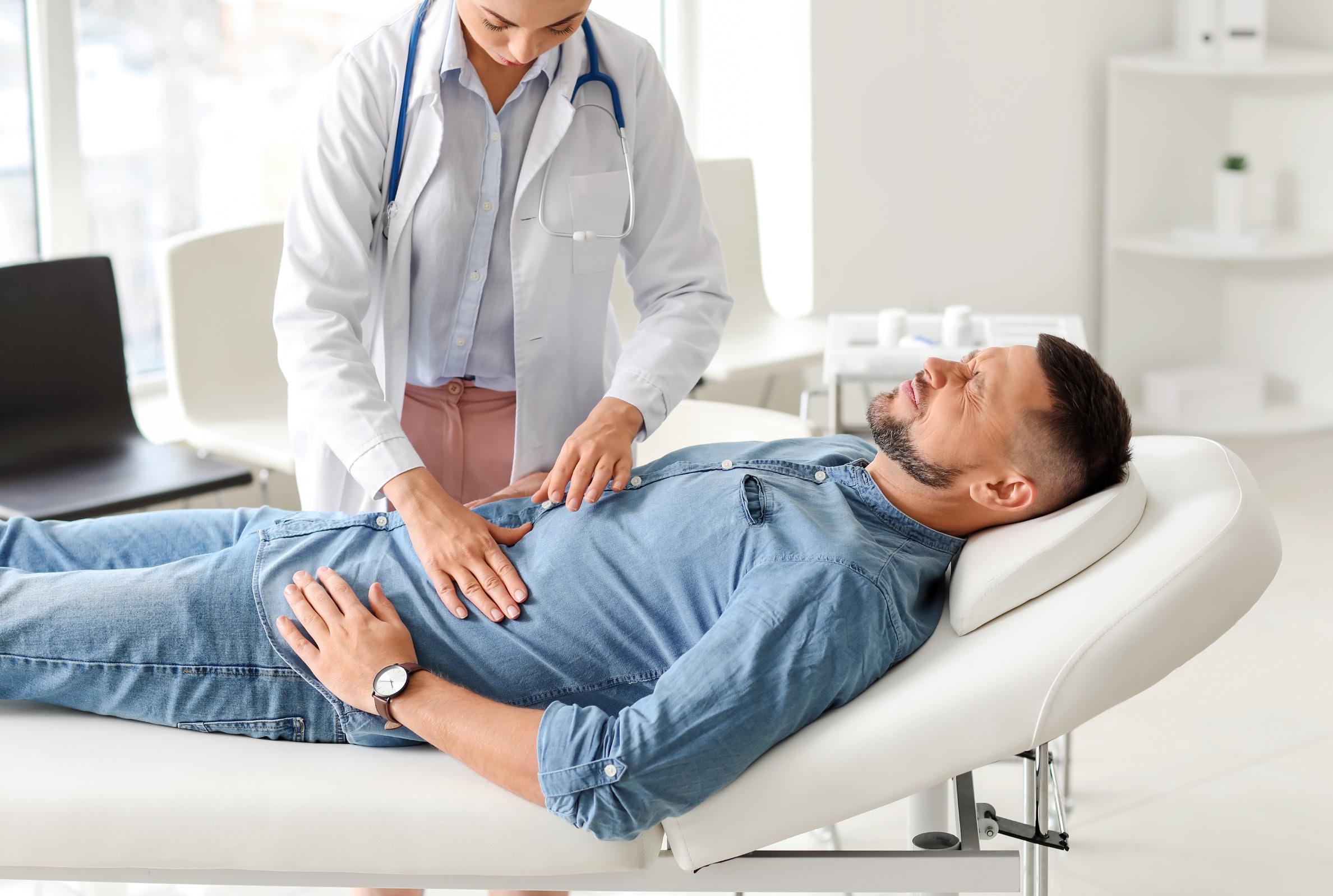
Palabras Clave: Enfermedad inflamatoria de intestino, cuidado preventivo, enfermedad de Crohn, colitis ulcerosa, vacunas, cernimiento de cáncer, osteoporosis, cáncer colorrectal, COVID-19
RESUMEN
El Colegio Americano de Gastroenterología (ACG por sus siglas en inglés) publicó unas guías para el cuidado preventivo requerido en pacientes con enfermedad inflamatoria de intestino (EII), que incluyen vacunas y cernimientos de cáncer, osteoporosis, e infecciones como tuberculosis latente. Debido al impacto que tiene esta condición crónica en la salud emocional de los pacientes con EII, las guías también
hacen un enfoque en el cernimiento para condiciones psiquiátricas como depresión mayor y ansiedad generalizada. Fumar activamente es un factor de riesgo para complicaciones y pobre respuesta a terapia, por lo cual también se recomienda un cernimiento cercano sobre el uso del cigarrillo. Ante la llegada de la pandemia y la severidad del coronavirus, el ACG también recomienda la vacuna contra
el COVID-19 de acuerdo con las recomendaciones del Centro para el Control y la Prevención de Enfermedades (CDC por sus siglas en inglés). Estas guías son una fuente de educación no solo para los gastroenterólogos sino para los médicos primarios con el fin de mejorar el cuidado preventivo y la calidad de vida de estos pacientes.
50 Revista Puertorriqueña de Medicina y Salud Pública
Karelys Burgos Irizarry
 Yue Sai Jao
Yue Sai Jao
La enfermedad inflamatoria de intestino (EII) es un término amplio que se usa para describir desórdenes que causan inflamación crónica del tracto digestivo, como colitis ulcerosa (CU) y enfermedad de Crohn (EC). EII es una de las 5 condiciones crónicas gastrointestinales más prevalentes en los Estados Unidos, con un costo total anual en servicios de salud que sobrepasa los $1.5 billones. Por la severidad de esta condición y la continuidad de cuidado que requiere, estos pacientes tienden a considerar su gastroenterólogo como su médico primario, lo cual lleva a una disminución de visitas al médico primario.
Hay una alta prevalencia de pacientes con EII moderada a severa que requiere tratamiento con terapia inmunosupresora, lo que conlleva un cuidado preventivo específico por las complicaciones asociadas a la inmunosupresión. Sin embargo, los estudios demuestran que estos pacientes no reciben cuidado preventivo con la
INTRODUCCIÓN DISCUSIÓN
Vacunas
Todo paciente con Enfermedad Inflamatoria del Intestino (EII) debe recibir las vacunas inactivadas sin importar su estado de inmunosupresión; estas vacunas incluyen, pero no se limitan a Influenza, Neumococo, Hepatitis A y B, Haemophilus influenza B, Virus del Papilloma Humano, tétano y pertussis. No obstante, la mayoría de las vacunas atenuadas también son parte del mantenimiento de salud y son recomendadas a aquellos pacientes
misma frecuencia que el resto de la población. Una de las razones para esta disparidad es que hay un enfoque primordial en control de síntomas por encima de medidas preventivas. La ausencia de cuidado preventivo en estos pacientes afecta tanto la calidad de cuidado como los resultados de los pacientes. Por lo tanto, no solo los gastroenterólogos, sino los médicos primarios deben crear un enfoque multidisciplinario y tomar un rol proactivo con la meta de empoderar al paciente a responsabilizarse y crear conciencia sobre su enfermedad y el cuidado preventivo que compele.
De acuerdo con las guías del Colegio Americano de Gastroenterología (ACG por sus siglas en inglés) para el cuidado preventivo de pacientes con EII, los asuntos de mantenimiento de salud incluyen: vacunas indicadas para pacientes con EII, cernimiento de tuberculosis latente, osteoporosis, cáncer cervical, melanoma y cáncer de piel no melanoma, cáncer de colon y una
evaluación para depresión, ansiedad y el hábito de fumar. Todos deben ser repasados en cada visita para asegurar la adherencia y el cumplimiento por parte de los pacientes.
Para el año 2013, la prevalencia de EII en Puerto Rico mostró un alza de hasta 4 veces el número de pacientes en comparación con la prevalencia del 2005. Desafortunadamente hay datos limitados sobre el conocimiento y percepción del cuidado preventivo de pacientes con EII en Puerto Rico. Actualmente se está conduciendo un estudio con el fin de evaluar el conocimiento y percepción tanto en los pacientes como en los gastroenterólogos acerca de las guías de cuidado preventivo con el fin de crear conciencia, aumentar el conocimiento y de esa manera mejorar el cuidado multidisciplinario de los pacientes con EII. Mediante este artículo les proveemos un resumen sobre los aspectos más importantes del cuidado preventivo de pacientes con EII.

con EII que no estén inmunosuprimidos (sea por medicamentos, cáncer o malnutrición severa) o que estén a un nivel mínimo de inmunosupresión. Sin embargo, existen excepciones. Hasta el momento, no tenemos evidencia de que las vacunas desencadenen una exacerbación de EII según varios estudios publicados.
La vacuna inactivada contra la influenza se recomienda anualmente.
Las vacunas contra el neumococo PCV13 y PPSV23 se deben administrar en pacientes inmunosuprimidos, pero no simultáneamente. Actualmente, existen dos formulaciones nuevas de la vacuna conjugada contra el neumococo (PCV15 o PCV20). El Centro para Control y Prevención de Enfermedades (CDC) recomienda que se les ofrezca estas alternativas a los adultos de 65 años o más que nunca han recibido la versión de PCV13 o
Revista Puertorriqueña de Medicina y Salud Pública 51
Programa de Adiestramiento en Gastroenterología, Escuela de Medicina, Recinto de Ciencias Médicas UPR
Fellow de Gastroenterología UPR Recinto de Ciencias Médicas
MD, fellow de Gastroenterología, Escuela de Medicina, Recinto de Ciencias Médicas
que su historial de vacunación sea desconocido. A adultos mayores de 50 años, que incluyen los pacientes con EII sin discriminación alguna por su estado de inmunosupresión, se les recomienda que se vacunen contra el Herpes Zoster. El estudio de VERVE, mejor conocido por sus siglas en ingles de “VaricElla ZosteR VaccinE”, demostró seguridad y eficacia de la vacuna en pacientes que fueron administrados la vacuna atenuada de Herpes Zoster mientras estaban recibiendo su tratamiento con biológicos (anti-TNF).
Se recomienda que se administre la vacuna de varicela antes de empezar terapia de inmunosupresión, verificando que el paciente no haya tenido exposición previa al virus o la vacuna. No existe contraindicación para que miembros del hogar que conviven con personas inmunosuprimidas reciban vacunas atenuadas como las vacunas contra paperas, Sarampión o Rubeola, Rotavirus, Varicela y Herpes Zoster, tomando las debidas precauciones para proteger a los pacientes vulnerables. En caso de que una persona saludable se vacune contra la varicela y luego presente una lesión sospechosa en piel, esta persona debe ser aislada para proteger al individuo inmunocomprometido. A los pacientes con EII se les recomienda ser evaluados por un infectólogo por
Prevención de cáncer
El mantenimiento de salud en los pacientes con EII no solo incluye la vacunación sino también los cernimientos adecuados para la detección temprana y correcta de distintos tipos de cáncer. De acuerdo con el Colegio Americano de Obstetricia y Ginecología (ACOG en inglés) y el CDC, se recomienda que se haga un cernimiento anual para la prevención de cáncer cervical con el uso de la prueba de Papanicolaou, ya que los pacientes con EII están predispuestos a tener mayor incidencia de displasia y cáncer cervical en comparación con la población general. A los pacientes con EII se les recomienda que rutinariamente usen bloqueador solar y se hagan cernimientos dermatológicos rutinarios para evaluación de cáncer de la piel incluyendo el melanoma. Se debe enfocar aún más la atención en los pacientes que estén usando medicamentos de inmunosupresión. Se ha identificado una relación entre
lo menos tres meses antes de que se vayan a viajar a países en donde haya diversidad de enfermedades infecciosas para poder administrar las vacunas correspondientes, de ser candidatos. La vacuna contra la fiebre amarilla es atenuada. A aquellos pacientes que por su inmunosupresión no puedan recibir la vacuna contra la fiebre amarilla, se les recomienda que eviten viajar a países endémicos donde puedan contraer esta condición y de ser inevitable el viaje tomar las precauciones necesarias para evitar la infección.
A los pacientes con EII se les recomienda la vacuna contra el meningococo sin importar su estado de inmunosupresión; la vacuna está específicamente destinada para las adolescentes y los adultos jóvenes entre 16 a 23 años que tienen mayor riesgo de infección. A todo paciente de EII se le recomienda recibir las vacunas que le correspondan de acuerdo con su edad, específicamente las vacunas atenuadas e inactivadas al menos 4 y 2 semanas antes de comenzar una terapia de inmunosupresión, respectivamente. Una vez que se le ofrezcan las vacunas inactivadas se recomienda verificar inmunidad en el tiempo de post vacunación para corroborar la necesidad de un refuerzo. La respuesta inmune a la vacuna disminuye a medida que aumente
el estado de inmunosupresión, pero se ha visto que, a pesar de lo antes mencionado, las vacunas siguen siendo beneficiosas y seguras en esta población, según lo descrito en literatura científica. Es importante resaltar que la responsabilidad de las recomendaciones de vacunas descritas en esta publicación debe ser compartida entre el gastroenterólogo y el médico primario para poder fomentar el cuidado preventivo necesario para los pacientes de Crohn y colitis ulcerosa. Por último, la Fundación de Crohn y Colitis, la cual aboga por prácticas seguras en pacientes con EII, apoya la vacunación contra el virus de COVID 19 en pacientes con EII. Ellos entienden que los pacientes con EII no tienen riesgo aumentado de enfermedad severa por coronavirus y no consideran que los pacientes con EII necesiten una dosis adicional de la vacuna de mRNA, ya que muchos no son considerados inmunosuprimidos. Deben seguir las recomendaciones de la población general. Las recomendaciones en cuanto a la vacuna de COVID por parte del ACG se basan en las guías del CDC, por lo cual hay que mantenerse al tanto a los cambios y las actualizaciones más recientes para poder proveerle al paciente la información adecuada.
los biológicos contra factor de necrosis tumoral y el riesgo de melanoma y el uso de tiopurina con el cáncer escamoso de la piel, el último mencionado en adultos mayores de 50 años según el estudio de CESAME.15 A los pacientes con EII se les recomienda cernimiento de cáncer colorrectal después de 8 años de enfermedad mediante evaluación por colonoscopia, dependiendo de la extensión de la enfermedad.
está en remisión.18,19 Hay un estudio en donde se observó una correlación inversa entre la presencia de depresión y la adherencia de medicamentos.20 Lo antes descrito resalta la importancia de identificar las condiciones sicológicas en los pacientes con EII para proveer un manejo completo de la enfermedad y sus complejidades.
Estrés, Depresión y Ansiedad Osteoporosis
Las enfermedades sicológicas son factores que añaden complejidad a los pacientes con Crohn y colitis ulcerosa, ya que afectan como el paciente percibe su condición y el manejo que está recibiendo. Hay diversas publicaciones que han determinado que hay mayor nivel de estrés, depresión y ansiedad en pacientes que tienen la enfermedad activa en comparación con aquellos en quienes su enfermedad
Los pacientes con EII están a mayor riesgo de tener osteoporosis.21 Los factores de riesgo para pérdida de densidad de masa ósea incluyen a mujeres deficientes de estrógeno/ postmenopausia, mujeres de bajo índice de masa corporal, hiperparatiroidismo primario e individuos que estén usando terapia prolongada de corticoesteroides definida como mayor o igual a 5 mg por día de prednisona o una dosis
52 Revista Puertorriqueña de Medicina y Salud Pública
equivalente por al menos 3 meses. A los pacientes que tengan los factores de riesgo antes mencionados se les recomienda el estudio para evaluar la densitometría ósea (DXA), el cual también es útil para mujeres de 65 años o más y hombres de 70 años o más según la Fundación Nacional de Osteoporosis.
Osteoporosis
Se ha identificado una asociación significativa entre el fumador activo y el desarrollo y progresión de la enfermedad de Crohn. Según diversos estudios publicados, los pacientes con
EII que se denominaban fumadores activos estaban relacionados con una alta frecuencia de uso de terapia de mantenimiento específicamente el uso de biológicos, pobre respuesta a terapia y alta frecuencia de intervenciones quirúrgicas en comparación con los no fumadores.
CONCLUSIONES
• Existen guías para el cuidado preventivo requerido para pacientes con EII que incluyen vacunas y cernimientos de cáncer, osteoporosis y tuberculosis latente, indicados para estos pacientes.
• Las guías también hacen enfoque en la evaluación de condiciones psiquiátricas como depresión mayor, ansiedad generalizada y abuso de sustancias, como el cigarrillo, que afectan la percepción y el manejo de la enfermedad.
• No solo los gastroenterólogos deben ser los responsables de inculcar el cuidado preventivo en pacientes con EII y el enfoque debe dirigirse al empoderamiento del paciente con respecto a su cuidado.
• Se debe recomendar a todos los pacientes la vacunación contra el virus de COVID 19 de acuerdo con las recomendaciones del CDC.
CUIDADO DE SALUD PREVENTIVO PARA PACIENTES CON EII: VACUNAS
* Frecuencias y dosis indicadas están disponibles en las recomendaciones del Comité Asesor sobre Prácticas de Inmunización (ACIP).
• Contraindicada en pacientes recibiendo tratamiento inmunosupresor.
† Depende del tipo de medicamento inmunosupresor.
Δ Las recomendaciones resumidas en esta tabla se basan en las guías de cuidado preventivo de EII del ACG y la lista de cotejo de mantenimiento de salud de la Fundación de Crohn y Colitis.
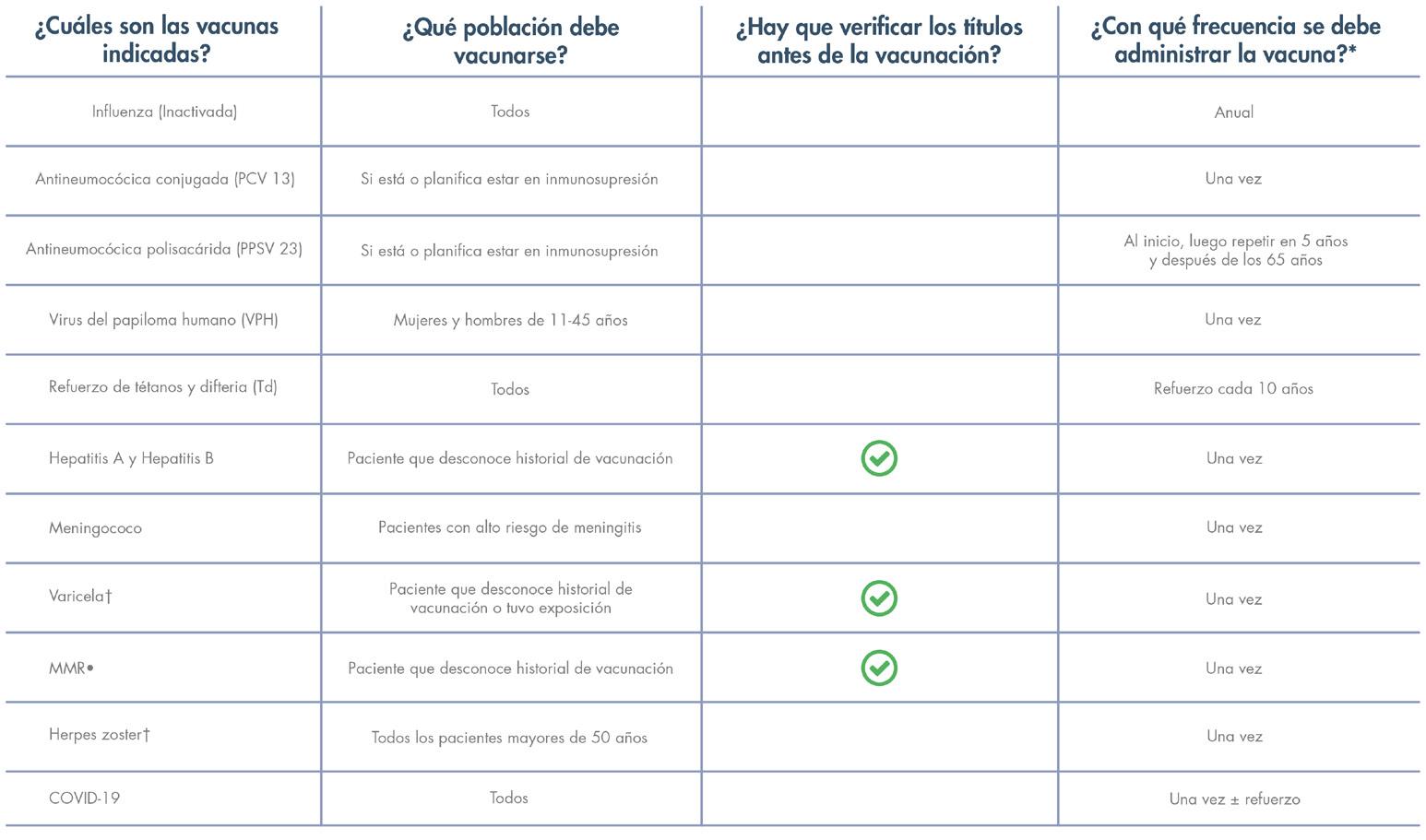
REFERENCIAS
Abegunde AT, Muhammad BH, Ali T. Preventive health measures in inflammatory bowel disease. World J Gastroenterol. 2016 Sep 14;22(34):7625-44. Doi: 10.3748/ wjg.v22.i34.7625.
Farraye F A, Melmed GY, Lichtenstein GR, Kane,SV. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. Amer J Gastroenterol: February 2017 – Volume 112 – Issue 2 – p 241258 doi: 10.1038/ajg.2016.537.
Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, Lindsay R; National Osteoporosis Foundation. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int. 2014 Oct;25(10):2359-81. Doi: 10.1007/s00198-014-2794-2. Epub 2014 Aug 15. Erratum in: Osteoporos Int. 2015 Jul;26(7):2045-7.
Kroger A, Bahta L, Hunter P. General Best Practice Guidelines for Immunization. Best Practices Guidance of the Advisory
Committee on Immunization Practices (ACIP). https://www.cdc.gov/vaccines/hcp/aciprecs/general-recs/index.html
Pillai N, Dusheiko M, Burnand B, Pittet V. A systematic review of cost-effectiveness studies comparing conventional, biological and surgical interventions for inflammatory bowel disease. PloS One. 2017 Oct 3;12(10):e0185500. Doi: 10.1371/ journal.pone.0185500.
Revista Puertorriqueña de Medicina y Salud Pública 53
Practice Bulletin No. 157: Cervical Cancer Screening and Prevention. Obstet Gynecol. 2016 Jan;127(1):e1-e20. Doi: 10.1097/ AOG.0000000000001263.
Torres EA, Torres-Cintron M, Velazquez S, Vendrell R, Del Valle A, Pérez CM. Prevalence of Inflammatory Bowel Diseases in Puerto Rico: A Health Care Claims Analysis of an Insured Population. P R Health Sci J. 2021 Sep;40(3):103-109.
Rahier JF, Papay P, Salleron J et al. H1N1 vaccines in a large observational cohort of patients with inflammatory bowel disease treated with immu- nomodulators and biological therapy. Gut 2011;60:456–62.
Dotan I, Werner L, Vigodman S et al. Normal response to vaccines in inflammatory bowel disease patients treated with thiopurines. Inflamm Bowel Dis 2012;18:261–8.
Agarwal N, Ollington K, Kaneshiro M et al. Are immunosuppressive medications associated with decreased responses to routine immuniza- tions? A systematic review. Vaccine 2012;30:1413–24.
Curtis JR, Cofield SS, Bridges SL, et al; The Safety and Immunologic Effectiveness of the Live Varicella-Zoster Vaccine in Patients Receiving Tumor Necrosis Factor Inhibitor Therapy: A Randomized Controlled Trial. Ann Intern Med.2021;174:1510-1518. [Epub 28 September 2021]. doi:10.7326/
M20-6928
Andrisani G, Frasca D, Romero M et al. Immune response to influenza A/ H1N1 vaccine in inflammatory bowel disease patients treated with anti TNFalpha agents: effects of combined therapy with immunosuppressants. J Crohn’s Colitis 2013;7:301–7.
Hagihara Y, Ohfuji S, Watanabe K et al. Infliximab and/or immunomodu- lators inhibit immune responses to trivalent influenza vaccination in adults with inflammatory bowel disease. J Crohn’s Colitis 2014;8:223–33.
Nguyen DL, Nguyen ET, Bechtold ML. Effect of immunosuppressive therapies for the treatment of inflammatory bowel disease on response to routine vaccinations: a metaanalysis. Dig Dis Sci 2015;60:2446–53.
Beaugerie L, Brousse N, Bouvier AM, Colombel JF, Lémann M, Cosnes J, Hébuterne X, Cortot A, Bouhnik Y, Gendre JP, Simon T, Maynadié M, Hermine O, Faivre J, Carrat F; CESAME Study Group. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet. 2009 Nov 7;374(9701):1617-25. doi:
10.1016/S0140-6736(09)61302-7. PMID: 19837455.
Farraye FA, Odze RD, Eaden J, Itzkowitz SH. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:746–774, 774.e1-4; quiz e12-13.
Clarke WT, Feuerstein JD. Colorectal cancer surveillance in inflammatory bowel disease: Practice guidelines and recent developments. World J Gastroenterol. 2019 Aug 14;25(30):4148-4157. doi: 10.3748/ wjg.v25.i30.4148. PMID: 31435169; PMCID: PMC6700690.
Persoons P, Vermeire S, Demyttenaere K et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment Pharmacol Ther 2005;22: 101–10.
Calvet X, Gallardo O, Coronas R et al. Remission on thiopurinic immunomodulators normalizes quality of life and psychological status in patients with Crohn’s disease. Inflamm Bowel Dis 2006;12:692–6.
Nigro G, Angelini G, Grosso SB et al. Psychiatric predictors of noncompli- ance in inflammatory bowel disease: psychiatry and compliance. J Clin Gastroenterol 2001;32:66–8.
Pollak RD, Karmeli F, Eliakim R et al. Femoral neck osteopenia in patients with inflammatory bowel disease. Am J Gastroenterol 1998;93:1483–90.
Nunes T, Etchevers MJ, Merino O et al. Does smoking influence Crohn’s disease in the biologic era? The TABACROHN study. Inflamm Bowel Dis 2013;19:23–9.
To N, Gracie DJ, Ford AC. Systematic review with meta-analysis: the ad- verse effects of tobacco smoking on the natural history of Crohn’s disease. Aliment Pharmacol Ther 2016;43:549–61.
Parsi MA, Achkar JP, Richardson S et al. Predictors of response to infliximab in patients with Crohn’s disease. Gastroenterology 2002;123:707–13.
Moss A, Farraye F, Gordon G, Vrabie R. Health Maintenance Checklist for Adult IBD Patients. Crohn’s & Colitis Foundation’s Professional Education Committee Sub-Group. https://www. crohnscolitisfoundation.org/sites/default/
files/legacy/science-and-professionals/ programs-materials/health-maintenancechecklist.pdf
54 Revista Puertorriqueña de Medicina y Salud Pública
Tuve suerte.
“Estaba ocupada – trabajando y disfrutando de la vida. Ya me tocaba hacerme la prueba de Papanicolaou para verificar si tenía cáncer de cuello uterino. Cuando me hicieron la prueba, creímos que podría tener cáncer de cuello uterino. Finalmente, me llegaron resultados buenos – ¡no tuve cáncer! Mujeres, por favor háganse la prueba para detectar el cáncer de cuello uterino. Infórmense sobre el cáncer ginecológico.”
Cote De Pablo, Actriz

Los cánceres ginecológicos incluyen: cáncer de cuello uterino, de ovario, de útero, de vagina y de vulva. Solo el cáncer de cuello uterino se puede detectar a través de una prueba.
Aprenda cuáles son los síntomas y qué puede hacer para prevenir los cánceres ginecológicos.
http://www.cdc.gov/spanish/ cancer/knowledge
1-800-CDC-INFO
LA DIABETES Y SU MANEJO CAMBIANTE
La diabetes es una condición crónica en la que se ve alterada la manera en que el organismo utiliza el azúcar ya sea por ausencia o resistencia a la insulina. Esto provoca niveles elevados de glucosa en la sangre (también llamado azúcar en la sangre). En Puerto Rico, la prevalencia de dicha enfermedad según datos del 2021 fue de un 16.8%1. Inclusive, de los años 2010 al 2016 fue la tercera causa de muerte en Puerto Rico2 por
lo que es nuestro deber ser proactivos y mantenernos a la vanguardia en cuanto a su manejo. Han sido muchos los grandes avances en herramientas para monitorear el azúcar en la sangre y medicamentos que ayudan a las personas con diabetes a mejorar en gran medida el control de sus niveles de azúcar en la sangre, el exceso de peso, la presión arterial alta y su calidad de vida.
Palabras Clave: Diabetes, Diabetes Mellitus, Manejo, Insulina
El cuidado de las personas con prediabetes y diabetes es una hazaña que incluye cambios en el estilo de vida con énfasis en la alimentación saludable, el ejercicio y ajustes en los hábitos de sueño por lo que merece un enfoque multidisciplinario siendo el paciente el centro del equipo y quien debe ayudar a tomar decisiones junto con sus médicos. Además de médicos, el equipo debe incluir: educadores de diabetes, enfermeras, dietistas,
56 Revista Puertorriqueña de Medicina y Salud Pública
Mónica A. Ortiz Rivera

Endocrinologa del hospital universitario de adultos
RESUMEN
El cuidado de las personas con diabetes incluye cambios en el estilo de vida con énfasis en la alimentación saludable y el ejercicio. Alcanzar los objetivos de azúcar en sangre, presión arterial, grasas como el colesterol y peso puede prevenir el daño de la diabetes en los ojos, los riñones, el corazón y el sistema nervioso. Han sido muchos los grandes avances en herramientas para monitorear el azúcar en la sangre y medicamentos que están ayudando a las personas con diabetes a mejorar en gran medida el control de sus niveles de azúcar en la sangre, el exceso de peso, la presión arterial alta y su calidad de vida. Es por esto que debemos mantenernos a la vanguardia con la meta de mejor proteger la salud de nuestros pacientes.

farmacéuticos, podiatra, psicólogos, entre otros especialistas. La terapia nutricional es parte integral del control de la diabetes, con el objetivo de promover y apoyar patrones de alimentación saludables. A su vez, estas intervenciones en el estilo de vida deben incluir una receta individualizada de actividad física que incluya ejercicios aeróbicos y de resistencia con una meta de reducción del sedentarismo. La recomendación inicial de actividad
física aeróbica requiere un aumento progresivo del volumen y la intensidad del ejercicio con el objetivo final de completar 150 min/semana de ejercicio moderado realizado en 3 a 5 sesiones por semana.
Cada paciente es un mundo distinto, así que los objetivos de glucosa deben individualizarse teniendo en cuenta la duración de la enfermedad, la presencia o ausencia de complicaciones micro y
TAMBIÉN SE DEBE TOMAR EN CONSIDERACIÓN EL ESTADO COGNITIVO Y PSICOLÓGICO DE LA PERSONA.
Revista Puertorriqueña de Medicina y Salud Pública 57
macrovasculares, los factores de riesgo de enfermedad cardiovascular (ECV), las condiciones comórbidas y el riesgo de hipoglucemia.

El fin de alcanzar estos objetivos de azúcar en la sangre, presión arterial, grasas como el colesterol y peso es prevenir el daño que puede ocasionar la diabetes en los ojos, los riñones, el corazón y el sistema nervioso a largo plazo. Se recomienda un nivel de A1C del 6.5% o menos para la mayoría de los pacientes adultos, si se puede lograr de manera segura. Sin embargo, hay casos en los que se deben adoptar metas glucémicas menos estrictas de una A1C de 7% a 8%.
El monitorear los niveles de azúcar en la sangre con meramente pinchazos en los dedos ha evolucionado a la implementación del uso de sensores colocados debajo de la piel (monitores continuos de glucosa), lo que hace que sea más fácil y seguro para las personas con diabetes evitar niveles altos y bajos de azúcar en la sangre. Estos valores de glucosa son vitales y nos ayudarán a guiar la terapia médica personalizada de nuestro paciente.
Son muchos los nuevos medicamentos con mayor índice de seguridad que han salido al mercado y ayudan a controlar
el azúcar en la sangre. Estos tienen como beneficio la reducción del riesgo de enfermedades cardíacas y renales. Algunos también reducen el colesterol y el peso.
El manejo farmacológico de diabetes depende primordialmente de las reservas de insulina que tenga el páncreas del paciente. En pacientes con historial de diabetes mellitus tipo 1 cuyo páncreas no produce insulina, pacientes con diabetes mellitus tipo 2 que apenas producen insulina ó tienen alta resistencia a la insulina, el uso de insulina es vital para lograr las metas en niveles de azúcar. Se suele administrar una insulina de duración larga (glargine, detemir, degludec) una vez al día ó una insulina de duración media (NPH) dos veces al día como insulina basal. Adicional a esto, se administra insulina previo a las comidas de acción ultra-rápida, accion rápida (lispro, aspart, glulisine) ó acción corta (regular).
La metformina ha sido la terapia inicial de elección por muchos años y los inhibidores de la dipeptidil peptidasa 4 (iDPP4) solían ser el segundo agente que se añadía a terapia si la meta glucémica del paciente no había sido lograda. Sin embargo, actualmente previo a comenzar un agente oral
Los agentes iSGLT-2 incluyen empagliflozin, canagliflozin, dapagliflozin y ertugliflozin. Entre ellos canagliflozin, dapagliflozin y empagliflozin han mostrado efectos protectivos para los riñones. Empagliflozin, dapagliflozin y canagliflozin han mostrado beneficios cardiovasculares específicos.
Ante la disponibilidad de este amplio repertorio farmacológico favorable para nuestros pacientes, se debe evitar la inercia terapéutica y clínica caracterizada por falta de intensificación del tratamiento cuando no se alcanzan los objetivos o metas. Dicha inercia también incluye la falta de desintensificación de la terapia cuando las personas reciben un tratamiento excesivo. En pacientes con episodios recurrentes de hipoglucemia severa o que tengan incapacidad de sentir episodios de hipoglucemia, se vuelve una prioridad el prevenir
eventos futuros. Las causas más comunes incluyen evitar comidas, mayor actividad física o tener dosis excesivas de medicamentos por lo que en estos pacientes debemos revisar su lista de medicamentos y de ser necesario desintensificar su terapia antidiabética.
Por esta razón debemos conocer sobre los beneficios y efectos secundarios de todos estos medicamentos para lograr un mejor control glucémico en nuestros pacientes y así evitar complicaciones a largo plazo.
debemos evaluar si en el paciente existe un riesgo alto o tiene enfermedad cardiovascular aterosclerótica establecida, insuficiencia cardíaca y/o enfermedad renal crónica (hacer referencia a Figura 1). En presencia de estas condiciones se debe comenzar un agonista del receptor del péptido similar al glucagón tipo 1 (agonista del receptor GLP-1) o un inhibidor del cotransportador sodio-glucosa tipo 2 (iSGLT-2) con eficacia comprobada para la(s) condición(es) específica(s) de la persona con diabetes mellitus tipo 2.
Las indicaciones aprobadas por la FDA para los agonistas del receptor del GLP-1 exenatida QW, lixisenatida y semaglutida oral son para mejorar el control glucémico en adultos con diabetes mellitus tipo 2. Dulaglutide tiene una indicación adicional para reducir eventos cardiovasculares adversos mayores en personas con diabetes mellitus tipo 2 con y sin enfermedades cardiovasculares establecidas. La liraglutida y la semaglutida subcutánea están aprobadas para reducir el riesgo de infarto de miocardio, accidentes cerebrovasculares o muerte cardiovascular en adultos con diabetes mellitus tipo 2 y enfermedad cerebrovascular. Estos agonistas GLP1 tienen como beneficio secundario la pérdida de peso.
58 Revista Puertorriqueña de Medicina y Salud Pública
ltrationrate;GLP-1RA,glucagon-likepeptide1receptoragonist;HF,heartfailure;HFpEF,heartfailure
Useofglucose-loweringmedicationsinthemanagementoftype2diabetes.ACEi,angiotensin-convertingenzymeinhibitor;ACR,albu- min/creatinineratio;ARB,angiotensinreceptorblocker;ASCVD,atheroscleroticcardiovasculardisease;CGM,continuousglucosemonitoring; CKD,chronickidneydisease;CV,cardiovascular;CVD,cardiovasculardisease;CVOT,cardiovascularoutcomestrial;DPP-4i,dipeptidylpeptidas e4
REFERENCIAS
1. Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Division of Population Health. BRFSS Prevalence & Trends Data [online]. 2015. [accessed Dec 01, 2022]. URL: https:// www.cdc.gov/brfss/brfssprevalence/.
2. Departamento de Salud (2019). Informe Anual Estadísticas Vitales Mortalidad 2015 - 2016.
Blonde L, Umpierrez GE, Reddy SS, McGill JB, Berga SL, Bush M, Chandrasekaran S, DeFronzo RA, Einhorn D, Galindo RJ, Gardner TW, Garg R, Garvey WT, Hirsch
IB, Hurley DL, Izuora K, Kosiborod M, Olson D, Patel SB, Pop-Busui R, Sadhu AR, Samson SL, Stec C, Tamborlane WV Jr, Tuttle KR, Twining C, Vella A, Vellanki P, Weber SL. American Association of Clinical Endocrinology Clinical Practice Guideline: Developing a Diabetes Mellitus Comprehensive Care Plan-2022 Update. Endocr Pract. 2022 Oct;28(10):923-1049.
Davies MJ, Aroda VR, Collins BS, Gabbay RA, Green J, Maruthur NM, Rosas SE, Del Prato S, Mathieu C, Mingrone G, Rossing P, Tankova T, Tsapas A, Buse JB. Management of Hyperglycemia in Type 2 Diabetes, 2022. A Consensus Report by the
American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2022 Nov 1;45(11):2753-2786.
Figura 1. Davies MJ, Aroda VR, Collins BS, Gabbay RA, Green J, Maruthur NM, Rosas SE, Del Prato S, Mathieu C, Mingrone G, Rossing P, Tankova T, Tsapas A, Buse JB. Management of Hyperglycemia in Type 2 Diabetes, 2022. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2022 Nov 1;45(11):2753-2786.
Revista Puertorriqueña de Medicina y Salud Pública 59
Figura 1. Recomendaciones de práctica clínica sobre el uso de medicamentos hipoglucemiantes en el tratamiento de la diabetes tipo 2.
Figure3
inhibitor;eGFR,estimatedglomerular
from http://diabetesjournals.org/care/article-pdf/doi/10.2337/dci22-0034/688913/dci220034.pdf by guest on 26 September 2022
fi
Downloaded
INNOVACIÓN Y EXPERIENCIA PARA TRATAR AL CÁNCER DE PULMÓN EN HOSPITAL DAMAS
La cirugía robótica implementada en procedimientos cardiotorácicos y colorrectales ahora es una realidad en la Isla.
Aunque hace varios años en Puerto Rico se comenzó a implementar la tecnología robótica para procedimientos quirúrgicos mediante la conocida plataforma Da Vinci, esta innovación no estaba disponible para otras especialidades como lo es la cirugía cardiotorácica y colorrectal.
Su uso se delimitaba a los procedimientos ginecológicos, cirugía general e intervenciones urológicas. Actualmente, el Hospital Damas en Ponce utiliza esta plataforma desarrollada específicamente para los pacientes con condiciones del pulmón, el esófago y la cavidad torácica.
Tecnología robótica para procedimientos pulmonares en Puerto Rico
El especialista pionero en traer esta tecnología a la Isla es el Dr. Alberto Maldonado Molina, cirujano cardiotorácico y especialista en procedimientos quirúrgicos del Hospital Damas, quien llevó a cabo con éxito, en diciembre del año anterior, la primera biopsia pulmonar por robótica en Puerto Rico, un hito sin precedentes en la medicina del país.
“Estuve en Estados Unidos desde el 2013 practicando cirugía cardiotorácica y cardiovascular y siempre me interesó la cirugía robótica. Uno de los sueños que tenía era volver
a la Isla y traer esa tecnología en la Isla que no existe en Puerto Rico, existe para otras especialidades, pero no para cirugía torácica”, mencionó el experto.
Entre las ventajas de esta innovación tecnológica, el especialista destaca que permite operar a distancia del paciente, es decir, el médico encargado opera el robot Da Vinci en la misma sala de operaciones desde un monitor externo. Esto brinda una mayor precisión en las incisiones que se realizan, además de proporcionar una intervención mínimamente invasiva.
60 Revista Puertorriqueña de Medicina y Salud Pública
Dr. Alberto Maldonado Molina
Cirujano cardiotorácico y especialista en procedimientos quirúrgicos del Hospital
Damas
“Eso hace una diferencia increíble en la recuperación del paciente, el dolor y la estadía en el hospital. Llevo realizando esta cirugía desde el 2013 y los pacientes se van más rápido hacia la casa, sin mayores complicaciones, se ven en una semana en la oficina y después pueden retomar sus labores diarias, eso con una cirugía abierta no se puede hacer”, destacó el Dr. Maldonado.
Intervenciones mínimamente invasivas y con mayor precisión, un beneficio para los pacientes
En el Hospital Damas la cirugía robótica se utiliza actualmente en todo el campo de la cirugía cardíaca y torácica, sin embargo, la principal prioridad es la atención de pacientes que padecen cáncer de pulmón, de esófago, o en la cavidad torácica, reconoció el galeno.
Al respecto, el especialista ahondó en los servicios que se pueden brindar con esta tecnología de última generación. “Se utiliza para lesiones en pulmón, cáncer de pulmón, enfermedades nódulo-linfáticas en el tórax, para cáncer de esófago y otros procedimientos que no son lesiones malignas en el esófago”, puntualizó.
Además, añadió que “también se utiliza para lesiones en el mediastino, que es un compartimiento de la cavidad torácica donde pueden salir tumores y se puede utilizar la tecnología del Da Vinci para resecar esos tumores sin tener que abrir el tórax, eso provee mucha más comodidad para el paciente, menos dolor, más rapidez en recuperarse, menos gasto para el hospital y menos incidencia de sangrado”.
Este tipo de tecnología se ha venido practicando en los Estados Unidos desde hace años y para los especialistas la cirugía robótica ha demostrado resultados favorables en la recuperación de los pacientes,

aunque como en cualquier modalidad quirúrgica, existen riesgos.
“Cabe señalar que existía una manera mínimamente invasiva de realizar estos procedimientos que era la cirugía toracoscópica, con un par de roticos, pero no ofrece la versatilidad que ofrece el robot cuando permite un campo amplio y con más movimiento de los brazos del robot. Esto provee mejores resultados que ambas operaciones: la toracoscopia y abierta”, resaltó el Dr. Maldonado.
El especialista también explicó que en el caso particular de un paciente que tiene cáncer de pulmón o una lesión posiblemente cancerosa en el pulmón, en el Hospital Damas cuentan con tecnologías recientemente desarrolladas, aparte del Da Vinci, para intervenir.
“Vamos a ser el primero de todos los lugares en Latinoamérica que va a tener esa tecnología que se llama VEGAN que nos permite guiado por CT, localizar una lesión en el tórax, hacer una biopsia percutánea guiado por el robot y luego de marcar la lesión, uno entra con cuatro incisiones pequeñas; se introducen unos accesos para que los brazos del robot se acoplen y el operador, en ese caso yo, opero desde una consola”.
Antes de intervenir a un paciente oncológico se realizan protocolos de evaluación para conocer si la lesión es robóticamente resecable, o si el paciente presenta comorbilidades asociadas o riesgo cardíaco.
Además de eso, se implementa un control de calidad y revisiones periódicas de cada uno de los operadores para asegurar el cumplimiento de los parámetros del robot.
Una de las ventajas más relevantes de esta tecnología es que permite diagnosticar y tratar al paciente en un mismo sitio especializado, así como también mejorar su tiempo de recuperación.
“Si el paciente tiene una lesión maligna, puede comenzar la quimioterapia o terapia adyuvante que requiera, más temprano que con la cirugía convencional”, afirmó.
Aparte de eso, el especialista reconoce que el uso de la tecnología robótica también es un beneficio para los planes médicos y los hospitales, debido a que los costos requeridos son menores y los planes médicos cubren estos procedimientos.
Revista Puertorriqueña de Medicina y Salud Pública 61
Los pacientes pueden ser diagnosticados y tratados por medio de la tecnología robótica
AGENDA MÉDICA 2023

EVENTOS Y CONVENCIONES
www.medicinaysaludpublica.com
25 al 28 de enero de 2023
Febrero 2022 Marzo 2023
16 al 19 de febrero de 2023
17 y 18 de febrero de 2023
24 al 25 de febrero de 2023
23 al 25 de febrero de 2023
2 al 4 de marzo de 2023
15 al 18 de marzo de 2023
19 de marzo de 2023
30 de marzo de 2023
1 al 2 de abril de 2023
21 al 22 de abril de 2023
21 al 22 de abril de 2023
28 al 29 de abril de 2023
28 al 30 de abril de 2023
20 de mayo de 2023
26 al 29 de mayo de 2023
1 al 4 de junio de 2023
9 al 10 de junio de 2023
1st Winter Congress & 1rst Pan American Pulmonary Hypertension Symposium
Diabetes University Online
70 th Convención Anual de la Sociedad Puertorriqueña de Pediatría
Puerto Rico Alzheimer’s Disease Research Symposium
Asociación Puertorriqueña de Gastroenterología Digestive Diseases of the Caribbean
Convención annual American College of Surgeons- PR Chapter
American College of Physicians PR Chapter
Convención anual #49 de la Academia Médica del Sur & Casa del Médico
Nephrology Symposium for Primary Physicians 2023
Convención anual de PIA
Academia de Patología y Medicina de Laboratorio de Puerto Rico
Convención de Médicos de Familia
Convención anual American College of Cardiology- PR Chapter
Convención anual Sociedad Radiológica de PR
Sociedad de Enfermedades Infecciosas de PR (SEINPR)
Caribbean Breast Symposium Sociedad Puertorriqueña de Senología
Convención Sociedad Puertorriqueña de Oftalmología
Convención Sociedad de Otorrinolaringólogos de Puerto Rico
Convención Semi anual de la Sociedad Puertorriqueña de Neumología
Puerto Rico Convention Center, San Juan, PR
VIRTUAL
Sheraton Convention Center Hotel San Juan, PR
Ponce Hilton Hotel & Casino Ponce, PR
Sheraton Convention Center Hotel San Juan, PR
23-RCM // 24-25 Embassy Suites Hotel, Carolina, PR
Hotel La Concha San Juan, PR
Ponce Hilton Hotel & Casino Ponce, PR
Antonio’s Restaurant, Condado, PR
IC Planners 787-504-3655 / ivettecolon@icplannerspr.com
SPED diabetesuniversityonline@gmail.com
SDMS Group Rosa Correa Lilly Rivera 787-731-3325 / 787-438-0671 www.pediatras.org
Lourdes Burgos 787-562-2932
Rivs Marketing 787-294-9185 / 787-409-6497 rinavega@rivsmarketing.com
Aixa Vélez 787-649-7681 / genteinc@gmail.com
Rivs Marketing 787-294-9185 / 787-409-6497 rinavega@rivsmarketing.com
Academia Médica del Sur 787-987-8301 / 787-843-0610 academiamedicadelsur@gmail.com
Business Planners - Merna Morales 787-645-9914 bplanner21@gmail.com
Sheraton Puerto Rico Hotel & Casino, San Juan Virginia de los Reyes
Caribe Hilton Hotel San Juan, PR
Marriot Resort, San Juan, Stelaris Casino, Condado, Puerto Rico
Royal Sonesta San Juan Carolina, PR
Sheraton Convention Center Hotel San Juan, PR
Embassy Isla Verde
Embassy Suites, Isla Verde, Puerto Rico
El Conquistador Resort, Fajardo, Puerto Rico
Hyatt Regency Grand Reserve PR Río Grande, PR
Sheraton Convention Center Hotel, Miramar
Serra & Serra 787 406-4571 / 787 640-5776 patologospr@serrayserra.com
Nelly Feliciano
Aixa Vélez 787-649-7681 / genteinc@gmail.com
Serra & Serra 787 406-4571 / 787 640-5776 info@socrad.com / info@serrayserra.com
Enid Rivera (804) 774-6326 / enidrm27@gmail.com
Priscilla González 787-615-6796 / sociedadsenologapr@gmail.com
Rivs Marketing 787-294-9185 / 787-409-6497
rinavega@rivsmarketing.com
Aixa Vélez 787-649-7681 / genteinc@gmail.com
Puerto Rico
Simposio
Educación Continua
Convención
Simposio
Convención
Convención
Convención
Convención
Simposio
Convención
Convención
Convención
Convención
Convención
Convención
Simposio
Convención
Convención
Business Planners - Merna Morales 787-645-9914
bplanner21@gmail.com
Convención
62 Revista Puertorriqueña de Medicina y Salud Pública
TIPO DE EVENTO
Fecha Actividad Lugar Contacto
ACTIVIDADES ESTÁN SUJETAS A CAMBIO
AGENDA MÉDICA 2023
EVENTOS Y CONVENCIONES
www.medicinaysaludpublica.com
9 al 11 de junio de 2023
9 al 11 de junio de 2023
22 al 24 de junio de 2023
24 de junio de 2023
Agosto (Fecha exacta pendiente)
11 al 13 de agosto de 2023
11 al 13 de agosto de 2023
19 de agosto de 2023
23 al 24 de agosto de 2023
23 al 27 de agosto de 2023
26 de agosto de 2023
18 al 20 de octubre de 2023
Convención Anual Asociación de Reumatólogos de Puerto Rico
Convención Anual Asociación de Reumatólogos de Puerto Rico
Asociación Puertorriqueña de Medicina Física y Rehabilitación
Convención Psiquiátrica anual Asociación de Psiquiatras de Bayamón (APREBA) - Conferencia sobre Salud Mental, “Psychiatry & Mental Health Updates 2022” cumplen 20 años este año
Convención Sociedad de Médicos Podiatras
Congreso Coalición de Asma y otras condiciones Respiraciones Crónicas de PR
13ma Convención Anual de la Academia de Médicos Generalistas y Primarios de PR
The St. Regis Bahia Beach Resort, Río Grande PR
Hyatt Regency Grand Reserve PR Río Grande, PR
The St. Regis Bahia Beach Resort, Río Grande PR
Por confirmar Hotel La Concha
Pendiente
Hotel Sheraton Convention Center, San Juan, PR
Hotel Sheraton Convention Center, San Juan, PR
Convención anual de la Asociación de Gastroenterología y Hepatología Pediátrica de PR Por confirmar
Convención Sociedad Puertorriqueña de Nefrología Royal Sonesta SJ Hotel, Carolina, PR
Convención anual del Colegio de Farmacéuticos de PR
Sheraton Convention Center, San Juan, PR
Simposio de alergistas e immunólogos del Oeste Rincón beach resort
Convención annual de la Asociación de Hospitales de PR
Sheraton Convention Center Hotel San Juan, PR
27 al 29 de octubre Caribe Gyn Ponce Hilton
26 al 28 de octubre de 2023
28 de octubre de 2023
3 al 5 de noviembre de 2023
17 al 19 de noviembre de 2023
10 al 12 noviembre de 2023
Convención anual Puerto Rico Urological Association (PRUA)
Congreso Anual Coalición de Asma y otras condiciones Respiraciones Crónicas de PR
Convención Sociedad de Cirugía Plástica de PR
Serra & Serra 787 406-4571 / 787 640-5776 reumatologospr@serrayserra.com
Serra & Serra patologospr@serrayserra.com 787 406-4571 & 787 640-5776
Serra & Serra 787 406-4571 / 787 640-5776 asocfisiatraspr@serrayserra.com
AMEC 787-289-8989 amec@amec-pr.com
Aixa Vélez 787-649-7681 / genteinc@gmail.com
IC Planners 787-504-3655 ivettecolon@icplannerspr.com
SR Consultants & Evets 939-292-4115 / 787-649-2367 srconsultants&events@gmail.com
IC Planners 787-504-3655 ivettecolon@icplannerspr.com
Business Planners - Merna Morales 787-645-9914 bplanner21@gmail.com
Colegio de Farmacéuticos de PR 787-753-7157
IC Planners ivettecolon@icplannerspr.com / 787-504-3655
Business Planners - Merna Morales 787-645-9914 bplanner21@gmail.com
Germaine Quiñones ahomprgq@gmail.com / 787-608-1477
Puerto Rico
Convención
Convención
Convención
Convención
Convención
Congreso
Convención
Convención
Convención
Convención
Convención
Convención
Sheraton Convention Center Hotel San Juan, PR
Sheraton Convention Center Hotel, San Juan PR
Hyatt Regency Grand Reserve PR Río Grande, PR
Convención Anual Sociedad Puertorriqueña de Neumología Por confirmar
Convención annual de la Asociación de Médicos Pediatras de la Región Este (AMPRE)
Noviembre ElectroCardio Workshops Conference Arrhytmia Group
Por confirmar
Por confirmar
Aixa Vélez genteinc@gmail.com / 787-649-7681
IC Planners ivettecolon@icplannerspr.com / 787-504-3655
Aixa Vélez genteinc@gmail.com / 787-649-7681
Business Planners - Merna Morales bplanner21@gmail.com / 787-645-9914
Business Planners - Merna Morales bplanner21@gmail.com / 787-645-9914
AMEC amec@amec-pr.com / 787-289-8989
Convención
Congreso
Convención
Convención
Convención
Conferencia
Revista Puertorriqueña de Medicina y Salud Pública 63
Fecha Actividad Lugar Contacto TIPO DE EVENTO
ACTIVIDADES ESTÁN SUJETAS A CAMBIO
PUEDES TOCARNOS EL CORAZÓN Y SALVARNOS LA VIDA.
PUEDES TOCARNOS EL CORAZÓN Y SALVARNOS LA VIDA.
Las mujeres tienen menos probabilidades de sobrevivir a un paro cardíaco fuera de un hospital, en parte porque las personas en la calle tienen temor de tocarlas. Los héroes valientes no tienen miedo; tocan corazones y salvan vidas.
Las mujeres tienen menos probabilidades de sobrevivir a un paro cardíaco fuera de un hospital, en parte porque las personas en la calle tienen temor de tocarlas. Los héroes valientes no tienen miedo; tocan corazones y salvan vidas.
Aprende RCP solo con las manos. Visita heart.org/rcpheroes

manos.
64 Revista Puertorriqueña de Medicina y Salud Pública
Aprende RCP solo con las
Visita heart.org/rcpheroes
Visita Nuestro Mundo Digital
Visita nuestra plataforma web, redes sociales y podcast ahora también disponibles en iTunes y Spotify, Suscríbete y no te pierdas el mejor contenido de Medicina y Salud Pública
Desde las instalaciones del Centro Cardiovascular de Puerto Rico y Del Caribe, la Revista Medicina y Salud Pública lideró el primer simposio cardiovascular del año, que contó con la masiva asistencia de profesionales del área de la salud y pacientes, quienes recibieron orientaciones sobre la prevención de las enfermedades cardiovasculares.


EXITOSA ALIANZA ENTRE PACIENTES Y EL CENTRO CARDIOVASCULAR DE PUERTO RICO Y EL CARIBE
Con la participación de reconocidos especialistas en cardiología se llevó a cabo el encuentro educativo dirigido a pacientes, médicos y profesionales de la salud.
“Hemos intervenido a pacientes desde una hora de nacidos, hasta noventa años de edad, y somos el único programa de cirugía cardiovascular pediátrica en Puerto Rico. El Centro Cardiovascular es un proyecto de todo el pueblo de Puerto Rico que ha logrado impactar a más de medio millón de pacientes”, destacó el Dr. Iván González Cancel, cirujano cardiovascular torácico y de trasplante de corazón.
El Hospital Cardiovascular es un centro médico histórico para la medicina especializada en Puerto Rico, ya que allí se llevó a cabo el primer trasplante de corazón en la Isla. Además, a través de los años sus especialistas han sido pioneros en liderar intervenciones cardiovasculares de alta complejidad para los pacientes boricuas.

Los pacientes también fueron protagonistas
El evento educativo contó con un elemento diferenciador; los pacientes que asistieron contaron en vivo sus testimonios y aprendizajes acerca de sus procesos en el Centro Cardiovascular de Puerto Rico y Del Caribe.

Revista Puertorriqueña de Medicina y Salud Pública 65
www.medicinaysaludpublica.com Web - Podcast - Video @revistaMSP @revistaMSP @revistaMSP
DR. CABANILLAS, FIGURA CLAVE DEL CENTRO AUXILIO DE CÁNCER

LA INSTITUCIÓN QUE HA MARCADO UN LEGADO EN LA ONCOLOGÍA DE PUERTO RICO
DESDE HACE 20 AÑOS EL CENTRO MÉDICO CAMBIÓ
EL
RUMBO DE LA SALUD EN PUERTO RICO.
Es imposible hablar del Centro de Cáncer del Hospital Auxilio Mutuo sin hacer referencia a su director y fundador, el Dr. Fernando Cabanillas, quien con férrea determinación ha luchado junto a un equipo sanitario abnegado para vencer el cáncer, tal como se ha evidenciado en estos veinte años de funcionamiento de dicho centro.
El Dr. Cabanillas se ha destacado como un prolífico científico, con conocimientos a la vanguardia en el área oncológica. Pero, ser médico e innovador no ha sido su única faceta, ya que, además de ser un excelente padre, es un hombre humanista, con una conducta impecable, siempre inclinado al servicio del pueblo puertorriqueño, el cual le reconoce como un recio patriota de la nación, cuyas ideas se pueden encontrar en diferentes medios comunicacionales que hacen eco de su enfadada voz la
cual, con palabra y acción, promueven el cambio hacia una mejor nación.
Los afectos y valores del Dr. Cabanillas hacia su tierra puertorriqueña se han manifestado a través del servicio y el cuidado de los pacientes que padecen cáncer, lo cual le ha llevado a entregar su vida abriendo caminos hasta entonces ignotos en la carrera investigativa. Respecto a los avances que ha presenciado e impulsado, hoy admite:
“Es tan brillante el futuro de la oncología. Se sabe que hay un avance acelerado de cosas que antes no se podían conseguir. El primer ejemplo de esto fue el medicamento conocido como Glivec, que se usó para tratar pacientes con leucemia crónica mielógena. Antes no había un tratamiento efectivo y hoy en día prácticamente nadie muere de esa enfermedad porque se descubrió cuál es el defecto molecular y con esa medicina se puede atacar ese defecto”.
66 Revista Puertorriqueña de Medicina y Salud Pública
MSP / MUNDO DIGITAL
Dr. Fernando Cabanillas, Director y Fundador del Centro de Cáncer del Hospital Auxilio Mutuo
LOS INICIOS DEL CENTRO DE CÁNCER DEL HOSPITAL AUXILIO MUTUO
Aprincipios de los años 2000, el Hospital Auxilio Mutuo vio la necesidad de tener un centro que generara un efecto importante en el tratamiento del cáncer en Puerto Rico.

En este sentido, la Sociedad Española de Auxilio Mutuo y Beneficencia estableció un plan para crear un centro que ofreciera manejo integrado y digno para el paciente con cáncer, esto de la mano del Dr. Fernando Cabanillas, oncólogo y actual director médico del Auxilio Centro de Cáncer.
“Llegando ya casi al 2000, como un par de años antes empezaron a desarrollar un interés aquí en el Hospital y en la administración del hospital, en promover el manejo del paciente con cáncer”, dijo el Dr. Cabanillas.
PIONEROS EN INVESTIGACIÓN PRIVADA EN PUERTO RICO
ENFOQUE HUMANO
Desde sus inicios, el Centro ha mantenido un enfoque humano con un equipo médico multidisciplinario y la más avanzada tecnología para ofrecer tratamientos más certeros.
“La idea fue reclutar personas comprometidas, no solamente que fueran peritos o especialistas en su campo, sino que fueran profesionales que brindaran servicio al paciente de forma especial y personalizada, haciendo que no sean un número más” expresó Gladys Urbina, gerente Centro de Cáncer 2002-2013.
El Centro también se caracteriza por respetar la dignidad y privacidad del paciente con cáncer en cada una de las etapas de su padecimiento.
“Lo más importante es que respetamos la dignidad del paciente, por ejemplo, tenemos una unidad de quimioterapia con habitaciones, en vez de poner a todos los pacientes uno al lado del otro”, explicó el Dr. Cabanillas.
Como director del Centro, el Dr. Cabanillas dirigió la investigación hacia el sector privado de Puerto Rico, demostrando que esta se puede realizar en un ambiente no universitario, justo como el del Centro.
El Centro está caracterizado por el trato humano del personal sanitario que labora en él. También, hacen presencia muchos voluntarios, quienes, con un espíritu humanitario, dedican tiempo y esfuerzo para el cuidado de los pacientes con cáncer.
Actualmente, el Centro Auxilio de Cáncer cuenta con más de 36.000 pacientes, y espera continuar dejando una huella en el cuidado, tratamiento e investigación del cáncer de Puerto Rico hacia el futuro.
PADRE DE DOS MUJERES EXITOSAS
Además de su intachable trayectoria como médico, el Dr. Fernando Cabanillas se destaca como padre de dos mujeres exitosas, quienes se inspiraron en su ejemplo para también hacerse un camino dentro del área médico-oncológica.
María Antonia, su hija mayor, sintió el llamado para desempeñarse en el campo de la salud, pero presidiendo una compañía de seguros médicos. En cuanto a su hija menor, la doctora María Eugenia Cabanillas, ella decidió formarse como endocrinóloga oncológica y labora en el MD Anderson Cancer Center en Texas, en donde además lidera un programa de investigación en cáncer de tiroides.
Respecto a esta decisión de su hija menor, el prominente oncólogo afirma: “Me sorprendió cuando me dijo que iba a estudiar medicina. No fue guiada por mí. De hecho, yo creo que ella trató de hacer exactamente lo que yo hice en el campo de linfoma, pero lo está haciendo muy bien, está haciendo algo bien innovador con el cáncer anaplásico”.
En conversación con la Revista MSP, el Dr. Cabanillas abordó el tema de los avances de la medicina oncológica en Puerto Rico, resaltando que él atiende mayormente casos de linfomas y está al tanto de las drogas disponibles capaces de curar el linfoma, pero hasta el momento no hay ninguna capaz de erradicar en un 100 % el tumor.
Por ejemplo, en casos hodgkin se cura un 85 % de los pacientes, mientras que
Revista Puertorriqueña de Medicina y Salud Pública 67
MSP / REVISTAMSP.COM
En el Centro se reúne un equipo multidisciplinario de profesionales de la salud, que analiza e investiga cada caso para lograr un diagnóstico completo y planificar un tratamiento acertado.
el porcentaje restante se puede rescatar en otros tipos de quimioterapias: “Cada vez estamos usando más la inmunoterapia cuando falla la quimioterapia. Lo negativo es que estimula las células del sistema inmune y ataca las células normales del cuerpo, pero no es raro que las ataquen”,
sostuvo.
“Cada vez estamos más cerca de la cura. Una de las metas es la posibilidad de curar los linfomas con tratamientos que no sean quimioterapias. MD Anderson sigue esa meta y hay otros tumores como el melanoma que se han
curado. Por ejemplo, el expresidente Jimmy Carter, cuyo tumor metastatizó al cerebro, y en el pasado en cuestión de tres meses ya hubiese muerto, ahora lleva más de tres años en remisión completa. O sea que estamos hablando que sí se pueden curar los pacientes y esos números están subiendo”.
Con el anuncio del entonces presidente Barack Obama sobre el indulto del exprisionero Oscar López, el científico Dr. Fernando Cabanillas logró un sueño de vida, en el que primeramente lo acompañó su familia desde la Plaza de Recreo de Ponce y que luego se convirtió en una lucha de todos los puertorriqueños y hermanos países a nivel mundial.
Fue en el año en el 2013, un Viernes Santo, cuando precisamente Óscar López, prisionero federal desde 1981, cumplía 32 años encarcelado. Como muestra de solidaridad, el Dr. Cabanillas y su esposa acordaron realizar la primera cárcel simulada en reclamo por su libertad.
Con cinco décadas al frente de los avances científicos, el Dr. Cabanillas hoy es testigo de cómo se pueden combinar fármacos que ayuden a los pacientes, sin que ellos tengan que sufrir efectos secundarios severos.
“Oscar López es un patriota puertorriqueño cuyo único crimen ha sido defender la independencia de Puerto Rico. Pienso que es una injusticia muy grande lo que le está haciendo. Un individuo que mata a alguien, el promedio que recibe de cárcel era de 10 años para esa época (principios del 1980)”, recalcó el Dr. Cabanillas durante el creciente apoyo que recibía de todos los sectores de la isla, incluyendo el gobierno de Puerto Rico.
Con cinco décadas al frente de los avances científicos, el Dr. Cabanillas hoy es testigo de cómo se pueden combinar fármacos que ayuden a los pacientes, sin que ellos tengan que sufrir efectos secundarios severos.
El Dr. Cabanillas continúa siendo una autoridad en el área oncológica. De la mano de su hija María Eugenia, continúa al tanto de los desarrollos en el campo, tal como él mismo sostiene: “Ahora se sienta conmigo a discutir algunos trabajos científicos, como en la parte de quimioterapia. Ella está sumergida en lo que es la terapia molecular, que es algo distinto, para mejorar el resultado del cáncer anaplásico de tiroides, que es horriblemente agresivo. Una de las veces que me conmovió fue verla como toda una profesional de la medicina brindando una conferencia ante sus colegas del Colegio de Médicos Cirujanos de Puerto Rico. Dije ‘wow’. Me quedé atónito”, dijo con voz entrecortada.

68 Revista Puertorriqueña de Medicina y Salud Pública
El Dr. Cabanillas ofreciendo una de sus charlas durante la segunda edición del evento educativo “Puerto Rico contra el Cáncer”.

Revista Puertorriqueña de Medicina y Salud Pública 69
EL DR. LUIS RENTA, SERÁ EL NUEVO
PRESIDENTE DE LA SOCIEDAD
PUERTORRIQUEÑA DE CARDIOLOGÍA
Durante la Asamblea General de la Sociedad Puertorriqueña de Cardiología se eligió al Dr. Luis Renta para presidir la corporación en el periodo 2023-2025. Anteriormente, el especialista era secretario de la Sociedad Puertorriqueña de Cardiología.
El Dr. Renta es graduado de la Universidad Central del Caribe y realizo su especialidad en Cardiología en el Hospital de Veteranos de San Juan. Además, es certificado por los Boards de Medicina Interna y Cardiología.

70 Revista Puertorriqueña de Medicina y Salud Pública
El especialista en cardiología estará al frente de la Sociedad en el periodo 2023-2025

A CASI SEIS MESES DEL PROCEDIMIENTO,
TODO UN ÉXITO LA PRIMERA ABLACIÓN EPICÁRDICA DE TAQUICARDIA VENTRICULAR EN PUERTO RICO
En el mes de Febrero, los especialistas cardiovasculares estamos de regocijo ya que se celebra el mes del Corazón. Queremos promover una buena salud cardiovascular, y sobre todo enfatizar en la prevención de enfermedades cardiovasculares. Además, queremos resaltar la excelente labor que realizamos día a día para ofrecerles a nuestros pacientes el mejor cuidado cardiovascular, mejorar la sobrevida de ellos y su calidad de vida.
Es por esta razón que quiero aprovechar este espacio para destacar y discutir los resultados de la primera ablación epicárdica de taquicardia ventricular, realizada el 14 de septiembre del 2022, en el Hospital Damas en Ponce, Puerto Rico. Este procedimiento complejo se realizó en un paciente con miocardiopatía isquémica luego de un infarto al miocardio y fallo cardiaco con presencia de un marcapasodesfibrilador, múltiples descargas del
desfibrilador a pesar dos ablaciones endocárdicas previas, y dosis máximas tolerables de medicamentos antiarrítmicos.
El racional de esta ablación epicárdica (desde la superficie externa del corazón), incluyendo también el abordaje endocárdico (desde la superficie interna del corazón), fue modificación del sustrato arritmogénico, con el fin de disminuir significativamente los eventos de arritmias ventriculares,
72 Revista Puertorriqueña de Medicina y Salud Pública
MSP / MUNDO DIGITAL
descargas del desfibrilador y mejorar la calidad de vida del paciente.
La ablación epicárdica se realizó en la sala de operaciones híbrida del Hospital Damas. El procedimiento se realizó bajo anestesia general y con monitoreo invasivo hemodinámico. El acceso epicárdico se obtuvo con mucha cautela y sin complicaciones. Una vez se estableció el acceso epicárdico, se atravesaron catéteres de mapeo y de ablación para identificar mediante un mapa tridimensional las áreas que provocaban las arritmias y así eliminarlas mediante la ablación por radiofrecuencia.
Durante el procedimiento, se identificaron áreas en el epicardio que provocaban dos de las arritmias clínicas del paciente. Estas áreas se mapearon cuidadosamente, utilizando el método de mapeo de sustrato y mapeo de activación en periodos donde el paciente se encontraba en arritmias sostenidas. La ablación de estas áreas resultaron en no-inducibilidad de estas arritmias, lo cual se consideró el punto final o “endpoint” del procedimiento. El paciente no tuvo complicaciones relacionadas a la intervención. A pesar que fue un procedimiento extenso (cerca de siete a ocho horas), el paciente toleró muy bien el mismo. Su estadía hospitalaria luego del procedimiento fue favorable, a pesar de presentar arritmias ventriculares, las cuales pudieran ocurrir secundario
Electrofisiólogo Hospital Damas de Ponce, Puerto Rico
al proceso de inflamación aguda de la ablación. El paciente se manejó con medicamentos anti-arrítmicos (Amiodarone y Mexiletene), y fue dado de alta en condición estable.
Posteriormente, el paciente se ha visto en la oficina en varias ocasiones, y a pesar de tener ciertas arritmias residuales, las mismas han disminuido significativamente en cantidad, comparado previo a la intervención. Estos episodios han sido tratados con terapia de estimulación antitaquicárdica del dispositivo, sin la necesidad de descargas eléctricas del desfibrilador. Actualmente, el paciente se encuentra en un solo medicamento anti-arrítmico (Sotalol), además de su terapia de fallo cardiaco. El paciente fue evaluado por última vez en la clínica el pasado viernes, 3 de febrero del 2023. El mismo se encontró estable, refirió sentirse mejor, con síntomas mínimos de fallo cardiaco y algunos eventos de arritmias tratadas con terapia de estimulación anti-taquicárdica pero ninguno requirió descargas eléctricas. Además, la función cardiaca del ventrículo izquierdo ha mejorado de un 20-25% previo a la ablación a un 35%, según reportado en un ecocardiograma reciente. La calidad de vida del paciente ha mejorado significativamente, lo cual nos llena de mucha alegría y orgullo.
Los resultados de la ablación en nuestro paciente son comparables a los resultados de estudios publicados
sobre el beneficio de la ablación de taquicardia ventricular en la prevención de arritmias ventriculares. Un meta-análisis1 publicado de los estudios SMASH VT, VTACH, VANISH y SMS, demostró el beneficio de la ablación de taquicardia ventricular en términos de disminución de eventos de arritmias (tormenta eléctrica), terapias del desfibrilador (terapia de estimulación anti-taquicárdica y descargas eléctricas), y disminución en hospitalizaciones relacionadas a eventos cardiovasculares.
Nuevamente, en celebración del mes del Corazón, queremos resaltar la labor encomiable de cada uno de los que estuvimos envueltos en este procedimiento complejo. El Hospital Damas cuenta con el personal y equipo especializado para realizar todo tipo de ablaciones complejas, incluyendo ablaciones epicárdicas de taquicardias ventriculares, de manera segura y eficaz.

Estamos sumamente felices y orgullosos de haber realizado esta intervención por primera vez en Puerto Rico, y de los resultados obtenidos en nuestro paciente. ¡Enhorabuena!
REFERENCIAS
Guolin et al. J Interv Card Electrophysiol (2021) 61:435–443.
Revista Puertorriqueña de Medicina y Salud Pública 73
Helder Hernández Rivera, MD, FACC, FHRS
MSP / REVISTAMSP.COM
La fortaleza se enciende de color verde azulado para concienciar sobre Cáncer Cervical
En conmemoración del mes nacional de la prevención del cáncer cervical, La Fortaleza en San Juan se enciende de color TEAL en solidaridad con las pacientes que enfrentan esta enfermedad.

La Fortaleza reconoce los esfuerzos de las fundaciones VOCES de PR & Las Voces de Rhaiza, por su labor fomentando la educación para combatir el cáncer cervical, así como sobre las medidas preventivas que se pueden tomar para evitarlo y detectarlo a tiempo.


El gobernador de Puerto Rico, Pedro Pierluisi, firma Proclama que declara el mes de enero y el día 27 de enero como el Mes y el Día de la Prevención del Cáncer Cervical.

74 Revista Puertorriqueña de Medicina y Salud Pública
MSP / MUNDO DIGITAL
5k Corre o camina por ellas de las Voces de Rhaiza

Por primera vez de manera presencial con el fin de concienciar a la población sobre el cáncer cervical se celebró el 5K Corre o Camina por Ellas de Las Voces de Rhaiza, doliendo de Ballajá y llegando a la Escuela de Artes Plásticas.





¡Misión cumplida! Lilliam Rodríguez se abraza a Myra Plumey en un hermoso acto de recordación a la memoria de Rhaiza Vélez Plumey.

Revista Puertorriqueña de Medicina y Salud Pública 75
Lilliam Rodríguez recibe el donativo de la cadena de supermercados SuperMax
Dra. Carmen Zorrilla dice presente corriendo el 5 k en solidaridad con todas las pacientes afectas por esta condición.
MSP / REVISTAMSP.COM
“Nuestra misión es ayudar a los pacientes”
FEMPR agradece masiva participación en el PR MS Walk 2023
La carrera 3k-5k de la Fundación Esclerosis Múltiple de Puerto Rico se llevó a cabo en el parque Luis Muñoz Rivera.
Por su parte, el Dr. Ángel Chinea, neurólogo y director médico de la Fundación de Esclerosis
Múltiple de Puerto Rico, agradeció el apoyo de especialistas, médicos y personas que acudieron a la carrera 3k-5k.



“Hacemos las cosas sin interés. Nuestra misión es ayudar a estos pacientes con esta condición crónica que aunque sabemos que no tiene cura, podemos trabajar y podemos
controlar la enfermedad”, afirmó.
El especialista también indicó que “cada año se diagnostican 150 casos nuevos de esclerosis múltiple, es decir, se diagnostican 3 a 4 casos nuevos en toda la Isla. Estamos teniendo un número de casos que es el más alto en el Caribe, Centro América y Latinoamérica”.
76 Revista Puertorriqueña de Medicina y Salud Pública
La Fundación de Esclerosis Múltiple de Puerto Rico (FEMPR) celebró el ‘PR MS Walk 2023’, un evento deportivo realizado para los pacientes con esclerosis múltiple, con el objetivo de sensibilizar sobre el impacto de esta enfermedad en los puertorriqueños.
Médicos, pacientes, fundaciones y organizaciones médicas se unen por los pacientes con esclerosis
Este evento deportivo de la Fundación de Esclerosis Múltiple de Puerto Rico tuvo como objetivo concienciar acerca de la enfermedad y contribuir a llevar una vida con la mayor calidad posible.





Más de 2.500 personas, entre pacientes, deportistas, aficionados y familiares de las personas con esclerosis múltiple, se dieron cita en el parque Luis Muñoz Rivera para realizar la caminata 3k y la carrera 5k.



Revista Puertorriqueña de Medicina y Salud Pública 77
El Dr. Ángel Chinea, neurólogo y creador de la Fundación de Esclerosis Múltiple de Puerto Rico, agradeció el apoyo de especialistas, médicos y personas que acudieron a la carrera 3k-5k.
Sociedad Puertorriqueña de Pediatría celebra su convención anual número 70 bajo el lema ‘Excelencia en Pediatría’
El Centro de Convenciones Sheraton en San Juan fue el escenario de esta importante actividad enfocada en brindar experiencias de aprendizaje para los médicos y profesionales de la salud acerca del manejo de las condiciones que más afectan a la población pediátrica.
Entre las temáticas se destacó el papel de la terapia nutricional para pacientes con alergia alimentaria;
las enfermedades raras en Puerto Rico, como el síndrome de distrofia muscular de Duchenne; el manejo del asma en los niños; la bronquiolitis; los tratamientos para trastornos de tics como el Síndrome de Tourette; y otros abordajes de las enfermedades psiquiátricas y discapacidades en los infantes.
El Dr. Gerardo Tosca, presidente de la Sociedad Puertorriqueña de
Pediatría, recibió máximos elogios por la organización de la convención de los especialistas en salud infantil.
“Hoy, una vez más, los pediatras y la población pediátrica de Puerto Rico cuentan con una Sociedad Puertorriqueña de Pediatría lista y preparada para decir presente en los tiempos en que vivimos”, destacó el v

78 Revista Puertorriqueña de Medicina y Salud Pública
MSP / MUNDO DIGITAL
Asociación Puertorriqueña de Gastroenterología celebra congreso enfocado en la endoscopia terapéutica
La Asociación Puertorriqueña de Gastroenterología llevará a cabo un congreso caribeño sobre la endoscopia terapéutica y otros avances en esta especialidad médica el 24 y 25 de febrero desde el Centro de Convenciones del Hotel Sheraton en San Juan.
Durante los dos días de capacitación, se reunirán especialistas en gastroenterología y hepatología de América Latina y el Caribe, quienes se han convertido en referentes en torno al manejo integral de las condiciones digestivas que más afectan a la población.
Allí se compartirán casos en vivo,
conferencias y laboratorios prácticos a los que podrán acceder los participantes y actualizarse en técnicas endoscópicas con tecnología moderna, usadas para el tratamiento de los trastornos gastrointestinales de los pacientes.
Los especialistas puertorriqueños que harán parte de este congreso son el Dr. Carlos Micames, pasado presidente de la Asociación Puertorriqueña de Gastroenterología; el Dr. Kermit Richiez, gastroenterólogo y catedrático en el Grupo de Interés en endoscopia terapéutica; la Dra. Priscilla Magno, profesora asistente de Medicina en la Universidad de Puerto Rico y otros gastroenterólogos de la Isla.

Entre las temáticas que abordarán se destaca la endoscopia bariátrica, las técnicas de miotomía endoscópica peoral, de Helicobacter Pylori a metaplasia intestinal, imagenología, endo-hepatología, trastornos pancreáticos, uso de inteligencia artificial para el manejo de estas enfermedades y los trastornos del colon.
La participación de especialistas de Colombia, Argentina, Estados Unidos, Perú, Uruguay, Ecuador y España en el cuarto congreso caribeño de Asociación Puertorriqueña de Gastroenterología, permitirá ampliar el conocimiento de los profesionales de la salud.
Revista Puertorriqueña de Medicina y Salud Pública 79
MSP / REVISTAMSP.COM
Dr. Federico Rodríguez, Presidente de la Asociación Puertorriqueña de Gastroenterología.
Puertorriqueña es aceptada en prestigioso programa de tecnología genética en San Francisco

Rachell Martínez Ramírez, estudiante del Recinto de Ciencias Médicas (RCM) de la Universidad de Puerto Rico, fue seleccionada para participar en el prestigioso y competitivo programa de internado postdoctoral de la Biofarmacéutica Genentech, ubicada en San Francisco.

Genentech es considerada como pionera en el éxito y expansión de la biotecnología a nivel global. La biofarmacéutica se dedica al desarrollo de ciencia innovadora y tratamientos para pacientes con enfermedades terminales, siendo reconocida por crear el primer anticuerpo contra el cáncer y el primer medicamento para tratar la esclerosis múltiple primaria progresiva.
Entregan proclama que designa a marzo como el mes de alerta de las enfermedades de la tiroides
El Dr. Félix Rodríguez Schmidt, subsecretario del Departamento de Salud de Puerto Rico, hizo entrega de la proclama del mes de alerta sobre estas condiciones a los líderes de la Sociedad Puertorriqueña de Endocrinología y Diabetología, pacientes, representantes gubernamentales y directores médicos de farmacéuticas.
"Con el propósito de orientar sobre el impacto adverso a la salud que pueden tener las enfermedades de la tiroides, el Gobierno de Puerto Rico, designa el mes de marzo de 2023 como mes de alerta sobre las enfermedades de la tiroides en Puerto Rico", informó Rodríguez Schmidt.
De acuerdo con las estadísticas presentadas por los especialistas, se estima que las enfermedades de la tiroides tienen una prevalencia de más del 20% en la población puertorriqueña y aún más preocupante es el hecho de que el 50% de las personas desconocen que padecen algún trastorno relacionado con la tiroides.
80 Revista Puertorriqueña de Medicina y Salud Pública
MSP / REVISTAMSP.COM

LEGADO DE UN HOMBRE DEDICADO AL SERVICIO Y A LA INVESTIGACIÓN MÉDICA
El doctor Norman Maldonado Simón, internista y hematólogo, exrector del RCM, expresidente de la UPR, natural de Adjuntas, cursó estudios en las escuelas públicas de Puerto Rico y en 1959 se graduó de la escuela de medicina tropical, ubicada en San Juan; sus prácticas de tercero y cuarto año fueron en el Hospital de San Juan.
Inició un programa de cirugía cardiovascular con el doctor Frank Fucci, y este fue muy exitoso. Otras áreas importantes para el doctor Maldonado fueron las de obstetricia y ginecología, desde las cuales tuvo la oportunidad, no solo de atender partos, sino de instruir a otros estudiantes provenientes de República Dominicana y de España, quienes venían a la Isla con muy poco conocimiento y experiencia práctica.
Un alumno destacado e intérprete
En Nueva York, el doctor Maldonado cursó un semestre de medicina interna, lo cual fue sencillo para él, pues su madre era de Rumanía y había crecido en Estados Unidos, por lo que el inglés nunca fue una barrera para el galeno. Más bien, él ayudaba, como intérprete, a sus compañeros para que pudiesen comprender qué expresaban los pacientes.
El doctor Maldonado fue parte del primer grupo que trabajó en el hospital universitario, allí comenzó su residencia, un lugar en donde atendió pacientes con fallo respiratorio y parálisis. Recuerda que allí había una máquina especial que respiraba por los pacientes, la cual era novedosa para él y sus compañeros.
Dr. Norman Maldonado

El doctor Maldonado es considerado gran especialista puertorriqueño, quien en diferentes facetas pudo hacer gala de sus conocimientos y por ello forma parte de nuestro primer A Ciencia Cierta 2023.
El doctor Maldonado y sus colegas inspiraron a otros jóvenes para que estudiaran hematología. Esta entrega y dedicación fueron trascendentales, porque contribuyó a que las enfermedades tropicales y las mauertes por esta causa disminuyeran.De igual modo, él y sus compañeros fueron los primeros en tratar a los pacientes con la medicina rusa Alkeran, la cual hizo que los afectados experimentaran mejorías increíbles. Los pacientes se aglomeraban y llegaban hasta 120 personas en busca de su ayuda.
Descubrieron un nuevo tratamiento para tratar el linfoma, se trataba de un anticuerpo que destruye las células malignas. Este fue uno de los hallazgos más importantes en la carrera del doctor Maldonado, quien regresó a su práctica y luego se jubió.
Al día de hoy, califica su trabajo como maravilloso. Entre tantas áreas en las que se destacó, también resalta su obra titulada: “On health in Puerto Rico”, una historia sobre unos 800 médicos que han tenido una labor destacable en el manejo del cáncer a lo largo de la historia puertorriqueña.
82 Revista Puertorriqueña de Medicina y Salud Pública MSP / A CIENCIA CIERTA
Internista y hematólogo, ex-rector del RCM, expresidente de la UPR.

Diagnosing
(HCM)
HCM shares symptoms with other common cardiovascular and pulmonary diseases.1-5
LOOK DEEPER
Reveal
the hidden impact of HCM.
ExposeHCM.com

References: 1. Argulian E, Sherrid MV, Messerli FH. Misconceptions and facts about hypertrophic cardiomyopathy. Am J Med. 2016;129(2):148-152. 2. Wexler R, Elton T, Pleister A, Feldman D. Cardiomyopathy: an overview. Am Fam Physician. 2009;79(9):778-784. 3. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121(7):749-770. 4. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020;142(25):e558-e631. 5. Feldman DN, Douglas JS Jr, Naidu SS. Indications for and individualization of septal reduction therapy. In: Naidu SS, ed. Hypertrophic Cardiomyopathy. Springer-Verlag London; 2015. 6. University of Maryland Medical Center. Hypertrophic cardiomyopathy types, symptoms and causes. Accessed June 14, 2021. https://www.umms.org/ummc/health-services/heart-vascular/services/hypertrophic-cardiomyopathy/types-symptoms-causes
© 2021 MyoKardia, Inc., a Bristol-Myers Squibb Company. All rights reserved. CV-US-2100769 09/21
hypertrophic cardiomyopathy
can be challenging.1
When patients experience fatigue, chest pain, dyspnea (especially exertional), palpitations, and/or syncope, consider the possibility of HCM.6
