
5 minute read
09. DEMENCIAS – ENFERMEDAD ALZHEIMER


Aspectos generales
Desde un punto de vista académico las demencias pueden clasificarse en 2 grupos.
Cortical: en este tipo de demencias predomina al inicio la clínica de afectación de funciones cerebrales superiores como la afasia, apraxia, agnosia, acalculia. Entre sus ejemplos tenemos a la enfermedad de Alzheimer (90%), demencia alcohólica (10%), demencia frontotemporal de Pick donde predomina los cambios de conducta, agresividad y edad joven de presentación.
(ENAM EXTRA 2021-I) y la enfermedad de Creutzfeldt-Jakob.
Subcortical: donde al inicio predomina clínica subcortical como retardo psicomotor, movimientos anormales, disartria, depresión endógena, parkinsonismo, entre otros. Algunos ejemplos son la enfermedad de Huntington, enfermedad de Parkinson, enfermedad de Wilson, demencia vascular (10%) (ENAM 2014-A), VIH, demencia pugilística o post traumática.
En cuando a la enfermedad de Alzheimer, el 95% de casos son espontáneos sin antecedentes familiares directos, se ha visto implicado la mutación del alelo E4 de APO E (cromosoma 19). Son factores de riesgo la edad mayor a 60 años, ser mujer, TEC previo. Se conocen factores protectores como la presencia del APOE2, uso crónico de AINES, exposición a estrógenos y educación permanente.
El aspecto patogénico característico de la esta enfermedad es la degeneración de las neuronas colinérgicas, sobre todo del hipocampo. Se encuentra en la anatomía patológica los ovillos neurofibrilares por depósito de la proteína tau dentro de los somas neuronales y también se detectan las placas beta amiloide por el depósito anormal de esta proteína en las sinapsis, esta placa activa el sistema inflamatorio que terminan destruyendo la sinapsis. Con el progreso de la enfermedad la degeneración se extiende a toda la corteza.
Diagnóstico
Para establecer el diagnóstico seguimos los criterios de la DSMV.
DSMV -DESORDEN NEUROCOGNITIVO MAYOR (DNM)
Deterioro cognitivo en 1 o más de:
♦ Aprendizaje y memoria.
♦ Lenguaje.
♦ Funciones ejecutivas.
♦ Atención compleja.
♦ Motor-percepción.
♦ Cognición social.
Dependencia en actividades diarias. No hay delirium o trastorno del sensorio. No hay otros trastornos mentales que expliquen el deterioro mental. Adicionalmente, para especificar que el DNM se debe a la enfermedad de Alzheimer se debe cumplir (ENAM 2017-B, ENAM 2015-B):
♦ Inicio gradual del cuadro y deterioro cognitivo continuo.
♦ No hay otra causa que lo explique mejor.
Tratamiento
Atrofia cerebral en la enfermedad de Alzheimer. (A y D: persona sana; C y D: persona con enfermedad de Alzheimer)

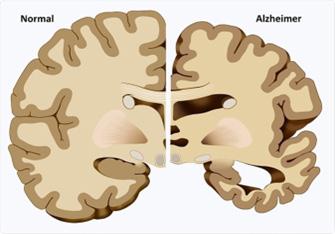
El manejo no altera el curso inexorable de la enfermedad, pero sí puede mejorar la calidad de vida. Se aplica inhibidores de la acetilcolinesterasa de acción central como la rivastigmina, donapezilo y galantamina. Para casos severos se recomienda memantina, un antagonista no competitivo del receptor NMDA.
Recientemente se aprobó el uso de aducanumab como tratamiento de Alzheimer en fases iniciales para pacientes selectos. Se vio que puede retrasar la progresión de la enfermedad.
Aspectos generales
La epilepsia es una enfermedad que afecta al 0.5 a 1% de la población. Se define como la presencia de 2 o más crisis espontáneas sin una causa desencadenante inmediata que las explique (no provocadas). Las epilepsias tienen una edad de presentación, una causa, factores de riesgo, desencadenantes, patrones de electroencefalograma entre otras características que permiten agruparlas como síndromes epilépticos. Así clasificamos estos síndromes epilépticos según su origen en:
Idiopáticos: cuando la causa de origen orgánica no es conocida ni se puede demostrar en imágenes. Sin embargo, se asocian familiarmente a un patrón hereditario de canalopatías y otros trastornos de conducción sináptica.
Sintomáticos: son los síndromes que ocurren con lesión orgánica evidente, la cual explica la epilepsia. Para ello se deben evidenciar las lesiones con neuroimágenes.
Criptogénicos: cuando el origen es muy probablemente orgánico por historia clínica pero aún no se puede demostrar la lesión con las neuroimágenes.
Diagnóstico
Para diagnosticar un síndrome epiléptico se requiere una adecuada historia clínica, un electroencefalograma y una resonancia magnética con contraste.
Una vez confirmado la epilepsia, el cuadro clínico y el patrón electroencefalográfico podemos clasificar los síndromes de forma más detallada según el siguiente cuadro de la Liga Internacional de lucha contra la Epilepsia (ILAE).
Las crisis de ausencia se caracterizan por presentar pérdida del sensorio sin alteraciones motoras. Ocurren en niños menores de 10 años y los episodios se repiten varias veces durante el día .
Clasificación de los síndromes epilépticos (ILAE 2010) - RESUMEN
Periodo neonatal
– Crisis neonatales benignas
– Epilepsia familiar neonatal benigna
– Síndrome de Ohtara
Lactancia
Crisis febriles/crisis febriles plus
– Epilepsia benigna de la infancia (de la lactancia)
– Epilepsia benigna familiar de la infancia
– Síndrome de West
Síndrome de Dravet
Epilepsia mioclónica de la infancia (de la lactancia
Infancia
– Crisis febriles/crisis febriles plus
– Epilepsia occipital de la infancia de inicio temprano (Síndrome de Panylotopoulos)
– Epilepsia con crisis mioclónicas atónicas (previamente astáticas)
Epilepsia en ausencia infantil
Epilepsia benigna con puntas centrotemporales
– Síndrome de Lennox-Gastaut
Adolescencia/edad adulta
– Epilepsia en ausencia juvenil
Epilepsia mioclónica juvenil
Epilepsia con crisis generalizadas tónico-clónicas solamente
– Epilepsia autosómico dominante con características auditivas
Edad de inicio variable
– Epilepsia focal familiar con focos variables
Epilepsia mioclónica progresiva
Constelaciones específicas/síndromes quirúrgicos
– Epilepsia temporal mesial con esclerosis del hipocampo
– Síndrome de Asmussen
– Crisis golásticas con hemartoma hipotalámica
– Epilepsia con hemiconvulsión-hemiplejia
Constelaciones específicas/síndromes quirúrgicos
Epilepsia temporal mesial con esclerosis del hipocampo
– Síndrome de Rasmussen
Epilepsias no sindrómicas
– Epilepsias atribuidas a causas estructurales-metabólicas y organizadas de acuerdo con ellas
• Malformaciones de desarrollo cortical (hemimegalencefalia, heterotópicas, etc.)
• Síndromes neuro cutáneos (complejo esclerosis tuberosa, Stuger Weber, etc.)
• Tumor, infección, trauma, angioma, lesiones prenatales y perinatales, accidente cerebrovascular, etc.
– Epilepsia de causa desconocida
F Rmaco Antiepil Tico
Valproato
Nauseas, vomitos, temblor, peso, alopecia.
Fenitoina Nistagmo, hipertrofia gingival, hipertricosis, rasgos faciales.
Carbamazepina Diplopía, hiponatrémia, neutropenia, rash
Fenobarbital (Inductor GABA) (ENAM 2013-A) Sedación, impotencia.
Etosuccimida
Tratamiento
Nauseas, vomitos, cefalea, sedación, agitación.
Hepatotoxicidad, pancreatitis, “penias”.
Discrasias, S. Johnson, hepatoxicidad, polineuropatías, ataxia, LES.
Agranulocitosis, anemia aplásica , S. Johnson, ataxia
Necrólisis epidermolítica tóxica, hepatotoxicidad, distrofia escapulohumeral.
Eritema multiforme, S. Johnson.
Recuerda
El efecto adverso mas frecuente del uso de carbamazepina es diplopía, hiponatrémia, neutropenia, rash; y los efectos adversos más raros son: agranulocitosis, anemia aplásica, S. Johnson, ataxia.
El manejo crónico de la epilepsia debe procurar administrar la dosis más baja de solo un fármaco antiepiléptico (FAE), es decir se prefiere la monoterapia siempre. Todo cambio o supresión de FAE debe hacerse de forma gradual. Recomendar siempre evitar el alcohol y mantener el sueño nocturno adecuado. (ENAM 2016-A)
En general los síndromes de epilepsia generalizada idiopática se tratan con: acido valproico, lamotrigina, topiramato o levetiracetam. De todos ellos el valproato es el más usado. Un caso especial es la crisis generalizada de ausencia, la cual puede tratarse con etosuximida que es un fármaco de espectro reducido (bloquea solo el canal de calcio) pero es eficaz en niños con crisis típicas, para crisis atípicas se debe usar valproato.
Para los síndromes epilépticos que cursan con crisis focales, los fármacos principales son: carbamazepina (ENAM 2012-B) y oxcarbacepina, fenitoína, o gabapentina.
Nomenclatura abreviada de fármacos antiepilépticos (FAE):
CBZ: Carbamazepina
VPA: Valproato
ETX: Etosuximida
LTG: Lamotrigina
LEV: Levetiracetam
TPR: Topiramato
PHT: Fenitoína
PB: Fenobarbital
GBP: Gabapentina
Clasificaci N De Fae Seg N Espectro
Amplio espectro: Clobazam - Felbamato –Lamotrigina –LevetiracetamPerampanel -Rufinamida – Topiramato – Valproato – Zonisamida
Espectro reducido: Carbamazepina – Gabapentina - Lacosamida –Oxcarbazepina – Fenobarbital – Fenitoína – Pregabalina – Tiagabina –Vigabatrina – Etosuximida (ausencia)
Aproximadamente un 20-30 % de los pacientes con epilepsia son resistentes al tratamiento médico y se procede a la cirugía. Seccionar el cuerpo calloso se ha mostrado eficaz en las convulsiones tónicas clónicas intratables.
Recuerda
Las crisis generalizada de ausencia, puede tratarse con etosuximida que es un fármaco de espectro reducido (bloquea solo el canal de calcio) pero es eficaz en niños con crisis típicas.






