JOURNAL OF PHARMACOLOGY AND THERAPY
Neurogene Detrusorüberaktivität: Hoher Druck in der Blase = hoher Leidensdruck für die Patienten
Bronchodilatatoren: Zweimalgabe bessert nächtliche und morgendliche COPD-Symptome Secukinumab bei Spondyloarthritiden – ein früher Behandlungsbeginn kann Therapiean sprechen verbessern ACP-Therapie in Orthopädie und Sportmedizin: Innovative Operationsstrategien mit AutoCart™
Kastrationsresistenz beim Prostatakarzinom vermeiden Upstaza™ – die erste krankheitsmodifizierende Gentherapie für den AADC-Mangel
Eptinezumab erweitert die Optionen zur Migräneprophylaxe
Atopische Dermatitis: JAK1-Inhibitor Abrocitinib verbessert Hautläsio nen und Juckreiz
Mittelschwere bis schwere Plaque-Psoriasis: Wechsel auf Bimekizumab kann sich lohnen
FÜR PHARMAKOLOGIE UND THERAPIE
JAHRGANG 31 HEFT 4 September 2022 ISSN 1432-4334
VERL AG PERFUSION
Gemeinsam bringen wir die Therapie Ihrer Patienten auf eine neue Ebene

VESOXX® (1 mg/ml Oxybutynin-HCl) wirkt durch seinen multimodalen Wirkmechanismus2–5 und schützt dadurch langfristig die Nieren.6, 7 Die Dosierung von VESOXX® wird entsprechend urodynamischer Parameter patientenindividuell festgelegt.1 Die direkten Wirkungen von VESOXX® in der Blase und die geringere Verstoffwechselung lassen eine hohe Effektivität und eine bessere Verträglichkeit im Vergleich zur oralen Therapie erwarten.1, 7–11
VESOXX (1 mg/ml) wird angewendet zur Unterdrückung einer neurogenen Detrusorüberaktivität bei Kindern ab 6 Jahren u. bei Erwachsenen, d. ihre Blase mittels sauberer intermittierender Katheterisierung entleeren und nicht adäquat mit oralen Anticholinergika eingestellt werden können.1
1. VESOXX® Fachinformation. 2. Murakami S. et al. Urol Int. 2003; 71(3):290–298; (präklinische Studie an isoliertem humanen Blasengewebe mit Antimuskarinika). 3. Chapple C.R. et al. Urology. 2002; 60(5 Suppl 1):82–88; discussion 88–89; (Übersichtsartikel). 4. Kim Y. et al. Urology. 2005; 65(2):238–242; (präklinische Studie an Ratten mit intravesikalen Antimuskarinika). 5. De Wachter S. and Wyndaele J.J. J Urol. 2003; 169(5):1892–1895; (präklinische Studie an Ratten mit intravesikalem Oxybutynin). 6 Pannek J. et al. Urology. 2000; 55(3):358–362; (prospektive Open-Label-Studie mit intravesikalem Oxybutynin in Kombination mit oralem Oxybutynin, n = 25). 7. Humblet M. et al. Neurourol Urodyn. 2015; 34(4):336–342; (retrospective Kohortenstudie mit intravesikalem Oxybutynin, n = 10 bei Re-Evaluation). 8. Krause P. et al. J Urol. 2013; 190(5):1791–1797; (prospektive, randomisierte Cross-Over Open-Label-Studie (Periode I und II: orales oder intravesikales Oxybutynin, Periode III: intravesikales Oxybutynin), n = 20). 9. Buyse G. et al. 1998; 160(3 Pt 2):1084–1087; discussion 1092; (prospektive Open-Label-Studie mit intravesikalem Oxybutynin, n = 15). 10. Oki T. et al. J Urol. 2004; 172(5 Pt 1): 2059–2064; (präklinische Studie an Ratten mit oralem und intravesikalem Oxybutynin). 11. Schröder A. et al. Neurourol Urodyn. 2016; 35(5):582–588; (randomisierte, prospektive, aktiv kontrollierte, multizentrische Open-Label-Studie mit intravesikalem Oxybutynin (n = 18) und oralem Oxybutynin (n = 17)).
VESOXX 1 mg/ml, Lösung zur intravesikalen Anwendung. Wirkst.: Oxybutyninhydrochlorid. Zus.: 1 ml Lös. enth. 1 mg Oxybutyninhydrochlorid; 1 skalierte Fertigspritze m. 10 ml Lös. enth. 10 mg Oxybutyninhydrochlorid. Sonst. Bestandt.: Salzsäure, Natriumchlorid, Wasser f. Inj.-zwecke. Anw.: Zur Unterdrück. einer neurogenen Detrusorüberaktivität (Neurogenic Detrusor Overactivity; NDO) b. Kdrn. ab 6 J. u. b. Erw., die ihre Blase mittels sauberer intermittierender Katheterisier. (CIC) entleeren, wenn sie durch eine Beh. m. oralen Anticholinergika aufgrund mangelnder Wirksamkeit und/oder unerträglicher Nebenwirk. nicht adäquat eingestellt werden können. Gegenanz.: Überempfindlichk. gg. d. Wirkst. od. sonst. Bestandt.; schwere gastrointest. Erkrank. (z. B. schwere Colitis ulcerosa u. tox. Megakolon); Myasthenia gravis; Engwinkelglaukom u. Pat. m. einem Risiko dafür; begleit. Sauerstoffther. Nebenwirk.: Harnwegsinfekt.; asymptomat. Bakteriurie; Hyperprolaktinämie; Prolaktin erhöht; Teilnahmslosigk.; Halluzinat.; kognitive Stör.; Hyperaktivität; Schlaflosigk.; Schlafstör.; Agoraphobie; Orientierungsstör.; Aufmerksamkeitsstör.; Schwindelgefühl; Kopfschmerz; Somnolenz; Erschöpf.; Dysgeusie; getrübter Bewusstseinszustand; Bewusstlosigk.; anticholinerges Syndr.; Krampfanfall; Vertigo; Trockenes Auge; anomale Sinnesempfind. d. Auges; Akkommodationsstör.; supraventrik. Tachykardie; Hypotonie; Gesichtsröt.; Obstipat.; Mundtrockenh.; abdominale Beschwerden; Schmerzen im Unter- od. Oberbauch; Übelk.; Dyspepsie; Diarrhö; Hypohidrose; Ausschlag; nächtl. Schwitzen; (verstärkter) Harndrang; Proteinurie; Hämaturie; Stör. b. d. Entleer. d. Harnblase; Schmerzen an d. Instill.-stelle; Durst; Brustkorbbeschwerden; Kältegefühl. Verring. Sauerstoffsätt. im Rahmen einer Sauerstoffther. Bek. NW einer anticholinergen Ther. (bisher b. intravesikaler Anw. v. Oxybutynin nicht beob.): Erbrechen; Anorexie; vermind. Appetit; Dysphagie; gastroösophag. Refluxkrankh.; Pseudoobstrukt. b. Risikopat. (ältere Personen od. Pat. m. Obstipat. u. b. Behandl. m. and., die intest. Motilität verring. AM); Verwirrth.-zustand; Agitiert.; Angst; Alpträume; Paranoia; Sympt. einer Depress.; Abhängigk. v. Oxybutynin (b. Pat. m. einer Vorgeschichte v. Drogen- od. Substanzmissbrauch); Arrhythmie; Hitzschlag; (Engwinkel-) Glaukom; Augeninnendruck erhöht; trockene Haut; Angioödem; Urtikaria; Photosensitivität; Überempfindlichk. Kdr. könnten empfindlicher f. d. Wirk. d. Produktes sein, insbes. in Hinblick auf psychiat. u. d. ZNS betreff. NW. Warnhinw.: Enth. d. sonst. Bestandteil m. bek. Wirk. Natrium (3,56 mg/ml). Weit. Angaben: s. Fach- u. Gebrauchsinfo. Verschreibungspflichtig.
FARCO-PHARMA GmbH, Gereonsmühlengasse 1-11, D 50670 Köln. Stand: 08/2022.
VESOXX® (1 mg/ml Oxybutynin-HCl) – die intravesikale Lösung zur Behandlung der neurogenen Detrusorüberaktivität1 Alle Informationen zu Verordnung und Erstattung finden Sie unter www.vesoxx.de
VESOXX® für NDO Patient*innen – unabhängig ihrer Grunderkrankung z. B. Spina bifida, Querschnittlähmung, Multiple Sklerose, Morbus Parkinson
NEU:
Dieses Editorial ist insofern ein No vum in der langen Geschichte die ses Journals, als es nahtlos an das Vorhergehende anschließt. Dieses endete mit der spekulativen Frage, ob die traditionelle Kurortmedizin mit ihrem regulationsmedizinischen Ansatz und ihrem staatlich prädi katisierten „Healing Environment“ für die an Long COVID Erkrankten sowie für die Volkswirtschaft einen segensreichen Beitrag leisten kann. In den vergangenen Monaten war ich an mehreren Studien zum Thema Long-COVID-/Post-COVIDSyndrom beteiligt. Logisch, dass da beständig vieles gedanklich vor sich hin schwelt, das durch eine „pas sende“ neue Veröffentlichung dann unmittelbar Feuer fängt.
Die Zeitschrift Lancet etwa veröf fentlichte bereits im Juni dieses Jah res eine Studie von Forschern des King’s College in London, in der In formationen von Nutzern einer weit verbreiteten Gesundheits-App ana lysiert wurden. Der Vergleich von gut 40.000 Teilnehmern, bei denen eine Infektion mit der Delta-Variante zu grunde lag, mit 56.000 Teilnehmern, die sich mit der Omikron-Variante infiziert hatten, ergab, dass die Häu figkeit einer Long-COVID-Symp tomatik nach Omikron-Infektion (4,5 % der Infizierten) nur etwa halb so hoch lag wie nach Infektion mit der Delta-Variante (10,8 %) – und das weitgehend unabhängig vom Alter bzw. dem Zeitraum seit der letzten Impfung [1].
Überträgt man diese Zahlen auf die Zahl der gemeldeten Infektionen in Deutschland, dann wären bis Ende 2021 etwa 650.000 der bis dahin dem RKI gemeldeten gut 6 Millionen COVID-19-Fälle von Long COVID betroffen. Ende August 2022 betrug die Zahl der insgesamt gemeldeten Fälle in Deutschland gut 32 Millio nen, also 26 Millionen gemeldete Fälle allein im Jahr 2022, wobei die meisten Epidemiologen davon aus gehen, dass die Omikron-Dunkel ziffer mindestens ebenso hoch sein dürfte. Demnach wäre mit 1,2 2,5
Back to the roots: Long-COVID-Therapie am Kurort
Millionen Long-COVID-Patienten nach Omikron-Infektion zu rechnen, die zu den 650.000 nach Infektion 2020 bzw. 2021 hinzukämen. Und das RKI meldet immer noch Inzi denzzahlen für Neuinfektionen um die 200 (Basis: ausschließlich PCRDiagnosen) und erwartet ein deutli ches Ansteigen im Herbst. Erkrankungen nach einer OmikronInfektion sind typischerweise kli nisch deutlich weniger schwerwie gend, was möglicherweise auch mit dem weithin besseren Immunstatus im Vergleich zu den Vorjahren zu tun haben könnte. Dass dies aber nicht notwendigerweise für die LongCOVID-Symptomatik gilt, darauf deuten u.a. die Ergebnisse einer so eben auf einem Preprint-Server vor veröffentlichten Studie hin [2]. Die Daten ließen auch erkennen, dass sich die bunte Vielfalt der Sympto me 3 Grundtypen („Clustern“) zu ordnen lässt: neurologische Symp tome (weiterhin die größte Gruppe), Schädigungen an den Atemwegen (abnehmende Tendenz assoziiert mit zunehmender Impfquote) so wie Störungen im Bereich des Im munsystems, verbunden mit einer breiten Vielfalt an Symptomen von abdominalen Beschwerden über muskuloskelettale Schmerzen bis hin zu Erschöpfungszuständen und unspezifischen Beeinträchtigungen des Befindens [3]. Dieses SymptomMuster entspricht weitgehend dem nach einer Infektion mit einer der vorher dominierenden Varianten [4]. Insbesondere in der sich inzwischen abzeichnenden Problematik eines aus dem Gleichgewicht geratenen Immunsystems könnte ein essenzi eller Schlüssel für die Behandlung von Long COVID liegen.
Prof. Dr. med. Karl-Ludwig Resch
Schon in den ersten Monaten der Pandemie wurde klar, dass nicht so sehr die viralen Attacken auf einzelne Zellen, sondern die über schießende Gegenreaktion des Im munsystems (Zytokinsturm) eine Infektion gefährlich machen. Folge richtig erwies sich die ex juvantibus angewandte Gabe von Kortison als vergleichsweise viel versprechend. Eine ebenfalls gerade zugänglich gemachte Vorveröffentlichung [5] beschreibt eine wissenschaftlich verlässliche Basis für diese plausib len Überlegungen. Spätestens hier kommt die oben erwähnte Kurortmedizin mit ihrem regulationsmedizinischen Ansatz ins Spiel. Über viele Jahrzehnte konn ten empirische Untersuchungen wie kontrollierte klinische Studien zeigen, dass der balneologische Ansatz auf eine intrinsische Neu ordnung/Adaptation des Immunsys tems wirkt. Exemplarisch sei hier der Kneipp’sche Ansatz genannt, insbe sondere aber die Radonbalneologie,

JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH
EDITORIAL
97
die mit minimalen Wirkdosen modu lierend auf die Langerhans-Zellen der Haut sowie auf dentritische Zel len im Darm, den Lungen und ande ren Körpersystemen einwirkt. Was diese Erkenntnisse besonders über zeugend macht, sind Untersuchun gen in einem völlig anderen Kontext wie die eben erschienenen Studien zur Wirksamkeit von BCG-Impfun gen zur Corona-Prävention [6] oder zur Langzeit-präventiven Wirksam keit einer gezielten Exposition mit Allergenen zur Prävention juveniler Allergien [7].
Karl-Ludwig Resch, Nürnberg
ÜBERSICHTSARBEIT
Neurogene Detrusorüberaktivität: Hoher Druck in der Blase = hoher Leidensdruck für die Patienten 100 Brigitte Söllner
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Bronchodilatatoren: Zweimalgabe bessert nächtliche und morgendliche COPD-Symptome 105
Secukinumab bei Spondyloarthritiden –ein früher Behandlungsbeginn kann Therapieansprechen verbessern 106
Quellen
1 Antonelli M, Pujol JC, Spector TD et al. Risk of long COVID associated with delta versus omicron variants of SARS-CoV-2. Lancet 2022;399:2263-2264. PMID: 35717982
2 Canas LS, Molteni E, Deng J et al. Pro filing post-COVID syndrome across dif ferent variants of SARS-CoV-2. https:// www.medrxiv.org/content/10.1101/2022 .07.28.22278159v1.full.pdf
3 https://www.medicalnewstoday.com/ articles/what-are-the-3-main-types-oflong-covid
4 Han Q, Zheng B, Daines L et al. Longterm sequelae of COVID-19: a systema tic review and meta-analysis of one-year follow-up studies on post-COVID sym ptoms. Pathogens 2022;11:269. PMID: 35215212
5 Klein J, Wood J, Jaycox J et al. Distingu ishing features of Long COVID identified through immune profiling. https://www. medrxiv.org/content/10.1101/2022.08.0 9.22278592v1.full.pdf
6 Faustman DL, Lee A, Hostetter ER et al. Multiple BCG vaccinations for the pre vention of COVID-19 and other infec tious diseases in type 1 diabetes. Cell Rep Med 2022:100728. PMID: 36027906
7 Strieker S, Weinmann T, Gerlich J et al. Farm living and allergic rhinitis from childhood to young adulthood: Pros pective results of the GABRIEL study. J Allergy Clin Immunol 2022;S00916749(22)00886-7. PMID: 35779667
PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG
ACP-Therapie in Orthopädie und Sportmedizin: Innovative Operationsstrategien mit AutoCart™ 108
Kastrationsresistenz beim Prostatakarzinom vermeiden 111
NEUE UND BEWÄHRTE ARZNEIMITTEL
Upstaza™ – die erste krankheitsmodifizierende Gentherapie für den AADC-Mangel 112
Eptinezumab erweitert die Optionen zur Migräneprophylaxe 114
Atopische Dermatitis: JAK1-Inhibitor Abrocitinib verbessert Hautläsionen und Juckreiz 115
Mittelschwere bis schwere Plaque-Psoriasis: Wechsel auf Bimekizumab kann sich lohnen 117
RUBRIKEN
Wissenswertes 104, 107, 110, 138 Kongresse 119
©
PERFUSION GMBH 98 INHALT
JOURNAL
VERLAG
*Zur Prophylaxe von Schlaganfällen und systemischen Embolien bei erwachsenen Patient:innen mit nicht-valvulärem Vorho immern und einem oder mehreren Risikofaktoren bietet ELIQUIS® (Apixaban) eine signifikant überlegene Wirksamkeit und signifikant weniger schwere Blutungen vs. Warfarin.1,2
Literaturangaben: 1. Granger CB, et al. N Engl J Med. 2011;365(11):981–92. 2. ELIQUIS® Fachinformation, aktueller Stand. Eliquis 2,5 mg Filmtabletten. Eliquis 5 mg Filmtabletten. Wirkstoff: Apixaban. Zusammensetzung: Wirksto : 2,5 mg bzw. 5 mg Apixaban. Sonst. Bestandteile: Lactose, Mikrokristalline Cellulose, CroscarmelloseNatrium, Natriumdodecylsulfat, Magnesiumstearat, Lactose-Monohydrat, Hypromellose, Titandioxid, Triacetin, Eliquis 2,5 mg zusätzlich: Eisen(III)-hydroxid-oxid x H2O; Eliquis 5 mg zusätzlich: Eisen(III)-oxid. Anwendungsgebiete: Prophylaxe v. Schlaganfällen u. systemischen Embolien bei erw. Pat. mit nicht-valvulärem Vorho immern u. einem o. mehreren Risikofaktoren, wie Schlaganfall o. TIA in der Anamnese, Alter ≥75 Jahren, Hypertonie, Diabetes mellitus, symptomatische Herzinsu zienz (NYHA Klasse ≥II). Behandlung v. tiefen Venenthrombosen (TVT) u. Lungenembolien (LE) sowie Prophylaxe v. rezidivierenden TVT und LE bei Erw. Eliquis 2,5 mg zusätzlich: Prophylaxe venöser Thromboembolien bei erw. Pat. nach elektiven Hü - o. Kniegelenksersatzoperationen. Gegenanzeigen: Überempfindlichkeit gg. den Wirksto o.e.d. sonst. Bestandteile; akute klinisch relevante Blutung; Lebererkrankungen, die mit einer Koagulopathie u. einem klinisch relevanten Blutungsrisiko verbunden sind. Läsionen o. klinische Situationen, falls sie als signifikanter Risikofaktor für eine schwere Blutung angesehen werden (z.B. akute o. kürzl. aufgetretene gastrointestinale Ulzerationen, maligne Neoplasien m. hohem Blutungsrisiko, kürzl. aufgetretene Hirn- o. Rückenmarksverletzungen, kürzl. erfolgte chirurgische Eingri e an Gehirn, Rückenmark o. Augen, kürzl. aufgetretene intrakranielle Blutungen, bekannte o. vermutete Ösophagusvarizen, arteriovenöse Fehlbildungen, vaskuläre Aneurysmen o. größere intraspinale o. intrazerebrale vaskuläre Anomalien). Gleichzeitige Anwendung anderer Antikoagulanzien z.B. UFH, niedermol. Heparine, Heparinderivate, orale Antikoagulanzien außer bei Umstellung der Antikoagulation oder mit UFH in Dosen um die Durchgängigkeit e. zentralvenösen o. arteriellen Katheters zu erhalten oder während einer Katheterablation. Nebenwirkungen: Häufig: Anämie, Thrombozytopenie; Blutungen am Auge (einschließlich Bindehautblutung); Blutungen, Hämatome, Hypotonie (einschließlich Blutdruckabfall während des Eingri s); Epistaxis; Übelkeit, Gastrointestinale Blutung, Blutung im Mundraum, Rektalblutung, Zahnfl eischblutung; erhöhte Gamma-Glutamyltransferase, erhöhte Alanin-Aminotransferase; Hautausschlag; Hämaturie; Abnormale vaginale Blutung, urogenitale Blutung; Kontusion. Gelegentlich: Überempfindlichkeitsreaktionen, allergisches Ödem, anaphylaktische Reaktion, Pruritus; Gehirnblutung; Intraabdominalblutung; Hämoptyse; Hämorrhoidalblutung, Hämatochezie; abnormale Leberfunktionstests, erhöhte Aspartat-Aminotransferase, erhöhte Blutwerte für alkalische Phosphatase, erhöhte Blutwerte für Bilirubin; Alopezie, Muskelblutung; Blutung an der Applikationsstelle; Okkultes Blut positiv; Postoperative Blutung (einschließlich postoperatives Hämatom, Wundblutung, Hämatom an Gefäßpunktionsstelle und Blutung an der Kathetereinstichstelle), Wundsekretion, Blutungen an der Inzisionsstelle (einschließlich Hämatom an der Inzisionsstelle), intraoperative Blutung, Traumatische Blutung. Selten: Blutung der Atemwege; Retroperitoneale Blutung. Sehr selten: Erythema multiforme. Nicht bekannt: Angioödem, kutane Vaskulitis. Weitere Hinweise: siehe Fachinformation. Verschreibungspflichtig. Pharmazeutischer Unternehmer: Bristol-Myers-Squibb/Pfizer
Corporate Park 2 - Dublin 15, D15 T867, Irland. v14
-




















































































































432-DE-2200140
EEIG, Plaza
MEINE PATIENT:INNEN MEINEN MENTOR MEINE MUTTER FÜR MICH SELBST Zur Schlaganfallprophylaxe bei VHF-Patient:innen* ELIQUIS®: WEIL MIR WIRKSAMKEIT UND SICHERHEI T*, 1, 2 WICHTIG SIND
254
Blanchardstown
Die neurogene Detrusorüber aktivität (Neurogenic De trusor Overactivity, NDO) aufgrund von Störungen des zen tralen und/oder peripheren Ner vensystems ist eine große Heraus forderung in der Urologie, da die zugrunde liegenden Pathomecha nismen komplex sind und die Be handlung der NDO in das Manage ment einer Multisystemerkrankung integriert werden muss. Verursacht werden kann die chronische Bla senfunktionsstörung durch eine Rückenmarksverletzung oder eine Spina bifida sowie unterschied liche Grunderkrankungen (z.B. Schlaganfall, Morbus Parkinson, Multiple Sklerose), die das Ner vensystem betreffen, wobei die Intensität von der Schädigung des zentralen oder peripheren Nerven systems abhängt.
Die Inzidenz- und Prävalenzraten der NDO sind schwer zu ermit teln, da es nur wenige valide epi demiologische Berichte gibt. Eine systematische Untersuchung der Epidemiologie zeigte z.B. durch schnittliche Prävalenzraten für die NDO von 49,7 % nach einer Rückenmarksverletzung, 58,2 % für Patienten mit MS, 64,7 % nach einem Schlaganfall und 58,6 % für Patienten mit Morbus Parkinson [1]. Nur ca. 12 % der Neugebore nen mit einer Meningomyelozele haben post partum keine neuroge nen Störungen des unteren Harn trakts [2].
Eine Erkrankung mit schwerwiegenden Folgen
Die NDO geht mit einem erhöhten Risiko schwerwiegender Folgeer krankungen einher [3]. Unregulier te, ungedämpfte Kontraktionen des Detrusormuskels und der Anstieg des intravesikulären Drucks verur
Neurogene Detrusorüberaktivität:
Hoher Druck in der Blase = hoher Leidensdruck für die Patienten
Brigitte Söllner, Erlangen
sachen die Kardinalsymptome der NDO: Pollakisurie, Nykturie und Urgency (häufiges bis ständiges und äußerst unangenehmes Harn dranggefühl). Harnwegsinfekte und unkontrollierter Harnabgang sind darüber hinaus die häufigsten klinischen Symptome einer NDO. Der erhöhte Blasendruck gefährdet den oberen Harntrakt, da er infolge eines vesikoureteralen Refluxes zu einem irreversiblen Nierenschaden bis hin zum Nierenversagen füh ren kann [4]. Für die Betroffenen bedeutet das eine signifikant redu zierte Lebensqualität und bei nicht optimaler Therapie auch eine ver kürzte Lebenserwartung [5].
Diagnose der NDO
Die Diagnostik umfasst neben der klinischen Untersuchung des Pa tienten die laborchemische Über prüfung der Nierenwerte, mikro skopische und mikrobiologische Urinkontrollen, Ultraschall des gesamten Urogenitaltrakts sowie eine Nierenfunktionstestung (Nie renszintigraphie). Wichtig ist der Ausschluss nicht neurogener Ursa chen der Blasenfunktionsstörung. Entscheidend für die Diagnose der NDO ist unter anderem die Bla
sendruckmessung, idealerweise mit simultaner röntgenologischer Darstellung des unteren Harntrakts (Videourodynamik) [6, 7]. Die urodynamische Untersuchung ist essenziell für den Nachweis einer Funktionsstörung und dient dazu, auf Basis der urodynamischen Zielgrößen die Therapie einzu stellen und den Therapieerfolg zu kontrollieren. Dabei gilt die Video urodynamik als Goldstandard und wird von den Leitlinien sowohl für die Diagnostik als auch für die Therapieeinstellung und Therapie kontrolle der NDO empfohlen [2, 6, 7, 8].
Empfehlungen der S2k-Leitlinie zur Therapie der NDO Therapieziele sind die Schaffung einer ausreichenden Speicherfunk tion, eine Reduzierung der Harn wegsinfekte sowie der Schutz des oberen Harntrakts mit Erhalt der Nierenfunktion [5]. Die Optimie rung der Blasenentleerung und der Harnkontinenz führt insgesamt zu einer deutlichen Verbesserung der Lebensqualität. Als Erstlinientherapie bei neuro gener Überaktivität des Detrusors empfiehlt die aktuelle S2k-Leit
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 100 ÜBERSICHTSARBEIT
Abbildung 1: Mögliches Vorgehen bei der Therapie der NDO im Einklang mit den Leitlini en (mod. nach [2, 7, 8]). * Voraussetzung für die intravesikale medikamentöse Therapie mit Vesoxx® ist der aseptische intermittierende Katheterismus [9].
linie [7] die orale Gabe von An timuskarinika (Anticholinergika) wie Oxybutynin, Propiverin oder Trospium (Abb. 1). Für diese Me dikamentengruppe gibt es eine lan ge Anwendungserfahrung. Werden die Therapieziele, wie Schutz des oberen Harntrakts, Verbesserung der Kontinenzsi tuation und der Lebensqualität, damit nicht zufriedenstellend er reicht oder treten nicht tolerierba re Nebenwirkungen auf, sollten Patienten, die ihre Blase mittels aseptischer intermittierender Ka theterisierung (ISK) entleeren, als Zweitlinientherapie intravesikales Oxybutynin (Vesoxx®) erhalten, bevor minimalinvasive und opera tive Verfahren eingesetzt werden (Abb. 1) [7]. Um den Therapierfolg zu überwachen, müssen urodyna mische Parameter in regelmäßigen Abständen gemäß den Vorgaben des behandelnden Urologen kont rolliert werden.
Die S2k-Leitlinie schätzt die Wirk samkeit und Sicherheit der intra vesikalen Gabe von Oxybutynin als erwiesenermaßen effizient, sicher und gut tolerabel ein. Die Dosierung soll sich laut Leitli nienempfehlung an der klinisch überprüften Wirksamkeit (z.B. Blasenentleerungsprotokoll) und/ oder der urodynamisch überprüf
ten Wirksamkeit sowie der indi viduellen Verträglichkeit orientie ren. Darüber hinaus empfiehlt die Leitlinie eine Kombination von Instillation und oraler antimuskari nerger Therapie, sofern sich diese als effektiv erweisen und weniger unerwünschte Nebenwirkungen zeigen [7].
Einschätzung der S2k-Leitlinie zur oralen vs. intravesikalen Gabe von Oxybutynin
Die Autoren der Leitlinie weisen in ihrem Report auf folgende Vor teile der intravesikalen im Ver gleich zur oralen Gabe von Oxy butynin hin:
• Unter der intravesikalen im Vergleich zur oralen Gabe zeig te sich eine signifikant höhere Bioverfügbarkeit (294 %; 90%KI: 211–408) bei geringerer Nebenwirkungsrate [10].
• Die höhere Bioverfügbarkeit des aktiveren Oxybutynin-REnantiomers kann zu einer hö heren Effizienz führen [11].
• Die Vorteile der intravesikalen Applikation sind durch die Um gehung des First-Pass-Effektes in der Leber und nachfolgend durch die geringere Konzentra tion des aktiven Metaboliten N-
Desethyloxybutynin (NDEO) im Serumplasma zu erklären, da dieser hauptsächlich für die Entstehung von Nebenwirkun gen verantwortlich gemacht wird [12].
Daher kann der frühzeitige Wech sel auf die intravesikale Therapie bei zu starken Nebenwirkungen oder nicht ausreichender Wirksam keit eine sinnvolle ZweitlinienOption darstellen [7].
Klinische Überlegenheit von intravesikalem Oxybutynin
Wirksamkeit, Sicherheit und Ver träglichkeit der intravesikalen Oxybutynin-Behandlung wurden im Vergleich zur oralen Gabe in einer randomisierten, prospekti ven, aktiv kontrollierten, offenen, multizentrischen Studie mit 35 erwachsenen Patienten mit NDO untersucht [13].
Die Patienten mit intravesikaler Behandlung (n = 18) erhielten 3 × täglich 10 ml 0,1 % Oxybutynin hydrochlorid (Vesoxx®) intravesi kal, die oral behandelten Patienten (n = 17) 3 × täglich 5 mg Oxybu tyninhydrochlorid oral über einen Zeitraum von 28 Tagen. Primärer Wirksamkeitsendpunkt war die Veränderung der maximalen Bla senkapazität zwischen dem Beginn der Studie und nach 4 Wochen, be wertet mittels urodynamischer Un tersuchungen.
Die intravesikale Applikation führte zu einer Steigerung der maximalen Blasenkapazität um 117 ml nach 28 Tagen Behand lungszeit versus 18 ml bei der oralen Applikation. Die Differenz erwies sich als statistisch signifi kant (p = 0,0086) (Abb. 2). Uner wünschte Ereignisse (UEs) wurden von 10 Patienten (55,6 %) mit int ravesikaler und von 14 Patienten

JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 101ÜBERSICHTSARBEIT
Signifikanter Unterschied der Blasenkapazität zwischen beiden Behandlungsarmen
Abbildung 2: Die intravesikale Applikation von Oxybutynin (Vesoxx®) war der oralen Gabe hinsichtlich des primären Wirksamkeitsendpunkts, der maximalen Blasenkapazität nach 28-tä giger Behandlung, signifikant überlegen (mod. nach [13]). # Differenz der maximalen Blasen kapazität von Visite an Tag 28 und Visite an Tag 0.
Intravesikales
Oxybutynin
0,1 % Oxybutynin-HCl (Vesoxx®) ist das erste in Deutschland zuge lassene Arzneimittel zur intravesikalen Therapie der neurogenen Detrusorüberaktivität infolge einer Rückenmarksverletzung oder Meningomyelozele (Spina bifida). Es wird angewendet bei Kindern ab 6 Jahren und Erwachsenen, die ihre Blase mittels aseptischer intermittierender Katheterisierung (ISK) entleeren und die nicht adäquat mit oralen Anticholinergika eingestellt sind. Intravesikales Oxybutynin verfügt über einen multimodalen Wirk mechanismus: Der Wirkstoff blockiert die Muskarinrezeptoren und verringert dabei auch die präsynaptische Ausschüttung von Ace tylcholin, was zu einer Entspannung der glatten Blasenmuskulatur führt. Außerdem wirkt Oxybutynin durch seinen kalziumantago nistischen Effekt spasmolytisch am Detrusormuskel, sodass sich die Anzahl der Kontraktionen und damit der Detrusordruck signifikant verringert. Indem es am Urothel afferente C-Fasern inhibiert, hat es auch eine lokalanästhetische Wirkung [9].

(82,4 %) mit oraler Verabreichung berichtet. Signifikante Unterschie de zugunsten der intravesikalen Instillation wurden bei UEs beob achtet, die das Sehvermögen (1/10 vs. 9/14), den Gastrointestinaltrakt (8/10 vs. 14/14), das Nervensys tem (2/10 vs. 8/14) und die Haut und Unterhaut (1/10 vs. 6/14) be trafen. Es traten keine schwerwie genden unerwünschten Arzneimit telwirkungen auf [13].
Wirksamkeit und Verträglichkeit von intravesikalem Oxybutynin bei Kindern

In einer prospektiven Open-LabelStudie mit 15 Kindern erzielte in travesikales Oxybutynin (0,1 %) eine signifikante Erhöhung der Blasenkapazität von 114 ± 15,2 ml auf 161 ± 26,6 ml nach 4 Monaten (p < 0,01) bzw. auf 214 ± 21,7 ml nach 24 Monaten (p < 0,01) [14]. Gute Ergebnisse bei pädiatrischen Patienten lieferte auch eine retro spektive Kohortenstudie mit 10 Kindern, die die Wirkung von in travesikalem Oxybutynin (0,1 %) nach 15-jähriger Therapie unter suchte. Die Kinder wurden vor Studienbeginn ±2 Jahre lang mit oralem Oxybutynin behandelt. Die Blasen-Compliance hatte sich nach 15 Jahren signifikant verbessert und der mittlere Blasendruck bei voller Blase verringerte sich von 52,5 ± 24 auf 24,5 ± 14,4 cmH2O. Die Behandlung wurde insgesamt gut vertragen und die jungen Pa tienten zeigten sich zufrieden mit ihrem Kontinenzstatus [15].
In einer Meta-Analyse wurde die Wirksamkeit von intravesikalem Oxybutynin bei 297 Kindern mit neurogener Blasenfunktionsstö rung überprüft, die refraktär ge genüber oralem Oxybutynin waren oder dieses nicht vertragen hatten.
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 102 ÜBERSICHTSARBEIT
Multimodaler Wirkmechanismus von intravesikalem Oxybutynin (mod. nach [18]).
3 2 1
In den 8 eingeschlossenen Studi en (2 prospektiv, 6 retrospektiv) hatten fast alle Kinder eine Me ningomyelozele und wurden mit einer durchschnittlichen Dosis von 10 mg/d Oxybutynin intravesikal behandelt. Die mittlere Verände rung der Blasen-Compliance mit intravesikalem Oxybutynin wurde lediglich in 2 Studien gemessen: jeweils 7,4 ml/cmH2O und 7,5 ml/ cmH2O, die gepoolte mittlere Dif ferenz der maximalen Blasenka pazität zeigte eine Verbesserung um 78 ml. Die gepoolte durch schnittliche Veränderung des Bla sendrucks bei terminaler Kapazität reduzierte sich um 16,4 cmH2O und die Inkontinenz verbesserte sich in den meisten Studien, mit Trockenheits-/Besserungsraten von 61 83 %. Die Inzidenz von Nebenwirkungen unter intravesi kalem Oxybutynin war geringer als die veröffentlichten Inzidenz werte unter Anwendung oraler An timuskarinika. Lediglich 9 % der Patienten brachen die Behandlung aufgrund von Nebenwirkungen ab [16].
Einfache Anwendung und Integration in die tägliche ISK-Routine
Patienten, die ihre Blase mittels aseptischer ISK entleeren, können die Behandlung mit intravesikalem Oxybutynin (Vesoxx®) einfach in ihren Alltag integrieren (Abb. 3). Das Anticholinergikum wird nach der Katheterisierung ein bis mehr mals täglich in die Blase instilliert. Dazu wird nach der vollständigen Blasenentleerung ein separater Stufenkegeladapter auf eine Fer tigspritze geschraubt, welche die Oxybutynin-Lösung enthält, und anschließend mit dem Katheter verbunden. Nun kann die erforder
Abbildung 3: Unkomplizierte Anwendung von intravesikalem Oxybutynin im Rahmen der täg lichen ISK-Routine. Mit einer Konzentration von 0,1 % Oxybutyninhydrochlorid kann durch die Instillation von nur 10 ml eine Dosis von 10 mg erreicht werden [9].
liche Dosis direkt in die Blase ap pliziert werden. Die einfache An wendung fördert die Therapietreue und damit den Therapieerfolg. Für die Patienten bedeutet eine erfolgreiche Therapie eine er hebliche Erleichterung ihres Alltags, vor allem durch gerin geren unwillkürlichen Urinver lust, weniger Harnwegsinfekte und größere Blasenvolumina. Intravesikales Oxybutynin hilft den Betroffenen nicht nur, die Blasenentleerung zu kontrollie ren, sondern reduziert auch das Schmerzempfinden in der Blase [17]. Durch die Unterdrückung der Detrusorüberaktivität wer den das Auftreten von Harn wegsinfektionen und das Risiko einer Nierenschädigung redu ziert – eines der wichtigsten Zie le im Therapiemanagement von Patienten mit neurogener Detru sorüberaktivität.
Literatur
1 Ruffion A, Castro-Diaz D, Patel H et al. Systematic review of the epidemiology of urinary incontinence and detrusor overac tivity among patients with neurogenic overactive bladder. Neuroepidemiol 2013;41:146-155
2 Stein R, Assion C, Geißler AB et al. S2k Leitlinie der AWMF: Diagnostik und Therapie der neurogenen Blasenfunkti onsstörungen bei Kindern und Jugendli chen mit spinaler Dysraphie. AWMF-Re gister-Nr. 043–047, Update 2019
3 Van Ophoven A. Diagnose und Therapie neurogener Blasenfunktionsstörungen. CME-Verlag 2019
4 Haab F. Chapter 1: The conditions of neurogenic detrusor overactivity and overactive bladder. Neurourol Urodyn 2014;33 (Suppl 3):S2-S5
5 Kurze I. Neurogene Blasenfunktionsstö rungen (nBFS) beiSpina bifida – aktuelle Behandlungsmöglichkeiten. Teil 1: Kon servative und minimal invasive Therapie. ASBH Brief 2012;04:34-41
6 Haensch CA, Jost W, Kaufmann A et al. Diagnostik und Therapie von neurogenen Blasenstörungen. S1-Leitlinie 2020. In: Deutsche Gesellschaft für Neurologie e.V. (Hrsg.): Leitlinien für Diagnostik und Therapie in der Neurologie. Online: Leitlinien Archiv (dgn.org)

JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 103ÜBERSICHTSARBEIT
7 Kutzenberger J et al. Medikamentöse Therapie der neurogenen Dysfunktion des unteren Harntraktes (NLUTD) Ent wicklungsstufe: S2k, DGU 2022. AWMF-Register-Nr. 043–053
8 EAU Guidelines on Neuro-Urology 2020. Online: https://uroweb.org/guidelines/ neuro-urology
9 Fachinformation Vesoxx®; Stand: Juli 2020
10 Krause P, Fuhr U, Schnitker J et al. Phar macokinetics of intravesical versus oral oxybutynin in healthy adults: results of an open label, randomized, prospective clinical study. J Urol 2013;190:17911797
11 Kretschmar M, Suleiman AA, Krause P et al. A population pharmacokinetic model of (R)- and (S-) oxybutynin and its active metabolites after oral and intravesical ad ministration to healthy volunteers. J Clin Pharmacol 2021;61:961-971
12 Buyse G, Waldeck K, Verpoorten C et al. Intravesical oxybutynin for neurogenic bladder dysfunction: less systemic side effects due to reduced first pass metabo lism. J Urol 1998;160:892-896
13 Schröder A, Albrecht U, Schnitker J et al. Efficacy, safety, and tolerability of intra vesically administered 0.1% oxybutynin hydrochloride solution in adult patients with neurogenic bladder: A randomized, prospective, controlled multi-center trial. Neurourol Urodyn 2016;35:582-588
14 Buyse G, Verpoorten C, Vereecken R et al. Intravesical application of a stable oxybutynin solution improves therapeutic compliance and acceptance in children with neurogenic bladder dysfunction. J Urol 1998;160:1084-1087
15 Humblet M, Verpoorten C, Christiaens MH et al. Long-term outcome of intrave sical oxybutynin in children with detru sor-sphincter dyssynergia: with special
reference to age-dependent parameters. Neurourol Urodyn 2015;34:336-342
16 Guerra LA, Moher D, Sampson M et al. Intravesical oxybutynin for children with poorly compliant neurogenic bladder: a systematic review. J Urol 2008;180:10911097
17 De Wachter S, Wyndaele JJ. Intravesical oxybutynin: a local anesthetic effect on bladder C afferents. J Urol 2003;169: 1892-1895
18 Reitz A, Schurch B. Intravesical therapy options for neurogenic detrusor overacti vity. Spinal Cord 2004;42:267-272
Anschrift der Verfasserin: Brigitte Söllner Medizinjournalistin und Wissenschaftliche Lektorin Lärchenweg 10 91058 Erlangen brigitte.soellner@online.de
Zulassungserweiterung für Enhertu® erlaubt früheren Einsatz beim HER2positiven metastasierten Mammakarzinom
Im Juli 2022 hat die Europäische Kommission die Zulassung für Enhertu® (Trastuzumab-Deruxte can, T-DXd) erweitert: Das gegen HER2 gerichtete Antikörper-Wirk stoff-Konjugat kann nun als Mo notherapie bei erwachsenen Pati entinnen mit nicht resezierbarem oder metastasiertem HER2-posi tivem Mammakarzinom, die zu vor mindestens eine gegen HER2 gerichtete Vortherapie erhalten ha ben, eingesetzt werden.
Die Zulassungserweiterung basiert auf den Ergebnissen der PhaseIII-Studie DESTINY-Breast03, in der T-DXd das Risiko für Krank heitsprogression oder Tod im Ver gleich zu Trastuzumab-Emtansin (T-DM1) um 72 % reduzierte (HR: 0,28; 95%-KI: 0,22 0,37; p < 0,000001). Untersucht wurden Patientinnen mit HER2-positivem
nicht resezierbarem und/oder me tastasiertem Mammakarzinom, die zuvor mit Trastuzumab und einem Taxan behandelt worden waren. Das mediane progressionsfreie Überleben wurde von den Patien tinnen unter T-DXd-Therapie nicht erreicht (95%-KI: 18,5 – n.e.), im Vergleich zum T-DM1-Arm mit ei nem PFS von 6,8 Monaten (95%KI: 5,6 8,2).
Da es trotz einer initialen Thera pie mit Trastuzumab, Pertuzumab und einem Taxan bei zahlreichen HER2-positiven metastasierten Patientinnen dennoch zu einer Krankheitsprogression kommt, bietet die erweiterte Indikation für die Betroffenen die Chance, noch früher im Krankheitsverlauf mit T-DXd behandelt zu werden, was ihre Chancen auf bessere Thera pieergebnisse erhöht.
Zusätzliche Auswertungen von se kundären Endpunkten von DESTI NY-Breast03 zeigten einen starken Trend hin zu einem verbesserten Gesamtüberleben im T-DXd-Arm (HR: 0,55; 95%-KI: 0,36 0,86) – diese Daten sind zum jetzigen Zeitpunkt allerdings noch nicht abschließend beurteilbar, und ein
PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG
weiteres Follow-up ist notwen dig. Nach 9 Monaten waren unter T-DXd noch nahezu alle Patien tinnen am Leben (96,1 %; 95%KI: 92,8 97,9), im Vergleich zu 91,3 % im T-DM1-Arm (95%-KI: 87,1 94,2). Die bestätigte ob jektive Ansprechrate war unter TDXd mehr als doppelt so hoch wie unter T-DM1 (79,7 % vs. 34,2 % [n = 208; 95%-KI: 74,3 84,4 vs. n = 90; 95%-KI: 28,5 40,3).
Auf Basis der DESTINYBreast03-Ergebnisse wurden die ESMO-Leitlinien im Oktober 2021 überarbeitet und empfehlen jetzt Enhertu® als die zu bevor zugenden Zweitlinientherapie für Patientinnen mit HER2-positivem metastasiertem Mammakarzinom, wenn diese nach einer Taxan- und Trastuzumab-Therapie progredient sind.
Als Bestandteil dieser Zulassung hat die Europäische Kommission den Patentschutz für Enhertu® in diesem Setting auf Basis des sig nifikanten klinischen Nutzens im Vergleich zu bestehenden zugelas senen Therapien um ein weiteres Jahr verlängert.
B. S.
©
PERFUSION GMBH 104 ÜBERSICHTSARBEIT
JOURNAL
VERLAG
Die chronisch obstruktive Lungenerkrankung (COPD)
äußert sich u.a. durch Hus ten, Dyspnoe und Fatigue. Die Symptome variieren im Tages verlauf und sind nachts sowie früh morgens meist schlimmer, insbesondere bei schwerer COPD – mehr als 70 % der COPD-Patien ten leiden daher unter Schlafprob lemen [1, 2]. Schlechter Schlaf ist jedoch mehr als ein „Luxuspro blem“, denn bei COPD-Patienten sind nächtliche Symptome prädik tiv für eine erhöhte Mortalität und häufigere Exazerbationen [1]. Au ßerdem sind sie mitverantwortlich für die frühmorgendlichen Symp tome, die besonders belastend für die Patienten sind, wenn es darum geht, ihre Tagesroutine zu bewäl tigen [2]. Wie Studien belegen, lassen sich die nächtlichen und morgendlichen Symptome gut mit einer 2 × täglichen Gabe von Bron chodilatatoren behandeln [3, 4].
Einnahmezeitpunkt entscheidet über Therapieerfolg
Da der zirkadiane Rhythmus bei COPD-Patienten gestört sein kann, ist der Einnahmezeitpunkt von Inhalativa, wie auch systemisch verabreichten Medikamenten, ent scheidend für eine wirksame The rapie. So konnte die Post-hoc-Ana lyse einer Phase-IIIb-Studie mit 277 symptomatischen Patienten mit moderater bis schwerer COPD zeigen, dass die 2 × tägliche Gabe des LAMA Aclidiniumbromid (z.B. Bretaris® Genuair®, 400 μg), besonders nachts eine bessere Bronchodilatation bewirkte als die 1 × tägliche Gabe von Tiotro pium [3]. Verbesserungen gab es ebenso bei den frühmorgendlichen Symptomen. Auch die 2 × tägliche Gabe der Fixkombination aus Ac
Bronchodilatatoren:
Zweimalgabe bessert nächtliche und morgendliche COPD-Symptome
lidiniumbromid und dem LABA Formoterolfumarat (z.B. Brimica® Genuair®) führte in der Phase-IIIStudie AUGMENT bei Patienten mit stabiler COPD im Vergleich zu Placebo zu einer Reduktion der nächtlichen und frühmorgendli chen Symptome [4].
Je schwerer die COPD, desto größer die tageszeitlichen Schwankungen
Wie stark sich tageszeitliche Schwankungen äußern, hängt auch vom Schweregrad der COPD ab: Je schwerer die COPD ist, des to variabler sind die Symptome bei Tag und Nacht. So beschrie ben 37 % aller COPD-Patienten ihre morgendliche Routine als beschwerlich; unter jenen mit schwerer COPD waren es sogar 73 % [1]. Eine Pilotstudie mit 30 COPD-Patienten mit moderater bis schwerer COPD untersuchte den Effekt von Aclidiniumbromid auf die Lungenfunktion, Schlafquali tät, COPD-Symptome und körper liche Aktivität [6]. Dazu wurden die Teilnehmer auf zwei aufeinan derfolgende 3-wöchige Behand lungsperioden randomisiert, in de nen sie Aclidiniumbromid 400 μg 2 × täglich oder Placebo erhielten (jeweils mit einer 10–14-tägigen Auswaschphase dazwischen). Ac lidiniumbromid führte zu einer
Reduktion der aktivitätseinschrän kenden Symptome gegenüber Pla cebo. Und dies sowohl was früh morgendliche als auch abendliche Aktivitäten betraf [6].
Die Prävalenz nächtlicher Sympto me steigt zudem mit dem GOLDStadium – der größte prozentuale Anstieg zeigt sich zwischen GOLD 2 und GOLD 3 [1]. Für Patienten mit schweren COPD-Symptomen (CAT ≥20) legt GOLD eine initi ale Dualtherapie mit LAMA und LABA nahe, da diese in Studien mit Patient-reported Outcomes bessere Resultate als Einzelsubs tanzen erzielten [7].
Elisabeth Wilhelmi, München
Literatur
1 Chron Obstruct Pulmon Dis 2020;15: 1269
2 Miravitlles M et al. Respir Res 2017; 18:67
3 Beier J et al. Int J Chron Obstruct Pulmon Dis 2017;12:1731
4 D’Urzo AD et al. Respir Res 2014;15:123
5 Sethi S et al. Int J Chron Obstruct Pul mon Dis 2019;14:667
6 Magnussen H et al. Eur Respir J 2017; 49:1700485
7 Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the dia gnosis, management and prevention of chronic obstructive pulmonary disease (Update 2022)
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 105AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Secukinumab bei Spondyloarthritiden – ein früher Behandlungsbeginn kann Therapieansprechen verbessern
Menschen mit axialer Spon dyloarthritis (axSpA) erhalten ihre Diagnose häufig erst spät – im Durchschnitt vergehen 7–10 Jahre [1], was in der Folge zu einem verzögerten Therapiebeginn führt und einen negativen Effekt auf das Thera pieansprechen haben kann. Für die Betroffenen ist das doppelt bitter, denn wie neueste Daten aus Klinik und Praxis zeigen, verbes sert gerade ein früher Beginn der Behandlung mit dem Interleukin (IL)-17A-Inhibitor Secukinumab (Cosentyx®) das klinische Anspre chen [2, 3].
Überzeugende Ergebnisse der PREVENT-Studie
Aktuelle Post-hoc-Analysen der randomisierten, doppelblinden und placebokontrollierten PREVENTStudie ergaben, dass die Wirksam keit von Secukinumab 150 mg bei jüngeren nicht röntgenologischen (nr)-axSPA-Patienten mit kürzerer Symptomdauer höher war als bei älteren Patienten: In Woche 104 erreichten 69,4 % der Studienteil nehmer in der Altersgruppe 18–33 Jahre ein ASAS40-Anspechen, bei Personen im Alter von ≥52 Jahren waren es dagegen 52,4 %. Neben dem Alter hatte auch eine kürze re Symptomdauer einen positiven
Effekt auf die Wirksamkeit der Therapie: 75,0 % der nr-axSpAPatienten mit einer Krankheitsdau er ≤2 Jahre zeigten ein ASAS40Ansprechen, bei den Teilnehmern mit einer Symptomdauer von >10 Jahren dagegen nur 51,3 % [2].
Beobachtungsstudie AQUILA stützt die Befunde
Dass die Dauer vom Symptombe ginn bis zur Diagnose einen deut lichen Einfluss auf den Therapieer folg haben kann, wurde auch in der multizentrischen Beobachtungs studie AQUILA deutlich, die die Wirksamkeit und Sicherheit von Secukinumab im Behandlungs alltag bei Patienten mit ankylosie
Secukinumab
render Spondylitis (AS) und Pso riasis-Arthritis (PsA) untersuchte [3]. Vorläufige Ergebnisse zeigen, dass Secukinumab in beiden Indi kationen die Krankheitsaktivität und das allgemeine Funktionsni veau im Praxisalltag verbesserte. Männer und Frauen mit AS spra chen tendenziell gleich gut auf die Behandlung an. Der Therapieer folg war jedoch besser, wenn die AS frühzeitig diagnostiziert und behandelt wurde.
Bei Männern mit einer späten ASDiagnose war zu Beginn wie auch während der Studie eine höhere Krankheitsaktivität festzustellen.
Bei den Patienten mit einer Dia gnose früh nach Symptombeginn (<1 Jahr) sank der Bath Ankylo sing Spondylitis Disease Activity
Secukinumab (Cosentyx®) ist ein vollhumaner, monoklonaler An tikörper, der direkt gegen Interleukin-17A gerichtet ist. Dieses Zytokin ist an Entzündungsprozessen und der Entstehung von Psoriasis (PsO), Psoriasis-Arthritis (PsA) und der axialen Spondyloar thritis (axSpA) beteiligt. Der IL-17A-Inhibitor ist zugelassen für die Behandlung von erwachsenen Patienten sowie von Kindern und Jugendlichen ab 6 Jahren mit mittelschwerer bis schwerer PlaquePsoriasis, die für eine systemische Therapie infrage kommen. Au ßerdem ist Secukinumab zugelassen für die Behandlung der akti ven Psoriasis-Arthritis sowie der ankylosierenden Spondylitis, wenn die Patienten unzureichend auf eine konventionelle Therapie mit krankheitsmodifizierenden Antirheumatika (DMARD) bzw. nicht steroidalen Antirheumatika (NSAR) angesprochen haben [4].
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 106 AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Index (BASDAI) bis Woche 52 vom Ausgangswert 4,7 auf 3,2. Bei Patienten, die die Diagnose erst nach einem Jahr oder später erhiel ten, sank der BASDAI bis Woche 52 von 5,3 auf 3,8. Auch bei den an AS erkrankten Frauen nahm der Therapieer folg mit ansteigender Dauer vom Symptombeginn bis zur Diagnose ab: Die mittlere Veränderung im BASDAI gegenüber dem Aus gangswert lag bei einer Diagno se <1 Jahr nach Symptombeginn bei –1,0, bei 2 bis <5 Jahren nach Symptombeginn bei –0,9 und bei >10 Jahren nach Symptombeginn bei –0,4 [3].
Fabian Sandner, Nürnberg
– das erste Medikament für die zielgerichtete Therapie des Mammakarzinoms im Frühstadium mit BRCA1/2Keimbahnmutation
Im August 2022 wurde der PARPInhibitor Olaparib (Lynparza®) in der EU als Monotherapie oder in Kombination mit einer endo krinen Therapie für die adjuvan te Behandlung von erwachsenen Patientinnen mit BRCA1/2Keimbahnmutationen zugelassen, die an einem humanen epiderma len Wachstumsfaktor-Rezeptor 2 (HER2)-negativen Mammakar zinom im Frühstadium erkrankt sind und ein hohes Rezidivrisiko aufweisen. Die Patientinnen müs sen zuvor mit einer neoadjuvanten oder adjuvanten Chemotherapie behandelt worden sein.
Die Zulassung durch die Europä ische Kommission stützt sich auf die Ergebnisse der Phase-III-Stu die OlympiA, die im Juni 2021 im New England Journal of Medicine veröffentlicht wurden, und folgt der Empfehlung des Ausschusses für Humanarzneimittel (CHMP) für die Zulassung von Olaparib in der EU in dieser Indikation.
Signifikante Senkung des Rezidivund Mortalitätsrisikos
die eine lokale Behandlung und eine neoadjuvante oder adjuvante Chemotherapie abgeschlossen hatten.
Die Patientinnen erhielten nach einer 1:1 Randomisierung über 12 Monate Olaparib oder Place bo. Patientinnen mit einem HR+/ HER2- Mammakarzinom konnten zusätzlich zum PARP-Inhibitor Olaparib bzw. Placebo eine endo krine Therapie erhalten. Der pri märe Endpunkt der Studie war das invasive krankheitsfreie Überle ben (iDFS), definiert als die Zeit von der Randomisierung bis zum Auftreten des ersten lokalen oder Fernrezidivs, eines neuen Tumors oder des Todes jeglicher Ursache. Wichtige sekundäre Endpunk te waren das Gesamtüberleben (OS) und das fernrezidivfreie Überleben (dDFS), definiert als Zeit von der Randomisierung bis zum dokumentierten Nachweis des ersten Fernrezidivs oder des Todes jeglicher Ursache.
In der Studie führte Olaparib zu einer statistisch signifikanten Ver besserung des invasiven krank heitsfreien Überlebens (iDFS).
Das Risiko für eine invasive Er krankung oder Tod wurde inner halb von 3 Jahren um 42 % gegen über Placebo reduziert (HR: 0,58; 99,5%-KI: 0,41 0,82; p< 0,0001).
Literatur
1 Lapane KL et al. BMC Fam Pract 2021; 22:251
2 Deodhar A et al. EULAR 2022, Abstract AB0759
3 Benesova K et al. EULAR 2022, Abstract AB0752
4 Fachinformation Cosentyx®; Stand: Feb ruar 2022
Die doppelblinde, placebokontrol lierte Studie OlympiA untersuchte die Wirksamkeit und Verträglich keit von Olaparib im Vergleich zu Placebo als adjuvante Thera pie für Patientinnen mit frühem HER2-negativem Mammakarzi nom mit hohem Rezidivrisiko und BRCA1/2-Keimbahnmutation,
Unter der Therapie mit Olaparib kam es außerdem zu einer statis tisch signifikanten und klinisch bedeutsamen Verbesserung des Gesamtüberlebens. Der PARPInhibitor verringerte das Sterberi siko gegenüber Placebo innerhalb von 4 Jahren um 32 % (HR: 0,68; 98,5%-KI: 0,47 0,97; p = 0,009).
Das Sicherheits- und Verträglich keitsprofil von Olaparib in dieser Studie entsprach dem, das in frühe ren klinischen Studien beobachtet wurde.
JOURNAL PHARMAKOL.
© VERLAG PERFUSION GMBH 107WISSENSWERTES
U. THER. 4/2022 · 31. JAHRGANG
Olaparib
B. S.
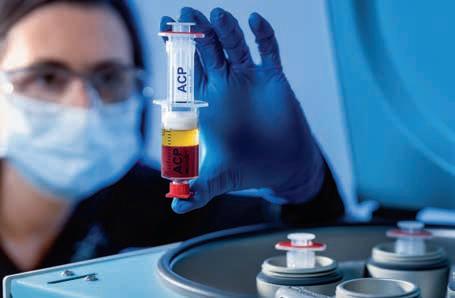
Die Behandlung mit autolo gem konditioniertem Plasma (ACP) ist inzwischen ein an erkannter Therapiebaustein und aus dem Alltag der Mannschaftsärzte von Bundesligavereinen sowie eu ropaweit in den Praxen der Sport ärzte nicht mehr wegzudenken [1].
Der Einsatz hat sich vor allem bei arthrotischen Erkrankungen sowie im Bereich von Sportverletzungen etabliert. ACP bietet eine langfris tig wirksame Alternative zu Thera pieansätzen mit Kortikoiden oder Analgetika. Die Eigenbluttherapie ist dabei nicht nur eine wichtige Säule der konservativen Therapi en, sondern wird auch im Rahmen innovativer, knorpelerhaltender Operationsstrategien eingesetzt (AutoCart™-Methode) [2].
Die ACP-Therapie basiert auf den Kenntnissen zu körpereigenen Hei lungsprozessen und unterstützt die Selbstheilungskräfte des Körpers mithilfe konzentrierter thrombozy tärer Wachstumsfaktoren [3]. Das Blutplasma fördert Regenerations prozesse und Wundheilungsakti vitäten und kann damit einen na türlichen sowie gut verträglichen Beitrag zur funktionellen Wieder herstellung verletzter Strukturen leisten [4].
Das benötigte PRP (platelet rich plasma) lässt sich mit der Arthrex ACP®-Doppelspritze in wenigen Minuten einfach und sicher her stellen, die Trennung des PRP von den anderen Blutbestandteilen er folgt dabei rein physikalisch (Abb. 1).
AutoCart™-Methode:
ACP als Bestandteil der modernen Knorpelchirurgie
Der Einsatz von ACP im Rah men innovativer, knorpelerhalten der Operationsstrategien mit der
ACP-Therapie in Orthopädie und Sportmedizin: Innovative Operationsstrategien mit AutoCart™
AutoCart™-Methode unterstreicht das regenerative Potenzial der in ACP enthaltenen Wachstumsfak toren. So können beispielsweise intraoperativ Knorpelschäden in Gelenken wie Knie oder Sprung gelenk behandelt werden. Knorpel besitzt keine eigenen Blutgefäße, sodass dort körpereigene Rege nerations- und Reparaturprozes se kaum vorhanden sind und das Beheben von Knorpeldefekten er schwert ist.
Bei der AutoCart™-Methode wer den mit Spezialinstrumenten Knor pelteile gewonnen und mit ACP aus dem Blut des Patienten zu ei ner Knorpelpaste gemischt. Durch die hohe Anzahl an Wachstums faktoren kann so an der schadhaf ten Stelle im Gelenk die Bildung von neuem Knorpelgewebe ange regt und ermöglicht werden.
Weniger Schmerzen und verbesserte Gelenkfunktion bei Arthrose
Im klinischen Alltag etabliert so wie durch eine gute Studienlage und Metaanalysen belegt ist die Anwendung von ACP vor allem bei Patienten mit leichter und mit telgradiger Arthrose (Grad 1–3). Insbesondere bei Gonarthrose hat
sich die gute Wirksamkeit von ACP hinsichtlich Schmerzreduktion und Gelenkfunktion auch in wissen schaftlichen Analysen gezeigt: So belegt ein systematisches Review von 6 Studien mit insgesamt 739 Patienten mit Kniegelenk-Arthro se, dass die Anwendung von PRPPräparaten wie ACP bei den Pro banden (817 Kniegelenke, 39 % männlichen Geschlechts, mittleres Alter 59,9 Jahre mit einem durch schnittlichen Follow-up über 38 Wochen) gegenüber einer Behand lung mit Hyaluronsäure allein zu signifikanten klinischen Verbesse rungen über einen Zeitraum von bis zu 12 Monaten nach der In jektion sowie zu einem insgesamt besseren Outcome führte [5].
Auch bei degenerativen, ent zündlichen Prozessen an spinalen Komponenten wie Wirbelkörpern, Facettengelenken oder Band scheiben kann ACP eingesetzt werden und akute oder chronische Rückenschmerzen lindern [6–9]. In einer auf 5 Monate angelegten Untersuchung bei 46 Patienten mit chronischen Rückenschmer zen infolge eines Facettensyn droms war die Therapie mit PRP ab einem Zeitraum von 3 Monaten der Behandlung mit einem Lokal anästhetikum und Kortikoiden überlegen [6].
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 108 AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Das ACP kann direkt aus der Spritze injiziert werden. Wird das ACP innerhalb von 30 Minuten nach Herstellung verwendet, ist kein Antikoagulans erforderlich.
Effektive Hilfe bei Verletzungen beim Freizeit- und Profisport
ACP ist seit Jahren auch fest im Behandlungsrepertoire von Sport medizinern integriert und wird bei akut oder chronisch entstandenen Funktionsdefiziten an Bändern, Sehnen oder Muskeln eingesetzt. Für Indikationen wie Epikondyli tis, Plantarfasziitis oder auch das Patellaspitzensyndrom belegen zahlreiche Studien die Wirksam keit der ACP-Therapie [10–13]. Wie eine systematische Über sichtsarbeit zeigt, ist die Injektion von PRP-Präparaten bei der Be handlung von Tendinopathien der Steroidinjektion überlegen [14]. Auch bei Patienten mit Epikondy litis führte die Injektion von Kor
tikoiden zu schlechteren Ergebnis sen als die ACP-Therapie.
Sichere und natürliche Hilfe –ohne synthetische Zusätze
Die ACP-Therapie ist zu 100 % biologisch und es sind weder kurz- noch langfristige Neben wirkungen bekannt [15–20]. Das Blutplasma fördert die Zell- und Geweberegeneration an den ver letzten oder erkrankten Strukturen auf zahlreichen Ebenen. Therapeu tisch wirksam sind dabei die hoch konzentrierten thrombozytären Wachstumsfaktoren und Zytokine, die zugleich antiinflammatorische und proliferative Effekte haben [2]. Neben der Schmerzlinderung




wird auch die Beweglichkeit der erkrankten Körperareale verbes sert.
ACP ist aufgrund seiner Wirk weise im Vergleich zu anderen in der Orthopädie und Sportmedi zin angewandten konservativen Behandlungsmethoden eine gut verträgliche Alternative [21]. Zu den etablierten Therapien gehö ren beispielsweise Kortikoide, die insbesondere bei Patienten mit Ko morbiditäten wie Diabetes, Blut hochdruck oder einem Glaukom zu Komplikationen führen können. Entzündungshemmende NSAR sind hingegen bei Patienten mit Niereninsuffizienz kontraindiziert [22, 23, 24].
Die Behandlung mit ACP kann ambulant durchgeführt werden
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 109AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Abbildung 1: Die Arthrex ACP®-Doppelspritze ermöglicht eine geschlossene, schnelle und einfache Herstellung von PRP (platelet rich plasma). 15 ml Vollblut ergeben etwa 5 6 ml PRP mit einer Thrombozytenkonzentration, die etwa um das Zweieinhalbfache höher ist als der Ausgangs wert (©
Zentrifugierung für 5 Minuten
15 ml Blut werden direkt in die Arthrex ACP-Doppelspritze entnommen
Das ACP wird in die innere Spritze gezogen
und dauert insgesamt 30 Minuten. Meist umfasst die Therapie mit ACP mehrere Intervalle und ist für Ärzte und Patienten gut planbar.
Brigitte Söllner, Erlangen
Follikuläres Lymphom:
Mosunetuzumab als Monotherapie ab der 3. Therapielinie zugelassen
Literatur
1 Rauch G. www.sportaerztezeitung.com/ rubriken/therapie/3224/ muskelverletzun gen-im-profihandball; Beitrag vom 21.01.2022
2 Schneider S et al. Arthrosc Tech 2020;10: e97-e101
3 Obata S et al. Arthritis Res Ther 2012; 14:R241
4 Hodgkinson T et al. JOR Spine 2019;2: e1045
5 Meheux CJ et al. Arthroscopy 2016;32: 495-505
6 Wu J et al. Pain Pract 2017;17:914-924
7 Mohammed S et al. J Spine Surg 2018;4:115-122
8 Tuakli-Wosornu Y et al. PM R 2016;8:110
9 Mazzocca A et al. Am J Sports Med 2012;40:1742-1749
10 Zayni R et al. Muscles Ligaments Ten dons J 2015;5:92-98
11 Murray DJ et al. J Hand Microsurg 2015; 7:320-325
12 Chiew SK et al. Res Med Sci 2016;14; 21:38
13 Shetty SH et al. J Foot Ankle Surg 2019; 58:42-46
14 Miller LE et al. BMJ Open Sport Exerc Med 2017;3:e000237
15 Smith PA. Am J Sports Med 2016;44: 884-891
16 Cerza F et al. Am J Sports Med 2012;40: 2822-2827
17 Cole BJ et al. Am J Sports Med 2017;45: 339-346
18 Ford RD et al. Hand (NY) 2015;10:285291
19 Lebiedzinski R et al. Int Orthop 2015;39: 2199-2203
20 Chew KT et al. PM&R 2013;5:10351043
21 Weber AE et al. Int Orthop 2021;45:335344
22 Radi ZA et al. Inflamm Res 2005;54:358366
23 Mehallo CJ et al. Sport Med 2006;16: 170-174
24 Dahners LE et al. Am Acad Orthop Surg 2004;12:139-143
Die Europäische Kommission hat Mosunetuzumab (Lunsumio®) als Monotherapie für die Behandlung erwachsener Patienten mit rezidi vierendem oder refraktärem fol likulärem Lymphom (r/r FL), die mindestens zwei vorherige syste mische Behandlungen erhalten ha ben, zugelassen. Mosunetuzumab repräsentiert als T-Zellen-rekrutie render Anti-CD20/CD3-Antikör per ein neues Wirkprinzip zur The rapie von B-Zell-Lymphomen und ist der erste und einzige bispezifi sche Antikörper zur Behandlung des FL.
Sehr gute Wirksamkeit: Primärer Studienendpunkt CRR erreicht
Im Rahmen der zulassungsrele vanten Studie GO29781 der klini schen Phase Ib/II wurde die sehr gute Wirksamkeit von Mosunetu zumab bestätigt: Unter einer Mo notherapie mit Mosunetuzumab zeigte sich ein objektives Anspre
Mosunetuzumab
chen (ORR) von 80 %, wobei 60 % der eingeschlossenen Patienten mit einer kompletten Remission (CR) auf die Therapie reagierten und da mit der primäre Studienendpunkt erreicht wurde. Ein tiefes Anspre chen auf die Behandlung wurde rasch erreicht (median 1,4 Monate bis zum ersten Ansprechen; medi an 3 Monate bis zum Erreichen ei ner CR) und dauerte durchschnitt lich 22,8 Monate an. In der Gruppe der Patienten mit kompletter Re mission zeigten 76 % auch nach 12 Monaten keinen Krankheitspro gress. Das mediane progressions freie Überleben (PFS) der Patien ten betrug 17,9 Monate (95%-KI: 10,1 nicht abschätzbar). Neben der hohen Wirksamkeit wurde für den Anti-CD20/CD3Antikörper ein gut handhabbares Nebenwirkungsprofil beschrieben. Nebenwirkungen vom Grad 3 oder 4 im Zusammenhang mit Mo sunetuzumab wurden bei 51,1 % der Patienten dokumentiert. Zu den am häufigsten berichteten Ne benwirkungen zählten Neutrope nie, Fieber und Hypophosphat ämie. Ein CRS trat bei insgesamt 44,4 % der Patienten auf und war in den meisten Fällen von milder oder moderater Ausprägung (Grad 1 oder 2) sowie überwiegend auf den ersten Zyklus beschränkt.
F. S.
Mosunetuzumab (Lunsumio®) ist ein vollständiger bispezifischer T-Zellen-rekrutierender Anti-CD20/CD3-Antikörper. Dieser wirkt als eine Art Brücke zwischen körpereigenen Abwehrzellen und bösartig veränderten B-Zellen und führt sie so zusammen. Das Wirkprinzip: Einer der beiden Antikörper-Arme bindet spezifisch an das B-Lymphozyten-Antigen CD20 auf der Oberfläche maligner B-Zellen. Über den anderen Antikörper-Arm werden durch spezifi sche Bindung an das CD3-Antigen zytotoxische T-Zellen gebunden. Dies bringt die körpereigenen Abwehrzellen in engen räumlichen Kontakt mit den malignen Zellen und begünstigt deren Abtötung.
JOURNAL PHARMAKOL.
© VERLAG PERFUSION GMBH 110 AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
U. THER. 4/2022 · 31. JAHRGANG
Für Patienten mit Prostatakarzi nom (PCa) spielen im Verlauf der Erkrankung zwei Ent wicklungen eine besondere Rolle, da sie die weitere Therapie und die Überlebenschancen maßgeblich beeinflussen: der Nachweis einer Metastasierung und die Entwick lung einer Kastrationsresistenz. Beide Ereignisse sind prognostisch ungünstig und für den betroffenen Patienten emotional stark belas tend [1]. Das Risiko einer Kast rationsresistenz lässt sich jedoch durch die Therapiewahl signifikant beeinflussen.
Neue Generation der Hormontherapie für Patienten mit mHSPC
Beim Entstehen der Kastrations resistenz verändert sich das Kar zinom in der Prostata derart, dass trotz nicht messbarem Testosteron spiegel im Blut aufgrund einer chi rurgischen oder medikamentösen Kastration die Erkrankung weiter voranschreitet – typischerweise steigt dann der Marker prostata spezifisches Antigen (PSA) im Blut an. Deshalb blicken Arzt und Patient im Rahmen der Nachsorge regelmäßig und mit Argusaugen auf den PSA-Wert, und dessen Übermittlung durch den Arzt ist für viele Patienten eine angsterfüll te Situation.
Wird bei einem Patienten eine Me tastasierung festgestellt und der Tumor spricht noch auf eine An
Kastrationsresistenz beim Prostatakarzinom vermeiden
drogendeprivationstherapie (ADT) an, liegt ein metastasiertes hor monsensitives PCa (mHSPC) vor. In dieser Situation steht seit mehr als 50 Jahren die ADT als Thera pieoption zur Verfügung. Zudem kann auch mit einer klassischen Chemotherapie behandelt werden. Seit mittlerweile einigen Jahren gibt es allerdings auch nicht steroi dale Androgenrezeptor-Antagonis ten der zweiten Generation („neue Generation der Hormontherapie“), die zusammen mit einer ADT ge geben werden können. Nun wurde in einem aktuellen Daten-Update der ARCHES-Studie gezeigt, dass genau diese Kombination aus Enzalutamid (Xtandi™, ein Ver treter der Androgenrezeptor-Anta gonisten der zweiten Generation) und ADT das Risiko für das Ein treten einer Kastrationsresistenz um 61 % reduzieren konnte [2].
Vorteile der Kombination aus Enzalutamid und ADT
In ARCHES wurden 1.150 Pati enten mit mHSPC randomisiert entweder mit Enzalutamid 160 mg 1 × täglich + ADT oder Placebo + ADT behandelt. Die Studie wurde in einem Cross-over-Design ange legt, wobei Patienten des PlaceboArms nach der geplanten Entblin dung in den Enzalutamid-Arm wechseln konnten. Neben dem primären Endpunkt, einer signifi kanten 43%igen Senkung des Ge samtsterberisikos unter Enzalut
amid (adjustiert nach Cross-overPatienten), zeigte sich vor allem, dass Patienten mit Enzalutamid signifikant seltener eine Kastrati onsresistenz entwickelten: Unter Placebo + ADT trat die Kastrati onsresistenz im Median nach 14,0 Monaten ein, unter Enzalutamid + ADT war der Median noch nicht erreicht, das Ereignis also noch nicht ausreichend häufig eingetre ten, um diese statistische Kennzahl zu berechnen (HR: 0,39; 95%-KI: 0,33 0,47) [2].
Fazit
Für die Patienten mit mHSPC be deuten diese Ergebnisse, dass die Kombination aus Enzalutamid + ADT und insbesondere der frühzei tige Enzalutamid-Einsatz das Risi ko für den Eintritt einer Kastrati onsresistenz und das Fortschreiten eines mHSPC zum mCRPC (me tastasiertes kastrationsresistentes PCa) signifikant reduzieren kann, sodass ihnen eine Veränderung bzw. Intensivierung der Therapie erspart bleibt [1, 2].
Fabian Sandner, Nürnberg
Literatur
1 Armstrong AJ et al. J Clin Oncol 2022; doi:10.1200/JCO.22.00193
2 S3-Leitlinie Prostatakarzinom; Version 6.2; Oktober 2021; AWMF-Registernum mer: 043/022OL
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 111AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Am 20. Juli 2022 erhielt Up staza™ (Eladocagene exuparvovec) von der Europä ischen Kommission die Markt zulassung für die krankheitsmo difizierende Behandlung des auf einem Gendefekt beruhenden Mangels an Aromatischer L-Ami nosäure-Decarboxylase (AADC), eines Enzyms, das L-Dopa in Do pamin umwandelt. Bei Eladocagene exuparvovec handelt es sich um eine Genthe rapie, bei der eine funktionsfähige Kopie des menschlichen AADCGens mithilfe eines Vektors gezielt in eine bestimmte Region des Ge hirns eingespritzt wird und dort die Dopaminsynthese in Gang setzt, sodass die Entwicklung motori scher Funktionen möglich wird. Upstaza™ wird nur einmal verab reicht und kann bei Patienten ab 18 Monaten angewendet werden [1].
Verheerende Erkrankung mit lebensbedrohlichen Komplikationen
Der AADC-Mangel ist eine tödlich verlaufende, sehr seltene genetisch bedingte Stoffwechselerkrankung, die schwere Entwicklungs- und Bewegungsstörungen verursacht [2]. Ursache ist eine Mutation des DopaDecarboxylase(DDC)-Gens, das die Aromatische L-Amino säure-Decarboxylase kodiert, die L-3,4-Dihydroxyphenylalanin (LDOPA) in Dopamin umwandelt.
Upstaza™ – die erste krankheitsmodifizierende Gentherapie für den AADC-Mangel
Die Mutationen am DDC-Gen führen zu einer Verringerung oder einem Ausbleiben der AADC-En zymaktivität, wodurch zu wenig Dopamin, Serotonin und Kate cholamine synthetisiert werden. Der Mangel an diesen Botenstof fen führt bereits in den ersten Le bensmonaten zu tiefgreifenden Entwicklungsverzögerungen und schweren Behinderungen infolge einer extrapyramidalmotorischen Bewegungsstörung mit Hypo- oder Akinesie, Dystonien und rumpfbe tonter muskulärer Hypotonie. Die an AADC-Mangel erkrankten Kin der müssen rund um die Uhr betreut
Eladocagene exuparvovec
werden und häufig ins Krankenhaus bzw. in die Notaufnahme eingelie fert werden, denn ihr Leiden wird häufig durch schwere Symptome verschlimmert: Episoden anfallsar tiger okulogyrer Krisen, die täglich auftreten und stundenlang andau ern können und dazu führen, dass die Augen krampfartig verdreht werden, häufiges Erbrechen, Ver haltensstörungen, Schlafprobleme, ständig verstopfte Nase und lebens bedrohliche Komplikationen wie Atemwegsinfektionen und MagenDarm-Probleme. Die Kinder be nötigen eine fortlaufende Physio-, Ergo- und Sprachtherapie sowie
Eladocagene exuparvovec (Upstaza™) ist eine Genersatztherapie, die für die Behandlung von Patienten ab 18 Monaten mit einer klinisch, molekularbiologisch und genetisch bestätigten Diagnose eines Aromatische L-Aminosäure-Decarboxylase (AADC)-Mangels mit einem schweren Phänotyp indiziert ist. Bei dem Genersatztherapeutikum handelt es sich um einen nicht replizierenden rekombinanten Vektor auf Grundlage des Adenoassoziierten Virus vom Serotyp 2 (AAV2), der die cDNA des huma nen Dopa-Decarboxylase (DDC)-Gens enthält und unter der Kon trolle eines Zytomegalievirus (CMV)-Promoters ist. Upstaza™ soll den zugrunde liegenden genetischen Defekt korrigieren, indem ein funktionierendes DDC-Gen direkt in das Putamen eingebracht wird, wodurch das AADC-Enzym exprimiert und die Dopaminpro duktion wiederhergestellt wird. Die einmalige intrazerebrale Applikation von Upstaza™ erfolgt durch einen stereotaktischen neurochirurgischen Eingriff mit bila teraler Platzierung zweier Kanülen in 2 Bereichen pro Putamen. Pro putaminaler Infusionsstelle werden 0,08 ml Upstaza™ infun diert, bei insgesamt 4 Infusionen mit einem Gesamtvolumen von 0,320 ml [1].
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 112 NEUE UND BEWÄHRTE ARZNEIMITTEL
Baseline Zeitintervall nach der Behandlung (Monate) Insgesamt (kumulativ) nach der Behandlung
Motorische Meilensteine Vor der Behandlung (n=20)
0–3 (n=20) 3–12 (n=17) 12–24 (n=17) 24–36 (n=13) 36–48 (n=8) 48-60 (n=6) 60 Monate (n=20)
Kopfkontrolle 0 1 5 6 2 0 0 14 (70%) Sitzen ohne Unterstützung 0 1 2 6 2 1 1 13 (65%)
Stehen mit Unterstützung 0 0 0 4 1 1 0 6 (30%) Gehen mit Unterstützung 0 0 0 0 2 0 0 2 (10%)
Tabelle 1: Ergebnisse der beiden zulassungsrelevanten Studien zu Upstaza™ [3, 4] für den primären Wirksamkeitsendpunkt, das Erreichen von mororischen Meilensteinen, gemessen mithilfe der Peabody Developmental Motor Scale, Version 2 (PDMS-2*). Angegeben ist jeweils die An zahl der Patienten, die nach der Behandlung mit Eladocagene exuparvovec neue motorische Meilensteine der PDMS-2 erreichten. Die kumulative Spalte umfasst alle Studienteilnehmer, die den bestimmten Meilenstein zu irgendeinem Zeitpunkt während der klinischen Studie innerhalb von 60 Monaten erreichten. Die Patienten mussten mit einem Score von 2 (Hinweis auf eine Beherrschung der Fähigkeit) in Bezug auf einen Meilenstein bewertet werden, um den Meilenstein erreicht zu haben [1].
eine Betreuung durch ein multidis ziplinäres Team hochqualifizierter Spezialisten. Der Erfolg der kon ventionellen, medikamentösen Be handlung mittels Kombinationen aus Vitamin B6, Dopaminagonisten und Monoaminoxidaseinhibitoren ist besonders bei schweren Verläu fen nur sehr limitiert, sodass viele Patienten bereits im Kindesalter versterben. Mit der intrazerebralen Applikation von Eladocagene exu parvovec steht erstmals ein kausaler Therapieansatz zur Verfügung.
* Die PDMS-2 ist eine Beurteilung der mo torischen Entwicklung des Kindes bis zum Entwicklungsalter von 5 Jahren und bewer tet sowohl die Grob- als auch die Feinmoto rik mit speziell auf das Erreichen motori scher Meilensteine bezogenen Aspekten. Die Aspekte der motorischen Fähigkeiten der PDMS-2 wurden ausgewählt, um die Anzahl der Patienten zu bestimmen, die mindestens die folgenden motorischen Meilensteine er reichten: 1. vollständige Kopfkontrolle, 2. Sitzen ohne Unterstützung, 3. Stehen mit Unterstützung und 4. Gehen mit Unterstüt zung [1].
Klinisch relevante Verbesserungen
Die Gentherapie mit Upstaza™ kann die Prognose für Kinder ver bessern, die mit einem AADCMangel geboren wurden: In den beiden zulassungsrelevanten kli nischen Studien mit Upstaza™ [3, 4] erreichten die Patienten bereits 3 Monaten nach der einmaligen Injektion von Upstaza™ klinisch relevante motorische Fähigkei ten (Tab. 1). Bei allen behandel ten Kindern verbesserten sich die kognitiven und sprachlichen Fä higkeiten. Upstaza™ reduzierte auch Symptome, die potenziell lebensbedrohliche und krankhafte Komplikationen verursachen. In einer weiteren Studie setzten sich die Verbesserungen sogar bis zu einem Beobachtungszeitraum von 10 Jahren fort [5]. Die beobachteten Nebenwirkun gen waren moderat. Fast alle Be
handelten litten nach dem Eingriff unter Dyskinesien, die nach eini gen Wochen nachließen.
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Uptaza™. https://ec. eu ropa.eu/health/documents/community-re gister/2022/20220718156036/anx_ 156036_de.pdf
2 Opladen T et al. Die intrazerebrale Gen therapie des Aromatischen-L-Aminosäu re-Decarboxylase-Mangels mit Eladoca gene exuparvovec. Monatsschr Kinder heilkd 2021;169:738-747
3 Hwu WL al. Improved motor function in children with AADC deficiency treated with eladocagene exuparvovec (PTCAADC): interim findings from a phase I/ II Study. Poster presented at the 23rd ASGCT Annual Meeting, May 12–15 2020
4 Tseng CH et al. Gene therapy improves brain white matter in aromatic l amino acid decarboxylase deficiency. Ann Neu rol 2019;85:644-652
5 Hwu WL et al. Gene therapy for aromatic L amino acid decarboxylase deficiency. Sci Transl Med 2012;4:134ra161
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 113NEUE UND BEWÄHRTE ARZNEIMITTEL
Migräne ist eine häufige neu rologische Erkrankung, die von einer episodischen Migräne (EM, <15 Kopfschmerz tage/Monat) zu einer chronischen Migräne (≥15 Kopfschmerztage/ Monat, davon ≥8 Migränetage/ Monat) fortschreiten kann und mit erheblichen Beeinträchtigun gen im Alltag verbunden ist. Am 1. September 2022 wurde mit Ep tinezumab (Vyepti®) ein weiterer Antikörper gegen das Neuropeptid Calcitonin Gene-Related Peptide (CGRP) für die Migräneprophyla xe von Erwachsenen mit mindes tens 4 Migränetagen pro Monat auf den deutschen Markt eingeführt.
Intravenöse Applikation führt zu schnellerem Wirkeintritt
Der humanisierte monoklonale IgG1-Antikörper wird als einziger der vier zugelassenen CGRP-Anti körper als intravenöse Kurzinfusi on über 30 Minuten appliziert. Ein
Eptinezumab erweitert die Optionen zur Migräneprophylaxe
spezifisches Monitoring ist nicht er forderlich. Der logistische Aufwand für die Praxis ist daher gering.
Die Bioverfügbarkeit beträgt 100 % und die maximale Plasma konzentration wird bereits am Ende der Infusion erreicht. Aufgrund der Eliminationshalbwertszeit von 27 Tagen ist die Verabreichung alle 12 Wochen möglich. Der Vorteil des intravenösen Applikationswe ges ist ein im Vergleich zu anderen CGRP-Antikörpern schnellerer Wirkeintritt [1] – und ein rasches Einsetzen der Wirkung ist wieder um das, was sich Migränepatienten wünschen.
Signifikante Reduktion der Migränetage
In den zulassungsrelevanten PhaseIII-Studien PROMISE-1 bei EM [2] und PROMISE-2 bei CM [3] führte Eptinezumab in den zuge lassenen Dosierungen von 100 mg und 300 mg alle 12 Wochen zu ei ner gegenüber Placebo signifikanten Reduktion der monatlichen Migrä netage (Tab. 1). Eine prophylakti sche Wirkung war sowohl bei EM als auch bei CM bereits an Tag 1 nach der ersten Gabe zu beobach ten [2, 3]. Von der i.v.-Prophylaxe können auch Patienten mit CM
Primärer Wirksamkeitsendpunkt in PROMISE 1 (episodische Migräne)
Eptinezumab 100 mg (n = 221) Eptinezumab 300 mg (n = 222) Placebo (n = 222)
Monatliche Migränetage (MMD) – Wochen 1–12 Baseline 8,7 8,6 8,4
Mittlere Veränderung –3,9 –4,3 –3,2 Unterschied zu Placebo –0,7 –1,1 95%-KI (–1,3 bis –0,1) (–1,7 bis –0,5) p-Wert vs. Placebo 0,0182 0,0001
Primärer Wirksamkeitsendpunkt in PROMISE 2 (chronische Migräne)
Eptinezumab 100 mg (n = 356) Eptinezumab 300 mg (n = 350) Placebo (n = 366)
Monatliche Migränetage (MMD) – Wochen 1–12 Baseline 16,1 16,1 16,2 Mittlere Veränderung –7,7 –8,2 –5,6 Unterschied zu Placebo –2,0 –2,6 95%-KI (–2,9 bis –1,2) (–3,5 bis –1,7) p-Wert vs. Placebo 0,0001 0,0001
Tabelle 1: Ergebnisse der Studien PROMIS-1 und -2 für den primären Wirksamkeitsendpunkt [1, 2, 3].
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 114 NEUE UND BEWÄHRTE ARZNEIMITTEL
Eptinezumab
Eptinezumab (Vyepti®) ist ein rekombinanter humanisier ter Immunglobulin-G1(IgG1)Antikörper, der an die α- und β-Formen des menschlichen Calcitonin-Gene-RelatedPeptide(CGRP)-Liganden mit niedriger pikomolarer Affini tät (4 bzw. 3 pM Kd) bindet. Er weist dabei keine signifikante Aktivität gegen andere Calci tonin-Rezeptoren auf.
Eptinezumab verhindert die Aktivierung der CGRP-Rezep toren und damit die nach geschaltete Kaskade physio logischer Ereignisse, die mit der Entstehung von Migrä neanfällen verbunden sind. Eptinezumab hemmt α- und β-CGRP-vermittelte neurogene Entzündungen und Vasodila tation. Der Wirkstoff wird alle 12 Wochen mittels intravenö ser Infusion in einer Dosis von 100 mg verabreicht. Falls er forderlich, kann die Dosis auf 300 mg gesteigert werden [1]. und MedikamentenübergebrauchKopfschmerz profitieren [5]. Epti nezumab war in den klinischen Stu dien gut verträglich. Die häufigsten Nebenwirkungen waren Nasopha ryngitis und Überempfindlichkeit [1]. Unerwünschte Ereignisse an der Infusionsstelle waren selten und ver gleichbar mit Placebo (<2 %) [1].
Abdol A. Ameri, Weidenstetten
Atopische Dermatitis: JAK1-Inhibitor Abrocitinib verbessert Hautläsionen und Juckreiz
Die schnelle und anhaltende Verbesserung der Hautsym ptomatik sowie die zügige Linderung des belastenden Juck reizes [1, 2] machen den innova tiven Januskinase 1 (JAK1)-In hibitor Abrocitinib (Cibinqo®) zu einer wichtigen neuen Therapie option bei erwachsenen Patienten mit mittelschwerer bis schwerer atopischer Dermatitis (AD), die für eine systemische Therapie in frage kommen. Dies zeigen die Er gebnisse der zulassungsrelevanten klinischen Studien.
Ein weiterer Pluspunkt: Der Ge meinsame Bundesausschuss (GBA) hat kürzlich einen Anhalts punkt für einen beträchtlichen Zusatznutzen für Abrocitinib ge genüber einer zweckmäßigen Ver gleichstherapie mit Dupilumab (ggf. in Kombination mit topi schen Glukokortikoiden und/oder topischen Calcineurininhibitoren) in der Endpunktkategorie Morbidi tät bei erwachsenen Patienten mit mittelschwerer bis schwerer AD festgestellt [3].
wurden die Wirksamkeit und Si cherheit von Abrocitinib 200 mg einmal täglich mit dem IL-4/IL13-Blocker Dupilumab (300 mg q2w) bei 727 erwachsenen Patien ten mit mittelschwerer und schwe rer AD verglichen. Nach 2 Wochen zeigte sich bereits ein hochsigni fikanter Unterschied bei der Juck reizlinderung zwischen JAK1-In hibitor und Biologikum. So wiesen zu diesem Zeitpunkt 48 % der Pa tienten unter Abrocitinib 200 mg ein Ansprechen im PP-NRS (Peak Pruritus Numerical Rating Scale, primärer Endpunkt) auf gegenüber 26 % unter Dupilumab. Die gute Wirksamkeit von Abrocitinib be züglich des Juckreizes setzte sich bis Woche 12 fort, danach war die Wirkung beider Substanzklassen in etwa vergleichbar und blieb bis in Woche 26 bestehen.
Literatur
1 Fachinformation Vyepti®; Stand: 1/ 2022
2 Ashina M et al. Cephalalgia 2020;40: 241-254
3 Lipton RB et al. Neurology 2020;94: e1365-e1377
4 Dodick DM et al. Headache 2020;60: 2220-2231
5 Marmura MJ et al. Headache 2021;61: 1421-1431
JADE DARE: Überlegenheit gegenüber Dupilumab
Grundlage der Nutzenbewertung durch den G-BA sind die Daten der Studie JADE DARE [4]. In der randomisierten, über 26 Wochen laufenden Head-to-head-Studie
Auch das ambitionierte Therapie ziel eines EASI (Eczema Area and Severity Index)-90-Ansprechens wurde in Woche 4 (primärer End punkt) und in Woche 16 (sekun därer Endpunkt) signifikant häu figer in der Abrocitinib-Gruppe erreicht als unter Dupilumab (Wo che 4: 29 % vs. 15 %; Woche 16: 54 % vs. 42 %). Die Patienten der Abrocitinib-Gruppe benötigten zu dem signifikant seltener topische Glukokortikoide als die mit Dupi lumab behandelten Studienteilneh mer (51 vs. 33 Tage ohne topische Hintergrundtherapie) [4].
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 115NEUE UND BEWÄHRTE ARZNEIMITTEL
JADE COMPARE: Signifikant bessere Linderung des Juckreizes ab Tag 4
Die schnelle Besserung von Juck reiz und Hautläsionen unter Ab rocitinib (hier in den Dosierun gen 100 mg und 200 mg einmal täglich) zeigte sich auch in der 16-wöchigen, randomisierten, doppelblinden, placebokontrol lierten Studie JADE COMPARE [5]. Hier wurde Dupilumab als ak tiver Kontrollarm mitgeführt, alle Patienten erhielten eine topische Hintergrundtherapie. Auch in dieser Studie zeigten die Ergebnisse für die ko-primären Endpunkte IGA (Investigator Global Assessment)-Ansprechen 0/1 (mit einer 2-Punkte-Verbesse rung) und EASI-75-Ansprechen in Woche 12 sowie dieselben (se kundären) Endpunkte zu Woche 16 eine überlegene Wirksamkeit des JAK1-Inhibitors: Die IGAAnsprechrate in Woche 12 betrug unter Abrocitinib 200 mg 48,4 %, das EASI-75-Ansprechen 70,3 % (Dupilumab: 36,5 % bzw. 58,1 %). Knapp die Hälfte der Patienten (46,1 %) erzielte unter Abrocitinib 200 mg in Woche 12 ein EASI90-Ansprechen (sekundärer End punkt), während dies unter Dupi lumab nur bei 34,9 % der Fall war [6].
Auch bei der raschen Linderung des Juckreizes überzeugte der neue JAK1-Inhibitor: So war Abrociti nib 200 mg bereits ab Tag 4 wirk samer als Dupilumab 300 mg und in Woche 2 erreichten 49,1 % der Patienten unter Abrocitinib 200 mg eine Verbesserung um mindestens
4 Punkte im PP-NRS (vs. 26,4 % unter Dupilumab) [1, 5].
Moderate Nebenwirkungen
Eine integrierte Sicherheitsanaly se der JADE-Studien zur Verträg lichkeit zeigte, dass die meisten unerwünschten Ereignisse mild bis moderat ausfielen und in der Regel nicht zum Therapieabbruch führten [7]. Am häufigsten traten dosisabhängig Übelkeit und Kopf schmerz auf, wobei die Übelkeit bei Einnahme der Tabletten mit einer Mahlzeit weitgehend vermie den werden konnte. Da unter der Therapie mit JAK-Inhibitoren ein erhöhtes Risiko für Infektionen mit Herpes zoster (HZ) besteht, ist es ratsam, vor allem ältere Patienten zuvor gegen HZ zu impfen.
Bedarfsgerechte Dosierung
Ein wichtiger Vorteil für die tägli che Praxis ist die Möglichkeit, den neuen JAK1-Inhibitor bedarfsge recht dosieren zu können. Abroci tinib steht als 50 mg-, 100 mg- und 200 mg-Tablette zur Verfügung [8]. Die empfohlene Anfangsdosis beträgt 200 mg einmal täglich, bei Patienten ab einem Alter von 65 Jahren wird eine Anfangsdosis von 100 mg einmal täglich empfohlen. Während der Behandlung kann die Dosis je nach Verträglichkeit und Wirksamkeit verringert oder erhöht werden. Für die Erhaltungs therapie sollte die niedrigste wirk same Dosis in Betracht gezogen werden.
Patienten mit atopischer Dermatitis vor Schub schützen
Um die Patienten vor einem Schub (Flare) zu schützen, sollte die Therapie mit Abrocitinib länger fristig erfolgen. Wie die Daten der 52 Wochen laufenden Studie JADE REGIMEN zeigen, haben nach einer 3-monatigen offenen Induktionstherapie mit Abrociti nib 200 mg über 80 % der Patien ten, die in der 40-wöchigen Er haltungsphase weiterhin 200 mg Abrocitinib täglich (verblindet) erhalten hatten, über den Zeitraum keinen Krankheitsschub (definiert als ≥50%iger Verlust des initialen EASI-Ansprechens zu Woche 12 und neuer IGA-Punktwert ≥2) er litten [9].
Elisabeth Wilhelmi, München
Literatur
1 Ständer S et al. Posterpräsentation. Amer ican Academy of Dermatology Associa tion Virtual Meeting Experience 2021; 23. – 25. April 2021
2 Reich K et al. Präsentation 523. Präsen tiert auf: Revolutionizing Atopic Derma titis Virtual Conference. 13. Juni 2021
3 https://www.g-ba.de/beschluesse/5517/
4 Reich K et al. Lancet 2022;400:273-282
5 Bieber T et al. N Engl J Med 2021;384: 1101-1112
6 Supplement to Bieber T et al. N Engl J Med 2021;384:1101-1112
7 Simpson EL et al. Am J Clin Dermatol 2021;22:693-707
8 Fachinformation Cibinqo®; Stand: De zember 2021
9 Blauvelt A et al. J Am Acad Dermatol 2022;86:104-112
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 116 NEUE UND BEWÄHRTE ARZNEIMITTEL
Patienten mit Plaque-Psoria sis haben mittlerweile hohe Erwartungen an die Thera pieergebnisse. Nach heutigen the rapeutischen Standards ist selbst eine nahezu bis vollständige er scheinungsfreie Haut (PASI 90 / PASI 100, Psoriasis Area and Se verity Index) ein erreichbares Ziel geworden. Vielversprechend sind die Studiendaten zu Bimekizumab (Bimzelx®), dem ersten in der EU zugelassenen Wirkstoff, der selek tiv und direkt sowohl Interleukin 17A (IL-17A) als auch Interleukin 17F (IL-17F) inhibiert [1].
Vorteile der dualen Hemmung von IL-17A und IL-17F
Der Plaques-Psoriasis liegt ein komplexes inflammatorisches Ge schehen zugrunde, an dem maß geblich die Zytokine Interleukin (IL)-17A und IL-17F beteiligt sind. Die Expression von IL-17A und IL-17F ist in psoriatischen Läsionen um das bis zu Achtfache erhöht [2]. Bimekizumab, ein hu manisierter monoklonaler IgG1Antikörper, konnte in den zulas sungsrelevanten Studien zeigen, dass eine duale Hemmung im Ver gleich zur alleinigen Blockade von IL-17A zu einer effektiveren Blo
Mittelschwere bis schwere Plaque-Psoriasis: Wechsel auf Bimekizumab kann sich lohnen
ckade der Entzündungsreaktionen bei der Psoriasis führt. Die Sicherheit und Wirksam keit von Bimekizumab wurde bei 1.480 Patienten mit mittelschwe rer bis schwerer Plaque-Psoriasis in 3 randomisierten, placebo- und/ oder aktiv kontrollierten Phase-IIIStudien untersucht: BE VIVID [3] verglich Bimekizumab mit Usteki numab und Placebo, BE READY [4] war placebokontrolliert und BE SURE [5] verglich Bimekizumab mit Adalimumab. Die Behandlung mit Bimekizumab war mit einem schnellen Wirkeintritt assoziiert: In allen Studien erreichten bereits nach der ersten Dosis Bimekizumab (nach 4 Wochen) mehr als 70 % der Patien ten einen PASI 75. Im Vergleich zu Placebo, Ustekinumab oder Ada limumab hatten sich in Woche 16 unter Bimekizumab auch die PASI90- und PASI-100-Ansprechraten signifikant verbessert (Tab. 1).
PASI 100 erreichen und aufrechterhalten
Für die Patienten ist mit dem schnellen Therapieansprechen auf Bimekizumab bereits ein relevan tes Behandlungsziel erreicht. Da rüber hinaus sollte die Haut mög lichst erscheinungsfrei sein und das gute Ansprechen langfristig erhalten werden. Dass auch das unter der Therapie mit Bimeki zumab möglich ist, belegen die Ergebnisse Phase-IIIb-Studie BE RADIANT [6], die Bimekizumab mit dem IL-17A-Inhibitor Secuki numab 48 Wochen lang verglich. In der anschließenden offenen Verlängerungsphase (OLE) wech selten die mit Secukinumab be handelte Patienten auf den dualen Inhibitor. Nach der Umstellung zeigte sich eine Verbesserung der Erscheinungsfreiheit der Haut: In Woche 48 lag der Anteil der Pa
PASI 90 in Woche 16 4,8% 85,0%*, # 49,7% 1,2% 90,8%* 86,2%* 47,2%
PASI 75 in Woche 4 in Woche 16 2,4% 7,2% 76,9%*, # 92,2% 15,3% 73,0 1,2% 2,3% 75,9%* 95,4% 76,5%* 92,5% 31,4% 69,2%
Tabelle 1: Klinisches Ansprechen in den zulassungsrelevanten Studien BE VIVID [3], BE READY [4] und BE SURE [5]. * p < 0,001 gegenüber Placebo (BE VIVID und BE READY) sowie gegenüber Adalimumab (BE SURE), auf Multiplizität adjustiert; # p < 0,001 gegenüber Usteki numab (BE VIVID), auf Multiplizität adjustiert.
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 117NEUE UND BEWÄHRTE ARZNEIMITTEL
BE VIVID BE READY BE SURE Placebo (n=83) (%) Bimeki zumab (n=321) (%) Ustekinumab (n=163) (%) Placebo (n=86) (%) Bimeki zumab (n=349) (%) Bimeki zumab (n=319) (%) Adalimumab (n=159) (%) PASI 100 in
16 0 58,6%* 20,9% 1,2% 68,2%* 60,8%* 23,9%
Woche
Bimekizumab
Bimekizumab (Bimzelx®) ist ein humanisierter monoklonaler IgG1Antikörper, der selektiv und direkt sowohl Interleukin 17A (IL-17A) als auch Interleukin 17F (IL-17F) inhibiert, zwei wichtige Zytoki ne, die Entzündungsprozesse steuern. Der Wirkstoff hemmt diese proinflammatorischen Zytokine, was zu einer Normalisierung der Hautentzündung und in der Folge zu einer Verbesserung der klini schen Symptome der Psoriasis führt.
Bimekizumab ist indiziert zur Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine syste mische Therapie infrage kommen. Es wird durch subkutane Injekti on verabreicht; die Injektionslösung ist in einer Fertigspritze und in einem Fertigpen erhältlich. Die empfohlene Dosis für Erwachsene mit Plaque-Psoriasis beträgt 320 mg Bimekizumab in Woche 0, 4, 8, 12, 16 und danach alle 8 Wochen. Bei einigen Patienten mit ei nem Körpergewicht ≥120 kg, die bis Woche 16 keine vollständige Symptomfreiheit der Haut erreicht haben, könnten 320 mg alle 4 Wochen nach Woche 16 das Ansprechen auf die Behandlung wei ter verbessern.
Cave: Bimekizumab kann das Risiko von Infektionen erhöhen. Bei Patienten mit einer klinisch relevanten aktiven Infektion darf die Behandlung mit Bimekizumab nicht eingeleitet werden [1].
tienten mit PASI 100 unter Secu kinumab bei 52,8 %, der Anteil erhöhte sich nach dem Wechsel zu Bimekizumab auf 76,1 % zu Wo che 96 [6].
Während der OLE-Phase waren die häufigsten unerwünschten Er eignisse unter Bimekizumab Na sopharyngitis (11,8/100 Patienten jahre), orale Candidose (7,8/100 Patientenjahre) und Harnwegsin fektionen (4,5/100 Patientenjah re). Die unerwünschten Ereignisse waren bei Patienten, bei denen der Wirkstoff angewendet wurde oder die von Secukinumab zu Bimeki zumab wechselten, vergleichbar [6].
G-BA bestätigt Zusatznutzen von Bimekizumab
Der Gemeinsame Bundesaus schuss hat einen Zusatznutzen des dualen Inhibitors gegenüber Secukinumab und Adalimumab festgestellt. Grundlage für den G-BA-Beschluss bildeten die Stu dien BE SURE [5] und BE RA DIANT [6], die Bimekizumab mit Adalimumab bzw. Secukinumab verglichen. Beide Studien wiesen in der Endpunktkategorie Remis sion unter kontinuierlicher Erhal tungsdosierung in Woche 16 einen PASI 90 und/oder PASI 100 und damit eine fast vollständige bis
vollständige Erscheinungsfreiheit der Haut auf:
• PASI-90-Ansprechen unter Bimekizumab im Vergleich zu Adalimumab: 86,2 % vs. 47,2 % (p < 0,0014)
• PASI-90-Ansprechen unter Bimekizumab im Vergleich zu Secukinumab: 85,5 % vs. 74,3 % (p < 0,0016)
Außerdem zeigte sich in Woche 24 gegenüber Adalimumab eine Verbesserung der gesundheitsbe zogenen Lebensqualität über den DLQI: 67,1 % der Patienten er reichten unter Bimekizumab ein DLQI 0/1, unter Adalimumab wa ren es 47,8 % [5].
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Bimzelx®; Stand: März 2022
2 Blauvelt A et al. Clin Rev Allergy Immu nol 2018;55:379-390
3 Reich K et al. Lancet 2021;397:487-498
4 Gordon KB et al. Lancet 2021;397:475486
5 Warren RB et al. N Engl J Med 2021; 385:130-141
6 Strober B et al. American Academy of Dermatology (AAD), March 25–29, 2022, Boston, USA; P34321
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 118 NEUE UND BEWÄHRTE ARZNEIMITTEL
Kongress aktuell: Berichte von der FOBI 2022
Effektive Narbenbehandlung –Strategien zu Prophylaxe und Therapie
Narben sind nach operativen Ein griffen, Unfällen und Verletzungen oft nicht zu vermeiden. Wenn bei der Erstversorgung oder vor dem Eingriff Strategien für eine minima le Narbenbildung beherzigt werden und eine entsprechende Nachsorge erfolgt, lassen sich ästhetisch anspre chende Behandlungsergebnisse er zielen (Abb. 1). Welche Optionen die aktuelle Leit linie zur Behandlung pathologischer Narben empfiehlt [1], diskutierten Experten auf einem von Merz ver anstalteten Symposium im Rahmen der 28. Fortbildungswoche für prak tische Dermatologie und Venerologie (FOBI).
Auf den richtigen Schnitt kommt es an
„Im Praxisalltag ist die Prävention von unschönen Narben das Ent scheidende – am besten, Sie entfer nen eine Läsion operativ so, dass die Narbe gar nicht erst nachbehan delt werden muss“, sagte der Der matologe Professor Gerd Gauglitz,
München. Und der Unfallchirurg und Orthopäde Dr. Dirk-Jonas Danneberg, Darmstadt, appellierte, bei jedem Zugang die Schnittfüh rung zu hinterfragen, die Inzision so klein wie möglich zu halten und so zu setzen, dass keine Kontrak turen entstehen. Bei der Schnitt führung helfe die Orientierung an den Spaltlinien, den Spannungsli nien der Haut, die durch Ausrich tung der Kollagenfaserbündel im Stratum reticulare entstehen. Eine atraumatische Wundrandbehand lung und ein Wundverschluss mit geringstmöglicher Spannung an den Wundrändern seien wichtig. Subkutannähte können die Wund spannung verringern, auch die Sta bilisierung mit Wundverschluss streifen könne die Wundränder weiter entlasten.
Präventive topische Narbenbehandlung
Wenn seine Patienten pathologi sche Narben entwickeln, bietet Danneberg ihnen aktiv Behand lungsmöglichkeiten an. Eine evi denzbasierte Option ist die Be handlung mit dem Narbengel Contractubex® mit den Wirkstoffen Extractum Cepae (Zwiebelextrakt),
Heparin und Allantoin [2], die nach erfolgtem Wundverschluss, im Allgemeinen 10 14 Tage nach der Operation, begonnen wird. Die Wirksamkeit bei rechtzeitiger An wendung von Contractubex® Gel bei bei hypertrophen und atrophen Narben sowie Keloiden wurde durch klinische Studien belegt [3, 4, 5]. So kam es in einer nicht in terventionellen Studie mit 1.268 Patienten in den mit Contractubex® Gel behandelten Arealen zu einer signifikant verbesserten Narben heilung gegenüber unbehandelten Verletzungsarealen. Narben-typi sche Symptome wie Spannung, Schmerz und Juckreiz besserten sich nach der 4- bis 5-monatigen Behandlung signifikant [4]. Die Anwendung von Contrac tubex® Gel führt zu einer Verbes serung der Wundheilung, einer Hemmung der Fibroblastenprolife ration, zur Reduktion von TGF-β1 und 2, zum Rückgang der Produk tion extrazellulärer Matrix, zur Bil dung Fibroblasten-senkender Fak toren und zum Remodelling von Kollagen [6]. Daher sieht Gauglitz gerade für die leitliniengerechte Prophylaxe der pathologischen Narbenbildung ein effektives Ein satzgebiet für das Narbentopikum Contractubex® Gel.
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 119NEUE UND BEWÄHRTE ARZNEIMITTEL
a) b)
Abbildung 1: Wundnaht nach Venenstripping für eine Bypass-Operation (a) und Narbe nach 6-monatiger Behandlung mit Contractubex® (b).
Effektive Penetration des Narbengels mithilfe von Ultraschall
In seiner Praxis setzt Danneberg ein Kombinationsverfahren aus Ultraschall und Narbengel nicht nur bei frischen, sondern auch bei älteren Narben ein [7]. Bei diesem Kombinationsverfahren zur Nach behandlung von Narben wird die Penetrationstiefe des Narbenspe zifikums Contractubex® Gel durch die Anwendung von Ultraschall (US) erhöht.
Laut Danneberg ist UST (Ultra sound Scar Therapy) für die Be handlung von Narben unterschied licher Genese, Größe und Alter geeignet – bis auf Narben von ma lignen Hautveränderungen. Er rät zu einer möglichst frühen An wendung, sobald der Wundschorf abgeheilt ist. Neben der zweimal täglichen Gel-Anwendung zuhau se wird alle 2 3 Tage therapeuti scher US eingesetzt. Nach der etwa 5-wöchigen Therapie empfiehlt Danneberg eine Gel-Anwendung mit sanfter Massage zuhause über weitere 6 Monate. „Das Ergebnis sind allgemein ästhetisch schönere Narben“, resümierte der Unfallchi rurg.
Leitliniengerechte Therapie pathologischer Narben
„Hypertrophe Narben und Keloide sind Ausdruck einer pathologisch veränderten Wundheilung mit ex zessiver Bildung von Narbengewe be“, erläuterte Gauglitz, Mitautor der Leitlinien [1]. Die Narbenrei fung kann durch Infektionen, über steigerte Fibroblastenbildung und patientenindividuelle Faktoren be einflusst werden. Auch ein auf die Wundränder wirkender Zug spielt eine Rolle. Daher rät Gauglitz sei
nen Patienten, unbedingt 6 Monate lang eine Dehnung der Wundrän der zu vermeiden. Denn in dieser Zeit reagieren die Fibroblasten auf einen Dehnungsreiz mit einer er höhter Kollagensynthese [8]. Be sonders gefährdet sind in dieser Hinsicht der Brust- und Schulter bereich.
Für die Behandlung von Keloiden sind intraläsionale Kortikostero ide (z.B. Triamcinolonacetonid [TAC]) und Kryotherapie, auch in Kombination, immer noch die Me thode der ersten Wahl. Bei nicht ausreichendem Ansprechen auf diese Therapie nach 8 12 Wochen empfehlen die Leitlinien eine Be handlung mit dem Zytostatikum 5-Fluorouracil (5-FU) plus TAC [1].
Laserbehandlung
Bei Keloiden und Narbenverfär bungen kommen auch Lasersys teme zum Einsatz. Die Leitlinie zur Lasertherapie der Haut geht zwar ausführlich auf diese Optio nen ein, formuliert aber mangels kontrollierter Studien nur zurück haltende Empfehlungen [8]. So könne zur farblichen Optimierung von Narben ein Farbstofflaser an gewandt werden und für die Fol gebehandlung hypertropher Nar ben der fraktionierte CO2-Laser. Dieser ist jedoch, wie Gauglitz betonte, bei Keloiden fast immer kontraindiziert. Wichtig sei, mit geringer Energie zu arbeiten, um eine Überhitzung des Gewebes zu vermeiden. „Weniger ist mehr, ge ringe Dichten, geringe Energien“, so sein Fazit, um eine erneute Ke loidbildung zu vermeiden.
Fabian Sandner, Nürnberg
Quellen
1 S2k-Leitlinie Therapie pathologischer Narben (hypertrophe Narben und Keloi de), AWMF-Register-Nr. 013-030, 2020
2 Fachinformation Contractubex®, Stand: Mai 2021
3 Maragakis M et al. Drugs Exp Clin Res 1995;21:199-206
4 Willital GH et al. J Drugs Dermatol 2013; 12:38-42
5 https://www.contractubex.de/studien/
6 Phan TT et al. J Trauma 2004;57:10321037
7 Danneberg DJ, Kosmetische Medizin 2007;28:133-138
8 S2k-Leitlinie Lasertherapie der Haut, AWMF-Register-Nr. 013-095, 20217
5 Jahre Kyntheum® Schnelle und langanhaltende Wirksamkeit bei Psoriasis
„Biologika haben die Behandlung der mittelschweren bis schwerer Psoriasis revolutioniert“, sagte Dr. Ralph von Kiedrowski, Sel ters, auf der Pressekonferenz von LEO Pharma im Rahmen der 28. Fortbildungswoche für praktische Dermatologie und Venerologie (FOBI). Mit seiner Zulassung vor 5 Jahren war Kyntheum® (Broda lumab) der erste Antikörper für die Behandlung der Psoriasis, der nicht einzelne Zytokine hemmt, sondern direkt den IL-17 Rezeptor blo ckiert. Seitdem hat sich viel getan: Langzeitdaten sowie Alltagserfah rungen zu Brodalumab bestätigen die schnelle und langanhaltende Wirksamkeit und das gute Sicher heitsprofil des Medikaments [1, 2, 3]. „Unter Kyntheum® erreicht ein hoher Anteil der Patienten eine er scheinungsfreie Haut“, berichtete Kiedrowski und ergänzte: „Über die Hälfte der Patienten kann die ses Ansprechen über 5 Jahre beibe halten – das ist ein großer Gewinn an Lebensqualität.“
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 120 KONGRESSE / FOBI 2022
andauernde 52-wöchige, offene, prospektive, multizentrische, nicht-interventionelle Studie mittelschwerer bis (sehr) schwerer Psoriasis aus 216 Zentren in Deutschland. Primärer
Therapieziel eines absoluten PASI-Scores von ≤ 3 Punkten unter Brodalumab. präsentiert die Baseline-Daten (n = 502) und Interimsdaten zu Woche 12 (n = 460).
Schneller Wirkeintritt
PASI-Wert in Woche 2, 4 und 12 nach Brodalumab („as observed“ Auswertung; kommastelle.)
64,9 % männlich, mittlerer BMI Psoriasis-Dauer 20 Jahre, mittlerer
eine schwere (PASI ≥ 20) bis (PASI ≥ 30).
Für die Patienten sind ein schneller Wirkeintritt und eine langanhal tende Linderung der mit Psoriasis assoziierten Symptome besonders wichtig. In der gepoolten Auswer tung der Phase-III-Studien AMA GINE-2 und -3 betrug die Zeit, bis 25 % der Patienten unter Broda lumab ein PASI 75-Ansprechen* erreicht hatten, 2,1 Wochen. Unter Ustekinumab dauerte es mit 4,8 Wochen mehr als doppelt so lan ge [4]. „Gehen die Hautläsionen schnell zurück, bessern sich in der Regel auch Begleiterscheinungen rasch“, betonte von Kiedrowski. Gemäß einer Netzwerk-Meta-Ana lyse ist Brodalumab eines der am schnellsten wirksamen Biologika für mittelschwere bis schwere Pso riasis [5].
Mittlerer absoluter PASI
18 16 14 12 10 8 6 4 2 0
16,9 9,1 4,8 2,9
Baseline (n = 502) ca. W2 (n = 471) ca. W4 (n = 477) ca. W12 (n = 460)
%) der Patienten waren mit einem %) oder mindestens drei (14,9 %) vor allem mit Adalimumab (45,3 %), Ustekinumab (24,4 %). sich ein schnelles und starkes Ansprechen über alle Subgruppen hinweg: PASI-Wert verringerte sich um fast die Hälfte von 16,9 zu Baseline auf weiter auf 2,86 Punkte in Woche 12 nach der Ein- oder Umstellung auf 1). erreichten 3 von 4 Patienten (77,2 %) in Woche 12 den primären observed“-Auswertung). Ein Drittel der Patienten (32,4 %) erreichte ein
Abbildung 1: Die noch andauernde 52-wöchige nicht interventionelle Studie LIBERO lieferte erstmals prospektive Daten zur Effektivität des vollhumanen monoklonalen Antikörpers Bro dalumab (Kyntheum®) bei nicht selektierten Patienten mit mittelschwerer bis schwerer Psori asis unter Alltagsbedingungen. Endpunkt ist das praxisrelevante Therapieziel eines absoluten PASI-Scores von ≤3 Punkten unter Brodalumab. Das Diagramm zeigt die Baseline- und Inte rimsdaten zu Woche 12 [1].
Langanhaltende Wirksamkeit
„Natürlich ist aber auch wich tig, dass das Ansprechen weiter hin aufrechterhalten wird“, führte von Kiedrowski weiter aus. „In den letzten 5 Jahren gab es hier zu vielversprechende Daten, die zeigen, dass Brodalumab auch in der Langzeitbehandlung wirksam ist.“ In einer gepoolten Auswer tung der offenen Langzeitver längerung der Phase-III-Studien AMAGINE-2 und -3 zeigte sich,
* Der Psoriasis Area and Severity Index (PASI) ist das am häufigsten eingesetzte Score-Verfahren zur klinischen Beurteilung einer Psoriasis. Er beurteilt sowohl den pro zentualen Anteil der betroffenen Kör peroberfläche als auch die Symptomstärke (Rötung, Dicke der Plaques und Stärke der Schuppenbildung) mit einer Skala von 0 72 Punkten. Basierend auf dem PASI lässt sich der Schweregrad der Psoriasis ermitteln. Das PASI-75-Ansprechen ist laut Psoriasis-Leit linie das aktuelle Therapieziel einer Psoria sis-Behandlung und entspricht einer 75%igen Verbesserung der Krankheits symptome.
dass 86 % der Patienten, die einen PASI 90 erreicht hatten, diesen bei kontinuierlicher Behandlung mit Brodalumab über 2 Jahre lang auf rechterhalten konnten. Bei PASI 100 waren es 74 % [6]. Darüber hinaus zeigte die offene Langzeit verlängerungsstudie der Phase II, dass über 80 % der Patienten unter kontinuierlicher Brodalumab-The rapie über 240 Wochen – also über 5 Jahre – ein nahezu erscheinungs freies Hautbild (PASI-90-Anspre chen) aufrechterhalten konnten. Außerdem erreichten mehr als die Hälfte dieser Patienten eine PASI100-Response und behielten diese über 240 Wochen bei [3].
Das Sicherheitsprofil war in beiden Langzeitstudien günstig und zeigte sich konsistent mit dem Profil der 12- und 52-Wochen-Phasen. Es fanden sich keine neuen Sicher heitssignale.
Fest im Praxisalltag etabliert
In der Real-World-Studie LIBERO wurde das Ansprechen auf Broda lumab unter Alltagsbedingungen geprüft [1]. Das Fazit: Auch in der allgemeinen klinischen Praxis wirkt Brodalumab schnell. Bereits 2 Wochen nach Therapiebeginn hatte sich der absolute PASI na hezu halbiert. Der Therapieerfolg war auch hier hoch: 77 % der Pa tienten hatten zu Woche 12 einen absoluten PASI von ≤3 erreicht (Abb. 1). Ein vergleichbar schnel les Ansprechen war auch bei den Patienten zu beobachten, die nach einer Biologika-Vortherapie mit TNFα- (47 %, v.a. Adalimumab) bzw. IL-17A-Inhibitoren (39 %, v.a. Secukinumab) auf Broda lumab umgestellt wurden [1]. Damit mehr Patienten von diesen Vorteilen profitieren können, ist
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 121KONGRESSE / FOBI 2022
praxisrelevante
Kyntheum® seit Mai innerhalb des DermaOne-Versorgungsvertrages als besonders wirtschaftliches Fo kusarzneimittel gekennzeichnet und als einziges Arzneimittel in nerhalb der IL-17-Klasse als be sonders wirtschaftliches Fokusarz neimittel anerkannt [7, 8].
Elisabeth Wilhelmi, München
Dr. Uwe Schwichtenberg, Bremen, auf einem Mittagsseminar anläss lich der FOBI 2022.
Quellen
1 von Kiedrowski et al. LIBERO: erste RWE Daten von Brodalumab, Posterprä sentation bei der Münchner Fortbildungs woche digital 2020; e-Poster #eP-66
2 Puig et al. J Am Acad Dermatol 2020;82: 352-359
3 Lebwohl et al. Am J Clin Dermatol 2019; 20:863-871
4 Blauvelt A et al. J Am Acad Dermatol 2017;77:372-374
5 Warren RB. et al. Dermatol Ther 2020; 10:73-86
6 Reich K et al. J Eur Acad Dermatol Vene reol 2022;36:1275-1283
7 Vertragssteckbrief DermaOne: Vertrag zur besonderen Versorgung nach §140A SGB V in den Indikationen Psoriasis und Neurodermitis. https://richtercareconsul ting
8 de/wp-content/uploads/2021/11/Vertrags steckbrief_Psoriasis_20220101.pdf
9 Gemäß Arzneimittelampel des DermaO ne-Vertrages der Vertragspartner: BVDD, DermaMed e.G., Richter Care Consulting GmbH und Techniker Krankenkasse
Haarausfall – Antworten aus der Haarsprechstunde
„Patienten mit Haarausfall zeigen oftmals einen extrem hohen Lei densdruck. Hier kann und muss die Dermatologie mit der entspre chenden Fachkompetenz zur Seite stehen. Es ist wichtig, im Rahmen einer speziellen Haarsprechstunde nachgewiesen wirksame Behand lungen zu empfehlen und den Be troffenen darüber hinaus zu helfen, den eventuellen Stellenwert von Shampoos oder topischen Kosme tika einordnen zu können“, betonte
Eine gut organisierte Haarsprech stunde besteht laut Schwichten berg aus einer Befragung zur Vorgeschichte (evtl. unter Einsatz eines Fragebogens, den die Patien ten möglichst schon zuhause aus gefüllt haben), aktivem Zuhören in der Praxis sowie einer Unter suchung (Haare anfassen, zupfen und vergrößern), einer Diagnose und einer möglichst wirksamen Behandlung.
Diffuses Effluvium oder androgenetische Alopezie?
„Typische Patientinnen sind die, die oberflächlich betrachtet noch viele Haare haben, aber um ihr Haarvo lumen fürchten; oft kommen sie mit den massiv ausgefallenen Haaren in die Sprechstunde“, berichtete Dr. med. Andreas M. Finner, Ber lin. Wichtig für die Diagnosestel lung sind Angaben zum Beginn und Verlauf sowie die Lokalisation des Haarausfalls, die Erfassung von Ereignissen vor Beginn des Effluvi ums wie Krankheiten, Medikamen ten-Einnahme oder Ernährungs umstellungen (z.B. Crash-Diäten). „Der geschulte Blick des Arztes ist wichtig bei der Frage, ob eine Haardichteminderung vorliegt und ob sie umschrieben oder diffus ist. Mittels Vergrößerung im Dermatobzw. Trichoskop, lässt sich schon eine Verdachtsdiagnose stellen“, er läuterte Finner.
Zur Bestimmung der Aktivität des Haarausfalls gehört der Zupftest, bei dem der Arzt ca. 60 Haare greift und wenn >6 davon klar po sitiv mit Wurzel ausgegangen sind, kann entweder ein diffuses Effluvi um vorliegen oder eine androgene tische Alopezie. Bei Letzterer zeigt
sich dann der Haarausfall insbeson dere am Oberkopf von Frau oder Mann. Bei diesem Krankheitsbild könnte z.B. Pantostin® mit dem Wirkstoff Alfatradiol 0,25 mg/ml als Lösung zum Auftragen auf die Kopfhaut geeignet sein. Es kann die verminderte Anagenhaar-Rate bei der androgenetischen Alopezie bei Männern und Frauen steigern. Die äußerliche Anwendung auf der Kopfhaut ist auch als gut oder sehr gut verträglich bewertet [1].
„Im Gegensatz zur androgeneti schen Alopezie, die ein klassisches Muster hat, findet sich bei einem diffusen Haarausfall kein Muster“, ergänzte Schichtenberg. Dauert er weniger als 6 Monate an, besteht ein akutes Telogeneffluvium, z.B. bei der Mutter nach der Geburt des Kindes oder nach sog. Crash-Diä ten oder aber nach einer COVID19-Infektion [2], also postinfek tiös. Besteht diffuser Haarausfall länger als 6 Monate, ist er chro nisch und es können Medikamen te, Grunderkrankungen, Eisen mangel oder psychogene Probleme als Ursachen vorliegen. In diesen Fällen kann es sinnvoll sein, die Haarwurzeln ausreichend mit den für das Wachstum wichtigen Auf baustoffen, z.B. enthalten in Pan tovigar® vegan, zu versorgen.
Wie können Betroffene gestärkt werden?
„Es geht nicht um den täglichen Haarausfall, sondern darum, ob die Haare, die ausfallen, in gleichem Maße wieder nachwachsen“, beton te Finner. Nachwachsende Haare kann man anhand einer Haarkarte sichtbar machen oder durch eine digitale Haarmessung, wobei die Untersuchung möglichst immer an derselben Kopfhautstelle erfolgen sollte.
JOURNAL
©
PERFUSION GMBH 122 KONGRESSE / FOBI 2022
PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG
VERLAG
Diätmanagement bei diffusem Haarausfall
Das Problem des diffusen Haarausfalls resultiert oft aus einer Unterversorgung der Haarwurzeln mit wichti gen Nährstoffen, sodass die Wachstumsphase des Haares verkürzt ist und Haare vor zeitig ausfallen. Die erfolgrei che Behandlung des diffusen Haarausfalls kann daher eine Supplementierung mit wich tigen Nährstoffen einschlie ßen. Betroffenen Patientin nen kann empfohlen werden, Pantovigar® vegan über einen Zeitraum von mindestens 3 Monaten einzunehmen. Pantovigar® vegan ist qualita tiv bewertet ein Lebensmittel für besondere medizinische Zwecke (bilanzierte Diät) und steht für das Diätmanage ment bei diffusem Haaraus fall bei Frauen zur Verfügung. Die Kapseln enthalten eine spezielle Nährstoffkombina tion aus Cystin, B-Vitaminen (B1, B5, B7), Vitamin H und Folsäure sowie Eisen und Zink.
„Bei der androgenetischen Alope zie ist das Therapieziel, dass sich der Haarausfall nicht weiter ver schlimmert, sondern das Niveau bleibt“, erinnerte Finner. Eine Haartransplantation kann kahle Stellen wieder auffüllen. Da an fangs häufig Mischformen mit diffusem Haarausfall auftreten, kann eine zusätzliche Nährstoff versorgung mit Pantovigar® vegan empfohlen werden. Die Kapseln enthalten Cystin, B-Vitamine (Thi amin, Pantothensäure, Biotin, Fol säure) sowie Mineralstoffe (Zink, Eisen) und sind laktose- und glu tenfrei.
Große Angst besteht bei den Be troffenen vor einer Glatzenbil dung. Ein Krankheitsbild, bei dem diese Furcht besteht, ist die fronta le fibrosierende Alopezie, bei der der Haaransatz immer weiter zu rückweicht. Sie wird antientzünd lich behandelt. In manchen Fällen kann eine Haartransplantation helfen. Auch hier kann eine ergän zende Supplementierung mit der Nährstoffkombination aus Cystin, B-Vitaminen und Mineralstoffen sinnvoll sein.
Haarausfall bei COVID-19-Infektion
Auch eine COVID-19-Infektion kann Ursache für einen vorüber gehenden Haarverlust (Zeitfenster 2 –9 Monate) sein [3]. „Das Telo geneffluvium tritt etwa 2 Monate nach einer COVID-19-Infektion auf“, berichtete Schwichtenberg. Meistens kommt es nach 4 Mona ten wieder zum Haarwuchs, aber die Zeit bis dahin ist von Angst und Sorge um die Haarpracht geprägt. „Haarausfall ist kein LifestyleThema, unsere Patienten leiden daran immens“, sagte Schwichten berg. Motivierend für den Patien ten ist es, ihm anhand eines Fotos und durch die computergestützte Trichoskopie eine neutrale Beur teilung seiner Haarstruktur zu lie fern.
Schwichtenberg organisiert we gen der Haarnotfälle Online-Vi deosprechstunden (versierte Ärzte können sich auf www.trichocare. de eintragen lassen).
Fabian Sandner, Nürnberg
Lasertherapie der Haut:
Zielgerechte Nachbehandlung mit Bepanthen® Wund-
und Heilsalbe
Laser kommen heute in einer gan zen Reihe dermatologischer In dikationen zum Einsatz. „Bei der Auswahl eines bestimmten La sers für eine definierte Indikation und deren Anwendung gibt die im März 2022 erschienene neue S2kLeitlinie Behandlerinnen und Be handlern mehr Sicherheit im Pra xisalltag“, erläuterte Professor Dr. Peter Arne Gerber, Düsseldorf, auf einem Seminar im Rahmen der 28. Fortbildungswoche für praktische Dermatologie und Venerologie (FOBI 2022).
„Zu den wichtigen Einsatzgebie ten ablativer Laser – zum Beispiel von CO2- oder Er:YAG-Lasern (Erbium:Yttrium-Aluminium-Gra nat-Lasern) – gehören die Behand lung aktinischer Keratosen sowie die Narbentherapie“, erklärte Ger ber und ergänzte: „Diese Laser schädigen die Hautbarriere und es entstehen Wunden. Um deren Ab heilung zu optimieren und Kom plikationen zu vermeiden, ist eine adäquate Nachsorge unbedingt zu empfehlen.“
Dexpanthenol erfüllt Anforderungen der Leitlinie an die Nachsorge
Quellen
1 2000;11:137 ff.
2 Starace M et al. JAAD Int 2021;5:11-18
3 Tosti A et al. JAAD Int 2022;7:67-77
Ziel der Nachbehandlung laser induzierter Läsionen ist es, die Wunden vor Infektionen und frei en Radikalen zu schützen, die Entzündung zu modulieren, die Zellproliferation und Migration zu fördern sowie das Remodelling zu unterstützen. Eine in der Leitlinie empfohlene Option ist die Anwen dung dexpanthenolhaltiger Salben [1].
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 123KONGRESSE / FOBI 2022
Bepanthen® Wund- und Heilsalbe beschleunigt die Wundheilung nach Lasertherapie
Die Evidenz für die Nennung dexpanthenolhaltiger Externa wie Bepanthen® Wund- und Heilsal be liefern klinische Studien. Bei spielsweise zeigte eine Untersu chung von Heise et al., dass nach einer ablativen Lasertherapie we gen einer aktinischen Keratose der relative Wunddurchmesser unter der Bepanthen® Wund- und Heil salbe in den ersten Tagen signifi kant geringer war als unter Vaseli ne [2].
„Um die biologischen Effekte von Lasersystemen und der topischen Nachbehandlung besser zu verste hen, führen wir Untersuchungen an humanen dreidimensionalen Hautmodellen durch. Diese Un tersuchungen bestätigten, dass Dexpanthenol die Abheilung nach einer Behandlung mit einem abla tiven CO2Er:YAG-Laser fördert“, berichtete Professor Dr. Jens Malte Baron, Aachen [3].
Tattoo-Entfernung: schnellere Abheilung mit Bepanthen® Wund- und Heilsalbe
„Neben ablativen Lasern kommt eine Reihe weiterer Laser-Systeme in der Dermatologie zum Einsatz wie beispielsweise der Picosekun den-Laser zur Entfernung von Tat toos“, sagte Baron und fügte hinzu: „Ebenfalls am Hautmodell konn ten wir demonstrieren, dass auch beim Einsatz dieses Laser-Typs eine einmal tägliche Nachbehand lung mit Bepanthen® Wund- und Heilsalbe über 5 Tage mit einer schnelleren Heilung assoziiert ist. Eine Genexpressionsanalyse be stätigte, dass das in der besonderen atmungsaktiven Galenik enthalte
ne Dexpanthenol regenerative Pro zesse wie Gefäßneubildung sowie Entwicklung anatomischer Struk turen fördert und Stressantwort und Zellwanderung reguliert.“
Ausblick: Laser-assistierte Narbenheilung
Erste Untersuchungen deuten da rauf hin, dass eine Nachbehand lung mit Bepanthen® Wund- und Heilsalbe auch bei der laserun terstützten Narbenbehandlung (LASH) sowie dem medizinischen Mikroneedling von Vorteil sein könnte. Bei einer LASH wird der unmittelbar postoperative Wund heilungsprozess durch eine Tem peraturerhöhung in der Haut modi fiziert. Dazu wird dem Hautmodell zunächst mit einem fraktionierten ER:YAG-Laser eine standardi sierte Wunde zugefügt und diese dann mit einem nicht ablativen Er:Glass-Laser oder einem Dio denlaser nachbehandelt. Bereits diese Nachbehandlung beschleu nigt den Heilungsprozess. Wird zusätzlich Bepanthen® Wund- und Heilsalbe angewendet, verbessert sich die Abheilung weiter. „Bereits 16 Stunden nach der Behandlung
mit einem Er:YAG-Laser oder ei ner LASH-Behandlung zeigten sich im Hautmodell Unterschie de im histologischen Bild: Un ter der Nachbehandlung mit der dexpanthenolhaltigen Salbe war der Wundverschluss weiter fort geschritten als unter der alleinigen Anwendung der nicht ablativen Lasertherapie.“
Baron resümierte: „Die Nachbe handlung mit der dexpanthenol haltigen Salbe kann die Abheilung von Hautbarriereverletzungen nach ablativer Lasertherapie, Ent fernungen von Tätowierungen, medizinischem Mikroneedling oder einer Laser-assistierten Haut heilung (LASH) beschleunigen ohne die Wirkeffekte der Behand lung aufzuheben.“
Elisabeth Wilhelmi, München
Quellen
1 S2k-Leitlinie Lasertherapie der Haut, AWMF-Registernummer 013-095, 2022; verfügbar unter: https://www.awmf.org/ uploads/tx_szleitlinien/013-095l_S2k_ Lasertherapie-der-Haut_2022-03.pdf
2 Heise R et al. Cutan Ocul Toxicol 2019;38:274-278
3 Huth S et al. Laser Med Sci 2021;36: 1117-1121
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 124 KONGRESSE / FOBI 2022
Abbildung 1: Durch die Nachbehandlung mit Bepanthen® Wund- und Heilsalbe heilen Haut barriereverletzungen nach einer ablativen Lasertherapie rascher ab (© Bayer).
Chronisch entzündliche Darmerkran kungen: Moderne
Kombinationstherapie bleibt vorerst Einzelfallentscheidung
Der Nutzen klassischer Kom binationstherapien, etwa von Tumornekrosefaktor-α-Inhibitoren (TNFi) plus Azathioprin, ist bei chronisch entzündlichen Darmer krankungen (CED) gut belegt. Er liegt vor allem darin, ein sekun däres Wirkversagen zu verhindern. „Moderne“ Kombinationen die verschiedene Biologika unterein ander oder mit Januskinase-Inhi bitoren (JAKi) kombinieren, sind Optionen für gut begründete Ein zelfälle, hieß es beim 33. Interdis ziplinären Kolloquium Chronisch entzündliche Darmerkrankungen der Falk Foundation e.V. Prof. Britta Siegmund, Berlin, er innerte daran, dass schon 2010 nachgewiesen wurde, dass Infli ximab (IFX) in Kombination mit Azathioprin signifikant häufiger zur steroidfreien Remission führt als die Monotherapien beider Prä parate. Wesentlich ist, dass sich durch die Kombinationen seltener Anti-Drug-Antikörper entwickeln. Das belegt eine Studie mit 1240 Morbus Crohn (MC)-Patienten, die IFX oder Adalimumab jeweils mit oder ohne klassisches Immun suppressivum erhielten.
Anders als bei TNFi bringt die Kombination von Ustekinumab (UST) oder Vedolizumab (VDZ) mit Thiopurinen oder Metho trexat keinen Mehrwert. Dafür werden zielgerichtete Therapien zunehmend untereinander kombi niert. Beispiele sind UST+VDZ, TNFi+VDZ/UST oder auch
VDZ+Tofacitinib. „Naheliegend ist immer eine Kombination mit Vedolizumab, weil es lokal wirkt und dadurch auch die Nebenwir kungen potenziell in Schach ge halten werden können“, erläuterte Siegmund. Ihr Fazit zu modernen Kombinationen: „Bei einigen Pa tienten funktioniert es, aber es ist keine Win-win-Situation. Letztlich fußt die Evidenz trotz Metaanalyse auf kleinen Fallserien – deswegen sind das im Moment Fall-basierte Entscheidungen.“
„Was in der Praxis neben Komor biditäten am häufigsten die In dikation für eine moderne Kom binationstherapie darstellt, sind die extraintestinalen Manifesta tionen“, so Siegmund. Also etwa Gelenk- oder Wirbelsäulenbe teiligung, Psoriasis, Uveitis und Pyoderma gangraenosum. TNFi bezeichnete Siegmund als Joker. „Damit liegen Sie eigentlich im mer richtig.“ Auch JAKi haben ein breites Wirkspektrum, Interleukin23-Blocker punkten bei Hautbe fall.
Fabian Sandner, Nürnberg
Multiple Sklerose: Früher Einsatz der oralen Impulstherapie lohnt sich
Aktuelle Daten vom ACTRIMS* Forum 2022 untermauern erneut die Wirksamkeit von CladribinTabletten (Mavenclad®). Professor Hans-Peter Hartung, Düsseldorf, verwies dazu auf die GLIMPSEStudie, eine longitudinale, retro spektive Analyse der Daten von 3.475 Patienten mit hochaktiver schubförmiger MS [1]. Die Studi
* Americas Committee for Treatment and Research in Multiple Sclerosis
enteilnehmer waren zwischen 2018 und 2021 entweder mit CladribinTabletten, Fingolimod (FTY), Di methylfumarat (DMF) oder Teri flunomid (TER) behandelt worden. Wie Hartung ausführte, blieben Patienten unter Cladribin-Tablet ten signifikant länger schubfrei als unter den übrigen oralen MSTherapien. Die Zeit bis zum ersten Schub war bei den mit CladribinTabletten behandelten Patienten um 40 %, 42 % bzw. 67 % länger als bei Patienten, die FTY, DMF bzw. TER erhalten hatten. Zudem war das Intervall bis zum Thera piewechsel unter Cladribin-Tab letten 4-mal länger als unter FTY, 7-mal länger als unter DMF und 6,5-mal länger als unter TER [1].
Diese Erkenntnisse werden durch Daten der Real-World-Studie MERLYN-MS gestützt, die eben falls auf dem ACTRIMS Forum 2022 präsentiert wurden. MER LYN-MS ist eine retrospektive Datenerhebung bei überwiegend deutschen Patienten mit hochaktiver schubförmiger MS im ersten Jahr einer Behandlung mit Cladribin-Tabletten bzw. FTY [2]. Die Wirksamkeit in Bezug auf die Reduktion der Schubrate war un ter beiden Therapien vergleichbar, allerdings kam es unter CladribinTabletten seltener zu einem Thera piewechsel [2].
„Hit smart and early“
Die Bedeutung des Konzepts „Hit smart and early“ verdeutlichte der Neurologe Michael Ernst, Sins heim, anhand von Fallbeispielen. „Je früher wir mit der Therapie beginnen, desto besser, um die Le bensqualität bestmöglich erhalten und die Erkrankung effektiv aus bremsen zu können.“ In der Praxis ist die Therapie sehr einfach zu
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 125KONGRESSE
initiieren und erfordert nur weni ge Kontrollen. Dabei überzeugt die orale Impulstherapie auch bei der SARS-CoV-2-Impfung durch hohe Antikörpertiter und eine voll ständige Impfantwort. Eine Fami lienplanung ist unter CladribinTabletten-Therapie grundsätzlich bereits 6 Monate nach der letzten Behandlungsphase möglich.
Dr. med. Kirsten Westphal, München
mit 1.610 Teilnehmern. Jetzt lie fert eine Subgruppenanalyse für Deutschland vergleichbare Ergeb nisse.
Subgruppenanalyse für Deutschland ...
... belegt deutliche Verbesserungen unter Safinamid
Quellen
1 Butzkueven H et al. ACTRIMS Forum 2022, Poster P342
2 Brownlee W et al. ACTRIMS Forum 2022, Poster P095
Morbus Parkinson: Safinamid überzeugt auchbei Älteren und Patienten mit Komorbiditäten
Auch ältere Patienten mit Morbus Parkinson (>75 Jahre) und solche mit psychiatrischen Erkrankungen oder anderen relevanten Komorbi ditäten können von der Sicherheit und Wirksamkeit einer Add-onTherapie mit Safinamid (Xadago®) profitieren. Dafür sprachen bereits die europäischen Real-WorldDaten der SYNAPSES-Studie
Professor Wolfgang Jost aus Or tenau stellte die Analyse bei einer Veranstaltung des Unternehmens Zambon vor. Von den in Deutsch land teilnehmenden 181 Patienten waren 28,7 % mehr als 75 Jahre alt. Das Durchschnittsalter lag bei 69 Jahren. 84,5 % wiesen relevan te Komorbiditäten auf und 38,1 % psychiatrische Erkrankungen. Pa tienten in diesem Alter oder mit solchen Komorbiditäten müssen natürlich ebenfalls behandelt wer den, sind aber in vielen Zulas sungsstudien unterrepräsentiert. Primäres Studienziel der einjähri gen Kohorten-Beobachtungsstudie war die Verträglichkeit der Addon-Gabe von Safinamid. Die Ta gesdosis betrug zu Beginn 50 mg; sie konnte später erhöht oder ver ringert werden (erhöht bei 62 %; gesenkt bei 6 %). Die sekundären Endpunkte umfassten unter ande rem motorische Funktionen.
Klinisch relevante Verbesserungen zeigten sich in der Verringerung der Unified Parkinson‘s Disease Rating Scale (UPDRS). Der mo torische Score nahm um 45 % (> –2,5 Punkte) ab und der Gesamt score um 50 % (> –4,3 Punkte). Motorische Fluktuationen, etwa am Morgen, gingen stark zurück. Unter Dyskinesien litten anfangs 27,6 % der Patienten, nach 12 Mo naten Safinamid-Behandlung nur noch 14,8 %. Wearing-off-Sym ptome trafen nur noch 55 % statt 78,5 % (Tab. 1). Unerwünschte Ereignisse gaben 62 % der Teilnehmer an; 16 % meldeten mindestens ein schweres unerwünschtes Ereignis. Wie Jost betonte, stand ein Großteil der un erwünschten Wirkungen nicht im Zusammenhang mit dem Prüfprä parat. So waren zum Beispiel Sym ptome wie Obstipation eine Folge der Grunderkrankung und nicht der Therapie.
Helga Brettschneider, Frankfurt
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 126 KONGRESSE
Tabelle 1: Ergebnisse der SYNAPSES-Studie für die teilnehmenden Parkinson-Patienten aus Deutschland (Quelle: Abbruzzese G et al. J Par kinsons Dis 2021;11:187-198). Motorische Kompli kationen Gesamtkohorte (n=181) Anzahl der Patienten nach 4 Monaten (n=160) Anzahl der Patienten nach 8 Monaten (n=147) Anzahl der Patienten nach 12 Monaten (n=149) Alle 181 (100,0%) 128 (80,0%) 115 (78,2%) 106 (71,1%) Wearing-off 142 (78,5%) 94 (58,8%) 86 (58,5%) 82 (55,0%) Unvorhersehbare Fluk tuationen 17 (9,4%) 9 (5,6%) 5 (3,4%) 10 (6,7%) Morgendliche Fluktu ationen 60 (33,1%) 40 (25,0%) 40 (27,2%) 36 (24,2%) Verspätetes ON 9 (5,0%) 3 (1,9%) 3 (2,0%) 4 (2,7%) Dyskinesien 50 (27,6%) 34 (21,3%) 25 (17,0%) 22 (14,8%) Andere 8 (4,4%) 3 (1,9%) 6 (4,1%) 3 (2,0%)
Rheuma und kardiovaskuläres Risiko: EULAR präzisiert Therapieempfehlungen bei der rheumatoiden Arthritis
Die Therapie der rheumatoiden Arthritis wird kontinuierlich wei terentwickelt. Neuere Erkenntnis se zur Verträglichkeit von bislang verordneten Rheuma-Medikamen ten haben nun zu einer Überarbei tung der Therapieempfehlungen der European Alliance of Associa tions for Rheumatology (EULAR) geführt: Um das kardiovaskuläre Risiko für Patienten zu minimie ren, sollen Glukokortikoide nur in sehr geringer Dosis und zeitlich begrenzt eingesetzt werden. Auch für die Medikamentengruppe der Januskinase-Inhibitoren gibt die EULAR neue Anwendungshinwei se. Die Deutsche Gesellschaft für Rheumatologie e.V. (DGRh) be grüßt die Anpassung der EULAREmpfehlungen.
Glukokortikoide so schnell wie möglich absetzen
Zum Zeitpunkt der Diagnose „rheumatoide Arthritis“ (RA) lei den die Betroffenen häufig bereits unter ausgeprägten Beschwer den: Gelenke im ganzen Körper sind schmerzhaft entzündet, die Lebensqualität ist deutlich ein geschränkt. Weil die Wirkung der Basistherapie mit konventio nellen, synthetisch hergestellten DMARDs (Disease Modifying Anti-Rheumatic Drugs) wie z.B. Methotrexat nur verzögert einsetzt, werden diese zunächst mit Gluko kortikoiden (Kortison-Präparate) kombiniert. Diese unterdrücken die Entzündung rasch, lindern die
Krankheitssymptome sofort und überbrücken somit die Zeit bis zur Wirkung der Basismedikamente. „Glukokortikoide haben jedoch ein breites Spektrum an möglichen unerwünschten Wirkungen, vor allem wenn sie über eine längere Zeit oder in hoher Gesamtmen ge eingenommen werden“, sagt Professor Andreas Krause, Präsi dent der DGRh. „Im Hinblick auf kardiovaskuläre Ereignisse zäh len u.a. Herzinfarkt, Schlaganfall, entgleister Bluthochdruck und Thrombosen dazu.“ Daher ach ten Rheumatologen sehr darauf, Glukokortikoide nur so kurz wie möglich und so niedrig wie nö tig zu dosieren. Dies wird in den EULAR-Empfehlungen nun noch einmal konkretisiert.
Die aktuelle Empfehlung der EU LAR (2022) zum Ausschleichen der begonnenen GlukokortikoidTherapie wurde gegenüber der vorherigen Empfehlung aus dem Jahr 2019 insofern verschärft, als dass Glukokortikoide nicht nur so schnell wie möglich reduziert, son dern reduziert und dann abgesetzt werden sollen. Diese überarbeitete Empfehlung stimmt mit der Leit linie der DGRh überein, welche bereits 2018 empfahl, dass die Glukokortikoid-Therapie auf 3 – 6 Monate beschränkt werden soll. „Dies impliziert, dass auch sehr ge ringe Dosen von weniger als 5 mg pro Tag demnach nicht als Dauer therapie gegeben, sondern weiter reduziert und abgesetzt werden sollen“, so Krause. Denn für das kardiovaskuläre Risiko sei nicht nur die tägliche, sondern auch die im Lauf der Zeit eingenommene Gesamtmenge an Glukokortikoi den ausschlaggebend.
Anwendungshinweise für JAK-Inhibitoren
Aktualisiert hat die EULAR auch die Empfehlungen zu den so ge nannten Januskinase-Inhibitoren (JAK-Hemmer). Diese noch jun ge Gruppe von synthetisch herge stellten DMARDs (tsDMARDs) blockiert die für Entzündungspro zesse wichtigen Enzyme der Fa milie der Januskinasen. „Zu diesen Wirkstoffen sind in den vergange nen Monaten teils widersprüchli che Studienergebnisse publiziert worden“, sagt Krause. Vor allem die von der US-amerikanischen Zulassungsbehörde FDA in Auf trag gegebene ORAL Surveillance Studie habe Fragen zur Sicherheit des JAK-Hemmers Tofacitinib bei Patienten mit einem erhöhten kar diovaskulären Risiko aufgeworfen. An der Studie hatten über 4300 Pa tienten, die an einer RA erkrankt waren und das 50. Lebensjahr überschritten hatten, teilgenom men. Bei den Studienteilnehmern, die Tofacitinib erhielten, traten geringfügig häufiger schwerwie gende kardiovaskuläre Ereignis se auf – etwa Herzinfarkte oder Schlaganfälle – als in einer Ver gleichsgruppe von Patienten, die mit einem TNF-Blocker behandelt wurde. Auch Lungenkrebserkran kungen waren unter Tofacitinib etwas häufiger. „Diese Effekte se hen wir in Registerdaten allerdings nicht“, betont Krause und verweist auf eine kürzlich publizierte Aus wertung des deutschen RABBITRegisters. Hier war die Einnahme von JAK-Hemmern nicht mit ei nem höheren Herz-Kreislauf-Risi ko verbunden. Auch die EULAR-Empfehlung rät von JAK-Inhibitoren daher nicht generell ab. In ihrem an gestammten Einsatzgebiet – als Zweitlinientherapie, wenn kon
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 127WISSENSWERTES
Quelle: Aletaha D et al. EULAR 2022; POS0676
Filotinib: effektiv und verträglich auch bei älteren RA-Patienten
Originaltitel: Efficacy and safety of Filgotinib in patients aged ≥ 75 years: a post hoc subgroup analysis of the FINCH 4 long-term extension (LTE) study
ventionelle DMARDs nicht aus reichend ansprechen – können sie weiterhin gegeben werden, so die EULAR. In der Abwägung gegen über dem Einsatz von Biologika müssen dabei die einschlägigen individuellen Risikofaktoren (u.a. die kardiovaskulären und infek tiologischen Risikofaktoren) und Begleiterkrankungen besonders berücksichtigt werden. „Patienten sprechen unterschiedlich auf medi kamentöse Therapien an, und auch die Nebenwirkungen unterschei den sich“, betont Krause. Daraus ergäben sich persönliche Präferen zen, die bei der Wahl der Behand lung berücksichtigt werden sollten – denn nicht zuletzt sei auch die Therapietreue entscheidend dafür, wie gut die Erkrankung und mög liche Komplikationen beherrscht werden könnten. Es sei daher sehr zu begrüßen, dass auch die neuen EULAR-Empfehlungen die The rapiefreiheit im Wesentlichen auf rechterhielten.
Der Januskinase-Inhibitor Fil gotinib (Jyseleca®, 200-mg- und 100-mg-Filmtabletten) ist zugelas sen zur Behandlung von erwachse nen Patienten mit mittelschwerer bis schwerer aktiver rheumatoider Arthritis (RA), die auf ein oder mehrere krankheitsmodifizierende Antirheumatika (DMARDs) unzu reichend angesprochen oder diese nicht vertragen haben. Eine aktuelle Post-hoc-Analyse der Studie FINCH 4 (eine Verlän gerungsstudie der zulassungsrele vanten Phase-III-Studien FINCH 1 bis 3) verglich Patienten, die zu Be ginn der Eingangsstudie ≥75 Jahre alt waren (n = 102) mit Patienten im Alter <75 Jahre* (n = 2.627).
Effektive und verträgliche Behandlungsoption auch für ältere RA-Patienten
Bis Woche 48 der Verlängerungs studie war Filgotinib in beiden Alterskohorten effektiv. Die Ver träglichkeit von Filgotinib war gut, bei Patienten ≥75 Jahre traten ge ringfügig mehr unerwünschte Er eignisse auf als bei Jüngeren. Als mögliche Gründe hierfür nennen die Autoren vermehrte Komorbidi täten bei Patienten ≥75 Jahre sowie die bei Älteren häufig reduzierte Nierenfunktion, die mit einer hö heren Wirkstoffexposition einher gehen kann.
S. M.
DGRh
* Bei RA-Patienten ≥75 Jahre wird eine An fangsdosis von 100 mg 1 × täglich emp fohlen, da die klinische Erfahrung begrenzt ist.
Jyseleca ® (Filgotinib 200-mg- und 100-mg-Filmtabletten) ist zugelassen zur Behandlung von erwachsenen Patienten mit mittelschwerer bis schwerer aktiver rheumatoider Arthritis (RA), die auf ein oder mehrere krankheitsmodifizierende Antirheumatika (DMARDs) unzureichend angesprochen oder diese nicht vertragen haben.1 Eine beim EULAR 2022 vorgestellte Posthoc-Analyse der Studie FINCH 4 – einer Verlängerungsstudie der zulassungsrelevanten Phase3-Studien FINCH 1 bis 3 – verglich Patienten, die zu Beginn der Eingangsstudie ≥ 75 Jahre alt waren (n=102) mit Patienten im Alter < 75 Jahre (n=2.627). a Bis Woche 48 der Verlängerungsstudie war Filgotinib (Jyseleca ®) in beiden Alterskohorten effektiv. Die Verträglichkeit von Filgotinib war gut, bei Patienten ≥ 75 Jahre traten geringfügig mehr unerwünschte Ereignisse auf als bei Jüngeren. Als mögliche Gründe hierfür nennen die Autoren vermehrte Komorbiditäten bei Patienten ≥ 75 Jahre sowie die bei Älteren häufig reduzierte Nierenfunktion, die mit einer höheren Wirkstoffexposition einhergehen kann. 2
Quelle: Aletaha D et al. EULAR 2022; POS0676 (Abstract) Abbildung
Filgotinib 200 mg LTE Baseline n=1.480 n=50 < 75 Jahre ≥ 75 Jahre Filgotinib 100 mg LTE Baseline n=1.147 n=52 Filgotinib 200 mg Woche 48 n=1.480 n=50 Filgotinib 100 mg Woche 48 n=1.147 n=52
Abb.: Anteil an Patienten, die in der Langzeitverlängerungsstudie FINCH 4 zu Beginn und nach 48 Wochen die Wirksamkeitskriterien erreichten (Non Responder Imputation). Zu den Schlüsselparametern gehörten die Ansprechraten gemäß des American College of Rheumatology (ACR 20/50), der CDAI (Clinical Disease Activity Index) und der DAS28 (Disease Activity Score 28) (mod. nach [2]).
a Bei RA-Patienten ab einem Alter von ≥ 75 Jahren
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 128 WISSENSWERTES
Anteil der Patienten, die
der Langzeitverlängerungsstudie
4 zu
und nach
Zu
100 90 80 70 60 50 40 30 20 10 0 Responder (%) < 75 Jahre ≥ 75 Jahre ≥ 75
≥ 75
≥ 75
< 75 Jahre < 75
<
ACR20
1:
in
FINCH
Beginn
48 Wochen die Wirksamkeitskriterien erreichten.
den Schlüsselparametern gehörten die Ansprechraten gemäß des American College of Rheumatology (ACR 20/50), der CDAI (Clinical Disease Activity Index) und der DAS28 (Disease Activity Score 28), LTE: Long Term Extension.
Jahre
Jahre
Jahre
Jahre
75 Jahre
ACR50 CDAI ≤ 10 DAS28 < 2,6
LTE: Long Term Extension (Langzeitfortsetzungsstudie)
DE-RA-JY-202207-00006 | Erstellt August 2022
Herausgeber: Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat: Prof. Dr. M. Alexander, Infektiologie, Berlin Prof. Dr. L. Beck, Gynäkologie, Düsseldorf Prof. Dr. Berndt, Innere Medizin, Berlin Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main Prof. Dr. K. M. Einhäupl, Neurologie, Berlin Prof. Dr. E. Erdmann, Kardiologie, Köln Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK Prof. Dr. K. Falke, Anästhesiologie, Berlin Prof. Dr. K. Federlin, Innere Medizin, Gießen Prof. Dr. E. Gerlach, Physiologie, München Prof. Dr. H. Helge, Kinderheilkunde, Berlin Prof. Dr. R. Herrmann, Onkologie, Basel Prof. Dr. W. Jonat, Gynäkologie, Hamburg Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
KRIEGEN SETZEN WIR HOFFNUNG ENTGEGEN
Mit Ihrer Spende rettet ÄRZTE OHNE GRENZEN Leben: Mit 52 Euro können wir zum Beispiel 40 Menschen auf der Flucht drei Monate lang mit den wichtigsten Medikamenten versorgen. Private Spender*innen ermöglichen unsere weltweite Hilfe –jede Spende macht uns stark
Spendenkonto: Bank für Sozialwirtschaft IBAN: DE72 3702 0500 0009 7097 00 BIC: BFSWDE33XXX www.aerzte-ohne-grenzen.de/spenden
Titelbild: Arthrex ACP®-Doppelspritze für die Eigenblutbehandlung (© Arthrex GmbH)
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt Prof Dr. J. Mann, Nephrologie, München Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim Prof. Dr. R. Nagel, Urologie, Berlin Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf Prof. Dr. P. Ostendorf, Hämatologie, Hamburg Prof. Dr. Th. Philipp, Innere Medizin, Essen Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf Prof. Dr. H. Rieger, Angiologie, Aachen Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen Prof. Dr. E. Rüther, Psychiatrie, Göttingen Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München Prof. Dr. E. Standl, Innere Medizin, München Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsfor schung gGmbH, Ossecker Str. 172, 95030 Hof E-Mail: info@d-i-g.org E-Mail persönlich: k.l.resch@d-i-g.org

Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrach ten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wis senschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, ins besondere das Recht der Vervielfältigung jeg licher Art, sowie die Übersetzung vor. Nach druck, auch auszugsweise, nur mit Genehmi gung des Verlages.
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfül lung von Aufträgen und Leistungen von Schadensersatz.
Satz: HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM GmbH In der Büg 8 91330 Eggolsheim
VERL AG PERFUSION
Verlag PERFUSION GmbH Storchenweg 20 90617 Puschendorf Telefon: 09101/990 11 10 Fax: 09101/990 11 19 www.Verlag-Perfusion.de E-Mail: perfusion@t-online.de
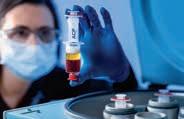
JOURNAL PHARMAKOL. U. THER. 4/2022 · 31. JAHRGANG © VERLAG PERFUSION GMBH 129
Journal
REPUBLIK MOLDAU: Raisa Pavlova flieht vor den Kämpfen in der Ukraine, unsere Mitarbeiterin Svetlana Bujac bietet ihr Hilfe an. © Peter Bräunig

MUSKELHYPOTONIE BEI KINDERN? KÖNNTE ES EIN AADC-MANGEL SEIN? Die häufigsten Symptome sind:1-3 Muskelhypotonie Entwicklungsverzögerungen Bewegungsstörungen Okulogyre Krise Nasale Kongestion Erfahren Sie mehr auf www.aadc-mangel.de und www.aadc-testen.de Referenzen: 1. Manegold C, Hoffmann GF, Degen I, et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis. 2009;32(3):371-380. 2. Wassenberg T, Molero-Luis M, Jeltsch K, et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis. 2017;12(1):12. doi: 10.1186/s13023-016-0522-z. 3. Brun L, Ngu LH, Keng WT, et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology. 2010;75(1):64-71. ©2022 PTC Therapeutics. PTC2105KK504

