Organizadores
Marilia Martins Guimarães
Mestre e Doutora em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Professora-associada da Faculdade de Medicina da UFRJ.
Professora do Instituto de Endocrinologia da Santa Casa da Misericórdia do Rio de Janeiro.
Maria Alice Neves Bordallo
Mestre em Endocrinologia pela Pontifícia Universidade Católica do Rio de Janeiro (PUC-RJ).
Doutora em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Professora Associada de Endocrinologia da Universidade do Estado do Rio de Janeiro (Uerj).
Kássie Regina Neves Cargnin
Mestre em Endocrinologia pela Universidade do Estado do Rio de Janeiro (Uerj).
Professora e Diretora do Instituto de Endocrinologia da Santa Casa da Misericórdia do Rio de Janeiro.
Honomar Ferreira de Souza
Mestre e Doutor em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Professor Titular da Faculdade de Medicina da Universidade Federal Fluminense (UFF).
Cláudia Braga Monteiro
Mestre e Doutora em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médica do Hospital Universitário Pedro Ernesto (Hupe) da Universidade do Estado do Rio de Janeiro (Uerj).
Colaboradores
Ana Paula Neves Bordallo
Mestre em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médica do Hospital Universitário Pedro Ernesto (Hupe) da Universidade do Estado do Rio de Janeiro (Uerj).
Médica Endocrinologista Pediátrica do Hospital Federal da Lagoa, RJ.
Bruna Santiago Pugliese
Mestre em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médica do Hospital Federal dos Servidores do Estado do Rio de Janeiro (HFSE‑RJ).
Daniel Luis Schueftan Gilban
Mestre em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médico da Universidade do Estado do Rio de Janeiro (Uerj).
Médico do Hospital Federal de Bonsucesso, RJ.
Gustavo Guida Godinho da Fonseca
Mestre em Medicina pelo Programa de Ciências Médicas da Universidade Federal do Rio de Janeiro (UFRJ).
Membro Titular da Sociedade Brasileira de Genética (SBG).
Consultor Médico do Diagnósticos da America S.A. (Dasa).
Isla Aguiar Paiva
Mestre em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médica do Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG) da Universidade Federal do Rio de Janeiro (UFRJ).
Médica Endocrinologista Pediatra do Instituto Estadual de Diabetes e Endocrinologia.
Izabel Calland Ricarte Beserra
Doutora em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Professora Associada da Faculdade de Medicina da UFRJ.
Vice diretora da Faculdade de Medicina da Universidade Federal do Rio de Janeiro (UFRJ).
Karina de Ferran
Mestre em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médica do Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG) da Universidade Federal do Rio de Janeiro (UFRJ).
Karinne Condack Mafort Branco
Mestre em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médica do Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG) da Universidade Federal do Rio de Janeiro (UFRJ).
Médica Pediatra Intensivista do Instituto Nacional de Traumato e Ortopedia (Into), RJ.
Marcus Vinicius Leitão de Souza
Mestre e Doutor em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Professor Adjunto da UFRJ.
Micheline Abreu Rayol de Souza
Mestre em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médica do Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG) da UFRJ.
Paulo Alonso Garcia Alves Junior
Mestre e Doutor em Medicina (Endocrinologia) pela Universidade Federal do Rio de Janeiro (UFRJ).
Médico Endocrinologista Pediátrico do Instituto Nacional de Câncer (Inca).
Triagem Neonatal
DeFINIção
Triagem neonatal refere‑se à pesquisa de doenças cujo diagnóstico é neces‑ sário em razão da ausência de sinais clínicos significativos nessa fase da vi‑ da, o que requer diagnóstico e tratamento precoce. Isso evita graves sequelas após o nascimento.
A idade ideal para a coleta de sangue é após 48h de vida, ainda na pri meira semana. Não há necessidade de aguardar ganho de peso ou melhora da icterícia para a coleta.
Os testes de triagem neonatal são realizados por vários serviços no Brasil desde a década de 1980 e, em junho de 2001, o Ministério da Saúde instituiu o Programa Nacional de Triagem Neonatal (PNTN). Sua meta é a triagem de 100% dos nascidos vivos em todo o território nacional para as seguintes doenças:
Hipotireoidismo congênito (HC).
Fenilcetonúria.
Fibrose cística.
Doença falciforme e outras hemoglobinopatias.
Em cada Estado existem serviços de referência de triagem neonatal (SRTN) credenciados pelo Ministério da Saúde para a realização dos exames, visan do à detecção precoce dos casos suspeitos, à confirmação diagnóstica, ao acompanhamento e ao tratamento dos casos identificados.
Em dezembro de 2012, o Ministério da Saúde ampliou o programa, e atualmente os serviços de referência devem estar habilitados para a realização
Cláudia Braga Monteiro
dos procedimentos também nos casos de hiperplasia adrenal congênita (HAC) e deficiência de biotinidase.
Em abril de 2021, o Plenário do Senado Federal aprovou o Projeto de Lei (PL) no 5.043/2020, que amplia o número de doenças rastreadas pelo Sistema Único de Saúde (SUS) por meio da utilização da espectrometria de massas em tandem e de outras tecnologias. Com a mudança o exame pas sará a contemplar 14 grupos de doenças cuja implementação será escalo nada da seguinte forma:
Primeira etapa: incluir a pesquisa da toxoplasmose congênita às doenças já contempladas.
Segunda etapa: incluir a pesquisa da galactosemia, aminoacidopatias, disúrbios da betaoxidação dos ácidos graxos e do ciclo da ureia.
Terceira etapa: pesquisa de doenças lisossômicas.
Quarta etapa: pesquisa de imunodeficiências primárias.
Quinta etapa: pesquisa da atrofia muscular espinhal.
A lista de doenças a serem rastreadas pelo teste deverá ser revisada perio ‑ dicamente, com base em evidências científicas e priorizando‑se as doenças com maior prevalência no país, com protocolo de tratamento aprovado e in‑ corporado ao SUS (Lei no 14.154 de 26 de maio de 2021). A implementação plena dessas etapas depende, entretanto, do financiamento público pleno, do treinamento e da capacitação das equipes dos SRTN, além da disponibi‑ lização de medicamentos e fórmulas metabólicas.
tRIAGeM NeoNAtAL PARA HIPotIReoIDISMo CoNGêNIto
O HC tem incidência de 1 em 3.500 a 4.000 nascidos vivos. A triagem neo natal e a reposição precoce com levotiroxina têm alta relação custo‑benefício pelo considerável impacto no desenvolvimento neurológico e na qualidade de vida dos pacientes acometidos.
Método
Dosagem do hormônio tireoestimulante (TSH) por método ultrassensível a partir do sangue total coletado do calcanhar e impregnado em papel‑filtro.
Ponto de corte para o TSH: o PNTN recomenda um valor de corte de 10mUI/L para o TSH em sangue total (método imunofluorimétrico). Entretanto, existe uma tendência atual em reduzir esse valor para 5 a 6µU/L, o que possibilita
Diferenças do Desenvolvimento Sexual
DIFeReNçAS No DeSeNVoLVIMeNto SexuAL
As diferenças no desenvolvimento sexual (DDS) comumente são evidenciadas ao nascimento pelo aspecto incomum da genitália externa (genitália atípi‑ ca). São consequentes a múltiplas causas (anomalias cromossômicas, mu tações gênicas, fatores ambientais, idiopática) que podem afetar uma das seguintes fases do desenvolvimento: determinação do sexo cromossômico, determinação das gônadas, diferenciação dos genitais internos e externos ou diferenciação sexual secundária (resposta dos tecidos aos hormônios produ zidos pelas gônadas).
Classificação
As DDS são classificadas em três categorias de acordo com a causa:
1. DDS 46,XX:
y Distúrbios do desenvolvimento ovariano: disgenesia gonadal, DDS ovotesticular e DDS testicular.
y Excesso androgênico: hiperplasia adrenal congênita (HAC), causas maternas (ingestão de progestágenos ou androgênios, doenças virili zantes), deficiência da aromatase placentária e deficiência da p450 oxidorredutase.
y Outros: p. ex., associados às síndromes e à agenesia mülleriana.
2. DDS 46,XY:
y Distúrbios do desenvolvimento testicular: disgenesia gonadal com‑ pleta ou parcial, DDS ovotesticular e síndrome da regressão testicular.
Izabel Calland Ricarte Beserra
y Defeitos na síntese ou ação dos androgênios: grupo mais extenso, com destaque para hipoplasia ou aplasia das células de Leydig, deficiência da enzima 5 alfarredutase tipo 2, formas raras de HAC e insensibili dade androgênica completa ou parcial.
y Outros: esses defeitos estão associados à síndrome da persistência dos ductos müllerianos e à regressão testicular ou síndrome dos tes tículos evanescentes.
3. DDS cromossomos sexuais:
y Síndrome de Turner (45,X).
y Síndrome de Klinefelter (47,XXY).
y Cariótipos mosaicos (p. ex., 45,X/46,XY, 46,XX/46,XY).
Os principais diagnósticos etiológicos e suas características são:
DDS ovotesticular: trata se da existência de ambos os tecidos, ovariano e testicular, no mesmo indivíduo, separados ou juntos (ovotestis). O diag‑ nóstico é histológico. Na maioria, o cariótipo é 46,XX (60%), seguido de mosaicismo contendo os cromossomos Y (25%) e 46,XY (15%).
DDS testicular: nesta condição, embora o cariótipo do paciente seja 46,XX, na maioria ocorre translocação do gene SRY para o braço curto de um dos cromossomos X durante a meiose paterna, acarretando diferenciação das gônadas em testículos e fenótipo semelhante à síndrome de Klinefelter. Em 10% dos casos o gene SRY não está presente (duplicação do gene SOX9).
HAC: é a causa mais comum de DDS no cariótipo 46,XX e deve ser a pri meira hipótese diagnóstica em pacientes com genitália ambígua e gônadas impalpáveis, não somente pela frequência, mas também pelo potencial de morbimortalidade (perda de sal). Três formas de HAC causam virilização no feto feminino: deficiência da enzima 21 hidroxilase (95% dos casos de HAC), deficiência da 11 beta hidroxilase e deficiência da 3 beta hidro xiesteroide desidrogenase. Outras formas mais raras de HAC podem cau sar ambiguidade genital no sexo masculino (mutação da proteína StAR; deficiência da p450scc, 3 beta hidroxiesteroide desidrogenase ou 17 alfa‑ hidroxilase/17,20‑liase).
Disgenesia gonadal 46,XY: o fenótipo depende do grau de desenvolvi mento gonadal e pode estar associada a vários distúrbios, como displasia campomélica, síndrome de Denys Drash e síndrome de Frasier.
y Completa: não ocorre desenvolvimento gonadal, e os indivíduos apre sentam fenótipo completamente feminino, estruturas müllerianas nor mais e gônadas em fita bilateralmente. Na adolescência, evoluem com
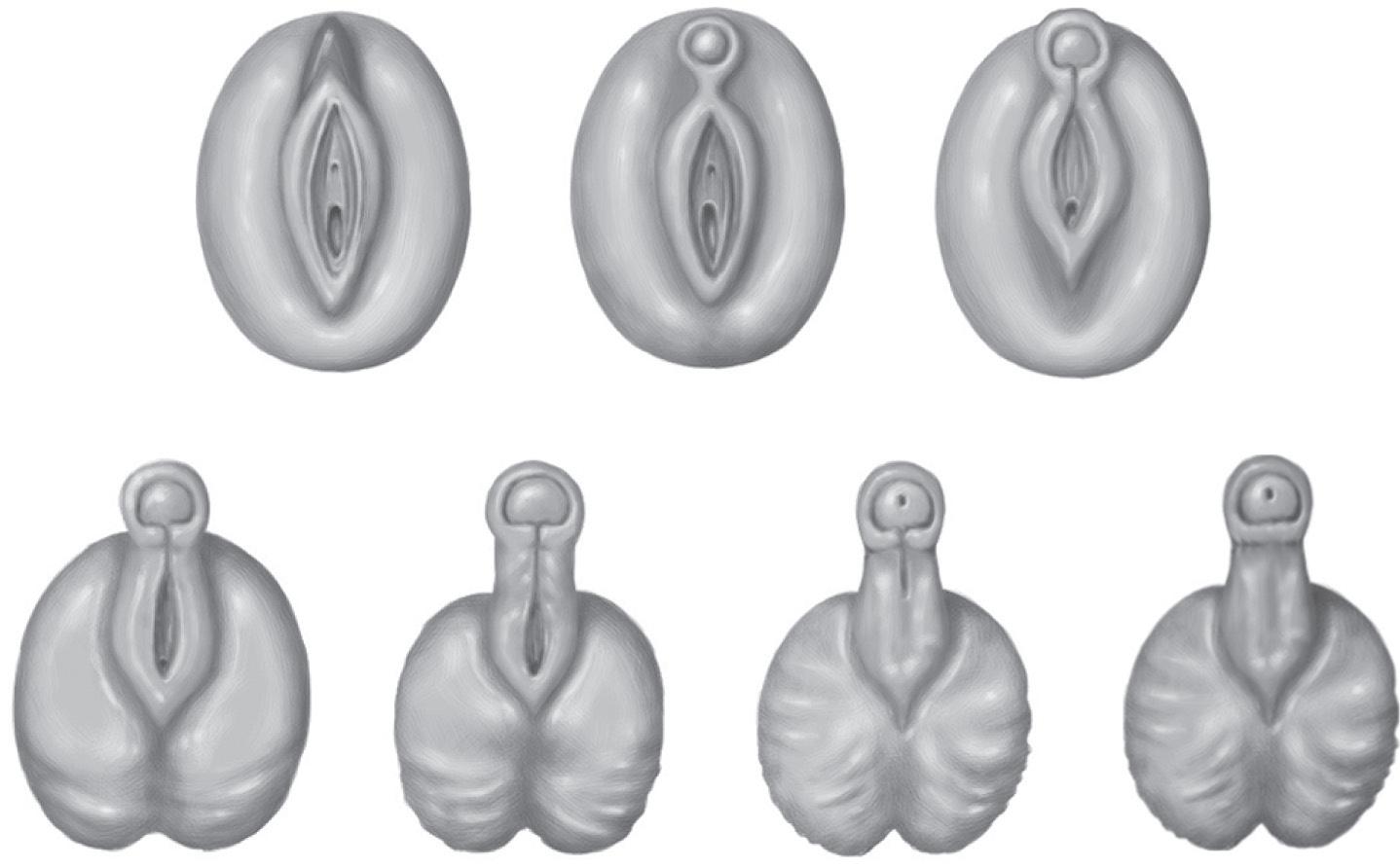
Figura 5.1 Classificação de Prader
Exames Complementares
Na primeira etapa da investigação, devem ser realizados os seguintes exames:
Cariótipo.
Ultrassonografia (USG) pélvica ou ressonância magnética (RM): são fun‑ damentais para a investigação da existência de estruturas müllerianas. Vale lembrar que a USG é um método que depende do operador. Assim, a experiência na realização desse exame é importante.
Dosagem sérica da 17 OH progesterona (diagnóstico de HAC por defi ciência da 21 hidroxilase).
Eletrólitos séricos: a hiponatremia ou a hiperpotassemia sugerem HAC, forma perdedora de sal.
O teste de estímulo com hormônio adrenocorticotrófico (ACTH) deverá ser realizado se os valores de 17 OH progesterona não forem conclusivos e permanecer a suspeita diagnóstica de HAC.
Dosagem sérica de hormônio luteinizante (LH), hormônio foliculoestimu lante (FSH), testosterona, Dihidrotestosterona (DHT), androstenediona e, quando possível, hormônio antimülleriano (HAM). A concentração de HAM no soro é um indicador da existência e da função dos testículos. Valores in‑ detectáveis indicam ausência de tecido testicular, enquanto valores normais
Grau 1
Feminina
Grau 2
Grau 3Grau 4Grau 5
Masculina
ou elevados sugerem defeito na síntese de androgênios ou um defeito do receptor. As dosagens de LH, FSH, testosterona, DHT e androstenediona somente têm valor quando realizadas até os 4 meses de vida, época de estimulação fisiológica do eixo hipotálamo hipófise gônadas.
Com os resultados dos exames laboratoriais, além das informações sobre a existência ou não de gônadas palpáveis e de estruturas müllerianas, nor‑ malmente já se tem um guia razoável sobre o manejo prático desses pacien‑ tes e a possibilidade de definição do sexo de criação. Os exames em uma segunda etapa, estabelecidos a partir dos resultados dos exames da primeira, servem para a definição etiológica, mas nem sem pre são necessários para determinar o sexo de criação. Nessa situação, são incluídos:
Teste de estímulo com gonadotrofina coriônica humana (hCG) para dosa gem de testosterona e DHT pré e pós estímulo (diagnóstico de deficiência de 5 alfarredutase tipo 2).
Dosagem sérica de outros precursores androgênicos (diagnóstico das for mas raras de HAC, como deficiências das enzimas 11 beta hidroxilase, 3 beta hidroxiesteroide desidrogenase tipo 2, 17 alfa hidroxilase/17,20 lia se, 17 beta hidroxiesteroide desidrogenase tipo 3 e p450 oxidorredutase).
Avaliação in vitro da ligação dos androgênios a seu receptor e análises genéticas e moleculares. Quando disponíveis, ajudam na confirmação diagnóstica de cerca de 20% dos casos de DDS.
Outros Exames Complementares
A genitografia é importante para a avaliação pré operatória e a programação cirúrgica. As informações obtidas a partir dela complementam as dos achados da genitália interna.
A avaliação histológica mediante biópsia de gônadas pode ser necessária para a confirmação diagnóstica das disgenesias gonadais e do DDS ovotes ticular. Além disso, a biópsia de pele com cultura de fibroblastos ajuda na confirmação de casos de insensibilidade androgênica e deficiência da 5 al farredutase tipo 2.
Índice
AAberrações cromossômicas, 21
Acetato de hidrocortisona, 64
Acidemias orgânicas, 8
Ácido
bempedoico, 209 ribonucleico interferente, 211 úrico, 219
Adeno hipófise, 57 Adenomas
foliculares, 97 hipofisários, 66
Agonista(s)
duplo GLP 1/GIP, 223 GLP 1, 223
Alelo, 251
Alta estatura, 19 causas de, 19
‑ familiar, 19
Alterações da função gonadal, 163 da função tireoidiana, 164 do crescimento, 162 do metabolismo ósseo, 165 epigenéticas, 251
Aminoacidopatias, 8
Androgênicos, 43
Aneuploidias, 251 do par sexual, 251
Anorexia nervosa, 227
Antagonistas beta‑adrenérgicos, 92
Anticorpos
‑ bloqueadores maternos, 80 monoclonais da proteína 3 semelhante à angiopoetina (ANGPTL3), 211
Antidepressivos, 223 Antiestrogênicos, 43
Atraso constitucional do crescimento e da puberdade, 15, 32, 34
Atrofia ovariana, 33
Aumento da secreção de hormônio tireoestimulante, 89
Autossomos, 251
Avaliação do crescimento, 13
Avanço constitucional do crescimento, 19
B Baixa
e alta dose de dexametasona, 261 estatura, 14 16 causas de, 16 familiar, 15
Bandeamento, 251
Bicarbonato de sódio, 181
Bloqueio de puberdade, 241
Bócio(s), 94 difuso(s), 95, 99 tóxico, 95 idiopático, 96 nodulares, 88, 96, 100
Bociogênicos, 96
Bulimia nervosa, 229
Burosumab, 141
C
Câncer de tireoide, 100, 102
Carcinoma(s) anaplásico, 103 diferenciados, 101 folicular, 103 indiferenciados, 103 insular, 103 medular, 103 metastático, 104 papilífero, 97, 102 pouco diferenciados, 103
Cariótipo(s), 47, 251 mosaicos, 47
Carta de orientação para paciente com insuficiência adrenal, 263
Centrômero, 252
Cetoacidose diabética, 179
Cirurgia bariátrica, 224
Cistinose, 139
Cistos da bolsa de Rathke, 67
Citogenética, 252
Citrato de clomifeno, 43
Classificação do tamanho ao nascer, 11
Codominância, 252
Códons, 252
Complexo de Carney, 104
Conduta diagnóstica, 15
Cortisol livre urinário, 219
Craniofaringioma, 67
Crescimento
‑ anormal, 14 de recuperação (catch-up growth), 13 fetal, 11
‑ normal, 11 pós‑natal, 12
Cretinismo endêmico, 81
Cromátides, 252
Cromossomo(s), 252, 253 derivativo, 252 em anel, 252 marcador, 253
‑ sexuais, 253
D
Danazol, 43
Defeitos da betaoxidação dos ácidos graxos, 8 da homeostase do fosfato, 138 metabólicos e funcionais da vitamina D, 138
Deficiência(s) da enzima 5 alfarredutase tipo 2, 47 de ACTH, 62
‑ de biotinidase, 9 de FSH e LH, 62 de GH, 62 e do eixo somatotrófico, 59
de gonadotrofinas, 59 de iodo, 96 de TSH, 62
familiar de glicocorticosteroide, 20 hormonais, 196
isoladas de ACTH e de TSH, 59 ou resistência aos hormônios sexuais, 20
Deleção, 253
Designação do sexo de criação, 51
Desvios fisiológicos do crescimento, 12
Diabetes insípido, 70 central, 73
‑ ‑ nefrogênico, 73 melito, 169 classificação do, 170 complicações crônicas, 182 critérios diagnósticos, 169 tipo 1, 171 tipo 2, 176
Diferenças no desenvolvimento sexual, 45, 46 46,XX, 45 46,XY, 45 cromossomos sexuais, 46 ovotesticular, 46 testicular, 46
Dihidrotestosterona, 43
Diploide, 253
Disbetalipoproteinemia, 207
Disforia/incongruência de gênero, 236
Disgenesia gonadal 46,XY, 46 tireoidiana, 79
Dislipidemias, 199
‑ avaliação diagnóstica das, 201
etiologia e classificação das, 205 secundárias, 207 tratamento, 208
Disormonogênese, 81, 96
Dissomia uniparental, 253
Distúrbios da puberdade, 25 do ciclo da ureia, 8 endócrinos, 19 metabólicos, 19 DNA (ácido desoxirribonucleico), 253
Doença(s) associadas ao carcinoma de tireoide, 104 da adrenal, 111 da hipófise, 57 59, 70 anterior, 59
‑ ‑ classificação quanto à função, 58 etiologia, 57 posterior, 70
‑ da tireoide, 79 de Dent, 139 de Graves, 95 na infância e na adolescência, 88, 89 neonatal, 88, 89 do metabolismo mineral, 127 metabólicas
‑ ‑ e genéticas intrínsecas do esqueleto, 142 ósseas, 137
EEdema cerebral, 182
Efeitos da radioterapia, 165 tardios do tratamento do câncer, 161
Esteroides sexuais, 23, 64
Estirão puberal, 12
Euploidia, 253
Evinacumabe, 211
Exoma, 253
Éxon, 253
F
Fator de crescimento semelhante à insulina tipo 1, 220
Fenilalanina, 7
Fenilcetonúria, 7
Fenótipo, 254
Feocromocitoma, 122
Fibrose cística, 9
FISH (hibridização in situ fluorescente), 254
G
Galactosemia, 8
Gene(s), 254 do Y, 254 selvagem, 254
SHOX, 254
Genética mendeliana, 254
Genótipo, 254
Germinomas, 66
Ginecomastia, 37 no recém nascido, 37 puberal, 37
Glicocorticosteroides, 64
Glicose, 219
H
Hamartomas, 66
Haploide, 254
Hemizigose, 254
Hemoglobina glicada, 219
Heptanoato de dihidrotestosterona, 43
Herança
‑ autossômica dominante, 254 recessiva, 255 ligada ao X, 255
‑ mendeliana, 255
Heterozigose, 255 composta, 255
Hiperaldosteronismo, 121
Hipercalcemia, 131
Hipercalciúria, 134
Hipercolesterolemia familiar, 206
Hiperinsulinismo, 190, 195
Hiperlipidemia(s) familiar combinada, 206 primárias, 206
Hipermagnesemia, 137
Hiperplasia adrenal congênita, 4, 19 forma clássica
perdedora de sal, 118 virilizante simples, 119 forma não clássica, 119
Hipertireoidismo, 86
Hipertrigliceridemia primária, 207
Hipoalfalipoproteinemias, 207
Hipocalcemia, 127
Hipofunção hipofisária, 59
Hipoglicemia, 178
‑ assintomática, 195 definição, 185 diagnóstico diferencial, 190 em crianças diabéticas em uso de insulina, 196 investigação diagnóstica, 189 quadro clínico, 188 sintomática, 195 tratamento, 190
Hipomagnesemia, 135
Hipoparatireoidismo, 129
Hipoplasia ou aplasia das células de Leydig, 47
Hipotireoidismo
adquirido, 82 congênito, 2, 82, 84 primário, 79, 81, 165 adquirido, 81 congênito, 79 secundário e terciário, 82 subclínico, 85
Homocistinúria, 21
Homozigose, 255
Hormônio(s) do crescimento, 64
‑ ‑ humano recombinante, 18 tireoestimulante, 219 tireoidianos, 64, 219
Hormonização cruzada, 243
Hot spots, 255
I
Idade
cronológica, 13 estatural, 14 gestacional, 11 óssea, 14 peso, 14
Identidade de gênero, 235
Inativação do cromossomo X, 255
Incidentalomas, 124 Índice de Capurro, 11
‑ de massa corporal, 215
Indução puberal, 53, 54
Inibidor(es)
Apo C3, 212
‑ da aromatase, 43
Insensibilidade androgênica, 47
Inserção, 256
Insuficiência adrenal, 111, 263 aguda, 112 crônica, 114 primária, 112
Insulina, 219
Insulinização, 173, 181 Íntron, 256
Inversão cromossômica, 256
Isocromossomo, 256
L
Leptina, 219, 223
Levotiroxina, 84
Linfoma, 104
Lipídios, 199, 201 de origem endógena, 201
Liraglutida, 222
Lojuxta, 212
Lomitapida, 212
M
Medida de pressão arterial, 283
Metabolismo dos lipídios, 200
Metformina, 223
Metimazol, 90
Métodos de aferição das medidas antropométricas, 13
Micropênis, 53
Monossomia, 256
Mosaico, 256
Mounjaro, 223
Mutação de ponto, 256 do gene
APC, 105
PPKAR1A, 104 PTEN, 104
frameshift, 256
‑ missense, 256 no receptor do hormônio tireoestimulante, 81 nonsense, 256 silenciosa, 257
N
Neoplasia endócrina múltipla tipo 2B, 21
Neuro hipófise, 57
Nulissomia, 257
O
Obesidade diagnóstico clínico, 217
‑ exame(s) físico, 217 complementares, 218 na infância, 215 prevalência, 216 tratamento, 220
Oftalmopatia de Graves, 94
Orlistat, 222
Osteogênese imperfeita, 144
Osteomalácia induzida por tumor, 138
Osteoporose, 142
Ozempic, 223
P
Padrão genético, 15
Parada ou diminuição fisiológica do crescimento pré‑puberal, 12
Paraganglioma, 122
Parâmetros auxológicos, 13
PCR, 257
Peso e comprimento ao nascer, 15
Polimorfismo de nucleotídio único, 257
Polipose adenomatosa familiar, 105
Propiltiouracil, 90
Proteínas, 199
Prova(s) da vasopressina, 261 de estímulo com análogo do hormônio liberador de gonadotrofinas, 259 do hormônio do crescimento, 259 de restrição hídrica, 260
Pseudo hipoparatireoidismo, 218
Pubarca precoce, 31
Puberdade atrasada, 32
‑ precoce, 19, 25‑27, 29 central ou completa ou dependente de hormônio liberador de gonadotrofina, 26
‑ ‑ dependente de hormônio liberador de gonadotrofina (GnRH), ou central, 25 heterossexual, 25
‑ ‑ independente de GnRH, ou periférica, 25 isossexual, 25 periférica ou incompleta ou independente de hormônio liberador de gonadotrofina, 26 sinais e sintomas da, 27 tratamento, 29
R
Radioiodo, 93
Radioiodoterapia, 106
Raloxifeno, 43
Raquitismo, 137
‑ hipofosfatêmico, 141
hereditário com hipercalciúria (HHRH), 139 por acelerada inativação da vitamina D, 141 por defeito no receptor de vitamina D, 141 por deficiência da 1‑alfa‑hidroxilase ou 25‑hidroxilase, 141
Recém‑nascidos PIG, 11 assimétricos ou desproporcionais, 12 simétricos ou proporcionais, 11
Região pseudoautossômica, 257
RNA (ácido ribonucleico), 257
S
Sanger (sequenciamento de), 257
Sibutramina, 222
Síndrome de Beckwith‑Wiedemann, 21 de Berardinelli, 21 de Cowden, 104 de Cushing, 115 de Down, 152 de Fanconi, 139 de Klinefelter, 21, 33, 47, 150 de Laurence Moon Biedl, 218 de Lowe, 139 de Marfan, 21 de McCune‑Albright, 139
‑ de Noonan, 154 de Prader‑Willi, 156, 218 de resistência hormonal, 81 de secreção inapropriada de hormônio antidiurético e de perda cerebral de sal, 73 de Silver‑Russell, 158 de Sotos, 20
‑ de Turner, 33, 47, 147
de Weaver, 21 do “desaparecimento de testículos”, 33 do X frágil, 21 genéticas com repercussões endócrinas, 147 hipofosfatêmicas, 138, 141 osteoporóticas, 142
SNP (snipes), 257
Splicing, 257
STKr, 257
Substâncias
antitireoidianas ou de iodo, 80 citotóxicas, 164
Supressão com hormônio tireoidiano, 106
T
Tamoxifeno, 43
Telarca precoce isolada, 31
Terapia afirmativa de gênero, 243
Teste
de estímulo com gonadotrofina coriônica, 262 hormônio adrenocorticotrófico sintético, 262 de supressão noturna com dexametasona, 261
Testotoxicose familiar, 27
Tionamidas, 90
Tireoidite
aguda supurativa, 95 autoimune, 81, 95 de De Quervain, 95 de Hashimoto, 95 subaguda, 95
Tireoidites, 89, 95
Tireotoxicose, 86 factícia, 89
Tiroxina livre, 219
Tirzepatida, 223
Topiramato, 223
Transgeneridade, 235 abordagens terapêuticas, 238 desafios, 247
estimativas populacionais, 238 intervenções cirúrgicas, 247
Transição social de gênero, 240
Translocação, 258
Tratamento da alta estatura familiar, 23 pré natal da hiperplasia adrenal congênita, 121
Triagem neonatal
‑ definição, 1 para deficiência de biotinidase, 9 para fenilcetonúria, 7 para fibrose cística, 9 para galactosemia, 8 para hiperplasia adrenal congênita, 4 para hipotireoidismo congênito, 2
para outras doenças, 7
Triploidia, 258
Trissomia, 258
Tumores da região hipofisária, 65
‑ funcionantes, 66
‑ não funcionantes, 67
‑ produtores de gonadotrofina coriônica humana, 27
VVariação na velocidade de crescimento, 12
Variantes da normalidade, 15 normais do desenvolvimento puberal, 31
Velocidade de crescimento, 15
Volanesorsen, 212
WWaylivra, 212
Mais de 20 anos se passaram desde o lançamento da primeira edição da obra Endocrinologia Pediátrica – Guia
Prático, em 2002, e 7 anos desde a última, publicada em 2017. Para esta nova edição, além de todos os capítulos terem sido revisados e atualizados, os editores sentiram a necessidade de acrescentar um novo capítulo sobre uma questão antiga, mas que vem ganhando destaque e importância nos dias de hoje: transgeneridade. Outra característica mantida nesta quarta edição foi a inclusão de uma nova geração de autores. Foram convidados a participar e enriquecer o livro com seus conhecimentos, quatro autores com ampla experiência na área.
Pediatras, endocrinologista e especialistas relacionados têm em mãos uma obra de fácil manuseio, acessível a qualquer momento e que servirá de grande auxílio no cotidiano.
Áreas de interesse