PHARMA R&D A POST-PANDEMIC RENAISSANCE


ISSUE 50 2023 www.pharmafocusasia.com
PROUDLY PRESENTING OUR 50 th ISSUE
Trials to Revolutionise Cancer Care
Real-World Evidence and the Drug Development Process Dealing with ‘Pipeline Vision’ Novel Approaches in Early Phase Oncology
UPGRADE YOUR MARKETING STRATEGY
Highly accountable marketing campaigns. Every dollar counts.
Digitally powered marketing campaigns may be cheaper than you thought... Ask us how?
Use the webinar as a platform to launch new products and services


Grow your audience with increased reach, impact and user-friendliness
Rise above geographical boundaries
Generate new business

Gain the strong web presence differentiating yourself from competitors
Connect and engage with your target audience

Give more exposure to industry specific people
Increase your brand profile and share your capabilities with leading industry professionals
Our recent successful partnerships:

Let the true “Digital Transformation” be the base of all your marketing campaigns
“Digital
is the way forward”
AFTER COVID-19 PANDEMIC
Transformation
Email : advertise@pharmafocusasia.com Website : www.pharmafocusasia.com
Pharma R&D

A post-pandemic renaissance
Welcome to the 50th edition of Pharma Focus Asia Magazine!
As the pharmaceutical industry continues to evolve, innovation in Research & Development (R&D) remains crucial to meet the demand for new and effective treatments.
The COVID-19 pandemic had a significant impact on the pharmaceutical industry accelerating the development of vaccines, therapies, and diagnostic tests to combat the virus. Many pharmaceutical companies have redirected their resources towards R&D activities, infectious diseases, and development of innovative medicines.
The pandemic highlighted the importance of diverse populations in clinical trials. As a result, there is likely to be increased emphasis on recruiting more participants from racial and ethnic minorities, to ensure that new drugs are safe and effective for everyone. Collaborations and data sharing between governments, academic institutions, and pharmaceutical companies have resulted in stakeholders accelerating drug development and responding more quickly to future public health crises.
The adoption of new technologies such as artificial intelligence, machine learning, blockchain and big data analytics is becoming increasingly prevalent in drug discovery and development.
I n this issue, we highlight some of the exciting advancements in pharmaceutical research and development. We explore the latest trends in drug discovery, including real-world evidence into the process of clinical development, as well as cutting-edge technologies such as blockchain, artificial intelligence, etc.
Cancer remains a major area of focus for pharma R&D, with many new treatments being developed to improve patient outcomes and quality of life. We also examine novel approaches in early phase oncology trials to revolutionise cancer care and 3D bioprinting of tissues/organs for biomedical applications. We take a closer look at the growing field of precision medicine and the potential impact it could have on the future of healthcare.
As always, we are committed to providing our readers with the latest trends and valuable business insights from the industry. We hope this edition of magazine inspires you and provides valuable information in the industry’s pursuit to contribute to better healthcare for all.
Prasanthi Sadhu Editor
06
CLINICAL TRIALS

44 Novel Approaches in Early Phase Oncology Trials to Revolutionise Cancer Care
Sowmya Kaur, Head APAC Navitas Clinical Research and Global Head Clinical Solutions, Navitas Life Sciences
Atul Gupta, Vice President-Medical & Scientific Affairs, Navitas Life Sciences
Akash Gadgade, Senior Manager, Medical Services, Navitas Life Sciences
48 Reinventing Patient Recruitment

Achieving accelerated clinical endpoints through a revolutionary patient recruitment model
Jeff Parke, Visionary Co-founder, P.A.C.E. (Project for Accelerating Clinical Endpoints)
Raman Sehgal, Clinical Research, PFC Pharma Focus India Pvt. Ltd.
INFORMATION TECHNOLOGY

52 Digital Innovation
How you and your teams can digitally innovate successfully in the Pharma ecosystem

Fausto Artico, Global R&D Tech Head and Director of Innovation and Data Science – GSK
Kevin Harrigan, Director of Innovation and Engineering – GSK
56 Demystifying the Potential Applications of Blockchain Technology in Pharma and Biopharma Industry
A bird’s eye view
Amita Puranik1, Prajakta Dandekar2 and Ratnesh Jain1
1 Department of Biological sciences and Biotechnology, Institute of Chemical Technology
2 Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology
2 PHARMA FOCUS ASIA ISSUE 50 - 2023
CONTENTS STRATEGY
to
The Glocalisation Choice Your first, most fundamental glocalisation decision is where
focus.
Winning in
can
to ace the game?
Director, Beigene Cover Story RESEARCH & DEVELOPMENT 16 Real-World Evidence and the Drug Development Process Dealing with ‘Pipeline Vision’
N Liebman, Managing Director, IPQ Analytics, LLC 22 Precision Medicine Needs Precision Drugs
Brian D Smith, Principal Advisor, PragMedic 13
Launch How Indian Generic Pharma
use forecasting
Sanobar Syed, Associate
Michael
Chief Scientific Officer, Nangiotx 25 Growing Complex Injectable Portfolio in the Indian Generic Industries
Karel Petrak,
Senior Scientist 1,
Ltd
Sivakumar Ramachandran,
Jodas Expoim Pvt
Chief Technology Officer (CTO), Jodas Expoim Pvt Ltd 29 Doing More with Less Leveraging model-based pharmacology for cost-effective drug discovery and development
Chakravarty, CEO, Fractal Therapeutics 33 Bacterial Cancer Therapy
Chen, Scientific Researcher, CancerCare Manitoba Research Institute 36 3D Bioprinting of Tissues/Organs for Biomedical Applications
Kathuria, Co-founder, Nusmetics Pte. Ltd Nileshkumar Dubey, Assistant Professor (tenure-track) in the Faculty of Dentistry, National University of Singapore 42 Advancing Towards a Silent Pandemic A call to action
Rana, Doctor of Medicine, Clinical Microbiology A SPECIAL ISSUE ON PHARMA
Tathagata Dutta,
Arijit
Yongqiang
Himanshu
Aditya

Advisory Board
Alessio Piccoli
Lead
Sales and Business Development Activities for Europe Aragen Life Science

Andri Kusandri
Market Access and Government & Public Affairs Director
Merck Indonesia
Brian D Smith
Principal Advisor PragMedic
Gervasius Samosir

Partner, YCP Solidiance, Indonesia

Hassan Mostafa Mohamed Chairman & Chief Executive Office ReyadaPro


Imelda Leslie Vargas Regional Quality Assurance Director Zuellig Pharma

Neil J Campbell Chairman, CEO and Founder Celios Corporation, USA


Nicoleta Grecu Director

Pharmacovigilance Clinical Quality Assurance Clover Biopharmaceuticals
Nigel Cryer FRSC
Global Corporate Quality Audit Head, Sanofi Pasteur
Pramod Kashid

Senior Director, Clinical Trial Management Medpace
Quang Bui
Deputy Director at ANDA Vietnam Group, Vietnam

Tamara Miller
Senior Vice President, Product Development, Actinogen Medical Limited
EDITOR
Prasanthi Sadhu
EDITORIAL TEAM
Grace Jones
Harry Callum
Rohith Nuguri
Swetha M
ART DIRECTOR
M Abdul Hannan
PRODUCT MANAGER

Jeff Kenney
SENIOR PRODUCT ASSOCIATES
Ben Johnson
David Nelson
John Milton
Peter Thomas
Sussane Vincent
PRODUCT ASSOCIATE
Veronica Wilson
CIRCULATION TEAM
Sam Smith
SUBSCRIPTIONS IN-CHARGE
Vijay Kumar Gaddam
HEAD-OPERATIONS
S V Nageswara Rao
Ochre Media Private Limited Media Resource Centre,#9-1-129/1,201, 2nd Floor, Oxford Plaza, S.D Road, Secunderabad - 500003, Telangana, INDIA, Phone: +91 40 4961 4567, Fax: +91 40 4961 4555 Email: info@ochre-media.com
www.pharmafocusasia.com | www.ochre-media.com


© Ochre Media Private Limited. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, photocopying or otherwise, without prior permission of the publisher and copyright owner. Whilst every effort has been made to ensure the accuracy of the information in this publication, the publisher accepts no responsibility for errors or omissions. The products and services advertised are not endorsed by or connected with the publisher or its associates. The editorial opinions expressed in this publication are those of individual authors and not necessarily those of the publisher or of its associates.
Copies of Pharma Focus Asia can be purchased at the indicated cover prices. For bulk order reprints minimum order required is 500 copies, POA.
Magazine Subscribe LinkedIn
leading brand name consisting of information with latest breakthroughs and trends in the industry, with huge subscriber database that leads you to best promotions in the market. uses on Strategy, Research & Development, Clinical Trials, Manufacturing, Information Technology. Available in online version. e-Book also available.

www.pharmafocusasia.com Hard-copy Subscriptions: Email: info@pharmafocusasia.com Tel: +91 40 4961 4567 Media Contact: Email: advertise@pharmafocusasia.com Tel: +91 40 4961 4567
We Brand the Future. We create Difference.
190,000 READERS
globe.
A
More than
in pharma industry across the
This article addresses the first, key question in glocalisation: How should we allocate effort and resources across multiple countries? It argues that most current practice, depending on population size or GDP, is simplistic and naive. The article proposes a sophisticated multifactorial targeting approach that creates a portfolio approach to country management, so allocating the right sort of resources in the right amount to the right countries. This article therefore sets up article three, which is concerned with adapting strategies to targeted countries.
Brian D Smith, Principal Advisor, PragMedic
The Glocalisation Choice
Your first, most fundamental glocalisation decision is where to focus
In the first article in this series, I began from the premise that as soon as any life sciences firm moves significantly outside its home market, it faces the glocalisation challenge, the dilemma between global opportunities and local needs. This dilemma simultaneously promises growth and economies of scale whilst threatening expensive failure if local needs and wants are not addressed. The glocalisation challenge can only be addressed by thoughtful, intelligent choices about where and how to compete globally. In that first article, I outlined the three major steps to glocalisation that all companies follow: targeting, tailoring and learning. I then contrasted their
execution in exemplary companies with that in most companies. In this second article in the series, I go into more depth about exemplary practice in the first and most fundamental step in the glocalisation process: targeting effort across countries.
The dangers of isomorphic targeting
If, as I do in my research, you ask many different life science companies about how they allocate effort between the 200 national markets they could target, you get near-identical answers. This is odd. It’s like every customer in a fashionable boutique store choosing the same outfit despite their hugely different body shapes,
occasions and budgets. This similarity of choices might be understandable if it were the outcome of a rigorous decision process but in general it is not. Instead, these near-identical choices typically result from a phenomenon that academics call isomorphism, the instinct to do what one’s peers do. And what most firms do is to target countries according the market size or, if that isn’t known, population or economy size.
This isomorphic, “follow the heard” approach to targeting reduces the effectiveness of glocalisation in a number of ways. First, it often doesn’t focus efforts on where the most business is; a large market may have only a small accessible
6 PHARMA FOCUS ASIA ISSUE 50 - 2023
STRATEGY
market and vice versa. Second, it often ignores the probability of winning; a medium sized market with weak competitors deserves more attention than a large market with a dominant incumbent. Third, it ignores interaction between markets; some markets influence whole regions’ clinical practice or market access. Despite these flaws, isomorphic targeting persists because it is easy and safe; in career terms, copying what most other people are doing is the low-risk option. Nobody was ever fired for doing what everyone else does. But if a life sciences firm wants to glocalise better than its rivals, it must glocalise differently from its rivals. This begins with allocating effort between countries differently from its rivals. Exemplary firms who target better than most companies do so in 6, sequential steps, as in figure 1.
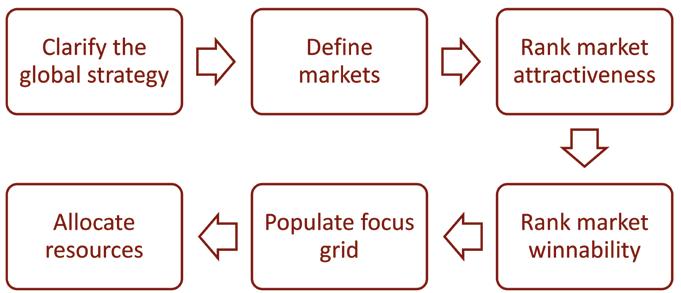
Step 1: Clarify the global strategy

Every disease area or brand has a core global strategy. Although it might be buried in a massive slide deck of a plan, the essence of that core global strategy is a statement of which customers to focus on and what kind of value to offer them. In pharma markets, the former is usually a context of patient, payer and professional and the latter usually a combination of clinical, economic and other customerperceived value drivers. Exemplary glocalisers begin targeting by being crystal clear
about what that core global strategy is. They then use it to guide the rest of the targeting process. Those companies that begin with an unclear global strategy inevitably lose sight of the benefits of globalisation.
Step 2: Define markets
The next difference between isomorphic and exemplary targeting is that the former targets countries whilst the latter targets markets. Often, countries are not homogeneous and contain two
7 www.pharmafocusasia.com
STRATEGY
Figure 1:The exemplary targeting process
or more distinct and different markets. Countries with basic state provision and developed private provision are an example of this. The process of targeting, as described in the following steps, works best when the inputs are homogenous markets and not heterogenous countries. So exemplary companies compile a list of targets that is not the same as, and is usually longer than, their list of possible countries. Each of these potential target markets is relatively homogenous (e.g., basic state market country X, private provision market country Y). Those companies that target countries rather than markets lose sight of the complexity within countries.
Step 3: Rank market attractiveness
The third difference between how most companies target and what exemplary companies do lies in their criteria for prioritisation. The many potential markets identified in step 2 are almost always too many for any company to
attack with sufficient resource, which implies prioritisation of markets. Isomorphic targeters prioritise on the basis of size but exemplary targeters’ criterion is multifactorial attractiveness. They decide what factors make a market attractive and weight those factors. Size is typically the most important but not market size; it is size of the target segment (as identified in the core global strategy, step 1) that is considered. Then other factors are considered: synergy with other markets, rate of growth and price levels are typical factors. Whatever the attractiveness criteria chosen, a simple calculation is all that is needed to rank the potential markets list from step 2 in order of relative multifactorial attractiveness, from most to least attractive. Companies that skip step 3 take too simplistic a view of what makes a market attractive.
Step 4: Rank market winnability
The fourth characteristic of exemplary targeting that distinguishes it from what
most companies do is relative competitive strength assessment. This takes the potential markets list from step 2 and, like step 3, puts them in order. But in step 4 the ordering is on the basis of the company’s ability to compete in each market, relative to all the other markets. Again, this assessment involves multiple, weighted factors. Typically, the performance of the product (or product range) relative to the local market leader is the most heavily weighted factor but price, distribution strength and brand reputation are also commonly considered competitive strength factors. As with step 3, a straightforward calculation is used to order the potential markets in a list from “Most likely to win” to “Least likely to win”. Companies that skip step 4 don’t allow for their own strengths and weaknesses.
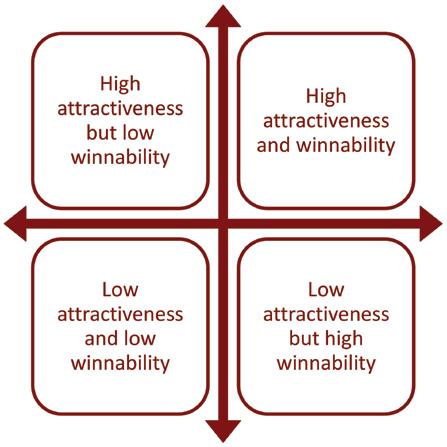
Step 5: Populate focus grid
The exemplary targeting step that distinguishes it most from isomorphic practice is the focus grid. As a result of steps 2, 3 and 4, exemplar companies are now equipped with two lists. Both lists include the names of all the potential markets to which they could allocate resources (the output of step 2) but each list has those markets in a different order. One list ranks the potential markets from most to least attractive on the basis of step 3’s multifactorial, weighted calculation of attractiveness factors. The other lists the same markets from most winnable to least winnable on the basis of step 4’s multifactorial, weighted calculation of competitive strength factors. Step 5 involves combining these two lists to create a focus grid that places each market according to both relative attractiveness and relative winnability, as in figure 2. In this way, the focus grid captures all of the analyses carried out in the preceding steps. Companies that don’t use it lack the critical insight it gives.
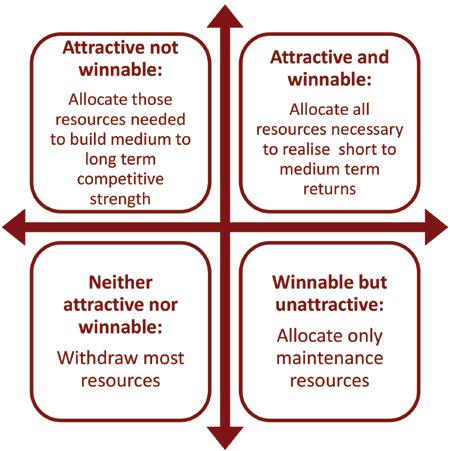
Step 6: Allocate resources
The culmination of exemplary targeting is how the analyses encapsulated in the
8 PHARMA FOCUS ASIA ISSUE 50 - 2023
STRATEGY
Figure 2: Focus grid
focus grid are used to guide resource allocation. Isomorphic target leads to binary decisions (e.g. enter or don’t enter market X) or graduated decisions (lots of resource here, some here, none there) about the quantity of resource allocated. But isomorphic targeting provides little or no guidance about the qualities or nature of the resource allocated. By contrast, the focus grid guides both how much resource and what kind of resource to allocate to each market. This makes the glocalisation process more effective. Attractive, winnable markets get lots of resource of the kind needed to create short term returns, such as investment in local sales and marketing. Attractive markets where the company is relatively weak get the amount and type of resources needed to build competitiveness with the aim of medium term returns, such as establishing local distribution and building brand reputation. Relatively unattractive
markets where the company is currently strong are allocated limited resources aimed at maintaining the current position, such as supporting current activity. Markets that are relatively unattractive and hard to win are treated opportun-
istically, with minimal resources allocated only to seize unexpected chances, such as competing for binding tenders. If unattractive markets are currently allocated large resources, these can be diverted to attractive markets. The guidance implied by the focus grid is summarised in figure 3.
Targeting is only the start
I hope it is obvious that the exemplary, six step process described in this article is significantly different from the isomorphic process that is typical of the life sciences industry. I hope too that the advantages of its more careful analyses and more structured approach are clear. Done well, this process avoids the three dangers of isomorphic targeting, described above. By intelligently allocating resources between markets, it gives exemplary companies a solid foundation on which to build their glocalisation strategy. But intelligent targeting of resources is only the first step in an effective glocalisation process. Exemplary practice also differs from isomorphic practice at the tailoring and learning stages. I will address those differences in the next two articles in this series.
Brian D Smith
University of Hertfordshire, UK, and Bocconi University, Italy and researches the evolution of business models and competitive strategy in the global life sciences industry. He has published over 300 papers, articles and books. www.pragmedic.com

AUTHOR BIO
STRATEGY
Figure 3: Resource allocation
Controlling nitrosamines impurities in pharmaceutical products
In 2018, N-Nitrosodimethylamine (NDMA) was identified in an active pharmaceutical ingredient (API). Since then, nitrosamines have been a pressing topic in the pharmaceutical world. With major regulatory updates introduced by the FDA and EMA, all pharmaceutical manufacturers have been instructed to conduct a risk assessment and proactively test their products for the presence of nitrosamines. It’s therefore crucial for pharmaceutical companies and manufacturers to get up to speed with these latest requirements
What are nitrosamines?
Nitrosamines are known environmental contaminants found in water and foods but have recently been identified in multiple drug products. They are a concern for pharmaceutical companies because they have even been identified as part of a group of high potency mutagenic carcinogens referred to as the “cohort of concern” (as per ICH’s M7 Mutagenic Impurities guidelines). Even at low levels, these genotoxic impurities pose a significant threat to human health.
Regulations to control nitrosamine contamination
International regulatory authorities have partnered to share information and publish guidelines for market authorisation holders (MAHs), including analytical methods to detect and identify nitrosamine impurities in drug products. Under the new regulations, almost all drug products are required to undergo nitrosamine testing. Even drug products which are planned for submission or have already been submitted require a risk assessment for the potential presence of nitrosamine impurities.
Regulatory agencies have set acceptable limits for nitrosamine impurities in relation to Angiotensin II receptor blockers (ARB). The FDA has determined that nitrosamines should not be present in the ARB API or DP. They have also published interim acceptable intake limits based on the maximum daily dose for NDMA, NDEA and NMBA.
These are:
• NDMA – 96 ng/day
• NDEA – 26.5 ng/day
• NMBA – 96 ng/day
The EMA has also published the same limits for NDMA and NDEA. Manufacturers may be required to conduct voluntary recalls if laboratory testing finds nitrosamine impurity levels exceed these limits.
The three-step risk evaluation
Both the FDA and EMA have described methods to identify the potential sources of nitrosamine contamination and formation as well as approaches to control or eliminate these sources during the manufacturing process. Accordingly, a three-step risk assessment has been proposed.
10 PHARMA FOCUS ASIA ISSUE 50 - 2023
STEP 1
Assess the risk of nitrosamine formation during Drug Substance (API) and drug product manufacturing (including products in development and approved or marketed products), taking into consideration potential root causes and sources of impurities formation.
STEP 2
Perform confirmatory testing (in case a risk of nitrosamine formation is confirmed).
STEP 3
Implement and report changes implemented to prevent or reduce nitrosamine impurities. The risk evaluation applies not only to finished products, but also New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs) that are planned for submission.

STEP 1: RISK ASSESSMENT Nitrosamines can be formed across all three stages of the manufacturing process (Figure 1). Common sources of nitrosamine formation are the solvents, reagents and catalysts used (especially amines) during API and drug manufacturing. Other sources include the presence of nitrites or nitrates in water, and contamination or cross-contamination of the equipment and starting materials used.
Manufacturers must consider the risk of nitrosamine formation at all stages of the manufacturing process from API synthesis to final product packaging.
The FDA instructed manufacturers to complete risk evaluations and report the outcome by March 31, 2021.
If a risk is identified: MAHs should submit the Step 1 response and proceed with Step 2 confirmatory testing of the drug or product.
If there is no risk: A risk evaluation of the finished product should be conducted and the outcome submitted once a final conclusion is reached.
STEP 2: CONFIRMATORY TESTING If a risk of nitrosamine impurities is identified during the risk assessment step, then MAHs must conduct confirmatory testing on the products identified to be at risk and report any confirmed presence of nitrosamines as soon as possible.
Acceptable limits to consider have been proposed, based on a product’s maximum daily dose (Table 1).
1. The AI limit is a daily exposure to a compound such as NDMA, NDEA, NMBA, NMPA, NIPEA or NDIPA that approximates a 1:100,000 cancer risk after 70 years of exposure. Appendix B includes a description of the AI derivation for NDMA, which is an example of how the FDA applied ICH M7(R1) to set a limit.
2. The conversion of AI limit into ppm varies by product and is calculated based on a drug’s maximum daily dose (MDD) as reflected in the drug label
11 www.pharmafocusasia.com
Advertorial
API
Packaged
Nitrosamine AI Limit (ng/day)1,2 NDMA 96 NDEA 26.5 NMBA 96 NMPA 26.5 NIPEA 26.5 NDIPA 26.5
Manufacturing Drug Product Manufacturing
Drug Product & Shelf life
Figure 1: Three stages of the manufacturing process
Table 1: Acceptable intake limits
(ppm = AI (ng)/MDD
(mg)). Control of Nitrosamine Impurities in Human Drugs | FDA
While there is no requirement to use the published testing and analytical methodologies described, any method used should be quantitative, adequately sensitive, validated and conducted at a GMP-compliant facility. Below are some key points to consider for confirmatory testing:
• The LOQs and LODs mentioned by the regulatory agencies should be met to ensure that the method used is equivalent to published methods.
• If a limit-based test is used, it must be accompanied by appropriate scientific justification in the risk assessment document, with evidence that there is no increase in the concentration of nitrosamine impurities over time.
• It’s recommended to use the drug product for the appropriate market for method validation and the choice of product strength should be described in case of the presence of multiple drug strengths.
When developing analytical methods, MAHs should consider interferences caused by the presence of trace amounts of nitrosamines in testing materials utilised such as water, plastic or rubber products. In situ formation of nitrosamines is also possible and should be accounted for, such as in the case of ranitidine in high-temperature conditions.
If nitrosamines are detected during testing: The root cause should be identified and stated in the report before moving to Step 3.
If no nitrosamines are detected: A report should be filed with (or be available to) the appropriate regulatory authority.
STEP 3: IMPLEMENTING AND REPORTING
CHANGES If the confirmatory tests in Step 2 confirm the presence of nitrosamine impurities, MAHs should implement changes to prevent or reduce nitrosamine impurities in APIs and drug products. The changes implemented should be reported to the appropriate regulatory agency.
If one or more nitrosamine impurities detected are below the interim acceptable limit, then steps should be taken to determine their origin as well as actions needed for respective batches. Root causes should be determined, and corrective and preventive actions should be implemented as well as a risk mitigation plan that ensures that levels will be consistently below interim acceptable limits at the end of the product’s shelf life.
COMPENDIAL CHAPTERS ON NITROSAMINES
In addition to the guidance from regulatory authori-
ties, compendial chapters have been proposed and/or published for publications USP Nitrosamine Impurities and EP 2.5.42. N-Nitrosamines in active substances. The compendial chapters are aligned with the guidelines in terms of the assessment and testing as well as the recommended acceptable levels. Several methods using GC-MS, GC-MSMS or LC-MSMS have been published for the detection of specific nitrosamine impurities.
SGS Nitrosamine Testing Capabilities at Navi Mumbai, India
SGS offers a complete range of services for Nitrosamine Testing at our campus in Navi Mumbai, India. Our cGMP compliant laboratories use stateof-the-art technologies that detect ultra-trace levels of leachables and nitrosamines at recommended LOD/LOQ levels and specifications. Our test method is based on LC-MS/MS, GC-MS/MS and LC-HRMS to detect the presence of nitrosamine traces in drug products, raw materials, process impurities and active pharmaceutical ingredients (APIs).
Sensitivity achieved for respective nitrosamine with this state-of-the-art equipment are at very low levels and depending on the specification of individual nitrosamine, number of methods can be developed and validated as per customer requirement.
SGS Solutions
SGS scientists deliver multifaceted, customercentric programs at local and international levels — supporting the delivery of high-quality, compliant biopharmaceutical and pharmaceutical drugs and medical devices. With a global network of 22 laboratories, we provide a diverse range of trusted analytical testing and clinical research solutions to help you navigate your journey to market and ensure patient safety.
About SGS
We are the world’s leading testing, inspection and certification company. We are recognised as the global benchmark for quality and integrity. Our 96,000 employees operate a network of 2,700 offices and laboratories, working together to enable a better, safer and more interconnected world. Wherever you are, whatever your industry, our experts worldwide provide specialised solutions to make your business faster, simpler and more efficient.
12 PHARMA FOCUS ASIA ISSUE 50 - 2023
WINNING IN LAUNCH How Indian Generic Pharma can use forecasting to ace the game?
In a pharmaceutical world that is changing rapidly, the only strategy that is guaranteed to fail is not taking risks. How do you quantify these risks? Forecasting & analytics is the modern day ‘crystal ball’ which is rescuing decision makers and how?.
In a pharmaceutical world that is changing rapidly, the only strategy that is guaranteed to fail is not taking risks. How do you quantify these risks? Forecasting and analytics is the modern day “crystal ball” which is rescuing decision makers and how?
Generic and innovator pharmaceutical companies have stark differences such as times, resources and costs allocated to launch products. Given longer clinical phases and regulatory approval periods in innovator companies, it takes a great deal of time, while such requirements are not mandatory within generic companies. The innovator companies have leapfrogged
in the use of technology in deriving key critical decisions. However, generic pharmaceutical companies still lag behind in adopting technology in commercial functions. The objective of generic companies is to get fastest to the market at the lowest cost. But the industry is consolidating and hence there is a need for 'differentiation’. This can be achieved by using the appropriate market research, forecasting and analytical techniques to drive uniqueness in the crowded generic market. If generic companies in pharma want to thrive it’s time to reimagine traditional business models and embrace new technologies that put the patient front and centre. Learning key best practices from
the innovator industry will not only let them thrive but also soar high.
India has grown into an epicentre of the generic industry and is rightfully known as the “pharmacy of the world” due to the low cost and high quality of its medicines.
According to InvestIndia.org, The Indian pharmaceutical industry is expected to reach US$65 billion by 2024 and to US$130 billion by 2030. The pharmaceutical industry in India is currently valued at US$50 billion. India supplies over 50 per cent of Africa’s requirement for generics, ~40 per cent of generic demand in the US and ~25 per cent of all medicine in the UK. India has

13 www.pharmafocusasia.com
STRATEGY
Sanobar Syed, Associate Director Beigene.
“The biggest risk is not taking any risk.”
– Mark Zuckerberg, CEO, Meta
the maximum number of pharmaceutical manufacturing facilities that are in compliance with the US Food and Drug Administration (USFDA) and has 500 API producers that make for around 8 per cent of the worldwide API market.
Generic companies aspiring to capture the growth opportunities must ensure they have functional and commercial excellence in place. They will need to master a staggering set of capabilities in product development, supply, and commercialisation. For some companies, achieving operational excellence is a matter of survival. Consider the Indian generics players that have transformed their manufacturing operating models and undertaken ambitious lean programs in recent years. The keys to success include having a clear operations agenda, building centres of competence, ensuring an open environment, creating opportunities for their best talent, and constantly searching for the next extraordinary goal. As a result, Indian companies are among the leaders in the generics industry, and their manufacturing plants are among the world’s top-performing facilities, with conversion costs per production unit of less than 10 percent of the industry median.
But how much have top Indian pharmaceutical companies adopted modern forecasting practices?
Before we deep dive into this it is important to understand the value of right launch.
Launch is one of the most critical moments in a product lifecycle. For 85 per cent of pharmaceutical launches, the product trajectory is set in the first six months. Especially in generic industry this is the time to penetrate the market reap the benefits of market share. Historically, limited real-life insights and an inflexible commercial model made it impossible for generic pharmaceutical companies to monitor performance dynamically and make timely course corrections.
What do the companies need to know? The wealth of data now available in the form of hospital data, clinical data and social media data, can boost the industry’s ability to respond to increased market complexity and enables the adoption of leading practice from other industries. These trends are shifting the basis for competition in pharmaceutical launch away from share of voice to share of insight. With this increased data transparency, winning launches will require pharma to harness new sources of information to develop superior real-time insights, and rapidly operationalise decisions based on these insights.
In the consumer packaged goods (CPG) industry, the first month of a product launch are critical in determining its success, brand teams monitor product launches and adjust their strategy in close to real-time. For pharma there are important questions: which doctor prescribed it and where? To which patient? What did the doctor and patient think about it?
Barring few leaders like Dr. Reddys Labs, Glenmark, or Cipla, the rest of the companies are still looking at traditional ways of predicting or forecasting their future sales. While this may have worked until now, will it really give them the edge in the growing consolidated generic space?
Most Indian players are using inorganic growth as their main strategy. More deals are happening in Indian companies acquiring assets in US/ Europe, rather than MNCs acquiring Indian companies. As a general principle (assuming management bandwidth is not constricted), Indian companies are better off buying companies overseas than in India for the following reasons: Financial reporting in US/Europe has higher compliance and balance sheets are cleaner (penalty for noncompliance is very high); cost of borrowing capital is 2-3X lower than in India; regulatory and quality compliance is higher (USFDA approved assets); valuations
are more reasonable and in sync with market realities.
The industry focus is shifting from ‘growth’ to ‘sustainability of growth’. Profitable growth for the Indian generic pharma industry has been fuelled primarily by exports to regulated markets like USA. Fortunes for the likes of Sun Pharma, Dr. Reddy’s, Aurobindo, Lupin etc. are determined primarily by what happens to their US business. True 'information integration’ will happen only when the following are evaluated and implemented. To caution that this is not an exhaustive list but an indicative suggestion to raise the decision-making bar high in the company.
Data galore but where are the insights?
At any point in a generic company (defined here as anywhere between 250+ products) multiple in-house projects are ongoing where their R&D data is being collected in multiple databases and warehouses. This data is valuable and needs to be consolidated for better analytics. One of the best uses is to push all the data into a data repository or a ‘lake’ and build applications. This can be used by cross-functional teams like R&D, data analysts, data scientists, and others. This data lake will eventually be used for analytics and driving efficiencies in various streams and enable to view information on the go. This also helps in record keeping and maintaining a data sequence especially when investment, budgeting & resource allocations decisions are to be taken.
Know thy customer
Better insight into patient behaviour to improve market trends and effectiveness and healthcare outcomes. At any time, there are multiple purchase orders being dispatched and shipped which generates greater amounts of data that companies can tap — coupled with advanced analytic and forecasting models, mean that pharmaceutical manufacturers can gain much greater insight into existing
14 PHARMA FOCUS ASIA ISSUE 50 - 2023
STRATEGY
patient and buying behaviour. The company can then use that information to create a targeted approach and rather educate their customer about their buying patterns. Key questions can be asked if there has been a sudden dip or rise in the supply or demand of any particular active pharmaceutical ingredient or formulation. Innovators are using modern technology to improve compliance and leading to uptake and higher revenues.
Linkage between strategic forecasting and sales performance
With increasing competition from generics, big pharma is getting smarter about analysing and driving effectiveness in its sales and marketing operations. New, niche and underserved markets may be spotted by analysing information from social media, demographics, electronic medical records and other sources of data. Equally, analysing the effectiveness
of sales efforts and capturing the feedback received by the sales force during client visits and using it effectively can help pharmaceutical companies get an edge on their competition. In theory, this will allow the companies to make better informed decisions, faster than ever before.
Rise with the industry
Being able to intelligently search vast data sets of patents, scientific publications, and clinical trials data should, in theory, help accelerate the discovery of new generic drugs by enabling researchers to examine previous results of tests. Applying predictive analytics to the search parameters should help narrow down on the relevant information and also get insight into which avenues are likely to yield best results. The industry is already starting to look at how it can get greater access to more data in order to help accelerate this
process. The generic industry can learn and implement avenues to share information to benefit the patient at large.
Conclusion
In order to effectively integrate meaningful forecasts, organisations need to tackle three areas: secure access to the most valuable data (including through collaboration with payers, providers, academics, and third parties), develop unique granular insights by combining advanced analytics with creativity and visualisation technologies, and create organisational flexibility including creating a 'Launch Situation Room’ to rapidly course correct launch plans. Predictive analytics and forecasting can revolutionise how the generic pharmaceutical industry will approach different functional areas like R&D, clinical, sales, marketing, supply chain and inventory management by providing a valuable tool for companies to optimise their processes, minimise the number of canceled orders, and maximise revenue opportunities.
Overall, adopting appropriate business strategy and forecasting process will be a game-changer for the generic pharmaceutical industry, providing valuable insights and enabling companies to make more informed decisions and grow to an unbeatable scale and might. This will accelerate Indian generic pharmaceutical industry to become truly holistically innovative.

Sanobar
is currently Associate Director at Beigene. She has over 14 years of proven achievements in establishing and leading business strategy and forecasting, with Top global pharmaceutical firms (AbbVie, Novartis, McKesson). With a master’s degree in Organic chemistry coupled with MBA, she is regularly published and invited to speak at reputed industry conferences across North America and EU. She is considered a subject matter expert, delivers guest lectures & has developed academic modules at TRIEC, Toronto Metropolitan University and Schulich University (Healthcare & Biotech) Canada. She is also on the advisory board of the prestigious CPHI conference board.
15 www.pharmafocusasia.com
AUTHOR BIO
STRATEGY
Syed
Real-World Evidence and the Drug Development Process Dealing with ‘Pipeline Vision’
Challenges in Drug Development

Drug development, today, is a risky and expensive business. Drug discovery and development exhibits a 90 per cent failure rate that as a process can take between 10-15 years and whose average cost is US$1-2 billion/newly approved drug. The appearance of toxic side effects and/or the lack of efficacy highlights the fact that human patients are different and more complex than the animal and cell models used in early development. Even for drugs that achieve regulatory approval, commercial success is not guaranteed, financially affecting both pharma and payers (public and private). Among the diverse challenges in this complex
process are those that involve technology, science, regulatory oversight, financial issues, and the sociology, culture and psychology of both the physician and the patient. Currently, significant efforts are underway to evaluate and incorporate the use of artificial intelligence and real world data/real world evidence to enhance the probability for success. One critical consideration is whether these approaches are actually attacking the root cause problems or are being constrained by ‘pipeline vision’, i.e. the need to continue to support the current drug development pipeline model.
Pharma, over many years and across the industry, has evolved and operates
common drug discovery and development ‘pipelines’ that influence internal organisation and infrastructure. In general, pharma adopts new technology in an effort to optimise performance while only. slowly evolving its infrastructure and culture to support it. Optimisation can be viewed as ‘attempting to improve efficiency (and effectiveness?) of the pipeline model by reducing the ‘entropy’ at specific points along the path, e.g. target selection, target validation, drug discovery, clinical trial design, patient recruitment, regulatory submission, etc. In addition, the FDA’s does not require understanding the mechanism of action for a drug that is submitted for regulatory

16 PHARMA FOCUS ASIA ISSUE 50 - 2023
Pharma has begun incorporating real-world data (RWD) and real-world evidence (RWE) into the process of clinical development, primarily to facilitate clinical trials and preparation for submission to regulatory agencies, e.g. FDA, EMEA. In a previous article, we pointed out the opportunity to enhance product value and now can show how this can be accomplished using novel analytics applied to real world data in both drug discovery and development.
provides a convenient visualisation, i.e. of a linear process, there are additional characteristics of real pipelines whose considerations in drug development could further benefit drug development beyond a visual model. A true pipeline has pumps, valves and control devices and is subject to leaks, blockages and contamination. These elements can also be mapped to drug development and can provide additional critical insights.
This article focuses on the issues of pipeline leaks and contamination, i.e. development of potentially good drugs but addressing the wrong target and inadequate understanding of the complexity of the patient, of the disease and of the practice of medicine. Some re-direction of the use of real-world data could contribute to closing these gaps.
Addressing this Reality
Currently, two somewhat divergent approaches to improve successful drug development have been adopted in large pharma: 1) internal investment in access and application of new technologies that result from exciting, new scientific breakthroughs, and 2) outsourcing/licensing/ investing involving small biotechnology and technology companies for early access to potential products to minimise dependence on the less efficient parts of the current discovery process.
approval. As a result, drug discovery can add ‘phenotypic discovery and validation’. to its traditional ‘target selection and validation’. Below, in this article, the opportunity for real world data to redefine ‘phenotype’ and improve target selection will be discussed.
The inefficiencies in the current process are not uniformly distributed, ranging from 3 per cent (target validation) to 6 per cent (compound screening) and 66.4 per cent (Phase I), 48.6 per cent (Phase II) and 59 per cent (Phase III), respectively. A recent analysis of the average cost for each stage of drug discovery and development and final cost suggests that:
~$1B drug discovery and lead optimisation, >$300M on preclinical studies and
> $50M (Phase I clinical trials)
>$100M (Phase II clinical trials)
>$300M (Phase III clinical trials).
< $10M regulatory review and approval
$1.7B average total cost (13.5 years and increased from $1.5B in 2018)
The Pipeline Model
Pharma has adopted the concept of a ‘pipeline’, borrowed from the petroleum industry, to describe the linear alignment of steps in drug discovery and development, through clinical trials and regulatory submission/approval. While this
-A potential challenge to implementing the first approach is that most new technologies are applied to the existing pipeline model rather than exploring whether ‘pipeline redesign’ might provide a better solution, because of ‘pipeline vision’. Re-engineering the pharma pipeline would require significant disruption to both its existing infrastructure but even more, its culture, i.e. its people. Improving efficiency is a valid target, but this focuses on speed, i.e. ‘fail fast’, and may not address the ‘leaks and contamination’ in the pipeline, discussed later in this article, which are lessons to be learned for further drug development.
-In the second approach, outsourcing is effectively carried out by modest
17 www.pharmafocusasia.com
Michael N Liebman, Managing Director, IPQ Analytics, LLC
A SPECIAL ISSUE ON PHARMA R&D
investment, e.g. <US$20M, in smaller, specialised companies who may specialise in developing/using these advanced technologies. Outsourcing early research with low risk also provides the potential for high stakes success payments to these small companies, e.g. ~US$1B. Initial research success can then benefit from pharma’s infrastructure and experience for carrying out validation, clinical trials and regulatory submission, all of which are typically beyond the financial constraints and expertise of a small company. This is a form of ‘insurance’ for big pharma, locking in and supporting the smaller biotechnology (or technology company) towards success in reaching milestones while retaining an ‘escape clause’. An analysis of such deals across the industry could be very revealing and provide such small companies with greater insight for forming such relationships…but this is not the topic at hand.
The Promise and Potential of RealWorld Data
The recognition of the potential value currently encapsulated in rich, real world data sources has evolved along with advances in artificial intelligence and machine learning that can manage large data sets in an automated manner to identify patterns that might be difficult for an individual researcher. The emerging field of Big Data analytics, using the aggregate of real world data as noted above, suggests potential benefits may include:
• new druggable targets may be found
• rapid computational screening of expanded small molecule libraries will be possible
• analyse and categorise patient behaviour patterns concerning adherence, etc
• patient recruitment for clinical trials may be enhanced
• digital twins may augment and shorten clinical trials leading to faster regulatory review -identification of new drug combinations, of population
groups who exhibit better responses to a specific drug, of physician’s actual practice patterns
Even this small subset of all potential uses of Big Data and analytics could provide great value to pharma/biotech industry, physician decision making and, most importantly, the patient, but even greater value might be recognised by addressing the challenges that remain. I am reminded of this quote (actually acknowledged by Laurie Anderson, 2020, as being borrowed)
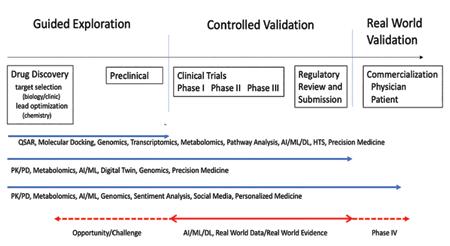
are addressing critical, underlying, and complex questions along the pipeline. The alignment of computational and informatic technologies along the pipeline are shown in Figure 1 which further delineates the pipeline in terms of three stages: guided exploration, controlled validation and real-world validation, where validation includes Verification. Currently the primary use of real-world data is in these latter two stages; the opportunity is to use it to address the first stage, hence enable ‘fail faster’ and increase the potential for greater success in drug discovery, regulatory approval and clinical acceptance.
Addressing Challenges in Guided Exploration
I would rephrase the last line to “you don’t fully understand the complexity of the problem and its underlying challenges”.
The poor success rate in drug development may be the impact of not addressing or even acknowledging these challenges without recognising that this may result from ‘pipeline vision’. With an infrastructure and culture that focuses on supporting and implementing the drug pipeline model, new technologies and new data are being applied to the existing model rather than exploring whether they
Drug Discovery typically begins with the biological identification and assessment of a target molecule, e.g. protein, receptor, RNA, DNA, etc., and progresses to either small molecule or biologic selection or development that exhibits selectivity and specificity for that target. The ideal target would represent an early step along the known mechanism of the disease but this is not typically well understood. Currently RWD/ RWE contributes indirectly to this by evaluating which drugs have been more effective or exhibit reduced side-effects in real world populations so that potentially relevant pathways and additional
18 PHARMA FOCUS ASIA ISSUE 50 - 2023
"If you think technology will solve your problems, — you don’t understand technology — and you don’t understand your problems."
Figure 1: The Current Drug Development Pipeline and Supporting Technologies
molecular targets can be identified. This approach is further supported by use of machine learning methods to analyse RWD/RWE.
The drug discovery component of drug development consists of two phases: the biology phase and the chemistry phase. The biology phase precedes the chemistry phase as it focuses on the identification and qualification of the target, i.e. biologic process and molecular entity, through biological (and clinical) analysis. While this requires a compre hensive understanding of the disease process, ideally it should also include understanding the complexity of the realworld patient and also guidelines and patterns of diagnosis and treatment. This is where RWD/RWE can contribute most significantly. Many of the computational approaches shown in Figure 1 (and also experimental approaches) involve chemical analysis and are applied to refine the lead compound, its physical, chemical and biochemical properties, and its potential selectivity and specificity for a target, i.e. lead optimisation.
Redefining disease as a process, not a state

It is critical to recognise and incorporate the reality that disease is a process and not a state. This means that disease progresses over time and actually in a high-dimensional space that includes both clinical, e.g. lab results, and non-clinical, e.g. diet, environment, lifestyle, factors. That very over time. Access to much of this data does not exist or is variable in quality, and
Which factors are most relevant for any given disease is also unknown. Ideally we should consider 3 key elements to diagnose disease more accurately than we do: 1) disease trajectory, what is the high-dimensional vector that defines how the patient is progressing over time; 2) disease staging, how far along that vector is the patient when presenting for diagnosis; and, 3) disease velocity, how rapidly is the patient progressing. The result of not having such ideal data is
a small segment of the real world patient population and may not lead to a commercially viable product. It is common that clinical trials utilise inclusion/ exclusion criteria that do not accurately reflect the real-world patient population, e.g. exclusion of women or lack of diversity, because their goal is to establish efficacy and safety. The drug discovery step presents the optimal opportunity to more broadly analyse (and understand) the real-world complexity of the disease, of the intended patient and even of the practice of medicine so that the drugs being developed can have a higher rate of success going from discovery to validation to regulatory approval and commercialisation, i.e. physician and patient acceptance and adherence. Real-world data and evidence, when appropriately aggregated and analysed, can significantly enhance the probability for success and provide additional verification. This first requires re-examining the definitions/usage of ‘disease’ and ‘phenotype’. Phenotype is commonly defined as the expression of one’s genomic makeup under the influence of environmental factors.
that 1) a patient coming in for diagnosis at different stages of the disease may be diagnosed differently, 2) two patients may appear to be identical in terms of lab results but actually have different diseases (and progressions), 3) two patients may appear different in their labs but have the same disease, just presented for diagnosis at different stages of the disease. The further reality is that an average patient has 5 co-morbid conditions and these may be previously diagnosed and treated, currently diagnosed and being treated, undiagnosed and as yet untreated. These co-morbidities can significantly impact
the disease trajectory and resulting diagnosis, treatment decision and response. These realities all present challenges, i.e. ‘leaks’ that impact our ‘pipeline vision’.
Redefining phenotype
Phenotype is commonly defined as the expression of one’s genomic makeup under the influence of environmental factors. The concept of ‘environmental influence’ needs to consider factors beyond the conventional definition of environment. As noted above, co-morbidities, poly-pharmacy, lifestyle, social determinants of health and cultural
19 www.pharmafocusasia.com
A SPECIAL ISSUE ON PHARMA R&D
determinants are examples of ‘environmental factors:’ that contribute to how genomic factors may or may not be expressed in an individual, and these factors may change over time. It is critical to re-examine current definitions of phenotype from seeking common observable factors in patients with the same diagnosis, to actually use the changes in these factors over time, which include clinical measurements, to define the ‘next generation phenotype’ and establish datadriven diagnoses of disease sub-types.
The lack of requirement for understanding mechanism of action for FDA approval, focusing on safety and efficacy, reinforces the use of correlative approaches for drug discovery and development rather than addressing the difficult study of causality. Many of the AI/ML methods currently used in early drug discovery further support these approaches utilising the increasing access to big data.
RWD and its challenges
Real world data can contribute significantly to supporting the critical evolution from correlation to causality and approaching better definitions of disease and mechanisms of action but there remain challenges. To understand how real-world data and evidence might be used to ‘seal leaks in the pipeline’ and enhance the efficiency and the effectiveness of drug development, the common sources of the data must be considered. In a recent industry survey where more than 70 per cent were strongly committed to its use already, it was noted that more than half of the organisations surveyed used disease and product registries and electronic health records, with patient
surveys, insurance claims, pharmacy records, digital health/monitoring/ wearables and imaging as data sources. Interesting was the observation that genomics data trailed these in terms of its use. It is also important to remember that a leak can impact a pipeline in at least two different ways: to lose materials, i.e. data, that results in inefficiency; and can contaminate the material, i.e. data, that remains within the pipeline because it is not aligned with what is needed. The application of AI/ML methods represents an opportunity for analysing large amounts of data and identifying critical patterns difficult to visualise, but the thirst for big data to support this needs to be cognisant of what the data actually does and does not represent.
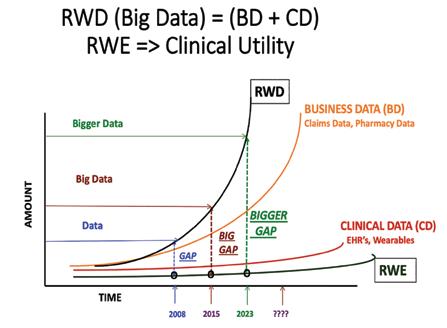
As shown in Figure 1, RWD comprises at least two major sub-groups, business data (BD) and clinical data
(CD). Each may be valid and useful but are developed to address different needs and this must be considered when using, and especially combining, them for analysis purposes. Is the goal to address the disease process at the molecular and clinical level or is it to examine associated business practices?
-Perhaps the most abundant data source in healthcare is claims data. This is acknowledged by physicians to not adequately represent the patient and their disease but rather what must be documented to support diagnostic testing, treatment procedures and drugs. In addition, it must bear some alignment with standard of care and clinical guidelines. As such, it is not a reliable source of data to define the true disease course, i.e. next generation phenotype and disease subtype, particularly as recent studies have shown that, on average, only just over 50 per cent of the time and their patients compliance with physician recommendations only achieves 54 per cent.
-this extends to the use of ICD-10 codes to define disease and disease progression as well
-even reliance on EHR data to define the patient journey can be inadequate, particularly when combining records, i.e.
20 PHARMA FOCUS ASIA ISSUE 50 - 2023
The pessimist sees difficulty in every opportunity. The optimist sees the opportunity in every difficulty.
– Winston Churchill
Figure 2: The Development of Big Data, Real-World Data and Real-World Evidence
the issue of interoperability of patient data. Many data scientists rely on matching of data fields and use of machine learning methods to assist in this process, to further support the development of big data sets for analysis. It is critical to understand that many of the data fields do not reflect the underlying complexity of the data entered, whose numerical value may fit well into use in algorithmic approaches. For example, blood pressure measurements are recorded but without definition of the method used to measure them, whether a patient had been resting for a period prior to measurement or just ‘run up the stairs to their appointment’, etc. It is important to understand that data quality needs to be examined and prioritised over data quantity even when using clinical records.
The increased access to wearable data and to patient records, to both claims data and to clinical (EHR) data, continues to expand the content of RWD but does not guarantee its conversion to RWE, particularly in light of the need to redefine the concept of phenotype and disease stratification to support and enhance both target selection and phenotypic screening for drug development remains a challenge. Figure 2.

The challenge is not just pharma’s While the development of drugs remains primarily a commercial activity, even if initiation may take place in an academic research laboratory, the ultimate beneficiary will always be the patient with the physician serving as the intermediary. Several studies point to significant errors in diagnosis, averaging 5 per cent (12 million patients) for all outpatients and 20 per cent for those with severe medical conditions and resulting in 40,00080,000 deaths. In addition, the FDA reports that more than 100,000 medication errors are reported annually.


Conclusion

Michael N

(theoretical chemistry and protein crystallography) is an Adjunct Professor of Pharmacology and Physiology at Drexel College of Medicine and Adjunct Professor of Drug Discovery, First Hospital of Wenzhou Medical University and also Fudan University. He serves on the Advisory Board for the International Park for Translational Biomedicine (Shanghai) and the Center of Biomedical and Health Research in Data Sciences, Univ Massachusetts(Lowell).
Addressing many of the issues raised above could serve to enhance the cooperation and collaboration between the physician, who deals with the real-world patient, and pharma to benefit all. The challenge is to recognise the truth in Anderson’s quote. Technology, alone, will not solve the complex problems in drug development and healthcare. It is critical to re-evaluate the perspectives that have evolved and expand drug discovery and development beyond the constraints of ‘pipeline vision’. This would seem to be scientifically, economically and humanitarily needed and valued. It is not likely that the technology will produce solutions to real-world problems if we do not first take the time to acknowledge the complexity of the problems, themselves. While it may seem simple to keep ‘rolling the rock up the hill’ as Sisyphus came to learn, ‘complexity keeps it from reaching the top’.
21 www.pharmafocusasia.com sales@sciencix.com LEARN MORE +1.800.682.6480 www.Sciencix.com EASY, FAST ORDERING KEY DIFFERENTIATORS MAJOR MARKETS SERVED • Pharmaceutical • Environmental • Bioanalytical Chemistry • Forensic & Toxicology • Service Companies • Academia • Food & Beverage Customers in 100+ countries High stock levels maintained Engineers with 135+ years of experience • HPLC & Mass Spec replacement parts • Customized PM Kits • Consumables
Warranty
¾ Tested & proven comparable to OEMs ¾ Same-day & international shipping ¾ Up to 30% less cost ¾ Lifetime
AUTHOR BIO
Liebman, Ph.D
A SPECIAL ISSUE ON PHARMA R&D
Precision Medicine Needs Precision Drugs
Precision medicine aims to tailor disease prevention and treatment to fit people’s genes, environments, and lifestyles, targeting the right treatments for the right patients at the right time. It needs precision drugs with defined and validated molecular structures that interact with a precisely defined disease target to achieve this.
Karel Petrak, Chief Scientific Officer, Nangiotx, Inc; Independent Consultant
Precision medicine relies on tailoring disease prevention and treatment according to differences in people’s genes, environments, and lifestyles, aiming to target the right treatments to the right patients at the right time. However, it has not yet been clearly stated how this concept and goals could be achieved. The availability of “precision drugs” that are essential to make precision medicine possible is frequently not considered.
Precision drugs
Precision drugs need to be defined and validated as having the molecular structure that would interact with a precisely
defined disease target. Further, it must exhibit the right pharmacokinetics to facilitate the drugs’ efficacy. Ideally, onand off-target interactions should not cause disabling adverse events.
Precision drugs are yet to be discovered and developed. Drug discovery often starts with a “shot in the dark.” A huge number of compounds are often “filtered” in silico before being examined using high throughput screening to
identify potentially relevant active compounds that are subsequently optimised and validated by establishing relationships between chemical structure and biological activity.
Once the disease indication has been selected, the process proceeds to identify the physiological mechanisms that need to be targeted and, ideally, a specific molecular ‘drug target.’ Drug discovery has largely relied on random in

22 PHARMA FOCUS ASIA ISSUE 50 - 2023
vitro screening of chemical compounds, in vivo animal studies, intuition, and serendipity.
Promising compounds are studied further for their toxicity and pharmacokinetics before clinical studies are conducted to generate the safety and efficacy needed before a drug can be approved for use in humans. However, the failure rate for compounds entering clinical studies to reach drug approval is greater than 90 per cent. So, perhaps, referring to the process as a “hit-and-miss” is not inaccurate. The above conventional activities do not provide the data needed for developing “precision drugs.”
The FDA approved several drugs described as “targeted”: Uptravi, Cosentyx, Cotellic, Odomzo, Xifaxan, Darzalex, Praxbind, Technivie, Opdivo, Alecensa, Empliciti, Keytruda, Ninlaro, Tagrisso, and Orkambi. None of these drugs act on a validated molecular target directly associated with the disease. Many “targeted” drugs have been approved to treat diseases. However, none of these drugs act on validated molecular structures responsible for the initiation and
progression of the disease. Invariably, such drugs act on an element in a pathway known to be involved with the disease. However, some progress has been made in this direction.
One form of “targeted therapy” uses drugs that attack specific cancer cells. Such therapies usually cause less harm to normal cells than chemotherapy or radiation therapy. For example, chronic lymphocytic leukemia (CLL) is now treated successfully using monoclonal antibodies that attach to a specific target on cancer cells. The antibodies are able to then kill the cancer cells, block their growth, or keep them from spreading. Rituximab, ofatumumab, and obinutuzumab alone and in combination with chemotherapy are used to treat symptomatic or progressive, recurrent, or refractory CLL, targeting CD20, a protein found on the surface of B lymphocytes.
Immunotherapy
Immunotherapy has been hailed as one of the most promising new cancer treatments. It is expected to turn the power of the immune system — more powerful
than any cancer drug —against cancer cells.
Immunotherapy is a treatment that uses a person’s immune system to fight cancer. It can boost or change how the immune system works to find and attack cancer cells. Substances made by the body or in a laboratory are used to boost, direct, or restore the body’s natural defenses against cancer. However, applying immunotherapy is not compatible with the precision medicine principle of “right dose for the right patient at the right time.”
The conventional drug discovery process has advanced our ability to treat many diseases. However, to advance to precision medicine, precision drugs are needed.

Immunotherapy is anything but precise. One of the reasons is the information a physician may have about the patient to treat.
While the premise of precision medicine requires detailed information about the biological mechanisms of the disease. At present, good biomarkers to predict a patient’s response to immunotherapy are not available. Further, good biomarkers to predict toxicities have not been identified.
Precision medicine requires the treatment to be applied at the right time. Adjuvant immunotherapy trials can last for 1–3 years, frequently not generating overall survival data. Such therapy puts an enormous therapeutic burden on too many patients for an unproven benefit.
The pharmacokinetics of precision drugs needs to be fully known and documented. Concerns have been expressed that patients receive unnecessarily high doses of immunotherapy. Several studies have shown that a dose-response relationship is often not established with immunotherapy. For instance, peripheral receptors may become saturated at a dose of 0.3 mg/kg of nivolumab. However, giving a lower dose is not in the financial interest of the healthcare business ecosystem, so a “personalised” weightbased dosing is replaced by a flat dose, irrespective of weight.
23 www.pharmafocusasia.com
A SPECIAL ISSUE ON PHARMA R&D
Similarly, the frequency of dosing is often not supported by data and may be too frequent. For example, during the pandemic, regimens were developed to allow patients to get the care they needed but at fewer intervals, limiting their contact with the healthcare system, for instance, changing from every three weeks to every six weeks. However, despite knowing there is no dose-response relationship with immunotherapy, the manufacturers decided to double the doses. Using precision drugs could make promising immunotherapy treatment more precise and effective.
New paradigm
Without recognising the need for precision drugs and exactly defining what is needed, precision medicine is either a fantasy or bad propaganda.
In developing disease-targeted drug therapies, it is critical to make the task very clear and define it in terms of unique molecular structures present in specific disease-associated cells. However, a question does remain: “Can this be done using the approaches employed so far?”
A new paradigm for developing truly disease-targeted, precision drugs needs to be developed and adopted to develop
• Approaches to identify any unique molecular structures present in a narrowly defined population of cells
• An effective algorithm for identifying such unique structures relevant to diseases (e.g., cancer).

Network pharmacology
Network pharmacology aims to understand the network interactions between a living organism and drugs that affect normal or abnormal biochemical function. It gathers data from pharmacology, network biology, systems biology, bioinformatics, and related sciences and uses the power of computers to identify possible interactions. This novel approach may be used to predict and identify multiple drug targets and interactions in disease.
Network pharmacology-based drug design integrates systems biology, metabolomics,
network analysis, and connectivity. This new paradigm enables the drugs to target several different proteins or networks implicated in a disease. The information generated by network pharmacology illustrates the complexity of diseases and could be employed in identifying molecular targets for precision-drug development.
Artificial intelligence
This process will inevitably demand a major input from Artificial intelligence (AI). Such AI will need to match human “brain power” in many respects. The input will need to provide guidance on
• How to get and curate relevant data (published and perhaps even unpublished; regardless, all should be validated)
• How to validate that the algorithm is performing as needed, and
• How to improve the approach to answer the initial questions.
All this will require solid guidance from human-level intelligence for the AI to assimilate all the existing data on the topic, all the current assumptions and theories about how disease originates and progresses, and how it could be cured. The process will need to go beyond extracting information and draw novel conclusions and recommendations on what steps need to be taken to reach the ultimate goal–understanding the disease
and defining unique molecular structures. The AI will need to acquire full capabilities in all aspects of human intelligence, such as being able to perceive the real world beyond the information given to it by its programmers. Further, it will need to have the capacity to determine the significance of various parts of the overall task and decide on which to focus. It will need to process information in human-thinking terms such as perception, abstraction, and memories, apply critical analysis of the information, and then remember and recall outcomes as and when needed to synthesize a new, more complex whole.
Ultimately, the AI will need to have the capacity to generate new knowledge and know-how. Will AI be able to speculate and imagine? It will need to. Will it be able to reason? It will have to. Will it be logical where humans often cannot be? Will it handle the “what if” questions, predict, and make decisions? All that and more. It would better be. However, intelligent human input will likely be needed all along the way to solve this challenging task.
References are available at www.pharmafocusasia.com
24 PHARMA FOCUS ASIA ISSUE 50 - 2023
AUTHOR BIO
Karel Petrak obtained a D. Phil. degree at the Imperial Cancer Research Fund in London, England. He joined Novartis to develop advanced drug-delivery systems. Subsequently, he led research groups in developing systems to deliver genes and proteins. He now advises developing precision drugs using new paradigms for disease-specific drugs and their delivery.
Promising compounds are studied further for their toxicity and pharmacokinetics before clinical studies are conducted to generate the safety and efficacy needed before a drug can be approved for use in humans.
Growing Complex Injectable Portfolio in the Indian Generic Industries
The Indian pharmaceutical industry has seen an exponential growth in the field of fill finished dosage forms, especially generics but the future lies beyond generics in the field of complex generics, biosimilairs, vaccines and New Chemical Entities (NCE)/New Biological Entities (NBE). Developing NCEs and NBEs will position Indian companies in the ivy league of global innovators. Risk adverseness, lack of perseverance and complex, long regulatory approval process are impeding Indian pharma companies to venture into NCE/NBE research. Product portfolio expansion into complex generic injectables is an attractive high return alternative for the Indian generic pharmaceutical industries.
Its biggest strength is its low cost manufacturing and thereby providing costeffective medications to the entire world. Further, the highly skilled talent pool produced by various National Institute of Pharmaceutical education and research (NIPERs) located in 7 cities across the country like Mohali, Ahmedabad, Hajipur, Hyderabad, Kolkata, Guwahati and Raebareli — providing the manpower needs of the Indian pharmaceutical Industry. These scientists and engineers possess excellent chemistry and process re-engineering skills required by the generic industry.
The Indian pharmaceutical industry was incepted as an API/bulk drug manufacturers which slowly diversified into fill-finish dosage form and witnessed an exponential growth in this sector for the last two decades. In the future, the focus of the Indian generic industry is to expand their portfolio into number of off-patent complex generics, bio-similar and vaccines which
are hard-to-make products with a higher entry barrier. Further, complex generic drugs deliver more value to the patients by addressing additional unmet medical needs and also enable the drug manufacturer to achieve market differentiation and an opportunity to earn higher margins.
The industry is analysed for its strength, weakness, opportunities and threats (SWOT Analysis) in Figure 1.
The main weakness of the Indian pharmaceutical industry is its relatively low investment in R&D and risk adverseness. Sun Pharma Advanced Research Company (SPARC) and Suven Life Sciences had 587 per cent and 304 per cent of R&D spending to revenue ratio in the Fiscal year 2020. Other pharma companies which also figured as the largest spenders of R&D in absolute terms are Alembic Pharma and Unichem Labs, who spent between 14 per cent and 31 per cent of their revenues on R&D. The R&D spending to revenue ratio for
25 www.pharmafocusasia.com
Sivakumar Ramachandran, Senior Scientist 1, Jodas Expoim Pvt Ltd
Tathagata Dutta, Chief Technology Officer (CTO), Jodas Expoim Pvt Ltd
A SPECIAL ISSUE ON PHARMA R&D
STRENGTHS
· Highest Growth Potential
Highly Skilled Workforce
· Cost of manufacturing less compared to other countries.
Posses excellent chemistry and process reengineering skills.
OPPORTUNITIES
· Growing opportunities for export in emerging markets.
· Large number of drugs going off-patent in US and Europe in the coming years.
Several US FDA approved plants with low cost of manufacturing
Growth opportunity in complex generics, vaccines and biosimilars
Dr Reddy's Lab, Cipla, Aurobindo and Sun Pharma were only 11.3 per cent, 8.9 per cent, 6.3 per cent and 9.8 per cent respectively. In addition, there is a lack of co-ordination between pharmaceutical Industry and academia.
There is a huge opportunity for Indian generic companies to export to both developed nations and emerging markets. There are several USFDA approved manufacturing plants that can produce high quality, low cost generic medicines in India. Further, Indian pharmaceutical companies can gain competitive advantage by expanding their portfolios into complex generics, biosimilar and vaccines.
One of the major threat for Indian generic companies is the patent regime followed by India. An interesting point to note is that the original Indian Patents Act, 1970, did not grant patent protection to pharmaceutical products to ensure that medicines were available to the masses at a low price. Patent protection of pharmaceuticals were re-introduced after the 2005 amendment to comply with TRIPS (Trade-Related Aspects of Intellectual Property Rights).
Gaining competitive advantage with complex generics
India enjoys a dominant position in global generics, it has the largest number of FDA approved manufacturing units
WEAKNESSES
· Low investment in R&D Lack of coordination between industry and academia
THREATS
· Product patent regimes poses serious problem.
· High threat to loss of Intellectual property impeding R&D investment by MNCs Drug price control order restricts drug prices in India
Threat of low cost manufacturing from other countries like China, Israel etc.
outside the US and accounts for around 30 per cent of US generics market (by volume). However, more recently, several new entrants (including companies from South Korea and China) are seeking to establish their presence in global generics. This intensifying competition combined with significant pricing pressure clearly indicates a red zone. Indian companies have strategically responded by developing greater strengths in complex generics (a propitious niche, a blue ocean strategy). Blue oceans are defined by untapped market space, demand creation, and the opportunity for highly profitable growth without a great deal of competition as seen in the red zone (fish eating fish: a highly competitive scenario). A propitious niche is where an organisation can use its core competencies to take advantage of a particular market opportunity and the niche is just large enough for the firm to satisfy the market demand. Complex
generics include complex injectable formulations (liposomal, microsphere based depot formulations), inhalation drugs (DPIs and MDIs), topical products and transdermal etc. Complex generics are products that have a complex active ingredient, complex dosage form, complex route of delivery or a complex drug-device combinations. (Figure 2)
Most of the existing drugs possess undesirable physico-chemical and pharmacokinetic properties like low solubility, short half-life, high protein binding, extensive first-pass metabolism or undue toxicity, thereby limiting their therapeutic potential and overall pharmaceutical application. In the past few decades, advanced drug delivery systems like nano-suspensions, nano-emulsions, and other carrier systems like liposomes and microspheres have been explored by pharmaceutical researchers to improve the clinical outcomes of therapeutic agents through parenteral route of administration.
Doxil, a pegylated liposomal Doxorubicin, originally approved in 1995, has been in short supply since the production facility of Johnson & Johnson’s contract manufacturer Ben Venue Laboratories in Ohio was closed in November 2011 due to ‘significant manufacturing and quality concerns’. To address the shortage of Doxil in US market, USFDA initially allowed temporary importation of Lipodox (Sun Pharma’s pegylated liposomal doxorubicin) from India. Later, in February 2013, FDA prioritised Sun Pharma’s abbreviated new drug application and approved it. The Sun Pharma product was also given RLD (Reference listed Drug) status then. Few
26 PHARMA FOCUS ASIA ISSUE 50 - 2023
Figure 1: SWOT Analysis of Indian Pharmaceutical Companies
Surfactants and
Liposomes
Auto-injectors - Pen Devices COMPLEX API DRUG-DEVICE COMBINATION PRODUCTS COMPLEX DOSAGE FORM COMPLEX EXCIPIENTS
Figure 2: Classification of Complex Injectable Products
Peptides and hormones
oils
& Polymeric particles
other generic companies like Dr. Reddy’s Laboratories Ltd and Zydus Worldwide DMCC have also received the approval for generic doxorubicin liposomes now. In recent years, few more products based on Stealth® Technology are either under development/approval stage or have seeked approval for commercial purpose. Ipsen Biopharmaceuticals Inc. received a








USFDA approval in 2015 for the irinotecan liposomal injection, Onivyde®, which is also based on similar stealth liposome technology.







Exparel®, approved in 2011 by USFDA, is a Multivesicular liposomal formulation (DEPOFOAM® technology) of Bupivacaine (a local anesthetic), reduces post-surgical pain up to 72 hrs






post infiltration into the surgical site. Vyxeos®, approved in 2017 by USFDA (Figure 3), for the treatment of Acute Myeloid Leukemia (AML) is a dual drug (Cytarabine and Daunorubicin) loaded into small unilamellar vesicular liposomal formulation presented to tumor cells at their therapeutically synergistic ratio. Many Indian generic companies are currently exploring opportunities to develop a generic version of these two liposomal drugs without much success.
Microspheres formulations are difficult to mimic or copy because of the limited reverse characterisation data available on polymers used in these formulation like PLGA molecular weight, lactide:glycolide ratio etc. and complex manufacturing process involved. Further, these microsphere formulation require strict aseptic manufacturing facilities to scale-up. For these reasons, till date there is not many generic microsphere products in US market even after the innovator patent has expired for several products like
27 www.pharmafocusasia.com Agência Nacional de Vigilância Sanitária ANVISA www.quantysclinical.com EXPERIENCE IN CONDUCTING BIOEQUIVALENCE STUDIES ON INJECTABLESLIQUIDSTRANSDERMAL PATCH TOPICAL CREAMS ORAL SOLIDSINHALERS, ORAL & NASAL SPRAYS ACCREDITED BY GCC Ph-II & Ph-III with INDICATIONS IN Cardiology 104 Beds 4 LCMS/MS Over 40 Methods Neuropsychiatry De-addiction Oncology OTHER SERVICES In-Silico studies Trial Monitoring Bioanalytical Services Medical Writing Email: info@quantysclinical.com Call: +91 2836 253330 Plot No. 668, 671, 672, New Area Kandla Special Economic Zone, Gandhidham, Kutch-270230, India
Drug Product Therapeutic Category Complex Dosage Form US Sales 2022 Number of Generics* Doxil® Anti-cancer Liposome $ 1,240 Mn 5 AmBisome® Anti-fungal Liposome $ 136 Mn 1 Onivyde® Anti-cancer Liposome € 122 Mn 0 Exparel® Local Anesthetic Multi Vesicular Liposome $ 59.1 Mn 0 Vyxeos® Anti-cancer Liposome $ 33.8 Mn 0 Sandostatin LAR® Acromegaly, Carcinoid tumors Microspheres $ 881 Mn 0 Lupron® Anti-cancer Microspheres $ 81 Mn 1 Eligard® Anti-cancer Microspheres $ 87 Mn 0 Abraxane® Anti-cancer Nano-particles $ 173 Mn 0 Invega Sustenna/ Trinza® Anti-psychotic Nano-Suspension $ 661 Mn 0 *As on 30th Nov 2022
Table 1: Net sales and generic availability information of Complex Injectable drug products.
A SPECIAL ISSUE ON PHARMA R&D
Lupron®, Sandostatin LAR® etc.(Table 1). Recently on 29th November 2022, Cipla launched the first generic for leuprolide acetate depot 3 months, 22.5 mg, in the US market for prostrate cancer.
Zydus pioneers a breakthrough in NCE research with LIPAGLYN®, India's First NCE to reach the market. Saroglitazar is branded as Lipaglyn and marketed in India since 2013. In December 2020, Zydus Lifesciences (formerly Zydus Cadila), saroglitazar (brand name, Lipaglyn) was given fasttrack designation by the US Food and Drug Administration (FDA) to treat individuals with Primary Biliary Cholangitis (PBC), a liver disorder due to progressive destruction of the bile ducts. The drug also received orphan drug designation in January 2021. High risk involved, lack of perseverance and complex, long regulatory approval process are impeding Indian pharma companies to venture into NCE/NBE research.
Today, India is the largest provider of generic drugs globally, ranked third in terms of pharmaceutical production by volume and 14th by value. The
Sivakumar Ramachandran has earned his PhD in Pharmaceutics and Pharmaceutical Chemistry from University of Utah, Salt Lake City, Utah, USA and an M Pharmacy from BITS, Pilani, India. He is currently pursuing his MBA in Business Analytics from BITS, Pilani, India. He was involved in teaching and academic research at NIPER, Mohali, India at initial part of his career. Later, he has 11 enriching years of Complex Injectable formulation development (NDDS) experience in various pharmaceutical industries like Zydus Cadila, Hetero, Mylan Laboratories etc. His research interests include self-assembled biomaterials based on lipids (liposomes) and peptides for drug delivery and tissue engineering applications along with microspheres (depot formulations) and nano-particulate drug delivery systems. He has 1 US patent and 7 international publications to his credit. He is currently working as senior scientist 1 at Jodas Expoim Private Limited, Hyderabad, India.


domestic pharmaceutical industry includes a network of 3,000 drug companies and around 10,500 manufacturing units. India enjoys a competitive advantage in the global pharmaceutical sector particularly in complex generics. India also has a large pool of scientists and engineers with a potential to transform and drive the growth of the industry to great heights. Complex generic delivery systems are the latest trend in the generic pharmaceutical industry and companies that can adapt to a more holistic complicated development process are positioned to benefit greatly in the coming years. Sustainable growth going forward for Indian generic pharmaceutical Industry will be from high-barrier complex generics.
References are available at www.pharmafocusasia.com
Tathagata Dutta has earned his M.Pharm and PhD in Pharmaceutical Sciences from Dr. Hari Singh Gour University, Sagar and completed his postdoctoral studies from School of Pharmacy & Diamantina Institute for cancer, Immunology and Metabolic Medicine, University of Queensland, Brisbane, Australia and was engaged in development of RNAi therapeutics for effective management of cancer. He has served as Chief Scientific Officer (CSO), Vice President (R&D), Executive Director, Managing Director & CEO of several reputed Multinational Pharmaceutical Companies for more than two decades. His research interests include novel drug delivery systems, dendrimers, liposomes, polymer bioconjugates, tissue targeted drug delivery systems, nano drug delivery systems, gene therapy, genetic immunisation, depot injections, gene delivery, and development of RNAi therapeutics. Presently, he is the Chief Technology Officer (CTO) of JODAS EXPOIM Pvt Ltd.
28 PHARMA FOCUS ASIA ISSUE 50 - 2023 AUTHOR BIO
Figure 3: Approvals of Complex Injectable products over the years
VYXEOS (Multidrug encapsulated liposome) 2017
INVEGA SUSTENNA (Nanosuspension) 2010
ABRAXANE (Nano-particle) 2005
DOXIL (liposome) 1995
LUPRON (Microspheres) 1989
DOING MORE WITH LESS
Leveraging model-based pharmacology for cost-effective drug discovery and development
Drug discovery and development consists of a series of choices, from programme inception to post-marketing. Every choice along the way can be decisive, and even more so when money is tight. This article shares how model-based drug discovery and development helps companies get their molecules to the finish line with fewer costly missteps along the way
Arijit Chakravarty, CEO, Fractal Therapeutics
For many of us in pharma and biotech R&D trained as academic scientists, having timelines associated with drug discovery and development projects can feel both unintuitive and bizarre at times. This is science, we find ourselves thinking. How can it fit on a GANTT chart?
Learning to corral the experimental vagaries of drug discovery and development into a timeline and a budget is a critical skill, both for an individual project lead’s career and for a company’s success. In this article, we’ll dig into how that’s even possible. To make things a bit concrete, we’ll use a specific scenario (small-molecule preclinical development for Oncology), but the general guidelines here are readily applied to nearly all therapeutic areas and modalities. Similar approaches can be used for more efficient and cost-effective screening, lead optimisation or early clinical development as well.
So, let’s take a closer look at what has to happen when a molecule is in the preclinical development stage. Typically, the project team has an identified mole-
cule in hand and now has to put together the IND-enabling package, which gates the entry of the molecule into the clinic. For the pharmacologist, this boils down to answering a number of questions that allow for the design of a science-driven Phase I trial:
• What is the right starting dose for the candidate drug?

• What is the dose escalation scheme?
• What is the right dose route and schedule?
• What are the right time points and dose levels for pharmacodynamic (PD) biomarker collection?
• Has the patient population been identified already, or will it be identified during Phase I?
These questions are critical for the IND package, because they will define the clinical strategy, and the right (or wrong) answers can be decisive for a programme’s fate.
These questions are also deceptively easy to answer, however, because they can often be brushed off with an opinion. Because there is a tendency to seek certainty as early in development as possible, there is a tendency for project teams to shoot from the hip, creating an artificial sense of progress by locking down answers to these questions with very little preliminary workup. Unfortunately, a lack of rigour in addressing these questions will usually lead to fatal consequences for a programme in early clinical development (see this article of mine for more details on that!).
Part of the issue lies with the approach taken in preparing for the IND
29 www.pharmafocusasia.com A SPECIAL ISSUE ON PHARMA R&D
application. Often, teams will work their way through a laundry list of studies that looks something like Figure 1. The problem with the “laundry list” approach is that a project team can easily execute on the studies without having them answer the questions above.
It takes careful planning to address the questions above, and often project teams can end up thrashing around without getting to the ‘killer’ slide necessary to
support a data-driven choice for each one. A better approach is to frame each clinical translation question in terms of a scientific hypothesis and then try to address that with a set of experiments. Easier said than done, right? Let’s dig into how to make this happen in practical terms.
At the heart of the development paradigm for a successful oncology drug lies the therapeutic window- the gap between
the efficacious and the toxic dose (Figure 2). (A closely related concept, the therapeutic index, is the ratio of the efficacious dose to the toxic dose). In oncology, for example, drugs that have a therapeutic index of one or more are likely to succeed in the clinic. Drugs with a therapeutic index of less than one will fail. (Every time. It’s as simple as that.)
So, it goes without saying that estimating the therapeutic index efficiently is the key to advancing molecules to the clinic. Many, if not most, design choices during preclinical development impact the therapeutic index- choice of formulation, dose schedule, dose route, dose escalation scheme and patient population, to name a few. If a molecule doesn’t have a therapeutic index of greater than one on the intended development path, then the team can iteratively refine the development path (changing one or more of the parameters mentioned above) to seek a wider therapeutic index. Ironically, many teams avoid making a rigorous assessment of a programme’s therapeutic index, as they are afraid of the consequences of the wrong answer. In reality, early and rigorous therapeutic window assessments maximise the chances of programme success.
The therapeutic window assessment also underpins many of the key questions for clinical trial design. Each of the critical questions above can be framed and answered rigorously, if you have the dose-response curves for efficacy and toxicity in hand:
What is the right starting dose for the candidate drug? Project the dosetoxicity relationship for humans, setting the starting dose based on toxicology (based on the no adverse effect level) or on PD (Minimal Anticipated Biological Effect Level).
What is the dose escalation scheme? Based on the steepness of the projected dose-toxicity relationship, decide whether a traditional 3+3 scheme is acceptable (you can usually do better). If not, use the projected dose-toxicity relationship as a Bayesian prior and design a dynamic

30 PHARMA FOCUS ASIA ISSUE 50 - 2023
Figure 1: studies required for an ind
dose escalation scheme that leverages this prior to escalate quickly and safely.
What is the right dose route and schedule? Estimate the pharmacokinetic (PK) parameter that is best correlated with efficacy (the PK “driver” of efficacy). Do the same for toxicity. Leverage this knowledge in identifying the dose route and schedule that provide the greatest therapeutic window. For example, if peak concentration (Cmax) drives toxicity, while total drug concentration over time (AUC, or area under the curve) drives efficacy, a once-weekly i.v. infusion may be a better idea than a single i.v. bolus delivered every three weeks. A difficult side effect profile will make it harder to achieve the full potential of the drug and will negate the added convenience of the single i.v. bolus dose.
What are the right time points and dose levels for PD biomarker collection? Build a similar PK/PD dose-response relationship for the biomarker(s). Leverage this PK/PD relationship to project the PD response over time in humans, by accounting for differences in PK between preclinical model species and humans. Has the patient population been identified already, or will it be identified during Phase I? If the patient population has already been identified, construct PK/ efficacy dose-response relationships in a range of translationally relevant animal models. (Usually, for Oncology, this can be done in patient-derived xenograft models, which have very high predic -
tive value for this sort of work). If the patient population hasn’t been identified, you can use the projected dose-response relationship for efficacy as a Bayesian prior in a retrospective analysis at the end of a Phase I/II trial to zero in on sensitive subpopulations. The plans for this should ideally be in place before the start of the Phase I trial.
This is a high-level overview of some very complex topics, so there’s lots more to say on each of the above points. (Check out this article by me for a little more detail, or head over to our webpage for white papers and peer-reviewed publications by us on these topics).
With that said, it should be clear at the thirty-thousand foot level that the therapeutic window forms the key to efficient preclinical development, and most of the critical-path questions on the road to the IND are dependent on this analysis. Estimating the therapeutic window requires us to project the doseresponse relationship for efficacy and for toxicity in the clinical setting. Here’s the funny thing- getting to a precise quantitative estimate of the therapeutic window is much easier said than done!
Let’s use a simulated in vivo efficacy study to explain why this is so tricky. For oncology in vivo pharmacology, the workhorse model is the mouse xenograft. Mice are injected in their flank with cancer cells, these cells grow after about a week or two into small tumours, which are then either allowed to grow untreated
or are treated over the study period. The impact on tumour growth is typically expressed as the ratio of treated/control tumour volume at the end of the study period (although there are better ways to do this, as this publication by us shows).
So, when running an in vivo efficacy study, you will start with an idea of what the maximum tolerated dose (MTD) of the drug is. Now, in a typical setting, you might do an initial efficacy study looking at the efficacy at MTD, ¾ MTD, ½ MTD and so on. The problem is that the relevant range for the dose-efficacy relationship can take many iterations to discover. In this simulated example, the underlying dose-efficacy relationship is steep. So, you run your first efficacy experiment starting from the MTD (40 mg/kg) and stepping down in ¼ intervals (Fig 3A). That didn’t go so well- it’s not a dose-response! So you try again, this time from 20 to 40mg/kg (Fig 3B)- still not a dose-response. Finally, you get it right on the third try (Fig 3C). This third run gives you the information you need to lock in the EC50 for the dose-response relationship for efficacy. But, unfortunately, each in vivo study cost (say) $65k, and took a month for the turnaround (optimistically). So that was ~$200k and 3 months of work.
Was there a better way? Yes, there was. Establishing the PK/PD relationship (which is also used in the IND) first with a mechanistically relevant biomarker (e.g. target occupancy) would have indicated that target occupancy saturates around 35 mg/kg, and most of the information is contained in the range between 28 and 35 mg/kg. A single PK/PD study (~$40k and 1 week of work if the assays are already in place), followed by a focused efficacy study would have gotten you the doseefficacy relationship for a total tab of ~$100k and one-and-a-half months. (Of course, this is not an apples-to-apples comparison because the PK/PD workup would need to be performed either way.) The big difference is this- using a modelbased approach gets you there reliably, while the trial-and-error method comes
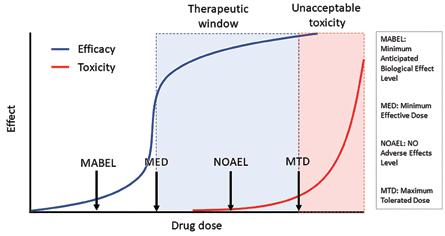
31 www.pharmafocusasia.com
A SPECIAL ISSUE ON PHARMA R&D
Figure 2: The therapeutic index
with open-ended timelines and budgets. This is a simplistic simulation-based example, but it reflects what I’ve seen in my decades of experience as a preclinical and translational pharmacologist: using PK/PD modelling allows for more focused experimental designs that can limit the ‘thrashing around’ that is so common in preclinical development.
A rigorous approach to preclinical development requires three things:
• commitment to focusing on the critical-path questions
• persistence in getting the data required to answer these questions quantitatively
• judicious application of PK/PD modelling to narrow down the search space and speed things up.
The end products are an IND application and a Phase I design that are both scientifically “tight” and cost-effective. While we focused here on small-molecule oncology, these approaches can be applied to almost any therapeutic area and modal-

ity. The details may change (for example, in other therapeutic areas the focus can end up being on the relationship between target occupancy and drug biology, rather than therapeutic index per se), but the strategy is almost identical.
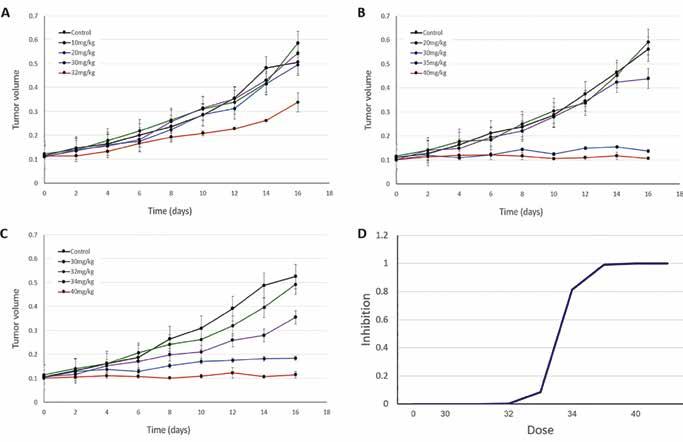
In lean times, the ability to execute reliably on time and under budget can make the difference between life and death for early-stage biotechs (and projects). Model-based pharmacology allows companies to derisk the road to the IND by allowing them to focus limited resources on the questions that matter most. This focus allows for a more robust and rigorous package and leaves room for recovery from unexpected developments on the way there.
This was a quick overview of a complex topic - there’s much more that can be said on many of the aspects covered here. Head over to our website for more white papers and publications, or feel free to ping me directly on LinkedIn if you have more questions!
Arijit is
32 PHARMA FOCUS ASIA ISSUE 50 - 2023
a seasoned drug hunter with two decades of industry experience as a project and team leader, spanning in vitro, in vivo and computational line functions in both Big Pharma and biotech settings. His company, Fractal Therapeutics, helps clients design modelbased discovery and development strategies and apply them to their projects.
AUTHOR BIO
BACTERIAL CANCER THERAPY
Bacteria was used to treat cancer decades ago. However, this method was mostly replaced by radiotherapy and chemotherapy. In recent years, research on bacterial cancer therapy has revived, and this article introduces the current studies in this field.


Cancer is a deadly disease featured by unchecked cell growth and the potential to spread to different body parts. Despite various therapies, such as surgery, radiation, chemotherapy, targeted therapy, and immunotherapy, cancer remains incurable for many patients, mainly due to treatment resistance and toxic side effects. Thus, complementary or alternative therapies, including bacteria-based cancer therapy, have been explored to overcome these limitations.
Bacterial cancer therapy began decades ago. In 1891, oncologist Dr. Willianm B. Coley (1862-1936) first injected live bacteria into a patient with inoperable cancer, leading to tumourshrinking (Coley 1891; McCarthy 2006). This success led to the development of "Coley's toxins," constituted by
Streptococcus pyogenes and Serratia marcescens, which were used to treat sarcomas, lymphomas, myelomas, and melanomas (Hernández-Luna et al., 2018) until the 1960s, with survival rates similar to those of patients diagnosed with cancer in 1983 (Richardson et al., 1999). However, this approach was mostly abandoned later due to the emergence and popularity of radiotherapy and chemotherapy. In recent years, progress in biotechnology, immunology, and molecular biology has led to a revival in bacterial cancer therapy.
The mechanisms of bacterial cancer therapy
Due to the rapid growth of tumour cells, the oxygen level becomes very limited, a condition called hypoxia, in the tumour microenvironment (TME). Severe hypoxia is detected in the TMEs
of nearly all solid tumours (Sedighi et al., 2019; Jing et al., 2019). Thus, many anaerobic bacteria, germs that can survive and grow under an environment lacking oxygen, for example, Salmonella, Clostridium, and Bifidobacterium, have been studied for cancer treatment due to their ability to multiply in the hypoxic tumour tissues preferentially (Sedighi et al., 2019).
Bacteria use two ways to suppress tumour cell growth and progression: stimulating the immune system and directly killing cancer cells. The bacteria-stimulated immune response is the mechanism for bacteria-based cancer immunotherapy (Tang et at., 2022). The immune system contains various types of immune cells, such as lymphocytes (B cells and T cells), natural killer cells, dendritic cells (DCs), macrophages, and
33 www.pharmafocusasia.com A SPECIAL ISSUE ON PHARMA R&D
Yongqiang Chen, Scientific Researcher, CancerCare Manitoba Research Institute
neutrophils. When bacteria invade the body, macrophages and neutrophils can attack and kill them. Bacteria that escape the attack by immune cells tend to move to the TME because the hypoxia condition in the TME could suppress the functions of local immune cells (Tang et at., 2022; Huang et al., 2021), which benefits the survival and growth of bacterial and tumour cells. However, bacteria-derived molecules, such as lipopolysaccharide (LPS), peptidoglycan, lipoteichoic acid, and flagella, can stimulate the immune system to kill tumour cells by mechanisms such as binding to pattern recognition receptors (PRRs) from DCs and macrophages and increasing the production of pro-inflammatory cytokines and chemokines in the body (Tang et at., 2022). Live or dead bacteria can be used in bacteria-based cancer immunotherapy. Live bacteria are injected into the body or tumours or taken up into the gut to stimulate the immune response. Dead bacteria or their components are directly injected into the tumours to elicit an increased immune response to attack tumour cells.
Bacteria can directly kill cancer cells by producing bacterial toxins (Huang et al., 2021; Duong et al., 2019; Zahaf et al. , 2017), such as diphtheria toxin and pore-forming toxins, or by entering cancer cells to undermine or exploit xenophagy, a type of selective macroautophagy (Uchugonova et al., 2015; Sui et al., 2017). Macroautophagy (autophagy) is a "self-digestion" and stress-responsive process in eukaryotic cells, the type of cells with a nucleus. During autophagy, materials (cargos) inside the cell are wrapped in autophagosomes, a structure with double membranes. An autophagosome fuses with the lysosome, a "suicidal bag" containing digestive enzymes to break down materials, to generate a new structure named autolysosome, where the cargo(s) are broken down into small molecules. Then these small molecules can be used to produce new components and energy to help the cell survive under stress, for example, starvation (Chen et
al. , 2022). Autophagy is called selective autophagy when it recognises and degrades a specific cargo. Xenophagy is a type of selective autophagy degrading microbes, such as bacteria and viruses.
When bacterial cells enter tumour cells, they can either be killed by xenophagy or survive to cause tumour cell death by undermining or exploiting the xenophagy pathway (Sui et al., 2017) (Figure 1). For example, Salmonella enterica serovar Typhimurium (SeT) (Sui et al. , 2017) and Streptococcus pyogenes (Nozawa et al., 2012) can be degraded by xenophagy. In contrast, Mycobacterium
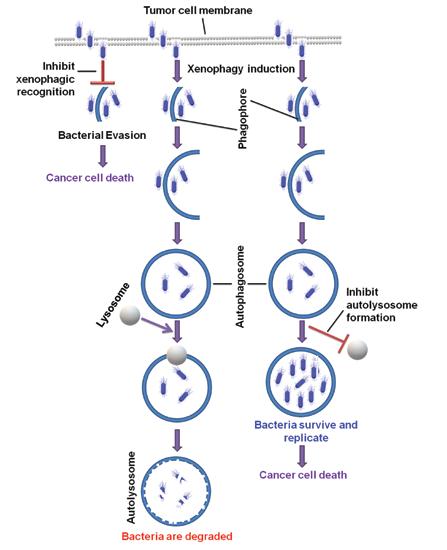
tuberculosis and Legionella pneumophila can escape clearance by xenophagy (Kimmey et al. , 2016). The detailed mechanisms of xenophagy and its blockage by bacteria are still largely unknown.
The challenges and perspectives of bacterial cancer therapy
Bacterial cancer therapy can be used as a complementary or alternative therapy to overcome treatment resistance and toxic side effects of radiotherapy, chemotherapy, and targeted therapy. However, it faces challenges (Rommasi 2022):
1. A systemic bacterial infection of the
34 PHARMA FOCUS ASIA ISSUE 50 - 2023
Figure 1: Bacteria escape the degradation by xenophagy inside the cancer cells (Modified from Sui et al., 2017).
body could render a big risk to a living organism.
2. Low cytotoxicity may require a low dose of bacteria, which might compromise therapeutic efficacy.
3. It is difficult to use bacteria to eradicate all cancer cells in the body, which might lead to cancer recurrence.
Instead of suppressing cancer, bacteria could help cancer cells grow and spread to other parts of the body, a process called metastasis (Galeano Niño et al., 2022). A cancer-suppressing bacterium might mutate to benefit cancer progression.
In recent years, genetically engineered bacteria (GEB) have been generated to express reporter genes, cytotoxic proteins, and tumour-specific antigens (Sedighi et al., 2019; Duong et al., 2019). These bacteria can be modified to have reduced pathogenicity to the host and increased multiplication and cytotoxicity in tumour cells (Low et al., 1999). The use of GEB renders a bright future for bacterial cancer therapy (Gurbatri et al. , 2022).

The roles of microbiota in cancer prevention and progression are catching increasing attention from researchers. Gut microbiota can inhibit or promote cancer progression depending on the context (Galeano Niño et al. , 2022; Cheng et al. , 2020). A recent study reports that the chemotherapeutic
drug 5-Fluorouracil (5-FU) inhibits the growth of the cancer-promoting bacterium Fusobacterium nucleatum in the tumours of colorectal cancer (CRC); however, members of the intratumoural microbiota of CRC can modify 5-FU into a nontoxic product that does not inhibit the growth of CRC cells and F. nucleatum cells (LaCourse et al. , 2022). This study's findings suggest that intratumoural microbiota's impact on cancer progression and therapy has been undervalued. Intratumoural microbiota likely affects any type of cancer treatment. A maximal therapy efficacy could be reached by using bacterial cancer therapy as a complementary therapy with other cancer treatments.
One promising area of research is using probiotic bacteria in cancer treatment. Probiotic bacteria are considered “good bacteria” to maintain a balanced gut microbiota for helping digestive health and boosting the immune system. They might be safer than other types of bacteria for cancer therapy. Current studies mainly focus on probiotic bacteria's roles in gut microbiota for preventing and treating CRC, breast cancer, and other cancers. Genetic engineering of probiotic bacteria could be an appealing strategy for improving bacterial cancer therapy. For example, the probiotic lactic acid bacterium Pediococcus pentosaceus was genetically engineered to inhibit CRC tumour growth (Chung et al., 2021).
Conclusions
The centuries-old bacterial cancer therapy has been revived in recent years. This treatment depends on two mechanisms to suppress cancer progression, including (1) using live or dead bacteria to stimulate the immune system and (2) facilitating bacteria to directly kill tumour cells via producing bacterial toxins or entering tumour cells to induce cancer cell death. The hurdles to using bacterial cancer therapy include the potential pathogenicity of bacteria to the host and the bacteria's low cytotoxicity in tumour cells. These issues might be overcome by generating GEB. The types of dominant bacteria in intratumoural microbiota can determine whether the microbiota will benefit or antagonise cancer therapy. Gut microbiota is known to affect cancer prevention and treatment. The uptake of probiotic bacteria can improve gut microbiota to help treat cancer, especially CRC. Since probiotic bacteria are generally safe for the body, their genetic engineering is a promising strategy for improving bacterial cancer therapy.
References are available at www.pharmafocusasia.com
Yongqiang Chen is a scientific researcher, writer, reviewer & editor. He completed B.Sc. (Huazhong Agricultural University) and Ph.D. (University of Manitoba) in microbiology and postdoc (CancerCare Manitoba & University of Michigan) in cancer biology and molecular biology. His current research focuses on cancer cell death, drug resistance, and autophagy.
35 www.pharmafocusasia.com A SPECIAL ISSUE ON PHARMA R&D
AUTHOR BIO
3D Bioprinting of Tissues/Organs for Biomedical Applications

Tissue/organ transplants are necessary and lifesaving in several medical conditions. However, donor scarcity and patient immune rejections are immense medical challenges. 3D bioprinting of tissues and organs might solve this challenge. Research shows that 3D-printed tissue can be used for transplantation, disease diagnosis, infection studies, and drug testing.
Tissue/organ transplants are necessary and lifesaving in several medical and disease conditions. However, donor scarcity and patient immune rejections are immense medical challenges. Additionally, approval from family, lack of proper organ storage/transport facility, and ethical concerns make it further challenging. Most transplants fail within 10 years, and the patient must take medication to suppress the immune response. 3D bioprinted tissues and organs might solve this challenge. Using bioprinters, the patients or donor cells can be used to print tissue, which can be used to test drugs for personalised treatment. In the future, the same technology and knowledge can be used to print functional organs for medical use. In animal models, it is challenging to mimic the human disease condition. The use of bioprinted tissue and their models can better mimic human disease conditions and biology. The use of printed tissue and organs from human cells in drug testing could decrease drug development time, fasten the drug discovery process, and increase the success rate. Moreover, bioprinted tissues could also replace or reduce the use of animals. With the recent ban by several countries on animal use for 3D bio-printed tissues,
36 PHARMA FOCUS ASIA ISSUE 50 - 2023
Himanshu Kathuria, Co-founder, Nusmetics Pte. Ltd Nileshkumar Dubey, Assistant Professor (tenure-track) in the Faculty of Dentistry, National University of Singapore
including skin, can be very useful in cosmetics and drug testing. Many recent research shows that 3D bioprinters can make tissue and organs, such as skin for grafting, ear membrane to repair eardrums, cancer diagnosis, infection studies, printing tissue directly on the human body, and safety and efficacy testing.
1.How tissue/organs are 3d printed?
There are several optimisation steps to print tissue constructs and organs. However, it commonly involves the 3D scanning of the body part, translation of scan to printable model, use of software, and printing of model using a bioprinter. Additionally, suitable biomaterials are also required in bioprinting. These printable biomaterials, when combined with cells for bioprinting are called bioinks, which mimic the biomatrix for organ/tissue production. The cells can be extracted from patient-specific biopsies for tissue production. Multiple cells could also be used to replicate organ biology. Further, tissue or organ repair can be done directly on patients in the operating room using hand-held bioprinters and bioinks. This technique is called in-situ bioprinting or bioprinting on body, which is a new frontier in personalised treatment.
2.Biomedical applications
Various tissue/organ-like structures of several organ systems have been bioprinted. This includes the nervous, cardiovascular, skeletal, integumentary, endocrine and exocrine, gastrointestinal, respiratory, and urinary systems. In the nervous system, the primary functional unit is the neurons that transmit signals in the body. In neurological diseases and neural damage due to trauma, the functionality of the nervous system is disturbed. For example, neurodegenerative diseases are progressive degeneration of neurons and neuronal death that cause loss of body functionality, hindering daily life activities. Implantation of stem cells and neural cell constructs could be a reliable therapy for these diseases. Bioprinting of brainlike multilayer structures helped neurons to grow and form neural networks like in the brain (Figure 1). These bioprinted structures could be used as models to study and understand brain functions, neural injuries, and neurogenerative disease.
Figure 1. 3D bioprinted neuronal tissues. A: Hand-held reactive bathless printing; B: Representation of the 6 layered brain structure found in the human cortex; C: A proposed design for optimal free-form artificial brain-like structures; D: 3D printed brain-like layered structure; E-J: Confocal microscope images of neural tissues stained with b-III tubulin (red) K: 3D printed hydrogel scaffold with aligned microchannels; L: (a,b) Schematic of 3DBP and scaffold design (c) FITC (green) with a gradient concentration (d-e) Traumatic injury and immunostaining of lesion (f-g) 3D printed scaffolds to match lesion.
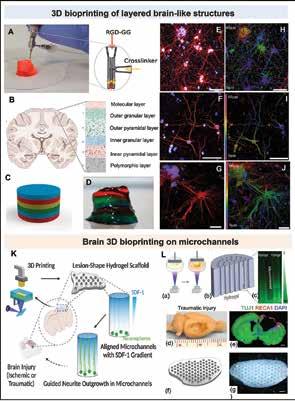
A novel cardiac patch consisting of stem cells was prepared using a microchannel-based 3D bioprinter. The patch helped in cardiac regrowth and prevented fibrosis (Figure 2). Likewise, other bio-printed micro-channeled hydrogel patch was studied to treat infracted patients. A study showed heart tissue printing using multicellular spheroids on needle array. It was used to test drugs, contractile force, and beating rate (Figure 2).
Figure 2. 3D bioprinted cardiac tissues using microchannels, microneedles, and hydrogel process. A: Schematic representation of 3D bioprinting using microchannels, B1-B2: hMSCs orientation induced by micro-channeled hydrogel, C1-C2: Immunofluorescence
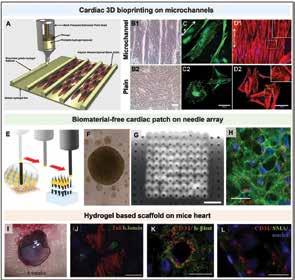
37 www.pharmafocusasia.com A SPECIAL ISSUE ON PHARMA R&D
Figure 1: 3D bioprinted neuronal tissues copy
Figure 2: 3D bioprinted cardiac tissues using microchannels, microneedles, and hydrogel process copy
staining of vinculin with alexafluor 488 secondary antibody (green) demonstrates the arrangement of focal adhesion, and the nuclei were stained with DAPI (blue). The double arrow indicates the pattern direction. D1-D2: Aligned F-actin fiber (red) E: Using vacuum suction, the printer selects cardio-spheres and puts them onto a needle array, F: Cardio-spheres formation, G: 3D bio-printed cardiac patch, H: Formation of sarcomeres, I: In-vivo engraftment of the printed scaffold on the infarcted area of the mice, J: CMPCs organized into sarcomeric structures, K: CMPCs formed tubular structures, and L: Vessels formation. Likewise, bones and cartilage have also been bioprinted using bone ceramics and biopolymer. Several disease conditions and traumatic injuries require personalised medical treatment. Further, the structural dimensions vary with the individual, and extent of traumatic injury. 3D bioprinted can solve this issue by providing a patient-specific implant for cartilage and bone regeneration. A study showed the extrusion bioprinting in presence of cations to prepare ear cartilage(Fig.3). Other 3D cartilage grafts, such as nose, and spinal disc transplants, have been produced as well. A clinical study showed auricular repair with the use of bioprinted personalised cartilage.Another study showed tracheal tissue implantation helped in the regeneration of tracheal cartilage and smooth muscle.
Figure 3. A, D: Schematic representation of the 3D bioprinting process; B: CAD file; C: 3D bioprinted ear; E: Process of printing different cartilage tissues. & Co. KGaA, Weinheim; F: CAD file; G: Mould; H: PLA scaffold; I: 3D bioprinted ear; J: Implanting bio-printed ear to the patient; K: skin graft is sutured onto retroauricular and mastoid regions; L-O: Post-implantation follow-ups.
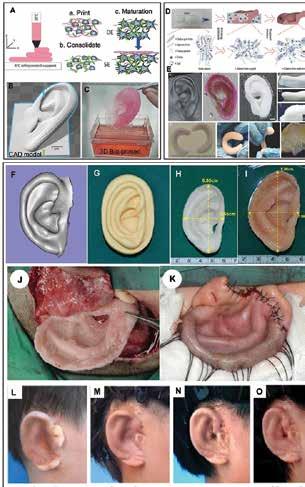
In acute traumas, skin can regenerate. However, its repair is impaired in chronic traumatic injuries, requiring a surgical procedure. This includes skin grafting and cell therapies, but it has a risk of infection, and immune rejection. In contrast, cell therapy does not provide structure,reducing its effectiveness. Perfusable vascularised 3D human skin has been developed using bioprinter (Figure 4), with distinct skin layers mimicking human skin. Furthermore, skin pigmentation has also been achieved. The RECELL device has been approved for burn treatment that helps regenerate the skin quickly.
Figure 4. 3D bioprinting process for fabrication of 3D skin model. A-B: Schematic of the fabrication process; C: Prototype of skin tissue; D: Schematic representation of skin tissue anatomy; E: Epidermis stratified (H&E staining) and stained with keratin 10 (K10) and filaggrin, to depict early and late epidermis differentiation; F: Protein markers representing the epidermal-dermal junction and ECM components; G: The existence of endothelial cells demonstrated by the vascular channel; H: Adipocyte lipid droplets are shown by hypodermis staining. I: Melanocytes stained and blue arrows indicate the areas of melanin pigmentation in epidermal junction.
Regeneration of body glands and reviving their functions could alleviate several diseases. A study showed bioprinting of sweat
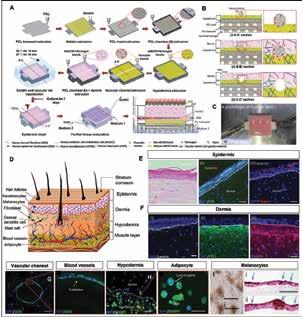
38 PHARMA FOCUS ASIA ISSUE 50 - 2023
Figure 3: A, D Schematic representation of the 3D printed ear
Figure 4: 3D bioprinting process for fabrication of 3D skin model
glands with cellular differentiation and regeneration capabilities. Similarly, another group bioprinted salivary gland using a magnetic bioprinter (Figure 5), which was able to secret amylase enzyme. Further, the thyroid gland with vascularisation was bioprinted (Figure 6). Its implantation in hypothyroid mice restored homeostasis, helped in neuronal development and growth hormones secretion. Likewise, parathyroid-like gland tissues were bioprinted using stem cells that regulate PTH hormone secretion, which have potential to treat osteoporosis.
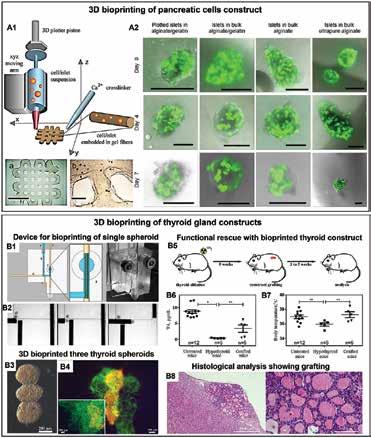
Figure 5.3 DBP of salivary and sweat glands. A:Engineering secretory epithelial organoids by magnetic 3DBP.
Figure 6. 3DBP of pancreatic and thyroid glands. A1:Schematic of 3D bioprinting of islet transplantation and bioprinted scaffold; A2: Fluorescence images of islets.B1-B2: Representation of device for printing spheroids; B3-B4: Bioprinted thyroid gland construct; B5: Illustration of thyroid ablation and monitoring (B6) blood levels and (B7) body temperature in hypothyroid mice; B8: Histological analysis after grafting.
Another emerging application is tissue printing directly onto body defects, which can also betailored to surgical needs. This is achieved with medical
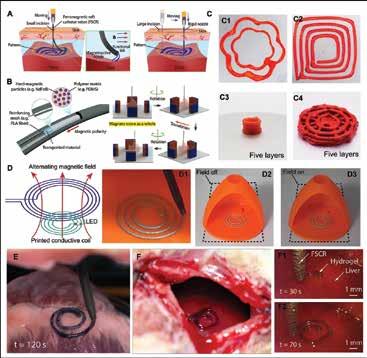
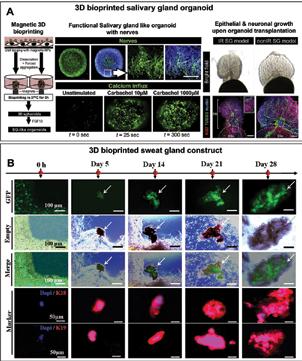
39 www.pharmafocusasia.com A SPECIAL ISSUE ON PHARMA R&D
Figure 5: 3DBP of salivary and sweat glands
Figure 6: 3DBP of pancreatic and thyroid glands
Figure 7: In-situ bioprinting using a novel ferromagnetic catheter in an animal model
imaging, allowing accurate bioprinting on body defects. This is more favourable because the patient's body provides an environment for nourishment to grow and heal. This technique needs lesser labor, is relatively low cost than other bioprinting, is simple, and saves time. For skeletal muscle regeneration, the handheld printer was used on mice models. The bioprinted materials had good adherence to the defective tissue which helps in repair and regeneration. In situ, bioprinter material had no sign of inflammation and showed myotubular formation, which is a sign of muscle formation. Another pre-clinical study showed that in-situ printed muscle improved its healing and physiological response compared to the untreated group, which shows the potential of in situ bioprinting in the treatment of soft tissue injuries. Devices such as Biopen have been used for in-situ bioprinting to treat skin repair. Recently, a study showed roboticbioprinting on organs using a ferromagnetic soft catheter with minimal invasion (Figure 7). This technique can reduce trained surgeon dependency and simple surgical procedures.
Figure 7. In-situ bioprinting using a novel ferromagnetic catheter in an animal model. A-B: Schematic representation of minimal
invasive 3DBP using magnetic particles; C-D: Direct printing of material in different shapes, multiple layers, and an electronic device; E: Invitro BP on porcine tissue; F: In-vivo BP on rat liver with minimal invasion.
In vitro models could help understand the disease pathophysiology, test drug safety or efficacy and mechanism of new therapeutics, helping in drug discovery processes. Several invitroorgan/tissue models have been developed as an alternative to animal models. In 2013, European Union banned animal use for cosmetics testing that urged industries to discover non-animal alternatives to test new cosmetics or skincare products. Nowadays, many industries are developing and using 3D bioprinting for model development. Organovo is developing 3D human tissues.Some companies are providing services for testing liver models. TissUse GmbH developed a model platform for many organs including skin, intestine, lung, and liver.Spheroids, a model to mimic cell aggregates,has been developed to mimic thebrain tumor. Likewise, several cancer models have been developed for various cancer or tumor types (Fig. 8). Glioblastoma-on-a-chip examines patient-specific drug responses. An OTMEChip model for ovarian cancercan assess antitumor-antiplatelet therapy.These platformscan be used to study mechanism of cancer spread within body and test drugs to stop spread. A patient-specific tumour on-chip model helped in understanding the drug resistance in a patient. Personalised cancer treatment could be designed in future with use of these models. Likewise, antibacterial, anti-infective models help study mechanism of infections and test drugs to treat infections (Figure 9).
Figure 8. 3D bioprinting of tumor model. A-B: Representation of bioprinted invitro tumour metastatic model; C: Image captured before capsule rupture by laser. D: 3D bioprinting of glioblastoma-on-a-chip.
Figure9. Microbial constructs for drug testingand disease modeling. A: Schematic representation of microbial bioprinting for engineered biofilms;B: Biofilm encapsulated E. coli and microbial adsorption and sensing;C: Representation of fungal braininfection model using the neurovascular chip;D: BBB penetration studies with four species of fungi;E:Immunofluorescence images of fungi crossing BBB.
40 PHARMA FOCUS ASIA ISSUE 50 - 2023
Figure 8: 3D bioprinting of tumor model
Himanshu is a co-founder of Nusmetics Pte. Ltd. He did his Postdoctorate at the National University of Singapore. His research interests are healthcare interventions, drug delivery, nanomedicine, and alternative and culinary medicine. He has published 26 peer-reviewed journal articles in leading journals such as Advanced Drug Delivery Review, Journal of Controlled release, Biomaterials, International Journal of Pharmaceutics, Applied Materials Today.


4.Clinical progress
3D printing technologies helped in fastening ideas testing and prototype developments, which included diagnostic kit development. Recently, France based organisation clinically tested bioprinted skin tissue for therapeutic use (NCT04925323). Likewise, the colorectal cancer model is developed for testing drugs (NCT04755907).TUMOVASC (NCT04826913), GLIOMANOID (NCT03971812), and ORGANOIDES (NCT04278326) are some of the organoid models under clinical investigation. National Cancer Institute prepared 3D organoid to study blood cancer and test relevant therapies for its treatment (NCT03890614). Similarly, a cardio device is being investigated in patients for thrombus aspiration (NCT03832153).The Food and Drug Administration (FDA) approved A bioresorbable tracheal splint to treat tracheomalacia in an infant, which improved the infant condition. AuriNovo, a patient-specific ear implant, helps in auricular reconstruction (NCT04399239). Similarly, National Dental Centre, Singapore developed 3D bioprinted scaffold for dental application (NCT03735199). Recently, the FDA approved 3D printed Phonograft for eardrum healing. Overall, 3D bioprinting is advancing but many challenges must be overcome, and it can provide creative solutions in revolutionising modern healthcare systems.
Nileshkumar Dubey is an Assistant Professor (tenure-track) in the Faculty of Dentistry, National University of Singapore. His lab is focused on Biofabrication and Electrospinning technology for tissue regeneration and drug delivery applications. Recently, he has been honoured with Interstellar initiative Award by the New York Academy of Sciences and the Japan Agency for Medical Research and Development that recognises and connects the world's most promising Early Career Investigators.
41 www.pharmafocusasia.com A SPECIAL ISSUE ON PHARMA R&D
AUTHOR BIO
Figure 9: Microbial constructs for drug testing and disease modeling
Advancing Towards a Silent Pandemic A call to action
The emergence of antibiotic resistance is becoming a serious threat to the therapeutic efficacy of antibiotics which have transformed medicine and saved millions of lives. Overuse and misuse of antibiotics has led to the antibiotic resistance crisis. Lack of research and development of newer antibiotics after 2000 is also a matter of concern. Multisectorial coordination to implement new policies, enhancing research efforts, and pursuing steps to manage this serious situation is the need of time.
Aditya Rana, Doctor of Medicine, Clinical Microbiology
The rising antibiotic resistance trend is a concern globally.
Antimicrobial resistance (AMR) resulted in 1.27 million deaths annually world over, leading towards a pandemic. This number is predicted to rise dramatically if radical actions are not taken in the near future. Resistance has acquired the iceberg phenomenon — the hidden part is leading toward one of the greatest threats to global health.
The importance of rapid development and judicious use of antibiotics has come under the limelight with the emergence of the upcoming silent pandemic of multi-drug resistance organisms. The SARS-CoV-2 pandemic has shown the significant urgency of the development of vaccines and drugs. The threat has been raised by the increasing resistance towards antibiotics.
History of Antibiotics
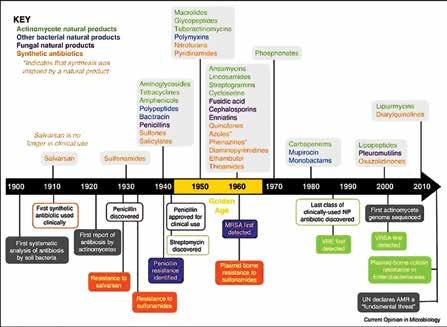
Antibiotics have been considered one of the wonder discoveries of the 20th century. Salvarsan, the first antibiotic, was deployed in 1910. The life span of human beings has been extended by 23 years after antibiotics were introduced, significantly helping modern medicine.
The golden era of antibiotics started with the discovery of penicillin in 1928 and various antibiotics were discovered till 1960 with remarkable pace and decline thereafter.
Development of resistance
With the development of newer methods of diagnosis and discoveries of infective
agents the use and evolution of antibiotics started. Simultaneously the uncontrolled use of antibiotics in the field of agriculture and animal husbandry started for better outcomes. These microbes possess extraordinary genetic ability to overcome the activity of antibiotics and development of resistance. Inappropriate and overuse of antibiotics in humans
42 PHARMA FOCUS ASIA ISSUE 50 - 2023
also contributed to the development of resistance.
Resistance towards penicillin developed within 20 years of its discovery followed by tetracycline and erythromycin in 1959 and 1968 and concurrently with time the resistance towards other antibiotics kept on rising.
Discovery of the newer antibiotics has declined remarkably after 2000. Many pharmaceutical companies withdraw the manufacturing of antibiotics due to their relatively shorter periods of use, antibiotics are not as profitable as drugs that treat chronic conditions, such as diabetes, psychiatric disorders and asthma.
Risks caused by raised antibiotic resistance

Resistance causes delay and failure during the treatment of infections this lead to overstay of patients in the healthcare setting causing economical burden to the patients. Limitation of the treatment options causes higher morbidity and mortality.
Resistance cause development of multidrug resistant organism (MDRO) also known as Superbugs, which are a potential threat to communities. These include multidrug resistant mycobacterium tuberculosis, extensively resistant Mycobacterium tuberculosis, methicillin resistant staphylococcus aureus, carbapenem resistant enterobacterales and MDRO acinetobacter species. Spread of these MDRO add to the financial burden of countries.
Action to be taken
Resistance is a serious matter of concern that requires a multisectoral approach. Multiple sectors should be brought together for the one health approach. This includes the healthcare system, animal husbandry, agriculture, pharmaceutical companies and government bodies that communicate and work together in the design and implementation of programmes, policies, legislation and research to attain better public health outcomes.
Emphasis should be given to innovation and investment for research and development of new antimicrobial medicines, vaccines, and diagnostic tools. Strict guidelines by the government should be made to restrict the over-the-counter availability and judicious use of antibiotics.
Antimicrobial stewardship should be incorporated in the health care setting for the cautious use of antibiotics. Six ‘D’ should be followed: right diagnosis, right drug, right dose, right duration, right direction (route) and right time of de-escalation.
Proper infection control policies should be framed by the health institutions. Periodic check should be kept for the changing trends of resistance in various microbes. Healthcare workers should be trained from time to time regarding the infection control measures.
References are available at www.pharmafocusasia.com
Aditya Rana is MBBS and Doctor of Medicine in Clinical Microbiology. Presently working as Clinical Microbiologist (Senior Resident) in the department of Microbiology, Dr.RPGMC Kangra Himachal Pradesh, India. His professional experiences include close work with Antimicrobial resistance, Infectious diseases, Infection control in health care setups, Molecular diagnostics and Medical writing in the field of Clinical Microbiology.

43 www.pharmafocusasia.com
AUTHOR BIO
A SPECIAL ISSUE ON PHARMA R&D
Novel Approaches in Early Phase Oncology Trials to Revolutionise Cancer Care

The pursuit for effective treatment modalities for cancer rides on sustained efforts with catalyzed adoption of technological advancements that surpass traditional paradigms. Cancer, with nearly 10 million deaths yearly, is the leading cause of death worldwide. However, the beam of comfort is the intensive research work being conducted in the oncology space. Cancer survivors living in the U.S. increased from 3.0 million to 15.5 million between 1971 and January 2016, with 70 per cent and more of the gains in cancer survival due to novel medicines, highlighting the importance of innovative clinical trials as vital tools in the pursuit of improved health.
Sowmya Kaur, Executive Vice President, Navitas Clinical Research, Navitas Life
Atul Gupta, Vice President, Medical and Client Solutions, Navitas Life Sciences Akash Gadgade, Senior Manager, Medical Services, Navitas Life Sciences
The pursuit for effective treatment modalities for cancer rides on sustained efforts with catalysed adoption of technological advancements that surpass traditional paradigms. Cancer, with nearly 10 million deaths 1 yearly, is the leading cause of death worldwide. Every year, there are 23.6 million2 new cancer cases, with an estimated 250 million disability-adjusted life years (DALYs) due to cancer.
However, the beam of comfort is the intensive research work being conducted in the oncology space. Cancer survivors living in the U.S. increased from 3.0 million to 15.5 million between 1971 and January 2016, with 70 per cent and more of the gains in cancer survival due to novel medicines, highlighting the importance of clinical trials as a vital tool in the pursuit of improved health.
1 https://www.who.int/news-room/fact-sheets/detail/cance
2 C:\Users\amrita.surendranath\AppData\Roaming\Microsoft\Word\ Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for Cancer Groups From 2010 to 2019 A Systematic Analysis for the Global Burden of Disease Study 2019 JAMA Oncology
Sciences
CLINICAL TRIALS
There have been sustained efforts to make clinical trials faster and efficient to facilitate augmented adoption of drug products. About 85 per cent of the oncology medicines in development are likely to be first-in-class, while more than 6,500 cancer drugs are in the R&D pipeline, with oncology investigational drugs accounting for 37 per cent of agents in clinical development, in line with the high unmet need.
Despite some significant success achieved, the challenges in cancer research are monumental. Cancer therapeutics currently have the lowest clinical trial success rate of all major diseases. Of the oncology agents that enter Phase I trials, only 3 per cent eventually receive U.S. Food and Drug Administration (FDA) approval. The answer to this critical problem lies in novel approaches to cancer clinical trials, which not only increases the success rate but also recognises failure in a faster time and lesser cost, to allow the resources and efforts to be directed in an optimised way. This need to be applied from the outset of the clinical development program, and this paper focuses on innovative designs in early phase oncology trials (Phase 1 and 2). While study designs are the focus area, innovation can be extended to other aspects as well like patient selection, endpoints as well as study logistics.
Why Traditional Designs Will Become Obsolete in Early Phase Oncology Trials

The traditional approach to clinical development follows a fixed straight-line path and tests a specific scientific assumption throughout the study. Whereas innovative designs are dynamic in nature and allow updation of the study protocol based on the data generated
Regulatory Stand on Innovative Designs
Although in theory, the concept of innovative designs was formed nearly two decades ago, there was gradual acceptance with increasing evidence-based proof.
The European Medicines Agency (EMA) in 2007 came up with guidelines
on the introduction of adaptive measures in trials. As a part of their Life Sciences Industrial Strategy in 2017, the U.K. Government committed investment towards clinical trials that incorporate “novel methodology”, in partnership with the pharmaceutical industry.
The U.S. Food and Drug Administration (FDA) released a draft guidance around master protocols and adaptive designs in 2018. The FDA also started a pilot program to support complex innovative design (CID) trials that accelerate drug development in areas of unmet need.
EMA on Complex Clinical Trials (2022): “The rationale for the complexity of the design and conduct of a complex clinical trial needs to be explained in clear terms and justified in the protocol(s) and related documentation; such information should be made available to investigators, regulators, and provided in lay language to clinical trial participants. It should also be explained why the same objectives are not pursued using more conventional, non-complex designs.”
FDA on Master Protocols in Oncology (2022): “The potential advantage of a master protocol is flexibility and efficiency in drug development, consistent with FDA’s goal of helping to make safe and effective drugs and drug combination treatments available to the public. A master protocol provides an opportunity to incorporate efficient approaches, such as a shared control arm and/or the use of centralised data capture systems to enhance efficiency. However, a master protocol can also create challenges in the conduct and analysis of the trial that, if not properly addressed, can increase risk to human subjects or delay the development of the drug”.
Dose Escalation: Finding the MAXIMUM TOLERATED DOSE (MTD) and Recommended Phase 2 Dose (RP2D)
MTD estimation in phase 1 dose-escalation trial can be based on either Rule Based Designs or Model Based Designs.
CLINICAL TRIALS
Rule-based design
Rule-based designs are commonly used and referred to as the 3+3 designs. Patients are enrolled into small cohorts (usually three patients), with a starting dose initially defined by the data from animal models. Provided that no predefined dose-limiting toxicities (DLTs) occur in the first three patients (i.e., <33 per cent in the first cycle), the dose is escalated to the next dose level using a predefined algorithm. Dose escalation proceeds until a DLT occurs; at this point, the dose level is either expanded (if ≤1 DLT) or patients are accrued to a lower dose level (if ≥2 DLT). If there is further DLT, then that is the Multiple Ascending Dose (MAD) and the dose level below is usually considered there commended Phase 2 dose (RPTD).
While this trial design continues to be the most commonly used , it has been criticised on the grounds of inefficiency, lack of statistical foundation, ethical considerations (the number of patients treated at low doses) and applicability to targeted agents where acute DLT in cycle one may not be relevant.
Newer designs, such as accelerated titration (AT), attempts to address this by enrolling a single patient to each early dose level until a prespecified level of toxicity (e.g., ≥ grade 2) is observed. Thereafter, three patients per dose level are accrued until the MAD/ RPTD is reached. Simon and colleagues demonstrated that AT designs have the potential to reduce the number of patients necessary to determine the RPTD and increase
the number of patients receiving a potentially therapeutic dose. Unfortunately, these theoretical advantages may not translate into clinical practice. In our view, between the traditional versus AT design, the proportion of patients that were treated below the MTD was lower for the AT design compared with the 3+3 design (58 vs 71 per cent). However, the total number of dose levels was higher for the AT design, while the length of study was similar for both designs
Model-based designs
Model-based designs adapt to data insights that become available during the clinical trial. Proponents consider

them to have the potential to increase efficiency, treat more patients at or near optimal doses and address several questions within the context of a single trial. Although there are no direct comparisons of efficiency between model-based designs and traditional designs, reviews suggest that the new trial designs might result in fewer cohorts, or fewer patients treated at lower dose level.
Potential reasons for this include the added complexity of the trial and the need for biostatistical support during the conduct of the study. Commonly discussed examples are the continual reassessment method (including trivariate continual reassessment model [Tri-CRM]) and escalation with overdose control (EWOC).
The continual reassessment method selects the first dose near the predicted RPTD based on statistical modeling. Toxicity data obtained from the first patient enrolled onto the trial are then used to reassess the probability of a DLT occurring at a specific dose level; this information is then used to select the next appropriate dose. Proponents of the model believe that it offers a more accurate and precise measurement of the MTD with fewer patients experiencing DLT. However, there are concerns that patients would be exposed to toxic doses of the experimental agent due to the rapid increase in dosing that the model proposes.
The EWOC design minimises the risk of overdose by specifically setting the probability of a dose above that desirable to a preset low level. The trial design then proceeds in a similar manner to approach the RPTD efficiently. Simulation studies have demonstrated that a greater proportion of patients are treated at optimal doses compared with rule-based design and has the potential additional safeguard over the continual reassessment method.
Common Approach May Not Be THE Best Approach
Traditional 3+3 method. Used most often, but lacks precision, yields an
46 PHARMA FOCUS ASIA ISSUE 50 - 2023
CLINICAL TRIALS
About 85 per cent of the oncology medicines in development are likely to be first-in-class, while more than 6,500 cancer drugs are in the R&D pipeline, with oncology investigational drugs accounting for 37 per cent of agents in clinical development, in line with the high unmet need.
overestimation or underestimation of the true MTD. It involves an excessive number of escalation steps, which results in a large proportion of patients who are treated at potentially subtherapeutic doses while few patients actually receive doses at or near the recommended dose for phase II trials.
Other Rule Based Designs (Accelerated titration, Pharmacologically Guided Dose Escalation etc.) These designs may be better than conventional 3+3 method, but still have the inherent disadvantage of rule-based designs. For example, these designs may be inefficient in establishing the dose that meets a specific target toxicity level. In addition, the decision of dose allocation for future patients as well as the definition of the RP2D rely on information from the current dose level and do not use all available information.
Modified continual reassessment method: This method has several advantages including precisely defining target toxicity level, faster dose escalation, utilising all available information from all patients, and accounting for late-onset toxicities.
The Continual Reassessment Method (CRM) is an adaptive method and can better identify the target dose. Unfortunately, many stakeholders are hesitant to use CRM as it is considered a completely unknown territory. The solution is to engage a CRO partner experienced in CRM at an early stage.
Biomarkers in Early Phase I Trial


Biomarkers, especially tissue-based ones, may be incorporated into Phase I clinical trials for a number of reasons. They may be used to confirm that the agent is achieving the desired molecular effect (proof of principle) or penetrating into the tumor. They may also be used to help define the recommended Phase 2 dose (RPTD), either by demonstrating an effect estimate, based on data obtained from preclinical studies, or by demonstrating a dose-response (or lack thereof).
This is especially useful for drugs that are associated with little toxicity. Finally, their use in the early clinical trials setting may allow early identification of subsets of patients most likely to benefit. This can be further explored in the Phase II/ III setting.
Biomarkers in early-phase clinical trials: Demonstrating their potential impact in the future.
In the development of the DNA repair protein polyADP ribose polymerase (PARP) inhibitor, Fong et al. not only utilised biomarkers in tumor and surrogate tissues to demonstrate proof of principle of the PARP inhibitor, but, based on preclinical data, enriched the trial with patients known to have germline BRCA mutations demonstrating an impressive relative risk (R.R) in this subset of patients. Such data provided insights for further development of this agent. Biomarkers were also of use in the development of other agents such
as bortezomib.
Innovations in early phase oncology trials design and in clinical drug development, moving towards the implementation of adaptive designs and master protocols, are encouraged by regulatory agencies.
The biggest advantages of adaptive designs and master protocol are flexible decision-making, accelerated timelines and cost efficiencies. The early phase adaptive trial design will also help in the later stages of clinical trial.
Early phase adaptive clinical trial success story continues to inspire and influence the development of nextgeneration trial designs in oncology as well as other therapeutic areas.
These flexible methodologies in adaptive clinical trial design are critical enablers of scientific breakthroughs that will aid in faster pace of drug development.
References are available at www.pharmafocusasia.com
Sowmya Kaur is Executive Vice President Navitas Clinical Research, Navitas Life Sciences, took on her new leadership role during the COVID-19 pandemic. With a career spanning over 22 years, Sowmya has worked across multiple aspects of the industry including operations, business development, and strategy with leading industry players like Cognizant, IQVIA, Kendle etc. She has a successful track record of building and leading Clinical Development engagements across Emerging Markets with successful delivery of a portfolio of projects.

Atul, Vice President-Medical & Scientific Affairs, Navitas Life Sciences has over 16 years’ experience in Clinical Research, Drug Development and clinical practice. Atul has worked as Global Medical Lead (USA, Europe and Asia Pacific) in more than 50 studies, and has been involved in apt designing of complex trials. Atul has experience in diverse therapeutic areas, in all the phases (I-IV), in both drugs as well as devices. Atul has multifaceted experience in medical writing related to clinical studies (Protocol, ICF, CSR etc..)
Akash Gadgade, Senior Manager, Medical Services, Navitas Life Sciences has over 10 years of experience in clinical research including Pharmaceutical Industry and CRO. He worked with various capacities as Principal/Co- Investigator, Medical Advisor, Medical Officer and Medical Monitor. Key expertise area includes Diabetes, Hypertension, Oncology, Immunology (Vaccines), Ophthalmology, Hepatology, Hematology, Devices, Dermatology, Infectious disease, Healthy Volunteer study for early phase trials. He has multiple publications in National and International Journals.
47 www.pharmafocusasia.com
AUTHOR BIO
CLINICAL TRIALS
Reinventing Patient Recruitment Achieving accelerated clinical endpoints through a revolutionary patient recruitment model
Retention and recruitment of participants in clinical trials is often the most labor-intensive and challenging component. Poor recruitment and retention frequently pose a major barrier to the successful completion of clinical trials. This article examines the effective strategies for the research industry to improve the success rate in recruiting research participants using innovative recruitment methods.
 Jeff Parke, Visionary Co-founder, P.A.C.E. (Project for Accelerating Clinical Endpoints) Raman Sehgal, Clinical Research, PFC Pharma Focus India Pvt. Ltd.
Jeff Parke, Visionary Co-founder, P.A.C.E. (Project for Accelerating Clinical Endpoints) Raman Sehgal, Clinical Research, PFC Pharma Focus India Pvt. Ltd.
It is widely recognised that clinical trial success is dependent on patient recruitment. Low enrolment rates have led to study times potentially increasing by up to two-thirds. Refusals to meet recruitment goals can have serious scientific, financial, and ethical implications. There are many intangible consequences that can be detrimental to investigators, sponsors, and participants. The most important aspect is that failure to recruit and achieve overall study goals can have a negative impact on patients. It hinders efforts to diagnose, treat or
prevent disease. Despite multiple decades of efforts to identify and enroll study participants, recruitment continues to be one of the largest barriers to clinical trial success.
Researchers have reviewed factors that could contribute to successful recruitment. They examined the design of trials, issues with study staff, recruitment strategies, and the need for revising recruitment targets and timelines. Other researchers have focused on increasing recruitment and retention by giving more consideration to participant contact, convenience,
financial support for patients, incentives, compensation for participation, as well as other human factors. The most important phase of a clinical trial's life cycle, the upstream planning and design phases, may be the most influential for positively impacting downstream recruitment efforts. Effective planning will require input not only from those who have traditionally led this effort but also from a range of stakeholders including patients and patient advocacy groups, sponsors, investors, site staff, and healthcare providers. Given the variety of factors that can affect recruitment for clinical trials, it is imperative to develop proactive and inclusive strategies that go beyond those that are specific to a particular study.
It will take more than simply reaching out to the community to find the solution. It is about understanding the complex motives and concerns of everyone. This requires the same level of individualised understanding that companies strive to attain, which taps into the personalities of their customers. Healthcare profes-
48 PHARMA FOCUS ASIA ISSUE 50 - 2023
CLINICAL TRIALS
sionals have not yet considered factors such as these because they are not visible to them. These factors are only available through self-reporting techniques and focus groups. They are not reliable for uncovering the true motivations of people. To make progress, this issue must be addressed.
Create relationships
In today’s research landscape, recruitment is seen as an end-to-end process rather than an experience. The industry is missing an opportunity to engage potential participants through a sense of community and purpose. This is what a clinical trial should be built upon. This challenge was further highlighted by the experience of pharmaceutical companies during the pandemic. COVID-19 required the industry to test different methods of conducting trials, and a few companies tested the possibility of remotely running them. Initial expectations were that this would lead to increased participation and better recruitment. This has not been the reality.
This teaches us a simple lesson: People want human contact and emotional connection. It is crucial to make this connection, both because trials can be difficult and because participants can gain a great sense of purpose from their roles. People should never underestimate the power of feeling able to give back to others who are suffering from similar conditions. Maybe reducing friction inadvertently reduces the meaning of potential participants. These interactions can be harder to quantify emotionally, but they are just as important as monetary incentives. People are not rational beings that are only motivated by money. Trial recruitment should take a holistic approach to human motivations and be as focused on the sense of belonging
and purpose that comes with being a participant. To help participants, find meaning in trials, this may require some friction and complexity.
The business case
Patients, as with all people, are predictably irrational. Although scientific and evidence-based understandings of human behaviour are crucial for driving sustainable behavioural change, it is not enough to know that people don't make decisions based only on rational information. Therefore, design and recruitment must consider both rational and emotional arguments. Even small changes can be difficult to implement, especially in large organisations that have been conducting trials in the same way for years. An outsider's perspective is crucial in this situation.
The industry must reevaluate its approach to trial design to address the urgent recruitment crisis. It must begin from the beginning, with how participants are understood. There is a business case to make that change. The industry can be helped by a facilitating new perspective.
Patients play an undisputed role in determining the care they receive. Since the days when patients were not included in the development of healthcare products was a controversial idea, we have made great strides. The healthcare ecosystem and the role of patients has been evolving in a promising direction in recent years. This has led to a deeper understanding of how the patient voice can and should impact healthcare.
The goal of including patients is now a top priority across the entire research landscape. This includes regulators, academia, and pharma. Patient partnerships have thrived because of the autonomy and confidence patients have
acquired. Patients who have experienced the disease firsthand are an invaluable resource. They can clarify their priorities and needs well before we begin to discover and develop new therapies.
Many initiatives, including those of the Clinical Trial Transformation Initiative (CTTI), as well as many others, like the Patient Centered Outcomes Research Institute (PCORI), have given us valuable insights into patient engagement. These initiatives give us a glimpse into the types of metrics and operating models that work best, as well as the skills and experience that are most effective.
We have all come together in these cases and found common ground. We should not rest on our laurels. These experiences can be used to help us make progress in areas where we have only made modest or no progress in giving patients the active role that they deserve.
How we treat our clients
The United States is recognised as a world leader in clinical research. However, there are sub-optimal rates of participation in both industry and investigator led clinical trials in the USA. More than ever, the industry needs to develop recommendations for optimising recruitment, which are broadly translational and applicable at the site level.
There are some common strategies that research organisations can use to recruit participants, regardless of whether they are conducting a clinical trial or a community trial.
Preparing for Success
There are many factors about the study and your participant criteria that can affect:
• Where you recruit
• How you communicate about your study
49 www.pharmafocusasia.com
Figure 1: Steps Associated with Successful Recruitment
you
Where you
recruit
How participants are
approached
How
communicate about the Study
CLINICAL TRIALS
The structure of the study team How participants are compensated
Age Communicating with an adolescent, an adult, or an older adult can be vastly different, and where you recruit these individuals varies based on age. For example, social media and other digital recruitment methods will likely be more effective with a younger population.
Language Language might seem like an obvious factor when developing a recruitment plan but there is much more to this than simply translating from English into another language. Spanish for example, uses many different terms for the same thing depending on the country. Literacy level is another very important component to keep in mind when translating. Even when using English, care must be taken to carefully define medical terms that the general population might not be familiar with or understand.
Cultural Norms Understanding the cultural norms of your participants is key in developing an effective recruitment strategy. For instance, within the population of interest, is the decision-making done as a family, a community or individually? In a study conducting phone surveys with adult Korean women, we found they were most responsive when an adult Korean person called and less responsive when a young person called.
Geography
Gender/ Gender Identity
Certain cities and towns have their own culture, popular locations, safe and unsafe areas of town and common hangouts for people of certain ages. Whether the region is urban or rural can also affect your recruitment strategy. For instance, in a rural farming area, recruiting during harvest season might not be effective as farmers and their families are working longer days.
The ways in which you communicate effectively with women can be different from the ways you communicate with men. Remember, women are from Venus and men are from Mars. What motivates one to participate can be different from the other. Therefore, what you highlight about the study and the images you use can draw more men or more women to your study. In addition, society is now beginning to recognize that gender is not always a binary identity – there can be fluidity between the two primary genders. Similarly, transgender identities are also gaining greater visibility in society. New terms that are particularly popular with youth populations, such as genderqueer and intersex, are also becoming more common. Therefore, researchers working with these populations need to be careful not make assumptions about gender and the ways in which someone selfidentifies.
Years Living in the U.S.
Socio Economic Status
This factor is often overlooked. Designing a recruitment strategy for a foreign-born person who has been living in the U.S. for 20 years versus someone who just arrived 5 years ago can be completely different, even if they are originally from the same country. Their understanding of and comfort with participating in anything official like research can be different. Their understanding of the English language will likely also vary.
Someone with a higher economic status most likely also has a higher level of formal education, which means they most likely already have an advantage to others in their understanding of research and access to research studies. It probably would not take as much effort to reach this population as it would to reach those of the working class and those in lower socio-economic status. It is also probable that those of a lower socio-economic status have less flexibility in their job for time off to par ticipate in a research study, which means your study will have to be more accommodating if you want to have a diverse population (e.g., scheduling at nights or other non-traditional hours). Likewise, transportation or the lack of it can also be an issue.
Those with no legal status in the U.S. are probably the most difficult group to reach. in the U.S.
Legal Status
They do not want to be identified or stand out in any way for fear of being deported. The simple term, “research” gives the idea of being investigated which can be a very scary thought. Finding other terms to explain research and the research process can be helpful. Also reinforcing the concept of anonymity or confidentiality can make people more open to participating.
Condition Being Studied
Study Participation Requirements
Are there any stigmas associated with the condition you are studying? For example, mental health and HIV issues can be stigmatized among family members and in the general community. Therefore, people with these conditions may not be as willing to participate for fear of others finding out.
The level of risk and type of involvement required to participate in a study is a big factor in the volunteer’s decision of whether to participate. Participating in a 10-minute survey, an interview, or a focus group is viewed differently than more invasive procedures, such as a blood draw, taking medication, or having to see a doctor for other medical procedures. A onetime participation versus a study that requires multiple months or years of commitment is also very different. The latter would require more time and effort on your part to explain the study process and benefits before getting someone to agree to participate.
• How you approach individuals
• How you compensate your participants
• The structure of your study team.
Figure 1
These factors will ultimately determine whether you will reach your recruitment goal. It is important to consider
every one of these factors when planning your recruitment protocol and as you confront recruitment challenges during your study. A variety of participant and study factors can alter your recruitment strategies; therefore, these factors should be considered in the development of your recruitment protocol (see table 1).
Key Takeaways
The industry faces challenges implementing novel technologies to engage participants and collect clinical data. However, research suggest that by developing strategies to maximise participant and partner involvement and reduce participant and staff burden by simpli-
50 PHARMA FOCUS ASIA ISSUE 50 - 2023
Table 1: Participant Characteristics to Consider for Developing a Recruitment Protocol
CLINICAL TRIALS
CLINICAL TRIALS
BARRIERS TO RECRUITMENT
• Awareness
• Poor Communication
• Mistrust of Research
• Lack of Cultural Training / Social Barriers
• Logistigal Issues
• Price = The Risk of Participating in a Study
Figure 2: Barriers to Successful
fying participant experiences and staff workflows — Artificial Intelligence (AI), Machine Learning (ML), Natural Language Processing(NLP), Gamification, Wearables, Direct-to-Patient, remote direct to patients (RDCTs), and Agile (Hybrid) approaches should maximise recruitment, engagement and retention and disrupt the standard and outdated methods. It's hard to believe that patient recruitment is still a challenge despite all the advancements in clinical research. Traditional clinical research models are a challenge at multiple levels, which ultimately hampers the efficiency of clinical trials.
There are many barriers that could prevent patients from participating. These include:
• Awareness - People are not made aware that clinical trials could be available to them
• Poor communication - Complex materials are shared with caregivers and patients that can be confusing. They don't help them make informed decisions
• Price of Participation: While this fear may be justified, it is often not clear what the risks and benefits are or how many procedures the protocol requires

• Mistrust of research, Pharma or Healthcare - Evidence shows that many people don't trust clinical trial administrators and don't believe their best interests are in the patients' hands.
Figure 2
Both at the site level and the sponsor, time and costs are often the greatest challenges in recruiting patients. The industry should be aware of the following aspects:
• Poor patient engagement - Not engaging with patients early enough or at all can lead to major problems with recruitment

• Complex protocol and demanding times - Patients and caregivers may be greatly affected by a study design too burdensome.
AUTHOR
• Exclusion and inclusion criteria that are too restrictive - The study population must reflect the intended treatment population. However, eligibility criteria should still allow diversity
• Poor or ineffective patient recruitment strategies - All studies must have a carefully considered patient recruitment strategy to avoid unnecessary delays and additional costs
• Cultural training is lacking - cultural and social barriers can hinder studies, particularly if you are trying to recruit a particular group of people. Sponsors and site staff can help overcome such barriers.
This might sound familiar to many in the industry. However, it is still amazed that these problems are not being addressed faster. To overcome these problems and increase patient recruitment, it is crucial to understand them fully.
Rebuilding trust with the public takes time. Strong campaigns and community engagement are necessary if we want to see industry improvements. There are also short-term solutions to the problems mentioned above. Patients and caregivers are the best place to begin. These insights can be used to inform patient recruitment strategies and study design.
References are available at www.pharmafocusasia.com
Jeff Parke is the visionary co-founder of P.A.C.E. (Project for Accelerating Clinical Endpoints). Jeff has developed numerous recruitment tools that have helped facilitate the development of operationally efficient recruitment tools for CROs, Pharma and Investigative Sites. P.A.C.E. establishes singular operational and technological advantages for an entire study planning and enrollment cycle and is the fulcrum by which any efficient and cost-effective modern patient recruitment effort should be mounted. For more information about the P.A.C.E. initiative, please contact via LinkedIn
51 www.pharmafocusasia.com
• Logistical problems - Having to travel to the investigator site for frequent appointments is a burden. This is especially true for people with care responsibilities or mobility restrictions BIO
Recruitment
Raman Sehgal is an experienced drug development professional with demonstrated leadership skills in clinical operations, global project/program management, strategy development and customer engagement. His experience spans the clinical development spectrum from pre-IND to NDA. He has managed multiple teams at small and large CROs delivering integrated solutions that meet patient needs and drive customer success. He also moderates the largest group of Clinical Research Professionals on LinkedIn.
DIGITAL INNOVATION How you and your teams can digitally innovate successfully in the Pharma ecosystem
Digital innovation provides Pharma companies with unique opportunities for creating long-term, competitive, sustainable advantage. However, Pharma companies aren’t tech companies; they are often not structured in ways that foster innovation, and their culture is very different from that of more agile and risk-taking BioTech companies. In this article, we will, therefore, cover some of the most important challenges you will face when you try to introduce and integrate digital innovation into the fabric of the Pharma ecosystem, as well as provide suggestions for solutions. – Fausto Artico: Global R&D Tech Head and Director of Innovation and Data Science, GSK.
Fausto Artico, Global R&D Tech Head and Director of Innovation and Data Science, GSK Kevin Harrigan, Director of Innovation and Engineering, GSK
Data is the new oil, or so they say. However, your ability to extract the oil and to ensure its quality are only two of the elements you need to create digitally innovative solutions. There are important technical activities that are also required in order to create a solid and reliable foundation for your

digital innovation journey. And lastly, there are aspects related to the organisational structure and the pervasive culture that characterises Pharma companies that must be addressed. These two additional aspects (i.e., organisational structure and culture) are critical to the success or failure of your digital innovation efforts and so
your capacity to create new digital solutions and have them adopted inside and outside your organisation.
Technology
Your existing systems determine the next phase of your digital innovation process. Where are you today in your journey?
52 PHARMA FOCUS ASIA ISSUE 50 - 2023
INFORMATION TECHNOLOGY
You need to build on strong foundations, and that requires the ability to correctly self-assess what infrastructure you, your teams and your organisation have. To start, are you sure you and your teams have the skills to correctly evaluate the reliability and quality of your company’s existing systems or do you need outside help? The reason I am asking is because I often see non-technical people who are very enthusiastic about what they and their teams have accomplished in the past three to five years or have available as existing systems, yet all too often the architectures and software components of the systems are not reliable. In fact, many times the architectures are the result of no more than activities executed to make many different legacy systems and vendors’ products able to “speak with each other.” In other cases, where new systems are created from scratch, all too frequently such systems are standalone and not really integrated and able to seamlessly communicate with existing ones, and so the value they generate is only a small fraction of what it could be. Correctly assessing your systems necessitates multiple teams (internal and external) to obtain a more reliable snapshot of the real situation and must be executed before starting or continuing your digital innovation initiatives.
Try not to take steps longer than the length of your legs (i.e., your capabilities). Often people rush forward and try to create and put into production Artificial intelligence (AI) and machine learning (ML) solutions that cannot succeed because they are designed incorrectly. This isn’t due to missing data, poor data quality and/or the lack of some system components that are foundational for the solutions but is due to a lack of capabilities. Asking for help to understand which capabilities you need to build or acquire for the different phases of your journey is money and time well invested and will avoid many headaches when you have to design, implement and productionalise multi-year transformational projects. Such projects are complex but can be de-risked in smart ways especially if you modular-
ise them, deliver incremental value, and design their architectures in ways that will hyper-automate activities. Remember also to embed security principles from day one. De-couple software components enough to avoid vendor lock-in. Enhance plug-and-play capabilities as much as possible so that you can swap architectural components as necessary if some technologies become mature much more quickly than others. Finally, to correctly scope and decide on future architectural choices, you will probably need to hire the right people (e.g., computer scientists) with the right skills from outside your company.
Don’t consider just some architectural aspects to the detriment of others during the creation of your solutions. Many times, solutions fail to be put into production because somebody overlooked some regulatory aspects, or the final systems aren’t secure, or some technologies cannot be validated for the production systems because they simply cannot be integrated with other production components that have already been approved for commercialising a drug. Creating new solutions requires much more than just the creation of new digital models (e.g., AI/ML).
Pharma is complex and the number of components that need to be considered to create new solutions goes well beyond the creation of such new models and their ability to correctly predict outcomes or generate new insights. Involve many teams as you design or acquire new solutions; you do not have all the pieces of the puzzle, and getting expert feedback greatly helps to de-risk the process.
Remember, you must at least have an accurate, ten-thousand-foot view of what is going on under the hood of your overarching system; you must correctly assess your capabilities; and you must make sure there aren’t overlooked aspects. Otherwise, the risk is that independently of how many skilled software engineers you have in your team and organisation, you will take decisions that are destined to fail (and some could consume years of work and a lot of money).
Prioritise your opportunities and focus on the ones on which you can already deliver today. They are easy to discover if you execute the previous activities. As you deliver on low-risk, but valuable, low-hanging fruits, continue in parallel to develop new capabilities for opportunities that are out of your reach now but tomorrow will become easy to deliver, thanks to the additional work you and you team are doing.
Organisational Structure
Pharma can be very conservative and risk averse. This is because patients’ lives are important, and over the last 200 years, regulations to approve and commercialise drugs have become more and more complex. Getting a new blockbuster drug approved can easily require more than 10 years. It can, therefore, be difficult to take risks, try new things or bet on new innovative technologies not yet regulated and approved. In addition, the binary nature of clinical trials (i.e., success or failure) makes it difficult for executives to approve the use of such emerging technologies in some verticals of the organisation (e.g., clinical development compared to earlystage discovery).
53 www.pharmafocusasia.com
INFORMATION TECHNOLOGY
Two additional aspects (i.e., organisational structure and culture) are critical to the success or failure of your digital innovation efforts and so your capacity to create new digital solutions and have them adopted inside and outside your organisation.
It is much easier for a Pharma company to acquire other companies than to create new ones. This is especially true after a new digital technology has been proven to be mature or a BioTech company has successfully completed the phase III of a clinical trial and therefore its drug can be commercialised. Pharma is often even willing to pay a premium for waiting so long (e.g., 7 to 10 years in the case of drug approval) just to de-risk the process. Pharma venture capital branches would be well advised to be more willing to use a fraction of the money they have to help digital companies in their early stages to survive. Furthermore, they should drive their product roadmaps, at least for the digital products that are not directly connected to critical and highly regulated aspects of drug approval but that can help to greatly accelerate other phases of the Pharma pipelines. This would allow Pharma companies to leverage external expertise to create new systems in ways impossible to accomplish using only their internal capabilities. It would also allow Pharma companies to create much more flexible digital ecosystems that are today impossible to deliver in any reasonable time by big vendors since big vendors are often forced to standardise their products and use cookie-cutter approaches because of the support they must provide to their many clients.
While Pharma executives may understand the importance of innovating, they are often unaware of the fact that the existing organisational structure threatens the efficacy of innovation teams and/or is setting them up for failure. For example, many of the non-technical challenges innovation teams face are mainly due to how they are funded, and this is directly connected to how the organisation is structured. Funding is typically provided by other teams that often impose many constraints. Instead, Pharma should create and set up innovation teams as independent units (i.e., independent companies, really). The innovation teams should receive enough money to execute twoto-three years of work and be tasked with delivering multiple projects and initiatives. This does not require a lot of money (just from a few million to a max of 10 million dollars). This amount of money is nothing compared to how much is usually invested in other large-scale initiatives and paid to big vendors (i.e., from tens to many hundreds of millions of dollars). The innovation teams should also be set free to design and implement whatever they deem necessary to prove their value and maximise the return on investment. They should set up their own systems and ways of doing things, independently from the existing IT systems and other teams’ ways of working (although, of course, co-development and collaborations need to be supported and incen-

tivised). This freedom might be scary for some executives to accept, but it would allow innovation teams to prove their value. Furthermore, freeing up innovation teams from as many constraints as possible provides them with the ability to turbocharge the transformational changes that senior executives need developed and deployed into the organisation in very short time frames (e.g., from 1-2 quarters to a max of 2-3 years).
Culture
Existing teams may unfortunately think of digital innovators as servants to their most pressing needs. Because of this, innovation teams often are told what to do instead of being free to explore. In addition, many members of existing Pharma teams do not have the technical background necessary to understand how to drive architectural choices and so create and inadvertently deliver digital transformation plans that generate only partial success, lots of frustration for tech people and make it difficult for an innovation team to prove its value. In addition, because the team members of an innovation team are usually very skilled, they are liable to be asked to put their critical long-term projects on hold again and again as they are continuously reassigned on demand to different teams to fix urgent problems that are affecting the day-to-day production system operations. Under such conditions, it is
54 PHARMA FOCUS ASIA ISSUE 50 - 2023
INFORMATION TECHNOLOGY
practically impossible for an innovation team to deliver on its projects. Overlaps and competition. Digital innovation is chic. If several innovation teams are created in different parts of an organisation to satisfy the requests of several high-level stakeholders, they may well find themselves competing against each other. This is a cultural alignment problem that can be solved only from the top layers of the organisation. In addition, some teams that are powerful enough could decide to create inside themselves an innovation team from scratch. I understand that existing teams might want to transition some of their people to do digital innovation work, but is this really a feasible option? Pharma people are usually biologists, statisticians, etc., and don’t come from a computer science background. Many times, what could seem easy to them to accomplish in the digital field is full of technical traps visible only to the eyes of a computer scientist. Furthermore, Pharma people usually don’t know programming, and Pharma, like Finance, is a heavily regulated sector full of legacy systems and complex interdependencies among many software components. Would it not be better to have innovation teams and existing teams co-develop solutions together? Each could add value through its domain of expertise and skillset (programming and architectural choices for computer scientists, drug discovery for biologists, etc.). Each should be considered a peer, with the equal possibility of proposing new solutions. Each should decide how to implement the parts of the solutions that fall inside its domain of expertise. Senior executives need to avoid competitive dynamics and generate as much alignment as possible. If this is impossible, they should at least make sure that in each vertical (e.g., early-stage drug discovery, clinical development, manufacturing, supply chain, etc.) there is no more than one innovation team working on each objective per geographical area / site and that it is considered a peer department, appropriately funded,
to the others that exist in the vertical. Finally, for executives, it is difficult to justify the creation of independent and peer-like innovation teams. They seem like big, ‘risky’ bets. But this is not true, especially if other initiatives can be used as comparison. However, such comparisons can be difficult to make because failures tend to be brushed under the carpet and/or presented and reframed as at least partial successes that were necessary to execute anyway. But without funding innovation teams appropriately, what kind of future can we create? Patients need our help, and so innovating is important. It cannot be dismissed or deprioritised. While people understand this, it is also important for executives to continue to educate their companies and C-level stakeholders that innovation initiatives need to be sufficiently funded, and that innovation teams need to be independent and considered peers by other teams and departments.
Summary
We have discussed how technical aspects are only some of the important components that are necessary to consider to successfully design, implement and deliver digital innovation. Organisational structure and culture are two other critical aspects that will determine the failure or success of your digital initiatives. With
the information in this article, you are better equipped to successfully start or continue your digital innovation journey.

Without digital innovation, your company will fall too far behind competitors and disappear. This may take a long time, but it will be inevitable if you do not successfully innovate in the digital field. Some people might argue that the Pharma sector is characterised by incredibly high barriers to entry, but 3D printing with other emerging technologies could destroy or at least lower considerably all such barriers in the next 5 to 10 years. It isn’t difficult to image a future where people can buy a 3D printer, install it at home as a plug-andplay device and automatically program it in a hyper-automated, push-button fashion and download recipes from Blockchain networks in a way that could be completely unregulated and/ or difficult to monitor for governments.
Being open-minded, creating partnership ecosystems and digitally innovating are some of the best means of continuing to create useful, life-saving products for people, align companies with great environmental and social goals, as well as personalize experiences in ways that will transform patients into even more confident consumers and expand opportunities to positively impact larger and emerging markets.
Fausto Artico has two PhDs. As a Physicist, Mathematician, Engineer, Computer Scientist, and High-Performance Computing (HPC) and Data Science expert, Fausto has worked on key projects at European and American government institutions and with key individuals, like Nobel Prize winner Michael J. Prather. After his time at NVIDIA corporation in Silicon Valley, Fausto worked at the IBM T J Watson Center in New York on Exascale Supercomputing Systems for the US government (e.g., Livermore and Oak Ridge Labs).
Kevin Harrigan graduated with a BSAE in aerospace engineering from Pennsylvania State University. During his collegiate career he gained experience in a co-opposition with Capital One Financial as a Data Analyst at in Richmond, Virginia. It was this experience that afforded him the opportunity to find passion in data munging, applied statistics, and programming. Following graduation, he accepted a full time offer in their newly formed Digital Enterprise Organization, expanding his technical and analytical knowledge in areas such as distributed computing, clickstream analytics, multivariate testing, anomaly detection, and propensity modeling

55 www.pharmafocusasia.com
AUTHOR BIO
INFORMATION TECHNOLOGY
Demystifying the Potential Applications of Blockchain Technology in Pharma and Biopharma Industry A bird’s eye view
After every few days, we all come across the headlines about rising or falling values of ‘Bitcoin’ followed by torrents of predictions about the global economy and future of financial market. Though his true identity is unknown, Satoshi Nakamoto – a mysterious inventor of Bitcoin, is a renowned figure in the world of cryptocurrency Bitcoin or cryptocurrency is just the tip of an iceberg. This technology was essentially developed to solve the huge problem associated with the centralisation of power which may become corrupt intentionally or unintentionally. Thus, blockchain technology, also known as distributed ledger technology (DLT), is a decentralised way of maintaining a track/ledger of every transaction associated with the system, which makes it practically impossible to falsify any information. As explained in simple words by Quazi Mamun, a blockchain is a time-stamped series of permanent data records managed by a collection of computers that are not owned by any entity.
Amita Puranik1, Prajakta Dandekar2 and Ratnesh Jain1
1 Department of Biological sciences and Biotechnology, Institute of Chemical Technology.
2 Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology.
This groundbreaking technology has enormous applications in almost every walk of life and it is not surprising that the healthcare industry, being a high-asset and knowledgeintensive industry, is amongst the top few sectors to experience its great potential and benefits. The ability to treat previously untreatable diseases, with remarkable efficacy and safety, marks biopharmaceuticals
as one of the most sophisticated achievements of modern science and technology. Biopharmaceuticals account for the rapidly growing segment of prescription drugs and are forecasted to reach the global market share of approximately US $400 billion by 2025. With the emergence of 3D bio-printing, stem cell technologies, CART cell therapy, etc., we are at the cusp to witness the next therapeutic
revolution that is striding from single cell component (current protein, mRNA based therapeutics) to a whole cell-based therapies. Apart from these scientific vertical of innovations, the healthcare industry is intensifying horizontally, especially due to integration of technological advancements such as machine learning, artificial intelligence, cloud computing, internet of things (IOT), quantum computing,
56 PHARMA FOCUS ASIA ISSUE 50 - 2023
INFORMATION TECHNOLOGY
virtual and augmented reality and blockchain technology.
Decentralised distribution of information can reshape the pharma and biopharma industry by offering streamlined processes, reduced operational costs, removal of fraud and duplicity, providing trust, and enabling data integration in a sharing-based economy. Blockchain technology can be applied to numerous facets of drug discovery and development, such as for clinical trials, data management, ensuring drug authenticity, intellectual property, licensing and royalties, supply chain compliance, etc.
The unprecedented global pandemic due to COVID-19 exposed some of the fragilities in healthcare ecosystem, especially in clinical trials and supply chain management. Many world class hospitals and vaccine manufacturers struggled due to shortages of basic personal safety equipment (such as masks, face shields, gowns, etc.), and more advanced requirements like diagnostic kits and essential therapeutics. This shortage led to pitching and distribution of many counterfeit drugs, fraudulent testing kits and other biomedical supplies. This situation was an eye-opener for the healthcare ecosystem to understand the urgent need to expedite the adoption of advanced technologies such as blockchain, automation, digitisation, etc., to improve the robustness of supply chain management. With blockchain technology, organisations with competing interests can safely work together in a shared permanent ledger, without giving up the system control or revealing the critical information. The cryptographic principles and codes of blockchain technology will enable organisations to work together as teams to expose frauds, reduce frictions in the raw material supplies and ensure product authenticity at a speed never experienced before.
In order to evaluate blockchain as a solution to meet the requirements of the ‘Drug Supply Chain Security Act of 2023’, a pilot project was supported by the United States food and drug
administration (US-FDA) in 2019. This consortium focused on pharma supply chains and included several global pharma leaders such as Genentech, Amgen, Pfizer, etc. Underlying blockchain technology was provided by Chronicled. The first solution that this consortium seeks is to establish a product verification system to verify the authenticity of the returned drug product. In an article published in Harvard business review, Alison McCauley has highlighted that approximately 60 million units of saleable drugs are returned annually as per a healthcare distribution alliance. Blockchain can also ensure the legitimacy of surplus drugs procured by wholesalers before they are re-sold. Currently, without a strong method to verify drug authenticity, these drugs are either returned to the manufacturing companies (which destroy them, resulting in a waste of resources) or the wholesalers contact manufacturers to
track down serial numbers. The whole process of tracing and tracking down the serial number of drugs has many practical flaws and can take up to 48 h. Using blockchain and digitisation, wholesale distributors can make use of barcode scanners to perform such verifications within fraction of seconds and quickly put back the product into commercial distribution. Manufacturers can also maintain control of their data simultaneously with such transparent and traceable transactions. This rapid system of product verification can be very helpful for hospitals and pharmacies as well. Counterfeiters could still copy barcodes in an attempt to pass drugs off as legitimate — but the ledger will flag and permanently record suspicious activity.
In clinical trials, blockchain can help handle the trial’s subject identity better, including electronic health records, without losing privacy or security. Blockchain
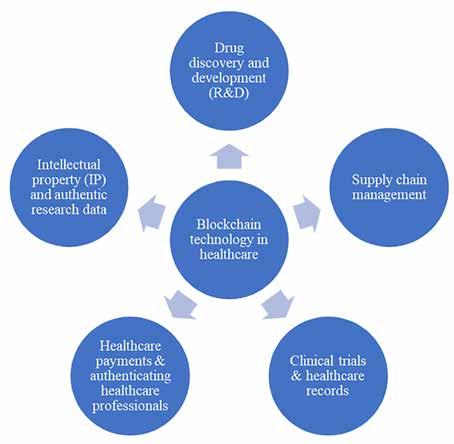
57 www.pharmafocusasia.com
INFORMATION TECHNOLOGY
Figure 1: Applications of block-chain technology in healthcare eco-system
technologies could translate into a set of solutions that may improve patient participation and relevant processes, leading to better health outcomes. All clinical trial participants: patients, clinical investigators, coordinators, and primary physicians would be touching different parts of the same ledger. Moreover, public registries could be created, which will provide an access to the results of clinical trials to all the stakeholders, and in turn could further re-shape drug R&D. This healthcare ecosystem of the future will thus have an access to the same, synchronous version of the truth.
A similar synchronous version of truth is essential for all research laboratories and commercial biopharmaceutical manufacturers. The journey of a prospective new drug begins from research laboratories, which generate raw (output files from the instruments) and sophisticated (human readable and interpretable) data, that serves as a proof of experimental findings. Cautious review of literature and citation of relevant data is very essential during drug discovery because loss of critical data may lead to huge loss of investments into new drugs. Thus, right from the clone section or sample
collection, every small or big process and analysis could be recorded by researchers with blockchain-enabled electronic laboratory notebooks (ELN). These records could be encrypted to avoid unauthorised access and obviate any conflicts about the date of creation of the material and about its creator. This traceability offered by blockchain can ensure authenticity of published data and help to protect the integrity of scientific information. This could increase the operational confidence and excellence, with additional benefits of saving a significant amount of time and resources around inoperable and/or deceitful reports/literature. Integrating other technologies, such as artificial intelligence, machine learning, internet of things, etc. will offer rapid data mining and analysis of existing literature. Thus, the future research and development (R&D) ecosystem will have advantages of intellectual property protection, with opportunities for most effective utilisation and revenue generation from new research findings.
Authenticating the experience of medical professionals can be a timeconsuming and wasteful endeavor by phone calls and snail mails.
Cryptographic protocols can help in tracking the experience of medical professionals, which will not only help to streamline the hiring process for healthcare organisations but also help patients to take decisions on where and from whom to receive treatment. Due to poor communication network and technology, healthcare payments by individuals or insurance companies are the major pain-points of healthcare ecosystem. Pre-authorisation process required by certain insurance companies and enquiries into a certain expense being covered by a member’s insurance policy can be extremely slow due to involvement multiple stakeholders and manual processes. Use of blockchain for pre-authorisation can offer various benefits such as improving cash flow due to faster claim settlements, appropriate payment to the healthcare providers, reduced administrative costs, timely patient treatments, etc.
Beyond these peripheral applications, blockchain technology can also be applied to understand and/or address fundamental biological issues. Alfred Chin in frontiers in blockchain journal has outlined the application of blockchain principles

58 PHARMA FOCUS ASIA ISSUE 50 - 2023
INFORMATION TECHNOLOGY
Biopharma and pharma giants will need to avoid operational silos and develop clear business implementation plans for new technologies in order to experience the benefits of large scale implementation of blockchain technology.
to study and model biological mechanism. He suggests that retrospectively reconstructing cell lineage information is valuable for understanding human diseases as experimental manipulation is not possible. Naturally occurring mutations, such as single nucleotide variants, LINE-1 transposition, copy number variation, microsatellite mutation, mitochondrial DNA mutation etc., can moonlight as endogenous lineage barcodes. These barcodes could serve as a starting point for reconstructing the cancer history blockchain. Incorporating current genetic methods that probe biological memory with a blockchain model may expand the extensiveness and efficacy of retrospective lineage tracing. ‘Smart contracts’ are the protocols that get automatically implemented upon accomplishment of certain conditions and enjoy all the prime features of blockchain technology. These smart contracts as well as Boolean logic gates are both based on the principle of conditionality. Trust in blockchain technology may develop confidence that the biological Boolean logic gates function appropriately, which may be collectively employed to create a ledger of state of a particular cell. This valuable fundamental understanding may further facilitate engineering of dynamic and multiplexed biochemical circuits.
Though this technology has numerous benefits to offer, there are significant research gaps and lack of attention on actual technological aspects since 2008. Lack of thorough knowledge and a legal framework to support smart contracts makes it difficult for companies to fully embrace and implement blockchain technology in business. In a global blockchain survey conducted by Deloitte in early 2019, 29% of the respondents identified lack of understanding of the underlying technology as a key obstacle in adopting blockchain for real-time applications. In fact majority of the research continued surrounding the Bitcoin rather than understanding the infrastructure that Bitcoin uses. The major factors behind reluctance for adopting blockchain in
healthcare ecosystem (including pharma and biopharma manufacturers) may include –i) lack of experts and extremely high cost to hire the available experts who can assist in implementation of this technology ii) building softwares capable of handling such great complexities may cost at least US $800,000 or more for the developers iii) linear flow of material and data across manufacturing and supply chain (i.e. data flows linearly one partner up and one partner down) iv) Powerful incumbents and off-chain transactions v) ideological push backs, privacy concerns and lack of faith such as thefear of losing proprietary data and trade secrets.
Biopharma and pharma giants will need to avoid operational silos and

develop clear business implementation plans for new technologies in order to experience the benefits of large scale implementation of blockchain technology. Associations among healthcare and life-science sector competitors and regulatory bodies (FDA) would be needed to foster mass adoption. To summarise in simple words, with an increasing interest of industry players and rapid growth in blockchain technology, it will be interesting to watch how the technology contributes to discovery and development process for biologics and pharmaceuticals in the upcoming years.
References are available at www.pharmafocusasia.com
Amita Puranik is PhD research scholar at Nano-medicine research group, Institute of chemical technology, Mumbai. She has received prestigious Prime-minister’s fellowship for the doctoral research from government of India, cosponsored by Lupin Ltd., a renowned pharmaceuticals and biopharmaceuticals manufacturing company. Her PhD research work is focused on development of LC-MS based multi-attribute-method (MAM) for characterisation of biopharmaceuticals. She has two first author research papers and two scientific review papers published in international peer reviewed journals.


Prajakta Dandekar Jain is UGC Assistant Professor at DPST, ICT, Mumbai. She completed her Ph.D. in Bioprocess Technology from the same department in 2010,following which she was the first women researcher to receive the RESPIRE long-term fellowship from the European Respiratory Society (ERS)-MarieCurie Co-fund, to conduct research in siRNA delivery systems for all eviating lung infections. She conducted this research at the Helmholtz Institute of Pharmaceutical Sciences, Saarland, Germany for two years. Currently, her research work is focused in the areas of nanofibers and tissue engineering, upstream bioprocessing, 3Dcell culture technology and green technology. She has guided 5 Ph.D. students and over 25 masters’ students.
Ratnesh Jain is Associate Professor in Department of Biological sciences and Biotechnology at Institute of Chemical Technology, Matunga, Mumbai. Dr. Jain is heading “National Facility for Analytical Characterization of Biopharmaceuticals” being setup by Department of Pharmaceuticals, Govt. of India in addition to the existing Biologics Characterization Lab at ICT. From 2012, Dr. Jain has taken various initiative towards biosimilar characterisation and educational initiative e.g. Biosimilar-Workshop to support biopharmaceutical sector related to skill development, infrastructure development and entrepreneurship. He has more than 55 publications and 150 presentations.
59 www.pharmafocusasia.com
AUTHOR BIO
INFORMATION TECHNOLOGY

60 PHARMA FOCUS ASIA ISSUE 50 - 2023 PRODUCTS & SERVICES STRATEGY AMI Polymer 03 Valsteam ADCA Engineering ............................................................. OBC RESEARCH & DEVELO PMENT Quantys Clinical Pvt. Ltd ...................................................................... 27 Sciencix 21 SGS 10-12 CLINICAL TRIALS Quantys Clinical Pvt. Ltd ...................................................................... 27 Sciencix ............................................................................................... 21 SGS 10-12 MANUFACTURING AMI Polymer 03 Valsteam ADCA Engineering OBC Sciencix ............................................................................................... 21 Company Page No. SUPPLIERS GUIDE AMI Polymer 03 www.amipolymer.com Quantys Clinical Pvt. Ltd ...................................................................... 27 www.quantysclinical.com Sciencix ............................................................................................... 21 www.Sciencix.com SGS 10-12 www.sgs.com Valsteam ADCA Engineering ............................................................. OBC www.valsteam.com Company Page No. To receive more information on products & services advertised in this issue, please fill up the "Info Request Form" provided with the magazine and fax it. 1.IFC: Inside Front Cover 2.IBC: Inside Back Cover 3.OBC: Outside Back Cover
Introducing a group of highly focussed magazines for the American and Asian markets.



Aspiring to be leading journals in the B2B landscape of Pharmaceutical-Industry, the magazines covers Medical Sciences, Business & Technology and all the latest innovations.
Our magazines bring a fresh outlook towards insightful and pragmatic Pharmaceutical-Industry reporting. Delightfully selected topics presented by the gurus of the industry comes packed with latest happenings, sharp analysis & deep insights. We strive to keep you engaged, knowledgeable & wanting for more.
61 PHARMA FOCUS ASIA ISSUE 50 - 2023 Introducing Advent of NEW-AGE PHARMACEUTICAL REPORTING From the house of Ochre Media: Automotive-technology.com | Defence-industries.com | Hospitals-management.com | Packaging-labelling.com Pharmaceutical-tech.com | Plantautomation-technology.com | Plastics-technology.com | Pulpandpaper-technology.com Sportsvenue-technology.com | Steel-technology.com | Asianhhm.com | Pharmafocusasia.com
Scan to check websites Scan to check websites www.ochre-media.com
















Pressure regulators Control valves Pipeline ancillaries Special equipment Steam traps Safety valves adca@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467 PRODUCTS MANUFACTURED IN PORTUGAL Guia PBL PORTUGAL HIGH PURITY. HIGH DEMANDS. H igh purity equipment for clean steam AV 001 AP E 01.20
