



SPONSORS:





Issue 03 | 2024 | www.pharmafocusamerica.com Twin-Screw Granulation An Alternative Continuous Granulation Approach 65 PAGE The Journey of Mass Spectormetry in Biopharmaceuticals CMC 69 PAGE Revolutionizing Drug Manufacturing with
3D Printing
ANNA WORSLEY CEO, FABRX-AI
HANNAH WATTON Account Manager FABRX
ALVARO GOYANES Co-Founder and Director FABRX
TWO POWERFUL DOSES PER YEAR

Every issue of Pharma Focus America magazine is a powerful dose of information and knowledge – filled with original and undiluted content.
Written by the best brains in pharma industry, the magazine offers timely business insights and articles on cutting-edge technologies.
Subscribe now to get your doses regularly.
Email: subscriptions@pharmafocusamerica.com
www.pharmafocusamerica.com

2 PHARMA FOCUS AMERICA ISSUE 03 - 2024
Exploring the Disruptive Force of 3D Printing in Pharmaceuticals
Welcome to the third edition of Pharma Focus America, where we delve into the intricate world of Pharmaceuticals, presenting insightful articles and expert interviews that provide a unique Industry perspective. Our spotlight theme, "Revolutionizing Drug Manufacturing with 3D Printing," offers key insights from influential figures shaping this transformation narrative in the Pharma Industry. We hope that the third edition of Pharma Focus America Magazine serves as a valuable resource for Pharmaceutical industry professionals globally.
Anna Worsley, CEO, FABRX-AI, Alvaro Goyanes, Co-Founder and Director, FABRX, Hannah Watton, Account Manager, FABRX. In this edition, we delve into the pioneering force that is pharmaceutical 3D printing—an innovation that extends its influence from personalized medicine to the intricacies of clinical trial workflows. This article offers a comprehensive overview, providing valuable insights into 3D printing as a pharmaceutical manufacturing tool and exploring its transformative impact on the field.
Introducing a new element in this edition, we proudly present the "Book Interview" featuring a comprehensive exploration into Clinical Trial Project Management. Authored by Ashok Kumar Peepliwa, Associate Professor at the School of Pharmaceutical Management, IIHMR University, and this book provides a detailed overview of the intricacies involved in conducting clinical trials on a global scale. Navigating the regulatory regimes of various nations, Peepliwa's guide offers a comprehensive understanding of the challenges associated with clinical trial management, from country-specific regulatory dossier submission to final study report preparation, all in compliance with ICH-GCP standards. This addition adds a valuable dimension to our exploration of pharmaceutical innovations.
We are pleased to present the expert panel discussion on "Next-Generation Vaccines," featuring
Aaron B. Cowley, Chief Scientific Officer at ReciBioPharm; Patrick Thiaville, Chief Technology Officer at Exothera; and Daniel Kavanagh, Senior Scientific Advisor, Gene Therapy, Vaccines and Biologics, WCG. Together, they explore the future landscape of next-generation vaccines, providing insights into discovering and developing manufacturing processes for enzymes, live biotherapeutics, and advancements in clinical manufacturing and infrastructure design.
Finally, we are honoured to present an expert interview with Dr. Courtney Noah, VP, BIOIVT. Explore the world of liquid biopsy research with insights from her, offering a glimpse into the advancements shaping disease detection, diagnosis, and treatment.
Thank you for being a vital part of the Pharma Focus America community. We eagerly anticipate the continued exchange of ideas and knowledge, as together, we navigate the ever-evolving global Pharmaceutical landscape.
If you have a perspective, an idea, or a story to share, we welcome your voice in our upcoming issues. Whether it's an article that sheds light on emerging trends, an interview with a thought leader, or a unique insight into the Pharmaceutical ecosystem, your wisdom can serve as a guiding beacon for others on their pharmaceutical journey. We want to hear from you via email: editorial@Pharmafocusamerica.com
Thank you for joining us on this exploration of Pharmaceutical innovations in Pharma Focus America.
Stay Engaged for Exclusive Insights in Upcoming Editions!
www. pharmafocusamerica.com 3
N D Vijaya Lakshmi Editor
STRATEGY
08 New MHLW Guidance Aims to Facilitate Inclusion of Japanese Participants in Global Clinical Trials
Mayumi Hasegawa, PhD Senior Director, Drug Development Solutions, Certara
RESEARCH & DEVELOPMENT
15 Beyond the Blueprint: mRNA’s in Promise Immunotherapy
Vidya Niranjan, Professor and Head of the Department, Biotechnology, Lead- Centre of Excellence Computational Genomics, R V College of Engineering
CLINICAL
26 Revitalizing Research: Prioritizing Participant-centric Strategies to Accelerate Clinical Trials
Amy Thue, Associate Director, Project Management, WCG
Shelby Ward, Site Services, WCG
33 Applications and the Limitations of RealWorld Data in Gene Therapy Trials
Karen Ooms, Joint Chief Operating Officer, Quanticate
MANUFACTURING
47 Data Exchange in Pharmaceutical Manufacturing
Balancing Collaboration and Competition
John Ward, Founder and CEO of ServBlock
Mick Cummins, Managing Director of Ingeniero Solutions
56 Digitalization of a Novel Advanced Modular Continuous Pharmaceutical Drug Substance Manufacturing Process
Ravendra Singh, Faculty of Chemical and Biochemical Engineering, Chemical and Biochemical Engineering, Rutgers University
65 Twin-Screw Granulation
An Alternative Continuous Granulation Approach
Sateesh Kumar Vemula, Department of Pharmaceutics and Drug Delivery, School of Pharmacy, The University of Mississippi
Michael A. Repka, Department of Pharmaceutics and Drug Delivery, School of Pharmacy, The University of Mississippi
69 The Journey of Mass Spectormetry in Biopharmaceuticals CMC development
Vesela Encheva, PhD, Associated Principal Scientist, R&D department at Lonza
Ian Anderson, PhD, Senior Technical Leader, Global Biologics Technical Development, Lonza
Revolutionizing Drug Manufacturing with 3D Printing



38
Anna Worsley CEO, Department: Management FABRX-AI
Alvaro Goyanes Co-Founder and Director FABRX
Hannah Watton Account Manager, Organisation: FABRX

Vikalp Khare, Director, Otsuka Pharmaceutical
4 PHARMA FOCUS AMERICA ISSUE 03 - 2024
CoverStory INFORMATION TECHNOLOGY
The Emerging World
Smart Pills, Patches, and Implants: Innovative AI-Enabled Drug Delivery Systems
Pharmaceutical
A Quick start Guide
Successful Cloud Migration
(GPV) IT Applications
CONTENTS
76
of
Prachi Khamkar, Director at Atlaas
81
for
of Global Pharmacovigilance
EXPERT TALK 88 Advances in Liquid Biopsy Research for Disease Detection, Diagnosis, and Treatment and What the Future May Hold Dr. Courtney
95 WHITEPAPER 97 INDUSTRY SENSE 106 THROUGH THE HOURGLASS 116 BOOK INTERVIEW 126 EVENT PREVIEW 132 EVENTS LIST 134 NEWS
Development & Commercialization
Noah, Vice President of Scientific Affairs, BioIVT.
TRIALS

EDITOR
Vijaya Lakshmi N D
EDITORIAL TEAM
Sarah Richards
Debi Jones
Harry Callum
Supraja BR
ART DIRECTOR
M Abdul Hannan
PRODUCT MANAGER
Jeff Kenney
ASSISTANT MANAGER
David Nelson
Peter Thomas
BUSINESS EVENTS
Sussane Vincent
CIRCULATION TEAM
Sam Smith
SUBSCRIPTIONS IN-CHARGE
Vijay Kumar Gaddam
HEAD-OPERATIONS
Sivala VNR




www. pharmafocusamerica.com 5
Ochre Digi Media www.ochre-media.com Magazine Subscribe ©Ochre Digi Media. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, photocopying or otherwise, without prior permission of the publisher and copyright owner. Whilst every effort has been made to ensure the accuracy of the information in this publication, the publisher accepts no responsibility for errors or omissions. The products and services advertised are not endorsed by or connected with the publisher or its associates. The editorial opinions expressed in this publication are those of individual authors and not necessarily those of the publisher or of its associates. Copies of Pharma Focus America can be purchased at the indicated cover prices. For bulk order reprints minimum order required is 500 copies, POA. LinkedIn www.pharmafocusamerica.com
Advisory Board





Amine Bekkali
Director chez MEDFIELDS United Arab Emirates

Alessio Piccoli
Director & Head, Business Development Europe, Aragen Life Sciences
Italy
David Contorno
Founder & CEO, E Powered Benefits, USA


Eiman Shafa
Medical Director, Spine Surgery, Abbott Northwestern Hospital USA

Hassan Mostafa Mohamed
Chairman & Chief Executive Officer at ReyadaPro, Saudi Arabia




Juris Hmelnickis
CEO, Grindeks, Latvia
Nicoleta Grecu
Director, Pharmacovigilance, Clinical Quality Assurance, Clover Biopharmaceuticals
Romania
Nigel Cryer FRSC
Global Corporate Quality Audit Head, Sanofi Pasteur, France
Pinheiro Neto Joao
Chief Executive Officer, Omnimed
Angola

Hector Alejandro Andonie
General Manager, Laboratorios Andifar
Honduras
Scott M. Wheelwright
Chief Operating Officer, BioInno Bioscience Co., Ltd. China

Hoda Gamal
Director of Regulatory and Corporate Affairs, Middle East and Africa (MEAC), Sirgio international, Egypt
Joaquin Campbell
Global Director, Managed Access Services, EarlyHealth Group
Spain
Josipa Ljubicic
QA Director/Principal GCP and GVP auditor, Proqlea Ltd
Croatia
Svetlana Busiguina
CEO, Bi-Connex BD Consulting, Spain


Tamara Miller
Senior Vice President, Actinogen Medical Limited, Australia
Thitisak Kitthaweesin
Chief of Phramongkutklao Center of Academic and International Relations Administration
Thailand
6 PHARMA FOCUS AMERICA ISSUE 03 - 2024
•
•
•
•
•
•
•
www. pharmafocusamerica.com 7 Custom Antibody Development and Production Accelerating Drug Discovery with One-stop CRO Services www.sinobiological.com Sino Biological US Inc. (U.S.A.) Tel: +1-215-583-7898 Email: cro_us@sinobiologicalus.com Sino Biological, Inc. (Global) Tel: +86-400-890-9989 Email: cro-service@sinobiological.com
Biological Europe GmbH (Europe) Tel: +49(0)6196 9678656 Email: cro-service@sinobiological.com
Hybridoma, single B cell discovery (Beacon® and FACS), phage display, and rabbit pAb platforms
Sino
•
Antibody humanization & AI-powered affinity maturation Comprehensive Antibody Development Technologies
Comprehensive services & reagents
Multi-dimensional efficacy evaluation system Solutions for In Vitro Efficacy Evaluation
Over 15 years of experience
14,000+ antibodies & 6,500+ recombinant proteins library Antibody Development & Protein Production Expert
1,000+ antibodies per batch
Flexible scales: µg - kg level
Diverse antibody formats
Fast delivery: as short as 2 weeks
Multiple expression systems High-throughput & Scale-up Recombinant Production
•
•
•
New MHLW Guidance Aims to Facilitate Inclusion of Japanese Participants in Global Clinical Trials
Previously, sponsors had to conduct additional phase 1 studies with Japanese participants before Japan could be included in global clinical trials. This approach delayed approval of new medicines in Japan. With its new guidance, MHLW is endeavoring to expedite drug development by removing this step for drugs where early clinical development is preceding outside Japan.
Mayumi Hasegawa, PhD Senior Director, Drug Development Solutions Certara

On September 13, 2023, the Ministry of Health, Labour and Welfare’s (MHLW’s) "Study Group on the Drug Regulation System to Enhance Drug Discovery and Ensure Stable Supply" agreed that "there is
no need to conduct additional phase 1 studies on Japanese [participants] before the start of global clinical trials" for products whose development has already been advanced overseas. However, the committee also notes that "it is desirable
8 PHARMA FOCUS AMERICA ISSUE 03 - 2024
STRATEGY
to collect information on pharmacokinetics (PK) in Japanese [participants] as much as possible.” There had been cases where Japan alone was required to conduct additional phase 1 trials, which proved disadvantageous because it prevented the country’s participation in some global joint clinical trials. To minimize these disadvantages, while also ensuring the safety of Japanese participants in international joint clinical trials, the Ministry issued a new notification on December 25, 2023, entitled “Basic principles for conducting phase 1 studies in Japanese prior to initiating multi-regional clinical trials [MRCTs] including Japan for drugs in which early clinical development is preceding outside Japan.”This announcement also repealed the 2014 notification entitled “Basic Policy on Conducting Phase 1 Clinical Trials in Japanese Patients Prior to the Start of Global Clinical Trials.” In addition, it removed the requirement to obtain both PK and safety data in Japanese participants to compare with non-Japanese before global clinical trials, which was described in the #Notification No.0928010 issued by MHLW in 2007. Furthermore, the notification "Basic Approach to International Clinical Trials (Reference Examples)" (MHLW, Pharmaceutical and Food Safety Bureau, September 5, 2012) was updated as follows:
1) What are the general points to consider in comparing PK data between different ethnicities?
In general, it is recommended that interethnic PK comparison is based on data collected
according to the same protocol including measurement methods etc. (also applies to studies conducted separately) to minimize variations caused by non-intrinsic ethnic factors. If genetic variation in metabolic enzymes or transporters is expected to affect the PK of the investigational drug, genetic tests should be performed in the clinical trial to examine the incidence of genetic variation in different ethnicities and the PK-genotype relationship. If no PK data are available from Japanese and non-Japanese participants included in studies conducted under the same protocol, collection of PK data is recommended in consideration of the characteristics of the drug at several time points in the major ethnic groups to be included in a confirmatory trial, at least before submission of [a new drug application] an NDA (previously “initiating a global confirmatory trial”).
Japan's MHLW aims to accelerate drug development by omitting extra phase 1 studies with Japanese participants, facilitating global trial inclusion, and expediting new medicine approvals.
www. pharmafocusamerica.com 9
STRATEGY
2) When only a monotherapy study of an investigational drug was conducted in Japan, is it possible for the drug to be used in an exploratory global clinical trial including Japan investigating its combined treatment with Drug A?
Previously, data on the investigational drug in Japanese participants who had received the combination therapy with Drug A was needed before their participation in a global clinical trial. In addition, only when the dose of Drug A had been used in patients in Japan and its safety had been established, could a global clinical trial for the combination therapy be initiated. This description has been replaced by the statement that “In principle, if it is considered that there is no risk of increased safety risks associated with the concomitant administration of Drug A based on the results of non-Japanese clinical trials, it is possible for Japan to participate in global clinical trials investigating its combined treatment with Drug A without conducting a study for the combination therapy in Japan prior to Japan’s participation.”
It should be noted that the new notification1 is intended for drugs in which early clinical development is preceding outside Japan. In recent years, there has been an increase in the number of innovative drugs, mainly those developed by emerging biopharmaceutical companies, which are in advanced clinical development overseas. To keep Japan up to speed with these developments, Japan's participation in MRCTs has been considered in

an increasing number of cases. The participation of Japanese patients in a global clinical trial has a significant impact on the drug’s potential approval in Japan.
The new guidance1 says: “In general, it is not mandatory to conduct a phase 1 study in each race/ethnicity or country/region before initiating MRCTs. In principle, an additional phase 1 study in Japanese is not needed unless it is deemed necessary after assessing whether the safety/tolerability of the dosage to be evaluated in the MRCTs in Japanese participants can be explained and the safety is clinically acceptable/ manageable based on the data available prior to Japan’s participation.”
However, the Pharmaceuticals and Medical Devices Agency (PMDA) recommends
10 PHARMA FOCUS AMERICA ISSUE 03 - 2024
STRATEGY

conducting a phase 1 study in Japanese participants when the number of target patients in Japan is large and there is sufficient time to conduct a phase 1 study in Japanese prior to the MRCTs.
The justification for the recommended dose regimen in Japanese participants for MRCTs should be established based upon pooled data from non-clinical studies, non-Japanese clinical studies, and ethnic sensitivity consideration.
Japan can participate in MRCTs without conducting a phase 1 study in Japanese provided appropriate informed consent is obtained for drugs with high unmet medical needs, such as drugs for rare diseases, diseases that are refractory and serious, or pediatric regardless of whether the drug is developed in adults, where
participation in planned or ongoing MRCTs are considered desirable to develop the drug in Japan.
Regulatory Approvals
In FY2022, 32 drugs containing new active ingredients (excluding vaccines) were approved in Japan. Of these, 21 products were approved based on an MRCT as a pivotal study, and there were two products (vosoritide and cabotegravir/ rilpivirine for chondrodysplasia and HIV-1 infection, respectively) where no Japanese phase 1 studies were conducted. In addition, there were three approved medicines in FY2022 whose pivotal studies were domestic studies and for which Japanese phase 1 studies were not conducted: pegvaliase for phenylketonuria, cholic acid for inborn errors of bile acid metabolism, and immunosuppressant antihuman thymocyte immunoglobulin for aplastic anemia.
The reasons why Japanese patients were recruited to the registrational clinical trials without first conducting Japanese phase 1 studies included the fact that the disease had a high medical need, there was a limited number of patients, and the phase 3 trials were planned to give sufficient consideration to the safety of the Japanese population. All five products had been designated as orphan drugs. For the 32 approved medicines, no clinically meaningful differences in PK between Japanese and non-Japanese participants have been reported based on phase 3 clinical data.
www. pharmafocusamerica.com 11
STRATEGY
However, this does not mean that all orphan drugs and pediatric drugs do not need to have Japanese phase 1 studies. That will ultimately be determined based on the characteristics of each drug and the available scientific data. For example, with anticancer drugs, which are expected to cause a high frequency of serious adverse events, have a narrow safety margin, and where there is limited safety information, (e.g., there is no experience of administration in Japanese patients of any age or indication), whether a Japanese phase 1 study is necessary should be judged more carefully.
What points should be considered to determine whether the safety of Japanese participants is clinically acceptable and manageable in MRCTs?
The MHLW Q&A says the risks of the study drug should be comprehensively examined (see points 1 and 2 below), to confirm whether there is a possibility that the risk for Japanese participants is greater than that for non-Japanese participants. If so, it must be determined whether the safety considerations for Japanese participants in the MRCT are clinically acceptable and manageable in the proposed dosing regimen.
1) Safety of study drug
• The results of non-clinical studies suggest no significant risk with an unclear mechanism of onset at the dose used in the MRCT.
• The maximum dose used in the MRCT has a sufficient safety margin, and no clinically
significant risks have been identified in the preceding foreign clinical trials.
• There are clear approaches and monitoring methods for mitigating potential risks and the potential risks are manageable by defining appropriate measures/monitoring in the MRCT.
• No clinically significant risks that increased in incidence or severity dose-dependently have been identified in the preceding foreign clinical trials.
• When there are similar drugs which can be used as a reference in the safety evaluation, no clinically significant risk of the study drug is anticipated from the safety data of those drugs.
2) Effect of ethnic factors on study drug
• Ethnic differences in PK are unlikely based on comprehensive considerations.
• The drug has characteristics that make the safety and PK unlikely to be affected by ethnic factors.
• There is no significant impact of ethnic factors such as race, region, body weight on the safety or PK based on previous clinical trial(s) in which the drug has been administered in multiple races/regions, or participants covering a wide range of body weight.
• When there are similar drugs which can be used as a reference in a safety evaluation, no clinically significant ethnic differences in the safety are observed with those drugs, and the same is anticipated for the study drug.
12 PHARMA FOCUS AMERICA ISSUE 03 - 2024
STRATEGY
Recruiting Japanese Patients
When sponsors are starting to recruit Japanese patients into a registrational MRCT without conducting a local phase 1 study in Japanese participants, what additional measures can be taken to ensure their safety? Q&A guidance says safety measures differ depending on the characteristics of each study drug. The appropriate safety measures should be selected based on prior information about the drug, the study design, and how those additional measures could affect the safety evaluation.
In many registrational MRCTs in oncology, a safety-lead-in cohort has been set up to evaluate the safety and intensive PK of a small number of Japanese patients (usually three or six participants) prior to the main part of the study. In these cases, this safetylead-in cohort serves as a “mini-phase-1-study in Japanese [participants] embedded into a MRCT” to compare both safety and PK data with non-Japanese participants before the main part of the MRCTs.
Sponsors should be aware that there is a possibility that the efficacy data from these safety-lead-in participants cannot be used in the primary endpoint analysis, while it could be used as supportive data because of the different setting from the main part of the MRCTs such as safety monitoring schedules and hospitalization conditions.
Other measures used to secure Japanese participants’ safety are 1) administer the drug to a small number of Japanese
participants (e.g., one participant at a time) with appropriate intervals between each administration, 2) execute safety monitoring with special attention to Japanese participants in an organization composed of third parties, such as an independent data monitoring committee, 3) Japanese participants will either be hospitalized or observed at the study site for a certain period, 4) Increase the frequency of visits and monitoring during the early stage of administration.
Monoclonal Antibody Review
When a systematic review of monoclonal antibodies (mAbs) approved in Japan from 2007 through 2023 was conducted, the difference in PK parameters between Japanese and non-Japanese patients was less than two times. According to PMDA’s review reports, it was concluded that the PK difference was not clinically significant because there was no apparent difference in safety or efficacy profile in Japanese patients compared with non-Japanese. A 20-30% difference in PK was typically explained by the difference in body weight of participants included in the clinical trials.
The mAbs review revealed that the observed PK differences between Japanese and non-Japanese populations were either due to differences in body weight or differences in receptor expression between the populations. If the difference is covered by the safety margin, dose adjustment might not be necessary. Other literature reports no
www. pharmafocusamerica.com 13
STRATEGY
essential differences between Japanese and non-Japanese participants, particularly in healthy volunteers, in the exposure of mAbs that have been approved in Japan. The doses selected in subsequent Japanese patient phases were the same as those stated on the approved US labels, even if there were differences in PK in the healthy volunteer studies.
It is recommended that the ethnic difference in PK is evaluated using Population PK modeling based upon pooled data from both non-Japanese and Japanese data, which would take other covariate effects into account. In addition, the impact of PK difference on the clinically recommended dose in patients should be evaluated through a thorough review of the clinical efficacy and safety profile, and the exposure-response analysis to evaluate the relationship between efficacy/safety and PK (estimated PK measures from the final Population PK model).

When it is difficult to determine PK differences due to small sample size, PMDA says that safety/efficacy should be monitored closely after approval and the label updated when needed.
Conclusion
Regardless of whether a Japanese phase 1 study is conducted, MHLW’s new announcement requests that the difference between Japanese and overseas patients be examined by collecting pharmacokinetic/pharmacodynamic data on Japanese patients prior to submission for marketing authorization approval. Considering that PMDA has been accepting modeling in NDAs and there are multiple cases where Japanese patients have been recruited into registrational MRCTs without conducting local Japanese phase 1 studies, this new guidance is not a surprise. But MHLW has made its opinion public to help diminish drug approval lag and encourage sponsors to include Japan in their global drug development programs as early as possible.
References are available at www.pharmafocusamerica.com
Mayumi Hasegawa, PhD, is Senior Director, Drug Development Solutions at Certara. Mayumi has more than 20 years of drug development experience focused on clinical pharmacology and pharmacometrics in the Asia-Pacific region (Japan-Korea-Taiwan). Her expertise includes early- and late-stage development programs in oncology/immunology, rheumatoid arthritis, and cardiovascular disease and she has supported multiple filings to the FDA, EMA and PMDA.
14 PHARMA FOCUS AMERICA ISSUE 03 - 2024
AUTHOR BIO
STRATEGY STRATEGY
Beyond Blueprintthe
mRNA’s in Promise Immunotherapy
As a researcher in the field of cancer computational immunotherapy, I have analyzed the potential of messenger RNA (mRNA) technology in revolutionizing cancer treatment. Through my research, I have identified the challenges and concerns related to mRNA-based immunotherapies, including the instability of mRNA and the efficient delivery of mRNA to target cells. However, ongoing research and development efforts are focused on addressing these concerns and further harnessing the capabilities of mRNA. I have also discussed the regulatory landscape surrounding mRNA-based therapies and the promising results of clinical trials evaluating the safety and efficacy of mRNA-based immunotherapies in cancer treatment. Overall, I believe that mRNA technology has the potential to revolutionize the way we diagnose, treat, and prevent a wide range of conditions, offering new hope for patients and healthcare advancements globally.
 Vidya Niranjan Professor and Head of the Department, Biotechnology, Lead- Centre of Excellence Computational Genomics, R V College of Engineering
Vidya Niranjan Professor and Head of the Department, Biotechnology, Lead- Centre of Excellence Computational Genomics, R V College of Engineering
RESEARCH & DEVELOPMENT
Introduction to mRNA Immunotherapy
The current landscape of cancer immunotherapy has seen significant advancements in recent years, with several immunotherapeutic agents receiving US Food and Drug Administration (FDA) approval for treating various cancer types, including breast cancer, melanoma, non-small-cell lung cancer (NSCLC), and genitourinary cancers. Key immunotherapeutic strategies include immune checkpoint blockade (ICB), adoptive cell therapies (ACTs), cancer vaccination, and oncolytic viruses.
ICB has achieved objective responses in patients with breast cancer, with higher rates seen when administered in earlier lines of therapy. Responses are durable for responding patients. ACTs involve identifying and isolating peripheral blood or tumor-activated and expanded cells ex vivo before transferring them back into the patient. Cancer vaccination aims to provide therapeutic immunity by stimulating the patient's immune system to target cancer cells. Oncolytic viruses can selectively infect and destroy cancer cells, bypassing the need for the immune system to recognize and attack them.
Despite these successes, challenges remain in the successful implementation of immunotherapy in managing breast cancer and other cancer types. These include incorporating immunotherapy into adjuvant and neoadjuvant cancer therapy, refining dose, schedule, and duration, and establishing robust predictive biomarkers for response
to ICB. Future opportunities and challenges in cancer immunotherapy include the development of combination therapy strategies, refining treatment combinations, and further understanding the tumor microenvironment and host factors to inform treatment decisions.
Messenger RNA (mRNA) has emerged as a groundbreaking approach with vast potential in medicine. mRNA technology has been at the forefront of vaccine development, particularly evident in the rapid creation of COVID-19 vaccines. Beyond infectious diseases, mRNA has shown promise in treating existing conditions such as cancer. Its flexibility allows for the creation of mRNA cancer vaccines, which activate the immune system to target cancer cells. mRNA vaccines have significant advantages over conventional cancer vaccines, including efficient production of protective immune responses, low side effects, and lower cost of acquisition. mRNA vaccines can also bypass the need for genomic/genetic engineering, which can lead to undesired immune responses. mRNA is used to induce the immune system to destroy tumor-associated antigens and growth factors. Despite the challenges of mRNA's fragility, ongoing research and development aim to address these challenges and further harness the capabilities of mRNA. The Nobel Prize in Physiology or Medicine 2023 was awarded for groundbreaking findings on mRNA, highlighting the transformative impact of this technology. As research and development in this field continue, mRNA
16 PHARMA FOCUS AMERICA ISSUE 03 - 2024
RESEARCH & DEVELOPMENT
stands poised to revolutionize the way we diagnose, treat, and prevent a wide range of conditions, offering new hope for patients and healthcare advancements globally.
Current status of mRNA research
The advent of mRNA technology has sparked a revolution in immunotherapy, offering unprecedented precision and versatility in combating a wide range of diseases. Its ability to deliver genetic instructions directly to cells has unlocked a new era of targeted immune responses.
Personalized Vaccines Encoding Tumor
Antigens: mRNA vaccines can be tailored to encode specific tumor antigens found in a patient's cancer, triggering a potent and personalized immune response that targets their unique cancer cells. For example, injecting mRNA blueprints for the KRAS mutation in a pancreatic tumor can train T cells and B cells to eradicate those specific cancer cells.
Engineering CAR T Cells with Exquisite Precision: mRNA technology is transforming CAR T-cell therapy by enabling the rapid and efficient engineering of T cells with tumor-targeting receptors, without the need for complex viral vectors. This empowers T cells to directly attack and destroy cancer cells with remarkable precision, acting like laser-guided missiles.
Modulating the Immune Landscape: mRNA can also be used to manipulate the broader immune environment to enhance anti-tumor activity. For instance, delivering mRNA
encoding IL-12 into dendritic cells amplifies their antigen-presenting function, leading to greater T cell activation. Conversely, mRNA targeting immunosuppressive factors like PD-1 can unleash the full killing potential of T cells, further strengthening the immune response.
Beyond Oncology-Infectious Diseases and Autoimmunity:
The impact of mRNA immunotherapy extends beyond cancer. It is being explored for infectious diseases like influenza and rabies, with promising efficacy and scalability compared to traditional vaccines. In autoimmune diseases, mRNA holds potential for inducing tolerance towards self-antigens, potentially halting the body's attacks on itself. For example, delivering mRNA encoding a modified autoantigen could train the immune system to recognize it as harmless, offering hope for diseases like rheumatoid arthritis and multiple sclerosis.
Precision Medicine Unleashed
The process of creating a personalized mRNA cancer vaccine involves several key steps. Firstly, tumor samples are collected from the patient, and these samples are then sequenced to identify specific tumor antigens or neoantigens that are unique to the patient's cancer cells. This sequencing helps in identifying the genetic mutations and the resulting abnormal proteins that can be targeted by the immune system. Once these unique antigens are identified, a personalized mRNA vaccine is developed to target them. The vaccine is customized to encode for the specific neoantigens identified
www. pharmafocusamerica.com 17
RESEARCH & DEVELOPMENT
in the patient's tumor. This customization is a crucial aspect of personalized mRNA vaccines, as it allows for the creation of a vaccine that is tailored to the genetic profile of each patient's tumor, enabling a precisely targeted immune response against the cancer cells. The development of the personalized mRNA vaccine involves the design and production of the mRNA sequence that encodes the identified neoantigens. This process is followed by the production of the vaccine and its administration to the patient. The vaccine is then used to prompt the patient's cells to produce the cancer-specific neoantigens, which in turn induce an immune response against the cancer cells. The personalized nature of mRNA vaccines facilitates a precisely targeted immune response, ensuring minimal impact on healthy cells. The ability to customize vaccines based on the individual genetic profile of each patient's tumor holds immense potential for personalized cancer therapy, harnessing the patient's own immune system to specifically target and eliminate the cancer cells. The process of creating a personalized mRNA cancer vaccine is a complex and tailored approach that has shown significant promise in the field of cancer immunotherapy.
One notable success story is a personalized mRNA vaccine against pancreatic cancer, which showed a strong anti-tumor immune response in half of the participants in a small study. The researchers used gene sequencing on tumor samples to find proteins that might trigger an immune response and then created a
personalized mRNA vaccine for each patient, targeting up to 20 neoantigens. Customized vaccines were successfully created for 18 of the 19 study participants, and the process has shown promising results.
Another example is a personalized mRNA cancer vaccine for melanoma, which combined with immunotherapy reduced the risk of recurrence by half. This randomized, controlled trial is the first to show a benefit from this type of cancer vaccine. The study's results are considered "exciting" and "an important advance in the field of cancer vaccines".
More than twenty immunotherapies based on mRNA have progressed to the clinical trial stage, and the outcomes of these trials have been promising in the treatment of solid tumors. mRNA vaccines provide a considerable
Messenger RNA (mRNA) tech promises to revolutionize cancer treatment, with ongoing research, clinical trials, and regulatory advancements offering hope and transforming healthcare.
18 PHARMA FOCUS AMERICA ISSUE 03 - 2024
RESEARCH & DEVELOPMENT

edge over anti-cancer immunotherapies when it comes to personalization and targeted delivery to specific tissues and cell types. In summary, the success stories and promising results in the field of mRNA-based immunotherapy demonstrate the potential of this technology to revolutionize cancer treatment and offer new hope for patients and healthcare advancements globally.
Overcoming Challenges
mRNA-based immunotherapies have shown significant promise in cancer treatment, but there are still some challenges and concerns that need to be addressed. One of the primary challenges is the instability of mRNA, which
can be degraded by nucleases and has a short half-life. This instability can limit the effectiveness of mRNA-based vaccines and therapies. Another challenge is the efficient delivery of mRNA to target cells, which can be difficult to achieve. The immune system can also recognize mRNA as foreign, leading to an immune response that can reduce the effectiveness of the therapy. Despite these challenges, ongoing research and development aim to address these concerns and further harness the capabilities of mRNA. Modifications to the mRNA structure and delivery methods have been made to improve stability and delivery efficiency. Additionally, the use of adjuvants and other immune-stimulating agents
www. pharmafocusamerica.com 19
RESEARCH & DEVELOPMENT
can help to enhance the immune response to mRNA-based vaccines and therapies. Several clinical trials are currently underway to assess the safety and efficacy of mRNAbased immunotherapies in cancer treatment. These trials are evaluating the pharmacological, dosing, and immunogenic features of mRNA vaccines, as well as larger-scale evaluations of their potential to mitigate tumor recurrence or enhance survival rates
Ongoing research efforts are focused on addressing the challenges and concerns related to mRNA-based immunotherapies. One of the primary challenges is the instability of mRNA, which can be degraded by nucleases and has a short half-life. Recent research has focused on chemical modifications of mRNA to improve stability, such as optimizing the 5′ cap structure and the 3′ poly(A) tail length, and regulatory regions.
Additionally, advanced delivery systems, such as cell-penetrating peptides, hydrogels, polymer-based nanoparticles, and dendrimers, have been investigated to increase the delivery efficacy and immunogenicity of mRNA.
Another challenge is the immune system's recognition of mRNA as foreign, leading to an immune response that can reduce the effectiveness of the therapy. Strategies to control the innate immune activity of conventional and self-amplifying mRNA therapeutics are being developed, such as modifications to the mRNA backbone itself, optimization of production and purification processes, and the combination of mRNA
with adjuvants and other immune-stimulating agents.
Clinical trials are currently underway to assess the safety and efficacy of mRNA-based immunotherapies in cancer treatment. These trials are evaluating the pharmacological, dosing, and immunogenic features of mRNA vaccines, as well as larger-scale evaluations of their potential to mitigate tumor recurrence or enhance survival rates.
Clinical Milestones and Breakthroughs
Personalized mRNA Cancer Vaccines take the form of tailor-made weapons, crafted with the patient's own tumor in mind. Imagine identifying the enemy's unique flags (tumorspecific antigens, TSAs) and forging vaccines encoded with their blueprints. BioNTech's BNT111, wielding up to 34 TSA flags, is already in Phase II trials against melanoma and colorectal cancer, a testament to the personalized approach.
Neoantigen mRNA Vaccines delve deeper, seeking out the enemy's hidden mutations –neoantigens. These are like secret insignia worn by individual cancer cells and targeting them can unleash a more potent immune response. Moderna's mRNA-4157, currently in Phase II for melanoma, exemplifies this strategy, promising to strike at the heart of each patient's unique tumor identity.
Beyond direct attacks, mRNA can also supercharge the immune system's own troops. Imagine delivering mRNA encoding cytokines,
20 PHARMA FOCUS AMERICA ISSUE 03 - 2024
RESEARCH & DEVELOPMENT
the immune system's chemical messengers, directly to the battlefield. Ziopharm's Zilucovax does just that, utilizing mRNA for IL-12 to rally the troops against melanoma and head and neck cancers, with promising early results.
CAR-T cell therapy, where immune T cells are engineered to recognize and attack cancer, receives a boost from mRNA as well. Platforms like CARsgen's deliver mRNA for IL-15, providing T cells with an extra energy pack to fight harder and longer. This combined approach, in Phase I/II trials for various cancers, promises to turn CAR-T into an even more formidable force.
But the most futuristic weapon in this arsenal might be mRNA for gene editing and repair. Imagine sniping out the very mutations that cause cancer or disabling the genes that fuel its growth. Editas Medicine's EDIT-201, designed to edit a gene causing Leber congenital amaurosis, is a glimpse into this future, with potential applications in some cancers.
Regulatory Landscape
The regulatory environment surrounding mRNA-based therapies is still evolving, with the FDA having approved the first mRNA vaccines for COVID-19 from Pfizer/ BioNTech and Moderna. However, the path to approval for upcoming mRNA therapeutics remains unclear, and biopharma companies face challenges in navigating the regulatory landscape. Over 250 potential mRNA therapies
are currently being investigated in cancer, with more than 500 in other indications.
Some recent regulatory approvals and their significance include:
COVID-19 vaccines: The approval of mRNA vaccines from Pfizer/BioNTech and Moderna marked a significant milestone in the field of mRNA-based therapies. These vaccines have demonstrated high efficacy and safety in preventing COVID-19 and have saved countless lives worldwide. Despite these approvals, there are still challenges and concerns related to mRNA-based therapies, such as stability, delivery, and immunogenicity.
Ongoing research efforts are focused on addressing these concerns and further harnessing the capabilities of mRNA-based therapies. The biopharma community remains uncertain about how mRNA therapies will be classified, and the lack of clarity in the regulatory environment can lead to delays in approval and higher drug prices. The fast rise in the development of mRNA therapies has left some concerned that therapeutic development is outpacing the regulatory environment. As more mRNA therapies enter clinical trials and their development progresses, the regulatory landscape will continue to evolve, and the approval process for future mRNA-based therapies will become more streamlined.
The significance of these approvals lies in the validation of mRNA-based therapies as a
www. pharmafocusamerica.com 21
RESEARCH & DEVELOPMENT
viable and effective treatment modality, paving the way for more mRNA therapies to reach the market and benefit patients worldwide.
Conclusion
In conclusion, mRNA immunotherapy represents a promising new frontier in cancer treatment. The ability to personalize vaccines and target specific tumor antigens has shown great potential in clinical trials for various cancer types. While there are still challenges to overcome, such as mRNA instability and efficient delivery, ongoing research and development efforts are focused on addressing these concerns and further harnessing the capabilities of mRNA. The recent regulatory approvals of mRNA vaccines for COVID19 have validated mRNA-based therapies as effective treatment modalities, paving the way for the development and approval of future mRNA therapies.
The potential of mRNA immunotherapy extends beyond cancer treatment, with ongoing

research exploring its use in infectious diseases, genetic disorders, and other conditions. The versatility and precision of mRNA technology offers new opportunities for healthcare advancements globally. However, navigating the regulatory landscape remains a challenge for biopharma companies, and the lack of clarity in the regulatory environment can lead to delays in approval and higher drug prices. As more mRNA therapies enter clinical trials and their development progresses, the regulatory landscape will continue to evolve, and the approval process for future mRNAbased therapies will become more streamlined.
Overall, the success stories and promising results in the field of mRNA-based immunotherapy demonstrate the potential of this technology to revolutionize cancer treatment and offer new hope for patients. With ongoing research and development efforts, the future of mRNA immunotherapy looks bright, and we can expect to see more breakthroughs in the years to come.
Vidya Niranjan, Ph.D. is a leading scientist and academic researcher excelling in computational biology. She has worked extensively on genome analysis, drug discovery, tools and database development. With extensive research experience of over 20 years, she has published over 100 research articles. She has bagged research funding worth 42 million USD from various government agencies and pharmaceutical companies. She is now a part of NVIDIA inception program on AI based drug discovery. She is currently working on project related to quantum computing and protein folding funded by the Ministry of Electronics and Information Technology (MEITy) is an executive agency of the Union Government of the Republic of India in collaboration with Amazon AWS.
22 PHARMA FOCUS AMERICA ISSUE 03 - 2024
AUTHOR BIO
RESEARCH & DEVELOPMENT
Recombinant Antibody Technology is Crucial to Past and Future Success in Biopharma
Recombinant antibody technology addressed many problems associated with hybridoma platforms, opening the door to a new class of biologic drugs. Today, this technology continues to contribute to the discovery and development of new antibody-based therapeutics and improve the performance of novel modalities.
Many Drivers for the Recombinant Approach
Early monoclonal antibody (mAb) therapies were developed using the hybridoma platform, which requires production of the target antigen or immunogen and involves animal immunizations. The hybridoma technology suffers from not only ethical issues but also batch-to-batch variability, antibody heterogeneity, loss of antibody productivity, and limitations with respect to scale and cost. In addition, the process is timeconsuming and typically takes multiple months from immunization to establishment of specific hybridoma clones and production of mAbs.
Recombinant antibodies are generated from host cell lines by recombinant DNA technology. This approach not only greatly reduces the reliance on animal use but also enables the production of large quantities of products much more quickly — within just a few weeks in most cases. Since recombinant antibodies are based on the known DNA sequences and can be exactly replicated, their production is highly controlled, reproducible, and consistent. Moreover, recombinant antibodies offer advantages including targeting hybridoma-refractory antigens and amenability to antibody engineering.
Many Advantages of Engineering
Recombinant antibody engineering provides many opportunities for creating unique molecules with optimal attributes for treating specific diseases. The fragment crystallizable (Fc) region can be modified to prevent the initiation of any undesired responses, while tiny variable regions can be designed to bind
to the target more effectively. The antibodies initially discovered in mice or other animals can be humanized to reduce potential immunogenicity.
In addition, years of experience inspires confidence about which changes are necessary to realize specific benefits. As artificial intelligence (AI) algorithms advance, the level of predictability will only increase. In fact, many drug developers are leveraging AI technologies to predict and analyze potential therapeutics from antibody sequences before production. Then high-throughput recombinant antibody production and characterization to validate the predictions in wet lab further accelerate the discovery and development. Platform processes, meanwhile, allow for rapid scale-up of recombinant antibody production to meet clinical and commercial demand.
Fragment Understanding
One of the important advantages of recombinant antibody technology is the ability to explore and understand many flavors of mAbs limited only by the imagination. Recent years have witnessed the rise of engineered antibody fragments as biopharmaceuticals, such as monovalent formats (e.g., Fab, scFv, and VHH) and bispecific constructs (e.g., BiTE and Diabody).
The conventional mAbs have certain limitations in clinical applications. Immunoglobulin G (IgG) molecules are relatively large (~150 kDa), which can prevent them from effectively penetrating into tissues. In addition, the presence of Fc region in the IgG molecule can mediate bystander activation of the immune system. However, antibody fragments not only retain the targeting specificity of intact mAbs but also possess superior properties for a range of therapeutic and diagnostic applications. Due to their small size, antibody fragments offer enhanced tissue penetration, the ability to bind to traditionally inaccessible epitopes, and reduced immunogenicity. Additionally, these fragments provide flexibility in structure, ease of production and engineering, and serve as building blocks for novel constructs.
Production and purification of antibody fragments require extensive expertise especially with the increase
www. pharmafocusamerica.com 23
in their design complexity and diversification in recent years. A combination of aspects should be carefully considered, including design strategies, how the upstream production can offer high productivity while suppressing the generation of impurities, and what downstream processing requirements will likely be involved in producing the desired fragment in a highly pure form.
Different Modifications for Research, Diagnostic, and Therapeutic Applications
While antibodies for research, diagnostic, and therapeutic applications must all exhibit high binding affinity and specificity, there are other aspects that are uniquely sought for each specific application. When biomedical researchers require antibodies that can penetrate different tissues to assess different molecular and behavioral characteristics, antibody fragments with better tissue penetration than the full-length IgG are ideal for these applications. In the field of diagnostics, engineered multivalent antibodies with enhanced antigen-binding avidity are promising agents. For therapeutic applications, recombinant antibodies can be modified in endless ways: to avoid cross-reactivity, to target effector cells more strongly, to enable sitespecific conjugation to small molecule payloads, and to achieve many other desired characteristics.
Significant Potential Yet to be Unlocked
While there is tremendous excitement about the therapeutic possibilities presented by cell and gene therapies and other novel modalities, recombinant antibodies still have significant potential going forward. Recombinant antibodies, for instance, will continue to play a critical role in advancing treatments for various human diseases, including cancers, autoimmune, metabolic and infectious diseases, because they enable targeted therapies with minimal side effects. Beyond mAbs, bispecifics, antibody fragments, antibody–drug conjugates, and other next-generation antibody therapeutics exhibit novel functionalities and have even greater targeting abilities and thus enhanced efficacy. From a diagnostic standpoint, recombinant antibodies allow the highly selective and sensitive detection of specific biomarkers present at varying disease stages and thus facilitate the development of more tailored treatments focusing on disease progression.
Innovations in Targets and Modifications Expected
The recombinant antibody sector will continue to grow and expand to include novel targets and modifications. With a deeper insight into genomic and
proteomic changes in diseases, many new targets have emerged, for which no antibody drugs are available. There’s a considerable gap between target discovery and corresponding therapeutic antibody development. To address the pressing need for antibodies to be developed for these novel entities, recombinant antibodies enabling precise design, powerful engineering, and efficient production have a widening application in medicine.
Greater understanding of the design landscape, investigating different known and unknown opportunities, and identifying the best possible engineering steps required are critical to provide the optimal affinity, specificity, and other properties necessary for a given target or application. In this way, we expect the near future to include advances that further revolutionize the recombinant antibody universe and antibody therapies will likely be available for targets that are unthinkable today.
The future of recombinant antibodies is exciting. There is, of course, always room for improvement. It is key to keep asking questions. Continuous inquiry leads to more information and increases our understanding of all aspects of recombinant antibody technology. At Sino Biological, we believe that this continual exploration will lead to compelling answers and ultimately novel technologies that facilitate the discovery, development, and production of novel recombinant antibodies that can change patient lives
The Role of Recombinant Antigens and Antibodies in the COVID-19 Pandemic Response
The COVID-19 pandemic represented a tragic time with respect to the loss of life around the world. It also, however, showed the public many positive aspects of the biopharma industry that are normally not apparent. Sino Biological remained very busy throughout the pandemic producing research reagents and providing contract research services for biopharma and
24 PHARMA FOCUS AMERICA ISSUE 03 - 2024
diagnostic industries to help advance therapeutics, vaccines, and immunodiagnostic assays for the SARS-CoV-2 virus. The company has produced a comprehensive collection of recombinant antigens and antibodies for SARS-CoV-2 and supported scientists worldwide for their SARS-CoV-2 research, which led to over 2,000 publications to date.
In the race against time to fight the pandemic, Sino Biological is extremely fast at developing reagents and implementing contract research services. For example, in January 2020, the company produced the key SARSCoV-2 spike protein reagents within a record 11 days; then, in December 2021, it developed the Omicron RBD protein in a new record of 6 days. They were developed and manufactured using the company’s proprietary recombinant platforms. Since the COVID19 outbreak, Sino Biological has been actively tracking the change of new variants to make sure products are "up to date" and its SARS-CoV-2 reagent portfolio fully covers the variants of interest (VOIs) and variants under monitoring (VUMs). Sino Biological has developed the world’s largest viral antigen bank, ProVir®, antibodies for almost all of these viral antigens, and even antibody pairs with high specificity and sensitivity, which have a significant impact on vaccine, therapeutic, and diagnostic research in areas like COVID-19, influenza, RSV, and many other fields of virology and immunology.
Sino Biological: Building Recombinant Antibody Expertise for Over 15 Years
At Sino Biological, recombinant technology has been not only leveraged from the outset to generate catalog protein and antibody products but also widely employed in the company’s contract development and

manufacturing business. For both applications, there is a powerful drive to continually fine-tune and advance the company’s capabilities. Sino Biological has been highly involved in supporting COVID-19 therapy and vaccine developers and continues to provide custom services for novel therapy and diagnostic product developers.
Sino Biological has comprehensive expression platforms in place to meet the diverse needs of customers, ranging from cell-based production platforms, such as mammalian cell and baculovirus–insect cell expression systems, too fast and efficient cell-free platform.
Customers can benefit from Sino Biological’s many years of experience working not only with IgGs but also dimeric IgAs, multivalent IgMs (pentameric or hexameric), antibody fragments, and bispecifics.
New US-based Center for Bioprocessing
Headquartered in Beijing, with subsidiaries in the United States, Europe, and Japan, Sino Biological has over 900 employees and serves researchers in academia and industry worldwide. In October 2023, the company announced the formal opening of its new Center for Bioprocessing (C4B) in Houston, Texas USA, which marks a significant milestone in Sino Biological’s global presence. The new center specializes in CRO services including custom recombinant protein and recombinant antibody development and manufacture. The C4B represents Sino Biological’s natural global expansion of its CRO service capabilities and extends upon the company’s already strong CRO service offering at its Beijing headquarters. The team is committed to delivering high-quality, custom recombinant proteins and antibodies, and looks forward to partnering with researchers and industry leaders worldwide to forge a brighter future in the life sciences.
With over a decade's expertise in molecular biology, Dr. Suranjana Sen, a Ph.D. holder from Illinois State University, excels in advancing scientific knowledge. Currently serving as a Technical Account Manager at Sino Biological US Inc., she strategically expands business opportunities through profound scientific knowledge, ensuring seamless collaboration with biopharmaceutical clients. Previously, as a Technical Services Scientist at Promega, Dr. Sen showcased exceptional problem-solving and customer service skills. During her postdoctoral tenure at Loyola Medical Center, she conducted groundbreaking research on HBV infection dynamics, utilizing experimental systems and computational models to advance our understanding of virus-associated liver disease.
www. pharmafocusamerica.com 25
Advertorial
Revitalizing Research
Prioritizing Participantcentric Strategies to Accelerate Clinical Trials
The future success of clinical research is contingent on the prioritization of the participant experience. From crafting protocols to engaging participants across multi-year studies, the participant experience must consistently be a central focus throughout the clinical trial journey. The "why" of clinical trials must remain centered on patients, influencing protocol development, and emphasizing participant education for optimal recruitment and retention.
We all know someone – a loved one, friend, or close acquaintance – who has struggled with a health condition that upended their life and changed their perception of “normal.” These individuals often endure great physical and/or mental pain daily.
Clinical trials offer a powerful approach; they can restore a sense of control to patients that may have
Amy Thue
Associate Director
Project Management, WCG
Shelby Ward
Manager, Site Services, WCG
26 PHARMA FOCUS AMERICA ISSUE 03 - 2024
CLINICAL TRIALS

previously felt unattainable. Not only can they provide a potential treatment, but equally important, they can be a source of hope. This is why clinical trial design needs to focus on the patient’s needs, and clinical trial execution must always center on the participant.
Clinical trial success begins with cultivating trust.
Cultivate Trust
Never underestimate the importance of public perception of clinical trials. It has a direct effect on people’s willingness to participate. Regulations, regulatory agencies, and institutional review boards ensure the safety and
ethics of these trials, but they merely establish the baseline. It falls upon the sites and sponsors to cultivate trust among potential participants, who stand to benefit most from the treatments being developed.
The COVID-19 pandemic serves as an excellent case study for how to effectively build trust. One could argue that during the pandemic, the public had greater visibility into clinical trials than at any other time in recent history. Also, during this period, public opinion was sharply divided. On one hand, the rapid development of COVID-19 vaccines demonstrated the value of clinical research and drug development in response to public health threats. On the other hand,
www. pharmafocusamerica.com 27
CLINICAL TRIALS
the circulation of misinformation, primarily through social media platforms, contributed to distrust of – and even active opposition to – clinical trials.
The clinical trial community has made significant advancements in patient engagement. Nevertheless, distrust persists, and those who would most benefit from clinical trials will suffer.
The 2023 WCG Avoca State of the Industry Report found that 75% of patient respondents had never participated in a clinical trial and didn’t know anyone who had. When the survey asked these research-naïve patients their opinion on “I believe clinical trials only benefit the pharmaceutical companies that run them,” 36% agreed, 36% disagreed, and 29% were neutral.
These findings underscore a clear need for the clinical research industry to prioritize patient education about clinical trials. Understanding the purpose, process, and results of these trials is crucial not only for the application of current treatments but also for addressing future diseases that lack effective standardized treatment options. Moreover, building trust through patient education can improve both recruitment and retention.
It begins with outreach.
Educate and Engage the Community
To cultivate trust and interest in clinical trials, community outreach and education are essential. Community engagement, which includes
teaching patients what it means to be part of a clinical trial, is crucial to recruitment, enrollment, and retention.
The first step is to identify individuals and organizations with strong ties to the local community. These trust bearers could be local physicians and clinics, but they might also be places of worship, community support groups, social workers, or community centers. Understanding where potential participants spend their time – barber shops, local libraries, colleges, or festivals – is also key to effective engagement.
The next step is reaching out to them. By partnering with these trusted networks and individuals, sponsors can share information with the community through a trusted source. But it’s not just about getting in front of an audience, talking about a trial, and departing with the assumption that people will reach out if they are interested; it’s about building relationships. Consistent engagement is vital. It’s unlikely that a community leader will provide access to potentially vulnerable individuals after just one short informational meeting.
Advocating for clinical research is a longterm investment. But it is worth it. The more time a sponsor and/or site invests, the more likely they are to build meaningful, lasting connections.
Successful outreach often results from longterm relationship building and collaborative efforts. When implemented with sensitivity, these relationships not only aid in the success
28 PHARMA FOCUS AMERICA ISSUE 03 - 2024
CLINICAL TRIALS
Revitalizing clinical trials means prioritizing a participant-centric approach, shaping protocols, and emphasizing education for optimal recruitment and retention.
of the current study but also lay a foundation for participant enrollment in future studies.
Engagement is a conversation: It requires listening to patients and their support networks, not just talking to them.
Put Patients First
Patients live and breathe their condition every day. That’s why protocol development, including the schedule of events and study assessment selection, should always focus on the patient. Sites know their patients and the obstacles they face. Considering their firsthand knowledge and close interaction with study participants, sites should be recognized as subject matter experts on their patients.
Patients should be viewed as providing critical input that can help sponsors, clinical research organizations (CROs), and other stakeholders keep those obstacles top of mind when developing protocols.
Factors such as finding daycare, arranging transportation, and loss of work hours are all issues patients might consider before consenting to a clinical trial. Sometimes a stipend helps, but it may be inadequate to compensate a patient for wages lost. Or perhaps the individual lacks the means to arrange for an eight-hour on-site screening visit. In the 2023 WCG Avoca State of the Industry Report, 57% of the trial-naïve patients cited virtual visits as a motivation to participate in a clinical trial. In addition, 71% cited transportation or transportation reimbursement as a motivation, and 45% cited childcare or childcare reimbursement.
Offering transportation assistance, childcare, meals during longer visits, reimbursement for parking costs, and other tactics can also lessen the burden for the patient and make participation possible. Lessening the financial and logistical burden on the patient goes a long way toward turning patients into clinical research participants.
Keep in mind that the barriers and burdens often vary by patient population; site teams have the experience and expertise to help sponsors understand patient needs.
Leverage Site Expertise from the Start
Sites understand their patient populations and they have insights into what might deter patients from or motivate them to consider clinical trial participation.
Collaborating with sites on study design is one way to incorporate the patient perspective
www. pharmafocusamerica.com 29
CLINICAL TRIALS
from the beginning. Site teams can help sponsors and CROs promptly identify and address protocol design issues. This is not a novel concept, but one that few sponsors and CROs have implemented.
Sponsors are missing a wealth of insight.
By integrating site expertise at the beginning of the protocol development process – and maintaining it throughout the trial – sponsors and CROs can learn about potential challenges to patient participation. For example, such collaboration allows for the creation of protocols and assessments that take into consideration the demands of patient-reported outcomes and the time intensity of patient assessments. This approach helps sponsors support patient convenience while still capturing the data they need to assess safety and efficacy to evaluate study endpoints.
Sponsors will be able to expedite enrollment because patients will see that the benefits of participation outweigh the burdens. The sites will have the opportunity to collaborate with the sponsor and vendors on trial design, helping them to feel more involved in the trial from the beginning. This, in turn, will increase enthusiasm for the trial and keep the study top of mind when speaking to potential participants.
Engage with the Site
Effective participant engagement begins with selecting the right sites. Only a small percentage of practicing physicians participate in clinical research. The most frequently cited figure is 3%, but even twice that would be inadequate.
Potentially more concerning, a 2021 analysis by the Tufts Center for the Study of Drug Development found that 66% of principal investigators leave clinical research after just one trial.
Clearly, there is a pressing need for new investigators and sites.
To reach the right participant populations, sponsors need to encourage research-naïve practices and centers to consider clinical research. Not only will this broaden the base of principal investigators taking part in clinical studies, but it will also increase participant diversity.
Support Site Recruitment and Retention Efforts
Whether a site has years of clinical trial experience or is just starting its first trial, it will undoubtedly have valuable knowledge of the motivations of their patient demographic to participate in clinical research. So just as sponsors should consult sites on protocol design, they should solicit site input on developing recruitment and retention plans.
Experienced sites bring an abundance of knowledge in patient motivation and demographic-specific strategies. Researchnaïve sites offer fresh perspectives and innovative approaches.
Both perspectives are invaluable. Integrating these diverse insights helps sponsors and sites develop effective recruitment and retention strategies. It also creates opportunities for sites to share best practices.
30 PHARMA FOCUS AMERICA ISSUE 03 - 2024
CLINICAL TRIALS
For example, sometimes sponsors ask their most senior research sites to support more novice ones as mentors or “site champions,” sharing their insights on a particular patient population and helping the smaller sites identify the most promising recruitment pathways.
Such an approach can boost the overall site engagement and morale. It also provides an opportunity for sponsors and CROs to identify and address any gaps, ensuring efficient site operation and goal achievement.
When sponsors invest in creating more collaborative platforms for discussions among sites, CROs and sponsors, they pave the way for success. This level of collaboration fosters better site engagement, boosts recruitment, supports more efficient study workflows from the start, and ultimately, accelerates research.
However, none of this happens if sites lack the support they need.
Bolster Site Capacity
Nearly half (48%) of respondents to the WCG 2023 Clinical Research Site Challenges Survey Report identified patient recruitment and enrollment as a top concern. This suggests sites and sponsors would benefit from proactively defining the requirements for recruitment and retention responsibilities on a study, creating the opportunity to identify capacity or operational gaps.
Most sponsors and CROs today understand that the success of their studies depends on the performance of their sites. But they may not fully appreciate how much sites are struggling with bandwidth issues. When sites lack the necessary overall team capacity to support a study, they may miss enrollment targets and important milestones.
Empowering patients with a sense of control and offering hope, clinical trials are a beacon of potential treatments.
Meanwhile, demand continues to outpace capacity. The number of clinical trials continues to grow exponentially, and they are increasingly complex. For example, Tufts Center for the Study of Drug Development noted a threefold rise in the number of data points collected in Phase III trials from 20132020.
WCG’s 2023 Site Challenges Survey found that 51% of respondents work on more than 26 studies at any given time, and 21% manage more than 150 trials at once. This portfolio of studies is in addition to their regular patient care responsibilities. Further,
www. pharmafocusamerica.com 31
CLINICAL TRIALS
63% of respondents to the survey identified staffing and retention as their most significant concern. That should come as no surprise: Nationwide, for every experienced clinical research coordinator seeking work, there are seven jobs posted. For clinical research nurses, the ratio is 1:10 and, for regulatory affairs professionals, 1:35.
These stats suggest that many sites lack the staffing capacities to fully dedicate team members to any one specific study. Instead, team members are likely juggling multiple studies, constantly shifting their priorities amongst varied complex projects.
Exacerbating the capacity issue, clinical trial protocols have become increasingly complex. For example, Phase II and III protocols average 263 procedures per patient, supporting approximately 20 endpoints. Many sites struggle to meet these escalating demands. For example, in the United States, more than 80% of clinical trials fail to achieve their enrollment targets, and 30% of study participants leave the trial.
AUTHOR BIO

Amy Thue, Associate Director, Project Management, at WCG has been in the life sciences industry for more than 15 years. She worked in clinic as a medical assistant and study coordinator prior to her role at WCG. Patient-focused care delivery has always been a passion of hers.
Sites need sponsor support. Research-naïve sites in particular may need experts to lean on while they get up and running. Working closely with sites allows sponsors to identify ways to help. Providing resources that allow for smooth and effective operations at sites will benefit the sponsor, site, and patients.
To Support Patients, Support Sites
Putting the patient’s perspective first is both the right thing to do and a smart strategy. It allows sites to recruit engaged participants, boosting enrollment and retention rates, and increasing the likelihood of a trial’s success. Site expertise is essential.
By integrating site insights into trial design and execution and ensuring they have the resources they need, sponsors and CROs can help sites improve efficiency, accelerating research and getting new therapies in the hands of patients sooner.
References are available at www.pharmafocusamerica.com

Shelby Ward is a Manager, Site Services for WCG, leading a team of professionals that are dedicated to propelling the clinical trial industry forward through partnering hand in hand with study sites and sponsors. Her dedication to the patient experience is at the core of everything Shelby does.
32 PHARMA FOCUS AMERICA ISSUE 03 - 2024
CLINICAL TRIALS
Applications and the Limitations of RealWorld Data in Gene Therapy Trials
In the quest to address rare diseases, one of the most complex and crucial areas in healthcare, the industry is constantly in search of innovative approaches. Enter Real World Evidence (RWE) studies, which are changing the clinical trial landscape in a big way. Contrasting with the controlled environment of Randomized Clinical Trials (RCTs), RWD is an integral part of a decentralised trial approach and offers a unique perspective on the effectiveness of Investigational Medicinal Products (IMPs) in diverse, real-world scenarios. This data, often overlooked in traditional research paradigms, provides crucial insights into patient outcomes and treatment effectiveness across various populations, driving a new era of data for researchers, medical professionals, and patients. Furthermore, RWD can reveal how an IMP interacts with other medications, a factor often overlooked in controlled trial settings. In this article, Karen Ooms, Joint Chief Operating Officer at Quanticate, examines the applications and the limitations of real-world data in gene therapy trials.

With an estimated 7,000 rare diseases touching the lives of over 350 million people globally, the challenge is immense. Traditional clinical trials, while valuable, often grapple with the intricacies of such conditions due to limited patient numbers, varied
www. pharmafocusamerica.com 33
Karen Ooms
Joint Chief Operating Officer Quanticate
CLINICAL TRIALS
symptomatology, and scarce historical data, leading to protracted drug development timelines. Increasingly, trials are adopting a hybrid model where they utilize a decentralised approach and collect new RWD from wearable devices and other smart technologies, plus tap into existing patient data from registries and other sources, all while still using traditional site data in their submissions.
This is why many organizations are incorporating RWE studies into their trials, drawing on the capability to gather a multitude of RWD sources such as electronic health records, insurance claims, patient registries, and first-hand patient accounts. This isn't just data; it’s a tapestry of human experiences, offering a deeper, multifaceted understanding of these rare conditions.
However, the path to realizing the full potential of gene therapy is fraught with challenges. Gene therapy, which uses genes to treat or prevent diseases at their most fundamental level, is gaining momentum with technological advancements such as CRISPR/ Cas9. This innovative approach offers potential cures by directly fixing the genetic abnormalities causing these diseases. The rarity of these conditions means smaller patient populations for clinical trials, a limited understanding of disease progression, and difficulty in defining suitable endpoints. These research pain points have led to the introduction of RWE into gene therapy trials, which signifies a pivotal shift in how researchers and clinicians approach the development and evaluation of new therapies.
The Bright Spots of Real World Data
Imagine being able to connect with patients scattered across the globe, to understand their unique stories and struggles. RWD makes this possible, breaking down barriers and bringing a wealth of diverse experiences into the spotlight. It’s not just about numbers and statistics; it’s about understanding the human side of disease progression and crafting treatments that resonate on a personal level. Real World Data is revolutionizing the way we approach gene therapy trials by offering unique advantages: Global Patient Access: One of the foremost benefits of RWE studies is the ability to tap into a vast and diverse patient base, far exceeding the scope of centralized clinical trials, allowing for a deeper dive into patient demographics, disease progression, and treatment outcomes. Utilizing RWD enables researchers to connect with international
Gene therapy trials leverage real-world data for transformative insights into diverse patient outcomes, driving a new era of research.
34 PHARMA FOCUS AMERICA ISSUE 03 - 2024
CLINICAL TRIALS
databases and patient registries, which helps in identifying sub-populations that might benefit most from the therapies, as well as understanding the natural history of diseases by overcoming geographical and logistical barriers, thus enriching data sets and enhancing the relevancy of findings.
Streamlining Drug Development: RWE studies are increasingly acknowledged by regulatory bodies for supplementing clinical trial data, including additional safety and efficacy evidence and identifying potential patient groups. This is especially beneficial for rare diseases, streamlining the development, evaluation, and approval of new treatments, thereby hastening access to novel therapies and enhancing post-marketing surveillance, ensuring that any long-term effects or rare side effects are quickly identified and addressed.
Patient-Centric Care and Cost Efficiency: RWE studies foster a more patient-focused approach to treatment and care. By understanding the real-life experiences of patients, healthcare providers can deliver more tailored and effective treatment strategies. Additionally, patients and caregivers are empowered to make informed decisions about their care, based on a broad spectrum of similar patient experiences. The use of wearable devices to gather RWD also improves patient adherence and reduces the need for travel to clinical sites. Conducting traditional clinical trials for rare diseases is typically costly due to extensive recruitment and study durations. RWE studies offer a cost-effective alternative

by leveraging existing data, accelerating the research process, and lessening the financial strain on healthcare systems and patients.
Real World Success Stories
The Impact of RWD in Gene Therapy: The application of RWD is particularly significant in the field of gene therapy. Understanding the long-term effects and variability of treatments is critical, and RWD offers longitudinal data that aids in tracking these outcomes and adapting strategies accordingly. For rare genetic disorders, this translates into more individualized and effective treatment approaches, informed by extensive real-world patient experiences. Several gene therapy trials have already benefited from the integration of RWD. For instance, in trials for rare genetic
www. pharmafocusamerica.com 35
CLINICAL TRIALS
disorders like Cystic Fibrosis and Hemophilia, RWD has been instrumental in understanding the disease's natural history, identifying patient subgroups, and tailoring interventions accordingly. These success stories underscore the potential of RWD to refine, accelerate, and enhance the development and delivery of gene therapies.
Response to Global Challenges: The COVID-19 pandemic highlighted the adaptability of RWD. Strict lockdowns, social distancing and self-isolation meant patients were unable to get to clinical sites. As the world struggled with the virus, RWD provided timely insights into its behavior, treatment responses, and vaccine effectiveness. This underlines the potential of RWD in tackling global health emergencies, offering valuable lessons applicable in the realm of rare diseases. The use of digital technologies helped overcome the challenges set out by the pandemic and have created new trends in the way Clinical Research Organizations (CROs) and pharma/biotech conduct clinical trial analysis.
Advancements in digital data-capturing technology have fundamentally transformed the landscape, steering the market away from conventional paper-based record-keeping. These technological innovations facilitate more patient-focused approaches in study conduct, as Clinical Research Organizations (CROs) and sponsors increasingly utilize these technologies to gather data beyond clinical sites. With the industry trending towards virtual, decentralized,
and hybrid trial methodologies, it's crucial for CROs and sponsors to gain a thorough understanding of these approaches, and be wellrehearsed in how to handle RWD, analyse this data type and design RWE trials. There are many sources of RWD such as longitudinal databases, medical records and data from other new device sources, so ensuring proficiency in how to design trials and handle these various data sources is key.
Navigating the Challenges
Admittedly, while the use of RWD is promising, it's not without limitations. There’s the challenge of making sure the stories we gather are accurate and not muddled by other factors. Ensuring the quality and privacy of this data is paramount, akin to protecting the most personal and sensitive aspects of a patient’s life journey. The representativeness can also vary significantly, potentially leading to biases or inaccurate conclusions. The potential for 'dirty data' – data not rigorously checked for inconsistencies or errors – is another issue. With the vast amount of data available, there is also risk of overanalysis, leading to conclusions that may not be supported by the data.
Technological advancements in data analytics, artificial intelligence, and machine learning are revolutionizing how we interpret vast datasets, but as we continue to embrace this wealth of information, it’s crucial to remember that at its core, it’s about people – their experiences, their struggles, and their requirements.
36 PHARMA FOCUS AMERICA ISSUE 03 - 2024
CLINICAL TRIALS
Regulatory acceptance of RWD is rapidly evolving, with authorities scrutinizing the methodologies and validity, and also emphasizing the need for stringent guidelines to ensure its ethical use. This involves clear protocols for data collection, usage, and sharing, ensuring that patient rights and confidentiality are never compromised. To overcome these challenges, stakeholders in the gene therapy field must advocate for standardization in data collection and analysis methods, invest in robust data infrastructure, and foster collaborations between academia, industry, and regulatory bodies.
It’s important to note that the integration of RWD into gene therapy trials doesn’t diminish the significance of traditional randomized, placebo-controlled trials (RCTs). Rather, it provides a complementary perspective, creating a richer, more nuanced understanding of treatments, their impacts on patients with rare diseases, and the application of therapeutic interventions. It's essential to strike a balance between the rigor of RCTs and the expansive insights offered by RWD. This approach allows for a more nuanced and comprehensive evaluation of IMPs, catering to the diverse needs and contexts of patients suffering from rare diseases.
The Road Ahead
As we look to the future, the story of RWD in gene therapy is one of promise and potential. With technological advancements in data collection and analytics, RWD will become
even more robust and insightful. It promises to not only enhance the design and execution of gene therapy trials but also ensure that therapies are safe, effective, and tailored to those who need them most. It's not just about embracing new technology or data; it's about reshaping our understanding and approach to treating rare conditions, with each patient's story adding a piece to the puzzle, moving us closer to more effective treatments and improved care.
As we continue to navigate this exciting era of genetic medicine, the journey of incorporating Real World Data into gene therapy trials is a testament to an evolving narrative in medical research and patient care. It represents a shift towards a more inclusive, comprehensive, and patientcentric approach to healthcare, blending the rigor of scientific research with the richness of human experiences. While challenges persist, the potential of RWD to enhance our understanding, optimize gene therapy trials, develop more effective treatments, and improve patient outcomes is immense.

AUTHOR BIO
Karen Ooms is a Chartered Fellow of the Royal Statistical Society and has a background in biostatistics spanning over 25 years. Prior to joining Quanticate in 1999 (Statwood), Karen was a Senior Statistician at Unilever.
www. pharmafocusamerica.com 37
CLINICAL TRIALS
Revolutionizing Drug Manufacturing with 3D Printing
Pharmaceutical 3D printing is a pioneering force in pharmaceutical engineering driving innovation, reshaping pharmaceutics from personalised medicine to clinical trial workflows. Providing a comprehensive overview, this article offers insight into 3D printing as a pharmaceutical manufacturing tool and explores its transformative impact in the field.
Anna Worsley CEO, FABRX-AI
Alvaro Goyanes Co-Founder and Director,
FABRX
Hannah Watton Account Manager, FABRX

Introduction to Pharmaceutical 3D Printing
Three-dimensional (3D) printing is an advanced additive manufacturing technique that constructs complex products by layering materials according to a computer aided design (CAD) 3D model. 3D printing has marked a
38 PHARMA FOCUS AMERICA ISSUE 03 - 2024
COVER STORY Cover Story
transformative paradigm shift in the healthcare sector since the 1990s, where it has played a crucial role in the fabrication of custom dental implants and personalised prosthetics, a development that even surprised innovator Charles Hull, cofounder of the pioneering company 3D Systems. 3D printing has also seen significant traction in tissue engineering research, enabling the creation of complex structures using innovative biomaterials to mimic natural tissues. The pharmaceutical sector witnessed the emergence of pharmaceutical 3D printing in the early 2010s, driven by pioneering academics striving to revolutionise medication forms to align with intricate and personalised patient needs (2). Its disruptive nature for pharmaceutical manufacturing holds immense promise for healthcare, poised to fundamentally transform the pharmaceutical industry.
Personalised Medicine and Automated Compounding
3D printing exploded as a manufacturing technique due to its ability to create bespoke objects with unparalleled precision and customisation. Unlike conventional methods, pharmaceutical 3D printing excels in creating complex drug-containing structures and combining multiple drugs into a single pill, unlocking new possibilities in drug development and advanced healthcare that caters to the unique needs of individual patients.
The current method to prepare personalised and non-commercially available prescriptions, pharmaceutical compounding, involves
Pharmaceutical
3D printing pioneers innovation, reshaping drug manufacturing for personalized medicine and streamlining clinical trial workflows.
some-what old fashioned and imprecise techniques, including the breaking of tablets by hand and weighing powders to hand-fill capsules. This not only creates risk but restricts personalised medicine to specific treatments and fewer pharmacies. Pharmaceutical 3D printing automates this process while offering more personalisation options such as shape, colour, flavour and drug combinations (polypills) to increase treatment adherence. Automation reduces specialist workload and human error, making compounding more accessible and easier to implement. With increasing stakeholder investment, this unlocks personalisation for more treatment pathways, moving away from conventional “one-size-fits-all” doses and towards truly personalised medicine. As such, 3D printing paves the way for a new era in personalised medicine, where treatments are prepared to meet the distinct needs of each patient. Imagine
www. pharmafocusamerica.com 39
COVER STORY
a world where a person with polypharmacy can take one multi-drug chewable polypill with a personalised dose rather than five different large capsules, with a colour and flavour that suits their particular preferences.
Pharma-ink refers to the feedstock formulation that includes excipients and drugs to be printed, and is used to print tablets, or printlets. There are multiple 3D printing technologies fit for precision medicine, each using different pharma-ink forms. Semisolid extrusion (SSE) is the most common technique being used by stakeholders. Pharmacists are able to use pastes or gels to print at room temperature, immediate release tablets, chewable tablets or fast disintegrating tablets. Fused deposition modelling (FDM), the most well-known technique, is also being
investigated for use, with researchers developing filaments that are deposited with melting, useful for improving drug bioavailability and complex release profiles. Finally, direct powder extrusion (DPE) was developed as an alternative to FDM. Essentially a mini hot melt extruder, DPE offers the same benefits as FDM while avoiding the difficulties with filament pharma-ink development as it prints straight from powder. The University Medical Center Hamburg-Eppendorf have described the successful development and analysis of Levodopa printlets with rapid dissolution, printed using DPE. In addition, they evaluated the integration of machine-learning assisted medicine 3D printing into their hospital’s workflows. An exciting step forward in the field. (Image 1)

40 PHARMA FOCUS AMERICA ISSUE 03 - 2024
COVER STORY
Image 1: The University Medical Center Hamburg-Eppendorf using a M3DIMAKER pharmaceutical printer with the DPE printhead
UK Company FABRX has played a significant role in the field by actively exploring and implementing 3D printing technology for personalised medicine, as well as the opportunities for more efficient batch manufacture in clinical trials. Notably, they were first mover in embracing this transformative technology, conducting the first clinical study in the field in 2018. Carried out at the Hospital Clinico Universitario de Santiago de Compostela, Spain, this study focused on paediatric patients with Maple Syrup Urine Disease (MSUD), a rare metabolic disorder. Chewable isoleucine tablets with personalised doses, flavours and colours demonstrated
improved patient acceptability and enhanced bioavailability when compared to standard filled capsules. This success led to the development of the M3DIMAKER, the first ever pharmaceutical 3D printer designed for personalised medicine and small-batch manufacture, launched in 2020. FABRX now has two printers on the market, is involved in over 8 clinical studies for personalised medicine and is in the process of implementing 3D printing for automated compounding, in collaboration with hospitals and pharmaceutical companies across the world. (Image 2)
Gustave Roussy Institute, recognised as the top oncology hospital in Europe, is
www. pharmafocusamerica.com 41
COVER STORY
Image 2: Chewable Printlets (3D printed tablets) can be printed in different flavours, colour, and doses

conducting a clinical study targeting earlystage breast cancer, involving over 200 patients and evaluating novel, personalised, multi-drug printed tablets (polypill printlets). Specifically, combining anti-cancer therapy with anti-side effect treatment into a single pill using semi-solid extrusion, for more reliable personalised doses, improved adherence rates and overall patient wellbeing. This innovative approach challenges conventional compounding and mass manufacturing methods, providing accurate personalised doses and drug combinations for improved cancer treatment outcomes. (Image 3)
Small Batch Manufacturing
Pharmaceutical 3D printing has emerged as a transformative force challenging typical mass manufacturing, revolutionising drug production processes and offering many advantages that contribute to operational efficiency, cost reduction, and waste minimisation. 3D printing produces small batches that cater to individual patient or clinical trial needs. Prototyping is made more efficient with rapid iterations and shorter development cycles, ultimately reducing time-to-market for new medications. The precision and small batch size of 3D printing minimises waste, depositing materials layer by layer and enhancing cost efficiency in drug
42 PHARMA FOCUS AMERICA ISSUE 03 - 2024
COVER STORY
Image 3: Pharmaceutical 3D printing using direct powder extrusion (DPE) on to blister packing
production. The varied technologies available increase manufacturing versatility, allowing different dosage forms to be prepared. In short, pharmaceutical 3D printing emerges as a catalyst for streamlining drug production, ushering in a new era of efficient, cost-effective pharmaceutical manufacturing for personalised medicine and clinical trials.
Mass Manufacturing
Although 3D printing is known for small, bespoke creations, pharmaceutical 3D printing also holds the potential for largescale production of novel dosage forms to unlock advanced release properties. Significant milestones for this mass manufacturing journey were made by companies Aprecia and Triastek.
Aprecia, based in the US, was the first company to enter the field for mass manufacturing. They implemented pharmaceutical binder jetting technology, leading to the first ever FDA approved 3D printed medication, SPRITAM® in 2016 (14, 15). This distinctive 3D printing method is a powder bed system. It uses a roller to move fresh powder mix over the printing area and a binder liquid to precisely define new layers, in line with the CAD 3D model. Binder jetting allows for the production of fast-dissolving oral dispersible tablets with high drug loading, especially good for Aprecia’s target use-case, epilepsy.
Triastek in China is another key player in pharmaceutical 3D printing, focusing on Melt Extrusion Deposition (MED®) for mass
production. This advanced technique involves melting powder feedstocks into softened states, allowing for deposition, similar to a hot melt extruder. This approach grants the mass manufacturing of complex oral solid dosage release profiles and can increase the bioavailability of drugs. Triastek gained FDA approval for their rheumatoid arthritis formulation for enhanced drug delivery control in 2021, securing its place as pioneers in the field.
Environmental impacts
Pharmaceutical 3D printing stands as a promising avenue for environmentally sustainable drug manufacturing. Unlike traditional methods, 3D printing allows for on-demand production of medications in small batches, minimising material waste and reducing the environmental impact associated with mass production and transportation. The decentralised approach of small-batch 3D printing when implemented at the point-of-care for personalised medicine reduces transportation requirements further. Additionally, the technology’s flexibility in design and the ability to utilise biodegradable materials contribute to environmentally friendly practices.
The precision and versatility of 3D printing technologies for complex release profiles and precision dosing, as well as improved treatment adherence from multidrug polypills and personalised flavours and colours, enable the creation of more effective healthcare. This, in theory, will lower the overall quantity
www. pharmafocusamerica.com 43
COVER STORY
of pharmaceuticals needed, reducing its environmental impact in new ways. While acknowledging the energy consumption challenges associated with 3D printing, ongoing research aims to optimise processes and reduce carbon emissions, aligning pharmaceutical 3D printing with the broader industry shift towards sustainable practices and environmental stewardship. Overall, the adaptability and efficiency offered by pharmaceutical 3D printing hold promise in fostering a more sustainable and eco-conscious approach to drug production.
Challenges and Future Directions
The utilisation of 3D printing in drug manufacturing presents both exciting opportunities and challenges that require careful consideration for the advancement of the field.
One key challenge lies in the need for standardisation and scalability of 3D printing processes to enable reliable production of pharmaceuticals. Establishing consistent quality control measures and regulatory frameworks is essential to ensure the reliability and safety of 3D printed medications. Additionally, the selection and optimisation of suitable printing materials for diverse drug formulations pose a significant challenge, demanding additional research into the compatibility, stability, and bioavailability of various pharmaceutical compounds within the 3D printing context. Unfortunately, there is no one pharma-ink that works with multiple drugs at clinically-relevant doses because the addition of different drugs effects excipient mixes differently, with varying
consistencies and release profiles created. Each drug formulation needs developing, optimising and testing separately to allow for useful drug concentrations fit for patient use. Novel pharma-ink for each drug needs to be manufactured, meaning significant time and effort from stakeholders. The good news is that more and more researchers in academia and industry are getting involved. Collaboration is key, with companies working together alongside universities and hospitals to reach exciting new milestones.
Regulatory attention focuses on the potential of 3D printing to revolutionise dosage form development. The FDA proposed guidance in 2017 for regulating 3D-printed medical devices, but challenges remain, especially for patient-specific products, as 3D printed pharmaceuticals are not counted as medical devices. Ongoing efforts within the FDA’s Office of Testing and Research indicate a push towards practical solutions. In fact, the FDA (US), EMA (EU) and MHRA (UK) regulatory agencies are all preparing new regulations for decentralised and point-ofcare manufacturing. All three agencies have described similar future frameworks, with hub sites acting as points of contact for the agencies to mass audit the spokes, pharmacies in hospitals and communities who 3D print. Pharmaceutical 3D printing for automated compounding and personalised medicine fits under this, meaning clearer guidelines will be published in the next year or so for larger scale implementation.
44 PHARMA FOCUS AMERICA ISSUE 03 - 2024
COVER STORY
3D printing revolutionizes pharmaceuticals, reshaping drug manufacturing for personalized precision and transforming clinical trial workflows for a groundbreaking future in pharmaceutics.
The 3D printing process involves several stages: modelling, slicing, printing, postprocessing and quality control. Technical challenges such as nozzle clogging, binder migration, and power feed differences, impact completion rates and formulation performance. Pioneering companies are investigating these problems to speed up real-world implementation. As an example, Aprecia has developed a powder recycling system for their mass manufacturing binder jetting machine, reducing waste. Additionally, FABRX have developed in-built near-infrared spectroscopy and a balance print-bed to enable in-line scanning and weighing respectively of each individual printlet post-printing for automated quality control.
Developing formulations for printing can be a lengthy trial and error process. Mechanical properties of dosage forms are a key consideration in ensuring quality control.
This is influenced by factors such as viscosity, surface tension, and nozzle size. Post-printing methods such as drying methods, drying time and drying temperature may also affect product appearance and quality. To help streamline the development process AI-driven software like M3DISEEN (available for free at M3DISEEN. COM) can be used to predict the 3D printability of pharma-inks. Despite advancements, challenges persist, such as the limited availability of suitable excipients, requiring accelerated research for broader pharmaceutical applications. Addressing these challenges is vital for realising the full potential of 3D printing in pharmaceuticals. New companies are working with pharmaceutical companies to test well-known and novel excipients that they supply for 3D printing. Universities are also getting involved, publishing new tried and tested formulations every year.
Closing Remarks
In summary, the strides made in pharmaceutical engineering herald a groundbreaking era for healthcare. Aprecia and Triastek exemplify the transformative potential in mass manufacturing, having navigated regulatory challenges for innovative formulations. The activities of world leading hospitals in collaboration with companies such as FABRX, particularly in paediatric care and oncology collaborations, highlight the profound impact of 3D printing in personalised medicine and small batch manufacturing. As challenges in standardisation and material optimisation persist, ongoing
www. pharmafocusamerica.com 45
COVER STORY
research initiatives signal a commitment to refining and expanding the applications of pharmaceutical 3D printing to bring it closer to wide-spread adoption.
Looking ahead, the long-term impact on healthcare for patient treatments and general wellbeing is exceptionally positive. This technology promises a future where medications are precisely tailored, enhancing treatment efficacy and minimising side effects. Patients stand to benefit from more accurate dosages, improved acceptability, and innovative drug delivery systems designed for their unique needs. Furthermore, the eco-friendly aspects of on-demand and small-batch production not only contribute to sustainable practices in drug manufacturing but also ensure a healthier and

Dr Anna Worsley is the CEO of FABRX-AI.
Prior to this, Anna was Director of Innovation at FABRX, FABRX-AI’s parent company. She completed her PhD in 2020 in Biomaterials for Diabetic Chronic Wounds at The Royal Veterinary College and University College London (UCL).
more environmentally conscious future for all.
To actively participate in this transformative journey, connecting with innovative companies becomes crucial. There is also a new consortium to get involved in, The International Pharmaceutical 3D Printing initiative (PHARMA3DPI.ORG), where all stakeholders can get together to discuss ideas and challenges. Collaboration and engagement within the evolving landscape of pharmaceutical 3D printing, as explored in this article, not only contribute to advancements but also play a pivotal role in shaping a future healthcare paradigm centred around personalised, efficient, and sustainable practices.
References are available at www.pharmafocusamerica.com

Alvaro is a Lecturer at the Faculty of Pharmacy - University of Santiago de Compostela (Spain), co-founder at FABRX (UK) and Honorary Lecturer at University College London (UK). Listed amongst the World's Most Highly Influential Researchers for five years by Clarivate. Recognized world expert in 3D printing of medicines.

Hannah holds a BSc in Chemistry (Northern Kentucky University) and an MSc in Pharmaceutical and Biotechnology Management (EADA Business School, Barcelona). As a previous Junior Consultant, she developed effective go-to-market strategies within the pharmaceutical sector whilst establishing valuable connections with industry leaders.
46 PHARMA FOCUS AMERICA ISSUE 03 - 2024 AUTHOR BIO
COVER STORY
Data Exchange in Pharmaceutical Manufacturing
Balancing Collaboration and Competition
 John Ward Founder and CEO, ServBlock
Mick Cummins Managing Director, Ingeniero Solutions
John Ward Founder and CEO, ServBlock
Mick Cummins Managing Director, Ingeniero Solutions
The Challenge of Managing Data, in Pharmaceuticals; Balancing Collaboration and Competition" delves into the equilibrium between sharing data and maintaining an edge within the pharmaceutical industry. This article explores the obstacles and advantages associated with manufacturing examining how the exchange of proprietary information impacts innovation, efficiency and adherence to regulations. It underscores the importance of data security measures and ethical sharing practices showcasing real life examples where successful collaboration resulted in expedited drug development and production. Ultimately this piece serves as a resource, for stakeholders navigating the realm of pharmaceutical manufacturing partnerships.
In the pharmaceutical industry, the importance of sharing proprietary information plays a crucial role in driving innovation. Developing new drugs and therapies is a complex and time consuming process that can be significantly expedited through data sharing. Collaborative efforts, such as joint research initiatives or platforms for sharing data, have the potential to accelerate the discovery of new drug candidates and treatment methods. Similarly, when sponsor companies outsource manufacturing to Contract Manufacturing Organizations (CMOs), they need to be willing to share trade secrets in order to maximize productivity, ensure product quality and minimize time before patients
www. pharmafocusamerica.com 47
MANUFACTURING
can benefit from these medications. However, concerns about intellectual property theft or loss of exclusivity may discourage companies from engaging in open collaborations, thereby potentially slowing down the pace of innovation and manufacturing output.
Sponsor companies that own drug formulas rely on CMOs to manufacture their pharmaceutical products while maintaining the same level of quality assurance as if they were producing them in house. This collaboration requires sharing essential information for the manufacturing process. CMOs play a vital role by utilizing their production capabilities and expertise to produce drugs at scale while adhering to quality standards and compliance regulations.
However, the mutually beneficial relationship between sponsor companies and CMOs becomes complex when it comes to managing sensitive proprietary data. If this data is compromised, it could have serious financial and reputational consequences for both parties involved.
In this article, we will delve into the intricacies of data management within the context of sharing information between sponsor companies and CMOs. We will explore the challenges and opportunities that arise when sponsor companies outsource their operations to CMOs. Our focus will be on understanding the extent of data exchanged between these entities, addressing the associated risks related to data security and exploring available options to mitigate these risks.
In pharmaceutical manufacturing, balancing collaboration and competition in data exchange is crucial for innovation, efficiency, and regulatory adherence, with success stories highlighting the benefits of secure and ethical sharing for industry stakeholders.
The Challenge
Sharing information plays a crucial role in driving innovation in drug manufacturing. When sponsor companies collaborate with Contract Manufacturing Organizations (CMOs), they can leverage the capabilities and technologies offered by these organizations. This collaboration aims to bring about advancements in drug manufacturing processes, enhance cost efficiencies and reduce time to patient.
However, outsourcing Good Manufacturing Practice (GMP) processes like this requires rigorous oversight by the sponsor company throughout the entire lifecycle of the process at the CMO manufacturing facility. This oversight
48 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
is made possible through data exchange between sponsors and CMOs. It allows sponsors to evaluate various aspects such as batch manufacture performance, batch release documentation, deviations in manufacturing processes, raw materials analysis, supply chain information and more.
Sharing real time production data is highly advantageous in refining manufacturing processes, ensuring the production of drugs that meet efficacy and safety standards. It helps save valuable time when scaling up production, which is crucial in an industry where every day counts. Furthermore, CMOs may possess production techniques that improve drug quality or minimize waste during manufacturing. By accessing such data, sponsor companies can
incorporate these advancements into their own products, resulting in better outcomes for end users and potentially accelerating the time to market for new drugs.
Regrettably, the pharmaceutical industry often relies on outdated methods of data exchange such as emails, spreadsheets and physical documents. These manual processes come with inherent challenges and risks. Sharing information through email or spreadsheets can lead to loss, misdirection or interception of confidential data while also potentially corrupting it. Such methods necessitate time consuming manual handling of data that is prone to errors due to the effort required for organization, sending and tracking. As the volume of data increases, these manual

www. pharmafocusamerica.com 49
MANUFACTURING
processes become increasingly burdensome and inefficient. Additionally, the absence of real time sharing capabilities can cause delays in decision making.
Data security is another critical aspect within the lifecycle of CMOs and sponsor companies that demands proper attention.
The concern over the public disclosure or unauthorized access to data by competitors is a significant issue. Intellectual property plays a vital role in the pharmaceutical industry and any leaks without permission could give competitors an advantage or even lead to losing market exclusivity. Therefore, it is essential to find a balance between sharing data for effective collaboration and protecting valuable intellectual property. To achieve this, it is crucial to use protocols that safeguard shared data through secure electronic transfer systems and controlled access measures. Regular audits should be conducted to ensure both parties adhere to the agreed upon standards of data management.
Fortunately, advancements in data management technology now enable secure sharing of information between organizations. These technologies streamline the transfer of sensitive data from one site to another while maintaining stringent security protocols for its protection.
The Evolution of Data Sharing
Technologies
The field of data management is evolving alongside the introduction of innovative
technologies like data spaces and unified name spaces. These concepts are more than mere buzzwords; they form part of an emerging framework designed to empower those who generate data by allowing them not only to share it but also control how it is consumed and used by their supply chain partners.
In this article, we will explore the practical applications of these concepts in various scenarios such as deviation management, troubleshooting, real time in process control monitoring, quality control (QC) results and investigations within the relationship between CMOs and sponsor companies.
Data Space
In the realm of managing data in today's world, several essential technical components play crucial roles in ensuring the integrity, security and efficient utilization of data. Firstly, we have the concept of a 'Data Space,' which refers to a structured digital environment that stores, processes and manages data from various sources. It's a scalable and flexible framework typically based on cloud technology that allows for the integration and compatibility of diverse datasets. Within this space, 'Identity Management' becomes a vital aspect involving processes and technologies that authenticate and authorize individuals and systems to access specific data. This ensures that each entity interacting with the data space is uniquely identified and granted access rights according to predefined policies.
50 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
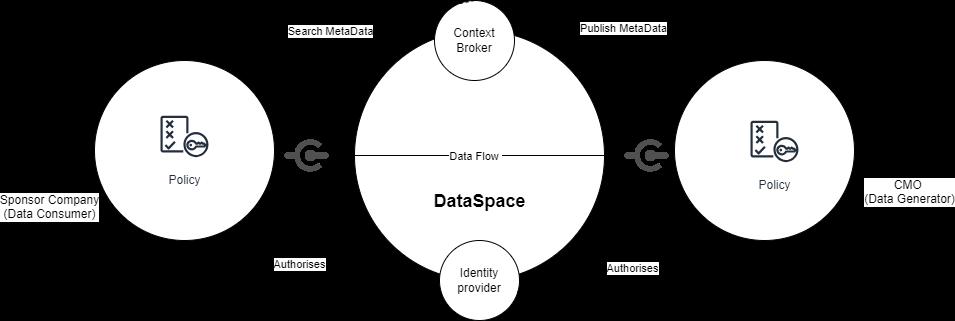
Data spaces serve as central hubs that enable multiple stakeholders to securely share and manage data within a governed environment. Data spaces act as central hubs where data creators have control over who can access their data, how it is used and the conditions under which it is utilized.
It's worth noting that data spaces can be designed to adhere to important standards like GDPR or HIPAA—which are vital within the pharmaceutical industry. Secure data sharing not only reduces access risks but also ensures compliance with industry regulations. (Figure1)
Data Sovereignty
When it comes to business, data sovereignty refers to a company's ability to retain control over its data regardless of where it is stored or processed. It plays a crucial role in data management by ensuring that valuable company information remains within the organization's boundaries and is used in alignment with its values, regulatory requirements and business objectives.
One effective approach to strengthen data sovereignty is through the implementation of “Policy Enforcement” within the data space. 'Policy Enforcement' encompasses setting up and managing rules and protocols governing how data is accessed, manipulated and shared within the data space.
Enforcing policies is crucial to ensure adherence to legal regulations, industry standards and internal guidelines. This usually entails the use of automated systems that oversee and regulate data interactions, ensuring that all activities within the data realm align with established policies. These components collectively establish a robust foundation for managing data in today's intricate and data centric environments.
Context Broker
Another key component is the 'Context Broker.' Acting as an intermediary layer within the data space, it collects and analyzes contextual information from different sources. By understanding
www. pharmafocusamerica.com 51
MANUFACTURING
Figure1: Visual representation of a Data Space
relationships between various data points, it enables more intelligent processing of information tailored to specific situations. This aspect holds great significance for applications such as smart cities or IoT environments where real time processing and decision making are critical.
Unified Namespace (UNS)
The concept of a UNS holds immense significance in industrial automation and data management, including within pharmaceutical manufacturing. It is revolutionizing how
these industries operate by centralizing and standardizing data. The UNS serves as a centralized hub that collects and stores information from various sources involved in the manufacturing process. This encompasses sensor generated information from the distributed control system (DCS), equipment related details from a computerized maintenance management system (CMMS), as well as system inputs from ERP (Enterprise Resource Planning), MES (Manufacturing Execution System), QMS (Quality Management System) and LIMS (Laboratory Information Management
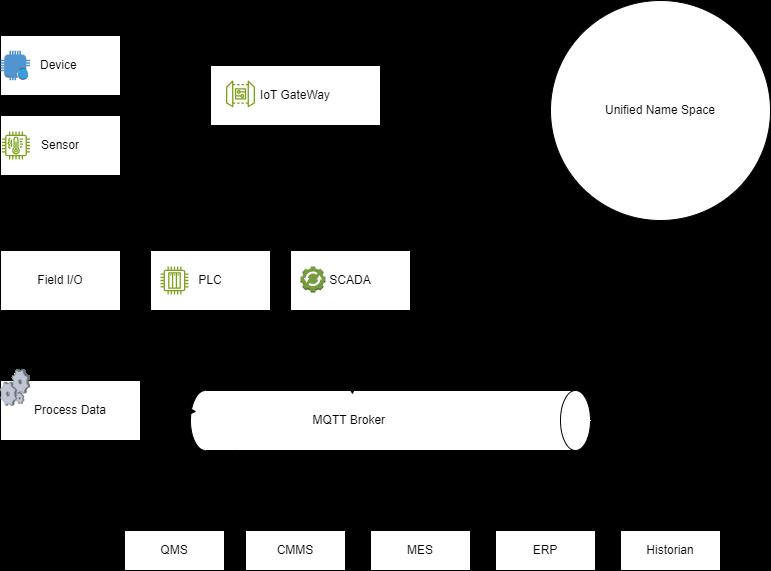
52 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
Figure 2: Reference Architecture of a Unified Name Space
System). By having a single point of access, it simplifies overall data management.
Unified name spaces establish an organized system for naming and retrieving information across different systems, making it easier to locate and access relevant data.
Just imagine navigating through a network of databases where each one has its own unique way of naming files. It can be quite complicated, right? Well, that's where a UNS comes in to simplify things. It's like having a common language specifically designed for retrieving information, making it easier to find what you need. This is particularly useful in troubleshooting scenarios, as engineers and technicians can quickly locate manufacturing information across different companies using this unified referencing system. It saves time and reduces errors. (Figure 2)
The Technological Building Blocks for Balancing Collaboration and Competition
Now, let's talk about the benefits of combining a UNS with a data space in the context of contract pharmaceutical manufacturing and its relationship with the sponsor company. This integration brings clear advantages such as enhanced collaboration, improved transparency and more effective decision making based on data.
In practical terms, the data space allows for controlled sharing of information between the contract manufacturer and the sponsor company. This seamless exchange ensures that both
Success stories highlight the transformative impact of strategic data sharing, an essential resource for pharmaceutical partnership stakeholders.
parties are aligned when it comes to production processes, quality control measures, quality assurance practices, supply chain requirements and timelines and technology transfers.
Examples of potential use cases for such data sharing technology in the area of pharmaceutical contract manufacturing are listed below. For these use case examples, data exchange is typically done in fragments through emails, drop boxes and sharepoints, among other methods. However, having a shared dataspace that brings together all the information related to a manufacturing process from various data sources within a manufacturing facility, with appropriate security measures in place, would eliminate the need for manual and inefficient knowledge transfer processes that are commonly observed in the pharmaceutical industry.
www. pharmafocusamerica.com 53
MANUFACTURING
Real Time Process Analysis
By utilizing a data space, the sponsor company gains real time access to CMO manufacturing process data. This level of transparency enables monitoring of production activities to ensure compliance with quality standards while ensuring the process is operating within established boundaries and meeting regulatory requirements.
Deviation Management
When there is a deviation from operating procedures or filed/qualified process parameters, it is important to understand why it occurred. Data spaces facilitated by context brokers allow controlled sharing of essential investigative data such as batch records, process control trending, historical deviations within the same system or category, machine logs and environmental monitoring data with external stakeholders. By granting access to this data, teams can effectively collaborate in identifying the root cause behind deviations without compromising data security.
Change Management
Sponsor companies will have on demand access to review change controls that impact regulatory filing of qualified processes at the CMO facility. Ease of access to such information enables and expedites informed decision making at sponsor company leadership level and ultimately will lead to a much speedier progression of the CMO change control process.
Supply Chain
Having a shared data space improves supply chain optimization. Both parties involved can manage raw material and consumable inventory, production schedules and logistics efficiently, resulting in fewer delays and cost savings.
Technology Transfer
Shared data spaces facilitate efficient and robust technology transfers between the sponsor company and the contract manufacturing organization (CMO) or vice versa. When introducing a new product at either facility, it is crucial to have comprehensive knowledge transfer between sites.
Equipment Reliability
When there are issues with the equipment that affects the production process at the CMO, the Sponsor company may want to conduct a joint investigation to determine the root cause. Engineers can make use of a data space that facilitates seamless data sharing across different companies within the CMO and Sponsor relationship. This allows for easier identification of problems and potential solutions.
Quality Control
QC testing and its results are highly sensitive and prone to human error. In cases where unusual or out of specification results occur, particularly in instances that affect batch release, a sponsor company may wish to have oversight on the testing methods and associated documentation. To ensure authorized personnel
54 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
can access QC data and any anomalies are investigated, a context broker can be used to facilitate machine to machine interactions within the Dataspace.
Conclusion
To sum up, the concepts of Data Spaces, The Unified Name Space and context brokers all contribute to improving data sharing practices while maintaining data ownership and ensuring ethical handling. These technologies give data creators control over how their data is used while also ensuring integrity. As these technologies become more integrated into data management practices, they have the potential


to significantly enhance efficiency and security in operations involving data.
It should be a goal of all pharmaceutical sponsor companies to embrace this new approach to data sharing, particularly interorganisational data sharing with CMOs. As the industry moves forward with advancements in technology and more stringent regulations, it becomes increasingly important for sponsor companies and CMOs to consistently enhance their practices of sharing data. By doing so, they can foster innovation, maintain operational efficiency, and uphold the standards of data security and ethical responsibility.
John Ward is founder and CEO of ServBlock, founded in early 2021, following John’s recognition of the potential for Blockchain and Data Space technologies to revolutionise the regulated manufacturing space. ServBlock focusses on trusted data exchange through distributed ledger and blockchain technologies. For over a decade, John has been at the forefront of the highly regulated pharmaceutical manufacturing industry, working alongside many of the world's leading biotech companies in his role as principal consultant within the validation space. Most recently as a product owner in Pfizer's global Smart Factory team. In addition to his work at ServBlock, John is an expert committee member for blockchain and distributed ledger technologies with the National Standards Authority for Ireland.
Mick Cummins has served as the Managing Director of Ingeniero Solutions for over 9 years, delivering technical and consulting services to numerous prominent biopharmaceutical clients throughout Ireland and mainland Europe. He possesses expertise in biopharmaceutical manufacturing, specifically in equipment design, qualification, and handover. Additionally, he provides manufacturing engineering and equipment ownership services within commercial GMP manufacturing sites. These services include change management, resolution of quality deviations, equipment redesigns and retrofits, technology transfer from contract manufacturing organizations (CMOs) to Sponsor company sites, and the introduction of new products to the client manufacturing site.
www. pharmafocusamerica.com 55 AUTHOR
BIO
MANUFACTURING
Digitalization of a Novel Advanced Modular Continuous Pharmaceutical Drug Substance Manufacturing Process

Currently, pharmaceutical industries are trying to digitalize their manufacturing processes to reduces the overall cost and enhance product quality with less time and resources. There are different ways to digitalize the liquid-based manufacturing processes (e.g. API synthesis) and one approach is to use computational fluid dynamics (CFD). However, because of different level of complexities, the development and implementation of CFD model to support pharma manufacturing is still a challenging task.
IBennie Anderson PhD student, Rutgers University, NJ, USA
n this work, the digital transformation process is demonstrated through a continuous pre-heating process which is a critical unit operation used in continuous API synthesis. CFD approach has been
56 PHARMA FOCUS AMERICA ISSUE 03 - 2024
Ravendra Singh Faculty of Chemical and Biochemical Engineering,
MANUFACTURING
employed. There are a variety of methods available for controlling the reaction rates in continuous API synthesis. One method used in the pharmaceutical industry involves temperature regulation of reactant streams prior to the reactor by introducing a heat transfer unit operation, such as a preheater. Preheating units have been studied in a variety of different geometries, but one geometry less common in scientific literature is the twisted helix, also known as a bent helix. Consisting of a standard helix twisted into a secondary helix, there are three different diameters present in this geometry that can influence heat transfer. In this article, a preheater of twisted helix geometry used in a continuous API synthesis process is modeled using computational fluid dynamics (CFD) to predict the influence of each diameter on heat transfer efficiency. Experimental data was first used to calibrate the model, then these three design parameters were varied in a standard 2 3 full factorial design (yielding eight different configurations) at two different flowrates. The efficiency of each configuration was calculated and used to fit empirical correlations between the different diameters and heat transfer efficiency. In addition to the main effects of primary diameter, secondary diameter, and inner diameter, the correlations incorporate the possibility of two-factor interactions between the design parameters. Effect estimates were generated using statistical analysis software to predict which parameters or interactions have
Digitalizing pharmaceutical manufacturing with CFD for cost reduction and quality improvement poses challenges in developing liquid-based process models.
the greatest influence on efficiency. Lastly, model adequacy of the developed correlations is supported through half-normal plots and analysis of variance (ANOVA) tables.
1. Introduction, background, objectives
In the pharmaceutical industry, temperature regulation is often considered a key or critical process parameter (CPP) in continuous API synthesis as it can influence reaction rates thereby desired product as well as the generation of unwanted byproducts from side reactions. As a result, the importance of temperature control has been widely studied utilizing a wide variety of methods. Mathematical models have been found to be useful tool to learn more about heat and mass transfer in manufacturing processes and can reduce the number of experimentations that may be time and resource intensive or infeasible to conduct.
www. pharmafocusamerica.com 57
MANUFACTURING
When considering heat transfer, helices are usually a superior geometry relative to straight tube configurations due to their compact nature and higher heat transfer coefficients, showing promise in multiple fields of process industry. Submerged standard helical coils have been widely analyzed in literature using both experimental and modeling approaches but is less common for the twisted helix geometry and its variants. However, heat transfer in even standard helices is difficult to characterize, much less the relatively complex geometry of a twisted helix. This is in part due to the Dean effect, which has the effect of changing the flow regime within helical tubes when operating at different flowrates. The Dean effect describes the development of secondary flows perpendicular to the primary flow, as well as imbedded vortex structures depending on the flow region and regime. As a result, a large amount of data is

often required at different flows to perform detailed statistical analysis, which makes CFD modeling an appealing alternative to cost intensive experiments.
In this work, the digital transformation process is demonstrated through a continuous pre-heating process which is a critical unit operation used in continuous API synthesis. This work help to increase understanding of which design parameters have the greatest influence on heat transfer for twisted helix geometries. In standard helix studies, many have reported empirical correlations for flow regime and heat transfer using the ratio of outer diameter to inner diameter, known as the curvature ratio. In the case of the twisted helix, there are three diameters to consider, and thus three possible ratios that could influence system characterization (see Figure 1).
The resultant variable under investigation is heat transfer efficiency and its relationship with the three diameters. A sensitivity analysis of these three geometric design diameters was performed using a mechanistic model built in COMSOL Multiphysics, calibrated using experimental data and used to perform a three-factor two-level factorial experiment, with analysis of the results performed using Matlab. Constructing a CFD model enables a great deal of data to be collected without costly experimentation, which allows a full factorial approach to be taken at each flowrate. These different flowrates were treated as blocks when generating empirical models to reduce the probability of the Dean effect acting as
58 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
Figure 1. The three diameters of a twisted helix geometry: primary (orange), secondary (red), and inner (blue)
a nuisance factor, which might otherwise cause these results to be valid at only certain flowrates.
To replicate the approach and analysis described in this article with an experimental setup, eight separate coil configurations of different diameter combinations would be necessary. This highlights the benefits of CFD when performing statistical analysis, especially for design parameters which cannot be changed without difficulty or cost. Oftentimes, to avoid the issue of increasing cost and investment, a fractional factorial approach will be used in experimental sensitivity analysis, which involves conducting experiments at specific combinations of parameters to effectively cover the entire design space with fewer experiments. However, this can come at the cost of accuracy and potentially missed interactions between the parameters under investigation.
2. Materials and Methods
The twisted-helix preheating unit is directly placed before a custom-built plug-flow reactor (PFR) used in the continuous synthesis of APIs. The unit consists of a steel tank with minimal
insulation filled with ethylene glycol and a submerged coil made of PFA tubing in twistedhelix geometry. This coil usually contains the reactant stream to be warmed prior to reaction to minimize variability in reaction rates and improve overall control of the process. The ethylene glycol within the tank is heated using a submerged immersion heater and is highly agitated with a mechanical mixer to ensure the thermal energy is distributed throughout the transfer fluid, aiming to minimize temperature gradients. Within the reactant coil, water was used in place of the usual reactants to focus on heat transfer characterization without the added complexity of reactions generating or absorbing heat. The experimental procedure used to generate calibration data for the CFD model involved varying the reactant stream flowrate at a constant rate of heating until a steady outlet stream temperature was achieved. The CFD model was created with COMSOL Multiphysics and statistical analysis performed using Matlab.
3. Model Development
The CFD model was calibrated by treating the
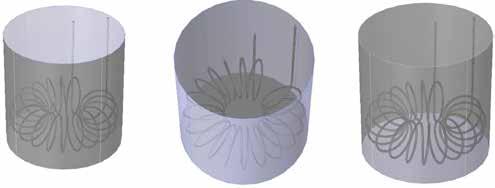
www. pharmafocusamerica.com 59
MANUFACTURING
Figure 2. The three sides of the model given different heat transfer coefficients
tank as if it were experiencing convection with different heat transfer coefficients on each side exposed to a different environment. The three “environments” were the bottom surface of the tank (which was resting on an aluminum surface), the top surface of the tank (which experiences increased heat transfer from currents induced by natural convection) and the sides of the tank with minimal insulation (see Figure 2).
During calibration, different heat transfer coefficients on each side were tested until the outlet stream temperature generated from the CFD model closely matched those gathered experimentally. Upon completion of calibration, the CFD model outlet temperatures at each flowrate differed by less than 5% from those gathered experimentally. Additionally, as COMSOL Multiphysics utilizes a finiteelement algorithm for mechanistic modeling, a mesh dependence study was conducted to confirm a fine enough mesh which was used during data generation.
The complete design matrix above was run at two different flowrates, which was treated
as a blocking variable to minimize nuisance factors. One factor in particular that comes to mind is the Dean effect, which can cause significant disturbances in flow regime (and thus heat transfer) depending on flowrate in helical systems. By treating the flowrate as a blocking variable, flowrate is separated from the parameters under investigation and minimizes the influence of differences in flow regime. This allows the empirical model to focus solely on the diameters of the twisted helix geometry.
4. Statistical Analysis Approach
Once calibrated, a low (-) and high (+) value of each diameter in the twisted-helix geometry was selected for use in the three-factor factorial experiment (see Table 1 for values).
Selecting these values required some heuristic approach, as only certain combinations of diameter values are feasible for achieving valid geometries. For example, if the outer diameter is too small relative to the secondary or inner diameter, the coils might overlap and create an impossible geometry.
60 PHARMA FOCUS AMERICA ISSUE 03 - 2024
Parameter Experimental Value (in.) High (+) Value (in.) Low (-) Value (in.) Primary Diameter (p) 8” 8” 7” Secondary Diameter (s) 4” 4” 3” Inner Diameter (i) 0.25” 0.35” 0.25” Flow Rate (ml/min) Various 150 100
Table 1. High and low values used in factorial experiment
MANUFACTURING
Alternatively, if the outer helix was set too high, it would protrude from the tank walls and invalidate any possible results. Running multiple different geometries in series (such as during a parametric analysis) frequently posed an issue, as there were cases where the model construction would fail when transitioning between configurations due to impossible overlap of system elements.
The complete design matrix (Table 2) was run at two different flowrates, which was treated as a blocking variable to minimize nuisance factors. By treating the flowrate as a blocking variable, flowrate is separated from the parameters under investigation and minimizes the influence of differences in flow regime. This allows the empirical model to focus solely on the diameters of the twisted helix geometry.
The outlet temperature of each run was collected and used to calculate a rate of energy transfer based on the reactant stream’s flow rate (V), density (ρ), specific heat capacity (cp) and temperature change (ΔT). Preheater efficiency is defined as the ratio of energy
absorbed by the fluid stream to the overall energy added to the system by the immersion
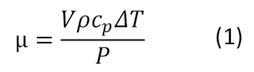
heater. Any energy that does not contribute to warming of the outlet stream is lost to the surroundings. Preheater efficiency (µ) was then found using the immersion heater’s power setting (P).
Once the data had been collected, it was used to fit a linear model with two-factor interactions between primary diameter (p), secondary diameter (s) and inner diameter (i) using Matlab’s fitlm() function. A non-interactive blocking term (β) for flowrate was added to account for different flowrates. The one possible three-factor interaction (ABC) was not considered as significance decreases as the term order increases. Inclusion of superfluous terms has been shown to improve fitting and decrease error relative to the dataset, but decreases the applicability of results outside of the specific datasets used [19]. This model is useful to predict the effect of pre-heater design parameters on its efficiency for a given material and heat source.
µ ~ 1+ p+s+i+ps+pi+si+ β (2)
Analysis was performed treating the data as a three-factor two-block full factorial experiment. Term significance was based on a half-normal distribution generated using Matlab’s probplot() function and ANOVA tables with associated p-values. The final step was checking model adequacy through
www. pharmafocusamerica.com 61
Run Primary Secondary Inner Diameter Value Diameter Value Diameter Value (In) (In) (In) 1 7 4 0.25 2 7 4 0.35 3 7 3 0.25 4 7 3 0.35 5 8 4 0.25 6 8 4 0.35 7 8 3 0.25 8 8 3 0.35
Table 2. Preheater Model Parameter Experimental Design Matrix
MANUFACTURING
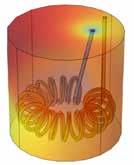
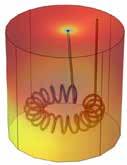
a normal probability plot and predicted vs. residuals plot, which checks for data normality and constant variance respectively (two assumptions required for this statistical approach).
5. Results and Discussions
Running a full 2 3 factorial with two blocks (flowrates) gave 16 efficiency datapoints with six possible significant terms as seen in Equation 2 above (excluding flowrate as a blocking variable). Figure 3 shows surface temperature profile plots generated by the CFD model at low (-) and high (+) values of each diameter as indicated by the design matrix. These profiles are useful for assessing the temperature distribution within the tank and where the majority of heat loss occurs. As shown in Figure 3, there is a visible shift in temperature between the upper half of the tank and the lower half where the coil resides due to the warming reactant stream.
The simulation has been used to identify the significant design parameters. In order to estimate term significance, two approaches were used. The first approach was a half-normal probability plot as shown in Figure 4, which plots the effect estimate of each term (p, s, and i) or two-factor interaction (p:s, p:i, s:i) against the term’s expected location assuming a normal distribution (the dashed line). Points that lie on or near the line are not likely to significantly impact preheater efficiency and do not deviate significantly from the expected normal values. Those further from normality are typically significant terms. In this case the primary diameter, secondary diameter, and the interaction between secondary and inner diameters (secondary: inner) are significant design parameters.
The second method was the traditional ANOVA (Analysis of Variance) table. While there are multiple reasons why ANOVA table is useful, the most salient aspect to this work
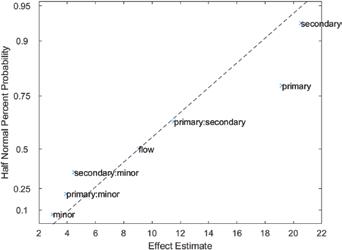
62 PHARMA FOCUS AMERICA ISSUE 03 - 2024
Figure 4. Half normal plot of preheater model efficiency results
MANUFACTURING
Figure 3. Simulated surface temperature profiles of preheater at (+, +, +) values (left) and at (-, -, -) values (right). Red shading indicates higher temperature while blue indicates lower temperature.
is the column of p-values used to evaluate the likelihood of these results occurring due to chance. A standard level of significance is 0.05, below which a term is deemed significant. Results indicate that the primary, secondary, and secondary: inner terms likely dominate efficiency, but the inner and secondary: inner terms are also relevant (Table 3). However,
the p-values for inner and secondary: inner terms are an order of magnitude larger than the other significant terms, and in the case of the inner effect, is barely < 0.05. Further testing and analysis would be useful in determining just how significant the inner diameter is, particularly at a larger range of diameters.
Taken as a whole, these statistical results indicate that the efficiency of the twisted-helix preheater design is heavily influenced by the primary and secondary diameters, which have a significant interaction effect. The inner diameter and its secondary interaction also have an effect on efficiency, but further testing is needed to determine to what extent. The ANOVA table
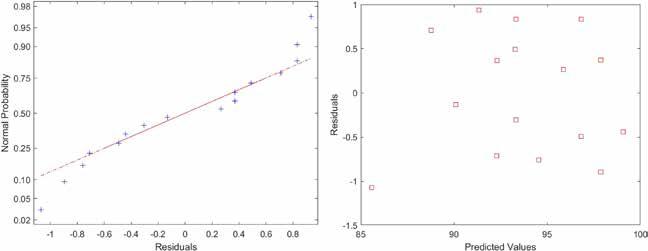
www. pharmafocusamerica.com 63
Figure 5. Normal probability plot (left) and predicted and residuals plot (right) of preheater model efficiency results
Sum of Squares DoF Mean Square F-Value p-Value Primary 60.38646 1 60.386462 69.66325 3.21E-05 Secondary 12.37601 1 12.376007 14.27725 0.0053995 Inner 4.659878 1 4.6598779 5.375745 0.04902911 Flow 82.09408 1 82.094077 94.70566 1.04E-05 Primary: Secondary 33.31287 1 33.312867 38.43051 0.00025964 Primary: Inner 3.868963 1 3.8689632 4.463327 0.06759307 Secondary: Inner 5.048834 1 5.0488338 5.824454 0.0422839 Error 6.934671 8 0.8668339 1 0.5
Table 3. Preheater Model Efficiency ANOVA
MANUFACTURING
suggests the effect of the inner diameter is significantly less than the other two.
Lastly, in terms of adequacy analysis, the predicted values vs residual plot indicates a relatively constant variance (Figure 5, right), one of the initial assumptions used when performing this statistical analysis. Normality was assessed using a normal probability plot (Figure 5, left), which suggest that while there are some outliers, the majority of data falls within normality.
6. Conclusions
In this work, the digital transformation process is demonstrated through a continuous pre-heating process which is a critical unit operation used in continuous API synthesis. A CFD model of a twisted helix geometry preheater designed for continuous API synthesis is used to evaluate design parameters influencing preheater efficiency through statistical analysis. CFD allowed a great
deal of data to be collected and analyzed without the need for time-intensive and costly experimentation, enabling a full factorial experiment to be conducted. The results indicate that in this particular geometry, the curvature ratio between the secondary and primary diameters plays a larger role in determining heat transfer efficiency than the inner diameter. This result could be applied in future works which develop more detailed correlations for twisted helix geometries or for designing preheater coils with greater efficiencies. Future work includes, further experimentation using CFD modeling to expand the results, such as repeating this experiment with an expanded factorial utilizing three or four different values for each diameter rather than simply one high and one low value.
Acknowledgements
This work is supported by the US Food and Drug Administration (FDA) under contract number 75F40121C00106.
AUTHOR BIO

References are available at www.pharmafocusamerica.com
Dr. Singh is the faculty of C-SOPS, Chemical and Biochemical Engineering Department, Rutgers University, NJ, USA. He is the recipient of prestigious EFCE Excellence Award from European Federation of Chemical Engineering. His research focus is continuous manufacturing of drug substance and product. He is PI/Co-PI of several projects funded by FDA, NSF, and companies. He has published more than 85 papers, edited one pharmaceutical system engineering book published by Elsevier, written more than 12 book chapters, and presented at over 150 conferences. He is actively serving as a Journal editorial board member, and conference session chair.
64 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
Twin-Screw Granulation An Alternative Continuous Granulation Approach
Academic scientists and pharmaceutical industries are interested in granulation using HME technology. Continuous manufacturing, such as twin-screw granulation (TSG), is favored over batch manufacturing in pharmaceutical production due to its advantages. Different techniques for twin-screw granulation, including dry, wet, and melt granulation, are being developed for robust and reproducible granulation processes. TSG is the technique where the material is conveyed to the mixing zone. The quality of granules is significantly affected by both process and formulation variables. The process parameters that need to be carefully controlled include screw configuration and speed, feed rate, barrel temperature, residence time, torque, and Liquid/Solid ratio (for wet granulation). In summary, the continuous production of granules using TSG is an emerging alternative to the traditional batch process of granulation.
Sateesh Kumar Vemula
Department of Pharmaceutics and Drug Delivery, School of Pharmacy, The University of Mississippi
Michael A. Repka
Department of Pharmaceutics and Drug Delivery, School of Pharmacy, The University of Mississippi
Melt granulation using hot-melt extrusion is one of the promising alternative approaches to producing granules. It is known as a solvent-free green technique, which is scalable utilizing continuous manufacturing techniques. The twinscrew extruder is a highly versatile and customizable mixer used for continuous granulation, well-known as
www. pharmafocusamerica.com 65
MANUFACTURING
twin-screw granulation (TSG). It achieves this using various conveying, kneading, and distributive mixing elements in different combinations. TSG is an innovative granulation method with advantages such as reduced processing time and cost, effective management of formulations with high drug loads and poor flow properties, less binder to produce suitable granules, minimized lot-to-lot variation, and compatibility with continuous manufacturing processes [1]. Based on temperature and binder utilization, twin-screw granulation is classified into twin-screw dry granulation (TSDG), twinscrew wet granulation (TSWG), and twin-screw melt granulation (TSMG). TSG can develop denser granules with improved homogeneity, compressibility, and flowability through all three types of granulations. Depending on equipment, formulation, and process, final granule properties may differ, and these variables are shown in Figure 1.
All three types of twin-screw granulation could be widely used for various pharmaceutical applications. TSG has several applications and benefits in the pharmaceutical industry. Some of the key applications of twin-screw granulation include improved granule properties in terms of Twin-screw granulation (TSG) is a promising alternative in continuous pharmaceutical manufacturing, offering advantages over traditional batch methods with precise control of parameters for highquality granules.
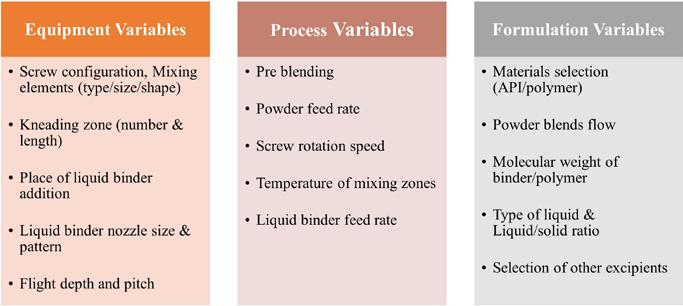
66 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
Figure 1: Equipment, process, and formulation variables in twin-screw granulation
Process Key Process Differences
• Liquid binder
TSWG
TSDG
TSMG
Melt extrusion
Applications
• Die is not required at the barrel end
• Dry binder in solid state
• Process Temp. < Tg or melting point of process ingredients
• Die not is required at the barrel end
• Meltable binder in solid state
• Process Temp. > Tg or melting point of process ingredients
• Die is not required at the barrel end
• Drug + Polymer + Additives
• Process temp > Tg or melting point of process ingredients
• Die is required at the barrel end
uniformity in size and shape, better control over the density of granules with enhanced dissolution rate, and reduced processing steps. TSG enables easy scalability, continuous
processing, and reduces material waste [2].
Table 1 and 2 shows a comparison of different types of TSG with conventional HME for its pharmaceutical applications.
www. pharmafocusamerica.com 67
TSWG TSDG TSMG Melt extrusion API stabilization No No Yes Yes Continuous process Yes Yes Yes Yes Fixed dose combination Yes Yes Yes Yes Heat sensitive API Yes Yes No No High mixing efficiency Yes Yes Yes Yes Improve API processibility Yes Yes Yes Yes Moisture sensitive API No Yes Yes Yes Solubility enhancement No No Yes Yes Taste masking Yes Yes Yes Yes
Table 2: Applications of the twin-screw extruder
Table 1: Processing differences among the twin-screw granulations
MANUFACTURING
Despite the above advantages and widespread applications, TSG has some significant challenges to overcome such as maintenance and cleaning of equipment, temperature control, dust control, and regulatory compliance. Granulation equipment components, such as screws, wear over time. Consistent performance requires maintaining tooling integrity to avoid costly replacements. Material feed rate and screw speed, and equipment size are the crucial factors in the scale-up and large-scale production. It is of utmost importance to meet regulatory


requirements in the pharmaceutical industry. Complying with Good Manufacturing Practice (GMP) standards by documenting and validating the granulation process can be a challenging and time-consuming task. In conclusion, twin-screw granulation is a highly effective and efficient alternative to traditional granulation methods in the pharmaceutical industry. Due to innovative TSE applications and extensive research in scaling up the process, there has been a rise in the number of approved HME products in the market. The authors anticipate this trend to continue.
References are available at www.pharmafocusamerica.com
Sateesh Kumar Vemula, Ph.D., is an accomplished researcher and teacher in Pharmaceutical Sciences. He has been working as a Postdoctoral Research Associate since January 2023 in the Department of Pharmaceutics and Drug Delivery, University of Mississippi, USA. He has extensive research experience in Colon Specific drug delivery, Fast Dissolving Tablets, and Melt Granulation (Hot-melt extrusion). He has published 80 research publications in international and national journals and serves on the Editorial Advisory Boards of three prominent journals.
Michael A. Repka, Ph.D., is a Distinguished Professor of the Department of Pharmaceutics & Drug Delivery and Director of the Pii Center for Pharmaceutical Technology at The University of Mississippi. Dr. Repka’s research primarily focuses on enhancing the solubility and bioavailability of poorly soluble drugs via hot-melt extrusion technology, with novel dosage forms as a common denominator. He has published over 230 peer-reviewed articles in prestigious pharmaceutical journals and serves on the Editorial Advisory Boards of six prominent journals. Recently, he received the Ralph Shangraw Memorial Award from the International Pharmaceutical Excipients Council.
68 PHARMA FOCUS AMERICA ISSUE 03 - 2024
AUTHOR BIO
MANUFACTURING
The Journey of Mass Spectrometry in Biopharmaceuticals CMC Development

Mass Spectrometry is increasingly popular as a tool for accelerating CMC development of biopharmaceuticals.Technological advances in mass spectrometry workflows for biopharmaceutical development have allowed faster speed to clinic. Here we discuss two of the most beneficial workflows: multi-attribute method and host cell protein analysis.
Mass Spectrometry (MS) is playing a bigger role than ever in supporting biopharmaceutical
Vesela Encheva
PhD, Associated Principal Scientist R&D Department, Lonza
Ian Anderson
PhD, Senior Technical Leader, Global Biologics Technical Development, Lonza
CMC development. This is in large part due to advances in MS instrumentation, more separation techniques becoming amenable
www. pharmafocusamerica.com 69
MANUFACTURING
to MS detection and advancements in data processing and system operation software. MS really has had a face lift. It was once thought of as niche technique, needing highly skilled operators to both operate the systems and also process the data, and only used for the extended characterisation of biopharmaceuticals and bioanalysis. Such limitations led to minimal use of MS in biopharmaceutical development and data was rarely included in regulatory dossiers. One of the biggest barriers to using MS data in a QC release environment is the requirement for FDA 21 CFR Part 11 compliance. FDA 21 CFR Part 11 governs the use of electronic records and signatures; MS techniques generate large data sets containing significant amounts of meta-data which all needs to be tracked.
In recent years, advances in this area have allowed the use of MS data in QC. Now multiple MS vendors offer 21 CFR Part 11 compliant solutions for both operating systems and data processing options.
New MS hardware options have led to an ever growing list of workflows that can be employed to support biopharma development. The introduction of softer ionisation techniques and native MS have expanded it usage in workflows such IEX (Ion exchange chromatography)-MS and SEC (Size exclusion chromatography)-MS. The key improvement being the speed at which the scientist gets the data required to make critical product development decisions. Workflows that used to require weeks of data analysis can be now
completed in minutes. Currently multiple vendors offer MS vendor neutral software enabling the harmonisation of workflows and offering further reductions in data processing timelines. These software solutions allow data from multiple workflows to be brought together enabling a holistic view of the molecule.
MS has increased in popularity due to its flexibility to work with multiple different product types from antibodies through to cell and gene therapies. The ability to setup platform workflows across modalities is advantageous compared to other analytical methods. The simplicity of both hardware and software allows workflows to become routine in non-specialist MS labs, i.e process development for PAT, process analytical and QC laboratories.
In this article we focus on two of the most significant areas of development in recent years for MS workflows; Multi Attribute Methods (MAM) and Host Cell Protein (HCP) analysis.
Multi-attribute method –
The term Multi-attribute method was first coined by Amgen and is now widely adopted in the biopharmaceutical industry. As originally proposed the MAM approach is in essence a peptide mapping analysis by LC MS/MS but the novelty is in its application to monitor multiple product attributes in a single analysis.
The idea of using a single method to monitor a range of attributes was born from the shift to (Quality by Design) QbD approach recommended by regulatory agencies.
70 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
The QbD approach includes establishing a quality target product profile (QTPP) that identifies potential critical quality attributes (pCQAs). Once determined the pCQAs need to be consistently monitored throughout development and production. For this reason, advanced analytical approaches are needed to ensure a CQA driven manufacturing process instead of final product testing. The MAM approach supported by recent advancement in MS has the potential to meet this need. Execution of MAM usually begins by digesting the protein of interest with sitespecific protease (trypsin is most commonly used) followed by chromatography separation of the resulting peptides and their detection by MS. The peptides undergo two stages of MS analysis: MS1, where their accurate mass is determined and MS2, where the peptides are fragmented to reveal their amino acid sequence. The MS data is analysed by specialised software to produce the primary amino acid sequence of the protein as well as any modifications (e.g. deamidation, oxidation, glycosylation). The MS1 data can be used to quantify the relative abundance of the detected modifications while the MS2 can provide site-specific localisation of the modification. Another advantage of MAM is that it can be used to detect unmonitored quality attributes (New Peaks) and thus identify potential process and product-related impurities. As post-translational modifications and impurities could have effect on product safety, activity or immunogenicity they are
potential CQAs and their detection and identification is essential throughout the product life cycle.
Advantages of MAM
Traditionally, a series of methods are used to characterise the product CQAs e.g. icIEF for changes in charge variants, 2-AB labelling and HILIC separation for glycans or Reverse Phase for oxidation etc. (see Table 1).
If we consider the time required to develop, validate and transfer these methods as well as execute them this approach is not supportive of accelerate product development. Moreover, the conventional analytical methods address categories of product related variants e.g. charge variants but cannot distinguish e.g. between different deamidation sites and their individual contribution to the observed variant. MAM provides site-specific information often down to specific amino acid thus allowing for enhanced product understanding.
MAM also outperforms other methods when it comes to detection of New Peaks. New Peaks can sometimes be observed by current purity tests, but as these are limited to UV based detection they can easily be misled by co-eluting components. MAM is more sensitive than UV based approaches and is able to further investigate and assign identity to the unknown components by applying the principles of “Error Tolerant Searching” and de novo sequencing. Overall, these unique capabilities make MAM an enabler of the QbD approach
www. pharmafocusamerica.com 71
MANUFACTURING

iMAM
Most publications refer to MAM as peptide level analysis following protease digestion. However, MAM can also be performed at the intact protein level (iMAM). In the recent years technological advances in the field of native MS have allowed this approach to become a reality. MS of intact proteins under native conditions used to be state-ofthe-art approach employed only by a few specialised laboratories around the world. Since instruments with extended mass range became commercially available this approach has become more accessible to any laboratory with expertise in MS. As a result native MS is now beginning to be applied
for the characterisation of biotherapeutics. The approach has the advantage of limited sample preparation where majority of the variability of MAM stems from. iMAM offers great potential for characterising the heterogeneity of glycosylated biotherapeutics and for supporting higher order structure and non-covalent interaction studies. Despite that this is a relatively new approach with only a few documented examples in the literature it is already being applied for product quality testing and to couple common separation methods such as SEC and IEX to MS.
MAM as a method for QC
MAM coupled to LC MS/MS has been
72 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
proposed as a replacement for several physicochemical QC methods used for batch release or stability testing. However, the intention is not to replace all QC assays with MAM. As shown in Table 1 detection of dimers and aggregates, higher order structure, biological activity and microbiological properties are outside the remit of MAM and will continue to be monitored using conventional methods. Despite that, there aren’t any regulatory impediments to introduce MAM in QC recent publications from FDA emphasize that majority of BLA application include MS data but none as part of release or stability testing. The main bottlenecks appear to be the need for bridging studies, the lack of MS expertise in QC labs and complexity of the validation. Perhaps the MAM approach will be most successful in the instances where no conventional methods have been applied to control the molecule. (Table 1)
Host cell protein (HCP) analysis
Host cell protein content is critical quality attribute that can impact product safety and efficacy and monitoring clearance is obligatory. Traditionally ELISAs have been the method of choice for measurement of HCP. But there has been a paradigm shift and last year the first ever HCP-MS method was approved for product release testing to support a phase III clinical trial (1st MS-based HCP analysis under GMP for release test (alphalyse. com)). The COVID pandemic accelerated CMC timelines for new medicine approval became the norm and this is a challenge when it comes to developing a product and process specific ELISA. Ease of development makes the use of a MS based method advantageous for supporting shorter CMC timelines. The appropriate method for host cell protein analysis needs to be considered as part of the overall process and analytical
www. pharmafocusamerica.com 73
Product Quality Attribute Conventional method Monitored by MAM Monitored by iMAM Charge variants icIEF Yes Potentially Size variants (fragments) CE SDS, SEC Yes Yes Glycosylation 2-AB HILIC Yes Yes Oxidation RP, HIC Yes Yes Identity Peptide Mapping by LC-UV Yes Yes Dimers, oligomers and aggregates SEC No No Higher order structure CD, FT IR, AUC, DSC No No Functional activity Cell based potency assay No No Microbiological Properties Bioburden And Endotoxin Testing No No MANUFACTURING
Table 1. Examples of product quality attributes and the corresponding conventional methods and if they can be monitored by MAM.
risk assessment, and there is now flexibility, due to improvements in MS hardware and software, to employ MS as the sole analytical method for control of HCP. This is no better illustrated in an update to the United States Pharmacoepia (USP) chapter 1132, there is a new chapter USP 1132.1; Residual Host Cell Protein Measurement in Biopharmaceuticals by Mass Spectrometry, that contains general guidance and best practices for MS-based identification and quantification of high-risk and high-abundance HCPs.
Enabling MS technolgies to support host cell protein analysis by MS
The MS requirements from a hardware and software perspective for HCP analysis are a different prospect compared to a MAM workflow. Whilst a high resolution and high sensitivity sensitive MS system is a definite benefit to a MAM workflow, it is essential for an MS HCP screening method. The advances in MS sensitivity have brought the levels of sensitivity to comparable levels of a HCP ELISA, in the order of magnitude of part per million (ppm). Such sensitivity was needed to enable to use of MS. It is also important to note that with any MS assay, as with any ELISA, the sensitivity is HCP dependent. For any HCP MS workflow the MS sensitivity and resolution is only one factor, it is a careful balance between separation technique and amount of sample loaded. Chromatographic separation is most commonly used prior to
Mass Spectrometry revolutionizes biopharmaceutical CMC development, speeding up the path to the clinic with impactful workflows and technological advances
MS HCP analysis but it is possible to use CE based separation methods. Advances in chromatography systems and columns have also significantly improved the repeatability and robustness of the HCP MS workflow.
Depending on the HCP MS workflow different softwares are required, but now there are multiple options for software depending on the workflow. The key is matching the HCP MS workflow and software with what is needed from an analysis perspective.
What method to choose for HCPMS
Advances in MS hardware and software in the last ten years have provided flexibility in terms of biopharma HCP MS workflows. Normally there will be a discovery or screening workflow to identify as many host cell proteins with relative quantitation, followed by a more targeted method that yields better quantitation and sensitivity. There is a trade of between
74 PHARMA FOCUS AMERICA ISSUE 03 - 2024
MANUFACTURING
a method with maximum sensitivity and one with the highest level of accuracy for quantitation, the decision on the workflow should always be based upon the question that needs to answered.
Conclusion
In summary the number of MS workflows in routine and ad hoc use for biopharmaceutical CMC development has never been higher and will only continue to grow given the increasing advances in MS instrumentation and associated separation techniques. Here we have illustrated through two of the most

Vesela Encheva, PhD is an Associated Principal Scientist at the R&D department at Lonza where she supports the development and implementation of mass spectrometry based methods for the characterisation of biotherapeutics. Prior to joining Lonza Vesela completed her Doctoral degree on the subject of Proteomics and spent several years at the Francis Crick Institute establishing the Mass Spectrometry Technology platform and providing analytical solutions to answer complex biological questions.
popular MS workflows MAM/iMAM and HCP-MS how far the use of MS has come in the last 10 to 15 years. The major challenge is still data processing given the large amount of high quality data produced by modern MS intruments, and how to interpret this, we are only really scratching the surface of utilising all the data is available. MS software vendors need to embrace AI and machine learning tools in order to help develop software that process MS data quicker, more easily and providing even more valuable insights to help accelerate biopharmaceutical CMC development.

Ian Anderson, PhD, is a Senior Technical Leader in the Global Biologics Technical Development team at Lonza. He has over 15 years’ experience in mass spectrometry, focusing on analytical characterisation strategies for numerous complex recombinant proteins, NME and AAV from pre-clinical through to commercial. Ian has previously worked for Allergan, AbbVie and Pharmaron gene therapy.
www. pharmafocusamerica.com 75 AUTHOR BIO
MANUFACTURING
The Emerging World of Smart Pills, Patches, and Implants
Innovative AI-Enabled Drug Delivery Systems
Personalized medicine aims to provide treatments unique to each individual for superior therapeutic outcomes. As computing technology and artificial intelligence capabilities rapidly advance, the future of individualized healthcare may be unlocked via intelligent drug delivery systems that can be honed to your body’s changing needs. Novel ingestible, wearable and implantable devices infused with AI offer self-tailored diagnostics and automated, timely interventions with minimal patient effort.

Prachi Khamkar serves as Director at Atlaas Pharmaceutical
Oral Smart Pills: Sensing and Intervening from the Inside
In the past, monitoring gastrointestinal status required invasive procedures or imaging tests external to the body. Now smart pills traversing the digestive tract can directly examine tissue microenvironments, capture disease signals and release targeted drugs in real-time through AI guidance (Nguyen
76
INFORMATION TECHNOLOGY
et al., 2022). Enabled by breakthroughs in material miniaturization and microelectronics integration, digital pills can autonomously regulate therapies based on dynamic personal biology.
The Philips IntelliCap pill senses biosignals via embedded electrodes as it travels through the GI tract, gathering temperature, pressure and transit metrics wirelessly transmitted to physicians (Philips, 2023). Future iterations envision the capsule releasing medications at ideal intestinal locations when antibiotic levels dip or inflammation markers spike.
Meanwhile, the SmartPill system utilizes a range of sensors including manometry, pH, pressure and temperature detectors to assess motility patterns, analyze gastric emptying functionality and pinpoint intestinal aberrations over 24-48 hours via location-stamped data (Rao & Yan, 2022). Dysmotility profiles help diagnose gastrointestinal disorders like gastroparesis or chronic constipation.
AI algorithms can continuously process streams of sensor measurements from the indigestible pills, modulating drug diffusion rates from reservoir wells on cue. This could enable insulin release spikes after measuring elevated post-prandial glycemia or anti-emetic discharge when vomiting motions are sensed. AI personalizes treatments to the host body’s momentary conditions for optimized, timely interventions.
Such smart pill pipelines herald a coming wave of automated, individualized medicine via miniaturized drug delivery systems responsive
to our dynamic inner physiology inaccessible before to external monitoring or control.
Wearable Drug Delivery: OnDemand Dosing Wherever You Go
Transdermal patches have long offered continuous drug input through the skin. Now wearable drug delivery platforms integrated with artificial intelligence (AI) analytics and connectivity establish closedloop systems modulating dosing based on the wearer’s changing biology and activities (Wang et al., 2022). Apps sync with patch data including sweat biomarkers, heart rate, oxygen saturation, sleep cycles and motion patterns. Cloud analytics inform AI models controlling drug diffusion from reservoirs via onboard micropumps, with doses personalized to the patient by continually sensing individual physiological responses. Source
www. pharmafocusamerica.com 77
image: www.ondrugdelivery.com INFORMATION TECHNOLOGY
of
For diabetics, a smart insulin-release patch could monitor interstitial glucose via sensors pricking dermal interstices along with tracking carbohydrate intake and exercise. AI then regulates insulin infusion rates to maintain euglycemic equilibrium. This avoids overcorrection extremes the way fixedschedule injections may induce occasionally. Meanwhile, neural stimulation patches help manage substance withdrawal syndromes by detecting craving biomarkers, physiological arousal or contextual addiction triggers. The patch intervenes with customized electric nerve stimulation for relieving these states based on the person’s unique characteristics.
Such self-regulating closed-loop drug delivery implants free patients from continual self-monitoring and complex manual self-dosing decisions. AI empowerment helps overcome the challenges of accounting for multidimensional lifestyle and health data by automating systemic, subcutaneous or nerve targeted drug input tailored to your changing biology. You simply live life while your intelligent patch therapeutically adapts as needed!
Implanted Microdevices: Automated Therapies Within
Active implantable medical devices like pacemakers have transformed patient outcomes for decades by electrically regulating organ activities. Now rapid advances in microelectronics, biossensors, wireless connectivity and AI enable more intelligent implants continually optimizing therapies
AI integration into devices revolutionizes healthcare, offering personalized medicine with self-tailored diagnostics and automated interventions for superior therapeutic outcomes.
aligned to your physiology’s 24-hour fluxes (Haddad et al., 2022). These embedded smart devices analyze biomarker patterns using machine learning algorithms to autonomously tune stimulation parameters, drug infusion or gene editing constructs (Eblin et al., 2022). Millimeter scale microchips integrated into miniaturized pumps, stimulator arrays or drug reservoirs provide persistent in vivo closed-loop relief without repeated interventions.
For instance, researchers have developed an implantable artificial pancreas with embedded sensors tracking interstitial glucose continuously together with an insulin reservoir that is algorithmically controlled (Seidman et al., 2023). This smart insulin pump autonomously learns each diabetic patient’s glycemic patterns to optimize hormone release timing and dosing for maintaining euglycemia while minimizing hypoglycemic risk day and night. Meanwhile, neural implants aim to treat refractory neurological illnesses by chronic deep brain
78 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INFORMATION TECHNOLOGY
stimulation (DBS) or optogenetic modulation guided by AI (Kernel, 2023). These protocols promise personalized therapies by tuning stimulation sites, durations and intensities to biomarkers and symptoms continually parsed by algorithms.
Driven by electrical or chemical cues, future cellular engineering chips may travel to diseased tissue for executing localized gene, RNA or chromatin editing. Still, ensuring longterm safety alongside potency remains vital before clinical adoption (Gelinas et al., 2021), given implants’ direct access to sustaining physiology difficult to retrieve or deactivate. But the promise of set-and-forget, perpetually vigilant intelligent implants enhancing health ominously also raises risks of potential hacking, glitches or uncanny side effects without easy resolution. And as autonomous AI systems replace patient oversight, how is responsibility shared for adverse outcomes?
Nonetheless, AI-guided therapy-secreting
miniaturized implants offer an exciting frontier this decade for perpetually responsive safeguards against destabilizing or painful states. Together with companion wearable pods tracking relevant lifestyle contexts, they support harmonized care rooted in your multifaceted biological truth.
The Optimizing Power of AI-Guided 3D Printing
Three-dimensional printing of pharmaceuticals allows creating personalized products like tablets, printlets or drug-integrated implants matching patients’ preferences and needs (Buanz et al., 2020). Now integration of big data inputs, predictive analytics and generative computational design by artificial intelligence can further enhance this manufacturing approach.
First, AI algorithms can trawl through electronic health records, pharmacogenomics data as well as lifestyle contexts to model

www. pharmafocusamerica.com 79
INFORMATION TECHNOLOGY
optimal treatment regimens tailored to someone’s cumulative health status and constraints. This forecasting then guides 3D printer parameters and materials selected. For instance, based on age, weight, disease severity and genomic enzyme profiles, the system can define the ideal dose of drug X to combine with a sustained release agent Y for a elderly patient with organ dysfunction.
Additionally, as the product blueprint is iteratively modeled, generative neural networks can construct parametric 3D architectures aligned to ease swallowing for this individual or medications adhering to intestinal mucosa targets. Tablet geometries, densities and polymer coatings are computationally evolved to provide desired diffusion rates through gastro intestinal terrain while avoiding sensitivities. Integrating sensor-laden digestible materials further enables functional smart pills.
Together with high resolution printers depositing pharmaceutical inks, the AI system guides fabrication of products with properties hopelessly challenging to conceptualize alone!
AUTHOR BIO

Hence, pairing strong predictive algorithms and simulations with rapid prototyping techniques support creation of revolutionary therapeutics attuned to your system’s intricate coordinating impulses. Conclusion
The integration of artificial intelligence into innovative drug delivery systems signals a paradigm shift towards highly personalized medicine. Intelligent pills, patches, and implants promise to revolutionize care by continually sensing physiological changes and providing tailored, real-time therapeutic interventions. Despite outstanding questions surrounding security and accountability, these AI-enabled treatments herald exciting new possibilities for predictive, preventative, and participatory care centered around the patient's unique biology. In essence, the future of healthcare may reside inside the body, as personalized "theranostic" systems another step closer to reality. References are available at www.pharmafocusamerica.com
Prachi Khamkar serves as Director at Atlaas Pharmaceutical, where she pursues her passions for pharmaceutical 3D printing and personalized medicine. Holding a pharmaceutical sciences degree with research specialization in topical drug delivery, she also holds an India Director position at ReachSci in University of Cambridge. Additionally, Khamkar studied business management in London. With articles and book chapters published internationally, Khamkar contributes valuable industry knowledge and leadership - furthering pharmaceutical innovation through cutting-edge technologies and patient-centered approaches.
80 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INFORMATION TECHNOLOGY
A Quick start Guide for Successful Cloud Migration of Global Pharmacovigilance (GPV) IT Applications

The article will talk about how to plan for migrating Global Pharmacovigilance (GPV) related IT applications which assists every pharma company to ensure patient safety as per global Health Authority requirements. It will have following subtopics.
Vikalp Khare Director, Otsuka Pharmaceutical Development & Commercialization
In the ever-evolving regulatory landscape of Global Pharmacovigilance (GPV) operations to ensure patient safety for a pharmaceutical company, the decision to migrate GPV specific IT applications to a cloud-based infrastructure is a pivotal strategy for
www. pharmafocusamerica.com 81
INFORMATION TECHNOLOGY
organizations seeking to enhance agility, reduce costs, and embrace transformative technologies. Cloud migration offers a pathway to a more flexible, scalable, and innovative IT infrastructure which is also easily compatible with new age Artificial Intelligence (AI) & Machine Learning (ML) platforms.
This journey to the cloud-based setup is not without its challenges and a well-thoughtout plan is paramount to success. This article aims to serve as a compass for Clinical Safety & Pharmacovigilance teams who are looking to seamlessly migrate their GPV-IT applications on to cloud. We will delve into the key considerations, recommendations, and crucial steps that will pave the way for a successful cloud migration.
Need for Cloud Migration
The journey to set up a cloud-based operations starts with identifying the need for change. Migrating to Cloud offers numerous advantages not only from Infrastructure operations perspective but will also enhance the scalability of GPV IT applications (like Safety Databases, Signaling solutions, etc.). Few basic questions that need an evaluation are:
• How the Cloud operations will be beneficial for GPV operations?
• How will they impact the GPV technology roadmap?
• How will it help the objective of being a globally compliant organization as per Global Health Authority requirements?
• Which Cloud Service Provider (CSP) aligns
with my organization’s future technology roadmap/strategy to ensure an efficient intra-company integration with other existing applications?
• Which cloud model deployment model (public, private or hybrid) is the best fit considering residency requirements, regulatory needs, scalability, and cost- effectiveness
A major driver for identifying the need is also the calculation of Return-On-Investment (ROI) from GPV system maintenance perspective. Migration to cloud will have its own effort requirements which should also be part of the organization’s ROI analysis as well.
Cloud migration strategy should consider the regulatory requirements, Geography location criteria (e.g. Europe as a strict General Data Protection Regulation (GDPR) requirement which may need especial arrangements), local/ global security & privacy consideration.
Overall need for cloud migration should focus on the improvisation such as data
Migrating Global Pharmacovigilance IT to the cloud enhances agility, reduces costs, and facilitates transformative technologies, requiring careful planning for compatibility with AI and ML platforms.
82 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INFORMATION TECHNOLOGY
accessibility, global technology integration, scalability, cost reduction, and disaster recovery capabilities.
Plan Cloud Migration Planning
The future state of GPV operations will rely heavily on how the cloud infrastructure is managed. Organizations may opt for a Software-as-a-service (SaaS) model OR can also opt to have an in- house team manage the applications & dependent cloud infrastructure. A hybrid setup which is a combination of SaaS & internally managed IT cloud applications is also a possibility. However, the network communications between SaaS & internal clouds needs a robust set-up to ensure reliant interactions. A well-defined strategy finalized post evaluation of the existing state of GPV IT applications, infrastructure & data will effectively manage potential challenges and risks of a Cloud migration. This strategic plan will also be the driver for your Post Go-Live maintenance activities.
Needless to highlight that a robust project plan which is tracked & controlled by strong program managers is critically important for

success. A cloud migration project plan should at least focus on the following:
• Risk assessments & regulatory compliance adherence at each step of migration.
• Internal organizational policy adherence in case there is a geographical change of location.
• Ensure Data Integrity is maintained to support the desired business operations.
• If opting for a hybrid operational model, Infrastructure integration should support information exchange in multiple formats with both in-house & outside applications.
• Comprehensive platform qualification & validation of applications as per the requirements.
• Security & vulnerability management during the migration. Strategy for Post Go-Live operations.
• Ensuring business continuity through Cloud High Availability & Disaster Recovery setups.
• Once the cloud migration is completed & Post migration business operations have been successfully established, decommissioning of your old infrastructure and application should also be part of the plan.
Apart from a robust project plan, Strong & proactive communication is also essential. Cloud migration will involve collaboration with multiple teams & hence collaboration with multiple stakeholders is needed to ensure success.
Data Migration & PV Application
Refactoring
GPV applications generally exchange Product
www. pharmafocusamerica.com 83
INFORMATION TECHNOLOGY

Safety data outside of the organization with Global Health Authorities, Licensed Partners, Clinical Research Organizations (CROs), etc. in predefined formats like Individual Case Safety Report (ICSR) R2 or R3. Hence, Data migration strategy will be critical to meet the Organizational compliance goals. Need for Data migration will also be governed by the need to perpetually maintain the data. Sometimes, data might not be needed for daily operational needs, but needs to be maintained for future references. Data migration strategy should be formulated to involve data cleansing, transformation, and validation to enhance operations currently managed through PV applications. Another critical driver for Data migration is the location of existing data center & the new target cloud data center.
Validation requirements, application setup changes & modification of company policies (both internal & external) would be driven by the location where the data for GPV Application is stored.
Organizations may need to revisit the existing policies to safeguard the data exchange as per the regional privacy requirements (For e.g.: Exchanging the patient safety information from and outside of Europe may need to adhere to
(1) GDPR regulations &
(2) The guidance from European Medicine Agency (EMA)). Cloud migration can be formalized through multiple ways:
“As-Is” Migration Approach
This is commonly also referred to as a ‘Lift & Shift’ approach, where the existing setup of GPV applications is re-installed on a cloudbased infrastructure with a copy of underlying data from the previous infrastructure. This normally will involve a lot of technology-related operations for data migration and application installation. This approach has almost no impact on existing business operations. All applications will continue to operate nearly the same manner as they were prior to cloud based operations. This approach does not need a lot of investment & has a quick turnaround too. Most of the time this approach is followed by a second wave of business process re-engineering, where the existing operation processes are further tuned to benefit from existing cloud capabilities.
Phased Approach
This approach is common for the initiatives where the cloud benefits are first to be evaluated on a smaller set of applications. Organizations may choose to first migrate only a couple of GPV applications and evaluate their operations
84 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INFORMATION TECHNOLOGY
& interactions with global applications. Once successful, other applications will follow the same approach till all are operational on cloud. It is normally preferred to prioritize the initial migration for the stand-alone applications which supports a part of business operations & have minimal interaction with other applications/ business operations. Transforming the business processes along with migration is optional and depends on the end objectives. Coz of the iterative nature of this migration, the risk associated is quite low, effort involved is high and can potentially take longer time to attain the final future state. In case, if the cloud migration does not align with expectations, it is easy to roll-back to the previous setup.
Transform/Upgrade with Migration
Each IT application needs a periodic upgrade or modification to ensure the GPV operational compliance in accordance with evolving global regulatory requirements. Each of these activities has a direct impact on the existing business processes. Hence, it is also a suggested approach to combine the cloud migration activity with your planned upgrade/modifications. This can be merged with both “Phased” or “Lift & Shift” approach. There is also a possibility that combining the application upgrade & cloud migration can result in better overall operational & cost benefits. Sometimes companies prefer to perform an upgrade of their safety database as they move to cloud. Performing an upgrade on existing platform & then migrating to cloud may result in added validation effort/cost as one
will need to re-certify the expected operations on cloud. Hence, it is suggested to migrate first and then upgrade/modify.
Validation & Deployment on Cloud
Any Pharmacovigilance system at a broad level comprises of 2 tiers – Application level & the underlying Infrastructure on which application runs. Validating a Pharmacovigilance system is a critical step in ensuring the safety and efficacy of pharmaceutical products. Usually, cloud platform/infrastructure validation is done first & is followed by application’s recertification for operating as per the requirements. It helps organizations meet regulatory obligations, maintain data integrity, and contribute to the overall improvement of drug safety practices.

www. pharmafocusamerica.com 85
INFORMATION TECHNOLOGY
Strategic cloud migration in Global Pharmacovigilance enhances patient safety, offering a flexible IT infrastructure aligned with AI and ML technologies.
Usually, companies rely on the standard internal tests conducted by various CSP for platform validation. The functionality requirements of the platform should be tied to the contractual agreements with CSP, if the company is relying heavily on the Out-of-Box functionality of the platform. Companies may also choose to audit the platform development procedure of the CSP to be sure of the services expected to be received in future from a CSP.
Validation of a Pharmacovigilance application involves ensuring that the system is designed, implemented, and maintained in a way that meets regulatory requirements, industry best practices, and organizational needs. Specifically for Cloud migrations, Validation of handshakes between applications on different platforms needs to be planned at early stages of project itself. This is because sometimes, the findings from these may result in changes on the other applications which may not be in original scope of migration.
Furthermore, Validation plan should specifically focus on meeting the regional & global data protection regulations if the data migration involves movement of data between continents. Any data migration pertaining to Global Safety Database should always account for notifying the Global Health authorities and certifying that the company’s data exchange gateway is able to exchange information back and forth with required Licensed Partners, Affiliates, CROs & Health Authority gateways. It may also be advantageous to leverage the outcome of Cloud platform to assist with applicationlevel validation activities.
Performance testing (to test response time & scalability) should also be considered at the application level to ensure that the underlying cloud platform is able to provide the necessary resources for application to work as desired.
Please note that validation of Global Pharmacovigilance systems should specifically consider testing the application accessibility from different geographic regions, to proactively highlight any network related issues, if any.
For assistance with deployment of applications on cloud, utilize cloud-native services for authentication, authorization, storage, and data processing to gain maximum advantage. Consider Deploying the PV application to the cloud using Infrastructure-ascode (IaC) tools for repeatability and scalability. IaC capabilities will save effort if opting for a phased migration approach. Utilize pre-existing cloud capabilities for application back-up, restoration & disaster recovery to ensure
86 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INFORMATION TECHNOLOGY
business continuity. Furthermore, Cloud’s auto-scaling and load balancing can also be effectively used to handle variable loads.
Trainings & Post Go-Live monitoring
As they say, “Change is the only constant”, Cloud operations will only result in achieving the desired ROI as envisioned at time of finalizing the strategy to migrate if the operating teams are trained to effectively harness the cloud capabilities both at application & infrastructure level. End user operations will significantly change based on the cloud capabilities and the opted data migration approach. Effective Change management strategies should be devised in advance to ease-in transition for teams.
Technical maintenance teams are recommended to go for certified training of cloud platforms. Monitoring alerts needs to be set up both for security vulnerabilities & cloud resource utilizations to achieve maximum cost

benefit with cloud operations. Native cloud capabilities can also be considered for this. It is strongly recommended to conduct periodic reviews of cloud resource utilization needs to ensure efficient cloud expenditure. If one continues to use the GPV applications with same static platform parameters like CPU, RAM, etc., then ROI realization may be difficult. Cloud computing cost is directly linked to the resource usage & it is strongly advisable to tweak these parameters based on the utilization trends of an application. In summary Cloud gives a lot of flexibility in ways of how we use the system.
Using these new flexible options will eventually lead to significant savings on a year-on- year basis along with increased reliability of the system.
With the guidance from this article, pharmaceutical organizations (Big/Small) can embark on a successful cloud migration journey for GPV-IT applications, realizing the full potential of cloud computing while maintaining compliance, data integrity, and operational efficiency.
Vikalp Khare i s a Director at Otsuka Pharmaceutical Development & Commercialization (OPDC). He has been implementing new age technology-based solutions to support Global Pharmacovigilance Operations. He has extensive experience in setting up Global Safety Database & related technology applications for Pharmaceutical Companies which are globally compliant to Health Authority Safety Health Authority regulations for ensuring Patient Safety. He currently oversees the Global technology & Business Intelligence (BI) operations as the US-Head of Safety Data Management, GPV at Otsuka Pharmaceutical Development & Commercialization (OPDC).
www. pharmafocusamerica.com 87 AUTHOR BIO
INFORMATION TECHNOLOGY
Advances in Liquid Biopsy Research for Disease Detection, Diagnosis, and Treatment and What the Future May Hold
 Dr. Courtney Noah Vice President of Scientific Affairs, BioIVT.
Dr. Courtney Noah Vice President of Scientific Affairs, BioIVT.
1. Can you elaborate on the transformative potential of liquid biopsy in revolutionizing disease diagnostics, especially in comparison to traditional biopsy methods?
Liquid biopsies offer several advantages over traditional biopsies. They are minimally invasive; the sample can be obtained from a simple blood draw and does not require an incision or tissue sample. Also, administering traditional tissue biopsies can lead to excessive bleeding, infection or less likely but possible spreading of cancer cells throughout the body.
EXPERT TALK
Liquid biopsies can provide more comprehensive information about the disease burden than tissue biopsies. They can detect circulating cancer cells or genetic signatures before they form a primary tumor. This allows doctors to learn more about the cancer and any mutations that may be present throughout the body, not just at one site, at a time when they would be difficult to detect with imaging techniques.
The data obtained from liquid biopsies can be used to inform diagnoses and provide more precise treatment options. Due to their ease of collection, liquid biopsies provide an opportunity for serial sampling, enabling ongoing monitoring of a patient’s response to treatment or disease recurrence.
Liquid biopsies also give researchers access to longitudinal data, allowing them to study the evolution of drug resistance and development of the metastatic state with the goal of improving future treatment options.
Compared to traditional biopsies, liquid biopsies also have a much shorter turnaround time. Traditional biopsies can take days or weeks to process and analyze, whereas liquid biopsies can provide results in as little as 24 hours. This shorter turnaround time can help physicians make quicker decisions about treatment options and sponsors decide whether to move forward with clinical trials for a new drug candidate.
2. How has the integration of nextgeneration sequencing (NGS) and other cutting-edge technologies improved the precision and accuracy of liquid biopsy, particularly in detecting rare genetic mutations?
The barriers to liquid biopsies clinical utility center on the low abundance of circulating tumor DNA (ctDNA), which causes many sensitivity and specificity challenges. However, researchers are seeing benefits from using a multi-analyte approach. By simultaneously targeting multiple biomarkers, studies have demonstrated improved assay sensitivity for the detection of mutations as well as the potential to uncover therapy-resistant genes.
Some recent examples of this multianalyte approach include the CancerSEEK platform, which screens for up to nine cancers by detecting eight unique proteins and oncogenic mutations using ctDNA. The OneTest platform uses machine learning and proteomics to screen for up to six cancers. Targeting multiple biomarkers has the potential to significantly enhance the efficacy of screening for cancers at an early, treatable stage.
Liquid Biopsy: Advances in Disease Detection, Diagnosis, and Treatment, Explores the Promising Future of Diagnostic Innovation.
www. pharmafocusamerica.com 89
EXPERT TALK
3. In your view, what specific advancements in liquid biopsy have shown promising results in the early detection of cancers, and how do these findings impact the overall diagnostic landscape?
One of the most exciting advancements pertains to increasing the sensitivity of liquid biopsies using a priming agent. False negatives have been a constant challenge for the industry due to the low levels of target analytes. Cell free assays must detect tiny amounts of target DNA or RNA, which may be as low as two or three molecules in each tube of blood. Researchers at MIT and the Broad Institute of MIT and Harvard have developed two types of injectable molecules or priming agents that can be used to temporarily slow down the clearing of tumor DNA from the bloodstream and effectively boost that signal. Their work is still in the early phases of testing and development, but it holds a lot of promise for improving early cancer detection.
As the volume of tumor-derived signals in a tube of blood is very small, techniques need to be developed that can increase the amount of information that can be gleaned from it. Recent approaches based on epigenetics, transcriptomics, and proteomics data are showing promising results that could lead to improved sensitivity and specificity of liquid biopsy tests in certain clinical contexts. By including additional patterns and insights identified by using artificial intelligence (AI), it is possible to get more answers from a single blood draw. By taking a multi-modal analytical approach to liquid biopsy develop-
ment, we can provide patients and healthcare providers with faster, less expensive, and more flexible approaches.
4. Could you discuss a notable example or case study where liquid biopsy played a pivotal role in monitoring treatment response and guiding the implementation of personalized therapeutic strategies?
Labcorp’s Plasma Focus liquid biopsy test was recently brought to market and helps oncologists to develop targeted, personalized treatment plans for their patients with advanced solid cancers. Requiring only a standard blood draw, this test compares cellfree DNA released by tumor cells against a highly curated gene panel that focuses on U.S. Food and Drug Administration (FDA-) approved biomarkers. It was developed to help improve outcomes for patients with non-small cell lung, colorectal, breast, esophageal, gastroesophageal junction, gastric cancers and melanoma.
5. What key challenges in terms of sensitivity and standardization must be addressed to bridge the gap between liquid biopsy research and widespread clinical implementation?
Members of academia, industry, and government agencies are all acutely aware of the gaps in standardization practices. As a result, many of them have joined consortiums and are working together to better define standards and improve the clinical utility of precision medicine.
One such group, BLOODPAC, recognized the challenges involved in liquid biopsy
90 PHARMA FOCUS AMERICA ISSUE 03 - 2024
EXPERT TALK
development and standardization. It has established Minimum Technical Data Elements (MTDEs), which reflect factors that influence the performance of a liquid biopsy test in the laboratory and may affect the results for different areas of test validation. These MTDEs enable researchers to compare data sets across studies, draw important conclusions about the impact of specific pre-analytical variables, and (not to be overlooked) present a common vocabulary to ensure terms are used consistently and with appropriate meaning.
In addition, industry stakeholders such as the International Society of Liquid Biopsy and the European Society for Medical Oncology have been establishing guidelines for specimen handling, processing, and storage. These guidelines aim to ensure that samples are collected and processed consistently across different laboratories, enabling results to be compared between studies.

Another challenge that has been raised in the development of liquid biopsies focuses on experimental design. When comparing study results, there is some evidence that prospective cases show higher sensitivity than retrospective studies. This finding has been attributed to there being a high proportion of false negatives in retrospective studies. As the industry endeavors to accelerate development in this area, it needs to obtain results from prospective studies that more closely mirror the real-world diagnostic workflow.
6. Looking ahead, how might liquid biopsy technologies evolve to address not only cancer but also other disease areas, and what implications could this have for preventive healthcare?
While it is still early days, there has been an uptick in discussions about the application of liquid biopsies for central nervous systemrelated diseases. There are many lessons learned from studying cancer biomarkers that can help in advancing the early detection of Alzheimer’s, Parkinson’s and other neurodegenerative diseases. Both early detection and surveillance of neurological disorders can benefit from the fast and non-invasive nature of liquid biopsy assays. As population’s worldwide age and the incidence of neurological diseases rises, identifying biomarkers that can help to diagnose those conditions and inform treatment options has become an urgent need.
Research into cerebrospinal fluid (CSF) biomarkers for neurological disease has been progressing steadily over the past decade. Some of these targets go beyond the traditional cell-free DNA and include
www. pharmafocusamerica.com 91
EXPERT TALK
extracellular vesicles (exosomes, microvesicles, and apoptotic bodies), microRNA, and other nucleic acid derivatives. Some researchers are investigating a combination approach with CSF and blood-based biomarkers. Roche, in collaboration with Eli Lilly, is developing a blood-based liquid biopsy test to facilitate earlier diagnosis of Alzheimer’s disease, the Elecsys Amyloid Plasma Panel.
As more effective treatment options become available for these neurodegenerative diseases, it is critical that improved diagnostics are brought to market to streamline the path to diagnosis.
7. How do you envision the ethical considerations surrounding liquid biopsy evolving, and what regulatory frameworks should be in place to ensure responsible and equitable deployment in clinical practice?
Research shows significant racial and income disparities with respect to access to genomic profiling for cancer treatment selection. However, liquid biopsies offer the future potential to reduce these gaps in usage and improve access due to their minimally invasive nature. They will enable improved testing compliance by minimizing the burden of repeat hospital/clinic visits and attendant travel expenses, requisite time off work, and childcare costs. Liquid biopsies also allow the use of alternative care delivery models, e.g., direct-to-patient via mobile phlebotomy services, or blood draws during doctor visits, which may eliminate barriers for patients who face transportation roadblocks. They could also help to reduce geographic healthcare challenges in rural communities.
In terms of regulatory issues, one of the greatest challenges facing liquid biopsy usage today is broadening accessibility through more consistent payer coverage. While some states have passed legislation requiring health plans to cover biomarker tests, including some liquid biopsy assays, that meet certain evidentiary requirements, many US states have not passed any minimum coverage laws. This problem is not specific to the US. In the European Union, the usage of liquid biopsies seems to be limited to clinical trials or research in highincome countries, and not in low- or middleincome countries. It is critical that advancements proceed at a rapid pace to continue to generate clinical evidence in support of the validity of these assays.
8. Can you share an example of a successful interdisciplinary collaboration that significantly advanced liquid biopsy research, and what lessons can be drawn from such collaborations for future endeavors?
One example is the ctDNA for Monitoring Treatment Response (ctMoniTR) project that was facilitated through Friends of Cancer Research. This project succeeded in bringing together data from multiple, independent studies (spanning non-profits, academia, industry, and government) which accelerated their ability to generate robust analyses as a collective organization. The groups came together to answer one principal question, “Do changes in ctDNA reflect response to treatment?”
They were able to harmonize data across eight clinical trials and observe an important
92 PHARMA FOCUS AMERICA ISSUE 03 - 2024
EXPERT TALK
clinical finding that for patients with non-small cell lung cancer (NSCLC), who were treated with immune checkpoint inhibitors, reductions in ctDNA levels on treatment (detected via liquid biopsy) were associated with improved overall survival. While this interdisciplinary collaboration initially focused on NSCLC, the group intends to extend its study to include additional tumor types, stages, and drug classes.
9. From your perspective, how has industry involvement accelerated the development and commercialization of liquid biopsy technologies, and what challenges persist in achieving widespread accessibility?
Accounting for diversity in clinical trials has taken center stage in the past few years.
Researchers and companies seeking approval for late-stage clinical trials are now required to submit a plan for ensuring diversity among trial participants to the FDA. This requirement highlights both the need and urgency to overcome barriers to ensure appropriate patient groups are participating in clinical studies. Although this has become an important policy priority, progress has largely stalled on improving the participation of minority population groups. We need to make inroads, particularly as liquid biopsies become more widely available, in designing equitable clinical research programs that match the demographics of the disease.
I believe diversity plans need to be included earlier in the diagnostic development workflow where there are opportunities to gain insights into demographicdriven factors. It is critical that researchers

assess the impact that diversity may have on their diagnostic’s sensitivity and accuracy during the preclinical phases of discovery and development, rather than at later-stage clinical validation.
As the population diversifies organically, there is a greater need and urgency to support generalization of findings. With enhanced preclinical data from diverse cohorts (that represent the disease incidence), there is more informed development of clinical guidelines.
The genetic, proteomic, and epigenetic profiles of populations can vary significantly with age, race and gender. Without earlystage assessment of these factors, unique targets can go unnoticed/undiscovered, minimizing the potential opportunities for precision medicine.
Finally, compiling representative data can help in gaining trust and educating the patients that are most critical for clinical trial
www. pharmafocusamerica.com 93
EXPERT TALK
enrollment. Presenting scientific evidence that is applicable and relevant can have a tremendous positive impact and highlight the value of participation.
10. In promoting public awareness and understanding, what key messages should be conveyed about the capabilities and potential impact of liquid biopsy, and what steps can be taken to ensure broader acceptance within the medical community and beyond?
Public awareness of liquid biopsies needs to improve in two areas. First, we must ensure that the patient populations which have the most to gain from these novel assays are educated on their availability and value. Patients need to understand the potential risks, benefits and common misconceptions surrounding these tests. Second, physicians must be informed about the availability and appropriate use of these tests and how to discuss them with their patients.
Physicians must continue to receive new evidence demonstrating the reliability of existing and emerging liquid biopsy technologies in cancer care. It is critical that they receive use cases and interpretation of data that can complement or provide alternatives to the standard of care. To achieve this, a coordinated effort needs to be made by assay developers, government, research associations and patient advocacy groups to provide guidance and education on the best approach for integrating liquid biopsy testing into traditional cancer treatment programs.
11. Considering the advancements, challenges, and future prospects we've discussed, what overarching message or insight would you like to leave our audience with regarding the role of liquid biopsy in transforming disease detection, diagnosis, and treatment, and its potential impact on the future of healthcare?
Given the enormous potential these assays hold for improving standard of care with a less invasive approach, we will continue to see rapid advances to broaden accessibility and use cases with the technology. As we are just scratching the surface regarding utilization of AI, I believe that will be the next big step forward with algorithms seeking out patterns and associations in multi-omics datasets. With this, I expect to see further adoption of liquid biopsies to improve cancer care and support personalized medicine approaches.

Dr. Courtney Noah is BioIVT's Vice President of Scientific Affairs. She leads a team that provides research solutions for BioIVT’s clients and business partners. Dr. Noah received her PhD in Molecular and Cellular Biology from Stony Brook University, and her BS is in Food Science from Cornell University.
94 PHARMA FOCUS AMERICA ISSUE 03 - 2024
AUTHOR BIO
EXPERT TALK

Determining the ability of a target to bind to a drug with high affinity to modulate its function with a therapeutic benefit could be termed Druggability. It needs a non-trivial amount of time, financial investment, and resources. An investment of US$2.6 billion (£1.9 billion) over 10-15 years for inventing a new drug from target identification to approval, which ultimately benefits the patients, underscores the importance of prioritizing the most potential druggable targets.
“Effective target identification lies at the core of drug discovery. In theory, it sounds simple — find a target, and match it with a game-changing therapy. In practice, confirming that match is a scientific quest in itself”
Comparable to the high cost, the success rate for drug development programs is substantially low with over 90% of the drug candidates failing in clinical trials, which puts the pharmaceutical industry under mounting
pressure. The biggest reason for drug attrition is mainly safety findings or lack of efficacy (Figure 1), nearly 60% of failures are attributed to inappropriate target identification and validation in drug discovery.2,3 To increase the chances of success and maximize return on investment, drug inventors must consider strategies focusing on the identification of the most biologically plausible druggable targets that effectively modulate the disease phenotype on a molecular level.
Selecting the right target early in the discovery R&D process is crucial in enabling the prospects of regulatory approval success. Prioritization of the most promising targets involves consideration of several key factors including disease relevance, role in underlying pathophysiology, specificity to the disease process or state, high frequency in the patient population, tractability (the possibility of finding small molecule compounds with high affinity), safety profile, novelty, as well as the
www. pharmafocusamerica.com 95
WHITEPAPER
competitiveness in the market. After target identification, the critical imperative is to assess target druggability which is integral to successful target validation and is a pivotal step in every drug design quest. Despite the known 3D protein structures surpassing 100,000 in number and ChEMBL database encompasses around 5,000 known proteins with bindable pockets, a mere 854 of these proteins have been recognized as established therapeutic targets for FDA-approved drugs. These data demonstrate that the eligibility of a protein, and consequently the protein pocket, for binding of a molecule and hence drug discovery is a formidable challenge. Additionally, because a substantial number of small-molecule drug discovery project failures are owing to poor bioavailability, careful evaluation of biological and molecular basis of target druggability and building a precise druggability prediction method (DPM) are extremely essential at the early stage of drug discovery programs.
A drug discovery program starts with the identification of novel and effective drug development technology with biological targets for the development of new drugs with unmet clinical needs. Discovering and evaluating the potential therapeutic benefit of a drug target is founded not only on experimental, mechanistic, and pharmacological studies but also on a theoretical molecular druggability assessment, an early evaluation of potential safety measures, and through considerations regarding opportunities for commercialization as well
as options for the generation of IP. Traditional approaches are inadequate for large-scale exploration of novel drug targets, as they are expensive, time-consuming, and laborious. In recent years, various computational strategies for predicting potential druggable proteins have emerged, which commonly use the sequence, structural, and functional features of proteins as input but also system-level properties such as network topological features. Despite tremendous success, unfortunately, many promising and experimentally validated targets are not within the scope of drug modifiability. The Discovery of next-generation technologies including targeting protein degradation, protein stabilizers (RESTORACs), excellent drug delivery systems, targeting PPI, targeting intrinsically disordered regions, as well as targeting protein-DNA binding may provide significant assistance in overcoming these undruggable targets.
Our latest whitepaper "Identifying Druggable Therapeutic Targets: Unveiling promising avenues in Drug Discovery" sheds light on transformative approaches to increase drug discovery and development success rates.

96 PHARMA FOCUS AMERICA ISSUE 03 - 2024
to Download the white paper CLICK TO DOWNLOAD THE WHITE PAPER
WHITEPAPER Scan
Next-Generation Vaccines Harnessing mRNA and Novel Platforms
Welcome to the PFAm Magazine's Panel Discussion on "Next-Generation Vaccines: Harnessing mRNA and Novel Platforms." We are honored to host three distinguished experts in the field, each bringing a unique perspective and wealth of experience to the conversation divided into three series, each exploring different facets of the transformative landscape of NextGeneration Vaccines.
Series 1 - Unveiling the Future of Vaccines
Explore the landscape of next-gen vaccines with experts Aaron B. Cowley, Patrick Thiaville, and Daniel Kavanagh as they discuss breakthroughs, challenges, and opportunities in mRNA and novel platforms.


Aaron B. Cowley
Chief Scientific Officer at ReciBioPharm with over 20 years of experience in discovering and developing manufacturing processes for enzymes and live biotherapeutics.
Patrick Thiaville
Chief Technology Officer – Nucleic Acids, Exothera renowned for his visionary leadership in GMP operations, Process Development, and MSAT, particularly in clinical manufacturing and infrastructure design.

Daniel Kavanagh
Senior Scientific Advisor, Gene Therapy, Vaccines and Biologics, WCG, an expert in IBC oversight of gene transfer trials with a rich background in infectious diseases and tumor immunology.
Can you provide an overview of your current role and the work you are involved in regarding next-generation vaccines?
Aaron B. Cowley : As Chief Scientific Officer at ReciBioPharm, my role largely encompasses spearheading innovation and staying at the forefront of advancements to deliver exciting new therapies to patients more effectively. This is best highlighted by our ongoing project collaborating with the Massachusetts Institute of Technology (MIT) to create a fully continuous and integrated
www. pharmafocusamerica.com 97
*This is 3 part series. Episode 2 will be published next month on our website.
OUR PANELISTS INCLUDE
INDUSTRY SENSE Industry
Sense
RNA manufacturing platform, supported by the FDA’s Center for Biologics Evaluation and Research (CBER) Advanced Technology Program.
We have come a long way since initiating the project around 1.5 years ago, and our artificial intelligence (AI)-enabled continuous manufacturing service is quickly becoming a reality. With a digitally controlled system relying on predictive modelling based on the sequence of RNA, formulation and other factors, the platform will help to expedite timelines to rapidly generate RNA clinical trial material.
With innovation in mind, we’ve also developed the platform to not only support messenger RNA (mRNA) but also other formats, including self-amplifying mRNA and circular RNA, which can offer enhanced antigen expression at lower doses and higher stability, respectively.
From your perspective, what are the key scientific breakthroughs that have enabled the development of nextgeneration vaccines, particularly those utilizing mRNA and novel platforms?
Patrick Thiaville: Two key breakthroughs should be highlighted for the advancement of next-generation vaccines. The first is the incorporation of modified uracils into mRNA sequences. These modifications play a critical role in enabling the mRNA to evade host immune responses, enhancing the stability and effectiveness of mRNA vaccines. The breakthrough was recognized with the
awarding of the Nobel Prize and Physiology and Medicine in 2023 to Katalin Karikó and Drew Weissman, for their work that paved the way for the widespread application of mRNA vaccines to target various diseases — with particular significance and success in the treatment COVID-19 during the pandemic.
The second breakthrough is the development of clinical-grade lipid nanoparticles (LNPs). Although these have been used for RNA-based therapeutics for several years, we are now seeing more widespread use of LNPs in clinical research. This utilization of LNPs represents a significant advancement in vaccine technology, allowing for targeted and controlled delivery of genetic material to host cells.
The enhanced versatility and scalability offered by both of these breakthroughs hold great promise for addressing emerging infectious diseases, advancing personalized medicine, and combating global healthcare challenges.
In what ways do you foresee mRNA and novel platforms revolutionizing the vaccine industry in the coming years?
Daniel Kavanagh: The first approved mRNA vaccines have demonstrated some of the potential of synthetic biology in medicine. These vaccines delivered a viral antigen primarily designed to stimulate B cells to generate neutralizing and protective antibodies. While these vaccines also engage the cellular arm of the immune system, outcomes such as induction of antiviral
98 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INDUSTRY SENSE
Next-gen vaccines revolutionize immunization, leveraging mRNA and novel platforms for a transformative future in vaccine development.
cytolytic “killer” T cells have received less attention. Holistic mobilization of the immune system may provide increased protection, especially for latent and endemic pathogens such as mycobacteria and herpesviruses.
Synthetic biology advances medicine by developing modular components that enable flexible combinatorial solutions. Antiviral vaccines induce immunity by allowing the immune system to select and refine antigen receptors in response to the vaccinal antigen.
A potential revolutionary novel platform starts with the genetic sequence for a known effective antibody or other antigen receptor and uses a gene transfer approach such as mRNA to deliver that receptor sequence to human cells in vivo. This approach bypasses vaccination and skips straight to antibody production. Components of a protective immune response can be expressed immediately upon dosing, without waiting for immunity to develop. In the future, this approach may be combined with vaccines.
What challenges do you anticipate in the widespread adoption of nextgeneration vaccines, and how can these be mitigated?
Aaron B. Cowley : Viruses that replicate in the human respiratory mucosa generally do not elicit complete and durable protective immunity by themselves. To date, viruses like influenza A, SARS-CoV-2, endemic coronaviruses and RSV have not been effectively controlled by vaccines. As a result, many next-generation vaccines are largely being developed to target mucosal respiratory viruses and other viruses of importance, with delivery directly to these surfaces via inhalation.
However, developing next-generation vaccines for mucosal respiratory viruses and encouraging their widespread adoption comes with several challenges. Firstly, the immune system has likely evolved to tolerate these viruses for very short intervals due to a higher frequency of natural infection, meaning expert vaccine design will be needed to elicit a durable immune response. Additionally, the mucosal and systemic immunity only partially protect against infection, and immunoprotection against these respiratory viruses is not completely understood.
As a result, additional research is required to achieve the level of effectiveness that has been reached using the mainstay vaccine technologies against polio, measles and other diseases.
www. pharmafocusamerica.com 99
INDUSTRY SENSE
Patrick Thiaville: Obtaining regulatory approval and effectively communicating the safety and efficacy of next-generation vaccines to the public are critical challenges. As with any next-generation type of medication, widespread adoption relies on effective education, communication, and collaboration. On one hand, regulatory agencies must ensure that these innovative vaccines meet rigorous safety and efficacy standards before approval. Simultaneously, public education campaigns are essential to address concerns, build trust, and dispel misconceptions surrounding novel vaccine technologies. Robust communication strategies that involve healthcare professionals, community leaders, and scientific experts can help foster public confidence in nextgeneration vaccines.
Aside from communication, the inherent transient nature of RNA poses a significant challenge to vaccine stability and durability. To address this issue, ongoing research focuses on modifying RNA molecules to enhance their stability and prolong their expression within the body. Innovations such as circular RNA and self-amplifying RNA offer promising avenues to augment the stability and duration of RNA-based vaccines. By optimizing RNA design and incorporating these modifications, we can overcome the limitations associated with RNA degradation and ensure sustained vaccine efficacy over time.
Another approach to solving this problem is to explore alternative formulations and dosage forms. Moving beyond tradi -
tional frozen liquid vaccines, to investigating the possibilities of powder and solid-dose formulations is key to extending the shelf life and improving the delivery of next-generation vaccines. By diversifying formulation options, vaccine manufacturers can address logistical challenges and broaden access to next-generation vaccines, particularly in resource-limited settings where cold chain infrastructure may be inadequate.
By proactively addressing these challenges through coordinated efforts and innovation, stakeholders can pave the way for the widespread adoption of next-generation vaccines, realizing their potential to revolutionize disease prevention and public health outcomes.
Daniel Kavanagh: Some practical challenges are centered on making a clear costbenefit case for adoption of a new vaccine. This can involve continued and expanded public discussions regarding what makes a vaccine “good enough” for general use or mass vaccination. What kind of safety and efficacy criteria can be defined in advance of the next pandemic emergency, and can we achieve a general consensus on acceptability criteria?
We cannot plan for widespread acceptance of new public health initiatives without accounting for the combined effects of artificial intelligence and social media, which have obviously produced very disruptive and unpredictable effects in recent years. I don’t have a ready answer to that challenge, other
100 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INDUSTRY SENSE
than to hold on and expect a bumpy ride. How does your experience in discovering and developing manufacturing processes for enzymes and live biotherapeutics contribute to the advancement of nextgeneration vaccines?
Aaron B. Cowley : Although I am a chemist by training, in practicality, I’m an enzymologist and microbiologist, which aligns closely with RNA therapy development and manufacturing.
Producing RNA relies heavily on enzymes to drive the process; for example, T7 polymerase is the mainstay enzyme used in the in vitro transcription (IVT) process. When working with catalysts that are biological in nature, it’s always important to remember the potential for variability. T7 polymerase from one organism will likely have different properties to a T7 polymerase sourced from another organism, such as different fidelities for binding, propensity for off-target binding, etc. All of these properties will determine
Next-gen vaccines, powered by mRNA and innovative platforms, revolutionize immunization, promising enhanced efficacy and adaptability for a transformative future.
the enzyme’s catalytic efficiency, and subsequently the yield and purity of the resulting RNA produced.
To deliver the next generation of vaccines, having an appreciation of how variation could impact the production process is critical to ensuring these drugs are safe and effective.
With your extensive experience in GMP operations and process development, how do you foresee the integration of good manufacturing practices in the production of mRNA-based vaccines?
Patrick Thiaville: Just like any pharmaceutical product, integrating good manufacturing practice (GMP) or GxPs into the production of mRNA-based vaccines is essential to ensure we are producing products that are safe for patients. Robust process development, equipment qualification, and validation are essential to design scalable and reproducible manufacturing processes that meet GMP standards. Applying a risk-based approach allows us to prioritize control measures and maintain critical quality attributes throughout production.
Additionally, reducing production steps and human intervention minimizes risk, while having only one clean room and a one-step process simplifies the path for implementing and adhering to GMP standards. This eases the path for the process validation for commercial products. As the same process is used from research use
www. pharmafocusamerica.com 101
INDUSTRY SENSE
only (RUO) production to commercial scale, results can be leveraged from the beginning, simplifying the chemistry, manufacturing, and control (CMC) process.
Could you elaborate on your role in providing IBC oversight for gene transfer trials and its relevance to mRNA-based vaccines?
Daniel Kavanagh: The federal requirement for IBC (Institutional Biosafety Committee) review of molecular biology research goes back to the publication of the original version of the NIH Guidelines for Research Involving Recombinant and Synthetic Nucleic Acid Molecules in 1975. The current version of the guidelines delineates the specific requirements for IBC review of clinical trials with genetically modified investigational products, including mRNA vaccines. IBCs provide advice and approval under NIH Guidelines for equipment, training, and procedures involved in the safe handling of drug and vaccine products at a site.
Most academic institutions in the U.S. maintain a locally administered IBC registered with the NIH to review research, including a lot of nonclinical or preclinical research. Most community hospitals, clinics, and site networks do not have locally administered IBCs. As a scientific advisor at WCG, I help sites access WCG’s centralized IBC review service for clinical trials. For multicenter vaccine trials, a central IBC service is generally the most efficient and uniform approach to securing IBC approval. This is true for both community clinics and large academic institutions.
From your respective backgrounds, how do bioassays, protein engineering, and structural elucidation play crucial roles in optimizing and qualifying nextgeneration vaccines?
Aaron B. Cowley: For any vaccine, the goal is to achieve a long-lasting and effective immune response; this relies on having the right protein or antigen delivered to the right cells in the body.
However, many next-generation vaccines currently in development are targeted towards mucosal respiratory viruses. A key challenge in vaccine development for mucosal immunisation is the barrier of the mucosal surfaces, which encompasses degrading enzymes, mucus and clearance mechanisms. As a result, these surfaces represent a significant hurdle for vaccine development, inhibiting delivery and immune stimulation.
With this challenge in mind, bioassays, protein engineering and structural elucidation will play an integral role in the development of next-generation vaccines. It will therefore not only be critical to select the right antigen, but also to know the antigen configuration and adjuventation, as well as the optimal route of administration.
Patrick Thiaville: Right now, we are using bioinformatics and AI to move forward in the structural elucidation of vaccine candidates.
102 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INDUSTRY SENSE
Next-Generation Vaccines redefine immunization, merging mRNA and innovative platforms to shape a future where prevention is personalized and adaptable.
These predictive methods allow us to rapidly design vaccines for both personalized and large-scale infectious diseases.
At Exothera, our discovery services work closely with biotech partners for a range of activities, including antigen definition, construct design, and optimization, which are facilitated by predictive methods. By leveraging these tools, we can efficiently identify and characterize vaccine candidates with the desired immunogenic properties, accelerating the development process and enhancing the likelihood of success in clinical trials.
Are there specific opportunities or areas of innovation within mRNA and novel platforms that you find particularly promising for future vaccine development?
Patrick Thiaville: One of the most promising areas for innovation in mRNA and novel platforms lies in developing standardized yet scalable processes for vaccine manufacturing. With the Ntensify TM manufactur-
ing technology, powered by Quantoom Biosciences and delivered by Exothera, we've achieved the capability to seamlessly scale from personalized cancer vaccines to pandemic-grade responses, all using the same process. This scalability, from producing a single dose to millions of doses per week, unlocks the potential for rapid responses to evolving health situations such as pandemics. Together with NcapsulateTM for mRNA-LNP encapsulate, the platform, delivers a suite of automated, small-footprint bioproduction technologies for RNA manufacturing and encapsulation, is also highly cost effective.
Additionally, innovations in the formulation space present exciting opportunities. One of our key funders is the Bill & Melinda Gates Foundation. During the 2023 Grand Challenges, the Foundation announced a new investment to advance access to mRNA research and vaccine manufacturing technology. As a result, we had the opportunity to receive a grant from the Grand Challenges to advance the commercialization of our formulation system. This aims to enhance vaccine stability and efficacy, ultimately improving vaccine delivery and accessibility worldwide.
Daniel Kavanagh: One important area is the design of new kinds of nanoparticles for nucleic acid delivery. More efficient delivery has the potential to increase vaccine potency and lower the cost per dose. New approaches should be able to target mRNA to designated cellular compart -
www. pharmafocusamerica.com 103
INDUSTRY SENSE
ments. Professional antigen-presenting cells such as dendritic cells are clearly a desirable target for vaccine delivery, but mRNA encoding instructions for many other components in an immune response may be targeted to other cells. In the future, rationally designed instructions delivered to specific cellular compartments can serve as molecular adjuvants to guide and enhance immune responses to achieve the desired outcome.
Aaron B. Cowley : As our understanding of RNA therapies has expanded and enabled the production of RNA with improved stability, shelf life and efficacy, many exciting innovations in the RNA therapy space have emerged. In particular, developers and manufacturers are exploring alternative methods of delivery to injection, including inhalation or transdermal patches, which could provide an enhanced patient experience with greater ease of use and comfort.
Although these administration routes could help to improve patient compliance and provide greater patient comfort, to make these options a reality for future vaccine development, there are still many challenges to overcome. Scaling the production of inhalation and transdermal patch RNA therapies to enable the global rollout of vaccines will likely be a significant hurdle. For global-scale vaccine production, millions of doses per day will need to be manufactured. At the moment, inhalation and transdermal patch production lines are not capable of meeting this demand.
But with continued innovation, we can expect to see developers and manufacturers break down these barriers and deliver these exciting advancements in RNA production to patients in the near future.

104 PHARMA FOCUS AMERICA ISSUE 03 - 2024
INDUSTRY SENSE
How do you see the role of collaboration and knowledge-sharing in advancing the field of mRNA-based vaccines, and are there emerging trends or innovations in the industry that you find particularly exciting?
Aaron B. Cowley : Although the success of RNA therapies is still relatively new, the field itself has been around for a long time. Realising the potential of this modality for treating patients for a wide variety of disease indications has relied heavily on collaboration and knowledge sharing.
As the RNA therapy space expands, the role of organisations like the Alliance for mRNA Medicines dedicated to the advancement of RNA therapeutics and vaccines will be increasingly important in accelerating progress in the space through collaboration and disseminating knowledge.
Leveraging expertise not only within the biotech industry but also within academia is critical for the advancement of RNA therapies. Teaming up with MIT has been instrumental in the development of our continuous RNA manufacturing platform, which is built on advances made within this institution.
Patrick Thiaville: Collaboration and knowledge-sharing are paramount in driving advancements in mRNA-based vaccines, particularly in addressing global health
challenges and fostering equitable access to vaccines. Effective collaborations, organized around pandemic response and decentralization of vaccine manufacturing, are instrumental in accelerating the development, production, and distribution of mRNA vaccines worldwide. Additionally, emerging trends such as the application of mRNA in personalized cancer vaccines hold immense promise for advancing precision medicine and tackling complex diseases. However, this presents new manufacturing challenges for the industry as a whole, necessitating continued collaboration and innovation to overcome barriers and realize the full potential of mRNA-based therapeutics.
Daniel Kavanagh: I am very enthusiastic about the potential for synthetic biology to produce a deep catalog of modular adaptable solutions to molecular engineering problems. One challenge, as always, is creating an environment where innovators can share information while maintaining patent positions and marketability. Another challenge is investing in discoveries with broad applicability that can be implemented in multiple applications and clinical contexts.
Thank you all for the invigorating & gripping session. Your insights contribute to both a concise magazine feature and in-depth online discussions! I am sure there is a lot for our readers to take back home.
www. pharmafocusamerica.com 105
INDUSTRY SENSE
"Innovation in the Shadows: Unveiling the Neglect of ICU and Rare Disease Therapeutics." Authored by Alexander J. Spicer, Head of Corporate De-velopment and Financial Operations at Ampleia, the article presents a visionary approach, offering diverse perspectives within the same piece.
"Trial Tribulations: Navigating the Complex Terrain of ICU and Rare Disease Research," Joab Williamson, Director of Clinical Operations at Faron Pharmaceuticals in the UK, contributes to this thought-provoking discourse. Together, these insightful perspectives shed light on the often-overlooked realms of ICU and rare disease therapeutics, providing a comprehensive and nuanced exploration of the challenges and innovations in these critical healthcare domains.


Innovation in the Shadows:
Unveiling the Neglect of ICU and
Rare Disease Therapeutics
This opinion piece explores the paradox of neglect in two critical areas of healthcare— ICU and rare dis-ease drug development. Despite the significant impact on patient populations, these fields suffer from a lack of innovation and funding.
Alexander J. Spicer
Head of Corporate Development and Financial Operations – Ampleia
As we journey through the 21st century, the pharmaceutical industry has achieved significant milestones, each marking a new era of medical innovation. The ascendancy of biologic drugs, encompassing monoclo-nal antibodies, vaccines, and gene therapies, represents a
106 PHARMA FOCUS AMERICA ISSUE 03 - 2024
THROUGH THE HOURGLASS THROUGH THE HOURGLASS
Alexander J. Spicer Head of Corporate Development and Financial Operations – Ampleia
Joab Williamson Director, Clinical Operations –Faron Pharmaceuticals. UK
revolution in patient care. Concurrently, the rise of big data has reinvented the drug repurposing landscape, while the industry's globalization, exemplified by China's emergence and the rise of contract research and manufacturing in costefficient regions, has reshaped its structure. Nonetheless, even these strides pale beside the industry's agile and innovative re-sponse to the COVID-19 pandemic, where pharmaceutical companies played a pivotal role, underlining their ability to mobilize quickly and effectively in a global health crisis.
Despite these advancements, two critical areas remain conspicuously underserved by the latest therapeu-tic breakthroughs: the intensive care unit (ICU) and orphan diseases. On the surface, these sectors may seem worlds apart, yet a deeper analysis reveals a surprising parallel in the challenges they face in drug development, hinting at a shared underlying complexity.
Where are the Patients?
A common misconception surrounding both the ICU and orphan diseases is their supposed small patient populations. The ICU in the United States serves approximately 5 million patients annually. To put this into perspective, consider that 5.8 million Americans are living with Alzheimer's Disease, 1.9 million are newly diagnosed with cancer each year, and 1.2 million Americans live with HIV. While these diseases are not deemed 'small,' the ICU's patient population is often underrated in comparison.
Orphan diseases are characterized by their limited patient numbers, a requirement for the FDA's Orphan Drug Designation being fewer than 200,000 affected individuals. Collectively, however, these diseases im-pact an estimated 25-30 million Americans, a figure that aligns with the number of patients with diagnosed diabetes, the prevalence of depression, and the incidence of asthma in the United States.
The significant patient populations of these categories offer vast opportunities for innovation and the abil-ity to impact many lives. Yet, this is where we encounter another layer of complexity: heterogeneity.
Heterogeneity
The term 'heterogeneity' barely scratches the surface when describing the diversity among patients with rare diseases or those admitted to the ICU. These individuals, while
Unseen challenges persist: ICU and orphan diseases, overlooked in drug development, share a complex narrative in the pharmaceutical journey.
www. pharmafocusamerica.com 107
THROUGH THE HOURGLASS
grouped together within their respec-tive categories, present with a staggering array of genetic, clinical, and demographic variables. This diver-sity is compounded by differing pathophysiology and the varied responses to existing treatments.
Consider the case of a patient presenting with acute respiratory distress syndrome (ARDS) in the ICU. The critical question is not just the diagnosis but the causation: Is the patient an end-stage cancer sufferer? Have they aspirated, leading to an obstruction? Are they battling a novel viral infection?
The etiologies of ARDS are as diverse as the patients themselves, necessitating highly individualized treatment plans. This complexity is further intensified by the life-and-death stakes that ICU clinicians navigate daily, making the application of approved therapies and the adherence to clinical trial criteria exceptionally challenging.
This multifaceted nature of ICU conditions parallels the complexities of rare diseases, where thousands of disorders emerge from genetic mutations, affecting everything from isolated tissues to entire organ sys-tems. These diseases require a collaborative, cross-disciplinary approach to treatment that is as nuanced and varied as the conditions themselves. Genetic crossover between diseases can sometimes allow for a broader application of drug development strategies, as seen with treatments like Ataluren from PTC Ther-apeutics. Initially conditionally approved for Duchenne muscular dystrophy, it has also
been trialed for cystic fibrosis. Such crossover potential is a beacon of hope, suggesting that advancements in one area can cascade to others, expanding therapeutic horizons across multiple rare diseases.
The latter part of the 20th century and the dawn of the 21st saw the rise of genetic sequencing technolo-gies, heralding a new frontier in understanding disease etiology. These advancements have made their way into academic laboratories worldwide, democratizing the research landscape and propelling our comprehension of disease mechanisms forward. Yet, the benefits of these technologies have not been evenly distributed; while some diseases like cancer have seen a deluge of research and development, rare diseases often wait longer for the trickle-down effect of these innovations.
Heterogeneity in ICU and rare diseases demands nuanced, cross-disciplinary breakthroughs for treatment complexities.
108 PHARMA FOCUS AMERICA ISSUE 03 - 2024
THROUGH THE HOURGLASS
As our grasp of genetic underpinnings in disease states has grown, regulatory bodies have begun to ap-prove more explorative and potentially life-altering therapies. These breakthroughs have transformed care for some, but many patients with rare diseases are still waiting for their breakthrough moment.
The ICU and Genetics: A Divergent Path
In contrast to rare diseases, the application of genetic approaches within the ICU has not yielded the same level of success. These technologies have been employed in the fight against sepsis, ARDS, and even COVID-19, among other conditions. Unfortunately, rather than facilitating a unified treatment approach, the results have often sown discord among clinicians, creating factions and debates over best practices. These methodologies have primarily been used in the ICU to predict patient responses to therapies and understand the fundamental mechanisms of the diseases and syndromes encountered.
Curative, Supportive, or Expedient Treatment Options
Curative therapies often entail dramatic interventions. The 2020s have seen a surge approvals of such with notable examples including Zolgensma (Novartis), Luxturna (Spark/Roche), Hemgenix (Uniqure/CSL Beh-ring), and Skysona (Bluebird Bio). The number of these therapies gaining approval grows annually, as do the clinical trials that
aim to expand the arsenal of curative options available to patients.
The ripple effects of such approvals can be profound. For instance, the approval of Herceptin (Roche) set off a cascade where, as the costs of antibody manufacturing and development decreased, a wave of com-panies entered the fray. These therapies, while among the most expensive on the market, have garnered attention not just for their price tags but for their potential to profoundly alter patients' lives, offering them a semblance of normalcy.
In the ICU, unlike the realm of rare diseases, there is no ready toolbox of curative therapies. The focus is instead on a plethora of supportive treatments. Similar supportive therapies are also found in the arsenal of rare diseases, albeit in various forms across different conditions. When developing drugs for the ICU, cost considerations are paramount, as ICU budgets are typically tighter than those of other departments, with significant funds allocated to life-saving equipment like ventilators and ECMO machines. The high cost of such equipment, coupled with the intensive human resources required to operate them (a single ECMO patient can incur costs upwards of $250,000 over ten days), necessitates a reliance on affordable and read-ily available drugs.
The 'Just Because' Approach
An often-unspoken aspect of medical practice is the 'just because' phenomenon. Dexamethasone,
www. pharmafocusamerica.com 109
THROUGH THE HOURGLASS
a cost-effective steroid, gained prominence during the COVID-19 pandemic for its efficacy in late-stage ven-tilated patients. Following this, it became commonplace for all hospital admissions to receive dexame-thasone, irrespective of the nuances of individual cases. Subsequent meta-analyses have suggested that such blanket applications may have led to unnecessary fatalities. While it's easy to point fingers, the re-sponsibility does not lie solely with the front-line medical staff; instead, it extends to organizations like the World Health Organization, which could have done more to safeguard patients against such indiscriminate treatment practices.
Regulatory Agencies: Catalysts for Change
At the crossroads of innovation for ICUs and rare diseases, regulatory agencies wield significant influence. The FDA has a checkered history, having been implicated in the opioid crisis and criticized for its delayed response to emergencies such as the Celecoxib controversy. However, past performance does not preclude the agency from driving future successes. The current drug approval framework is undergoing scrutiny by industry insiders, advocating for a reevaluation that could better address the unique challenges presented by both ICUs and rare diseases. This period of introspection and potential reform presents an opportunity for the FDA to redefine its role and reassert its commitment to facilitating medical advancements.
The Potential for Breakthroughs in ICU and Rare Disease Treatment
Despite the obstacles, the prospect of groundbreaking therapies entering the market for both ICUs and rare diseases remains a potent possibility. Such advancements will require more than theoretical discus-sions; they demand actionable strategies that can navigate the complexities of drug development in these nuanced fields.
Strategic Approaches for Advancing Treatment
1. Real-World Clinical Trials: The traditional model of drug development could benefit from an inver-sion, beginning with small-scale safety studies that include both healthy volunteers and patients who match the therapy's target profile. By scaling up based on accumulating realworld data, re-searchers can return to regulatory bodies with a robust dataset that reflects a wider range of pa-tient experiences and outcomes. This approach could streamline the approval process and acceler-ate the availability of new treatments.
2. Informed Application of Technology: The incorporation of genetic and epigenetic data into drug development has the potential to revolutionize treatment, but only if it is applied judiciously. Re-search hypotheses must be grounded in a clear understanding of how this information will enhance patient care. When technology genuinely augments the data, it can provide a more targeted and effective use of resources, potentially leading
110 PHARMA FOCUS AMERICA ISSUE 03 - 2024
THROUGH THE HOURGLASS
to more efficient clinical trial development and more personalized patient care.
3. Breaking Down Silos in Medicine: The challenges encountered in the treatment of ICU patients and those with rare diseases are not unique; similar issues have been addressed in fields such as oncology, cardiology, and neuroscience. The lessons learned in these disciplines must be leveraged and applied to other areas of medicine. By fostering a collaborative environment where physicians and researchers can share insights and expertise, the pace of drug development can be significantly increased, ultimately benefiting a broader spectrum of patients.
Conclusion
In conclusion, the path to enhancing treatment options for rare diseases and ICU patients is fraught with significant challenges, yet it is also laden with opportunities for innovation. By adopting a more pragmatic and collaborative

approach to clinical trials, embracing the judicious use of technology, and encouraging cross-disciplinary dialogue, the pharmaceutical industry can overcome existing hurdles. It is essential to not only consider the potential of novel therapies but also to ensure their practical application in re-al-world settings.
The future of drug development in these critical areas hinges on the willingness of regulatory bodies to adapt and embrace new methods that can keep pace with the evolving landscape of medicine. With a concerted effort that aligns regulatory frameworks with contemporary medical needs, the pharmaceutical industry can deliver on the promise of its innovations, ensuring that patients in ICUs and those with rare diseases receive the cuttingedge treatments they deserve.
As this discussion continues, it is imperative for all stakeholders, from researchers to regulators, to remain committed to the shared goal of advancing healthcare. It is through such commitment that the industry can fulfill its highest purpose: improving and saving lives with every new discovery.
Alexander J. Spicer: Head of Corporate Development and Financial Operations
– Ampleia, Recently joined Ampleia, a venture studio in Paris as Head of Corporate Development after spending nearly five years within Faron, an immuno-oncology biotech as their Business Development Director. Alex has developed a rounded understanding of the biotechnology business and continues to develop research within the eco-nomics of the sector.
www. pharmafocusamerica.com 111
AUTHOR BIO
THROUGH THE HOURGLASS
Trial Tribulations: Navigating the Complex Terrain of ICU and Rare Disease Re-search
This article examines the operational challenges of clinical trials for ICU and rare diseases, highlighting the need for global collaboration, regulatory flexibility, and innovative trial designs. It advocates for integrated research and patientcentered approaches to ensure equitable advancements in medical treatments for all patients.
Joab Williamson Director, Clinical Operations – Faron Pharmaceuticals. UK
In the realm of healthcare, where the pursuit of ground-breaking medical treatments is ceaseless, certain areas remain starkly underexplored, hidden in the shadows of broader research priorities. Among these, the Intensive Care Unit (ICU) and rare diseases represent sectors where the disparity between the potential for impact and the reality of research attention is profound. Following the insights from our initial explora-tion of the funding challenges highlighted in Alexander Spicer’s article, it becomes evident that beyond financial constraints, a myriad of operational hurdles further complicates the path to innovation in these critical fields. The challenges faced in conducting clinical trials for ICU and rare disease conditions are not merely procedural but are emblematic of the systemic barriers that
hinder equitable progress in medical research. This follow-up piece seeks to unravel these complex operational challenges, offering a lens into the intricate world of clinical trial management within these specialized areas. By dissecting the unique hurdles encountered, from logistical to ethical, this article aims to illuminate the path towards a more in-clusive and effective research paradigm. Our goal is to foster a dialogue that transcends traditional boundaries, advocating for systemic changes that empower researchers to bring life-saving treatments to all patients, especially those whose needs are greatest yet often overlooked.
Rare Diseases: A Logistical Puzzle
Embarking on clinical trials for rare diseases presents a logistical labyrinth, where the scarcity of patients turns the recruitment process into a global expedition. The rarity of these conditions means that potential participants are often scattered across vast geographical distances,
112 PHARMA FOCUS AMERICA ISSUE 03 - 2024
THROUGH THE HOURGLASS
posing significant challenges for trial managers tasked with assembling a viable study cohort. This dispersion necessitates an international col-laboration that, while enriching the research through diverse patient data, introduces complex regulatory and logistical hurdles. Each country comes with its own regulatory landscape, patient privacy laws, and healthcare systems, making the coordination of multi-center trials a daunting task.
Moreover, the heterogeneity inherent in many rare diseases complicates trial design and the establish-ment of uniform endpoints. Disease manifestations can vary dramatically between patients, even within the same condition, requiring trials to adopt highly flexible and adaptive designs. This flexibility must ex-tend to outcome measures, which need to capture the nuances of rare diseases in ways that are meaning-ful to patients' lives. Developing such patient-centred outcome measures often involves pioneering new methodologies, further stretching the capabilities and resources of research teams.
ICU Trials: Ethical and Practical Dilemmas
The ICU presents a unique clinical trial environment, marked by patients in critical conditions, making the ethical considerations and logistical execution of trials exceptionally challenging. Informed consent, a cor-nerstone of ethical clinical research, becomes a complex issue in the ICU, where patients may be incapac-itated or under severe distress. This
situation necessitates a delicate balance between advancing medical knowledge and respecting patient autonomy, often requiring surrogate decision-making by family mem-bers or legal representatives. The emotional and ethical weight of these decisions cannot be understated, requiring clear communication and sensitivity from research teams.
Similarly, as most drug developers know, a comparator-blinded trial is the most effective design at demonstrating the value of a new product. However, for patients in the ICU, a question is raised around the ethicality should the new product demonstrate substantial benefits compared to standard of care in earlier phase studies, or even worse, if no comparator is available and placebo should be used. Is there any case where a placebo-comparator trial is ethical in the ICU setting? I believe not.
From a practical standpoint, the dynamic nature of ICU patients' health status demands trial designs that can adapt to rapid changes.
ICU trials walk an ethical tightrope, balancing medical advancement with patient autonomy, requiring clear communication and sensitivity from research teams.
www. pharmafocusamerica.com 113
THROUGH THE HOURGLASS
Traditional fixed trial protocols struggle to accommodate the fluidity required in the ICU, where a patient’s condition can shift dramatically in a matter of hours. Moreover, the polypharmacy prevalent in ICU settings introduces variables that can confound the assessment of a new treatment's efficacy and safety. Designing trials that can isolate the effects of the investigational treat-ment from those of other concurrent therapies requires innovative statistical and methodological ap-proaches.
Integration with Clinical Practice
Successfully integrating clinical trials into the routine care of rare diseases and ICU settings demands not only logistical finesse but also a cultural embrace of research within clinical settings. For rare diseases, this integration often means forging partnerships with specialized centres that can offer the expertise and re-sources necessary for managing complex conditions, or decentralised site networks to spread the study as wide as possible to allow for recruitment across diverse regions. This integration also involves engaging with patient communities to ensure that research priorities align with patient needs and that trials are de-signed with patient accessibility in mind.
In the ICU, the integration of research activities challenges the traditional workflow(s) of critical care teams. The addition of trialrelated procedures must be seamlessly woven into the tapestry of critical care, ensuring that research does not detract from the primary
objective of patient care, or bring unnecessary procedures that could take valuable time. Achieving this integration requires a collaborative effort be-tween researchers and clinical staff, fostering a mutual understanding of the value and logistics of clinical research.
Regulatory Navigation
Both rare disease and ICU research navigate a regulatory landscape that, while designed to ensure patient safety and data integrity, often imposes additional barriers to trial execution. The complexity of obtaining multi-national approvals for rare disease trials and the ethical quandaries of consent in the ICU necessitate a regulatory framework that is both robust and adaptable. Encouragingly, regulatory agencies have begun to recognize these challenges, offering pathways such as orphan drug designations and adaptive trial guidelines to facilitate research. However, there remains significant room for improvement in creating a regulatory environment that fully supports the unique needs of ICU and rare disease trials.
Overcoming The Hurdles: A Path Forward
The complexities of clinical trials in ICU and rare diseases necessitate a multifaceted strategy for im-provement. Central to overcoming these hurdles is the spirit of innovation, collaboration, and pa-tient-centeredness.
1. Fostering Global Collaboration: Establishing international consortia can mitigate the recruitment challenges in rare disease trials
114 PHARMA FOCUS AMERICA ISSUE 03 - 2024
THROUGH THE HOURGLASS
by pooling resources, standardizing protocols across borders, and facilitating data sharing. For ICU trials, collaboration can help harmonize ethical standards and consent processes, ensuring respect for patient autonomy while advancing critical research.
2. Embracing Regulatory Flexibility: Advocacy for adaptive regulatory policies that recognize the unique challenges of ICU and rare disease trials is essential. This includes streamlined processes for cross-border studies, flexible trial designs that can adjust to the dynamism of ICU settings, and al-ternative endpoints that reflect meaningful outcomes for rare diseases.
3. Innovating Trial Design: Leveraging technology and novel methodologies, such as virtual trials, re-al-world evidence, and patient-reported outcomes, can address both logistical and ethical chal-lenges. These innovations offer the potential to reduce the
AUTHOR BIO

Joab is the Director, Clinical Operations at Faron Pharmaceuticals, a clinical stage biotech focusing on building the future of immune-oncology. He has a vast amount of experience in clinical operations and program/project management and is also focused on continuing academic pharmacoeconomic research.
burden of trial participation on patients and families, especially in dispersed rare disease populations and critical care contexts.
4. Integrating Research and Clinical Care: Building a culture where clinical research is viewed as an integral component of patient care can enhance trial integration. Education and training for healthcare providers on the importance and logistics of clinical trials can foster a more re-search-positive environment, improving recruitment and ensuring that trials complement standard care practices.
Conclusion
The journey to advance clinical trials in ICU and rare disease research is fraught with challenges but also ripe with opportunity. By addressing the operational, ethical, and regulatory hurdles with innovative and collaborative solutions, we can pave the way for a future where groundbreaking treatments are accessible to all patients, regardless of the rarity or complexity of their condition. This article underscores the necessity of a collective effort among researchers, healthcare professionals, regulators, and, importantly, pa-tients and their advocates, to reshape the landscape of clinical trials. Together, we can surmount the bar-riers that have historically impeded progress, ensuring that the promise of medical research is realized for every patient in need. As we move forward, let our actions be guided by the principles of equity, innova-tion, and compassion, heralding a new era of healthcare where no patient is left behind.
www. pharmafocusamerica.com 115
THROUGH THE HOURGLASS
Clinical Trial Project Management

Book Description: This book provides a detailed overview of how to conduct clinical trials, in an international context. The process of conducting clinical studies across nations is based on a set of regulatory regimes developed by respective regulatory agencies like USA, Europe, Brazil, China, India, Singapore, Canada, Australia, and JAPAN. This book aims to be your comprehensive guide to understanding the intricacies and challenges of managing the clinical trials at global level from country specific regulatory dossier submission to final study report preparation in compliance of ICH-GCP.
1. Could you briefly introduce yourself and share a personal anecdote or experience that particularly shaped your perspective on clinical trial management and influenced your decision to write this book. Regarding clinical trials, my journey began in 2004 when I first had an opportunity to work in the clinical research profession. Since then, I've been involved in various clinical trial projects, spanning from dossier submission to the preparation of clinical trial study reports. Along the way, I noticed the absence of a comprehensive resource covering the entire process of clinical trial project management. Learning the fundamentals of clinical trials

116 PHARMA FOCUS AMERICA ISSUE 03 - 2024
Know your Book, Know your Author
Ashok Kumar Peepliwal Associate Professor, School of Pharmaceutical Management, IIHMR University
from the ground up proved to be challenging. This challenge motivated me to create a manuscript, and after almost 8 years of hardship, I successfully completed this book scripture.
2. In a nutshell, how does your book enhance readers' understanding of clinical trials, and what core message do you hope they will grasp? Can you highlight specific chapters or examples from your book that you believe will significantly contribute to readers' comprehension of clinical trials?
The chapter on the historical development of clinical trials aims to inspire the reader to comprehend the imperative need for conducting clinical research with a strong ethical foundation, ensuring the protection of patients' rights, safety, and well-being. The book enriches the reader's insight into the historical evolution of clinical research, illustrating the development of various clinical research guidelines in a chronological order. Notable events such as the Tuskegee experimentations, the Nuremberg code, Nazi experiments, and the thalidomide tragedy are explored, showcasing their contributions to shaping ethical standards in clinical research.
Section 1.2 delves into the consequences of unethical experimentation during the Nazi era, revealing that individuals involved faced execution. Such penalties set an example for the world to execute the trials in an ethical manner. This is discussed in detail under the heading "Nuremberg Code, Germany, and Ethical Concerns."
3. In addressing the challenges of international clinical trials, can you provide a specific case study where these challenges were particularly pronounced, and your book offers practical solutions?
There is a challenge (patient retention, data collection, execution, monitoring, and other logistic issues) of smooth functioning of clinical trials at various locations in international context. A solution to such challenges is provided with the help of decentralized clinical trials (DCT) which enable broader access to clinical trials for participants worldwide. Individuals from diverse geographical locations can participate without the need to travel to a specific study site, promoting inclusivity and diversity in clinical trial populations.
DCTs facilitate the collection of real-world data by allowing participants to be monitored in their natural environments. This can provide a more comprehensive understanding of a treatment's effectiveness and safety in diverse populations and settings.
By reducing the burden of travel and frequent site visits, DCTs can enhance participant engagement. Participants are more likely to remain committed to the trial, leading to improved retention rates and more reliable data. The ability to enroll participants from various regions helps overcome recruitment challenges often faced in traditional, site-centric trials. This can expedite the recruitment process, reducing trial timelines.
DCTs can result in cost savings for sponsors and participants alike. Reduced travel
www. pharmafocusamerica.com 117
BOOK IN THE SHELF - A REVIEW
expenses, site infrastructure costs, and the need for physical space can contribute to more efficient and cost-effective trial conduct.
The decentralized model often involves simplified logistics, reducing the time needed for study start-up activities. This can be especially advantageous in international trials where coordinating various site-related activities may pose challenges.
Advances in technology (artificial intelligence, machine learning and blockchain) enable remote monitoring and data management, allowing for real-time oversight of the trial. This can enhance data quality, protocol compliance, and the overall efficiency of the trial.
4. Could you delve deeper into how this specific feature highlighted in your book revolutionizes clinical trial success? Perhaps share a case where its implementation made a significant difference.
A case of Pfizer incorporated in the book where it conducted the first fully webbased trial, known as REMOTE (Research on Electronic Monitoring of Overactive Bladder Treatment Experience), in 2011 under an Investigational New Drug application. The study investigators employed online recruitment, online questionnaires, electronic diaries, and home delivery of the investigational drug during REMOTE, with no in-person site visits conducted at all.
The COVID-19 pandemic posed unprecedented challenges to traditional clinical trial methodologies due to lockdowns, travel restrictions, and safety concerns. In response, a pharmaceutical company initi-
DCTs monitor participants in real-world settings for a comprehensive understanding of treatment effectiveness and safety.
ated a decentralized clinical trial to evaluate the efficacy and safety of a novel antiviral drug for COVID-19.
However, there are the challenges (data quality, patients, site and investigators, regulators and internal change management) in operation of DCTs too, but this book provides the solutions of such challenges in section 18.5 management of decentralized clinical trials.
5. Clarify whether the skill mentioned for navigating regulatory landscapes is technical, interpersonal, or regulatory in nature. Provide an example from the book illustrating its pivotal role in ensuring success.
Many times, a clinical research professional struggles with the regulatory understanding of countries like USA, European Union, Brazil, Canada, Singapore, India, Japan, Australia, and China for clinical trial approval and its conductance. In the book, such country specific regulatory requirements of clini-
118 PHARMA FOCUS AMERICA ISSUE 03 - 2024
BOOK IN THE SHELF - A REVIEW
cal trials are described as more technically oriented. Chapter 3 clinical trial approval in world encompasses these procedures and clinical trial approval requisite in these jurisdictions.
As an example, one can understand a uniform approach of clinical trial information system (CTIS) for approval of clinical trials in Europe and its member states. A more robust method of approvals saving time, and cost of a clinical trial project as illustrated in section 3.4.1.
6. Elaborate on how the statistical tools or methodologies discussed in your book contribute to the reliability of clinical trial data analysis. Provide a case study where their application led to notable improvements.
In the book, the statistical tools (Basic and advanced) presented play a pivotal role in bolstering the reliability of clinical trial data analysis with appropriate clinical trial-based outcomes and conclusions. These tools (One-sample t-test, Two-sample t-test, One-way ANOVA, Two-way ANOVA, Repeated measure analysis, crossover design and analysis, linear regression method, ANCOVA, and Chi-square test) are specifically tailored to address the inherent complexities of clinical research, offering a robust framework for the interpretation and validation of results.
A case of antihypertensive bioequivalence study described in section 13.2 crossover design and analysis. The outcome of the study is based on the clinical data collected from 20 patients showing the significant difference between two treatments. Such
more explanatory examples are taken in every statistical tool to make the audience more understandable about the study outcomes and its approvals from regulatory authorities.
7. Could you offer more insight into how the integration of PERT and CPM in clinical trial project management specifically enhances efficiency and success? Share practical examples or case studies from the book.
The integration of PERT and CPM in clinical trial project management provides a comprehensive and dynamic framework. This integration enhances efficiency by facilitating thorough planning, resource optimization, risk management, and adaptability, all of which contribute to the successful execution of clinical trials within established timelines and budgets.
A clinical trial project case study described in section 4.2.1.2. PERT (Estimating the duration of each task involved in the clinical trial. By considering optimistic, pessimistic, and most likely time estimates, PERT provides a more realistic assessment of the project timeline) and 4.2.1.3. CPM (CPM helps identify the critical path, which is the sequence of tasks that determines the overall duration of the trials)
Integrating PERT with CPM ensures that the project plan is comprehensive, accounting for uncertainties and dependencies.
Apart from PERT and CPM, earned value analysis (EVA) also described in more practically way to determine the project status at specific time of interval whether the running project is meeting the timelines with the
www. pharmafocusamerica.com 119
BOOK IN THE SHELF - A REVIEW
budgeted cost or not; if its not, then what strategic decision can be taken to meet the project timelines under the control budget.
8. Specify the emerging trends covered in your book and explain why they are considered transformative. Provide examples or instances where these trends have already started shaping the landscape.
Electronic Clinical Outcome Assessment (eCOA) and Electronic Patient-Reported Outcomes (ePRO) are described in the book. It plays crucial roles in modern clinical trials by leveraging digital technology to collect patient-reported data efficiently and accurately. These are included in the manuscript because of following reasons.
eCOA: It ensures that clinical outcome assessments are collected directly from patients electronically, minimizing the risk of data transcription errors or inaccuracies. The use of electronic devices, such as smartphones or tablets, makes it more convenient for patients to participate in assessments. This often leads to higher compliance rates and increased patient engagement. Realtime data collection allows for immediate access to patient-reported information. This is especially valuable for monitoring patient symptoms or treatment effects promptly. Electronic data collection eliminates the need for manual data entry and associated verification processes, saving time and reducing the likelihood of errors. Electronic data collection platforms implement robust security measures to protect patient information and maintain data integrity. Enables remote monitoring of patients' health and
treatment outcomes, reducing the need for frequent site visits.
ePRO: Patients can directly input their responses to questionnaires or surveys, reducing the potential for errors introduced during manual data entry. Patients can conveniently report their symptoms, experiences, or outcomes from the comfort of their homes, promoting consistent participation throughout the trial. Researchers and clinicians can access up-to-date patientreported data, enabling them to make informed decisions quickly and potentially improving patient safety. By streamlining the data collection process, ePRO contributes to overall trial efficiency, potentially accelerating study timelines and reducing costs associated with traditional paper-based methods. Patient-reported data is securely transmitted and stored, ensuring compliance with data privacy regulations, and safeguarding sensitive patient information. Facilitates ongoing assessment of patient-reported outcomes without requiring patients to travel to study sites regularly.
Three case studies are described in chapter 19. Electronic Clinical Outcome Assessment (eCOA) and Electronic PatientReported Outcomes (ePRO) where these modern transformative technological approaches are adopted.
9. Can you detail specific proactive measures that professionals can take to ensure smooth audits and inspections?
Share practical advice based on your experiences or examples from the book. To ensure smooth audits and inspections in clinical trials, professionals can implement
120 PHARMA FOCUS AMERICA ISSUE 03 - 2024
BOOK IN THE SHELF - A REVIEW
PERT and CPM ensure a comprehensive plan for successful clinical trial execution within set timelines and budgets.
specific proactive measures. These measures are essential for maintaining compliance, data integrity, and the overall success of the trial.
Standard Operating Procedures (SOPs), Document Management Systems,Mock Audits, Regular Internal Audits, Source Data Verification (SDV, Data Management Procedures, Site Inspection Readiness Plan, Site Training and other Corrective and Preventive Actions (CAPA).
During the audits certain documents (Study Protocol, EC related documents, Patient diaries, source documentation, case record forms, agreements, investigator files, correspondences, recruitment advertisements, safety monitoring plans and compensation, laboratory related documents and patient examination reports, study personnel logs and other essential trial related documents etc.) of clinical study mut be verified from the scratch. An auditor must mention their observation as major and minor
observations in the audit report, so issues prioritized accordingly to address the it.
By implementing these proactive measures, clinical trial professionals can contribute to a culture of compliance, transparency, and preparedness, fostering smooth audits and inspections throughout the trial lifecycle.
10. Reflecting on your career, what singular insight or lesson do you want readers to remember after finishing your book? Provide an example or anecdote that encapsulates this key takeaway. One singular insight or lesson learned from “clinical trial project management” book is the critical importance of meticulous planning and effective communication throughout the entire clinical trial lifecycle.
Clinical trial project management involves intricate coordination of various stakeholders, compliance with regulatory standards, and adherence to rigorous timelines. A key insight from a clinical trial project management book is that a well-structured project plan, developed before the trial initiation, serves as the backbone for successful execution. This includes detailed planning for study related documents development like protocol, informed consent form, investigator brochure, and case record forms etc., country specific regulatory submissions, patient identification, screening, enrolment and recruitment, data generation, recording and data collection, monitoring, and statistical analysis.
Moreover, the book underscores the significance of adaptability in project management, stakeholder’s role, budget
www. pharmafocusamerica.com 121
BOOK IN THE SHELF - A REVIEW
preparation, steps of its conductance, monitoring, data management, SAE reporting, risk-based monitoring, statistical tools, and others.
By focusing on meticulous planning, effective communication, and adaptability, clinical trial project management can navigate the complexities of the drug development process, ensuring the delivery of reliable results within established timelines and budgets. This singular insight highlights the pivotal role of project management principles in the success of clinical trials is the takeaway anecdote.
11. Discuss specific unexplored areas in clinical trial management that you believe will become crucial. Mention whether you have plans for future publications to delve into these areas. Generative artificial intelligence (AI) represents a cutting-edge and relatively unexplored frontier in clinical trial management. While traditional AI applications in clinical trials focus on data analysis, patient recruitment, and predictive modeling, generative AI introduces innovative possibilities that can transform various aspects of trial management. Here are some unexplored areas where generative AI can make significant contributions:
Generative AI has the potential to assist in the creation of trial protocols by analyzing existing data, scientific literature, and regulatory requirements. This could lead to the generation of more robust and efficient protocols, optimizing study design and objectives. Generative AI, powered by Natural Language Generation (NLG), can
eCOA and ePRO use digital tech for efficient patientreported data collection, enhancing compliance and engagement in clinical trials.
create personalized and easily understandable informed consent forms. This could improve patient comprehension, leading to better engagement and adherence throughout the trial.
It can analyze historical trial data to identify potential risks and predict areas prone to challenges. This could enable proactive risk mitigation strategies and enhance overall trial management. It can contribute to the development of dynamic adaptive trial designs that evolve in real-time based on ongoing data. This approach allows for more efficient decision-making, such as early termination of ineffective arms or adjustments to sample sizes. It can assist in site selection by analyzing diverse factors such as patient demographics, investigator expertise, and logistical considerations. This could lead to more informed decisions on optimal site placement. It predicts the patient enrollment rates and potential varia-
122 PHARMA FOCUS AMERICA ISSUE 03 - 2024
BOOK IN THE SHELF - A REVIEW
tions, assisting in optimizing drug supply chain logistics. This could minimize wastage, reduce costs, and ensure a steady and timely supply of investigational products. Generative Adversarial Networks (GANs), a type of generative AI, can be employed to augment limited datasets, improving the robustness and diversity of training data for machine learning models. This is particularly relevant for scenarios with small or imbalanced datasets. Generative AI can assist in creating models that generate synthetic patient-reported data, aiding in scenarios where real patient data may be scarce or challenging to collect. Generative AI, leveraging Natural Language Processing (NLP), can assist researchers in reviewing vast amounts of scientific literature to extract relevant insights, potentially speeding up the literature review process during trial planning. Generative AI can streamline the creation of clinical study reports by synthesizing and summarizing key findings, reducing the time and resources required for manual report writing. While these applications remain largely unexplored in clinical trial management, the integration of generative AI has the potential to revolutionize processes, enhance efficiency, and contribute to more successful and streamlined clinical trials. As the field continues to evolve, exploration and innovation in these areas are likely to bring about transformative changes in how clinical trials are designed, conducted, and managed.
Yes, I have plans to add models of generative AI in my upcoming manuscripts so pharmaceutical industry can leverage the models and minimize the cost, and time for functioning of trials smoothly.
12. How does your book specifically address challenges and opportunities related to diversity and inclusion in clinical trials? Share practical advice for professionals in navigating these aspects effectively.
The book delves into the distinct approach to clinical trial conductance, providing valuable insights for clinical research professionals. It systematically explores opportunities, offering a step-by-step guide through the unique process. Chapter 4 focuses on practical solutions for budget optimization, utilizing Earned Value Analysis (EVA). This chapter equips clinical research professionals with the knowledge to comprehend intermittent expenditures related to trial activities. Highlighting diversity in clinical study dossier submission, the book navigates through various regulatory processes in countries such as the USA, Japan, India, Singapore, Canada, China, Australia, Brazil, and Europe. From initial submissions to final report writing, the book incorporates cited examples for statistical analysis, enhancing its practical applicability. Professionals are encouraged to refer to this book for country-specific regulatory references. It serves as a valuable resource for designing clinical studies, understanding randomization procedures, implementing pharmacovigilance practices, navigating project management tools, utilizing statistical tools for outcome assessments, staying updated on ICH-GCP developments, and embracing technological advancements in eCOA/ePRO. In summary, the book offers comprehensive guidance for
www. pharmafocusamerica.com 123
BOOK IN THE SHELF - A REVIEW
clinical research professionals, combining theoretical insights with practical applications. Its extensive coverage makes it a go-to reference for a myriad of aspects in the field, providing professionals with the knowledge needed to excel in their roles.
13. In conclusion, considering the extensive insights shared in your book, what do you envision as the broader impact of your work on the field of clinical trial management? Additionally, is there any final message or encouragement you would like to convey to readers who are navigating the complexities of clinical trials based on your experiences and expertise?
The book encapsulates the expertise and wisdom of seasoned professionals in the field, providing a comprehensive roadmap for navigating the complex terrain of regulatory, study designs, statistics, pharmacovigilance, clinical data management, audits/inspection, project management, report writing etc. Whether you are a novice entering the field or a seasoned professional seeking to enhance your skills, this book offers invaluable insights, practical tools, and strate -
gic approaches to ensure the success of clinical trials in an ever-evolving healthcare landscape.
In the dynamic landscape of healthcare and pharmaceutical research, the successful execution of clinical trials is pivotal for the development and approval of new treatments. "Book: Clinical Trial Project Management" serves as an indispensable resource for professionals navigating the intricate and multifaceted realm of clinical trial management. This book is meticulously crafted to provide a thorough understanding of the key principles, best practices, and strategic insights essential for orchestrating successful clinical trials.
The book is designed to equip professionals with the tools and insights necessary to unravel intricacies and streamline the path toward successful trial outcomes. By addressing the core elements of trial management with practical solutions, this book not only demystifies complexity but empowers professionals to transform hurdles into steppingstones toward groundbreaking discoveries. Embrace the journey, master the complexities, and contribute to the advancement of medical science with confidence and competence.
Dr. Ashok Kumar Peepliwal (PhD, M.Pharm, LLB, MBA) played former roles in clinical research at PharmaNet, Integrated Clinical Research, Lambda Therapeutic, and Torrent Pharmaceuticals. He vast therapeutic experience in Oncology, Hypertension, Psychiatric, Ophthalmology, Gynaecology, Rheumatoid Arthritis, Nephrology, Hematology, Respiratory, Immunology, Dermatology, Hyperlipidaemia, and Gastroenterolog for Pfizer Ltd., Fresenius Kabi, Himalaya Drugs, Panacea Biotec Ltd., Dr. Reddy’s Limited, Intas Pharmaceuticals Limited etc. Experience in patient based pharmacokinetic studies for different regulatory submissions like US-FDA, EMEA, TGA, ANVISA and DCGI etc. Currently he is a member of various hospital ethics committees to contribute to new drug developments.
124 PHARMA FOCUS AMERICA ISSUE 03 - 2024
AUTHOR BIO
BOOK IN THE SHELF - A REVIEW
UPGRADE YOUR MARKETING STRATEGY
Let the true “Digital Transformation” be the base of all your marketing campaigns

“Digital Transformation is the way forward”
Highly accountable marketing campaigns. Every dollar counts. Digitally powered marketing campaigns may be cheaper than you thought...
Ask us how?
Use our guaranteed ROI programs for reposition, penetrate or launch of your new products or services.
Grow your audience with increased reach, impact and user-friendliness
Rise above geographical boundaries
Generate new business
Gain the strong web presence differentiating yourself from competitors
Connect and engage with your target audience
Give more exposure to industry specific people
Increase your brand profile and share your capabilities with leading industry professionals
Our recent successful partnerships:

Email : advertise@pharmafocusamerica.com
Website : www.pharmafocusamerica.com



pharmafocusamerica.com 125







126 PHARMA FOCUS AMERICA ISSUE 03 - 2024 EVENT PREVIEW BOOK NOW at www.terrapinn.com/fobbsandiego/pfa San Diego Convention Center, CA April 15-17th, 2024 America's Most Exciting Biologics Event
Embark on a Biologics Journey at the Festival of Biologics 2024
Get ready for an unparalleled experience in the heart of innovation as the Festival of Biologics returns to the San Diego Convention Center, CA, from April 15th to 17th, 2024. This premier event is set to be America's most exciting biologics gathering, bringing together the entire spectrum of the life science community.
Bridging the Gaps in Biologics: From Discovery to Market
The Festival of Biologics serves as the ultimate meeting point for pharma and biotech leaders, academics, research institutes, regulators, patient groups, and payers, alongside their partners across the value chain. Delve into the intricacies of the biologics journey, from discovery to market, as the event unfolds the start-to-finish narrative of the biologics value chain.
A Convergence of Minds: Inspire Change, Champion Innovation
Join industry luminaries in a vibrant exchange of knowledge, ideas, and conversations aimed at inspiring change and championing innovation. The Festival's agenda spans the entire biologics value chain, providing a holistic understanding of the industry's challenges and opportunities.
What's New for 2024: Expanding Horizons
Building on the success of previous editions, the Festival of Biologics introduces new dimensions to its program. This year, the focus extends to the critical stages of Fill and Finish, Supply Chain, and Logistics. Explore breakthroughs in dry and wet filling solutions, cold storage transportation, and more through engaging presentations, panels, and an exhibi-
tion floor featuring cutting-edge solutions. Features that Define the Festival of Biologics 2024:
• Over 350 World-Leading Expert Presentations: Immerse yourself in the latest insights from renowned experts shaping the biologics landscape.
• 5 Main Channels: Explore the diverse realms of antibodies, immunotherapy, biosimilars, clinical trials, and manufacturing and bioprocessing through dedicated channels.
• 2000+ Attendees: Network with a vast and diverse community of professionals, fostering collaboration and partnerships.
• Customized Lead Generation Solutions: Experience tailored opportunities for networking and collaboration with the world's leading industry partners.
• 100 Start-Ups Showcasing Innovation: Witness the cutting edge of research and development with dynamic presentations from 100 innovative start-ups.
Don't miss out on this unique opportunity to be part of the Festival of Biologics 2024, where the industry's thought leaders converge to shape the future of biologics. Secure your spot and be at the forefront of the dynamic advancements propelling the biologics sector into a new era.
www. pharmafocusamerica.com 127 EVENT PREVIEW







128 PHARMA FOCUS AMERICA ISSUE 03 - 2024 EVENT PREVIEW APRIL 16-18, 2024 | JAVITS CENTER, NYC BRIDGING THE GAP BETWEEN SCIENCE AND BUSINESS CLAIM YOUR FREE EXHIBIT HALL PASS INTERPHEX24.COM/AHHM
EVENT PREVIEW
INTERPHEX 2024
Event Preview: INTERPHEX 2024
When: April 16-18, 2024
Where: Javits Center, NYC
Registration Open!
Join us at INTERPHEX, the premier global pharmaceutical and biotechnology event, returning to New York City on April 16-18, 2024. Immerse yourself in the leading platform that navigates all stages of the pharmaceutical product development lifecycle.
Key Features:
2024 INTERPHEX Conference in the Learning Lab:
• An all-new, transformative event enhancing the meeting experience.
• Industry-current technical and scientific sessions by experts in key areas.
• Hosted in the exclusive INTERPHEX Learning Lab.
• Limited registration and early sign-up are recommended.
Conference Format:
• Quick Fires, Keynote, Panel Discussion, Late Breaking sessions.
• Engaging formats covering the latest scientific topics and innovations.
Tracks and Themes:
• Track 1: Non-Sterile Product Development and Manufacturing.
• Track 2: Recent Advances in Sterile Product Manufacturing.
Special Pavilions:
• AI in Pharma Pavilion: showcasing cuttingedge AI solutions in pharmaceuticals
• Innovation Technology Gateway Pavilion: Global pharmaceutical technologies in sequential order of development.
• New Exhibitor Pavilion: Explore the newest
products, services, and solutions from new vendors.
Additional Features:
• Tech Theater Stage: Live presentations and demonstrations from leading exhibitors.
• Interphex Learning Lab: Exclusive education space for conference participants.
About the Show
INTERPHEX 2024, the global pharmaceutical and biotechnology event at Javits Center, NYC from April 16-18, is the epitome of industry innovation. The event features an expansive exhibition floor, showcasing cutting-edge products and services. Attendees engage in expert-led seminars, explore technology showcases and benefit from unmatched networking opportunities. INTERPHEX 2024 is the platform to stay abreast of advancements in pharmaceutical manufacturing, process development, and regulatory compliance, fostering collaborations and elevating industry knowledge. Join us to connect, innovate, and lead in pharmaceutical excellence.
Don't miss the chance to be part of INTERPHEX 2024 - Register today and experience the future of pharmaceutical development!
www. pharmafocusamerica.com 129
EVENT PREVIEW



130 PHARMA FOCUS AMERICA ISSUE 03 - 2024
Biomarkers 2024: Unveiling the Future of Biomarker Research
Join us at Europe's premier Biomarkers Congress, taking place from February 29 to March 1, 2024, in the vibrant city of London, UK. This flagship event is a convergence of global pharmaceutical leaders, groundbreaking biotech innovators, and esteemed academic professionals, all contributing to the dynamic landscape of biomarker research.
Explore the Forefront of Biomarker Trends and Tools
Engage with the latest trends in transforming biomarker and translational research across diverse therapeutic areas and stages of drug development. The 19th Annual Biomarkers Congress promises an immersive experience designed to address challenges in the biomarkers discovery and development landscape.
Key Highlights of the Event:
1. Cutting-Edge Presentations: Immerse yourself in over 90 cutting-edge presentations and interactive discussions led by top experts. Learn about the integration of biomarkers in drug development with case studies covering immuno-oncology, neuroscience, NASH, and immunology.
2. Novel Insights: Gain novel insights into key biomarkers for detection, monitoring, and diagnosis. Explore innovative applications of digital markers and learn about early cancer detection.
3. Comprehensive Discussions: Discuss the use of biomarkers in preclinical and clinical development, including trial design and data analysis, assay development, patient stratification, and selection, and the crucial role of safety and efficacy markers.
Tracks to Tailor Your Experience:
Track 1: Biomarkers - Identification & Validation in Oncology, Immuno-Oncology & Immunology
Track 2: Biomarkers for Diagnostics & Precision Medicine
Track 3: Biomarkers for Clinical Development
Track 4: Digital Pathology & Imaging and AI for Biomarker Research
Networking Opportunities:
• Engage with 500 leading pharma, biotech, and academic delegates.
• Engage in 20 hours of presentations, discussions, and interactive content.
• Benefit from 9 hours of networking breaks, including speed networking sessions and refreshments.
• Participate in 200 hours of pre-arranged 1-2-1 meetings, fostering business growth.
Event Features:
Engaging Thought Leadership: Over 80 dynamic interactive discussions, roundtables, and presentations curated with insights from top industry leaders and experts.
Global Technological Showcase: An international exhibition featuring cutting-edge technologies and services from leading providers in the scientific field.
Networking Emphasis: Unparalleled networking opportunities, including speed networking sessions, a vibrant drinks reception, and informal gatherings to foster meaningful connections.
Innovative Poster Displays: Explore inspiring poster displays and presentations unveiling the latest breakthroughs from emerging biotech, pharma, and academic institutions.
Why Attend?
Biomarkers 2024 promises a unique opportunity to stay at the forefront of biomarker research, network with industry leaders, and explore cutting-edge technologies shaping the future of drug development. Don't miss this chance to be part of the conversation that is driving innovation in biomarker discovery and development.
Secure your spot today!
www. pharmafocusamerica.com 131 EVENT PREVIEW
EVENTS LIST
DNA Process Development & Manufacturing Summit
5 - 7 March, 2024 | Boston, USA
https://dna-process-development-manufacturing. com/
About the Event: The Inaugural DNA Process Development and Manufacturing Summit is the only forum solely dedicated to increasing plasmid and cell-free based DNA production while reducing costs, time, and ensuring regulatory compliance. Meet 80+ DNA production experts over 3 days packed with deep-dive workshops, data-led presentations, and dedicated discussion and networking sessions to address the most pressing challenges of producing DNA, brought together by our 18+ expert speakers from leading companies like Sanofi, Roche, Genentech, Pfizer and more.
Listed Under: Manufacturing
25th Annual Clinical Trial Supply Europe 2024
6 - 7 March, 2024 | Barcelona, Spain
https://www.arena-international.com/event/ ctseurope/
About the Event: 25th Annual Clinical Trial Supply Europe 2024 2024 will see the return of the Clinical Trial Supply Europe conference to Barcelona where pharma, large and small, alongside biotechs will have the opportunity to discuss, debate and consider new technologies and processes to streamline supply chain operations.
Listed Under: Clinical Trials
2nd ACE Drug Discovery Summit
20 - 21 March, 2024 | London, UK
https://acxpo.com/ace-drug-discovery-summit/
About the Event: 2nd ACE Drug Discovery Summit, 2024 provides a platform for scientists, researchers and decision makers from all over the world to debate on the latest scientific advances, trends, current challenges and futuristic advancements in Drug Discovery. The two day conference is designed to maximize collaborations and innovation with pharma and technology presentations, interactive sessions by professionals from industry and academia.
Listed Under: Research & Development
iPharma Expo 2024
21- 22 March, 2024 | San Francisco, USA
https://www.ipharmaexpo.com/
About the Event: The iPharma Expo will showcase the latest trends and technologies in pharmaceuticals, drugs, and formulations. The expo is expected to witness approx. 150 exhibitors and 1500- 2000 visitors footfall from pharma industry & management. Direct access to highly targeted senior pharma executives, buyers, procurement managers, contract manufacturers, hospital administration, and many more Meetings with manager and business development managers who are looking for new supplies, building strategic partnerships, or entering into new ventures.
Listed Under: Manufacturing
World Vaccine Congress Washington
1 - 4 April, 2024 | Washington,United States
https://www.terrapinn.com/template/live/add2diary. aspx?e=10771

About the Event: The World Vaccine Congress is an award-winning series of conferences and exhibitions that have grown to become the largest and most established vaccine meetings of their kind across the globe since 2000. Following a record attendance in 2023 of 3000+ attendees, the World Vaccine Congress Washington will be back in April
132 PHARMA FOCUS AMERICA ISSUE 03 - 2024
2024 bringing together the most senior and informed industry experts across the entire vaccine landscape.
Listed Under: Research & Development

Pharmaceutical Compliance Congress
16 - 18 April, 2024 | Virginia, United States
https://informaconnect.com/pharmaceuticalcompliance-congress-pcc/
About the Event: The Pharmaceutical Congress Event Experience Hundreds of professionals from the compliance, ethics and legal community unite under one roof for compelling discussions, industry benchmarking and extended networking.
Listed Under: Strategy

INTERPHEX
16 - 18 April, 2024 | New York, United States
https://www.interphex.com/en-us.html
About the Event: INTERPHEX is the leading global pharmaceutical and biotechnology event that fuses industry innovation with expert-led conference. It’s where the newest ideas are shared, technology is unveiled, and the power of science though commercialization comes to life. No matter where you are in the pharmaceutical development lifecycle, INTERPHEX delivers relevant solutions to drive growth and fuel scalability for your business.
Listed Under: Manufacturing
5th Annual Pharmacovigilance & Drug Safety Summit
18 - 19 April, 2024 | Berlin, Germany
https://globalbsg.com/events/5th-annualpharmacovigilance-drug-safety-summit/
About the Event: This event will be focusing on trends on Regulatory challenges, Signal detection, Data management, Risk management, Audit & Inspection and New technology applied to Pharmacovigilance procedures.
Listed Under: Research & Development
3rd Annual LNP Formulation & Process Development Summit
April 29 - May 2, 2024 | Boston, USA
https://lnp-formulation-process-developmentpharma.com/
About the Event: Join the ever-growing community of LNP pioneers as they tackle the challenges associated with developing safe, compliant and efficacious LNPs. This starts with the in vitro discovery of LNPs through to the manufacturing processes, with end-to-end synergy fundamental.
Listed Under: Research & Development
US Pharma and Biotech Summit
16 May, 2024 | Downtown, New York
https://uspharma.live.ft.com/
About the Event: The Financial Times US Pharma and Biotech Summit returns to New York in May 2024, partnered with Endpoints News for the first time. The event will gather biopharma leaders, regulators, investors and scientists to share unique insights about the year ahead and discuss the most important trends affecting the industry, and the country.
Listed Under: Strategy

www. pharmafocusamerica.com 133
EVENTS LIST
ACQUISITIONS
Jazz Pharma Secures Global Rights to Redx's KRAS Inhibitor Program
Jazz Pharmaceuticals and Redx Pharma plc have officially inked a definitive agreement, paving the way for Jazz's acquisition of Redx's KRAS (Kirsten rat sarcoma virus) inhibitor program.
The collaboration entails a joint effort to progress candidates through IND-enabling studies, with Jazz taking the lead on clinical development, regulatory matters, manufacturing, and commercialization.


To secure all rights, patents, and interests tied to Redx's proprietary KRAS inhibitor program, Jazz will make an upfront payment of US$10 million. This program encompasses G12D selective and panKRAS molecules.
The agreement outlines potential milestone payments amounting to US$870 million for Redx, contingent on development, regulatory achievements, and commercial success. The initial milestone hinges on IND clearance from the U.S. Food and Drug Administration. Additionally, Redx stands to receive tiered, mid-single-digit percentage royalties based on future net sales.
In a parallel collaboration agreement, Jazz will compensate Redx for research and preclinical development activities.
EXPANSIONS
AstraZeneca Boosts US Presence for Cell Therapy Innovations
AstraZeneca is investing US$300 million into a state-of-the-art facility in Rockville, MD, marking a significant stride in launching its life-saving cell therapy platforms in the United States.
This strategic move aims to support critical cancer trials and future commercial supply endeavors. The investment will not only facilitate the manufacturing of T-cell therapies but will also spawn over 150 new highly skilled jobs. These positions will initially focus on producing T-cell therapies to pave the way for global clinical trials, with the potential to broaden the facility's scope to address other disease areas in the future.
Situated less than five miles away from one of AstraZeneca's global R&D centers, the Rockville facility is strategically located within Maryland's thriving life sciences corridor. The region's proximity to universities enhances the potential for attracting new and experienced talent to contribute to AstraZeneca's endeavors in the field of cell therapy.
This venture aligns with AstraZeneca's broader commitment to cell therapy, evidenced by previous collaborations with Quell Therapeutics, AbelZeta, Cellectis, and the acquisition of Neogene Therapeutics. The Rockville facility will integrate into AstraZeneca's global manufacturing and supply network, which encompasses nearly 30 sites across 16 countries.
AstraZeneca's US manufacturing sites, with a focus on small molecules and biologics, contribute significantly to the pharmaceutical landscape, employing over 2,600 full-time professionals and delivering more than 9 billion doses of medicines annually.
134 PHARMA FOCUS AMERICA ISSUE 03 - 2024
NEWS
ACQUISITIONS
Novo Nordisk Secures Fill-Finish Sites in Catalent Deal
Novo Nordisk has confirmed the acquisition of three fill-finish sites from Novo Holdings A/S as part of a larger transaction involving the acquisition of Catalent, Inc. by Novo Holdings. The deal aligns with Novo Nordisk's strategic goal of broadening access to diabetes and obesity treatments.
The move facilitates an expansion of manufacturing capacity, emphasizing scalability and speed, while providing flexibility for Novo Nordisk's existing supply network. Anticipated to enhance Novo Nordisk's filling capacity from 2026 onward, the acquisition targets an increased outreach to individuals living with diabetes and obesity.
The three specialized manufacturing sites, situated in Anagni (Italy), Brussels (Belgium), and Bloomington (Indiana, US), focus on the sterile filling of drugs. Employing over 3,000 individuals, these sites already have established collaborations with Novo Nordisk. The upfront payment for the acquisition amounts to US$11 billion.


The agreement outlines that the acquisition is likely to have a low single-digit negative impact on operating profit growth in 2024 and 2025, contingent upon the timing of closing.
Given the predominantly debt-financed nature of the acquisition, Novo Nordisk assures that its communicated share buyback program of DKK 20 billion remains unaffected.
EXPANSIONS
WuXi Biologics Hits Success with Massive 16,000L Run in Ireland


WuXi Biologics, a leading global Contract Research, Development, and Manufacturing Organization (CRDMO), has achieved a significant milestone with the successful completion of the inaugural manufacturing run at its drug substance facility MFG7 in Ireland. This accomplishment sets the stage for large-scale commercial manufacturing initiatives at the site.
The manufacturing run, conducted at a pioneering 16,000-liter scale, involved the integration of four 4,000-liter single-use bioreactors. This achievement not only signifies the first successful manufacturing run at the MFG7 facility but also represents the largest manufacturing scale to date for WuXi Biologics. Importantly, the Cost of Goods (COGS) from this run aligns with that of a 16,000-liter traditional stainless-steel bioreactor. This underscores the comparable cost efficiency demonstrated in over 100 runs at the 12,000-liter scale with single-use bioreactors in other WuXi Biologics facilities when compared to traditional stainless steel bioreactors.
WuXi Biologics' Ireland site, recognized with the Facility of the Year Award (FOYA) in the Operations category by ISPE in 2023, has swiftly gained regulatory approvals. Within nine months of commencing operations, it secured the first GMP certificate from the Irish Health Products Regulatory Authority (HPRA).
www. pharmafocusamerica.com 135
NEWS
FDA APPROVAL
AlphaMedixTM Earns FDA Breakthrough Status for Neuroendocrine Tumor Treatment
Clinical stage radiopharmaceutical companies RadioMedix, Inc. and Orano Med have achieved a significant milestone with the United States Food and Drug Administration (FDA) granting Breakthrough Therapy Designation (BTD) to AlphaMedixTM (212Pb-DOTAMTATE). This breakthrough status is for the treatment of adult patients facing unresectable or metastatic, progressive somatostatin receptorexpressing gastroenteropancreatic neuroendocrine tumors (GEP-NETs) who are new to peptide receptor radionuclide therapy (PRRT).
AlphaMedixTM, currently in Phase 2 clinical development, stands out as a Targeted Alpha Therapy. It comprises an SSTR-targeting peptide complex radiolabeled with lead-212 (212Pb), functioning as an in vivo generator of alpha particles. Notably, AlphaMedixTM is the first Targeted Alpha Therapy to secure Breakthrough Therapy Designation, a recognition of its potential impact on patient outcomes.
The Breakthrough Therapy Designation is grounded in the compelling results from both the phase 1 and ongoing phase 2 clinical trials evaluating the safety and efficacy of AlphaMedixTM. The phase 1 study demonstrated favorable tolerability, with a notable response rate of 62.5% for GEP-NET patients who had not previously undergone PRRT with LutatheraTM, based on the beta-particle emitter Lutetium-177. Moving into the phase 2 trial, the target response rate has already been achieved ahead of top-line data, anticipated in mid-2024.
COLLABORATIONS
Pfizer and Saama Boost AI Integration in R&D
Saama has entered into an expanded, multi-year agreement with global biopharmaceutical leader Pfizer to further enhance clinical research processes using AI-driven solutions. Originating from their 2020 partnership, the collaboration initially aimed to automate Pfizer's data review procedures with AI, resulting in the creation of Smart Data Quality (SDQ).

This innovative solution substantially reduced the time required for database lock. Building on this success, the new agreement extends beyond SDQ to incorporate Saama's advanced Biometrics Research and Analysis Information Network. This expansion will enable Pfizer to expedite regulatory submissions across its global study portfolio.
Saama's next-generation solution enhances statistical programming and biostatistics workflows, digitizes study specifications, and generates submission-ready tables, listings, and figures (TLF) artifacts. By leveraging Saama's technology, Pfizer anticipates a reduction in regulatory submission timelines, fostering efficiency across diverse global studies. The collaboration sets the stage for ongoing automation and innovation, providing a pathway to further accelerate future trial submissions.
Saama's AI-driven SaaS products and solutions, utilizing cutting-edge technology, empower study teams to navigate the complexities of contemporary clinical trial data.
136 PHARMA FOCUS AMERICA ISSUE 03 - 2024
NEWS



Introducing a group of highly focussed magazines for the Europe and Asian markets.
Aspiring to be leading journals in the B2B landscape of Pharmaceutical-Industry, the magazines covers Medical Sciences, Business & Technology and all the latest innovations.
Our magazines bring a fresh outlook towards insightful and pragmatic Pharmaceutical-Industry reporting. Delightfully selected topics presented by the gurus of the industry comes packed with latest happenings, sharp analysis & deep insights. We strive to keep you engaged, knowledgeable & wanting for more.
www. pharmafocusamerica.com 137 Introducing Advent of NEW-AGE PHARMACEUTICAL REPORTING
Automotive-technology.com |
| Hospitals-management.com |
Pharmaceutical-tech.com |
|
|
|
|
From the house of Ochre Media:
Defence-industries.com
Packaging-labelling.com
Plantautomation-technology.com
Plastics-technology.com
Pulpandpaper-technology.com Sportsvenue-technology.com
Steel-technology.com
Asianhhm.com | Pharmafocusasia.com
Scan to check websites Scan to check websites www.ochre-media.com

Transporting critical pharma and medical devices across 6 continents. THE WORLD WORKS BETTER WITH EMIRATES SKYCARGO SKYCARGO.COM
