8 minute read
DIAGNÓSTICO


DERMATOMIOSITIS ASOCIADA A ANTICUERPOS ANTI-MDA5.
Discusi N
La DM asociada a anticuerpos anti-MDA5 es una miopatía inflamatoria que se caracteriza por una llamativa afectación cutánea en ausencia, o con mínima, afectación muscular.
Las manifestaciones cutáneas más frecuentes son las úlceras cutáneas y orales, la alopecia, las pápulas palmares y el engrosamiento de la cara lateral de los dedos¹. Puede producir también afectación articular.
Estos pacientes presentan un riesgo elevado de desarrollar enfermedad pulmonar intersticial difusa (EPID) que típicamente es rápidamente progresiva (EPID-RP). Radiológicamente se caracteriza por áreas de consolidación y vidrio deslustrado, compatibles con un patrón de neumonía organizada².
El neumomediastino es una complicación rara de la DM asociada a anti-MDA5, cuya frecuencia estimada está en torno al 2% y que conlleva una alta tasa de mortalidad. El mecanismo generalmente aceptado por el que se produce es por el aumento de la presión intraalveolar o la formación de blebs por la distorsión de la arquitectura pulmonar. No obstante, existen también casos descritos en los que se produce neumomediastino a pesar de una mejoría en la afectación intersticial, por lo que se ha planteado el papel deletéreo que puedan tener los esteroides sobre el tejido conectivo y la asociación entre el neumomediastino y la vasculopatía cutánea³,⁴
Dada la potencial gravedad de la enfermedad, sobre todo en los casos que desarrollan EPID-RP, es fundamental realizar un diagnóstico precoz y un tratamiento agresivo. Como en nuestro caso, existen pacientes que presenta anticuerpos anti-MDA5 positivos siendo los ANA y el resto de anticuerpos negativos5 por lo que, ante una sospecha clínica, deberemos solicitar determinaciones de inmunología complementarias para descartar una miopatía inflamatoria.
Los esquemas más comunes de tratamiento en los casos con afectación intersticial incluyen corticoides y otros fármacos inmunosupresores como ciclofosfamida, inhibidores de la calcineurina o rituximab. Se recomienda un manejo agresivo desde el principio con una terapia combinada con esteroides, tacrolimus y/o ciclofosfamida6
Bibliograf A
1. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: A concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018 Apr;78(4):776-785.
2. Laporte A, Mariampillai K, Allenbach et al. Idiopathic inflammatory myopathies: CT characteristics of interstitial lung disease and their association(s) with myositis-specific autoantibodies. Eur Radiol. 2022 May;32(5):3480-3489.
3. Kono H, Inokuma S, Nakayama H et al. Pneumomediastinum in dermatomyositis: association with cutaneous vasculopathy. Ann Rheum Dis. 2000 May;59(5):372-6.
4. Chan CWS, Chung HY, Lau CS, Tsang HHL. Spontaneous pneumomediastinum in a dermatomyositis patient with anti-melanoma differentiation-associated gene-5 antibody and interstitial lung disease despite an initial response to immunosuppressant. Int J Rheum Dis. 2019 Mar;22(3):521-524.
5. Yamaguchi K, Yamaguchi A, Kashiwagi C et al. Differential clinical features of patients with clinically amyopathic dermatomyositis who have circulating anti-MDA5 autoantibodies with or without myositis-associated autoantibodies. Respir Med. 2018 Jul;140:1-5.
6. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol 2020;72:488-498.
Figuras

El D A A D A En La Consulta De Epid
A PROPÓSITO DE UN CASO: PACIENTE CON CRITERIOS DE IPAF
Autor
Dr. Pablo Mariscal Aguilar
Hospital Universitario La Paz. Madrid
Anamnesis
Presentamos el caso de varón de 77 años con antecedentes personales de tabaquismo (ex fumador índice acumulado paquete-año de 5), dislipidemia, cardiopatía isquémica crónica (último ecocardiograma con FEVI 67%). En cuanto a la información relativa a su situación basal, se trataba de un paciente que no tenía animales y estaba jubilado, trabajó de informático en oficina.
Acudió a la consulta de Neumología por tos seca de un año de evolución y síndrome de Raynaud intermitente de tres años de evolución. En la anamnesis se interrogó al paciente por clínica sistémica (fotosensibilidad, xeroftalmia, xerostomía, artritis, clínica neurológica, rash, etc.) y refería que no había tenido ninguno de estos síntomas.
Exploraci N F Sica
Presentaba aspecto cianótico de piel y mucosas. La auscultación pulmonar revelaba crepitantes secos. La tensión arterial era de 132/73 mmHg y la saturación de oxígeno medida por pulsioximetría era de 94%. No tenía acropaquias y el resto de la exploración física no mostraba alteraciones significativas.
Pruebas Complementarias
Los análisis de sangre mostraron un hemograma, bioquímica (incluidas hormonas tiroideas y vitamina D) y coagulación sin hallazgos patológicos. El panel autoinmune completo que se solicitó tenía unos anticuerpos anticelulares (ANA) positivos (1/640) con patrón homogéneo y moteado. El resto de la inmunología junto con la velocidad de sedimentación glomerular (VSG) fueron negativas.
Las pruebas de función respiratoria fueron: volumen espiratorio en el primer segundo (FEV1) 3,90 litros (77%) capacidad vital forzada (FVC) 1,79 litros (78%); FEV1/FVC 76%; capacidad de difusión del monóxido de carbono (DLCO) 7,49 mmol/min/kPa (72%) y difusión del monóxido de carbono/volumen alveolar (DLCO/VA) 1,20 mmol/min/kPa/l (87%), capacidad pulmonar total (TLC) 6,20 (91%) y volumen residual (RV) 2,24 (94%).
La radiografía de tórax reflejaba un patrón alveolo intersticial en bases por lo que se decidió realizar una tomografía computarizada de alta resolución (TCAR) de tórax.
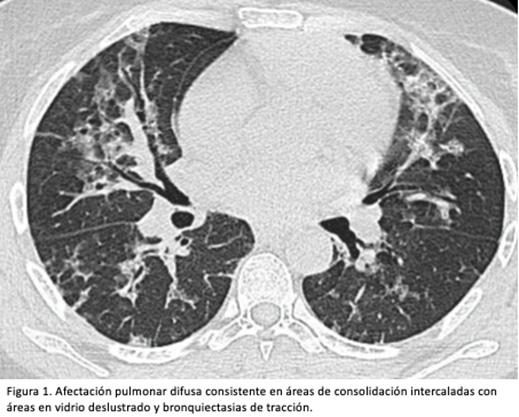
La TCAR mostró de tórax revelaba engrosamiento de septos en las bases con pérdida de volumen, bronquiectasias de tracción y vidrio deslustrado en ambos hemitórax (figura 1).
Ante estos hallazgos se practicó una broncoscopia, obteniendo muestras de broncoaspirado (BAS), lavado broncoalveolar (LBA) y biopsia transbronquial (BTB). El recuento celular del LBA fue: linfocitos 29%, macrófagos 80% neutrófilos 80% y eosinófilos 2%. El análisis anatomopatológico de la muestra extraída por BTB no presentaba alteraciones patológicas, aunque el patólogo responsable justificaba este hallazgo con la escasa cantidad de tejido obtenido. La tinción de Ziehl-Neelsen y los cultivos del LBA para bacterias y micobacterias fueron negativos.
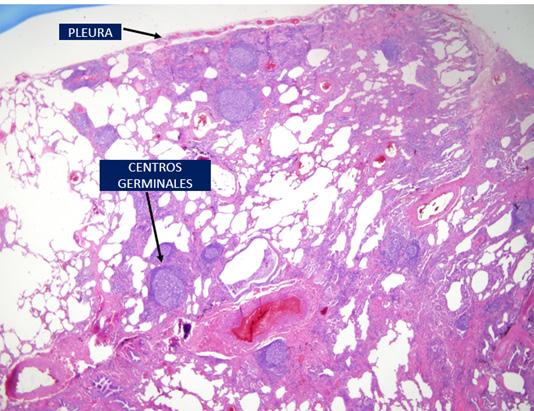
Dada la escasa cantidad de parénquima pulmonar analizado se decidió efectuar una biopsia pulmonar mediante segmectentomía donde se objetivó un parénquima pulmonar con patrón histopatológico de neumonía intersticial no específica (NINE) con infiltrados linfoplasmocitarios y numerosos centros germinales (figura 2).
Tras la reunión del comité multidisciplinar se concluyó que el paciente tenía una neumonía intersticial con características autoinmunes (IPAF).
Diagn Stico
NEUMONÍA INTERSTICIAL CON CARACTERÍSTICAS AUTOINMUNES (IPAF).
Tratamiento Y Evoluci N
Una vez que el paciente volvió a la consulta tras el diagnóstico, se inició tratamiento empírico con prednisona 0,5 mg/kg, (30 mg).
Cuatro meses más tarde se citó al enfermo para revisión. Clínicamente había mejorado la tos y el fenómeno de Raynaud. En cuanto a las pruebas complementarias, se ejecutó otra TCAR de tórax donde había una discreta mejoría de las zonas de vidrio deslustrado con persistencia del resto de rasgos radiológicos. Por otro lado, las pruebas de función respiratoria fueron las siguientes FEV1 4,03 L (82%); FVC 2,85 L (83%); FEV1/ FVC 76%; DLCO 7,57 mmol/min/kPa (74%) y DLCO/VA 1,20 mmol/min/kPa/l (88%). Por tanto, se decidió comenzar pauta descendente de prednisona hasta 7,5 mg. En la siguiente revisión el enfermo continuaba estable clínicamente sin deterioro radiológico ni funcional.
Discusi N
La neumonía intersticial con características autoinmunes (IPAF) es una entidad definida en 2015 por un consenso de expertos ante la necesidad de reunir bajo una misma entidad a aquellas enfermedades pulmonares intersticiales difusas (EPID) que, si bien se asocian a algunas características autoinmunes, no reúnen los criterios necesarios para ser definidas como parte de las enfermedades sistémicas autoinmunes¹.
Los criterios para definir una IPAF se dividen en tres dominios: clínico serológico (cualquier anticuerpo específico positivo) y morfológico.
En este caso se trata de una paciente con patrón radiológico e histológico de NINE, ANA positivos (1/640) homogéneo y moteado con fenómeno de Raynaud. No reúne criterios para ser diagnosticado de una conectivopatía, ya que a parte del fenónmeno de Raynaud no tiene clínica compatible con ninguna enfermedad autoinmune y tampoco presenta anticuerpos específicos de ninguna patología.
No obstante sí que tiene un criterio clínico (fenómeno de Raynaud), serológico (ANA positivos 1/640) y morfológico (patrón NINE tanto histológico como radiológico).
En cuanto al tratamiento indicado, no hay actualmente evidencia suficiente para validar una estrategia las IPAF. Sin embargo, dado que se trata de una entidad de índole autoinmune, es fundamental valorar terapia con corticoides e inmunosupresores². De hecho, en este caso el enfermo evoluciona favorablemente con prednisona en pauta descendente. Además, si la EPID asociada desarrollase criterios de fibrosis pulmonar progresiva, también se debería valorar terapia antifibrótica, aunque, no es el caso que nos ocupa ya que tanto la clínica, función respiratoria y radiología se estabilizan a medio plazo³
El pronóstico en pacientes con IPAF no está bien definido, ya que depende en gran medida de las características concretas de cada caso. De hecho, en muchas ocasiones con el paso del tiempo estas IPAF pueden evolucionar a enfermedades autoinmunes bien definidas.
Bibliograf A
1. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976-87.
2. Vacchi C, Sebastiani M, Cassone G, Cerri S, Della Casa G, Salvarani C, et al. Therapeutic Options for the Treatment of Interstitial Lung Disease Related to Connective Tissue Diseases. A Narrative Review. J Clin Med. 2020;9.
3. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205:e18-e47.
4. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med. 2019;381:1718-27.
Figuras
El D A A D A En La Consulta De Epid
Osificaci N Pulmonar
Dendriforme En Fibrosis
Pulmonar Familiar
Autora
Dra. Celia Montaño Montaño
Hospital Universitario de Bellvitge. Hospitalet de Llobregat, Barcelona
Anamnesis
Se trata de un hombre de 63 años con antecedentes de tabaquismo durante unos 5 años y sin historia de exposición ambiental. Trabajó de chapista de coches (contacto con pinturas y amianto). Presenta apnea obstructiva del sueño en tratamiento con CPAP y talasemia minor como antecedentes personales. Como antecedente familiar de interés, su hermana mayor falleció de fibrosis pulmonar y otro hermano presenta fibrosis pulmonar idiopática. Por este motivo, comienza estudio en la Unidad Funcional de Intersticio Pulmonar (UFIP).
Se encuentra asintomático desde el punto de vista respiratorio y niega síntomas y signos de enfermedad sistémica. Se auscultan mínimos crepitantes en bases pulmonares.
Pruebas Complementarias
La radiografía de tórax muestra un patrón retículo-nodular periférico bilateral. La analítica sanguínea con hemograma, coagulación, función renal y hepática no presenta alteraciones. La evaluación serológica (anticuerpos antinucleares, anticuerpos anticitoplasma de neutrófilos, factor reumatoide y anticuerpos antipéptido cíclico citrulinado) y las IgG específicas a diferentes antígenos (aves y hongos) son negativas. El paciente y su hermano tienen una longitud telomérica por debajo del percentil 1 (acortamiento telomérico severo). Las pruebas de función pulmonar revelan un patrón ventilatorio restrictivo leve con capacidad vital forzada (FVC) de 3.830 ml (79%), capacidad pulmonar total (TLC) de 6.430 ml (85%), volumen espiratorio forzado en el primer segundo (VEMS) de 3.050 ml (82%), y la capacidad de difusión de monóxido de carbono (DLCO) de 96% con un coeficiente de transferencia de CO (KCO) de 125%. En el test de la marcha de 6 minutos no se detecta desaturación de oxígeno al esfuerzo. En la TC de tórax de alta resolución (figura 1) se objetiva engrosamiento septal difuso de predominio subpleural y en lóbulos inferiores asociado a calcificaciones pulmonares reticulares. Se realiza biopsia pulmonar quirúrgica en el lóbulo superior e inferior izquierdo cuya histología (figura 2) muestra numerosos focos de tejido óseo ramificados, algunos de ellos con elementos medulares asociados, y fibrosis intersticial focal de predominio subpleural con actividad fibroblástica leve (focos fibroblásticos). No se observa inflamación significativa, granulomas ni material extrínseco. Todo ello compatible con osificación pulmonar dendriforme y patrón histológico de neumonía intersticial usual (NIU).








