ISSN 1432-4334 JAHRGANG 29 HEFT 1 Januar 2020
FÜR PHARMAKOLOGIE UND THERAPIE
JOURNAL OF PHARMACOLOGY AND THERAPY
Nicht interventionelle Studie mit Femannose® N zur Untersuchung von Verträglichkeit, Lebensqualität und Symptomverlauf bei akuter unkomplizierter Harnwegsinfektion Fortschritte in der Therapie des Prostatakarzinoms CIDP: Vorteile der Erhaltungstherapie mit subkutanem Immunglobulin Fortgeschrittenes Leberzellkarzinom: Für die Zweitlinientherapie ist jetzt auch Ramucirumab zugelassen Rezidiviertes Ovarialkarzinom: Niraparib ist eine gut verträgliche Option zur Langzeit-Erhaltungstherapie Multiple Sklerose: Real-World-Daten können bei der Therapieentscheidung unterstützen Neue hochdosierte Mesalazin-Formulierung optimiert Basistherapie der Colitis ulcerosa Colitis ulcerosa: JAK-Inhibitor Tofacitinib erweitert die Therapieoptionen
VERLAG
PERFUSION
Neues Therapieprinzip beim vorbehandelten follikulären Lymphom1,2
GEMEINSAM
REV
LIM
ID ®
R ituxim
STARK
ab
NEU
ZULASSUNG
• 3,3 Jahre medianes PFS3 • Bewährte Substanzen • Bekanntes Sicherheitsprofil
REVLIMID® 2,5 mg / 5 mg / 7,5 mg / 10 mg / 15 mg / 20 mg / 25 mg Hartkapseln. Wirkstoff: Lenalidomid. Zusammensetzung: Jede Kapsel enth. 2,5 mg / 5 mg / 7,5 mg / 10 mg / 15 mg / 20 mg / 25 mg Lenalidomid; sonst. Bestandteile: Kapselinhalt: Lactose, mikrokrist. Cellulose, Croscarmellose-Natrium, Magnesiumstearat; Kapselhülle: Gelatine, Titandioxid (E171), Indigocarmin (E132) (nur 2,5/10/15/20 mg), Eisen(III)-hydroxid-oxid x H2O (E172) (nur 2,5/7,5/10/20 mg); Drucktinte: Schellack, Propylenglycol, Kaliumhydroxid, Eisen(II,III)-oxid (E172). Anwendungsgebiete: Als Monotherapie für die Erhaltungstherapie v. erwachsenen Pat. mit neu diagnostiziertem multiplem Myelom nach einer autologen Stammzelltransplantation. Als Kombinationstherapie mit Dexamethason, o. Bortezomib u. Dexamethason, o. Melphalan u. Prednison für die Behandl. v. erwachsenen Pat. mit unbehandeltem multiplem Myelom, die nicht transplantierbar sind. In Komb. mit Dexamethason für die Behandl. d. multiplen Myeloms bei erwachsenen Pat., die mind. eine vorausgegangene Therapie erhalten haben. Als Monotherapie für die Behandl. v. erwachsenen Pat. mit transfusionsabh. Anämie infolge myelodysplast. Syndrome mit Niedrig- o. Intermediär-1-Risiko in Verb. mit isolierter del(5q) als zytogenet. Anomalie, wenn andere Behandlungsopt. nicht ausreichend o. nicht angemessen sind (MDS). Als Monotherapie für die Behandl. v. erwachsenen Pat. mit rezidiviertem o. refraktärem Mantelzell-Lymphom (MCL). In Komb. mit Rituximab (Anti-CD20-Antikörper) für die Behandlung v. erwachsenen Pat. mit vorbehandeltem follikulärem Lymphom (Grad 1 – 3a). Gegenanzeigen: Schwangerschaft; gebärfähige Frauen, außer alle Bed. d. Schwangerschaftsverhütungsprogramms werden eingehalten; Überempf. gegen d. Wirkstoff o. einen d. sonst. Bestandteile. Nebenwirkungen: Schwerwiegende NW: Neutropenie; Anämie, Thrombozytopenie, Thrombose. Sehr häufig: Anämie; Hautausschläge, Juckreiz, Muskelkrämpfe, -schwäche, -schmerzen, Rücken-, Glieder-, Gelenk-, Knochenschmerzen, Myalgie, Arthralgie, Ödeme einschl. periphere Ödeme; Schwäche, Müdigkeit; Fieber u. grippeart. Symptome; Parästhesie, Hyperästhesie, Schwindel, Tremor; Vermind. Appetit, Störung d. Geschmacksempfindung; Zunahme von Schmerzen, Tumorgröße o. Rötung um den Tumor; Gewichtsabnahme; Obstipation, Diarrhoe, Nausea, Erbrechen, Magenschmerzen, Sodbrennen; Hypokaliämie, -kalzämie, -natriämie; Hypothyreose; venöse Thromboembolien, vorw. tiefe Venenthrombose u. Lungenembolie; Infektionen aller Art, einschl. Nasennebenhöhlen, Lunge u. d. oberen Atemwege, Dyspnoe; verschwomm. Sehen; Katarakt; Nierenversagen; abnormale Leberwerte; erhöhte Leberwerte; Vaskulitis; Diabetes; Hypoglykämie; Kopfschmerzen; Nasenbluten; trockene Haut; Depression, Veränd. d. Stimmungslage, Schlafstörungen; Husten; Hypotonie; unklare körperliche Beschwerden, Unwohlsein; Stomatitis, Mundtrockenheit; Dehydrierung. Häufig: Hämolytische Anämie; Basalzellkarzinom, Plattenepithelkarzinom d. Haut Zahnfleischbluten, gastrointest. Blutungen (einschl. rektale Blutungen); Hypertonie, Bradykardie; Bilirubin im Blut erhöht; C-reaktives Protein erhöht; Hyperpigmentierung d. Haut, Prellungen, Hämatom; Hyperurikämie; Ekzem, Erythem, rissige Haut, Abschuppen o. Schälen d. Haut, Urtikaria; Pruritus, vermehrtes Schwitzen, Nachtschweiß; Dysphagie, Halsschmerzen, Schwierigkeiten mit d. Stimmqualität o. Stimmveränderung; laufende Nase; Harnverhalt, Harninkonsistenz; Hämaturie; Herzinsuffizienz; erektile Dysfunkt.; Schlaganfall, Ohnmachtsanfall, Vertigo, Synkope; Myokardinfakt; Muskelschwäche, Asthenie; Nackenschmerzen, Brustschmerzen; Schüttelfrost; Gelenkschwellung; verlangsamter o. blockierter Gallenfluss aus d. Leber; Hypophosphatämie o. -magnesemia; Schwierigkeiten beim Sprechen; Leberschädigung; Gleichgewichtsstörungen, Ataxie; Taubheit, Tinnitus; Neuralgie, Dysästhesie; Eisenüberladung; Durst; Verwirrtheit; Zahnschmerzen; Sturz. Gelegentlich: Intrakranielle Blutungen; Kreislaufstörungen; Verlust an Sehvermögen; Libidoverlust; Fanconi-Syndrom; Leberversagen; Colitis o. Typhlitis; renale Tubulusnekrose; Hautverfärbung, Lichtempfindlichkeitsreakt.; allerg. Reaktion; Tumorlyse-Syndrom; Arzneimittelreakt. mit Eosinophilie u. system. Sympt (DRESS). Selten: Stevens-Johnson-Syndrom (SJS), toxische epidermale Nekrolyse (TEN). Nicht bekannt: Pankreatitis; interstitielle Pneumonitis; seltene Fälle v. Rhabdomyolyse, einige wenn Lenalidomid mit einem Statin angewendet wurde; leukozytoklast. Vaskulitis; gastrointest. Perforation; Virusinfekt. (einschl. Reaktivierung v. Herpes-Zoster u. Hepatitis-B-Virus-Infekt.); Abstoßung eines transplant. soliden Organs. Warnhinweise: Risiko für schwere, angeborene Fehlbildungen, deshalb während der Schwangerschaft kontraindiziert. Bedingungen d. Schwangerschaftsverhütungsprogramms müssen erfüllt werden (männl. Pat.: Verwendung v. Kondomen; gebärf. Patientinnen: zuverl. Empfängnisverhütung; nicht-gebärf. Patientinnen: zuverl. Nachweis d. Nicht-Gebärfähigkeit). Stillen sollte während d. Behandl. abgebrochen werden. Erhöhtes Risiko f. venöse u. arterielle Thromboembolien. Pat. bzgl. sekundärer Primärmalignome (SPM) sorgfältig überwachen. Regelm. Blutbildkontr. notwendig. Vorsicht bei Pat. mit eingeschr. Nierenfunkt. Pat. mit anamnestisch bek. schwerw. Hautausschlag unter Thalidomid nicht mit Lenalidomid behandeln. Bei Verdacht auf SJS, TEN und DRESS Behandl. absetzen. Lenalidomid wird nicht für die Behandl. v. MCL-Pat. mit hoher Tumorlast empf., wenn alternative Behandlungsopt. z. Verf. stehen. Pat. mit unbehandeltem MM sind auf ihre Eignung, eine Lenalidomid-Kombinationstherapie zu tolerieren, sorgfältig zu beurteilen. Enthält Lactose. Weitere wichtige Informationen entnehmen Sie der Zusammenfassung d. Merkmale d. Arzneimittels (Fachinformation). Wenn Lenalidomid in Komb. mit anderen Arzneimitteln gegeben wird, müssen vor Beginn der Behandlung die entspr. Fachinformationen berücksichtigt werden. Darreichungsform u. Packungsgröße: REVLIMID® 2,5 mg / 5 mg / 7,5 mg / 10 mg / 15 mg / 20 mg / 25 mg Hartkapseln - Packung mit 7 oder 21 Hartkaps. (N1). Verschreibungspflichtig. Pharmaz. Untern.: Celgene Europe B.V., Winthontlaan 6 N, 3526 KV Utrecht, Niederlande. Stand d. Inf.: Dezember 2019 Celgene GmbH 81829 München info@celgene.de www.celgene.de Tel.: 089 / 451519-010
PM-DE-REV-00041
1 Aktuelle REVLIMID® Fachinformation. 2 Chiu H et al. Br J Haematol. 2019;185(2):240–253. 3 Leonard JP et al. J Clin Oncol. 2019;37(14):1188–1199. PFS = Progressionsfreies Überleben R² = REVLIMID® (Lenalidomid) + Rituximab
EDITORIAL
Asklepius hieß bekanntermaßen der griechische Gott der Heilkunst, was ein untrügliches Indiz dafür liefert, dass die Medizin schon damals einen bedeutenden Beitrag zum Wohl der Menschheit leistete, zumindest was den Bereich der damaligen griechischen Stadtstaaten anbelangt. Sein bildliches Symbol, der Schlangenstab, ist bis heute mit der latinisierten Form seines Namens als Äskulapstab bekannt, das Symbol für den ärztlichen Stand. Dabei wäre es wohl viel gerechter, statt der Schlange dem Apfelbaum symbolisch Dank und Ehre zu erweisen. Schließlich war es das Malheur mit dem Apfel, das der Überlieferung nach Adam und Eva aus dem Paradies trieb und Krankheit und Leid über die Menschheit brachte. Mit Bitte um Nachsicht für die jetzt folgende Plattheit sei auf die ebenfalls überlieferte Weisheit verwiesen, wonach des einen Leid des anderen Freud ist – in diesem Fall die bis heute auskömmliche Existenzgrundlage für den ärztlichen Stand. Ausgerüstet mit diesem Wissen um unsere originären Wurzeln wird plausibel, dass sich (frühe) Kulturen Krankheit und Gebrechen, um die epochale Definition für Gesundheit der WHO aus dem Jahr 1946 zu bemühen, zwanglos als Strafe der Götter erklären konnten und dies gerne auch taten. Konsequent zu Ende gedacht, würde der erfolgreiche Heiler den Göttern relevant ins Handwerk gepfuscht – und damit deren Zorn auch auf sich gezogen haben. Vor diesem Hintergrund ist es nur folgerichtig, dass Schamanen seit Äonen ihren Erfolg über den Umweg über die Götter suchen nach dem Motto: Götter beschwichtigen, dann nehmen die den Fluch vom Kranken. Der für den Erfolg dieser Strategie hilfreiche gute Draht zu den Göttern war als angenehmer und persönlich nützlicher Nebeneffekt auch hilfreich fürs eigene Image. So einfach erklärt sich die bis heute beobachtbare herausragende gesellschaftliche Stellung der Schamanen. Parallel dazu fällt es nicht schwer sich vorzustellen, dass irgendwann irgendwo irgendeinem professionellen Götterbesänftiger die zeitliche Nähe des Genusses einer Kalebasse Weiden-
1
Eminenz-basierte EBM
rindentee (willkürliches Beispiel ohne konkreten historischen Bezug) zur Verbesserung seiner Zahnschmerzen auffiel. Und dass dann eine Art urtümlicher Verifizierung des kausalen Zusammenhangs zunächst intraindividuell (Selbstversuch) wie interindividuell (Versuch am Patienten) erfolgte. Jetzt fehlt eigentlich nur noch ein einziger Schritt, um die Heilkunst multiplizierbar zu machen: die Weitergabe dieses Wissens – die Geburtsstunde der Schulmedizin, wenn wir den ersten Teil des Begriffs definieren als „lehrbar und lernbar“. Jetzt müssen wir nur noch, und dazu sollte das obige Beispiel dienen, den Begriff Wissen gleichsetzen mit Erfahrung, um mit mathematischer Logik zur folgenden Gleichung zu kommen: Schulmedizin = Erfahrungsmedizin bzw. Erfahrungsmedizin = Schulmedizin. Um die hier skizzierte Entwicklung der Medizin über die Ebene der Spekulation hinaus zu erheben, seien an dieser Stelle nur zwei historisch verbriefte Fakten angeführt. So belegen Skelettfunde eindeutig, dass schon vor 5000 bis 10000 Jahren Schädeltrepanation und Amputationen von Gliedmaßen so fachkundig durchgeführt wurden, dass die so Behandelten die Behandlung langfristig überlebten. Im (übrigens in Leipzig verwahrten) ägyptischen „Papyrus Ebers“ aus der Zeit um 1500 vor Christus sind mehr als 700 Arzneipflanzen mit Anwendungsregeln und Indikationen beschrieben. Das kann sich kein einzelner Heilkundiger ausgedacht und noch weniger „ausgetestet“ haben! Das Motiv der schriftlichen Dokumentation kann wohl auch nur die Weitergabe dieses Wissens gewesen sein. Inhärent verbunden mit dieser Berufsgrundlage der Heilkundigen war implizit, dass das Individuum mit der meisten Erfahrung auf das größte Arsenal an medizinisch hilfreichen Ansätzen zurückgreifen konnte, oder,
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Prof. Dr. med. K.-L. Resch, Bad Elster
wie zeitgenössische Kritiker abfällig und Nase rümpfend feststellen, auf das System der „Eminenz-basierten Medizin“ als böser Schwester der Evidenz-basierten Medizin (EBM). Die Evidenz-basierte Medizin steht für die Integration einer neuen Dimension in die Heilkunst. Ihr Urvater, der britische Epidemiologie Archie Cochrane, beklagte zu recht, dass wissenschaftliche Erkenntnisse, die nicht zugänglich (besser noch: jederzeit abrufbar) sind, für die Therapie in der Praxis nicht zur Verfügung stehen. Die Väter der EBM stellten aber auch unmissverständlich klar: „Die Praxis der EBM bedeutet die Integration individueller klinischer Expertise mit der bestmöglichen externen Evidenz aus systematischer Forschung“ [1]. Darin stecken zwei Forderungen, die einem zunehmenden Anteil der besonders lauten Protagonisten der Evidenz-basierten Medizin abhanden gekommen zu sein scheinen: Zuerst einmal die Forderung nach der „bestmöglichen externen Evidenz“. Die kommt sicher nicht aus den vielen Evidenzfabriken, die typischerwei© VERLAG PERFUSION GMBH
INHALT
2
se hirnlos ein paar Begriffe medlinen (Wort kreiert in Anlehnung an das bekannte googeln), das Gefundene ohne Sinn und Verstand in einen EBM-Thermomix stopfen und dann, wenn nichts Vernünftiges rauskommt, messerscharf schließen: Diese Therapie kann nicht empfohlen werden! Zum Zweiten die Einbeziehung „individueller klinischer Expertise“, was nichts anderes bedeutet als klinische Erfahrung. Und die sollte nicht mit polemischem Verweis z.B. auf das jahrhundertelange, ignorante Beharren auf die exzessive Anwendung des Aderlasses pauschal diskreditiert, sondern bestenfalls in gleicher Weise professionalisiert werden wie die „bestmögliche externe Evidenz“. Dass dies möglich ist (vgl. dazu auch [2]), zeigen eindrucksvoll z.B. bevölkerungsrepräsentative Surveys wie formal gut (etwa in Form eines DelphiProzesses oder eines formalen Gruppenkonsens) durchgeführte Konsensus-Initiativen. Vor allem aber wäre zu wünschen, dass die Evidenz-basierte Medizin nicht nach dem Vorbild der gerade aus dieser Ecke so vehement kritisierten Eminenz-basierten Medizin in absolutistischer Weise die Existenz der Heilkunst gefährdet. Karl-Ludwig Resch, Bad Elster
ORIGINALARBEIT Nicht interventionelle Studie mit Femannose® N zur Untersuchung von Verträglichkeit, Lebensqualität und Symptomverlauf bei akuter unkomplizierter Harnwegsinfektion 4 Florian Wagenlehner, Ludwig N. Baumgartner, Bettina Schopf, Jens Milde
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS Fortschritte in der Therapie des Prostatakarzinoms
10
CIDP: Vorteile der Erhaltungstherapie mit subkutanem Immunglobulin 16 Fortgeschrittenes Leberzellkarzinom: Für die Zweit linientherapie ist jetzt auch Ramucirumab zugelassen
19
Rezidiviertes Ovarialkarzinom: Niraparib ist eine gut verträgliche Option zur Langzeit-Erhaltungstherapie
23
Multiple Sklerose: Real-World-Daten können bei der Therapieentscheidung unterstützen
24
NEUE UND BEWÄHRTE ARZNEIMITTEL
Quellen: 1 Sackett DL et al. Evidence based medicine: what it is and what it isn’t. BMJ 1996;312:71-72 2 Resch KL. Anwendungsbeobachtung reloaded. J Pharmakol Ther 2014;2:37
Neue hochdosierte Mesalazin-Formulierung optimiert Basistherapie der Colitis ulcerosa
26
Colitis ulcerosa: JAK-Inhibitor Tofacitinib erweitert die Therapieoptionen
28
RUBRIKEN Wissenswertes 21, 35 Kongresse 31
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
Der neue CGRP-Antikörper für die spezifische Migräneprophylaxe1
Weniger Migräne. Mehr vom Leben.
Jakobs Geburtstag Nein, wegen meiner Migräne brauche ich absolute Stille Ja, ich kann mit ihm feiern!
Stark & schnell – Schnell weniger Migränetage1–3 Verträglich – Verträglichkeitsprofil auf Placebo-Niveau1–3 Flexibel – Flexibles Injektionsintervall: subkutane Gabe 1 x/Monat oder 1 x/Quartal1
NEU
Sie möchten mehr über AJOVY® erfahren? Über den QR-Code gelangen Sie auf unser Fachkreis-Portal unter www.moretomigraine.de
DE/FRE/19/0140
1. Fachinformation AJOVY®, Stand: 03/2019. 2. Dodick DW et al. JAMA 2018; 319(19): 1999–2008. 3. Silberstein SD et al. N Engl J Med 2017; 377(22): 2113–2122. AJOVY® 225 mg Injektionslösung in Fertigspritze Wirkstoff: Fremanezumab. Zusammensetzung: Eine Fertigspritze enth. 225 mg Fremanezumab. Fremanezumab ist ein humanisierter monoklonaler Antikörper, der mittels rekombinanter DNA-Technik in Eizellen des chinesischen Hamsters (Chinese Hamster Ovary, CHO) hergestellt wird. Sonst. Bestandt.: Histidin, Histidinhydrochlorid-Monohydrat, Sucrose, Natriumedetat (Ph.Eur.), Polysorbat 80, Wasser für Injektionszwecke. Anwendungsgebiete: Migräneprophylaxe bei Erw. mit mind. 4 Migränetagen pro Monat. Gegenanzeigen: Überempfindlichkeit gg. den Wirkstoff od. einen der sonst. Bestandt. Warnhinw.: AM enth. weniger als 1 mmol Natrium (23 mg) pro Dosiereinheit, d. h. es ist nahezu „natriumfrei“. Schwangerschaft/Stillzeit: Anwend. währ. der Schwangerschaft vermeiden. Anwend. währ. der Stillzeit nur in Betracht ziehen, falls diese klinisch erford. ist. Nebenwirkungen: Schmerzen an der Injektionsstelle. Verhärtung an der Injektionsstelle. Erythem an der Injektionsstelle. Juckreiz an der Injektionsstelle. Ausschlag an der Injektionsstelle. Immunogenität. Dosierung: Es stehen zwei Dos.optionen zur Verfügung: 225 mg einmal monatlich od. 675 mg alle drei Monate. Status: Verschreibungspflichtig. Stand: 3/19. TEVA GmbH, Graf-Arco-Str. 3, 89079 Ulm, Deutschland. Weitere Informationen siehe Fachinformation. ▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
ORIGINALARBEIT
4
ZUSAMMENFASSUNG Hintergrund: Diese Beobachtungsstudie untersuchte die Anwendung des Medizinprodukts Femannose® N mit dem Wirkstoff D-Mannose bei Patientinnen mit einer akuten unkomplizierten Harnwegsinfektion, entweder allein oder in Kombination mit Antibiotika oder anderen Medikamenten/Maßnahmen. Patienten und Methodik: 103 erwachsene Patientinnen mit der Diagnose einer akuten, unkomplizierten Harnwegsinfektion wurden in die Studie eingeschlossen. Während der Anwendung von Femannose® N wurden Wirkung, Sicherheit und Verträglichkeit von den Ärzten sowie anhand von Patiententagebüchern ermittelt. Ergebnisse: Von 97 auswertbaren Patientinnen (mittleres Alter 53,6 Jahre) waren 86,6 % am letzten Tag der Dokumentation geheilt. Etwa 2 Drittel aller Patientinnen (64,9 %) hatten dieses Behandlungsziel bereits nach 3 Tagen erreicht. Für 7 leichte oder mäßige unerwünschte Ereignisse wurde gemäß Einschätzung des Arztes ein Kausalzusammenhang mit der Anwendung des Mannoseprodukts als möglich erachtet. Schlussfolgerung: Das Medizinprodukt Femannose® N ist aus Sicht der Patientinnen und der behandelnden Ärzte ein wirksames Mittel zur Behandlung von unkomplizierten, bakteriellen Harnwegsinfektionen. Die Anwendung ist auf Basis der erhobenen Daten als sicher und allgemein gut verträglich einzustufen. Schlüsselwörter: Beobachtungsstudie, akute unkomplizierte Harnwegsinfektion, D-Mannose, Trinkgranulat, Femannose® N
Nicht interventionelle Studie mit Femannose® N zur Untersuchung von Verträglichkeit, Lebensqualität und Symptomverlauf bei akuter unkomplizierter Harnwegsinfektion Florian Wagenlehner1, Ludwig N. Baumgartner2, Bettina Schopf3, Jens Milde3 Klinik und Poliklinik für Urologie, Kinderurologie und Andrologie, Gießen 2 Privatpraxis für Frauenheilkunde, Freising 3 Pharmalog Institut für klinische Forschung GmbH, Ismaning 1
U
nkomplizierte bakterielle Harnwegsinfektionen gehören zu den am häufigsten vorkommenden bakteriellen Infektionen bei ambulanten Patientinnen, sind häufig wiederkehrend und einer der häufigsten Gründe für eine Arztkonsultation. Gemäß Leitlinien wird für deren Behandlung üblicherweise eine antibiotische Therapie empfohlen. Jedoch haben Resistenzen von Krankheitserregern gegen häufig verordnete Antibiotika bei unkomplizierten Harnwegsinfektionen in den vergangenen Jahren deutlich zugenommen [1]. Vor diesem Hintergrund und der Tatsache, dass eine antibiotische Therapie von den betroffenen Patientinnen nicht immer gewünscht wird, gewinnen andere Behandlungsoptionen zunehmend an Bedeutung. D-Mannose ist ein Epimer der Glukose, das die Anhaftung von Pathogenen an das Epithel der Harnwege wirksam
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
herabsetzt und dazu beiträgt, ein erneutes Auftreten von Infektionen physikalisch zu verhindern. D-Mannose ist als CE-zertifiziertes Medizinprodukt unter dem Handelsnamen Femannose® N in mehreren europäischen Ländern zur Prävention und Behandlung von bakteriellen Harnwegsinfektionen auf dem Markt. Die Wirkung von D-Mannose wurde in mehreren klinischen Studien untersucht [2, 3, 4]. Zur therapeutischen Anwendung werden beim akuten Infekt gemäß Gebrauchsanweisung in den ersten 3 Tagen jeweils 3 und an den folgenden beiden Tagen jeweils 2 Portionsbeutel à 2 g Mannose über den Tag verteilt empfohlen [5]. Die vorgestellte Studie wurde konzipiert, um klinische Nachweise zur Verwendung und zum potenziellen Nutzen bzw. zu den Risiken des Produkts basierend auf Daten aus der täglichen Anwendungspraxis zu generieren. © VERLAG PERFUSION GMBH
ORIGINALARBEIT
Methodik
Diese multizentrische, prospektive, nicht interventionelle Studie wurde im Zeitraum von Januar bis Juli 2019 durchgeführt. An der Studie nahmen 18 Arztpraxen der Fachrichtungen Urologie, Gynäkologie und Allgemeinmedizin in Deutschland teil. Insgesamt 103 Patientinnen im Alter von mindestens 18 Jahren mit einer akuten unkomplizierten Harnwegsinfektion wurden in die Studie eingeschlossen. Diese hatten zuvor ihr schriftliches Einverständnis zur Teilnahme gegeben. Vor Beginn der Studie erfolgte eine Beratung durch die zuständige Ethikkommission. Die Studie ist im Deutschen Register Klinischer Studien unter der Nummer DRKS00016632 registriert. Die Diagnose einer akuten, unkomplizierten Harnwegsinfektion stellte der Arzt gemäß seiner üblichen Routine, d.h. klinisch in Verbindung mit einer Urinuntersuchung (Urinanalyse mittels Teststreifen und/oder Urinkultur). Der Arzt empfahl im Rahmen seiner ärztlichen Routine das Medizinprodukt entweder als alleinige Therapie oder zusammen mit einem Antibiotikum oder anderen Medikamenten/Maßnahmen, unabhängig von seiner Entscheidung, die Patientin in die Studie einzubeziehen. Datenerhebung Folgende Daten wurden über Tagebucheinträge generiert: Die Patientinnen bewerteten die folgenden 5 vorgegebenen Symptome einer Harnwegsinfektion bis zur subjektiven Symptomfreiheit, jedoch maximal bis Tag 7: • Brennen/Schmerzen beim Wasserlassen (numerische Skala
•
• • •
von 0 = keine Schmerzen/Brennen bis 5 = extreme Schmerzen/ Brennen) Häufigkeit der Blasenentleerung (nicht häufiger / kaum häufiger / etwas häufiger / sehr viel häufiger) Kleine Mengen Urin (ja / nein) Ständiger Harndrang (ja / nein) Veränderungen des Urins (ja inkl. Spezifikation / nein)
Heilung wurde definiert als Fehlen von Brennen/Schmerzen beim Wasserlassen (Score 0 oder 1 an Tag 7 bzw. am letzten Tag der Dokumentation). Als symptomfrei wurde eine Patientin definiert, die zur Frage nach Brennen/Schmerzen einen Score von 0 oder 1 hatte, auf die Frage nach der Häufigkeit mit „nicht häufiger“ oder „kaum häufiger“ antwortete und die letzten 3 Fragen mit „nein“ beantwortete. Ferner beantworteten die Patientinnen folgende Fragen: • Zusätzliche Behandlung der Harnwegsinfektion mit Antibiotikum oder anderen Medikamenten/Maßnahmen (inkl. Spezifikation) • Einschränkung der täglichen und gesellschaftlichen Aktivitäten (Lebensqualität) aufgrund der Harnwegsinfektion (ja sehr / ja ziemlich / ja ein wenig / nein) • Beurteilung einer ersten Symptomverbesserung (ja / nein) • Anzahl der eingenommenen Portionsbeutel • Sonstige Beschwerden/Nebenwirkungen (unerwünschte Ereignisse)
5
SUMMARY Background: This observational study examined the use of the product Femannose® N with the active ingredient D-mannose in patients with an acute uncomplicated urinary tract infection, either alone or in combination with antibiotics or other drugs/ measures. Patients and methods: 103 adult patients diagnosed with an acute, uncomplicated urinary tract infection were included in the study. While using the product, the effects, safety and tolerability were determined by the doctors and on the basis of patient diaries. Results: Of 97 evaluable patients (mean age 53.6 years), 86.6 % were cured on the last day of the documentation. About two thirds of all patients (64.9 %) had achieved this treatment goal after only 3 days. For 7 mild or moderate adverse events, a causal relationship with the use of the mannose product was considered possible by the doctor. Conclusions: From the point of view of the patient and the treating physician, Femannose® N is an effective product for uncomplicated, bacterial urinary tract infections. On the basis of the data collected, the application can be classified as safe and generally well tolerated. Key words: observational study, acute uncomplicated urinary tract infection, D-mannose, drink granules, Femannose® N
Am Tag der Symptomfreiheit, aber spätestens am Tag 7 bewerteten die Patientinnen die Anwendung des Produkts:
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
ORIGINALARBEIT
6
• Zufriedenheit • Bereitschaft zur künftigen Anwendung zur Behandlung einer Harnwegsinfektion • Verständliche Formulierung der Gebrauchsanweisung • Anwendung als Pulver zum Auflösen • Geschmack Nach dem letzten Eintrag gaben die Patientinnen das Tagebuch an den behandelnden Arzt während eines optionalen Besuchs zurück oder sendeten es postalisch zu. Nach Abschluss der Behandlung evaluierte der behandelnde Arzt die globale Wirkung und Verträglichkeit der Behandlung sowie unerwünschte Ereignisse (optionale Visite oder telefonisch). Auswertung Die Auswertung der Daten erfolgte mittels deskriptiver Statistik. Für die Patientinnen, die die Tagebuchdokumentation vor Tag 7 beendet hatten, wurden die letzten Einträge für Parameter mit täglicher Dokumentation nicht für die folgenden Tage fortgeschrieben. Vielmehr wurden diese Einträge zusammen mit den Einträgen der Patientinnen mit Dokumentationsende an Tag 7 zusätzlich als „letzter Tag“ ausgewertet. Subgruppenanalysen wurden für die Endpunkte zur Wirkung durchgeführt. Für die Vergleiche der Subgruppen „Kombination mit Antibiotikum“ bzw. „Kombination mit anderen Medikamenten/Maßnahmen“ versus „Monotherapie“ wurden adäquate statistische Tests angewendet (exakter Fisher-Test, Wilcoxon-Rangsummentest, Log rank-Test). Gemäß Fallzahlberechnung waren 95 Patientinnen erforderlich,
um eine angenommene Heilungsrate von 80 % mit 95%iger Wahrscheinlichkeit zu erzielen. Bei einer vermuteten Abbruchquote von ungefähr 5 % waren insgesamt 100 Patientinnen in die Studie aufzunehmen. Ergebnisse
6 der 103 aufgenommenen Patientinnen waren nicht auswertbar (v.a. wegen nicht erfolgter Rückgabe des Tagebuchs). Das mittlere Alter der verbleibenden 97 Patientinnen lag bei 53,6 Jahren. Die Einschlussdiagnose wurde mittels Urinteststreifenuntersuchung (92,8 %) und/oder Urinkultur (26,8 %) bestätigt. Gemäß Tagebuchdokumentation wendeten mehr als 2 Drittel der Patientinnen (71,1 %) eine Kombination entweder mit Antibiotikum (54,6 %) oder anderen Medikamenten/Maßnahmen (16,5 %) an. Am häufigsten wurde hier ein Nieren- und Blasentee genannt (25,8 % bei Behandlungsbeginn). Der Anteil der Patientinnen, die ihren akuten Harnwegsinfekt ausschließlich mit dem Mannoseprodukt behandelten, lag bei 28,9 %, wohingegen die behandelnden Ärzte die Monotherapie 47,4 % der Patientinnen empfohlen hatten. Die Behandlungsdauer variierte zwischen 3 und 7 Tagen (Median: 5 Tage). Die Einnahme-Compliance mit der empfohlenen Standarddosierung lag im Mittel bei 95,9 %. Wirkung Die Heilungsrate stieg im zeitlichen Verlauf an und erreichte 86,6 % am letzten Tag der Dokumentation. Etwa 2 Drittel aller Patientinnen (64,9 %) hatten dieses
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Behandlungsziel bereits nach 3 Tagen erreicht. In der Subgruppe mit Monotherapie war die Erfolgsquote zu diesem Zeitpunkt höher als unter der Kombinationsbehandlung mit einem Antibiotikum bzw. anderen Medikamenten/ Maßnahmen (85,7 % vs. 56,6 % bzw. 56,3 % an Tag 3), während sie am letzten Tag in den Subgruppen weitgehend vergleichbar war (92,9 % vs. 83,0 % bzw. 87,5 %, Abb. 1). Die Heilungsraten waren am letzten Tag der Dokumentation etwas niedriger als an den Tagen 5 bzw. 6 – 7, da 7 Patientinnen (7,2 %) die Eintragungen in das Tagebuch bereits vor Eintritt der Heilung beendet hatten. Die Unterschiede in der Heilungsrate zwischen einer Kombinationsbehandlung mit einem Antibiotikum bzw. anderen Medikamenten/Maßnahmen versus Monotherapie waren statistisch nicht signifikant. Der Anteil an symptomfreien Patientinnen erhöhte sich im Studienverlauf und erreichte 70,1 % am letzten Studientag. Analog zur Heilungsrate war der Anteil der Patientinnen, die am Studienende keine typischen Symptome einer Harnwegsinfektion hatten, unter Monotherapie (78,6 %) höher als bei der kombinierten Anwendung mit einem Antibiotikum (67,9 %) bzw. anderen Medikamenten/Maßnahmen (62,5 %) (Abb. 2). Der Anteil der symptomfreien Patientinnen war am letzten Tag der Dokumentation etwas niedriger als an den Tagen 4 bzw. 5 – 7, da 17 Patientinnen (18,3 %) ihre Tagebucheinträge schon vor Erreichen der Symptomfreiheit beendeten. Die Gruppenunterschiede hinsichtlich der Symptomfreiheit erreichten keine statistische Signifikanz. Eine erste Verbesserung der Symptome einer Harnwegsinfektion wurde im Mittel nach 2,8 Tagen er© VERLAG PERFUSION GMBH
ORIGINALARBEIT
7
Abbildung 1: Geheilte Patientinnen (%) nach Subgruppe, Studientag und letztem Tag der Dokumentation.
Abbildung 2: Symptomfreie Patientinnen (%) nach Subgruppe, Studientag und letztem Tag der Dokumentation. JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
ORIGINALARBEIT
8
Zeitpunkt
Monotherapie
vor Therapie
3,8
Dokumentationsende
Kombination mit einem Antibiotikum
Kombination mit anderen Medikamenten/ Maßnahmen
2,6
2,4
5,0
2,1
4,5
Tabelle 1: Summenscore aus Einschränkungen in den täglichen und gesellschaftlichen Aktivitäten.
reicht mit nur geringen Unterschieden in den Subgruppen (2,6 bis 3,0 Tage). Die Zeit bis zum Erreichen einer Heilung war unter Monotherapie etwas kürzer im Vergleich zu einer Kombinationsbehandlung mit einem Antibiotikum bzw. anderen Medikamenten/Maßnahmen (Mittelwert 4,8 Tage vs. 5,2 bzw. 5,4 Tage). Patientinnen unter Monotherapie wurden im Mittel nach 3,9 Tagen und unter Kombinationsbehandlung nach 4,9 (Kombination mit Antibiotikum) bzw. 4,6 Tagen (Kombination mit anderen Medikamenten/Maßnahmen) symptomfrei. Die behandelnden Ärzte beurteilten die Wirkung der Behandlung als „ausgezeichnet“ oder „gut“ bei fast allen Patientinnen unter Monotherapie (96,0 %) und der Kombination mit anderen Medikamenten/Maßnahmen (92,9 %). Die Kombinationsbehandlung mit einem Antibiotikum wurde für 76,6 % dieser Patientinnen entsprechend bewertet. Sicherheit und Verträglichkeit 7,2 % aller Patientinnen berichteten insgesamt 10 unerwünschte Ereignisse leichten oder mäßigen Schweregrades. Für 7 Ereignisse wurde gemäß Einschätzung des Arztes ein Kausalzusammenhang mit der Anwendung des Mannoseprodukts als möglich erachtet bzw. konnte nicht ausgeschlossen werden. Am häufigsten handelte es sich dabei um gastrointestinale Beschwerden (Blähungen, Bauchschmerzen, Durchfall, Übelkeit,
Gastroenteritis). Darüber hinaus wurde für eine Patientin ein Hautauschlag berichtet. 6 der 7 Ereignisse mit potenziellem Kausalzusammenhang zum Produkt traten unter kombinierter Anwendung mit einem Antibiotikum auf, eines (Blähungen) zusammen mit anderen Medikamenten/Maßnahmen. Die Verträglichkeit bewerteten die Ärzte als „ausgezeichnet“ oder „gut“ bei 87,6 % aller Patientinnen und als „mäßig“ bzw. „sehr schlecht“ bei jeweils 1 Patientin (1,0 %). Für 10,3 % der Patientinnen lag keine Einschätzung des Arztes vor. Patienteneinschätzung Gemäß der Einschätzung der Patientinnen hinsichtlich ihrer Lebensqualität waren 80,4 % in ihren täglichen und 69,1 % in ihren gesellschaftlichen Aktivitäten vor Beginn der Behandlung aufgrund der Harnwegsinfektion eingeschränkt. Der Anteil dieser Patientinnen reduzierte sich bereits bis Tag 3 auf 47,4 % bzw. 42,3 %. In den Subgruppen ging der Summenscore aus Einschränkungen in den täglichen und gesellschaftlichen Aktivitäten bis zum Dokumentationsende zurück, gleichbedeutend mit einer Verbesserung der Lebensqualität bzw. Abnahme der Beeinträchtigungen in den täglichen und gesellschaftlichen Aktivitäten (Tab. 1). Die Unterschiede in der Score-Reduktion zwischen den Gruppen waren statistisch nicht signifikant.
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Der Anteil der Patientinnen mit der Bereitschaft, das Produkt künftig wieder zur Behandlung einer Harnwegsinfekten anzuwenden, war am höchsten unter Kombination mit anderen Medikamenten/ Maßnahmen (93,8 %), gefolgt von der Monotherapie (82,1 %) und der zusätzlichen Behandlung mit einem Antibiotikum (74,0 %). 7,4 % aller Patientinnen würden das Medizinprodukt in Zukunft nicht mehr anwenden, die übrigen 12,8 % waren unentschieden. Die Gebrauchsanweisung für Femannose® N schätzten 94,7 % aller Patientinnen als verständlich formuliert ein, 2,1 % verneinten dies und 3,2 % waren in der Beurteilung unschlüssig. Die Anwendung als Pulver zum Auflösen beurteilte die überwiegende Mehrzahl der Patientinnen (je nach Subgruppe 85,7 – 94,0%) als (sehr) angenehm/einfach. 7,4 % waren in ihrer Einschätzung unentschieden und eine Patientin (1,1 %) empfand die Zubereitung als unangenehm. Den Geschmack des Produkts bewerteten je nach Subgruppe zwischen 75,0 % und 88,0 % der Patientinnen als (sehr) angenehm. 11,7 % waren in ihrer Einschätzung unentschieden und 4,3 % empfanden ihn als unangenehm. Sehr zufrieden oder zufrieden mit dem Produkt waren fast alle Patientinnen unter Monotherapie bzw. in Kombination mit anderen Medikamenten/Maßnahmen (92,9 % bzw. 93,8 %), während nur 74,0 % der Patientinnen durch die Kombination mit Antibiotikum zur © VERLAG PERFUSION GMBH
9
ORIGINALARBEIT
gleichen Einschätzung gelangten. 14,9 % aller Patientinnen waren unentschieden und 2,1 % gaben Unzufriedenheit mit dem Produkt an. Diskussion
Die große Mehrzahl (86,6 %) aller Patientinnen war am Ende des Beobachtungszeitraums geheilt ohne größere Unterschiede zwischen den Subgruppen. Symptomfrei wurden jedoch 78,6 % der Patientinnen unter Monotherapie gegenüber 67,9 % unter der Kombination mit einem Antibiotikum oder 62,5 % unter der Kombination mit anderen Medikamenten/Maßnahmen. Die Monotherapie führte im Mittel auch früher zur Heilung und Symptomfreiheit im Vergleich zur kombinierten Behandlung. Für diese Ergebnisse könnte von Bedeutung sein, dass Patientinnen unter Kombinationsbehandlung möglicherweise eine schwerere Harnwegsinfektion hatten, worauf eine im Vergleich zur alleinigen Therapie höhere Gesamtpunktzahl bei den Beeinträchtigungen im täglichen und gesellschaftlichen Leben aufgrund der Harnwegsinfektion vor Behandlungsbeginn hindeutet. Die positive Wirkung des Mannoseprodukts wird in dieser Studie gestützt durch einen hohen Anteil an Patientinnen, die mit der Behandlung zufrieden waren und das Produkt bei einer Harnwegsinfektion künftig wieder an-
wenden würden. Die statistischen Tests im Hinblick auf die Unterschiede zwischen kombinierter Anwendung und Monotherapie bezüglich Heilung, Symptomfreiheit und Lebensqualität waren zwar statistisch nicht signifikant, jedoch war diese Beobachtungsstudie nicht konzipiert, um Signifikanzen auf der Basis von Hypothesen aufzuzeigen. Die wenigen unerwünschten Ereignisse mit potenziellem Kausalzusammenhang zum untersuchten Produkt sind auch typische Nebenwirkungen einer Antibiotikagabe, die fast alle betroffenen Patientinnen zusätzlich anwendeten. Eine abschließende kausale Zuordnung zum jeweiligen Produkt ist daher nicht möglich. Schlussfolgerungen
Femannose® N ist aus Sicht der Patientinnen und der behandelnden Ärzte ein wirksames Produkt zur unterstützenden Behandlung von unkomplizierten bakteriellen Harnwegsinfektionen. Zudem konnte im Rahmen dieser Studie gezeigt werden, dass viele Patientinnen das Produkt auch ohne Antibiotika anwenden (entweder als Monotherapie oder in Kombination mit anderen Medikamenten/ Maßnahmen) und zumindest bei leichten bis mittelschweren Harnwegsinfektionen ebenfalls sehr gute Heilungserfolge erzielen. Die positive Einschätzung der Anwen-
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
dung und des Geschmacks des Produkts sowie der Verständlichkeit der Gebrauchsanweisung durch die Patientinnen sprechen für dessen Einsatz bei Harnwegsinfektionen. Die Anwendung von Femannose® N ist auf Basis der erhobenen Daten als sicher und allgemein gut verträglich einzustufen.
Literatur 1 nterdisziplinäre S3 Leitlinie Epidemiologie, Diagnostik, Therapie, Prävention und Management unkomplizierter, bakterieller, ambulant erworbener Harnwegsinfektionen bei erwachsenen Patienten, Registernummer 043-044; letzte Aktualisierung 04/2017 2 Domenici L, Monti M, Bracchi C, Giorgini M, Colagiovanni V, Muzii L, Benedetti Panici P. D-mannose: a promising support for acute urinary tract infections in women. A pilot study. Eur Rev Med Pharmacol Sci 2016;20:2920-2925 3 Kranjčec B, Papeš D, Altarac S. D-mannose powder for prophylaxis of recurrent urinary tract infections in women: a randomized clinical trial. World J Urol 2014; 32:79-84 4 Porru D, Parmigiani A, Tinelli C, Barletta D, Choussos D, Di Franco C, Bobbi V, Bassi S, Miller O, Gardella B, Nappi RE, Spinillo A, Rovereto B. Oral D-mannose in recurrent urinary tract infections in women: a pilot study. J Clin Urol 2014;7:208-213 5 Gebrauchsanweisung Femannose® N; Stand: Juni 2017
Für die Verfasser: Dr. med. Ludwig N. Baumgartner Wissenschaftlicher Studienkoordinator Facharzt für Frauenheilkunde und Geburtshilfe Privatpraxis für Frauenheilkunde Marienplatz 3 85354 Freising
© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
10
Fortschritte in der Therapie des Prostatakarzinoms
D
as Prostatakarzinom gilt hängigkeit vom Rezidivrisiko in heute als eine der wichtigs- Tumoren mit niedrigem, intermeten Erkrankungen des männ- diärem und hohem Risiko. Das lolichen Geschlechts. Bei den tödlich kal fortgeschrittene Prostatakarziverlaufenden Tumorerkrankungen nom umfasst die Stadien T3-4 N0 bei Männern steht es hierzulande M0. Die Stadien N1 und/oder M1 an zweiter Stelle – im Jahr 2014 werden als fortgeschrittenes bzw. verstarben ca. 13.700 Patienten an metastasiertes Prostatakarzinom seinen Folgen [1]. bezeichnet (Abb. 1) [2]. Etwa 3 von 4 Tumoren werden in einem frühen Stadium (T1 oder Die Stadien des T2) diagnostiziert. Die relative 5-Jahres-Überlebensrate für Pros Prostatakarzinoms und ihre tatakrebs liegt bei 91 % [1]. Die Therapieoptionen Stadieneinteilung basiert auf der UICC-Klassifikation (Union Inter- Lokal begrenztes nationale Contre le Cancer) bzw. Prostatakarzinom TNM-Klassifikation (T = Tumor, N = Nodes bzw. Lymphknoten, Beim lokal begrenzten ProstataM = Metastasen). In den Stadien karzinom (T1-2 N0 M0), das sehr Stadien T1-2 N0 M0des ist dasProstatakarzinoms Prostatakar- günstig, aber auch sehr aggressiv zinom lokal begrenzt. Hier erfolgt verlaufen kann, empfiehlt die akeine weitere Unterteilung in Ab- tuelle S3-Leitlinie der Deutschen
Gesellschaft für Urologie (DGU) für die Patienten, die für eine kurativ intendierte Behandlung in Frage kommen, die radikale Prostatektomie, die perkutane Strahlentherapie und – je nach Risikoprofil – die LDR- oder HDRBrachytherapie. In Abhängigkeit vom Risikoprofil kann ergänzend zur perkutanen Strahlentherapie eine antihormonelle (hormonab lative) Therapie erfolgen, da das Wachstum des Tumors durch An drogene gefördert wird. Diese Androgendeprivationstherapie besteht aus der Gabe eines LHRHAgonisten bzw. -Antagonisten oder einer Orchiektomie, also einer medikamentösen oder chirurgischen Kastration [2]. Eine weitere Option, die der Charité über UROLOGIE Patient laut DGU informiert werden sollte, ist die Strategie der
C
Nicht metastasiert Neu diagnostiziertes mHSPC
Metastasiert Erstdiagnose Prostatakarzinom
Primär progredientes mHSPC Lokal begrenztes oder lokal fortgeschrittenes Prostatakarzinom
Biochemisches Rezidiv
mCRPC 1st
mCRPC 2nd
Tod
M0CRPC ADT
Abbildung 1: Stadien des Prostatakarzinoms. M0CRPC = nicht metastasiertes kastrationsresistentes Prostatakarzinom, mHSPC = metastasiertes hormonsensitives Prostatakarzinom, mCRPC = metastasiertes kastrationsresistentes Prostatakarzinom, ADT = Androgendeprivationstherapie.
•
Damit Patienten möglichst lange profitieren, erfolgt die moderne Therapie des kastrationsresistenten PCa in der Sequenz.
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
11
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Active Surveillance. Dabei sollen die unerwünschten Wirkungen und Therapiefolgen einer sofortigen lokalen Therapie gegen das Risiko einer nicht rechtzeitigen Behandlung abgewogen und die aktive Behandlung bis zu einem Zeitpunkt aufgeschoben werden, an dem sich Hinweise auf eine Progression ergeben oder der Patient die Therapie wünscht. Voraussetzungen für eine Active Surveillance sind ein PSA-Wert ≤10 ng/ml, ein Gleason-Score ≤6, die Kategorie cT1 bzw. cT2a und Tumornachweis in der Biopsie in höchstens 2 Stanzen bei leitliniengerechter Entnahme von 10–12 Stanzen sowie höchstens 50 % Tumor pro Stanze. Zudem sollten bei der Indikationsstellung Alter und Komorbidität berücksichtigt werden [2]. Lokal fortgeschrittenes Prostatakarzinom Beim lokal fortgeschrittenen Prostatakarzinom (T3-4 N0 M0) und geplanter lokaler Therapie sollte der Patient gemäß S3-Leitlinie über Vor- und Nachteile sowohl einer radikalen Prostatektomie mit Lymphadenektomie als auch einer perkutanen Strahlentherapie mit ggf. zusätzlicher antihormoneller Therapie über 2, besser 3 Jahre aufgeklärt werden. Bei Patienten, die eine mutmaßliche Lebenserwartung unter 10 Jahren haben, empfiehlt die S3-Leitlinie, anstelle der kurativen Behandlung ein Watchful Waiting in Erwägung zu ziehen, wobei erst bei einer symptomatischen Progression eine palliative Intervention erfolgt. Eine weitere Option, die in dieser Situation besprochen werden sollte, ist die sofortige Androgendeprivationstherapie [2].
Nicht metastasiertes kastrations resistentes Prostatakarzinom (M0CRPC) Wenn unter der Androgendeprivationstherapie (ADT) trotz eines Serum-Testosteronspiegels auf Kastrationsniveau (<50 ng/dl) ein biochemisches Rezidiv auftritt, sich aber noch keine Fernmetastasen nachweisen lassen, liegt ein nicht metastasiertes kastrationsresistentes Prostatakarzinom (M0CRPC) vor. Nach mehreren übereinstimmenden Studien zum natürlichen Verlauf des M0CRPC haben die Betroffenen ein besonders hohes Risiko, einen Progress zu erleiden oder zu versterben, wenn ihr PSA-Wert schnell steigt, d.h. wenn ihre PSA-Verdopplungszeit bei 10 Monaten oder darunter liegt [3]. In der deutschen S3-Leitlinie wurde bei Patienten mit M0CRPC über viele Jahre hinweg ein abwartendes Vorgehen unter Beibehaltung der klassischen ADT empfohlen [2]. Demnach mussten sich die Betroffenen und ihre Ärzte lange Zeit damit abfinden, bis zum Nachweis von Metastasen nur regelmäßig zu Kontrolluntersuchungen zu gehen, ohne effektiv in das Krankheitsgeschehen eingreifen zu können. Dies hat sich erst in jüngster Zeit geändert, nachdem neue Wirkstoffe für die Therapie des HochrisikoM0CRPC, wie z.B. Apalutamid (Erleada®), verfügbar wurden (s.u.) [4, 5, 6]. Metastasiertes hormonsensitives Prostatakarzinom (mHSPC) Auch beim metastasierten Prostatakarzinom, bei dem lokale Maßnahmen nicht mehr angezeigt sind, erfolgt eine palliative antihormo-
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
nelle Therapie. Lange Jahre wurde die ADT beim hormonnaiven oder hormonsensitiven metastasierten Prostatakarzinom (mHNPC, mHS PC) zunächst allein eingesetzt. Seit 2016 wird basierend auf damals neuen Daten für viele Patienten in dieser Situation in nationalen und internationalen Leitlinien eine Kombination aus ADT und Chemotherapie mit Docetaxel empfohlen, die allerdings außerhalb der Zulassung von Docetaxel erfolgen muss [6]. Seit der entsprechenden Indikationserweiterung 2017 basierend auf der LATITUDE-Studie [7] stellt für Patienten mit neu diagnostiziertem Hochrisiko-mHSPC Abirateronacetat (Zytiga®) plus Prednison/Prednisolon in Kombination mit einer ADT eine Alternative dar, die in den aktuellen Leitlinien ebenfalls empfohlen wird (s.u.) [2]. Metastasiertes kastrations resistentes Prostatakarzinom (mCRPC) Auf die alleinige ADT spricht das Prostatakarzinom in den meisten Fällen (80 – 90 %) zwar zunächst an, bemerkbar an einem Rückgang der Beschwerden, einer Verkleinerung des Primärtumors bzw. der Metastasen, einem Abfall des Serum-Testosteronspiegels (auf Kastrationsniveau <20 – 50 ng/dl) und einem Rückgang des PSAWertes. Nach einigen Monaten bis mehreren Jahren schreitet die Erkrankung jedoch in der Regel trotz weiterer ADT fort, was sich am Wiederanstieg des PSA-Werts, einer radiologischen Progression und/oder an Symptomen wie Schmerzen infolge von Knochenmetastasen zeigt [6]. Nach ADT-Versagen war zunächst nur Docetaxel als einzige Erstli© VERLAG PERFUSION GMBH
12
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
nientherapie zugelassen, mittlerweile stehen auch antihormonelle (Abirateronacetat, Enzalutamid), andere zytostatische (Cabazitaxel) und weitere Therapiestrategien (Radium-223) zur Verfügung [2, 4, 5, 6]. Apalutamid und Abirateron als wichtige Säulen der modernen Prostatakarzinomtherapie
Sowohl der selektive Androgenrezeptor-Inhibitor Apalutamid (Erleada®) als auch der Androgenbiosynthese-Inhibitor Abirateron (Zytiga®) spielen heute eine zentrale Rolle bei der Behandlung des fortgeschrittenen Prostatakarzinoms. Erleada® ist indiziert zur Therapie des nicht metastasierten kastrationsresistenten M0CRPCStadiums [8], Zytiga® ist in Verbindung mit Prednison/Prednisolon unter anderem zugelassen für das neu diagnostizierte HochrisikomHSPC und das nicht oder mild
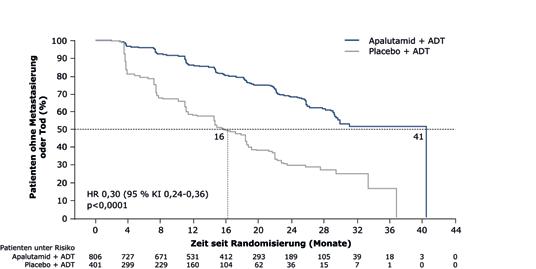
symptomatische mCRPC [9]. Beide Medikamente zeigten in ihren Zulassungsstudien eine signifikante Überlegenheit gegenüber dem jeweiligen Vergleichsarm [7, 10, 11, 12]. Effektive Therapie des Hochrisiko-M0CRPC mit Apalutamid Die Zulassung von Apalutamid basierte auf Ergebnissen der ersten Interimsanalyse der Phase-IIIStudie SPARTAN, in der der moderne Androgenrezeptor-Inhibitor in Kombination mit einer Androgendeprivationstherapie (ADT) bei M0CRPC-Patienten mit einer PSA-Verdopplungszeit von ≤10 Monaten im Vergleich zu Placebo plus ADT eine gute Wirksamkeit und Verträglichkeit erzielte [10]. Das metastasenfreie Überleben (primärer Endpunkt) lag unter Apalutamid bei fast 3,5 Jahren, sodass Patienten aus der Verum-
Gruppe im Vergleich zur PlaceboGruppe (je plus ADT) mehr als 2 zusätzliche Jahre metastasenfrei lebten (41 Monate vs. 16 Monate; HR: 0,30; p < 0,0001) (Abb. 2). Außerdem war – neben anderen sekundären Endpunkten (Tab. 1) – die mediane Zeit bis zum symptomatischen Progress unter Apalutamid signifikant verlängert (HR: 0,45; p < 0,0001). Dies ist für die Patienten von besonderer Bedeutung, weil die Symptomverschlechterung einen erheblichen Einfluss auf die Lebensqualität der Patienten hat [10]. PFS2 weist auf den Erhalt der Wirksamkeit von Folgetherapien hin Aufgrund der Fortschritte der letzten Jahre und der damit einhergehenden Verfügbarkeit verschiedener Behandlungsoptionen erhalten Männer mit CRPC heute moderne antihormonelle
Patienten unter Risiko Apalutamid + ADT 806 727 671 531 412 293 189 105 39 18 3 Placebo + ADT 401 299 229 160 104 62 36 15 7 1 0
0 0
Abbildung 2: Ergebnisse der SPARTAN-Studie für den primären Endpunkt: Das metastasenfreie Überleben (MFS) war unter der Therapie mit Apalutamid (Erleada®) um mehr als 2 Jahre länger als unter Placebo (jeweils plus ADT) [10]. JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
13
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Mediane Zeit bis zur symptomatischen Progression Progressionsfreies Überleben (PFS) Mediane Zeit bis zur Metastasierung (TTM) Gesamtüberleben (OS)
HR (95%-KI) 0,45 (0,32 – 0,63) 0,30 (0,25 – 0,36) 0,28 (0,23 – 0,34) 0,70 (0,47 – 1,04)
p-Wert <0,0001 <0,0001 <0,0001 0,0742
Tabelle 1: Sekundäre Endpunkte der SPARTAN-Studie: Apalutamid schnitt bei allen sekundären Endpunkten besser ab als Placebo [10].
Therapien meist nacheinander. Dabei kann die Wahl der richtigen Sequenz für die Dauer und den Erfolg der gesamten Therapie ausschlaggebend sein. Damit Patienten möglichst lange profitieren, sollte demnach neben der Wirksamkeit und Verträglichkeit einer anfänglichen Therapie auch die Wirksamkeit von Folgetherapien betrachtet werden. Das progressionsfreie Überleben in der zweiten Therapielinie (PFS2) ist ein neuer Endpunkt in klinischen Studien zum fortgeschrittenen Prostatakarzinom, der
Hinweise auf die Wirksamkeit einer Systemtherapie über 2 Therapielinien gibt. Es ist definiert als die Zeit zwischen Randomisierung und Krankheitsprogression oder Tod in der ersten Folgetherapie, die unter anderem mit Abirateron plus Prednison/Prednisolon erfolgte. Anfang 2019 wurde ein Jahr nach der zulassungsrelevanten Interimsanalyse, welche bereits die Überlegenheit von Apalutamid plus ADT bezüglich des PFS2 (explorativer Endpunkt) gezeigt hatte, eine aktualisierte Auswertung präsentiert. Hierbei
bestätigte sich die signifikante Verlängerung des PFS2 unter Apalutamid plus ADT gegenüber Placebo plus ADT (Median nicht erreicht vs. 39,3 Monate; HR: 0,5; p < 0,0001; Abb. 3) [13]. Die positiven PFS2-Ergebnisse werden durch die im Zuge der SPARTANStudie erhobenen Resistenzdaten ergänzt. So war unter Apalutamid im Vergleich zu Placebo (jeweils plus ADT) die Rate von Androgenrezeptor-Anomalien nicht erhöht, die die Wirksamkeit späterer Therapielinien einschränken können [14].
Patienten unter Risiko Apalutamid + ADT 806 779 760 730 693 637 525 398 289 191 117 44 13 0 Placebo + ADT 401 386 362 331 285 243 182 124 75 45 26 9 1 0
Abbildung 3: Ergebnis der SPARTAN-Studie für das progressionsfreie Überleben 2 (PFS2) [13]. JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
14
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Abirateron/P hat sich im Versorgungsalltag bewährt Im Verlauf einer Androgendeprivationstherapie entwickeln die meisten Patienten mit der Zeit eine Kastrationsresistenz. Die nachlassende Wirksamkeit der konventionellen antihormonellen Therapie ist auf unterschiedliche Resistenzmechanismen zurückzuführen, von denen viele die Signalkaskade des Androgenrezeptors betreffen. Zu ihnen gehört unter anderem die Überexpression des Androgenrezeptors. Dies hat zum Beispiel zur Folge, dass der Androgenrezeptor sensitiver auf Androgene reagiert, zudem erlangen die Tumorzellen oft selbst die Fähigkeit, Androgene zu produzieren [15]. Außerdem unterdrückt die ADT nur die Androgenproduktion in den Hoden, nicht jedoch in Nebennieren und Tumorzellen [15]. Mit Abirateronacetat (Zytiga®) steht für die betroffenen mCRPCPatienten eine antihormonelle Therapie zur Verfügung, die in diesem Stadium noch wirken kann. Denn Abirateron, der aktive Metabolit von Abirateronacetat, hemmt die CYP17-Enzyme und unterdrückt
Empfehlungen für die Erstlinientherapie des nicht / mild symptomatischen mCRPC 2019
C
Charité UROLOGIE
7.32
Evidenzbasierte Empfehlung
Empfehlungsgrad
Patienten mit metastasierter, kastrationsresistenter, asymptomatischer oder gering symptomatischer und progredienter Erkrankung sollte (alphabetische Reihenfolge) Abirateron (in Kombination mit Prednison / Prednisolon) oder Enzalutamid als Erstlinientherapie angeboten werden.
7.33
Evidenzbasierte Empfehlung
Empfehlungsgrad
Patienten mit metastasierter, kastrationsresistenter, asymptomatischer oder gering symptomatischer und progredienter Erkrankung kann Docetaxel als Erstlinientherapie angeboten werden.
B
0
Abbildung 4: Empfehlungen der S3-Leitlinie für die Erstlinientherapie des nicht/mild symptomCRPC, metastasiertes kastrationsresistentes Prostatakarzinom. matischen mCRPC [2]. UNIVERSITÄTSMEDIZIN BERLIN
Mod. nach S3-Leitlinie Prostatakarzinom, Version 5.1 – Mai 2019, AWMF-Registernummer: 043/022OL: www.leitlinienprogramm-onkologie.de/leitlinien/prostatakarzinom/; letzter Abruf: 2.9.19.
auf diese Weise – im Sinne einer Androgeneliminationstherapie – die Androgen-Biosynthese nicht nur in den Hoden, sondern auch in den Nebennieren und im Tumorgewebe [9]. Seit einer Zulassungserweiterung im Jahr 2012 kann der orale Androgenbiosynthese-Inhibitor in Kombination mit Prednison/Prednisolon in der Erstlinientherapie bei nicht oder mild symptomatischen mCRPCPatienten nach ADT-Versagen eingesetzt werden, bei denen eine Chemotherapie noch nicht klinisch indiziert ist (Abb. 4). Seitdem hat
Primäre Endpunkte Kombiniertes PSA-PFS über beide Therapielinien Median (Monate) PSA-Ansprechrate unter 2. Therapielinie Sekundäre Endpunkte Kombiniertes PFS über beide Therapielinien Median (Monate) PFS unter 2. Therapielinie (seit Cross-over) Median (Monate) PSA-PFS unter 2. Therapielinie (seit Cross-over) Median (Monate) Gesamtüberleben seit Beginn Erstlinientherapie Median (Monate)
sich Abirateron/P in der Praxis bewährt und es liegen neben den zulassungsrelevanten klinischen Studiendaten zunehmend Versorgungsdaten vor, die seine Wirksamkeit und Verträglichkeit auch unter Alltagsbedingungen untersucht haben [16, 17]. Wirksamkeit von Abirateron/P in der Sequenz Für die Therapie des nicht oder mild symptomatischen mCRPC nach Versagen einer vorherigen
Abirateron/P → Enzalutamid
Enzalutamid → Abirateron/P
p-Wert
13,6
11,9
0,106
31 %
4 %
<0,001
13,6
10,0
0,220
2,7
1,6
<0,001
2,7
1,3
<0,001
n.e.
24,3
0,378
Tabelle 2: Ergebnisse der prospektiven Phase-II-Studie zur Therapiesequenz bei Chemotherapie-naiven Patienten mit mCRPC, die in der Erstlinie randomisiert Abirateron plus Prednison/Prednisolon (Abirateron/P) oder Enzalutamid erhalten hatten und bei PSA-Progress oder inakzeptabler Toxizität auf die jeweils andere Therapie umgestellt worden waren [18]. PSA = prostataspezifisches Antigen, PFS = progressionsfreies Überleben, n.e. = nicht erreicht. JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
ADT wird von der aktuellen S3Leitlinie neben Abirateron/P auch Enzalutamid bevorzugt empfohlen (Docetaxel hat nur eine „Kann“Empfehlung; Abb. 4) [2]. In die Therapieentscheidung sollte hier ebenfalls die Wirksamkeit der Folgetherapie einfließen. Wie eine aktuelle prospektive Phase-II-Studie [18] zeigt, lässt sich durch die Therapiefolge Abirateron/P → Enzalutamid im Vergleich zur Sequenz Enzalutamid → Abirateron/P ein signifikant besseres PSA50-Ansprechen in der zweiten Therapielinie erreichen (31 % vs. 4 %; p < 0,001). Auch das PSA-progressionsfreie Überleben (PSAPFS) in der zweiten Therapielinie war signifikant länger bei Therapiestart mit Abirateron/P (median 2,7 vs. 1,3 Monate; p < 0,001) (Tab. 2). In die Studie waren Chemotherapienaive Männer mit mCRPC eingeschlossen worden, die in der Erstlinie randomisiert Abirateron/P oder Enzalutamid erhielten (je n = 101) und bei PSA-Progress oder inakzeptabler Toxizität auf die jeweils andere Therapie umgestellt wurden (n = 65 bzw. 71) [18].
Fazit für die Praxis
Mit Apalutamid und Abirateron/P stehen CRPC-Patienten im Rahmen der jeweiligen Indikation 2 wirksame und verträgliche Substanzen zur Verfügung, die zudem die Wirksamkeit von entsprechend untersuchten Folgetherapien nicht einzuschränken scheinen. Seit der Zulassung von Apalutamid im Januar 2019 wird die CRPC-Sequenztherapie daher um eine weitere Option erweitert, von der viele Patienten profitieren können. Brigitte Söllner, Erlangen Literatur 1 Robert Koch-Institut, Gesellschaft der epidemiologischen Krebsregister in Deutschland e. V. (Hrsg.). Krebs in Deutschland 2013/2014. 11. Ausgabe, Berlin, 2017 2 S3-Leitlinie zum Prostatakarzinoms. Version 5.1, Mai 2019. AWMF-Registernummer: 043/022OL. Im Internet: www.leitlinienprogramm-onkologie.de/leitlinien/ prostatakarzinom 3 Saad F et al. J Urol 2018;199:e229 & PD10-04 4 AUA Guideline 2018: Castration-Resistant Prostate Cancer. Im Internet: http:// www.auanet.org/guidelines/castration-resistant-prostate-cancer-(2013-amended-2018)
15
5 NCCN Guidelines Prostate Cancer. Version 2.2019 – April 17, 2019. Im Internet: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf 6 EAU Guidelines Prostate Cancer 2019. Im Internet: uroweb.org/guideline/prostate-cancer 7 Fizazi K et al. N Engl J Med 2017;377: 352-360 8 Fachinformation Erleada®; Stand: Januar 2019 9 Fachinformation Zytiga® 500 mg Filmtabletten; Stand: November 2017 10 Smith MR et al. N Engl J Med 2018; 378:1408-1418 11 Rathkopf DE et al. Eur Urol 2014; 66:815-825 12 Ryan CJ et al. Lancet Oncol 2015; 16:152-160 13 Small EJ et al. J Clin Oncol 2019; 37(Suppl 7S): abstr 144 & Poster, GU ASCO 2019 14 Smith MR et al. Androgen receptor anomalies and efficacy of apalutamide in patients with nonmetastatic castration-resistant prostate cancer from the phase 3 SPARTAN study. AACR Annual Meeting 2018 (abstr 2605 & Poster) 15 Merseburger AS et al. Oncologist 2013;18: 558-567 16 Boegemann M et al. BMC Cancer 2019; 19: 60 17 Suttmann H et al. Prospektive, clusterrandomisierte Real-World Studie zum Einfluss von Adhärenzmaßnahmen auf die Therapie von Abirateron Acetat plus Prednisolon bei Patienten mit metastasiertem, kastrationsresistentem Prostatakarzinom (mCRPC). 71. Kongress der DGU 2019 (Vortrag) 18 Khalaf D et al. J Clin Oncol 2018;36(15, Suppl); abstr 5015 & Poster, ASCO Annual Meeting 2018
© Fathema Murtaza
Ihr neueR ArbeitsPLATZ
WIR SUCHEN QUALIFIZIERTE MITARBEITERINNEN UND MITARBEITER, DIE MIT UNS IN DEN BEREICHEN MEDIZIN, TECHNIK, ADMINISTRATION UND PROJEKTKOORDINATION HUMANITÄRE HILFE LEISTEN.
Unsere Teams sind in rund 60 Ländern im Einsatz. Werden Sie ein Teil davon! Informieren Sie sich online: www.aerzte-ohne-grenzen.de/mitarbeiten
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Träger des Friedensnobelpreises
© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
16
B
ei der chronischen inflamma torischen demyelinisierenden Polyneuropathie (CIDP) handelt es sich um eine Autoimmunerkrankung, bei der primär die Myelinscheiden im peripheren Nervensystem von Antikörpern, Zytokinen oder Lymphozyten oder Lymphozyten und Makrophagen angegriffen werden [1]. Folgen der entzündlichen Reaktion sind eine segmentale Demyelinisierung sensibler und motorischer Nerven sowie eine axonale Degeneration. Die Erkrankung verläuft progressiv oder schubförmig und ist klinisch durch proximale und distale symmetrische Paresen der Arme und Beine sowie Sensibilitätsstörungen charakterisiert [2]. Die Prävalenz liegt bei bis zu 9 Fällen pro 100.000 Einwohner [3]. Die CIDP sollte so früh wie möglich diagnostiziert werden, um rechtzeitig eine adäquate Behandlung einleiten zu können. Nur so lassen sich irreversible Schädigungen der Axone verhindern. Zur Erhaltungstherapie steht CIDP-Patienten, die bereits auf intravenöses Immunglobulin (IVIg) angesprochen haben, seit März 2018 mit Hizentra® alternativ das erste und einzige subkutane Immunglobulin (SCIg) zur Verfügung [4]. Therapie so früh wie möglich beginnen
Die aktuelle DGN-Leitlinie zu immunvermittelten Neuropathien empfiehlt für die Akuttherapie 3 ähnlich wirksame Optionen: intravenöse Immunglobuline (IVIg), Glukokortikosteroide und Plasmapherese [5]. In der Langzeittherapie sind Steroide allerdings meist keine Option und gerade bei Begleiterkrankungen wie Diabetes oder Osteoporose problematisch.
CIDP: Vorteile der Erhaltungstherapie mit subkutanem Immunglobulin
Die Plasmapherese ist für die Langzeitanwendung nicht geeignet – bleibt also die Therapie mit Immunglobulinen. Alternativ zu IVIg kann eine wirksame Symptomkontrolle auch durch eine subkutane Ig-Applikation erzielt werden. Der Vorteil: Das SCIg Hizentra® kann – nach einer entsprechenden Schulung – zuhause vom Patienten selbst appliziert werden [4]. Überzeugende Daten zur Wirksamkeit des SCIg
Die Zulassung des 20%igen SCIg Hizentra® basiert auf den Ergebnissen der PATH-Studie [6]. Mit 172 eingeschlossenen Patienten ist sie die bisher größte internationale, doppelblinde Phase-III-Studie zur Therapie der CIDP. Untersucht wurden 2 verschiedene Hizentra®Dosierungen (0,2 g/kg und 0,4 g/ kg KG pro Woche subkutan). Nach Stabilisierung mit IVIg erhielten die Patienten über einen Behandlungszeitraum von 24 Wochen die Erhaltungstherapie mit Hizentra® oder Placebo (Abb. 1). Primärer Endpunkt war der prozentuale Anteil der Patienten, die eine Verschlechterung im adjustierten INCAT (Inflammatory Neuropathy Cause and Treatment) Disability Score erfuhren oder aus anderen Gründen aus der Studie ausschie-
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
den. Sekundäre Endpunkte waren die Zeit bis zum Erreichen des primären Endpunkts, der INCATScore, die mittlere Griffstärke der Hände sowie der MRC-Score (MRC = Medical Research Council). Nach 24 Wochen waren beide SCIg-Dosierungen der Placebogabe in Bezug auf den primären Endpunkt signifikant überlegen: Die absolute Risikoreduktion betrug 30 % in der 0,4 g/kg-Dosisgruppe und 25 % in der 0,2 g/kg-Dosisgruppe). Umgekehrt war der Anteil der Patienten, bei denen sich der INCAT-Score nicht verschlechterte, im Vergleich zu Placebo hochsignifikant höher und betrug unter 0,2 g/kg/Woche 67 %, unter 0,4 g/ kg/Woche 81 % und 44 % in der Placebogruppe (Abb. 2). Auch bei den sekundären Endpunkten (I-RODS, Griffstärke der Hände und MRC-Summen-Score) waren beide SCIg-Dosierungen dem Placebo deutlich überlegen. Dazu passend zeigte der QoL-Fragebogen EQ-5D eine Steigerung der Lebensqualität der Patienten unter SCIg in den Dimensionen Mobilität, Selbstfürsorge, Angst/ Depression, Alltagsaktivitäten und Schmerzen/Unwohlsein im Vergleich zur Placebogruppe (Abb. 3). Die SCIg-Therapie wurde gut vertragen. 24 % der SCIg-Patienten entwickelten lokale, überwiegend © VERLAG PERFUSION GMBH
17
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
geringgradig ausgeprägte Reaktionen an der Infusionsstelle (95 % leicht, 5 % mäßig), die im Verlauf der Studie zurückgingen [6]. Die Studiendaten zeigen, dass das SCIg Hizentra® zur Stabilisierung der Erkrankung beiträgt und als wirksame und gut verträgliche Erhaltungstherapie zur Schub-Prophylaxe bei Patienten mit CIDP eingesetzt werden kann. Während IVIg alle 3 – 4 Wochen in einer Dosis von 1 g/kg KG stationär oder ambulant in einem Therapiezentrum intravenös infundiert wird, kann Hizentra® in der Regel wöchentlich und dann in niedrigerer Dosierung bei gleichbleibender monatlicher Gesamtdosis zuhause vom Patienten selbst appliziert werden [4]. Die wöchentliche SCIg-Gabe führt zu gleichmäßigeren IgG-Spiegeln, wodurch der unter IVIg am Ende des Dosierungsintervalls mögliche Wear-off-Effekt (Nachlass-Effekt) vermieden werden kann [7].
Ausschluss, Ausschluss, falls nach falls nach 12 Wochen keine 13 Wochen nicht Verschlechterung restabilisiert *2 g/kg KG Anfangsdosis, 1 g/kg KG Erhaltungsdosis
CDIP-Patienten ohne Verschlechterung des INCAT-Scores in %
Abbildung 1: Design der PATH-Studie [6].
Placebo
0,2 g/kg KG Hizentra® wöchentlich
p=0,012 (vs. Placebo)
0,4 g/kg KG Hizentra® wöchentlich
p=0,001 (vs. Placebo)
Abbildung2: Ergebnis der PATH-Studie: Unter der Therapie mit Hizentra® war mit beiden Dosierungen eine signifikante Zunahme der Patienten ohne Verschlechterung des INCAT-Scores zu verzeichnen [6].
Abbildung 3: Die große Mehrheit der Patienten konnte unter der Behandlung mit Hizentra® hinsichtlich der Lebensqualität profitieren (gemessen mittels EQ-5D im Vergleich zum Ausgangswert und Placebo) [6]. JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Umstellung von IVIg auf SCIg
Eine Umstellung von IVIg auf SCIg empfiehlt sich z.B. bei Patienten mit Wear-off-Effekt, ausgeprägten Nebenwirkungen unter IVIg-Gabe oder schlechtem Venenstatus [7]. Neben medizinischen können auch persönliche Gründe des Patienten für eine Umstellung sprechen, so vor allem „Krankenhausaufenthalte und lange Anreise vermeiden“. Die Therapie mit Hizentra® wird 1 Woche nach der letzten IVIgInfusion begonnen. Die empfohlene subkutane Dosis beträgt 0,2 – 0,4 g/kg KG einmal pro Woche. Abhängig vom erreichten IgG-Spiegel und dem klinischen Ansprechen kann das Dosierungsintervall aber auch von den Patienten individuell z.B. mehrmals wö© VERLAG PERFUSION GMBH
18
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
chentlich oder zweiwöchentlich, gewählt werden. Wichtig ist dabei, dass die kumulative Gesamtdosis pro Woche gleich bleibt [8]. Hizentra® kann an verschiedenen Stellen wie Bauch, Oberschenkel, Oberarm und lateraler Hüfte verabreicht werden. Bei höheren Dosen (mehr als 25 ml) ist es ratsam, die Applikation auf mehrere Stellen zu verteilen. Dabei sollte zwischen den Injektionsstellen ein Mindestabstand von 5 cm eingehalten werden [4]. Brigitte Söllner, Erlangen
Literatur 1 Mäurer M. Immunneuropathien. In: Stangel M, Mäurer M (Hrsg.): Autoimmunerkrankungen in der Neurologie: Diagnostik und Therapie. Berlin: Springer, 2018 2 Grimm A et al. Fortsch Neurol Psychiatr 2018;86:439-452 3 Hughes RA, J Clin Immunol 2010;30 (Suppl 1):70-73 4 Fachinformation Hizentra®; Stand: Februar 2019 5 Sommer C. et al. Therapie akuter und chronischer immunvermittelter Neuropathien und Neuritiden, S2e-Leitlinie, 2018. In: Deutsche Gesellschaft für Neurologie (Hrsg.), Leitlinien für Diagnostik und Therapie in der Neurologie. Online: www.dgn.org/leitlinien 6 van Schaik IN et al. Lancet Neurol 2018; 17:35-46 7 Aktuelle Neurologie, Blickpunkt Medizin: Therapie mit Immunglobulinen bei CIDP. Empfehlung zum Vorgehen bei der Umstellung von IVIg auf SCIg in der klinischen Praxis, Heft 8, Oktober 2018 8 Sidhu J et al. Biol Ther 2014;4:41-55
Fortgeschrittenes Leberzellkarzinom: Für die Zweitlinientherapie ist jetzt auch Ramucirumab zugelassen
D
as Leberzellkarzinom (hepatozelluläres Karzinom, HCC) ist die häufigste primäre maligne Erkrankung der Leber und entwickelt sich wahrscheinlich aus hepatischen Stammzellen. Die Ausbreitung des aggressiven Tumors kann über die lokale Expansion, eine intrahepatische Ausbreitung sowie über Fernmetastasen erfolgen [1]. Das HCC ist eine der wenigen Krebsarten mit steigender Inzidenz und Mortalität, die relativen 5-Jahres-Überlebensraten liegen bei Männern bei etwa 14 % und bei Frauen bei etwa 11 % [2]. Neue Perspektiven für Patienten mit fortgeschrittenem oder inoperablem HCC ergeben sich aus der
Zulassungserweiterung für Ramucirumab (Cyramza®), die die Europäische Kommission dem monoklonalen Antikörper 2019 erteilt hat: Ramucirumab ist nun – zusätzlich zur Behandlung des Magen-, Kolorektal- und nicht kleinzelligen Lungenkarzinoms – auch als Monotherapie zur Behandlung von erwachsenen Patienten mit fortgeschrittenem oder inoperablem HCC indiziert, die ein Serum-Alpha-Fetoprotein (AFP) von ≥400 ng/ml aufweisen und die zuvor mit Sorafenib behandelt wurden [3]. Basis für die Zulassungserweiterung waren die Ergebnisse der REACH-Studien [4, 5].
Ramucirumab Ramucirumab (Cyramza®) ist ein humaner, monoklonaler Immunglobulin (Ig)G1-Antikörper, der spezifisch an die extrazelluläre Domäne des VEGF (Vascular Endothelial Growth Factor)-Rezeptors 2 bindet und so die die Andockstelle für die Signalmoleküle VEGF-A, VEGF-C und VEGF-D blockiert. Dadurch verhindert Ramucirumab die Aktivierung des VEGF-Rezeptor-2 und der nachgeordneten Signalkaskaden, die für die Bildung neuer Blutgefäße und damit für die Versorgung des Tumors, das Tumorwachstum und die Metastasierung eine wichtige Rolle spielen. Dass dies funktioniert, hat der VEGFR-2-Antagonist Ramucirumab bereits bei der Behandlung des Adenokarzinoms des Magens, des Kolorektalkarzinoms und des fortgeschrittenen nicht kleinzelligen Lungenkarzinoms (NSCLC) unter Beweis gestellt. Mit der Zulassung für die Behandlung des HCC steht Ramucirumab nun als Behandlungsoption für die Zweitlinientherapie bei 4 Tumorentitäten zur Verfügung.
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
19
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Überlebensverlängerung bei HCC-Patienten mit erhöhtem Alpha-Fetoprotein
Wahrscheinlichkeit für das Gesamtüberleben
In der Phase-III-Studie REACH [4] wurde geprüft, ob Patienten mit einem fortgeschrittenen HCC nach Sorafenib-Versagen in der Zweitlinie von Ramucirumab plus beste supportive Behandlung (Best Supportive Care, BSC) profitieren können. In der Studie erhielten insgesamt 560 Patienten mit HCC 1 : 1 randomisiert Ramucirumab plus BSC oder Placebo plus BSC. Das primäre Studienziel, die Verlängerung des Gesamtüberlebens unter Ramucirumab, wurde nicht erreicht. In einer präspezifizierten Subgruppenanalyse fiel jedoch eine Patientengruppe besonders auf: Die Studienteilnehmer mit einem Alpha-Fetoprotein (AFP)Spiegel von ≥400 ng/ml wiesen eine Überlebensverlängerung von median 4,2 auf 7,8 Monate auf (HR: 0,67; 95%-KI: 0,51 – 0,89; p = 0,0059). Für Patienten mit einem AFP-Wert <400 ng/ml wurde dagegen kein Unterschied zwischen den Behandlungsarmen gefunden (Ramucirumab 10,1 vs. Placebo 11,8 Monate; HR: 1,09; 95%-KI: 0,84 – 1,44; p = 0,5059). Dieses Ergebnis wurde in der Folgestudie REACH-2 [5] näher untersucht. Dafür wurden nur HCC-Patienten mit einem AFP ≥400 ng/ml eingeschlossen, deren Erkrankung nach oder während einer Erstlinientherapie mit Sorafenib fortgeschritten war. Die 292 Teilnehmer erhielten 2 : 1 randomisiert in einem zweiwöchigen Zyklus entweder eine Therapie mit Ramucirumab plus BSC oder Placebo plus BSC. Unter Ramucirumab wurde im Vergleich zum Kontrollarm eine signifikante Verlängerung des medianen Gesamtüberlebens (8,51 vs. 7,29 Monate;
1,0 HR = 0,710 95%-KI: 0,531 – 0,949 p = 0,0199
0,8 0,6 0,4 0,2 0
Cyramza® + BSC Placebo + BSC 0 3 6 9 12 15 18 21 24 27
Zeit seit Randomisierung (Monate)
Anzahl unter Risiko: Cyramza® + BSC 197 172 121 87 56 37 26 14 4 0 Placebo + BSC
95 76 50 36 19 12 4 1 0 0
Abbildung 1: Kaplan-Meier-Kurve des Gesamtüberlebens für Ramucirumab (Cyramza®) versus Placebo in der REACH-2-Studie [5].
HR: 0,71; 95%-KI: 0,53 – 0,95; p = 0,0199; Abb. 1) und eine signifikante Verlängerung des medianen progressionsfreien Überlebens (2,83 vs. 1,61 Monate; HR: 0,45; 95%-KI: 0,34 – 0,60; p < 0,0001) erzielt. Die Krankheitskontrollrate (komplette und partielle Remissionen sowie stabile Erkrankung) war im Ramucirumab-Arm mit 59,9 % versus 38,9 % signifikant höher als im Placebo-Arm (p = 0,0006) [5]. In einer Metaanalyse [6] wurden die Patienten beider REACH-Studien mit AFP ≥400 ng/ml gemeinsam ausgewertet. Es zeigte sich eine signifikante Verlängerung des Gesamtüberlebens um mehr als 3 Monate unter Ramucirumab im Vergleich zum Placebo-Arm. Das Mortalitätsrisiko wurde um 31 % gesenkt. 5,4 % der Patienten sprachen auf die Therapie mit Ramucirumab an gegenüber 0,9 % unter Placebo und 50,9 % (versus 36,3 %) der Patienten zeigten zudem eine Stabilisierung der Erkrankung [6]. Ramucirumab zeigte auch in den beiden REACH-Studien bei Patienten mit fortgeschrittenem HCC die bereits bekannte, gute Verträg-
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
lichkeit. Diese wichtigen Ergebnisse führten zur vierten in der EU zugelassenen Tumorindikation für Ramucirumab. Wobei hervorzuheben ist, dass sich die Zulassung beim HCC erstmals auf einen prädiktiven Biomarker (AFP ≥400 ng/ ml) stützt, der durch die REACHStudien für die Therapie mit Ramucirumab etabliert wurde. Elisabeth Wilhelmi, München
Literatur 1 Alison MR Stem Cell Rev 2005;1:253260 2 Robert Koch-Institut. Krebs in Deutschland für 2013/2014. Publikation 2017 3 Fachinformation Cyramza®; Stand: August 2019 4 Zhu AX et al. Lancet Oncol 2015;16:859870 5 Zhu AX et al. J Clin Oncol 2018;36:Abstr. 4003 6 Zhu AX et al. J Clin Oncol 2019;20:282296 © VERLAG PERFUSION GMBH
Anzeige
Das Besondere, Einzigartige, Ungewöhnliche erleben: magische Momente im alpinen Lifestyle Hideaway Hotel Goldener Berg in Oberlech am Arlberg
Ein Winter voller Golden Moments
Die Weiten des Arlbergs mit ihrer glitzernden Schneelandschaft liegen unter mir. Mein Herzschlag wird übertönt vom Rauschen der Rotorblätter. Schon bald ziehe ich die ersten Schwünge in den Pulver am Mehlsack. Dieses einzigartige Erlebnis „Heli Flug & Pulverabfahrt“ ist nur einer der speziellen Golden Moments für die Gäste des Hotels Goldener Berg. Wenn Sie Glücksmomente lieben, hochwertiges Design mit stylischen Akzenten schätzen und noch dazu an einem ganz besonderen Ort Ihren Urlaub verbringen möchten, dann ist das Hotel Goldener Berg genau das Richtige für Sie. Hier verbinden Gäste die wohlverdiente Flucht aus dem Alltag mit „Quality-Time“ vom Feinsten. Stilvolle Winterferien abseits des Mainstreams, mit absoluter Top-Lage und Ski-in & Ski-out-Vergnügen de luxe – ein alpines Lebensgefühl mit dem weltbekannten „Arlberg-Flair“. Die gesunde Mischung macht‘s
Auch die eigene Gesundheit findet im himmlischen Oberlech Beachtung. „Gezielte Bewegung, gesunde Ernährung und ausreichend Schlaf sind im Urlaub die drei wichtigsten Stützen, um die leeren Batterien wieder aufzuladen“, bringt es Gastgeberin Daniela Pfefferkorn auf den Punkt. Und das funktioniert hier oben auf 1750 Metern Höhe, weit weg von Erdstrahlung, Elektrosmog und Stress besonders gut. Für Bewegung und frische Luft sorgen die einzigartige Bergwelt und die Piste direkt vorm Haus. Danach darf in einem der vier Gourmetrestaurants geschmaust werden. Das Küchenteam setzt auf Regionalität und bringt täglich kreative und biologische Gerichte auf den Tisch. Auch vegetarische und vegane Speisen werden angeboten. „Wir wollen unseren Gästen ein neues Lebensgefühl bieten“, so Daniela Pfefferkorn. Dazu zählt auch die Ernährung, bei der das sogenannte Glyx-Prinzip ein Baustein im Angebot ist. Das Credo: eine Lebensweise schaffen, die gesund ist, schmeckt, glücklich macht – und zwar ein Leben lang, ohne den gefürchteten Jojo-Effekt. Dieses Konzept hat die Küche des Hotels aufgenommen und überlässt den speisenden Gästen die Wahl. Your perfect Golden Moments Noch einzigartiger wird der Winterurlaub am Arlberg mit den buchbaren „Golden Moments“. Es warten außergewöhnliche Erlebnisse auf Sie, wie Helikopter-Rundflüge, Champagner & Austern auf der privaten Terrasse oder Skitouren mit Privateguide. Gemeinsam besondere Momente erleben und unvergessliche Erinnerungen schreiben. Entdecken Sie Ihre perfect Golden Moments für ein Urlaubsglück, das Sie sich verdient haben und buchen Sie diese gleich mit! Kontakt: Hotel Goldener Berg, Oberlech 117, A-6764 Oberlech am Arlberg, Tel.: +43 (0)5583 22050, happy@goldenerberg.at | www.goldenerberg.at
WISSENSWERTES
Follikuläres Lymphom: Kombination aus Lenalidomid und Rituximab in der EU zugelassen Die Europäische Kommission (EC) hat Lenalidomid (Revlimid®) in einer neuen Indikation zugelassen: Zur Behandlung erwachsener Patienten mit vorbehandeltem follikulärem Lymphom (FL) des Schweregrads 1 – 3a steht Lenalidomid nun in Kombination mit dem Anti-CD20-Antikörper Rituximab zur Verfügung. Die als R² bezeichnete Kombination ist das erste chemotherapiefreie Kombinationsregime, das von der EC für Patienten mit FL zugelassen wurde. Komplementärer Wirkmechanismus
Das follikuläre Lymphom ist ein Subtyp des indolenten Non-Hodgkin-Lymphoms (NHL), bei dem die Funktion des Immunsystems beeinträchtigt ist. Infolge dieser Dysfunktion kann das Immunsystem Krebszellen nicht erkennen oder nicht angreifen. Standardbehandlung ist die Chemotherapie. Dennoch kommt es bei den meisten Patienten im Krankheitsverlauf zu einem Rezidiv oder sie werden refraktär. Da es sich beim FL um eine unheilbare Erkrankung handelt, besteht ein hoher Bedarf an Medikamenten, die auf Basis eines neuen Wirkmechanismus und eines verträglicheren Sicherheitsprofils dazu beitragen können, das progressionsfreie Überleben zu verbessern. Mit der Kombination aus Lenalidomid und Rituximab ist nun das
21
erste Regime verfügbar, das keine Chemotherapie enthält. Die Wirkweise der R2-Therapie beruht auf komplementären Mechanismen, die das Immunsystem des Patienten bei der Erkennung und Bekämpfung der Krebszellen unterstützen: • Der monoklonale Antikörper Rituximab richtet sich gegen das CD20-Antigen auf der Oberfläche von pre-B- und reifen B-Lymphozyten. Bei seiner Bindung an CD20 löst Rituximab die B-Zell-Lyse aus. • Lenalidomid ist eine immunmodulierende Substanz, die die Anzahl und Aktivierung von TZellen und natürlichen Killerzellen (NK-Zellen) fördert, was wiederum zur Lyse der Tumorzellen führt.
Studie war das progressionsfreie Überleben (PFS), definiert als Zeitraum von der Randomisierung bis zur ersten Beobachtung der Krankheitsprogression oder bis zum Tod (alle Ursachen). Die mit R2 behandelten Patienten zeigten ein signifikant längeres PFS als die Patienten der Placebogruppe (HR: 0,46; 95%-KI: 0,34 – 0,62; p < 001): Das PSF betrug unter der R2-Therapie im Mittel 39,4 Monate und im Kontrollarm 14,1 Monate. B. S.
Wegweisende Ergebnisse der AUGMENT-Studie
Ob Ernährung, Freizeit, Sozialleben oder Kinderwunsch – Urtikaria schränkt Betroffene in nahezu jedem Lebensbereich ein. Dennoch befinden sich fast 2 Drittel der Patienten (60,3 %) nicht in ärztlicher Versorgung – so das Ergebnis der DERMLINE-Umfrage mit 1.037 Urtikaria-Patienten, die auf dem EADV-Kongress in Madrid präsentiert wurde**. Jeder zweite Patient (52 %) ist mit seiner derzeitigen Therapie unzufrieden – der am häufigsten genannte Grund: Die Therapie hilft nicht. Jeder Fünfte gibt sogar an, er erhalte aus Kostengründen nicht die bestmögliche Therapie. Viele Patienten wünschen sich außerdem mehr Zeit für das Arzt-Patienten-Gespräch. Bei einem Großteil der Patienten besteht also das Potenzial, die Versorgung deutlich zu verbessern. Damit hat sich die Situation der
Die Zulassungsempfehlung für R2 basiert primär auf den Ergebnissen der randomisierten, doppelblinden Phase-III-Studie AUGMENT*, in der die Wirksamkeit und Sicherheit der R²-Kombinationstherapie im Vergleich zu Rituximab plus Placebo bei 295 Patienten mit vorbehandeltem FL Grad 1, 2 oder 3a untersucht wurden. Die Patienten hatten zuvor bereits mindestens eine systemische Therapie und 2 Zyklen Rituximab erhalten und mussten nach der systemischen Therapie rezidiviert, refraktär oder progredient sein, waren jedoch nicht Rituximab-refraktär. Primärer Endpunkt der AUGMENT* Leonard JP, Trneny M, Izutsu K et al. AUGMENT: a phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol 2019; 10;37:1188-1199
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Urtikaria-Patienten weiterhin unterversorgt:
Leitlinie empfiehlt bei nicht ausreichender Kontrolle Omalizumab
** Wagner N et al. EADV 2019. Abstract P1958 © VERLAG PERFUSION GMBH
WISSENSWERTES
22
Patienten in den letzten 5 Jahren kaum verändert – in der Vorgängerstudie ATTENTUS waren fast ebenso viele Urtikaria-Patienten nicht ärztlich versorgt oder unzufrieden mit ihrer Behandlung. Fehlende Kontrolle, belastende Symptome, Einschränkungen im Alltag
Die DERMLINE-Umfrage zeigte, dass die Urtikaria bei 80 % der Betroffenen gemäß Urtikariakontrolltest (UCT) weiterhin aktiv war: 9 von 10 Patienten litten unter Jucken (91 %), fast ebenso viele unter Hautnesseln (86 %), 43 % der Patienten klagten über entstellende und schmerzhafte Ödeme. Betroffen waren alle Körperregionen, bei einem Großteil der Patienten auch gut sichtbare Bereiche wie Gesicht und Hände, 15 % bzw. 12 % der Betroffenen berichteten von besonders schmerzhaften und belastenden Hautnesseln bzw. Ödemen im Genitalbereich. Bei etwa einem Viertel (28 %) traten die Beschwerden spontan auf, 43 % gaben an, dass die Symptome sowohl spontan auftraten als auch durch bestimmte Auslöser induziert wurden – darunter auch Stress in Alltag und Beruf. Jede fünfte Betroffene verschiebt sogar einen Kinderwunsch
Die schwerwiegenden und oftmals unberechenbar auftretenden Symptome beeinträchtigen das Leben der Betroffenen in nahezu allen Bereichen: 52 % meiden stressige Situationen, 42 % ändern ihre Ernährung, jeder Dritte (33 %) reduziert sportliche Aktivitäten oder
Omalizumab Omalizumab (Xolair®) ist seit 2014 zur Behandlung der therapierefraktären chronischen spontanen Urtikaria zugelassen, seit Dezember 2018 zur Selbstapplikation. Der humanisierte monoklonale Antikörper bindet an das Urtikaria-treibende IgE und reduziert die Menge an freiem IgE sowie die dadurch bedingten Effekte auf die zellulären Aktivierungsmechanismen. Damit unterdrückt es die durch Histamin induzierten Hautreaktionen und tieferen Schwellungen.
verzichtet auf Partys und Feiern (29 %), etwa ein Viertel (26 %) versucht die entstellenden Hautveränderungen unter langer Kleidung zu verbergen – auch bei Sonne oder Hitze. Jeder Fünfte meidet den körperlichen Kontakt zu Freunden oder trifft sich seltener mit ihnen. Besonders bedauerlich ist, dass fast jede fünfte betroffene Frau (18 %) wegen der Urtikaria sogar ihren Kinderwunsch verschiebt. Es erstaunt daher nicht, dass sich mehr als 2 Drittel (68 %) der Befragten aufgrund der Erkrankung niedergeschlagen fühlen. Fast ebenso viele (56 %) gaben an, weniger Freude an eigentlich spaßigen Unternehmungen zu haben. Einzig positiv: 9 von 10 Betroffenen gaben an, dass ihr Partner sie und ihre Erkrankung verstehe. Therapie rechtzeitig intensivieren, leitliniengerecht behandeln
Um eine jahrelange Leidenszeit zu verkürzen und den Patienten Frust und Resignation zu ersparen, sollte die Therapie rechtzeitig intensiviert und als Behandlungsziel die Beschwerdefreiheit angestrebt werden. Für die Behandlung der chronischen spontanen Urtikaria sieht die aktuelle Leitlinie* ein 4-Stufen-Schema vor:
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
1. Therapie der ersten Wahl sind H1-Antihistaminika der zweiten Generation. 2 Führt die kontinuierliche Einnahme über 2 – 4 Wochen zu keiner ausreichenden Kontrolle der Beschwerden, empfiehlt die Leitlinie eine Höherdosierung bis auf das Vierfache der Standarddosierung. 3. Bei nicht ausreichender Kontrolle nach 2 – 4 Wochen durch hochdosierte Antihistaminika ist eine zusätzliche Therapie mit Omalizumab anzuraten. Omalizumab (Xolair®) ist ein rekombinanter humanisierter Anti-IgE-Antikörper, der in der empfohlenen Dosis von 300 mg alle 4 Wochen als subkutane Injektion verabreicht wird. Die meisten Patienten sprechen bereits vor der zweiten Behandlung und meist vollständig auf diese Therapie an. In einer Meta-Analyse mit mehr als 60 Studien erreichten 72,2 % der mit Omalizumab behandelten Patienten Symptomfreiheit. 4. Tritt nach einer 6-monatigen Omalizumab-Therapie kein Therapieerfolg eintritt, wird eine Off-Label-Behandlung mit Ciclosporin empfohlen. F. S. * Zuberbier T et al. Allergy 2018;73:13931414
© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
I
n der NOVA-Studie bei Patientinnen mit rezidiviertem, platinsensitivem high-grade serösem Ovarialkarzinom verlängerte die Erhaltungstherapie mit dem PARPInhibitor Niraparib (Zejula®) das mittlere progressionsfreie Überleben (mPFS) unabhängig vom BRCA-Status der Patientinnen: Die Frauen mit BRCA-Keimbahnmutation erzielten unter Niraparib ein mPFS von 21,0 Monaten gegenüber 5,5 Monaten in der Placebo-Gruppe (HR: 0,27; p < 0,001), bei den Frauen ohne BRCA-Keimbahnmutation betrug das mPFS 9,3 Monate unter Niraparib gegenüber 3,9 Monaten unter Placebo (HR: 0,45; p < 0,001). Während der Erhaltungstherapie ergaben sich hinsichtlich der Lebensqualität keine Unterschiede zwischen den mit Niraparib behandelten Patientinnen und den Frauen, die Placebo erhalten hatten [1]. Da die Langzeitverträglichkeit für die Patientenakzeptanz bei einer länger währenden Erhaltungstherapie essenziell ist, wurden in der NOVA-Studie auch Daten zu unerwünschten Ereignissen unter der Therapie mit Niraparib erhoben, wobei teilweise ein Therapiezeitraum von mehr als 13 Monaten erfasst wurde. Die Ergebnisse wurden auf dem ESGO-Kongress 2019 in Athen präsentiert [2]. Sicher auch über lange Zeiträume
Am häufigsten traten Nebenwirkungen im ersten Therapiezyklus auf. Danach sank die Inzidenz und blieb dann auf einem niedrigen Niveau. Dosisreduktionen aufgrund von Nebenwirkungen waren im ersten Therapiemonat am häufigsten (34 %), während im 5. Monat nur noch bei 7 % der Frauen eine Dosisreduktion notwendig war.
23
Rezidiviertes Ovarialkarzinom: Niraparib ist eine gut verträgliche Option zur Langzeit-Erhaltungstherapie
Während der ersten 6 Monate sank die Inzidenz hämatologischer und symptomatischer Nebenwirkungen wie Übelkeit stetig und blieb dann auf diesem niedrigen Niveau. Blutbildveränderungen gehörten zu den häufigsten Nebenwirkungen unter Niraparib. Thrombozytopenien ≥ Grad 3 traten im ersten Therapiemonat bei 28 % der Frauen auf, gingen aber in Monat 2 auf 10 % und in Monat 3 auf 5 % zurück. Von Vorteil für das Nebenwirkungsmanagement ist, dass sich die Dosis von Niraparib individuell modifizieren lässt: Falls erforderlich, kann sie von 1 × 300 mg/d auf 200 mg/d und ggf. auf 100 mg/d reduziert werden [3]. Aufgrund der nur 1 × täglichen Gabe kann die Patientin die Einnahme zudem an ihren persönli-
chen Alltagsablauf anpassen, was dazu beiträgt, Schlaflosigkeit und Übelkeit – bekannte Symptome bei Patientinnen mit fortgeschrittenem Eierstockkrebs – besser zu kontrollieren. Dies erhöht auch die Compliance und die Therapieadhärenz während der längerfristigen oralen Erhaltungstherapie mit dem PARP-Inhibitor. Elisabeth Wilhelmi, München Literatur 1 Mirza MR et al. N Engl J Med 2016;375: 2154-2164 2 Mirza MR et al. Long-term safety assessment of niraparib in patients with recurrent ovarian cancer: results from the ENGOT-OV16/NOVA trial. Presented at the ESGO Annual Meeting 2019 in Athens, Greece 3 Fachinformation Zejula® 100 mg Hart kapseln; Stand: Juni 2019
Niraparib Niraparib (Zejula®) ist ein oraler, einmal täglich einzunehmender Poly(ADP-Ribose)-Polymerase (PARP) 1/2-Inhibitor. In der EU ist Niraparib zugelassen als Monotherapie zur Erhaltungstherapie bei erwachsenen Patientinnen mit einem Rezidiv eines Platin-sensiblen, gering differenzierten serösen Karzinoms der Ovarien oder der Tuben sowie bei Patientinnen mit einer primären Peritonealkarzinose, die sich nach einer Platin-basierten Chemotherapie in Remission (komplett oder partiell) befinden.
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
24
D
ie Auswahl der passenden Therapieoption bei schubförmig remittierender Multipler Sklerose (RRMS) erfolgt auf Basis einer Reihe praxisrelevanter Kriterien. So spielen neben dem zu erwartenden Therapieerfolg, den Nebenwirkungen und Abbruchraten der individuelle Krankheitsverlauf sowie die Lebensplanung des Patienten in der Therapieentscheidung eine wichtige Rolle. Ergänzend zu den Ergebnissen klinischer Studien liefert heute eine steigende Zahl von Daten direkt aus der Versorgungspraxis, sogenannte RealWorld-Daten, belastbare Informationen. Eine aktuelle Auswertung von Behandlungsdaten aus dem dänischen MS-Register zeigt einen Vergleich von Therapieergebnissen und Abbruchraten zwischen Dimethylfumarat (Tecfidera®, DMF) und Teriflunomid. Demnach blieb die jährliche Schubrate unter DMF um 42 % signifikant niedriger als unter Teriflunomid [1]. Vor der Therapie die zu erwartenden Ergebnisse abschätzen
Bei fehlender oder unzureichender Therapie führt die RRMS langfristig fast immer zu einer Zunahme der Behinderung. Ziel der Behandlung ist es daher, die Langzeitprognose bezogen auf die Behinderung maßgeblich zu verbessern [2]. Im Rahmen der Auswahl der individuell passenden Therapieoption gilt es daher, mehrere praktische Fragen zu bedenken. Besonders wichtig für Arzt und Patient ist dabei eine Einschätzung der kurz-, mittel- und langfristig zu erwartenden Therapieergebnisse. Auch unerwünschte Ereignisse spielen in Bezug auf die Patientensicherheit und -adhärenz eine wichtige Rolle.
Multiple Sklerose: Real-World-Daten können bei der Therapieentscheidung unterstützen Hier gilt es, mögliche Nebenwirkungen und deren Schweregrad in der Therapieentscheidung zu berücksichtigen. Da die MS-Behandlung immer eine langfristige sein sollte, ist auch die Wahrscheinlichkeit eines Therapieabbruchs wegen unzureichender Wirkung bzw. nicht tolerierter Nebenwirkungen ein wichtiges Kriterium. Nicht zuletzt sollte gemeinsam mit dem Patienten entschieden werden, welches Präparat bei seinem individuellen Krankheitsverlauf, seinen persönlichen Bedürfnissen und seiner Lebensplanung am besten geeignet ist. Dänisches Register ermöglicht Vergleich zwischen DMF und Teriflunomid
Neben den Ergebnissen klinischer randomisierter Studien kann der Arzt bei der Therapieplanung heute auf eine Reihe von Daten direkt aus der ärztlichen Praxis – RealWorld-Daten – zurückgreifen. Die gemeinsame Betrachtung der verfügbaren Daten ermöglicht es, zahlreiche praxisrelevante Fragen zu beantworten. So lassen die Ergebnisse einer aktuellen Auswertung des dänischen MS-Registers wertvolle Rückschlüsse auf die Therapieergebnisse im Behandlungsalltag unter DMF im Vergleich zu Teriflunomid zu [1].
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Anders als in Deutschland werden alle dänischen Patienten mit MS (inkl. klinisch isoliertem Syndrom, KIS) landesweit in insgesamt 14 spezialisierten MS-Kliniken behandelt. Die Patientendaten werden dabei vom jeweiligen behandelnden Neurologen zu Therapiebeginn sowie nach 3 bzw. 6 Monaten und danach alle 6 Monate in ein großes Register eingepflegt. Analysen dieses Registers spiegeln daher die gesamte dänische MS-Population wider [1]. In die aktuelle Auswertung flossen Daten von 2.236 erwachsenen Patienten ein, die zwischen Oktober 2013 und Mai 2018 DMF oder Teriflunomid erhalten hatten. Ausgeschlossen waren u.a. Patienten mit vorheriger Behandlung mit hochwirksamen krankheitsmodifizierenden Therapien (Disease Modifying Therapy, DMT), mehr als 2 unterschiedlichen DMTs in der Vorgeschichte oder einer Behandlungsdauer von über 8 Jahren. Als primäre Endpunkte wurden die jährliche Schubrate (ARR) sowie das Verhältnis der Schubraten unter DMF vs. Teriflunomid definiert. Sekundäre Endpunkte waren die Zeit bis zum ersten Schub, die Zeit bis zur Behinderungsprogression (bestätigt nach 6 Monaten), die kumulative Inzidenz ursachenspezifischer Therapieabbrüche bzw. Therapiewechsel aufgrund von Krankheitsaktivität oder uner© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Dimethylfumarat (DMF) Dimethylfumarat (Tecfidera® 120 mg/240 mg magensaftresistente Hartkapseln) ist zugelassen zur oralen Behandlung von erwachsenen Patienten mit schubförmig remittierender Multipler Sklerose. Auch wenn der exakte Wirkmechanismus derzeit nicht bekannt ist, ist Dimethylfumarat bisher der einzige in klinischen Studien untersuchte Wirkstoff für die Behandlung von MS, für den angenommen wird, dass er den Nrf2-Signalweg aktiviert. Der Nrf2-Signalweg ist ein körpereigener Abwehrmechanismus, der Zellen vor potenziell schädlichen Einflüssen wie Entzündungen und oxidativem Stress schützt, die unter anderem bei der MS-Pathophysiologie eine Rolle spielen [4].
wünschten Ereignissen. Die Auswertung erfolgte auf Basis einer Gewichtung der Therapieeffekte (Inverse Probability of Treatment Weights; IPTW) sowie der Propensity-Score-Methode [1]. Die Ergebnisse zeigen eine über 4 Jahre um 42 % signifikant niedrigere jährliche Schubrate unter DMF verglichen mit Teriflunomid (0,09 vs. 0,16; p < 0,001). Zudem blieben etwa 76 % der Patienten unter DMF über 4 Jahre schubfrei, verglichen mit ca. 65 % unter Teriflunomid. Die Abbruchraten aufgrund unerwünschter Ereignisse betrugen 18,0 % unter DMF und 18,5 % unter Teriflunomid – und waren damit nach 4 Jahren vergleichbar hoch. Dagegen brachen unter Teriflunomid verglichen mit DMF etwa doppelt so viele Patienten die Therapie aufgrund mangelnder Wirksamkeit ab (22,4 % vs. 10,7 %). Bei DMF führten vor allem folgende Nebenwirkungen zu Therapieabbrüchen: gastrointestinale Ereignisse, Flush-Symptomatik, Reduktion der Lymphozytenzahl. Bei Teriflunomid waren es ein Anstieg der Leberwerte, gastrointestinale Ereignisse und Haarausdünnung [1]. Als Limitation der Studie führten die Autoren an, dass aus den Daten keine Informationen zur Fa-
milienplanung bei den dänischen MS-Patientinnen hervorgehen. In dänischen Leitlinien wird empfohlen, Frauen, die im nächsten Jahr eine Schwangerschaft planen, mit DMF anstelle von Teriflunomid zu behandeln. Dazu passe auch, dass Frauen häufiger mit DMF behandelt wurden als Männer. Es sei zudem nicht auszuschließen, dass Frauen, die eine Schwangerschaft in Betracht ziehen eine geringere Krankheitsaktivität aufweisen. Dies könnte einen positiven Einfluss auf die Therapieergebnisse von DMF mit sich bringen. Die Autoren gehen allerdings davon aus, dass Frauen mit objektiven Anzeichen hoher Krankheitsaktivität ihre Therapie eher mit einem hocheffektiven DMT beginnen und nicht mit Teriflunomid. Eine weitere Limitation der Auswertung war der Ausschluss von Patienten, die zwischen Teriflunomid und DMF wechselten [1]. Moderne statistische Verfahren schaffen zusätzliche Evidenz
MS-Patienten im Wartezimmer des Neurologen unterscheiden sich von Studienpatienten [3]. So ist der durchschnittliche MS-Patient oft älter und weist eine längere
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
25
Krankheitsdauer auf als Patienten aus Phase-III-Studien. Zudem sind körperliche Funktionsbeeinträchtigungen häufig deutlicher ausgeprägt als bei Patienten im Rahmen klinischer Studien. Real-WorldDaten bieten hier eine wertvolle Ergänzung mit Daten direkt aus dem Versorgungsalltag. Die so gewonnenen Ergebnisse erreichen zwar nicht den Evidenzgrad der RCTs, beschreiben aber den Nutzen eines Arzneimittels in der klinischen Routine. Um die Vergleichbarkeit der z.B. in Beobachtungsstudien und Behandlungsregistern beobachteten Therapieeffekte in diesen (meist größeren) gemischten Patientenkollektiven herzustellen, sind statistische Verfahren unverzichtbar. Mithilfe beispielsweise des Propensity Score Matching (PSM), einer Gewichtung der Therapieeffekte (Inverse Probability of Treatment Weights, IPTW) sowie rigoroser Sensitivitätsanalysen können die Therapieeffekte in den untersuchten Patientenkollektiven angeglichen und Therapieeffekte eingeschätzt werden. Fabian Sandner, Nürnberg
Literatur 1 Buron M et al. Neurology 2019; 92:e1811-e1820 2 Gold R et al. Nervenheilkunde 2015;34: 915-922 3 Pellegrini F et al. ECTRIMS/ACTRIMS 2017; P352 4 Fachinformation Tecfidera®; Stand: Februar 2018 © VERLAG PERFUSION GMBH
26
NEUE UND BEWÄHRTE ARZNEIMITTEL
Colitis ulcerosa: Neue hochdosierte Mesalazin-Formulierung optimiert Basistherapie
T
herapie-Goldstandard bei leichten bis mittelschweren Krankheitsschüben der Colitis ulcerosa (CU) ist Mesalazin (5-Aminosalicylsäure, 5-ASA), das eine antientzündliche Wirkung auf die Darmschleimhautzellen ausübt. In den meisten Fällen lässt sich damit eine Remission erreichen und lange aufrechterhalten. Voraussetzung dafür ist, dass die Erhaltungstherapie langfristig fortgesetzt wird und die Patienten die verschriebene Medikation in der vereinbarten Dosierung konsequent einnehmen. In der Behandlungsrealität neigen jedoch bis zu 60 % der Patienten dazu, ihre Medikamente nicht mehr wie verschrieben zu nehmen, wenn die CU-Beschwerden nachlassen [1]. Damit steigt die Wahrscheinlichkeit für erneute Krankheitsschübe – nicht therapieadhärente* Patienten haben ein mehr als fünffach höheres Rezidivrisiko im Vergleich zu adhärenten Patienten [2]. Mesalazin-Präparate sollten bevorzugt als tägliche Einmalgabe verabreicht werden [3], weil sich dies positiv auf die Therapietreue und damit den Behandlungserfolg auswirkt: In Studien zur Therapie mit Mesalazin war die 1 × tägliche Einnahme der 2 × täglichen An-
* Nicht adhärent war definiert als die Einnahme von weniger als 80 % der verschriebenen Medikamente.
wendung bei CU-Patienten mit aktiver Erkrankung und in Remission überlegen [4, 5]. Zu einer besseren Adhärenz kann auch die Verordnung des Medikaments in einer vom Patienten präferierten Darreichungsform beitragen. So ergab eine kürzlich veröffentlichte Studie bei 380 Patienten mit leichter bis mittelschwerer CU, dass 67 % der Befragten Tabletten bevorzugten, 29 % würden sich für Granulat und weitere 4 % für andere Darreichungsformen entscheiden [6]. Beide Voraussetzungen für eine optimale Adhärenz erfüllt eine innovative Mesalazin-Formulierung, die mit 1600 mg Wirkstoff die bislang höchste Mesalazin-Einzeldosis in einer geschmacksneutralen Tablette enthält, was die 1 × tägliche Gabe erleichtert. Asacol® 1600 mg ist seit 1. Oktober 2019 zur Behandlung akuter Schübe mit leichtem bis mittelschwerem Krankheitsverlauf sowie zur Langzeitbehandlung zur Vermeidung eines Rezidivs der leichten bis mittelschweren CU verfügbar [7].
COlonic Release und basiert auf einer innovativen Beschichtung (Abb. 1). Der aus 1600 mg Mesalazin bestehende Tablettenkern ist von einem mehrschichtigen Überzugssystem ummantelt, das so konzipiert ist, dass die Freisetzung von Mesalazin verzögert wird, bis die Tablette den Dickdarm erreicht hat. Dort wird über verschiedene Trigger ein dualer Freisetzungsmechanismus in Gang gesetzt: Der erste wirkt mithilfe von Eudragit S chemisch ab einem pH-Wert von 7 im terminalen Ileum und im Kolon, der zweite nutzt Enzyme von Darmbakterien, die die in der äußeren Tablettenschicht enthaltene Stärke zersetzen. Die Wirkstofffreisetzung beginnt im terminalen Ileum und setzt sich im Caecum/Colon ascendens bis zum Colon sigmoideum fort (Abb. 2). Beschleunigt wird sie zusätzlich durch eine alkalische Pufferschicht [8].
Dualer Freisetzungsmechanismus
Die randomisierte, aktiv kontrollierte, verblindete Studie zur Zulassung von Asacol® 1600 mg ist mit 817 Patienten in Europa und Kanada die größte jemals durchgeführte Studie zur Remissionsinduktion mit Mesalazin bei leichter
Um eine gezielte Wirkstofffreisetzung im gesamten Kolon zu gewährleisten, nutzt die neue Tablette die OPTICORE™-Technologie. OPTICORE steht für OPTImized
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Sehr gutes Wirkungs- und Nebenwirkungsprofil
© VERLAG PERFUSION GMBH
27
NEUE UND BEWÄHRTE ARZNEIMITTEL
Intakte Tablette im distalen Ileum
Initiale Mesalazin-Freisetzung im Caecum/Colon ascendens
Weitere Freisetzung im Colon ascendens und transversum
Weitere Freisetzung im Colon sigmoideum
Abbildung 1: Bei der Asacol® 1600 mg-Tablette wird der MesalazinKern von einer innovativen, mehrschichtigen Ummantelung umhüllt, die über einen dualen Freisetzungsmechanismus eine gezielte Wirkstoffabgabe im gesamten Kolon (einschließlich Rektum) ermöglicht (© Tillots Pharma).
Abbildung 2: Die Wirkstoff-Freisetzung beginnt im terminalen Ileum und setzt sich im Caecum/Colon ascendens bis zum Colon sigmoideum fort (szintigraphische Daten nach Nüchtern-Applikation von Smmarkierten Tabletten; © Tillots Pharma).
bis mittelschwerer CU [9]. Außerdem handelte es sich um die erste und bisher einzige Studie, die individuelle Mesalazin-Dosierungen anhand von Dosis-Eskalation und Dosis-Deeskalation untersucht hat. Neben der klinischen Dokumentation wurden in der Studie auch endoskopische Befunde erhoben und zentral über eine anonymisierte Videobeurteilung ausgewertet [9]. Nach achtwöchiger Behandlung mit 1 × täglich 2 Tabletten Asacol® 1600 mg (3,2 g Mesalazin/Tag) erreichten 22 % der Patienten sowohl eine klinische als auch eine endoskopische Remission. Bei primären Non-Respondern in der Schubphase wurde nach Woche 8 die Ausgangsdosis von 3,2 g Mesalazin/Tag auf eine Tagesdosis von 4,8 g Mesalazin erhöht (3 Tabletten Asacol® 1600 mg als tägliche Einmalgabe). Nach dieser einfachen Dosis-Eskalation erreichten 75 % der primären Non-Responder
bis Woche 16 ebenfalls ein klinisches Ansprechen auf Mesalazin. Die Dosis-Eskalation war möglich, ohne dass die Nebenwirkungsrate anstieg [9]. Daten aus der Open-Label-Extensionsphase zeigen, dass die Deeskalation und Fortsetzung der Behandlung mit 1 × täglich 1 Tablette Asacol® 1600 mg (1,6 g Mesalazin/ Tag) wirksam in der Aufrechterhaltung der klinischen Remission war: Nach 38 Therapiewochen befanden sich 70 % der erfolgreich behandelten Patienten weiterhin in Remission. Fazit
Die tägliche Einmalgabe von Asacol® 1600 mg ist sowohl bei der Remissionsinduktion (1 × täglich 2 – 3 Tabletten) als auch bei der Erhaltungstherapie (1 × täglich 1 Tablette) wirksam und gut verträg-
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
lich. Damit lässt sich die Dosierung einfach auf die Patientenbedürfnisse abstimmen – für eine effektive, individuelle und alltagstaugliche Therapie der CU mit Mesalazin. Fabian Sandner, Nürnberg
Literatur 1 Kane SV et al. Aliment Pharmacol Ther 2010;3:1051-1058 2 Kane SV et al. Am J Med 2003;114:3943 3 Kucharzik T et al. Z Gastroenterol 2018; 56:1087-1169 4 Barth R. Ars Medici 2016;23:1075-1076 5 Dignass A et al. Clin Gastroenterol Hepatol 2009;7:762-769 6 MacKenzie-Smith L et al. Inflamm Intest Dis 2018;3:43-51 1600 mg; 7 Fachinformation Asacol® Stand: Juni 2019 8 Varum F et al. American Association of Pharmaceutical Scientists (AAPS) Annual Meeting, 13.–17. Nov. 2016, Denver, USA; Poster 02W0200 9 D’Haens GR et al. Aliment Pharmacol Ther 2017;46:292-302 © VERLAG PERFUSION GMBH
NEUE UND BEWÄHRTE ARZNEIMITTEL
28
Z
um Zeitpunkt der letzten Aktualisierung der S3-Leitlinie Colitis ulcerosa der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten im Mai 2018 war Tofacitinib noch nicht in Europa zugelassen. Da sich wichtige Neuerungen in der Diagnostik und Therapie der Colitis ulcerosa (CU) ergaben, ist im November 2019 ein Update der Leitlinie erschienen [1]. In die aktualisierte S3-Leitlinie Colitis ulcerosa hat der Januskinase-Inhibitor Tofacitinib (Xeljanz®) Einzug der gehalten. Demnach wird Tofacitinib – als zusätzliche verfügbare Therapie gleichberechtigt neben den Biologika – für CUPatienten mit steroidrefraktärem und steroidabhängigem Erkrankungsverlauf, bei nicht ausreichendem Ansprechen auf Thiopurine sowie bei primärem oder sekundärem Therapieversagen unter Tumornekrosefaktor-Inhibitoren empfohlen (Abb. 1) [1]. Durch das OCTAVE-Studienprogramm [2] liegt für Tofacitinib Evidenz zur Wirksamkeit und Sicherheit vor – mit Daten von mittlerweile bis zu 5,5 Jahren [3].
Colitis ulcerosa: JAK-Inhibitor Tofacitinib erweitert die Therapieoptionen tion sowohl bei steroidrefraktärem als auch bei steroidabhängigem Verlauf empfohlen. Als „Ergänzung im Hintergrund“ wird dazu in der Leitlinie auf die Daten der 3 OCTAVE-Studien verwiesen: Hier konnte die Wirksamkeit von Tofacitinib in den beiden 8-wöchigen Induktionsstudien Induction 1 und 2 (jeweils 10 mg Tofacitinib 2 × täglich) sowie in der über 52 Wochen laufenden Erhaltungsstudie OCTAVE Sustain (5 oder 10 mg Tofacitinib 2 × täglich) gezeigt werden (Abb. 2) [2]. Mindestens 70 % der dort eingeschlossenen Patienten hatten einen steroidrefraktären Krankheitsverlauf [6].
Gleichzeitig wiesen aber auch viele Patienten einen steroidabhängigen Verlauf auf: Beim Studieneinschluss wurden noch über 45 % der Patienten mit Steroiden behandelt. Bei der Aktualisierung der S3Leitlinie der DGVS bestand starker Konsens, dass bei Ansprechen auf die Tofacitinib-Therapie, diese weiter fortgesetzt werden sollte. Knapp 60 % der Patienten, die zu Beginn der Erhaltungsstudie nach der Induktion in Remission waren, erreichten nach 52 Wochen unter 10 mg Tofacitinib 2 × täglich eine steroidfreie Remission, die laut S3-Leitlinie ein erklärtes Therapieziel der CU ist [7].
Empfehlung bei steroidrefraktärem und bei steroidabhängigem Erkrankungsverlauf
Unter einer Steroidtherapie bei CU kann es zu einer Glukokortikoid (GC)-Abhängigkeit kommen, das heißt, nach initialem Ansprechen tritt unter der Dosisreduktion von GC ein erneuter Schub auf. Andere Patienten weisen einen steroidrefraktären Erkrankungsverlauf auf – bei ihnen kommt es trotz intensiver GC-Therapie nicht zu einer klinischen Remission [4, 5]. Tofacitinib wird nun als eine Op-
Abbildung 1: Optionen für die Therapie der Colitis ulcerosa nach Versagen der konventionellen Therapie [1].
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
29
Patienten in Remission (%)
NEUE UND BEWÄHRTE ARZNEIMITTEL
100 90 80 70 60 50 40 30 20 10 0
Differenz 10,3 Prozentpunkte p=0,007
Differenz 13,0 Prozentpunkte p<0,001
18,5 8,2 Placebo (n = 122)
16,6 3,6
Tofacitinib 10 mg (n = 476)
OCTAVE Induction 1
Placebo (n = 112)
Tofacitinib 10 mg (n = 429)
100 90 80 70 60 50 40 30 20 10 0
Differenz 29,5 Prozentpunkte p<0,001 Differenz 23,2 Prozentpunkte p<0,001 34,3
40,6
11,1 Placebo (n = 198)
OCTAVE Induction 2
Tofacitinib 5 mg (n = 198)
Tofacitinib 10 mg (n = 197)
OCTAVE Sustain
Abbildung 2: Ergebnisse der OCTAVE-Studien für den primären Endpunkt: In OCTAVE Induction 1 und 2 erreichten in Woche 8 signifikant mehr Patienten unter 2 × täglich 10 mg Tofacitinib als unter Placebo eine Remission. Patienten, die eine der Induktionsstudien über 8 Wochen abgeschlossen und klinisch angesprochen hatten, wurden für die Studie OCTAVE Sustain erneut randomisiert. Von den 593 Patienten befanden sich 179 (30,2 %) in Remission. Im Vergleich zu Placebo erreichte auch hier ein signifikant größerer Anteil an Patienten sowohl in der Behandlungsgruppe mit zweimal 2 × 5 mg als auch 2 × täglich 10 mg Tofacitinib in Woche 52 eine Remission [2].
Empfehlung bei nicht ausreichendem Ansprechen auf Thiopurine
Auch die Empfehlung, bei Patienten mit leichter bis mittelschwerer CU, die nicht ausreichend auf die Behandlung mit Thiopurinen ansprechen, Tofacitinib als eine von verschiedenen Optionen einzusetzen, wird durch Studienergebnisse untermauert: In den beiden Induktionsstudien Induction 1 und Induction 2 waren auch Patienten eingeschlossen, die auf eine vorangegangene Therapie mit Azathioprin nicht angesprochen hatten – ca. 70 % der Patienten in den OCTAVE-Studien zeigten ein Therapieversagen auf Azathioprin [2]. Empfehlung bei primärem und sekundärem Versagen auf eine TNFi-Therapie
Bei 19 – 58 % der mit TNFi behandelten Patienten kommt es zu einem primären Therapieversagen,
bei 17 – 22 % zu einem sekundären Wirkverlust, hier vor allem durch die Entwicklung von Anti-DrugAntikörpern. Bei 19 – 40 % der Patienten muss im Therapieverlauf die TNFi-Dosis erhöht werden [8, 9]. Tofacitinib wird nun auch bei primärem und sekundärem Versagen auf die Behandlung mit einem TNFi empfohlen. In beiden Situationen hat der oral zu applizierende JAK-Inhibitor seine Wirksamkeit unter Beweis gestellt: In den Studien OCTAVE Induction 1 und 2 hatten jeweils über 50 % der mit Tofacitinib behandelten Patienten eine vorangegangene TNFi-Therapie erhalten. Bei diesen schwer zu behandelnden Patienten führte die Behandlung mit Tofacitinib bei signifikant mehr Patienten zur Remission als Placebo. TNFi-naive Patienten profitierten noch etwas stärker von der Tofacitinib-Therapie als TNFi-erfahrene (Abb. 3) [2].
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Daten zur Wirksamkeit und Sicherheit über 5,5 Jahre
Um die Sicherheit und Verträglichkeit von Tofacitinib in der Langzeittherapie der mittelschweren bis schweren CU zu evaluieren, wurde im Oktober 2012 die noch laufende Verlängerungsstudie OCTAVEOpen initiiert [3]. Aktuell liegen für die Verlängerungsstudie Daten von 944 CU-Patienten mit 2.004 Patientenjahren und einem Behandlungszeitraum von bis zu 5,5 Jahren vor. Die Interimsanalyse zeigt keinen Anstieg der Inzidenzraten gegenüber früheren Auswertungen bezogen auf die unerwünschten Ereignissen von besonderem Interesse [10, 11]. Insgesamt erhielten 944 Patienten mindestens eine Dosis Tofacitinib. Über 80 % der Patienten wurden mit 10 mg Tofacitinib zweimal täglich behandelt. Die häufigsten therapiebedingten Nebenwirkungen umfassten Nasopharyngitis (20,3 %), eine Verschlechterung © VERLAG PERFUSION GMBH
NEUE UND BEWÄHRTE ARZNEIMITTEL
30
OCTAVE Induction 1
Prozent der Patienten mit Remission
60
OCTAVE Induction 2
Placebo (n=122) Tofacitinib 10 mg 2x tgl. (n=476)
Placebo (n=112) Tofacitinib 10 mg 2x tgl. (n=429)
50 40
∆=9,4
30
10
22,1
15,8
12,6
12,0
1,5
8,5
0
0 TNFi-behandelt n N
∆=12,0
25,2
∆=11,1
20
∆=13,5
1 65
TNFi-naiv 32 254
8 57
TNFi-behandelt 56 222
0 65
28 234
TNFi-naiv 4 47
43 195
Abbildung 3: Ergebnisse der Studien OCTAVE Induction 1 und 2 je nach vorheriger TNFi-Behandlung [2]. * p≤0,05 vs. Placebo [2].
der CU (19,5 %) sowie eine Erhöhung der Kreatinkinase (10,8 %). Bei den unerwünschten Ereignissen von besonderem Interesse lagen die Inzidenzraten für schwere Infektionen bei 1,6 % und für Herpes zoster bei 3,35 %, für schwere kardiovaskuläre Ereignisse (MACE) betrugen sie 0,15 % und für Malignome (ausgenommen nicht melanozytärer Hautkrebs) 0,85 % [3]. Auch Daten zu venösen Thromboembolien liegen vor: Im klinischen Entwicklungsprogramm von Tofacitinib bei CU, zeigten sich bei
1.157 untersuchten CU-Patienten eine Inzidenzrate für tiefe Ve nenthrombosen (TVT) von 0,04 % und von Lungenembolien (LE) von 0,16 % pro 100 Patientenjahre. 4 Patienten entwickelten eine LE und 1 Patient eine TVT. Die Fälle betrafen Patienten, die überwiegend eine Dosierung von 10 mg zweimal täglich erhalten hatten und begleitend zur CU weitere Risikofaktoren für VTE aufwiesen* [12]. Fabian Sandner, Nürnberg
* Nachdem in einer Post-Marketing-Safety-Studie (ORAL Surveillance) bei Patienten (≥50 Jahre) mit rheumatoider Arthritis und mindestens einem begleitenden kardiovaskulären Risikofaktor eine erhöhte Inzidenz von Lungenembolien unter Tofacitinib 2 × 10 mg täglich im Vergleich zu TNF-Inhibitoren beobachtet wurde, gab es eine Überprüfung durch die europäische Arzneimittelagentur EMA. Anhand dieser Daten empfiehlt der Ausschuss für Humanarzneimittel (CHMP) in seiner finalen Stellungnahme, dass Tofacitinib – unabhängig von der Dosis und Indikation – bei Patienten mit hohem Risiko für thromboembolische Ereignisse mit Vorsicht angewendet werden sollte. Darüber hinaus empfiehlt die EMA, dass Patienten im Alter von über 65 Jahren aufgrund eines erhöhten Infektionsrisikos nur dann mit Tofacitinib behandelt werden sollten, wenn es keine geeignete alternative Behandlung gibt. Bei Patienten mit Colitis ulcerosa, die ein hohes Risiko für Blutgerinnsel haben, soll die Erhaltungsdosis von 10 mg 2 × täglich nicht verwendet werden, es sei denn, es gibt keine geeignete Behandlungsalternative [13].
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Literatur 1 Kucharzik T, Dignass AU, Atreya R et al. Aktualisierte S3-Leitlinie Colitis ulcerosa; August 2019. AWMF-Registriernummer: 021-009. Z Gastroenterol 2019;57: 1321-1405 2 Sandborn WJ et al. N Engl J Med 2017; 376:1723-1736 3 Lichtenstein CR et al. United European Gastroenterology Week (UEGW), Barcelona, 2019: OP213 4 Gomollón F et al. J Crohns Colitis 2017; 11:3-25 5 Harbord M et al. J Crohns Colitis 2017; 11:769-784 6 Lichtenstein et al. United European Gastroenterology Week (UEGW), Barcelona, 2019: Poster 0386 7 Feagen GB et al. World Congress of Gastroenterology at the American College of Gastroenterology Annual Scientific Meeting, Orlando, 2017: Poster 416 8 Gordon P et al. Eur J Gastroenterol Hepatol 2015;27:804-812 9 Gisbert JP Aliment Pharmacol Ther 2015;41:613-623 10 Sandborn WJ et al. Digestive Disease Week (DDW), Orlando, 2019: TU1717 11 Lichtenstein GR et al. Am J Gastroenterol 2018;113 (Suppl 1): Abstract 571 12 Sandborn WJ et al. Aliment Pharmacol Ther 2019;50:1068-1076 13 https://www.ema.europa.eu/en/documents/referral/xeljanz-article-20-procedure-ema-confirmsxeljanz-be-used-caution-patients-high-risk-blood-clots_en.pdf
© VERLAG PERFUSION GMBH
KONGRESSE
ADHS bei Erwachsenen: Langfristig effektiv behandeln, Unfälle vermeiden Zahlreiche epidemiologische Studien zeigen, dass etwa 2 – 4 % der Erwachsenen von einer Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS) betroffen sind. Jedoch wird bei erwachsenen Patienten mit ADHS die Erkrankung noch immer zu oft nicht erkannt und noch seltener leitliniengerecht therapiert. Das ist nicht nur im Hinblick auf die Belastung durch die ADHS-Kernsymptomatik, sondern auch aufgrund des deutlich erhöhten Unfallrisikos ein Problem. Die im Rahmen des DGPPN-Kongresses präsentierte PRADA-Studie zeigt jetzt auch für die deutsche Versorgungslandschaft, dass ADHS-Patienten bei Unfallopfern im Krankenhaus überrepräsentiert sind. Zudem belegen die Langzeitauswertung der COMPAS-Studie sowie deutsche Versorgungsdaten aus der IDEAKohorte, dass die medikamentöse ADHS-Therapie bei Erwachsenen mit Methylphenidat (z.B. Medikinet® adult) langfristig effektiv ist. PRADA-Studie: Unter Unfallopfern ist ADHSPrävalenz doppelt so hoch
„Dass ADHS-Patienten in jedem Alter ein deutlich erhöhtes Unfallrisiko aufweisen, wurde in den letzten Jahren immer besser untersucht und beschrieben“, sagte Professor Sarah Kittel-Schneider, Würzburg. Sie berichtete unter anderem über eine dänische, populationsbasierte Kohorte, in der ge-
zeigt werden konnte, dass das Unfallrisiko bei 10-jährigen Kindern mit ADHS um rund ein Drittel erhöht war. Eine niederländische Studie hat zudem kürzlich gezeigt, dass bei Erwachsenen mit ADHS das Risiko, 3 oder mehr Unfälle zu erleiden, fast dreimal so hoch ist wie bei Erwachsenen ohne ADHS. Kittel-Schneider und Professor Andreas Reif, Frankfurt/Main, haben in der PRADA (Prevalence of ADHD in Accident Victims)Studie jetzt erstmals untersucht, wie häufig eine ADHS bei erwachsenen Menschen ist, die sich aufgrund von Unfällen in Unfallkliniken behandeln lassen müssen. Die Expertin berichtete darüber bei der DGPPN-Jahrestagung in Berlin: „Zu den Besonderheiten unserer Studie gehört, dass wir Unfälle jeglicher Art berücksichtigt haben, nicht nur Verkehrsunfälle.“ Insgesamt füllten 905 Unfallopfer ASRS (Adult Self Report Scale)-Screening-Fragebögen für die ADHS-Diagnostik aus. Bei Nutzung der Kurzform ASRS-SF (Adult Self-Report Scale-short form) zeigte sich eine ADHS-Prävalenz von rund 6,2 %, bei Nutzung der Langversion ASRS-18 waren es sogar 8,3 %. „Die Prävalenz der ADHS bei Unfallopfern ist etwa doppelt so hoch wie in der Allgemeinbevölkerung“, so KittelSchneider. Tatsächlich diagnostiziert war die Erkrankung aber nur bei 0,7 % der Studienteilnehmer bzw. bei jedem fünften mit pathologischem ASRS-SF, und von diesen Patienten erhielt nur jeder dritte eine Pharmakotherapie mit einem Stimulans. Kittel-Schneider stellte vor diesem Hintergrund die Frage zur Diskussion, ob Unfallopfer unter bestimmten Umständen auf ADHS gescreent werden sollten. „Zumindest könnte man darüber nachden-
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
31
ken, ob bei wiederholten Unfällen ein Screening veranlasst werden sollte.“ So könnten Patienten identifiziert werden, denen ggf. mit einer zielgerichteten Therapie geholfen werden kann. Denn epidemiologische Untersuchungen deuten darauf hin, dass eine adäquate ADHS-Therapie mit Stimulanzien dazu beiträgt, das Unfallrisiko der Patienten zu verringern. So war das Unfallrisiko in der dänischen Kohorte in Phasen, in denen die Kinder medikamentös behandelt wurden, um ein Drittel geringer. Darüber hinaus wurde auf Basis von US-amerikanischen Versicherungsdaten berechnet, dass sich durch eine medikamentöse Therapie 22 % aller Autounfälle bei erwachsenen ADHS-Patienten verhindern lassen könnten. COMPAS-Studie: Methylphenidat ist nachhaltig effektiv über 2,5 Jahre
Wie langfristig eine Behandlung mit Methylphenidat bei erwachsenen ADHS-Patienten wirkt, untersuchte die deutsche COMPAS (Comparison of Methylphenidate and Psychotherapy in Adult ADHD Study)-Studie, für die aktuell die Ergebnisse des abschließenden Langzeit-Follow-up über zweieinhalb Jahre publiziert wurden. An der Studie hatten 433 erwachsene ADHS-Patienten teilgenommen, die nach Art eines 2x2-Faktorialdesigns mit Methylphenidat (Medikinet® adult) oder Placebo sowie Gruppenpsychotherapie oder klinischem Management mit Einzelgesprächen behandelt wurden. In der Primärauswertung hatte sich bereits gezeigt, dass durch die medikamentöse Therapie im Vergleich zu Placebo eine statistisch signifikant bessere Kontrolle der © VERLAG PERFUSION GMBH
KONGRESSE
32
ADHS-Kernsymptome über 3 und 12 Monate erreicht wird. Für das 2,5-Jahres-Follow-up, über das Professor Wolfgang Retz, Mainz, berichtete, standen knapp 60 % der ursprünglichen Patienten noch zur Verfügung. Für diese hatte es ab Woche 52 keinerlei therapeutische Vorgaben mehr gegeben. Trotzdem war der signifikante Unterschied zwischen den ursprünglichen MPH- und Placebo-Gruppen auch im Langzeit-Follow-up erhalten geblieben. Patienten, die MPH eingenommen haben, profitierten langfristig von dieser Behandlung, und zwar unabhängig davon, ob sie nach Studienende weiter MPH eingenommen hatten oder nicht. „Die deutlichste Symptomreduktion fand sich bei den Patienten, die von Baseline an mit MPH behandelt wurden und die bei Studienende in Woche 130 weiterhin MPH einnahmen“, berichtete Retz. IDEA-Studie: Rascher Wirkeintritt in der realen Versorgung
Die Frage „lassen sich die randomisierten Daten zur Effektivität der Methylphenidat-Therapie bei Erwachsenen auch auf die reale Versorgung in Deutschland übertragen?“ beantwortete Retz mit „eindeutig ja“ und verdeutlichte dies anhand der Beobachtungsstudie IDEA, an der 468 erwachsene ADHS-Patienten im Alter von 18 – 71 Jahren teilgenommen hatten. Die Patienten wurden erstmals oder nach Vordiagnose in der Kindheit erneut auf Methylphenidat (Medikinet® adult) eingestellt. Jeder zweite Patient hatte neben der ADHS eine weitere neuropsychiatrische Begleiterkrankung, mehrheitlich affektive Störungen. Nach durchschnittlich 3,3 Mona-
ten Behandlung wurden die Patienten anhand der Clinical Global Impression (CGI, Arzturteil) evaluiert. Während zu Studienbeginn rund 2 Drittel der Patienten vom Arzt als deutlich, schwer oder extrem schwer krank eingestuft wurden, war es 3 Monate nach Beginn der MPH-Therapie nur noch jeder vierte. Dies wurde mit einer durchschnittlichen MPH-Dosis von 35,8 mg/dl (Median 40 mg/ dl) erreicht. „Auch in der Patientenselbstbeurteilung gab es in allen Bereichen einen Rückgang der Symptomatik“, so Retz. Dies betraf die ADHS-Kernsymptome, aber auch Dimensionen wie funktionelle und emotionale Dysregulation und Desorganisiertheit. Insgesamt nahm der Gesamt-Score beim WR-SB-Fragebogen (WenderReimherr-Selbstbeurteilungsfragebogen) im Mittel von 203 Punkten bei Visite 1 auf 153 Punkte bei Visite 2 ab. „Insgesamt zeigen die IDEA-Daten, dass Medikinet® adult auch in der realen Versorgung bei Patienten mit zahlreichen Komorbiditäten sehr effektiv ist“, schlussfolgerte Retz und ergänzte: „Die Wirkung tritt zudem innerhalb der ersten Monate ein, sodass Ärzte relativ rasch, bereits innerhalb eines Quartals, beurteilen können, ob die Therapie anschlägt oder nicht. Komorbide neuropsychiatrische Störungen sollten nicht ignoriert, sondern mitbehandelt werden, um einen bestmöglichen Gesamttherapieerfolg zu erzielen.“ Sibylle Michna, Puschendorf
Quelle: Lunchsymposium „ADHS im Erwachsenenalter: Neue Daten – neue Situation“ am 28.11.2019 im Rahmen des DGPPNKongresses in Berlin, Veranstalter: MEDICE Arzneimittel Pütter GmbH.
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
ZEDEBAC-Studie liefert wertvolle Erkenntnisse zum Einsatz von Eslicarbazepinacetat in der klinischen Praxis Die nicht interventionelle Studie ZEDEBAC dokumentiert die Behandlungsverläufe erwachsener Epilepsie-Patienten mit fokalen Anfällen unter der Gabe des Antiepileptikums Eslicarbazepina® cetat (Zebinix ) in 43 deutschen Studienzentren. Im Rahmen einer von der Eisai GmbH und der BIAL Deutschland GmbH veranstalteten Fachpressekonferenz schilderte Dr. Florian Weissinger, Berlin, welchen Nutzen Beobachtungsstudien wie ZEDEBAC bieten. „Nicht interventionelle Studien bilden den klinischen Alltag und eine breitere Patientenpopulation ab als Zulassungsstudien. Durch die Untersuchung von Behandlungsszenarien im ‚real life‘ lassen sich vertiefende Informationen gewinnen, mithilfe derer eine Therapie besser auf die individuellen Bedürfnisse der Patienten angepasst werden kann“, sagte der Epileptologe und veranschaulichte dies am Beispiel der ZEDEBAC-Studie. Eslicarbazepinacetat überzeugt in der Monotherapie
In der multizentrischen, prospektiven Beobachtungsstudie wurde die Effektivität von Eslicarbazepinacetat (ESL) in Abhängigkeit von den Therapiebedingungen in der klinischen Praxis evaluiert. Hierzu wurden erwachsene Patienten (n = 237) mit einer gesicherten Diagnose fokaler epileptischer Anfälle mit oder ohne sekundärer Generalisierung in die Studie eingeschlossen und einer von 3 Beobachtungsgruppen zugeordnet: Die © VERLAG PERFUSION GMBH
33
KONGRESSE
Eslicarbazepinacetat Eslicarbazepinacetat (Zebinix®) ist in der EU zugelassen zur Monotherapie fokaler epileptischer Anfälle mit oder ohne sekundärer Generalisierung bei Erwachsenen mit neu diagnostizierter Epilepsie sowie als Zusatztherapie bei Erwachsenen, Jugendlichen und Kindern über 6 Jahren mit fokalen epileptischen Anfällen mit oder ohne sekundärer Generalisierung. Seit dem 1. November 2018 steht Zebinix® als Suspension zum Einnehmen zur Verfügung. Die neue Darreichungsform zur einmal täglichen Einnahme wurde speziell zur Behandlung besonderer Patientengruppen wie Kindern und Jugendlichen ab 6 Jahren, älteren Patienten sowie Patienten mit Schluckbeschwerden entwickelt.
Patienten erhielten ESL entweder als Monotherapie (n = 35), als einzige Zusatztherapie zu einem weiteren Antikonvulsivum (n = 114) oder als Zusatztherapie zu 2 oder mehr AEDs (n = 88). Der Beobachtungszeitraum betrug 6 Monate, als primärer Endpunkt war die Retentionsrate definiert, also der Anteil von Patienten, die das Medikament nach 6 Monaten weiterhin einnahmen. Sekundäre Endpunkte waren unter anderem die Wirksamkeit (definiert als Anfallssituation in den 3 Monaten vor Follow-up vs. 3 Monate vor Baseline), Verträglichkeit sowie die Anfallsfreiheitsund Ansprechrate. Von allen in die Studie eingeschlossenen Patienten verblieben nach 6 Monaten 79,3 % weiterhin auf der Therapie mit Eslicarbazepinacetat. Hohe Retentionsraten wurden auch in den einzelnen Beobachtungsgruppen festgestellt, wobei der Patientenanteil unter ESLMonotherapie mit 94 % besonders hervorsticht. In der Gruppe, die ESL als Zusatztherapie zu einer bestehenden AED-Monotherapie erhielt, betrug die Retentionsrate 78 %. Bei den Patienten, die ESL zusätzlich zu 2 oder mehr AEDs einnahmen, betrug der Anteil rund 75 %. Gleichzeitig verzeichneten die Patienten unter ESL-Monothe-
rapie mit 90,5 % die höchste Ansprechrate (1 AED+ESL: 77,6 %; ≥2 AEDs+ESL: 48,3 %). 81,5 % blieben unter ESL-Monotherapie anfallsfrei (1 AED+ESL: 47,9 %; ≥2 AEDs+ESL: 23,4 %). Gute Verträglichkeit begünstigt Einsatz in der Praxis
Neben der guten Wirksamkeit überzeugte ESL in der Studie auch durch sein günstiges Verträglichkeitsprofil. Die häufigsten unerwünschten Ereignisse waren Schwindel (5,1 %), Müdigkeit (3,8 %), Hyponatriämie (3,4 %) sowie Übelkeit (2,5 %). „Die belegte Sicherheit ermöglicht einen Einsatz in der klinischen Praxis sowohl in der initialen Behandlung als auch bei komplexeren Behandlungssituationen mit antikonvulsiver Polytherapie“, resümierte Weissinger. Dies bestätigten auch Kasuistiken, die Dr. Martin Hirsch, Freiburg, präsentierte: Patienten, die nach Vorbehandlung mit einem oder mehreren AEDs auf ESL umgestellt wurden, zeigten eine hohe Ansprech- und Anfallsfreiheitsrate bei guter Verträglichkeit. Fabian Sandner, Nürnberg
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Hepatozelluläres Karzinom: Lenvatinib ist eine wertvolle Option in der Systemtherapie Seit einem Jahr ist Lenvatinib (Lenvima®) ist als Monotherapie für die Behandlung von erwachsenen Patienten mit fortgeschrittenem oder inoperablem hepatozellulärem Karzinom (HCC) zugelassen, die zuvor noch keine systemische Therapie erhalten haben. Im Rahmen der Jahrestagung der Deutschen, Österreichischen und Schweizerischen Gesellschaften für Hämatologie und Medizinische Onkologie (DGHO) in Berlin gab Professor Henning Wege, Hamburg, einen aktuellen Überblick über die Therapie des HCC. Sein Fazit: Lenvima® ist auch im klinischen Alltag ein wirksames und gut handhabbares Medikament. Zusätzlich haben sich die Chancen auf eine Tumorkontrolle durch weitere neue Substanzen in der Zweitlinie verbessert. Der Stellenwert der Systemtherapie beim HCC nimmt zu. Rechtzeitig zur systemischen Therapie wechseln
Das Staging des HCC erfolgt in Europa nach dem Barcelona-Clinic-Liver-Cancer-System (BCLC). Die Patienten werden anhand von Tumorlast, Leberfunktion (ChildPugh-Score) und Allgemeinzustand in ein sehr frühes, frühes, intermediäres, fortgeschrittenes und terminales Stadium stratifiziert. Therapie der Wahl im intermediären BCLC-Stadium B ist die transarterielle Chemoembolisation (TACE). Das Gesamtüberleben (OS) variiert unter diesem lokoregionären Ansatz zwischen ca. 20 und über 40 Monaten.„Das liegt daran, dass es sich im BCLC-Sta© VERLAG PERFUSION GMBH
KONGRESSE
34
dium B um ein sehr heterogenes Patientenkollektiv handelt“, erläuterte Wege. „Die Therapieentscheidung muss daher immer individuell erfolgen. Im Falle einer TACE sollte die Indikation hierfür immer wieder überprüft werden, um die Leberfunktion zu erhalten. Dies ist wichtig für den Einsatz von Folgetherapien. In der Praxis gilt es, den Zeitpunkt für einen Wechsel von der lokoregionären zur systemischen Therapie nicht zu verpassen. Denn mit modernen MultikinaseInhibitoren wie Lenvatinib stehen gute Optionen zur Verfügung.“ Wichtige Bereicherung der Erstlinientherapie
Die Wirksamkeit von Lenvatinib wurde in der randomisierten kontrollierten Phase-III-Studie REFLECT bei Patienten mit nicht resezierbarem, fortgeschrittenem HCC im Vergleich mit der bis dahin einzigen Standardtherapie Sorafenib evaluiert. Der MultikinaseInhibitor war hinsichtlich des OS Sorafenib statistisch nachweisbar nicht unterlegen. Im Median betrug das OS unter Lenvatinib 13,6 und unter Sorafenib 12,3 Monate (HR: 0,92; 95%-KI: 0,79 – 1,06; NichtUnterlegenheitsgrenze definiert bei 1,08). Darüber hinaus zeigte sich eine statistisch signifikante und klinisch relevante Überlegenheit von Lenvatinib bei allen sekundären Wirksamkeitspunkten: Das mediane progressionsfreie Überleben verdoppelte sich mit Lenvatinib im Vergleich zu Sorafenib von 3,7 auf 7,4 Monate gemäß Prüfarztbeurteilung mittels mRECIST (HR: 0,66; 95%-KI: 0,57 – 0,77; p < 0,0001). Hinsichtlich der Zeit bis zur Progression (TTP) und der objektiven Ansprechrate (ORR) schnitt Lenvatinib ebenfalls besser ab als
Sorafenib: Die mediane TTP war nahezu 2,5-fach länger, die ORR 2,6-fach höher. In den Leitlinien der European Association for the Study of the Liver (EASL) und European Society of Medical Oncology (ESMO) ist Lenvatinib als Erstlinientherapie bereits verankert. „Es gab in den letzten 10 Jahren viele Versuche, die Wirksamkeit von Sorafenib in dieser Indikation zu erreichen. Lenvima® ist der einzige Multikinase-Inhibitor in der systemischen Therapie, mit dem das in einer Phase-III-Studie gelungen ist“, konstatierte Wege. „Dies zeigt aber auch, wie schwierig es ist, in dieser Indikation neue Therapien zu etablieren. Mit der Zulassung von Lenvatinib und neuen zielgerichteten Zweitlinientherapien können wir unseren Patienten eine erweiterte systemische Sequenztherapie anbieten, wenn die lokalen Maßnahmen ausgeschöpft sind. Das ist eine neue, erfreuliche Entwicklung in dieser schwierigen Indikation.“ Wie Wege am Beispiel einer seiner Patientinnen zeigen konnte, bestätigt sich die Wirksamkeit von Lenvatinib auch im klinischen Alltag. „Ich setze Lenvatinib seit dem ersten Tag der Zulassung ein und meine Erfahrungen decken sich mit den Daten der REFLECT-Studie. Unsere Patienten sprechen in der Regel gut auf die Therapie an. Dies ist relevant, weil wir aus Post-hocAnalysen wissen, dass das Ansprechen offenbar mit einem längeren Gesamtüberleben korreliert.“ Häufige Nebenwirkungen wie zum Beispiel Diarrhö lassen sich Wege zufolge mit einem proaktiven und therapiebegleitenden Management gut handhaben. Gestützt wird dies durch Subgruppen-Analysen der REFLECT-Studie. Hier verschlechterte sich die Lebensqualität in Hinblick auf Diarrhö im
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Sorafenib-Arm eher als unter Lenvatinib. Da sich die Therapietreue positiv auf den Therapieerfolg auswirken kann, sollte eine gute Aufklärung der Patienten zu Therapiebeginn über das mögliche Auftreten von Nebenwirkungen sowie das rechtzeitige Einleiten entsprechender Behandlungsmaßnahmen erfolgen. Interessant sind in diesem Zusammenhang auch retrospektive Daten, die darauf hindeuten, dass das Auftreten von Hypertonie, Diarrhö, Proteinurie und Hypothyreose mit einem signifikant längeren OS assoziiert sein kann. Sequenztherapie mit Lenvatinib beginnen
„Um das bestmögliche Therapieergebnis zu erzielen, müssen Überlegungen zur Sequenztherapie in die Behandlungsplanung mit einfließen“, betonte Wege. Prospektive Daten liegen hierzu nicht vor. Jedoch weisen Ergebnisse einer weiteren Post-hoc-Analyse der REFLECT-Studie darauf hin, dass die Patienten ein längeres OS erreichen können, wenn nach Lenvatinib als erste systemische Therapie eine weitere Therapie gegeben werden kann. Demnach betrug das mediane OS bei Patienten mit initialer Lenvatinib-Therapie gefolgt von jeglicher Anti-Tumormedikation 20,8 Monate vs. 17,0 Monate im Sorafenib-Arm. Am häufigsten handelte es sich bei der Zweitlinientherapie um Sorafenib. Bei Patienten, die auf Lenvatinib in der Erstlinie angesprochen hatten (n = 35) und anschließend Sorafenib bekamen, wurde sogar ein medianes OS von 26,2 Monaten erreicht. „Anhand der Subgruppenanalysen aus der REFLECT-Studie sehen © VERLAG PERFUSION GMBH
KONGRESSE / WISSENSWERTES
35
wir, dass eine Sequenztherapie im BCLC Stadium B/C, die mit Lenvatinib beginnt, zu einem Gesamtüberleben von über 20 Monaten führen kann. Das ist in dieser Indikation eine sehr ermutigende Entwicklung, weil sie zeigt, dass sich die Prognose unserer Patienten durch die erweiterten Therapieoptionen verbessert“, betonte Wege.
beschränken sich gemäß der aktuellen Datenlage auf die Sequenztherapie nach der Vortherapie mit Sorafenib. Zur Verfügung stehen Regorafenib, Cabozantinib und Ramucirumab, wobei der Einsatz von Ramucirumab an einen hohen Wert des prognostischen Markers Alpha-Fetoprotein (AFP) gekoppelt ist.
Neue Entwicklungen in der Zweitlinientherapie
Ausblick: Kombinationstherapien
Wie Wege ausführte, deuten sich auch beim fortgeschrittenen HCC neue Therapieansätze mit immunonkologischen Substanzen an. In den USA sind die PD-1-Antikörper Nivolumab und Pembrolizumab bereits als Zweitlinientherapien nach Sorafenib zugelassen. Der Stellenwert von PD-L1 als Biomarker für das Ansprechen auf eine Immuntherapie ist derzeit noch
unklar. Darüber hinaus sind insbesondere Kombinationstherapien von Tyrosinkinase-Inhibitoren oder monoklonalen Antikörpern mit Checkpoint-Inhibitoren interessant. Geprüft werden derzeit in Phase-III-Studien u.a. Lenvatinib plus Pembrolizumab (LEAP-002), Cabozantinib mit Atezolizumab (COSMIC-312) sowie Atezolizumab und Bevacizumab (IMbrave-150). Des Weiteren sind Studien mit Checkpoint-Inhibitor-Kombinationen in der klinischen Entwicklung, wie zum Beispiel Nivolumab und Ipilimumab (CheckMate 9DW) sowie Durvalumab und Tremelimumab (HIMALAYA). Ein weiterer Schwerpunkt der Forschung liegt darauf, prognostische und prädiktive Biomarker zu identifizieren. Fabian Sandner, Nürnberg
BPLS ist die häufigste peripher vestibuläre Schwindelursache. Hervorgerufen wird er durch abgelöste Otolithen aus der gelartigen Matrix des Utrikulus im Gleichgewichtsorgan, die in einen der drei Bogengänge gelangen und dort zu einer Irritation führen können. Die Folgen sind kurze Drehschwindelattacken, die durch Veränderungen der Kopfposition ausgelöst werden, etwa beim Umdrehen im Bett, Hinlegen oder Aufrichten.
Zur Diagnostik eines BPLS stehen verschiedene Lagerungsmanöver zur Verfügung. Die dabei auftretenden Nystagmen geben Aufschluss darüber, welche Seite und welcher Bogengang betroffen sind. Anschließend können durch spezifische Befreiungsmanöver die Otolithen aus dem Bogengang wieder zurückbefördert werden. Eine medikamentöse Begleitung kann den Therapieerfolg und die Lebensqualität erhöhen. Denn auch nach erfolgreicher Anwendung der Manöver sind nicht alle Patienten sofort beschwerdefrei. Hier hat sich vor allem das FirstLine-Therapeutikum Arlevert® mit der Fixkombination aus Cinnarizin und Dimenhydrinat bewährt, das zu einer deutlichen Linderung der Residualsymptome führt.
Durch die Zulassung neuer Wirkstoffe in der Zweitlinientherapie hat sich der Stellenwert der systemischen Behandlung beim fortgeschrittenen HCC zusätzlich erhöht. Zugleich spiegeln sich darin die Fortschritte in der Erstlinientherapie wider, weil zunehmend Patienten für eine Folgetherapie infrage kommen. Die Zulassungen
Lagerungsschwindel:
Neue Broschüre zur Diagnostik und Therapie bei BPLS Hennig Arzneimittel hat in Zusammenarbeit mit Professor Dr. med. Frank Schmäl die Broschüre zum benignen paroxysmalen Lagerungsschwindel (BPLS) komplett überarbeitet. Neu sind ein Leitfaden zur Diagnostik und Therapie sowie anschauliche Grafiken, die Schritt für Schritt die verschiedenen Lagerungsmanöver und die auftretenden Augenbewegungen zur Diagnostik sowie die spezifischen Befreiungsmanöver zur Therapie zeigen. Für Hals-Nasen-Ohren-Ärzte und Neurologen stellt die Broschüre eine wertvolle Hilfe bei der Durchführung der Manöver dar.
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
© VERLAG PERFUSION GMBH
WISSENSWERTES
36
Avelumab in Kombination mit Axitinib jetzt auch zur Erstlinientherapie des fortgeschrittenen Nierenzellkarzinoms zugelassen Die Europäische Kommission hat den Checkpoint-Inibitor Avelumab (Bavencio®) in Kombination mit dem Tyrosinkinase-Inhibitor Axitinib als Erstlinientherapie bei erwachsenen Patienten mit fortgeschrittenem Nierenzellkarzinom (RCC) zugelassen. Die Zulassungserweiterung für Bavenico® basiert auf positiven Zwischenergebnissen der Phase-III-Studie JAVELIN Renal 101, die die Kombination aus Avelumab und Axitinib bei unbehandelten Patienten mit fortgeschrittenem Nierenzellkarzinom gegenüber Sunitinib untersucht. Die primären Endpunkte waren das mediane progressionsfreie Überleben (mPFS) sowie das mediane Gesamtüberleben (mOS) bei Patienten mit PD-L1-positiven Tumoren (PD-L1 = programmierter Zelltod-Ligand 1). Signifikante Verlängerung des progressionsfreien Überlebens
In der Gesamtpopulation betrug das mPFS unter Avelumab plus Axitinib 13,3 Monate und war damit um 5,3 Monate länger als unter Sunitinib (8,0 Monate; HR: 0,69; 95%-KI: 0,574 – 0,825; p < 0,0001; Abb. 1). Damit einher ging ein um 31 % geringeres Risiko von Krankheitsprogression oder Tod unter der Kombination (HR: 0,69; 95%KI: 0,57 – 0,83; p < 0,0001). Die objektive Ansprechrate war mit 52,5 % für Avelumab in Kombination mit Axitinib fast 2 × so hoch wie für Sunitinib mit 27,3 %, unabhängig vom PD-L1-Status.
Abbildung 1: Ergebnis der Studie JAVELIN Renal 101 für das mediane progressionsfreie Überleben (Quelle: Fachinformation Bavencio®; Stand: September 2019).
Avelumab Avelumab (Bavencio®) ist ein humaner Antikörper, der gegen den programmierten Zelltod-Liganden 1 (PD-L1) gerichtet ist. Durch die Blockierung der Interaktion von PD-L1 mit PD-1-Rezeptoren hat Avelumab in präklinischen Modellen die Unterdrückung der T-Zell-vermittelten AntitumorImmunabwehr aufgehoben.
Die häufigsten Nebenwirkungen waren Diarrhö (62,8 %), Hypertonie (49,3 %), Fatigue (42,9 %), Übelkeit (33,5 %), Dysphonie (32,7 %), verminderter Appetit (26,0 %) und Hypothyreose (25,2 %). Die Kombination aus Avelumab und Axitinib zeichnete sich durch eine flexible Dosierung und eine niedrige Therapie-Abbruchrate aus. F. S.
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Oncotype DX® Brustkrebstest wird von allen deutschen Kassen erstattet Nach einem Beschluss der Kassenärztliche Bundesvereinigung und des GKV-Spitzenverbandes können seit dem 01.01.2020 die Leistungen für den Oncotype DX Breast Recurrence Score® Test innerhalb der gesetzlichen Krankenversicherung über den EBM abgerechnet werden. Den Biomarkertest können all diejenigen Patientinnen in Anspruch nehmen, bei denen ein Hormonrezeptor-positives, HER2/neu-negatives und nodal-negatives Mammakarzinom im Frühstadium diagnostiziert wurde und eine Entscheidung für oder gegen eine Chemotherapie alleine aufgrund der klassischen klinisch-pathologischen Parameter nicht möglich ist. Der Oncotype DX Test identifiziert die etwa 80 % aller Brustkrebspa© VERLAG PERFUSION GMBH
37
WISSENSWERTES
tientinnen mit einem RecurrenceScore-Ergebnis von ≤25, denen eine Chemotherapie erspart werden kann, ohne den Behandlungserfolg zu gefährden. Er identifiziert aber auch die kleine, wichtige Gruppe von Frauen mit einem RecurrenceScore-Ergebnis von 26 – 100, für die eine Chemotherapie lebensrettend sein könnte. Das geht aus den Ergebnissen der TAILORx-Studie hervor, der weltweit größten Brustkrebsstudie im adjuvanten Setting mit über 10.000 Patientinnen mit HR+, HER2-negativem frühem Brustkrebs ohne Lymphknotenbeteiligung.
Der Gemeinsame Bundesausschuss (G-BA) hatte den Oncotype DX bereits am 20. Juni 2019 als einzigen Biomarkertest bei Brustkrebs in die Regelversorgung aufgenommen, nachdem das deutsche Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) bestätigt hatte, dass ausreichende Evidenz für eine Entscheidung über den Nutzen einer adjuvanten Chemotherapie anhand des Oncotype DX Tests vorliegt. Der GB-A geht davon aus, dass die behandelnden Ärzte bei geschätzt jährlich 20.000 Frauen mit frühem Brustkrebs allein aufgrund der
klinisch-pathologischen Kriterien keine eindeutige Therapieempfehlung für oder gegen eine adjuvante Chemotherapie geben können. Der Test verringert die Anzahl der unnötigen Chemotherapien und die damit einhergehenden Kosten für das Gesundheitswesen signifikant. Eine deutsche Analyse aus dem Jahr 2017 schätzt die möglichen Einsparungen bei den gesamtgesellschaftlichen Kosten durch den Test auf mehr als 253 Millionen Euro pro Jahr.
B. S.
Titelbild: Innovative Katheter-Lösungen vereinfachen minimalinvasive Eingriffe (Quelle: BVMed).
Herausgeber: Prof. Dr. med. Karl-Ludwig Resch, FBK Deutsches Institut für Gesundheitsforschung gGmbH, Kirchstraße 8, 08645 Bad Elster Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin Wissenschaftlicher Beirat: Prof. Dr. M. Alexander, Infektiologie, Berlin Prof. Dr. L. Beck, Gynäkologie, Düsseldorf Prof. Dr. Berndt, Innere Medizin, Berlin Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main Prof. Dr. K. M. Einhäupl, Neurologie, Berlin Prof. Dr. E. Erdmann, Kardiologie, Köln Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK Prof. Dr. K. Falke, Anästhesiologie, Berlin Prof. Dr. K. Federlin, Innere Medizin, Gießen Prof. Dr. E. Gerlach, Physiologie, München Prof. Dr. H. Helge, Kinderheilkunde, Berlin Prof. Dr. R. Herrmann, Onkologie, Basel Prof. Dr. W. Jonat, Gynäkologie, Hamburg Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt Prof Dr. J. Mann, Nephrologie, München Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim Prof. Dr. R. Nagel, Urologie, Berlin Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf Prof. Dr. P. Ostendorf, Hämatologie, Hamburg Prof. Dr. Th. Philipp, Innere Medizin, Essen Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf Prof. Dr. H. Rieger, Angiologie, Aachen Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen Prof. Dr. E. Rüther, Psychiatrie, Göttingen Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München Prof. Dr. E. Standl, Innere Medizin, München Prof. Dr. W. T. Ulmer, Pulmologie, Bochum Schriftleitung: Prof. Dr. med. Karl-Ludwig Resch, FBK Deutsches Institut für Gesundheitsforschung gGmbH, Kirchstraße 8, 08645 Bad Elster Telefon: 037437 557-0 Bibliothek: 037437 2214 [Library] E-Mail DIG: info@d-i-g.org E-Mail persönlich: k.l.resch@d-i-g.org
JOURNAL PHARMAKOL. U. THER. 1/2020 · 29. JAHRGANG
Die Zeitschrift erscheint 6mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag. Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Satz: Rolf Wolle, Schwabacherstr. 117 90763 Fürth
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Druck und Verarbeitung: DRUCK_INFORM GmbH In der Büg 8 91330 Eggolsheim
Herstellung/Layout: HGS5 – Rolf Wolle Schwabacherstr. 117 90763 Fürth Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
VERLAG
PERFUSION Verlag PERFUSION GmbH Storchenweg 20 90617 Puschendorf Telefon: 09101/990 11 10 Fax: 09101/990 11 19 www.Verlag-Perfusion.de E-Mail: perfusion@t-online.de
Journal
© VERLAG PERFUSION GMBH
HIZENTRA – das einzige subkutane Immunglobulin zur Behandlung der CIDP ®
• Durch Heimtherapie flexibel in den Alltag integrierbar • Freiheit und Lebensqualität für Ihre CIDP-Patienten
Hizentra® 200 mg/ml Lösung zur subkut. Injektion. Wirkstoff: Normales humanes Immunglobulin. Zusammensetzung: 1 ml Hizentra® enth. 200 mg normales humanes Immunglobulin (Reinheitsgrad mind. 98% IgG). Verteilung der IgG-Subklassen: IgG1 ca. 69%, IgG2 ca. 26%, IgG3 ca. 3%, IgG4 ca. 2%. IgA max. 50 µg/ml. Sonst. Bestandteile: L-Prolin, Polysorbat 80, H2O. Anwendungsgebiete: Substitutionstherapie bei prim. Immundefizienz mit verminderter Antikörperprodukt., bei chron. lymphat. Leukämie oder multiplem Myelom mit Hypogammaglobulinämie und rezidiv. Infekt. sowie bei Hypogammaglobulinämie vor und nach allogener HSCT. Immunmodulationstherapie bei chron. inflammator. demyelinisierender Polyneuropathie (CIDP) als Erhaltungstherapie nach Stabilisierung mit IVIg. Gegenanzeigen: Überempfindlichk. gg. den Wirkstoff oder einen der sonst. Bestandteile, Hyperprolinämie Typ I od. II. Nebenwirkungen: Sehr häufig: Kopfschmerzen (einschl. Migräne), Hautausschlag, Schmerzen im Bewegungsapparat (einschl. Muskelspasmen und -schwäche), Reakt. an der Infusionsstelle. Häufig: Schwindel, Hypertonie, Durchfall, Bauchschmerzen, Übelk., Erbrechen, Pruritus, Urtikaria, Arthralgie, Fieber, Müdigk. (einschl. Unwohlsein), Schmerzen im Brustraum, grippeähnl. Symptome, Schmerzen. Gelegentl.: Überempfindlichk., Syndrom der asept. Meningitis, Tremor (einschl. psychomotor. Hyperaktivität), Tachykardie, Hautrötung, Schüttelfrost (einschl. Unterkühlung), erhöhtes Kreatinin, niedriger Blutdruck, Rückenschmerzen. In Einzelfällen: Anaphylakt. Schock. Häufigk. nicht bekannt: Anaphylakt. Reakt., Brennen, embol. und thrombot. Ereignisse, Ulkus an der Infusionsstelle. Verschreibungspflichtig. Pharmazeutischer Unternehmer: CSL Behring GmbH, Emil-von-Behring-Str. 76, 35041 Marburg. Stand: Februar 2019.