Reacción en Cadena de la Polimerasa
 Dra. Gabriela Jaramillo Koupermann
Dra. Gabriela Jaramillo Koupermann




PRODELAB Docente :
Introducción: Reacción en Cadena de la Polimerasa 2
Principio del método 2 Componentes de una PCR 3 Etapas del proceso 3 Diseño de cebadores 6
Ejemplo del diseño básico de una PCR y su protocolo 9 Variantes del método 10
PCR con transcriptasa inversa (RT PCR) 10

PCR Multiplex 13
PCR en tiempo real 13 Tipos de fluorescencia en la Q PCR 14
SYBR Green: 14 Sondas 15 Principios de la Q PCR 17 Aplicaciones de la pcr en tiempo real 18
Cuantificación 18
Cuantificación absoluta: 19 Cuantificación relativa: 19
Genotipificación 19 Genotipificación con sondas: 19 Genotipificación por Melting (HRM): 20
Referencias 22 Libros 22 Papers 22 Videos 22
Introducción: Reacción en Cadena de la Polimerasa
La reacción en cadena de la polimerasa (PCR) fue descubierta en 1985 por Kary Mullis, que recibió el premio Noble de química por ello en 1993. Esto marcó el inicio de la revolución genética y genómica que vivimos hoy en día.
Es una técnica in vitro utilizada para amplificar enzimáticamente, mediante una polimerasa, una región específica de ADN/ADNc (complementario), situada entre dos regiones de ADN cuya secuencia se conoce. La PCR es hoy día una herramienta imprescindible en los laboratorios de biología molecular y su objetivo es obtener sintéticamente, millones de copias del fragmento de interés para estudio. Toda muestra de ácido nucleico, no importa la especie, puede ser amplificada por PCR y su aplicación es infinita.
Antes de comenzar con la metodología se recomienda revisar el siguiente video: https://youtu.be/TNKWgcFPHqw
Algo más informal pero muy ilustrativo: https://youtu.be/Qqe4thU os8
Principio del método
La PCR no es una técnica analítica per se, sino más bien una metodología, resultante de la aplicación práctica de tres conceptos básicos:
1. Desnaturalización del DNA para dar moléculas monocatenarias las cuales serán “copiadas.
2. Hibridación específica de la molécula monocatenaria con un cebador sintético para lo cual se debe conocer la secuencia blanco.
3. Replicación de la molécula monocatenaria mediante una polimerasa de ADN que utiliza el cebador como extremo 3’dotador de OH. Este concepto también se puede denominar elongación o extensión del cebador, o polimerización.
Mediante la aplicación en forma cíclica de estos tres procesos se consigue un número muy alto de copias del fragmento de ácido nucleico en un corto espacio de tiempo.
En la práctica, se requiere un control preciso de las variables que condicionan este triple proceso (secuencia diana, cebadores, enzimas, buffer, coenzimas y otros componentes), además de instrumentos adecuados denominados termocicladores para establecer con precisión las condiciones de cada etapa y repetirlas cíclicamente (tiempo, temperatura, número de ciclos, etc.). Se consigue de esta forma amplificar secuencias de tamaños
diversos, comprendidos entre 50 pb y 2,5 kb dependiendo de la utilidad y origen de la muestra.
Componentes de una PCR
1. Dideoxinucleóticos (dNTPs). Adenina, Timina, Citocina y Guanina. Los cuatro dNTP, en exceso, sirven como sustratos para la síntesis de las copias de ADN. Para ser reconocidos por la polimerasa, deben ir acompañados de Mg2+ (generalmente en forma de cloruro) que es, además, una coenzima requerida por la polimerasa.
2. Dos oligonucleótidos monocatenarios, cebadores o “primers” (forward y reverse), de 18 a 30 nucleótidos. Sus secuencias son diseñadas al fin de ser complementarias, respectivamente, a los dos extremos 3’ de la región den ADN en estudio.
3. Un ADN polimerasa termoestable, es decir, enzimáticamente activa a temperaturas relativamente altas (habitualmente en torno a 75 94C). Esto contribuye así a la especificidad y rendimiento del proceso. Por otra parte, la estabilidad de la polimerasa a temperaturas de hasta 95C (aunque no mantenga su actividad, no se desnaturaliza) permite que recupere su actividad al enfriarse de nuevo y evita así la necesidad de reponer la enzima en sucesivos ciclos. La enzima más empleada se denomina polimerasa Taq, por proceder de la bacteria Thermus aquaticus, que vive en manantiales de agua caliente. Existen varios tipos de polimerasa las cuales se adaptan a las aplicaciones específicas y nivlel de resolución requerida.
4. Buffer o amortiguador que garantiza la estabilidad de la reacción en medio óptimo para la acción de la enzima sin frangmenar el ADN.
Etapas del proceso
La PCR, en su diseño original, es una técnica de punto final, es decir, analiza el producto obtenido tras finalizar la reacción, es un método cualitativo donde se observa únicamente la presencia o ausencia de la región genética amplificada (que puede ser endógena o exógena). Esta metodología es útil para preparar muestras que van a ser posteriormente estudiadas por otros métodos como la restricción enzimática o secuenciación o para definir, por ejemplo, una infección (presencia o ausencia de un microorganismo patógeno en una muestra biológica). El resultado es estudiado mediante geles de electroforesis donde se observan las clásicas bandas.
Con el fin de amplificar el fragmento de interés, se requiere una sucesión de ciclos (en general entre 20 y 40, de 1,5 a 5 minutos de duración cada uno). La duración promedio de una PCR es de aproximadamente 2 horas, aunque esto depende de las condiciones experimentales concretas. No se recomienda ampliar el número de ciclos debido a que esto provoca amplicones inespecíficos.
Cada ciclo de una PCR consta, de tres etapas:
1. Desnaturalización: calentamiento para la separación de las dos hebras del DNA, mediante una incubación breve (30 120 s) a una temperatura comprendida entre 68 y 97C. que debe ser superior a la temperatura de fusión o temperatura de melting (Tm) de la región de DNA que quiere amplificarse. La Tm es la temperatura a la cual el 50% del ADN se encuentra desnaturalizada y el oro 50% aun unido, esta temperatura es dependiente del % de AT y CG del fragmento de ADN de interés.
2. Hibridación: enfriamiento rápido por debajo de Tm, de forma que se permite el emparejamiento del ADN desnaturalizado con los cebadores. Esta temperatura depende del fragmento y puede estar comprendida entre 37 y 65C por entre 10 y 120 s.
3. Elongación (o replicación): etapa de amplificación propiamente dicha (72 75C, 1 3 min), en la que la polimerasa termoestable elonga los cebadores, empleando como molde ambas hebras originales. La replicación transcurre en dirección 5’ a 3’ a partir del extremo 3’ OH de cada cebador, empleando como sustrato los cuatro dNTP, hasta terminar la lectura del molde o hasta que se eleve la temperatura en una nueva etapa de desnaturalización (siguiente ciclo).
4. Además de las 3 etapas de cada ciclo, suele añadirse una etapa previa y una final al conjunto de todos los ciclos. La previa, a elevada temperatura (incluso superior a la de las etapas de desnaturalización), sirve principalmente para inactivar proteasas y nucleasas de la muestra, y deendiento de la polimeras, activarla. La etapa final, por su parte, consiste en una prolongación de la última elongación, para permitir que se completen todos los fragmentos.
Esquema de las etapas de una PCR estándar. Tomado de Herráez, Á. (2012). Biología Molecular e Ingeniería Genética. Elsevier Health Sciences, (p.203).


Esquema de la amplificación de un fragmento de ADN por PCR. Tomado de https://www.genome.gov/es/genetics glossary/Reaccion en cadena de la polimerasa
Diseño de cebadores
El diseño adecuado de los cebadores es básico para garantizar la especificidad de la PCR. Un primer debe cumplir con los siguientes parámetros:
Especificidad: Reconocer una secuencia única dentro del ADN templado. Así un cebador no puede unirse a más de una región del ADN y debe hacerlo en la dirección correcta para la síntesis (5’a 3’).
http://bioweb.uwlax.edu/GenWeb/Molecular/seq_anal/primer_design/primer_design .htm

Longitud: es recomendable que los primers tengan entre 18 y 30 nucleótidos. Esto garantiza la especificidad.

http://bioweb.uwlax.edu/GenWeb/Molecular/seq_anal/primer_design/primer_design .htm
Contenido de CG: Se recomienda que el contenido de citocinas y guaninas en la secuencia sea de entre 40 y 60%. Se debe evitar tramos de polipurinas o polipirimidinas. PoliC, PoliG o PoliG/C provoca unión más fuerte por lo tanto hibridación no específica. PoliT, PoliA o PoliA/T provoca unión más débil, por lo tanto se puede separar el complejo primer templado en esa zona.
Temperatura de hibridación (temperatura de annealing Ta): lo óptimo es que sea de entre 55 y 80C a menor tempreatura menor especificidad, a mayor temperatura menos rendimiento de la PCR. Depende de la longitud y composición del primer.
La Ta depende de la Temperatura de melting o fusión (Tm) de los primers. La Tm es la temperatura a la cual la mitad de las moléculas de primer están apareadas con el ADN molde y se calcula con la siguiente fórmula Tm= 2 (A+T) + 4(G+C). Los primers no deben tener Ta muy diferentes entre sí, no más de 5°C.

Composición de las regiones terminales 3': se deben evitar secuencias de 3 o más Cs o Gs en las regiones terminales 3' ya que pueden provocar amplicones inespecíficos.
Complementariedad de secuencia: las terminaciones 3' no deben ser complementarias, ya que esto puede llevar a la formación de dímeros. Con complementariedad en más de 3 pb el primer se pliega sobre sí mismo en una estructura doble cadena (hairpin).
http://bioweb.uwlax.edu/GenWeb/Molecular/seq_anal/primer_design/primer_design .htm

Correcto diseño de un par de primers:
Tamaño del amplicón
>chr7:44188824 44189018 195bp TCCACTTCAGAAGCCTA TCAGATTCTGAGGCTCA TCCACTTCAGAAGCCTActggggaaggctgaggggtcccagctccccacgctggctgctgtgcagat gctggacgacagagccaggatggaggccgccaagaaggagaaggtatctcgccctccattgggcatt ctgggagtgtttgcttgcctgtccccaacattccatggtttgttTGAGCCTCAGAATCTGA
> HYPERLINK "https://genome.ucsc.edu/cgi bin/hgTracks?hgsid=683063475_EKUAdqu5vj2Rm2P8Ohh6PtOGCHG1&db=hg 38&position=chr7:44188824 44189018&hgPcrResult=pack" chr7:44188824 44189018 195bp TCCACTTCAGAAGCCTA TCAGATTCTGAGGCTCA

Forward: 49.4 C tccacttcagaagccta Reverse: 50.1 C tcagattctgaggctca
Primer Fw Región de interésPrimer Rv Secuencia y longitud de los cebadores Temperatura de hibridación
TCCACTTCAGAAGCCTActggggaaggctgaggggtcccagctccccacgctggctgctg tgcagatgctggacgacagagccaggatggaggccgccaagaaggagaaggtatctcgccctc cattgggcattctgggagtgtttgcttgcctgtccccaacattccatggtttgttTGAGCCTC AGAATCTGA
Forward: 49.4 C tccacttcagaagccta Reverse: 50.1 C tcagattctgaggctca
Ejemplo del diseño básico de una PCR y su protocolo
La PCR, en su diseño original, es una técnica de punto final, es decir, analiza el producto obtenido tras finalizar la reacción, es un método cualitativo donde se observa únicamente la presencia o ausencia de la región genética amplificada (que puede ser endógena o exógena). Esta metodología es útil para preparar muestras que van a ser posteriormente estudiadas por otros métodos como la restricción enzimática o secuenciación o para definir, por ejemplo, una infección (presencia o ausencia de un microorganismo patógeno en una muestra biológica). El resultado es estudiado mediante geles de electroforesis donde se observan las clásicas bandas.
Nombre Primer Secuencia
Forward CACCCGTCCTGCCCCTTCACCTT
Reverse GGCTTCCCACGTGCGCAGCAGGA
Secuencia blanco y región de amplificación:
Amplicón esperado: 193bp
5’CACCCGTCCTGCCCCTTCACCTTCCAGCTCCGCCTCCTCCGCGCGGACCCCGCCCCGTCCCGA CCCCTCCCGGGTCCCCGGCCCAGCCCCCTCCGGGCCCTCCCAGCCCCTCCCCTTCCTTTCCGCG GCCCCGCCCTCTCCTCGCGGCGCGAGTTTCAGGCAGCGCTGCGTCCTGCTGCGCACGTGGGA AGCC3’
Componentes: Concentracion inicial Volumen Concentración final H2O 11,4ul
Buffer 10x 1x 2ul MgCl2 50mM 0.6ul 2mM dNTP`s 10mM 0.4ul 20mM Primer mix (F+R) 2ul 200nM each Taq 0,1ul 1,25U
DNA genómico 2ul 0.1 0.5ug/ul
Programa:
Activación 95ºC x 10min
Desnaturalización 94ºC x1min Hibridación 58.5ºC x1min 35 ciclos Elongación 72ºC x1min
Extensión final 72ºC x 9min Mantenimiento 4ºC hold ∞
Variantes del método
Existen un sinnúmero de variantes de la PCR las cuales podrán ser aplicadas según la necesidad de cada laboratorio. Realizaremos un resumen de las variantes más utilizadas.
Algunas variantes de la PCR. Tomado de https://microbenotes.com/types of pcr/ PCR con transcriptasa inversa (RT-PCR)

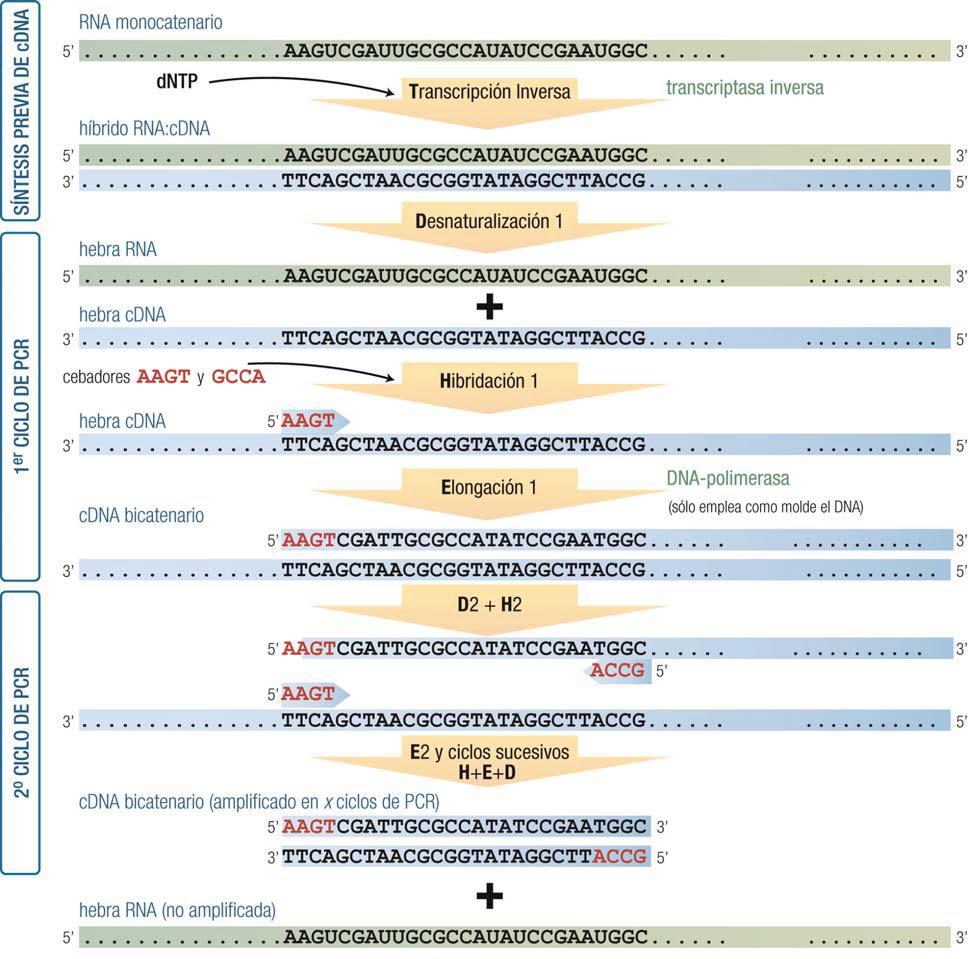
El nombre, "PCR con transcriptasa reversa" (iniciales de reverse transcriptase), indica que se trata de una amplificación de ARN (especialmente mensajero) a través de la síntesis previa de su cDNA (ADN complementario al ARN el cual es más estable), que después se amplifica por PCR.
Es el método más utilizado para determinar la expresión génica in vitro. Se utiliza para detectar el ARN mensajero (proveniente del ADN en momentos normales o patológicos de la célula), para el análisis de transcritos de genes (proveniente de fusiones génicas
anormales como en las leucemias y otros tumores), o para el estudio de patógenos (RNA viral).
La mezcla inicial contiene todos los componentes básicos: muestra de ARN, transcriptasa inversa, ADN polimerasa, cebadores universales y dNTP. El proceso comienza con la síntesis de una hebra de cDNA por la acción de la transcriptasa inversa, una polimerasa de ADN dirigida por ARN, permaneciendo el cDNA unido al molde como molécula bicatenaria híbrida RNA:cDNA. En una segunda etapa desnaturaliza la molécula bicatenaria y comienza la amplificación según el mecanismo de una PCR normal: la hebra de cDNA liberada actúa como molde para una segunda hebra de ADN y luego la molécula bicatenaria se amplifica en sucesivos ciclos.
PCR con retrotranscriptasa. Tomado de Biología Molecular e Ingeniería Genética. Elsevier Health Sciences, (p.207).
Existen tres tipos de primers para la reacción RT y todos tienen sus pros y sus contras:
Primer oligo(dT): Este cebador se une a la cola de poliA característica del mRNA, de manera que la retrotranscripción se inicia siempre por el extremo 3 del mRNA.
Primers específicos (SSPs, Sample Specific Primers): se unen de manera específica al mRNA en aquella zona de su secuencia donde esté el gen que queremos estudiar.
Random primers: Es un cocktail de oligonucleótidos (generalmente, de 6 nt) de secuencia variada que se unen de manera aleatoria a lo largo de la secuencia de RNA.
Tipos de primers utilizados para la RT PCR. Tomado de https://www.ucm.es/data/cont/docs/

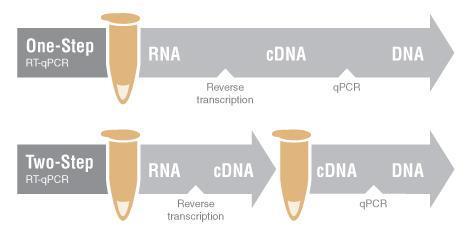
Una vez obtenido el cDNA este podrá ser utilizado como material genético para la PCR. La RT PCR puede ser de un paso, donde la PCR se realiza en la misma reacción que la síntesis de cDNA o dos pasos, donde la PCR se realiza en un segundo momento mediante una segunda amplificación.
https://international.neb.com/applications/dna
amplification pcr and qpcr/choice of one step rt qpcr or two step rt qpcr
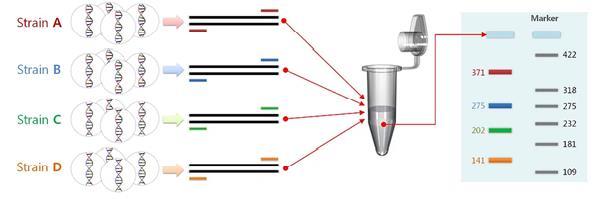
PCR Multiplex
La PCR multiplex es una técnica se utiliza para la amplificación de múltiples dianas en una única prueba de PCR. En la PCR Multiplex, se utilizan múltiples cebadores y una ADN polimerasa mediada por temperatura para la amplificación de varios fragmentos de ADN al mismo tiempo. Todos los pares de cebadores diseñados para la PCR Multiplex deben optimizarse para que la misma temperatura de hibridación sea óptima para todos los pares durante la PCR. Cuando se apuntan múltiples secuencias a la vez, se puede generar información adicional a partir de una sola ejecución de prueba que, de lo contrario, requeriría una mayor cantidad de reactivos y mucho tiempo y esfuerzo para realizarla.
Esta tecnología se ha aplicado en muchas áreas como genotipado, pruebas de paternidad, análisis de mutaciones y polimorfismos, análisis STR de microsatélites, detección de patógenos (paneles moleculares), etc.
Diseño de una PCR Multiplex. Tomado de https://www.dnasoftware.com/what is multiplex pcr/
PCR en tiempo real
El mismo principio de amplificación de la PCR se emplea en la PCR en tiempo real (Q PCR). Pero en lugar de mirar las bandas en un gel al final de la reacción, el proceso se monitorea en “tiempo real” por la adición de floresencia a la reacción. La reacción se coloca en un termociclador en tiempo real que observa cómo ocurre la reacción con una cámara o detector. Esta técnica puede ser aplicada para cuantificar o para caracterizar una muestra (genotipificación) y a su vez puede ser monoplex o multiplex.
Aunque se utilizan muchas técnicas diferentes para controlar el progreso de una reacción de PCR, todas detectan la fluorescencia emitida durante cada ciclo de PCR. Por
lo tanto, a medida que aumenta el número de copias de genes durante la reacción, también lo hace la fluorescencia, lo que indica el progreso de la reacción.
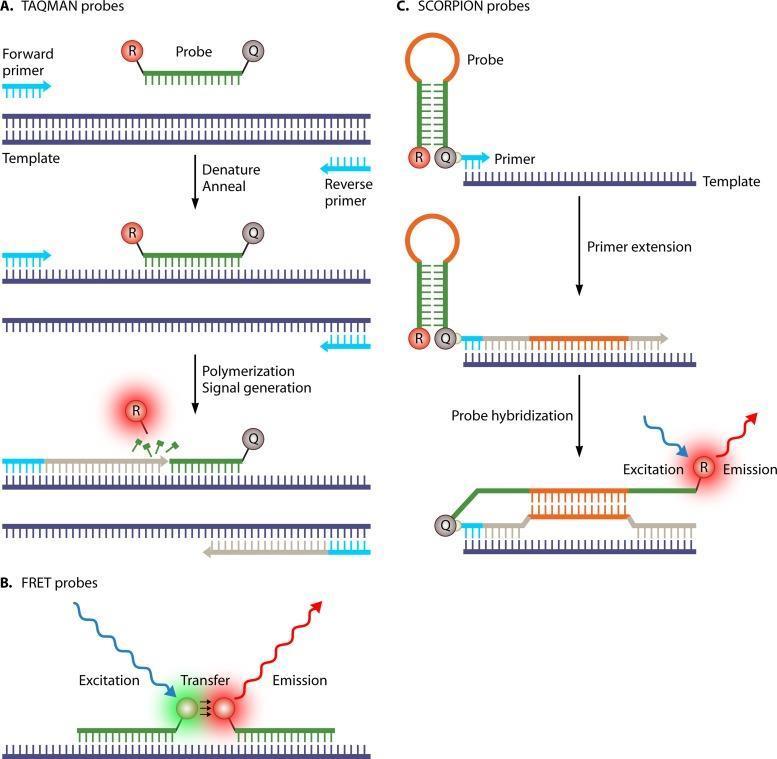
Las etapas de la Q PCR son las mismas que la PCR convencional, sin embargo, hay que considerar que dentro de cada ciclo existe una etapa de detección de señal la cual depende de la tecnología de fluorescencia sea esta un florocromo intercalate de ADN como el SYBR Green o una sonda como por ejemplo las sondas Taqman que son las más utilizadas. La señal emitida es captada por un detector y enviada a la computadora después de la conversión a una señal digital que se muestra en la pantalla. La señal se puede detectar cuando sube el nivel del umbral (nivel de detección más bajo del detector o CT). Esta metodología puede partir de ADN o de ARN mediante una previa conversión a ADN complementario mediante retrotranscripción (RT QPCR).
Ejemplo de los dos métodos de fluorescencia más usados en Q PCR. Tomado de https://microbenotes.com/real time pcr principle process markers advantages applications/

Tipos de fluorescencia en la Q-PCR
- SYBR Green:
Este es un colorante intercalante de ADN tinte que emite una señal fluorescente prominente cuando se une al surco menor del ADN bicatenario, de manera inespecífica. También se pueden utilizar otros tintes fluorescentes como el bromuro de etidio o el naranja de acridina, entre otros, pero es mejor utilizar SYBR Green por su mayor intensidad de señal y menor toxicidad.
El SYBR Green es más versátil y económico, sin embargo, no tiene la misma especificidad que presentan las sondas y su fluorescencia puede ser detectada de manera inespecífica.
Esquema de la detección con SYBR Green o similares. Tomado de Biología Molecular e Ingeniería Genética. Elsevier Health Sciences, (p.206).

- Sondas
Existen varios tipos de sondas utilizadas en Q PCR, las más populares son las sondas de hidrólisis o sondas Taqman que llevan un colorante indicador (Reporter), a menudo fluoresceína (FAM) en su extremo 5 'y un inhibidor (Quencher) de tetrametilrodamina (TAMRA), unido al extremo 3' del oligonucleótido.
En condiciones normales, la sonda permanece enrollada sobre sí misma llevando el reporter cerca del quencher, que inhibe o apaga la señal fluorescente del tinte. El quencher trabaja a modo de interruptor del reporter, cuando están juntos, la señal está “apagada”.
La sonda tiene una región homóloga con el gen diana y, por tanto, cuando la secuencia diana está presente en la mezcla se hibridan. A medida que la taq polimerasa comienza a formar una nueva cadena de ADN en la etapa de extensión, provoca la degradación de la sonda por la actividad nucleasa del extremo 5 'y el reporter de fluoresceína se separa del quencher como resultado de lo cual se genera la señal de fluorescencia.
A medida que continúa este procedimiento, en cada ciclo aumenta el número de moléculas de señal, provocando el aumento de la fluorescencia que se relaciona positivamente con la amplificación de la diana.
Fundamento de las sondas Taqman. Tomado de https://microbenotes.com/real time pcr principle process markers advantages applications/

Existen otros tipos de sondas como las SCORPION y BACON las cuales son autocomplementarias y las FRET que actúan de forma paralela.
Tipos de sondas de Q PCR. Tomado de Verweij JJ, Stensvold CR. Molecular testing for clinical diagnosis and epidemiological investigations of intestinal parasitic infections. Clinical Microbiology Reviews. 2014 Apr;27(2):371 418. DOI: 10.1128/cmr.00122 13.
Principios de la Q-PCR
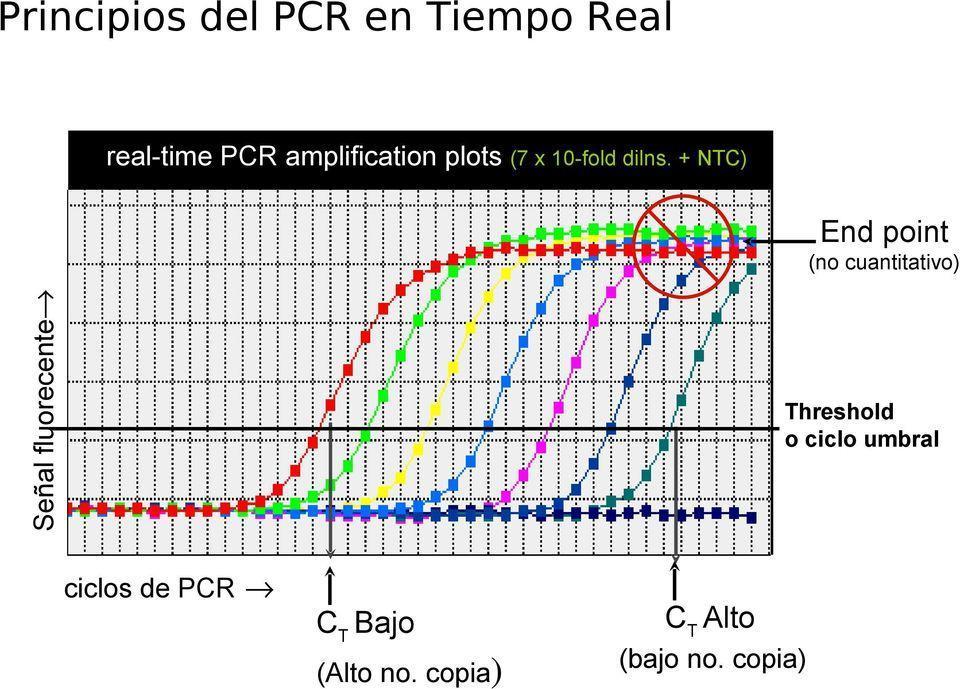
Una curva típica de PCR en tiempo real debe tener las siguientes partes:
• Línea base (Background): son los niveles de señal de fluorescencia durante los primeros ciclos (lo que se conoce como ruido, pues no pasa el umbral).
• Ct= threshold cycle: es el primer incremento significativo en la cantidad de producto de PCR, medido por el aumento de fluorescencia. No significa que no exista fluorescencia antes, solo que no es detectada por el termociclador.
• Fase logarítmica: incremento exponencial del producto de PCR.
• Meceta: Tope de amplificación.
Curvas de Q PCR. Tomado de https://docplayer.es/6771258 Experiencias en q pcr para la aplicacion y diagnostico en medicina humana.html
Aplicaciones de la pcr en tiempo real
La Q PCR es una técnica extremadamente versátil cuya utilización es muy amplia. Existen 2 aplicaciones principales para la Q PCR: la cuantificación (absoluta y relativa) y la genotipificación (con sondas o por High Resolution Melting HRM).
Cuantificación
Esta aplicación es muy útil para saber cuánto ADN o ARN (cDNA) existe en una muestra. Es la aplicación más utilizada para análisis de expresión génica (para la cual se parte de un ARN mensajero). Se utiliza una curva estándar con concentraciones conocidas frente a la cual se comparará la muestra en estudio.
Como control del ensayo es importante tener siempre un control interno de amplificación (así se garantiza de que la muestra no esté degradada y se evita un falso negativo). Este control generalmente es un gen presente en todo tipo de célula (gen endógeno) como por ejemplo el gen G6PDH, ABL1, actina, etc. Se recomienda correr también un blanco, un control positivo, un control negativo y un calibrador con el fin de normalizar el análisis.
a) Cuantificación absoluta:
Se realiza una cuantificación total sin compararlo con ningún gen de referencia. Un ejemplo de esta aplicación sería la cuantificación de la carga viral de VIH o de SARS CoV2, etc. o la cuantificación total de un ARN mensajero para saber su expresión absoluta en la muestra.
b) Cuantificación relativa:
Cuando se requiere conocer la proporción de un gen frente al total de ADN o ARN de una muestra, es decir, es una cuantificación diferencial. Como ejemplo de esta aplicación podemos citar el estudio transcrito BCR ABL en leucemia mieloide crónica, donde se reporta el % del RNA mensajero de BCR ABL frente al de un gen de referencia como el ABL1 (esto sería “equivalente”al % de células tumorales mutantes frente al total de células de una muestra).
En ambos casos se debe correr una curva estándar para utilizarla como base de la cuantificación.
Curva estándar en Q PCR. Tomado de http://sgpwe.izt.uam.mx/files/users/uami/fierrof/8 ANALISIS_DE_LA_EXPRESION_GENICA_1.pdf

- Genotipificación
a) Genotipificación con sondas:
Es muy utilizada, dependerá de la sonda para su interpretación. Se pueden diseñar sondas específicas (generalmente, FRET o BACON) de diferentes colores para los alelos de tipo salvaje y mutante (por ejemplo, verde y roja) donde los resultados se segerarán por interpretación de los colores (por ejemplo homocigoto normal emitirá una única señal verde, un heterocigoto una señal mixta y un homocigoto mutante una única señal roja).
https://www.nature.com/articles/s41598 019 45884 8
O también puede emplearse una sonda única (generalmente Taqman) que, conjuntamente con cebadores específicos, tenga afinidad únicamente a uno de los dos alelos (por ejemplo, el mutante), en este caso se interpreta por la cantidad de fluorescencia emitida (por ejemplo una señal nula significa un homocigoto normal, una señal a intensidad 1 significa heterocigoto y una señal al doble un homocigoto mutante). Es muy útil para la detección de variantes génicas en porcentajes muy bajos (1 entre 1000 células, por ejemplo).
a) Genotipificación por Melting (HRM):
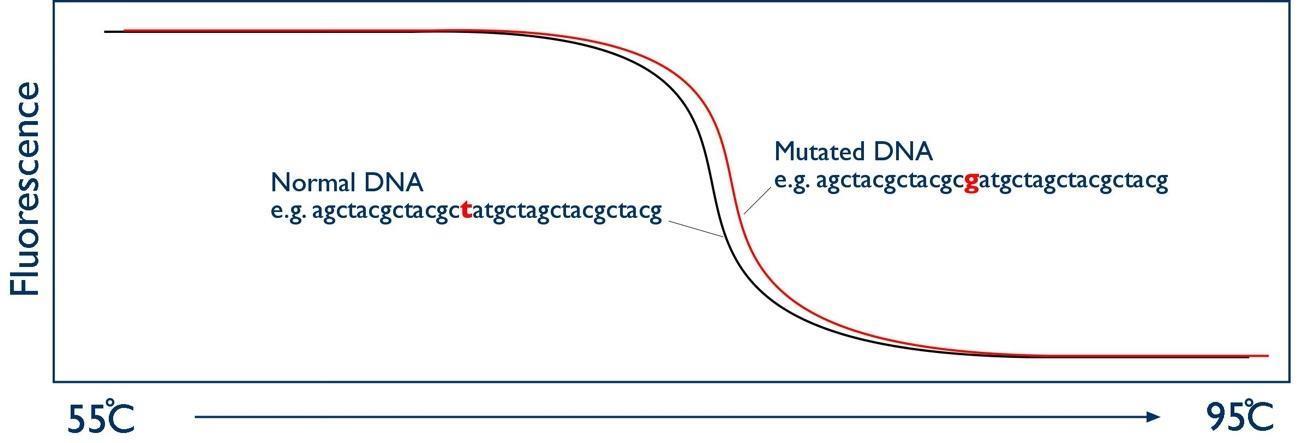
Esta técnica es un análisis post PCR que se basa en la comparación de la energía de disociación entre los pares de bases de una cadena bicatenaria de ADN. Así la energía requerida para el par CG es mayor al del par AT y esto puede ser medido con la adición de fluorescencia tipo SYBR Green, SYTO Dye, entre otros. Mediante un calentamiento gradual, el ADN bicatenario se desnaturaliza y emite cada vez menos fluorescencia (debido a que los colorantes usados se unen solo a ADN bicatenario), esta técnica es tan sensible que puede detectar las pequeñas variaciones entre la temperatura a la que se disiocian las bases.

Ejemplo de una curva de desnaturalización en HRM, se puede ver como la temperatura a la que se desnaturaliza el alelo T (normal) es menor al alelo G (mutante). Tomado de https://commons.wikimedia.org/wiki/File:DNA_melting_scematic_curve_2.jpg
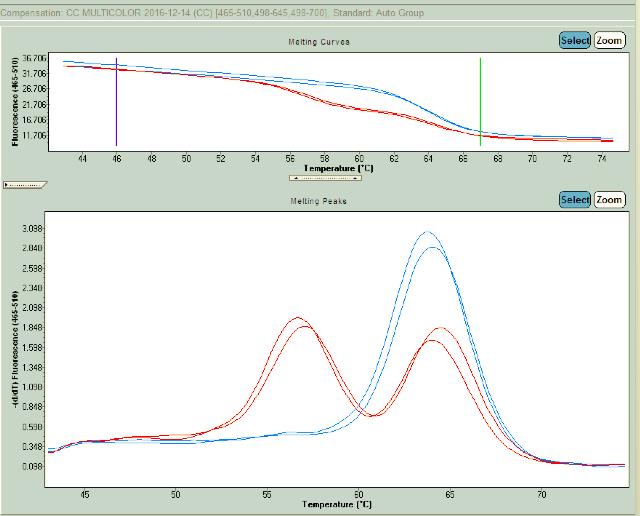
Se puede aplicar también a esta metodología, sondas específicas. Las sondas son una herramienta excelente para el genotipado de SNP y la detección de mutaciones porque identifican fácilmente muestras de tipo salvaje, mutantes y heterocigotas con una sola sonda corta.
Homocigoto normal C/C
Heterocigoto C/T
Homocigoto normal C/C
Heterocigoto C/T
Se puede observar la curva de melting (arriba) y los picos respectivos (abajo) donde el alelo T muestra un pico a 56C y el alelo C a 65C. Imagen propia, ensayo para MTHFR C677T.
Referencias
Libros
Herráez, Á. (2012). Biología Molecular e Ingeniería Genética. Elsevier Health Sciences. Capítulo 14 https://www.elsevier.com/books/texto ilustrado e interactivo de biologia molecular e ingenieria genetica/herraez sanchez/978 84 8086 647 7
Park, D (Ed). (2011). PCR Protocols. Humana Press. https://www.springer.com/gp/book/9781607619437
Hu, P., Hegde, M., & Lennon, P. A. (Eds.). (2012). Modern clinical molecular techniques. Springer Science & Business Media. https://www.springer.com/gp/book/9781461421696
Papers
Verweij JJ, Stensvold CR. Molecular testing for clinical diagnosis and epidemiological investigations of intestinal parasitic infections. Clinical Microbiology Reviews. 2014 Apr;27(2):371 418. DOI: 10.1128/cmr.00122 13. https://europepmc.org/article/PMC/3993103
Brattås, M.K., Lilleeng, K., Hovland, R. et al. Philadelphia chromosome positive AML arising from JAK2 positive myelofibrosis. Biomark Res 6, 33 (2018). https://doi.org/10.1186/s40364 018 0147 6 https://biomarkerres.biomedcentral.com/articles/10.1186/s40364 018 0147 6#Fig2
Sastre, D. A., Argaraña, C. E., Heller, V. B., Gallo, M., Fernández, E. N., & Rodríguez, C. M. (2007). An analysis of multiplex PCR in the detection of BCR ABL transcripts in hematological disorders. Genetics and Molecular Biology, 30(3), 520 523. http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1415 47572007000400003
Chirinos Arias, M. 2015. Guía práctica de PCR en tiempo real. Figshare. http://dx.doi.org/10.6084/m9.figshare.1417599 https://www.researchgate.net/publication/283090415_Guia_de_PCR_en_tiempo_real
Videos
Reacción en cadena de la polimerasa https://youtu.be/TalHTjA5gKU
PCR en tiempo real (qPCR): Conceptos Básicos https://youtu.be/C_luFY8mG2g
PCR digital (dPCR): Conceptos Básicos https://youtu.be/0Ta37aecY I
Enzimas de restricción https://youtu.be/a5TMHlRxwFA
