
15 minute read
Tumor derived exosomes regulate dendritic cell maturation and activation


Rosh Bharthi a , Carolina M. Gorgulho a , Nils Ludwig b,c , Theresa L. Whiteside e , Michael T. Lotze a, c-f a DAMP Laboratory, UPMC Hillman Cancer Center, Pittsburgh, PA, b Department of Pathology, c UPMC Hillman Cancer Center, Pittsburgh, PA, d Department of Immunology, e Department of Pathology, f Department of Bioengineering, University of Pittsburgh, Pittsburgh, PA
Rosh Bharthi
Rosh Bharthi is a bioengineering undergraduate at the Swanson School of Engineering. His current research interests include exosomes and how they play a role in tumor immunology. After graduating, he plans on attending medical school while still continuing his experience in bioengineering and research.
Michael T. Lotze, MD, is Professor of Surgery, Immunology, and Bioengineering; Vice Chair of Research within the Department of Surgery; Director of the DAMP Laboratories at the UPMC Hillman Cancer Center. His research work includes modern immunotherapy and gene therapy, dendritic cell and cytokine therapies, and investigation of the role of mitochondria, metabolism, and unscheduled cell death in cancer. Dr. Lotze is the co-inventor of 10 patents in dendritic cell vaccines and antigen discovery and serves as associate editor of the Journal of Immunotherapy; he is also an award-winning NCI-trained scientist (1978-1990), the inaugural Director of Surgical Oncology at Pitt (1990-2000), former Vice President of Research at GlaxoSmithKline (2001), founding director of the UPCI Academy, former Chief Scientific Officer at Lion/Iovance Biotherapeutics and innovative educator as a prolific scientist/ tumor immunologist with over 500 publications and several books. Michael T. Lotze
Significance Statement
It was initially unknown whether HMGB1-containing exosomes could trigger a dendritic cell response, potentially causing chronic inflammation. Understanding this would explain another mechanism for how cancer and chronic inflammation are related. From this study, exosomes do not contain HMGB1 nor do they stimulate dendritic cells. While this disproves the idea of tumor-derived exosomes causing chronic inflammation, the fact that tumor-derived exosomes did not activate dendritic cells could imply that exosomes are a potential vehicle for delivering antigens without stressing the immune system.
Abstract
Exosomes are nanovesicles ranging from around 50nm150nm that can carry various types of molecules, such as signaling molecules, lipids, and nucleic acids. In the context of cancer, exosomes are responsible for modulating the tumor microenvironment and signaling nearby cells to allow tumor development. HMGB1 is a damage-associated molecular pattern (DAMP) molecule known to be secreted by tumor cells and promote chronic inflammation when continually present in the body; some studies have also shown that HMGB1 is contained in exosomes. It has not been shown whether HMGB1 contained in exosomes can be responsible for causing dendritic cells to become activated, and thus starting the inflammation pathway. Exosomes were isolated from cell culture supernatants and characterized. Afterwards, Western blots were run to examine protein cargo, including but not limited to HMGB1, and flow analysis was used to determine whether exosomes from tumors might activate or suppress dendritic cells. HMGB1 was not present in exosomes, but RAGE and SQSTM-1/p62 was found to be present. Furthermore, exosomes inhibit dendritic cells, but not to the same magnitude that their corresponding tumor cells do.
1. Introduction
Exosomes are nanovesicles secreted by cells. They are formed by the cell membrane involuting and forming a vesicle inside the cytoplasm. This vesicle is then involuted once more, forming a multivesicular body (MVB), which contains many vesicles within it [1]. The MVB then fuses with the cell membrane, releasing the numerous exosome vesicles within. This pathway is mainly to package ubiquitinated proteins in exosomes and release them from the cell [2]. While exosomes were initially considered as a waste secretion system, research has shown that exosomes can carry a wide variety of cargo, such as lipids, proteins, mRNA, and miRNA which can then be taken up by nearby cells, thus acting as a signaling system [2]. The implications of this signaling system could be generalized to various physiological phenomena, such as angiogenesis.
Exosomes play an important role in the context of the immune system and tumor immunology. Research has shown that exosomes secreted by antigen-presenting cells (APCs) are able to activate immune responses based on MHC-peptide complexes found on the surface of exosomes [3]. Additionally, Regulatory T cells (Tregs) release exosomes that suppress production of T cells [4]. Tumor-derived exosomes (TEX) can both stimulate and inhibit an immune response. These exosomes carry inhibitory proteins typically released by the tumor cells which can prevent activation of T cells [5]. On the other hand, TEX contain factors capable of stimulating APCs [5]. From these examples, it is clear that exosomal cargo plays a role in modulating the immune response, and understanding how this signaling works or could be utilized is crucial.
It is known that HMGB1, a notable example of a damageassociated molecular patterns or DAMP, released from tumor cells can lead to chronic inflammation and that chronic inflammation
can promote tumors to further secrete HMGB1, creating a positive cycle [6]. Additionally, it is also known that TEX can play a role in tumor development and signaling; for instance, a study has shown that TEX from head and neck squamous cell carcinoma signal surrounding endothelial cells to promote angiogenesis [7]. This study sought to bridge these two ideas and determine whether TEX might be another form of delivering HMGB1 to surrounding cells. Doing so would show how exosome signaling plays a role in the development of chronic inflammation and cancer. In addition to finding what type of cargo is carried in TEX, the study also shows whether TEX activate dendritic cells (DCs). If TEX were to contain HMGB1 and activate DCs, it would further show evidence of a pathway for HMGB1 from exosomes to DCs. Lastly, in order to see if apoptotic deficiency leads to differences in exosome cargo or secretion, wild type HCT 116 human colorectal tumor cells were compared to a p53 KO, PUMA KO, and BAX KO (inhibiting apoptosis promotes autophagy, a degradation pathway that allowing molecules in the cell to be broken down into constituents and repurposed for other molecules; we believed that enhanced autophagy may thus reduce the cargo released in exosomes, hence why this was one of our goals).
2. Methods 2.1 Exosome Isolation
HCT 116 human colorectal cancer cells were used for these experiments; in particular, wild type, p53 knockout, BAX knockout, and PUMA knockout cell types were used. 6 million cells were seeded in T125 flasks, with two flasks per HCT 116 cell variant. 25 mL RPMI cell media (Corning) with 10% exosome-depleted fetal bovine serum (FBS, Gibco) per flask was used as media; to make exosome-depleted FBS, 1x FBS was ultracentrifuged at 28,000 rpm to pellet exosomes, and the supernatant was used. After 72h, culture supernatant was collected and centrifuged for 2000xg for 10min at room temperature to pellet large debris. Supernatant was centrifuged again at 10,000xg for 30min at 4°C and filtered with .22μm filter to remove larger vesicles remaining. Supernatant was concentrated to 1mL using Vivacell 20 concentrators spun at 2000xg. Mini-SEC (size exclusion chromatography) columns were made by adding 10mL sepharose (GE Healthcare Bio-Sciences) to columns. An upper filter was added to the column, and the column was flushed with 10mL phosphate-buffered saline (PBS). Afterwards, the concentrated supernatant was added to the column. The fourth fraction was collected, using PBS to elute the fractions. Exosome characterization was performed by transmission electron microscopy (TEM) imaging, protein estimation via bicinchoninic acid assay (BCA), and Western blot testing for TSG101. 2.2 Western blot
Using BCA to determine protein concentration, 10μg protein was loaded into each well. Bio-Rad TGX pre-made gels were used. TEX samples were concentrated using 0.5mL 100K Amicon Ultra centrifugal filters to reduce the sample volume needed to load 10μg protein. Cell lysates were used as a comparison for the TEX samples. TSG101 was used as a control molecule to test, since most exosomes contain this molecule. We tested for HMGB1, along with RAGE, LC3, PD-L1, and SQSTM-1/p62, which are other common molecules involved in tumor and immune system biology, to see if their presence could provide any insight on what role these exosomes may play in signaling. 2.3 TEX-Dendritic cell co-incubation
Dendritic cells were isolated from buffy coats. Ficoll gradient centrifugation separated blood into plasma, RBCs, and PBMCs. PBMCs were extracted and washed 3 times with RPMI to remove platelets. RBC lysis buffer (Sigma Aldrich) was added during sequential washing to lyse any remaining RBCs. PBMCs were plated in 6 well plates in AIM V media (Gibco). After 2hr, nonadherent lymphocytes were removed, and RPMI with 10% FBS was added to the adherent monocytes along with 50ng/mL GM-CSF and IL-4 to differentiate monocytes into immature dendritic cells after 5 days. 12μg TEX protein was added to each well on either the fifth day (48hr incubation) or sixth day (24 hr incubation). On the seventh day of monocyte culture, the cells were stained with antibodies for flow analysis. Negative controls received no antigen, and positive controls received 2μg LPS. DCs co-incubated with tumor cells served as comparison for the corresponding DC-TEX co-incubation. 2.4 Flow cytometry
Raw flow cytometry data was gated based on granularity, Zombie Live/Dead dye, and CD11c, a marker expressed by dendritic cells. Proportion of cells expressing marker of interest was obtained; markers of interested included HLA-DR, CD80, CD86, CD40, CD14, CCR7, CD25, CD274, and CD83. Data was plotted as mean±SD; ANOVA with Tukey post-test was performed on data.
3. Results 3.1 Exosome Characterization
Figure 1: TEM Images – Transmission electron microscopy of isolated exosome samples from each cell line. Exosomes are characterized by round vesicles ranging from around 50nm-150nm; they contain a dark, distinct outer border and may appear folded inwards, like a “crushed ping-pong ball” (the center cluster in the p53 KO image is a good example of this).
Exosomes were present in all samples; they are identified by circular structures ranging from 50-150nm with distinct darker borders. BCA protein estimation showed that protein was present in extracted exosome samples, around 100μg protein for each sample. Furthermore, Western blot confirmed the presence of TSG101 in all TEX samples.
3.2 Western Blot
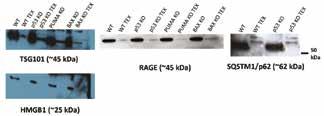
Figure 2: Western Blot – Membrane images of TSG101, HMGB1, RAGE, and SQSTM1/p62. Presence of SQSTM1/p62 was only tested for WT and p53 KO, but was not yet tested for PUMA KO and BAX KO. Blots were used to indicate the presence or absence of the molecules tested.
TSG101, the control marker, was shown consistently for all Western blots, confirming that the exosome isolation technique worked. When testing for HMGB1, PD-L1, and LC3, these markers were only present in cell lysates but not exosomal protein. However, the presence of RAGE, a receptor for HMGB1, was found in exosome samples, as well as SQSTM-1/p62 in WT and p53 KO exosomes. 3.3 Flow cytometry

The positive and negative controls showed a small difference in cell proportions for all markers tested. There was no significant difference between groups. However, the means for TEX-DC coincubation were higher than the means for tumor-DC co-incubation for all markers regardless of the cell type. Figure 3: Flow cytometry data – Data indicates the percent population containing DC markers. Data gated by granularity, Zombie L/D, and CD11c. “WT,” “p53,” “PUMA,” and “BAX” refer to DCs co-incubated with whole tumor cells from the respective cell line. Data is presented as mean±SD. ANOVA was run with Tukey post-test. Orange line shows mean of control group. Tumor cells appear to drastically inhibit expression of these markers compared to negative control. TEX from all samples also inhibit but not with the same magnitude as the tumor cells. Other markers tested did not yield meaningful, significant data.
4. Discussion
The lack of HMGB1 in TEX shows that exosome release is not a pathway HMGB1 takes to signal other cells. This contradicts the results of a study showing that HMGB1 is carried in gastric cancer TEX [8]. However, this and other studies showing the presence of HMGB1 in TEX have used ultracentrifugation to pellet out TEX; this technique is not a reliable method of extracting exosomes due to the potential contamination of soluble protein [9]. This method and the use of exosome isolation kits have this risk, and the soluble form of HMGB1 released by the cells may have produced a false positive for these studies. Use of size-exclusion chromatography has been shown to be the optimal exclusion technique, which elutes a fraction enriched in exosomes with limited soluble protein [9].
According to the flow data, TEX co-incubation inhibited dendritic cell markers compared to the negative control but less so than their respective tumor cells. The data shows that TEX prevent differentiation of monocytes to DCs (indicated by lowered %CD14 expression, compared to negative control), increase DCs’ ability to bind IL-2 (indicated by increased %CD25 expression, compared to negative control), and prevent T cell communication (indicated by decreased %CD80 and %CD86, compared to negative control). However, there was a large variance in the data, and in order to reduce this variance, a larger sample size will be needed. The amount of treated protein for all groups might need to be increased to show a distinct difference between the positive and negative controls and among the four cell lines. If this pattern were to still hold true, this could imply that tumor-derived exosomes do not activate an immune response; if a therapeutic aimed to deliver a certain antigen to dendritic cells, exosomes may provide a safe way of doing so without initiating an adverse immune response.
When using protein estimation to confirm the presence of exosomes and as preparation for the Western blot, the amount of exosomal protein extracted from each cell group (WT, p53 KO, PUMA KO, BAX KO) was similar across groups. This could indicate that there is no connection between apoptotic deficiency and exosome production. However, using a nanoparticle counter to directly count the number of exosomes extracted rather than use a protein estimation would provide direct results as to whether the exosome count differs across groups, making this a future direction to pursue.
The presence of SQSTM-1/p62 is interesting in that it is involved in autophagy, a process in which large molecules are broken down in order for monomers to be recycled for cell processes. SQSTM-1/p62 is known to bind ubiquitinated protein [10]. Additionally, proteins that package exosomal cargo bind to ubiquitinated protein to release them from the cell. Because both pathways tend to target proteins marked with ubiquitin, the presence of SQSTM-1/p62 may show a possible relationship between autophagy and exosome release.
The purpose of RAGE is to act as an embedded membrane receptor expressed on immune cells which causes the cells to release inflammatory cytokines when RAGE activated, though the purpose may change for other cell types [11]. However, soluble
RAGE (sRAGE) may play a role in inhibiting the inflammatory response; a study has shown that sRAGE acts as a decoy receptor in the context of AT1R binding to RAGE (AT1R activated by RAGE is responsible for downstream effects leading to inflammation, oxidative stress, and other effects) [12]. This same idea of sRAGE being a decoy receptor may also occur with TEX; TEX containing sRAGE may be taken up by DCs, and the sRAGE within exosomes may bind to DAMPs (damage associated molecular patterns) absorbed by DCs, which prevents RAGE from binding to DAMPs and thus prevents inflammation from occurring. However, it is unknown whether the RAGE in exosomes are soluble or membrane-bound, making this a possible future direction.
5. Conclusions
SQSTM-1/p62 and RAGE were present in TEX derived from HCT 116 human colorectal cancer cells but HMGB1, PD-L1, and LC3 were not observed. The presence or absence of these molecules provides some insight on pathways used for packaging exosome content. Functional experiments will need to be run to see whether SQSTM-1/p62 and RAGE serve a meaningful purpose in signaling. It will also be worthwhile to test other tumor cell lines or even patient samples to see whether the presence and absence of molecules from the HCT 116 cell line can be generalized to other tumor sources.
TEX are able to suppress DCs but not to the same degree as tumor cells, and we know from the Western blot that this is not due to HMGB1. This is shown by the increase in percent expression of CD14, CD25, CD80, and CD86 from tumor-dendritic cell coincubation to TEX-dendritic cell co-incubation. Tumors suppress the presentation of these molecules when compared to the negative control; TEX show a weaker suppression since most of the average values still remain lower than the control group. Other markers tested did not yield meaningful, significant data. This shows that TEX may be involved in modulating cells of the immune system. Future experiments could involve isolating TEX from other tumor sources or from tumors grown in different culture conditions to see if the produced TEX show similar patterns in modulating dendritic cells. Co-cultures of TEX with other antigen-presenting cells may also be worth considering.
6. Acknowledgements
Funding was provided by University of Pittsburgh Swanson School of Engineering, the DAMP Laboratory at UPMC Hillman Cancer Center, and the Office of the Provost at the University of Pittsburgh. Microscopy resources were provided by University of Pittsburgh Center for Biologic Imaging, funded as Core within the P30 of the Hillman Cancer Center of UPMC.
7. References
[1] R. Kalluri, The biology and function of exosomes in cancer, Journal of Clinical Investigation. 126 (2016). 1208-1215.
[2] J.R. Edgar, Q&A: what are exosomes, exactly?, BMC Biology. 14 (2016). 1-7.
[3] D.W. Greening et al, Exosomes and their roles in immune regulation and cancer, Seminars in Cell & Developmental Biology. 40 (2015). 72-81.
[4] Q Li et al, Exosomes: versatile nano mediators of immune regulation, Cancers. 11 (2019). 1-21.
[5] T.L. Whiteside, The effect of tumor-derived exosomes on immune regulation and cancer immunotherapy, Future Oncology. 13 (2017). 2583-2592.
[6] C.M. Gorgulho et al, Johnny on the spot- chronic inflammation is driven by HMGB1, Frontiers in Immunology. 10 (2019). 1-18.
[7] N. Ludwig et al, Exosomes from HNSCC promote angiogenesis through reprogramming of endothelial cells, Mol Cancer Res. 16 (2018). 1798-1808.
[8] X. Zhang et al, Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration, Molecular Cancer. 17 (2018). 1-16.
[9] N. Ludwig et al, Optimization of cell culture conditions for exosome isolation using mini-size exclusion chromatography (miniSEC), Experimental Cell Research. 378 (2019). 149-157.
[10] S. Pankiv et al, p62/SQSTM1 bids directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy, J. Biological Chemistry. 282 (2007). 24131-24145. [11] K. Kierdorf, G. Fritz, RAGE regulation and signaling in inflammation and beyond, J. Leukocyte Biology. 94 (2013). 55-68. [12] J. Jeong et al, Soluble RAGE attenuates AngII-induced endothelial hyperpermeability by disrupting HMGB1-mediated crosstalk between AT1R and RAGE, Experimental and Molecular Medicine. 51 (2019). 1-15.








