


del
Prima edizione novembre 2020
© 2020 Il Pensiero Scientifico Editore
Il Pensiero Scientifico Editore
Via San Giovanni Valdarno 8, 00138 Roma
Tel. (+39) 06 862821 - Fax (+39) 06 86282250 pensiero@pensiero.it www.pensiero.it - www.vapensiero.info www.facebook.com/PensieroScientifico twitter.com/ilpensiero www.pinterest.com/ilpensiero
Tutti i diritti sono riservati per tutti i Paesi
Stampato in Italia da Ti Printing S.r.l Via delle Case Rosse 23, 00131 Roma
Impaginazione e copertina Doppiosegno S.n.c. Immagine in copertina © iStock by Getty Images
Coordinamento editoriale Alessio Malta
ISBN 978-88-490-0701-5
Il progetto è stato realizzato con il contributo di

CURATORI
Nicola Normanno
SC Biologia Cellulare e Bioterapie
Istituto Nazionale Tumori
IRCCS Fondazione G. Pascale Napoli
Paolo Antonio Ascierto
SC Oncologia Medica e Terapie
Innovative, Dipartimento Melanoma, Istituto Nazionale dei Tumori IRCCS, Fondazione
G. Pascale, Napoli
Francesca Castiglione
Istologia Patologica e Diagnostica Molecolare, Azienda Ospedaliero Universitaria Careggi, Firenze
Chiara Cremolini
Dipartimento di Ricerca Traslazionale e delle Nuove Tecnologie in Medicina e Chirurgia, Università di Pisa
Alessia Di Lorito
Centro di Diagnostica Molecolare Avanzata e Terapie Innovative, Università di Chieti-Pescara
Lucia Festino
SC Oncologia Medica e Terapie
Innovative, Dipartimento Melanoma, Istituto Nazionale dei Tumori IRCCS, Fondazione G. Pascale, Napoli
Antonio Marchetti
Centro di Diagnostica Molecolare Avanzata e Terapie Innovative, Università di Chieti-Pescara
Carmine Pinto
UO di Oncologia Medica
Clinical Cancer Centre
AUSL-IRCCS di Reggio Emilia
Daniela Massi
Sezione di Anatomia Patologica, Dipartimento di Scienze della Salute, Università degli Studi di Firenze
Istologia Patologica e Diagnostica Molecolare, Azienda Ospedaliero Universitaria Careggi, Firenze
Silvia Novello
Dipartimento di Oncologia, Università degli Studi di Torino, Ospedale San Luigi Gonzaga, Orbassano (TO)
Filippo Nozzoli
Sezione di Anatomia Patologica, Dipartimento di Scienze della Salute, Università degli Studi di Firenze
Chiara Pisano
Dipartimento di Oncologia, Università degli Studi di Torino, Ospedale San Luigi Gonzaga, Orbassano (TO)
Fabrizio Tabbò
Dipartimento di Oncologia, Università degli Studi di Torino, Ospedale San Luigi Gonzaga, Orbassano (TO)
Inibitori di BRAF 36
Vemurafenib 36
Dabrafenib 38
Encorafenib 38
Resistenza primaria 39
Resistenza acquisita 40
Associazione di inibitori di BRAF e inibitori di MEK 42
Vemurafenib e cobimetinib 42
Dabrafenib e trametinib 43
Encorafenib e binimetinib 45
Altre combinazioni 47
Combinazione di target therapy e immunoterapia 49
Acquisizioni e prospettive 53
4. BRAF come bersaglio terapeutico nel carcinoma del polmone non a piccole cellule 59
Fabrizio Tabbò, Chiara Pisano, Silvia Novello
Introduzione 59
BRAF: il gene e la sua via di segnalazione 60
Mutazioni di BRAF: frequenza, classificazione e prognosi 60
Caratteristiche clinico-patologiche dei tumori del polmone BRAF mutati 62
Identificazione delle mutazioni di BRAF 63
Farmaci per il trattamento del tumore polmonare BRAF mutato 64
Vemurafenib 65
Dabrafenib e trametinib 67
Molecole per i pazienti con mutazioni non-V600E 68
Immunoterapia 68
Meccanismi di resistenza ai trattamenti a bersaglio molecolare 69
Acquisizioni e prospettive 70
5.
Le mutazioni di BRAF nel carcinoma del colon-retto 73
Chiara Cremolini
Introduzione 73
Caratteristiche e prognosi del carcinoma colorettale metastatico BRAF mutato 73
Trattamento di prima linea del carcinoma colorettale metastatico BRAF mutato 75
L’inibizione di BRAF come strategia terapeutica innovativa 76
Acquisizioni e prospettive 78
6.
Biopsia liquida e prospettive future 83
Nicola Normanno, Carmine Pinto
Introduzione 83
Biopsia liquida 83
Acquisizioni e prospettive 88
Comprendere il significato biologico e clinico di alterazioni geniche non per singola sede tumorale, ma con una visione ampia in più patologie neoplastiche rappresenta oggi un’importante sfida della Oncologia. Qual è il valore prognostico e/o predittivo di un’alterazione genetica e come interviene nelle complesse inter-relazioni delle molteplici alterazioni genetico-molecolari che caratterizzano i tumori solidi degli adulti? È possibile che un’alterazione genica actionable identificata in un singolo specifico tumore possa rappresentare un bersaglio terapeutico anche in altre neoplasie, ovvero avere un significato “agnostico”, indipendente dal tipo istologico? Per rispondere a queste domande risulta indispensabile incrementare le conoscenze biologiche, definire e standardizzare i test e le piattaforme di analisi molecolare, avere solide metodologie nella ricerca traslazionale e negli studi clinici. Nell’evoluzione dell’Oncologia sempre di più identificheremo nelle diverse patologie neoplastiche sottogruppi di pazienti sulla base di specifiche alterazioni geniche e il marcatore genetico-molecolare potrebbe diventare l’elemento nosologico che li definisce al di là della origine istologica del tumore. In quest’ambito i moderni studi clinici stanno sempre di più valutando l’attività di nuovi farmaci a bersaglio molecolare in pazienti che presentano il potenziale target molecolare indipendentemente dalla sede di origine del tumore.
Per realizzare questo nuovo approccio diagnostico e terapeutico dei tumori è indispensabile una continua informazione e condivisione tra ricercatori e clinici che rappresenta l’obiettivo di questo secondo volume “Mutazioni del gene BRAF nei tumori solidi” della collana “Test e terapie target in Oncologia”, in cui verranno presentati lo stato dell’arte e i potenziali sviluppi che derivano dall’identificazioni delle mutazioni del gene BRAF.
Punto di partenza è il tessuto tumorale e verranno passate in rassegna tutte le più recenti acquisizioni nella gestione del campione biologico e sulle tecnologie disponibili per
i test, considerando le differenti sensibilità e specificità. I campioni rappresentano un patrimonio di informazioni, ed è quindi indispensabile una standardizzazione di tutti i percorsi e le attività, dalla raccolta dei materiali biologici alla loro processazione, fino all’estrazione e conservazione dei campioni di RNA o DNA, procedure tutte che possono sensibilmente condizionare i risultati dei successivi test. I referti di conseguenza dovranno contenere tutte le informazioni utili che ne permettano l‘interpretazione, dalle caratteristiche quali-/quantitative del materiale esaminato, alla metodica (con indicazione della sensibilità) utilizzata, fino alle precise alterazioni molecolari rilevate e correlabili con un utilizzo clinico.
Il “modello” per la ricerca e l’utilizzo clinico del target rappresentato dalle mutazioni di BRAF è stato il melanoma. Analizzare e testare le mutazioni in questa neoplasia ha permesso di conoscerne il significato biologico e di sviluppare efficaci terapie. L’evoluzione dei trattamenti che riguardano i singoli farmaci e le combinazioni di farmaci anti-BRAF e anti-MEK che si sono sviluppate vengono passate in rassegna confrontandone efficacia e tossicità. Uno sguardo verrà poi dato al futuro con la presentazione delle possibili combinazioni di terapie a target molecolare e immunoterapia.
Partendo dal melanoma, le mutazioni di BRAF sono state successivamente identificate come possibile bersaglio di terapie a target molecolare anche nell’adenocarcinoma del polmone. In questa patologia neoplastica vengono definite le caratteristiche biologiche e cliniche e l’impatto nella terapia dell’introduzione della combinazione di farmaci a bersaglio molecolare. Terapie efficaci che vanno inserite nella strategia e nella sequenza terapeutica dei pazienti con adenocarcinoma del polmone BRAF mutati. La determinazione delle mutazioni di BRAF è poi un elemento imprescindibile per la scelta del trattamento nei pazienti con carcinoma del colon-retto avanzato. La conoscenza dello stato mutazionale di BRAF rappresenta infatti un fattore prognostico in questa patologia oncologica e ne indirizza anche le scelte terapeutiche. Le acquisizioni raggiunte in merito al significato delle diverse mutazioni di BRAF e alle possibilità di trattamento disponibili, anche con l’introduzione di farmaci a bersaglio molecolare, rappresentano un progresso importante nella terapia di questo tumore.
Le prospettive che riguardano lo studio delle mutazioni di BRAF vedono l’introduzione della biopsia liquida e la determinazione di queste mutazioni in altri tumori. La biopsia li-
quida permette di ottenere un profilo genetico-molecolare complessivo della neoplasia nonché di monitorare la sua evoluzione nel tempo. Impiegando questa tecnica, potrebbe essere possibile selezionare e targettizzare più precisamente pazienti sensibili alla terapia mirata e quindi valutarne l’evoluzione con l’individuazione di cloni tumorali resistenti. La determinazione delle mutazioni di BRAF con finalità terapeutica in altri tumori può rappresentare infine un importante e ulteriore sviluppo, e gli studi già in corso ne definiranno il reale impatto.
Tutti questi argomenti e criticità verranno sviluppati nel secondo volume di questa collana, che ha l’obiettivo di implementare le conoscenze e un percorso comune sia nella ricerca traslazionale e clinica che nella best practice di tutti i giorni.
Nicola Normanno, Carmine Pinto
ANTONIO MARCHETTI, ALESSIA DI LORITO
Nel corso dell’ultimo decennio, l’introduzione nella pratica clinica di numerosi farmaci a bersaglio molecolare ha rivoluzionato la terapia oncologica. L’identificazione di specifiche alterazioni molecolari alla base dei processi neoplastici si è accompagnata allo sviluppo di metodiche sempre più performanti per la caratterizzazione dei tumori volte all’individuazione di target per la terapia farmacologica mirata.1
Questi sviluppi hanno progressivamente portato a una vera e propria rivoluzione anche nella diagnostica anatomopatologica introducendo la necessità di correlare i referti morfologici con l’assetto immunofenotipico e molecolare per permettere ai pazienti di ricevere un trattamento in linea con i nuovi standard terapeutici.2
Fra i driver molecolari che possono permettere una terapia mirata, le mutazioni del gene BRAF hanno assunto una primaria importanza soprattutto in pazienti affetti da melanoma, da cancro del colon-retto e da tumori polmonari non a piccole cellule, in stadio avanzato. L’identificazione di mutazioni che possano essere target di farmaci a bersaglio molecolare è diventata cruciale, non solo per il trattamento di questi pazienti, ma anche per definirne la prognosi.3
Sulla base delle linee guida dell’ESMO Clinical Practice, il test di BRAF deve essere effettuato in pazienti con melanoma resecabile o non resecabile, in stadio III o IV, mentre è strettamente raccomandato in pazienti al II stadio, ad alto rischio. In caso di metastasi, è preferibile effettuare il test molecolare sulla lesione metastastica, in quanto rappresenta la lesione più recente. Se non è disponibile o sufficiente il tessuto della metastasi, può essere utilizzata la metastasi linfonodale o il tumore primitivo. Numerosi studi, infatti, hanno dimostrato l’alta concordanza dello stato del gene BRAF rilevato nella lesione primitiva e nelle lesioni metastastiche. Inoltre, siti di metastasi superficiali possono anche essere sottoposti ad agoaspirazione per cui si può disporre di campioni citologici.4 5
Nel caso di pazienti affetti da carcinomi del colon retto, il campione a disposizione proviene spesso da biopsie endoscopiche o da resecati chirurgici (lesioni primitive o sedi di metastasi). In rari casi si dispone di campioni citologici che possono essere impiegati quando non si ha a disposizione altro materiale e quando un sito di metastasi può essere facilmente raggiunto senza arrecare notevole disagio al paziente.6 7
Nel tumore del polmone, le mutazioni del gene BRAF sono molto rare (circa 1,5-3,5%), tuttavia, di recente, National Comprehensive Cancer Network (NCCN), College of American Pathologists (CAP), International Association for the Study of Lung Cancer (IASLC), Association for Molecular Pathology (AMP) e American Society of Clinical Oncology (ASCO) hanno raccomandato lo studio dello stato mutazionale del gene BRAF per pazienti in stadio avanzato. Il tessuto tumorale, quando disponibile, resta di primaria importanza, quale substrato per la caratterizzazione immunofenotipica e molecolare.8-10
Il campione tessutale, per il paziente affetto da NSCLC, si ottiene al momento della diagnosi (biopsia) o dell’intervento chirurgico (campione resecato). I campioni istologici, in anatomia patologica, vengono fissati in formalina e inclusi in blocco di paraffina. Il materiale citologico, ottenuto spesso mediante agoaspirazione, lavaggi, brushing o prelievi di essudati (ad esempio, pleurici/peritoneali, ecc.) può essere una valida alternativa al materiale istologico. Inoltre, la possibilità di ottenere citoinclusioni a partire da campioni citologici permette di avere un materiale efficace su cui determinare alterazioni molecolari.8-10
Il patologo, prima di procedere a qualsiasi analisi molecolare, è tenuto ad esaminare il campione di partenza. Gli esami molecolari sono condizionati dalla percentuale di cellule neoplastiche, pertanto solo una accurata analisi del campione al microscopio permette di analizzare la qualità e la quantità delle cellule neoplastiche infiltranti presenti nel preparato.
Soprattutto nel tumore del polmone, il campione tessutale presenta una vasta eterogeneità, ovvero possono essere presenti zone ricche di infiltrato infiammatorio, vaste aree di necrosi, tessuto normale accanto ad aree neoplastiche infiltranti. L’osservazione al microscopio di questi aspetti consente una accurata selezione delle aree tumorali e la scelta della metodica molecolare più idonea all’analisi richiesta.8
Inoltre, nella pratica clinica routinaria il patologo molecolare deve gestire spesso piccole biopsie, campioni con una bassa percentuale di cellule neoplastiche o prelievi con molta melanina o melanomi cutanei superficiali, difficili da dissezionare.
Metodiche e test disponibili per l’analisi del tessuto tumorale 3
L’analisi molecolare di campioni con basso contenuto di cellule neoplastiche potrebbe portare a falsi positivi per la presenza di artefatti e a falsi negativi legati ai limiti di sensibilità delle tecnologie impiegate.
Altro aspetto di fondamentale importanza riguarda le modalità con cui il campione viene gestito dal momento del prelievo fino alla fissazione in formalina. Infatti il tessuto risente fortemente della fase pre-analitica che, se non eseguita correttamente, comporta il rischio di non adeguatezza del campione o di risultati inaffidabili.8-10
Inoltre, è stato riportato che per circa il 30% dei pazienti affetti da NSCLC in stadio avanzato non si dispone di materiale sufficiente o qualitativamente/ quantitativamente idoneo per le analisi molecolari. In questi casi, la possibile alternativa è effettuare esami molecolari su plasma o su altri liquidi biologici, ovvero utilizzare la biopsia liquida. Come per il test EGFR, l’utilizzo del ctDNA è stato riportato in letteratura come utile anche per la valutazione dello stato mutazionale del gene BRAF.11
Per quanto concerne le metodiche a disposizione del patologo molecolare, la ricerca di alterazioni molecolari comporta la valutazione diretta di mutazioni geniche o indiretta attraverso i loro prodotti proteici, in termini di alterazioni qualitative e quantitative dell’espressione. Per queste determinazioni, le tecniche utilizzabili su tessuto si distinguono classicamente in metodiche in situ e metodiche non-in situ.
Le tecniche in situ vengono direttamente effettuate su sezioni di tessuto e pertanto rientrano tra le competenze del patologo, trattandosi di una valutazione di preparati al microscopio. In questo ambito, i principali settori tecnologici sono rappresentati dall’immunoistochimica (IHC), dall’immunofluorescenza e dall’ibridazione in situ (la cui trattazione specifica esula dallo scopo di tale capitolo).
Le metodiche in situ hanno come principale vantaggio la possibilità di confrontare la caratterizzazione molecolare con la morfologia, ovvero di poter osservare direttamente al microscopio in quali cellule si verificano le alterazioni molecolari. Inoltre, queste metodiche possono essere applicate con successo anche su piccoli campioni bioptici contenenti rare cellule neoplastiche non coese (il cut off prevede un minimo di 50 cellule tumorali valutabili), difficilmente analizzabili con approcci non-in situ. Tuttavia, la bassa sensibilità e il numero ristretto di marcatori analizzabili simultaneamente rendono queste metodiche talora non sufficienti da sole per caratterizzare l’assetto molecolare ai fini del trattamento.8 12
Tra le metodiche in situ, l’IHC è la più diffusa nella pratica clinica e si fonda sulla reazione antigene-anticorpo, permettendo di valutare l’espressione di specifiche proteine nei tessuti e la natura di strutture cellulari nei casi in cui la pura morfologia risulti insufficiente a caratterizzare la lesione. L’IHC consente di evidenziare la presenza/assenza della proteina che si sta valutando nel campione in esame ed eventualmente la localizzazione (nucleo, citoplasma, membrana cellulare, tessuti extracellulari). Inoltre, l’IHC permette non solo di stabilire l’immunofenotipo di un tumore, ma anche l’espressione di biomarcatori prognostici e predittivi di risposta a terapie a bersaglio molecolare (immunoistochimica predittiva).12
In ambito oncologico, l’immunoistochimica permette al patologo primariamente di identificare la natura di tumori maligni poco differenziabili con la sola morfologia (ad esempio, linfomi, carcinomi, melanomi), la caratterizzazione di neoplasie del sistema emo-linfopoietico (ad esempio, linfomi, leucemie), l’identificazione dell’origine di una metastasi e l’identificazione di agenti infettivi (ad esempio virus, batteri, protozoi). Per quanto concerne le applicazioni in ambito predittivo del metodo, l’esame risulta più difficoltoso e richiede una maggiore competenza sia nell’allestimento che nella lettura del preparato da parte del patologo molecolare. L’IHC predittiva può essere realizzata mediante In vitro Companion Diagnostics Devices/companion diagnostic test (CDx) o test sviluppati in laboratorio (LDT). Nel primo caso, si tratta di test specifici, sviluppati per la diagnostica in vitro (IVD) in parallelo con i farmaci target, testati in trial clinici e approvati in genere negli Stati Uniti D’America dalla Food and Drug Amministration (FDA) insieme al farmaco. Il test, così sviluppato, è in grado di predire l’outcome in termini di sicurezza, efficacia e tollerabilità del farmaco. I test IVD sono in genere comprensivi di sistemi di amplificazione del segnale. Nel caso dei test LDT, l’utilizzo di anticorpi ben studiati può rappresentare un’alternativa possibile, in grado di fornire risultati adeguati. Si richiede, tuttavia, che il laboratorio attui un rigoroso percorso di sviluppo e validazione del test, comprensivo anche di un controllo della sua efficienza nel tempo. Inoltre, in considerazione della bassa concentrazione di alcuni biomarcatori nelle cellule neoplastiche, nell’utilizzo di un test LDT si consiglia l’impiego di sistemi di rivelazione in grado di amplificare il segnale.13
Tra i principali biomarcatori predittivi e analizzabili mediante IHC, nella pratica clinica il BRAF è stato testato ampiamente per il melanoma e il carcinoma della tiroide. L’applicazione di questo biomarcatore si è estesa ad altre patologie oncologiche quali il tumore del colon-retto e più recentemente del polmone. 14
Al momento, l’IHC con l’anticorpo monoclonale VE1 è l’unico test anticorpale disponibile e si basa sulla determinazione delle mutazioni a carico dell’esone 15
Metodiche e test disponibili per l’analisi del tessuto tumorale 5
p.V600E. Alcuni studi hanno mostrato elevata specificità con una positività citoplasmatica. I vantaggi di questa metodica sono il basso costo, il tempo d’analisi (il test richiede circa 48 ore) e la diffusione capillare della tecnologia nelle anatomie patologiche. L’esame, tuttavia, può fornire falsi negativi dovuti all’eterogeneità di espressione e alla bassa concentrazione della mutazione p.V600E. Inoltre non è possibile identificare mutazioni come p.V600K o altre varianti. Infine, l’IHC risente delle variabili della fase preanalitica, soprattutto di una fissazione subottimale dei campioni.15
In letteratura sono riportati diversi studi che dimostrano una elevata sensibilità (85-98%) e specificità (98-100%) del test immunoistochimico, in confronto ad approcci DNA-based quali pirosequenziamento o altre metodiche in Real-Time PCR sia nei melanomi che nei NSCLC. Una serie di studi ha riportato una sensibilità e specificità rispettivamente pari a 90-96% e 98-100% nella valutazione della mutazione puntiforme p.V600E, studiata in IHC utilizzando il clone VE1. In studi retrospettivi, effettuati mediante sequenziamento massivo parallelo (MPS) e/o pirosequenziamento, l’IHC ha identificato correttamente tutti i pazienti con mutazione BRAF, a carico dell’esone 15, p.V600E e non ha mostrato falsi positivi tra pazienti wild type e con mutazione BRAF non p.V600E.16 17
In sintesi, al momento, il test immunoistochimico con il clone VE1 consente di selezionare rapidamente i tumori positivi per mutazione V600E del gene BRAF, da destinare a conferma del dato con esame su DNA e pertanto potrebbe di fatto entrare nella pratica clinica come test di screening.17
L’introduzione in ambito oncologico di nuovi farmaci a bersaglio molecolare ha determinato la necessità di analizzare dapprima singoli marcatori (analisi monomarker) e di recente quella di valutare, in una stessa seduta, marcatori multipli (analisi multimarker). Di conseguenza le metodiche a disposizione nei laboratori si sono dovute adattare alle necessità cliniche.
Tra le metodiche “monomarker” non-in situ, ci sono tecniche, quali il sequenziamento secondo Sanger, il pirosequenziamento e la Real-Time PCR, che richiedono l’estrazione del DNA dal campione tumorale.
L’estrazione e la purificazione del DNA possono essere eseguite su campioni citologici, tessuto congelato o paraffinato, utilizzando vari kit commerciali basati prevalentemente su tecniche cromatografiche o di separazione magnetica. Il vantaggio principale nell’utilizzo dei kit è che si favorisce la standardizzazione della metodica con una netta riduzione dei tempi di esecuzione. In seguito all’estrazione, il DNA viene valutato quantitativamente e qualitativamente con lo spettrofotometro prima di procede con la fase analitica.
Il sequenziamento genico è una metodica che permette il sequenziamento del DNA, dopo l’estrazione del campione in esame, ottenendo l’esatta sequenza del tratto genomico d’interesse. Il primo modello fu sviluppato negli anni Cinquanta dal biochimico Sanger. Il sequenziamento “secondo Sanger”, o metodo enzimatico, a terminazione di catena, utilizza dinucleotidi modificati, bloccanti (dideossitrifosfato, ddNTPs) che vengono introdotti dall’enzima DNA polimerasi per interrompere la reazione di sintesi del DNA in posizioni specifiche lungo la sequenza. Si ottengono in questo modo filamenti di diversa lunghezza, in base al punto del DNA in cui i bloccanti vengono incorporati, marcati con radioattivi o fluorocromi, separati in base alla loro lunghezza e visualizzati su lastra fotografica o gel di agarosio (a seconda della marcatura). La sequenza dei nucleotidi e la stringa del DNA stampo da sequenziare vengono identificati in base alla disposizione dei frammenti. Questo primo modello è stato successivamente modificato con l’introduzione dell’elettroforesi capillare (in cui il gel di separazione è inserito in un tubo capillare) e la marcatura dei ddNTPs con quattro diverse molecole fluorescenti. Ogni ddNTP presenta un colore diverso per cui un raggio laser permette di stabilire l’identità del nucleotide che viene progressivamente inserito nel filamento sintetizzato dalla PCR. Un rilevatore capta le emissioni fluorescenti e le informazioni vengono integrate e trasformate in picchi di colore diverso, con aree proporzionali all’intensità di emissione. Si ottiene così un elettroferogramma, ovvero una sequenza di picchi di quattro colori diversi.18
In passato, il sequenziamento secondo Sanger ha rappresentato la metodica gold standard per l’individuazione di mutazioni geniche note e non note su campioni tumorali. I vantaggi di questa tecnologia sono una elevata specificità e bassi costi per lo studio di un basso numero di target genici. Tuttavia i numerosi svantaggi hanno portato al superamento di questa tecnologia.
Il sequenziamento di Sanger, infatti, ha una bassa sensibilità e permette di individuare mutazioni quando le copie di DNA mutato rappresentano il 20% circa del totale. È necessario, pertanto, disporre di una quantità elevata di DNA tumorale per rilevare una mutazione nel tratto genico da studiare. Da un punto di vista tecnico, la metodica è poco processiva, richiede molto tempo per sequenziare lunghi tratti di DNA e permette di analizzare un solo campione e un solo gene o tratto genico per scorrimento elettroforetico.
Per quanto riguarda il melanoma, sono stati condotti degli studi di comparazione tra test mutazionali effettuati con il Sanger e con una metodica allelespecifica basata su PCR (Cobas 4800 V600 mutation test). I risultati ottenuti sono stati simili e l’overall agreement riportato era del 95,2%. In altri studi è stato riportato che il Sanger identificava più mutazioni del gene BRAF nei melanomi rispetto alla metodica sopra citata. Il Sanger, pertanto, può trovare impiego in casi in cui il test Cobas fornisca un risultato negativo, per evitare di escludere pazienti da trattamenti con farmaci target.19 20
Metodiche e test disponibili per l’analisi del tessuto tumorale 7
Un altro metodo di sequenziamento è il pirosequenziamento o sequenziamento mediante sintesi. La DNA polimerasi reagisce con un nucleoside trifosfato, lega un nucleotide a una catena polinucleotidica in accrescimento e viene liberato il pirofosfato. La liberazione del pirofosfato viene rilevata. Un segmento di DNA a singolo filamento funge da templato per la sintesi di una nuova catena polinucleotidica ad opera della DNA polimerasi. La sintesi del DNA viene monitorata in tempo reale grazie al rilevamento della bioluminescenza prodotta al termine di una cascata di reazioni enzimatiche innescata dall’incorporazione di un nucleotide. Alla fine si ottiene un pirogramma, ovvero una sequenza di basi con dei picchi di segnale. Questa metodica è stata automatizzata e implementata con l’uso di sequenziatori che consentono determinazioni molecolari in parallelo di diversi campioni. Inoltre, si dispone di un software che rileva, analizza ed elabora i risultati, riportando i pirogrammi e i dati di ogni singolo campione, permettendo di avere sequenze geniche di milioni di paia di basi in tempi rapidi. Si può anche fare contemporaneamente un’analisi comparativa di DNA di origine diversa, e rilevare con facilità mutazioni di singole basi.21
Tra i vantaggi di questa metodica si annoverano la maggiore sensibilità rispetto al sequenziamento di Sanger (riportata tra il 5 e il 10%), la possibilità di effettuare diagnosi del tipo specifico di mutazione rilevata e la possibilità di sequenziare frammenti piuttosto corti di DNA, superando in tal modo eventuali problematiche legate alla frammentazione del DNA nel materiale fissato e incluso in paraffina. Negli anni passati, il pirosequenziamento si è diffuso molto, soprattutto per l’analisi di delezioni geniche e mutazioni puntiformi dei geni KRAS, NRAS e BRAF nel carcinoma del colon retto, di EGFR nel tumore del polmone e di BRAF nel melanoma. Nello specifico, ci sono kit commerciali come il therascreen BRAF Pyro kit comunemente in uso in molti laboratori. Rispetto al sequenziamento diretto, è una metodica più rapida e più sensibile per la quantizzazione delle mutazioni V600. Studi di letteratura riportano sensibilità del 90-100% e specificità del 95-100%. Attualmente, questo test può essere effettuato quando il risultato dell’IHC non è certo.22
Diverse metodiche basate sull’impiego della Real-Time PCR possono essere utilizzate per l’individuazione di alterazioni geniche nei tumori solidi. La RealTime PCR consente simultaneamente di amplificare grazie all’enzima DNA polimerasi e quantificare il DNA estratto dopo ogni amplificazione. Spesso questa metodica si combina con la PCR Retro Trascrizionale (RT-PCR) che permette di quantificare i livelli di espressione di specifici tratti di RNA. Mediante la retro-
trascrizione (o trascrizione inversa) viene prodotta una molecola di DNA complementare a singolo filamento detto cDNA (complementary DNA), mantenendo inalterati i rapporti relativi di concentrazione delle diverse specie degli RNA. Applicazioni di questa metodologia sono kit commerciali CE-IVD basati sulla tecnologia ARMS/Scorpion in grado di individuare le mutazioni di BRAF.23
La metodica permette di identificare una mutazione se nel campione sono presenti almeno 5-10 copie di DNA mutato, la sensibilità teorica è pertanto tale da rilevare mutazioni quando le copie di DNA mutato rappresentano circa l’1% del DNA totale, con differenze tra le varie mutazioni identificate. L’analisi, però, può dare adito a falsi negativi quando viene condotta su un numero esiguo di cellule neoplastiche. In commercio, sono disponibili diversi kit, alcuni approvati dall’FDA come companion diagnostics, che consentono di individuare mutazioni a carico di diversi geni driver, basati su metodiche di Real-Time PCR e con sensibilità teorica variabile tra lo 0,1% e il 5% a secondo della tecnologia impiegata. Uno svantaggio di tali tecnologie è che evidenziano solo mutazioni hot spot, previste a priori, ma questi kit commerciali presentano per contro vari vantaggi: dispongono, infatti, di controlli interni negativi e positivi alla reazione, la procedura è standardizzata ed è più rapida rispetto al sequenziamento diretto. Per il melanoma, l’FDA ha approvato il Cobas 4800 BRAF V600 mutation test e il THXID-BRAF kit per le determinazioni delle mutazioni di BRAF a carico dell’esone 15 p.V600. Il principale limite di queste metodiche è che sono state ottimizzate per identificare solo le mutazioni note e più comuni. In particolare, il THxID-BRAF kit è stato approvato per le mutazioni V600E e V600K con una sensibilità analitica rispettivamente del 96% e del 92% e per una quantità di DNA tra 10 e 350 ng/μl (bioMérieuxCorporate). Al contrario, il Cobas è stato approvato solo per la mutazione V600E e ha una sensibilità analitica del 97%, partendo da una quantità minima di DNA di 125 ng (Drugs@FDA: FDA-ApprovedDrugs). Rispetto al Cobas 4800 test, il test THxID mostra una sensibilità più elevata per la determinazione delle mutazioni V600K come mostrato nella tabella 1.1.24 25
Di recente sono stati sviluppati altri test CE-IVD Real-Time PCR quale il Peptide Nucleic Acid (PNA)-mediated PCR clamping, un metodo Real-Time PCR che utilizza i PNAs per legare normali sequenze di DNA, per cui si amplifica solo il DNA mutato. La metodica ha elevate sensibilità e specificità (99,5% e 100%), rappresentando una valida alternativa al pirosequenziamento. La PCR basata su PNA clamp in real-time identifica circa lo 0,5% di BRAF V600E mutato in un background di DNA wild-type in quanto si basa sul principio per cui i PNAs inibiscono il DNA wild type attraverso l’ibridizzazione delle sequenze normali per cui il DNA mutato viene amplificato preferenzialmente.26
Per il tumore del colon-retto sono attualmente disponibili test in Real-Time PCR approvati da FDA per le mutazioni dei geni RAS e BRAF. In particolare, il TheraScreen KRAS Mutation Detection Kit (DxS-Qiagen) e il cobas® KRAS Mutation Test (Roche Molecular Systems) sono metodiche ad elevata sensibilità che
Metodiche e test disponibili per l’analisi del tessuto tumorale 9
Tabella 1.1
Comparazione tra due metodi RT-PCR approvati FDA per l’analisi delle mutazioni BRAF
TEST
CDx o LDT
Sensibilità, %
THxID BRAF Kit
CDx
96 per V600E 92 per V600K
Cobas 4800 BRAF
CDx
97 per V600E 66-70 per V600K
Specificità, % 100 98
Limite detezione 5 per V600E, V600K 5-7 per V600E, >35 per V600K
Mutazioni evidenziate Approvato per V600E, V600K Approvato solo per V600E
Tipologia campione DNA tumorale DNA tumorale
Costo Medio Medio
Modificato da: http://www.fda.gov.companiondiagnostics
consentono di identificare le principali mutazioni del gene KRAS. Per le determinazioni mutazionali dei geni NRAS e BRAF sono disponibili diversi kit in commercio quali, ad esempio, Therascreen® NRAS Pyro® e Therascreen BRAF RGQ kit (Qiagen), il kit COBAS® KRAS/NRAS (LightMix KRAS/NRAS) e Cobas® 4800 BRAF V600 Mutation Test.
Più recentemente è stato commercializzato l’Idylla™, test rapido e completamente automatizzato che permette l’identificazione delle principali varianti mutazionali negli esoni 2, 3 e 4 di KRAS (IdyllaTM KRAS Mutation Test), 18 mutazioni negli esoni 2, 3 e 4 di NRAS e 5 mutazioni di BRAF nel codone 600 (IdyllaTM NRAS-BRAF Mutation Test).
Per i pazienti affetti da melanoma, tra i kit in Real-Time PCR ricordiamo inoltre il Therascreen BRAF RGQ PCR Kit e l’IdyllaTM BRAF Mutation Test.27 28
Altre tecnologie disponibili in diagnostica sono quelle di ibridazione molecolare su supporti solidi del tipo monomarker quali il Southern Blot e il Northern blot, che al momento attuale trovano impiego soprattutto in ambito di ricerca, o tecnologie multimarker come il Nanostring e l’ibridazione su microchip. In alcuni laboratori si è diffusa anche la tecnologia Maldi-TOF per l’analisi simultanea multigenica. Infine il sequenziamento massivo parallelo (MPS) che nel tempo sta diventando una tecnologia di riferimento per l’analisi massiva delle alterazioni genetiche in funzione dei trattamenti a bersaglio molecolare.
Un test multimarker basato su ibridazione molecolare, sviluppato di recente, è l’IntelliplexTM system, che prevede una duplice strategia: amplificazione selettiva e πCode (PlexBio/Menarini). La prima, definita Locked Nucleic Acid, blocca l’amplificazione mediante PCR delle sequenze wild type, consentendo un incremento della sensibilità e della specificità della tecnologia. La strategia πCode, al contrario, si basa su micro-dischi magnetici di 50 μm che hanno impressi sulla superficie fino a 16.000 configurazioni differenti, ognuna delle quali si associa a una specifica coppia sonda-analita. Su questi dischetti virtualmente possono essere coniugati tutti i tipi di sonde: DNA, RNA, anticorpi, antigeni, ecc. L’interazione e l’ibridazione specifica tra analiti e sonde molecolari consentono una elevatissima sensibilità (compresa tra 0,1% e 0,01%).
Inoltre si possono studiare in parallelo campioni multipli e geni diversi, con una bassa quantità di acidi nucleici estratti dal campione (10-100 ng) in un turnaround time inferiore alle 6 ore. I kit a disposizione prevedono dei pannelli multipli includenti le alterazioni geniche a carico dei principali geni di interesse clinico nei tumori solidi.23
Un approccio tecnologico a parte è la spettrometria di massa (Matrix-Assisted Laser Desorption/Ionization – time of flight, MALDI-TOF) con tecnologia primer extension. Si tratta di una tecnologia multimarker per l’analisi di alterazioni hot-spot che permette la genotipizzazione del campione mediante l’amplificazione del DNA e una reazione di estensione a singola base di primer adiacenti ai siti polimorfici di interesse. Per ciascun sito polimorfico si ottengono uno o più analiti di massa nota, ciascuno corrispondente al genotipo wildtype oppure mutato del campione analizzato. La sensibilità analitica del metodo può variare dal 2,5% al 10% in base alla mutazione testata. Ad oggi sono disponibili in commercio kit validati per l’uso diagnostico basati sulla spettrometria di massa MALDI-TOF che permettono, mediante l’utilizzo di PCR multiplex, la rivelazione delle principali mutazioni dei principali geni di interesse. Negli anni, questa tecnologia è stata implementata permettendone l’utilizzo in molti laboratori nella diagnostica di routine e rappresentando un ponte tra le metodiche tradizionali quali Sanger e Real Time-PCR e il sequenziamento massivo parallelo. In questo contesto, sono disponibili vari kit commerciali come il TheiPLEX HS Melanoma panel (Agena Bioscience Inc., San Diego, CA) che identifica 97 varianti clinicamente rilevabili in 11 geni driver per il melanoma, incluso il BRAF da tessuto in paraffina, a una frequenza allelica inferiore all’1%. In letteratura, studi di comparazione della metodica con il pirosequenziamento riportano nel melanoma un tasso di concordanza del 99,3%, una sensibilità e specificità del 97,6% e 100%. Il MALDI-TOF MS, pertanto, è una tecnologia molto robusta che consente la genotipizzazione di mutazioni note con elevata sensibilità e specificità, permette la creazione di custom assays, ha il vantaggio di una multiplex come l’MPS. Tuttavia richiede piattaforme dedicate, ha costi abbastanza elevati e richiede expertise soprattutto tecnica.23 29
Metodiche e test disponibili per l’analisi del tessuto tumorale 11
L’introduzione del sequenziamento massivo parallelo, MPS, nella diagnostica molecolare è stato il progresso più importante verificatosi negli ultimi anni. La scoperta di nuove alterazioni molecolari e farmaci target ha portato all’acquisizione di nuove competenze e alla richiesta crescente da parte degli oncologi di determinare l’assetto molecolare del tumore prima della terapia. Inoltre, in seguito alle nuove acquisizioni scientifiche sulle alterazioni geniche che conferiscono al tumore resistenza alle terapie molecolari, è diventata fondamentale anche la rivalutazione dei tumori dopo la terapia target. Questo ha portato alla necessità di valutare l’assetto molecolare completo del campione tumorale in esame, sia esso primitivo o metastatico, includendo nell’analisi le variazioni di base (single nucleotide variants, SNV), le inserzioni/delezioni di basi (INDEL), la variazione del numero di copie (copy number variation, CNV) e la presenza di riarrangiamenti o fusioni geniche. A questo scopo il sequenziamento massivo parallelo sta diventando sempre più importante rispetto ad altri approcci tecnologici.30
In breve, sebbene in commercio ci siano piattaforme diverse, il workflow dell’MPS è caratterizzato da 4 fasi: costruzione della libreria, amplificazione clonale, sequenziamento massivo parallelo e analisi dei dati.
La strategia MPS consente il sequenziamento dell’intero genoma (whole genome sequencing), di regioni geniche codificanti (whole exome sequencing) e in contemporanea di regioni specifiche del genoma (targeted sequencing). La sensibilità teorica della metodica è tale da individuare mutazioni quando le copie di DNA mutato rappresentano circa lo 0,01% del DNA totale, con differenze tra le varie mutazioni identificate.
Tra gli approcci MPS, quello che ha trovato un impiego maggiore nella pratica clinica è il TS (targeted sequencing) che ha pannelli genici comprendenti i principali oncogeni e anti-oncogeni actionable o con un ruolo predittivo e/o prognostico. Le tecnologie a disposizione utilizzano strategie diverse per il metodo di sequenziamento e per la preparazione delle librerie genomiche da sequenziare.
Per quanto concerne la chimica di reazione, il sequenziamento mediante sintesi utilizza i terminatori con coloranti reversibili che consentono di identificare le singole basi introdotte nel DNA. Al contrario, il sequenziamento con semiconduttori ionici rileva gli ioni idrogeno rilasciati in seguito alla polimerizzazione del DNA.
Per la produzione di librerie genomiche è possibile ricorrere all’amplificazione selettiva delle sequenze target (ampliconi) o utilizzare l’ibridazione e cattura dei frammenti di acidi nucleici che devono essere sequenziati (figura 1.1). Questi due approcci, sono molto diversi tra di loro in termini di strategie analitiche e applicazioni diagnostiche, ma una descrizione più profonda di questi due approcci tecnologici va al di là dello scopo del presente capitolo. Per quanto concerne le tecnologie di sequenziamento, i pannelli spesso permettono di analizzare sia il DNA (per l’analisi delle varianti nucleotidiche e, a seconda dell’approccio tecno-
Basato su ampliconi
PCR e arricchimento
Preparazione libreria
Targeted Sequencing
DNA estratto
RNA➞cDNA
Basato su ibridazione e cattura
Frammentazione DNA Genomico
Preparazione libreria
Ibridazione e cattura
Sonde specifiche
Figura 1.1
Strategie MPS per la preparazione di librerie
logico, anche altre alterazioni genomiche) sia l’RNA messaggero (per identificare con maggiore accuratezza alterazioni più complesse inclusi i riarrangiamenti genici). Le piattaforme più diffuse sul territorio nazionale sono Ion Torrent (Thermofisher), Illumina e con minor frequenza GeneReader (Qiagen) (dati della survey nazionale del Gruppo Italiano SIAPEC-PMMP).
I pannelli disponibili sono diversi, alcuni dei quali già validati e disponibili come prodotti per la diagnostica in vitro (in vitro diagnostics, IVD), basati sia sulla chimica ad ampliconi sia su quella a cattura. I pannelli consentono di rilevare la presenza di SNV, CNV, INDEL e riarrangiamenti genici, caratterizzandone le varianti di fusione. I tipi e il numero di varianti di fusione studiate sono correlati al tipo di pannello.31
Le tecnologie MPS possono dare informazioni anche sui meccanismi di resistenza correlati ai farmaci a bersaglio molecolare. Sono stati sviluppati pannelli genici anche molto grandi, da centinaia di geni, che danno informazioni anche sul carico mutazionale del tumore (tumor mutation burden, TMB). Tra i pannelli più diffusi negli USA si annoverano TruSight Tumor 15 (specifico per alterazioni molecolari per pazienti affetti da NSCLC, melanoma e colonretto) 170 o 500 geni (Illumina), Oncomine Comprehensive™ (Thermofisher), Oncomine Tumour Mutational Load Assay™ (Thermofisher), FoundationOne CDx™ (Roche), MSKIMPACT (Memorian Sloan Kettering), Molecular Intelligence® (CARIS), Tempus xT (TEMPUS), ACE ImmunoID (PersonaLis), Cancer-Plex (KEW). Alla data attuale i pannelli con approvazione FDA sono il FoundationOne e il MSK-IMPACT. Il
Metodiche e test disponibili per l’analisi del tessuto tumorale 13
tipo e la quantità di campioni che possono essere analizzati, il numero di geni valutabili, il tipo di alterazioni evidenziabili e i tempi della metodica dipendono dai pannelli utilizzati. Nelle tabelle 1.2 e 1.3 sono riportati rispettivamente i pannelli genici piccoli e grandi più comuni utilizzati in diagnostica oncologica nel nostro paese.
In linea generale, l’elevata sensibilità e specificità, la possibilità di testare contemporaneamente più pazienti e diversi geni, l’abbattimento dei costi in virtù della maggiore diffusione della tecnologia sono tra i vantaggi più importanti dell’utilizzo della metodica MPS nella diagnostica molecolare.
La sensibilità dei test MPS dipende dal tipo di pannello, dalla strategia di cattura dei target, dalla piattaforma e dal software integrato per l’analisi dei risultati. In considerazione della sensibilità di queste tecnologie (detection rate di 0,1/0,01 di alleli mutati), la quantità minima di DNA/RNA richiesta per l’analisi permette
Tabella 1.2
Principali pannelli genici commerciali per il sequenziamento massivo parallelo (<100 geni)
Fornitori Nome commerciale del kit Campioni Numero geni testati
for Illumina® CancerHotSpot Panelv2
for Illumina® Focus Panel
Diatech Myriapod NGS Cancer Panel DNA
Diatech Myriapod NGS 56G OncoPanel
Agilent SureSelect Cancer All-In-One Lung Assay
Thermo Fisher Ion AmpliSeq™ Cancer panel v2/ Oncomine Dx Target Test
Thermo Fisher Ion AmpliSeq™CancerHotSpotv2 Panel
Solid Tumor Kit
DX
Oncology Research Kit
CTL Kit
Qiagen GeneRead QIAact Actionable Insight
GeneRead QIAact Lung UMI Panel
Roche AvenioTumor Tissue/ ctDNA Targeted/ Expanded Kit
biopsy
biopsy
biopsy
Tabella 1.3
Principali pannelli genici commerciali per il sequenziamento massivo parallelo (> 100 geni)
Fornitori Nome commerciale del kit Campioni Numero geni testati DNA/RNA
TruSight Oncology 170-5001
Illumina
TruSight RNA Pan-Cancer Panel
TruSight RNA Fusion Panel
AmpliSeq™ for Illumina® CancerHotSpot Panelv3
Roche Avenio Tumor Tissue Analysis/ctDNA Surveillance Kit
Thermo Fisher Ion AmpliSeq™ Comprehensive Cancer Panel
SureSelect Cancer All-In-One Solid Tumor assay
Agilent
biopsy
biopsy
di valutare mediante MPS non solo i campioni tessutali ma anche quelli di biopsia liquida. Per questa tipologia di campioni ci sono dei pannelli dedicati che variano per il tipo di alterazioni dimostrabili, la qualità e la quantità del campione di partenza richiesto.
Per quanto concerne il melanoma, sono stati condotti numerosi studi che suggeriscono alla data attuale l’utilizzo di questo approccio in casi particolari, di negatività con metodiche in Real-Time PCR allele-specifiche.32-34 Questo approccio consente di risparmiare sui costi e al contempo di evitare che casi positivi possano essere misdiagnosticati con metodiche meno sensibili. Inoltre, l’MPS nel melanoma trova indicazione per l’identificazione di mutazioni actionable rare. Come riportato in un lavoro, la metodica MPS è stata in grado di identificare una variante rara della mutazione dell’esone 15 di BRAF p.V600E (c.1799_1800TG >AA) che non era stata messa in evidenza con metodiche di Real-Time PCR.35 In questa fase, si consiglia l’utilizzo concomitante di una tecnologia MPS e di una metodica ortogonale, soprattutto quando si devono analizzare campioni di scarsa qualità o si hanno risultati dubbi. Infatti, l’elevata sensibilità della tecnologia può portare a falsi positivi. Inoltre per l’analisi dei dati si richiede la presenza di personale specificamente formato con competenze bioinformatiche. Per quanto concerne la scelta dei pannelli, questa dipende dalle esigenze del centro, tuttavia è preferibile ricorrere a pannelli CEIVD ed eventualmente approvati dall’FDA.36
Metodiche e test disponibili per l’analisi del tessuto tumorale 15
In termini tecnologici gli sviluppi futuri della caratterizzazione molecolare dei tumori riguardano sostanzialmente il sequenziamento massivo parallelo che permette la profilazione genomica del tumore tramite la sequenza contemporanea di numerosi geni. Questa tecnologia ha permesso di fare importanti passi in avanti nella caratterizzazione dei tumori in funzione del trattamento. È auspicabile che questa nuova strategia tecnologica si diffonda rapidamente in modo da poter essere estesa alla caratterizzazione di tutte le forme tumorali sia al momento della diagnosi che nel corso della progressione neoplastica. Ciò è fondamentale per monitorare i pazienti e poterli indirizzare ai trattamenti mirati. I risultati complessi ottenuti dal sequenziamento massivo parallelo devono essere sottoposti ad analisi bioinformatiche e gestiti da gruppi multidisciplinari, i Molecular Tumor Board (costituiti da oncologi, patologi, biologi molecolari, bioinformatici, genetisti, farmacologi, ecc.) per la scelta dei trattamenti più opportuni, utilizzando piattaforme analitiche in cui sono raccolti tutti i dati emersi dalla profilazione genomica assieme ai dati clinico-patologici del paziente. Le più importanti prospettive di sviluppo in questo settore prevedono la diffusione del sequenziamento massivo parallelo in centri di eccellenza diagnostica sul territorio che permettano la centralizzazione dei test, la riduzione dei costi e l’incremento della qualità delle analisi. Una stretta connessione tra centri di diagnostica molecolare riconosciuti dalle regioni, i Molecular Tumor Board e le reti oncologiche regionali, all’interno di Percorsi Diagnostico-Terapeutici (PDTA) sempre più definiti, rappresenterà la principale sfida per l’oncologia di precisione nel prossimo futuro.
1. Wakai T, Prasoon P, Hirose Y. et al. Next-generation sequencing-based clinical sequencing: toward precision medicine in solid tumors. Int J Clin Oncol 2019; 24: 115-22.
2. Rosas D, Raez LE. Review of the agnostic-type treatment approach: treating cancer by mutations, not by location. Oncol Ther 2020; 8(1): 59-66.
3. Malapelle U, Rossi G, Pisapia P, et al. BRAF as a positive predictive biomarker: focus on lung cancer and melanoma patients. Critical Reviews in Oncology/ Hematology 2020; 156: 103118.
4. Michielin O, van Akkooi ACJ, Ascierto PA, Dummer R, Keilholz U. ESMO guidelines committee. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2019; 30(12): 1884-901.
5. Eriksson H, Zebary A, Vassilaki I, Omholt K, Ghaderi M, Hansson J. BRAFV600E Protein Expression in Primary Cutaneous Malignant Melanomas and Paired Metastases. JAMA Dermatol 2015; 151(4): 410-6.
7. Sanz-Garcia E, Argiles G, Elez E, Tabernero J. BRAF mutant colorectal cancer: prognosis, treatment, and new perspectives. Ann Oncol 2017; 28(11): 2648-57.
8. Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the college of american pathologists, the international
6. Strickler JH, Wu C, BekaiiSaab T. Targeting BRAF in metastatic colorectal cancer: maximizing molecular approaches. Cancer Treat Rev 2017; 60(11): 109-19.
association for the study of lung cancer, and the association for molecular pathology. Arch Pathol Lab Med 2018; 142(3): 321-46.
9. Kalemkerian GP, Narula N, Kennedy EB, et al. Molecular testing guideline for the selection of patients with lung cancer for treatment with targeted tyrosine kinase inhibitors: American Society of Clinical Oncology Endorsement of the College of American Pathologists/ International Association for the Study of Lung Cancer/ Association for Molecular Pathology Clinical Practice Guideline Update. J Clin Oncol 2018; 36(9): 911-9.
10. Ettinger DS, Wood DE, Aggarwal C, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 1.2020. J Natl Compr Canc Netw 2019; 17(12): 1464-72.
11. Vitiello PP, De Falco V, Giunta EF, et al. Clinical Practice Use of Liquid Biopsy to Identify RAS/BRAF Mutations in Patients with Metastatic Colorectal Cancer (mCRC): a single institution experience. Cancers (Basel) 2019; 11(10): 1504.
12. Troxell ML, Fulton RS, Swanson PE, et al. Predictive markers require thorough analytic validation. Arch Pathol Lab Med 2019; 143(8): 907-9.
13. Thunnissen E. How to validate predictive immunohistochemistry testing in pathology? A Practical approach exploiting the heterogeneity of programmed death ligand-1 present in nonsmall cell lung cancer. Arch Pathol Lab Med 2019; 143(1): 11-2.
14. Hung YP, Sholl LM. Diagnostic and predictive immunohistochemistry for Non-Small Cell Lung Carcinomas. Adv Anat Pathol 2018; 25(6): 374-86.
15. Kwon JH, Jeong BK, Yoon YS, Yu CS, Kim J. Utility of BRAF VE1 Immunohistochemistry as a Screening Tool for Colorectal Cancer Harboring BRAF V600E Mutation. J Pathol Transl Med 2018; 52(3): 157-63.
16. Ilie M, Long E, Hofman V, et al. Diagnostic value of immunohistochemistry for the detection of the BRAFV600E mutation in primary lung adenocarcinoma caucasian patients. Ann Oncol 2013; 24(3): 742-8.
17. Gow CH, Hsieh MS, Lin YT, Liu YN, Shih JY. Validation of Immunohistochemistry for the Detection of BRAFV600E-Mutated Lung Adenocarcinomas. Cancers (Basel) 2019; 11(11): 866.
18. Verma M, Kulshrestha S, Puri A. Genome Sequencing. Methods Mol Biol 2017;1525: 3-33.
19. Qu K, Pan Q, Zhang X, et al. Detection of BRAF V600 mutations in metastatic melanoma: comparison of the Cobas 4800 and Sanger sequencing assays. J Mol Diagn 2013; 15(6): 790-5.
20. Mourah S, Denis MG, Narducci FE, et al. Detection of BRAF V600 mutations in melanoma: evaluation of concordance between the Cobas® 4800 BRAF V600 mutation test and the methods used in French National Cancer Institute (INCa) platforms in a real-life setting. PLoS One 2015; 10(3): e0120232.
21. Harrington CT, Lin EI, Olson MT, Eshleman JR. Fundamentals of pyrosequencing. Arch Pathol Lab Med 2013; 137(9): 1296303.
22. Taniguchi H, Okamoto W, Muro K, et al. Clinical validation of newly developed multiplex kit using luminex xMAP technology for detecting simultaneous
RAS and BRAF mutations in colorectal cancer: results of the RASKET-B Study. Neoplasia 2018; 20(12): 1219-26.
23. Vanni I, Teresa Tanda E, Spagnolo F, et al. The current state of molecular testing in the BRAF-mutated melanoma landscape. Front Mol Biosci 2020; 7: 113.
24. Marchant J, Mange A, Larrieux M, Costes V, Solassol J. Comparative evaluation of the new FDA approved THxID™-BRAF test with High Resolution Melting and Sanger sequencing. BMC Cancer 2014; 14(7): 519.
25. http: //www.fda.gov/ companiondiagnostics
26. Kwon MJ, Lee SE, Kang SY, Choi YL. Frequency of KRAS, BRAF, and PIK3CA mutations in advanced colorectal cancers: comparison of peptide nucleic acid-mediated PCR clamping and direct sequencing in formalinfixed, paraffin-embedded tissue. Pathol Res Pract 2011; 207(12): 762-8.
27. Bradish JR, Richey JD, Post KM, et al. Discordancy in BRAF mutations among primary and metastatic melanoma lesions: clinical implications for targeted therapy. Mod Pathol 2015; 28(4): 480-6.
28. Huang H, Springborn S, Haug K, et al. Evaluation, validation, and implementation of the Idylla system as rapid molecular testing for precision medicine. J Mol Diagn 2019; 21(5): 862-72.
29. Bonaparte E, Pesenti C, Fontana L, et al. Molecular profiling of lung cancer specimens and liquid biopsies using MALDI-TOF mass spectrometry. Diagn Pathol 2018; 13(1): 4.
30. Cheng L, Lopez-Beltran A, Massari F, MacLennan GT, Montironi R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: a move toward
precision medicine. Mod Pathol 2018; 31(1): 24-38.
Metodiche e test disponibili per l’analisi del tessuto tumorale 17
31. Wakai T, Prasoon P, Hirose Y, et al. Next-generation sequencing-based clinical sequencing: toward precision medicine in solid tumors. Int J Clin Oncol 2019; 24: 115-22.
32. Ma W, Brodie S, Agersborg S, Funari VA, Albitar M. Significant improvement in detecting BRAF, KRAS, and EGFR mutations using nextgeneration sequencing as compared with FDA-Cleared Kits. Mol Diagn Ther 2017; 21(5): 571-9.
33. Serratì S, De Summa S, Pilato B, et al. Next-generation sequencing: advances and applications in cancer diagnosis. Onco Targets Ther 2016; 9: 7355-65.
34. Zhu ML, Zhou L, Sadri N. Comparison of targeted next generation sequencing (NGS) versus isolated BRAF V600E analysis in patients with metastatic melanoma. Virchows Arch 2018; 473(3): 371-7.
35. roietti I, Michelini S, Di Fraia M, et al. A rare BRAF V600E mutation detected by
next-generation sequencing in a superficial spreading melanoma: case report and potential diagnostic implications. J Eur Acad Dermatol Venereol 2020; 34(8): e393-e395.
36. Bruno, W, Martinuzzi, C, Andreotti, V, et al. Heterogeneity and frequency of BRAF mutations in primary melanoma: comparison between molecular methods and immunohistochemistry. Oncotarget 2017; 8(5): 8069-82.
FILIPPO NOZZOLI, DANIELA MASSI, FRANCESCA CASTIGLIONE
Il melanoma cutaneo costituisce il 4-5% di tutti i tumori cutanei, la sua incidenza è costantemente aumentata negli ultimi decenni e i tassi di mortalità risultano anch’essi incrementati, specialmente nella popolazione caucasica di sesso maschile.1 2 La mortalità è più elevata nei pazienti in stadio avanzato, in condizioni di incompleta rimozione chirurgica e la tradizionale chemioterapia è caratterizzata da scarso beneficio oncologico.3
A partire dai primi anni Duemila, a seguito degli avanzamenti conseguiti nel campo delle tecniche di sequenziamento, vennero descritte mutazioni puntiformi nel gene BRAF in un sottogruppo di tumori, di cui il melanoma rappresentava quello maggiormente coinvolto.4 Il gene BRAF, localizzato sul cromosoma 7q34, codifica una proteina che media la trasmissione di segnali chimici dall’esterno al nucleo delle cellule. Questa proteina fa parte di una via di segnalazione nota come RAS/MAPK, deputata al controllo di diverse importanti funzioni cellulari quali proliferazione, differenziazione, migrazione e apoptosi. La regolazione fisiologica della segnalazione MAPK può risultare alterata in molti processi patogenetici, primo fra tutti quello neoplastico. Nelle cellule umane esistono tre proteine RAF: ARAF, BRAF e CRAF (nota anche come RAF1). Sebbene tutte e tre le chinasi RAF abbiano un ruolo importante nella normale fisiologia cellulare, BRAF è la chinasi RAF predominante che viene modificata in molti diversi tipi di cancro. La specificità biologica di BRAF, non condivisa dalle altre proteine appartenenti alla famiglia RAF, non è interamente compresa. Possibili spiegazioni includono la natura specifica del tessuto, il contesto di espressione di BRAF e le sue capacità di attivare il substrato MEK, rispetto a CRAF e ARAF che sembrerebbero invece assumere più in generale una funzione di geni costitutivi.5
La via di segnalazione RAS-RAF e la sua attivazione oncogena sono riportate nella figura 2.1. I membri della famiglia RAF sono composti da tre domini con-
Dabrafenib
Vemurafenib
Encorafenib
Sorafenib
Dasatinib
Belvarafenib
Ulixertinib LY3214996
Figura 2.1
Trametinib
Cobimetinib
Binimetinib
Vie di segnalazione, fisiologica e patologica, coinvolgenti BRAF
servati: le regioni CR1, CR2 e CR3. CR1 è un dominio di autoregolazione della proteina; CR2 costituisce una regione cerniera ricca di serina, mentre CR3 presenta una conformazione a doppio anello che accoglie il dominio catalitico della serina/treonina chinasi in grado di innescare, a seguito del legame con GTP, una serie di eventi di fosforilazione a cascata a livello di substrati proteici.6-8
A supporto della classificazione di BRAF come oncogene, è ormai conoscenza consolidata il fatto che le mutazioni a suo carico sono in grado di produrre una costitutiva attivazione del dominio chinasico della proteina, determinando una stimolazione incontrollata della proliferazione cellulare, evento chiave nel processo di cancerogenesi. Sono stati identificati circa 200 alleli mutanti di BRAF nei tumori umani e, finora, quasi 30 mutazioni distinte di BRAF sono state funzionalmente caratterizzate. È interessante notare che le mutazioni nel gene BRAF possono essere classificate in tre classi in base al loro effetto sull’attività della proteina, come riportato nella tabella 2.1. Le mutazioni BRAF di classe 1 sono indipendenti dal legame con la proteina RAS e funzionano come un monomero attivo. Il BRAF mutante di classe 2 funziona come un dimero attivo. Le proteine
Tabella 2.1
Classificazione delle maggiori mutazioni nel gene BRAF nei tumori solidi
CLASSE RAS-dipendenza Mutazioni Missenso
1 No V600E, V600K, V600D, V600R, V600M, ecc.
2 No K601E, K601N, K601T, L597Q, L597V, G469A, G469V, G469R, G464V, ecc.
3 Sì D287H, V459L, G466V, G466E, G466A, S467L, G469E, N581S, N581I, D594N, D594G, D594A, D594H, F595L, G596D, ecc.
chimeriche RAF di Classe 1 e Classe 2 diventano quindi in gran parte svincolate rispetto al regolatore inibitorio della loro attività. Al contrario, le proteine BRAF mutanti di classe 3 dipendono strettamente dalla segnalazione mediata dal legame con RAS al fine di andare incontro a un’ottimale attivazione.9
Nel melanoma, il gene BRAF è mutato in oltre il 50% dei casi; la mutazione più diffusa (85-90% dei casi) è rappresentata dalla sostituzione di una valina con acido glutammico al codone 600 (V600E). Le rimanenti mutazioni BRAF si verificano spesso nello stesso codone: V600K (la più frequente; <10% dei casi), V600D e V600R; le mutazioni in codoni diversi da V600 non sono comuni (tra questi, K601 è quella prevalente).
Il melanoma mutato in BRAF tende a esibire segni distintivi, peculiari caratteristiche epidemiologiche e cliniche ed è caratterizzato da un comportamento biologico più aggressivo rispetto ai melanomi wild-type (WT). I melanomi con mutazione di BRAF insorgono in pazienti più giovani con elevato numero di nevi in sedi non cronicamente fotoesposte quali tronco e arti, e in prevalenza mostrano istotipo a diffusione superficiale.10 Inoltre, è stato riportato che i melanomi BRAF mutati possano metastatizzare a livello cerebrale più frequentemente rispetto ai tumori BRAF WT.11 Il melanoma BRAF mutato è stato ricondotto a una più ridotta sopravvivenza globale in pazienti con stadio IV rispetto a quelli con malattia BRAF WT.10-12
Sebbene la mutazione BRAF V600 sia oncogenica in relazione alla sua capacità di promuovere in maniera incontrollata la proliferazione cellulare, è considerata insufficiente per indurre l’insorgenza di melanoma in assenza di altre anomalie citogenetiche. È infatti noto da tempo che le mutazioni BRAF V600 si possano dimostrare frequentemente in nevi melanocitici benigni e displastici, indicando la necessità di modifiche aggiuntive per la trasformazione maligna.13 Questi cambiamenti – inclusa la perdita di soppressori tumorali, l’attivazione del promotore TERT, l’inattivazione di geni coinvolti nella riparazione del DNA e l’attivazione di altre proteine chinasi e cascate di segnalazione – consentono di rilasciare i freni inibitori, permettendo la proliferazione cellulare alla base della cancerogenesi. Geni comuni in cui possono verificarsi mutazioni con perdita di funzione comprendono CDKN2A, PTEN e BAP1. La mutazione di CDKN2A, ad esempio, rimuo-
ve un elemento chiave della via di soppressione p53 del tumore. La disfunzione di questo percorso elimina un meccanismo che normalmente consegnerebbe cellule precancerose con mutazioni germinali preesistenti o locali, somatiche, di apoptosi o senescenza. Quindi, tessuti cutanei composti da cellule recanti una mutazione driver in BRAF, combinata con la perdita di un gene oncosoppressore, sono pronti a subire la trasformazione maligna e l’evoluzione in melanoma. Date queste funzionalità e le effettive terapie disponibili, è fondamentale determinare rapidamente se nei tessuti di melanoma siano presenti mutazioni di BRAF al fine di selezionare l’ottimale iter terapeutico. Attualmente, lo stato di mutazione di BRAF è infatti l’unico biomarcatore predittivo di risposta terapeutica nel melanoma avanzato.14 Test molecolari per l’identificazione di mutazioni in BRAF nei pazienti con melanoma avanzato sono quindi diventati una procedura standard per determinare il percorso terapeutico appropriato. Miglioramenti costanti nella sopravvivenza in questo sottogruppo di pazienti sono stati ottenuti con l’introduzione, avvenuta negli anni recenti, di terapie mirate e immunologiche. La mutazione BRAF V600 ha oggi infatti un significato prevalentemente predittivo nella gestione del paziente con melanoma, in quanto identifica una potenziale sensibilità al trattamento con la combinazione di BRAF e MEK inibitori (target therapy). In particolare, nei pazienti in stadio IV o III non operabile, in presenza di mutazione BRAF V600, qualora indicata target therapy, il trattamento con BRAF inibitore + MEK inibitore dovrebbe essere preso in considerazione come prima opzione. È importante sottolineare inoltre che, per quanto riguarda i pazienti con melanoma in stadio III, recenti studi randomizzati hanno dimostrato l’efficacia dei nuovi trattamenti (target therapy e immunoterapia) in termini di miglioramento della relapse free survival (RFS). La durata delle risposte nel melanoma mutato da BRAF è tuttavia limitata dall’insorgenza di resistenza ai farmaci. La sfida nei prossimi anni risiede nel determinare come impiegare inibitori della RAF su più tipi di tumore per ottenere il massimo beneficio clinico immediato, prevenendo contemporaneamente l’emergenza di meccanismi di resistenza alla terapia farmacologica a bersaglio molecolare.
Nel melanoma delle mucose, differentemente rispetto a quanto detto per il melanoma cutaneo, vi è una bassa frequenza di mutazione del gene BRAF o NRAS, mentre può essere più frequente la mutazione del gene c-KIT, presente fino al 25% dei casi. Nel caso dei melanomi mucosali viene suggerito tuttavia di procedere dapprima con la determinazione della mutazione BRAF e successivamente, in caso di negatività, con la ricerca della mutazione di c-KIT. È importante tuttavia sottolineare che essendo c-KIT mutato nell’1% dei casi in tumori localizzati in sedi non cronicamente esposte al sole, quando abbiamo un paziente con melanoma metastatico a partenza acrale o mucosale, lo studio mutazionale di c-KIT diventa mandatorio. Indipendentemente dallo stato mutazionale per i melanomi mucosali è disponibile il trattamento con immunoterapia.
Per quanto riguarda invece il melanoma dell’uvea, dal punto di vista molecolare, è distinto da quello cutaneo per la pressoché generale assenza di mutazioni nei geni BRAF, NRAS, c-KIT e NF1. Questo tipo di melanoma è caratterizzato dalla presenza di mutazioni nei geni GNAQ (frequenza del 28-50%) e GNA11 (frequenza del 32-50%), che codificano due subunità di proteine G (G/q alpha subunits), capaci di attivare la via MAPK e sono mutualmente esclusive. Altri eventi mutazionali sono rappresentati da monosomie del cromosoma 3, amplificazione del cromosoma 8q e, soprattutto, inattivazione del gene BAP1 (frequenza del 18-45%).15 Le mutazioni di GNAQ e GNA11 sono considerate eventi precoci nello sviluppo della malattia. Invece, le mutazioni somatiche di BAP1 sembrano caratterizzare la fase di progressione di malattia, essendo più frequentemente associate alla formazione di metastasi. Tuttavia, le mutazioni di BAP1 possono essere presenti anche a livello germinale, conferendo una predisposizione a particolari sindromi neoplastiche (aumentato rischio di tumori diversi, incluso il melanoma dell’uvea e di altre sedi). Al momento, la determinazione dell’assetto mutazionale, al di fuori della possibile, seppur rara, evidenza di una mutazione BRAF V600 che si consiglia comunque di determinare, non riveste un ruolo nella scelta terapeutica per questa malattia. Non sono infatti disponibili farmaci a bersaglio molecolare per le mutazioni più frequenti nel melanoma uveale; da segnalare comunque che alcuni studi hanno valutato la potenziale attività di un trattamento con inibitori multichinasici (sunitinib) o MEK inibitori (selumetinib) in questa malattia. Il melanoma congiuntivale si comporta invece come i melanomi che insorgono su cute non cronicamente fotoesposta, con frequenza di mutazioni in BRAF del 40% circa.
Lo stato mutazionale di BRAF può essere valutato mediante metodiche aventi diverso grado di sensibilità e specificità. L’analisi mutazionale può essere infatti affrontata tramite metodiche di screening a livello proteico, quali il test immunoistochimico, e metodiche di tipo molecolare su DNA genomico, quali: il sequenziamento nucleotidico mediante approccio convenzionale basato su metodo Sanger, il pirosequenziamento, la spettrometria di massa (Sequenom), la Real-Time PCR e mediante un approccio innovativo di Next-Generation Sequencing (NGS).
Basandosi sulle recenti linee guida ESMO, il test mirato a identificare un’eventuale mutazione di BRAF è obbligatorio nei pazienti con melanoma resecabile o non resecabile in stadio III o IV, ed è altamente raccomandato in pazienti con malattia resecata ad alto rischio in stadio IIC.16 Nell’ambito della malattia metastatica, si consiglia inoltre di eseguire analisi molecolari su campioni tessutali metastatici, se disponibili, in quanto essi sono rappresentativi della lesione più
recente e sono spesso composti da una vasta maggioranza di cellule neoplastiche. Quando il campione di tessuto metastatico non è disponibile, l’analisi può comunque essere eseguita su campioni ottenuti anche dal tumore primitivo a causa di un’elevata concordanza dello stato mutazionale BRAF tra i melanomi primari e le loro lesioni metastatiche.17 È opportuno sottolineare che le informazioni sullo stato mutazionale di BRAF devono essere fornite tempestivamente al fine di non provocare ritardi nell’avviamento della terapia.
Immunoistochimica
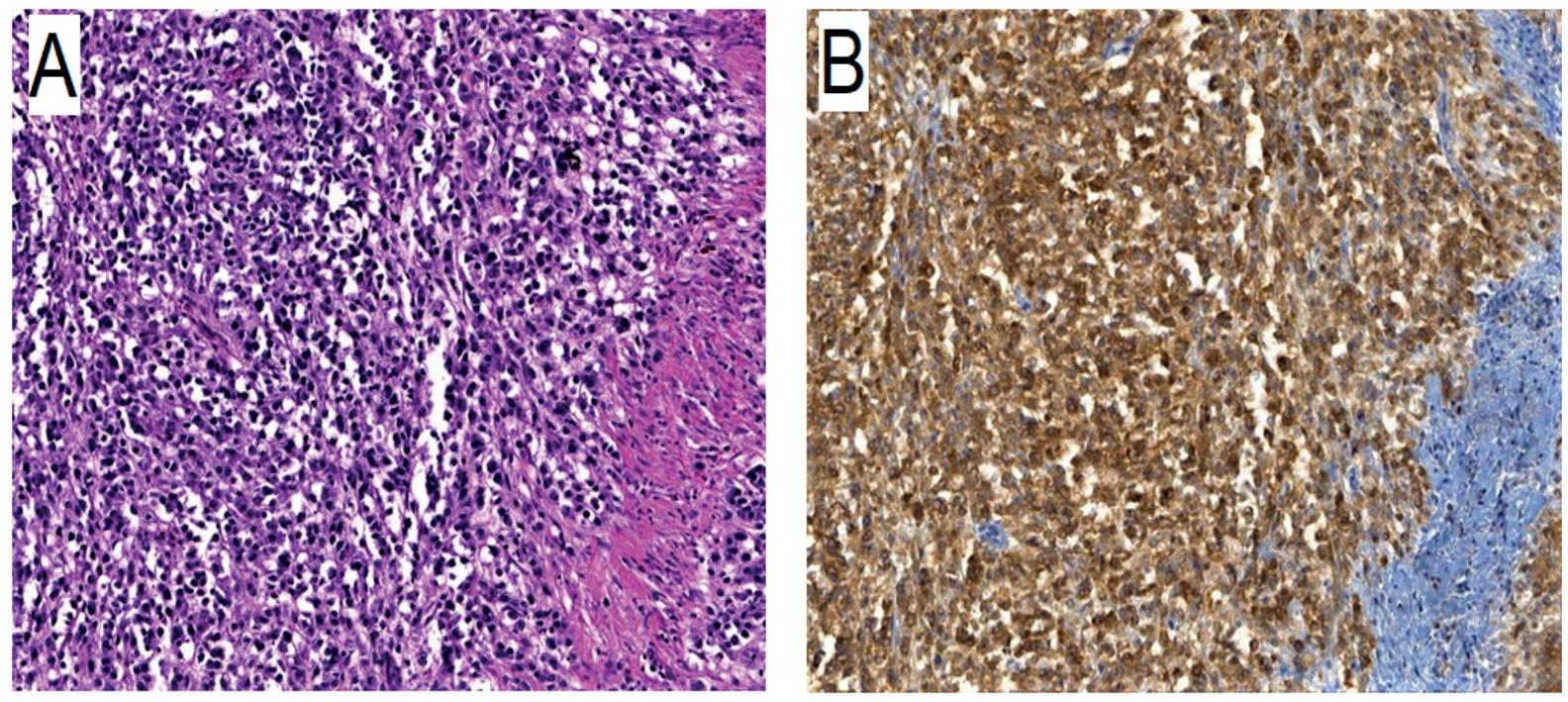
L’immunoistochimica (IHC) si configura come un’opzione affidabile per valutare la mutazione BRAF nell’esone 15 p.V600E nei pazienti con melanoma. Nell’indagine è comunemente impiegato l’anticorpo monoclonale VE1, che mostra una colorazione tipicamente citoplasmatica.18 I principali vantaggi dell’IHC sono associati alla sua semplicità, ai bassi costi, alla sua rapidità di esecuzione, alla sensibilità e specificità relativamente elevate e alla possibilità di valutare la distribuzione delle proteine mutanti a livello di una singola cellula.19 D’altra parte, i suoi principali svantaggi sono correlati alla possibilità di risultati falsi negativi dovuti all’eterogeneità o alla bassa concentrazione dell’esone 15 p.V600E di BRAF e all’impossibilità di identificare l’esone BRAF 15 p.V600K o altre varianti.19 Confrontando i risultati ottenuti dal clone VE1 e da un approccio basato sulla PCR, è stata raggiunta una concordanza complessiva dell’88% tra le due metodiche.20 Long et al hanno riportato alti tassi di sensibilità e specificità (rispettivamente 97% e 98%) confrontando il clone VE1 in IHC con un approccio basato sul DNA che suggerisce la possibilità di adottare l’IHC per lo screening dei pazienti con melanoma avanzato.21 Sensibilità e specificità simili sono state osservate per l’IHC quando i risultati sono stati confrontati con il pirosequenziamento (85% e 100%, rispettivamente) e approcci basati sulla PCR (98,6% e 97,7%).22 23 Generalmente, sono sottoposti ad analisi mutazionale campioni di tessuto fissati in formalina e inclusi in paraffina, previa sparaffinatura e purificazione del DNA genomico mediante protocolli standardizzati. Fondamentale è l’arricchimento del campione tessutale, considerando inoltre che la percentuale di cellule neoplastiche presenti nel tessuto da inviare all’analisi molecolare non dovrebbe mai essere inferiore al 50%. In caso di melanoma associato a nevo è cruciale che nella fase di arricchimento del campione sia posta attenzione nell’isolare una popolazione pura di cellule di melanoma, in quanto i nevi melanocitici possono essere portatori di mutazioni nel gene BRAF, con la stessa frequenza riscontrata nei melanomi. Dati recenti indicano l’appropriatezza della combinazione di una valutazione con immunoistochimica per il riconoscimento della mutazione BRAF V600E, quale metodo di screening rapido e affidabile, seguita da un test mutazionale di tipo molecolare, che rimane la metodica standard di riferimento in grado di fornire il massimo livello di sensibilità e specificità nell’identificazione dei casi mutati.40 Un esempio di campione istologi-
co colorato con ematossilina-eosina e la rispettiva reazione immunoistochimica ottenuta con l’utilizzo di un anticorpo monoclonale anti-BRAF (VE1), sono mostrate nella figura 2.2. Nel caso di discordanza tra le due metodiche utilizzate, viene effettuata un’ulteriore analisi con una metodica molecolare aggiuntiva.
Metodiche molecolari
Il sequenziamento Sanger è stato a lungo considerato il metodo di riferimento per l’identificazione delle mutazioni acquisite nei tumori. Questa metodica richiede un’alta percentuale di cellule tumorali all’interno dei campioni, requisito non sempre possibile da soddisfare nei test diagnostici di routine. In questo contesto, i patologi svolgono un ruolo fondamentale nel triage di campioni di melanoma da affidare ai test molecolari poiché devono stimare la cellularità del tumore all’interno della regione designata, al fine di ottenere materiale sufficiente a garantire la corretta sensibilità analitica del test molecolare richiesto, e al contempo cerchiare su vetrino l’area da sottoporre alle procedure di laboratorio, qualora nello stesso campione coesistano aree di lesione e zone fibrotiche o necrotiche. Per questo motivo può essere consigliabile eseguire la macro-dissezione da parte del patologo prima del test di valutazione dell’assetto mutazionale, quando la percentuale di cellule tumorali è <50%. Inoltre, la sensibilità del sequenziamento Sanger per il rilevamento della mutazione BRAF V600E è del 92,5%;18 ciò significa che, utilizzando questa tecnica da sola, il 7,5% dei pazienti potenzialmente eleggibili per il trattamento con inibitori di BRAF e MEK andrebbe perso.
Il pirosequenziamento è un metodo enzimatico completamente automatizzato, che permette il sequenziamento della catena complementare; differisce dal meto-
do Sanger per la capacità di identificare il rilascio di pirofosfato e la generazione di luce corrispondente all’incorporazione nucleotidica e non alla terminazione di catena con l’impiego di dideossiribonucleotidi. Questa tecnica è comunemente impiegata per rilevare mutazioni BRAF e numerosi sono i kit presenti in commercio a questo scopo. La metodica non è in grado di identificare accuratamente varianti all’interno di omopolimeri di più di 5-6 bp e può generare pattern difficili da interpretare e risolvere senza ulteriori indagini, nel caso di mutazioni complesse.18
Il pirosequenziamento permette l’analisi di polimorfismi di singoli nucleotidi o mutazioni puntiformi in una singola corsa, dato che la lunghezza della sequenza è usualmente <200 bp. Tuttavia, questa metodica presenta una maggiore sensibilità (limite di rilevazione pari a 5-8%) e una elevata copertura mutazionale rispetto all’ormai subottimale metodo Sanger (specificità pari a 90%; Near Comprehensive), presentandosi inoltre come un test di più rapida esecuzione.18
La spettrometria di massa (Sequenom) consente la genotipizzazione multiplex ed è un metodo sensibile, affidabile, veloce e conveniente. Il principio generale di MALDI-TOF MS si basa sull’amplificazione del DNA mediante PCR, risultante in copie sia degli alleli mutanti sia wild-type. Essa offre netti vantaggi rispetto al metodo Sanger sia in termini di sensibilità, con un limite di rilevazione ridotto al 2,5-7,5%, sia in termini di spettro d’azione, essendo in grado di rilevare un numero più ampio di mutazioni puntiformi nel gene BRAF. Sequenom ha ottimizzato le reazioni di estensione dei primer utilizzando terminatori di base aciclici modificati in massa che raggiungono spazi di almeno 16 Dalton tra le quattro possibili basi incorporate nell’estensione di una singola base. Questo approccio innovativo ha portato all’implementazione di MALDI-TOF nella patologia molecolare predittiva di routine come metodo genotipico basato su polimorfismi a singolo nucleotide (SNP) ad alto rendimento, rappresentando una tecnica a ponte tra metodi convenzionali, come il sequenziamento di Sanger, e metodi innovativi come RT-PCR e Next-Generation Sequencing (NGS) .
La Real-Time PCR, come regola generale, adotta un set di primer che mirano specificamente alle mutazioni BRAF e un altro in grado di identificare la sequenza wild-type. Ad oggi, il test di mutazione cobas® 4800 BRAF V600 e il kit THxIDBRAF hanno ottenuto l’approvazione della Food and Drug Administration (FDA) in pazienti affetti da melanoma per l’individuazione di mutazioni dell’esone 15 p.V600 di BRAF al fine di somministrare trattamenti mirati in pazienti con melanoma in stadio avanzato. Nell’esperienza di Anderson et al., il test di mutazione cobas® 4800 BRAF V600 è stato confrontato con il sequenziamento Sanger in 477 campioni di melanoma. Nel complesso, tutti i casi sono stati testati con successo con il test di mutazione cobas® 4800 BRAF V600, mentre per il sequenziamento Sanger è stato riportato un tasso di fallimento del 9,2%.24 Mourah et al. hanno confrontato i risultati ottenuti utilizzando il test di mutazione cobas® 4800 BRAF V600 con varie altre tecniche (tra cui sequenziamento Sanger, pirosequenziamento, PCR allele-specifica, SNaPshot e analisi di fusione ad alta risoluzione
(HRM)). Nel complesso, la frequenza delle mutazioni BRAF nell’esone 15 p.V600 rilevate è stata leggermente superiore per le altre tecniche rispetto al test di mutazione cobas® 4800 BRAF V600 (35,7% vs 34,0%, rispettivamente), mentre i casi wild-type sono stati rilevati con una frequenza più alta con il test di mutazione cobas® 4800 BRAF V600 (63,3% vs. 62,9%).25 Questi dati confermano l’elevata efficacia clinica del test di mutazione cobas® 4800 BRAF V600. La Real-Time PCR presenta infatti la più elevata sensibilità (limite di rilevazione pari a 2-3%) ma riesce a identificare un numero limitato di mutazioni nelle regioni geniche di interesse, dipendentemente dal set di primers utilizzati nella PCR (specificità variabile tra 90% e 98%). Un esempio di curva di amplificazione relativa alla rilevazione di mutazione BRAF V600E con Real-Time PCR è mostrato nella figura 2.3.
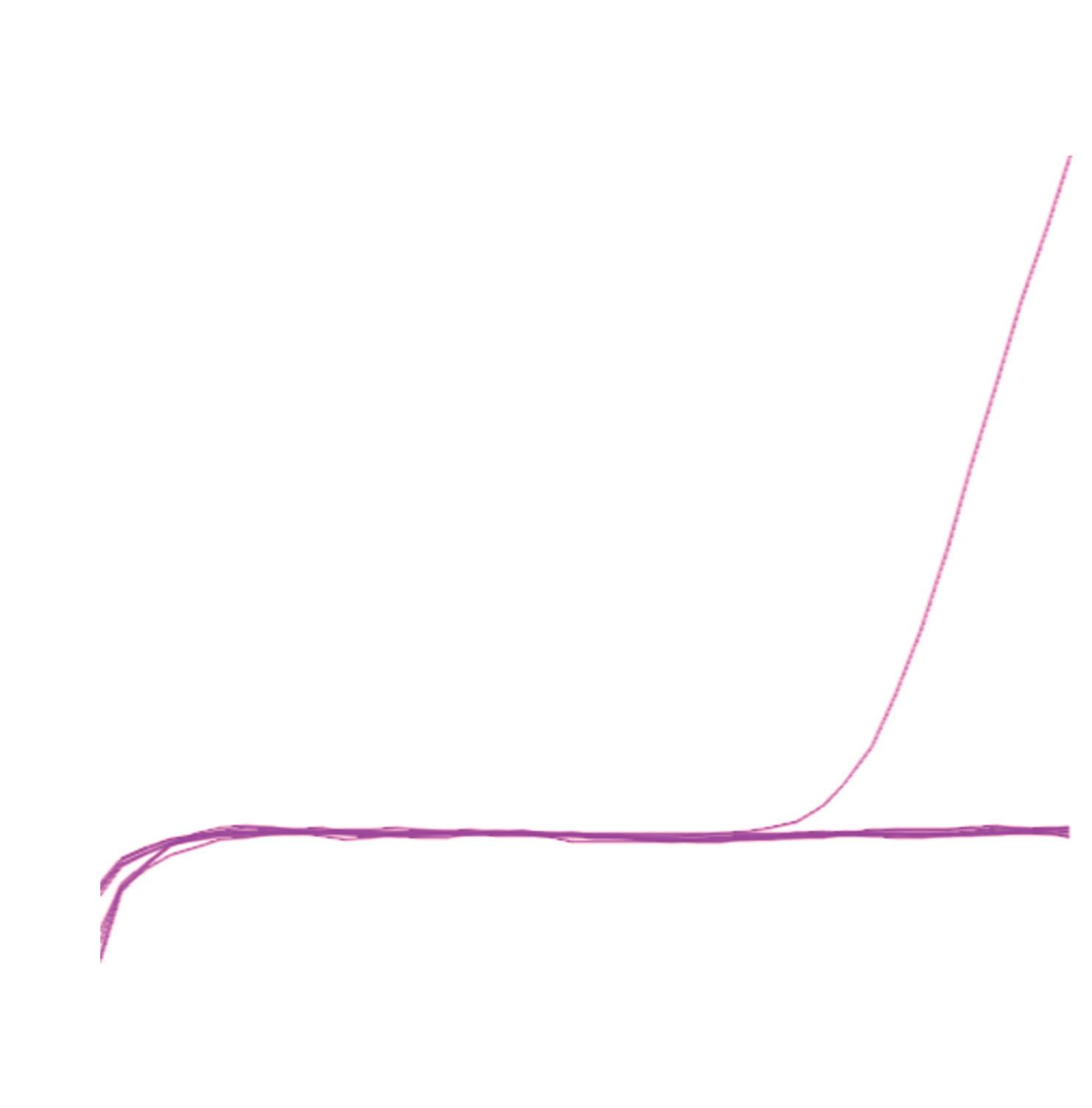
Figura 2.3
Curva esemplificativa di positività per mutazione genica BRAF V600E al test molecolare in Real-Time PCR
Il sequenziamento mediante Next-Generation Sequencing (NGS) è una tecnologia affascinante in grado di analizzare più mutazioni per diversi pazienti, contemporaneamente.26 Sebbene siano attualmente disponibili in commercio piattaforme diverse, l’iter di valutazione dello stato mutazionale tramite NGS è comunque caratterizzato da quattro fasi sequenziali: generazione di librerie, amplificazione clonale, sequenziamento parallelo massiccio e analisi dei dati.27 A causa dei costi più elevati e dei tempi di consegna della risposta più lunghi, Zhu et al. hanno suggerito di adottare NGS per melanomi avanzati che mostrano un risultato negativo con la PCR BRAF allele-specifica 15 p.V600E/K.28
NGS è generalmente basato sull’impiego di pannelli multigenici che consentono il rilevamento simultaneo di migliaia di varianti nei principali geni candidati, utilizzando quantità limitate di DNA, con una elevata sensibilità diagnostica (limite di rilevazione pari a 1-2%) e con la capacità di identificare tutte le mutazioni presenti nelle regioni genomiche analizzate (specificità pari a 100%; Comprehensive). NGS fornisce molte più informazioni genetiche rispetto alle metodiche precedentemente descritte: oltre alle mutazioni puntiformi, questa tecnica consente la rilevazione di ulteriori tipi di varianti e un’analisi simultanea di più geni. L’importanza dell’analisi multi-genetica nel melanoma è prevista in aumento, dato che maggiori saranno le mutazioni oncogeniche scoperte e, conseguentemente i trattamenti farmacologici disponibili. La tecnica di NGS è più sensibile anche quando il DNA del tumore rappresenta meno del 10% del DNA totale. In uno studio che confrontava metodi diversi, NGS aveva il 100% specificità e sensibilità del 99%, con la capacità emersa di fornire un’attendibile valutazione anche in campioni con tessuto tumorale limitato.29 Recentemente, 21 campioni di melanoma sono stati analizzati per confrontare le prestazioni di NGS con il sistema di spettrometria di massa Sequenom, con il sequenziamento Sanger e con la Real-Time PCR allele-specifica. NGS ha identificato correttamente due mutazioni a frequenza molto bassa in BRAF sfuggite sia alla spettrometria di massa che al sequenziamento Sanger, confermando l’elevata sensibilità del metodo NGS.30 D’altra parte, NGS richiede adeguata formazione ed elevate competenze nell’analisi dei dati e nella loro applicazione nel contesto clinico, necessita di una catena di lavoro strettamente organizzata per ottenere un sequenziamento affidabile, ha bisogno di più tempo ed è più costoso rispetto alla maggior parte delle altre tecniche molecolari.
Algoritmo diagnostico
Nel complesso, nella diagnostica quotidiana e nella pratica clinica, può essere suggerita un’analisi sequenziale condotta attraverso l’utilizzo di due metodi, con rilevamento iniziale di mutazioni BRAF all’immunoistochimica, insieme a una positività rilevata con una tecnica molecolare, come Real-Time PCR o NGS. In effetti, l’uso della sola immunoistochimica comporterebbe un rischio significa-
tivo di risultati falsi negativi e non dovrebbe essere considerato a meno che la resa del DNA del campione non sia sufficiente per condurre l’analisi molecolare. D’altra parte, un risultato positivo di una tecnica molecolare, seppur sensibile, può riflettere la presenza di un subclone minore di BRAF mutato in maniera predominante in un tumore wild-type, e questo non è quindi clinicamente rilevante in termini di risposta alla terapia mirata.
La biopsia liquida è una tecnica diagnostica non invasiva per la valutazione dello stato genetico del tumore basata sull’analisi di DNA libero circolante (cellfree DNA, cfDNA) da diversi fluidi corporei, come plasma e siero. Poiché i campioni di sangue sono facilmente ottenibili, plasma o siero-biopsia sono state a lungo considerate come promettenti metodi non invasivi da integrare nelle tradizionali tecniche di biopsia per l’analisi molecolare. Un modo attraverso cui i tumori forniscono informazioni sotto forma di biomarcatori, come il DNA tumorale circolante (circulating-tumour DNA, ctDNA), si verifica mediante la necrosi della cellula tumorale con successivo rilascio di cellule morte o detriti cellulari. Successivamente, i fagociti incorporano queste cellule che vengono processate ed elaborate, per poi rilasciare il ctDNA nel sangue sotto forma di piccoli frammenti. Il ctDNA consiste in cfDNA che viene rilasciato dalle cellule tumorali ed esso può quindi essere analizzato per la ricerca delle stesse alterazioni genetiche riscontrate nel tumore.31-34 Oltre al ctDNA, le cellule circolanti del tumore (CTC) intatte o gli esosomi, ovvero vescicole di membrana extracellulari con una dimensione compresa tra 40 e 100 nm, possono essere anch’esse utilizzate come fonte di DNA, RNA e proteine tumorali da valutare previa esecuzione di prelievo per biopsia liquida.
Uno studio35 mirato a dimostrare la fattibilità dei due diversi approcci tecnici per rilevare la mutazione BRAF V600E nella biopsia liquida è stato eseguito su una casistica di 20 pazienti con melanoma metastatico utilizzando sia la frazione CTC arricchita che il cfDNA circolante proveniente dallo stesso campione di sangue. È stata rilevata con un metodo di filtrazione la variante BRAF V600E su CTC e cfDNA mediante un saggio qPCR allele-specifico; i campioni mutati sono stati confermati mediante PCR ICE-COLD seguita da sequenziamento Sanger. Sono state riscontrate CTC nel 70% dei campioni e identificata la variante BRAF V600E. I risultati sono stati correlati con quelli ottenuti su cfDNA dallo stesso prelievo di sangue. Sono così emersi alcuni risultati discordanti tra CTC e cfDNA, a sottolineare l’importanza di indagare sia le CTC che il cfDNA nell’approccio con biopsia liquida in pazienti con melanoma.35
L’uso della biopsia liquida nella diagnostica di routine nel melanoma rimane attualmente frenato a causa dei seguenti limiti, emersi da un altro studio:36
barriere biologiche e tecnologiche, che portano a una lievitazione dei costi; dati insufficienti provenienti da studi clinici prospettici randomizzati che possano dimostrare la superiorità rispetto alle tecniche standard di rilevazione; l’origine ancora poco chiara di CTC/ctDNA (primario, metastatico, o lesione precancerosa); eterogeneità clonale, ad esempio il CTC/ctDNA rilevato potrebbe non rappresentare il clone tumorale predominante, quindi la progressione del clone principale potrebbe essere trascurata e, infine, l’utilizzo di altri campioni clinici prontamente disponibili, come saliva, espettorato, urina e campioni di feci potrebbero portare a campioni identici o forse risultati superiori rispetto a quelli ottenuti tramite rilevamento CTC/ctDNA nel sangue.
Mutazioni rare
Nel corso degli anni, oltre alla più classica V600E, sono state descritte varianti mutazionali rare di BRAF V600; ad esempio V600K, V600R e V600M che hanno dimostrato di essere associate a caratteristiche clinico-patologiche distinte, comprese differenze nella distribuzione per età (tassi più elevati nei pazienti anziani), localizzazione (tassi più elevati di presentazione a livello del distretto di testa e collo) e a peggiore sopravvivenza libera da metastasi a distanza.37 A seconda dello studio, tra il 6% e il 30% di tutte le mutazioni di BRAF è stato descritto come distinto dal genotipo V600E. Lovly et al.38 in uno studio condotto su 150 casi di melanoma hanno rilevato che tra le mutazioni BRAF V600, il 79%, il 12%, il 5% e il 4% erano rispettivamente V600E, V600K, V600R e V600M.
A causa dei progressi metodologici raggiunti nel corso degli ultimi anni, sono state inoltre rilevate sempre più mutazioni BRAF non-V600, oltre a fusioni coinvolgenti BRAF. Poiché queste mutazioni sono responsabili solo di una minoranza dei casi segnalati, dipendiamo strettamente dai case report per la conoscenza di nuove mutazioni, poiché ampi studi su questo argomento non esistono ancora, pertanto risulta urgentemente necessario ampliare la conoscenza del panorama dello stato mutazionale di BRAF per determinare la strategia ottimale di trattamento in tali pazienti.
Al fine di facilitare le opzioni di trattamento individualizzato per i melanomi con mutazioni rare, è importante attribuire un’alta priorità alla progettazione e allo sviluppo di pannelli per test molecolari diversi dal sequenziamento dell’intero esoma (whole exome sequencing, WES), ma in modo tale da includere almeno gli esoni 11 e 15 del gene BRAF.
Un utile elemento procedurale per caratterizzare le mutazioni rare di BRAF risulta nella loro classificazione37 in base al grado di attività chinasica indotta dall’evento mutazionale; è possibile così distinguere mutazioni ad alta, bassa e silente attività (dead-activity). Le prime sono sicuramente quelle di maggiore interesse, per la loro più alta probabilità di risposta al trattamento con MEK/RAF-inibitori.
Le mutazioni BRAF non-v600 ad alta attività fino ad oggi più frequentemente segnalate comprendono BRAF K601E39, BRAF L597Q/R39, BRAF E586K39 e BRAF G469A.39
Inoltre, la co-occorrenza di mutazioni BRAF non-V600 a bassa attività con mutazioni NRAS suggerisce che ogni paziente con una mutazione nel gene RAS dovrebbe essere testato per mutazioni di BRAF e viceversa. Se le chinasi BRAF a bassa attività rispondessero al trattamento con MEK/RAF-inibitori, le mutazioni rare di BRAF avrebbero la necessità di essere valutate in studi clinici su più ampia scala al fine di fornire migliori opzioni terapeutiche per i pazienti con mutazioni BRAF non-V600.
Infine, i pazienti con melanoma pan-negativo dovrebbero essere testati per la presenza di fusioni BRAF poiché potrebbero rispondere all’inibizione del MEK.40
L’identificazione, avvenuta fin dai primi Duemila, di mutazioni puntiformi di BRAF in campioni di melanoma, ha contribuito a rendere questa neoplasia un campo fondamentale di sperimentazione di tecniche sempre più attendibili per una rapida identificazione anche di altri tumori solidi BRAF-mutati. Sulla base di questo presupposto è possibile quindi affermare il ruolo paradigmatico del melanoma e di come esso possa essere applicato nella valutazione di altri tumori solidi. La costituzione di un algoritmo diagnostico affidabile e altamente riproducibile, passante per uno screening con immunoistochimica, da proseguire tramite approfondimenti molecolari dotati di alta specificità, basati sull’analisi del DNA genomico, rappresenta infatti un modello in grado di ampliare in maniera sistematica la conoscenza dello spettro di neoplasie solide che esprimono mutazioni nel gene BRAF. Partendo dall’identificazione di mutazioni BRAF nel melanoma, l’obiettivo della medicina di precisione in ambito oncologico risiede nell’acquisizione su scala genomica di un numero sempre più ampio di tumori mutanti in BRAF e della loro relazione con l’eventuale presenza di coesistenti instabilità a carico di altri geni. La completa comprensione della biologia dei tumori è diventata pertanto un prerequisito cruciale per prevedere la traiettoria evolutiva dalla neoplasia e la risposta alla terapia a bersaglio molecolare così come per l’implementazione di nuove terapie antitumorali di successo.
1. Cossu A, Casula M, Cesaraccio R, et al. Epidemiology and genetic susceptibility of malignant melanoma in North Sardinia, Italy. Eur J Cancer Prev 2017; 26: 263-7.
2. SEER Cancer statistics review 1975-2015.
3. Hung CY, Chang JWC. Chemotherapy and biochemotherapy for melanoma. In: Riker A, ed. Melanoma. A modern multidisciplinary approach. Basel: Springer International Publishing AG, 2018; 525-31.
4. Kim HS, Minna JD, White MA. GWAS meets TCGA to illuminate mechanisms of cancer predisposition. Cell 2013; 152: 387-9.
5. Desideri E, Cavallo AL, Baccarini M. Alike but different: RAF paralogs and their signalling outputs. Cell 2015; 161: 967-70.
6. Mark GE, Rapp UR. Primary structure of v-raf: relatedness to the src family of oncogenes. Science 1984; 224: 285-9.
7. McKay MM, Freeman AK, Morrison DK. Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small GTPases 2011; 2: 276-81.
8. Odabaei G, Chatterjee D, Jazirehi AR, et al. Raf-1 kinase inhibitor protein: structure, function, regulation of cell signalling, and pivotal role in apoptosis. Adv. Cancer Res 2004; 91: 169-200.
9. Bivona TG. Dampening oncogenic RAS signalling. Science 2019; 363: 1280-1.
10. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 2011; 29: 1239-46.
11. Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol 2011; 24: 426-33.
12. Hugdahl E, Kalvenes MB, Puntervoll HE, et al. BRAFV600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br J Cancer 2016; 114: 801-8.
13. Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet 2003; 33: 19-20.
14. Dummer R, Hauschild A, Lindenblatt N, et al. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment, and follow-up. Ann Oncol 2017; 26: v126-v132.
15. Harbour JW. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res 2012; 25(2): 171-81.
16. Michielin O, van Akkooi ACJ, Ascierto PA, et al. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2019; 30: 1884-901.
17. Colombino M, Capone M, Lissia A, et al. BRAF/ NRAS mutation frequencies among primary tumours and metastases in patients with melanoma. J Clin Oncol 2012; 30: 2522-9.
18. Eriksson H, Zebary A, Vassilaki I, et al. BRAFV600E protein expression in primary cutaneous malignant melanomas and paired metastases. JAMA Dermatol 2015; 151: 410-6.
19. Colomba E, Hélias-Rodzewicz Z, Von Deimling A, et al. Detection of BRAF p.V600E mutations in melanomas:
comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn 2013; 15: 94-100.
20. Hugdahl E, Kalvenes MB, Puntervoll HE, Ladstein RG, Akslen LA. BRAF-V600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br J Cancer 2016; 114: 801-8.
21. Long GV, Wilmott JS, Capper D, et al. Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am J Surg Pathol 2013; 37: 61-5.
22. Pearlstein MV, Zedek DC, Ollila DW, et al. Validation of the VE1 immunostain for the BRAF V600E mutation in melanoma. J Cutan Pathol 2014; 41: 724-32.
23. Manfredi L, Meyer N, Tournier E, et al. Highly Concordant results between immunohistochemistry and molecular testing of mutated V600E BRAF in primary and metastatic melanoma. Acta Derm Venereol 2016; 96: 630-4.
24. Anderson S, Bloom KJ, Vallera DU, et al. Multisite analytic performance studies of a real-time polymerase chain reaction assay for the detection of BRAF V600E mutations in formalin-fixed, paraffin-embedded tissue specimens of malignant melanoma. Arch Pathol Lab Med 2012; 136: 1385-91.
25. Mourah S, Denis MG, Narducci FE, et al. Detection of BRAF V600 mutations in melanoma: evaluation of concordance between the Cobas® 4800 BRAF V600 mutation test and the methods used in French National Cancer Institute
(INCa) platforms in a real-life setting. PLoS One 2015; 10(3): e0120232.
26. Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011; 475: 348-52.
27. Vigliar E, Malapelle U, De Luca C, Bellevicine C, Troncone G. Challenges and opportunities of nextgeneration sequencing: a cytopathologist’s perspective. Cytopathology 2015; 26: 271-83.
28. Zhu ML, Zhou L, Sadri N. Comparison of targeted next generation sequencing (NGS) versus isolated BRAF V600E analysis in patients with metastatic melanoma. Virchows Arch 2018; 473: 371-7.
29. Ihle MA, Fassunke J, König K, et al. Comparison of high-resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer 2014; 14: 13.
30. Mancini I, Simi L, Salvianti F, et al. Analytical evaluation of an NGS testing method for
routine molecular diagnostics on melanoma formalin-fixed, paraffin-embedded tumourderived DNA. Diagnostics 2019; 9: 117.
31. Lipson EJ, Velculescu VE, Pritchard TS, et al. Circulating tumour DNA analysis as a realtime method for monitoring tumour burden in melanoma patients undergoing treatment with immune checkpoint blockade. J Immunother Cancer 2014; 2: 42.
32. Knol AC, Vallée A, Herbreteau G, et al. Clinical significance of BRAF mutation status in circulating tumour DNA of metastatic melanoma patients at baseline. Exp Dermatol 2016; 25: 783-8.
33. Gangadhar TC, Savitch SL, Yee SS, et al. Feasibility of monitoring advanced melanoma patients using cell-free DNA from plasma. Pigment Cell Melanoma Res 2018; 31: 73-81.
34. Long-Mira E, Ilie M, Chamorey E, et al. Monitoring BRAF and NRAS mutations with cell-free circulating tumour DNA from metastatic melanoma patients. Oncotarget 2018; 9: 36238-49.
35. Salvianti F, Massi D, De Giorgi V, et al. Evaluation of the liquid biopsy for the detection of BRAFV600E mutation
in metastatic melanoma patients. Cancer Biomark 2019; 26(3): 271-9.
36. Gaiser MR, von Bubnoff N, Gebhardt C, Utikal JS. Liquid biopsy to monitor melanoma patients. J Dtsch Dermatol Ges 2018; 16(4): 405-14.
37. Menzies AM, Haydu LE, Visintin L, et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res 2012; 18 (12): 3242-9.
38. Zheng G, Tseng L-H, Chen G, et al. Clinical detection and categorization of uncommon and concomitant mutations involving BRAF. BMC Cancer 2015; 15: 779.
39. Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrolment in genotype-driven therapeutic trials. PLoS ONE 2012; 74: e35309.
40. Richtig G, Hoeller C, Kashofer K, et al. Beyond the BRAFV600E hotspot: biology and clinical implications of rare BRAF gene mutations in melanoma patients. Br J Dermatol 2017; 177(4): 936-44.
PAOLO ANTONIO ASCIERTO, LUCIA FESTINO
Il melanoma è al secondo posto tra i tumori in cui più frequentemente si osservano mutazioni BRAF (60% dei casi), preceduto solo dalla leucemia a cellule capellute (100%). La frequenza delle mutazioni di BRAF nel melanoma varia in base a vari fattori, in particolare il sottotipo istologico, la posizione anatomica del tumore e il modello di esposizione al sole. Più specificamente, oltre la metà dei melanomi a diffusione superficiale è mutata in BRAF, mentre tra i melanomi nodulari la frequenza è del 43%, del 15% tra gli acrali lentigginosi e del 14% nella lentigo maligna. È stato osservato che le mutazioni di BRAF sono molto meno frequenti nei melanomi mucosali (6%), e sono essenzialmente assenti nei melanomi uveali. 1 Nel melanoma sono state descritte più di 20 mutazioni di BRAF; tra queste, la mutazione BRAF V600E è stata identificata in circa il 50% dei melanomi e rappresenta il 90% di tutte le mutazioni BRAF note. Altre mutazioni di BRAF si verificano spesso nello stesso codone. In particolare, la sostituzione della lisina con la valina (V600K) è la seconda più frequente e rappresenta circa il 5-6% dei casi. Altre mutazioni osservate nei melanomi sono la p.V600K (con una prevalenza del 7,7%), la p.V600R (1%), la p.V600M (0,3%) e la p.V600D (0,1%), che risultano tutti in mutanti ad alta attività (tabella 3.1). 2 Le mutazioni si verificano all’interno del sito di attivazione nel CR3 e promuovono la conformazione attiva della proteina. Il BRAF V600 mutato è in grado di attivare a sua volta la via MAPK a valle, indipendentemente dall’attivazione RAS a monte, attraverso la fosforilazione diretta di MEK, determinando un aumento della trascrizione dei geni per fattori di trascrizione, l’upregolazione dei segnali pro-sopravvivenza e l’evasione della risposta immunitaria.
Tabella 3.1
Mutazioni di BRAF [80,81]
Classe Tipo di mutazione Frequenza nel melanoma1 2
I V600E 20,38-79%
I V600K 2,95-12%
I V600R 0,43-5%
I V600M 0,25-4%
I V600E2 NR (<1%)
I V600D NR (<1%)
II K601E 0,71%
II L597R/Q/S 0,25-0,18-0,28%
II G464E/V/R NR (<1%)
II G469A/V/S
III D594
Vemurafenib
NR (<1%)
NR (<1%)
Vemurafenib è un inibitore di tipo I competitivo, a basso peso molecolare, della serina/treonina chinasi che funziona legandosi al dominio di legame ATP dei mutanti V600E, V600D e V600R BRAF.
Lo studio di fase I BRIM1 è stato svolto per valutare il ruolo di vemurafenib nel melanoma in stadio avanzato con mutazione BRAF V600. Gli obiettivi dello studio erano di valutare la farmacocinetica e la sicurezza dei farmaci e di caratterizzare l’efficacia clinica precoce dei farmaci attraverso la valutazione del tasso di risposta, durata e fallimento del tumore. Sono stati arruolati in totale 55 pazienti. Nella fase di escalation è stata stabilita la dose massima di 960 mg due volte al giorno. Il tasso di risposta globale (ORR) nella fase di aumento della dose è stato del 69%, mentre la durata delle risposte variava da 2 mesi a più di 18 mesi. Nella fase di estensione l’81% dei pazienti ha avuto una risposta globale con sopravvivenza libera da progressione mediana superiore a 7 mesi.
I più comuni effetti collaterali di grado 2 o 3 sono stati artralgie, rash, nausea, fotosensibilità, affaticamento, carcinoma cutaneo a cellule squamose, prurito e disestesia palmo-plantare. Già in questo primo studio sono stati evidenziati effetti collaterali di tipo iperproliferativo, che sono poi stati confermati in tutti gli studi con BRAFi in monoterapia. Essi vanno da ipercheratosi e cheratoacantomi
BRAF come bersaglio terapeutico nel melanoma 37
(KA) a carcinomi a cellule squamose (SCC) e nuovi melanomi primari e sono dovuti alla iperattivazione paradossa del pathway di MAPK che si verifica nelle cellule BRAF wt che presentano iperattivazione di RAS, dovuta a eccessiva stimolazione ligando-dipendente o alla presenza di mutazioni attivanti di RAS. In tali condizioni, l’inibizione di BRAF comporta la formazione di dimeri BRAF-CRAF portando all’attivazione paradossale di MAPK attraverso la transattivazione del protomero CRAF non inibito.4
Nello studio BRIM2 sono stati arruolati 132 pazienti con melanoma metastatico, inclusi pazienti con metastasi cerebrali. LA ORR ottenuta è stata del 53%, mentre il 6% dei pazienti ha ottenuto una risposta completa (CR). La durata mediana della risposta è stata di 6,7 mesi e la sopravvivenza libera da progressione mediana (mPFS) è stata di 6,8 mesi. Il profilo di tossicità era sovrapponibile a quello evidenziato nello studio di fase I. Nel corso dello studio è emerso inoltre il valore prognostico negativo di livelli di LDH riscontrati al basale. Infatti, nei pazienti con LDH elevato, la risposta complessiva era solo del 33%.5
Lo studio internazionale multicentrico di fase III BRIM3 ha confrontato l’efficacia clinica di vemurafenib rispetto alla dacarbazina. Lo studio ha arruolato e randomizzato 680 pazienti nei bracci di trattamento con vemurafenib o dacarbazina. La sopravvivenza globale a 6 mesi è stata valutata per 672 dei pazienti trattati e ha rivelato un aumento della sopravvivenza con vemurafenib (84%) rispetto alla dacarbazina (64%). La mPFS è stata di 5,3 mesi nel gruppo vemurafenib e 1,6 mesi nel gruppo dacarbazina. La differenza nella sopravvivenza globale (OS) tra i due bracci di trattamento ha raggiunto un significato molto elevato già durante l’analisi intermedia predefinita; pertanto, lo studio è stato modificato per consentire ai pazienti con DTIC di passare al trattamento con vemurafenib. Al momento del crossover, lo studio ha dimostrato una OS mediana significativamente più lunga per vemurafenib (13,2 mesi; IC 95%: 12,0-15,4) rispetto a DTIC (9,7 mesi; IC 95%: 7,9-12,8).6 Ad un follow-up di 12,5 mesi la mOS è stata di 13,6 mesi con vemurafenib vs. 10,3 mesi con dacarbazina (HR 0,70, IC 95%: 0,60-0,93).7
Un grande studio internazionale è stato condotto al fine di studiare la sicurezza e l’efficacia di vemurafenib nella popolazione generale. Lo studio è stato condotto in 44 paesi, esclusi gli Stati Uniti. Lo studio ha arruolato 3226 pazienti con diagnosi di melanoma metastatico avanzato BRAF V600 (comprese metastasi cerebrali) indipendentemente dallo stato ECOG e dai livelli sierici di LDH. Complessivamente, i pazienti hanno tollerato bene il trattamento, sebbene il 95% abbia avuto almeno un evento avverso con una maggioranza (93%) di grado 1 e 2 secondo RECIST. Il profilo di tossicità confermava quello evidenziato nei precedenti studi. Una CR al trattamento è stata osservata solo nel 3% dei soggetti, mentre una risposta parziale (PR) è stata dimostrata nel 31% e una malattia stabile è stata osservata nel 55%. La mPFS è stata di 5,6 mesi con una sopravvivenza globale a 6 mesi del 75%, del 52% a 12 mesi e del 36% a 18 mesi dopo l’inizio del trattamento.8
Dabrafenib
Dabrafenib è un inibitore della chinasi di tipo I delle proteine mutate BRAF V600E/K/D. La sua approvazione da parte di FDA ed EMA è arrivata nel 2013 sulla base dei risultati dello studio BREAK. La fase 1 dello studio ha coinvolto 184 pazienti, di cui 156 affetti melanoma e 28 da altri tumori solidi e ha consentito di identificare la dose raccomandata di dabrafenib a 150 mg per via orale due volte al giorno. Nello studio di fase II BREAK-2 sono stati arruolati 76 pazienti con mutazione V600E e 16 con V600K di BRAF. Nel gruppo con mutazione V600E, il 59% dei pazienti ha ottenuto una risposta, inclusi cinque pazienti (7%) con risposte complete. Il 13% dei pazienti con mutazione V600K hanno ottenuto una risposta parziale confermata. La PFS mediana è stata di 6,3 mesi (V600E9 e 4,5 mesi (V600K) e l’OS mediana di 13,1 mesi e 12,9 mesi, rispettivamente. Gli eventi avversi più comuni sono stati artralgia (33%), ipercheratosi (27%) e piressia (24%). Complessivamente, 25 pazienti (27%) hanno manifestato un evento avverso grave e nove pazienti (10%) hanno avuto un carcinoma a cellule squamose.9
Nell’ambito dello studio di fase III (BREAK-3) dabrafenib è stato confrontato con la dacarbazina. La ORR ottenuta con dabrafenib è del 50% vs. il 6% con la dacarbazina. Dabrafenib ha mostrato risultati similari al vemurafenib in termini di PFS (5,1 vs. 2,7 mesi) e di mOS (20,0 vs. 15,6 mesi).10 I tassi di risposta (RR) e la PFS dei pazienti con mutazione di BRAF V600K sono risultati significativamente inferiori rispetto a quelle di pazienti con mutazione V600E. Eventi avversi correlati al trattamento si sono verificati nel 53% dei pazienti che hanno ricevuto dabrafenib ed erano principalmente rappresentati da effetti collaterali di tipo cutaneo, febbre, affaticamento, artralgia e mal di testa.11 Sulla base dei risultati dello studio di fase 1 che aveva evidenziato una risposta delle metastasi cerebrali in nove pazienti su dieci, quattro dei quali avevano ottenuto una remissione completa, è stato progettato lo studio di fase 2 BREAK-MB per valutare l’efficacia di dabrafenib nei pazienti con melanoma BRAF V600 mutato con metastasi cerebrali, sia pretrattati sia naïve. Una risposta intracranica complessiva è stata raggiunta nel 39,2% dei pazienti naïve al trattamento e nel 30,8% dei pazienti pretrattati con differenze significative nella durata mediana della risposta in relazione al tipo di mutazione. mediana di 20,1 mesi (naïve al trattamento) e 28,1 mesi (pretrattati). Inoltre, in questo sottogruppo, il profilo di sicurezza era accettabile, con eventi avversi simili a quelli precedentemente osservati.12
Encorafenib
Encorafenib è un inibitore selettivo di BRAF competitivo dell’ATP e appartiene alla seconda generazione di inibitori di BRAF. La differenza più significativa tra encorafenib e gli altri inibitori del BRAF di seconda generazione è la sua emivita
BRAF come bersaglio terapeutico nel melanoma 39
di dissociazione che è di 30 ore, rispetto a dabrafenib (2 ore) e vemurafenib (0,5 ore). Ciò significa che encorafenib manterrà il suo effetto inibitore più lungo rispetto agli altri inibitori del BRAF.13 Encorafenib ha inoltre dimostrato anche una potenza maggiore con una concentrazione inibente semi-massima (IC50) <40 nmol/l, rispetto ai più alti IC50 di dabrafenib (<100 nmol/l) e vemurafenib (<1 μmol/l). L’emivita di dissociazione prolungata e la potenza possono tradursi in effetti terapeutici prolungati e in minore tossicità.
Nello studio di fase I sono stati arruolati 54 pazienti, naïve o pretrattati con altri inibitori di BRAF. Nei pazienti naïve agli inibitori del BRAF, l’ORR era del 60% e la PFS mediana era di 12,4 mesi (n=18; IC 95%: 7,4 - non raggiunto). Nei pazienti pretrattati con inibitore di BRAF, l’ORR e la PFS mediana erano pari a 22% e 1,9 mesi (IC 95%: 0,9-3,7). Gli effetti avversi più comuni sono stati eritrodisestesia palmo-plantare (PPED), ipercheratosi, artralgia, nausea e prurito. Di tutti, il 9,3% (5/54) dei pazienti ha manifestato effetti avversi che hanno portato all’interruzione del trattamento (PPED, mal di testa, ipercheratosi e nevralgia). Nella fase di espansione è stato osservato solo un caso di carcinoma a cellule squamose (34% dei pazienti), con un’incidenza inferiore ai tassi osservati con la terapia con vemurafenib o dabrafenib.14
Nonostante le grandi risposte iniziali, il problema principale riguardante l’uso degli inibitori di BRAF è che mediamente entro 6-8 mesi la maggior parte dei pazienti non risponde più al trattamento.
Circa il 20% dei pazienti che presentano mutazioni attivanti nel BRAF presenta resistenza intrinseca e non risponde ab initio agli inibitori di BRAF,15 il che limita l’efficacia di questo approccio terapeutico. Infatti, già i primi studi hanno indicato che circa il 20% dei pazienti con melanomi BRAF V600E mutati non risponde agli inibitori di BRAF.16 Le cause della resistenza primaria sono state identificate in alterazioni genetiche presenti già prima dell’inizio del trattamento. Di seguito sono elencate le cause più frequenti di resistenza intrinseca ai BRAFi.
• Perdita di PTEN. PTEN è un soppressore cruciale del pathway PI3K/AKT: la perdita di PTEN porta all’attivazione costitutiva della cascata e consente la proliferazione cellulare anche in presenza di inibizione di BRAF.
• Mutazione RAC1P29S. Se mutata la proteina RAC1P29S determina l’attivazione della cascata di MAPK anche in presenza di inibitori di BRAF.
• Sovraespressione di MAP3K8. MAP3K8 codifica la proteina COT. La COT può attivare in modo indipendente la via MAPK/ERK, quindi un aumento dei livelli di COT promuove la proliferazione cellulare nonostante l’inibizione di BRAF.
• Secrezione del fattore di crescita degli epatociti (HGF) da parte delle cellule stromali. La secrezione di HGF da parte delle cellule stromali porta all’attivazione
del MET, un recettore per l’HGF, che è in grado di attivare il pathway MAPK/ ERK e PI3K/AKT determinando resistenza dell’inibitore BRAF.
• Perdita dell’oncosoppressore NF1. Il gene NF1 è un regolatore negativo della segnalazione RAS. La perdita di NF1 per mutazione consente l’aumento dell’attività di RAS, la successiva attivazione CRAF, portando all’attivazione della via MAPK, anche in presenza di inibizione BRAF
• Amplificazione di CCND1. CCND1 codifica per la ciclina D1, un regolatore chiave del ciclo delle cellule, la sua iperespressione può aiutare le cellule tumorali a bypassare l’inibizione della proliferazione da parte degli inibitori di BRAF.
• MicroRNA. i microRNA costituiscono una classe complessa di regolatori posttrascrizionali dell’espressione genica coinvolti nel controllo di numerosi processi fisiologici e patologici. Il loro meccanismo d’azione si basa principalmente sull’inibizione della traduzione dell’mRNA e alla fine alla sua degradazione. I singoli miRNA sono in grado di legarsi a diversi mRNA e di influenzare la funzione degli stessi mRNA. Negli ultimi anni stanno emergendo reti di miRNA in grado di controllare le vie di segnalazione chiave responsabili della crescita tumorale dello sviluppo della resistenza alle terapie farmacologiche mirate. In particolare la deregolamentazione dei miRNA è fortemente responsabile dello sviluppo della resistenza alle terapie target. Ad esempio miR-200c e miR-579-3p sono due potenti oncosoppressori che agiscono apparentemente su percorsi differenti e complementari. Il ripristino della loro espressione potenzia l’effetto dei farmaci inibitori della via MAPK e compromette l’instaurarsi della resistenza.17
• Aurora chinasi A (AURKA). Uno dei regolatori chiave della progressione della fase M, ha dimostrato di essere espressa ad alti livelli nel melanoma. AurkA è un membro di una famiglia di serina/treonina chinasi composta da tre classi (Aurk A, B e C) che sono componenti essenziali della via mitotica. L’inibizione di AurkA ha dimostrato di limitare la crescita del tumore, compromettere la mitosi e indurre senescenza nel melanoma, suggerendo un potenziale ruolo nel trattamento di questi tumori.18 19
Nella maggior parte dei casi, la resistenza agli inibitori di BRAF è correlata alla riattivazione del pathway di MAPK in seguito al trattamento, il che evidenzia ulteriormente il ruolo centrale svolto da questa cascata di segnalazione nei melanomi BRAF mutati. In alternativa, sono stati descritti meccanismi di resistenza indipendenti dalla riattivazione della via MAPK. L’analisi dettagliata dei melanomi dei pazienti con recidiva ha rilevato che nel 70% dei casi differenti meccanismi consentono alle cellule di melanoma di bypassare l’inibizione di BRAF mantenendo l’attivazione di MEK in modo indipendente da BRAF.20 Sebbene le mutazioni di RAF e RAS siano mutuamente esclusive nei
BRAF come bersaglio terapeutico nel melanoma 41
pazienti naïve, la mutazione/sovraespressione di NRAS in corso di trattamento costituisce uno dei meccanismi meglio conosciuti di riattivazione della cascata proliferativa. Anche la perdita della proteina di attivazione della GTPasi NF1 porta all’attivazione di MEK indipendentemente da BRAF. Inoltre, è stata riscontrata anche una maggiore attività di MEK in seguito all’attivazione di altri recettori tirosin chinasi (RTK) come EGFR, IGF-1R o PDGFR nei melanomi recidivanti.21 A livello delle chinasi RAF, la sovraespressione di CRAF, l’amplificazione del gene BRAF e la presenza di troncamenti di BRAF (come conseguenza dello splicing alternativo) consentono il mantenimento dell’attivazione di ERK.22 A valle di BRAF, la sovraespressione o la mutazione dello stesso MEK o anche dei suoi attivatori, COT/TPL2 MAP3K8 o MLKs, sono state descritte come meccanismi di riattivazione della via di trasmissione del segnale.23 Al di là della riattivazione di ERK attraverso la via RAS-CRAF-MEK-ERK, l’attivazione parallela della via PI3K/AKT è responsabile, in alcuni di casi, di resistenza acquisita. La resistenza a BRAFi può essere associata alla sovraregolazione della via PI3K/AKT, risultante da mutazioni AKT1/324 e mutazioni nei geni regolatori positivi (PIK3CA, PIK3CG) e negativi (PIK3R2, PTEN e PHLPP1) della cascata. Il MITF è uno dei principali regolatori della proliferazione dei melanociti e della biologia del melanoma. Livelli elevati di MITF consentono alle cellule di melanoma di eludere la morte cellulare innescata dagli inibitori di BRAF (e MEK) anche quando la via MAPK appare completamente bloccata. L’amplificazione del gene MITF si trova in una piccola percentuale di melanomi avanzati, tuttavia, in seguito al trattamento con inibitori di BRAf e/o di MEK, il MITF è frequentemente sovraregolato attraverso un ricablaggio dipendente da MAPK del controllo trascrizionale dell’espressione del MITF. Questo ricablaggio sembra avvenire nelle fasi iniziali del trattamento del paziente, perché circa l’80% dei melanomi mostra un’induzione significativa dell’espressione di MITF e questo consente alle cellule di melanoma di entrare in una fase di tolleranza al farmaco nelle prime fasi del trattamento con inibitori MAPK.25
D’altraparte i livelli di MITF sono stati inversamente correlati e hanno influenzato l’espressione di RTK attivati, ovvero EGFR e PDGFRβ, che sono correlati alla resistenza BRAFi/MEKi. Nella resistenza secondaria, la perdita di MITF è stata inversamente correlata al guadagno di espressione di questi RTK, in particolare AXL. Infatti, un basso rapporto MITF/AXL può predire una resistenza precoce alla terapia mirata.26 Quindi il ruolo del MITF nella risposta delle cellule di melanoma agli inibitori di MAPK è complesso; sebbene la sua sovraespressione conferisca resistenza agli inibitori di MAPK, livelli molto bassi di MITF quando coesistono con alti livelli di RTK AXL proteggono le cellule di melanoma dalla citotossicità indotta da inibitori di BRAF.27
La pletora di meccanismi di resistenza intrinseca/innata e acquisita sopradescritti limitano l’efficacia delle terapie a base di inibitori di BRAF nei pazienti con melanoma. L’individuazione di strategie per superare tale resistenza costituisce uno dei principali obiettivi nell’ambito della ricerca oncologica nel melanoma.
Associazione di inibitori di BRAF e inibitori di MEK
Considerato che il meccanismo centrale per la resistenza acquisita agli inibitori di BRAF è la riattivazione della via MAPK, l’associazione degli inibitori della chinasi BRAF con gli inibitori delle chinasi a valle nella via della fosforilazione MAPK, in particolare gli inibitori MEK, rappresenta la principale strategia a oggi disponibile. Inoltre, questa combinazione fornisce una serie di vantaggi aggiuntivi, tra cui una risposta clinica più potente e più duratura, insieme a una riduzione delle tossicità cutanee di tipo proliferativo, che vanno da ipercheratosi e cheratoacantomi (KA) a carcinomi a cellule squamose (SCC) e nuovi melanomi primari, che sono relativamente comuni con la monoterapia. La ridotta tossicità rispetto alla monoterapia con inibitori di BRAF è correlata all’assenza di attivazione paradossa del pathway di MAPK dovuta alla doppia inibizione di BRAF e MEK.
Vemurafenib e cobimetinib
Cobimetinib è un inibitore reversibile e altamente selettivo di MEK1 e MEK2. Cobimetinib è stato approvato per il trattamento di pazienti con melanoma non resecabile o metastatico con una mutazione BRAF V600E/K, in associazione con vemurafenib al dosaggio di 60 mg al giorno per 21 giorni ogni 28.
L’efficacia e la sicurezza di cobimetinib nell’uomo sono state inizialmente valutate nello studio clinico BRIM-7.28 I pazienti arruolati in questo studio hanno ricevuto vemurafenib orale 720 o 960 mg due volte al giorno in associazione con vari regimi orali di cobimetinib. È stata osservata una risposta obiettiva confermata nell’87% dei pazienti naïve agli inibitori di BRAF, con una PFS mediana di 13,7 mesi (IC 95%: 10,1-17,5), mentre nei pazienti pretrattati con vemurafenib in monoterapia il 15% ha avuto una risposta obiettiva, con una PFS mediana di 2,8 mesi (IC 95%: 2,6-3,4). Dopo un follow-up esteso, l’OS mediana ha raggiunto 28,5 mesi nei pazienti naïve agli inibitori di BRAF, con una percentuale di OS a 2 anni del 61%.29 L’associazione di vemurafenib e cobimetinib è stata approvata da FDA nel novembre 2015 sulla base dei risultati dello studio clinico di fase 3 co-BRIM (NCT01689519).30 In questo studio, 495 pazienti naïve con melanoma mutato BRAF V600 localmente avanzato o metastatico sono stati randomizzati a ricevere vemurafenib e cobimetinib o vemurafenib e placebo, con l’obiettivo primario di valutare la PFS. Sia il tasso di risposta che la PFS erano significativamente più alti nella combinazione rispetto al gruppo placebo (rispettivamente 68% vs. 45% e 9,9 vs. 6,2 mesi), con un tasso del 10% di risposta completa nei pazienti trattati con l’associazione vs. 4% nel gruppo di controllo. L’analisi di efficacia aggiornata pubblicata nel 201631 ha confermato i benefici clinici della combinazione, mostrando una sopravvivenza globale mediana di 22,3 mesi (IC 95%: 20,3 - non stimabile) per cobimetinib e vemurafenib rispetto a 17,4 mesi (IC 95%: 15,0-19,8) per placebo e vemurafenib (HR
0,70, IC 95%: 0,55-0,90; p=0,005). Non sono state osservate differenze significative negli eventi avversi di gradi 3 e 4 tra i due bracci di trattamento (65% vs. 59%), con una minore incidenza di tumori cutanei con la combinazione rispetto a vemurafenib in monoterapia.32 L’analisi finale è stata presentata al congresso 2019 della Society for Melanoma Research (SMR) e ha confermato la sostanziale superiorità della combinazione rispetto alla monoterapia.33 Ad un follow-up ha 5 anni la OS è stata del 30,8% con la combinazione contro il 26,3% con solo vemurafenib, mentre i tassi di PFS a 5 anni erano rispettivamente del 14% e del 10%. I pazienti che hanno risposto alla combinazione cobimetinib/ vemurafenib avevano una OS mediana superiore (32,1 vs. 9,7 mesi) così come una maggiore percentuale di sopravvivenza a 5 anni (37% vs. 13%). Allo stesso modo, quei pazienti avevano sia una PFS mediana (16,6 vs. 3,7 mesi) che una OS a 5 anni superiori (18% vs. 4%). Dall’analisi per sottogruppi è emerso che la combinazione è superiore alla monoterapia in tutti i sottogruppi di pazienti, compresi quelli con i pazienti con prognosi sfavorevole M1c, i pazienti con performance status ECOG 1, i pazienti che avevano la mutazione V600K e, naturalmente, le popolazioni di pazienti LDH. L’OS mediana per i pazienti con LDH normale era di 38,5 mesi. Al contrario, l’OS mediana era di soli 14,8 mesi (IC 95%: 11,3-18,6) nei pazienti con LDH elevata. La combinazione ha anche prodotto una migliore PFS mediana (15,0 vs. 8,6 mesi) e una percentuale di PFS a 5 anni superiore (18% vs. 7%) nei pazienti con LDH normale al basale rispetto a quelli con LDH elevata.34
Dabrafenib e trametinib
Trametinib (GSK1120212, CID PubChem: 11707110) è un inibitore reversibile DI MEK1 e MEK. Trametinib è stato in un primo momento valutato in monoterapia nello studio di fase 1 NCT00687622, che ha portato all’identificazione della dose giornaliera di 2 mg in quanto associata alla migliore risposta e maggiore sicurezza.35 In questo studio, le tossicità dose-limitanti erano rappresentate da rash, diarrea e retinopatia, mentre gli eventi avversi più comunemente riportati erano rash cutaneo, diarrea, edema, affaticamento e nausea. Sono state osservate risposte obiettive in circa il 10% dei pazienti, mentre nello studio di fase 2 la percentuale di risposte è aumentata al 25%.36 Nello studio di fase 3 METRIC con PFS come obiettivo primario e OS come endpoint secondario, 322 pazienti con melanomi metastatici mutati con BRAF V600E/K sono stati assegnati in modo casuale a ricevere trametinib (2 mg al giorno) o chemioterapia (DTIC per via endovenosa 1000 mg/m2 o paclitaxel 175 mg/m2 ogni 3 settimane) in un rapporto 2:1. I pazienti arruolati nel gruppo con chemioterapia al momento della progressione sono stati autorizzati a crossover per ricevere trametinib. Trametinib ha dimostrato di migliorare sia la PFS che la OS, rispetto alla chemioterapia convenzionale (PFS 4,8 vs. 1,5 mesi e a 6 mesi OS 81% vs. 67%).37
I risultati più incoraggianti sono stati ottenuti con l’associazione di trametinib a dabrafenib. In un trial di fase 1/2, pazienti affetti da melanoma avanzato BRAFmutato, pretrattati, hanno ricevuto la combinazione dabrafenib e trametinib. Questo studio ha identificato la dose di combinazione in 150 mg bid per dabrafenib e 2 mg/die per trametinib.37 La PFS mediana dell’associazione di BRAF e MEK inibitori è risultata 9,4 mesi, significativamente superiore ai 5,8 mesi della monoterapia con dabrafenib, con un HR di 0,39 (IC 95%: 0,25-0,62; p=0,03). La terapia di combinazione, oltre a prevenire i meccanismi di resistenza acquisiti, riduce la tossicità indotta da inibitori di BRAF secondaria all’attivazione paradossa del MAPK pathway. La combinazione di trametinib e dabrafenib ha determinato una ridotta incidenza di carcinoma cutaneo a cellule squamose (7% vs. 19%), ma più alti tassi di piressia (71% vs. 26%) rispetto al solo dabrafenib. L’update di questo trial ha visto importanti progressi in termini di OS con l’80% dei pazienti vivi a 12 mesi, il 51% a 24 mesi e il 38% a 36 mesi. I pazienti che hanno ottenuto il maggiore beneficio dal trattamento sono risultati quelli con livelli normali di lattico deidrogenasi (LDH), con basso carico tumorale, con meno di tre siti metastatici. L’efficacia di dabrafenib più trametinib nel melanoma avanzato è stata confermata nello studio di fase 3 COMBI-D38 in cui 432 pazienti precedentemente non trattati sono stati assegnati in modo casuale a ricevere la combinazione di dabrafenib e trametinib (BID 150 mg più 2 mg una volta al giorno) o dabrafenib e placebo. Nei pazienti trattati con la terapia di combinazione, sia la percentuale di PFS che quella di OR erano più elevate rispetto al gruppo trattato con dabrafenib da solo (9,3 vs. 8,8 mesi e 67% vs. 51%, rispettivamente). L’incidenza di eventi avversi era simile nei due gruppi, ma nel gruppo di trattamento in associazione l’incidenza dei carcinomi cutanei a cellule squamose era inferiore, anche se la piressia era più frequente e grave rispetto al solo trametinib. L’analisi dei dati del COMBI-D ad un follow-up di 3 anni ha mostrato una PFS a 3 anni del 22% con dabrafenib e trametinib rispetto al 12% con la monoterapia e una OS a 3 anni del 44% vs. 32%. La sopravvivenza a 3 anni nel sottogruppo a buona prognosi (LDH nella norma, e meno di tre sedi metastatiche) è stata del 62% vs. il 25% del sottogruppo a prognosi peggiore (LDH elevato).39 Nello studio COMBI-V la combinazione di dabrafenib e trametinib è stata confrontata con vemurafenib in monoterapia e ha dimostrato anche in questo caso vantaggi in termini di PFS e OS della terapia di combinazione). L’HR per morte è risultata 0,69 (IC 95%: 0,53-0,89; p=0,005) e la PFS mediana 11,4 mesi per la terapia di combinazione vs. 7,3 mesi per il vemurafenib in monoterapia (HR 0,56; IC 95%: 0,46-0,69; p<0,001). Il tasso di risposte ottenuto è stato del 64% (IC 95%: 59-69%) nella coorte dabrafenib-trametinib e del 51% (IC 95%: 46-57%) nella coorte vemurafenib monoterapia. Lo studio ha anche confermato la tossicità significativamente più bassa della terapia di associazione rispetto a vemurafenib.40 Da questi risultati positivi, la combinazione dabrafenib/trametinib è stata approvata per il trattamento dei melanomi metastatici BRAF V600E/K mutati dalla FDA nel gennaio 2014 e dall’EMA nell’agosto 2013.
BRAF come bersaglio terapeutico nel melanoma 45
Una pooled analysis condotta sui pazienti arruolati negli studi di fase III con dabrafenib + trametinib (COMBI-D e COMBI-v) ha valutato 563 pazienti che hanno ricevuto D+T (COMBI-d, n=211; COMBI-v, n=352). I tassi di PFS e OS a 4 e 5 anni erano simili, suggerendo una stabilizzazione della curva (PFS a 4 e 5 anni, 21% [IC 95%: 17-24%] e 19% [IC 95%: 15-22%, rispettivamente]; OS a 4 e 5 anni, 37% [IC 95%: 33-42%] e 34% [IC 95%: 30-38%], rispettivamente). Nei pazienti con livelli normali di LDH al basale la percentuale di PFS a 5 anni era del 25% vs. 8% dei pazienti con livelli elevati di LDH al basale. Allo stesso modo, il tasso di OS a 5 anni era considerevolmente più alto nei pazienti con livelli di LDH normali al basale rispetto a quelli con livelli di LDH elevati al basale (43% vs. 16%). Tra i pazienti con livelli normali di LDH al basale e meno di tre siti di organi con metastasi, le percentuali di PFS e OS a 5 anni erano rispettivamente del 31% e del 55%.41 Nel novembre 2017, Long, et al.42 hanno pubblicato i risultati dello studio di fase III COMBI-AD in doppio cieco, controllato con placebo (NCT01682083), che ha valutato il ruolo della terapia di associazione dabrafenib-trametinib come trattamento adiuvante per pazienti in stadio IIIA-IIIB-IIIC. Dopo un follow-up mediano di 2,8 anni, il tasso stimato di sopravvivenza libera da recidiva (RFS) a 3 anni era del 58% nel gruppo trattato e del 39% nel gruppo placebo, e la OS a 3 anni dell’86% nel gruppo di trattamento e del 77% nel gruppo placebo (IC 95%: 0,42-0,79; p=0,0006). Sulla base di questi dati la combinazione di dabrafenib e trametinib è stata approvata dall’FDA il 30 aprile 2018 per il trattamento adiuvante di pazienti adulti con melanoma in stadio 3 positivo alla mutazione BRAF V600, dopo resezione completa. L’update a 60 mesi presentato all’ASCO 2020 ha confermato il beneficio del trattamento in adiuvante: la mRFS non è ancora stata raggiunta nel braccio di combinazione mentre nel braccio del placebo era 16,6 mesi. La RFS a 4 e 5 anni è stato rispettivamente del 55% e 52% con la combinazione vs. 38% e 36% col placebo.43
Encorafenib e binimetinib
La combinazione di encorafenib e binimetinib è stata approvata dalla FDA il 27 giugno 2018, per il trattamento di pazienti con melanoma non resecabile o metastatico con mutazione BRAF V600E o V600K. L’approvazione si basava sullo studio COLUMBUS, uno studio di fase 3 randomizzato, in due parti, in aperto, che ha confrontato encorafenib più binimetinib con encorafenib e vemurafenib in monoterapia.44 Nella prima parte dello studio, 577 pazienti sono stati randomizzati a ricevere encorafenib 450 mg una volta al giorno (QD) e binimetinib 45 mg BID (COMBO450), encorafenib 300 mg (ENCO300) QD o vemurafenib 960 mg BID (VEM). La mPFS era significativamente più alta nei gruppi COMBO450 (14,9 mesi) ed ENCO (9,6 mesi) rispetto al gruppo VEM (7,3 mesi). L’ORR confermato dalla revisione centrale è stato rispettivamente del 63%, 51% e 40% per COMBO450, ENCO e VEM, mentre la durata della risposta è stata rispettivamente di 18,6 mesi,
15,2 mesi e 12,3 mesi. A un follow-up di 60 mesi la PFS era 14,9 mesi nel braccio COMBO450, 9,6 mesi nel braccio ENCO300, 7,3 mesi nel braccio VEM. La OS a 4 anni è del 39%, 37% e 26% rispettivamente nei bracci COMBO450, ENCO300 e VEM.45 Nella coorte COMBO450, eventi avversi di grado ≥3 si sono verificati nel 58% dei pazienti, rispetto al 66% e al 63% nei gruppi ENCO300 e VEM. Il tempo mediano al primo evento di grado ≥3 AE era di 2,5 mesi per COMBO450, 0,4 mesi per ENCO300 e 1,3 mesi per VEM. Gli eventi avversi gravi erano principalmente correlati all’inibitore di MEK (incremento creatinchinasi e tossicità oculare). Questi si sono verificati meno frequentemente rispetto alla monoterapia con binimetinib. L’8% dei pazienti COMBO450 ha interrotto il trattamento a causa di eventi avversi, rispetto al 12% e al 14% nei gruppi ENCO300 e VEM.44
La seconda parte dello studio COLUMBUS ha valutato il contributo di binimetinib alla terapia di associazione confrontando lo stesso braccio monoterapia ENCO300 con encorafenib 300 mg QD più binimetinib 45 mg BID (COMBO300). I pazienti sono stati randomizzati (3:1) a ricevere encorafenib 300 mg una volta al giorno in associazione con binimetinib 45 mg due volte al giorno (n=258) o encorafenib 300 mg monoterapia una volta al giorno (n=86). La PFS mediana era di 12,9 mesi (IC 95%: 10,1-14,0) nel braccio di combinazione rispetto a 9,2 mesi (IC 95%: 7,4-11,0) nel braccio con encorafenib in monoterapia (HR 0,77, p=0,029). La combinazione di encorafenib più binimetinib ha mostrato un buon profilo di sicurezza e tollerabilità nei pazienti con melanoma non resecabile o metastatico. Gli eventi avversi erano in gran parte simili a quelli osservati con le altre combinazioni di inibitori di BRAF-MEK, con una minore incidenza di iperpiressia rispetto a dabrafenib più trametinib e una minore incidenza di fotosensibilità rispetto a vemurafenib più cobimetinib. Gli eventi avversi più frequenti (≥25% dei pazienti) sono stati nausea, diarrea, vomito, affaticamento e artralgia, nella maggior parte dei casi di grado 1-2. In generale, la combinazione sembrava avere un miglior profilo di tollerabilità rispetto alla monoterapia con minore incidenza di molti eventi avversi, in particolare gli eventi cutanei; la comparsa di SCC cutanei e di carcinomi a cellule basali è stata riportato solo nel 2,6% e nell’1,6% di pazienti trattati con encorafenib più binimetinib, rispettivamente.
Gli eventi avversi che si sono verificati con maggior frequenza nel gruppo di combinazione comprendevano diarrea, vomito, costipazione, visione offuscata, aumento della CPK e dolore addominale. Il 20% dei pazienti trattati con encorafenib più binimetinib nella parte 1 dello studio COLUMBUS ha riportato retinopatia sierosa e il 4% ha riferito uveite. Disfunzione ventricolare sinistra è stata riportata nel 7,8% dei pazienti nel braccio di combinazione, con 1,6% di eventi di grado 3. Questo era generalmente reversibile con l’interruzione o la riduzione della dose. L’epatotossicità di grado 3-4 può verificarsi anche durante il trattamento con encorafenib più binimetinib. Un incremento G3-4 di AST, ALT e fosfatasi alcalina si è verificato rispettivamente nel 6%, 2,6% e 0,5% dei pazienti nel
BRAF come bersaglio terapeutico nel melanoma 47
braccio encorafenib più binimetinib. Altri eventi avversi gravi o potenzialmente gravi o fatali che sono stati riportati con la combinazione includono tromboembolia venosa, polmonite interstiziale, rabdomiolisi e prolungamento dell’intervallo QT. Eventi avversi gravi (G3-G4) si sono verificati in proporzioni simili di pazienti trattati con l’associazione (34%), encorafenib da solo (34%) e solo vemurafenib (37%). Si è verificato un unico caso di morte correlata al trattamento. Una riduzione di dose è stata necessaria meno frequentemente nel gruppo della combinazione rispetto quello di encorafenib e di vemurafenib (45% vs. 70% vs. 61%, rispettivamente), mentre l’interruzione del trattamento per tossicità si è verificata nel 13% vs. 14% e nel 17% dei pazienti.44
Altre combinazioni
Considerata la presenza riconosciuta di svariati meccanismi di resistenza al trattamento con inibitori di BRAF, negli ultimi anni sono stati avviati una serie di studi clinici al fine di identificare ulteriori strategie in grado di potenziare e prolungare il beneficio ottenuto con i BRAFi. Diversi studi clinici hanno dimostrato che i processi epigenetici hanno un ruolo fisiologicamente cruciale nella regolazione dell’espressione genica e del fenotipo cellulare. La comprensione di tale fenomeno ha portato allo sviluppo di potenziali nuovi agenti, come gli inibitori dell’istone deacetilasi (HDAC). Le deacetilasi istoniche sono regolatori critici dell’espressione genica attraverso la deacetilazione degli istoni. Nelle cellule tumorali, queste proteine sono aberranti e possono portare a una sottoregolazione dell’espressione dei geni oncosoppressori, danni al DNA da parte di agenti genotossici e interferenze con la riparazione del danno al DNA. Esistono diverse classi di HDAC (I, II e IV) e diversi tipi di inibitori HDAC sono attualmente in fase di studio.46
Uno studio clinico sta valutando vorinostat, un inibitore HDAC di classe I e II, nel melanoma avanzato mutato BRAF V600 resistente (NCT02836548). I dati sugli effetti collaterali degli inibitori HDAC derivano dall’analisi di studi di fase I-II su tumori maligni ematologici e tumori solidi. Gli eventi avversi più comuni colpiscono il tratto gastrointestinale e includono nausea, vomito e anoressia. Sono stati riportati anche affaticamento, alterazioni ematologiche come trombocitopenia e alterazioni dell’ECG.47 Vorinostat sembra anche essere in grado di regolare gli antigeni tumorali, aumentando l’MHC di classe I, per cui sono in corso anche studi di associazione con l’immunoterapia.48 HSP90 è una proteina chaperone ampiamente espressa in diverse cellule tumorali a causa di alti livelli di oncoproteine, ipossia e instabilità genomica. La proteina BRAF attivata è una delle proteine regolate da HSP90.49 XL888 è un inibitore HSP90 che è stato valutato in uno studio di fase I in combinazione con vemurafenib in 21 pazienti con melanoma mutante BRAF V600 avanzato.50 In un’analisi aggiornata il tasso di risposta globale (ORR) è stato del 75% con 3 risposte
complete e 12 risposte parziali. La PFS mediana era di 9,2 mesi (3,8 mesi - non raggiunta) e l’OS mediana era di 34,6 mesi (6,2 mesi - non raggiunta). La tossicità principale era correlata alla pelle.51 È in corso uno studio clinico di XL888 in combinazione con vemurafenib e cobimetinib nel melanoma BRAF mutato in stadio III/IV non resecabile (NCT02721459). Oltre a XL888, onalespib (AT13387), un inibitore HSP90 di piccole molecole biodisponibile per via orale, è in fase di valutazione in combinazione con dabrafenib e trametinib in uno studio di fase I (NCT02097225).
È noto da tempo che la sovraregolazione del percorso PI3K/AKT è uno dei principali processi coinvolti nello sviluppo di diversi tumori, in particolare il melanoma, e l’alterazione di questo percorso è un meccanismo riconosciuto di resistenza alla target.52 BK120 è un inibitore PI3K di classe I pan che inibisce PI3K wild-type e mutante ed esercita un effetto antitumorale inducendo l’apoptosi in diversi tipi di tumore. Gli studi clinici di fase I mostrano che BKM120 ha un buon profilo di sicurezza generale in pazienti con diversi tumori solidi. Gli eventi avversi più frequenti includono eruzione cutanea, iperglicemia, diarrea, anoressia, alterazioni dell’umore, nausea, affaticamento e mucosite.53 Tuttavia, l’inibizione di PI3K in associazione con l’inibizione di BRAF o MEK potrebbe provocare un aumento della tossicità.54 55
Le mutazioni geniche del checkpoint del ciclo cellulare (CDKN2A, CDK4) si verificano frequentemente nel melanoma e possono rappresentare una causa di resistenza al trattamento con inibitori di BRAF. Di conseguenza, sono stati valutati studi di fase 1/2 che combinano inibitori di BRAF e MEK con inibitori
CDK4/6. Ribociclib è un inibitore CDK4/6, che è stato valutato in uno studio di fase Ib/2 in associazione a encorafenib più binimetinib (NCT01543698). L’associazione ha determinato un aumento di tossicità rispetto alla terapia di associazione doppia. In particolare, si è verificata tossicità epatica da moderata a grave che ha portato all’interruzione del trattamento in quasi il 26% dei pazienti. Le transaminasi elevate sono state la ragione più comune per l’interruzione, in linea con il profilo di tollerabilità noto di ribociclib.56 INC280 (capmatinib) è un piccolo inibitore di recettore c-MET che rappresenta un altro meccanismo di resistenza alla terapia mirata.57 Questo agente sembra avere una tossicità gestibile ed è caratterizzato da lieve edema periferico, nausea, aumento della creatinina, vomito, affaticamento, diminuzione dell’appetito e diarrea e prolungamento dell’intervallo Q.58 Nello studio in corso di fase II LOGIC 2 (NCT02159066) in pazienti con melanoma BRAF-mutante avanzato, BGJ398, ribociclib e INC280 sono tutti in fase di studio in combinazione con encorafenib e binimetinib. Il disegno dello studio consente l’aggiunta di uno di questi agenti sperimentali dopo la progressione alla terapia mirata. La data stimata di completamento dello studio LOGIC-2 è il 2022.
Un altro approccio è basato sull’inibizione dei recettori tirosin chinasici (RTK) che sono coinvolti nella carcinogenesi di più tipi di tumori e nei comportamenti
BRAF come bersaglio terapeutico nel melanoma 49
immunitari e biologici all’interno dei tumori. AXL è una proteina RTK che appartiene alla grande famiglia dei recettori dei macrofagi associati al tumore (TAM). È implicato nella progressione tumorale e nell’immunosoppressione.59 Una maggiore espressione di AXL svolge un ruolo chiave nella regolazione della resistenza acquisita agli inibitori di BRAF nelle cellule di melanoma. L’uso di BGB324, un inibitore di AXL, in combinazione con pembrolizumab o dabrafenib/trametinib è in fase di studio in un trial clinico (NCT02872259). Itacinib è un inibitore proteico della tirosin chinasi, JAK1, che si ritiene sia coinvolto nelle metastasi del cancro. Questo composto è stato studiato in associazione con la chemioterapia e ha mostrato efficacia e un profilo di sicurezza gestibile. Itacinib è oggetto di ulteriori studi clinici in combinazione con dabrafenib e trametinib in uno studio di fase I (NCT03272464).
La combinazione dell’immunoterapia con gli inibitori di BRAF/MEK può produrre un effetto sinergico, superando la durata limitata delle risposte e aumentando i tassi di risposta e la sopravvivenza a lungo termine rispetto alle singole strategie da sole. I dati dei modelli preclinici e dei tessuti dei pazienti mostrano che la terapia mirata con inibitori di BRAF e MEK può influenzare il microambiente tumorale e l’immunogenicità nel melanoma in diversi modi, tra cui una maggiore infiltrazione e attività delle cellule T, un’espressione e una presentazione migliorate di MDA (antigeni di differenziazione del melanoma), e un cambiamento favorevole nell’equilibrio tra citochine immunostimolanti e soppressive. Lo sviluppo della resistenza immuno-mediata alla terapia mirata ha fornito ulteriore supporto per lo studio delle combinazioni con l’immunoterapia.60-63
Il primo tentativo di combinazione è stato effettuato con vemurafenib più ipilimumab, che tuttavia ha mostrato una tossicità epatica inaccettabile.64 Dabrafenib e ipilimumab sono stati ben tollerati insieme; tuttavia, l’aggiunta di trametinib alla combinazione è stata associata a eventi gastrointestinali gravi, con due pazienti che hanno sviluppato colite complicata da perforazione.65
Diversi studi di fase I sono stati condotti per valutare differenti triplette di farmaci, costituite da anti-PD1/PD-L1 in associazione alle diverse doppiette di inibitori BRAF/MEk. La combinazione di dabrafenib, trametinib e pembrolizumab è stata valutata su 15 pazienti nell’ambito di uno studio di fase I (NCT02130466) con risultati incoraggianti. Su 15 pazienti trattati, 11 hanno manifestato eventi avversi di grado 3/4 treatment-related, i più frequenti dei quali erano l’incremento delle transaminasi e la febbre. L’ORR è stato del 73% (11/15), con una risposta completa (CR) e 10 risposte parziali (PR), la durata mediana della risposta è stata di 19,4 mesi. 11 (73%) dei pazienti hanno manifestato eventi avversi correlati al trattamento di grado 3/4, portando all’interruzione del trattamento per 6 pazienti (40%). Non sono stati segnalati decessi correlati al trattamento.
Uno studio di fase Ib è stato condotto per valutare l’efficacia e l’attività della combinazione di atezolizumab con vemurafenib e cobimetinib. Atezolizumab viene somministrato dopo 28 giorni di assunzione di dabrafenib e trametinib. A fronte di un profilo di tossicità maneggevole, il tasso di ORR è stato del 71,8%.
La safety della tripla combinazione di dabrafenib, trametinib con o senza spartalizumab è stata valutata nella parte 1 dello studio COMBI-I. Lo studio era articolato in tre parti: la parte 1 dello studio di fase III era una fase di safety, la parte 2 era una coorte di biomarcatori, mentre la parte 3 era randomizzata. I primi risultati della parte 1 e 2, presentati all’ASCO 2018 sui primi 9 pazienti che avevano completato le 8 settimane di terapia hanno mostrato un profilo di tossicità maneggevole e un’attività promettente; tutti i pazienti avevano ottenuto una risposta, con 3 risposte complete (335) e 6 risposte parziali (67%).66 I risultati aggiornati presentati all’SMR 2019, dal totale di 36 pazienti (9 dalla parte 1 e 27 dalla parte 2) hanno mostrato un tasso di PFS a 12 mesi del 66,7% (IC 95%: 49-80) e un DOR a 12 mesi di 80,3% (59-91). I pazienti con LDH elevata (n=15) avevano un tasso di DOR a 12 mesi del 64,8% (IC 95%: 25-87), un tasso di PFS a 12 mesi e un tasso di OS del 33,3% (IC 95%, 12-56) e 66,7% (IC 95%: 37,5-85) rispettivamente. Nei pazienti con malattia M1c in stadio IV (n=20), il tasso di DOR è stato del 67,7% (IC 95%: 35-86,5) a 12 mesi, con un tasso di PFS a 12 mesi del 50,0% (IC 95%: 27-69) e un tasso di OS del 75,0% (IC 95%: 5089). I pazienti con elevato DNA tumorale circolante, basso carico mutazionale del tumore (TMB) e alti livelli di fattori immunosoppressivi nel microambiente tumorale (TME) avevano maggiori probabilità di sperimentare un evento di PFS nei primi 12 mesi (n=5; 23%). Per quanto riguarda il profilo di sicurezza, piressia (n=32), tosse (n=18), artralgia (n=18), eruzione cutanea (n=17), brividi (n=17) e affaticamento (n=16) sono stati i più comuni eventi avversi (AE) riportati. Eventi avversi gravi correlati al trattamento si sono verificati nel 36% (n=13) dei pazienti. Non si sono verificati decessi correlati al trattamento.67 Nei dati presentati all’ASCO 2020, relativi alle parti 1 e 2 dello studio, con un followup mediano di 24,3 mesi, la tripletta era associata a un ORR del 78% (28 su 36 pazienti) e una risposta completa (CR) nel 44% (16/36). La durata mediana della risposta (DOR) è stata di 20,7 mesi. La tripletta ha mostrato un’efficacia promettente nei pazienti con malattia M1c e livelli elevati di lattato deidrogenasi (LDH) che generalmente hanno una prognosi sfavorevole. Le percentuali di PFS e OS a 24 mesi erano rispettivamente del 41% e del 74%. Gli eventi avversi erano coerenti con i profili di tossicità individuali di ciascun farmaco in studio. Il profilo di tossicità della tripletta era gestibile e solo il 17% dei pazienti ha interrotto tutti e tre i farmaci in studio. I limiti di questi risultati includono il numero limitato di pazienti e l’assenza di un comparatore attivo.68
Un altro studio di fase 1 (NCT02027961) sta valutando la sicurezza e l’efficacia di MEDI4736 (durvalumab) a 3 o 10 mg/kg e.v. ogni due settimane in combina-
BRAF come bersaglio terapeutico nel melanoma 51
zione con dabrafenib 150 mg due volte al giorno con o senza trametinib 2 mg al giorno, in pazienti con entrambi melanoma BRAF mutato e BRAF wild-type avanzato. I pazienti con BRAF mutato sono trattati nella coorte A con dabrafenib, trametinib e durvalumab, mentre i pazienti con BRAF wild-type sono trattati con trametinib e durvalumab, contemporaneamente nella coorte B (M+T) o in modo sequenziale nella coorte C (T→M). I risultati dei primi 50 pazienti hanno mostrato un tasso di risposta del 76% (16/21) nei pazienti con mutazioni BRAF trattati con la tripletta, un tasso di risposta del 21% (3/14) nei pazienti BRAF wild-type in terapia di combinazione, e il 50% (3/6) nei pazienti BRAF wild-type in terapia sequenziale. Due pazienti hanno manifestato tossicità dose-limitanti (trombocitopenia e versamento coroideale). Gli eventi avversi correlati al farmaco più frequenti sono stati: piressia (63%) e affaticamento (54%) nella coorte A, diarrea (30%) ed eruzione cutanea (25%) nella coorte B e vomito (67%) nella coorte C.69 In un altro studio di fase Ib, i pazienti con melanoma metastatico, sia BRAF mutato che wild-type, hanno ricevuto cobimetinib (60 mg al giorno, 21 giorni ogni 28) in concomitanza con atezolizumab 800 mg e.v. q2w. L’ORR era simile al 45,0% per i pazienti con mutante BRAF e BRAF wild-type. Eventi avversi di grado 3 o 4 si sono verificati nel 54,5% dei pazienti. Diarrea ed eruzione cutanea sono state le tossicità più comuni segnalate.70 Sulla base dei risultati positivi degli studi di fase I, sono stati disegnati e condotti studi di fase II e III per valutare l’efficacia delle varie triplette.
Lo studio di fase KEYNOTE-022, dopo i risultati positivi della fase I, è proseguito con una fase II randomizzata che sta valutando la combinazione di dabrafenib, trametinib e pembrolizumab in pazienti precedentemente non trattati con melanoma avanzato mutato in BRAF.71 Sono stati recentemente riportati i risultati della parte di fase II randomizzata. I pazienti con melanoma avanzato mutato in BRAF sono stati randomizzati a ricevere dabrafenib più trametinib con pembrolizumab o placebo, con 60 pazienti in ciascun braccio. Stranamente il tasso di risposta è stato inferiore nel braccio pembrolizumab (63% vs. 72%). La PFS mediana con pembrolizumab era di 16,0 mesi rispetto a 10,3 mesi con placebo, tuttavia la PFS non ha raggiunto la soglia di significatività statistica per progetto di studio. In particolare, il maggior numero di pazienti che hanno ricevuto pembrolizumab ha avuto risposte che durano più di 18 mesi rispetto a quelli che hanno ricevuto placebo (60% vs. 28%).72 Ad un followup di 2 anni, la mPFS è stata di 16,9 mesi con pembrolizumab e 10,7 mesi con placebo. I tassi di sopravvivenza libera da progressione a 24 mesi sono stati del 41,0% con pembrolizumab rispetto al 16,3% con placebo. La mOS non è stata raggiunta con pembrolizumab mentre è stata di 26,3 mesi con placebo. I tassi di sopravvivenza globale a 24 mesi sono stati del 63,0% vs. il 51,7%. Il tasso di risposta obiettiva per RECIST v1.1 è stato del 63,3% con pembrolizumab e del 71,7% con il placebo, tuttavia la durata mediana della risposta è stata di 25,1 mesi e 12,1 mesi rispettivamente con pembrolizumab e placebo. Purtroppo la
combinazione ha determinato un significativo incremento degli eventi avversi. In particolare le tossicità di grado 3-5 si sono verificate in 56 (93,3%) pazienti trattati con pembrolizumab e 15 (25,0%) pazienti nel braccio placebo. Un paziente è deceduto per polmonite correlata a pembrolizumab. Gli eventi avversi più comuni (≥5% dei pazienti) di grado 3-5 emergenti dal trattamento sono stati piressia (10,0% [n=6] vs. 3,3% [n=2]), aumento dell’aspartato aminotransferasi (6,7% [n=4] vs. 3,3% [n=2]) e aumento della γ -glutamil transferasi (6,7% [n=4] vs. 5,0% [n=3]), rispettivamente.73
Lo studio di fase III IMspire150 TRILOGY (NCT02908672) ha raggiunto l’endpoint primario, mostrando un miglioramento della sopravvivenza libera da progressione (PFS) con la tripletta atezolizumab, cobimetinib e vemurafenib rispetto a cobimetinib e vemurafenib in pazienti con melanoma avanzato mutato BRAF V600 non trattato in precedenza. In questo studio sono stati arruolati 514 pazienti naïve con età mediana di 50 anni; l’81% dei pazienti aveva un LDH normale e l’83% un performance status ECOG di 0. Lo stadio della malattia variava da IIIC (27%) a IV M1c (20%). Durante un periodo di run-in di 28 giorni, vemurafenib è stato somministrato a 960 mg due volte al giorno nei giorni da 1 a 21. Dopo il run-in, la dose di vemurafenib è stata ridotta a 720 mg due volte al giorno e atezolizumab è stato somministrato a 800 mg ogni due settimane. Cobimetinib è stato somministrato alla dose di 60 mg al giorno nei giorni da 1 a 21 in entrambe le fasi dello studio.74 A un follow-up mediano di 18,9 mesi, la sopravvivenza libera da progressione è stata significativamente prolungata con atezolizumab rispetto al controllo (15,1 vs. 10,6 mesi). Il profilo di sicurezza del regime a tre farmaci era coerente con ciascun agente da solo Gli eventi avversi correlati al trattamento comuni in entrambi i gruppi erano aumento della CPK ematica (51,3% vs. 44,8%), diarrea (42,2% vs. 46,6%), eruzione cutanea (40,9%, entrambi i gruppi), artralgia (39,1% vs. 28,1%), febbre (38,7% vs. 26,0%), aumento di ALT/AST (33,9% vs. 22,8%) e aumento della lipasi (32,2% vs. 27,4%). A causa di eventi avversi, il trattamento è stato interrotto rispettivamente nel 13% dei pazienti nel gruppo atezolizumab e nel 16% nel gruppo di controllo.75
Nonostante i risultati positivi ottenuti nella prima parte dello studio COMBI-I, ad agosto 2020 Novartis ha annunciato, sulla base dei dati relativi alla parte 3 dello studio, che non è stato raggiunto un vantaggio in termini di PFS, che costituiva l’obiettivo primario dello studio.76 All’Esmo 2020 sono stati presentati i dati preliminari. L’incremento in termini di PFS è stato di 4,2 mesi a favore del braccio sperimentale (16,2 vs. 12 mesi, HR 0,82) ed è risultato non significativo nella popolazione generale (il disegno dello studio prevedeva un incremento minimo in PFS di 5 mesi per stabilire la superiorità della tripletta rispetto alla doppietta). Dallo studio dei biomarker è emerso un vantaggio significativo in PFS nel sottogruppo con high tumor mutational burden (TMB) (23,9 vs. 11,8 mesi, HR 0,703) a differenza del sottogruppo con low TMB, in cui la PFS è stata sovrapponibile in entrambi i bracci (12,8 vs. 12 mesi, HR 0,907). Sebbene la sopravvivenza globale non sia stata formalmente
BRAF come bersaglio terapeutico nel melanoma 53
testata, è stato osservato un HR di 0,785 a favore della tripletta e la sopravvivenza globale mediana non è stata raggiunta in nessuno dei due bracci di trattamento.
Nei pazienti trattati con Sparta-DabTram è stato osservato un numero maggiore di modifiche della dose (riduzioni/interruzioni) e interruzioni, suggerendo un aumento della tossicità.77
In uno studio di fase II TRIDeNT (studio di fase II della combinazione TRIplet di dabrafenib, nivolumab e trametinib in pazienti con melanoma metastatico), un totale di 14 pazienti con melanoma avanzato mutato in BRAF sono stati arruolati per ricevere contemporaneamente dabrafenib, trametinib e nivolumab. Sono stati ammessi pazienti con metastasi cerebrali non trattate, asintomatiche o lievemente sintomatiche. L’ORR è stata del 91%, 3 pazienti hanno interrotto lo studio a causa di eventi avversi correlati al farmaco, tra cui epatite e nefrite immuno-mediata.78
Nello studio multicentrico, in aperto, randomizzato di fase III IMspire170, i ricercatori hanno valutato l’efficacia, la sicurezza e la farmacocinetica di cobimetinib più atezolizumab rispetto a pembrolizumab in 450 pazienti naïve al trattamento con melanoma BRAF V600 wild-type avanzato. Nello studio, la dose di cobimetinib è stata aumentata da 20 mg a 40 mg a 60 mg. È stato somministrato una volta al giorno per 21 giorni seguiti da 7 giorni di riposo. Atezolizumab è stato somministrato 800 mg ogni due settimane e.v. Nel braccio pembrolizumab, l’inibitore del PD-1 è stato somministrato e.v. a 200 mg ogni 3 settimane. Il profilo di sicurezza osservato con il regime di associazione era coerente con i profili di sicurezza noti di ciascun agente da solo. Eventi avversi di grado 3/4 correlati al trattamento si sono verificati nel 54,5% dei pazienti, comprendenti diarrea (13,6%) e dermatite acneiforme (9,1%). Eventi avversi gravi correlati al trattamento si sono verificati nel 13,6% dei pazienti, tutti risultati gestibili.79 In un recente aggiornamento dello studio è stato riportato che la combinazione di atezolizumab + cobimetinib ha mancato l’endpoint PFS nel melanoma BRAF V600 wild-type.
La terapia con inibitori di BRAF in associazione con gli inibitori di MEK costituisce, insieme all’immunoterapia, un cardine nel trattamento del melanoma. Nel setting metastatico le tre combinazioni al momento approvate vemurafenib/cobimetinib, dabrafenib/trametinib ed encorafenib/cobimetinib hanno mostrato efficacia sovrapponibile con un profilo di tossicità maneggevole. La popolazione che maggiormente beneficia del trattamento è quella con LDH nella norma al basale, meno di tre siti metastatici e con buon performance status. La combinazione di dabrafenib e trametinib è efficace e approvata anche come trattamento adiuvante dei melanomi in stadio III radicalmente operati. Nonostante i buoni risultati raggiunti, la maggior parte dei pazienti con melanoma metastatico trattati con BRAF/MEKi manifesta
una progressione di malattia dopo 12-15 mesi di trattamento. Per tale ragione sono in corso studi di associazione delle varie doppiette con altri agenti a bersaglio molecolare o con anticorpi anti-PD1, con risultati preliminari incoraggianti. Un campo di estremo interesse, inoltre, è rappresentato dalle sequenze terapeutiche. A oggi, infatti, tutti gli agenti terapeutici per il melanoma metastatico sono disponibili sia in prima linea che nelle successive e la scelta della sequenza da utilizzare è a completa discrezione del clinico. Si attendono i risultati finali di diversi studi clinici che si spera possano fare chiarezza anche su questo aspetto. In conclusione, nonostante i grandi progressi ottenuti, restano ancora molti i quesiti aperti nell’ambito del trattamento del melanoma e in particolare sulla migliore strategia di utilizzo della target therapy nel trattamento del melanoma metastatico.
1. Candido S, Rapisarda V, Marconi A, et al. Analysis of the B-RafV600E mutation in cutaneous melanoma patients with occupational sun exposure. Oncol Rep 2014; 31 (3): 1079-82.
2. Catalogue of Somatic Mutations in Cancer (COSMIC) http: //www.sanger. ac.uk/cosmic
3. Wan PT, Garnett MJ, Roe SM, Lee S, et al. Oncogenic mutations of B-RAF. Cell 2004; 116(6): 855-6
4. Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KS. Paradoxical oncogenesis--the long-term effects of BRAF inhibition in melanoma. Nat Rev Clin Oncol 2013; 10(7): 390-9.
5. Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012; 366(8): 707-14.
6. Chapman PB, Hauschild A, Robert C, et al. BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364 (26): 2507-16.
7. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib
in BRAF(V600E) and BRAF(V600K) mutationpositive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, openlabel study. Lancet Oncol 2014; 15(3): 323-32.
8. Larkin J, Del Vecchio M, Ascierto PA, et al. Vemurafenib in patients with BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol 2014; 15(4): 436-44.
9. Ascierto PA, Minor D, Ribas A, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 2013; 31(26): 3205-11.
10. Hauschild A, Grob JJ, Demidov LV, et al. An update on overall survival (OS) and follow-on therapies in BREAK-3, a phase III, randomized trial: Dabrafenib (D) vs. Dacarbazine (DTIC) in patients (pts) with BRAF V600E mutation-positive metastatic melanoma (MM). Poster presented at ESMO Annual Meeting, 29 September, 2014; Madrid.
11. Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma,
untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012; 379(9829): 1893-901.
12. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol 2012; 13(11): 1087-95.
13. Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer 2017; 17(11): 676-91.
14. Delord JP, Robert C, Nyakas M, McArthur GA, et al. Phase I dose-escalation and -expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic <i>BRAF</i>-Mutant melanoma. Clin Cancer Res 2017; 23(18): 5339-48.
15. Turajlic S, Furney SJ, Stamp G, et al. BRAF Whole-genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann Oncol 2014; 25(5): 959-67.
16. Fedorenko IV, Paraiso KH, Smalley KS. Acquired and intrinsic BRAF inhibitor
resistance in BRAF V600E mutant melanoma. Biochem Pharmacol 2011; 82: 201-9.
17. Fattore L, Costantini S, Malpicci D, et al. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017; 8(13): 22262-78.
18. Caputo E, Miceli R, Motti ML, et al. AurkA inhibitors enhance the effects of B-RAF and MEK inhibitors in melanoma treatment. J Transl Med 2014;12: 216.
19. Caputo E, Wang E, Valentino A, et al. Ran signaling in melanoma: implications for the development of alternative therapeutic strategies. Cancer Lett 2015; 357(1): 286-96.
20. Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014; 4(1): 94-109.
21. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468(7326): 973-7.
22. Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011; 480(7377): 387-90.
23. Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA 2009; 106(48): 20411-6.
24. Shao Y, Aplin AE. Aplin Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res 2010; 70(16): 6670-81.
25. Smith MP, Brunton H, Rowling EJ, et al. Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 2016; 29(3): 270-284.
26. Müller J, Krijgsman O, Tsoi J, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014; 5: 5712.
27. Wellbrock C, Arozarena I Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res 2015; 28(4): 390-406.
28. Ribas A, Gonzalez R, Pavlick A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: A phase 1b study. Lancet Oncol 2014; 15: 954-65.
29. Pavlick AC, Ribas A, Gonzalez R, et al. Extended followup results of phase Ib study (BRIM7) of vemurafenib (VEM) with cobimetinib (COBI) in BRAF-mutant melanoma. J Clin Oncol 2015; 33 (Suppl 15): 9020-9020.
30. Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-] mutated melanoma. N Engl J Med 2014; 371: 1867-76.
31. Ascierto PA, McArthur GA, Dreno B, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)mutant melanoma (coBRIM): Updated efficacy results from a randomised, doubleblind, phase 3 trial. Lancet Oncol 2016; 17: 1248-60.
32. Dreno B, Ribas A, Larkin J, et al. Incidence, course, and management of toxicities associated with cobimetinib in combination with vemurafenib in the coBRIM study. Ann Oncol 2017; 28: 1137-44.
33. Society for Melanoma Research Congress (2019). Available online at: http// societymelanomaresearch.org/ congress
34. Society for Melanoma Research Congress
(2019). Available online at: https: //www. societymelanomaresearch.org/ congress
35. Infante JR, Fecher LA, Falchook GS, et al Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol 2012; 13(8): 773-81.
36. Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol 2013; 31: 482-9.
37. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAFmutated melanoma. N Engl J Med 2012; 367: 107-14.
38. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014; 371: 1877-88.
39. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study Ann Oncol 2017; 28 (7): 1631-9.
40. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015; 372 (1): 30-9.
41. Nathan PD, Robert C, Grob JJ, et al. Five-year analysis on the long-term effects of dabrafenib plus trametinib (D + T) in patients with BRAF V600–mutant unresectable or metastatic melanoma. J Clin Oncol 2019; 37 (suppl 15): 9507-9507.
42. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med 2017; 377: 1813-23.
43. Hauschild A, Dummer R, Santinami m et al. Longterm benefit of adjuvant dabrafenib + trametinib (D+T) in patients (pts) with resected stage III BRAF V600–mutant melanoma: Five-year analysis of COMBI-AD. J Clin Oncol 2020; 38 (suppl 15): 1000110001.
44. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018; 19: 603-15.
45. Gogas H, Ascierto PA, Flaherty K, et al. Update on overall survival in COLUMBUS: A randomized phase III trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in patients with BRAF V600mutant melanoma. J Clin Oncol 2020; 38 (suppl 15) 10012-10012.
46. Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med 2019; 25: 403-18.
47. Kumagai T, Wakimoto N, Yin D, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer 2007; 121: 656-65.
48. Khan ANH, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother 2008; 57 (5): 647-54.
49. Wu K, Liu T, Rios Z, et al. Heat shock proteins and cancer. Trend Pharm Sci 2017; 38: 226-56.
50. Acquaviva J, Smith DL, Jimenez JP, et al. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther 2014; 13(2): 353-63.
51. Paraiso KH, Haarberg HE, Wood E, et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res 2012; 18(9): 2502-14. Eroglu Z, Chen YA, Gibney GT, et al. Combined BRAF and HSP90 Inhibition in Patients with Unresectable BRAF V600E-Mutant Melanoma. Clin Cancer Res 2018; 24(22): 5516-24.
52. Davies MA. The role of the PI3K-AKT pathway in melanoma. Cancer J 2012; 18(2): 142-7.
53. Bendell JC, Rodon J, Burris HA, et al. Phase I, doseescalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012; 30: 282-90.
54. Greger JG, Eastman SD, Zhang V, et al. Combinations of BRAF, MEK, and PI3K/ mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther 2012; 11(4): 909-20.
55. GrilleyOlson JE, Bedard PL, Fasolo A, et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/ mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Invest New Drugs 2016; 34(6): 740-9.
56. Ascierto PA, Bechter O, Wolter P, et al. Phase Ib/II doseescalation study evaluating triple combination therapy
with a BRAF (encorafenib), MEK (binimetinib), and CDK 4/6 (ribociclib) inhibitor in patients (Pts) with BRAF V600mutant solid tumors and melanoma. J Clin Oncol 2017; 35 (suppl 15): 9518-9518.
57. Demkova L, Kucerova L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma Mol Cancer 2018; 17: 26.
58. Wolf J, Seto T, Han JY, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J Clin Oncol 2019; 37 (suppl 15).
59. Gay MC, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer 2017; 116(4): 415-23.
60. Ascierto PA, Dummer R. Immunological effects of BRAF+MEK inhibition. Oncoimmunology 2018; 7(9): e1468955.
61. Wilmott JS, Haydu LE, Menzies AM, et al. Dynamics of chemokine, cytokine, and growth factor serum levels in BRAF-mutant melanoma patients during BRAF inhibitor treatment. J Immunol 2014; 192(5): 2505-13.
62. Liu C, Peng W, Xu C, Lou Y, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res 2013; 19(2): 393-40.
63. Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013; 19(5): 1225-31.
64. Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib
and ipilimumab. N Engl J Med 2013; 368 (14): 1365-6.
65. Minor DR, Puzanov I, Callahan MK, Hug BA, Hoos A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res 2015; 28(5): 611-2.
66. Dummer R, Fernández AMA, Hansson J, et al. Preliminary findings from part 1 of COMBI-i: A phase III study of anti–PD-1 antibody PDR001 combined with dabrafenib (D) and trametinib (T) in previously untreated patients (pts) with advanced BRAF V600-mutant melanoma. J Clin Oncol 2018; 36 (5): 189-189.
67. Dummer R et al. Spartalizumab (S) + dabrafenib (D) + trametinib (T) in first-line treatment (tx) of BRAF V600mutant melanoma: clinical outcomes and biomarker analyses from COMBI-1 parts 1 and 2. Presented at: 16th International Congress of the Society for Melanoma Research; November 20-23, 2019; Salt Lake City, UT.
68. The Anti-PD-1 Antibody Spartalizumab in Combination With Dabrafenib and Trametinib in Advanced BRAF V600–Mutant Melanoma: Efficacy and Safety Findings From Parts 1 and 2 of the Phase III COMBI-i TrialASCO 2020.
69. Ribas A, Butler M, Lutzky J, et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J Clin Oncol 2015; 33: 3003-3003.
70. Miller WH, Kim TM, Lee CB, et al. Atezolizumab (A) + cobimetinib (C) in metastatic melanoma (mel): updated safety and clinical activity. J Clin Oncol 2017; 35: 3057-3057.
71. Ribas A, Hodi FS, Lawrence D, et al. 1216OKEYNOTE-022 update: phase 1 study of firstline pembrolizumab (pembro)
plus dabrafenib (D) and trametinib (T) for BRAFmutant advanced melanoma. Ann Oncol 2017; 28: mdx377.003. Ribas A, Hodi FS, Lawrence DP, et al. Pembrolizumab (pembro) in combination with dabrafenib (D) and trametinib (T) for BRAF-mutant advanced melanoma: phase 1 KEYNOTE-022 study. J Clin Oncol 2016; 34: 3014-3014.
72. Ascierto PA, Ferrucci PF, Fisher R, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 2019; 25(6): 941-6.
73. Society for Melanoma Research Congress (2019). Available online at: https: //www. societymelanomaresearch.org/ congress
74. A Study of Atezolizumab Plus Cobimetinib and Vemurafenib Versus Placebo Plus Cobimetinib and Vemurafenib in Previously Untreated BRAFv600 Mutation-Positive Patients With Metastatic or Unresectable Locally Advanced Melanoma. ClinicalTrials. gov. https: //bit.ly/2LS2IGG. Accessed December 13, 2019] [Roche announces positive Phase III study results for Tecentriq plus Cotellic and Zelboraf in people with previously untreated BRAF V600 mutation-positive advanced melanoma [press release]. Basel, Switzerland: Roche; December 13, 2019. https: //bit.ly/2RNTyyE. Accessed December 13, 2019
75. Gutzmer R, Stroyakovskiy D, Gogas H, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF V600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, doubleblind, placebo-controlled, phase 3 trial. Lancet 2020; 395(10240): 1835-44.
76. Novartis provides update on Phase III study evaluating
investigational spartalizumab (PDR001) in combination with Tafinlar® + Mekinist® in advanced melanoma [news release]. Basel. Published August 22, 2020. Accessed August 24, 2020. https: //www.novartis. com/news/media-releases/ novartis-provides-updatephase-iii-study-evaluatinginvestigational-spartalizumabpdr001-combination-tafinlarmekinist-advanced-melanoma
77. Nathan PD, Dummer R, Long GV, et al Spartalizumab plus dabrafenib and trametinib in patients with previously untreated BRAF V600–mutant unresectable or metastatic melanoma: results from the randomized part 3 of the Phase III COMBI-i trial Ann Oncol 2020; 31 (suppl 4): S1142-S1215.
78. Tawbi HAH, Amaria RN, Glitza IC, et al. Safety and preliminary activity data from a single center phase II study of triplet combination of nivolumab (N) with dabrafenib (D) and trametinib (T) [trident] in patients (Pts) with BRAF-mutated metastatic melanoma (MM). J Clin Oncol 2018; 36: 9560-9560.
79. Miller WH, Kim TM, Lee CB, et al. Atezolizumab (A) + cobimetinib (C) in metastatic melanoma (mel): Updated safety and clinical activity. J Clin Oncol. 2017; 35 (suppl): abstr 3057).
80. The AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discovery 2017; 7(8): 818-831. Dataset Version 6.
81. Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012; 7 (4): e35309.
FABRIZIO TABBÒ, CHIARA PISANO, SILVIA NOVELLO
La profonda comprensione dei meccanismi patogenetici alla base della trasformazione neoplastica nel tumore del polmone e l’avvento di raffinate ed efficaci tecnologie diagnostiche hanno permesso, negli ultimi anni, di indentificare sottogruppi di pazienti affetti da carcinoma del polmone non a piccole cellule ( non small cell lung cancer – NSCLC) caratterizzati da alterazioni molecolari peculiari. Parallelamente si è assistito al rapido sviluppo, preclinico e all’interno di studi clinici, di molecole capaci di colpire le cellule tumorali caratterizzate da questi difetti genetici. Tra questi ultimi, nello specifico per il NSCLC, vanno annoverate le mutazioni del gene EGFR e le traslocazioni dei geni ALK e ROS1. Recentemente, sono stati sviluppati numerosi farmaci capaci di inibire altre alterazioni molecolari, alcune delle quali a bassissima prevalenza. Per questo, diversi panel di esperti nonché società medico-scientifiche (CAP, AMP, IASLC, ESMO) hanno stilato linee-guida che meglio codificano i test molecolari da effettuare al momento della diagnosi per un paziente con NSCLC avanzato, particolarmente nel caso di istologia non-squamosa. 1
Tra i geni che hanno assunto rilevanza clinica rientra BRAF e in particolare la mutazione V600E. Quest’alterazione rara (presente all’incirca nel 2% dei NSCLC avanzati a istologia non-squamosa), se identificata, permette la corretta somministrazione di terapie mirate volte a bloccare la proliferazione BRAFdipendente delle cellule tumorali, consentendo migliori risultati clinici per il paziente rispetto al trattamento con chemioterapia standard. Nel presente capitolo tratteremo il ruolo delle mutazioni di BRAF, in termini di incidenza, diagnosi e trattamento, nel carcinoma polmonare.
BRAF è un proto-oncogene localizzato sul braccio lungo del cromosoma 7 (7q34), che codifica per la serina/treonina chinasi BRAF. Quest’ultima appartiene alla famiglia delle RAF chinasi, che svolgono un ruolo fondamentale nella via di segnalazione delle proteine MAPK (mitogen-activated protein kinases).2 In condizioni fisiologiche, l’attivazione di BRAF per omo- o etero-dimerizzazione è regolata dal signalling a monte da parte di RAS. Una volta fosforilato il dominio di attivazione, BRAF, a sua volta, fosforila e attiva la chinasi MEK1/2 che, a cascata, fosforila e attiva la chinasi ERK1/2. Infine, quest’ultima trasloca nel nucleo della cellula dove innesca fattori di trascrizione come FOS, TP53 e ELK12 (figura 4.1). In questo modo, il segnale di attivazione che origina dai recettori tirosin-chinasici presenti sulla superficie cellulare viene trasmesso al DNA della cellula, consentendo l’attivazione di geni coinvolti nella crescita, proliferazione e sopravvivenza della cellula stessa. Nei tessuti sani, BRAF è inattivato con meccanismo a feedback negativo, una volta che il segnale procede oltre nella sua trasduzione intra-cellulare. Le mutazioni del gene BRAF comportano un cambiamento strutturale della proteina responsabile dell’attivazione costitutiva della via di segnalazione a valle e, di conseguenza, della proliferazione incontrollata della cellula.3 Le mutazioni attivanti di BRAF, riscontrate sia nei tumori ematologici (come la leucemia a cellule capellute) che nei tumori solidi, possono svolgere il ruolo di driver oncogenico anche nel tumore al polmone.
Le mutazioni somatiche del gene BRAF sono state identificate nell’8% dei tumori solidi, tra cui il carcinoma colo-rettale, il melanoma, il carcinoma papillare della tiroide e il tumore del polmone. Nel NSCLC le mutazioni di BRAF sono rare e svolgono la funzione di driver oncogenico nel 2,5-3,5% dei casi.4 5 Quasi la metà di queste alterazioni corrisponde a mutazioni puntiformi a carico dell’aminoacido valina nel codone 600 (V600) a livello del dominio chinasico della proteina. Tra queste, la mutazione più comune è la V600E, che comporta la sostituzione dell’aminoacido valina con l’acido glutammico.
La difficoltà nell’identificare terapie targeted per le alterazioni di BRAF risiede nell’ampio repertorio mutazionale possibile. Infatti, le varianti mutazionali di BRAF possono essere distinte in tre categorie funzionali:6
• Classe 1: include le mutazioni a carico del codone 600, di cui la V600E è la più frequente, ma sono incluse nel gruppo anche altre tipologie di sostituzioni (ad esempio V600K, V600D). Tali mutazioni rendono la chinasi BRAF costitutivamente attiva, in grado di fosforilare e attivare gli effettori della segnalazione a valle, indipendentemente dall’attivazione di RAS e senza che sia necessaria la dimerizzazione.
BRAF come bersaglio terapeutico nel carcinoma del polmone non a piccole cellule 61
Dabrafenib
Vemurafenib
Encorafenib
Sorafenib
Dasatinib
Belvarafenib
Trametinib
Cobimetinib
Binimetinib
Ulixertinib LY3214996
Figura 4.1
La via di segnalazione di RAF, le molecole che la inibiscono e i meccanismi di resistenza. La dimerizzazione e autofosforilazione dei recettori tirosin-chinasici di mebrana (receptor tyrosine kinases – RTK) determina l’attivazione del complesso GRB2-SOS; quest’ultimo a sua volta determina attivazione di RAS, inducendo il passaggio dalla sua forma inattiva -GDP a quella attiva legata al -GTP. Una volta attivato, RAS fosforila le proteine della famiglia RAF (ARAF, BRAF e CRAF), riverberando il segnale a valle tramite l’attivazione a cascata delle proteine MEK1/2 ed ERK1/2. Quest’ultima, traslocando nel nucleo, provoca l’attivazione di fattori trascrizionali che regolano la proliferazione e la sopravvivenza cellulare. Le mutazioni di BRAF mantengono costitutivamente attiva questa via di segnalazione. Gli inibitori capaci di agire a livello di diversi effettori della via delle MAPK sono indicati. Parallelamente possono insorgere meccanismi di resistenza al trattamento: mutazioni (indicate da stelle rosse) nei geni RAS e MEK1/2 e anche di AKT e PTEN che stimolano la trasduzione del segnale, o attivazione e cross-talk con altre vie di segnalazione, come quella di PI3K, indotti dal reclutamento ligandodipendente di altri RTK. Illustrazione creata con BioRender.com
• Classe 2: queste varianti colpiscono codoni al di fuori del 600 (ad esempio K601E, L597V/Q/R, G469V/S/R/E/A, G464V). Anch’esse comportano un’iperattivazione della chinasi BRAF indipendentemente dall’azione di RAS ma, a differenza delle precedenti, necessitano di dimerizzazione per essere attive.
• Classe 3: include mutazioni inattivanti la chinasi BRAF (ad esempio G596R, D594Y/N/G/E, N581Y/S/I, G466V/L/E/A, D287Y), che quindi funzionerà meno o per nulla rispetto alla forma wild-type. L’attivazione costitutiva della via è quindi possibile per la presenza di mutazioni attivanti a monte (ad esempio a carico del recettore tirosin-chinasico di membrana o di RAS), che consentono la dimerizzazione di BRAF.
Le varianti di classe 2 e di classe 3 corrispondono alle mutazioni non-V600E. Il significato prognostico delle mutazioni di BRAF nel NSCLC è ancora poco chiaro: i risultati di piccole casistiche retrospettive sono talvolta in contraddizione tra loro, anche in ragione della bassa incidenza dell’alterazione indagata. Nella malattia in stadio precoce resecata, i pazienti con mutazioni BRAF V600E hanno una peggiore prognosi in termini di sopravvivenza libera da malattia (disease free survival - DFS, 12,5 vs. 52,1 mesi) e sopravvivenza globale (overall survival - OS, 29,3 vs 72,4 mesi) rispetto ai soggetti wild-type; tali differenze, invece, non emergono se si confrontano tutti i pazienti BRAF mutati (V600E e non-V600E) con la popolazione non mutata.4 Nel setting metastatico, in epoca antecedente all’introduzione degli inibitori di BRAF, non sono emerse differenze in termini di sopravvivenza libera da progressione (progression free survivalPFS) e OS tra i NSCLC BRAF-mutati (sia V600E che non-V600E) e le forme wildtype. Tuttavia, anche se non statisticamente significativa, è stata evidenziata una più breve PFS dopo chemioterapia a base di platino per i pazienti BRAFV600E+ rispetto ai non-V600E.7 Anche una casistica retrospettiva più recente non ha evidenziato differenze significative in termini di OS per le due classi di mutazioni di BRAF.8
Diversi studi sono stati condotti con lo scopo di valutare se vi siano caratteristiche clinico-patologiche associate alle mutazioni del gene BRAF. Tuttavia, la scarsa incidenza di queste ultime, unita alla variabilità delle evidenze emerse dagli studi, non consente di delineare delle specifiche caratteristiche associate alla categoria. Le alterazioni a carico di BRAF sono globalmente più frequenti nel sesso femminile, anche se le mutazioni non-V600E sembrano verificarsi con una maggiore frequenza negli uomini.5 Alcune evidenze suggeriscono che la maggior parte dei pazienti affetti da NSCLC con mutazioni di BRAF ha fumato o è ancora fumatore, ma con una diversa prevalenza rispetto al tipo di mutazione: in particolare, il 20-30% circa dei pazienti con mutazione V600E non ha mai fumato, mentre quasi tutti i pazienti con mutazioni non-V600E sono forti fumatori.9
Dal punto di vista istologico, le mutazioni di BRAF vengono rilevate quasi esclusivamente negli adenocarcinomi.5 Meno frequentemente, tali alterazioni possono essere reperite nei carcinomi polmonari di tipo squamoso, sarcomatoide o neuroendocrino (sia a grandi che a piccole cellule).10-12 A sua volta, l’istologia degli adenocarcinomi BRAF mutati è diversa per le mutazioni V600E rispetto alle non-V600E: i primi si presentano prevalentemente come istotipo non mucinoso con pattern di crescita micropapillare ed elevata espressione di TTF-1, mentre i secondi possono presentarsi con varie morfologie, compresa quella mucinosa.4
BRAF come bersaglio terapeutico nel carcinoma del polmone non a piccole cellule 63
Infine, vi sono iniziali evidenze circa una lieve tendenza alla maggiore espressione di PD-L1 e ad un maggior carico mutazionale (tumor mutational burdenTMB) per gli NSCLC BRAF-mutati rispetto ai non BRAF-mutati (sia wild-type che risultati positivi per altre mutazioni drivers), soprattutto per il sottogruppo con mutazioni non-V600E, suggerendo quindi un’ipotetica migliore risposta all’immunoterapia13 (come verrà successivamente discusso). Contrariamente a quanto accade nel tumore del colon, i tumori del polmone con mutazioni di BRAF non si associano a instabilità dei microsatelliti.13
La diagnosi di tumore al polmone è frequentemente ottenuta tramite piccole biopsie, essendo pertanto di fondamentale importanza saper gestire la quantità di tessuto a disposizione per svolgere sia la diagnosi istopatologica che le successive analisi molecolari. Analogamente a quanto avviene per altre alterazioni genetiche, le mutazioni di BRAF sono di solito identificate mediante metodiche estrattive e successivo sequenziamento del DNA. Il sequenziamento di Sanger rappresenta attualmente il gold standard per l’identificazione delle mutazioni puntiformi che rappresentano un target terapeutico nei tumori solidi. Per implementare la sensibilità della metodica, e ridurre così il tasso di falsi negativi per il rilevamento delle mutazioni di BRAF, così come per altre alterazioni, possono essere sfruttate le tecniche quantitative di reazione polimerasica a catena (RTPCR).14 Più recente è l’introduzione della Next Generation Sequencing (NGS), tecnologia che, tramite il sequenziamento simultaneo su larga scala di molteplici regioni del DNA, è in grado di ricercare alterazioni genetiche che possono rappresentare potenziali target terapeutici. L’utilizzo di pannelli genici allargati in NGS al momento della diagnosi consente una più completa caratterizzazione molecolare della malattia, identificando quei pazienti che possono beneficiare di terapie a bersaglio molecolare.
Per la mutazione V600E di BRAF l’immunoistochimica rappresenta una valida alternativa. Il clone VE1 è un anticorpo monoclonale (prodotto dalla Spring Bioscience Corporation) in grado di identificare con alta sensibilità e specificità il prodotto della mutazione V600E di BRAF nei tessuti, con un tasso di rilevamento sovrapponibile alle metodiche estrattive convenzionali.15 Lo stesso anticorpo non è invece in grado di identificare le mutazioni di BRAF non-V600E. L’eterogeneità delle mutazioni di BRAF nelle cellule tumorali e la piccola quantità di tessuto disponibile non consentono però a tale metodica di sostituire i metodi di sequenziamento.
Tutte le tecniche sopracitate richiedono una biopsia tessutale per poter essere applicate. Tuttavia, com’è noto, la biopsia polmonare è una procedura invasiva con un’elevata possibilità di complicanze maggiori e minori. Inoltre, l’eteroge-
neità intra-tumorale è elevata e il DNA utilizzato per le indagini genetiche non sempre adeguato. Per tutte queste ragioni la biopsia liquida, che valuta i campioni di sangue per la presenza di DNA tumorale circolante, rappresenta una promettente alternativa alla classica biopsia tessutale per effettuare le analisi mutazionali del tumore. Attualmente, l’unico utilizzo approvato della biopsia liquida per il NSCLC è la ricerca delle mutazioni a carico del gene EGFR, compresa la mutazione di resistenza T790M,16 mentre le evidenze per quanto riguarda la sua applicazione per la ricerca delle mutazioni di BRAF e il monitoraggio della terapia sono ancora limitate. Una recente revisione sistematica ha valutato se la biopsia liquida possa al giorno d’oggi rappresentare un’alternativa adatta per l’analisi mutazionale rispetto alla biopsia tessutale, quando entrambe disponibili, in pazienti con NSCLC in stadio avanzato. Nonostante i numerosi vantaggi a favore della biopsia liquida, i dati attuali suggeriscono che tale metodica non sia ancora in grado di sostituire appieno la sua controparte tessutale nel rilevamento delle mutazioni target, a prescindere dal gene testato, fatta eccezione per il gene EGFR, per il quale si ottengono risultati soddisfacenti. In particolare, per il gene BRAF la percentuale di concordanza tra biopsia tessutale e liquida è solo del 53,9%.17 Sono quindi necessari ulteriori studi per valutare il ruolo della biopsia liquida per quanto concerne la diagnosi e il monitoraggio dei pazienti affetti da NSCLC BRAF-mutati.
Diversi tentativi volti a bloccare la via di segnalazione delle MAPK sono stati messi in pratica. I primi dati riguardano i pazienti affetti da melanoma trattati con sorafenib, un inibitore tirosin-chinasico (tyrosine kinase inhibitor – TKI) multi-target diretto contro CRAF, BRAF, nella sua forma wild-type e mutata, e verso recettori di membrana. Tuttavia, i risultati clinici si sono dimostrati insoddisfacenti, in parte per la limitata attività inibitoria di questa molecola nei confronti della proteina mutata di BRAF. Per questo motivo, altre molecole più selettive sono state sviluppate e fanno parte oggi dell’armamentario farmaceutico. In particolare, vemurafenib, dabrafenib ed encorafenib sono TKI disegnati appositamente per legarsi alla tasca dell’ATP di BRAF, nella sua conformazione attiva, e in particolar modo della forma mutata, aumentando così la specificità per la proteina generata da BRAF-V600E.18 19 I primi risultati positivi, nuovamente osservati nei pazienti affetti da melanoma, hanno dimostrato come la monoterapia con un inibitore di BRAF (BRAFi) sia in grado di indurre un tasso di risposte obiettive (objective response rate – ORR) e una OS migliori di quelli osservati con la classica chemioterapia.20
BRAF come bersaglio terapeutico nel carcinoma del polmone non a piccole cellule 65
Nonostante questi traguardi positivi, quasi la metà dei pazienti trattati con BRAFi va incontro a recidiva di malattia, sviluppando resistenza alla terapia, dovuta a meccanismi di riattivazione del signalling delle MAPK. Inoltre, la somministrazione di questi farmaci può causare lo sviluppo di alterazioni cutanee iperproliferative, come il carcinoma squamocellulare, fenomeno questo osservato anche in altre patologie BRAF-guidate (ad esempio pazienti con leucemia a cellule capellute BRAF+).21 Poiché, sia lo sviluppo di resistenze, sia quest’ultimo fenomeno cutaneo avverso possono essere ricondotti a un’incompleta inibizione della via molecolare delle MAPK, la co-somministrazione di BRAFi e inibitori di MEK1/2 (MEKi) ha dimostrato di massimizzare il controllo della crescita tumorale e rallentare la comparsa di resistenza. Ancora una volta le prime evidenze di efficacia della combinazione (combo) di BRAFi e MEKi sono state ottenute nella cura del melanoma,22 portando le autorità regolatorie all’approvazione di tre differenti combinazioni di farmaci nelle neoplasie BRAF+: dabrafenib/trametinib, vemurafenib/cobimetinib ed encorafenib/binimetinib.
Vemurafenib
Il primo inibitore che ha riportato dati solidi di attività nel NSCLC è vemurafenib, la cui efficacia è stata indagata inizialmente all’interno di un basket trial (VE-BASKET) che valutava pazienti con neoplasie, non melanoma, V600 mutate. Il report finale di questo studio ha descritto una PFS mediana (mPFS) di 12,9 mesi e una OS mediana (mOS) non raggiunta nella popolazione non pre-trattata e una mPFS di 6,1 e una mOS di 15,4 mesi nella popolazione pre-trattata.23 Un altro studio, questa volta retrospettivo (EURAF), ha riportato i dati di attività di questo farmaco. Il gruppo di pazienti includeva 34 NSCLC avanzati (28 pazienti con mutazioni V600E e 6 con non-V600E): 5 hanno ricevuto il trattamento come prima linea, mentre 29 in linee successive. Son stati riportati una ORR del 54% e un tasso di controllo di malattia (disease control rate – DCR) del 96% per i 25 pazienti con mutazioni V600E trattati con vemurafenib. Da sottolineare come un paziente con una mutazione non-V600E (G596V) abbia ottenuto una risposta parziale; ciononostante i benefici per questa popolazione non-V600E sono risultati scarsi.24 Anche Mazieres e colleghi hanno valutato l’attività di vemurafenib a 960 mg bis in die in una coorte comprendente pazienti con NSCLC del programma francese Acsé. I pazienti, pre-trattati, hanno riportato una ORR media di 44,9% e una mPFS di 5,2 mesi, con assenza di beneficio clinico per il sottogruppo non-V600E. Questi dati sono stati recentemente aggiornati dai medesimi autori, riportando casi sporadici di risposta nel gruppo non-V600E: un paziente con mutazione V600K ha ottenuto una risposta parziale di malattia e altri due pazienti, con V600M e V600D, hanno ottenuto una stabilità di malattia (tabella 4.1).25
Tabella 4.1
Dati clinici di attività di molecole target ed ICI nel NSCLC BRAF+
Farmaco Studio Mutazioni di BRAF
Vemurafenib fase II
Vemurafenib retrospettivo V600E non-V600E
Vemurafenib fase II V600E non-V600E
Dabrafenib fase II V600E prima
+ trametinib fase
+ trametinib fase II V600E
ORR: objective response rate; DCR: disease control rate; PFS: progression free survival; OS: overall survival; ICI: immune checkpoint inhibitors.
Note: * Risultati per i pazienti con mutazioni V600E che hanno ricevuto vemurafenib ^ Dei 6 pazienti trattati in prima linea, 4 hanno riportato una risposta parziale § La popolazione di pazienti è stata considerata in modo omogeneo; la mPFS differisce tra il gruppo V600E (1,8 mesi) e quella del gruppo non-V600E (4,1 mesi)
In quest’ultimo lavoro gli Autori hanno anche riportato gli eventi avversi farmaco-relati: astenia, iporessia, dermatite acneiforme, nausea e diarrea di qualsiasi grado. Tossicità di grado 3 o più elevato sono state osservate: astenia, carcinoma cutaneo epidermoide, dermatite e aumento dei livelli di gamma glutamil-transferasi.25 Inoltre, benché non indagato sistematicamente, vemurafenib sembrerebbe essere attivo nel controllare la malattia localizzata al sistema nervoso centrale (SNC), particolarmente nel caso di interessamento lepto-meningeo.
BRAF come bersaglio terapeutico nel carcinoma del polmone non a piccole cellule 67
Dabrafenib e trametinib
L’attività di dabrafenib, come agente singolo, è stata inizialmente testata in uno studio di fase II multicentrico non randomizzato. Nel braccio A dello studio, 78 pazienti V600E+ pre-trattati e 6 naïve da trattamenti hanno ricevuto dabrafenib. Nel primo gruppo sono state osservate una ORR del 33% e una DCR del 58%. Entrambi gli outcome si sono dimostrati migliori nei pazienti che avevano ricevuto una sola linea di terapia precedente; la mOS si è attestata a 12,7 mesi.26 La coorte B dello stesso studio ha valutato la combo di dabrafenib e trametinib in pazienti pre-trattati. Nei 57 pazienti che hanno ricevuto il trattamento è stata registrata una ORR del 67% e un DCR dell’81% con una mPFS di 10,2 mesi e una durata della risposta (DoR) media di 9,8 mesi.27 Nella coorte C, invece, è stata presa in considerazione la stessa strategia di trattamento, ma come approccio di prima linea in pazienti non trattati. In presenza di risultati simili, gli outcome sono apparsi migliori: mPFS di 10,9 e DoR di 14,6 mesi, con una mOS di 24,6 mesi (follow-up mediano di 15,9 mesi)9 (tabella 4.1). L’attività di questa associazione BRAFi + MEKi nei pazienti con mutazioni non-V600E rimane appannaggio di rari casi: un paziente, il cui tumore riportava una differenziazione neuroendocrina e una mutazione G469R, ha ottenuto una stabilità di malattia, mentre una risposta parziale è stata osservata in un paziente con doppia mutazione di BRAF (G469A + W604C).28 Sulla base dei dati di questi studi le agenzie regolatorie hanno approvato il trattamento combo (dabrafenib + trametinib) per i pazienti con NSCLC avanzato V600E positivi, indipendentemente dalle linee di terapia precedenti. In tutte e tre le coorti del sopra citato studio di fase II, i pazienti trattati con localizzazioni a carico dell’SNC costituivano la minoranza. Benché i dati negli studi registrativi siano limitati, si può desumere un certo grado di controllo della malattia a livello centrale, basandosi sull’attività della combo nei pazienti con melanoma metastatico al SNC.
Gli eventi avversi ai trattamenti con TKI sono largamente noti e descritti per quanto riguarda i pazienti con NSCLC positivi a un oncogene. Tuttavia, l’associazione di dabrafenib + tramentinib ha aspetti peculiari: nello studio di fase II, gli eventi avversi più comunemente riportati erano febbre (che causava la maggior parte delle riduzioni di dose o interruzioni del trattamento), fatigue, nausea e cute secca.9 Altri eventi, seppur rari, erano alterazioni cutanee, oculari, emorragiche o muscolari.
Altri trial clinici in corso stanno valutando trattamenti di combinazione alternativi per pazienti affetti da NSCLC e mutazioni V600E classiche. Lo studio ARRAY-818-022 (fase II) sta valutando sicurezza, tollerabilità ed efficacia di encorafenib + binimetinib (NCT03915951). Lo studio B-FAST, invece, nella sua coorte E sta arruolando questo tipo di pazienti per sottoporli alla tripletta di vemurafenib + cobimetinib + atezolizumab, dopo una fase iniziale di sola associazione di BRAFi+MEKi (NCT03178552).
Molecole per i pazienti con mutazioni non-V600E
Altre strategie sono state prese in considerazione, nel tempo, per trattare pazienti con NSCLC caratterizzati da mutazioni di BRAF (sia V600E sia non-V600E). Sorafenib, già valutato nello studio EURAF, ha determinato risposte cliniche rilevanti in due pazienti con mutazioni non-V600E: G469R e G469V.29 30 L’esperienza di questi singoli casi suggerisce la necessità di ulteriori indagini in questo ambito. Anche dasatinib ha indotto una risposta di malattia prolungata in un paziente con una mutazione inattivante, Y472C, capace probabilmente di sensibilizzare le cellule tumorali a questo trattamento.31
Una chiara spiegazione ai bassi tassi di risposta dei pazienti con mutazioni non-V600E al trattamento combinato BRAFi+MEKi non esiste ancora. Alcune ipotesi suggeriscono una minor oncogenicità di queste mutazioni o una minor attività delle molecole disponibili nei confronti di queste ultime. Una recente analisi, che ha preso in considerazione l’andamento clinico dei pazienti con diverse tipologie di mutazioni di BRAF, ha sottolineato come le mutazioni di classe II e III siano gravate da comportamenti clinici nettamente più aggressivi.32 Conseguentemente, i ricercatori stanno valutando approcci alternativi. Le mutazioni di classe II sembrerebbero essere più dipendenti dalla via di segnalazione delle MAPK; perciò l’utilizzo di inibitori di MEK o, più a valle nella cascata, di inibitori di ERK, rappresenta una strategia promettente.32 In uno studio di fase I, un inibitore di ERK, ulixertinib, ha indotto una risposta parziale in due pazienti con mutazioni non-V600E (inclusa l’alterazione L579Q), oltre che in due pazienti V600E “classici”.33 Un altro inibitore di ERK1/2, LY3214996, è oggetto di sperimentazione, somministrato da solo o in combinazione, in uno studio di fase I (NCT02857270).
Inoltre, una terza generazione di inibitori di RAF è in corso di sviluppo, appositamente creata per evitare una riattivazione paradossa della cascata delle MAPK. Dati interessanti stanno emergendo da uno studio di fase I (NCT03118817), che sta valutando belvarafenib, un inibitore pan-RAF, in pazienti con tumori solidi BRAF+ o con mutazioni di K/NRAS, sia in monoterapia che in associazione a cobimetinib. LY3009120 è un altro inibitore pan-RAF, la cui sperimentazione di fase I (NCT02014116) è stata precocemente terminata per risultati clinici contrastanti.34
Immunoterapia
L’utilizzo di approcci immunoterapici nei tumori BRAF mutati è supportato dalle evidenze accumulatesi riguardo la capacità degli inibitori di BRAF/MEK di indurre un rimodellamento del microambiente tumorale. In particolare, sono stati osservati: un ridotto rilascio di citochine immunosoppressive, un aumentato infiltrato di linfociti T (CD4+ e CD8+) e un’aumentata espressione della protei-
BRAF come bersaglio terapeutico nel carcinoma del polmone non a piccole cellule 69
na PD-L1.35 Conseguentemente, un effetto sinergico tra BRAFi+MEKi e molecole anti-PD1 può essere ipotizzato ed è alla base del disegno di studi clinici, tuttora in corso, sia in pazienti con melanoma (COMBI-i and TRILOGY) che in pazienti con NSCLC avanzato.
Ad oggi, le evidenze cliniche di efficacia degli inibitori dei checkpoint immunitari (ICI) nei pazienti con NSCLC e mutazione di BRAF derivano soltanto da casistiche retrospettive, per definizione non conclusive. In particolare, due studi retrospettivi hanno valutato i segni di attività degli ICI in questi pazienti. Nel primo studio, in 22 pazienti (12 con mutazione V600E e 10 con mutazione non-V600E) è stata osservata una ORR del 25% e del 33% e una mPFS di 3,7 e 4,1 mesi nei due gruppi, rispettivamente.13 Il secondo studio, condotto in pazienti che avevano ricevuto ICI in seconda linea, ha riportato una ORR del 28%, una OS di 13,6 mesi e una PFS di 3,1 mesi36 (tabella 4.1). Apparentemente i tumori del polmone BRAF-mutati sembrano essere caratterizzati da un elevato livello di espressione di PD-L1 e un discreto carico mutazionale, considerando anche che originano per la maggior parte in pazienti fumatori e questo supporterebbe l’uso di ICI in questa popolazione. Tuttavia, recenti analisi dimostrano come l’utilizzo di ICI in seconde linee o oltre, in pazienti con alterazioni geniche specifiche, produca risultati comparabili rispetto a una popolazione non selezionata.37 Pertanto, studi clinici prospettici sono necessari per corroborare le evidenze preliminari, valutando l’opportunità di schemi chemio-immunoterapici a progressione dopo strategie mirate.
Meccanismi di resistenza ai trattamenti a bersaglio molecolare
Similmente ad altri scenari di neoplasie polmonari dipendenti da un oncogene, anche i pazienti con NSCLC BRAF-V600E+ tendono, in corso di terapia con dabrafenib + trametinib, a sviluppare resistenze e ad andare incontro a progressione di malattia. Se la maggior parte delle conoscenze nel campo è stata acquisita studiando campioni tumorali di melanoma, anche nel campo del tumore polmonare iniziano ad accumularsi dati consistenti.
I fenomeni di resistenza intrinseca, seppur rari, sono dovuti alla presenza de novo di alterazioni molecolari (come mutazioni di CDK4 o attivazione della via di segnalazione di Hippo) che causano resistenza primaria ai BRAFi/MEKi.13 I meccanismi di resistenza acquisiti possono essere, invece, suddivisi in: a) riattivazione della segnalazione di ERK attraverso la via delle MAPK; b) vie alternative che vicariano il segnale. Le mutazioni di K/N-RAS costituiscono la stragrande maggioranza (20-30%) dei meccanismi alla base del primo gruppo, causando una stimolazione BRAF-indipendente della via delle MAPK. Più raramente descritte sono le mutazioni di MEK1/2 (7%) (figura 4.1).38 In un lavoro recente di Facchi-
netti e colleghi, tra gli otto pazienti progrediti al trattamento combo dabrafenib/ trametinib, tre hanno riportato alterazioni nei suddetti geni: KRAS p.Q61R, NRAS p.Q61R e MEK1 p.K57N.39 Anche se rari, altri meccanismi in grado di ri-attivare la via delle MAPK esistono: le varianti di splicing di BRAF, le amplificazioni o i prodotti di fusione del gene BRAF stesso. L’attivazione di vie alternative, che costituiscono il secondo gruppo, può essere causata da diversi meccanismi. Tra gli altri si annoverano la stimolazione ligando-dipendente di recettori tirosin-chinasici di membrana, come il coinvolgimento della via dell’EGFR tramite attivazione di c-Jun, o il reclutamento della via di PI3K per mutazioni di AKT o perdita di funzione della proteina PTEN40 (figura 4.1). Nello studio precedentemente citato, un paziente progredito alla monoterapia con dabrafenib aveva sviluppato una mutazione frameshift di PTEN.39 Come già espresso, per superare queste limitazioni, altre strategie (ad esempio inibitori pan-RAF o inibitori di ERK1/2) sono in corso di valutazione, con l’obiettivo di aumentare gli algoritmi terapeutici disponibili per i pazienti con NSCLC BRAF+.
Le mutazioni del gene BRAF rappresentano un evento raro nei pazienti con NSCLC; ciononostante l’alterazione V600E rappresenta un meccanismo molecolare trainante per la trasformazione neoplastica. Per questo è raccomandata la ricerca di queste alterazioni, all’interno di una più ampia caratterizzazione molecolare del paziente con NSCLC avanzato a istologia non-squamosa. Ad oggi, le evidenze supportano in questo setting di pazienti il trattamento combinato orale con dabrafenib 150 mg per due volte e trametinib 2 mg una volta al giorno. Per quanto riguarda i pazienti non-V600E, invece, questi dovrebbero essere trattati come la restante popolazione, che non presenta mutazioni in oncogeni noti. Altre opportunità terapeutiche potenzialmente si potranno presentare sulla base delle ricerche pre-cliniche e cliniche attualmente in corso. Inoltre, l’utilizzo degli ICI sembra avere una non trascurabile applicazione clinica nei pazienti BRAFmutati, più che in presenza di altri oncogeni. Tuttavia, rimane ancora da chiarire il corretto momento e schema di somministrazione dell’immunoterapia (se in mono-somministrazione o in associazione ad altri farmaci).
Importante in ultimo menzionare, benché ancora rappresenti un fenomeno marginale e di rilevanza clinica da definire, la comparsa di mutazioni di BRAF in pazienti EGFR+ a progressione dopo terapia con osimertinib in prima linea.41 Ricerche future chiariranno la possibilità di implementare schemi terapeutici complessi volti a inibire entrambe gli oncogeni coinvolti (inibitori di EGFR + combo BRAFi/MEKi).
1. Mosele F, Remon J, Mateo J, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann Oncol 2020 Jul 28.
2. Pritchard AL, Hayward NK. Molecular pathways: mitogenactivated protein kinase pathway mutations and drug resistance. Clin Cancer Res 2013; 19(9): 2301-9.
3. Amaral T, Sinnberg T, Meier F, et al. The mitogen-activated protein kinase pathway in melanoma part I - Activation and primary resistance mechanisms to BRAF inhibition. Eur J Cancer 2017; 73: 85-92.
4. Marchetti A, Felicioni L, Malatesta S, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 2011; 29(26): 3574-9.
5. Cui G, Liu D, Li W, et al. A meta-analysis of the association between BRAF mutation and nonsmall cell lung cancer. Medicine (Baltimore) 2017; 96(14): e6552.
6. Lokhandwala PM, Tseng L-H, Rodriguez E, et al. Clinical mutational profiling and categorization of BRAF mutations in melanomas using next generation sequencing. BMC Cancer 2019; 19(1): 665.
7. Cardarella S, Ogino A, Nishino M, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res 2013; 19(16): 4532-40.
8. Tan I, Stinchcombe TE, Ready NE, et al. Therapeutic outcomes in non-small cell lung cancer with BRAF
mutations: a single institution, retrospective cohort study. Transl Lung Cancer Res 2019; 8(3): 258-67.
9. Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 2017; 18(10): 1307-16.
10. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489(7417): 519-25.
11. Schrock AB, Li SD, Frampton GM, et al. Pulmonary sarcomatoid carcinomas commonly harbor either potentially targetable genomic alterations or high tumor mutational burden as observed by comprehensive genomic profiling. J Thorac Oncol 2017; 12(6): 932-42.
12. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015; 524(7563): 47-53.
13. Dudnik E, Peled N, Nechushtan H, et al. BRAF mutant lung cancer: programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitors. J Thorac Oncol 2018; 13(8): 1128-37.
14. Gao J, Wu H, Shi X, Huo Z, Zhang J, Liang Z. Comparison of next-generation sequencing, quantitative pcr, and sanger sequencing for mutation profiling of EGFR, KRAS, PIK3CA and BRAF in clinical lung tumors. Clin Lab 2016; 62(4): 689-96.
15. Leonetti A, Facchinetti F, Rossi G, et al. BRAF in non-small cell lung cancer (NSCLC):
pickaxing another brick in the wall. Cancer Treat Rev 2018; 66: 82-94.
16. Planchard D, Popat S, Kerr K, et al. Metastatic nonsmall cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018; 29(Suppl 4): iv192-237.
17. Esagian SM, Grigoriadou GI, Nikas IP, et al. Comparison of liquid-based to tissue-based biopsy analysis by targeted next generation sequencing in advanced non-small cell lung cancer: a comprehensive systematic review. J Cancer Res Clin Oncol 2020; 146(8): 2051-66.
18. Rheault TR, Stellwagen JC, Adjabeng GM, et al. Discovery of dabrafenib: a selective inhibitor of raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med Chem Lett 2013; 4(3): 358-62.
19. Koelblinger P, Thuerigen O, Dummer R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr Opin Oncol 2018; 30(2): 125-33.
20. Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380(9839): 358-65.
21. Tiacci E, Pettirossi V, Schiavoni G, Falini B. Genomics of hairy cell leukemia. J Clin Oncol 2017; 35(9): 1002-10.
22. Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367(18): 1694-703.
23. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma
cancers with BRAF V600 mutations. N Engl J Med 2015; 373(8): 726-36.
24. Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015; 10(10): 1451-7.
25. Mazieres J, Cropet C, Montané L, et al. Vemurafenib in non-small-cell lung cancer patients with BRAFV600 and BRAFnonV600 mutations. Ann Oncol 2020; 31(2): 289-94.
26. Planchard D, Kim TM, Mazieres J, et al. Dabrafenib in patients with BRAF(V600E)positive advanced non-smallcell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016; 17(5): 642-50.
27. Planchard D, Besse B, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016; 17(7): 984-93.
28. Reyes R, Mayo-de-LasCasas C, Teixidó C, et al. Clinical benefit from BRAF/ MEK inhibition in a double non-V600E BRAF mutant lung adenocarcinoma: a case report. Clin Lung Cancer 2019; 20(3): e219-23.
29. Casadei Gardini A, Chiadini E, Faloppi L, et al. Efficacy of sorafenib in BRAF-mutated non-small-cell lung cancer (NSCLC) and no response in
synchronous BRAF wild typehepatocellular carcinoma: a case report. BMC Cancer 2016; 16: 429.
30. Sereno M, Moreno V, Moreno Rubio J, et al. A significant response to sorafenib in a woman with advanced lung adenocarcinoma and a BRAF non-V600 mutation. Anticancer Drugs 2015; 26(9): 1004-7.
31. Sen B, Peng S, Tang X, et al. Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci Transl Med 2012; 4(136): 136ra70.
32. Dagogo-Jack I, Martinez P, Yeap BY, et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant lung cancer. Clin Cancer Res 2019; 25(1): 158-65.
33. Sullivan RJ, Infante JR, Janku F, et al. First-in-Class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose-escalation and expansion study. Cancer Discov 2018; 8(2): 184-95.
34. Sullivan RJ, Hollebecque A, Flaherty KT, et al. A phase I study of LY3009120, a pan-RAF inhibitor, in patients with advanced or metastatic cancer. Mol Cancer Ther 2020; 19(2): 460-7.
35. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med 2006; 203(7): 1651-6.
36. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol 2019; 30(8): 1321-8.
37. Guisier F, Dubos-Arvis C, Viñas F, et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET tanslocation: GFPC 01-2018. J Thorac Oncol 2020; 15(4): 628-36.
38. Johnson DB, Menzies AM, Zimmer L, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer 2015; 51(18): 2792-9.
39. Facchinetti F, Lacroix L, Mezquita L, et al. Molecular mechanisms of resistance to BRAF and MEK inhibitors in BRAFV600E non-small cell lung cancer. Eur J Cancer 2020; 132: 211-23.
40. Obaid NM, Bedard K, Huang W-Y. Strategies for overcoming resistance in tumours harboring BRAF mutations. Int J Mol Sci 2017; 18(3).
41. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer 2019; 121(9): 725-37.
CHIARA CREMOLINI
La caratterizzazione molecolare del carcinoma colorettale ha portato negli anni recenti alla frammentazione della patologia in diverse entità nosologiche e ha permesso di identificare le peculiarità di tumori con caratteristiche molecolari differenti, nella auspicabile prospettiva di sfruttare tali conoscenze per offrire ai pazienti trattamenti mirati.
La storia degli avanzamenti delle conoscenze sull’importanza e il ruolo delle mutazioni di BRAF è un ottimo esempio di come gli sforzi di ricerca traslazionale abbiano consentito di trasferire conoscenze preliminari sull’impatto biologico di una specifica alterazione molecolare in nuove opzioni terapeutiche per i pazienti affetti. Ovvia conseguenza di tale evoluzione è la necessità di conoscere lo stato mutazionale di BRAF laddove questa informazione incida in modo concreto sulla strada terapeutica dei pazienti e sulle strategie che verranno adottate.
Oggi la conoscenza dello stato mutazionale di BRAF è fondamentale, accanto a quella di altre caratteristiche molecolari (KRAS/NRAS e instabilità microsatellitare in primis) per definire le migliori opzioni terapeutiche per i pazienti affetti da carcinoma colorettale metastatico, su cui pertanto ci concentreremo in questo capitolo.
Caratteristiche e prognosi del carcinoma colorettale metastatico BRAF mutato
Le mutazioni di BRAF si riscontrano nell’8-12% dei carcinomi colorettali metastatici e più del 95% di tali alterazioni consiste in una trasversione T>A nell’esone 15 risultante in una sostituzione aminoacidica da valina ad acido glutammico (V600E), capace di incrementare l’attività chinasica della proteina che ne deriva
di circa dieci volte rispetto alla sequenza wild-type.1 2 Il trattamento del carcinoma colorettale metastatico BRAF V600E mutato si è da subito rivelato una sfida difficile della moderna oncologia a causa dello spiccato impatto prognostico sfavorevole determinato da questa mutazione. La sopravvivenza complessiva (OS) di questi pazienti non superava in casistiche da studi controllati e da dati di real life i 12 mesi, durata vicina a quella della sopravvivenza libera da progressione (PFS) in prima linea dei pazienti BRAF wild-type, la cui aspettativa di vita oggi si assesta invece sui 25-30 mesi.3
I tumori BRAF V600E mutati sono più frequentemente a origine dalle porzioni prossimali del colon (a monte rispetto alla flessura splenica), presentano istologia mucinosa, sono più frequenti in soggetti più anziani e di sesso femminile, e metastatizzano più spesso a linfonodi e peritoneo. Dal punto di vista molecolare in questo sottogruppo si riscontrano più spesso instabilità microsatellitare e ipermetilazione delle isole CpG.4-6 In particolare, l’associazione con l’instabilità microsatellitare, caratteristica che si riscontra in circa il 5% di tutti i tumori colorettali metastatici mentre arriva al 30% circa nei tumori BRAF V600E mutati, ha una duplice ripercussione: da un lato impone una riflessione sulla miglior gestione terapeutica di questi tumori, che approfondiremo in seguito; dall’altro evidenzia il ruolo della determinazione dello stato mutazionale di BRAF nello screening per la sindrome di Lynch.
Infatti, l’analisi dell’instabilità dei microsatelliti su campione tessutale è oggi raccomandata da linee-guida nazionali e internazionali per identificare quei casi di carcinomi colorettali, indipendentemente dallo stadio di presentazione e dalla familiarità dei soggetti che ne sono portatori, associati appunto alla sindrome di Lynch. L’instabilità dei microsatelliti, spia di un deficit nei sistemi di riparo del DNA che determina l’accumularsi di mutazioni, può tuttavia essere un evento germinale o somatico. Solo nel primo caso è possibile fare diagnosi di sindrome di Lynch, individuando pertanto un maggior rischio di patologie neoplastiche oltre al carcinoma colorettale, tra cui carcinoma gastrico, tumori uroteliali e tumori ginecologici femminili in quei soggetti che già hanno sviluppato un carcinoma colorettale, e ponendo la necessità di un counselling genetico specifico per i loro familiari. La compresenza di instabilità microsatellitare e mutazione di BRAF V600E identifica forme sporadiche, non riconducibili alla sindrome di Lynch, per cui in caso di mancata espressione della proteina MLH1 è consigliabile ricercare la mutazione di BRAF che consente di escludere la sindrome di Lynch. In caso contrario, l’instabilità microsatellitare andrà testata anche su DNA germinale (da prelievo ematico) per approfondire la presenza o meno di questa sindrome familiare.7 8
Altri codoni di BRAF oltre al 600 possono presentare alterazioni genomiche molto più rare. Studi funzionali hanno permesso di classificare tali mutazioni in due classi (classi 2 e 3, mentre le alterazioni del codone 600 appartengono alla classe 1) in base al loro effetto sull’attività chinasica di BRAF. Mentre le mutazioni di classe 2 determinano un modesto incremento dell’attività chinasica di BRAF
Le mutazioni di BRAF nel carcinomadel colon-retto 75
rispetto alla proteina wild-type, quelle di classe 3 presentano un’attività addirittura inferiore. Coerentemente con tali osservazioni, i tumori BRAF mutati ma con alterazioni diverse da quelle a carico del codone 600 sono molto diversi dai tumori BRAF V600E mutati sia rispetto alle caratteristiche clinico-patologiche che alla prognosi. Tutte le considerazioni relative al trattamento dei tumori BRAF V600E mutati che seguiranno non si applicano pertanto a quelli che presentano altre mutazioni a carico dello stesso gene.9 10
L’esperienza clinica e le analisi post-hoc di studi controllati mostrano chiaramente come i trattamenti convenzionali, comunemente indicati nella prima linea della malattia metastatica, diano risultati del tutto insoddisfacenti in presenza della mutazione BRAF V600E, con durate di PFS di 6-7 mesi con le doppiette di chemioterapia (FOLFOX/XELOX/FOLFIRI) in combinazione a bevacizumab o a farmaci anti-EGFR.11 12 Si è lungamente dibattuto rispetto all’utilità degli antiEGFR in questo sottogruppo e, pur senza una dimostrazione formale del potere predittivo negativo della mutazione di BRAF rispetto all’efficacia di tali farmaci, il modesto impatto della loro aggiunta alla chemioterapia convenzionale ne ha fortemente limitato l’impiego nella pratica clinica.
L’analisi post-hoc di sottogruppo di uno studio di fase III randomizzato, TRIBE, che dimostrava l’efficacia dell’intensificazione della chemioterapia di prima linea dalla doppietta standard FOLFIRI alla tripletta FOLFOXIRI in combinazione con bevacizumab, metteva in evidenza come il sottogruppo di pazienti BRAF mutati sembrasse beneficiare maggiormente dell’approccio intensificato upfront, pur in assenza di un significativo test di interazione.3 Sulla base di tali dati, che corroboravano i precedenti risultati di un’esperienza retrospettiva13 e di uno studio prospettico di fase II,14 FOLFOXIRI +/- bevacizumab è stato raccomandato come opzione preferibile in prima linea dalle linee-guida della Società europea di oncologia medica (ESMO).15 Tale indicazione si basava certamente su un numero limitato di pazienti trattati ma era fondata sul razionale biologico di mettere in campo d’emblée una strategia più aggressiva per provare a contrastare l’aggressività biologica intrinseca di questi tumori, in virtù anche della limitata percentuale di casi in cui è possibile proporre un trattamento dopo l’eventuale progressione di malattia. Allo stesso tempo non tutti i pazienti con malattia BRAF mutata sono in grado di ricevere un trattamento intensificato, gravato per definizione da un profilo di tossicità più impegnativo, in ragione anche dell’alto carico di malattia, delle condizioni di partenza più compromesse e dell’età più avanzata alla diagnosi, comuni in questo sottogruppo molecolare.
Peraltro, la suggestione di una maggiore efficacia della tripletta FOLFOXIRI nel sottogruppo BRAF mutato non è stata confermata dal successivo studio di fase III TRIBE2 in cui FOLFOXIRI/bevacizumab, seguito dopo la progressione dalla reintroduzione dello stesso regime, era confrontato con la sequenza di doppiette FOLFOX/bevacizumab seguita da FOLFIRI/bevacizumab dopo la progressione.16 Analogamente, una recente metanalisi di dati individuali da cinque studi randomizzati, comprendente TRIBE, TRIBE2 e tre studi di fase II in cui FOLFOX/bevacizumab era il braccio di controllo, conferma come tra i 115 pazienti con tumore BRAF mutato non si osservi particolare beneficio dall’intensificazione della chemioterapia in termini di OS (HR: 1,11; IC: 95% 0,75-1,73). Sebbene non siano chiare le ragioni della discordanza dei risultati dell’analisi di sottogruppo dello studio TRIBE rispetto alle esperienze successive (diversa efficacia di irinotecano rispetto a oxaliplatino? Eterogeneità dei sottogruppi inclusi?), il valore di FOLFOXIRI/bevacizumab in questo sottogruppo è certamente messo in dubbio in virtù dell’assenza di chiaro beneficio a fronte dell’atteso incremento delle tossicità chemio-correlate.17
Evidenze retrospettive sembrano anche indicare un minor impatto della chirurgia resettiva secondaria sulle metastasi epatiche nei pazienti con malattia BRAF mutata.18 19 Tali suggestioni tuttavia non sono state confermate in tutte le serie analizzate.20 21 Resta comunque evidente come la frequente diffusione extraepatica della malattia BRAF mutata, le condizioni spesso precarie di questi pazienti, e l’intrinseca aggressività di questa patologia non rendano i soggetti con carcinoma colorettale metastatico BRAF mutato, ancorché limitato al fegato, i migliori candidati per i trattamenti locoregionali, pur nell’estrema variabilità ed eterogeneità dei quadri clinici.
Numerosi sforzi sono stati profusi allo scopo di trasformare la mutazione di BRAF da punto di forza a tallone d’Achille per le cellule di tumore colorettale analogamente a quanto ottenuto in altri tumori solidi, in primis il melanoma. Tuttavia, i risultati iniziali con i farmaci target anti-BRAF si sono rivelati inaspettatamente insoddisfacenti con percentuali molto basse di risposte obiettive.22 Nel tentativo di individuare le ragioni di tale fallimento, il ritorno dalla clinica al laboratorio ha consentito di identificare nell’iperattivazione di EGFR capace di riattivare il segnale veicolato dalle MAPKinasi a valle di BRAF in risposta alla sua inibizione un meccanismo di resistenza intrinseca delle cellule tumorali di carcinoma colorettale. Tale fenomeno non si verificava, viceversa, nelle cellule di melanoma. L’inibizione di EGFR in contemporanea a quella di BRAF determinava invece morte cellulare.23
Sulla scorta di tali evidenze, la combinazione di BRAF inibitori (vemurafenib o dabrafenib) e anticorpi anti-EGFR (cetuximab o panitumumab) è stata valutata
Le mutazioni di BRAF nel carcinomadel colon-retto 77
in successivi studi di fase I e II che hanno confermato l’indicazione preclinica mostrando tassi di risposte decisamente più promettenti rispetto a quanto riportato dagli inibitori di BRAF in monoterapia, pur con risultati eterogenei nelle varie esperienze.24
Studi preclinici avevano anche mostrato come una più profonda inibizione dell’asse di trasduzione del segnale si potesse ottenere con la simultanea inibizione di BRAF e MEK. Anche tale strategia terapeutica, pur consentendo di ottenere tassi di risposta migliori rispetto ai BRAF inibitori in monoterapia (circa 20%) con una durata mediana di PFS di 4-5 mesi, non si avvicinava tuttavia all’attività dimostrata nel melanoma metastatico,25 evidenziando la necessità di un ulteriore passo in avanti per l’identificazione di un’opzione target efficace in questo setting.
Sulla base dei promettenti risultati di uno studio di fase II che valutava la combinazione di encorafenib, un nuovo BRAF inibitore ATP-competitivo con emivita di dissociazione di 10 volte più lunga rispetto a dabrafenib e vemurafenib, e cetuximab,26 è stato condotto lo studio di fase III randomizzato BEACON, il primo sforzo globale in questa specifica popolazione molecolare.27 L’efficacia della combinazione di encorafenib e cetuximab con o senza l’aggiunta del MEK inibitore binimetinib è stata confrontata con quella di cetuximab con FOLFIRI o irinotecano in monoterapia (braccio di controllo) a scelta degli sperimentatori in pazienti con carcinoma colorettale metastatico BRAF V600E mutato già pretrattati con almeno una linea di terapia sistemica. Una fase iniziale di safety lead-in ha permesso di valutare la tollerabilità e il profilo di tossicità della tripla combinazione target, oltre a ottenere preliminari indicazioni di attività, peraltro estremamente promettenti.28 Si registravano, infatti, un tasso di risposte del 48%, una PFS mediana di 8 mesi e una OS mediana di 15,3 mesi.
Lo studio ha quindi raggiunto i suoi due endpoint co-primari, dimostrando un vantaggio sia in OS che in tasso di risposte obiettive per la tripletta target rispetto al controllo (9 vs. 5,4 mesi, p<0,001; 26% vs. 2%, p<0,001). Anche la doppietta determinava risultati significativamente migliori rispetto al controllo (OS mediana: 8,4 vs. 5,4 mesi, p<0,001; tasso di risposte: 20% vs. 2%, p<0,001) e anche a un follow-up più maturo, successivo alla pubblicazione dei risultati, si manteneva la medesima entità di beneficio. Sebbene lo studio non fosse formalmente disegnato per confrontare la tripletta con la doppietta target, l’aggiunta del MEK inibitore non determinava un vantaggio rilevante né in OS (mediana: 9,3 vs. 9,3 mesi) né in risposte obiettive (27% vs. 20%), a fronte di un incremento di alcune tossicità specifiche.29 Dal punto di vista della safety, entrambe le strategie a bersaglio molecolare erano gravate da una minore percentuale di eventi avversi di grado 3 e 4 rispetto al trattamento standard che consistevano prevalentemente in diarrea (10% con la tripletta vs. 2% con la doppietta vs. 10% nel braccio di controllo), nausea (5% vs. <1% vs. 1%) e anemia (10% vs. 5% vs. 4%). Il MEK inibitore non pareva in grado di ridurre la tossicità del BRAF inibitore, in particolare il verificarsi di tumori cutanei secon-
dari e di altri eventi avversi cutanei, dovuti all’attivazione paradossa della via delle MAPKinasi a valle attraverso vie di trasduzione capaci di bypassare BRAF, come invece si osserva con altre combinazioni di BRAF e MEK inibitore (vemurafenib/ cobimetinib e dabrafenib/trametinib).30 31 Una possibile spiegazione risiederebbe nella più efficace inibizione di BRAF da parte di encorafenib, che determinerebbe una minore attivazione paradossa delle MAPK.
Sulla base dei risultati dello studio BEACON sia l’ente regolatorio statunitense (FDA) che quello europeo (EMA) hanno approvato l’utilizzo di encorafenib e cetuximab come trattamento di pazienti affetti da carcinoma colorettale metastatico BRAF V600E mutato che abbiano già ricevuto almeno una linea di terapia. La strategia a target molecolare è pertanto ad oggi considerata la freccia più importante per il trattamento di questa patologia, sebbene una critica condivisibile al disegno dello studio BEACON sia quella relativa alla scelta del braccio di controllo. Come precedentemente accennato, infatti, l’utilità degli anticorpi monoclonali anti-EGFR da soli o in combinazione con la chemioterapia in questo sottogruppo è molto discutibile mentre l’aggiunta dei farmaci antiangiogenici pur in campioni di dimensione limitata sembrerebbe determinare una migliore efficacia.32 33
Le analisi di sottogruppo dello studio BEACON non permettono di identificare caratteristiche associate a maggiore o minore beneficio dal trattamento. Va tuttavia osservato come la quota di pazienti con instabilità microsatellitare inclusi nello studio (il 5-10% del totale della popolazione) sia molto inferiore rispetto a quanto atteso sulla base dei noti dati di prevalenza (circa il 30%). Tale riscontro pare conseguenza dell’incremento nell’uso dell’immunoterapia, strategia terapeutica dimostratasi estremamente efficace nei tumori con instabilità microsatellitare indipendentemente dall’istologia di origine, durante la conduzione dello studio BEACON.
In effetti, il vantaggio dall’uso dell’immunoterapia pare indipendente dallo stato mutazionale di BRAF34-36 e la durata del beneficio osservata sia con pembrolizumab che con nivolumab associato o meno a ipilimumab suggerisce di privilegiare questo trattamento rispetto alla terapia target in prima battuta.
La combinazione di encorafenib, cetuximab e binimetinib è stata valutata anche in uno studio di fase II in prima linea, ovvero in pazienti metastatici non precedentemente trattati. Il disegno dello studio prevedeva l’arruolamento di due coorti successive di pazienti, l’una di 40 e l’altra di 50 pazienti. I risultati sui primi 40 pazienti arruolati mettono in evidenza un tasso di risposte del 50% e una durata di PFS mediana di circa 5 mesi37 che certamente necessitano di ulteriore conferma e di confronto randomizzato con l’attuale standard terapeutico prima di poter aprire le porte della prima linea alla strategia target.
Le mutazioni di BRAF nel carcinomadel colon-retto 79
Negli ultimi anni non solo la mutazione V600E di BRAF si è affermata come biomarker identificativo di una specifica sottopopolazione con approccio terapeutico mirato, ma sono largamente incrementate le conoscenze sull’eterogeneità clinica38 e molecolare39 di questo sottotipo. In considerazione dell’effetto pro-immunogenico dell’inibizione di BRAF, la combinazione di terapia target e inibitori dei checkpoint immunologici nei tumori microsatelliti stabili, intrinsecamente resistenti all’immunoterapia, è attualmente oggetto di diversi studi clinici.
Nel frattempo le evidenze fin qui raccolte hanno permesso di definire un nuovo algoritmo terapeutico personalizzato per i pazienti con carcinoma colorettale metastatico BRAF V600E mutato (figura 5.1).
Carcinoma colorettale metastatico BRAF V600E mut
MSI-high/dMMR (30%)
1a linea
2a linea
3a linea
4a linea
Immunoterapia
Encorafenib + Cetuximab
Chemioterapia
Chemioterapia o FTD/TPI o Rego**
MMS/pMMR (70%)
FOLFOX(IRI) + Bev*
Encorafenib + Cetuximab
Chemioterapia o FTD/TPI o Rego**
* Considerare la reintroduzione dopo la PD in caso di ottima durata della risposta (PFS >12 mesi)
** In base ai trattamenti precedenti e alla loro efficacia/tossicità
Figura 5.1
Algoritmo terapeutico basato sulle attuali evidenze da studi clinici sul trattamento del carcinoma colorettale metastatico BRAF V600E mutato.
Bev: bevacizumab; FTD/TPI: tifluridina/tipiracil; Rego: regorafenib; PD: progressione di malattia
1. Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res 2014; 20(20): 5322-30.
2. Safaee Ardekani G, Jafarnejad SM, Tan L, Saeedi A, Li G. The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PloS One 2012; 7(10): e47054.
3. Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol 2015; 16(13): 1306-15.
4. Schirripa M, Cremolini C, Loupakis F, et al. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. Int J Cancer 2015; 136(1): 83-90.
5. Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011; 117: 4623-32.
6. Tie J, Gibbs P, Lipton L, et al. Optimizing targeted therapeutic development: analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer 2011; 128: 2075-84.
7. Sepulveda AR, Hamilton SR, Allegra C, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American Society for Clinical Pathology, College of American Pathologists,
Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol 2017: 35 (13): 1453-86.
8. NICE dg27. Molecular testing strategies for Lynch syndrome in people with colorectal cancer. Nice.org.uk/guidance/ dg27
9. Schirripa M, Biason P, Lonardi S, et al. Class 1, 2, and 3 BRAF-mutated metastatic colorectal cancer: a detailed clinical, pathologic, and molecular characterization. Clin Cancer Res 2019; 25(13): 3954-61.
10. Cremolini C, Di Bartolomeo M, Amatu A, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol 2015; 26(10): 2092-7.
11. Stintzing S, Miller-Phillips L, Modest DP, et al. Impact of BRAF and RAS mutations on first-line efficacy of FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab: analysis of the FIRE-3 (AIO KRK-0306) study. Eur J Cancer 2017; 79: 50-60.
12. Innocenti F, Ou FS, Qu X, et al. Mutational analysis of patients with colorectal cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol 2019; 37(14): 1217-27.
13. Masi G, Loupakis F, Salvatore L, et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: a phase 2 trial. Lancet Oncol 2010; 11(9): 845-52.
14. Loupakis F, Cremolini C, Salvatore L, et al. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant
metastatic colorectal cancer. Eur J Cancer 2014; 50(1): 57-63.
15. Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016; 27(8): 1386-422.
16. Cremolini C, Antoniotti C, Rossini D, et al. Upfront FOLFOXIRI plus bevacizumab and reintroduction after progression versus mFOLFOX6 plus bevacizumab followed by FOLFIRI plus bevacizumab in the treatment of patients with metastatic colorectal cancer (TRIBE2): a multicentre, openlabel, phase 3, randomised, controlled trial. Lancet Oncol 2020; 21(4): 497-507.
17. Cremolini C, Antoniotti C, Stein A, et al. Individual patient data meta-analysis of FOLFOXIRI plus bevacizumab versus doublets plus bevacizumab as initial therapy of unresectable metastatic colorectal cancer [published online ahead of print, 2020 Aug 20]. J Clin Oncol 2020.
18. Schirripa M, Bergamo F, Cremolini C, et al. BRAF and RAS mutations as prognostic factors in metastatic colorectal cancer patients undergoing liver resection. Br J Cancer 2015; 112(12): 1921-8.
19. Yaeger R, Cercek A, Chou JF, et al. BRAF mutation predicts for poor outcomes after metastasectomy in patients with metastatic colorectal cancer. Cancer 2014; 120(15): 2316-24.
20. Cremolini C, Casagrande M, Loupakis F, et al. Efficacy of FOLFOXIRI plus bevacizumab in liver-limited metastatic colorectal cancer: a pooled analysis of clinical studies by Gruppo Oncologico del Nord Ovest. Eur J Cancer 2017; 73: 74-84.
21. Margonis GA, Buettner S, Andreatos N, et al. Association of BRAF mutations with survival and recurrence in surgically treated patients with metastatic colorectal liver cancer. JAMA Surg 2018; 153(7): e180996.
22. Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 2015; 33(34): 4032-8.
23. Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483(7387): 100-3.
24. Yaeger R, Cercek A, O’Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res 2015; 21(6): 1313-20.
25. Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol 2015; 33(34): 4023-31.
26. Corcoran RB, André T, Atreya CE, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600Emutant colorectal cancer. Cancer Discov 2018; 8(4): 428-43.
27. Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med 2019; 381(17): 1632-43.
28. Van Cutsem E, Huijberts S, Grothey A, et al. Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAF V600E-mutant
metastatic colorectal cancer: safety lead-in results from the phase III BEACON colorectal cancer study. J Clin Oncol 2019; 37(17): 1460-9.
29. Kopetz S, Grothey A, Van Cutsem E, et al. Encorafenib plus cetuximab with or without binimetinib for BRAF V600E metastatic colorectal cancer: updated survival results from a randomized, three-arm, phase III study versus choice of either irinotecan or FOLFIRI plus cetuximab (BEACON CRC). J Clin Oncol 2020; 38(suppl; abstr 4001).
30. Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAFmutated melanoma. N Engl J Med 2014; 371(20): 1867-76.
31. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014; 371(20): 1877-88.
32. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 2012; 30(28): 3499-506.
33. Tabernero J, Yoshino T, Cohn AL, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol 2015; 16(5): 499-508.
34. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017; 18(9): 1182-91.
35. Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol 2018; 36(8): 773-9.
36. André T, Shiu K, Kim TW, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: the phase 3 KEYNOTE-177 study. J Clin Oncol 38: 2020 (suppl; abstr LBA4).
37. Grothey A, Tabernero J, Taieb J, et al. LBA-5 ANCHOR CRC: a single-arm, phase 2 study of encorafenib, binimetinib plus cetuximab in previously untreated BRAF V600E-mutant metastatic colorectal cancer. Ann Oncol 2020; 3(s): S242243.
38. Loupakis F, Intini R, Cremolini C, et al. A validated prognostic classifier for V600EBRAFmutated metastatic colorectal cancer: the ‘BRAF BeCool’ study. Eur J Cancer 2019; 118: 121-30.
39. Barras D, Missiaglia E, Wirapati P, et al. BRAF V600E mutant colorectal cancer subtypes based on gene expression. Clin Cancer Res 2017; 23(1): 104-15.
NICOLA NORMANNO, CARMINE PINTO
I capitoli precedenti hanno fornito dettagli sugli attuali approcci diagnostici per l’identificazione delle mutazioni di BRAF nei tessuti tumorali e sul ruolo di queste alterazioni genetiche come fattori predittivi di risposta alle terapie target nei carcinomi del colon e del polmone e nel melanoma, le tre neoplasie per le quali sono disponibili terapie anti-BRAF approvate dagli enti regolatori. In questo capitolo, illustreremo gli approcci diagnostici futuri, con particolare riguardo alla biopsia liquida, nonché le prospettive di sviluppo delle terapie anti-BRAF in altre patologie neoplastiche.
L’analisi del tessuto tumorale rappresenta l’approccio standard per l’individuazione di alterazioni genetiche driver che sono spesso anche fattori predittivi di risposta alla terapia a bersaglio molecolare. Tuttavia, nei casi in cui non è disponibile tessuto tumorale, le linee-guida nazionali e internazionali suggeriscono di ricorrere alla biopsia liquida. Il termine biopsia liquida indica genericamente lo studio di biomarcatori presenti nei liquidi biologici. In realtà, l’unico approccio attualmente impiegato nella pratica clinica è rappresentato dall’analisi del DNA tumorale circolante (ctDNA), che viene rilasciato dalle cellule tumorali in seguito a fenomeni di apoptosi e di necrosi, ma anche per secrezione diretta.1 2 Il ctDNA rappresenta tuttavia solo una frazione del DNA libero circolante (cfDNA) che è isolato dai liquidi biologici dei pazienti neoplastici, i quali contengono anche DNA rilasciato da tessuti non-tumorali. Infatti, la quantità di ctDNA presente nel cfDNA totale può variare da <1% a oltre il 90% ed è generalmente correlata al carico tumorale, sebbene la localizzazione del tumore e il tipo istologico ne possa-
no influenzare il rilascio.3 Il cfDNA può essere isolato da diversi liquidi biologici come sangue, urine, versamenti o anche liquido cerebrospinale.4 Nella pratica clinica attuale, il cfDNA analizzato a scopo diagnostico viene in genere isolato dal plasma ottenuto da prelievi di sangue periferico. Le modalità di prelievo di sangue e la sua corretta e tempestiva manipolazione per l’isolamento del plasma sono cruciali per il successo dell’analisi. In particolare, il plasma deve essere isolato entro massimo tre ore dal prelievo, se sono impiegate provette standard contenenti EDTA per la raccolta del sangue.4
Poiché il DNA tumorale può essere estremamente diluito nel DNA normale, sono necessarie per l’analisi del cfDNA metodiche altamente sensibili, in grado di rilevare una bassa frequenza di alleli mutati (mutant allelic frequency, MAF). A tale riguardo, sono state sviluppate diverse metodologie per rilevare e quantificare varianti rare nel sangue di pazienti oncologici, con sensibilità analitica fino allo 0,001%.5 6 Queste tecnologie possono essere classificate in tre gruppi: la PCR quantitativa (qPCR), la PCR in emulsione e il sequenziamento massivo parallelo, più comunemente definito sequenziamento di nuova generazione (NGS).
Sebbene la qPCR e la PCR basata su emulsione abbiano un’elevata sensibilità e specificità, entrambe queste metodologie hanno il limite di poter interrogare pochi loci per analisi, ovvero di poter identificare poche mutazioni.5 Inoltre, esse sono limitate all’analisi delle sole mutazioni note. Pertanto, queste tecnologie risultano utili per l’identificazione delle varianti più comuni, ma non sono in grado di fornire un quadro complessivo delle alterazioni genetiche della neoplasia. Inoltre, esse forniscono informazioni limitate sull’evoluzione genetica del tumore che potrebbero essere rilevanti per l’intervento terapeutico, come ad esempio l’identificazione di meccanismi di resistenza non precedentemente descritti. Molte di queste limitazioni vengono superate dalla NGS, che è un metodo di analisi che consente il sequenziamento simultaneo di più geni e il rilevamento, quindi, di tutte le alterazioni genetiche, comprese le nuove mutazioni, nel tratto sequenziato. Tuttavia, la relativa bassa sensibilità ha rappresentato per lungo tempo un limite all’utilizzo della NGS per la valutazione del cfDNA. A questo proposito, sono state recentemente sviluppate nuove tecnologie di NGS per l’analisi specifica del cfDNA che raggiungono livelli di sensibilità comparabili a quelli della qPCR e della PCR in emulsione.5 Alcune di queste metodiche sono disponibili sotto forma di service, come ad esempio i pannelli di Guardant Health e di Foundation Medicine, mentre altri pannelli commercializzati da Thermofisher, Illumina e altre aziende sono disponibili per laboratori indipendenti. I pannelli NGS per biopsia liquida variano da pochi geni a centinaia di geni, e sono quindi in grado di fornire un profilo genomico complessivo del tumore, al pari di quanto accade per il tessuto tumorale.
Diversi studi hanno dimostrato una buona concordanza tra test del cfDNA e analisi del tessuto tumorale per l’identificazione di mutazioni driver, incluse le mutazioni di BRAF. In particolare, alcuni studi rivolti in maniera specifica a de-
Biopsia liquida, altre patologie e prospettive future 85
terminare la sensibilità e la specificità di test su cfDNA per le mutazioni BRAF V600 sono stati condotti in pazienti con melanoma metastatico, la neoplasia con la più elevata frequenza di questa specifica alterazione genetica. Questi studi hanno dimostrato che tecniche di qPCR sono in grado di identificare le varianti BRAF V600 nel cfDNA, in percentuali che vanno dal 50% circa a oltre l’80% dei pazienti con melanoma avanzato che hanno la medesima alterazione nel tessuto tumorale.7 Le tecniche di PCR basate su emulsione hanno una sensibilità ancora più elevata, che può anche superare l’80%.
La percentuale di pazienti per i quali non è disponibile tessuto tumorale per l’analisi dei biomarcatori e che, pertanto, potrebbero beneficiare del test su cfDNA varia a seconda del tipo di neoplasia. Si stima che circa il 20% dei pazienti con carcinoma del polmone avanzato potrebbero non avere tessuto sufficiente per analisi farmacogenomica, mentre il tessuto tumorale è in genere sempre disponibile per pazienti con carcinoma del colon e melanoma. Tuttavia, dati recenti suggeriscono che l’analisi del cfDNA potrebbe fornire informazioni complementari a quelle derivanti dalla valutazione del tessuto tumorale.
Diversi studi hanno infatti dimostrato che le mutazioni driver, incluse le mutazioni di BRAF, possono essere in alcuni casi sub-clonali, anche nei tumori del polmone e del colon e nel melanoma cutaneo.8 Nel carcinoma del colon-retto, le mutazioni di BRAF sembrano essere più frequentemente sub-clonali rispetto a quanto avviene per le mutazioni di KRAS e NRAS.9 L’esistenza di mutazioni sub-clonali implica la possibilità che queste varianti siano presenti solo in alcune aree del tessuto tumorale oppure solo in alcune lesioni neoplastiche in pazienti con metastasi multiple. A tale riguardo, è stato dimostrato che lo stato mutazionale di BRAF può essere differente tra melanoma primitivo e metastasi e tra le diverse metastasi dello stesso paziente che si ripresentano nel tempo.10 11 Nei casi con eterogeneità tumorale, l’analisi del cfDNA può quindi meglio rappresentare lo stato mutazionale complessivo del tumore e dovrebbe essere sempre eseguita in caso di recidiva della malattia a distanza di tempo o dopo interventi terapeutici sistemici.
La determinazione dei livelli di ctDNA prima del trattamento in pazienti con malattia metastatica ha inoltre un importante valore prognostico. Diversi studi hanno dimostrato che la presenza di elevati livelli di ctDNA correla con una peggiore sopravvivenza in pazienti affetti da carcinoma del colon e del polmone o da melanoma in stadio avanzato di malattia.4 6 12 In pazienti con melanoma avanzato questa correlazione è stata dimostrata utilizzando in molto casi il test specifico per le mutazioni BRAF V600 per rivelare la presenza di ctDNA. L’informazione prognostica derivante dall’analisi del cfDNA è probabilmente dovuta alla forte correlazione che esiste tra livelli di ctDNA e il carico di malattia presente nel paziente. È interessante sottolineare che i livelli di ctDNA al basale presentano una migliore correlazione con il carico di malattia misurato mediante FDG-PET rispetto ai livelli di LDH in pazienti affetti da melanoma.13
Il ctDNA potrebbe quindi essere un indicatore non solo del carico di malattia ma anche della presenza di malattia metabolicamente attiva e, quindi, potenzialmente aggressiva.
Una delle più interessanti applicazioni dell’analisi del cfDNA nei pazienti con malattia metastatica è la possibilità di monitorare la risposta alla terapia. Per quanto riguarda le neoplasie con mutazioni di BRAF, la maggioranza delle evidenze deriva soprattutto da studi condotti in pazienti con melanoma metastatico che hanno ricevuto terapie con inibitori di BRAF da soli o in combinazione con farmaci anti-MEK. Sebbene questi studi abbiano arruolato un numero limitato di pazienti e abbiano impiegato differenti tecniche di qPCR o di PCR in emulsione,7 essi hanno dimostrato in maniera consistente che la risposta alla terapia a bersaglio molecolare è associata a una significativa diminuzione dei livelli di mutazioni BRAF nel cfDNA, mentre un aumento del DNA mutato in BRAF è stato osservato alla progressione di malattia. In questo ultimo caso, l’aumento dei livelli di ctDNA ha preceduto la progressione clinica della malattia in una frazione significativa di pazienti, con un anticipo diagnostico fino a 110 giorni.14 In uno studio di fase II di combinazione di dabrafenib più trametinib in pazienti con melanoma avanzato BRAF V600 mutato pretrattato con terapia target, è stato dimostrato che una riduzione precoce dei livelli di ctDNA, nel primo mese di trattamento, è associata a un più elevato tasso di risposte e a una più lunga PFS.15
L’analisi del cfDNA potrebbe anche fornire importanti informazioni sulla valutazione della risposta agli inibitori del checkpoint immunitario. Infatti, la valutazione della risposta alle terapie immunitarie è talvolta difficile, a causa del possibile aumento transitorio delle dimensioni delle lesioni tumorali, dovuto alla reazione immunitaria che simula una progressione della malattia. Tale pseudo-progressione potrebbe portare alla sospensione di un trattamento attivo. A tale riguardo, il valore predittivo del monitoraggio del ctDNA è stato esplorato in una coorte di pazienti con melanoma metastatico con mutazioni di BRAF, NRAS e cKIT, che hanno ricevuto un trattamento con pembrolizumab o nivolumab in monoterapia o in combinazione con ipilimumab.16 Questo studio ha dimostrato che il tasso di risposta nei pazienti con ctDNA rilevabile al basale ma non dopo 12 settimane di terapia era simile a quello dei pazienti con ctDNA non rilevabile al basale e dopo 12 settimane (77% e 72%, rispettivamente) e molto più alto rispetto ai pazienti con ctDNA rilevabile sia al basale che dopo 12 settimane (6%). Inoltre, i primi due gruppi avevano una sopravvivenza libera da progressione (progression free survival, PFS) e una sopravvivenza globale (overall survival, OS) significativamente più lunghe rispetto al terzo gruppo. Il valore predittivo del monitoraggio del ctDNA a 12 settimane è stato confermato all’analisi multivariata, suggerendo così che la clearance del ctDNA sia un marcatore predittivo rilevante di risposta agli inibitori del checkpoint immunitario nel melanoma metastatico. Lo stesso gruppo di ricerca ha anche dimostrato che l’analisi del ctDNA consente di distinguere con elevata precisione la pseudo-progressione dalla vera
Biopsia liquida, altre patologie e prospettive future 87
progressione di malattia in pazienti trattati con immunoterapia.17 La dinamica dei livelli di ctDNA nel primo mese dopo il trattamento ha fornito informazioni precoci di progressione di malattia o di risposta alla terapia in un recente studio che ha arruolato pazienti con melanoma in stadio III/IV che hanno ricevuto immunoterapia o terapia a bersaglio molecolare.18 Risultati simili sono stati, infine, ottenuti in altri studi che hanno monitorato differenti mutazioni nel cfDNA di pazienti con neoplasie di diversa origine istologica trattati con immunoterapia.
Il campo di applicazione in cui la biopsia liquida può fornire informazioni non ottenibili con l’analisi del tessuto tumorale è sicuramente rappresentato dallo studio dei meccanismi di resistenza acquisiti alle terapie target. Infatti, la resistenza acquisita ai farmaci a bersaglio molecolare è spesso multiclonale ed è quindi caratterizzata da una estrema eterogeneità, con differenze di quadro mutazionale in aree diverse della stessa lesione e tra differenti lesioni metastatiche. A supporto di questa ipotesi, uno studio recente ha dimostrato che l’analisi del cfDNA con tecnologie di NGS in pazienti con neoplasie gastrointestinali diventati resistenti a terapie target è in grado di individuare alterazioni genetiche che nel 78% dei casi non possono essere rivelate dall’analisi di una singola biopsia del tessuto tumorale.19 In particolare, un paziente con carcinoma del colon-retto metastatico e mutazione BRAF V600E trattato con farmaci a bersaglio molecolare presentava in tutte le lesioni metastatiche alterazioni molecolari distintive e differenti dalle altre localizzazioni di malattia.19 Evidenze a supporto dell’importanza dell’uso della biopsia liquida nello studio della resistenza alle terapie target provengono anche da diverse altre neoplasie, con particolare riguardo ai tumori polmonari.6
Sebbene la malattia metastatica rappresenti attualmente il maggiore settore di applicazione della biopsia liquida, l’analisi del cfDNA potrebbe fornire informazioni prognostiche importanti anche nella malattia iniziale resecabile. In particolare, l’analisi peri-operatoria del ctDNA in pazienti con tumore resecabile chirurgicamente può offrire informazioni prognostiche differenti e rilevanti: 1) il rilevamento del ctDNA prima dell’intervento chirurgico potrebbe fornire informazioni su estensione ed aggressività della malattia; 2) il test del ctDNA dopo l’intervento chirurgico e la eventuale terapia adiuvante potrebbe indicare la presenza di malattia minima residua; 3) testare il ctDNA durante il follow-up dopo una resezione chirurgica a intento curativo potrebbe rivelare una recidiva del tumore a livello molecolare, anticipando così la recidiva clinica o radiologica. Infatti, data la possibilità di effettuare campionamenti ripetuti per le sue caratteristiche di minima invasività, il test del ctDNA può consentire il monitoraggio in tempo reale del decorso della malattia. Tutte queste ipotesi sono state confermate in numerosi studi in pazienti con neoplasie iniziali del colon, della mammella e del polmone, che hanno dimostrato una forte correlazione tra presenza di ctDNA prima e/o dopo l’intervento chirurgico e la probabilità di recidiva della malattia.20-22 Inoltre, la individuazione di mutazioni
Paziente con malattia resecabile
Paziente con malattia metastatica
Figura 6.1
n I livelli di ctDNA prima della chirurgia hanno valore prognostico
n La individuazione di ctDNA dopo chirurgia/terapia adiuvante indica la presenza di malattia minima residua
n I livelli di ctDNA prima della terapia sistemica hanno valore prognostico
n L’analisi del cfDNA consente di individuare biomarcatori per farmaci a bersaglio molecolare e fornisce informazioni sulla eterogeneità tumorale
n Il monitoraggio dei livelli di ctDNA permette di valutare la risposta alle terapie sistemiche
n L’analisi del cfDNA consente la individuazione di meccanismi di resistenza alle terapie target
Principali campi di applicazione della analisi del cfDNA nel paziente con malattia in fase iniziale o metastatica
nel plasma di pazienti operati ha in molti casi anticipato di settimane o mesi la progressione clinica o radiologica. Il valore prognostico dell’analisi del cfDNA è stato anche confermato in pazienti con melanoma allo stadio II-III utilizzando come biomarcatori le mutazioni di BRAF, NRAS o cKIT.23 24 L’utilizzo di tecnologie di NGS può notevolmente aumentare la sensibilità e la specificità del test per il ctDNA in pazienti con neoplasia in fase iniziale di sviluppo.
I farmaci anti-BRAF sono al momento approvati per il trattamento di pazienti con melanoma metastatico e con neoplasie avanzate del polmone e del colonretto con mutazioni BRAF V600. Tuttavia, mutazioni BRAF V600, sebbene a bassa frequenza, sono state identificate in numerose altre neoplasie, suggerendo che esse potrebbero rappresentare un bersaglio terapeutico indipendentemente dal tipo istologico del tumore, ovvero tumor agnostic. Questa ipotesi necessita però di una verifica sperimentale, dato che i farmaci anti-BRAF hanno mostrato attività nel carcinoma del colon-retto solo se associati ad anticorpi monoclonali antiEGFR. A tale riguardo, i risultati recentemente pubblicati del sotto-protocollo dello studio NCI MATCH dedicato ai tumori con mutazioni BRAF V600 sembrano confermare un valore predittivo trans-patologia di queste varianti.25 Questo studio ha arruolato pazienti con neoplasie di diversa origine istologica con mutazioni BRAF V600 precedentemente trattati con 3 o più linee di terapia. I pazienti sono stati trattati con una combinazione di dabrafenib e trametinib. Il tasso di
Biopsia liquida, altre patologie e prospettive future 89
risposta è stato del 38% con 7 pazienti che hanno avuto una durata della risposta >12 mesi. Risposte durature sono state osservate in pazienti con neoplasie di diversa origine istologica, tra cui l’adenocarcinoma papillare del polmone, il carcinoma ovarico sieroso di basso grado, l’adenocarcinoma mucinoso-papillare del peritoneo, il sarcoma istiocitico cerebrale, lo xanto-astrocitoma pleiomorfico del lobo parietale e il colangiocarcinoma. Sebbene preliminari, questi risultati confermano evidenze raccolte in altri studi su specifiche istologie, come ad esempio quelli condotti nei tumori biliari, e sicuramente suggeriscono che le terapie antiBRAF potrebbero rappresentare un importante approccio terapeutico anche per neoplasie rare, per le quali non esistono valide alternative terapeutiche.
L’emergenza di fenomeni di resistenza acquisita rappresenta un limite per tutte le terapie a bersaglio molecolare, ivi incluse quelle indirizzate contro BRAF. Lo studio dei meccanismi di resistenza acquisita potrebbe consentire lo sviluppo di strategie terapeutiche innovative basate sull’impiego di combinazioni di farmaci a bersaglio molecolare. Tale strategia terapeutica non può tuttavia prescindere dalla determinazione del profilo genetico molecolare complessivo della neoplasia, soprattutto per valutare l’eventuale ruolo della eterogeneità tumorale nella resistenza intrinseca ed acquisita.
Lo sviluppo di nuove strategie terapeutiche nei pazienti con mutazioni di BRAF richiede una evoluzione anche degli approcci di diagnostica molecolare. In particolare, l’integrazione dello studio della biopsia liquida nelle diverse fasi di evoluzione della malattia potrebbe consentire una migliore stratificazione dei pazienti in base al loro profilo di rischio, la individuazione di fenomeni di resistenza precoce e la scoperta dei meccanismi di resistenza acquisita. Studi clinici prospettici randomizzati basati sulla biopsia liquida saranno tuttavia necessari per validare queste nuove strategie terapeutiche. Infatti, sarà necessario dimostrare che l’individuazione precoce della progressione della malattia resa possibile con l’analisi del cfDNA risulti effettivamente in un vantaggio di sopravvivenza per i pazienti. Infine, sebbene più rare, mutazioni BRAF non-V600 sono coinvolte nella patogenesi di diverse neoplasie umane. Per queste varianti è necessario raccogliere maggiori informazioni, al fine di determinare il loro ruolo prognostico e, soprattutto, sviluppare approcci terapeutici basati su farmaci specifici o combinazioni di inibitori della trasduzione del segnale.
1. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancergenetics in the blood. Nat Rev Clin Oncol 2013; 10: 472-84.
2. Corcoran RB, Chabner BA. Application of Cell-free DNA Analysis to Cancer Treatment.
N Engl J Med 2018; 379: 1754-65.
3. Wan JCM, Massie C, GarciaCorbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017; 17: 223-38.
4. Normanno N, Denis MG, Thress KS, et al. Guide to detecting epidermal growth factor receptor (EGFR) mutations in ctDNA of patients with advanced non-small-cell lung cancer. Oncotarget 2017; 8: 12501-116.
5. Normanno N, Cervantes A, Ciardiello F, et al. The liquid biopsy in the management of colorectal cancer patients: Current applications and future scenarios. Cancer Treat Rev 2018; 70: 1-8.
6. Esposito Abate R, Pasquale R, Fenizia F, et al. The role of circulating free DNA in the management of NSCLC. Expert Rev Anticancer Ther 2019; 19: 19-28.
7. Herbreteau G, Charpentier S, Vallee A, Denis MG. Use of circulating tumoral DNA to guide treatment for metastatic melanoma. Pharmacogenomics 2019; 20: 1259-70.
8. McGranahan N, Favero F, de Bruin EC et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015; 7: 283ra254.
9. Normanno N, Rachiglio AM, Lambiase M et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann Oncol 2015; 26: 1710-4.
10. Yancovitz M, Litterman A, Yoon J, et al. Intra- and inter-tumor heterogeneity of BRAF(V600E))mutations in primary and metastatic melanoma. PLoS One 2012; 7: e29336.
11. Heinzerling L, Baiter M, Kuhnapfel S, et al. Mutation landscape in melanoma patients clinical implications of heterogeneity of BRAF mutations. Br J Cancer 2013; 109: 2833-41.
12. Santiago-Walker A, Gagnon R, Mazumdar J, et al. Correlation of BRAF Mutation
Status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin Cancer Res 2016; 22: 567-74.
13. Wong SQ, Raleigh JM, Callahan J, et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precision Oncology 2017; 1-14.
14. Schreuer M, Meersseman G, Van Den Herrewegen S, et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J Transl Med 2016; 14: 95.
15. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAF(V600)-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol 2017; 18: 464-72.
16. Lee JH, Long GV, Boyd S, et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann Oncol 2017; 28: 1130-6.
17. Lee JH, Long GV, Menzies AM, et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti-programmed cell death 1 antibodies. JAMA Oncol 2018; 4: 71721.
18. Braune J, Keller L, Schiller F, et al. Circulating tumor DNA allows early treatment
monitoring in BRAF- and NRAS-mutant malignant melanoma. JCO Precision Oncology 2020; 20-31.
19. Parikh AR, Leshchiner I, Elagina L, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 2019; 25: 1415-21.
20. Garcia-Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 2015; 7: 302ra133.
21. Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 2016; 8: 346ra392.
22. Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017; 7: 1394-403.
23. Lee JH, Saw RP, Thompson JF, et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann Oncol 2019; 30: 815-22.
24. Tan L, Sandhu S, Lee RJ et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann Oncol 2019; 30: 804-4.
25. Salama AKS, Li S, Macrae ER, et al. Dabrafenib and Trametinib in patients with tumors with BRAF(V600E) mutations: results of the NCI-MATCH Trial Subprotocol H. J Clin Oncol 2020; JCO2000762.
Finito di stampare nel mese di novembre 2020 da Ti Printing S.r.l. Via delle Case Rosse 23, 00131 Roma per conto de Il Pensiero Scientifico Editore, Roma