

ISSUE 54 2024 www.pharmafocusasia.com Advanced Drug Delivery Systems to Address Unmet Medical Needs Enhancing Pharmacovigilance through the Scope of Artificial Intelligence Sponsors Innovations and Transformative Trial Designs Clinical research evolves

Transporting critical pharma and medical devices across 6 continents. THE WORLD WORKS BETTER WITH EMIRATES SKYCARGO SKYCARGO.COM
Innovations and Transformative Trial Designs Clinical research evolves
In recent years, the landscape of clinical trials has undergone a remarkable evolution, propelled by groundbreaking innovations and transformative trial designs. These advancements have not only reshaped the traditional paradigm but have also ushered in an era of unprecedented opportunity and progress in the field of healthcare.
The impact of this transformation is remarkable. With the emergence of innovative trial methodologies and state-of-the-art technologies, researchers now possess an extensive toolkit that empowers them to address intricate challenges with unprecedented accuracy and effectiveness. This evolution reflects a shared dedication to pushing the limits of scientific exploration and an increasing awareness of the imperative to expedite the creation and distribution of life-saving treatments for patients in critical need.
A prominent trend is the emergence of innovative trial designs. While traditional methods in clinical research have proven effective in numerous instances, they have frequently been linked with inefficiencies, delays, and substantial costs. Yet, with the introduction of adaptive trials, platform trials, and other inventive methodologies, researchers are now equipped with the ability to adjust and enhance their protocols in real-time, guided by emerging data and insights. This not only simplifies the trial process but also bolsters its scientific integrity and credibility, ultimately enabling smoother, more dependable outcomes.
Furthermore, the incorporation of digital technologies and data analytics has transformed every facet of
the clinical research process. From patient recruitment and interaction to data gathering and analysis, these tools present unparalleled chances to capitalise on the potential of big data and artificial intelligence, facilitating the emergence of fresh discoveries and understandings at an unmatched speed. By utilising real-world evidence and advanced analytics, researchers delve deeper into disease mechanisms, treatment effectiveness, and diverse patient demographics, laying the groundwork for more tailored and efficient therapies.
Article on ‘Innovative Trial Designs - Revolutionising Clinical Research by Tihomira Nikolova from PSI CRO AG in this issue explores various innovative trial designs, real-world applications, and their impact on patient recruitment.
Other interesting articles include Next-Generation Approach through Advanced Research and Engineering Techniques in Cell Based Therapy, Advanced Drug Delivery Systems to Address Unmet Medical Needs, Enhancing Pharmacovigilance through the Scope of Artificial Intelligence and many more. Hope you will enjoy reading this issue.
Thank you for your support!
 Prasanthi Sadhu Editor
Prasanthi Sadhu Editor
1 www.pharmafocusasia.com
Innovative Trial Designs
Revolutionising clinical research
Tihomira

STRATEGY
06 Cell and Gene Therapy (CGT)
Key regulatory insights
Ghulam Moinuddin, A Scientific Regulatory Professional
09 The JAPAC Drug Development Market
Understanding the regulatory landscape and cultural nuances
Hye Jin Choi, Senior Director, Regulatory Affairs, IQVIA Asia Pacific
RESEARCH &
13 What do We Know About Albumin Formulations?
Luke Condon1#, Miral Suthar2#, Sara Woytowicz1, Gagan Kaushal2 and Ankit Rochani1*
1. Wegmans School of Pharmacy, St John Fisher University
2. Jefferson College of Pharmacy, Thomas Jefferson University
18 Cocktail Nanoparticles
A precision drug delivery system to cancer cell
Sumel Ashique, Department of Pharmaceutical Sciences, Bengal College of Pharmaceutical Sciences & Research
Radheshyam Pal, Assistant Professor, Pandaveswar School of Pharmacy
22
Next-Generation Approach through Advanced Research and Engineering Techniques in Cell Based Therapy
Sibi Raj, Dhruv Kumar, School of Health Sciences and Technology (SoHST), UPES
26 Biomarkers for Cardiovascular diseases
A brief insight
Vignesh M, Rao SR, Agiesh B, MGM-Advanced Research Institute, Sri Balaji Vidyapeeth Deemed to be University
29 Drug Analysis in Sports
Shobha Ahi, Deputy Director, Drug Control Centre, Department of Analytical, Environmental and Forensic Sciences, School of Cancer and Pharmaceutical Sciences, King's College London
MANUFACTURING
41 Ensuring Safety in Geriatric Medicine Use
Explicit criteria to avoid potentially inappropriate medication
Laksmi Maharani, Lecturer and Researcher, Jenderal Soedirman University
44 Nanoparticle Biomimetics as an Avenue to Personalised and Precision Medicine
Stefano Giovagnoli, Dept. of Pharmaceutical Sciences, University of Perugia
INFORMATION TECHNOLOGY
47 Enhancing Pharmacovigilance through the Scope of Artificial Intelligenceo
Aditya Dilipkumar Patil, Assistant Professor, Department of Homoeopathic Pharmacy, Noble Homoeopathic Medical College and Research Institute, Noble University
Sargam Ramesh Singh, Assistant Professor, Department of Gynaecology & Obstetrics, Noble Homoeopathic Medical College and Research Institute, Noble University



50 Advanced Manufacturing
An answer to supply chain woes?
Atul Dubey, Director, Pharmaceutical Continuous Manufacturing (PCM) at the United States Pharmacopeial Convention (USP)
54 Advanced Drug Delivery Systems to Address Unmet Medical Needs
Yogeshwar Bachhav, Director (Consultant), AiCuris Anti-infective Cures AG
2 PHARMA FOCUS ASIA ISSUE 54 - 2024
CONTENTS
Nikolova Clinical
PSI CRO AG
Research Associate,
CoverStory 33
DEVELOPMENT
















3 www.pharmafocusasia.com Pressure regulators Control valves Pipeline ancillaries Special equipment Steam traps Safety valves geral@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467 PRODUCTS MANUFACTURED IN PORTUGAL Guia PBL PORTUGAL HIGH PURITY. HIGH DEMANDS. High purity equipment for clean steam AV 001 AP E 02.20
Advisory Board













Alessio Piccoli
Lead, Sales and Business Development Activities Europe Aragen Life Science
Andri Kusandri
Market Access and Government & Public Affairs Director Merck Indonesia
Brian D Smith Principal Advisor PragMedic
Gervasius Samosir Partner, YCP Solidiance, Indonesia
Hassan Mostafa Mohamed Chairman & Chief Executive Office ReyadaPro
Imelda Leslie Vargas Regional Quality Assurance Director Zuellig Pharma
Neil J Campbell Chairman, CEO and Founder Celios Corporation, USA
Nicoleta Grecu Director Pharmacovigilance Clinical Quality Assurance Clover Biopharmaceuticals
Nigel Cryer FRSC
Global Corporate Quality Audit Head, Sanofi Pasteur
Pramod Kashid
Senior Director, Clinical Trial Management Medpace
Quang Bui
Deputy Director at ANDA Vietnam Group Vietnam
Tamara Miller
Senior Vice President, Product Development Actinogen Medical Limited
Vivek Ahuja
Senior Vice President, Global Delivery Excellence Strategy & Growth Pharmacovigilance Quality and Regulatory Services
EDITOR
Prasanthi Sadhu
EDITORIAL TEAM
Grace Jones
Harry Callum
Rohith Nuguri
Swetha M
ART DIRECTOR
M Abdul Hannan
PRODUCT MANAGER
Jeff Kenney
SENIOR PRODUCT ASSOCIATES
Ben Johnson
David Nelson
John Milton
Peter Thomas
Sussane Vincent
PRODUCT ASSOCIATE
Veronica Wilson
CIRCULATION TEAM
Sam Smith
SUBSCRIPTIONS IN-CHARGE
Vijay Kumar Gaddam
HEAD-OPERATIONS
S V Nageswara Rao
Ochre Media Private Limited Media Resource Centre,#9-1-129/1,201, 2nd Floor, Oxford Plaza, S.D Road, Secunderabad - 500003, Telangana, INDIA, Phone: +91 40 4961 4567, Fax: +91 40 4961 4555 Email: info@ochre-media.com
www.pharmafocusasia.com | www.ochre-media.com
© Ochre Media Private Limited. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, photocopying or otherwise, without prior permission of the publisher and copyright owner. Whilst every effort has been made to ensure the accuracy of the information in this publication, the publisher accepts no responsibility for errors or omissions. The products and services advertised are not endorsed by or connected with the publisher or its associates. The editorial opinions expressed in this publication are those of individual authors and not necessarily those of the publisher or of its associates.
Copies of Pharma Focus Asia can be purchased at the indicated cover prices. For bulk order reprints minimum order required is 500 copies, POA.


4 PHARMA FOCUS ASIA ISSUE 54 - 2024
Magazine Subscribe LinkedIn



5 www.pharmafocusasia.com Use Code "PFA15" for 15% Off Tickets World's Top Summit For Life Science Supply Chain Leaders Learn more: TO LEARN FROM, NETWORK AND BENCHMARK

CELL AND GENE THERAPY (CGT)
Key regulatory insights
The rapidly advancing field of cell and gene therapy (CGT) presents transformative solutions for a multitude of previously untreatable diseases. Over the last few years, numerous CGT products have successfully obtained clinical approval, with over 75 such therapies that are approved by Health Agencies now available for treatment worldwide. It underscores the immense potential of these modalities to treat or even cure otherwise intractable diseases. However, alongside its immense promise lies the critical need for a robust and adaptable regulatory framework to ensure patient safety and efficacy. Understanding the Regulatory landscape is paramount in the dynamic and groundbreaking field of Cell and Gene Therapy, where scientific innovation meets therapeutic potential.
This article delves into the intricacies of CGT Regulations and hurdles surrounding Cell/Gene Therapy, exploring the challenges and opportunities that this transformative field presents for researchers, clinicians, Pharmaceutical/Bio Pharmaceutical industry professionals, and Health Agencies.
Ghulam Moinuddin, A Scientific Regulatory Professional
CGT stands at the forefront of personalised medicine, revolutionising healthcare by modifying patients' own cells or genes for therapeutic purposes. From tackling cancers and genetic disorders to potentially treating neurodegenerative conditions, CGT offers unprecedented avenues for medical intervention. However, this transformative potential is inextricably linked to the establishment of a robust and adaptable regulatory framework.
Matching up to the requirements and synchronising with technological advancements, the regulatory environment for Cell and Gene Therapy is rapidly evolving. Health authorities, including the FDA, EMA, and other global Regulatory bodies, are pivotal in creating a scientific and structured landscape.
Key Regulatory Insights
Some of the key and recent insights from the FDA include:
Expected Upsurge in CGT IND Filings
As per the statistics disclosed by the Director of the FDA’s Office of Tissues and Advanced Therapies (OTAT), there
6 PHARMA FOCUS ASIA ISSUE 54 - 2024
STRATEGY
seems to be a drop in the number of Investigational New Drug (IND) filings for Cell and Gene Therapies (CGT) from 350 in 2020 to 299 in 2021. Despite this fact, these numbers are anticipated to rise again in the forthcoming years. Additionally, OTAT highlighted a noticeable rise in Breakthrough (BT) and Regenerative Medicine Advanced Therapy (RMAT) designation requests. These requests are typically submitted concurrently with IND filings or during an existing IND filing, with a simultaneous increase in accepted RMAT requests.
Bespoke Gene Therapy Consortium (BGTC)
It is well known that the FDA has launched a new initiative called the Bespoke Gene Therapy Consortium (BGTC) under its NIH Accelerating Medicines Partnership Program. The consortium is designed to simplify the development of small-batch gene therapies by addressing key therapeutic challenges. It will guide basic and clinical research, manufacturing, production, and Regulatory requirements for these therapies.
INTERACT Meetings
A relatively recent FDA initiative is the "INTERACT" informal meeting program initiated by CBER. The INTERACT program aims to address the questions and needs of the Cell and Gene Therapy (CGT) industry within existing clinical frameworks. It involves informal meetings between CBER staff and researchers or sponsors who are in the pre-Investigational New Drug (pre-IND) stage of development. The INTERACT meetings do not incur a Prescription Drug User Fee Act (PDUFA) fee, and their scheduling depends on CBER's availability and resources. It's important to note that these meetings do not replace other formal meetings. Therefore, it is advisable to request a pre-IND meeting before submitting an IND to initiate the first-in-human Phase I study, especially when seeking guidance on toxicology study designs.
STRATEGY
Gene Therapy Pilot Program
The Gene Therapy Pilot Program, a recent initiative, includes the FDA giving sponsors immediate feedback during clinical development. This program works in conjunction with new Regulatory pathways like the Regenerative Medicine Advanced Therapy (RMAT) designation. The aim is to speed up the development process and reduce review times for submissions. Collectively, these initiatives provide sponsors and regulators with more opportunities for regular communication and discussion about technological advancements.
With ongoing rapid progress in this domain, plan sponsors can anticipate notable changes in the evolving landscape of the U.S. healthcare system. Emerging studies indicate a probable surge in the availability of gene therapies, with projections estimating that the count could surpass 60 by the year 2030.
Casgevy is the first FDA-approved therapy utilising CRISPR/Cas9, a type of genome editing technology. Patients’ hematopoietic (blood) stem cells are modified by genome editing using CRISPR/ Cas9 technology.
In addition to the FDA, EMA has also come up with key steps to accelerate Cell and Gene Therapy production. It includes:
Orphan Status to CGT Drugs
European authorities are eager to support novel treatments focusing on currently neglected disease areas. Consequently, the European Medicines Agency (EMA) has awarded orphan status to the majority of Cell and Gene Therapy (CGT) drugs in the developmental stage. Additionally, the agency has conducted expedited assessments for several therapies.
For e.g., The European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) has recommended the authorisation of Glybera (alipogene tiparvovec) for marketing in the European Union. Glybera is a designated orphan medicine. The orphan designation was granted by the
European Commission in March 2004. It is intended to treat lipoprotein lipase (LPL) deficiency in patients with severe or multiple pancreatitis attacks despite dietary fat restrictions.
Adhering to GMO Regulations
Within the EU, companies are obligated to adhere to an environmental risk assessment and comply with genetically modified organism (GMO) requirements. This is to ascertain whether their Cell and Gene Therapies (CGTs) contain any substances that may be harmful to patients, animals, plants, or micro-organisms and to evaluate their overall impact on the environment.
Despite the guidance provided by the EMA on GMO requirements, it is crucial for companies to comprehend the distinct requirements for clinical trials, as these may differ across EU countries.
Challenges and Hurdles
Global Health Authorities are taking a few key steps to streamline the production of CGTs. Alongside the initiatives, there exist a few challenges that have to be addressed at the forefront. These include the following.
Balancing Innovation and Safety
The core challenge lies in achieving a delicate balance between fostering innovation to swiftly bring life-altering therapies to patients and ensuring rigorous assessments that guarantee their long-term safety and efficacy. Expedited approval pathways, like the FDA's Breakthrough Therapy Designation, hold immense promise in accelerating access for patients in dire need (US Food and Drug Administration, 2023). However, this must be counterbalanced by well-defined clinical trial designs and robust post-market surveillance mechanisms, such as the European Medicines Agency's Pharmacovigilance Risk Assessment Committee (PRAC) (European Medicines Agency, 2023).
Clinical Trial Design and Oversight
Initiating the regulatory pathway hinges on robust clinical trials that uphold the
7 www.pharmafocusasia.com
highest ethical standards. Regulatory agencies play a pivotal role in overseeing these trials, meticulously scrutinising protocols to ensure patient safety, informed consent, and scientific integrity. This oversight becomes even more crucial for CGT due to the inherent complexity of these interventions (Cantor et al., 2023).
Regulatory Challenges
Despite the immense promise, CGT faces a spectrum of Regulatory challenges. Understanding long-term safety and efficacy remains a significant hurdle. Post-market surveillance emerges as a cornerstone in addressing this, necessitating seamless collaboration between manufacturers, healthcare providers, and regulatory agencies (U.S. Food and Drug Administration, 2019). Additionally, logistical complexities surrounding manufacturing, storage, transport, and administration demand innovative solutions and harmonisation among industry stakeholders (Alliance for Regenerative Medicine, 2023).
Consistency in Global Regulations
Achieving harmonisation across global regulatory standards remains a hurdle. Divergent requirements and expectations among different regulatory bodies can complicate the development and approval process for multinational clinical trials and global market access.
Manufacturing Standards
Ensuring consistent and high-quality manufacturing processes for Cell and Gene Therapies is a critical challenge. Strict adherence to Good Manufacturing Practices (GMP) is essential, and any deviations may impact regulatory approval.
Long-Term Follow-Up
The durability of therapeutic effects and long-term safety of Cell/Gene Therapies require extensive follow-up data. Establishing and maintaining comprehensive post-marketing surveillance systems is a regulatory challenge that ensures continued patient safety.
Best Practices to Take Up
Amidst the burgeoning innovation in virology and genetic engineering, the future of gene therapies appears promising. To keep pace with the market trends, it is important for Regulatory bodies and manufacturers as well to keenly evaluate every aspect that they take up. Some of the proposed best practices can be:
Robust Communication
As these groundbreaking therapeutics explore new treatment areas and cater to broader patient populations, regulatory bodies must remain responsive to ongoing advancements. The key to ensuring the safety, efficacy, and quality of approved gene therapies lies in regulatory guidance that mirrors these transformative changes.
Achieving the ambitious approval goals necessitates not only adjustments on the part of Regulatory bodies to minimise delays but also adaptations from drug developers and manufacturers to align their processes with these evolving standards. Given the complexity of these dynamics, robust communication between gene therapy producers and regulatory bodies is anticipated to become increasingly vital.
Proposed Regulatory Strategies
Toward Collaboration and Efficiency: Embracing innovative strategies is crucial for navigating this evolving landscape. Risk-based assessments, as endorsed by the European Medicines Agency (European Medicines Agency, 2022), empower Regulatory agencies to tailor oversight based on individual therapies' specific risks. This allows for a more efficient and streamlined process without compromising robustness, particularly for therapies with lower risk profiles.
Real-World Evidence Integration
Integrating real-world evidence (RWE) into Regulatory decision-making represents a paradigm shift. RWE offers valuable insights into a therapy's performance in real-world settings beyond the controlled environment of clinical trials.
Collaborative efforts between Regulatory bodies, healthcare providers, and data scientists are essential to harness the power of RWE effectively, allowing for continuous refinement of regulations based on real-world experiences.
Conclusion
The promise of CGT necessitates a collaborative and adaptable regulatory ecosystem. By bridging the gap between innovation and safety, fostering open communication, and leveraging RWE, we can pave the way for these transformative therapies to reach patients in need while upholding the highest ethical and scientific standards. This ongoing dialogue between researchers, clinicians, and Regulatory authorities is crucial to ensure that the future of medicine is shaped by both scientific advancement and responsible governance.

A Scientific Regulatory Professional with 17 years of extensive Regulatory Affairs and 5 years of academic experience, with a track record of providing leadership for Quality-CMC, Non-Clinical and Clinical Regulatory Projects at Amgen Inc., LG Life Sciences, Intas Bio-Pharmaceuticals, Dr. Reddy’s Biologics, Biocon and Recon. Demonstrated ability to strategise, author, compile, and review regulatory submissions to Health Authorities and customers. Actively participated in New Drug Development; Clinical Trial Approvals; Marketing Authorisations and Commercialisation of 14 Biologics, 1 NCE, 5 New Biologicals in India; SAARC; RoW; and 1 in Europe.
Extensive experience in CMC; with a strong background in Non-clinical Pharmacological and Toxicological studies; and Clinical Trials for small molecules and biologics. Efficiently managed Product Life Cycle, PAC, RTQ; Tech Transfer; Due-Diligence, Licensing. Participated in GMP audits by Benchmark HA and Global Pharmaceuticals.
8 PHARMA FOCUS ASIA ISSUE 54 - 2024
AUTHOR BIO STRATEGY
The JAPAC Drug Development Market
Understanding the regulatory landscape and cultural nuances

There are specific nuances that emerging biotechs and biopharmas (EBPs) need to take note of when navigating the rapidly expanding JAPAC drug development landscape. In this article, we will delve deeper into the regulatory landscape and trends in JAPAC and how to effectively navigate the ever-changing landscape.
Hye Jin Choi, R. Ph., Senior Director, Regulatory Affairs, IQVIA Asia Pacific
Overview of the JAPAC regulatory landscape
Regulatory authorities are critical to the development and commercialisation of drugs as they govern the safety, efficacy, and quality of pharmaceutical products. For emerging biotech and biopharma companies (EBPs), working with regulatory authorities is crucial to ensure that their products can be brought to market
9 www.pharmafocusasia.com
STRATEGY
efficiently and effectively.
The Japan and Asia-Pacific (JAPAC) drug development market is rapidly expanding, with EBPs accounting for around 75 per cent of the total clinical trial volume in the Asia-Pacific region in 2020. However, successful navigation of the JAPAC market requires a unique understanding of its regulatory nuances and safety strategies. In JAPAC countries, multilingual language proficiency is a necessity due to the processing of adverse events in a bilingual database. Additionally, individual case safety reports and other forms must be submitted in the local language throughout a product’s lifecycle. There are also specific pharmacovigilance reporting requirements that demand full compliance.
Before trying to navigate the JAPAC regulatory space effectively and efficiently, it is also important to understand how the regulatory trends have evolved.
Priorities of regulatory authorities in this rapidly evolving regulatory landscape
Regulatory authorities in JAPAC are faced with various drug development priorities with the increasing demand for innovative therapies and treatments. They need to develop new guidelines and standards quickly to address an everevolving drug development landscape. Hence, the following will be crucial for the development of new drugs:
1. Promote innovation while prioritising patient safety
Regulatory authorities have introduced new guidelines to streamline the regulatory process for EBPs. These guidelines encourage innovation by allowing for flexible and adaptive trial designs, where companies can modify their clinical trials as they evolve, based on new information or feedback from regulators. This can also speed up the drug development process and reduce costs. For instance, Singapore’s Health Science Authority (HSA) has established a regulatory sandbox programme that allows companies to test new healthcare products and services
in a controlled environment. This helps to identify potential risks and benefits before the products are released to the market. The HSA also offers a priority review scheme for medical devices and drugs that address unmet medical needs.
Such programmes encourage innovation and provide faster pathways to market, while ensuring that rigorous testing and evaluation criteria are met, bringing new and innovative treatments to patients in a timely and safe manner.
2. Refine guidelines to ensure the quality and safety of pharmaceutical products
This involves establishing standards and guidelines for the manufacturing, testing, and distribution of drugs, as well as pharmacovigilance, which focuses on monitoring, evaluating, and preventing adverse effects of drugs. The Pharmaceuticals and Medical Devices Agency (PMDA) and National Medical Products Administration (NMPA), regulatory bodies that oversee the safety, efficacy, and quality of pharmaceuticals and medical devices in Japan and China, respectively, have introduced new guidelines for pharmacovigilance to ensure patient safety.
The PMDA's guidelines emphasise the importance of risk management plans and post-marketing surveillance, while the NMPA's regulations require companies to report adverse events within 24 hours, supporting the establishment of pharmacovigilance standards in drug develop-
ment and post-marketing surveillance.
3. Adapt to the post-pandemic landscape
a. Misinformation
Misinformation is a growing concern across industries, and its significant impact has been witnessed since the start of the COVID-19 pandemic. Regulatory bodies in JAPAC recognised the need to tackle this and have been focusing on strengthening public communications. Regulatory bodies, such as HSA, PMDA and South Korea's Ministry of Food and Drug Safety (MFDS), monitor advertising and promotional content to ensure that information is truthful and accurate. In addition, these regulatory bodies are quick to address concerns from healthcare professionals and the public about drugs and medical devices. They have also been collaborating with other organisations to promote accurate information.
Education and awareness campaigns can also help to counter misinformation in drug development. For instance, the PMDA has launched campaigns to raise awareness of the risks of over-thecounter painkillers and encourage proper disposal of unused medications. It also shares educational content with healthcare professionals on topics such as drug interactions and adverse events.
b. Emphasis on Patient-Centred Drug Development (PCDD)
To support industries driving constant innovation and new technology in new drug development, health authorities in JAPAC need to monitor/adapt new global regulatory standards and requirements to address local specific needs to stay aligned with evolving global drug development landscape.
Drug development is traditionally focused on developing safe and effective treatments for patients. However, there has been a growing emphasis on proactive patient involvement during the drug development process to meet patients’ needs.
With this shift, new guidelines and regulations are being developed to promote patient participation in clinical trials and other aspects of drug development. Industry organisations, such as the International Society for Medical Publication Professionals (ISMPP), have created guidelines to ensure that patients are provided with accurate and reliable information about the drugs.
10 PHARMA FOCUS ASIA ISSUE 54 - 2024
STRATEGY
In JAPAC, regulatory authorities such as the NMPA, HSA, PMDA and MFDS dictate that patients must be fully informed about the purpose of the trial, the risks and benefits of participation, and their right to withdraw at any time. The guidelines also require patients to be given adequate time to consider their participation and that they provide informed consent before participating. The guidelines also encourage the recruitment of a diverse patient population, including women, minorities, and elderly patients, per Good Clinical Practice (GCP) standards.
c. Increased adoption of virtual and digital approaches
The pandemic has pushed the biopharmaceutical industry to adapt to new ways of working, with the biggest shift being the adoption of virtual and digital technologies in clinical trials. This has resulted in the development of new regulations and guidelines to ensure patient safety and data integrity. PMDA, NMPA, MFDS, Australia's Therapeutic Goods Administration (TGA) and HSA have issued guidance to ensure data quality and integrity when using electronic data capture and other digital technologies during virtual and remote clinical trials.
As the drug development landscape in JAPAC mirrors the rapid changes in global trends, regulatory authorities have had to pivot quickly to ensure that patients have access to safe and effective healthcare products, while also promoting innovation in the EBP industries.
The adoption of ICH guidelines in China and evolution of ICH guidelines in Japan
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), an international non-profit association, brings together regulatory authorities and the pharmaceutical industry to coordinate scientific and technical aspects of drug registration to ensure the safety, efficacy, and quality of
The adoption of ICH guidelines has helped China and Japan to join global clinical program more actively minimising drug lag time and eventually accelerating the launch of innovative drugs locally. It is paramount for regulatory authorities in JAPAC to explore various ways to streamline the drug registration process to help patients in need of new innovative treatments.
pharmaceutical products. Japan is one of the founding regulatory members, while South Korea, China and Singapore are members of the ICH.
International collaboration through the ICH has been at the forefront of global drug development, especially in the post-pandemic era. Both China and Japan have adopted ICH guidelines, which have accelerated the launch of innovative drugs locally.
A deeper dive into the adoption of ICH guidelines in China
China entered ICH regulatory membership in 2017 with a pledge to gradually transform its regulatory science ecosystem. As of December 2023, China has implemented 62 out of 63 ICH guidelines .
The NMPA in China actively participates in the coordination and implementation of the ICH guidelines. Clinical trial design guidance, electronic data capture guidelines, inspection and auditing, and training and education are just a few examples of how the NMPA has implemented the ICH guidelines in China. The adoption of ICH guidelines has accelerated drug development growth in China and globally, promoting the alignment of technical requirements with global standards, which support:
• Multinational companies to introduce early-stage research and development into China; and
• Global drug development with new drugs that are developed by local pharmaceuticals.
The impact of ICH guideline adoption in China can be seen in the significantly reduced review and approval timelines for Investigational New Drug (IND) Application, Biologics License Applications (BLA) and New Drug Application (NDA).
IND application approvals have been shortened to 60 days from 27 months, while BLA and NDA applications have been cut from 26 months to 60 days and 12 to 18 months respectively. In addition, the time lag between China and the United States for innovative drug development was between 5 to 7 years in 2015 but is being gradually synchronised.
The adoption of the ICH guideline has therefore made it easier for sponsors to conduct clinical trials in China and seek approval for their drugs. This has led to increased investment in the country's biopharmaceutical industry, which has the potential to drive economic growth and improve public health.
Ongoing development of the evolution of ICH guidelines in Japan
The evolution of Japan’s Multi-Regional Clinical Trials (MRCTs) has been significant.
In 2007, the PMDA developed a “Basic Principles on Global Clinical Trials (GCTs)” guideline that stated the requirements for conducting an MRCT in Japan. In 2012, they expanded it to offer points of consideration for countries in East Asia looking to conduct MRCT , covering jurisdictions of China, Japan, and South Korea. The “Basic Principles for Conducting Phase 1 Trials in the Japanese Population Prior to GCTs ” guidance was subsequently released in 2014 to address the requirements of phase 1 clinical trials in Japan on the Japanese population. This has increased MRCTs conducted in Japan.
11 www.pharmafocusasia.com
STRATEGY
As an ICH Rapporteur of the E17 Guideline, which provides guidance on the design, conduct, and analysis of clinical trials that are conducted in multiple regions of the world, Japan has been in a transition phase to achieve complete implementation of ICH E17 since 2018 . The E17 guideline is intended to help ensure that trials conducted are scientifically valid and ethically sound, while also taking into account the cultural and regulatory differences in different regions. The ICH E17 guideline provides recommendations on the trial design, trial conduct, data analysis and ethical considerations related to MRCTs.
An analysis done across 167 MRCTs of 130 drugs found that more than 75 per cent of MRCTs are well-considered in GCP compliance. However, there are several areas which are less adequately addressed, such as the use of relevant guidelines for disease and primary endpoint definitions, standardisation of efficacy/safety information, sample size allocation, and the training/validation on subject selection and primary endpoint.
For future MRCTs, it is recommended that the analysis strategy for consistency evaluation, including the use of pooled regions or subpopulations, is pre-specified. More clinical experiences and data will be necessary to pool Asia as a single region for efficient clinical development and appropriate consistency assessment in treatment effects.
South Korea’s regulatory update
In line with other countries' efforts to speed up the regulatory process, Korea introduced the Global Innovative Product on Fast Track (GIFT) in 2020. GIFT is a supporting programme for accelerating the regulatory review of “global innovative products”, aimed at facilitating the market launch of innovative products intended for life-threatening or serious diseases to ensure faster access for patients. With GIFT, the regulatory review process can be shortened by 75 per cent when compared to the standard review cycle.
Since 2020, GIFT has conducted expedited review designation on 23 products and expedited review after designation on 17 products. Anti-cancer products account for the largest portion of products designated for expedited review, followed by COVID-19 vaccines.
Navigating the regulatory landscape in JAPAC: A China case study
How can EBPs navigate the diverse regulatory landscape in JAPAC? Some of the major nuances in the JAPAC market that EBPs should be aware of include language barriers and differing regulatory guidelines. For instance, when adopting ICH guidelines in China, EBPs should note the additional requirements when conducting stability studies for formulations that are prone to phase separation, reduced viscosity, precipitation, or aggregation. Furthermore, consideration should be given to low temperature or freeze-thaw testing. Additionally, there are minor differences between Chinese Pharmacopoeia requirements and ICH Q4 (Pharmacopoeias). Chinese Pharmacopoeia places more emphasis on traditional Chinese medicine (TCM) than ICH Q4 as TCM is an important part of the healthcare system in China, and many drugs derived from TCM are regulated under the Chinese Pharmacopoeia.
Another factor to consider is the Ethnic Factors (E5) in the ICH guideline , which states that EBPs need to ensure that bridging clinical studies include Chinese population data, even if there is clinical data from outside of China as the data might prove insensitive to ethnic factors. However, products for critical diseases, rare diseases, paediatrics, and the products listed in the “Marketed overseas and urgently needed in China” are exempted. In compliance with ICH E5 3.2.2, the regulatory authorities may still request a bridging study for approval even for compounds insensitive to ethnic factors for regions with little experience with registration based on foreign clinical data.
Understanding these requirements can be challenging, but leveraging strategic tools and technology can simplify the process. Bilingual safety databases, robotic process automation, workflow management tools, and automated narrative tools are some examples that can help EBPs navigate the JAPAC regulatory landscape.
The best way forward in JAPAC
For EBPs looking to enter JPAC, they need to understand the regulatory environment, acknowledge cultural nuances when conducting preclinical and clinical studies, and prepare high-quality submissions in local languages.
They can also consider partnering with a company that has the expertise and experience to navigate the drug regulatory landscape in JAPAC. New technology could also help expedite the drug approval process, especially around data collection and submission preparations.
As the biopharmaceutical industry continues to evolve, it is essential to stay alert and understand the latest regulations and guidelines to ensure compliance and patient safety.

12 PHARMA FOCUS ASIA ISSUE 54 - 2024
Hye Jin Choi provides regional leadership of Regulatory Affairs & Drug Development Solutions (RADDS) business expansion across JAPAC. Based in South Korea, she has over 29 years of experience across the various healthcare industry sectors and leads the overall client engagement and integrating solution efforts interacting with various JAPAC biopharma companies seeking global drug development.
AUTHOR BIO
STRATEGY
What do We Know About Albumin Formulations?

Albumin is a natural material that covers nearly 60 per cent of the human proteome and is utilised as a buffering protein that helps transport various xenobiotics in the body. Pharmaceutically, it is used for the treatment of hypovolemia in patients with chronic conditions. Albuminbased Abraxane® (paclitaxel-albumin nanoparticles) is a billion-dollar drug for treating various cancer conditions. It helped to provide an organic solvent-free formulation of a hydrophobic drug (paclitaxel). There has been an exponential rise in the research to explore various applications of albumin as a biomaterial for therapeutic and formulation development applications. As a pharmaceutical biomaterial, albumin can be moulded to form nanoparticles, hydrogels, nanofibers, and films. This article highlights key aspects, such as the synthesis and application of albumin as a pharmaceutical excipient in developing dosage forms.
Luke Condon1#, Miral Suthar2#, Sara Woytowicz1, Gagan Kaushal2 and Ankit Rochani1*
1. Wegmans School of Pharmacy, St John Fisher University
2. Jefferson College of Pharmacy, Thomas Jefferson University
#: They have contributed equally *: Corresponding author. arochani@sjf.edu
1. Pharmaceutical Dosage Forms Using Albumin
Albumin is a globular protein soluble in water and coagulable by heat. It is found in egg white, blood serum, milk, and other animal and plant tissues and fluids. Human Serum Albumin (HSA) is the most abundant protein in human plasma. The use of colloidal albumin for therapeutic purposes dates back more than 50 years. Purified albumin solution is used clinically in the treatment of chronic illnesses dealing with hypovolemia (5 per cent or 25 per cent w/v). The extraction of albumin has been optimised and perfected over time. Being a natural xenobiotic career in the body, albumin (as a biomaterial) was used to synthesise nanoparticles. These are tiny particles (~200 nm size) for nanomedicine applications. HSA shows reversible binding capabilities with a range of substances, such as fatty acids, hormones, and metal ions. This makes
13 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
it an ideal biomaterial for developing novel drug delivery and biomedical imaging therapeutics. This subsection narrates the uses of albumin materials in developing nanomedicines and semi-solid dosage forms.
1. 2 Nano formulations vs nanoparticles for therapeutic applications.
Albumin nanoparticles are typically prepared using various methods, including desolvation, self-assembly, thermal gelation, spray-drying, emulsification, nanoparticle albumin-bound (Nab) technology, and pH coacervation. Each of these methods has its advantages and disadvantages, and the choice of method depends on the specific requirements of the drug delivery system. The desolvation method, for example, involves the removal of solvent from a solution, causing the albumin to precipitate and form nanoparticles. On the other hand, the self-assembly method relies on the natural tendency of albumin molecules to aggregate into nanoparticles under certain physical or chemical stress conditions.
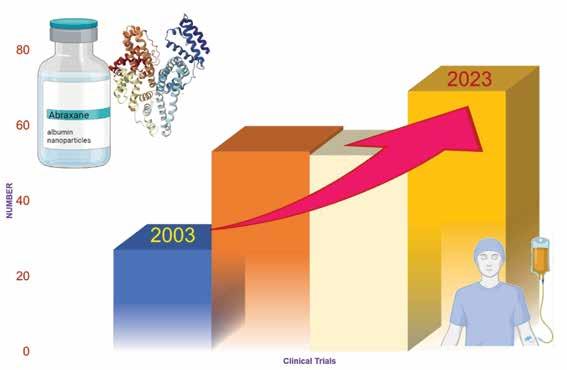
One of the most well-known commercialised albumin nanoparticles
is Abraxane®, which uses a technology known as Nab technology. This technology takes advantage of the natural properties of albumin and its accumulation in tumours. The drug paclitaxel is bound to albumin and delivered to tumours without the use of toxic solvents. Abraxane® binds to the albumin receptor of the endothelial cells, activating a process called caveolae-mediated transcytosis, which enhances the delivery of the drug to the tumour. Currently,>200 clinical trials are being conducted to explore the use of Abraxane® as a single or combination therapy to develop various therapeutic options (Figure 1). Moreover, many other particles are being produced using albumin nanoparticles as a delivery system, as shown in Table 1.
Looking at the success of albumin as a nano delivery system, this biomaterial was used to develop theranostic nanomedicines for various anticancer drugs. Following are some examples:
a) Photo thermal for melanoma: A newly synthesised near-infrared dye IR-817 was combined with bovine serum albumin (BSA) to create BSA@IR-817 nanoparticles (120 to 200 nm). These
nanoparticles were assessed for their fluorescence imaging (due to IR-817 dye) and photothermal (due near IR 808 irradiation) therapy potential against melanoma.
b) Photothermal therapy combined with MRI imaging of cancer: A selfassembly method of albumins with small molecules was used to encapsulate IR780 and super paramagnetic iron oxide (SPIO) nanoparticle, that forms IR780@BSA@SPIO nanoparticles. They provided NIR-II and MRI dual-mode imaging, that can accurately find cancer and guide photothermal therapy to eliminate tumours.
c) Albumin-coated or functionalised quantum dots were developed for bioimaging applications. These albuminbased nanoparticle designs were evaluated under in vitro or in vivo conditions. However, more data related to pharmacokinetics, biodistribution, dose safety and toxicity are required for their clinical testing.
Besides, microparticle designs like albumin microbubbles have been explored for ultrasound-mediated diagnostic applications. These microbubbles are typically
14 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
Figure 1: Consistent increase in the number of clinical trials with albumin nanoparticles for the treatment of cancers in the last two decades. Each block represents a span of approximately five years.
1 Fyarro® Sirolimus Malignant perivascular epithelioid cell tumours (PEComa)
2 Idelvion® Coagulation Factor IX Hemophilia B
3 Abraxane® Paclitaxel Indicated for breast cancer, pancreatic cancer, and non-small cell lung cancer
https://www.fyarro.com/
https://www.idelvion.com/
https://www.abraxanepro.com/
Note: Insulin products (Tresiba®, Victoza®, and Levemir®) interact with the albumin for their biodistribution and targeted pharmacological responses.
0.5–10 μm in size, which makes it easier for medication delivery mechanisms to navigate the body. Microbubbles made of albumin are effective vehicles for delivering photodynamic agents—drugs used in advanced anti-cancer therapy that avoid the side effects of chemo- and radiation therapy. Moreover, microbubbles have been utilised as contrast agents during ultrasound procedures. However, there has been a recent focus on exploring their potential as drug-delivery systems that respond to specific stimuli. The original microbubbles, known as Albunex, which consisted of a protein shell made from human serum albumin, were developed and successfully adopted in clinical practice in 1993. As a final observation, albumin microbubbles have shown significant potential in the clinical field, particularly in drug delivery and disease treatment. Their unique properties allow
for targeted and efficient delivery of therapeutic agents, making them a promising tool in modern medicine.
From a formulation perspective, several challenges need to be addressed. These include issues with reproducibility, proper characterisation, and biological evaluations. For example, the size and shape of the nanoparticles can affect their behaviour in the body, and these properties can be challenging to control. In addition, the interaction of the nanoparticles with the immune system is not fully understood and can lead to unexpected side effects. Therefore, rigorous studies alongside stringent guidelines for effective and safe nanomedicine development and use are still called for.
1.3. Semi-solid dosage forms made from albumin.
Hydrogels are a semisolid dosage
form. Pharmaceutical gels are primarily prepared from semi-synthetic or synthetic materials with polysaccharides as their backbone. Examples include methyl cellulose, hydroxyethyl cellulose (HEC), hydroxypropyl methylcellulose (HPMC), and others. Methylcellulose (water soluble) is a cellulose derivative used for bowel irregularity and is available over the counter. All these derivatives have variable water solubility and thickening behaviour. This also makes these materials being explored as suspending agents in the formulation development. Since these materials can produce viscoelastic gels, the cellulose-based systems provide an essential foundation for using sustainable green material for semi-solid formulation development. Significant research has been done to understand the sustainability of green materials for developing semisolid preparations.
5
8
15 www.pharmafocusasia.com
Table 1: Clinically approved drug products, where albumin as a delivery system
No. Product Name Drug Application Reference
No. Condition Criteria 1 Processing and Pasteurisation NLT 10min and NMT 11 hours at 60+/0.50C 2 Stabiliser 0.08 mM of sodium caprylate per gram of protein 3 Protein concentration 4 ± 0.25 per cent w/v, 5±0.30 per centw/v, 20± 1.2 per cent w/v and 25±1.5 per centw/v
Protein composition 96 per cent of total protein shall be albumin
4
pH 6.9 ± 0.5 (measured in
solution
protein with 0.15 M sodium chloride)
Sodium concentration 130 to 160 milliequivalents per litre
Potassium concentration 2 milliequivalents per litre
a
of the final product diluted to a concentration of 1 per cent
6
7
Heat stability Albumin should remain unchanged (by visual inspection) after heating at 570C for 50 hours, compared to a sample from the untreated lot.
RESEARCH & DEVELOPMENT
Table 2: General specification of albumin solution for IV infusion
Protein is one of the natural building blocks, and it was observed that the albumin-protein solution can produce a transparent hydrogel. Since then, researchers have been creating these albumin hydrogels for a potential real-world application. Over the years, research has been conducted to understand the gelling behaviour of albumin-based or albumin. Chemical crosslinking of albumin or albumin-based gels can be achieved through disulfide cross-linking under acidic pH. Solvents like ethanol and a composite of albumin-succinimidyl succinate-modified polyethylene glycol (PEG) provided gels with injectable behaviour. Using chemical denaturants in gel formation can provide gels, improper for utility due to toxic residues. Alternatively, the physical crosslinking phenomenon of albumin solutions to create hydrogel, is another method that is explored by some researchers, including our lab. The drug-binding nature of albumin combined with the fact that it can become a gel has led people to believe that albumin hydrogels could be a new form of drug delivery system. The hydrogels can be loaded with the drug and then injected into the body, where they slowly release the drug over
Unveiling albumin's potential: Discover how this natural biomaterial is not just a carrier for drugs, but a key player in the development of sustainable, effective pharmaceutical formulations, promising breakthroughs in medicine's future.
time. This allows for a more targeted and controlled drug delivery, which can improve its efficacy and reduce side effects.
The formation of albumin hydrogels creates a biomaterial that shows various biological and mechanical properties, making it a promising substance for drug delivery. The unique mechanical properties of the gel allow it to have a range of tensile strengths, leading to its ability to form hydrogels suitable for drug delivery (low strength) and hydrogels
that aid in tissue regeneration, providing structural scaffolding for new growth (high strength). Moreover, albumin-based gels can provide a supportive scaffold for new tissue growth. They can also be loaded with growth factors or other bioactive molecules to promote tissue regeneration. It could also be used as a cartilage replacement material. A firm but elastic gel for a knee replacement has the potential to give the knee the cushion that normal cartilage provides that no ceramic or metal material could provide. Albumin hydrogels can potentially have significant uses in medicine and other areas of life. The situation is ever evolving, and innovative ideas and discoveries are made every day. Hopefully, albumin as a biomaterial and therapeutic agent could answer many more questions than we even realise.
2. Albumin as a Pharmaceutical Excipient
No. Source Albumin Grade
1 Bovine Heat shock fraction
Heat shock fraction, protease-free, essentially globulin free
Fraction V-Fatty acid-free
Fraction V-low electrolyte
2 Human Fraction V serum albumin
Recombinant expressed in rice
Fatty acid-free
Globulin free
3 Chicken Albumin from albumin chicken egg white
From the excipient point of view, bovine serum albumin (BSA) is the most explored material by various research laboratories. From the clinical perspective, USP supplies detailed monograph data about recombinant human albumin. The monograph supplies details about the formulation of human serum albumin for intravenous use. FDA gives a manufacturing procedure for the fractionation of human serum albumin and a comparison of various products to provide clinical usefulness of albumin solution. The Code of Federal Regulations (CFR) includes information about the fractionation of serum albumin for making albumin solutions in clinical use. Still, the standard chemical signatures of albumin as material necessary to prevent batch-to-batch variation is unclear. However, the data provided by CFR, USP, and FDA (Table 2) can lay the foundation for creating material-specific monographs of albumin for its use in formulation development.
Albumin fractionation from various sources (bovine, human, egg, chicken, and others) dictates the quality of biomaterial isolated. The process of fractiona-
16 PHARMA FOCUS ASIA ISSUE 54 - 2024
Table 3: Albumin grades available from bovine (having ~80 per cent homology to human albumin) and human sources for research and development. All albumin forms listed into the table are available in the lyophilised form.
RESEARCH & DEVELOPMENT
tion of human albumin was set up nearly 60 years ago and is still the method that is being explored for isolating human albumin for clinical use. Various grades of albumin being synthesised (Table 3) are constantly researched in literature to create advanced controlled delivery formulation.
Each supplier company (like Sigma, Fisher Scientific, Bio World, and others) provides technical specifications of bulk material. However, no standardised criteria or standards are available to avoid inter and intra-company variability for the formulation development. This highlights the potential challenge of envisioning a biological product as a pharmaceutical excipient for formulation development.
3. Conclusion
Albumin is a pivotal and versatile material in pharmaceutical science, highlighting its
AUTHOR BIO

Luke Condon, a prehealth undergraduate student at St. John Fisher University, he is pursuing biology (with honors), and chemistry. He is a certified NYS EMT-B, volunteering in Rochester, Luke works under the supervision of Dr. Rochani on developing semisolid dosage forms for drug delivery applications. Specifically, he contributed to the refining of albumin hydrogel formulation methods. He conducts qualitative and quantitative analyses, focusing on hydrogel stability and the mechanism of gel formation.
significance in diverse dosage forms and drug delivery systems. It is a sustainable green biomaterial for advanced delivery systems. The evolution of albumin-based formulations, exemplified by Abraxane®, has revolutionised cancer treatment, offering a solvent-free platform for hydrophobic drugs and prompting numerous clinical trials exploring its therapeutic potential. The use of albumin in nanomedicine has opened new avenues, with albumin nanoparticles (shown in Table 1) showing promise in drug delivery and potential theranostic systems. Applications such as enhanced imaging and photothermal therapy underscore albumin's diverse roles in advancing medicine. Exploring semi-solid dosage forms, particularly albumin hydrogels, adds another dimension to albumin's pharmaceutical versatility. Albumin hydrogels offer a potential platform for controlled drug delivery and tissue regeneration engineering.

Miral Suthar, completed his MS in Pharmaceutical Sciences, at Thomas Jefferson University Philadelphia. He has worked on the formulation and characterisation of drug-loaded albumin hydrogel. His research focused on HPLC and LCMS method development for small molecules, stability study, pharmacokinetic study, rheological characterisation, and physical evaluation of the formulation.

Sara Woytowicz, an undergraduate student at St. John Fisher University, is on the path to earning a Doctor of Pharmacy at Wegmans School of Pharmacy. She stands out as an emerging researcher and scholar. Sara is an integral part of the team exploring hydrogel formulation, utilising sustainable green materials like albumin for innovative therapeutic applications.
As a pharmaceutical excipient, albumin, mainly bovine serum albumin, is commonly explored in laboratorybased-academic research. Challenges such as the need for standardised criteria to ensure consistency across formulations. Addressing these challenges is crucial for proving albumin as a reliable pharmaceutical excipient. Groundbreaking achievements and ongoing challenges mark Albumin’s journey in pharmaceuticals. As research continues, albumin holds promise not only in cancer treatment but also in diversified applications, pushing the boundaries of drug delivery and formulation development, potentially addressing unmet medical needs, and providing innovative solutions in various domains of medicine.
References are available at www.pharmafocusasia.com

Gagan Kaushal, a professor at Jefferson College of Pharmacy, earned his pharmacy degree from Panjab University and a doctorate in Industrial Pharmacy from St. John’s University. With expertise in drug delivery systems and formulation development, he conducts research funded by NIH and the Gates Foundation. Dr. Kaushal has extensive experience in academia, the pharmaceutical industry, and clinical trials, with over 40 published manuscripts.

Ankit Rochani, a leading pharmaceutical scientist with a PhD in Bio Nanotechnology, specializes in LCMS and HPLC studies. Currently an Assistant Professor at Wegmans School of Pharmacy, he explores sustainable materials for advanced biomimetic drug delivery formulations. His research encompasses stability of compounded formulations and pharmacokinetics in clinical trials, contributing significantly to the field.
17 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
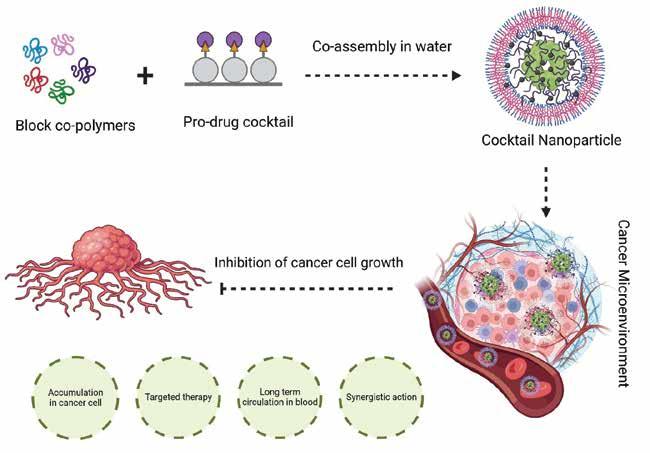
COCKTAIL NANOPARTICLES
A precision drug delivery system to cancer cell
Cocktail nanoparticles are a type of nanoparticles that are composed of a mixture of different materials. These nanoparticles have unique properties and are often used in various applications such as drug delivery, imaging, and sensing. The term ‘cocktail’ refers to the combination of different materials in the nanoparticle composition. These materials can be metals, metal oxides, polymers, or organic compounds. By combining different materials, cocktail nanoparticles can exhibit enhanced functionalities and performance compared to single-component nanoparticles in a precise drug delivery system. In drug delivery applications, cocktail nanoparticles can be engineered to carry multiple therapeutic agents simultaneously. This enables the delivery of combination therapies, where different drugs are released at specific locations and time points, enhancing treatment efficiency and reducing side effects.
Sumel Ashique
Department of Pharmaceutical Sciences, Bengal College of Pharmaceutical Sciences & Research
Radheshyam Pal Assistant Professor
Pandaveswar School of Pharmacy
Cocktail nanoparticles are also valuable in imaging applications, such as in cancer diagnosis and monitoring. By incorporating different contrast agents, these nanoparticles can provide multi-modal imaging capabilities, allowing for more accurate and comprehensive imaging of tissues
18 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
and organs. One major advantage of using cocktail nanoparticles is the ability to fine-tune their properties. By adjusting the composition and ratio of the different materials, researchers can tailor the nanoparticles' size, shape, surface chemistry, and optical properties to meet specific application requirements. This versatility allows for the design of nanoparticles with improved stability, biocompatibility, and targeting capabilities. These tiny particles hold immense potential in providing targeted and effective treatments for cancer patients. By combining different types of nanoparticles, researchers are able to create a ‘cocktail’ that can simultaneously target multiple aspects of cancer cells, enhancing the overall effectiveness of treatment. Each nanoparticle within the cocktail can perform a specific function, such as delivering
drugs directly to the tumour, enhancing immune response, or inhibiting cancer cell growth. By combining these different functionalities into one treatment approach, researchers hope to overcome some of the challenges of traditional cancer therapies, such as drug resistance and off-target effects. Cocktail nanoparticles hold immense potential in revolutionising cancer therapy. By combining different types of nanoparticles into one treatment approach, researchers can enhance the effectiveness of cancer treatment and overcome some of the limitations of traditional therapies.
Mechanism against Cancer cell
Cocktail nanoparticles work by combining the strengths of different nanoparticles into one treatment approach. Each nanoparticle within the cocktail
is designed to perform a specific function, such as targeting the tumour, delivering drugs, or enhancing immune response. By simultaneously targeting multiple aspects of cancer cells, cocktail nanoparticles have the potential to increase treatment efficacy and overcome some of the limitations of traditional cancer therapies. They can attack cancer cells from different angles, making it harder for the cells to develop resistance or evade treatment. In a study, cocktail nanoparticles were used to enhance the effectiveness of immunotherapy, a type of cancer treatment that harnesses the body's immune system to fight cancer. By combining nanoparticles that enhance immune response with nanoparticles that deliver immunotherapeutic agents, researchers were able to significantly improve the response rate and overall survival of cancer patients.
19 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
Opportunities and bottlenecks
The versatility of cocktail nanoparticles allows for the synergistic action of different therapeutic agents, increasing the chances of successfully eliminating cancer cells and improving patient outcomes. One of the primary advantages of using cocktail nanoparticles is their ability to target multiple aspects of cancer cells simultaneously. Cancer is a complex disease, and it often requires a multi-faceted approach to effectively treat it. Cocktail nanoparticles allow for the synergistic action of different therapeutic agents, increasing the chances of successfully eliminating cancer cells and preventing relapse. Furthermore, cocktail nanoparticles can be functionalized with various ligands or antibodies on their surfaces, enabling targeted delivery to specific cells or tissues. This targeted approach increases drug efficacy and reduces off-target effects. While cocktail nanoparticles offer great promise in cancer therapy, there are several challenges and limitations that need to be addressed. One challenge is the complexity of designing and synthesising nanoparticles with multiple functionalities. Each nanoparticle
Cocktail
nanoparticles offer a versatile and customisable platform for various applications, allowing researchers to explore new possibilities in nanomedicine, imaging, and sensing.
within the cocktail must be carefully engineered to perform its specific function while maintaining compatibility with the other nanoparticles. Another challenge is the potential for off-target effects. Since cocktail nanoparticles are designed to target multiple aspects of cancer cells, there is a risk of affecting healthy cells or tissues. Researchers need to ensure that the nanoparticles deliver their cargo precisely to the tumour site
and minimise any potential damage to healthy cells. Furthermore, the longterm safety and toxicity of cocktail nanoparticles need to be thoroughly evaluated. While nanoparticles have shown great potential in cancer therapy, their impact on the body's organs and immune system is still not fully understood. It is crucial to conduct rigorous preclinical and clinical studies to ensure the safety and effectiveness of cocktail nanoparticle therapy.
Future window
Despite the challenges, the future of cocktail nanoparticle therapy looks promising. Researchers are continuously exploring new ways to improve the design and functionality of nanoparticles, making them more effective and safer for cancer treatment. Advances in nanotechnology and material science are opening up new possibilities for creating nanoparticles with enhanced targeting abilities and controlled drug release properties. The use of cocktail nanoparticles in cancer therapy is still in its early stages, but there are already promising case studies and success stories that highlight their potential. For example, researchers have successfully used cocktail nanoparticles to deliver multiple chemotherapy drugs directly to the tumour site, resulting in improved treatment outcomes and reduced side effects. Moreover, the field of personalised medicine is expected to greatly benefit from cocktail nanoparticles. As our understanding of cancer biology improves, researchers will be able to tailor the composition of the cocktail to match the specific characteristics of each patient's cancer, maximising treatment efficacy and minimising side effects. In addition, advancements in imaging technologies will play a crucial role in the development of cocktail nanoparticle therapy. By combining nanoparticles with imaging agents, researchers can track the distribution of the nanoparticles in real-time, ensuring that they reach their intended targets and monitor the treatment response.
20 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
Conclusion
The use of cocktail nanoparticles in cancer treatment raises important ethical considerations. As with any new therapy, it is crucial to ensure that the benefits outweigh the potential risks and that patient autonomy is respected. Patients should have access to accurate information about the treatment, including its potential benefits, risks, and alternatives. In summary, cocktail nanoparticles offer a versatile and customisable platform for various applications, allowing researchers to explore new possibilities in nanomedicine, imaging, and sensing. Their unique combination of properties makes them a promising tool in advancing healthcare and diagnostics. Moreover, cocktail nanoparticles offer the potential to personalise cancer treatment. Researchers can tailor the composition of the cocktail based on the specific characteristics of the patient's cancer, ensuring a more targeted and effective therapy. This personalised approach has the potential to revolutionise cancer treatment and improve patient outcomes. With continued advancements in nanotechnology and personalised medicine, cocktail nanoparticles have the potential to transform the landscape of cancer treatment, offering new hope to patients and their families.
References are available at www.pharmafocusasia.com


Sumel Ashique has been working in the Department of Pharmaceutical Sciences at Bengal College of Pharmaceutical Sciences & Research, West Bengal, India. He has 3 years of teaching experience. He has achieved 50+ publications of International and national accredited reputed journals (Scopus, UGC). He has knowledge in drug delivery, nanotechnology and targeted treatment strategy. He has also 4 granted patents from IP and Australia, 12 published book chapters in International Books and 15 book chapters have been submitted to well-known publishers like Springer, Elsevier, Bentham and Taylor & Francis. Currently he is editing 6 books under Springer, Wiley & Taylor and Francis. He is serving as a potential reviewer in various International Journals.
Radheshyam Pal is an Assistant Professor at Pandaveswar School of Pharmacy. His areas of expertise in Neuropharmacology. He specifically works on the role of natural products in the CNS. He has 2 years of teaching experience. He has 2 patents and also 10 book chapters submitted in international publisher.
Drug Resistance in Cancer: Mechanisms and Strategies

Editors: Sameer Khan, Fayaz Malik
Date of Publishing: 26 May 2024
No of pages: 400
Description: This book tries to emphasize the mechanisms associated with the resistance towards various anti-cancer therapies The focus has been given to the role of cancer stem cells, immune cells, and the multiway impact of tumor microenvironment in drug resistance. The book delves into the role of epigenetic alterations, autophagy, intracellular compartments, and the impact of gut microbiome on therapeutic resistance. Each chapter of the book has elaborated on these aspects that are exclusively or mutually driving the therapeutic non-responsiveness towards various current clinical candidates. In addition to that the book has also discussed novel strategies to overcome the therapeutic challenge by employing Combinatorial therapies that can prove to be useful and effective. Overall the book reflects on the current treatment challenges, futuristic strategies and new research initiatives that explore novel treatment options.
21 www.pharmafocusasia.com
AUTHOR
RESEARCH & DEVELOPMENT
BIO
Book Shelf
Next-Generation Approach through Advanced Research and Engineering Techniques in Cell Based Therapy
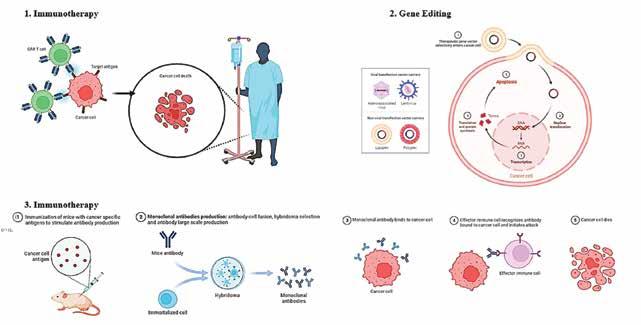
Through their incredibly effective mechanisms of action, cell-based therapies are an emerging technique that hold the promise of treating a wide range of diseases that are now incurable. Cell-based therapies continue to face many obstacles that prevent their widespread translation and commercialisation, despite notable recent clinical and commercial successes. These obstacles include identifying the right cell source, developing sufficiently viable, potent, and safe products that meet patient- and diseasespecific needs, and creating scalable manufacturing processes. Advanced fundamental research, fueled by next-generation engineering techniques, such as genome and epigenome editing, synthetic biology, and the use of biomaterials, is being used to overcome these obstacles.
Sibi Raj, Dhruv Kumar School of Health Sciences and Technology (SoHST), UPES
Biological therapy uses the body's immune system to combat cancer, using cells, antibodies, and organs to protect against invaders and differentiate between healthy and cancerous cells, potentially eliminating them. A few treatments have cleared regulatory obstacles and been available for purchase, which has increased public awareness and enthusiasm. Among these is the effective treatment of lymphoid cancers through genetically reprogrammed T cell adoptive cell transfer, which led to the FDA approving tisagenlecleucel and axicabtagene ciloleucel in 2017 for the treatment of large B cell lymphoma (LBCL) and acute lymphoblastic leukaemia (ALL), respectively. In the 1950s, bone marrow transplants were introduced as cell-based
22 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
therapies for patients with blood-borne cancers. Other notable achievements in recent times include the authorisation of the use of adult stem cells to treat fistulas connected to Crohn's disease and patient-derived limbal stem cells to restore damaged corneal epithelium. These innovations were the result of decades of fundamental study, and their successes—along with those of other cutting-edge treatments—have greatly increased interdisciplinary interest from a wide range of so far disconnected basic biological research and engineering disciplines.
Such continued excitement surrounding cell-based therapeutics stems from the possibility of rerouting natural cellular processes to allow for safety and effectiveness profiles that surpass those of other, more proven techniques. Biologics, which include recombinant proteins and other cell-derived biomolecules, can achieve high target specificity by utilising macromolecules' recognition capabilities, but their safety and efficacy may be limited by unfavourable pharmacokinetic (PK) and pharmacodynamic (PD) properties. With therapeutic transgene delivery, often via a viral vector, gene treatments provide the possibility of rectifying cellular genotype. Cell-based therapies, despite facing translational barriers like tumorigenicity and high manufacturing costs, possess unique intrinsic features such as migration, localisation and proliferation that could enhance efficacy against disease. Despite advancements in cellderived therapies for various purposes, commercialisation remains challenging due to the need for readily available cell sources.
Biological medicines often function by various mechanisms including inducing an immunological response against cancerous cells. Treatments using biological therapy can do this in a number of ways. Introducing substances into the body that stimulate the immune system is one method. Another involves reintroducing a person's immune system cells into their body after they have been
Cell-based therapies, with their potent mechanisms of action, hold promise in treating currently incurable diseases, but face obstacles in translation and commercialisation; advanced research utilising next-generation engineering techniques aims to overcome these challenges.
trained to combat cancer cells in-vitro. Targeting the cancer cells with biological treatment allows them to switch on or off cell signals that aid in their immune system evasion. Immune checkpoint inhibitors, for instance, are medications that specifically target certain receptors on the surface of cancer cells. There, they obstruct the signals that cancer cells transmit in an effort to hide from the immune system.
Adoptive cell transfer
The process of adoptive T cell transfer entails isolating and reintroducing T cells into patients in order to cure illnesses. The process's ultimate goal is to stimulate and expand powerful, antigen-specific T cell immunity, which is theoretically identical to the goal of a successful T cell immunisation. One of the major drawbacks of vaccine-based approaches, which is the need to de-novo activate and expand a tumour antigen-specific T cell response in patients who are frequently immunocompromised and highly tolerant to cancer antigens or antigens expressed during chronic infection, may also be addressed through adoptive T cell transfer. More than 50 years ago, adoptive cell transfer with the purpose of targeting illness was initially documented in rodent models. Expanding ex vivo large numbers of T cells for adoptive
immunotherapy has been made easier by advances in T cell biology, including mechanisms for T cell activation and target recognition, the function of accessory surface molecules and signal transduction pathways involved in the regulation of T cell function and survival, and the identification and cloning of soluble T cell growth factors.
Chimeric antigen receptor (CAR) T-cell therapy
Treatment for cancer using chimeric antigen receptor (CAR)-T cell therapy is a ground-breaking novel approach. CAR molecules are primarily composed of four components: (i) an intracellular signalling domain that facilitates T cell activation (CD3 ); (ii) a hinge region; (iii) a transmembrane domain; and (iv) an extracellular target antigen-binding domain, which is often an antibodyderived single-chain variable fragment (scFv). Third-generation CARs have two costimulatory domains, whereas second-generation CARs only contain one co-stimulatory intracellular domain (often CD28 or 4-1BB). The production of secreting molecules (cytokines, T cell engagers, agonists or inhibitors of various cell receptors, etc.) or membrane receptors (such as chemokine receptors) is another characteristic of fourth generation CAR-T cells that modifies their efficiency. Currently, relapsed or refractory (r/r) haematological malignancies are the only conditions for which CAR-T cell treatments have been given FDA and EMA approval for commercialisation. Tisagenlecleucel (KymriahTM), the first CAR-T treatment, was permitted by the FDA in 2017 to treat r/r acute lymphoblastic leukaemia (ALL), and in 2018 it was also licenced to treat r/r diffuse large B cell lymphoma (DLBCL). Lisocabtagene maraleucel (BreyanziTM) was approved in 2021 for r/r large B cell lymphoma, primary mediastinal large B cell lymphoma, DLBCL, and grade 3B follicular lymphoma. The same year, idecabtagene vicleucel (AbecmaTM) for r/r multiple myeloma was commercialised.
23 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
Ciltacabtagene autoleucel (CarvyktiTM) was the latest CAR-T cell treatment to get FDA and EMA approval in 2022 for multiple myeloma. Furthermore, in 2021, the Spanish Agency of Medicines and Medical Devices (AEMPS) authorised for ALL, under a unique local authorisation known as a "hospital exemption," the product ARI-0001, a CAR-T cell therapy developed at the Hospital Clínic of Barcelona that is not industrially manufactured. This is the first medicine of its kind to be approved in all of Europe. The therapeutic efficiency of CAR-T cells in solid tumours and haematological malignancies is limited by several obstacles, despite the fact that therapy with CAR-T cells has achieved significant clinical responses with some subsets of B cell leukaemia or lymphoma. Severe, potentially fatal toxicities, weak antitumor activity, antigen evasion, restricted trafficking, and restricted tumour invasion are some of the obstacles to successful CAR-T cell treatment. Furthermore, interactions between CAR-T cells and their host and tumour microenvironment significantly modify CAR-T cell activity. To address these major obstacles, new methods and techniques for creating more potent CAR-T cells with enhanced anti-tumor activity and reduced toxicity are required.
Cytokine therapy
Because of their critical functions in immunity, cytokines are appealing as treatments for a range of immune-related conditions. However, the short blood half-lives and serious side effects brought on by low specificity and unfavourable biodistribution of cytokines have prevented their broad clinical usage. Bioengineering advances have produced new technology for cytokine engineering as well as advances in our understanding of cytokine biology. During immunological reactions, information is exchanged between cells directly or by the release of biomolecules, the most significant of which are tiny proteins called cytokines. Based on their roles, many
primary cytokine classes can be differentiated. Pro-inflammatory cytokines, which include tumour necrosis factor (TNF), IL-1 , IL-6, IL-17, and IL-22, activate antimicrobial and immunostimulatory processes. Anti-inflammatory cytokines, such transforming growth factor- (TGF ) and IL-1 receptor antagonist (IL-1RA), reduce inflammation and accelerate wound healing. Another type of cytokines that control immune cell movement are chemokines, which include IL-8 and CC-chemokine ligand 2 (CCL2). Antiviral immunity relies on interferons, and homeostasis and the proliferation of immune cell progenitor cells are regulated by colonystimulating factors such as granulocyte–macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and macrophage colony-stimulating factor (M-CSF). The majority of cytokines are produced by immune cells, however non-immune cells and tissues can also release cytokines. Improvements in bioengineering have led to new understandings of cytokine biology, including information on the structure, function, and binding processes of receptors as well as cytokine sequence and structure. When cytokine production is increased, as in autoimmune and inflammatory illnesses, blocking cytokine function with monoclonal antibodies or receptor blockers is very effective. Treatment for Crohn's illness and rheumatoid arthritis that works well is blocking TNF31. Suppressing IL-17 or IL-23 can be used to treat psoriasis. COVID-19 can be treated with IL-6 and IL-1 antagonists. It is also possible to use cytokines therapeutically to control immunological reactions. Nevertheless, creating medicines based on cytokines is still difficult. The limited therapeutic range of cytokines is attributed to their short blood half-lives, pleiotropism, and poor tissue distribution.
Gene therapy
Many promising therapies are available through the developing science of cancer
gene therapy. In order to assist effect a cure, a variety of treatment modalities that all involve genetic material to change cells (either in vitro or in vivo) are collectively referred to as gene therapy. Extensive in vitro and preclinical animal models have demonstrated outstanding success in assessing a broad range of gene therapy drugs. For instance, using gene therapy to produce cancer vaccines, target viruses to cancer cells for lysis and death, cut off the blood supply to the tumour, and introduce genes that either kill the cancer cells or return them to their normal cellular phenotype has been shown to improve survival in lung cancer models. Preclinical gene therapy investigations have been conducted on several malignancies, including gliomas, pancreatic cancer, liver cancer, and many more. There are significant safety concerns, as there are with any novel form of therapy. The death of a patient in a 1999 dose-escalation gene therapy experiment put an end to the initial fervour surrounding gene therapy as a therapeutic approach. All gene therapy studies were reevaluated for safety, even though this one involved using gene therapy to treat ornithine transcarbamylase deficiency, a metabolic disorder, rather for cancer. Thirteen Since then, hundreds of cancer patients worldwide have taken part in gene therapy studies with very few treatment side effects, and safer and newer gene therapy delivery methods have been developed.
Immunoconjugates
The effective introduction of chimeric and genetically altered human immunoglobulin proteins has made monoclonal antibodies (mAbs) a proven treatment option for cancer. The distinct characteristics of monoclonal antibodies (mAbs) such as their elevated affinity and specificity, coupled with the differential expression of target antigen in tumour cells as opposed to normal cells, render them appealing tools for cancer immunotherapy. The goal of the field of immunoconjugate development is to
24 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
bring together the best features of these two distinct modalities—the cytotoxic and radionuclide compounds, and the selectivity of monoclonal antibody therapy. Gemtuzumab ozogamicin, a drug compound, and 90Y-ibritumomab tiuxetan, a radiolabeled monoclonal antibody, have been authorised for the treatment of cancers. Preclinical and clinical trials are being conducted on other conjugates that carry toxic payloads of peptide exotoxins, maytansinoids, geldanamycin, and calicheamicin.
Oncolytic virus therapy
A new class of cancer therapeutics called oncolytic viruses (OVs) has several advantages over existing ones, including the ability to deliver multiple eukaryotic transgene payloads, induce immunogenic cell death, promote antitumor immunity, and have a tolerable safety profile that largely does not overlap with other cancer therapeutics. It is possible to equip modified OVs with desired foreign genes that may use several methods to produce significant anticancer effects. Though OVs have similar anticancer processes, distinct virus types or subtypes are being investigated for their ability to treat diverse clinical diseases. Depending on the kind of nucleic acid, OVs can be single- or double-stranded DNA or RNA viruses. The first phase is the transformation of wild-type viruses into tumourspecific Ovs. This can occur during infection or replication. It is necessary to conduct the procedure in accordance with the characteristics of the viruses and tumour cells. While genetically modified oncoviruses (OVs) are developed for improved targeting selectivity, various virus types exhibit varying natural affinities and preferred replication tendencies in distinct tumour cells.
Though talimogene laherparepvec (T-VEC) is still the sole generally authorised medication, four oncolytic viruses and one non-oncolytic virus have received approval for the treatment of cancer worldwide so far. T-VEC was first licenced in 2015 and is designed for the
treatment of individuals with recurrent melanoma following first surgery. Data from clinical studies of many different drugs are also becoming accessible, along with an increasing amount of information on the clinical experience of patients receiving T-VEC.
Conclusion
As chemotherapy's efficacy and toxicity to normal tissues reach their limitations, researchers are searching for novel treatment plans based on combination and personalised therapy, which will likely shape medical practice in the future. Modern molecular biology techniques, like the NGS approach, assure the evolution of cancer therapeutics by providing a deeper understanding of the biology of cancer and identifying a suitable therapeutic target. This advancement is also made feasible by the application of more


sophisticated bioinformatics techniques, which allow for exact medication modification to the therapeutic target.
It is reasonable to predict that biological therapy in general will become more crucial in the management of neoplasms. We provide many strategies for enhancing the bioavailability, binding strength, or stability of CAR T cell treatment, anticancer vaccinations, and antibody structures in the evaluation of a few research. Furthermore, research possibilities that might enhance the efficacy of biological treatment were highlighted. It has been shown that there are advantages to combining different biological therapies, such as immune checkpoint inhibitors with oncolytic viruses and anti-cancer vaccinations.
References are available at www.pharmafocusasia.com
Dhruv Kumar is currently serving as a Professor and Head of Allied Health Sciences at The School of Health Sciences and Technology (SoHST), UPES, Dehradun. He has completed B.Sc. in Chemistry from Banaras Hindu University (BHU), Varanasi, and M.Sc. in Bioinformatics with Distinction from the University of Allahabad (AU), Prayagraj. He obtained Ph.D. in Cellular, Molecular, and Industrial Biology from the University of Bologna (UNIBO), Italy, under the esteemed fellowship of the Ministry of Human Resource and Development (MHRD), Indo-Italian Government fellowship, and completed Postdoctoral training from the University of Kansas Medical Center, Kansas City, USA. His expertise extends to various types of cancer, including Prostate, Pancreatic, Brain, Breast, Gallbladder, Endometrial, and Head and Neck Cancer. His research interest lies in understanding the molecular mechanisms behind Autophagy and Apoptosis regulation in Cancer Stem Cells and exploring the Autophagy-Metabolic axis in cancer cells, particularly those affecting the head and neck, oral, breast, and brain. Furthermore, his extensive research work has been published in numerous peer-reviewed high-impact journals. He has been recognised as one of the top 2% SCOPUS cited scientists in the fields of Cancer Biology, Oncology and Bioinformatics.
Dr. Sibi Raj has done PhD in Biotechnology from UPES, Dehradun. She is specialised in the area of Head and neck cancer metabolism under the guidance of Dr. Dhruv Kumar (Professor). Her research involved the area of cancer metabolism, drug designing, especially targeting the c-Met receptor. She published several high-quality research and review articles in prestigious journals such as Molecular Cancer, Seminars in Cancer Biology, Cancers, Antioxidants, and many more. She also received the ICMR-SRF fellowship and even filed a patent based on the groundbreaking work during her doctoral studies.
25 www.pharmafocusasia.com
AUTHOR BIO
RESEARCH & DEVELOPMENT
Biomarkers for Cardiovascular diseases
A brief insight
Cardiovascular disease (CVD) is the leading cause of death on a global scale due to the consistent rise in mortality rates. As such, biomarkers are the utmost priority within the medical domain for discerning early warning signs of CVDs and delivering an evidence-based delineation of heart failure. This overview highlights the variety of promising biomarkers that provide the need for risk stratification, diagnosis, prognostication, and decision-making regarding the management of cardiovascular diseases.
Vignesh M, Rao SR, Agiesh B
MGM-Advanced Research Institute, Sri Balaji
Cardiovascular disorders (CVDs), including peripheral vascular disease, ischemic heart disease, and stroke, are the leading causes of death globally, contributing to 17.9 million deaths in 2016. Recently, it has been predicted that 23.6 million people would have died from CVD, with heart disease and stroke accounting for the majority of deaths by 2030. Though non-pharmacological prevention of CVD is important for public health initiatives to avoid the onset and consequences of CVD, the use of pharmaceutical therapies is still a fundamental pillar for preventing and managing CVD. This is supported by a notable increase in drug development for CVD between 1995 and 2020. However, the major concerns in the advancement of novel medications are lack of efficacy and safety, which account for 52 per cent and 24 per cent of drug failures, respectively. In this context, biomarkers could serve as a powerful tool for the enhanced identification of high-risk patients, thereby facilitating prompt and precise diagnoses of diseases and effectively prognosticating and treating patients.
Why biomarkers for cardiovascular diseases?
Biomarkers are specific molecules or substances found in the body that can indicate the presence of risk or progression of a disease. Technically biomarkers are observable characteristics that possess the capability of being objectively evaluated and furnish measurable indications pertaining to physiological, pathological
26 PHARMA FOCUS ASIA ISSUE 54 - 2024
Vidyapeeth Deemed to be University.
RESEARCH & DEVELOPMENT
and pharmacological responses to an intervention. Investigating the role of biomarkers in causing disease manifestation can (i) Impart etiological knowledge, which can enhance our comprehensive understanding of the molecular mechanisms of disease pathology (ii) provide the comparative role of altered traits implicated in disease etiology during therapeutic intervention and (iii) contribute to the development of public health strategies in response to environmental exposure.
Cardiac biomarkers refer to biomolecular constituents released into circulation following cardiac injury or strain. These biomarkers aid in assessing an individual's susceptibility to cardiac diseases and serve as a means for monitoring and treating individuals suspected of suffering from various CVDs. In this context, this
Markers
Carbonic Anhydrase
Function
write up provides an overview of existing biomarkers that consistently aid in the prognosis of early CVD.
Biomarkers in CVDs
Cardiac Troponin
The regulation of cardiac troponin is a calcium-dependent process that governs the contraction and relaxation of the heart muscle. They are widely utilised as the gold standard biomarker for myocardial infarction (MI). Troponin T (TNT), troponin I (TNI), and troponin C (TNC) are the three components that make up the cardiac troponin complex, which are currently used to detect the cardiacspecific TNT and cardiac-specific TNI by immunoassays utilising monoclonal antibodies.
CVD
Brain natriuretic peptide (BNP and NT-pro BNP)
BNP and NT-proBNP have been shown in studies to be higher in patients with heart failure, making them an important biomarker for CVDs. BNP is also involved in the renin-angiotensin-aldosterone system and has been linked to a number of physiological processes, such as diuresis, natriuresis, vasodilation, and the inhibition of the sympathetic nervous system. Furthermore, a mid-regional sequence of pro-a-type natriuretic peptide has been shown to be a useful biomarker for the diagnosis and prognosis of acute heart failure in clinical settings.
Glycogen phosphorylase BB (GPBB)
Because of high oxygen demand, the heart is particularly sensitive to carbon
Regulates gluconeogenesis, acid– base homeostasis, adipogenesis, and calcification
Higher in MI patients compared to traumatic patients and exercise subjects
Endothelin-1 (CT-proET-1) Potent vasoconstrictor and sympathetic neurohormones Strongly connected with arterial hypertension
Creatinine Kinase – MB
Hydroxybutyrate dehydrogenase
Pregnancy- associated plasma protein- A
Copeptin
Suppression of Tumorigenicity (ST2)
Galectin-3
Neuregulin-1
Myeloperoxidase
Trimethylamine n-oxide
Neutrophil gelatinaseassociated lipocalin
Uric Acid
Retinol binding protein 4
Table 1
Predominantly found in heart tissues
Cardiac enzyme found in heart, red blood cells, kidney and brain
Zinc-binding matrix metalloproteinase that triggers insulin-derived growth factor
Stand-alone marker for myocardial damage
Elevated in non-ischemic dilated cardiomyopathy
Higher in patients with and without CAD
Activates the hypothalamus-pituitary- adrenals axis Considered as marker for cardiovascular events
A cytokine produced by heart under metabolic stress
Secreted by activated cardiac macrophage and plays a role in atherogenesis
Paracrine growth factor that promotes cell survival, growth and maintenance
Produced by macrophage and polymorphonuclear neutrophils during inflammation
Dietary metabolite produced by liver enzymes
Acute phase lipocalin protein widely used as a kidney indicator of nephrotoxic and ischemic damage
Independent predictor of HF mortality
Predicts cardiovascular death patients with CAD
Strongly correlated with advanced stages of HF
Elevated in CAD patients
Predicts cardiovascular mortality in peripheral artery disease
Elevated in chronic HF patients compared to healthy subjects
An end product of purine metabolism in human Independent risk factor for CVD morbidity
A lipocalin protein that acts a vector for vitamin A
Increased in CVD patients compared to healthy subjects
27 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
monoxide (CO)-induced hypoxia. For this reason, patients with cardiovascular issues may not be diagnosed with CO poisoning or receive treatment due to absence of obvious symptoms or distinct electrocardiogram alteration. Nevertheless, glycogen phosphorylase BB isoenzyme (GPBB) is one of the emerging and potential prognostic biomarkers for myocardial hypoxia. Moreover, investigations have shown that GPBB may be a useful cardiac marker in the diagnosis of acute coronary syndrome and myocardial ischemia.
Soluble CD40 Ligand (sCD40L)
TNF superfamily member CD40L is found in a variety of cellular types, including immune cells (lymphocytes, dendritic cells, neutrophils, and macrophages) and nonimmune cells (endothelial cells, vascular smooth muscle cells, and epithelial cells). Further, the interaction between CD40L and the receptor CD40 is crucial for immunomodula -



tory properties and is associated with atherosclerosis and plaque instability. Clinical trials have demonstrated the predictive capacity of sCD40L as a biomarker for determining CVD risk in patients with CAD. Another trial demonstrated that elevated levels of sCD40L could reliably predict the risk of stroke in patients who had previously experienced a minor stroke or transient ischemic attack.
Heart-fatty acid binding protein (H-FABP)
Heart-fatty acid binding protein (H-FABP) is a diminutive cytosolic protein found in cardiac tissues that serves as a mediator for the transportation of fatty acids from the plasma membrane to oxidation sites in both mitochondria and peroxisomes. Recently, studies have observed a rise in serum levels of H-FABP as soon as 30 min after MI. Additionally, H-FABP is widely employed as a prognostic biomarker for HF mortality.
Agiesh kumar Balakrishna Pillai received his Ph.D. in Biotechnology in 2008 from Pondicherry University, Puducherry. His postdoctoral research was conducted in virology (arboviral) at the University of Georgia and the Texas Tech University. His academic career continued with position as Senior Scientist and Associate professor at the Mahatma Gandhi Medical Advanced Research Institute (MGMARI), Sri Balaji Vidyapeeth. Currently, he is actively involved in various research projects in the field of arboviral disease, respiratory infection and heart failure.
Vignesh Mariappan recently received his Ph.D. in Interdisciplinary Research from Sri Balaji Vidyapeeth. During this period, he worked in dengue disease pathogenesis with a special focus on characterisation and validation of endothelial and macrophage released molecules for the early prognosis of dengue disease outcome.
S R Rao, former Senior Advisor in the Department of Biotechnology (Govt. of India), is a renowned expert in promoting R&D across various fields of biotechnology. Currently, he is serving as the Vice President of Sri Balaji Vidyapeeth University Pondicherry and committed to promoting research, innovation, and development ecosystems. Also, he is the founder of the Global Alliance for Pandemic Preparedness and Response and the RISAsian Biotechnology Development Review.
Growth differentiation factor-15 (GDF-15)
Growth differentiation factor-15 (GDF-15) is primarily generated by the placenta and plays a significant role in the activation of pathways linked to apoptosis, inflammation, and tissue damage. Prior research has demonstrated that raised GDF- 15 levels above a particular threshold may serve as a reliable marker for identifying individuals who are at a high risk of heart disease. Furthermore, in individuals with chronic heart failure, increased levels of GDG-15 have been linked to adverse remodelling and hypertrophy.
Lipoprotein-associated phospholipase A2 (Lp-PLA2) Lp-PLA2 is mostly produced by monocytes and macrophages and is a member of the phospholipase A2 superfamily, also known as platelet-activating factor acetylhydrolase. Fascinatingly, the first report from the West Scotland Coronary Prevention Study presented the main evidence supporting a link between elevated Lp-PLA2 levels and cardiovascular events. Also, further studies have demonstrated that Lp-PLA2 activity may function as a stand-alone predictor of CAD and stroke in the general population.
Apart from the commonly available proteins described above, some of the potential biomarkers that are associated with CVD are listed in Table 1
Conclusion and Future Perspectives
Though the existence of emerging and innovative biomarkers in clinical these markers into clinical practice. For instance, (i) the functions and physiological CAD, as well as their clinical usefulness, have to be thoroughly explained; (ii) the be defined; and (iii) since CVD is a multi-factorial disease, further multicentric and well as to understand the molecular pathology of CVD. Addressing these may lead to management of CVD.
References are available at www.pharmafocusasia.com
28 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
AUTHOR BIO

DRUG ANALYSIS IN SPORTS
The aim of sports is to provide equal opportunities to all athletes to seek victory and fame. Hence, the use of performance-enhancing substances and methods has been prohibited not only to ensure fairness, but also to protect athletes ‘health and safeguard the spirit of sport. Analytical chemistry is the backbone of the drug analysis methods being used in anti-doping, and has the core function to have the most sensitive testing methods being specific at the same time. In addition, these drug analysis procedures are also designed to differentiate endogenous substances from their exogenously derived counterparts. The ongoing research on drug analysis in sports is about advancing the technical capabilities of the anti-doping laboratories. Advancements in instrumental sensitivity and capability to screen increasing numbers of compounds in a single method has also played a pivotal role in anti-doping laboratories.
Shobha Ahi, Deputy Director, Drug Control Centre, Department of Analytical, Environmental and Forensic Sciences, School of Cancer and Pharmaceutical Sciences, King's College London
29 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
Performance enhancement has featured in sports since ancient times. Doping seems to be as old as sports itself, although the word "doping" was introduced in the English dictionary for the first time in 1889.
According to the International Olympic Committee (IOC), doping is the administration of or use by a competing athlete of any substance foreign to the body or any physiological substance taken in abnormal quantity or taken by an abnormal route of entry into the body with the sole purpose of increasing in an artificial and unfair manner his/her performance in competition.
The drug analysis methods are designed for routine doping control to determine whether a prohibited substance is present in a doping control sample.
From an outsider’s perspective, the process of doping control, spanning everything from receiving a urine or blood sample to reporting a violation of anti-doping rules, seems relatively straightforward. Nonetheless, a lot of
brainpower goes into developing the process of drug analysis that the athletes’ samples go through.
Generally, urine samples are homogeneous mixtures containing many different substances, including water, urea, salt, and other electrolytes. The presence of other chemicals depends on a person’s diet, medication or supplements taken. The testing methods designed for drug analysis in an athlete’s sample are governed by the World AntiDoping Agency (WADA). The main activities of WADA include scientific research, education, development of anti-doping capacities and monitoring the compliance with the World AntiDoping Code. The World Anti-Doping Code includes the prohibited drugs list, which covers all the substances and methods banned from sports inand out of competition. The different compounds are divided in several classes which are forbidden either out-/and in-competition or only during competition.
For athletes, samples are analysed at one of 30 WADA accredited laboratories which covers a list of over 350 prohibited substances and methods in sports — updated every year — that samples are screened for. Doping control relies on the separation and identification of all of the substances present in a sample, as per the WADA prohibited list, whether they might be metabolites of a performance-enhancing drug, or the confounding factors which can affect the results. To ensure reliability, repeatability and clean separations, different screening methods have been developed for each of the substances on the list. Most of the banned substances require qualitative analysis. For some specific compounds, it is difficult to distinguish between the social or therapeutic use and the misuse, and, therefore, threshold concentrations have been established. In other cases, the threshold is used to differentiate between physiological values and exogenous administration of the compound.

30 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
The analytics are divided into screening procedures and methods of confirmatory analysis. The aim of screening is to isolate suspicious samples for further analysis. In confirmation, samples are analysed with methods that provide unequivocal identification of the substances. Because of the large array of target compounds, many different analytical methods are used. Most methods are based on chromatography combined with mass spectrometry (MS). Analytical procedures have to be constantly improved and updated in order to keep pace with trends in substance abuse and to fulfill increasing quality requirements.
The work of anti-doping laboratories is regulated by WADA, which ensures global harmonisation of the anti-doping procedures/guidelines. The List of Prohibited Substances and Methods includes hundreds of chemically and pharmacologically diverse compounds from different classes. The List is updated annually, which means that laboratories have to constantly update their methods and to catch up with the ever-growing selection of drugs. This imposes a considerable demand on laboratories to be able to identify suspicious samples in a screening process with only a limited sample amount in a short period of time for a more specific conformation analysis. In the case of in-competition substances, the analyte selection is the broadest and the results from screening sometimes have to be reported in just 24 hours, emphasising the need for high throughput methods. Every presumptive analytical finding in screening has to be confirmed with a more specific method optimised for the target analyte. The results are compared with reference standards, and if WADA ‘s identification criteria, such as diagnostic ions, ions ratios and retention times (RT) are fulfilled, an adverse analytical finding is reported.
The analysis of prohibited substances and methods in the athlete biological samples mainly urine and
For athletes, samples are analysed at one of 30 WADA accredited laboratories which covers a list of over 350 prohibited substances and methods in sports — updated every year — that samples are screened for.
blood is a highly challenging task. Mostly because of limited sample volumes, faster turn-around time and analysis of compounds with a wide range of physico-chemical properties and molecular weight. Hence, it is always required from an anti-doping analytical method to be highly sensitive to be able to detect at very low levels and highly selective to be able to conclude that the signals are not arising from any interference. There are several issues that have to be considered before establishing an analysis method for a doping agent which involves studying the metabolism of the drug, its physicochemical properties, availability of reference standard, probability for inclusion in the existing test method as an extension etc. The approach for extension of detection windows by inclusion of drug metabolites also increases the number of target compounds. The analysis is predominantly performed on a urine sample, although few of the analytes are tested in blood viz. continuous erythropoietin receptor activator (CERA), haemoglobin-based oxygen carriers (HBOCs), human growth hormone (hGH) or blood transfusions. Other specimens such as hair and saliva have been proposed but are not used as
authenticated tests in doping control. However, urine is still the specimen of choice since the collection is noninvasive, the volume available is quite large, the concentrations of drugs are higher than in blood, and since hydrophilic metabolites are also excreted in urine, thus enlarging the detection time window.
The main tools for anti-doping analysis have always been chromatography and mass spectrometry. Gas Chromatography Mass Spectrometry (GC-MS) was the gold standard technique until early 2000. The combination of GC with MS was first reported in 1958. Since then, it has become increasingly utilised in doping control. The GC-MS can separate the volatile components of complex mixtures and can record a mass spectrum of each component. This hybrid instrument provides two separate dimensions of information about the components in the sample, GC retention times and electron ionisation (EI) mass spectra. GC retention time is related to specific chemical properties of the molecules in question (e.g. volatility, polarity, presence of specific functional groups) while molecular weight (derived from the mass spectrum) is indicative of atomic composition. The limitation of GC-MS includes that only volatile substances can be measured and extensive derivatisation steps. In GC-MS techniques, sensitivity may be lost due to chemical oxidation or derivatisation and are limited to volatile, non-polar and thermally stable compounds.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is a well-established technique for quantitative and qualitative analyses in the field of doping control. Availability of new generation LC-MS instruments have provided significant advancement in the detection of prohibited substances. The technique of liquid chromatography used in concert with (tandem) mass spectrometry has complemented sports drug testing strategies ever since
31 www.pharmafocusasia.com
RESEARCH & DEVELOPMENT
soft ionisation interfaces such as ESI or APCI became commercially available. Commonly no or little derivatisation is required while preparing samples for LC-MS analysis. Numerous applications have been developed that allow the determination of prohibited therapeutics that are barely detectable or undetectable with conventional GC-MS and comprehensive summaries of the methods commonly employed in doping controls have been published in the past. Due to the progressive nature of doping controls, numerous new applications and drug-testing strategies based on LC–MS/MS are frequently developed in order to improve the portfolios of drugtesting laboratories. With the advancements in the testing technology and invention of the LC-High Resolution Mass Spectrometers (HRMS), the identification of analytes and unknown structure elucidation can be facilitated. The untargeted approach has become a promising tool in doping control.
However, for some prohibited compounds and methods neither GC– nor LC–MS(/MS) techniques are suitable to report a positive finding. This led to enforcement of few alternative strategies for detection in doping control which includes immunoassay techniques, setting up of thresholds, inclusion of Isotope Ratio Mass Spectrometry (IRMS) to differentiate between endogenous and exogenous analogs of steroids. Immunoassays or electrophoretic methods are usually employed for macromolecules. In recent years, few indirect approaches have also been established as an advancement in the doping control detection methods. For example, the monitoring of biomarkers in case of human growth hormone (hGH) to discriminate between its endogenous and exogenous administration. The monitoring of an athlete's biological passport for monitoring intra-individual variation can be utilised as an indirect marker to conclude to administration of a banned substance. For example, erythropoietin
(EPO) misuse is indirectly monitored by the measurement of blood parameters, such as haemoglobin, hematocrit, ferritin, soluble transferrin receptor or reticulocyte.
Sample preparation remains the backbone of any analytical test procedures and has evolved in recent years from liquid-liquid extraction to solid phase extraction involving various chemistries. Various methods have been developed in recent years with use of reduced sample and solvent volume. The automation in sample preparation has also helped doping control laboratories in reducing their turn-around-times.
Recently, the Dried Blood Sample (DBS) methodology is also being explored in various anti-doping laboratories to enhance their technical capabilities. The literature suggests that the use of dried blood spots as a method for sample collection for anti-doping is a highly promising avenue of research, offering several advantages over existing methodology such as urine collection. which include reduced costs and ease of sample collection. The DBSs testing refers to a biosampling technique where a small volume of whole blood, typically in the 5-100 μ L range (i.e. from a finger “prick”) is “spotted” and dried onto a piece of filter paper. This technique has recently been adopted to test samples from approximately 70 athletes during the Tokyo Olympic Games 2021.
Anti-doping research is ever-evolving, as new performance-enhancing drugs and derivatives keep on emerging during the process of drug discovery. Following the inclusion of a new compound on the WADA prohibited list, a screening method for it needs to be developed and validated alongside the over 350 other compounds while keeping up with the turn-around times for reporting samples. This can be especially tricky at huge sporting events, which leaves the Drug Control Centre with upward of 200 samples a day to
analyse within a 24-hour turn-around.
My take on it: “The job of an antidoping scientist is for a lifetime; there are constantly new things to learn and explore. My quest for knowledge always finds its way because of the ever-improving scientific progress in the field of anti-doping, and that’s what I love about my work. This passion is reflected in my ongoing research projects: investigating flagged, suspicious peaks revealing possibly new drugs, finding analysis methods with lower limits of detection and more efficient sample preparation, and giving inspiring talks to aspiring chemists.
References are available at www.pharmafocusasia.com
AUTHOR BIO

Shobha Ahi is Deputy Director at the Drug Control Center, King's College London. She has over 17 years of experience as an anti-doping scientist and was serving the National Dope Testing Laboratory (NDTL), Govt. of India before joining the Drug Control Centre at King’s College London in 2021. She has developed and led several research projects pertaining to anti-doping science, drug metabolism and supplements in sports.
32 PHARMA FOCUS ASIA ISSUE 54 - 2024
RESEARCH & DEVELOPMENT
Innovative Trial Designs
Revolutionising clinical research
Clinical research has evolved significantly in recent years, with innovative trial designs transforming the traditional paradigm. These modern approaches expedite drug development and prioritise patient-centred research.
In this article, we will explore various innovative trial designs, real-world applications, and their impact on patient recruitment. While these innovations offer benefits like accelerated drug development and improved patientcentricity, they come with challenges, including regulatory complexity and data security. Additionally, platforms like Find Me Cure are bridging the gap between patients and
researchers, enhancing access and diversifying the participant pool.
Tihomira Nikolova, Clinical Research Associate, PSI CRO AG
Clinical research has always been at the forefront of medical advancements, but the methods for conducting trials have undergone significant transformations in recent years. Innovative trial designs have emerged as a powerful force, revolutionising the way we approach clinical studies. These modern approaches not only expedite the drug development process but also ensure that patients are at the centre of the research. In this article, we will explore various innovative trial designs, their advantages, challenges, and real-world applications, shedding some light on the dynamic landscape of clinical research.
The Traditional Clinical Trial Paradigm
Clinical trials have historically followed a fixed and sequential structure. In such trials, researchers define a study protocol
Cover Story CLINICAL TRIALS
and then enrol participants following those strict criteria. This rigid design often makes it challenging to adapt to new information, resulting in inefficiencies, wasted resources, and delays in bringing effective treatments to patients.
In recent past, the landscape of clinical trials looked significantly different. Paper Case Report Forms (CRFs) were the norm, with research data painstakingly recorded by hand. This paper-based approach, while reliable, was not without its challenges. It often resulted in cumbersome data entry, transcription errors, and delays in data collection and analysis. Furthermore, monitoring the progress of trials involved on-site visits by the Clinical Research Associates (CRAs), which could be resource-intensive and time-consuming for all parties involved. Whoever hasn't transported a pile of "data collected" CRFs in their car trunk may not understand the monitoring era I'm referring to. Data management was also a labour-intensive process, often requiring meticulous attention to detail and multiple rounds of data reconciliation.
The Rise of Innovative Trial Designs Innovative trial designs encompass a range of methodologies that break away from the traditional mould. These approaches embrace flexibility, adaptability, and efficiency, allowing researchers to learn from each patient's experience, update trial parameters, and optimiseo the research process.
The shift to innovative trial designs represents a departure from these traditional practices, embracing electronic data capture, real-time monitoring, and adaptive strategies that streamline the research process, enhance data quality, and bring us closer to the goal of more patient-centric and efficient clinical trials.
Here are some brilliant examples:
1. Adaptive Trials
Adaptive trials are designed to evolve in real-time based on the data collected.
CLINICAL TRIALS
They allow for modifications to key aspects of the trial, such as the sample size, randomisation ratios, or even the treatment arms, as new information becomes available. This adaptive approach enables researchers to make informed decisions and refine the trial as it progresses.
2**. Umbrella Trials**
Umbrella trials, on the other hand, focus on a single disease or condition but test multiple treatments within that umbrella. This design is particularly effective when dealing with diseases that are highly heterogeneous, enabling researchers to identify the most suitable treatment for individual patients.
3**. Basket Trials**
Basket trials focus on specific genetic mutations or biomarkers rather than the location of the tumour. This approach allows researchers to test multiple treatments simultaneously on patients with different cancer types but the same genetic mutation. Basket trials provide a more patient-centred approach, as they match treatment to the genetic makeup of the patient.
4. Platform Trials
Platform trials are master protocols designed to investigate multiple treatments for a specific disease. They have the advantage of continuously enrolling patients and testing multiple treatments concurrently. As one treatment arm concludes, a new one can begin, ensuring that the trial remains dynamic and efficient.
Real-World Applications
The impact of innovative trial designs is best understood through real-world applications. Several notable examples have showcased the power of these modern methodologies. Each of these trials aims to address specific medical challenges, introduces innovative approaches, and offers unique benefits, along with their own set of challenges:
Paper CRFs were the norm, with research data painstakingly recorded by hand. Whoever hasn't transported a pile of "data collected" CRFs in their car trunk may not understand the monitoring era I'm referring to.
1. The I-SPY 2 Trial ClinicalTrials.govIdentifier: NCT01042379
See Figure: 1
Aims: The I-SPY 2 trial, designed for breast cancer, aims to accelerate the development of effective treatments for breast cancer by evaluating multiple treatment regimens in the neoadjuvant setting. The primary goal is to identify and advance treatments that demonstrate the most promise.
Innovation: The trial's adaptability is its hallmark innovation. It allows for realtime assessment of treatment efficacy, enabling the removal of less effective treatments and the introduction of promising ones as the trial progresses. This approach reduces the time required to bring effective treatments to patients.
Benefits: Patients receive potentially more effective treatments in a timelier manner. The trial optimises resource utilisation by minimising the need for lengthy, separate trials for each treatment, and it accelerates decision-making for drug development.
Challenges: The adaptability of the trial necessitates rigorous statistical methods to account for changing treatment arms. Ensuring informed consent and ethical considerations regarding changes in treatment plans are ongoing challenges.
34 PHARMA FOCUS ASIA ISSUE 54 - 2024
2. The National Lung Matrix Trial
ClinicalTrials.gov Identifier: NCT02664935
Aims: This umbrella trial for non-small cell lung cancer aims to match patients to targeted therapies based on specific genetic mutations. It personalizes treatment by identifying and offering treatments tailored to each patient's unique genetic profile.
Innovation: The trial's innovation lies in its personalised medicine approach, which maximises the likelihood of therapeutic benefit for patients. It shifts the focus from one-size-fits-all treatments to precision medicine.
Benefits: Patients benefit from treatments that are more likely to be effective for their specific genetic makeup. The trial minimises exposure to ineffective treatments, reducing potential side effects and optimising resource allocation.
Challenges: Identifying and enrolling patients with specific genetic mutations
can be challenging. Coordinating a trial with multiple targeted treatment arms can be complex.
3. The REMAP-CAP Trial
ClinicalTrials.gov Identifier: NCT02735707
See Figure: 2
Aims: During the COVID-19 pandemic, the REMAP-CAP trial aimed to improve outcomes for critically ill COVID-19 patients. The trial's adaptability allowed it to quickly test multiple treatments, including novel therapies, as the pandemic evolved.
Innovation: The trial's innovation lies in its rapid adaptation to the evolving needs of the pandemic. It provides a platform for testing a wide range of treatments simultaneously.
Benefits: The trial helps identify effective treatments and improves the chances of survival and recovery for critically ill
COVID-19 patients. It reduces the time needed to assess the effectiveness of potential treatments.
Challenges: Adapting the trial to a rapidly changing situation requires swift decision-making and coordination. The large amount of data generated and the need for real-time analysis can be challenging.
4. The NEJM Catalyst Innovations in Care Delivery
Aims: The NEJM Catalyst Innovations in Care Delivery initiative aims to showcase innovative trial designs and their impact on healthcare delivery and patient outcomes. It serves as a platform for sharing and disseminating insights into new approaches to care.
Innovation: The initiative itself is innovative in that it regularly highlights novel approaches and methodologies that enhance patient care delivery. It acts as a bridge for sharing best practices and innovative strategies.
35 www.pharmafocusasia.com
CLINICAL TRIALS
Figure:
1
Benefits: The initiative benefits healthcare professionals by providing a platform for learning about and adopting innovative approaches to care delivery. It contributes to the dissemination of best practices and innovations in the field.
Challenges: While the initiative doesn't conduct clinical trials itself, it faces challenges related to the evaluation and validation of the approaches it showcases.
Ensuring that featured innovations are evidence-based and effective is crucial.
These real-world applications of innovative trial designs illustrate the diverse aims and innovations that can be achieved through modern clinical research methodologies. While they offer significant benefits, they also come with unique challenges, underscoring the need for ongoing adaptation and innovation in the field of clinical trials.
Benefits of Innovative Trial Designs
The adoption of innovative trial designs
offers a multitude of benefits, transforming the clinical research landscape:
1. Accelerated Drug Development
Innovative trial designs enable faster decision-making by allowing for adaptive changes. This acceleration of the drug development process is critical, especially in the context of emerging diseases or urgent medical needs.
2. Resource Optimisation
Traditional trials often suffer from resource waste due to inefficiencies. Innovative designs allow for efficient resource allocation, reducing the overall cost of research.
3. Enhanced Patient-Centricity
Innovative trials prioritise patient needs by matching treatments with genetic profiles or adapting to patient experiences. This patient-centric approach contributes to better outcomes and improved patient satisfaction.
A great example of patient-centred approach are the Decentralised Clinical Trials. DCTs are a modern approach to clinical research that leverages technology to conduct trials with minimal reliance on physical sites, such as traditional hospitals or clinics. These trials aim to bring the research directly to patients, often in the comfort of their homes. There are some undeniable key benefits of DCTs, particularly in the context of a patient-centred approach, such as DCTs prioritiseprioritise patient convenience and comfort by allowing participants to engage from their homes, reducing the need for frequent site visits. DCTs also improve access and inclusivity by removing geographical barriers, enabling individuals in remote areas or with limited mobility to participate. They attract a more diverse pool of participants, leading to a better understanding of treatment effectiveness in different demographics. Patients in DCTs experi-
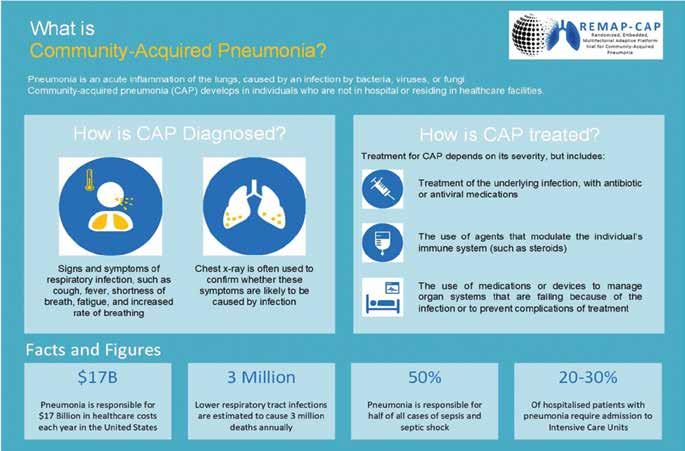
36 PHARMA FOCUS ASIA ISSUE 54 - 2024
CLINICAL TRIALS
Figure: 2
CLINICAL TRIALS

ence reduced travel, shorter wait times, and a flexible schedule, which is especially beneficial for those with chronic illnesses or ongoing treatment. Real-time data collection through remote monitoring enables researchers to access timely information on participants' health, treatment adherence, and potential side effects. DCTs also optimise resource utilisation by requiring fewer physical infrastructure and staff, resulting in cost savings and more efficient trial execution.
Despite all the benefits, there are still some challenges that need to be addressed in order to make the modern conduct of clinical research even more efficient. These challenges include ensuring data security and privacy in a decentralised environment, addressing technology access and literacy disparities, navigating regulatory hurdles, ensuring the quality of data collected remotely, maintaining participant engagement and adherence to trial protocols, addressing ethical considerations in decentralised trials, and implementing robust risk management strategies. As the field of clinical research continues to evolve, overcoming these challenges is essential to fully leverage the potential of DCTs.
4. Improved Data Quality
Real-time adaptability allows researchers to refine trial parameters, resulting in more precise and valuable data.
Challenges and Considerations
While innovative trial designs offer many advantages, they also come with their own set of challenges and considerations:
1. Regulatory Complexity
Regulatory bodies are still adapting to these new trial designs, which can create hurdles in approvals and compliance.
2. Statistical Rigour
Innovative designs require robust statistical methodologies to account for adaptations and multiple treatment arms.
3.
Data Sharing
As these designs involve larger and more complex datasets, data sharing and transparency become increasingly important.
4. Ethical Considerations
Patient consent and the potential for changes in treatment arms pose ethical considerations that must be addressed.
A Promising Future: ELEM Biotech S.L.
A significant step towards fostering innovation in clinical research is the recognition by regulatory bodies, such as the FDA, that in some cases, animal testing may no longer be necessary. This paradigm shift acknowledges the ethical concerns surrounding animal testing and the advancement of alternative methodologies, such as in vitro testing and computational modelling. By reducing the reliance on animal models, clinical research can become more ethical, efficient, and patient-centric. This shift not only accelerates the drug development process but also aligns with the principles of innovative trial designs by prioritising the welfare of both patients and animals. It represents a promising future where scientific progress and ethical considerations can coexist harmoniously in the pursuit of medical advancements.
Pioneering the Future of Clinical Trials
In the dynamic realm of clinical research, ELEM Biotech S.L. is pushing the boundaries of innovation with a groundbreaking approach that introduces virtual humans into the world of clinical trials. Their innovative methodology leverages predictive modelling and simulation, powered by supercomputers in the cloud, to create virtual populations that replicate human cells, tissues, and organs, mirroring the diversity and pathologies of society. This revolutionary technology is designed to address the entire spectrum of product development, from discovery to preclinical and clinical phases.
Benefits and Potential:
The integration of virtual humans offers a plethora of benefits to the clinical research landscape. It accelerates the drug development process, enhances resource efficiency, and prioritises a patient-centric approach. These virtual human models showcase how organs function in diseased states and predict the efficacy of treatments in restoring vital functions. They serve as
37 www.pharmafocusasia.com
surrogates in virtual patient trials, assist clinicians in making informed decisions, and contribute to the development of innovative treatments. ELEM's technology is designed to tackle complex physiological systems, including the cardiovascular and respiratory systems.
Challenges and Considerations:
While the prospect of virtual humans in clinical trials is immensely promising, it comes with its set of challenges and considerations. Regulatory bodies are adapting to these new methodologies, and the integration of predictive modelling into clinical trial approval processes can be complex. Robust validation of these virtual models is essential to ensure their accuracy and reliability. Ethical considerations regarding the use of virtual humans, especially in scenarios involving the prediction of treatment outcomes, must be addressed. Additionally, it's crucial to maintain transparency and data sharing standards in the application of this technology.
Impact on Revolutionising Clinical Trials:
In the ever-evolving landscape of clinical research, the integration of innovative trial designs represents a significant step toward revolutionising the field of clinical trials. These modern approaches, including adaptive trials, platform trials, and other innovative methodologies, collectively drive a transformation in the way clinical research is conducted.
By reducing reliance on animal testing and introducing advanced methodologies that prioritise patient welfare and resource efficiency, these innovative trial designs accelerate decision-making and bring a more ethical, efficient, and patient-centric approach to the development of medical treatments.
This pioneering work showcases how the convergence of advanced technology, predictive modelling, and clinical research can reshape the field of clinical trials. While challenges exist, the benefits are clear – a more efficient, ethical, and patient-focused approach to bringing
CLINICAL TRIALS
innovative medical treatments to the world. The path to revolutionising clinical trials is now not only patientcentric but also highly technologically driven, offering a promising future for the healthcare industry.
Innovative Trial Designs and Patient Recruitment: Impact, Changes, Benefits, and Challenges
Innovative trial designs have introduced a paradigm shift in the way clinical trials are conducted, including their impact on patient recruitment. These novel approaches have transformed the traditional recruitment landscape, ushering in significant changes, benefits, and challenges.
Thus, this may not be entirely applicable for all countries. For example, in Bulgaria or eastern Europe in general, many strategies for online patient recruitment, digitalisation, or marketing cannot be implemented. This is simply because there is still a very big social stigma, born out of common misconceptions and distrust in the pharmaceutical industry and clinical trials.
Moreover, the lack of awareness and education about the benefits of participating in clinical trials further hinders the implementation of these strategies. Without proper understanding and knowledge, potential participants may hesitate or refuse to engage in these activities.
In addition, the limited access to technology and internet infrastructure in certain regions also poses a challenge for the successful execution of online strategies. Without reliable internet connectivity and access to digital platforms, reaching out to potential patients and conducting effective marketing campaigns becomes increasingly difficult.
Therefore, it is crucial for stakeholders in these regions to address these barriers and work towards creating a more supportive environment for online patient recruitment, digitalisation, and marketing efforts. This can be achieved
through targeted awareness campaigns, education initiatives, and building trust within the community. Doctors can serve as a bridge in this process, as patients trust and highly regard them as key opinion leaders. Therefore, the role of physicians in this process is crucial. By involving doctors, the potential for successful implementation of these strategies can be increased, resulting in improved clinical trial participation and overall improved quality of life via patient education.
Impact of Innovative Trials Designs on Patient Recruitment:
1. Greater Access and Inclusivity: Innovative trial designs have broadened access to clinical trials. Patients who were previously excluded due to geographic barriers, limited mobility, or other constraints now have the opportunity to participate.
2. Enhanced Patient-Centric Approach: These designs prioritise the patient experience, making trials more convenient and comfortable. As a result, patient recruitment and retention rates have improved.
3. Improved Diversity: Innovative trial designs often attract a more diverse participant pool, ensuring that research outcomes are more representative of real-world populations. This diversity enhances the generalisability of trial results.
4. Accelerated Recruitment: With more patient-friendly protocols and adaptive designs, recruitment timelines are shortened. Real-time data collection enables swift identification of eligible participants.
Changes in Patient Recruitment:
1. Digitalisation:
Innovative trial designs utilise digital platforms for recruitment, significantly transforming the recruitment landscape. Online platforms, social media, and
38 PHARMA FOCUS ASIA ISSUE 54 - 2024
mobile apps have emerged as crucial channels for patient engagement. However, these methods are only effective for specific target groups, and the elderly population often faces challenges in using them. Consequently, this limits the potential indications for the particular disease being studied in the clinical trial, as these approaches are primarily applicable to younger individuals.
2. Patient Empowerment:
Patients are now better informed and empowered to seek out clinical trial opportunities independently, reducing their reliance on healthcare providers.
Benefits of Innovative Trial Designs in
Patient Recruitment:
1. Efficient Recruitment:
Faster patient recruitment results in shorter trial timelines, reducing costs and enabling quicker access to potentially life-saving treatments.
2. Increased Engagement:
The patient-centric approach enhances engagement and willingness to participate, leading to higher retention rates.
3. Real-Time Data:
Real-time data collection and remote monitoring provide a wealth of information, helping identify eligible patients more quickly.
4. Greater Diversity:
A more diverse participant pool improves the applicability of trial results to a broader population.
Challenges in Patient Recruitment:
1. Digital Divide:
Not all patients have equal access to the digital platforms used for recruitment, leading to disparities in trial participation.
2. Data Security:
The digitalisation of patient recruitment requires robust data security and privacy measures.
CLINICAL TRIALS
ELEM
Biotech S.L.
is pushing the boundaries of innovation with a groundbreaking approach that introduces virtual humans into the world of clinical trials.
3. Regulatory Compliance:
Complying with evolving regulations, especially in the context of decentralised trials and remote recruitment, can be complex.
4. Ethical Considerations: Ensuring informed consent and ethical conduct in digital recruitment channels is essential.
Here are some good examples of changes that emerged in patient recruitment brought about by innovative trial designs and digitalisation:
1. Online Patient Portals: Many clinical trials now offer online patient portals that allow potential participants to easily access trial information, complete eligibility questionnaires, and express their interest in joining a trial. Patients can browse trial details, eligibility criteria, and frequently asked questions, all from the comfort of their homes.
The Role of the the “Find Me Cure” platform:
Find Me Cure is a Bulgarian startup, which created a platform dedicated to connecting patients with clinical trials. It serves as a bridge between patients and researchers, helping potential participants find suitable trials while assisting researchers in locating eligible candidates.
Find Me Cure plays a significant role in transforming the landscape of clinical trials by enhancing access to clinical trial information, improving patient engagement, streamlining the recruitment process for researchers, and contributing to diversifying the participant pool.
Find Me Cure is bridging the gap between potential participants and researchers. By providing easy access to clinical trial information, Find Me Cure empowers patients to proactively seek trials that match their specific conditions and preferences. This enhanced access enables patients to take a more proactive role in their healthcare journey, empowering them with the knowledge and resources to make informed decisions about their treatment options.
Moreover, Find Me Cure contributes to improved patient engagement in clinical trials. The platform helps patients navigate the often complex landscape of clinical trials, providing them with the necessary information and resources to understand the trial process and requirements. This increased engagement not only leads to higher participation rates but also ensures that patients are actively involved throughout the trial, ultimately improving the quality and reliability of the research outcomes.
In addition, Find Me Cure streamlines the recruitment process for researchers. By providing a centralised platform for researchers to identify and engage with potential participants, the platform simplifies the often time-consuming and resource-intensive process of finding eligible candidates for clinical trials. This efficiency in recruitment allows researchers to quickly and effectively identify suitable participants, accelerating the overall trial timeline and facilitating faster access to potentially life-saving treatments.
1. Social Media Campaigns:
Researchers and organisations use social media platforms like Facebook, Twitter, and Instagram to run targeted ad campaigns that reach a broader audi-
39 www.pharmafocusasia.com
CLINICAL TRIALS
ence. Patients can come across trial opportunities while scrolling through their feeds, and these campaigns can provide direct links to trial websites or registration pages.
2. Mobile Apps:
Clinical trial sponsors have developed mobile apps that enable patients to search for trials, receive notifications about new studies, and complete prescreening questionnaires. These apps facilitate seamless interaction between patients and clinical trial teams.
3. Email and SMS Alerts:
Patients can subscribe to receive email or SMS alerts about clinical trial opportunities. These alerts notify them of new trials that match their profiles and preferences, reducing the need to actively seek out trials.
4. Virtual Recruitment Events:
Virtual events, including webinars and information sessions, have become popular for recruiting patients. These events allow potential participants to learn about the trial, ask questions, and express their interest, all without the need to attend in-person meetings.
5. Online Patient Communities:
Online communities and forums dedicated to specific medical conditions or diseases have emerged as valuable recruitment tools. Researchers can engage with these communities to inform patients about relevant trials and provide study details.
6. Remote Screening and Telemedicine: Digital recruitment goes beyond just finding trials. Remote screening and telemedicine enable initial assessments, eligibility checks, and consultations to occur online, minimising the need for patients to travel to physical sites for these processes.
7. Web-Based Pre-Screening Tools:
Many trials offer web-based pre-screening tools that allow patients to quickly
check their eligibility by answering a series of questions. If they meet the criteria, they can express their interest in participating.
8. Patient Advocacy Groups:
Advocacy groups often collaborate with researchers to promote trial opportunities within their networks. This approach is especially effective in recruiting patients who are already engaged with the advocacy group's activities.
9. Patient Registries:
Many diseases and conditions have patient registries that collect and store information about individuals interested in participating in research. These registries are valuable resources for identifying potential trial participants.
These changes in patient recruitment reflect the shift toward a more patientcentric and digitalised approach. They make it easier for patients to access trial information, express interest, and actively engage with the clinical trial process. However, they also bring challenges related to data privacy, digital literacy, and the need for clear and ethical communication throughout the recruitment process.
Innovative trial designs have transformed patient recruitment by increasing inclusivity, enhancing patient-centric approaches, and accelerating recruitment timelines. However, digitalisation, data security, regulatory compliance, and ethical considerations pose challenges. Platforms like Find Me Cure play a crucial role in making clinical trials more
Tihomira Nikolova is an experienced Clinical Research Associate at PSI CRO AG, with over 7 years of experience in the clinical trials industry. She has a background in pharmacy from Sofia Medical University and a B.A. in International Healthcare Management from the IU of Applied Science. Tihomira is also a prolific writer on various clinical trial topics, creator of the community Clinical Research Bulgaria, and the founder of "The CRA Wizard," a Clinical Research Associates academy.
accessible and efficient, ultimately benefiting both patients and researchers.
Conclusion
In light of the numerous benefits and despite the inherent challenges and areas that require improvement, I is my belief that innovation is not only welcome, but a crucial necessity for the advancement of medicine. The landscape of clinical trials has evolved significantly, thanks to innovative trial designs, which offer the potential for accelerated drug development, enhanced patient-centricity, and improved data quality.
While it's true that these innovations come with their set of issues, it's essential to emphasise that the pharmaceutical and clinical trial industry is deeply committed to safety and rigour. Stringent testing, adherence to multiple guidelines, and rigorous oversight are intrinsic to this field. Mistakes are highly consequential, and as a result, ensuring the safety of participants is paramount. It's a testament to the industry's dedication to patient welfare that, despite the complexity of clinical trials, adverse events are exceedingly rare. These innovations not only drive the medical field forward but also align with the broader goals of improving patient care and advancing healthcare outcomes.
References are available at www.pharmafocusasia.com

40 PHARMA FOCUS ASIA ISSUE 54 - 2024
AUTHOR BIO
Ensuring Safety in Geriatric Medicine Use Explicit criteria to avoid potentially inappropriate medication
The ageing population is at risk of developing multiple chronic diseases leading to polypharmacy. Preventing harm from medications used in the elderly can be promoted by using explicit PIM criteria. Beer’s criteria, STOPP/ START, and FORTA list were popular explicit criteria that were adopted into various criteria in Asian countries.
Laksmi Maharani, Lecturer and Researcher, Jenderal Soedirman University

Higher life expectancy among Asian people leads to a high number of elderly aged 60 and over nowadays. This phenomenon is called the "ageing population,” where one of the negative impacts is higher costs in healthcare services. In 2002, the United Nations (UN) stated the "Madrid International Plan of Action on Ageing," which includes: i) reducing the cumulative effects of factors that increase the risk of disease and consequently potential dependence in older age; ii) developing policies to prevent ill health among older persons; and iii) to provide access to
food and adequate nutrition for all older persons. The pan has been set for over 20 years, and countries have put various efforts in place to avoid healthcare risks for older people.
An epidemiological transition in all countries indicated a shift in the predominance of infectious diseases to chronic and degenerative diseases. The ageing population was at risk of developing chronic diseases such as diabetes, hypertension, obesity, and various mental health issues such as depression and dementia. Elderly or geriatric usually require age-related procedures and treatments that increase
the cost of long-term care. This problem highlights the need for healthcare and education reforms to promote healthy ageing.
Rational Use of Medicine
In promoting healthy ageing, the rational use of medicine is crucial. Rational use is defined as correct/ proper/ appropriate use of medications that refers to suitable selection, dose, duration, cost, and patients, and this medicine should be dispensed correctly and appropriately. Medicine against this principle will be considered irrational, incorrect, improper, or inappropriate.
Patient safety in the treatment process is one of the critical elements of qualified healthcare systems worldwide. Elderly patients undergo a series of pharmacological changes. The ability to maintain normal drug absorption was reduced in the elderly because of a rise in gastric pH, reduction in gastrointestinal motility and splanchnic blood flow. Drug distribution will also shift because of reduced lean body mass and increased body fat, which increase the distribution and prolong the half-life elimination of fat-soluble drugs. Metabolite clearance of hepatic and renal eliminated drugs usually decreases with age, slowing drug clearance and increasing some drugs' toxicity. In addition, the elderly also undergo pharmacodynamic changes because of the presence of medical illness and an increase in age. Many drugs are contraindicated for the elderly and require much caution.
41 www.pharmafocusasia.com
MANUFACTURING
Advanced age, along with multiple morbidity and polypharmacy, were predisposing factors for the occurrence of adverse effects. Multimorbidity in the elderly can impair drug handling and administration in chronic conditions. Multiple chronic conditions burdened in people with multimorbidity increase the risk of functional impairment, deterioration in quality of life, and increased mortality. Multimorbidity also increases the risk of polypharmacy caused by the coexistence of many diseases which require pharmacological treatments.
Potentially Inappropriate Medication (PIM) and Explicit PIM Criteria
Inappropriate use of medications, especially in the elderly population, is well known by the term PIMs (Potentially Inappropriate Medications). The risk of PIM use increased as the number of drugs consumed increased. The presence of mental/behavioural disorders influences the increased risk of developing PIM besides polypharmacy.
Many policies were established to prevent the rational use of medicine; some policies that were essentially adopted by developed countries were policies to reduce costs and develop national guidelines. Standard treatment guidelines should be evidence-based and unbiased. Guidelines developed to avoid potentially inappropriate medications are introduced as explicit or PIM criteria in geriatric medicine. These criteria focus on PIMs among older people and are usually developed based on evidence from clinical trials, experts’ opinions, and Delphi methods. Explicit criteria differed from implicit PIM criteria, focusing primarily on the patient rather than drugs or
disease. Implicit PIM criteria were timeconsuming and depended on the prescriber's knowledge and expertise. In contrast, explicit PIM criteria clearly defines which drugs cause PIMs in particular clinical circumstances.
Three popular explicit PIM criteria widely used worldwide are: Beers Criteria, STOPP/START Criteria, and FORTA list. Beer’s criteria were first published in 1991 and have undergone several updates. In 2011, the American Geriatric Society (AGS) took responsibility for updating and maintaining Beers Criteria. The recent update of Beers Criteria is the 2023 version. These criteria explain PIM into five categories: drugs to avoid in older adults, drugs to avoid in certain diseases or syndromes in older adults, drugs should be used with caution, drugs should be avoided or adjusted in older adults with renal disease, and critical drug-drug interactions to avoid in older adults.
The Screening Tool for Older Persons Potentially Inappropriate Prescriptions (STOPP) and the Screening Tool to Alert Doctors to the Right Treatment (START) were developed in 2008. The second version appeared in 2014, and the recent version was published in 2023. The STOPP criteria were organised according to the physiological system, and the START was designed to screen PIMs under-prescribing. In comparison, the FORTA (Fit for the Aged) list was introduced in 2008 and updated in 2015. This list is widely used across European countries based on data gathered from six regions. This list includes 264 medications/classes according to diagnosis or clinical syndrome. FORTA list graded the drugs into four levels of expected clinical benefit for geriatric populations.
Polypharmacy
Development of Explicit PIM Criteria in Asia Countries
Most explicit PIM criteria were based on medications marketed in Europe or the United States, making the applicability of those criteria in Asian countries relatively low. Asian countries have regional variations in drug formularies and health systems that differ from Western countries. This limitation led to the development of various similar criteria across Asian countries, listed below:
1) Lists of Risk Drugs for Thai Elderly (LRDTE): developed from 2012 Beers Criteria and 2008 STOPP in Thailand. This list considered the age group in the elderly population and the severity of medication risk. This list also covered standard treatment guidelines and hospital formulary for Thai elderly.
2) Guidelines for the Safety of Pharmacotherapy in the Elderly (GL2015) and STOPP-Japan: GL2015 were developed in 2015 while STOPP-Japan in 2016, both directed to the elderly aged ≥75 years. The GL2015 contains a list of medications that require prudent administration and a list of medications that should be considered for administration. At the same time, STOPP-Japan uses the terms "drugs to be prescribed with caution" and "drug to consider starting" to explain PIM.
3) Japan FORTA: developed in 2020 using EURO-FORTA, OAC-FORTA (Oral Anticoagulants for Long-Term Treatment of Atrial Fibrillation in Older Patients), and LUTS-FORTA (Lower Urinary Tract Symptoms-FORTA) list followed by two Delphi rounds by 13 experts in geriatric pharmacotherapy. This list consists of 210 PIMs across 24 indications.
4) The Chinese Criteria: Criteria of potentially inappropriate medications for older adults in China published in 2017. This criterion includes medication risk and medication risk under a morbid state. According to the expert panel evaluation, the Chinese criteria were divided into high-risk and low-risk medications and categorised according to defined daily doses.
42 PHARMA FOCUS ASIA ISSUE 54 - 2024
Multimorbidity Potentially Inappropriate Medication (PIM) Eldery (60 years and over)
Geriatric Medicine
MANUFACTURING
Figure
1: Ensuring Safety in
Use
MANUFACTURING
Clinical decision by healthcare professional
The use of explicit PIM criteria
Interviews with Patient/Caregiver
Medication Reconcilliation
Patients' Medication History
Rational use of medicine in eldery
Medicine Use
5) PIM-China: These criteria was developed in 2018 and categorises PIMs into general PIM and disease-specific PIMs.
6) PIM-Taiwan: developed in 2019 using pre-existing standards and categorises the PIM into General PIMs and Disease-specific PIMs.
7) STOPP-START Singapore NUH In-house Guidelines: developed in 2015 based on STOPP/START Criteria. This guideline includes 89 criteria consisting of 55 STOPP and 34 START criteria.
8) STOPP-Indonesia: This tool was developed in 2020 using STOPP/START as standard. STOPP-Indonesia consists of 81 PIM Criteria grouped into 13 sections based on organs/systems.
9) PIM-Korea: There are four versions of PIM-Korea: 2010, 2015, 2018, and 2022. Different researchers developed all of them. The 2022 version includes PIMs in general, potential drug interactions, disease-specific PIMs, dose adjustment and potential omissions.
10) PIM-Pakistan: developed in 2018 using Beers Criteria 2015 and STOPP/ START Criteria version 2. This list contains 32 PIMs categorised pharmacologically.
11) STOPP/START Sri Lanka: Sri Lanka's version of STOPP/START Criteria was developed in 2019 and includes 105 criteria.
12) PIM-Hong Kong: developed in 2021 using the PRISCUS list, STOPP
version 2, and Beers 2015 Criteria. This list categorised PIMs into general and disease-specific PIMs.
The Benefit of Explicit Criteria
Explicit PIM criteria simplify the medication optimisation process by alerting prescribers of potential irrational medicine use in specific circumstances. Some criteria have an RCT (Randomised Controlled Trial)-proven clinical benefit that increases patient-related outcomes. Intervention using explicit PIM criteria is proven to reduce adverse drug reactions, incidents of falls, medication costs, and PIMs. These criteria should be applied in routine medication review and prescribing practice for multimorbid older people, especially those who are exposed to polypharmacy.
Previous research involved various healthcare professionals in implementing explicit PIM criteria. Physicians, hospital or clinical pharmacists, geriatricians, nurses, physical therapists, psychologist dietitians, occupational therapists, and speech therapists participated in research about explicit PIM criteria implementation. Hospitals or clinical pharmacists conduct most studies. The application of explicit PIM criteria ranged from outpatients, hospitalised patients, and at-patient discharge.
In order to avoid PIM using explicit PIM criteria, healthcare professionals need some reliable source of patient information that can support their clinical deci-
sions. Besides medication history, healthcare professionals may need medication reconciliation, interviews with the patient or caregiver, and contact with pharmacists/ physicians. Reduction of PIM after intervention using explicit PIM criteria was found most successful through interprofessional collaboration by pharmacists and geriatricians. PIM use in older adults requiring long-term care should be reviewed from a multidimensional perspective.
Conclusion
Healthy ageing in the ageing population nowadays could be achieved by ensuring drug safety use in the elderly. Some components that affect the safety of drugs in geriatric patients are changes in the pharmacological and pharmacodynamics of drugs in the elderly. These changes lead to the occurrence of some potentially inappropriate medications if patients also consume polypharmacy due to multimorbidity. Rational use of medicine can be achieved by applying explicit PIM criteria in the prescribing process. Various explicit criteria were developed globally, and some Asian countries adopted some. This criterion has reduced PIMs in outpatients, hospitalised, and discharged elderly. Healthcare professionals must implement explicit PIM criteria in treating geriatric patients to avoid PIMs.

Laksmi Maharani is a lecturer and researcher at the Pharmacy Department, Jenderal Soedirman University, Indonesia since 2012. She received her MSc in Clinical Pharmacy at Gadjah Mada University, Indonesia, and currently is undergoing her doctoral program at the same university. Her current research focuses on drug safety and geriatric medicine.
43 www.pharmafocusasia.com
AUTHOR BIO
Figure 2: Ensuring Safety in Geriatric
Nanoparticle Biomimetics as an Avenue to Personalised and Precision Medicine
The rush towards a potential breakthrough
The past two decades have witnessed mind blowing progress in concepts, strategies as well as technologies that are about to revolutionise the nanomedicine landscape. Since the 80s-90s, nanocarriers have been continuously evolving to face the hurdles of delivering drugs more and more precisely to a specific target. Such advances have promoted the rise of innovative therapies for a broad range of diseases. For instance, cancer treatments have benefited from the enhanced ability to localise drug action to a malignant cell neighbouring space leaving the healthy ones nearly untouched. An increased understand -
Biomimetic nanoparticles are emerging as next generation nanotherapeutics deemed to overcome the known limitations of previous generations. Their biological origin and customisable nature can promote the process of therapy personalisation and enable new precise delivery approaches. Albeit affected by yet unmet gaps, biomimetic nanoparticles possess all requisites for a near to come successful clinical application..
Stefano Giovagnoli, Dept. of Pharmaceutical Sciences, University of Perugia, Perugia
ing of the multiple barriers and the fate of nanoparticles in the organism fostered new approaches in nanoparticle manufacturing technology leading to new generations of carrier engineering strategies. Today, at least two generations of nanoparticles are being recognised. The first are biologically inert particles able to ensure drug accumulation based on an extended physiological stability. Long-circulating liposomes that have also been the first and one of the few nanocarrier products to reach the market, belong to this class. The second-generation nanoparticles are instead designed to target specific organs, tissues, or cells through moieties, such as lipoproteins or antibodies, that confer high selectivity.
Though engineered to meet specific requirements, nanoparticles can be brutally transformed by surface interactions in the physiological environment. This is a well-recognised yet not fully understood phenomenon named
protein corona effect, by which proteins present in the biological fluids bind the nanoparticle surface mutating the nanoparticle native identity into a rather inconstant biological one. This poses numerous challenges, as such a phenomenon, although partially preventable, is unpredictable, being prone to the intrinsic variability of biological systems. Age, sex, health conditions, lifestyle, environment, food, and inner physiology all together contribute to a change in the protein corona properties and the biological identity of the nanoparticle therein. Needless to say, this phenomenon hampers nanoparticle targeting capability and physiological stability.
Nevertheless, the whole scenario seems to be changing since the biomimetic nanoparticles’ first appearance in early 2010s. These conceptually new nano-systems can be considered third-generation nanoparticles (Figure 1). Their main feature is their biological origin, as they are generated from cell
44 PHARMA FOCUS ASIA ISSUE 54 - 2024
MANUFACTURING
membranes or assembled using hybrid biomaterials. These nanoparticles are engineered to recapitulate physiological and target features. This approach is thought to underpin the evasion of the organism engagement rules to ensure higher targeting efficacy and consistency. Albeit in their infancy, these systems may open new and unexplored possibilities in the years ahead.
Blowing away the traditional concept of drug targeting
Hitting a therapeutic target is a rather complicated task, which lays on the intrinsic anatomical and physiological complexity of the organism. Drug targeting is accomplished by ensuring progressive accumulation of the carrier to the site of action through a mix of physical and chemical processes. Stability, diffusivity, and affinity must team up to allow the carrier to successfully engage the target over a required time frame. These three dimensions of drug targeting can exploit passive or active mechanisms by which the nanoparticle can either permeate physiological barriers or recognise specific target features that enhance selectivity and specificity of treatment. To this picture, nanoparticle biomimetics can be accounted as a fourth dimension of drug targeting. In fact, biomimetic nanoparticles can evade the known constraints by paving new avenues. The possibility to adapt and indulge environmental and
target features represents a pivotal crossroad that may foster new and modern patient-centred therapeutic approaches. After the advent of RNA nanotherapeutics, which could not have been possible without the development of suitable nanocarriers, biomimetic nanoparticles may represent the next breakthrough as they may allow to circumvent physiological barriers and exploit processes and mechanisms previously inaccessible. Indeed, biomimetic nanoparticles may promote therapeutic effects by triggering biological cascade processes. Drug-free nanocarriers enabling the targeting of so far undruggable pathways may be envisaged in the near future. Research is rapidly moving forward in this field, nourishing the slow but relentless evolution of nanoparticles from simple carriers into precise functional therapeutic systems.
From nanocarriers to personalised nanotherapeutics
Nanoparticle technology evolution is benefiting from the increased knowledge and understanding of the complexity of biological systems. An example in this sense is the fast-growing research around extracellular vesicles as potential carriers and therapeutic systems that may indeed represent a quantum leap in the progress of medical science. In a way, biomimetic nanoparticles are close relatives to extracellular vesicles as they can share similar basic features and origin as well
as an intrinsic versatile and tailorable nature. Such a potentially customisable nature of biomimetic nanoparticles makes them an attractive platform for a broad range of applications. They can be conceived as entities recapitulating all the wanted biological features through specific engineering and assembly of biological materials. Customisation may depend upon specific therapeutic requirements. Similarly to other scientific areas, technologies already exist that can be employed to engineer cells to express specific factors and membrane composition that, through proper manipulation, can be translated to nanoparticles that then will replicate the same features. Therefore, nanoparticle functional personalisation based on disease and patient conditions can be conceived by exploiting the patient’s autologous cells as a source. Thus, next generation nanoparticles are on their way to become novel customisable and precise therapeutic systems.
Implications for the patient and the Pharma industry
Recent research trends and clinical evidence suggest that therapy personalisation is a key process that may contribute to a remarkable improvement of therapeutic outcomes. It is today clear that providing the right treatment to the right patient at the right time is an avenue to clinical success. Nevertheless, personalisation requires proper tools for
45 www.pharmafocusasia.com
MANUFACTURING
Figure 1. Schematic representation of nanoparticle generations. Generation I are biologically inert particles with an extended physiological stability. Generation II are nanoparticles designed with selective moieties able to recognise selectively specific organs, tissues, or cells. Generation III are biomimetic nanoparticles that can be assembled by employing whole cell membranes or specific components.
optimal choice of interventions. Several approaches are currently adopted for treatment suitability testing, and some of them are already clinically employed to address issues in the personalisation of interventions. Other emerging strategies rely on a patient’s genetic profiling, health monitoring, or, where feasible, the generation of a patient’s cellular avatars. The latter is a strategy that consists in harvesting the patient’s own cells and, using stem cell technology, producing other cell types. As an example, the organoid technology is already employed in oncology and cystic fibrosis for clinical drug screening to address suitability of treatment. However, such a huge personalisation effort would be in vane if not supported by the development of proper delivery approaches. Biomimetic nanoparticles can be pivotal in such a process allowing to model the delivery strategy upon individual peculiarities.
Beyond the obvious impact of therapy personalisation on the patient’s qualityof-life, especially in chronic conditions, the change is expected to also affect the Pharma industry whose market strategies and investments require rethinking. In part, this is an already ongoing process, as companies started to scale down their production system and develop proper control strategies and enabling technologies. Personalised medicine has already ended the pharma blockbuster era by parting the drug market into smaller defined niches. A big role in this regard is being played by the orphan drug market, which has become a driving force in the development of personalised medicine due to intrinsic unmet needs that demand precise and specific therapeutic approaches and the dedicated and expedited market authorisation procedures promoted by regulatory agencies.
The digital-AI wave, clinical and regulatory cliffs
Huge change lies ahead for the industry driven by a digitalisation and the inroads made by artificial intelligence (AI) into R&D and pharmaceutical production.
Digitalisation is rapidly gaining ground for several pharmaceutical products and medical devices. In particular, orally inhaled and nasal drug products start taking advantage of advanced electronic devices that facilitate monitoring and control over patient’s self-medication. In general, digitalisation underpins improved performances and patient-centred therapy assessment. In addition, AI has already permeated large areas of fundamental and applied research and is dramatically imposing into clinical research as well. The power of AI algorithms is enabling progress towards an insightful understanding of complex systems, such as living organisms. Today, AI is already employed for patients’ health monitoring and profiling to lay the foundations for therapy personalisation. Indeed, the digitalisation and AI waves can promote the transition to personalisation of therapies and products by enabling the assessment of the best conditions for the precise and customised delivery of treatment. Approaches such as machine learning and deep learning are already employed in R&D to help the development of optimised therapeutic systems.
The above discussed biomimetic nanoparticle peculiarities can affect this transition process. Unfortunately, a setback to this fast-moving scenario is that regulatory bodies are playing catch-up with scientific progress. Such a situation is producing an understandable yet arguable bottleneck to the translation of novel therapeutic strategies to the clinic. When dealing with systems of biological origin such a gap broadens, owing to the lack of information on their fate and toxicity. Aspects that restrain the rush to the clinic deal with instability concerns in the first place, as nanoparticle size, composition, and physical–chemical properties change significantly according to source and preparation methods. Moreover, largescale production is today still far from being achieved and significant endeavours are required to address purity, component as well as drugs loading, and process yield issues. Linked to the
above considerations, safety of biomimetic nanoparticles remains unclear in humans and additional efforts are demanded to assess their biochemical properties, pharmacodynamics, and pharmacokinetics.
Remarks
Undoubtedly, nanoparticle biomimetics can unleash the huge potential of nanotherapeutics, albeit their journey as enabling tools to precision and personalised medicine has just begun. Indeed, biomimetic nanoparticles have the power to contribute to the transformation involving R&D and production environments even thanks to digitalisation and AI support. Unexplored scenarios are opening ahead with likely unprecedented clinical impact. However, barriers exist that can considerably slow down this yet irreversible process. Although biomimetic nanoparticles are still behind the establishment of proper clinical settings, the progress made in the development and approval of new cell and gene therapies indicate that reaching the clinic for biomimetic nanoparticles is only a matter of time.
References are available at www.pharmafocusasia.com

Stefano Giovagnoli is Associate Professor in Biotechnology and Drug Delivery at the University of Perugia, Perugia, Italy. Personal research interests encompass bioinspired drug delivery approaches through micro- and nanoparticles with a special focus on pulmonary drug delivery and host-directed therapies against infectious diseases.
46 PHARMA FOCUS ASIA ISSUE 54 - 2024
AUTHOR BIO
MANUFACTURING
Enhancing Pharmacovigilance through the Scope of Artificial Intelligence
Pharmacovigilance, the science and activities related to the detection, assessment, understanding, and prevention of adverse effects or any other drug-related problems, plays a crucial role in ensuring the safety of pharmaceutical products. In recent years, the pharmaceutical industry has witnessed a surge in the volume and complexity of available health data, necessitating innovative approaches to optimise pharmacovigilance processes. This abstract explores the integration of Artificial Intelligence (AI) in pharmacovigilance as a transformative paradigm.
Aditya Dilipkumar Patil, Assistant Professor, Department of Homoeopathic Pharmacy, Noble Homoeopathic Medical College and Research Institute, Noble University
Sargam Ramesh Singh, Assistant Professor, Department of Gynaecology & Obstetrics, Noble Homoeopathic Medical College and Research Institute, Noble University
AI, with its advanced analytical capabilities, has shown promising potential in enhancing pharmacovigilance throughout the drug life cycle. This abstract delves into the various applications of AI, including machine learning algorithms, natural language processing, and data mining techniques, in automating the identification and evaluation of adverse drug reactions. By leveraging largescale healthcare data, AI can facilitate real-time monitoring of drug safety, early detection of potential risks, and efficient signal detection.
Furthermore, the abstract highlights the role of AI in improving the

INFORMATION TECHNOLOGY
efficiency of reporting systems and the analysis of unstructured data sources, such as social media and electronic health records. The integration of AI-driven tools in pharmacovigilance not only expedites the identification of safety signals but also enables a more proactive and personalised approach to drug safety monitoring.
Challenges and ethical considerations surrounding the implementation of AI in pharmacovigilance are also discussed, emphasising the importance of transparent algorithms, data privacy, and collaboration among stakeholders. Additionally, the abstract explores the potential benefits of AI in predicting patient-specific responses to medications, paving the way for personalised medicine and tailored therapeutic interventions.
In conclusion, this abstract provides an overview of the transformative impact of Artificial Intelligence on pharmacovigilance, presenting a compelling case for the adoption of AI-driven solutions to enhance the safety and efficacy of pharmaceutical products in an everevolving healthcare landscape.
In the dynamic landscape of pharmaceuticals, ensuring the safety and efficacy of drugs is of paramount importance. Pharmacovigilance, the science and activities related to the detection, assessment, understanding, and prevention of adverse effects or any other drugrelated problems, plays a crucial role in maintaining public health. With the advent of cutting-edge technologies, the integration of Artificial Intelligence (AI) has opened new frontiers in pharmacovigilance, revolutionising the way adverse events are detected, assessed, and managed.
The Role of AI in Pharmacovigilance
Artificial Intelligence, encompassing machine learning and natural language processing, is proving to be a gamechanger in pharmacovigilance. The technology brings about efficiency,
Delve into the promising future where intelligent systems contribute to more efficient and robust pharmacovigilance practices, ensuring patient safety remains at the forefront of drug development and healthcare initiatives.
accuracy, and agility in the monitoring of drug safety, offering a proactive approach to identifying potential risks.
1. Early Detection of Adverse Events: AI algorithms can analyse vast amounts of healthcare data in real-time, identifying patterns and trends that might go unnoticed through traditional methods. This enables early detection of adverse events, allowing pharmaceutical companies and regulatory authorities to respond swiftly and effectively.
2. Automated Signal Detection: Traditional pharmacovigilance methods often involve manual review of individual case reports. AI automates this process, sifting through massive datasets to identify potential signals and patterns indicative of adverse reactions. This automation not only accelerates the detection process but also reduces the likelihood of oversight.
3. Data Mining and Surveillance: AI excels in data mining and surveillance, actively monitoring electronic health records, social media, and other sources of health-related information. By analysing this diverse data, AI systems can provide a more comprehensive understanding of drug safety profiles and potential risks.
4. Enhanced Risk Prediction: Through predictive analytics, AI can assess the likelihood of adverse events for specific patient populations. This enables healthcare professionals and regulatory bodies to implement targeted interventions and personalised risk mitigation strategies.
5. Improved Case Triage and Workflow Optimisation: AI-driven automation streamlines case triage by categorising and prioritising reports based on severity and relevance. This not only accelerates the review process but also allows pharmacovigilance teams to focus their efforts on the most critical cases.
Challenges and Considerations
While the integration of AI in pharmacovigilance holds tremendous promise, it is not without challenges. Ensuring the quality and reliability of AI algorithms, addressing ethical considerations, and establishing clear regulatory frameworks are crucial aspects that need careful attention.
1. Algorithm Transparency and Interpretability: Understanding and interpreting the decisions made by AI algorithms is essential for gaining trust and acceptance. Ensuring transparency in algorithmic processes and outcomes is crucial for pharmacovigilance professionals, healthcare practitioners, and regulatory authorities.
2. Data Quality and Standardisation: The effectiveness of AI in pharmacovigilance is heavily reliant on the quality and standardisation of the data it processes. Establishing robust data governance practices and ensuring interoperability between different systems are vital for the success of AI applications in this field.
3. Ethical Considerations: The use of AI in pharmacovigilance raises ethical concerns related to patient privacy, consent, and the responsible use of technology. Striking a balance between harnessing the benefits of AI and safeguarding patient rights is essential for the ethical deployment of these technologies.
48 PHARMA FOCUS ASIA ISSUE 54 - 2024
INFORMATION TECHNOLOGY
4. Data Quality and Bias:
• Incomplete or Biased Data: If the training data used to develop AI models is incomplete or biased, it can lead to inaccurate predictions or overlook certain patterns.
• Data Imbalances: AI models may struggle if there are imbalances in the data, such as a disproportionate number of reports for certain drugs or adverse events.
5. Lack of Understanding:
1. Black Box Nature: Deep learning models, in particular, are often considered as "black boxes" because their decision-making processes are not easily interpretable. This lack of transparency can make it difficult to understand how and why a specific decision was made.
6. Regulatory Compliance:
1. Regulatory Challenges: Meeting regulatory requirements can be challenging as the use of AI in pharmacovigilance must adhere to strict guidelines. Ensuring that AI models comply with regulations and are accepted by regulatory bodies is an ongoing concern.
7. Integration with Existing Systems:
1. Integration Challenges: Integrating AI systems with existing pharmacovigilance processes and databases can be complex and time-consuming. Ensuring seamless collaboration between AI and human analysts is crucial for effective pharmacovigilance.
8. Ethical Concerns:
1. Patient Privacy: Handling sensitive patient data raises privacy concerns. Proper measures must be in place to protect patient confidentiality and adhere to data protection regulations.
2. Ethical Use: There are ethical considerations surrounding the use of AI in healthcare. Ensuring that AI is used responsibly and ethically in pharmacovigilance is essential.
9. Technical Limitations:
1. Limited Generalisation: AI models trained on specific datasets may struggle to generalise well to new or unforeseen situations. This limitation can impact the ability to detect rare or emerging adverse events.
2. Dependency on Data Quality: The effectiveness of AI models heavily relies on the quality and relevance of the training data. Inaccurate or outdated data can lead to suboptimal performance.
10. Human-AI Collaboration:
1. Trust Issues: Building trust between human analysts and AI systems is crucial. Over-reliance on AI or distrust in its capabilities may hinder collaboration and the overall effectiveness of the pharmacovigilance program.
11. Costs and Resources:
1. High Initial Costs: Implementing AI systems can involve significant upfront costs for development, training, and infrastructure. Organisations may need to carefully weigh these costs against the potential benefits.
Despite these drawbacks, ongoing research, advancements in AI technology, and thoughtful implementation strategies can help address many of these challenges, allowing AI to enhance pharmacovigilance efforts effectively.
Conclusion:
The integration of AI into pharmacovigilance represents a significant leap forward in ensuring drug safety and enhancing public health outcomes. By leveraging the capabilities of AI, pharmaceutical companies and regulatory authorities can proactively identify, assess, and manage adverse events, ultimately contributing to a safer and more efficient healthcare ecosystem. As technology continues to advance, the synergy between pharmacovigilance and AI is poised to redefine the landscape of drug safety, ushering in a new era of proactive risk management and improved patient care.
Reference are available at www.pharmafocusasia.com
AUTHOR BIO

Aditya Dilipkumar Patil
Author received his Bachelor of Homoeopathic Medicine and Surgery and Doctor of Medicine in Homoeopathic Pharmacy from Bharati Vidyapeeth University, Pune, Maharashtra, India. He has received Bamra Arogya Trust (Lippe Shield Award) and is also a Gold Medal rank holder with more than 20 research paper published and indexed in Scopus, PubMed, Web of Science, Google Scholar and UGC Journals. He was working as Senior Research Fellow in Central Council for Research in Homoeopathy (CCRH) under Ministry of AYUSH, New Delhi, India. He has also worked as AYUSH Medical Officer in National Rural Health Mission (NRHM), Maharashtra, Kolhapur Division in pandemic of Covid-19.

Sargam Ramesh Singh
Author received her Bachelor of Homoeopathic Medicine and Surgery from Shree Kamaxidevi Homoeopathic Medical College and research Institute, Shiroda, Goa. She received her Doctor of Medicine in Organon of medicine and philosohy from National Institute of Homoeopathy (NIH), Kolkata, West Bengal, India. Currently she is working as National Accredited Board of Hospital consultant for Noble Homoeopathic College and Research Institute, Junagadh, Gujarat, India.
49 www.pharmafocusasia.com
INFORMATION TECHNOLOGY
 ATUL DUBEY Director, Pharmaceutical Continuous Manufacturing (PCM) at the United States Pharmacopeial Convention (USP)
ATUL DUBEY Director, Pharmaceutical Continuous Manufacturing (PCM) at the United States Pharmacopeial Convention (USP)
Advanced Manufacturing
An answer to supply chain woes?
Advanced Manufacturing technologies (AMTs) in the pharmaceutical industry have increasingly gained attention in the last decade. Amongst the AMTs, continuous manufacturing (CM) has had the most prominent impact so far. International efforts to define manufacturing approaches, terms, regulatory expectations in a harmonised manner have been made. In many cases, manufacturers have benefited due to early adoption of CM. As the interest grows in the areas of API and drug product as well as biologics, it is important to understand the barriers and challenges for a broader worldwide adoption. USP has been working in different ways to lower some of these barriers. These include facilitating open dialog, supporting and advancing research, advocacy at various forums and contributing to the development of standards and guidelines.
How is automation impacting advanced manufacturing, and what benefits does it bring to businesses?
The general trend in manufacturing has been to eliminate intermediate stops between processing steps as much as possible to reduce the overall processing time. To accompany these advanced processing techniques, new process analytical technology (PAT) tools have been developed to allow quick sampling, automated processing/recording of data into computers that can talk to control systems. This has dual benefits — it reduces the chances of human error, and helps economies where human capital is scarce. Furthermore, faster automated analysis, even when not as deep as a manual, offline one, can preemptively alert the operator if the process is veering towards the outer limits of safe performance.
50 PHARMA FOCUS ASIA ISSUE 54 - 2024
EXPERT TALK
How can contract manufacturers maintain flexibility to adapt to changing requirements from their clients?
One approach is to modularise the manufacturing set-up as much as possible. This can involve individual unit operations, such as, reactive crystallisation mounted on skids that can be rolled in and out of the process train. New equipment is being designed with interoperability and flexibility in mind. Furthermore, the manufacturers can plan ahead and schedule campaigns for products requiring similar unit operations together. This can reduce time and effort required in switchover and intermediate cleaning.
On the personnel front, the big challenge for contract manufacturers is to keep abreast of the latest technologies, methodologies and procure equipment that can maximise flexibility to meet diverse customer requirements. USP has created the USP CM Knowledge Center as a free resource to share open-source knowledge and get the community to engage.
One strategy that could help would be to ensure that their staff is in a continuous learning mode, and that they are able to devote some time to self-development as opposed to being fully absorbed into customer projects. They can aim to find common elements in their projects and use time and material saving techniques such as validated models. Learnings from one customer project can be applied to speed up another.
Can you discuss the role of real-time release testing (RTRT) in advanced manufacturing, and how it is changing traditional testing approaches?
RTRT refers to use of (a combination of)
Advanced manufacturing technologies associated with modalities like continuous manufacturing and 3D printing are evolving quickly.
measurements and calculations based on science- based validated models and PAT tools to determine the product quality. This determination can be an alternative to traditional release testing. RTR can be used for both batch and continuous manufacturing processes. In a fast-moving continuous manufacturing scenario, especially in a small footprint-high usage facility, RTR can help manufacturers maintain the production schedule flexibility. Being able to finish the manufacturing sooner can help improve the plant efficiency, workforce schedules, reduce laboratory waste, and enhance regulatory compliance.
However, achieving RTR requires additional experimentation which can lead to a significant added cost. It is not necessary to file for RTR along with a CM application. There are examples of manufacturers filing for it a few years later, after receiving the regulatory approval for the main CM process.
What are the current trends in advanced manufacturing, and how are they shaping the industry?
The industry is responding to a deepening supply chain crisis. Many supply
routes are now disrupted or are in danger of being so. With the uncertainty in demand and availability of key raw materials, the industry is looking to boost efficiency in operations.
Advanced manufacturing is an umbrella term that includes other modalities as well, in addition to CM. For example, a process can be an advanced-batch process, or a semicontinuous process with additive manufacturing (3D printing) as the final step. While this is not an exhaustive list, here are some trends we are observing:
• Finding new synthesis routes and techniques such as flow chemistry for small molecule APIs to build efficiency as well as flexibility to use raw materials that are more readily available
• New techniques in drug product manufacturing such as melt granulation to reduce complexity and risk. Aiming for direct compression whenever possible
• Near-shoring or on-shoring of manufacturing facilities which require higher automation, smaller footprint, deeper process insight and greater environment friendliness
• Semi-continuous manufacturing in biologics and biosimilars
• Increasing role and reliability of process models
• Advanced-batch and mini-batch manufacturing for lower risk
• Distributed manufacturing i.e. producing medicines closer to the patient.
In the era of digital transformation, how are pharmaceutical companies ensuring data integrity and traceability in advanced manufacturing processes?
This is best answered by the industry. However, we note that there is an FDA guidance on available on this topic
51 www.pharmafocusasia.com
EXPERT TALK
(Data Integrity and Compliance With Drug CGMP: Questions and Answers | FDA) which should help the readers familiarise themselves with the regulatory expectations. There are a variety of vendors and platforms available for manufacturers for acquiring, processing, storing and retrieving data (for example, from secure cloud storage).
Can you share examples of successful implementation of robotics in advanced manufacturing?
There has been an increase in the use of robotics and automation in all types of manufacturing industries. This is done with the aim of increasing productivity as well as reducing error, and hence, the risk.
This can include simple pick-andplace types of robotic arms, used in operations such as packaging, transport, and inspection. These operations, however, may not be regarded as a part of the manufacturing process. Many modern quality control laboratories deploy robots for tasks such as sample preparation and automated syringe filling. In the early stages of drug discovery, many companies use high-throughput screening, which utilizes robots for sample preparation, dosing, cell seeding, etc.
In the pharmaceutical advanced manufacturing context, however, I would focus more on the automation aspect. For example, in a continuous process for producing drug products, loss-inweight feeders are deployed. These are sophisticated devices that measure the weight of powder in their hopper with high sensitivity and precision. The weight measurement is relayed to a computer, and data is continuously recorded during the process. This ensures precise control over the formulation. In case of any unacceptable deviation from the set feed rate, an alarm is raised immediately. This is
an example of increasing automation on the manufacturing line.
What role does additive manufacturing play in the advanced manufacturing landscape, and how is it transforming traditional production methods?
While there are only a few products approved for additive manufacturing (AM) or 3D printing (3DP), there is great potential. The techniques are still under development and may not be suitable for all types of formulations. However, they offer great flexibility in both manufacturing and personalisation. A few things required for AM:
• Selection of the right AM approach among the many that are under development. This selection can depend on the product and its formulation
• Selection of appropriate ingredients and understanding their behaviour when subjected to the 3D printer conditions
• Determination of operational procedures, cleaning routines, etc. to complete the control strategy
• Development of printers to be distributed to the manufacturing or compounding locations, ensuring favourable economics
• Monitoring of their performance, collecting data and analysing it continuously.
How is advanced manufacturing contributing to the integration of supply chains, and what advantages does this offer?
Advanced manufacturing allows for:
• More sources of raw materials to be used, since there are more process controls available that equip the manufacturer to handle deviations
Since a lot of equipment used in advanced manufacturing is different from batch equipment, and some designs are still evolving, there can be challenges in procuring it in a timely and costeffective manner.
better, at times even proactively
• Smaller manufacturing facilities, which can make it easier to install plants in different geographies depending on market demand
• More transparency in process due to PAT, modelling, and control loops which can make it easier to transfer the process to a contract manufacturer. The same goes for scaling up or scaling down.
• All of these can help make more quality medicines in more places, which will strengthen supply chain resiliency
What efforts are being made in advanced manufacturing to promote sustainability and reduce environmental impact?
As an example, in our collaborative work with Medicines for All Institute, our scientists are developing new synthesis routes for essential APIs that are also at the risk of shortage. These methods prioritise the use of starting materials and reagents to reduce environmental burden significantly, using methods such as solvent recovery and recycling.
In the drug product side too, the increased use of in-line PAT aided by
52 PHARMA FOCUS ASIA ISSUE 54 - 2024
EXPERT TALK
process models reduces wastage. When developing a process, the sheer number of experiments in a design of experiments (DoE) can lead to tremendous amount of material usage. With AM, the process designer gains the capability to vary multiple parameters in a single continuous run, which can reduce the environmental burden significantly. The flexibility in production quantity (scaling up/down by time as opposed to equipment size) can also reduce environmental impact.
Are there challenges associated with supply chain integration in advanced manufacturing?
It depends on the type of advanced manufacturing being used. The materials for many continuous manufacturing operations are not significantly different from batch manufacturing. Hence, we don’t expect any additional supply chain related challenges for them beyond those already present for batch manufacturing. For newer modalities such as 3D printing, the materials and ancillary requirements can differ depending on the product and the chosen printing platform. These can include special type of excipients, polymers, ancillary equipment, software, etc.
Since a lot of equipment used in advanced manufacturing is different from batch equipment, and some designs are still evolving, there can be challenges in procuring it in a timely and cost-effective manner. Ongoing global supply chain disruptions impact both raw material as well as equipment supplies.
11. What do you foresee as the next wave of advancements in manufacturing standards for the pharmaceutical industry?
Advanced manufacturing technologies associated with modalities like
continuous manufacturing and 3D printing are evolving quickly. Creating a standard typically requires a certain level of technological maturity and widespread usage. Quite a number existing USP general chapters standards will continue to remain relevant for advanced manufacturing as well.
Given the fast-moving nature of technologies, we at USP have adopted a different approach to arrive at advanced manufacturing standards. In addition to our existing standards setting approaches, our series of Technical Guides on continuous manufacturing present detailed explanations of relevant topics such as development of control strategies, process models, dissolution modeling, and techniques such as residence time distribution. These are freely available for download.

Atul Dubey earned his Ph.D. in Mechanical Engineering from Rutgers University, NJ, USA. Dr. Dubey has carried out research in pharmaceutical manufacturing processes using modelling and simulation to optimise unit operations such as continuous mixing, granulation and pan coating. He has authored several journal articles and book chapters and is the editor of a recent book titled “Continuous Pharmaceutical Processing and Process Analytical Technology”, Taylor & Francis.
In his current role as Senior Principal Scientist in the Global Science and Standards Division (GSSD), he is leading the development of new Technical Guides on PCM while also engaging with Expert Panels and Subcommittees of the Physical Analysis, Chemical Analysis, and Chemometrics. His more recent contributions are USP’s first Technical Guide on Control Strategy and the USP CM Knowledge Center. Atul.Dubey@USP.org
In addition, our expert committees, consisting of volunteer experts from industry, regulatory bodies, nonprofits and academia, regularly publish thought-provoking publications known as stimuli articles. These are designed to illicit responses from the wider user community on specific topics. For example, the article New Approaches to Product Performance Testing describes new and upcoming methods for performance testing. The responses we get are very valuable and they play a crucial role in the direction that our expert committees take while creating a new standard.
Digital standards are another way in which process and material quality checks can be made quickly. USP is developing these standards such that they can be digitally delivered and be machine readable for quick, in-process decision making.
Several new advancements are being made by USP to help with the adoption of advanced manufacturing. Among these are:
• New general chapters on PAT for small molecules and biologics
• New and upcoming chapters on powder electrostatics property measurement, powder wettability, and tablet compaction simulation
• Technical Guides on Control Strategy (published), Process Modeling and Dissolution prediction (upcoming)
Any concluding remarks?
Significant amount of work is being carried out globally to reduce any perceived regulatory hurdles to the adoption of advanced manufacturing. Implementations such as continuous direct compression for oral solid dose drug products are considered mature and are no longer emerging methods. Technological challenges in other implementations are quickly being overcome. We are poised to see a significant shift in the use of technology in pharmaceutical manufacturing.
53 www.pharmafocusasia.com
AUTHOR BIO EXPERT TALK
Advanced Drug Delivery Systems to Address Unmet Medical Needs
Attaining effective concentration drugs at the site of action is of paramount importance to combat any disease. Suboptimal concentrations will lead to poor therapeutic response in chronic diseases and emergence of multidrug resistant strains in the case of infections. Advanced drug delivery systems such as lipid based systems (liposomes, microemulsions, mixed micelles), cyclodextrin based complexes, nanoparticulates (solid lipid nanoparticles, nanosuspension and polymeric nanoparticles) can be explored to address the challenges associated with the poor delivery of the active to obtain the desired therapeutic response.
Yogeshwar Bachhav, Director (Consultant), AiCuris Anti-infective Cures AG
What is the significance of Advanced Drug Delivery Systems (ADDS) in modern medicine?
Progress in the field of molecular pharmacology and improved understanding of disease pathophysiology have highlighted the importance of targeting specific cells involved in disease initiation and progress. For life-threatening diseases such as cancer, the use of therapeutic agents often leads to adverse events compromising patient compliance and resulting in the discon-

54 PHARMA FOCUS ASIA ISSUE 54 - 2024
EXPERT TALK
tinuation of the therapy. Anti-infective therapies dealing with bacterial and viral infections necessitate attaining therapeutic concentrations at the site of the infections. Sub-optimal concentrations of the antiinfective agents at the target site led to the emergence of the multi-drug resistant strains which has become a global concern. Modern medicines are formulated using advanced drug delivery systems to achieve drug delivery at the site of action (specific target site), thus improving therapeutic efficacy and minimising the off-target accumulation of the drug. This plays an important role in the management and treatment of the disease.
technology or functional excipients to enhance solubility, stability or cell/tissuespecific targeting of the drug.
Following are some of the examples of the ADDS (but not limited to)
• Solid dispersions
• Cyclodextrin complexes
• Mesoporous silica particles
• Nanoparticles (Nanosuspensions, polymeric nanoparticles, solid lipid nanoparticles)
Advanced drug delivery systems such as nanoparticulate systems (nanoparticles, solid lipid nanoparticles, nanosuspensions), solid dispersion, mesoporous silica particles, lipid-based systems (liposomes, intralipid emulsions, self-emulsifying/ micro emulsifying drug delivery system, mixed micelles) provide multiple advantages including biocompatibility, cellspecific targeting, extended shelf life, higher solubilisation and stabilisation potential of the drugs. These factors lead to superior performance, precise target delivery, and improved efficacy.
How do ADDS differ from traditional drug delivery methods?
Advanced drug delivery systems offer numerous advantages over traditional dosage forms such as tablets, capsules, and oral solutions/suspensions. Conventional oral drug delivery systems were developed using standard excipients to ascertain the

Advanced drug delivery systems are of immense importance to ensure the delivery of the active in the desired concentration and correct form at the disease site.
shelf life during storage, ease of administration, and achieve desired dissolution at the site of absorption. Hence, these systems have their limitations such as lower patient compliance, high levels of variations in the plasma concentrations, increased incidence of adverse events, and more frequency of drug administration. The cumulative effect of these drawbacks has resulted in increased toxicity, decreased efficiency, and unpleasant side effects. As the pharmaceuticals industry has evolved, advanced drug delivery systems were developed to address the shortcomings of the conventional dosage forms.
The basis for developing advanced drug delivery systems is to ensure the stabilisation of the drug in the dosage form and at the site of action (e.g. pH sensitive drugs), enhancing the bioavailability of the poorly soluble drugs (BCS Class-II) and achieving cell-specific targeting. Hence, advanced drug delivery systems provide advantages such as better patient compliance, less variability in the plasma concentrations, reduced incidence of side effects, and lesser frequency of drug administration.
Please discuss a few specific types of ADDS, such as nanoparticles, liposomes, and implants.
Advanced drug delivery systems are composed of dosage forms using specific
• Lipid-based systems (microemulsions, intralipid emulsions, mixed micelles, liposomes)
• Implants
• Specific advanced drug delivery systems such as nanoparticles, liposomes, and implants are discussed below.
Nanoparticles:
Nanotechnology deals with the conversion of particulate matter into a physical state of between 1 to 100 nm. This particulate matter can be rearranged into nano-systems with improved function. Nanoparticles are increasingly being explored for their potential applications as a drug delivery system. Specifically, nanoparticles are investigated as carriers to deliver drugs to specific cells or tissues in the body. The engineering of nanoparticles allows specific surface properties that allow them to selectively target diseased cells and sparing healthy cells. This phenomenon leads to increased efficacy and reduced side effects. Sustained release of the drug is also possible using nanoparticle-based formulation since they can release their cargo in a controlled manner. The field of diagnostics also finds applications in nanoparticles.
Liposomes:
Liposomes are delivery systems comprised of self-assembled phospholipid-based drug vesicles that form a bilayer (unilamellar) and/or a concentric series of multiple bilayers (multilamellar) enclosing a central aqueous compartment. This delivery system represents a size range from 30nm to micrometer scale and phospholipid bilayer
55 www.pharmafocusasia.com
EXPERT TALK
thickness is 4-5 nm. Liposomes have been considered promising and versatile drug vesicles. In comparison to the conventional drug delivery systems, liposomes exhibit superior properties such as site targeting, sustained or controlled release, avoidance of drug degradation, superior therapeutic efficacy, and decreased incidence of adverse events. Due to the unique features of these delivery systems, several liposome-based products have been successfully approved for use in the clinic.
Implants
This type of delivery system provides an extended release of the drug for the desired duration, usually over months or years. Implants are fabricated using a broad range of materials both non-degradable and biodegradable. The choice of the fabrication material depends on the drug to be delivered, the type and duration of the drug release required, and whether the device will remain in place permanently or not. Most of the implants are exposed to tissues for prolonged periods and hence each material must be biocompatible with reduced cytotoxic effects.
A wide variety of implantable devices are in clinical use including subdermal implants, vaginal rings, intrauterine devices and ocular implants, and intracerebral implants.
How do these systems work, and what are their advantages?
Advanced drug delivery systems comprise a wide range of dosage forms as listed above. Depending on the rationale, composition, and process used to prepare these formulations the mechanism of these systems differs as explained below.
Solid dispersions: These delivery systems are prepared using either spray drying or hot melt extrusion. Polymeric excipients such as HPMC, PVP, PEG, Eudragits, and Poloxamers are used to convert the crystalline drug into the amorphous form and thereby enhance the dissolution rate and bioavailabil -
ity. Amorphous forms are known to dissolve faster than the corresponding crystalline forms.
Mesoporous silica particles:
This system works on a similar principle as that of solid dispersion, however for this type of dosage form nanosized mesoporous silica is explored as an excipient, which has unique features like stability, adjustable pores (pores with a diameter ranging from 2 to 50 nm) and large surface area. These features make them advantageous over solid dispersion with higher drug loading capacity for poorly soluble drugs.
Cyclodextrin complexes:
Drugs with poor solubility pose a significant challenge for formulation especially at high doses.
Anti-infective drugs (antibacterials, antivirals, and antifungals) are often administered at high doses in hospital settings. Cyclodextrins are multifunctional pharmaceutical excipients able to form water-soluble host-guest inclusion complexes with poorly soluble drugs (hydrophobic drugs) and thus improve their apparent water-solubility, chemical stability, and bioavailability to obtain the formulations suitable for parenteral administration.
Nanoformulations:
Nanocrystalline ingredients (e.g. nanosus-
pensions) and drug-loaded nanocarriers (polymeric nanoparticles, solid lipid nanoparticles) have overcome the challenges associated with conventional therapies such as limited bioavailability, poor patient compliance, and adverse drug reactions. Different synthesis methods are available for the preparation of nano-formulations. The size of Nanoformulations plays an important role in their biodistribution and clearance. The typical challenge is to have a longer circulation time and thereby have a higher half-life by achieving a size between 1 to 100 nm. The smaller size allows reduced hepatic filtration and higher intracellular uptake compared to microparticles.
Lipid-based formulations:
This class of delivery systems comprises formulations such as microemulsions, emulsions, liposomes, and mixed micelles. Drugs with high log P (lipophilic/poorly water-soluble drugs) are encapsulated/ solubilised in oils/lipids and the oil phase is dispersed in the aqueous phase using surfactants. It is also possible to explore these delivery systems for water-soluble drugs where the aqueous phase-containing drug is emulsified in the external oily phase using surfactants.
The major advantages of the advanced drug delivery systems are summarised below.
• Higher bioavailability and reduced intra-individual variability of the
56 PHARMA FOCUS ASIA ISSUE 54 - 2024
EXPERT TALK
poorly soluble drugs.
• Reduced incidence of adverse drug reactions
• Site-specific delivery (targeted drug delivery)
• Higher patience compliance
• Avoidance of multidrug resistance
• Stabilisation of the labile molecules such as peptides, proteins, and antibodies
What is targeted drug delivery, and why is it considered a major advancement in drug delivery technology?
Targeted drug delivery systems (TDDS) are the dosage forms that are used to deliver a drug to a specific site rather than the entire body or organ. TDDS are designed using knowledge from diverse disciplines such as polymer science, molecular biology, pathophysiology of disease, and drug-receptor interactions.
TDDs are mainly designed to control pharmacokinetics and pharmacodynamics, undesired toxicity, immunogenic reactions, and specific recognition of the drug at the receptor. These systems differ from the conventional dosage forms that achieve site-specific delivery of the drug while the latter relies on the drug absorption through the biological membranes.
TDDs are gaining immense importance in advanced drug delivery systems owing to
• Unmet medical needs due to poor performance of the conventional dosage forms in terms of pharmacodynamic, and pharmacokinetic effects
• Selective targeting of drugs to particular disease organs is important to enhance therapeutic effectiveness but also to reduce the toxicity associated with the smaller therapeutic window and use of high-dose drugs
• Targeting of drugs results in increased efficacy, modulated pharmacokinetics, controlled biodistribution, specific delivery of the drug, decreased toxicity, reduced dose, and patient compliance
• Use of TDDS leads to overall cost reduction due to simpler adminis-
tration procedures, and reduced drug quantity needed for treatment.
What are some challenges in developing and implementing advanced drug delivery systems?
Advanced drug delivery systems have been used successfully, and there is growing demand to deliver drugs to the target site at the desired concentration. However, there are numerous limitations and challenges associated with these systems which are discussed below.
Limited amount of literature: A major challenge that limits the advancement of drug delivery systems is the limited amount of literature and discrepancies reported in the literature. Literature serves as important information for the advancement of the research especially for technologies such as nanomedicines. The discrepancy in the literature impedes the translation of the research from the laboratory to the bedside (clinical application). The safety of nanoparticles is a heavily discussed subject due to the variations in the reported literature.
Large-size particles: Few delivery systems use large particles as carriers which are not suited for the treatment because of the associated challenges such as poor absorption, low solubility and poor bioavailability, in vivo stability, absence of target-specific delivery, and higher incidence of adverse events.
Complications associated with target-specific delivery:
Although target-specific delivery is reported to reduce toxicity and show higher efficacy, the efficacy cannot be assured until the drug is delivered at the target site in a sufficient amount. Especially for nucleic acid molecules like siRNA targeted delivery is impacted by the heavy degradation of the active in the systematic circulation. Lipid-based systems such as micelles and liposomes are used for targeted delivery, but their efficacy is limited by physiological processes such as phagocytic absorption and hepatic filtration
Toxicity of the particles:
There are reports about the toxicity risk of the particles used in advanced drug delivery systems, notably some of the nanomaterials used could be harmful to human health and the environment too. Several in vitro and in vivo studies reported adverse effects of the metallic nanoparticles (silica, silver, gold, and titanium).
Biocompatibility: An important challenge faced by the advanced drug delivery system is biocompatibility (ability to function with the physiological system in the disease condition) and acceptability (acceptance of the delivery system by the body without inciting an immune response). The body may react differently to biological materials compared to synthetic materials and hence this phenomenon could cause serious implications.
Can you discuss any recent innovations or breakthroughs that have addressed these challenges?
The field of advanced drug delivery systems is progressing rapidly, and the continued efforts by researchers have led to possible solutions to some of the challenges listed before.
Industry-academic collaborations: Collaborations across academic institutions and the pharmaceutical industry have helped to align academic theory, and laboratory experiments followed by translation to the clinical site. Typically, basic research at the academic institution has helped to address key topics such as the passage of the particles across the membranes, the internalisation of the particles by the cells, and the safety of the nanoparticles through numerous experiments to understand the in vivo fate of nanomaterials.
Size of the carriers: Problems related to the use of large size particles can be addressed using the much smaller particles for delivery of the drugs. Nanosizing of the drugs has helped immensely to increase the bioavailability of the poorly soluble drugs.
57 www.pharmafocusasia.com
EXPERT TALK
Target-specific drug delivery: Labile molecules are prone to degradation in the systemic circulation. Hence it's important to deliver these molecules at the site of action in a sufficient concentration. Localisation and accumulation of the liposome-loaded drugs in the tumour tissues has been achieved using targeted liposomes. Improvement of the drug efficacy and reduction of the side effects is achieved using the actively targeted liposomes. This effect is mainly achieved through two strategies a) targeting the overexpressed surface receptors of the cancer cells and b) targeting the tumour microenvironment. The ligands used for actively targeted liposomes are antibodies, proteins, peptides, vitamins, growth factors, and aptamers.
Toxicity of the particles:
Nanoparticles with a size below 100 nm are reported to show major toxicological concern. Internationalisation of the smallsize nanoparticles (100nm) by the cells via the process of pinocytosis is proposed to be the reason for toxicity. The increase in size of the particles to 200nm results in the internalisation by macrophages which causes limited cell toxicity.
Biocompatibility:
Biocompatibility of the delivery system is key for their acceptance in clinical use and hence researchers are continuously striving to come up with biocompatible formulations. PLGA represents a family of FDA-approved biodegradable polymers that are physically strong and biocompatible excipients. These polymers have been extensively used as delivery vehicles for drugs, proteins, and various other macromolecules such as DNA, RNA, and peptides. Another such example from the recent reports is the use of the Metal–organic frameworks (MOFs) as carriers or vehicles which are excellent platforms for a sustained and controlled release of the drugs (particularly for the poorly soluble drugs). Metal-organic frameworks are crystalline materials containing cationic metal nodes and anionic/neutral organic linkers connected through coordination
Minimising the off-target accumulation of the drug is very important to rule out the incidence of adverse events in chronic disease conditions such as cancer. Precise selection of advanced drug delivery systems can help to achieve this objective.
bonds. These systems attracted the attention of the researchers because of their biocompatibility, biodegradability, and non-toxicity.
How can ADDS contribute to the concept of personalised medicine? Personalised medicine uses information derived from the genetic and genomic data of the patient to tailor the decisions related to diagnosis, treatment, and prevention of the disease. This concept allows physicians to make effective and more informed decisions about patient care based on the patient’s genes and genome. Personalised medicine is advantageous over conventional medicine which uses one strategy across the patient population without relying on the patient’s genetic makeup.
Advanced drug delivery systems are investigated for the personalised treatment of a broad range of highly prevalent diseases (e.g., cancer and diabetes). Typically, pH and tempera -
ture- sensitive polymers are explored for this purpose. For example, thermoresponsive polymers (poloxamers, poly (N-isopropylacrylamides)) can be mixed with drugs at room temperature and then injected into the body. When the temperature increases to 37°C, the polymer forms a gel for sustained release of the drug. pH-sensitive polymers (chitosan, alginate, hyaluronic acid) can be used to synthesise block copolymers which will self-assemble and can be used as nanocarrier systems for anti-cancer drugs. These carriers will release the drug when triggered by the acidic nature of the tumour microenvironment, endosomal compartment, or specific organs. 3D printing has been explored to develop controlledrelease dosage forms. This technique allows to production of personalised or unique dosage forms and thereby complex drug release profiles. This approach helps to address the issue of inter-individual variability while treating patients of different ages, races, genders, pharmacogenetics, and pharmacokinetic characteristics. Additional novel approaches are proposed to enable a flexible and patient-appropriate therapy such as the use of various dispensers for multiparticulate drug delivery systems.
Simple approaches are also explored to contribute to personalised medicine. Dosing of the liquid formulations can be accurately achieved by using novel dropping tubes or oral syringes. For the tablet dosage forms, breaking the scored tablets into fragments presents limitations such as inaccurate dosing, formation of potent dust, and stability of the residual segments.
What are the emerging trends in the field of advanced drug delivery systems? How might these advancements shape the future of medicine and patient care?
The COVID-19 outbreak caused a severe public health crises across the globe. Use of the nucleic acids as
58 PHARMA FOCUS ASIA ISSUE 54 - 2024
EXPERT TALK
potential drugs to treat this epidemic was the need of the hour. However, delivery of the mRNA is a challenge because the positive charge and hydrophilic nature of the nucleic acid hinder their diffusion across the cell membrane. Lipid nanoparticles have shown a great potential to deliver nucleic acids including the mRNAs resulting in the commercialisation of the Covid-10 vaccines. These lipid nanoparticles are continuously being investigated for the delivery of nucleic acids to treat life-threatening diseases such as cancer.
Digitalisation is being widely used across all industries. Keeping pace with the digital era, the pharmaceutical industry is working hard to develop smart drug systems using small medical technologies. Standard components of the drug delivery systems can be converted into smart components using state-of-the-art sensors and electronics.
Smart drug delivery systems offer the following advantages
• Improved patient-friendliness
• Higher functional response
• In-use collection and evaluation of the data
• Aiding in the monitoring of vital functions and measured data
The new drug delivery systems coupled with digitalisation will play a significant role in improving the quality of healthcare. The input from the potential use of the drug delivery systems will help to further adapt the formulations to improve the overall efficacy, safety, and patient compliance.
How do advanced drug delivery systems impact healthcare costs and accessibility? Are there challenges in making these technologies widely available?
Advanced drug delivery systems utilise complex processes and excipients, and it would result in increased costs for the patients compared to the conventional dosage forms. The impact of the higher cost of medication on acces -
sibility is heavily discussed across the globe including in the United States. It covers various aspects such as the pricing strategies of pharmaceutical companies, coverage by insurance, public policies, and existing healthcare systems.
The high cost of the medication is a complex challenge that would need a comprehensive approach. Policymakers, healthcare professionals, pharmaceutical companies, and advocacy groups must work together to propose sustainable solutions to ensure accessibility of essential medications (comprising advanced drug delivery systems) without exposing patients to financial burden.
Are there any ethical concerns or societal implications related to the use of advanced drug delivery systems? How can these be addressed?
Advanced drug delivery systems such as nanoparticles/vesicles are associated with structures and components that exhibit novel physical, chemical, and biological properties due to the specific (nano) size. The ethical considerations related to nanomedicine are mainly related to risk assessment in general, the effect on somatic cells versus germline cells, the enhancement of human capabilities, research on human embryonic stem cells, toxicity, uncontrolled function, and self-assembly of nanoparticles. Ethical concerns for advanced drug delivery systems, specifically nanoparticles are more complex than those for general medicine and biotechnology. This is mainly because of the toxicity of the nanoparticles due to their particle size in the nanoscale. General ethical principles such as respect for human anatomy, beneficence, nonmaleficence, and justice are at stake.
To address these issues, a reasonable sound knowledge base acquired in the field of bioethics can be applied to advanced drug delivery systems such as nanomedicine.
Any other comments?
The advanced drug delivery systems field has a bright future. Drug delivery systems continue to evolve because of the shift of the existing treatment landscape from small molecules to biologics.
It would be expected to see breakthrough technologies that can enhance stabilisation and targeting of the biologics as well as sustained release over a prolonged period. Efficient delivery of the molecules across complex biological barriers is also a key challenge to be addressed using the new delivery systems. The future of the drug delivery system would also rely on application of the materials that can effectively target specific biology and can be adapted to the disease pathophysiology yet remain simple for clinical translation. Future drug delivery systems will certainly impact global healthcare by improving the efficacy of treatments and making them more affordable and easier to use. Currently, only a certain population can afford the novel treatments. A decrease in the cost of novel drug delivery systems will be of great help compared to cheap generic versions for patients with limited resources. Affordable access to the novel medications, the future would demand a series of innovations, joint efforts in drug delivery technologies, and highly automated low-cost manufacturing platforms.
AUTHOR BIO
Yogeshwar Bachhav has pursued B.S.(Pharm) from North Maharashtra University (M.S.India). and M.S. (Pharm) and PhD from Institute of Chemical Technology, Mumbai (india) He has around 17 years of Post PhD experience in Europe in the field of Pharmaceutical Development of investigational drugs. Currently he is working as Director (Consultant) at AiCuris Anti-infective Cures AG Germany and responsible for Pharmaceutical Development of investigational drugs in the domain of innovative antiviral and antibacterial drugs. He has also started a consultancy firm called Adex Pharma which deals with solving complex issues in the pharmaceutical development of new and approved drugs since 2016.
59 www.pharmafocusasia.com
EXPERT TALK

60 PHARMA FOCUS ASIA ISSUE 54 - 2024 PRODUCTS & SERVICES STRATEGY Emirates Sky Cargo............................................................................. IFC LogiPharma Asia.................................................................................. 05 Novo Nordisk Pharmatech A/S OBC Qatar Cargo IBC Valsteam ADCA Engineering. 03 RESEARCH & DEVELO PMENT Novo Nordisk Pharmatech A/S .......................................................... OBC MANUFACTURING Novo Nordisk Pharmatech A/S OBC Valsteam ADCA Engineering 03 Company ............................................................. Page No. SUPPLIERS GUIDE Emirates Sky Cargo IFC www.skycargo.com LogiPharma Asia 05 https://logipharmaasia.wbresearch.com/ Novo Nordisk Pharmatech A/S OBC http://novonordiskpharmatech.com/ Qatar Cargo IBC https://qrcargo.com Valsteam ADCA Engineering 03 www.valsteam.com Company ............................................................. Page No. To receive more information on products & services advertised in this issue, please fill up the "Info Request Form" provided with the magazine and fax it. 1.IFC: Inside Front Cover 2.IBC: Inside Back Cover 3.OBC: Outside Back Cover


 by PHARMA
by PHARMA
Pharmaceutical grade Benzalkonium Chloride

Minimize your risks and get peace of mind



You can ensure optimal processes and even better medicines by prioritizing high quality, consistency and reliable GMP manufactured raw materials.
Our Benzalkonium Chloride is manufactured following cGMP guide ICH Q7 for APIs, the highest available quality standard in the industry.
Get peace of mind with:
• Full traceability
• High product purity
• Full pharmacopoeia compliance (PhEur, USP/NF, JP, ChP)
• Audit access
• Full regulatory documentation package
• Manufacturing under fully validated processes
• 30+ years of experience in cGMP manufacturing
Learn more about Benzalkonium Chloride at novonordiskpharmatech.com

