10 minute read
The Role of Toll Like Receptor 4 in Mediating Gut Brain Axis Inflammation and Pathologies Seen in Parkinson’s Disease


Kung Min Kim
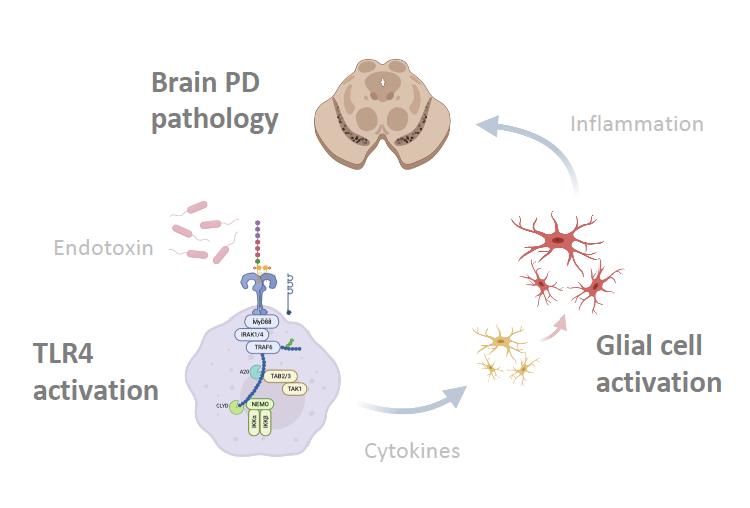
The gut-brain axis is suggested as the important contributor to the neurodegenerative diseases such as Parkinson’s disease (PD). It has been proposed that perturbation of intestinal environment activates enteric glial cells, whose inflammatory response propagates the pathophysiology from periphery to central nervous system (CNS) via the vagus nerve (Sampson et al. 2016, 1475). However, despite neuroinflammation being the common hallmark of neurodegenerative diseases, there is much to learn about the precise mechanism in which the inflammatory cascade is triggered. Perez-Pardo et al. studied the colon biopsy samples of PD patients to examine the molecular profiles and abnormalities. Compared to healthy controls, PD patients were found to have dysfunctional and inflamed intestinal wall. More importantly, it was noted that molecular markers pertaining to the immune activation of Toll-like receptor 4 (TLR4) by bacterial endotoxin were elevated. These findings prompted the authors to hypothesize that a leaky gut resulting from dysbiosis allows the invasion of gram-negative bacteria into the mucosa. Consequently, the pattern recognition receptor TLR4 is activated and proinflammatory cascade is initiated. To test, TLR4 knock-out (KO) mice and wildtype (WT) were treated with daily dose of rotenone to induce PD-like symptoms. Whereas the WT mice exhibited the similar pathophysiology and molecular profiles as the human PD patients, TLR4-KO mice were partially protected from the pathogenesis. They showed less epithelial disruption, low level of inflammation, superior motor functions, and healthier neurons. This translational study is highly relevant to the future direction of developing early diagnostic tools and treatment options for PD and other neurodegenerative conditions associated with TLR4 and the gut-brain axis.
Advertisement
Background
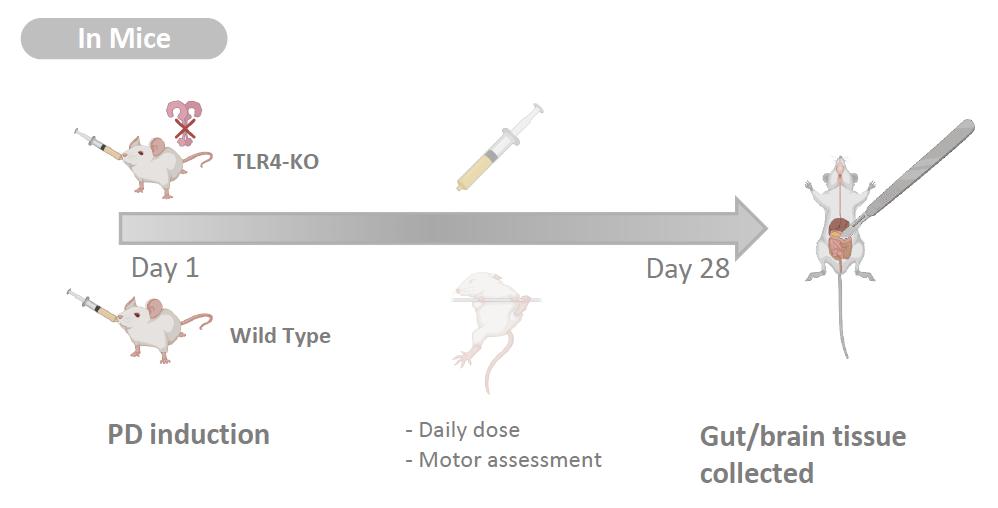
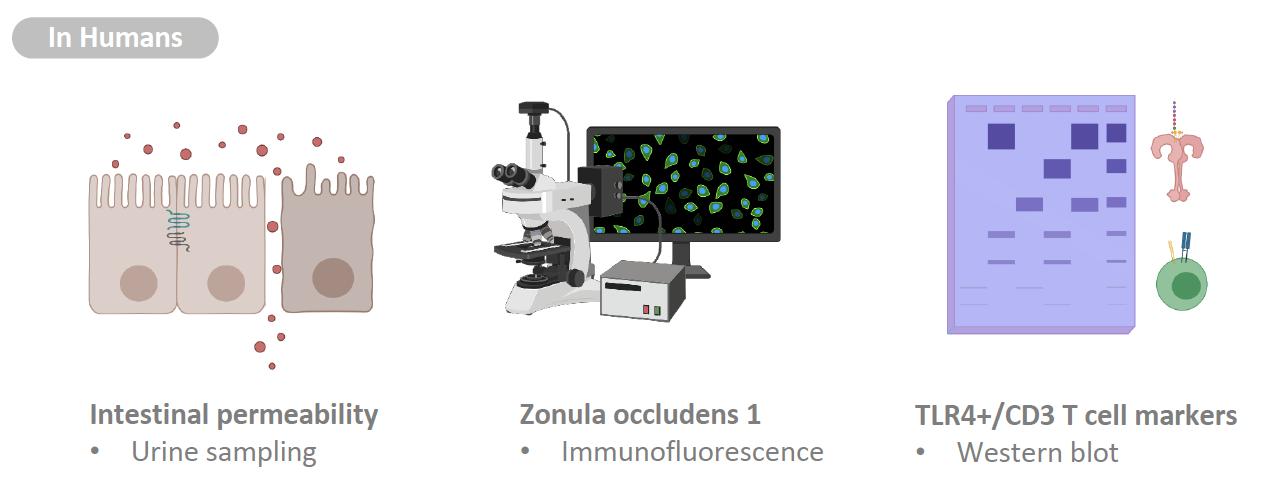
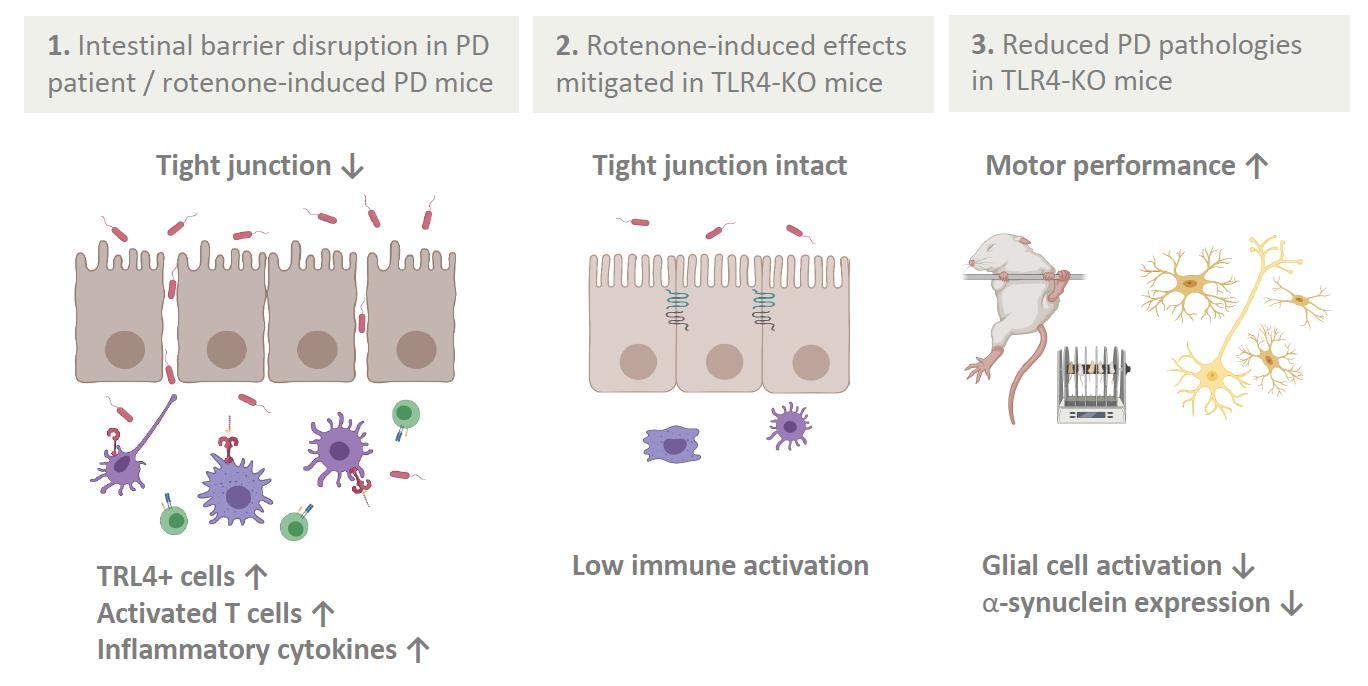
It has been more than 200 years since James Parkinson first described the ‘shaking palsy’ patients with slowness of movement and resting tremor (Parkinson 2002, 224). However, much about the disease including its etiology, genetics, preventive measures, and cures are not clearly known. Given that it’s the second most common neurogenerative disease that affects 572 per 100,000 individuals aged over 45 in North America (Marras et al. 2018, 2), the lack of definitive understanding of this disease is frustrating to the patients and to the researchers alike. Although many benefits from the dopamine replacement therapy, long term complication such as dyskinesia is common, and we currently do not have an effective way to fundamentally treat PD by stopping and reversing the neuropathy (Tambasco, Romoli, and Calabresi 2017, 1241). The misfolded alpha-synuclein protein aggregates known as the Lewy bodies are one of the pathological hallmarks and thought to be contributing to neuronal death. Yet, except for the few rare autosomal mutations that account for less than 10% of the cases, sporadic PD is believed to have multifactorial causes and there are numerous risks associated with the toxic oligomerization (Rocha, Miranda, and Sanders 2018, 250). A breakthrough came when Braak et al. (2002) published the finding that symptoms arising from dysbiosis such as constipation precedes the motor dysfunction by nearly a decade. More studies followed, solidifying the association between gut microbiota, intestinal health, and PD. And a new hypothesis was proposed: the altered gut microbiota activates neuroglial cells to launch immune responses through short chain fatty acid (SCFA) signaling (Sampson et al. 2016, 1472) and the oxidative stress from neuroinflammation and associated cytokines contribute to the onset of PD (Garcia-Esparcia et al. 2014, 584) in the genetically susceptible individuals. With works of the forerunners as the foundation, the authors further examined the nature of the immune responses that initiate neuroinflammation. First, they examined the colon biopsies of PD patients and noted that the integrity of gut barrier was significantly disrupted as measured by the degree of urinary excretion of ingested sugar. Specifically, the tight junctions which create an impermeability barrier were compromised. Immunocytochemistry analysis showed the elevated level of molecular markers for TLR4 and activated CD3+ T cells, indicative of active innate and adaptive immune responses against bacterial pathogens. Colonic gene expression analysis further supported the findings: the mRNA level of inflammatory cytokines and LPS binding protein (LBP) were indeed higher in the PD patients. LBP is an acute-phase protein that plays an integral role in TLR4 activation and its level is directly correlated. The second part of the study was designed as a translational study to replicate the findings in mice. The goal was to test whether TLR4 activation plays a significant role in triggering the inflammatory cascade in the gut-brain axis. Rotenone, a mitochondrial Complex 1 inhibitor, is a neurotoxin known to induce PD-like symptoms by causing the death of dopaminergic neurons. Adult TLR4-KO mice and the WT were fed rotenone daily for 4 weeks and their gut and brain tissue samples were collected. Like the human counterpart, the WT had altered gut microbiota, hyperpermeable epithelium, and higher degree of enteric glial cell-mediated inflammation. On the other hand, the TLR4-KO mice were found to be partially protected from the rotenone-induced effects in terms of intestinal disruption, immune activation, deterioration of motor functions, and neuropathy. This novel knowledge that silencing TLR4 pathway serves as a preventative and protective measure against PD is highly relevant to the current field of research, as it may be investigated as a potential therapeutic target.
Major Results
Human colon biopsy studies Compared to healthy subjects, PD patients showed disruption in their gut barrier even in the absence of clinical gastrointestinal dysfunction and the expression of tight junction protein ZO1 was found to be significantly reduced. When stained with antibodies, it was evident that PD subjects have greater TLR4 and CD3+ T cells immunoreactivity, which can be translated as the result of frequent invasions by gram-negative bacteria. Indeed, the mRNA expression level of LBP and associated proinflammatory cytokines were also elevated in the lamina propria (LP). These results precisely replicate the observations made by Forsyth et al. (2011) who had additionally singled out E. coli as the major contributor, as its presence in LP was 4 times higher in PD cases. Defining the intestinal characteristics of PD patients is highly important, as they can serve as biological markers for early diagnosis.
TLR4-KO mice study
Both TLR4-KO and WT mice experienced a change in their microbiota composition in response to daily dose of rotenone.
Specifically, there was a significant decrease in Bifidobacteria which is believed to be beneficial, while endotoxin-producing bacteria increased in abundance. Again, this data fits what has been routinely observed in the human PD patients as reported 92
by Gerhardt and Mohajeri (2018). However, TLR4-KO mice were partially protected from the detrimental effects of dysbiosis and PD-like pathologies induced by rotenone. Not only was the integrity of gut epithelium preserved with functional tight junctions, less signs and molecular markers of inflammation and TLR4 activation were observed. It was also demonstrated for the first time that the absence of TLR4 pro-inflammatory signaling is associated with a significantly lower level of enteric alphasynuclein expression and microglial activation. More importantly, same trend was observed in the CNS. This suppressive effect is highly relevant, for it can be further studied as a potential treatment to slow down the progression of PD at an early stage. not explored in this experiment despite it being extremely important for the risk of developing the disease. A future study in which alpha-synuclein overexpressing (ASO) transgenic animal model is incorporated would be a great addition to gather more applicable results. In addition, nowhere in the paper did the authors explore the deleterious effects of silencing TLR4 receptor which is an integral part of our innate immune defense. Mutations to TLR4 are highly associated with atherosclerosis, asthma, and Crohn’s disease (Lin, Verma, and Hodgkinson 2012, 635) not to mention the greatly increased susceptibility to infection that can be life-threatening. At least mention this aspect would have added more scientific objectivity and credibility.
Discussion
In this paper, Perez-Pardo et al. demonstrated that TLR4- mediated immune activation in the gut is a crucial contributor to neuroinflammation characterized in PD. By blocking the TLR4 signalling pathway, the development of PD and associated symptoms were largely mitigated in the animal model treated with rotenone. As such, the authors concluded by supporting the gut-brain axis model of pathogenesis in PD. Although there are number of other pattern recognition receptors such as TLR2, 5, and NLRP3 known to be associated with gut dysbiosis (Jin, Henao-Mejia, and Flavell 2013, 879), this study is first to clearly show the causal link between TLR4 activation and PD pathologies through an experimental manipulation.
Future Directions
The next suggested step involves repeating the experiment with ASO mice to test the effects of TLR4 activation in neuroinflammation and alpha-synuclein pathology. Two groups of ASO mice and a control WT will receive fecal transplant from a PD patient and only one ASO group will also receive a pharmacological intervention to inhibit TLR4. After few weeks, the gut and brain tissues will be analyzed for epithelial integrity, proinflammatory markers, microglial activation, and presence of alpha-synuclein pathology. It is expected that ASO mice will develop PD-like pathology as demonstrated by Sampson et al. (2016) following a dysbiotic microbiota transplant. However, the ASO group treated with the TLR4 inhibitor is expected to be partially protected as in the case for the TLR4-KO mice. The control WT may also suffer from dysbiosis, inflamed gut, and increased microglial activation, yet they are not expected to develop PD-related symptoms due to lack of genetic predisposition. The result may not match the expectation, that is, the ASO group treated the TLR4 inhibitor may still develop similar degree of neuroinflammation and alpha-synuclein pathology. If so, the ASO mice may have a lower threshold for initiating oligomerization compared to the TLR4-KO mice. Other modes of activating microglia, such as through SCFAs and TLR2 (Caputi and Giron 2018, 1689) may be suffice for those with strong genetic disposition to develop PD even in the absence of TLR4 signaling.
Critical Analysis
The authors used rotenone as the method of quickly inducing PD in mice. While highly effective, it is not the most relevant model of PD pathogenesis; exposure to the environmental neurotoxins is much more subtle and chronic in the most sporadic PD cases. In addition, the element of genetic predisposition was
10.
11.
12. Caputi, Valentina, and Maria Giron. “Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease.” International Journal of Molecular Sciences 19, no. 6 (2018): 1689. https://doi.org/10.3390/ijms19061689. Forsyth, Christopher B., Kathleen M. Shannon, Jeffrey H. Kordower, Robin M. Voigt, Maliha Shaikh, Jean A. Jaglin, Jacob D. Estes, Hemraj B. Dodiya, and Ali Keshavarzian. “Increased Intestinal Permeability Correlates with Sigmoid Mucosa AlphaSynuclein Staining and Endotoxin Exposure Markers in Early Parkinson's Disease.” PLoS ONE 6, no. 12 (2011). https:// doi.org/10.1371/journal.pone.0028032. Garcia-Esparcia, Paula, Franc Llorens, Margarita Carmona, and Isidre Ferrer. “Complex Deregulation and Expression of Cytokines and Mediators of the Immune Response in Parkinson's Disease Brain Is Region Dependent.” Brain Pathology 24, no. 6 (2014): 584–98. https://doi.org/10.1111/bpa.12137. Gerhardt, Sara, and M. Mohajeri. “Changes of Colonic Bacterial Composition in Parkinson’s Disease and Other Neurodegenerative Diseases.” Nutrients 10, no. 6 (2018): 708. https://doi.org/10.3390/nu10060708. Jin, Chengcheng, Jorge Henao-Mejia, and Richard A. Flavell. “Innate Immune Receptors: Key Regulators of Metabolic Disease Progression.” Cell Metabolism 17, no. 6 (2013): 873–82. https://doi.org/10.1016/j.cmet.2013.05.011. Lin, Yi-Tzu, Amanda Verma, and Conrad P. Hodgkinson. “Toll-Like Receptors and Human Disease: Lessons from Single Nucleotide Polymorphisms.” Current Genomics 13, no. 8 (2012): 633–45. https://doi.org/10.2174/138920212803759712. Marras, C., J. C. Beck, J. H. Bower, E. Roberts, B Ritz, G. W. Ross, R. D. Abbott, et al. “Prevalence of Parkinson’s Disease across North America.” npj Parkinson's Disease 4, no. 1 (2018). https://doi.org/10.1038/s41531-018-0058-0. Parkinson, James. “An Essay on the Shaking Palsy.” The Journal of Neuropsychiatry and Clinical Neurosciences 14, no. 2 (2002): 223–36. https://doi.org/10.1176/jnp.14.2.223. Perez-Pardo, Paula, Hemraj B Dodiya, Phillip A Engen, Christopher B Forsyth, Andrea M Huschens, Maliha Shaikh, Robin M Voigt, et al. “Role of TLR4 in the Gut-Brain Axis in Parkinson’s Disease: a Translational Study from Men to Mice.” Gut 68, no. 5 (2018): 829–43. https://doi.org/10.1136/gutjnl-2018-316844. Rocha, Emily M., Briana De Miranda, and Laurie H. Sanders. “Alpha-Synuclein: Pathology, Mitochondrial Dysfunction and Neuroinflammation in Parkinson’s Disease.” Neurobiology of Disease 109 (2018): 249–57. https://doi.org/10.1016/ j.nbd.2017.04.004. Sampson, Timothy R., Justine W. Debelius, Taren Thron, Stefan Janssen, Gauri G. Shastri, Zehra Esra Ilhan, Collin Challis, et al. “Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease.” Cell 167, no. 6 (2016): 1469-1480. https://doi.org/10.1016/j.cell.2016.11.018. Tambasco, Nicola, Michele Romoli, and Paolo Calabresi. “Levodopa in Parkinson’s Disease: Current Status and Future Developments.” Current Neuropharmacology 16, no. 8 (2018): 1239–52. https:// doi.org/10.2174/1570159x15666170510143821.








