17 minute read
The Therapeutic Potential of the Novel Player Kcnn2 in Fetal Alcohol Syndrome Disorder Pathogenesis


Sofia Tiu
One of the leading contributors to disability within fetal development is alcohol, affecting 3% of all newborn children. 1 Fetal alcohol spectrum disorders (FASD) is an umbrella term that is used for a set of disorders that arise by prenatal alcohol exposure (PAE) administered by a pregnant mother. 2 Main FASD treatments currently rely on cognitive-behavioural treatments due to the lack of medications. 3 Although the effects of these therapies have been well studied, there is ongoing research for more biological advances into discovering FASD pathogenesis. Currently, FASD pathogenesis has observed that PAE affects certain downstream neurodevelopmental pathways which lead to amplified consequences, such as excessive cell death and proliferation and modified cell signaling affecting gene expression. 4 A recent study by Mohammad et al. indicates that increased expression of Kcnn2 is associated with motor learning disability in CD-1 FASD mice models. 5 Furthermore, Kcnn2- knockdown PAE mice express higher levels of motor learning in earlier adulthood compared to PAEmice with increased Kcnn2 expression. 5 Investigation into this Kcnn2-motor pathway has indicated that inhibiting Kcnn2 channels increases motor cortex activation and decreases motor symptoms in a FASD mouse model, indicating that Kcnn2 inhibition may be a novel route for treating FASD motor progression in humans. 5
Advertisement
Key words: Fetal Alcohol Syndrome Disorder (FASD), prenatal alcohol exposure (PAE), Kcnn2, motor learning, CD-1 mice model
Introduction
Fetal Alcohol Syndrome Disorders (FASD) are caused by maternal administration of alcohol during gestation leading to prenatal alcohol exposure (PAE). 2 They are often characterized by various combinations of facial deformations, developmental deficits and neurocognitive deficiencies that gradually worsen overtime. 2 No single cause of FASD has been identified, and it is likely that several factors contribute to its presentation. Research indicates that different genetic mutations can contribute to different aspects of the abnormalities present in FASD. Due to this, there are no effective treatments for all individuals. 5 Additionally, research has linked infantile motor skills to early brain development. 6 Through focusing on motor skill development as one of the main indices of cognitive development, FASD research suggests that motor improvements in early infants can lead to progressive cognitive improvement in early childhood. 6
Previous studies have shown that heat shock signaling (HS) may contribute to the complex pathogenesis of FASD. 7,8,9 Evidence from mouse models show repetitive exposure of ethanol (EtOH) activates transcription factor heat-shock-factor-1 (HSF1) in a spontaneous manner. 7,8 Further investigation of the HSF1 pathway suggests that activation of HSF1 within the embryonic mouse brain in response to PAE may contribute to FASD consequences. 9 This research displayed that HSF1 deficiencies in cortical cells in utero could lead to higher variability in neural progenitor cells (NPC), not only in mice, but in humans as well. 10 Therefore, it is possible that HSF1 and its downstream pathways in NPCs are important therapeutic targets for FASD.
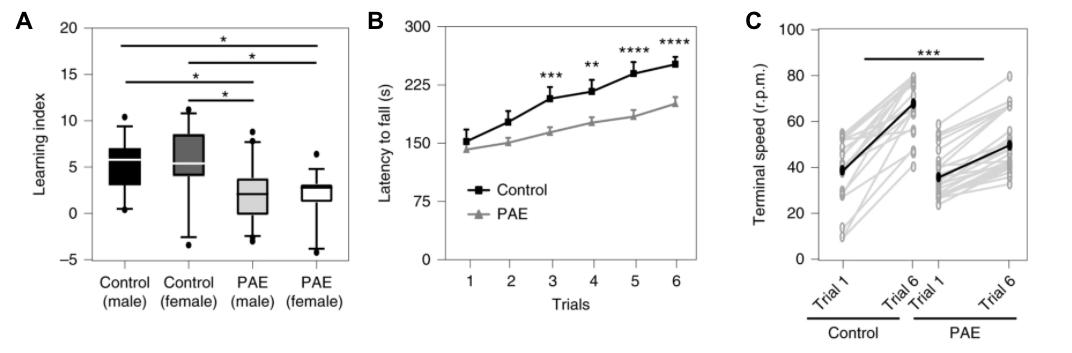
Further investigation of HSF1 pathways derived multiple targets that are responsible for mature brain abnormalities caused by HSF1. Although many of these targets remain unknown, few studies aim to investigate and analyze their role in FASD mice models. Mohammad et al., show that tracing HSF1 lineage leads to Kcnn2, which increased expression causes deficits in motor skills without affecting overall motor function. 5,11 Ultimately, this study demonstrates that decreased activity of Kcnn2 channels improves motor function in mice and provides therapeutic potential for humans with FASD due to its ability to decrease motor FASD symptoms. 511 embryonic (E) days 16 and 17. These days correlate with human embryonic motor cortical development. 5 Using EtOH or phosphate-buffered saline (PBS), fetal mice were administered EtOH/PBS via intraperitoneal injection. To prevent confounds, ratios of EtOH and PBS were created by comparing overall bodyweight at postnatal (P) day 0 for consistency. At P28, mice underwent open-field testing to measure locomotion and agility. PAE and control mice failed to show locomotor deficits and anxious behaviour. Thus, suggesting that PAE does not affect overall motor ability, only the learning of motor ability. Accelerated rotarod tests were performed at P30-31. Initially, PAE-mice did not display significant differences. However, PAE-mice exhibited an inability to learn as the rotarod accelerated and increased susceptibility to falling (Figure 1B). Additionally, PAE-mice had lower learning indices compared to the control-mice. (Figure 1A).
Figure 1.) A The learning indices of the control mice are significantly higher than PAE-mice, who are generally not capable of completing the accelerated rotarod task. Control mice withstand the acceleration of the rotarod more successfully than the PAE-induced mice and undergo motor learning (A-C). Mohammad, Shahid, Stephen J. Page, Li Wang, Seiji Ishii, Peijun Li, Toru Sasaki, Aiesha Basha, et al. “Kcnn2 Blockade Reverses Learning Deficits in a Mouse Model of Fetal Alcohol Spectrum Disorders.” Nature Neuroscience 23, no. 4 (April 2020): 533–43. https://doi.org/10.1038/s41593-020-0592-z.
Major Results
Mohammad et al. discovered that predisposed CD-1 mice models of FASD express higher genetic expression of Kcnn2 in cortical layers II and III of the motor cortex (M1). 5 Further analysis suggests that decreased activity of Kcnn2 channels has a causal effect on decreasing FASD motor symptoms and increasing cognitive capabilities, making it a potential target for FASD therapeutics. 5
PAE Consequences in FASD-Mice Models
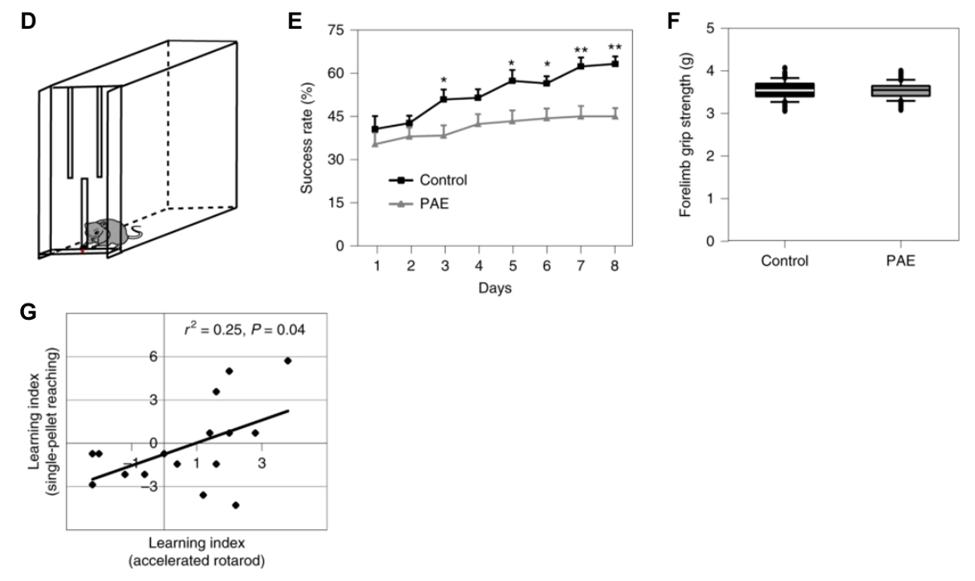
Previous literature has indicated that self-feeding behaviour is characteristic of FASD 12 . In the single-pellet reaching task, a successful trial consists of the mouse grasping the pellet and placing it in their mouth without dropping/fumbling. After several trials, control-mice increased their success rate by 25%. PAE-mice exhibited less than an 8% increase in their success rates (Figure 2E). Comparing the forelimb grip strength of PAE and control was similar suggesting failed trials was not due to muscular weakness, but the impaired learning of grasping the pellet (Figure 2F). Lastly, learning indices of the accelerated rotarod and single pellet reaching tests were compared and Researchers administered EtOH to fetal mice during
indicated a significant correlation (Figure 2G).
Figure 2.) An example of the single-pellet reaching box is pictured in D. Those treated with PAE were less likely to be successful in reaching the pellet with only slight increases in success after several days . This failure is not due to muscular weakness, as seen in E-F. Graphical correlation of the learning indices of the accelerated rotarod and single pellet reaching test signifies a connected pathway that controls the motor deficits seen in both tasks in G. Mohammad, Shahid, Stephen J. Page, Li Wang, Seiji Ishii, Peijun Li, Toru Sasaki, Aiesha Basha, et al. “Kcnn2 Blockade Reverses Learning Deficits in a Mouse Model of Fetal Alcohol Spectrum Disorders.” Nature Neuroscience 23, no. 4 (April 2020): 533–43. https://doi.org/10.1038/ s41593-020-0592-z. To investigate the inhibition of Kcnn2 channels, researchers administered tamapin, a Kcnn2 channel blocker. 14 Kcnn2+ neuron activity in the M1. Results showed that tamapin
Acute Activation of HS Signaling on PAE-Mice Models
Mohammad et al. utilized an HSE-RFP reporter system to lineage trace the cells that are responsible for motor deficits in the cortex and its downstream effects. Constructs were also developed with GFP to indicate successful electroporation in the M1. Constructs were delivered using electroporation at E15, 24-hours prior to PAE. In PAE-mice, most GFP+ neurons (in layers II and II of M1) were RFP+, indicating HS-signaling activation had occurred upon PAE. 7 In contrast, there was less than 2% of co-expression of GFP/RFP in control-mice. Researchers used unicellular RNA sequences to identify differentially expressed genes in PAE-mice to avoid masking Ishii, Peijun Li, Toru Sasaki, Aiesha Basha, et al. “Kcnn2 Blockade Reverses Learning Deficits in a Mouse Model of Fetal Alco
other genes. Principal component analysis (PCA) showed genes specific to RFP+ neurons were associated with learning, plasticity, and long-term potentiation (LTP).
Kcnn2 in PAE-Mice Models
Among 37 genes, Kcnn2 was the only gene that encoded a small-conductance calcium-activated potassium channel in the RFP+ group. Previous literature has shown that the Kcnn2 is influential to learning and memory. 13 Immunohistochemical analysis of PAE-mice neuronal cells demonstrate increased activation in the M1 at P30. In addition, researchers confirmed Kcnn2 is endogenous by using HSF1-knockout (KO) mice, where Kcnn2 failed to increase in response to PAE without HS-signaling activation,
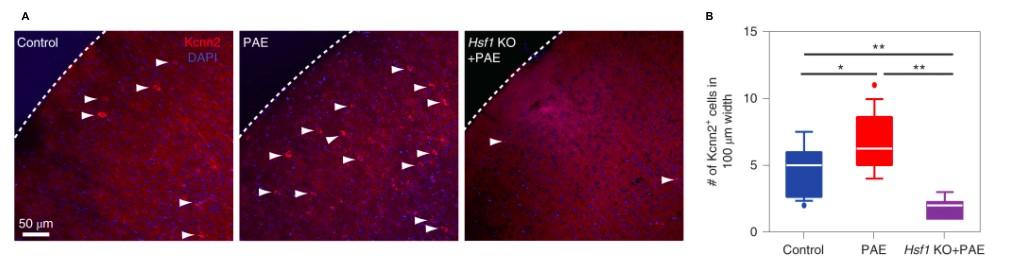
Figure 3.) Immunohistochemical analysis at P30 of PAE mice demonstrates the number of Kcnn2+ neurons are increased in PAE mice compared to the control. More so, this increase is not observed in HSF1-KO mice, seen in A-B. Mohammad, Shahid, Stephen J. Page, Li Wang, Seiji Ishii, Peijun Li, Toru Sasaki, Aiesha Basha, et al. “Kcnn2 Blockade Reverses Learning Deficits in a Mouse Model of Fetal Alcohol Spectrum Disorders.” Nature Neuroscience 23, no. 4 (April 2020): 533–43. https:// doi.org/10.1038/s41593-020-0592-z.
Kcnn2 Blockage and Learning in PAE-Mice
Intraperitoneal injection of labeled-tamapin at P30 decreased significantly increased motor learning in PAE-mice compared to con- trols. 3
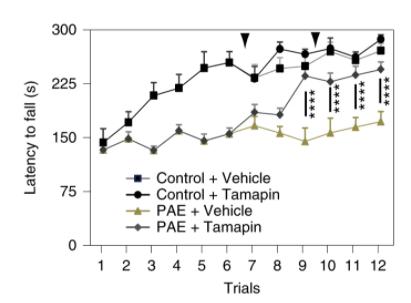
Figure 4.) Tamapin administration to PAE-mice increases time spent on the accelerated rotarod, indicating increased motor learning. Mohammad, Shahid, Stephen J. Page, Li Wang, Seiji thereby not a consequence of RFP+ cells (Figure 3A-B).
hol Spectrum Disorders.” Nature Neuroscience 23, no. 4 (April 2020): 533–43. https://doi.org/10.1038/s41593-020-0592-z.
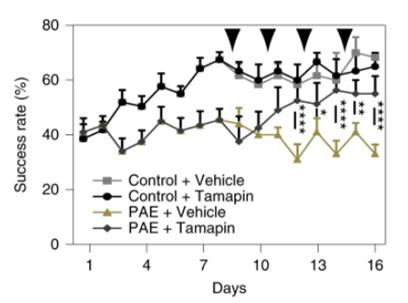
Figure 5.) Tamapin administration to PAE-mice increases the success rate of the single-pellet reaching test, indicating increased motor learning. Mohammad, Shahid, Stephen J. Page, Li Wang, Seiji Ishii, Peijun Li, Toru Sasaki, Aiesha Basha, et al. “Kcnn2 Blockade Reverses Learning Deficits in a Mouse Model of Fetal Alcohol Spectrum Disorders.” Nature Neuroscience 23, no. 4 (April 2020): 533–43. https://doi.org/10.1038/s41593-020 -0592-z.
Similarly, researchers created a tamoxifen-inducible Kcnn2 short-hairpin RNA (shRNA) and control shRNA into M1 at E15. Rotarod and single-pellet reaching tasks before and after tamoxifen-injection allows Kcnn2 blockage after 30 minutes of exposure. Increased motor learning skills of the rotarod and single-pellet reaching tasks demonstrated that Kcnn2- knockdown in the M1 was sufficient to improve learning deficits in PAE-mice.
Conclusions
The data presents evidence of a causal relationship between Kcnn2 expression and the motor-learning deficiencies of FASD in a mouse model. It was determined that Kcnn2 levels are increased in PAE-mice. Additionally, motor deficits are alleviated by decreased Kcnn2 activity via tamapin or tamoxifeninducible Kcnn2-shRNA. These findings suggest that the blockage of Kcnn2 shows great potential in future FASD therapies. The discovery of FASD treatments is important due to the lack of effective therapies. 3 Although Kcnn2 is new in FASD research, further investigation can determine whether Kcnn2 blockers are eligible for human intervention. Previous studies that investigated HS-signaling pathways target other genes that improvements in FASD adults. 3,6,10
Mohammad et al. conclude that blocking Kcnn2 expression in mice should be transferred over to human studies in order to investigate its true effectiveness. Although the evidence for Kcnn2 blockers is strong in mouse models, research is uncertain if these results would directly translate to human models. 5 Despite the strong findings in the mouse model, further research is required to ensure efficiency and safety in for future therapeutic uses. ers on older mice (P180+) to examine human adult equivalent effects. 16 By doing so, the experimental conditions will increase in external validity and representation of FASD medications being given to all age groups. Additionally, the researcher confirms that Kcnn2 was 5 th out of 37 possible genes that demonstrated interactions with EtOH in the cortex. Despite Mohammad et al. indicating that Kcnn2 and FASD have a causal relationship in the M1, there are other genes indicated by RFP+ neurons that may interfere with FASD pathogenesis. In order to increase the internal validity, future research should ensure there is a precise qualitative measure between all RFP+ genes that are being manipulated. Therefore, stating that Kcnn2 is the cause of motor deficiency in the mice may not necessarily be true due to confounding genes.
Furthermore, the discussion states that Kcnn2 channels were not limited to layers II and III of the M1 which decreases the internal validity that Kcnn2 are solely responsible in motor deficiencies. Kcnn2 channels are found in various parts of the are responsible for cortical deficits, but have yet to see any
central and peripheral nervous system and are largely known for neurodegeneration in Alzheimer’s disease (AD). 17 Previous research has noted that Kcnn2 channels inhibit synaptic plasticity, therefore, inhibiting Kcnn2 increases synaptic plasticity. However, many studies on Kcnn2 channels are not human studies and fail to administer specific neuronal Kcnn2 blockers without transgenic mice. 18 Human studies of AD have shown that humans cannot survive with minimal Kcnn2 channel activity due to their ability to modulate glutamate intake. 19 Research by Allen et al. argue against Kcnn2 blockers, as pharmacological Kcnn2 activation can increase neurorestorative properties and decrease neurodegeneration in AD. 19 Therefore, Kcnn2- blockage in humans may cause different consequences than in mice, especially in different areas that express Kcnn2. Lastly, the quantity of the drug and its effects are left inconclusive. Mohammad et al. based their drug concentrations on mouse body weight and only gave a singular dose. Although this singular dose improves motor learning in PAE-mice, these improvements diminished quickly with a two-week maximum. The research failed to expand on how often this drug would need to be administered. Other pharmacological questions that were not investigated were the dose-dependent consequences, blocking efficiency per dosage given and/or evaluating the margin of safety (MOS). Overall, these are all factors that should be considered in future research.
Critical Analysis
The data collected by Mohammad et al. provides evidence for a promising therapeutic approach to FASD. However, several aspects of this study require improvement. Firsly, this study only investigated the effects of Kcnn2 blockade in mice up to P35. Although the most studied age groups of FASD are children, evidence from literature displays that FASD-adults exhibit different biological and behavioural symptoms. 15 By only examining mice at date P35, concluding Kcnn2 blockers are an eligible FASD medication only applies to one age group. This can be prevented by examining the effects of Kcnn2 block
Future Directions
Future endeavors should begin by studying the significance of Kcnn2 in comparison to the other genes that are functionally relevant to FASD motor deficiencies. This would be done by examining other RFP+ genes which are activated in PAE response and present in HS-signaling of NPCs. If the results demonstrate that Kcnn2 is not affected by coinciding genes activated in HS-signaling, then it would increase causation of Kcnn2. If Kcnn2 results to act comorbidly with another gene, then this would contradict the conclusions of Mohammad et
al., potentially guiding researchers to investigate the new gene of interest’s downstream effects on motor learning. Another note of importance is investigating whether potential genes activate other brain regions than layers II and III of the M1 via HSF1-lineage trace. As seen in the present study, Kcnn2 upregulation was not exclusive to the M1 layers. If Kcnn2 and the new gene active other areas that influence motor learning, this would prompt a reevaluation of their findings. In the case of a multiple affected regions of FASD, this would prohibit the development of human studies as the FASD pathogenesis in mice remains unclear. In order to progress to human models, future research must produce a drug that minimizes negative side effects that may cause further motor and cognitive decline. The drug must target M1 Kcnn2 channels without transgenic humans and not target peripheral Kcnn2 that contribute neuroprotective properties. 19 Experiments that address these issues should include testing the long-term effects of these drugs and Kcnn2 regulation within the M1. One way to mimic the effects of Kcnn2 blockers and their specificity is to use transgenic-FASD and wild -type mice to analyze which Kcnn2 channels are the ultimate targets for Kcnn2 blockers. To understand the level of Kcnn2 responsiveness and MOS, another factor to consider is the dose-dependency. If the drug is required in higher doses to effectively inhibit, then the MOS decreases and individuals are more prone to negative side effects. Beyond this, testing Kcnn2 blockers on wild-type mice would examine the consequences of Kcnn2 inhibition that are currently unaccounted for. There are still many conflicts in confirming Kcnn2 is a therapeutic drug to combat FASD in humans. Currently, the research that has been conducted highlights promising genes that aid in creating future therapies for FASD in humans.
10.
11.
12.
13.
14.
15. Popova, Svetlana, Shannon Lange, Kevin Shield, Alanna Mihic, Albert E Chudley, Raja A S Mukherjee, Dennis Bekmuradov, and Jürgen Rehm. “Comorbidity of Fetal Alcohol Spectrum Disorder: A Systematic Review and Meta-Analysis.” The Lancet 387, no. 10022 (March 5, 2016): 978–87. https://doi.org/10.1016/S0140-6736(15)01345-8. Mukherjee, Raja A S, Sheila Hollins, and J Turk. “Fetal Alcohol Spectrum Disorder: An Overview.” Journal of the Royal Society of Medicine 99, no. 6 (June 1, 2006): 298–302. https://doi.org/10.1177/014107680609900616. Doig, Jenna, John D. McLennan, and W. Ben Gibbard. “Medication Effects on Symptoms of Attention-Deficit/Hyperactivity Disorder in Children with Fetal Alcohol Spectrum Disorder.” Journal of Child and Adolescent Psychopharmacology 18, no. 4 (May 9, 2007): 365–71. https://doi.org/10.1089/cap.2007.0121. Sulik, Kathleen K. “Chapter 26 - Fetal Alcohol Spectrum Disorder: Pathogenesis and Mechanisms.” In Handbook of Clinical Neurology, edited by Edith V. Sullivan and Adolf Pfefferbaum, 125:463–75. Alcohol and the Nervous System. Elsevier, 2014. https://doi.org/10.1016/B978-0-444-62619-6.00026-4. Mohammad, Shahid, Stephen J. Page, Li Wang, Seiji Ishii, Peijun Li, Toru Sasaki, Aiesha Basha, et al. “Kcnn2 Blockade Reverses Learning Deficits in a Mouse Model of Fetal Alcohol Spectrum Disorders.” Nature Neuroscience 23, no. 4 (April 2020): 533–43. https://doi.org/10.1038/s41593-020-0592-z. Streissguth, A. P., H. M. Barr, and P. D. Sampson. “Moderate Prenatal Alcohol Exposure: Effects on Child IQ and Learning Problems at Age 7 1/2 Years.” Alcoholism, Clinical and Experimental Research 14, no. 5 (October 1990): 662–69. https:// doi.org/10.1111/j.1530-0277.1990.tb01224.x. Ishii, Seiji, Masaaki Torii, Alexander I. Son, Meenu Rajendraprasad, Yury M. Morozov, Yuka Imamura Kawasawa, Anna C. Salzberg, et al. “Variations in Brain Defects Result from Cellular Mosaicism in the Activation of Heat Shock Signalling.” Nature Communications 8 (2017). https://doi.org/10.1038/ncomms15157. Torii, Masaaki, Masanori Sasaki, Yu-Wen Chang, Seiji Ishii, Stephen G. Waxman, Jeffery D. Kocsis, Pasko Rakic, and Kazue Hashimoto-Torii. “Detection of Vulnerable Neurons Damaged by Environmental Insults in Utero.” Proceedings of the National Academy of Sciences of the United States of America 114, no. 9 (February 28, 2017): 2367–72. https:// doi.org/10.1073/pnas.1620641114. El Fatimy, Rachid, Federico Miozzo, Anne Le Mouël, Ryma Abane, Leslie Schwendimann, Délara Sabéran-Djoneidi, Aurélie de Thonel, et al. “Heat Shock Factor 2 Is a Stress-Responsive Mediator of Neuronal Migration Defects in Models of Fetal Alcohol Syndrome.” EMBO Molecular Medicine 6, no. 8 (August 2014): 1043–61. https://doi.org/10.15252/ emmm.201303311. Hashimoto-Torii, Kazue, Masaaki Torii, Mitsuaki Fujimoto, Akira Nakai, Rachid El Fatimy, Valerie Mezger, Min J. Ju, et al. “Roles of Heat Shock Factor 1 in Neuronal Response to Fetal Environmental Risks and Its Relevance to Brain Disorders.” Neuron 82, no. 3 (May 7, 2014): 560–72. https://doi.org/10.1016/j.neuron.2014.03.002. Lam, Jenny, Nichole Coleman, April Lourdes A. Garing, and Heike Wulff. “The Therapeutic Potential of Small-Conductance KCa2 Channels in Neurodegenerative and Psychiatric Diseases.” Expert Opinion on Therapeutic Targets 17, no. 10 (October 2013): 1203–20. https://doi.org/10.1517/14728222.2013.823161. Amos-Kroohs, Robyn M., Birgit A. Fink, Carol J. Smith, Lyanne Chin, Sandra C. Van Calcar, Jeffrey R. Wozniak, and Susan M. Smith. “Abnormal Eating Behaviors Are Common in Children with Fetal Alcohol Spectrum Disorder.” The Journal of Pediatrics 169 (February 2016): 194-200.e1. https://doi.org/10.1016/j.jpeds.2015.10.049. Lin, Mike T., Rafael Luján, Masahiko Watanabe, John P. Adelman, and James Maylie. “SK2 Channel Plasticity Contributes to LTP at Schaffer Collateral-CA1 Synapses.” Nature Neuroscience 11, no. 2 (February 2008): 170–77. https://doi.org/10.1038/ nn2041. “Scorpion Venom Research Around the World: Indian Red Scorpion | SpringerLink.” Accessed June 10, 2020. https:// link.springer.com/referenceworkentry/10.1007%2F978-94-007-6647-1_5-1. Moore, Eileen M., and Edward P. Riley. “What Happens When Children with Fetal Alcohol Spectrum Disorders Become Adults?” Current Developmental Disorders Reports 2, no. 3 (September 2015): 219–27. https://doi.org/10.1007/s40474- 015-0053-7.
16.
17.
18.
19. Hagan, Catherine, D.V.M., and Ph.D. “When Are Mice Considered Old?” The Jackson Laboratory. Accessed June 11, 2020. https://www.jax.org/news-and-insights/jax-blog/2017/november/when-are-mice-considered-old. Kuiper, Els F. E., Ad Nelemans, Paul G. M. Luiten, Ingrid M. Nijholt, Amalia M. Dolga, and Uli L. M. Eisel. “KCa2 and KCa3 Channels in Learning and Memory Processes, and Neurodegeneration.” Frontiers in Pharmacology 3 (2012). https:// doi.org/10.3389/fphar.2012.00107. Stackman, Robert W., Rebecca S. Hammond, Eftihia Linardatos, Aaron Gerlach, James Maylie, John P. Adelman, and Thanos Tzounopoulos. “Small Conductance Ca2+-Activated K+Channels Modulate Synaptic Plasticity and Memory Encoding.” Journal of Neuroscience 22, no. 23 (December 1, 2002): 10163–71. https://doi.org/10.1523/JNEUROSCI.22-23- 10163.2002. Allen, Duane, Bernd Fakler, James Maylie, and John P. Adelman. “Organization and Regulation of Small Conductance Ca2+ -Activated K+ Channel Multiprotein Complexes.” Journal of Neuroscience 27, no. 9 (February 28, 2007): 2369–76. https:// doi.org/10.1523/JNEUROSCI.3565-06.2007.








