Die MR-Angiographie im Kontext konkurrierender Verfahren*
Thomas Störk1, 2, Ragnar Gareis3, Knut Kröger4, Thomas J. Vogl5
1 CardioPraxis Staufen, Göppingen
2 Kardiologie/Angiologie, Universitätsklinik Ulm
3 Cardiologicum Stuttgart
4 Helios Klinikum, Krefeld
5 Radiologie, Universitätsklinikum Frankfurt/Main
PERFUSION 2023; 36: 4 – 9
Die MR-Angiographie (MRA) nutzt magnetische Effekte zur Bildgebung. Sie wird mittlerweile in allen Körperregionen angewandt und profitiert von der fehlenden Strahlenexposition. Mit diesem Verfahren werden dreidimensionale Bilder erzeugt, wobei auch eine Beurteilung des umgebenden Parenchyms möglich ist. Im Wesentlichen kommen 3 verschiedene MRA-Verfahren zur Anwendung (Tab. 1). Die gute Kontrastauflösung sowie die gute räumliche und zeitliche Auflösung machen die MRA neben der Duplexsonographie und der CT-Angiographie (CTA) zu einem festen Bestandteil der Gefäßdiagnostik.
Abgrenzungen zu konkurrierenden bildgebenden
Verfahren
Farbkodierte Duplexsonographie (FKDS): Die FKDS ist preiswert, schnell verfügbar und (prinzipiell) gut wiederholbar. Das kann und wird die MRA in nächster Zeit nicht bieten können.
* Für die Sektion MR-Angiographie der Deutschen Gesellschaft für Angiologie
Zusammenfassung
Die MR-Angiographie (MRA) nimmt in der klinischen Gefäßdiagnostik einen zentralen Platz ein. Weitere technische Entwicklungen werden das Einsatzgebiet der MRA erweitern. Gleichwohl sind die hohen Anschaffungskosten und die Komplexität der Durchführung zu bedenken. So werden vorgeschaltete kleinere Verfahren, aber auch die farbkodierte Duplexsonographie (FKDS), ihren Stellenwert behalten. Gleiches gilt für die CT-Angiographie (CTA), die in einigen Anwendungen bereits zur Erstdiagnostik eingesetzt wird.
Schlüsselwörter: Angiologie, MR-Angiographie, CT-Angiographie, farbkodierte Duplexsonographie
Summary
MR angiography (MRA) is nowadays a critical tool in the diagnosis of vascular diseases. New techniques will continue to increase the value of MRA. However, the high acquisition costs and the complex procedure have to be taken into account. Bedside techniques, especially color-coded duplex sonography (CCD), will therefore remain highly important. And, last but not least, CT angiography (CTA) emphasizes its importance in vascular diagnostics.
Keywords: angiology, MR angiography, CT angiography, color-coded duplex sonography
Invasive Angiographie (Angio, DSA): Mit der konventionellen Angiographie als bewährtes Verfahren der Gefäßdiagnostik lassen
sich Verdachtsfälle auf pathologische Gefäßveränderungen wie z.B. Stenosen, Angiome oder Aneurysmen zuverlässig und detailliert
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
T. Störk, R. Gareis, K. Kröger, T. J. Vogl: Die MR-Angiographie im Kontext konkurrierender Verfahren
4
ORIGINALARBEIT
Verfahren Prinzip Darstellung
Time of Flight-MRA (TOF)
Phasenkontrast-MRA (PC-MRA)
Kontrastmittelverstärkte MRA (CE-MRA)
abklären. Bei der Durchführung einer Angiographie stehen nach heutigem Stand neben der konventionellen Angiographie auch die Techniken der digitalen Subtraktionsangiographie (DSA) zur Verfügung. Die DSA ist ein invasives Verfahren, das im Vergleich zu den nichtinvasiven Techniken mit einem höheren Risiko behaftet ist, weshalb aktuell vermehrt nichtinvasive Modalitäten zum Einsatz kommen.
CT-Angiographie (CTA): Die CTA ist eine Untersuchungstechnik mit hoher räumlicher, aber begrenzter zeitlicher Auflösung. Auch wenn eine Applikation von i.d.R. Jod-haltigem Kontrastmittel (KM) notwendig ist und eine gewisse Strahlenexposition vorliegt, bietet die CTA die Möglichkeit der gleichzeitigen Erfassung von Verkalkungen des Gefäßes, der Beurteilung der Wandstruktur und der genauen Bestimmung der Lumenweite.
Darstellung hirnversorgender Gefäße
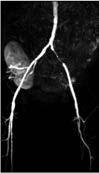
Extrakraniell: Das Verfahren der ersten Wahl ist sicher die FKDS (Abb. 1a–c). Als weitere Verfahren kommen dann die MRA (Abb. 2a, b) und gelegentlich auch die CTA zum Einsatz.
Frisch einströmendes Blut weist eine höhere Magnetisierung auf
Darstellung von Phasenunterschieden in aufeinanderfolgenden Bildern
Verwendung von T1-verkürzenden Kontrastmitteln, mit deren Hilfe in T1-gewichteten Aufnahmen die Gefäße signalreicher dargestellt werden
Frisch einströmendes Blut wird signalreich dargestellt
Bewegungen von fließendem Blut, analog zur Duplexsonographie
Standardverfahren für (fast) alle Anwendungen
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
5
T. Störk, R. Gareis, K. Kröger, T. J. Vogl: Die MR-Angiographie im Kontext konkurrierender Verfahren
Tabelle 1: Verfahren der MR-Angiographie.
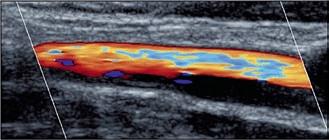
Abbildung 1a–c: Darstellung verschiedener hirnversorgender Gefäße mittels farbkodierter Duplexsonographie (FKDS) (a: A. temporalis, b: A. carotis) sowie mittels Power Mode (c: A. carotis) bei Riesenzellarteriitis.
Abb. 1a
Abb. 1b
Abb. 1c
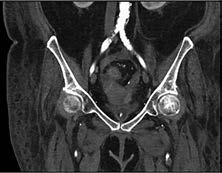
Intrakraniell: Die intrakranielle Gefäßdarstellung ist ein wichtiges diagnostisches Tool in der Therapieplanung und der Behandlung von Gefäßverschlüssen oder Gefäßmissbildungen (Aneurysmen, Angiome). Aktuell stützt sich die Bewertung von intrakraniellen atherosklerotisch-arteriellen Stenosen auf Luminalmessungen mithilfe von DSA, CTA und MRA. Gelegentlich kommt auch der transkranielle Ultraschall (TCD) zum Einsatz. Hierbei stellt die DSA weiterhin den Goldstandard dar. Die MRA wird aufgrund ihrer geringeren Invasivität jedoch wesentlich häufiger eingesetzt. Dabei ist die Timeof-Flight-MRA (TOF-MRA) die am häufigsten verwendete MRATechnik (Abb. 3a), insbesondere zum Ausschluss von Aneurysmen. Bei der intrakraniellen Stenosediagnostik mittels MRA kann infolge von Dephasierungsartefakten der Stenosegrad fälschlicherweise zu hoch eingeschätzt werden. Wegen dieser Einschränkungen hat die TOF-MRA als primäre neurovaskuläre Bildgebungsmodalität an Bedeutung verloren. Außerdem ist ihre räumliche Auflösung im Vergleich zu den anderen MRA-Techniken geringer, sodass die CTA bei der Beurteilung kleinerer Arterienabschnitte der TOF-MRA überlegen ist (Abb. 3b).
Zwar ist der Patient bei der CTA ionisierender Strahlung ausgesetzt und muss sich zudem einer intravenösen Kontrastmittelapplikation unterziehen, jedoch definiert die CTA die anatomische Konfiguration und die Beziehung der Pathologie zur Umgebung besser. Verschiedene Studien in
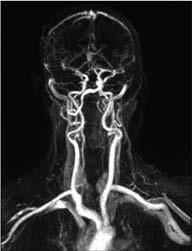
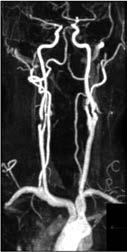
Abb. 2a
Abb. 2b
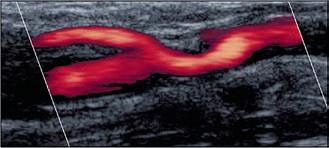
Abbildung 2a, b: Kontrastverstärkte MR-Angiographie (CE-MRA) der hirnversorgenden Gefäße.
a: Nachweis einer signifikanten ACI-Stenose rechts.
b: Ausschluss einer Dissektion der A. vertebralis bei C2-Fraktur.
Abb. 3a
Abb. 3b
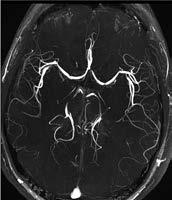
Abbildung 3a, b: Intrakranielle Gefäßdarstellung.
a: Mittels Time-of-Flight-MRA (TOF-MRA) erhobener Normalbefund (Arterien sind hell dargestellt).
b: In der CT-Angiographie zeigt sich ein Verschluss der A. cerebri media rechts.
der Vergangenheit haben jedoch gezeigt, dass es keinen signifikanten Unterschied hinsichtlich der
Genauigkeit der Gefäßdarstellung zwischen CTA und MTA gibt (Literatur beim Verfasser).
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
6
T. Störk, R. Gareis, K. Kröger, T. J. Vogl: Die MR-Angiographie im Kontext konkurrierender Verfahren
Kardiale Bildgebung
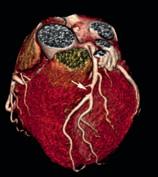
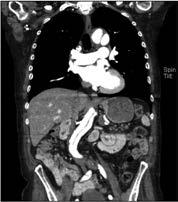
Kardio-CT: Auch im Bereich der kardialen Gefäßdarstellung ist nach wie vor die invasive Koronarangiographie in der Diagnose der koronaren Herzkrankheit der aktuelle Goldstandard. Jedoch hat die koronare CTA aufgrund ihrer hohen zeitlichen und räumlichen Auflösung einen wachsenden Stellenwert bezüglich der Diagnostik einer bestehenden koronaren Herzerkrankung, aber auch hinsichtlich eines sicheren Ausschlusses einer bestehenden revaskularisationsbedürftigen Stenose. Zudem vermeidet die CTA die mit einem invasiven Verfahren verbundenen Risiken und bietet darüber hinaus eine schnellere und möglicherweise kostengünstigere Möglichkeit zur Beurteilung von Patienten mit mittlerem Risiko für eine koronare Herzerkrankung. Die nichtinvasive anatomische Visualisierung mittels koronarer CTA ermöglicht eine direkte Darstellung des Ausmaßes und der Lage von Koronararteriosklerose und Koronarstenosen (Abb. 4).
Des Weiteren bietet die kontrastfreie CTA die Möglichkeit zur Berechnung eines Koronararterienkalk-Scores, wobei es sich um ein flächendeckend verfügbares, konsistentes und reproduzierbares Verfahren zur Bewertung des Risikos schwerer kardiovaskulärer Folgen handelt, das insbesondere bei asymptomatischen Personen für die Planung von Primärpräventionsmaßnahmen angewendet wird. Moderne CT-Untersuchungen (MultiDetektor CT, MDCT) können mit einer Gesamtdauer von 10 – 15 Minuten bei einer Strahlenbelastung von etwa 1 mSv durchgeführt werden.
Die CTA kann auch zur Diagnose von Koronaranomalien eingesetzt werden. Wie bei der koronaren Herzkrankheit war die invasive Koronarangiographie bisher auch hier der diagnostische Goldstandard. In den Leitlinien des American College of Cardiology (ACC) und der American Heart Association (AHA) wird die koronare CTA als Klasse-I-Indikation für das Erstscreening erwachsener Patienten mit Verdacht auf angeborene anomale Koronararterien aufgeführt. Im Rahmen der technischen Entwicklung wird die Anwendung der CTA auch für den Bereich der Perfusion und fraktionierten Flussreserve vorbereitet.
MR-Angiographie des Herzens: Die MRA hat im Vergleich zur CTA eine geringere räumliche Auflösung, und auch hier kann es aufgrund der bereits beschriebenen Flussdephasierungsartefakte zu einer falsch-negativen Einschätzung von Stenosen kommen, was die Beurteilbarkeit von Koronarstenosen einschränkt und die CTA als überlegende diagnostische Modali-
tät stehen lässt. Laut Expertenmeinungen, die durch klinische Leitlinien unterstützt werden, wird vom Einsatz der MRA zur Beurteilung von kardialen Stenosen abgeraten. Jedoch zeigt die MRA hinsichtlich der Beurteilung und Diagnostik von Koronaranomalien und Koronaraneurysmen eine hohe Sensitivität und Spezifität. Die MRA ist zudem in Bezug auf die Darstellung und Erfassung von funktionellen Parametern der CTA überlegen. Daher wird die MRA zunehmend zur Beurteilung von stressinduzierten Ischämien und der Myokardvitalität eingesetzt.
Darstellung der Aorta
Farbkodierte Duplexsonographie: Die FKDS steht als Bedside-Verfahren zur Verfügung und ist sicher die Methode der ersten Wahl. Zentral ist sie im präventiven Bereich, im Screening des Bauchaortenaneurysmas und in der Nachsorge nach Operation oder Intervention der Aorta.
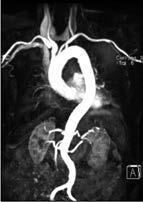
MR-Angiographie: Die MRA der Aorta bietet eine hervorragende Übersicht (Abb. 5a) und ist hier der FKDS überlegen. Die fehlende ionisierende Strahlung macht die Methode attraktiv auch gegenüber der CTA.
CT-Angiographie: Die CTA ist in der Regel das bildgebende Verfahren der Wahl, wenn der dringliche Verdacht auf eine akute Aortendissektion besteht (Abb. 5b). Sie ist mit einer Sensitivität und Spezifität von nahezu 100 % sehr genau. Zudem ist sie in den meisten medizinischen Einrichtungen
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
7
T. Störk, R. Gareis, K. Kröger, T. J. Vogl: Die MR-Angiographie im Kontext konkurrierender Verfahren
Abbildung 4: Mittels CT-Angiographie des Herzens nachgewiesene Stenose des Ramus interventricularis anterior (Pfeil).
leicht zugänglich sowie sicher und schnell durchführbar. Auch bei der Therapieplanung von Stentgrafts in der Aorta (endovascular aortic repair, EVAR) sowie zur Beurteilung des Zugangsweges für einen Aortenklappenersatz – TranskatheterAortenklappenimplantation (TAVI) oder chirurgischer Aortenklappenersatz (AKE) – wird sie regelhaft eingesetzt.
Periphere Gefäßdarstellung (pAVK-Diagnostik)
Kleine Verfahren und Ultraschall: Die gebräuchlichsten Verfahren für die Diagnostik einer peripheren arteriellen Verschlusskrankheit (pAVK) sind weiterhin Klinik, Pulsstatus, Knöchel-ArmIndex (ABI), Oszillographie sowie, nicht zuletzt aufgrund ihrer guten Verfügbarkeit, die farbkodierte Duplexsonographie (FKDS).
MR- und CT-Angiographie: In den letzten Jahren hat sich die nichtinvasive Bildgebung mittels MRA und CTA auch im Bereich der Darstellung der peripheren Arterien zu einer äußerst zuverlässigen Alternative zur DSA entwickelt. Die CTA und MRA sind beides hochpräzise Methoden, die eine genaue, detailgetreue Darstellung der arteriellen Anatomie, der artherosklerotischen Plaques und der Verengung des peripheren Gefäßsystems von der Aorta bis hinunter zu den Füßen ermöglichen (Abb. 6a, b). Mit beiden Verfahren lässt sich eine pAVK nicht nur zuverlässig darstellen, auch der Stenosegrad kann bestimmt werden. Beide Verfahren können zur Behandlung des gesamten Spektrums von Patienten mit
Abb. 5a Abb. 5b
Abbildung 5a, b: Darstellung der Aorta.
a: Mit MR-Angiographie erhobene Übersicht.
b: CTA-Darstellung einer langstreckigen Aortendissektion Typ A.
Abb. 6a
Abb. 6b
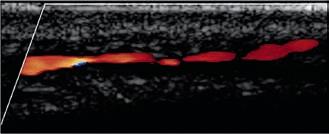
Abbildung 6a, b: Darstellung peripherer Arterien.
a: Die MR-Angiographie der distalen Aorta bis zum Femoralbereich zeigt einen Verschluss der A. femoralis superficialis (AFS) links sowie einen iliakalen Stent links.
b: CT-Angiographie der Bifurkation der Aorta abdominalis.
vermuteter oder bekannter pAVK eingesetzt werden. Hinsichtlich der diagnostischen Genauigkeit und dem Nachweis sowie der Einstufung einer peripheren Arterienerkrankung ähneln sich die MRA und CTA in Sensitivität und Spezifität, die bei beiden 90 – 100 % betragen. Die am häufigsten verwendete
MRA-Methode ist aktuell die kontrastmittelverstärkte MRA, aber auch das nicht kontrastmittelverstärkte Verfahren wird zunehmend eingesetzt, da hierbei kein Kontrastmittel appliziert werden muss.
Multidetektor-CTA: Die MDCTA hat aufgrund ihres größeren volu-
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
8
T. Störk, R. Gareis, K. Kröger, T. J. Vogl: Die MR-Angiographie im Kontext konkurrierender Verfahren
metrischen Erfassungsbereichs, der schnelleren Aufnahmegeschwindigkeit und der höheren räumlichen Auflösung die vaskuläre Bildgebung überholt und ist aufgrund der isotropen Bildgebung im Submillimeterbereich mit dem Goldstandard der DSA vergleichbar.
Venendarstellung
Klinische Standardverfahren:
Die Venendiagnostik beruht apparativ im Wesentlichen auf der Sonographie (farbkodierte Duplexsonographie, KompressionsUltraschall), aber auch auf kleineren Verfahren wie z.B. der Licht-Reflexions-Rheographie (LRR).
MR-Angiographie: Die MRA hat intrakraniell einen hohen Stellenwert, insbesondere bei der Sinusvenenthrombose. Außerdem können damit Basilaris-Thrombosen und auch Karotis-Dissektionen hervorragend dargestellt werden. Eine zentrale Rolle spielt die MRA bei der Beurteilung der intrathorakalen Venen, z.B. der Pulmonalvenen vor Pulmonalvenenisolation, aber auch von fehleinmündenden Lungenvenen. In der Peripherie kommt die MRA zum Einsatz, wenn die Ultraschalluntersuchung nicht zielführend ist oder die CTA aufgrund der Strahlenexposition schwierig zu indizieren (Schwangere, Kinder).
MITTEILUNGEN „Stumme
Hirninfarkte“: Unbemerkt, aber nicht harmlos
Sprach- und Sehstörungen, Lähmungserscheinungen – die Folgen eines Schlaganfalls sind häufig gravierend. Der Hirninfarkt ist in der Regel ein lebensveränderndes, einschneidendes Erlebnis, kann aber auch unbemerkt verlaufen. Solche „stummen Ereignisse“ sind dennoch nicht ungefährlich. Nach mehreren Ereignissen dieser Art kann die Gedächtnisleistung des Betroffenen stark leiden. Zudem steigt nach einem stummen Hirninfarkt das Risiko erheblich, erneut eine Durchblutungsstörung zu erleiden. Experten der Deutschen Schlaganfall-Gesellschaft (DSG) machen darauf aufmerksam, dass solche Ereignisse ernst genommen und professionell behandelt werden müssen.
Lähmungen und Gedächtnisverlust als mögliche Folgen
anfallsymptome wahrgenommen. Manchmal spüren die Betroffenen auch gar keine Symptome, vor allem wenn diese nur kurzzeitig auftreten oder während des Schlafes.“ Häufig werden stumme Hirninfarkte erst als Zufallsbefund bei einer Computertomographie oder bei einer Magnetresonanztomographie des Kopfes entdeckt.
Trotzdem ist so ein stummer Schlaganfall gefährlich, denn auch dieser erhöht das Risiko für einen weiteren Hirnschlag. „Stumme Hirninfarkte sind gar nicht so selten“, warnt der 1. Vorsitzende der DSG, Professor Darius Nabavi. Neben dem Alter gelten vor allem Bluthochdruck und Vorhofflimmern, aber auch Rauchen, ungesunde Ernährung, mangelnde Bewegung, Übergewicht, Diabetes und erhöhte Cholesterinwerte als Risikofaktoren.
Für die Verfasser:
Prof. Dr. med. Thomas Störk
CardioPraxis Staufen
Friedrichstraße 36
73033 Göppingen
E-Mail:
thomas.stoerk@cardiopraxis-staufen.de
„Wenn das Sprachzentrum oder das Areal im Gehirn, das für die Bewegung verantwortlich ist, von einem Schlaganfall betroffen sind, kommt es häufig zu massiven Folgen wie Sprach- oder Lähmungserscheinungen“, erläutert Professor Schäbitz, Pressesprecher der DSG. „Trifft ein Hirninfarkt aber einen unauffälligeren Bereich im Gehirn, dann können die Symptome viel unspezifischer sein. Dazu gehören etwa diffuser Schwindel, KribbelMissempfindungen und Koordinationsstörungen. Häufig werden diese Beschwerden gar nicht als Schlag-
„Gerade das Vorhofflimmern ist ein ernster Risikofaktor für einen Schlaganfall, weil hierbei leicht kleine Blutgerinnsel entstehen, die dann im Gehirn einen Schlaganfall auslösen können. Hier ist ein verlängertes Rhythmusmonitoring notwendig, auch wenn keine verdächtigen Beschwerden vorliegen“, rät Nabavi.
Studien zeigen auch, dass nach stummen Schlaganfällen die intellektuellen Leistungen des Betroffenen abnehmen. „Es kann bei wiederholten Schlaganfallereignissen im schlimmsten Falle zur sogenannten vaskulären Demenz kommen“, erklärt Schäbitz. „Sie hat andere Ursachen als die Alzheimer-Demenz, aber auch in diesem Fall kommt es zu Konzentrationsschwierigkeiten und anderen verminderten kognitiven Leistungen.“ DSG
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH Mitteilungen 9
Knapp 14 Millionen Erwachsene sind in Deutschland von der chronischen Erkrankung Adipositas betroffen [1]. Sie ist mit etwa 200 Komorbiditäten und Komplikationen assoziiert [2], die von Typ-2-Diabetes und Herzerkrankungen bis hin zum obstruktiven SchlafapnoeSyndrom und bestimmten Krebsarten reichen [3]. Zusätzlich zu Ernährungsumstellung, begleitenden Bewegungsprogrammen und Verhaltenstherapie (Basistherapie) werden auch bariatrische und medikamentöse Behandlungen eingesetzt, um eine Gewichtsabnahme bei Menschen mit Adipositas zu erreichen [4, 5].
Eine etablierte medikamentöse Therapieoption ist das GLP-1-Analogon Liraglutid (Saxenda®). Das Inkretin-Mimetikum ist effektiv in der Langzeitbehandlung der Adipositas und hat ein gut dokumentiertes Sicherheitsprofil. So erreichten Early Responder* unter Liraglutid (3 mg) nach einem Jahr Therapiedauer einen Gewichtsverlust von 11,2 % [6].
Aus für Amfepramon-haltige Arzneimittel
Amfepramon-haltige Arzneimittel, die zur kurzfristigen, unterstützenden Adipositasbehandlung eingesetzt wurden, sind nicht mehr auf dem EU-Markt erhältlich. Dies gaben die Herstellerunternehmen zusammen mit der Europäischen Arzneimittelagentur (EMA) und dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) kürzlich bekannt [7]. Mit dem entsprechenden Rote-Hand-Brief ist der EMA-Reviewprozess nun abgeschlossen, indem zuvor Bedenken hinsichtlich Sicherheit und Nutzen-Risiko-Profil des Wirkstoffs überprüft worden waren. Die Behörde hatte sich nach eingehender Prüfung dafür ausgesprochen, die EU-Zulassung für Amfepramon-haltige Arzneimittel zu widerrufen [8]. Eine Unterversorgung von Menschen mit Adipositas in Deutschland droht dank guter Alternativen dennoch nicht.
Wirksame Medikamente mit etabliertem Sicherheitsprofil für die langfristige Adipositastherapie
Adipositas bedarf als chronische Erkrankung einer langfristigen Behandlung und lässt sich keinesfalls
in wenigen Monaten effektiv in den Griff bekommen. Schon allein deshalb können Amfepramon-haltige Medikamente, die laut ihrem Zulassungstext nicht länger als 3 Monate angewendet werden dürfen, in der nachhaltigen Adipositasbehandlung keine Rolle spielen. In Deutschland stehen somit zur Behandlung der allgemeinen Adipositas nun noch der Lipasehemmer Orlistat und das GLP-1-Analogon Liraglutid zur Verfügung [9]. Doch was bedeutet das für die Versorgungssituation hierzulande?
Das Ende der Amfepramon-Zulassung ist für die Versorgungslage von Menschen mit Adipositas kein Grund zur Besorgnis. Ärzte können Patienten, die bislang mit Amfepramon behandelt wurden, auf geeignete alternative Behandlungsmöglichkeiten zur Adipositastherapie aufmerksam machen. Liraglutid bietet ein gut dokumentiertes Sicherheitsprofil und ermöglicht eine effektive Langzeitbehandlung der chronischen Erkrankung [6, 10].
Auf dem deutschen Markt ist es der einzige zur Gewichtsabnahme zugelassene GLP-1-Rezeptoragonist, der als langfristige Ergänzung zu einer kalorienreduzierten Ernährung und verstärkter körperlicher Aktivität verfügbar ist [9].
In der Zulassungsstudie SCALE™
Obesity and Prediabetes [10] erreichten die Teilnehmer nach 56
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 10 FORUM ADIPOSITAS
GLP-1-Rezeptoragonist Liraglutid ermöglicht eine langfristige und effektive Adipositastherapie
* Early Responder: Patienten, die in den ersten 12 Wochen mit der 3,0-mg-Erhaltungsdosis eine Gewichtsabnahme ≥5 % erzielten.
Wochen eine durchschnittliche Gewichtsabnahme von 8,0 % gegenüber 2,6 % unter Placebo (p < 0,0001). Etwa ein Drittel der Patienten (33,1 %) nahm mehr als 10 % des Ausgangsgewichts ab, in der Placebo-Gruppe waren es 10,6 % [10].
Darüber hinaus zeigt Liraglutid vorteilhafte pleiotrope Effekte, die in einer Studie mit Typ-2-Diabetikern mit hohem kardiovaskulärem Risiko mit einer Verbesserung des kardiovaskulären Risikoprofils und einer Senkung der kardiovaskulären Mortalität einhergingen [11].
Außerdem hat Liraglutid in der Anwendung für die unterstützende Therapie der Adipositas ein gut untersuchtes Sicherheitsprofil, belegt in einem umfangreichen Studienprogramm mit über 5.000 Patienten [11].
Medikamentöse Adipositastherapie in Deutschland –Past, Present & Future
Bei der Behandlung chronischer Krankheiten gibt es immer wieder Medikamente, die vom Markt genommen werden. Dies war und ist auch bei der Adipositastherapie der Fall. Erst in den letzten 2 Jahrzehnten ist das Verständnis der molekularen Mechanismen, die den Appetit steuern, so weit fortgeschritten, dass die Entwicklung von gezielt wirkenden Arzneimitteln sinnvoll
betrieben und echte Erfolge erzielt werden konnten. Die klinische Forschung führte schließlich 2014 zur Zulassung von Saxenda® (Liraglutid 3 mg) in den USA, dem damals ersten GLP-1-Rezeptoragonisten zur Behandlung von Adipositas. In Deutschland ist er seit 2016 erhältlich. Der Wirkstoff bindet an spezifische Rezeptoren in Gehirn, Pankreas und Magen-Darm-Trakt, was eine Reduktion des Hungergefühls sowie eine Steigerung des Sättigungs- und Völlegefühls bedingt. Dies führt zu einem verminderten Wunsch nach Nahrungsverzehr und letztlich zu einer geringeren Nahrungsaufnahme [12].
Mit seiner Bereitstellung und weiterer Forschungstätigkeit zu noch wirksameren Medikamenten in der Zukunft unterstreicht die Novo Nordisk Pharma GmbH ihren Anspruch, die Partnerin in der Adipositastherapie zu sein. Das Unternehmen unterstützt außerdemdem die Patienten mit Webinaren und dem umfangreichen Onlineangebot auf „mein-weg-zum-wunschgewicht.de“. Für die Ärzte stehen z.B. CME-zertifizierte Schulungen unter www.novoakademie.de und das Onlineportal www.rethink-obesity.de bereit. Novo Nordisk engagiert sich mit dieser und weiteren Maßnahmen dafür, eine nachhaltige Infrastruktur zur Behandlung von Menschen mit Adipositas in Deutschland aufzubauen.
Fabian Sandner, Nürnberg
Literatur
1 Statista GmbH. de.statista.com/statistik/ daten/studie/760961/umfrage/anzahl-erwachsene-in-deutschland-nach-bmi2014-und-2025
2 Yuen M et al. A systematic review and evaluation of current evidence reveals 195 Obesity-Associated Disorders (OBAD). The Obesity Society. 2016 Abstract Book:92
3 WHO 2020. Obesity and overweight United Nations. www.who.int/newsroom/fact-sheets/detail/obesity-andoverweight#:~:text=39%25%20of%20 adults%20aged%2018,overweight%20 or%20obese%20in%202020
4 DAG. Interdisziplinäre Leitlinie der Qualität S3 zur „Prävention und Therapie der Adipositas“. www.awmf.org/ uploads/tx_szleitlinien/050-001l_S3_ Adipositas_Pr%C3%A4vention_ Therapie_2014-11-abgelaufen.pdf
5 Engeli S. Ausblick auf neue Arzneimittel zur Gewichtsreduktion. Adipositas. 2022;16:7-11
6 Fujioka K et al. Early weight loss with liraglutide 3.0 mg predicts 1-year weight loss and is associated with improvements in clinical markers. Obesity 2016;24:2278-2288
7 Rote-Hand-Brief zu amfepramonhaltigen Arzneimitteln: Verzicht und Widerruf der Zulassungen. www.bfarm.de/ SharedDocs/Risikoinformationen/Pharmakovigilanz/DE/RHB/2023/rhb-amfepramon.html
8 EMA recommends withdrawal of marketing authorisation for amfepramone medicines. www.ema.europa.eu/en/ news/ema-recommends-withdrawalmarketing-authorisation-amfepramonemedicines
9 Engeli S. Ausblick auf neue Arzneimittel zur Gewichtsreduktion. Adipositas 2022;16:7-11
10 Pi-Sunyer X et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015;373:11-22
11 Marso SP et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311-322
12 Fachinformation Saxenda® (aktueller Stand)
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 11 FORUM ADIPOSITAS
Vorhofflimmern (VHF) ist mit einer Prävalenz von 2 – 4 % der Erwachsenen die häufigste anhaltende Herzrhythmusstörung und geht mit einem 5-fach erhöhten Schlaganfallrisiko und einer erhöhten Mortalität einher [1]. Allerdings sind die Symptome oft unspezifisch oder das VHF tritt asymptomatisch auf. Die Folge: Das VHF bleibt bei vielen Betroffenen unentdeckt und sie erhalten keine effektive Schlaganfallprophylaxe. In diesen Fällen wird das VHF meist erst dann diagnostiziert, wenn ein Ereignis eingetreten ist: 50 – 87 % der Betroffenen weisen zu Beginn keine Symptome auf und bei 10 % aller ischämischen Schlaganfälle wird ein zuvor undiagnostiziertes VHF entdeckt [1]. Ein wichtiger VHF-Risikofaktor ist das Alter. Aufgrund des demografischen Wandels ist daher eine Zunahme der VHF-Fälle zu erwarten. Bei den über 65-Jährigen in der EU wird ein Anstieg von etwa 8 Millionen Fällen im Jahr 2020 auf etwa 14 Millionen Fälle im Jahr 2060 prognostiziert [1]. Um möglichst viele undetektierte VHF-Betroffene rechtzeitig diagnostizieren und prophylaktisch behandeln zu können, sollte vor allem in dieser Altersgruppe ein Screening auf ein behandlungsrelevantes VHF erfol-
erreichen
ety of Cardiology (ESC) und der European Atherosclerosis Society (EAS). Bei den Empfehlungen von ESC und EAS determiniert das absolute kardiovaskuläre Risiko die Behandlungsziele: Ein LDLC-Wert <55 mg/dl (<1,4 mmol/l) sowie eine Senkung des LDL-CAusgangswertes um ≥50 % wird für Menschen mit einem sehr hohen kardiovaskulären Risiko empfohlen – beispielsweise Patienten mit einer schweren Nierenfunktionsstörung (eGFR [geschätzte glomeruläre Filtrationsrate] <30 ml/min/1,73 m2) [8].
Werden die Zielwerte trotz Lebensstilmaßnahmen, einer maximal tolerierten Statintherapie und Ezetimib nicht erreicht, sieht die Leitlinie die additive Gabe eines PCSK9Inhibitors wie Alirocumab vor [8]. Haben die Betroffenen ein sehr hohes bzw. extrem hohes kardiovaskuläres Risiko – z.B. nach einem akuten Koronarsyndrom (ACS), kritischer Extremitätenischämie oder einem ischämischen Schlaganfall – geht es darum, das LDL-C rasch auf den Zielwert von <55 mg/ dl (1,4 mmol/l) zu senken. In diesen Fällen sollte die lipidsenkende Therapie direkt mit einer maximal tolerierten Statindosis begonnen und dann weiter bis zum möglichen Einsatz von PCSK9-Inhibitoren eskaliert werden [9].
Zielwerterreichung bedeutet Reduktion des kardiovaskulären Risikos
Warum sich eine konsequente Umsetzung der Leitlinienempfehlungen von ESC und EAS lohnt, zeigt die Studie ODYSSEY OUTCOMES [6]. Eingeschlossen waren 18.924 Patienten mit einem hohen kardiovaskulären Risiko – sie hatten 1 – 12 Monate vor der Randomisierung ein ACS erlitten und erreichten trotz hochdosiertem bzw. maximal toleriertem Statin* (± anderen lipidsenkenden Therapien) keine ausreichende Kontrolle ihrer Lipide**. Sie wurden auf Alirocumab alle 2 Wochen oder Placebo randomisiert und im Median über 2,8 Jahre behandelt. Primärer Endpunkt war das Auftreten kardiovaskulärer Ereignisse (4-PunktMACE***) [7]. Wurde unter dem
* Patienten konnten auch mit niedriger Dosierung teilnehmen, wenn Unverträglichkeiten nachgewiesen und dokumentiert waren [7].
** LDL ≥70 mg/dl (≥1,8 mmol/l) oder non-HDL ≥100 mg/dl (≥2,6 mmol/l) oder Apolipoprotein B ≥80 mg/dl [7].
*** 4-Punkt-MACE (Major adverse cardiovascular events) bestehend aus KHK-bedingtem Tod oder nicht tödlichem Myokardinfarkt oder tödlichem bzw. nicht tödlichem ischämischem Schlaganfall oder instabiler Angina pectoris, die eine Hospitalisierung erforderte [7].
PCSK9-Hemmer ein LDL-CZielwertbereich von 25 – 50 mg/dl (0,6 – 1,3 mmol/l) erreicht, sank das Risiko für kardiovaskuläre Ereignisse signifikant (p < 0,001). Außerdem kam es in der Gesamtstudienpopulation numerisch zu weniger Todesfällen [7].
Alirocumab ist gut verträglich: Die Häufigkeit unerwünschter Ereignisse und Laboranomalien war in der Alirocumab- sowie der Placebogruppe ähnlich. Unter dem PCSK9-Inhibitor traten lediglich mehr Reaktionen an der Einstichstelle auf als unter Placebo [7]. Elisabeth Wilhelmi, München Literatur
1 Libby P et al. Nat Rev Dis Primers 2019;5:56
2 Ference BA et al. J Am Coll Cardiol 2018;72:1141-1156
3 https://www.destatis.de/DE/Themen/ Gesellschaft-Umwelt/Gesundheit/Todesursachen/_inhalt.html
4 Misselwitz B et al. Nervenarzt 2020; 91:484-492
5 Malyar N et al. Eur Heart J 2013;34: 2706-2714
6 Preiss et al. J Am Coll Cardiol 2020; 75:1945-55
7 Schwartz GG et al. N Engl J Med 2018; 379:2097-2107
8 Mach F et al. Eur Heart J 2020;41:111188
9 Mach F et al. Eur Heart J 2020;41:111188; modifiziert durch AG Südniedersachsen
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 15 FORUM LIPIDSENKER
Mit jährlich mehr als 41.000 neuen Fällen weltweit und einer Inzidenz von etwa 160 Fällen pro Jahr in Deutschland ist der Typ-2-Diabetes bei jungen Menschen auf dem Vormarsch. Treibende Faktoren sind Bewegungsmangel, ungesunde Ernährung und Übergewicht. Da bisher weltweit nur sehr wenige orale Medikamente für Kinder und Jugendliche zugelassen sind, besteht ein wachsender, bislang ungedeckter Bedarf an weiteren, vor allem oralen Arzneimitteln für diese Altersgruppe. Diese Behandlungslücke könnte der hochselektive Natriumglucose-Cotransporter 2 (SGLT2)-Inhibitor Empagliflozin (Jardiance®) in naher Zukunft füllen, der in der Phase-III-Studie DINAMO im Vergleich zu Placebo in der Studienpopulation von Kindern und Jugendlichen im Alter von 10 – 17 Jahren nach 26 Wochen eine statistisch signifikante Senkung des Blutzuckerspiegels (HbA1c) um 0,84 % bewirkte [1].
Empagliflozin ist das erste moderne Antidiabetikum, dessen Wirksamkeit im Rahmen von Typ2-Diabetes hinsichtlich der kardiovaskulären Risikoreduktion in die Fachinformation aufgenommen wurde [2].
Empagliflozin senkt HbA1c bei
Kindern und Jugendlichen mit Typ-2-Diabetes signifikant
bo auf den Blutzuckerspiegel von Kindern und Jugendlichen im Alter von 10 – 17 Jahren mit Typ-2-Diabetes (HbA1c ≥6,5 % und ≤10,5 %). Die 158 Teilnehmer wurden randomisiert einer Behandlung mit Empagliflozin (10 oder 25 mg; n = 52), Linagliptin (5 mg) (n = 53) oder Placebo (n = 53) einmal täglich zugewiesen. Alle Patienten wurden mit Diät und einem Bewegungsplan begleitet und mit Metformin und/oder Insulin behandelt. Die Studie erreichte in Woche 26 ihren primären Endpunkt: Empagliflozin konnte den Langzeitblutzucker signifikant reduzieren. In Kombination mit Diät, Bewegung,
Metformin und/oder Insulin betrug die durchschnittliche Senkung des HbA1c gegenüber dem Ausgangswert durch die Behandlung mit Empagliflozin –0,84 % im Vergleich zu Placebo (95%-KI: –1,50 bis –0,19; p = 0,012; Abb. 1). Unter der Behandlung mit Linagliptin kam es mit –0,34 % dagegen zu keiner statistisch signifikanten Abnahme des HbA1c im Vergleich zu Placebo (95%-KI: –0,99 bis 0,30; p = 0,29) [1].
Ein sekundärer Endpunkt der Studie zeigte, dass unter der Therapie mit Empagliflozin die NüchternPlasmaglukose um –35,2 mg/dl in Woche 26 abnahm (p = 0,0035).
Veränderungdes HbA1c gegenüber dem Ausgangswert in Woche 26
DINAMO-Studie überzeugt beim primären Endpunkt
Die randomisierte, doppelblinde, placebokontrollierte klinische Studie DINAMO untersuchte über 26 Wochen als primären Endpunkt die Wirkung von Empagliflozin und Linagliptin im Vergleich zu Place-
Unterschied zu Placebo: –0,34 (95% KI: –0,99, 0,30; p=0,2935)
Unterschied zu Placebo: –0,84 (95%-KI: –1,50, –0,19; p=0,0116)
Abbildung 1: Ergebnisse der DINAMO-Studie für den primären Endpunkt, die Wirkung von Empagliflozin und Linagliptin im Vergleich zu Placebo auf den Blutzuckerspiegel von Kindern und Jugendlichen im Alter von 10 – 17 Jahren mit Typ-2-Diabetes [1].
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 16 FORUM DIABETICUM
-0,6 -0,4 -0,2 0 0,2 0,4 0,6 0,8 1 Durchschnittliche (SE) Senkung des HbA 1c gegenüber dem Ausgangswert (%)
Placebo Linagliptin 5 mg Empagliflozin gepoolt
Keine neuen Sicherheitssignale
Die Daten zur Sicherheit waren mit früheren Ergebnissen bei Erwachsenen mit Typ-2-Diabetes vergleichbar. Bis Woche 26 traten Nebenwirkungen bei 77 % der mit Empagliflozin, bei 71 % der mit Linagliptin und bei 64 % der mit Placebo behandelten Patienten auf. Schwere Nebenwirkungen wurde berichtet bei jeweils 1 Teilnehmer (2 %) in der Empagliflozin- und der Linagliptin-Gruppe sowie 2 (4 %) Teilnehmern in der Placebo-Gruppe. Die häufigste Nebenwirkung war Hypoglykämie mit höheren Raten bei Patienten unter aktiver medikamentöser Behandlung im Vergleich zu Placebo. Es wurden
MITTEILUNGEN
Neue Webinar-Reihe: Diabetes-Docs erklären Technik
Diabetestechnologien wie kontinuierliche Glukosemessmethoden (rtCGM, isc-CGM), Insulinpumpen und Smart-Pens haben in den letzten Jahren zunehmend Einzug in die diabetologische Versorgung gehalten. Kaum eine andere Erkrankung verlangt sowohl vom Ärzteteam als auch von Menschen mit Diabetes mellitus so viel Kenntnisse von Medizintechnik wie die chronische Erkrankung Diabetes mellitus. Welche Vorteile bringt die Diabetestechnologie und ist sie für
keine schweren Hypoglykämiefälle gemeldet [1].
Fazit
Der SGLT2-Hemmer Empagliflozin ist derzeit zugelassen zur Behandlung von Erwachsenen mit Typ-2-Diabetes sowie für Erwachsene mit symptomatischer, chronischer Herzinsuffizienz (NYHAKlasse II–IV) [2]. Da es bisher weltweit nur sehr wenige orale Medikamente für Kinder und Jugendliche gibt, sind die Ergebnisse der DINAMO-Studie für das Management von Typ-2-Diabetes von jungen Menschen von besonderer Relevanz. Angesichts der überzeugenden Daten wird die Allianz von
Boehringer Ingelheim und Lilly nun Gespräche über die Zulassung von Empagliflozin zur Behandlung von Kindern und Jugendlichen mit Typ-2-Diabetes im Alter von 10 – 17 Jahren aufnehmen.
Brigitte Söllner, Erlangen
Literatur
1 Laffel LM, Danne T, Klingensmith GJ et al; DINAMO Study Group. Efficacy and safety of the SGLT2 inhibitor empagliflozin versus placebo and the DPP-4 inhibitor linagliptin versus placebo in young people with type 2 diabetes (DINAMO): a multicentre, randomised, double-blind, parallel group phase 3 trial. Lancet Diabetes Endocrinol 2023:S2213-8587(22)00387-4. doi: 10.1016/S2213-8587(22)00387-4. Epub ahead of print. PMID: 36738751
2 Fachinformation Jardiance®; Stand: Juli 2022
jedermann sinnvoll und leicht zu nutzen? Diesen und anderen Fragen stellt sich die neue Webinar-Reihe „Diabetes-Docs erklären Technik“, die von Diabetes-Anker.de (MedTriX Group) gemeinsam mit der Gesundheitsorganisation diabetesDE – Deutsche Diabetes-Hilfe entwickelt wurde. Dr. Jens Kröger, diabetesDE-Vorstandsvorsitzender, und weitere führende Diabetologen klären darin ganz praxisnah und verständlich über moderne Diabetes-Technologien auf. Die Reihe ist auf der Online-Plattform https:// diabetes-anker.de/diabetes-videoanker/ abrufbar.
In der ersten Folge mit dem Titel „Diabetes-Diagnose – und jetzt?“ geht es darum, wie Technik nach einer Diabetes-Typ-2-Diagnose eine
Lebensstilintervention unterstützen kann. Diabetologe Dr. med. Oliver Schubert-Olesen erklärt gemeinsam mit seinem Patienten Torsten von Elling (57) aus Hamburg eindrucksvoll, wie sehr das CGM dazu beitragen kann, dass sich die Glukosewerte verbessern.
Weitere Folgen der Webinar-Reihe sind:
• Folge 2: „Welche DiabetesTechnologie gibt es?“
• Folge 3: „Wie und von wem bekomme ich Diabetestechnologie verordnet?“
• Folge 4: „Wie kann ich mit Technologie den Diabetes verhindern?“
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 17 FORUM DIABETICUM
E. W.
Bei schwangeren Frauen mit Diabetes ist das A und O mit Blick auf die Gesundheit von Mutter und Kind eine normnahe Stoffwechseleinstellung, die allerdings durch hormonelle Schwankungen und unterschiedliche Insulinbedarfe des Körpers erschwert wird. Die kontinuierliche Glukosemessung in Echtzeit (real-time Continuous Glucose Monitoring, rtCGM) kann Schwangeren helfen, die Therapieziele zu erreichen [1]. Die S2e-Leitlinie „Diabetes in der Schwangerschaft“ [2] empfiehlt, Frauen mit einem Risiko für schwere Hypoglykämien bereits vor der Schwangerschaft mit einem Sensor auszustatten und verweist auf deutliche Vorteile hinsichtlich des neonatalen Outcomes bei Nutzung der rtCGM-Technologie im Vergleich zur konventionellen Blutzuckermessung [1, 2].
Diabetes in der Schwangerschaft:
Risiken des Fetus steigen linear mit HbA1c-Werten
Ein unzureichend behandelter Diabetes während der Schwangerschaft birgt ein erhöhtes Risiko für Fehlbildungen, intrauterinen Fruchttod, Schwangerschaftskomplikationen, aber auch die Gefahr von Diabetes-assoziierten kardio-
vaskulären Folgeerkrankungen [2, 3] sowie neonataler Mortalität [4]. Bei normnahen HbA1c-Werten sind die Risiken nur geringfügig erhöht, steigen mit höheren HbA1c-Werten aber linear an [2]. Frauen mit Typ-2-Diabetes sind ebenso betroffen wie Frauen mit Typ-1-Diabetes [5]. Auch Hyperglykämien aufgrund eines Gestationsdiabetes haben Einfluss auf den Schwangerschaftsverlauf und das fetale Outcome [2].
Normnahe Glukosewerte ab Kinderwunsch als Ziel
Da die kindliche Entwicklung bereits maßgeblich von der Qualität der Stoffwechseleinstellung in der Phase der Konzeption mitbestimmt wird, ist bereits vor der Empfängnis eine Optimierung der Stoffwechseleinstellung anzustreben [2, 5]. Laut Leitlinie sollte der HbA1c unter Berücksichtigung (weiterer) individueller Therapieziele vor und während der Schwangerschaft unabhängig vom Diabetestyp unter 7 % liegen, idealerweise sogar unter 6,5 % [2]. Heute ist mit guter Evidenz belegt, dass sich dieser Parameter der Stoffwechseleinstellung mit (rt) CGM-Systemen optimieren lässt [6, 7]. Weitere dokumentierte Vor-
teile gegenüber der herkömmlichen Blutzuckermessung ergeben sich hinsichtlich des Hypo- und Hyperglykämierisikos [7, 8], der Zeit im Zielbereich (Time in Range, TIR), des HbA1c-Werts [2, 6, 7] sowie der glykämischen Variabilität [8].
rtCGM kann Stoffwechselentgleisungen reduzieren ...
Eine gute Stoffwechselführung in der Schwangerschaft wird erschwert durch hormonelle Schwankungen und schwankende Insulinbedarfe, die klinisch relevante Auswirkungen auf den Glukosestoffwechsel haben. Die rtCGMSysteme von Dexcom können diese Glukoseschwankungen sichtbar machen. Hierzu wird ein Sensor etwa am Bauch oder an der Oberarmrückseite platziert. Alle 5 Minuten wird der aktuelle Glukosewert auf ein Smartphone oder den optionalen Empfänger übertragen und kann dort abgelesen werden (Abb. 1). Außerdem bieten die Systeme der Schwangeren die Möglichkeit, sich rechtzeitig und mit zeitlichem Vorlauf warnen zu lassen, wenn sich die Glukosewerte in Richtung hypo- bzw. hyperglykämischer Schwellenwerte bewegen, sodass
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 18 FORUM DIABETICUM
S2e-Leitlinie empfiehlt die kontinuierliche Glukosemessung in Echtzeit
Abbildung 1: Das Dexcom G6-System zur kontinuierlichen Glukoseüberwachung in Echtzeit besteht aus dem Applikator , der den kleinen Sensor mit nur einem Knopfdruck direkt unter die Haut einführt. Dieser misst den Gewebeglukosespiegel kontinuierlich und sendet die Daten drahtlos über einen Transmitter an ein mobiles Gerät mit iOS oder Android , das die Gewebeglukosedaten in Echtzeit anzeigt (© Dexcom).

Alarme bieten zusätzliche Sicherheit
Bei den rtCGM-Systemen von Dexcom ermöglichen individuell einstellbare Warnungen für hohe und tiefe Werte das rechtzeitige Eingreifen bei zu stark steigenden oder sinkenden Werten.
• Hypo-Vorwarnung: Sieht das System voraus, dass der Gewebezucker in den kommenden 20 Minuten auf oder unter die kritische Schwelle von 55 mg/dl (3,1 mmol/l) fallen könnte, ertönt die Hypo-Vorwarnung und kann der Trägerin so helfen, Unterzuckerungen und Gewebezuckerschwankungen vorzubeugen (bei sofortigem Handeln).
• Hypo-Sicherheitsalarm für den Notfall: Ertönt bei 55 mg/dl (3,1 mmol/l) und lässt sich weder abschalten noch deaktivieren.
rechtzeitig gegengesteuert werden kann. Dieser Vorteil der smarten Glukosesensoren wird auch in der S2e-Leitlinie ausdrücklich genannt [2].
... und das neonatale Outcome verbessern
In einer randomisierten, kontrollierten Studie an insgesamt 325
Schwangeren mit Typ-1-Diabetes konnte gezeigt werden, dass sich mithilfe der rtCGM-Technologie im Vergleich zur konventionellen Blutzuckerbestimmung in der Kontrollgruppe signifikante günstige Effekte bezüglich des Outcomes des Kindes erzielen lassen [1]: In der rtCGM-Gruppe kam es seltener zu neonatalen Hypoglykämien, die Kinder hatten im Mittel ein höheres Geburtsgewicht und mussten weniger häufig intensivmedizinisch betreut werden. Diese positiven Effekte waren assoziiert mit einer optimierten Stoffwechseleinstellung der Schwangeren: Die Time in Range lag in der rtCGM-Gruppe bei 68 % versus 61 % in der Kontrollgruppe, die Hyperglykämierate betrug 27 % versus 32 % [1].
Share-Funktion und Sicherheitsalarm helfen Hyper- und Hypoglykämien zu vermeiden
Eine weitere Verbesserung der Stoffwechseleinstellung lässt sich durch das Teilen der Glukosedaten und Warnungen z.B. mit Angehörigen erzielen. Bei den rtCGMSystemen von Dexcom ist dies über die Share-Funktion mit bis zu 10 Personen möglich. So kann im Vergleich zu rtCGM ohne ShareFunktion nicht nur der HbA1c-Wert weiter gesenkt werden, auch hyperglykämische Ereignisse lassen sich zuverlässiger verhindern, wie eine andere Studie zeigte [9]. Der wichtigste Risikofaktor für schwere Hypoglykämien im ersten Trimenon der Schwangerschaft ist eine diesbezüglich positive Ana-
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 19 FORUM DIABETICUM
Smarte Kombi auch in der Schwangerschaft: Glukosesensor und Insulinabgabe
Auch die vielfältigen Kombinationsmöglichkeiten werden in der S2e-Leitlinie angesprochen [2]. Die smarten Glukosesensoren dienen dabei stets als Grundlage für verschiedene Therapiestrategien. So können sie nicht nur als Stand-alone-System, sondern (je nach Bedarf) auch in Kombination mit weiteren Bausteinen genutzt werden.
Bei schwangeren Frauen mit Diabetes und Insulinpumpentherapie kann das Dexcom G6 aufgrund seiner Möglichkeit zur interoperablen Nutzung das geeignete rtCGM-System sein. So lässt es sich heute bereits mit verschiedenen Insulinpumpen – namentlich der t:slim X2 mit Control IQTM Algorithmus und der mylife YpsoPump mit der mylife CamAPS FX App – zu einem Hybrid-Closed-Loop vernetzen. Der Dexcom G6-Sensor kann außerdem mit dem DBLG1® Algorithmus von diabeloop kommunizieren. Darüber hinaus ist es möglich, sich mit Glooko die Glukosedaten des Dexcom G6 zusammen mit den Insulindaten bestimmter Smartpens (aktuell NovoPen® 6, NovoPen Echo® Plus und Tempo Pen® von Lilly) anzeigen zu lassen. Diese Kopplungen aus smartem Glukosesensor und Insulinabgabesystem ermöglichen somit eine (weitere) Individualisierung und Anpassung der Therapie an die Bedürfnisse und Behandlungsziele.
Für Schwangere mit intensivierter konventioneller Insulintherapie (ICT) kann das neue Dexcom G7 rtCGM-System eine gute Wahl sein. Die jüngste rtCGM-Generation zielt insgesamt auf noch mehr Diskretion, Nutzungseffizienz und Bedienungsfreundlichkeit ab als das Dexcom G6, etwa durch die um 60 % reduzierte Sensorgröße, die schnelle Aufwärmphase von unter 30 Minuten [12] oder die zusammenfassende Darstellung der Zeit im Zielbereich in leicht verständlicher Ampel-Farbcodierung.
mnese in den letzten 4 Monaten vor der Konzeption [2]. Ist das der Fall, sollten die Frauen laut Leitlinie vor oder während der Schwangerschaft mit einem CGM-System ausgestattet werden, um Hypoglykämien zu vermeiden (Empfehlungsgrad A).
Die rtCGM-Systeme von Dexcom verfügen als einzige über einen voreingestellten Hypoglykämie-Sicherheitsalarm sowie eine prädiktive Hypoglykämie-Vorwarnung, die die Anzahl und Dauer von Hypoglykämien nachweislich reduzieren kann [10].
Durch das Vermeiden von Hypoglykämien mithilfe dieser Vorwarnung lassen sich auch mögliche Rebound-Hyperglykämien verhindern [11], da es gar nicht zu einer Unterzuckerung kommt. Angesichts der genannten Vorteile empfiehlt die Leitlinie, dass auch schwangeren Diabetespatientinnen ohne Risiko für schwere Hypoglykämien ein kontinuierliches Glukosemonitoring in Echtzeit zum Selbstmanagement angeboten werden sollte (Empfehlungsgrad B) [2].
Elisabeth Wilhelmi, München
Literatur
1 Feig DS et al. Lancet 2017;390:23472359
2 S2e-Leitlinie „Diabetes in der Schwangerschaft“ 2021. Online verfügbar unter https://www.deutsche-diabetes-gesellschaft.de/fileadmin/user_upload/05_ Behandlung/01_Leitlinien/Evidenzbasierte_Leitlinien/2021/057-023_LL_Diabetes_und_Schwangerschaft.pdf
3 Arnaout R et al. Open Heart 2019;6: e000927
4 Battarbee AN et al. J Perinatol 2020;40: 232-239
5 Kleinwechter H et al. Diabetes und Stoffwechsel 2020;15(Suppl. 1):S93-S100
6 Šoupal J et al. Diabetes Care 2020;43: 37-43
7 Martens T et al. J Am Med Ass 2021; 325:2262-2272
8 Heinemann L et al. Lancet 2018;391: 1367-1377
9 Polsky S et al. PLoS One 2020;15: e0230476
10 Puhr S et al. J Diabetes Sci Technol 2020;14:83-86
11 Acciaroli G et al. J Diabetes Sci Technol 2022;16:677-682
12 Garg SK et al. Diabetes Technol Ther 2022;24:373-380
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 20 FORUM DIABETICUM
Die Europäische Kommission hat Dapagliflozin (Forxiga®), einen Inhibitor des Natrium-GlukoseCotransporters-2 (SGLT-2i), am 06.02.2023 die Zulassungserweiterung zur Therapie der symptomatischen chronischen Herzinsuffizienz (HF) unabhängig von der Ejektionsfraktion erteilt. Ab sofort können nun auch Patienten mit mäßig reduzierter und erhaltener Ejektionsfraktion (HFmrEF und HFpEF) mit Dapagliflozin behandelt werden. Damit steht mit Dapagliflozin die erste und einzige Herzinsuffizienztherapie mit nachgewiesenem signifikantem Mortalitätsvorteil für das gesamte Spektrum der linksventrikulären Ejektionsfraktion (LVEF) zur Verfügung [1]. Für die Therapie der HF mit reduzierter Ejektionsfraktion (HFrEF) ist Dapagliflozin bereits seit 2020 zugelassen.
Signifikanter Überlebensvorteil
über das gesamte LVEFSpektrum
Mit der Zulassungserweiterung folgt die Europäische Kommission der positiven Stellungnahme des Ausschusses für Humanarzneimittel (CHMP) vom Dezember 2022, die sich auf die Ergebnisse der Phase-III-Studie DELIVER stützt [2]. In dieser Studie, an der 6.263 Patienten teilnahmen, verringerte die Behandlung mit Dapagliflozin bei Herzinsuffizienzpatienten mit einer LVEF >40 % nach einer medianen Nachbeobachtungszeit von 2,3 Jahren das Risiko für den zusammengesetzten primären Endpunkt aus
Herzinsuffizienz: Dapagliflozin jetzt auch für die Behandlung der HFmrEF und HFpEF zugelassen
kardiovaskulärem Tod oder Verschlechterung der Herzinsuffizienz (Krankenhausaufenthalt oder ein gleichwertiges Ereignis, d.h. ein dringender HF-Besuch) signifikant um 18 % im Vergleich zu Placebo (16,4 % unter Dapagliflozin vs. 19,5 unter Placebo; HR: 0,82; 95%-KI: 0,73 – 0,92; p < 0,001; absolute Risikoreduktion [ARR]: 3,1 %). Dabei war der Behandlungseffekt von Dapagliflozin über den gesamten LVEF-Bereich hinweg konsistent, einschließlich der HF mit mäßig reduzierter oder erhaltener Ejektionsfraktion. Auch Patienten mit einem manifesten Typ-2-Diabetes (HR: 0,83; 95%-KI: 0,70 – 0,97) profitieren im selben Maß wie Betroffene ohne Diabetes (HR: 0,81; 95%-KI: 0,68 – 0,96) [2]. Untermauert wurden die Befunde zur LVEF-übergreifenden Wirkung von Dapagliflozin durch die Ergebnisse der präspezifizierten, auf Patientenebene durchgeführten gepoolte Analyse der Phase-IIIStudien DELIVER (bei HFrEF) und DAPA-HF (bei HFmrEF und HFpEF) [1]: Nach einer medianen Nachbeobachtungszeit von 22 Monaten verringerte sich unter
der Therapie mit Dapagliflozin das Risiko eines kardiovaskulären Todes um 14 % (HR: 0,86; 95%-KI: 0,76 – 0,97; p = 0,01; ARR: 1,5 %), die Gesamtmortalität um 10 % (HR: 0,90; 95%-KI: 0,82 – 0,99; p = 0,03; ARR: 1,5 %) und die Gesamtzahl der initialen und wiederholten Krankenhausaufenthalte wegen HF um 29 % (HR: 0,71; 95%-KI: 0,65 – 0,78; p < 0,001, ARR: 6 %) [1]. Diesen Ergebnissen zufolge war Dapagliflozin das erste und einzige HF Medikament, das einen Überlebensvorteil über das gesamte LVEF-Spektrum zeigen konnte. Durch die nun erfolgte Zulassungserweiterung kann mit Dapagliflozin ein neuer Standard in der Therapie der Herzinsuffizienz gesetzt werden.
Literatur
1 Jhund P et al. Nat Med 2022;28:19561964
2 Solomon S et al. N Engl J Med 2022; 387:1089-1098
3 Dunlay SM et al. Nat Rev Cardiol 2017;14:591-602
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 21 FORUM CARDIOLOGICUM
Brigitte Söllner, Erlangen
Die paroxysmale nächtliche Hämoglobinurie (PNH) ist eine seltene, komplementvermittelte hämatologische Erkrankung. Ursache der PNH ist eine erworbene somatische Mutation im PIG-A-Gen in einer oder mehreren pluripotenten hämatopoetischen Stammzellen des Knochenmarks. Diese produzieren infolge der Mutation rote Blutkörperchen, die keine komplementregulierenden Proteine auf ihrer Oberfläche tragen und damit anfällig für eine vorzeitige Zerstörung durch das Komplementsystem sind. Folge ist eine intravasale und extravasale (vor allem in der Milz und der Leber) Hämolyse, die wiederum zu einer Anämie, thromboembolischen Komplikationen, Fatigue und anderen, die Lebensqualität der Betroffenen beeinträchtigenden Symptomen führt. Typisches klinisches Zeichen der PNH ist der dunkelbraune (Morgen-)Urin, der aber nur bei etwa einem Viertel der PNH-Patienten zum Zeitpunkt der Erstdiagnose nachweisbar ist [1, 2]. Schätzungsweise leiden weltweit etwa 10 – 20 Menschen pro 1 Million Einwohner an PNH. Obwohl die Erkrankung in jedem Alter auftreten kann, wird sie häufig bei Menschen im Alter von 30 – 40 Jahren diagnostiziert [3].
Der einzige potenziell kurative Therapieansatz bei PNH ist die allogene Stammzelltransplantation, die aber mit einer erheblichen Transplantations-assoziierten Morbidität und Mortalität einhergeht. Behandlungsstandard ist eine zielgerichtete medikamentöse Therapie, bei der das terminale Komplementsystem durch monoklonale
Paroxysmale nächtliche Hämoglobinurie:
Faktor-B-Inhibitor Iptacopan ist Anti-C5-Therapie überlegen
Antikörper (Eculizumab oder Ravulizumab) inhibiert wird, indem diese den Komplementfaktor C5 binden, wodurch die intravasale Hämolyse verringert wird. Jedoch bleibt trotz der Behandlung mit Anti-C5-Therapien ein großer Teil der PNH-Patienten anämisch, erschöpft und auf Bluttransfusionen angewiesen, sodass nach wie vor ein ungedeckter Bedarf an Therapieoptionen besteht.
Klinisch relevante transfusionsfreie Erhöhung der Hb-Werte durch Iptacopan
Ein in der klinischen Entwicklung befindliches, vielversprechendes Prüfpräparat ist Iptacopan. Indem der Wirkstoff selektiv, kompetitiv sowie reversibel den Faktor B inhibiert, der Teil des alternativen Signalwegs im Komplementsystem ist, wirkt er bereits im proximalen Abschnitt des Signalwegs, der dem terminalen C5-Weg vorgeschaltet ist, und verhindert nicht nur die intravasale, sondern auch die extravasale Hämolyse bei PNH [4]. Dass sich dadurch ein klinisch relevanter Anstieg des Hb-Spiegels erreichen lässt, zeigen die Ergebnisse der Phase-III-Studie APPLY-PNH, die
auf der 64. Jahrestagung der American Society of Hematology im Dezember 2022 präsentiert wurden [5].
Die randomisierte, kontrollierte, offene und vergleichende PhaseIII-Studie untersuchte über 24 Wochen die Wirksamkeit und Sicherheit der zweimal täglich oral verabreichten Monotherapie mit Iptacopan (200 mg) bei Patienten mit PNH. Ziel war es, die Überlegenheit von Iptacopan gegenüber Anti-C5-Therapien (Eculizumab oder Ravulizumab) bei erwachsenen Patienten zu zeigen, die trotz einer stabilen Anti-C5-Therapie in den 6 Monaten vor der Randomisierung eine Restanämie (Hb <10 g/ dl) aufwiesen [5]. Als primäre Endpunkte wurden ein Anstieg des HbSpiegels von ≥2 g/dl bzw. ≥12 g/dl gegenüber dem Ausgangswert definiert, ohne dass Bluttransfusionen erforderlich waren.
Iptacopan erreichte beide primären Endpunkte [5]:
• Einen Anstieg des Hb-Spiegels von ≥2 g/dl gegenüber dem Ausgangswert, ohne dass Bluttransfusionen erforderlich waren, erreichten 82,3 % der mit Iptacopan behandelten Patienten (95%-KI: 73,4 – 90,2), aber nur 2,0 % der Patienten, die die An-
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 22 FORUM HAEMATOLOGICUM
ti-C5-Therapie erhielten (95%KI: 1,1 – 4,1) – ein Unterschied von 80,3 Prozentpunkten (95%KI: 71,3– 87,6; p < 0,0001). Diesen primären Endpunkt erreichten unter Iptacopan 51 von 60 und unter Anti-C5-Therapie 0 von 35 Patienten.
• Einen Anstieg des Hb-Spiegels von ≥12 g/dl gegenüber dem Ausgangswert, ohne dass Bluttransfusionen erforderlich waren, erreichten 68,8 % der mit Iptacopan behandelten Patienten (95%-KI: 58,3 – 78,9), aber nur 1,8 % der Patienten, die die Anti-C5-Therapie erhielten (95%KI: 0,9 – 4,0) – ein Unterschied von 67,0 Prozentpunkten (95%KI: 56,3 – 76,9; p < 0,0001). Diesen primären Endpunkt erreichten unter Iptacopan 42 von 60 und unter Anti-C5-Therapie 0 von 35 Patienten.
Auch bei 5 von 7 sekundären Endpunkten erwies sich Iptacopan der Anti-C5-Therapie als signifikant überlegen. Dazu zählten die Unabhängigkeit von Bluttransfusionen, die Veränderung der Hb-Werte gegenüber dem Ausgangswert, die Messung der Veränderung der von den Patienten angegebenen Fatigue (gemessen anhand des FACIT-FScores) und der absoluten Retikulozytenzahl (unreife rote Blutkörperchen) sowie die Rate an klinischen Durchbruchhämolysen (BTH) [5]. Nur bei 2 sekundären Endpunkten ergab sich kein signifikanter Unterschied zwischen der Iptacopan-Monotherapie und der Anti-C5-Therapie: bei der Häufigkeit schwerwiegender unerwünschter
vaskulärer Ereignisse und der Veränderung der Laktatdehydrogenase (LDH)-Werte im Vergleich zum Ausgangswert, wobei Letzteres die Kontrolle der intravasalen Hämolyse anzeigte [5].
Günstiges Sicherheitsprofil
Das Sicherheitsprofil von Iptacopan entsprach in der APPLY-PNHStudie den zuvor ermittelten Daten, wobei keine schwerwiegenden Infektionen durch bekapselte Bakterien auftraten. Die am häufigsten gemeldeten unerwünschten Wirkungen von Iptacopan waren Kopfschmerzen (16,1 % vs. 2,9 % unter Anti-C5) und Durchfall (14,5 % vs. 5,7 % unter Anti-C5), die ohne weitere Behandlung abklangen. Unter Anti-C5-Therapie wurden am häufigsten COVID-19 (25,7 % vs. 8,1 % unter Iptacopan) und BTHEreignisse (17,1 % vs. 3,2 % unter Iptacopan) gemeldet [5].
Bei 2 Patienten mit Anti-C5-Therapie trat eine Hämolyse als schwerwiegendes unerwünschtes Ereignis auf, unter Iptacopan wurde kein solches Ereignis beobachtet. Weder die Behandlung mit Iptacopan noch die Anti-C5-Therapie wurde aufgrund von Nebenwirkungen abgebrochen [5].
Fazit
Die Ergebnisse der Phase-III-Studie APPLY-PNH zeigen, dass der oral verabreichbare Faktor-B-Inhibitor Iptacopan den intravenösen Anti-C5-Therapien überlegen ist,
d.h., eine stärkere Hemmung der extravasalen Hämolyse bewirkte und auch die intravasale Hämolyse unter Kontrolle hielt. Die 24-wöchige Therapie mit Iptacopan ermöglichte dem Großteil (60 von 62) der Patienten eine transfusionsfreie Erhöhung der Hb-Werte, bei der Behandlung mit einer Anti-C5Therapie war dies nur bei 14 von 35 Patienten der Fall. Unter Iptacopan wurden 96,7 % der Patienten unabhängig von Bluttransfusionen, außerdem verbesserte sich die von den Patienten angegebene Fatigue um 8,59 Prozentpunkte. Es wurden keine Fälle von Durchbruchhämolysen berichtet.
Iptacopan hat von der FDA den Status Breakthrough Therapy Designation bei PNH, von der FDA und der EMA den Orphan-Drug-Status bei PNH und C3G, von der EMA den PRIME-Status für C3G und den Orphan-Drug-Status bei IgAN erhalten. Eine Zulassung steht noch aus.
Brigitte Söllner, Erlangen Literatur
1 Cançado RD et al. Hematol Transfus Cell Ther 2021;43:341-348
2 Dingli D et al. Ann Hematol 2022; 101:251-263
3 Schrezenmeier H et al. Ann Hematol 2020;99:1505-1514
4 Schubart A et al. Proc Natl Acad Sci 2019;116:7926-7931
5 Peffault de Latour R et al. Results from the randomized, active-comparatorcontrolled,open-label, multicenter, phase III APPLY-PNH study. Presented at: 64th American Society of Hematology Annual Meeting & Exposition (ASH); December 10–13, 2022; New Orleans
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 23 FORUM HAEMATOLOGICUM
Seit Jahren steigt die Anzahl übergewichtiger und fettleibiger Kinder in Deutschland an. Laut den Ergebnissen der zweiten Welle der KIGGS-Studie des Robert KochInstituts sind derzeit 9,5 % der Kinder und Jugendlichen (3 – 17 Jahre) übergewichtig, 5,9 % sogar adipös. Das entspricht etwa 2 Mio. übergewichtigen und ca. 800.000 adipösen Kindern [1].
Mediziner und Ernährungswissenschaftler machen dafür unter anderem die intensive Vermarktung von Süßigkeiten und Softdrinks durch die Lebensmittelwirtschaft verantwortlich. Zahlreiche wissenschaftliche Studien geben ihnen Recht! Kürzlich erschienen zwei von der WHO beauftragte systematische Übersichtsarbeiten [2, 3]. Beide belegen eindeutig, dass Werbung für ungesunde Lebensmittel das Ernährungsverhalten (Vorlieben, Auswahl, Kaufentscheidung) von Kindern prägt und den Verzehr von Süßigkeiten und Softdrinks erhöht. So zeigt die von Boyland et al. durchgeführte Metaanalyse einen statistisch hoch signifikanten Zusammenhang zwischen Lebensmittelwerbung und Nahrungsaufnahme bei Kindern und Jugendlichen: Eine Verstärkung der Werbung verursacht eine Zunahme des Konsums (standardisierte Mittelwertdifferenz 0,25;
Werbeverbot für mehr Kindergesundheit
95%-KI: 0,15 – 0,35; p < 0,001). Der Gesamteffekt war robust gegenüber zahlreichen Störgrößen, wie in Sensitivitäts- und GOSHAnalysen gezeigt werden konnte [3]. Auf Basis der vorliegenden wissenschaftlichen Evidenz sprechen sich die Autoren beider Reviews einhellig für staatliche Maßnahmen zur Einschränkung der an Kinder gerichteten Werbung für ungesunde Lebensmittel aus.
Gesetzesvorhaben für mehr Kinderschutz
Die bereits 2007 im „EU-Pledge“ versprochene freiwillige Selbstverpflichtung der weltweit führenden Lebensmittelunternehmen, nur noch Produkte für Kinder zu bewerben, die die von der WHO empfohlenen Nährwertanforderungen erfüllen, wurde bis heute nicht umgesetzt [4]. Daher besteht Handlungsbedarf auf staatlicher Ebene. Entsprechend hat Bundesernährungsminister Cem Özdemir, wie bereits im Koalitionsvertrag vereinbart, am 27.03.23 einen Gesetzentwurf zur Einschränkung der an Kinder (unter 14-Jährige) adressierten Werbung für Lebensmittel mit hohem Zucker-, Fett- oder Salzgehalt vorgelegt [5]. Wesentliche Inhalte sind:
• Werbeverbote in „allen für Kinder relevanten Medien“ zwischen 6 und 23 Uhr
• Verbot der Außenwerbung auf Plakaten für ungesunde Produkte im Umkreis von 100 Metern um Schulen, Kitas, Spielplätze und Freizeiteinrichtungen für Kinder
• Verbot eines an Kinder gerichteten Sponsoring für Lebensmittel mit hohem Zucker-, Fett- oder Salzgehalt
Zukünftig dürfen also nur noch solche Lebensmittel für Kinder beworben werden, die den Nährwertkriterien der WHO entsprechen [6]. Für das geplante Gesetz ist eine Übergangsfrist von 2 Jahren vorgesehen [5].
Süßwarenlobby zeigt wenig Bereitschaft
Die Reaktion der Süßwarenlobby hat nicht lange auf sich warten lassen. In der Pressemitteilung des Bundesverbands der Deutschen Süßwarenindustrie (BDSI) vom 28.02.23 behauptet deren Hauptgeschäftsführer Dr. Carsten Bernoth, es existierten keine wissenschaftlichen Untersuchungen zur Wirksamkeit von Werbeverboten auf die Entwicklung von kindlichem Übergewicht [7]. Diese Äußerung ist zynisch und unrichtig
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 24 FORUM ERNÄHRUNG
– und zeigt die geringe Bereitschaft der Lebensmittelwirtschaft, die Rezepturen zu verbessern. Die deutsche Süßwarenindustrie ist nicht gerade dafür bekannt, gesunde Produkte herzustellen. Süßwaren zeichnen sich i.d.R. durch einen hohen Gehalt an reinem Zucker, industriell verarbeiteten Fetten sowie zahlreichen Zusatzstoffen aus. Diese hochverarbeiteten Fertigprodukte sind nach Ansicht zahlreicher medizinischer und ernährungswissenschaftlicher Fachgesellschaften nicht für den täglichen Verzehr geeignet, da sie zur Entstehung von Übergewicht und ernährungsmitbedingten Erkrankungen, wie z.B. Adipositas und Diabetes beitragen [4]. Somit ist es eigentlich an der Süßwarenindustrie, die gesundheitliche Unbedenklichkeit ihrer Produkte nachzuweisen (Beweislastumkehr).
Mit der geplanten Werbeeinschränkung werden die ungesunden Süßwaren nicht vom Markt verschwinden, ihr Konsum könnte dadurch aber auf ein vertretbares Maß reduziert werden.
matische Produkte an Kinder zu vermarkten [9]. Kanada hat bereits 1980 ein Gesetz erlassen, das Werbung für Fast Food und Spielzeug an Kinder unter 13 Jahren in gedruckten und elektronischen Medien verbietet [10]. Laut einer Studie der University of British Columbia wurden infolge des Werbeverbots 13 % weniger Fast Food konsumiert [11]. In Chile gibt es seit 2016 strenge gesetzliche Auflagen zur Kennzeichnung und Vermarktung von Lebensmitteln. Danach müssen alle Lebensmittel mit zu viel Zucker, Fett, Kalorien oder Salz ganz deutlich auf der Vorderseite des Produktes gekennzeichnet werden. Diese Produkte dürfen nicht mehr in Schulen verkauft werden und zwischen 6 und 22 Uhr darf dafür keine Fernsehwerbung mehr ausgestrahlt werden. Auch gezielte Werbung für unter 14-Jährige ist verboten. Sämtliche Comic-Helden und Zeichentrickfiguren sind von den Verpackungen verschwunden. Dank dieser Maßnahmen geht die Fettleibigkeit in Chile kontinuierlich zurück [12].
Literatur
1 Schienkiewitz A et al. Übergewicht und Adipositas im Kindes- und Jugendalter in Deutschland – Querschnittergebnisse aus KiGGS Welle 2 und Trends. J Health Monitoring 2018;3:6-23
2 Boyland E et al. Systematic review of the effect of policies to restrict the marketing of foods and non-alcoholic beverages to which children are exposed. Obesity Rev 2022; 23:e13447
3 Boyland E et al. Association of Food and Nonalcoholic Beverage Marketing With Children and Adolescents’ Eating Behaviors and Health. A systematic review and meta-analysis. JAMA Pediatr 2022;176:e221037
4 https://www.foodwatch.org/fileadmin/_ migrated/content_uploads/2015-08-24_ DAG_DDG_dDE_Positionspapier_ EU_Pledge.pdf
5 https://www.bmel.de/SharedDocs/Pressemitteilungen/DE/2023/024-lebensmittelwerbung-kinder.html
6 WHO 2012: Set of recommendations on the marketing of foods and non-alcoholic beverages to children. https:// www.who.int/publications/i/item/ 9789241500210
7 https://www.bdsi.de/pressemeldungen/ details/plaene-von-bundesminister-oezdemir-bedeuten-ein-totalverbot-von-suesswarenwerbung/
8 https://www.presseportal.de/pm/58227/ 5412648
9 Théodore FL et al. Pitfalls of the selfregulation of advertisements directed at children on Mexican television. Pediatr Obes 2017;12:312-319
10 Clark CR. Advertising restrictions and competition in the children’s breakfast cereal industry. https://www.journals. uchicago.edu/doi/10.1086/519820
Vorbilder in aller Welt
In Deutschland hat der Discounter Lidl angekündigt, ab dem 1. März 2023 die auf Kinder abzielende Werbung für ungesunde Süßwaren einzustellen. Ausgenommen davon bleiben lediglich Aktionsartikel zu Weihnachten, Ostern und Halloween [8].
Andere Länder greifen härter durch. In Mexiko ist es seit 2014 nicht zulässig, in TV und Kino proble-
Fazit
Es besteht also kein Zweifel daran, dass Werbung das Konsumverhalten und damit die Gesundheit von Kindern nachhaltig beeinflussen kann. Der deutsche Gesetzgeber sollte sich der vorliegenden Evidenz anschließen und dem guten Beispiel anderer Staaten folgen, zum Wohl unserer Kinder.
Franz-Werner Dippel, Hohen Neuendorf
11 Dhar T et al. Fast-Food consumption and the ban on advertising targeting children: The Quebec experience. https://journals.sagepub.com/doi/10.1509/ jmkr.48.5.799
12 Dillman Carpentier FR et al. Evaluating the impact of Chile’s marketing regulation of unhealthy foods and beverages: pre-school and adolescent children’s changes in exposure to food advertising on television. Public Health Nutr 2020;23:747-755
Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH 25 FORUM ERNÄHRUNG
KONGRESSE
Patientenversorgung verbessern: Eiseninfusionen und Kaliumbinder in der Intensivmedizin
Die Sicherheit der Patienten vor, während und nach Operationen sowie die Vermeidung von Komplikationen in der Versorgung vulnerabler Patientengruppen sind weiterhin beherrschende Themen in der Intensivmedizin. Welche Möglichkeiten zur Optimierung der Therapieergebnisse bei Operationen die präoperative Gabe von Eisencarboxymaltose (Ferinject®) bietet und wie die Versorgung von kardiorenalen Patienten mit Hyperkaliämie mit dem Kaliumbinder Patiromer (Veltassa®) verbessert werden kann, erörterten Experten anlässlich des 33. Symposiums Intensivmedizin und Intensivpflege in Bremen.
Eisenmangelmanagement senkt Bedarf an Blutkonserven
Die abnehmende Spendebereitschaft führt bundesweit zu einer zunehmend kritischen Versorgungslage mit Blutkonserven für Transfusionen. Ein Umdenken ist gefragt, wobei das Patient Blood Management (PBM) einen möglichen Beitrag leisten kann, um den Bedarf an Blutprodukten zu senken. Ziel dieses multidisziplinären Behandlungskonzepts ist es, durch die Reduktion und Vermeidung von Anämie, Blutverlust und unnötigen Transfusionen bessere Behandlungsergebnisse zu erreichen. Eine der 3 Säulen, auf denen das PBM-Konzept beruht, ist das
prä- und postoperative Eisenmangelmanagement. Anämie und Eisenmangel sind Risikofaktoren für ein schlechtes klinisches Outcome. Präoperativ zeigen bis zu 40 % der Patienten eine Anämie, die bei fast der Hälfte der Betroffenen auf einem Eisenmangel beruht.
Eisenmangel frühzeitig identifizieren und leitliniengerecht behandeln
Die Folgen einer präoperativen Anämie verdeutlichte Professor Jochen Renner, Kiel, anhand eines Patienten, bei dem trotz Anämie eine aortokoronare Bypassoperation durchgeführt wurde. Der präoperative Hämoglobin-Wert lag unterhalb des Schwellenwerts. Daher erhielt der Patient präoperativ 2 Erythrozytenkonzentrate. Intraoperativ und nach Verlegung auf die Intensivstation war die Gabe von jeweils 2 weiteren Erythrozytenkonzentraten erforderlich. Im weiteren Verlauf erkrankte der Patient an einer Pneumonie, sodass sich der Weaning-Prozess und der Aufenthalt auf der Intensivstation verlängerten. Wie Renner ausführte, hätte das relativ einfach vermieden werden können, wenn – wie in den Leitlinien der European Society of Anaesthesiology (ESA) empfohlen – der Eisenstatus durch Bestimmung von Serumeisen, Ferritin und Transferinsättigung (TSAT) spätestens 30 Tage vor dem Eingriff erhoben und der Eisenmangel präoperativ ausgeglichen worden wäre. Dazu empfiehlt die ESA-Leitlinie die intravenöse anstelle der oralen Eisensubstitution, insbesondere wenn das Intervall zwischen der Diagnose einer Anämie und der
Operation weniger als 6 – 8 Wochen beträgt.
Eine bedarfs- und leitliniengerechte Option zur Korrektur des Eisenmangels ist die intravenöse Gabe von Eisencarboxymaltose (z.B. Ferinject® *). Wie die randomisierte, doppelt verblindete, parallel kontrollierte Studie von Spahn et al. (Lancet 2019;393:2201-2212) zeigte, lässt sich durch i.v. Eisencarboxymaltose in Kombination mit Erythropoetin, Vitamin B12 und Folsäure eine präoperative Eisenmangelanämie wirksamer beheben als durch orales Eisensulfat. Die Kombination ermöglicht selbst bei sehr kurzen Zeitfenstern bis zu 24 Stunden vor dem Eingriff eine Ultrakurzzeitbehandlung. Deren Wirkung hielt in der Studie auch 90 Tage nach der Operation noch an und konnte den Bedarf an Erythrozytentransfusionen im Vergleich zur Placebo- und Eisensulfat-Gruppe deutlich reduzieren. Darüber hinaus ermöglicht die präoperative Gabe von Ferinject® auch erhebliche Kosteneinsparungen, wie eine explorative Analyse australischer Patientendaten (n = 72) ergab. Adaptiert auf ein deutsches Kostenmodell können im Vergleich zur Standardbehandlung (d.h. fortgesetzte Beobachtung, orale Eisenempfeh-
* Ferinject® ist indiziert zur Behandlung von Eisenmangelzuständen, wenn orale Eisenpräparate unwirksam sind, orale Eisenpräparate nicht angewendet werden können und die medizinische Notwendigkeit einer raschen Eisengabe besteht. Die Diagnose eines Eisenmangels muss durch geeignete Laboruntersuchungen bestätigt sein.
Bei Patienten mit hämodialysepflichtiger chronischer Nierenerkrankung darf eine einmalige Dosis von 200 mg/Tag Ferinject® nicht überschritten werden. Kinder unter 14 Jahren dürfen kein Ferinject® erhalten. Für eine genauere Dosierung beachten Sie bitte die Fachinformation.
26 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH Kongresse
lungen und allogene Bluttransfusion) pro Patient Kosten in Höhe von ca. 786 € eingespart werden.
Notfallmanagement der Hyperkaliämie
Eine unerkannte Hyperkaliämie (Serumkalium >5,0 mmol/l) ist eine der Hauptursachen für Morbidität und Mortalität. Patienten mit einer chronischen Nierenerkrankung, Diabetes mellitus oder Herzinsuffizienz tragen ein besonders hohes Risiko für eine Hyperkaliämie, denn sie werden meist mit ReninAngiotensin-Aldosteron-SystemInhibitoren (RAASi) behandelt. Da Aldosteron die Ausscheidung von Kaliumionen in der Niere anregt, führt die Blockade des RAAS zu einer Erhöhung der Kaliumkonzentration und damit auf kurz oder lang zu einer behandlungsbedürftigen Hyperkaliämie. Weil diese lebensbedrohliche Arrhythmien auslösen kann, empfehlen die Leitlinien des European Resuscitation Council (ERC) eine Akutbehandlung zum Schutz des Myokards mit Calciumgluconat, das die Zellmembranen stabilisiert. Danach gilt es, die intrazelluläre Kaliumaufnahme zu stimulieren, wobei intravenöses Insulin und Glukose zum Einsatz kommen können und in einigen Fällen Beta-2-Rezeptor-Agonisten zur Inhalation.
Moderne Kaliumbinder empfohlen
Im Anschluss an die Akuttherapie empfehlen die ERC-Leitlinien bei moderater und schwerer Hyperkaliämie zur Kaliumeliminierung den
Einsatz von modernen Kaliumbindern wie z.B. Patiromer (Veltassa®) mit einer einmal täglichen Gabe. Patiromer ist ein nicht resorbierbares Polymer, das Kalium im distalen Kolon im Austausch gegen Kalzium bindet und so die die fäkale Kaliumausscheidung erhöht. Wie die Real-World-Studie von Di Palo et al. (JAMA Network Open 2022;5:e2145236) zeigte, konnte Patiromer bei Einmalgabe bereits im ersten Messzeitraum gemittelt nach 2,9 Stunden den Kaliumspiegel signifikant senken. Hierzu erhielten 82,3 % der Patienten nur eine einzige Dosis. Bei herzinsuffizienten Patienten ist nach Initiierung der leitliniengerechten Therapie eine langfristige Kaliumkontrolle essenziell. Wenn sich eine Hyperkaliämie entwickelt hat, sollte eine prognoserelevante Medikation wie z.B. ein RAASi nicht nicht dauerhaft abgesetzt, sondern nur pausiert werden, bis sich unter der Gabe moderner Kaliumbinder der Kaliumwert wieder normalisiert hat. Patiromer ist der einzige in Deutschland erhältliche Kaliumbinder, der in 4 klinischen randomisierten placebokontrollierten Studien gezeigt hat, dass der Einsatz der individuell bestmöglichen Herzmedikation mit RAASi und/oder Mineralkortikoid-Rezeptorantagonisten (MRA) möglich ist, wenn begleitend ein Kaliumbinder verabreicht wird. Die damit erreichte Kaliumabsenkung muss dann mit einer entsprechenden Erhaltungstherapie aufrechterhalten werden.
Für das langfristige Kaliummanagement kardiorenaler Patienten hat ein anwenderfreundlicher und gut verträglicher Kaliumbinder wie Patiromer relevante Vorteile: Das
geschmacksneutrale Pulver zur einmal täglichen Gabe erfordert keine tägliche Kaliumbestimmung. Es wird mit Wasser oder Fruchtsaft gemischt und kann auch mit verschiedenen weichen Lebensmitteln wie Joghurt, Apfelmus, Vanille- und Schokoladenpudding eingenommen werden. Patiromer ist natriumfrei und daher auch für Patienten geeignet, die selbst einen geringen Anstieg der Natriumbelastung nicht tolerieren können.
Fabian Sandner, Nürnberg
Quelle: Symposium „Eisen rauf – Kalium runter: Wieso, weshalb, warum? – Interdisziplinäre Therapieoptimierung“, veranstaltet von CSL Vifor im Rahmen des 33. Symposiums Intensivmedizin + Intensivpflege am 16. Februar 2023 in Bremen.
TransthyretinAmyloidose mit Kardiomyopathie –eine noch immer unterdiagnostizierte Erkrankung
Die Transthyretin-Amyloidose mit Kardiomyopathie (ATTR-CM) ist eine unterdiagnostizierte Erkrankung. Trotz leitlinienbasierter Diagnose-Algorithmen – wenige, gängige Methoden tragen entscheidend dazu bei, den Anfangsverdacht zu stärken – sind die bei der Diagnose wegweisenden Symptome ebenso wie die Erkrankung selbst noch zu wenig bekannt. Dabei steht mit Tafamidis 61 mg (Vyndaqel®) erstmals eine kausale Therapieoption zur Verfügung, die auch in der aktuellen ESCLeitlinie einen hohen Stellenwert bei den Therapieempfehlungen zur ATTR-CM einnimmt. Wie Exper-
27 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
ten im Rahmen eines von Pfizer veranstalteten Pressegesprächs anlässlich des Welt Amyloidose Tag am 26. Oktober 2022 erläuterten, können bestimmte Symptome und Symptomkonstellationen wichtige Anzeichen für die Erkrankung sein. Die Ärzte sind daher aufgefordert genau hinzuschauen, denn wer die relevanten Krankheitszeichen kennt, kann die ATTR-CM leicht erkennen.
Epidemiologie der ATTR-CM: Eine Krankheit wird sichtbar
Professor Roman Pfister, Köln, stellte eine epidemiologische Untersuchung vor, an der er entscheidend mitgewirkt hatte. Ziel war, die Gesamtheit der diagnostizierten Fälle der ATTR-CM abzubilden – mit Fokus auf Erkrankte ab 60 Jahren, um die Epidemiologie der ATTRwt-CM abzubilden. Die Resultate zeigen, dass die Erkrankung bereits vermehrt Aufmerksamkeit erhält. Zwischen 2009 und 2018 kam es zu einem Anstieg der Prävalenz von 15,5 auf 47,6/100.000 Personen. Die Inzidenz stieg von 4,8 auf 11,6/100.000 Personenjahre an. Zudem konnte ermittelt werden, dass Prävalenz und Inzidenz sowie deren Anstiege bei Männern und älteren Menschen über 80 Jahren besonders ausgeprägt waren. Die Untersuchung bestätigte darüber hinaus die schlechte Überlebensprognose. Pfister zeigte anhand einer MetaAnalyse, dass sowohl bei HFpEF (Herzinsuffizienz mit erhaltener Ejektionsfraktion)-Patienten mit ungeklärter LV-Hypertrophie als auch bei Patienten mit (beidseitigem) Karpaltunnelsyndrom, die
Prävalenz für die ATTR-CM erhöht war. „Wir müssen letztendlich interdisziplinär arbeiten, um diese Patienten zu erkennen”, schlussfolgerte Pfister. Wie eine gute interdisziplinäre Zusammenarbeit vom Initialverdacht auf eine ATTR-CM zur gesicherten Diagnose führen kann, erläuterte Professor Wilhelm Haverkamp, Berlin. „Zwar ist die Erkrankung von einer hohen Dunkelziffer gekennzeichnet, doch handelt es sich dabei vermutlich um ein Krankheitsbild, dessen Häufigkeit wir erheblich unterschätzen“, so der Experte.
Bei Herzinsuffizienz mit erhaltener Ejektionsfraktion genau hinschauen
Die beiden häufigsten Formen der kardialen Amyloidose sind die Leichtketten-Amyloidose (AL) und die Transthyretin-Amyloidose (ATTR-CM). Aufgrund der unterschiedlichen Ätiologien und Therapiebedürfnisse ist bei Verdacht auf eine ATTR-CM stets die Abklärung auf freie Leichtketten in Serum oder Urin notwendig. Während die AL-Amyloidose auf fehlgefaltete monoklonale ImmunglobulinLeichtketten zurückgeht, lagern sich bei der ATTR-CM fehlgefaltete Transthyretin-Monomere in Form von Fibrillen ab. Diese Ablagerungen treten nicht nur kardial, sondern systemisch auf. Hinweise auf eine ATTR-CM können auch die Hypertrophie und Versteifung des Herzmuskels sein, die durch extrazelluläre Ablagerungen von Amyloidfibrillen hervorgerufen wird und sich in Form einer HFpEF darstellt.
Wie eine Meta-Analyse zeigte, leidet mehr als jeder 10. Patient (11 %) mit HFpEF an einer ATTRCM. „Das ist ein Grund, bei dieser Ausprägung der Herzinsuffizienz genau hinzuschauen, zumal die ATTR-CM mit Tafamidis 61 mg ursächlich behandelt werden kann. Dass uns damit eine effektive Therapie zur Verfügung steht, ist ein Sachverhalt, der gewürdigt werden muss“, betonte Haverkamp.
Frühe Diagnose als Chance für die Erkrankten
„Es ist nicht schwer, eine ATTRCM zu erkennen“, so Haverkamp. Red Flags leiten durch die Diagnose der ATTR-CM. Ein möglicher Hinweis ist die relative Niedervoltage im EKG, bei der die QRS-Voltage nicht zur echokardiografisch gemessenen linksventrikulären (LV)-Dicke von ≥12mm zu passen scheint. Sind des Weiteren die Laborwerte von NTproBNP und Troponin erhöht, sollte rasch die weitere Abklärung erfolgen. Extrakardiale Symptome wie eine Spinalkanalstenose und ein (beidseitiges) Karpaltunnelsyndrom gehören zu den frühen Manifestationen der kardialen Amyloidose, weshalb insbesondere ältere Patienten im Anschluss an diese orthopädischen Symptomatiken regelmäßig auf eine ATTR-CM hin untersucht werden sollten.
Für eine klare Diagnose ist stets eine enge interdisziplinäre Zusammenarbeit nötig. „Vermehrt wird die nichtinvasive Skelettszintigrafie eingesetzt, bei der die kardialen Amyloidablagerungen als „Schwarzes Herz” (Perugini-Score von 2 oder 3) visualisiert werden
28 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
können. Das ist beweisend für eine ATTR-Amyloidose des Herzens“, erläuterte Haverkamp. Die genetische Abklärung zur Unterscheidung zwischen der altersbedingten Wildtyp-Variante (ATTRwt-CM) und der erblichen Form (ATTRvCM) ist nicht zuletzt für das enge familiäre Umfeld relevant. Alternativ zur Szintigrafie kann auf die kardiale Biopsie zurückgegriffen werden.
Eindringlich wies der Experte auf die Notwendigkeit der frühen Diagnosestellung und Therapie hin. Gestützt wird sein Statement von den Ergebnissen einer aktuellen modellbasierten Untersuchung, die den Nutzen der frühen Diagnose und Behandlung mit Tafamidis berechnete. Bei der ATTRwt-CM wurde bei früher Diagnose eine mittlere verbleibende Lebenserwartung von 11,65 Jahren ermittelt. Erfolgte die Diagnose der Erkrankung hingegen verspätet, blieben den Patienten im Schnitt 6,19 Jahre – ein Verlust von beinahe 5 ½ Lebensjahren. Lag den Schätzungen eine ATTRv-CM zugrunde, betrug der Gewinn an verbleibender Lebenszeit durch eine frühe Diagnosestellung sogar 7,76 Jahre (12,62 vs. 4,86 Jahre).
Langzeitdaten untermauern die klinische Bedeutung von Tafamidis
In der zulassungsrelevanten ATTRACT-Studie zeigte sich ein statistisch signifikanter Behandlungseffekt im Hinblick auf die Endpunkte Gesamtmortalität und kardiovaskuläre Hospitalisierung für Patienten mit Herzinsuffizienz der NYHAKlasse I und II. In dieser Studie wurden Tafamidis-Meglumin 80 mg
(4 × 20 mg), Tafamidis-Meglumin 20 mg und Placebo bei Patienten mit ATTR-CM verglichen. Die Behandlungsdauer betrug 30 Monate, die beiden Tafamidis-Studienarme wurden gepoolt analysiert. Tafamidis-Meglumin 80 mg ist dosisäquivalent zu Tafamidis 61 mg, einer Weichkapsel, die entwickelt wurde um die Einnahme komfortabler zu machen.
In einer sich an die ATTR-ACTStudie anschließenden Long-TermExtension-Studie erhielten jene Patienten, die in der ATTR-ACTStudie mit Tafamidis behandelt worden waren, weiterhin Tafamidis (= kontinuierlicher Behandlungsarm). Die Patienten im diskontinuierlichen Arm hatten zunächst Placebo erhalten und wurden bei Re-Randomisierung 20 mg oder 80 mg Tafamidis zugewiesen. Im Laufe der LTE-Studie wurden alle Patienten auf Tafamidis 61 mg umgestellt. „Die Überlebenszeitanalyse ist überaus eindrucksvoll, weil sie eben noch einmal den Effekt der primären Studie widerspiegelt”, so Pfister zur Tatsache, dass Patienten im kontinuierlichen Arm einen Vorteil gegenüber der diskontinuierlichen Behandlung hatten. Deutlich wird der Überlebensvorteil der Patienten an der Gesamtmortalität, die 44,9 % im kontinuierlichen bzw. 62,7 % im diskontinuierlichen Arm betrug. Doch auch die Patienten, die zunächst Placebo erhalten hatten, profitierten wahrscheinlich von der Umstellung auf Tafamidis, wie Pfister durch einen Vergleich der Kaplan-Maier-Kurven mit dem extrapolierten Placebo-Arm erläuterte. Die 5-Jahres-Überlebensdaten legen nahe, dass die langfristige kontinuierliche Gabe von Tafamidis auch in den untersuchten Subgrup-
pen, ATTRwt-CM bzw. ATTRvCM sowie NYHA-Klasse III, die bisherigen Beobachtungen aus der Zulassungsstudie übertreffen. Wie morphologische Untersuchungen bestätigen, kann Tafamidis die Progression der Amyloidablagerungen verzögern. Neue Sicherheitssignale wurden nicht beobachtet. „Die Nebenwirkungsrate war unter der Langzeittherapie eher etwas niedriger als in der frühen Studientherapie”, bestätigte Pfister.
ATTR-CM häufiger erkannt –Kapazitäten zur Versorgung ausbauen
Doch allein mit der frühen Diagnose und dem raschen Beginn einer Tafamidis-Therapie ist es nicht getan. Zu den notwendigen Maßnahmen in der Folgeüberwachung zählen zum Beispiel Bluttests (Troponin und NTproBNP) und ein EKG alle 6 Monate. Einmal jährlich kommen die echokardiografische Überwachung und ein 24-Stunden-EKG hinzu. Bei der ATTRv-CM sollten neurologische und ophthalmologische Untersuchungen ergänzt werden.
„Es wurde schon sehr viel in kurzer Zeit erreicht, dennoch gibt es Potenzial für Verbesserungen, z.B. beim Ausbau der aktuellen Versorgungskapazitäten und hinsichtlich der Kriterien für die Deeskalation der Therapie. Außerdem scheinen niedergelassene Ärzte zurückhaltend zu sein, was die Verordnung von Tafamidis angeht, obwohl die Therapie mittlerweile als nationale Praxisbesonderheit anerkannt ist”, schloss Pfister.
29 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
Elisabeth Wilhelmi, München
Kongresse
Mineralokortikoidrezeptor-Antagonist
Finerenon – die neue Therapiesäule bei CKD und T2D
Die chronische Nierenerkrankung (CKD) ist eine der häufigsten Komplikationen bei Typ-2-Diabetes (T2D) und trägt beträchtlich zur Morbidität und Mortalität bei. Die Lebenserwartung von CKD-Patienten mit T2D ist im Vergleich zu Gesunden um bis zu 16 Jahre verkürzt. Ohne adäquate Therapie kann die Verschlechterung der Nierenfunktion in einer terminalen Niereninsuffizienz mit Dialysepflicht, Nierentransplantation oder Tod enden. Außerdem erhöht sich das kardiovaskuläre Risiko. Trotz bestehender Therapien kommt es bei den T2D-Patienten oftmals zur Progression der CKD. Welche neuen Therapieansätze bei diesen Patienten dazu beitragen können, das Restrisiko für kardiorenale Ereignisse zu reduzieren, diskutierten Experten auf einem von Bayer veranstalteten Symposium anlässlich der 16. Herbsttagung der DDG in Kooperation mit der 51. Jahrestagung der DGA.
Innovativer Wirkmechanismus zum Schutz von Nieren und Herz
Lange Zeit zielten die Therapien für CKD bei T2D vorrangig auf metabolische und hämodynamische Faktoren ab. Die Mineralokortikoidrezeptor-(MR-)Überaktivierung, zu der es bei T2D kommt, konnte nicht adressiert werden. In diese therapeutische Lücke bei CKD und T2D stößt nun der erste und einzige in dieser Indikation zugelassene
Finerenon erhält Zulassungserweiterung
Basierend auf den positiven Ergebnissen der Phase-III-Studie FIDELIO-DKD wurde Finerenon (Kerendia®) im Februar 2022 in der EU erstmals zugelassen für erwachsene Patienten mit chronischer Nierenerkrankung (CKD-Stadium 3 und 4 mit Albuminurie) in Verbindung mit Typ-2-Diabetes (T2D). Die Europäische Kommission hat am 10. Februar 2023 die Zulassung für Finerenon auf die Behandlung von frühen Stadien der CKD in Verbindung mit T2D erweitert. Grundlage dafür waren die Erkenntnisse aus den kardiovaskulären Ergebnissen der Phase-III-Studie FIGARO-DKD. Diese Studie umfasste etwa 7.400 Patienten über ein breites Spektrum an Schweregraden der Erkrankung, einschließlich der CKD Stadien 1 – 4 mit Albuminurie in Verbindung mit T2D. Wie die Ergebnisse zeigen, senkte Finerenon im Vergleich zu Placebo das Risiko von kardiovaskulären Ereignissen bei erwachsenen Patienten mit CKD und T2D signifikant. Kerendia® ist jetzt für die Behandlung der chronischen Nierenerkrankung (mit Albuminurie) im Zusammenhang mit Typ-2-Diabetes bei Erwachsenen indiziert.
nichtsteroidale MR-Antagonist Finerenon (Kerendia®), der seit 2022 zur Behandlung der chronischen Nierenerkrankung (Stadium 3 und 4 mit Albuminurie) bei Erwachsenen mit Typ-2-Diabetes zugelassen ist.
„Das Fortschreiten der CKD bei T2D wird durch die Kombination metabolischer, hämodynamischer sowie entzündlicher und fibrotischer Faktoren beeinflusst. Angetrieben werden Entzündung und Fibrose durch die Überaktivierung des Mineralokortikoidrezeptors“, erklärte Dr. Inga-Nadine Kummer, Aschaffenburg. Inflammation und Fibrose können unbehandelt bei CKD und T2D zu irreparablen Zellveränderungen führen, die sich nicht nur auf die Nieren beschränken.
Das Management von T2D und CKD fußt nun mit Finerenon auf einer weiteren Therapiesäule, wie Kummer erläuterte. So wird Finerenon bereits in internationalen Leitlinien und im ADA/KDIGOKonsensus-Report 2022 zum Dia-
betes-Management bei CKD empfohlen: für Patienten mit T2D, einer eGFR (geschätzte glomeruläre Filtrationsrate) ≥25 ml/min/1,73m2 , normaler Serumkaliumkonzentration und einem Urin-Albumin-Kreatinin-Quotient (UACR) ≥30 mg/g.
Patienten profitieren zusätzlich von Finerenon
Die zulassungsrelevante klinische Phase-III-Studie FIDELIO-DKD untersuchte die Wirksamkeit und Sicherheit von Finerenon bei Patienten mit CKD bei T2D. Im kombinierten primären renalen Endpunkt (Zeit bis zum Nierenversagen, anhaltende ≥40 % Abnahme des eGFR vs. Baseline oder Tod durch Nierenversagen) zeigte Finerenon eine signifikante relative Risikoreduktion von 18 % gegenüber Placebo (HR: 0,82; 95%-KI: 0,73 – 0,93; p = 0,001). „Zusätzlich zur einer optimierten RAS-Blockade senkte Finerenon auch das Risiko für den kombinierten kardiovaskulä-
30 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH Kongresse
ren Endpunkt signifikant um 14 %. Dabei konnten die Patienten von Finerenon zusätzlich profitieren – unabhängig davon, ob sie einen SGLT2-Hemmer oder GLP-1-Rezeptorantagonisten erhielten“, erläuterte Dr. Ludwig Merker, Haan. Dieser wichtige sekundäre Endpunkt war eine Kombination aus der Zeit bis zum kardiovaskulären Tod, dem Auftreten nicht tödlicher Myokardinfarkte, einem nicht tödlichen Schlaganfall oder Krankenhausaufenthalt wegen Herzinsuffizienz. Die mediane Dauer der Nachbeobachtung betrug 2,6 Jahre.
Nebenwirkungen mit Finerenon weitgehend auf Placebo-Niveau
Die Ergebnisse von FIDELIODKD belegen auch das günstige Sicherheitsprofil von Finerenon: Therapiebedingte unerwünschte und schwerwiegende unerwünschte Ereignisse lagen weitgehend auf Placebo-Niveau. Der Blutdruck sank durchschnittlich um ca. 3 – 4 mmHg. Es gab kaum geschlechtshormonelle Nebenwirkungen. „Unter der Therapie mit Finerenon kann es zu einer leichten Zunahme des Serumkaliums kommen. Die klinischen Auswirkungen waren allerdings gering. Die Hyperkaliämien konnten im Rahmen von routinemäßigen Kontrollen des Kaliumspiegels und mit kurzen Therapiepausen gut beherrscht werden“, berichtete Merker.
Professor Thomas Ebert, Leipzig, empfahl, entsprechend der Fachinformation das Serumkalium und eGFR vor Beginn der Therapie mit Finerenon zu bestimmen. Die Anfangsdosis beträgt 10 mg Finerenon einmal täglich. Nach 4 Wo-
chen sollten die Kaliumkonzentration im Serum und die eGFR erneut bestimmt werden, um dann die Therapie auf die Standarddosis von 20 mg Finerenon täglich anzupassen, wenn es das Serumkalium erlaubt. „Wenn der SerumkaliumWert über 5,5 mmol/l steigt, sollte die Behandlung ausgesetzt werden. Eine Wiederaufnahme mit 10 mg Finerenon einmal täglich, kann erwogen werden, wenn das Serumkalium unter 5 mmol/l gesunken ist“, ergänzte Merker.
Diagnostik und RisikoKlassifikation der CKD
Eine CKD kann lange Zeit asymptomatisch verlaufen und wird bei T2D in vielen Fällen nicht frühzeitig erkannt. Um die Patienten mit einem erhöhten Risiko frühzeitig zu identifizieren, ist eine Früherkennung mittels UACR bzw. Albuminurie-Messung und eGFR-Bestimmung wichtig. Denn der Albuminurie-Wert ist ein wichtiger Marker zur Früherkennung von strukturellen Nierenschäden. Internationale Leitlinien empfehlen bei T2D ab Diagnosestellung mindestens einmal jährlich UACR und eGFR zu ermitteln. „Trotz der Empfehlungen in den Leitlinien ist CKD bei Patienten mit T2D unterdiagnostiziert, und die kombinierte Diagnostik mit eGFR plus UACR erfolgt zu selten. Die eGFR allein liefert ein unvollständiges Bild des kardiovaskulär bedingten Sterberisikos bei CKD-Patienten. Erst die kombinierte Betrachtung von eGFR und UACR ergibt eine genauere Risikoprognose der Mortalität“, erläuterte Kummer abschließend.
Fabian Sandner, Nürnberg
Verstärkte Menstruationsblutungen – eine Blutgerinnungsstörung könnte die Ursache sein
Verstärkten Menstruationsblutungen sollten Frauen, Mädchen sowie Ärzten stets Aufmerksamkeit schenken. Denn sie sind ein ernst zu nehmender Indikator für eine mögliche Blutgerinnungsstörung. Nicht nur Männer, auch Mädchen und Frauen können angeborene oder erworbene Gerinnungsstörungen haben, die speziell abgestimmte Therapien erfordern. Problematisch ist, dass Blutgerinnungsstörungen bei vielen Frauen erst spät oder gar nicht erkannt und behandelt werden. Bei einem von Novo Nordisk veranstalteten virtuellen Roundtable berichteten Expertinnen über ihre Erfahrungen und stellten die Ergebnisse einer großen Umfrage zu Diagnose und Versorgungslage bei Frauen und Mädchen mit Gerinnungsstörungen vor.
Eine verstärkte Menstruationsblutung hat vielfältige Auswirkungen
Wann ist eine Menstruationsblutung „normal“ und wann wird sie als übermäßig stark eingestuft? Stephanie Seremetis von Novo Nordisk, erklärte dazu: „Eine verstärkte Menstruationsblutung dauert mehr als 7 Tage an, geht mit einem Blutverlust von etwa 80 ml pro Menstruation und großen Blutklumpen einher. Binden/Tampons müssen alle 1 – 2 Stunden und während der Nacht gewechselt werden. Zudem ist ein doppelter Schutz aus Bin-
31 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
den und Tampons notwendig.“ Verstärkte Menstruationsblutungen haben erhebliche Auswirkungen auf das Leben der betroffenen Frauen, so Seremetis weiter. „Neben körperlichen Problemen, wie Menstruationsschmerzen und Eisenmangel mit all seinen Folgen, haben verstärkte Menstruationsblutungen einen immensen Einfluss auf die Psyche, die Lebensqualität und das soziale Leben.“ Häufig kommt es zu Fehlzeiten in der Arbeit bzw. der Schule. Auch Auswirkungen auf einen Kinderwunsch sowie Depression, Angst, soziale Isolation und Scham sind mögliche Folgen.
Verbesserungsbedarf bei Diagnose und Behandlung
Warum mehr Bewusstsein für verstärkte Menstruationsblutungen geschaffen werden muss, verdeutlichte Professorin Rezan Abdul-Kadir, London. „Verstärkte Menstruationsblutungen sind ein weit verbreitetes gynäkologisches Problem, das rund 30 % der Frauen irgendwann einmal während ihrer reproduktiven Jahre betrifft. Doch aufgrund der Stigmatisierung des Themas Menstruation – bei betroffenen Frauen und sogar bei Ärztinnen –kommt es oft zu Verzögerungen bei der Diagnose und Behandlung der Erkrankung. Außerdem denken viele Ärzte bei verstärkten Menstruationsblutungen oder anderen starken gynäkologischen Blutungen nicht an eine Blutgerinnungsstörung als Ursache.“ Weitere Gründe für eine (zu) späte Diagnose sind laut Abdul-Kadir, dass der Zugang zu geeigneten Laboruntersuchungen oft eingeschränkt ist oder ganz fehlt und zu wenig Patientinnen an ein
spezialisiertes Zentrum überwiesen werden, wo sie eine angemessene Behandlung erhalten können. „Laut einer niederländischen Studie“, so Abdul-Kadir weiter, „traten bei Männern und Frauen mit autosomal vererbten Blutgerinnungsstörungen die ersten Blutungen im gleichen Alter auf, die Diagnose wurde bei Frauen jedoch erst 6 Jahre später gestellt. Bei der Diagnose waren die Frauen im Schnitt 22,5 Jahre alt – über 10 Jahre nach der ersten Menstruation.“
Blutgerinnungsstörungen – bei Frauen häufiger als vermutet
„Überdies liegt bei 15 – 30 % der Frauen mit verstärkten Menstruationsblutungen eine nicht diagnostizierte angeborene Blutgerinnungsstörung zugrunde“, berichtete die Gynäkologin. „Viele Menschen glauben irrtümlicherweise, dass Hämophilie – die bekannteste, wenn auch nicht die häufigste – Blutgerinnungsstörung ausschließlich Männer betrifft, da sie X-chromosomal vererbt wird, und Frauen daher nur die genetische Mutation übertragen könnten, aber nicht erkranken würden.“ Tatsächlich kommen auf jeden Mann mit Hämophilie 2 – 5 Überträgerinnen, von denen 30 % symptomatische Blutungen aufweisen. Andere Gerinnungsstörungen, wie das von-Willebrand-Syndrom und angeborene Thrombozytenstörungen, betreffen Frauen und Männer sogar gleichermaßen.
Gerade bei Frauen und Mädchen haben Blutgerinnungsstörungen besonders starke Auswirkungen: Bei ihnen treten wie bei Männern z.B. Zahnfleisch- und Nasenbluten, Gelenkblutungen und Blutungen
nach Verletzungen und Operationen auf, aber hinzu kommen spezifische Blutungen, die mit Menstruation, Schwangerschaft und Geburt einhergehen.
Umfrage zeigt Möglichkeiten zur Verbesserung der Versorgung
Was sind die Ursachen für die unzureichende Diagnose von verstärkten Menstruationsblutungen und Blutgerinnungsstörungen bei Frauen und Mädchen und wie kann dies verbessert werden? Diesen Fragen ging eine große multinationale Umfrage bei 6.099 Frauen im Alter von 16 – 60 Jahren, 353 Allgemeinärzten sowie 426 Geburtshelfern/Gynäkologen nach. Professorin Roshni Kulkarni, East Lansing/USA, fasste die eindrücklichen Ergebnisse zusammen: „Mehr als ein Viertel der befragten Frauen und Mädchen gab an, Symptome zu haben, die als Risikoindikatoren für verstärkte Menstruationsblutungen gelten. 61 % berichteten von häufigen Beeinträchtigungen durch verstärkte Menstruationsblutungen bei Arbeit, Ausbildung oder alltäglichen Aktivitäten.“ Die Umfrage machte zudem deutlich, dass Allgemeinärzte und Geburtshelfer/Gynäkologen einige wichtige Indikatoren für eine zugrunde liegende Blutgerinnungsstörung nur bedingt kennen. „Man sollte meinen, dass dieser Personenkreis an eine mögliche Gerinnungsstörung denkt, wenn eine Patientin etwa Verwandte mit einer solchen Erkrankung, verstärkte Menstruationsblutungen, übermäßige postoperative Blutungen, Blutungen nach Zahnbehandlungen oder starke postpartale Blutungen aufweist. Doch bei den Befrag-
32 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH Kongresse
ten waren es je nach Indikator nur 20 – 56 %“, so Kulkarni. Die Umfrage zeigte auch, dass nicht immer alle wesentlichen Untersuchungen bei verstärkten Menstruationsblutungen durchgeführt werden. Weniger als 10 % der Ärzte suchen mit dem Blutungsscore nach anderen Blutungssymptomen und weniger als 15 % verwenden Menstruationsblutungsscores.
Ein weiteres Problem ist laut Kulkarni das Bewusstsein bei den Frauen selbst: Von den 76 % der befragten Frauen und Mädchen, die glauben, verstärkte Menstruationsblutungen gut zu erkennen, würden 22 % bei einer mehr als 8 Tage anhaltenden Menstruation nicht zum Arzt gehen. Die Umfrage hat außerdem ergeben, dass die große Mehrheit der Mädchen (74 %) zur Menarche Informationen und Ratschläge von der Mutter erhält. Das könnte bedeuten, dass die Vorstellung einer jungen Frau von einer normalen Menstruation stark von der Sichtweise ihrer Mutter beeinflusst wird. Kulkarni berichtete von Familien, bei denen Mütter während der Menstruation 15 – 20 Tage lang bluteten und dies als normal ansahen.
„Es muss einfach unter Frauen, Mädchen, Familienmitgliedern, Medizinstudenten, Ärzten und anderen Mitarbeitern des Gesundheitswesens bekannt werden, dass eine Menstruation, die länger als 7 Tage andauert, nicht normal ist und medizinisch abgeklärt werden muss“, betonte Kulkarni abschließend. Denn nur so können eine frühe Diagnose und Behandlung der zugrunde liegenden Ursache gewährleistet werden.
Elisabeth Wilhelmi, München
ESC-LeitlinienUpdate: Implikationen für das Management des plötzlichen Herztods mit der LifeVest®
Die European Society of Cardiology (ESC) hat mit dem kürzlich veröffentlichten Update der Guidelines* für Kammertachykardien (VT) und Kammerflimmern (VF) ihren Leitlinienansatz nochmals weiterentwickelt. So werden Therapiestrategien für bestimmte Indikationen, wie Myokardinfarkt oder Herzinsuffizienz, heute in verschiedenen Leitlinien adressiert. 2021 waren bereits die ESC Heartfailure (HF) Guidelines** überarbeitet worden. Die Leitlinien empfehlen nach wie vor eine Wartezeit von mindestens 3 Monaten bis zur ICDImplantation, da sich die Pumpleistung des Herzens unter optimaler medikamentöser Therapie verbessern und ein ICD obsolet werden kann. Um die vulnerable Phase der Wartezeit zu überbrücken und medikamentösen Therapien Zeit zu geben, um ihre Wirkung zu entfalten, kann ein tragbarer Cardioverter Defibrillator (WCD, LifeVest® Defibrillatorweste) eingesetzt werden. Dieser schützt Patienten dann bestmöglich vor dem plötzlichen Herztod (sudden cardiac death, SCD). Bei Post-Myokardinfarkt-Patienten hat der WCD in den aktuellen VT/ VF Guidelines* eine IIb-Empfeh-
* Zeppenfeld K. et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 2022;43:3997-4126
** McDonagh TA et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021;42:3599-3726
lung mit Evidenzgrad B erhalten. In den ESC HF Guidelines** besteht für Patienten mit ischämischer und nicht ischämischer Kardiomyopathie (ICM bzw. NICM) ebenfalls eine IIb-Empfehlung mit Evidenzgrad B. Vor diesem Hintergrund diskutierten internationale Kardiologie-Experten auf einer von ZOLL ausgerichteten Presseveranstaltung in Gießen über die Implikationen für das Management von SCD-Risikopatienten mit dem WCD.
WCD für die Wartezeit bis zur ICD-Implantation empfohlen
Der plötzliche Herztod zählt zu den häufigsten Todesursachen in der westlichen Welt: Allein hierzulande versterben daran pro Jahr über 100.000 Menschen. Entsprechend wichtig ist eine aktuelle und evidenzbasierte Entscheidungsgrundlage, anhand derer eine individuelle, möglichst optimale Therapiestrategie für den Patienten abgeleitet werden kann. „Modernen Diagnostiken zur SCD-Risikostratifizierung, wie etwa genetische Testungen, wird in den neuen VT/VF Guidelines ein zunehmend größerer Stellenwert beigemessen. Dennoch bleibt die linksventrikuläre Ejektionsfraktion (LVEF) ein entscheidender Parameter dafür, ob ein ICD nach 3 Monaten Wartezeit unter optimaler medikamentöser Therapie (OMT) infrage kommt“, sagte PD Sven Reek, Aarau. Für Patienten mit entsprechend erhöhtem SCD-Risiko empfiehlt die ESC in ihren Leitlinien während der Wartezeit weiterhin den Einsatz der LifeVest® (IIb, Evidenzgrad B). Diese Herangehensweise unterstützt auch Professor Frank
33 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
Breuckmann, Kitzingen: „Trotz extrem guter medikamentöser Therapie, die mittlerweile auch die sehr potenten Sodium-Glucose-CoTransporter-2(SGLT2)-Inhibitoren umfasst, haben wir immer noch viele Patienten, die in der frühen Phase nach einem kardialen Ereignis am SCD versterben.“ Dass bei der OMT nicht bloß reine Titrationseffekte zum Tragen kommen, lässt sich am Beispiel der SGLT2-Inhibitoren zeigen, deren Gabe direkt auf der Zieldosis erfolgt. „Aus den EMPEROR-Reduced und DAPAHF-Studien wissen wir, dass auch diese Medikamente ihre Wirkung erst nach einer Wartezeit entfalten“, berichtete PD Carsten Israel, Bielefeld. „Bei 40 % der Patienten lässt sich durch OMT die LVEF so weit verbessern, dass keine ICD-Indikation mehr besteht. Bis dahin können wir diese Menschen effektiv, und nicht invasiv mit der LifeVest® schützen. Das ist essenziell, gerade in der Zeit der Therapieeinleitung“, ergänzte Breuckmann.
Nicht ischämische Kardiomyopathie –Implikationen der ICDGuideline-Abwertung
Doch wie sieht es mit dem SCD-Risiko bei Patienten mit NICM aus? Bereits 2016 hatte die DANISHStudie ergeben, dass bei nicht ischämischer Herzinsuffizienz eine ICD-Prophylaxe nicht mit einer signifikant niedrigeren langfristigen Sterblichkeitsrate einherging als die klinische Standardversorgung. Dieser Erkenntnis wurde im letzten Update der ESC HF Guidelines Rechnung getragen und die Empfehlung für die primärprophylakti-
sche ICD-Implantation bei NICMPatienten vom Empfehlungsgrad I auf IIa (Evidenzgrad A) herabgestuft. Dr. Christian Ebner, Linz, betonte angesichts dessen, dass es sich bei dem austherapierten Patientenkollektiv der DANISHStudie ausdrücklich nicht um das LifeVest®-Patientenkollektiv gehandelt hat, denn „die in die DANISH-Studie eingeschlossenen Patienten hatten die Phase mit dem höchsten Risiko für den SCD ja bereits überlebt“. Wichtig ist hingegen, dass der WCD eine sichere und wirksame Option ist, um Denovo-Herzinsuffizienzpatienten mit einer NICM so lange zu schützen, bis die medikamentöse Therapie optimiert ist. Diese Schlussfolgerung spiegelt sich auch in den ESC HF Guidelines wider, in der die LifeVest® entsprechend mit einer IIb-Empfehlung (Evidenzgrad B) versehen ist.
hatte während des gesamten Follow-up keiner lebensbedrohliche Arrhythmien und keiner verstarb an einem SCD. Der Initiator der PROLONG-II-Studie, Professor David Duncker, Hannover, brachte die Ergebnisse auf den Punkt: „Es lohnt sich zu warten, um unnötige ICD-Implantationen zu vermeiden. Es lohnt sich aber auch zu schützen, denn auch in der verlängerten Wartezeit benötigen die Patienten Schutz vor dem plötzlichen Herztod. Die mittlere Tragezeit der Patienten in der PROLONG-Studie in Hannover war daher 101 Tage.“
WCD In der Praxis: Viel mehr als SCD-Schutz
Warten lohnt sich, schützen auch
Die HF-Leitlinienempfehlungen werden auch von der PROLONGII-Studie gestützt, in der untersucht wurde, welche Prognose Patienten mit neu diagnostizierter Herzinsuffizienz mit reduzierter LVEF (64 % NICM, 36 % ICM) haben, nachdem sie lebensrettende WCD-Schocks erhalten hatten. Sowohl 4 % der NICM- als auch 4 % der ICM-Patienten erhielten adäquate Schocks bei hämodynamisch relevanten VTs oder VFs. Bei 53 % der mit einer LifeVest® versorgten Patienten verbesserte sich nach einer verlängerten Tragezeit von bis zu 6 Monaten die LVEF auf >35 %, sodass bei ihnen keine ICD-Indikation mehr gegeben war. Von diesen Patienten
Die LifeVest® hat seit der Einführung vor 10 Jahren über 80.000 Patienten in Deutschland vor dem SCD geschützt. Dass sie ein wichtiges Tool in einem integrierten Gesamtkonzept zum Management von Herzinsuffizienzpatienten sein kann, erläuterte Dr. Stefan Winter. Als Oberarzt und Leiter für den Schwerpunkt Devicetherapie der Klinik für Kardiologie & Rhythmologie, St. Vinzenz-Hospital Köln kennt er den stationären Sektor der Patientenversorgung. In seiner weiteren Tätigkeit als niedergelassener Kardiologe im Medizinischen Versorgungszentrum St. Marien ist er aber auch für die Patienten im ambulanten Bereich verantwortlich. Nicht zuletzt vor dem Hintergrund einer sektorübergreifenden Betreuung bezeichnete er die Aufklärung und Schulung der Patienten als „das A & O“ in der Betreuung neu diagnostizierter herzinsuffizienter Patienten: „Die LifeVest® hilft auch dabei, die Patienten aufzuklären,
34 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
Kongresse/Mitteilungen
und unterstützt sie beim Selbstmanagement.“
Über die telematische Infrastruktur lässt sich außerdem während der WCD-Tragezeit jederzeit auf aktuelle Gesundheitsdaten des Patienten zugreifen. Das ZOLLPatient-Management-Network ermöglicht Einsicht in Elektrokardiogramme, Compliance-Reports, arrhythmische Ereignisse und optional in zusätzliche Trend-Daten, wie z.B. die Herzfrequenz, um eine sich ändernde Gesundheitssituation des Patienten frühzeitig zu erkennen. So kann die LifeVest® helfen, den Therapiefortschritt zu überwachen. „Wir sehen, dass das datengestützte Management bei Herzinsuffizienzpatienten zunehmend an Bedeutung gewinnt, und sind gespannt, wie sich das Thema zukünftig weiterentwickeln wird“, ergänzte Winter.
Brigitte Söllner, Erlangen
MITTEILUNGEN
Lyumjev® auch für Kinder und Jugendliche mit Typ1-Diabetes zugelassen
Die Europäische Arzneimittelagentur hat die Zulassung des Mahlzeiteninsulins Lyumjev®, der weiterentwickelten Formulierung von Insulin lispro, um die Anwendung bei Kindern und Jugendlichen erweitert. Damit kann Lyumjev® jetzt in der Konzentration U100 bei Erwachsenen, Jugendlichen und Kindern mit Typ-1-Diabetes ab einem Alter von 1 Jahr verordnet werden.
Lyumjev® wurde mit dem Ziel entwickelt, der Insulinwirkung stoffwechselgesunder Menschen noch näher zu kommen als bisherige Mahlzeiteninsuline. Es zeichnet sich durch einen besonders schnellen Wirkeintritt und eine verkürzte Wirkdauer aus. Dadurch können postprandiale Blutzuckerspitzen effektiver abgefangen werden, sodass die Patienten von einer verbesserten glykämischen Kontrolle profitieren. Ein weiterer Vorteil ist, dass kein Spritz-Ess-Abstand mehr eingehalten werden muss, was den Patienten eine größere Flexibilität bei den Mahlzeiten ermöglicht.
Studie PRONTO-Peds belegt Wirksamkeit und Verträglichkeit auch bei Kindern
Die EMA hat die pädiatrische Zulassungserweiterung von Lyumjev® auf Basis der Ergebnisse der Treatto-Target-Studie PRONTO-Peds ausgesprochen. In dieser Studie erhielten 716 Kinder und Jugendliche mit Typ-1-Diabetes im Alter zwischen 1 und 17 Jahren für einen Zeitraum von 26 Wochen neben einem Basalinsulin entweder Lyumjev® (n = 280) oder herkömmliches Insulin lispro (n = 298) vor der Mahlzeit; eine kleinere Gruppe erhielt Lyumjev® 20 Minuten nach der Mahlzeit (n = 138). Primärer Endpunkt der Studie war die Nichtunterlegenheit der Veränderung des HbA1c-Wertes von Lyumjev® vs. Insulin lispro während des Beobachtungszeitraums.
Die Auswertung ergab eine Nichtunterlegenheit von Lyumjev® – sowohl zur Mahlzeit als auch danach verabreicht – bezüglich der HbA1cVeränderung gegenüber Insulin lispro von Baseline bis Woche 26.
Somit wurde der primäre Endpunkt der Studie erreicht. Der geschätzte Behandlungsunterschied gegenüber dem Vergleichspräparat betrug bei Lyumjev®-Gabe zur Mahlzeit –0,02 % (95%-KI: –0,17 bis 0,13) bzw. –0,23 mmol/mol (95%-KI: –1,84 bis 1,39) und bei Lyumjev®Gabe nach der Mahlzeit –0,02 % (95%-KI: –0,20 bis 0,17) bzw. –0,17 mmol/mol (95%-KI: –2,15 bis 1,81).
Dem Vergleichspräparat überlegen zeigte sich Lyumjev® in Bezug auf die Reduktion der postprandialen Glukosewerte. Zur Mahlzeit appliziert, erzielte Lyumjev® niedrigere Durchschnittswerte des eine Stunde nach einer Mahlzeit gemessenen täglichen Glukosewerts als herkömmliches Insulin lispro (p = 0,001). Außerdem reduzierte Lyumjev® zur Mahlzeit die täglichen Glukoseexkursionen in der Zeit vor einer Mahlzeit bis eine Stunde danach signifikant stärker als Insulin lispro (p < 0,001). Im Hinblick auf das Auftreten schwerer Hypoglykämien ergaben sich keine signifikanten Unterschiede zwischen den Patientengruppen. Auch die Häufigkeit von unerwünschten Nebenwirkungen war vergleichbar. Lediglich Reaktionen an der Injektionsstelle wurden unter Lyumjev® zur Mahlzeit mit 7,9 % häufiger beobachtet als in der Vergleichsgruppe mit 2,7 %.
Lyumjev® steht für Kinder ebenso wie für Erwachsene als U100er Konzentration im KwikPen und Junior KwikPen sowie in Patronen und Durchstechflaschen zur Verfügung. Für Erwachsene ab 18 Jahren gibt es Lyumjev® zusätzlich als U200er Konzentration im KwikPen.
B. S.
35 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH
TactiFlex™ Ablationskatheter in der EU zugelassen
Mit der CE-Kennzeichnung für den TactiFlex™ Ablationskatheter, Sensor Enabled™ von Abbott steht seit Februar 2023 ein innovativer Katheter für die Behandlung von Herzrhythmusstörungen wie z.B. Vorhofflimmern zur Verfügung. Der weltweit erste Ablationskatheter mit einer einzigartigen flexiblen Spitze und Anpressdruckmessung reduziert die Verfahrensdauer und die Strahlenbelastung der Patienten im Vergleich zur Standard-Ablation.
In der TactiFlex AF DIE-Studie erzielte der Katheter überzeugende klinische Ergebnisse bei der Behandlung mit einer High-Power-Ablation (zwischen 40 und 50 Watt). Die Studie zeigte, dass der Katheter schnelle, sichere Läsionen erzeugt und die Arrhythmie bereits bei der erstmaligen Behandlung mit über 99 % Behandlungserfolg beendet.
Neu bei Arrhythmien: Flecainid-Tabletten 50 mg und 100 mg von Micro Labs
gert, wird die Herzmuskelaktivität gedämpft, die hohe Herzfrequenz verlangsamt und der unregelmäßige Herzschlag gebremst.
Flecainid Micro Labs wirkt schnell, denn der Organismus kann auf die volle Dosis des Arzneimittels zugreifen, eine Umwandlung des Wirkstoffs innerhalb des Körpers ist nicht erforderlich. Das Medikament wird rasch vom Blut über den Darm aufgenommen, die höchste
Klasse-Ic-Antiarrhythmika
TactiFlex™ Ablationskatheter, Sensor Enabled™ (© Abbott).
Nach Integration des TactiFlex™
Katheters in das EnSite™ X EP System von Abbott, mit dem sich Bereiche im Herzen identifizieren lassen, die eine Ablationsbehandlung erfordern, gibt der Katheter High-Power-Radiofrequenzenergie ab, wobei er sich dank seiner speziellen Spitze, die sich bei Kontakt mit der Herzwand biegt, besser an das Herzgewebe anpasst als herkömmliche Katheter. Darüber hinaus verkürzt sich bei Verwendung des TactiFlex™ Katheters die Behandlungszeit im Vergleich zu den Kathetern der vorherigen Generation von Abbott.
Flecainid ist ein Wirkstoff aus der Gruppe der Antiarrhythmika der Klasse Ic. Es wird angewendet zur Behandlung und Rezidivprophylaxe von symptomatischen und behandlungsbedürftigen tachykarden supraventrikulären Herzrhythmusstörungen und ist außerdem angezeigt zur Therapie und Rezidivprophylaxe von tachykarden ventrikulären Herzrhythmusstörungen, wenn diese nach Beurteilung des Arztes lebensbedrohend sind und wenn andere Therapieformen unwirksam sind oder nicht vertragen werden.
Die antiarrhythmischen Effekte von Flecainid Micro Labs beruhen auf der Blockade kardialer Natriumkanäle an der Herzmuskelzelle. Diese verzögert die Weiterleitung der vom Sinusknoten ausgehenden elektrischen Impulse – die Schrittmacherzellen im Sinusknoten des rechten Herzvorhofs bestimmen die Herzpumpgeschwindigkeit. Da der Wirkstoff die intraventrikuläre Erregungsleitung signifikant verlän-
Antiarrhythmika der Klasse Ic (Flecainid oder Propafenon) führen zu einer ausgeprägten Natriumblockade, haben aber keine Auswirkung auf das QT-Intervall. Die Medikamente sind sinnvoll für die Behandlung von Patienten ohne strukturelle Herzerkrankung oder ischämische Herzkrankheit, die eine symptomatische supraventrikuläre Tachykardie (SVT) aufweisen und keine Kandidaten für eine Katheterablation sind. Darüber hinaus können Klasse Ic Antiarrhythmika auch für die pharmakologische Kardioversion von Vorhofflimmern eingesetzt werden.
Konzentration entsteht nach rund 3 Stunden. Der Wirkstoff wird in der Leber abgebaut und über die Nieren ausgeschieden.
Für eine gleichbleibende Konzentration im Blut werden die Tabletten in der Regel 2 × täglich zu oder nach den Mahlzeiten mit ausreichend Flüssigkeit eingenommen.
S. M.
36 Perfusion 1/2023 36. Jahrgang © Verlag PERFUSION GmbH Mitteilungen
F. S.








