Adopting the Power of AI to Drug Development Projects



ISSUE 52 2023 www.pharmafocusasia.com
Long-term Success
to look for in a cell culture
supplier
Challenge Trials
early risk-benefit in development of vaccines and
for infectious diseases
Securing
What
media
Human
Establishing
therapies
NICK BROWN Associate Director of Marketing Certara
MATT ZIERHUT Vice President, Integrated Drug Development Certara
Your air cargo partner of choice































ETIHADCARGO.COM Choose reliable
Vaccine Development The rise of human challenge trials
Vaccines and antivirals have been the fundamental pillars in the battle against infectious diseases. Nonetheless, their development is fraught with substantial hurdles, necessitating a protracted and costly journey, frequently marred by a notable rate of failure. The onset of the COVID-19 pandemic brought Human Challenge Trials (HCTs) showcased the significant potential of this research approach in advancing our understanding and assessment of infectious diseases and expediting vaccine development.
“With small biotechs, these human challenge trials offer clear proof-of-concept data, much better than data from animal studies, which can be plagued with translation issues on how it would perform in humans. As for big pharma, human challenge trial data can be used to argue their case for internal funding over other assets in the pipeline,” said Open Orphan’s chief scientific officer Andrew Catchpole.
Human challenge studies have made a remarkable contribution to speeding up the progress of treatments for various diseases, such as malaria, typhoid, cholera, norovirus, and influenza in the past few decades. These trials have also been instrumental in aiding researchers determine which candidate vaccines are most promising for advancement to phase 3 clinical trials—a crucial step that typically entails the involvement of thousands of volunteers.
In the realm of medical research, where science and ethics intersect, HCTs stand as a powerful symbol of collective pursuit of knowledge and commitment to improving public health.
All risks in human challenge studies ought to be minimised to the greatest extent possible, while focusing on not excessively compromising the potential research benefits. This requires consultation with scientific experts, prospective participants, and the broader community. Through these consultations, it becomes possible to assess and determine the acceptable level of residual risks, ensuring that they are justified by the expected research benefits. This collaborative
and ethical approach helps uphold the principles of transparency, informed consent, and the responsible advancement of scientific knowledge in the context of human challenge studies.

Despite the significant inroads made by human challenge trials (HCTs) in infectious disease research, however, we do not yet have codified regulations pertaining to HCTs. Furthermore, there is a visible lack of regulatory guidance related to standardising approaches to HCTs among different regulatory bodies.
As indicated by a study published in The Lancet Infectious Diseases journal, recent assessments of the ethical frameworks concerning human challenge studies have emphasised on striking a balance between the acceptable risk associated with exposing individuals to experimental interventions or pathogens for vaccine development and the social value or public health benefits. This delicate equilibrium ensures that the ethical conduct of these studies prioritises the wellbeing of participants while advancing vital research with the potential to benefit public health and combat infectious diseases effectively.
The article in this issue on “Human Challenge TrialsEstablishing early risk-benefit in development of vaccines and therapies for infectious diseases” by Bruno Speder, VP Regulatory Affairs, Poolbeg Pharma explains the role of HCTs in the testing and development of novel antivirals and vaccines.
Keep reading!
Prasanthi Sadhu Editor
CONTENTS STRATEGY
06 The Glocalisation Classroom Astute glocalisers learn from their successes and failures
Brian Smith, Principal Advisor, PragMedic
14 Accelerating Cell and Gene Therapy Treatments to Patients with the Right CDMO Outsourcing Model

Andy Whitmoyer, VP Supply Chain, Center for Breakthrough Medicines
Emily Moran, SVP, Vector Manufacturing, Center for Breakthrough Medicines
18 Securing Long-term Success
What to look for in a cell culture media supplier

Chad Schwartz, Senior Manager, Global Product Management, Thermo Fisher Scientific
RESEARCH & DEVELOPMENT
27 Emerging Tools Shaping Drug Discovery and Development Landscape

Qasem Ramadan, Alfaisal University
Adopting the Power of AI to Drug Development Projects

CLINICAL TRIALS
54 Human Challenge Trials
Establishing early risk-benefit in development of vaccines and therapies for infectious diseases
Bruno Speder, VP, Regulatory Affairs, hVIVO plc
36 An introduction to FDA’s Human Factors Engineering Requirements for DrugDevice Combination Products
Allison Strochlic, Global Leader – Human Factors, Emergo by UL,
Yvonne Limpens, Managing Human Factors Specialist, Emergo by UL,
Katelynn Larson, Technical Writer, Emergo by UL
42 A Step-By-Step Strategy for Designing A Meta-Analysis
Ramaiah M, Manager, Freyr solutions
Balaji M, Deputy Manager, Freyr solutions
49 A Plant Virus Nanoparticle Toolbox to Combat Cancer
Mehdi Shahgolzari, Department of Medical Nanotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences
Afagh Yavari, Department of Biology, Payame Noor University
Kathleen Hefferon, Virology Laboratory, Department of Cell & Systems Biology, University of Toronto
MANUFACTURING
58 Respirable Engineered Spray Dried Dry Powder as a Platform Technology
Aditya R Das, Founder and Principal, Pharmaceutical Consulting LLC
61 Just in Time Manufacturing
Creating more sustainable supply chains
Lyn McNeill, JTM Supply Chains Solutions Manager, Almac Clinical Services
2 PHARMA FOCUS ASIA ISSUE 52 - 2023
Cover Story 22
Matt Zierhut, Vice President, Integrated Drug Development, Certara Nick Brown, Associate Director of Marketing, Certara

Advisory Board
Alessio Piccoli
Lead
Sales and Business Development Activities for Europe Aragen Life Science

Andri Kusandri
Market Access and Government & Public Affairs Director
Merck Indonesia
Brian D Smith
Principal Advisor PragMedic
Gervasius Samosir

Partner, YCP Solidiance, Indonesia

Hassan Mostafa Mohamed Chairman & Chief Executive Office ReyadaPro


Imelda Leslie Vargas Regional Quality Assurance Director Zuellig Pharma


Neil J Campbell Chairman, CEO and Founder Celios Corporation, USA



Nicoleta Grecu Director
Pharmacovigilance Clinical Quality Assurance Clover Biopharmaceuticals

Nigel Cryer FRSC
Global Corporate Quality Audit Head, Sanofi Pasteur
Pramod Kashid
Senior Director, Clinical Trial Management Medpace
Quang Bui
Deputy Director at ANDA Vietnam Group, Vietnam

Tamara Miller
Senior Vice President, Product Development, Actinogen Medical Limited
EDITOR
Prasanthi Sadhu
EDITORIAL TEAM
Grace Jones
Harry Callum
Rohith Nuguri
Swetha M
ART DIRECTOR
M Abdul Hannan
PRODUCT MANAGER
Jeff Kenney
SENIOR PRODUCT ASSOCIATES
Ben Johnson
David Nelson
John Milton
Peter Thomas
Sussane Vincent
PRODUCT ASSOCIATE
Veronica Wilson
CIRCULATION TEAM
Sam Smith
SUBSCRIPTIONS IN-CHARGE
Vijay Kumar Gaddam
HEAD-OPERATIONS
S V Nageswara Rao
Ochre Media Private Limited Media Resource Centre,#9-1-129/1,201, 2nd Floor, Oxford Plaza, S.D Road, Secunderabad - 500003, Telangana, INDIA, Phone: +91 40 4961 4567, Fax: +91 40 4961 4555 Email: info@ochre-media.com
www.pharmafocusasia.com | www.ochre-media.com


© Ochre Media Private Limited. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, photocopying or otherwise, without prior permission of the publisher and copyright owner. Whilst every effort has been made to ensure the accuracy of the information in this publication, the publisher accepts no responsibility for errors or omissions. The products and services advertised are not endorsed by or connected with the publisher or its associates. The editorial opinions expressed in this publication are those of individual authors and not necessarily those of the publisher or of its associates.
Copies of Pharma Focus Asia can be purchased at the indicated cover prices. For bulk order reprints minimum order required is 500 copies, POA.
Magazine Subscribe LinkedIn
















5 www.pharmafocusasia.com Pressure regulators Control valves Pipeline ancillaries Special equipment Steam traps Safety valves adca@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467 PRODUCTS MANUFACTURED IN PORTUGAL Guia PBL PORTUGAL HIGH PURITY. HIGH DEMANDS. H igh purity equipment for clean steam AV 001 AP E 01.20
THE GLOCALISATION CLASSROOM Astute glocalisers learn from their successes and failures
Done well, this process avoids the three dangers of isomorphic targeting.
Brian D Smith, Principal Advisor, PragMedic
In the first article in this series (“The Glocalisation Challenge”), I differentiated between astute and naïve glocalisers and, in this fourth and final article, I describe the most undervalued difference between them. For astute glocalisers, local execution of global strategies goes further than allocating effort between national affiliates (see article two, “The Glocalisation Choice”). It also goes beyond how those affiliates adapt global strategy to local market conditions (article three, “The Glocalisation Change”). They
also sustain and expand their success by using glocalisation as a classroom. They deliberately learn lessons from their affiliates and spread them around the firm. This article describes how they do that in practice.
Let’s assume that you have assimilated and acted on the lessons from the first three articles in this series. You have understood that there is a capability chasm between those life science companies that are astute and those that are naïve. You have learned not to target countries
naively, by market size, but astutely, according to the winnable segment size. And you have learned not to adapt to local markets by naively making only the essential and easy changes but by astutely building augmented value propositions around contextual segments. If you have done all that, then your results will be more like those of the astute companies I studied and less like the naïve ones. But you will still have an important question to answer: how do we sustain our glocalisation success?
The best practice lllusion
Sustaining success is difficult because market change is a constant and what worked yesterday may not work tomorrow. That’s why the respected academics
Sparrow and Hodgkinson said that, in a changing market, a firm’s only sustainable competitive advantage is its ability to learn. But how do firms learn and, in particular, how do pharma, medtech and other life science firms learn from and about glocalisation? Let’s begin with how naïve glocalisers try and fail to learn.
You might be familiar with the following scenario from your present or past firm. Key people from each affiliate in a region, or globally, are invited to a meeting. Most of the agenda is HQ communicating the global strategy to the

6 PHARMA FOCUS ASIA ISSUE 52 - 2023
STRATEGY
affiliates. But there’s also a session about sharing best practice. In that session, the affiliates are asked to share their successes and failures, although naturally they focus on the former. These successes are held up as “best practice” and the other affiliates are encouraged to copy them. It has the obvious credibility of being a proven, real-world case study in your own company, analogous to learning from a sibling. For naïve glocalisers, this imitation of perceived best practice of friends and colleagues is the default approach to learning and adapting to market changes.
But this kind of best practice is illusory and it hinders learning and adaptation as much as it helps. It is illusory because it is built on the assumption that what works in one country will work with only minor adaptation in another because the company and its products are the same. This is often a false assumption because even affiliates in the same region can be working in markets that differ in important ways. I mentioned some of those important differences between markets, such as competitive activity and segmentation patterns, in article two, “The Glocalization Choice”. And this kind of best practice imitation can hinder adaptation because mimicry drives value proposition design instead of market needs.
So if sharing and copying illusory best practice is the default of naïve glocalisers, how do astute glocalisers learn from and about glocalisation? In my research,
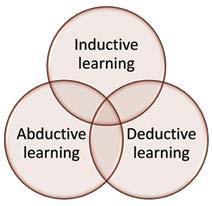
I observe three approaches to learning. The most astute glocalisers use all three in a complementary manner, as summarised in figure 1.
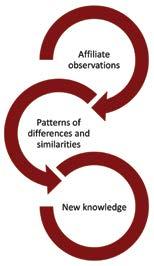
Inductive learning
The most prevalent approach to learning among astute glocalisers is the inductive approach. This is a form of “pattern seeking”, as shown in figure 2. It involves searching across a range of national affiliates for similarities and differences in what works and what does not and then drawing a conclusion about the lessons to be learned. In companies I’ve worked with, inductively-drawn lessons have included the heterogeneity of payer behaviours and the role of communities of practice in influencing new product adoption. Inductive learning works best when there is someone, typically with regional responsibility, who has an open mind and looks at affiliates objectively. That objectivity allows them to be receptive to the successes and failures of the affiliates and to find the patterns in their activity and outcomes. Induction fails when there is no one in that role or when that person comes with fixed opinions. In that case, the process is thwarted by the cognitive bias that psychologists call “confirmation bias” and inductive learning becomes no more effective than the best practice illusion. Typically, inductive learning is used most when a firm is unclear about both what it needs to learn and how to learn.
Deductive learning
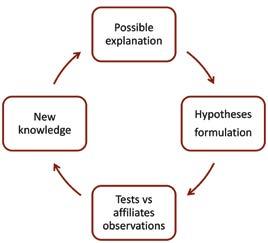
Deductive learning is more methodical and less prevalent than inductive learning. It parallels the scientific method that evaluates a theory, via a pair of hypotheses, to learn something new, as shown in figure 3. Deductive learning is best explained by one real world example I observed. The company believed, but did not know, that uptake of a women’s health product in Asia Pacific was hindered by cultural attitudes towards this therapy
area. From that putative explanation of why uptake varied, they created a pair of hypotheses to test:
• H1 = If our putative explanation is correct, uptake will vary between affiliates with different cultural beliefs
• H0 = If our putative explanation is incorrect, uptake will not vary between affiliates with different cultural beliefs. By comparing uptake across a number of affiliates, they found H0 was better supported by the real world data. Although this did not give them a new explanation of uptake, it did remove an old, incorrect explanation, which is just as useful. Both inductive and deductive learning need a thoughtful person in a regional role to own the learning process. Unlike inductive learning, deductive learning needs a working theory, testable hypotheses and sufficient data for evaluating them. Deductive learning fails when firms cannot make their working theories explicit, or if hypotheses are poorly designed or if they misinterpret data. As you can imagine, deductive learning processes are most effective when the firm has a scientific, data-driven culture.
Abductive learning
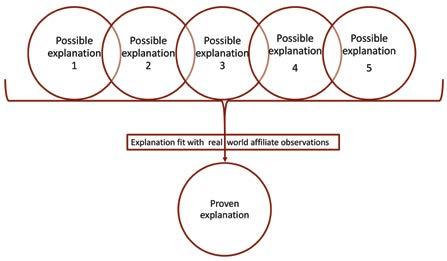
The least common but often most effective way that glocalisers learn is by abduction. This combines elements of induction and deduction, although it is a distinct approach in its own right. Abductive learning works by finding which of several explanations best explain the real world, as shown in figure 4. In my research, I found the Asia-Pacific women’s health company, mentioned above, used abduction to build on their deductive process. They had five putative explanations of why uptake varied between their affiliates in the Asia-Pacific region:
• Cultural beliefs regarding women’s health
• Level of economic development
• Attitudes toward traditional medicine
• Education level of women in the target age group
7 www.pharmafocusasia.com
STRATEGY
Figure 1: How Glocalisers Learn
• Commercial resources in each country
They then evaluated the five possible explanations against the quantitative data, looking for correlations. They complemented this with a qualitative research programme of focus groups, which looked for causal mechanisms. They found that the level of economic development was the dominant influence on uptake but it was significantly moderated by women’s education level, with the other three factors being relatively unimportant. This practical, useful finding strongly shaped their strategy.
As well as needing someone in a regional role who is skilled and commit-
ted to drive it, abductive learning works best when there are multiple competing explanations for the business issue you are interested in, such as uptake, market share or customer attitudes. It fails when a single reason is taken for granted or when firms cannot identify alternative possible explanations for the issue. Abductive learning works best in firms with a pragmatic culture who are not too obsessed with numerical analysis.
Challenge, choice, change and classroom
As always, the inspiration for my glocalisation research was curiosity. Global life
science companies are often very similar in their size, structure and strategy. Yet they differ greatly in how well they translate core, global strategies into local ones and how much value their national affiliates create. As I studied these companies, the difference between astute glocalisers and their naïve peers emerged. As I described in “The Glocalisation Challenge”, they differed in how they targeted, tailored and learned. Astute targeting is the result of clever competitive analysis, super segmentation and prioritising differently between countries, as I described in “The Glocalisation Choice”. Then, in “The Glocalisation Change”, I looked closely into astute tailoring and saw the building of layers of value around contextual market segments. And finally, in this article, I unpicked the ways that astute companies learn from glocalisation. They use a mixture of induction, deduction and abduction, making the global operations a classroom in which they learn to sustain their success.
Despite the complexity of my research and the richness of its findings, there is one salient difference between astute, successful glocalisers and their naïve, less successful rivals. It is their attitude towards strategy in general and glocalisation in particular. The naïve think it is unimportant and treat local affiliates as a little more than local concierges. Astute, exemplary companies see glocalisation of strategy as critical and view effective affiliates as vital to it.

8 PHARMA FOCUS ASIA ISSUE 52 - 2023
AUTHOR BIO
Professor Brian D Smith works at the University of Hertfordshire, UK, and Bocconi University, Italy and researches the evolution of business models and competitive strategy in the global life sciences industry. He has published over 300 papers, articles and books. www.pragmedic.com
Figure 2: Inductive learning
Figure 3: Deductive learning
Figure 4: Abductive learning
STRATEGY
From curiosity
to cure
Curia is a Contract Development and Manufacturing Organization with over 30 years of experience, an integrated network of 29 global sites and over 3,500 employees partnering with customers to make treatments broadly accessible to patients.
Our biologics and small molecule offering spans discovery through commercialization, with integrated regulatory and analytical capabilities.
Our scientific and process experts and state-of-the-art facilities deliver best-in-class experience across drug substance and drug product manufacturing. From curiosity to cure, we deliver every step to accelerate and sustain life-changing therapeutics.
To learn more visit us at curiaglobal.com

© 2023 Curia Global, Inc. All rights reserved.
SM
Shaping the Future of Pharmaceutical Manufacturing
Trends, Technologies and Beyond at ISPE Singapore

International and Regional Bio/Pharma meets online / in-person at Suntec Singapore this month.
Following its biggest gathering post-pandemic, ISPE Singapore Affiliate’s highly anticipated annual meeting reflects the energy and positive growth for the industry in Asia Pacific. Each year, the show provides an extraordinary opportunity for professionals in the bio/pharmaceutical and related industries to gather, learn, and exchange insights on the latest trends and advancements. The Conference runs alongside the sold-out Exhibition, packed with over 90 leading companies offering the spectrum of expertise from aseptic to digital solutions!
Annual Symposium + Main Conference
Now into its 23rd edition, the 3 day event kicks off with an online pre-Conference Symposium on 23 August. This is followed by the Main Conference, held in-person from 24-25 August at Suntec Singapore. True to its mission of ‘Connecting Pharmaceutical Knowledge’, the conference will also be livestreamed and available on-demand for 6 months afterwards.
Under the theme of "Shaping the Future of Pharmaceutical Manufacturing: Trends, Technologies and Beyond," the conference features over 90 distinguished speakers including 12 Keynotes.
10 PHARMA FOCUS ASIA ISSUE 52 - 2023
Spanning global to regional biologics, vaccines and pharmaceutical manufacturers, CDMOs, regulatory authorities and industry leaders, highlights include:
• Latest updates from 8 Regulatory agencies: EMA, PIC/s, WHO, HSA, ANSM, Philippine FDA, DRAP Pakistan & Malaysia’s NPRA
• Case study insights from Bio/pharma manufacturers including Pfizer, Amgen, Merck & Co, MSD, Lonza Biologics, Bayer, LOTTE BIOLOGICS, AstraZeneca, Genentech, WuXi Biologics, Moderna, Takeda, Thermo Fisher, GSK and more.
ISPE Singapore Conference & Exhibition
23-25 August | Online & In-person @ Suntec Singapore

Full programme & registration details: www. ispesingapore.org1
Attendance is by registration and open to ISPE Members and non-Members alike. This year, non-Members benefit from 1 year’s ISPE membership included when they register!
This year’s symposium & programme tracks comprise extensive in-depth coverage on digital transformation / pharma 4.0, digital CQV, regulatory & compliance, quality, advanced manufacturing and ATMPs, Annex 1, CCS, GAMP5, sustainability and supply chain amongst others. Extended panel discussions revolve around hot topics, including innovating for speed-to-market, tackling drug shortages, challenges and opportunities working with CDMOs, mRNA vaccines development and beyond.
Regulators & Industry on Drug Shortages
5, yes 5 Keynote speakers open the online Pre-conference Symposium; from both regulatory agencies and industry. Hear from Pfizer, Amgen & Merck and regulators from ANSM/PIC/s & EMA. Coming together in a live Global Panel discussion, they will address ‘Regulatory, Policy & Industry Approach to Combat Drug Shortages’. Diane L Hustead, Executive Director, Regulatory Affairs, Merck & Co, Inc, & Drug Shortage Initiative Team Chair, ISPE, USA will moderate this online session.
Industry Best Practices
Following the plenary, two concurrent tracks focus on A: Industry Best Practice, sponsored by Fedegari and B: Digital Transformation, sponsored by Lachman Consultants.
Keynote speaker, Dr Joerg Block, GMP Compliance Engineering, Bayer AG Pharmaceuticals,
1 http://www.ispesingapore.org/
Germany will present on ‘Good Engineering Practice: Accelerating Project Delivery‘. Case studies range from HPAPI biomanufacturing, CIP Operation and single-use for mRNA to good engineering practice. Discussion on ‘Applying Good Practices for the Project Life Cycle’ conclude this track.
Visionary Panel: CDMOs, Regulatory & Compliance
ISPE President & CEO, Thomas B Hartman will grace ISPE Singapore Affiliate’s annual conference for the first time this month. He will moderate the Opening VISIONARY PANEL: Biotechnology Industry Challenges and Opportunities Working with Contract Development & Manufacturing Organizations (CDMO’s). Global and regional regulators are well represented, culminating with the in-person ASEAN REGULATORY ROUNDTABLE: How Regional Regulatory Agencies are Adopting Global Standards.
Global & Regional Regulatory Agencies:
Keynote Speakers:
Jacques Morenas, Chair, Training sub-committee, PIC/s & Technical Advisor, Inspection Division, ANSM, France
Andrei Spinei, Manufacturing Team Lead, Inspections Office, Quality and Safety of Medicines Dept, European Medicines Agency, Netherlands
11 www.pharmafocusasia.com 11
Chong Hock Sia, Director (Quality Assurance) & Senior Consultant (Audit & Licensing), Health Products Regulation Group, Health Sciences Authority, Singapore
Vimal Sachdeva, Senior GxP Inspector, WHO, Switzerland
Jesusa Joyce N. Cirunay, Director IV, FDA Center for Drug Regulation and Research (CDRR), Philippine.
Abdul Mughees Muddassir, Assistant Director to CEO, Drug Regulatory Authority of Pakistan (DRAP)
Dr Noraida Mohamad Zainoor, Deputy Director, Centre of Compliance and Quality Control, National Pharmaceutical Regulatory Agency (NPRA), Malaysia
Manufacturing Trends
On 24th August, Keynote speaker Takashi Kaminagayoshi, Head, Biotherapeutics Process Development, Pharmaceutical Sciences, Takeda Pharmaceutical Co Ltd, Japan will expand on ‘Global Engineering Standards for Biopharma Manufacturing’. Additional highlights include: CDMOs outlook, aseptic filling platform technology and a discussion on ‘Innovating to Gain Speed to Market’.
Held in-person, this track in the Main Conference is sponsored by Yokogawa, which hosts a deep-dive ‘Lunch n’ Learn’ workshop on ‘Digital Transformation Toward Smart Manufacturing’ earlier in the day.
Digital Transformation
This year’s call for presentations yielded a majority under Digital Transformation. Unsurprisingly, given the prevalence and influence of technology in both our work and personal lives. Undoubtedly spurred by necessity during the pandemic, it is significantly impacting and expediting this industry into digitalisation and virtual work practices.
Fundamental to this evolution, ISPE’s GAMP® CoP2 (Community of Practice) has been prolific in disseminating pragmatic guidance on the use of computerised systems in regulated industries without which DT could not happen. Instrumental in this arena is Charlie Wakeham, Global Head of Quality and Compliance, Magentus & Co-Chair, ISPE GAMP Global Steering Committee and Lead, ISPE GAMP CSA Special Interest Group. Her online discussion entitled ‘How does GAMP5 Revision Impact the Industry?’ appears in the ‘Digital Transformation’ track. Held online, presentations such as ‘No-Code GMP Apps: Has Paper Finally Met its Match?’, ‘The Nexus: Data Integrity & Pharma
2 https://cop.ispe.org/forums/community-home?CommunityKey=ed72d220-cd9e4ba9-87c2-a4b3623e5ca5
4.0’, and ‘Digitalisation in Predictive Maintenance’ are discussed.
Charlie will be onsite in Singapore for the launch of the GAMP® South Asia CoP. and lead an interactive workshop on ‘Applying Critical Thinking in Computerised Systems Projects’. Key supporting companies, CAI, NNIT, No deviation and PQE will host an onsite GAMP® Quiz in conjunction with recruitment of members to this new Community of Practice.
The importance of Digital CQV is reflected as a track, sponsored by No deviation. Featuring presentations by experts on ‘Transition & Implementation Strategies for Digitising C&Q’, Digital CPV and a discussion on ‘C&Q Digitalisation’.
Hosted by NNIT on 25 August, the Digital Transformation/Pharma 4.0 theme continues with sessions encompassing innovation, advanced therapies, smart manufacturing, digital twins and sustainability. Keynote speakers Richard Lee, Chief Executive Officer and Member of the Board, LOTTE BIOLOGICS, Korea & Meli Gallup, Director, Global MSAT Commercial Product, Genentech, USA precede a Digital Pharma Discussion tackling ‘Challenges to Digital 'Emancipation' (Freedom)’.
Advanced Manufacturing / ATMPs
Rounding off the Main Conference, the Advanced Manufacturing/ATMPs track opens with a Keynote by Dr Jerry Yang, CTO & Head of TechOps, Fosun Kite Biotechnology Co Ltd, China. This is complemented by talks on QdD, multimodal ATMP facilities etc are discussions on ‘Reliability in Continuous Manufacturing’ and ‘Key Strategies in Supply Chain Resiliency’.
View all Speakers & the full Symposium/ Conference Programme on www.ispesingapore.org3
Over the 3 days, spanning from aseptic / sterile, single-use, multi-modal, flexible or continuous manufacturing and more, speakers will share their experience alongside the latest updates and developments.
Whether in-person or virtually, the ISPE Singapore Annual Meeting offers a great opportunity to network with peers, engage in meaningful discussions, and explore innovative solutions in the pharmaceutical landscape.
On-Site Exclusives Open to all: Free Workshops & ‘Power Hour’ Talks
Taking place alongside the main conference and exclusively for all who join in-person, 22 open-access satellite workshops will further facilitate knowledge
3 http://www.ispesingapore.org/
12 PHARMA FOCUS ASIA ISSUE 52 - 2023
sharing among attendees. Each hour-long session provides in-depth coverage on a wide range of topics, complementing and adding to the conference. Each led by industry subject matter experts, workshops are open to both conference attendees and visitors.
Concurrently, this year’s expanded exhibition showcases the largest gathering of leading product, technology and service providers. Visitors may also attend the ‘Power Hour’ programme of 32 talks held daily at participating exhibitors’ stands. The schedule of workshops, Power Hour talks and associated onsite events can be viewed on the event site www.ispesingapore.org4
Outreach & Networking Opportunities
ISE’s community of volunteers reaches across manufacturing industry professionals, with student engagement to executive mentoring programmes. The Singapore affiliate encourages students and Emerging Leaders locally and from the region, with the Hackathon competition held online, resulting onsite with final presentations. Results are announced during Pharmanite on 24th August.
Women in Pharma are well represented, gathering for a Roundtable Lunch discussion on multiple topics, each table featuring an inspiring host. This takes place on 25th August, with the Emerging Leaders Networking reception providing an informal celebration to conclude the event.
Pre-event Commissioning & Qualification Training & Site Tours
ISPE Commissioning and Qualification Training Course – 22 & 23 August 2023
For the first time in this region, ISPE Global training offers participants an accessible and affordable onsite course on:
Science and Risk-based Commissioning and Qualification - Applying the ISPE Baseline Guide, Volume-5, Second Edition: Commissioning and Qualification (T40)
Worldwide Regulatory expectations and guidance as led by FDA and the EU have stated that all Pharmaceutical Quality Systems should apply a QRM (Quality Risk Management) approach. Through interactive workshops, this course will explain and apply the science and risk-based approach to integrated lifecycle Commissioning & Qualification by conducting verification of systems, equipment and facilities in accordance with the
recently issued 2nd Edition Guide, ICH documents Q8 (R2), Q9, and Q10, current Regulatory Guidance, industry best practices, and ASTM E2500.
Registration & details can be found at www.ispesingapore.org to Schneider Electric Singapore’s Innovation Hub & Merck’s M Lab™ Collaboration Center provide a great opportunity to view state-of-the-art facilities and experience technology hands-on.
Last year’s ISPE Singapore affiliate Conference & Exhibition drew a record 1,300+ participants, with returning regional delegates and visitors expected to boost this year’s attendance.
With a packed schedule of events lined up, the stage is set to maximise learning and collaboration among regulatory agencies, manufacturers, and technology and service providers. The committee appreciates the support from the community and looks forward to welcoming the industry to Singapore this August!
Exclusive Offer for readers: Register for the Symposium/Conference with discount code PHARM10 to save an extra 10% off prevailing rates.
ABOUT ISPE
The International Society for Pharmaceutical Engineering is the world’s largest not-for-profit association serving its Members by leading scientific, technical and regulatory advancement throughout the entire pharmaceutical lifecycle. www.ispe.org
ISPE Singapore Affiliate since 2000 has over 300 Active Members and over 3,000 supporters predominantly in Singapore and the region.

13 www.pharmafocusasia.com 13
4 http://www.ispesingapore.org/
Advertorial
Accelerating Cell and Gene Therapy Treatments to Patients with the Right CDMO Outsourcing Model
As global supply chains continue to restabilise, innovator companies must carefully manage resources to reach key milestones. Traditional CDMO models with booking, reservation, and other fees devour critical development runway. The new generation of CDMO offers flexible terms, transparent access, and collaboration to ensure success without compromising timelines or budgets.
therapies. In April, Breyanzi (lisocabtagene maraleucel) by Juno Therapeutics, Inc., a Bristol-Myers Squibb Company, obtained approval in the EU. Novartis's Kymriah and Breyanzi also achieved approvals for additional indications, further expanding the reach and potential of these therapies.
The global need for cell & gene therapy CDMOs
The promise of cell and gene therapies

Approximately 400 million people worldwide are living with a rare disease, and about half of those affected by rare diseases are children. Cell and gene therapies have the potential to revolutionise the treatment of many diseases, including rare genetic disorders and cancer.
In July of last year, PTC Therapeutics achieved a significant milestone when the European Commission (EC) granted authorisation for Upstaza. This treatment marks the first-ever approved therapy for aromatic L-amino acid decarboxylase (AADC) deficiency, a rare genetic disorder affecting the nervous system. Following this, BioMarin received marketing authorisation from the EC in August for Roctavian, the world's first gene therapy developed to treat Hemophilia A, a hereditary bleeding disorder. In the same month, the FDA approved betibeglogene
autotemcel (beti-cel; Zynteglo) by bluebird bio for the treatment of beta thalassemia in both adults and children. This therapy had previously obtained approval in the EU back in 2019. Another notable approval came in September when the FDA granted its authorisation for elivaldogene autotemcel (eli-cel; Lenti-D), also developed by bluebird bio. This therapy offers a solution for early and active cerebral adrenoleukodystrophy (CALD) patients without an HLA-matched donor. Lastly, in November, the FDA approved a Hemophilia B treatment developed through the collaboration of uniQure and CSL Behring.
In the realm of chimeric antigen receptor (CAR) T-cell therapies, two notable approvals took place in 2022. In February, Carvykti (ciltacabtagene autoleucel) by Legend Biotech and Janssen received approval from the FDA, marking a significant milestone in CAR-T
Despite the clinical promise, getting these treatments to patients quickly and efficiently is a significant challenge. Bringing cell and gene therapies to market is a complex process that requires a significant investment of time and resources from both a developmental and manufacturing perspective.
The clinical development process for these treatments involves several phases of testing, including preclinical studies, phase 1 safety studies, and larger phase 2 and phase 3 studies to evaluate efficacy and safety in larger patient populations. The process of clinical development can take many years and require significant investment. For diseases with small patient populations with no other treatment options (requiring production of small batches), incentives are often not aligned for large pharmaceutical companies and mediumsized biotech companies to invest in.
Developing cell and gene therapies also requires manufacturing processes that can be scaled up to meet the needs of patients. This can be a challenge, particularly for therapies that involve complex manufacturing processes or require specialised facilities.
14 PHARMA FOCUS ASIA ISSUE 52 - 2023
Andy Whitmoyer, VP Supply Chain, Center for Breakthrough Medicines Emily Moran, SVP, Vector Manufacturing, Center for Breakthrough Medicines
STRATEGY

Additionally, the global supply chain for cell and gene therapies can be complex, as starting materials and other components may need to be sourced from multiple locations around the world. While bottlenecks from pandemic lockdowns have improved since 2021, certain equipment and consumables used in cell and gene therapies continue to have long lead times and unreliable availability.
Contract development and manufacturing organisations (CDMOs) play a crucial role in accelerating the availability of cell and gene therapies to patients by absorbing and mitigating risk, simplifying supply chains with starting material on-hand, and accelerating development with processes and platforms.
Limitations with traditional CDMO models
However, traditional CDMO models charge fees that can consume critical development runway and negatively impact the financial resources of innovator companies. While fees for traditional CDMO services can vary widely, they can be a significant financial burden for innovator companies. Common charges have included suite reservations and holding, booking, project initiation, tech transfer, onsite accommodation, and programme management. These fees can consume critical development runway and negatively impact the financial resources of innovator companies, which can delay the development of cell and gene therapies.
Another common occurrence as the cell and gene industry evolved was the need to work with multiple providers of steps within the process. For example, an innovator company may need to work with one CDMO for plasmid manufacturing, another for vector manufacturing, and yet another for release testing and analytics. This has led to common challenges in:
Coordination and communication: One of the main challenges of working with multiple CDMOs is coordinating and communicating effectively between the different organisations, in addition to overall vendor management. It can be
challenging to ensure that each CDMO is aware of the project’s overall goals and timelines, and that all parties are working together to achieve these goals. This can be particularly challenging when multiple CDMOs are located in different countries or time zones, making it difficult to coordinate schedules and communication.
Quality control: Another challenge with using multiple CDMOs is ensuring consistent quality control across all aspects of the development and manufacturing process. Different CDMOs may have different quality control standards and procedures, making it challenging to maintain consistent quality throughout the process. This can be particularly problematic when there are handoffs between different CDMOs during the manufacturing process.
Intellectual property & licensing: Another challenge with using multiple CDMOs is protecting intellectual property. Developing cell and gene therapies often involves proprietary technology and processes, and it can be challenging to protect this information when working with multiple CDMOs. There may be concerns around confidentiality and the risk of intellectual property theft, which can hamper coordinating work with multiple CDMOs. Furthermore, development using standard materials e.g., (off-the-shelf plasmid or gene editing nuclease) may surprise an innovator company when the CDMO provider is litigated in court.
A new generation of single-source, flexible CDMO
To address these challenges, a new generation of CDMOs has emerged. These CDMOs offer flexible solutions—both in terms of contracting as well as facilities, equipment, and platforms. As early stage clinical and scale-up clients face capital headwinds, models being tried include: Modular contracting: Allows innovator companies to select specific services from a menu. For example, a CDMO might offer separate modules for process development, analytical development, and manufacturing, and innovator companies
can choose some or all of the modules they need based on their in-house talent. Pay-for-performance: Under this approach, payment is contingent on specific performance metrics, such as meeting project milestones e.g., (vial thaw, successful batch) or achieving specific quality standards such as yield. This approach can help incentivise the CDMO to perform at the highest level possible and can help mitigate risk for the innovator company.
Flexible pricing: Some CDMOs are offering more flexible pricing models to meet the changing needs of innovator companies. For example, a CDMO might offer a fixed-price contract for a specific scope of work, or a cost-plus pricing model where the CDMO is paid for the actual costs incurred plus a percentage markup. Increasingly, as programs are rationalised and at the mercy of clinical data readouts, recent contracts have offered parking lots and offramps for innovators to manage their pipeline.
In terms of facility design, fit-forpurpose cell therapy CDMOs have re-engineered and decoupled unit operations to eliminate process and operator bottlenecks. As an example, early-generation autologous cell therapy suites manufacture a patient dose in a singular Grade B environment with operators responsible for the entire process. This requires both time-intensive gowning for current staff as well as lengthy training and focused retention efforts. These headwinds lead to both lower throughput due to nonprocess time and space utilisation, as well as risk to output from attrition.
In next-generation suites, process steps with that can be contained (cell selection, activation, transduction, expansion, and harvest) are performed in closed Grade C space while those that require access to the (formulation, filling) are performed in stricter Grade B to enable quick movement of personnel materials across the GMP facility as operators do not have to gown/de-gown as often as in Grade C as they would in a Grade B environment. This enables seamless processing steps to handle higher throughput.
16 PHARMA FOCUS ASIA ISSUE 52 - 2023
STRATEGY
For vector manufacturing, process steps are broken down into seed, upstream processing, and downstream processing. The seed and upstream processing steps are performed in a contained area to ensure high levels of purity, while downstream processing steps that require access to the open environment are performed in a less strict environment. Additionally, this approach enables the manufacturing team to become highly specialised in their respective roles making it more conducive to larger batches and pooling strategies with multiple upstream bulks to one downstream run. Efficiency and batch success metrics are optimised, leading to lower COGS per dose of vectorbased therapy.
Collaboration in the new generation of CDMO
In light of increasingly complex therapy development, the pace at which cell and gene therapies on accelerated approval pathways speed through clinical trials, and patients with no other choice waiting for therapies, tighter collaboration between innovators and CDMOs is key. A more collaborative relationship enables speed and increases chances of first-time success as sponsor companies share more information and expertise with the CDMO, and the CDMO takes a more active role in risk mitigation and resolution beyond simply executing an initial scope of work.
At the Center for Breakthrough Medicines (CBM), we enable tighter collaboration through programmes such as Partner-in-Plant (PIP) and digitallyenabled suites and infrastructure. Unlike audit-only models, PIP allows a sponsor company access to their product with office space and access to manufacturing suites (with appropriate GMP training) at CBM. Sponsor company scientists can work hand-in-hand in our process development labs to work through parameters that influence batch success.
For partners that are unable or choose not to be onsite, CBM’s digitally native facilities enable access globally 24/7. Live 360-degree cameras are thoughtfully located in suites to capture a complete
view of the production environment. The cameras capture video footage of the manufacturing process from all angles, providing a comprehensive view of the production line. This technology can be used for process improvement and employee training. By having a real-time view of the manufacturing process, operators can identify bottlenecks, optimise workflows, and improve production efficiency.
Sensor-enabled equipment can also provide real-time visibility into a manufacturing process. These sensors collect data on temperature, pressure, flow rates, and other critical parameters. The data collected by these sensors can be analysed to predict and mitigate risk and manage deviations, improving overall product quality and reducing waste.
Managing resources through change

As cell and gene therapies continue to evolve and mature in an uncertain global macroeconomic environment, the new CDMO model of flexible collaboration and access is critical to managing scarce resources and constrained timelines to deliver product globally to the patients that need them.
CBM provides faster turnaround times for client projects by having all development, testing, and manufacturing capabilities connected digitally on shared platforms and physically on one campus, and by having invested working capital in consumable materials and critical supplies for cell and gene therapies. This approach reduces the risk of supply chain disruptions and improves overall supply chain resilience. Our proactive inventory strategy has enabled the company to establish strong relationships with strategic suppliers and secure favourable pricing agreements. By anticipating client needs for consumable materials and supplies and investing our working capital accordingly, the company can quickly respond to client requests thereby accelerating the path to product manufacturing. This approach has helped clients focus their time and energy on value-added activities
aimed at delivering advanced therapies to patients rather than being consumed with the complexities associated with building bespoke supply chains in today’s highly complex global sourcing environment.
To discuss how to collaborate with CBM on an ideal outsourcing model or supply chain strategy, reach out to expert@cbm.com
Andy has over 20 years of experience in industries such as Pharma / Consumer Healthcare, Fast-Moving Consumer Goods, and Agricultural Chemicals across North America, South America, and Europe. His professional background includes progressive leadership roles within Supply Chain Management, Production Operations, and General Operations Management of strategic cGMP supply centers. Prior to joining The Center for Breakthrough Medicines Andy led a 500 FTE supply center with annual net sales over 300 million Euro as the Head of Consumer Health and Pharma Product Supply Argentina within Bayer AG. Andy’s educational background includes a B.S. in Supply Chain Management from the Pennsylvania State University and an MBA from the University of North Carolina at Chapel Hill.
Emily Moran is Senior Vice President of Vector Manufacturing at Center for Breakthrough Medicines. She is an experienced leader in cell and gene therapy and biologics manufacturing with a focus on commercial readiness, industrialisation, and manufacturing stabilisation. She most recently served as Head of Viral Vector Manufacturing at Lonza in Houston Texas.

17 www.pharmafocusasia.com
Andy Whitmoyer, VP of Supply Chain at CBM
AUTHOR BIO STRATEGY
Securing Long-term Success

What to look for in a cell culture media supplier
Minimising costs and lead times, while also maximising supply security, is essential when choosing a cell culture media supplier; however, this must be balanced with robust quality standards, optimal consistency, and experienced support. This article outlines the key capabilities that developers should look for in a supplier to help achieve long-term manufacturing success.
Cell culture media is a significant driver of productivity within biopharmaceutical manufacturing processes. As it provides the essential components for cell growth and function, finding a suitable media formulation is a
critical step during process development, and fundamental to achieving high titres and optimal product quality.
During media selection, there are two main choices for developers to consider: an off-the-shelf, catalogue product or a
custom solution—either a customised catalogue product or a fully proprietary formulation. Although many media manufacturing considerations will be similar, if a custom solution is chosen, developers need to pay specific attention to considering how it will be manufactured at a commercial scale.
This can require finding a suitable external media manufacturer, as well as possibly qualifying a secondary or even tertiary supplier to proactively prevent any potential disruptions in supply. There are several options that developers utilising a custom solution can choose from—ranging from smaller, local suppliers to larger, global vendors with multiple manufacturing sites around the world.
When making this decision there are several factors that need to be considered—in particular, cost and lead times. While it is desirable to minimise these factors where possible, it is essential to do so without compromising on quality and consistency standards, supply chain reliability, and even provisions for the protection of intellectual property (IP). Choosing a supplier that has the
18 PHARMA FOCUS ASIA ISSUE 52 - 2023
Chad Schwartz, Senior Manager, Global Product Management, Thermo Fisher Scientific
STRATEGY
experience and capabilities to efficiently resolve any media-related challenges can also help facilitate long-term manufacturing success.
To determine if a supplier can provide the required support and help minimise risks, there are several key areas to assess, including specific capabilities, infrastructure, and processes. By choosing the right supplier, developers can establish a dependable media supply, supporting them to scale up with confidence and mitigate the risk of costly delays arising in the future.
Starting with the basics
While there are many areas to evaluate during the selection process, there are a few that are pivotal to long-term success. In particular, ensuring regulatory compliance and safeguarding IP should be primary considerations for all developers.
Adhering to regulatory requirements is not only crucial to protect patient safety, but also to avoid incurring costly fines for non-compliance. Consequently, developers should prioritise choosing a supplier that has accredited manufacturing facilities and can provide documentation to validate compliance. When choosing a global supplier, it is particularly important for developers to verify that the supplier can meet the relevant regulatory requirements of the countries they are operating in.
IP protection should be another fundamental consideration. Proprietary formulations and other key process and product details are valuable information and keeping them confidential is essential for developers to protect their innovations and achieve their commercial goals. As a result, a supplier’s data security infrastructure should be a key topic of discussion during the selection and qualification process. This should include a dedicated cybersecurity programme and associated policies designed to mitigate data risks and respond to any threats.
Maintaining quality and lot-to-lot consistency, now and into the future
Given the foundational role of cell culture media in biopharmaceutical production, maintaining quality and consistency is vital—particularly as production volumes increase.
To support seamless scale-up, it is paramount that a supplier has the necessary infrastructure, facilities, and procedures in place to help ensure quality and consistency are prioritised at all scales. Without these systems, inconsistency can lead to several challenges for developers, including reduced productivity, wasted resources, and manufacturing delays. Although many different factors can give rise to media and process variability, by carefully evaluating four key areas, the risks can be greatly reduced, and challenges more quickly resolved.
1) Raw material sourcing and characterisation
Cell culture media formulations consist of a wide range of components that are sourced from suppliers around the world. As a result, maintaining the overall quality and consistency of media requires ensuring the quality of every raw material used. In particular, identifying the presence of trace element impurities should be prioritised. As key drivers of cell metabolism, even small amounts of trace elements—such as copper and manganese—can have detrimental effects on overall process
productivity by impacting critical protein quality attributes. To minimise this risk, developers should look for a manufacturer that has an in-depth procurement programme and only sources raw materials from reputable and strictly qualified suppliers.
Once a supply of high-quality raw materials is secured, media suppliers should also have rigorous internal testing processes in place to help maintain quality standards. This includes measures such as accurately assessing all incoming raw materials. The use of techniques such as inductively coupled plasma mass spectrometry (ICP-MS) can help vendors identify and quantify any potential raw material impurities. In tandem with this, digital inventory management systems can be used to quickly highlight any variability. This can help enable the supplier’s quality teams to rapidly respond and prevent out-of-specification raw material lots from being introduced into the media manufacturing process.
Alongside routine raw material testing, analytical technology can also be leveraged to help optimise the media manufacturing process. Specifically, some suppliers will be able to offer services to help developers understand which trace elements their process or cell line is most sensitive to, and the optimal concentrations of these elements. Using these insights, developers can work with the supplier to implement proactive steps, such as dedicated raw material screening protocols, to control variability, lowering the risk of inconsistency and optimising productivity.
2) Contamination mitigation measures
In addition to raw material impurities, contaminants can also be introduced into media during the manufacturing process itself. These unexpected contaminants are particularly critical to monitor as, in addition to causing process variability, they can also present potential safety concerns for the final biopharmaceutical product.
19 www.pharmafocusasia.com
STRATEGY
Diligent testing and sterilisation are especially crucial process steps that can help reduce the risk of variabilitycausing contamination being introduced.
One potential source that should be considered is the contamination of animal origin-free (AOF) products with animal origin (AO) material. To reduce this risk, any sites that are manufacturing both AO and AOF products should have dedicated mitigation protocols in place. These should include measures such as maintaining a unidirectional flow of raw materials, material airlocks, integrated operating systems for realtime monitoring, and dedicated QC labs with separate material and waste flows.
Outside of AO/AOF cross-contamination, microbial testing is also central to controlling cell culture media quality—in particular, bioburden and endotoxin management. This is an area where choosing an established supplier can be advantageous, as it can leverage its expertise to detect and minimise endotoxin and bioburden risks. Developers should look for a supplier that can demonstrate that all raw materials are validated to their appropriate specifications and that the equipment utilised for production has been subjected to the necessary procedures. Diligent testing and sterilisation are especially crucial process steps that can help reduce the risk of variability-causing contamination being introduced.
3) Site-to-site equivalency
Alongside carefully evaluating a supplier’s quality systems and manufacturing protocols, considering the level of supply assurance it can provide is also essential. To reduce the risk of supply chain challenges and any associated costly production delays, it can be beneficial to choose a supplier with a multi-site network that has built-in manufacturing redundancy. However, it is key that all processes across its locations are harmonised to enable equivalent products to be manufactured at each site. Site harmonisation encompasses the entire production workflow, so there are several areas where equivalency should be implemented.
Within a harmonised site network, raw material management is key. As
previously mentioned, inconsistent raw materials can have significant effects on media performance. To mitigate this risk, suppliers should implement equivalency in their raw material processes, harmonising both sourcing specifications and supplier review protocols.
Process and equipment equivalency is another area developers should verify with the supplier. In particular, suppliers should be able to demonstrate that manufacturing steps are mirrored, and that all manufacturing equipment can deliver comparable performance and is validated to the same standards. Quality assurance, control frameworks, and operating infrastructure should also be harmonised to help verify the same standards are achieved across locations. This includes utilising identical testing methods, equipment, cell lines, and specifications at each site.
4) Analytical capabilities
Outside of media manufacturing, considering the overall cell culture knowledge and analytical capabilities of the supplier can also be advantageous. When issues such as batch-to-batch inconsistency arise during production it can often be difficult to identify the exact causes. However, an experienced vendor will often have the knowledge and the tools to carry out detailed investigations to help resolve any media-related challenges as efficiently as possible.
Support through manufacturing stability studies can also help give developers confidence in their formulation. Real-time stability testing of cGMPmanufactured media in the desired packaging configuration should be offered as a customisable service conducted under conditions and time points determined by the developer.
In addition to stability studies, an experienced media manufacturer may also be able to offer in-depth investigative analytical services to help developers resolve more complex challenges such as precipitates, extractables, and leachables. Using the outcomes of these investigations, developers can take steps to reduce
the risk of similar challenges arising in the future—for example, by modifying their formulation or optimising media storage.
Mitigating risks by choosing a knowledgeable and experienced supplier
From regulatory compliance and IP protection to raw material sourcing and site-to-site harmonisation, there are many considerations developers need to take into account when selecting a media manufacturer. By working with the supplier to carefully evaluate and validate each of these areas, developers can determine if the supplier can meet their needs and begin the process of building a long-term collaborative relationship.
Although the evaluation process can require an initial investment of time and money, this can often be offset by the considerable benefits of working with a dependable media manufacturer. Through supplying high-quality and consistent media, alongside dedicated and responsive support, an experienced supplier can help developers streamline scale-up and accelerate their speed to market. Moreover, using its knowledge and capabilities, the supplier can also help quickly resolve any challenges that arise during manufacturing, helping developers maintain commercial success into the future.
After completion of his PhD at the University of Kentucky and a postdoctoral appointment at UC Davis, Chad has had professional stints across the bioprocessing industry. As a Senior Product Manager at Thermo Fisher Scientific, Chad is responsible for the Gibco™ Efficient-Pro™ medium and feed system and the customer-owned formulation product line.

20 PHARMA FOCUS ASIA ISSUE 52 - 2023
STRATEGY
AUTHOR BIO








































Adopting the Power of AI to Drug Development Projects


The top challenge today for life sciences and healthcare organisations is effectively extracting and operationalising information from complex sources for decision-making. Certara’s deep-learning platform and modelbased meta-analysis services offer integrated solutions for researchers seeking to enhance their R&D programs by leveraging all available clinical trial data sources.
Matt Zierhut, Vice President, Integrated Drug Development, Certara
 Nick Brown, Associate Director of Marketing, Certara
Nick Brown, Associate Director of Marketing, Certara
1. How can artificial intelligence (AI) and deep learning help in integrating and analysing data in clinical trials? Can you share any examples?
NICK: A tremendous amount of documentation is created throughout the clinical trial lifecycle. These documents include recruitment forms, trial summaries, scientific publications, clinical study reports, drug labels and post-marketing materials; all of which hold valuable data points that can influence future trials.
The challenge with these documents is that it’s incredibly time consuming to review and extract relevant data points from this content. That’s where AI comes in.
22 PHARMA FOCUS ASIA ISSUE 52 - 2023
AI in the form of generative pre-trained transformers (GPTs) and large language models (LLMs) are uniquely adept at searching and “understanding” complex content.
By applying these AI models to clinical trial documents, researchers can accelerate screening and data extraction workflows enabling them to collect highly-relevant information in a format that complements their analytics needs. For example, at Certara.AI we have a dataset creation tool that researchers use to take large corpuses of clinical trial documents and leverage AI to extract the relevant data points they need directly into a structured format. These newly structured datasets can then be used for expanding clinical outcomes databases or fed into visualisation tools to run a variety of different analyses.
2. How does deep learning benefit small compound analysis and generating new drugs? Can you highlight any advancements or breakthroughs in this area?
NICK: Just as LLMs and GPTs can be trained to “understand” language and then leveraged to analyse documents, they can also be trained on SMILES and SELFIES strings. SMILES and SELFIES are sequences of numbers and letters that represent molecules and in simple terms can be considered the “language of compounds.”
Models trained on SMILES or SELFIES can perform exciting tasks such as property prediction and de novo compound generation which complement the existing analytics workflows of chemists and discovery scientists. For example, the Certara.AI team is collaborating closely with our
colleagues in medicinal chemistry, to develop these models. To date we’ve successfully deployed models to help users predict the toxicity, blood brain barrier permeability and lipophilicity of structures which can enhance which molecular properties to prioritise. In addition, we also have made tremendous progress in models for de novo compound generation which will deliver highly-relevant structures to complement existing discovery workflows.
3. What factors should organisations consider when selecting a deep-learning platform for R&D in life sciences and healthcare?
NICK: Flexibility, data access and model training are all factors organisations should consider when selecting a deep learning platform for R&D in life sciences. The AI landscape is rapidly evolving, so having a platform with the flexibility to handle multiple types of models will enable your team to shift as new innovative models hit the market. Secondly, data access will always be a challenge in AI. A platform that allows you to securely connect your internal data and apply AI models onto those assets enables you to create an expansive environment for leveraging AI across multiple data types and teams within your organisation. Last, but certainly not least, is the base training of the AI models. In many cases, AI models are trained on broad datasets that enable an expansive, but top-level understanding of concepts. At Certara.AI, we focus specifically on developing models that are trained specifically on life sciences content. This enables our customers to leverage these models with confidence knowing that they understand the unique complexities of the life sciences industry.

4. What is Model-Based Meta-Analysis (MBMA), and how does it impact drug development? Can you provide examples of how it optimises clinical trial outcomes and informs regulatory decisions?
MATT: Model-based meta-analysis utilises study results at the summary-level (aggregate data) to gain a deeper understanding of the landscape of both available treatments and treatments currently in development for any given indication or therapeutic area. MBMA is a broad term that can encompass almost all types of meta-analyses. These meta-analyses can range from simple pairwise meta-analyses of two treatments based on multiple similarly-designed studies (testing the same compounds), to network meta-analyses that compare multiple treatments using connected networks of studies (allowing for indirect comparisons of treatments without direct comparison data), and to full model-based meta-analyses that can add complex model structures to account for variability from dose-ranging data, longitudinal data, and data from studies with different populations or designs. MBMA can provide detailed context around any drug development program by enabling an appropriate comparison across all relevant trials, thus providing insight into the likelihood any drug in development would be able to successfully compete with other treatment options. This insight can be appreciated by all parties involved: sponsors, regulatory bodies, patients, and others.
23 www.pharmafocusasia.com
5. How does MBMA help understand covariates in clinical trials? What methodologies are employed to unravel these factors and their implications for decision-making?
MATT: MBMA uses modeling techniques that are common to the fields of pharmacometrics and biostatistics to help explain variability in trial results that may be due to a variety of sources and factors. Usually, covariate effects are explored and described using additive, proportional, and power/ exponential terms, but more complex functions can also be used. For example, to capture the influence of dose on outcome, a sigmoidal or Emax function can be applied. Exponential decay is also commonly used to explain changes in outcomes over time. Additionally, MBMA can capture the influence of both prognostic and predictive covariates, a difference that is critical to development decisions. Prognostic covariates are factors that influence trial outcomes at the general treatment arm level, not relative to any other treatments. Prognostic covariates can be thought of as covariates that effect populations receiving placebo, or control treatment, or they can help explain differences in disease progression. Predictive covariates, on the other hand, help to explain differences in relative treatment effects, and relative effects are ultimately what involved parties are interested in. The key question being: does this drug elicit a larger benefit than the control or competitor treatment?
6. What limitations and considerations should researchers and stakeholders keep in mind
when applying MBMA and AI-powered analytics? Are there potential biases, challenges in data interpretation, or regulatory considerations?
NICK: Data quality is one of the top considerations to keep in mind. MBMA requires highly-specific datasets to ensure accuracy for any statistical analyses taking place. As discussed earlier, AI can accelerate the creation of these datasets. However, it’s critical that validation workflows are in place to ensure AI predictions are accurate. This is a key focus for us at Certara. AI. We’ve developed human-in-theloop validation capabilities that enable users to review AI predictions and adjudicate their accuracy and relevance before the dataset is finalised and used by MBMA experts.
7. What are the common challenges in applying MBMA to gain insights into disease, trial characteristics, and investigational drugs? How does Certara overcome these challenges and develop innovative solutions?

MATT: Each MBMA project presents its own unique challenges; however, the most common challenges typically involve data availability. This could be high-level data availability like the number of trials that have published results in the indication of interest. MBMA in rare diseases is not frequently done due to the typically
low number of drugs in development for a specific rare disease, although MBMA has been utilised in multiple rare disease cases – again it depends on the unique case and questions being asked. Other data challenges can also exist in more popular indications that have many published trial results to work with. Trials in these indications may not always publish the same details around population or trial characteristics. If these missing characteristics are potentially influential to trial outcomes, analysts would have to choose between excluding these trials or imputing the missing data. The most common solution is data imputation, thus keeping trials with other potentially relevant information. However, sensitivity analyses are always conducted to ascertain the potential influence of data imputation techniques. Other data challenges can
24 PHARMA FOCUS ASIA ISSUE 52 - 2023
exist in how published results define important variables and even endpoints. If a common definition is not used across all studies, the definition may be a significant covariate itself. Finally, there are general challenges in transferring data from external (and internal) sources to an analysis-ready database, typically due to human error. These variables make it incredibly challenging to effectively collect data through manual efforts. Fortunately, advances in AI are a key solution. At Certara, we leverage advanced large language models trained on life science concepts to help our teams extract data from unstructured documents, identify semantically relevant content that is at risk of being overlooked and deliver results in a structured spreadsheet. Automating this first step with AI allows our team of expert curators to focus on data quality. With a low code AI validation tool, the team can more efficiently conduct QC workflows enabling them to be more productive while mitigating some of the tedious tasks that often lead to human error.
8. What are the benefits of conducting a meta-analysis by systematically gathering data from maintained databases? How does this approach enhance reliability and aid evidence-based decision-making in drug development?
MATT:As any modeller would (or should) tell you: garbage in, garbage out. The benefits of using systematically gathered data can be directly observed in the quality of the model output. Additionally, by maintaining these systematically developed databases, one can quickly jump into any
new analysis within an indication that has an already existing database. If you can trust that existing databases accurately portray all the relevant and available information, you can not only quickly initiate work to inform critical decisions, but you can also be confident that those decisions were properly informed. The highest priority of the Data Science team at Certara is to develop and maintain accurate databases using a systematic review and quality control process.
9. How does Certara's advanced deep-learning platform benefit R&D programmes in life sciences and healthcare? Can you share success stories or use cases?
NICK: Certara’s AI analytics platform helps life science R&D teams solve two key challenges — provide a solution for applying LLMs and GPTs to structured and unstructured files and make those multiple data sources searchable and accessible in a single platform. This combination improves collaboration and accelerates insight discovery that can inform a number of go/no-go decisions across the drug discovery pipeline.
As previously mentioned, so much of the data needed for R&D efforts resides in literature-based documents. The Certara AI platform's strength in analysing and bringing new value to this content enables it to be leveraged in a number of use cases. For example, using LLMs to accelerate large scale systematic literature reviews that are used to inform trial design and development, GPTs assist in the creation of regulatory documents and concept tagging to improve insight discovery and data standardisation.
10. How have AI-powered analytics improved decision-making in drug development through model-based metaanalysis?
MATT: Effective MBMA starts with high quality data. The area that we see the greatest promise in AI complementing MBMA is in the identification and collection of relevant data needed to effectively conduct analyses. As mentioned earlier in this interview, much of the data we need to understand a clinical landscape comes from unstructured documents. AI’s ability to “comprehend” this content enables us to effectively identify relevant insights, and curate more accurate datasets that can improve our understanding of the given area we’re studying.
11. What motivated Certara to incorporate deep learning expertise? How has this collaboration benefited Certara's clients and drug development initiatives?
Can you share specific success stories?
NICK: AI holds tremendous promise in life sciences and drug discovery. Certara AI's focus area of AI development complements Certara’s product portfolio. By integrating this technology into the Certara product portfolio, we’re able to add new predictive and generative AI capabilities to drug discovery workflows that arm our clients with the ability to easily access insights that can improve go/no go decisions.
25 www.pharmafocusasia.com
12. What tools and services does Certara offer to help life science companies analyse and interpret data for drug development decisions? Can you provide examples of their impact on decisionmaking?
MATT: Certara offers many more tools and services than what we have discussed today. We are a full-service company that can improve drug development at all stages. To demonstrate our size and success, in each of the last nine years, 90 percent of new drug approvals by the U.S. Food and Drug Administration’s (FDA) Center for Drug Evaluation and Review (CDER) were received by Certara’s customers.
Focusing on Certara AI, Data Sciences, and MBMA consulting services, we offer essential tools to identify, curate, and analyse both public and proprietary data sources. Our recently developed and newly improved software is available to provide all available relevant information to the decision maker. This information could come in the form of a comprehensive database curated from multiple sources, or it could
come in the form of simulations based on a model that was developed using the comprehensive database. Certara provides the tools and the recommendations to make better decisions.
13. How does Certara handle the limited sharing of individual-level data from in-house trials? How does it
MATT: Successful drug development depends on making wise decisions about portfolios, clinical trials, marketing, etc. We are continuously faced with the challenge of deciding whether to continue development or stop it. To support those decisions, we gather data, typically through clinical trials. We analyse the data from those clinical trials, and then we use these analyses to build models that we then use to predict what may happen in the next trial. The data collected from these in-house trials are “internal data” or “proprietary data.” Companies rarely share individual level data. But, they all publish most of their aggregate level trial results.
Before publishing results from an internal study, sponsors are in a unique position where they have access to all published competitor data, and are the only ones with access to their own proprietary data. Certara utilises MBMA to put this proprietary data into the proper context of the published summary-level trial results, thus enabling the sponsor to make critical decisions before others have seen their new trial results. By fully understanding the landscape, now including their drug, they are able to make fully-informed decisions about the best next steps for their compound, whether it’s advancing to the next stage or shifting resources to a different compound, with a better predicted probability of success.
14. How does the integration of AI in Certara's CODEx platform enhance MBMA capabilities? In what ways does AI empower the analysis of complex datasets and enable informed decision-making in drug development?
Dr. Matt Zierhut advances the integration of external aggregate clinical trial data into development decisions and commercial and regulatory strategy via modelbased meta-analysis (MBMA) at Certara. Matt works closely with clinical development teams to ensure MBMA is leveraged for optimal impact when making the most critical decisions. Previously, Matt was at Janssen (J & J) where over the past 5+ years he has led the development of their global MBMA capability and has worked together with many clinical development teams to ensure optimal impact of MBMA into the decision making in their programs.
https://www.certara.com/teams/matthew-zierhut/
Nick Brown is associate director of marketing at Certara AI. He manages customer and partner communication and engagement for the Layar platform and AI integrations across the Certara product portfolio. Nick has over 10 years of experience in AI marketing where he focuses on educating a variety of audiences on complex technology across deep learning, AI and big data.


https://www.certara.com/teams/ nick-brown/
NICK & MATT: High-quality clinical outcomes data is at the core of the CODEX platform. By adding AI into CODEX, we’re able to provide our customers with an intuitive solution for custom dataset curation that mixes our CODEX indication databases with other relevant data sources, including customer’s proprietary data. As a result, our customers can dynamically expand the datasets they’re analysing to quickly provide them greater insights and results that impact their most critical drug development decisions.
26 PHARMA FOCUS ASIA ISSUE 52 - 2023
leverage internal data to enhance research and development capabilities?
AUTHOR BIO
Emerging Tools Shaping Drug Discovery and Development Landscape

Current advancements in bioengineering, AI, supercomputing, and ML are reshaping the landscape of the pharmaceutical industry. They are anticipated to make an even greater impact and draw a new vision to guide pharmaceutical research and innovation in the coming years.
Qasem Ramadan, Alfaisal University
Rapid scientific and technological innovations have enabled us to manipulate complex living systems and expanded our understanding of human biology. Additionally, the unprecedented investment in pharmaceutical research and development, enormously increased the identification of novel targets and their associated modalities. However, drug development is still a costly undertaking that involves a high risk of failure during clinical trials. The number of new molecular entities annually approved by the Food and Drug Administration (FDA) remains low, the likelihood of success continues to fall, and the number of drug withdrawals
has shown historic increase. Therefore, it is time to transform the traditional approaches of drug development to reduce the development costs and allow medicines to reach patients faster. In addition, pharmaceutical companies are facing increasing public and political pressure to reduce the prices of drugs, which may imply lower returns. In response, large investments are directed towards translational research to find innovative tools that accelerate the drug discovery and development processes. The success of one drug would stimulate reinvestment of drug sales into the development of new drugs. This innovation cycle is essentially impacted positively/negatively by the success/fail-
ure of the initial development of a drug [Saadi and White, 2014]. In fact, the impact of innovation on dug development within the pharmaceuticals industry has not been fully realised the same way in other industries [https://druginnovation. eiu.com/, The economist]. This can be attributed to three limitations factors: 1) technical constrains that is associated with lack of understanding of the underlying science; 2) financial constrains that caused by technical risk which leads drug makers to favour safer investment, hence less innovation; 3) regulatory constraints which make it difficult to approve new drugs due to uncertainty about new ways to combat disease.
Drug discovery and development processes are complex and lengthy. Progress and evolution in the way new drugs are discovered is the need of the hour; and this evolution benefits from the emergence of new technologies. Over the past few years, there have been numerous technological advancements within the pharmaceutical industry that display immense potential for expanding the boundaries of pharmaceutical research. Specifically, the integration of various (bio-)engineering disciplines, computing, and data science into the medical and pharmaceutical fields has enabled the application of engineering principles to tackle challenges in biology and medicine, leading to a new era of technological breakthroughs in the detection and manipulation of a wide spectrum of biological entities.
Harnessing artificial intelligence (AI), machine learning (ML) and supercomputing in drug discovery and development
We are witnessing a leap in data science and technology where the intersection of
27 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
data generation, automation, and supercomputing offer unprecedented advances for the pharmaceutical industry. The current drug development process is inherently sequential, and success is defined based on data, which is usually unshared, from unrepresentative models. These processes are associated with tremendous waste of resources and cost. Implementing smart technologies is anticipated to be the most impactful strategy that will revolutionise the existing drug development paradigm. In silico methods, digitalisation, AI, and ML are proposed to play an important role in the drug development cycle, from R&D stages through the clinical trials, and in commercial manufacturing. For instance, digitalisation allows connecting disparate data sets from data management systems across the whole processes which significantly enhance the workflow efficiency. Smart drug development allows design and validation early in parallelised processes and quickly identifies dead-end outcomes before investing in heavy experimentation which save costs and time. Digitalisation converts analog data to be used in complex analysis and ML,which in turn empowers the prediction capability. Moreover, this enables researchers to match the shared data from partners across the ecosystem including academia, contract research organisations (CROs), and contract development and manufacturing organisations (CDMOs) which save time and costs [Mirasol, 2023].
Unsurprisingly, Besides automation and digitalisation, AI and ML techniques are increasingly used in drug development. For instance, AI and ML can be used to generate compound hits for a given target by predicting the likelihood that a particular compound from billions of potential molecules will interact with the target in a desirable way [Olivecrona, 2017]. This is done by: 1) virtual screening of large compound libraries to identify compounds that are likely to bind to the target of interest; 2) designing new compounds based on the structural features of known ligands that are known to bind to the target; 3) designing new compounds based on the 3D structure of the target and
its binding site, and 4) generating novel compounds that have not been previously synthesised or tested for their ability to interact with the target. AI and ML can develop models that can predict the mechanism of action of a compound by analysing its chemical structure and its interactions with biological targets. These models can be trained on large datasets of known compounds with known mechanisms of action to identify patterns and predict the mechanism of action of new compounds and their possible toxicity [Cholleti, 2018]. Furthermore, AI and ML techniques can be used to understand how drug candidate efficacy may vary in different patient populations by analysing large-scale patient data, such as electronic health records, clinical trial data, and genomic data [Liu et al, 2019]. In June 2021, NVIDIA launched its supercomputer to support AI and ML research in life sciences, including drug discovery and development, genomics, and medical imaging. Following the launch of Cambridge-1, NVIDIA announced a series of collaborations with pharmaceutical companies including Astrazeneca, GSK, Novartis and Oxford Nanpore to accelerate the discovery of new drugs [NVIDIA. (2020, October 21)]. Fugaku is a supercomputer developed by Fujitsu and RIKEN, a Japanese research institute in 2020 which is currently the fastest supercomputer in the world. Fugaku is equipped with AI accelerators that enable it to perform AI and ML tasks at a high level of performance. Fugaku was used extensively in COVID-19 research, including drug discovery and development, epidemiological modelling, and medical imaging analysis [Ota, 2021]. Another supercomputing power is quantum computing which can be used to simulate the behaviour of complex molecules, such as proteins and enzymes, and predict how they will interact with potential drug compounds [McArdle, S. 2021].
In September 2018, Boehringer Ingelheim launched a collaboration with Google Quantum AI to explore the potential of quantum computing in drug design and in silico modelling. The collaboration aims
to leverage Google's expertise in quantum computing and Boehringer Ingelheim's expertise in drug discovery to accelerate the discovery and development of new drugs. However, it should be noted that quantum computing is still in its early stages, and there are significant technical and practical challenges that must be overcome before it can be widely adopted in drug discovery. The combination of AI and supercomputing enables the simulation of more complex molecular interactions and the analysis of larger amounts of data than either technology alone. While machine learning allows optimising the performance of supercomputing simulations and identify the most promising drug candidates for further study.
Another exciting emerging technology in healthcare is gene editing which enables the insertion, deletion, modification, or replacement of DNA in a genome. Tessera Therapeutics develops gene editing platform based on the CRISPR-Cas technology and uses a new approach called "gene writers" to edit genes. This platform involves cutting and pasting DNA in the genome and can selectively activate or deactivate genes by adding or removing small chemical tags, epigenetic modifications at specific locations in the genome [https://www.tesseratherapeutics.com/ gene-writing]. Tessera has partnerships with several pharmaceutical companies, including Novartis and Biogen, to develop gene editing therapies for various diseases.
Human multi-cellular models
Animal models are still the gold standard for drug research to evaluate the potential efficacy and toxicity of drug candidates before they are tested in humans. In fact, most drugs entering clinical trials are tested with limited human genetic backgrounds before they are given to people. But the results obtained from animal studies may not always accurately predict their effect in humans. Animals and humans may differ in their physiology, metabolism, and immune response, which can affect how they respond to drugs. Furthermore, the use of animal models is based on the assumption that
28 PHARMA FOCUS ASIA ISSUE 52 - 2023
RESEARCH & DEVELOPMENT
FROM THE ONLINE PRE-CONFERENCE SYMPOSIUM ON 23 AUGUST TO IN-PERSON CONFERENCE & EXCLUSIVE WORKSHOPS, MEET AND EXCHANGE UPDATES WITH YOUR INDUSTRY PEERS.


KEYNOTE SPEAKERS INCLUDE:
12 KEYNOTES | 22 WORKSHOPS | 90 EXHIBITORS Head of Asia Pacific Regional Supply Chain, Amgen, Japan

Lee Chief Executive O cer and Member of the Board, LOTTE BIOLOGICS, Korea




China

23-25
2023 | SUNTEC SINGAPORE & ONLINE
Use discount code
AUG
Shaping the Future of Pharmaceutical Manufacturing: Trends, Technologies and Beyond
Richard
Meli Gallup Director, Global MSAT Commercial Drug Product, Genentech, USA
Dr Jerry Yang CTO & Head of TechOps, Fosun Kite Biotechnology,
the underlying biology of the disease being studied is similar in animals and humans. However, this assumption may not always hold true, particularly for complex diseases such as cancer or neurological disorders. In December 2022, the US House of Representatives passed the FDA Modernization Act of 2022, which eliminates the animal-testing mandate for drug development and replaces this strategy with 21st-century methods that focus on Replacing, Reducing and Refining (3Rs) the use of laboratory animals through the adoption of New Alternative Methods grounded on human biology.
The infusion of bioengineering disciplines, such as biomaterials science, tissue engineering, and nanotechnology in drug discovery involves applying engineering principles and techniques in drug discovery. Tissue engineering allows scientists to move from simple single-cell models to larger multicellular structures and to explore the intra- and extracellular environment, especially the extracellular matrix (ECM) or niche.
Organoids: from 2D to 3D biology
Organoids are 3D structures composed of self-organising cells that recapitulate key features of tissues or organs in vitro
These 3D tissue models transform biology from 2D to 3D space by providing a more physiologically relevant environment for studying complex biological processes.
According to Roche, human model systems like organoids could revolutionise the way data is gathered in drug development, leading to more accurate predictions of patient responses to new molecules paving the way for more effective drug discovery and development. Therefore, the company believes that it is the opportune time to invest heavily in this cutting-edge technology. This strategy represents a significant shift towards a more humancentric approach to drug development. The Swiss drugmaker has set up a new research entity, the Institute of Human Biology (IHB), in 2021. The rebranded, Basel-based institute is the new name for Roche’s Institute for Translational Bioengineering and led by Matthias
Lutolf, a pioneer professor of bioengineering at the Swiss Federal Institute of Technology/Lausanne. Roche plans to grow the IHB to around 250 scientists and bioengineers to harness the organoid technology in drug discovery and development process from target identification and target validation through preclinical safety and efficacy to stratification in clinical trials. Human stem cell-derived organoids enable researchers to identify human-specific drug targets, disease mechanisms and toxicity which mice may miss. Researchers can even develop patient-specific organoids to determine the best treatment for an individual patient. Unlike iPSC-derived organoids, patientderived organoids require no reprogramming steps of stem cells to develop in fully functional 3D tissue structures, hence, preserve the genetic characteristics of the original tissue and the pathophysiology of the patient’s disease. Organoid Technology is exponentially growing, and we have seen several companies working on developing and commercialising this technology. For instance, Hubrecht Organoid Technology has developed a platform for generating and characterising organoids from healthy and diseased tissues based on the Lgr5+ stem cells in the adult intestine by the Hans Clever´s lab [Barker et al, 2007], which subsequently led to the development of the fist “mini-gut in a dish”. Organoid Therapeutics is another biotechnology company that is focused on developing organoid-based therapies for various diseases, including cystic fibrosis and inflammatory bowel disease. Some of the exciting development is using the
iPSCs to generate insulin-producing pancreatic islet cells and incorporating it in pancreatic organoids. The technology also promotes microvasculature formation within the organoids by integrating micro-vessel fragments in the 3D cellular structure, leading to the formation of a functional vascular network.
As this field continues to grow, we can expect to see more companies developing and commercialising organoid-based products and services.
Organ(s)-on-a-chip (OOCs)
Multiple cell types/organs, originated from humans, are now being grown onto single chips demonstrating promising possibilities for assessing drug toxicity and mechanisms of action. The target of the OOC technology is to create effective and translatable integrated microphysiological models which recapitulate the key structure and functions of a specific human tissue or a network of functional organs in vitro for investigating the physiological events that characterise the interaction between organs, immune system, and exogenic substances in health and disease states. OOC devices are created by integrating microfabrication and microfluidic techniques with heterogeneous cellular structures and ECM by incorporating complex microfluidic networks that link various cell types from different organs onto a single tiny chip.
While both organoids and OOC technologies aim to mimic the architecture and function of human organs, there are key differences between them. Organoids have a relatively simple structure that can be generated from iPSCs or tissue-specific adult stem cells and are capable of self-organising into 3D structures with multiple cell types. OOCs, on the other hand, enable more complex integration of cellular/tissue components thanks to the microfluidic and microfabrication techniques which provide superior cell-cell or tissue-tissue interfacing. OOCs represent a sophisticated form of cell culture architecture that provide precise cellular positioning and in vivo-like cell polarisation by using
30 PHARMA FOCUS ASIA ISSUE 52 - 2023
RESEARCH & DEVELOPMENT
Integrating powerful technologies is expected to revolutionise the pharmaceutical industry by accelerating the drug discovery and development process.
microfabricated tailored fluidic templates on which cells can reproduce a complex assembly and mimic the actual tissue organisation. Therefore, OOC devices can provide a more physiologically relevant environment for testing drug toxicity and efficacy. Researchers are constantly developing new organ-on-a-chip using relatively similar device architecture with many attempts to integrate these devices with other techniques such as sensing and high-throughput screening. The OOC technology is still in the early stage, but its technology readiness level is increasing rapidly due to the considerable interest among researchers in academia and industry. In recent years, a number of start-ups have introduced devices or tools based on OOC technology for in vitro modelling. Of these products, the OrganoPlate® platform by MIMETAS has demonstrated a wide range of applications. TissUse GmbH (Berlin, Germany) introduced a variety of HUMIMIC Chips for different in vitro modelling purposes. The organ models are connected by microfluidic channels, which are covered with human dermal microvascular endothelial cells. Emulate Inc. (Boston, USA) is currently commercialising a variety of organ chips based on the lung-on-a-chip which was developed by the Wyss Institute at Harvard University [Huh et al 2010].
Regulatory agencies, such as the FDA, are currently considering the use of OOC devices in drug development and toxicity testing. Guidelines and standards are being developed to ensure the reliability and reproducibility of data generated from these devices. Overall, OOC is a rapidly evolving field that has the potential to transform drug discovery and development. As technology continues to advance, we can expect to see more widespread adoption and commercialisation of OOC devices, as well as the development of more complex and physiologically relevant models.
3D bioprinting
Bioprinting originated from the more established 3D printing but uses bioinks (cells and gels) as printing materials.
Bioprinting uses a digital file as a blueprint to print the tissue or organs using cells as a building block which involves printing cells layer by layer to create a functional tissue [Ramadan 2021]. Like 3D printing, 3D bioprinting has the ability to control cell deposition in the x, y and z axes to create tissue-specific patterns with in vivo-like architecture which exhibit tissue-like density with highly organised cellular features, including intercellular tight junctions and microvascular networks. 3D bioprinting has great capability to produce complex 3D microfluidic architectures with multi-compartment which enables growing various cell types arranged in specific, discrete positions that enhance tissue-tissue crosstalk. This approach enables the creation of complex tissue structures with highly organised cellular features that closely resemble those found in native tissues and encourage the formation of cell-cell crosstalk and signalling pathways that are critical for the proper function of tissues and organs.
The first 3D bioprinter was developed by Organovo® in 2009. Today, there are several bioprinter platforms available for tissue and organ printing, each with their own unique features and capabilities for example, NovoGen MMX Bioprinter from Organovo, BIOXTM from CELLINK, EnvisionTEC 3D-Bioplotter from 3DSMAN, Rgen200 from RegenHU, NGB-R bioprinter from Poietis and Allevi 2 from Allevi3D. In 2021, CELLINK announced a collaboration with researchers at the University of British Columbia to develop a 3D bioprinted model of a functional kidney tissue that can be used for drug testing and disease modelling. Earlier, the company developed 3D bioprinted liver, heart and skin tissue.
The FDA recognises the potential of 3D bioprinting for regenerative medicine and tissue engineering and is taking a proactive approach to regulating this technology. The agency is working to establish regulatory frameworks and guidelines to ensure that 3D bioprinted products are safe, effective, and of high quality.
An integrated approach
Integrating these powerful technologies is expected to revolutionise the pharmaceutical industry by accelerating the drug discovery and development process, enabling the development of more effective and personalised treatments, and reducing the time and cost of bringing new drugs to market. OOCs allow testing the safety and efficacy of drugs in an accurate way and bioprinting technology allow creating customised tissues and organs that are specific to an individual patient while AI and ML can be used to analyse the data generated by these models, helping to identify potential drug candidates more quickly.
More technological advances will continue to infuse into pharmaceutical research and development, and it is exciting to see how drug discovery and development will be reshaped to make drugs more affordable for patients.
References are available at www.pharmafocusasia.com
Qasem Ramadan received his Ph.D. from Nanyang Technological University/Singapore. He was a Senior Scientist at the Agency for Science, Technology and Research/ Singapore and the Swiss Federal Institute of Technology. In 2019, he joined Alfaisal University/ KSA as an assistant professor of research. His current research lies at the interface of engineering and biology focusing on developing technologies for interfacing living cells with synthetic systems

31 www.pharmafocusasia.com
AUTHOR BIO RESEARCH & DEVELOPMENT
Nitrosamine Control and Prevention
A comprehensive approach
What are nitrosamines?
Nitrosamine describes a class of compounds with a nitroso group's chemical structure bonded to an amine. They are common in water and foods, including cured and grilled meats, dairy products, and vegetables. Everyone is exposed to some level of nitrosamines. However, the Medicine Regulatory Authorities first became aware of the presence of the nitrosamine impurity, N-nitrosodimethylamine (NDMA), in products containing valsartan in July 2018. Subsequently, nitrosamine impurities were detected in other medicines belonging to Sartan Family. In September 2019, it was detected in antacids such as Ranitidine and Nizatidine, and in May 2020, detected in metformin extended-release formulation, etc1,2,3 Nitrosamine impurity has led to more than 1500 voluntary recalls of several anti-diabetic (metformin, pioglitazone), hypertensive (valsartan, losartan), and histamine blocker (ranitidine, nizatidine) drugs and their combination.
1. https://www.who.int/news/item/20-11-2019-information-note-nitrosamine-impurities
2. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m7r1assessment-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit_en.pdf
3. https://www.fda.gov/media/141720/download Control of Nitrosamine Impurities in Human Drugs - Guidance for Industry
Nitrosamine impurities constitute a significant concern because of their impact at the genetic level: (Figure 1)
Regulatory Requirement:
Owing to this, the regulators demanded detailed risk assessment and clear communication from the drug manufacturers on the presence of nitrosamine impurities in the application with the agency. So that the quality, efficacy of the drug, and patient safety are not compromised.
The risk assessment should not be limited to drug substances/API's but also requires evaluating starting materials, solvents, reagents, and catalysts. Awareness of the supply chain of raw materials became an essential factor in assessing and controlling the impurities levels. The drug manufacturer and API producers may not be aware of nitrosamine contamination in raw or starting materials they have sourced from vendors. As illustrated in the diagram, sources of nitrosamines could be through a.) Primary source – drug substances b.) Secondary source –excipients, solvents, packaging, etc. c.) Formation through a mechanism other than drug substance degradation. (Figure 2)
32 PHARMA FOCUS ASIA ISSUE 52 - 2023
DNA damage & DNA mutation (potentially leading to cancer)
N-nitroso compound
Drug specific nitrosamine
Formation of DNA-reactive metabolites
Figure 1
Nitrosamines in Drug product
The nitrosamines impurities can also form during the manufacturing process of drug substances/ products or the shelf-life storage period of the final drug substances/products.
Based on the above, the drug manufacturer/ formulator is responsible for a comprehensive risk assessment for their final drug product and reports to regulatory authorities. The drug manufacturer should consider and apply the below 3-way approach in the final formulation risk assessment. (Figure 3)
Step 1: Assessment: Risk mitigation of all the input sources of the drug formulation and appropriate action.
Step 2: Testing:Below is the FDA's recommendation for acceptable intake (AI) limits for the nitrosamine impurities in drug products:(Figure 4)
Most methods used are Quantitative on LC-MS/ MS method using APCI in positive MDDe and GC-MS /MS with Liquid extraction for Nitrosamine.
Role of excipient manufacturer
As stated in the IPEC federation position paper published in March 2022, Excipient manufacturers are diverse. As a proactive approach, excipient manufacturers could include the relevant and available information on nitrosamine impurities for an excipient
in the form of a declaration to customers to mitigate the risk in drug product development. However, the responsibility for overall risk assessment for the presence of nitrosamines in a drug product lies with the MAH or the drug product manufacturer, depending on the region4.
Excipient manufacturers can support drug manufacturers with the following:
• Understanding the manufacturing process and the basic chemistry of the raw materials used. This

33 www.pharmafocusasia.com 33
Packaging b Drug substance a Solvents/water b Formation During storage c Excipients b Manufacturing process b
Figure 2
Nitrosamine AI limit (ng/day) N-nitrosodimethylamine (NDMA) 96 N-nitrosodiethylamine (NDEA) 26.5 N-nitroso-N-methyl-4-aminobutanoic acid (NMBA) 96 N-nitrosoisopropylethyl amine (NIPEA) 26.5 N-nitrosomethylphenylamine (NMPA) 26.5 N-Nitrosodiisopropylamine (NDIPA) 26.5
Figure 3
Figure 4 STEP01 ASSESSMENT Assess the all probable sources for risk of Nitrsoamine Impurities REPORTING Changes implemented to prevent or reduce Nitrsoamines TESTING Presence of Nitrosoamine impurities
02
03
4. https://www.ipec-europe.org/uploads/publications/202203-if-nitrosamines-position-paperfinal-1646123452.pdf
STEP
STEP
LoD/LoQ is reasonably practical for products MDD is high (>1 g). If >1 nitrosamine is listed, the method LOQ should be <0.03 ppm. If MDD >1 g (e.g., 1200 mg), LOQ should be below 0.02 ppm
LoD or LoQ <10% of the limit of Al
LoQ should be < 30 ppb (0.03 ppm)
Testing type Quantitative Quantitative Quantitative Quantitative Quantitative Sensitivity of the method based on the relevant acceptable intake (Al) for the respective Nitrosamine impurity
LoQ should be <= to the acceptable limit for the most potent Nitrosamine detected in an API or drug product.
EMA
the LoQ of the analytical method employed should be < 10% of the acceptable limit based on the Al
the LoQ of the analytical procedure employed should be 30% of the acceptable limit based on the Al
the LoQ should be <= of the acceptable limit based on the relevant acceptable intake (Al) for the respective nitrosamine impurity
FDA
Alternate approaches (e.g., an upstream test of an intermediate) should be supported by sufficient process understanding and evidence of adequate statistical control and should be submitted to FDA prior to implementation.
If a nitrosamine impurity is detected above the LOQ
ANVISA Admitted the absence of nitrosamines when <10% of the Al limit
The control must be included if results are >10% of the Al limit. Other approaches can be justified, not exceeding the 30% limit. If the >1 nitrosamine is to be controlled, the limits must be adjusted to ensure negligible risk maintenance.
SWISS MEDIC
The detection of every nitrosamine impurity must lead to an investigation of the causes, and appropriate CAPA's should be taken in accordance with GMP. As with any case of an identified problematic risk, companies must follow the standard procedure and inform Swiss medic immediately if nitrosamines are detected in APls or medicinal products - regardless of the quantities- and submit a risk evaluation.
The API specification should include a test and acceptance criterion for each nitrosamine impurity when the risk for nitrosamine presence is considered to be high and/or when the concentration of any nitrosamine is found to be at significant levels (e.g., greater than 30% of the acceptable intake) during confirmatory testing.
5. https://www.ipa-india.org/wp-content/themes/ipa/pdf/session-4-bm-rao.pdf
34 PHARMA FOCUS ASIA ISSUE 52 - 2023 Testing EMA FDA ANVISA Swiss Medic Health Canada Method Validated
method Validated sensitive method Validated sensitive method Validated sensitive method Validated sensitive method
sensitive
Table 1: Region-specific testing requirements and expectations from the leading countries5
Testing Omission Skip testing Routine control
Table 2: Testing requirements of nitrosamines5:
HEALTH CANADA NA NA
potentially rules out the likelihood of nitrosamines. • IPEC questionnaires that identify checkpoints, namely the manufacturing process, vulnerable amines, and nitrosating agents to be assessed.
Furthermore, excipients are also discussed a lot for their nitrites - common precursors for nitrosating agents- reported in many excipients at ppm levels. There is a lot of information on nitrite data in excipients in the public domain.
Therefore, as an excipient manufacturer, Ashland continuously monitors and controls the Nitrite levels in all our excipients and ensures the consistency of nitrite levels in all the lots. We have tested all excipients products for simple nitrosamines, such as N-Nitrosodimethyl amine (NDMA), N-Nitrosodiethylamine (NDEA), N-NitrosoN-dipropylamine (NDPA), and N-Nitroso-Nbutylamine (NDBA), the same are not present in our experiments.
Low Nitrite Benecel™ XRF HPMC for controlled release formulation:
Metformin HCl ER Tablets are one of the most recalled formulations from many pharmaceutical manufacturers because of nitrosamine issues.
In view of the same, Ashland tested lots of low Nitrite Benecel K100M XRF in Metformin HCl ER Tablets. All tablet formulations with low Nitrite Benecel K100M XRF showed unchanged NDMA levels even at 40°C/75%RH for 3 months.
Typical values of nitrites were tested using Ashland's in-house "post-column derivatisation HPLC with UV detection method with a detection limit of 6 ppb. Nitrates were tested using in-house "Ion exchange HPLC with UV detection method" with a detection limit of 100 ppb. NDMA was tested using the "GC/MS-SIM method" with a detection limit of 5 ppb.
Ashland is committed to supporting our customers on upcoming regulatory challenges and technical queries by providing product-related risk assessments and technical discussions. Abbreviation:
1. DNA - Deoxyribonucleic acid
2. API - Active pharmaceutical ingredient

3. FDA - Food and Drug Administration
4. LC-MS/MS - Liquid chromatography-mass spectrometry
5. APCI -Atmospheric pressure chemical ionization
6. GC-MS /MS - Gas chromatography-mass spectrometry
7. LoD – Limit of Detection
8.
9.
10.
11.
12.
13.
14.
15.
17.
19.
35 www.pharmafocusasia.com 35
LoQ – Limit of Quantitation
MDD - Minimum Daily Dose
CAPA - Corrective and preventive action
NA - Not Available
GMP - Good manufacturing practice
ANVISA - Brazilian Health Regulatory Agency
EMA - European Medicines Agency
IPEC - International Pharmaceutical Excipients Council
16. RH - Relative humidity
HPLC - High-performance liquid chromatography
18. UV - Ultra Violet
GC/MS-SIM - Gas Chromatography-Mass SpectrometrySelected ion monitoring Advertorial
An introduction to FDA’s Human Factors Engineering Requirements for Drug-Device Combination Products
Human Factors Engineering
Product Development
This article goes beyond published guidance documents of the US Food & Drug Administration (FDA) and outlines the latest trends in its feedback that must be considered when applying human factors engineering (HFE) during combination product development. The authors outline key research and analysis activities and nuanced scoping considerations for the HF validation test required for FDA approval
Allison Strochlic, Global Leader – Human Factors, Emergo by UL, Yvonne Limpens, Managing Human Factors Specialist, Emergo by UL, Katelynn Larson, Technical Writer, Emergo by UL
Although less well known than other drug development phases, human factors engineering (HFE) is critical to meeting regulatory expectations for drug-device combination products. Many markets, including the European Union and China, have standards and guidelines for applying HFE. However, this article focuses on the HFE requirements of the FDA (sometimes referred to hereafter as “the Administration”), which are currently considered to be more rigorous.
36 PHARMA FOCUS ASIA ISSUE 52 - 2023
RESEARCH & DEVELOPMENT
HFE is a multi-disciplinary field that involves engineering, psychology and design and is focussed on ensuring that a product’s design is well-matched to the product’s intended users. Specifically, products should be safe, usable and satisfying while accounting for intended users’ skills, knowledge, abilities and limitations.
HFE activities occur at many stages of product development. They can include user research, development of use-related risk documentation, formative evaluations, and a human factors (HF) validation test, also known as a summative usability test, among others. HFE activities help fulfill regulatory requirements associated with product safety and help designers identify specific product requirements, such as an appropriate force required to activate an auto-injector’s drug delivery.
The FDA has established HFE guidelines for drug-device combination products, which include injection devices (e.g., pre-filled syringe, auto-injector, on-body injector), inhalation devices (e.g., inhalers, nebulisers) and other devices (e.g., nasal delivery devices). The Administration is particularly concerned with these products’ usability and use safety, partly because many of these products are used by laypeople, such as patients and caregivers, without medical training. While this article focuses on drug-device combination products,
the FDA might also expect HFE to be applied to oral medications and/or drug products with unique treatment regimens and/or packaging.
The FDA expects all materials the user interacts with (i.e., the user interface) will be subject to HFE activities. Per the Administration, “user interface” refers to more than just the physical product, such as a pre-filled syringe’s plunger, stopper, barrel, flanges and needle. This term also includes the packaging, labelling and any instructional materials. When discussing “combination products” or “product,” we are referring to this comprehensive user interface (UI) definition.
Key HFE activities
HFE efforts can be subdivided many ways, but we typically distil the process into six activities. Notably, the scope of these activities depends on the intended users, indications for use, associated userelated risks and the ability to leverage prior HFE work. For example, pharmaceutical companies may be able to leverage the platform manufacturers’ early-stage HFE work upon selecting a platform drug-delivery device. (Figure 1).
Research users and use environments
User research activities, including contextual observations, interviews with intended users and literature reviews, help inform the product design, deepen
understanding of user needs and determine the intended use environments’ key characteristics. These activities generate insights needed to develop user profiles, use environment descriptions and provide context for the product’s use.
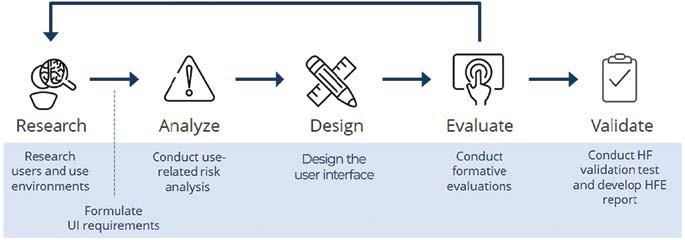
Formulate UI requirements
UI requirements describe components, features and characteristics you consider ‘must haves’ in your final design. These requirements are derived from user research, the use-related risk analysis (URRA) and knowledge about existing products. They describe how the new product should be designed to enable safe and effective use.
Conduct use-related risk analysis
The URRA helps to identify possible use errors and resulting potential harm, such as injury, death and compromised medical care. Using a task analysis, known problems analysis and/or hazard analysis, you can identify more potential use errors, describe the resulting hazardous situation and categorise the severity of the potential resulting harm(s) in an itemised table.
Conduct formative evaluations iteratively Formative evaluations help assess the usability and use safety of an in-progress design. Examples include an expert review, such as a product critique performed by HFE practitioners and/ or designers, a cognitive walkthrough
37 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
Figure 1. User-centred process, including the HFE key activities
during which an intended user shares feedback while interacting with the product’s prototype and a formative usability test involving simulated product use and related interviews. Formative evaluations are technically optional but can be valuable activities highly encouraged by the FDA.
Conduct HF validation test
The FDA requires an HF validation test to demonstrate that the product is safe and effective for use. Such HF validation testing is needed for new products and marketed products that have been changed in a way that results in new or impacted critical tasks. Typically, the HF validation test simulates actual product use (e.g., injecting into a cushion rather than administering a real injection), a distinct difference from clinical trials. We discuss this pivotal task in more detail below.
Develop HFE report
Assuming favourable HF validation test results, the HFE report asserts that the product is safe and effective to use and provides a comprehensive overview of all HFE activities conducted throughout the product’s development. The FDA provides a detailed outline for this report and covers the intended users, use environments and training, if applicable; known use problems with previous or similar products; the URRA; formative evaluations and any subsequent changes made to the product design; HF validation test method, including any changes implemented based on feedback from the FDA; and HF validation test results and residual risk analysis.
Key HF validation testing considerations
The HF validation test is the HFE version of a pivotal trial and can require rigorous planning, data collection and analysis. The test involves a production-equivalent product and representative users to serve as participants.
Identifying evaluation activities
Evaluation activities are the core of the usability test, occurring after informed consenting and an introduction and before open-ended, debriefing questions. Based on FDA guidance, evaluation activities include all critical tasks. The FDA defines these as “user tasks that, if performed incorrectly or not performed at all, would or could cause harm to the patient or user, where harm is defined to include compromised medical care.” Evaluation activities are both hands-on, simulated product use scenarios and knowledge-based assessments to check participants’ understanding of essential steps that cannot be easily evaluated through observation, such as storage, preparation and disposal. Each scenario should represent actual use, including the use environment and setup.
Defining and recruiting representative users
Per the FDA, an HF validation test requires a minimum of 15 participants per user group. The general expectation is that a single user group will have shared characteristics, including backgrounds, abilities and experience with similar devices. Your product might have one user group (e.g., patients) or many (e.g., adolescent patients, adult patients, lay caregivers and healthcare professionals). Your user research findings and the product’s indications for use inform the type and number of user groups required.
The FDA expects HF validation test participants will represent actual, intended users. However, recruiting people diagnosed with rare medical conditions can be challenging. Recruiting “proxy” (i.e., surrogate) participants with similar backgrounds and characteristics to the target population might be acceptable in these cases. When taking this approach, it is suggested that you thoroughly document your attempts to recruit users and submit a convincing justification regarding why these proxy participants are appropriate. As an added assurance, you can submit your HF validation test protocol to the FDA before conducting the HF validation test to get the Administration’s feedback on the test approach.
Conducting test sessions
Typically, two HFE practitioners conduct each test session in person. One individual administers the session and leads interviews while the other records test data. While many variables affect the duration and scope of a test session, a combination product test session might last one to two hours and usually involves a single participant.
Data collection and reporting
Each task, or use step, should have clear pass/fail criteria, and the HFE practitioners conducting the test should assess participants’ performance. Participant performance on each task should be categorised as one of the following:
• Success: The participant completed the task as intended
• Use error: The participant performed the task in an incorrect manner that is different from what the manufacturer expects or required assistance to complete the task
• Close call: The participant almost committed a use error but selfcorrected before any harmful consequences
• Difficulty: The participant encoun-
38 PHARMA FOCUS ASIA ISSUE 52 - 2023
RESEARCH & DEVELOPMENT
With proper execution, HFE work can enable compliance with regulatory expectations, help ensure the product is safe and usable, and facilitate a positive user experience, promoting commercial success.
tered notable difficulties in completing the task as intended.
Per the FDA, test personnel should perform a root cause analysis to determine why each use error or interaction difficulty occurred by considering a participant-reported root cause and their HFE expertise. As such, the moderator interviews the participant about what led to a particular decision, action or difficulty toward the test session’s end.
The HF validation test report summarises observed test data and describes all use errors, close calls, difficulties and root causes. Rather than relying on quantitative analysis, a product has passed or failed the HF validation test based on reviewing all use errors and interaction difficulties in the context of the URRA. Determining if the HF validation test findings do not pose an unacceptable risk to users (the residual risk analysis) is a key component of the HFE report.
HF validation test timing
Conducting an HF validation test before a clinical trial can provide added assurance of patient safety and confidence in trial results. If clinical trial participants are not using a product correctly — for example, if a participant lifts an auto-injector from the injection site prematurely — clinical data regarding the drug’s efficacy will be compromised.
When conducting a pre-clinical trial HF validation test, it can reflect the clinical trial conditions. For example, if healthcare professionals (HCPs) will use the product during a clinical trial, the pre-trial HF validation test could include only HCPs as opposed to also including other identified user groups. Nonetheless, the FDA expects HF validation data based on the production-equivalent product, meaning a second (full-scope) HF validation test is likely necessary.
Key takeaways
Scoping and planning for HFE activities can seem daunting. However, with proper execution, HFE work can enable compliance with regulatory expectations, help ensure the product is safe and usable and facilitate a positive user experience, promoting commercial success. To streamline processes and help establish a smooth trajectory of HFE activities, consider the following:
• Plan for and integrate HFE activities throughout your development process
• Conduct research to understand your users, which will help enable you to create safe, usable, satisfying products that meet users’needs
• Develop a thorough and comprehensive URRA to base the scope of other HFE activities
• Seek FDA feedback on your HF validation test protocol to ensure approval of your test method, including user group selection and critical task identification, before conducting the HF validation test.
References are available at www.pharmafocusasia.com
Allison Strochlic has spent nearly 20 years applying HFE principles to medical and pharmaceutical product development. She delivers HFE workshops and advises clients on strategies to HFE to meet the FDA’s and other regulators’ expectations and leads key HF meetings with regulators. Strochlic has undergraduate and graduate degrees in HF, is a Certified HF Professional, and is coauthor of the book Usability Testing of Medical Devices.
Yvonne Limpens has 10 years of experience delivering human factors (HF) services to the medical, pharmaceutical, and IVD industries. Limpens delivers HFE workshops, advises on HFE strategy, leads research and analysis activities and develops key deliverables to support FDA and international regulatory submissions. Limpens holds a bachelor’s degree in industrial design and a master’s in human technology interaction.
Katelynn Larson has five years of experience supporting technical, regulatory and other forms of documentation for the insurance and human factors industries. She supports developing assets regarding human factors activities for a variety of medical device and pharmaceutical product manufacturers.



39 www.pharmafocusasia.com
AUTHOR BIO RESEARCH & DEVELOPMENT
Getting Back to Business Let’s meet in Bangkok!
MEDICAL FAIR THAILAND 2023 | 13-15 September


Preparations are in full swing as MEDICAL FAIR THAILAND makes its way to Bangkok once again in 2023. After a three-year break, the 10th edition of the exhibition will run its physical edition from 13 to 15 September at BITEC, followed by a 7-day digital extension where exhibitors and visitors can engage further online through its AI-powered businessmatching system until 22 September. This is the first time MEDICAL FAIR THAILAND will be held in a ‘phygital format’.
Highlights this year include signature showcases such as the Community Care Pavilion and Start-Up Park, and also the introduction of the Medical Manufacturing pavilion. As the region’s leading specialist trade fair for the medical and healthcare sectors for the past two decades, MEDICAL FAIR THAILAND serves the full value chain and end-to-end needs of the medical and healthcare sectors. From diagnostics, wearable technology, connected healthcare solutions, rehabilitation and therapy equipment, 3D printing technology, and now - medical technology (MedTech) components, processes and solutions - the exhibition offers the ideal destination for medical and healthcare buyers and professionals looking to meet their sourcing objectives, gain industry insights and to share best practices.
“We have been waiting for three years so we are excited and are gearing up for a big comeback for MEDICAL FAIR THAILAND 2023. With the positive feedback, industry commitment, and almost 80% bookings received for 2023, we should be on track to reach close to pre-pandemic levels by next year. On the back of a highly successful and wellreceived phygital edition of MEDICAL FAIR ASIA that was held in Singapore earlier this year, and as we navigate further in a post-pandemic landscape, we are confident by this year the industry will be more than ready to move into high gear and Thailand will be an ideal location.”
Gernot Ringling, Managing Director, Messe Düsseldorf Asia
40 PHARMA FOCUS ASIA ISSUE 52 - 2023
MEDICAL FAIR THAILAND 2023 comes against a strengthening backdrop where Thailand continues to firm its position as a medical hub of the region with its supportive government policies and incentives, making it a model investment destination for a wide range of medical and healthcare service sectors. In line with Thailand’s 4.0 policy, the Thai government considers the healthcare industry to be a priority sector for investment, thus the staging of MEDICAL FAIR THAILAND 2023 is well-positioned.

New! Medical Manufacturing Pavilion

A special themed pavilion focused on medical manufacturing processes and componentsfrom new materials, intermediate products, packaging and services, to microprocessors and nanotechnology. With Thailand’s growing reputation as a production and distribution base of medical devices both within and outside Thailand, it has become a natural market for medical devices.

According to data from the Office of Industrial Economics, Ministry of Industry (Medical Devices Intelligence Unit), there is much potential for investment opportunities in sophisticated medical devices particularly due to Thailand’s reliance on imports for this segment.

Community Care Pavilion Special Focus on Mental Health
With a special spotlight on mental health with a showcase featuring digital mental health technologies, from smart medicine to therapeutic medical equipment. Its mainstay of addressing the needs of ageing societies on the back of rising chronic diseases and an ageing population, the pavilion will also feature a full suite of geriatric medicine, rehabilitative equipment, assistive technology, and mobility products.
Thailand’s proportion of citizens aged over 60 years, is forecast to be one of the highest in ASEAN by 2045, and will also exceed countries such as Europe and the United States. Thailand’s fast-increasing ageing population and the estimated more than three million Thais suffering from poor mental health, is expected to further drive the demand for related healthcare services.
Start-up Park
A strategic platform for companies with ready-to-market healthcare solutions to meet relevant buyers and partners, industry influencers, experts, and potential investors. From innovative healthcare industry solutions, health apps and new tools for gathering and AI-supported analysis of health data, to robotic assistance systems and new approaches in diagnostics – the Start-Up Park is a must-attend for SMEs looking to scale-up their business.
The Start-Up Park plays a significant role as an enabler of the entrepreneurial ecosystem that encourages life sciences and medical and health innovation in Thailand. With the country’s vibrant start-up landscape propelled further by the government’s numerous grants and new regulations as part of Thailand’s ambitious plans to be a start-up-based country, the start-up scene has grown systematically over the years and is considered one of Asia’s hidden gems. At the last edition of MEDICAL FAIR THAILAND held in 2019, a total of 11 start-up companies participated from Singapore, Japan, South Korea, Hong Kong, Taiwan and Thailand.
41 www.pharmafocusasia.com 41 To find out more, go to www.medicalfair-thailand.com
A Step-By-Step Strategy for Designing A Meta-Analysis
Meta-analysis is a subset of systematic reviews that combines pertinent qualitative and quantitative study data from several selected studies to develop a single conclusion with greater statistical power. In this article we focus more on general framework of meta-analysis and a detailed perspective on designing meta-analysis including the five-step process.
Ramaiah M, Manager, Freyr solutions Balaji M, Deputy Manager, Freyr solutions
Meta-analysis is a subset of systematic reviews that combines pertinent qualitative and quantitative study data from several selected studies to develop a single conclusion with greater statistical power. This conclusion is statistically more significant than the analysis of any single research due to increased numbers of subjects, greater diversity among subjects, or accumulated effects and results. In simple words, metaanalysis is the statistical combination of results from two (02) or more separate studies.
Studies comparing healthcare interventions, notably randomised trials, use the outcomes of participants to compare the effects of different interventions. Meta-analyses focus on pairwise comparisons of interventions. The contrast between the outcomes of two (02) groups treated differently is known as the ‘effect’ - the ‘treatment effect’ or the ‘intervention effect.’ The analysis of the included studies is either narrative or quantitative.
The general framework for metaanalysis may be provided by considering the following four (04) questions:
1. What is the direction of the effect?
2. What is the size of the effect?
3. Is the effect consistent across studies?
4. What is the strength of evidence for the effect?
Meta-analysis provides a statistical method for questions 1 to 3. Assessment of question 4. relies additionally on judgments based on assessments of study design and risk of bias, as well as statistical measures of uncertainty.
On the other hand, narrative synthesis uses subjective (rather than statistical) methods to follow through questions

42 PHARMA FOCUS ASIA ISSUE 52 - 2023
RESEARCH & DEVELOPMENT
1 to 4 for reviews where meta-analysis is either not feasible or not sensible.
Purpose
• To establish statistical significance with studies that have conflicting results
• To develop a correct estimate of the effect magnitude
• To provide a more complex analysis of harms, safety data, and benefits
• To examine subgroups with individual numbers that are not statistically significant.
Advantages
• Improved precision in convincing evidence about the intervention effects when the studies are too small
• Greater statistical power and confirmatory data analysis
• A good source to respond to conflicting studies to generate new hypothesis
• Answer the unanswered or unmentioned questions in the research or a study
• Considered an evidence-based resource.
Disadvantages
• Complex and time-consuming to identify appropriate studies
• Not all studies provide adequate data for inclusion and analysis
• Requires advanced statistical techniques
• Heterogeneity of study populations.
The five-step process of designing meta-analysis
Step 1: Define the research question and eligibility criteria
A clinical research question is identified, and a hypothesis is proposed. The likely clinical significance is explained, and the study design and analytical plan are justified.
Usually, two (02) standard tools are used: Patient, Intervention, Comparison, Outcome (PICO) or Sample, Phenomenon of Interest, Design, Evaluation, and Research type (SPIDER).
PICO is primarily used in quantitative evidence synthesis. The authors demonstrated that the PICO holds more sensitivity than the more specific SPIDER approach. The latter was proposed as a method for qualitative and mixed method searches.
PICO is typically used for systematic review and metaanalysis of clinical trial studies.
PICO stands for:
P – Population: patient, or problem: How do you describe the patients, people, or problems you are looking at?
I – Intervention: What is considered an intervention, exposure, or a factor?
C – Comparison: Do you have something to compare to the intervention, exposure, or factor you are considering?
O – Outcome: What is hoping to measure, improve, affect, or accomplish?
Step 2: Protocol for the Search Process
The protocol “outlines how the review authors will handle the review process and the challenge they are addressing. The procedure describes how the studies in the review were identified, assessed, and summarised. The protocol serves as a public record of how the review authors aim to address their research question by making this information available.”
In addition to serving as a road map for the research question, protocols also allow the individual to comprehend what type of research is performed and helps avoid duplication of research.
Step 3: Search for studies
a) Identification of Literature Search Database (Registries, Repositories, or Libraries)
Most frequently used databases are
as follows:
• PubMed
• Scopus
• Web of Science
• EMBASE
• MEDLINE
• HINARI
• Cochrane
• Google Scholar
• Clinicaltrials.gov
• mRCTs
• POPLINE
• SIGLE
This list covers almost all the published articles in tropical medicine and other health-related fields.
b) Search for Relevant Literature (Reported and New Studies) Using a String-based Search on the Research Question
The search process needs to be documented in enough detail to ensure that it can be reported correctly in the review to the extent that all the databases’ searches are reproducible. The search strategies will need to be copied and pasted exactly as run and included in full, together with the search set numbers and the number of records retrieved. The search strategy should emphasise on:
• Searching previous studies
• Identification of new studies via databases and registers
• Identification of recent studies via other methods.
c) Collection of All the Retrieved Literature Using Reference Management Tools
Specially designed bibliographic or reference management software such as Mendeley, EndNote, ProCite, Reference Manager, and RefWorks are helpful and relatively easy to use to keep track of references and report studies.
d) Determination of Inclusion and Exclusion Criteria Based on Eligibility Criteria
The PICO strategy, study design, and deadline determine eligibility criteria. Most exclusion criteria are irrelevant, duplicate, unavailable, or abstract-only
43 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
papers. These exclusions should be specified in advance to prevent bias from the researcher. The inclusion criteria would include publications containing the target patients, researched interventions, or comparing two (02) evaluated interventions.
In brief, they would contain material pertinent to the study subject. Most importantly, the information should be clear and sufficient to answer the positive or negative issue.
e) Identification of Supporting Studies and Finalising the Articles to be Included
For many authors, the appearance of a diamond (statistical analysis) at the bottom of a plot is an exciting moment, but the results of meta-analyses can be extremely misleading if adequate attention is not compensated to formulating the review question, specifying eligibility criteria, identifying, selecting, and critically evaluating studies, collecting
Step 4: Data extraction
Once the studies are selected for inclusion in the meta-analysis, summary data or outcomes are extracted from each study. In addition, sample sizes and measures of data variability for both intervention and control groups are required. Depending on the study and the research question, outcome measures could include numerical or categorical measures. For example, differences in
appropriate data, and deciding what would be meaningful to analyse.
f)
Reporting the Search Process
The search process must be recorded in
scores on a questionnaire or measurement level, such as blood pressure, would be reported as a numerical mean. However, differences in the likelihood of being in one (01) category versus another (e.g., vaginal birth versus cesarean birth) are usually reported in terms of risk measures such as odds ratio or relative risk.
a) Data extraction for dichotomous outcomes: It is most reliable to collect dichotomous outcome data as the number of individuals in each group who did and did not experience the result. Although, in theory, this is comparable to collecting both the total number of individuals and the number of individuals sharing the outcome and it is not always apparent if the total number of individuals reported is the number of individuals on whom the outcome was assessed. Occasionally, the numbers incurring the event need to be derived from percentages.
b) Data extraction for continuous outcomes: Due to inadequate and inconsistent reporting, it may be difficult or impossible to obtain the required infor-


precise detail to ensure that it can be reported accurately in the review, to the extent that all searches of all databases can be reproduced. The search
mation from the provided data summaries. Additionally, they differ in the scale used to analyse the data. In a research report, standard deviations and standard errors are occasionally conflated, and the nomenclature needs to be more consistently applied. When necessary, the authors must always request missing information and clarification about the reported statistics. Nevertheless, there is an approximate or direct algebraic link between numerous variance measures and the standard deviation.
c) Data extraction for ordinal outcomes: The retrieved data for ordinal outcomes depends on whether the ordinal scale will be dichotomised for analysis, treated as a continuous outcome, or analysed directly as ordinal data. In turn, this choice will be influenced by how the authors of the studies analysed their data. The strategy of capturing all the categorisation is also useful when studies utilise somewhat different short ordinal scales. Whether a consistent cutpoint can be used for dichotomisation across all studies is uncertain.
RESEARCH & DEVELOPMENT
d) Data extraction for counts: Count data can be analysed in various ways. The crucial option is whether the interesting outcome should be dichotomous, continuous, time-to-event, or a rate. A typical error is treating counts directly as dichotomous data, considering the total number of participants or person-years of follow-up as sample sizes. Though it is preferred to decide how count data will be analysed in advance, the option is frequently driven by the structure of the available data and cannot be made until most studies have been reviewed.
e) Data extraction for time-to-event outcomes: Meta-analysis of time-toevent data typically involves obtaining individual patient data from the original investigators, reanalysing the data to estimate the log hazard ratio and its standard error, and then conducting a meta-analysis. Whether individual patient or aggregate data are used, there are two
(02) approaches to get estimates of log hazard ratios and associated standard errors for inclusion in a meta-analysis that employs generic inverse variance methods.
f) Data extraction for effects estimates: When extracting data from non-randomised studies and some randomised trials, it may be possible to obtain adjusted effect estimates. The process of data extraction and analysis using the generic inverse variance approach is identical to that for unadjusted forecasts; however, the variables that have been corrected must be noted. The disadvantage of this approach is that the estimates and standard errors for the same effect measure must be produced for every other study included in the same meta-analysis, even if they provide summary data per intervention group.
Heterogeneity: A systematic review will assemble studies with varying results.
Heterogeneity is a term for any variation between research in a systematic review. Differentiating between various types of heterogeneity (clinical, methodological, and statistical) can be beneficial. Specifically, heterogeneity related only to methodological variety would indicate the studies are biased to varying degrees. Explorations of heterogeneity designed after identifying heterogeneity can only result in the development of hypotheses. They should be evaluated with much greater caution and normally should not be included among the review findings.
Methods for tackling clinical heterogeneity should be mentioned, along with how the authors will assess whether a meta-analysis is acceptable. Methods for spotting statistical heterogeneity should be described (e.g., visually, utilising I, etc.) using the chi-squared test.
Publication bias: The publication or non-publication of research find -
45 www.pharmafocusasia.com
Figure 1: PRISMA 2020 Flow Diagram Template for Systematic Reviews or Meta-analysis
RESEARCH & DEVELOPMENT
ings may be influenced by publication bias based on the type and direction of the results. There are two (02) types of scientific studies that investigate the existence of publication bias - indirect and direct evidence. As the proportion of all hypotheses tested for which the null hypothesis is false is unknown, surveys of published results such as those mentioned above, can only give indirect evidence of
publication bias. There are also considerable direct indications of publishing bias.
Publication bias should be viewed as one of the potential sources of ‘smallstudy effects’ — the tendency for intervention effect estimates to be more positive in smaller trials. Using funnel plots, review authors can visually determine whether small-study effects may be present in a meta-analysis.
true treatment effects in the individual studies may be different from each other’ and attempts to allow for this additional source of interstudy variation in Effect Sizes. Whether this latter source of variability is likely important is often assessed within the meta-analysis by testing for ‘heterogeneity.’
Forest plot
Funnel plots: A funnel plot is a basic scatter plot of the intervention effect estimates from individual studies versus a measure of each study's size or precision.
A PRISMA flowchart template is presented, which can be adjusted based on whether the systematic review or a meta-analysis is original or updated (figure 1).
techniques must be carefully copied and pasted, together with the search set numbers and the total number of records retrieved.
Step 5: Final estimates of the effect
The final stage is to select and apply an appropriate model to compare Effect Sizes across different studies. The most common models used are Fixed Effects and Random Effects models. Fixed Effects models are based on the ‘assumption that every study is evaluating a common treatment effect.’ This means that the assumption is that all studies would estimate the same Effect Size were it not for different levels of sample variability across various studies. In contrast, the Random Effects model ‘assumes that the
The final estimates from a meta-analysis are often graphically reported as a ‘Forest Plot.’ A forest plot displays effect estimates and confidence intervals for individual studies and meta-analyses. The standard method for illustrating individual research outcomes and metaanalyses uses forest plots. These can be generated with the Review Manager software, and a selection of them can be chosen for inclusion.
Forest plots and funnel plots from the ‘data and analysis’ section may be chosen as figures for inclusion in an integrated section. Forest plots describe all the studies, and study data for the principal outcomes will be presented as figures. A funnel plot for one (01) or more key outcomes may be a vital contributor to these forest plots if there are sufficient studies.
In the hypothetical forest plot shown in Figure 2, for each study, a horizontal line indicates the standardised Effect Size estimate (the rectangular box in the center of each line) and 95% CI for the risk ratio used. For each of the studies, drug X reduced the risk of death (the risk ratio is less than 1.0). However, the first study was larger than the other
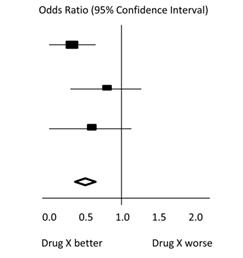
two (the size of the boxes represents the relative weights calculated by the meta-analysis). Perhaps, because of this, the estimates for the two (02) smaller studies were not statistically significant (the lines emanating from their boxes include the value of 1.0). When all three (03) studies were combined in the meta-analysis as represented by the diamond, we get a more precise estimate of the drug’s effect, where the diamond represents both the combined risk ratio estimate and the limits of the 95 per cent CI.
Manuscript drafting and submission to a Journal
The drafting of a manuscript is based on the guidelines of the ICMJE, i.e., the IMRaD model’s four (04) scientific sections: introduction, methods, results,

46 PHARMA FOCUS ASIA ISSUE 52 - 2023
RESEARCH & DEVELOPMENT
Figure 2: Hypothetical Forest Plot

and discussion, mainly with a conclusion. Performing a characteristic table for study and patient characteristics is a mandatory step that includes a detailed search strategy for database searches. Figure 2
After completing the manuscript draft, characteristics table, and PRISMA flow diagram, the draft should be sent out for review. Finally, a suitable journal with a significant impact factor and a relevant field should be chosen for the manuscript. Before submitting the manuscript, we must pay close attention to the author guidelines of the journals.
Estimated timelines for a systematic review
The time required to complete a systematic review is highly variable. However, considering the tasks and the time required for each of these might aid the authors in estimating the amount of time needed. Tasks include protocol development, searching for studies, evaluating citations, and full-text reports of studies for eligibility, assessing the risk of bias in included
Meta-analyses focus on pair-wise comparisons of interventions. The contrast between the outcomes of two (02) groups treated differently is known as the ‘effect’ - the ‘treatment effect’ or the ‘intervention effect.’
and validating it, forming criteria, developing a search strategy, searching databases, importing all results to a library and exporting them to an excel sheet, protocol writing and registration, title and abstract screening, full-text screening, manual searching, extracting data and assessing its quality, data checking, conducting statistical analysis, double data checking, manuscript writing, revising, and submission to a journal. Nevertheless, this is an important outcome that could impact the current practice and promote higher-quality future studies to address evidence gaps.
References are available at www.pharmafocusasia.com
AUTHOR BIO
Ramaiah M is a Manager, Scientific Writing with 14+ years of experience in Scientific Writing and CER Writing. He has a proven track record in developing highly complex manuscripts reporting safety and efficacy data from pivotal clinical studies for publication in highimpact journals as per ICMJE, GPP, EQUATOR, EASE, AMWA, STM, and applicable ethical regulations (COPE).


1–11
12
Table
studies, collecting data, pursuing missing data and unpublished studies, analysing the data, interpreting the results, and writing the review, as well as keeping the review up to date as shown in Table 1.
Conclusions
The reliability of meta-analysis findings is mainly determined by the quality of the data used for the compilation. The steps in this process include developing a research question
Balaji. M is a Deputy Manager, Scientific Writing with 12+ years of experience in Scientific Writing. He has a recognized background in developing complex manuscripts in biomedical engineering, drug delivery, biomaterials, and nanotechnology with novel therapeutics to bring advanced medication systems for various unmet medical needs. He has published numerous articles following publication guidelines (ICMJE, GPP, EQUATOR, EASE, AMWA, STM).
48 PHARMA FOCUS ASIA ISSUE 52 - 2023
MONTH ACTIVITY
Preparation of protocol 3–8 Searches for published and unpublished studies 2–3 Pilot test of eligibility criteria 3–8 Inclusion assessments 3 Pilot test of risk of biased assessment 3–10 Validity assessments 3 Pilot test of data collection 3–10 Data collection
Data entry
Follow-up on missing information
Analysis
1–2
3–10
5–11
8–10
Preparation of review report
Keeping the
to date
review up
1: Estimated Timelines for a Systematic Review
RESEARCH & DEVELOPMENT
A Plant Virus Nanoparticle Toolbox to Combat Cancer
Plant viruses are nano-sized particles with the natural capacity to transfer and release nucleic acids into eukaryotic cells. Plant virus-based nanoparticles (PVNPs) refer to plant viruses or virus-like particles (VLPs) and spherical nanoparticles (SNPs) which originated from plant viruses and have medical applications. With rapid developments in viral nanotechnology research, PVNPs are emerging as a beneficial nano-toolbox for cancer treatment through their ability to carry anticancer components and activate an anti-tumour immune system in vivo. Herein we briefly review the application of PVNPs as vehicles of therapeutic agents, immunotherapeutic agents, and as direct immunomodulators.
Mehdi Shahgolzari, Department of Medical Nanotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences
Afagh Yavari, Department of Biology, Payame Noor University
Kathleen Hefferon, Virology Laboratory, Department of Cell & Systems Biology, University of Toronto
Nowadays, nanoparticles are a popular toolbox used in cancer research for improving the pharmaceutical capacity of therapeutic agents. Viral nanoparticles (VNPs) are naturallyoccurring NPs that have emerged as key players. VNPs are structures between 30 -200 nm in diameter, having unique sizeand shape-dependent physicochemical and biological properties. Depending on their origin, VNPs can belong to mammalian viruses, bacteriophages, or plant viruses. While VNPs exist with differences in shape, structure, and molecular components, they can share similar applications
in cancer therapy. VNPs offer powerful platforms for vaccines, immunotherapy, and delivery of theranostic payloads. Due to cytotoxicity, unwanted immunologic reactions and adverse side effects, mammalian viruses influence VNP technology’s cost, complexity, and safety. With regard to these limitations, plant viruses or plant viral nanoparticles (PVNPs) have emerged as an alternative platform. The many properties of PVNPs make them good candidates for developing tumour therapy. These features include their lack of pathogenicity, biocompatibility and biodegradability properties in mammalian
systems, stability in rigid environment conditions, simple external functionalisation by either single or multiple functional group expression, payload loading capacity and their inherent immunostimulatory effect. PVNPs include Brome mosaic virus (BMV), Red clover necrotic mosaic virus (RCNMV), Cowpea chlorotic mottle virus (CCMV), Cowpea mosaic virus (CPMV) Potato virus X (PVX), Tobacco mosaic virus (TMV), Alfalfa mosaic virus (AMV) that have been used as scaffolds in cancer research. Here, we present the functions and applications of PVNPs in cancer treatment.
Plant virus nanoparticle-based tumour therapies
PVNPs are potent platforms from the nano-toolbox for the treatment of cancer. Similar to synthetic NPs, the nature, structure, and physicochemical features of PVNPs determine their medical application. PVNPs are selfassembled protein coat (CP) units that form a hollow structure for entrapping nucleic acids. In the presence of nucleic acid, CPs self-assemble in icosahedral, filamentous, rod-like, and bacillus morphologies. In contrast, in the deprivation of nucleic acids, CPs self-assemble as hollow virus-like particles (VLP), or spherical nanoparticles (SNP). PVNPs used to combat cancer can manifest as two strategies:
1) as protein nanovehicles for loading anticancer therapeutic agents to increase their therapeutic efficacy, and
49 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
2) as immunomodulatory agents for enhancing anti-tumour immune responses.
The unique structural and chemical properties of PVNPs make them ideal nanocarriers. PVNPs’ empty internal cavities, surface groups, and open/closed conformations enable cargo to be loaded. Therapeutic cargos are usually loaded in PVNPs via noncovalent (i.e., selfassembly, infusion, and charge/ ionic interaction) and covalent (i.e., genetically and chemically) mechanisms (reviewed by refs). Genetic manipulation is often used for displaying target ligands, antigenic structures, or specific amino acids or small peptides as tags (e.g. SNAP-tag) for further functionalisation. The selfassembly process is based on the caging of coat protein around a cargo. PVNP chemical modifications can be achieved through conjugation reactions (bioconjugate chemistry, click chemistry). Some PVNPs can trap cargo via a pore-opening mechanism in response to environmental conditions (i.e., pH and salt concentrations). PVNPs possess a negative/positive charge within a biological pH, and thus, they load charged cargos via electrostatic interactions.
More importantly and for addressing human solid tumours, PVNPs can target and transfer their cargo into the tumour
microenvironment (TME) and within tumour cells themselves. Accumulation of PVNPs in TME is determined by size and blood distribution. PVNPs tend to accumulate in TME much more than in normal tissue, because of leaky vasculature and poor lymphatic drainage, as well as enhanced permeability and retention (EPR) effect. For targeting tumour cells, PVNPs don't require specific ligands by nature, however, they could be manipulated or engineered to display agonist ligands of tumour cell-membrane receptors, in order to directly deliver and transfer their cargo. Generally, structural properties (e.g., size, charge, and shape), various methods of cargo loading, and bioengineering all provide PVNPs for non-targeted and targeted delivery of therapeutic agents (e.g., small molecule drugs, nucleic acids, peptides, and proteins) and immunotherapeutic agents for cancer treatment (Figure 1).
PVNPs for the delivery of therapeutic agents
Therapeutic agents consisting of chemical and biological drugs can target and kill tumour cells via specific mechanisms. Traditional therapeutic drugs are highly effective to kill cancerous cells. However, their systemic administration have certain
disadvantages such as lower bioavailability, minimal effectiveness, and severe side effects. Therefore, PVNP-based formulations have been designed as nano vehicles toward improving the pharmacological profiles of therapeutic drugs (Table 1). As mentioned above, PVNPs can load therapeutic agents via the exterior and/ or interior of the capsid surface using covalent or non-covalent interactions. The nanoparticulate features (size, shape, charge, and surface functionalities) of PVNPs, and leaky nature of the vasculature (or EPR effect) of tumours lead to the accumulation of loaded PVNPs with therapeutic agents in the TME.
For example, charge-driven drug loading strategies were applied for encapsulating mitoxantrone (MTO) into TMV, a 300 × 18 nm nanorod containing a 4 nm-wide channel, and lined with glutamic acids. The negative charge of the glutamic acids allows for electrostatic interactions with the positively charged MTO, thus allowing for pH dependent drug-loading and release. In vitro and in vivo results confirmed that MTO maintained its efficacy when delivered by TMV in a panel of cancer cell lines in addition to a triple negative breast cancer mouse model. Another drug for loading into the nano-channel of TMV has been the active dictation
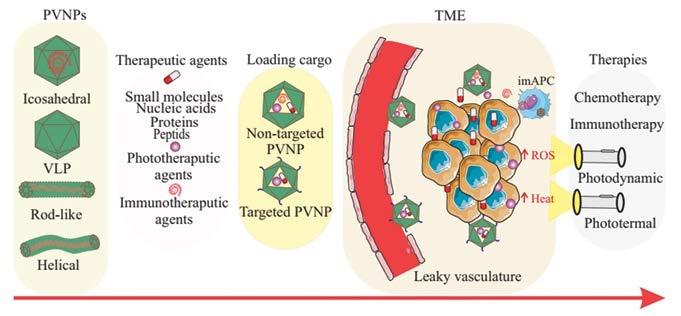
50 PHARMA FOCUS ASIA ISSUE 52 - 2023
RESEARCH & DEVELOPMENT
Figure 1. Loaded and functionalized PVNPs with therapeutic agents for tumour therapy
form of cisplatin (cisPt2+), making use of the negatively charged Glu acid side chains that line the interior channel of TMV. TMV-cisPt exhibited superior efficacy vs free cisPt in ovarian tumour mouse models.
The greatest challenge in PVNP -based therapeutic agents is the low efficiency of delivery to tumour cells. PVNP-based targeted delivery is designed using an overexpression of tumour cell biomarkers -based agonist ligands for targeting, binding and delivering the payload to tumour cells. For example, display of (((S)-5-amino-1-carboxypentyl) carbamoyl)-L-glutamic acid (DUPA), a specific ligand to prostate-specific membrane antigen (PSMA), in TMV and loaded with MTO increased cytotoxicity of PSMA+ prostate cancer threefold, and is a promising therapeutic strategy. PVNPs displaying folic acid, GE11 (a small peptide with 12 amino acids), HER2 ligands (trastuzumab, CH401 epitope), tumour-homing peptides (THPs, IR780 iodide, F3), TRAIL (tumour-necrosis factor related apoptosisinducing ligand), arginine–glycine–aspartate (RGD) peptide, and Asp-Gly-Glu-Ala (DGEA) peptide, have been shown to selectively target tumour cells.
PVNPs for the delivery of phototherapy agents
Photodynamic therapy (PDT) and photothermal therapy (PTT) are promising avenues for improving the efficacy of cancer treatment. PVNPs can deliver photo activated therapeutic agents for inducing cytotoxicity via PDT and PTT. In this process, PD-PVNP accumulates in TME, and then is activated by light to generate reactive oxygen species (ROS) such as hydrogen peroxide, hydroxyl radicals, superoxide anions or singlet oxygen, which consequently cause cytotoxicity to tumour cells. For example, the cationic photosensitiser Porphyrin encapsulated the interior channel of TMV via electrostatic interactions to improve cell uptake and efficacy compared to free photosensitisers in a melanoma model. In another study, the incorporation of Zn-EpPor PS
through electrostatic interactions with the carboxyl dendron attached to CPMV improved 2-fold the uptake efficacy and cell death in a B16F10 melanoma cell line compared to free PS. Free Zn-Por3+ displayed the greatest cell toxicity and the loading of Zn-Por into TMV and TMGMV resulted in slightly decreased cell toxicity in vitro. In contrast, targeted Zn-Por -TMV (with the nucleolin-specific F3 peptide accumulating at the cancer cell surface) can increase tumour cytotoxicity fivefold.
Photothermal therapy (PT) includes a PVNP associated with a photothermal agent. In this process, PT-PVNP accumulates in TME, and becomes activated by light to generate heat. Gd-TMV– polydopamine (PDA), with a strong near-infrared absorption and a high photothermal conversion efficiency (28.9%), offer promising results for effectively killing PC-3 prostate cancer cells in vivo and in cancer models. Coating of TMV with polydopamine (PDA), as a PTT agent was demonstrated to
increase anti-tumour efficacy based on PTT- immunotherapy in B16F10 dermal melanoma in C57BL/6 mice. Targeted Cowpea chlorotic mottle virus (CCMV) capsids with tumour homing peptide F3 (via genetic engineering), and loaded with near-infrared fluorescent dye IR780 iodide, (F3-CCMV-IR780 NPs), displayed excellent molecular targeting PTT to nucleolin receptor over-expressed on the surface of MCF-7 tumour cells.
PVNPs for the delivery of immunotherapeutic agents
Nanocarrier properties of PVNPs have prepared them for the delivery of immunotherapeutic agents to the target site (e.g. TME, antigen presenting cells (APCs), and other components of the immune system) to improve the therapeutic index. Toward this goal, encapsulation of oligodeoxynucleotides (CpG ODNs, ODN1826), as agonists of Tolllike receptor 9 (TLR 9) into CCMV and TLR 7 agonist (1V209) into TMV has been shown to slow tumour growth
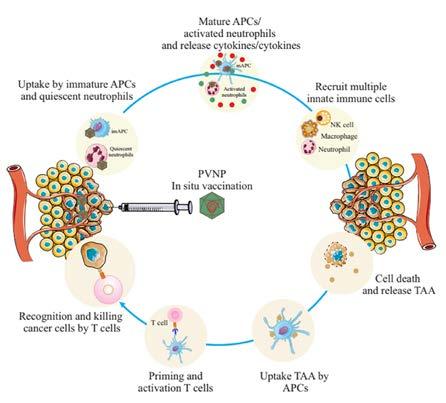
51 www.pharmafocusasia.com
Figure 2. PVNPs for in Situ vaccination. Intratumoural injection of PVNP active innate immune cells (e.g. APCs) in TME, and recruitment of other innate immune cells to the tumour that are cytotoxic to cancer cells, take up, process and priming tumour antigens lead to recognition and killing tumour cells by T cells.
RESEARCH & DEVELOPMENT
and prolong survival in mouse models of colon cancer and melanoma. This demonstrated that bioconjugation with the anti-PD-1 peptide SNTSESF (AUNP) into CPMV (the CPMV-AUNP formulation) increased the anti-tumour efficacy of AUNP into ovarian cancer cells compared to free peptide.
PVNPs structural properties (i.e., size, shape and rigidity) can transfer and stimulate APCs within the lymph node. For example, to overcome immunological tolerance against HER2-positive tumour cells, integrated HER2 epitopes loaded onto Potato virus X (PVX), and CPMV acted as vaccines without requiring additional adjuvants to induce a strong and sustained anti-HER2 immune response. Similarly, a VLP-based vaccine has been
designed via click chemistry with the attachment of the HER2-derived CH401 peptide epitope into PhMV. Results have shown that PhMV-based vaccine enhanced anticancer immunity by high titers of HER2-specific immunoglobulins, increased the toxicity of antisera to DDHER2 tumour cells, and prolonged survival of the vaccinated vs. naïve BALB/C mice. Testis antigen NY-ESO-1 is an attractive antigenic target for cancer vaccines. Displaying multiple copies of human HLA-A2 restricted peptide antigen NY-ESO-1157–165 into CPMV enhances uptake and activation of APCs and stimulates a potent CD8+ T cell response. This study shows the potential of CPMV-NY-ESO-1 vaccine against NY-ESO-1+ malignancies. These studies
also explain how PVNP formulations are expected to exhibit prolonged tumour residence and favorable intratumoural distribution. (Table 1)
PVNPs for in situ vaccination (ISV)
The inherent immunogenicity of PVNPs have provided them with tremendous potential as direct immunomodulators to activate the innate immune response. The function of PVNPs in immunomodulation depends on their structural components, capsid protein and genome. They can act as non-self (foreign), or danger signals, and active pattern recognition receptors (PRRs) on immune cells carrying Toll like receptors (TLRs). For example CPMV (virion) and empty CPMV (eCPMV,
52 PHARMA FOCUS ASIA ISSUE 52 - 2023
Therapeutic agent PVNP Loading strategies Targeted ligand Therapy Ref Chemotherapeutic DOX CPMV Chemical conjugation Chemotherapy [43] CMV Pore opening Chemotherapy [44] JgCSMV Pore opening Chemotherapy [45] AMCV Pore opening Chemotherapy [46] TBSV Pore opening Chemotherapy [47] RCNMV Pore opening Chemotherapy [48] TMV Chemical conjugation Chemotherapy [49] HCRSV Self-assembly Folic acid Chemotherapy [20] MTO CPMV Pore opening Chemotherapy [50] TMV Charge interactions Chemotherapy [14] TMV Charge interactions DUPA Chemotherapy [19] Cisplatin TMV Charge interactions Chemotherapy [51] Proflavine CPMV Infusion Chemotherapy [12] vcMMAE TMV Bioconjugation Chemotherapy [52] MMAE PVX Chemical conjugation Chemotherapy [51] Phototherapeutic Zn-EpPor TMV Charge interactions Nucleolin- F3 Photodynamic therapy [31] PhMV Infusion Photodynamic therapy [53] Polydopamine (PDA) TMV Chemical conjugation Photothermal therapy [32] Immunotherapeutic ODN1826 CCMV Self-assembly Immunotherapy [35] 1V209 TMV Chemical conjugation Immunotherapy [33]. AUNP CPMV Chemical conjugation Immunotherapy [36] Trastuzumab PVX Chemical conjugation Immunotherapy [54]
RESEARCH & DEVELOPMENT
Table 1. PVNPs of cargo-loaded and functionalized for tumour therapy.
without the nucleic acid) capsids are recognised by MyD88-dependent TLR2 and TLR4, and the release of the ssRNA contained within CPMV and PapMV is recognized by TLR7. It was demonstrated that PVNPs can act as pathogenassociated molecular patterns (PAMPs) for TLR of the surface (1, 2, 4, 5, and 6) or the endosome (TLRs 3, 7, 8, and 9) on APCs.
In situ vaccination (ISV) uses intratumoural injection of PVNPs to activate the innate and adaptive immune system in TME. Virions with active or deactivated nucleic acids and VLP can be used for ISV. PVNPs-ISV therapy leads to changes in cytokine levels within the TME, and reprogram and repolarise suppressed innate immune cells toward an anti-tumour phenotype. They also induce the recruitment of innate immune cells that are cytotoxic to cancer cells. PVNPs can induce proinflammatory cytokine production such as interleukin (IL)-1, IL-6 and IL-12, interferon (IFN)- , and IFNthat potentiate to induce an adaptive immune response (Figure 2). A typical PVNP used for ISV is CPMV. The CPMV capsid triggers TLRs 2, 4 and the ssRNA within CPMV activates TLR 7, and receptor signalling cascades lead to the release of immunostimulatory cytokines such as IL-1 , IL-12, IFN- , chemokine ligand 3, macrophage inflammatory protein-2, and granulocyte-macrophage colonystimulating factor (GM-CSF).
The tumour acts as a resource of antigens in ISV, and upon tumour antigen release in the TME, processing and priming by the APCs leads to the activation of the induction of systemic and tumourspecific immune responses. To date, the in situ injection of CPMV, Cowpea severe mosaic virus (CPSMV) and Tobacco ring spot virus (TRSV), Cowpea Chlorotic mottle virus (CCMV), Physalis mosaic virus (PhMV), and Sesbania mosaic virus (SeMV), TMV, PVX, Papaya mosaic virus (PapMV), Alfalfa mosaic virus (AMV) have demonstrated anti-tumour potentials in mouse models.
PVNP-mediated ISV is generally effective only against small tumours, and most patients do not respond to PVNP monotherapy. Combining multiple treatment regimens with PVNP -based ISV could form the basis for success. For example, immune checkpoint therapy (ICT) has the potential to treat cancer by removing the immunosuppressive brakes on T cell activity. It is shown that combined treatment with CPMV and selected checkpoint-targeting antibodies, specifically anti-PD-1 antibodies, or agonistic OX40-specific antibodies, reduced tumour burden, prolonged survival, and induced tumour antigen-specific immunologic memory to prevent relapse in mouse tumour models. In addition, CPMV-based in situ vaccination combined with systemic low-dose CPA chemotherapy achieved impressive synergistic efficacy against 4T1 tumours. Low doses of CPA induce pro-immunogenic activity in tumour cells, including the hallmarks of immunogenic cell death (ICD). Activated APCs with CPMV ISV induce IL-12, IFN- , and IFN- and potentiated to induce an adaptive immune response. Data indicated that the combination of RT + CPMV enhanced efficacy over RT alone, and that this may be attributed to an expansion of T cells within the tumours. Many investigations examined the combination of PVNP in situ vaccination with chemotherapy, radiation therapy, checkpoint immunotherapies. Overall, multifunctional PVNPs can combine multiple treatment modalities into a single platform with ISV.
Conclusions
The development of plant viruses as expression vectors for pharmaceutical production has played an integral role in the emergence of plants as inexpensive and facile systems for the generation of therapeutic proteins. More recently, plant viruses have been designed as nontoxic nanoparticles which can target a variety of cancers via loading conventional therapeutic agents, tumour antigens, and immunotherapeutic agents.
The tendency of PVNPs to interact with and become phagocytosed by innate immune cells can empower the immune system to slow or even reverse tumour progression.
References are available at www.pharmafocusasia.com
Kathleen Hefferon received her PhD in Medical Biophysics at the University of Toronto and currently is on faculty in the Department of Microbiology, Cornell University. Kathleen also has a visiting professor appointment in the Department of Cell and Systems Biology, University of Toronto. Kathleen's research interests include global health, food insecurity and plant biotechnology.

Mehdi Shahgolzari obtained his B.Sc. (Biology) from Azad University of Borujerd, Iran, and his M.Sc. (plant biology) from Bu-Ali Sina University, Hamedan, Iran. He is Medical Nanotechnology Ph.D. He works on nanoimmunotherapy, and in situ vaccination for cancer immunotherapy.

Afagh Yavari obtained her B.Sc. (plant Biology) from Alzahra University, Tehran, Iran, and her M.Sc. (plant biology) from Bu-Ali Sina University, Hamedan, Iran. She is a plant physiology Ph.D., assistant professor of the Department of Biology, at Payame Noor University, Tabriz, Shabestar, Iran. She works on plant stresses, and seed priming.

53 www.pharmafocusasia.com
AUTHOR BIO RESEARCH & DEVELOPMENT
Human Challenge Trials
Establishing early riskbenefit in development of vaccines and therapies for infectious diseases
Human challenge trials are a useful approach in the establishment of early efficacy for vaccines / therapeutics for infectious diseases. This article will describe how they can be used as a strategic tool for establishing an early risk-benefit and guide and enhance the success rate of future clinical development.
 Bruno Speder, VP, Regulatory Affairs, hVIVO plc
Bruno Speder, VP, Regulatory Affairs, hVIVO plc
Although slowly fading away from the collective memory, the SARSCoV-2 pandemic that started in early 2020 and the unprecedented health crisis on a global scale that followed, reminded us about the necessity of pandemic preparedness.
Pandemics are not new to humanity. Throughout history, mankind has been confronted with pandemics on a regular basis. Two well-known examples are the bubonic plague, better known as the Black Death, that ravaged large parts of Europe in the 1300s killing between 25 and 75 million people, and the “Spanish flu” influenza pandemic killing 40–70 million people worldwide shortly after the First World War.
CLINICAL TRIALS
In the second half of the 20th century, a number of less severe pandemic influenzas emerged in 1957–58, 1968, and again in 2009. In each instance, influenza vaccines targeting specifically the circulating virus were developed, although the scientific community is still debating how effectively the vaccines curtailed the disease spread. Also tropical diseases can cause a pandemic, as illustrated by the 2014–2016 Ebola outbreak in west Africa caused over 28,000 cases and 11,000 deaths.
Vaccines and antivirals are a critical part of our arsenal against infectious diseases,Their development however is a challenging, lengthy and expensive process with a high attrition (Gouglas et al., 2018).
Establishing an early risk-benefit profile for a vaccine or antiviral under development is essential to guide the future strategy and speed up time to market. Human challenge studies (HCT) or Controlled Human Infection Models (CHIM) are useful tools to assess early efficacy.
Human challenge trials
In human challenge studies, healthy volunteers are administered a well-characterised pathogenic or virulent strain of a challenge agent. This can be a virus (i.e. influenza), bacteria (i.e. cholera) or a parasite (i.e. malaria) depending on the trial.
Challenge studies are not a novel concept. Already in the 19th century, Louis Pasteur conducted challenge trials where chickens were challenged with a weakened bacteria causing chicken cholera, immunising them from further chicken cholera infection. In the United Kingdom, HCTs have been performed since 1946 when the Medical Research Council established the Common Cold Unit (CCU) — also known as the Common Cold Research Unit (CCRU) — at Salisbury, Wiltshire. This unit was set-up to research common colds in view of reducing their human and
economic costs (Metzger et al., 2019; Roestenberg et al., 2018).
Since the 1990s HTCs have found their way into clinical development, and are used to provide early performance data through proof-of-concept (PoC) trials and to establish the mode of action (MoA) of vaccines and antivirals. Governmental and commercial organisations are developing various challenge models to accelerate vaccine development programmes in respiratory tract infections like influenza and RSV. They are also used to gain a better understanding of the underlying pathological processes that drive immune responses. HCTs were first mentioned in regulatory guidance in 2010.
Challenge trials have been performed in a great variety of infectious diseases as illustrated in the table below, including, recently, a COVID-19 challenge model (Killingly et al. 2022). (Figure 1)
In a CHIM trial a well-characterised strain of an infectious agent (virus / bacteria / parasite) is given to carefully selected adult volunteers after they have been vaccinated or before they receive
the antiviral to be tested. They follow a traditional double-blind design comparing the treatment group with a placebo control group. These trials are performed in specialised quarantine units, where the inoculated volunteers are under 24/7 medical supervision. A schematic overview of HCTs involving a vaccine and an antiviral can be found below: (Figure 2) (Figure 3)
In current drug / vaccine development, human challenge trials can be used in a variety of ways, but the most common is for proof-of-concept (PoC) studies. Due to the reduced number of subjects needed for a challenge trial (40-60 subjects) compared to a classic `field` PoC trial (180-600 subjects), HCTs provide a cheaper alternative. In addition, their shorter duration allows for a quicker availability of efficacy data.
The primary objective of POC trials is to provide early evidence about efficacy and to establish the likelihood of a drug being successful in later clinical trial phases. Hence, PoC studies allow drug developers to make smarter and
55 www.pharmafocusasia.com
CLINICAL TRIALS
Figure 1 Overview of Human Challenge Trials per Pathogen (Adams-Phipps et al., 2023)
data-driven "go or no-go" decisions about continuing with the clinical development and start with larger, more expensive clinical trials.
They can also be used as dose-finding studies, testing different dosing regimens or vaccine / adjuvant combinations to decide which combination to move forward to field trials. This approach is being used extensively in the downselection of malarial vaccine candidates and is now an integral part of the malaria vaccine development cycle (Sauerwein et al..2011) — In the development of Mosquirix (RTS,S) for example, a phase 2 malaria human challenge trial was used not only to show early efficacy, but also to test different vaccine adjuvant regimens. (rationale: a new malaria vaccine has been approved recently) This enabled the initiation of phase 3 field trials conducted in over 15,000 infants and young children in Africa (RTS SCTP, 2015) and the ultimate market approval of the vaccine.
The results of point of care (PoC) studies are pivotal for strategic decisions in early drug clinical trial development and help the design of later stage field trials (Baay et al., 2019; Wildfire, 2021). Being able to establish an early risk-benefit profile based on phase 1 safety data (`risk`) and HCT efficacy data (`benefit`) can guide further finetuning by taking a data driven approach, and deciding which candidates or treatment regimens to bring to field trials. This reduces the risk of `late stage` failures and focuses time and resources on vaccines and treatments that are more likely to show success in phase 3 trials.
Regulatory acceptance
Data from CHIM trials have been
accepted by regulators as supportive for the following:
• As PoC studies for influenza and other upper respiratory tract infections. In such early phase studies, protective (vaccine) or curative (vaccine/drug) efficacy is being assessed. There are currently no specific guidelines regarding efficacy markers (correlates of protection) for PoC in CHIM studies, although the US FDA guideline on influenza studies mentions haemagglutination and tissue culture infective dose 50 per cent (TCID50)
• As a method for determining optimal dosage (to identify the correct individual dose, dose range, or schedule for field studies)
• `preliminary clinical evidence` in the framework of Fast Track / Breakthrough designation by the US Food and Drug Administration
• Acceptance as a pivotal efficacy trial: The Vaxchora vaccine — aimed at preventing cholera in travellers — received marketing authorisation approval based on a pivotal efficacy part. Vaxchora received marketing authorisation valid throughout the EU in April 2020 and was approved by the FDA in June 2016. In the US, the marketing authorisation holder is PaXVax; in Europe it is Emergent BioSolutions. The challenge trial was supported by a large safety immunogenicity trial. The main reason for both the FDA and the EMA to have agreed with a CHIM trial as a pivotal efficacy trial is that it would have been extremely difficult to perform a meaningful Phase III trial that would give conclusive results in this indication (FDA, 2016; EMA, 2020).
The World Health Organization (WHO) has published guidance on the use of challenge trials in vaccine development (WHO, 2016; WHO, 2017; WHO, 2020a; WHO, 2020b). The guidance also specifies that if they are performed, human challenge trials may be of particular use:
• When there is no appropriate nonclinical model (eg, when a candidate vaccine is intended to protect against an infectious disease that is confined to humans)
• When there is no known immune correlate of protection (ICP)
• When vaccine efficacy field trials are not feasible.
HCTs can play a unique role in resolving a number of the issues faced in the development of a future `pandemic vaccine`, like the recent SARS-CoV-2 pandemic.
The first advantage is that fewer subjects are needed for a CHIM (80 –120) than for a classic clinical field trial (200-400). Also the inclusion criteria for the study group can be limited to subjects at the lowest possible risk, ie, healthy 18 to 25-year-olds with no comorbidities. Secondly, challenge trials can be used to establish the infectious dose of the pandemic virus, starting from a very low dose and uptitrating in a classic Single Ascending Dose design. Additionally, CHIMs can be used for side-by-side comparison of vaccines, limiting the need for placebo controls, and therefore reducing potential ethical burden. They can also be used to determine correlates of protection, which could in turn be used to better design Phase III studies. Finally, if the CHIM results are favourable, such a trial can be used to support
56 PHARMA FOCUS ASIA ISSUE 52 - 2023
Figure 2 Schematic overview HCT with vaccine (green: quarantine phase / blue: outpatient phase)
Follow-up Discharge Quarantine Challenge Antiviral Screening Admission Quarantine Follow-up Discharge Quarantine Challenge Screening Admission Quarantine Vaccine CLINICAL TRIALS
Figure 3 Schematic overview HCT with antiviral (green: quarantine phase / blue: outpatient phase)
an emergency use authorisation application, for example for use in high-risk populations (Baay et al., 2021; Rapeport et al., 2021).
Early interaction with regulatory agencies including the European Medicines Agency (EMA), Food and Drug Administration (FDA), etc, via a scientific advice meeting or a pre-IND meeting is strongly recommended to discuss the implementation of a Human Challenge Trial in any development programme.
Performing a human challenge trial
Another important issue are the regulatory and operational aspects of running a human challenge study itself. It is extremely important to avoid crosscontamination between the patients infected with the virus on one side and the study staff on the other side. The aim is to avoid the virus from being spread to the “outside world” and to avoid infected study staff infecting the patients and potentially jeopardising the study results by infecting placebo patients. Before enrolling into a HCT, volunteers undergo a battery of screening test, including a serosuitability test to ensure potential volunteers don’t already have antibodies against the challenge agent being used.
Participants are isolated in a specifically designed quarantine unit and will be treated according to the principle of “reversed-barrier nursing.” This method is very similar to barrier nursing used in an intensive care unit (ICU) setting, where the aim is to keep pathogens away from the ICU patients by creating a barrier between the outside world and inside a patient room by using gloves, masks, gowns, and disinfectants. The same principle also confines the challenge agent to the facility and prevents it from spreading into `the outside world`. Depending on the pathogen used subjects can remain in quarantine for up to two weeks.
Conclusion
HCTs play a very important and support-
Human challenge studies (HCT) or Controlled Human Infection Models (CHIM)
ive role in both our understanding of disease and the testing of novel antivirals and vaccines. Animal models for many diseases are poor at predicting disease pathogenesis especially for human host restricted diseases. Properly designed and ethically conducted CHIM studies
AUTHOR BIO

Bruno brings a wealth of drug development and regulatory strategy experience to hVIVO (formerly Open Orphan). He has an extensive expertise in supporting global drug development programs and guiding biotech / pharma companies in their interactions with global regulators, including EMA & FDA. He is currently advising a broad range of organisations (non-profits, biotechs, large pharma) on the regulatory aspects of their drug/vaccine development, from early development to commercialisation stage. Before joining hVIVO, Bruno had a variety of roles at SGS Life Sciences, where in his last position he managed the global regulatory consultancy and modelling & simulation teams. Bruno is a bio-engineer in cell- & genetechnology from the University of Ghent in Belgium, and holds a postgraduate degree in Health Economics (EHSAL Management School, Belgium).
have tremendous potential to improve our understanding of pathogenesis, help design better vaccine candidates, reduce the costs and timelines of vaccine / drug development by providing early insights in the efficacy and dosing / administration regimens.
In combination with phase 1 safety data, this establishes an early risk-benefit profile to further guide the development of the vaccine / antiviral.
As illustrated by the Vaxchora approval, human challenge trials can be used as the `pivotal efficacy` efficacy element in a marketing authorization application. A similar approach can be envisaged in a regulatory pathway for accelerated approval for any future pandemic vaccine.
References are available at www.pharmafocusasia.com
are useful tools to assess early efficacy.
CLINICAL TRIALS
Respirable Engineered Spray Dried Dry Powder as a Platform Technology

Respirable Engineered Spray Dried Dry Powder represents a technology to generate a low bulk density dry powder that can be inhaled via slow and deep inhalation using a simple to use, low cost Dry Powder Inhaler such as the Plastiape RS00 Mod 2 Low Resistance Device for therapeutics delivered both regionally to the lungs as well as to the systemic vasculature using the lungs as a portal. The particles are entirely homogeneous in composition and allow the pharmaceutical actives to be stabilised in an amorphous glassy state which results in stability at ambient conditions. Case Studies for Budesonide, Leuprolide and Tobramycin Sulfate are discussed along with the Regulatory Strategy along the 505(b) 505(b)(2) regulatory route pathway..
Aditya R Das, Founder and Principal, Pharmaceutical Consulting LLC
The dichotomous branching of the lungs from the 0th generation (the trachea) to the 23rd generation of the lungs offers a surface area of 110m2 of the alveolar region of the 23rd generation where it takes a red blood cell approximately 0.25 seconds to traverse the entire network where gas exchange occurs and the RBC actually travel in single file. This deep lung membrane provides a half life of delivery for a hydrophobic molecule of <30 seconds and allows hydrophilic molecules also to penetrate to the systemic vasculature hence allowing the lungs to be used either for regional delivery or as an entryway to the systemic vasculature depending on the disease being treated and provides an efficient bypass to the “first pass” effect through the liver as well as access to the central nervous system.
The bulk density of spray dried powders may be engineered to be 0.6-0.8g/cc or 0.06-0.08g/cc which allows delivery to the local lung area (mid lungs) for the treatment of diseases such as asthma or infectious diseases versus using the ultra-low density bulk powder for delivery to the deep lungs and thence to the systemic vasculature.
When slowly and deeply inhaled (inhalation flow rate of 25-35LPM), the particles of homogeneous composition are able to traverse the bend in the throat and upon reaching the 3rd generation of the lungs, gravitational sedimentation takes over (where the Reynolds number of the flow is ~0) to bring the particles to the deep lung as a natural consequence.
One of the most simple devices (a thermal aerosol vapor inhaler) using a
58 PHARMA FOCUS ASIA ISSUE 52 - 2023
MANUFACTURING
coated API without any excipients using an airflow trigger (at 20 LPM) to actuate the burning of a fuel coated inside a steel box housed in a polycarbonate housing allows a hydrophobic API such as Loxapine to be inhaled directly through the lungs to the systemic vasculature with a Tmax of 2 minutes.
Similarly, tobramycin sulfate which was used as a treatment for pseudomonas aeruginosa infection in the lungs of cystic fibrosis patients resulted in the patients sitting in front of a table-top Nebulizer inhaling a 300mg dose over a period of 30 minutes with approximately 9% of the dose delivered to the whole lung. A new delivery method using the ultra-low density spray dried dry powder resulted in a reduction of the dose to 112mg taken over 5 minutes with an active content of 28mg tobramycin in each of 4 capsules (50mg bulk powder fill, inserted into a DPI, pierced and orally inhaled).
Details around Regulatory Strategy of the 505(b) 505(b)(2) regulatory route pathway in the US or also known as a Hybrid Application in Europe have already been discussed previously in an invited paper including the clear differences shown by gamma scintigraphy studies of whole lung deposition comparing the Pulmicort pMDI, Pulmicort Turbuhaler and Inhale Therapeutic Systems first generation spray dried calcitonin powder.
Case studies
1. Leuprolide
Luteinizing hormone releasing hormone (LH-RH) agonists are currently used for treatment of disease states susceptible to excessive or detrimental concentrations of steroid sex hormones, such as endometriosis or prostate cancer. Dosing is limited to parenteral or nasal administration due to the low oral bioavailability of LH-RH agonists, which is secondary to extensive first pass metabolism. The major products, such as Zoladex® (AstraZeneca Pharmaceuticals LP, Wilmington, DE)
and Lupron (TAP Pharmaceutical Products Inc., Lake Forest, IL) are available as depot parenteral formulations that require dosing once every one to three months, depending on the formulation and indication. The use of monthly depot products greatly increases patient convenience and compliance and reduces the frequency of painful injections, but the irretrievable and long-term nature of these formulations limits the physician’s ability to adjust dosing to manage adverse events. Product labelling indicates that chemical castration results in most patients receiving the recommended dose, leading to a significant incidence of vasomotor symptoms (hot flashes), headache, and to a six-month limit of therapy secondary to clinically significant loss of bone mineral density.
In the case of LH-RH agonists, pulmonary delivery offers a stable, reproducible drug input profile similar to that of oral dosing, free from changes in drug input (such as initial drug bursts) which occur with time following administration of depot formulations, and the ability to adjust the dose in response to the clinical situation, or stop therapy if indicated or desired by the patient. Thus pulmonary delivery of leuprolide is expected to improve patient care by providing reversible, adjustable, efficacious treatment of endometriosis using a proven LH-RH agonist, but using a dosing procedure in the patient’s home that is little more complex than oral administration.
The PulmoSphere® particle engineering technology produces ultralow density powders for inhalation using portable, passive dry powder inhalers, without the need for chlorofluorocarbon propellants. Proof-of-concept clinical studies have been completed with both the corticosteroid budesonide and the anti-infective tobramycin sulfate. In the budesonide study, 67 per cent% of the emitted dose was deposited in subject’s lungs independent of peak inspiratory flow rate, while the tobramycin clinical study illustrated that powder doses as large as 25mg could be delivered efficiently in a single inhalation.
Fivemg of the leuprolide PulmoSphere powder (0.75mg leuprolide acetate) was hand-filled into size #2 hydroxypropylmethylcellulose (HPMC) capsules (Shionogi, Nara, Japan). The capsules
59 www.pharmafocusasia.com
Mean Budesonide Deposition Pulmosphere Eclipse Pulmicort Turbuhaler N=8 29 LPM 44LPM 30 LPM 60 LPM Device (%) 13.6 13.0 11.2 12.8 Oropharynx (%) 29.4 29.3 79.2 69.8 Lung (%) 57.0 57.7 9.1 16.8
LPM = Liters per minute (Inhalation Flow Rate) Results show that in the case of Budesonide Pulmospheres, the lung deposition (% of nominal dose) of ~57 per cent is independent of flow rate
1, 0.5x Oropharyngeal Deposition
1 2 MANUFACTURING
2, 2.1x Lung Deposition from ~1/2 the dose
were then loaded into the Turbospin® dry powder inhaler (PH&T, Milan, Italy) for aerosol administration to subjects. The Turbospin (now referred to as the Inhale T-326 dry powder inhaler) is a portable passive dry powder with a device resistance of 0.09 (cm H2O1/2)/(L·min-1).
Leuprolide acetate for injection (Lupron®, TAP Pharmaceutical Products Inc., Lake Forest, IL) was supplied in vials containing 2.8ml of leuprolide acetate (5mg/ml). The formulation also includes sodium chloride for tonicity adjustment, and 9mg/ml of benzyl alcohol (preservative). The leuprolide concentration was diluted to 0.71mg/ml with normal saline prior to injection of a 1 ml bolus.
• The bioavailability of leuprolide
PulmoSphere powder for inhalation relative to an intravenous dose was 16.5 %, with mean peak serum concentrations of 4.0ng/ml observed 1 hour after inhalation
• Therapeutic serum leuprolide concentrations, comparable to those reported for the TAP MDI, were achieved with the leuprolide PulmoSphere formulation delivered from a portable, passive, dry powder inhaler
• The single 0.75mg dose of leuprolide
PulmoSphere was safe and well tolerated in this study of 12 healthy male volunteers.
2. Budesonide Pharmacoscintigraphy of Budesonide Powders
Dose Pulmosphere Eclipse: 372 μg Budesonide
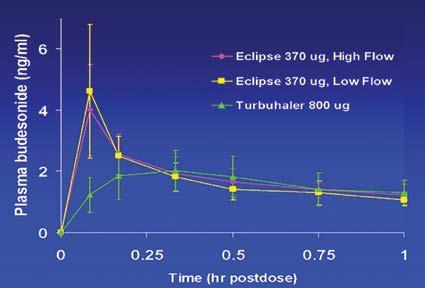
Dose Pulmicort Turbuhaler: 800 μg Budesonide (Micronized Blended Lactose)
Conclusions
A platform technology comprising a respirable engineered spray dried dry powder demonstrates flexibility and diversity in the development of a flow rate independent whole lung drug delivery of a homogeneous, low bulk density and room temperature stable product that can incorporate up to 3 APIs (e.g. ICS, LABA and LAMA of Astra Zeneca’s Breztri based on the Pulmosphere Platform-Pearl Therapeutics a spinoff of Inhale Therapeutics who were licensed to use the Pulmosphere Technology in pMDIs was bought by Astra Zeneca). This results in a low cost, self administered medication and is amenable to a large variety of APIs from small molecules, peptides, globular proteins, monoclonal antibodies, siRNA, mRNA, DNA and whole killed vaccines all of which are stabilised in an amorphous glassy state when spray dried due to the fact that the water evaporation from the drying droplet in <300ms, results in a Joule Thomson cooling effect that is instantaneous. The 505(b)(2) regulatory route offers a rapid entry to market-typically a Phase 1 for Safety followed by a Pivotal Phase 3 for both Intranasal as well as Pulmonary Delivery.
References are available at www.pharmafocusasia.com
AUTHOR BIO
30 years work experience in the US Biopharmaceutical Industry focused on Combination Product Therapeutic Strategies for the treatment of Infectious, Genetic, Allergic, Metabolic, Oncologic and Cardiovascular Disease. Assisted in the successful submission and marketing of 3 Products-Dry Powder Inhaled Insulin (Exubera, approved by EMA and FDA in Jan 2006), Thermal Aerosol Vapor Staccato Loxapine (Adasuve approved by FDA and EMA Dec 2012 and Jan 2013 respectively) and Dry Powder Inhaled Tobramycin Sulfate (Tobi PodHaler, approved by FDA on March 2013). Very strong background in all aspects of Drug Product Development and CommercializationNonclinical, Clinical and CMC Strategies and Submissions involving Global Regulatory Agencies in addition to US FDA and European EMA include Health Canada, China CFDA, Japanese PMDA, Indian CDSCO and DCGI, UK MHRA and Australian TGA, Brazilian ANVISA and South African SAHPRA.

60 PHARMA FOCUS ASIA ISSUE 52 - 2023
MANUFACTURING
Drug development has transformed immeasurably over the past decade. Even before the Covid19 pandemic and the conflict in Ukraine, which have contributed to raw material shortages and unprecedented supply chain disruption, the pharma sector’s tectonic plates were shifting. With the emergence of high-value, low-yield biologics — coupled with increasing competition — the concept of ‘waste’ has been brought into sharper focus.
Just in Time Manufacturing Creating more sustainable supply chains
Rising to the challenges of modern drug development, while effectively responding to growing pressure to operate more sustainably is dependent on a fresh approach - one where waste is no longer tolerated. According to Almac’s Lyn McNeill, Just in Time Manufacturing is a supply chain strategy that can help Sponsors achieve just that.
In today’s geopolitical climate, where demand outstrips supply, key resources — including APIs, comparators, co-therapies, and clinical packaging materials - are more difficult to come by and more expensive to procure. The cost of raw materials for the pharmaceutical sector has increased by up to 160 per cent and therefore waste can no longer be afforded. And while the commercial impact of waste remains a key driver for sponsors to embrace ways to limit it,
pressure to operate more sustainably is emerging as another influencing factor for change.
With growing numbers of sponsors scrutinising clinical trial operations against corporate Environmental, Social, and Governance (ESG) frameworks, sustainability is no longer a buzzword in the background. Instead, sustainability is rapidly becoming a key performance indicator and a mechanism to lower the environmental impact of bringing
61 www.pharmafocusasia.com
Lyn McNeill, JTM Supply Chains Solutions Manager, Almac Clinical Services
MANUFACTURING
new drugs to market, ensuring limited resources are ethically utilised to safeguard patient access, and drive more cost-effective operations.
Just in Time (JIT) manufacturing has an important role to play in supporting sponsors to meet these objectives by injecting flexibility into supply chains, preserving precious clinical trial material (CTM), and significantly reducing product waste.
The push for sustainability
To keep global warming to no more than 1.5°C – as called for in the Paris agreement Paris – emissions must be reduced globally by 45 per cent by 2030, and we must collectively reach net zero by 2050.
Conversely, the pharma industry’s CO2 footprint is set to triple by 2050. If mitigation measures are not widely adopted , the need to urgently and meaningfully seek out opportunities to operate clinical trials more sustainably is clear.
A break from tradition
One way to reduce waste arising from clinical trial operations — and deliver enhanced sustainability — is to replace traditional drug production approaches with LEAN methodologies, including JIT.
JIT can range from partial late-stage customisation, where an auxiliary label is added at the time of dispatch, to full latestage customisation, where drug supply is stored in its primary pack, such as naked vials, ampules & syringes, until there is an actual patient need at a clinical site.
Just in Time Labelling (JTL) is ideal for quick and simple additions to an already clinically labeled kit and is typically best suited to small molecule operations involving wallets and bottles. For biologics, Just in Time Manufacturing (JTM) can help create optimised and adaptive supply chains that promote enhanced product viability and reduced product waste.
With a JTM model, the process of label printing and kit assembly for full late-stage customisation is initiated
by drug order forms. This introduces key advantages for sponsors looking to optimise the use of clinical supplies while maintaining viability. Firstly, this approach provides sponsors with the ability to utilise single panel, country-specific labels. Incidentally, this has been proven to decrease study start-up timelines by up to 50 per cent, helping sponsors meet key study milestones faster and more costeffectively.
Secondly, it empowers sponsors to print study-specific labels with the most current label variables, such as the most recently approved expiry date. This helps to reduce waste and improve biggerpicture efficiency. Finally, by delaying labelling and kit assembly until clinical need arises, supplies remain usable for any country or study, which drastically reduces the risk of costly stock outages that can negatively impact patients and overall trial performance.
Prevention over cure
A common scenario where JTM’s role in delivering more sustainable clinical supply chains is evident relates to the traditional method of site seeding.
When this is the case, one of two scenarios unfold. In the first, CTM remains on the shelf at the clinical site before being returned, reworked, and redistributed to fulfill anticipated needs elsewhere at a significant cost to
the sponsor and the environment. The second scenario involves unused CTM that has already been produced, transported, and stored at the site — within specific temperature ranges that bump up energy demand and environmental impact — being returned to the sponsor for reconciliation and destruction.
In contrast, JTM allows sponsors to prevent the added financial and environmental impact of sites failing to recruit patients. With a JTM approach, drug is only sent to sites once patients are recruited and demand is known. This is achieved by storing bulk inventory that can be packaged and labelled on demand, resulting in highly customised patient kits. This also removes the need to forecast large quantities of bulk drug for packaging, which typically see errors of +50 per cent - 200 per cent. With JTM, drugs do not gather dust on site shelves or require rework, return, or destruction, which contributes to less waste and more sustainable supply chains.
A greener response to mid-study changes
Of course, site seeding is not the only scenario that JTM delivers on its waste limiting potential. It is standard for studies to be changed significantly or stopped all together mid-stream. Whether stemming from emerging study dose information, adverse events, recruitment challenges, regulatory hurdles, protocol design discrepancies, or the addition of new territories, the impact is the same. If supply is produced in bulk utilising a traditional approach, all will require return, reconciliation, rework, or destruction at significant financial and environmental cost.
Contrastingly, with JTM, only the clinical supplies that have been distributed are lost. The rest remains unpackaged and unlabelled in inventory and can be utilised in another configuration for the same or a different study altogether.
This is particularly pertinent for biologics and next generation cell and gene therapies (CGT) that require precise handling and extremely limited time out
62 PHARMA FOCUS ASIA ISSUE 52 - 2023
JTM’s ability to expedite key study milestones, promotes increased supply chain agility, and reduces the need for accountability and destruction activity.
MANUFACTURING
of conditions. These compounds require significant energy to manufacture, transport, and store at ultra-low and even cryogenic temperature ranges. JTM can help to offset some of this environmental impact by extending the lifecycle of the drug and mitigating product waste that would have a substantial impact on both ESG credentials and a sponsor’s bottom line.
As these compounds are more expensive to manufacture and have extremely limited stability, product conservation is paramount. Advanced therapeutics especially are high value commodities, with the cost for manufacturing one batch of CGT product typically varying between US$500,000 and US$1m. Considering the proportion of the batch that will need to be allocated for samples, stability testing and so forth, the number of units that can be utilised for clinical trial use is already low; resulting in little margin for error. As such, a traditional production approach – that necessitates time out of conditions to manually rework products (that have been pre-packaged and labelled) in the event of a mid-study change - threatens product integrity and sustainability objectives.
However, by harnessing JTM, sponsors can better conserve high cost, low yield, low stability, energy intensive supply by eliminating the need to remove naked vials etc. from appropriate conditions to rework it. Instead, sponsors can manage changes, such as expiry updates, within systems and without ever physically touching the bulk drug product. This increases the longevity of the drug’s lifespan, promotes enhanced product integrity, and removes the need to exert additional energy reworking an entire batch.
Considerations for adopting JTM

The limited stability, high value, low yield nature of large molecule compounds also goes hand in hand with unpredictable recruitment, especially where next generations CGTs are concerned. Being patient-specific and primarily used to
target rare diseases or last-lines of therapy, CGT serve smaller patient populations due to low disease prevalence and/or strict eligibility criteria. This serves to cloud visibility for forecasting and heightens the risk of product waste. Limited stability and short expiry dates combine with unpredictable recruitment to create a perfect storm where inefficiency, risk and waste can escalate. And with the increase in CGT trials, more sponsors will need to develop effective mitigations to address the associated issues and ensure sustainable, cost-effective supply chain practice prevails.
Despite the added complexity of bringing all drug compounds to market in a post-pandemic clinical trials’ landscape, drug development continues to boom. There are now more ongoing clinical trials than ever before, with the number of initiated trials increasing by 59 per cent from 2012 to 2021 .
However, while JTM is well established in other sectors, pharma has taken longer to embrace its potential. This is owed in part to misconceptions surrounding costs, with many sponsors assuming it to be a more expensive method of assuring supply to patients compared with traditional approaches. This supposes that running smaller operations more frequently chips away at economies of scale yet fails to acknowledge the bigger picture. While initial overheads of JTM can be higher than traditional manufacturing approaches, the financial and environmental savings delivered via a substantial reduction in product waste soon justify the investment. As does JTM’s ability to expedite key study milestones, promotes increased supply chain agility, and reduces the need (and financial burden and environmental impact) for accountability and destruction activity.
Another important consideration to bear in mind when weighing up the merits of JTM in relation to waste reduction is that it does not have to be all or nothing, and it is not just capable of adding value to small studies. Many
sponsors are now harnessing JTM at the start of a study when demand is less predictable, and the location of patients remains unclear. Then, once enrolment stabilises and demand is less of an unknown, switching to traditional production approaches becomes feasible. Likewise, a hybrid model featuring both JTM, and a traditional approach is also an option. This can be ideal in trials with multiple arms, where some treatments lend themselves better to JTM than others, in terms of high unit price and availability.
Drug development will continue to evolve and the push for more sustainable supply chain operations is only going to increase, along with the commercial and ethical pressure to mitigate waste in a competitive market. While JTM by no means represents the silver bullet in the quest to establish carbon neutral operations, it does have proven potential to drastically reduce waste and support sponsors to uphold their responsibility to patients and profit margins.
References are available at www.pharmafocusasia.com
63 www.pharmafocusasia.com
AUTHOR BIO
MANUFACTURING
Lyn McNeill is the Just in Time Manufacturing Solutions Manager at Almac Clinical Services in Northern Ireland. Lyn joined Almac in 2016 and has transitioned through roles in IRT and business Project Management before taking on responsibility for service implementation, stakeholder engagement and development of Almac's Just in Time service suite in August 2021.

64 PHARMA FOCUS ASIA ISSUE 52 - 2023 PRODUCTS & SERVICES STRATEGY DUPHAT 2024 ...................................................................................... 47 Etihad IFC ISPE 10-13, 29 Invest Selangor Berhad........................................................................ 15 MasterControl 03 Medical Fair Thailand ............................................................... 21, 40-41 Qatar Cargo IBC Turkish Cargo .................................................................................... OBC Valsteam ADCA Engineering 05 RESEARCH & DEVELOPMENT Ashland India .................................................................................. 32-35 Curia Global Inc 09 Invest Selangor Berhad 15 MasterControl ...................................................................................... 03 MANUFACTURING Ashland India 32-35 Curia Global Inc ................................................................................... 09 Invest Selangor Berhad 15 MasterControl ...................................................................................... 03 Valsteam ADCA Engineering 05 Company ............................................................. Page No. SUPPLIERS GUIDE Ashland India .................................................................................. 32-35 www.ashland.com Curia Global Inc 09 curiaglobal.com DUPHAT 2024 47 https://duphat.ae/ Etihad IFC www.etihad.com ISPE .......................................................................................... 10-13, 29 https://ispe.org/ Invest Selangor Berhad........................................................................ 15 www.investselangor.my MasterControl 03 www.mastercontrol.com Medical Fair Thailand 21, 40-41 www.medicalfair-thailand.com Qatar Cargo IBC www.qrcargo.com Turkish Cargo .................................................................................... OBC www.turkishcargo.com Valsteam ADCA Engineering ................................................................ 05 www.valsteam.com Company ............................................................. Page No.
receive more information on products & services advertised in this issue,
fill up the "Info
Form"
with the magazine and fax it. 1.IFC: Inside Front Cover 2.IBC: Inside Back Cover 3.OBC: Outside Back Cover
To
please
Request
provided
Ensuring lifelines.

Our Pharma product is designed to transport your pharmaceutical and healthcare cargo safely and efficiently. We guarantee a seamless cool chain for your temperature-sensitive goods.
 by GENERATION
by GENERATION
65 PHARMA FOCUS ASIA ISSUE 52 - 2023

