
ISSUE 51 2023 www.pharmafocusasia.com Opportunities & Challenges Real-world Evidence Measuring Vaccine Efficacy The right endpoints are key to success Revolutionising Pharma Six game-changing ways to excel with integrated quality solution

NIPRO PHARMAPACKAGING INTERNATIONAL Blokhuisstraat 42, 2800 Mechelen, Belgium | pharmapackaging@nipro-group.com | www.nipro-group.com with anti-counterfeiting technology GLASS AMPOULES Additional feature to prove drug product authenticity • Invisible to the naked eye • Compatible with existing ampoule types • Low regulatory impact • Economical solution
Real-World Evidence Opportunities & challenges

Real-world evidence (RWE) is increasingly valued by pharmaceutical companies for its ability to complement and enhance the evidence generated from traditional clinical trials. RWE provides insights into how drugs and treatments perform in routine clinical practice, considering diverse patient populations, healthcare settings, and treatment patterns. However, persistent challenges have, and continue to impact implementation, especially around data.
RWE can support regulatory decision-making processes, such as drug approvals, label extensions, and post-approval commitments. By demonstrating the real-world effectiveness and safety of a drug, pharmaceutical companies can improve the efficacy of their submissions to regulatory agencies. RWE allows for the comparison of different treatment options and interventions in real-world populations. Monitoring and analysing adverse events and other safety outcomes in real-world settings, pharmaceutical companies can identify potential risks, implement risk management strategies, and ensure patient safety.
A recent McKinsey report estimated that, over the next three to five years, an average top-20 pharmaceutical company could unlock more than US$300 million a year by adopting RWE across its value chain.
RWE plays a critical role in unlocking the real-world benefits and value of drugs to payers and reimbursement authorities. By leveraging RWE, pharmaceutical companies can strengthen their case for market access, negotiate favourable pricing and reimbursement agreements, and overcome barriers to formulary inclusion. It also helps pharmaceutical companies conduct post-marketing studies to further evaluate drug safety, efficacy, and effectiveness in real-world populations. These studies can provide additional data to support label expansions, identify new patient populations, optimise dosing regimens, and inform clinical practice guidelines.
In real-world settings, RWE allows pharmaceutical companies to understand patient needs, preferences, and treatment outcomes. This patient-centric approach enables the development of more patient-centered therapies, improved treatment guidelines, and better-informed decision-making throughout the drug development and commercialisation process.
RWE is essential in conducting Health technology assessment (HTA), which evaluate the clinical and economic value
of healthcare technologies, treatments, and interventions. By analysing real-world data on outcomes, costs, and patient experiences, policymakers can make evidence-based decisions on the adoption, reimbursement, and utilisation of healthcare technologies.
Nevertheless, implementation challenges abound. Realworld data is subject to various sources of bias and confounding factors. These include, selection bias, treatment allocation bias, and confounding variables that can affect the accuracy and reliability of RWE studies. Integrating data from various sources and systems with different formats and structures is also a significant challenge. Demonstrating the reliability, validity, and generalisability of RWE studies to regulatory and reimbursement authorities is crucial to support decision-making and gain acceptance. Respecting patient privacy, ensuring informed consent, and maintaining data confidentiality are critical ethical considerations when working with real-world data. Collaboration among stakeholders, including pharmaceutical companies, healthcare providers, regulatory bodies, and patient advocacy groups can address these challenges and unleash the full potential of RWE.
The cover story “Real-world Evidence - How patientled insights are transforming the way we develop health policy”, Julie Cini, CEO, Advocacy Beyond Borders and Ruth Kuguru, Executive Director, Communications & Patient Engagement, Novartis Asia Pacific, Middle East, and Africa, highlights why the Healthcare leaders in the Asia-Pacific region are keen to address the perception gap that exists around RWE. To bridge the current gap, the article offers two perspectives on the relevance of RWE to patient communities and strategies to foster greater multidisciplinary participation in RWE collection. How patient organisations use RWE to gain disease awareness traction and shape health policy and the importance of collaboration among multiple stakeholders.
Prasanthi Sadhu Editor
CONTENTS STRATEGY
06 The Glocalisation Change
Your second important glocalisation decision is how to adapt to local conditions
Brian Smith, Principal Advisor, PragMedic
28 Pharmaceutical Business strategy and Forecasting

Creating insights from market research
Sanobar Syed, Associate Director, Beigene
31 Revolutionising Pharma

Six game-changing ways to excel with integrated quality solution
Jasmin Kumar, Digital Marketing Expert
RESEARCH & DEVELOPMENT
36 Towards Precision Medicine for Cancer Pain Treatment in Asian Populations
Yow Hui-Yin, Pharmacy Lecturer, Department of Pharmaceutical Life Sciences, Faculty of Pharmacy, Universiti Malaya
Shobha Elizabeth Satkunananthan, Master student, School of Pharmacy, Taylor's University
Vijayaprakash Suppiah, Senior Lecturer in Pharmacy, Clinical & Health Sciences at the University of South Australia
Toh Gaik-Theng, Senior Lecturer, School of Medicine, Taylor's University
Real-world Evidence
How patient-led insights are transforming the way we develop health policy
Julie Cini, CEO, Advocacy Beyond Borders
Ruth Kuguru, Executive Director, Communications & Patient Engagement, Novartis Asia Pacific, Middle East, and Africa
INFORMATION TECHNOLOGY
56 Challenges and Advantages of Using Artificial Intelligence in Pharma Fausto Artico, Global R&D Tech Head and Director of Innovation and Data Science, GSK

Kevin Harrigan, Director of Innovation and Engineering, GSK
60 Blockchain
Mandatory in Constructing a High-fidelity Database Readying for The Downstream Disruptive Technologies, such as for The Artificial Intelligence Training.
Frank Leu, Founder and managing member, BioPharMatrix LLC
ExpertTalk
22 Revolutionising the Pharma Supply Chain
42 A Step-By-Step Strategy for Designing A MetaAnalysis
Ramaiah M, Manager, Freyr solutions
Balaji M, Deputy Manager, Freyr solutions
48 Measuring Vaccine Efficacy
The right endpoints are key to success
Paul Gillard, Vice President, Medical and Scientific Strategy, Vaccines Therapeutic Area
Delphine Saragoussi, Executive Director, Epidemiology and Scientific Affairs, Post-Approval Studies and Real-World Evidence
Sarah Rosen, Senior Director Project Management, Peri- and Postapproval Studies and Real-World Evidence PPD clinical research business of Thermo Fisher Scientific
Blockchain, Digitalisation, and Collaboration
John Ward, Founder, and CEO, ServBlock
40 Preformulation Studies
Its importance in drug development
Yogeshwar Bachhav, Founder and Director, Adex Pharma consultancy Services
52 Adaptive Clinical Trial Designs
Esther Mahillo, Vice President, Operational Strategy and Feasibility, Precision for Medicine
2 PHARMA FOCUS ASIA ISSUE 51 - 2023
Cover Story 14
















3 www.pharmafocusasia.com Pressure regulators Control valves Pipeline ancillaries Special equipment Steam traps Safety valves adca@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467 PRODUCTS MANUFACTURED IN PORTUGAL Guia PBL PORTUGAL HIGH PURITY. HIGH DEMANDS. H igh purity equipment for clean steam AV 001 AP E 01.20
Advisory Board
Alessio Piccoli
Lead
Sales and Business Development Activities for Europe Aragen Life Science

Andri Kusandri
Market Access and Government & Public Affairs Director
Merck Indonesia
Brian D Smith
Principal Advisor PragMedic
Gervasius Samosir

Partner, YCP Solidiance, Indonesia

Hassan Mostafa Mohamed Chairman & Chief Executive Office ReyadaPro


Imelda Leslie Vargas Regional Quality Assurance Director Zuellig Pharma

Neil J Campbell Chairman, CEO and Founder Celios Corporation, USA


Nicoleta Grecu Director

Pharmacovigilance Clinical Quality Assurance Clover Biopharmaceuticals
Nigel Cryer FRSC
Global Corporate Quality Audit Head, Sanofi Pasteur
Pramod Kashid

Senior Director, Clinical Trial Management Medpace
Quang Bui
Deputy Director at ANDA Vietnam Group, Vietnam

Tamara Miller
Senior Vice President, Product Development, Actinogen Medical Limited
EDITOR
Prasanthi Sadhu
EDITORIAL TEAM
Grace Jones
Harry Callum
Rohith Nuguri
Swetha M
ART DIRECTOR
M Abdul Hannan
PRODUCT MANAGER

Jeff Kenney
SENIOR PRODUCT ASSOCIATES
Ben Johnson
David Nelson
John Milton
Peter Thomas
Sussane Vincent
PRODUCT ASSOCIATE
Veronica Wilson
CIRCULATION TEAM
Sam Smith
SUBSCRIPTIONS IN-CHARGE
Vijay Kumar Gaddam
HEAD-OPERATIONS
S V Nageswara Rao
Ochre Media Private Limited Media Resource Centre,#9-1-129/1,201, 2nd Floor, Oxford Plaza, S.D Road, Secunderabad - 500003, Telangana, INDIA, Phone: +91 40 4961 4567, Fax: +91 40 4961 4555 Email: info@ochre-media.com
www.pharmafocusasia.com | www.ochre-media.com


© Ochre Media Private Limited. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, photocopying or otherwise, without prior permission of the publisher and copyright owner. Whilst every effort has been made to ensure the accuracy of the information in this publication, the publisher accepts no responsibility for errors or omissions. The products and services advertised are not endorsed by or connected with the publisher or its associates. The editorial opinions expressed in this publication are those of individual authors and not necessarily those of the publisher or of its associates.
Copies of Pharma Focus Asia can be purchased at the indicated cover prices. For bulk order reprints minimum order required is 500 copies, POA.
Magazine Subscribe LinkedIn







































5 www.pharmafocusasia.com
The Glocalisation Change
The limits of naivety
This third article in the series addresses the second, key question of glocalisation: How should we adapt our global strategy to our local markets? Current practice, which defaults to change only what must be changed, is naïve, undervalues local affiliates and fails to create competitive advantage. This article identifies the three flaws in that approach and the three habits that characterise more astute glocalisers. It then sets up article four, which looks at how life science firms learn from their glocalisation.
Brian Smith, Principal Advisor, PragMedic
The first article in this series began from the premise that exploiting global markets creates a dilemma for pharmaceutical and other life sciences companies, which I called the glocalisation challenge. This is the need to adapt to local market conditions whilst adhering to a global strategy. That first article identified
the three parts of that challenge and the second article addressed the first of those: how to allocate the right amount of the right kind of resources to the right markets. This article, the third of the series, addresses the second part of the glocalisation challenge: how to adapt to the market conditions of the targeted countries.
The most striking finding of my research is that most pharmaceutical companies take an “isomorphic” approach to glocalisation; i.e. they copy what they see others doing and call it “adopting best practice”. In fact, there is rarely any evidence that what they are emulating is best practice and, even if it were, this isomorphic behaviour ignores the important differences between different companies’ goals, strategies and cultures. When executing global strategies in local markets, isomorphic behaviour is typically minimalist. It begins with making essential changes, for example to meet local language, regulatory, reimbursement and other market access requirements. It ends with making minor tweaks to marketing communications, such as adapting the mix to local media or changing imagery to reflect local use. In other words, isomorphic adaptation of global strategy is usually limited to the 2 Es: The essential and the easy.
This minimalist approach is necessary but not sufficient for success. It limits the ability of the firm to create compelling customer preference by neglecting the three key factors that make each market different from other, even neighbouring, markets. It neglects the local competitive environment, it neglects market heterogeneity and it neglects the “beyond the pill” needs of the targeted market segments, as discussed in article 2, The Glocalisation Choice.
But whilst most companies are naïve in this way, not all are. In my research, I’ve found that a few exemplary companies, those I refer to as astute globalisers,

6 PHARMA FOCUS ASIA ISSUE 51 - 2023
STRATEGY
Your second important glocalisation decision is how to adapt to local conditions
attend to these three neglected factors with marketing processes that are quite different from those of their naïve peers. They have different, more sophisticated habits for competitive analysis, market segmentation and value proposition design. In figure one and in the next three sections, I’ll describe those three important differences and explain how they enable more effective glocalisation.
Habit 1: Pooling competitors
When I observe naïve glocalisers, their definition of competitors is “any firm selling similar products”. That is, competitors are those who compete for market share. This is an operationally useful way of framing proximal competitors but it is strategically myopic. By contrast, astute glocalisers take a much wider view. To them, a competitor is anyone who want to drink from the same pool of profit associated with the market. In other words, they frame competition in much the same way as Porter did in his 1980 seminal work “Competitive Strategy”. In that framing, competitive pressures can come from many directions. For example, in many AsiaPacific primary care markets, traditional remedies are as much of a rival as more obvious, direct competitors. And in mature markets, large customers such as state and private insurers or healthcare providers may be the most significant rivals for the profit pool.
What makes this habit valuable is that whilst a global strategy probably usually includes positioning against direct, similar competitors, it may disregard the blend of substitutes, new entrants, buyers and suppliers that create the unique competitive conditions of the targeted market. The habit of thinking of your competitors as those who drink from your market’s profit pool, not just those who compete for market share, is, therefore, the first big difference between naïve and astute glocalisers. It is a necessary precursor to adapting the global strategy to local markets in a way that creates competitive advantage.
Habit 2: Contextualising decisions
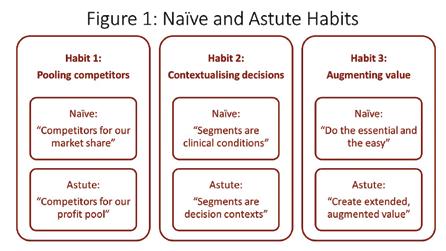
All markets are heterogeneous, meaning that even patients with same condition, prescribers with the same role and payers in the same kind of organisation differ significantly in their needs, motivations, attitudes and, consequently, value-seeking preferences. Understanding and adapting to this three-dimensional heterogeneity is critical to effective glocalisation and it is the second big difference between naïve and the astute glocalisers.
Naïve glocalisers are often driven by global brand plan templates and other aspects of an inflexible, top-down culture. As a result, they tend to mimic the market segmentation described in global plans with little or no adjustment for local conditions. This has the benefits of simplicity and compliance
with global views but those benefits come at the cost of reduced competitiveness. That’s because they discount the subtle differences in local market heterogeneity that appear trivial from Paris, Chicago or Tokyo but are commercially consequential in Shanghai, Mumbai, or Kuala Lumpur. In other words, the brand plans of naïve globalisers often use global market segments even when they know them to be crude approximations of local reality.
Astute glocalisers do not the segmentation that comes with global plans but neither do they follow it slavishly. Instead, they use it as a starting point. In particular, they build on the often onedimensional global segmentation that is usually expressed as a disease condition. Onto this they add non-clinical patient attributes, such as adherence behaviours or engagement with treatment decisions. Then they add in the heterogeneity of prescribers and professionals, usually also created by a combination of tangible and intangible drivers. Finally, they overlay the heterogeneity of local payers, which often exists at both national level (for example, insured vs out of pocket) and account level (for example value- and price-driven local formularies). Collectively, this approach is called contextual segmentation because it describes the complete context of the prescribing or usage decision.
Using the global view of market segmentation as starting point for contextual segmentation, rather than applying it locally without adaptation, has a cost. This habit requires deep market insight and the courage to tell head office that their view is insufficiently nuanced. But paying those costs gives astute glocalisers an advantage over their naïve rivals. Their richer, deeper understanding of the local market’s contextual segmentation puts astute glocalisers in a position to create much stronger and more compelling value propositions.
Habit 3: Augmenting value
The minimalist, “do only what’s essential and easy” approach of naïve globalisers
7 www.pharmafocusasia.com
STRATEGY
is most visible in the multitude of value creating activities that are involved in strategy execution. Naïve globalisers focus on the essentials of gaining regulatory and reimbursement access and the easy of adapting marketing communications to local media and culture. In an increasingly digital world, even the latter is becoming a minor task. Fundamentally, the naïve habit of executing strategy involves adoption of the global strategy’s core value proposition with minimal change. Typically, this core offer is centred on a clinical value proposition of efficacy compared to direct rivals. In other words, naïve glocalisers do little more than translate the global strategy’s clinical claims into local language. This minimalist-adaptation habit is shaped by the first two habits of defining competition narrowly and understanding market segmentation simplistically. The cumulative outcome of all three naïve habits is that, unless the clinical claims are outstandingly differentiated (a rare and even temporary situation in today’s markets), the added value perceived by patients, prescribers and payers is small. Perceiving low added value, payers then do what they are paid to do; play rivals against each other to put price pressure on all manufacturers.
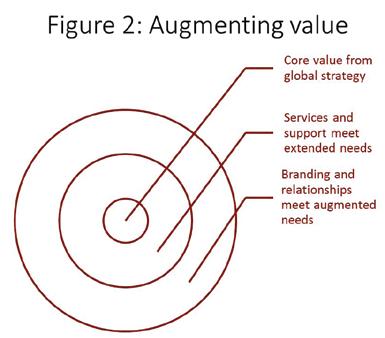
The astute glocalisers’ strategy execution habit is also shaped by how they have defined competition (widely) and market segmentation (contextually). Because, unlike their naïve comparators, they have a multidimensional understanding of the competitive environment, they have a better appreciation of their customers’ framing of relative value. And because they have a richer, deeper and contextualised understanding of patients’, prescribers’ and payers’ motivations, they are clearer about how to create value. Together, these advantages allow them to build on the “core” value proposition from the global strategy in two steps, as shown in figure 2. First, they add a layer of extended value; for example risk-sharing price deals that address payers’ conservativeness, or education
programmes that address prescribers’ fears of misprescribing. Then they add a layer of augmented value; for example, their opinion leader programmes address the self-actualisation needs of key professionals and branding messages address the reassurance needs of “laggard” segments. Their target segments’ perception of superior value, based on everything they experience, allows astute firms to avoid the price pressure faced by naïve glocalisers.

The sum of our habits
To steal from Aristotle, glocalisers are the sum of their habits. Naïve glocalisers’ habits of limiting themselves to the “essential and easy” means they take a narrow view of competition, adopt global segments without local adaptation and
simply echo the core value proposition that comes with the global strategy. Astute glocalisers’ habits of using global strategy as a starting point means they compete for the profit pool, not market share. To do so, they work hard to understand the rich, multidimensional, contextual segmentation that makes their market unique. They use this to build extended, augmented value propositions that create added value in the eyes of their customers.
These differences have three consequences that astute leaders in the life sciences industry grasp but naïve glocalisers don’t. First, it means that astute glocalisers create competitive advantage and shareholder value. Second, it means that local affiliates have a much bigger, more value adding role than merely executing headquarters’ instructions. Finally, it means that local affiliates can be a rich source of organisational learning for the wider, global organisation. It is to that last point that I will turn to in article four.
Brian D Smith works at the University of Hertfordshire, UK, and Bocconi University, Italy and researches the evolution of business models and competitive strategy in the global life sciences industry. He has published over 300 papers, articles and books. www.pragmedic.com
8 PHARMA FOCUS ASIA ISSUE 51 - 2023
AUTHOR BIO
STRATEGY
EXTRACTABLE AND LEACHABLE TESTING
Insights into Risk Assessment, Method Validation and Study Design in Bio/Pharmaceuticals
Bio/Pharmaceutical products encounter a wide range of polymeric materials on their journey from the production line to the patient, with plastic and rubber surfaces present at almost every stage of a product’s lifecycle. While these materials are extensively used in the manufacture and storage of medicines, the leaching of these potentially hazardous substances into the products themselves can pose a risk to human health.
Extractable & Leachable studies enable drug and device manufacturers to identify and quantify the risks of potentially toxic impurities migrating into a drug product from container closure systems, processing equipment or medical devices. In this article, we take a closer look at Extractable and Leachable species and the design of E&L studies as well as test methods which are deployed for accurate determination of E&L contamination.
Understanding Extractables and Leachables.
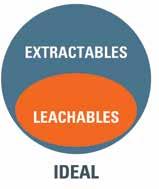
Extractables: Organic and inorganic chemical species that can be released from the surfaces of components used in the manufacture and storage of drug products under laboratory conditions (accelerated or exaggerated temperatures, solvents or surface exposure).

Leachables: Organic and inorganic chemical species that can be released from the surfaces of components used in the manufacture and storage of drug products under conditions of normal use.
Extractables represent the worst-case scenario regarding release of mobile chemical species from manufacturing and packaging components during forced extraction. Generally, leachables should then comprise a sub set within this pool of mobile chemical species, released under the gentler conditions of on-shelf storage or use.
Rigorous testing workflows must be employed to make sure extractable and leachable compounds are fully understood and do not impact patient safety or

9 www.pharmafocusasia.com
pharmacological activity. The need to detect a wide variety of compounds necessitates the application of a range of different analytical techniques and testing protocols, making E&L studies often lengthy and complex.
In practice, however, the leachables study may identify additional species that were not observed during the preceding extractables study. Thus, the set of leachable species is not wholly included within

leachable species, and some common examples are detailed below:
Leachable species to consider as part of an E&L study include:
• Antioxidants and stabilisers
• Anti-static coatings
• Lubricants, slip agents and emulsifiers
• Dyes and colourants
• Vulcanising agents
• Residual monomer, polymer and oligomer species
• Phthalates, nitrosamines and polyaromatic hydrocarbons (PAHs)
• Toxic elements – e.g. mercury, lead, arsenic, cadmium
The extensive array of materials employed in the production and preservation of therapeutic goods, along with the considerable range of leachables that could potentially migrate into the final product, necessitates the deployment of structured and thorough E&L studies.
E&L studies for toxicological evaluation
the set of extractables, but there is strong overlap between both sets (figure 1).
There are multiple sources which can contain
Leachables that migrate into pharmaceutical products from manufacturing and packaging systems require identification and monitoring over the shelf-life of the product. The collated data permits a toxicological assessment to be made with respect to any extractables and leachables found, ensuring patient safety.
One must also determine the Safety Concern Threshold (SCT) for the product under investigation.
The concept of Safety Concern Thresholds (SCTs) was first introduced by the Leachables and Extractables Working Group of the Product Quality Research Institute (PQRI). SCTs are defined as the threshold dose at which an individual leachable would not pose a safety concern, including potential effects such as carcinogenicity.
Two factors which have a significant impact on the SCT are the route of administration and the degree of product-packaging contact. To assign
Topical powders; oral powders
Oral tablets and oral (hard and soft gelatin) capsules
10 PHARMA FOCUS ASIA ISSUE 51 - 2023
Figure 1: Relationships between extractables and leachables
Manufacturing Primary packaging - External components Primary packaging - Internal components Secondary and tertiary packaging Filters Container-closure system (vials, caps, lids and foils) Gaskets and O-rings Labels and adhesives Single-use bags Single-use systems (syringes and intravenous bags) Valves Inks and colourings Tubing Springs
Degree of concern associated with the route of administration Likelihood of packaging component-dosage form interaction High Medium Low Highest Inhalation aerosols and solutions; injections and injectable suspensions Sterile powders and powders for injection; inhalation powders High Ophthalmic solutions and suspensions; transdermal onasal aerosols and sprays Low Topical solutions and suspensions; topical and lingual aerosols; oral solutions and suspensions
risk from leachable species to the pharmaceutical product under investigation, the USFDA developed the following matrix:
Products such as aerosols and injectables have the highest associated risk, whilst oral tablets and capsules have the lowest risk.
The PQRI have recommended that the high risk SCT is set at 0.15 µg/day, whilst the low risk SCT is set at 1.5 µg/day, both having been justified from toxicological and safety perspectives.
the number of components and material types that are to be tested, and the solvents with which to perform the extractions.
Simple storage systems, e.g. glass ampoules or plastic bottles with screw caps, will have a limited number of components. However, more intricate units, e.g. pump dispensers containing O-rings and springs, will contain multiple components which all require investigation. Secondary and tertiary packaging also needs to be considered at this stage.
To ensure a comprehensive representation of both organic and inorganic extractable species, it is imperative to perform extractions utilising a diverse range of solvents possessing distinct solvating capacities. In the context of liquid formulations, it is advisable to opt for solvents that closely mimic the pharmaceutical product's final composition, thereby facilitating the generation of an extractables profile that reflects a worst-case scenario. However, caution must be exercised to prevent the utilisation of solvents with excessive potency, as their aggressive nature may induce material degradation and consequently lead to an artificially inflated array of extractable species.
Two or three solvents are typically chosen, but more can be used if considered appropriate. Common examples include:
• Water (neutral, and acidic or basic if pH ≠ 7)
• Organic solvent (ethanol, isopropanol or n-hexane)
Under certain conditions, such as short-term exposure or in the treatment of a life threatening condition, the SCT can be raised above 1.5 µg/day.
Designing an E&L study
An E&L study comprises two interlinked yet distinct projects. The extractables study aims to identify any substances originating from manufacturing components (wherever relevant) and the packaging system that could potentially migrate into the pharmaceutical product during storage or use under typical conditions. This establishes a foundational assessment for any subsequent leachables study, which includes a series of tests carried out at predefined time-points on the pharmaceutical product for the duration of its shelf-life.
Extractables study
The manufacturing, delivery and storage components under investigation are often extracted in isolation of the pharmaceutical product. Key points to consider are
The material type under investigation is an important consideration. It is necessary to perform extractions using all selected solvents on plastics and rubbers. However, conducting organic solvent extraction on metal springs would primarily yield inorganic impurities, resulting in limited usefulness. Therefore, optimising the extraction process requires thoughtful assessment of solvent compatibility with the specific components under investigation.
Leachables study
In an E&L study, predefined time-points are established prior to initiation, which can be conducted in parallel with a stability trial. Samples obtained at these timepoints undergo targeted screening with validated methods for leachables previously identified from the extractables study and untargeted screening for any newly discovered species during the leachables study. If any new leachables are found to exceed the Safety Concern Threshold (SCT), they are identified and subjected to toxicity assessment.
Crucial to a successful study is the creation and storage of appropriate controls and samples. The controls should be stored in such a fashion that there
11 www.pharmafocusasia.com
is minimal risk of leachable ingress, and carefully labelled avoiding the use of inks and adhesives directly on the container. Leachable samples may be stored inverted as well as upright (e.g. bottles fitted with caps or lids), and specific storage conditions (e.g. 4°C, 25°C/60% RH, 40°C/75% RH) should be carefully considered.
Analysing E&L samples
In general, extractables and leachables can be divided into three broad groups:
• Non-volatile
• Volatile and semi-volatile
• Inorganic / elemental.
Well established analytical methods are required to analyse all samples and can be used across both studies. Figure 2 exemplifies the typical analytical strategy employed.
Liquid chromatography–mass spectrometry (LC-MS) analysis permits the analysis of larger, non-volatile species. Direct injection gas chromatography mass spectrometry (GC-MS) analysis permits analysis of both volatile and semivolatile species. Head-space GC-MS analysis is an option when significant volatile species are expected. Elemental impurities are analysed and quantified by inductively coupled plasma mass spectrometry (ICPMS).
Well established analytical methodologies are a critical prerequisite prior to initiating research, although they can be customised to accommodate the diverse array of pharmaceutical products. For leachable studies, feasibility is a necessary prerequisite for validation of the method to ensure that the pharmaceutical product under investigation does not exhibit inhibitory matrix effects allowing successful recovery of extractables from the sample matrix, thus avoiding the necessity for method redevelopment and subsequent revalidation.
The complex nature of the E&L studies requires thorough planning, access to a variety of complex analytical hardware and expertise running validated methods. The investigative nature of the work
demands a team of analysts with capabilities across method development & validation, molecular identification and toxicological evaluation, to ensure that the study is run in a smooth and efficient manner and data is interpreted accurately.
With thorough planning, appropriate analytical hardware and in-depth knowledge in place from the beginning, an E&L study can be run efficiently and successfully, ultimately ensuring that patient safety is maintained.
SGS Capabilities in E&L Testing
SGS Health Science enables the medical and health innovators of the world to deliver life-changing solutions in the fastest, safest and most efficient way, helping improve the lives of many. We provide the highest quality services, reliable expertise and guidance through our network of laboratories located around the globe.
SGS has a global expert network with more than 20 years of experience in E&L studies:
The SGS India Centre of Excellence in Navi Mumbai is a USFDA approved facility and can undertake complex E&L studies, which are an important part of drug product development and materials qualification.
SGS capabilities for E&L studies include:
• Extractables on Container Closure Systems (Glass vials with rubber stoppers, prefilled syringes, IV plastic bags, plastic bottles with caps, tubes, transdermal patches)
• Extractables on Secondary Packaging (Pouches, label, ink migration, etc.)
• Extractables on Single Use Systems and Multiple Use Systems (Bioprocess bags, tubing, connectors, filters, gaskets, etc.)
• Leachables Screening
• Leachables Method Development & Validation
• Leachables Stability Studies
• Impurity Unknown Identification
• Toxicological Reports Assessment (Optional)
• Consultancy E&L Strategies
12 PHARMA FOCUS ASIA ISSUE 51 - 2023
LC-MS GC-MS (Direct Injection / Head space) ICP-MS Extractables Sample Leachables Sample ANALYSIS Non-volatile leachables Volatile and semi-volatile leachables Elemental / Inorganic leachables
Figure 2: Generalised approach to analysis of E&L samples
This state-of-the-art facility is equipped with high-end instruments including HRMS, LCMS/MS, GCMS/MS, ICPMS which are extensively used for conducting extractable and leachable analyses.
The extractables and leachables testing and assessment strategy, followed by SGS, includes:
• Extractable Study: Container closure extraction study done by using varying polarity aqueous and non-aqueous solvents
• Method Development and Validation: Well developed and validated method as per analytical evaluation threshold (AET) derived for volatile organic compounds, semi-volatile organic compounds, non-volatile organic compounds and elemental impurities.
• Leachable contact part study: Manufacturing components such as, filters, tubing’s, gasket’s etc can be executed on a product-specific basis or as a standalone study to support a basket of finished product to be exposed to these manufacturing components.
The SGS laboratory is equipped to conduct comprehensive study design and testing for extractables and leachables in finished pharmaceutical packaging, process equipment, and
medical devices. Additionally, it undertakes analysis for leachables in final products. These services can be customised based on specific requirements or aligned with the guidelines provided by the Product Quality Research Institute (PQRI).
Our experts can be reached at: in.pharmaqc@sgs.com
About SGS
SGS is world’s leading Testing, Inspection and Certification company. We are recognised as the global benchmark for sustainability, quality and integrity. Our 97,000 employees operate a network of 2,650 offices and laboratories, working together to enable a better, safer and more interconnected world.
Wherever you are, whatever your industry, our experts worldwide provide specialised solutions to make your business faster, simpler and more efficient.
13 www.pharmafocusasia.com
STRATEGY
Real-world Evidence
How patient-led insights are transforming the way we develop health policy


Healthcare leaders in the Asia-Pacific region are keen to address the perception gap that exists around real-world evidence (RWE). While they recognise the significant advantages of RWE, patients are not aware of its actual or potential use. To bridge the current gap, the article offers two perspectives on the relevance of RWE to patient communities and strategies to foster greater multidisciplinary participation in RWE collection. How patient organisations use RWE to gain disease awareness traction and shape health policy and the importance of collaboration among multiple stakeholders are discussed in the article.
Julie Cini, CEO, Advocacy Beyond Borders
Ruth Kuguru, Executive Director, Communications & Patient Engagement, Novartis Asia Pacific, Middle East, and Africa
Hearing the diagnosis from the doctor that your child has spinal muscular atrophy (SMA), a severe genetic condition that leads to muscle wastage and often life changing complications, is something no parent should have to experience. Hearing those words with a second child goes beyond the unthinkable. Eighteen years ago, this was my reality. That is when I vowed that what was ultimately a terminal diagnosis for my two children, wouldn’t be for many others who would come after.
One in 35 people in Australia unknowingly carry the faulty SMA gene which can be passed down to newborns. The disease is cruel and can often lead to the most basic everyday actions that many of us take for granted, becoming an impossible challenge without the help of external care. Despite its brutal implications, treatment options and wider support services were rarely available in Australia at the time. As such, I recognised that the only path to meaningful change was if we as a patient community could demonstrate the burden of the disease, and the urgent need for radical improvements in the care available. Thus, I unknowingly began collecting what would turn out to be the largest set of privately held SMA Real World Evidence (RWE) records in Australia, and in parallel, started a patient organisation named SMA Australia, to enhance the quality of life for anyone associated with this unforgiving condition. Today, RWE is being utilised across a variety of conditions and it is my hope that other patient organisations can make use of this approach to help raise their voice in advocacy opportunities.
What Is RWE?
RWE are the clinical insights that are derived from a patient’s practical health status. RWE can help us to understand how patient individuality and habits impact health outcomes — thus helping predict the progression of a disease, a patient’s responses to a therapy, or the risk of adverse events. Use of RWE is by no means restricted to SMA — this approach can be applicable to a variety of conditions where patient organisations want to highlight the lived experience of their members. Unlike clinical trials, whose cohorts and conditions are often very restrictive, RWE can be sourced from a large number of individuals living in the population, sometimes only restricted by the number of patients living with a condition. By gathering wider context around how someone lives, we are able to understand the more personal and
deeper consequences of a condition on both patients, as well as their families.
RWE is not a new phenomenon but with the advent of new digital and advanced analytics, the opportunities for enhanced utilisation across a variety of diseases is improving. Presently, the information plays a critical role in the development of treatments, the creation of health policy, and in understanding the overall lived experience for patients with a number of conditions. While advances in modern medicine have helped us understand the components of many diseases, the implications on a patient’s day-today experience still needs to be derived from them. The expansion of online patient organisations and communities has provided a platform for this evidence to be collected and shared, meaning that we now have a much clearer picture of the impact on quality of life across
multiple populations. At the outset of my research, my data was focused largely on the epidemiology of the condition: How old was the individual? What SMA condition type did they have? Were they male or female? While this was hugely valuable for mapping the condition across Australia, many of the unique findings around quality of life came much later.
Using RWE to strengthen the patient voice
Despite securing some valuable SMA data, the perennial battle that I, and many other patient organisations face, is being included in the meaningful discussions that will ultimately shape your members’ lives, and the lives their families. Throughout my 18 years of being involved in the SMA community, I have often encountered shut doors where my opinions and data have been disregarded.
15 www.pharmafocusasia.com
Collaboration between all stakeholders is vital for unleashing the potential of real-world evidence (RWE), addressing hurdles that impede access for patients, and driving us towards being healthier as a region. With strong support from passionate patient leaders like Julie, APPIS continues to partner with communities in this region to share practical knowledge on how to effectively collect RWE and how to include patients at the core of healthcare decision-making processes in the country.
STRATEGY
Ruth Kuguru, Executive Director, Communications & Engagement, Novartis Asia Pacific, Middle East and Africa, and APPIS 2023 Council Member.
The reality of engaging in a medical field is that it can be a tightly knit group that is often difficult to participate in if you don’t have the relevant experience. Well, my experience was that I had two children who lived with SMA and had been collecting data across the country for years, and in my mind, that made me and others like me, qualified to join the discussion.
However, I am pleased to say that the tide is changing, through platforms like The Alliance & Partnerships for Patient Innovation & Solutions (APPIS), for which I am a 2023 Council Member and I was also a speaker at the recent APPIS 2023 Summit. APPIS is a platform that is co-created by the APPIS Council, a panel of patient and healthcare leaders in Asia Pacific, Middle East and Africa, and organised and funded by Novartis. Since 2021, the platform has convened patient advocates and healthcare stakeholders from more than 60 countries to share their experience of a variety of conditions alongside a network of experts including: leading clinicians, healthcare providers, health policy experts, health tech companies and industry representatives, and more. APPIS provides opportunities to exchange ideas and contribute towards key discussions around the themes of health literacy, healthcare policy shaping and digital health and communications. The goal of APPIS is to help accelerate access for patients by bringing together the members of the health community to work together to create solutions. The APPIS platform and community helps empower and inspire patient organisations to think that they too can achieve progress using RWE in their countries by arming them with guidance reports and the right tools they need to expand their impact for the community.
The broader recognition for the power of RWE data has coincided with new collection processes. What used to take three to four years to collect can now be gathered in 12-to-18 months. RWE can help to provide quick analysis that can complement the existing
scientific research and potentially address the unknown surrounding patient experience. For SMA Australia, much of our value was centred around highlighting the level of carer assistance often required by patients. This sort of detail is often outside the remit of clinical trials or health policy research, but coordination of these services is a significant part of the lived experience of patients and their families. By understanding this logistical challenge, it gave us a strong platform to call for improved access to treatments that could help to provide patients with greater personal autonomy.
Increasing support is being given to the notion that we cannot expect policy initiatives to have positive outcomes if we haven’t consulted with the group that it directly relates to. For SMA Australia, this was one of our key messages and when a platform was provided to us, we could accurately demonstrate the unmet need we had, and work with all stakeholders to push towards practical solutions.
Building for the future
While partnerships with all stakeholders are essential for securing long-term
policy change, there are some actions that patient organisations can take to positively position themselves to highlight the unmet needs of their communities. Here are some of the lessons I’ve learnt from nearly two decades in this space:
Start early
When I first started SMA Australia, it felt like an uphill battle. It was a oneperson band where I was responsible for bookkeeping, marketing, copywriting, and all the rest. However, it was during this time that I started to engage with the community, to understand the hopes and trials of patients and families, and what more could be done to help them. At this critical juncture, treatment access and policy change felt like a lifetime away. However, with each connection, the ball starts to roll and our voice gets louder and louder. Soon you start to get traction and people sit up and listen. Ultimately, I knew that if the data was of value for me, it had the potential to be valuable for other stakeholders, which is why I encourage other patient organisations to collect early and broadly.
Educate your community
I encourage patient groups to gather data early, and to focus on the daily lived experience of the patients. While the epidemiology data is useful, there are now large datasets available within government public health organisations. However, they do not always have access to the day-to-day practical insights that truly indicate the quality of life of a patient. For example, when thinking about how the treatment will help the patient, it’s important to think about enhancing the quality of patients’ everyday lives. It’s up to patient groups to communicate patients’ unmet needs to stakeholders, while communicating to patients about treatment and care options. This is where patient groups have significant value and by grouping our voices, we can provide robust reasoning for why we need advancements in access. Further, when engaging with the community, try to
16 PHARMA FOCUS ASIA ISSUE 51 - 2023
STRATEGY
RWE is not a new phenomenon but with the advent of new digital and advanced analytics, the opportunities for enhanced utilisation across a variety of diseases is improving.
provide regular updates, as this is when people become frustrated, when they feel that stakeholders are unresponsive.
Be personal
It is easy to assume that governments want to protect budgets and are not interested in hearing about the need for enhanced resources for patients. However, through my time working at SMA Australia, we approached a number of policymakers directly and personably, and managed to secure advocates who became champions for our cause. In many cases, they found the work to be deeply rewarding and one of the key reasons for engaging in public service. Setting the tone by highlighting the quality of life for patients is crucial, followed by having a clear ask around what they can do to help. I encourage patient organisations to cast a wide net and think about who can support on your journey. I have seen first-hand how patient representatives can emphasise the harsh reality of disease and highlight the need for improved access to care and treatment to improve the quality of life of patients. If we don’t tell our story, politicians may not appreciate the scale of the problem.
Build on the work of others
Utilising RWE effectively can be an

overwhelming experience for patient organisations, but we don’t need to start from scratch or do it alone. Being part of the APPIS community can help you gain access to helpful resources and a network of members who can share best practices around amplifying the patient’s voice to secure access to healthcare and policy change. By engaging with a broad community of stakeholders we can exchange ideas and build comprehensive policy asks that
can help to deliver improvements in the care and quality of life of patients.
Call to Action
Although the burden of diseases like SMA is something no family wants to endure, the strength of the patient community is hugely powerful and can be a valuable support in tough circumstances. We now have an opportunity in an increasingly connected world to interact with each other and amplify our advocacy. APPIS brings together stakeholders from across the healthcare landscape and provides a unique platform to share best practice around how we improve the lives of patients. Patient groups should be consulted throughout the disease cycle if we are to develop impactful and longlasting health policy. One thing is certain, everyone has a role to play and by engaging with all stakeholders, we can ensure that care for patients with SMA and other conditions can be levelled up.
You can find out more about APPIS 2023 here 1, where you will be able to rewatch the insightful presentations, download useful reports from the APPIS Resource Centre and join our growing community.
Ruth Kuguru is the Executive Director, Communications & Patient Engagement for Novartis Asia Pacific, Middle East, and Africa. In her role, she leads her team to foster open, inclusive, and strong partnerships with patient communities, media, and healthcare leaders in Asia Pacific, Middle East and African countries. She is passionate about the impact strong collaboration can have in accelerating access and developing innovative solutions for patients.

Julie Cini founded Spinal Muscular Atrophy Australia in 2005, and had grown a successful non-profit patient advocacy group. For 18 years under Julie’s leadership in this organisation has seen her successfully campaign for increased awareness, diagnosis and better access to treatment for SMA patients. Julie’s consumer led research has had a great impact on the Pharmaceutical Benefits Advisory Committee (PBAC) and highlighting the severe unmet need within the SMA community for treatment. She continues to work with the policy makers to ensure patient evidence is valued and addressed as part of the submissions.
1, https://www.appisinitiative.com/appis-summit-2023

17 www.pharmafocusasia.com
AUTHOR BIO
STRATEGY
FUTURE OF CLINICAL TRIALS IN APAC
Oracle’s initiative
Jeyaseelan Jeyaraj (Jey), Senior Director, Solutions Consulting, Asia Pacific, Oracle Life Sciences Global Business Unit

What will the future of clinical trials be in the next few years?
We are seeing pharma shift to explore newer clinical trial models and technologies. For example, many are looking at decentralised trials as an option to open the pool of potential candidates and to make trials more accessible and equitable for more people. In the US, the retail pharmacy model is also expanding according
to Forbes1, which notes during the pandemic retail pharmacies provided vaccinations and has opened the door for more services.
We continue to see the expansion of the use of sources such as electronic health records (EHR) and connected devices that will enable companies to collect larger quantities of anonymous, highquality real-world evidence. Applying AI and machine learning to this data will have a tremendous impact on the market, especially in areas like pharmacovigilance. As new treatments come to market faster, patients and providers will further utilise these advancements to have more accurate safety information.
We expect the emergence of virtual CROs and their use of platform technology to address the demands of many new clinical trials contracted in Asia. Overall we see clinical trial recruitment in the region increasing faster than in Europe, North America, and Australia. According to “Clinical trials in Asia: A World Health Organization database study” 2there has recently been a seven-fold increase in the number of registered clinical trials in Asia. More trials were registered in Japan than in any other Asian country (30.8 per cent). Lower trial costs and a large patient pool may be key contributors to this increase – technology will also be a major factor in the success of these trials.
Organisations that strike the balance of futureforward technology and patient-centric models will
1 Retail pharmacies add new services as specialty drugs take greater role, Forbes May 2, 2023 - https://www.forbes.com/sites/brucejapsen/2023/05/02/retail-pharmacies-add-newservices-as-specialty-drugs-take-greater-role/?sh=45e0beead2bb
2 “Clinical Trials in Asia: A World Health Organization database study” 2019 - https:// www.ncbi.nlm.nih.gov/pmc/articles/PMC6647899/. Oracle is referencing this study under the Creative Commons License: https://creativecommons.org/licenses/by-nc-sa/3.0/igo/
have a greater chance of success and in taking a leading position in the industry.
How will real-world data (RWD) impact clinical trials?
Real-world data (RWD) will be critical in decisionmaking. The insights learned from this data combined with clinical research data can help shape drug companies’ approach to all aspects of the business including discovery, understanding safety in real-world conditions, go-to-market strategies, and how they run their clinical trials. As noted previously, sources like EHRs and connected devices will also enable companies to collect larger quantities of high-quality real-world evidence to better inform clinical trials.
What industry trends will we see specifically in APAC?
According to recent reports34, APAC continues to be a promising region for clinical trials. This is an opportunity for sponsors who are looking to capitalise on growth and for CROs who are emerging in the market. We expect that as this growth continues companies will turn to innovative platform technologies that can support all aspects of clinical trials and provide cost-effective models that bring value. This growth could also bring more cross-border studies and collaborations as sponsors look for larger pools of participants. According to “Growth opportunities: The clinical trials landscape of Asia-Pacific”5 in APAC, decentralised Phase I trials grew 60 per cent between 2017 and 2022, compared to an overall average of 10-20 per cent. APAC also accounted for the highest usage of in-home devices for oracle in any region in 2022.
What do companies need to do to adopt a more “consumer-centric” approach to patient care and engagement?
There needs to be a shift change within organisations to move toward a consumer-centric model where the patient has options relative to their participation in a trial. Some examples include enabling patients to remotely provide intake information, wearing sensor
3 APAC As a Clinical Trial Powerhouse, Informa, November 2022https://invivo.pharmaintelligence.informa.com/IV146738/APAC-As-A-Clinical-TrialPowerhouse
4 “Growth opportunities: The clinical trials landscape of Asia-Pacific” Clinical Trials Arena, January 2023 - https://www.clinicaltrialsarena.com/sponsored/growth-opportunities-theclinical-trials-landscape-of-asia-pacific/
5 “Growth opportunities: The clinical trials landscape of Asia-Pacific” Clinical Trials Arena, January 2023 - https://www.clinicaltrialsarena.com/sponsored/growth-opportunities-theclinical-trials-landscape-of-asia-pacific/
devices to convey clinical data, and sharing feedback via tele-visits. Leadership has to be willing to consider a new way of doing things and support these types of changes. COVID-19 was a changing force for many organisations to conduct trials differently.
What is the biggest roadblock to achieving better patient engagement?
One of the biggest challenges with patient engagement is accessibility. Many patients don’t have proximity to the trial site which creates both recruitment and engagement issues. However, this is being addressed in many ways with the new models of decentralised clinical trials. For example, electronic patient-recorded outcomes via a wearable device make it easier to collect data from a patient when they are at home, and electronic engagement tools such as chatbots make it very easy and efficient for patients to engage with their caregivers.
The pandemic accelerated the adoption of several different approaches to clinical trials. In APAC, which of these approaches is taking hold? What is the biggest trend, challenge, or
market factor that you are tracking?
The past several years challenged the status quo and catalysed the adoption of a technology-enabled ‘patient-centric’ decentralised clinical trials model. The reality of this shift has led to most of the industry planning on continuing to implement newer clinical trial models and technologies. Patient optionality now rules the day, and organisations must plan on giving patients the choice about how they participate in clinical trials – such as in-person, in a decentralised model, or a hybrid of both. This will continue to drive the changes in clinical trials and clinical trial models in the future.
Can you address the growth in CROs and the clinical trial market in Asia i.e. what is driving this growth and how will technology meet the demand?
As we have discussed publicly6, several factors are fueling this growth: local regulations in APAC countries continue to evolve and drive the need for “globallyaccepted” solutions to conduct clinical trials; growing
6 “The CRO market in APAC is heating up”, Oracle blog, February 2022 - https://blogs. oracle.com/health-sciences/post/the-cro-market-in-asia-pacific-is-heating-up-clinixir-is-ready-
19 www.pharmafocusasia.com 19
for-growth-with-clinical-one
health challenges in APAC, such as diabetes and hepatitis, are an impetus for more trials to be conducted; government funding is rising, and APAC is emerging as a cost-effective destination to conduct clinical research trials. Forward-thinking CROs are adopting purposebuilt technology that utilises a uniform platform to address the challenges and complexities of running clinical trials in a global economy. These will improve operational performance and differentiate themselves as the market becomes more competitive.
giving researchers access to a much more diverse pool of participants, and further information about these populations.
Access to trials may be hampered in many communities by factors like distance. Do you see the integration of remote trials as a feasible opportunity?
Diversity in clinical trials is a complex issue. However, the more diverse the population pool, the more that diverse data could be used to inform researchers overall about differences amongst the various populations. Ensuring people from diverse backgrounds join clinical trials is key to advancing health equity. Participants in clinical trials should represent the patients that will use the medical products. This is important because people of different ages, genders, races, and ethnicities may react differently to certain medical products. If the pool of trial participants does not represent the individuals most affected by a particular disease, condition, or behavior, there is an increased likelihood of creating gaps in our understanding of diseases and conditions, preventive factors, and treatment effectiveness across populations.
In terms of diversity and its effect on the regulatory framework, it will be interesting to see how governing bodies choose to address it. During the COVID-19 pandemic, Oracle worked with the University of Oxford on a system that helps identify variants early on, so that healthcare providers can learn whether current vaccines are working against those strains and public health officials can better determine if hospitals will be overrun with new patients. In general, better information will contribute to a better public health policy. If new policies around public health are put into place, there will likely be changes made to the existing regulatory framework as it relates to clinical trials.
We will also see more data-based communication, so that data from wearable devices and home monitoring systems can be communicated to healthcare facilities. This two-way channel will also allow clinical trial researchers to expand their reach to communities that aren’t close to a clinical site,
Yes, not only is it feasible but the onset of the COVID19 pandemic quickly established that some aspects of decentralised clinical trials could, in fact, work. With the right technology and infrastructure in place, patients can participate in trials without having to visit the site every time the site needs to collect a data point. As we’ve discussed elsewhere, the emergence of electronic patient-reported outcomes, such as with wearables and other devices, paired with better communication infrastructure such as chatbots can provide a better and more efficient experience than patients have under traditional clinical trial models.
Another current issue, health equity, has diverse definitions around the world. What is health equity, and how is Oracle Life Sciences defining and pursuing it across the Asia Pacific?
Health equity in clinical trials refers to the fair and just inclusion of diverse populations in the design, implementation, and analysis of clinical research studies. Historically, clinical trials have not always included diverse populations, particularly people from underrepresented communities, which can limit the generalisability and applicability of the research findings.
There are several reasons why health equity in clinical trials is important. First, diverse populations may have different responses to medical interventions and treatments, and by not including them in clinical trials, we may miss important differences that could impact patient outcomes. Additionally, by excluding underrepresented populations, we may perpetuate health disparities and worsen existing health inequities.
To promote health equity in clinical trials, researchers can take several steps. One key strategy is to recruit intentionally a diverse participant population, including people of different ages, genders, races/ ethnicities, socioeconomic backgrounds, and health conditions. Researchers can also consider the cultural and linguistic needs of participants and provide appropriate accommodations to help ensure that everyone can participate fully.
20 PHARMA FOCUS ASIA ISSUE 51 - 2023
Speaking of clinical progress, diversity is a subject that hasn't been on the health agenda for very long. How do you see this affecting the regulatory framework and being more than simply a talking point?
In addition to recruitment, researchers can also work to ensure that their study designs and protocols are inclusive and accessible to all participants. For example, they may need to consider factors such as transportation, childcare, and language barriers when designing study procedures. They may also need to be flexible in their approach to accommodate individual participant needs.
With smaller biotech companies now receiving the majority of FDA approvals and maybe having fewer resources to commit to issues like clinical trial diversity, how much of an impact can Oracle Life Sciences initiative really have?
Ensuring clinical trial diversity is critical to advancing healthcare equity and providing the best possible care for all patients. Biotech companies, regardless of their size, have a responsibility to prioritise diversity in their clinical trials.
Here are some ways to overcome the challenge of limited resources:
Collaborate with patient advocacy groups: Biotech companies can partner with patient advocacy
groups to identify diverse patient populations and recruit them for clinical trials. These groups can also help companies understand the unique healthcare needs and experiences of underrepresented populations.
Leverage technology: Biotech companies can use technology to streamline the clinical trial process, reduce costs, and reach a more diverse patient population. For example, virtual clinical trials, which allow patients to participate in trials from their homes, can help overcome geographical barriers and increase participation from underrepresented populations.
Incorporate diversity into trial design: Biotech companies can design their clinical trials with diversity in mind. This means considering factors such as race, ethnicity, age, gender, sexual orientation, and socioeconomic status when selecting study participants.
How much is the industry's effort to increase the number of clinical trials hampered by issues with pricing and market access for novel products?
AUTHOR BIO
Jeyaseelan Jeyaraj (Jey) is a recognised Digital Health Strategist on addressing the challenges facing the global Healthcare Life Sciences industry and prospects for improving patient outcomes through better use of data and information. Jey brings his 20 plus years of broad consulting experience with Healthcare Providers, Public Health Pharmaceutical R&D that spans biotech, pharma and CRO.

Jey is the President of the Healthcare Information and Management Systems Society (HIMSS) India chapter. He was the member of the drafting committee for Digital Health Standards, National Accreditation Board for Hospitals Healthcare Providers (NABH) in India. He has published several papers and articles on Digital Health technologies. Jey has completed Post Graduate Program in Management from University of California (ULCA) Anderson School of Management. Jey is a frequent speaker about healthcare, biopharmaceutical trends and strategy in industry conferences. Jey is the recipient of CXO Health Excellence Award 2021 and recognised as the Top 50 Strategists in Healthcare.
The industry’s effort to increase the number of clinical trials is not hampered but rather evolving. We are already seeing a greater interest from pharmas, CROs, and sites for a technology platform that can enable trials to be set up and conducted more quickly and efficiently. Over time, at Oracle, we hope our technology will contribute to the number of trials increasing. We recognise that healthcare, including clinical trials, is very complex, but with easier-to-use systems, we believe we can increase participation rates which can help lead to better outcomes7 .
There is a lot of anticipation and excitement surrounding CAR-T, but how can you balance patients' expectations with the current scientific landscape?
These are complex problems that healthcare professionals and researchers are trying to solve. We support various discovery processes with our technologies. We are hopeful that as technology evolves and access to data increases, therapies and drugs will come to fruition more quickly to help those in need.
7 “Larry Ellison outlines how Oracle can help shape a new future in healthcare” Oracle Blog, June 2022 - https://blogs.oracle.com/healthcare/post/larry-ellison-oracle-shape-futurehealthcare
21 www.pharmafocusasia.com 21
In this article, John Ward discusses the impact of blockchain, digitalisation, and collaboration on the pharmaceutical supply chain. Addressing vulnerabilities, fostering partnerships, and embracing technology can lead to a more resilient, patientcentric industry. Ward highlights the importance of ethical considerations, workforce development, and strategic alliances for the sector's future success.
John Ward, Founder and CEO, ServBlock

Revolutionising the Pharma Supply Chain Blockchain, Digitalisation, and Collaboration
1. What are the major changes you see affecting today’s pharmaceutical supply chain?
The continuous digital transformation, including blockchain technology for greater traceability, increasing attention to sustainability, and a move towards localised production to reduce risks are the main developments I see affecting today's pharmaceutical supply chain. The necessity for a resilient and adaptable supply chain has been further underscored by the COVID-19 pandemic.
Technology frontiers have witnessed some quick recent breakthroughs that will help with the digital transition.
My personal opinion and the main objective of ServBlock is that the pharma supply chain will undergo significant changes as a result of blockchain technology. Blockchain technology can increase supply chain traceability, security, and trust by offering a decentralised, tamper-proof ledger. It might help stop the sale of fake medications, speed up recalls, and enable stakeholders to share information securely.
Pharma firms are now giving environmental friendly activities, s uch as decreasing waste, increasing energy efficiency, and adopting circular economy ideas into their operations, top priority due to an ever-increasing focus on sustainability.
STRATEGY
ExpertTalk
Business organisations are looking into localised production and distribution solutions to lessen supply chain vulnerabilities. By using this strategy, disruptions brought on by geopolitical variables or transportation problems can be reduced.
Companies increasingly rely on third-party providers for manufacturing, packaging, and other supply chain activities to streamline operations and cut costs as outsourcing and contract manufacturing gain dominance. Once more, Servblock has positioned itself to further decentralised manufacturing made possible by distributed ledger technologies. Our mission is to assist businesses in making sure that outsourced production runs just like it does in-house.
2. What are the top trends shaping pharma supply chains?
Digitalisation and advanced analytics: Businesses may streamline operations, improve teamwork, and facilitate better decision-making throughout the supply chain by leveraging the power of data and technology.
Blockchain technology: This cuttingedge innovation provides a safe, transparent, and traceable answer for handling pharmaceutical supply chain data, confirming the genuineness of the products, and enhancing patient safety in general.
When it comes to personalised medication, supply chains must be more adaptable and agile to suit more focused, smaller manufacturing runs and a range of distribution requirements.
Sustainability: Businesses are embracing greener practices to lessen their influence on the environment, such as minimising waste, implementing renewable energy sources, and making sure that raw materials and packaging materials are sourced ethically. This emphasis on sustainability can also improve brand perception and assist businesses in complying with legal regulations. Approaching
3. How can pharma companies improve supply chain resilience?
Diversifying suppliers and manufacturers can help manage risks associated with relying too heavily on a single source, lessening the effects of disruptions brought on by geopolitical reasons, natural catastrophes, or other unforeseen events.
Strong risk management: Using all-encompassing risk management techniques, such as contingency planning, supplier audits, and scenario analysis, can assist businesses in proactively identifying and addressing potential supply chain weaknesses.
Investing in sophisticated analytics allows businesses to improve decision-making, streamline operations, and spot potential dangers before they materialise into major disruptions.
Implementing digital technologies can enhance transparency, traceability, and collaboration throughout the entire supply chain. Examples of these technologies include blockchain, IoT, and AI. By utilising these technologies it can supercharge businesses in improving inventory management, streamlining logistics, and guaranteeing product quality.
Establishing good relationships with stakeholders, such as governments, business leaders, and nonprofit groups, can promote a collaborative environment that makes it easier to share information, work on problems together, and plan reactions to disruptions. This collaboration may improve the pharmaceutical supply chain's overall resilience.
Constant improvement and learning: To increase supply chain resilience, businesses should keep an eye on continuous improvement, draw lessons from previous disruptions, and put best practices into practice. Frequent evaluations and benchmarking against industry standards can help pinpoint problem areas and promote continuous growth.
Pharma businesses can improve the resilience and agility of their supply chains by concentrating on these tactics, better preparing them to meet the challenges of a globally connected and complicated market.
the market collaboratively can result in shared innovations, resources, and risk mitigation techniques. Partnerships can be formed with suppliers, contract manufacturers, technology providers, and even rival companies.
Improved traceability and visibility throughout the whole supply chain are essential for locating possible bottlenecks and guaranteeing product quality. These levels of visibility and traceability are being made possible by technologies like IoT, blockchain, and cloud computing.
4. What are your thoughts on ongoing digitalisation of the pharma supply chain industry?
Several factors contribute to the pharmaceutical supply chain industry's ongoing digitalisation, which has far-
reaching repercussions and a transformational nature.
Demand planning, and decisionmaking, artificial intelligence (AI) and machine learning (ML) technologies are now routinely harvested to spot patterns and trends in supply chain data. Inventory management can be improved by AI and ML, resulting in fewer stockouts and overstocks.
Automation and robotics: Automating repetitive operations, including labelling and packaging, can assist lower human error and boost overall productivity. This could result in shorter production periods, lower prices, and better-quality goods.
Blockchain technology: With its tamper-proof, decentralised ledger, blockchain can improve trust, traceability, and security by enabling safe data exchange and product
23 www.pharmafocusasia.com
authentication along the whole supply chain. Blockchain can help patients stay secure by preventing the sale of fake medicines.
Sophisticated analytics and data visualisation: By utilising these tools and approaches, pharmaceutical organisations can acquire a deeper understanding of their supply chains. This may increase the danger.
5. What are the key challenges for pharma companies in 2023?
Managing complex supply chains: As personalised medicine and specialised therapies gain prominence, supply chains must become more flexible and adaptable to accommodate these changes. This requires the development of new manufacturing processes and logistics strategies to meet the specific demands of such therapies.
Securing raw materials: The increasing demand for pharmaceuticals has led to concerns about the availability and cost of raw materials, particularly those sourced from a limited number of suppliers or countries. Ensuring a stable supply of raw materials may require diversification of sources, vertical integration, or strategic partnerships with suppliers.
Regulatory compliance: Pharma companies face stringent regulations that vary across countries and regions. Adapting to new or changing regulations, such as serialisation requirements or environmental standards, can be time-consuming and resource-intensive.
Cybersecurity and data protection: As companies embrace digital technologies and data sharing, they must also prioritise securing their systems and protecting sensitive information from cyber threats.
Talent management and workforce development: With the rapid advancement of technology and changing industry dynamics, pharma companies need to attract and retain skilled talent. This involves investing in employee
training, fostering a culture of innovation, and embracing diversity and inclusion.
Cost containment and pricing pressure: Rising R&D costs, coupled with increasing pressure from payers and governments to reduce drug prices, present challenges for maintaining profitability. Companies may need to explore innovative pricing models, optimise internal processes, and adopt cost-effective technologies to stay competitive.
Patient engagement and satisfaction: As patients become more empowered and knowledgeable about their health, pharma companies must prioritise patient-centricity, focusing on improving patient outcomes, ensuring timely access to therapies, and addressing unmet medical needs.
6. What disruptions are global pharma supply chains vulnerable to that can significantly impact their operations?
Limited suppliers and manufacturing facilities: When suppliers and manufacturing facilities are clustered in a narrow area, the supply chain is more susceptible to risks such as regional political turmoil, natural disasters, and transportation disruptions.
Pandemics and global health crises: As was the case with the COVID-19 pandemic, a sharp rise in demand for a given medication or piece of medical equipment can strain the supply chain and result in shortages.
Extreme weather events and climate change are ever-increasing through global warming. As a result of climate change, natural disasters like hurricanes, floods, and droughts are occurring more frequently and with greater severity. This can have direct and indirect consequences for the supply chain such as impeding transportation and production, which may result in supply chain bottlenecks.
Cybersecurity risks: As the pharmaceutical supply chain digitises, it is more vulnerable to intrusions. These attacks have the potential to steal private information, halt operations, and erode stakeholder confidence.
Fostering collaboration amongst many stakeholders, including governments, significant players in the business, and nonprofit organisations, will be crucial to building a pharmaceutical supply chain that is more strong and flexible.
7. Any other thoughts that you would like to share?
The pharmaceutical business must negotiate the opportunities and challenges that lie ahead as it finds itself at a critical juncture.
Here are some further ideas for the future of the sector:
Strategic alliances and collaborations between the sector and external parties including technology suppliers, governments, and nonprofit groups will be advantageous for the industry. Working together can encourage innovation, allow for resource sharing, and strengthen supply chains.
Pharma companies must be open to investigating and implementing cuttingedge business strategies if they are to keep up with the changing environment. Direct-to-consumer channels, digital therapies, and value-based pricing are a few examples of these.
The business must concentrate on putting patients at the core of its plans as personalised medicine develops popularity. This may entail making use
24 PHARMA FOCUS ASIA ISSUE 51 - 2023
STRATEGY
Pharma companies must be open to investigating and implementing cuttingedge business strategies if they are to keep up with the changing environment.
of data that patients create themselves, creating patient support initiatives, and guaranteeing quick access to life-saving treatments.
It is crucial to spend on staff training and development given the rising usage of digital technology and decisionmaking based on data. An educated workforce will be better able to meet the demands of the digital age and advance the sector.
The industry must keep ethical considerations at the forefront as it innovates and adopts new technologies. This includes a dedication to minimising environmental effects, protecting privacy, and treating data responsibly.










In conclusion, to assure the delivery of life-saving medications, the pharmaceutical business must address supply chain vulnerabilities, integrate digital technologies like blockchain, and work together throughout the sector. By









confronting these problems head-on


With the increasing adoption of digital technologies and data-driven decision-making, it is essential to invest in employee training and development. A skilled workforce will be better equipped to handle the challenges of the digital era and drive the industry forward.
As the industry continues to innovate and embrace new technologies, ethical considerations must remain at the forefront. This includes responsible data handling, privacy protection, and a commitment to reducing environmental impact.
In summary, the pharmaceutical industry must confront supply chain vulnerabilities, adopt digital technologies like blockchain, and collaborate across the sector to ensure the delivery of life-saving medicines. By addressing these challenges head
John Ward is founder and CEO of ServBlock, founded in early 2021, following John’s recognition of the potential for Blockchain and Data Space technologies to revolutionise the regulated manufacturing space. ServBlock focusses on trusted data exchange through distributed ledger and blockchain technologies. For over a decade, John has been at the forefront of the highly regulated pharmaceutical manufacturing industry, working alongside many of the world's leading biotech companies in his role as principal consultant within the validation space. Most recently as a product owner in Pfizer's global Smart Factory team.

25 www.pharmafocusasia.com
AUTHOR BIO
STRATEGY Agência Nacional de Vigilância Sanitária ANVISA www.quantysclinical.com EXPERIENCE IN CONDUCTING BIOEQUIVALENCE STUDIES ON INJECTABLESLIQUIDSTRANSDERMAL PATCH TOPICAL CREAMS ORAL SOLIDSINHALERS, ORAL & NASAL SPRAYS ACCREDITED BY GCC Ph-II & Ph-III with INDICATIONS IN Cardiology 104 Beds 4 LCMS/MS Over 40 Methods Neuropsychiatry De-addiction Oncology OTHER SERVICES In-Silico studies Trial Monitoring Bioanalytical Services Medical Writing Email: info@quantysclinical.com Call: +91 2836 253330 Plot No. 668, 671, 672, New Area Kandla Special Economic Zone, Gandhidham, Kutch-270230, India
Break with the past: Know how Single use Assembly components and bags Ease the productivity of the Bioprocess Applications
It was almost 10 hours for a line changeover to around 3 weeks of downtime for preparing a Stainless steel bioreactor for the upstream process. This stainless steel system required huge maintenance, cleaning and sterilisation after a batch of drugs were produced. This process always was costlier, time consuming and gave a sense of addition fear of cross contamination to the manufacturers.

FDA|USP CLASS VI|CLASS 10000| BPOG STUDIES|E&L
Aswathy Nair, Asst. Manager International Marketing, Ami Polymer
To overcome the challenges of cross contamination and provide an aseptic environment at each stage of production with Increased Productivity the innovation of single use bags and components adoption came into huge demand.
Maintain the GMP: Adopt the highest standard of the aseptic processing

Current GMP requires all kinds of safety and quality standards. Ami Polymer manufactures all the Single use components, bags, tubes and hoses in ISO Clean room class 7, with all the products gamma irradiated prior to their usage. It is ensured that the products meets the most important factors like the extractable and leachable (Bpog) and the biocompatibility USP 87, 88 and ISO 10993.
Insights on how single use bioprocess benefits the biopharma:-
1)Increase the Productivity:- It eliminates the downtime of cleaning and validation between two stages of production.
2)Minimises the risk of Cross Containmination:- the traditional method had the risk of cross containmination between batches and products. Current method simplifies the process by disposing off the assemblies or bags after a batch.
3)Cost and Time:- The complexity involved CIP & SIP process, required more in the traditional stainless steel vessels, it got remarkably decreased with the
new single use process. It thereby reduced cost of labour and material. The downtime also reduced and changeover is now reduced to 2-3 hours.
4)Scalable:- The single use products can be scalable cale from prototype to production, available with different sizes and volumes and easy to handle, with less labour or resources.
Aswathy Nair is a highly accomplished international sales professional with a proven track record and expanding global market presence. She travels parts of South East Asian Countries like Singapore, Malaysia, Thailand, South Korea, Vietnam, etc. to meet our pharma/biopharma customers and provide them best fluid transfer solutions, offering wide variety of single use consumables to the industry. There are more than hundreds of customers delighted and enjoying our services in these regions.

By Continuesouly monitoring the customer needs we manufacture the right and hygiene components for the pharma and biopharma applications? Ami Polymer adheres to provide solutions globally, we have built a team of expertise working on innovative new designs in the single use assemble products 24x7.
Stay connected for more updates:www.amipolymer.com
27 www.pharmafocusasia.com 27
Pharmaceutical Business Strategy and Forecasting Creating insights from market research
In the increasing uncertainties and volatile market, Pharma companies have to rethink their approach to forecasting in oncology. When projections are misaligned, pharma companies will have to answer shareholders. But when forecasters get it right, manufacturers can optimise resources to fit the opportunity, and produce plans that deliver innovative medicines to patients who need them. Oncology is moving away from the old approaches and is embracing a new set of forecasting tools built specifically for its complexity.
Sanobar Syed, Associate Director, Beigene
The pharma sector’s ongoing transformation has prompted most pharma and biotech players to focus on launching new products, developing innovative technologies, virtual clinical trials, and rapid digitalisation. These initiatives are proving to be quite challenging.
To develop a revenue forecast for a new pharmaceutical product in development, primary research is conducted in which physician respondents are shown a target product profile and asked how

28 PHARMA FOCUS ASIA ISSUE 51 - 2023
STRATEGY
much they would use “Product X” if it was available. Anyone who has experience doing physician market research will be quick to point out that one must apply a standard research “haircut” to account for bias. Take 50 per cent off of physician-reported responses, they say. Or potentially chop based on a top two box score. Either way, it’s acknowledged common practice, everyone does it. But an arbitrary standard haircut may not be appropriate for every market research study. Physicians have a general idea of their historical prescribing but asking them to estimate future prescribing share without putting their responses in the context of a structured process may result in a significant margin of error. Different biases may cause physicians to over-estimate stated prescribing share (HCP-Reported Adoption), particularly for new novel products.
Physician responses also typically do not account for other stakeholders such as payers and patients. Payers may manage access to certain products by implementing restrictions or placing them on non-preferred tiers (Payer Access). Patients are bearing an increased burden of pharmaceutical costs, as more and more patients are enrolled in highdeductible or high out-of-pocket maximum insurance plans. Determining patient “fill rates”, or what percentage of patients pick up the prescription at the pharmacy, is key — particularly in conditions with very expensive or noncovered therapies (patient fill rates). Lastly, because of patient volume or previous prescribing behaviour, some physicians may never be detailed on a new product, and, therefore, should be accounted for differently in terms of commercial forecasting (Manufacturer Detailing Reach Adjustment). It is imperative to find the impact of each factor that can be identified and analysed to both arrive at the most accurate forecast and identify potential levers to optimise launch uptake.
Physicians do not treat all patients with a condition with the same thera -
peutic option. When asking physicians about the potential uptake of a new product, it is important to not only focus on the relevant patient segments but also to consider all current and future treatment options.
An example line of questioning may be:
1. For your last 10 patients, how many would receive (currently available) Competitor A? How about Competitor B?
2. For your next 10 patients, how many would receive Product X (product being forecasted) vs. currently available options (Competitor A and B)?
3. For your first 10 patients in 2 years, after Product Y enters the market and Competitor B goes generic, how would you allocate treatment across all different options?
Using this methodology, the physician is forced to think about the tradeoffs of allocating patients to each of the potentially available products within a relevant patient segment, instead of simply estimating the share of patients

that would receive the novel product that is being studied. This approach also allows pharma manufacturers to have visibility into which competitors they may be gaining or losing a share against. The physician is ultimately asked to consider how the market might evolve as products become generic and new products enter, generating a more dynamic and informed forecast estimate.
Payer Access and Competition Payers determine the level of access (e.g., covered/not covered, preferred/not preferred) and restriction (e.g. prior authorisation, step edit, new to market block) that a product will receive. Novel products that are classified as preferred or nonrestricted may be prescribed more often than products with significant restrictions. Forecasters can assess likely formulary coverage by conducting primary research with payer decisionmakers as well as exploring coverage for analog products. In addition to payer research, discussions with physicians should be designed to understand prescribing behaviour given different
29 www.pharmafocusasia.com
STRATEGY
levels of access. An understanding of the potential “lift” these strategies offer can help the manufacturer determine their attractiveness.
Patient fill rates
For many therapeutic areas, the pharmaceutical industry has long been considered a price inelastic market, where the price of therapy does not significantly impact fill volumes. However, over the last decade, average deductibles and out-of-pocket maximums have steadily increased and a growing number of patients are enrolled in high-deductible plans. As a larger portion of the pharmaceutical cost-sharing burden is placed on the patient, patients are becoming increasingly sensitive to price. Other initiatives that may impact prescription volumes are direct-to-consumer or advocacy campaigns. These initiatives can impact the ‘top of the funnel’, prompting patients to ask their physician about a specific product; or at the end of the process, pushing the patient ‘over the hump’ to fill a prescription. In many cases, forecasters would be wise to either identify fill rates for analog products or conduct patient research to understand the willingness to pay under different out-of-pocket cost scenarios. The inclusion of patient input into product forecasting can often lead to more robust and reliable results.
Physicians cannot prescribe a novel new treatment if they do not know about it. When conducting primary research to assess a new product opportunity, one must account for the eventual breadth of detailing upon launch. Forecast uptake should only account for physicians who will be detailed or become familiar with the product. However, there will still be a portion of non-detailed physicians who will not become aware of a new product, at least early in the product’s lifecycle.
It is imperative to have an overview of the strategic imperatives to drive the effectiveness of the above tactics. Few of the ways to do that effectively can be as follows:
Identify consumer needs
Big pharma doesn’t just serve patients, it also serves a host of other types of consumers, from clinicians who prescribe drugs and the hospitals that buy and administer them to the distributors and pharmacies that stock and deliver them. With so many customer types, it’s paramount that drug companies learn as much as possible about each one so that they can identify the areas and people with the most profitable issues to address. They can do this through a variety of research measures. They can use established secondary research literature or conduct primary research (such as ethnographic studies and satisfaction surveys with physicians, hospitals and/ or patients) themselves.
Assess the market
Picking the right environment (both physically and generally) in which to market a drug is critical. Disease patterns, political turmoil, economic stability, as well as market size/segmentation, and operating conditions, are just a few of the key factors affecting a company’s overall success with a product. Pharmaceutical market research can highlight any obstacles or opportunities that might disturb or encourage favourable outcomes.
Provide a competitive analysis
Emerging new products to the market are only one type of threat. Generic products can also severely impact a company’s bottom line. Thus, pharmaceutical companies must keep on top of the products that their competitors are developing, as well as improving. They can utilise SWOT analyses and sales force assessments to diagnose and treat deficiencies. Focus groups and interviews can be used to expose untapped technological innovations and other resources. Competitive analysis can save companies from reinventing the wheel. They can observe and use others’ successes for their benefit instead.
Address compliance and other regulatory concerns
The pharmaceutical industry is strictly regulated. It must deal with patient and data privacy issues and comply with FDA and patent regulations. Pharmaceutical market research can help companies monitor developing patent laws and any changes to FDA legislation and gather consumer feedback so that they can protect or modify their practices as needed.
Conclusion
A good pharmaceutical market research project can reveal industry trends and help forecast market growth. It can describe how well a company is (or is not) doing with its consumer encounters, marketing efforts, and distribution methods. It can provide the raw data needed for accurate benchmarking, and it can uncover unmet needs. These facts assist shareholders, who use them to focus and direct decisions, shaping their business’s future.
With over 14 years of deep expertise in pharmaceutical business strategy and forecasting. Sanobar has led business strategy and forecasted multi-million dollar brands across her career in pharmaceutical giants like AbbVie, and Novartis to name a few. With a master's in organic chemistry and MBA, she has solidified her knowledge of long-range planning, business strategy, analytics, and forecasting. She is a subject matter expert and regularly speaks in this field at various national and international conferences globally.

30 PHARMA FOCUS ASIA ISSUE 51 - 2023
AUTHOR BIO STRATEGY
Revolutionising Pharma Six game-changing ways to excel with integrated quality solution
In an era of heightened regulatory scrutiny and patientcentric care, the pharmaceutical industry is on the brink of a game-changing transformation where integration of quality solutions has become the cornerstone, propelling life sciences industry to achieve unprecedented levels of success.
Jasmin Kumar, Digital Marketing Expert
The pharmaceutical industry is known for its complexity and strict regulations, making it challenging for companies to maintain high-quality standards while still remaining competitive. From research and development to the production of the final product, quality of the product is of utmost importance to ensure its safety and efficacy for patients. Hence, Integrated quality solutions can play a crucial role in ensuring that pharmaceutical manufacturing meets the required standards and regulations.
Integrated quality solutions refer to the use of advanced technologies and tools that are integrated into the manufacturing process to monitor and control the quality of the product at every stage. These solutions can help to identify potential issues early on in the manufacturing process, allowing for quick resolution and reducing the risk of errors and defects.
Let’s understand how.
Why do Pharma Companies require Integrated Quality Management Solutions?
Integrated quality solutions in the pharmaceutical industry refer to the combination of various quality management tools and systems into a unified platform. This integrated platform provides a comprehensive approach to managing and improving quality processes throughout the entire manufacturing process, from development to distribution.
The integrated quality solutions can include various tools such as Quality Management Systems (QMS), Document Management Systems (DMS), Electronic Logbooks (ELOGBOOK), Learning Management Systems (LMS), Annual Product Quality Review (APQR), and more. These tools help to automate processes, ensure compliance with industry regulations, and improve overall efficiency and productivity.
By integrating these tools and systems into a unified platform, pharmaceutical companies can ensure that all processes are closely monitored and controlled, from raw material sourcing to final product release. This not only helps to ensure product quality and patient safety but also helps to reduce the risk of product recalls, penalties, and fines.
Overall, integrated quality solutions in the pharmaceutical industry provide companies with a comprehensive approach to maintaining high-quality standards, ensuring compliance, and improving overall efficiency and productivity throughout the manufacturing process.
Let’s take examples of integrated quality solutions of QMS and DMS products and their the ability to manage and control documentation in real-time. With a DMS solution, documents such as Standard Operating Procedures (SOPs), batch records, and other critical documents can be stored,
31 www.pharmafocusasia.com
STRATEGY
managed, and controlled in a centralised location. By integrating the DMS with the QMS, the company can ensure that these critical documents are managed in a way that complies with industry regulations and standards.
For example, with this integrated solution, a document control process can be initiated through the QMS, which would then trigger the creation and approval of the necessary documents through the DMS. The documents could then be stored and managed in the DMS, with version control and access control features ensuring that the correct document versions are being used, and only authorised personnel have access to them.
The integrated QMS and DMS solution can also provide real-time visibility into the status of document control, allowing for efficient decisionmaking and continuous improvement. Any changes or updates made to documents can be immediately tracked and recorded in both systems, ensuring that there is a complete audit trail of all document changes.
Pharmaceutical manufacturing without integrated quality solutions can lead to several challenges, including:

Inconsistent quality: In the absence of integrated quality solutions, it is difficult to maintain consistent product quality throughout the manufacturing process. This can lead to variability in the final product, which can affect its efficacy and safety.
Increased risk of errors: Without integrated quality solutions, there is a higher risk of errors in the manufacturing process, such as incorrect formulation, incorrect labelling, or contamination. These errors can compromise the safety and efficacy of the final product.
Compliance issues: Pharmaceutical manufacturing is subject to strict regulations and guidelines. Without integrated quality solutions, it can be challenging to ensure compliance with these regulations, which can result in regulatory actions, such as warning letters, fines, or product recalls.
Higher costs: The absence of integrated quality solutions can result in
additional costs, such as rework, scrap, or product recalls, which can impact the overall profitability of the manufacturing process.
Limited visibility: Without integrated quality solutions, it can be difficult to get a complete view of the manufacturing process and identify areas for improvement. This can lead to missed opportunities for optimisation and increased efficiency.
How can integrated solutions give competitive edge to pharma companies?
Modern
manufacturing integrations in the pharma industry involve the integration of various processes, technologies, and systems to improve efficiency, productivity, and product quality. These integrations enable real-time monitoring, predictive maintenance, data-driven decision-making, compliance with regulatory requirements, and innovation, providing a competitive edge to pharma companies.
Some of the modern manufacturing integrations in the pharma indus -
32 PHARMA FOCUS ASIA ISSUE 51 - 2023
STRATEGY
try include: Manufacturing execution systems (MES), Enterprise resource planning (ERP), Computerised systems validation (CSV), Internet of Things (IoT), Artificial intelligence (AI) and machine learning (ML), Robotics etc.
Modern manufacturing integrations are giving a competitive edge to pharma companies in several ways:
• Improved product quality: Modern manufacturing integrations enable realtime monitoring and quality control, leading to improved product quality and a lower risk of product recalls
• Increased efficiency: Integrating manufacturing processes and equipment can increase efficiency, reduce downtime, and reduce production costs
• Enhanced data analytics: Manufacturing integrations enable the collection and analysis of data across the manufacturing process, allowing for data-driven decision-making and continuous process improvement
• Compliance with regulatory requirements: Integrating compliance requirements into the manufacturing process can reduce the risk of regulatory violations, such as warning letters, fines, and product recalls
• Supply chain management: Integrating supply chain management into the manufacturing process can improve inventory management, reduce lead times, and improve order fulfilment
• Innovation: Integrating emerging technologies, such as artificial intelligence, can improve product development, manufacturing processes, and quality control, leading to innovative and more effective products.
How can Integrated quality solutions help pharmaceutical companies thrive?
1. Improved Compliance
Integrated quality solutions such as QMS, DMS, APQR, LMS, Elogbook, softwares etc. can help companies maintain strict compliance with regulations and standards. These solutions enable
companies to document and track processes, detect deviations in real-time, and take corrective action before they lead to non- compliance issues.
2. Enhanced Quality Control
In the pharmaceutical industry, quality control is critical to ensure patient safety and product efficacy. Quality control software solutions can help companies to automate and streamline processes, ensuring that products meet the highest quality standards. Realtime monitoring and reporting can help detect and address issues quickly, further improving quality control.
3. Simplified Training and Development
Training and development are essential in the pharmaceutical industry. With the help of Learning Management Systems (LMS), companies can ensure that their employees are continuously improving their skills and knowledge. LMS solutions can also help companies keep track of employee progress, ensuring that they are meeting their training objectives.
4. Improved Collaboration and Communication
Effective communication and collaboration are vital in the pharmaceutical industry. DMS and ELOGBOOKs can help companies manage and share data, facilitate communication between departments, and reduce the likelihood of miscommunication and errors.
5. Better Data Management and Analysis
Pharmaceutical companies generate large amounts of data that can be challenging to manage and analyse. Integrated quality solutions such as QMS and APQR can help companies collect and manage data, providing insights that can inform decisionmaking and improve processes continually.
6. Increased Agility and Adaptability
The pharmaceutical industry is constantly evolving, and companies must be able to adapt to stay competitive. Integrated quality solutions provide companies with the flexibility
and scalability necessary to adapt to changing circumstances quickly. This enables companies to respond to market trends and regulatory changes, staying ahead of the curve.
The pharmaceutical industry is at the forefront of a revolution driven by integrated quality solutions. By embracing these game-changing approaches, companies can elevate their operations to new heights of success. From ensuring patient safety and regulatory compliance to optimising manufacturing processes and fostering collaboration, the power of integration is reshaping the industry landscape.
As we move forward, it is crucial for pharmaceutical companies to recognise the transformative potential of integrated quality solutions and seize the opportunity to revolutionise their operations. By doing so, they will not only thrive in a competitive market but also contribute to the advancement of healthcare and the well-being of countless patients worldwide.
The future of pharma is here, and it belongs to those who dare to transform with integrated quality solutions.
Jasmin Kumar is an award winning professional and a digital marketing expert. Her experience of 15+ years spans across multiple industries, making her a versatile and an accomplished individual. Her passion for writing has led her to become a Co-author of an international bestselling book.

33 www.pharmafocusasia.com
STRATEGY AUTHOR BIO
Getting Back to Business Let’s meet in Bangkok!
MEDICAL FAIR THAILAND 2023 | 13-15 September


Preparations are in full swing as MEDICAL FAIR THAILAND makes its way to Bangkok once again in 2023. After a three-year break, the 10th edition of the exhibition will run its physical edition from 13 to 15 September at BITEC, followed by a 7-day digital extension where exhibitors and visitors can engage further online through its AI-powered businessmatching system until 22 September. This is the first time MEDICAL FAIR THAILAND will be held in a ‘phygital format’.
Highlights this year include signature showcases such as the Community Care Pavilion and Start-Up Park, and also the introduction of the Medical Manufacturing pavilion. As the region’s leading specialist trade fair for the medical and healthcare sectors for the past two decades, MEDICAL FAIR THAILAND serves the full value chain and end-to-end needs of the medical and healthcare sectors. From diagnostics, wearable technology, connected healthcare solutions, rehabilitation and therapy equipment, 3D printing technology, and now - medical technology (MedTech) components, processes and solutions - the exhibition offers the ideal destination for medical and healthcare buyers and professionals looking to meet their sourcing objectives, gain industry insights and to share best practices.
“We have been waiting for three years so we are excited and are gearing up for a big comeback for MEDICAL FAIR THAILAND 2023. With the positive feedback, industry commitment, and almost 80% bookings received for 2023, we should be on track to reach close to pre-pandemic levels by next year. On the back of a highly successful and wellreceived phygital edition of MEDICAL FAIR ASIA that was held in Singapore earlier this year, and as we navigate further in a post-pandemic landscape, we are confident by this year the industry will be more than ready to move into high gear and Thailand will be an ideal location.”
Gernot Ringling, Managing Director, Messe Düsseldorf Asia
34 PHARMA FOCUS ASIA ISSUE 51 - 2023
MEDICAL FAIR THAILAND 2023 comes against a strengthening backdrop where Thailand continues to firm its position as a medical hub of the region with its supportive government policies and incentives, making it a model investment destination for a wide range of medical and healthcare service sectors. In line with Thailand’s 4.0 policy, the Thai government considers the healthcare industry to be a priority sector for investment, thus the staging of MEDICAL FAIR THAILAND 2023 is well-positioned.

New! Medical Manufacturing Pavilion

A special themed pavilion focused on medical manufacturing processes and componentsfrom new materials, intermediate products, packaging and services, to microprocessors and nanotechnology. With Thailand’s growing reputation as a production and distribution base of medical devices both within and outside Thailand, it has become a natural market for medical devices.
According to data from the Office of Industrial Economics, Ministry of Industry (Medical Devices Intelligence Unit), there is much potential for investment opportunities in sophisticated medical devices particularly due to Thailand’s reliance on imports for this segment.


Community Care Pavilion Special Focus on Mental Health
With a special spotlight on mental health with a showcase featuring digital mental health technologies, from smart medicine to therapeutic medical equipment. Its mainstay of addressing the needs of ageing societies on the back of rising chronic diseases and an ageing population, the pavilion will also feature a full suite of geriatric medicine, rehabilitative equipment, assistive technology, and mobility products.
Thailand’s proportion of citizens aged over 60 years, is forecast to be one of the highest in ASEAN by 2045, and will also exceed countries such as Europe and the United States. Thailand’s fast-increasing ageing population and the estimated more than three million Thais suffering from poor mental health, is expected to further drive the demand for related healthcare services.
Start-up Park
A strategic platform for companies with ready-to-market healthcare solutions to meet relevant buyers and partners, industry influencers, experts, and potential investors. From innovative healthcare industry solutions, health apps and new tools for gathering and AI-supported analysis of health data, to robotic assistance systems and new approaches in diagnostics – the Start-Up Park is a must-attend for SMEs looking to scale-up their business.
The Start-Up Park plays a significant role as an enabler of the entrepreneurial ecosystem that encourages life sciences and medical and health innovation in Thailand. With the country’s vibrant start-up landscape propelled further by the government’s numerous grants and new regulations as part of Thailand’s ambitious plans to be a start-up-based country, the start-up scene has grown systematically over the years and is considered one of Asia’s hidden gems. At the last edition of MEDICAL FAIR THAILAND held in 2019, a total of 11 start-up companies participated from Singapore, Japan, South Korea, Hong Kong, Taiwan and Thailand.
35 www.pharmafocusasia.com 35 To find out more, go to www.medicalfair-thailand.com
Towards Precision Medicine for Cancer Pain Treatment in Asian Populations

Cancer pain is one of the most debilitating symptoms and opioids are the cornerstone of therapy in its management. However, opioid treatment outcomes are unpredictable due to inter-individual variability. Single nucleotide polymorphisms in genes encoding drug-metabolising enzymes (CYP2D6, UGT2B7), neurotransmitter metabolising enzymes (COMT, ABAT), drug transporters (ABCB1) and drug targets, including receptors (OPRM1) and ion channels (P2RX7) have been reported to be associated to inter-individual variability of opioid responses among patients of Asian ethnicities.
Yow Hui-Yin, Pharmacy Lecturer, Department of Pharmaceutical Life Sciences, Faculty of Pharmacy, Universiti Malaya
Shobha Elizabeth Satkunananthan, Master student, School of Pharmacy, Taylor's University
Vijayaprakash Suppiah, Senior Lecturer in Pharmacy, Clinical & Health Sciences at the University of South Australia
Toh Gaik-Theng, Senior Lecturer, School of Medicine, Taylor's University
The prevalence of cancer pain is high among patients with advanced cancer, affecting up to 96 per cent of patients, which significantly impacts their quality of life. However, inadequate pain relief remains a serious health concern for cancer patients, particularly in Asia, where the prevalence of undertreated cancer pain is relatively high. Strong opioids, such as morphine, have been the mainstay of treatment for cancer pain based on three-step analgesic ladder of the World Health Organization (WHO). However, inter-individual differences in analgesic response pose a major disadvantage for the clinical use of these opioids. Pharmacogenomics,
36 PHARMA FOCUS ASIA ISSUE 51 - 2023
RESEARCH & DEVELOPMENT
study of a person’s genetic make-up and its effect on treatment response, provide a reasonable explanation for the inter-individual variability observed in analgesic response, particularly in long-term opioid use. While genetic variants associated with opioid treatment outcomes in patients with cancer pain have been identified, there have been inconsistent findings between studies. This review aimed to summarise current evidence in the association of genetic variants with treatment responses in Asian patients treated with opioids for cancer pain.
Materials and methods
A literature search was conducted in Medline and Embase using free-text and MeSH terms grouped into four main subject groups: cancer pain, pharmacogenetic, opioids and treatment outcome. Only original research articles published in English from the year 2000 to November 2020 investigating the associations between single nucleotide polymorphisms (SNPs) and treatment outcomes in cancer pain patients treated with opioids for pain were included. A total of 658 records were screened and 337 articles were shortlisted after removing duplicates and those published before 2000. Furthermore, a full text review resulted in 14 articles that fulfilled the inclusion criteria. The included studies assessed pain control and opioid dosage as treatment outcomes, and findings were presented according to the opioids studied in included articles, namely morphine, fentanyl, oxycodone, tramadol, codeine, hydrocodone, hydromorphone, levorphanol, methadone, and oxymorphone.
Results
Seventeen SNPs from eight genes were significantly associated with opioid dosage, pharmacokinetics, and pain control among Asian populations (Table 1).
37 www.pharmafocusasia.com Gene Polymorphism Parameters with significant association Population Opioid CYP2D6 *10 Opioid dose requirement, pain control Chinese Tramadol, fentany *2, *5, *10, *14 Pharmacokinetics Japanese Tramadol CYP3A5 *3 Pharmacokinetics Japanese Fentany UGT2B7 rs7439366 Pharmacokinetics, pain control Han Chinese Morphine Pain control Chinese Oxycodone OPRM1 rs1799971 Opioid dose requirements Lebanese Morphine Pain control Chinese Tramadol/ paracetamol combination Opioid dose requirement Han Chinese Sufentanil rs1323040 Opioid dose requirement Han Chinese Sufentanil COMT rs4680 Opioid dose requirements, Pharmacokinetics Japanese Morphine Opioid dose requirement, pain control Japanese Morphine ABAT rs1641025 Opioid dose requirement, pain control Japanese Combination of opioids ABCB1 rs1128503 Opioid dose requirement Japanese; Han Chinese Fentanyl; sufentanil rs2032582 Opioid dose requirements Han Chinese Sufentanil rs1045642 Opioid dose requirements Han Chinese Various opioids P2RX7 rs1718125 Opioid dose requirement Han Chinese Hydromorphone the
RESEARCH & DEVELOPMENT
Table 1: List of genes reported with significant associations for opioid treatment response in Asian patients with cancer pain
Discussion
This review focused on the impact of genetic variations on opioid responses in Asian patients with cancer pain. The study identified genetic variants in drug-metabolising enzymes (CYP2D6, CYP3A5, UGT2B7), neurotransmittermetabolising enzymes (COMT, ABAT), drug transporters (ABCB1), and drug targets such as receptors (OPRM1) and ion channels (P2RX7) as contributing factors to the observed inter-individual variability in treatment outcomes.
Of these genetic variants, CYP2D6 polymorphisms were the most extensively studied, with the CYP2D6* 10 allele being associated with higher tramadol dosage requirements and reduced analgesic effects due to decreased plasma concentration of metabolites. Additionally, the CYP2D6*10 variant was also associated with higher fentanyl dose and pain scores, despite not being involved in fentanyl’s metabolism. This CYP2D6*10 allele is present in up to 50 per cent of individuals of Asian ethnicities. The CYP3A5*3 allele was found to impact fentanyl’s pharmacokinetics as CYP3A4 and CYP3A5 are responsible for its metabolism. Further research investigating the potential relationship between CYP2D6*10 and fentanyl pharmacokinetics may explain the observed association.
Genetic variations in UGT2B7 affect morphine glucuronidation in forming its metabolites - morphine3-glucuronide (M3G) and morphine-6 glucuronide (M6G). The rs7439366*C allele was observed to enhance analgesic efficacy and was significantly associated with higher levels of M3G and M6G. The T allele carriers were observed to have a reduced response to morphine and prolonged-release oxycodone. Additional studies are required to investigate this relationship as the level of evidence for this SNP is low (between levels 3 to 4 in PharmGKB) as this is currently insufficient to be used as a biomarker for drug responses.
Opioids bind to the μ-opioid receptor to bring about opioid-induced analgesia. The OPRM1 G472A (rs1799971) SNP, which is present at a high frequency in Asian populations, is associated with reduced analgesic efficacy of morphine requiring higher doses in affected individuals.
The G allele of the SNP is thought to reduce signalling efficacy and expression of the μ-opioid receptor, leading to decreased binding potential resulting in increased morphine requirement. On the other hand, heterozygous patients for both rs1799971 and rs1323040 SNPs required a higher dose of sufentanil, suggesting of presence of a variant that reduced opioid analgesic efficacy.
The catechol-O-methyltransferase (COMT) enzyme is responsible for breaking down dopamine in the prefrontal cortex, and the COMT G472A (rs4680) SNP had been linked to various neurological conditions, including pain modulation and opioid-related disorders. Carriers of the AA genotype of this SNP were found to have lower plasma concentrations and dose requirements for morphine, while the carriers of the GG genotype had higher dose requirements. However, this finding was inconsistent with studies associat -
ing the A allele with reduced analgesic efficacy. The A allele carriers may have a higher sensitivity to pain but require less morphine doses. Pooled data from a meta-analysis found that the opioid consumption of patients who were COMT rs4680 AA and GG homozygotes was not significantly different in the first 24 hours of opioid treatment, while AG genotypes required significantly lower doses of opioids.
The rs1641025 SNP corresponds to a substitution from a T to C nucleotide within ABAT, which encodes GABA transaminase. Studies suggested that the ABAT rs1641025*C allele was associated with lower pain scores and enhanced opioid efficacy. While there is a lack of studies to show the clinical significance of this association, it is thought to modulate pain perception through the GABAergic synapse pathway by regulating physiological functions including pain perception. Further studies are needed to explore the potential pharmacogenetic role of this SNP in pain control and opioid analgesic responsiveness and possibly its usefulness as a biomarker.
The bioavailability of opioids is affected by P-glycoprotein (P-gp), a family of efflux transporters encoded by ABCB1. Three polymorphisms, namely rs1045642, rs2032582, and rs1128503 in ABCB1 have been extensively studied. Patients carrying the rs1045642*T allele have been found to have reduced expression of P-gp, potentially resulting in reduced efficacy or higher toxicity from opioids treatments. Studies on the three SNPs in patients receiving multiple opioids including fentanyl and sufentanil have produced mixed results, with some suggesting enhanced analgesic efficacy in carriers of the T allele while others showing reduced efficacy. This warrants further investigations to elucidate the impact of these SNPs on opioid effectiveness and toxicity.
The purinergic receptor P2X7 was associated with chronic inflammatory and neuropathic pain. In patients with lung cancer, the P2RX7 rs1718125 SNP
38 PHARMA FOCUS ASIA ISSUE 51 - 2023
RESEARCH & DEVELOPMENT
Compared to pharmacogenetic studies in Caucasian and European populations, the sample sizes in Asian populations tend to be smaller and focused on a limited number of genes and SNPs.
was significantly associated with postoperative pain and fentanyl dose, with A allele homozygotes requiring the highest dose for pain control and having significantly higher pain scores than the GG genotype group. However, this finding contradicted a recent study that reported that carriers of the A allele required lower fentanyl and had better pain scores than those who were allele G homozygotes. The association of P2RX7 polymorphisms and opioid dose requirements and pain control needs further investigation, as preclinical studies suggested this gene may contribute to pain modulation both
in peripheral tissue as well as processing in the central nervous system.
Most of the pharmacogenetic studies in Asian patients with cancer pain treated with opioids have focused on patients with East Asian ancestries, which may not be representative of other Asian populations. There were also differences in outcome measures used in these studies, making comparison of findings challenging. Compared to pharmacogenetic studies in Caucasian and European populations, the sample sizes in Asian populations tend to be smaller and focused on a limited number
Yow Hui-Yin is a Pharmacy Lecturer from Department of Pharmaceutical Life Sciences, Faculty of Pharmacy, Universiti Malaya. She obtained her Pharmacy Bachelor’s and PhD’s degrees from Universiti Kebangsaan Malaysia in 2009 and 2012, respectively. Her research interests are in pharmacogenomics, epilepsy research, molecular pharmacology and pharmacy practices.
of genes and SNPs. Nevertheless, this review highlighted the findings on the association of genetic variants with treatment responses in Asian patients treated with opioids for cancer pain. The CYP2D6* 10 SNP was found to be relevant to tramadol pharmacokinetics and opioid dose requirement. SNPs in UGT2B7, ABAT, and P2RX7 have demonstrated potential clinical relevance, while SNPs in OPRM1, COMT , and ABCB1 were found to have low clinical significance due to inconsistent findings. Several ABCB1 SNPs have been reported to be associated with opioid treatment outcomes, but further investigations are required to understand their role.
Conclusion
Shobha Elizabeth Satkunananthan is currently pursuing her MPhil in Pharmaceutical Sciences under the School of Pharmacy of Taylor’s University, Malaysia. Her research focuses on the pharmacogenomics of morphine treatment outcomes among Malaysian patients with cancer pain. Her background is in BSc (Hons) in Biotechnology from UCSI University, Malaysia.
Variability in opioid responses was linked to genetic polymorphisms in drug-metabolising enzymes, neurotransmitter-metabolising enzymes, drug transporters, receptors, and ion channels. The clinical significance of OPRM1 rs 1799971, COMT rs4680, and ABCB1 SNPs need further exploration in Asian patients with cancer pain.
Citation
Vijay Suppiah is a Senior Lecturer in Pharmacy at the University of South Australia. Trained in Pharmacy and genetics, his research interests are in pharmacogenomics of chronic medical conditions. Additionally, he is also interested in the use of and long-term effects of psychotropic medications across the lifespan.

Satkunananthan, S.E.; Suppiah, V.; Toh, G.-T.; Yow, H.-Y. Pharmacogenomics of Cancer Pain Treatment Outcomes in Asian Populations: A Review. J. Pers. Med. 2022, 12, 1927. https://doi. org/10.3390/jpm12111927

Funding
Toh Gaik Theng, PhD is a Senior Lecturer in Physiology at Taylor’s University. Prior to this, she was an acting project leader at the Cancer Research Malaysia where she worked closely with breast surgeons and cancer patients. Her current research interests are in molecular signaling, hereditary cancer and regenerative medicine.


This research was funded by the Ministry of Higher Education (MOHE) Malaysia for Fundamental Research Grant Scheme (FRGS/1/2019/SKK09/ TAYLOR/03/1).
Competing interests
The authors declare no conflicts of interest.
References are available at www.pharmafocusasia.com
39 www.pharmafocusasia.com AUTHOR BIO
RESEARCH & DEVELOPMENT
Preformulation Studies Its importance in drug development
Preformulation study forms an important part of the drug development process as it helps formulation scientist to build the foundation of the drug product to be used in the preclinical safety/efficacy studies and early human trials. A precise and well-planned formulation study not only helps to reduce the overall timelines for drug development process but also reduces the chance of failure of clinical trial owing to CMC issues.
Yogeshwar Bachhav, Founder and Director, Adex Pharma consultancy Services
1. What is the impact of preformulation on drug development?
In the preformulation phase of drug development, medicinal chemists investigate the important aspects of new chemical entity such as ionisation constant (pKa), partition coefficient (log P). Formulation scientists also take advantage of these intrinsic parameters of active moiety to plan the preformulation studies. These physicochemical parameters determine the stability of the active in the final formulation and drive the bioavailability (upon oral administration). The preformulation studies also involve studying the stability of the active in solid state as well in dissolved state. Furthermore, drug-excipient interactions are studied in this phase to guide the selection of excipients in the final formulation (to be used in the
clinical phases followed by commercial launch). For the complex formulations such as injectables (i.v, s.c and i.m.) and those containing poorly soluble drugs, the preformulation studies are of prime importance as it forms foundation of the drug product development. A precise design and execution of the preformulation studies reduces the risk of the failure of the drug development owing to CMC issues.
2. Is preformulation a proactive phase?
Planning preformulation studies is actively initiated as soon as the lead candidate is nominated for preclinical studies followed by phase 1 clinical (First in Man studies). Any red flags for the drug under development arising from the physicochemical properties of the active moiety as well as
its interaction with the excipient and associated issues with the attainment of the bioavailability could be detected at the earliest due to careful planning of the preformulation studies.
The Big Pharma companies have in-house set ups to plan these preformulation studies to gain time. However, start-up and mid-size pharma companies often need to rely on external partners to execute these studies. Careful selection of third party CROs is very important as specific skillsets are required to execute these studies due to the less amount of API available at this stage and timelines and budget consideration also play an important role.
3. What are the techniques used for preformulation evaluation parameters of a drug?
40 PHARMA FOCUS ASIA ISSUE 51 - 2023
RESEARCH & DEVELOPMENT
Yogeshwar Bachhav is Pharmacist by training and PhD in Advanced drug delivery systems from ICT, Mumbai (India). He has around 20 16 years of Post PhD experience in Europe in the field of Pharmaceutical Development of investigational drugs. Currently he is working as Director (Consultant) at AiCuris Anti-infective Cures AG Germany and responsible for Pharmaceutical Development of investigational drugs in the domain of innovative anti-viral and anti-bacterial drugs. He has also started a consultancy firm called Adex Pharma which deals with the solving complex issues in the Pharmaceutical development of new and approved drug since 2016.
As indicated above, the preformulation studies aim to study the physicochemical parameters of the drug such as pKa, Log P, permeability as well as stability of API in solid and solution state, polymorphism, drug excipient-interactions etc. The techniques/methods used to perform the preformulation studies are as below
• Polymorphism: X ray diffraction
• Hygroscopicity: DVS, Karl Fischer Titration
• Permeability: Caco-II cell line
• Stability of API and Drug-excipient interaction: HPLC analysis, differential scanning calorimetry and FITR.
ExpertTalk
drugs, excipients must be selected to enhance the chemical stability of the drug in the final formulation. Regulatory authorities are also keen to understand the rationale behind the selection of type and concentration of an excipient and hence drug-excipient interactions must be planned carefully.
5. What are the key challenges to preformulation studies?
The amount of API available at the preclinical or early clinical phase of the drug development process is always a major challenge and hence formulation scientist must strive to design the preformulation studies at microscale (microformulation studies) rather than conventional preformulation screening. This will help economize the API consumption and yield the speedy outcome. Often at this stage of drug development there are multiple compounds to be screened, hence preformulation studies must be able to rule out the compounds which would be difficult to develop from the Pharmaceutical development point of view.
4. What are the key areas to focus in the preformation of a drug?
The most important aspect of the preformulation studies is to focus on solid state and solution stage stability of API. If there are any bioavailability issues for the API to be developed (linked to BCS-II category), preformulation studies must aim to identify the excipients which can improve the dissolution rate followed by bioavailability. For BCS class IV compounds, the focus should be to select the excipients and optimise the formulation accordingly to improve permeability in vivo. For poorly stable
Start-up and mid-size pharma companies need to identify the right partner to perform these studies. Lack of knowledge and non-availability of the required techniques will significantly jeopardise the outcome of the preformulation studies which would hard the preclinical safety and early clinical studies.

6. Any other comments?
There is abundant literature available and significant work has been done related to the preformulation studies for small molecules. However, these days the pipeline of drug development is heavily dominated by large molecules (peptides, proteins, nucleic acids, and antibodies). Hence, having a flowchart or guidance to execute the preformulation studies of biomolecules will be of great importance to speed up the drug development process.
41 www.pharmafocusasia.com AUTHOR BIO
A Step-By-Step Strategy for Designing A Meta-Analysis
Meta-analysis is a subset of systematic reviews that combines pertinent qualitative and quantitative study data from several selected studies to develop a single conclusion with greater statistical power. In this article we focus more on general framework of meta-analysis and a detailed perspective on designing meta-analysis including the five-step process.
Ramaiah M, Manager, Freyr solutions Balaji M, Deputy Manager, Freyr solutions
Meta-analysis is a subset of systematic reviews that combines pertinent qualitative and quantitative study data from several selected studies to develop a single conclusion with greater statistical power. This conclusion is statistically more significant than the analysis of any single research due to increased numbers of subjects, greater diversity among subjects, or accumulated effects and results. In simple words, metaanalysis is the statistical combination of results from two (02) or more separate studies.
Studies comparing healthcare interventions, notably randomised trials, use the outcomes of participants to compare the effects of different interventions. Meta-analyses focus on pairwise comparisons of interventions. The contrast between the outcomes of two (02) groups treated differently is known as the ‘effect’ - the ‘treatment effect’ or the ‘intervention effect.’ The analysis of the included studies is either narrative or quantitative.
The general framework for metaanalysis may be provided by considering the following four (04) questions:
1. What is the direction of the effect?
2. What is the size of the effect?
3. Is the effect consistent across studies?
4. What is the strength of evidence for the effect?
Meta-analysis provides a statistical method for questions 1 to 3. Assessment of question 4. relies additionally on judgments based on assessments of study design and risk of bias, as well as statistical measures of uncertainty.
On the other hand, narrative synthesis uses subjective (rather than statistical) methods to follow through questions 1 to 4 for reviews where meta-analysis is

42 PHARMA FOCUS ASIA ISSUE 51 - 2023
RESEARCH & DEVELO PMENT
either not feasible or not sensible.
Purpose
• To establish statistical significance with studies that have conflicting results
• To develop a correct estimate of the effect magnitude
• To provide a more complex analysis of harms, safety data, and benefits
• To examine subgroups with individual numbers that are not statistically significant.
Advantages
• Improved precision in convincing evidence about the intervention effects when the studies are too small
• Greater statistical power and confirmatory data analysis
• A good source to respond to conflicting studies to generate new hypothesis
• Answer the unanswered or unmentioned questions in the research or a study
• Considered an evidence-based resource.
Disadvantages
• Complex and time-consuming to identify appropriate studies
• Not all studies provide adequate data for inclusion and analysis
• Requires advanced statistical techniques
• Heterogeneity of study populations.
The Five-Step Process of Designing Meta-analysis
Step 1: Define the Research Question and Eligibility Criteria
A clinical research question is identified, and a hypothesis is proposed. The likely clinical significance is explained, and the study design and analytical plan are justified.
Usually, two (02) standard tools are used: Patient, Intervention, Comparison, Outcome (PICO) or Sample, Phenomenon of Interest, Design, Evaluation, and Research type (SPIDER). PICO is primarily used in quantitative evidence synthesis. The authors demon-
strated that the PICO holds more sensitivity than the more specific SPIDER approach. The latter was proposed as a method for qualitative and mixed method searches.
PICO is typically used for systematic review and metaanalysis of clinical trial studies.
PICO stands for:
P – Population: patient, or problem: How do you describe the patients, people, or problems you are looking at?
I – Intervention: What is considered an intervention, exposure, or a factor?
C – Comparison: Do you have something to compare to the intervention, exposure, or factor you are considering?
O – Outcome: What is hoping to measure, improve, affect, or accomplish?
Step 2: Protocol for the Search Process
The protocol “outlines how the review authors will handle the review process and the challenge they are addressing. The procedure describes how the studies in the review were identified, assessed, and summarised. The protocol serves as a public record of how the review authors aim to address their research question by making this information available.”
In addition to serving as a road map for the research question, protocols also allow the individual to comprehend what type of research is performed and helps avoid duplication of research.
Step 3: Search for Studies
a) Identification of Literature Search Database (Registries, Repositories, or Libraries)
Most frequently used databases are as follows:
• PubMed
• Scopus
• Web of Science
• EMBASE
• MEDLINE
• HINARI
• Cochrane
• Google Scholar
• Clinicaltrials.gov
• mRCTs
• POPLINE
• SIGLE
This list covers almost all the published articles in tropical medicine and other health-related fields.
b) Search for Relevant Literature (Reported and New Studies) Using a String-based Search on the Research Question
The search process needs to be documented in enough detail to ensure that it can be reported correctly in the review to the extent that all the databases’ searches are reproducible. The search strategies will need to be copied and pasted exactly as run and included in full, together with the search set numbers and the number of records retrieved. The search strategy should emphasise on:
• Searching previous studies
• Identification of new studies via databases and registers
• Identification of recent studies via other methods.
c) Collection of All the Retrieved Literature Using Reference Management Tools
Specially designed bibliographic or reference management software such as Mendeley, EndNote, ProCite, Reference Manager, and RefWorks are helpful and relatively easy to use to keep track of references and report studies.
d) Determination of Inclusion and Exclusion Criteria Based on Eligibility Criteria
The PICO strategy, study design, and deadline determine eligibility criteria. Most exclusion criteria are irrelevant, duplicate, unavailable, or abstract-only papers. These exclusions should be specified in advance to prevent bias from
43 www.pharmafocusasia.com
RESEARCH & DEVELO PMENT
the researcher. The inclusion criteria would include publications containing the target patients, researched interventions, or comparing two (02) evaluated interventions.
In brief, they would contain material pertinent to the study subject. Most importantly, the information should be clear and sufficient to answer the positive or negative issue.
e) Identification of Supporting Studies and Finalising the Articles to be Included
For many authors, the appearance of a diamond (statistical analysis) at the bottom of a plot is an exciting moment, but the results of meta-analyses can be extremely misleading if adequate attention is not compensated to formulating the review question, specifying eligibility criteria, identifying, selecting, and critically evaluating studies, collecting appropriate data, and deciding what would be meaningful to analyse.
Step 4: Data Extraction
Once the studies are selected for inclusion in the meta-analysis, summary data or outcomes are extracted from each study. In addition, sample sizes and measures of data variability for both intervention and control groups are required. Depending on the study and the research question, outcome measures could include numerical or categorical measures. For example, differences in scores on a questionnaire
f) Reporting the Search Process
The search process must be recorded in precise detail to ensure that it can be reported accurately in the review, to the extent that all searches of all data -


or measurement level, such as blood pressure, would be reported as a numerical mean. However, differences in the likelihood of being in one (01) category versus another (e.g., vaginal birth versus cesarean birth) are usually reported in terms of risk measures such as odds ratio or relative risk.
a) Data Extraction for Dichotomous Outcomes: It is most reliable to collect dichotomous outcome data as the number of individuals in each group who did and did not experience the result. Although, in theory, this is comparable to collecting both the total number of individuals and the number of individuals sharing the outcome and it is not always apparent if the total number of individuals reported is the number of individuals on whom the outcome was assessed. Occasionally, the numbers incurring the event need to be derived from percentages.
b) Data Extraction for Continuous Outcomes: Due to inadequate and inconsistent reporting, it may be difficult or impossible to obtain the required information from the provided data summa-
bases can be reproduced. The search techniques must be carefully copied and pasted, together with the search set numbers and the total number of records retrieved.
ries. Additionally, they differ in the scale used to analyse the data. In a research report, standard deviations and standard errors are occasionally conflated, and the nomenclature needs to be more consistently applied. When necessary, the authors must always request missing information and clarification about the reported statistics. Nevertheless, there is an approximate or direct algebraic link between numerous variance measures and the standard deviation.
c) Data Extraction for Ordinal Outcomes: The retrieved data for ordinal outcomes depends on whether the ordinal scale will be dichotomised for analysis, treated as a continuous outcome, or analysed directly as ordinal data. In turn, this choice will be influenced by how the authors of the studies analysed their data. The strategy of capturing all the categorisation is also useful when studies utilise somewhat different short ordinal scales. Whether a consistent cut-point can be used for dichotomisation across all studies is uncertain.
RESEARCH & DEVELOPMENT
d) Data Extraction for Counts: Count data can be analysed in various ways. The crucial option is whether the interesting outcome should be dichotomous, continuous, time-to-event, or a rate. A typical error is treating counts directly as dichotomous data, considering the total number of participants or person-years of follow-up as sample sizes. Though it is preferred to decide how count data will be analysed in advance, the option is frequently driven by the structure of the available data and cannot be made until most studies have been reviewed.
e) Data Extraction for Time-toEvent Outcomes: Meta-analysis of time-to-event data typically involves obtaining individual patient data from the original investigators, reanalysing the data to estimate the log hazard ratio and its standard error, and then conducting a meta-analysis. Whether
individual patient or aggregate data are used, there are two (02) approaches to get estimates of log hazard ratios and associated standard errors for inclusion in a meta-analysis that employs generic inverse variance methods.
f) Data Extraction for Effects
Estimates: When extracting data from non-randomised studies and some randomised trials, it may be possible to obtain adjusted effect estimates. The process of data extraction and analysis using the generic inverse variance approach is identical to that for unadjusted forecasts; however, the variables that have been corrected must be noted. The disadvantage of this approach is that the estimates and standard errors for the same effect measure must be produced for every other study included in the same meta-analysis, even if they provide summary data per intervention group.
Heterogeneity: A systematic review will assemble studies with varying results. Heterogeneity is a term for any variation between research in a systematic review. Differentiating between various types of heterogeneity (clinical, methodological, and statistical) can be beneficial. Specifically, heterogeneity related only to methodological variety would indicate the studies are biased to varying degrees. Explorations of heterogeneity designed after identifying heterogeneity can only result in the development of hypotheses. They should be evaluated with much greater caution and normally should not be included among the review findings. Methods for tackling clinical heterogeneity should be mentioned, along with how the authors will assess whether a meta-analysis is acceptable. Methods for spotting statistical heterogeneity should be described (e.g., visually, utilising I, etc.) using the chi-squared test.
45 www.pharmafocusasia.com
Figure 1: PRISMA 2020 Flow Diagram Template for Systematic Reviews or Meta-analysis
RESEARCH & DEVELOPMENT
Publication Bias: The publication or non-publication of research findings may be influenced by publication bias based on the type and direction of the results. There are two (02) types of scientific studies that investigate the existence of publication bias - indirect and direct evidence. As the proportion of all hypotheses tested for which the null hypothesis is false is unknown, surveys of published results such as those mentioned

above, can only give indirect evidence of publication bias. There are also considerable direct indications of publishing bias.
Publication bias should be viewed as one of the potential sources of ‘smallstudy effects’ — the tendency for intervention effect estimates to be more positive in smaller trials. Using funnel plots, review authors can visually determine whether small-study effects may be present in a meta-analysis.
within the meta-analysis by testing for ‘heterogeneity.’
Forest plot
The final estimates from a meta-analysis are often graphically reported as a ‘Forest Plot.’ A forest plot displays effect estimates and confidence intervals for individual studies and meta-analyses. The standard method for illustrating individual research outcomes and metaanalyses uses forest plots. These can be generated with the Review Manager software, and a selection of them can be chosen for inclusion.
Funnel plots: A funnel plot is a basic scatter plot of the intervention effect estimates from individual studies versus a measure of each study's size or precision.
A PRISMA flowchart template is presented, which can be adjusted based on whether the systematic review or a meta-analysis is original or updated (figure 1).
boxes include the value of 1.0). When all three (03) studies were combined in the meta-analysis as represented by the diamond, we get a more precise estimate of the drug’s effect, where the diamond represents both the combined risk ratio estimate and the limits of the 95 per cent CI.
Manuscript Drafting and Submission to a Journal
Step 5: Final Estimates of the Effect
The final stage is to select and apply an appropriate model to compare Effect Sizes across different studies. The most common models used are Fixed Effects and Random Effects models. Fixed Effects models are based on the ‘assumption that every study is evaluating a common treatment effect.’ This means that the assumption is that all studies would estimate the same Effect Size were it not for different levels of sample variability across various studies. In contrast, the Random Effects model ‘assumes that the true treatment effects in the individual studies may be different from each other’ and attempts to allow for this additional source of interstudy variation in Effect Sizes. Whether this latter source of variability is likely important is often assessed
Forest plots and funnel plots from the ‘data and analysis’ section may be chosen as figures for inclusion in an integrated section. Forest plots describe all the studies, and study data for the principal outcomes will be presented as figures. A funnel plot for one (01) or more key outcomes may be a vital contributor to these forest plots if there are sufficient studies.
In the hypothetical forest plot shown in Figure 2, for each study, a horizontal line indicates the standardised Effect Size estimate (the rectangular box in the center of each line) and 95% CI for the risk ratio used. For each of the studies, drug X reduced the risk of death (the risk ratio is less than 1.0). However, the first study was larger than the other two (the size of the boxes represents the relative weights calculated by the meta-analysis). Perhaps, because of this, the estimates for the two (02) smaller studies were not statistically significant (the lines emanating from their
The drafting of a manuscript is based on the guidelines of the ICMJE, i.e., the IMRaD model’s four (04) scientific sections: introduction, methods, results, and discussion, mainly with a conclusion. Performing a characteristic table for study and patient characteristics is a mandatory step that includes a detailed search strategy for database searches. Figure 2
46 PHARMA FOCUS ASIA ISSUE 51 - 2023
RESEARCH & DEVELOPMENT
Figure 2: Hypothetical Forest Plot
After completing the manuscript draft, characteristics table, and PRISMA flow diagram, the draft should be sent out for review. Finally, a suitable journal with a significant impact factor and a relevant field should be chosen for the manuscript. Before submitting the manuscript, we must pay close attention to the author guidelines of the journals.
Estimated Timelines for a Systematic Review
The time required to complete a systematic review is highly variable. However, considering the tasks and the time required for each of these might aid the authors in estimating the amount of time needed. Tasks include protocol development, searching for studies, evaluating citations, and full-text reports of studies for eligibility, assessing the risk of bias in included studies, collecting data, pursuing missing data and unpublished studies, analysing the data, interpreting the results, and writing the review, as well as keeping the review up to date as shown in Table 1.
data checking, conducting statistical analysis, double data checking, manuscript writing, revising, and submission to a journal. Nevertheless, this is an important outcome that could impact the current practice and promote higher-quality future studies to address evidence gaps.
References are available at www.pharmafocusasia.com
Ramaiah M is a Manager, Scientific Writing with 14+ years of experience in Scientific Writing and CER Writing. He has a proven track record in developing highly complex manuscripts reporting safety and efficacy data from pivotal clinical studies for publication in highimpact journals as per ICMJE, GPP, EQUATOR, EASE, AMWA, STM, and applicable ethical regulations (COPE).


CONCLUSIONS
The reliability of meta-analysis findings is mainly determined by the quality of the data used for the compilation. The steps in this process include developing a research question and validating it, forming criteria, developing a search strategy, searching databases, importing all results to a library and exporting them to an excel sheet, protocol writing and registration, title and abstract screening, full-text screening, manual searching, extracting data and assessing its quality,
Balaji. M is a Deputy Manager, Scientific Writing with 12+ years of experience in Scientific Writing. He has a recognized background in developing complex manuscripts in biomedical engineering, drug delivery, biomaterials, and nanotechnology with novel therapeutics to bring advanced medication systems for various unmet medical needs. He has published numerous articles following publication guidelines (ICMJE, GPP, EQUATOR, EASE, AMWA, STM).
47 www.pharmafocusasia.com
Meta-analyses focus on pair-wise comparisons of interventions. The contrast between the outcomes of two (02) groups treated differently is known as the ‘effect’ - the ‘treatment effect’ or the ‘intervention effect.’
AUTHOR BIO
MONTH ACTIVITY 1–2 Preparation of protocol 3–8 Searches for published and unpublished studies 2–3 Pilot test of eligibility criteria 3–8 Inclusion assessments 3 Pilot test of risk of biased assessment 3–10 Validity assessments 3 Pilot test of data collection 3–10 Data collection 3–10 Data entry 5–11 Follow-up on missing information 8–10 Analysis 1–11 Preparation of review report 12 Keeping the review up to date
Timelines for a Systematic
Table 1: Estimated
Review
RESEARCH & DEVELOPMENT
Measuring Vaccine Efficacy The right endpoints are key to success
Endpoints are needed throughout the vaccine development life cycle. Their selection are critically important and impact study outcomes. Each development stage needs a careful assessment of fit-for-purpose endpoints. In this article we will discuss how to develop, assess and select endpoints at each development step.
Paul Gillard, Vice President, Medical and Scientific Strategy, Vaccines Therapeutic Area
Delphine Saragoussi, Executive Director, Epidemiology and Scientific Affairs, Post-Approval Studies and Real-World Evidence
Sarah Rosen, Senior Director Project Management, Peri- and Post-approval Studies and RealWorld Evidence PPD clinical research business of Thermo Fisher Scientific
Development of pharmaceutical products requires generation of data that enable a comprehensive evaluation of benefit against potential risks. Prophylactic vaccines are no exception. The benefit component must be accurately measured throughout the product development life cycle to determine vaccine efficacy (VE). Measuring VE is accomplished with disease- and/or pathogen-specific clinical study endpoints. These endpoints are carefully tracked, captured, evaluated in detail and sometimes adjudicated to assess if study efficacy objectives are being met. This comprehensive approach is critical for regulators, public health experts, prescribers and patients to make informed decisions on licensing, guideline recommendations, clinical use, and patient adoption and adherence. A vaccine benefit-risk profile relies on conclusions drawn from the analysis of VE endpoints.
Identifying appropriate and valid endpoints
Measuring efficacy endpoints is required for all novel vaccines and for existing vaccines used in new target populations, unless there are established and validated surrogates of efficacy.
From polio and influenza — against which the first vaccines demonstrated their efficacy decades ago — to more recent disease targets such as SARS-CoV-2 and RSV, the use of carefully defined endpoints is a fundamental part of assessing VE in both large, prospective, randomised, placebo-controlled and real-world studies. The type of endpoints of interest varies throughout the clinical development life cycle.
Epidemiologists and clinicians are instrumental in assessing the key medical needs that a novel vaccine intends to address, as well as the targeted outcomes
48 PHARMA FOCUS ASIA ISSUE 51 - 2023
RESEARCH & DEVELOPMENT
it hopes to achieve. Endpoints typically are considered across four dimensions:
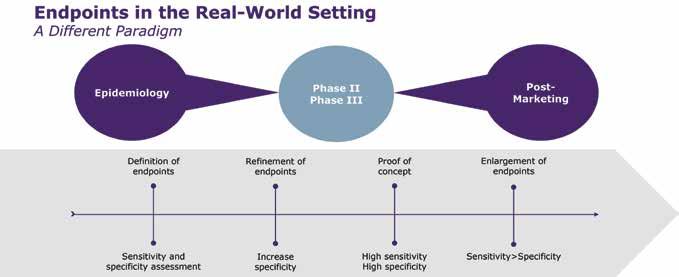
1. Clinical triggers—The symptoms of the disease or ailment that will trigger further diagnostic procedures (clinical trial setting) or may form the basis for a diagnosis based on clinical judgment or other circumstantial evidence (realworld setting).
2. Pathogen identification—Currently dominated by high-throughput polymerase chain reaction (PCR) based on assays but, depending on the pathogen, other lab tests may play an important role, such as virological, microbiological cultures.
3. Geography and seasonality—Factors that influence when and where a disease outbreak is likely and how this impacts the positive and negative predictive values of any assays.
4. Population specifics— How different populations (e.g., infants vs. the elderly) express signs or symptoms differently, and how acceptability and application of diagnostic procedures may vary between these populations. Throughout a clinical development plan, endpoints must be refined to strike the right balance between diagnostic sensitivity and specificity (Figure 1). When a test’s sensitivity is high, it is more likely to give a true positive result and correctly detect the presence of disease or illness. When specificity is high, it is
more likely to give a true negative result and correctly identify the lack of disease or illness. Each has its place in the vaccine development life cycle, so both sensitivity and specificity should be assessed and their roles defined at the outset. Phase III studies require the highest sensitivity to ensure the entire disease burden is assessed for efficacy but also specificity as the vaccines are pathogen specific and sometimes restricted to strains or serotypes. (Figure 1)
Pre-clinical phases
The pre-clinical phase requires that experts assess the epidemiology of the disease in terms of incidence, prevalence, severity, risk factors, outcomes and other elements. Where feasible, disease burden and medical need are measured or assessed across different health care systems and geographies. These are typically assessed in non-interventional epidaemiologic studies, often called natural history or descriptive studies. The main objectives of a natural history study are to describe the following:
Disease identification
• Geography, time period and setting
• Source population
• Clinical diagnosis
• Biological diagnosis
• Patient characteristics
• Age
• Comorbidities
Disease outcomes
• Symptoms, including variance by age or other factors
• Hospitalisation
• Persisting symptoms or disability
• Death
Other endpoints
• Economic burden, including cost of care
• Workplace absenteeism
Humanistic burden
• Quality of life
Natural history studies necessitate data that reflect the real-world management of patients and that their methodologies are different from clinical trials. There are many different data sources that can be used for such studies. Data can be generated for the purpose of the research in a specific setting or extracted from existing administrative data collection systems. Between these two extremes there is a continuum of different data sources, whose choice (based on granularity, quality, reliability) is also key to determine endpoints (Figure 2). Primary data collection is at one end of the spectrum and is generally time and cost intensive, representing a high burden with high specificity. Secondary data are a lower burden with respect to cost and
49 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
Figure 1. Drug development life cycle: Endpoint, sensitivity and specificity considerations
time. However, the specificity is lower compared with primary data. (Figure 2)
When a new pathogen emerges, primary data need to be collected via a hospital- or site-based approach obtaining data from patients and their doctors, usually via questionnaires.
Primary data collection also can be useful for studying known pathogens, such as different pneumococcal serotypes in pneumococcal pneumonia CAP. In this case, as serotyping is not part of any existing surveillance system and is not routinely assessed in clinical practice, the information cannot be found in secondary data sources making primary data collection the best way to identify and track these specific patients.
At the other end of the spectrum, secondary data provide the opportunity
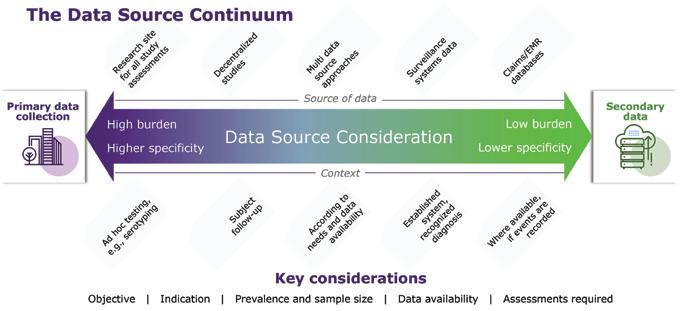
to use existing data, such as insurance claims or infectious diseases surveillance systems.
Given all the factors that continually affect the generation of real-world data, it is important to create a nimble fit-forpurpose study design and to anticipate how the pathogen, the subjects, and any testing and treatment may change over time.
Phase II–III
Endpoints captured in clinical development are precise endpoints with high specificity and high sensitivity across Phases II and III, such that precision peaks with highly targeted and reliable endpoints in Phase III.
Consider how one might track endpoints for an influenza vaccine. In
Phase III, a diagnosis would be made according to a strictly defined set of criteria (e.g., cough, fever, shivering, general malaise, myalgia), but, to increase sensitivity of results, a PCR test would then be deployed to confirm viral presence. In this way, sensitivity (anyone who meets the symptom requirements) is combined with specificity (lab test) for optimal results.
Regulators also play an important role in ensuring optimal endpoints are used to properly reflect VE. Regulatory agencies publish general guidelines such as “Considerations for Developmental Toxicity Studies for Preventative and Therapeutic Vaccines for Infectious Disease Indications ” (2006). Guidance also may be much more targeted with pathogen-specific guidance, such as those
50 PHARMA FOCUS ASIA ISSUE 51 - 2023
Laboratory-Confirmed Influenza Pooled vaccine groups Relative efficacy N= 31,983 Respiratory symptoms,at least one General symptoms, at least one n (%) % (95% CI) ≥1 of: sore throat, cough AND ≥1 of: fever [> 37.2°C] 217 (0.7%) 20.6 (−4.6 to 39.9) ≥1 of: sore throat, cough, sputum production, wheezing, difficulty breathing AND ≥1 of: fever [> 37.2°C], chills, tiredness, headache, myalgia 529 (1.7%) 24.2 (9.7 to 36.5) ≥1 of: sore throat, cough, sneezing, stuffy/runny nose, sputum production, wheezing, difficulty breathing(none) 703 (2.2%) 18.3 (5.0 to 29.8)
Table 1: Defining endpoints requires finding the optimal balance to identify the presence of virus – operationalising the definition of disease.
RESEARCH & DEVELOPMENT
Figure 2: Continuum of data sources for VE studies
developed for COVID-19, “Development and Licensure of Vaccines to Prevent COVID-19 ” (2020). While those examples come from the U.S. Food & Drug Administration other regions issue guidance similarly.
Post-marketing phase
As a novel prophylactic vaccine moves into a post-marketing phase, endpoints of interest may shift to accommodate changing circumstances. With the experimental phases of development essentially over and with subjects no longer carefully selected according to stringent protocol defined criteria, the real-world setting brings new levels of complexity. For example, comorbidities tend to take on a much more significant role and endpoint sensitivity (increasing probability of detection) may take preference over specificity.
As more and more real-world evidence is generated, research experts will further use and adapt the endpoints to measure real-world vaccine effectiveness or post-marketing safety. It is also worth noting that less stringent endpoints—though still clinically relevant— also may be required to deliver meaningful insights and to inform vaccine policies. It is crucial that these endpoints also reflect standard of care and therefore the real-world methodology used to detect diseases/pathogens. For example, a recent study of the effectiveness of a bivalent COVID19 vaccine booster was conducted in the Netherlands using self-reported COVID-19 as an endpoint. This example also reflects the fact that the timing of the study plays an important role in endpoint definition as pathogendetection testing evolves over time.

Critical choices: Balancing signs and symptoms in case definitions

Defining and measuring endpoints for novel prophylactic vaccine trials and real-world studies is a complicated but critically important piece of the development process. Case definitions that
are overly restrictive may significantly impact incidence rates and detrimentally affect efficacy and success criteria. On the other hand, if case definitions are overly accommodating, they may introduce excess noise into the efficacy signal. From the outset, it is important to engage with the right experts and develop a thorough understanding of the targeted disease in order to accurately trace efficacy of the test solution.
For example, Table 1 (adapted from “Efficacy of High-Dose Versus StandardDose Influenza Vaccine in Older Adults” ) shows results from a Phase III trial for an influenza vaccine. Here, a different combination of signs and symptoms leads to a dramatically different number of efficacy endpoints, all confirmed by lab testing. The figures shown here are all from the same study and show how by changing the qualifying symptoms has led to a threefold increase in case counts, and also impacted the lower
bound of the confidence interval of VE. (Table 1)
Conclusion
Endpoint election is critically important and has a major impact on study outcomes and the assessment of VE. Selecting, defining and evaluating the endpoints to be used throughout the life cycle of vaccine development require a broad range of expertise from clinicians and epidemiologists. Optimal study development requires prospective planning to precisely define the disease and factors that may impact VE, from geography and seasonality to age, population and socioeconomic factors, as well as diagnostic methods. The success of a vaccine program will be determined by the endpoint measures and vaccine efficacy. A continuous dialogue with regulators on the endpoint selection is highly beneficial and increases the likelihood of positive regulatory reviews.
Paul Gillard serves as vice president and a medical lead for the vaccines therapeutic area within the PPD clinical research business of Thermo Fisher Scientific. In this role, he provides medical and scientific expertise with a focus on vaccine development strategies across various therapeutic areas. He joined the business in 2022 with training in internal medicine and more than 18 years of scientific leadership expertise in the clinical development of prophylactic vaccines.
Delphine Saragoussi serves as an executive director of the epidemiology and scientific affairs team of the real-world evidence (RWE) group within the PPD clinical research business of Thermo Fisher Scientific. As a physician, she specialises in public health and social medicine and has more than 20 years of applied experience in epidemiology and pharmacoepidemiology. Before joining the business, she served as a global therapy area lead in RWE and epidemiology at Lundbeck.
Sarah Rosen serves as a director within non-interventional studies, which is part of the RWE/peri- and post-approval practice within the PPD clinical research business of Thermo Fisher Scientific. Prior to joining the business, she was a project manager within lifecycle management at a global clinical research organisation and served as the observational subject matter expert. She has more than 15 years’ experience in project management and has successfully conducted a number of full-service noninterventional studies.

51 www.pharmafocusasia.com AUTHOR BIO
RESEARCH & DEVELOPMENT
Adaptive Clinical Trial Designs
Adaptive clinical trial designs are a preferred approach when certain circumstances concur, like rare diseases, a need to optimise the dose finding process in patients, or a medical open need lacking a treatment. We discuss in this interview the utility, level of implementation, and future trends in adaptive clinical trial designs.
Esther Mahillo, Vice President, Operational Strategy and Feasibility, Precision for Medicine
How would you define adaptive clinical trials?
The definition, concept, and scope of controlled clinical trials in human beings is a relatively new one and has rapidly evolved during the last 100 years. Regulations and guidance on the conduct of clinical research worldwide, are based on three principles:

1) a prospective and predetermined assignment of medical intervention in human beings
2) balance between efficacy and safety of the medical intervention and
3) the voluntary and informed acceptance of subjects to participate in the investigation.
The main objective of clinical trials is to generate scientific evidence to support relevant decisions related to therapies for human beings. The strength of scientific evidence relies on two principles: the strength of the study design, and the fortress of endpoints measured. At the basis of the pyramid of scientific evidence
we find the expert opinion. At the top, the paradigm of best scientific evidence is data generated on randomised, double-blind, placebo-controlled (when possible) clinical trials (RCT), aimed to demonstrate with the lower possible biases, that a new therapeutical approach is better (safer, more efficacious) than the standard available treatment. There are different guidelines on scientific evidence ranking, from the one published in United States by its Preventive Services Task Force (USPSTF) to the Oxford Center for Evidence-Based Medicine (EBM) Levels of Evidence.
The Grading of Recommendations Assessment, Development, and Evaluation (GRADE) has been endorsed
52 PHARMA FOCUS ASIA ISSUE 51 - 2023
CLINICAL TRIALS
ExpertTalk
by over 100 organisations worldwide (gradeworkinggroup.org).
Adaptive trials differ from the classical ones in the fact that therapeutic and business decisions are taken during the study conduct, based on ongoing analysis of data. This approach is based on statistical designs which are well validated and use predefined algorithms for decisions (Krendyukov, A. et al. Value of Adaptive Trials and Surrogate Endpoints for Clinical Decision-Making in Rare Cancers. Front. Oncol., 08 March 2021).
Which are the advantages or disadvantages of adaptive clinical trials?
Some of the benefits of conducting clinical trials with adaptive designs are:
• Reduce sample sizes and total cost while maintaining statistical power
• A higher number of patients treated at optimal dose levels
• Quick and efficient identification of patient sub-populations who may better benefit from investigational treatment
• Early stop of clinical trials with ineffective and/or unsafe treatments
• Acceptance by a majority of regulatory authorities of these adaptive designs to grant conditional accelerated approval to certain drugs.
However, its counterpart is the additional level of organisational and logistical effort required, including the role of subject matter experts and the establishment of different data review committees. Most of the adaptive designs require ongoing or multiple interim analyses of data, the establishment and management of Safety Review Committees (SRC), formed by members related to the research; and/ or Data Safety Monitoring Boards (DSMB), embodied by independent subject matter experts.
Additionally, more frequent and fluent communication with the regulatory authorities is needed to guarantee that the designs and study endpoints are adequate to the patients and investigational needs, the study hypothesis remains
valid, and the decisions taken throughout the study are adequate.
Why is there a shift from the classical drug development pathway to adaptive clinical trials?
The clinical development plan for a new drug is generally based on traditional study designs, which first need to demonstrate that safety administration in human beings is feasible (phase I, optimal safer dose level finding); shows signals of efficacy to guarantee further investigation (phase II, proof of concept); and renders a superior result than the standard therapy (phase III, pivotal clinical trials). This classical drug clinical development plan is a lengthy process that can take as long as 10 years, and it is expensive, with a median cost of $985 million, and an average cost of $1.3 billion (Woters J. JAMA. 2020;323(9):844-853).
However, generating top-level scientific evidence is not the end of the process, but the starting point for the regulatory agencies to assess the data, and integrate all other criteria which may influence their decision-making, like quality of life, availability of similar drugs, pharmacoeconomy data, etc. At this stage, a paradox can happen that a new therapy has met the golden criteria for scientific evidence level I, but the data, though statistically significant, may not be clinically meaningful. For example, a phase III, RCT comparing the investigational product to the best standard of care, proves superior efficacy with a longer median overall survival in a cancer population. The design and the primary endpoints are the best to guarantee the highest level of evidence. But the magnitude of improvement between earlier median overall survival and new one is in the range of a few days… In this context, the level of recommendation for the new drug is expected to be low, the same as the probability to get this drug approved by the regulatory authorities.
The average time for review of the informative dossier for a new therapy varies depending on the country or
region involved and may range from 6 to 12 months in US or European Union, (health.ec.europa.eu; www.drugwatch.co), and has been historically longer in some Asian countries, like China.
Under certain circumstances, all sponsors, patients, and society may benefit from a more rapid and efficient approach to classical drug development.
How are adaptive designs used to shorten clinical drug development?
The classical clinical development pathway may become suboptimal in certain scenarios. For example:
• Patients are in a dire need for a therapy, in diseases where no efficacious treatment has yet become available
• Rare indications, with a small number of subjects affected globally. The epidemiology data may not support a classical study design which may require hundreds or even thousands of patients
• Patients with potentially deadly diseases, who need new therapies to change expectancy of life, etc.
Regulatory authorities have embraced the commitment to accelerate the approval process of new drugs in such cases. For example:
• FDA, through its Center for Drug Evaluation and Research’s (CDER)
• EMEA, thought the Medicine´s Adaptive Pathways to Patients (MAPP) initiative
• Australian Therapeutic Goods Administration (TGA) through its Priority Review Pathway; etc.
However, we still need scientific evidence based on study design and strength of study endpoints. The response is an “intermediate step” in the clinical development pathway, where we seek a conditional marketing authorisation to allow early access of patients to more efficacious drugs, while the mature data is obtained.
Adaptive designs in clinical trials are part of these solutions, and aimed to shorten drug clinical development, while minimising the number of subjects exposed during the research.
53 www.pharmafocusasia.com
Are adaptive clinical trial designs beneficial in proof-of-concept (phase II) studies?
Phase II studies may benefit from an adaptive design under certain circumstances such as in biomarkerdriven pathologies, where the presence of a biomarker implies a worse prognosis and/or can be predictive of response to targeted therapies. Certain study designs are aimed to select only those patients with a biomarker-positive disease, to test the potential efficacy of targeted therapies against this biomarker. A similar principle applies also in the research of gene therapies with genetic mutations.
Some of the more frequent study designs used in this context are:
UMBRELLA STUDIES: evaluation of multiple investigational products in patients with same primary disease, characterised by expression of different biomarkers. For example, patients diagnosed with non-small cell lung cancer (NSCLC), can present different altered genes, like EGFR, ALK, ROS1, BRAF, NTRK, MET, or RET. A phase II umbrella study in NSCLC would be based on genotyping of patients, and assignment to biomarker-driven targeted therapies: (Figure 2)
Are adaptive clinical trial designs used in early phase studies?
Adapted study designs in early phase studies are broadly accepted and implemented, with oncology leading the ranking of planned or ongoing clinical trials, as per Citeline data (search date: April 2023) (Figure 1)
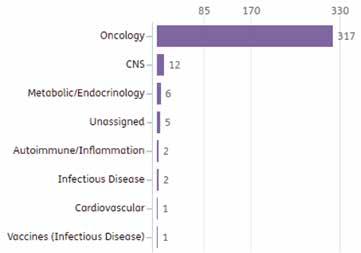
In oncology, the classical 3+3 dose escalation design was originally used with cytotoxic drugs, and the principle to make decisions on escalating or de-escalating the dose is based on maximum toxicity in 33 per cent of patients deemed as acceptable. This model has evolved with alternative algorithm-based designs such as accelerated titration, Bayesian optimal interval (BOIN), continual reassessment method, escalation with overdose control, or modified toxicity probability interval (mTPI-2), among others (doi: 10.3390/ cancers14061566).
A combined phase I (dose escalation)-phase Ib (selection of target subpopulations)-phase II (efficacy data obtained) is a relatively frequent approach in certain therapeutic areas. Approximately 1 per cent to 2 per cent of all ongoing or planned clinical trials in phase I, phase I/II, or phase II are
using this approach (Citeline search date: April 2023).
FDA has released a guidance on this topic which may be used as a reference: “Expansion Cohorts: Use in FirstIn-Human Clinical Trials to Expedite Development of Oncology Drugs and Biologics (August 2018)”.
54 PHARMA FOCUS ASIA ISSUE 51 - 2023
Figure 2: Example of umbrella clinical trial design
Phase Il Umbrella CT NSCLC Patients
Treatment arms with targeted therapies
EGFR ALK MEK ROS1 BRAF
Next Generation Sequencing
CLINICAL TRIALS
Figure 1. Number of ongoing or planned early phase clinical trials with adaptive design. Citeline Database (Pharma Intelligence)
Are adaptive clinical trial designs used in pivotal (phase III) studies?
In later phases of development, an adaptive design may serve different objectives, like sample-size re-estimation, dynamic modification of the primary endpoint, closing or maintaining study arms, or setup of multiple investigational combinations, during the conduct of the study. There are varied examples of these approaches, and some of them are considered as standard in some areas.
What is the present (and future) of adaptive clinical trials?
Adaptive clinical trial designs in an early phase of development (phase I, phase I/II, phase II) are consolidated in some therapeutic areas, where sponsors and patients need rapid decisions based on
critical safety and efficacy data, which can be obtained with a lower number of subjects exposed at a more optimal dose level.
Oncology and rare diseases have been the areas where adaptive trials have been more frequently used to support accelerated conditional approval of new drugs. A certain number of them have been obtained with results of single-arm studies, utilising intra-arm or historical data as controls. Whereas this approach seems to be still valid and well received by the regulatory authorities for some rare diseases, the FDA released in March 2023 a new guidance for oncology research (Clinical Trial Considerations to Support Accelerated Approval of Oncology Therapeutics Guidance for Industry), which supports the RCT design as the golden rule to obtain scientific level 1 data, and therefore, facilitate accelerated approval granted.
BASKET STUDIES: evaluation of one targeted therapy in patients with different indications, which have in common the expression of the same biomarker. For example, overexpression of HER2 has been observed in different tumour types, like breast, gastric, and colorectal cancers, etc. A basket study would test one single targeted anti-HER2 drug in multiple arms enrolling different patient populations to test individual efficacy on each one of them and make decisions on study design for phase III RCT. (Figure 3)
Among the different adaptive approaches, bridging clinical trials (phase I/II, phase II/III), allow for ongoing re-definition of the target patient population and decision making (stop or continue).
In conclusion, the arena of adapted clinical designs is a dynamic environment and close attention should be paid to this evolving field to obtain the maximum benefits of them.
Mahillo has 30 years of experience in clinical research. She is a post-grade magister at 4 Spanish Universities and the author of 3 books, 25 scientific articles, and 27 contributions to congresses. She obtained the first prize from the SEOM for her work on enhancing clinical research awareness among patients

55 www.pharmafocusasia.com
Figure 3: example of basket clinical trial design
Phase Il Basket Clinical Trial
Patients with tumors harboring same mutation or biomarker
Breast Cancer Gastric Cancer Colorectal Cancer Ovarian Cancer
AUTHOR BIO
CLINICAL TRIALS
Same Investigational Product
Challenges and Advantages of Using Artificial Intelligence in Pharma

The use of Artificial Intelligence (AI) in Pharma can greatly accelerate activities. However, Pharma companies today face many tough challenges in creating reliable AI models for the complex environments that characterise the sector. This is because, given the soon-to-be pervasive presence of AI models in our lives, you must consider many more factors besides the usual regulatory ones when you want to create and deploy AI models into Pharma workflows. In this article, we therefore discuss some of the most critical challenges you face when you need to create reliable AI models for Pharma. We also explain how you can solve, or at least ameliorate, some of these challenges. Finally, we provide advice on how you can deploy AI models at scale and explain the advantages that you will garner in so doing.
Fausto Artico, Global R&D Tech Head and Director of Innovation and Data Science, GSK Kevin Harrigan, Director of Innovation and Engineering, GSK
Introduction
Many advanced, large-scale AI initiatives still need to be proved successful in Pharma. This is because Pharma is a highly regulated sector. The discovery and approval processes for a drug can easily take 12+ years, and advanced AI is still a field in formation. It is important, then, to design the future advanced AI systems focusing on certain critical elements. This is because not integrating them into the designs can completely invalidate otherwise technically sound advanced AI solutions. We will therefore discuss three principal issues, mainly: ethical concerns surrounding the objective of the advanced AI systems; the ability to explain the AI models in layman’s terms; and the dangers of training the models using data sources that contain biases.
56 PHARMA FOCUS ASIA ISSUE 51 - 2023
INFORMATION TECHNOLOGY
Ethics
What is the final objective of the advanced AI system you are creating? The answer to this question is very important. Different final objectives imply the need to ponder different ethical matters during the creation of such systems. When you design an advanced AI system, you should at least ask yourself the following questions: Is the goal of the advanced AI system the optimisation of some processes? Is it to automate them? Is it to generate new income streams? Is it to create a better user experience? In addition, how can we trust the actions that the advanced AI models propose? Will they really be for the greatest good of everybody? How can we verify this? Could an advanced AI system be considered a form of intelligence? And if so, how should we treat AI? Where is the point at which we should worry whether an advanced AI system is complex enough that it could generate unintended consequences?
Try to limit the scope and decision power of advanced AI systems. For example, you can create advanced AI systems to solve “narrow” problems that require the optimisation of a small number of equipment settings in a manufacturing train at a site. Doing so, you will feel more comfortable trying the recommendations the systems propose because the consequences of erroneous decisions will be very limited and not critical. This is especially true if you execute Proof of Values on just a piece of equipment for a very limited time. Such design and method of operating make it possible for you to start to build trust in the models’ decisions and later, when you have become confident enough, to scale them up into production on multiple manufacturing trains and sites. And make sure to have a monitoring system in place so so that you can continuously assess whether the environmental conditions in which the models operate are the same as those used for their training and thus safe to continue to use.
Advanced AI systems are there to help humans to take decisions, not to
supplant them. The more important and the more critical the consequences of the decisions an advanced AI system needs to make, the more important it is that you have humans assess them. In addition, the final approval for the important decisions should always involve a series of steps and activities, and not just one decision maker or single point of failure. Furthermore, it is unlikely that future regulators will approve the use of completely automated advanced AI systems for Pharma processes. And complete digital twins will probably not wholly obviate, if at all, the need for tests on animals first and later clinical trial phases on humans. Therefore, advanced AI systems will accelerate some activities and increase the probability of success of some objectives (e.g., the discovery of new drugs), but they will only be enhancements to existing procedures and processes and not substitutes for them. It is also impossible to take an advanced AI system to court. Regulators hold humans accountable for the decisions taken by the systems they design. So, it is more important than ever to simplify the models as much as possible to allow humans to understand them and to double-check the sugges -
tions and actions the systems propose. This is especially true if we want to develop swarm systems composed of many models, each solving or improving human activities on very narrow tasks but with the need to communicate with each other and to feed each other inputs and outputs.
Explainability
Many non-AI Pharma domain knowledge experts need to interact with advanced AI systems. It is, therefore, important that they understand why the models propose certain actions or why they “think” they have discovered important insights they deem appropriate to flag for human attention. This is especially true if the models’ actions and insights are counterintuitive. Answers on why and how models work that are purely based on statistical principles are not going to be understood by many people. If people cannot understand why the models make certain choices they will see them as black boxes, will not trust them and/or resist using and integrating them in their standard ways of working. I have seen many situations in which domain knowledge experts in non-tech domains (e.g., biologists) simply refused to believe the models’ choices were correct because they did not understand how such choices were generated. It is difficult for a person to accept such choices if their validity can only be proved after years of testing and long processes, as is the case in clinical trials. Compare this to other situations such as in manufacturing, for example. There you can just execute a “quick,” albeit not always easier, Proof of Value using existing equipment and a lot of historical data related to mechanical engineering processes that we know very well in contrast to our more limited knowledge of the biology and working mechanisms of a human body.
Hybrid models will probably be the way of the future. Domain knowledge experts and data scientists need to work together to get the best of both worlds (i.e., life science and data
57 www.pharmafocusasia.com
INFORMATION TECHNOLOGY
In Pharma, considering the importance of many decisions related to the preservation and betterment of human lives, we should use advanced AI systems to accelerate and enhance human activities and ways of working, but not think of them as r.eplacements for our higher cognitive functions.
science domains). We cannot just use models that are purely statistical. While such models could be great to discover correlations and therefore tell us how to achieve a specific objective, they are very limited in their ability to explain why such phenomena happen from the scientific point-of-view. Combining science with more purely statistical methods will drive research and discovery in ways that can be understood and used by many domain knowledge experts and so advance science much more rapidly than today. This is critical considering that we finally have enough data and computational power to analyse processes and activities in ways that are impossible for humans but easy for machines. People can continue to develop science but leverage machines and algorithms to more efficiently verify hypotheses, search for patterns and more generally liberate themselves from tedious activities like sifting through enormous amounts and types of data.
Most data is and will remain unstructured. Data types such as images, long-form text and the like, for example, contain many more bits of data than what is captured and stored in tables and/or other structured data formats. What this implies is that many models in future will have to be able to approximate cognitive capabilities to interact with humans. In fact, it is not far-fetched to imagine that more and more advanced AI solutions will be able to interact with humans in a way that is more humanlike than what is possible today. Our ability to ask questions to advanced AI systems, interact with them and, more generally, design them in ways that make this possible will only increase over time. The fact that you will not need to be a data scientist able to code to interact with such advanced AI systems will speed up our ways of working and make our life easier.
Personalisation will be important too. It is not difficult to imagine that models will be able to interact with a variety of stakeholders who have a
variety of interests and use a variety of lenses to interpret the world. Already today, we have Large Language Models (LLM) that can generate text and be trained on various corpora. Training them on the corpora in which different professionals were educated will make it possible for such models to use the language of various professions and so to interface with diverse types of people (e.g., lawyers, biologists, HR people, engineers in manufacturing, etc.). Layering speech and voice capabilities on top of the LLMs and adding personalisation features that are related to the personality and attitude style of each individual they interact with will open possibilities that are difficult to image today but that will greatly enhance our human-machine interactions. This is especially true if we also design empathic capabilities into such future advanced AI systems as well as other soft skills that would enable them to take into account feelings/emotions, mental thought patterns and social dynamics.
Biases
Datasets used for training should be representative of the environmental conditions in which the models will operate. This is easier said than done. You need to be careful about this
because it is easy to make the erroneous assumption that the models can generalise well enough. Typical examples are: recommendations on how to select a cohort of people for a clinical trial without realising that minorities will not be sufficiently represented because of a lack of data related to them in the datasets used to feed the model, and corpora or databases that are thought to contain high quality data and to be perfectly valid and curated but instead contains mistakes and errors.
Augment context to allow people to understand how activities were executed and what was done. Today, many people do not trust models because they cannot retrieve enough information on why they were created in some ways and not others, or why some data sources were used or chosen and others not. Essentially, it is important that during model creation, manually or automatically you enrich the datasets and the protocol with metadata that explains and helps to contextualise the choices the designers made to create the model. Therefore, you should design, implement and deploy as many automated checks as possible for the models and the datasets used for training them. The reason for doing so is that in this way you can at least log and track what was decided, why, the problems that you are aware of and that affect the datasets and models, etc. In this way, if in future somebody wants to verify the protocol that was used to create the models and the datasets, he/she will be able to do so quickly and will be able to add additional checks to verify if something was omitted in the protocol or a new hypothesis on the datasets and/or model needs to be tested. This is important for reassessing the model (to invalidate and retrain it or recreate it) if for any reason you become aware of new conditions that are critical to how it should have been created as well as in which environmental conditions it is supposed to operate. Software makes it possible to automate many such activities. And since many choices
58 PHARMA FOCUS ASIA ISSUE 51 - 2023
INFORMATION TECHNOLOGY
Advanced AI systems are there to help humans make decisions, not to supplant them. The more important and critical the consequences of the decisions an advanced AI system needs to make, the more important it is that you have humans assess them.
could be questioned at a later time by other people, it is important to generate and save a history of all these choices. It will be impossible, otherwise, to execute any further assessment, validate or disproof the model, or add new checks at a later time in reliable ways that augment the analyses previously executed. The alternative of recreating them from scratch would not only be prohibitively time consuming, it may not even be possible.
Also, beware of the fact that problems can arise because some environmental conditions were thought unimportant and are not captured by any sensor or change very infrequently or slowly. They can unexpectedly generate strong nonlinear dynamics that have never been assessed or discovered because they never manifested before and so never caused any issues till now. You cannot always solve this problem (i.e., understand and discover what you do not know you do not know). However, with the right monitoring systems, you can at least verify whether the environmental baselines related to the things that you are tracking have changed or not, and if the answer is no, be more quickly able to execute root cause analyses. That is to say, you know that it is better to focus your attention on something that you are not logging and monitoring yet because all the other things you have always logged and monitored seem to be working as usual in ways that never generated any problem before. Conversely, the other issue that you could face is when things that you are logging and monitoring really are the root cause of the problems because, for the first time ever, they combined and interacted in ways they never have before (even if they still satisfy all the requirements). You could use clustering techniques in these situations to verify what is different and has never happened before even if everything looks in spec. (Admittedly, this could be difficult to execute if the sampling frequency of your monitoring systems is not high enough).
Summary
Advanced AI systems can greatly enhance existing Pharma activities. This is because, with the right computational power available and thanks to their ability to process enormous amounts of data, we can start to solve more global problems connecting different verticals and business units in ways that are much more holistic than ever before. However, we need to be careful how we design, test and continuously monitor such systems. This is because they have a tendency to overfit the datasets used for their training and are usually not able to strongly generalise as conditions in the environments in which they operate change in ways that are not represented and captured in the datasets. In addition, there are important ethical components that are going to become more prominent as we start to build advanced AI systems able to mimic broader cognitive human capabilities. In Pharma, considering the importance of many decisions related to the preservation and betterment of human lives, we should use advanced AI systems to accelerate and enhance human activities and ways of working, but not think of them as replacements for our higher cognitive functions. Hybrid models have great advantages and will be the model of choice more readily adopted by non-data science domain knowledge experts. Such experts will be reassured using such models because they will understand that they, the experts, will remain critical for the design, use and interaction with such models. Finally, from the regulatory point-of-view, advanced AI systems will probably always be seen as just tools and not fully sentient. Humans will therefore remain fundamental in using such systems, will have to continue to validate their choices, and will leverage them to accelerate and improve many important and complex activities impossible to tackle exclusively by humans.
Fausto has two PhDs (Information and Computer Science respectively), earning his second master’s and PhD at the University of California, Irvine. He also holds multiple certifications from MIT. As a Physicist, Mathematician, Engineer, Computer Scientist, and HighPerformance Computing (HPC) and Data Science expert, Fausto has worked on key projects at European and American government institutions and with key individuals, like Nobel Prize winner Michael J. Prather. After his time at NVIDIA corporation in Silicon Valley, Fausto worked at the IBM T J Watson Center in New York on Exascale Supercomputing Systems for the US government (e.g., Livermore and Oak Ridge Labs).

Kevin graduated with a BSAE in aerospace engineering from Pennsylvania State University. During his collegiate career he gained experience in a co-opposition with Capital One Financial as a Data Analyst at in Richmond, Virginia. It was this experience that afforded him the opportunity to find passion in data munging, applied statistics, and programming. Following graduation, he accepted a full time offer in their newly formed Digital Enterprise Organization, expanding his technical and analytical knowledge in areas such as distributed computing, clickstream analytics, multivariate testing, anomaly detection, and propensity modelling.

59 www.pharmafocusasia.com
AUTHOR BIO
INFORMATION TECHNOLOGY
Blockchain
Mandatory in Constructing a High-fidelity Database Readying for The Downstream Disruptive Technologies, such as for The Artificial Intelligence Training
Blockchain is the foundation layer for the data procured during drug development that are transparent, secure, and traceable. This tamper-proof and decentralised ledger also ensures that data generated during drug development is both robust and accessible to all stakeholders while allowing machine learning and artificial intelligence to harness data effectively and provide accurate solutions.
Frank Leu, Founder and managing member, BioPharMatrix LLC
Ablockchain is a simple digital serial ledger of tamper-proof data blocks that are cryptographically protected via peer-to-peer networks (known as validators and/or nodes) and is distributive and decentralised by nature. It contains a record of transactions in a chain of data blocks in a decentralised form with each validator/node storing a copy, meaning with multiple validators/nodes operating in a blockchain making that no single entity or authority could have
control over the entire blockchain data and its network in a centralised fashion.
Another important aspect is its immutability, which makes it virtually impossible for malicious actors to alter it. Bitcoin, Ethereum, and many other cryptocurrencies made it well known to the masses. However, its applications have extended far beyond cryptocurrency. Its ability to offer a superior architecture for efficient data management and secure transactions has attracted interest across
several sectors, including healthcare and pharmaceutical drug development.
Currently, blockchain has the potential and is on its course to disrupt the drug development landscape profoundly. Blockchain should no longer be referred to as a “future technology”, rather it is the technology that has already been implemented in various business sectors, such as supply chain logistics, product storage management, real estate, and decentralised finance.

Artificial intelligence (AI) can be trained on broad and various databases within the ecosystem of drug development and then allow AI to solve many complicated problems quickly and accurately. Blockchain technology constructs a trustless database, which is highly trustworthy with incredibly high intrinsic fidelity. When AI is trained with a database with low intrinsic fidelity, there is a serious risk that AI would provide solutions that are overly biased, error-prone, and vulnerable to security threats, which can diminish its solutions’ and renders them useless and unreliable. Blockchain provides a secure, transparent means for recording and verifying each transaction and interaction, thus allowing AI to derive machine intelligence from its training through data compiled by blockchain and thus greatly reducing the risk of generating faulty solutions for complicated problems. Blockchain-based solutions can, for example, be used to verify the integrity of each data block added that is used to train AI models, ensuring accuracy, unbiasedness, and representativeness.
Therefore, the blockchain must serve as the foundation layer for the pyramid of downstream disruptive technologies, providing a reliable information source for machine learning and AI training. By achieving high data fidelity using blockchain architecture, hyper-efficient drug development is likely to be achievable. By doing so, AI systems will be functioning more accurately and fairly, and the risk of errors or biases will be reduced. Moreover, blockchain can
60 PHARMA FOCUS ASIA ISSUE 51 - 2023
INFORMATION TECHNOLOGY
UPGRADE YOUR MARKETING STRATEGY
Highly accountable marketing campaigns. Every dollar counts.
Digitally powered marketing campaigns may be cheaper than you thought... Ask us how?
Use the webinar as a platform to launch new products and services
Grow your audience with increased reach, impact and user-friendliness
Rise above geographical boundaries
Generate new business
Gain the strong web presence differentiating yourself from competitors
Connect and engage with your target audience

Give more exposure to industry specific people
Increase your brand profile and share your capabilities with leading industry professionals

Email : advertise@pharmafocusasia.com


Website : www.pharmafocusasia.com

61 www.pharmafocusasia.com
Let the true “Digital Transformation” be the base of all your marketing campaigns
PANDEMIC
AFTER COVID-19
“Digital Transformation is the way forward”
Our recent successful partnerships:
provide individuals with control over their personal information and ensure that sensitive data is not compromised or misused, thereby ensuring both data security and privacy.
However, while blockchain technology can be an important tool in building a trusted AI system to enhance the process of drug development, it is not the only technical requirement to ensure AI works robustly and accurately. Other technologies and processes, such as robust data governance, explainability, and ethical frameworks, are also important components needed to implement to ensure the trustworthiness and reliability of the data collected during drug development.
In the hope to achieve the overall best efficiency to reduce both drug developmental costs and timespan. An overview of a few specific areas during drug development of how blockchain could impact drug development positively and provide the high fidelity required for AI to be accurately and unbiasedly trained and allow it to best perform is listed below in random order:
1.Data Governance and Assurance. By ensuring that data is managed, stored, and shared consistently and transparently across all parties concerned, blockchain's decentralised nature and consensus mechanisms help improve the governance of data. Because blockchain technology is immutable, it assures the accuracy, reliability, and trustworthiness of the data it records. For pharmaceutical companies, where data quality is crucial to regulatory compliance and decision-making, this characteristic is particularly important.
2.Enhanced Stakeholder Collaboration with Efficient Inventory Management. As blockchains offer real-time visibility into stock levels, expiration dates, and product locations, pharmaceutical inventory management can be optimised. By increasing transparency to all stakeholders, better forecasting is possible, stockouts and waste are reduced, and essential medicines are available to patients in a better way. Blockchain can facilitate
secure data sharing, intellectual property management, and collaborative efforts among pharmaceutical companies, research institutions, and regulators. Consequently, new treatments are more likely to be brought to market faster with a lower cost, which promotes innovation, accelerates drug discovery, and accelerates drug discovery.
3.Pharma Supply Chain and Pharmaceuticals Turnover Monitoring. Pharma supply chains can be complex, involving many intermediaries and crossing international borders. As a result of this complexity, transparency can be compromised, logistical bottlenecks can occur, and fraud and theft can be more common, ultimately impacting the availability and affordability of medicines. Pharma supply chains can be significantly more transparent and traceable with blockchain technology. In addition to enabling stakeholders to track and authenticate drugs from their point of origin to their destination, blockchain also prevents counterfeit drugs and ensures patient safety by creating secure, tamper-proof records of sales and product movements. As a result of optimising supply chain processes, reducing waste, and ensuring efficient distribution, blockchain can assist pharmaceutical companies in reducing turnover. Pharma companies can save money and ensure patients have access
to medicines on time as a result. This use of blockchain technology could also ultimately enable better monitoring and analysis of prescription drug dispensing patterns, drug utilisation, and concerns about the potential abuse of prescription drugs.
4.Anti-counterfeiting and Safe Plus Secure Distribution of Pharmaceutical Products. As blockchain creates a secure, tamper-proof record of drug provenance and transactions, it will be able to effectively prevent pharmaceutical counterfeiting, preventing patients and manufacturers from receiving unsafe medications. Pharmaceutical products can be accurately tracked and traced using blockchain in real-time, so that they can be tracked from the manufacturing facility to their destination. The product’s whereabouts must be transparent to verify its authenticity and prevent counterfeits from entering the marketplace. An immutable record of transactions and data provided by blockchain technology also improves the safety and security of the pharmaceutical industry, making it nearly impossible to conceal unauthorised alterations. As a result of using blockchain, more efficient allocation of resources will ultimately lead to cost reductions.
5.Clinical Trials and Patient Data Management. Streamlining clinical trial processes with blockchain can be a major benefit for patients by ensuring data integrity and transparency through the secure storage and sharing of trial data. In addition to improving regulatory compliance, this enables faster and more efficient drug development, which ultimately benefits patients. Various sensitive data, including clinical trial results, patient information, and intellectual property, are generated by the pharmaceutical industry. Despite highly fragmented systems, outdated technologies, and evolving cyber threats, secure storage, retrieval, and sharing of this data remains a significant challenge. Through the use of blockchain technology, patient data management can be revolutionised
62 PHARMA FOCUS ASIA ISSUE 51 - 2023
INFORMATION TECHNOLOGY
Blockchain-based solutions can, for example, be used to verify the integrity of each data block added that is used to train AI models, ensuring accuracy, unbiasedness, and representativeness.
by providing a secure, decentralised way to store and share sensitive patient information. As a result, clinical trial stakeholders and healthcare providers can collaborate better between or among themselves, allowing for more personalised and effective clinical trial execution and/or patient care, meanwhile ensuring data privacy and security.
In a lacklustre drug development data management system, it is understood that blockchain technology may not be necessarily required for downstream analysis in a decentralised drug developmental ecosystem. Downstream analytics refers to the analysis of data generated during drug development, such as using machine learning and training AI to better understand and make the best decisions based on the clinical trial data gathered for drug safety and efficacy. The FDA recognises the current limitations and challenges that exist in the current drug development and has initiated exploratory projects to implement blockchain in drug development. Below are a couple of examples of FDA-sponsored pilot initiatives:

1) To track prescription drugs through the supply chain, the FDA announced an investigation into blockchain technology in February 2019 with Merck, KPMG, Walmart, and IBM. From manufacturer to patient, this pilot study evaluated the effectiveness of blockchain technology in improving traceability and security in drug supply chains. An important focus of the pilot study was the use of blockchain technology to track prescription drugs throughout the production process, dispensing phase, and beyond, particularly drugs at risk of counterfeiting, theft, or diversion. As part of the pilot study, the IBM Blockchain Platform was used to track drug movements securely and transparently throughout the supply chain using a blockchain-based platform. To monitor and verify the movement of drugs, the platform combined blockchain technology with other advanced technologies such as the Internet of Things (IoT)
sensors and AI algorithms. There was an improvement in efficiency, security, and transparency in the drug supply chain following the pilot study, which was considered a success. Blockchain technology is a tamper-proof and secure way to track drug movement, enabling all parties to have access to real-time information about drug status and location. In general, the FDA pilot study involving Merck, KPMG, Walmart, and IBM demonstrated the potential of blockchain technology for improving drug supply chain traceability and security. The study may lead to the wider adoption of blockchain-based solutions in the pharmaceutical industry in the future.
2) The FDA has piloted a programme called the MediLedger Project to enhance the security and traceability of the pharmaceutical supply chain through blockchain. Through this pilot programme, blockch ain will be evaluated for its potential to enhance security and traceability in pharmaceutical supply chains. Blockchain will be used to track and verify the movement of pharmaceutical drugs from the point of manufacture to the point of dispensing as part of the pilot program. Several major pharmaceutical companies are participating in the pilot programme, including Pfizer, McKesson, and AmerisourceBergen. Furthermore, the FDA has been exploring the use of blockchain in drug development with other organisations, in addition to its pilot programme with MediLedger Project. To develop standards and protocols for the use of blockchain technology in the development of drugs, the FDA has been working with the Institute of Electrical and Electronics Engineers (IEEE). It is evident from the FDA's involvement in blockchain initiatives in drug development that blockchain technology could contribute to improving drug development security, transparency, and efficiency. Besides facilitating innovation in the pharmaceutical industry,
the FDA also ensures the safety and efficacy of new drugs by supporting the development of blockchain-based solutions for drug development.
While blockchain can offer advantages such as data immutability and transparency, other approaches can be used to manage and analyse the data generated during drug development securely with elevated efficiency. For example, building a distributed database can provide a similar level of data integrity and security through replication and distributed consensus mechanisms. However, this is limited in its efficiency when compared to using a blockchain data architecture.
Currently, blockchain technology adoption may still depend on the specific requirements and goals of the existing drug developmental sponsor. Overall, blockchain technology has the potential to significantly improve the drug development process by increasing transparency, reducing fraud, and improving access to new therapies. However, implementing these solutions will require collaboration between pharmaceutical companies, regulators, and technology providers to ensure that these systems are effective, secure, and compliant with regulatory requirements.
63 www.pharmafocusasia.com
AUTHOR BIO
INFORMATION TECHNOLOGY
Frank Leu is the Founder and managing member of BioPharMatrix LLC, providing intelligence in the areas of innovative processes and platforms for biotech, pharmaceutical, and life sciences. Frank has combined around a hundred speaking presentations and publications on disruptive technologies and processes in drug development.

64 PHARMA FOCUS ASIA ISSUE 51 - 2023
STRATEGY AMI Polymer ...................................................................................26-27 eppendorf IBC Medical Fair Thailand 05, 34-35 Nipro IFC Oracle 18-21 Turkish Cargo OBC Valsteam ADCA Engineering 03 RESEARCH & DEVELO PMENT Quantys Clinical Pvt. Ltd ...................................................................... 25 SGS .................................................................................................09-13 CLINICAL TRIALS Quantys Clinical Pvt. Ltd 25 SGS 09-13 MANUFACTURING AMI Polymer 26-27 eppendorf IBC Nipro IFC Valsteam ADCA Engineering 03 INFORMATION TECHNOLOGY Oracle .............................................................................................18-21 Company ............................................................. Page No. SUPPLIERS GUIDE AMI Polymer ...................................................................................26-27 www.amipolymer.com eppendorf........................................................................................... IBC www.eppendorf.com/DASbox Medical Fair Thailand 05, 34-35 www.medicalfair-thailand.com Nipro IFC www.nipro-group.com Oracle 18-21 www.oracle.com Quantys Clinical Pvt. Ltd 25 www.quantysclinical.com SGS 09-13 www.sgs.com Turkish Cargo OBC www.turkishcargo.com Valsteam ADCA Engineering ................................................................ 03 www.valsteam.com Company ............................................................. Page No. To receive more information on products & services advertised in this issue, please fill up the "Info Request Form" provided with the magazine and fax it. 1.IFC: Inside Front Cover 2.IBC: Inside Back Cover 3.OBC: Outside Back Cover
PRODUCTS & SERVICES
Most Compact
QbD-driven process development with the DASbox® Mini Bioreactor System
With working volumes of 60 – 250mL the DASbox is the optimal tool for advanced cell culture and microbial process development and Design of Experiments (DoE) applications. All critical parameters can be precisely controlled.
> Parallel set-up of up to 24 bioreactors
> Perfectly suited for microbial and cell culture applications

















> Liquid-free exhaust condensation



> Fully mass flow-controlled gas mixing
> Available with single-use vessels
Designed for stem cell process development: BioBLU 0.3sc with 8-blade impeller



65 PHARMA FOCUS ASIA ISSUE 51 - 2023
www.eppendorf.com/DASbox Eppendorf ® and the Eppendorf Brand Design are registered trademarks of Eppendorf SE, Germany. DASbox ® is a registered trademark of DASGIP Information and Process Technology GmbH, Juelich, Germany. All rights reserved, including graphics and images. Copyright ©2022 by Eppendorf SE.

