
7 minute read
Artículo


EL PASO DE SUSTANCIAS POR EL ORGANISMO
MODELOS COMPARTIMENTALES Y FARMACOCINÉTICA
Advertisement
Mucho de lo que conocemos del organismo humano ha sido debido a nuestra constante necesidad de querer conservarlo saludable o remediar aquellas condiciones que lo alejan de dicho estado. Durante milenios el hombre a recurrido a diversas técnicas para llevar a cabo este particular, una de dichas técnicas es la medicación y los elementos ejecutores de dicha técnica son los compuestos activos o fármacos –últimante también llamados API’s (Active Pharmaceutal Ingredient, del idioma inglés).
ARTÍCULO
Laura Jiménez Ramírez
Estudiante de 8vo semestre de Químico Farmacobiologo Universidad de Guadalajara
laura.laboratorista@gmail.com
Para que un fármaco pueda tener una acción farmacológica en un sitio específico del cuerpo humano, es necesario que éste alcance los niveles terapéuticos y llegue al sitio diana; de igual manera, para que el fármaco no cause toxicidad, es necesario que sea eliminado gradualmente. Al comportamiento de un fármaco y su evolución durante la liberación, absorción, distribución, metabolismo, y eliminación (LADME) se le conoce como farmacocinética.
Existen diversos factores que pueden influenciar la farmacocinética, entre ellos, los grupos etarios, los grupos étnicos, el padecimiento de enfermedades crónico degenerativas, entre otros. Es decir, el comportamiento de un fármaco en un paciente anciano caucásico con insuficiencia renal puede no ser el mismo que el de un paciente afrodescendiente de mediana edad que goza de buena salud; por lo tanto, los estudios farmacocinéticos son fundamentales en la investigación y el desarrollo de nuevos fármacos y formas farmacéuticas, ya que permiten establecer la dosis adecuada para cada paciente.
Para conocer el comportamiento de un fármaco -aunque técnicamente para cualquier sustancia- en el cuerpo humano, se hace uso de modelos matemáticos basados en la monitorización de los niveles séricos de los fármacos en función del tiempo como una técnica de control terapéutico y la individualización posológica en diferentes poblaciones de pacientes.
La distribución de los fármacos se establece como un equilibrio reversible entre el fármaco libre y el que se encuentra unido a tejidos o proteínas. Puede ser simple, o bien muy compleja, dependiendo de factores propios del fármaco como el peso molecular o grado de ionización de la molécula y por factores fisiológicos como el flujo sanguíneo, la capacidad de fijación a las proteínas plasmáticas y tisulares, a la permeabilidad de las membranas, entre otras.
Respecto al metabolismo de los fármacos, los polimorfismos genéticos que manifieste una población pueden MODELOS COMPARTIMENTALES
Gibaldi, M.; Perrier, D. Farmacocinética. Ed. Reverte
Los compartimientos no son entidades fisiológicas o anatómicas reales, sino modelos de estudio basados en similitudes de irrigación sanguínea, cantidad de masa o volumen.
Modelo Monocompartimental
• A pesar de su sencillez, ayuda a explicar el paso de muchos fármacos por el organismo.
• Asume que el organismo es un recipiente con plasma y sangre.
• Considera que la eliminación es constante y directa.
• Explica el comportamiento de fármacos vía intravenosa que no experimentan absorción ni distribución.
La cantidad de fármaco en sangre decae de forma lineal conforme pasa el tiempo.

Modelo Bicompartimental
Tras una administración oral, un fármaco pasa de un compartimiento periférico a uno central.
Compartimento periférico Compartimento Central
Vía de eliminación
ABSORCIÓN. El paso del compartimiento periférico al central. DISTRIBUCIÓN. Cuando el compartimiento central ya no puede alojar más fármaco sin ELIMINARLO o
METABOLIZARLO.
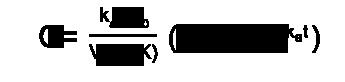
La concentración en plasma (C) en cualquier tiempo (t) después de la administración de la dosis inicial (X0), distribuida en el volumen (V), es igual a la fracción absorbida (F), de la dosis administrada la cual es influenciada por las constantes de absorción (ka) y de eliminación (K). influir en la funcionalidad de las enzimas responsables de la biotransformación de los fármacos y, por lo tanto, en su farmacocinética.
La excreción del fármaco inalterado permite su eliminación, siendo la excreción renal la principal vía de eliminación, más no la única, ya que también puede eliminarse por excreción biliar, sudorípara, salivar o mamaria, contribuyendo a la eliminación de los fármacos del organismo de manera secundaria.
En estudios farmacocinéticos, una vez que se obtiene la información sobre los niveles del fármaco en los fluidos o tejidos del organismo debe plantearse un modelo matemático que permita interpretar los procesos LADME implicados. Los parámetros a utilizarse en un modelo matemático se estiman a partir de las curvas de nivel del fármaco y sus metabolitos en plasma y otros tejidos mediante métodos de regresión no lineal. Los métodos de análisis matemáticos más utilizados son los modelos compartimentales. Éstos se basan en considerar al organismo un conjunto de compartimentos que comparten una distribución homogénea. La transferencia del fármaco entre los diferentes compartimentos suele ser mediante procesos lineales y de primer orden.
Modelo monocompartimental
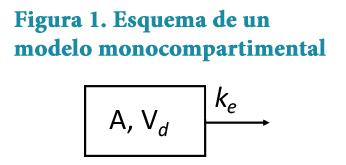
Un modelo monocompartimental es aquel que considera al organismo como un solo contenedor en el que el fármaco es distribuido instantáneamente de forma homogénea a todos los órganos. Un ejemplo de éste es la administración de un bolus intravenoso, que se caracteriza por ser eliminado con una cinética de primer orden; es decir, que la eliminación del fármaco es proporcional a su concentración, obedeciendo a una linea recta cuando se grafican los datos de concentración contra tiempo. El esquema de un modelo monocompartimental se muestra en la Figura 1, dónde A representa la cantidad de fármaco; Vd el volumen de distribución y k e la constante de velocidad de eliminación de primer orden desde ese compartimento.
Sin embargo, en el modelo monocompartimental existe una variación en el tiempo de equilibrio de concentración entre la sangre y los órganos que, en ocasiones, puede llegar a ser significativa. Para evitar esta variación es preciso recurrir a modelos más complejos en los que se contempla más de un compartimento.
Modelo bicompartimental.
El modelo bicompartimental se basa en la interacción entre dos contenedores relacionados entre sí; un contenedor de rápido acceso, o compartimento central, y un contenedor secundario denominado compartimento periférico. Los órganos y tejidos que cuentan con un buen flujo sanguíneo son considerados parte del compartimento central, mientras que los órganos menos irrigados y en los que, consecuentemente, el fármaco tarda más en acceder, se consideran parte del compartimento periférico. La Figura 2 representa un modelo bicompartimental en el que V c y V p representan el volumen de distribución de cada compartimento, A c y A p la concentración del fármaco de cada compartimento y k12 y k21 las constantes de velocidad de transferencia del fármaco entre los compartimentos central y periférico.
Figura 2. Esquema de un modelo bicompartimental
Ac’ Vc
K10 K12 K21
Ap’ Vp
Cuando un fármaco es administrado por vía intravenosa, éste es distribuido inmediatamente de forma homogénea en el compartimento central y, moderado por la constante de velocidad k12, el fármaco pasa a distribuirse al compartimento periférico logrando así que ambos contenedores logren el equilibrio en la concentración. Asimismo, la constante k21 permite el retorno del fármaco al compartimento central para ser eliminado posteriormente bajo el régimen de la constante k10.
Como se mencionó en un inicio, es importante resaltar que la farmacocinética depende también de las propiedades del fármaco; además, la vía de administración también se considera como un factor influyente. Existen numerosas vías de administración que implican la absorción de un fármaco antes de entrar al torrente sanguíneo; ya sea oral, intramuscular, cutánea, rectal, entre otras. A éstas se les conoce como extravasales o extravasculares.
Modelos bicompartimentales extravasales
En un modelo extravasal se requiere contemplar la absorción del fármaco, por lo tanto, un nuevo compartimento es añadido, mismo que será moderado por una constante que definirá el paso de la sustancia del sitio de absorción hasta la circulación sanguínea como se esquematiza en la Figura 3.
En este modelo se contempla el paso del fármaco a través de una membrana para posteriormente ser distribuido de manera homogénea, a partir de entonces, se considera que continúa con una distribución al compartimento periférico para su posterior retorno
al compartimento central para que, finalmente, sea eliminado.
Así como los modelos compartimentales, existen otros métodos matemáticos para evaluar la farmacocinética, ya sea en suero, orina, leche materna, entre otras secreciones, y que sirven como herramientas clave para la formulación y dosificación de nuevos principios activos y formas farmacéuticas. Con el empleo de estos modelos no sólo se logra mejorar la relación costo-beneficio para los pacientes, sino que a su vez permiten que tengan una mejor calidad de vida, el conocimiento de la farmacocinética en las poblaciones especiales y una reducción significativa en la tasa de mortalidad de los pacientes.
Absorción K01
Compartimento central K10
K12 K21
Compartimento periférico







