PHARMACEUTICAL 3D PRINTING
 Alvaro Goyanes CEO and Co-founder, FABRX
Anna Worsley Director of Innovation, FABRX
Alvaro Goyanes CEO and Co-founder, FABRX
Anna Worsley Director of Innovation, FABRX
Gene Therapy for Cancer & Immunology
Transforming the Pharma Industry Issue 01 | 2023 | www.pharmafocusamerica.com
How AI is


www. pharmafocusamerica.com 2 TWO POWERFUL DOSES
YEAR Every issue of Pharma Focus America magazine is a powerful dose of information and knowledge – filled with original and undiluted content. Written by the best brains in pharma industry, the magazine offers timely business insights and articles on cutting-edge technologies. Subscribe now to get your doses regularly. Email: subscriptions@pharmafocusamerica.com Tel: +91 40 4961 4567 Fax +91 40 4961 4555 www.pharmafocusamerica.com
PER
Welcome & Introduction
As a publishing house there can be no other joy for us than to announce the launch of our very first issue of Pharma Focus America magazine 2023. I take this opportunity to congratulate my team, the panel of knowledgeable advisory board and all the authors who with their tenacious efforts believed in our vision of relevancy with uncompromising quality.
Backed by the team which powers Pharma Focus Asia and Pharma Focus Europe, Pharma Focus America is the newest kid in the block and strategically positioned to cater to the Pharma professionals across the American subcontinent. Fostering quality and timely delivery of trending topics relevant to the industry, Pharma Focus America magazine is committed to be its readers delight…leaving them in awe.
Pharmaceutical 3D Printing
Alvaro Goyanes, CEO and Co-founder at FABRX treads the path and unfolds the latest trend in 3D Printing that the Pharma sector is witnessing. Pharmaceutical 3D printing is a rapidly growing field, with more clinical studies starting every year. Able to manufacture novel dosage forms with advanced release profiles personalised to the patient, this technique is set to bring massive changes.
The pharmaceutical manufacturing industry has until now, followed a batch processing scenario to ensure that each batch was produced efficiently and effectively. Hassan Mostafa Mohamed, Chairman & CEO at ReyadaPro explains the latest trends in pharmaceutical manufacturing with the adoption of continuous processing with its impact on time taken and resources used.
Pharma industry seems to be ripe for AI transformation from drug discovery and the performance of trials, to remote patient monitoring, medication adherence tools and beyond. This article from Lydia Torne, Partner at Simmons & Simmons LLP explains some of the potential legal considerations when entering into licensing collaborations for the use of AI in drug discovery.
In the pharmaceutical industry Artificial Intelligence (AI) and Machine Learning (ML) models are being applied to improve the pharmacovigilance process. Ryanka Chauhan, Product Manager, Datafoundry – covers how these advancements have the potential to increase efficiency, accuracy, and consistency in pharmacovigilance, as well as reduce costs and delivery timelines.
Piet van der Graaf, SVP, QSP at Certara details in his article how Model Informed Drug Development (MIDD) continues to be a game changer in accelerating drug development. Quantitative Systems Pharmacology (QSP) is one such technology, which employs virtual patients in virtual clinical trials, allowing more approaches to be investigated than would be possible in the real world.
If you have any views or ideas or if you want to share your feedback with Pharma Focus America, you are more than welcome. You may also connect with us on our social media handles for exquisite stories, articles and much more.
N D Vijaya Lakshmi Editor
www. pharmafocusamerica.com 3
CONTENTS
RESEARCH & DEVELOPMENT
06 Advances in mRNA Therapeutics and Vaccines
Daniel Kavanagh, PhD RAC, Senior Scientific Advisor, Gene Therapy, Vaccines, & Biologics, WCG
13 Using Quantitative Systems Pharmacology Modelling to Accelerate Drug Development
Piet van der Graaf, Senior Vice President, Quantitative Systems Pharmacology, Certara
19 IgA-Mediated Inflammatory Disorders
Autoantigen-specific IgA, a biomarker with strong effector functions
Louis Boon, CSO & Board Member, JJP Biologics
CLINICAL TRIALS
30 Decentralization - The Future of Clinical Trials
Nicoleta Grecu, Director, Pharmacovigilance, Clinical Quality Assurance, Clover Biopharmaceuticals
39 Gene Therapy for Cancer & Immunology
Josipa Ljubicic, QA Director/Principal GCP and GVP auditor, Proqlea Ltd
MANUFACTURING
53 Choose Carefully: The Potential of Continuous Flow Chemistry in API Synthesis Charles Johnson, Senior Director, Commercial Development, Lonza
59 Viral Gene Therapy – How Can the Industry Drive Down the Cost of Goods to Better Serve the Patients?
Emmanuelle Cameau, Strategic Technology Partnership Leader - Cell and Gene Therapy, Pall Corporation
66 Adapting Containment Strategies to Futureproof the Manufacture of Sterile Drug Products
Ben Wylie, Head of Product Management & Marketing, ChargePoint Technology
71 Advanced Model Predictive Control System of Continuous Biopharmaceutical Manufacturing Process

Ravendra Singh, C-SOPS, Department of Chemical and Biochemical Engineering, Rutgers, The State University of New Jersey
79 Continuous Vs. Batch Manufacturing
Hassan Mostafa Mohamed, Chairman & Chief Executive Officer at ReyadaPro
PHARMACEUTICAL
Alvaro Goyanes
CEO and Co-founder, FABRX
Anna Worsley
Director of Innovation, FABRX
INFORMATION TECHNOLOGY
90 How AI is Transforming the Pharma Industry
Lydia Torne, Partner, Simmons & Simmons LLP
96 Use of Artificial Intelligence in Automation of Pharmacovigilance
Ryanka Chauhan, Product Manager, Datafoundry
EXPERT TALK
104 Next Generation Sequencing - Genomics
Ravi Dashnamoorthy, Senior Scientist, Genosco
EVENT PREVIEW
112 Interphex Event
WEBINAR REVIEW
114 Developability and Immunosafety: Don’t let them threaten your drug development plans
115 Redefining Liquid Formulation Technology in Hard Capsules
4 PHARMA FOCUS AMERICA ISSUE 01 - 2023
Cover
Story
122 NEWS
116 EVENTS LIST
46 3D PRINTING
EDITOR
Vijaya Lakshmi N D
Advisory Board
Alessio Piccoli
Director & Head, Business Development
Europe, Aragen Life Sciences
Italy
Hassan Mostafa Mohamed
Chairman & Chief Executive Officer at ReyadaPro Saudi Arabia
Hector Alejandro Andonie
General Manager, Laboratorios Andifar Honduras
Hoda Gamal
Director of Regulatory and Corporate Affairs, Middle East and Africa (MEAC), Sirgio international Egypt






Joaquin Campbell
Global Director, Managed Access Services, EarlyHealth Group
Spain
Josipa Ljubicic
QA Director/Principal GCP and GVP auditor, Proqlea Ltd
Croatia
Nicoleta Grecu
Director, Pharmacovigilance, Clinical Quality Assurance, Clover Biopharmaceuticals Romania

Pinheiro Neto Joao
Chief Executive Officer, Omnimed Angola
Thitisak Kitthaweesin
Chief of Phramongkutklao Center of Academic and International Relations Administration Thailand

Eiman Shafa
Medical Director, Spine Surgery, Abbott Northwestern Hospital USA


Amine Bekkali
Director chez MEDFIELDS
United Arab Emirates
David Contorno
Founder & CEO, E Powered Benefits, USA


EDITORIAL TEAM
Sarah Richards
Debi Jones
Swetha M
Harry Callum
Supraja B R
ART DIRECTOR
M Abdul Hannan
GRAPHIC DESIGNER
Akhilesh
PRODUCT MANAGER
Jeff Kenney
SENIOR PRODUCT ASSOCIATES
Ben Johnson
David Nelson
John Milton
Peter Thomas
Sussane Vincent
PRODUCT ASSOCIATE
Veronica Wilson
CIRCULATION TEAM
Sam Smith
SUBSCRIPTIONS IN-CHARGE
Vijay Kumar Gaddam
HEAD-OPERATIONS
Sivala VNR
www.pharmafocusamerica.com
Ochre Media Group info@ochre-media.com www.ochre-media.com




©Ochre Media Private Limited. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, photocopying or otherwise, without prior permission of the publisher and copyright owner. Whilst every effort has been made to ensure the accuracy of the information in this publication, the publisher accepts no responsibility for errors or omissions.
The products and services advertised are not endorsed by or connected with the publisher or its associates. The editorial opinions expressed in this publication are those of individual authors and not necessarily those of the publisher or of its associates.
Copies of Pharma Focus America can be purchased at the indicated cover prices. For bulk order reprints minimum order required is 500 copies, POA.
Magazine Subscribe
LinkedIn
www. pharmafocusamerica.com 5
Advances in mRNA Therapeutics and Vaccines

mRNA is a type of nucleic acid molecule that plays an essential role in the natural processing of genetic information. Using modern tools of molecular biology and genetic engineering, it is now possible to design synthetic mRNA molecules capable of executing an astonishing array of therapeutic functions. Advances in nanotechnology are starting to enable safe and effective delivery of therapeutic mRNA for the treatment of acquired and inherited conditions and for prevention of infectious disease.
Daniel Kavanagh PhD RAC, Senior Scientific Advisor, Gene Therapy, Vaccines, & Biologics, WCG
Prophylactic Vaccines
In the field of infectious disease, a prophylactic vaccine is a product that prevents disease by inducing an immune response against an infectious agent or pathogen. Prophylactic vaccines have played a critical role in the public health response to diseases such as smallpox, polio, measles, and many others – most recently COVID-19. The coordinated response by government, academic, and commercial research teams to the COVID-19 epidemic raised public awareness of the potential for mRNA vaccines to play a significant role in public health and resulted in the
RESEARCH & DEVELOPMENT
first two FDA approvals for mRNA products. These results helped to reinvigorate ongoing efforts to develop prophylactic mRNA vaccines for a broad variety of infectious diseases. In the coming years, mRNA vaccines will play an important and complementary role alongside other approaches, such as those involving recombinant proteins, live attenuated viruses, and engineered viral vectors in protecting public health.
For pathogens that cause cancer, such as human papilloma virus (HPV), a vaccine against that infectious agent can also contribute to protection against cancer. HPV prophylactic vaccination programs have provided significant public health benefits by reducing the occurrence of cervical cancer.
The molecular target of an induced immune response is called an antigen. Many investigators are interested in developing prophylactic vaccines that may prime an immune response directly against a cancer antigen rather than an infectious agent; however, development of such prophylactic cancer vaccines is still in early stages.
Therapeutic vaccines
Building on the science of prophylactic vaccines, much recent effort has focused on therapeutic vaccines. A therapeutic vaccine is intended to treat an existing disease by inducing an immune response against a disease-associated antigen. Some of this effort involves infectious disease, but most of the research is focused on treatment of cancer.
A defining feature of both prophylactic and therapeutic vaccines is the targeted induction of an immune response against one or more antigens associated with the disease. Most therapeutic drug products are not vaccines. However, modern gene transfer techniques provide shared platforms that are being used to develop (i) prophylactic vaccines, (ii) therapeutic vaccines and (iii) non-vaccine therapies across a broad range of therapeutic areas.
Gene transfer research
Gene transfer research constitutes a rapidly growing sector of drug/biologic product development and encompasses the closely related fields of cell and gene therapy (CGT), gene editing, molecular medicine, genetic medicines, and advanced therapy medicinal products.
In every nucleated cell, genetic information is stored as DNA in chromosomes in the nucleus, where that information is transcribed into RNA, which then exits the nucleus and passes into the cytoplasm. In the cytoplasm, information from this RNA is translated into proteins, which act as essential structural and enzymatic components of cells. RNA performing this function of carrying the message encoding instructions for the protein sequence from the nucleus to the cytoplasm is called messenger RNA (mRNA). Molecular interventions that add or alter genetic information in DNA or mRNA have the potential to affect almost any biological
www. pharmafocusamerica.com 7
RESEARCH & DEVELOPMENT
process controlling human health and disease. The key challenges are making the correct genetic changes and targeting them to the correct cells.
Transient nature of mRNA
As part of the natural process of gene expression, individual mRNA molecules are generally short-lived in the cytoplasm. The transient nature of mRNA is an essential aspect of dynamic gene regulation. For some applications utilizing engineered mRNA, highly transient expression is desirable. For other applications, investigators have developed modifications to the basic mRNA design that result in mRNA persistence and prolonged translational activity. Some such modifications are based on naturally occurring sequence variations that signal the cell to protect and preserve the mRNA molecule. Other approaches use chemical modifications of the building blocks of mRNA not found in nature to protect the mRNA product from rapid degradation. Discovery, testing, and implementation of these modifications to basic mRNA chemistry have been critical steps in enhancing the efficacy of mRNA products destined for commercialization and clinical use.
Introducing genetic information into a cell
One way to deliver new genetic information to a cell is by adding genetically modified DNA. For most systems, the engineered DNA must cross both the outer plasma membrane of the cell and
the nuclear membrane to reach the nucleus, where the genetic information can be expressed. Often the engineered DNA is delivered by means of a viral vector—a biological construct derived from a wildtype virus that has been extensively modified to carry recombinant genetic material and not cause disease. Viral vectors are the key components in successful products including both prophylactic vaccines and gene therapies for cancer and other diseases. Many different families of wild viruses serve as the basis for engineered viral vectors in clinical and preclinical development. Various FDA-approved products utilize viral vectors based on adenoviruses, adeno-associated viruses (AAV), and herpes simplex viruses. Compared to most small molecules and many biologic products, viral vector products involve complex and resource-intensive efforts for design, testing, manufacturing, and quality control.
Rather than directing DNA to the nucleus, another way to deliver genetic information to a cell is to introduce synthetic mRNA into the cytoplasm. In contrast to most DNA-based approaches, genetic information encoded in synthetic mRNA can be expressed immediately upon entering the cell and does not need to cross the nuclear membrane to enter the nucleus, be transcribed, and then exit the nucleus to reach the cytoplasm as a translatable message. Thus, functional gene delivery by synthetic mRNA is simpler than that required for DNA in significant ways. Rather than viral vectors, many approaches
8 PHARMA FOCUS AMERICA ISSUE 01 - 2023
RESEARCH & DEVELOPMENT
to mRNA delivery utilize nanoparticles. The simplest nanoparticles for gene transfer use lipid or lipidoid compounds to form selfassembling nanoscale spheres around mRNA cargo. These particles serve the dual function of protecting mRNA from destructive enzymes in the environment and allowing the mRNA to cross the plasma membrane and enter the cell.
The DNA in our chromosomes is chemically very stable, with information stored in two complementary strands assembled as a double-helix (double-stranded DNA). mRNA
is a single stranded molecule, inherently less stable and suitable for transient functions in the cell. Partly as a mechanism for protection against dangerous viruses, our bodies produce an excess of RNA-degrading enzymes, called RNAses. For these reasons, in the past, mRNA molecules were often regarded as too unstable to be very useful for drug development. Recent experience has shown that RNA prepared under strict RNAse-free conditions and packaged appropriately, as with lipid nanoparticles, is quite stable and very much suitable for use as a drug substance in many applications.
Manufacturing biological products
Non-biologic drugs such as small molecules can often be manufactured via an easily routinized process that reliably yields a product with predictable CMC (Chemistry, Manufacturing, and Control) output. In contrast, biological products in the CGT category often pose severe challenges for design, testing, and CMC, due to the complexity of products comprised of nucleic acids, proteins, lipid membranes, and in some cases living cells. With synthetic mRNA, manufacturing considerations are largely independent of the sequence of the genetic code, meaning that an established manufacturing process for one mRNA
product can often be implemented broadly in many different applications. In this way, the production of mRNA drug products is more like non-biologic chemical drugs than most CGT products. Using advanced sequencing and bioinformatics techniques, it is possible to acquire genetic sequences from biological samples and generate new mRNA products within a very short timeframe – within days of sample acquisition for preclinical research. This allows mRNA products to be incorporated into rapid-response plans for public health emergencies for example. This flexibility also makes mRNA approaches suitable for personalized therapeutic cancer vaccines.
www. pharmafocusamerica.com 9
RESEARCH & DEVELOPMENT
Commercialization Challenges
As discussed above, DNA-based gene transfer approaches usually rely on a viral vector for gene delivery. One of the most popular platforms uses AAVs as vectors for DNA transfer. An important issue with these vectors is that for any natural AAV serotype, a significant percent of the population will have circulating antibodies that neutralize the respective AAV vector. Seronegative individuals usually seroconvert after a single dose of AAV gene therapy, and thereby become ineligible for future treatment with the
same AAV serotype. As a result, most AAV products are designed to be one-and-done treatments, intended to provide lifelong therapeutic effects without the possibility of re-dosing. The one-time nature of AAV dosing poses significant challenges for dose-finding studies, as well as for longterm follow up to assess safety and efficacy. This treatment approach is also a challenge for commercialization strategies given that payment models in many therapeutic areas are built around the assumption that the same drug will be administered in multiple doses over time.
Advantages for clinical applications and commercialization
In contrast to AAVs, mRNA lipid nanoparticles currently in use do not induce an immune response to the particle that interferes with redosing. Furthermore, as explained above, after administration, synthetic mRNA products are usually short-lived in the cell and result in transient protein expression. In combination, these factors make it easier to optimize dosefinding approaches with mRNA compared to AAVs. The ability to apply repeated doses allows for serial boosting in vaccination regimens. As a therapeutic approach for chronic disease, mRNA treatment plans would potentially allow continuous administration of the therapeutic in multiple doses for as long as clinically necessary; they would also allow for rapid
cessation of treatment in case of unwanted side effects and adverse events. It is likely that commercialization plans for mRNA-based therapies would also be more compatible with standard reimbursement plans for the cost of drugs. While AAVs and other vectorbased approaches will certainly be the basis for many valuable new therapies for the indefinite future, mRNA presents an opportunity to develop complementary modalities in many therapeutic areas.
Targeting neoantigens
One area where mRNA therapeutics show particular promise is the development of personalized vaccines. An essential challenge in cancer immunology comes from the fact that cancer cells originate from the patient’s
10 PHARMA FOCUS AMERICA ISSUE 01 - 2023
RESEARCH & DEVELOPMENT
own tissue. This means that most potential antigens found in tumor cells are shared with normal healthy tissue and cannot be the basis of a useful immune response. Over the course of oncogenesis, tumors usually undergo extensive mutations in tumor cell chromosomal DNA. Many of these changes encode proteins that contain altered, tumorspecific antigens, also known as neoantigens. A key feature of neoantigens is that they are sufficiently distinct from antigens in normal tissue to serve as targets for an immune response. For certain cancer types, immune responses directed against neoantigens have been shown to provide significant clinical benefits in preclinical and clinical testing. For most tumor types, the majority of neoantigens detected from a biopsy are “private” and specific to the respective individual patient.
Creating personalized cancer vaccines
As noted above, new mRNA products can be rapidly designed and manufactured from any genetic sequence. This makes mRNA a promising platform for personalized cancer vaccines. To create a personalized vaccine, investigators sequence genetic content of a tumor biopsy and use bioinformatic methods to identify potential neoantigen targets –representing protein sequences present in the tumor but not in healthy tissue. mRNA encoding those neoantigen targets can then be rapidly synthesized and administered to the originating patient in an immunogenic
formulation to induce a protective immune response against the tumor.
As with any new treatment approach, development of neoantigen vaccines involves challenges and potential pitfalls. If the identified neoantigens are too similar to native host proteins, then the immune system may fail to see the neoantigens as “non-self,” and thus may fail to mount an effective response. Alternatively, if the neoantigens are too similar to native host proteins, an aggressive immune response could be primed against both the tumor antigen and normal healthy tissue, resulting in potentially serious autoimmune disease. Another potential issue is that neoantigens are often the result of extreme genetic instability of tumors. Therefore, as effective immune responses start to suppress the tumor, the tumor may rapidly evolve to cease expression of the most effective neoantigen targets. This process is called
www. pharmafocusamerica.com 11
RESEARCH & DEVELOPMENT
antigenic escape. One approach to minimize antigenic escape is to vaccinate with many neoepitopes at once. At the time of this writing, news reports indicate very promising results in a Phase 2b clinical trial of a personalized mRNA vaccine in combination with an anti-PD-1 immunotherapy, in participants with Stage III/IV melanoma.
To date, the majority of clinical trials of mRNA products have been for prophylactic vaccines for infectious disease. Most of the remaining trials are for the development of therapeutic cancer vaccines. Nevertheless, there are many preclinical and clinical efforts to apply mRNA in the area of nonvaccine therapeutics. One promising approach is a gene replacement therapy for inherited rare disease.
Rare disease applications
There are more than 7,000 rare diseases recognized, with a large percentage being congenital conditions affecting individuals that received two defective versions of the same gene, one from each parent (i.e., through autosomal recessive inheritance). In theory, each of these conditions should be treatable via molecular gene replacement therapy. Gene replacement refers to a variety of therapeutic approaches whereby the missing function of the defective gene is restored by delivering a corrected gene to some key tissue in the body. To date, most gene replacement therapy approaches have utilized AAV, an approach with great potential and significant drawbacks as described above. Some of those drawbacks
may be avoided with mRNA-based gene replacement, and ongoing efforts in this area include clinical trials in cystic fibrosis and a variety of inborn errors of metabolism.
Aside from gene replacement, mRNA can potentially be utilized for the delivery of a next generation of products developed with synthetic biology. Synthetic biology uses systematic modular engineering approaches to create novel biomolecules, some of which perform new functions distinct from any natural molecules. Some products of synthetic biology may enable white blood cells to recognize and attack tumor cells. Some are designed to detect and correct hormonal imbalances in vivo in real time. For clinical purposes, the underlying genetic constructs may be delivered by a variety of gene transfer techniques, including mRNA. Thus, we can expect mRNA to be an important part of the drug development landscape in the coming decades.

12 PHARMA FOCUS AMERICA ISSUE 01 - 2023 AUTHOR BIO
Daniel Kavanagh, PhD RAC, Senior Scientific Advisor, Gene Therapy, Vaccines, & Biologics, WCG, where he engages with sponsors, CROs, and research institutions on topics related to human gene transfer research. He was previously an Assistant Professor of Medicine at Harvard Medical School, and a member of the Executive Committee of the Harvard Center for AIDS Research.
RESEARCH & DEVELOPMENT
Using Quantitative Systems Pharmacology Modelling to Accelerate Drug Development
Model informed drug development (in silico modelling) continues to be a game changer in accelerating drug development. Quantitative Systems Pharmacology (QSP) is one such technology, which employs virtual patients in virtual clinical trials, allowing more approaches to be investigated than would be possible in the real world. Examples described here demonstrate the impact on some of today’s most urgent medical needs, including evaluating a potential treatment for Alzheimer’s disease, paediatric dosing for a COVID-19 vaccine, and evaluation of novel therapies for irritable bowel disease.
Piet van der Graaf
Senior Vice President, Quantitative Systems Pharmacology, Certara
Quantitative systems pharmacology (QSP) combines computational modelling and experimental methods to explore the relationship between a drug, human biology, and the disease
process. QSP modelling leverages large quantities of biological and pharmacological data to increase scientific knowledge of disease pathophysiology and facilitate the investigation of different therapeutic
RESEARCH & DEVELOPMENT
approaches in virtual patients in virtual clinical trials. As virtual patients are used instead of real people, QSP modelling saves time and money and allows more drug doses and drug combinations to be investigated than could be practically and ethically evaluated in real life.
QSP modelling can help to identify a new drug target or biomarker, select the optimal drug dose for a vulnerable patient population or an individual patient, repurpose an existing on-market drug for a different indication or develop a new combination therapy.
QSP is a relatively new discipline, but it is already having a profound impact on decision making at many stages along the drug development continuum. In a CPT:
Treating Alzheimer’s Disease
Eisai and Biogen Inc. announced positive results from their large, global, Phase 3 confirmatory Clarity AD clinical study of lecanemab on November 29, 2022. Lecanemab is an investigational antiamyloid beta (Aβ) protofibril antibody for the treatment of mild cognitive impairment due to early Alzheimer's disease (AD) with confirmed presence of amyloid pathology in the brain. Clinical results were presented at the 2022 Clinical Trials on Alzheimer's Disease conference, in San Francisco, California.
Pharmacometrics & Systems Pharmacology paper published in November 2022, U.S. Food and Drug Administration (FDA) staff reported that there has been a notable increase in the number of regulatory submissions that contain QSP, including Investigational New Drug Applications (INDs), New Drug Applications (NDAs), and Biologics License Applications (BLAs) during the past several years. Their report concluded that QSP is increasingly applied to model and simulate both drug effectiveness and safety throughout the drug development process across disease areas.
The following case studies illustrate what all the excitement is about and demonstrate QSP’s potential to advance drug development.
Lecanemab treatment showed 31% lower risk of converting to next stage of disease by Global CDR assessment (Hazard Ratio: 0.69). A slope analysis using CDR-SB based on observed data and extrapolation to 30 months showed that lecanemab takes 25.5 months to reach same level as placebo at 18 months, indicating a 7.5 month slowing of progression.
The Certara team had started to work with Eisai about two years before these results were announced and developed a QSP model to predict and help interpret the outcomes of the trial. In fact, Certara’s
14 PHARMA FOCUS AMERICA ISSUE 01 - 2023
RESEARCH & DEVELOPMENT
Studying Amyloid Aggregation
AD pathogenesis is widely believed to be driven by the production and deposition of the Aβ peptide. Amyloid aggregates are classified as monomers, oligomers, protofibrils and fibrils based on their size and solubility. As antiamyloid monoclonal antibodies have different affinities for these species, it could lead to different levels of standardized uptake value ratio (SUVr) reduction, and potentially different clinical outcomes.
Certara created an amyloid QSP model to study lecanemab. The model included all known features of the Aβ aggregation process, together with the mechanisms of action employed by drug candidates endeavouring to treat Alzheimer’s disease by modifying Aβ pathology.

The model was validated by demonstrating that it could accurately replicate the clinical pharmacokinetic/pharmacodynamic (PK/ PD) data for several monoclonal antibodies – lecanemab, gantenerumab, solanezumab, bapineuzumab, and crenezumab. The model was then used to investigate 18 months of treatment with those antibodies, followed by 2.5 years of observation. The platform revealed significant differences in oligomer, protofibril, and plaque reduction between the monoclonal antibodies. At clinical doses, lecanemab produced pronounced reductions in protofibrils and the SUVr, which served as encouraging precursors to its phase 3 clinical results.
Other examples of using QSP modelling to predict clinical outcomes are included below.
COVID-19 Pediatric Vaccine Dosing
QSP model predicted the positive outcome of this trial about one year early, as presented at the AD PD Annual Meeting in March 2022.
At the start of the COVID-19 pandemic, Certara quickly realized that the QSP model of the human immune system that it had developed to predict immunogenicity with protein-based therapeutics, such as antibodies, could be repurposed as a vaccine model.
Immunogenicity, or the tendency to trigger an unwanted immune response, is a major problem with biological therapies because it can lower the patient’s exposure to the medicine and reduce its efficacy. Immunogenicity can also cause immune-related adverse events. While the goal of the original model was to identify drugs that generated low levels of immunogenicity, the goal of the new model was
www. pharmafocusamerica.com 15
RESEARCH & DEVELOPMENT
the opposite, to find vaccines that produced high levels of immune response to provide protection against the SARS-CoV-2 virus. Part of the COVID-19 virus sequence was input into the new vaccine model to demonstrate that it would generate an immune response. Then, the model was calibrated using the structure of actual COVID-19 vaccines and validated by replicating their published clinical data. The resulting COVID-19 vaccine simulator was able to predict with a virtual trial the outcomes of actual clinical trials using those vaccines.
The vaccine simulator was initially used to determine the optimal timing between COVID19 vaccine doses and whether vaccines could be combined during a shortage. Daiichi Sankyo employed Certara’s vaccine model to optimize the phase 1 study design for its COVID-19 vaccine and to predict immune responses in specific populations, such as Japanese people and older adults.
The focus later shifted to paediatrics where the simulator was used to guide dose selection in young children. That is one of the biggest challenges in vaccine development. When working with vulnerable populations, such as the elderly, young children, or babies, it is even more imperative to use exactly the right dose and not a little too much or too little. It is a delicate balancing act, selecting a dose that is high enough to be sufficiently efficacious but low enough to produce minimal side effects. The situation is more complex in children because their physiology, enzyme and transporter levels change as they mature. One
of the many advantages of QSP modelling is that it allows vaccine doses and other variables to be adjusted and the results observed virtually without the need to conduct clinical trials.
Pfizer-BioNTech’s rollout of its COVID19 vaccine was extremely successful. But even Pfizer-BioNTech had to redo its first paediatric COVID-19 vaccine trial and change the dosing regimen because two 3 µg doses of its vaccine did not produce a sufficiently protective immune response in children aged two to four years. They ultimately added a third 3 µg vaccine dose, administered at least two months after the second dose, for that population.
Certara’s COVID-19 vaccine simulator, which now includes 32 clinical datasets, three vaccines, eight dosing regimens, and 10 biomarkers, predicted that response. Its QSP model confirmed that two 30 µg doses of Pfizer-BioNTech’s COVID-19 vaccine would be effective in children aged 16 to 25 years (the same as an adult), two 10 µg doses (one-third the adult dose) would be effective in children aged 5 to 11 years, but two 3 µg doses (one-tenth the adult dose) would be ineffective in children aged 2 to 4 years and they would require a third 3 µg dose.
Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) refers to two conditions, Crohn’s disease (CD), and ulcerative colitis (UC), which are characterized by chronic inflammation of the gastrointestinal (GI) tract. Prolonged inflammation results in damage to
16 PHARMA FOCUS AMERICA ISSUE 01 - 2023
RESEARCH & DEVELOPMENT
the GI tract. IBD symptoms include persistent diarrhoea, abdominal pain, rectal bleeding/ bloody stools, weight loss, and fatigue.
In 2015, an estimated 1.3% of US adults (three million) reported having received a diagnosis of IBD. The prevalence of IBD varies based on a person’s age, race/ethnicity, education level, employment status, nativity, poverty status, and urbanicity. IBD occurs more often in non-Hispanic White persons than in other racial/ethnic groups.
The impact of medication on IBD symptoms has traditionally been hard to measure. When a patient with IBD meets with their clinician, they are often asked to rate how they are feeling on a scale of 1 to 5 with five representing the worst/most severe symptoms. But how do you rate “not great”? How do you represent biology on a questionnaire?
Further compounding the complexity of the situation, different hospitals use different disease activity scores to measure
disease severity and therapeutic outcomes. For example, the Mayo Clinic developed the Mayo Score/Disease Activity Index for Ulcerative Colitis to assess the initial disease severity, change in activity over time, and response to treatment. This scoring system factored in four elements: rectal bleeding, stool frequency, physician assessment, and endoscopic appearance. Each element was rated from 0 to 3, giving a maximum total score of 12. The higher the score, the more severe the ulcerative colitis.
But the FDA is now accepting a modified Mayo Score, which no longer includes mucosal friability in the endoscopic score or the physician’s global assessment in the composite index, and has a maximum total score of 9.
Clinicians prefer to use these scores rather than levels of biomarkers, such as fecal protectin (FCP) or serum c-reactive protein (CRP), when evaluating a patient’s progress because they also factor in clinical observations such as intestinal bleeding and ulceration.
The primary challenge in this field is how to link biological mechanisms to these subjective endpoints, which are the actual clinical development scores that clinicians use.
To achieve that goal, Certara combined QSP with model-based meta-analysis (MBMA), another in-silico technology, to create QSP models which can predict those subjective clinical endpoints in IBD. Essentially, the team used artificial intelligence and machine learning to link mechanistic QSP to clinical endpoint scores.
www. pharmafocusamerica.com 17
RESEARCH & DEVELOPMENT
QSP combines computational modelling and experimental methods to explore the relationship between a drug, human biology, and the disease process
QSP can model the biological effects of drugs up to the biomarker level, i.e., it can determine whether cytokine levels are going up or down. Then, MBMA uses data reported from clinical trials and in the scientific literature to correlate the tissue biomarker measurements from the QSP model with disease activity scores for UC and CD. It determines in a purely statistical manner what cytokine levels correlate with specific scores on the Mayo Scale.
The Certara team included inflammatory cytokines and therapeutic antibodies in a mechanistic, multistate QSP model. Then they added published baseline and patient Mayo Score, Crohn’s Disease Active Index (CDAI), CRP, and FCP data to enable correlation between the biomarker levels and clinical measurements. A virtual patient population was created with tissue cytokine, cell density, and gut measurements to match an existing clinical patient population. The tissue biomarkers used were Treg, TNFα, neutrophil, and Th17. An algorithm then assigned the appropriate clinical score to the virtual patients based on their Mayo, CDAI score distribution in relation to FCP or CRP. The resulting virtual population helped to train a multinomial logistic regression model which was able to successfully predict clinical response and remission rates for patients prescribed anti-TNFα (adalimumab), anti-IL-23 (mirikizumab) or a combination of the two drugs in clinical trials.
The resulting predictive model can now be used to guide dose selection for IBD drug candidates in Phase I/II clinical trials.

Prior to this breakthrough, most QSP model predictions ended at the biomarker level with no quantitative linkage to actual patient outcome.
Conclusion
QSP modelling is a powerful tool that can be applied throughout the drug development continuum from identifying a new drug target in the earliest stages through to predicting clinical outcomes. These advances all serve to accelerate the drug development process safely and efficiently.
Professor Piet van der Graaf is Senior Vice President, Quantitative Systems Pharmacology at Certara and Professor of Systems Pharmacology at Leiden University in the Netherlands. Before joining Certara, he was the Director of Research (CSO) of the Leiden Academic Centre for Drug Research and held various research leadership positions at Pfizer across discovery and clinical development.
18 PHARMA FOCUS AMERICA ISSUE 01 - 2023
AUTHOR BIO
RESEARCH & DEVELOPMENT
IgA-Mediated Inflammatory Disorders

Autoantigen-specific IgA, a biomarker with strong effector functions
Autoantigen-specific IgA autoantibodies closely correlate with symptoms severity in a subgroup of patients in multiple autoimmune diseases. High autoantigen-specific IgA serum levels will be used as companion diagnostic to stratify patients for personalized treatment with JJP-1212. Interfering with the IgA/ CD89 axis by JJP-1212, resolves IgA/ autoantigen-induced inflammation and tissue damage in autoimmune diseases.
Louis Boon
CSO & Board Member, JJP Biologics
In current drug development, we are educated and trained to follow identical development strategies that have been proven to be successful in the past and assume a guaranteed success for the future. However, as we see with the stock market, past successes do not guarantee anything for the future and a case-by-case customized approach is essential.
For example, consider the biology around IgA-IgA receptor (CD89 or FcαR1) interaction in which there are significant differences in species. Ample information has been published over the last 3 decades to show that levels and appearance of autoantigen specific IgA antibodies correlate very well with disease severity in over a dozen of different clinical
www. pharmafocusamerica.com 19
RESEARCH & DEVELOPMENT
autoimmune indications in humans. For this reason, it is difficult to understand why absolutely no therapeutic development efforts have been initiated to target the interaction between IgA and CD89. It becomes even more surprising when one considers the significant body of data that proves the functionality of the IgA-CD89 biology using human patient cells in in vitro experiments. It becomes even further surprising when you realize that auto-antigen IgA responses are already measured in a clinical setting and reported to for over thirty years to correlate with disease severity. The auto-antigen IgA antibody response, measured as antigen-specific IgA antibodies in patient serum, is a ready-made companion diagnostic (CDx) that provides the opportunity to stratify patients for interference of IgA-CD89. The question remains why in spite of all the evidence no development to target this pathway has been initiated? In the case of CD89, this is most probably due to the fact that the CD89 receptor is completely absent in murine models and therefore impossible to perform preclinical in vivo studies in mice or rat disease models, a presumed essential part of current drug development. Apparently, this has been viewed as a major roadblock, because the supportive human in vitro data has not resulted in the development of a specific drug for these patients with high IgA autoantibodies. Blindly follow the identical route without considering a detour may end up at a roadblock, like with geese that colliding with the wind turbines and are dying because
the always follow the identical route. In my view, not a major issue, since geese that fly into a stationary wind turbine can be categorized into the group of “stupid geese” because they always follow the same route, and ultimately we can let natural selection take care of it since there is a group of “smart geese” that exist, which are fast, flexible, creative and adapt easily to new situations that will escape a tragic death following a collision with a wind turbine. For the development of a drug like anti-CD89 a detour around the roadblock of preclinical in vivo evidence is needed and behavior like a fast, flexible and creative smart goose is needed. We have faced many examples where accumulating evidence proceeds protective action. With age, we can become more nostalgic, how many of us have heard elderly parents lament the current state of society and refer to the GOOD OLD DAYS when life was so much better or so it seemed. Wouldn’t it be interesting to go back in time and be a fly on the wall during the so called GOOD OLD DAYS to witness what was so good. For those of us of a certain age, we can recall jumping into a car without seatbelts, flying through the streets on motorbikes without a helmet and not a care in the world. The tobacco industry
20 PHARMA FOCUS AMERICA ISSUE 01 - 2023
RESEARCH & DEVELOPMENT
was able to convince people of the benefits of smoking, and whether you were convinced by that or not, you were subjected to other people’s smoke in close confinement such as aircraft cabins or entertainment venues. Alcohol was seen as a tonic, in particular doctors would encourage pregnant ladies to take a Guinness a day and excessive consumption was seen as a virtue. There was no understanding of the link between LDL cholesterol and heart disease, the so called benefits of fatty foods outweighed any negative impacts. Many of the chemicals that we came into contact with on a day to day basis were deemed to be safe and useful, asbestos, in particular, had a place in many homes across Europe.
All of this flies in the face of what we currently understand, and it is only through a scientific endeavour that enabled a change in public health. Let’s remember, the pioneering epidemiologists, Doll and Hill first published a series of ground breaking papers on the impact of environmental and lifestyle factors on health in 1954, yet a complete change in perception of these practices was not fully accomplished until the 1990s, and in some nations, the dated perceptions linger.
The historical resemblance of the involvement of IgA-autoantibodies in autoimmune disease, especially in rheumatoid arthritis (RA) and the serum levels of IgA autoantibodies (e.g., IgA-rheumatoid factor (IgA-RF)), is remarkable. It is curious to note that the link between high serum levels of anti-RF IgA antibodies and the severity
of RA was established over 30 years ago, and yet no one has really exploited this in delivering therapeutic interventions. Strong neutrophil-driven and CD89-mediated effector functions of IgA antibodies have been reported. The activation of neutrophils through IgA autoantibody-CD89, resulting in an excessive innate immunity response and has pathological consequences in autoimmunity. This justifies antagonizing the potent damaging and pathogenic effector functions of autoantigen specific IgA antibodies in autoimmunity by an antagonist anti-CD89.
RA is the most common autoimmune disease, affecting 0.5 to 1.0% of the world population, it is characterized by chronic inflammation in the symmetric joints and by the appearance of autoantibodies and infiltration of inflammatory cells. Blocking TNFα in RA patients has remarkably improved treatment outcomes for RA patients. Still, about 25-30% of anti-TNFα treated RA patients, do not effectively respond to treatment and for this non-responsive group, RA is a highly personalized disease with regard to its flare ups and periods of remission. Each patient's unique disease pathology requires a high level of personalization in terms of treatment, which can be achieved by stratifying patients according to biomarker profiles through the use a CDx. Precision medicine treatments are rapidly shifting the treatment of many diseases from a ‘one-size-fits-all’ to a ‘targeted testing and treatment’ approach. Since the determination of various isotypes
www. pharmafocusamerica.com 21
RESEARCH & DEVELOPMENT
of autoantibodies (e.g., IgM-RF, IgG-RF and sometimes IgA-RF) in RA patients is included in current standard diagnostic practice, potential effector cells and mechanisms responding to autoantibody complexes can be predicted and novel therapeutics developed. Local autoantibody complexes provide a highavidity matrix for homing of cells expressing the corresponding Fc-receptor. These cells subsequently locally get activated inducing collateral tissue damage and aggravating and broadening the inflammatory response. Interestingly, high pre-treatment levels of IgA RF are associated with a poor clinical response to TNFα inhibitors. Whereas patients with low IgA-RF and those with negative IgA-RF had a good response rate, patients with high positive IgA-RF were poor responders to anti-TNF treatments. This perfectly shows how already available CDx (anti-RF IgA) and clinical unresponsiveness to current treatment can enable a personalized medicine approach for patients receiving JJP-1212. The absence of IgA autoantibodies renders JJP-1212 into a non-relevant treatment in these specific IgA negative patients. By excluding these IgA negative patients, potential adverse events of the negative group will be excluded and potential adverse events are limited to the IgA positive patients in which JJP-1212 can have its clinical efficacy. Such a personalized medicine approach, supported by the failure to respond to a current treatment and a relevant CDx, provides a strategy that optimizes clinical development with respect to efficacy, risk and
costs. For note in the realm of personalized medicine, costs are associated with failure to effectively treat patients such that the symptoms of a chronic disease have to be continually treated palliatively, additionally giving a targeted treatment to a biologically irrelevant individual in which a CDx does not support its use, can cause more damage than simply administering a placebo. The massive and impressive amount of data which associates IgA autoantibodies to severity of disease in RA, makes it hard to understand why no therapeutic interventions were designed to antagonize this pathway. The considerable species differences in the IgA-CD89 axis may have contributed to this, since in vivo evidence is hard to obtain when biology is completely different between species. So despite having evidence of the causal link between IgA autoantibodies and disease severity for many years, little has been done with this data to address and treat the progression of disease in this cohort of patients. It seems therefore inevitable to subdivide high titre IgA autoantibody RA patients in a new clinical indication: IgA-mediated RA and subsequently customized their treatment protocol to their disease specifically supporting a personalized medicine approach.
With all these positive points it is incomprehensible why targeting this pathway has been completely ignored. The answer to this lies in the fact that there is considerable species difference in the biology and that mice do not have CD89. Hence it becomes difficult to get in vivo proof of concept data for this pathway in
22 PHARMA FOCUS AMERICA ISSUE 01 - 2023
RESEARCH & DEVELOPMENT
mouse models. Since both venture capital funds and large pharma due diligence teams stipulate that in vivo proof of concept is essential to support their investment analysis, the absence of animal models to support IgA-CD89 as a target has been drowned by development dogma. Targeting the CD89 receptor needs an unconventional approach in which animal experiments can only be done using transgenic animals delivering limited information, and therefore a custom approach is obligatory that needs fast and flexible adaptation and creativity in development.
The parallel between the situation with our bad habits from the GOOD OLD DAYS, for which healthcare professionals were able to initiate changes in public health with massive and impressive amounts of data which conclusively demonstrated causal links, is remarkable. They succeeded to discourage and reduce smoking, reduce consumption of alcohol and junk food and obliged the use of safety belts and helmets over time. Improvements in current diagnosis, application of RF-IgA as CDx and treatment in RA using JJP-1212 in the future will make us smile retrospectively and make us conclude that the GOOD OLD DAYS when anti-TNFα therapies and small molecule treatments prevailed in the treatment of severe RA and were not always that good.
By following a standard, so called tried and tested drug development route, new therapies with significant potential such a targeted antagonist anti-CD89 antibody are at risk of being lost. The IgA-CD89 example

shows that drug developers that blindly follow the same established pathway without adapting diverting onto an alternative route are consigning themselves to the same fate of as the “stupid goose” that is found lying dead under a turbine. It’s up to you to decide which category of geese you would like to belong! Unevidentely, this will result in joining and supporting the “SMART GOOSE” community.

www. pharmafocusamerica.com 23
AUTHOR BIO
RESEARCH & DEVELOPMENT
Louis is CSO and Management Board Member of JJP Biologics, an innovative Polish Company backed by the Starak family. JJP Biologics develops novel personalized medicine and companion diagnostics. Louis is an author of over 350 publications and an inventor at more than 20 patent applications.
Recombinant Antibody Production in Drug Discovery
Therapeutic antibodies are one of the best-selling drug classes in the pharmaceutical market, and the advent of antibody engineering and recombinant production dramatically improved the arsenal of therapeutics against acute and chronic diseases.
Li Na, Technical Account Manager, Sino Biological
Antibody engineering and production are essential tools in drug discovery, providing powerful and specific therapeutics and diagnostic or prognostic materials for various diseases. Antibodies are proteins produced by the immune system in response to foreign invaders,
Antigen
such as bacteria or viruses. They can bind specifically to these invaders, neutralizing them and preventing them from causing harm. Over the last few decades, scientists have developed methods for engineering and producing antibodies that can be used as drugs.
Myeloma cells
Mouse
B cells
24 PHARMA FOCUS AMERICA ISSUE 01 - 2023
Monoclonal antibody Hybridoma
Figure 1. Hybridoma technology
This article will provide an overview of the process of antibody engineering and production as well as their applications in drug discovery.
History
The history of antibody engineering and production in drug discovery can be traced back to the 1970s and 1980s, when scientists first began to explore the potential of using antibodies as therapeutics. The first-generation antibodies were derived from natural sources, such as animal blood, and were used primarily as diagnostic reagents.
One of the key early milestones in the history of antibody engineering was the development of hybridoma technology in 1975 by César Milstein and Georges Köhler. This allowed for the production of monoclonal antibodies (mAbs) by fusing a B cell, which produces a single antibody type, with a myeloma cell that can divide indefinitely (Figure 1). This made it possible to produce large quantities of specific antibodies. However, the production of early mAbs was limited by the availability of suitable myeloma cell lines (usually mouse or rat) and the fact that hybridomas can be low-yielding or genetically unstable. (Figure 1)
In the 1980s, scientists began to develop methods to produce recombinant antibodies, which are made using genetic engineering techniques. These methods allowed scientists to produce large quantities of pure and consistent antibodies in a controlled environment. This was a significant
step forward, as it made it possible to manufacture antibodies on a large scale for use in preclinical and clinical trials.

Then, about a decade later, the development of display technologies, such as phage display, yeast display, and ribosome display, made it possible to screen large libraries of antibodies for specific binding properties. This quickened the process of identifying antibodies that bind to specific antigens. This also made it possible to optimize the structure of antibodies to improve their binding properties using mutant libraries. Such display technologies, particularly phage display, are a major driving force in the discovery of numerous antibodies that can be engineered, optimized, and recombinantly produced. Phage display antibody library technology involves multiple steps, including the construction of phage display antibody libraries, panning, mAb identification, and recombinant expression of positive clones (Figure 2). Sino Biological is specialized in recombinant protein production and antibody development. Based on its expertise in the construction and screening of phage display antibody libraries, Sino Biological provides efficient antibody discovery services. (Figure 2)
The first licensed mAb drug was muromonab-CD3 (1986), a hybridom-derived antibody that was used for immunosuppression during organ transplantation. Rituximab (Rituxan), approved by the FDA in 1997, targeted the CD20 protein in B-cell non-Hodgkin’s lymphoma. It marked a significant milestone in the

www. pharmafocusamerica.com 25
RNA isolation Tissues VL/VH scFv Cloning Phagemid Electro-transformation Phage Library Sequencing Phage Amplification Hits Expression Clones Analysis Phage Elution Washing Panning 2~4 Rounds Target Binding Phage Library Amplification Flexible screening strategies Supernatant screening Multiple screening methods 1 2 3 4 5 6
Figure 2. Technical route of phage display library construction and screening
field of antibody engineering and production as it was the first antibody drug made with recombinant technology. Rituximab was also the first monoclonal antibody approved for cancer treatment, followed by a number of other mAbs in the following years. Since then, approximately 170 antibodies and antibody-based therapeutics (including antibodydrug conjugates) have been approved by authorities worldwide.
Antibody optimization
Besides the ability to produce large quantities of antibodies with a controlled quality, benefits of recombinant antibody production include: through antibody engineering, allowing for the generation of different antibody structures and derivatives (Figure 3), such as bispecific antibodies (bsAbs) and Fc-fusion proteins; and enabling the optimization of antibody structure, which can be done by introducing mutations into antibody genes, in order to improve the clinical effectiveness of antibodies. (Figure 3)
BsAbs, which are designed to bind two different antigens or epitopes at the same time, have a wide range of applications, including redirecting specific immune effector cells to tumour cells and blocking two different pathways. Fc-fusion proteins, composed of the Fc region of an IgG antibody and a desired linked protein, such as receptor extracellular domains, enzymes, cytokines, and active peptides, have additional beneficial biological and pharmacological properties. The primary reason for fusing a biologically active protein of interest with Fc is that the Fc domain contributes to plasma half-life extension. Sino Biological has extensive experience in producing recombinant antibodies in diverse formats
and provides a variety of service packages to meet different research and drug discovery needs.
Antibody humanization is one of the key antibody optimization technologies. Humanized antibodies represent a major type of antibody drug. The clinical application of mouse mAbs is limited by the human anti-mouse antibody (HAMA) response, which not only neutralizes these therapeutic antibodies but also leads to an allergic response in patients. Humanization of mouse mAbs by genetic engineering can minimize the heterologous nature of mouse mAbs while maintaining their specificity and affinity. Humanized antibodies also improve the safety and therapeutic efficacy of mAbs in clinical applications. Sino Biological provides high-quality mAb humanization services using complementaritydetermining region (CDR) grafting technology and computer-aided molecular modeling (Figure 4).
More optimization methods are evolving as the field of antibody engineering and production continues to expand. For instance, scientists can optimize the way an antibody binds to its antigen using techniques such as artificial intelligence (AI)-driven affinity maturation performed in silico instead of relying on in vivo affinity maturation that requires animal use. Sino Biological provides an AI-driven affinity maturation service, which is a computational and experimental service aimed at improving the affinity and specificity of mAbs using machine learning algorithms to predict the effect of mutations on antibody-antigen binding and then validating the predictions in a wet lab (Figure 5). This service is useful for researchers who are developing mAbs for therapeutic or diagnostic applications, making their candidate antibodies more effective. (Figure 5)
26 PHARMA FOCUS AMERICA ISSUE 01 - 2023 VL CL CH1 VH CH2 CH3 Fab Fc IgG Nanobody Bispecific scFv Bispecific F(ab)2 Fc-fusion Protein ScFv Fab
Figure 3. Examples of recombinant antibody formats
Process
Production method
Once the desired antibody sequences are obtained, they must be produced in large quantities for use in drug development. This is typically done by growing large numbers of cells, such as Human Embryonic Kidney 293 (HEK293) cells or Chinese hamster ovary (CHO) cells, which have been genetically modified to produce the desired antibodies. CHO and HEK293 cells are advantageous over other host cells, such
as E. coli and yeasts, due to their high capacity for post-translational modifications, which are important for the proper folding and activity of antibodies. The cells are grown in large bioreactors and are given the nutrients and growth factors they require. Then, the antibodies are purified from the culture media with various available purification techniques and are ready for use in in vitro, preclinical,
clinical studies.
www. pharmafocusamerica.com 27
and
FR1 CDR1 CDR2 CDR3 FR2 FR3 FR4 FR1 CDR1 CDR2 CDR3 FR2 FR3 FR4 FR1 CDR1 CDR2 CDR3 FR2 FR3 FR4 CDR Identification & 3D Structural Modeling CDR Grafting Mouse Antibody Human Framework Region Humanized Antibody Back Mutations
Figure 4 depicts the antibody humanization and CDR grafting technology processes
Antibody
Chimeric Antibody Humanized Antibody Mouse Antibody CDR Grafting Technology
of
Humanization
Lead Time 4 weeks 103 increase High Affinity 15% Hit Rate Screen up to 1010 sequence space Direct access to sequence No animal use Cost effective Can be combined with “humanization” module Higher hit rate will be achieved on subsequent computation by incorporating wet-lab data
Figure 5. AI-powered affinity maturation platform advantages
Future perspectives
Antibodies have become one of the most successful classes of therapeutics used to treat various diseases, including cancer, autoimmune diseases, and infectious diseases. They are also essential tools for biomarker detection in the process of drug discovery.
Researchers are still working on improving the manufacturing process and developing new ways of engineering and high-throughput (HTP) procedures to produce antibodies that can be used during drug discovery. Sino Biological is continually working on developing new workflows and methods to improve the efficiency and reduce the cost of antibody production. Sino Biological currently offers HTP recombinant antibody production services, providing the most cost-effective solution for the rapid production of a large number of antibodies. In response to the rapidly increasing therapeutic needs, Sino Biological combines its expertise in gene synthesis, vector design, and transient antibody expression technology to produce highly efficient antibodies in HEK293 and CHO cells in as little as 2 weeks (Figure 6).
The future of recombinant antibody production is expected to be driven by several factors, including the development of new technologies, increased demand for therapeutics, and the need for more affordable and efficient production methods. The field is likely to continue to evolve and expand, providing new opportunities for the development of more effective and accessible therapeutics.
1. Marks L. The birth pangs of monoclonal antibody therapeutics: the failure and legacy of Centoxin. MAbs. 2012;4(3):403-412. doi:10.4161/mabs.19909
2. Liu JK. The history of monoclonal antibody developmentProgress, remaining challenges and future innovations. Ann Med Surg (Lond). 2014;3(4):113-116. Published 2014 Sep 11.
doi:10.1016/j.amsu.2014.09.001
3. Geyer CR, McCafferty J, Dübel S, Bradbury AR, Sidhu SS. Recombinant antibodies and in vitro selection technologies. Methods Mol Biol. 2012;901:11-32. doi:10.1007/978-1-61779931-0_2
4. Wang SS, Yan YS, Ho K. US FDA-approved therapeutic antibodies with high-concentration formulation: summaries and perspectives. Antib Ther. 2021;4(4):262-272. Published 2021 Nov 18. doi:10.1093/abt/tbab027
5. The Antibody Society. Therapeutic monoclonal antibodies approved or in regulatory review. (Accessed on January 10, 2023); www.antibodysociety.org/antibody-therapeutics-product-data
Na Li (Lina), Ph.D., QIHCCM, is a technical account manager at Sino Biological. She has completed her Ph.D. research at the Singapore-MIT Alliance for Research and Technology. She has multiple years of experience as an antibody-based assay development scientist and has led pre-clinical projects involving multiple research areas, including cancer therapy, toxicology, and infectious diseases. Lina joined Sino Biological in 2022 as a technical account manager, helping clients from various biotechs, academic research groups, and pharmaceutical companies with protein and antibody development.

28 PHARMA FOCUS AMERICA ISSUE 01 - 2023 AUTHOR BIO
References
Sequence Synthesis Vector Transient Expression Purification & QC Fast Delivery Framework & Sequence Check Codon Optimization Multiple Isotypes & Species Available HEK293/CHO Cells Optimized Culture Conditions to Increase Yield Guaranteed 100% Success Rate Flexible Scales Guaranteed Purity >90% or Higher SDS-PAGE, Endotoxin Additional Characterization Tools (ELISA, Octet, FC, Biacore, SEC, etc.) High-copy Expression Vector
Figure 6. Technical workflow of Sino Biological’s high-throughput recombinant production platform
“Digital Transformation is the way forward”

Highly accountable marketing campaigns. Every dollar counts. Digitally powered marketing campaigns may be cheaper than you thought... Ask us how?
Use our guaranteed ROI programs for reposition, penetrate or launch of your new products or services.


Grow your audience with increased reach, impact and user-friendliness
Rise above geographical boundaries
Generate new business
Gain the strong web presence differentiating yourself from competitors
Connect and engage with your target audience

Give more exposure to industry specific people
Increase your brand profile and share your capabilities with leading industry professionals

pharmafocusamerica.com 29
Let the true “Digital Transformation” be the base of all your marketing campaigns
UPGRADE YOUR MARKETING STRATEGY
Email : advertise@pharmafocusamerica.com Website : www.pharmafocusamerica.com Our recent successful partnerships:
Decentralization - The Future of Clinical Trials
 Nicoleta Grecu Director, Pharmacovigilance, Clinical Quality Assurance, Clover Biopharmaceuticals
Nicoleta Grecu Director, Pharmacovigilance, Clinical Quality Assurance, Clover Biopharmaceuticals
The COVID-19 pandemic made the regulatory authorities recognize that there is a need for change in the conduct of clinical trials. Clinical trial management guidelines were released to adjust to the hindrances that this pandemic brought but also to support pharmaceutical innovation, especially for COVID19 vaccines and treatments. These changes led to decentralized clinical trials. Currently, the regulatory authorities are working to strengthen the system and the regulatory framework for decentralization. The first steps in this direction were taken, but more changes are about to come.
Decentralization was a necessity for the conduct of the clinical trials during the Coronavirus Disease 2019 (COVID-19) pandemic. This new way of managing clinical trials became a more recognized concept during the past few years that needs further development and a regulatory framework.
The decentralized clinical trial (DCT) is defined by the Food and Drug Administration (FDA) as "a clinical investigation where some or all of the trial-related activities occur at a location separate from
30 PHARMA FOCUS AMERICA ISSUE 01 - 2023
CLINICAL TRIALS
the investigator’s location." DCTs are divided into two categories based on the degree of decentralization, which can range from fully decentralized to partially decentralize. In the case of a fully DCT approach, all trial procedures are conducted remotely through digital means and supply delivery with no in-person visits required to the trial site, whereas a hybrid approach may include some decentralized trial activities combined with traditional trial procedures such as in-person visits via mobile clinicians or alternative sites.

Clinical trials are essential for drug development and improving the health of patients. The remote DCT approach was recommended for adoption during the pandemic, with the scope to facilitate participation in trials by offering maximum flexibility and convenience. Nevertheless, the DCT approach has been recognised to bring benefits, and there is an increased demand for conducting clinical trials remotely. Some of the benefits DCTs provide are presented in Image 1.
Hereinafter, the background of clinical trial decentralization, the current knowledge, and the future of DCTs will be described.

The desire to conduct fully or partially remote clinical trials has been supported by continuous technological advances despite the lack of a regulatory framework on this topic. The industry’s limited experience and the absence of corresponding infrastructure and regulatory guidelines slowed down DCT adoption.
Industry recommendations and best practices
The benefits of conducting remote clinical trials have been recognized over the last few years, especially during the COVID-19 pandemic. However, the challenges of implementing this approach compelled pharmaceutical companies, clinical research organizations, investigators, and technology companies to collaborate in order to accelerate the development of tools and methods for working with DCTs.
The first to issue relevant recommendations on DCTs is an organization that was co-founded by Duke University and the FDA, the Clinical Trials Transformation Initiative (CTTI). CTTI sought to develop and drive adoption of DCT practices by releasing their recommendations in 2018. These recommendations covered DCT approaches and protocol design in brief, as well as telemedicine state licensing issues, drug supply chain, mobile healthcare providers,

www. pharmafocusamerica.com 31
Participants Investigators Stakeholders Easier access to Clinical Trials Faster recruitment Less burdensome trial participation Better retention and engagement Potential for real-time feedback Less manual data entry Cost-effective Sense of ownership Real-time safety and monitoring Faster access to trial outcomes Trial data generated in real-world settings Agile and resilient trials Increased diversity of trial populations Greater geographical reach CLINICAL TRIALS
Image 1. Benefits of clinical trial decentralization
investigator delegation and oversight, and safety monitoring.
The opportunities brought by DCTs were further explored by a multi-stakeholder consortium supported by EU/EFPIA Innovative Medicines Initiatives (IMI), namely Trials@ Home, which had as common goal the development of in-depth recommendations and pilot tools supporting widespread acceptance and use of remote DCTs in Europe. Trials@Home issued their first deliverables in 2020, and since then they have completed several reports that are spread throughout six work packages, each of which focused on investigating solutions and developing practical recommendations for the adoption of DCTs on the following six topics: (1) best practices in DCTs; (2) technologies—barriers, enablers, and data management; (3) a pan-EU remote DCT pilot; (4) ethical, regulatory, good clinical practices (GCP), and legal aspects; (5) communication, dissemination, and stakeholder engagement; and finally, (6) project management and synthesis.
The Association of Clinical Research Organizations (ACRO) is another organization dedicated to promoting clinical trial innovation and efficiency. It brings together CROs and technology companies to support optimization and development of new methodologies affecting DCTs. In 2020, ACRO developed a DCT toolkit in order to support and advance the adoption of decentralized trials, which includes five resources: (1) a detailed Quality by Design (QbD) manual for DCTs; (2) an
accessible, quick-reference QbD manual; (3) a risk assessment considerations template; (4) DCT data flow maps; and (5) a change management question-and-answer (Q&A) resource. In addition to the DCT Toolkit, ACRO’s White Paper provides an overview of key issues in the decentralization of clinical trials and includes case studies from ACRO members.
One last organization worth mentioning is the Digital Medicine Society (DiMe), which tackles digital medicine involving the different fields of medicine, healthcare, and technology by generating best practices and guiding principles for accelerating clinical research and enhancing clinical care. They promote the use of digital medicine through the development of tools for measurement and intervention to support the practice of medicine broadly. Their library includes a large number
32 PHARMA FOCUS AMERICA ISSUE 01 - 2023
CLINICAL TRIALS
Decentralization has been used in a few trials because of the difficulties in making the quick transition from on-site to remote trial activities
of projects and resources categorized by four topics: digital measures; health care and public health; regulatory science; and publications.
DiMe’s Playbook of Digital Clinical Measures addressed the processes for digital clinical measures and technologies used for remote monitoring in clinical trials, clinical care, and in public health settings.
Regulatory framework
The existing legal framework on clinical trials with medicinal products and associated guidelines do not prohibit the conduct of decentralized trials in general, but few of them provide advice on the use of specific decentralized elements (e.g., remote recruitment, digital endpoint selection) and trial-related interventions outside the trial site (e.g., digital consent, electronic data collection systems, electronic consultations).
The first regulations to provide recommendations for performing remote activities during the conduct of a clinical trial were issued during the COVID-19 pandemic. In March 2020, the World Health Organization (WHO) declared a COVID-19 pandemic for which no specific treatment existed at that time. COVID-19 had a large impact on the performance of traditional clinical trials due to quarantine measures, travel restrictions, and supply chain interruptions that led to patient trial participation discontinuation or withdrawal, unavailability of the clinical trial staff, site closures, and shortages of investigational medicinal products (IMPs).
The degree of impact was different between the countries and regions, as this was depended on the implementation of the pandemic prevention measures at the national level as a response to the public health emergency. Thus, the decentralization of clinical trials started to gain attention and needed to be adopted within the regulatory framework. Given the impact of COVID-19 on traditional clinical trials, as well as the critical need for developing new COVID-19 treatments, regulatory agencies were forced to issue guidelines on clinical trial management during this time period.
Under these circumstances, the European Medicines Agency (EMA) released the Guidance on the Management of Clinical Trials during the COVID-19 (coronavirus) Pandemic to limit the disruption of clinical research during COVID19 in the European region, which supported a harmonized approach among European (EU) Member States (MS) even though the local or national requirements prevailed in case of conflicting requirements. DCT-related elements introduced by this guideline were: conversion of trial participants’ physical visits into phone or video visits; contacting the trial participants via phone or video calls; obtaining written consent or approvals by email or mail; direct distribution of IMP to trial participants’ homes; transfer of trial participants to another trial site closer to their homes; trial participants testing in local laboratories; remote centralized and site monitoring activities; remote source data verification; and remote audits. The FDA also issued guidance for the conduct
www. pharmafocusamerica.com 33
CLINICAL TRIALS
of clinical trials of medical products during the COVID-19 Public Health Emergency, with recommendations similar to the EMA guidance and additional details such as practice examples and Q&A that provide readers with more clarity.
The COVID-19-related guidelines and the specific exemptions granted during this pandemic are effective until the COVID-19 outbreak has passed. Moreover, EMA advised sponsors managing clinical trials impacted by the war in Ukraine to use the experience gained during the COVID-19 pandemic and apply the approaches and flexibilities agreed in this context.
Although the EU guideline on the management of clinical trials during COVID19 is still effective, a few EU agencies issued national guidance on the DCT implementation to cover the continuous conduct of remote activities and to provide guidance on the safe implementation of DCT elements without compromising the safety and rights of trial participants or the integrity of the clinical trial. At the end of 2021, the Danish regulatory agency issued a guidance on the implementation of decentralized elements in clinical trials based on the knowledge gained from the close collaboration with clinical trial sponsors which conducted a small number of pilot projects with sub-elements of decentralized clinical trials that challenged the traditional framework. At the beginning of 2022, the Swedish regulatory agency published its recommendations on the decentralized trials
based on the experience gained during a pilot project where five interventional clinical trials, representing all phases and both academic and commercial sponsors, were approved.
The recent and most-awaited joint EMA/ European Commission (EC)/Heads of Medicines Agencies (HMA) Recommendation Paper on Decentralized Elements in Clinical Trials was released in December 2022. The recommendation paper addressed the roles and responsibilities of the sponsor and investigator, electronic informed consent, IMP delivery, trialrelated procedures at home, data management, and monitoring in a decentralized clinical trial setting. Additionally, an overview of the current national requirements applicable in each relevant EU country has been provided in relation to the general recommendations of this paper.
The future of clinical trials decentralization
As noted above, COVID-19 has increased awareness of DCT, and the use of digital
Likelihood of increasing DCT within next two years
34 PHARMA FOCUS AMERICA ISSUE 01 - 2023
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Very unlikely Unlikely Neutral Likely Certain Row 13% 13% 53% 20% North America 10% 35% 56% 2% 1% Europe 12% 37% 48% 3% 1% Average 12% 38% 46% Image 3.
of
DCT CLINICAL TRIALS
Likelihood
increasing
Biggest challenges to overcome for running DCTs
methods was a necessity for many trials to allow their continuation. This experience increased knowledge of using DCT elements, on which the industry and regulatory current recommendations presented above were based. This situation led to an increased interest in using DCT elements in clinical trials.
In May 2020, Informa Connect surveyed clinical trial professionals from different types of organizations on the adoption of DCT and related technologies, indicating that DCT adoption will increase faster in North America than elsewhere, as shown in Image 3.
Although the DCT approach provides several benefits compared to conventional
trials, there are still disadvantages and challenges to be considered when this new approach is considered. Currently, full remote trials are rarely conducted, and some of the DCT's elements may not be suitable for adoption in some trials. Nevertheless, the majority of trials can benefit from digitalizing trial components and switching to off-site or remote activities. However, the ability to adapt to the technological advances and to manage them should be considered, along with the recognition of the decentralized elements that are likely to have a significant impact on scientific validity, data integrity, the benefit-risk ratio, or
the
protection of
www. pharmafocusamerica.com 35
0% Other 6% 17% 27% 34% 38% 39% 43% 44% Staff training Proportion of responsers Ease of use for patients Stakeholder buy in Technology functionality Data protection and privacy Regulatory acceptance Quality of data 5% 10% 15% 20% 25% 30% 35% 40% 45%
CLINICAL TRIALS
Image 4. DCTs biggest challenges
perceptions around DCT data challenges
address the challenges DCTs bring as presented above.
trial participants’ rights. The key challenges facing DCT adoption are presented in Image 4 as per the Informa Connect 2020 survey. Among these challenges, the Informa Connect 2020 survey showed that the data protection and privacy challenge is hindering DCTs, especially in Europe as the GDPR framework is more restrictive than other data practices and that the quality of data is a more frequent challenge in the rest of the world as presented in Image 5.
Industry recommendations and best practices
Decentralization is the future of clinical trials, and as this is getting more attention, there is a need for clear and in-depth recommendations for implementing DCT elements, especially to
CTTI has developed a vision for how clinical trials should be done in 2030, following the following five views: (1) clinical trials are patient-centered and easily accessible; (2) clinical trials are fully integrated into health processes; (3) clinical trials are designed with a quality approach; (4) clinical trials maximally leverage available clinical and nonclinical data, including data collected via digital technologies, to minimize the collection of necessary trialspecific data; (5) clinical trials contribute knowledge about how to prevent, diagnose, and treat disease, and clinical trials are one of many sources of information that can be acted upon to improve population health. The ways of reaching this vision will need to be further investigated, and CTTI recognizes that some aspects will likely happen sooner than others, particularly in some countries.
Trials@Home is a five-year project, and the Trials@Home consortium continues to define and identify best practices for the DCT's conduct by completing the ongoing or not yet started deliverables contained in their six work packages. Based upon project activities that explore the methodological, technical, ethical, legal, regulatory, and practical aspects of implementing remote DCTs, they will publish their final recommendations in 2024.
Regulatory framework
Although COVID-19 has increased awareness of DCT, there is still a gap in understanding,
36 PHARMA FOCUS AMERICA ISSUE 01 - 2023
Proportion of responces
Regional
50% 40% 30% 20% 10% 0% 43% 34% 52% 37% 34% 29% 38% 50%
Data Protection and privacy Quality of data
CLINICAL TRIALS
Image 5. Regional perceptions around DCT data challenges
experience, and familiarity with this concept. Different regulatory requirements at the national level also complicate the submission of multi-state trial applications. To support the industry, regulatory authorities would need to develop a long-term regulatory framework to address the complexity of DCTs and the continuous technological advances.

The International Council for Harmonization (ICH) GCP guideline update to revision 3 (R3) is a work in progress for which a draft version has been made available. The revision addresses the application of GCP principles to the new diverse trial types and data sources, and facilitates the use of technological innovations in clinical trials. To the ICH GCP (R3) will be added Annex 1 to address interventional clinical trials, and Annex 2 to provide any needed additional considerations for non-traditional interventional clinical trials. The adoption of the technical document is anticipated for December 2023.
Accelerating Clinical Trials in the EU (ACT EU), a joint EMA/EC/HMA clinical trials transformation initiative, was launched in January 2022 with the goal of better integrating clinical research in the European health system and strengthening the European environment for clinical trials, while maintaining a high level of data protection and robustness.
EMA has issued a draft Guideline on computerized systems and electronic data in clinical trials that will replace the current guideline Reflection paper on expectations for electronic source data and data transcribed to electronic data collection tools in clinical trials. A new guideline has been needed to address the complexity of computerized systems that have rapidly evolved in the clinical research environment during the last few years, from eCRFs and ePROs to various wearable devices (including instruments, software, and services) that are used in the creation and capture of electronic clinical data and in the control of
www. pharmafocusamerica.com 37
CLINICAL TRIALS
other processes in the conduct of a clinical trial. The use of artificial intelligence (AI) will be further elaborated on in a future annex to this guideline.
The FDA Oncology Center of Excellence (OCE) supports efforts to modernise clinical trials through their collaboration with CTTI to conduct sponsor surveys to understand the prevalence of remote trial modifications for trials that provided evidence supporting the approval of supplemental or new drug or biologic applications for oncology indications.

FDA provided draft guidance for industry, investigators, and other stakeholders on digital health technologies for remote data acquisition in clinical investigations. This guidance outlines recommendations for use in clinical investigations that address the following topics: selection of digital health technologies (DHTs); verification and validation of DHTs; use of DHTs to collect data for trial endpoints; identification of risks associated with the use of DHTs; and management of risks related to the use of DHTs. The contents of this document intend to facilitate the use of DHTs in a clinical trial.
Conclusion
Clinical trial decentralization was not a widely implemented approach before the COVID-19 pandemic, despite the benefits this may bring. The transition from traditional clinical trials to DCTs has been forced in the context of the pandemic in order to support further clinical research on the IMPs. Decentralization has been adopted in a few trials where the partially
remote approach has been preferred due to the challenges faced by the quick shift from on-site to remote trial activities. The current industry and regulatory framework provide guidance in support of decentralization, and it is expected that more companies involved in clinical trial research will implement decentralized elements and practices. To facilitate the employment of decentralization, the industry is encouraged to share their real-world experiences with DCTs and to closely collaborate with regulatory authorities to address both the successes and the challenges they faced. Decentralization of clinical trials is rapidly developing, and the continuous improvement of best practices promotes DCT adoption.
References are available at www.pharmafocusamerica.com
38 PHARMA FOCUS AMERICA ISSUE 01 - 2023
AUTHOR BIO
CLINICAL TRIALS
Nicoleta Grecu has over 13 years of experience in the pharmaceutical industry with a broad expertise in human medicinal products, combination products, biologics, and vaccines. She is currently the Director of Pharmacovigilance Clinical Quality Assurance at Clover Biopharmaceuticals.
Gene Therapy for Cancer & Immunology
Gene therapy, in general, encompasses a wide range of treatments that all use genetic material to modify cells to aid in healing. To understand the long-term benefits, it is necessary to be aware of the obstacles to therapeutic intervention and to have a developed approach to overcome them. In addition to, for example, failures in targeting metastatic cells, obstacles also include ethical issues, since approaches in gene therapy result in integration into the individual’s genome and thus the possibility of transmitting genetic changes to the patient’s offspring. As with any new type of therapy, there are serious safety concerns. On the other hand, when compared with the side effects of chemotherapeutic treatments, the side effects of gene therapy are minimal. Therefore, gene therapy needs to be explained through the treatment itself, that is, in the context of immunotherapy, oncolytic viral therapy, and gene transfer.
Josipa Ljubicic
QA Director, Principal GCP and GVP auditor, Proqlea Ltd
After the 1970s and the development of recombinant DNA techniques, tests showed that foreign genes are able to correct disease phenotypes and genetic defects in mammalian cells, while the use of gene transfer methods has facilitated the effective demonstration of phenotype correction in vitro and in vivo. Thus, gene therapy has justified studies with human patients and has been widely accepted. Both the positive and negative results of the tests so far in
www. pharmafocusamerica.com 39
CLINICAL TRIALS
the field of immunotherapy have provided scientists with a better understanding of the immune reactions to cancer, which will hopefully lead to the improvement of the next generation of cancer vaccines. Some clinical trials have already shown a positive effect of immunotherapy on survival and have the potential to become part of standard treatment and/or be useful as adjuvant therapy to remove remaining cancer cells. All in all, the initial stages of vaccine development are nearing completion, and it is very likely that they will soon lead to the existence of cancer treatments with vaccines included in the therapeutic regimen.
Introduction:
The human genome contains about 19,000 genes that encode a wide range of proteins whose roles range from building elements of the cell all the way to the main stakeholders in all biological processes necessary for life. Although the genetic code remains largely unchanged throughout the generations, errors may occur in the form of mutations, deletions, or disorders in gene order. These genetic changes lead to the altered appearance and function of a protein, resulting in disease. The term "gene therapy" includes all procedures during which the cells of someone in the organism modify the genetic material in a targeted manner with the aim of achieving a therapeutic effect or disease prevention. The great potential and interest in the development of this therapy are based on the fact that it acts
on the very cause of the pathophysiological disorder, correcting mistakes at the gene level.
The history of the development of this type of therapy has been going on for more than sixty years, and the foundation for the beginning research was about the possibilities of gene identification and transfer. The application of gene therapy in the treatment of life-threatening diseases (e.g., cystic fibrosis, cancers) that did not have adequate conventional treatment at the time was the basis of its development in the beginning. Nowadays, we are witnessing that research into the effect of gene therapy is also turning towards those diseases that are not life-threatening, in this case with the purpose of improving the quality of life of the affected person.
Gene therapy is a rapidly evolving field of medicine that involves the use of genetic material to treat or prevent disease. One area of significant interest in gene therapy is the treatment of cancer, as well as immunology.
In this article, we will explore the basic concepts of gene therapy for cancer and immunology, summarize recent scientific research in this area, and discuss current trends and future directions for this promising field of medicine.
Gene therapy for cancer is a type of medical treatment that utilizes genetic material to specifically target and eliminate cancer cells while sparing healthy cells. One of the most promising techniques within this field is the use of viral vectors, which are modified forms
40 PHARMA FOCUS AMERICA ISSUE 01 - 2023
CLINICAL TRIALS
of viruses, to directly deliver therapeutic genes to cancer cells. Adenoviruses and lentiviruses are common examples of viral vectors that are used in gene therapy for cancer.
Scientists can genetically modify these viral vectors to carry specific therapeutic genes, so that when the vectors infect cancer cells, the therapeutic genes can then be expressed and exert their therapeutic effects. This approach has shown promise in treating a wide variety of cancer types, including leukemia, lymphoma, and solid tumors.
The important concept to highlight here is the specificity of this approach. Scientists have been able to design these viral vectors to be attracted to certain characteristics of cancer cells; once the vector finds these characteristics, it will infect and deliver the therapeutic gene inside the cancer cell, and then the therapeutic gene will do its job of either killing the cancer
cell or slowing down its growth. The specificity of this approach ensures that healthy cells are left unharmed. Another approach that shows promise in gene therapy for cancer is the use of CAR-T cells. CAR-T cells are immune cells that have been genetically modified to specifically target and eliminate cancer cells while leaving healthy cells unharmed. T cells are a type of white blood cell that play a critical role in the immune system's response to cancer cells.
CAR-T cells are a form of T cells that have been genetically altered to express a special protein called a chimeric antigen receptor (CAR). This CAR protein acts as a sort of "homing device" that allows the CAR-T cell to recognize and bind to specific proteins, called antigens, found on the surface of cancer cells. This binding allows the CAR-T cell to then launch an attack on the cancer cell, effectively destroying it.
www. pharmafocusamerica.com 41
CLINICAL TRIALS
This approach has shown significant promise in the treatment of blood cancers such as leukemia and lymphoma. It works by extracting T cells from the patient's own blood, genetically modifying them to produce the CAR protein, and then infusing them back into the patient. The genetically modified T cells can then recognize and attack the cancer cells in the patient's body. This is a very specific form of treatment, and it has shown a very high rate of success in these types of blood cancer.
Research on this approach is ongoing, and it is being tested in a number of solid tumor types as well. There are ongoing clinical trials, and researchers are working on developing new ways to improve the efficacy of this approach and extend its application to other types of cancer.
Gene therapy is also being used to enhance the immune system's capacity to combat diseases in the field of immunology. One way this is being done is by using genetic engineering to create improved versions of T cells, a type of white blood cell that plays a crucial role in the immune response. By genetic engineering, these T cells can be enhanced with new abilities, such as better recognition and elimination of pathogens— the microorganisms or substances that cause disease.
Another way gene therapy is used in immunology is by creating new vaccines. Vaccines are biological preparations that can help the immune system identify and attack
disease-causing organisms, such as viruses or bacteria, without causing the disease itself. Genetic engineering can be used to create vaccines that are more potent at stimulating the immune system to recognize and combat these organisms.
Gene therapy in immunology can be used to improve the immune system's response in various ways. Researchers are currently investigating different methods to enhance T cells, such as by increasing the number of receptors on their surface that can recognize pathogens. Additionally, they are working on creating new vaccines that use genetic engineering to make them more effective at activating the immune system. This field of research is important as it can help create new ways to treat and prevent a wide range of diseases.
Recent scientific research in the field of gene therapy has yielded many promising results, particularly in the areas of cancer and immunology. One example is the use of CAR-T cell therapies, which have shown significant promise in treating blood cancers such as leukemia and lymphoma.
CAR-T cells are a form of immunotherapy that involves collecting a patient's own T cells, genetically modifying them to produce chimeric antigen receptors (CARs), and then infusing the modified cells back into the patient. CAR-T cells are specially designed to recognize and attack cancer cells by binding to specific proteins found on the surface of the cancer cells.
42 PHARMA FOCUS AMERICA ISSUE 01 - 2023
CLINICAL TRIALS
Clinical trials have yielded promising results for CAR-T cell therapies, with remission rates as high as 90% in some cases. This has led to the FDA approving several CAR-T cell therapies for the treatment of certain types of leukaemia and lymphoma.
Here are some examples of peer-reviewed journal articles that provide an overview of recent research on CAR-T cell therapies:
1. Title: "Chimeric antigen receptor T cells in cancer therapy" Authors: Rossi JF, et al. Source: Nature Reviews Cancer, Volume 18, Issue 3, March 2018, Pages 142-158
2. Title: "Recent advances in CAR T-cell therapy for solid tumors"
Authors: Chen L, et al. Source: Journal of Hematology & Oncology, Volume 13, Issue 1, June 2020, Pages 68.
Additionally, researchers have made significant progress in developing new viral vectors for use in gene therapy for a wide range of diseases. Viral vectors are modified forms of viruses that can be used to deliver therapeutic genetic material directly to cells in the body. New viral vectors are being developed with improved properties such as increased targeting specificity, improved durability, and lower toxicity; these new vectors have the potential to be used for a wider range of diseases and for a longer period of time.
This is an active area of research, and there are many studies and articles that have been published on this topic. It's good to check different sources and look for articles with the most recent publication date in order to have the most up-to-date information on this field.
The field of gene therapy is expected to continue to grow and evolve rapidly in the future as researchers gain a deeper understanding of the genetic basis of cancer and other diseases. This increased understanding is expected to lead to the development of new therapies and treatment strategies that will be more effective at treating a wide range of diseases.
One area that is expected to drive this growth is the field of gene editing. Gene editing is a powerful tool that allows scientists to precisely target and manipulate the genetic material of cells. Advances in this field have led to the development of new methods, such as CRISPR-Cas9, that make gene editing more accurate and efficient. With the use of these new methods, scientists are able to more precisely target and manipulate the genetic material of cells, which opens up new possibilities for the treatment of a wide range of diseases.
Here are some examples of peerreviewed journal articles that provide an overview of recent research on gene editing and its potential future applications:
www. pharmafocusamerica.com 43
CLINICAL TRIALS
3. Title: "The future of genome editing: emerging technologies
and applications" Authors:
Gaudelli NM, et al. Source: Nature Reviews Genetics, Volume 21, Issue 7, July 2020, Pages 351-366
4. Title: "The future of genome editing: promise
and challenges"
Authors: Liu Y, et al.
Due to the widespread spread of adenovirus among people, a large part of the population has antibodies from previous infections, which neutralize the effect of adenovirus and reduce the success of gene therapies and vaccines. An attempt was made to overcome this problem by using chimp adenoviruses, which are similar enough to humans to infect human cells but, on the other hand, successfully avoid the action of existing antibodies.
Despite this, over 500 clinical trials using gene therapy have been conducted to date. A 16-month trial of lentivirus-based gene therapy for -thalassemia in France revealed that one patient did not require a blood transfusion. Another successful use of a lentiviral vector in clinical practice has been a recorded trial for the treatment of X-linked adrenoleukodystrophy due to ABCD1 gene deficiency. In this study, progressive cerebral demyelination in two patients was successfully blocked during the 14–16 months after lentiviral delivery of wild-type ABCD1 to CD34+ cells ex vivo. These results provided
a good basis for the implementation of more clinical trials in the use of lentiviral vectors for gene therapy, but the limited duration of the therapy effect warranted further attempts to understand and improve it.
The first gene therapy drug, Vitravene (fomivirsen), was approved by the US Food and Drug Administration (eng. Food and Drug Administration, FDA) in 1998.It is a type of antisense oligonucleotide for local treatment of cytomegalovirus-induced retinitis in immunocompromised patients that is administered intraocularly by injection. Due to low demand for the drug, the manufacturer withdrew it from the European market in 2002. Year, and in the USA in 2006.
One of the first approved gene therapies in the world and the first approved in Europe, Glybera, rests on AAV. AAV vectors are currently used in more than 200 clinical studies for the treatment of a wide range of diseases and disorders worldwide. Recently, the treatment of genetically caused blindness has also started using the AAV-based medicine, Luxturna, and it is expected that the market will soon see the entry of another AAV-mediated gene therapy drug, also for the treatment of ophthalmological disorders, whose clinical trials are in the final phase. In recent years, the AAV-CRISPR/Cas9 system for in vivo genome editing has been developed, expanding therapeutic possibilities even further.
In conclusion, gene therapy is a rapidly advancing field that has the potential to
44 PHARMA FOCUS AMERICA ISSUE 01 - 2023
CLINICAL TRIALS
revolutionize the way we treat a wide range of diseases, including cancer and immunological disorders. The use of viral vectors and CAR-T cells are some examples of promising therapies that are being developed in this field. Viral vectors, such as adenoviruses and lentiviruses, are a type of vehicle that scientists use to deliver therapeutic genetic material directly to cells in the body. Through the use of these vectors, researchers are developing new therapies that have the potential to specifically target cancer cells while leaving healthy cells unharmed.
On the other hand, CAR-T cell therapies are a type of immunotherapy that involve genetically modifying a patient's own T cells to recognize and attack cancer cells. This

approach has shown significant promise in the treatment of blood cancers such as leukemia and lymphoma, with remission rates as high as 90% in some cases.
Conclusion:
The ongoing research in this field is expected to bring new and more advanced therapies for different diseases and to improve the lives of patients suffering from cancer and immunological disorders. The continued advancements in genetic editing technology will also enable scientists to target and manipulate genetic material with more precision, which would open up new possibilities for the treatment of a wide range of diseases.
www. pharmafocusamerica.com 45
Josipa is QA Director and Principal GCP and GVP Auditor at Proqlea Ltd. She has over 18 years of experience in the pharmaceutical industry and is an acknowledged expert in the QA (Quality Assurance) field.
AUTHOR BIO
CLINICAL TRIALS
PHARMACEUTICAL 3D PRINTING

Pharmaceutical 3D printing is a rapidly growing field, with more clinical studies starting every year. Able to manufacture novel dosage forms with advanced release profiles personalised to the patient, this novel manufacturing technique is set to massively disrupt the pharmaceutical industry.
 Alvaro Goyanes
CEO and Co-founder, FABRX
Anna Worsley Director of Innovation, FABRX
Alvaro Goyanes
CEO and Co-founder, FABRX
Anna Worsley Director of Innovation, FABRX
3D printing is a well-known additive manufacturing technique of the modern age. Since the first documentation of the process in 1980s Japan, 3D printing has entered many different fields, such as building construction, the arts and sports equipment. In healthcare, 3D printing has been used since the 1990s to manufacture dental implants and prosthetics, allowing for custom shape and size for each individual patient. More recently, 3D printing has entered the field of tissue engineering to build complex geometries out of novel
46 PHARMA FOCUS AMERICA ISSUE 01 - 2023
Pharmaceutical 3D printing using semi-solid extrusion (Fabrx image, permission to use image granted).
biomaterials to mimic tissues. The use of 3D printing to manufacture medication, or pharmaceutical 3D printing, started growing in the early 2010s, pushed by leading academics attempting to make novel dosage forms fit complex and personalised patient needs. This new, disruptive approach to manufacture medication brings many advantages to healthcare and is set to revolutionise the pharmaceutical industry.
Uses in the real world
Pharmaceutical 3D printing has many different benefits over standard manufacturing techniques for medicines. Pharmaceutical 3D printing allows for the easy creation of hard-to-reach or complex release profiles. For example, fast oral disintegrating printlets (3D printed tablets) or multi-release profile polypills. Pharmaceutical 3D printing can be used to personalise medication, printing small batches of patient-specific prescriptions at a time with personalised dosages, colours, flavours, release profiles, shape and drug combinations as potential options. The small batch manufacture can also benefit clinical trials, reducing costs and waste associated with clinical trial batch manufacture and allowing for the rapid alteration of dosage and formulation when required.
There are many different 3D printing technologies being investigated and utilised to manufacture medication, each with different benefits and target uses. Below we take a closer look at small batch and mass manufacturing
3D printing as well as the techniques currently being exploited for them.
Personalised Medicine And Small Batch Manufacture
The World Health Organisation, European Medical Agency, and US National Institute of Health have all recognised the need for personalised dosage forms, tailored using patient data (age, weight, co-morbidities, gender, ethnicity, etc). $528 billion could be saved annually in the US through the increased treatment efficacy and reduced side effects that would be obtained with personalised medicine. The current method to personalise medicine, compounding, is slow, costly and prone to errors. An average of 41 patients are affected negatively for every compounding error, with the most infamous being the Framingham contamination error medication in 2012, effecting 13,534 patients with 753 documented instances of patient harm and 64 deaths, leading to regulatory reform in the US. However, personalised medicine preparation practices themselves are still outdated and often based on inaccurate dosing and carried out by hand. Pharmaceutical 3D printing could bring the compounding of oral medication into the modern, automated age. Money and time could also be saved if pharmaceutical 3D printing was implemented in clinical trial batch manufacture, allowing for the rapid production of novel dosage forms in smaller more manageable batches than currently feasible without preparation by hand.
www. pharmafocusamerica.com 47
Currently, pharmaceutical 3D printing can be used as a compounding manufacturing method, in line with compounding regulation. Leading regulatory agencies, including the FDA and MHRA, are now setting up legislation to allow for pharmaceutical 3D printing to be used for personalised medicine and small batch manufacture on a wider scale. Often termed point-of-care-manufacturing, once finalised this legislation will regulate hub and spot style systems with multiple printers in multiple hospitals and pharmacies being regulated by single hub organisations.
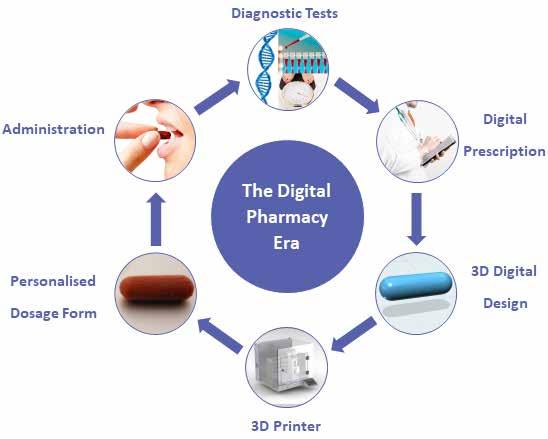
Extrusion based 3D printing technologies are best suited for personalised medicine and small batch manufacture, due to the versatility these techniques offer in terms of
personalisation options. You can easily change the formulation colour, flavour and printed shape to fit the patients’ needs, or combine multiple formulations into a multi-layered, multi-drug polypill. For this reason, these technologies are the ones entering clinical studies for personalised medicine, but other techniques are also being pushed by eager academics looking to expand the field further.
Fused deposition modelling
Fused deposition modelling is one of the most well-known extrusion-based 3D printing technologies. A thermoresponsive filament made up of excipients and drugs, manufactured using hot melt extrusion, is fed through the printhead, melted and deposited
The virtuous cycle of personalised medicine (Fabrx image, permission to use image granted).
48 PHARMA FOCUS AMERICA ISSUE 01 - 2023
on the build plate. As the layers are set on cooling, the 3D model can be built up. Fused deposition modelling allows for increased personalisation options compared to vat or bed techniques described above. The popularity of fused deposition modelling also adds to its attractiveness, having a wealth of literature behind the technique. The downside of fused deposition modelling is that drugs need to be thermally stable at temperatures used, which tend to be over 80 °C. Also, sometimes filament creation via hot melt extrusion can be tricky, with mechanical property issues preventing pharmaceutical filaments from entering the market. To tackle this, M3DISEEN.com was launched in 2021 by FABRX as a researchled, freely available, AI-driven, 3D printability prediction tool to aid filament creation and formulation development for fused deposition modelling.
Direct Powder Extrusion
Direct powder extrusion is another extrusionbased 3D printing technology and is one of the more novel approaches for 3D printing. Based on hot melt extrusion technology, a powder mix (drug and excipients) is fed directly into a hopper on the printhead. The powder mix is melted and pulled down via a rotating screw system to be deposited on the build plate, skipping the filament manufacturing step of fused deposition modelling. Because of this, direct powder extrusion is often seen as superior compared to fused deposition modelling as it shortens and simplifies the
manufacture of printlets. Thermally stable drugs and excipients are required as melting is involved, but the simplicity of this system is proving attractive to pharmacists and formulation scientists entering the field. Direct powder extrusion can be used for personalised medicine on smaller scales or converted into a mass manufacturing method. FABRX is pushing ahead with the former with their M3DIMAKER pharmaceutical 3D printer series with some exciting hospital collaborations across Europe.
Semi-solid Extrusion
Semi-solid extrusion is another popular extrusion-based 3D printing technology. As the name suggests, semisolid extrusion uses gels or pastes containing the drug to build a 3D model. The semisolid mixture is pushed through the nozzle and deposited layer by layer on the build plate. Different methods of pushing the semi-solid through the printhead can be used, such as a stepper motor or compressed air. The semi-solid then sets via cooling or drying depending on the developed formulation. Published in 2019, the first and so far, only published clinical study for 3D printed personalised medicine in a hospital setting used this method. Led by a collaborative team at University College London, UK, the Universidade de Santiago de Compostela, Spain, and start-up FABRX, chewable isoleucine tablets with personalised dose were compared to standard treatment in paediatric patients with Maple Syrup Urine
www. pharmafocusamerica.com 49
Disease at the Hospital Clinico Universitario de Santiago de Compostela, Spain. The study proved successful with improved patient adherence rates, and as a consequence, improved bioavailability. This exciting study propelled the field forward, allowing FABRX to build the first desk-top pharmaceutical 3D printer for personalised medicine and small batch manufacture, the M3DIMAKER.
Vat photopolymerisation
In addition to extrusion-based techniques, researchers are tackling novel formulations to make vat photopolymerisation techniques active players in the race to personalise medicine with 3D printing.
There are many different vat photopolymerisation techniques, defined by their light source. All are based on a bath, or vat, of photosensitive resin containing the drug of choice, that uses photons to polymerise the resin together into the desired 3D model. Stereolithography (SLA) and Direct Light Processing (DLIP) are some favourable techniques in pharmaceutical 3D printing R&D. They use a laser and projector for light sources respectively, directed into the vat of resin as a layer, building the polymerised resin layer by layer. Volumetric 3D printing, a more recently developed 3D printing technique, uses multiple light beams to create a 3D model as a whole object, printing in seconds. In fact,

50 PHARMA FOCUS AMERICA ISSUE 01 - 2023
The University Medical Center Hamburg Eppendorf using FABRX’s M3DIMAKER printer with Direct powder extrusion printhead (The University Medical Center Hamburg Eppendorf image, permission to use image granted with reference).
a research group led by start-up company FABRX printed multiple paracetamolcontaining tablets in seconds using a lab-based prototype printer. FABRX has also developed a mini vat photopolymerisation 3D printer, using the light from a smartphone to manufacture warfarin printlets, opening up the possibility of printing medicines at home in the future.
Vat photopolymerisation techniques offer high resolution prints and are often used for microneedle mould manufacture for medical devices. It is also used in orthopaedics and dentistry. For pharmaceutics, vat photopolymerisation is useful for drugs with poor thermal stability as printing is carried out at room temperature. Unfortunately, there are only a few non-toxic excipients available that can be used for pharmaceutical 3D printing, limiting use to early research and development in the pharmaceutical field. The resins that are biocompatible tend to have less favourable mechanical properties for pharmaceutically useful disintegration times. More research is needed to fully exploit this exciting manufacturing technique.
Mass manufacturing
Pharmaceutical 3D printing can be scaled up for the mass manufacture of novel dosage forms with advanced release properties. Two companies are making strides in this area, Triastek in China with their Melt Extrusion Deposition technology and Aprecia in the US with their binder jetting, ZipDose, technology.
Binder jetting is based on a powder bed system, with a roller to move fresh powder mix over the printing area while a binder liquid is deposited where the 3D model is being built. Gradually, the model is built from layers of powder glued together, and the final tablets are sieved out of the powder mass. This type of 3D printing is commercially used for the production of fast dissolving oral dispersible tablets with high drug loading. It is also useful for drugs with poor thermal stability as heat is not used. The downside of binder jetting is the some-what wasteful nature of the powder bed and difficulty maintaining cleanliness as a result. Pharmaceutical company Aprecia have managed to overcome this hurdle with an in-built powder recycling system, allowing binder jetting to become the first pharmaceutical 3D printing technique with a regulatory approved medication on the market. Aprecia gained FDA approval in 2015 for their epilepsy medication Spritam (levetiracetam), quoting the high drug loading ability and fast oral dispersion of tablets as benefits from using this manufacturing technique. Their ZipDose technology is now being expanded for more drugs and treatment pathways.
On the other side of the world, Triastek are exploring Melt Extrusion Deposition for mass production, a scaled-up version of the direct powder extrusion technique mentioned earlier. Triastek recently gained FDA approval for their first mass manufactured formulation for rheumatoid arthritis, listing advanced drug
www. pharmafocusamerica.com 51
delivery control as the main benefits versus other medication available.
What’s happening now and what’s next?
The field at large
The pharmaceutical 3D printing field is now growing exponentially, with more and more stakeholders getting involved. As mentioned previously, US Company Aprecia is leading the way for mass manufactured, fast dissolving 3D printed formulations. Triastek in China is making bounds with mass manufactured 3D printed formulations with advanced drug delivery control. UK Company FABRX is pushing the personalised medicine and small batch manufacture front. This is alongside hospitals from around the world setting up clinical studies to make this a reality. An example is Gustave Roussy, the leading hospital for cancer research in Europe, who is setting up clinical studies involving hundreds of people to capture data showing the benefits of 3D printed personalised medicine. Big pharmaceutical companies are also embracing the opportunities that this new technology could offer. More and more companies are entering the race to develop 3D printable formulations for personalised medicine and to implement 3D printing in their clinical trial workflows, for cheaper and more efficient batch manufacture.
Closing remarks
The field now needs new clinical researchers and pharmaceutical industry groups to get
involved to help push the advances further, allowing healthcare to fully take advantage of this game-changing technology. By getting involved in clinical studies for more treatment pathways, patients can feel the benefits sooner rather than later.
References are available at www.pharmafocusamerica.com
Alvaro Goyanes is the CEO and co-founder at FABRX. He holds a PhD in pharmaceutical technology and has over 10 years of experience in Pharmaceutics spanning Industry and Academia. He was one of the first researchers investigating the use of 3D printing using Fused Deposition Modelling (FDM) to manufacture oral dosage forms and medical devices.

Anna Worsley is a Director of Innovation at FABRX. Prior to this, she gained experience in multiple healthcare and biomaterial start-ups after completing her PhD in Biomaterials for Diabetic Chronic Wounds at The Royal Veterinary College and University College London (UCL).

52 PHARMA FOCUS AMERICA ISSUE 01 - 2023
AUTHOR BIO
Choose Carefully: The Potential of Continuous Flow Chemistry in API Synthesis
Charles Johnson Senior Director, Commercial Development, Lonza Small Molecules
Flow chemistry has huge potential in API manufacture, particularly for reactions that would be overly hazardous when run at scale as batch processes. This article will look at the mechanics behind flow chemistry, and give examples of where it has been successfully employed to make APIs on a large scale.
The batch reactor has long been the mainstay of API manufacture for good reason. They are well understood, and many processes work very successfully in batch mode. Yet there are some reactions where running a batch process at a very large scale is less than ideal. High up the ‘best avoided’ list are reactions that are extremely exothermic, where running them on a large scale risks explosion in the event of a thermal runaway. But, equally, one of the reaction intermediate might be explosive, demonstrate auto-catalytic behaviour, or perhaps extremely toxic or otherwise hazardous, and handling large volumes might be considered unwise. In such cases, the answer might lie in flow
chemistry. The concept is not new, and has been touted as the potential future for the manufacture of APIs for at least a couple of decades now without making widespread inroads in replacing batch vessels. But, given the right choice of project, flow chemistry has huge potential, and has already been used at commercial scale in the manufacture of APIs and intermediates.
The key difference between a continuous flow reactor and a batch reactor is, of course, the volume of reagents that reside within the vessel at any one time. Batch reactors with a 10,000L capacity are not uncommon in a commercial environment, whereas the volume within a flow reactor might be as small as a
www. pharmafocusamerica.com 53
MANUFACTURING
few millilitres. In a batch process, the reaction mixture will usually remain within the reactor for several hours, whereas in a flow reactor, it will pass through quickly – the residence time is commonly a handful of seconds or a few minutes – and heat exchange is drastically increased. This short residence time also means the product is far less likely to decompose, or to over-react and form side products.
Work-up can also be carried out in flow if another device is connected in series, with the reaction mixture flowing from the reactor device directly into a work-up device to quench the reaction. Multiple reaction steps can even be done in sequence, by dint of connecting another reactor (or reactors) after the first one.
Scaling up can be achieved simply by leaving a continuous flow reactor running for longer, and, if necessary, running multiple reactors in parallel. This represents a further advantage: when scaling up a batch process, it is normal to sequentially scale up through multiple, increasingly large, vessels, with tweaks to the reaction conditions commonly being necessary. It is, of course, also possible to create a production-scale reactor that is a little larger than the one used in development to reduce the need for parallelisation.
With a flow process, the reaction conditions for an experimental scale will likely be identical to those for pilot scale and full-scale commercial production. This will speed up the overall development process, as much of the time required for optimisation is eliminated.

Reaction types
As a rule-of-thumb when trying to decide what type of reactor to use for a particular process when switching from batch to continuous, the nature of the reaction should be considered carefully. Reaction characteristics can be split into three types, depending on their reaction rates and behaviour under standard operating conditions. Type A reactions proceed extremely quickly – they will have a half-life shorter than one second, and the rate will be controlled by mixing or mass transfer.
A type C reaction is much slower, typically taking 10 minutes or more to go to completion. The timescale can even be an hour or more. These slow, kinetically controlled reactions might benefit from being carried out in flow if there were safety considerations in play, or if they are amenable to process intensification at higher temperature, pressure, reagent concentration, or a combination of these factors.
54 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
Between the two lies Type B, with the reaction proceeding in seconds or minutes. These reactions are, predominantly, kinetically controlled, but it may be that they are limited by mass transfer. Efficient heat removal and good mixing will both be necessary but in many cases a good control of residence time may also be required at time scale that is not achievable in a batch reactor. (It is often possible to speed them up by increasing pressure, temperature, reagent concentration, or a combination of these factors).
Reactor types
There are several basic types of flow reactors. Coil reactors – essentially a long tube, usually coiled up to resemble a spring – are most suitable for homogeneous processes. These are most likely to be appropriate for Type C reactions, and some Type B reactions, and can improve safety via process intensification.
Type A reactions are more likely to be successful if they are run in some form of plate reactor, where the reaction takes place in a channel cut into the plate. This may be a good option if mixing control is beneficial, and the reaction time ranges from milliseconds to seconds. It should be possible to increase the yield with better mixing or improved heat exchange, or both, and the correct design of reactor can prevent the reaction from overcooking. Some Type B reactions are also best carried out in a plate reactor.
If the reaction is heterogeneous, then a packed bed reactor may be appropriate; metal
catalysts, for example, can be coated onto a support within the reactor. These vessels are usually made from stainless steel, or even Hastelloy, enabling reactions to be carried out at higher pressures. And then there is the continuous stirred tank reactor, which uses active mixing such as an impeller, which can also be useful for heterogeneous processes.
The physical form of the reagents has an impact on reactor design, too. Liquid–liquid reactions benefit from some form of mixing strategy within the reactor; gas–liquid reactions will need some form of pressurised reactor. But in both cases, either a plate or a coil design could be used, with the precise selection depending on the reaction type.
Reaction condition choices
Mixing is an important consideration when designing a flow process. If active mixing is not used, then some form of passive mixing will be required to ensure the different components of the reaction come into proper contact with each other. This could be as simple as two streams of reactants entering the reactor at steady flow rates via a T-piece, but if more effort is required to make them mix effectively, then structures within the reactor that cause turbulence may be required. Options include a tangential design, SZ-shaped curves, or a more complex liquid-liquid design for multiphase applications.
Temperature and pressure also need to be considered. Temperature is far easier to control within a flow reactor than it is in a large batch
www. pharmafocusamerica.com 55
MANUFACTURING
vessel, with the larger surface area giving better heat transfer. This is particularly important when thermal runaway is a possibility, one of the reasons why some reactions are particularly hazardous in batch.
Pressure can be important, too. If one of the reagents is a gas, for example, then it can increase its solubility, or its speed of dissolution, in the liquid reaction mixture. Pressure can be increased via a back pressure regulator.
With a pump of some form used to introduce the reagents into the reactor on a continuous basis, the flow rate is an important factor in controlling the reaction. This, and the length of the path through the reactor, will affect the residence time – the amount of time the reaction mixture spends within the reactor before it emerges from the other end. The time
needs to be sufficiently long that the reaction goes to completion, but not so long that sideproducts start to form or the product begins to decompose in some way, for example.
Hazardous reactions
Of course, before any process is moved from batch to flow, a comprehensive assessment of the reagents, reactor type and reaction conditions will have to be carried out to determine the optimal design. A great example of a study on continuous flow in hazardous chemistry comes in the form of a recent collaboration between Lonza experts and scientists from the Center for Continuous Synthesis and Processing and the University of Graz in Austria. The aim was to see how effectively sulfonyl chlorides could be made using N-chloroamides, and a
56 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
variety of reagents and reaction conditions were investigated.
Organic sulfonyl chlorides are the starting point for a variety of different sulfonyl derivatives including sulfonamides, which are of particular importance in the pharmaceutical industry. These groups are common in drug structures, featuring in medicines as diverse as antibacterials, HCV polymerase inhibitors, beta-3 receptor agonists, and even carbonic anhydrase inhibitors.
Typically, sulfonyl chlorides are made by the oxidative chlorination of either a thiol or a disulfide, a process that normally uses chlorine gas in the presence of an acid. Using chlorine gas rather than a chlorinating reagent is extremely atom-efficient, but its hazardous and gaseous nature means great care is required in handling it, so alternative chlorinating agents are commonly used, but many of these have limited functional group tolerance, and they are often poorly selective.
N-Chloroamides represent another alternative. These reagents, such as N-chlorosuccinimide (NCS), 1,3-dichloro5,5-dimethylhydantion (DCH) and trichloroisocyanuric acid (TCCA), are milder, and tend to make fewer side products. But they have a downside, particularly at scale: they are thermally unstable, and if reactions are carried out at elevated temperature, they can be explosive. They therefore represent an ideal candidate for continuous flow chemistry, with microreactors ensuring that only small volumes of reaction mixture are present at any one time, alongside a high surface to volume ratio.
Could we create a flow chemistry protocol to make sulfonyl chlorides using N-chloroamides?
The simple answer is: yes we could. We selected DCH as this gave a homogeneous solution throughout, preferable for flow reactions, and found that in the reaction with diphenyl disulfide, 2.5eq of the reagent were required to make the reaction go to completion. Our initial studies on the reaction suggested that it could be considered either Type A or Type B, depending on the reaction conditions selected. We therefore chose a microstructured plate flow reactor that was designed to give efficient heat exchange and good plug-flow mixing, via an SZ mixing structure.
We found that increasing the residence time from 24s to 48s upped the yield from 45% to 87%. We also realised that results from the SZ mixing plate showed the reaction could actually be categorised as Type B, so in order to overcome the corrosion issue observed with metallic plates (even Hastelloy C showed corrosion at higher temperature), a simple T-piece followed by a coil out of polymer material ought to suffice. Using this system, and increasing the residence time further to 58s, gave close to a quantitative yield. Similar protocols also worked successfully with a variety of other disulfide substrates, and also with thiols.
Temporal kinetic profiling was then used to pinpoint the auto-catalytic nature of the reaction and appropriate operating conditions for a preparative scale process (the batch results had shown a reaction runaway even at laboratory scale). These were checked out
www. pharmafocusamerica.com 57
MANUFACTURING
with a four-hour run, and a throughput of 4.3g/h was achieved, with benzendulfonyl chloride isolated in 81% yield, and with a purity above 98%. We even managed to telescope the reaction into an amination reaction, by adding a T-piece to introduce diethylamine in acetonitrile, plus a second coil where the amination reaction took place. This successfully formed a sulfonamide, in 94% isolated yield with a 156s residence time.
Towards a larger scale
These experiments clearly show the potential for the synthesis of sulfonamides via continuous flow to be scaled up and produce large quantities of material. By tweaking multiple parameters, including the reactor style and set-up, reagent stoichiometry, temperature and residence time, very high yields of pure products can be achieved for reactions that present significant hazards when run in batch, including the avoidance of thermal runaway.
As an example, we scaled up the continuous flow manufacture of an indole compound. This was required on a large scale as a starting material for the synthesis of an API. The reaction was run at very high temperature and pressure – 285°C and 80 bar – with a coil-shaped reactor enclosed within a thermal fluid calendar. Two of these were connected in series, and we managed to produce 2,125kg of the compound, with an 81% yield, in 32 days. The material was in spec, and was used successfully in the next step of the reaction sequence to make the API.
In a many cases batch processing remains the method of choice for many reactions where the chemistry is straightforward and the reagents, solvents and products do not present particular hazards. But for the right process, flow chemistry represents a simple way to make and scale up a reaction, especially those that are, one way or another, hazardous, but also increasingly more standard chemistries where sufficient process intensification can be achieved.
References are available at www.pharmafocusamerica.com
Charles Johnson is currently working as Senior Director, Commercial Development at Lonza Small Molecules. Charles has over 20 years of experience in the pharmaceutical sector and specializes in Antibody Drug Conjugates. Prior to joining Lonza, Charles was CEO of ADC Biotechnology which focuses on innovative immobilization technologies for protein conjugation and purification applications.

58 PHARMA FOCUS AMERICA ISSUE 01 - 2023
AUTHOR BIO
MANUFACTURING
Viral Gene Therapy –How Can the Industry Drive Down the Cost of Goods to Better Serve the Patients?
Recent approvals of viral gene therapies have reached records in cost of dose, up to the most recent approval of HEMGENIX priced at 3.5M$ per dose. In this article we look at how process development, cost modelling and process intensification can help drive cost of dose down.
Emmanuelle Cameau Strategic Technology Partnership Leader - Cell and Gene Therapy, Pall Corporation
Gene therapy is a fast-growing sector in the life sciences industry, and there are countless reasons to be enthusiastic about it. The fact that we can use specific gene-modifying technology to amend genetic disorders and provide patients with such impactful treatment is amazing. We have seen an increase in drugs approved over 2021 and 2022, and there are several others pending approval in 2023 - 2024. When we look at the clinical trial pipeline, one can only wonder whether we could reach 10 to 25 approvals
per year by 2025. The idea of having all these approved potentially curative therapies on the market is certainly exciting. However, the question of the accessibility to these drugs by the wider population is still unanswered. Indeed, the last 4 drugs approved by health authorities range between 2.5 and 3.5 million dollars per dose (respectively Roctavian®, Zynteglo ®, Skysona ® and Hemgenix ® approved by the FDA just end of 2022). In some cases, regulatory agencies have even withdrawn approval as a direct result of the pricing such
www. pharmafocusamerica.com 59
MANUFACTURING
Healthy Gene
as the case of Skysona in EU, withdrawn at the request of the manufacturer for commercial reasons, or Zynteglo who had to be pulled out of Germany due to the government officials refusing to pay the listed price.
If we compare the cost per dose of these approved drugs with a lifetime of treatment, the cost is usually justified. In a recent article, the example of the recent Hemgenix® approval was taken. The $3.5 million single dose treatment would replace a lifetime treatment up to $20 million for Hemophilia B patients. However, in the healthcare system we live in, these single dose prices are not compatible with giving the wider population access to all these potentially curative therapies almost at the same time. This is even more true when we think of ultra-rare diseases, as the amount of research and development necessary is usually
comparable but the patient population, being much smaller, leads to a much lower drive for return. Something has to change.
There are many angles from which we could tackle this. Manufacturers could be pressured to decrease the prices. But why would they do that? Of course, these drugs are the fruits of years of R&D, and the investment in R&D is clearly a large part of the cost, as it includes accounting for the risks associated with developing a drug that will take a lot of effort to reach the market and might not get approved in the end. A recent study published in 2021 estimated that “R&D costs per new medicine (accounting for the cost of failure) ranged from $944m to $2,826m (adjusted to 2019 prices)”. The current cost of manufacture is far harder to define. Even if the manufacturing platforms were identical, just differences in the dose
60 PHARMA FOCUS AMERICA ISSUE 01 - 2023
Virus Vector
MANUFACTURING
Virus Vector with Healthy Gene
required could mean more than a 1000-fold difference. While this cost is smaller than the R&D costs, it still has an impact and this is a part of the cost that we, as industrialists, can help to reduce. If successful this helps the manufacturer’s lower costs, while still being profitable.
When we look at that last part, the high cost of manufacturing and the direct cost of a dose is a combination of many factors, but I believe there are two main ones. The first one is the actual quantity of product that is going into the patient compared to the total quantity of material produced. Indeed, some case studies outline that for a phase 3 clinical trial, only 2% of the product that is manufactured actually goes into the patient (Blue, 2022, 16:56). This percentage might increase slightly once the product moves into routine production, but
there is still a large part of the manufactured product that is used for analytical testing, comparability studies, assay controls, stability testing, device losses… the list of points where losses occur goes on. Optimizing the methods around analytics, stability testing, and material requirements overall will help increase the
www. pharmafocusamerica.com 61
Gene therapies for ultrarare diseases will never be commercially viable unless costs are driven down.
MANUFACTURING
Innovation is needed to help shift the paradigm of costs of dose
quantity of product produced and that can be administered to the patient. This ultimately decreases cost per dose. The second one is that we are manufacturing these therapies, these viral vectors, with technologies that have mostly been developed for the mAb and recombinant protein industry. They are ok and have enabled more and more approved products to reach the market. But they could be better if specially developed for viral vectors. There is a major lack of process maturity and adapted technologies. This is an area where solution providers will have a major role to play.
Let’s keep focus on that last point. Solution and technology providers are constantly looking to drive innovation in the way we manufacture products, and drug developers are constantly seeking higher performing processes. Closing the gap on the technologies for viral vector production can only be achieved through close collaboration between drug manufacturers and technology providers. This is the only way we can maximize the chances of developing the right product that the industry needs, in the shortest time. By using more adapted technologies specially developed for these specific products, we will be able to increase the total productivities, increase the recovery and ultimately leading to a decrease in the manufacturing cost per dose. Keeping close contact with regulatory experts through this process will also help making sure the improved technologies will not raise more concerns that they bring solutions.
When we look at the manufacturing process for a viral vector (e.g. adeno-associated virus or AAV, lentivirus or LV) the process can usually be broken into two parts: the upstream process (USP) and the downstream process (DSP). USP productivity can be optimized through bioprocess development/optimization, cell line optimization, plasmids and transfection reagent optimization, and in some cases the use of stable cell lines. To increase the DSP yield, the main levers are process optimization and better recovery at every step of the process. In the case of AAV manufacturing for example, DSP recovery is often around 25 – 30%, with a few achieving up to 40%. Therefore, in a worst-case scenario (25% yield), all the improvements we could think of such as better affinity ligands, innovative solutions to better purify these viral vectors, we can perhaps expect to improve the yield to 80% delivering a 3X yield improvement, and a probable decrease in cost per dose between 1.5 and 2X.
Most experts in this area align on the fact that we need to improve the yields at a factor between 10X and 100X in order to decrease the cost of these therapies significantly to facilitate their access and make them economically viable. So, the question is: how do we go from a 3X improvement to something that is closer to a 100X increase in yield? To do this we need to work on the USP. This includes the use of better plasmids and transfection reagents, stable producer cell lines, USP intensification (e.g. engineering cell lines to excrete AAV for
62 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
example to make processing easier), improving the AAV full:empty ratio out of the cell and the infectious titer among others. USP optimization is the real cost lever that will help making these therapies more accessible.
What are the direct benefits of increased USP productivity? First, it means more doses per batch. This means more patients treated and thus a reduced direct cost per dose. Secondly, it means fewer batches are required to manufacture the target quantity of doses. This means less starting material, less labor and operational expense (OPEX) in general. Alternatively, it could mean same number of batches but enable a reduction of the bioreactor size required. This means, fewer skids, a reduction in consumables cost, and capital expenditure (CAPEX) investment and a smaller manufacturing footprint.
Many development scientists are focused on the day-to-day challenges and may not naturally look forwards to the future challenge of full-scale manufacture. But the fact remains that the molecule in development will need to be scaled up at some point to deliver a product to the market. We often encounter inefficient, unoptimized, poorly scalable and poorly manufacturable processes as a result. Hindsight will always highlight the importance of developing a process to manufacture a therapeutic molecule, such as a viral vector, with the final scale in mind. When this knowledge is available before development commences, this allows for the right choices in terms of technology to be made as early as
possible in the drug development journey. Of course, one could argue that there are process changes that are allowed between Clinical phase 1 and 2 for example. But starting the clinical journey with the process that will be the closest to the final manufacturing sequence ensures process robustness with enhanced knowledge and a more reliable Chemistry, Manufacturing and Control (CMC) package. By starting with a process that is close to the final full-scale process, drug developers enter the clinic with substantial volume of historical data showing process consistency and reproducibility. To generate the most cost-efficient process, one should not only screen, test and choose the right technology available, but every step needs to be optimized.
Several published examples have shown how taking some time to screen reagents such as cell culture media or even transfection reagents in the case of a transient process can have a major impact on viral vector yield and therefore overall cost. Some companies have also been looking at ways to improve cell line productivity, either through cell line engineering and/or stable cell line establishment, others have been looking at innovative ways to improve productivity. Some recent studies have shown that by improving the USP productivity by 50%, the USP manufacturing costs would decrease by 33%.
Tools such as cost modelling can also help understand the impact on the cost of batch or dose of specific process optimizations, or
www. pharmafocusamerica.com 63
MANUFACTURING
even just pinpoint the main cost drivers of the process. This can help process developers tackle the right parameters to further focus optimization efforts in a more efficient way.
Whatever the process development strategy or process optimization we choose to improve USP productivity, it is highly likely this will be accompanied by an increase in total process and product related impurities and contaminants. This increase will likely have an impact on the DSP: An extra purification step might be required; the size of chromatography columns or filters might need to be increased. It is therefore important to constantly balance the yield improvement on the USP side with the extra cost required on the DSP side to ensure the cost improvement is still valid.
Another strategy that can help optimizing the cost of dose is what we call process
intensification (PI). Process intensification looks at minimizing the sources of waste, called Muda by lean process practitioners. Waste is usually attributed to one of 7 different categories: Transportation, Inventory, Motion, Waiting, Overprocessing, Overproduction and Defects. Some business drivers for process intensification are cost of production, footprint reduction, manufacturing flexibility, time to market, facility use, scalability, ease of use… Process intensification has recently been used during the ChAdOx-1 chimpanzee adenovirusvectored SARS-CoV-2 vaccine development. In this paper, Joe et al. illustrate two examples of process intensification that have a direct effect on the cost without affecting product quality. The first example is the optimization of the upstream process with a lower multiplicity of infection (MOI), leading to a reduced virus seed (1 order of magnitude) keeping the same productivity, same critical quality attributes (CQAs) and same production time. The second example relies on the downstream process, where the team have proven that the pre-established process contained an unnecessary step for this particular virus purification. Thus, by removing this step before chromatography, the process was shortened while keeping similar CQAs (within specifications) and similar yield.
While the industry is moving towards the application of scientific and technical platforms to decrease time to market and
64 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
Gene therapy is a method of delivering DNA sequences directly into cell bodies using viruses as drug-delivery vehicles
costs as outlined above, it is essential that we keep in mind the importance of combining it with process intensification. As outlined in the SARS-CoV-2 example, for every new molecule developed, the platform should be reassessed to make sure it is as cost efficient as possible.
So, can we decrease the cost of dose of gene therapies? I believe the answer is yes. It will however require a combined effort of:
• The technology providers and developers to find levers to increase upstream productivity by 10 to 100 times through cell line engineering, capsid engineering, etc.;
• The technology providers and developers to develop new manufacturing and characterization tools to make the processes more efficient;
• The broadening of process intensification and process optimization from the early stages of the drug development.
• Scientists developing processes in research need to start thinking manufacturability from the very early stages, leveraging suitable platforms to avoid slowing the development of suitable processes.

• The health authorities, governments and manufacturers to rethink the way a treatment is invoiced to the social security and patients;
Gene therapies for ultra-rare diseases will never be commercially viable unless we drive these costs down. There is a need of real innovation to help shifting the paradigm of the cost of dose.
AUTHOR BIO
Emmanuelle has more than 15 years of experience in Biotechnology Process Development and GMP production. She is highly skilled in the field of cell culture applications, she has joined Pall Biotech almost 11 years ago first as a Bioprocess Specialist (BPS) during 4 years. She then transitioned to the Bioreactor Applications team, where she was Principal Bioreactor Specialist during 5 years. She then moved to the newly formed Gene Therapy Business unit of PALL where she has been Strategic Technology Partnership Leader for the past +1.5 years. Prior to Pall, she has worked as Biotech Process Sciences Upstream Process development engineer at Merck Serono for 4 years. She received her Biotechnology Engineer diploma from the former Ecole Supérieure d’Ingénieurs de Luminy (now Polytech Marseille). Emmanuelle is a keen horse rider and enjoys spending time with her husband and two daughters.
www. pharmafocusamerica.com 65
MANUFACTURING
Adapting Containment Strategies to Future-proof the Manufacture of Sterile Drug Products
Preparing for the Future: Upgrading Cleanroom Containment Strategies to Ensure Long-term Drug Product Sterility.
ChargePoint Technology’s Ben Wylie, Head of Product Management & Marketing, explores the latest advancement in containment technology and shares his advice on how pharma companies can thrive in a fast-evolving environment.
Ben Wylie Head of Product Management & Marketing, ChargePoint Technology
The aseptic pharmaceutical processing market is growing at a rapid rate - its value is currently projected to increase from $62.2 billion in 2020 to $73.6 billion by 2027.
A key driver of this growth comes from heightened demand across the generic sterile injectable market. This market was worth $434.7 billion in 2022 and is expected to reach $930.3 billion by 2030, growing at a CAGR of 10%.
With this in mind, it is essential for manufacturers to take the necessary steps to ensure their facilities are equipped to handle heightened demand for sterile drug products. Containment is essential during aseptic processing, and cleanrooms and sterile processing technologies serve as critical tools to ensure that drug products remain free from contamination and safe for patient use.
But there are significant challenges for pharmaceutical manufacturers to contend with, as they must expand their capacity to meet market demand, as well as implement advanced containment strategies across their cleanrooms to maintain the integrity of their drug products and comply with rigorous new regulatory requirements.
66 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
To overcome these obstacles, manufacturers must explore innovative solutions to futureproof their investments in 2023 and beyond.
Key challenges facing sterile integrity.
Operating in such a highly regulated environment, manufacturers of sterile drug products face the challenge of ensuring that their cleanrooms and processes remain compliant with ever-evolving regulatory requirements.
One of the most significant of these regulations is Annex 1 of the Good Manufacturing Practice (GMP). These
guidelines outline the principles of aseptic processing and detail the design, operation and maintenance requirements for cleanroom facilities. This includes the use of appropriate equipment and materials, as well as the proper training and gowning of the personnel handling them.
Manufacturers must follow specific guidelines, which includes the implementation of a Contamination Control Strategy (CCS), to prevent contamination. This formally documented action plan examines and identifies the critical control points (CCPs) across a production line where contamination may occur and establishes a strategy to eliminate the risk at each of these CCPs through the use of appropriate equipment, PPE and operator behaviours.
Under Annex 1, this strategy should be informed by a living document that is subject to regular review to ensure a high degree of contamination control across all cleanroom processes. It should evaluate various aspects of contamination control, such as environment and process design, facility equipment, personnel training and gowning procedures, cleaning and validation procedures, environmental monitoring and prevention testing through process simulations.
Additionally, it is crucial for manufacturers to ensure that all cleanroom team members are fully trained and up to speed with the requirements of the CCS in order to effectively carry them out and ensure they meet Annex 1 requirements.

www. pharmafocusamerica.com 67
MANUFACTURING
Ensuring containment across cleanroom designs
To preserve sterile integrity and adhere to Annex 1 regulations, it is crucial for manufacturers to implement closed systems throughout their process.
Manufacturers must design their cleanroom systems to maintain sterile integrity and minimise manual interventions across aseptic manufacturing processes. Part of this includes performing comprehensive risk assessments and implementing new methods for monitoring integrity.
Traditionally, ensuring this integrity requires manufacturers to incorporate open and closed restricted access barrier systems (RABS) or isolator technologies across their cleanroom spaces. Acting as sealed cabinets, these technologies allow for interaction with drug transfer processes while keeping a physical barrier between operators and their production environments.
While isolators and RABs are highly effective at ensuring sterility there are additional factors for manufacturers to consider. These include the cost and time it takes to install, maintain, clean, and validate them after each use, which can result in production downtime and reduced productivity.
To ensure proper operation of RABS and isolator systems in cleanrooms, manufacturers must adhere to the following procedures:
• Use of properly designed equipment

• Provide management oversight of all processes
• Use a high-quality system that adheres to ISO 5 classifications in critical zones, which is the highest level of containment (equivalent
68 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
to EU GMP grade A/B or US Fed. Standard 209 class 100)
• Adhere to proper gowning practices
• Allow only fully trained operators to use the RABS
• Perform high levels of disinfection
• Follow SOPs and document any cases of human intervention.
Containment technology evolving to keep up
However, recent advancements in containment technology are acknowledged by Annex 1 regulations as enabling stringent sterile integrity while eliminating the need for the strictest cleanroom standard in many production processes or the inclusion of RABs or isolators.
One of these solutions increasingly used across cleanrooms is the aseptic split butterfly valve (SBV) - a stainless steel component that consists of two components: an "active" half that connects to production line equipment and a "passive" half that attaches to the container or primary packaging.
When connected, these two halves form a single plate that allows the drug product to flow into the container from the production line, maintaining a closed environment. SBVs offer a range of key benefits for manufacturers, both in terms of improving efficiencies of RABs and isolators, as well as eliminating the need for their use altogether, in some circumstances.
For example, the integration of aseptic SBVs into isolators or RABS systems and processes
has been vital in achieving high-performance containment and improving sterility assurance. Adopting this technology allows for closed handling, reducing manual intervention and resources required for cleaning and validation, while also increasing flow and yield from product transfers.
Additionally, as the latest revision to Annex 1 stipulates, drug product transfers may occur in cleanroom environments lower than a Grade B, provided that the connection device has been validated to prevent microbial contamination and has undergone a CCS review. As a result, this can streamline processes and aid in smallbatch manufacturing due to their ease of use and flexibility.
Single-use variants of aseptic components such as the SBV are also becoming more common in sterile processing. Offering the same level of sterility as its stainless steel counterpart, these innovations streamline
www. pharmafocusamerica.com 69
MANUFACTURING
Innovations such as smart factory technology can help factories control cleanrooms and operations more efficiently and precisely
drug transfer processes further by removing the requirement for lengthy cleaning and validation procedures after use.
Similarly, chargebags, which are single use alternatives to reusable bottles, are increasingly being used across cleanroom facilities for the storage and transport of drug product materials. These bags feature a valve that connects to the oppsing half of an SBV, allowing for the transfer of the drug product without exposure to the outside environment. Like the single-use SBV, these chargebags are intended for disposal after use, ensuring the continued sterility and integrity of the drug product material, while minimising cleaning and validation requirements.
Future-proofing containment strategies for sterile drug product manufacture.

As the industry evolves, manufacturers of sterile drug products must stay informed not only of current regulatory requirements but also the latest advancements in technology to ensure that their manufacturing processes are prepared for the future.
While technologies such as RABs and isolators will continue to be used in cleanrooms for many years, they have certain limitations. Advances in drug product transfer equipment, such as SBVs, mean that in a growing number of situations they can eliminate the need for RABs and isolators while reducing the stringency of the surrounding cleanroom environment.
As we look ahead to the cleanrooms of the future, manufacturers will be required to explore the latest innovations to ensure the longevity of their processing equipment.
Innovations such as smart factory technology can play a critical role in this process, providing real-time monitoring and control of cleanroom environments and allowing for more efficient and precise operations.
These technologies can also provide manufacturers with predictive maintenance, reducing downtime and ensuring equipment continues to operate at optimal levels.
By incorporating these technologies, drug manufacturers are better equipped to futureproof their cleanrooms and ensure their ability to meet the ever-increasing demands of the industry by producing safe and effective medications for patients.
Ben Wylie is the Senior Product Manager at ChargePoint. He has joined ChargePoint in 2005 and is responsible for the day-to-day and strategic management of the product portfolio, including the development of the ChargePoint single use solutions. Ben has 14 years of experience in Pharma with a focus on marketing and product management of powder processing and containment handling
70 PHARMA FOCUS AMERICA ISSUE 01 - 2023
AUTHOR BIO
MANUFACTURING
Advanced Model Predictive Control System of Continuous Biopharmaceutical Manufacturing Process
The progressive acceleration of continuous bioprocessing method has become the focal point of advanced research and study for the biotech companies in recent years. Implementation of this method is a tedious and lengthy process but has been proven beneficial down the line. Advancement of this methodology is initiated by the U. S. Food and Drug Administration (FDA) through introduction of Quality by Design (QbD) approach. In parallel, sophisticated bioreactors are now available in the market with various applications to support the continuous upstream process. In motion, monoclonal antibodies (mAb’s) are produced by studying the behavior of CHO cell culture, affected by different media and controlled variables. However, the control of continuous biomanufacturing process is still a challenging task because of different level of complexities.
In this work, an advanced model predictive control (MPC) system has been developed for continuous biomanufacturing process. The control relevant process models have been developed using experimental data. A PID based feedback control system has been also developed. The performance of MPC based control system has been compared with PID based control system.
Ravendra Singh
C-SOPS, Department of Chemical and Biochemical Engineering, Rutgers, The State University of New Jersey
Currently, the bio pharmaceutical industry is going under a paradigm shift from batch to continuous manufacturing (CM). One of the main advantages of CM is that it provides a suitable manufacturing platform to enable
real time monitoring and control of critical process parameters (CPP’s) and critical quality attributes (CQA’s). In the last decade, the PAT tools are studied in detail for pharmaceutical manufacturing. Different types of chromatography and spectroscopy
www. pharmafocusamerica.com 71
MANUFACTURING
technologies are used to monitor the CQA’s of biopharmaceuticals. However, much less attention has been paid to the real time control of CPP’s and CQA’s using the feedback model predictive control mechanism.
The objective of this work is to demonstrate the design and development of a model predictive control (MPC) strategy for continuous biomanufacturing processes.
Design and evaluation of the control system
This study is focused on design and evaluation of advanced control system for upstream biomanufacturing process. The manufacturing process has been previously described. For
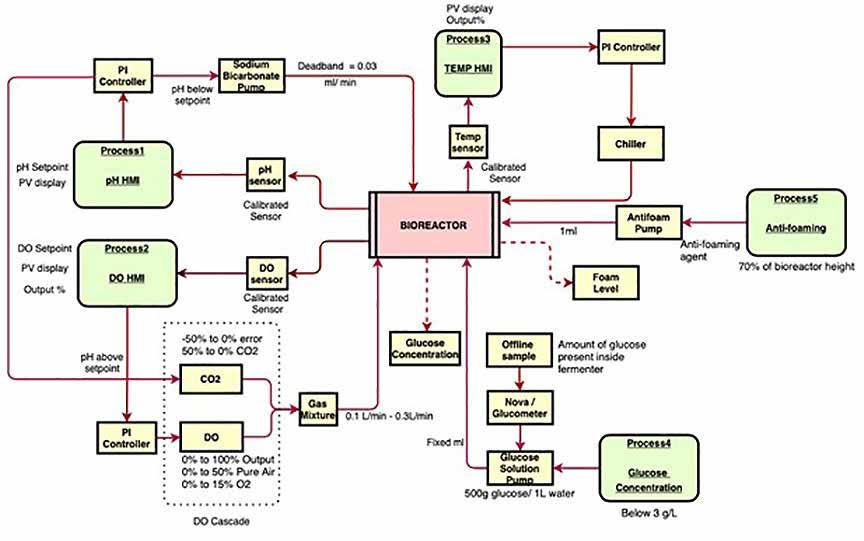
control of pH, dissolved oxygen (DO) and temperature, the bioreactor uses inbuilt Proportional-Integral (PI) controller. In addition to that, a supervisory control system has been developed for control of any additional parameters required for the process. The supervisory control system provides the setpoint of the local level control system and thereby linking the CQA’s with operating parameters.
The control architecture of the bioreactor is shown in Figure 1. In the first layer of the control system, the pH is monitored using an inbuilt electrode sensor and controlled utilizing a PI controller. The actuator, in this case, is the speed of the pump that feeds base
72 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
Figure 1: Conventional control strategy of bioreactor
materials. For this loop, the controller acts according to two different conditions i.e., whether the pH is below the setpoint or pH above the setpoint. When the measured pH is below the setpoint, the base (e.g., sodium bicarbonate) is added into the system via inbuilt pump. On the other end, when the measured process pH value is above the setpoint, CO2 gas is added to the system via gas chamber. The next control variable dissolved oxygen (DO) is monitored using an inbuilt ‘polarographic sensor’ and is controlled using the PI-based controller by manipulating the gas flow rate. In DO cascade, air and O2 gas are added into the system via the gas chamber. The amount of these two gases added into the system depend on the DO controller output percentage (%).
The total gas mixture of all the 3 gases (air, O2, CO2) combined should be 100%. Once the gas mixture is occupied in the gas chamber, gases are then entered into the system via an inbuilt multi flowmeter. DO and pH control loops are highly interactive. The temperature is monitored using the inbuilt platinum resistance temperature detector (RTD) sensor and controlled through a PI controller by manipulating the temperature of fluid flowing through the jacket of the bioreactor. The closedloop model has been developed and apply to evaluate the performance of these control loops. The advanced model predictive control (MPC) system has been developed and implemented into the model for performance evaluation.
Glucose concentration and foam level are conventionally controlled manually. Offline sample from the bioreactor was analyzed using nova analyzer/ glucometer (glucose oxidase method) to find out the concentration of glucose. If the glucose concentration was less than the desired setpoint, mass balance was carried out to find out the amount of glucose solution needed for the total volume at that time. Simultaneously, fixed volume of glucose solution would be added to the system via inbuilt pump at a fix flowrate. For foam level control, in person observation is currently needed to constantly keep a track on the level which is resource intensive. Whenever the foam level increases above 70% of the bioreactor’s height, immediately 1ml of antifoaming agent is added to the system via inbuilt pump. Pictorial representation of this

www. pharmafocusamerica.com 73
MANUFACTURING
strategy is reflected in Figure 1. A novel control strategy has been developed and evaluated for controlling of glucose concentration and foal level as discussed in next section.
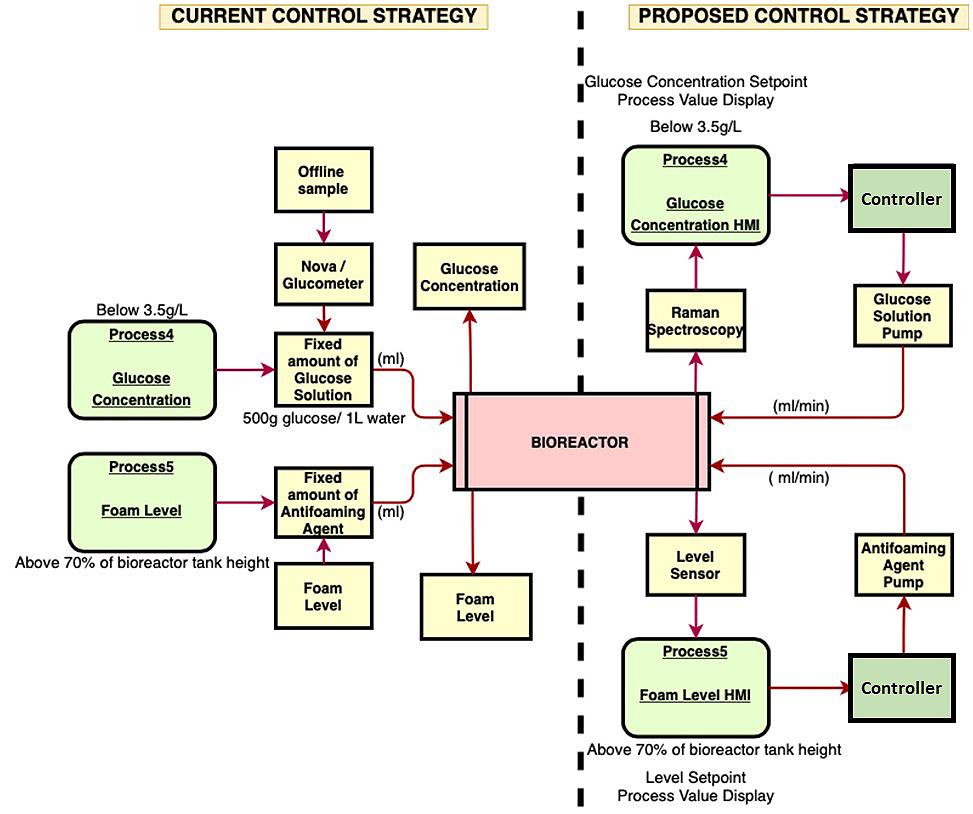
Proposed strategy for Glucose Concentration and Foam Level control
Control strategy for glucose concentration and foam level has been proposed in this work to
convert them into feedback control loops for the aim of achieving the complete automated upstream bioprocess. For glucose, on-line monitoring of glucose concentration can be achieved using Raman spectroscopy. Once the process value of glucose concentration is generated from Raman spectroscopy, the data can be transferred to the HMI where the error would be calculated in reference to the desired setpoint. If the glucose concentration
74 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
Figure 2: Current vs Proposed control strategy for glucose concentration and foam level
is less than the given setpoint, controller will automatically turn ON the pump to release the required amount of glucose solution into the system by manipulating the pump flowrate. The actuator in this case is the flowrate of the pump. The foam level control will follow the same steps. The only difference here would be that the level indicator probe will be inserted into the bioreactor via head plate port for in-line monitoring of the level. In Figure 2, the pictorial representation of the current versus proposed control strategy of glucose concentration and foam level is displayed.
Integrated closed loop flowsheet model of bioreactor
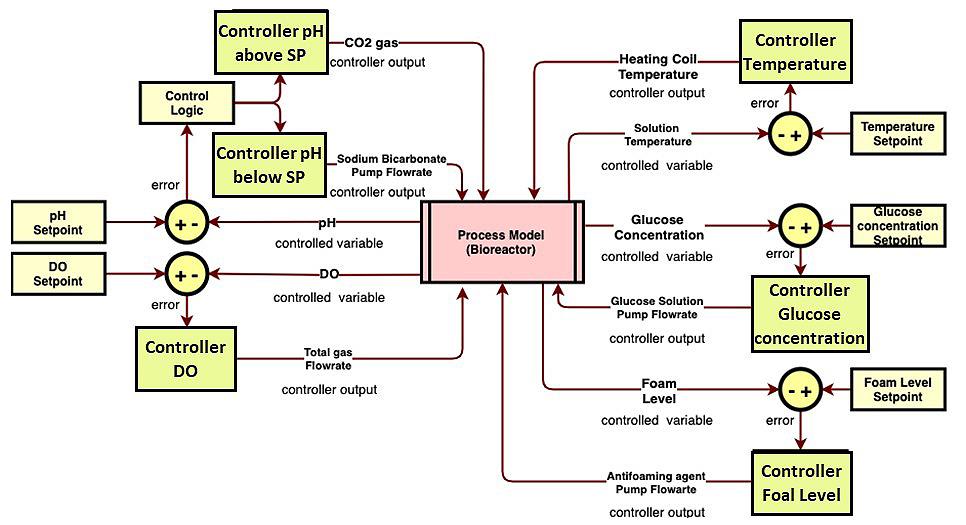
The integrated closed loop flowsheet model of bioreactor has been developed as shown in Figure 3. The pH feedback control loop has
been developed using two process models. One process model has been used when the CO2 is the input (pH is above the setpoint) and other process model has been used when the sodium bicarbonate is the input (pH is below the setpoint). For each of these models, one controller has been added. A control logic has been developed using math function in Simulink (Mathworks) which states that when the error is positive, the sodium bicarbonate pump will be active for manipulating the flowrate. On the other hand, when the error is negative, the flow rate of CO2 gas flowing through the gas chamber will be manipulated. Both these models have been added using an addition block and further divided by two to get the actual value of the controlled pH which is further compared by the setpoint value to generate the error. The DO feedback control
www. pharmafocusamerica.com 75
MANUFACTURING
Figure 3: Integrated closed loop flowsheet model of bioreactor
loop is comparatively more complicated than the pH loop. Currently the closed loop has been designed in a way where the input to the process model is the total gas flowrate which include O2 and air gas and the output is the controlled DO values. The temperature feedback control loop follows the same design as shown in figure 3. The feedback control loop design for glucose concentration and foam level has been also shown in Figure 3. Both, PID and MPC controller has been implemented in the model to evaluate its performance.
Implementation of the control system via distributed control system (DCS)
For implementation of the control system, the pilot-plant need to be integrated with the distributed control system. The integration
strategy of the bioreactor with distributed control system (DCS) is shown in Figure 4. For this integration, there is a need for multi-layer communication between different software and hardware. In Figure 4, the integration of DeltaV (Emerson), a DCS platform with the bioprocess is shown. The first step is the communication of DeltaV PC with an OLE process control (OPC) Kepware server 1 via OPC data access (DA) connection. Kepware OPC is a connectivity platform enabling communication between two systems. DA connection provides data from different data sources to the server client. Kepware server 1 is connected with another Kepware server 2 via OPC Unified Architecture (UA) connection. OPC UA is a standard that ensures open connectivity, security, and reliability of automated systems.
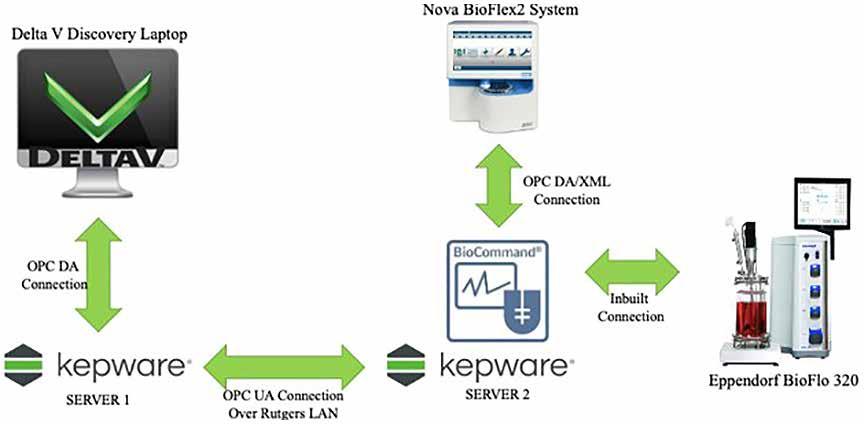
76 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
Figure 4: A systematic framework for the integration of bioreactor with the distributed control system (DCS)
OPC server 2 essentially communicates with bioreactor (Eppendorf) command PC which is a communication platform to operate the bioreactor. Eppendorf itself communicates with the Nova BioFlex2 Embedded PC via DA/ Extensible Markup Language (XML) connection. The focus of developing this integration strategy is to implement the
control strategy of the bioreactor in DCS to control the CPP’s and CQA’s via this multilayer communication protocol
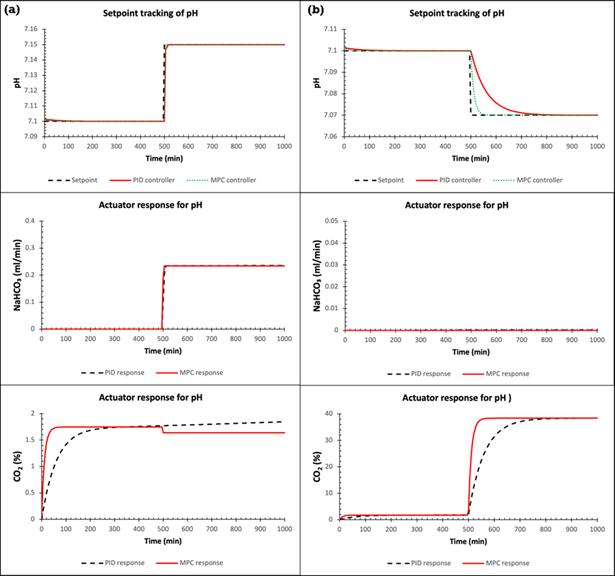
Results and Discussions
The performance of the control system has been evaluated for set point tracking and disturbance rejection. The pH control loop
www. pharmafocusamerica.com 77
MANUFACTURING
has been considered here for demonstration purposes. In the Figure 5, the performance of the PID and MPC controller is compared where in one case (a), the pH is changed from 7.10 to 7.15 pH. In this case, the PID controller takes approximately 35 minutes to reach to the new setpoint whereas the MPC controller takes 15 minutes to reach to the new assigned setpoint. The flowrate of the pump for both controllers remains
steady at 0.235 ml/min after reaching the setpoint. The value of flowrate can vary depending on the step change provided to the system. In the other case (b), the pH is changed from 7.10 to 7.07. In this case, the CO2 gas needs to be added to the system to decrease the value of pH. The Figure 4 shows that the MPC completes the step change action in 5.5 times lesser time in compared to PID controller. As expected, the sodium bicarbonate pump remains off throughout the operation. The response of CO2 gas (actuator) for both controllers is also shown in the figure. Similarly, the performance of other control loops has been evaluated.
Conclusions
Ravendra Singh is faculty at C-SOPS, Department of Chemical and Biochemical Engineering, Rutgers University, NJ, USA. He is the recipient of prestigious EFCE Excellence Award from European Federation of Chemical Engineering. His research focus is continuous manufacturing of drug substance and product. He is PI/Co-PI of several projects funded by FDA, NSF, and companies. He has published more than 75 papers, edited one pharmaceutical systems engineering book published by Elsevier, written more than 12 book chapters, and presented at over 122 conferences. He is actively serving as a Journal editorial board member, and conference session chair.

An advanced model predictive control (MPC) system has been developed for bioreactor used in continuous manufacturing. A validated integrated closed-loop flowsheet model has been developed as well. The performance of the PID and MPC for setpoint tracking and disturbance rejection has been compared and it has been observed that the MPC perform better. A systematic framework to implement the control system in the biomanufacturing process has been proposed.
Acknowledgements
This work is supported by the U. S. Food and Drug Administration (FDA) through FDA-CBER Award Number1R01FD006588.
78 PHARMA FOCUS AMERICA ISSUE 01 - 2023
AUTHOR BIO
MANUFACTURING
Continuous Vs. Batch Manufacturing
The pharmaceutical manufacturing industry is different from other industries in that it is concerned with human health and follows a highly sensitive and critical operating scenario, aiming to produce pharmaceutical drugs for public treatment. The pharmaceutical manufacturing industry has until now, followed a batch processing scenario to ensure that each batch was produced efficiently and effectively, conforming to its pre-planned specifications and in compliance with cGMP and regulatory requirements.
A particular quantity of a medication or other substance that is produced in accordance with a single production order and is produced during the same manufacturing cycle is referred to as "batch processing" in regulations like the FDA's Code of Federal Regulations (21 CFR) 210.31. To put it another way, it entails the step-by-step assembling of various product components to produce the finished manufactured product.
Hassan Mostafa Mohamed Chairman & Chief Executive Officer at ReyadaPro
Introduction
In batch processing, a subsequent batch can be processed only after the current batch has been completed. Continuous pharmaceutical manufacturing [CM] has become increasingly popular in recent years, but the transition from batch to continuous manufacturing is still under evaluation.
Continuous manufacturing [CM] is a production scenario that uses the continuous
supply of raw materials directly within the manufacturing process at the same facility without stopping and/or shutting down until completion.
In this article, we shed light on a new recent scenario for producing pharmaceutical products and the one currently in place, discuss the challenges and pros and cons of each, and make comments on them.
www. pharmafocusamerica.com 79
MANUFACTURING
Continuous Manufacturing vs. Batch Processing
Pharma manufacturing industries are complicated and depend on several factors, such as capital expenditures ("CAPEX") that include infrastructure, supplied resources, buildings, equipment, machines, etc., in addition to giving a great deal of attention to operation expenditures ("OPEX") to assure the effect of planning, doing the proper feasibility studies, and analyzing the relevant market trends, including competent personnel contributions as decision makers to ensure successful production operations.
Pharma manufacturing needs to include consumer demands, industry competitions, manufacturing legal requirements, and the correct implementation of a quality management system (QMS) to ensure the smooth running of manufacturing processes

and relevant integrated processes, aiming to get the product as per the approved specifications, fit for its intended use, and attaining the best possible return on investment (ROI) for the sake of the stakeholders.
As per recent market trends, the pharmaceutical industry loses about $50 billion due to the shortcomings of batch processing as a result of time constraints, delivery issues, damage, or the cost of a review.
Senior management should pay close attention to the analysis of the factors and attributes controlling the manufacturing processes before deciding which manufacturing method is reliable and appropriate for their manufacturing methodology, taking into account applying and adhering to all relevant pharma legislation and regulation and coming in at the bottom of the list.
80 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
Continuous manufacturing and batch processing are two sides of the same coin. Both are used in different manufacturing industries, "especially pharmaceutical manufacturing," provided that they consider the relevant regulations and requirements aiming to produce pharmaceutical drugs as per the approved specifications, in compliance with cGMP regulations, and fit for their intended use.
Manufacturers have been producing pharmaceutical products using batch processing for many years. A batch is defined in the FDA’s Code of Federal Regulations (21 CFR) 210.3 as a certain amount of a drug or other material that is intended to have a uniform character and quality within defined limits and is manufactured according to a single production order during the same manufacturing cycle.
Multiple components of a drug are mixed in a single step during batch processing. Each
stage of the creative process requires a break from batch processing. It takes multiple stages [in terms of time, money, and labour] as the materials go from step to step to process the current batch before the next batch can be processed.
The European Medicines Agency's EMA ICH Q7 defines a batch in batch processing as a homogeneous material that falls within specific parameters. According to EMA ICH Q7, a batch can correspond to a specific percentage of the production in the event of continuous manufacturing. The batch size can be defined both as a fixed quantity and as a fixed time interval.
In continuous manufacturing, where a batch can be based on a defined amount of product or raw material, a fixed time period, or a timeframe in production, the FDA has declared that the batch and batch definitions from 21 CFR 210.3 apply.
www. pharmafocusamerica.com 81
MANUFACTURING
Due to factors like COVID-19 PANDEMIC, which highlighted the vulnerability of global supply chain networks, including those supporting the pharmaceutical industry, to large external shocks, in addition to Brexit and a push for sustainability, some manufacturers have transitioned to continuous manufacturing.
Early in the crisis, labour and shipping problems forced nations to scramble to maintain access to life-saving medications, raising concerns about the pharmaceutical industry's long-term viability as climate change and mounting geopolitical tensions weigh heavily on the world.
The phrases "continuous processing," "continuous manufacturing," and similar expressions are employed. However, they cannot be used interchangeably because they have different meanings. The phrase "continuous production" refers to a production schedule that operates constantly throughout the whole week. A single-unit operation is referred to as "continuous processing" if raw materials are continually loaded, processed, and discharged without pause.
Batch manufacturing (BM), the primary method of production, uses separate unit operations with offline quality testing and storage between each stage. It is characterized by the sequential management of the arrangement of tanks for each process. It is used in the pharmaceutical and fine chemical industries.
While manufacturers are willing to maximize profits, their main reason for using continuous
manufacturing is its potential to reduce manufacturing costs, making them more competitive in the market and increasing their ROI.
A comparison of the net present values of investing in CM versus batch manufacturing in a research study funded by the US Food and Drug Administration (FDA) provides the most comprehensive analysis to date on this issue. The analysis demonstrates that the economics of investing in CM versus batch facilities is not only a winning investment strategy but also one that can reduce disruptions in the flow of pharmaceutical products from global risk events.
Continuous manufacturing is a recent and growing model used in the pharmaceutical manufacturing industry. It has recently gone from being a trendy idea to a reality. Given that the number of continuous manufacturing facilities under review has expanded in the last five years, see the table below. Continuous manufacturing (CM) is the use of a continuous flow of raw materials via continuous manufacturing processes using a single unit operation to produce huge quantities of products without stopping. It is distinct from batch processing, which employs starting materials in a sequential process with breaks in between batches. CM processes, which are used for facilities producing oral solid dosage (OSD) pharmaceutical products, will replace the current batch processing technology that is more labor-intensive, less efficient, and more likely to result in product defects.
82 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
Continuous manufacturing (CM) is a process that combines a number of unit operations together. It is continuously processing raw materials to create the finished product via a series of procedures that maximize drug production to maintain a steady stream of medications.
Within continuous manufacturing [CM], it is important to be able to systematically understand what is happening in your process, find out what is happening at your continuous manufacturing process, and be ready for an explanation at the time of an authority inspection.
To satisfy regulatory bodies concerning continuous manufacturing, especially at an elevated level of traceability, companies need to establish smart factories for continuous manufacturing using AI, industry 4, pharma 4, and machine learning (ML) with data from IIoT sensors, planning to satisfy customer needs for their standard and customized products that enable companies to improve their bottom line by becoming more productive, competitive, and profitable. Continuous manufacturing will play a significant role in the years to come. Despite continuous manufacturing's slow acceptance, the FDA is in support of a wider adoption of these technologies.
Manufacturing processes and process analytical technologies are the two main components of continuous manufacturing that require control (PAT). What aspects of the manufacturing processes are essential, and what particular difficulties arise in the ongoing
production of medicinal substances? Important process parameters (CPPs), important material attributes (CMAs), and important quality parameters (CQAs) are monitored and regulated by them. By eliminating work-up unit operations, it streamlines manufacturing processes. It is usually housed in a single building and has smaller hardware.
Small molecules used in pharmaceuticals are subject to continuous manufacturing (CM), which is also applicable to personnel medications. Maintaining quality throughout an operational line in continuous manufacturing enables rapid response to alerts or recommendations. It makes sure that the control, monitoring, and output are all consistently within specs.
For companies to be transition from batch processing to continuous manufacturing, they need to have extensive manufacturing knowledge and equip their facilities with competent production, engineering, maintenance, financial, quality, and safety staff who will play crucial roles in continuous manufacturing operations. They need to keep an eye on the method of linking the used technology, software automation, and smart automated equipment during the implementation duration. They need to focus on the optimization of the production process by saving the required, proper, efficient, and sufficient resources to create, store, track, and check asset data during continuous manufacturing operations. Continuous manufacturing makes effective use of the
www. pharmafocusamerica.com 83
MANUFACTURING
resources within the manufacturing facility, such as machines, energy consumption, and infrastructures such as utilities (such as HVAC systems and water treatment systems), which are major utilities.
In continuous manufacturing (CM), you need to allocate the proper starting materials all the time to meet the quality or potency requirements, always considering risk management assessment, doing the necessary validation, and controlling product quality attributes throughout all the operation phases. You need to adapt and use Effective,
accurate, precise, and efficient planning for an alternate route is needed in case there is equipment downtime and/or failure, which is considered a primary route need during implementation to avoid loss and failure of manufacturing continuous operation and to keep manufactured product specifications within the acceptance criteria.
In continuous manufacturing (CM), employees are less involved in tracking procedures and guaranteeing quality, so you need to have the skilled and qualified personnel required during production operations, which
84 PHARMA FOCUS AMERICA ISSUE 01 - 2023
In Batch Processing Flow
MANUFACTURING
Continuous Manufacturing Flow
costs more in personnel fees. Use cutting-edge AI-based solutions that enable facilities to use continuous manufacturing while still customdesigning the product.
Batch processing, which enables product tracing to the original batch, is considered a valuable tool in product tracing and tracking for investigation and the recall process, if required. In continuous manufacturing, to track and trace items, you need to use batch numbers to track items and, if required, recall them by creating a paper trail for them.
Challenges of Continuous manufacturing:
• Use smart regulating gauges and/or devices, which enable businesses to track batches more effectively, recall fewer goods, and reduce waste, in addition to employing multiple criteria to guarantee that product definition.
• Required approval for each lot for each regulatory affairs body one by one, which raises the cost of the process and delays the release time for selling purposes.
• Currently, manufacturers tend to follow and use the current batch processing used in the manufacturing of pharmaceutical drugs to avoid installing new equipment with PAT and automation software, which is expensive for the majority of them.

• Although there is a significant cost reduction in continuous manufacturing trials, they prefer to avoid continuous manufacturing for high demand generic products and
medications in order to keep their profit at an acceptable level.
• By outweighing the initial excessive cost of CM implementation and the cost reductions. Only, companies can achieve long-term profits through continuous manufacturing.
Examples of Continuous Manufacturing
There are many industries that use continuous manufacturing to reduce costs and get more concise finished products, such as automotive, metals and mining, biotechnology, and pharmaceuticals.
Pros of continuous manufacturing:
• Constant production efficiency improvements saving time by eliminating the need for each batch processing to start the machine. Save plant energy by avoiding shutting down and restarting machines repeatedly.
• Manufacturing processes are simplified by eliminating preparation and arrangement steps from batch to batch, resulting in faster
www. pharmafocusamerica.com 85
MANUFACTURING
and more efficient manufacturing processes. Getting reliable quality and consistency product with the same stable attributes during the operating duration, assuring constant production processes and the same product specifications. Reduce labour costs by eliminating the time and personnel required to perform line start-up and between batches, as well as their interaction among processes within the same batch, resulting in lower overhead costs lead to focusing on increasing process optimization and automation. Eliminating built-in production gaps and reducing manufacturing timelines Use all their production lines' capacity. Integrate quality testing. Respond to changes in demand more quickly. Producers can respond to altering markets more rapidly with a continuous line.
• Used in producing generics and biosimilars, their lower operational costs and lower capital expenditures lead to elevated revenue for the company.
Cons of Continuous Manufacturing
• Required a higher initial investment compared to batch processing. Companies must meet product specifications over a long period of time; they require high-capacity machines that are also durable and dependable for continuous manufacturing. Need to carefully consider and plan preventive maintenance is used to keep machines from breaking down or malfunctioning. Require a high investment in
setting the machine properly for continuous manufacturing processes. Reduce the possibility of a customer's modification request after production has begun. Shutdowns and reconfiguration cost a lot of money, resulting in an expensive operation.
Pros of Batch Processing
• Initially lower costs, a simpler process, and a specific and/or certain type of product with a dedicated product formula Facilitate responding to market demands; enable good mixing for batch starting materials as all starting materials are mixed in a specific tank following the product formula; enable regulating, managing, and monitoring the production processing steps, which satisfy all quality requirements; use simpler, lowercost equipment, suitable for small companies and/or investors; allow manufacturers to produce different batches of new products, ideal for seasonal products and limited production runs;
Cons of Batch processing
• Processing SDF typically takes longer due to the presence of several steps such as grinding, mixing, seizing, sieving, compression, coating, and storing, as well as stopping at different intervals as per processing instructions, resulting in low utilization rates and difficult process scheduling before the next batch enters the processing phase. Request testing between each stage in the lab.
86 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
• When quality has been established, most of the ongoing work is stored before materials are passed on to the following phase, consuming storage space in the production quarantine area and/or warehouse quarantine location until further disposition and decision in the case of non-conforming specifications. This increases the possibility of making mistakes, errors, and mix-ups due to repetitive personnel interaction and the long time between possessing phases and getting the finished product. In order to enhance product quality, reduce product faults, and ease shortages, FDA officials in the United States have been urging firms to convert from batch production to continuous manufacturing for years. According to the findings of a recent FDA audit, applications relying on continuous manufacturing methods received approval more quickly and generated more income.
The International Council for Harmonization (ICH) has adopted its guidelines on continuous manufacturing (CM), embracing more modern modes of manufacturing. In the last version of guidance, ICH acceded to the industry’s request to clarify the state of control and process dynamics.
ICH announced the final version of the Q13 guidance and releases on November 16, 2022. To transition from batch processing to continuous manufacturing, manufacturers must first understand the differences between the two and the benefits and drawbacks of each. They must also consider the following
factors before moving forward with continuous manufacturing:
You need to define the development consideration with a concentration on process description. Control strategy includes state of control, starting materials, equipment, uniform quality and character of product, product collection or rejection, process monitoring and sampling, risk assessment, failure modes, scale-up, and specifications. When the process is used to make clinical, bioequivalence, or registration stability, the terms comparability and bridging are used. Process Validation and Verification and Lifecycle Management Stability considerations include representative stability batching, stability consideration at scale-up, and site changes or technology transfer in addition to considering the location of information in a regulatory submission.
High and thorough coordination is required for quality and cGMP considerations, including batch release, start-up and shutdown
www. pharmafocusamerica.com 87
MANUFACTURING
Continuous manufacturing is a new idea in the pharmaceutical manufacturing business that has gone from being trendy to practical
procedures, state of control (product collection and in-process sampling), process validation and continuous process validation, materials traceability in continuous train, stability of starting materials and in-process materials, detection and treatment of non-conformity, personnel procedures and training, materials carry-over, diversion and yield calculation, and production floor product monitoring. Starting materials variability, cleaning validation, equipment breakdown, and failure.
Relevant regulation supporting continuous manufacturing current situation:
ICH Guidelines: International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use On November 16, 2022, the ICH harmonized guideline for continuous manufacturing of drug substances and drug products Q13 final version was adopted, with the goal of
• Harmonization: Capture key technical and regulatory considerations that promote harmonization, including certain cGMPs specific to continuous manufacturing.
• Manufacturing flexibility for new and existing products: Allow drug manufacturers to adapt flexible approaches to develop, implement, or integrate continuous manufacturing for the drug manufacturing of small molecules and therapeutic proteins for new and existing products.
• Regulatory Expectation: Provide guidance to industry and regulatory
agencies regarding regulatory expectations on the development, implementation, and assessment of continuous manufacturing technologies.
US FDA Recommendations: The FDA Guidance for Industry PAT-A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance clearly notes that the adoption of a scientific risk-based approach to process design may result in the introduction of continuous processing as one of the results. Control procedures that incorporate real-time quality evaluation that is at least equivalent to, or better than, laboratory-based testing on collected samples are made possible by process understanding, control strategies, and on-line, in-line, or at-line monitoring of essential quality attributes.
FDA Guidance on Process Validation and Continuous Verification: Continuous Verification, processes that involve process validation are in line with a product lifecycle paradigm. At every level of the lifespan of the manufacturing process, the advice promotes the application of contemporary pharmaceutical development principles, quality risk management, and quality systems. The lifecycle idea ties together the development of products and processes, the certification of commercial manufacturing processes, and the upkeep of processes in a controlled state during ordinary commercial production. This advice uses strong science to promote process innovation and development, including continuous manufacturing.
88 PHARMA FOCUS AMERICA ISSUE 01 - 2023
MANUFACTURING
ASTM Standard: ASTM E2537 Validation: Continuous Quality Verification (CQV) is an approach to process validation where the performance of a manufacturing process (or that of a supporting utility system) is continuously monitored, assessed, and adjusted, as necessary. This approach is described in the ASTM Standard: ASTM E2537 Validation: Standard Guide for the Application of Continuous Quality Verification to Pharmaceutical and Biopharmaceutical Products. An approach based on science is used to confirm that a process can create products that consistently meet certain critical quality requirements (CQAs). With real-time quality assurance (which CQV will offer), manufacturing is continuously assessed to guarantee the appropriate quality attributes. Data from production batches can support validation with each manufacturing batch by reflecting the overall system design idea and serving to validate the process.

EU Guidelines: In the European Union, the ICH Guidelines previously mentioned are applicable. The Guidelines for Process Validation, which introduce the idea of continuous process verification, the Guidelines on NIR because it is frequently used as a Process Analytical Technology (PAT) tool for process monitoring and/or control, and the Guidelines on Real-Time Release Testing are three EU Guidelines that may be particularly pertinent to continuous manufacturing. Realtime release testing is frequently combined with continuous manufacturing, although
it is not necessary (RTRT). A PAT team was also established by the European Medicines Agency in 2003 to support PAT and QbD initiatives inside the EU. The teams serve as a venue for communication between the Good Manufacturing Practice/Good Distribution Practice Inspectors’ Working Group, the Quality Working Party, and the Biologics Working Party.
Conclusion: In summary, global and regional regulations, guidelines, and standards are supportive of innovative pharmaceutical development and manufacturing approaches and the consideration of continuous manufacturing operations as experience is gained. References are available at www.pharmafocusamerica.com
www. pharmafocusamerica.com 89 AUTHOR BIO
Dr. Hassan Mostafa Mohamed, Chairman & Chief Executive Officer at ReyadaPro is an entrepreneurial and growth-driven executive with more than 25 years of experience in pharmaceutical industries, including, Technical, sales & marketing, Production, Supply chain, Engineering/utilities, Quality, and regulatory issues. Mr. Hassan is an expert in driving pharmaceutical facilities to accomplish corporate goals, building and leading technical & quality aspects with market consideration for rapid growth, and efficient operational excellence.
MANUFACTURING
How AI is Transforming the Pharma Industry

In many ways, every aspect of the pharma industry is ripe for AI transformation from drug discovery and the performance of trials, to remote patient monitoring, medication adherence tools and beyond. This article considers some of the potential legal considerations when entering into licensing collaborations for the use of AI, in particular for drug discovery.
Lydia Torne Partner, Simmons & Simmons LLP
The global pharmaceutical industry is currently facing many, wide ranging, challenges, including an aging population, increased life expectancy, a rise in chronic conditions, reduced funding for treatments, reduced numbers of clinical staff, the ever increasing cost of drug development
and raw materials, and supply chain issues. Consequently, the pharmaceutical industry is increasingly looking at how a wealth of data, including compound libraries, trial data, and patient data, can be used and reused by artificial intelligence (“AI”) to alleviate these challenges and improve patient care.
90 PHARMA FOCUS AMERICA ISSUE 01 - 2023
INFORMATION TECHNOLOGY
The opportunities for AI in the pharmaceutical industry are vast: in many ways, there is no area which is not ripe for AI transformation. For the purposes of this article, we will focus on drug development using AI, though many of the issues discussed may apply to other AI applications in the pharma industry.

Drug development
Developing a new pharmaceutical product is currently estimated to cost in the region of US$944m to US$2,826bn taking approximately ten years from discovery to approval. Soaring costs reduce the number of drug products being developed (with particular hesitancy in developing drug products for rare diseases where, due to the limited patient population, recovery of development costs may be challenging). The use of “computer aided drug design” (“CADD”) to reduce these metrics by accelerating the identification and early stage discovery and assisting with the development of a new drug product is therefore of great interest.
CADD can be broadly described as the use of AI to identify and/or develop a potential lead drug candidate. For example, to review and assess a small molecule library against a defined target to identify potential “hits”, or to assist with “molecular docking” (e.g. software which predicts both the binding affinity between ligand and protein and the structure of protein–ligand complex) to develop product candidates. Another AI predicts characteristics
of the product, e.g. predicting the absorption, distribution, metabolism, excretion, and toxicity properties of the product candidate thereby allowing scientists to better understand the product’s safety and efficacy profile before it ever reaches a trial subject. Likewise, the use of AI can be hugely helpful in repurposing an existing or an abandoned drug product. For example, using an AI model to map an existing drug product onto huge patient data sets to identify whether it might be viable as a drug product for another indication; or to look into why a particularly promising drug product when tested in animals, fails once used in-human.
Once identified, AI can then be deployed to accelerate and improve the outcomes of clinical trials. Increasingly pharmaceutical companies are applying AI to patient data to identify the optimal trial subject, most likely to have the best response to the relevant product.
www. pharmafocusamerica.com 91
INFORMATION TECHNOLOGY
The use of in silico trials continues to attract attention, “testing” a drug product against data sets for theoretical clinical trial subjects. While, as yet, it is no replacement for in-human studies, it may offer an effective “screening” tool for a product to ensure that a Phase I trial is as positive as possible; it may also be used to complement a clinical trial (reducing the number of enrolled patients and improving statistical significance), and/or support clinical decisions. For example, the PreDICT project used patient data models to assess the cardio toxicity of new drugs.
Legal considerations
While many pharmaceutical companies are trying to hire talented software developers to create and/or oversee the use of complex AI, this is often not an easy task due in part to a global lack of resources, industry culture considerations, and traditionally significant remuneration in the tech industry. Other pharmaceutical companies are entering into collaboration agreements with software companies either for the development of bespoke models or to use “off the shelf” technology.
When entering into these types of collaborations there are specific legal issues which should be considered carefully and addressed. These include the software developer’s rights in and to any product its software identifies or refines, how the software developer should be remunerated and how liability for the product should be apportioned.
Co-developer and co-owner?
If AI is crucial to the discovery of a new drug product, should the software developer (or more controversially, the AI itself) constitute a co-inventor of the resulting drug product pursuant to patent laws? Even if listed as a co-inventor, should it co-own the relevant product patent?
The courts have recently begun considering the issue of AI as an inventor of a patented technology. In Thaler v. Comptroller General of Patents Trade Marks and Designs, Dr Thaler invented DABUS - an AI "creativity machine" which Dr Thaler included as the inventor on a number of patent applications. In the UK, the High Court and the Court of Appeal considered whether AI software could constitute an inventor recognising that the most advanced AI systems operate in a “black box” where their decisions and actions are outside the comprehension and control of the human software developer. The UK courts held that an "inventor" under English law must be a natural person, such that DABUS could not be an inventor. The European Patent Office ("EPO") and the US Patent and Trademark Office ("USPTO") have ruled similarly. While, the Australian Federal Court initially considered that AI, such as DABUS, was capable of being an inventor under Australian patent laws, this has since been overturned on appeal. At the time of writing the only known jurisdiction to have granted patent rights to an AI inventor is South Africa.
92 PHARMA FOCUS AMERICA ISSUE 01 - 2023
INFORMATION TECHNOLOGY
Notably, the courts are not empowered to determine whether AI should be an inventor; only whether AI can be an inventor as a matter of statutory interpretation in the relevant jurisdiction. As a result, governments have begun to intervene to question the policy considerations of whether AI should be an inventor. The UK government’s recent consultation on the impact of AI on intellectual property law suggests that respondents are split with many believing AI should not own intellectual property rights in their own name, but the AI developer should logically be a co-owner of any patent for an AI-assisted invention. In view of diverging opinion, the UK government is considering whether AI-devised inventions should be protected through a new form of IP protection.
To best protect their interests, pharmaceutical companies using AI via a licensing collaboration should ensure that the contractual terms of their licensing agreement provide for any inventions and intellectual property rights resulting from the use of the AI to be solely owned by the pharmaceutical company, including appropriate assignment provisions and obligations requiring the software developer to assist in procuring IP protection for the relevant invention and perfecting title to that IP right.
Remuneration structures
Software licences often rely on a one-off or periodic licence fee structure for a certain number of users to use the software. However,
if licensed AI is integral to the development of a drug product, which may generate billions of dollars of revenue for the pharmaceutical company, software developers may explore alternative remuneration structures.
While no-one disputes that a pharmaceutical company will need to expend extensive further resource developing and commercialising any drug product identified or refined by AI, there is an argument that the value of the AI software used to make that discovery is far greater than a limited licence fee. The interest in seeking an uplift in remuneration will be intensified if the AI system (or an element of it) has been developed or adapted specifically for that pharmaceutical company, recognising the additional expenditure the developer has incurred to develop a specific program, the potentially limited ability to re-use such specific model, and the market advantage afforded to the pharmaceutical company as a result of having exclusive access to such a bespoke software model. However, applying a usual royalty structure is likely to be considered excessive for the contribution.
Alternative remuneration might be structured by way of milestones payable if/ when the AI identifies a potential drug product, and if such drug product achieves certain stages of development and is ultimately authorised. If a royalty is considered appropriate (perhaps, for example, if the developer is providing a broad package of assistance incorporating discovery, CADD modelling, and even in silico testing) this might be structured as a small capped
www. pharmafocusamerica.com 93
INFORMATION TECHNOLOGY
royalty if sales exceed a certain threshold or addressed via commercial milestone payments.
Liability
In direct correlation with claims for additional remuneration based on the material contribution of the AI system in drug development, is the extent to which the software developer should be liable for the resulting product. If the software is truly crucial to creation, should the developer not also bear some liability?
A usual contractual tool to apportion liability is indemnification. Careful thought will need to be given to the triggers for indemnification. In particular, requiring indemnification from a software developer for issues with the drug product (e.g. defective product) is challenging given the extensive further development performed by the pharmaceutical company and is likely to be resisted. Instead, you might apportion liability slightly differently. For example, providing for indemnification
from the AI software developer in connection with issues specific to the software model (e.g. third party IP infringement or misuse of data claims or bias in the algorithm), coupled with a claw back right in respect of milestone payments made (so, if a milestone payment is made for commencing a Phase I trial using the AI generated drug product, which then subsequently materially fails, a portion of the milestone payment would be repayable). If the AI software developer is unwilling to bear any risk and liability for the resulting product this is a strong counterargument for the provision of increased remuneration.
In addition to contractual liability, thought should be given to the AI developers possible statutory liability for any resulting product and/or the software it provides, pursuant to a country’s product liability regime (with those regimes in the EU and the UK currently changing to provide for product liability for software). The EU’s proposed AI Liability Directive will also make it easier for claims to be brought arising out of harm caused by defective AI (including class actions, which are expressly referred to in the Directive).
Pharmaceutical companies should also ensure that their contractual indemnification provisions include recovery of any losses suffered as a result of the AI model being in breach of applicable AI regulations. In April 2021, the EU published its draft Regulation on AI. In its present form, the regulation applies to (among others) providers and users of AI, prohibits certain AI uses and imposes
94 PHARMA FOCUS AMERICA ISSUE 01 - 2023
INFORMATION TECHNOLOGY
Artificial Intelligence and Machine Learning can change the focus of the pharmacovigilance function by improving product quality, streamlining treatment regimens, reducing costs, and enhancing patient safety
additional obligations in respect of "high-risk AI systems" including increased record keeping, oversight, governance and reporting measures. Currently, AI aimed at drug development is neither prohibited, nor constitutes a “high-risk AI system” (unlike AI driven medical devices). However, as the use of AI in drug development increases, this may change. Likewise other uses of AI by the pharmaceutical industry (e.g. trial subject/patient monitoring) may become considered “high risk” for much the same reasons as AI driven medical devices are. The draft AI Regulation proposes that non-compliance should result in significant financial penalties of up to 2%, 4% or 6% of worldwide annual turnover depending on the nature of the infraction. The UK (and other jurisdictions) is also considering introducing its own legislation in respect of AI. The pharmaceutical industry should, therefore, continue to monitor whether the development and use of AI software in its business is likely to trigger additional regulatory obligations, not least because the use of AI in the context of healthcare and life sciences is likely to attract the attention of regulators due to the material physical harm which could potentially result from an issue with such AI. If so, obligations for ensuring compliance with (and corresponding liability for) these regulatory obligations should be contractually passed to the AI developer.
Conclusion
The above reflects a fraction of the legal considerations associated with the use of AI in
the pharmaceutical industry. Further detailed thought should be given to data protection compliance, cybersecurity and regulatory and ethical compliance issues, among many more. While there are many legal challenges to consider, with more to surely reveal themselves in the coming months and years, it should not be forgotten that AI presents a tremendous opportunity for the pharmaceutical industry to improve and accelerate its development pathway and, most importantly, to deliver a material difference to patients.
References are available at www.pharmafocusamerica.com
Lydia Torne is a Partner at international law firm Simmons & Simmons LLP. Her specialisation is in life sciences licensing transactions including, research, and development and commercialisation agreements for early stage, pre-clinical and clinical assets, and related collaboration and consortia arrangements. Lydia has a particular focus on biotechnology assets and digital health.

www. pharmafocusamerica.com 95 AUTHOR BIO
INFORMATION TECHNOLOGY
Use of Artificial Intelligence in Automation of Pharmacovigilance
Ryanka Chauhan Product Manager, Datafoundry
The pharmaceutical industry is expected to be worth $1.5 trillion by 2023, and the field of pharmacovigilance plays a crucial role in ensuring the safety of drugs. Artificial Intelligence (AI) and Machine Learning (ML) models are being applied to improve the pharmacovigilance process, including case intake using Optical Character Recognition (OCR) and Natural Language Processing (NLP), natural language generation (NLG) for narrative writing, robotic process automation (RPA) for dynamic case workflow, AI-based signal detection, and AI-based adverse event prediction. These advancements have the potential to increase efficiency, accuracy, and consistency in pharmacovigilance, as well as reduce costs and delivery timelines for pharmaceutical organizations.
Introduction
With rapid digitalization and increased research in the sector, the pharmaceutical industry has experienced a remarkable development in the last three years, opening doors for innovative routes of therapy for humankind. The pharmaceutical industry is expected to be worth $1.5 trillion by 2023, according toestimates.
According to a research published in the Clinical Pharmacology and Therapeutics Journal, AI/ML approaches are being used in multiple areas to support drug development
and regulatory submissions as well as facilitating in review and research.
Pharmacovigilance is an essential component of the drug development cycle that ensures pharmaceutical products meet the required safety profile before they are approved for market. It is vital to do post-marketing safety and efficacy studies for most products.
With organizations seeking to optimize their spend through the entire drug life cycle, experts are looking at the use of AI/ML to make the Pharmacovigilance process more effective and efficient.
96 PHARMA FOCUS AMERICA ISSUE 01 - 2023
INFORMATION TECHNOLOGY
The following are the nine areas in which AI/ ML technologies have been utilized to improve the entire pharmacovigilance process:
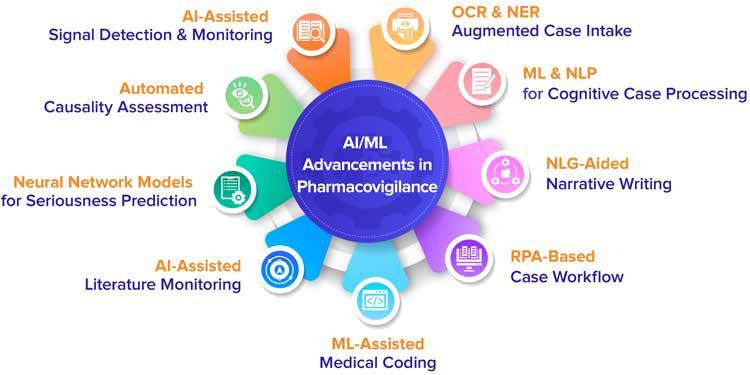
1. OCR & NER Augmented Case Intake
In pharmacovigilance, case intake refers to the process of collecting and recording information about adverse events associated with the use of a medication or medical device. Case intake is one of the most common applications of AI.
OCR & NER has the potential to extract data quickly and efficiently with the expanding volume of information and multiple data sources like Medical Literature, Social Media, Electronic Health Records, Regulatory Reporting, News Websites and Papers, Mobile Applications, etc. containing primarily unstructured data including free text narratives
and scanned PDFs, avoiding the massive amount of time required for human data entry.
OCR is a technology to recognize and extract text from images and scanned documents, such as PDFs or scanned reports. This can be particularly useful in pharmacovigilance, as many adverse event reports are submitted in paper format, and OCR can be used to digitize these reports and make the information they contain accessible to automated systems.
In addition to OCR, NER (Named Entity Recognition) is another technique used in natural language processing that can be applied to case intake to automatically identify and classify relevant entities, such as the name of the drug, the symptoms experienced by the patient, and the outcome of the event. This can help to streamline the case intake process and make it more efficient.
www. pharmafocusamerica.com 97
INFORMATION TECHNOLOGY
There are various NER techniques and models that can be used for this purpose, such as rule-based systems, statistical models, and neural network-based models.
When using OCR for case intake, the first step is to convert the scanned document or image into a digital format that the OCR software can process. Once the image is in a digital format, OCR software can analyse it and recognize the text it contains. The
2. ML & NLP for Cognitive Case Processing
Machine learning based cognitive case processing is a method of automating the analysis and triage of adverse event reports in pharmacovigilance using ML algorithms. It involves the use of ML techniques to analyse large amounts of text data, such as patient narratives or adverse event reports, to extract relevant information and classify the reports into different categories.
In cognitive case processing, an ML model is trained on a large dataset of labelled adverse event reports. The model learns to identify patterns and relationships in the data that correspond to different categories
3. NLG-Aided Narrative Writing
Narrative writing plays a key role in pharmacovigilance by providing a detailed account of an adverse event, enabling better understanding and communication of the event, and helping to identify patterns or trends that may indicate a safety issue.
software will then output the recognized text in a machine-readable format, such as a plain text file or a structured data format like XML or JSON.
The next step is to use NER to extract the named entities such as drug name, symptoms and patient details, etc. from the recognized text. This information can then be stored in a structured format for further analysis and reporting.
of reports, such as serious adverse events or non-serious events.
Once the model is trained, it can be applied to new adverse event reports to automatically classify them into the appropriate categories. This can help to streamline the case processing process and make it more efficient, as well as increasing the consistency and accuracy of the classification.
Additionally, this approach can be enhanced with the use of natural language processing (NLP) techniques to extract entities such as drug names, symptoms and patient details from unstructured narratives which can further improve the accuracy and speed of case processing.
According to an ICSR research article in their publication, Perspectives in Clinical Research, resourcing, consistency, timeliness with highquality and potentially various data sources utilised as input, and considerable variation in the templates are all challenges with narrative writing.
98 PHARMA FOCUS AMERICA ISSUE 01 - 2023
INFORMATION TECHNOLOGY
Furthermore, in situations with several follow-ups, there is a high possibility that the story might become fragmented and confusing.
Using Natural Language Generation (NLG) to automate narrative writing increases the quality and consistency of the narratives while decreasing the time required to construct and complete the narrative. The model can extract all necessary information quickly and effectively, produce the narrative, and place it in the required format or template.
Built-in audit trails and version control can ensure that each version of the narrative is logged and saved for easy access and comparison.
4. RPA-Based Case Workflow
Robotic Process Automation (RPA) is one of the emerging forms of business process automation technology based on the notion of software bots or artificial intelligence.
5. ML-Assisted Medical Coding
Medical coding is the classification of a number of similar verbatim phrases using a certified medical lexicon that is either given by the client or granted permission by the relevant licencing authorities, in order to provide a statistically quantifiable count of all related terms.
The process of matching a reported term to a dictionary entry can be aided by machine learning. The verbatim words that
According to one study published in the Applied Clinical Informatics Journal of the American Medical Informatics Association (AMIA), workflow automation, which entails finding sequences of processes that may be optimised by leveraging technology and modern computing, provides potential to solve quality, safety, and efficiency concerns.
Automation is something pharmaceutical companies are eager to implement to save costs and delivery times. Robotic processes that perform repetitive operations can greatly minimise the need for manual effort by enhancing productivity, compliance, and overall quality while also immediately boosting efficiency.
For case workflows in pharmacovigilance, bringing in rule-based automation, robotic process automation (RPA) or automation based on regular expressions could help improve compliance significantly.
have a precise match in the dictionary will be automatically coded as the programme runs validation during this process.
ML can reduce the risk of coding errors, as it is not subject to the same types of mistakes that humans can make when coding manually. By automating the coding process, it is possible to reduce the effort and cost associated with manual coding.
www. pharmafocusamerica.com 99
INFORMATION TECHNOLOGY
6. AI-Assisted Literature Monitoring
Pharmacovigilance professionals seek for indications of drug safety risks in research findings, case studies, and other publications. This process typically takes a long time and is prone to human error. AI-enabled solutions could be used to automate the process by ingesting data from an increasing number of scientific sources and automatically flagging possible drug safety risks.
Literature monitoring in pharmacovigilance could use Artificial Intelligence (AI) and Natural Language Processing (NLP) techniques to automate the process of monitoring scientific literature for new information related to the safety and efficacy of drugs and medical devices. The process typically involves the use of AI algorithms to search and extract relevant information from scientific journals, conference proceedings, and other sources. This can include identifying new studies related to a specific drug or medical device, tracking changes in the safety profile of a drug over time, and identifying emerging safety concerns.
One of the main benefits of AI-assisted literature monitoring is the ability to quickly and efficiently process large amounts of information from a wide range of sources. This can help to identify potential safety issues early on and can be used to inform the development of new safety measures or to make regulatory decisions about a drug or device.
Additionally, AI-assisted literature monitoring can be used to prioritize articles for review by human experts, focusing on the
most relevant studies and publications for a given topic, which can save time and resources for the pharmacovigilance teams.
7. Neural Network Models for Seriousness Prediction
The seriousness of adverse events is a critical component in determining reporting timelines, and it is usually handled manually by pharmacovigilance professionals. Because of the huge growth in the amount of safety reports, it has become necessary for pharma companies to utilize AI/ML enabled scalable solutions that also fulfil reporting timeline requirements.
A neural network approach can provide a precise and scalable solution for potentially enhancing the assessment of the seriousness of adverse events in spontaneous, solicited, and medical literature reports, according to the findings of a research study that was published in Drug Safety, the official journal of the International Society of Pharmacovigilance (ISoP).
Neural networks can be used to predict the seriousness of AEs reported in pharmacovigilance. These models can take a variety of inputs, such as the patient's demographic information and the details of the AE and use that information to make a prediction about the seriousness of the event.
There are different types of neural networks that can be used for this task, such as feedforward neural networks and recurrent neural networks. In some cases, a combination
100 PHARMA FOCUS AMERICA ISSUE 01 - 2023
INFORMATION TECHNOLOGY
of both can be used to improve the performance of the model. These neural networks can be trained on large amounts of historical data on AEs to learn the patterns that are associated with serious events.
Once trained, the model can then be used to predict the seriousness of new AE reports as they come in. This can help to quickly identify and prioritize serious events, which can be important for ensuring the safe and effective use of medications.
8. Automated Causality Assessment
In pharmacovigilance, causality assessment is the process of determining whether an adverse event is causally related to the use of a specific drug or medical device. Machine Learning (ML) models can be used to automate this process, making it more efficient and accurate.

The WHO UMC causality assessment and the Naranjo causality assessment are two widely accepted and utilised methods for assessing causality worldwide.
Some other specific ML models that can be used for causality assessment in pharmacovigilance include:
• Decision Trees: Decision trees are a type of ML model that can be used to classify adverse events based on a set of predefined criteria. The model is trained on a dataset of labelled adverse event reports and learns to identify patterns and relationships in the data that correspond to different causality categories.
• Random Forest: Random Forest is an ensemble of decision trees, which can be used to improve the performance and generalization of the model. It generates multiple decision trees and combines them to make a final decision.
• Neural networks: Neural networks are a type of ML model that can be used to classify adverse events based on a large number of input features. Neural networks can handle high-dimensional data and can be trained to recognize complex patterns in the data.
www. pharmafocusamerica.com 101
INFORMATION TECHNOLOGY
• Logistic Regression: Logistic Regression models the probability of a certain class or event existing such as the causality of a specific adverse event. It can take multiple features into account and can be useful for large datasets.
• SVM (Support Vector Machine): SVM is a type of supervised learning algorithm that can be used for classification and regression. It can be used for causality assessment by training the model on a labelled dataset of adverse events and then applying the model to new data to classify the events as causally related or not. The models should be trained with a high-quality, representative, and wellannotated data, and should be validated with independent datasets to ensure the performance and generalization.
9. AI-Assisted Signal Detection & Monitoring
The increasing complexity of data reporting and regulators' expectations of being more proactive in detecting adverse events is transforming how signals are gathered and managed.
Proactive signal detection approaches are considered as a part of Good Pharmacovigilance Practices (GVP). More significantly, by including signal detection into their PV monitoring, pharma organizations lower their risks and raise the likelihood of positive outcomes - even when adverse events are identified.
There are several ways that AI and ML can be used to improve signal detection in pharmacovigilance:
• Natural Language Processing (NLP): NLP algorithms can be used to extract information from unstructured text data, such as patient reports of adverse events. This can help to identify patterns and trends that may indicate a potential safety issue.
• Predictive Modelling: Machine learning algorithms can be used to predict the likelihood of a particular adverse event occurring based on a patient's characteristics and their use of a medication or vaccine. This can help to identify high-risk populations and prioritize safety monitoring.
• Data Visualization: AI and ML can be used to create visualizations of data, such as graphs and maps, to help identify trends and patterns that may indicate a safety issue.
• Automated Analysis: Machine learning algorithms can be used to automatically analyse substantial amounts of data, reducing the need for manual review, and allowing for more efficient signal detection.
Challenges with AI/ML in Pharmacovigilance
There are some challenges with using AI/ML in pharmacovigilance which include:
• Data Quality and Availability: The quality and quantity of data can affect the performance of AI/ML models. In pharmacovigilance, data may be incomplete, inconsistent, or biased, which can lead to inaccurate or unreliable results.
102 PHARMA FOCUS AMERICA ISSUE 01 - 2023
INFORMATION TECHNOLOGY
• Explainability: AI/ML models can be difficult to interpret and explain, which can be a problem in a regulated field such as pharmacovigilance where decisions may have significant consequences.
• Legal and Ethical Considerations: AI/ML models in pharmacovigilance may raise legal and ethical concerns, such as inadvertent bias and liability concerns.
• Integration with existing systems: AI/ML models may need to be integrated with existing systems and processes, which can be a challenging task.
• Validation and Regulatory Approval: AI/ ML models in pharmacovigilance may need to be validated and approved by regulatory bodies before they can be used in practice. Artificial Intelligence and Machine Learning have a lot of potential for safety and pharmacovigilance. These technologies have the ability to change the focus of the pharmacovigilance function from data collecting and reporting to assisting in improving product quality, streamlining treatment regimens, reducing costs, and enhancing patient safety.
Agile pharmaceutical organisations may be able to provide enticing alternatives to current procedures and workflows as a result of the shift to AI/ML-based pharmacovigilance solutions. The future of pharmacovigilance is in digitalization, AI analytics, and patient-centred data collection, all of which are expected to increase overall medication safety. To ensure performance and generalisation, the AI/ML
models should be trained on high-quality, representative, and well-annotated data and evaluated on separate datasets.
The advantages of implementing AI/ML are visible in the long term. It's time for the pharmaceutical business to advance by fast adapting, creating AI/ML use cases, and implementing them at scale. References are available at www.pharmafocusamerica.com
Ryanka Chauhan serves as a Product Manager at Datafoundry for DF mSafety AI, a Safety platform that leverages AI/ML for Safety Vigilance automation. She works closely with customers and business leaders across the organisation, being involved in multiple functional areas including technology, marketing and sales for delivering transformational new solutions to organizations in the life sciences and healthcare industry.

www. pharmafocusamerica.com 103
AUTHOR BIO
INFORMATION TECHNOLOGY
NEXT GENERATION SEQUENCINGGENOMICS
With advances in genomic technology, precision medicine continues to gain traction in oncology. Developing a structured approach for navigating Charlotte's web of cancerous pathways allows precision medicine to maximize the potential of targeted drugs by setting a firm foundation through clear illustration of the biological dynamics.

EXPERT TALK
Ravi Dashnamoorthy Senior Scientist, Genosco
1. It seems like genomic sequencing is getting more attention. What distinguishes genomic sequencing from conventional cancer genetic testing?

The genomic sequencing approach allows for a comprehensive analysis of human DNA for gene sequence changes, enabling the detection of cancer associated novel genetic changes in unexpected genes. Conversely, cancer genetic testing is oriented toward preselected target genes, generally chosen by family history, inheritance and susceptibility risk. Further, with less than 10% of cancers being inheritable, genetic testing may miss identifying cancers that are not inheritable, which make up the majority of all cancers. Most importantly, genomic sequencing provides a means of identifying potential drug targets and allows physicians to prescribe precision medicine and facilitate providing personalized treatment. Finally, with the completion of the human genome map and the availability of affordable sequencing technology, genomic sequencing is becoming an increasingly acceptable diagnostic tool.
2. Why do you think we are seeing an increase in the demand for genetic data in clinical development?
Cancer treatment was traditionally dominated by treatments such as chemotherapy, radiation, and surgery until the early part of this century. In the United States and around the world, these treatments continue to be the firstline option for many types of cancer. In recent years, genomic sequencing has enabled the discovery of novel targets and the development of targeted drugs that match these targets. This has resulted in improved treatment efficacy and a reduction in side effects. These newly developed targeted drugs were also formulated for oral consumption, making them very attractive options for patients who wish to reduce the frequency of hospital visits. Consequently, healthcare providers who opt to provide personalized care with tailored therapy began integrating genetic abnormalities into their treatment decision-making processes. As a result, genetic datasets are in high demand in the healthcare sector.
www. pharmafocusamerica.com 105
EXPERT TALK
3. Could you explain precision oncology's full range of applications and how they affect clinical outcomes?
The treatment outcome of patients with the same type of cancer tends to vary widely from patient to patient; this has remained a challenge that is biologically unresolved. Through precision oncology powered by genomics, new molecular subtypes have been identified, allowing for improved disease stratification, more accurate patient selection for clinical trials, and providing better personalized treatment. Because targeted therapeutics aim to inhibit the biological activity of key genetic drivers of the disease, administering these treatments results in better clinical outcomes. As a result of improved outcomes, healthcare providers are also able to grow profitably. Overall, precision oncology is becoming a crucial part of diagnostics, drug discovery, and development, enabling clinical trials and facilitating treatment decisions.
4. Is there any particular genomic development that you find fascinating at the moment?
The use of spatiotemporal resolution, single-cell RNA sequencing platforms provides a fascinating new insight into the microenvironment of cancerous tissues containing different types of cells. Targeted therapeutics, whether small molecules, biologics, or cell-based therapies, require a thorough understanding of the biological
background of the disease. This is in part due to the fact that some drugs target cancerous cells whereas others work indirectly through normal cells located in the vicinity. It is also imperative to note that cancer is intrinsically a biologically complex disease. Thus, such technologies provide precision medicine with a better basis for providing personalized treatment by resolving the complexities of these diseases. Oxford Nanopore is another exciting development in genomics, which is a pocket-sized, affordable, portable sequencing system that can be used by anyone anywhere without requiring any technical skills or training. This platform can be effectively integrated into any small healthcare facility without a significant investment in capital, and eventually could evolve into a useful tool appropriate for self-diagnosis at home.
106 PHARMA FOCUS AMERICA ISSUE 01 - 2023
EXPERT TALK
Precision medicine is becoming more popular in cancer thanks to developments in genomic technology. Without understanding the biological mind of cancer, it cannot be established
5. Could you explain the Abacus Strategy and how it is applied to genomics?
The conventional wisdom holds that genetic changes are unpredictable and random. Genetic changes can be inherited (germline, which is passed from parent to child) or acquired (somatic, which is not associated with reproduction). If the changes are consequential, germline and somatic genomic changes can cause cancer. These genetic and biological changes are dynamic and evolve with disease progression, which presents a significant challenge for precision medicine applications. In essence, these changes in genomic and biological dynamics mimic the "Charlotte's Web" effect, which enables cancer cells to evade the therapeutic effects of a drug intended to block a key biological regulator associated with malignancy by redistributing the control to a different biological factor. For this reason, predicting cancer's next genetic move is an effective method for developing smart strategies to prevent cancer progression. We therefore, developed an Abacus strategy based on primitive learning to convert RNA sequencing datasets into linear biological paths, then overlay genomic data to predict genetic changes over time, published in Biomedicines 2022, 10(11), 2720; doi. org/10.3390/biomedicines10112720. As a result of this research, a systematic method has been developed to construct biological GPS maps of cancer progression patterns. This will eventually allow us to predict
future biological states and genetic changes associated with cancer.
6. How would you rank the USA's competitiveness given the global boom in the genomics industry?
As a nation that has sequenced the entire human genome, developed and introduced several sequencing platforms, integrated artificial intelligence into genomics, incorporated genomics into clinical and translational research, and facilitated the discovery of many new drugs, the USA has always been a leader in genomics, ranked number one in this field.
7. The promise of genomics for personalized and preventative medicine has lagged behind, according to several experts. What obstacles do you observe?
There is no doubt that genomics has fallen short of expectations when it comes to the development of personalized and preventive medicine. The former US president Barack Obama expressed frustration about the slow advancement of genomics in medicine. According to him, the healthcare system isn't keeping up. In reality, it's different. The results of several highly-profile genomicsguided precision medicine trials (SHIVA, MOSCATO-01, Copenhagen Prospective Personalized Oncology (CoPPO), MAST, PERMED 01, PREDICT, etc.,) indicate that only 10% of patients improved their progression-free survival through drug-target
www. pharmafocusamerica.com 107
EXPERT TALK
matching. As a result, precision medicine fails to help many cancer patients. Integration of genomics into healthcarehas several obstacles. The process of making therapeutic recommendations based on genomic data is complex. Physicians may be hesitant to accept target-drug match recommendations based on genomic analysis. Precision medicine cannot be established without understanding the biological mind of cancer. We also do not have the ability to create drug combination cocktails based on patient biology instantly, nor do we have access to matching drugs for many actionable targets. Implementing the proper framework by integrating biology and dynamics, defining guidelines for conducting clinical genomics investigations, ensuring data security, creating a database of clinical experiences using target-drug matching treatments, integrating intelligent decisionmaking algorithms and establishing principles and practices for precision medicine all these are necessary to ensure patient and oncologist confidence.
8. What would you say about the state of translational research in the USA, especially in oncology?
Translational research aims to turn bench research into clinical practice at the bedside. Academic centers and hospitals are utilizing advances in genomic research to foster research activities. This is done through the establishment of clinical and translational research institutes for training and attracting
grant funding from government organizations and private sources. These initiatives have been widely adopted in the United States, which is certainly commendable. Academic institutions primarily provide seed funding so that investigators can secure larger grant funding, thereby allowing them to realize significant benefits from indirect costs, several successful start-ups, enabling academic-industry collaborations grew out of such initiatives. More emphasis must be placed on identifying and solving unmet needs in the local community, and visionary leadership is essential to develop and grow and establish a long-term research resources that will benefit patients in the long run. Recent debate suggesting that cancer arises from bad luck rather than genetic changes (Source: Scientific American, March 2017 and other outlets) not only confuses the public, but also suggests that our understanding of cancer biology is incomplete. Translating oncologists are responsible for ensuring that the biological foundations for the understanding of cancer and its nature are solidly established. If translational oncology research adopts the rocket science mentality of tackling and achieving a single mission, single goal attitude, it may be able to achieve remarkable results for every cancer.
9. What do you think the future of biomarkers is?
Biomarkers are rapidly gaining in popularity as non-invasive diagnostic tools. As an example, colon cancer can now be detected
108 PHARMA FOCUS AMERICA ISSUE 01 - 2023
EXPERT TALK
through DNA analysis of stool specimens, eliminating the need for uncomfortable colonoscopies. It is becoming more common to perform liquid biopsies, as the sequence of circulating tumors and tumorderived materials from blood is becoming a valuable tool for detecting cancer early, tracking prognosis, and tracking recurrence. Simplicifying sequencing will lead to faster biomarker assessment and will enable at-home "glucometer-like" systems with smart technology and AI. Biopsies of tumors are ideal specimens for the assessment of biomarkers, however, repeat biopsies from the same patient may not be possible. Additionally, for certain types of cancer, biopsies may be difficult to obtain due to anatomical limitations, making liquid biopsies the only option for planning any precision medicine therapy.
10. What are the most serious issues that cancer patients in the US are currently facing?
There is limited access to advanced cancer therapies. Therefore, patients must either accept what is locally available or may be required to travel a long distance or across states to receive such treatment. The cost of cancer treatment is skyrocketing, and patients without access to comprehensive health insurance suffer the most from physical, mental, and financial hardships. In some cases, patients who have exhausted all other options, financially broke resort to risky unproven alternates in the hope of a miracle. In the case of uneducated or low-income individuals, disease awareness, treatment options, and the importance of follow-up are relatively low, and some people do not seek medical help until the disease becomes terminal. Celebrities, community leaders, and foundations can play a significant role in bringing awareness and providing guidance regarding cancer to the community.
11. What innovations do you anticipate taking place in the next 12 or 5 years?
During the next decade, genomic sequencing will be an affordable, simple and user-friendly diagnostic tool that will be seamlessly integrated into clinical settings. As a result of the success of the mRNA-based COVID vaccines, a mRNA-based cancer vaccine will be developed based on targets derived from genomics. Genomic sequencing is utilized
www. pharmafocusamerica.com 109
EXPERT TALK
NGS is a massively parallel sequencing technology used to determine the order of nucleotides in entire genomes or targeted regions of DNA or RNA
to assess tumor mutation burden for cellbased immunotherapy, the presence of tumor reactive immune cells, and therapeutic immune cells, such as CAR-T cells. Increasing FDA approvals for immunotherapy and targeted therapy will lead to the development and marketing of targeted genetic screening with digital PCR and portable next generation sequencing platforms, ultrafast innovative biosensor chips, digital wearables or stripbased detection kits along with AI integration will be the future.
The "omics era" presents us with unprecedented opportunities for scientific advancement because of our ability to integrate assessments from genomics (DNA), transcriptomics (transcriptomics), protein (proteomics), and metabolomics (metabolites derived from biochemical reactions). The development of fundamental knowledge, early detection, diagnosis, prediction of prognosis, and drug discovery and development have enabled newer approaches to arriving at treatment decisions that provide unparalleled opportunities. In recent years, advancements have already resulted in a reduction in cancer death rates, but much remains to be done. The incidence of cancer is on the rise worldwide, and the number of cases and deaths are expected to nearly double by 2040. Increased healthcare costs and inadequate expansion will lead to
an unimaginable tragedy. As a comparison, COVID killed 6.6 million people in 3 years, while cancer kills 9.96 million each year. With an estimated 30 million new cases per year by 2040, 17 million people will die from the disease. In the absence of quick action, our entire world will face far reaching misery worse than COVID. Public-private partnerships that promote science advocacy, establish a basic science foundation, raise awareness, invest in technology and drug development, share genomic data and experience from precision therapeutics, and increase intellectual cooperation, although they are already present, require more intensified efforts to strengthen our fight against cancer.
AUTHOR BIO
Ravi Dashnamoorthy is a Ph.D cancer biologist who studies molecular pathways of cancer progression. As a Senior Scientist at Genosco, Billerica, MA, with 25 years of academic research experience, published 40+ articles and 75+ abstracts in basic and translational oncology. Genosco is a clinical stage biotech company specializing in discovering novel kinase inhibitors for patients with unmet medical needs.

110 PHARMA FOCUS AMERICA ISSUE 01 - 2023
12. Do you have any more information to share with our international audience?
EXPERT TALK



www. pharmafocusamerica.com 111 www.pharmafocusamerica.com Scan the QR-Code and start with your Authority Journey. Alternatively you may visit: https://www.pharmafocusamerica.com/get-published PUBLISH IN MAGAZINE Submission Deadline: July 2023 PUBLISH ON OUR PORTAL Submission Deadline: Anytime Be heard… Stay relevant… Get published. Ask us how? Opportunity to share industry insights and thoughts with our readers.







112 PHARMA FOCUS AMERICA ISSUE 01 - 2023 EVENT PREVIEW EXPERIENCE SCIENCE THROUGH COMMERCIALIZATION APRIL 25 –27, 2023 JAVITS CENTER, NYC REGISTER FOR FREE AT INTERPHEX23.COM/OCHRE_MEDIA
Experience science through commercialization
April 25 - 27, 2023 at New York, NY, USA
The International Pharmaceutical Expo (INTERPHEX), the premier event in the USA dedicated to pharmaceutical and biotechnological innovation from development to marketing, is scheduled to take place at the end of April at the Javits Center in New York, NY.
INTERPHEX is the leading global event that fuses industry innovation with expert-led technical conference. It’s where the newest ideas are shared, technology is unveiled, and the power of science through commercialization comes to life. No matter where you are in the pharmaceutical supply chain, INTERPHEX delivers relevant solutions through curated education sessions, networking and over 500 global suppliers to source quality products and services. The future of pharma, through the power of experience.
Who can attend?
Industry professionals from pharmaceutical, biotech and device facilities and service providers involved in specifying, recommending or purchasing technologies/products/services for the develop and manufacture of cost effective, quality products.
Benefits of attending INTERPHEX –
• Find new, more cost-effective alternatives to existing processes and procedures
• Discover highly efficient new technologies to increase productivity
• Meet partners who can provide instant access to cutting-edge technologies
• Acquire new skills and insights from qualified experts
• Evaluate the full range of competing solutions in every product category
• Learn from industry experts who share effective strategies to maximize efficiency, enhance product quality, and ensure regulatory compliance and more
• Upgrade your skills, your knowledge and your on-the-job effectiveness
• Network with peers from across the country and around the world
• Hear free keynotes by industry thought leaders and innovators
Registration includes access to:
INTERPHEX exhibit hall and technical conference badge offers access to the latest in technology, innovation, device development and manufacturing to:
Learn
• 3-Day technical conference
• Interphex live
• Keynote series with pharmaceutical technology and biopharm int’l
• Ips technologies tours
• Contract-specific sessions
Discover & explore
• 3-Day access to exhibit hall
• New exhibitor zone
• Contract services pavilion
• Interphex exhibitor award winners
• Access to leading suppliers
• Technology launches
• Poster hall
• New! Innovatech gateway pavilion
Network
• Technology-based networking events
• Conference recommendations
• Exhibitor recommendations
• New exhibitor networking happy hour
• Ips reception
• Exhibitor in-booth events
Interphex 2023
Date: April 25 - 27, 2023
Location: Javits Center, NYC, USA
Website: https://www.interphex.com/
Email: inquiry@interphex.com
www. pharmafocusamerica.com 113 EVENT PREVIEW
Developability and Immunosafety: Don’t let them threaten your drug development plans
This webinar was presented by Dr. Yvette Stallwood from Lonza.
Advancing your drug or vaccine candidate from latestage discovery into the clinic is one of the most critical steps in development. To maximize your chances of success, it is essential to de-risk your drug candidates as early as possible. This webinar will provides insights and solutions on how to de-risk your development and how to accelerate your path to FIH (First-in-Human) clinical studies. You will also learn how in silico and in vitro protein design and optimization tools can help you to identify and mitigate manufacturing, development, and immunogenicity liabilities and how to improve the quality and safety of your therapeutic proteins.
Dr. Yvette Stallwood
Designation: Head of Early Development Services
Organization: Lonza
Yvette Stallwood completed her Ph.D. at the University of Birmingham (UK) and has a background in Virology, Cell & Molecular Biology. She joined Lonza in 2007, initially leading the cell and molecular biology expression group in the Applied Protein Services department, and is currently Head of Early Development
Services and Head of the Cambridge site. The Early Development Services team is focused on the development and provision of services to support the development of new biotherapeutic proteins and vaccines with a particular focus on immunogenicity, manufacturability, and protein expression.
Founded in 1897 in the Swiss Alps, today, Lonza is the preferred global partner in the pharmaceutical, biotech, and nutrition markets. We focus on enabling treatments that prevent illness and support healthier lifestyles. We optimize scientific innovation and manufacturing technology to enable our customers to serve their patients and consumers.
We provide a wide range of services and products from early-phase discovery to custom development and manufacturing of active pharmaceutical ingredients to innovative dosage forms for the pharma and consumer health and nutrition industries.
https://www.pharmafocusasia.com/lonza-webinar-feb14

Get direct access by scanning the QR Code

114 PHARMA FOCUS AMERICA ISSUE 01 - 2023 WEBINAR REVIEW
Redefining Liquid Formulation Technology in Hard Capsules
This webinar was presented by three industry experts - Justin Kalafat, Anthony Drager, Andrey Lopes.

The experts tread through the path on how the use of Self-Emulsifying Drug Delivery Systems (SEDDS) enables oral bioavailability of poorly soluble drugs (BCS Class II/IV), and how to formulate the different types of SEDDS formulations in pharmaceutical product development.
This webinar provided insights and solutions on why there is an increased emphasis on liquid filling to enhance certain product lines, brand highend ingredients, and the use of combination filling to separate products from a crowded market. It also takes the viewer through the importance and techniques of banding liquid-filled capsules & the versatility & advantages of liquid filling compared to similar dosage forms

Justin Kalafat, Anthony Drager, Andrey Lopes.
Designation:
Justin Kalafat, Head of International Scientific Business Development, ACG Capsules
Anthony Drager, Senior Director, Pharmaceutical Development, Venatorx Pharmaceuticals
Andrey Lopes, Business Development Manager
LATAM, ACG Capsules
Organization: ACG
Founded in Mumbai in 1961, ACG now serves pharmaceutical and nutraceutical companies all over the world, touching almost every aspect of solid dosage manufacturing.
Our holistic view of manufacturing is based on a conviction that this is the best approach to creating seamless production married with fully accountable service. Ultimately, it is the only way to, literally, ‘make it better’.
ACG believes that everyone on Earth deserves access to good medicine, and is focused on working with its partners to create a healthier world.
https://www.pharmafocusamerica.com/promotion/ acg-world-capsules-webinar
www. pharmafocusamerica.com 115 WEBINAR REVIEW
direct access by scanning the QR Code
Get
AI in Drug Discovery
March 13-14, 2023
London, United Kingdom
https://www.smgconferences.com/ pharmaceuticals/uk/conference/drug-discovery
About Event: About the Event: With the recent pandemic highlighting the need for rapid drug discovery, AI has become an area of increased interest. This is driven by the ability to discover drugs through the use of machine and deep learning. The current challenges within the drug discovery industry include the significant time consumption and expenses involved. This conference will discuss the solutions to these problems with presentations and updates from leading industry experts.
Listed Under: Research & Development.
4th International Conference on Pharmacology and Toxicology
March 15–16, 2023
Dubai, UAE
https://clinicalpharmaforum.com/
About Event: Exploring New Research and Innovative Development in Pharmacology and Toxicology.
Listed Under: Research & Development.
Clinical Trial Supply Europe 2023

March 15-16, 2023
Milano, Italy
https://www.arena-international.com/event/ ctseurope/
About Event: Clinical Trial Supply Europe is the meeting place for the pharmaceutical and biotechnology community to discover how to excel in clinical supply strategy as well as form key connections for long-term success.
Listed Under: Clinical Trials.
Outsourcing In Clinical Trials
Southeast 2023
March 28-29, 2023
North Carolina, United States
https://www.arena-international.com/event/ octsoutheast/
About Event: The must attend clinical outsourcing event in the Southeast region where industry professional can encounter a platform to explore new solutions to common issues within their clinical trial. This is the leading industry event in the Southeast for outsourcing specialists’ that brings together industry leaders, solution providers and professionals into the same rooms, allowing them to interact in ways which can simplify processes and maximise their effort to get drugs to patients in a timely fashion.
Listed Under: Clinical Trials.
116 PHARMA FOCUS AMERICA ISSUE 01 - 2023
EVENTS LIST
3rd International Congress on Advances in Clinical Research and Trials
March 20-21, 2023
London, United Kingdom
https://clinicalresearch. peersalleyconferences.com/

About Event: The future of clinical trials How cutting-edge technologies could make patient-centered drug development cheaper, faster, effective for maintaining the integrity of science and ensuring patient safety.
Listed Under: Clinical Trials.
Pharma USA 2023
March 28–29, 2023

Philadelphia, USA
https://events.reutersevents.com/pharma/pharmausa
American Manufacturing Summit
March 28-29, 2023
Illinois, United States
https://manusummit.com
About Event: Pharma USA 2023 is where 1,200+ pharma leaders and critical stakeholders will unite to create an agile commercial model, learn how to be truly customer centric and create a unified and company-wide value-proposition that all stakeholders yearn for.
Listed Under: Research & Development.
About Event: The American Manufacturing Summit serves as an annual platform to exchange ideas surrounding the impact of market dynamics and new technologies for current and future manufacturing leaders. This year’s Summit creates an opportunity to examine key case studies around how workforce management, lean manufacturing, process improvement and automation are being rolled out in the world’s best facilities. Network with over 200+ of your peers, connect with exhibitors and learn from the top industry influencers as we explore strategies to optimize, modernize, train and innovate across multiple manufacturing facilities.
Listed Under: Manufacturing.
American Biomanufacturing Summit

April 12-13, 2023
California, USA
https://biomanamerica.com/
About Event: The American Biomanufacturing Summit is one of the most senior-level conferences and networking events in the industry. It is designed to provide biopharmaceutical executives with current trends, strategic insights and best practices in manufacturing, outsourcing, capacity management, quality assurance, quality control, regulatory compliance, operational excellence, supply chain and logistics. Network with over 150+ of your peers as we explore strategies to maximize efficiency while remaining compliant in an ever-evolving environment.
Listed Under: Manufacturing.
www. pharmafocusamerica.com 117
EVENTS LIST
Global Pharma & Drug Delivery Summit
April 24-26, 2023
Frankfurt, Germany
https://worldpharmasummit.com/
About Event: This Pharma Summit 2023 revolves around the theme – “Together with Pharma Masterminds: Opportunities, Technology, and Challenges”. The Pharma Summit 2023 will help the delegates to establish their research or business relations as well as to make international linkage for future collaborations in their career path. Your presence will be much appreciated.
Listed Under: Research & Development
Pharmaceutical Microbiology USA

April 26-27, 2023
Boston, USA
https://www.smgconferences.com/pharmaceuticals/ northamerica/conference/PharmaceuticalMicrobiology-East-Coast
About Event: Microbiology remains an essential tool in reducing microbial growth in the manufacture of pharmaceuticals, to detect and eliminate microorganisms that would pose a risk to patients and jeopardise product batches
Listed Under: Research & Development
Pharmaceutical Compliance Congress
April 25 – 27, 2023
Virginia, United States
https://informaconnect.com/pharmaceuticalcompliance-congress-pcc/
About Event: The Premier Education Hub for Mitigating Risk and Delivering Novel, Allencompassing Ethics & Compliance Programs.
Listed Under: Research & Development
European Clinical Trial Supply Forum
April 18-19, 2023
Germany
https://worldbigroup.com/3rd-Clinical-Trial-Supply/ About Event: World BI takes pride in organizing 3rd Clinical Trial Supply Forum – a unique platform to meet your clinical trial supply requirements and solutions. With the COVID-19 led disruptions around the globe, attendees will get a chance to meet and network in a safe, learning environment to discuss, debate and consider new technologies and processes to streamline supply chain operations.
Listed Under: Clinical Trials
European Pharma & Device Packaging and Labelling Forum
April 04-05, 2023
Frankfurt
https://worldbigroup.com/5th-PharmaPackaging/
About Event: Packaging innovation has had to accelerate at a faster rate than perhaps ever before in recent years to optimize drug delivery for pharma, healthcare professionals and patients alike – but it's necessary to keep up with medical advances, stay ahead of competition and accommodate the unique and differentiated needs of combination products, biologics, vaccines and other specialty medicines.
Listed Under: Manufacturing
118 PHARMA FOCUS AMERICA ISSUE 01 - 2023
EVENTS LIST
Precision in Drug Discovery & Preclinical Summit
April 03-04, 2023
Amsterdam, Netherlands
https://events.precision-globe.com/single-event/ precision-in-drug-discovery-preclinical-summitamsterdam-april-3rd-4th-2023
About Event: 18th PDDP Europe for a twoday journey into the insightful world of drug discovery and preclinical drug development. You’ll connect with an incredible gathering of experts and uncover practical advice from industry-leading scientists and executives. Get ready for an event designed to deliver real value-packed insights, connections with fellow professionals and learn strategies you can put to use the very next day.
Listed Under: Research & Development
American Supply Chain Summit
May 16-17, 2023
USA
https://supplychainus.com/
About Event: The American Supply Chain Summit is a leadership focused meeting designed around improving supply chain and procurement strategy across the globe. The Supply Chain Summit serves as an annual platform to exchange ideas and collaborate on the impact of market dynamics and new technologies for current and future supply chain and operations leaders. This year's event creates an opportunity to examine key case studies on how to navigate supply chain disruptions as well as how workforce management, advanced analytics, process improvement and automation are being rolled out in the world's best facilities. Join the in-depth discussions on achieving innovation, maximizing supply chain profitability and increasing visibility and flexibility to mitigate risk.
Listed Under: Manufacturing
Precision in Clinical Trials Summit
May 01-02, 2023
USA
https://events.precision-globe.com/single-event/ precision-in-clinical-trials-summit-boston-may-1st2nd2023
About Event: The Precision in Clinical Trials summit will be a unique platform for the largest bio-pharmaceutical hub in the region to network and discuss collaboration in order to achieve their outsourcing and operational strategies.
Listed Under: Clinical Trials
European Pharma Supply Chain & Logistics Innovation Programme


May 25-26, 2023
Switzerland
https://worldbigroup.com/9th-Pharma-SupplyChain/
About Event: Pharma Supply Chain & Logistics
Innovation Programme is a prominent platform that encourages you to network with the most vibrant and influential leaders in the global supply chain sector. This event is dedicated to exchanging information on every aspect of the supply chain and logistics, especially digitization using multiple approaches, i.e., Blockchain, Machine learning, and Artificial Intelligence. Which will have Pharma industry experts sharing various challenges faced, new strategies, case studies, and the use of innovative ideas, the conference will also offer opportunities to encourage partnerships and collaborations.
Listed Under: Information Technology
www. pharmafocusamerica.com 119
EVENTS LIST
iPharma Expo 2023
July 20 - 21, 2023
USA
https://www.ipharmaexpo.com/
About Event: The expo will showcase the latest trends and technologies in pharmaceuticals, drugs, and formulations. The expo is expected to witness approx. 150 exhibitors and 1500- 2000 visitors footfall from pharma industry & management. Direct access to highly targeted senior pharma executives, buyers, procurement managers, contract manufacturers, hospital administration, and many more Meetings with manager and business development managers who are looking for new supplies, building strategic partnerships, or entering into new ventures.
Listed Under: Strategy
Highly Potent Active Pharmaceutical Ingredients

May 10-11, 2023
London, United Kingdom
https://www.smgconferences.com/ pharmaceuticals/uk/conference/Highly-PotentActive-Pharmaceutical-Ingredients
About Event: The Global High Potency API/ HPAPI Market is projected to reach 37 billion USD by 2027, growing at an ever-increasing CAGR of 8.43%. The expanding production of high potency APIs, along with the increase in associated toxicities, are driving requirements for more effective containment solutions and much-needed guidelines to ensure worker safety. The conference will encompass the critical topics including risk assessment, occupational toxicology, engineering controls for containment, facility design, and the impact of pharmaceutical manufacture on the environment.
Listed Under: Research & Development
5th International Conference on Proteomics, Genomics and Molecular Medicine
July 10-12, 2023
Italy
https://proteomics.alliedacademies.com/2022/
About Event: Proteomics Congress works on the theme “Current Trends and Innovations in Proteomics, Genomics and Molecular Medicine” which encompasses Keynote presenters, establishers, Delegates, and Speakers. Reach out to potential experts and break through the excellence of understanding the innovations in Proteomics, Genomics, and Molecular Medicine.
Listed Under: Clinical Trials
120 PHARMA FOCUS AMERICA ISSUE 01 - 2023
EVENTS LIST
COLLABORATION
Moderna to Acquire Oriciro Genomics for US $ 85 million

Moderna has announced it will acquire Japanbased DNA supplier OriCiro Genomics K.K (OriCiro) for $85m, marking the US Company’s first acquisition since its 2010 launch.
Through the acquisition, Moderna will get access to OriCiro's equipment for cell-free plasmid DNA synthesis and amplification, a form of DNA molecule used in the production of mRNA.
FDA APPROVAL

According to the firm, the medicines and vaccines in Moderna's portfolio will be supported by OriCiro's synthetic biology and enzyme technology.
The development of therapeutics and vaccines for infectious diseases, immunooncology, rare diseases, cardiovascular diseases, and autoimmune diseases has been made possible by Moderna's mRNA platform, which is built on ongoing advances in basic and applied mRNA science, delivery technology, and manufacturing. For the previous eight years, Moderna has been recognised by Science as a top biopharmaceutical employer.
The mission of OriCiro Genomics, established in December 2018, is to develop and commercialise cell-free plasmid DNA synthesis and amplification methods for use in gene/cell-based therapeutics and synthetic biology.
Read the complete post
TScan Therapeutics Announces FDA Approvals for Solid Tumor Treatment in the United States
The U.S. Food and Drug Administration (FDA) has approved TScan Therapeutics' investigational new drug (IND) applications for T-Plex, TSC204-A0201, and TSC-204-C0702.
T-Plex will allow patients to get individualized combinations of T cell receptor (TCR)-engineered T cell therapies (TCR-T) based on the HLAs and targets expressed in their tumours. It will serve as the principal IND for TScan's solid tumour programme.
Each patient's individual TCRs will be chosen from the ImmunoBank of TScan. Each distinct TCR-T will be submitted as a secondary IND with a reference to the T-Plex primary IND.
In addition to the T-Plex IND, TScan also submitted secondary INDs for the original TCR-T products, TSC-204-A0201 and TSC-204-C0702.
With the approval of these INDs, TScan is now working to begin a multicenter Phase I clinical trial in order to determine the preliminary efficacy, safety, and viability of repeating multiplexed TCR-T doses.
Patients with melanoma, non-small cell lung cancer, head and neck cancer, ovarian cancer, and cervical cancer will participate in this experiment.
Read the complete post


www. pharmafocusamerica.com 121 NEWS
COLLABORATION

Boehringer Ingelheim and 3T Biosciences Collaborate to Develop Next-Generation Cancer Immunotherapies
Read

Boehringer Ingelheim and 3T Biosciences recently announced a strategic collaboration to address unmet patient needs.
3T Biosciences is combining with Boehringer Ingelheim's best-in-class 3T-TRACE (T-Cell Receptor Antigen and Cross-Reactivity Engine) discovery platform to significantly enhance the pipeline. The 3T platform intends to find novel common T-cell receptor (TCR) targets for effective immune responses and screen them thoroughly for specificity and cross-reactivities.
The aim is to discover the most frequent and immunogenic targets in solid tumours by combining highdiversity target libraries with active machine learning. The end result would be more targeted, high-dose medicines that can safely attack specific tumours.
Boehringer Ingelheim will contribute patient-derived TCR data to support 3T's target discovery activities using its 3T TRACE discovery platform.
Boehringer will pay 3T a total of US$268 million, including an upfront payment & ongoing research and development assistance.
Boehringer Ingelheim is qualified to collect royalties on product sales made by 3T Biosciences.
122 PHARMA FOCUS AMERICA ISSUE 01 - 2023
the complete post NEWS
COLLABORATION
Anima Biotech and AbbVie Collaborate to Discover and Develop mRNA Biology Modulators
FDA APPROVAL
New Treatment for Adults with Relapsed or Refractory MCL Has Been Approved
Jaypirca (pirtobrutinib) has been approved by the U.S. Food and Drug Administration for adult patients with relapsed or refractory mantle cell lymphoma (MCL).
The approval follows after at least two lines of systemic therapy, including a BTK inhibitor, was announced by Loxo@Lilly, the oncology division of Eli Lilly and Company.
AbbVie and Anima Biotech (Anima) have formed a partnership, to find and develop mRNA biology modulators for three oncology and immunology targets.
The partners will find novel mRNA biology modulators for the cooperation targets using Anima's mRNA Lightning platform, with AbbVie obtaining exclusive rights to license, further develop, and market the programmes.
As part of the deal, Anima will receive an upfront payment of US $42 million. In addition, Anima may be eligible to receive up to $540 million in option fees, research and development milestones, commercial milestones, and tiered royalties on net sales for the three objectives.
Although AbbVie will also have the choice to extend the agreement with up to three more targets under the same conditions, possibly raising the partnership’s worth.

Modulation of mRNA biology with small molecules is a novel strategy that has the potential to address "undruggable" targets and have ramifications for numerous therapeutic fields. Phenotypic screening and AIdriven elucidation of the mechanisms of action are combined in "Anima's differentiating approach in the realm of small molecule mRNA medicines."
The first and only non-covalent (reversible) BTK inhibitor to receive FDA approval is Jaypirca. It extends the benefit of addressing this pathway by re-establishing BTK inhibition in MCL patients who had previously received treatment with a covalent BTK inhibitor (ibrutinib, acalabrutinib, or zanubrutinib).
Warnings for infections, bleeding, cytopenias, atrial fibrillation and flutter, second primary malignancies, and embryo-fetal toxicity are included in the labeling for Jaypirca.
Data from a sample of patients in the BRUIN Phase 1/2 trial served as the basis for the FDA's clearance. The effectiveness evaluation was based on 120 MCL patients who received Jaypirca 200 mg once daily until the disease progressed or the toxicity became intolerable. Jaypirca received FDA approval under the accelerated approval process. Patients are now enrolled in the confirmatory Phase III trial (NCT04662255; BRUIN MCL-321).
Within the next several weeks, Jaypirca is anticipated to become accessible in the US.


www. pharmafocusamerica.com 123 Read the complete post Read the complete post
NEWS
Eli Lilly to invest US $450 million in Research Triangle Park's Manufacturing Site
Eli Lilly & Co. announced that it intends to invest an additional US
$450 million in a manufacturing facility in North Carolina's Research Triangle Park.


Eli Lilly said that the investment will lead to the creation of at least 100 new jobs, and added that the expansion will improve parenteral filling, device assembly, and packaging capacity in response to rising demand for Lilly's incretin-based diabetes treatments.

This project phase is anticipated to produce the majority of manufacturing personnel when it is fully operational in 2027. This team will create incretin therapies and medical equipment using cutting-edge technology.
In North Carolina, Lilly has invested almost US $4 billion since 2020, including US $1.7 billion for the development and expansion of its site in Research Triangle Park. Initial manufacturing at Research Triangle Park is expected to start this year, and the FDA inspections are currently being prepared for.
Since 2014, Lilly has introduced 18 new drugs to help people with illnesses including cancer and diabetes live better lives, and the company intends to introduce a number of potential new drugs in 2023. This growth will be crucial in maintaining the supply of existing Lilly medications while laying the groundwork for distributing the following generation of medicines to patients all across the world.
FDA APPROVAL
Evaxion's Personalised Cancer Therapy has been Granted FDA Fast-Track Status
The U.S. Food and Drug Administration has granted KEYTRUDA® and EVX-01, tailored cancer treatments from Evaxion, fast-track classification.
Evaxion acquired FDA approval in December of last year to proceed with its Phase 2b clinical research, in which patients with metastatic melanoma are administered EVX-01 and KEYTRUDA.
Additionally, Evaxion received the fast track designation for the vaccine candidate on January 17, 2023.
With this fast-track designation, the regulatory body will assess new medications more quickly that showcase the potential to address an unmet medical requirement.
The most cutting-edge clinical tool available to Evaxion is EVX-01, a peptide-based cancer immunotherapy. According to the programme, each patient receives a customized medication based on the analysis of their tumors’ genes and a match with their immune systems. The Company's in-house AI platform, PIONEER, enables this approach.
Clinical sites in the United States, Europe, and Australia are participating in the ongoing Phase 2b study. It is being done in association with Merck, which is providing the PD-1 inhibitor KEYTRUDA for the study.
In September of last year, the first patient in Australia was enrolled in the trial.
124 PHARMA FOCUS AMERICA ISSUE 01 - 2023 Read the complete post Read the complete post EXPANSION
NEWS

Introducing a group of highly focussed magazines for the Europe and Asian markets.



Aspiring to be leading journals in the B2B landscape of Pharmaceutical-Industry, the magazines covers Medical Sciences, Business & Technology and all the latest innovations.
Our magazines bring a fresh outlook towards insightful and pragmatic Pharmaceutical-Industry reporting. Delightfully selected topics presented by the gurus of the industry comes packed with latest happenings, sharp analysis & deep insights. We strive to keep you engaged, knowledgeable & wanting for more.
126 PHARMA FOCUS AMERICA ISSUE 01 - 2023 Introducing Advent of NEW-AGE PHARMACEUTICAL REPORTING From the house of Ochre Media: Automotive-technology.com | Defence-industries.com | Hospitals-management.com | Packaging-labelling.com Pharmaceutical-tech.com | Plantautomation-technology.com | Plastics-technology.com | Pulpandpaper-technology.com Sportsvenue-technology.com | Steel-technology.com | Asianhhm.com | Pharmafocusasia.com
Scan to check websites Scan to check websites www.ochre-media.com















