
34 minute read
Aβ aggregation and Tau phosphorylation suggest PhIP correlation to Alzheimer’s disease


Bhavya Patel
PhIP, 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine, is a dietary heterocyclic aromatic amine that is chemically released when cooking meat at increased temperatures. Syeda et al. (2020) examines the correlation between PhIP exposure and the neuropathology of AD. The authors experimented on C57BL/6 mice and divided them into three groups that experienced differing levels of PhIP exposure. A 6-7 week old male and female group of mice were treated with 100/200 mg PhIP/kg for 8 hours once a week. The next group consisted of 8-9 week old male mice exposed to 75mg PhIP/kg for a period of 4 weeks(3x/week) and the last group was injected with 75mg PhIP/ kg for 16 weeks(3x/week). The control for each group was a vehicle treated group injected only with corn oil. Mice were then decapitated, brains were divided into two halves and stored in liquid nitrogen for further processing. Assessments included measuring oxidative damage through ChAT positive cells in the striatal region, synaptic alterations in the hippocampus, and inflammation via GFAP intensity. BACE1, APP, Aβ and tau protein phosphorylation was compared between each group of mice and control. An accumulation of Aβ and tau-phosphorylation are major characteristics of AD and are prominently seen in the 16-week group. Furthermore, oxidative stress is increased in the hippocampus suggesting that the mechanism of Aβ aggregation via PhIP may be a result of BACE1 and APP upregulation. A summary of results is indicated in Figure 1.
Advertisement
Introduction: PhIP is found in beer, wine, smoke from cigarettes and produced when cooking meat at high temperatures. Being a carcinogen, it has increased exposure to humans and harmful consequences. After oxidising to cytochrome p450, PhIP leads to neurotoxicity. AD consequently is the leading cause of Dementia and leads to loss of declarative memory and cognition. Initial symptoms include poor retention of memory that ultimately intensifies to impaired speech and difficulty in processing thought. There are two subtypes of this disease namely the early-onset AD and the late-onset AD. Likely caused by a combination of genetics and campus is susceptible to oxidative damage in both positive and
environmental factors, APP is one gene that is identified to be associated with early-onset AD. Hence, it is clinically important to assess the neurogenerative and neurotoxic properties of AD for future treatments.
Furthermore, amyloid plaques are a characteristic of AD. BetaAmyloid is formed by the proteolytic cleavage of Amyloid Precursor Protein, a type I transmembrane glycoprotein, by enzymes β-secretase and γ-secretase. Increased formation of Aβ Synaptic Proteins such as V-GLUT1 and PSD-95 increase is seen
is seen with increased
BACE1, the β-site cleaving enzyme. One of the major characteristics of brain lesions of AD involves the abnormal phosphorylation of tau protein. Firstly, phosphorylated tau will tangle and subsequently cause aggregation in the CA3 regions of the hippocampus that are associated with declarative memory. The CA1 and CA3 regions of the Hippocampus are essential for spatial learning and long-term spatial memory acquisition. This mechanism of phosphorylation is responsible for a cytoskeleton In Mann-Whitney test, Kruskal-Wallis nonparametric test and Dunn’s post-hoc test, p < 0.05 is considered statistically significant.
Oxidative damage in ChAT positive cells and negative cells
Nikon A1R inverse scanning confocal microscopes were used to measure oxidative damage in the striatal regions of the Hippocampus. Nitrotyrosine intensity was measured in ChAT positive and negative cholinergic cells to assess for oxidative damage. Statistical significance compared to control shows the hipponegative cholinergic ChAT cells post PhIP exposure. (Figure 2)
Increased Synaptic Proteins
disruption and microtubule disassembly. only in the 8hr group of mice. 4 weeks and 16 weeks had no effect on the upregulation or downregulation of the synaptic proteins in the Hippocampus.
Syeda et al. (2020) assessed whether differing levels of PhIP leads to correlation between exposure intensity and symptomatic AD. PhIP effects on the brain are not well studied and thus, this article brings forth some light as to possible mechanisms of impact and related consequences.
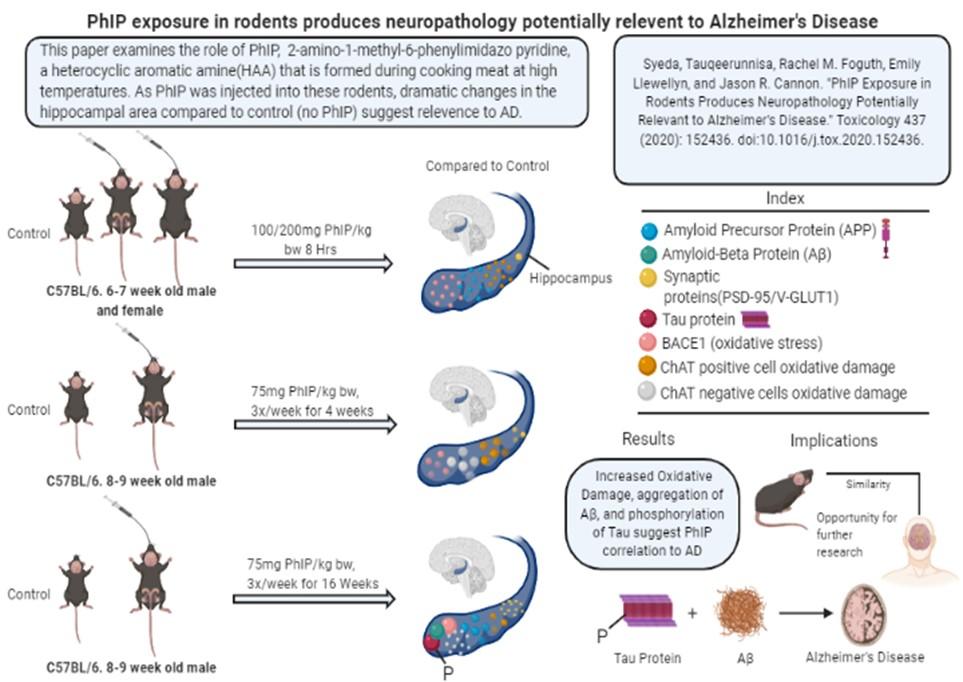
Fig.1. A detailed visual summary of the primary research article Fig.2. Nitrotyrosine staining assessing for oxidative damage in ChAT positive and negative cells of the dorsal striatal region. (A) PhIP exposure to 8hr group, p < 0.01, dose dependent increase in oxidative damage for ChAT positive cells. No difference in ChAT negative cells. (B) PhIP exposure to 4 weeks group, p < 0.01 for ChAT positive cells. In ChAT negative cells, p < 0.001. (C) PhIP exposure to 16 weeks group, p < 0.001 for ChAT posi
by Syeda et. al. (2020). tive neurons. In ChAT negative cells, p <0.001. No Inflammation Microglia activation was studied using GFAP intensity markers at the CA1 hippocampal regions. No signifi
No significant changes in microglia activation and no inflammation is seen in hippocampal regions pertaining to all 3 groups.
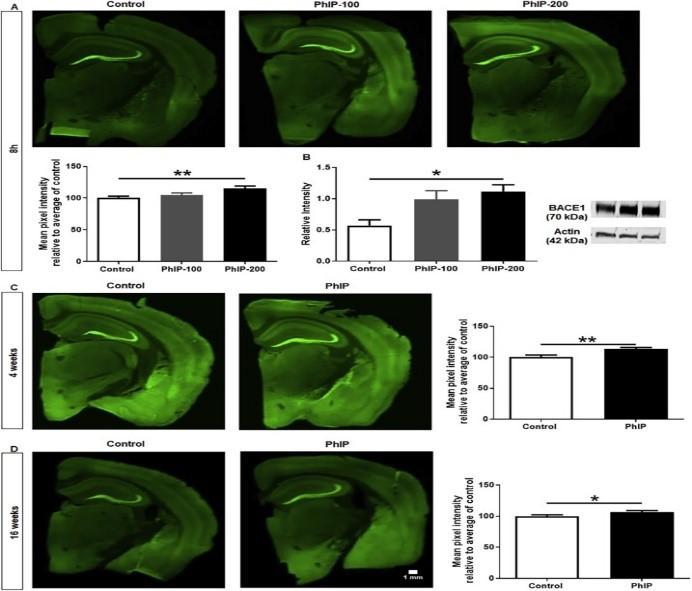
BACE1 upregulation
Mossy fiber projections were quantified in the hippocampus to assess for BACE1 changes. BACE1 is linked to oxidative stress and is positively correlated with PhIP exposure and AD.
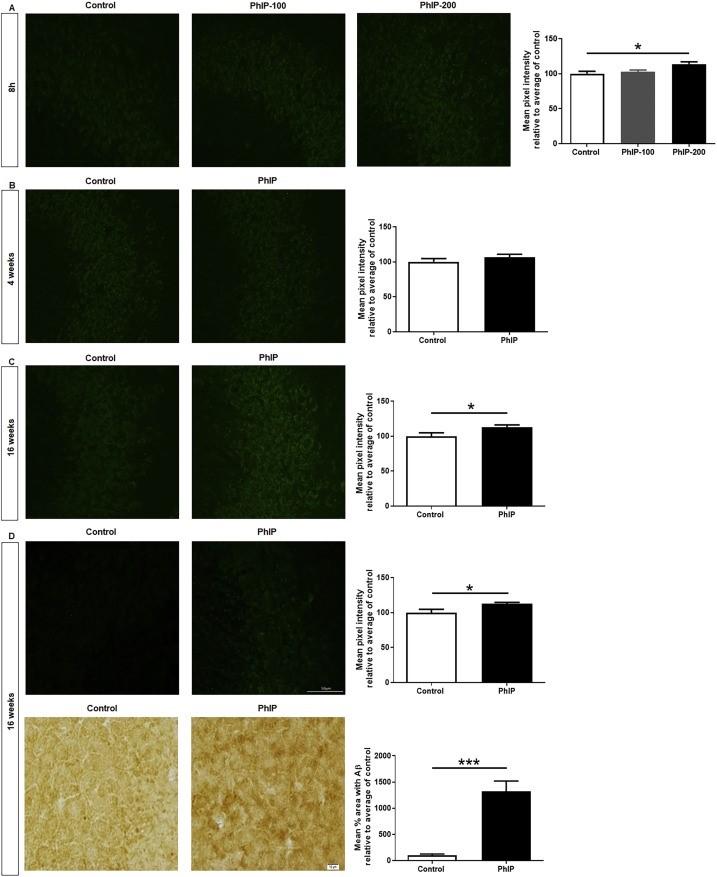
Fig. 9. ROI’s drawn around CA3 regions for quantification of APP and Aβ 1-42. (A) 8hrs, PhIP-200, p < 0.05 for APP staining. (B) In
significant results. (C) 16 weeks, p < 0.05 for APP staining; p < 0.05 for Aβ 1-42 staining. (D) DAB staining, p < 0.001; Immunofluorescence staining p < 0.05 for Aβ 1-42 .
Tau Phosphorylation
An increase in Tau Phosphorylation in the CA3 St. Pyramidale region of the Hippocampus was seen in the 16 weeks group. Abnormal tau-protein phosphorylation can also affect normally functioning tau proteins. This directly interacts with microtubules causing disassembly of tubulin in AD patients.
Fig.7. BACE1 assessments by drawing ROIs around mossy fiber regions of the hippocampus. Western Blot analysis for each group were completed to relate BACE1 upregulation with PhIP exposure. (A) 8hrs, PhIP-200, p < 0.01. (B) 4 weeks, p < 0.01. (C) 16 weeks, p <0.05.
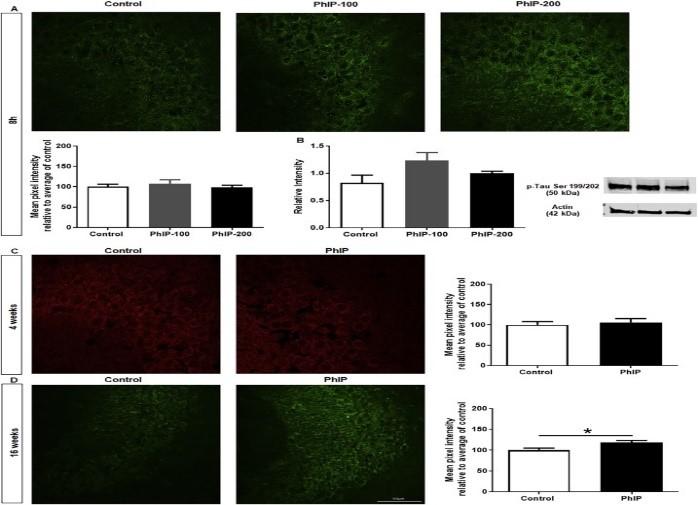
Fig.8. BACE1 activity measured in three groups: 1) CTR- control, 2) NA- cognitively normal subjects, 3) Sporadic Alzheimer’s Disease. Results indicate 47% increase of BACE1 activity in AD.
Upregulation of APP and Aβ
The St. Pyramidale region in the CA1 and CA3 regions were assessed for APP changes while the St.Pyramidale region of CA3 was only imaged for Aβ changes. There was an increase in APP for both 8 hrs and 16 weeks as predicted by BACE1 upregulation. 16 weeks also resulted in an increase of Aβ 1-42 aggregation. Aβ formation leads to sequential activation of BACE1 by an increase in oxidative stress that positively feedbacks to increase Aβ aggregation. Previous studies conclude that increased Aβ plaques result in neuronal hyperactivity in transFig. 10. Staining for p-Tau Ser 199/202 to assess tau phosphorylation in the hippocampus. (A, B) No change. (C) 16 weeks, p < 0.05 for p-tau in hippocampal region; no change in DG.
Conclusion
PhIP shows relevance to AD neuropathology by inducing oxidative damage in ChAT positive neurons, Aβ aggregation and tauphosphorylation. Amyloid plaques are a predominant charac-
teristic of AD that is characterised by its insolubility and fibrillar structure. The correlation of PhIP with neurotoxicity is also seen in prior reports that suggest downregulation of Dopaminergic neurons in its presence.. Furthermore, the authors agree that PhIP selfmetabolizes to N-OH- PhIP and 4’-OH-PhIP resulting in oxidative stress as seen is Chen et al. (2007). Oxidative stress without inflammation and microglia activation suggest these are early neurotoxic events in the presence of PhIP. PhIP exposure leading to aggregation of Aβ and tau phosphorylation at 16 weeks suggest exposure time is positively correlated with enhanced symptoms of AD. Assessment of PhIP and its effects on glutaminergic synapses creased oxidation and BACE1 upregulation may be better inter
was studied via measuring levels of PSD-95 and VGLUT-1. Staand Glutamatergic neurons by staining with their respective injected mice should be kept alive while the other decapitated
tistical significance is quite low, but a mild increase concludes that acute PhIP exposure leads to increased glutaminergic signalling. BACE1 cleavage of APP leads to the formation of Aβ. BACE1 upregulation was seen in acute, sub-acute and sub-chronic exposure to PhIP. Lastly, Tau phosphorylation was dominant in sub-chronic exposure levels of PhIP but the mechanisms of PhIP induced activation remains unknown. Syeda et al. (2020) also conclude that PhIP is sensitive to CA3 regions of the hippocampus whereas previous studies indicated variations in CA1 region and the Dentate Gyrus.
Critical Analysis
Most findings of Syeda et. Al are consistent with previous records; however, many causal relations have not been established. Exact mechanisms of tau-phosphorylation, BACE1 upregulation and direct AD related neurotoxicity are not confirmed. Previous papers mention similar ways of PhIP mediated neurotoxic events, however, the authors have found no results that accurately prove AD relation to PhIP. Increased studies must be done to form a causal relationship between HAAs, and AD. Limitations of this study include experimentation on mostly male mice and a smaller group of mice models. Previous studies of rat embryos suggest PhIP inducing harm to dopaminergic cells via mediating oxidative stress. A decrease in neurite length is also seen in the presence of PhIP and N-OHPhIP(Griggs et al, 2014). Furthermore, PhIP also decreases dopamine metabolite turnover in striatal regions of the hippocampus and increases oxidative damage in the Substantia Nigra. Such results are key to understanding exact mechanisms ied. It is important to include female neuropathology in the sub -acute and sub-chronic levels of exposure as well as females are more susceptible to AD. Furthermore, the time frame of exposure should be increased with increasing groups of experimental models. Natural exposure to PhIP is higher for humans and thus, longer time periods of study would convey naturally occurring results. For accurate results, the time should be 24 hrs, 2 weeks, 4 weeks, 8 weeks,16 weeks and 32 weeks. With 6 groups of models containing both male and female, this type of experimental set-up would create more efficient analysis of results. Furthermore, the mechanisms of Aβ aggregation, tau phosphorylation, inpreted with group specificity. Dopaminergic neurons were not assessed in this literature. Furthermore, significances of studying synaptic proteins were briefly mentioned. Evaluations of both Dopaminergic neurons antibodies should be completed. Newer findings may have arisen if this had been done. If PhIP induces neurotoxicity, there should be a decline in dopaminergic cells and a rise in glutaminergic cell activation. AD results in hyperactivity of synapses before hypoactivity. Lastly, learning and memory were not assessed in this paper. The authors should have included behavioural tests for the rodent models to assess for cognition and memory as disturbances to these are seen in AD positive individuals. A group of
of AD neuropathology and thus, should also have been studied. to assess both behavioural changes and molecular changes. A swim test, adhesive removal test, reach and grasp test, corner test, cylinder tests or open-field test could be used to assess sensorimotor stability in PhIP-injected mice vs. control. A decline in learning and memory is associated with decreased sensorimotor function. Therefore, the mice injected with PhIP will take longer time to complete tasks compared to control. E.g. PhIP injected mice would take longer times to remove adhesive stickers from their paws in the Adhesive Removal Test compared to control. Overall, this study will include behavioural tests, assess for both sexes, and allows a longer time frame to gather results. This experimental setup will allow for increased statistical significance to study the correlation of PhIP exposure and AD neuropathology.
Future Directions
The authors have created a valid technique to study the effect of PhIP exposure by creating three groups of C57BL/6 mice. PhIP injected for 8hrs, 4 weeks and 16 weeks exposure are appropriate models to study exposure effects. However, female exposure to PhIP was only measured in the acute exposure 8hr group and long-term effects on female rodents were not stud
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16. “2-Amino-1-Methyl-6-Phenylimidazo[4,5-b]Pyridine (PhIP) Is Selectively Toxic to Primary Dopaminergic Neurons In Vitro |
Toxicological Sciences | Oxford Academic.” Accessed June 6, 2020. https://academic.oup.com/toxsci/ article/140/1/179/1673985.
Agim, Zeynep Sena, and Jason R. Cannon. “Alterations in the Nigrostriatal Dopamine System after Acute Systemic PhIP
Exposure.” Toxicology Letters 287 (May 1, 2018): 31–41. https://doi.org/10.1016/j.toxlet.2018.01.017. Alonso, A. C., T. Zaidi, I. Grundke-Iqbal, and K. Iqbal. “Role of Abnormally Phosphorylated Tau in the Breakdown of Microtubules in Alzheimer Disease.” Proceedings of the National Academy of Sciences 91, no. 12 (June 7, 1994): 5562–66. https://doi.org/10.1073/pnas.91.12.5562. Bekris, Lynn M., Chang-En Yu, Thomas D. Bird, and Debby W. Tsuang. “Review Article: Genetics of Alzheimer Disease:” Journal of Geriatric Psychiatry and Neurology, November 2, 2010. https://doi.org/10.1177/0891988710383571. Bharadwaj, Prashant, and Ralph Martins. “A Rapid Absorbance-Based Growth Assay to Screen the Toxicity of Oligomer A
Beta(42) and Protect against Cell Death in Yeast.” Neural Regeneration Research 15, no. 10 (October 2020): 1931–36. https://doi.org/10.4103/1673-5374.280318. Blazquez-Llorca, Lidia, Virginia Garcia-Marin, Paula Merino-Serrais, Jesús Ávila, and Javier DeFelipe. “Abnormal Tau Phosphorylation in the Thorny Excrescences of CA3 Hippocampal Neurons in Patients with Alzheimer’s Disease.” Journal of
Alzheimer’s Disease 26, no. 4 (January 1, 2011): 683–98. https://doi.org/10.3233/JAD-2011-110659. Borghi, Roberta, Stefania Patriarca, Nicola Traverso, Alessandra Piccini, Daniela Storace, Anna Garuti, Gabriella Cirmena,
Patrizio Odetti, and Massimo Tabaton. “The Increased Activity of BACE1 Correlates with Oxidative Stress in Alzheimer’s
Disease.” Neurobiology of Aging 28, no. 7 (July 1, 2007): 1009–14. https://doi.org/10.1016/ j.neurobiolaging.2006.05.004. Busche, Marc Aurel, Xiaowei Chen, Horst A. Henning, Julia Reichwald, Matthias Staufenbiel, Bert Sakmann, and Arthur Konnerth. “Critical Role of Soluble Amyloid-β for Early Hippocampal Hyperactivity in a Mouse Model of Alzheimer’s Disease.” Proceedings of the National Academy of Sciences 109, no. 22 (May 29, 2012): 8740–45. https://doi.org/10.1073/ pnas.1206171109. Chen, Chi, Xiaochao Ma, Michael A. Malfatti, Kristopher W. Krausz, Shioko Kimura, James S. Felton, Jeffrey R. Idle, and Frank
J. Gonzalez. “A Comprehensive Investigation of 2-Amino-1-Methyl-6-Phenylimidazo[4,5-b]Pyridine (PhIP) Metabolism in the Mouse Using a Multivariate Data Analysis Approach.” Chemical Research in Toxicology 20, no. 3 (March 1, 2007): 531–42. https://doi.org/10.1021/tx600320w. Florian, Cédrick, and Pascal Roullet. “Hippocampal CA3-Region Is Crucial for Acquisition and Memory Consolidation in Morris
Water Maze Task in Mice.” Behavioural Brain Research 154, no. 2 (October 5, 2004): 365–74. https://doi.org/10.1016/ j.bbr.2004.03.003. Griggs, Amy M., Zeynep S. Agim, Vartika R. Mishra, Mitali A. Tambe, Alison E. Director-Myska, Kenneth W. Turteltaub,
George P. McCabe, Jean-Christophe Rochet, and Jason R. Cannon. “2-Amino-1-Methyl-6-Phenylimidazo[4,5-b]Pyridine (PhIP) Is Selectively Toxic to Primary Dopaminergic Neurons In Vitro.” Toxicological Sciences 140, no. 1 (July 1, 2014): 179–89. https://doi.org/10.1093/toxsci/kfu060. Jarrett, Joseph T., and Peter T. Lansbury. “Seeding ‘One-Dimensional Crystallization’ of Amyloid: A Pathogenic Mechanism in
Alzheimer’s Disease and Scrapie?” Cell 73, no. 6 (June 18, 1993): 1055–58. https://doi.org/10.1016/0092-8674(93)90635 -4. Nunan, Janelle, and David H Small. “Regulation of APP Cleavage by α-, β- and γ-Secretases.” FEBS Letters 483, no. 1 (October 13, 2000): 6–10. https://doi.org/10.1016/S0014-5793(00)02076-7. Probst, Alphonse, Dominique Langui, and Jürg Ulrich. “Alzheimer’s Disease: A Description of the Structural Lesions.” Brain
Pathology 1, no. 4 (1991): 229–39. https://doi.org/10.1111/j.1750-3639.1991.tb00666.x. Seabrook, Guy R., and Thomas W. Rosahl. “Transgenic Animals Relevant to Alzheimer’s Disease.” Neuropharmacology 38, no. 1 (January 1, 1999): 1–17. https://doi.org/10.1016/S0028-3908(98)00170-1. Syeda, Tauqeerunnisa, Rachel M. Foguth, Emily Llewellyn, and Jason R. Cannon. “PhIP Exposure in Rodents Produces Neuropathology Potentially Relevant to Alzheimer’s Disease.” Toxicology 437 (May 15, 2020): 152436.
Apelin-36: An Overlooked Peptide with Promising Effects on Parkinson’s Disease
Andrea Pinto
Parkinson’s Disease (PD) is the second most prevalent progressive neurodegenerative disease. It is attributed to a gradual loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) due to the accumulation of misfolded α-synuclein proteins.As a result, common symptoms of PD include rigidity of the muscles, a resting tremor, a shuffling gait and bradykinesia, all of which vary in severity among diagnosed individuals. This variance produces a lack of understanding on how to reverse the disease pathology. Zhu et al. (2019), attempts to study the mechanism by which Apelin -36 prevents endoplasmic reticulum stress (ERS) to ameliorate the common symptoms associated with PD. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridin (MPTP) was injected into C57BL/6 mice to induce neurotoxicity mirroring the hallmark effects of PD. Injection of Apelin-36 into the SNpc of MPTP-treated mouse models improved motor deficits. The treatment also decreased levels of αsynuclein expression but rescued the progressive loss of dopaminergic neurons, SH-SY5Y cells, and tyrosine hydroxylase (TH) expression. Furthermore, a suppression of GRP78, CHOP and the cleaved caspase-12 is detected, indicating an aversion of ERS.Considering these results, Apelin-36 establishes as a promising treatment for PD by improving the major clinical symptom, motor disturbances. This approach can also be considered as a treatment option for other neurodegenerative disorders with similar symptoms such as Alzheimer's Disease and amyotrophic lateral sclerosis. Moreover, the results reveal a greater understanding of the mechanism by which Apelin-36 can induce neuroprotective effects. Key words: Parkinson’s disease, neurodegeneration, MPTP-induce mouse models, SH-SY5Y cells, substantia nigra, endoplasmic reticulum stress (ERS), Apelin-36, treatment
INTRODUCTION
Parkinson’s Disease (PD) is becoming an increasingly concerning neurodegenerative disease due its forecasted prevalence. The disease is further compounded by its complexity, involving a multitude of risk factors as well as affecting both motor and psychiatric function at varying severities (Bietz, 2014). Due to this complexity and variability in symptoms, there has been a low yield in promising treatments. Motor dysfunctions such as bradykinesia, have been associated with the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) as result of α-synuclein aggregation. Zhu et al. (2019) attempts to inhibit endoplasmic reticulum stress (ERS) to alleviate both the loss of dopaminergic neurons and α-synuclein aggregation, thus improving motor deficits. The mechanism by which this is achieved, involves examining the neuroprotective effects of Apelin-36 on MPTP-treated mice, which reveals its potentiality in treating PD (Zhu et al., 2019). An arduous, yet crucial step to developing a treatment, is to establish a model for the disease. A common chemically induced model for PD is an intranigral injection of MPTP into C57BL/6 mice. MPTP is capable of crossing the blood brain barrier due to its lipophilic properties, where it is then modified into its toxic form, 1-methyl-4-phenylpyridinium, also known as MPP+. The toxic metabolite has a strong selectivity for dopamine receptors, causing an efficient uptake into dopaminergic neurons. Upon its uptake, MPP+ concentrates within the mitochondria to produce reactive oxygen species (ROS), becoming toxic to the neurons (Langston, 2017). Loss of dopaminergic neurons, consequently decreases levels of tyrosine hydroxylase (TH), a rate-limiting enzyme involved in the synthesis of dopamine (Wang et al., 2017). These effects are also demonstrated in SH-SY5Y cells, a neuroblastoma cell line commonly used to study neurodegenerative diseases (Ko et al., 2018; Xicoy et al., 2017). Similarly to the development of PD, MPTP conveniently targets dopaminergic neurons of the SNpc exclusively (Langston, 2017). Although this selectivity remains unclear, it is apparent that MPTP-induced mice models are a valuable model for PD by mirroring its pathophysiology. Another significant contributor to the loss of dopaminergic neurons, are the consequences of endoplasmic reticulum stress (ERS) (Martinez et al., 2017). ERS is triggered by an accumulation of α-synuclein within the ER which activate ER transmembrane protein kinases, PERK and IRE1α (Ryu et al., 2002). A signalling cascade is then initiated, producing transcription factors to promote apoptosis. ERS serves an important interference point to which loss of dopaminergic neurons can be averted, thus decelerating neurodegeneration within individuals. To reverse the effects of neurodegeneration, Apelin-36 emerges as a potential treatment option. Apelin-36 is a neuropeptide derived from Apelin, an endogenous ligand of a Gprotein coupled receptor (GCPR) (Sakamoto et al., 2016). Most studies are currently examining the ability of Apelin to protect brain injury caused by ischemia. Apelin is naturally produced within the nervous system and has associated neuroprotective properties (Ishimaru et al., 2017). Examination of these neuroprotective aspects highlights its importance in reversing Parkinsonian symptoms. The study of Zhu et. al consisted of four groups, which received injections of saline, MPTP and/or Apelin-36 for 5 days. The control group was injected with saline. The Apelin-36 group was injected with Apelin-36 and saline. The MPTP group was injected with MPTP and saline. Finally, the Apelin-36+MPTP group was injected with Apelin-36 and MPTP. Motor disturbances were measured using a rotarod test. Immunohistochemistry and immunofluorescence were used to evaluate loss of dopaminergic neurons. Western Blotting was conducted to detect expression of TH and α-synuclein in SNpc. Lastly, cell viability was assessed using CCK-8 assay whereas cell apoptosis was measured by flow cytometry and TUNEL staining. The results of these experiments indicated, intranigral injection of Apelin-36 was capable of dissipating neurotoxic effects by promoting survival of MPP+- treated SH-SY5Y cells, preventing further loss of dopaminergic neurons, restoring TH expression, and improving motor functions of MPTP -induced mice models. Apelin-36 also had the added benefit of inhibiting α-synuclein expression thus preventing further neurodegeneration. Furthermore, a mitigation of ERS was observed in Apelin-36 treated MPTP-induced mouse models. Effects on ERS were concluded by noting a decrease in GRP78, CHOP and cleaved caspase-12 in SH-SY5Y cells, which overall inhibited apoptosis (Zhu et al., 2019).
MAJOR RESULTS
Dissipation of Neurotoxicity
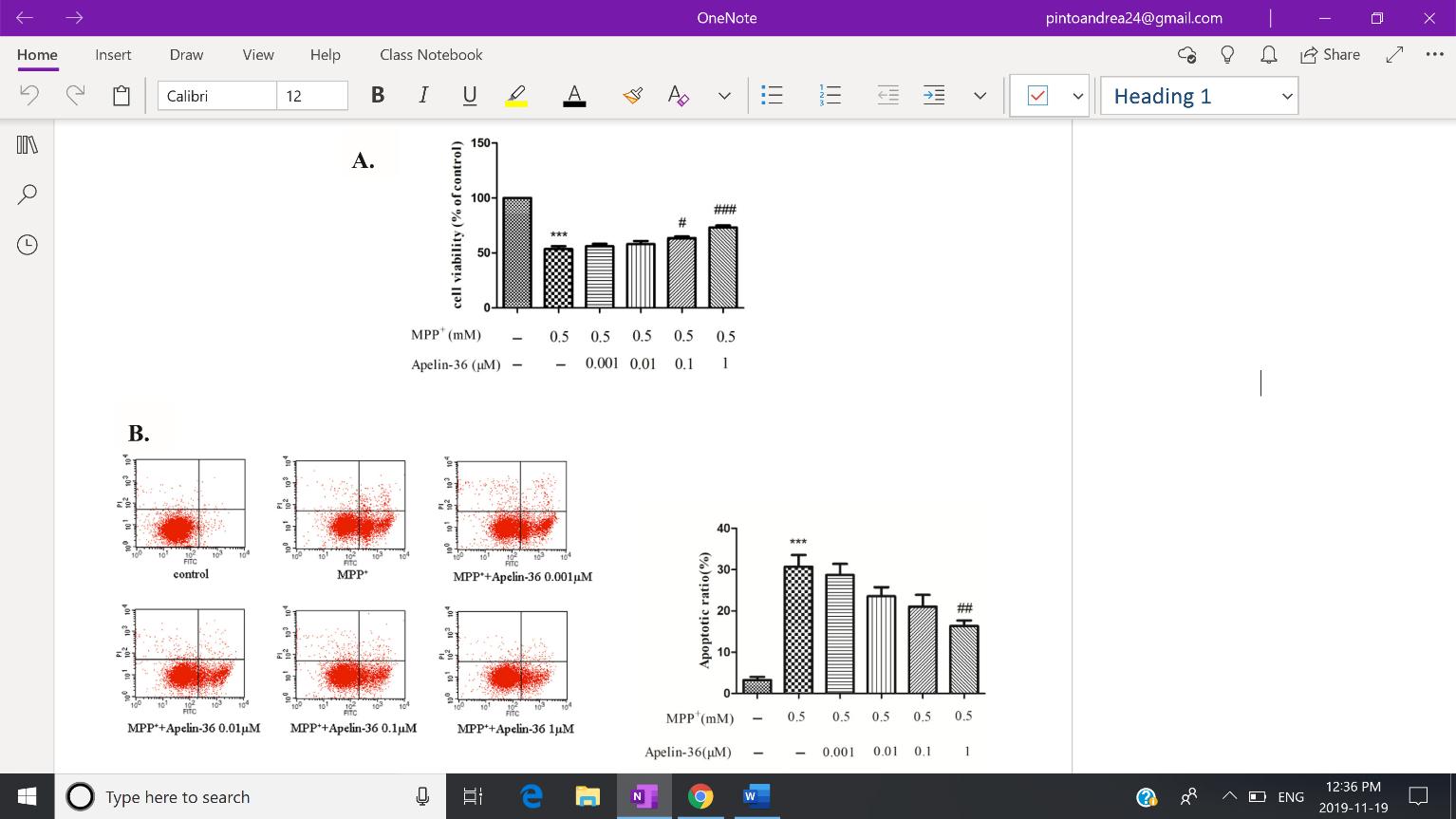
Apelin-treated MPTP-induced mouse models displayed a dissipation in neurotoxicity, as measured by the survival of SH-SY5Y cells, dopaminergic neurons, TH expression, and motor function. SH-SY5Y cells were initially treated with Apelin-36 followed by MPP+. CCK-8 assay shows higher dosages of Apelin-36 (1µM) is correlated with a greater cell viability. On the other hand, quantitative analysis of flow cytometry (Fig. 1) reveals decreased levels of cell apoptosis, corroborating the increased cell viability revealed by the CCK-8 assay. Although the effects of Apelin-36 on SH-SY5Y cells have yet to be studied by other groups, the study of Jiang et al. (2018) also noted a gradual increase in the survival of MPP+-treated SH-SY5Y cells after treatment with its isoform Apelin-13.
Fig. 1: Quantitative analysis of flow cytometry indicates a decrease in cell apoptosis in MPP+ treated SH-SY5Y cells, occurring in a dose-dependent manner. Figure Adapted from Zhu et al. (2019) Brain Research, 1721.
An attenuation of neurotoxicity is also indicated by an increase in dopaminergic neurons in the Apelin-36+MPTP mice group
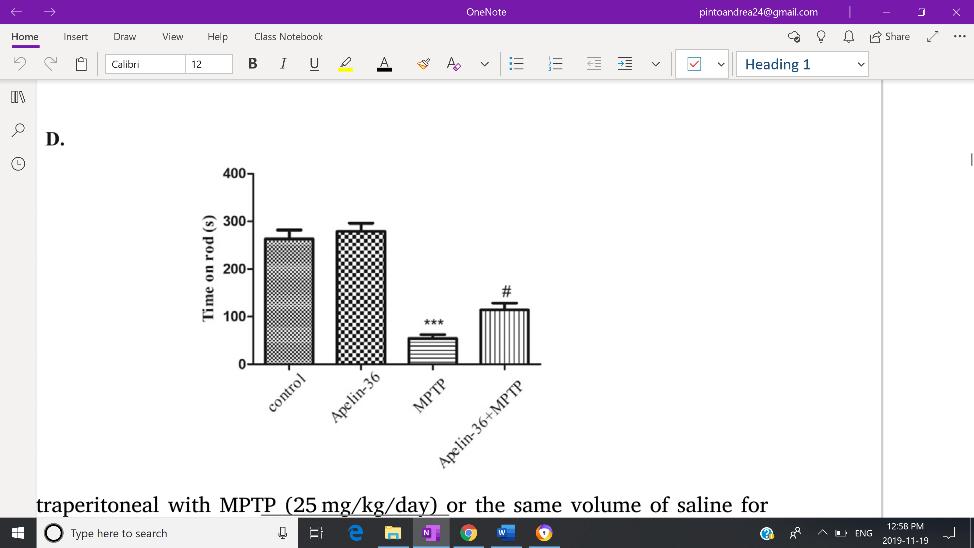
compared to the MPTP group, as presented by immunohistochemistry and immunofluorescence of TH. Increased levels of TH expression reflects increased prevalence of dopaminergic neurons. Likewise to the previous experiment, Apelin-13 treatment on 6-hydroxydopamine treated mice also revealed an attenuation of dopaminergic neuron death (Haghparast et al, 2019). Apelin-36+MPTP mice further demonstrated increased TH expression compared to the MPTP mice group. Due to the lack of experiments conducted using Apelin-36, these results are unable to be confirmed by current literature. However, the results align with the improved motor function of Apelin36+MPTP group, as a significantly longer time was spent on the rotarod compared to MPTP group (Fig. 2). Considering these results, Apelin-36 displays an ability to dissipate neurotoxicity and improve Parkinsonian symptoms.
er, this study involved Apelin-13 treatment as opposed to the Apelin-36. Nevertheless, a reduction in GRP78, CHOP and cleaved caspase-12 mitigated ERS, emphasizing the relevance of the ER in cell viability.
Fig 2: MPTP group shows significant loss in motor function, which was reversed after Apelin-36 treatment, as seen by an increased time spent on the rotarod. Figure Adapted from Zhu et al. (2019) Brain Research, 1721.
Regulation of α-synuclein A decelerating loss in dopaminergic neurons suggests an involvement between Apelin-36 and α-synuclein. Western blotting reveals increased α-synuclein in the MPTP group which was significantly repressed after Apelin-36 treatment in the mice models and SH-SY5Y cells. This repression in α-synuclein is Figure Adapted from Zhu et al. (2019) Brain Research, 1721.
consistent with a previous study conducted by Zhu et. al. (2019) using identical experimental procedures. The ability for Apelin-36 to prevent α-synuclein aggregation in SNpc, suggests ERS may also be mitigated.
Mitigation of ERS
DISCUSSION
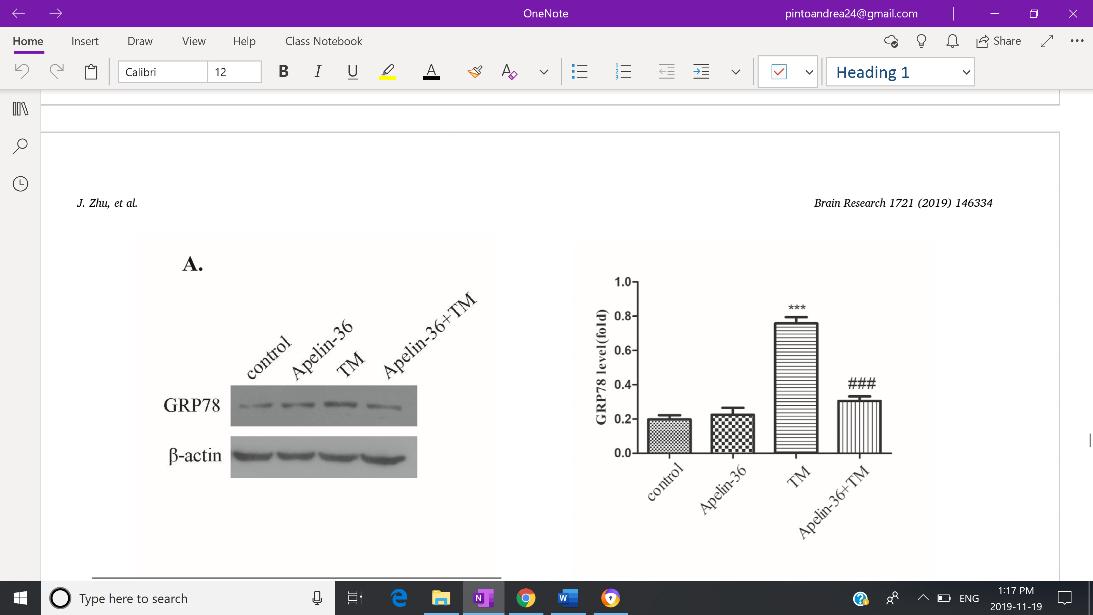
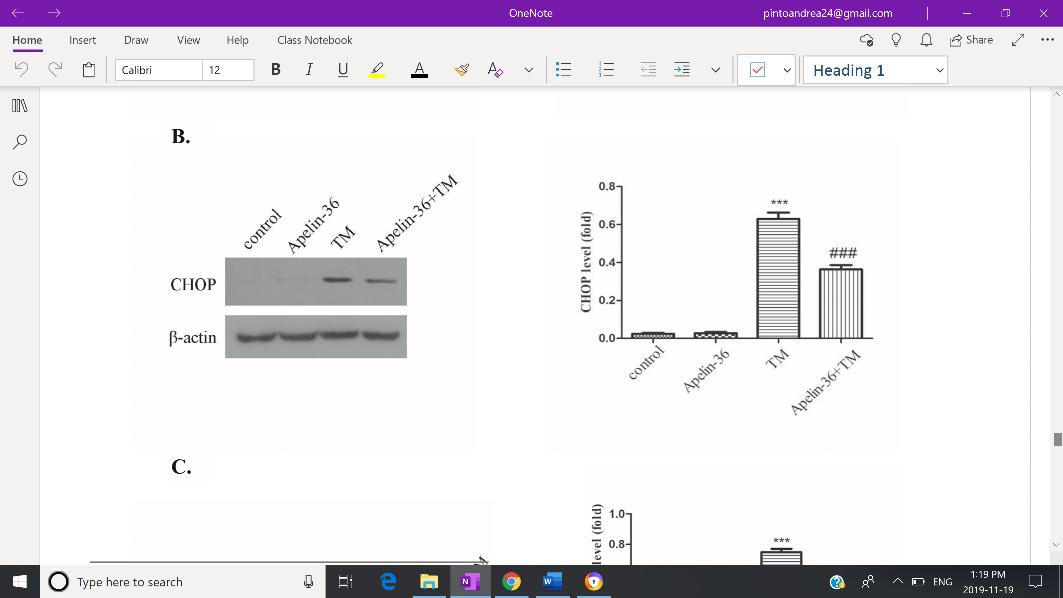
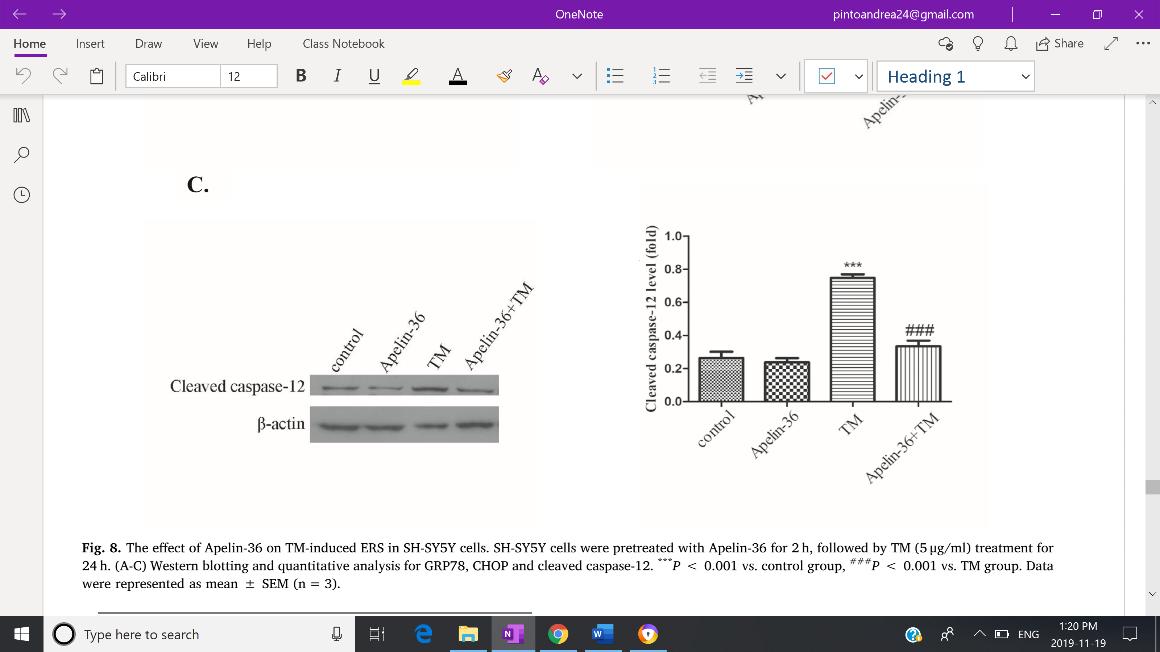
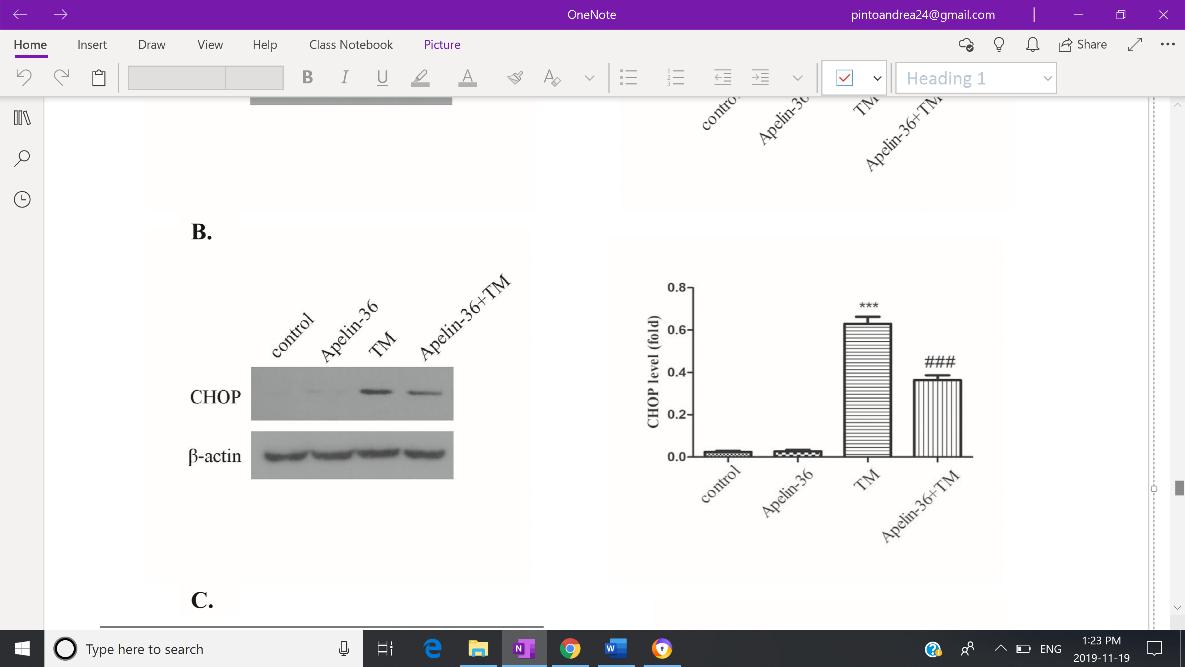
Zhu et al. (2019), as it suggests to be the main target in preventing progressive neurodegeneration. Expression of GRP78, CHOP and cleaved caspase-12 were used as to reflect the severity of ERS. Both Western Blotting and immunofluorescent staining revealed increased expression of GRP78 (Fig. 3-A), CHOP (Fig.3-B), and cleaved capse-12 (Fig. 3-C) in the MPTP group, but expression of all three indicators were inhibited by Apelin-36 treatment. Jian et. al. (2017) also found decreased levels of GRP78, CHOP and cleaved caspase-12 in MPTP-treated mice which was associated with an attenuation of ERS. HowevFig 3: Quantitative analysis of the Western Blotting reveals decreased expression of GRP78 (A), CHOP (B), and cleaved capase -12 (C) in SH-SY5Y cells treated with MPP+, indicating an overall mitigation of ERS.
Effects of Apelin-36 on ERS was the focus in the study of
From the experiments conducted by Zhu et. al. (2019), intranigral injection of Apelin-36 into MPTP-treated mouse models was capable of dissipating neurotoxicity by promoting survival of SH-SY5Y cells, presence of dopaminergic neurons, expression of TH, and motor function. Furthermore, the treatment prevented aggregation of α-synuclein in not only the mouse models but as well as the MPP+ treated SH-SY5Y cells. Most importantly, Apelin-36 mitigated ERS by decreasing expression of its indicators, GRP78, CHOP, and cleaved caspase12. Taking these results into consideration, the authors highlight the neuroprotective abilities of Apelin-36 as well as the
inhibition of ERS, causing an overall reversal of Parkinsonian symptoms. Previous studies have only investigated the role of Apelin (Ishimaru et. al, 2017) or Apelin-13 (Jiang et. al, 2018) in neurodegenerative disorders. Ishimaru et al. (2017) reveal an acceleration in retinal neuronal degeneration in Apelin deficient mice, as a result of downregulation in its GCPR signaling. On the other hand, Jiang et al. (2018), demonstrates the protective effects of Apelin-13 on MPP+-treated SH-SY5Y cells by promoting its survival. Both studies strengthen the correlation between presence of Apelin and the prevention of neuronal death. However, there exists a lack of knowledge whether the success of Apelin and Apelin-13 in decelerating neurodegeneration also extends to Apelin-36. Zhu et al. (2019) attempt to fill this gap by introducing Apelin-36 as a possible treatment option for PD. The treatment has produced promising results both in vitro using MPP+-treated SH-SY5Y cells and in vivo using MPTP-induced mouse models. This neuropeptide sets itself apart from other treatments such as Amantidine, by its ability to combat several mechanisms involved PD pathobiology, as opposed to a single target (Paquette et al., 2013). This is significant considering individuals diagnosed with PD experience several symptoms at varying severities. Although significant research has been published on the molecular pathway of ERS (Ryu et al., 2002) and its consequences (Martinez et al., 2017), Apelin-36 appears to be one of the few drugs able to attenuate this response. As a result, this study creates an opportunity to identify certain targets to suppress within the ERS. Additionally, it shifts the focus of researchers to an entirely unexplored area of neuroscience requiring further attention to the specific molecular mechanisms by which Apelin-36 induces these neuroprotective effects. By considering this avenue for treatment, it generates a new perspective through which researchers can approach other neurodegenerative diseases as well.
CRITICAL ANALYSIS
As alluded to previously, the results of the experiment examining the effects of Apelin-36 on dopaminergic neurons, α -synuclein, TH levels, motor function, SH-SY5Y cells and ERS, are all heavily supported by existing literature. This becomes extremely crucial as Zhu et al. (2019) are one of the few groups investigating Apelin-36 treatment through the lens of PD. By performing experiments on already heavily researched topics, the authors rationale for the peptide’s neuroprotective effects becomes increasingly plausible. Another favourable aspect of ed both in vitro and in vivo. An in vitro experiment performed on MPP+-treated SH-SY5Y cells, allows to examine the effects of the treatment on a molecular basis as well as its relevance to human cells (Xicoy et al., 2017). An in vivo experiment was also performed using the MPTP-induced mouse models, allowing to observe the effects of the treatment in the context of the whole organism. Furthermore, the experiment itself involved a localized injection of Apelin-36 into the SNpc, ensuring only the cells of the SNpc are affected (Machado et al., 2011). Contrastingly, MPTP was systemically introduced via an intraperitoneal injection. As a result, the neurotoxin is able to naturally reproduce the pathophysiology similarly displayed in individuals diagnosed with PD. This includes the aggregation of αsynuclein, the pattern of dopaminergic neuron loss, and some behavioural deficits (Meredith et al., 2011). Despite the benefits of the MPTP-induced mouse model, much controversy persists surrounding its use to accurately recapitulate PD pathology as observed in humans. Zeng et. al (2018) contest the benefits of this model due to its inability to generate Lewy-body inclusions, a major hallmark of PD. This issue becomes most prominent if MPTP is introduced intraperitoneally, as performed by the Zhu et al (Gibrat et al, 2009). Beyond this, PD is often diagnosed in humans approximately at the age of 60 years or older. Unfortunately, the life span of mice make it challenging to replicate this onset naturally and is not feasible for researchers to experiment using aged mice (Potashkin et al, 2011) To overcome this issue, a future study should examine alternative models that are more representative of human PD pathophysiology. A major limitation in the study of Zhu et. al, was the lack of reasoning for the use Apelin-36 over other isoforms of Apelin such as Apelin-12, Apelin-13, or Apelin-17 (Luo et al, 2019). Ishimaru et al. (2017), specifically emphasizes the biological potency of Apelin-13 in comparison to Apelin-36. This claim suggests if Apelin-13 demonstrates identical neuroprotective effects, then a smaller dose of Apelin-13 could be equivalently effective as a larger dose of Apelin-36. If true, Apelin-13 can be a more beneficial treatment in terms of cost-effectiveness and the safety of the patient. Additionally, to improve the plausibility of the results from Zhu et al. (2019), the timeline of the study must be extended. Effects of the Apelin-36 treatment models were only studied 5 days after the first injection on mouse models and a 24h incubation period on SH-SY5Y cells, making it unclear if there was a retention in neuroprotective effects. Future investigations should consider monitoring the consequences of Apelin -36 on mice and SH-SY5Y cells periodically over months as op
the study was that its experimental procedures were conductposed to days. Another potential dilemma is the method of administration. Although no significant concerns arise from intranigral injections into mouse models, this method lacks specific targeting of dopaminergic neurons. Accordingly, Apelin -36 is also taken up by surrounding cell types such as astrocytes and microglia. Considering the potency Apelin isoforms, this form of administration may lead to potential off-target effects (Ishimaru et al., 2017). Therefore, alternative techniques of administration must be explored to ensure only dopaminergic neurons are targeted.
FUTURE DIRECTIONS
Using the appropriate mouse model is an ongoing and heavily debated issue. Commonly used mouse models are treated with environmental toxins such as 6-hydroxydopamine (6-OHDA), MPTP, paraquat or rotenone (Potashkin et al., 2011).
As mentioned earlier, MPTP is most capable, amongst these options, of reflecting PD pathophysiology as seen in humans.
However, they lack the ability of reproducing an important hallmark of PD, Lewy Body inclusions. A study conducted by Gibrat et al (2009), revealed a 14-day chronic intraperitoneal infusion of MPTP into mice is able to generate these inclusions. Therefore, identical experimental procedures can be conducted on chronically administrated MPTP-induced mouse models to de- 129
termine if Apelin-36 is also capable of attenuating Lewy Body inclusions. As for the other limitations such as age of onset, nonhuman primates appear to be a more representative model. These primates not only have similar brain anatomies to humans but also display the most accurate pathophysiology for PD, especially when administered MPTP. Nonhuman primates clearly exhibit muscular rigidity and bradykinesia, behavioural deficits not demonstrated in mouse models or other simple organisms (Zeng et al., 2018). Due to the astounding success of Apelin-36 in MPTP-induced mouse models, this treatment should also be investigated in nonhuman primates to study its extent in alleviating behavioural deficits. To compare the efficacy of neuroprotective effects between Apelin-36 and Apelin-13 a study must be conducted such that it is identical to the standards of Zhu et al. (2019). All experimental procedures will be identical, except MPTP-induced mouse models and SH-SY5Y cells will be treated with Apelin-13 as opposed to Apelin-36. From the results presented by Jiang et al., Apelin-13 is expected to reverse Parkinsonian symptoms in a similar manner to Apelin-36. Accordingly, it is reasonable to expect the dissipation of neurotoxicity as demonstrated by an increased survival of SH-SY5Y cells, appearance of dopaminergic neurons, expression of TH, and improved motor function. An enhanced regulation of α-synuclein should also be expected to be displayed by both the mouse model and SH-SY5Y cells. Additionally, Apelin-13 will likely downregulate the expression of GRP78, CHOP, and cleaved caspase-12, thus mitigating ERS. As mentioned earlier, Ishimaru et al. (2017) showed Apelin-13 is biologically more potent compared to Apelin-36, suggesting these results would be amplified under identical dosage levels. Although these results are foreseeable, a possibility exists where contradictory results are received. If this is the case, the results then suggest Apelin-13 activates an alternative pathway that does not contribute to the neuroprotective effects of neurons in SNpc. In order to accurately examine the efficacy of the Apelin36 treatment, a similar study to Zhu et al. (2019) must be conducted but with an extended timeline. This prospective study would involve injecting Apelin-36 or saline once during the beginning of the experiment and monitoring the appropriate markers for approximately 2 months. Due to the potency of Apelin-36, it suggests a single injection produces long-lasting effects (Ishimaru et al, 2017). Therefore, it would be expected that there will be a significant increase in neuroprotective effects before reaching a plateau. On the other hand, a decline in survival of dopaminergic neurons, TH expression, and motor function, or an increase in α-synuclein aggregation, GRP78, CHOP, and cleaved caspase-12, may also occur. These results imply a continuous re-administration of the treatment is required in order to maintain the neuroprotective effects. The method of administration is also another point of contention, as intranigral injections lacks specificity to dopaminergic neurons. Introduction of cre-dependent adenosineassociated virus (AAV) vectors in MPTP-induced mouse models may be able to overcome this hurdle. The gene encoding for Apelin-36 will be initially inverted and be flanked by 2 loxP sites orientated towards each other. Only cells exposed to a Crerecombinase will recognize the orientation of the loxP sites, reverting the position of the gene such that it is no longer inverted, thus resulting in its transcription driven under the elongation 1α promoter. This construct will be introduced by AAV serotype 9 by intranigral injection. The purpose of this injection is to avoid possible transduction of the AAV9 into cells of other brain regions, minimizing off-target effects. A major advantage of this administration, is the high tropism AAV9 vectors innately possess for the SNpc. To ensure the vector is expressed in dopaminergic neurons of the SNpc, only these neurons will exclusively contain a Cre-recombinase capable of inverting the gene to allow for its transcription, which can be achieved by developing Cre-transgenic animals (Grames et al., 2018). Before Apelin36 can be introduced, transfection into dopaminergic neurons can be confirmed by replacing the Apelin-36 gene with a green fluorescent protein (GFP) gene, in a separate mice group. The results can then by analyzed by immunohistochemistry. Other factors studied by Zhu et al. can also be examined to determine if this method alters the effectiveness in the reversal of PD symptoms. Due to the similarity of the experiment performed by Grames et al. (2018), it is expected that the immunohistochemistry reveals localized expression of GFP and Apelin-36 in the mice receiving AAV9 vectors to exclusively dopaminergic neurons of SNpc. The MPTP group will not show any expression as no treatment was introduced. Based on the results presented by Zhu et al. (2019), it would be hypothesized that the MPTPinduced mice models treated with the AAV9, will have increased TH expression but decreased GRP78, CHOP and cleaved caspase-12 expression. Also, these groups will show improved motor function as indicated by longer times spent on the rotarod. On the other hand, the MPTP group will expect to show contrasting results in all experiments performed. If these expectations are not observed, it suggests other cells of the SNpc are also contributing to the neuroprotective effects through its interaction with Apelin-36. As a result, subsequent studies can then examine the involvement of other cells to provide a more reliable and effective treatment for the future.
1. 2.
3.
4.
5.
6.
7.
8.
9. 10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21. Beirtz J. M. (2014) Parkinson’s disease: a review. Front Biosci (Schol Ed), 1(6), 65-74 Gibrat, C., Saint-Pierre, M., Bousquet, M., Levesque, D., Rouillard, C., Cicchetti, F. (2009). Differences between subacute and chronic MPTP mice models: investigation of dopaminergic neuronal degeneration and α‐synuclein inclusions. J Neurochem, 109(5), 1469-82 doi:10.1111/j.1471-4159.2009.06072.x Grames, M. S., Dayton, R. D., Jackson K. L., Richard, A. D., Lu, X., Klein, R. L. (2018) Cre-dependent AAV vectors for highly targeted expression of disease-related proteins and neurodegeneration in the substantia nigra. FASEB J, 32(8), 4420-4427 doi:10.1096/fj.201701529RR Haghparast, E., Sheibani, V., Abbasnejad, M., Esaeili-Mahani, S. (2019) Apelin-13 attenuates motor impairments and prevents the changes in synaptic plasticity-related molecules in the striatum of Parkinsonism rats. Peptides, 117, 170091 Ishimaru, Y., Sumino, A., Kajioka, D., Shibagaki, F., Yamamuro, A., Yoshioka, Y., Maeda, S. (2017) Apelin protects against NMDA-induced retinal neuronal death via an APJ receptor by activating Akt and ERK1/2, and suppressing TNF-α expression in mice. J Pharmacol Sci, 133(1), 34-41 doi:10.1016/j.jphs.2016.12.002 Jian, Q., Wang, X., Wu, F., Wan, L., Cheng, B., Wu, Y., Bai, B. (2017). Low Dose of Apelin-36 Attenuates ER StressAssoicated Apoptosis in Rats with Ischemic Stroke. Front Neurol, 8, 556 doi:10.3389/fneur.2017.00556 Jiang, Y., Liu, H., Bingyuan, J., Wang, Z., Wang, C., Yang, C., Pan, Y., Chen, J., Cheng, B., Bai, B. (2018) Apelin‑13 attenuates ER stress‑associated apoptosis induced by MPP+ in SH‑SY5Y cells. Int J Mol Med, 42(3), 1732-1740 doi:10.3892/ ijmm.2018.3719 Ko, J. H., Lee, J. H., Choi. B., Park, J. Y., Kwon, Y. W., Jeon, S., Park, S. D., Kim, S. N. (2018). Neuroprotective Effects of Gagam-Sipjeondaebo-Tang, a Novel Herbal Formula, against MPTP-Induced Parkinsonian Mice and MPP+-induced Cell Death in SH-SY5Y Cells. Evid Based Complement Alternat Med doi:10.1155/2018/2420809 Langston, J. W. (2017). The MPTP Story. J Parksinons Dis, 7 (S1), S11-S19 doi:10.3233/JPD-179006 Luo, H., Han, L., Xu, J. (2019). Apelin/APJ system: A novel promising target for neurodegenerative diseases. J Cell Physiol. 1 -20 Machado, A., Herrera, A. J., Venero, J. L., Santiago, M., de Pablos, R. M., Villaran, R. F., Espinosa-Oliva, A. M., Arguelles S., Sarmiento, M., Delgado-Cortes, M. J., Maurino, R., Cano, J. (2011). Inflammatory Animal Model for Parkinson's Disease: The Intranigral Injection of LPS Induced the Inflammatory Process along with the Selective Degeneration of Nigrostriatal Dopaminergic Neurons. ISRN Neurology Martinez, B. A., Petersen, D. A., Gaeta, A. L., Stanley, S. P., Caldwell, G. A., Caldwell, K. A. (2017) Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J Neurosci, 37(46), 11085-11100 doi:10.1523/JNEUROSCI.1294-17.2017 Meredith, G. E., Rademacher, D. J. (2012). MPTP Mouse Models of Parkinson’s Disease: An Update. J Parkinsons Dis, 1(1), 19-33. doi:10.3233/JPD-2011-11023 Paquette, M. A., Martinez A. A., Macheda T., Meshul, C. K., Johnson, S. W., Berger, S. P., Giuffrida, A. (2013) Antidyskinetic mechanisms of amantadine and dextromethorphan in the 6-OHDA rat model of Parkinson’s disease: role of NMDA vs. 5-HT1A receptors. Eur J Neursci, 36(9), 3224-3234 doi:10.1111/j.1460-9568.2012.08243.x Potashkin, J. A., Blume, S. R., Runkle, N. K. (2011). Limitations of Animal Models of Parkinson's Disease. Parkinsons Dis, doi: 10.4061/2011/658083 Ryu, E. J., Harding, H. P., Angelastro, J. M., Vitolo, O. V., Ron, D, Greene, L. A. (2002) Endoplastic Reticulum Stress and he Unfolded Protein Response in Cellular Models of Parkinson’s Disease. J Neurosci, 22(24), 10690-10698 doi:10.1523/ JNEUROSCI.22-24-10690.2002 Sakamoto, K., Murakami, Y., Sawada, S., Hiroko U., Mori, A., Nakahara, T., Ishii, K. (2016) Apelin-36 is protective against Nmethyl-D-aspartic-acid-induced retinal ganglion cell death in the mice. Eur J Pharmacol, 791, 213-220 doi:10.1016/ j.ejphar.2016.08.036 Wang, Q., Ren, N., Cai, Z., Lin, Q., Wang, Z., Zhang, Q., Wu, S., Li, H. (2017). Paraquat and MPTP induce meurodengeneration and alteration in the expression profile of microRNAs: the role of transcription factor Nrf2. NPJ Parkinsons Dis, 3, 31 doi:10.1038/s41531-017-0033-1 Xicoy, H., Wieringa, B., Martens, G. J. (2017). The SH-SY5Y cell line in Parkinson’s disease research: a systematic review. Mol Neurodegener, 12(1), 10 doi:10.1186/s13024017-0149-0 Zeng, X. S., Geng, W. S., Jia, J. J. (2018) Neurotoxin-Induced Animal Models of Parkinson Disease: PathogenicMechanism and Assessment. ASN Neuro, doi:10.1177/1759091418777438 Zhu, J., Dou, S., Wang, C., Jiang, Y., Wang, C., Cheng, B. (2019). Apelin-36 mitigates MPTP/MPP+-induced neurotoxicity: Involvement of α- synuclein and endoplasmic reticulum stress. Brain Res, 1721, 146334 doi:10.1016/ j.brainres.2019.146334








