12 minute read
Restoration of the UPS through enhancement of UBA1 levels improves SMA models outcomes


Restoration of the UPS through enhancement of UBA1 levels
Elaine Pityn
Advertisement
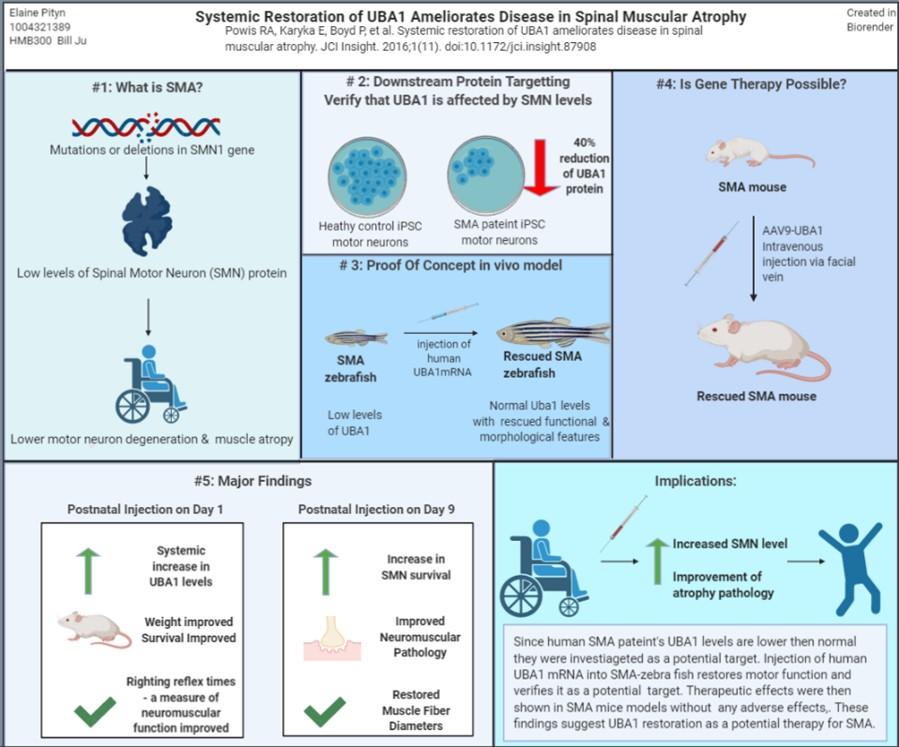
Spinal Muscular Atrophy (SMA) is a disease caused by mutations in survival motor neuron 1 gene (SMN1), inherited through an autosomal recessive pattern. Mutations in SMN1 reduce levels of survival motor neuron (SMN) protein, which causes muscle atrophy and death in severe cases. The reduction of SMN results in aberrant molecular pathways that may be targeted for therapeutic approaches like the ubiquitin pathway. Research shows the depletion of ubiquitin activating enzyme 1 (UBA1) across all SMA models and patient’s cells (Powis et al. 2016). Replenishment of UBA1 prevents SMA pathogenesis; for instance in SMA zebrafish, motor axon and motor performance were improved (Powis et al. 2016) . UBA1 levels were replenished by a gene therapy approach with adeno-associated virus serotype 9-UBA1 (AAV9-UBA1). In mouse models it was shown that mice could withstand AAV-UBA1 levels over long periods and secondly, systemic restoration of UBA1 resulted in less organ pathology and improved neuromuscular outcomes, overall weight, survival, and motor performance (Powis et al. 2016). The findings suggest UBA1 restoration has potential as a therapy for SMA.
3. Major Results:
This study verifys targetting UBA1 in SMA models. They showed that manipulating levels of this enzyme had therapeutic effects and offers a potential therapeutic approach.
3.1 UBA1 is a valid target for SMA
The neurological disorder SMA has been investigated using disease relevant phenotype for iPSC-derived motor neurons. The same approach was used to verify UBA1 levels. In Figure #2 the UBA1 levels were found to be reduced by 40% in Type I SMA patient derived iPSC compared to control patients UBA1 levels (Powis et al. 2016). Figure 2 also shows that decreasing in motor axon growth, like the pathology of SMA (Wishart et al. 2014, McWhorter et al. 2003) . Powis et al. quantified protein
Figure 1: Visual Summary for the paper under review (Powis et al. 2016)
2. Introduction:
Spinal Muscular Atrophy is a disorder in which genetic mutations of motor neurons lead to a decrease in the amount of SMN protein (Chang et al 2004). This disorder is a leading cause in infant deaths, occurring about 1 in 6,000 to 1 in 10,000 live births (Bowerman et al. 2017). SMA results in deterioration of the lower motor neuron which leads to muscle weakness, atrophy and progressive decline in motor function. SMN has ubiquitous expression, across all cells and tissue types (Chaytow et al. 2018). It is also important for mRNA trafficking, cytoskeleton dynamics, endocytosis and autophagy (Chaytow et al. 2018). SMN levels have pathological consequences for organs like the heart and liver. SMA may range in severity as modulated by a second duplicate gene SMN2 which can modify severity levels. In 95% of SMA cases mutations in SMN1 are the cause, but there are forms of X-linked SMA which result from mutations in the ubiquitin-like modifier activating enzyme (UBA1) (Dlamini et al. 2013, Chang et al. 2004). Since the UBA1 pathway has been associated with X-linked SMA it may be implicated in other forms of SMA pathology, as theorized in this paper. The UBA1 enzyme is necessary for protein homeostasis; it is the initial step of the ubiquitin cascade which results in protein catabolism. Since the ubiquitin proteasome system (UPS) marks proteins for degradation, modifications to this pathway can have pathological consequences. This enzyme has not only been connected with x-linked SMA, but has also been implicated in other neurodegenerative conditions (Groen and Gillingwater 2015). The paper by Powis et al. entitled Systematic restoration of UBA1 ameliorates disease in SMA demonstrates that UBA1 is a target for SMA therapy. The authors show that UBA1 is not only implicated in the x-linked SMA, but also reduces levels in other types of SMA. They used an iPSC from a SMA patient to verify decreased UBA1 levels in humans. Then with SMA-zebra fish they showed restoration of UBA1 levels that could decrease pathology. Finally, in SMA mouse models they injected AAV9- UBA1 in mice to replenish UBA1 levels. This rescued mice from declining organ pathology of the heart, liver, promoting better levels of UBA1 in healthy zebrafish will produce abnormalities
survival, overall health outcomes and weight. These findings levels using western blot analysis and found a reduction in SMN levels. They verified this in both SMA zebrafish and SMA mice by demonstrating UBA1 and SMN were reduced and resulted in SMA pathology.

Figure 2:Treatment of Zebrafish with MO, a pharmacological inhibitor of UBA1 produced abnormal axon growth, a pathological feature of SMA (Wishart et a. 2014)`
3.2 UBA1 restorations will recover SMA models
The UBA1 levels were recovered in both SMA models for zebrafish and mice via an AAV9-UBA1 injection. In zebrafishSMA the motor phenotype and motor axon morphology was rescued (Powis et al. 2016). Various other authors have shown that inhibition of downstream UBA1 targets in SMA mice will produce similar neuromuscular outcomes (Shorrock et al. 2018, 1; Wishart et al. 2014). In mouse models, AAV9- UBA1 injection via the facial vein produced systemic improvements. Organ outcomes, weight, survival, and motor performance all improved. Studies showed a reversal of the neuromuscular pathology and restored muscle fibre diameter. The work of Shorrock et al. 2018 in figure 3 shows AAV9-UBA1 injection increased UBA1 levels which later rescued sensory neurons.
3.2 UBA1 restorations will recover SMA models
The UBA1 levels were recovered in both SMA models for zebrafish and mice via an AAV9-UBA1 injection. In zebrafishSMA the motor phenotype and motor axon morphology was rescued (Powis et al. 2016). Various other authors have shown that inhibition of downstream UBA1 targets in SMA mice will produce similar neuromuscular outcomes (Shorrock et al. 2018, 1; Wishart et al. 2014). In mouse models, AAV9-UBA1 injection via the facial vein produced systemic improvements. Organ outcomes, weight, survival, and motor performance all improved. Studies showed a reversal of the neuromuscular pathology and restored muscle fibre diameter. The work of Shorrock et al. 2018 in figure 3 shows AAV9-UBA1 injection increased UBA1 levels which later rescued sensory neurons.
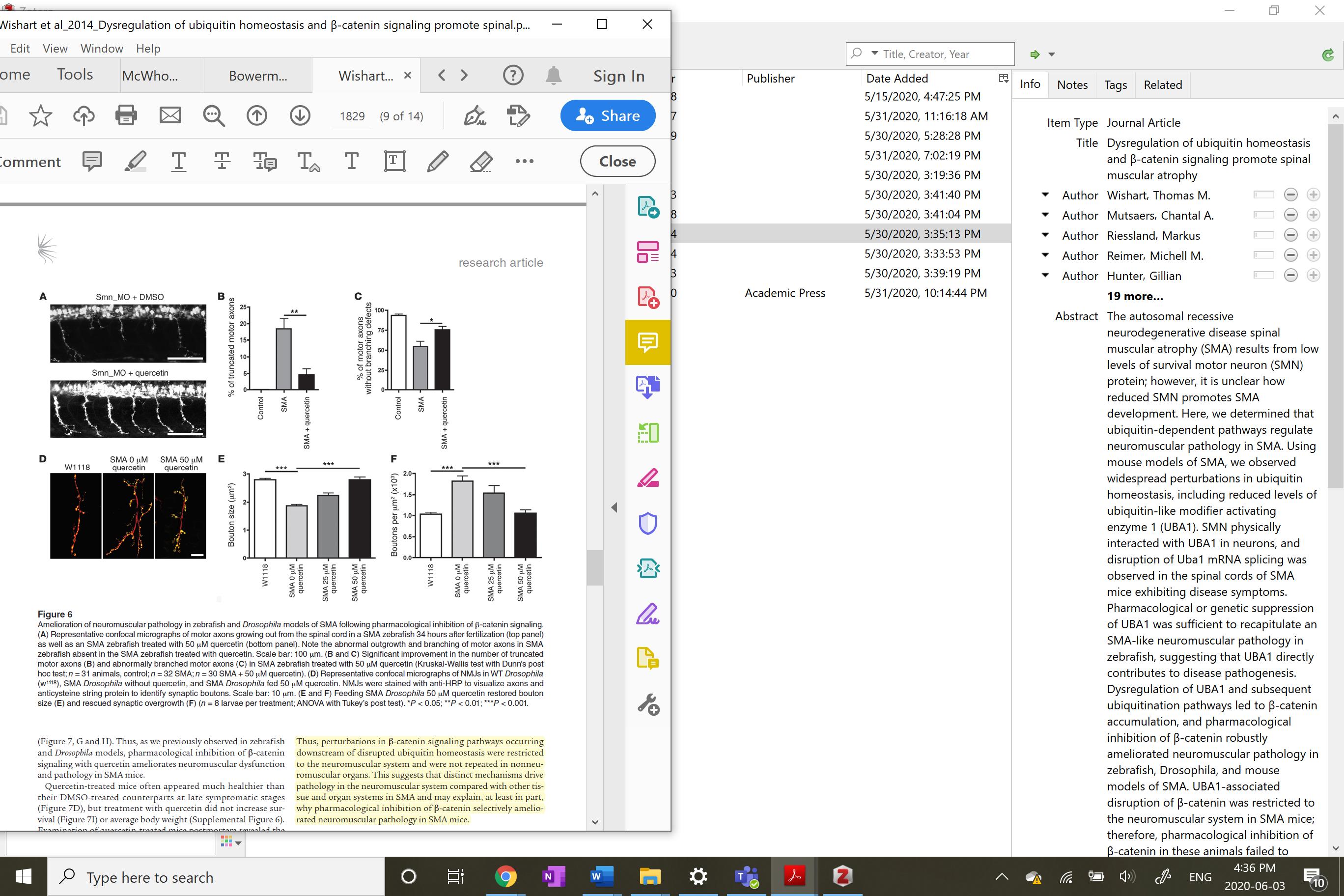
Figure 4 Rescuing neuromuscular pathology in SMA Zebrafish though pharmacological inhibition of B-catenin a downstream effect of UBA1 upregulation by quercetin (Wishhart et al. 2018)
3.3 a UBA1 safe ther

and survival. Powis et al. conclude that UBA1 is necessary for maintenance of the neuromuscular system and can be used as a therapeutic target to treat neuromuscular and systemic issues of SMA. UBA1 may be implicated in other neurodegenerative conditions by disruption of protein degradation (Groen and Gillingwater 2015). This paper is critical for the advancement of new therapies treating SMA. Although there are already drugs and gene therapies, there are none that target the UBA1 system (David et al. 2020). UBA1 restoration whether through AAV mediated injection or pharmacological intervention by increasing UBA1 levels improve SMA models (Shorrock et al. 2018; Wishart et al. 2014).
5. Critical Analysis
Figure 3 Protein Levels of SMN, UBA, and GARS a downstream UBA1 target in SMA and SMA+AAV9-UBA1 treated mice. The authors used the same type of treatment. (Shorrock et al. 2018) The authors conducted extensive research on the influence of the UBA1 protein and mediating SMA progression. The authors three groups of mice, but does not include the true control group. The caption refer back to a different figure for the conshould be presented one graph. In figure 5D the control was
apeutic approach
Powis et al. monitored adverse effects of UBA1 systemic increase, through human UBA1 mRNA levels. Using western blot analysis the levels of UBA1 and SMN were increased in both control and SMA mice. They found no adverse responses to the protein levels. The authors suggest that UBA1 is in the top 2% of cellular proteins; cells may have capacity to withstand high levels. In earlier studies, one of which appears in figure 4, SMA progression could be ameliorated by changing UBA1 downstream targets (Wishart et al. 2014).
4. Discussion
UBA1 levels are significantly reduced in SMA models which implies that changes to this system contribute to SMA progression. Replenishing UBA1 levels and a subsequent decrease in SMA pathology provides substantial verification. UBA1 influ
5.1 Powis et al. limitations and success
were correct in their conclusion that UBA1 is a valid target for SMA treatment since other papers produced similar findings (Powis et al. 2016). Although this paper recognizes UBA1 mediated SMA progression, it did not address the mechanisms in which UBA1 is implicated, rather it showed changes in levels of the enzyme; further investigators researched this. The authors reported a conserved response of UBA1 decline among SMA patients and models. The only issue in this paper was evident in Figure 5 (figure 4 in their paper), which compared survival, motor performance and body weight of mice. This figure compares trol’s results. Since the groups are being compared they all ences organ pathology, muscle atrophy, overall size, health,
injected with AAV9-UBA in the original caption, but does not contain the information in the figure.
5.2 Why this treatment works –at a molecular level 5.2.1 UBA1/GARS Pathway
The levels of UBA1 in SMA models effect the levels of GARS, an important enzyme for protein synthesis (Shorrock et al. 2018). This target underlies the molecular pathways for the deafferentation of motor neurons in SMA (Shorrock et al. 2018). They employed the same technique with AAV-9-UBA1 injection systemic restoration of SMN and UBA1 levels. If the injection
which corrected the sensory-motor connectivity at the level of the spinal cord, verifying this method of gene therapy and mo
ditions
A hallmark of neurodegenerative conditions is protein clumping. UBA1 marks protein for degradation, therefore it may contribute to disease progression. For instance, UBA1 is implicated in polyglutamine protein toxicity in mice with Huntington’s Disease (HD) (Groen and Gillingwater 2015). Therefore, using the systemic increase of UBA1 in different mouse models of neurodegenerative conditions like HD may reveal a novel treatment. If administration of UBA1 blocks disease progression this means it could be a broader therapeutic approach than just SMA. Neurodegenerative conditions are complex disease though, so if the UBA1 administration does not work, it may indicate different enzyme in the UPS system that would need further investigation.
lecular targets like the original paper.

Figure 5 Comparing outcomes between treatment groups for SMA. A:Weight, B: Survival, C: Behavioural reflex. D: Appearance (Powis and see subsequent responses. The combined therapeutic approach should improve the outcomes of SMA models through a fails to ameliorate disease this would indicate that packaging of the AAV does not work or there are competing protein interactions. They could also deliver two separate injections and compare to the single injection to verify that packaging is not the issue, rather than upregulation of the gene and enzyme.
5.3.2 UBA1 can be used to treat other neurodegenerative con
et al. 2016)
5.2.2 UBA1 and B-catenin
Disruption of UBA1 in SMA models is due interactions in the cytoplasm involving physical interactions between SMN and UBA1 proteins, and splicing of UBA1 mRNA (Wishart et al. 2014). Accumulation of B-actin resulted from UBA1 dysregulation and blocking B-catenin reduced SMA pathology (Wishart et al. 2014). B-catenin signalling pathways regulate motor neuron differentiation, stability, synaptic function, and structure. Lower levels of UBA1 contribute to neuromuscular issues by ubiquitin impairment and subsequent B-catenin signalling; this information re-enforces the main objectives by Powis et. al that enhancing UBA1 will ameliorate neuromuscular issues.
5. 3 Future Directions 5.3.1 UBA1 as a combined therapeutic approach
UBA1 replenishment has been proposed as a novel therapeutic approach for SMA. Current treatments use AAV9 to delivery intact copy SMN1, referred to as Zolgenesma (Schorling, Pechmann, and Kirschner, 2020). A combined therapeutic approach which uses SMN1 and UBA1 levels should be tried in future. The authors should conduct an experiment which delivers both UBA1 mRNA and SMN1 in one injection to mice and assess therapeutic effects. The authors could use the technique of packaging recombinant AAV vector with a cleavage site placed between the coding sequence of UBA1 and SMN1, for both to be expressed (Foti, Samulski, and McCown 2009). The authors could then administer the virus via the facial vein to SMA mice 135
10.
Shorrock, Hannah K, Dinja van der Hoorn, Penelope J Boyd, Maica Llavero Hurtado, Douglas J Lamont, Brunhilde Wirth, James N Sleigh, et al. “UBA1/GARS-Dependent Pathways Drive Sensory-Motor Connectivity Defects in Spinal Muscular Atrophy.” Brain 141, no. 10 (October 2018): 2878–94. https:// doi.org/10.1093/brain/awy237 Bowerman, Melissa, Catherina G. Becker, Rafael J. Yáñez-Muñoz, Ke Ning, Matthew J. A. Wood, Thomas H. Gillingwater, and Kevin Talbot. “Therapeutic Strategies for Spinal Muscular Atrophy: SMN and Beyond.” Disease Models & Mechanisms 10, no. 8 (August 1, 2017): 943–54. https://doi.org/10.1242/ dmm.030148.
McWhorter, Michelle L., Umrao R. Monani, Arthur H.M. Burghes, and Christine E. Beattie. “Knockdown of the Survival Motor Neuron (Smn) Protein in Zebrafish Causes Defects in Motor Axon Outgrowth and Pathfinding.” The Journal of Cell Biology 162, no. 5 (September 1, 2003): 919–32. https://doi.org/10.1083/jcb.200303168. Wishart, Thomas M., Chantal A. Mutsaers, Markus Riessland, Michell M. Reimer, Gillian Hunter, Marie L. Hannam, Samantha L. Eaton, et al. “Dysregulation of Ubiquitin Homeostasis and β-Catenin Signaling Promote Spinal Muscular Atrophy.” The Journal of Clinical Investigation 124, no. 4 (April 1, 2014): 1821 –34. https://doi.org/10.1172/JCI71318. Chang, Hui-Chiu, Wen-Chun Hung, Yen-Ju Chuang, and Yuh-Jyh Jong. “Degradation of Survival Motor Neuron (SMN) Protein Is Mediated via the Ubiquitin/Proteasome Pathway.” Neurochemistry International 45, no. 7 (December 1, 2004): 1107–12. https://doi.org/10.1016/j.neuint.2004.04.005. Groen, Ewout J.N., and Thomas H. Gillingwater. “UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration.” Trends in Molecular Medicine 21, no. 10 (October 2015): 622–32. https:// doi.org/10.1016/j.molmed.2015.08.003. Foti, SB, RJ Samulski, and TJ McCown. “Delivering Multiple Gene Products in the Brain from a Single Adeno-Associated Virus Vector.” Gene Therapy 16, no. 11 (November 2009): 1314–19. https:// doi.org/10.1038/gt.2009.106. C, David C., Astrid Pechmann, and Janbernd Kirschner. “Advances in Treatment of Spinal Muscular Atrophy –New Phenotypes, New Challenges, New Implications for Care.” Journal of Neuromuscular Diseases 7, no. 1 (2020): 1–13. https://doi.org/10.3233/JND-190424. Dlamini, Nomazulu, Dragana J. Josifova, Simon M. L. Paine, Elizabeth Wraige, Matthew Pitt, Amanda J. Murphy, Andrew King, et al. 2013. “Clinical and Neuropathological Features of X-Linked Spinal Muscular Atrophy (SMAX2) Associated with a Novel Mutation in the UBA1 Gene.” Neuromuscular Disorders 23 (5): 391–98. https://doi.org/10.1016/j.nmd.2013.02.001. Chaytow, H., Huang, Y. T., Gillingwater, T. H., & Faller, K. (2018). The role of survival motor neuron protein (SMN) in protein homeostasis. Cellular and molecular life sciences : CMLS, 75(21), 3877–3894. https://doi.org/10.1007/s00018-018-2849-1








