





In today’s rapidly evolving landscape, the integration of data and clinical practice is redefining how we understand and enhance patient outcomes. We are excited to present this issue of Pharma Focus America Magazine, which explores the transformative impact of Real-World Data (RWD) and Real-World Evidence (RWE) in shaping clinical insights and advancing medical innovation.
While clinical trials have traditionally been essential for evaluating new treatments and providing valuable insights through controlled experimental conditions, they often fail to capture the full complexity of everyday patient experiences. RWD and RWE provide a broader perspective by reflecting on how treatments perform across diverse, real-world settings. This approach enhances traditional clinical trial findings and supports more informed regulatory decisions and healthcare interventions.
We are honoured to feature insights from Dr. Yun Lu as we explore how RWD and RWE are reshaping clinical research and practice. This issue highlights innovative methodologies and discusses the regulatory considerations necessary for integrating RWE into clinical decision-making.
As you turn the pages, this edition covers a range of groundbreaking topics that are reshaping the pharmaceutical industry:
Bridging Boundaries in Cell and Gene Therapy Regulations: Joab Williamson highlights the need for global regulatory harmonization to speed up the delivery of innovative therapies.
Navigating Biotech Innovation: Kishore outlines strategies for accelerating drug development and commercialization.
AI’s Impact on Pharmaceuticals: Gaurav Jaggi discusses how artificial intelligence is enhancing
drug discovery and development, offering insights into real-world applications within big pharma.
Predictive Maintenance in Sterile Manufacturing:
Mostafa Essam illustrates how AI and machine learning are revolutionizing equipment maintenance to ensure higher quality and efficiency in drug production.
Patient-Centered Drug Repurposing: Vidya Niranjan explains how patient insights and advanced technologies are driving personalized medicine through innovative drug repurposing.
Virtual Clinical Trials: Sowmya Kaur shares insights into virtual clinical trials and their implications for patient care and research methodologies.
Pharmaceutical 3D Printing: Dr. Anna Worsley, Dr. Alvaro Goyanes, and Dr. Khalid Garba Mohammed present their pioneering work on 3D printing applications within the pharmaceutical industry.
We hope you enjoy this latest edition of Pharma Focus America. If you have ideas for an article or would like to be featured in an interview for our upcoming issues, please reach out to us at editorial@ pharmafocusamerica.com.
Stay connected with us on social media to keep up with the latest industry trends and engage with our community. We look forward to your insights and to continuing our journey together in the pharmaceutical world.
Stay Tuned!
N D Vijaya Lakshmi Editor
08 Bridging Boundaries toward Global Harmonization of Cell and Gene Therapy Regulations
Joab Williamson, Director, Clinical Operations, Faron Pharmaceuticals
18 Driving Biotech Innovation
Navigating Fast-to-Clinic and Fast-toMarket Strategies from Discovery to Commercialization
Kishore Kumar Hotha, Global Vice President, Veranova
28 Rethinking Evidence Strategy in HEOR Budget Planning
Nicole Betor, Consultant II, Avalere
Nancy El Hoyek, Associate Principal
Laura Housman, Practice Director, Avalere
Josie Lloyd, Consultant II, Avalere
Taylor Schwartz, Principal, Avalere
32 Nanorobotics in Drug Delivery Targeting A Review on Future of Advanced Pharmaceutical Technology
Mostafa Essam Eissa, Independent Researcher and Consultant, Bioinformatics and Biometry Department, Pharmaceutical Research Facility, Cairo
39 Beyond the Bench Patient-Centered Drug Repurposing Initiatives and the Future of Personalized Medicine
Vidya Niranjan, Professor and Head of the Department, Department of Biotechnology
Lead-Centre of Excellence Computational Genomics, R V College of Engineering
44 Enhancing Sterile Manufacturing with AI and Machine Learning for Predictive Equipment Maintenance
Mostafa Essam Eissa, Independent Researcher and Consultant, Bioinformatics and Biometry Department, Pharmaceutical Research Facility, Cairo
51 Pharmaceutical 3D Printing Small Batches Making Big Waves
Anna Worsley, CEO, FABRX-AI
Dr Alvaro Goyanes, Co-Founder, Director, FABRX
Dr Khalid Garba Mohammed, Senior Formulation Scientist, FABRX

Dr Yun Lu Chief Scientific & Innovation Officer, Navitas Life Sciences
58 How AI is Transforming the Pharmaceutical Industry
Gaurav Jaggi, PhD, Director of Strategic Insights and Analytics, Bayer Oncology
66 AI's Game-Changing Role in Drug Discovery: Insights from Dr. Samanta's Research Team
Dr Samanta, Assistant Professor, in IIIT Allahabad
Ananya, PhD, Biomedical Engineering, Department of Applied Sciences, IIIT-Allahabad
Vidushi, Project Research Fellow, Department of Applied Sciences, IIIT-Allahabad
77 Future-Ready Pharma: The Transformative Role of GenAI
Tarun Mathur, Chief Technology Officer, Indegene
84 Exploring the Impact of Virtual Clinical Trials on Patient Care
Sowmya Kaur, Executive Vice President, Navitas Life Sciences

EDITOR
Vijaya Lakshmi N D
EDITORIAL TEAM
Sarah Richards
Debi Jones
Harry Callum
Supraja BR
ART DIRECTOR
M Abdul Hannan
PRODUCT MANAGER
Jeff Kenney
ASSISTANT MANAGER
David Nelson
Peter Thomas
BUSINESS EVENTS
Sussane Vincent
CIRCULATION TEAM
Sam Smith
SUBSCRIPTIONS
Vijay Kumar Gaddam
HEAD-OPERATIONS
Sivala VNR









Amine Bekkali
Director chez MEDFIELDS
United Arab Emirates

Alessio Piccoli
Director & Head, Business Development Europe, Aragen Life Sciences Italy
David Contorno
Founder & CEO, E Powered Benefits, USA


Eiman Shafa
Medical Director, Spine Surgery, Abbott Northwestern Hospital USA
Hassan Mostafa Mohamed
Chairman & Chief Executive Officer at ReyadaPro, Saudi Arabia




Hector Alejandro Andonie
General Manager, Laboratorios Andifar Honduras



Hoda Gamal
Director of Regulatory and Corporate Affairs, Middle East and Africa (MEAC), Sirgio international, Egypt
Joaquin Campbell
Global Director, Managed Access Services, EarlyHealth Group Spain
Josipa Ljubicic
QA Director/Principal GCP and GVP auditor, Proqlea Ltd Croatia
Juris Hmelnickis
CEO, Grindeks, Latvia
Nicoleta Grecu
Director, Pharmacovigilance, Clinical Quality Assurance, Clover Biopharmaceuticals Romania
Nigel Cryer FRSC
Global Corporate Quality Audit Head, Sanofi Pasteur, France
Pinheiro Neto Joao
Chief Executive Officer, Omnimed Angola
Scott M. Wheelwright
Chief Operating Officer, BioInno Bioscience Co., Ltd. China
Svetlana Busiguina
CEO, Bi-Connex BD Consulting, Spain


Tamara Miller
Senior Vice President, Actinogen Medical Limited, Australia
Thitisak Kitthaweesin
Chief of Phramongkutklao Center of Academic and International Relations Administration Thailand
Let the true “Digital Transformation” be the base of all your marketing campaigns

Highly accountable marketing campaigns. Every dollar counts. Digitally powered marketing campaigns may be cheaper than you thought...
Ask us how?
Use our guaranteed ROI programs for reposition, penetrate or launch of your new products or services.
Grow your audience with increased reach, impact and user-friendliness
Rise above geographical boundaries
Generate new business
Gain the strong web presence differentiating yourself from competitors
Connect and engage with your target audience
Give more exposure to industry specific people
Increase your brand profile and share your capabilities with leading industry professionals

Email : advertise@pharmafocusamerica.com
Website : www.pharmafocusamerica.com



This article explores the need for global harmonization in cell and gene therapy regulations. It discusses the challenges and benefits of synchronized regulatory frameworks, emphasizing improved market access and patient outcomes, and proposes strategies for enhancing international regulatory cooperation to accelerate the availability of innovative therapies worldwide.
Joab Williamson Director, Clinical Operations, Faron Pharmaceuticals

In the rapidly evolving field of medicine, cell and gene therapies represent a frontier replete with promise and potential. These therapies, leveraging the capacity to replace, engineer, or regenerate human cells, tissues, or genes, offer revolutionary treatments for a wide range of diseases, including many that were previously untreatable. However, as the
technologies underpinning these therapies advance, regulatory frameworks across different regions struggle to keep pace. This discordance between technological advancements and regulatory updates poses significant challenges not only to biotechnology and pharmaceutical companies but also to healthcare providers and patients awaiting new therapies. The global
harmonization of regulations governing cell and gene therapies is not merely beneficial but necessary to streamline development processes, reduce costs, facilitate international collaborations, and, most importantly, hasten patient access to life-saving treatments.
The regulatory frameworks governing cell and gene therapies vary significantly across the globe, each shaped by unique regional medical, ethical, safety, and economic considerations. These variations affect how therapies are developed, approved, and brought to market, impacting the speed and availability of innovative treatments.
The United States stands out with its progressive policies designed to facilitate the rapid introduction of regenerative medicines. The Food and Drug Administration (FDA) plays a pivotal role, primarily through the Regenerative Medicine Advanced Therapy (RMAT) designation, established under the 21st Century Cures Act. This designation aims to expedite the development and review process for therapies showing potential to address severe health conditions. The FDA also provides extensive guidance for industry stakeholders on how to expedite clinical development and ensure compliance with regulatory standards, reflecting a proactive approach to fostering medical innovation
while safeguarding patient safety. Canada, through Health Canada, has similarly embraced innovative regulatory frameworks to support cell and gene therapies. Initiatives such as the Advanced Therapeutic Products Pathway are designed to provide flexible regulatory oversight that adapts to the novel nature of these therapies, ensuring timely access while maintaining high safety standards.
The European Union’s European Medicines Agency (EMA) coordinates the regulatory processes in the European Union. The PRIME (PRIority MEdicines) designation is a key initiative, assisting developers of promising medicines by offering enhanced support during the licensing process, particularly for treatments targeting diseases with significant
unmet medical needs. This system emphasizes early dialogue and continuous support, facilitating a smoother regulatory review process that can lead to earlier market entry.
Post-Brexit, the United Kingdom has tweaked its regulatory pathway through the country’s regulator, Medicines and Healthcare products Regulatory Agency (MHRA). The Innovative licensing and Access Pathway (ILAP) aims to accelerate the time to market for innovative medicines, focusing on enhanced collaboration and support throughout the development process.
Japan is a leader in the approval of regenerative therapies within Asia, known for its proactive regulatory stance. The Pharmaceuticals and Medical Devices Agency (PMDA) encourages the rapid approval of therapies based on preliminary evidence of safety and efficacy. This approach often results in earlier patient access to new treatments compared to Western counterparts and reflects Japan's national strategy to be at the forefront of regenerative medicine.
China’s National Medical Products Administration (NMPA) has also been making significant strides in adapting its regulatory framework to accommodate advanced therapies. Recent reforms have introduced accelerated approval pathways and a more transparent review process, aiming to stimulate innovation and reduce time-to-market for critical therapies.

South Korea’s Ministry of Food and Drug Safety (MFDS) has established the Advanced Biopharmaceuticals Act, which includes provisions for expedited approval and market entry for regenerative therapies. This regulatory environment supports the rapid development and commercialization of innovative treatments.
In Australia, the Therapeutic Goods Administration (TGA) oversees the regulation of cell and gene therapies. The TGA has introduced the Priority Review Pathway and the Provisional Approval Pathway to expedite the approval process for promising therapies addressing serious conditions, thereby ensuring timely patient access.
Latin American countries, such as Brazil and Argentina, are also evolving their regulatory frameworks to support advanced therapies.
Brazil’s National Health Surveillance Agency (ANVISA) has implemented special procedures for the evaluation of cell and gene therapies, aiming to streamline the approval process while ensuring safety and efficacy.
The lack of regulatory harmonization poses multifaceted challenges. For global pharmaceutical companies, divergent regulatory paths entail duplicative clinical trials, increased development costs, and elongated timelines to market entry. This fragmentation is particularly cumbersome for cell and gene therapies, which often target rare diseases with small patient populations; conducting multiple, separate trials to meet different regional standards is not only inefficient but also ethically questionable, as it may unnecessarily expose patients to placebo or less effective treatments.
Furthermore, regulatory discrepancies impact multinational clinical trials, which are vital for gathering diverse data and improving the generalizability of findings. Companies often face logistical nightmares in aligning trial designs to meet the standards of multiple regulatory bodies, which can differ in requirements for trial phases, patient enrolment criteria, and endpoints. For instance, a therapy may qualify for an accelerated pathway in one region but require a full set of phase III trials in another, complicating strategic planning and execution.
These challenges are not just bureaucratic
hurdles but have real-world implications on patient access to therapies. Delays in the development and approval process mean delays in treatment for patients who have few or no alternatives. The case of Glybera, the first gene therapy approved in the European Union in 2012 for the treatment of lipoprotein lipase deficiency, illustrates this point vividly. Despite its approval, Glybera was ultimately withdrawn from the market due to high costs and the complex reimbursement landscape across different countries, which were partly attributable to the regulatory challenges of establishing its value proposition uniformly across markets.
Zynteglo, a gene therapy for betathalassemia, offers a poignant example of the delays and complications arising from non-harmonized regulatory environments. After its approval in the European Union in 2019, Zynteglo faced significant setbacks in other major markets, including the United States. These challenges were not unique to Zynteglo; similar issues have plagued other therapies. For instance, Strimvelis, another
As diseases know no borders, the solutions to them must transcend national boundaries, requiring a more integrated global health approach.
gene therapy for ADA-SCID (a rare immune disorder), approved in Europe in 2016, has yet to gain approval in the United States. These delays stem from differing regulatory standards and risk assessments, which can vary dramatically across regions. As a result, potentially lifealtering treatments are restricted by borders, limiting patient access based on geographic location rather than medical need.
Another illustrative case is the approval process of Novartis’s CAR-T cell therapy, Kymriah. While swiftly approved in both the U.S. and EU in 2017 for certain cancer treatments, regulatory hurdles in other parts of the world, such as Asia and South America, have slowed its availability. Each country's unique requirements for clinical data and manufacturing standards can delay launch dates by years, despite clear global needs. These examples underscore a fragmented approach to regulatory approval that can delay or even prevent access to critical therapies for patients worldwide.
The harmonization of regulatory frameworks across regions can offer substantial benefits, chief among them being accelerated patient access to new therapies. By aligning approval standards and procedures, regulators can reduce redundancy, minimize delays, and facilitate the smoother transition of therapies from clinical trials to market. Additionally, harmonization supports greater international collaboration in research and development. When scientists and
companies can plan multi-center trials with a unified set of standards, the global sharing of data and resources becomes more feasible, driving faster innovation and broader therapeutic applications.
Economically, the pharmaceutical industry stands to gain significantly from reduced development costs and simplified logistic requirements. A unified regulatory landscape would allow companies to streamline operations and leverage efficiencies that lower the financial barriers to entry for new treatments, particularly important in a field as resource intensive as regenerative medicine. Moreover, harmonization can lead to more predictable market conditions, encouraging investment in innovative therapies by reducing the regulatory risk associated with developing treatments for diverse markets.
Achieving global regulatory harmonization requires concerted efforts from all stakeholders, including regulatory agencies, pharmaceutical companies, academic researchers, and patient advocacy groups. One effective strategy could involve the expansion of international consortia and working groups, such as the International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), which has successfully aligned technical requirements among regulators in the United States, Europe, and Japan.
Regulators should consider mutual recognition agreements that allow data from clinical trials
conducted in one region to be accepted in others. This approach not only reduces duplication of clinical trials but also expedites the review process by pooling expertise and resources. For instance, the European Union’s reliance on a centralized approval process through the EMA enables member states to benefit from a single scientific evaluation that applies uniformly across all countries.
Furthermore, developing common guidelines for emerging scientific and ethical issues in gene and cell therapy could help standardize safety and efficacy assessments. These guidelines should be adaptable and responsive to the rapid advancements in technology and scientific understanding that characterize regenerative medicine.
As we look towards the future, the landscape of global healthcare, especially in the realm of innovative therapies like gene and cell treatments, is poised at a crucial juncture. The need for global regulatory harmonization has never been more urgent. Technological advancements are progressing at a rapid pace, outstripping the ability of current regulatory frameworks to adapt swiftly. This mismatch not only hampers the deployment of new therapies but also affects international health equity. The challenge ahead lies not just in aligning regulatory standards but also in fostering a global regulatory ethos that embraces flexibility, transparency, and collaboration. As diseases know no borders, the solutions to them must










also transcend national boundaries. This requires a paradigm shift towards a more integrated global health approach, one that leverages digital health data, real-world evidence, and advanced analytics to inform and harmonize regulatory decisions.
Moreover, the COVID-19 pandemic has underscored the critical importance of agility in health regulations. The rapid development and approval of vaccines through unprecedented global cooperation provide a blueprint for what is possible when the world unites towards a common health goal. Learning from this, future regulatory frameworks for cell and gene therapies should aim for similar agility, balancing speed with scientific rigor to respond to global health crises effectively.
Sustainability is another crucial aspect. As we advance, ensuring that these innovative treatments are not only approved quickly but are also accessible and affordable to populations worldwide remains a pivotal challenge. This will require innovative financing models and partnerships between public and private sectors to support the widespread adoption and manufacturing of gene and cell therapies.
In conclusion, the journey towards global regulatory harmonization in cell and gene therapy is fraught with challenges yet filled with immense potential. The vision for the future is clear establish a globally integrated regulatory framework that not only expedites the availability of innovative therapies but does so
without compromising on safety and efficacy. It's about creating a system where scientific breakthroughs can reach all corners of the globe, ensuring that no patient is left behind due to regulatory discrepancies.
As stakeholders in the global health landscape, it is incumbent upon us regulatory bodies, healthcare providers, pharmaceutical companies, and policymakers to forge pathways that facilitate not just the swift approval of therapies but their ethical distribution and responsible use. By championing collaborative efforts and embracing adaptive regulations, we can catalyze the kind of transformative change that makes universal health access a reality. Let this be the moment we prioritize health equity and innovation equally, driven by a unified commitment to enhancing patient care on a global scale.

Joab Williamson is the Director, Clinical Operations at Faron Pharmaceuticals, a clinical stage biotech focusing on building the future of immune-oncology. He is also a PhD researcher with work focusing on how regulatory processes influence investment decisions and development timelines through the pharmaceutical/biotech industry.
Antibody humanization enhances the adaptability and efficacy of therapeutic antibodies derived from non-human sources, promoting safer and more effective applications in human medicine. This article explores the necessity of antibody humanization, reviews the evolutionary progression and development requirements of humanized antibodies, and examines various humanization strategies.
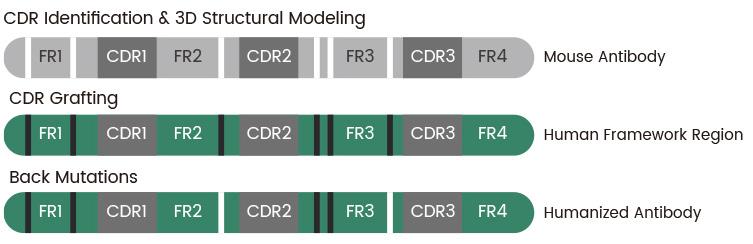
The development of therapeutic antibodies has transitioned from mouse monoclonal antibodies to fully humanized forms, marking a significant advancement in biotechnology. Initially, antibodies were directly
derived from mice and exhibited considerable immunogenicity in humans. The first generation of humanized antibodies, known as chimeric antibodies, involved replacing the constant regions of a mouse antibody with those of a human antibody, significantly lowering immunogenicity while retaining the mouse antibody’s high specificity. Subsequent generations, such as CDR-grafted antibodies, involve transplanting only the complementarity-determining regions (CDRs) of a mouse antibody onto a human framework, further reducing immunogenicity while aiming to maintain binding affinity. Surface-reshaped antibodies have been engineered by altering the surface amino acids of mouse antibodies to minimize epitope recognition by the human immune system, thus decreasing immunogenicity without compromising the antigenbinding site. Finally, fully human antibodies, generated either through phage display technology or from human B cells, are entirely derived from human sequences, providing the lowest immunogenicity risk and are optimal for repeated or long-term treatments.
Humanized antibodies must adhere to stringent development standards to guarantee safety and efficacy. Essential criteria include minimal
immunogenicity, high binding affinity, and substantial in vivo stability. These antibodies should also be producible in large quantities under current Good Manufacturing Practices (cGMP) to maintain consistent quality and therapeutic effectiveness. Furthermore, extensive biocompatibility testing is required to verify that these antibodies do not elicit unintended biological responses.
Antibody humanization involves several sophisticated techniques, each designed to reduce the immunogenicity of non-human antibodies while preserving or enhancing their therapeutic efficacy. These methods are essential for developing antibodies that can be used safely and effectively in human medicine.
CDR grafting is the predominant method of humanization. This technique involves transferring the CDRs from a non-human antibody to a human antibody
framework. CDRs are crucial for antigen binding, and maintaining their integrity is essential for retaining the antibody’s specificity and affinity. The primary challenge of CDR grafting is to incorporate these regions into a human framework without altering their structure. Techniques such as advanced molecular modeling and X-ray crystallography are frequently used to ensure and validate the structural compatibility of the humanized CDRs within the human framework.
SDR grafting refines the focus to the most critical parts within the CDRs—specifically, the amino acids that directly interact with the antigen. By modifying only these key areas, SDR grafting minimizes disruptions to the overall antibody structure and function, thus preserving the original binding properties more effectively than broader CDR grafting approaches.
The resurfacing method alters the antibody’s external amino acids to reduce its immunogenicity without
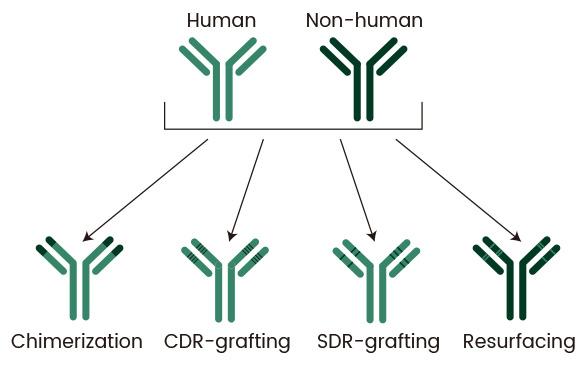
1. Evolution of Humanization Methods: Starting with chimerization, the goal has been to minimize non-human elements without compromising functionality. This includes CDR grafting, where non-human CDR loops are grafted onto a human framework, SDR grafting, which involves grafting only the binding residues of the CDRs onto the framework, and resurfacing, where exposed non-human residues are replaced with human ones.

affecting the antigen-binding sites. This is achieved by replacing surface amino acids with variants more common in humans, thereby diminishing recognition by the human immune system. This approach is especially beneficial when it is vital to preserve the integrity of CDRs to maintain binding affinity. Resurfacing can also be integrated with other grafting techniques to optimize outcomes.
Antibody library technologies utilize extensive collections of human antibodies to find sequences that serve as effective scaffolds for humanization. These libraries are generated either from B cells of immunized humans or by synthetic means such as phage display technology. Through meticulous screening, antibodies that exhibit desirable traits like high affinity, low immunogenicity, and specific effector functions are selected. This method is highly versatile and facilitates the rapid evaluation and optimization of numerous variants. Additionally, the use of transgenic mice engineered to produce human antibodies upon antigen exposure represents another innovative approach. These mice harbor human immunoglobulin genes, producing antibodies that are inherently human and typically require minimal, if any, further humanization, offering a robust platform for generating fully human antibodies for therapeutic applications.
Nanobodies, or VHH segments derived from camelid antibodies, are single-domain antibodies significantly smaller than conventional antibodies. Their compact size and unique structure enable access to cryptic epitopes inaccessible to conventional antibodies. Humanizing these nanobodies can substantially enhance their therapeutic potential by reducing
immunogenicity and improving tissue penetration, making them particularly beneficial in targeted cancer therapies and diagnostic applications.
Humanized antibodies have transformed the treatment landscape for various diseases, especially in oncology and autoimmune disorders. These antibodies are engineered to target specific antigens, such as tumors or pathological immune cells, with high specificity, thereby minimizing side effects associated with traditional treatments. Additionally, humanized antibodies play a crucial role in the diagnostic process, where their specificity enables the detection of trace amounts of biomarkers.
Sino Biological provides alpaca nanobody and murine monoclonal antibody humanization services using complementarity-determining region (CDR) grafting technology and computer-aided molecular modeling, boasting a success rate of 100% and >95% sequence homology compared to human antibody frameworks.
The future of antibody humanization looks promising, driven by continued advances in genetic engineering, molecular biology, and immunology. Emerging techniques such as next-generation sequencing are poised to further refine the precision and efficacy of humanized antibodies. The development of universal platforms for rapid antibody humanization is expected to reduce both the time and costs associated with antibody development, enabling quicker responses to emerging health challenges.
References are available at www.pharmafocusamerica.com

Kishore Hotha Global Vice President, Veranova.
In the dynamic landscape of pharmaceutical research, small- to medium-sized biotech firms, many of which operate virtually, play a crucial and integral role in early drug development. These firms focus on reaching key milestones, such as submitting investigational
This article explores biotech's fast-to-clinic/market strategy, focusing on the role of CDMOs. It emphasizes the strategic decision-making and phase-appropriate approach that streamlines R&D, the importance of flexibility and agility in adapting to regulatory and market changes, and the significant role of CDMOs in enhancing product development speed without sacrificing quality.
new drug (IND) applications or leveraging intellectual property for acquisition deals. Given their unique goals, constraints, and regulatory requirements, their approach to Chemistry, Manufacturing, and Controls (CMC) must be highly customized. The strategic use of external advisors and contract manufacturing organizations (CMOs) is critical in crafting CMC strategies tailored to each company's needs.
Transitioning from discovery to market is a complex journey, with only about 6% of molecules progressing to Phase III clinical trials. This high early attrition rate, often due to CMC complexities, underscores the need for strategic planning to navigate the CMC landscape from development to commercialization effectively. A well-thoughtout strategy can provide reassurance and confidence in facing these challenges. The rapid development of COVID-19 vaccines, achieved in less than a year, serves as a beacon of hope, demonstrating the potential to expedite development phases while upholding safety and compliance standards.
The 'Fastlane' strategy, designed to reduce market entry times, involves proactive planning and resource allocation to promising drug candidates. However, the need for speed must be carefully balanced with the responsibility to ensure patient safety and
regulatory compliance. Diligent attention to these aspects is crucial to minimize financial risks and enhance the likelihood of clinical success. (Figure 1)
The FDA's five special designations—Orphan, Fast Track, Accelerated Approval, Priority Review, and Breakthrough Therapy—accelerate the development and approval of medications for severe medical conditions with unmet needs, benefiting healthcare providers and patients. Orphan Designation targets drugs for rare diseases affecting fewer than 200,000 people in the U.S., offering benefits like tax credits and extended exclusivity. Fast Track facilitates faster development for severe conditions lacking therapies by enabling more frequent FDA interactions and a rolling review process. Accelerated Approval allows drugs
that improve on existing treatments through surrogate endpoints to enter the market sooner, pending confirmation in post-marketing studies. Priority Review shortens FDA review times from ten months to six for drugs with significant treatment advances, while Breakthrough Therapy offers all Fast Track benefits plus intensive FDA guidance for drugs that markedly improve on existing therapies, ensuring rapid and efficient market entry.
The requirements for an Investigational New Drug (IND) submission are critical in the fast-to-clinic approach, demanding a comprehensive dossier that aligns with the strict regulatory standards of the U.S. Food and Drug Administration (FDA). This dossier includes detailed preclinical study data such as pharmacological, toxicological, and pharmacokinetic evaluations to affirm the investigational product’s safety and justify human trials [8,9]. It also incorporates Chemistry, Manufacturing, and Controls (CMC) information detailing the synthesis, characterization, and quality control of the drug substance and product, ensuring their consistency and safety. Moreover, the submission must present a proposed clinical trial protocol that outlines the study design, objectives, and methodology, along with credentials of clinical investigators and informed consent forms for participants, making it a pivotal step in advancing to clinical trials.
The 'Fastlane' strategy balances proactive planning and resource allocation with patient safety and regulatory compliance.
Accelerating the drug development process involves optimizing supply chains, refining project management, implementing automation, outsourcing, and efficiently managing inventory. Emphasizing early coordination between biotech companies and Contract Development and Manufacturing Organizations (CDMOs) is vital. This strategy leverages existing data, enhances process optimization, and promotes early engagement with regulatory bodies. Early attention to CMC activities, streamlined synthesis, and formulation development are essential to tackle manufacturing challenges. Adaptive trial designs and strategic regulatory pathways like Fast Track or Priority Review can substantially shorten development timelines. Robust project management and outsourcing to CDMOs expedite progress by drawing on external expertise, ensuring that development plans are robust, scalable, and comply with regulations, thus accelerating the journey from discovery to clinical trials.
Following IND approval, phase 1 clinical studies assess human safety and efficacy, involving healthy volunteers and requiring a strategy finely tuned to drug development specifics. This initial phase focuses on process optimization and control within the CMC sections, including revisiting existing data, assessing manufacturing capabilities, leveraging technical expertise, optimizing processes, and implementing impurity controls. These activities must be meticulously calibrated, considering clinical significance, drug designation, and the urgency of a fast-to-clinic approach, with essential coordination with CDMOs.
Safety and efficacy are paramount at all stages, necessitating clear expectations and prioritizing streamlined activities to align with regulatory standards and ensure the delivery of safe, effective therapeutics. A focused review of CMC development across phases is vital from IND to NDA. Early phase requirements establish a foundation for safe drug development, while later phases demand rigorous validation and scalability to meet regulatory standards for widespread use.
For emerging biotech companies, mastering the complex landscape of Chemistry, Manufacturing, and Controls (CMC) development is pivotal in transitioning from early-stage innovation to commercial
success. Early phases focus on establishing a solid foundation through meticulous risk mitigation and adherence to regulatory and industry standards without compromising essential CMC activities. These stages are crucial for accurate toxicology assessments and a thorough understanding of the Active Pharmaceutical Ingredient (API) solid-state properties. Scalability in formulating processes is essential to ensure seamless transitions from clinical to commercial stages.
As development progresses to late-stage and commercial phases, the need for scalability and advanced analytical methods becomes more pronounced. Transitioning from disposable to multi-product, commercial-scale process trains accommodate growing production demands. This stage requires evolving analytical techniques to be robust and stable, indicating protocols crucial for maintaining regulatory compliance and ensuring product stability. Comprehensive documentation throughout this phase facilitates smoother regulatory reviews and approvals, streamlining the transition into commercial manufacturing.
A strategic partnership with a Contract Development and Manufacturing Organization (CDMO) is a cornerstone of the CMC development journey. Selecting a CDMO that aligns well with the molecule's requirements from early to late phases can significantly enhance operational speed, reduce overhead, and ensure streamlined knowledge transfer. These collaborations are tailored to address the unique challenges of each phase, recognizing
that a one-size-fits-all approach is insufficient. By planning early, meticulously documenting development processes, and customizing CMC activities to fit specific timelines, funding limitations, and regulatory frameworks, biotech companies can navigate the path to commercialization with greater assurance and efficiency.
Strategic financing in biotech development critically shapes the operational decisions of small biotech companies. These organizations must navigate the delicate balance between essential development activities that cannot be postponed and those that can be temporarily deferred when funding is limited. This balancing act is crucial as securing adequate financing supports ongoing development and ensures that regulatory activities, which are vital for long-term success, are not compromised. Practical strategies might include prioritizing core development activities directly impacting regulatory success and exploring alternative funding sources, such as partnerships or venture capital, to sustain other necessary operations without delay.
Navigating regulatory landscapes for drug approval involves regulators requiring comprehensive data detailing the synthesis or fermentation of the drug substance, along with its characterization, testing methodologies, and stability. Drug product formulations and phase-appropriate stability tests must also be established and documented. In practice, biotech companies must invest in thorough
early-phase testing and data collection to build a robust foundation for regulatory submissions, ensuring that each step of the process is well-documented and meets the stringent requirements of regulatory bodies.
Optimizing CMO partnerships is crucial for small biotech firms that rely on these organizations to handle their CMC activities. This outsourcing demands meticulous management to keep the development timeline on track. This involves establishing clear communication channels and setting precise expectations with CMOs to ensure all parties understand the timelines and critical milestones. Managing these relationships effectively is crucial, as delays or miscommunications can directly impact the supply of clinical trial materials and the support of subsequent applications.
Tailoring development strategies to clinical phases and balancing speed and quality are crucial in the competitive and fast-paced pharmaceutical industry. Biotech companies sometimes need to accept calculated quality risks to accelerate development phases. However, by gathering comprehensive data before critical stages such as Phase III trials, companies can improve the efficiency of these trials and potentially reduce overall development time. This involves strategic decision-making to determine when accelerated timelines are worth the risk and when more thorough data collection will benefit the project in the long run. (Table 1)
DECISION MAKING
API PROPERTIES
MATERIALS & VENDORS
REGULATORY COMPLIANCE
CDMO PARTNERSHIP
MANUFACTURING ADAPTABILITY
ANALYTICAL METHOD EVOLUTION
DOCUMENTATION
PROACTIVE COMMERCIAL PLANNING
TAILORED CMC PROGRAMS
QUALITY VS. SPEED
RESOURCE ALLOCATION
TECHNOLOGY TRANSFER
INTEGRATED SOLUTIONS
Risk-based decisions to balance short-term goals with long-term planning.
Initial understanding of polymorphs and crystallinity for scale-up.
Selection of raw materials and vendors based on compatibility and availability.
Adherence to basic regulatory guidelines for early development.
Aligning with CDMOs that understand the molecule's earlyphase needs.
Single-use systems or flexible process trains are used for initial production.
Developing phase-appropriate analytical methods.
Documenting decision-making and risk mitigation strategies.
Consideration for future commercialization in formulation and process design.
Custom CMC strategies to meet the company’s specific goals.
Balancing rapid development with maintaining product quality.
Strategic use of funding and expertise to meet development milestones.
Disciplined approach to technology transfer to late-phase CDMO partners.
Seeking CDMOs with integrated early-phase solutions.
Decisions must maintain the required CMC work for regulatory compliance.
Detailed characterization to set API specifications.
Considerations for scalability and long-term supply agreements.
Increasing substantiation and detail as development progresses.
Seamless transition and knowledge transfer between phases.
Transition to scalable, multiproduct trains.
Adaptation and validation of methods for later stages.
Detailed development history for regulatory purposes.
Addressing scale-up and technology transfer.
Adjusting programs based on the molecule’s progression.
Mitigating risks that could lead to delays or increased costs.
Efficient use of resources to avoid unnecessary expenditure.
Addressing potential issues in technology transfer to avoid delays.
Leveraging CDMO expertise to reduce risks during phase transitions.
Table-1: Bridging the Gap: Key Considerations for Early to Late-Phase Pharmaceutical Development Key Area
Commercial Strategy
Strategic planning for IP sale or company acquisition.
Contingency plans for rework or redevelopment of methods.
Ensuring quality and uninterrupted supply for commercialization.
Comprehensive documentation for full regulatory approval.
Integrated services for consistent quality throughout the market.
Readiness for full-scale commercial manufacturing.
Development of robust, commercial-grade analytical methods.
Maintaining a comprehensive knowledge base for the product.
Strategy for market access and distribution.
Adapting CMC strategies for large-scale operations.
Quality assurance in line with accelerated timelines.
Optimal investment in critical areas for market readiness.
Ensuring smooth scale-up to commercial manufacturing.
Utilizing comprehensive CDMO services for market launch.
The development timing and requirements for progressing a pharmaceutical product from preclinical stages through commercialization are meticulously planned to ensure efficiency and compliance with quality standards. Initially, the process involves a familiarization run-through on a small scale, typically less than 10 grams, which requires about one week per step across different development stages. As the scale increases, from kilo scale preparation (up to 100 liters) in both GMP and non-GMP environments to plant manufacturing, additional time is allocated for scaling up and ensuring quality, with increased time needed for plant manufacturing processes across all stages.
In preclinical and Phase I, the approach maintains a high-risk tolerance, emphasizing
Define Shared Objectives
Leverage Expertise
Streamline Communication
QC Synergy
Adaptive Project Plan
identifying key quality attributes of raw materials and tracking critical impurities. The process is designed to ensure purity through chromatography and stability under standard conditions. The requirements become more stringent as the product moves to Phase II and III. The focus shifts to optimizing the process through experimental verification, vendor qualification, and detailed design of experiments (DOE). The process parameters are increasingly optimized, and major impurities are managed with strategic hold points and assessments. By the commercial phase, the process is fully optimized to ensure robustness, minimal waste, and optimal yield, with all impurities identified and controlled, providing the drug’s stability and efficacy for market release.
This structured approach ensures compliance with regulatory standards and maximizes efficiency and productivity (Figure 2).
Establish clear, mutual goals ensuring both parties are aligned.
Utilize the CDMO's specialized knowledge in ADCs
Regular meetings and collaborative tools
Integrate biotech innovation with CDMO's quality systems
Allow for flexibility to innovative changes and unforeseen challenges
Performance Metrics Set up KPIs to measure progress and drive continuous improvement
Risk Management
Regulatory Alignment
Post-launch Review
Figure 2: Integration of CDMO & Biotech Partners
Identify potential risks and proactive mitigation strategies
Coordinate with CDMOS to navigate the complex regulatory landscape
Key learnings and areas for improvement for future projects
A strategy focusing on rapid progression from initial development to clinical stages emphasizes clarity and cost control. It involves specific plans for the scope of work (SoW), including timelines and pricing, to ensure projects stay within budget. By adopting flexible approaches in the discovery phase and selecting the most viable formulations early on, the process seeks to minimize later modifications, speed up the development, and ensure intellectual property can be appropriately secured.
The approach leverages different costing models that clarify deliverables, timelines, and costs, thereby maintaining budget adherence. It features flexible solid form exploration early in the discovery phase, accelerating familiarization and scale-up processes. This strategy ensures a smoother regulatory development trajectory and caters to specific program needs through customizable analytical and chemical development options, enhancing cost-effectiveness.
The commercial strategy is designed to attract early-phase clients through a well-structured proposal review and pricing strategy. It introduces a standardized template for SoW and a pricing model to leverage the project's total cost to provide more attractive earlyphase proposals. It incorporates comprehensive
Early planning, meticulous documentation, and customized CMC activities help biotech companies navigate commercialization efficiently.
chemical and analytical development strategies that streamline processes, ensure quality control from the onset, and facilitate a faster transition from development to clinical stages (Figure 3).
The holistic offering is designed to expand the customer base and build opportunities through a phase-appropriate approach. It integrates a standard proposal review process to ensure alignment with strategic objectives, optimized early developability assessment to lock in preferred forms, streamlined development to reduce process times, and tiered analytical development options that align with client needs. The production strategies are clarified early to ensure quality assurance and process control, complemented by a fixed fee pricing model that provides clear cost expectations (Figure 3).
Establish a CMC strategy early in development for readiness for scale-up and regulatory scrutiny.
Choose CDMOs that align with development needs and understand early- to late-phase transitions.
Designing a phase-appropriate CMC (Chemistry, Manufacturing, and Controls) strategy is crucial for the success of emerging biotech companies. These strategies must be tailored to balance the company's unique drivers and goals with the specific product requirements, as the "one-size-fits-all" model does not apply. Each CMC program must be individually crafted to navigate the complex

Maintain comprehensive documentation for regulatory filings and ensure knowledge transfer.
Customize each CMC program to the unique drivers of timing, funding & regulatory guidance. Avoid one-size-fits-all approach.
interplay of funding, regulatory expectations, and the practicalities of drug development. This strategic approach is essential as the pharmaceutical industry evolves, playing a critical role in a company’s ability to bring new therapies to market successfully. Emerging biotech companies face unique challenges that require bespoke CMC strategies. By focusing on a phase-appropriate approach and emphasizing careful planning and partnerships, these companies can navigate drug development complexities swiftly and successfully, bringing their innovations to market.
References are available at www.pharmafocusamerica.com
Dr. Kishore Hotha is a scientific & business leader in the pharmaceutical biotech & CDMO. He contributed commercialization of several IND, NDA & ANDAs of drug substances and products in small and large molecules, ADCs, Oligos, and peptides. He holds a Ph.D. from JNT University and an MBA from SNHU. Currently serving as Global VP at Veranova, his career includes pivotal roles at Lupin and Dr. Reddy’s, and he has contributed to over 50 publications and serving editorial boards.



For those who focus on generating value propositions through health economics and outcomes research (HEOR), the fall season is a key time period for budget planning for the upcoming fiscal year. With so many changes influencing the healthcare landscape, revisiting and advancing an organization’s evidence needs is necessary to more accurately build an effective value-based commercialization strategy.
Nicole Betor
Consultant II, Avalere
Nancy El Hoyek
Associate Principal
Laura Housman
Practice Director, Avalere
Josie Lloyd
Consultant II, Avalere
Taylor Schwartz
Principal, Avalere
Health economics and outcomes research (HEOR) can no longer be approached as a “one-and-done,” cookie-cutter process. The dynamic policy environment in the U.S., the evolving value assessment landscape, and the continued shift toward patient-centered care are impacting the evidence-generation needs of healthcare decision makers in a new and advanced way. In response to the changing marketplace, life sciences companies are proactively

utilizing HEOR to drive broader business decisions such as market access strategy, field and marketing strategy, clinical development and patient-level interventions, and to support program development. Now, more than ever, manufacturers must demonstrate both the traditionally expected and novel value of their products to a variety of stakeholders — such as commercial payers, providers, patients, and the Centers for Medicare & Medicaid Services (CMS).
In January of this year, the International Society for Pharmacoeconomics Outcomes Research (ISPOR) released the publication of its 2024-2025 Top 10 HEOR Trends Report. The report, which is based on input from ISPOR members and its Health Science Policy Council, provides insights into topics that are re-shaping how companies approach HEOR, such as real-world evidence, value assessment, artificial intelligence, health equity, and drug pricing.
One of the biggest trends discussed in the report is how the Inflation Reduction Act (IRA) is influencing HEOR. The IRA’s introduction of medicare price negotiation will formalize value assessment during later stages of the product life cycle and require manufacturers to develop an evidence-generation program to support the shifting value requirements. Value assessment informs coverage, reimbursement, utilization management decisions, and the continued shift toward patient-centered care.
Now, more than ever, manufacturers must demonstrate both the traditionally expected and novel value of their products to stakeholders…
The evidentiary needs of specific assets at different points in the product life cycle can vary significantly based on a variety of factors, including market dynamics, the policy environment, the targeted disease itself, and its impact on patients.
As life sciences companies develop their HEOR budgets, leaders can opportunistically revisit their portfolio’s research and evidence needed to support their company’s commercialization strategy. To ensure a robust HEOR strategy that prepares a manufacturer for the coming year, there are four specific elements that should be considered. Incorporating these elements offer a “PEEC” into ways to position the company for success in the current and future years to come.
As life sciences companies develop their HEOR budgets, leaders can opportunistically revisit their portfolio’s research and evidence needed to support their company’s commercialization strategy.
1. Perspective. Examine plans for evidence generation from a stakeholder’s point of view. Evaluating and updating evidence generation plans to reflect the latest dynamics and stakeholder priorities is a strategic imperative. Consider the important stakeholders (e.g., payers, providers, and patients), understand what each stakeholder expects and needs from the evidence, and incorporate those evidence needs into the research plans. Stakeholders increasingly expect manufacturers to infuse the patient perspective into evidence-generation strategies. Evidence can be used to better understand the experiences of patients with a particular condition, for example, or to describe how innovative treatments can directly and indirectly impact that experience. Conducting
patient preference studies and exploring health disparities research can highlight existing barriers to care or provide insights to determine what patients need for successful care. Ensuring evidence generation plans incorporate the patient perspective will ensure their needs can be communicated to healthcare decisionmakers.
2. Evaluation. Assess the value assessment needs based on each product’s stage in the life cycle.
Value assessment in the U.S., such as Institute for Clinical and Economic Review (ICER), the IRA, and other policy changes are shifting evidence needs across the product life cycle. Pre-launch assets may require economic modeling and value assessment planning to prepare for a potential review by ICER and other health technology assessment bodies. In-line products, meanwhile, will need to generate evidence to prepare for potential Medicare price negotiation late in a product’s life cycle, or the negotiation of competitor products in the same therapeutic area(s).
3. Expansion. Extend capabilities with innovative data partnerships and data sources.
Consider how evidence generation capabilities can be improved through partnerships and new data sources. Data on social determinants of health, for example, may uncover details of patient access challenges, or biomarker lab data can be used to identify and target specific patient sub-populations.
4. Connection. Consider HEOR’s integral relationship to other aspects of the business. Research agendas are traditionally intended to support market access, but a well-planned strategy considers the potential uses by other aspects of the business. With forethought, research can have greater impact and produce a higher return on investment if used to support field teams speaking to providers





about improving treatment protocols, marketing teams seeking to reach a target patient population, or patient support programs that need to understand patient needs and barriers to access. Incorporating a broader set of business needs in the research agenda can improve business efficiency and provide additional value across the organization.
Nicole Betor is a Consultant II at Avalere who support clients by conducting research and evaluating evidence in value assessment, HEOR, and disease burden, particularly through a patient-centered lens. In recent projects, Betor has helped design and execute value and evidence strategies, including preparation for Medicare drug price negotiations under the Inflation Reduction Act.
Nancy El Hoyek is an Associate Principal who advises life sciences companies on their HEOR, market access, and evidence generation strategies with particular focus on oncology and rare disease. El Hoyek also served as HEOR field director at Exact Sciences, where she led external HEOR engagements with health systems, policymakers, and payers.
Laura Housman is a Practice Director at Avalere and leads the Evidence & Strategy practice, applying her background in evidence generation, commercialization, and strategy to a broad range of projects. Before joining Avalere, Housman held a variety of leadership positions in industry and academia, including at Novartis, Exact Sciences, and BlueCross BlueShield of Massachusetts.
Josie Lloyd is a Consultant II who supports clients in developing and executing research projects across a range of therapeutic areas, with a focus on value strategy and HEOR. Lloyd also worked as a senior at Inovalon, where she helped clients develop evidence-generation strategies and articulate value propositions for their products.
Taylor Schwartz is a Principal who works collaboratively with clients to develop, design, and execute epidemiological and HEOR studies in a variety of therapeutic areas, in addition to health policy analyses and medical innovation value assessment studies. His expertise is in epidemiological methods, survey research methods, health, policy, and value assessment and frameworks.
Traditional drug delivery struggles with targeting and side effects. Nanorobots offer precise delivery, overcoming barriers and protecting drugs. Different types of nanorobots are being developed, but safety and ethical concerns need to be addressed.
Independent Researcher and Consultant, Bioinformatics and Biometry Department, Pharmaceutical Research Facility, Cairo

Drug delivery systems (DDS) are technologies that improve how medicine works in the body. They can target specific areas, release drugs slowly over time, and reduce side effects. DDS are
crucial for many diseases but face challenges like biological barriers and a lack of standard testing methods. More research is needed to overcome these hurdles and unlock the full potential of DDS to revolutionize medicine.
Conventional drug delivery struggles with poorly absorbed drugs, short lifespans, and unintended side effects. Oral drugs face stomach acid, enzymes, and first-pass liver metabolism. Many drugs can't reach their targets or require frequent dosing. They also struggle to deliver delicate molecules or target specific locations within cells.
Nanorobotics stands at the forefront of a groundbreaking shift in drug delivery and targeting. With the capability to precisely navigate within the body, these minuscule machines hold the promise of transforming the way drugs are delivered and targeted. From enhanced targeting abilities to on-demand release and real-time monitoring, nanorobotics offers a spectrum of advantages that could revolutionize the effectiveness of therapeutic treatments. Some of the major highlights for the potential of nanorobotics in revolutionizing drug delivery and targeting could be in the following areas:
1. Enhanced targeting ability: nanorobots can be precisely guided to target tissues, organs or cells. They can incorporate targeting ligands to home in on biomarkers overexpressed on diseased sites. This allows the selective accumulation of drugs at areas of interest.

2. Ability to cross biological barriers: The small size of nanorobots allows them to extravasate from leaky vasculature and potentially traverse barriers like the bloodbrain and blood-tumour barriers, facilitating delivery to previously inaccessible sites.
3. Improved intracellular delivery: Nanorobots may be capable of transporting therapeutic payloads across cell membranes and navigating within cells and organelles. This enables the targeting of intracellular pathogens or the delivery of drugs directly to the site of action.
4. On-demand and controlled release: Nanorobots can be designed to only release their payload upon receiving external commands or stimuli like temperature, specific enzymes or reductive environments at target sites. This ensures drugs are released only when and where needed.
5. Sustained dosing: Using refillable reservoirs or the production of drugs on-site, nanorobots may provide sustained release
Nanorobots offer unprecedented control over drug delivery for maximizing therapeutic benefits.
of drugs over extended periods, improving therapeutic compliance and outcomes.
6. Real-time monitoring and feedback: Nanorobots can incorporate diagnostic sensors to monitor physiological parameters, release profiles and therapeutic responses in real time. This enables closed-loop, personalized delivery based on individual patient needs.
7. Protection from degradation: The protective chassis of nanorobots can shield fragile cargo from degradation in circulation and at sites of delivery, improving pharmacokinetics.
8. Targeting of multiple biomarkers: Multiple targeting moieties can facilitate homing to tissues expressing a combination of biomarkers for enhanced specificity. So, in summary, nanorobotics offers unprecedented control over drug delivery for maximizing therapeutic benefits. In the future, it is possible to co-administer a miscellaneous group of nanobots that could serve complementary functions in the drug treatment targeting approach and working
in harmony through the control of Artificial Intelligence (AI). Table 1 provides comparison between nanorobots with other nanocarriers for drug delivery. (Table 1)
of nanorobots and their design
Scientists are developing tiny robots for drug delivery. These come in different forms, like DNA or protein-based, and each has its strengths. They can target specific areas and respond to body signals for precise drug release (Figure 1-3). Advancements allow these robots to handle multiple tasks and hold promise for personalized medicine. The future of drug delivery might involve these robots working with AI for real-time treatment.
Nanorobotics and its key principles in drug delivery applications: Targeting strategies
Nanorobotics involves the use of specific ligands or antibodies to guide the nanorobots to their intended targets within the body. These targeting strategies ensure that the drugs are delivered directly to the affected areas, minimizing side effects and increasing treatment efficacy (Table 2). Additionally, nanorobotics also allows for the controlled release of drugs, where the nanorobots can be programmed to release the drugs at specific times or in response to certain stimuli, further enhancing their precision and effectiveness in drug delivery. (Table 2)
Tiny robots are being designed to travel through the body, acting like doctors on a
other nanocarriers for drug delivery
MATERIAL DNA, proteins, synthetic materials Phospholipids Surfactants
TARGETING Highly specific (DNA/ protein sequences, external control)
DRUG RELEASE Controlled by triggers, environment, or external signals
MULTIFUNCTIONALITY
BIOCOMPATIBILITY
Yes (can combine drug delivery, imaging, sensing)
Highly variable depending on materials
COST High research and development costs
REGULATORY APPROVAL
None yet approved for clinical use
ADVANTAGES High targeting specificity, controlled release, potential for multifunctional therapy
DISADVANTAGES
Complex design and development, high cost, limited in vivo studies
Passive targeting (size, charge)
Passive diffusion, triggered release
Limited
Generally high
Moderate
Several approved for clinical use
Biocompatible, wellestablished technology, passive targeting
Limited drug loading capacity, potential stability issues
Passive targeting, some active targeting strategies
Passive diffusion, triggered release
Limited
Generally high
Moderate
Several approved for clinical use
Biocompatible, wellestablished technology, solubilize hydrophobic drugs
Limited drug loading capacity, potential stability issues
Additional notes: This is a rapidly evolving field, so staying updated on the latest advancements and potential applications is mandatory.
Polymers, metals, inorganic materials
Passive targeting, some active targeting strategies
Passive diffusion, triggered release
Some types (e.g., theranostic nanoparticles)
Variable depending on materials
Variable depending on material and complexity
Several approved for clinical use
Versatile, diverse range of materials and properties
Potential toxicity, safety concerns for some materials
Table 2. Key features of each type of nanorobotics for drug delivery side-by-side comparison
DRUG
ADVANTAGES
Triggered by environment
Controlled by signals Magnetic/light control
Biocompatible, programmable Responsive, controlled release Multifunctional, versatile
DISADVANTAGES Limited payload, complex design Immunogenicity, limited customization
Potential toxicity, complex control
DNA-based nanorobot
Recognition of target cells
Targeting site Drug
Flowchart 1: These diagrams can visualize the mechanism of action for each type of nanorobot, highlighting key steps
Figure 2. Schematic diagrams simplify illustrations that can depict the basic structure and components of protein-based nanorobot.
Figure 3. Schematic diagrams simplify illustrations that can depict the basic structure and components of synthetic nanorobot.
microscopic scale. These "nanorobots" can deliver drugs directly to diseased cells, treat blood clots, and even fight infections. This technology has the potential to revolutionize medicine by offering more precise and effective treatments.
Design considerations for nanorobots include biocompatibility, which ensures that the materials used in their construction do not cause harm or elicit an immune response when introduced into the body. Maneuverability is another important factor, as nanorobots need to be able to navigate through complex biological environments to reach their target. Payload capacity refers to the number of therapeutic agents or drugs that can be carried by the nanorobot, allowing for effective treatment. Lastly, drug release mechanisms are crucial for controlling the timing and dosage of medication delivery, ensuring optimal effectiveness and minimizing side effects. These considerations are essential for developing safe and efficient nanorobots for treatment and control of various medical conditions.
Nanorobots use various methods to target drug delivery. Special coatings or attached molecules help them find and bind to diseased cells. Doctors can also use magnets or sound
Nanorobots offer precise targeting, overcome biological barriers, enable intracellular delivery and provide ondemand drug release.
waves to guide them. This improves treatment effectiveness and avoids harming healthy cells. The potential of nanorobots for overcoming biological barriers and reaching specific diseased tissues
Tiny robots could deliver drugs directly to diseased areas by navigating through our body's barriers. These nanorobots would be controlled to reach specific targets, improving treatment effectiveness. Challenges remain in guiding them precisely, but success could revolutionize medicine.
One potential benefit of nanorobotic drug delivery is the ability to precisely target and deliver medications to specific cells or tissues within the body. This targeted approach can potentially minimize side effects and maximize the therapeutic effects of drugs. Additionally, nanorobots can be programmed to release drugs in a controlled manner, allowing for sustained release over a desired period.
However, there are also limitations to consider. One limitation is the potential for immune system response or rejection of the nanorobots. The body's immune system may recognize these foreign particles as threats and attempt to eliminate them, which could hinder their effectiveness. Another limitation is the current limitations in the manufacturing and scalability of nanorobots. Producing large quantities of nanorobots with consistent quality and functionality can be challenging.
Overall, while nanorobotic drug delivery holds great promise for revolutionizing medical treatments, further research and development are needed to address these limitations and ensure their successful implementation in clinical settings.
Nanorobotic drug delivery is promising but expensive due to development costs, complex manufacturing, and regulations. However, potential benefits like reduced side effects and personalized medicine could lead to long-term cost savings. Public and private investments, collaboration, and value-based pricing models
are needed to overcome economic challenges and make this technology more affordable.
Nanorobotic drug delivery promises medical advancements, but safety and ethics are crucial. Ensuring these tiny robots don't harm patients and addressing privacy concerns are key. Thorough testing, clear guidelines, and open communication are needed before widespread use.
Nanorobots hold promise for precise drug delivery and personalized medicine. Overcoming safety, manufacturing, and regulation hurdles is crucial. Integrating them with other technologies like biosensors could revolutionize healthcare by enabling real-time monitoring and improved patient outcomes.
Mostafa
has over 25 years of experience in the pharmaceutical and medical field embracing multiple projects. He has published more than 150 articles on various scientific subjects with a keen interest in AI applications in sciences that revolutionize human life and protect the environment. Former inspector in the Ministry of Health

In this article discuss about the trends, innovations, constraints and future of patient-centered drug repurposing initiatives and their impact on personalized medicine. By harnessing patient insights and cutting-edge technologies like genomics and big data analytics, we are revolutionizing the way treatments are developed for unmet medical needs. Collaboration and data sharing are pivotal in driving this transformative journey.
Vidya Niranjan
Professor and Head of the Department, Department of Biotechnology Lead- Centre of Excellence Computational Genomics, R V College of Engineering

In recent years, the pharmaceutical landscape has been rapidly evolving, driven by innovative approaches that transcend traditional drug development. One of the most promising and transformative trends is drug repurposing, particularly when guided by patient-centered initiatives. This strategy not only accelerates the availability of treatments but also aligns with the broader movement towards personalized medicine, offering a beacon of hope for patients with unmet medical needs.
Drug repurposing, or repositioning, involves finding new therapeutic uses for existing drugs. This approach leverages the known safety profiles and pharmacokinetics of approved drugs, significantly reducing the time and cost associated with bringing a new treatment to market. Historically, serendipitous discoveries—such as sildenafil's transition from an angina treatment to Viagra for erectile dysfunction— highlight the potential of repurposing. Today, systematic and patient-centered approaches are poised to take this concept to new heights. Traditional drug development has often been a top-down process driven by pharmaceutical companies and regulatory agencies. However, the advent of patientcentered drug repurposing initiatives marks a significant shift. By placing patients at the core of the research process, these initiatives aim to identify and prioritize unmet medical needs directly from those who experience them.
Patient-centered drug repurposing initiatives typically involve collaborations between patients, clinicians, researchers, and pharmaceutical companies. Patients contribute valuable insights into their conditions, experiences with current treatments, and the symptoms that impact their quality of life the most. This collaborative approach ensures that research efforts are aligned with the real-world needs of patients, potentially leading to more effective and meaningful treatments.
Several patient-centered drug repurposing initiatives have already shown promising results. The Cure Parkinson's Trust, for instance, has been instrumental in driving research on repurposing drugs for Parkinson's disease. By engaging patients and leveraging their experiences, the Trust has identified several candidate drugs that are now undergoing clinical trials. Another notable example is the Cures Within Reach initiative, which focuses on funding and supporting pilot clinical trials for repurposed drugs, particularly for rare and neglected diseases. In addition to these examples, the growing availability of big data and advanced computational tools has further empowered patient-centered approaches. Platforms that integrate patientreported outcomes, electronic health records, and genomic data enable researchers to identify potential repurposing opportunities with greater precision and efficiency. The synergy between
Drug repurposing or repositioning involves finding new therapeutic uses for existing drugs. This approach leverages the known safety profiles and pharmacokinetics of approved drugs, significantly reducing the time and cost associated with bringing a new treatment to market.
drug repurposing and personalized medicine is particularly exciting. Personalized medicine aims to tailor treatments to individual patients based on their genetic, environmental, and lifestyle factors. Drug repurposing can accelerate this vision by providing a diverse arsenal of therapeutics that can be matched to specific patient subgroups. For instance, cancer treatment is witnessing a revolution through the combination of precision oncology and drug repurposing. By identifying genetic mutations and molecular pathways driving a patient's cancer, researchers can repurpose existing drugs that target these specific abnormalities. This approach not only enhances treatment efficacy but also minimizes adverse effects by avoiding the one-size-fits-all model of traditional chemotherapy.
The integration of genomics and big data analytics plays a crucial role in the success of personalized medicine and drug repurposing. By analyzing large datasets, researchers can uncover patterns and associations that might be missed in smaller studies. For example, the use of machine learning algorithms to sift through genomic data can identify potential drug targets and repurposing opportunities with unprecedented speed and accuracy. One notable project in this domain is the National Institutes of Health's (NIH) All of Us Research Program, which aims to collect
health data from one million participants to accelerate research and improve health outcomes. This vast dataset provides a valuable resource for identifying repurposing opportunities tailored to specific genetic profiles.
Despite its promise, patient-centered drug repurposing faces several challenges. Regulatory hurdles, intellectual property concerns, and the need for robust clinical evidence can impede progress. The traditional regulatory framework is often geared towards novel drug development and adapting it to accommodate repurposed drugs requires careful consideration. Regulatory agencies like the FDA and EMA need to develop streamlined pathways for approving repurposed drugs to ensure they reach patients more quickly. Intellectual property issues also pose significant challenges. Since repurposed drugs are often off-patent or nearing the end of their patent life, there is limited financial incentive for pharmaceutical companies to invest in repurposing efforts. New models of intellectual property management, such as patent extensions or market exclusivity for repurposed uses, may be necessary to encourage investment in this area.
Fostering effective collaboration between diverse stakeholders—including patients,

researchers, and pharmaceutical companies— requires careful coordination and sustained effort. Successful patient-centered initiatives rely on transparent communication and shared goals. Collaborative platforms and consortia, such as the Open-Source Pharma Foundation, aim to create open-source models for drug discovery and development, promoting collaboration and data sharing across the pharmaceutical ecosystem.
Data sharing is another critical component. To maximize the potential of big data and computational tools, researchers must have access to comprehensive datasets. Policies that promote data sharing while protecting patient privacy are essential. Initiatives like the Global Alliance for Genomics and Health (GA4GH) work towards developing frameworks and standards for data sharing in genomics and health research.
Several case studies illustrate the potential of drug repurposing in addressing unmet medical needs. For instance, thalidomide, originally
developed as a sedative and later infamous for causing birth defects, has been repurposed for the treatment of multiple myeloma and certain complications of leprosy. Its antiangiogenic properties, discovered through research into its mechanisms, have made it a valuable drug in oncology.
Similarly, metformin, a common diabetes medication, is being investigated for its potential use in cancer prevention and treatment. Studies have shown that metformin may inhibit cancer cell growth by affecting metabolic pathways, leading to ongoing research into its repurposed applications.
Looking ahead, several innovations and strategies can further enhance the impact of patient-centered drug repurposing. One such approach is the use of artificial intelligence (AI) to predict drug repurposing opportunities. AI algorithms can analyze vast amounts of biomedical data to identify potential drug-disease connections, accelerating the discovery process. Another promising area
is the use of organ-on-a-chip technology, which allows researchers to model human organ systems on microchips. This technology can be used to test the effects of repurposed drugs on specific tissues or disease models, providing valuable insights into their potential efficacy and safety.
To overcome the challenges facing patientcentered drug repurposing, a multi-faceted approach is needed. Policymakers must develop frameworks that facilitate the approval and reimbursement of repurposed drugs. This includes creating regulatory pathways that recognize the unique aspects of repurposing and providing incentives for companies to invest in this area. Increased funding for repurposing research, particularly for rare diseases, is also crucial. Governments and philanthropic organizations can play a key role in supporting pilot studies and clinical trials for repurposed
drugs. Public-private partnerships, such as the Accelerating Medicines Partnership (AMP), can also help bridge funding gaps and drive collaborative efforts.
The intersection of drug repurposing and personalized medicine represents a transformative frontier in healthcare. By placing patients at the heart of the research process, we can accelerate the development of effective treatments and move closer to a future where medicine is truly personalized. As we continue to explore and expand these initiatives, the promise of better, faster, and more tailored healthcare becomes an increasingly attainable reality. The journey beyond the bench, driven by patient-centered innovation, is paving the way for a new era in medicine—one where the needs and voices of patients are paramount and the possibilities for improving human health are boundless. The combined efforts of researchers, clinicians, patients, and policymakers will be essential in realizing this vision, ensuring that drug repurposing and personalized medicine fulfill their potential to transform lives.
Vidya Niranjan, Ph.D., is a leading computational biologist with over 20 years of experience. She has published 124 research articles and secured $42 million in funding. She is part of NVIDIA's AI-based drug discovery program and is working on a quantum computing and protein folding project funded by the Ministry of Electronics and Information Technology, in collaboration with Amazon AWS. Her expertise spans genome analysis, drug discovery, and tool/database development, making her a prominent figure in her field.

This article discusses how AI can improve maintenance in sterile drug manufacturing. Traditional methods can be inefficient and disrupt production. AI can predict equipment failures before they happen, leading to fewer disruptions and higher quality drugs. The article explores the benefits, challenges, and future potential of AI in this field.
Independent Researcher and Consultant, Bioinformatics and Biometry Department, Pharmaceutical Research Facility, Cairo
Sterile pharmaceutical manufacturing is a complex process governed by stringent regulations to ensure product sterility and patient safety. The success of this process hinges on the reliable performance of sophisticated equipment like filling lines, isolators, and sterilizers. Equipment failures
can disrupt production, lead to product recalls, and pose significant financial and reputational risks. Traditional Preventive Maintenance (PM) programs involve scheduling routine maintenance tasks based on manufacturer recommendations or predetermined operating hours. While PM is essential, it has limitations:
• Inefficiency: Scheduled maintenance can occur unnecessarily, leading to production downtime and wasted resources.
• Rigidity: PM schedules may not account for actual equipment usage or degradation,

potentially leading to missed failures.
• Reactive Approach: PM addresses issues after they occur, increasing the risk of product contamination and production delays.
Predictive maintenance (PdM) offers a paradigm shift from reactive to proactive equipment management. PdM leverages data analytics to predict equipment failures before they occur, enabling targeted interventions to optimize productivity and ensure sterility. Artificial Intelligence (AI) and Machine Learning (ML), subfields of AI, hold immense potential for PdM in sterile manufacturing. AI algorithms
can analyze vast amounts of data from sensors embedded within equipment, including vibration, temperature, pressure, and power consumption. By identifying subtle changes in these parameters that precede failures, AI/ML models can predict equipment issues with high accuracy. The application of AI/ML for PdM is gaining traction across various industries, with demonstrably positive outcomes. Table 1 summarizes relevant fields showcasing the effectiveness of AI/ML in diverse industrial settings.
Industry Application Benefits
Automotive Anomaly detection in engines
Aerospace Predicting component failures in aircraft
Oil & Gas Monitoring pipeline integrity
Reduced downtime by 50%
Improved safety and reduced maintenance costs
Early detection of leaks and corrosion
Nevertheless, AI/ML demonstrated limited application in pharmaceuticals. AI/ML While AI/ML demonstrates success in other sectors, its application in sterile pharmaceutical manufacturing remains nascent. This is partly due to the stringent regulatory environment, the complexity of sterile processes, and the historical reliance on traditional PM practices. However, recent advancements indicate growing interest in AI/ML for pharmaceutical PdM.
Implementing AI/ML for PdM in sterile manufacturing requires a structured approach. This framework outlines the key steps involved:
The foundation of any AI/ML system is highquality data. Data for PdM can be sourced from various sensors embedded within equipment, including Vibration sensors: Detect changes in bearing health and potential imbalances. Temperature sensors: Monitor critical process parameters and identify potential overheating issues. Pressure sensors: Track pressures within isolators and sterilizers to ensure containment and proper sterilization cycles. Power consumption sensors: Analyze fluctuations in power usage that may indicate equipment malfunctions.
Raw sensor data often requires preprocessing to ensure its suitability for AI/ML models. This may involve:
• Cleaning: Removing outliers, inconsistencies, and missing values.
• Feature Engineering: Extracting relevant features from the data that can be used for model training.
• Normalization: Scaling data to a common range for optimal model performance.
Choosing the appropriate AI/ML model for
PdM depends on the specific equipment and desired outcomes. An overview of popular choices as a general guiding principle: Supervised Learning: These models require labeled historical data, where each data point is associated with a known outcome (e.g., equipment failure or normal operation). Common supervised learning algorithms for PdM include:
• Decision Trees: These algorithms create a tree-like structure to classify data points based on a series of decision rules. They can be effective for identifying root causes of equipment failures.
• Support Vector
(SVMs): SVMs create a hyperplane that separates data points belonging to different classes (e.g., normal operation vs. failure). They are well-suited for high-dimensional data and can handle limited datasets.
• Random Forests: These ensemble methods combine multiple decision trees to improve prediction accuracy and robustness. Unsupervised Learning: These models can be used when labeled data is scarce. They identify patterns and anomalies in the data to predict potential failures. Common unsupervised learning algorithms for PdM include:
•K-Means Clustering: This method groups data points into clusters based on their similarity. It can be used to identify deviations from normal operating conditions.
•Principal Component Analysis (PCA): PCA reduces data dimensionality by identifying
the most significant features, allowing for efficient model training.
Once a model is trained, it needs to be evaluated on unseen data to assess its generalizability and effectiveness. Commonly used evaluation metrics for PdM models include:
• Accuracy: The proportion of correctly predicted outcomes (e.g., failures).
• Precision: The proportion of true positives among all predicted positives.
• Recall: The proportion of true positives identified by the model.
• F1 Score: A harmonic mean of precision and recall, balancing both metrics.
Following successful evaluation, the model can be deployed for real-time PdM. This involves integrating the model with the existing data acquisition system and production line controls. The model continuously analyzes sensor data and triggers alerts when it predicts a potential failure. Continuous monitoring of the model's performance is crucial. Over time, equipment behavior and failure patterns may change. The model needs to be periodically retrained with new data to maintain its accuracy and effectiveness.
Several challenges need to be addressed for successful implementation of AI/ML-based PdM in sterile manufacturing:
• Data Quality: High-quality, well-labeled data is essential for training effective AI/ML models. Ensuring data integrity and consistency across diverse equipment types is critical.
• Regulatory Considerations: The pharmaceutical industry operates under stringent regulatory guidelines. Regulatory bodies like the FDA may require clear documentation and validation of AI/ML models used for PdM to ensure product quality and patient safety.
• Ethical Implications: AI/ML algorithms are susceptible to bias present in the training data. It is crucial to ensure fairness and explainability of the models to avoid unintended consequences.
• Infrastructure and Expertise: Implementing AI/ML requires investments in data infrastructure, computational resources, and specialized expertise in data science and machine learning.
AI/ML has the potential to revolutionize PdM in sterile manufacturing. With continued advancements in AI technology, data collection capabilities, and regulatory frameworks, there some positive outcomes that could be expected:
• Improved Accuracy and Reliability: AI/ML models will become more sophisticated, leading to more accurate and reliable predictions of equipment failures.
• Enhanced Process Optimization: PdM can be integrated with other manufacturing processes to optimize overall production efficiency and resource utilization.
• Real-time Decision Making: AI-powered systems can enable real-time decision making, allowing for immediate intervention and mitigation of potential problems.
• Advanced Analytics: Integration of AI/ML with other data analytics tools can provide deeper insights into process performance and equipment health.
To further solidify the concepts presented, exploring illustrative examples with data and tables that showcase the potential of AI/ ML for PdM in sterile manufacturing would demonstrate the benefits.
Aseptic filling lines are critical equipment for sterile product manufacturing. Early detection of potential failures in this equipment is crucial to prevent product contamination and ensure sterility.
Data Acquisition: Sensor data can be collected from various points within the filling line, including:
• Peristaltic pump vibration sensors: Monitor for potential bearing wear or imbalances that could affect filling accuracy. (Table 2)
• Filling head pressure sensors: Track pressure fluctuations during product dispensing, potentially indicating blockages or leaks. (Table 3)
• Temperature sensors: Monitor critical parameters within the filling chamber to ensure aseptic conditions. (Table 4)
Table 2: Sample Vibration Sensor Data for Aseptic Filling Line
Table 3: Sample Filling Head Pressure Sensor Data for Aseptic Filling Line
Table 4: Sample Temperature Sensor Data for Aseptic Filling Line
A supervised learning model like Random Forest can be trained using historical data labeled with equipment status (normal, warning, alert, failure). The model learns to identify patterns
in sensor data that precede failures, enabling prediction before issues escalate.
Benefits:
• Early detection of potential filling line failures allows for preventive maintenance, minimizing production downtime and waste.
• Proactive interventions can prevent product contamination and potential product recalls, ensuring patient safety and product quality.
Sterilization is a critical step in ensuring product sterility. Anomaly detection using AI/ML can be applied to monitor sterilizer performance and predict potential malfunctions.
Data Acquisition:
• Temperature sensors: Track temperature profiles within the sterilizer chamber during sterilization cycles. (Table 5)
Table 5: Sample Temperature Sensor Data for Sterilizer
Table 6: Sample Pressure Sensor Data for Sterilizer
• Pressure sensors: Monitor pressure fluctuations that could indicate leaks or malfunctioning valves. (Table 6)
An unsupervised learning model like K-means clustering can be used to identify deviations from the expected temperature and pressure profiles during sterilization cycles. The model can detect anomalies that may indicate potential equipment malfunctions before they compromise product sterility.
Benefits:
• Early detection of sterilizer anomalies allows for corrective actions before a failed sterilization cycle, preventing wasted product and ensuring sterility assurance.
• Proactive maintenance based on AI/ML insights can extend the lifespan of sterilizers and optimize equipment utilization. These examples demonstrate how AI/ ML can be used with sensor data to predict equipment failures and anomalies in sterile manufacturing. The specific data points, models, and benefits will vary depending on the equipment and desired outcomes.
While the potential of AI/ML for PdM in sterile manufacturing is promising, several areas require further exploration:
• Integration with Existing Systems: Seamless integration of AI/ML models with existing Manufacturing Execution Systems (MES) and Process Control Systems (PCS) is crucial for real-time decision making and automated responses to predicted failures.
• Standardization and Interoperability: Standardization of data formats and communication protocols across diverse equipment types would facilitate wider adoption of AI/ML in the industry.
• Cybersecurity Considerations: Robust cybersecurity measures are essential to protect sensitive data from cyberattacks that could disrupt operations or compromise product quality.
• Explainable AI: Developing explainable AI models can enhance trust and transparency in the decision-making process, addressing concerns about potential bias in AI algorithms.
Traditional PM methods are being challenged by the transformative potential of AI/ML for PdM in sterile manufacturing. By leveraging AI/ML, pharmaceutical companies can achieve proactive equipment management, optimize production processes, and ensure the highest standards of product quality and patient safety. While challenges exist, ongoing research and collaboration between industry stakeholders and regulatory bodies can pave the way for a future where AI/ML empowers a more efficient, reliable, and data-driven sterile manufacturing environment.
Mostafa Essam Eissa has over 25 years of experience in the pharmaceutical and medical field embracing multiple projects. He has published more than 150 articles on various scientific subjects with a keen interest in AI applications in sciences that revolutionize human life and protect the environment. Former inspector in the Ministry of Health

Pharmaceutical 3D printing in hospitals and pharmacies is revolutionizing medicine manufacturing. Pioneering pharmacists in hospitals have started 3D printing medicine on-site to treat their patients, using pharmaceutical 3D printers designed for precision medicine. This innovative technique enhances patient clinical outcomes, reduces waste, and optimizes the supply chain, disrupting healthcare with a rapidly evolving industry.
The concept of pharmaceutical 3D printing is not new, with the phenomenon gripping the attention of the scientific community for the past years. However, only recently has this exciting new technology started to reach patients. In the 2010s, its potential in medicine mass manufacturing for complex release profiles had been taking centre stage, with real-world
Anna Worsley CEO, FABRX-AI
Dr Alvaro Goyanes
Co-Founder, Director, FABRX
Dr Khalid Garba Mohammed
Senior Formulation Scientist, FABRX
translation being led by companies including Aprecia Pharmaceuticals. More recently, the field has been turning to the clinical implementation of smaller scale pharmaceutical 3D printing in hospital and community pharmacy settings to benefit patients in what is commonly referred to as on-demand or pointof-care manufacturing. In this article, we discuss the progress of small-batch pharmaceutical 3D printing as a point-of-care manufacturing platform, its potential in various use cases and highlights on the evolving position of regulatory agencies to support the innovation.
It is evident that one size does not fit all when it comes to drug dosing. 40-70% of

off-the-shelf medicine are not effective for patients and 7% of UK hospital admissions result from related adverse drug reactions. Medical organizations and regulatory bodies around the world are calling for personalised medicine, and for children, age-appropriate medications fit-for-purpose. In fact, studies show that only 48% to 54% of all approved medicines are commercially available for pediatrics, leading to 50% of pediatric patients receiving an unlicensed or off-label prescription. The common practice to prepare these non-commercially available prescriptions is to manually prepare them via compounding. Also used for medicine shortages, compounding involves the breaking of commercially available tablets, hand-filling capsules with powders or manually measuring out liquids. This practice
is associated with a myriad of problems such as over or under-dosing, instability of liquid medications, arduous frequent refill processes for long-term treatment and pour patient treatment adherence. In fact, it has been reported that 35% of pediatric non-adherence to compounded treatment is directly caused by these old-fashioned techniques.
It is easy to see how small batch pharmaceutical 3D printing can address these pressing medication problems, automating the compounding process to prepare easy-totake exact dosages. This automation increases accessibility to truly personalized medicine on a wider scale by reducing issues surrounding compounding. Namely, by reducing risk for the pharmacist and patient while improving patient acceptability and adherence to their medicines.
There are two scenarios for pharmaceutical 3D printing at the point-of-care, focusing on where the pharma-ink (the mixture of drug and excipients used to print medicines) is prepared. Scenario 1 is similar to standard pharmaceutical compounding workflows, preparing the drug loaded pharma-ink in the compounding pharmacy itself. Scenario 2 is when the pharmaink preparation is outsourced, being supplied to pharmacies as pre-filled cartridges containing
the drug-loaded pharma-ink, manufactured by pharmaceutical companies and CDMOs. Scenario 1 is the easier approach to implement clinically, already being used in European compounding pharmacies for patients as standard care. Pharmacists follow normal compounding workflows and regulation, preparing the pharma-ink from scratch in the compounding pharmacy using raw drug and excipient materials. The pharmacist then fills the printing cartridge themselves, inserts it into the printer, and selects the prescription and
pharma-ink protocol being used in the software and prints. Prior to use as standard care under compounding regulation, many pharmacies opt for clinical studies to be able to publish their formulation development journey and retain a research active output. These range from small paediatric studies in Spain for rare diseases to a breast cancer multi-drug polypill study involving over 200 patients in Europe’s top oncology hospital, Gustave Roussy Institute in France. Those already published have reported successful pharmacy implementation, positive staff engagement and improved patient treatment acceptability, supporting the benefits pharmaceutical 3D printing companies advertise.
Scenario 2 offers many benefits over Scenario 1, going a step further with reducing pharmacist workload, pharmacist drug exposure risk and dosing inaccuracies. Essentially the pharmacist would only need to scan the cartridge that contains the drug loaded pharmaink, insert the cartridge into the printer, input the prescription requirements and press print. All of the pharma-ink specific activities would be outsourced or automated further. Although futuristic right now, these workflows are being investigated by pharmaceutical companies around the world, working with pharmaceutical 3D printing companies to smooth out the clinical translation process. One such project was published recently, with pharmaceutical company Losan Pharma working with pharmaceutical 3D printing company FABRX, University College London and the University
of Santiago de Compostela. The team simulated scenario 2 successfully with Losan Pharma’s pre-prepared Efavirenz-loaded granulates, providing a vision of our not-too-distant future.
Pharmaceutical companies globally are exploring advanced workflows, collaborating with 3D printing firms to streamline the clinical translation process.
Pharmaceutical 3D printing enables customized drug formulations tailored to specific animals' needs. Reflecting human medicine, it allows precise dosing, unique shapes, and flavours particular to specific animals to enhance treatment compliance; everyone with a pet knows how difficult this can be. Pharmaceutical 3D printing improves treatment effectiveness and accessibility by producing on-demand, patient-specific medications, revolutionizing veterinary care with personalized therapies for various species and animal sizes.
Space...the final frontier, and another future use case for pharmaceutical 3D printing. As far off as it sounds, more stakeholders engage in this discussion every year. NASA’s Chief Medical Officer Dr J.D Polk and Dr. Neal Zapp, the Manager of NASA’s Health and Medical Technical Authority only recently visited the world leading University College London’s Pharmaceutical 3D printing research group, The Basit Group, earlier this year. Steps to make this a reality are in discussion in order to reap the benefits on offer in time for incoming missions, for example reduced supply chain dependency and rapid, dynamic pharmaceutical preparation for the hard-to-predict future needs of astronauts on long-haul missions.
Pharmaceutical 3D printing has great potential for clinical and pre-clinical trials. It allows fast production of small batches containing personalized drug loading. This speeds up the process of testing multiple drug formulations and dosages while only using small quantities of experimental drug, leading to cost savings and reduced waste. There are also opportunities to bring clinical trial batch manufacture ‘closer-to-the-patient’, improving access for participants, making recruitment easier and therefore increasing representation of diverse populations. Large, international pharmaceutical companies have already started to get involved, working with pharmaceutical 3D printing experts to help
implement 3D printing into their complex clinical trial workflows.
Pharmaceutical 3D printing at the point-ofcare can already be used as a compounding technique for personalised, non-commercially available prescriptions. Compounding regulation is how pharmacies in Europe have already started printing dosage forms for standard practice in scenario 1. However, the novelty of this innovative technology is restricting the implementation of scenario 2 where the pharma-ink is outsourced and not prepared at the point-of-care. Furthermore, there must be clear guidelines and standards to encourage pharmacists and ensure high quality of produced dosage forms. Recently, leading stakeholders in the field set-up the non-profit The International Pharmaceutical 3D Printing Initiative to help solve these issues, alongside encouraging field growth and collaboration. The initiative includes people from across the healthcare sector, including academia, hospitals, industry and regulatory bodies.
As the field grows, additional legislation will be needed to regulate the large number of printing sites popping up globally. Key regulatory authorities have already started preparing for this, with new legislation expected in the next few years from the FDA as well as the EMA in the EU, and MHRA in the UK, all with similar proposals. This
point-of-care manufacturing legislation describes hub-and-spoke style systems. One hub site will regulate multiple spokes (3D printing pharmacies and hospitals) in a geographical region or commercial network and the governing regulatory body will regulate the hubs, reducing their workload. Some companies have already started to prepare for this. In fact, FABRX has built software
directly mirroring this new legislation. Sharing the same name, their M3DIMAKER Studio Hub & Spoke software acts as hub and spoke access points, automatically saving every printing session at spoke sites and allowing for remote auditing for hubs.
Small batch pharmaceutical 3D printing is a
rapidly developing field with many exciting collaborations and projects starting in the past few years. With international pharmaceutical companies, key regulatory bodies and worldrenowned research organisations getting involved, major advancements are being achieved. American organizations are beginning to catch up to the fast-moving European players who are already integrating pharmaceutical 3D printing into standard patient care. However, it is crucial that American stakeholders act swiftly to stay competitive in this rapidly evolving sector. There are ample opportunities to be had, in regular compounding workflows able to implement immediately and in the future, truly personalised medicine as part of advanced healthcare offerings. To get involved, it is advisable to reach out to stakeholders who are already active in the field. These include members of The International Pharmaceutical 3D printing Initiative (www.pharma3dpi.org)

Dr Anna Worsley Is the CEO of FABRX-AI. Prior to this, Anna was Director of Innovation at FABRX, FABRX-AI’s parent company. She completed her PhD in 2020 in Biomaterials for Diabetic Chronic Wounds at The Royal Veterinary College and University College London (UCL).
and pharmaceutical 3D printing companies. Collaborative efforts can offer valuable insights and guidance for practical implementation.

Dr Alvaro Goyanes Goyanes Is a Lecturer at the Faculty of Pharmacy- University of Santiago de Compostela (Spain), co-founder at FABRX (UK) and Honorary Lecturer at University College London (UK). Listed amongst the World's Most Highly Influential Researchers for five years by Clarivate. Recognized world expert in 3D printing of medicines.

Dr Khalid Garba Mohammed Is a Senior Formulation Scientist at FABRX. He holds PhD from University of Milan, Italy, where he developed personalized dosage forms using 3D printing. He was Postdoc Research Fellow at Queen’s University Belfast UK, and Lecturer at Bayero University Kano, Nigeria. Khalid is experienced in academia, medicines supply chain, hospital and community pharmacy.
Gaurav Jaggi PhD, Director of Strategic Insights and Analytics, Bayer Oncology
AI is transforming the pharmaceutical industry by streamlining and enhancing various aspects of drug discovery, development, and regulatory, marketing and patient care. These advancements have a vast potential to revolutionize the pharmaceutical industry, leading to faster, more efficient drug discovery & development, improve patient management and outcomes. With so much hype around AI, it is important to be grounded by being cognizant of some actual use cases being implemented in big pharma. This article aims to highlight some real applications of AI being implemented and used within the pharma industry (Note: these stem from the author being directly involved/ leading these various AI driven projects and approaches)

AI in Pharmacovigilance (Adverse Event Identification, Validation and Reporting):
Pharma companies are pivoting towards beyond the pill solutions by harnessing the broad capabilities of digital and data analytics to research, develop, and craft experiences in new ways across entire value chain. Some pharma companies are using digitally native solutions that integrate with consumer/clinician activities and workflows to improve potential adverse events detection, accelerating potential

remediation and improved outcomes. Identifying and reporting AEs is an extremely labor intensive process for pharma companies requiring significant manual effort and is prone to errors. Enormous number of potential AEs are reported annually however only a small fraction of them (5-20%: source FDA MedWatch System) end up being actually confirmed post investigation as an AE and linked to the use of pharma companies’ drug. Each potential AE needs to be assessed and adjudicated carefully to assess if it’s a true AE or not. Therefore, it is a perfect use case for using the power of AI to automate the
process and make it more efficient/less prone to errors.
Solutions out there include chatbots/ apps on social media and other channels which consumers/ clinicians can interact with directly, and report their potential AEs that are linked to use of pharma companies’ drugs. These inputs are assessed by AI algorithms to ascertain if the symptoms being experienced were indeed linked to the manufacturer’s drug or potentially something else. This assessment needs intricate algorithms and some manual involvement initially by the AE teams to validate what the AI is adjudicating is correct.
The system/data from several AE adjudication cases feeds self-learning whereby the manual verification overrides the AI recommendation and eventually the AI is optimized by being trained on data that becomes more available over time. If classified as an AE, it is sent to the company's Drug Safety database and eventually reported to the FDA. Interestingly these clinician/patient facing chatbots can also be used in a compliant fashion to gather valuable patient/caregiver/HCP data that can inform commercial teams with several key metrics
Outcome: Using AI in AE identification, validation and reporting helps differentiate true AEs from potential AEs, filter out false positive AEs in a highly efficient fashion by reducing millions of dollars of overhead costs linked to manually performing these processes. It also helps leverage end user inputs for improved clinical and commercial insights generation, and enhanced overall consumer experience.
AI-based drug discovery in oncology has made significant advancements in recent years, transforming the way new cancer treatments are being developed. Drug screening, repurposing and target identifications are three common use cases. One of them which the author intends to share here was the topic of his PhD dissertationthe use of in silico/ AI based drug design to
Pharma companies are pivoting towards beyond the pill solutions by harnessing the broad capabilities of digital and data analytics to research, develop, and craft experiences in new ways across entire value chain.
characterize and find ligands binding to tumor suppressor protein p53, a pioneering approach which is now being followed by multiple pharma companies to identify oncology therapeutics, one of which is currently in Phase 2 trials and granted FDA Fast Track Designation. p53 is a key protein in cell cycle regulation, the defect in which causes unchecked growth of more 50% of cancers in humans. In many tumors, p53 is inactivated directly by destabilizing mutations. The aim of the project was to rescue the function of p53 by binding of small-molecule compounds. A lot of efforts have been made in past to target p53 however none have ended up in the clinic. The strategy used here was to screen and design compounds which could bind to cavities on the mutated p53 protein surface, and thus shift the folding-unfolding equilibrium toward the native folded state.
It is a very elegant and unique mechanism of action whereby the aim is to re-activate a mutated protein, and thus very different to traditional mechanisms of blocking protein targets which is more or less the standard in the industry.
In-silico/ AI based screening of small molecules that might have a stabilizing effect is a viable and attractive strategy to minimize the number of compounds to a few, which can then be experimentally tested. Using this philosophy, over 2.5 million compounds were computationally screened by employing several filters such as the Lipinski's rule of 5, pharmacophore models, small molecule docking and manual analysis of the resulting high-scoring small molecules. For the pharmacophore model, the structure of p53 core DNA binding domain with Y200C mutation (reported to affect 100,000 human cancers every year) was used as a starting point to identify small molecule compounds that bind to a the Y220C cavity. The small molecules were then tested experimentally using a battery of biophysical experimental assays ranging from the less sensitive to the most sensitive ones. This included Calorimetry, Fluorimetry, Ultracentrifugation and NMR. The study identified a few compounds that were shown to bind p53 Y220C crevice. A lead compound Phi Kan 83 was found to bind to the Y220C cavity tightly enough with a potential to stabilize the protein. The same tumor agnostic approach targeting the identical crevice in
the Y220C mutated p53 is being leveraged by pharma companies with potential impact on patients with various cancers, one of which is currently in Phase 2 trials and granted FDA Fast Track Designation. If this is approved it would be a paradigm shifting proof of concept for targeting tumor suppressor p53, by activation vs. inhibition, and a good validation of in silico-based drug discovery.
Phi Kan 83 was discovered 16 years ago. The computation capacity has significantly improved. The ligand docking simulations which took days now take hours in processing, cutting time by years in the discovery process. Insilico Medicine, a budding AI based drug discovery company combines AI with bioinformatics to identify novel drug candidates and biomarkers for aging and age-related diseases. In silico has two drug candidates identified through AI based/ computational workflows within Idiopathic Pulmonary Fibrosis (IPF). If marketed it will be another proof of concept which cut the cost of identification and commercialization by a fraction of the time to the traditional drug discovery process, and an industry changing event. Google Deep Mind's Alphafold 3 (from Isomorphic labs) released a few weeks back has the power to predict the structure and interactions of all life's molecules with unprecedented accuracy. It has opened a plethora of opportunities to maximize discovery efforts in this golden age of tech and science convergence. AlphaFold 3 is already helping Isomorphic design new

drugs for Eli Lilly and Novartis. There are other players like Benevolent, Exscientia and more who are following suit.
Another use case is a new platform being developed at University of California (San Diego), called POLYGON, is unique among AI tools for drug discovery in that it can identify molecules with multiple targets, while existing drug discovery protocols currently prioritize single target therapies. Multitarget drugs are of major interest to doctors and scientists because of their potential to deliver the same benefits as combination therapy, in which several different drugs are used together to treat cancer, but with fewer side effects (https://www.sciencedaily.com/ releases/2024/05/240506131601.htm). The possibilities are virtually endless. No wonder this use case of AI in drug discovery has
received the most attention/ funding in the last year
Pharmaceutical marketing teams are increasingly leveraging Generative AI (Gen AI) to enhance their strategies and achieve more personalized and effective engagements with healthcare professionals (HCPs) and patients. Some use cases being used summarized below:
Gen AI tools can generate tailored content for different segments of healthcare professionals and patients. This includes creating personalized emails, brochures, and educational materials that address specific
needs and preferences. Field force reps can upload an HCP profile and based on their therapeutic inclinations can carve out messages that would be better suited for them in order to ascertain positioning of different therapy options
AI models analyze vast amounts of data to predict which physicians or patients are most likely to respond to specific treatments or messages. This helps in targeting advertising efforts more precisely, improving the return on investment (ROI) for marketing campaigns:
Early patient identification: Tools/ vendors out there using labs/EHR/Claims data are able to find patients as soon as they have a diagnosis and target their HCPs to educate them about potential therapeutic options they have that could help their patients. Each of these 3 datasets have slightly nuances offerings – while labs is great in cutting the data lag by 1-2 mths, EMR helps confirm a diagnosis but has limitations to be used for any targeting purposes. It is also highly unstructured/ difficult to decipher and relatively smaller. Claims has very rich data (both breadth and depth) however often comes with a time lag linked to the claims processing. Some vendors out there are using proprietary clinical algorithms to extract the medical diagnosis from the labs data and offer a much earlier patient identification which is a very novel use case. An application
of this could be finding patients as soon as they turn metastatic and targeting them with therapies approved for metastatic disease Predict chances of metastasis based on longitudinal therapy history: Another novel approach is by using patient’s historical drug use, diagnosis, and social data spanning several years to predict when the patient will progress/ turn metastatic. The results have been surprisingly impressive with > 90% accuracy
based on their prescribing
A more common approach within the industry has been to alert the field force based on HCP prescribing patterns of a specific drug or its competitor e.g. going after HCPs who are recent decliners/ have reduced prescribing, or growers/increased in order to reinvigorate the decliners and encourage the growers.
AI can analyze market trends and competitor activities providing strategic insights that inform product positioning and marketing strategies.
HCP interviews conducted as part of the market research projects can be uploaded to company specific Chat GPT like tools in an audio format. These can be easily converted into word documents and used to synthesis/summarize the key findings pertaining to the HCP preferences, unmet needs and how different therapies address those unmet needs
AI can analyze market trends and competitor activities, providing strategic insights that inform product positioning and marketing strategies.”
Behavioral Analysis: AI tools analyze digital behavior patterns of HCPs and patients to understand their interests, preferences, and pain points. This insight allows for more effective segmentation and targeting

Sentiment Analysis: By analyzing social media, forums, and other online discussions, AI can gauge public sentiment towards certain drugs or treatments. This feedback is invaluable for adjusting marketing strategies and addressing concerns
Omni-Channel Campaigns: AI systems orchestrate and optimize campaigns across multiple channels (email, social media, webinars, etc.), ensuring a cohesive and effective marketing strategy that reaches customers wherever they are This is just tip of the iceberg. There are ‘n’ number of uses cases that will be developed which will redefine the way pharmaceutical industry operates at every functional level. The end beneficiary would be all the players in the value chain- pharma companies who will benefit from more efficient and effective processes, HCPs who will get a personalized messaging for their needs at the right time, but most importantly patients who will get more superior treatments earlier especially with AI based drug discovery promising to cut the development and approval times by >70%. The golden dawn of life sciences made possible by merging of sciences and technology has only begun.
Gaurav Jaggi, PhD is a Director of Strategic Insights and Analytics at Bayer Oncology. He leads the market research, forecasting, secondary and AI/ML focused predictive analytics, strategic insights and launch preparation efforts for Oncology brands. He has a strong scientific background with a PhD in AI based Oncology drug discovery from Cambridge University, and a passion for improving patient outcomes.

Artificial intelligence (AI) is transforming drug discovery by accelerating target identification, enhancing molecular simulations, and enabling de novo drug design. Dr. Samanta’s research team explores AI's impact, highlighting platforms like AtomNet and Insilico Medicine that are pioneering new approaches to drug development. Their research focuses on AI-assisted target identification and molecular simulations, leading to significant advancements in combating antibiotic-resistant bacteria. While AI offers immense potential, challenges like data quality and ethical concerns must be addressed. The team is optimistic about AI’s future in creating more personalized and effective treatments through continued collaboration and innovation.

Dr Samanta
Assistant Professor, in IIIT Allahabad
Ananya
PhD, Biomedical Engineering, Department of Applied Sciences, IIIT-Allahabad
Vidushi
Project Research Fellow, Department of Applied Sciences, IIIT-Allahabad.
In recent years, artificial intelligence (AI) has emerged as a transformative force in various industries, and the field of drug discovery is no exception. As members of Dr. Samanta’s research team, we frequently discuss the latest advancements in AI and their implications for drug discovery. This article provides a glimpse into our conversations, highlighting the potential, challenges, and future directions of AI-powered drug-discovery platforms.
The traditional drug discovery process is notoriously lengthy and expensive, often taking
over a decade and billions of dollars to bring a single drug to market. However, AI is poised to change this paradigm. By leveraging machine learning algorithms, deep learning models, and large-scale data analytics, AI-powered platforms are accelerating the identification of potential drug candidates, reducing costs, and improving the accuracy of predictions. Some of the key goals that can be achieved with the help of AI are as follows:
One of the most critical steps in drug discovery is the identification and validation of drug targets—proteins or genes essential for disease intervention. AI technologies are significantly improving this process by analyzing vast datasets, including genomic and clinical data, to swiftly pinpoint novel targets. Platforms like AtomNet, for example, utilize structurebased drug design to predict interactions between drug molecules and their targets with remarkable precision. This advancement not only accelerates the discovery process but also enhances the accuracy of identifying viable drug candidates.
AI is increasingly employed to conduct highfidelity molecular simulations, allowing researchers to predict key properties such as
toxicity, bioactivity, and pharmacokinetics without relying on extensive physical testing. This capability is particularly valuable in reducing the costs and time associated with traditional chemistry methods. By simulating how molecules behave in different biological environments, AI-powered platforms provide critical insights that guide the development of safer and more effective drugs.
De novo drug design, the technique of creating completely novel therapeutic compounds from scratch, is one of the most intriguing advancements in AI-driven drug discovery. Comparing this to the conventional method of screening already-existing chemical libraries is a paradigm shift. AI programs like Alphafold, and platforms with integrated AI-based tools like PepFold and AntiBP2 make it simple and quick to obtain structure and associated data about the characteristics and functions of peptides. By using data from studies such as structure-activity relationships and ML models, researchers can create new leads or design new drugs based on peptide-based drug candidates.
After identifying promising drug candidates, the next challenge lies in efficiently prioritizing them for further development. AI algorithms excel in this area, utilizing advanced ranking techniques to assess the potential of each
compound based on a variety of factors, including efficacy, safety, and manufacturability. This prioritization process significantly improves the efficiency of the drug development pipeline, ensuring that the most promising candidates advance quickly through the stages of research and clinical trials.
AI is also making its mark beyond drug design by optimizing the synthesis pathways for new compounds. This involves generating practical, cost-effective methods for manufacturing drugs at large-scale. AI-driven platforms can suggest modifications to synthesis pathways, enhancing feasibility and reducing the environmental impact of drug production. This capability is essential for bringing new therapies to market in a sustainable and economically viable manner.
As part of Dr. Samanta's research team, we have been actively contributing to the field of AI-driven drug discovery through our own research initiatives. Our team has focused on integrating AI with computational chemistry and molecular biology to identify novel drug targets and optimize drug design.
• AI-Assisted Lead Identification: In a recent study, we analyzed metagenome data from the human microbiome for the identification of powerful antimicrobial peptides. In addition to demonstrating the way AI can speed up
target discovery, this study showcased new avenues for the development of specific treatments against infections that are resistant to drugs.
• Predicting toxicity and generating analogues: Before going to the testing stage, we make all efforts to improve our lead candidate based on available data, such as with the help of AI-assisted toxicity prediction tools like ToxinPred. ToxinPred is AI-based model that helps predict the bioactivity and toxicity of novel chemicals. We have identified numerous potential peptides as a result of our investigation in this field.
• Creating ML models with larger datasets: Additionally, our group is working on development of AI and ML models for identification of potent peptide inhibitors against targets in drug-resistant bacteria. The uniqueness of our efforts is underscored by the exhaustive data curation which is the main focus of our team.
While the potential of AI in drug discovery is immense, our team acknowledges several challenges that must be addressed. The quality and diversity of data used to train AI models are critical; biased or incomplete data can lead to inaccurate predictions. Additionally, there are ethical considerations, such as data privacy and the transparency of AI decision-making processes that must be carefully managed to ensure the responsible use of AI in drug discovery.
Looking ahead, our team is optimistic about the future of AI in drug discovery. We believe that as AI technologies continue to evolve, they will enable more personalized and precise treatments for a wide range of diseases. Collaborative efforts between AI developers, pharmaceutical companies, and academic researchers will be key to realizing this potential.
The integration of AI into drug discovery is not just a trend but a fundamental shift in how we approach the development of new therapies. As Dr. Samanta’s research team continues to explore these advancements, we are excited about the possibilities that lie ahead. By harnessing the power of AI, we are moving closer to a future where life-saving drugs are discovered faster, more efficiently, and with greater accuracy.
We encourage our peers and fellow researchers to engage in discussions about AI in drug discovery. By sharing knowledge and insights, we can collectively advance this exciting frontier and contribute to the development of better treatments for patients worldwide.

Dr Samanta is working as an Assistant professor, in IIIT Allahabad, India. He is working in the area of Biochemistry and Bioinformatics. His research group is exploring the antimicrobial peptides to combat antibiotic resistance in bacteria. He did his Ph.D. and post-doctoral research from IIT Kharagpur and IISc Bangalore, India respectively. He has published research works in several international journals and has filed two Indian patents.

Ananya is pursuing PhD in Biomedical Engineering from the Department of Applied Sciences, IIITAllahabad. She completed her M. Sc. in Molecular and Cellular Biology from M. S. Ramaiah University of Applied Sciences, Bengaluru. She is currently working on the identification of antimicrobial peptides against multi-drug resistant bacteria and deciphering the underlying molecular mechanism of action.

is currently working as a Project Research Fellow at the Department of Applied Sciences, IIITAllahabad. She completed her M. Sc. In Bioinformatics from BHU, Banaras. She is currently working on the evolution of Beta-lactamases across different bacteria.

Explore the use of real-world data (RWD) and realworld evidence (RWE) in generating clinical insights, supporting regulatory decisions, and informing healthcare
interventions. Identify challenges and opportunities in leveraging RWD/RWE to complement traditional clinical trial data.
Dr Yun Lu VP & Chief Scientific & Innovation Officer, Navitas Life Sciences
Can you describe your expertise in integrating real-world data (RWD) and real-world evidence (RWE) into clinical research and decision-making processes?
Real world data and real world evidence (RWD/ RWE) and being patient centric continues to gain importance in drug development, clinical research, healthcare and regulatory decision making. Navitas Life Sciences has been leveraging our experience and expertise in RWD/ RWE through comprehensive capabilities in implementing and managing clinical registry programs.
The purpose of implementing a registry program is to characterize a disease and its progress, to define patient population and sub-populations, and to examine the use of certain approved drug or medical devices. A registry can be very valuable for informing future clinical trial design(s), for targeting possible participants for recruitment, and for care cost, and policy decisions.
The clinical registry programs provide valuable insights into the effectiveness and safety of medical interventions in real-world settings. It is essential to utilize the collected data efficiently by implementing clinical registry programs that support data aggregation and analysis. The efforts in real-world data (RWD) and real-world evidence (RWE) also empower scientists and researchers, creating incentives for centers of excellence and sites to partner with the government, disease foundations, and collaborate with pharmaceutical and biotech companies. Most importantly, clinical registry programs connect with patients, caregivers, and patient advocacy groups. While gathering patient data, it is crucial to protect patients' privacy and provide them with insightful information using the right tools to support and engage them. Leveraging our capabilities in clinical registries with RWD and RWE is essential for informing treatment decisions, optimizing patient outcomes, and ultimately improving the quality of healthcare delivery.
What motivates you to utilize RWD and RWE, and how do these data sources complement traditional clinical trial data?
Navitas Life Sciences’ years of experience in implementing RWD/RWE registry programs
motivate us to support sponsors and their stakeholders in planning programs with scientific rigor. Our scientific leads and subject matter experts direct and oversee landscape analysis and study plan development. We possess the necessary skills and experienced team to support registry operations, manage sites both in the US and internationally, provide regulatory support, and offer data coordinating center services.
Properly assessing the registry data sources is crucial for running a successful registry program. Registries typically include RWD/ RWE and other data from EHR/EMR systems, as well as data collected directly from patients, such as patient-reported outcomes, imaging data, and genetic data. Data standardization and common data elements, including HL7, CDISC, and disease-specific controlled terminologies, are vital for harmonizing and integrating multiple data sources and metadata definitions. Data quality control, management approaches, and software choices can differ significantly from those in clinical trials. A one-stop clinical registry repository powered by AI/ML becomes a valuable tool for efficiently and effectively handling the five Vs of data: volume, variety, velocity, veracity, and value. RWE and ‘Patient Centric’ are all hot topics and in high demand these days. Many organizations rush to start a registry program without enough strategic planning and careful evaluation. The common pitfall we have observed is that before starting a registry program, some organizations did not have a clearly defined purpose and goals. We have also observed that some of the registry programs may attempt to include too much data into the registry program and eventually lose focus and must then face the scope and
cost constraints. Navitas understands that it is critical to start with a defined list of questions and find the answers. Some registry programs start with limited registry stakeholder consideration and a lack of registry community consideration (for example, patient engagement, participation and involvement of clinicians and researchers).
Could you discuss specific examples where RWD and RWE have led to significant advancements in healthcare interventions or improved patient outcomes?
A disease-specific registry study was awarded to Navitas Life Sciences as a "rescue" project, previously managed by another CRO. The project goals for Navitas were to provide registry operation and regulatory support to the sponsor and the US and ex-US sites, and to serve as the Data Coordinating Center. We leveraged our deep experience in managing registry programs, building registry platforms in less than three
months, and supporting metadata-driven data harmonization and system integration to develop and implement this registry program. This registry project serves as a model for our clinical registry coordinating center and Clinical Study Rescue services, which have become a major marketing thrust for Navitas to future clients. The project also demonstrated the Navitas team’s capability to rapidly respond to evolving client requirements and tight deadlines, further refining our approach to providing timely "rescue" of clinical registry studies that require highquality, customized deliverables to meet the needs of the sponsor and multiple stakeholders.
What are the primary challenges you face when harnessing RWD and RWE for clinical insights, and how do you address them?
Clinical trials and clinical registry studies based on RWD/RWE) include patients ‘data from standard care, patient reported
outcomes, medical history and disease and treatment, and other information. Some trials and registries also include data from caregivers. A lack of clear objectives can result in poor quality or less usable data due to improper protocol design or insufficient scientific rigor. RWD/RWE-specific considerations should include:
• Protocol design and strategy
• IRB and regulatory consideration
• Data and data sources
• eSolution choices, system integration and inter-operability, especially EHR/EMR data vs. clinical research data; stakeholders
• Data sharing
• Data insights and secondary uses, and
• Sustainability
A large Disease Foundation has a multicenter community and offers genetic testing to create genetic data and sample repository. This registry program also provides genetic counselling to the participants. The foundation required the services of a Registry Coordinating Center to support their large genetic registry program. We overcame challenges and streamlined processes across 100 US and ex-US sites, implemented robust data management, successfully integrated clinical and genetic data for over 15,000 patients, and facilitated data sharing with influential global entities and government agencies and other organizations.
What role do technological advancements, such as AI and machine learning, play in analyzing and interpreting RWD and RWE for clinical insights?
Increasingly, clinical research involves RWD/
RWE collected from various sources and systems, with disparate data types and formats. Validating RWD sources and dealing with RWD data quality can be challenging. Harmonizing, integrating and interpreting such data requires clinical data scientists with a more complex background and skillset. Generative AI in healthcare and clinical research is expanding rapidly by generating content and results through analyzing large data and training examples. eSolutions empowered by AI/ML can deliver a more streamlined and automated solution for executing clinical data by using AI to integrate and automate processes from data aggregation to the generation of validation-ready datasets for data mining and insights.
More than ever, advanced statistical models and AI/ML algorithms are required to monitor data continuously throughout a trial, including RWD/RWE, as the complexity and volume of data expands. Modern technology and system integrated Modeling and Analysis Programming (MAP) offering allows data scientists and biostatisticians to seamlessly develop data models using SAS, R, and Python, with no data transfers or exports required. Models can be developed using real-time data curated in as well as imported in external historical and RWD/RWE data sets. Data Scientists can use pre-built models to perform correlation analyses, identify duplicate patients, and uncover digit preferences, or they can create new models to perform predictive analytics. Statistical models can also be created to test hypotheses, set benchmarks, and monitor data throughout the trial.
How important is collaboration among stakeholders in leveraging the full potential of RWD and RWE, and what strategies do you employ for effective collaboration?
It is very important to identify the stakeholders as early as possible; this will determine and drive the study plan, governance and policy, and maximize the potential of RWD/ RWE. A team of experts in scientific, clinical research and standard care should serve as the Scientific Lead and oversee the development of the scientific study plan and the landscape analysis. The scientific lead will interact with the principal investigator and the external experts on any relevant scientific issues regarding RWD/RWE. They will also direct RWD/RWE data and other data to be collected and scientific questions that need to be answered, help establish inclusion and exclusion criteria; strategize recruitment and retention efforts for facilities, providers and patients. Collaboration with patients’ partners and caregivers can result in efficient and effective collection of RWD, produce valuable RWE and ultimately gain useful data insights.
A fully functioning Coordinating Center to provide support for clinical study and research operations, regulatory support, and data coordination is another crucial element to the success in RWD/RWE programs. A successful Registry Coordinating Center is made up of a team of experts that ensures that the data collected across multiple sources is accurate, adheres to appropriate local and federal regulations, and has been quality reviewed. This team should have the knowledge and experience to maintain high-

quality systems and platforms throughout the RWD/RWD program life cycle and implement advancements to adapt to research and technology advancements in the field. Our unique effective collaboration approach and effort includes supplying longterm support to various National Institute of Health (NIH) Institutes and Centers (ICs), Department of Defense (DoD), and the Centers for Disease Control and Prevention (CDC), as well as disease focused foundations, commercial biopharmaceutical and medical device companies, and academic institutions. In addition, Navitas has been partnering with various organizations, vendors, networks and center of excellence. Together, our team is very agile and responsive, and able to supply the depth of RWD/RWE capability, network of subject matter experts (SMEs), and trusted experience necessary to fully implement requests and future needs.
What are your views on data sharing and privacy concerns related to RWD and RWE, and how do you ensure ethical considerations are met?
RWD/RWE is based on access to patient data and is essential for the advancement of medical research and the improvement of healthcare outcomes. To address data sharing and privacy concerns, ethical considerations must include obtaining informed consent from patients, implementing robust data anonymization techniques to protect patient identities, ensuring data security through advanced protection measures, maintaining transparency about data usage policies, and adhering to regulations like The General Data Protection Regulation (GDPR) and The Health Insurance Portability and Accountability Act (HIPAA). These measures collectively ensure the ethical use of patient data while protecting data from piracy.
Where do you see the future of RWD and RWE headed in terms of their impact on clinical research and healthcare interventions?
The future of RWD/RWE is poised to significantly impact clinical research and healthcare interventions. These data sources will enhance clinical research by providing valuable insights that lead to more effective and personalized treatments. They can expedite drug approvals as regulatory bodies increasingly consider RWE in their decision-making processes. Additionally, RWD and RWE will improve
patient outcomes by enabling healthcare providers to tailor interventions to individual needs. The integration of innovative technologies like AI and machine learning with RWD will further uncover previously unattainable patterns and predictions, revolutionizing healthcare delivery.
Can you share insights on the strategic integration of RWD and RWE into clinical decision-making processes and their contribution to improved patient outcomes?
Clinical registries play a vital role in collecting RWD and RWE, reflecting patient experiences in everyday clinical settings. This data provides insights into treatment effectiveness, safety, and quality of care. By leveraging clinical registries, healthcare professionals can discern patterns in patient outcomes and develop personalized treatment plans. Integrating clinical registries with RWD/ RWE enhances patient safety, quality of care, and evidence-based decision-making. These registries help identify knowledge gaps, optimize treatment options, and inform clinical trial designs. Additionally, they aid in policy making and cost management by providing a comprehensive understanding of medical practices and outcomes.
What advice would you give to organizations looking to integrate RWD and RWE into their clinical research and decision-making processes effectively? RWD/RWE integrate more closely with today’s clinical research. We would advise other organizations that it is important to
consider the following points when planning and implementing a clinical research program:
o Start a clinical study and/or registry with a landscape analysis and study plan, incorporating scientific rigor to inform the registry development and road map.
o Identify the stakeholders as early as possible as this will determine and drive your governance and policy; in addition, focus on patient centric needs.
o Evaluate the operation skills required to implement a clinical registry program: define resources and specialties to run a registry program including balanced consideration of the internal expertise and capacity, and the need of an experienced CRO to serve as the coordinating center.
o Consider RWD/RWE data and other data sources, platform selection and modern technology and AI/ML to streamline the process and automation.
o Work closely with the regulatory authorities, develop public-private partnerships to support ongoing activities, engage patient advocate groups, and appropriate use and share data.
Is there anything else you would like to share about your work or insights on harnessing RWD and RWE for clinical advancements?
Over the years Navitas Life Sciences has provided clinical trial support to over 100 institutions from the biopharmaceutical and medical device industries, academic institutions, disease research-based foundations, and the U.S. Federal Government. Navitas has been providing high QUALITY deliv -
erables and FLEXIBLE and RESPONSIVE “High-Touch” services to the client, resulting in both significant praise for the team as well as continued requests to perform additional services in support of the client’s RWD/RWE programs.

Dr. Lu, VP and Chief Science Officer at Navitas Life Sciences, leads global efforts in clinical data science and eSolution, with 20+ years of experience in RealWorld Data (RWD) and Evidence (RWE). Specializing in data standardization and system interoperability, she drives innovation across Phase I-IV clinical trials and disease registries. Dr. Lu plays a pivotal role in project governance, financial management, and business development within Navitas's leadership team.

With pharma companies ramping up their GenAIbased experiments, how can commercial, medical and clinical leaders in life sciences organizations make the right GenAI bets? In an interview with Pharma Focus America, Tarun Mathur, CTO, Indegene, sheds light on why GenAI is here to stay, and areas with maximum business impact.
Tarun Mathur Chief Technology Officer Indegene
1. Can you explain how GenAI is driving significant advancements in drug discovery and the specific technologies or methodologies that have been most effective in this area?
GenAI is revolutionizing the drug discovery process - which traditionally takes years and has a high attrition rate, given only 10% of candidate molecules advance to
clinical trials, per industry research. We have seen GenAI add value across multiple stages of the discovery process including target identification and validation to drug interaction prediction and lead optimization. A particularly interesting development is the use of Deep Generative Models (DGM) to optimize chemical structures based on target-derived 3D models. With large, high-quality training datasets, DGMs can generate a diverse array of valid small and macromolecules, accelerating the discovery process.
2. In what ways can GenAI contribute to an increase in productivity and efficiency within the life sciences sector, particularly over the next five years?
GenAI's capacity to process large and diverse data sets and generate preliminary reports has increased its adoption across the life sciences industry. In drug discovery, for example, GenAI improves efficiency by employing predictive and classification models to generate new molecules. Beyond discovery, GenAI's impact extends across the entire drug development value chain, notably in commercial, medical, and clinical functions. Over the next five years, we anticipate significant advancements in automation and personalization within these areas, driving a substantial increase in productivity and efficiency.
3. How does GenAI enhance the drug development process, and what are the key factors that enable it to make more accurate predictions of molecular behavior?
GenAI enhances the drug development process through its unique approach to integrating domain knowledge with AI models. Its predictive capabilities rest on three core pillars: big data, advanced algorithms, and sophisticated feature engineering. By leveraging large, domain-specific datasets - both structured and unstructuredGenAI models can learn intricate molecular patterns and relationships. Utilizing deep neural networks and embeddings to capture critical molecular features, these models produce highly reliable predictions of molecular behavior. That said, iterative refinement with real-world data and expert validation is essential to ensure the accuracy and robustness of these predictions.
4. Discuss the impact of GenAI in reducing the time-to-market for new treatments. What specific stages of drug development benefit the most from this technology?
GenAI can significantly reduce the time-tomarket for new treatments by streamlining processes across the drug development value chain. Large language and multimodal models, which process a diverse array of data including unstructured text, images, patient information, and omics data, play a pivotal role especially when implemented in agentic workflows. The most transformative impact of GenAI is seen in the commercial and clinical development stages.

In clinical development, GenAI accelerates the creation of dossiers and regulatory documents, while in commercial operations, it enables the rapid generation of personalized content for healthcare professionals and other stakeholders.
5. How are life sciences companies leveraging GenAI to offer highly personalized treatment plans, and what has been the observed impact on patient outcomes?
We are seeing an emergent use case of GenAI impacting personalized treatment plans leading to a direct effect on improving patient outcomes. At its core, GenAI enhances patient access to tailored care by increasing the efficiency of patient support programs (PSPs). These programs often involve patient education, financial assistance, disease management tools, and various administrative tasks. The impact is particularly noticeable in specialty medicines and access to rare and orphan drugs, where
benefits verification and prior authorization are traditionally time-consuming and laborintensive. GenAI's ability to process text, audio, and images enables the automation of these processes, significantly reducing wait times for patients requiring urgent treatment. Additionally, while early in development, the ability to have personal GenAI-based agents that have access to key information enables the possibility of near real-time guidance and interventions from this technology. This has the potential to make a major impact on personalized wellness and treatment plans on an individual basis.
6. In terms of reducing healthcare costs, what role does GenAI play in the personalization of patient care, and how significant are the cost savings?
GenAI can play a vital role in reducing healthcare costs by enhancing personalized patient care and streamlining operational
processes across providers, payers, and manufacturers. By automating tasks such as patient scheduling, medical coding, benefits verification, and prior authorization, analysts anticipate GenAI could deliver immediate cost savings of up to 10%, with potential reductions of up to 50% in the long term. Particularly within patient support programs (PSPs), the automation of labor-intensive tasks and the generation of actionable insights is expected to lead to cost savings of up to 75%, minimizing the need for human intervention while improving service efficiency.
7. What are the major challenges faced by life sciences companies in integrating GenAI into their existing drug development pipelines, and how are these challenges being addressed?
We see several challenges regularly flagged, primarily related to data and IT readiness, regulatory compliance, and the explainability of AI-generated insights. Many life sciences companies are actively upgrading their data and IT infrastructure and partnerships with hyperscalers to support the computational demands of GenAI applications. Regulatory compliance poses a challenge due to GenAI’s potential for generating hallucinated responses. To address this, companies are adopting a three-pronged approach: architecting GenAI-based systems with fit-for-purpose agents and risk-mitigating workflows, incorporating a human-in-the-loop mechanism
to review content and eliminate bias and inaccuracies, and implementing quality control software that flags deviations and potential hallucinations. The issue of explainability is another significant hurdle. Organizations are working to increase transparency around what GenAI models can and cannot do, while also developing rules-based mechanisms and algorithms that trace the origin and logic of AI-generated information.
8. Can you provide examples of specific cases where GenAI has led to breakthroughs in drug discovery or development, and what were the critical success factors in these cases?
Google DeepMind has had some major developments specifically targeting drug discovery. An example is a new fine-tuned model called Tx-LLM. This model is specifically tuned to solve many of the complex, data-intensive tasks involved in the development pipeline. Earlier this year, a research paper on this model was shared and it demonstrated that the model was able to outperform the existing
Many life sciences companies are actively upgrading their data and IT infrastructure and partnerships with hyperscalers to support the computational demands of GenAI applications.
state-of-the-art approaches for around 33% of the tasks involved. This will have a significant impact on the cost and timelines involved during the development process.
Critical to this is the careful fine-tuning of the model and the fact that the model is specifically targeting this application. DeepMind partnered with a deep domain expertise organization and curated data for this use case and developed a testing approach just for this.
As the GenAI landscape evolves, we expect to see a growing ecosystem of highly specialized models that are tuned and managed by deep domain experts.
9. How do life sciences companies ensure the accuracy and reliability of GenAI predictions in drug development, and what methodologies are used to validate these predictions?
We have seen several approaches to addressing this challenge. A human-in-the-loop approach is one of the most effective strategies to achieve this. By integrating human oversight at various stages - from design through to commercialization - companies can validate GenAI outputs, ensuring they meet the highest standards of accuracy and reliability. This approach is particularly crucial in use cases such as regulatory report automation, drug discovery predictions, and clinical trial design, where precision is paramount.
Another approach that is emerging - but rapidly growing - is using AI Feedback. We
have seen that GenAI models can be specifically tuned to critique and evaluate the output of other models. This AI feedback loop can then deliver feedback to the creator models. Some advanced systems could even include both human-inthe-loop and AI feedback mechanisms.
10. Discuss the role of GenAI in identifying potential side effects and adverse reactions earlier in the drug development process. How does this improve overall patient safety?
GenAI can play an important role in identifying potential side effects and adverse reactions earlier in the drug development process, thereby enhancing overall patient safety. With the proliferation of digital channels, patients and healthcare stakeholders now have multiple avenues to report adverse events. GenAI excels at processing unstructured data from diverse sources such as social media, call centers, audio files, and images. It consolidates and identifies adverse events efficiently, streamlining the quality assurance and validation processes. This not only saves time but also enables a more granular and comprehensive search across all available data sources, ensuring that adverse events are detected and managed promptly.
11. What are the strategic priorities for life sciences companies in implementing GenAI technologies, and how do these priorities align with broader industry trends?
In 2023, almost 50% of major biopharma firms surpassed industry SG&A averages, making cost reduction crucial amid a patent cliff and new regulations.
We see life sciences companies aligning their priorities closely with broader industry trends, particularly the need to reduce costs and keep pace with digital transformation. With nearly 50% of large global biopharma companies exceeding industry averages in SG&A expenses in 2023, cost reduction has become a critical focus, especially as the industry faces a looming patent cliff and new legislative pressures. Additionally, the evolving technology landscape and the shift toward digital content consumption by healthcare stakeholders, including HCPs, have necessitated a more omnichannel approach to commercialization. GenAI is well-suited to support these strategies, offering optimized, cost-effective outreach solutions that meet the demands of a digital-first market.
12. How does GenAI facilitate collaboration between different stakeholders in the life sciences sector, such as researchers, clinicians, and regulatory bodies?
With GenAI impacting the entire healthcare ecosystem and publicly available use cases
like ChatGPT, there has been a general awareness among healthcare stakeholders about the disruptive potential of the technology. GenAI, in many ways, has the potential to strengthen collaboration due to the very nature of its technology. Being a unified platform for accepting virtually all forms of data (structured, unstructured, language, images, video) allows researchers, clinicians and life sciences companies to analyze and assess data at the same table, almost simultaneously - creating a unified collaboration ecosystem. In addition, GenAI’s ability to accept information in natural language allows various stakeholders to interact in plain language, that is understood by clinicians, technologists and regulatory experts alike. The FDA too has taken steps in this regard by notifying a framework for AI in drug manufacturing. This not only shows regulatory bodies’ inclination to adopt transformational technologies but also their willingness to get manufacturers to participate in this transformation in a safe and regulated environment.
13. In what ways is GenAI expected to transform personalized medicine in the coming years, and what are the key innovations driving this transformation?
GenAI combined with other technologies such as digital health tools and wearable devices, is helping to redefine personalized medicine. Highly capable models that can interpret and reason over complex personal information
are one of the major continuous innovation areas that are fueling this transformation. Additionally, new algorithms that are making these models more efficient and can run on personal devices and protected environments with lower inference costs, will dramatically impact adoption - and ultimately transformation. We are already seeing the emergence of highly capable models that can run on smartphones and we expect to see these models continue to evolve in their advanced reasoning capabilities. This type of technological innovation will enable new patient-centric use cases at scale and transform personalized medicine.
14. What measures are being taken by life sciences companies to manage the ethical and regulatory considerations associated with the use of GenAI in drug development and patient care?

AUTHOR BIO
As companies evaluate their digital maturity and readiness to adopt AI, they are increasingly recognizing the need for a robust AI governance framework. In response, many pharma companies are establishing roles dedicated to AI compliance and governance, implementing processes such as AI risk management and third-party AI risk assessment.
Organizations are also embedding principles of ethical AI into their broader corporate Codes of Ethics, with oversight at the highest levels. To ensure these ethical standards are met, companies are also adopting quality control technologies that monitor and flag issues like hallucinations and bias in GenAI models, thereby maintaining the integrity and reliability of AI-generated insights.
Enterprises are increasingly setting up centralized governance teams and processes that not only establish and audit GenAI usage for ethics and compliance, but also establish the parameters for GenAI to reflect the voice of the brand(s), rules for explainability, auditing, and safety. To support this, many enterprises are establishing enterprise-wide technology enablers that include GenAI-based agents, testing and evaluation tools, and federated enterprise data catalogs that are GenAI-focused.
Tarun Mathur has close to three decades of experience. He has been associated with Indegene for nearly two decades. He leads the Technology domain at Indegene and his responsibilities include the development of technology-based solutions focused exclusively on the healthcare industry. His strengths lie in his technological expertise and business acumen, which help in developing various platforms and next-generation tech solutions.
Virtual clinical trials represent a paradigm shift in clinical research, leveraging remote technologies to decentralize trial operations. The discussion explores the benefits, challenges, and regulatory considerations of virtual trials, along with strategies for enhancing patient engagement, ensuring data quality, and shaping the future of decentralized research methodologies.

Sowmya Kaur Executive Vice President, Navitas Life Sciences
1. Can you elaborate on the concept of virtual clinical trials and how they differ from traditional trials in terms of design and execution?
Virtual clinical trials leverage digital tools to conduct the trial remotely, including patient recruitment, data collection, and monitoring.
Unlike traditional trials where patients often visit sites for assessments, virtual trials let patients participate from their homes or other locations using digital methods. This allows for greater flexibility, convenience, and potentially broader participant demographics. Virtual trials also often employ decentralized or hybrid designs, where data collection may occur at various
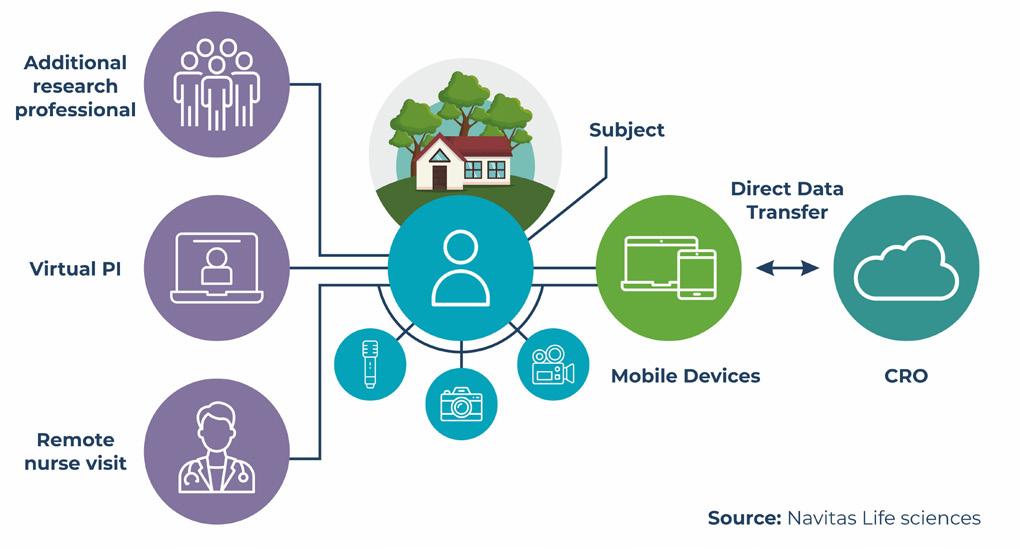
locations, including patients' homes, local clinics, or through mobile devices, rather than solely at centralized trial sites.
We made a significant move towards virtual clinical trials during the initial stages of the COVID-19 pandemic. We have always consistently worked towards enhancing our systems and technological solutions, including electronic Data Capture (eDC), imaging, Laboratory Information Management Systems (LIMS), and statistical analyses, together with their backup and Data Recovery methods. This helped in the transition to remote working.
While we had the necessary technological advancements prior to the pandemic, it served as the impetus for widespread adoption. As one of the few CROs to transition seamlessly, we played a pivotal role in supporting clinical research and medical advancement during this critical phase.
2. What are the key benefits that virtual trials offer compared to traditional models, particularly in terms of patient recruitment, retention, and overall trial efficiency?
One of the biggest benefits is that virtual trials can reach a wider pool of patients by reducing geographical barriers and offering more flexible participation options, driving patient centricity as a core value. This results in greater patient retention as well, as it reduces the burden of frequent site visits. Remote data collection and monitoring enables real time data review, reducing overall trial timelines and costs. Also, virtual trials can enhance data quality. Digital data collection tools used in virtual trials often lead to higher data quality and accuracy compared to traditional paper-based methods. Real-time data monitoring and
automated validation checks help identify and address data discrepancies promptly.
3. One of the challenges often discussed with virtual trials is ensuring data quality and integrity. How do you address this challenge, and what technologies or methodologies are crucial in maintaining high data quality standards?
To maintain high data quality standards in virtual trials, rigorous protocols, and technologies are essential. These include ensuring robust systems for secure data collection, and utilizing the right tool and AI platform, like One Clinical Analytics, to get near real time data insights. We also need to ensure data validation checks and quality control measures throughout the trial. CROs should develop standardized procedures and protocols for data collection, entry, and validation. Also, digital signatures and timestamps to authenticate data entries and ensure their integrity.
4. Patient engagement is a critical aspect of successful virtual trials. Could you share some strategies or best practices for enhancing patient engagement in decentralized research settings?
Strategies for enhancing patient engagement in virtual trials include providing clear communication and educational materials about the trial. Offering remote support services, such as telehealth consultations and virtual support groups also helps improve patient interest. Another important strategy is to incorporate patient feedback into the trial design and processes to improve the participant experi-
ence. Our personal investigators work closely with patients to ensure their interests are met and that they are comfortable throughout the study period.
5. Regulatory considerations play a significant role in the adoption of virtual trial methodologies. How do you navigate regulatory complexities to ensure compliance while leveraging the benefits of virtual trials?
To ensure compliance with regulatory requirements while leveraging the benefits of virtual trials, Navitas Life Sciences employs a proactive approach by engaging with regulatory agencies early in the trial planning process to address concerns and ensure alignment. We adhere to Good Clinical Practice (GCP) guidelines and relevant regulatory frameworks while ensuring robust data security and privacy measures to protect patient information.
6. Could you discuss the role of digital health technologies, such as wearables, remote monitoring devices, and telehealth platforms, in facilitating virtual trials and improving patient outcomes?
Digital health technologies play a crucial role in facilitating virtual trials and improving patient outcomes by enabling remote monitoring of patients’ vital signs, medication adherence, and symptoms using wearable devices and remote monitoring platforms. Facilitating telehealth consultations allows patients to communicate with healthcare providers remotely.
Moreover, providing real-time data insights to researchers allows proactive intervention and personalized care for participants. These approaches collectively contribute to the successful implementation of virtual trials while maximizing patient engagement, data quality, and regulatory compliance.
7. Collaboration and partnerships are often essential to the success of virtual trials. Can you share insights into how collaborations between pharmaceutical companies, technology providers, and healthcare organizations contribute to the advancement of virtual trial capabilities?
Collaboration between pharmaceutical companies and technology providers is crucial for accessing cutting-edge digital tools and platforms specifically designed for virtual trial implementation. By partnering with technology providers, pharmaceutical companies gain access to expertise in developing and integrating digital solutions tailored to the unique needs of clinical research.
Partnerships with healthcare organizations play a key role in enhancing participant engagement and adherence in virtual trials. This integration not only facilitates participant recruitment and retention efforts but also ensures that virtual trial protocols align with clinical practice standards, ultimately improving the overall quality of trial data.
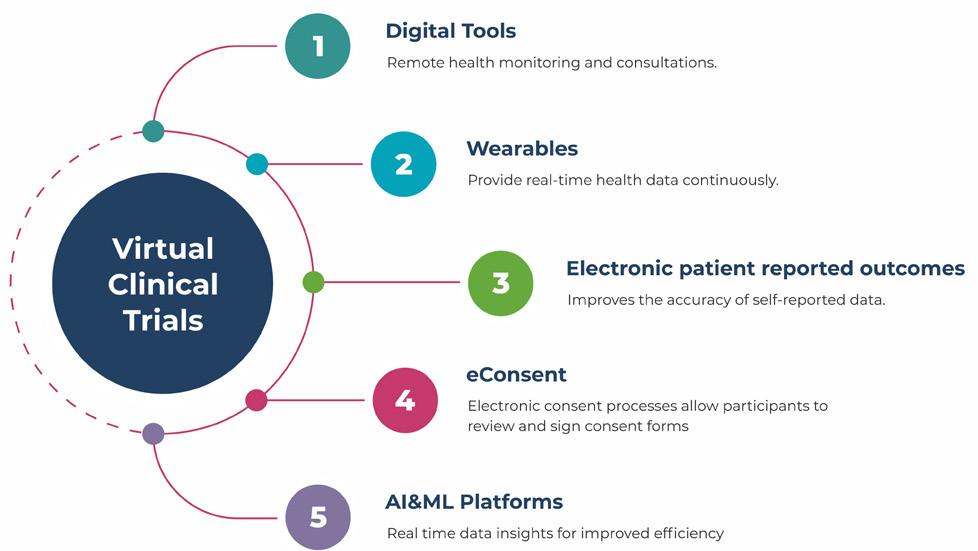
Collaborative efforts among pharmaceutical companies, technology providers, and healthcare organizations are essential for ensuring regulatory compliance and data security in virtual trials. By working together, stakeholders can navigate complex regulatory requirements and standards for virtual trial conduct. Technology providers contribute expertise in implementing robust data security and privacy measures throughout the trial process.
Partnerships streamline trial operations by leveraging the strengths of each stakeholder involved. Pharmaceutical companies provide clinical expertise and trial oversight, while technology providers offer solutions for remote data collection, monitoring, and analysis. Healthcare organizations facilitate access to patient data and medical expertise, supporting efficient trial execution.
Partnerships enable the scalability and generalizability of virtual trial models across diverse therapeutic areas and patient populations. By collaborating across sectors, pharmaceutical companies, technology providers, and healthcare organizations can adapt virtual trial strategies to varying clinical contexts and geographical locations. This collaborative approach ensures that virtual trials are accessible and applicable across a wide range of healthcare settings, ultimately maximizing their impact on improving patient outcomes.
8. How do you see the future of virtual clinical trials evolving, particularly in terms of scalability, adoption across different therapeutic areas, and integration with real-world evidence generation?
Combining data from virtual trials with real-world data sources such as electronic health records and wearable devices, will provide deeper insights into treatment effectiveness, safety profiles, and patient outcomes in real-world Settings.
The future of virtual clinical trials holds significant promise, with several key trends shaping their evolution is scalability. Virtual trials will become more scalable as technology advances and becomes more accessible. Cloud-based platforms, wearable devices, and remote monitoring tools will enable larger and more diverse patient populations to participate in trials, leading to faster recruitment and more comprehensive data collection.
Virtual trials will increasingly span across different therapeutic areas beyond traditional fields like oncology and rare diseases. Advancements in digital health technologies will enable remote monitoring of various health parameters, making virtual trials feasible for a broader range of conditions, including chronic diseases and mental health disorders.
Virtual trials will be integrated more seamlessly with Real-World Evidence (RWE) generation efforts. By combining data from virtual trials with real-world data sources such as electronic health records and wearable
devices, researchers can gain deeper insights into treatment effectiveness, safety profiles, and patient outcomes in real-world settings.
9. Are there specific case studies or success stories from Navitas Life Sciences' experience in implementing virtual trials that you can share to highlight the impact and outcomes achieved?
Certainly, it's an exciting time as we continue to push the boundaries of traditional clinical trial methodologies through the implementation of virtual trials. While it's important to acknowledge that this approach is still evolving and we're refining our strategies along the way, there have been notable successes in various aspects of several trials. Here are a few examples:
o Remote Patient Monitoring in Allergic Broncho Pulmonary Trial: In this recent trial, we successfully integrated remote patient monitoring device, Peak expiratory flow meter to track participants' respiratory function through peak expiratory flow rate (PEFR). The Investigators got real time alerts for significant decline in PEFR in real time. While this was a significant advancement, other aspects of the trial still required in-person visits, indicating a partial implementation of virtual trial elements.
o Smartphone App for a Type 2 Diabetes Mellitus trial (T2DM): Participants were provided with a smartphone app specifically designed for diabetes self-management. The app was developed in collaboration with a technology provider specializing in digital health solutions. Key features of the smartphone app included Remote Symptom Monitoring and Medication Adherence
Tracking.:
Participants used the app to regularly log their symptoms related to hypoglycemia like dizziness, sweating, palpitations etc. This allowed study coordinators to remotely monitor participants' well-being and disease progression over time.
The app also included a medication adherence feature that reminded participants to take their diabetes medication according to the prescribed schedule. Participants could also log when they took their medication, providing researchers with insights into medication adherence patterns.
As we continue to refine our processes and technologies, we aim to expand the scope of virtual trials to maximize their benefits for participants, sponsors, and research stakeholders alike.
10. In conclusion, what do you believe are the key factors that will drive the continued growth and innovation in virtual clinical trials, and what advice would you give to organizations considering adopting virtual trial methodologies?
Several key factors will continue to drive the growth and innovation in virtual clinical trials like
a. Advancements in Technology: Continuous advancements in digital health technologies, such as wearables, remote monitoring devices, and telehealth platforms, will enable more efficient and patient-centric trial processes.
b. Effective Collaborations: Collaborations between pharmaceutical companies, technology providers, and healthcare
organizations will lead to greater innovation and facilitate the development of scalable and effective virtual trial solutions.
c. Patient-Centric Approach: Emphasizing patient engagement and participation will be crucial for the success of virtual trials.
Strategies that prioritize patient convenience, communication, and feedback will enhance recruitment, retention, and overall trial outcomes.
d. Real-World Evidence Integration: Integrating virtual trial data with real-world evidence sources will enrich the understanding of treatment effectiveness and safety profiles, driving evidence-based decision-making in drug development.
For organizations considering adopting virtual trial methodologies, they should consider beginning with hybrid approach to assess the feasibility and effectiveness of virtual trial approaches within their organization. This allows for iterative learning and optimization before scaling up.

Strategic partnerships with virtual clinical trial specialists will help leverage collective expertise and resources in implementing virtual trial methodologies successfully
They should invest in technology and infrastructure to support virtual trial operations securely and efficiently. They must stay informed about regulatory requirements and seek guidance from regulatory experts to ensure compliance with relevant guidelines and standards throughout the trial process. They should develop strategies to engage patients effectively throughout the trial, including clear communication, remote support services, and incorporating patient feedback into trial design and processes. The ideal step would be to collaborate between stakeholders. Strategic partnerships with virtual clinical trial specialists will help leverage collective expertise and resources in implementing virtual trial methodologies successfully.
Sowmya Kaur, Executive Vice President at Navitas Life Sciences, has worked across multiple aspects of the industry including operations, business development, and strategy with leading industry players. With a career spanning over 2 decades, she has a successful track record of building and leading Clinical Development engagements across Globe and Emerging Markets with successful delivery of a portfolio of projects.
The current landscape of psychedelics research shows great promise. FDA guidance presents a thorough consideration, yet these trials face substantive obstacles. Appreciating the range of psychedelic options and the perspectives of those conducting the trials as well as those participating is needed to fully understand how to move forward.
Sara Reed
Documentary Producer and Clinical Research Participant
Currien MacDonald
Medical Chair Director, WCG
Psychedelics as a class are not just another study drug. They have recently drawn intense scrutiny for being potentially effective in treating serious mental health conditions that lack good treatment options. However, they have a host of unique associations and face immense obstacles to achieving regulatory approval. They are classified as Schedule I drugs, without current accepted medical use and with high abuse potential. Further complicating matters, psychedelic compounds may not have similar effects or side effects even if
they have a similar structure or produce a similar subjective experience. Taking a closer look at the participant experience in a clinical study with a psychedelic can help the research community understand the journey, and how to help the journey be the best it can.
The general population is somewhat aware of psychedelics and has already established opinions about them. A U.S. survey of 1,800 adults in 2022 showed that only 15% of respondents had a positive attitude toward
psychedelics. Of that 15%, only 24% were in favor of using psychedelics for mental health conditions, although that number rose to 36% if they had seen a therapist recently. Another U.S. survey of 1,500 adults conducted one year later revealed that 61% of respondents supported legalizing regulated therapeutic access to psychedelics with 35% expressing strong support. However, 61% of that sample thought that psychedelics were not good for society and 69% said that those drugs were not for someone like me.
If that is public opinion, what does data from clinical research into common and serious mental health conditions such as depression, anxiety, and post-traumatic stress disorder (PTSD) show? Systematic reviews conducted from 2018 to 2022 of psilocybin, LSD, and ayahuasca clinical trials, to pick three of the many products being studied, showed strong evidence of improvement in generalized anxiety disorder, alcohol dependence, major depressive disorder including treatment-resistant depression, and cancer-related anxiety. These included studies of varying quality, and a growing body of well-designed clinical trials. These effects included results from single treatments, with immediate, significant and enduring effects. Of note, the studies also showed these treatments to be feasible and with limited side effects. Moreover, the rate and pace of clinical trials in psychedelics has increased exponentially, from fewer than 50
published articles in 1990 to more than 700 in 2020.
Most importantly, these products are showing promise for significant diseases without good treatment pathways. Two examples are major depressive disorder (MDD) and PTSD. From a simplistic economic viewpoint, MDD has an annual $93 billion impact on U.S. health and society with about $44 billion of that being treatment resistant disease. PTSD has a staggering and underappreciated social and economic impact of about $232 billion per year.
While some researchers are going through the required clinical trials to develop an FDA-approved medicine, other avenues are also being taken, including several U.S. states introducing their own measures. For example, Oregon’s Psilocybin Services Act requires a trained and licensed facilitator to administer psilocybin at a state facility and does not specify the purpose of the use. Two of Utah’s largest healthcare systems now include psilocybin and MDMA [3, 4-Methylenedioxymethamphetamine] treatment as options if they are provided in a medical facility. California
Psychedelics are classified as Schedule I drugs, without current accepted medical use and with high abuse potential.
bill 1012 is in review, which would establish a board of regulated psychedelic facilitators to license and systematize control of the use of psychedelic substances.
While this background provides context, we can learn a deeper truth by listening to someone who has been there. Sara is brave and generous enough to share her story.
“I’d never been part of a clinical study, so I was walking into an entirely new world,” says Sara. “Before this, I had spent a lot of time trying to find alternative ways to deal with my anxieties or depression. By the time I found the research group, my psychiatrist had deemed me treatment resistant.”
“The first study was using an esketamine nasal spray. Esketamine is the S enantiomer of ketamine. I knew of ketamine! I had tried IV ketamine, but I unfortunately wasn’t able to complete the six to eight treatment sessions, due to work taking me out of the city and it was an expensive endeavor that wasn’t covered by insurance. Esketamine was essentially ketamine, but it would be administered differently this time, and I wouldn’t have a therapist by my side. I was hopeful that it would help me and that my participation would help in getting this treatment more widespread.”
“The paperwork, blood draws, urine samples, and questionnaires, all made sense. When the study coordinator pulled out this mess of cords with nodes attached to them to place on my head, it made me feel like I was in some sort
of sci-fi movie. I kept wondering if what was being measured looked ‘normal.’”
“For treatment, the doctor/therapist came in, asked a few questions, and pointed to a camera in the room so I could simply raise my hand, and they’d see it and come in. Then the nurse came in the room and handed me the nasal spray, which I’d administer myself. They walked me through it; one spray in each nostril, and wait 10 minutes to repeat the process. Then the nurse turned the lights off and left me to go on my little journey. I was terrified, being in a dark room alone, but I was open to the process, so why the hell not? I did raise my hand once or twice in that dark room. The doctor came in and grounded me until I returned to calm, and they’d leave me to journey. It’s hard to sum up my journeys, but here’s a bit from a journal entry after my first treatment:”
It feels like I’m going into a 2D world. Like I’m becoming the static you’d see on an old TV. Difficult to describe.
I started to cry. A heavy cry. Sobbing, you know, where you have trouble catching your breath.
The staff came in and helped me. We’d talked about setting an intention at the beginning. He asked me to remember what my intention was. Trust myself. My decisions. Just trust. “What does that look like?” he asked...
“The ocean,” I responded. Why the ocean? I don’t know. It’s vast. It’s deep. Has a lot of weight. It takes trust to dive in. It’s vast and unknown, like putting trust in someone
In a U.S. survey of 1,500 adults, 61% of respondents supported legalizing regulated therapeutic access to psychedelics.
else. Another person’s life is vastly different from my own. I don’t know their thoughts or how they think. And I love that. I love that difference. That variety.
But it’s also risky, incredibly risky to “dive in” with another person. What’s under the surface? I can handle it at first. It may even be adrenalin pumping, exciting. But then I become so exhausted. How much longer can I swim like this before I drown? Is it worth it?
How can I free myself to let myself go in the water? How can I take that dive and trust that it will be there and will provide safety? I just do. My trust is the most important. I spend most of my time worrying about pleasing others. But my approval or trust is vital. I know this deep down. I can do this.
“In the last session I had, I reached a black wall and fought going beyond it. I felt as if I’d die if I approached it. I surprised myself when I thought, ‘Well, if I die, I die!’ I can’t tell you how freeing that was. I then slid down this slide into an abyss that went beyond the black wall which I could have never fathomed.
I think this was a big turning point for me. I finally let go and let myself release my iron grip on what I thought was living. Turns out, there’s a lot more out there. I still feel sad this was my last session. But life got in the way.”
“My second study was with LSD. Again, I did my best to clear any expectations that showed up, especially as there was a chance that I could receive all placebo pills. My family had a lot of hesitation. They had been informed their entire lives of the dangers of LSD and other psychedelics. They thought that people who did LSD wore colorful clothes, were free loving, didn’t respect our country, and many of them went mad! The going mad part seemed to be weighing the heaviest. I could see it in my parents’ eyes, the concern that they might lose me if I took part in this study. I felt some support, but it was more of a hesitant support.”
“During the study, I was poked about the amount I expected but questioned a lot more. I’d see someone on a video call to answer a series of questions about anxieties and depression. It wasn’t unusual for me to cry. Then, the coordinator would bring me to another room to answer another computer’s questions. Often, the questions were similar if not the exact questioning I had just gone through. This process was quite exhausting.”
“Dosing day was an adventure. I tried to put myself in the most comfortable place I could be, to clear my mind, to be open to whatever would come my way. The two Dosing Session Monitors (DSMs) met with me once prior
to dosing day, which was good, but I still felt like they were strangers on dosing day. I wished one of the DSMs could be someone I’d been pouring my soul out to with those questionnaires.”
“I could talk for days about the various experiences I had during dosing day. Some experiences I am still processing. A common theme throughout was that I saw externally what I was feeling internally. When I wasn’t sure of the DSMs, their faces turned scary, edges of the room were harsh and sharp. Unfriendly. In another moment, I felt safe and calm, the DSMs’ faces looked almost angelic, and the room was soft, glowing, and shimmery even. I was safe and comfortable to reflect on so many deep and meaningful things in my life. Honestly, time didn’t make any sense and I couldn’t tell you how things happened linearly. But I think initially unsettling, scared, then feeling in awe of some of the realizations of what my mind is capable of. Then bringing myself back time and time again to when I felt as calm as I have ever felt. In a field. Gently swaying grasses. A feeling of childlike wonder and contentment for my life.”
Traveling through the layers in which a clinical study with a psychedelic exists, from historical to public opinion, clinical data to one person’s experience is quite a journey. Everyone, including the sponsor, researcher, institutional review board (IRB), and participant, needs
to be mindful of how the layers can impact each perspective. Doing this will maximize the potential benefits from the knowledge gained and minimize the risks of employing these powerful products. References are available at www.pharmafocusamerica.com

Sara Reed has an MFA in Documentary Media from Northwestern University and has a decade of freelance video production experience, from directing to editing, spanning North, Central/South America, and Africa, and renowned clients such as Discovery, HBO, and NASA. Her research advocacy is driven by her participation in multiple trials.

Currien MacDonald , MD, CIP, is Medical Chair Director at WCG and contributes to biosafety reviews for WCG’s institutional biosafety committee. Prior to joining WCG, Dr. MacDonald served for four years as the medical director for Aspire IRB, and vicechair of Canadian REB.

Book Description: Most Pharmacology taught in medical schools refers to the use of drugs, not how they are made, evaluated and improved. This book does that with a compilation of the latest research as it is applied to drug discovery and evaluation. Pharmacology is a unique science having scales of drug activity that transcend the assays where they are measured to enable prediction of activity in all (including the therapeutic) systems. This book breaks down complex discovery techniques on binding, functional, orthosteric and allosteric assays to quantify drug effect and then goes on to consider the use of such molecules in vivo for therapeutic advantage.
Thus, Pharmacokinetics (the delivery of drugs to whole body systems) and early safety (new drugs must cause no harm) are then considered along with the scales of drug activity in whole body systems aimed for therapy. Pharmacology is a fast paced and changing science relying on the latest technology; this 6th edition considers the latest cutting edge technology as it is applied to drug research.
1. Dr. Kenakin, your extensive background in both academic and industrial pharmacology research uniquely positions you to write a book like "A Pharmacology Primer." Can you share what inspired you to write this book and how your experiences influenced its content?
Actually, the beginnings of this book were instigated by a need to educate newly hired biology and chemistry staff who had no background in pharmacology when they joined the

Terry Kenakin Professor, University of North Carolina School of Medicine
company where I worked, GlaxoSmithKline. We realized there was no source of information and I wrote the course which then became a book.
Other books simply discussed the use of drugs for treatment of various ailments but not how to make and improve them.
2. In your book, you emphasize the application of pharmacology in drug discovery. What are some of the most significant changes you’ve observed in this field over the years, and how does the 6th edition address these changes? Pharmacologists are almost always working in systems they do not fully understand… physiology and Nature still hold many mysteries. This being the case, pharmacology is uniquely based on new technologies to unveil these new secrets of physiological system and thus, it is a fast paced field of endeavor requiring regularly updated information.
Genomics, bio and chemoinformatics, structural biology, highthroughput technologies, and virtual screening have transformed pharmacology in the past few years and technologies such as AI promise to do more of this.
3. One of the key themes in your book is the breakdown of complex discovery techniques. Could you elaborate on how binding, functional, orthosteric, and allosteric assays contribute to quantifying drug effects and their therapeutic applications?
These are the building blocks of Pharmacologic research that form the
framework of discovery. Two fundamentally different approaches to the study of drug-target interaction are through binding (physical measurement of molecules binding to protein) and function (measuring the cellular consequences of drugs binding to targets). Each of these have their strengths and limitations and yield complimentary information.
Two other fundamentally different approaches measure the actual interactions of molecules and targets: orthosteric and allosteric. Orthosteric basically measures the ‘steric hindrance’ of bodies interfering with each other as they compete for a common binding site. Allosteric is where drugs bind to a separate site on the target to modify physiology through changing the shape of the protein.
4. The book discusses the use of new technologies for screening, such as virtual, DNA-encoded libraries, and fragment-based approaches. How have these technologies revolutionized the process of drug discovery, and what future advancements do you foresee in this area?
Basically all of these technologies have drastically increased the scope and capacity to test vast numbers of chemical structures. Before the advent of these technologies, libraries of thousands of compounds could be tested with robotic screening; with these new approaches, millions to billions of new structures can now be tested and this radically enhances the chances of finding fruitful binding ‘hits’.
It is defensible to say that probably now any target in physiology can be ‘drugged’ (i.e. a molecule found that binds to it).
5. You’ve highlighted phenotypic (target agnostic) screening for new leads and the determination of drug targets in this edition. Can you explain the importance of this approach in modern drug discovery and its potential impact on the industry?
Two basic approaches to screening are ‘targetbased’ (where a biological target is identified and then molecules tested for binding) and ‘systems-based’ where a complex system is exposed to a myriad of molecules to explore the impact on the system.
The aims of these approaches are different; target-based approaches are designed to yield new drug molecules while systems-based approaches may do this but really are primarily designed to explore new mechanisms in physiology that can be manipulated chemically with molecules. Thus, systems screening can yield new approaches to therapy that would not be evident with target-based approaches.
6. Pharmacokinetics and early safety are crucial aspects of drug development. How does your book address these topics, and what are some key considerations for researchers in ensuring new drugs cause no harm while being effective?
If a biologically active molecule cannot access the tissue in the body where the therapeutic need resides or if, when it does, it causes more harm than good, then it is useless as a therapeutic drug. Thus, pharmacokinetics
(the study of drug absorption, distribution, metabolism and excretion) are critical to the discovery process.
Similarly, although drug safety takes the fore in late stage drug candidate development, it also plays a pivotal role in early development.
For example, in a recent program for HIV-1 treatment for AIDs, the candidate chemical scaffold was tested in the hERG assay and found to have fatal cardiac activity. There would be no sense in further developing this scaffold until this harmful property was eliminated through medicinal chemistry and to the program did this before continuing to optimize the primary effects on HIV-1.
7. The concept of receptor theory and allosteric protein function is central to your research interests. How do these concepts play a role in the strategic drug discovery techniques presented in your book?
Drug mode of action is determined mainly by comparing drug effect to mathematical models. These explicitly define how the drug works and also provide theoretical predictions that can be tested with further experimentation.
This is especially important in pharmacology since drugs are discovered and studied in test systems (not the therapeutic ones) and predictions of activities in other systems (including the therapeutic one) must be inferred until testing in humans is done.
An especially fruitful area of such modeling is in the allosteric nature of proteins since these complex effects have the to potential to provide molecules with a wide range of effects not possible through simple steric hindrance systems.
Our view of what is happening in our experiment is obtained through our assay, and as the Nobel Laureate Sir James Black wrote “.The prismatic qualities of the assay distort our view in obscure ways and degrees.” (1993; Nobel Lectures: Physiology and Medicine).
8. Your book includes new material on target engagement and demonstrating the physical interaction of molecules with drug targets. Could you provide an example of how this process works and its significance in the context of drug discovery?
In the beginnings of Pharmacology, much was inferred from the exposure of drugs to tissues, observing a response and then having to assume that a specific interaction took place to cause that response.
With modern techniques to actually prove a physical interaction, a greater detailed knowledge can be gained which can be used to optimize such interactions and make better drugs. Often, the target for a molecule may not be known and thus isolated test -
ing of the interaction of a molecule with the isolated target could be useful.
Thus, techniques such as CETSA (Cellular Thermal Shift Assay) or Isothermal Titration
Calorimetry can identify specific interactions and allow more detailed analyses of drug binding.
9. With advancements in genomics, CRISPR, and mRNA therapies, how has the landscape of drug discovery evolved, and what role do these technologies play in the strategies outlined in your book?
As stated previously, pharmacologists are often working in uncharted physiological territory and specific modification of that territory through techniques such as PROTAC or CRISPR to remove components of systems or mRNA to add components can be immeasurably valuable.
Thus, the observation of the modification of a particular part of a physiological system (leaving the rest of the system intact) on drug effect can go a long ways in identifying mechanism of action and indicating further avenues of improving drug effect.
10. "A Pharmacology Primer" features full-color illustrations and new examples to aid understanding. How important is visual representation in conveying complex pharmacological concepts, and what approach did you take to ensure clarity and comprehension for readers?
The depiction of complicated schemes visually is critically important to the understand-
ing of pharmacologic effect on physiology. Oftentimes a drug will affect numerous parts of a system and it is important to see the array of outcomes in a concerted way; diagrams and flowcharts can do that.
One of the most versatile and important tools in pharmacology is the ‘dose response’ curve which functions as compact statement about drug action, potency and efficacy; variation in dose-response curves thus becomes a universal language for expressing drug effect.
11. The reorganization of content for better flow and clarity in this edition is a notable feature. What motivated these changes, and how do they enhance the reader's learning experience?
Such reorganizations basically reflect changing technology and emphasis in drug discovery as new techniques yield new information. Twenty years ago, the cutting edge may have been robot highthroughput screening whereas now genomics, structural biology and virtual screening have taken the fore.
Hopefully, this book will reflect the changing times in discovery and guide readers who may not have the time to keep up with then changes. In addition, the new ideas presented are put into historical perspective where readers can see their development and from that, also see their potential.
12. As a professor at the University Of North Carolina School Of Medicine, how do you integrate the principles and techniques from your book into your teaching and research?
In years past, there was a student perception that scholarly activity could only be pursued in an academic environment and thus pharmacology and drug discovery took a secondary career choice role.
Now it is realized that industrial drug discovery is every bit as academically challenging and rewarding; drugs do not automatically appear from screening and industrial pharmacologists need to be practicing scientists in the pharmacologic community at the cutting edge to apply new knowledge to the discovery process.
Moreover, the face of drug discovery has changed and now it is pursued in academia with equal vigor as it is in in industry. At UNC we offer specific courses in drug discovery to expose students to careers in discovery utilizing pharmacology since this, to a large extent, is where the new opportunities lie. Such courses are increasingly being developed in other universities as well.
13. Given the rapid pace of change in pharmacology and drug discovery, what advice would you give to students and early-career researchers entering this field?
I tell students to imagine the ‘pure’ sciences such as genetics, chemistry, physiology, biochemistry as purebred dogs; in this setting, a pharmacologist would be represented by the greatest cross-bred mix (‘mutt’ if you will) since we take from all of these disciplines towards a single pursuit, the chemical control of physiology.
So I council students to ‘think big’ and see the big picture of utilizing all of the sciences toward the discovery of new thera-
pies. Given this, they should not hang their star on the latest fashionable technology but rather incorporate what it brings to the field and then think how it can be extended.
14. Lastly, what are your hopes for the future of pharmacology and drug discovery, and how do you envision your book contributing to the ongoing advancements in this critical field? The expansion of understanding of physi -
ological systems and pathological processes has been, and still is, remarkable. Even as little as twenty years ago it would not have been imagined that we would have the means to manipulate systems chemically in the way we do so today.
Given this, the horizon is very bright indeed for new drug discovery and this book is privileged and honored to be riding that train; there will always be a need for revision as the field never stops progressing.
Terry Kenakin received a BS in Chemistry and PhD in Pharmacology from the University of Alberta, Canada and then went on to a post-doctoral fellowship in London, UK. He worked 32 years in industrial drug research (7 yrs. Burroughs Welcome, 25 yrs. GlaxoSmithKline) and currently is a professor of Pharmacology at University of North Carolina School of Medicine, Chapel Hill NC. His interests are in receptor theory and allosteric protein function as applied to new drug discovery.


Brought to you by


Industry 4.0, also known as Smart Industry, is becoming prevalent across various sectors, including the pharmaceutical industry. Technologies like the Internet of Things (IoT), cloud computing, and automated data exchange are now integral to the industry. This white paper explores
the implications of Industry 4.0 for pharma, highlighting the benefits of adopting pharma 4.0 technologies and strategies to avoid common Challenges. It emphasizes the need for the (bio) pharmaceutical industry to produce high-quality medicines consistently. The paper introduces smart pharma technologies to achieve this,
focusing on our innovation: a voice-controlled electronic Batch Record system.
The International Society for Pharmaceutical Engineering (ISPE) is advancing the adoption of Industry 4.0 in the pharmaceutical industry through the concept of pharma 4.0. The ISPE developed business cases to demonstrate the benefits and challenges of implementing automation and digitalization technologies in pharma, especially concerning health regulations. Pharma 4.0 aims to connect all aspects of digitalized plant operations, enhancing transparency, speed, decision-making, and real-time control over processes, operations, and quality. Advances in AI, IIoT, VR, cloud computing, advanced analytics, and machineto-machine communication have significantly impacted pharmaceutical manufacturing, quality control, equipment maintenance, and supply chain documentation. However, the increased connectivity also raises security vulnerabilities, necessitating enhanced security measures in pharma 4.0.
1. Accelerate Your Operations: Technological advancements, such as tailor-made Manufacturing Execution Systems (MES) and personalized electronic Batch Record (eBR) systems, ensure precise operational control and compliance with regulations, thereby speeding up processes.
2. Increase Operational Reliability: Realtime working instructions and smart sensors
enhance process control and reliability. These technologies also improve the reliability of outsourced processes and reduce falsification risks using real- time remote control and RFID technology.
3. Save Time and Reduce Risk: Automating batch record completion ensures complete traceability, cuts operational costs, reduces the risk of contamination, and eliminates the need for manual entry, thus saving time.
4. Accelerate the Go-to-Market Process: Automatic generation of batch records, highlighting non-conformities and critical steps, speeds up and improves the medication release process.
Step 1: Choose Your Investments Wisely To successfully implement digital transformation through smart pharma technologies, it's crucial to analyze businesscritical processes carefully. Identifying which processes need technological support can be challenging, leading to the risk of investing in popular but unsuitable Pharma 4.0 technologies. Organizations often end up adapting their processes to fit new technologies instead of choosing technologies that align with and solve their specific business needs. This is particularly relevant for supply chain control and data recording processes. Companies, especially startups and those in Advanced Therapy Medicinal Products (ATMPs), must assess their true needs for optimizing processes.
The type and size of the pharmaceutical company determine which processes require attention. Small companies producing earlyphase clinical trial medications have different needs than large companies manufacturing millions of drug products. Each company needs a unique approach tailored to its business objectives and expectations.
However, all (bio) pharmaceutical companies share common goals: reducing operational supply chain costs and relieving administrative burdens. Smart pharma technologies help tackle these goals effectively.
Smart pharma technology focuses on processing large amounts of data to enhance the quality and efficiency of supply chain processes like manufacturing, filling, quality control, and shipping, while adhering to pharmaceutical safety regulations. It primarily controls Critical Process Parameters (CPPs) and achieves Critical Quality Attributes (CQAs) without setting them. To gain the full benefits, companies must carefully analyze and optimize their processes and pain points before adopting Pharma 4.0 technology.
Streamlining existing processes can be done through traditional quality improvement techniques or by automating repetitive tasks. Quality by design tools help set CQAs and CPPs, while risk management tools like Failure Modes and Effects Analysis (FMEA) reduce risks associated with supply chains, quality, and safety. Automating routine tasks with robots or
computer programs eliminates human variability and enhances process efficiency.
Pharmaceutical companies must ensure complete traceability of each drug batch for patient safety, which involves creating and completing batch records that document various aspects of production. Manual generation of Master Batch Records (MBRs) and Batch Records (BRs) is cumbersome and inefficient, requiring constant operator presence and offering no added value to medication quality. Automation of these processes can significantly reduce costs and shorten time-tomarket. Many companies still use paper-based MBRs and BRs, so transitioning to electronic
Batch Records (eBRs) is the first step towards automation, offering numerous advantages. Despite the benefits of electronic Batch Records (eBRs), the aseptic manufacturing industry hesitates to adopt the technology due to regulatory requirements and a lack of real innovation. Current eBR solutions are more efficient than paper records but still fall short of customer needs, especially in aseptic processes. Operators must manually read instructions and enter data, which is timeconsuming and increases the risk of human error and contamination.
SmartReg, a voice-controlled eBR bot developed by salamanderU
Addresses these issues by automating the recording process. This innovation minimizes human contact, reduces contamination risks, and enhances productivity by allowing operators to focus on their tasks without constant interruptions. SmartReg aims to ensure patient safety and optimal manufacturing traceability with minimal inconvenience for operators.
SmartReg, designed by pharmaceutical experts for the industry, is the world’s first patented, web-based electronic Batch Record (eBR) system featuring voice recognition and a voice synthesizer. This innovative system simplifies and streamlines manufacturing and recording operations by guiding operators through work processes, verifying correct execution, and alerting them to any mistakes. SmartReg records audio of the operator’s actions, eliminating the need for additional
personnel to document the process, thereby allowing the operator to manage everything independently.
It goes without saying that a voice-controlled eBR retains all the advantages of other eBRs, some of which have already been mentioned above. SmartReg, however, also offers other useful attributes such as easy BR and MBR accessibility, easy MBR preparation and validation, easy BR creation and approval, as explained in the white paper.
Industry 4.0, or the Smart Industry, offers numerous advantages to the pharma industry, such as optimized production processes, realtime control, and reliable manufacturing. The voice-controlled electronic Batch Record system, SmartReg, reduces human error and contamination, saves time and costs, boosts productivity, and simplifies data management.
Pharma 4.0 ensures patient safety by providing clear traceability, better control, and optimized processes, enabling faster and more effective decision-making. However, each organization requires a customized approach. SalamanderU tailors solutions like SmartReg to meet specific needs and challenges and ensuring optimal process improvement.


Scan the QR code to access the comprehensive guide that can revolutionize your approach to Industry 4.0.
Dear Readers,
Welcome to our panel discussion on “The Promise and Challenges of Gene Editing in Drug Development.” We are honored to host two distinguished experts in the field, each bringing a unique perspective and wealth of experience to the conversation, each exploring different facets of the transformative landscape of Gene Editing in Drug Development.


Chris Bohl
PhD:
Senior Manager Technical Support, Products at BioIVT / XenoTech, United States
Daniel Kavanagh
PhD, RAC: Senior Scientific Advisor, Gene Therapy, Vaccines, and Biologics, WCG, United States
1. How can conventional ADME test systems be adapted to support gene editing technologies?
Dr. Chris Bohl: Conventional ADME test systems can support gene editing technologies in a couple of ways. The first is most advantageous for groups that
are specifically targeting the liver for their gene editing treatment. Using cryoplateable primary hepatocytes, scientists can assess the efficiency of targeted changes made to the delivery system, editing machinery, and/ or hepatocyte genotype/phenotype changes due to the editing. Utilizing various experimental designs, each of the editing steps can be assessed independently.
The second is advantageous to all gene editing programs. Long-term, micro-patterned co-cultures using primary hepatocytes (HEPATOPAC®) can give insights into potential off-target toxicity caused by gene editing platform(s). Many gene editing delivery approaches utilize or lead to systemic dispersion of the therapy throughout the body. Even if the liver is not specifically targeted, it is very likely that the therapy will encounter the liver and other highly vascularized tissues. This exposure has the potential to inadvertently
edit hepatocytes. This may lead to unforeseen off-target hepatic toxicity. Utilizing primary hepatocyte cultures that stay viable and active for many weeks, scientists can utilize the extended time in culture to examine the effect of chronic exposure of the gene editing platform and phenotype changes associated with off-target editing. This data may provide insight into predicting hepatic risks involved with their gene editing programs. These long-term cultures can also be built with additional cell types, such as Kupffer cells (HEPATOMUNE®), which might be useful as they add the resident hepatic macrophages back into the test system. This could help them understand potential risks of negative effects on co-administered treatments due to immune cell activation, cytokine release, and decreases in hepatic drug metabolizing enzymes.
In addition to hepatocytes, conventional tissue subcellular fractions and plasma/serum are used in cell and gene therapy programs to assess the stability of the platform while in circulation, much like their use in traditional small molecule assays. These in vitro test systems will help scientists test and optimize their platforms as well as reduce the amount of in vitro studies.
2. What are the main regulatory challenges associated with gene editing in clinical trials, and how can they be addressed?
Dr. Daniel Kavanagh: Major regulatory issues for this class of products include the potential for off-target changes in the
chromosome, potential for inadvertent germline modification affecting future generations, and the need for long-term follow-up to monitor for safety and efficacy.
An additional concern is biosafety. For viruses and nanoparticles capable of making permanent alterations in a human chromosome, biosafety guidance and oversight for shipping, storage, administration, and disposal are critical. Addressing these challenges requires continuous cooperation and communication among statutory regulators and funding agencies. This includes maintaining appropriate risk-based oversight by Institutional Biosafety Committees (IBCs).
3. How have recent advancements in gene editing technologies, such as CRISPR, influenced drug development?
Dr. Chris Bohl: I think it has generated a lot of excitement and energy in the drug development field. It has opened a new avenue to explore in the effort to treat disease indications that were thought to be
Gene editing has
also
been tremendously useful for creating cells lines for high throughput small molecule screening.
untreatable using other strategies/modalities. It has led biotech and pharma companies to open new programs and lines of research, brought new technologies and new scientists with different types of training into the drug development field, and created new bioprocesses and challenges for engineers to solve. But most importantly, these new technologies have given hope to families that there could be new therapies, or even functional cures, on the horizon that could increase the quality of life for their loved ones.
Dr. Daniel Kavanagh: Most public attention may be focused on medicines for which the mechanism of action is based on genetic modification of human chromosomes. Gene editing has also been tremendously useful at the preclinical discovery stage, for creation of cells lines for highthroughput small molecule screening. Gene editing can also be used to create better and more relevant animal model systems for preclinical testing.
4. How do you foresee gene editing impacting the pharmaceutical market in the next decade?
Dr. Chris Bohl: This new technology has the potential to positively impact a lot of families, but I expect the impact of these new therapies will not be fully realized market-wide for some time. I believe that
adoption beyond those that are afflicted by rare, monogenetic diseases where there are no current or foreseeable treatments will take significant effort. I think it will take considerable time and coordinated effort to convince people that these treatments exhibit positive long-term safety profiles and have acceptable risks. The difficulty in attesting to safety is that we simply don’t have enough data points from the clinic nor full understanding of which in vitro safety tests/ assays will be needed to predict safety in the clinic. Development of predictive in vitro test systems will be a crucial step towards developing safe and efficacious gene editing therapies.
Dr. Daniel Kavanagh: Some gene editing approaches will result in curing otherwise incurable conditions, potentially via a oneand-done treatment. This creates challenges for manufacturers and payers working to fairly address the needs of commercialization, reimbursement and accessibility. We are already seeing proposals with a lot of innovative suggestions, and the final implementation of specific reimbursement models is likely to have a large effect on this sector in the next decade.
5. How important is collaboration between research institutions, pharmaceutical companies, and regulatory bodies in advancing gene editing technologies?
Dr. Chris Bohl: I think collaborations between research groups and regulatory bodies are essential for the development, advancement, and wide acceptance of these new types of therapies. Even though these technologies have been in use in research labs for more than a decade, their use as disease therapies is so novel that they generate many new questions. Open and transparent discussions between experts in the field can only be advantageous for moving more gene editing therapies into the clinic and the aim of helping as many people as possible. Open and frank discussions between groups, combined with data sharing, would go a long way toward calming fears and reducing hesitancy regarding wider use and accelerated development.
Dr. Daniel Kavanagh: These collaborations are certainly extremely important. Originally CRISPR DNA sequences were discovered through curiosity-driven basic
science, and this type of basic research must be preserved. At the same time, scientists at research institutions can be better educated about the path from discovery to commercialization for potentially useful new technologies. Development of cell and gene therapy products at institutions can benefit from a clear understanding of commercial and regulatory requirements for manufacturing and distribution at scale. Collaborative guidance from companies and regulators is a critical aspect of this process.
6. Are there notable case studies or real-world applications of gene editing that highlight its potential or challenges?
Dr. Chris Bohl: I feel that the general population’s mood towards acceptance and trust in therapies that utilize gene modifying components will be a major challenge in achieving widespread acceptance and use. In addition, costs and ensuring availability will be major challenges that governments and private health insurance companies will have to work through to ensure that these therapies reach those who need them.
A real-world challenge affecting any treatment that changes DNA is longterm follow-up and safety monitoring.
Dr. Daniel Kavanagh: We are already hearing personal stories from patients experiencing tremendous lifechanging benefits from an FDA-approved product. There is obviously a potential to see this success repeated many times in a broad array of therapeutic areas. A realworld challenge affecting any investigational
or commercialized treatment that changes DNA—especially genome editing—is longterm follow-up and safety monitoring.
7. What future trends or technologies in gene editing are you most excited about?
Dr. Chris Bohl: I think the natural evolution of these therapies is to expand to complex diseases that have polygenic/multi-factorial etiologies. As our understanding of genetic variability and its contribution to disease expands, new targets or groups of targets will emerge. This will hopefully lead to treatments that can help increase the quality of life for broader populations and more diverse disease indications.
Dr. Daniel Kavanagh: One thing that I find especially exciting is the potential for incorporation of gene editing into every aspect of medicine, in combination with small molecules, biologics, and devices. Years ago, computer science was a discrete endeavor with specialized applications, but today silicon chips are routinely incorporated into industrial and consumer products in every sector. DNA has always been the core determining element in human health and disease. The ability to precisely alter DNA has almost unlimited potential to enhance the effectiveness of other classes of medical products.
8. What key message would you like to convey about the future of gene editing in drug development?
Dr. Chris Bohl: Even with all the challenges facing the field, I believe that science will develop the tools needed to accelerate the development of gene editing therapies that will have the potential to bring life changing treatments to patients and their families. It opens the possibility of one-time treatments, functional cures, and therapy for diseases where no specific treatments are currently available. While setbacks are to be expected, I am confident that the field will be successful in developing the tools needed to bring more of these therapies to the market.
Dr. Daniel Kavanagh: I think the key message is always to look for appropriate technology. Gene editing is not a single tool. It is a set of related approaches that will evolve and expand over time. When one product succeeds, that does not mean that that exact same technology can be replicated for other applications. If one product fails, that does not imply a broad challenge to the validity of gene editing as an approach. I recommend hopeful enthusiasm without irrational exuberance.
Thank you, panelists, for your valuable insights on “Gene Editing in Drug Development.” Your expertise and perspectives have greatly enhanced our understanding of this complex field. We appreciate your time and the engaging information you provided.
Welcome to today’s panel discussion on “Drug Repurposing: Unlocking New Therapeutic Possibilities.” We are delighted to have two experts in the field: Dr. Adekemi Taylor, Vice President of Quantitative Science Services at Certara, and Joab Williamson, Director of Clinical Operations at Faron Pharmaceuticals.
Happy Reading!



Joab Williamson
Director of Clinical Operations, Faron Pharmaceuticals.
Dr. Adekemi Taylor
PhD: Vice President, Quantitative Science Services at Certara, United States
Q1. How do you address the main challenges faced in the drug repurposing process?
AI will provide general answers to address common questions for broad understanding, while our expert panel will offer detailed, indepth responses on the issues for advanced and seasoned readers seeking more specialized insights.
Joab Williamson: One of the biggest hurdles in drug repurposing is convincing people—whether they’re investors, partners, or even physicians—that a drug with a previous life (and possibly a few bumps along the way) deserves a second chance. This can be tackled by passionately communicating the potential of the repurposed drug, backed by solid science, and ensuring everyone is on board from the get-go. On the legal side, the maze of patents and regulations should be followed with meticulous planning, making sure there are no unexpected roadblocks.
AI's POV: “Drug repurposing faces several challenges, including identifying new therapeutic uses for existing drugs, ensuring efficacy and safety in novel applications, and navigating regulatory hurdles. Researchers often struggle with limited data on off-label uses and must address the complexities of adapting existing treatments to new conditions. Additionally, securing funding and demonstrating commercial viability can hinder progress, despite the potential for significant benefits and cost savings in drug development.”
Q2. Can you share an example of a successful drug repurposing project and its impact?
Dr. Adekemi Taylor: Ketamine is a drug repurposing success story. The FDA approved it in 1970 as an intravenous anesthetic. Ketamine is a racemic mixture of Rand S-enantiomers. Esketamine, ketamine’s S-enantiomer, has a higher affinity than R-ketamine for the NMDA receptor, which is involved in the etiology of depression. In 2019, the FDA approved intranasal esketamine to be used with an oral antidepressant for treatment-resistant depression. Esketamine represents a much-needed treatment option for an underserved condition. Research on esketamine’s mechanism of action is also expanding our understanding of depression.
Q3. What regulatory hurdles have you encountered in drug repurposing, and how have you navigated them?
Joab Williamson: Regulatory challenges can sometimes feel like threading a needle, especially when you’re repurposing a drug that didn’t hit the mark in its original use. It can be expected that the key to navigating this is early and open dialogue with regulators. By clearly outlining why this time is different and how the new indication stands on its own merits, potential hurdles can be turned into manageable steps. And of course, it is crucial to keep a close eye on the patent situation to ensure smooth sailing.
AI's POV: “Regulatory hurdles in drug repurposing often include proving the new use is both safe and effective, which can require additional clinical trials and data. Navigating these challenges involves rigorous research to meet regulatory standards, engaging with regulatory agencies early to understand requirements, and potentially working with specialized consultants. Clear documentation and robust clinical evidence are crucial for securing approvals and ensuring the repurposed drug meets all safety and efficacy standards.”
Q4. How do you see drug repurposing evolving in the future, and what factors will drive this evolution?
Dr. Adekemi Taylor: In the past, drug repurposing largely relied on serendipitous identification of new indications. The most famous example is sildenafil citrate (Viagra), which was originally developed for treating angina.
The availability of high throughput screening methods and increasingly powerful high-performance computing, as well as the accumulation of -omics data and knowledge about pathways and networks, will facilitate identification of more new indications for existing drugs.
Further advances in mathematical modeling of animal and clinical data will also save money by streamlining clinical development. For example, the FDA originally approved eculizumab for the rare disease paroxysmal nocturnal hemoglobinuria (PNH). Population pharmacokinetic (PK) and pharmacodynamic (PD) modeling leveraged data from patients with PNH to inform clinical study design and dose selection for a new indication with a shared disease mechanism, atypical hemolytic uremic syndrome (aHUS). Results from this model supported eculizumab’s subsequent approval for aHUS.
Q5. What role does interdisciplinary collaboration play in successful drug repurposing?
Joab Williamson: Interdisciplinary collaboration isn’t just a buzzword—it’s the lifeblood of successful drug repurposing. When you bring together minds from clini -
cal research, regulatory, business development and legal fields, you get a 360-degree view of the challenges and opportunities. This teamwork ensures that sponsors are not only finding innovative ways to breathe new life into old drugs but are also ready to address any concerns from stakeholders before they even arise. It’s like having an all-star team where everyone plays their part to perfection.
Q6. In what ways have new technologies influenced your approach to drug repurposing?
Dr. Adekemi Taylor: Today, drug developers are using AI technology widely. For example, drug developers often spend an inordinate amount of time searching and summarizing their field’s biomedical research literature. AI algorithms can accelerate this process by curating and summarizing biomedical publications to identify new drug and disease targets.
In later drug development, mathematical/statistical modeling methods such as population PK/PD, physiologically-based pharmacokinetics (PBPK), and quantitative systems pharmacology modeling can integrate previously obtained preclinical and clinical data with data from studies for the new indication to support dose selection. This technology can even be used to replace some clinical studies.
AI's POV: “New technologies, such as advanced data analytics, AI, and
AI algorithms can accelerate drug development by curating and summarizing biomedical publications to identify new drug and disease targets.
high-throughput screening, have significantly influenced drug repurposing. AI helps identify potential new uses by analyzing vast datasets, while high-throughput screening accelerates the evaluation of drug candidates. Additionally, bioinformatics tools facilitate the integration of complex biological data, enhancing the precision of target identification and improving the overall efficiency of the repurposing process.”
Q7. How do you prioritize which existing drugs to repurpose, and what criteria do you use for this decision-making process?
Dr. Adekemi Taylor: Several factors determine which candidate drugs to prioritize for repurposing. These include unmet medical need, financial, and commercial viability, the pharmacokinetics of the drug, as well as safety profiles. For instance, an
older drug that is vulnerable to drug-drug interactions (DDIs) could be a poor repurposing candidate as it will potentially interact with co-administered drugs.
Joab Williamson: Answering from a personal perspective, I would take a twopronged approach. First, I would look for drugs where the science and mechanism of action (MOA) are well-understood, ideally with someone on our team—like our CMO— having hands-on experience with the drug in a previous trial. This familiarity gives us confidence in the drug’s potential. On the business side, we would assess the market landscape, patent situation, and partnering potential to ensure the project is commercially viable. It’s all about finding that balance between scientific promise and business opportunity. A drug is not feasibly without both.
Q8. Can you discuss how patient involvement or feedback influences the drug repurposing process?
Dr. Adekemi Taylor: Patient feedback is crucial for ensuring that the treatment meets their needs. Patient reports of side effects can also inform the drug repurposing process. For example, a drug that causes hypoglycemia as a side effect may be repurposed for treating diabetes. Furthermore, different drugs having similar side effect profiles can signify shared pharmacological targets which can help identify candidates for repurposing.
Joab Williamson: Patient involvement is important in many areas of drug repurposing. Their feedback helps focus on what truly matters in treatment, guiding decision-making and trial design. But it goes beyond feedback—patient participation in clinical trials is essential, as it allows the gathering of critical data, including samples that inform the repurposing process. This data helps understand how the drug behaves in the new indication, ensuring that the approach is grounded in real-world evidence. Finally, how the drug sponsor communicates externally is important, if a patient Googles the name of a medicine, headlines of previous failures may be alarming, so patient-facing communication is crucial to enabling the engagement in the drug.
Q9. How can we balance the thorough clinical testing required for new drugs with the efficiency and costeffectiveness of repurposing existing ones?
Dr. Adekemi Taylor: Repurposing drugs, while time- and cost-effective, has its limits. For example, repurposing small molecule drugs for progressive or genetic diseases may merely treat symptoms, whereas new modalities such as gene therapy have the potential to be curative. Furthermore, other new modalities such as antibody-drug conjugates and targeted
radiopharmaceuticals target cancer cells directly, minimizing damage to healthy tissues, unlike traditional chemotherapy. Of course, these emerging therapies may be candidates for repurposing in the future.
Joab Williamson: Balancing thorough testing with the efficiencies of drug repurposing is like walking a tightrope— you need precision, but also a bit of agility. Sponsors should leverage adaptive trial designs that allow real-time responses to data readouts, making the process both rigorous and flexible. By using existing data where possible, sponsors are able to cut down on time and costs, which makes the project more appealing to everyone from investors to regulators. It’s all about working smart while maintaining the highest standards.
AI's POV: “Balancing thorough clinical testing with cost-effective repurposing involves leveraging existing data to streamline trials, focusing on targeted studies that address new indications. Utilizing adaptive trial designs and leveraging real-world evidence can reduce time and costs. By building on established safety profiles and using advanced data analytics to guide trial design, researchers can efficiently bridge the gap between thorough testing and the benefits of repurposing existing drugs.”
Q10. What strategies do you employ to overcome potential intellectual property
and patent issues when repurposing a drug?
Dr. Adekemi Taylor: Old drugs often have run out of patent life. Establishing a new indication with an already approved drug or drug combination can be used to acquire new intellectual property. For example, Red Hill Biopharma obtained a new patent in the US for the fixed-dose combination of the antibiotics, clarithromycin, clofazimine, and rifabutin, for treating pulmonary non-tuberculous mycobacterial infections. These antibiotics were originally approved in the 1980s and early 1990s.
Joab Williamson: Intellectual property issues can be the elephant in the room when it comes to drug repurposing. Sponsors should address this head-on with a thorough patent analysis right from the start. If conflicts are found, they can be seen as opportunities explore licensing deals or collaborations with the original patent holders to create a win-win scenario. Plus, by securing new patents for the drug’s new use, it is not only protecting the research but also to add value to the overall project, making it more attractive to investors and partners.
AI's POV: “To address intellectual property and patent challenges in drug repurposing, employ strategies such as
conducting comprehensive patent landscape analyses to identify relevant patents and assess potential infringements. Engage with IP attorneys to evaluate the scope of existing patents and explore opportunities for securing patents on novel indications. Consider negotiating licensing agreements or strategic partnerships with patent holders to mitigate legal risks and facilitate access to critical IP while preserving the integrity of innovation.”
Q11. How do you assess and mitigate risks associated with repurposing drugs, especially regarding unforeseen side effects or interactions?
Dr.Adekemi Taylor: Highthroughput screening and in silico methods used to identify candidates for drug repurposing can also reveal off-target activities that could lead to adverse effects. If not done at the time of initial approval, in vitro experiments can identify the risk of DDIs.
If the new indication requires higher doses than previously approved, relevant animal experiments should be performed to assess safety with the increased drug exposures, and clinical studies should be designed to assess the safety of the higher doses.
In addition, population PK and PK/PD modeling can aid in selecting the dose regimen that balances safety and efficacy. This approach is also helpful for assessing the need for dose adjustment in different patient populations. Similarly, PBPK modeling can help assess the risk of DDIs and the impact of renal/hepatic impairment on drug exposure.
Joab Williamson: In drug repurposing, mitigating risks is all about being proactive. Alongside the patent exploration, the clinical team should be deep diving into the drug’s existing safety profile and investigating how it might behave in the new context. But it’s not just about the science— it is important to stay ahead of potential regulator concerns by maintaining open lines of communication and being transparent about the existing data. By addressing issues early and being ready to adapt, you can minimize surprises and keep the project on track.
AI's POV: “To effectively address risks in drug repurposing, perform in-depth preclinical and clinical studies to uncover any unexpected side effects or interactions. Utilize advanced predictive analytics and simulation techniques to forecast potential adverse reactions. Continuously refine safety measures based on the latest findings, maintaining thorough oversight throughout the repurposing process to ensure patient safety and maximize therapeutic benefits.”
Q12. In your opinion, how can industry stakeholders better support drug repurposing initiatives to maximize their success and impact?
Dr. Adekemi Taylor: Regulatory
agencies can encourage drug repurposing by removing existing barriers and providing accelerated pathways. Pharma and biotech leaders can also prioritize drug repurposing as an efficient, cost-effective means for more quickly delivering medicines that fulfill unmet medical needs. For venture capital firms, drug repurposing is an attractive lower-risk prospect given the established safety profiles and drug knowledge. Industry scientists should also continue developing methods to accelerate the identification of new indications for existing drugs.
Joab Williamson: For drug repurposing to really take off, the industry needs more than just good ideas—we need a concerted effort from everyone involved. Industry stakeholders can play a huge role by fostering collaboration and regulators offering incentives that make these projects viable. Simplifying regulatory pathways and offering clearer guidelines on intellectual property issues would go a long way in encouraging innovation. Additionally, larger pharmaceutical companies which often hold left-over patent rights from failed projects, or acquisitions could be more open to partnerships with smaller companies. Ultimately, it’s about creating an ecosystem where repurposed drugs can thrive and make a real difference in patients’ lives.
Thank you for joining us for this insightful discussion on drug repurposing. We’ve explored key questions with our panelists, Adekemi Taylor and Joab Williamson. We appreciate your engagement and look forward to the continued progress in this exciting field.

Dr. Jeannie Lee
Appointed as Non-Executive Director at GSK


David Habiger
Appointed as Board of Directors at Boston Scientific


Roopal Thakkar
Appointed as Executive Vice President, Research & Development and Chief Scientific Officer at AbbVie


Roxanne S. Austin
Appointed as Lead Independent Director at AbbVie


Laura Hamill
Appointed as Board of Directors at Jazz Pharmaceuticals


Samantha Pearce
Appointed as Chief Commercial Officer at Jazz Pharmaceuticals

Dr. Oliver P. Kronenberg
Appointed as Chief Legal Officer and Company Secretary at Santhera


Robert A. Michael
Appointed as Chief Executive Officer at AbbVie

Bret DiMarco
Appointed as Chief Legal Officer at Agilent


Dr. Martin Stumpe
Appointed as Chief Data & Artificial Intelligence Officer at Danaher


Dr. Cheryl Pegus
Appointed as Board of Directors at Boston Scientific

Joins Board of Directors at Parexel


Melissa Seymour
Appointed as Executive Vice President of Global Quality at Eli Lilly and Company


Matthew Shields
Appointed as Executive Vice President, Global Operations at Teva Pharmaceutical Industries Ltd.


Christoph Funke
Appointed as Chief Technical Operations Officer at Lupin


Guy Oliver
Appointed as General Manager at Bristol Myers Squibb


Audrey Duval Derveloy
Appointed as Global Head of Corporate Affairs at Sanofi


Sandi See Tai
MD, has been promoted to Chief Development Officer at Lexeo Therapeutics

Biocytogen Pharmaceuticals (Beijing) Co., Ltd., a leading biotech firm specializing in novel antibody and ADC therapeutics, has announced a significant option and license agreement with IDEAYA Biosciences, Inc., an oncology-focused precision medicine company.
The agreement pertains to a groundbreaking B7H3/PTK7 bispecific antibody-drug conjugate (BsADC) program, which is potentially the first of its kind.
This deal provides IDEAYA with the option to secure an exclusive worldwide license from Biocytogen for the B7H3/PTK7 topo-I-payload BsADC program. B7H3 and PTK7 are co-expressed in several solid tumors, including lung, colorectal, and head and neck cancers, among others.
Under the terms of the agreement, Biocytogen will receive an upfront payment and, should IDEAYA exercise its option, will also be entitled to additional fees, including option exercise, development and
Cellectis, a clinical-stage biotech company specializing in gene-editing technologies, has announced that the U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation (ODD) to its investigational product CLLS52 (alemtuzumab).
This drug is used as part of a lymphodepletion regimen in conjunction with UCART22, a treatment being tested in the BALLI-01 clinical trial for relapsed/ refractory B-cell acute lymphoblastic leukemia (ALL).
Cellectis pioneered the use of CD52 knockout UCART cells combined with a lymphodepleting regimen that includes an anti-CD52 antibody like alemtuzumab.
This approach aims to make UCART cells resistant to alemtuzumab through TALEN® gene editing technology.

regulatory milestones, and commercial milestone payments.
Biocytogen will also earn single-digit royalties on net sales. The total potential financial value of the agreement, including all milestones, is US$406.5 million, with US$100.0 million allocated to development and regulatory milestones.
Preclinical data suggests that the B7H3/PTK7 topo-I-inhibitor-payload BsADC has strong potential as both a monotherapy and in combination with other programs in IDEAYA’s pipeline, such as the DDR-based PARG inhibitor IDE161.
A development candidate nomination for this BsADC program is expected in the second half of 2024.

The FDA’s Orphan Drug Designation is awarded to treatments for rare diseases affecting fewer than 200,000 individuals in the U.S., potentially accelerating development, reducing costs, and expediting the approval and commercialization process.

BridGene Biosciences, Inc. has announced an expanded strategic collaboration with Galapagos NV to advance the development of a selective oral SMARCA2 PROTAC for precision oncology.
This expansion builds on their existing partnership, which began in January 2024, focusing on creating a highly selective small molecule proteolysis targeting chimera (PROTAC).
Under the enhanced agreement, BridGene will utilize its PROTAC discovery platform alongside Galapagos’ expertise in selective ATPase small molecules.
The goal is to advance the PROTAC molecule into preclinical development, with Galapagos retaining exclusive rights for further development and commercialization.
The deal includes upfront and preclinical milestone payments, with additional payments tied

BostonGene has announced a new partnership with Takeda to advance immuno-oncology research using AI-powered molecular profiling.

to clinical and commercial milestones, potentially totaling US$159 million. BridGene is also set to receive tiered royalties on net sales from any resulting products.
This expanded collaboration underscores the success of BridGene’s innovative approach and the value of strategic partnerships in advancing drug development.
Both companies are dedicated to improving patient outcomes by developing cutting-edge precision medicines to address high unmet medical needs through targeted protein degradation technology.


This collaboration aims to identify critical molecular drivers and predictive markers for treatment efficacy and adverse effects, ultimately enhancing clinical solutions and patient outcomes.
Under the agreement, Takeda will utilize BostonGene’s AI-driven multiomics platform in selected early-stage clinical trials. This will help optimize trial design, refine indication selection, and uncover biomarker signatures related to treatment response and toxicity.
BostonGene will apply its advanced multiomic analytics and bioinformatics expertise to analyze clinical and laboratory data provided by Takeda, including flow cytometry, RNA-seq, and proteomics data.
Agilent Technologies Inc. has announced that the U.S. Food and Drug Administration (FDA) has approved its MAGE-A4 IHC 1F9 pharmDx (SK032) as a diagnostic tool for identifying patients with synovial sarcoma who may be eligible for treatment with TECELRA® (afamitresgene autoleucel, also known as afami-cel or ADP-A2M4).


TECELRA® is a MAGE-A4-directed engineered TCR T-Cell therapy.

MAGE-A4 (melanoma-associated antigen A4) is a cancer-testis antigen that is overexpressed in various cancers, including synovial sarcoma.
The MAGE-A4 IHC 1F9 pharmDx is an immunohistochemistry (IHC) assay designed to detect MAGE-A4 expression in formalinfixed paraffin-embedded (FFPE) tissue from synovial sarcoma.
Detection of MAGE-A4 positivity in these tissues indicates eligibility for TECELRA® treatment. This assay is the first in vitro diagnostic (IVD) test for MAGE-A4 available on the market.
TECELRA® is approved for use in treating advanced MAGE-A4 positive synovial sarcoma in adults with specific HLA types who have previously undergone chemotherapy. It is the first FDA-approved engineered TCR T-Cell therapy for a solid tumor cancer.
The Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has recommended expanding the indication for RYBREVANT® (amivantamab) to include its use in combination with chemotherapy for adult patients with advanced EGFR-mutated non-small cell lung cancer (NSCLC) who have failed prior treatments.
Amivantamab, used alongside carboplatin and pemetrexed, demonstrated a significant 52% reduction in the risk of disease progression or death compared to chemotherapy alone in the MARIPOSA-2 study.
This combination also showed a median progression-free survival (PFS) of 6.3 months versus 4.2 months with chemotherapy alone (hazard ratio [HR] = 0.48; 95% CI, 0.36–0.64; P<0.001).
The objective response rate (ORR) was 64% for the amivantamab combination, compared to 36% with chemotherapy alone.
Additionally, the combination therapy exhibited notable intracranial activity, reducing the risk of intracranial progression or death by 45% compared to chemotherapy alone, with a median intracranial PFS of 12.5 months versus 8.3 months (HR=0.55; 95% CI, 0.38–0.79; P=0.001).
The safety profile of amivantamab plus chemotherapy was consistent with the individual components, though 72% of patients experienced Grade 3 or higher adverse events, mainly hematologic toxicities, compared to 48% with chemotherapy alone.
Serious adverse events occurred in 32% of patients receiving the combination therapy, compared to 20% with chemotherapy alone.

03 – 05 September 2024|Boston, USA
https://bispecific.com/
About Event: The 15th World Bispecific Summit has established itself as the key biopharma directed event to gain clear insights into the latest advancements and future directions amongst the mass of bispecific antibodies currently being developed and how to address associated development challenges encountered within this therapeutic modality.
24 – 26 September 2024|Boston, USA
https://mashdrugdevelopmentsummit.com/
About Event: The 8th MASH Drug Development Summit unites 120+ industry leaders to address the most pressing challenges in MASH and metabolic diseases. Harnessing FGF21s, THR-β Agonists, GLP-1 Agonists, FXR & PPAR Agonists to propel your MASH therapeutic pipelines. This is your opportunity to access cutting-edge R&D insights that can super-charge your end-to-end metabolic
21 – 22 October 2024|San Diego, USA
https://events.precision-globe.com/single-event/ Precision-in-pharma-supply-chain-logistics-summitsan-diego
About Event: The PPSL Summit for a two-day journey into the insightful world of Pharmaceutical Supply Chain & Logistics. You’ll connect with an incredible gathering of experts and uncover practical advice from industry-leading researchers and executives.
05 – 06 November 2024|Pennsylvania, USA
https://www.arena-international.com/event/ ctseastcoast/
About Event: The Clinical Trial Supply East Coast is the meeting place for the pharmaceutical and biotechnology community to discover how to excel in clinical supply strategy as well as form key connections for long-term success. It’s time to build relationships and take away valuable lessons to for clinical trial success moving forward so we look forward to seeing you there!

28 – 29 November 2024|Barcelona, Spain
https://pgsolx.com/supplychain/CTSLC/
About Event: Pharma Supply and Logistics Conference bring together industry leaders,
innovators, and experts to unravel the intricacies of pharmaceutical logistics. Explore the latest trends, strategies, and technologies driving efficiency in the supply chain. Join us in fostering collaboration and shaping the future of pharmaceutical logistics for a resilient and agile industry. Get ready to dive into insightful discussions, gain actionable insights, and forge connections that will elevate your understanding of the evolving dynamics in pharma supply chain management.
03 – 05 December 2024|Barcelona, Spain
https://www.drugdiscoverychemistry.com/europe
About Event: Drug Discovery Chemistry is a dynamic conference and networking event for drug discovery scientists working in pharma, biotech, and academia. One of the few events that reflects the new cross-functional and collaborative nature of drug discovery, our speakers and attendees are comprised primarily of pharma and biotech scientists, as well as key academic thought leaders working in translational research.

30 - 31 January 2025|Dublin, Ireland
https://sairap.org/conf/index.php?id=2627426
About Event: World Summit on Pharmaceutics and Drug Designs centered around addressing the key challenges faced by various industries, including engineering, medicine, social science, applied science, management, and others. The aim is to bring together organizations and professionals from these fields to identify bottlenecks and collaborate on finding solutions.

10 - 11 February 2025 London |United Kingdom
https://www.smgconferences.com/pharmaceuticals/ uk/conference/rna-therapeutics
About Event: The RNA therapeutics is addressing novel delivery methods and extra-hepatic delivery solutions, applications of AI for RNA sequence optimization and case studies of novel modalities progressing through the pipeline including circRNA, siRNA, miRNA, lncRNA and more. Further challenges that will be addressed include the lack of effective assays for in vivo analytical testing, including toxicity and immune-modulation considerations. We will also examine the rapidly changing regulatory landscape, identifying how the industry can adapt to meet changing guidelines and regulations.

"41%
of event professionals are participating on more events in 2024 than they originally planned."
Don't be left behind... We are here to help!



Date: 16-17 September 2024
Location: San Diego
The American Drug Delivery & Formulation Summit returns for its 14th year, showcasing the latest advancements in drug delivery, biologics, and device development. With over 300 scientific leaders expected, this event promises an extensive program covering critical issues and industry trends.
Highlights Include:
Case Studies on:
• Enabling Bioperformance Prediction Through Data Analytics
• Engineering Prodrug Therapies for Infectious Disease and Cancer Therapy
• The Port Delivery System with Ranibizumab: A New Paradigm for Long-Acting Retinal Drug Delivery
• Development of High-Volume Wearable Drug Delivery Systems and more.
Why Attend?
High-Quality Content featuring four parallel streams: Small Molecules, Biologics, Tech & Innovation, and Devices & Combination Products.
Invaluable Networking with numerous opportunities to connect with senior industry professionals and explore cross-disciplinary learning. Audience Breakdown: Senior pharmaceutical development and formulation scientists from pharma, biotech, and drug delivery industries. Expand your network, enhance your expertise, and engage with industry leaders at both of these premier events!
Date: November 18-19, 2024
Location: Marina Bay Sands Hotel, Singapore
Join us for the inaugural Asian Drug Delivery & Formulation Summit, expanding from our successful editions in Europe and America to Asia. This summit will explore cutting-edge advancements in drug delivery and formulation technologies across three content streams: Technology & Innovation, Small Molecules, and Biologics.
Highlights Include:
Case Studies on
• Enabling Bio performance Prediction Through Data Analytics
• Engineering Pro-drug Therapies for Infectious Disease and Cancer Therapy
• Predictive High-Throughput Screening of Oligonucleotide Lipid Nanoparticles for Gene Silencing
• Development of High-Volume Wearable Drug Delivery Systems
• Advanced TPPs for Biosimilars and more.
Why Attend?
• High-Quality Content with tailored agendas across three streams, featuring keynotes, case studies, and panel discussions.
• Invaluable Networking opportunities through numerous breaks, receptions, and roundtable discussions.
Industries in Attendance: Pharmaceuticals, Biotechnology, Generics, Higher Education, Non-Profit.







Event: Advanced Therapies USA 2024
Dates: November 12-13, 2024
Location: Pennsylvania Convention Center, Philadelphia, USA
Theme: Advancing Cell and Gene Therapies
Event Overview:
The Advanced Therapies Congress is set to be the premier gathering for innovators and leaders in the cell and gene therapy fields. Scheduled for November 12-13, 2024, at the Pennsylvania Convention Center in Philadelphia, this event promises a comprehensive exploration of the latest advancements and challenges in these transformative therapies.
Conference Highlights:
Diverse Content: The Congress features 8 tracks spanning two days, addressing critical topics from manufacturing and supply chain logistics to pricing, market access, and cutting-edge developments in therapeutic areas such as cell therapy, gene therapy, gene-modified cell therapy, and gene editing.
Expert Speakers: Gain insights from a broad spectrum of industry leaders, including forward-thinking pharma and biotech companies, researchers, clinicians, and regulators. The event offers a platform to explore challenges and solutions that accelerate the development of next-generation therapies.
Exhibition:
Solution Providers: Engage with 50 leading solution providers showcasing the latest technologies and services across all stages of cell and gene therapy development. This exhibition provides a unique opportunity to connect with vendors and discover innovative solutions tailored to your needs.
Networking Opportunities:
1-2-1 Meetings: Benefit from over 12 hours of dedicated networking time, connecting with thousands of experts in the field. The event's built-in networking sessions are designed to facilitate meaningful connections and collaborations.
Key Topics:
Evidence, Pricing & Access: Explore strategies for ensuring clinical success, navigating large-scale regulatory challenges, and establishing frameworks for reimbursement and market access.
Gene Modified Cell Therapy: Delve into topics such as autologous vs. allogeneic approaches, CAR-T, NK, and Gamma-Delta cells, and patient stratification.
Cell Therapy: Learn about stem cells, regenerative medicine, MSCs vs. iPSCs, and advancements in cord blood.
Gene Therapy: Focus on next-generation therapies, gene delivery systems, and addressing immunogenicity.
Cell Therapy Manufacturing: Examine automation, scaling up production, and cost management.
Viral Vector Manufacturing: Discuss continuous manufacturing, scaling up, and in-house vs. outsourcing considerations.
Supply Chain & Logistics: Address challenges in logistics, patient experience, and point-of-care delivery.
Innovation Showcase: Discover the latest academic research and cutting-edge technologies.
• Industry Leaders: CEOs, CSOs, SVPs, and VPs of Pharma & Biotech
• Business Development & Regulatory Professionals
• Academia & Research Heads
• Patient Groups & Associations
• Investors & Start-ups
• Manufacturing & Process Development Heads
• Clinicians, Researchers, Scientists, and More Join us at Advanced Therapies USA 2024 to be at the forefront of cell and gene therapy innovation. Secure your spot to engage with industry experts, explore new technologies, and shape the future of advanced therapies.



Hal Stern VP & CIO, Pharmaceutical R&D Janssen

Anne Heatherington Head, Data Sciences Institute and R&D Chief Data & Technology Officer Takeda

Jason Lott Vice President of Global Integrated Evidence generation Bayer

Ziv BarJoseph Head, R&D Data and Computational Sciences Sanofi


22-25 OCTOBER 2024 | FIRA DE BARCELONA MONTJUIC
The World Orphan Drug Congress Europe is the largest and most established orphan drug & rare disease event worldwide.
Meet over 2000 attendees, hear from 250 leading speakers, and connect with 130 exhibitors as we bring together experts from the start-to-finish of orphan drugs From regulation and policy, to global pricing and gene therapy
Sponsors Include









Supporting Partners Include












Introducing a group of highly focussed magazines for the Europe and Asian markets.
Aspiring to be leading journals in the B2B landscape of Pharmaceutical-Industry, the magazines covers Medical Sciences, Business & Technology and all the latest innovations.
Our magazines bring a fresh outlook towards insightful and pragmatic Pharmaceutical-Industry reporting. Delightfully selected topics presented by the gurus of the industry comes packed with latest happenings, sharp analysis & deep insights. We strive to keep you engaged, knowledgeable & wanting for more.