ISSN 1432-4334 JAHRGANG 30 HEFT 3 Juni 2021
FÜR PHARMAKOLOGIE UND THERAPIE
JOURNAL OF PHARMACOLOGY AND THERAPY
Inanspruchnahme osteopathischer Medizin bei chronisch und Langzeit-Erkrankten Exzellente Sicht auf die Mukosa – eine unabdingbare Voraussetzung für die Darmkrebsvorsorge Therapie der Plaque-Psoriasis: Secukinumab-Fertigpen überzeugt durch Wirksamkeit und Patientenadhärenz Morbus Parkinson: Rechtzeitig Chancen der Add-on-Therapie mit Safinamid nutzen Asthma-Therapie in Zeiten von COVID-19: Mit fixer Dreierkombination zu einer besseren Therapiekontrolle – Interview mit Herrn Dr. Justus de Zeeuw, Köln Isatuximab in zweiter Indikation zur Behandlung des rezidivierten multiplen Myeloms zugelassen Guselkumab – ein selektiver Interleukin-23-Hemmer zur Behandlung der aktiven Psoriasis-Arthritis Kinasehemmer Pemigatinib – die erste Option zur Zweitlinienbehandlung des Gallengangkarzinoms
VERLAG
PERFUSION
WIR LIEBEN GEBURTSTAGE!
Und zwar möglichst viele davon.
Seit 2011
Jahre ZYTIGA®
In den letzten 10 Jahren konnten über 700.000 Patienten mit metastasiertem Prostatakarzinom mit ZYTIGA®* behandelt werden.** Somit konnten wir gemeinsam Geburtstage ermöglichen und Lebenszeit schenken. Das ist mehr als ein Grund zum Feiern: Auf 10 Jahre ZYTIGA®. Auf 10 Jahre erfolgreiche Partnerschaft. Auf 10 Jahre gemeinsame Therapie für unsere Patienten. www.zytiga.de Janssen-Cilag GmbH www.janssen.com/germany
* ZYTIGA® ist indiziert mit Prednison oder Prednisolon zur Behandlung des neu diagnostizierten Hochrisikometastasierten hormonsensitiven Prostatakarzinoms (mHSPC) bei erwachsenen Männern in Kombination mit Androgenentzugstherapie (androgen deprivation therapy, ADT); zur Behandlung des metastasierten kastrationsresistenten Prostatakarzinoms (mCRPC) bei erwachsenen Männern mit asymptomatischem oder mil symptomatischem Verlauf der Erkrankung nach Versagen der Androgenentzugstherapie, bei denen eine Chemotherapie noch nicht klinisch indiziert ist; und zur Behandlung des mCRPC bei erwachsenen Männern, deren Erkrankung während oder nach einer Docetaxelhaltigen Chemotherapie progredient ist. ** Anzahl der weltweit behandelten ZYTIGA®Patienten (Stand Dezember 2020). ZYTIGA® 500 mg Filmtabletten. Wirkstoff: Abirateronacetat. Zusammensetz.: Jede Filmtabl. enth. 500 mg Abirateronacetat. Sonst. Bestandt.: Siliciumdioxid-beschichtete mikrokristalline Cellulose, Croscarmellose-Natrium, Hypromellose 2910 (15 mPa.S), Lactose-Monohydrat, Magnesiumstearat, hochdisperses Siliciumdioxid, Natriumdodecylsulfat; Filmüberzug: Eisen (II,III)-oxid (E172), Eisen(III)-oxid (E172), Macrogol 3350, Poly(vinylalkohol), Talkum, Titandioxid. Anw. geb.: Zusammen m. Prednison od. Prednisolon; z. Bhdlg. des neu diagnostiz. Hochrisikometastasierten hormonsensitiven Prostatakarzinoms (mHSPC) b. erwachs. Männern in Komb. m. Androgenentzugsther. (androgen deprivation therapy, ADT) u. des metastasierten kastrationsresistenten Prostatakarzinoms (mCRPC) b. erwachs. Männern m. asympt. od. mild sympt. Verlauf d. Erkr. nach Versagen d. Androgenentzugsther., b. denen e. Chemother. noch nicht klin. indiz. ist sowie z. Bhdlg. d. mCRPC b. erwachs. Männern, deren Erkr. währ. od. nach e. Docetaxel-halt. Chemother. progredient ist. Gegenanz.: Überempfindl. gg. Abirateronacetat od. einen d. sonst. Bestandt.; schwere Leberfunkt.störg. (Child-Pugh-Klasse C); Kombinat. m. Ra-223; nicht z. Anw. b. Frauen sowie b. Kindern u. Jugendl.. Nebenwirk.: Sehr häufig (≥ 1/10); Häufig (≥ 1/100) bis < 1/10); Gelegentlich (≥ 1/1.000 bis < 1/100); Selten (≥ 1/10.000, < 1/1.000); Nicht bekannt (Häufigk. auf Grundlage d. verfügb. Daten nicht abschätzbar). Sehr häufig: Harnwegsinfekt., Hypokaliämie, Hypertonie, Diarrhö, periph. Ödeme, erhöhte Alaninaminotransferase u./ od. erhöhte Aspartataminotransferase (ALT, AST, abnorm. Leberfunkt.). Häufig: Sepsis, Hypertriglyceridämie, Herzinsuff. (auch kongest. Herzinsuff., linksventrik. Dysfunkt. u. vermind. Ejektionsfraktion), Angina pect., Vorhofflimmern, Tachykardie, Dyspepsie, Hautausschlag, Hämaturie, Frakturen (Osteoporose u. alle Frakturen m. Ausn. d. patholog. Frakturen). Gelegentlich: Andere Arrhythmien, Nebenniereninsuff., Myopathie, Rhabdomyolyse. Selten: allerg. Alveolitis, fulminante Hepatitis, akut. Leberversagen. Nicht bekannt: anaphylakt. Reakt., Myokardinfarkt, QT-Verlängerung. Warnhinw. u. Vorsichtsmaßn. für d. Anw.: Arzneim. f. Kdr. unzugängl. aufbewahren; b. Geschlechtsverkehr m. e. Schwangeren ist ein Kondom erforderl.; b. Geschlechtsverkehr m. e. Frau im gebärfähigen Alter ist ein Kondom u. gleichz. e. and. zuverlässige Verhütungsmethode erforderl.; bes. Vors. bei: Pat. m. Hypertonie, Herzinsuff., Hypokaliämie (QT-Verlängerung wurde b. Pat. m. Hypokaliämie unter ZYTIGA beob.) od. kardiovask. Erkr. i. d. Anamnese, b. Pat. m. mäßiger Leberfunkt.störg.: nach Markteinf. selt. Berichte üb. akut. Leberversagen u. fulminante Hepatitis, einige m. tödl. Ausg.; b. Pat., d. währ. d. Bhdlg. e. schwere Hepatotoxizität entwickeln (ALT od. AST 20-fach üb. d. ULN) muss d. Bhdlg. abgebr. u. d. Pat. dürfen nicht erneut bhdlt. werden; b. Pat. m. schwerer Nierenfunkt.störg., beim Absetzen v. Prednison od. Prednisolon. B. Männern m. metastasiertem Prostatakarzinom können sex. Funkt.störg. u. Anämien auftreten (jeweils einschl. derer unter Bhdlg. m. ZYTIGA); ZYTIGA darf nicht zusammen m. Nahrungsmitteln eingenommen werden (Einn. mind. 1 Std. vor od. frühest. 2 Std. nach d. Essen) u. kann d. Vermind. d. Knochendichte verstärken; b. Vorbehdlg. m. Ketoconazol könnten gering. Response-Raten auftreten; Vors. b. Pat. m. Hyperglykämie od. gleichz. Bhdlg. m. Pioglitazon od. Repaglinid (Hypoglykämien mögl.); b. Pat., d. gleichz. m. Arzneim. bhdlt. werden, die m. d. Entstehung v. Myopathie/Rhabdomyolyse assoziiert sind. Vors. b. gleichz. Anw. v. Arzneim., d. durch CYP2D6 od. CYP2C8 aktiviert od. metabolisiert werden; starke CYP3A4-Induktoren vermeiden; Vors. b. gleichz. Anw. v. Arzneim., d. bek.maßen d. QTIntervall verlängern; gleichz. Anw. m. Spironolacton nicht empf.. Verschreibungspflichtig. Pharmazeut. Unternehmer: Janssen-Cilag International NV, B-2340 Beerse, Belgien. Örtlicher Vertreter für Deutschland: JanssenCilag GmbH, Johnson & Johnson Platz 1, 41470 Neuss. Stand d. Inform.: 10/20.
EDITORIAL
Im alten Griechenland war Chaos das Gegenteil von Kosmos, der (Welt-)Ordnung, also das vollständige Durcheinander. Moderne Chaos forschung fokussiert dagegen darauf, die Unvorhersagbarkeit von Prozessen wissenschaftlich in den Griff zu bekommen. Obwohl die politischen Aktivitäten, die Pandemie unter Kontrolle zu bringen, immer mehr an das Gegenteil von Kosmos erinnern, ist die Unvorhersehbarkeit von Prozessen mitnichten gegeben. Wohl aber eine zunehmende Unterordnung der Corona-Agenda unter politische und wirtschaftliche Interessen bei gleichzeitig fortschreitender Ignoranz gegenüber der sich konsolidierenden wissenschaftlichen Erkenntnis bezüglich SARS-CoV-2 und COVID-19. Hauptleidtragende sind derzeit Kinder und Jugendliche. Da stellt sich ein Gesundheitsminister hin und verkündet, dass jetzt alle ab 12 Jahren geimpft werden sollen, wohl in der Hoffnung, dicke Punkte bei Eltern und Lehrern einzufahren, aber ohne jede Rücksicht auf das Wohl der jungen Generation. Die Entscheidung der Ständigen Impfkommission (STIKO), Stand Ende Juni 2021 keine „allgemeine Freigabe“ ab 12 Jahren zu empfehlen, ist zunächst einmal nur logische Konsequenz aus der Zusammenschau der wissenschaftlichen Evidenz [1]. Denn für eine positive Empfehlung ist nicht nur die Frage der Wirksamkeit positiv zu beantworten (dazu geben z.B. die kürzlich publizierten Ergebnisse der ergänzenden Biontech-Pfizer-Studie tatsächlich Anlass [2]), sondern auch die der Sicherheit, unter dem Strich also das Verhältnis von Nutzen und Risiko. Und dazu ist eben diese Studie völlig unbrauchbar, schlimmer noch, die Ankündigung im Titel, eine Aussage zur Sicherheit ableiten zu können, unwissenschaftlich und damit unseriös. Bei einer Zahl von gerade einmal 1131 Jugendlichen in der Impfstoff-Gruppe lassen sich lediglich vorsichtige Schlussfolgerungen bezüglich häufig vorkommender Impfreaktionen ziehen. Seltenere, schwerere Impfreaktionen und mehr noch, schwere, lebensbedrohliche oder gar tödliche Nebenwirkungen brauchen, und das lehren die konkre-
65
Chaos Corona Club Berlin: Kinder im Visier ten Erfahrungen mit großen Kohorten geimpfter Erwachsener, um Dimensionen größere Fallzahlen. Die dürfen aber nur dann „generiert“ werden (also eine allgemeine positive Impfempfehlung ausgesprochen werden), wenn realistische Aussichten auf eine positive Nutzen-RisikoRelation erwartbar sind. Das war nach Lage der Dinge im Frühjahr bei der Diskussion um die Freigabe von „AstraZeneca“ klar der Fall [3], aber bei Kindern und Jugendlichen ist die Realität eine ganz andere! Die Hälfte aller dem Robert-Koch-Institut (RKI) bis 22.6.2021 gemeldeten 90.310 Corona-Toten war 84 Jahre und älter, 89.278 (97,0 %) waren 50 Jahre und älter. Demgegenüber sind dem RKI im gesamten Zeitraum insgesamt „nur 22 validierte COVID19-Todesfälle bei unter 20-Jährigen übermittelt worden. Diese Kinder und Jugendlichen waren zwischen 0 und 19 Jahre alt, bei allen 17 Fällen mit Angaben hierzu, sind Vorerkrankungen bekannt“ [4]. Selbst im schlimmsten Fall, nämlich dem, dass die restlichen 5 Todesfälle unter 20 Jahren nicht mit einschlägigen Vorerkrankungen in Zusammenhang gebracht werden könnten, ergäbe sich rechnerisch weniger als 1 Todesfall pro 3 Millionen Kinder und Jugendliche in Deutschland. Und das bei inzwischen 587.927 dem RKI gemeldeten Infektionen in dieser Altersgruppe und einer mutmaßlichen Dunkelziffer von etwa 1:3 bis 1:4 [5]. Zum Vergleich: Allein in den 12 Monaten des Jahres 2019 starben 229 Kinder und Jugendliche unter 20 Jahren im Straßenverkehr! Umgekehrt unken Gesundheitsminister und RKI schon jetzt, dass Maskenpflicht und weitere Restriktionen wohl über das Ende 2021 hinaus in den Schulen unvermeidbar seien. Einspruch! Die Pandemiestatistiken in Deutschland und anderen Län-
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Prof. Dr. med. Karl-Ludwig Resch
dern, namentlich die Altersverteilung der Belegung von Intensivstationen und die letalen Ausgänge, sind ein nicht weg zu diskutierender, schlagender Beweis, dass die Maßnahmen in den Schulen nicht die Kinder selbst (sieht man von inzwischen gut definierbaren Vorerkrankungen ab), sondern ausschließlich Eltern und andere Erwachsene vor einer Infektion und dem konsekutiven Risiko eines schweren Verlaufs schützen konnten. Dies war vor dem Hintergrund, dass es für weite Teile der (älteren) Bevölkerung keine andere, ähnlich wirksame Schutzmaßnahme gab und die Gefahr lebensbedrohlicher Nebenwirkungen des Tragens von Masken im Klassenzimmer wenig real erscheint, vielleicht noch tolerabel. Kinder nach den Sommerferien, wenn absehbar jedem Bürger ein Impfangebot gemacht werden konnte, entsprechende Restriktionen auch nur anzudrohen, ist unverantwortlich, ja ein ethisch-moralischer Skandal. Es hat sich ja inzwischen wohl herumgesprochen, dass jeder, der sich © VERLAG PERFUSION GMBH
INHALT
66
aus eigener, freier Entscheidung nicht impfen lassen möchte, sich perspektivisch mit SARS-CoV-2 infizieren wird. Masken im Klassenzimmer etc. würden da grundsätzlich nichts ändern, sondern bestenfalls dazu beitragen, dass die „Durchseuchung der Verweigerer“ etwas länger dauert. Dafür Kinder und Jugendliche pauschal „bluten“ zu lassen, ist keine Lösung. Wohl aber, ab Herbst überall (aber wirklich überall!) in öffentlich zugänglichen Räumlichkeiten und lückenlos „geimpft, genesen oder getestet“ zu fordern für alle Personen, die älter als z.B. 30 Jahre sind und keines der gut definierbaren „medizinischen Ausschlusskriterien“ für eine Impfung amtsärztlich bestätigt vorweisen können. Die genaue Altersgrenze muss sich dabei natürlich an der Nutzen-Risiko-Bewertung der Impfung auf Basis aller verfügbaren Daten orientieren. Karl-Ludwig Resch, Nürnberg
ORIGINALARBEIT Inanspruchnahme osteopathischer Medizin bei chronisch und Langzeit-Erkrankten André-Michael Beer, Pascal Kiseier, Hanna Weber
68
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS Exzellente Sicht auf die Mukosa – eine unabdingbare Voraussetzung für die Darmkrebsvorsorge
76
Therapie der Plaque-Psoriasis: Secukinumab-Fertigpen überzeugt durch Wirksamkeit und Patientenadhärenz 79 Morbus Parkinson: Rechtzeitig Chancen der Add-on-Therapie mit Safinamid nutzen
82
INTERVIEW Asthma-Therapie in Zeiten von COVID-19: Mit fixer Dreierkombination zu einer besseren Therapiekontrolle Interview mit Herrn Dr. Justus de Zeeuw, Köln
Quellen 1 Lavine JS et al. Vaccinating children against SARS-CoV-2. Hard to justify right now for most children in most countries. BMJ 2021;373:n1197 2 Frenck RW Jr et al. Safety, Immunogenicity, and Efficacy of the BNT162b2 Covid-19 Vaccine in Adolescents. N Engl J Med 2021 May 27; doi: 10.1056/NEJMoa2107456 3 Resch KL. Wer schützt die Alten vor der Stiko? https://www.aerzteblatt.de/ forum/140666#entry140666 4 https://www.rki.de/DE/Content/InfAZ/N/ Neuartiges_Coronavirus/Situationsberichte/Jun_2021/2021-06-22-de.pdf?__ blob=publicationFile 5 Hippich M et al. A public health antibody screening indicates a marked increase of SARS-CoV-2 exposure rate in children during the second wave. Med (N Y). 2021;2:571-572
83
NEUE UND BEWÄHRTE ARZNEIMITTEL Isatuximab in zweiter Indikation zur Behandlung des rezidivierten multiplen Myeloms zugelassen
86
Guselkumab – ein selektiver Interleukin-23-Hemmer zur Behandlung der aktiven Psoriasis-Arthritis
88
Kinasehemmer Pemigatinib – die erste Option zur Zweitlinienbehandlung des Gallengangkarzinoms
90
RUBRIKEN Wissenswertes 74, 78, 80, 92, 95 Kongresse 93
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
© VERLAG PERFUSION GMBH
J E TZ T ZU G E L A S S E N : SA R C LI SA ® + Kd
NEU – in der 2L beim rezidivierten Multiplen Myelom
IN DER TIEFE LIEGT DIE KRAFT Tiefes Ansprechen und kurze Infusionszeiten in der IKEMA-Studie mit SARCLISA® + Kd1 nur
effektiv bis zu
46
%
CR-Rate möglich*,1
30%
MRDNegativität#,1
Infusionszeit verkürzbar auf bis zu
75 Min.+
,1
SARCLISA® + Kd wird in den aktuellen EHA-ESMO Leitlinien mit der höchsten Evidenzstufe (I,A) innerhalb der Zweitlinien-Therapie empfohlen.2
* vs. 28 % mit Kd allein; 39,7 % CR-Rate mit SARCLISA® + Kd vor Bereinigung der Interferenz von SARCLISA® mit der Bestimmung des kompletten Ansprechens. In massenspektrometrischer Untersuchung zeigten 15 der Non-CR-Patienten laut IRC kein nachweisbares residuales Myelom M-Protein. Unter diesen 15 Patienten hatten 11 Patienten < 5 % Plasmazellen im Knochenmark. Dies deutet darauf hin, dass 11 (6,1 %) weitere der 179 mit SARCLISA® + Kd behandelten Patienten eine CR als bestes Ansprechen erzielt haben könnten, was eine mögliche CR-Rate von 45,8 % bedeuten würde.1 # vs. 13 % mit Kd allein; Intention-To-Treat-Population, Next-Generation-Sequenzierung, Sensitivität 10–5. + Ab der 3. Infusion – eine schrittweise Erhöhung der Infusionsgeschwindigkeit sollte nur nach Ausbleiben von infusionsbedingten Reaktionen in Betracht gezogen werden. CR = komplettes Ansprechen; EHA = European Hematology Association; ESMO = European Society for Medical Oncology; IRC = Independent Response Committee; Kd = Carfilzomib und Dexamethason; MRD = minimale Resterkrankung 1. Fachinformation SARCLISA® (Stand: April 2021). 2. Dimopoulos MA, et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Mar; 32(3): 309–322. Sarclisa 20 mg/ml Konzentrat zur Herstellung einer Infusionslösung. Wirkstoffe: Isatuximab. Zusammens.: Arzneil. wirks. Bestandt.: 1 Durchstechfl. m. 5/25 ml Konzentrat enth. 100/500 mg Isatuximab, entspr. 20 mg/ml. Sonst. Bestandt.: Sucrose, Histidinhydrochlorid-Monohydrat, Histidin, Polysorbat 80, Wasser f. Injektionszwecke. Anw.-geb.: In Kombination m. Pomalidomid u. Dexamethason z. Behandl. d. rezidivierten u. refraktären Multiplen Myeloms b. Erwachsenen, d. mind. 2 vorausgegangene Ther., darunter Lenalidomid u. e. Proteasom-Inhibitor, erhalten haben u. unter d. letzten Ther. e. Krankheitsprogression zeigten. In Kombination m. Carfilzomib u. Dexamethason z. Behandl. des Multiplen Myeloms b. Erwachsenen, d. mind. 1 vorausgegangene Ther. erhalten haben. Gegenanz.:. Überempfindlichk. ggü. d. Wirkstoff od. e. d. sonst. Bestandt. Warnhinw. u. Vorsichtsm.: Nicht schütteln. Nebenw. Isatuximab m. Pomalidomid: Infekt. u. parasit. Erkr.: Sehr häufig: Pneumonie, Infekt. d. ob. Atemw., Bronchitis. Gutart., bösart. u. unspez. Neubild.: Häufig: Plattenepithel-Ca d. Haut. Blut u. Lymphsyst.: Sehr häufig: Neutropenie, febrile Neutropenie. Stoffw. u. Ernähr.-stör.: Häufig: vermind. Appetit. Herz: Häufig: Vorhofflimmern. Atemw., Brustr., Mediast.: Sehr häufig: Dyspnoe. GIT: Sehr häufig: Diarrhö, Übelk., Erbrechen. Untersuchungen: Häufig: Gewichtsabnahme. Verletz., Vergift. u. durch Eingriffe bedingte Komplikat.: Sehr häufig: infusionsbedingte Reaktion. Nebenw. Isatuximab m. Carfilzomib: Infekt. u. parasit. Erkr.: Sehr häufig: Pneumonie, Infekt. d. ob. Atemw., Bronchitis. Gefäßerkr.: Sehr häufig: Hypertonie. Gutart., bösart. u. unspez. Neubild.: Häufig: Hautkrebs, solide Tumore außer Hautkrebs. Blut u. Lymphsyst.: Häufig: Neutropenie. Atemw., Brustr., Mediast.: Sehr häufig: Dyspnoe, Husten. GIT: Sehr häufig: Diarrhö, Erbrechen. Allg. Erkr. u. Beschw. am Verabreichungsort: Sehr häufig: Fatigue. Verletz., Vergift. u. durch Eingriffe bedingte Komplikat.: Sehr häufig: infusionsbedingte Reaktion. Verschreibungspflichtig. Sanofi-aventis groupe, 54 rue La Boétie, 75008 Paris, Frankreich Stand der Information: April 2021 Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
Mit wegweisenden Therapien komplexen Erkrankungen begegnen.
2103_ISA_C_MAT-DE-2102281v1.0 - 05/2021
SARCLISA® ist in Kombination mit Carfilzomib und Dexamethason zur Behandlung des Multiplen Myeloms bei Erwachsenen indiziert, die mindestens eine vorausgegangene Therapie erhalten haben.
ORIGINALARBEIT
68
ZUSAMMENFASSUNG Hintergrund: Die Osteopathie gilt in Deutschland noch immer als therapeutisches Randgebiet. In der Klinik für Naturheilkunde in Hattingen-Blankenstein, in der vor allem chronisch- und langzeiterkrankte Patienten stationär behandelt werden, zeigt sich, dass viele der Behandelten im Vorfeld des Aufenthalts bereits mit Osteopathie in Kontakt gekommen sind. Um diese Beobachtung zu verifizieren, wurden über einen Zeitraum von 7 Monaten systematisch Daten hierzu erfasst. Methoden: Im Zeitraum von Mai bis Dezember 2020 wurden 158 stationäre Patienten zur Inanspruchnahme osteopathischer Therapien im ambulanten Bereich befragt. Fragen hierzu wurden bei der physiotherapeutischen Befunderhebung gestellt. Dabei wurden vor allem Informationen zur ambulanten Vorbehandlung mit Osteopathie, zu den Indikationen sowie der Beschwerdeeinschätzung erhoben. Ergebnisse: Bei den 158 teilnehmenden Patienten (Durchschnittsalter: 55 Jahre; 150 Frauen, 8 Männer) stellten Krankheiten des Muskel-Skelett-Systems die primäre Behandlungsindikation dar (n = 146). Mehr als 2 Drittel der behandelten Patienten waren länger als 1 Jahr erkrankt (n = 129), davon wiederum 86 seit mehr als 5 Jahren. Die physiotherapeutischen Maßnahmen führten bei fast allen Patienten zur Verbesserung des Zustands (n = 117, rund 89 %). Ein Drittel der Patienten (n = 49, 31 %) gab an, bereits ambulante Vorerfahrungen mit Osteopathie gemacht zu haben. 83 (52,5 %) der behandelten Patienten kamen während des stationären Aufenthalts zum ersten Mal mit Osteopathie in
Inanspruchnahme osteopathischer Medizin bei chronisch und LangzeitErkrankten André-Michael Beer, Pascal Kiseier, Hanna Weber Klinik für Naturheilkunde, Klinik Blankenstein, Hattingen
D
ie osteopathische Medizin und ihre Reputation in Deutschland durchlaufen derzeit eine langsame Entwicklung [1]. Ursprünglich in den Nachbarländern Frankreich und Belgien verwurzelt, begann sich die Osteopathie als medizinischer Behandlungsansatz vor rund 25 Jahren auch in Deutschland zu etablieren. Lange Zeit in Eigenleistung durch die Betroffenen finanziert, werden die Kosten osteopathischer Behandlungen seit Kurzem zumindest in begrenztem Umfang, jedoch zu heterogenen Bedingungen, auch von den gesetzlichen Krankenkassen übernommen. Wenngleich die Osteopathie also eine Integration in bestehende medizinische Infrastrukturen erfährt, ist die osteopathische Medizin in Deutschland im Gegensatz zu anderen westlichen Ländern wie Frankreich, England, Australien oder Neuseeland dadurch charakterisiert, dass es hierzulande kein offiziell anerkanntes und einheitliches Berufsbild des Osteopathen gibt. Wo in anderen Ländern also uniforme Ausbildungsstrukturen der Osteopathie eine Koexistenz mit anderen Heilberufen in der medizinischen Versorgung der Bevölkerung ermöglichen, herrscht
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
in Deutschland eine Pluralität an sogenannten Bildungsgängen, die weder vereinheitlicht noch verstaatlicht und damit in ihrer Qualität nicht kontrolliert sind. Auch eine akademische Verankerung der Osteopathie in Deutschland oder innerhalb Europas hat sich nicht etablieren können. Hinsichtlich ihrer wissenschaftlich untersuchten Evidenz ist die osteopathische Medizin anderen universitär verankerten Disziplinen gegenüber in Verzug [1]. Aus Sicht des Patienten besteht somit in Deutschland keinerlei verlässliche Sicherheit bezüglich der Qualifikation des Behandlers. Auch die Initiative und das Engagement vorwiegend praktisch ausgebildeter „Osteopathen“ in Deutschland [2] und anderswo [3] können nicht darüber hinwegtäuschen, dass klinische und Grundlagenforschung bislang nicht mit den klassischen Bereichen der universitär verankerten Medizin Schritt halten konnten. Eine wissenschaftliche Auseinandersetzung mit der Osteopathie durch die Bundesärztekammer erfolgte erstmals im Jahr 2009, wobei auch standespolitische Implikationen mit in den Blick genommen wurden [4, 5]. Initiiert © VERLAG PERFUSION GMBH
ORIGINALARBEIT
durch die A.T. Still University, Gründungsuniversität der Osteopathie in den USA, wird seit etwa dieser Zeit die Akademisierung der osteopathischen Medizin in Europa gefördert. In Kooperation mit dem Verband der Osteopathen Deutschlands (VOD) ist aus diesen Strukturen in jüngerer Vergangenheit das Forschungsnetzwerk DOTouch.Net hervorgegangen, für das der VOD federführend mit der Initiierung, der Organisation, der Durchführung von Forschungsprojekten und Mitgliedertreffen sowie der Erhebung von Daten beauftragt ist [6, 7]. Resch et al. haben äquivalent eine Erhebung für Deutschland vorgenommen, deren Ergebnisse darauf hindeuten, dass osteopathische Maßnahmen bei den Betroffenen gleichermaßen beliebt und hoch wirksam sind [8]. Seitdem haben sich weitere Untersuchungen zum Ziel gesetzt, insbesondere für Deutschland gültige empirische Aussagen zur Inanspruchnahme osteopathischer Leistungen zu generieren, um daraus Anforderungen an Bedarfe und Qualität abzuleiten. Auch die Diskussion der Etablierung eines eigenständigen und durch qualifizierte Ausbildung vereinheitlichten Berufsbildes des Osteopathen ist in diesem Zusammenhang von Relevanz. Osteopathische Verfahren im Allgemeinen nehmen die interdisziplinäre Anwendung ihrer diagnostischen und therapeutischen Techniken zur Erkennung und der Behandlung gestörter Funktionen des Bewegungssystems und der davon ausgehenden Beschwerden in den Fokus. Osteopathische Verfahren werden auch innerhalb der stationären naturheilkundlichen Komplexbehandlung in der Klinik Blankenstein, Hattingen angeboten. Dazu gehören insbesondere
kraniosakrale Techniken. Sie bilden neben den konventionellen krankengymnastischen Interventionen (manuelle Therapie, funktionelle Weichteiltechniken/FWTT, aktive Bewegungsübungen, propriozeptive neuromuskuläre Fazilitation/PNF, Extensionsbehandlung/ Schlingentisch, Bindegewebsmassage) einen elementaren Baustein der Behandlung. Chronisch- und langzeiterkrankte Patienten [9, 10, 11] gaben immer wieder an, während ihrer mitunter jahrelangen „Patientenkarriere“ bereits mit Osteopathie in Kontakt gekommen zu sein. Um diesen Eindruck zu verifizieren, wurde die bereits etablierte physiotherapeutische Befundung genutzt und ausgeweitet. Dadurch konnte auch erfasst werden, ob es durch die jeweilige physiotherapeutische Maßnahme zu einer Änderung der Beschwerdesymptomatik kam. Auch vor dem Hintergrund der aktuellen Diskussionen zur Reputation und der generellen osteopathischen Daseinsberechtigung innerhalb der medizinischen Infrastruktur, primär jedoch zur Verifizierung des Eindrucks, dass viele der hochchronifizierten Patienten bereits Berührungspunkte mit der Osteopathie in der ambulanten Versorgung erfahren hatten, wurden die behandelten Patienten hierzu befragt. Kollektiv und Methodik
Im Zeitraum von Mai 2020 bis Dezember 2020 wurden 158 Patienten (150 weiblich, 8 männlich; Durchschnittsalter 55 Jahre) während ihres stationären Aufenthalts in der Klinik für Naturheilkunde in Hattingen-Blankenstein mittels physiotherapeutischem Befundbogen befragt (Abb. 1). Dabei wurden
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
69
Kontakt, 26 (16,5 %) machten keine Angabe dazu. Diskussion: Ein Drittel der chronisch- und langzeiterkrankten Patienten (31 %) hatte bereits ambulante Vorerfahrungen mit osteopathischen Maßnahmen gemacht. Damit bestätigte sich, dass Patienten im Rahmen ihrer „Patientenkarriere“ die Osteopathie bereits in Anspruch genommen hatten. Weil auf diesem Gebiet eine Qualitätssicherung noch immer nicht sichergestellt ist, ist daher einerseits über die Implementierung der Osteopathie in die bestehende medizinische Versorgung zu diskutieren. Andererseits ergibt sich daraus auch die Forderung nach einer Kon trolle der Ausbildungsqualität mit dem Ergebnis eines eigenständigen Berufsbildes. Schlüsselwörter: Osteopathie, demoskopische Analyse, chronisch Erkrankte, Epidemiologie, Qualitätssicherung, bestehende Versorgungsstruktur
SUMMARY Background: Up to now, osteopathy can be characterised as fringe area in medical treatment. In a hospital specialised in natural therapies located in HattingenBlankenstein („Klinik für Naturheilkunde“), especially long-term and chronically ill patients are treated. Regarding osteopathy’s fringe character, observating that a great amount of the patients nevertheless had made experience with osteopathic treatment appeared even more remarkable. In order to verify this observation, data was recorded systematically during a period of 7 months. © VERLAG PERFUSION GMBH
ORIGINALARBEIT
70
Methods: 158 in-door-patients (female: 150, male: 8; average age: 55 years) were interviewed from May 2020 to December 2020 concerning their treatment indication, their individual suffering and several other anamnestic information at the time of first contact. Up to the time of last contact, the interviewed were questioned with regard to an eventual change in symptoms. In addition, the patients were questioned concerning their prior experience with outdoor osteopathic treatment. Results: Most common treatment indications were diseases of the musculoskeletal system and connective tissue (n = 146). Most of the recipients were suffering from symptoms since several years (n = 129), 86 since more than 5 years. Regardless the technique, physical treatment in general provoked a decrease of physical complaints (n = 117, about 89 %). Although 83 patients (52.5 %) were undergoing osteopathic treatment for the first time, about one third (31 %) had experienced osteopathy before. 26 (16.5 %) did not answer. Discussion: One third of the interviewed were attached to osteopathy in a pre-ward context (31 %). In conclusion, it could be considered that quiet a lot of patients had claimed for osteopathic treatment before. Altogether, the results give valuable suggestions for further research initiatives, and substantiate the necessity of legal recognition of a sensibly tailored profession of osteopaths in Germany, in particular with regard to the patient’s protection. Key words: osteopathy, demoscopic survey, chronic illness, epidemiology, quality assurance, current health care structure
Abbildung 1: Fragebogen.
neben der Erfassung der Behandlungsindikationen in Haupt- und Nebendiagnose auch Angaben zum Behandlungszeitraum und der subjektiven Beschwerdeeinschätzung des Patienten vor und nach Abschluss der physiotherapeutischen Behandlung mittels visueller Analogskala (VAS) erhoben. Außerdem wurden die Patienten nach der Beschwerdedauer, den angewandten physiotherapeutischen Maßnahmen und den Auswirkungen der Behandlung auf Schmerz, Tonus und Bewegung gefragt. Aufgrund der unterschiedlichen Behandlungsindikationen und den daraus auf die Bedürfnisse der Patienten zugeschnittenen therapeutischen Interventionen wurden die Patienten krankengymnastisch entweder mit oder ohne osteopathischen Schwerpunkt behandelt. Bei der konventionellen Krankengymnastik kamen Behandlungen wie manuelle Therapie, funktionelle Weichteiltechniken (FWTT), aktive Bewegungsübungen, proprio-
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
zeptive neuromuskuläre Fazilitation (PNF), Extensionsbehandlung/ Schlingentisch und Bindegewebsmassage zur Anwendung, als osteopathische Maßnahmen im engeren Sinne wurden kraniosakrale Techniken eingesetzt. Jeder Teilnehmer wurde zudem zu seiner ambulanten Vorerfahrung bezüglich einer osteopathischen Behandlung befragt. Bei denjenigen Patienten, die angaben, bereits über eine ambulante Vorerfahrung mit Osteopathie zu verfügen, wurden weitere Angaben zum Behandlungserfolg, zum Behandlungszeitraum und zur durchschnittlichen Anzahl der Behandlungen pro Monat erfragt. Angaben zum behandelten Beschwerdebild und dem möglichen Grund für die Beendigung der Behandlung wurden mittels freier Antwortmöglichkeit erhoben. Die strukturierten Antwortmöglichkeiten des Erhebungsbogens sowie die freien Antwortmöglichkeiten wurden deskriptiv erfasst, wobei © VERLAG PERFUSION GMBH
71
ORIGINALARBEIT
letztere erst als Freitexteingaben übernommen wurden. Nach kompletter Dokumentation wurden diese gesichtet, wo inhaltlich sinnvoll zusammengefasst und schließlich ebenfalls kodiert. Die Freitexteingaben zu den Diagnosen wurden äquivalent zur jeweiligen ICD10-Diagnose kodiert. Die Rohdaten wurden mit Microsoft Excel 2016 erfasst und ausgewertet.
100 90
Beschwerdedauer Insgesamt 129 Patienten gaben an, seit mehr als 1 Jahr an den Beschwerden zu leiden, wobei davon mehr als die Hälfte (n = 86; 54,4 %) wiederum seit mehr als 5 Jahren erkrankt war. 43 Befragte (27,2 %) gaben Beschwerden seit 1 – 5 Jahren an, 26 (16,5 %) seit 6 – 12 Monaten; 3 Patienten (1,9 %) machen keine Angaben dazu (Abb. 2). Osteopathische Behandlungen Von den insgesamt 158 Befragten gab fast ein Drittel (31 %) osteopathische Vorerfahrungen im ambulanten Bereich an, 83 (52,5 %) waren zuvor noch mit keiner osteopathischen Behandlung in Kontakt gekommen, 26 (16,5 %) konnten aufgrund von Verständnisproblemen dazu keine Aussage treffen (Abb. 3). Bewertung der Behandlung Von den Teilnehmern, die bereits im Vorfeld des stationären Aufenthalts mit Osteopathie in Kontakt gekommen waren, bewerteten 10 die prästationäre Behandlung als „sehr gut“, 20 als „gut“, 4 als „wenig hilfreich“ und 3 als „gar nicht
n=86
80 70 60 50 40 30
n=43 n=26
20 10 0
Ergebnisse
keine Angabe: n=3 (1,9%)
16,5%
27,2%
6-12 Monate
1-5 Jahre
54,4% > 5 Jahre
Abbildung 2: Beschwerdedauer. 90
n=83
80 70 60 50
n=49
40
n=26
30 20 10 0
31%
52,5%
16,5%
Ja
Nein
keine Angabe
Abbildung 3: Ambulante osteopathische Vorbehandlungen.
hilfreich“. Eine Person konnte keine Aussage treffen („weiß nicht“), 11 der Befragten mit osteopathischen Vorerfahrungen machten keine Angabe. Behandlungsdauer und -häufigkeit Die meisten der Patienten, die von der Osteopathie profitieren konnten, waren bereits über Jahre (n = 12) bzw. über Monate (n = 10) in Behandlung, 11 gaben hinsichtlich des Behandlungszeitraumes „über Wochen“ an. Der Großteil der Befragten wurde 1 × (n = 17) oder 2 × (n = 9) pro Monat osteopathisch behandelt; eine höhere Behandlungsfrequenz war seltener (3 ×: n = 4, 4 ×: n = 6, 5 ×: n = 1, 6 ×: n = 2).
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Osteopathisch behandelte Krankheitsbilder Ähnlich wie im stationären Setting wurde osteopathische Hilfe vor allem wegen Wirbelsäulenbeschwerden in Anspruch genommen (n = 28), Hüftbeschwerden und Beschwerden aufgrund eines Fibromyalgie-Syndroms waren weniger häufig Grund für eine osteopathische Behandlung (n = 3), ebenso Kniebeschwerden (n = 2). Grund für die Beendigung der Behandlung 14,2 % der Befragten mit ambulanter osteopathischer Vorerfahrung befanden sich sowohl vor als auch nach dem stationären Aufenthalt in der Klinik für Naturheilkunde in einem osteopathischen Behand© VERLAG PERFUSION GMBH
ORIGINALARBEIT
72
lungssetting („Behandlung nicht beendet“, n = 7). 14 Teilnehmer gaben hingegen an, nicht mehr an eine osteopathische Behandlung angebunden zu sein. Als Begründung dafür wurden zeitliche (n = 6) und finanzielle Gründe (n = 1) angeführt, jeweils 2 Teilnehmer gaben an, keine neue Verordnung erhalten bzw. keinen Erfolg gehabt zu haben. Wiederum 2 Befragte konnten so gut von der osteopathischen Behandlung profitieren, dass sie die Maßnahme aufgrund des Behandlungserfolges beenden konnten. 28 der Befragten mit osteopathischer Vorerfahrung machten aufgrund von sprachlichen bzw. anderen Verständnisproblemen keine Angaben. Physiotherapeutische Maßnahmen/Behandlungsarten bei muskuloskeletalen Erkrankungen Bei 112 Patienten (70,9 %) wurden muskuloskeletale Erkrankungen mit konventioneller Krankengymnastik behandelt, bei 46 (29,1 %) mit osteopathischem Schwerpunkt. Indikationen für die Behandlung 146 Patienten wurden wegen Erkrankungen des Muskel-SkelettSystems und Bindegewebes („MDiagnosen“) behandelt, 8 Patienten wegen Erkrankungen des Nervensystems und 4 Patienten aufgrund von sonstigen Erkrankungen. Unter den M-Diagnosen dominierten als primäre Behandlungsindikation Rücken- bzw. Wirbelsäulenbeschwerden (n = 55) sowie Krankheiten der Weichteilgewebe (n = 37), etwas seltener waren Arthropathien (n = 31); sonstige Beschwerden hatten 23 Patienten (Abb. 4).
60
n=55
50 40
n=37 n=31
30
n=23
20 10 0 Arthropathien Krankheiten der Wirbelsäule und des Rückens Krankheiten der Weichteilgewebe Sonstiges
Abbildung 4: Diagnoseverteilung innerhalb der M-Diagnosen.
Behandlungsentwicklung während des stationären Verlaufs Die meisten Patienten (n = 46) gaben eine Verbesserung der Beschwerden im Sinne eines Rückgangs um 2 bzw. 3 Punkte (n = 31) auf der visuellen Analogskala (VAS) an. 19 Befragte berichteten eine Beschwerdereduktion um 1 VAS-Punkt, bei 14 verringerten sich die Beschwerden um 4 VAS-Punkte und bei 6 um 5 VASPunkte. Der maximale Beschwerderückgang wurde von einem Teilnehmer mit minus 7 VAS-Punkten angegeben. Der geringste Anteil der Behandelten (n = 14) konnte keine Veränderung der Symptomatik feststellen, bei 27 Teilnehmern waren Angaben unvollständig oder aus Verständnisgründen nicht erfolgt. Vergleicht man den Behandlungsverlauf im Hinblick auf die Veränderung der Beschwerden unter den physiotherapeutischen und osteopathischen Maßnahmen, wird ersichtlich, dass sich mit beiden Techniken ähnlich gute Besserungen erzielen ließen. Während krankengymnastische Maßnahmen öfter zu einer Verringerung der Symptome um 1 (n = 16) bzw.
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
2 (n = 33) VAS-Punkte führten, hatten osteopathische Techniken im Verhältnis zur Größe der Patientengruppe häufiger positivere Auswirkungen auf die Beschwerdebewertung. So sind höhere Einschätzungen des Beschwerderückgangs ab einer Reduktion um 3 VAS-Punkte mehrheitlich den osteopathischen Techniken zuzuschreiben (n = 16). Verbesserungen der Symptome um 4 bzw. 5 Punkte auf der VAS wurden mit jeweils 7 bzw. 3 Nennungen gleich häufig genannt. Unter Bezug auf die Größe der jeweiligen Patientengruppe ergibt sich, dass sich bei 15 % der Befragten, die osteopathisch behandelt wurden, die Beschwerden um 4 VAS-Punkte reduzierten, bei den konventionell krankengymnastisch Behandelten lag der Anteil hingegen bei 8 %. Ähnliches zeigte sich bei der Beschwerdeverringerung um 5 VAS-Punkte: bei 6 % mit osteopathischem Behandlungsschwerpunkt versus 3 % mit konventioneller Krankengymnastik. Auch die größte genannte Verbesserung der Symptome, eine Reduktion um 7 Punkte auf der VAS-Skala, wurde durch den Einsatz osteopathischer Techniken erzielt. © VERLAG PERFUSION GMBH
ORIGINALARBEIT
Insgesamt scheinen osteopathische Techniken wirksamer zu sein als krankengymnastische Maßnahmen. Während 11 Befragte, die Krankengymnastik erhalten hatten, keine Beschwerdeveränderung angaben, berichteten nur 3 Befragte Ähnliches in Bezug auf die osteopathischen Techniken. Diskussion
In der Klinik für Naturheilkunde in Hattingen-Blankenstein werden chronische Patienten behandelt, die bereits die üblichen Angebote des Gesundheitssystems in Anspruch genommen haben [9, 10, 11]. Ziel unserer Erhebung war es, herauszufinden, inwieweit osteopathische Behandlungen, die immer noch als Randgebiet der Medizin gelten, von dieser Klientel bereits ambulant nachgefragt wurden. Die Ergebnisse unserer physiotherapeutischen Patientenbefundung zeigen den Nutzen osteopathischer Maßnahmen in der stationären und eine hohe Nachfrage nach osteopathischen Behandlungsmaßnahmen in der ambulanten osteopathischen Versorgung. Daraus lässt sich schließen, dass Patienten mit chronischen Schmerzen Ärzte und Therapeuten brauchen, die mit der osteopathischen Behandlung nicht nur praktische Erfahrung haben, sondern zusätzlich auch über eine umfassende theoretische medizinische Ausbildung in diesem Bereich verfügen, wie das zum Beispiel in der Physiotherapie und anderen medizinischen Bereichen üblich ist. Fakt ist aber, dass bis heute die Ausbildung zum Osteopathen nicht einheitlich geregelt und staatlich anerkannt ist. Wenngleich für die stationäre Versorgung gezeigt werden konnte, dass der Einsatz osteopathischer
Techniken insbesondere für Patienten mit stark chronifizierten Beschwerden hilfreich ist [8], konnten wir feststellen, dass ein Großteil der Befragten zuvor nicht mit osteopathischen Maßnahmen in Kontakt gekommen ist. Aufgrund der Tatsache, dass die meisten der befragten Patienten bereits seit Jahren unter chronischen Beschwerden leiden, weist dies auf eine Versorgungslücke im deutschen Gesundheitswesen hin. Bis heute gibt es in der Osteopathie keine einheitliche Ausbildung, außerdem ist sie als Berufsbild nicht gesetzlich geschützt. Daher bestehen leider häufig große qualitative Unterschiede hinsichtlich der medizinischen und therapeutischen Kenntnisse der Osteopathen und der Patient ist nicht vor einer Fehlbehandlung geschützt. Für chronisch Erkrankte ist das besonders fatal, weil jeder frustrane Therapieversuch die Chronifizierung befördert. Erhebliche Differenzen zeigen sich außerdem bei der Dauer der osteopathischen Behandlungen und deren Abrechnung – so, wie es bislang keine einheitliche Ausbildungsordnung gibt, so fehlt es auch an verbindlichen Behandlungsleitlinien und Abrechnungskatalogen. Auch die Fort- und Weiterbildungsmöglichkeiten zum Osteopathen sind uneinheitlich: Zum einen gibt es Osteopathen, die beispielsweise als staatlich anerkannte Physiotherapeuten eine 5-jährige Osteopathieweiterbildung absolviert haben und dann als Osteopath tätig sind. Zum anderen gibt es aber auch Osteopathen, die lediglich in einer einwöchigen Fortbildung die entsprechenden Behandlungen kennenlernen. Sie alle können aber nach derzeitigem Stand in Deutschland Osteopathie am Patienten durchführen.
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
73
Im Sinne einer Nutzen-Risiko-Bewertung der osteopathischen Tätigkeit lässt sich daher konstatieren: Eine Chance auf Beschwerdebesserung für Langzeit-Erkrankte besteht unter den derzeitigen Bedingungen nicht, da die Osteopathie für viele Patienten schwer zugänglich ist. Für viele potenzielle Profiteure scheint darüber hinaus die Finanzierung über die gesetzlichen Leistungen hinaus ein Hindernis darzustellen. Auch wenn aufgrund dieser Piloterhebung keine generalisierte Aussage über den Nutzen von osteopathischen Anwendungen getroffen werden kann, lassen sich die Ergebnisse mit anderen Untersuchungen vergleichen [1, 8]. Jüngere Studien weisen zudem darauf hin, dass das Wirkspektrum der Osteopathie sich nicht nur auf die Behandlung Erwachsener beschränkt, sondern bereits im Säuglings- und Kleinkindalter bei heterogenen Beschwerdebildern grundlegende Verbesserungen erzielt werden konnten [12]. Vieles spricht dafür, die formulierte Not der Patienten als Notwendigkeit zu verstehen, die Osteopathie künftig in die Infrastruktur der medizinischen Grundversorgung zu implementieren. Die Kostenübernahme durch die gesetzlichen Krankenkassen kann dabei nur einen ersten Schritt darstellen. Neben dem Bedarf an empirischen Basiserhebungen [1], die – auch auf Grundlage größerer Datensätze – das Potenzial der osteopathischen Medizin wissenschaftlich eruieren, bedarf es gleichzeitig einer Reform zur Etablierung adäquater, staatlich gesicherter Ausbildungsstandards, die dem Anspruch nach Qualität und Professionalität gerecht werden. Eine adäquate Definition des Berufsbildes „Osteopath“ steht nicht zuletzt © VERLAG PERFUSION GMBH
74
ORIGINALARBEIT / WISSENSWERTES
auch im Interesse des gesundheitlichen Verbraucherschutzes. Literatur 1 Steel A, Sundberg T, Reid R et al. Osteopathic manipulative treatment: a systematic review and critical appraisal of comparative effectiveness and health economics research. Musculoskelet Sci Pract 2017;27:165-175 2 http://www.german-afo.de/research.html 3 httw://www.osteopathic-research.htm 4 Wissenschaftlicher Beirat der Bundesärztekammer: Wissenschaftliche Bewertung osteopathischer Verfahren. Dtsch Ärztebl 2009;106:A-2325-A-2334 5 Resch KL. Gutachten zur Fragestellung „Osteopathie und Evidenz“. 2009. Im Internet: http://www.bundesaerztekammer. de/fileadmin/user_upload/downloads/ StellOVLiteraturgutachtenResch.pdf 6 http://www.do-touch.net 7 http://www.osteopathie.de/home-do_ touch.net 8 Resch KL: Osteopathische Medizin – Inanspruchnahme von Gesundheitseinrichtungen in Deutschland: eine prospektive Patientenbefragung, Journal Pharmakol Ther 2018;27:3-13 9 Beer AM, Ostermann T, Matthiessen PF. Evaluation stationärer Naturheilkunde. Das Blankensteiner Modell. Teil I: Patientenklientel und therapeutische Konzepte. Forsch Komplementärmed 2001;8:613 10 Ostermann T, Beer AM, Matthiessen PF: Evaluation stationärer Naturheilkunde. Das Blakensteiner Modell. Teil II: Effektstärken und Gesundheitsstatus der Patienten im zeitlichen Verlauf. Forsch Komplementärmed 202;9:269-276 11 Wiebelitz KR, Teske W, Henke T et al. Naturheilkundliche und orthopädische stationäre Behandlung bei chronischen Rückenschmerzen. Eine Vergleichsstudie. MMW Fortschr Med Originalia 2011; 153:47-55 12 Schwerla F, Daakeb B, Moeckelc E et al. Osteopathic treatment of infants in their first year of life: a prospective multicenter observational study (OSTINF Study). Complement Med Res 2021 Feb 18;1-12. doi: 10.1159/000514413 (online ahead of print)
Für die Verfasser: Prof. Dr. med. André-Michael Beer Direktor der Klinik für Naturheilkunde, Klinik Blankenstein, Hattingen, Im Vogelsang 5–11 45527 Hattingen E-Mail:andre.beer@klinikum-bochum.de
Ofatumumab – eine neue Erstlinientherapie für Erwachsene mit aktiver schubförmiger MS Vor Kurzem hat die Europäische Kommission Ofatumumab (Kesimpta®) zur Behandlung von erwachsenen Patienten mit schubförmig verlaufender Multipler Sklerose mit aktiver Erkrankung (definiert durch klinischen Befund oder Bildgebung) zugelassen. Ofatumumab ist damit der erste und einzige zielgerichtete Anti-CD20Antikörper, der durch eine monatliche (ab Woche 4*) subkutane Applikation mit dem Sensoready® Fertigpen von den Patienten selbst verabreicht werden kann. Der vollständig humane Antikörper bindet zielgerichtet an das CD20-Molekül auf der Oberfläche von B-Zellen, wobei es auf einem anderen Epitop andockt als andere Anti-CD20-Antikörper und dadurch eine B-Zell-Lyse und Depletion induziert. Der selektive Wirkmechanismus und die subkutane Verabreichung ermöglichen eine spezifische Wirkung auf die B-Zellen, die vor allem im peripheren lymphatischen Gewebe angesiedelt sind und eine wichtige Rolle in der MS-Pathogenese spielen. Überlegene Wirksamkeit gegenüber Teriflunomid
Die EU-Zulassung von Ofatumumab basiert auf den beiden identischen, doppelblinden, randomisierten Phase-III-Studien ASCLEPIOS I und II, in denen die Sicherheit und Wirksamkeit von Ofatumumab 20 mg (monatliche
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
[ab Woche 4] subkutane Injektionen) im Vergleich zu Teriflunomid 14 mg (1 × täglich orale Tabletten) bei Erwachsenen mit einer bestätigten aktiven schubförmigen MS untersucht wurden. Eingeschlossen in die Studien waren 1.882 Patienten im Alter zwischen 18 und 55 Jahren mit einem EDSS-Wert (Expanded Disability Status Scale) zwischen 0 und 5,5. Die Behandlung mit Ofatumumab bewirkte im Vergleich zu Teriflunomid: • eine signifikante Reduktion der jährlichen Schubrate (ARR, primärer Endpunkt) um 51 % bzw. 59 % (p < 0,001 in beiden Studien), • eine signifikante Reduktion der bestätigten Behinderungsprogression nach 3 Monaten um 34,4 % (p = 0,002) sowie • eine signifikante Reduktion der Anzahl der Gadoliniumanreichernden (Gd+) T1-Läsionen um bis zu 94 % bzw. 98 % (p < 0,001) und neuer oder sich vergrößernder T2-Läsionen um bis zu 82 % bzw. 85 % (p < 0,001). Die jährliche Rate des Hirnvolumenverlustes unterschied sich nicht signifikant zwischen der Ofatumumab- und der TeriflunomidGruppe. Beide Medikationen zeigten ein vergleichbares Sicherheits- und Verträglichkeitsprofil. Die wichtigsten und am häufigsten gemeldeten Nebenwirkungen unter Ofatumumab waren Infektionen der oberen Atemwege (39,4 %), systemische injektionsbedingte Reaktionen (20,6 %), Reaktionen an der Injektionsstelle (10,9 %) und Harnwegsinfektionen (11,9 %). F. S. * Nach einer Initialdosierung in den Wochen 0, 1 und 2 (bei jeder Applikation 20 mg). © VERLAG PERFUSION GMBH
Richtig zupacken.
FÜR MICH EIN TRIUMPH.
#
IM LEBEN.
NEU
PsA b ei
*
1. reiner IL-23 Inhibitor bei Psoriasis-Arthritis* # Signifikante Überlegenheit vs. Placebo in Bezug auf ACR20 (64% vs. 33%, p<0,0001; Non Responder Imputation) nach 24 Wochen in der 8-Wochen-Dosierung (n=248) in bionaiven Patienten mit aktiver PsA.2 * TREMFYA® ist indiziert: 1) allein oder in Kombination mit MTX für die Behandlung der aktiven Psoriasis-Arthritis bei erwachsenen Patienten, wenn das Ansprechen auf eine vorherige nicht-biologische krankheitsmodifizierende antirheumatische (DMARD-)Therapie unzureichend gewesen ist oder nicht vertragen wurde; 2) für erwachsene Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Therapie in Frage kommen.1 1. Aktuelle Fachinformation TREMFYA®.
2. Mease P et al. The Lancet 2020; https://doi.org/10.1016/S0140-6736(20)30263-4 (Supplementary)
TREMFYA® 100 mg Injektionslösung in einer Fertigspritze/ in einem Fertigpen. Wirkstoff: Guselkumab. Zusammensetz.: Fertigspritze/Fertigpen enth. 100 mg Guselkumab. Sonst. Bestandt.: Histidin, Histidinmonohydrochlorid-Monohydrat, Polysorbat 80, Sucrose, Wasser f. Injektionszw.. Anw.geb.: Für d. Bhdlg. erw. Pat. m. mittelschwerer bis schwerer Plaque-Psoriasis indiziert, d. für e. syst. Therapie in Frage kommen. Als Monotherapie od. in Komb. m. Methotrexat für d. Bhdlg. erw. Pat. m. Psoriasis-Arthritis indiziert, d. auf e. vorherige nicht-biolog. kranheitsmodifiz. antirheumat. (DMARD)-Therapie unzureich. angesprochen od. diese nicht vertragen haben. Gegenanz.: Schwerwieg. Überempfindl. gg. Guselkumab od. e. d. sonst. Bestandt., klin. relev. aktive Infektionen (einschl. aktive Tuberkulose), Schwangersch., Stillzeit. Bes. Warnhinw. u. Vorsichtsmaßn.: Um d. Rückverfolgbark. b. biolog. Arzneim. zu verbessern, sollten Name u. Ch.-Bez. d. verabreich. Prod. deutl. protokoll. werden. Vors. b. Infektionen, Tuberkulose, Impfungen (vor Anw. v. Lebendimpfst. muss d. Bhdlg. m. Tremfya nach d. letzt. Gabe f. mind. 12 Wo. ausgesetzt werden). B. Erhöh. v. Leberenzymwerten (ALT/AST) u. Verdacht auf arzneimittelinduz. Leberschädig. sollte d. Bhdlg. vorüberg. unterbr. werden. B. schwerwieg. Überempfindl.reakt. sollte d. Anw. v. Tremfya unverzügl. abgebrochen u. e. geeign. Bhdlg. eingel. werden. Frauen im gebärfäh. Alter sollen währ. u. f. mind. 12 Wo. nach d. Bhdlg. e. zuverläss. Verhütgs.meth. anw.. Arzneim. f. Kdr. unzugängl. aufbewahren. Nebenwirk.: Sehr häufig (≥ 1/10), Häufig (≥ 1/100 bis < 1/10), Gelegentlich (≥ 1/1.000 bis < 1/100). Sehr häufig: Atemwegsinfekt.. Häufig: Kopfschm., Diarrhoe, Arthralgie, Reakt. a. d. Injektionsst., Transamin. erhöht. Gelegentlich: Herpes-simpl-Infekt., Tinea-Infekt., Gastroenteritis, Überempfindl.reakt., Anaphylaxie, Urtikaria, Hautausschlag, Neutrophilenzahl erniedr.. Verschreibungspflichtig. Pharmazeut. Unternehmer: JANSSEN-CILAG International NV, Turnhoutseweg 30, B-2340 Beerse, Belgien. Örtl. Vertreter für Deutschland: Janssen-Cilag GmbH, Johnson & Johnson Platz 1, D-41470 Neuss. Stand d. Inform.: 12/2020.
www.tremfya.de
CP-180011
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Daher ist es wichtig, jeden Verdacht auf Nebenwirkungen in Verbindung mit diesem Arzneimittel zu melden.
76
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Exzellente Sicht auf die Mukosa – eine unabdingbare Voraussetzung für die Darmkrebsvorsorge
I
n Deutschland erkranken jährlich rund 37.000 Menschen an Kolonkarzinomen, den häufigsten der malignen Darmtumoren [1]. Darmkrebs gehört zu den Krebsarten, die sich häufig und unbemerkt über Jahre hinweg aus benignen Vorstufen (den Adenomen) zu malignen Adenokarzinomen entwickeln. Daher können die Vorstufen im Rahmen einer Untersuchung zur Früherkennung rechtzeitig identifiziert und der Ausprägung des Kolonkarzinoms vorgebeugt werden. Die Darmkrebsvorsorge wird von den gesetzlichen Krankenkassen mit einem umfassenden Screening-Angebot unterstützt. Dieses beinhaltet einen jährlichen Test auf fäkales okkultes Blut ab dem 50. Lebensjahr sowie eine Koloskopie ab 50 Jahren. Sofern der erste Befund unauffällig war, kann die Koloskopie nach 10 Jahren wiederholt werden [2]. Bei Patienten und Patientinnen mit erhöhtem Darmkrebsrisiko wird eine Koloskopie auch in jüngeren Jahren von der gesetzlichen Krankenkasse übernommen [3]. Für die Darmkrebsvorsorge stehen laut S3-Leitlinie verschiedene Untersuchungen zur Verfügung: Stuhltests, radiologische sowie endoskopische Verfahren. Als Goldstandard gilt die Koloskopie [4]. Dabei wird – anders als bei einer Sigmoidoskopie – nicht nur der letzte Abschnitt des Dickdarms untersucht, sondern auch der Bereich
vom Kolon bis zum terminalen Ileum. Vor der Behandlung wird der Darm mithilfe von Darmspülpräparaten gereinigt, sodass Polypen, Adenome oder Karzinome zuverlässig entdeckt werden können. Darmreinigung: vom Einlauf bis zu modernen Spüllösungen
Für die Darmreinigung sind heute viele verschiedene Präparate und Lösungen verfügbar. Welche Form für die jeweiligen Patienten und Patientinnen am geeignetsten ist, ist abhängig von verschiedenen Faktoren wie dem Allgemeinzustand oder der zu erwartenden Therapietreue. Bevorzugt werden aber in der Regel Darmspüllösungen mit einem geringen Trinkvolumen, unterschiedlichen Geschmacksrichtungen und einer einfachen Zubereitung. Nachdem über lange Zeit vor allem Einläufe in Kombination mit diätetischen Einschränkungen zur Darmreinigung eingesetzt worden waren, etablierten sich in den 1980er Jahren PolyethylenglykolLösungen (PEG), d.h. osmotisch ausgewogene Elektrolytspüllösungen zur oralen Einnahme, die sich schnell zum Goldstandard der Darmreinigung vor Koloskopien entwickelten [5]. Denn die PEGLösungen wirkten sehr effektiv: Das PEG in der Darmspüllösung wird praktisch nicht resorbiert
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
und bindet durch seine osmotische Wirkung Wasser in Form von Wasserstoffbrücken. Das Wasser passiert anschließend den Gastrointestinaltrakt, ohne resorbiert zu werden [6]. Ein Nachteil früherer PEG-Lösungen war jedoch, dass die Patienten 4 Liter der Lösung innerhalb weniger Stunden zu sich nehmen mussten. Neben dem hohen Trinkvolumen erschwerte auch ein salziger Geschmack die Einnahme. Daher wurden die PEG-Lösungen durch den Zusatz von Ascorbinsäure/ Natriumascorbat (ASC) ergänzt, wodurch sich das Trinkvolumen auf 2 Liter reduzieren ließ [7]. Durch die osmotische Wirkung gelangt zusätzlich Wasser in den Darm und induziert einen laxativen Effekt. Ein positiver Nebeneffekt von ASC ist der angenehme Geschmack. Mittlerweile gibt es die PEG+ASC-Lösungen auch mit einem geringeren Trinkvolumen und sie werden außerdem in unterschiedlichen Geschmacksrichtungen angeboten. Plenvu® schreibt die Innovationen fort
Heute steht mit Plenvu® eine Darmspüllösung zur Verfügung, die in besonderer Weise die Wirkmechanismen von PEG und ASC kombiniert. Dadurch wird es möglich, das Trinkvolumen der Spül© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
77
lösung auf 1 Liter* zu reduzieren. Zusätzlich enthaltene Elektrolyte wie Natrium- und Kaliumchlorid unterstützen durch ihre ebenfalls osmotischen Effekte die Wirkung von PEG und ASC. Die Elektrolyte verhindern, dass klinisch signifikante Konzentrationsänderungen von Natrium, Kalium oder Wasser auftreten – außerdem wird das Dehydrierungsrisiko reduziert [8]. Dieser Effekt wird dadurch unterstützt, dass zu beiden Dosen der Trinklösung zu je 500 ml zusätzlich mindestens 500 ml klare Flüssigkeit (z. B. Wasser, klarer Apfelsaft) eingenommen werden sollen. Dosis-Splitting Im Idealfall sollten die Patienten die Trinklösung in 2 Dosen (mit nächtlicher Pause dazwischen) zu sich nehmen. Findet die Koloskopie z.B. am Vormittag statt, wird die Trinklösung am Vorabend und am Morgen des Untersuchungstages getrunken. Durch das Dosis-Splitting verbessert sich nicht nur die Adhärenz der Patienten, sondern es verlängert sich auch der Reinigungsprozess und die Reinigungsleistung erhöht sich um 15 – 20 % im Vergleich zur Einnahme der gesamten Lösung am Vortag [9]. Bei einer Koloskopie am Nachmittag sollte als adäquate Alternative zum Dosis-Splitting die Einnahme beider Dosen am Untersuchungstag – im Abstand von mindestens 1 Stunde – erfolgen. Eine gesplittete Einnahme wird sowohl von der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS) als auch von der Europäischen Gesellschaft für gastrointestinale Endoskopie (ESGE) empfohlen [10, 11]. * Zusätzlich muss 1 Liter frei wählbare klare Flüssigkeit getrunken werden.
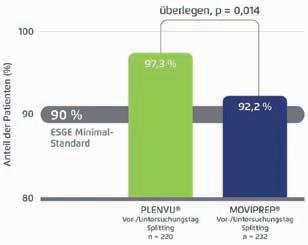
Abbildung 1: In der MORA-Studie war Plenvu® der 2-Liter-PEG+ASC-Lösung Moviprep® bei gesplitteter Einnahme hinsichtlich der Reinigung des gesamten Kolons überlegen (mod. nach [6]).
Abbildung 2: Im kritischen Segment des rechten Kolons bewirkte Plenvu® bei doppelt so vielen Patienten eine exzellente und gute Reinigung wie Moviprep® (mod. nach [6]).
Wie die MORA-Studie belegt, ist Plenvu® trotz des niedrigen Trinkvolumens bei der Gesamtreinigung des Darms nicht unterlegen
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
[6]: Mit der Einnahme der 1-Liter-PEG+ASC-Lösung* erzielten 97,3 % der Patienten bei zweigeteilter Trinkmenge eine erfolg© VERLAG PERFUSION GMBH
78
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
reiche Darmreinigung gegenüber 92,2 % nach gesplitteter Einnahme der 2-Liter-PEG+ASC-Lösung Moviprep® (Abb. 1). Auch bei der Reinigung des kritischen rechten Kolons zeigt sich eine signifikante Überlegenheit von Plenvu® (Abb. 2). Durch die die exzellente und gute Reinigungsleistung von Plenvu® bei 31,6 % der Patienten musste während der Koloskopie zudem weniger Spülflüssigkeit abgesaugt werden, wodurch sich die Untersuchungszeit verringerte [6]. Fabian Sandner, Nürnberg
Literatur 1 International Agency for Research on Cancer, 2019. Im Internet: http://gco.iarc. fr/today/data/factsheets/populations/276germany-fact-sheets.pdf 2 Positionspapier Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselerkrankungen zur Darmkrebsvorsorge, 2019 Im Internet: www. dgvs.de/wp-content/uploads/2018/03/ Positionspapier_Organisiertes-DK-Screen ing_M%C3%A4rz 2018_final.pdf 3 Kassenärztliche Bundesvereinigung. Richtlinien des Gemeinsamen Bundesausschusses über die Früherkennung von Krebserkrankungen (Krebsfrüherkennungs-Richtlinien). Bundesanzeiger; 2009; 148a 4 Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften. S3-Leitlinie 021-007OL: Kolorektales Karzinom, 2017 5 Wexner SD et al. Surg Endosc 2006;20: 1147-1160 6 Bisschops R et al. Endoscopy 2019;51: 60-72 7 Bitoun A et al. Aliment Pharmacol Ther 2006;24:1631-1642 8 Fachinformation Plenvu®; Stand: Oktober 2019 9 Martel M et al. Gastroenterol 2015;179: 79-88 10 Ell C et al. Z Gastroenterol 2007;45: 1191-1198 11 Hassan C et al. Bowel preparation for colonoscopy: ESGE Guideline – Update 2019. Endoscopy 2019;51:775-794
Aktualisierte S3-Leitlinie Rauchen und Tabakabhängigkeit:
Vareniclin erhält höchsten Empfehlungsgrad Für den Einsatz zur medikamentösen Tabakentwöhnung erhielt der partielle Nikotinrezeptorblocker Vareniclin (Champix®) in der im Januar aktualisierten Leitlinie „Rauchen und Tabakabhängigkeit: Screening, Diagnostik und Behandlung“ erstmals eine starke Empfehlung. Er hat somit den höchsten Empfehlungsgrad für eine medikamentöse Behandlung von entwöhnungswilligen Rauchern und Raucherinnen. Darüber hinaus wurden zum ersten Mal auch Smartphone-gestützte Entwöhnungsverfahren in die Leitlinie mit aufgenommen. Effektiver Wirkmechanismus
Gemäß der aktualisierten S3Leitlinie soll Vareniclin eingesetzt werden, wenn eine medikamentöse Behandlung mit einer Nikotinersatztherapie (NET), wie z.B. Nikotinpflaster oder -kaugummi, nicht ausreichend wirksam ist. Grund für die starke Empfehlung von Vareniclin sind die guten Ergebnisse der EAGLES-Studie, die den effektiven dualen Wirkmechanismus von Vareniclin bestätigt. Dabei reduziert Vareniclin das mit dem Nikotinkonsum einhergehende Belohnungs- und Genussgefühl und verringert gleichzeitig die Freisetzung von Dopamin. Dies führt zu einem schwächeren Rauchverlangen und weniger starken Entzugssymptomen. In einer Metaanalyse von insgesamt 27 Studien wurde eine hohe
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Evidenz für die Wirksamkeit von Vareniclin im Vergleich zu Placebo festgestellt (RR: 2,24, 95%-KI: 2,06 – 2,43). In weiteren Studien und Auswertungen der Behandlungsarme war Vareniclin auch effektiver als Bupropion (RR: 1,39, 95%-KI: 1,25 – 1,54; 5 Studien; n > 5.800) und als NET (RR: 1,25, 95%-KI: 1,14 – 1,37; 8 Studien; n > 6.200). Zudem konnte bei Vareniclin, im Vergleich zu Placebo, kein erhöhtes Risiko für neuropsychiatrische Nebenwirkungen festgestellt werden. Neu: Empfehlung für digitale Verfahren zur Tabakentwöhnung
Zusätzlich zu den etablierten Empfehlungen für verhaltenstherapeutische Interventionen plus medikamentöse Unterstützung spielen auch digitale Optionen – online bzw. durch Apps – eine wichtige Rolle. Hierdurch wird der Weg zu breiteren Zielgruppen geöffnet. Gerade die einkommensschwächeren abhängigen Raucher erhalten so Zugang zu niederschwelligen Unterstützungsangeboten. Aufgrund der weiten Verbreitung von Mobiltelefonen stellen diese Entwöhnungsverfahren eine günstige Rauchentwöhnungsoption für weniger motivierte Raucher dar. Zu den Smartphone-gestützten Verfahren zählen auch „Rauchfrei-Apps“. Da mobile Selbsthilfeprogramme, elektronisch oder neuerdings per App, eine sehr viel breitere Gruppe von Rauchern ansprechen, die sich für Gruppenprogramme nicht bereit finden würden oder diese nicht bezahlen können, sollten Ärzte auch diese Entwöhnungsmöglichkeiten in die Therapie integrieren. E. W.
© VERLAG PERFUSION GMBH
79
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
B
ei der Therapie der PlaquePsoriasis ist neben der Wirksamkeit und Sicherheit eines Medikaments auch das Thema Therapietreue wichtig für den Behandlungserfolg. Seit Mitte Februar 2021 steht der Interleukin (IL)-17A-Inhibitor Secukinumab (Cosentyx®) als 300-mg-Fertigpen zur Verfügung. Basis der Zulassung war die Phase-IIIb-Studie MATURE. Die Studiendaten zeigen bei guter Wirksamkeit und einem günstigen Sicherheitsprofil eine hohe Patientenzufriedenheit bei der Anwendung des 300-mgFertigpens [1]. Fast 40 % der Patienten sind bislang nicht adhärent
Dank innovativer Therapieoptionen können viele Psoriasis-Patienten heute dauerhaft ein PASI-90bzw. PASI 100-Ansprechen und häufig auch eine von der Erkrankung unbeeinträchtigte Lebensqualität erreichen. Dennoch haben
Therapie der Plaque-Psoriasis: Secukinumab-Fertigpen überzeugt durch Wirksamkeit und Patientenadhärenz Untersuchungen gezeigt, dass die durchschnittliche Adhärenz bei einer systemischen Psoriasis-Therapie bei etwa 62 % liegt – damit sind fast 40 % der Patienten nicht adhärent [2]. Besonders stark sinkt die Therapieadhärenz innerhalb der ersten 2 Jahre nach Therapiebeginn [3]. Die neue Applikationsform des IL-17A-Inhibitors Secukinumab als 300-mg-Fertigpen ermöglicht es, diese Dosierung in nur einer Injektion zu verabreichen, was die Handhabung für Patienten deutlich erleichtert und somit auch die Therapietreue positiv beeinflussen kann.
Hohe Ansprechraten und Patientenzufriedenheit nach der 300-mg-Injektion
Zur Untersuchung der Wirksamkeit, Sicherheit und Verträglichkeit sowie der Patientenzufriedenheit der 300-mg-Injektion von Secukinumab wurde die randomisierte, verblindete und placebokontrollierte MATURE-Studie durchgeführt [1]. In der Studie wurde die Anwendung des 300-mg-Fertigpens mit der zweimaligen Anwendung des 150-mg-Fertigpens bzw. Placebo verglichen. Die beiden coprimären Endpunkte in Woche 12 wurden erreicht: 95,1 % der Patien-
Interleukin-17A – Zentrale Rolle im Entzündungsgeschehen bei Psoriasis, PsA und axSpA Ausschüttung aus mehreren Quellen
Unmittelbarer Einfluss
auf die Entzündungsprozesse bei Psoriasis, PsA und axSpA
TH17-Zellen
Haut
Mastzellen, Makrophagen und Neutrophile Zytotoxische T-Zellen und γδ-T-Zellen
IL-23-abhängig
Nägel
IL-17A
IL-23-unabhängig
entzündungsfördernd
Periphere Gelenke Wirbelsäule Herz-Kreislauf
Mechanischentzündlicher Stress
Abbildung 1: Secukinumab (Cosentyx®) ist ein 1.vollhumaner, monoklonaler der 9:direkt IL-17A gerichtet ist. Dieses ist Quellen: Brembilla NC, Senra L, Boehncke WH.Antikörper, Front Immunol 2018; 1682. 2.gegen Schön MP, Erpenbeck L. Front Immunol 2018; 9: Zytokin 1323. 3. Papotto et al. Nature Immunology 2017; 18(6): 604–611. Beringer et al. Trends in Molecular Medicine 2016; 22(3): 230–241. an Entzündungsprozessen und der Entstehung derPHPsoriasis, Psoriasis-Arthritis (PsA)4.und derA axialen Spondyloarthritis (axSpA) beteiligt [4]. 5. Kirkham BW, Kavanaugh A, Reich K. Immunology 141(2): 133–142. 6. Boehncke WH. Front Immunol 2018; 9: 579. PsA = Psoriasis-Arthritis; axSpA = axiale Spondyloarthritis; IL-23: Interleukin-23
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
© VERLAG PERFUSION GMBH
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
80
ten, die mit dem 300-mg-Fertigpen behandelt wurden, erreichten ein PASI-75-Ansprechen und 75,6 % ein IGA von 0/1. Auch die PASI90- bzw. PASI-100-Ansprechraten konnten überzeugen: 75,6 % hatten unter der Behandlung mit dem 300-mg-Fertigpen fast keine, 43,8 % hatten überhaupt keine Hautsymptome mehr. Die guten Ergebnisse setzten sich bis in Woche 16 fort (PASI 75: 95,1 %, PASI 90: 80,5 %, IGA 0/1: 80,5 %). Die Studienergebnisse belegen auch, dass 92,1 % der Patienten mit der Anwendung des 300-mgPens zufrieden oder sehr zufrieden waren. Zudem zeigte sich an der Einstichstelle nur eine geringe Injektionsreaktion. Das Sicherheitsprofil beider Applikationsformen war allgemein günstig und vergleichbar mit den Ergebnissen früherer Studien. Fazit
Die Studiendaten zeigen, dass die 300-mg-Dosis von Secukinumab bei hoher Patientenzufriedenheit, guter Wirksamkeit und einem günstigen Sicherheitsprofil mit dem 300-mg-Fertigpen in nur einer Injektion verabreicht werden kann. Die vereinfachte Handhabung des Biologikums könnte die Therapietreue positiv beeinflussen und ermöglicht damit eine umfassende Therapieoption für Patienten mit Plaque-Psoriasis. Fabian Sandner, Nürnberg Literatur 1 Sigurgeirsson B et al. AAD Virtual Meeting 2021, P27599 2 Dommasch et al. J Am Acad Dermatol 2018;79:1061-1068.e1 3 Egeberg et al. BJD 2018;178:509-519 4 Brembilla NC et al. Front Immunol 2018; 9:1682
xiskapazitäten angepasst werden kann. Multiple Sklerose:
Natalizumab jetzt auch zur subkutanen Applikation zugelassen Natalizumab (Tysabri®) ist seit März 2021 in der EU auch zur s.c. Injektion bei erwachsenen Patienten mit (hoch-)aktiver schubförmig remittierender Multipler Sklerose (RRMS) zugelassen. Die neue Verabreichungsform vereinfacht die Applikation in der Praxis und bietet außerdem mehr Flexibilität sowie Zeitersparnis in der MSTherapie. Natalizumab kommt bereits seit fast 15 Jahren bei RRMS zur Anwendung. Aktuelle 10-JahresDaten des Tysabri Observational Program (TOP) untermauern das Effektivitäts- und Sicherheitsprofil von Natalizumab auch in der Langzeittherapie. Verabreichung in kürzerer Zeit
Natalizumab s.c. liegt in Form von 2 Fertigspritzen vor, für eine vollständige Dosis werden 2 Spritzen verabreicht. Wie die bisherige i.v. Applikationsform wird auch Natalizumab s.c. alle 4 Wochen injiziert. Im Gegensatz zur einstündigen Infusion bei Natalizumab i.v. ermöglicht die s.c. Applikationsform eine kürzere Verabreichungsdauer. Nach den ersten 6 Applikationen ist eine Nachbeobachtungszeit von 1 Stunde nötig, bei allen weiteren Applikationen liegt die Nachbeobachtung im Ermessen des Arztes. Ein Infusionsplatz ist für die subkutane Verabreichung von Natalizumab nicht erforderlich, sodass die Therapie besser an die Pra-
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Wirksamkeits- und Sicherheitsprofil mit i.v. Applikationsform vergleichbar
Die Zulassung von Natalizumab s.c. erfolgte auf Basis der Ergebnisse der beiden Studien REFINE und DELIVER. Primärer Endpunkt der exploratorischen Dosis- und Frequenz-verblindeten, prospektiven, randomisierten DosisfindungsStudie REFINE war die kumulative Anzahl aktiver Läsionen nach der Gabe von Natalizumab in verschiedenen Dosierungen, Intervallen und Verabreichungswegen. Eingeschlossen waren Patienten, die mindestens 11 i.v. Infusionen von 300 mg Natalizumab in den 12 Monaten vor der Randomisierung erhalten hatten. Primärer Endpunkt der offenen, randomisierten Studie DELIVER war der Vergleich von 300 mg Natalizumab über 8 Wochen nach der s.c., i.m. oder i.v. Verabreichung einer Einzeldosis. Eingeschlossen waren Patienten ohne Natalizumab-Vorbehandlung. In beiden Studien zeigten sich unter Natalizumab 300 mg s.c. und i.v. alle 4 Wochen eine vergleichbare α4-Integrin-Sättigung und vergleichbare pharmakokinetische Eigenschaften. Die Aussagekraft der Ergebnisse für die jährliche Schubrate und MRT-Parameter ist zwar in beiden Studien limitiert, Unterschiede wurden jedoch nicht festgestellt. Das galt auch für das Sicherheitsprofil: In beiden Studien wurden keine unerwarteten unerwünschten Ereignisse beobachtet. E. W.
© VERLAG PERFUSION GMBH
Look
BETTER#
Move
BETTER§
Feel
BETTER‡
PSORIASIS GEHT TIEFER Mit dem umfassenden Cosentyx®-Ansatz behandeln! #, §, ‡
DIREKT.1 WIRKSTARK.2 LANGANHALTEND.3
EINFACH 1-MALIG –
NEU!
Cosentyx® 300 mg
Nur eine Injektion im Monat ▲ ▲
In der Erhaltungsphase gemäß Fachinformation
DIREKT: Jeder zweite Plaque-Psoriasis-Patient erreicht PASI 75 bereits in Woche 4.° Thaçi D et al. J Am Acad Dermatol 2015; 73(3): 400 – 409. 2. WIRKSTARK: Über 8 von 10 Plaque-Psoriasis-Patienten haben einen PASI < 3 in Woche 24.° Thaçi D et al. J Eur Acad Dermatol Venereol 2019; doi: 10.1111/jdv.15962. 3. LANGANHALTEND: Fast 7 von 10 Plaque-Psoriasis-Patienten halten PASI 90-Ansprechen über 5 Jahre.° Bissonnette R et al. J Eur Acad Dermatol Venereol 2018; 32(9): 1507 – 1514. ° Daten für die Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Therapie in Frage kommen. 1.
# LOOK BETTER: Erwachsene: Fast 8 von 10 Patienten erreichen PASI 90 in W16 (vs. Ustekinumab). Kinder und Jugendliche: 6 von 10 Patienten erreichen PASI 100 in W12.*, ** § MOVE BETTER: Erwachsene: Hemmung der strukturellen Schädigung bei 88,5 % der Psoriasis-Arthritis-Patienten in W24 (vs. Placebo). Kinder und Jugendliche: 7 von 10 Patienten erreichen CDLQI von 0/1 in W24.*, ** ‡ FEEL BETTER: Erwachsene: Bis zu -11,6 DLQI-Reduktion nach W12. Kinder und Jugendliche: 7 von 10 Patienten erreichen CDLQI 0/1 in W24.*,** * Fachinformation Cosentyx®. ** Alle Zahlenwerte sind aus der offenen, zweiarmigen multizentrischen Studie CAIN457A2311 (Niedrig-Dosis-Arm: Dosierung 75 mg bei einem Körpergewicht < 50 kg und 150 mg bei einem Körpergewicht ≥ 50 kg). Cosentyx® 150 mg Injektionslösung in einer Fertigspritze, Cosentyx® 150 mg Injektionslösung in einem Fertigpen, Cosentyx® 150 mg Pulver zur Herstellung einer Injektionslösung, Cosentyx® 300 mg Injektionslösung in einer Fertigspritze, Cosentyx® 300 mg Injektionslösung in einem Fertigpen. Wirkstoff: Secukinumab (in Ovarialzellen d. chinesischen Hamsters [CHO-Zellen] produzierter, gegen Interleukin-17A gerichteter, rekombinanter, vollständig humaner monoklonaler Antikörper der IgG1/k-Klasse). Zus.-setz.: Arzneil. wirks. Bestandt.: 1 Fertigspritze/Fertigpen enthält 150 mg Secukinumab in 1 ml bzw. 300 mg Secukinumab in 2 ml. Sonst. Bestandt.: Trehalose-Dihydrat, Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 80, Wasser f. Inj.-zwecke. Eine Durchstechflasche mit Pulver enthält 150 mg Secukinumab (nach Rekonstitution enthält 1 ml Lösung 150 mg Secukinumab). Sonst. Bestandt.: Sucrose, Histidin, Histidinhydrochlorid-Monohydrat, Polysorbat 80. Anwend.: Behandl. erw. Pat. mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine system. Ther. in Frage kommen. Für Behandl. von Kdr. und Jugendl. ab einem Alter von 6 J. mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Ther. in Frage kommen. Behandl. erw. Pat. mit aktiver Psoriasis-Arthritis, allein od. in Kombination mit Methotrexat (MTX), wenn das Ansprechen auf eine vorhergehende Ther. mit krankheitsmodifizierenden Antirheumatika (DMARD) unzureichend gewesen ist. Behandl. erw. Pat. mit aktiver ankylosierender Spondylitis, die auf eine konventionelle Ther. unzureichend angesprochen haben. Behandl. erw. Pat. mit aktiver nicht-röntgenologischer axialer Spondyloarthritis mit objektiven Anzeichen der Entzündung, angezeigt durch erhöhtes C-reaktives Protein (CRP) und/oder Nachweis durch Magnetresonanztomographie (MRT) die unzureichend auf nichtsteroidale Antirheumatika (NSAR) angesprochen haben. Gegenanz.: Überempfindlichkeitsreakt. gegen d. Wirkstoff od. einen d. sonst. Bestandt. Klinisch relevante, aktive Infekt. (z. B. aktive Tuberkulose). Nebenw.: Sehr häufig: Infekt. d. oberen Atemwege. Häufig: Oraler Herpes, Tinea pedis. Kopfschmerzen. Rhinorrhö. Diarrhö, Übelkeit. Ermüdung. Gelegentl.: Orale Candidose, Otitis externa, Infekt. d. unteren Atemwege. Neutropenie. Konjunktivitis. Entzündl. Darmerkrankungen. Urtikaria. Selten: Anaphylakt. Reakt. Exfoliative Dermatitis. Häufigkeit nicht bekannt: Mukokutane Candidose (einschl. ösophageale Candidose). Verschreibungspflichtig. Weit. Angaben: S. Fachinformationen. Stand: Februar 2021 (MS 04/21.16). Novartis Pharma GmbH, Roonstr. 25, 90429 Nürnberg. Tel.: (09 11) 273-0, Fax: (09 11) 273-12 653. www.novartis.de
82
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
W
ährend in der frühen Phase des idiopathischen Parkinson-Syndroms gute Erfolge mit der Levodopa-Therapie erzielt werden, nimmt die Komplexität der Erkrankung im Verlauf zu, wodurch auch die Anforderungen an die Therapie steigen. Neuere Medikamente, die als Add-on zu Levodopa gegeben werden, haben die Therapieoptionen in den letzten Jahren erweitert und damit die Möglichkeiten zur Kontrolle der sich ausweitenden Symptomatik verbessert. So kann mit Safinamid (Xadago®) nicht nur Wearing-offBeschwerden begegnet werden, sondern auch nicht motorischen Symptomen wie Schmerzen und depressiven Verstimmungen. Um den maximalen Nutzen für die Patienten zu erreichen, ist es wichtig, das Add-on-Präparat frühzeitig in die Therapie zu integrieren. „Honeymoon“ endet oft rasch
Die „Honeymoon-Phase“, in der die Behandlung mit Levodopa dem Patienten ein relativ beschwerdearmes Leben ermöglicht, ist je nach Subtyp der Krankheit von unterschiedlicher Dauer. Während beim Tremor-dominanten Typ oft eine mehrjährige Stabilität beobachtet wird, kommt es beim Rigor-Akinese-Typ und beim Äquivalenztyp, bei dem alle Kardinalsymptome auftreten, oft schon nach 1 – 2 Jahren zu Verschlechterungen der Motorik, zu einer Wearing-off-Symptomatik sowie zu nicht motorischen Beschwerden wie Schmerzen, Depressionen, Müdigkeit und Störungen des autonomen Nervensystems. Dann ist es an der Zeit, nach einer genauen Analyse des Beschwerdebildes die Therapie in Abhängigkeit von der individuellen Symptomatik sinnvoll zu
Morbus Parkinson: Rechtzeitig Chancen der Add-on-Therapie mit Safinamid nutzen erweitern, sodass dem Patienten weiterhin eine akzeptable Lebensqualität geboten werden kann. Besserung motorischer und nicht motorischer Beschwerden
Hier haben Neueinführungen die Behandlungsmöglichkeiten deutlich verbessert. Für Patienten, bei denen es nach der HoneymoonPhase zu ersten motorischen Komplikationen oder leichten Dyskinesien kommt, stellt Safinamid als Add-on zu Levodopa eine Therapieoption dar, die dem Patienten vielfältige Chancen bietet. Denn Safinamid bewirkt nicht nur eine reversible MAO-B-Hemmung, sondern reguliert außerdem die Überaktivität der glutamatergen Neuronen, die eine schädigende Wirkung auf die motorischen Schleifen im Gehirn ausüben. In Studien hat Safinamid eine sehr konstante und signifikante Verbesserung von Off- und On-Zeiten gezeigt [1]. Aufgrund seiner antiglutamatergen Wirkung hat das Medikament außerdem positive Effekte auf nicht motorische Beschwerden wie Schmerzen und die emotionale Befindlichkeit [2, 3]. Wichtig ist, nicht zu lange mit dem Beginn der Add-on-Therapie zu warten. Safinamid sollte nicht erst nach mehreren Jahren verordnet werden, sondern bereits wenn der Patient nicht mehr so gut durch den Tag kommt und sich motorisch vor der nächsten Levodopa-Dosis
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
verschlechtert und/oder leichte Überbeweglichkeit auftritt. Denn damit ist das Wearing-off milder oder verschwunden, leichte Überbeweglichkeiten gehen zurück und der Patient ist alerter, auch Schmerzen und Müdigkeit bessern sich. Einsparmöglichkeiten bei Levodopa
Ein nicht zu vernachlässigender Aspekt ist auch die Möglichkeit, mit der Gabe des Add-onMedikaments die Steigerung der Levodopa-Dosen zu vermeiden. Denn durch die Zugabe von Safinamid lässt sich über längere Zeit Levodopa einsparen und auch das Entstehen von Dyskinesien verhindern. Von Vorteil sind nicht zuletzt die einfache Anwendbarkeit durch eine einmalige tägliche Dosierung sowie die gute Verträglichkeit von Safinamid – die Nebenwirkungsrate ist überdurchschnittlich niedrig und die Vorteile der Zusatztherapie müssen nicht mit unerwünschten Wirkungen erkauft werden. Elisabeth Wilhelmi, München Literatur 1 Borgohain R et al. Mov Disord 2014;29: 229-237 2 Cattaneo C et al. J Parkinson’s Dis 2017; 7:95-101 3 Cattaneo C et al. J Parkinson’s Disease 2017;7:629-634 © VERLAG PERFUSION GMBH
83
INTERVIEW
Asthma-Therapie in Zeiten von COVID-19: Mit fixer Dreierkombination zu einer besseren Therapiekontrolle Interview mit Herrn Dr. Justus de Zeeuw, Arzt für Innere Medizin, Pneumologie und Schlafmedizin, Medizinisches Versorgungszentrum im Rolshover Hof, Köln
S
ARS-CoV-2 sorgt derzeit bei vielen Menschen für Verunsicherung. Besonders beunruhigt sind Asthmatiker, da das Coronavirus vor allem die Lunge schädigt und sie befürchten, aufgrund ihrer Vorerkrankung ein höheres Risiko zu haben, schwer an COVID-19 zu erkranken. Viele treibt zudem die Sorge um, dass die verordneten inhalativen Kortikosteroide das Immunsystem herunterregulieren, und setzen die Medikamente deshalb eigenmächtig ab – und riskieren damit eine Verschlechterung des Asthmas. Wie gefährlich die Situation für Menschen mit Asthma derzeit wirklich ist, war Thema auf dem Welt-Asthma-Tag am 5. Mai 2021. In einem Gespräch erläuterte Dr. Justus de Zeeuw, Facharzt für Lungen- und Bronchialheilkunde in Köln, die besonderen Herausforderungen, die durch die COVID19-Pandemie bei der Behandlung von Patienten mit Asthma entstanden sind. Außerdem warf er einen Blick auf die Ursachen für den hohen Anteil an unzureichend kontrollierten Asthma-Patienten in Deutschland sowie die Möglich-
keiten, die Behandlung durch innovative Fixkombinationen und digitale Unterstützung zu optimieren. Herr Dr. de Zeeuw, inzwischen begleitet uns die COVID-19-Pandemie seit über einem Jahr. Welchen Einfluss hat Corona auf Ihren Alltag in der pneumologischen Praxis? Dr. de Zeeuw: Beim unmittelbaren Patientenaufkommen spüren wir keine allzu großen Veränderungen. Es gibt allerdings Patienten, die aufgrund der Sorge vor einer Ansteckung seltener oder gar nicht mehr in die Praxis kommen. Dadurch gewinnen zwar digitale Angebote wie die Videosprechstunde durchaus an Bedeutung, diese hat jedoch in der Pneumologie gewisse Limitationen. Wir müssen die Patienten abhören oder auch eine Lungenfunktionsprüfung durchführen können, um eine adäquate Diagnostik und Behandlung zu gewährleisten – dafür ist ein persönlicher Besuch in der Praxis unabdingbar. Hinzu kommen naturgemäß viele Rückfragen von Patienten, die wir rund um die COVID-19-Pandemie im Hinblick
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
auf Risikofaktoren, die jeweilige medikamentöse Therapie und eine potenzielle Impfung täglich beantworten müssen. Leider kommt es im Zuge der intensiven Berichterstattung der Medien mitunter vor, dass beispielsweise auch Daten aus sehr kleinen und begrenzt aussagekräftigen Studien in einen missverständlichen Kontext gesetzt werden und dadurch unter Umständen zu einer gewissen Verunsicherung der Patienten beitragen. Wie beurteilen Sie die wissenschaftlichen Erkenntnisse, was das Risiko für Asthma-Patienten durch eine COVID-19-Erkrankung betrifft? Dr. de Zeeuw: Die Datenlage ist mit zahlreichen Studien und MetaAnalysen inzwischen sehr eindeutig und lässt den Schluss zu, dass bei Menschen mit Asthma ein geringeres Risiko für eine Infektion mit dem Corona-Virus besteht [1] und die Erkrankung keinen unabhängigen Risikofaktor für einen schweren COVID-19-Verlauf darstellt [2]. Die vermutliche Erklärung ist eine verminderte Expression des für die Aufnahme von © VERLAG PERFUSION GMBH
INTERVIEW
84
SARS-CoV-2 verantwortlichen ACE-2-Rezeptors in den Atemwegen. Möglicherweise spielt auch die Therapie mit inhalativen Kortikosteroiden (ICS) eine protektive Rolle, denn auch diese führen zu einer verminderten Expression von ACE-2-Rezeptoren [2]. Dieser Umstand mag daher ein wenig im Widerspruch zur Priorisierung der Asthma-Patienten im Zuge der Impfverordnung stehen, dennoch empfehlen wir in der Praxis jedem Betroffenen, sich so schnell wie möglich gegen COVID-19 impfen zu lassen. Unabhängig von einer potenziellen Impfung sollten gut eingestellte Asthma-Patienten zudem ihre inhalative Therapie unbedingt weiterhin beibehalten, um die Krankheitskontrolle nicht zu gefährden. Das gilt auch für Patienten mit schwerem Asthma, die ein Biologikum erhalten – es gibt derzeit keine wissenschaftlichen Anhaltspunkte dafür, dass diese mit dem Wirkmechanismus der verfügbaren Impfstoffe in irgendeiner Weise interagieren [3]. Registerdaten zeigen, dass in Deutschland ein Großteil der Patienten unter einer Kombination aus ICS und langwirksamen Beta2-Agonisten (LABA) weiterhin unzureichend kontrolliert ist [4]. Welche Ursachen sind dafür aus Ihrer Sicht ausschlaggebend? Dr. de Zeeuw: Grundsätzlich stehen uns heute von der inhalativen Bedarfs- und Erhaltungstherapie mit fixen Zweier- und Dreierkombinationen bis hin zum Biologikum viele Behandlungsoptionen zur Verfügung, um Menschen mit Asthma ein nahezu beschwerdefreies Leben zu ermöglichen. In der Realität besteht jedoch eine erhebliche Diskrepanz zwischen dem, was dank Präzisionsmedizin möglich ist, und dem, was Be-
troffene im Alltag akzeptieren. So gibt es in der Praxis immer wieder Patienten, die subjektiv über eine gute Krankheitskontrolle berichten, nach objektiven Kriterien aber lediglich teilweise oder unzureichend kontrolliert sind. Wichtigster Faktor im Hinblick auf eine gute Krankheitskontrolle ist dabei die Therapietreue: Mittels optimierter Inhalatoren und Fixkombinationen bestehen dabei inzwischen Möglichkeiten, eine Verbesserung der Adhärenz mit einer adäquaten Pharmakotherapie bestmöglich zu kombinieren. Welche therapeutischen Möglichkeiten bestehen, wenn Patienten unter LABA/ICS unkontrolliert sind und wonach richtet sich die Entscheidung, die Behandlung entsprechend umzustellen? Dr. de Zeeuw: Wenn die bisherige LABA/ICS-Therapie nicht zu einer ausreichenden Asthma-Kontrolle führt, lohnt sich zunächst ein Blick auf das Device bzw. die Inhalationstechnik. Wenn ein Patient beispielsweise mit seinem bisherigen Inhalator nicht zurechtkommt, kann sich das negativ auf die Krankheitskontrolle auswirken und daher einen Wechsel des Devices notwendig machen. Auch eine mehrfach tägliche Anwendung ist im Behandlungsalltag häufig eine Herausforderung und kann zu einer verminderten Adhärenz führen, sodass Patienten in diesem Fall von einer einmal täglichen Fixkombination profitieren. Darüber hinaus besteht die Möglichkeit, die medikamentöse Behandlung durch eine Umstellung auf eine Dreierkombination aus LABA, ICS und einem langwirksamen Muskarinrezeptor-Antagonisten (LAMA) zu optimieren [5]. Voraussetzung dafür ist eine unzureichende Krankheitskontrolle un-
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
ter vorheriger Erhaltungstherapie mit LABA und hochdosiertem ICS sowie mindestens eine Exazerbation im Vorjahr [6]. Mit Indacaterol/Glycopyrronium/ Mometasonfuroat (IND/GLY/MF, Enerzair® Breezhaler®) ist seit letztem Jahr erstmals eine einmal täglich anzuwendende fixe Dreierkombination für erwachsene Asthma-Patienten verfügbar, die mit einer Kombination aus einem LABA und einer hohen Dosis eines ICS als Erhaltungstherapie nicht ausreichend kontrolliert sind und bei denen im Vorjahr eine oder mehrere Exazerbationen aufgetreten sind [6]. Wie beurteilen Sie den Stellenwert dieser Therapieoption in der Praxis? Dr. de Zeeuw: Die fixe Dreierkombination hat den Vorteil, dass sich durch die einmal tägliche Gabe und die Charakteristika des Inhalators nicht nur die Adhärenz und Inhalationstechnik verbessern lassen, sondern eben auch eine Optimierung der Pharmakotherapie für unter LABA/ICS unzureichend kontrollierte Patienten möglich ist. Zuvor stand für die potenzielle Umstellung auf eine LABA/LAMA/ICS-Therapie lediglich eine freie Kombination zur Verfügung, die die parallele Anwendung mehrerer Inhalatoren notwendig machte. Daher stellt die fixe Dreierkombination IND/GLY/ MF eine wichtige Erweiterung des Therapiespektrums dar. Zudem kommt der Breezhaler® mit anderen Wirkstoffkombinationen auch auf weiteren Behandlungsstufen zum Einsatz, sodass sich Patienten bei einer Umstellung der Therapie nicht an einen neuen Inhalator gewöhnen müssen. In der fixen Dreierkombination ist mit Mometason ein ICS enthalten, © VERLAG PERFUSION GMBH
85
INTERVIEW
das bislang eine vergleichsweise untergeordnete Rolle im Bereich der Asthma-Therapie spielte. Wie würden Sie dessen Potenzial einordnen, gerade auch in Kombination mit Indacaterol und Glycopyrronium? Dr. de Zeeuw: Mometasonfuroat ist ein interessantes Kortikosteroid mit einem guten therapeutischen Quotienten: Es ist durch seine hohe Affinität zum Glukokortikoid-Rezeptor gekennzeichnet und weist gleichzeitig eine niedrige orale Bioverfügbarkeit auf, sodass die systemische Verfügbarkeit und das Nebenwirkungsrisiko gering sind [7, 8]. Indacaterol wiederum bewirkt eine über 24 Stunden lang anhaltende Bronchodilatation und effektive Symptomkontrolle bei guter Sicherheit sowie Verträglichkeit [9]. In Kombination mit Glycopyrronium und Mometasonfuroat sind ein schneller Wirkungseintritt sowie ein anhaltender Effekt über das gesamte Dosierungsintervall charakteristisch [6]. IND/GLY/MF ist mit einem optionalen Sensor (vom Unternehmen Propeller Health) und dazugehöriger App erhältlich [10]. Welche Patienten kommen aus Ihrer Sicht für den Einsatz infrage? Dr. de Zeeuw: Sensor und App eignen sich besonders für Patienten, die aktiv am Behandlungsmanagement mitwirken wollen. Ihnen wird durch Funktionen wie dem automatisierten Inhalationstagebuch die Möglichkeit geboten, sich einen Einblick in den Therapieverlauf zu verschaffen, die eigene Adhärenz zu kontrollieren und die erhobenen Daten mit dem behandelnden Arzt zu teilen. Darüber hinaus gibt es Situationen, in denen der Sensor und die dazugehörige App besonders für die Angehörigen aus dem familiären
Umfeld eine Hilfestellung bei der Frage sein kann, ob der Betroffene seine Medikamente wirklich zuverlässig einnimmt. Insgesamt bietet der Sensor jedoch besonders für die Patienten selbst ein hilfreiches Instrument zur Selbstkontrolle, mit dem das persönliche Therapiemanagement erleichtert werden kann. Im letzten Jahr habe ich beispielsweise eine 72-jährige Patientin in unserer Praxis auf IND/GLY/MF eingestellt, die daraufhin von einer Verbesserung ihrer Erkrankung berichtete. Im zweiten Schritt haben wir die medikamentöse Therapie um den optionalen Sensor ergänzt. Obwohl die Patientin zuvor bereits durchaus adhärent gewesen ist, hilft ihr die Erinnerung durch den Sensor, die regelmäßige Einnahme im Alltag nicht zu vergessen und die Krankheitskontrolle dadurch noch einmal spürbar zu optimieren. Wie gehen Sie vor, wenn Patienten auch unter inhalativer Maximaltherapie keine ausreichende Krankheitskontrolle erreichen? Dr. de Zeeuw: Bereits während der potenziellen Umstellung der inhalativen Therapie von LABA/ ICS auf eine Dreierkombination prüfen wir, ob perspektivisch auch ein Biologikum infrage kommen könnte. Wenn beim IgE-Spiegel oder der Anzahl der Bluteosinophilen entsprechende Auffälligkeiten bestehen, haben wir für den Fall, dass auch die inhalative Maximaltherapie keine ausreichende Krankheitskontrolle bewirkt, bereits eine potenzielle Eignung für die verfügbaren Biologika abgeklärt und verlieren bei der späteren Umstellung keine Zeit. Für das schwere allergische Asthma ist mit dem Anti-IgE-Antikörper Omalizumab (Xolair®) beispielsweise ein Biologikum verfügbar, mit dem
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
wir in der Praxis als Zusatztherapie bei vielen Patienten über Jahre hinweg eine erhebliche Verbesserung der Erkrankung erzielen konnten. Seit dem vergangenen Jahr können wir auch Betroffenen mit schwerer chronischer Rhinosinusitis mit Nasenpolypen diese Option als Zusatztherapie anbieten, wenn unter intranasalen Kortikosteroiden keine ausreichende Krankheitskontrolle besteht [11]. Herr Dr. de Zeeuw, wir danken Ihnen für das informative Gespräch. E. W.
Literatur 1 Sunjaya AP et al. J Asthma 2021;1:1-14 2 Lommatzsch M et al. Pneumologie 2021; 75:19-30 3 Idzko M et al. Deutsche Gesellschaft für Pneumologie und Beatmungsmedizin e.V. Im Internet: https://pneumologie.de/fileadmin/user_upload/COVID-19/ 20210125_DGP_AsthmaBiologika_Covid-19.pdf 4 Kondla A et al. Respir Med 2016;118: 58-64 5 Nationale VersorgungsLeitlinie Asthma, 4. Auflage 2020. Im Internet: https:// www.leitlinien.de/nvl/asthma 6 Fachinformation Enerzair® Breezhaler® 7 Daley-Yates PT. Br J Clin Pharmacol 2015;80:372-380 8 Ye Q et al. Pulm Ther 2017;3:1-18 9 Beeh KM et al. Eur Respir J 2007;29: 871-878 10 Propeller Health®. QR Rationale for EU Classification-2017-B. 2019-07-24 Update 11 Fachinformation Xolair® © VERLAG PERFUSION GMBH
86
A
m 15. April hat die Europäische Kommission den CD38-Antikörper Isatuximab (Sarclisa®) in Kombination mit Carfilzomib und Dexametha son für die Behandlung von Erwachsenen mit rezidiviertem multiplem Myelom zugelassen, die mindestens eine vorausgegangene Therapie erhalten haben [1]. Damit kann Isatuximab in der EU nun in Kombination mit 2 Standardregimen eingesetzt werden, nachdem es im Juni 2020 in Kombination mit der Standardtherapie aus Pomalidomid und Dexamethason (POMDEX) für die Behandlung des rezidivierten und refraktären multiplen Myeloms bei Erwachsenen zugelassen wurde, die mindestens 2 vorausgegangene Therapien, darunter Lenalidomid und einen Proteasom-Inhibitor, erhalten haben und unter der letzten Therapie eine Krankheitsprogression zeigten. Die neue Dreierkombination aus Isatuximab plus Carfilzomib und Dexamethason (Kd) bietet Patienten mit rezidiviertem multiplem Myelom jetzt bereits früher im Verlauf der Krankheitsprogression eine weitere Behandlungsoption und hat das Potenzial, eine Standardtherapie bei der Erkrankung zu werden, zumal das Kd-Regime in der Rezidivsituation bereits häufig eingesetzt wird. Risiko für Krankheitsprogression oder Tod um fast die Hälfte reduziert
Grundlage für die zweite Zulassung von Isatuximab sind die Ergebnisse der randomisierten, offenen Phase-III-Studie IKEMA [2, 3], an der insgesamt 302 Patienten mit rezidiviertem multiplem Myelom teilnahmen, die nach 1 – 3 vorangegangenen Therapielinien
NEUE UND BEWÄHRTE ARZNEIMITTEL
Isatuximab in zweiter Indikation zur Behandlung des rezidivierten multiplen Myeloms zugelassen einen Rückfall erlitten hatten. Die Studienteilnehmer wurde 3 : 2 auf Isatuximab plus Kd (Isa-Kd; n = 179) und Kd (n = 123) randomisiert. Die Isa-Kd-Gruppe erhielt einmal wöchentlich 10 mg/kg Isatuximab i.v. für 4 Wochen, danach alle 2 Wochen. Beide Gruppen bekamen 2 × wöchentlich 20 mg/m2 Carfilzomib an den Tagen 1 – 2 und danach 56 mg/m2 für 3 von 4 Wochen sowie 2 × wöchentlich 20 mg Dexamethason. Primärer Studienendpunkt war das progressionsfreie Überleben (PFS). Während der Median des PFS, also der Zeit bis zur Krankheitsprogression oder bis zum Tod des Patienten, unter der Behandlung mit dem Kd-Regime 19,15 Monate betrug, war er bei den Patienten, die Isatuximab in Kombination mit Carfilzomib und Dexamethason erhielten, zum Zeitpunkt der vorab festgelegten Zwischenanalyse noch nicht erreicht. Die Isa-KdTherapie verringerte im Vergleich zur alleinigen Standardtherapie mit dem Kd-Regime das Risiko für Krankheitsprogression oder Tod um 47 % (HR: 0,531, 99%-KI: 0,318 – 0,889; p = 0,0007). Dieser Vorteil der Triplett-Kombination wurde über alle Subgruppen hinweg beobachtet, auch bei schwer behandelbaren Patienten mit höherem Alter, einer Hochrisiko-Zytogenetik oder Nierenstörungen.
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Die Tiefe des Ansprechens auf die Therapie wurde mithilfe von sekundären Endpunkten untersucht. Dazu gehörten die Gesamtansprechrate (ORR), eine komplette Remission (CR), eine sehr gute partielle Remission (VGPR) und die Negativität in Bezug auf eine minimale Resterkrankung (MRD). Die ORR war mit 86,6 % unter der Isa-Kd-Therapie und 82,9 % unter dem Kd-Regime in den beiden Gruppen vergleichbar (p = 0,1930). Die CR-Rate betrug in der Isa-KdGruppe 39,7 % versus 27,6 % in der Kd-Gruppe. Nach Korrektur einer möglichen Antikörperinterferenz bei der Bestimmung der CR-Rate durch massenspektrometrische Verfahren wurde effektiv eine mögliche CR-Rate von bis zu 45,8 % im Isa-Kd-Arm bestimmt. Ein deutlicher Vorteil für die Isa-Kd-Therapie zeigte sich hinsichtlich des Erreichens einer mindestens sehr guten partiellen Remission: Die VGPR-Rate war mit 72,6 % in der Isa-Kd-Gruppe signifikant höher als in der KdGruppe mit 56,1 % (p = 0,0011). Auch die minimale Resterkrankung war in der Isa-Kd-Gruppe bei signifikant mehr Patienten negativ (29,6 % vs. 13 %; p = 0,0004). Das bedeutet, dass fast 30 % der mit dem Isatuximab-Regime behandelten Patienten ein derartig tiefes Ansprechen zeigten, dass bei ihnen © VERLAG PERFUSION GMBH
87
NEUE UND BEWÄHRTE ARZNEIMITTEL
Isatuximab Isatuximab (Sarclisa®) ist ein monoklonaler IgG1-Antikörper, der gegen das CD38-Molekül gerichtet ist [1]. Bei CD38 handelt es sich um ein auf den Tumorzellen des multiplen Myeloms durchgängig und in großen Mengen exprimiertes transmembranes Glykoprotein, das sowohl als Rezeptor als auch als Ektoenzym aktiv ist. Dadurch bietet sich das CD38-Molekül als Zielstruktur für Antikörperbasierte Therapieansätze beim multiplen Myelom an. Isatuximab bindet an ein spezifisches Epitop des humanen CD38Oberflächenantigens, d.h. an einen definierten Molekülabschnitt, der eine spezifische Immunantwort triggern kann. Dadurch werden mehrere Folgereaktionen ausgelöst: • die Induktion der Antikörper-abhängigen zellulären Zytotoxizität, der Komplement-abhängigen Zytotoxizität und der Antikörper-abhängigen zellulären Phagozytose, • die Hemmung der ektoenzymatischen Aktivität von CD38, • eine Immunmodulation sowie • die direkte Induktion der Apoptose, d.h. des programmierten Tumorzelltodes.
mittels Next-Generation Sequencing mit einer Sensitivität von 10-5 keine Myelomzellen mehr nachweisbar waren. Die Daten für das Gesamtüberleben waren zum Zeitpunkt der Zwischenanalyse noch nicht vollständig [2, 3].
rapiebedingten Nebenwirkungen war in beiden Armen ähnlich. Von den mit der Isatuximab-Kombinationstherapie behandelten Patienten brachen weniger die Therapie aufgrund von Nebenwirkungen ab als im Kd-Arm (8,5 % vs. 13,9 %).
Durch die nach der Zulassung der Kombination aus Isatuximab plus Pomalidomid und Dexamethason für die Drittlinienbehandlung nun bereits für die Zweitlinie zugelassene Addition des CD38-Antikörpers zu Carfilzomib und Dexamethason erhöht sich die Chance, das Ansprechen und damit die Prognose der refraktären bzw. rezidivierten Patienten weiter zu verbessern. Da die Kombination von Isatuximab mit Carfilzomib und Dexamethason in der IKEMA-Studie das progressionsfreie Überleben verbesserte und zu tieferen Remissionen führte als Carfilzomib plus Dexamethason, könnte sich diese Triplett-Therapie zum neuen Standard bei rezidivierten/refraktären Patienten mit multiplem Myelom entwickeln. Brigitte Söllner, Erlangen
Fazit Daten zur Sicherheit belegen gute Verträglichkeit
Die häufigsten Nebenwirkungen (≥20 %) waren Infusionsreaktionen (45,8 %), Hypertonie (36,7 %), Diarrhö (36,2 %), eine Infektion der oberen Atemwege (36,2 %), Pneumonie (28,8 %), Fatigue (28,2 %), Dyspnoe (27,7 %), Schlaflosigkeit (23,7 %), Bronchitis (22,6 %) und Rückenschmerzen (22,0 %). Die Rate an schweren (59,3 % unter Isa-Kd vs. 57,4 % unter Kd) und tödlichen (3,4 % vs. 3,3 %) the-
Obwohl durch die Entwicklung von Immunmodulatoren, Proteasom-Inhibitoren sowie myelomspezifischen Antikörpern bereits erhebliche Fortschritte in der Behandlung erzielt wurden, bleibt das multiple Myelom bislang nicht heilbar. Insbesondere für die Zweit- und Drittlinie besteht ein hoher Bedarf an neuen Therapieoptionen, da intensiv vorbehan delte Patienten oft refraktär gegenüber den Standardtherapeutika sind.
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Literatur 1 European Medicines Agency. Sarclisa (isatuximab). Im Internet: https://www. ema.europa.eu/en/medicines/human/ EPAR/sarclisa 2 Moreau P et al. Isatuximab plus carfilzomib and dexamethasone vs carfilzomib and dexamethasone in relapsed/refractory multiple myeloma (IKEMA): Interim analysis of a phase 3, randomized, open label study. EHA25 virtual 2020, Abstract LB2603 3 Moreau P et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, openlabel, randomised phase 3 trial. Lancet online first, June 04, 2021. DOI: https:// doi.org/10.1016/S0140-6736(21)00592-4
© VERLAG PERFUSION GMBH
88
NEUE UND BEWÄHRTE ARZNEIMITTEL
Guselkumab – ein selektiver Interleukin-23-Hemmer zur Behandlung der aktiven Psoriasis-Arthritis*
D
ie Psoriasis-Arthritis (PsA) manifestiert sich häufig in Form einer peripheren Arthritis, Spondylitis, Daktylitis und Enthesitis sowie Plaque-Psoriasis und Nagelveränderungen. Viele Patienten leiden zudem an Fatigue, reduzierter körperlicher Funktionsfähigkeit, Schlafstörungen sowie Einschränkungen der Arbeitsfähigkeit sowie in ihrem Sozialleben [1, 2]. Eine maßgebliche Rolle in der Pathogenese der PsA kommt dem Schlüsselzytokin Interleukin(IL)-23 zu [3, 4]. Guselkumab (Tremfya®) ist der erste zugelassene Antikörper, der selektiv IL-23 hemmt. Er ist seit November 2020 in der BiologikaErstlinien-Therapie allein oder in Kombination mit Methotrexat (MTX) für die Behandlung der aktiven PsA bei erwachsenen Patienten indiziert, wenn das Ansprechen auf eine vorherige nicht biologische krankheitsmodifizierende antirheumatische (DMARD-)Therapie unzureichend gewesen ist oder diese nicht vertragen wurde [5]. Darüber hinaus kann Guselkumab bei Erwachsenen mit mit* G uselkumab (Tremfya®) ist als Monothe rapie oder in Kombination mit MTX für die Behandlung der aktiven PsA bei erwachsenen Patienten indiziert, die auf eine vorangegangene krankheitsmodifizierende antirheumatische (disease-modifying antirheumatic drug, DMARD-) Therapie unzureichend angesprochen oder diese nicht vertragen haben [5].
telschwerer bis schwerer PlaquePsoriasis verabreicht werden, die für eine systemische Therapie infrage kommen [5]. Wirkmechanismus von Guselkumab
Guselkumab ist ein voll humaner, monoklonaler IgG1λ-Antikörper, der selektiv gegen die p19-Untereinheit von IL-23 gerichtet ist. Guselkumab verhindert, dass IL-23 an dessen Rezeptor bindet und blockiert so den IL-23-Signalweg. In der Folge können sich die TH17Zellen nicht weiter teilen und ihre Anzahl reduziert sich. Damit greift Guselkumab sehr früh in die Pathogenese der PsA ein. Indem Guselkumab die Produktion verschiedener nachgelagerter proinflammatorischer Zytokine hemmt, reguliert es die überschießende Immunantwort [4, 5].
Daten zur Wirksamkeit in den Zulassungsstudien
Die Wirksamkeit von Guselkumab wurde in den randomisierten, doppelblinden, dreiarmigen und placebokontrollierten Phase-III-Studien DISCOVER-1 und DISCOVER-2 bei Studienteilnehmern untersucht, die eine aktive PsA trotz Standardtherapien aufwiesen (Einzelheiten zum Studiendesign in Tab. 1) [6, 7]. Während für DISCOVER-1 (n = 381) etwa 30 % der Studien teilnehmer mit einem oder zwei Tumornekrosefaktor-alpha-Inhibitoren vorbehandelt waren, wurden in DISCOVER-2 (n = 739) ausschließlich Biologika-naive Patienten eingeschlossen. In beiden Studien wurden die Studienteilnehmer im Verhältnis 1:1:1 randomisiert und bis Woche 24 mit Guselkumab (100 mg s.c. in Woche 0 und 4, gefolgt von einer Erhaltungsdosis alle 4 Wochen
Interleukin-23 als Angriffspunkt der PsA-Therapie IL-23 kommt die Rolle eines regulatorischen Schlüsselzytokins zu, das früh in der Entzündungskaskade Weichen stellt. Es wird vor allem von dendritischen Zellen und Makrophagen produziert. Indem es an den IL-23-Rezeptor der TH17-Zellen bindet, stimuliert es deren Überleben und Proliferation. Die TH17-Zellen sezernieren daraufhin weitere proinflammatorische Zytokine wie IL-12, IL-17, IL-22 und TNF-α. Bei PsA-Patienten infiltrieren aktivierte T-Zellen und Makrophagen das Synovium von Gelenken und sezernieren proinflammatorische Zytokine [3, 4].
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
© VERLAG PERFUSION GMBH
89
NEUE UND BEWÄHRTE ARZNEIMITTEL
Studie
Patienten
Medikation
DISCOVER-1 [6, 8]
• 3 81 Patienten mit aktiver PsA, definiert als mindestens jeweils 3 geschwollene und druckschmerzhafte Gelenke sowie als erhöhter Wert des C-reaktiven Proteins (CRP) ≥0,3 mg/dl zu Studieneinschluss, die unzureichend auf Standardtherapien (konventionelle DMARDs, Apremilast, nicht-steroidale Antirheumatika [NSAR]) angesprochen oder diese nicht vertragen haben. • R und 30 % waren zuvor mit 1 oder 2 TNF-aInbitoren behandelt worden.
Die Patienten erhielten 1:1:1 randomisiert: • G uselkumab q4w (n = 128): 100 mg s.c. alle 4 Wochen bis Woche 52 oder • G uselkumab q8w (n = 127): 100 mg s.c. in den Wochen 0, 4 und anschließend alle 8 Wochen bis Woche 52 oder • P lacebo (n = 126) bis Woche 24, danach Crossover in den Guselkumab q4w-Arm
DISCOVER-2 [7, 9]
• 7 39 Biologika-naive Patientienten mit aktiver PsA, definiert als mindestens jeweils 5 geschwollene und druckschmerzhafte Gelenke sowie als erhöhter CRP-Wert ≥0,6 mg/ dl zu Studieneinschluss, die unzureichend auf Standardtherapien (konventionelle DMARDs, Apremilast, NSAR) angesprochen oder diese nicht vertragen haben. Die Patienten erhielten 1:1:1 randomisiert: • G uselkumab q4w (n = 246): 100 mg s.c. alle 4 Wochen bis Woche 52 oder • G uselkumab q8w (n = 248): 100 mg s.c. in den Wochen 0, 4 und anschließend alle 8 Wochen bis Woche 52 oder • P lacebo (n = 247) bis Woche 24, danach Crossover in den Guselkumab q4w-Arm
Tabelle 1: Design der Zulassungsstudien DISCOVER-1 und DISCOVER-2 [8, 9].
[q4w] oder alle 8 Wochen [q8w]) oder Placebo behandelt. Nach Woche 24 wechselten alle Patienten der Placebogruppe in den Guselkumab q4w-Arm [6, 7]. Die Daten der anschließenden aktiven Behandlungsperiode bis Woche 52 wurden in beiden Studien deskriptiv ausgewertet [8, 9]. Der primäre Endpunkt, ein mindestens 20%iges Ansprechen nach den Kriterien des American College of Rheumatology (ACR20) wurde in beiden Studien erreicht [6, 7]: In DISCOVER-1 wiesen signifikant mehr
Patienten (%)
80 60 40
Patienten unter Guselkumab q8w (52 %) ein ACR20-Ansprechen zu Woche 24 auf als unter Placebo (22 %); in DISCOVER-2 waren es 64 % versus 33 % (jeweils p<0,0001; Non-Responder-Imputation [NRI]). Das ACR20-Ansprechen konnte in beiden Studien unter Guselkumab q8w-Gabe über ein Jahr aufrechterhalten werden. In DISCOVER-1 nahm der Anteil der Patienten mit einem ACR20-Ansprechen zu Woche 52 numerisch auf 59,8 % zu, in DISCOVER-2 auf 74,6 % (Abb. 1) [8, 9].
74,6 %
64 %
ACR20
Primärer Endpunkt p<0,0001 vs. Placebo (33%)
20 0 0 4 8 12 16 20 24 28 Woche
36
44
52
Abbildung 1: DISCOVER-2: ACR20-Ansprechen unter Guselkumab 100 mg q8w (n = 248; NRI) [7, 9]. JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Zu den wesentlichen sekundären Endpunkten zählte in DISCOVER-1 und -2 u. a. ein ACR50/70Ansprechen zu Woche 24 [6, 7]. In DISCOVER-1 erreichten signifikant mehr Patienten unter Guselkumab q8w (30 %) ein ACR50Ansprechen zu Woche 24 als unter Placebo (9 %; p<0,0001, NRI). Ein ACR70-Ansprechen wiesen 12 % der Patienten unter Guselkumab q8w bzw. 6 % unter Placebo auf [6]. Aufgrund des hierarchischen Testverfahrens ließen sich hierzu keine Aussagen zur statistischen Signifikanz treffen, was ebenso auf die entsprechenden Ergebnisse zum ACR50/70-Ansprechen in Woche 24 in DISCOVER-2 zutraf. In DISCOVER-2 ergaben sich Hinweise auf ein nominell besseres ACR50/70-Ansprechen unter Guselkumab q8w: 32 % der Patienten im Guselkumab-Arm erreichten ein ACR50- sowie 18 % ein ACR70Ansprechen zu Woche 24. Bei den Patienten im Placeboarm lagen diese Ansprechraten bei 14 % bzw. 4 % [7]. In den deskriptiven Auswertungen zu Woche 52 konnte in beiden Studien eine Fortsetzung © VERLAG PERFUSION GMBH
NEUE UND BEWÄHRTE ARZNEIMITTEL
90
dieser positiven Tendenz unter Guselkumab q8w beobachtet werden [8, 9]. Daten zur Verträglichkeit
In den Zulassungsstudien DISCOVER-1 und -2 erwies sich Guselkumab ab Woche 24 bis Woche 60 bzw. 52 als gut verträglich [8, 9]. In einer gepoolten Analyse der beiden Studien DISCOVER-1 und -2 weist Guselkumab bei PsA eine Verträglichkeit auf Placeboniveau auf: Der Anteil schwerer unerwünschter Ereignisse (≥1) lag bis Woche 24 bei 1,9 % unter Guselkumab q8w (n = 375) und bei 3,2 % unter Placebo (n = 372). Unerwünschte Ereignisse wurden in der gepoolten Analyse bis Woche 24 unter Guselkumab bei 48,5 % und unter Placebo bei 47,3 % der Patienten beobachtet [8]. Elisabeth Wilhelmi, München
CP-229292
Literatur 1. Ogdie A et al. Rheumatology 2020;59: i37-i46 2. Ritchlin CT et al. N Engl J Med 2017; 376(21):2095-2096 3. Coates LC et al. Sem Arth Rheum 2016; 46:291-304 4. Gooderham MJ et al. JEADV 2018;32: 1111-1119 5. Fachinformation Tremfya®, Stand: De zember 2020 6. Deodhar A et al. Lancet 2020;395: 1115-1125 (incl. Supplementary Appendix) 7. Mease PJ et al. Lancet 2020;395:11261136 (incl. Supplementary Appendix). 8. Ritchlin CT et al. RMD Open 2021; 7:e001457 9. McInnes IB et al. Arthritis Rheum 2021; doi:10.1002/art.41553 10. Rahman P et al. EULAR 2020; Poster FRI0359 Mit freundlicher Unterstützung der Janssen-Cilag GmbH
Kinasehemmer Pemigatinib – die erste Option zur Zweitlinienbehandlung des Gallengangkarzinoms
S
eit Mai steht in Deutschland mit Pemigatinib (Pemazyre®) ein neuer oraler Kinasehemmer für die zielgerichtete Krebstherapie auf dem deutschen Markt zur Verfügung. Zugelassen ist er zur Monotherapie von Erwachsenen mit lokal fortgeschrittenem oder metastasiertem Cholangiokarzinom (CCA), das nach mindestens einer vorherigen systemischen Therapielinie weiterhin progredient ist und bei dem Fusionen oder Rearrangements des Fibroblasten-WachstumsfaktorRezeptors 2 (Fibroblast Growth Factor Receptor 2, FGFR2) nachweisbar sind [1]. Eine operative Entfernung des CCA ist derzeit die einzige potenziell kurative Option. Die palliative Erstlinie sieht eine Chemotherapie auf Basis von Gemcitabin und Cisplatin vor, eine Standardtherapie, die darüber hinausgeht, gibt es bislang nicht, sodass Patienten mit fortgeschrittenem CCA eine sehr ungünstige Prognose haben. Die nun verfügbare zielgerichtete Therapie mit Pemigatinib stellt zumindest für die Gruppe der Patienten mit nachgewiesenen FGFR2Fusionen oder -Umlagerungen einen erheblichen Fortschritt dar, denn in der Zulassungsstudie wurde in vielen Fällen ein Ansprechen erreicht [2].
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Genetische Veränderungen als onkogene Treiber
Das CCA entwickelt sich im Gallengang und wird nach seinem Ursprung in 2 Typen unterteilt: Das intrahepatische Cholangiokarzinom (iCCA) tritt im Gallengang innerhalb der Leber auf, das extrahepatische Cholangiokarzinom (eCCA) im Gallengang außerhalb der Leber. Bei beiden Formen sind oft genetische Veränderungen nachweisbar (Abb. 1), so auch Fusionen oder Rearrangements des FGFR2, die fast ausschließlich beim iCCA beobachtet werden und bei 10 – 16 % der Patienten nachweisbar sind [3, 4, 5, 6]. Sie gelten als starke onkogene Treiber, da sie eine ligandenunabhängige Rezeptor-Dimerisierung fördern. Die FGFR2-Dimerisierung aktiviert den FGFR-Signalweg, der an zellulären Prozessen wie Proliferation, Überleben, Migration und Angiogenese beteiligt ist [1]. FGFR-Inhibition als Wirkprinzip
In diese Signalkette greift Pemigatinib ein: Als selektiver Hemmer der FGFR-Isoformen 1, 2 und 3 inhibiert er die FGFR-Phosphorylierung und -Signalübertragung. Um Patienten zu identifizieren, die von © VERLAG PERFUSION GMBH
NEUE UND BEWÄHRTE ARZNEIMITTEL
RISIKOFAKTOREN UND MOLEKULARE VERÄNDERUNGEN MALIGNER BILIÄRER TUMOREN
91
iCCA Hauptrisikofaktoren: Cholangitis, Adipositas, Diabetes mellitus, chronische Hepatitis B/C, Hepatolithiasis, Lynch-Syndrom Mutationen, die zu molekularen Alterationen führen: FGFR 1-3, IDH 1/2, BAP1, ARID1A-Alteration, EPHA2
eCCA Hauptrisikofaktoren: Cholangitis, Lynch-Syndrom, Gallensteine Mutationen, die zu molekularen Alterationen führen: PRKACA- oder PRKACB-Fusion, ELF3, ARID1B-Mutation Subtyp mit schlechter Prognose und hoher ImmunCheckpoint-Aktivität: TP53, BRCA 1/2, PIK3CA-Mutation
Gallenblasenkarzinom Hauptrisikofaktoren: Adipositas, Diabetes mellitus, chronische Cholezystitis, Gallensteine Mutationen, die zu molekularen Alterationen führen: EGFR, ERBB3, PTEN, ARID2, MLL2, MLL3, TERT-Promoter-Mutation, APOBEC-Signatur
iCCA und eCCA (gemeinsame Risikofaktoren): KRAS, SMAD4,
ARID1A, GNAS
Abbildung 1: Risikofaktoren und molekulare Veränderungen maligner biliärer Tumoren. FGFR2-Fusionen oder -Rearrangements treten fast Mod. Ghidini et al., Cancer Manag Res. 2019; 11: 379–388. ausschließlich bei intrahepatischen Cholangiokarzinomen (iCCA) auf. eCCA = extrahepatische Cholangiokarzinome (mod. nach Ghidini4et al. Cancer Manag Res 2019;11:379-388. © Incyte).
Pemigatinib profitieren könnten, sind molekularbiologische Untersuchungen des Tumors notwendig, mit denen sich FGFR2-Fusionen und -Rearrangements mit bekannten und neuartigen Partnergenen nachweisen lassen. Dies ist mit einem von der FDA zugelassenen Test (Next-Generation-Sequenc ing, NGS) möglich, der auch in der Zulassungsstudie von Pemigatinib angewendet wurde und nun im klinischen Alltag implementiert werden soll. Ansprechrate von 37 Prozent
Die EU-Zulassung basiert auf den Daten der unverblindeten einarmigen Studie FIGHT-202, in der die Sicherheit und Wirksamkeit von Pemigatinib bei Erwachsenen (Alter ≥18 Jahre) mit vorbehandeltem, lokal fortgeschrittenem oder metastasierendem Cholangiokarzinom mit dokumentiertem FGF/FGFRStatus geprüft wurde [1]. Alle Patienten hatten mindestens 1 vorherige systemische Therapielinie erhalten (bei 27,1 % waren 2 und bei 12,1 % 3 oder mehr Therapien vorausgegangen).
Die Patienten wurden einer von 3 Kohorten zugeteilt: Kohorte A mit FGFR2-Fusionen oder -Rearrangements, Kohorte B mit anderen FGF/FGFR-Genveränderungen oder Kohorte C ohne FGF/ FGFR-Genveränderungen. Alle Patienten erhielten einmal täglich oral die empfohlene Dosierung von 13,5 mg Pemigatinib in einem 21-Tage-Zyklus (14 Tage mit/7 Tage ohne Medikation) bis Endpunkte
zur radiologisch festgestellten Krankheitsprogression oder inakzeptablen Toxizität. Die mediane Beobachtungsdauer betrug 17,8 Monate. Primärer Endpunkt der Studie war die Gesamtansprechrate (ORR) in Kohorte A (n = 108). Zu den sekundären Endpunkten zählten die ORR, das progressionsfreie Überleben (PFS), das Gesamtüberleben (OS), die Dauer der Remission Kohorte A (FGFR2-Fusion oder -Rearrangement) Hinsichtlich der Wirksamkeit auswertbare Population (n = 108)
Gesamtansprechrate (95%-KI)
37,0 % (27,94 – 46,86)
• Vollständiges Ansprechen (n)
3,7 % (4)
• Teilweises Ansprechen (n) Mediane Dauer des Ansprechens (95%KI)
33,3 % (36) 8,08 Monate (5,65 – 13,14)
Kaplan-Meier-Schätzungen der Dauer des Ansprechens (95%-KI) • 3 Monate
100,0 (100,0 – 100,0)
• 6 Monate
66,0 (48,0 – 79,1)
• 9 Monate
47,6 (30,2 – 63,1)
• 12 Monate
37,5 (21,3 – 53,7)
Tabelle 1: Ergebnisse der Studie FIGHT-202 zur Wirksamkeit von Pemigatinib (Pemazyre®) bei vorbehandelten CCA-Patienten mit FGFR2-Fusionen oder -Rearrangements [1].
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
© VERLAG PERFUSION GMBH
NEUE UND BEWÄHRTE ARZNEIMITTEL
92
(DOR), die Krankheitskontrollrate (DCR) und die Sicherheit in allen 3 Kohorten. Bei den 108 Patienten mit FGFR2Fusionen oder -Rearrangements führte die Monotherapie mit Pemigatinib zu einer Gesamtansprechrate von 37 % (primärer Endpunkt), wobei es sich meistens um ein teilweises und nur selten um ein vollständiges Ansprechen handelte (Tab. 1). Die mediane Ansprechdauer betrug 8 Monate, das mittlere progressionsfreie Überleben 7 Monate. Informationen zur Sicherheit
Die Therapie mit Pemigatinib wurde in den Studien im Allgemeinen
Meilenstein in der HIV-Forschung:
Erste langwirksame Kombinationstherapie zur intramuskulären Injektion Seit dem 1. Mai 2021 steht für virologisch supprimierte Menschen mit HIV-1 in Deutschland die erste langwirksame Injektionstherapie zur Verfügung. Die Kombinationstherapie aus Rekambys® (Rilpivirin-Depot-Injektionssuspension, Janssen) und Vocabria® (Cabotegravir-Depot-Injektionssuspension, ViiV Healthcare) wird ab der Erhaltungsphase alle 2 Monate injiziert – statt einer bisher notwendigen täglichen Tabletteneinnahme. Die Relevanz eines langwirksamen Regimes wird aus den Bedürfnissen und Hoffnungen der Patienten ersichtlich: Positive Perspectives, die weltweit größte Studie, die sich auf Aussagen von HIV-Patienten stützt und von ViiV Health-
gut vertragen. Das Nebenwirkungsprofil von Pemigatinib erscheint handhabbar und entspricht dem, was bei dieser Substanzklasse zu erwarten ist. Die häufigsten schwerwiegenden Nebenwirkungen waren Hyponatriämie und ein Anstieg des Kreatinins im Blut. Die häufigsten Nebenwirkungen waren Hyperphosphatämie, Alopezie, Diarrhö, Nageltoxizität, Ermüdung, Übelkeit, Geschmacksstörung, Stomatitis, Obstipation, Mundtrockenheit, trockenes Auge, Arthralgie, Hypophosphatämie, trockene Haut und palmar-plantares Erythrodysästhesiesyndrom. Als Warnhinweise und Vorsichtsmaßnahmen für Pemazyre® nennt die Fachinformation hohe und niedrige Phosphatwerte im Blut,
Seh- oder Augenprobleme, ein Anstieg des Blut-Kreatinin-Wertes und bei schwangeren Frauen das Risiko einer Schädigung des Fetus [1]. Brigitte Söllner, Erlangen
care durchgeführt wurde, zeigt, dass 54,7 % der befragten Patienten einer langwirksamen Therapie offen gegenüber sind. Die tägliche Medikamenteneinnahme hingegen empfinden 58,4 % als eine tägliche Erinnerung an die eigene HIV-Erkrankung und 37,9 % befürchten, dass Menschen in ihrem Umfeld dadurch von ihrer HIV-Infektion erfahren könnten. Vocabria® + Rekambys® ist indiziert für die Behandlung erwachsener Patienten mit dem humanen Immundefizienz-Virus-1 (HIV-1), die unter ihrem stabilen aktuellen antiretroviralen Therapieschema virussupprimiert sind (HIV-RNA <50 Kopien/ml). Die Patienten dürfen keine derzeitigen oder früheren Hinweise auf Virusresistenzen gegen nichtnukleosidische Reverse-Transkriptase-Inhibitoren (NNRTI) oder Integrase-Inhibitoren (INI) aufweisen. Außerdem
darf es unter diesen Therapien zu keinem virologischen Versagen gekommen sein. Die Behandlung sollte mit einer Einleitungsphase von mindestens 28 Tagen beginnen, bei der die Wirkstoffe Cabotegravir und Rilpivirin jeweils täglich oral eingenommen werden. Im zweiten und dritten Monat folgt die Initiierungsphase, in welcher die Vocabria® + Rekambys®-Injektionen intramuskulär in je eine Gesäßseite durch medizinisches Fachpersonal appliziert werden. Ab dem fünften Monat erfolgen die Injektionen alle 2 Monate. Die Zulassung für Vocabria® + Rekambys® basiert auf den Ergebnissen der 3 Phase-III-Studien ATLAS-2M, ATLAS und FLAIR mit mehr als 1.200 Probanden in 16 Ländern. F. S.
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Literatur 1 Fachinformation Pemazyre®; Stand: März 2021 2 Abou-Alfa GK et al. Lancet Oncol 2020; 21:671-685 3 Lowery MA et al. Clin Cancer Res 2018; 24:4154-4161 4 Graham RP et al. Hum Pathol 2014; 45:1630-1638 5 Ang CJ. Gastroenterol Hepatol 2015; 30:1116-1122 6 Ross JS et al. The Oncologist 2014; 19:235-242
© VERLAG PERFUSION GMBH
KONGRESSE
Gar nicht übel:
Therapie der Emesis gravidarum mit Cariban® Übelkeit ist oft eines der ersten Anzeichen, mit dem Frauen ihre Schwangerschaft wahrnehmen: Im ersten Drittel leiden 80 % aller Frauen an Übelkeit und Erbrechen (Emesis gravidarum). Beim FOKO Fortbildungskongress 2021 diskutierten Experten aus Klinik und Praxis Sicherheitsdaten und praktische Erfahrungen beim Einsatz von Cariban®, der spezifischen Wirkstoffkombination aus 10 mg Doxylamin (als Doxylaminsuccinat) und 10 mg Pyridoxin (als Pyridoxinhydrochlorid), die weltweit seit mehr als 50 Jahren in mehr als 33 Millionen Schwangerschaften eingesetzt wird und seit September 2019 in Deutschland verfügbar ist. „Mehr als eine Belästigung“
Der Blick auf Inzidenzdaten zeigt, dass viele Schwangere betroffen sind: „Übelkeit und Erbrechen in der Schwangerschaft sind mehr als eine Belästigung“, erläuterte Dr. Matthias Krick, Moers. Bei etwa 10 % der Frauen dauern die Beschwerden während der gesamten Schwangerschaft an. „Emesis gravidarum ist trotz der Häufigkeit noch nicht im Fokus“, konstatierte der niedergelassene Frauenarzt. Er grenzte Emesis gravidarum klar von der selteneren Hyperemesis gravidarum (Inzidenz 0,3 – 3 %) ab: „Die Hyperemesis-Patientin gehört klassischerweise stationär behandelt. Die Mehrzahl der Frauen mit Emesis gravidarum wird in der Niederlassung mit einer
Vielzahl an Therapieansätzen behandelt. Es gibt keine echte deutsche Leitlinie – jeder macht was er will.“ Seit September 2019 ist mit Cariban® eine neue Therapieoption in Deutschland verfügbar. Krick beschrieb die Kombination aus Doxylamin, einem Antihistaminikum der ersten Generation, und Pyridoxin, einem wasserlöslichen Vitamin B6: „Cariban® ist für uns eine neue Therapiemöglichkeit, letztendlich aber eine alte, im besten Sinne des Wortes – eine bekannte und langjährig überprüfte Therapieoption.“ Mit Cariban® haben Mediziner im Praxisalltag nun ein zugelassenes Therapeutikum an der Hand, das in klinischen Studien eine bestätigte Wirksamkeit gezeigt hat. Welche Therapieleitlinien gibt es?
Ein wichtiges Ziel all der Maßnahmen bei Schwangerschaftsübelkeit ist es, die schwere Form der Hyperemesis gravidarum zu verhindern, erläuterte Dr. Wolfgang Paulus, Ulm: „Die Empfindlichkeit des Embryos gegenüber toxischen Einflüssen hängt von seinem Entwicklungsstadium ab. Die empfindlichste Phase der Organogenese ist ausgerechnet die Phase, in der wir mit dem Problem der Hyperemesis zu kämpfen haben und in der wir die Schwangeren mit Übelkeit betreuen und behandeln müssen. Deshalb ist es genau die Phase, für die wir besonders kritisch hinterfragt werden.“ In Deutschland gibt es hier aktuell keine Leitlinie. Paulus berichtete, dass eine Übersichtsarbeit zur Arzneimittelsicherheit im Jahr 2015 Medikamente zum Einsatz bei Übelkeit in der Schwangerschaft analog zu den damals üblichen
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
93
US-amerikanischen FDA-Kriterien eingestuft hat: Doxylamin und Pyridoxin (Cariban®) wurden in der Kategorie A aufgeführt, bei der aus kontrollierten Studien weder Hinweise eines Risikos für den Fötus im ersten Trimenon noch in späteren Schwangerschaftsphasen vorliegen. Dimenhydrinat, Diphenhydramin, Meclozin wurden mit Kategorie B als tolerabel eingestuft – allerdings auf deutlich geringerer Datenbasis. Für die Anwendung von Metoclopramid und Ondansetron gibt es von den Zulassungsbehörden inzwischen Vorbehalte. Wie Paulus ausführte, empfiehlt die Leitlinie der US-amerikanischen Fachgesellschaft ACOG (American College of Obstetricians and Gynecologists) die Kombination aus Doxylamin und Pyridoxin als First-Line-Präparat bei persistierender Übelkeit und Erbrechen in der Schwangerschaft, wenn nichtpharmakologische Methoden keinen ausreichenden Erfolg haben. „Damit sind wir jetzt in der Lage, leitliniengerecht – was die amerikanischen Leitlinien betrifft – vorzugehen.“ Die Arzneimittelsicherheit von Doxylamin wird in 12 Kohorten- und 5 Fall-KontrollStudien mit über 200.000 Patientinnen dokumentiert. Der PUQE-Score – Unterstützung in der täglichen Praxis
Die Schwangerschaftsübelkeit könnte in der Frühschwangerschaft ein Warnsignal für Frauen sein, auf ihre Ernährung zu achten und potenziell schädliche Speisen zu meiden, vermutete Dr. Susanne Hampel, Berlin. „An sich ist es ja ein gutes Zeichen, dass ihnen übel ist. Sie merken am steigenden Schwangerschaftshormon, dass © VERLAG PERFUSION GMBH
KONGRESSE
94
ihre Schwangerschaft gut verläuft.“ Hampel appellierte an die niedergelassenen Kollegen, die Schwangerschaftsübelkeit auch zu dokumentieren: Die ICD-10-Codierung O21.- beschreibt „Erbrechen, das die Schwangerschaft verkompliziert“ bis hin zu „übermäßige[m] Erbrechen während der Schwangerschaft“. Eine weitere Möglichkeit ist, Schwangerschaft und Übelkeit separat zu codieren. Weil Beschwerden rund um Übelkeit und Erbrechen sehr subjektiv wahrgenommen werden, kann es schwierig sein, den Schweregrad der Symptomatik zu erfassen. „Viele der betroffenen Frauen haben nicht nur am frühen Morgen Symptome, sondern sind über den gesamten Tag damit belastet“, sagte sie. Zur Diagnostik nutzt sie im Praxisalltag auch den von kanadischen Gynäkologen entwickelten PUQE-Score (Pregnancy Unique Quantification of Emesis and Nausea), den Schwangere schon im Wartebereich ausfüllen können: Mit 3 einfachen Fragen werden die Symptome Übelkeit, Erbrechen und Würgereiz in 3 Schweregraden abgefragt und ergeben einen Score von leicht über mittelschwer bis schwer. An 2 Fallbeispielen verdeutlichte Hampel ihre positiven Erfahrungen mit Cariban® bei mittelschwer bis schwer betroffenen Patientinnen: „Wir sehen bereits nach einer Woche der Einnahme eine Abnahme des PUQE-Scores.“ Individualisierte Therapie – für Schwangere rezeptierbar auf rosa Kassenrezept
Dank der Retardformulierung von Cariban® lässt sich mit der abendlichen oder morgendlichen Gabe der Wirkzeitraum steuern und die
individuelle Dosierung bedarfsgerecht auf die Symptomatik der Patientin im Tagesverlauf einstellen. „Je nach Ausprägung der Beschwerden kann man bei einer Besserung etwa die nachmittägliche oder abendliche Kapsel langsam ausschleichen“, erläuterte Hampel und ergänzte: „Wenn es den Frauen besser geht, kann man sagen, sie freuen sich jetzt über den glücklichen Verlauf.“ Auch Dr. Erwin Göckeler-Leopold, Geseke, betonte, wie wichtig es ist, herauszufinden, wo eine Patientin gerade steht, um sie auf ihrem Weg zu begleiten: „Beim Angebot von Hilfestellungen müssen wir ihr die Wahl lassen, auch Widersprüche aufzeigen und sie immer wieder motivieren.“ „Hier gibt die spezifische Zulassung von Cariban® in der Indikation vor allem niedergelassenen Ärzten eine Verordnungssicherheit“, fasste Krick zusammen. „Cariban® ist das einzige Arzneimittel mit der Zulassung zur symptomatischen Behandlung von Übelkeit und Erbrechen in der Schwangerschaft bei erwachsenen Frauen, die nicht auf konservatives Management reagieren. Das heißt, wir können es ganz normal mit einem rosa Kassenrezept verordnen, müssen uns keine Sorgen machen, dass es aus dem Ausland kommt, dass es off label ist, dass es einen Rote-Hand-Brief gibt. Das gibt Sicherheit für den Arzt und die Patientin.“ Cariban® ist im Rahmen der GKVVerordnung erstattungsfähig: Basis ist hier das Sozialgesetzbuch (SGB V). Das Medikament fällt unter den § 24 c – Leistungen bei Schwangerschaft und Mutterschaft –, der explizit Leistungen bei Schwangerschaft und hier auch Arzneimittel einschließt. Martina Freyer, München
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
IncobotulinumtoxinA zur Behandlung der pädiatrischen Sialorrhö IncobotulinumtoxinA, das aufgereinigte, komplexproteinfreie Botulinum Neurotoxin Typ A, wird in der Praxis zur Behandlung von Bewegungsstörungen, z.B. aufgrund von Spastik oder Dystonien, sowie der chronischen Sialorrhö bei Erwachsenen eingesetzt. Auf einem virtuellen Symposium im Rahmen des Kongresses für Parkinson und Bewegungsstörungen stellte Professor Steffen Berweck, Vogtareuth, neue wissenschaftliche Erkenntnisse zur Anwendung von IncobotulinumtoxinA (Xeomin®) bei Kindern und Jugendlichen mit chronischer Sialorrhö aufgrund neurologischer Grunderkrankungen und/oder geistiger Behinderung vor, die dessen Wirksamkeit und gute Verträglichkeit auch in der Therapie der pädiatrischen Sialorrhö bestätigen. SIPEXI-Studie liefert neue Erkenntnisse
Die randomisierte, doppelblinde, placebokontrollierte Phase-IIIStudie SIPEXI wurde mit dem Ziel durchgeführt, Wirksamkeit und Sicherheit von IncobotulinumtoxinA zur Behandlung der chronischen Sialorrhö bei Kindern und Jugendlichen zur Reduktion der Speichelflussrate und Verbesserung der Symptomatik zu untersuchen. In die Studie eingeschlossen wurden 256 Kinder und Jugendliche im Alter von 2 – 17 Jahren, die aufgrund einer neurologischen Erkrankung (z. B. Zerebralparese oder Schädel-Hirn-Trauma) und/oder einer geistigen Behinderung eine seit mindestens 3 Monaten bestehende Sialorrhö aufwiesen. Die Schwere © VERLAG PERFUSION GMBH
KONGRESSE / WISSENSWERTES
des unkontrollierten Speichelflusses als weiteres Einschlusskriterium wurde von den Eltern der Patienten anhand der Investigator’s modified Teacher’s Drooling Scale mit einem Punktwert von mindestens 6 bewertet. Ausschlusskriterien waren unter anderem eine andere Ursache der Sialorrhö als eine neurologische Erkrankung und/ oder geistige Behinderung, eine moderate bis schwere Schluckstörung, ein Gewicht unter 12 kg sowie eine Vorbehandlung mit Botulinumtoxin innerhalb des vorangegangenen Jahres. Die Patienten erhielten in der 16-wöchigen Hauptphase sowie in der offenen Verlängerungsphase der Studie eine ultraschallgestützte bilaterale gewichtsadaptierte Injektion in die großen Speicheldrüsen im fixen Dosis-VolumenVerhältnis 3 : 2 mit IncobotulinumtoxinA (25 Einheiten pro ml) oder Placebo mit dem Ziel, die Ruheund Reizsekretion der Speicheldrüsen zu reduzieren. Analgetika und/oder Sedativa wurden nach Bedarf vor oder während der Injektion verabreicht. Primäre und coprimäre Wirksamkeitsparameter der Studie waren zum einen die Veränderung der nicht stimulierten Speichelflussrate und zum anderen die Einschätzung der allgemeinen Veränderung der „Hypersalivation“ durch pflegende oder betreuende Personen anhand der Global Impression of Change Scale (GICS) – einer Skala, mit der Patienten ihren klinischen Gesamteindruck einschätzen – jeweils 4 Wochen nach der ersten Behandlung gegenüber dem Ausgangswert. In der Kohorte der 6bis 17-jährigen Patienten wurde jeweils der Vergleich von Verum zu Placebo gemessen. Unerwünschte Ereignisse wurden durchgehend aufgezeichnet.
Ergebnisse bestätigen Wirksamkeit und gute Verträglichkeit
Botulinum Neurotoxin Typ A hemmt nach Injektion in die Speicheldrüsen die cholinerge neuro glanduläre Signalübertragung und damit die Aktivität der Speicheldrüsen lokal und reversibel. In beiden coprimären Endpunkten, der unstimulierten Speichelflussrate (uSFR) und im Wirksamkeitsurteil der Eltern oder betreuenden Personen zur allgemeinen Veränderung der Sialorrhö (GICS), zeigte sich 4 Wochen nach der Injektion von IncobotulinumtoxinA eine signifikante Reduktion des Speichelflusses gegenüber Placebo. Die Effekte hielten bis zu 16 Wochen nach der ersten Behandlung an. Mit der wiederholten, kontinuierlichen Injektion in 3 weiteren Behandlungszyklen konnte ein anhaltender Effekt der Wirksamkeit sowohl anhand der uSFR sowie des Urteils durch die betreuenden Personen belegt werden. Darüber hinaus zeigen die Studiendaten eine gute Verträglichkeit ohne unerwartete oder ohne im Zusammenhang mit der Behandlung stehende schwerwiegende unerwünschte Ereignisse im Verlauf der Studie. Insgesamt belegen die Ergebnisse der SIPEXI-Studie, dass IncobotulinumtoxinA bei Kindern und Jugendlichen mit chronischer Sialorrhö aufgrund neurologischer Grunderkrankungen und/oder geistiger Behinderung wirksam und gut verträglich ist. In den USA führten die Studienergebnisse inzwischen zur Zulassung von IncobotulinumtoxinA bei chronischer Sialorrhö bei Kindern und Jugendlichen ab dem Alter von 2 Jahren. Die Zulassungserweiterung für Europa ist bereits beantragt. Fabian Sandner, Nürnberg
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
95
Zulassungserweiterung für Kaftrio® plus Kalydeco® zur Behandlung der zystischen Fibrose Die Europäische Kommission hat die Zulassung von Kaftrio® (Ivacaftor/Tezacaftor/Elexacaftor) in Kombination mit Kalydeco® (Ivacaftor) zur Behandlung der zystischen Fibrose (CF, Mukoviszidose) erweitert. Die Indikation umfasst nun alle Betroffenen ab einem Alter von 12 Jahren, die mindestens eine F508del-Mutation im CFTR-Gen (Cystic Fibrosis Transmembrane Conductance Regulator) aufweisen, unabhängig davon, welche Mutation das zweite Allel aufweist. Damit kommen nun unter anderem auch alle CF-Patienten ab 12 Jahren für die Tripel-Kombinationstherapie infrage, die heterozygot für die F508del-CFTR-Mutation sind und eine CFTR-Gating-Mutation (F/G) oder eine mit Restfunktion assoziierte Mutation (F/RF) auf dem anderen Allel aufweisen. Insgesamt können damit 85 % aller CF-Betroffenen ab 12 Jahren von der Dreifachkombination profitieren. Die Zulassungserweiterung basiert auf den positiven Ergebnissen dreier internationaler Phase-IIIStudien mit CF-Patienten ab 12 Jahren: • einer Phase-III-Studie über 24 Wochen (Studie 445-102) an 403 Patienten mit einer F508del-Mutation und einer Minimalfunktions-Mutation (F/ MF), • einer Phase-III-Studie über 4 Wochen (Studie 445-103) an 107 Patienten mit zwei F508del-Mutationen (F/F) und © VERLAG PERFUSION GMBH
WISSENSWERTES
96
Zystische Fibrose Die zystische Fibrose ist eine seltene, progrediente, genetisch bedingte Krankheit, die etwa 6.500 Menschen in Deutschland betrifft. Ursache ist ein defektes oder fehlendes Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)-Protein, das in der Mem bran von Epithelzellen als Inonenkanal fungiert und das Ein- und Ausströmen von Chlorid-Ionen und Wasser reguliert. Ist die CFTRFunktion infolge einer Mutation im CFTR-Gen gestört oder fehlt sie ganz, kommt es zu massiven Störungen des Salz-Wasser-Haushalts und Körpersekrete, wie z.B. der Schleim in der Lunge, werden dickflüssig und zäh, was schließlich zum Tod führen kann. Um an CF zu erkranken, muss ein Kind 2 defekte CFTR-Gene – jeweils eines von beiden Elternteilen – geerbt haben. Es sind etwa 2.100 Mutationen im CFTR-Gen bekannt, die Mehrheit aller an CFPatienten weist mindestens eine F508del-Mutation auf. Kaftrio® (Ivacaftor/Tezacaftor/Elexacaftor) als Kombinationsbehandlung mit Kalydeco® (Ivacaftor) wurde zur Behandlung der zystischen Fibrose bei Patienten ab 12 Jahren entwickelt, die mindestens eine F508del-Mutation im CFTR-Gen aufweisen, um die Menge und Funktionsfähigkeit von F508del-CFTR-Protein an der Zelloberfläche zu erhöhen.
• einer Phase-III-Studie (Studie 445-104) an 258 Patienten, die heterozygot für die F508delCFTR-Mutation sind und eine CFTR-Gating-Mutation (F/G) oder eine mit Restfunktion assoziierte Mutation (F/RF) tragen. In allen Studien zeigte die Kombination aus Ivacaftor/Tezacaftor/ Elexacaftor plus Ivacaftor signifikante und klinisch relevante Verbesserungen des Gesundheitszustands bei Menschen mit zystischer Fibrose, die mindestens 12 Jahre alt sind und mindestens eine Kopie der häufigsten Gen-Mutation F508del haben. Die Studienergebnisse belegen zudem einen klinischen Nutzen für Betroffene, die zusätzlich eine CFTR-GatingMutation (F/G) oder eine mit Restfunktion assoziierte Mutation (F/ RF) auf dem anderen Allel aufweisen. F. S.
Oncotype DX Test sagt Nutzen von Chemo therapien bei nodalpositivem Brustkrebs voraus Das Mammakarzinom ist in Europa die unter Frauen meistverbreitete Krebsart. Rund 25 % der Patientinnen mit HR-positivem, HER2-negativem Brustkrebs im Frühstadium haben befallene Lymphknoten, 2 von 3 dieser Patientinnen sind postmenopausal. Während den Patientinnen die Chemotherapie routinemäßig angeboten wird, zeigen Studien, dass nur eine Minderheit der Patientinnen mit Brustkrebs im Frühstadium tatsächlich davon profitiert. Der einzige genomische Test, der sowohl für eine sichere Aussage über den zu erwartenden Nutzen einer Chemotherapie als auch über das Rückfallrisiko bei Brustkrebs im Frühstadium validiert ist, ist der Oncotype DX Breast Recurrence
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Score® Test. Umfassende Evidenz dazu liegt aus der Studie RxPONDER vor, die vom unabhängigen Krebsforschungsnetzwerk SWOG durchgeführt und vom National Cancer Institute (NCI) finanziert wurde. Als eine der größten klinischen Studien bei Patientinnen mit nodal-positivem, Hormonrezeptor(HR)-positivem, HER-2-negativem Brustkrebs im Frühstadium schloss sie über 5.000 Frauen mit bis zu 3 positiven Lymphknoten ein. 2 Drittel der Frauen waren postmenopausal und etwa 85 % davon hatten ein Recurrence Score®Ergebnis von 0 bis 25. Ziel der Studie war es, zu definieren, unter welchen Voraussetzungen eine Chemotherapie bei Patientinnen mit einem Recurrence Score®-Ergebnis von 0 bis 25 von zusätzlichem Nutzen ist oder nicht. Dazu wurden die Frauen auf eine Behandlung mit Hormontherapie allein bzw. Chemotherapie mit anschließender Hormontherapie randomisiert und nach Recurrence Score®-Ergebnis, Menopausenstatus und der Art der Lymphknotenoperation stratifiziert. Postmenopausale Patientinnen mit einem Recurrence Score® von 0 bis 25 brauchen keine Chemotherapie
Die Auswertung von RxPONDER zeigte basierend auf den Recurrence Score®-Ergebnissen einen unterschiedlichen Nutzen der Chemotherapie bei postmenopausalen und prämenopausalen Frauen. Für postmenopausale Frauen mit einem Recurrence Score®-Ergebnis von 0 bis 25 wurde kein Nutzen einer Chemotherapie beobachtet – und zwar unabhängig von der Anzahl der betroffenen Lymphkno© VERLAG PERFUSION GMBH
97
WISSENSWERTES
ten, dem Grading oder der Größe des Tumors. Daraus lässt sich schließen, dass diese Patientinnen auf eine Chemotherapie verzichten und ihnen die damit verbundenen Nebenwirkungen erspart werden können. Dagegen zeigen die Ergebnisse nach einer medianen Nachbeobachtungsdauer von 5 Jahren bei prämenopausalen Frauen mit einem Recurrence Score®-Ergebnis von 0 bis 25 einen statistisch signifikanten Nutzen der Chemotherapie mit einer durchschnittlichen Verbesserung der Fernrezidivraten nach 5 Jahren um 2,9 %.
Test von NCCN-Leitlinien empfohlen
Der Oncotype DX Breast Recurrence Score® Test kann bei nodalpositiven, postmenopausalen Patientinnen die Frauen identifizieren, die nicht von einer Chemotherapie profitieren können – nämlich diejenigen mit einem Recurrence Score®-Ergebnis von 0 bis 25, und das sind etwa 85 % dieser Patientengruppe. Dadurch lassen sich die Anzahl unnötiger Chemotherapien und die damit einhergehenden Kosten für das Gesundheitswesen signifikant verringern. Angesichts
ihrer hohen Relevanz wurden die Erkenntnisse der RxPONDERStudie umgehend in den aktualisierten Brustkrebsleitlinien des National Comprehensive Cancer Network (NCCN) berücksichtigt. Hier wird der Oncotype DX® Test als einziger Test anerkannt, der zur Nutzenvorhersage von Chemotherapien bei Patientinnen mit Brustkrebs im Frühstadium und 1 bis 3 positiven axillären Lymphknoten, einschließlich Mikrometastasen, eingesetzt werden kann. F. S.
Titelbild: Viele Patienten profitieren von einer osteopathischen Behandlung, insbesondere bei Wirbelsäulenbeschwerden (Quelle: Pixabay). Herausgeber: Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin Wissenschaftlicher Beirat: Prof. Dr. M. Alexander, Infektiologie, Berlin Prof. Dr. L. Beck, Gynäkologie, Düsseldorf Prof. Dr. Berndt, Innere Medizin, Berlin Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main Prof. Dr. K. M. Einhäupl, Neurologie, Berlin Prof. Dr. E. Erdmann, Kardiologie, Köln Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK Prof. Dr. K. Falke, Anästhesiologie, Berlin Prof. Dr. K. Federlin, Innere Medizin, Gießen Prof. Dr. E. Gerlach, Physiologie, München Prof. Dr. H. Helge, Kinderheilkunde, Berlin Prof. Dr. R. Herrmann, Onkologie, Basel Prof. Dr. W. Jonat, Gynäkologie, Hamburg Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt Prof Dr. J. Mann, Nephrologie, München Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim Prof. Dr. R. Nagel, Urologie, Berlin Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf Prof. Dr. P. Ostendorf, Hämatologie, Hamburg Prof. Dr. Th. Philipp, Innere Medizin, Essen Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf Prof. Dr. H. Rieger, Angiologie, Aachen Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen Prof. Dr. E. Rüther, Psychiatrie, Göttingen Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München Prof. Dr. E. Standl, Innere Medizin, München Prof. Dr. W. T. Ulmer, Pulmologie, Bochum Schriftleitung: Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof E-Mail: info@d-i-g.org E-Mail persönlich: k.l.resch@d-i-g.org
JOURNAL PHARMAKOL. U. THER. 3/2021 · 30. JAHRGANG
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag. Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Satz: HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Druck und Verarbeitung: DRUCK_INFORM GmbH In der Büg 8 91330 Eggolsheim
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
VERLAG
PERFUSION Verlag PERFUSION GmbH Storchenweg 20 90617 Puschendorf Telefon: 09101/990 11 10 Fax: 09101/990 11 19 www.Verlag-Perfusion.de E-Mail: perfusion@t-online.de
Journal
© VERLAG PERFUSION GMBH
1× TÄGLICH
Indacaterol /Glycopyrronium /Mometasonfuroat (Ph.Eur.)
Asthma strukturiert therapieren.
1×
täglich
Zur Erhaltungstherapie bei Asthma für Erwachsene und Jugendliche ab 12 Jahren, die mit ICS und SABA nicht ausreichend kontrolliert sind. Wirkstoffe: Indacaterol / Mometasonfuroat (Ph.Eur.)
Zur Erhaltungstherapie bei Asthma für Erwachsene, die mit LABA und Hochdosis ICS nicht ausreichend kontrolliert sind und bei denen im Vorjahr eine oder mehrere Asthmaexazerbationen aufgetreten sind. 1 Kapsel
Erhältlich in drei Dosierungen: niedrig
mittel
150 µg / 80 µg*
Wirkstoffe: Indacaterol / Glycopyrronium / Mometasonfuroat (Ph.Eur.) Erhältlich in einer Dosierung:
hoch
150 µg / 160 µg* 150 µg / 320 µg*
24h
Wirkung1
hoch 150 µg / 50 µg / 160 µg*
* in Kapsel enthaltene Füllmenge Literatur: 1. s. Fachinformation Atectura®Breezhaler® & Enerzair®Breezhaler® Atectura® Breezhaler® 125 Mikrogramm/62,5 Mikrogramm Hartkapseln mit Pulver zur Inhalation (Pulver zur Inhalation) Atectura® Breezhaler® 125 Mikrogramm/127,5 Mikrogramm Hartkapseln mit Pulver zur Inhalation (Pulver zur Inhalation) Atectura® Breezhaler® 125 Mikrogramm/260 Mikrogramm Hartkapseln mit Pulver zur Inhalation (Pulver zur Inhalation) Wirkstoffe: Indacaterol (als Acetat), Mometasonfuroat (Ph.Eur.) Zus.setzung: Eine 125 Mikrogramm/62,5 Mikrogramm- bzw. 125 Mikrogramm/127,5 Mikrogramm- bzw. 125 Mikrogramm/260 Mikrogramm Kapsel enthält 150 µg Indacaterol (als Acetat) und 80 µg bzw. 160 µg bzw. 320 µg Mometasonfuroat (Ph.Eur.), entsprechend über das Mundstück abgegebene Dosis 125 µg Indacaterol (als Acetat) und 62,5 µg bzw. 127,5 µg bzw. 260 µg Mometasonfuroat (Ph.Eur.). Sonst. Bestandteile: Kapselinhalt: etwa 25 mg Lactose-Monohydrat, Kapselhülle: Gelatine, Drucktinte. Anwend.: Erhaltungstherapie bei Asthma bei Erw. u. Jugendl. ab 12 Jahren, die mit inhalat. Kortikosteroiden u. inhalat., kurzwirks. Beta2-Agonisten nicht ausreichend kontrolliert sind. Geg.-anz.: Überempfindl. gegen d. Wirkstoffe od. einen d. sonst. Bestandteile. Nebenw.: Die Angaben zur Häufigkeit beruhen auf der PALLADIUM-Studie. Sehr häufig: Nasopharyngitis. Asthma (Exazerbation). Häufig: Infektion der oberen Atemwege. Überempfindlichkeit. Kopfschmerz. Schmerzen im Oropharynx, Dysphonie. Schmerzen des Muskelund Skelettsystems. Gelegentlich: Candidose. Angioödem. Hyperglykämie. Verschwommenes Sehen, Katarakt. Tachykardie. Ausschlag, Pruritus. Muskelkrämpfe. Warnhinw.: Enthält Lactose. Verschreibungspflichtig. Weit. Hinweise: Siehe Fachinformation. Stand: April 2021 (MS 05/21.3). Novartis Pharma GmbH, Roonstr. 25, 90429 Nürnberg. Tel.: (0911) 273-0, Fax: (0911) 273-12 653. www.novartis.de
www.zusammen-gesund.de
Enerzair® Breezhaler® 114 Mikrogramm/46 Mikrogramm/136 Mikrogramm Hartkapseln mit Pulver zur Inhalation (Pulver zur Inhalation) Wirkstoffe: Indacaterol (als Acetat), Glycopyrroniumbromid (Ph.Eur.), Mometasonfuroat (Ph.Eur.) Zus.setzung: 1 Kapsel enthält 150 µg Indacaterol (als Acetat), 63 µg Glycopyrroniumbromid (Ph.Eur.), entsprechend 50 µg Glycopyrronium und 160 µg Mometasonfuroat (Ph.Eur.), entsprechend über das Mundstück abgegebene Dosis 114 µg Indacaterol (als Acetat), 58 µg Glycopyrroniumbromid (Ph.Eur.), entsprechend 46 µg Glycopyrronium und 136 µg Mometasonfuroat (Ph.Eur.). Sonst. Bestandteile: Kapselinhalt: etwa 25 mg LactoseMonohydrat, Magnesiumstearat (Ph.Eur.) [pflanzlich], Kapselhülle: Hypromellose, Drucktinte. Anwend.: Erhaltungstherapie für d. Behandlung v. Asthma bei erw. Patienten, die mit einer Kombination aus einem langwirks. Beta2-Agonisten u. einer hohen Dosis eines inhalat. Kortikosteroids als Erhaltungsther. nicht ausreichend kontrolliert sind u. bei denen im Vorjahr eine od. mehrere Asthmaexazerbationen aufgetreten sind. Geg.-anz.: Überempfindl. gegen d. Wirkstoffe od. einen d. sonst. Bestandteile. Nebenw.: Die Angaben zur Häufigkeit beruhen auf der IRIDIUM-Studie. Sehr häufig: Nasopharyngitis. Asthma (Exazerbation). Häufig: Infektion der oberen Atemwege, Candidiasis, Harnwegsinfektion. Überempfindlichkeit. Kopfschmerz. Tachykardie. Schmerzen im Oropharynx, Husten, Dysphonie. Gastroenteritis. Schmerzen des Muskel- und Skelettsystems, Muskelkrämpfe. Fieber. Gelegentlich: Hyperglykämie. Katarakt. Mundtrockenheit. Ausschlag, Pruritus. Dysurie. Warnhinw.: Enthält Lactose. Verschreibungspflichtig. Weit. Hinweise: Siehe Fachinformation. Stand: April 2021 (MS 05/21.2). Novartis Pharma GmbH, Roonstr. 25, 90429 Nürnberg. Tel.: (0911) 273-0, Fax: (0911) 273-12 653. www.novartis.de