Introducing a group of highly focussed magazines for the American and Asian markets.
Aspiring to be leading journals in the B2B landscape of Pharmaceutical-Industry, the magazines covers Medical Sciences, Business & Technology and all the latest innovations.
Our magazines bring a fresh outlook towards insightful and pragmatic Pharmaceutical-Industry reporting. Delightfully selected topics presented by the gurus of the industry comes packed with latest happenings, sharp analysis & deep insights. We strive to keep you engaged, knowledgeable & wanting for more.
The Journey of Drug Discovery Past lessons, future innovations
The field of drug discovery is a testament to human ingenuity and perseverance. Over the years, extraordinary breakthroughs have revolutionised healthcare, saved innumerable lives, and expanded the horizons of medical science. However, the path from identifying a therapeutic target to bringing a safe and effective drug to market has always been riddled with challenges— scientific, regulatory, and financial.
Reflecting on the history of drug discovery, we see a narrative of relentless progress, defined by milestones that have transformed our approach to treating disease. From the accidental discovery of penicillin to the development of high-throughput screening and the emergence of biotechnology, each era has introduced new tools and methodologies that have driven the industry forward. We've witnessed the rise of personalised medicine, the growing influence of big data and artificial intelligence, and a shift towards more targeted therapies, offering the promise of greater efficacy with fewer side effects.
But as we reflect on past achievements, we must also acknowledge the challenges that lie ahead. The demands on the pharmaceutical industry have never been higher, with a pressing need for faster, more cost-effective drug development, and an intensified focus on addressing unmet medical needs. The landscape is growing increasingly complex, marked by regulatory challenges, pricing pressures, and a heightened emphasis on sustainability and ethical considerations.
Looking ahead, the future of drug discovery will depend on our capacity to innovate and adapt. Breakthroughs in
genomics, proteomics, and systems biology are unlocking new possibilities, while digital technologies are revolutionising every stage of the process, from early research to clinical trials. Collaboration will be crucial—between academia and industry, across various disciplines, and spanning international borders.
In this issue, Mahesh Narayan FRSC Biophysicist Chief, Biochemistry Division, Department of Chemistry and Biochemistry, University of Texas elaborates on the cusp of a revolution in drug-discovery, driven by the AI/ ML catalysed the marriage between the prediction of biological structures (proteins, DNA, RNA and heteromorphs thereof) across the biome and the techniques for accelerating the discovery, synthesis and development of small-molecule agonists and antagonists against them.
As we navigate this rapidly changing landscape, one thing remains clear: the quest to discover new therapies is as vital as ever. The challenges are significant, but so too are the possibilities. Together, we have the opportunity to shape a future where innovative medicines continue to improve and save lives.
Prasanthi Sadhu Editor
RESEARCH & DEVELOPMENT
06 The Significance of microRNAs in Cancer
A brief overview
Ancuta Jurj, Department of Translational Molecular Pathology, The University of Texas MD Anderson Cancer Center
George A Calin, Center for RNA Interference and Non-Coding RNAs, The University of Texas MD Anderson Cancer Center
12 Advanced Drug Delivery Strategies for Inflammatory Bowel Disease
Julia Mantaj, Lecturer, Biomedical Science, Anglia Ruskin University
22 Drug Discovery Today
Looking back, looking forward
Mahesh Narayan, Biophysicist, The University of Texas
CLINICAL TRIALS
24 Nanocarriers in Breast Cancer Therapy
Myth or miracle?
Nalla Usha Kumari, Neelesh Kumar Mehra Department of Pharmaceutics, National Institute of Pharmaceutical Education and Research
27 Biomarkers in Drug Discovery and Clinical Trials
Kirti Singh, Postdoctoral Scientist, Eli Lilly and Company
MANUFACTURING
30 CDMO Considerations for CGT/ATMP Companies
Christine Feaster, SVP, Strategic Operations, Quality Executive Partners, Inc. (QxP®)
Copies of Pharma Focus Asia can be purchased at the indicated cover prices. For bulk order reprints minimum order required is 500 copies, POA.
Our high-quality, animal-free, and reliable Recombinant Insulin is manufactured by the world’s largest insulin manufacturer, Novo Nordisk. It is specifically manufactured for use in cell culture media, supported by a dedicated team of insulin experts, and securely supplied around the globe.
The Significance of microRNAs in Cancer
A brief overview
In recent decades, microRNAs (miRNAs) have become pivotal in cancer therapeutic strategies, serving as compelling tools and biomarkers. With dual roles as oncogenes or tumour-suppressors, miRNAs play key functions in cancer biology. Several miRNA-targeted therapeutics gaining attention have advanced to clinical development. This surge is reflected in increased miRNA integration across oncology trials, screening, diagnostics, and drug testing. Consequently, this mini-review aims to provide a succinct overview of the evolving landscape of miRNA applications in oncology research, shedding light on their potential as a therapeutic modality.
Ancuta Jurj
Department of Translational Molecular Pathology, The University of Texas MD Anderson Cancer Center
George A Calin
Center for RNA Interference and Non-Coding RNAs, The University of Texas MD Anderson Cancer Center
Decades ago, microRNAs (miRNAs) were first identified in Caenorhabditis elegans (C. elegans) by Ambros and Ruvkun’s groups in 1993 and subsequently linked to cancer by Calin et al in 2002. This discovery sparked a surge in miRNArelated research, highlighting their pivotal
role in tumourigenesis and leading to diverse applications, from biomarker development to potential RNA therapeutics. Structurally, miRNAs are small noncoding RNAs (ncRNAs), composed of single-stranded RNA molecules typically 19-24 nucleotides (nt) in length. Their biogenesis initiates in the nucleus, where primary miRNAs (pri-miRNAs) are transcribed from RNA transcripts and cleaved by RNA polymerase into precursor miRNAs (pre-miRNAs), which mature into single-stranded miRNAs approximately 22 nt long. Each step of miRNA biogenesis relies on essential
components such as RNA polymerase II (Pol II) for transcription, RNase III enzymes (DROSHA and DICER1), and members of the Argonaut family (AGO2).
miRNAs play crucial roles in modulating various biological processes, exhibiting distinct spatial and temporal expression patterns. In addition to their conventional function in posttranscriptional repression of specific target proteins by promoting mRNA decay or inhibiting translation, miRNAs have been observed to influence transcriptional activation, epigenetic regulation, and upregulation of translation. In this mini-review, we aim to provide a concise overview of the role of miRNAs in cancer biology.
miRNA and cancer – a glimpse behind these two pioneers
As is widely recognized, cancer constitutes a genetic ailment affecting a growing number of individuals worldwide. It is characterised by a cascade of mutations occurring in tumour suppressor genes, facilitating uncontrolled cell proliferation, and impeding cell death. Recent findings have unveiled the role of miRNA dysregulation in bolstering cancer progression. Intriguingly, a single miRNA has the capacity to bind to numerous targets, potentially exceeding several hundreds, encompassing diverse functionalities such as transcription factors, receptors, and vectors. Ample evidence underscores the marked differences in miRNA expression between normal and tumour tissues, strongly implicating their involvement in tumour progression, development, invasion, and metastasis.
Do miRNAs play a dual role?
As mentioned previously, miRNAs are implicated in modulating all cancer hallmarks, functioning either as oncogenes or tumour suppressors. However, certain miRNAs exhibit context-dependent roles, acting as oncomiRs in some malignancies while serving as tumour suppressors in others. For instance, in
certain cancers, members of the let-7 family function as tumour-suppressor miRNAs, impeding invasion, metastasis, epithelial-to-mesenchymal transition (EMT), and self-renewal. Conversely, when their expression profile is elevated in the tumour microenvironment, let-7 family miRNAs can adopt oncogenic roles. Another example is miR-146a-5p, which demonstrates a dual role as both an oncomiR and a tumour suppressor in several cancers. Similarly, miR-186 exhibits a dual function, serving as both a diagnostic and therapeutic target, and playing pivotal roles in oncogenesis, cell migration, invasion, cell death, metastasis, and drug resistance. In gastrointestinal cancers, miR-9 has been identified to play a dual role as well. These observations underscore the heterogeneity of cancer and the dual role of miRNAs in malignant pathophysiology. However, further studies are imperative to elucidate the specific roles of miRNAs and the underlying mechanisms governing their regulation.
microRNAs and cancer as a full body disease
There are studies supporting the hypothesis that miRNAs can directly modulate genes encoding hormones or enzymes responsible for hormone maturation and metabolism. Furthermore, miRNAs indirectly modulate hormone-mediated cell signaling transmission by targeting hormone antagonists or receptors.
Recent evidence suggests an important network of interactions between miRNAs and ER (estrogen receptor), coordinating cellular responses to estrogen. Certain miRNAs modulated by ER , such as (pri-) miR-17-92 and miR-206a-363, have been identified as targets that downregulate ER expression at the protein translational level. Another study highlights the significance of miR-149-5p, which inhibits the regulatory activity of the transcription factor SP1 under 17 -estradiol therapy, while its decreased profile promotes the expression of hnRNPA1, regulating the load-
ing of let-7 miRs into EVs. Regarding androgen receptors (AR), miR-let-7c indirectly suppresses receptor activity by targeting c-Myc. Direct associations between miR-185 and miR-205 with AR expression have been described. In prostate cancer, AR regulates the transcription of miR-21, enhancing tumour cell growth, while a confirmed connection exists between miR-185-5p and AR in clear cell renal cell carcinoma. miR-21 and miR-206 show increased expression profiles in hormone-dependent cancers. miRNAs also play a crucial role in regulating stress responses. Hyperthermia, among the stress-induced factors, suppresses the expression of miR-23a, leading to elevated levels of the proapoptotic protein NOXA and subsequent apoptosis. Furthermore, a downregulated profile of miR-23a is associated with CDK5 inhibition, resulting in increased expression of HSP70 in stressed cells. In response to environmental stress, the tumour suppressor p53 modulates the expression of miRNAs at both transcriptional and processing levels. Consequently, an increased number of cancers harbouring p53 mutations exhibit specific alterations in miRNA processing and transcriptional activity. Loss of p53 function facilitates tumour progression by impeding stress response mechanisms. Interestingly, despite their increased expression, some of these miRs have been identified as oncomirs but paradoxically act as tumour suppressors, thereby inhibiting cancer progression.
Extracellular microRNAs as biomarkers for therapeutic clinical trials
Extracellular vesicles (EVs) are small bilipid entities secreted by various cell types and can be detected in body fluids such as blood, milk, urine, cerebrospinal fluid, and saliva, typically ranging in size from 50 to 200 nm. While EVs serve as a general term, they can be categorised into exosomes, microvesicles, and apoptotic bodies based on their distinct biogenesis pathways. Exosomes originate
from inward budding of multivesicular bodies (MVBs), microvesicles originate from the outward budding of the plasma membrane, while apoptotic bodies result from cell membrane fragmentation during cell death. Recent attention has been focused on these vesicles, particularly exosomes, revealing that critical bioactive molecules such as nucleic acids (DNA, mRNA, miRNA), specific proteins, and lipids are packaged into EVs during biogenesis. These molecules are subsequently transported from donor cells to target cells, underscoring the importance of EVs as natural transporters facilitating cell-to-cell communication. In this context, EVs possess the potential to influence the phenotype of target cells, whether normal or tumour, and contribute to processes associated with cancer hallmarks. Numerous diseases are linked to dysregulated expression of miRs, and importantly, the presence of miRs in bodily fluids is associated with disease progression. Exosomal miRs, in particular, hold promise as biomarkers for various pathological conditions, offering insights into disease pathogenesis and potential mechanisms of repair. However, a critical question arises from the limited understanding of the miRNA content packaged into EVs, highlighting the need for further investigations.
Exosomal miRNAs show potential as biomarkers for early detection and prognosis across a spectrum of cancers. In non-small cell lung cancers, the miRNAs let-7b-5p, let-7e-5p, miR-23a-3p, and miR-486-5p have been identified for early diagnosis, while exosomal miR451a shows promise as a prognostic marker for patients across stages I to III. Additionally, exosomal miR-4257 and miR-21 serve as recurrence-specific biomarkers in non-small cell lung cancers. In glioblastoma, the presence of exosomal miR-320 and miR-574-3p distinguishes patients from healthy individuals, with exosomal miR-301a correlating with pathological grades and recurrence. In prostate cancer, exosomal miR-182 and miR-183 are mark -
edly expressed, while miR-1290 and miR-375 show promise for prognosis. In breast cancer, miR-223-3p is associated with detection of biomarkers and is linked to histological types, nuclear grade, pathological stages, and lymphatic invasion. Moreover, exosomal miR-373 holds potential as a biomarker in triplenegative breast cancer.
miRNAs have emerged as promising biomarkers for prognostic and diagnostic purposes across various types of cancers. Achieving a comprehensive understanding of miRNA-based diagnostic approaches entails delineating tumour
subtypes with similar phenotypes and uncovering new malignant sub-entities. To enhance the specificity of diagnostic tools, it is imperative to elucidate the mechanisms governing miRNA circulation in bodily fluids, including their associations with proteins, lipids, and exosomes. Moreover, exploring chemical modifications within miRNA structures holds potential for identifying novel molecules serving as biomarkers. IsomiRs, which represent miRNA variants resulting from post-maturation editing events such as modifications at the 3’-end facilitated by nucleotide
Table 1. Clinical trials investigating the involvement of miRNAs in cancer
NCT04305366 MicroRNA Markers in Head and Neck Cancers Squamous Cell Carcinoma of Head and Neck
NCT02243592 Molecular Profiling in Tissue Samples from Patients with Cancer Who Are Exceptional Responders to Treatment
not recruiting
not recruiting
NCT02366494 Micro RNAs to Predict Response to Androgen Deprivation Therapy Prostate cancer Active, not recruiting
NCT03953443 Expression & Epigenetic Silencing of MicroRNA for Predicting Therapeutic Response and Prognosis of HPV-negative HNSCC
NCT04100811 Identification of Clinically Insignificant or Significant Prostate Cancer With the miR Scientific Sentinel™ Platform
NCT00900224 Studying Tissue and Blood Samples from Patients with Acute Myeloid Leukaemia
Head and Neck Squamous Cell Carcinoma Active, not recruiting
Prostate cancer Active, not recruiting
Leukaemia Active, not recruiting
NCT01231386 MIRNA Profiling of Breast Cancer in Patients Undergoing Neoadjuvant or Adjuvant Treatment for Locally Advanced & Inflammatory Breast Cancer Breast Cancer Completed
NCT00806650 Anti-IMP3 Autoantibody and MicroRNA Signature Blood Tests in Finding Metastasis in Patients with Localised or Metastatic Kidney Cancer
Kidney Cancer Completed
NCT01595139 MicroRNAs in Patients with Neurofibromatosis Type 1 Glioma Completed
NCT04720430 Blood Sample Collection for Experimental Blood Test to Track Liver Cancer
Hepatocellular Carcinoma Completed
NCT03452514 Addition of microRNA Blood Test to Lung Cancer Screening Low Dose CT Lung Cancer Completed
NCT00909350 Micro-RNA (miR) Expression in Upper Gastrointestinal Mucosal Tissue
NCT02412579 Genetic Profiling of Liver Cancer in Patients Undergoing Liver Transplantation
Esophageal Adenocarcinoma Completed
Hepatocellular Carcinoma Completed
transferase and 3’-exonuclease processes, offer promising avenues for enhancing tumour type specificity. By leveraging the specificity of isomiRs, more refined distinctions between tumour types can be achieved, thereby advancing miRNA biomarker development. Nonetheless, further investigations are warranted to refine and optimise cancer diagnostics.
miRNAs and small molecule inhibitors therapeutics
miRNA therapy holds promise as an adjunct to standard therapies such as chemotherapy, radiotherapy, or immunotherapy. However, its success hinges on effective delivery systems capable of transferring miRNAs into target cells. While existing delivery systems possess certain limitations, a promising strategy involves targeting miRNAs that are selectively expressed in malignant cells while remaining absent in normal cells. This approach aims to mitigate toxicity, enhance therapy specificity, and simplify the quantification of specific miRNAs, thereby optimising therapeutic outcomes.
Small molecules have garnered significant attention in the medical field, emerging as pivotal elements in drug development. Particularly noteworthy is the increasing interest in discovering small molecule inhibitors targeting noncoding RNA (ncRNA), driven by the presence of modifications in RNA molecules that render them attractive drug targets. In this regard, small molecules can be tailored to selectively target miRNAs both before and after their maturation process. A recent study offers valuable insights into small molecule inhibitors, including the identification of structures capable of binding to the tertiary folded structures of miRNAs. Understanding this concept sheds light on how stem-loop hairpin secondary structures can fold into tertiary configurations, thereby forming bulges or pockets crucial for small molecule interactions. These findings underscore the potential for developing targeted therapies that leverage small molecules to modulate miRNA function effectively.
According to ClinicalTrials.gov, there are currently 417 ongoing clinical trials investigating the use of miRNAs in cancer. Among these trials, 16 focus on assessing miRNAs' prognostic significance, while 148 are specifically exploring their diagnostic potential in cancer. Notably, there are no ongoing clinical trials that directly combine miRNA research with cancer treatment using small molecule inhibitors. However, it's worth noting that the field of miRNA-based cancer therapy is continually evolving, and future trials may explore this approach.
Table 1 provides a snapshot of selected ongoing clinical trials in this area. Further research and clinical investigation are needed to fully understand the potential of miRNAs and small molecule inhibitors in cancer treatment.
Conclusions
miRNAs serve as pivotal regulators in various aspects of cancer biology. Further research is essential to fully understand and decipher the intricate processes governed by miRNAs, including their biogenesis, modulation of cancer hallmarks, and diverse functions. These complex processes offer valuable insights into strategies for combating cancer. To date, mounting evidence highlights the potential of miRNAs as biomarkers for prognosis and diagnosis across different pathologies. Moreover, the design of molecules specifically targeting miRNAs shows promise in counteracting various cancer mechanisms and ultimately improving the survival rates of patients diagnosed with various forms of malignancies.
Acknowledgements
G.A.C. is the Felix L. Haas Endowed Professor in Basic Science. Work in G.A.C.’s laboratory is supported by NCI grants 1R01 CA182905-01 and 1R01CA222007-01A1, NIGMS grant 1R01GM122775-01, DoD Idea Award W81XWH-21-1-0030, a Team DOD grant in Gastric Cancer W81XWH-211-0715, a Chronic Lymphocytic Leukemia Moonshot Flagship project, a CLL Global
Research Foundation 2019 grant, a CLL Global Research Foundation 2020 grant, a CLL Global Research Foundation 2022 grant, The G. Harold & Leila Y. Mathers Foundation, two grants from Torrey Coast Foundation, an Institutional Research Grant and Development Grant associated with the Brain SPORE 2P50CA127001.
References are available at www.pharmafocusasia.com
Adrian
research delves into the roles of microRNAs and other non-coding RNAs in cancer initiation, progression, immune disorders, and cancer predisposition mechanisms. Additionally, he investigates body fluid microRNAs as potential biomarkers and explores RNA therapeutic options for cancer patients.
Ancuta Jurj, Ph.D., is a postdoctoral fellow at M. D. Anderson Cancer Center, Houston. Her research focuses on identifying microRNAs and other noncoding RNAs as potential biomarkers associated with solid tumours. Additionally, she investigates the role of the tumour microenvironment in modulating cancer progression and therapy response.
George
Calin, M.D., Ph.D., is a Professor at M. D. Anderson Cancer Center, Houston. His
Elevating Healthcare Innovation MEDICAL FAIR ASIA
As the healthcare and medical sectors continue to advance at a rapid pace, MEDICAL FAIR ASIA 2024, along with its co-located exhibition MEDICAL MANUFACTURING ASIA, is not only a hub for industry-leading products and technologies but also a pivotal platform for thought leadership through its comprehensive conference programming. From 11 to 13 September 2024 at Marina Bay Sands, Singapore, the co-located events will bring together over 1,000 exhibitors from 62 countries and an array of highimpact conferences designed to address the most pressing issues and innovations in healthcare today.
Conference programming that complements the exhibition experience
The dynamic line-up of co-located conferences at MEDICAL FAIR ASIA 2024 underscores the event’s role as more than a trade exhibition. The carefully curated sessions are integral to the event’s mission, offering deep insights, expert knowledge, and strategic discussions that complement the sourcing platform of medical technologies and solutions. These conferences are poised to drive forward industry dialogue and innovation, making them an essential component for attendees who seek not only to explore the latest products but also to gain a competitive edge through knowledge.
Highlights of the conference programmes include:
WT
|
Wearable
– 11 September:
Technologies
Conference
As part of Asia's leading WT conference series, this event will focus on the latest developments in wearable technology, particularly in health and sports. Co-organised with WT | Wearable Technologies AG, the conference will offer a platform for industry leaders to share insights and discuss trends that are shaping the future of wearable tech. The full-day event is an unparalleled opportunity for networking and exploring new market opportunities.
MEDICINE + SPORTS CONFERENCE
– 12 September:
A flagship event within MEDICAL FAIR ASIA’s conference lineup, this full-day conference will gather an interdisciplinary array of experts in sports medicine, sports science, and healthcare. The discussions will focus on critical areas such as enhancing athlete performance, injury prevention, and the integration of digital innovations in sports healthcare. Recognised for its thought leadership, the conference has been a key training event for professionals over the past decade.
GITEX DIGI HEALTH 5.0 SINGAPORE Conference
– 11-12 September:
Bringing together digital health innovators and industry pioneers, this conference is dedicated to accelerating the technological transformation of healthcare across Asia. Over two-half day sessions, the conference will explore the latest advancements in digital health and offer strategic insights into how technology can enhance well-being in the region.
3rd Paradigm Shifts in Healthcare Symposium –
12 & 13 September:
This two-part symposium will address the evolving landscape of healthcare delivery, focusing on the well-being of patients, healthcare staff, and the environment. The sessions will cover vital topics such as sustainability in healthcare operations and innovative care models, making it an essential forum for professionals committed to advancing holistic healthcare solutions.
Start-Up Podium® Programme
– 11 to 13 September:
A central feature of the Start-Up Park is the Start-Up Podium®, which spans three days and includes a series of dynamic presentations, panel discussions, and fireside chats. The programme covers a wide array of topics crucial for start-ups, such as navigating healthcare regulations, digital health innovation, and the role of women in MedTech. This curated content is delivered by thought leaders and stakeholders from various sectors, including hospitals, academia, government agencies, and the pharmaceutical industry.
This programming not only supports the growth and visibility of emerging healthcare companies but also complements the broader offerings of MEDICAL FAIR ASIA by fostering collaboration and innovation within the industry. It positions the event as not just a showcase for products but as a critical forum for advancing the future of healthcare through start-up innovation.
Strategic Importance of Conference Programming
“The conference programming at MEDICAL FAIR ASIA is an essential complement to the exhibition, providing a platform where industry professionals can engage in meaningful dialogue, share knowledge, and explore innovations that will shape the future of healthcare,” said Gernot Ringling, Managing Director of Messe Düsseldorf Asia. “These conferences not only enhance the value of the exhibition but also position MEDICAL FAIR ASIA as a platform for thought leadership and industry collaboration.”
For more information on the exhibitions and to pre-register, please visit: www.medicalfair-asia.com and www.medmanufacturing-asia.com.
Advanced Drug Delivery Strategies for Inflammatory Bowel Disease
This article explores innovative drug delivery strategies for the treatment of inflammatory bowel disease (IBD). Novel approaches based on enteric-coated microneedle pills, nanoparticles, liposomes, prodrugs, hybrids, and biological systems will be discussed with regards to improving therapeutic outcomes and reducing side-effects in patients with IBD.
Julia Mantaj, Lecturer, Biomedical Science, Anglia Ruskin University
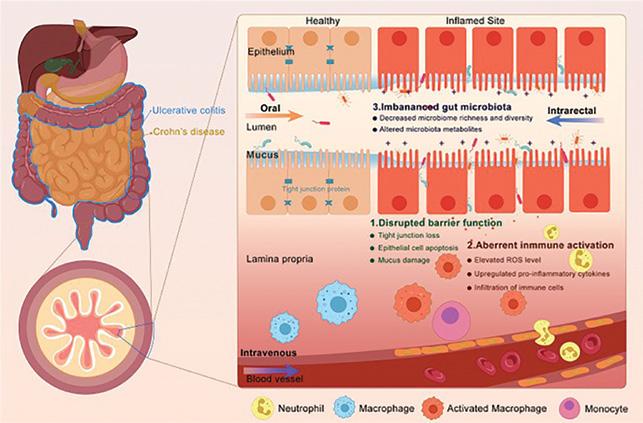
Crohn’s Disease (CD) and Ulcerative Colitis (UC) are the two main forms of IBD affecting more than 10 million people worldwide. These are chronic and progressive conditions, characterised by destructive inflammation of the intestinal tract. With time, the intestinal wall in IBD patients becomes ‘leaky’, and harmful bacteria from the gut microbiota can enter the blood circulation triggering an immune response and intensifying gut inflammation (Figure 1).
Symptoms of IBD include fever, anaemia, weight loss, diarrhoea, rectal bleeding, abdominal pain, and the urgency to evacuate the bowels. IBD is recognised to significantly impact the quality of life (QoL) of patients. Presently, IBD is not curable. Much effort has been made in developing therapeutic strategies for the treatment of IBD aiming to reduce symptoms, maintain clinical and
endoscopic remission, and prevent longterm disability. These therapies target the intensified inflammation process by decreasing the body’s immune response (i.e., immunomodulators azathioprine and methotrexate) or blocking the inflammation (i.e., biologics infliximab and ustekinumab). Despite the significant advancements in disease treatment, conventional drugs have major limitations such as severe adverse reactions (e.g., nausea, fever, higher risk of infections, diabetes) due to systemic absorption leading to therapy failure or ineffectiveness. Furthermore, a considerable proportion of IBD patients are unresponsive to the advanced therapies (biologics) or lose the response over the course of the disease. A study carried out by Gibble et al revealed that over 60 per cent of IBD patients responded inadequately to their first advanced therapies within 1 year after initiation. Consequently, over
time, surgical removal of the inflamed bowel part is necessary. Approximately 23-45 per cent of UC and up to 80 per cent of CD patients will require colon surgery at some point during their disease.
The major challenge in developing IBD therapeutic strategies is the delivery of therapeutics directly to the inflamed colon site. Traditional intestinal delivery systems are stimuli-responsive meaning that drugs are released following certain stimuli i.e., changes in pH, temperature, transit time, or presence of enzymes under healthy physiological conditions. However, these stimuli are different in IBD, and it is still unclear how they contribute to disease pathophysiology.
In recent years, a plethora of innovative strategies have been reported for the treatment of IBD aiming to achieve site-specific drug delivery to the inflamed tissue, thereby reducing adverse drug effects and improving the efficacy.
Enteric-coated microneedle pills
Initially developed for the transdermal delivery of small drug molecules and
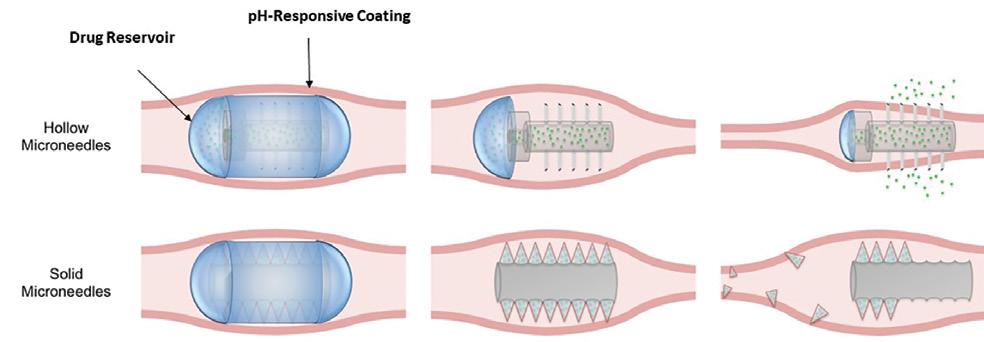
macromolecules, enteric-coated microneedle pills have gained attention in recent years as oral dosage forms. A research team at the Massachusetts Institute of Technology and Massachusetts General Hospital Harvard Medical School developed a capsule coated with drug-loaded microneedles capable of injecting drugs directly into the intestinal lining following ingestion (Figure 2).
Animal studies demonstrate that the dosage form delivered insulin more effi-
ciently compared to subcutaneous administration with no harmful side-effects observed. The microneedle capsules safely passed and were excreted from the gastrointestinal tract (GIT) making it suitable for use in the inflamed GIT in IBD. The incorporation of enteric coating onto microneedles enables the pills to protect incorporated drugs from the harsh acidic environment of the stomach and deliver them directly to the site where they are required. In a pre-clin -
ical study, Rani Therapeutics tested an enteric-coated microneedle pill loaded with the TNF- blocker adalimumab. The study results showed that the technology protects adalimumab from the acidic and enzymatic degradation of the GIT and directly delivers it to the site of inflammation where it slowly releases the drug. In summary, enteric-coated microneedle pills represent a promising drug delivery strategy, especially biological compounds for the therapy of IBD. However, further rigorous research is necessary to evaluate their clinical long-term effectiveness and safety in the management of IBD.
Nano-delivery system strategies
Nanoparticulate (NP) systems represent a promising approach to delivering drugs to inflamed colonic tissues, thereby offering the advantage of reducing the drug dose and systemic side-effects. Macrophages, neutrophils, and M cells that are present in inflamed intestinal regions can easily take up the NPs due to their small size (1-100 nm). Triggered by inflammatory cytokines (e.g., TNF- , IL-1 , IL-6), in the inflamed colon, the intestinal permeability is increased due to the loss of integrity of intestinal tight junctions, cellto-cell contacts, and immune cell infiltration that is also referred to as ‘leaky gut’. The loss of intestinal integrity enables the
Figure 2: Therapeutic use concept of the microneedle pill. Both hollow and solid microneedles could be used. The pill’s needles are initially coated by a pH-responsive coating to aid in ingestion (left). When the pill has reached the desired location in the GI tract, the coating dissolves, revealing the microneedles (middle). In the case of hollow microneedles (top right), the drug reservoir is compressed through peristalsis, releasing the drug through the needles. In the case of solid microneedles (bottom right), the drug is formulated into the microneedles. The microneedles penetrate the tissue and break off from the pill, leaving the needle to release the drug in a controlled manner, based on the needle formulation.
Figure 1: Affected locations of ulcerative colitis and Crohn’s disease (left) and a pathological microenvironment of the IBD (right).
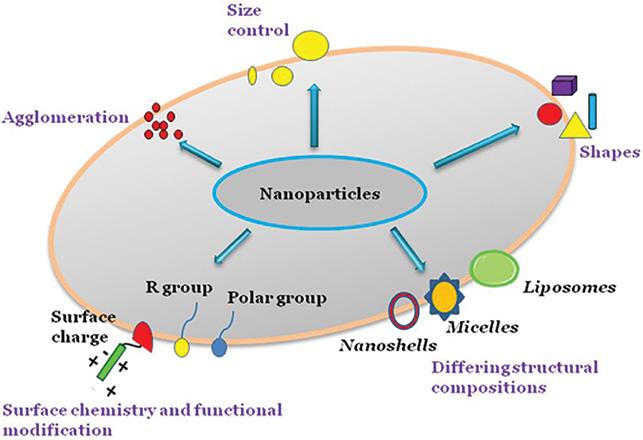
3: Physicochemical properties of nanoparticles. The surface chemistry of nanoparticles can be modified by adding reactive groups or molecules such as antibodies to surfaces in targeted drug delivery systems.
NPs to easily penetrate mucosal tissues and transport drugs to the inflamed colon sites. The NPs can be designed to display distinctive attributes and physicochemical properties (size, surface charge, ligands, targeting moieties) that can contribute to the extended retention of drugs at inflammation sites due to e.g., nanoparticle adhesion to mucus (Figure 3).
Over the past decade, considerable research efforts have been made to advance nano-delivery system strategies for the treatment of IBD. For example, a study by Ali et al revealed a significant accumulation of 3 nm poly(lactic-coglycolic acid) (PLGA) NPs on the colonic mucosal surface in mice compared to 250 nm NPs. To date, the data obtained is predominantly based on in vitro, ex vivo tissue binding studies, or in vivo studies following rectal administration using animal models. To further evaluate the NP behaviour in humans, further comprehensive studies are necessary for a definitive conclusion.
Prodrugs
Prodrug systems consist of a pharmacologically active agent that is protected in a temporarily inactive form that becomes
bioactive upon specific stimuli, in the case of IBD these are enzymes that are overexpressed in inflammatory tissues. Most studied prodrug systems employ 5-ASA derivates (mesalamine/5-aminosalicylic acid) that are metabolised to the active form by the N-acetyltransferase overexpressed in many IBD patients. Additionally, amino acids and carbohydrates (e.g., dextrans, cyclodextrins) have been studied as carrier systems for 5-ASA and dexamethasone. The drugs bind to the conjugate via glycosidic bonds that are cleaved by the colonic glucanase and esterase releasing the free active drug in the colon, thereby achieving target-specific drug delivery. Despite
representing one of the most practical approaches for targeted drug delivery treatment in IBD, the development of novel prodrug delivery is challenging. For instance, only a few studies are reporting the utilisation of prodrug delivery systems for the treatment of IBD highlighting the immaturity of the development of these systems. Additionally, extensive safety and stability in vivo evaluation of the systems is required.
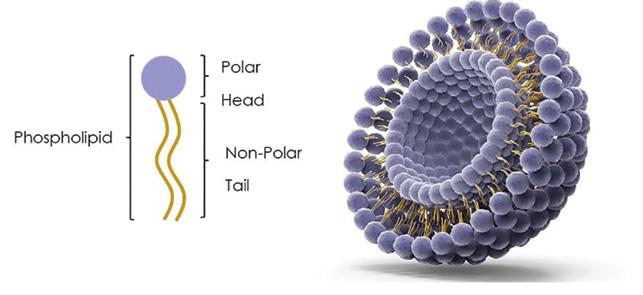
Liposomes
Liposomes are nanosized bubble structures consisting of one or more phospholipid layers designed to deliver hydrophilic and lipophilic drugs (Figure 4).
The hydrophilic drugs are encapsulated in the aqueous liposome core, whereas hydrophobic compounds can dissolve into the lipid membrane. A wide range of modified liposomal drug systems are being investigated in experimental colitis and the accumulation efficacy and the improvement of clinical IBD symptoms are being intensively evaluated. One option to specifically deliver drugs to the inflamed colon in IBD is the modification of the liposome surface with cationic lipids which results in liposome binding to mucosal tissues. For example, Myers et al developed a cationic liposome complex loaded with antisense TNFoligonucleotide that demonstrated a decrease in TNF- mRNA expression in in vitro studies. These observations were further supported by in vivo experiments where the administration of the liposome complex resulted in reduced Disease
Figure
Figure 4: Structure of liposomes.
Activity Index scores in a murine model of colitis. Furthermore, the prolonged circulation of liposomes in the body can be achieved via PEGylation (covalent linking of polyethylene glycol chains) that can be functionalised with various drugs for targeted drug tissue accumulation. However, cationic liposomes have shown toxicity when administered systemically due to their abnormal aggregation with proteins in the circulation. Hence, further research into liposome functionalisation to reduce systemic toxicity is required.
Hybrid delivery systems
Hybrid drug delivery systems combine the advantages of multiple carriers within a single structure. The drugs are encapsulated in a carrier, and these drug-loaded carriers are then embedded in an additional protective external compartment that degrades in inflamed tissues. This external compartment selectively dissolves or degrades in the inflamed intestinal region, releasing the embedded drugcarrier complex specifically at the target site. Xiao et al designed a hydrogel hybrid carrier system that releases CD98 antibodies incorporated in NPs. The systems demonstrated a reduction in disease severity in animal models. Furthermore, Li et al fabricated structures consisting of hyaluronic acid-modified porous silicon NPs loaded with budesonide encapsulated in a pH-responsive matrix. After oral administration, in vivo studies revealed decreased expression of proinflammatory cytokines and enhanced therapeutic efficacy.
Biological delivery systems
This novel approach exhibits significant therapeutic potential in IBD treatment involving bacteria, cells, and other biosystems for targeted drug delivery.
Bacteria
The advancement of genetically engineered probiotic bacteria offers a novel approach to utilising probiotics as drug carrier systems for the delivery of IBD medications to inflamed colon sections. Several research groups investi-
Advanced drug-delivery systems are promising tools for targeted therapy of Inflammatory Bowel Disease. They can reduce systemic toxicity, and improve drug release kinetics and efficacy leading to improved patient outcomes and quality of life.
gated the delivery of anti-inflammatory cytokine IL-10, anti-TNF- nanobodies, and immunomodulatory proteins by Lactococcus lactis. The study findings demonstrated reduced levels of proinflammatory cytokines after oral administration. Despite the advantages of administration of genetically engineered Lactococcus lactis expressing anti-inflammatory molecules (noninvasive, allochthonous, endotoxin-free) biological containment poses a risk of transgene escape remains. Furthermore, drug delivery systems based on bacteria probably have a limited effect on IBD due to colonisations only at specific niches in the colon.
Cells
Red blood cells (RBC) offer an advantage in delivering drugs to target sites due to their features such as high biocompatibility and long circulation time in blood. A clinical trial conducted by Castro et al investigated the safety and efficacy of autologous RBCs infusion loaded with dexamethasone 21-phosphate in the maintenance of long-term remission in children with steroid-dependent CD. The endoscopic results showed a significant therapeutic effect revealing 44 per cent of disease remission in patients. Furthermore, subsequently, a sustained efficacy and safety of this treatment was reported during the next six years in 50
per cent of the steroid-dependent CD patients who continued the study. Despite these encouraging results, it should be noted that drug-loading strategies can pose a risk to the structure and function of RBCs. Additionally, further research is necessary to improve drug stability during delivery and the release of drugs immediately after reaching the inflamed tissues. Lastly, the safety of the drugloaded RBCs in vivo necessitates extensive evaluation.
Conclusion
Novel targeted drug delivery systems not only enhance therapeutic efficacy by delivering drugs in a targeted and controlled manner directly to the affected regions of the inflamed colon in IBD but also reduce the systemic side-effects associated with conventional IBD treatment. A plethora of drug delivery systems are currently under investigation and further research is required to evaluate their efficacy and safety. Despite the challenges in delivering drugs to the inflamed colon in IBD, the new strategies hold great promise in improving IBD symptoms and the quality of life of patients.
References are available at www.pharmafocusasia.com
development of physiological 2D models based on human-derived organoids and the development of hydrogel-based systems for the rectal delivery of cells in inflammatory bowel disease.
AUTHOR BIO
Julia obtained her PhD in Pharmacy from King’s College London. She holds a position as a Lecturer in Biomedical Science at Anglia Ruskin University. Julia’s research focuses on the
Biologics and Biosimilars Global Market Cytiva’s strategies
Manoj Panicker Commercial General Manager India, Cytiva
1. Given your extensive experience in the life sciences industry, what do you perceive as perceive the global growth trajectory of biosimilars and biologics, and what factors contribute to their increasing prominence?
The global growth trajectory of biosimilars and biologics is set to continue expanding. Biologic drugs have changed the treatment landscape for various diseases, such as cancer, autoimmune disorders, and chronic conditions. Their efficacy and specificity make them highly sought after. As the demand for biologics continues to rise, biosimilars offer an opportunity to increase patient access to these life-changing therapies. Several factors drive their increasing prominence, including patent expirations, supportive healthcare policies, regulatory frameworks, and manufacturing advancements.
Firstly, many biologic drugs have reached or are approaching the expiration of their patents in several years. This opens up opportunities for the development
and commercialization of biosimilars, which are highly similar versions of the original biologic drugs. As patents expire, biosimilar manufacturers can compete and drive growth.
Secondly, many countries and healthcare systems globally are implementing policies and initiatives to promote the use of biosimilars as they offer cost savings compared to their reference biologic counterparts. These policies include incentives for healthcare providers, reimbursement adjustments, and educational campaigns aimed at raising awareness about biosimilars. Such supportive policies contribute to the increasing prominence of biosimilars globally.
Thirdly, regulatory agencies around the world have established robust frameworks for the approval and commercialization of biosimilars. These frameworks provide clear guidelines and pathways for biosimilar development, ensuring efficacy and quality standards. The establishment of these regulatory frameworks has instilled confidence in biosimilars, encouraging their adoption and growth.
Lastly, advances in bioprocessing and manufacturing technologies have made the production of biosimilars more efficient and cost-effective. Improved manufacturing processes, analytics, and quality control measures have enhanced the ability to develop highquality biosimilars, further fueling their growth.
Now, what does this mean for India? Research by Tuffs highlights that India currently contributes 3-4% of the world's biosimilars output today. According to Cytiva’s 2023 Global Biopharma Resilience Index, 65% of biopharma executives in India say that the domestic manufacturing of biologics is set to significantly increase until 2026. This is 15% higher than the global average, and resonates with India’s National Biopharma Mission to make India a global biomanufacturing hub.
At Cytiva, we’re excited that India will be able to capture the growth,
building on its established reputation as the world’s largest producer of generic drugs. India can replicate its success in pharmaceutical generics and harness a combination of regulatory enhancements, therapeutic innovation, and robust R&D initiatives to seize the opportunity in biologics and biosimilars.
2.
Could you discuss the strategic initiatives that Cytiva is undertaking to support the development and commercialisation of biosimilars and biologics on a global scale? Please provide specific examples or case studies related to these initiatives.
At Cytiva, we support our customers at every stage of biomanufacturing from discovery to delivery. In region for region manufacturing has been, and will continue to be, Cytiva’s strategy globally, and in India, in line with the post-COVID shift towards onshoring production.
To support this, we focus on our strengths in fostering collaborations, as well as offering advanced bioprocessing solutions, process development,
trainings, regulatory support, and a global network of expertise.
First of all, Cytiva collaborates with academic institutions to support the development of biosimilars and biologics and empower growth within the biotech ecosystem. These collaborations often involve knowledge sharing, technology transfer, and joint research projects to accelerate the development process.
Here in India, we have meaningful collaborations with incubation centers supported by Biotechnology Industry Research Assistance Council (BIRAC) and Department of Biotechnology (DBT), and we’re active supporters and mentors for the next wave of biotech innovators. Our collaboration with translation centers like C-CAMP (Centre for Cellular and Molecular Platforms) exemplifies our dedication to guiding new startups. By offering mentorship and support, we help emerging companies solidify their footing in the biotech world.
Furthermore, as an active member of the Life Sciences Council of the Confederation of Indian Industry (CII), we contribute to skill development discussions, most recently engaging with DBT to highlight the significant strides being made by the government in this area.
Secondly, since integrating with Pall Life Sciences in 2023, Cytiva has
the depth and breadth of experience globally, and advanced bioprocessing solutions, to enhance the efficiency and productivity of biologics manufacturing. These solutions include innovative bioreactors, chromatography systems, filtration technologies, and purification resins, which enable streamlined production processes and improved product quality.
We also have the expertise in process development and optimization for biosimilar and biologic manufacturing, available through our six Fast Trak™ Centers globally. By leveraging our vast experience in bioprocessing, we support our customers in refining their manufacturing processes, optimizing yields, and ensuring compliance with regulatory requirements. Our Fast Trak ™ Center in Bangalore, India, represents one of the three centers in Asia Pacific.
Thirdly, Cytiva provides onsite and virtual training programs and workshops to support the knowledge and skills development of talent working in the biosimilars and biologics space.
These educational initiatives aim to enhance the understanding of bioprocessing techniques, quality control, and regulatory compliance, ultimately facilitating the successful development and commercialization of these products.
Fourthly, we understand that navigating the regulatory landscape can be challenging. By providing guidance and support in meeting regulatory requirements, ensuring compliance, and addressing any challenges that may arise during the approval process, Cytiva’s Fast Trak ™ validation services helps customers mitigate risks and accelerate time to market. Process and product specific validation of sterile filters and single use-components are handled in the lab in Bangalore, India.
In addition, as a global company with nearly 15,000 associates in 40 countries and more than 300 years’ heritage of expertise, Cytiva is better positioned to help customers solve biotechnology challenges in an era of emerging modalities. We now have the scale, talent, and incredible footprint to
help our customers advance therapeutics for the benefit of patients around the world. In India alone, we have 50 field service and service sales associates to support our customers.
3. What are some specific market dynamics that have a significant impact on Cytiva's strategies in the biosimilars and biologics space? How have these dynamics influenced your approach?
At Cytiva, we aim to respond to the needs of our customers and the industry. As mentioned earlier, a few factors can have a significant impact on companies in this industry.
Firstly, the regulatory landscape in the biosimilars and biologics space is complex and can vary across different
At Cytiva, we’re excited that India will be able to capture the growth, building on its established reputation as the world’s largest producer of generic drugs.
regions. Cytiva's strategies take into account the regulatory requirements for product development, approval processes, and access which our customers face. Adapting to and complying with these regulations is crucial for success.
Secondly, patents and intellectual property rights play a significant role in the biosimilars and biologics sector. We observe developments in existing patents for reference biologics, which can impact the industry’s ability to develop biosimilar products.
Thirdly, pricing and reimbursement policies set by healthcare systems and insurance providers can significantly impact the commercial viability and access of biosimilars and biologics. Manufacturers need to consider factors such as cost-effectiveness, and reimbursement negotiations to ensure acceptance and viability.
Hence, our approach to these dynamics is to play a bigger role in India’s “Make in India” vision and align with the Department of Biotechnology’s strategies. That means focusing on our long-term strategy of regional manufacturing.
In October last year, we inaugurated our manufacturing facility and customer experience centre in Pune in anticipation of the growth in demand for biosimilars and biologics manufacturing from our customers.
Before we added these capabili -
ties, Cytiva already has established a strong presence in India with facilities in Mumbai, Delhi, Ahmedabad, Hyderabad, Chennai, Bangalore, and Kolkata.
Our Bangalore site includes a Fast Trak Center that offers training as well as validation and bioprocessing services, and a well-established center for research and development across bioprocess, discovery, medical, and genomic medicine businesses.
4. Can you elaborate on the regulatory challenges and opportunities faced by the industry in navigating the global landscape for biosimilars and biologics, particularly in different regions? How does Cytiva adapt its strategies to address these challenges effectively?
In my view, the industry can reframe challenges in navigating regional regulatory challenges as opportunities.
For example, the regulatory approval process for biosimilars and biologics can be complex and time-consuming. Companies seeking to produce biosimilars need to navigate through stringent regulatory requirements, including demonstrating similarity to reference biologics, conducting comprehensive analytical and clinical studies, and addressing regulatory concerns.
Secondly, regulatory frameworks for biosimilars and biologics can vary across different regions. Companies producing biosimilars need to adapt their strategies to comply with and meet the specific regulatory require-
ments of each country. This can involve additional resources, costs, and complexities in gaining regulatory approvals in different countries. By staying informed about the latest regulatory developments, Cytiva can help customers adapt their strategies accordingly to navigate the global landscape for biosimilars and biologics successfully.
5. In the competitive landscape of the global biopharma industry, how does Cytiva utilise advancements in bioprocessing, discovery, medical, and genomic medicine
to pioneer innovation and carve out a distinctive position in the market?
The biopharma industry is in constant movement and needs a strong collaborator that is capable of creating and innovating the tools needed to advance the next generation of medicines. Following our integration with Pall Life Sciences, we now have the scale to do this at every phase – from discovery to delivery. Our portfolio now includes product brands such as Allegro, Supor, iCELLis, Kleenpak, and Pegasus, in addition to ÄKTA, Amersham, Biacore, FlexFactory, HyClone, MabSelect, Sefia, Whatman, Xcellerex, and Xuri.
Recently, we launched innovative solutions like Supor Prime sterilizing grade filters to address the needs of customers manufacturing highconcentration biologic drugs, Xcellerex magnetic mixer to address challenges faced by customers engaged in largescale mAb, vaccine, and genomic medicine manufacturing processes, and Sefia cell therapy manufacturing platform to increase efficiencies and accelerate speed to clinical
milestones, among others. All solutions are aimed at solving our customers’ distinct needs.
We’ll continue being that strong partner to our customers by focusing on innovation and digitalization, especially in data management. I’m excited about the adoption of digital solutions for our customers to enable flexible, reliable bioprocess development. Through the Bioreactor Scaler tool, our customers can get their cell culture process right the first time for optimal predictions. Through GoSilicoTM process development and intensification, they can speed up high performance and flexible processes. There’s also so much potential around data sharing. We’re exploring how we can help customers save time, money, and increase output by understanding what happened in every stage of a process.
6. How does Cytiva utilise academic, industry, and technology partnerships to advance transformative technologies for global biopharmaceutical research, manufacturing, and diagnostics? Could you highlight successful collaborations that have bolstered Cytiva's role in the biopharma ecosystem and spurred innovation?
As mentioned earlier, Cytiva collaborates with academic institutions to support the development of biosimilars and biologics and empower growth within the biotech ecosystem. These collaborations often involve knowledge
sharing, technology transfer, and joint research projects to accelerate the development process.
Here in India, we have meaningful collaborations with incubation centers supported by Biotechnology Industry Research Assistance Council (BIRAC) and Department of Biotechnology (DBT), and we’re active supporters and mentors for the next wave of biotech innovators. Our collaboration with translation centers like C-CAMP (Centre for Cellular and Molecular Platforms) exemplifies our dedication to guiding new startups. By offering mentorship and support, we help emerging companies solidify their footing in the biotech world.
Furthermore, as an active member of the Life Sciences Council of the Confederation of Indian Industry (CII), we contribute to skills development discussions, most recently engaging with DBT to highlight the significant strides being made by the government in this area.
Looking ahead, we’re excited to deepen our collaborations with academic institutions and industry bodies in India, aiming to advance the skills of R&D talent in the biotech sector. By sharing our knowledge on the latest bioprocessing techniques, we’re not just anticipating the future; we’re helping to create it.
7. What is Cytiva doing to adapt to shifting customer demands in the dynamic global biopharma sector, particularly in
light of industry changes? Could you discuss specific strategies or initiatives Cytiva has implemented in response to customer feedback and market trends?
Looking ahead, we’re excited to deepen our collaborations with academic institutions and industry bodies in India. By sharing our knowledge on the latest bioprocessing techniques, we’re not just anticipating the future; we’re helping to create it.
It’s an exciting time to work in the biopharma industry, with the growth of genomic medicine especially in RNA technologies and cell and gene therapies globally and in Asia Pacific. We understand that startups, and R&D institutions compared to big biopharma companies in Asia Pacific, are struggling to compete for mature talent, and to train fresh hands.
As a Danaher OpCo, we adopt the mindset of Customer Talk, We Listen. To meet their demands in talent training, we’ll continue to provide Fast Trak Education and Training Program at three centers based in Asia Pacific namely India, South Korea, and China.
Fostering strong collaborations between academia and the industry can be transformative. By looking at global success stories, like Sweden's Testa Centre which offers an open door to innovators for accessing complete lab setups, we can envision a similar pathway for India. Such collaborative environments could address the current gaps in academia-industry interactions and the challenges of scaling up, paving the way for a thriving ecosystem that supports innovative breakthroughs.
8. How does Cytiva incorporate sustainability into its
biopharmaceutical manufacturing processes, given the industry's growing focus on eco-friendly practices? Can you outline specific initiatives aimed at reducing environmental impact and promoting sustainability?
Sustainability is a key priority for Cytiva and we’re excited to see that this has emerged as a focus area for India’s biopharma industry in this year’s budget announcement. During her 2024-2025 Budget address, Finance Minister of India Nirmala Sitharaman discussed the introduction of a fresh initiative focused on bio-manufacturing and bio-foundry. This initiative aims to offer eco-friendly substitutes like biodegradable polymers, bio-plastics, bio-pharmaceuticals, and bio-agricultural inputs. The announcement aims to boost the bio-economy's contribution to India's economy, reaching $300 billion by 2030 and $1 trillion by 2047. Bio-products are vital for India's sustainability and green economy goals.
At Cytiva, “designing in sustainability’ is our plan to make a tangible impact on people, and the planet, and to build the foundation for a resilient company. We have ambitions to 2030 and targets that we’re working to achieve by 2025.
Our six focus areas are: reducing carbon footprint, using water responsibly, evolving our use of plastics, rethinking packaging, driving community impact, and increasing diversity and inclusion. As you can see, four focus on making a positive impact on our environment and our 2025 goals include
• Zero polystyrene, replaced with reusable solutions or that are widely acceptable for recycling.
• 35% Reduction in absolute CO2e emissions in operations.
• 100% of sites powered exclusively by renewable electricity.
• 100% of district steam, heating, and cooling generated by renewable sources.
I’m happy to also share that our latest manufacturing facility in Pune uses GreenPro-certified insulation, cement, plywood, and steel along with water efficient fixtures that reduce water demand by 46%. Additionally, 100% of the effluent water from the facility is treated and reused for gardening, and 95% of the facility’s footprint will be air-conditioned, with a BEE5 star rating to improve energy efficiency.
Most recently, we launched a fruit seed collection drive at our manufacturing facility in Pune, to commemorate World Environment Day 2024. Our associates brought in over 450 fruit seeds that have been washed and dried. These seeds were then handed over to an organisation called Forrest (Forest Regeneration and Environmental Sustainability Trust), where they will be sown before being moved to appropriate locations as soon as they reach the seedling stage. This is just one of the many examples of initiatives that we participate in for the good of People and Planet.
9. What are your predictions for the future of the global biopharma sector, specifically concerning the adoption and impact of biosimilars and biologics? What challenges lie ahead, and how does Cytiva plan to sustain its leadership amidst these advancements?
Manoj Panicker is a dynamic business leader with a product development and marketingvbackground. Based in Bengaluru, he is currently serving as the General Manager, India at Cytiva. He is responsible for shaping the vision and growth path of Cytiva’s business and overseeing commercial operations in India and neighbouring countries Bangladesh, Sri Lanka, and Nepal across Bioprocess, Discovery & Medical, and Genomic Medicine businesses. Manoj is a member of National Biotechnology Council-CII and Association of Biotechled Enterprises.
We anticipate the adoption of biosimilars to grow in light of the supportive policies by many countries and healthcare systems globally, regulatory frameworks, manufacturing advancements. as well as cost savings compared to their reference biologic counterparts.
As mentioned earlier, by keeping abreast with the latest regulatory developments, Cytiva can help customers adapt their strategies accordingly to navigate the global landscape for biosimilars and biologics successfully.
Interested in visiting the Cytiva Experience Centre? Please contact us here.
Looking back, looking forward Drug Discovery Today
This op-ed elaborates on the cusp of a revolution in drug discovery, driven by the AI/ML marriage between the prediction of biological structures (proteins, DNA, RNA and heteromorphs thereof) across the biome and the techniques for accelerating the discovery, synthesis and development of small-molecule agonists and antagonists against them.
Mahesh Narayan, Biophysicist, The University of Texas
While recently attending, by invitation, a state-of-theart ChimeraX workshop on molecular modelling at the Harvard Medical School I could not but reflect on the times that we live in with respect to our understanding of biological processes, the chemistry and physics that drive the workings of proteins and enzymes, the regulation of DNA and the ability to interfere a-la a pen striking out a typo
via RNA interference. The acceleration in the field of CRYO-EM techniques, and the ability to accurately predict protein structures has now fully matured. As a result, magnum molecular machines whose structures lay within the confines of our imagination have not only been solved but resolved with sub-angstrom clarity and reside on the computer screen of many graduate students.
As a corollary, this is a heady era for drug-development. In the days of ‘yore’ solving protein structures had their parallels in drug-discovery with the common denominator being months to years from initiation to product realisation. While the former necessitated either X-ray or NMR techniques coupled with tools including ‘hanging-drop’, molecular replacement, site-directed mutagenesis, H- D exchange, isotope labelling, the latter relied on bottom-up synthesis, natural products screens, SARS, enzyme inhibition kinetics, IC50s et al.
By contrast, as aforementioned, the last couple of years has witnessed a ‘renaissance’ in both areas. The inclusion of AI/ML techniques and predictive algorithms such as Alpha-fold (which is now coupled to visualisation programmes such as ChimeraX) have narrowed the gap between the previously vast ‘sequence’ vs ‘structure’ divide. Alpha-fold itself has a worthy rival(s). Its 220M predicted structures have been eclipsed by the ‘efforts’ of Meta AI. This ‘bot’ has crawled the ocean, the soil and in fact the entire terristrome and known oceanome to reveal the structure of over 600M proteins found in microbes deep in the soil, in the depths of the oceans and even within us. This ‘revelation’ vastly outnumbered our knowledge base from 5 years ago.
This trove of structures forms the basis for today's drug-discovery which had in effect transitioned into biologicals over the last 10-15 years for not only cancer but also autoimmune disorders such as arthritis, psoriasis, colitis, and degenerative disorders including MS and AD.
The chance to revive small-molecule based drugs has never heretofore been more compelling. AI/ML methods have
The AI/ML-officiated wedding between biological structures and drugdevelopment is set to revolutionise the healthcare industry, impact quality of life and influence longevity.
been used for predicting retrosynthetic techniques, accelerating bottom-up procedures by harnessing their ability to ‘invoke’ appropriate reagents and catalysts at each step; they have been instrumental in the design of de novo entities. For example, platform discovery is now highly facile with AI coupling databases of known compounds with biomolecular structures (proteins, enzymes, NA, ribosomal complexes) to identify lead candidates, fine-tune their optimization, make accurate ADME and tox. predictions, control selectivity, simplify the synthetic route by reducing steps, and embrace one-pot techniques to improve
yields. The inroads include target predictions, improvements on repurposing existing drugs, reductions in off-target hits (thereby helping attrition side-effects), predictive costing, embracing of green synthesis, addressing scale- up and essentially creating an algorithm for almost any synthetic route which can be coupled a robotic microfluidic platform to execute synthesis. Effectively, classical protocols in drug-discovery that are labour and time-intensive can be shunted.
During the COVID pandemic, the undersigned has previously commented in Chemical and Engineering News (https://cen.acs.org/physical-chemistry/ modeling/Reactions/99/i37) on the interplay between the principles of evolution and the drug discovery (vaccine development) to effectively one-up any virus by generating vaccines against future variants. This was followed by the potential of ChatGPT to influence Chemistry (https://cen.acs.org/physical-\ chemistry/computational-chemistry/ReactionsChatGPT-means-chemistry/101/i9).
Today, just two years downstream, the author is confident that the vast playground of structures lying around waiting to be tapped coupled with the power of AI/ML in intervening in their function is a celestial wedding waiting to happen. The invitees to this grand and virtual event, you the reader and hopefully I the writer, are bound to have unforeseen benefits in healthcare, quality of life and longevity. It is this authors’ sincere hope that the benefits percolate to the have-nots at a speed even greater than Bill Gates’ benevolent funding machine.
Mahesh Narayan, FRSC, is a thinker and a biophysicist with broad interests spanning protein folding and misfolding, neurodegenerative disorders, development of prophylatics and therapeutics using both natural products and carbon based nanomeric platforms, and halogen bonding. He is an exponent of back-of-the-envelope approximations to solving a multitude of problems. He enjoys authorship and co-authorship of over 130 research articles, op-editorials, book chapters, reviews and educational works.
AUTHOR BIO
Nanocarriers in Breast Cancer Therapy Myth or miracle?
Breast cancer (BC) is a genetically and clinically heterogeneous cancer with diverse molecular, immunochemical, morphological, and clinicopathological features with different responses to therapy. Cautious harmonisation of nanocarriers can improve BC therapy by reducing the adverse effects, metastasis, and relapse. Nevertheless, how far this advanced technology serves the challenges posed by the heterogeneity of BC is still a matter of ongoing research and development. This article emphasises the current position of nanocarriers in BC therapy and provides future suggestions for the clinical translation of nanocarriers.
Nalla Usha Kumari, Neelesh Kumar Mehra Department of Pharmaceutics, National Institute of Pharmaceutical Education and Research
Breast cancer is a formidable public health challenge in the world.
Based on origin, BC is divided into carcinomas (which develop from the terminal ducts and lobules of the
breast) and sarcomas (which develop from connective tissues that support ducts and lobules). Adenocarcinomas represent 95 per cent of BC cases, and triple negative BC is the most molecularly diverse and
biologically aggressive subtype of BC with the lack of ER, PR, and HER2 overexpression. It affects younger and premenopausal women and has high metastatic potential. Recent advancements in BC therapy, such as radiotherapy, endocrine treatment, surgical techniques, and chemotherapy, have improved the survival of patients. Surgery is ineffective in case of recurrence and metastasis. Conventional chemotherapy is unable to offer selective therapy, leading to off-target effects and toxicity, whereas radiotherapy affects neighbouring healthy cells. 20-30 per cent of the patients develop metastatic cancer, and One of the main reasons are tumour heterogeneity, drug resistance, and chemotherapy failure. Keeping these in mind, novel approaches are required to tackle BC effectively.
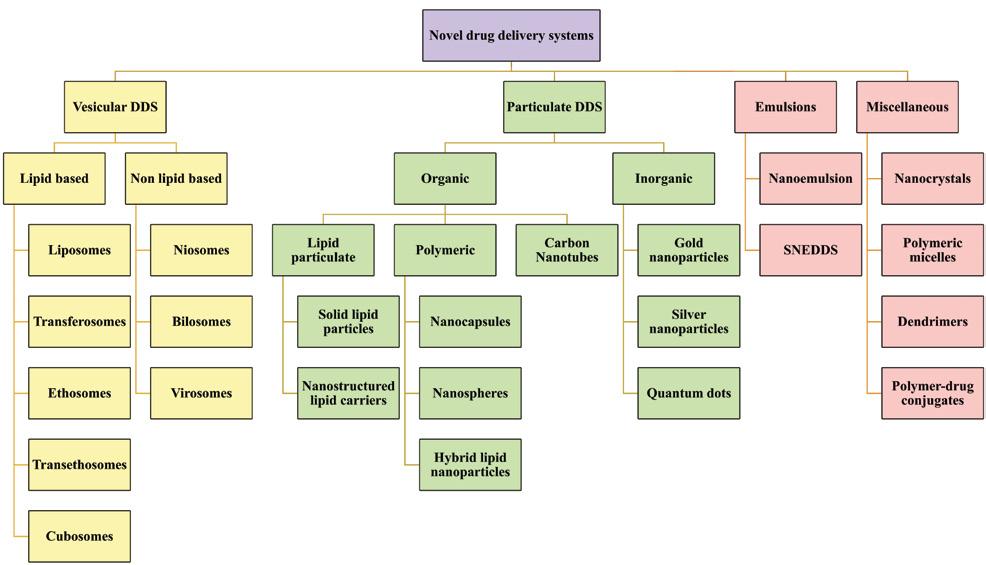
Nanocarriers are colloidal dispersions of drug carriers in the submicron range with exclusive properties like a high surface area to volume ratio, favourable pharmacokinetics, etc., that increase the efficacy and reduce the toxicity of the chemotherapeutics while conquering biological barriers and overcoming drug resistance. They are of vesicular, particulate, emulsion, and miscellaneous systems. The detailed classification of nanocarriers is depicted in Figure 1. Vesicles are typically made up of a polymer backbone with properties allowing for self-assembly into
vesicular structures. These drug delivery systems include nanocarriers based on lipids and non-lipids, capable of loading hydrophilic and lipophilic substances for controlled drug delivery. The term "particulate system" encompasses a broad range of nanoscale structures, including polymeric, lipid-based, micellar nanoparticles, dendrimers, quantum dots, carbon nanotubes, nanorods, and nanoparticles made of silver, gold, or iron. These systems, composed of natural and synthetic polymers or other biomaterials, offer significant benefits across various applications in regenerative medicine and drug delivery. The nanocarriers exhibit advantages like reduced drug dose, offtarget effects, degradation and improved solubility, pharmacokinetic parameters, therapeutic efficacy, increased stability, controlled drug release, etc. Various chemotherapeutics, including natural products, vaccines, proteins, mRNA, siRNA, DNA, diagnostic agents, etc., can be delivered to the targeted site using nanocarriers. These nanocarriers provide targeted therapy and protect the encapsulated components from
degradation. Further, targeted therapy with spatial and temporal control on the drug release decreases the drug dose and improves therapeutic efficacy. (Figure 1)
Nanocarriers: Myth or Miracle?
With the advances in nanotechnology, nanocarriers are being tailored to obtain optimal therapeutic properties while reducing unwanted side effects. Nanocarriers exhibit accumulation at the tumour site by extravasating leaky vasculature of the tumour (EPR effect) and reducing the off-target effects on healthy cells. The EPR effect is expected to increase the accumulation of nanocarriers by 20-30 per cent, and transcellular pathways also improve the nanoparticle's extravasation into the tumour site. They reduce the system toxicity of the drug; for instance, liposomal doxorubicin exhibited lower cardiotoxicity than free doxorubicin. The physicochemical properties of the nanoparticles, as well as the pathophysiological and pathoanatomical features of the tumours, influence the extent of tumour accumulation.
However, the heterogeneity of the vascular architecture, multidrug resistance etc., remain challenging. More strategies are required to improve the effectiveness of the nanocarriers by pharmacological and physical co-treatments through active targeting/stimuli-responsive nanocarriers or a combination of nanocarriers with immunotherapy. Active targeting uses the interaction between surfacemodified nanocarriers that bind with the receptors overexpressed at the target site. FunctionaliSation of nanocarriers with ligands like folic acid, hyaluronic acid, antibodies, aptamers, etc., whose receptors are overexpressed or selectively expressed at tumor site can improve the internaliSation of the nanocarriers into the cancer cells. Investigations have revealed that tumour cells exhibit greater uptake of hyaluronic acid-coated nanoparticles than normal cells. Considering the BC subtype, various active ligands can be used to modify the nanocarrier surface to enable active tumour targeting. Epidermal growth factor receptor (EGFR) targeted nanoparticles displayed greater uptake of
Figure 1: The detailed classification of nanocarriers is depicted
nanocarriers by EGFR-expressing BC cells than the EGFR-negative BC cells. The folate receptor is overexpressed in TNBC, and the folate-conjugated microspheres exhibited a significant reduction in the tumour volume in the BALB/C mice model compared to the non-targeted microspheres.
Over the past two decades, extensive research has focused on stimuli-responsive nanocarriers that are responsive to intrinsic (pH, redox, hypoxia, temperature, etc.) or extrinsic (ultrasound, light, magnetic, etc.) stimuli and can produce a rapid and controlled release of the drug upon the stimulus. Contrary to the normal tissue, the tumour environment has lower pH, overexpressed enzymes, high glutathione concentration, etc., which are being exploited to rationally design different responsive structures, from polymers to hybrid nanocarriers. Upon exposure to the stimuli, these modified systems release the drug through various mechanisms. Hybrid nanocarriers are nanocomposites of two or more different materials (polymer and inorganic- or organic-based systems) that have superior properties combining the advantages of the individual components while overcoming the individual shortcomings. In our lab, we have fabricated ligand-conjugated pH-responsive chitosan-lecithin hybrid nanocarriers for stimuli-responsive targeted delivery of palbociclib to breast cancer. The hybrid nanoparticles exhibited pH-dependent drug release, greater endocytosis, and hindered cellular migration. Notably, hybrid nanocarriers are being investigated for theranostic purposes to achieve more significant therapeutic potential.
Despite the preclinical success of nanocarriers in BC therapy, their translation has exhibited limited success. Despite the advantages of improved solubility, permeability, bioavailability, circulation time, targeted delivery, etc., nanocarriers still have limitations like complex manufacturing procedures, challenges with quality control, scalability, reproducibility, characterisation tech -
niques, nanocarrier stability, etc., which pose challenges in translation to market. Furthermore, the opsonisation of nanocarriers, immune reactions, drug leakage, toxicity of excipients, etc., are some of the other challenges. One of the reasons for in vitro–in vivo discrepancy is the change in the physicochemical properties of nanocarriers after entering the biological system due to their interaction with the biomolecules. These interactions give a new biological identity different from the pristine nanocarriers, which impacts the nanocarriers' biodistribution, release kinetics, targeting ability, stability, etc.
The current limitations, such as stability, limited translational potential, in vitro-in vivo discrepancy, and knowledge gaps, must be addressed for the clinical translational of nanocarriers. Carrier-free nanocarriers are one option for minimising polymer toxicity, and they have high drug loading with low systemic toxicity. The balance of nanocarrier properties and tumour conditions is essential for delivering the loaded cargo to the intended site. A thorough understanding of nanocarrier properties is required for rational nanocarrier design and structural optimisation. The biomolecular interactions of nanocarrier with the biological milieu determine its fate in the body. Similarly, pathoanatomical features, tumour microenvironment, physiological process, etc., should be considered while designing nanocarriers for accurate delivery to the tumour site. One practical approach for replicating a tumour microenvironment involves utilising patient-derived xenografts to investigate how nanocarriers accumulate, extravasate, and selectively target tumours.
In summary, nanomedicine has technical limitations and is in its infancy. This novel technology is expected to transform BC therapy and improve the survival of patients. The balance between conducting thorough research on nanocarriers to ensure their efficacy and safety is crucial for facilitating regulatory approval processes. A deeper under-
standing of the physicochemical nature of nanocarriers and physiological conditions may result in fruitful outcomes in the clinical translation of nanocarriers and BC treatment.
References are available at www.pharmafocusasia.com
Neelesh Kumar Mehra is currently working as an assistant professor in the Department of Pharmaceutics, National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad, Telangana, India. He has authored >120 peer-reviewed publications in reputed international journals and >30 book chapter contributions and has been the editor of three international books. He guided more than >30 MS students and 10 PhD students and 03 granted patents. His research group is working on novel drug delivery systems for the treatment of cancers, rheumatoid arthritis, wound healing, and ophthalmic diseases.
Usha Kumari is a Ph.D. scholar working under Dr. Neelesh Kumar Mehra at the Pharmaceutical Nanotechnology Research lab, Department of Pharmaceutics, National Institute of Pharmaceutical Education And Research (NIPER), Hyderabad. She completed her Master's in Pharmaceutics under the guidance of Dr. Neelesh Kumar Mehra at NIPER Hyderabad and her Bachelor's in Pharmacy from Andhra University. Her research work is focused on the development of novel drug delivery systems in the management of cancer.
AUTHOR BIO
Nalla
Dr.
Biomarkers in Drug Discovery and Clinical Trials
Novel robust biomarkers identification during drug development posess immense potential for treating incurable diseases. The increased popularity of biomarkers in pharmaceutical discovery is attributed to a rapidly evolving utilisation of biomarkers from early to late-phases drug discovery. This overview highlights biomarker categories based on clinical drug development, in addition to addressing the challenges in biomarker research.
Kirti Singh, Postdoctoral Scientist, Eli Lilly and Company
Biomarkers are biological markers in the body that can be utilised to predict the presence or absence of abnormal biological processes. Biomarkers play a crucial role in disease prediction, detection, prognosis, and monitoring. Furthermore, biomarkers are also utilised to assess and monitor the response to a pharmacological intervention. Human body is composed of a plethora of biomolecules which can serve as biomarkers, including but not limited to DNA, mRNA, carbohydrates, lipids and proteins. Generally, protein and gene-based biomarkers are commonly utilised for diagnostic purposes. These biomarkers can be detected in body fluids, such as blood, urine, saliva, sweat, milk, cerebrospinal fluid or tissues. For example, blood glucose or haemoglobin A1c (HbA1c) is commonly used as a diagnostic biomarker for diabetes. Apart from complex biomolecules detected in body fluids and tissues, functional measurements to gauge physiological responses can also be categorised as biomarkers. For instance, heart rate and blood pressure are non-invasive functional biomarkers used to monitor cardiovascular diseases.
Any biomarker must possess few characteristics to be reliably used for monitoring and detection. Firstly, the biomarker must be safe and easy to measure. It should be reproducible, reliable, sensitive and specific to accurately reflect the outcome. In addition, for research purposes, it should be cost- effective and should be in the quantifiable detection range by current tools and technologies for diagnosis and monitoring of a particular disease. Biomarkers have been broadly classified by the US Food and Drug Administration (FDA) into seven different categories based on clinical application.
Risk/ Susceptibility Biomarker
A risk biomarker can be used to predict the increased or decreased likelihood of disease development in future, for an
individual who yet does not have a medical condition. These could be genetic mutations that makes certain individuals more susceptible to developing a disease and can be detected years or decades before disease manifestation, so that preventative measures could be employed. For instance, mutation in breast cancer genes 1 and 2 (BRCA1/2) is used to identify individuals predisposed to develop breast cancer. Elevated low-density lipoproteins (LDL) blood cholesterol could increase the susceptibility to cardiovascular diseases.
Diagnostic Biomarkers
As the name suggests, diagnostic biomarkers are used for accurate disease diagnosis by confirming the presence of biomarker. For example, glomerular filtration rate (GFR) is used as a diagnostic biomarker for chronic kidney conditions. Elevated chlorine content in sweat is used to confirm the presence of cystic fibrosis. A reliable and disease selective diagnostic marker is crucial for not only accurate diagnosis but also for clinical assessment. Further, an ideal diagnostic biomarker candidate should be highly specific and sensitive, which means 100 per cent positive result in diseased population, and 100 per cent negative result in nondiseased subset to minimise both falsepositive (non- diseased individual wrongly diagnosed) and false-negative (diseased individual not diagnosed) outcomes. Diagnostic biomarkers are often used to determine the inclusion and exclusion eligibility criteria for a target population in clinical trials.
Monitoring Biomarkers
Biomarkers which are repeatedly measured over a period of time to monitor disease progression, such as alterations in disease severity, occurrence of other associated co-morbidities, worsening of the previously determined diseased state are defined as monitoring biomarkers. These biomarkers could also be utilised to gauge the response to a treatment regimen on the disease state. These biomark-
Rapidly evolving utilisation of biomarkers from early to late-phases of drug discovery is accelerating innovation by novel target identification and improving drug efficacy, quality and safety ensuring better patient outcomes
of prostate-specific antigen (PSA) and Gleason score are used to predict the likelihood of cancer progression in prostate cancer patients. The correlation of prognostic indicators with the likelihood of future event is highly context specific and may vary depending upon patient history, severity of disease, the strength of prognostic indicator along with other comorbid conditions etc. Hence, caution must be exercised before establishing any strong correlation.
Predictive Biomarkers
ers usually project a rate of change in disease characteristics over time. In clinical trials, it is extremely important to establish biomarker monitoring during a patient's clinical course to assess the effect of intervention as the pre-treatment baseline characteristics may vary from patient to patient. Continuous monitoring during the course of treatment is necessary to detect when and how early therapeutic effects are observed. On the other hand, monitoring biomarkers could help identify non-responders with aggressive rate of disease progression or to detect toxicity.
Prognostic Biomarkers
Prognostic biomarkers are utilised to indicate the increased or decreased probability of a clinical event, such as disease reoccurrence or progression in diseased population. For example, mutation in tumour suppressor gene, tumour protein 53 (p53) and chromosome 17p deletion are used as prognostic biomarkers to assess the likelihood of mortality in patients suffering from chronic lymphatic leukaemia. Another example, expression
Predictive biomarkers help identify individuals who are more likely to respond, either favourably or unfavourably upon clinical intervention as compared to similar individuals without the biomarker. In clinical settings, predictive biomarkers are used either as eligibility criteria or to stratify the study population into biomarker positive and negative groups to identify the subset of diseased population for which the intervention is most effective. Furthermore, predictive biomarkers could also be patient characteristics, such as renal or hepatic function, cytochrome P450 mutation which makes a patient more likely to respond unfavourably due to toxicity.
Pharmacodynamic Biomarkers
Pharmacodynamic biomarkers serve as molecular indicators of biological response upon exposure to clinical intervention. The expected biological response could be beneficial or harmful. These biomarkers are critical for dose-selection, proof-of-concept studies, and early go/ no-go decisions in clinical trials. For instance, in patients with systemic lupus erythematosus, circulating B lymphocytes could serve as a pharmacodynamic biomarker to evaluate the effect of Belimumab, a monoclonal neutralising antibody against B-cell survival factor. Sweat chloride levels were utilised as clinical end point to determine the effect of Ivacaftor, a cystic fibrosis transmembrane conductance regulator (CFTR) channel modulator in cystic fibrosis patients.
Safety Biomarkers
Approximately, 30 per cent drugs fail in clinical trials due to toxicity-related adverse effects. Safety biomarkers are used to indicate the likelihood and extent of toxic effect due to drug administration and hence are crucial in clinical trials. Safety biomarker is a type of monitoring biomarker as repeated measurements are necessary to detect and manage potential toxicity by treatment interruption or dose modification. For example, in conjugation to serum creatinine levels, urinary biomarkers, such as total protein, albumin, Kidney injury molecule-1 (Kim-1), urinary clustarin, cystatin C and Trefoil factor 3 are used to monitor drug-induced nephrotoxicity. In addition, bilirubin and hepatic aminotransferase are commonly employed to assess hepatotoxicity.
Challenges in biomarker research and drug development
Over the past five decades, the applications of biomarkers in drug discovery and all phases of development have exponentially skyrocketed. The increased popularity of biomarkers in pharmaceutical discovery is attributed to a rapidly evolving utilisation of biomarkers from early to late-phases drug discovery. Over time the definition
of biomarkers rapidly evolved and till date there is no standardised definition of biomarkers. Although, FDA has categorised biomarkers in 7 different categories, many other classification systems and sophisticated biomarker categories are emerging in literature, such as complex biomarkers, digital biomarkers etc. The explosive growth
Kirti Singh is a postdoctoral scientist at Eli Lilly and Company, Indianapolis. She completed her Ph.D. in molecular and cellular pharmacology from Mercer University, Atlanta. Her doctoral research focused on studying the effects of reactive oxygen species on β2AR in airway obstructive disorder, particularly asthma. She has published her work on GPCRs in many highimpact factor, peer-reviewed journals.
and integration of artificial intelligence (AI) and deep learning in various stages of drug development have accelerated drug-discovery efforts. The compound effect of AI-based algorithms sorting through huge human omics-datasets and the power of high-throughput screening (HTS) have expanded the horizons for biomarker discovery efforts. However, one of the major challenges is biomarker qualification and validation of analytical methods for biomarker quantification. Conventional immunoassay-based methodologies and histopathological assessments are used for biomarker discovery, however very low concentration biomarkers remain undetected with current tools and technologies. Further development and validation of analytical methods with lower limits of detects will not only accelerate drug-discovery efforts but constant innovation in the biomarker field will ultimately benefit patient and healthcare outcomes. References are available at www.pharmafocusasia.com
CDMO Considerations for CGT/ATMP Companies
Converging naturally are the worlds of Cell & Gene Therapies and the CDMO. This convergence is necessary and timely. Given the exciting times of the growing biotechnology there are fewer and fewer options to make these products at scale to meet clinical needs much less commercial. This article takes the reader through the product lifecycle and helps the CGT companies make good decisions around their regulatory strategy, decision to build or buy, and then ultimately to whom and why.
Christine Feaster, SVP, Strategic Operations, Quality Executive Partners, Inc. (QxP®
We are in an exciting time where biotechnology products are growing at such a rapid pace. Cell & gene therapies (CGT), Tissue engineered products and Advanced Therapy Medicinal Products (ATMPs) offer several new benefits. These benefits range from potential cures for genetic disorders, better more effective treatment options for what was once deemed incurable diseases, personalised medicines tailored to individual patients, and the possibility of long-lasting or even permanent treatment effects. These therapies represent a significant advancement in the field of medicine with the potential to revolutionise the treatment of various diseases.
In addition, the global cell and gene therapy market is expected to grow from US$6.58 billion in 2021 to US$8.57 billion in 2022 at a compound annual growth rate (CAGR) of 30.2 per cent. The cell and gene therapy market is expected to reach US$21.33 billion in 2026 at a CAGR of 25.6 per cent. 1 The outsourcing market is the largest, according to the report, remains North America. Despite current geopolitical ‘frictions,’ reshoring and the like, Asia is still estimated to have the highest growth during the next five years. This will be something to consider in your selection of CDMOs in the future.
From true end-to-end development through scale up and commercialisation,
New CGT/ATMP therapies are looking at speed to market, while managing risks, technical expertise and regulatory known how.
there are many unique considerations when starting out on this journey that can have a major impact on a make-orbreak approval pathway of your advanced therapy. This includes deep technical and regulatory know-how, but also how to generate buy-in from investors and the board members. This becomes paramount as you have received or are looking to receive new funding for future growth of your company. In the very early stages of discovery and development for a new CGT/ATMP therapy and/or its delivery, it is typical for small quantities of investigational drug substance to be produced in a development lab within your company or even through an academic collaboration within a university lab setting.
This early material serves various purposes, including characterisation, small-scale animal studies for proof of concept and safety, analytical method development, and excipient uses for ideal stability. Data from this initial work may be used to demonstrate the potential of the technology or substance being developed, with the intention of attracting investment funds or collaborating partners to invest in the future commercial product.
After proof of concept (PoC) and funding, scaling up becomes necessary. This requires generating more material to demonstrate process and analytical controls, including the following:
• Adequate suitability of analytical methods
• Robust manufacturing processes
• Next stage stability studies and stabil-
ity indicating methods
• Safety and efficacy in the clinical trial stages
• Proof of phase appropriate GMP controls where you are looking to prove consistency
Effectively managing the decisionmaking process involves balancing risk and speed to market, especially as you prepare for pre-clinical stages and thoroughly vet potential partners. Here’s how you can navigate the decisions about whether to outsource and the next steps once you've made that decision.
Smaller companies with a new product’s promise of a cure, may need to hire a Contract Development and Manufacturing Organization (CDMO) to start the journey. To make a build or buy decision, the following questions needs to be asked:
• Do you, as the CGT/ATMP company,
Corporate and Quality Culture
have a facility already, or a plan to build a facility and have the time necessary (2-3 years) before you start?
• Do you have the financial support (stakeholders, VCs, etc.) to engage a CDMO?
In the broadest sense, the MarketsAndMarkets report estimates that the global pharmaceutical contract manufacturing market was worth $176.5 billion in 2023 and is projected to reach $258.3 billion by 2028, growing at a CAGR of 7.9 per cent.2 CDMOs provide an excellent match with the projected growth trajectory of CGT/ATMP companies.
There are specific reasons for this outlook, including the fact that CDMOs continue to grow at a record pace every day. Essentially, with new CGT/ATMP therapies you are looking at speed to market balanced with risk, technical
• What does the corporate culture reflect? Good collaboration from the top down?
• What will the Project Management function provide, and does it match with the corporate culture?
• How do the experts making your product reflect that culture? Do not assume that what is conveyed at the beginning of the journey is reflected the same with the folks who you will be working with every day.
• Does CDMOs have consulting-like services to guide you through the process?
• What is their track record for meeting timelines and budgets?
• How do they handle intellectual property rights related to the project?
• What is their regulatory history and track record?
Pre-Clinical Considerations
• Do they offer CMC support and guidance?
• What experience does the CDMO have with CGT products at the early stage?
• Do they have a compatible Adeno-Associated Virus (AAV) platform that can be used as a vector for gene therapy applications to deliver genetic material into target cells?
• What experience do they have in Formulation – from pre-formulation to formulation design?
• How do they scale-up and optimise product development?
• What experience do they have in packaging design (primary and secondary)?
• Can they scale up to meet your needs throughout the process?
• Do they offer analytical method development and validation?
• Do they have quality control methodologies for potency and yield analysis that work for your product?
• Do they have a Drug Master File (DMF) that the agency is familiar with?
experience, and regulatory know-how. In many cases, the CDMO choice becomes an easy one to make for new companies who have never commercialised a product in the past. First, you need to find a CDMO that will focus development on iterative changes and focus manufacturing
Clinical Phase Considerations
on process control and consistency. Next, you must choose the one that will balance speed to market and risk properly and have the technical and regulatory experience to take your product through the right stages. Below are some tips across the product lifecycle to consider.
• For your investigational product manufacturing, do they have phase appropriate GMPs (from early to late stage)?
• What visual inspection guidance do they follow for your geographic market of choice?
• Do they offer the guidance on packaging and labeling requirements?
• How do they manage release and stability testing?
• Can they label for randomised and blinded studies?
• How do they transfer analytical methods from pre-clinical to clinical to commercialisation?
• How do they transfer and scale the manufacturing process? Can they? Or do they transfer back to you or another site or company?
• CTD preparation?
1. The answers to these questions will help you and your company make the right choice for the five basic stages of lifecycle support population, geographic targets, potency of product, and the number of doses needed for launch and thereafter. Additionally, consider the growth trajectory in this area for future production.
Commercial and Post approval Considerations?
• Do they partner with you on regulatory correspondences and interactions?
• Will the CDMO be available to speak to your shareholders during the journey to commercialisation?
• Will they be able to provide you the volumes and potency needed for test batches, validation batches and launch? (PLEASE DO NOT SKIP TEST or ENGINEERING BATCHES)
• How can you support compliance? Afterall, you are responsible for the product being launched? Person in Plant (PIP) during manufacturing? For cause or periodic audits during commercial manufacturing?
• Product shipment and storage?
• Sample planning and storage?
• Preparation of annual report?
2. You are the product expert. Your experience is necessary for the CDMO to be successful. Be available and provide product guidance throughout all stages.
3. You should have a Quality Agreement specifically outlining the requirements and expectations to help the 2 sides maintain clarity and trust. Most sponsor companies will utilise the services of a CDMO at some point in the timeline of product development through commercialization and launch, as the CDMO plays a vital role in the cell therapy industry. Developing a successful CDMO relationship involves navigating a complex road forward, building a robust contract,
and adopting a collaborative approach based on shared goals, open communication, and unwavering commitment to product quality and patient health. The success of the Sponsor/CDMO relationship heavily depends on selecting the right partner initially (using some of the guidelines above) and on a clear understanding of what both companies need and bring to the table. Asking the right questions and providing truthful, transparent answers will result in choosing the right partners. Your CDMO will be an extension of your company. Remember that you are both on the same side and that open and timely communication throughout the journey will build trust and most likely lead to success in the relationship and increase the odds for commercialization success.
References are available at www.pharmafocusasia.com
pharmaceutical executive focused on quality manufacturing and new manufacturing modalities to ensure access and affordability of life saving medicine and treatments around the world. With 30 years’ experience in leadership roles in Quality, Quality Control and Analytical Methods development, Christine has worked for top 50 pharmaceutical companies and start-ups in creating robust quality systems that have withstood international inspections successfully. Working for USP for 11 years, Christine has had executive roles of increasing responsibility starting in Quality, Global Public Health and then transitioning her career into the Commercial side of the business.
Christine Feaster has been a
Genomics in Precision Medicine
Precision or targeted medicine is an approach that uses a patient’s genetic and lifestyle information to obtain appropriate disease treatment and prevention strategies. Genomics research has facilitated precision medicine by providing improved genetic testing as well as drug target discovery.
Precision medicine can be described as treatment options optimised to take into account the patient’s genetic information and lifestyle. Traditional model of drug development assumes that a drug will show a similar response in all patients afflicted with the same disease. But it is seen that most drugs show a positive response on a subset of the population. Another subset may develop adverse reactions to the drugs while others may not show any response at all. The underlying reason for this variability is the differences in genetic makeup between individuals. Precision medicine aims to understand and utilise these genetic differences to provide more effective therapies, which improve treatment outcome & prevent adverse effects while excluding the need of unnecessary treatments or diagnostic testing.
The first reference for precision medicine can be found in 1892 writing of Dr. William Osler “It is more important to know what kind of a patient the disease has, than to know what kind of a disease the patient has.” In more recent times, the term personalised medicine was first described in 1999 as performing a simple blood test to find out which patients will show positive response to a drug and which ones might show an adverse
response. The basis of the diagnostic test would be the minute differences in the genetic makeup of individuals.
Applications of precision medicine
The field of oncology has been the earliest adapter of targeted treatment. The first drug prescribed on the basis of a genetic test was Trastuzumab, approved by USFDA in 1998, which is used for treatment of patients with metastatic breast cancer whose tumours overexpress the HER-2 protein. This was followed by regulatory approval of Imatinib, which inhibits the BCR-ABL protein tyrosine kinase, which is present in BCR-ABL gene fusion positive chronic myeloid leukaemia (CML). Later on, drugs targeting ALK, ROS1, BRAF V600E mutant melanoma and MET-mutant lung cancer were also developed. Multigene panels for diagnostic testing in several types of cancers have been approved by FDA.
In 2017, US Food and Drug Administration (FDA) approved the chimeric antigen receptor T-cell (CART) for treatment of refractory pre-B cell acute lymphoblastic leukaemia and diffuse large B-cell lymphoma. Chimeric antigen receptors (CAR) are patient’s own T-cells that are engineered to express fusion proteins which directs the T-cells to cancer-specific antigens, causing destruction of cancer cells. In 2020, 28 targeted therapies were approved by the FDA in patient populations defined by specific molecular biomarkers.
Cystic fibrosis is caused by one of several defects in the CFTR gene. Majority of cystic fibrosis patients have F508del mutation. Ivacaftor, which works on 5 per cent of patients who carry the G551D mutation, is ineffective for the majority of patients, for whom a combination of Lumicaftor with Ivacaftor is prescribed. In 2021, Evinacumab was approved by USFDA as an add-on treatment with cholesterol lowering agents (for example, Statins) for homozygous familial hypercholesterolemia (HoFH). HoFH patients have two mutations in genes responsible for clearing excess cholesterol from the body. Evinacumab is an
angiopoietin-like protein 3 (ANGPTL3) inhibitor. ANGPTL3 slows the function of certain enzymes that break down fats in the body.
Role of genomics in precision medicine
In precision medicine, sequencing and analysis of the patient’s genetic data play a crucial role in disease diagnosis and tailoring treatment. Technologies like Sanger sequencing, real -time PCR and Microarrays were the pioneering techniques utilised for DNA sequencing. Next generation sequencing technologies have enabled fast and efficient DNA sequencing, leading to more biomarkers being identified which can help to classify patients into different subtypes and provide treatment accordingly.
Precision medicine utilises genetic information to predict which treatment is more suitable for a patient. The confluence of Artificial Intelligence and genomic data with precision medicine will increasingly play a significant role in improving disease outcomes.
The process of determination of variants from sequencing data is called variant calling. The output of DNA sequencing is recorded in the form of reads, which contain the sequence of a DNA fragment along with the sequence quality scores of each nucleotide base. Any sequencing adaptors or bases with low quality are trimmed before using the reads for analysis. Trimmed reads are aligned to the Human reference genome (GRCh38 is the latest version), and the
location in the reference genome where each read aligns is determined. Based on the pileup of all reads overlapping at each nucleotide position, the most likely genotype at that position is determined. Any difference in genotype as compared to the reference sequence is called a variant. There are many classes of variants like Single nucleotide polymorphism (SNP), Insertion/deletion, Structural variation, Copy number variation. All classes of variants can have distinct impacts on disease development and drug metabolism.
Targeted gene panels, which sequence a select set of genes which are known to be associated with the disease being investigated, are the most widely used in genomic testing. However, increasing reduction in sequencing costs have generated interest in the use of Whole Exome Sequencing (WES) and whole genome sequencing (WGS) for clinical diagnostic applications.
WES includes enrichment and sequencing of all the protein coding regions of the genome. Since 85 per cent of the disease-causing variants are located in the protein coding regions, WES covers most of the actionable regions of the genome to identify disease causing variants. WES has been used to discover genes associated with Mendelian as well as multigene disorders.
WGS provides the most comprehensive details about the genome. In WGS, a single test can provide the complete genome sequence, including non-coding regions. It also provides better determination of Copy number variations, Structural variants and chromosomal rearrangements as compared to WES. However, WGS has a higher cost of sequencing and data storage.
Whole genome sequence data aids in improved profiling of genetic variants. Genome wide association study (GWAS) is the study of genome-wide genetic variants in a set of individuals to determine if any variant is found to be associated with a trait or disease. In 2005, GWAS found two variants associated with Age-related macular degeneration. Since then, GWAS
has identified associated variants for various diseases like coronary heart disease, obesity, type-2 diabetes and schizophrenia. Till date, GWAS have identified more than hundreds of thousand SNP-trait associations, which help to identify newer targets for targeted treatment.
Integration of genomic data with multiomics
Though every cell of an organism has identical DNA, each type of cell performs different functions. This difference in function is resultant of different gene expression patterns in each cell type. Similarly, gene expression can be different between diseased and healthy states. Also, epigenetic modifications (for example, histone modification, DNA methylation) also affect gene expression without changing the DNA sequence. Studying the effect of these changes provides additional layers of understanding about cellular mechanisms.
Apart from genome sequencing, genomics has also facilitated the generation of vast quantities of transcriptomic, epigenomic, proteomic, metabolomic and microbiome data. Integration of these multiomics approaches helps create a deeper understanding of disease mechanisms and opens the door for elucidation of innovative treatments.
Artificial Intelligence and Machine learning in PM
The process of identifying drug targets from large multiomics datasets needs sophisticated computational techniques which can analyse and identify patterns in the data. Artificial intelligence (AI) tools like machine learning, deep learning, natural language processing and network-based approaches are being increasingly used for analysis of genomic datasets. Development of Evinacumab as treatment for HoFH, is an example of AI being utilised to analyse genetic and biochemical information to identify the specific targets for cholesterol regulation. Other examples of AI optimised drugs are Pembrolizumab and Sotorasib, used for cancer treatment.
Gene therapy
The huge amount of available genomic data and advances in genome editing tools (like CRISPR) have also facilitated development of novel gene therapies for diseases with genomic basis, like cystic fibrosis, adenosine deaminase deficiency, familial hypercholesterolemia, cancer, and severe combined immunodeficiency (SCID) syndrome. Gene therapy aims to replace a faulty, mutation-containing gene with a normal, healthy gene within an affected individual’s genome, to cure a disease. Cell and gene therapy can be used for diseases where the causal gene is well known and well- characterised. In 2023, two cell-based gene therapies— Casgevy and Lyfegenia—received FDA approval for treating sickle cell disease.
Future perspectives
Research in precision medicine has huge potential benefits. The insights obtained from multiomics data can have a big impact on prevention, diagnosing and treatment of diseases. Numerous challenges are also evident in attainment of these goals. One of the biggest bottlenecks
AUTHOR BIO
Gulnaz Zaidi is an experienced researcher in biotechnology industry, and is currently working as Bioinformatics Scientist at Mibiome Therapeutics, Mumbai, India. Her current passion is development of computational analysis workflows for high throughput sequencing genomic, epigenomic and metagenomics datasets
in implementing precision medicine is lack of genomic data from developing countries. Creation of variant databases catering to all ethnic subpopulations in the world is critical to wider usage of precision medicine. Development of decision support tools which empower physicians to use genetic data for treatment will be an important step. Concerns about data storage, privacy and willingness of individuals to undergo screening tests need to be addressed. Regulatory frameworks that prevent unethical use of medical and genomic data also need to be in place to safeguard patients’ rights.
The confluence of AI and genomic data with Precision medicine will increasingly play a significant role in improving disease treatment. The use of advanced computational tools has the potential to generate useful insights from genomic research, which aid in precise and timely clinical decision making, leading to improved patient outcomes.
References are available at www.pharmafocusasia.com
How Industry 4.0 Technologies Improve Supply Chain Visibility and Enable Accurate Demand Forecasting in Pharmaceutical Manufacturing
In the rapidly evolving landscape of pharmaceutical manufacturing, the integration of Industry 4.0 technologies has emerged as a transformative force. To enhance supply chain visibility and refine demand forecasting, pharmaceutical companies are embracing digital innovations to optimise their operations. In this article, we explore the transformative power of Industry 4.0 in enhancing supply chain visibility and enabling accurate demand forecasting within the pharmaceutical sector.
The term "Industry 4.0" refers to the fourth industrial revolution, characterised by the integration of smart technologies, data exchange, and automation into manufacturing processes. In the pharmaceutical sector, this shift has been gradual but profound, driven by the need for
increased efficiency, reduced costs, and enhanced quality control.
Industry 4.0 and Supply Chain Visibility Enhancement
Industry 4.0 technologies, including the internet of things (IoT), blockchain, and advanced analytics, are revolutionising
supply chain visibility in pharmaceutical manufacturing.
1. Internet of things (IoT)
IoT sensors deployed throughout the supply chain provide real-time data on inventory levels, temperature, humidity, and other critical parameters. Continuous monitoring allows for proactive decisionmaking, minimising disruptions and ensuring product quality and safety.
2. Blockchain technology
Blockchain technology offers a transparent and immutable ledger for tracking product movement, ensuring authenticity, and combating counterfeit drugs. By leveraging blockchain, pharmaceutical companies enhance traceability, regulatory compliance, and trust across the supply chain.
3. Advanced analytics
Advanced analytics tools analyse vast amounts of supply chain data, uncovering insights, patterns, and trends. Predictive analytics enable proactive risk management, demand forecasting, and optimisation of inventory levels and distribution channels.
Enhanced Supply Chain Visibility
Traditionally, pharmaceutical supply chains have grappled with opacity and fragmentation, hindering realtime monitoring and decision-making. However, with the advent of Industry 4.0 technologies like IoT sensors, blockchain, and advanced analytics, stakeholders now have unprecedented visibility across the entire supply chain network. By harnessing these technologies, pharmaceutical companies can miti-
gate risks, optimise inventory management, and uphold regulatory compliance standards with greater efficacy.
Accurate Demand Forecasting
Effective demand forecasting is paramount for pharmaceutical manufacturers to streamline production processes, minimise stockouts, and meet customer demands promptly. Yet, traditional forecasting methods often fall short due to their reliance on historical data and limited predictive capabilities. Industry 4.0 ushers in a new era of demand forecasting by leveraging artificial intelligence (AI), machine learning algorithms, and big data analytics. These advanced tools analyse vast datasets encompassing market trends, patient demographics, and consumption patterns to generate precise demand forecasts. By discerning subtle patterns and correlations, AI-driven forecasting models empower pharmaceutical companies to anticipate fluctuations in demand, optimise resource allocation, and reduce wastage. This enables proactive decision-making, facilitating agile responses to changing market dynamics and emerging trends.
Accurate demand forecasting is critical for pharmaceutical manufacturers to optimise production, minimise inventory costs, and meet customer demand effectively. Industry 4.0 technologies enable precise demand forecasting through:
1. Big Data Analytics
Industry 4.0 enables the aggregation and analysis of diverse datasets, including sales data, market trends, and customer preferences. By harnessing big data analytics, pharmaceutical companies gain actionable insights into demand drivers, enabling informed decision-making and strategic planning.
2. AI and machine learning:
AI and machine learning (ML) algorithms analyse historical data, identify patterns, and predict future demand with greater accuracy. These algorithms continuously learn and adapt, improving forecasting
accuracy and responsiveness to market dynamics.
3. Collaborative planning, forecasting, and replenishment (CPFR):
Industry 4.0 facilitates collaboration among supply chain partners through CPFR initiatives. By sharing real-time data and insights, stakeholders can align forecasts, optimise inventory levels, and improve supply chain efficiency.
Integration of industry 4.0 technologies: Seamless integration of Industry 4.0 technologies is imperative for unlocking the full potential of supply chain visibility and demand forecasting in pharmaceutical manufacturing. Cloud computing platforms serve as the backbone for storing, processing, and disseminating data across the supply chain ecosystem in a secure and scalable manner. Moreover, edge computing capabilities enable real-time data processing at the point of origin, minimising latency and enhancing responsiveness. Collaborative robotics (cobots) automate repetitive tasks in manufacturing facilities, augmenting workforce efficiency and productivity. Additionally, digital twins—a virtual representation of physical assets—facilitate simulation-based optimisation and predictive maintenance, enhancing operational resilience and agility.
Embracing sustainability in pharma manufacturing
Industry 4.0 technologies present opportunities for pharmaceutical companies to enhance sustainability. By optimising logistics, reducing energy consumption, and minimising waste, companies can mitigate environmental impact while improving efficiency. For instance, IoT sensors and advanced analytics optimise transportation, reducing fuel usage and greenhouse gas emissions. Additionally, transitioning to paperless operations minimises waste and streamlines document management, showcasing a commitment to environmental stewardship.
Benefits
for pharmaceutical manufacturers
The adoption of Industry 4.0 technologies in pharmaceutical manufacturing brings about a multitude of benefits, ranging from enhanced operational efficiency to improved patient outcomes.
1. Reduced lead times and costs
Real-time visibility and predictive analytics enable manufacturers to optimise production schedules, reducing lead times and minimising production costs. This efficiency is crucial in meeting urgent demands for critical medications.
2. Improved quality control
IoT sensors and data analytics contribute to improved quality control by continuously monitoring production parameters. Any deviations from set standards trigger immediate alerts, allowing for swift corrective actions and ensuring the production of high-quality pharmaceuticals.
3. Enhanced regulatory compliance
The pharmaceutical industry is subject to strict regulatory requirements. Industry 4.0 technologies aid in maintaining compliance by providing a transparent and auditable record of every step in the manufacturing and distribution process.
4. Supply chain resilience
The ability to anticipate and respond to disruptions enhances the resilience of the pharmaceutical supply chain. Whether facing unexpected demand surges, supply chain interruptions, or regulatory changes, Industry 4.0 technologies empower manufacturers to adapt swiftly.
Challenges and opportunities
While the adoption of Industry 4.0 technologies offers immense benefits, pharmaceutical companies must navigate certain challenges on their digital transformation journey. Data privacy and security concerns necessitate robust cybersecurity measures to safeguard sensitive information and intellectual property. Moreover, interoperability issues may
Industry 4.0 ushers in a new era of demand fore casting by leveraging artificial intelli gence (AI), machine learning algorithms, and big data analytics.
arise due to the heterogeneity of legacy systems and disparate data formats across supply chain partners. Nonetheless, by addressing these challenges proactively and fostering a culture of innovation, pharmaceutical manufacturers can capitalise on the opportunities afforded by Industry 4.0 to drive sustainable growth and competitive advantage.
Siddharth Singhal, an accomplished business leader with an MBA from Manchester University, stands at the helm of Vibcare Healthcare Pvt Ltd as the Co-Founder and Managing Director. With a passion for innovation and a profound vision to revolutionise the pharmaceutical industry, Siddharth has been a driving force behind the company's remarkable journey.
Insights by Siddharth Singhal, Cofounder & MD of Vibcare Pharma
Siddharth Singhal is a visionary leader and co-founder of Vibcare Pharma, a top pharma PCD franchise company. With a passion for revolutionising the pharmaceutical industry, Siddharth's strategic leadership drives Vibcare's growth, fosters innovation, and ensures a strong ethical foundation. Under his guidance, Vibcare continues to achieve unprecedented success, delivering value to customers while upholding its core values.
Conclusion
Industry 4.0 technologies are reshaping the pharmaceutical supply chain, driving efficiency, transparency, and resilience. By harnessing IoT, blockchain, AI, and advanced analytics, pharmaceutical companies can optimise operations, enhance decision-making, and deliver value to patients and healthcare providers. As the industry continues to embrace digital transformation, proactive collaboration, and strategic investments in technology will be essential for staying ahead in an increasingly complex and dynamic landscape.
Industry 4.0-Revolutionising Hot Melt Extrusion
Smart manufacturing for enhanced efficiency and quality
Incorporating Industry 4.0 into Hot Melt Extrusion (HME) processes offers improved efficiency and quality in pharmaceutical manufacturing. Automation, data exchange, and AI enablereal-time optimisation, while digital twins foster innovation. This synergy promotes smarter manufacturing, ensuring competitiveness and high-quality products.
1 Department of Pharmaceutics, Sri Adichunchanagiri College of Pharmacy, Adichunchanagiri University
Hot Melt Extrusion (HME) has emerged as a groundbreaking technology in numerous industrial sectors, with a particular emphasis on its transformative impact within pharmaceutical research and manufacturing. Through its seamless integration with Industry 4.0 principles—namely, automation, data exchange, and Artificial Intelligence (AI)—HME has redefined the landscape of production processes.
In recent years, HME has ascended as a cornerstone technology for drug delivery applications, owing to its automated and cost-effective scale-up properties. By significantly reducing labour costs and capital investment, HME has become an indispensable tool for pharmaceutical scientists across the globe. Moreover, the ongoing advancements in HME, bolstered by a comprehensive understanding of material science and process engineering, empower pharmaceutical scientists to craft highly efficient and durable products.
One of HME's most noteworthy achievements lies in its ability to address prevalent challenges faced by pharmaceutical scientists, including issues related to poor water solubility, bioavailability, and the physical and chemical stability of active pharmaceutical ingredients (APIs), thereby surpassing conventional techniques. At the heart of HME lies the extrusion process, a fundamental element that propels its efficacy. This process commences with the meticulous preparation of solid raw materials, encompassing polymers and pharmaceutical compounds, which undergo melting within an extruder barrel. Through a meticulously controlled environment, these materials are uniformly mixed to form a molten mass. Following this stage, the molten material undergoes a cycle of cooling and solidification, culminating in further refinement processes such as cutting and finishing to yield the final product. Regulatory bodies, recognising the transformative potential of HME, fervently advocate for the implementation of Quality by Design (QbD) and Process Analytical Technology (PAT) within this domain. These strategic tools, which include Raman and Near-infrared (NIR) spectroscopy, play a pivotal role in enabling real-time quality evaluation and process monitoring, thus harmoniously aligning with the principles of Industry 4.0.
The integration of HME with Industry 4.0 principles— automation, AI, and data exchange—has transformed pharmaceutical manufacturing, driving realtime process monitoring and continuous optimisation.
Smart Manufacturing technology in HME Process
Smart manufacturing technologies are transforming HME, revolutionising efficiency, quality control, and productivity in pharmaceutical research and manufacturing. Machine Learning (ML), among these technologies, offers significant benefits in HME processes but also presents challenges, particularly concerning model transferability. Achieving good transferability, as observed with Partial Least Squares (PLS) and Linear Discriminant Analysis (LDA) models, ensures effective moni-
toring of filler particle size across various grades of Polylactic acid (PLA). Similarly, soft sensor models exhibit promising transferability in monitoring melt viscosity across diverse materials and equipment. Challenges arise, however, when monitoring physicochemical properties like API content, where direct relationships with real-time process measurements may be limited. Despite these challenges, integrating machine learning into HME holds promise for optimising process parameters, predicting product quality, and facilitating real-time decision-making in pharmaceutical manufacturing. Ongoing research endeavours aim to overcome these challenges, fostering broader adoption of machine learning in HME and industrial processes.
Digital twin is also one of the smart technologies related to HME process where HME utilises co-rotating twin screw extruders like blending Active Pharmaceutical Ingredients (API) and polymers. This process occurs in heated barrels, where mechanical and thermal energy facilitates polymer plasticisation around API particles. Temperaturecontrolled zones ensure uniformity, with screw configurations for feeding,
Figure 1. Revolutionising HME
Revolutionising Hot Melt Extrusion
kneading, dispersion, conveying, and metering. Gravimetric feeders ensure precise material delivery. HME is a wellestablished process increasingly adopted in pharmaceutical continuous manufacturing. It achieves efficient mixing, enhancing product uniformity and bioavailability, with advantages including solvent-free processing, fewer steps, lower costs, scalability, and consistency. Challenges include compound degradation and recrystallisation, hindering continuous processing adoption due to investment costs and changeovers. Digital twin technology offers a solution by creating virtual replicas of HME processes. This enables real-time monitoring, predictive modelling, and scenario analysis, optimising manufacturing and predicting API manufacturability. Ultimately, digital twins enhance efficiency, quality control, and decision-making in pharmaceutical manufacturing, aligning with industry innovation goals.
The combination of HME with 3D printing, particularly Fused Deposition Modelling (FDM), offers a promising method for producing controlled-release tablets. In this research, a co-rotating
twin-screw extruder with 11-mm diameter screws was utilised to create fused filaments suitable for 3D printing. With precise temperature control facilitated by the extruder's design, which includes an L/D ratio of approximately 40 and eight electrically heated zones, the study aimed to assess the suitability of various pharmaceutical polymers for 3D printing. By integrating FDM-based 3D printing with HME, the researchers aimed to develop controlled-release tablets with customised drug delivery profiles [Figure 1]. Furthermore, comparing the drug release profiles of 3D printed tablets to conventional tablets provided valuable insights into the performance of 3D printed pharmaceutical formulations, highlighting the potential of this approach for advancing pharmaceutical manufacturing and drug delivery systems. (Figure 1)
Enhancing efficiency through automation and robotics in HME
The synergy between HME and Industry 4.0 enables real-time data exchange and process optimization, resulting in superior product quality and a more resilient pharmaceutical manufacturing process.
Automation and robotics play a crucial role in advancing HME processes, particularly in pharmaceutical manufacturing. In HME, automation technologies streamline the production process by precisely controlling parameters such as temperature, pressure, and material feeding. This automation ensures consistent product quality and reduces the need for manual intervention, enhancing efficiency and productivity. Robotic systems complement automation by performing various tasks within the HME workflow. These tasks include material handling, loading and unloading of raw materials, and operating the extruder itself. Robots execute these tasks with speed and accuracy, minimising errors and optimising the production process. Moreover, automation enables real-time monitoring and control of the HME process. Advanced sensors and monitoring systems continuously collect data on key parameters, allowing for immediate adjustments to maintain optimal conditions and product quality. This real-time feedback loop ensures
that the HME process operates at peak efficiency. Furthermore, automation facilitates seamless integration with other manufacturing processes, such as downstream packaging and quality control. Automated systems can transfer extruded products to packaging lines, conduct quality inspections, and ensure batch traceability, thereby streamlining the entire production workflow. Overall, automation and robotics in HME enable pharmaceutical manufacturers to achieve higher levels of productivity, reduce costs, and improve product quality. These technologies pave the way for advanced manufacturing techniques, driving innovation and competitiveness in the pharmaceutical industry.
Technical challenges
Several factors significantly influence HME implementation rates.
Figure 2. Technical challenges for implementation of HME
Technological complexity, constituting 18 per cent, encompasses the intricacies of HME machinery and the sophistication of control systems required for precise operations. Algorithm development and tuning, also at 18 per cent, are crucial for optimising process parameters to ensure efficient material flow and consistent product quality [Figure 2]. Testing and validation, making up 15 per cent, are paramount for verifying product integrity and adherence to regulatory standards, essential for market approval and consumer safety. These factors collectively shape the landscape of HME adoption, highlighting the importance of technological advancements, process optimisation, and regulatory compliance in driving its widespread implementation. (Figure 2)
Future Perspectives and Conclusion
Looking ahead, the integration of Industry 4.0 principles into Hot Melt Extrusion (HME) processes signals
a significant shift towards smarter manufacturing for improved efficiency and quality. Combining automation, data exchange, AI within HME holds immense potential for revolutionising pharmaceutical manufacturing and beyond. Industry 4.0 technologies will empower HME processes to become more autonomous and adaptable, continuously optimising parameters in real-time to ensure consistent product quality and operational efficiency. AI algorithms will analyse extensive data sets, offering valuable insights into process optimisation and predictive maintenance, ultimately reducing downtime and expenses. Moreover, the introduction of digital twins will enable the creation of virtual replicas of HME equipment and processes, facilitating simulation, optimisation, and scenario analysis. This digital representation of the production environment will expedite prototyping and foster the development of new formulations, accelerating innovation in drug delivery
Dr. N Raghavendra Naveen, an esteemed Associate Professor in the Department of Pharmaceutics at Adichunchanagiri University, brings a decade of rich academic experience highlighted by groundbreaking research. With 67 publications to his credit, 53 indexed in leading databases like Web of Science and Scopus, his scholarly impact is truly global. Over the last three years, he has authored 40 publications, achieving an impressive cumulative impact factor of 148.776. His contributions extend to 7 book chapters and the authorship of a book with renowned publishers such as CRC Press, Wiley, Taylor and Francis, and Elsevier. Holding 4 patents, he demonstrates a deep commitment to innovation and practical solutions. Dr. N Raghavendra Naveen's research excellence earned him a prestigious research grant of 15 lacs under the KFIST level 1 from VGST and has garnered over 600 citations, showcasing his profound influence. Additionally, he plays a key role in academic leadership as a Member of the Board of Studies, while guiding 11 M.Pharma projects and mentoring Ph.D. and M.Pharma students, nurturing their research skills and academic growth.
systems. As pharmaceutical companies transition towards continuous manufacturing, Industry 4.0-enabled HME processes will play a central role in meeting the industry's evolving demands. Continuous monitoring and control, facilitated by advanced sensors and analytics, will ensure compliance with regulatory requirements and enable realtime product release. In summary, the integration of Industry 4.0 principles into HME processes stands poised to transform hot melt extrusion into a smarter, more efficient, and agile manufacturing process. By embracing smart manufacturing technologies, pharmaceutical companies can strengthen their competitiveness, drive innovation, and deliver top-tier products to satisfy the needs of patients and regulatory bodies alike. The evolution of HME lies at the intersection of technology and pharmaceutical science, where smart manufacturing illuminates the path towards enhanced efficiency and quality in drug delivery.
Dr. Prakash Goudanavar is a distinguished academician and researcher with over 18 years of teaching experience and 6 years in the pharmaceutical industry. He serves as Vice Principal, Professor, and Head of the Department of Pharmaceutics & Regulatory Affairs at Adichunchanagiri University. Dr. Goudanavar has an impressive publication record with 65 national and 33 international papers, and he has authored three textbooks. He has guided 7 Ph.D. scholars to completion and mentors 6 more, while also fostering the research development of 45 postgraduate students. He has received several awards for his research and presentations and actively contributes to scientific journals as a reviewer and board member. He has chaired scientific sessions and served as a keynote speaker for numerous international conferences.
Srikruthi KS, a driven Postgraduate scholar at Adichunchanagiri College of Pharmacy, excels in pharmaceutics under Dr. N Raghavendra Naveen's mentorship. Scholarly impact spans academic literature and research, with two standout articles in Oral Oncology Reports and IJPER. Specialising in Nanotechnology, Optimisation, and Polymer Technology, work embodies a meticulous approach to advancing pharmaceutical sciences. Passionate about addressing healthcare challenges, strives to inspire future researchers through innovative contributions in pharmaceutics.
Personalised Medicine A new era for women's health
The rise of personalised medicine heralds a paradigm shift in women's healthcare. This review explores how advancements in genomics, biomarkers, and targeted therapies are tailoring treatments for common and complex women's diseases, including breast cancer, endometriosis, and autoimmune disorders. We discuss the potential benefits and challenges associated with personalised approaches and their impact on improving treatment efficacy and patient outcomes..
Mukhabbatkhon Mirzaolimova, Pharmaceutical Scientist, Formulation science, Drug Analysis, and Regulatory Affairs
Women's healthcare has been based on a ‘one-size-fits-all’ strategy for many decades. Age, symptoms, and general health were frequently taken into account while choosing a course of treatment, but individual differences in biology and disease processes were rarely taken into account. Personalised medicine, on the other hand, is changing healthcare and providing a new perspective on women's health. This article examines how developments in targeted medicines, biomarkers, and genetics are changing the way we identify, manage, and prevent common and complicated diseases that impact women. This essay will examine the possible advantages and difficulties of personalised medicine strategies as well as
how they might enhance patient outcomes and treatment efficacy.
Genomics: Unveiling the Blueprint of Health and Disease
A key component of personalised therapy is genomics. Through genetic analysis, differences (mutations) in genes that might affect a person's reaction to a therapy or make them more susceptible to certain diseases can be identified. For example, it is well known that mutations in the BRCA1 and BRCA2 genes greatly increase a woman's risk of breast cancer. When these mutations are found, preventative treatments including genetic counselling and prophylactic surgery can be used, as well as early identification and risk classification.
Biomarkers: Guiding Diagnosis and Treatment Decisions
Biological indications known as biomarkers provide important information about a patient's state of disease. These compounds may be found in tissues, blood, or imaging results. Utilising developments in biomarker research, personalised medicine creates focused treatments with low side effects.
One biomarker present in some breast tumours, for instance, is the HER2 receptor. The results of patients with breast cancer who have HER2-positive tumours have improved dramatically thanks to trastuzumab, a targeted treatment created especially to inhibit HER2 activity. Similar to this, hormone profiles can influence the choice of treatment for breast tumours that are hormone receptor positive, opening the door to treatments like tamoxifen that target estrogen receptors.
Tailoring Therapies: Precision Medicine for Women's Health
Personalised medicine goes beyond diagnostics. It gives medical practitioners the ability to customise treatment regimens according to each patient's own genetic and molecular profile. This strategy has a number of benefits:
• Enhanced efficacy: By focusing on the fundamental causes of a certain person's illness, personalised medicines have the potential to increase success rates and enhance disease control.
• Lessened side-effects: Many patients experience avoidable side-effects from traditional treatment methods because of their wide-ranging impacts. Personalised treatments reduce this risk by concentrating on certain disease pathways.
• Improved risk stratification: Personalised medicine enables proactive treatments and preventative actions for women
who are more susceptible to specific diseases by detecting genetic predispositions.
Personalized Medicine in Action:
Examples from Women's Health
There are several facets of women's health where customised therapy has the potential to be revolutionary:
• Breast cancer: As previously indicated, targeted treatments such as trastuzumab for HER2-positive tumours and genetic testing for BRCA mutations are prime examples of tailored techniques in the treatment of breast cancer.
• Endometriosis: Personalised medicine has the potential to treat endometriosis, a chronic illness that results in infertility and pelvic discomfort. Finding particular biomarkers linked to certain endometriosis subtypes can help tailor treatment regimens, which may enhance symptom management and improve reproductive results [4].
• Autoimmune disorders: Women are disproportionately affected by a number of autoimmune disorders, including lupus and rheumatoid arthritis. Strategies for personalised medicine that make use of certain biomarkers can help with risk assessment, early diagnosis, and the choice of tailored immunomodulatory treatments for better disease control [5].
Challenges and Considerations in
Personalized Medicine
Personalised medicine for women's health has enormous promise, but it also has certain drawbacks.
• Cost: Targeted medicines and genetic testing can be costly, which some patients may find unaffordable. To guarantee that everyone has fair access to these developments, healthcare systems must address insurance coverage and cost-effectiveness.
• Data security and privacy: A lot of patient data is necessary for personalised medication. Strong data security protocols and unambiguous ethical standards are necessary to safeguard
patient confidentiality and stop the improper use of genetic data.
• Limited access to tests and therapies: Although progress is being made, many women's health concerns still lack tailored treatment methods. To increase the number of tests and tailored medicines available, more funding and research are required.
The Future of Women's Health: A Personalized Approach
Personalised medicine offers a revolutionary road map for women's health in the future. Through the utilisation of genomes, biomarkers, and targeted medicines, scientists may progress toward a future in which healthcare is customised to the individual biology and disease profile of every woman. It will be essential to tackle the obstacles related to expenses, confidentiality of data, and accessibility of diagnostics and treatments to guarantee fair participation and optimise the advantages of customised medicine for every woman.
Empowering Women in Personalized Medicine
Women's participation in clinical trials and research studies is essential to advancing scientific understanding of the ways in which illnesses impact women differently. Due to historical underrepresentation of women in clinical research, there is now a knowledge gap on how treatments and drugs affect the female body. This can have serious repercussions since women may be more susceptible to adverse effects or have different treatment outcomes than males.
Researchers may:
• Close the gender gap in medical knowledge by increasing the number of women participating in research. This is because data from a more representative population will provide scientists a more thorough understanding of how illnesses appear, develop, and react to therapy in women.
• Create more effective treatments: Studies focusing on the unique biology of
women may result in the creation of medicines with fewer adverse effects and greater efficacy for females.
• Empower women to make knowledgeable healthcare decisions: Increasing women's involvement in research cultivates a feeling of agency and gives them the ability to make knowledgeable healthcare decisions based on the most recent developments in customised medicine.
Conclusion
Beyond the one-size-fits-all paradigm, personalised medicine provides a novel approach to women's health. Healthcare may be customised to a woman's specific biology by utilising genomes, biomarkers, and targeted medications. This might result in faster diagnosis, more effective treatments, and fewer side effects. There are still issues with cost, data privacy, and restricted test availability, but further research and solutions to these problems are needed. Increasing the number of women in medical research is essential to narrowing the gender gap in medical knowledge and creating more potent medicines. The field of personalised medicine has great potential to improve the health of women in the future by enabling them to actively participate in their own healthcare decisions and get specialised care.
References are available at www.pharmafocusasia.com
Mukhabbatkhon Mirzaolimova is a Pharmaceutical Scientist with expertise in formulation science, drug analysis, and regulatory affairs. She is currently working at the Pharmaceutical Technical University. Her research focuses on personalized medicine approaches for women's health, leveraging her background in protein target discovery and advanced drug delivery systems.
AUTHOR BIO
Integrating Artificial Intelligence to Enhance Advanced Therapy Medicinal Product Manufacturing in Academic Medical Centers
The paper discusses integrating Good Manufacturing Practices and Artificial Intelligence within hospitals to enhance Advanced Therapy Products production. It addresses challenges in complying with standards, improving efficiency, and overcoming resource limitations. The focus is on AI's role in streamlining therapy development while ensuring safety, quality, and regulatory compliance.
Cristobal Aguilar-Gallardo, Ana Bonora-Centelles
La Unidad de Terapias Avanzadas. Instituto Investigación Sanitaria La Fe. Av.
Advanced therapy medicinal products (ATMPs) are innovative therapeutic approaches that modify patients' genes or cells to treat disease. 'Academic advanced therapy' refers to those ATMPs developed within hospitals, in contrast to industrial development by pharmaceutical companies. Academic institutions play a unique role in translating discoveries into practical therapies, but face challenges establishing
in-house good manufacturing practices (GMP) facilities. This article explores how emerging technologies like artificial intelligence (AI) could enhance academic ATMP manufacturing.
Key Advantages of Hospital Facilities
On-site hospital facilities provide major advantages for autologous/allogenic ATMP manufacturing. Proximity allows rapid production and direct delivery to patients, avoiding transportation risks. Alignment with clinical schedules enables flexible batch planning accommodating dynamics or urgent needs. After demonstrating safety in trials, hospital facilities can continue patient access through expanded programs. However, hospitals face spatial constraints retrofitting manufacturing into existing infrastructure. Strategic facility design, isolators, and closed systems can limit cleanroom requirements. Multi-product facilities segregating therapies like viral vectors or gene-modified cells facilitate flexibility within the space available.
Implementing GMPs in Hospitals
Implementing pharmaceutical GMPs within health centres poses challenges. Hospital staff lack familiarity with GMP documentation, training, and oversight. Quality systems in healthcare focus on services, unlike GMPs ensuring consistent product quality. Extensive GMP training and specialised education are essential to bridge this gap. Cross-departmental coordination fosters awareness of GMPs for staff unfamiliar with pharmaceutical standards. A risk-based approach concentrates quality efforts on critical factors affecting products and patients, facilitating GMP translation while ensuring safety.
Investment in Infrastructure and Personnel
Considerable investment is required to establish in-house facilities, including cleanrooms, manufacturing equipment, and recruiting qualified personnel.
Discover how artificial intelligence is transforming the production of ATMPs within academic medical centers, enhancing efficiency, consistency, and quality while overcoming resource limitations and regulatory challenges.
Cell therapy process development, GMP manufacturing, quality control, quality assurance, and regulatory experience is crucial. Strategies maximising operator productivity allow hospitals to meet clinical demand within personnel constraints. Financial planning and multi-department coordination are critical for transitioning therapies from research to clinical use.
Emerging Technologies for Hospital Facilities
Advanced technologies like process analytical technology (PAT), continuous manufacturing, and AI present opportunities to enhance hospital capabilities within ever-present constraints. PAT employs in-line monitoring and automated feedback control to optimise manufacturing. By tracking parameters like pH, nutrients, metabolites, and cell growth in real- time, bioreactors can be automatically controlled, reducing manual operations while ensuring consistency between batches. Continuous manufacturing in an uninterrupted flow, rather than individual batches, can increase productivity. AI and machine learning facilitate analysing the vast data from PAT monitoring to identify patterns, predict deviations, and guide actions. AI automation of manufacturing tasks, sample tracking, contamination control, and batch record review increases oversight. Altogether, these technologies
allow hospitals to boost productivity and quality within limited space, time, costs, and staffing.
Analysis of AI's Role and Impact
AI provides key advantages for academic ATMP manufacturing:
Enhanced Process Monitoring: AI algorithms enable improved real-time tracking and oversight to maintain consistency and quality. Multivariate models integrate sensor data streams to identify relationships between parameters and product attributes. This allows earlier deviation detection and automated corrections compared to periodic manual sampling.
Automation of Tasks: Increases efficiency, precision, scalability, and reduces workload through robotic automation of repetitive activities. AI scheduling of maintenance procedures minimises disruptions. Environmental monitoring prevents contamination. Overall, AI-driven automation addresses hospitals’ spatial and staffing constraints.
Dynamic Control: Adaptive AI control systems recalculate optimal parameters in real time based on multivariate data. This maximises batch-to-batch consistency and robustness compared to basic feedback loops, critical for variable autologous products. Hybrid models combine mechanistic and machine learning algorithms for sophisticated adaptive optimisation tailored to each batch.
Data Management: AI techniques like natural language processing and dimensionality reduction effectively consolidate the disparate data generated throughout manufacturing and clinical care. This contextualised data enhances process comprehension and real-time decision making.
Systems Biology Integration: AI modelling of complex biological relationships from multi-omics data provides key insights, furthering predictive and personalised medicine.
However, AI integration also faces challenges: data privacy risks, high
upfront investment, limited data availability in early stages, scarcity of qualified personnel, and regulatory ambiguity. Overall, AI offers transformative capabilities to enhance process monitoring, automation, control, and data analysis to produce personalised ATMPs. But it requires extensive oversight across technology, data, people, and regulations for responsible adoption.
Scaling Gene and Cell Therapies from Research to Clinic
A major obstacle in developing gene and cell therapy ATMPs is scaling from small research batches to the larger volumes needed for patient treatments. Differences in growth kinetics and product quality attributes often emerge during scale-up as research protocols fail to directly translate. Traditionally, extensive empirical optimisation was required to adapt processes. AI technologies can accelerate scale-up by revealing stress factors affecting cells through integrating multivariate data. AI control systems then dynamically optimise parameters based on continuous metabolite monitoring. Cellular metabolism provides key indicators of gene engineering effects and culture environment. AI modelling of highthroughput metabolomics data clarifies metabolic shifts responsible for reduced clinical-scale productivity. Tracking disrupted pathways highlights target areas for supplementation or gene modifications to restore favourable metabolism. Overall, AI-driven process understanding, and control maintain cultures in ideal metabolic regimes, enhancing robustness during scale-up.
Enhanced Process Monitoring and Control with AI in ATMP Manufacturing:
The integration of AI into the monitoring processes within ATMP facilities marks a significant advancement in maintaining product quality and consistency. AI algorithms have proven to be instrumental in real-time tracking and oversight, enabling the early detection of deviations and ensuring that each batch meets the
Explore the future of personalized medicine with AI-driven advancements in hospital-based ATMP production, ensuring real-time control, superior data management, and improved patient outcomes through innovative automation and dynamic process optimisation.
stringent quality standards required. For instance, in certain academic centres, AI-driven systems have successfully identified patterns in cell growth and metabolite concentrations, allowing for timely adjustments and reducing the incidence of batch failures. These systems integrate sensor data streams, employing multivariate models to analyse relationships between various parameters and product attributes. This capacity for real-time analysis surpasses traditional periodic manual sampling, allowing for more dynamic and responsive process control.
Addressing Manufacturing Challenges through AI-driven Automation:
Automation, driven by AI, is transforming ATMP manufacturing by improving efficiency, accuracy and scalability. Robotic systems and artificial intelligence software have been deployed to automate repetitive tasks such as maintenance scheduling and environmental monitoring, significantly reducing manual workload and addressing spatial and staffing limitations prevalent in hospital environments. By automating these supplementary manufacturing operations, including supply chain management and batch record review, AI-driven systems help academic medical centres to overcome the challenges associated with limited space and human
resources, ensuring more streamlined and error-free operations.
Dynamic Control Systems Tailored by AI for ATMP Production:
Dynamic control systems, facilitated by AI, represent a pivotal innovation in the manufacturing of ATMPs, particularly autologous therapies. These adaptive control systems leverage real-time data to continuously recalculate and optimise bioprocess parameters, accommodating the inherent variability between individual patient samples. By employing hybrid models that combine traditional mechanistic understanding with machine learning algorithms, AI permits the development of sophisticated control strategies. These strategies adapt to the unique characteristics of each batch, improving the consistency and quality of the final product. This approach exemplifies how AI can address one of the most challenging aspects of ATMP manufacturing: ensuring batch-to-batch consistency despite biological variability.
Advanced Data Management for Informed Decision-Making:
Effective management of data is vital in ATMP manufacturing, due to the volume of information generated across different stages of the production processes. AI technologies, employing techniques such as natural language processing and dimensionality reduction, which play a key role in organising and interpreting this diverse data. By converting datasets into insights AI aids in making better decisions improving manufacturing efficiency and enhancing patient care quality. This robust data management is essential for the personalised nature of ATMPs, ensuring that each therapy is optimally tailored to the individual patient’s needs.
Leveraging Systems Biology through AI for Personalised Medicine:
Thru AI application in the modelling of complex biological systems offers deep and well-structured insights into
Integrated AI-Enhanced ATMP Production and Healthcare Ecosystem in Academic Medical Centers
the interactions and dynamics within biological networks, significantly advancing the field of personalised medicine. By analysing multi-omics data, AI models have the ability to discover key connections and pathways which can help in shaping better and personalised therapies. For instance, AI-driven systems biology approaches have been instrumental in identifying biomarkers and predicting patient responses to specific therapies, thereby enhancing the personalisation and effectiveness of ATMPs.
Key Elements for AI Integration in ATMP Manufacturing
Effective AI integration requires addressing several key elements, including process monitoring, automation, dynamic control, data management, and systems
biology. Each plays a crucial role in overcoming the specific challenges of ATMP manufacturing.
Process Monitoring: The variability between autologous cell samples makes achieving batch consistency difficult. Conventional periodic sampling is limited. AI enables improved real-time oversight through algorithms continuously analysing patterns in data from advanced sensors and probes. This allows earlier deviation detection and automated adjustments to maintain quality.
Automation: AI automates supplementary manufacturing operations like supply chain management, tracking, maintenance, contamination control, and batch record review. This enhances precision, oversight, efficiency, costeffectiveness, and reduces workload.
Adaptive AI systems recalculate optimal parameters in real-time based on multivariate data analysis, maximising consistency between autologous batches compared to basic feedback loops. Hybrid models combine mechanistic and machine learning algorithms for sophisticated optimisation tailored to each batch.
Data Management: AI techniques consolidate disparate datasets from manufacturing, clinical care, and other sources into contextualised knowledge enhancing process understanding and decision-making. This is critical for synthesising the data underlying personalised therapies.
Systems Biology: AI modelling of multi-omics data provides key insights into complex biological relationships,
Dynamic Control:
furthering predictive and personalised medicine.
Advanced Control Systems: Building upon other AI capabilities can help implement sophisticated adaptive and model predictive control algorithms that continuously re-optimise parameters based on hybrid mechanistic and machine learning models customised to each batch.
Responsible AI Adoption
The responsible adoption of AI in ATMP manufacturing necessitates a synchronised approach across technology, data, personnel, and regulations. Ensuring data integrity, training staff in AI applications, and adhering to regulatory standards are essential for harnessing AI's full potential while maintaining patient safety and product quality. Thus, successful integration relies on synchronised management across technology, data, people, regulations, and healthcare objectives:
Data: Quality data and governance systems ensuring privacy are essential before launching initiatives. Data provenance, security, and integrity are critical qualifiers for infrastructure.
People: Personnel fluent in AI applications and limitations are crucial, especially for regulated manufacturing. Cross-functional collaboration between domains promotes effective adoption. Academic curriculums must also evolve to integrate data science.
Regulation: Continuous communication, documented risk management, and quality focus help ensure regulatory compliance.
Healthcare: Product quality and safety improvements must be prioritised over purely economic incentives. AI can enhance consistency when appropriately designed.
Conclusions
ATMPs stand at the lead of innovative treatment modalities. However, their manufacturing presents challenges significantly diverged from traditional pharmaceutical paradigms. The advent of emerging technologies, notably
Process Analytical Technology, continuous production, and AI, offers new pathways to overcome Hospital-based facilities limitations. Byenhancing process monitoring, automating routine tasks, enforcing dynamic control, and streamlining data management, AI technologies are pivotal in transforming ATMP manufacturing into a more efficient, and reliable process. Nonetheless, the integration and responsible AI adoption within ATMP manufacturing necessitate a balanced and synchronised approach, considering various facets such as technological innovation, data integrity, human capital, regulatory compliance, and overarching healthcare objectives. Collaborative efforts among biotechnologists, AI specialists, clinicians, and regulatory bodies are essential to ensure that AI is implemented responsibly and effectively. Employing the full potential of AI, we can significantly advance the field of regenerative medicine, translating groundbreaking scientific discoveries into life-enhancing treatments. This concerted effort mitigates the inherent challenges associated with ATMP production but also maximises the therapeutic impact, delivering on the promise of personalised healthcare and improving patient outcomes.
Key words: Bioprocess; Advanced Therapy Medicinal Products (ATMPs); Artificial Intelligence (AI); Cell and Gene Therapies; Process Analytical
Technology (PAT); Personalized Medicine; Data Integration and Analysis.
References are available at www.pharmafocusasia.com
AUTHOR BIO
Cristóbal Aguilar Gallardo, with over 15 years in the biotechnology and biomedicine field, has specialized in bioprocess engineering and cellular production, focusing on stem cell bioprocessing, tissue engineering, and cell and gene therapy. His work has led to innovative cell therapies and GMPcompliant processes. With numerous publications and three patents, Dr. Aguilar is a leading figure, currently heading cellular manufacturing at the Advanced Therapy Unit in the research institute La Fe.
Ana Bonora has over 15 years of experience in biotechnology and regulatory compliance, specialising in GMP regulations and ATMP development. Since 2017, as lead of the Advanced Therapy Unit at the research institute La Fe, she has significantly advanced the field of cell and gene therapies. Her extensive expertise in quality management and cleanroom operations has positioned her as a key figure in biopharmaceuticals, contributing to innovative therapeutic approaches.
1. In your opinion, what are some factors that have shifted the consumer preference towards home-based treatments for chronic conditions?
Looking at the global healthcare landscape there is a clear drive from hospital to home. The home-based healthcare market is expanding, driven by patient demand for more convenient treatment options. Patients are interested in using advanced technologies like self-injectable drugs and on-body drug delivery systems (OBDS)– and are even excited about the convenience and autonomy they could gain by administering the medications themselves rather than having to travel to a doctor’s office or infusion centre.
Pharmaceutical innovations have significantly improved the treatment of many diseases, including cancer and chronic inflammatory diseases such as rheumatoid arthritis. By attacking the root biological underpinnings of disease, these treatments have greatly improved the prognosis for patients, bringing symptoms well under control, and in some cases, extending lives. But patients often must travel to hospitals or clinics to have these
PHARMACEUTICAL TRENDS
In this interview, Nilesh Shah shares his insights into the shift for more hospital to home treatment options for chronic conditions, and how health care providers and pharmaceutical companies can collaborate to address any potential gaps to enhance patient care at home.
Nilesh Shah, Vice President and General Manager of Emerging Markets, West Pharmaceutical Services
medications infused intravenously or injected by health care providers (HCPs) an onerous process that can be timeconsuming, costly, and inconvenient. That’s why both patients and HCPs express a strong need for alternative ways of receiving these medicines.
Among the most innovative drugdelivery options are self- administered injectable medicines and OBDS. These technologies enable patients to administer the medicines they need themselves, whether they’re at home, work or on vacation.
For a patient suffering from chronic conditions, home-based treatments offer comfort and familiarity that hospital or institutional treatments often cannot provide. This is a very personal decision that some patients may make, in hopes that they can feel more comfortable at home, and in turn, develop a better mental outlook outside of stressful hospital settings. For some patients, they may also enjoy a faster recovery trajectory in a comfortable, familiar place.
Thanks to the advancement of pharmaceutical technologies, over the years, there have been a number of improvements in therapies and medications that can be self-administered by a caregiver at home or even the patient themselves. Sometimes, these technologies also come at a lower cost than in the hospitals, which is a key driver of this trend.
For example, glucose monitoring devices for chronic diabetes patients have improved to offer real-time measurements in continuous glucose monitoring devices that can be worn for longer time periods through a sensor under the skin. This sensor transmits real-time data to a handheld monitor, reducing the chances of long-term complications associated with diabetes. These reduce any need for the traditional prick of the finger to draw blood or for patients to seek out hospital or out-of-home support to help manage their blood sugar levels.
This greater access to devices and quality treatments at home developed by
pharma companies have thus empowered more patients to select home-based care for chronic conditions.
2. What role does technology play in enabling this transition from hospital to home in patient care?
From providing new systems to track medication and health statuses to establishing a direct line to hospital systems in case of emergencies, technology has played a pivotal role in enabling this transition from hospital to home. Simultaneously, finding effective ways to deliver new technologies to patients is equally important to innovating them. For example, hospitals must have access to these tools to supply them to patients going home and have adequately trained staff to teach patients and their caregivers on using them. User-friendly interfaces and other technical integrations with existing healthcare systems must also be applied.
Our industry is getting increasingly more sophisticated with new technology and connected medical devices. This requires leadership that dares to be different and building innovation around these unmet needs. This means leaders who dare to challenge the norms and are unafraid of pushing for more patients to be able to access what they need through partnerships and collaborations.
Aside from the technologies that drive the innovation behind medical devices and therapies, technology has allowed for the rise of telemedicine where patients can seek medical help from home through virtual check-ups and consultations. These devices are also able to connect to one’s smartwatch and health apps, helping patients track their progress and care. Furthermore, they act as medication management systems to ensure that one takes their medication on time.
There are also other forms of technology that can be tapped into by doctors and medical officers to help analyse patient data, even while they are home. Any needed intervention is then quickly flagged.
COVID-19 highlighted many benefits of home-based healthcare, as well as the growing pressures on our global healthcare systems for adaptability to changes. Technologies such as smart pill containers and wearable sensors quickly became a lever that allowed more patients to lean into taking care of themselves at home.
Of course, the effectiveness of technology in patient care still remains dependent on patients’ acceptance and comfort with these tools, as well as the integration with local healthcare data and systems. If patients can trust technology and use its integrations properly, it can help bring them closer to being in touch with their health rather than just a tool that operates independently without patient interaction.
3.
Where do pharmaceutical companies come into the picture when it comes to ensuring the effectiveness of homebased healthcare?
There is little doubt that home-based healthcare depends very greatly on pharmaceutical companies. From the development of the medication itself to the packaging and development of medical devices, pharmaceutical companies, and their packaging partners can contribute greatly to both medical professionals in their work and the care and quality of life for patients that medical professionals serve.
Most pharmaceutical companies are in the drug development space, where active and robust research is accompanied by the formulation of medications that can be safely and effectively administered at home. These companies typically work closely with medical
professionals and pharmaceutical packaging companies to ensure the effectiveness and safety of the treatments.
For home-based healthcare especially, the key lies in being able to deliver medicine easily and with as little safety risk for the patient as possible. Hence, it is also the responsibility of pharmaceutical companies to be on top of global, regional, and local legislation and regulatory requirements that ensure the safety of the treatments and therapies to be used at home.
A great pharmaceutical firm, no matter where they sit in the drug development supply chain, would take extra care to ensure that the development process and packaging are well documented and assured to protect the sterile drug quality and patient safety. It is only when a pharmaceutical firm recognises its integral role in enabling better patient safety at home, with devoted resources and time towards strict adherence to compliance rules, can home-based healthcare truly be effective and useful for all patients.
4. What are the gaps for patient care from hospital
to home?
From hospital to home, there are differences in patient care, especially in the pharmaceutical aspect.
Knowledge on administering medication is one of the major gaps wherein patients or their caregivers at home now have to learn new medication regimes and applications, which, in a hospital setting, would be managed by trained medical professionals. Thus, patients and caregivers have to invest the time and resources needed to learn to administer the treatments and prepare their home environment accordingly.
For patients, relevant device support or follow-up care schedules are needed to accompany the initial transition from hospital to home. The preparation work before the transition from hospital to
All of us in the industry must be open to sharing our knowledge to help ensure every patient is able to receive the best quality care they require.
home is also another gap to fill, ahead of an effective home-based treatment.
The good news is that resources and efforts from medical officers and pharmaceutical companies to help patients fill in these gaps continue to evolve. Examples of these resources include detailed notes, webinars, workshops and more on home-based medication, as well as built-in applications, platforms to track progress or medication dosage, and open communication lines to support patients at home. Given how the home-based healthcare market in Asia Pacific is due to grow at a CAGR of 11.48% from now until 20291, we can expect more of these investments and resources to be available for patients.
5. How can healthcare providers and pharmaceutical companies collaborate to address these gaps?
To best serve patients around the world – whether they are in hospital or now, increasingly at home, healthcare providers must work closely with pharmaceutical companies to develop solutions to meet patient needs. The solutions will vary according to each
patient, depending on their comfortability using self-administration, their age range, and where they live. Knowing that the relationships with healthcare providers and health care practitioners is critical, we have to make sure that providers are aware of the concerns of their patients.
Essentially, a close partnership between healthcare providers and pharmaceutical companies can develop a strong feedback loop that spurs greater innovation for patients, and improves their outcomes transitioning from hospital to home. All of us in the industry must be open to sharing our knowledge to help ensure every patient is able to receive the best quality care they require.
It’s critical that we all work together to ensure that the patients who could most benefit from these technologies have all the resources they need to get comfortable with using them. In addition, given the significant interest from patients and providers in selfadministration methods for managing chronic conditions, industry leaders and research groups have a prime opportunity to conduct research. Evaluating the potential of these technologies to advance healthcare access, reduce health disparities, and improve chronic disease management is essential. Documenting their impact will be a pivotal step toward their broader adoption and the optimisation of these innovative solutions.
Overseeing the Asia Pacific and South America regions, Nilesh is responsible for growing the West's presence and leadership in emerging markets. He joined West in 2023 with more than 25 years in the medical devices industry and global experience while residing in the USA, Europe and most recently in Singapore.
AUTHOR BIO
UPGRADE YOUR MARKETING STRATEGY
Let the true “Digital Transformation” be the base of all your marketing campaigns
AFTER COVID-19 PANDEMIC
“Digital Transformation is the way forward”
Highly accountable marketing campaigns. Every dollar counts. Digitally powered marketing campaigns may be cheaper than you thought... Ask us how?
Use the webinar as a platform to launch new products and services
Grow your audience with increased reach, impact and user-friendliness
Rise above geographical boundaries
Generate new business
Gain the strong web presence differentiating yourself from competitors
Connect and engage with your target audience
Give more exposure to industry specific people
Increase your brand profile and share your capabilities with leading industry professionals
Email : advertise@pharmafocusasia.com
Website : www.pharmafocusasia.com
Unlocking the Potential of Hydrophobic Ion Pairing in Pharmaceutical Formulations
Hydrophobic ion pairing (HIP) involves the electrostatic interaction between a hydrophilic drug molecule and a hydrophobic counterion, reducing its solubility in water and forming stable complexes. HIP is crucial in pharmaceutical science, especially in mRNA delivery, where it enhances control over encapsulation and release. This innovative approach improves drug properties, holding promise for small molecule, protein and peptide drug delivery and enhanced therapeutic outcomes across diseases.
Dimple Modi, Investigator, Drug Development Department, GlaxoSmithKline
What is hydrophobic ion pairing? What are the advantages of using HIP in various drug delivery?
Hydrophobic ion pairing (HIP) represents a promising avenue within pharmaceutical science, offering a solution to the challenges posed by poorly bioavailable drugs. Fundamentally, HIP is a meticulously designed technique aimed at improving the solubility, stability, and bioavailability of compounds that exhibit limited dissolution in non-aqueous environments. The mechanism underlying HIP involves the formation of intimate bonds between hydrophobic counterions and the charged functional groups of drug molecules by long range electrostatic interactions, resulting in the creation of a more lipophilic and stable complex. This intricate process delves into the
fundamental components and mechanisms orchestrating HIP's efficacy. By modifying the solubility extent of poorly permeable drugs, HIP provides relief to a common challenge in pharmaceutical formulation. The amphiphilic nature of hydrophobic counterions enables them to navigate the molecular site, bridging the gap between hydrophilic and hydrophobic realms. The formation of ion pairs, where oppositely charged species interact through ionic interactions, leading in a new era of solubility and stability. Moreover, HIP extends its impact beyond solubility enhancement by stabilising drug molecules against degradation, ensuring the longer shelf life and efficacy of pharmaceutical formulations. Various formulation techniques like co-precipitation, complexation, or integration into lipidbased delivery systems are emerging as preferred methodologies for HIP.
The versatility of HIP finds applications across various dosage forms, including oral tablets, capsules, injectables, and lipid nanoparticles, with particular efficacy demonstrated for drugs with poor bioavailability.
What has led to the increased interest in mRNA-based technology?
The heightened interest in mRNAbased technology has surged following the authorization by the U.S. Food and Drug Administration (FDA) of two mRNA vaccines targeting SARS-CoV-2 on an emergency basis. Once delivered intracellularly, mRNA demonstrates the remarkable capability to stimulate the production of various therapeutic proteins, enabling the treatment of a wide range of diseases,
including infectious diseases, cancers, and genetic disorders. Consequently, mRNA holds significant therapeutic potential and presents an attractive avenue for addressing historically challenging medical conditions. Ongoing clinical initiatives utilising mRNA technology encompass vaccination, cancer immunotherapy, protein replacement therapy, and genome editing. The clinical translation of mRNA technology has been facilitated by the utilisation of nanoparticle delivery methods. However, the effective utilisation of mRNA for therapeutic purposes is impeded by the necessity for customised, effective, and safe delivery systems.
How is Ion pairing used in lipid nanoparticle (LNP) formulations for mRNA delivery?
The integration of ion pairing into lipid nanoparticle (LNP) formulations for mRNA delivery stands as a transformative advancement, providing meticulous control over mRNA payload encapsulation and release. LNPs composed of an ionisable lipid, a helper lipid, cholesterol, a PEG lipid, and therapeutic nucleic acids have demonstrated potent efficacy and safety as both prophylactic vaccines and therapeutic delivery carriers. Ion pairing is intricately woven into LNP formulations through molecular interactions, with the overarching goal of enhancing stability, solubility, and efficacy of mRNA therapeutics. At its core, this integration hinges on the selection of hydrophobic counterions or lipids, which form stable complexes with the negatively charged phosphate groups of mRNA molecules, shielding them from enzymatic degradation and prolonging their stability in physiological environments. Additionally, cationic components facilitate efficient mRNA encapsulation within LNPs through electrostatic interactions, ensuring their integration into the lipid bilayer. This
strategic incorporation of ion pairing enables precise modulation of mRNA payload release kinetics, tailored to specific therapeutic needs by adjusting counterions, lipid constituents, and formulation techniques. The versatility of ion pairing enables the customization of LNPs to accommodate diverse mRNA payloads and delivery requirements, with optimised characteristics such as size, surface charge, and stability. In essence, leveraging ion pairing in LNP formulations represents a sophisticated strategy for enhancing the efficacy and feasibility of mRNA therapeutics, facilitating their translation into clinical practice.
What are the challenges and restrictions emerging in mRNA delivery using ion pairing?
While ion pairing offers considerable advantages in mRNA delivery, several challenges and considerations necessitate careful attention during its application. One such challenge involves the potential for off-target effects resulting from nonspecific interactions between ion-paired mRNA and cellular constituents, leading to unintended cellular uptake or immune activation. Furthermore, the selection of suitable counterions or lipids for ion pairing demands meticulous optimization to ensure compatibility with mRNA molecules and minimise cytotoxicity. The dynamic nature of ion pairing interactions underscores the need for precise control over formulation parameters to achieve consistent outcomes and mitigate batch-to-batch variability. Additionally, premature release of mRNA payloads from ion-paired complexes presents a concern, potentially limiting efficacy or inducing systemic toxicity. Moreover, the scalability and manufacturing complexity of ion pairing-based delivery systems pose obstacles for large-scale production and clinical translation. Addressing these challenges entails interdisciplinary collaboration and
ongoing research endeavours to refine ion pairing strategies for safe and effective mRNA delivery, thereby unlocking its full therapeutic potential.
What
regulatory considerations or limitations exist to facilitate mRNA delivery support? What areas of improvement are necessary to expedite the deployment of this drug delivery method to patients?
Regulatory considerations and constraints exert a significant influence on the advancement and approval process of mRNA delivery systems, influencing the pace at which these pioneering therapies can be made available to patients. A pivotal aspect of regulatory scrutiny revolves around substantiating the safety, efficacy, and quality of mRNA delivery systems through meticulous preclinical and clinical investigations, aligning with guidelines established by regulatory bodies such as the Food and Drug Administration (FDA) in the United States and the European Medicines Agency (EMA) in Europe. Furthermore, regulatory agencies mandate exhaustive data regarding the pharmacokinetic, pharmacodynamic, and immunogenic properties of mRNA therapeutics, coupled with comprehensive characterization of the delivery system encompassing its composition, manufacturing procedures, and stability attributes. Additionally, regulatory endorsement hinges upon the demonstration of a favourable benefit-risk profile, necessitating an evaluation of potential adverse effects and the implementation of corresponding management strategies.
Despite the potential of mRNA delivery systems to revolutionise medical treatment paradigms, numerous challenges persist, necessitating improvements to expedite their integration into clinical practice. Enhanced delivery
Lipophilic/hydrophobic compounds are trending for their bioavailability, achievable through hydrophobic ion pairing, which not only promises more efficient delivery systems but also enhances the stability and cellular uptake of mRNA molecules, improving the efficiency and effectiveness of mRNA-based therapeutics, though broader applications are needed to address formulation consistency and stability challenges.
system design optimization is imperative to augment delivery efficiency, target specificity, and tissue penetration. Strategies aimed at bolstering mRNA stability, diminishing immunogenicity, and amplifying cellular uptake are indispensable for maximising therapeutic efficacy while minimising off-target effects. Advancements in manufacturing technology and scalability are indispensable to facilitate large-scale production of mRNA delivery systems, ensuring uniform quality and supply chain reliability. Moreover, innovative approaches geared towards refining dosing regimens, augmenting patient adherence, and mitigating treatment expenses are pivotal for widening patient access and affordability. Collaborative endeavours among researchers, clinicians, regulators, and industry stakeholders are essential to tackle these challenges, expediting the development and regulatory approval of mRNA delivery systems across a broad spectrum of therapeutic applications. By surmounting regulatory obstacles and fostering innovation, mRNA delivery
technologies stand poised to revolutionise treatment landscapes and enhance patient outcomes across diverse disease contexts.
Is HIP useful in protein
and peptide delivery?
Provide recent research examples.
HIP has attracted considerable attention in the field of protein and peptide delivery, with recent investigations shedding light on its effectiveness and diverse applications. By creating stable complexes between hydrophobic counterions and charged functional groups on proteins or peptides, HIP enhances their solubility and stability, thus addressing challenges associated with their delivery. Recent studies underscore HIP's potential in enhancing the bioavailability and therapeutic efficacy of proteins and peptides, especially those with poor solubility or susceptibility to enzymatic degradation. Furthermore, advancements in nanoparticle-based delivery systems have enabled the formulation of HIP-mediated formulations with precise control over drug release kinetics, further augmenting therapeutic outcomes. Researchers have also explored innovative approaches to optimise HIP formulations, including the use of novel hydrophobic counterions and the incorporation of targeting ligands to achieve site-specific delivery. Overall, recent research underscores the promising role of HIP in advancing protein and peptide delivery, offering prospects for the development of effective therapies across various diseases.
In recent applications of HIP techniques, insulin has been a subject of investigation to enhance its stability and bioavailability, particularly for diabetes treatment. Studies have explored the formation of complexes between insulin and hydrophobic counterions like long-chain fatty acids or bile salts, resulting in improved solubility and stability, thereby enhancing delivery and
therapeutic outcomes. Furthermore, the utilisation of HIP has been innovatively applied to augment the oil solubility of challenging drugs like phenytoin, by transforming them into more lipophilic form. This adaptation facilitates their incorporation into nanoemulsion formulations, which can significantly mitigate the side effects associated with the in vitro precipitation of phenytoin. A quaternary ammonium compound serves as a hydrophobic counterion, enhancing the drug's lipophilicity by up to eightfold when combined with hydrophobic counter ion. This modification not only boosts the drug's lipophilicity but also allows for its use in sustained-release therapeutic applications. This exemplifies how HIP can ingeniously modify the properties of a drug molecule without altering its structural integrity, thereby enhancing its therapeutic efficacy and application scope.
How is hydrophobic ion
pairing utilised in the development of small molecule formulations?
Hydrophobic ion pairing (HIP) emerges as a transformative innovation in the realm of small molecule drug development, addressing formidable challenges in formulation and delivery by intricately modifying drug characteristics to enhance solubility, stability, and bioavailability. This method holds particular significance in augmenting oral bioavailability through the elevation of molecular lipophilicity, facilitating integration into lipid-based delivery systems like micelles and nanoparticles, thus improving intestinal absorption. Moreover, HIP serves as a protective shield, safeguarding molecules against degradation mechanisms such as hydrolysis and oxidation, especially crucial for drugs vulnerable to the harsh gastrointestinal environment. Furthermore, HIP enables targeted delivery by strategically modulating drug hydrophobicity, facilitating precise drug release at specific
anatomical sites, thereby optimiszing therapeutic efficacy while mitigating adverse effects. The versatility of HIP in formulation design opens novel pathways for solid dispersions, nanoparticles, and other sophisticated delivery modalities, catering to drugs with complex physicochemical properties. This may lead to formulations necessitating reduced dosing frequency, enhancing patient adherence, and significantly augmenting solubility and dissolution rates, pivotal for oral drug effectiveness. Additionally, HIP's potential to bypass hepatic first-pass metabolism may
elevate systemic drug levels, while its compatibility with combination therapies allows for the co-encapsulation of diverse drugs, enhancing therapeutic targeting. Nonetheless, harnessing
the advantages of HIP necessitates meticulous counterion selection, consideration of the drug's physicochemical profile, and the intended route of administration, alongside rigorous evaluation of safety and biocompatibility, ensuring
Dimple Modi, PhD, is currently employed as an investigator in the Drug Development Department at GlaxoSmithKline, Pennsylvania. She previously served as a senior scientist at Lupin Pharmaceuticals, New Jersey. With a wide range of experience in various drug formulation techniques and dosage forms such as oral solid, parenteral, and topical, Dimple has been crucial as a lead in drug product development. Her expertise encompasses the full spectrum of development from early to late phases, including lab to pilot plant scale-up, and the development and optimization of formulations and processes. As US lead for Extemporaneous compounding at GSK, Dimple has also made significant contributions in implementation of innovative technologies and strategies.
Nanocarrier Vaccines
BiopharmaceuticsBased Fast Track Development
both therapeutic efficacy and patient well-being.
References are available at www.pharmafocusasia.com
Date of Publishing: 5 March 2024
Editors: Vivek P Chavda, Vasso Apostolopoulos
Book Description: This book details the benefits, restrictions, and types of nanoparticles used in the creation of vaccines for the treatment and prevention of illnesses. This book aims to be a single source material for understanding all the current and novel advancements in the field of nanotechnology.
In this groundbreaking book the reader will find:
• biodegradable and non-biodegradable formulations and properties such as size, shape, charge, inertness, efficacy, morphology, and more;
• show how different nanoparticles, such as lipid-based, viral vectorbased, and metal, uphold very significant properties individually, suggesting applicability in various management tactics;
• examines how genetic information-carrying entities are becoming the norm for eradicating some diseases;
• gathers an exhaustive amount of information on routes of administration such as the oral route, mucosal immunity, intramuscular, subcutaneous, and intradermal;
• explores the legal regulations for nanotechnology-based approaches.
Asia-Pacific Trials
Ding Ming
Senior Vice President, China, PPD clinical research business
Thermo Fisher Scientific
What are some of the key trends impacting clinical trials in the Asia-Pacific region?
The Asia-Pacific (APAC) region is becoming increasingly influential in the global clinical trials landscape due to several key trends.
First, there is a notable surge in Phase I trials, with the region accounting for a significant global share. This growth is fueled by the region's ability to rapidly enrol patients and its focus on key therapeutic areas.
Second, the expansion of clinical trial infrastructure presents opportunities for multi-country trial growth.
Third, supportive government initiatives and regulatory frameworks are fostering a positive environment for clinical research.
Fourth, the adoption of virtual and decentralised trials, accelerated by the COVID-19 pandemic, is another trend enhancing the region's efficiency and patient diversity in trials.
Fifth, increased R&D investments by local pharmaceutical and biotech companies are driving the need for more clinical trials.
Sixth, the region's attractiveness for global clinical trials—due to its diverse patient population, skilled professionals and cost-effectiveness—is positioning it as a hub for complex clinical research. These trends collectively contribute to the APAC region's significant growth and development in the clinical trials sector.
What is your outlook on the potential impact of the evolving regulatory landscape in key AsiaPacific countries on the efficiency and execution of clinical trials for pharmaceutical and biotech companies operating in the region?
The changing regulatory landscape in the APAC region will have a significant impact on the efficiency and execution of clinical trials conducted by pharmaceutical and biotech companies. Notable trends include the harmonisation of regulatory frameworks, which simplifies the process of conducting trials across multiple countries by reducing variations in requirements. Streamlined approval processes, as observed in countries like Singapore and Australia, are expected to expedite the timeline for clinical trials, making the region more appealing for conducting trials. Additionally, the adoption of international standards is enhancing the quality and reliability of clinical trial data, thereby increasing global confidence in the research capabilities of the region.
Regulatory bodies also are focusing on diseases prevalent in the region, leading to trials that are more relevant to the local population and potentially faster patient recruitment. The emergence of new regulatory challenges, particularly in response to public health emergencies such as COVID-19, is prompting the development of more flexible and agile regulatory mechanisms. These changes aim to balance expedited approval pathways with maintaining rigorous safety and efficacy standards. Thanks to governments incentivising local and foreign investment in innovation and R&D, the overall environment is much more supportive of clinical trials and expanding the region’s clinical research capacity.
Lastly, with the increasing global nature of clinical trials, robust data protection and sharing regulations are being prioritised to ensure privacy and security while facilitating cross-border collaboration. In conclusion, regulatory changes in the region are likely to make the conduct of clinical trials more efficient and effective, positioning the region as a hub for innovative clinical research and development. Companies operating in the region will need to adapt to these changes to fully leverage the opportunities they present.
What effect will the implementation of digital health technologies have on the future of clinical trials in the region?
The integration of digital health technologies is reshaping clinical trials in the
Asia-Pacific region, offering new opportunities for efficiency and innovation. Decentralised trials facilitated by remote monitoring and telemedicine can reach a wider patient population, enhancing trial diversity and reducing the burden on participants. Real-time data collection through wearable devices and mobile apps improves data quality and allows for continuous health monitoring, which can accelerate the trial process. Artificial intelligence and machine learning can address large datasets to optimise trial design and patient selection, potentially leading to more effective treatments. These technologies also support virtual consultations, making trials more accessible and cost effective by reducing the need for physical site visits. Pharma/biotech and their clinical research service providers need to have clear strategies to manage the challenges that can accompany the adoption of digital health technologies, particularly in ensuring regulatory compliance, data privacy and security.
The integration of these technologies is vital for expanding clinical trial access and improving health care services, particularly in underserved areas. Bottom line, digital health technologies will play a pivotal role in the future of clinical trials in APAC, driving progress toward more efficient, patient-centric and data-driven research processes.
Which disease areas in the APAC region have the most potential for growth in clinical trials over the next few years?
The APAC region has seen significant growth in clinical trials across various disease areas in recent years and there are several that have the potential for further growth. Some of these include oncology (particularly lung, liver and gastric cancers), infectious diseases (such as dengue fever, tuberculosis and HIV), cardiovascular diseases (including hypertension and coronary artery disease), and neurological disorders (such as Alzheimer's disease and Parkinson's disease). Additionally, rare diseases and
AUTHOR BIO
Ding is a seasoned pharmaceutical executive with more than 25 years of pharmaceutical industry operational leadership experience. He joined the PPD clinical research business in 2019 to oversee the China operations, which include more than 1,500 clinical development and analytical services professionals. Prior to that, he served as vice president and R&D head for Emergent Biosolutions. He has worked at a variety of pharmaceutical companies, including GSK, Boehringer Ingelheim, Pfizer and Novartis.
orphan indications are gaining attention in the region. It is important to note that the potential for growth in clinical trials can vary based on factors such as disease prevalence, emerging therapies, regulatory environment, and investment in research and development.
What models of collaboration and partnership are emerging in Asia-Pacific clinical trials?
As a global clinical research services provider, we have seen some emerging models of collaboration and partnership
o Functional service provider (FSP ) collaboration is surging ahead of full-service outsourcing (FSO) among companies of all sizes globally in the APAC region. Drug developers increasingly favour FSP partnerships and hybrid FSP/FSO models for their ability to deliver enhanced resource flexibility, global talent acquisition and efficient access to specialised skills.
o The number of multi-regional clinical trials (MRCTs) in APAC has been steadily increasing in recent years due to the regulatory harmonisation, large patient populations, cost effectiveness, etc. Particularly in China, we have seen rapid growth in MRCTs trials, with more emerging China biotechs going abroad for their global development.
o APAC, particularly China and South Korea, has emerged as a hub for biotech innovation. Local biopharma companies are developing novel therapies and technologies, leading to an increase in licence-out deals to Western markets. On the other hand, favourable regulatory changes in countries like China and Japan have facilitated faster approval processes for imported drugs, encouraging more licence-in deals to bring foreign innovations to local markets. The licensing landscape in APAC demonstrates a two-way flow of innovation, with companies both importing and exporting technologies and products to meet global health care demands. This trend underscores the region's growing importance in the global pharmaceutical ecosystem and its transition from being primarily a recipient of licensed technologies to a key player in the development and dissemination of innovative therapies worldwide.
What is the role of multinational collaborations in clinical trials in the Asia-Pacific region?
Multinational collaborations play a vital role in clinical trials in APAC. These collaborations involve partnerships between pharmaceutical companies, contract research organisations (CROs), study sites and regulatory authorities. Mainly reflected in following aspects:
o Increasing diversity and inclusion
o Promoting innovation and expertise sharing in the region
o Improving efficiency and speed
o Strengthening the region’s influence in global R&D
o Promoting the coordination of regulations in the region
o Improving the quality of clinical trials. Particularly for those innovative emerging biotech companies with global ambitions, working with a global CRO with a local presence could bring global expertise and experience, as well as local operational delivery, to shorten timelines and support cost-effective clinical development.
Supply Chain Planning for Clinical Trials A Practical Guide
Date of Publishing: 13 August 2024
Author: Ryan Mills
Book Description: Supply Chain Planning for Clinical Trials offers a practical introduction to this process for researchers and industry professionals. Beginning with the basics of clinical trial supply chain management, it proceeds step by step through all aspects of demand and supply planning for clinical trials. The result is a thorough overview that also offers practical examples of how to plan supply for clinical trials.
Supply Chain Planning for Clinical Trials
readers will also find:
• Tools for minimizing risk and expense by optimizing the relationship between supply and demand
• Detailed discussion of topics including quality and regulatory considerations and the business processes that support clinical trial supply chain management
• Spreadsheet-based models to illustrate key concepts, adaptable to the readers' specific scenarios
• Supply Chain Planning for Clinical Trails is ideal for pharmaceutical industry professionals involved in clinical trial supply planning, as well as academics and researchers interested in the pharmaceutical industry and its logistics.
Comprehensive Assessment of Risk-based Quality Management Adoption in Clinical Trials
The FDA and EMA have encouraged the use of risk-based monitoring (RBM) and risk-based quality management (RBQM) to mitigate risks related to essential safety and efficacy data in clinical trials. Insights from the Tufts Center for the Study of Drug Development’s comprehensive assessment of RBQM adoption inform adoption levels, barriers, and industry perceptions.
Abigail Dirks, Data Scientist, Tufts Center for the Study of Drug Development
1. What is the current state of RBQM adoption in the industry? Many companies have implemented some components of RBQM in some trials, but full implementation of RBQM across a portfolio is rare. In the average company, a little more than half of clinical trials are utilising RBQM. Certain components are widely adopted, including risk assessment and risk control planning at 79 per cent of trials, while others, like the solicitation of input from the patient community to support optimal trial design
are only being used in 27 per cent of trials on average. Companies expect to increase usage of all components, besides those related to the documentation of SDV and SDR.
2. How has RBQM adoption been measured in the past, and how is it measured in this recent study?
Other assessments of RBQM are important foundational tools for understanding RBQM implementation. In one study, about 6000 clinical trials were assessed for the implementation of eight main components. RBQM adoption was quantified as the percent of those trials that had adopted at least one of the 8 components. CROs reported usage of each compo
clinical trials. Each respondent was asked to estimate the proportion of trials within their company that had been implemented across 32 discrete RBQM components, gaining insight into the extent to which adoption is occurring as well as more granularity into where there are gaps in adoption, based on which components are more widely adopted.
3. How does the current assessment and results align with regulatory guidance, mainly FDA’s ICH E6 (R3)?
ICH E6 (R3) emphasises the adoption of a risk-based approach to the identification, assessment, control, and review of risks throughout a clinical trial, aligning with compo
of centralised monitoring, remote monitoring, and advanced statistical methods. The study found that on average, a centralised monitoring plan was developed and the deviations or updates made to the plan were documented in about half of clinical trials. Remote monitoring was utilised in 60 per cent of trials on average and statistical data monitoring is reported to be utilised in 49 per cent of trials on average.
The use of source data verification (SDV) was also assessed and is encouraged in the guidance. Looking forward to 2027, companies reported that they expect to reduce SDV or target SDV in about 50 per cent more trials than currently. 56 per cent of trials currently reduce or target SDV during Execution. They also report that the documentation of SDV will decrease by 13 per cent less trials by 2027, presumably as more trials reduce SDV or conduct more targeted SDV. Currently, 76 per cent of trials are documenting SDV.
Although not explicitly mentioned, the most recent guidance outlines quality tolerance limits (QTLs) as acceptable ranges that are set to detect deviations in support of risk mitigation. While 48 per cent of trials per company identify QTLs, 46 per cent utilise QTLs during clinical trial execution, 46 per cent identify important QTL deviations, and 44 per cent evaluate, follow-up, and resolve each important QTL deviation.
Although the industry has implemented some components of RBQM, compliance with ICH E6 (R3) will still require further adoption within companies, and many expect this to happen, reporting increased adoption by 2027.
4. What challenges is the industry experiencing in adopting RBQM?
The top challenge across stages, within Planning & Design, Execution, and Documentation & Resolution, is the lack of
knowledge and awareness of RBQM, although 76 per cent of respondents did feel their organisation understands the meaning of RBQM somewhat or very well. A key theme from a roundtable of industry experts was the importance of change management strategies to ensure cross-functional understanding of RBQM, especially the purpose of RBQM. A meaningful change management plan that includes a variety of functions is an important step to implementation.
Other challenges include lack of technology and skills. For instance, many companies report using Microsoft Excel for risk assessment. It appears that RBQM professionals are looking for more advanced technology to handle advanced statistical methods for risk assessments. Respondents also report challenges with skills required for RBQM. Industry experts emphasised the need for analytical and critical thinking skills, along with communication skills. These skills may need to be further developed, as they were not critical for those conducting traditional monitoring.
5. Do companies trust RBQM and how can trust be increased?
Almost 80 per cent of companies trust RBQM to improve the overall quality of clinical research, while 63 per cent believe it will increase efficiency and cost savings, and a little over half trust RBQM to reduce study timelines. Respondents felt that within their organisations, site management/ site monitoring, clinical development, and clinical operations/study management have the lowest trust in RBQM. The functional areas that were least mentioned for low trust in RBQM were data management/data sciences and biostatistics. When asked how trust can be increased, 75 per cent of respondents chose “More experience
with successful implementations”. As mentioned, a top challenge is lack of knowledge and awareness, emphasising the need for change management strategies. A key part of a change management strategy is cross-functional education. Case studies from other companies may be a great way to spread knowledge to all employees on how RBQM may work and in turn, increase trust across an organisation.
6. What is the current level of understanding of RBQM in the industry?
As noted above, a key challenge for companies is the lack of knowledge and awareness of RBQM in the industry. Definitions of RBQM varied, although these definitions were collected before the release of ICH E6 R3. Regulatory guidance may help companies further define RBQM. The study also asked each respondent about their organisation’s level of familiarity with RBQM, understanding of what RBQM means, and commitment to RBQM.
7. What are the expected outcomes from implementation of RBQM?
A vast majority of respondents (91 per cent) noted that RBQM would improve reliability of trial outcomes and improve patient safety. Regarding cost savings, 61 per cent expected lower monitoring costs, while only 28 per cent expected
lower data management costs. In terms of speed, 60 per cent felt that RBQM would cause faster time to database lock, but only 18 per cent felt that RBQM would cause faster patient enrollment.
8. Does your study cover insights into how RBQM adoption typically unfolds?
A deep dive into the data collected revealed certain subgroups that represent different stages of adoption, based on the number of components that have been implemented and the average percent of trials across the company that they are implemented within. Those who are more advanced in adoption tend to be larger companies, conducting 100+ trials per year. The most advanced are conducting duplicate patients detection in RBQM execution. About 70 per cent of the lowest adoption grouping is those conducting less than 25 trials annually. Most of the least advanced groups are soliciting input from the investigative research community to support optimal trial design, conducting risk assessment and risk control planning, identifying risks detected, documenting data SDV’ed, and documenting updates to and/or deviations from the site monitoring plan. Throughout the adoption process, companies begin to incorporate components like criticalto-quality (CTQ) factors, key risk indicators (KRIs), quality tolerance limits (QTLs), remote site monitoring, evaluation, follow-up and resolution of each risk, and eventually implement all 32 components.
Drug
where
in analysing large datasets pertaining to industry drug development performance, including risk-based quality management (RBQM) adoption, investigative site burden and experience, impact of decentralised clinical trials (DCT), and clinical trial participation. She recently received her MS in Health Informatics and Analytics from Tufts University School of Medicine, Boston, MA and has a BS in Data Science and Mathematics from St. Michael’s College, Colchester, VT.
AUTHOR
BIO
Abigail Dirks is a Data Scientist at the Tufts Center for the Study of